Область техники, к которой относится изобретение
Настоящее изобретение относится к фармацевтической композиции для лечения дефицита гормона роста, содержащей чГР-hyFc (GX-H9), который представляет собой рекомбинантный гормон роста человека, полученный путем слияния гибридного Fc с человеческим гормоном роста чГР. Более конкретно, настоящее изобретение относится к соответствующему способу применения рекомбинантного чГР, который эффективен для лечения дефицита гормона роста, и к фармацевтической композиции для лечения дефицита гормона роста, включающей рекомбинантный чГР GX-H9 и фармацевтически приемлемый носитель, где рекомбинантный чГР GX-H9 вводят один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента, или два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента. Кроме того, настоящее изобретение относится к способу лечения дефицита гормона роста, включающему введение рекомбинантного чГР GX-H9 пациенту с дефицитом гормона роста один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента или дважды в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента.
Уровень техники
Гормон роста представляет собой гормон, который секретируется из передней доли гипофиза как одномолекулярный полипептид, состоящий из 191 аминокислоты. Инсулиноподобный фактор роста-1 (ИФР-1) экспрессируется в сочетании с рецептором гормона роста, который участвует в росте и регенерации клеток. Известно, что гормон роста вырабатывается в гипофизе нормального человеческого организма, и его продукция постепенно увеличивается до подросткового возраста и постепенно уменьшается с возрастом.
Наиболее распространенным дефицитом гормона роста является дефицит гормона роста взрослых (AGHD) и дефицит гормона роста у детей (PGHD). Дефицит гормона роста у взрослых возникает, когда гипофиз пациента повреждается под действием радиации или хирургического вмешательства при лечении опухолей головного мозга и кровоизлиянии в мозг, или по неустановленным причинам. Если секреция гормона роста не осуществляется должным образом, то развиваются симптомы, включающие потерю массы тела, снижение минеральной плотности кости, увеличение жира, снижение уровня ЛПВП, увеличение ЛПНП, снижение мышечной силы и тому подобное и, таким образом, ухудшается качество жизни. У взрослых пациентов с дефицитом гормона роста концентрация ИФР-1 в сыворотке составляет по коэффициенту стандартного отклонения (SDS) -2 или менее (<-2 SDS) или в пределах 2,5 перцентиля (<2,5 перцентиля) по сравнению с нормальными людьми в той же возрастной группе. Значение ответа гормона роста в крови можно измерить с помощью тестов на стимуляцию, таких как тест на толерантность к инсулину (ITT), тест на аргининовую нагрузку (GHRH + ARG), тест на глюкагон, тест на L-DOPA и клонидиновый тест. Когда пик GH гормона роста составляет 11,0 мкг/л или менее у пациентов с индексом массы тела (ИМТ) менее 25 кг/м2, 8,0 мкг/л или менее у пациентов с ИМТ от 25 до 30 кг/м2 или 4,0 мкг/л или менее у пациентов с ИМТ более 30 кг/м2, пик GH считается дефицитным (Guidelines for Use of Growth Hormone in Clinical Practice, Endocr. Pract. 2009; 15 (Suppl 2)).
Дефицит гормона роста у детей возникает при повреждении гипофиза или нарушениях развития. Расстройство секреции гормона роста проявляется как низкорослость и увеличение роста менее чем на 3% или 5 см или менее в год по сравнению с кривой роста того же возраста, и проявление таких симптомов, как гипогликемия, ухудшение выносливости, депрессия и психическая незрелость. В случае, когда рост ниже среднего на 3 SD или более для того же возраста; в случае, когда рост ниже среднего роста родителей на 1,5 SD или более; в случае, когда рост ниже среднего роста на 2 SD или более, и ниже, чем рост в том же возрасте, на 1 SD или более в течение 1 года или более; в случае, когда рост ниже, чем рост в том же возрасте, на 0,5 SD или более в течение 2 лет и более; или в случае, когда хотя симптомы низкого роста не проявляются, рост поддерживается на уровне менее 2 SD в течение 1 года или более, или 1,5 SD в течение 2 лет или более, этот симптом может быть определен как детский дефицит гормона роста (Consensus guideline for the diagnosis and treatment of GH deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society, J. Clin. Endocrinol. Metab., 2000 Nov; 85(11): 3990-3).
В случае дефицита гормона роста у взрослых дозировку препарата выбирали исходя из массы тела пациента в соответствующей области, но в последнее время пациентов лечили с индивидуальной дозировкой. То есть, лечение начинают с дозы ниже ожидаемой терапевтической оптимальной дозы, и корректируют дозу методом увеличения или уменьшения дозировки от 0,1 до 0,2 мг/сутки в зависимости от клинического ответа, побочных эффектов (глюкозы натощак) или уровня ИФР-1. Пол, эстрогеновый статус и возраст пациента необходимо учитывать при выборе дозировки для лечения гормоном роста. Цель лечения у пациентов с дефицитом гормона роста взрослых заключается в улучшении нормализации обмена веществ и качества жизни. С этой целью уровень ИФР-1 в крови необходимо оптимизировать от середины (50-го перцентиля или 0 SDS) до 1 SDS нормального диапазона (от -2 SDS до 2 SDS) дозы в зависимости от возраста и пола.
В случае детского дефицита гормона роста рекомендуется начинать лечение как можно скорее после постановки диагноза пациенту. Как правило, используют способ подкожного введения гормона роста каждую ночь, и рекомендуемая доза составляет от 25 до 50 мкг/кг массы тела в сутки. Как правило, скорость роста периодически проверяют в течение 3 месяцев или 6 месяцев, и рекомендуется анализировать нежелательные явления для проверки увеличения роста, изменения скорости роста, индивидуального соблюдения режима лечения пациентом и безопасности, а также анализировать уровни ИФР-1 или ИФРСБ-3 в сыворотке крови. Цель лечения у пациентов с дефицитом гормона роста детского возраста - достичь нормального увеличения роста, и гормон роста необходимо вводить таким образом, чтобы уровни ИФР-1 в крови были близки к среднему уровню того же возраста (50-й перцентиль или 0 SDS).
Когда лечение гормоном роста впервые начали в 1950-х годах, гормон роста извлекали из гипофиза трупа, и предложение было очень ограниченным, а стоимость очень высокой, поскольку количество гормона роста, экстрагированного у одного человека, очень мало. С развитием технологий рекомбинантных генов был получен гормон роста, синтезированный в Escherichia coli (Соматропин, 1981, Genentech Corporation в США). Препараты рекомбинантного гормона роста, которые в настоящее время продаются в США, включают генотропин от Pfizer, хуматроп от Eli Lilly, нутропин от Genentech, нордитропин от Novo Nordisk и тому подобное.
Однако препараты рекомбинантного гормона роста являются препаратами для ежедневного применения, требующими введения 6 раз или 7 раз в неделю. При дефиците гормона роста у взрослых хуматроп используют в дозе 0,2 мг/сутки (диапазон от 0,15 до 0,30 мг/сутки). Когда применение нутропина не основано на массе тела и устанавливается как дозировка, исходная доза составляет 0,2 мг/сутки (диапазон от 0,15 до 0,3 мг/сутки) и может быть изменена до 0,1-0,2 мг/сутки в течение цикла от 1 до 2 месяцев. Когда дозировку нутропина устанавливают на основе массы тела, исходную дозу используют так, чтобы не превышать 0,005 мг/кг/сутки или более. Если необходимо увеличить дозировку, дозу повышают так, чтобы она не превышала 0,01 мг/кг/сутки после 4 недель применения. Когда применением нордитропина не основано на массе тела и устанавливается как дозировка, исходная доза составляет 0,2 мг/сутки (диапазон от 0,15 до 0,3 мг/сутки) и может быть изменена до 0,1 и 0,2 мг/сутки в течение цикла от 1 до 2 месяцев. Когда дозировку нордитропина устанавливают на основе массы тела, исходную дозу используют так, чтобы она не превышала 0,004 мг/кг/сутки или более. Если необходимо увеличить дозировку, то её увеличивают, чтобы не превышать 0,016 мг/кг/сутки или более спустя 6 недель применения. В случае дефицита гормона роста у детей генотропин используют в дозе от 0,16 до 0,24 мг/кг/неделю, а хуматроп используют в дозе 0,026-0,043 мг/кг/сутки. Нутропин используют с дозой 0,3 мг/кг/неделю, а нордитропин используют с дозой 0,024-0,034 мг/кг/сутки.
В настоящее время препараты гормона роста являются однодневными препаратами, и в частности, в случае педиатрических пациентов неудобно вводить препарат каждый день в течение длительного периода лечения от 3 до 4 лет; и известно, что психический стресс, вызванный инъекцией, снижает качество жизни пациента. Кроме того, соблюдение режима лечения пациентом становится самым большим фактором, препятствующим лечению. Далее, также известно, что количество неудач применения значительно возрастает по мере увеличения продолжительности лечения (Endocrine practice, 2008 Mar; 14 (2): 143-54). Приблизительно 2/3 пациентов по умолчанию проявляют низкое соблюдение режима лечения, и фактически, как известно, это ведет к снижению скорости прибавки в росте (PloS one, 2011 Jan; 6 (1): e16223).
Из-за этих проблем были предприняты попытки разработать гормон роста длительного действия с использованием различных методик, но до сегодняшнего времени разработка прошла успешно, и в качестве продукта, имеющегося в продаже, состав длительного действия является уникальным. Депо нутропина было разработано Genentech в США как состав для ежемесячного применения, но из-за сложности производства он был отозван с рынка. Eutropin Plus/Declaging от LG Life Sciences был разработан как состав для еженедельного применения с использованием гиалуроновой кислоты (ГК), но из-за увеличения размера иглы по сравнению с иглой для препарата в первом поколении, он имеет неудобства.
Таким образом, необходимо разработать безопасный и эффективный гормон роста длительного действия, удобный для пациента, позволяющий преодолеть снижение соблюдении режима лечения. GX-H9 (чГР-гибридный Fc) является препаратом гормона роста длительного действия, находящимся в клинической разработке. В патенте США № 7,867,491 гибрид Fc типа, способный преодолевать проблемы комплемент-зависимой цитотоксичности и антитело-зависимой цитотоксичности, которые являются проблемами существующей технологии Fc-гибридизации, был получен путем комбинации иммуноглобулина IgD и иммуноглобулина IgG4. Впоследствии в патенте США № 8,529,899 рекомбинантный чГР (чГР-hyFc, GX-H9), который является материалом, способным заменить существующую композицию гормона роста для ежедневного применения, получали путем слияния гибридного Fc с человеческим гормоном роста чГР. Однако фактический период полужизни в организме и терапевтическая доза Fc-слитого белка сильно изменяются в зависимости от того, какой фармакологически активный ингредиент связан с Fc. Эффективные и безопасные дозы и частота их применения для лечения дефицита гормона роста с использованием GX-H9, в котором человеческий гормон роста чГР слит с hyFc, еще не установлены.
Таким образом, в настоящем изобретении были проведены клинические испытания с участием 32 здоровых взрослых индивидуумов (2013-002771-18) и 45 взрослых пациентов с дефицитом гормона роста (2014-002698-13), (EudraCT), чтобы разработать дозировку и частоту дозы, демонстрирующую оптимальный эффект рекомбинантного гормона роста, GX-H9. В результате настоящее изобретение было завершено подтверждением дозировки, частоты применения дозы и безопасности GX-H9, поддерживающих значение SDS ИФР-1 в нормальном диапазоне с минимальным количеством побочных эффектов, вызванных ростом.
Техническая проблема, решаемая изобретением
Целью настоящего изобретения является обеспечение способа лечения дефицита гормона роста, включающего введение рекомбинантного чГР, GX-H9, пациенту с дефицитом гормона роста один раз с интервалом в течение по меньшей мере недели и с дозой по меньшей мере 0,1 мг на килограмм массы тела пациента.
Целью настоящего изобретения является обеспечение способа лечения дефицита гормона роста с использованием рекомбинантного чГР GX-H9 путем определения дозы и частоты применения дозы рекомбинантного чГР GX-H9, эффективных для лечения дефицита гормона роста.
Один из аспектов настоящего изобретения представляет собой фармацевтическую композицию для лечения дефицита гормона роста, включающую рекомбинантный гормон роста человека, GX-H9 и фармацевтически приемлемый носитель, где рекомбинантный чГР вводят один раз с интервалом в течение по меньшей мере недели и в дозе по меньшей мере 0,1 мг на кг массы тела пациента.
Техническое решение проблемы
Один из аспектов настоящего изобретения обеспечивает фармацевтическую композицию для лечения дефицита гормона роста, включающую рекомбинантный гормон роста человека, GX-H9 и фармацевтически приемлемый носитель, где рекомбинантный чГР вводят один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента.
Другой аспект настоящего изобретения обеспечивает фармацевтическую композицию для лечения дефицита гормона роста, включающую рекомбинантный чГР GX-H9 и фармацевтически приемлемый носитель, где рекомбинантный чГР вводят дважды в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента.
Еще один аспект настоящего изобретения обеспечивает способ лечения дефицита гормона роста, включающий введение рекомбинантного человеческого гормона роста GX-H9 пациенту с дефицитом гормона роста один раз в неделю с дозой от 0,1 до 0,3 мг на кг массы тела пациента.
Еще один аспект настоящего изобретения обеспечивает способ лечения дефицита гормона роста, включающий введение рекомбинантного гормона роста человека GX-H9 пациенту с дефицитом гормона роста два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента.
В соответствии с настоящим изобретением, когда рекомбинантный человеческий гормон роста GX-H9 вводят пациенту с дефицитом гормона роста один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента или два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента, уровень гормона роста в организме можно поддерживать в течение более длительного периода, а в отношении уровня чГР значение SDS ИФР-1 можно поддерживать в нормальном диапазоне в течение более длительного времени. Таким образом, обеспечивается возможность лечить дефицит гормона роста путем введения гормона роста один раз в неделю или два раза в месяц без необходимости ежедневного введения.
Краткое описание фигур
Вышеупомянутые и другие аспекты, признаки и другие преимущества настоящего изобретения будут более понятны из следующего подробного описания, взятого в сочетании с прилагаемыми чертежами, на которых:
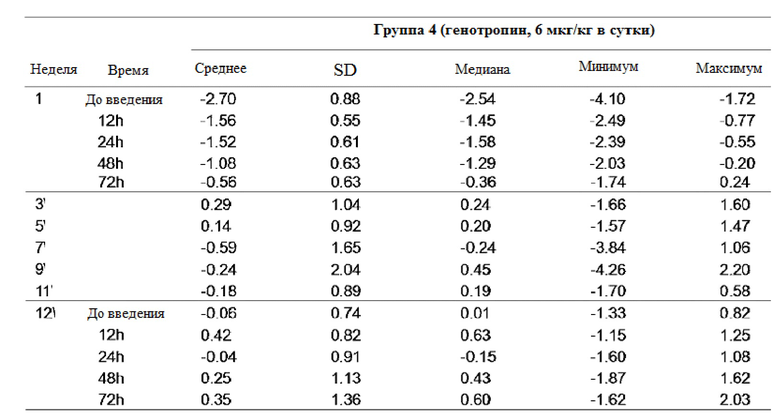
фиг. 1 иллюстрирует результат определения аффинности связывания Fcγ-рецептора FcγR I для рекомбинантного чГР (GX-H9);
фиг. 2 иллюстрирует результат аффинности связывания C1q для рекомбинантного чГР (GX-H9);
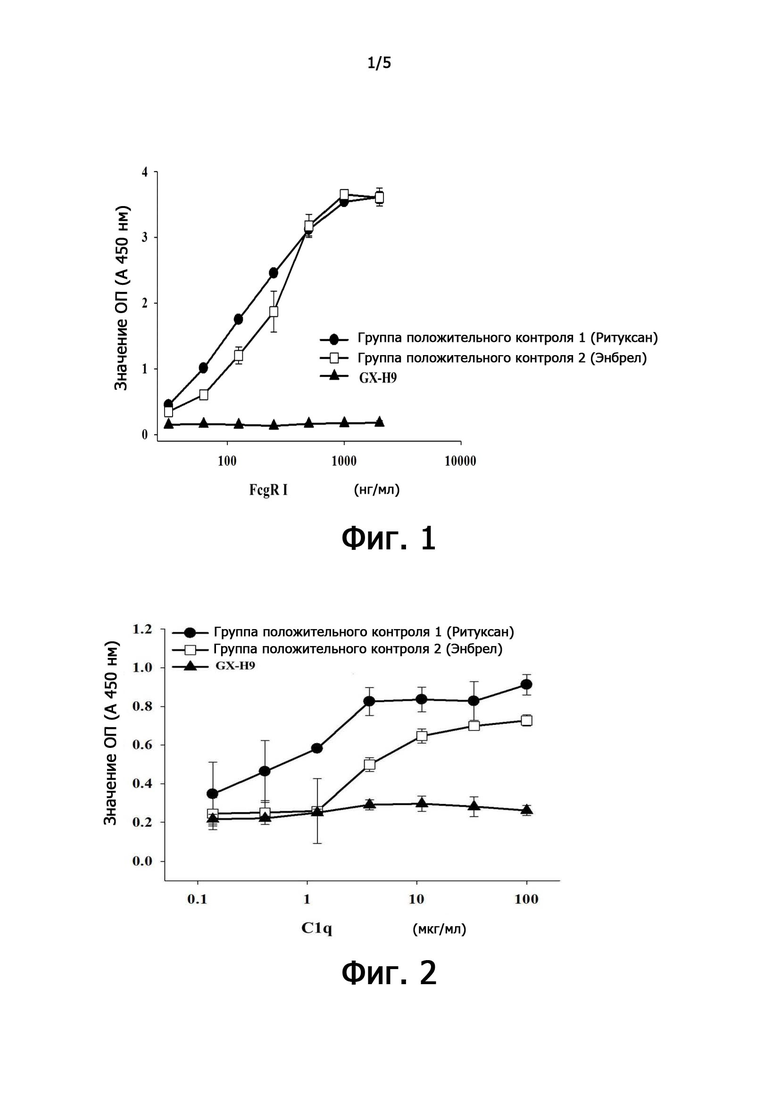
фиг. 3 - демонстрирует результат увеличения массы тела у крыс с гипофизэктомией;
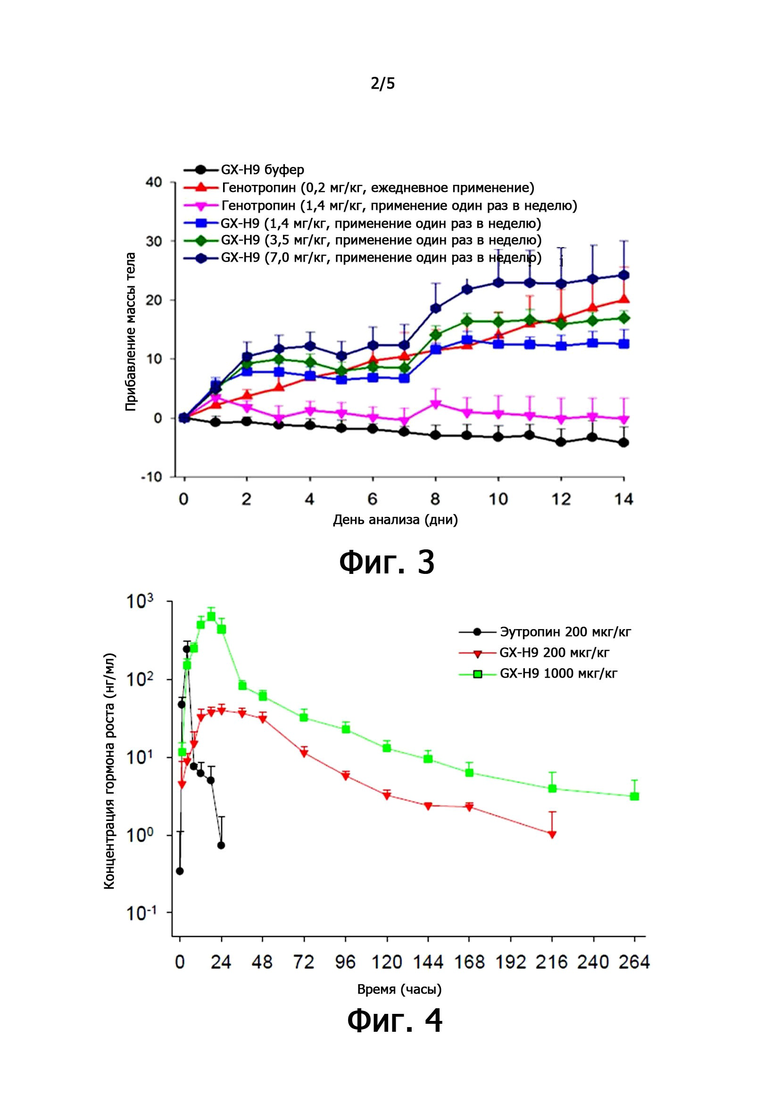
фиг. 4 иллюстрирует характеристики фармакодинамики при единичном подкожном введении рекомбинантного чГР (GX-H9) у крыс;
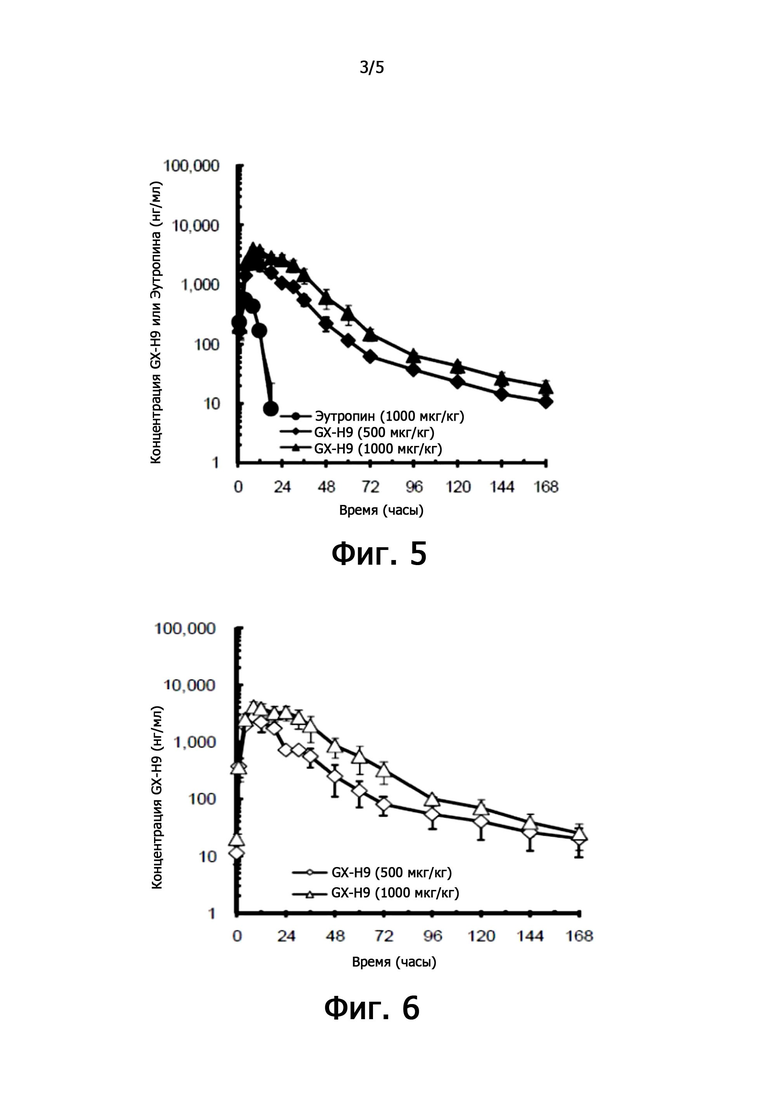
фиг. 5 иллюстрирует характеристики фармакодинамики при единичном подкожном введении рекомбинантного чГР (GX-H9) у макак;
фиг. 6 иллюстрирует характер фармакодинамики при повторном подкожном введении рекомбинантного чГР (GX-H9) у макак;
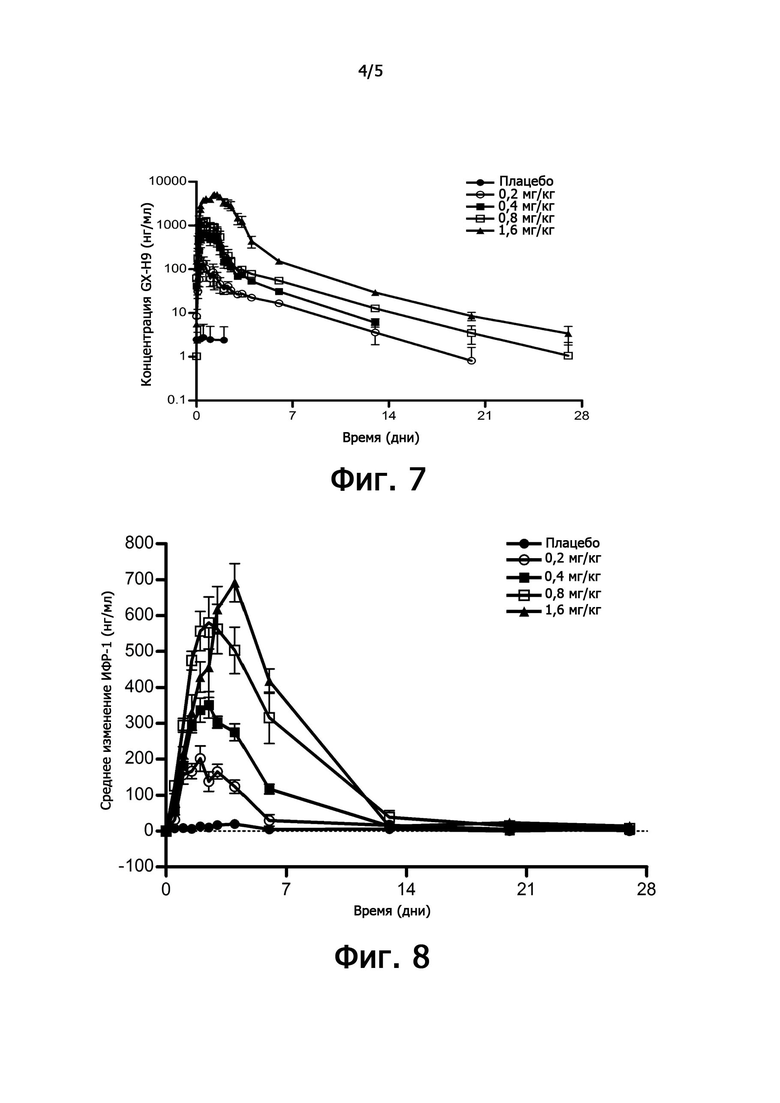
фиг. 7 иллюстрирует характеристики фармакодинамики рекомбинантного чГР (GX-H9) в клиническом испытании 1 фазы;
фиг. 8 иллюстрирует характеристики фармакокинетики (SDS ИФР-1) рекомбинантного чГР (GX-H9) в клиническом испытании 1 фазы;
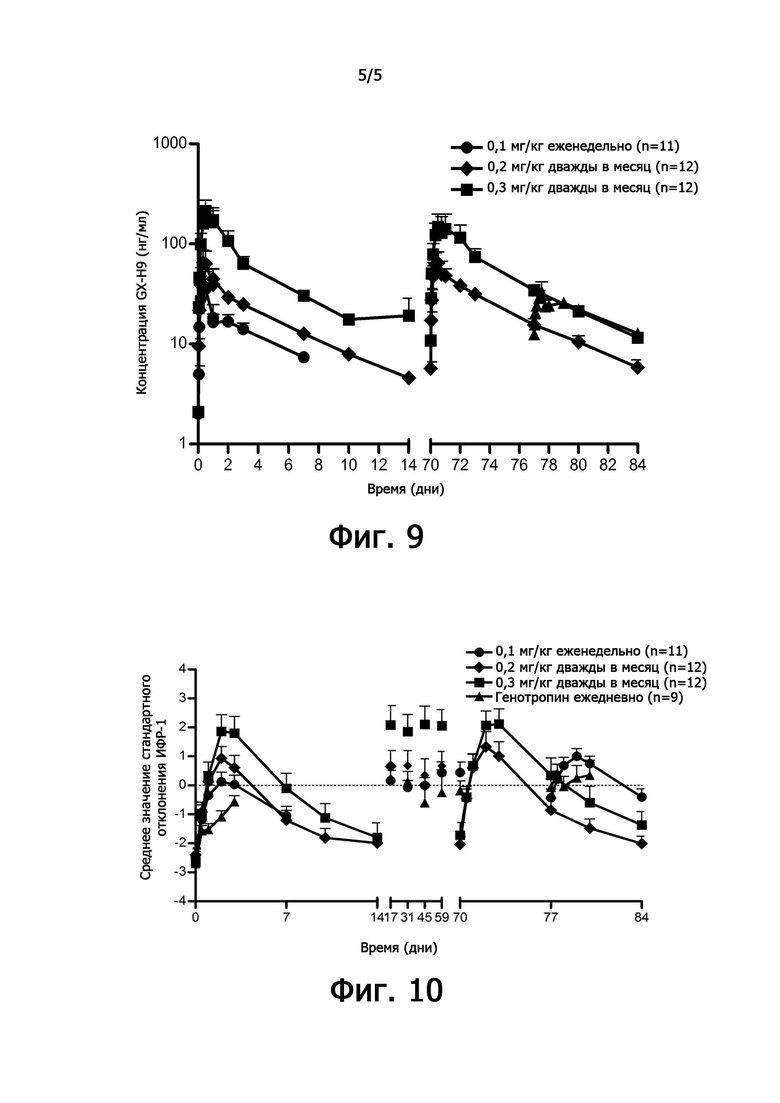
фиг. 9 иллюстрирует характеристики фармакодинамики при повторном введении рекомбинантного чГР (GX-H9) в клиническом испытании 2 фазы; и
фиг. 10 иллюстрирует характеристики фармакокинетики при повторном введении рекомбинантного чГР (GX-H9) в клиническом испытании 2 фазы.
Осуществление изобретения
Эффективная доза и частота применения дозы рекомбинантного человеческого гормона роста GX-H9, которые способствуют фактическому росту у человека, пока не установлены.
Авторы изобретения провели клинические испытания с целью разработки дозировки и частоты применения дозы, способной проявлять оптимальный эффект GX-H9, путем использования у 32 здоровых взрослых индивидуумов (2013-002771-18) и 45 взрослых пациентов с дефицитом гормона роста (2014-002698-13, EudraCT). В результате было отмечено, что в случае введения рекомбинантного чГР, GX-H9 один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента или два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента, гормон роста в организме сохраняется в течение более длительного периода времени, а значение SDS ИФР-1 можно поддерживать в нормальном диапазоне.
Один аспект настоящего изобретения относится к фармацевтической композиции для лечения дефицита гормона роста, содержащей рекомбинантный чГР (GX-H9) и фармацевтически приемлемый носитель, где рекомбинантный чГР вводят один раз с интервалом не менее недели и в дозе по меньшей мере 0,1 мг на кг массы тела пациента.
Один аспект настоящего изобретения относится к фармацевтической композиции для лечения дефицита гормона роста, содержащей рекомбинантный чГР (GX-H9) и фармацевтически приемлемый носитель, где рекомбинантный чГР вводят один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента. В частности, настоящее изобретение относится к фармацевтической композиции, где рекомбинантный чГР вводят один раз в неделю с дозировкой от 0,1 до 0,2 мг на кг массы тела пациента.
Кроме того, еще один аспект настоящего изобретения относится к фармацевтической композиции для лечения дефицита гормона роста, содержащей рекомбинантный белок чГР GX-H9 и фармацевтически приемлемый носитель, где рекомбинантный чГР вводят дважды в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента. В частности, настоящее изобретение относится к фармацевтической композиции, где рекомбинантный чГР вводят один раз в две недели в дозе от 0,15 до 0,4 мг на кг массы тела пациента.
В фармацевтической композиции из настоящего изобретения рекомбинантный чГР, GX-H9 может включать аминокислотную последовательность SEQ ID NO: 1. Фармацевтическая композиция из настоящего изобретения может быть введена подкожно.
Кроме того, еще один аспект настоящего изобретения обеспечивает способ лечения пациентов с дефицитом гормона роста, включающий введение рекомбинантного чГР GX-H9 пациенту с дефицитом гормона роста один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента.
Кроме того, еще один аспект настоящего изобретения обеспечивает способ лечения пациентов с дефицитом гормона роста, включающий введение рекомбинантного чГР GX-H9 пациенту с дефицитом гормона роста два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента.
Рекомбинантный чГР «GX-H9», используемый в настоящей заявке, упоминается как чГР-hyFc, который является человеческим гормоном роста, слитым с гибридным Fc, и может иметь аминокислотную последовательность SEQ ID NO: 1. Рекомбинантный чГР GX-H9 может быть получен способом, раскрытым в патенте США № 8,529,899.
Согласно настоящему описанию фармацевтическую композицию, содержащую рекомбинантный чГР GX-H9, можно вводить взрослым с дефицитом гормона роста.
Дефицит гормона роста у взрослых означает случай, когда коэффициент стандартного отклонения (SDS) по сравнению с нормальными людьми в той же возрастной группе у взрослых составляет -2 или менее (<-2 SDS) или в пределах 2,5 перцентиля (<2,5 перцентиля). У взрослых с заболеваниями гипофиза и гипопитуитаризмом дефицит гормона роста обычно связан с физическими расстройствами и может вызвать психические расстройства, а также нарушения состава организма и обмена веществ. Дефицит гормона роста у взрослых можно разделить на три категории. Дефицит гормона роста у взрослых можно разделить на первый, развивающийся в детском возрасте дефицит гормона роста; дефицит гормона роста, обусловленный гипоталамо-гипофизарными нарушениями, и идиопатический дефицит гормона роста.
Фармацевтическая композиция по настоящему изобретению может включать фармацевтически приемлемый носитель, который может представлять собой любой нетоксичный материал, подходящий для доставки рекомбинантного чГР пациенту. В качестве носителя могут быть включены дистиллированная вода, спирт, жиры, воски и инертные твердые вещества. Фармацевтически приемлемые адъюванты, такие как буфер, диспергатор и разбавитель, например, бактериостатическая вода для инъекций (BWFI), фосфатно-солевой буферный физиологический раствор, раствор Рингера, раствор декстрозы, сахароза, полоксамер и тому подобное, могут быть включены в фармацевтическую композицию из настоящего изобретения.
В настоящем изобретении рекомбинантный чГР, GX-H9 может быть введен один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента и, например, применяемая доза может меняться, составляя 0,1; 0,15; 0,2; 0,25 или 0,3 мг на кг массы тела, в зависимости от возраста, пола и эстрогенного статуса у пациентов. Предпочтительно, рекомбинантный чГР GX-H9 можно вводить один раз в неделю в дозе от 0,1 до 0,2 мг на кг массы тела пациента. Кроме того, рекомбинантный чГР GX-H9 можно вводить дважды в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента и, например, можно вводить один раз в две недели с дозой 0,1; 0,15; 0,2; 0,25; 0,3; 0,35 или 0,4 мг на кг массы тела пациента, в зависимости от возраста, пола и эстрогенного статуса. Предпочтительно, рекомбинантный чГР GX-H9 можно вводить дважды в месяц в дозе 0,15-0,4 мг на кг массы тела пациента. В частности, предпочтительная доза рекомбинантного чГР, GX-H9 составляет 0,1 мг/кг при введении один раз в неделю, 0,2 мг/кг при введении дважды в месяц, или 0,3 мг/кг при введении дважды в месяц. Кроме того, в некоторых случаях рекомбинантный чГР можно вводить дважды в месяц, один раз в три недели или ежемесячно с 0,3 до 0,6 мг/кг в зависимости от возраста, пола и эстрогенного статуса.
Дозировка рекомбинантного чГР может быть скорректирована в зависимости от возраста, пола и эстрогенного статуса пациента, и может быть увеличена или уменьшена при мониторинге прогресса применения. Доза рекомбинантного чГР, вводимого впоследствии, может быть выше или ниже начальной дозы, или равна начальной дозе, в зависимости от уровня изменений SDS ИФР-1. На начальном этапе небольшое количество рекомбинантного чГР следует безопасно вводить, а затем постепенно повышать после проверки отсутствия нежелательной реакции. Кроме того, дозировку рекомбинантного чГР можно регулировать при осуществлении контроля уровня SDS ИФР-1 в образце плазмы или сыворотки пациента. Доза рекомбинантного чГР, подходящая для каждого отдельного пациента, может меняться в зависимости от возраста, пола, конституции, массы тела пациента.
Фармацевтическую композицию по настоящему изобретению, содержащую рекомбинантный чГР, GX-H9, можно вводить субъекту различными способами. Например, фармацевтическую композицию можно вводить парентерально, и например, подкожно, внутримышечно или внутривенно. Композицию можно стерилизовать в соответствии с общеизвестным способом стерилизации. Композиция может включать фармацевтически приемлемые вспомогательные вещества и адъюванты, модификаторы токсичности и их аналоги, необходимые для регуляции физиологических условий, таких как установление рН; и например, может включать ацетат натрия, хлорид натрия, хлорид калия, хлорид кальция, лактат натрия и тому подобное. В композициях концентрация рекомбинантного чГР может быть очень разной, и может быть выбрана предпочтительно на основе объема жидкости в организме, вязкости и тому подобного в соответствии с выбранным конкретным способом введения.
В дальнейшем настоящее изобретение будет описано более подробно посредством примеров. Эти примеры являются лишь иллюстрацией настоящего изобретения, и для специалистов в данной области техники очевидно, что объем настоящего изобретения не ограничивается этими примерами.
Пример 1. Приготовление рекомбинантного чГР, GX-H9.
Рекомбинантный чГР, GX-H9 может быть приготовлен в соответствии со способом, раскрытым в патенте США № 8,529,899.
Во-первых, последовательность нуклеиновых кислот чГР-hyFc, в которой hyFc слит с гормоном роста человека (чГР), кодирующая аминокислотную последовательность SEQ ID NO: 1, была вставлена в вектор экспрессии pAD15 для получения клеточной линии, экспрессирующей чГР-hyFc. Для получения вектора, включающего структурный ген чГР-hyFc, последовательность GenBank AAA98618.1 использовали для гена чГР гормона роста человека, а ген hyFc был получен путем слияния последовательностей GenBank P01880 (IgD) и GenBank AAH25985 (IgG4). Гены, полученные от производителя генов, вводили с использованием специфических рестрикционных ферментов в качестве векторов экспрессии для получения продукции клеточной линии.
Вектор экспрессии, обеспечиваемый описанным выше способом, трансфицировали в клетки CHO DG44 (Колумбийский университет, США) способом с фосфатом кальция. Через 6 часов после трансфекции трансфицированные клетки промывали фосфатным буфером, а затем среду заменяли на 10% dFBS (диализованной эмбриональной телячьей сыворотки) (Gibco, США, 30067-334), MEM альфа (Gibco, 12561, США, Кат. №. 12561- 049) и HT+ (Gibco, США, 11067-030) (гипоксантин-тимидиновой) среды. Через 48 часов после трансфекции НТ-селекцию проводили путем непрерывного разбавления трансфицированных клеток на 100 мм планшете с использованием 10% dFBS среды + MEM-альфа среды без HT. В то время как среду заменяли два раза в неделю, трансфицированные клетки оставляли до образования единственной колонии. После этого для усиления производительности с использованием DHFR-системы (с дигидрофолатредуктазой) была выполнена МТХ-амплификация по отношению к клонам из HT селекции. После амплификации метотрексатом проводили субкультивирование 4-5 раз для стабилизации клеток для оценки производительности, и оценивали удельную производительность. Был получен клон, подходящий для продукции целевого белка.
Для получения единственного клона по отношению к наиболее продуктивным клонам было выполнено клонирование методом предельных разведений (LDC). LDC инокулировали в 96-луночный планшет, разбавляя клетки культуральной средой с концентрацией 1 клетка/лунку. После 10-14 дней инокуляции получали только клетки из лунок с одиночными клонами с помощью микроскопа, и культивировали во флаконе T25, достаточном для оценки производительности, и затем обеспечивали клеточную линию с высокой продуктивностью.
После того как культуральный раствор был получен из обеспеченной клеточной линии, целевой белок очищали из культурального раствора. Образец раствора белковой культуры подвергали анализу связывания образца, используя раствор белковой культуры с применением Prosep® Ultra Plus (Merck), уравновешенный с использованием буфера из 50 мМ фосфата натрия, 150 мМ хлорида натрия и с pH 7,0. Использовали колонку XK16/20 (GE Healthcare), и белок элюировали с применением буфера из 100 мМ цитрата натрия, 200 мМ L-аргинина и с рН 3,1.
Пример 2. Анализы антитело-зависимой клеточно-опосредованной цитотоксичности (АЗКЦ) и комплемент-зависимой цитотоксичности (КЗЦ) рекомбинантного чГР GX-H9
Чтобы убедиться, что гибридный Fc домен GX-H9 не индуцирует антитело-зависимую клеточно-опосредованную цитотоксичность (АЗКЦ) и комплемент-зависимую цитотоксичность (КЗЦ), был проведен иммуноферментный анализ (ИФА).
В качестве групп положительного контроля использовали ритуксан (от Roche Corporation, Швейцария) и энбрел (от Amgen Corporation, США), которые, как известно, имели очень высокую аффинность связывания с Fcγ -рецепторами FcγR I, II и III. GX-H9, ритуксан и энбрел наносили на 96-луночный планшет, а затем проводили реакцию с серийными разведениями Fcγ-рецептора I. По истечении времени реакции планшеты промывали буферным раствором для удаления Fcγ-рецептора I, который не связался с анализируемыми веществами. После этого силу связывания между Fcγ-рецептором I и анализируемым веществом измеряли с использованием биотинилированного антитела против FcγRI и стрептавидина, конъюгированного с пероксидазой хрена.
Силу связывания между C1q и GX-H9, индуцирующим клеточно-опосредованную цитотоксичность, также измеряли с использованием вышеуказанного ИФА метода. В качестве групп положительного контроля использовали ритуксан (от Roche Corporation, Швейцария) и энбрел (от Amgen Corporation, США), а силу связывания между испытуемыми веществами измеряли с использованием конъюгированного с пероксидазой хрена анти-C1q-антитела.
В результате было подтверждено, что GX-H9 имеет низкую силу связывания с Fcγ-рецептором I, индуцирующим антитело-зависимую клеточно-опосредованную цитотоксичность, как показано на фиг. 1, и низкую силу связывания с C1q, индуцирующим комплемент-зависимую цитотоксичность, как показано на фиг. 2.
Пример 3. Результаты доклинических испытаний рекомбинантного чГР GX-H9
3,1. Анализ эффективности при повторном подкожном введении GX-H9 у крыс с гипофизэктомией.
Эффективность GX-H9 тестировали с использованием гипофизэктомированных крыс в качестве животной модели болезни. В качестве контрольной группы использовали генотропин (Pfizer Corporation, США) в качестве агента для ежедневного применения, а GX-H9 вводили один раз в неделю, а затем сравнивали эффективность.
Испытание проводили для достижения целевого увеличения массы тела на 10% или менее в течение приблизительно 1 недели после гипофизэктомии. Группа 1 была группой отрицательного контроля с подкожным введением только буфера для композиции в течение двух недель. Группе 2 подкожно вводили генотропин по 0,2 мг/кг каждый день. Группе 3 вводили один раз в неделю генотропин 1,4 мг/кг, что составляло недельную дозу. Группе 4 вводили один раз в неделю GX-H9 1,4 мг/кг (что соответствовало недельной дозе генотропина). Группе 5 вводили один раз в неделю GX-H9 3,5 мг/кг (что соответствовало 1/2 молярного количества недельной дозы генотропина). Группе 6 вводили один раз в неделю GX-H9 7,0 мг/кг (что соответствовало тому же молярному количеству, что и недельная доза генотропина. После введения препарата наблюдали симптомы и измеряли массу тела каждый день.
В результате, как показано на фиг. 3, когда генотропин вводили один раз в сутки, масса повышалась в среднем примерно на 20 г/кг при введении 0,2 мг/ г в сутки, но когда еженедельную дозу (1,4 мг/кг) вводили сразу один раз в неделю, не было увеличения массы тела. Когда GX-H9 вводили в дозе 7 мг/кг один раз в неделю (группа 6), прирост массы был больше, чем у генотропина (группа 3), где вводили одинаковое количество молей. Кроме того, когда GX-H9 вводили в дозе 3,5 мг/кг (группа 5), была показана та же эффективность, как в случае, когда генотропин вводили по 0,2 мг/кг в сутки (группа 2).
3,2. Фармакодинамический анализ после единственного подкожного введения рекомбинантного чГР GX-H9 крысам.
Чтобы проанализировать фармакокинетику GX-H9, GX-H9 вводили крысам один раз подкожно. В качестве контрольной группы эутропин (LG Life Sciences, Inc., Корея) вводили один раз подкожно, и сравнивали эффективность. Группе 1 вводили одну дозу 200 мкг/кг эутропина подкожно, а группе 2 вводили одну дозу 200 мкг/кг GX-H9 подкожно. Группе 3 вводили одну дозу 1000 мкг/кг GX-H9 подкожно.
Кровь брали до введения, и спустя 1, 4, 8, 12, 18, 24, 36, 48, 72, 96, 120, 144, 168, 216, 264 и 336 часов после подкожного введения. Концентрацию каждого вещества в крови измеряли посредством специфического биоанализа (ИФА).
Результаты испытаний были проиллюстрированы на фиг. 4, и фармакокинетика после однократного подкожного введения GX-H9 в дозе 200 или 1000 мкг/кг достигала самой высокой концентрации в крови спустя 17 или 24 часа (Tmax), и GX-H9 был обнаружен в крови до 9 дней и 11 дней. По мере увеличения дозы, системное воздействие также увеличилось.
По сравнению с группой с введением 200 мкг/кг эутропина в качестве контрольного вещества в крови, тестируемое вещество было обнаружено в течение более продолжительного времени (эутропин 24 часа против GX-H9 9 суток). В случае подкожного введения 200 мкг/кг GX-H9 разница времени достижения максимальной концентрации в крови (Tmax) составляла около 20 часов (эутропин 4 часа против GX-H9 24 часа). Из приведенных выше результатов было отмечено, что GX-H9 непрерывно воздействует на систему у крыс в течение длительного времени по сравнению с контрольным препаратом, эутропином. Кроме того, по мере увеличения дозы GX-H9 системное воздействие после подкожного введения пропорционально увеличивалось.
3.3. Фармакодинамический анализ после единственного подкожного введения рекомбинантного чГР GX-H9 у макак
Фармакокинетику GX-H9 и эутропина в качестве контрольного вещества анализировали у макак-крабоедов. У самцов макак (по 3 на группу) GX-H9 подкожно вводили повторно четыре раза в неделю с дозами 500 мкг/кг и 1000 мкг/кг, а эутропин в качестве контрольного вещества подкожно вводили один раз в дозе 1000 мкг/кг.
В группе, которой вводили GX-H9, образцы крови брали до 1-го и 4-го введения (введение 0 и 21 день) и спустя 1, 4, 8, 12, 18, 24, 30, 36, 48, 60, 72 , 96, 120, 144 и 168 часов после введения.
В группе, в которой вводили эутропин, образцы крови брали перед однократным введением и через 1, 4, 8, 12, 18, 24, 30, 36, 48, 60, 72, 96, 120, 144 и 168 часов после введения.
Концентрацию в крови измеряли с помощью специфического биоанализа (ИФА) для GX-H9 и эутропина, и результаты были проиллюстрированы на фиг. 5 и 6. Когда GX-H9 подкожно вводили в дозе 500 или 1000 мкг/кг, было обнаружено, что системное воздействие увеличивается в зависимости от увеличения дозы после однократного введения (0 дней) и после повторного введения (4 недели).
По сравнению с эутропином (1000 мкг/кг, однократное подкожное введение), GX-H9 (500 или 1000 мкг/кг) (от 12 до 18 часов после введения эутропина против 168 часов после введения GX-H9) обнаруживается в течение более длительного времени в крови. То есть, когда GX-H9 вводили подкожно, было обнаружено, что системное воздействие было более стойким, чем у контрольного лекарственного средства, эутропина. Кроме того, по мере увеличения дозы GX-H9 от 500 до 1000 мкг/кг наблюдалось, что системное воздействие после подкожного введения увеличивается пропорционально увеличению дозы.
Пример 4. Клинические испытания 1 фазы для рекомбинантного чГР GX-H9
4,1. Фармакокинетические характеристики рекомбинантного чГР GX-H9 у здоровых взрослых людей.
Рандомизированное, двойное слепое, плацебо-контролируемое клиническое испытание 1 фазы с однократной восходящей дозой проводили с привлечением здоровых добровольцев. Целью клинического испытания 1 фазы была оценка безопасности, переносимости и фармакодинамических/фармакокинетических характеристик после однократного подкожного введения GX-H9. После того, как здоровые добровольцы были произвольно распределены в исследовательскую группу или группу плацебо, оценку проводили в течение 56 дней после однократного подкожного введения GX-H9 в четырех группах с разными дозами (0,2; 0,4; 0,8 и 1,6 мг/кг).
В группе, которой вводили GX-H9, образцы крови брали перед однократным введением и спустя 0,25; 1, 2, 4, 6, 8, 12, 16, 24, 28, 32, 36, 40, 48, 54, 60, 72, 80, 96, 144, 312, 480, 648 и 1320 часов после введения.
Концентрацию в крови измеряли с помощью специфического биоанализа (ИФА) GX-H9, и результаты показаны в таблице 1 ниже и на фиг,7.
Таблица 1
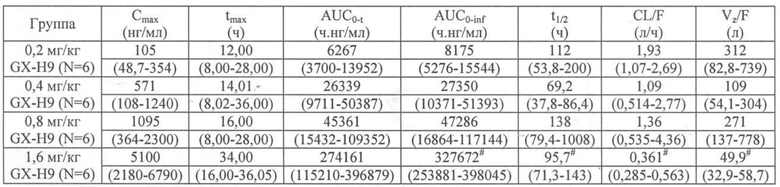
tmax оценивали в медианном диапазоне.
# n=5 (полученный параметр значения t1/2 от одного добровольца не был достоверно определен)
Пик с геометрической средней концентрацией наблюдался примерно через 12 часов (8-16 часов) после однократного подкожного введения GX-H9, а второй пик при более низкой концентрации наблюдался примерно через 32 часа (28-32 часа) после введения. При максимальной дозировке второй пик соответствовал Cmax (см. фиг. 7). Cmax и AUC возрастали вместе с дозировкой для всех доз. Период полувыведения (t1/2) составлял от 69,2 до 138 часов, и была индивидуальная вариабельность.
4.2. Фармакокинетические характеристики рекомбинантного чГР GX-H9 у здоровых взрослых людей.
В группе, которой вводили GX-H9, кровь брали перед однократным введением и спустя 12, 24, 36, 48, 60, 72, 96, 144, 312, 480, 648 и 1320 часов после введения. Величина изменения проиллюстрирована на фиг. 8 путем установки концентрации ИФР-1 в крови, измеренной до введения в качестве базовой линии.
Фиг. 8 иллюстрирует изменение концентрации (нг/мл) ИФР-1 в крови по сравнению с исходным уровнем у групп, которым вводили плацебо, и 0,2; 0,4; 0,8 и 1,6 мг/кг GX-H9. После однократного введения GX-H9 подкожно в дозе 0,2; 0,4; 0,8 и 1,6 мг/кг концентрация ИФР-1 в крови увеличивалась пропорционально дозе. Среднее максимальное увеличение (% изменения от исходного уровня) составило 81%, 157%, 301% и 349% при дозе 0,2; 0,4; 0,8 и 1,6 мг/кг, соответственно. Время достижения максимальной концентрации ИФР-1 в крови составляло от 48 до 96 часов и увеличивалось пропорционально дозе. Было подтверждено, что средняя концентрация ИФР-1 восстанавливалась до исходного уровня на 7-й день после введения в дозе 0,2 мг/кг и на 14-й день для других доз.
4.3. Результаты анализа безопасности рекомбинантного чГР GX-H9 у здоровых взрослых людей
В таблице 2 ниже приведены результаты нежелательных явлений в результате лечения, возникших после применения у субъектов в зависимости от вводимого препарата, соотношения между лекарственным средством и нежелательными явлениями, и интенсивность нежелательных явлений.
Таблица 2
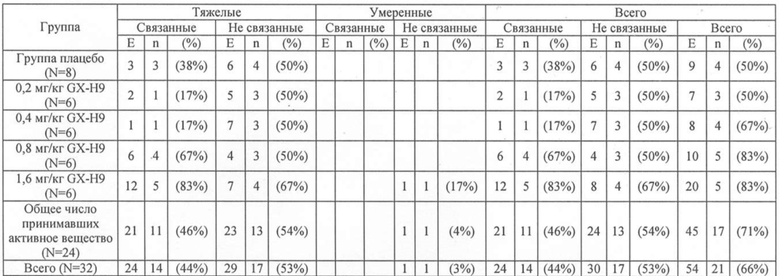
N = Число индивидуумов, принимавших лекарство
n = Число индивидуумов, у которых отмечены нежелательные явления
E = Число нежелательных явлений
(%) = Отношение индивидуумов, у которых отмечены нежелательные явления, в соответствии с лечением (n/N)*100
Серьезные нежелательные явления или слабые побочные явления не отмечались.
Как показано в таблице 2, у 21 индивидуума отмечено в целом 54 нежелательных явления. Не отмечено смерти или серьезных нежелательных явлений. Тяжелое нежелательное явление отмечено у одного испытуемого, но было установлено, что это не было нежелательным явлением, связанным с лекарственным средством. Тяжесть всех нежелательных явлений, за исключением вышеуказанного нежелательного явления, была легкой. Наиболее частыми нежелательными явлениями были расстройства опорно-двигательного аппарата и соединительной ткани (19 случаев), системные расстройства и реакции в месте инъекции (11 случаев) и неврологические расстройства (10 случаев). Отмеченными случаями проявления трех или более неблагоприятных явлений были: миалгия (7 случаев), реакция на месте введения катетера (6 случаев), головная боль (5 случаев), назофарингит (5 случаев), артралгия (4 случая) и мелагра (3 случая).
Между тем, образование антитела против GX-H9 анализировали у субъектов, которым вводили GX-H9. Противолекарственное антитело (ADA) анализировали перед введением, а на 28-й и 56-й день. В результате ни у одного пациента не выявлено антитела, образованного в ответ на GX-H9.
Пример 5. Результаты клинического испытания 2 фазы для рекомбинантного чГР GX-H9
5.1. Фармакокинетические характеристики GX-H9 у взрослых пациентов с дефицитом гормона роста.
Для оценки безопасности, переносимости, эффективности и фармакодинамических/фармакокинетических характеристик GX-H9 у взрослых пациентов с дефицитом гормона роста (таблица 3) проводили рандомизированное, контролируемое по активному препарату открытое клиническое испытание 2 фазы. Дозу повторно вводили в течение 12 недель с 0,1 мг/кг в неделю (группа 1), 0,3 мг/кг дважды в месяц (группа 2) или 0,2 мг/кг дважды в месяц (группа 3). Кроме того, в качестве активного контрольного препарата ежедневно вводили генотропин 6 мкг/кг (G: группа 4).
Таблица 3

В группе, которой вводили GX-H9 один раз в неделю, кровь брали во время первого введения (1-я неделя) и последнего введения (12-я неделя), и брали спустя 1, 2, 4, 8, 12, 18, 24, 48, 72 и 168 часов после введения.
В группе, получавшей GX-H9 два раза в месяц, кровь брали до первого введения (первая неделя) и последнего введения (11-я неделя), и брали спустя 1, 2, 4, 8, 12, 18, 24, 48, 72, 168, 240 и 336 часов после введения.
Концентрацию GX-H9 в крови измеряли в полученных образцах крови, и результаты проиллюстрированы в таблицах 4-6 и на фиг. 9.
Таблица 4
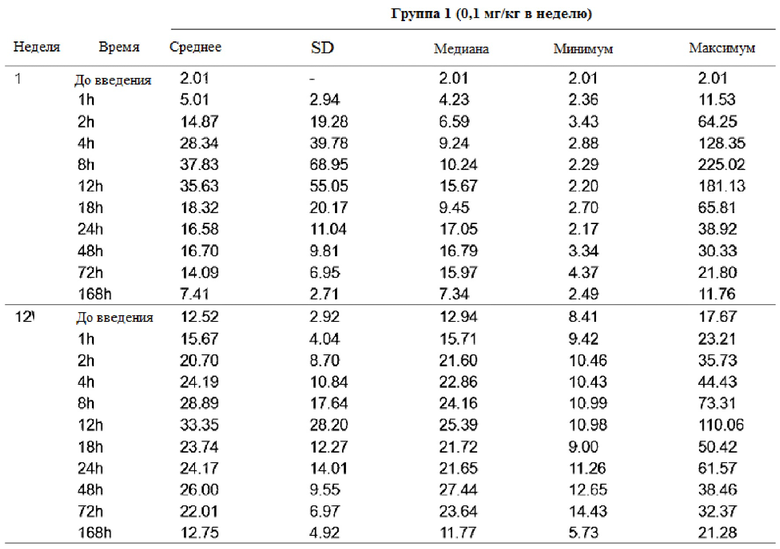
Таблица 5
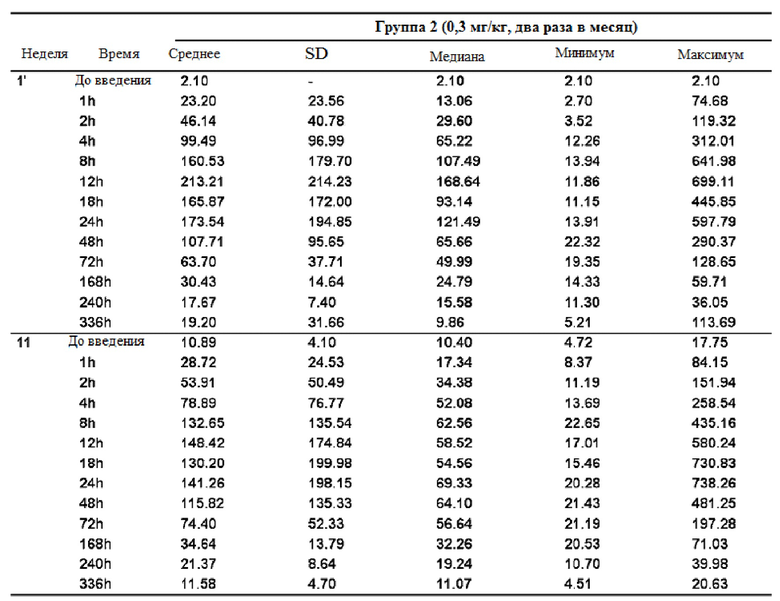
Таблица 6
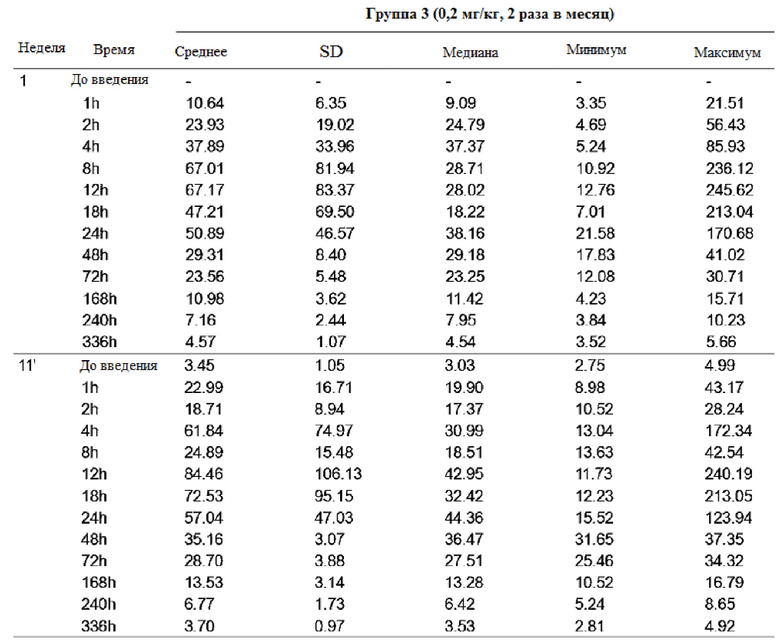
После повторного введения GX-H9 с 0,1 мг/кг в течение 12 недель изменение фармакокинетики анализировали при первом введении (1-я неделя) и последнем введении (12-я неделя), перед введением и спустя 1, 2, 4, 8, 12, 18, 24, 48, 72 и 168 часов после введения. Было подтверждено, что среднее значение (стандартное отклонение) концентрации GX-H9 от введения (ниже предела количественного определения, BLQ) до 168 часов (7 дней) после введения для первого (1-я неделя) и последнего введения (12-я неделя) составили 7,41 (2,71) и 12,75 (4,92), соответственно. Кроме того, после повторного введения GX-H9 с 0,3 мг/кг и 0,2 мг/кг в течение 12 недель изменение фармакокинетики анализировали при первом введении (1-я неделя) и последнем введении (11-я неделя), до введения и спустя 1, 2, 4, 8, 12, 18, 24, 48, 72, 168, 240 и 336 часов после введения. Было установлено, что среднее значение (стандартное отклонение) концентрации GX-H9 от введения (ниже предела количественного определения, BLQ) до 336 часов (14 дней) после введения для первого (1-я неделя) и последнего введения (12-я неделя) составило 19,20 (31,66) и 11,58 (4,70) для 0,3 мг/кг/два раза в месяц и 4,57 (1,07) и 3,70 (0,97) для 0,2 мг/кг/ два раза в месяц, соответственно.
Таким образом, было показано, что когда GX-H9 вводили еженедельно или дважды в месяц в диапазоне концентраций от 0,1 до 0,3 мг/кг, концентрация GX-H9 поддерживалась на нормальном уровне без какого-либо накопления в организме.
5.2. Фармакокинетические характеристики рекомбинантного чГР GX-H9 у взрослых пациентов с дефицитом гормона роста.
В группе, в которой вводили GX-H9 один раз в неделю, кровь брали во время первого введения (1-я неделя) и последнего введения (12 недель), и брали через 12, 24, 48, 72 и 168 часов после введения. Чтобы оценить изменение коэффициента стандартного отклонения ИФР-1 (SDS ИФР-1) по сравнению с нормальными индивидуумами по отношению к времени и дозе, анализировали SDS ИФР-1 через 4 дня после введения на 3, 5, 7, 9 и 11-й неделе. В группе, которой вводили GX-H9 два раза в месяц, кровь брали во время первого применения (1-я неделя) и последнего применения (11 недель), и брали спустя 12, 24, 48, 72, 168, 240 и 336 часов после применения. Чтобы оценить изменение SDS ИФР-1 по отношению к времени и дозе, изменение SDS ИФР-1 анализировали через 4 дня после введения на 3, 5, 7 и 9-й неделе.
Результаты характеристик фармакодинамики GX-H9 показаны в таблицах 7-10 и на фиг. 10.
Таблица 7
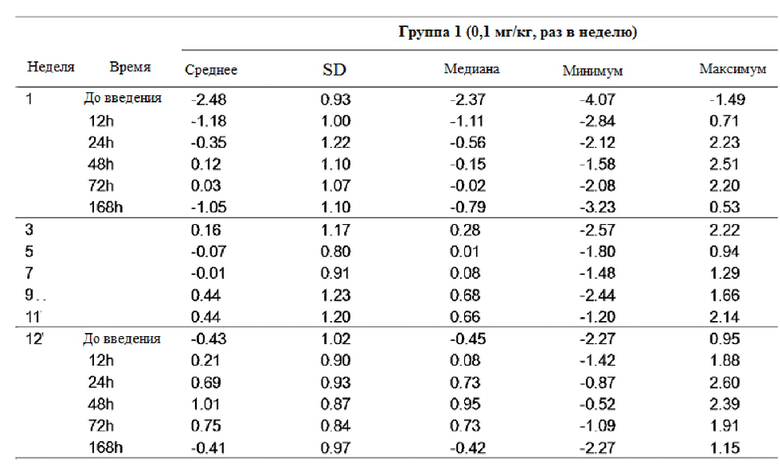
Таблица 8
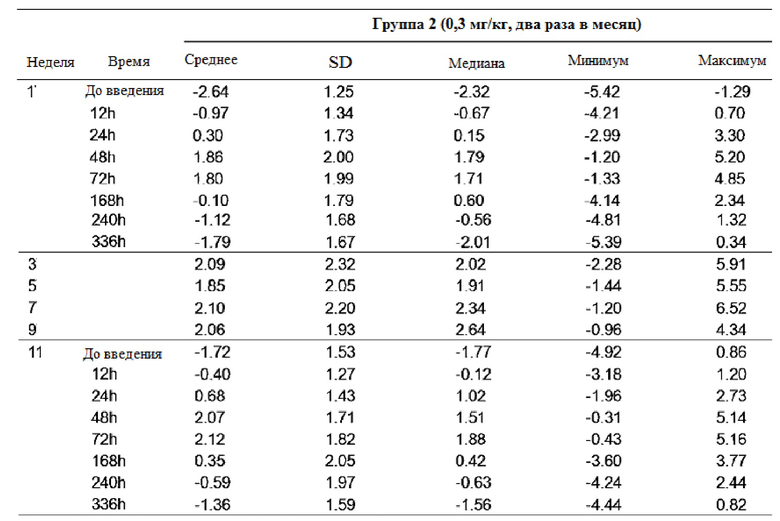
Таблица 9
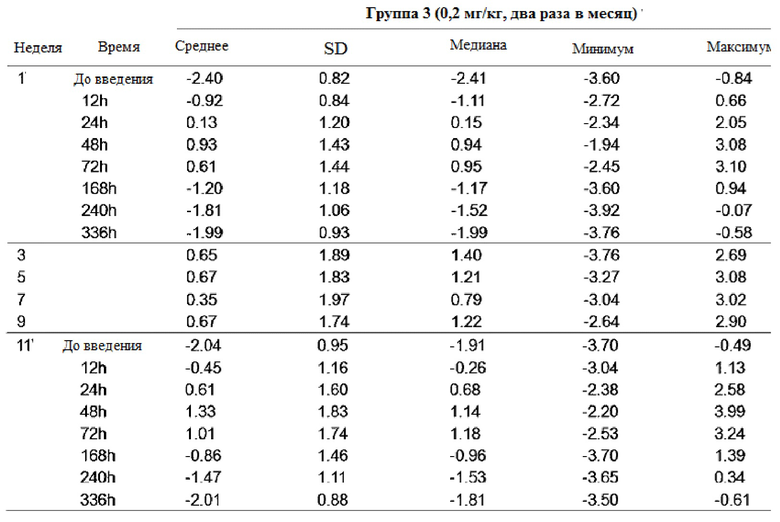
Таблица 10
После повторного введения GX-H9 в дозе 0,1 мг/кг в течение 12 недель, изменение фармакодинамики анализировали при первом введении (1-я неделя) и последнем введении (12-я неделя), перед введением и спустя 12, 24, 48, 72 и 168 часа после введения. У пациентов с дефицитом гормона роста среднее значение (стандартное отклонение) SDS ИФР-1 -2,48 (0,93) до введения показало изменение с минимумом -0,41 (0,97) и максимумом 1,01 (0,87) в течение 7 дней после последнего введения (12-я неделя). Кроме того, после повторного введения GX-H9 в дозе 0,3 мг/кг и 0,2 мг/кг в течение 12 недель изменение фармакодинамики анализировали при первом введении (1-я неделя) и последнем введении (11-я неделя), до введения и спустя 12, 24, 48, 72, 168, 240 и 336 часов после введения. У пациентов с дефицитом гормона роста среднее значение (стандартное отклонение) SDS ИФР-1 -2,64 (1,25) и -2,40 (0,82) до введения показало изменение с минимумом -1,36 (1,59) и -2,01 (0,88) и максимумом 2,12 (1,82) и 1,33 (1,83) в течение 14 дней после последнего введения (11-я неделя). Кроме того, после повторного введения генотропина, активного препарата для контроля в дозе 6 мкг/кг в сутки, изменение фармакодинамики было проанализировано при первом введении (1-я неделя) и последнем введении (12-я неделя), до введения и спустя 12, 24, 48 и 72 часа после введения. У пациентов с дефицитом гормона роста среднее значение (стандартное отклонение) SDS ИФР-1 -2,70 (0,88) до введения показало изменение с минимумом и максимумом 0,42 (0,82) в течение 3 дней после последнего применения (12-я неделя).
Максимальное среднее изменение SDS ИФР-1 GX-H9 было показано от 48 часов до 72 часов после введения 0,1 мг/кг в недельном интервале или 0,3 мг/кг с интервалом в два месяца, а минимальное среднее изменение было показано в течение 168-336 часов после введения. Между тем, для генотропина, активного контрольного препарата, который вводили ежедневно, максимальное среднее изменение было показано в течение 12 часов после введения, а минимальное среднее изменение было достигнуто в течение 24 часов после введения.
Цель лечения для взрослых пациентов с дефицитом гормона роста заключается в улучшении нормализации обмена веществ и качества жизни. Поскольку имеются отличия по возрасту и полу пациентов, уровень ИФР-1 в крови после введения GX-H9 должен быть оптимизирован до середины (50-й перцентиль или 0 SDS) до диапазона 1 SDS в пределах нормального диапазона (-2 SDS до 2 SDS). Соответственно, в случае введения GX-H9 один раз в неделю было установлено, что диапазон SDS ИФР-1 получавшего лечение пациента можно поддерживать на нормальном уровне, регулируя дозировку лечения в диапазоне концентраций от 0,1 мг/кг до 0,2 мг/кг в соответствии с уровнем ИФР-1. Кроме того, в случае введения GX-H9 два раза в месяц было подтверждено, что диапазон SDS ИФР-1 пациента, получавшего лечение, можно поддерживать на нормальном уровне, регулируя дозировку лечения в диапазоне концентраций от 0,2 мг/кг до 0,4 мг/кг в соответствии с уровнем ИФР-1.
5.3. Результаты анализа безопасности рекомбинантного чГР GX-H9 у пациентов с дефицитом гормона роста.
Результаты анализа нежелательных явлений, наблюдаемых у субъектов в соответствии с применяемым лекарством, и соотношения между лекарством и нежелательными явлениями представлены в таблице 11 ниже.
Таблица 11
N = Число индивидуумов, принимавших лекарство
n = Число индивидуумов, у которых отмечены нежелательные явления
E = Число нежелательных явлений
(%) = Отношение индивидуумов, у которых отмечены нежелательные явления в соответствии с лечением (n/N)*100
Серьезные нежелательные явления или слабые нежелательные явления не наблюдались.
Отмечено в целом 11 нежелательных явлений у 5 пациентов, получавших GX-H9 один раз в неделю. Не отмечено смерти или серьезных неблагоприятных явлений. Тяжесть всех нежелательных явлений была легкой. Наиболее частыми нежелательными явлениями были 2 случая скелетно-мышечных нарушений и расстройств соединительной ткани, и 3 случая расстройств крови и лимфатической органов. Сообщалось, что 3 нежелательных явления были связаны с приемом лекарства. Все отмеченные нежелательные явления обычно наблюдались при обычной гормональной терапии.
5.4. Выработка антител (ADA) в ответ на рекомбинантный чГР GX-H9 у пациентов с дефицитом гормона роста.
Чтобы проверить фармакологический эффект при непрерывном введении, исследовали, индуцируется ли образование антител против GX-H9. В случае одного пациента (образец 1502-001) было показано, что результат анализа был положительным, поскольку антитело присутствовало до начала клинических испытаний. Кроме того, GX-H9 не увеличивал величину антител даже в дни 1, 32 и 106-й дней после применения. Далее, в результате изучения фармакодинамики у пациентов было подтверждено, что уровни ИФР-1 были значительно увеличены, и поэтому предполагается, что существующие антитела против чГР не связаны с терапией GX-H9. В результате введение GX-H9 не вызывало ответной выработки антител у всех пациентов.
Таблица 12
1) Если образец не давал реакции, в отчете это отмечали как <20; если давал реакцию, оценивали специфичность.
2) Специфичность: процент ингибирования определяли как 100 x (1-(среднее значение ОП с добавкой GX-H9/ среднее значение ОП образца без добавки)); если отмечалось ≥ 17% ингибирование, оценивали титр образца.
3) Значение титра: титр образца определяют как величину, обратную разведению, которая генерирует среднее значение ОП, равное или превышающее точку разделения ОП плато, где последующие разведения в серии приводят к среднему значению ОП, меньшему, чем точка разделения ОП.
НР: образец не дает реакции, дополнительный анализ не требуется.
Известно, что рекомендуемая дозировка чГР первого поколения (ежедневное введение) для лечения дефицита гормона роста у взрослых составляла от 6 до 12 мкг/кг. Дозировка препарата GX-H9 составляет 0,21-0,42 мг/кг один раз в неделю и 0,42-0,88 мг/кг дважды в месяц при пересчете на то же самое число молей, что и 7-дневное количество чГР первого поколения. Однако было показано, что эффективные дозировки, подтвержденные клинически, отличаются от тех, которые были прогнозированы простыми расчетами. Из результатов, полученных на основе имитации и моделирования фармакокинетики и фармакодинамики в фактических клинических испытаниях, было прогнозировано, что оптимальная доза для взрослых пациентов с дефицитом гормона роста составляет от 0,1 мг/кг до 0,2 мг/кг один раз в неделю или от 0,2 мг/кг до 0,4 мг/кг два раза в месяц. То есть было отмечено, что эффективная доза для GX-H9 была ниже, чем дозировка, прогнозируемая из существующего ежедневного количества чГР.
Кроме того, было установлено, что, когда дозу 0,1 мг/кг вводили взрослым пациентам один раз в неделю в течение 12 недель, не было значительных побочных эффектов и не было ответа антител при введении лекарственного средства. В результате применения дозы 0,3 мг/кг дважды в месяц было подтверждено, что не было никаких побочных эффектов или выработки антител. Таким образом, GX-H9 показал эквивалентную эффективность по сравнению с гормоном роста в организме или продуктом гормона роста первого поколения, и имел улучшенный период полувыведения, и, таким образом, удобство лечения было существенно повышено, а безопасность была подтверждена.
Хотя специфическая часть настоящего раскрытия описана подробно, специалистам в данной области техники очевидно, что такое конкретное описание является лишь предпочтительным вариантом осуществления, и объем настоящего изобретения не ограничен. Таким образом, существенный объем настоящего изобретения будет определяться прилагаемой формулой изобретения и ее эквивалентами.
--->
SEQUENCE LISTING
<110> GENEXINE, INC.
HANDOK INC.
<120> PHARMACEUTICAL COMPOSITION COMPRISING RECOMBINANT HGH FOR THE
TREATMENT OF GROWTH HORMONE DEFICIENCY
<130> PF-B2077
<140> PCT/KR2017/001726
<141> 2017-02-16
<150> 10-2016-0018695
<151> 2016-02-17
<160> 1
<170> PatentIn version 3.5
<210> 1
<211> 436
<212> PRT
<213> Artificial Sequence
<220>
<223> GX-H9
<400> 1
Phe Pro Thr Ile Pro Leu Ser Arg Leu Phe Asp Asn Ala Met Leu Arg
1 5 10 15
Ala His Arg Leu His Gln Leu Ala Phe Asp Thr Tyr Gln Glu Phe Glu
20 25 30
Glu Ala Tyr Ile Pro Lys Glu Gln Lys Tyr Ser Phe Leu Gln Asn Pro
35 40 45
Gln Thr Ser Leu Cys Phe Ser Glu Ser Ile Pro Thr Pro Ser Asn Arg
50 55 60
Glu Glu Thr Gln Gln Lys Ser Asn Leu Glu Leu Leu Arg Ile Ser Leu
65 70 75 80
Leu Leu Ile Gln Ser Trp Leu Glu Pro Val Gln Phe Leu Arg Ser Val
85 90 95
Phe Ala Asn Ser Leu Val Tyr Gly Ala Ser Asp Ser Asn Val Tyr Asp
100 105 110
Leu Leu Lys Asp Leu Glu Glu Gly Ile Gln Thr Leu Met Gly Arg Leu
115 120 125
Glu Asp Gly Ser Pro Arg Thr Gly Gln Ile Phe Lys Gln Thr Tyr Ser
130 135 140
Lys Phe Asp Thr Asn Ser His Asn Asp Asp Ala Leu Leu Lys Asn Tyr
145 150 155 160
Gly Leu Leu Tyr Cys Phe Arg Lys Asp Met Asp Lys Val Glu Thr Phe
165 170 175
Leu Arg Ile Val Gln Cys Arg Ser Val Glu Gly Ser Cys Gly Phe Arg
180 185 190
Asn Thr Gly Arg Gly Gly Glu Glu Lys Lys Lys Glu Lys Glu Lys Glu
195 200 205
Glu Gln Glu Glu Arg Glu Thr Lys Thr Pro Glu Cys Pro Ser His Thr
210 215 220
Gln Pro Leu Gly Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu
225 230 235 240
Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser
245 250 255
Gln Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu
260 265 270
Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr
275 280 285
Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn
290 295 300
Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser
305 310 315 320
Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln
325 330 335
Val Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val
340 345 350
Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val
355 360 365
Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro
370 375 380
Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr
385 390 395 400
Val Asp Lys Ser Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val
405 410 415
Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu
420 425 430
Ser Leu Gly Lys
435
<---
Предложенная группа изобретений относится к области медицины. Предложены применение рекомбинантного гормона роста человека (чГР) GX-H9 для лечения дефицита гормона роста и способ лечения дефицита гормона роста, включающий введение чГР GX-H9 пациенту с дефицитом гормона роста один раз в неделю или два раза в месяц в дозе по меньшей мере 0,1 мг на кг массы тела пациента. Предложенная группа изобретений обеспечивает эффективное лечение дефицита гормона роста. 2 н. и 8 з.п. ф-лы, 10 ил., 12 табл., 5 пр.
1. Применение рекомбинантного гормона роста человека (чГР) GX-H9 для лечения дефицита гормона роста путем его введения, где рекомбинантный чГР применяют один раз в неделю или два раза в месяц в дозе по меньшей мере 0,1 мг на кг массы тела пациента.
2. Применение по п. 1, где рекомбинантный чГР применяют один раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента.
3. Применение по п. 2, где рекомбинантный чГР применяют один раз в неделю в дозе от 0,1 до 0,2 мг на кг массы тела пациента.
4. Применение по п. 1, где рекомбинантный чГР применяют два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента.
5. Применение по п. 4, где рекомбинантный чГР применяют два раза в месяц в дозе от 0,15 до 0,4 мг на кг массы тела пациента.
6. Применение по любому из пп. 1-5, где рекомбинантный чГР включает аминокислотную последовательность SEQ ID NO:1.
7. Применение по любому из пп. 1-5, где рекомбинантный чГР вводят подкожно.
8. Способ лечения дефицита гормона роста, включающий введение рекомбинантного гормона роста человека (чГР) GX-H9 пациенту с дефицитом гормона роста один раз в неделю или два раза в месяц в дозе по меньшей мере 0,1 мг на кг массы тела пациента.
9. Способ по п. 8, в котором введение рекомбинантного чГР GX-H9 пациенту с дефицитом гормона роста осуществляют раз в неделю в дозе от 0,1 до 0,3 мг на кг массы тела пациента.
10. Способ по п. 8, в котором введение рекомбинантного чГР GX-H9 пациенту с дефицитом гормона роста осуществляют два раза в месяц в дозе от 0,1 до 0,4 мг на кг массы тела пациента.
| WO 2016011281 A1, 21.01.2016 | |||
| HOYBYE C | |||
| et al | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Growth Horm IGF Res | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |

