Настоящее изобретение относится к соединениям, ингибирующим или регулирующим активность киназы p70S6, фармацевтическим композициям, содержащим указанные соединения и вариантам терапевтического применения указанных соединений.
Уровень техники
Фермент киназа p70S6 (также известная как p70S6K, p70S6K1, pS6K, S6K, S6K1) представляет собой серин-треонин киназу и является представителем семейства AGC. Она представляет собой нижележащий эффектор сигнального пути фосфатидилинозитол-3 киназы (PI3K)/AKT/ мишени для рапамицина у млекопитающих (mTOR), и p70S6K подвергается фосфорилированию и активации в ответ на факторы роста, такие как ИФР-1, ЭФР, ТФР-[альфа] и ФРГ.
Активация p70S6K, в свою очередь, фосфорилирует ряд белков, участвующих в трансляции белка, включая рибосомальный белок S6 (RPS6), eIF4B и eEF2K. Результатом является усиление трансляции, ведущее к увеличению синтеза белка в клетке. Высокие уровни синтеза белка необходимы для клеточной пролиферации. Также было показано, что p70S6K играет важную роль в митотическом цикле клетки (Lane et al, Nature, 1993, 363(6425):170-2).
Было показано, что киназа p70S6K конститутивно активируется в опухолевых клетках человека, что приводит к пролиферации опухолевых клеток. Также было показано, что ингибирование пути mTOR/p70S6K приводит к снижению пролиферации опухолевых клеток и увеличивает апоптоз опухолевых клеток (Реnе et al (2002) Oncogene 21, 6587 and Le et al (2003) Oncogene 22, 484). Ингибирование активности p70S6K, таким образом, представляет собой перспективный подход в лечении рака.
Было показано, что путь mTOR/p70S6K активируется при почечно-клеточной карциноме и ингибируется CCI-779 (Robb, V.A.; Karbowniczek, М.; Klein-Szanto, A.J.; Henske, Е.P. J Urol 2007, 177, 346-52). Кроме того, было обнаружено, что лечение CCI-779 дает положительный результат у пациентов, страдающих мультиформной глиобластомой, опухоли которых экспрессируют повышенные уровни фосфорилированной p70S6K (Galanis et al. J. Clin. Oncol. 2005, 23, 5294-304).
Кроме того, в работе Peralba et al сообщалось о значительной линейной зависимости между временем прогрессирования заболевания и ингибированием активности p70S6K в мононуклеарных клетках периферической крови (МКПК) вследствие введения ингибитора mTOR CCI-779 у пациентов, страдающих почечно-клеточной карциномой [(2003) Clinical Cancer Research 9, 2887]. Это означает, что активность p70S6K при этом является фактором, влияющим на развитие заболевания, и потенциально активность p70S6K может быть использована в качестве клинического биомаркера.
Ген RPS6KB1, кодирующий p70S6K, локализуется в участке хромосомы 17q23, и указанный участок амплифицируется при раке молочной железы (Cancer Res. (1999) 59: 1408-11 Localization of pS6K to Chromosomal Region 17q23 and Determination of Its Amplification in Breast Cancer). Это приводит к избыточной экспрессии белка p70S6K, и у пациентов, страдающих от рака молочной железы, наблюдается статистически значимая связь между амплификацией и неблагоприятным прогнозом (Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J. Natl. Cancer Inst. 2000; 92: 1252-9).
Кроме того, в работе Belletti et al сообщалось, что S6K1 опосредует выживаемость и повторное проявление рака молочной железы после хирургической операции (Mol Oncol. 2014 May; 8(3):766-80).
P70S6K участвует в миграции и инвазии рака яичников (р70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells, Oncogene (2011), 1-13). Кроме того, было выявлено, что p70S6K принимает участие в обеспечении инвазии, миграции и метастазирования высоко агрессивных клеток трижды негативного рака молочной железы (Targeting p70S6K Prevented Lung Metastasis in a Breast Cancer Xenograft Model, Akar et al, Molecular Cancer Therapeutics (2010), 9 (5), 1180 and Hung et al, S6K1 promotes invasiveness of breast cancer cells in a model of metastasis of triple-negative breast cancer, Am. J. Transl, Res. 2014 Jul 18; 6(4):361-76).
Кроме того, лимфангиолейомиоматоз (ЛАМ) представляет собой заболевание, характеризующееся избыточной активацией оси PI3K/Akt/mTOR/p70S6K вследствие инактивации мутации репрессороного комплекса, Комплекса Туберозного Склероза (КТС). Клетки ЛАМ также являются метастатическими, вызывающими метастазы в легких.
ЛАМ представляет собой редкое деструктивное заболевание легких, поражающее в основном только женщин, связанное с метастазами туберин-нулевых клеток (Taveira-DaSilva et al. (2006). Cancer Control. 2006; 13:276-285). Метастазы развиваются в отдаленных органах, включая легкие, почки и лимфатические узлы, представляя собой тяжелое и изнуряющее заболевание.
ЛАМ возникает либо спорадически, либо как проявление Комплекса Туберозного Склероза (КТС), доминантного аутосомного наследственного нарушения (Экспертный обзор представлен на http://www.orpha.net). Нарушения ЛАМ и КТС характеризуются исключающими мутациями опухолевых супрессоров TSC1 или TSC2, ведущими к избыточной активации mTOR и S6K1. Это, в свою очередь, стимулирует рост и пролиферацию клеток ЛАМ (Holz et al. (2014), Cell Cycle 2014; 13:371-382). Также известно, что S6K1 усиливает метастазирование других раковых заболеваний: рака молочной железы (Akar et al. (2010), Mol Cancer Ther; 9(5)) и рака яичников (Wong et al. (2011), Oncogene (2011) 30, 2420-2432). Из-за зависимости клеток ЛАМ от S6K1, а также возможного участия S6K1 в процессе метастазирования, предполагается, что ингибитор S6K1 будет обладать свойствами, изменяющими течение заболевания в отношении ЛАМ.
Распространенность спорадического ЛАМ составляет приблизительно 1 из 125000 новорожденных, тогда как распространенность легочного ЛАМ, возникающего при КТС, составляет приблизительно 1 из 15000 новорожденных (данные из базы данных редких заболеваний, представленных в интернете, http://www.orpha.net). Для ЛАМ не существует одобренных средств лечения, и поэтому в настоящее время ЛАМ классифицируется как орфанная болезнь.
С учетом того, что p70S6K усиливает трансляцию, известно, что p70S6K играет ключевую роль в патологии заболеваний, обусловленных избыточным синтезом белка (например, синдром фрагильной Х-хромосомы, Genetic Removal of р70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Klann et al, Neuron, Volume 76, Issue 2, p 325-337, 18 October 2012). Кроме того, p70S6K играет роль в патологии раковых заболеваний, включающих синтез онкогенных белков, таких как с-Мус, например, рака поджелудочной железы (The mTORC1/S6K1 Pathway Regulates Glutamine Metabolism through the elF4B Dependent Control of c-Myc Translation, Blenis et al, Current Biology, Volume 24, Issue 19, p 2274-2280, 6 October 2014). Для лечения указанных состояний было бы целесообразно применять перорально биодоступный ингибитор p70S6K с целью устранения избыточного синтеза белка.
P70S6K участвует в патологии ряда раковых заболеваний головного мозга. Такие состояния включают, но не ограничиваются указанными:
• Метастазы в головном мозге, возникающие вследствие наличия рака в других частях организма, например, метастазы в головном мозге, возникающие вследствие рака молочной железы, такого как трижды негативный рак молочной железы (Distant metastasis in triple-negative breast cancer. Neoplasma 2013; 60: 290-294)
• Метастазы в головном мозге, возникающие вследствие метастатического рака молочной железы (метастазы в центральной нервной системе или в головном мозге обычно встречаются у 10-16% пациентов, страдающих метастатическим раком молочной железы и обычно связаны с неблагоприятными прогнозами - см Breast Dis. 2006-2007; 26:139-47.)
• Глиомы и Глиобластомы (S6K1 играет ключевую роль в глиозной трансформации, Cancer Research (2008), 68(16), 6516-6523)
Кроме того, ингибитор киназы p70S6K может быть особенно подходящим для лечения следующих видов рака, зависящих от сигналинга p70S6K:
• Рак мочевого пузыря
• Рак молочной железы
• Колоректальный рак (CRC)
• Диффузные крупноклеточные В-клеточные лимфомы (DLBCL)
• Рак желчного пузыря
• Глиомы и глиобластомы
• Рак головы и шеи
• Гепатоцеллюлярная карцинома
• Ольфакторная нейробластома человека
• Лейкозы
• Лимфомы
• Назофарингиальная карцинома
• Нейроэндокринный рак
• Немелкоклеточный рак легких (NSCLC)
• Мелкоклеточный рак легких
• Рак яичников
• Рак поджелудочной железы
• Феохромоцитома
• Почечно-клеточная карцинома (RCC)
• Плоскоклеточный рак
• Метастазы, например, метастазы в костях и метастазы в легких
P70S6K также играет ключевую роль в ряде расстройств нервно-психического развития (много упоминаний в The Autistic Neuron: Troubled Translation?. Bear et al, Cell 135, October 31, 2008). В частности, такие расстройства вызваны избыточным синтезом белка, обусловленным P70S6K. Такие состояния включают, но не ограничиваются указанными:
• Синдром фрагильной Х-хромосомы, редкое расстройство нервно-психического развития, вызванное избыточными уровнями активности p70S6K
• Аутизм и заболевания аутического спектра
• Тремор/атаксия, ассоциированная с фрагильной Х-хромосомой (FXTAS)
• Синдром Ангельмана
• Туберозный склероз
• Синдром PTEN-гарматомы
• Синдром дупликации гена МЕСР2
• Нейрофиброматоз
• Болезнь Альцгеймера (см. (1) Oddo et al, Reducing Ribosomal Protein S6 Kinase 1 Expression Improves Spatial Memory and Synaptic Plasticity in a Mouse Model of Alzheimer's Disease, The Journal of Neuroscience, October 14, 2015, 35(41): 14042-14056 и (2) Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer's disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature, Journal of Neuroscience (2014), 34(23), 7988-7998)
• Синдром Дауна (mTOR Hyperactivation in Down Syndrome Hippocampus Appears Early During Development, Journal of Neuropathology & Experimental Neurology (2014), 73(7), 671-683)
Синдром PTEN гамартомы
Синдром опухоли PTEN-гамартомы (PHTS) охватывает четыре основных клинически различных синдрома, связанных с герминативными мутациями опухолевого супрессора PTEN. Эти аллельные нарушения, синдром Коудена, синдром Банаяна-Райли-Рувалькаба, синдром Протея, Протея-подобный синдром связаны с нерегулируемой клеточной пролиферацией, ведущей к образованию гарматом (доброкачественных и злокачественных опухолей щитовидной железы, молочной железы и эндометрия) (Genetics in Medicine (2009) 11, 687-694). Отсутствие PTEN приводит к потере понижающей регуляции фосфорилированной Akt, что, в свою очередь, обеспечивает бесконтрольное выживание, рост и пролиферацию указанных клеток. Поскольку S6K1 представляет собой ключевой эффектор Akt, ингибитор S6K1 может быть подходящим для контролирования роста рака. В настоящее время распространенность PHTS неизвестна.
Нейрофиброматоз 1 типа
Нейрофиброматоз 1 типа представляет собой состояние, характеризующееся изменениями окраски кожи (пигментации) и ростом опухолей, распространяющихся по нервам в кожу, головной мозг и другие части тела. Признаки и симптомы этого состояния широко варьируются у пораженных им людей. У большинства взрослых, страдающих нейрофиброматозом 1 типа, развиваются нейрофибромы, которые являются нераковыми (доброкачественными) опухолями, обычно расположенными на коже или непосредственно под кожей. Эти опухоли также могут возникать в нервах около спинного мозга или вдоль нервов в других частях организма. У некоторых людей, страдающих нейрофиброматозом 1 типа, развиваются раковые опухоли, которые растут вдоль нервов. Такие опухоли, которые обычно развиваются в подростковом или зрелом возрасте, называют периферическими периневральными злокачественными опухолями. Люди, страдающие нейрофиброматозом 1 типа также подвержены повышенному риску развития других видов рака, включая опухоли головного мозга и рак кроветворной ткани (лейкоз).
Нейрофиброматоз 1 типа встречается у 1 из 3000-4000 людей по всему миру и в настоящее время основным способом лечения является хирургическая операция; поскольку средств направленной терапии не существует, он классифицируется как орфанное заболевание (http://ghr.nlm.nih.gov/condition/neurofibromatosis-type-1).
Мутации в гене NF1 вызывают нейрофиброматоз 1 типа. Ген NF1 кодирует белок нейрофибромин. Этот белок продуцируется во многих клетках, включая нервные клетки и специальные клетки, окружающие нервы (олигодендроциты и Шванновские клетки). Нейрофибромин действует как опухолевый супрессор. Мутации в гене NF1 приводят к получению нефункционального варианта нейрофибромина, неспособного регулировать рост и деление клеток. В результате вдоль нервов по всему организму могут образовываться опухоли, такие как нейрофибромы. Ингибитор S6K1 способен контролировать рост клеток, экспрессирующих мутантный ген NF1 путем подавления продуцирования белка нейрофибромина и других белков, необходимых для роста опухоли.
Роль P70S6 в неврологических заболеваниях
P70S6K также играет ключевую роль в патологии ряда расстройств нервно-психического развития (много упоминаний в The Autistic Neuron: Troubled Translation?. Bear et al, Cell 135, October 31, 2008). В частности, такие расстройства вызваны избыточным синтезом белка, обусловленным P70S6K.
Хорошо известно, что точный контроль трансляции (синтеза белка) совершенно необходим для неврологических процессов, проходящих в головном мозге, таких как длительная синаптическая пластичность и формирование долговременной памяти. Кроме того, изменения в контроле трансляции являются общим патофизиологическим признаком неврологических нарушений человека, включая нарушения развития, нейропсихиатрические нарушения и нейродегенеративные заболевания. Кроме того, известно, что механизмы контроля подвержены модификации малыми молекулами, проникающими в головной мозг (Klann and Santini, Dysregulated mTORC1-dependent translational control: from brain disorders to psychoactive drugs, Front. Behav. Neurosci., 08 November 2011, doi: 10.3389/fnbeh.2011.00076).
S6K1 хорошо известен как главный регулятор биосинтеза белка благодаря его роли в инициации трансляции, а также фосфорилирования и активации различных субстратов, которые приводят к продуцированию белка (eIF4B, PDCD4, SKAR, eEF2K, RPS6 - обзор представлен в Ma and Blenis, Nature Reviews Molecular Cell Biology 10, 307-318 (May 2009), doi:10.1038/nrm2672).
Следующие нарушения характеризуются основными отклонениями в регуляции трансляции белка, ведущим к наблюдаемым патологиям. Ингибитор S6K1, действие которого заключается в снижении избыточной трансляции белка, таким образом, может являться подходящим лекарственным средством для таких нарушений.
Определенные нарушения могут быть классифицированы в подгруппы: (1) Расстройства нервно-психического развития (2) Нейродегенеративные заболевания. Внутри каждого подкласса указанные нарушения связаны общими мотивами:
1. Расстройства нервно-психического развития
Расстройства нервно-психического развития определяются как заболевания, вызванные аномальным развитием головного мозга в течение первых двух десятилетий жизни. Возможно определить подгруппу указанных нарушений, характеризующихся одногенными мутациями. Общей молекулярной аномалией нескольких из указанных нарушений являются мутации, обеспечивающие потерю функции и/или делецию генов, кодирующих белки, обычно подавляющие сигнальный путь mTORC1. Указанные нарушения перечислены ниже.
Синдром фрагильной Х-хромосомы
Синдром фрагильной Х-хромосомы (FXS) представляет собой генетическое состояние, вызывающее ряд сложностей, связанных с развитием, включая трудности в обучении и когнитивные нарушения. Обычно на мужчин это нарушение оказывает более серьезное воздействие, чем на женщин, поскольку указанное состояние наследуются через X-хромосому. Пораженные индивиды обычно характеризуются задержкой развития речи и языка к возрасту 2 лет. Большинство мужчин, страдающих FXS, характеризуются умственными нарушениями от легкой до умеренной степени, тогда как примерно треть пораженных женщин являются интеллектуально недееспособными. У детей, страдающих FXS, также может наблюдаться тревожное и гиперактивное поведение, которое проявляется в беспокойных движениях или импульсивных действиях. Они могут страдать синдромом дефицита внимания (СДВ), который включает нарушение способности удерживать внимание и трудности при сосредоточении на конкретных задачах. Примерно треть индивидов, страдающих FXS, имеют признаки заболеваний аутического спектра, влияющие на общение и социальное взаимодействие. Примерно у 15 процентов мужчин и 5 процентов женщин, страдающих FXS, случаются пароксизмальные судороги. Большинство мужчин и примерно половина женщин, страдающих FXS, имеют характерные физические признаки, которые с возрастом становятся более заметными. Такие признаки включают длинное узкое лицо, большие уши, выступающие челюсть и лоб, необычно гибкие пальцы рук, плоскостопие и увеличенные семенники у мужчин (макроорхидизм) после половой зрелости. FXS встречается у приблизительно 1 из 4000 мужчин и 1 из 8000 женщин.
FXS вызывают мутации гена Fmr1. Ген Fmr1 обеспечивает образование белка, называемого белком умственной отсталости, вызванной фрагильной Х-хромосомой (fragile X mental retardation 1), или FMRP. Этот белок помогает регулировать продукцию других белков и играет роль в развитии синапсов, которые представляют собой специализированные связи между нервными клетками. Синапсы играют решающую роль в передаче нервных импульсов.
Почти все случаи FXS вызваны мутацией в сегменте ДНК, известной как CGG триплетный повтор, в гене Fmr1. Обычно этот ДНК-сегмент повторяется в диапазоне от 5 до 44 раз (чаще всего либо 29, либо 30 раз). Однако у людей, страдающих FXS, сегмент CGG повторяется более 200 раз. Аномально расширенный сегмент CGG выключает (подавляет) ген Fmr1, что предотвращает продуцирование FMRP указанным геном.
FMRP является репрессором трансляции белка. В случае пациентов, страдающих FXS, которые испытывают потерю или нехватку FMRP, репрессии трансляции не происходит, что ведет к избыточному продуцированию массива белков, обычно контролируемых FMRP. Ряд таких белков экспрессируется в нейронах и контролирует синаптическую пластичность (формирование памяти, обучение, способность к хранению информации). Бесконтрольное продуцирование этих белков ведет к нейропатологическому состоянию, наблюдаемому у пациентов, страдающих FXS. В работе Klann et al сообщалось, что S6K1 играет центральную роль в избыточной трансляции указанных белков, и генетический нокаут S6K1 приводил к устранению клинических признаков мышиной модели FXS (Genetic Removal of р70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron 76, 325-337, 2012). Было определено, что ингибиторы S6K1, описанные в настоящей заявке, также обладают способностью подавлять синтез белка в нейронах, что приводит к устранению не соответствующих норме клинических признаков в мышиной модели FXS.
Кроме того, в работе Tassone et al (Genes, Brain and Behavior (2012), doi: 10.1111/j.1601-183X.2012.00768.x) было опубликовано, что лимфолиты, выделенные из крови пациентов-людей, страдающих FXS, демонстрировали более высокие уровни фосфорилированной (активированной) p70S6K, а также более высокие уровни RPS6, непосредственного субстрата S6K1. Полученные данные подтверждают, что p70S6K у пациентов-людей с FXS активирован сильнее и подтверждает концепцию об ингибировании активности p70S6K с целью купирования заболевания. Кроме того, это является возможным клиническим биомаркером для оценки фармакодинамического эффекта ингибитора p70S6K в клинической практике.
Тремор/атаксия, ассоциированная с фрагильной Х-хромосомой (FXTAS)
Тремор/атаксия, ассоциированная с фрагильной Х-хромосомой (FXTAS), представляет собой редкое нейродегенеративное заболевание, характеризующееся приобретенной прогрессирующей интенцией и тяжелой атаксией. Она представляет собой Х-сцепленное генетическое заболевание и указанному заболеванию в первую очередь подвержены мужчины (База данных орфанных редких заболеваний, http://www.orpha.net/consor/cgi-bin/index.php?lng=EN).
Распространенность оценивается в 1-9 из 100000 человек. Возраст возникновения тремора и/или атаксии у мужчин составляет приблизительно 60 лет. Клиническая картина может быть различной, с доминантными проявлениями, включающими: интенцию, прогрессирующую мозжечковую атаксию походки, нарушение исполнительной функции лобной доли, снижение когнитивных функций, периферическую нейропатию и вегето-сосудистую дистонию. Другие признаки включают умеренный паркинсонизм и психиатрические проявления (депрессию, тревожность и ажитацию), возможно прогрессирующие до деменции. Женщины-носители обычно имеют менее выраженные проявления, чем мужчины, однако они также имеют повышенный риск развития синдрома истощения яичников, хронической мышечной боли и гипотиреоза. FXTAS вызвана экспансией тринуклеотидных повторов CGG (55-200 повторов) в диапазоне пермутаций гена Fmr1. Не существует конкретных средств лечения FXTAS, нацеленных на основной патологический механизм; в связи с этим FXTAS классифицируется как орфанное заболевание. Экспансия тринуклеотидных повторов CGG часто приводит к пониженным уровням белка FMRP, репрессора трансляции белка. Это ведет к избыточной трансляции белка, чему можно препятствовать путем применения ингибитора S6K1.
Аутизм и нарушения аутического спектра
Нарушения аутического спектра (НАС) и аутизм являются терминами, характеризующими группу комплексных нарушений развития головного мозга. Указанные нарушения характеризуются трудностями в социальном взаимодействии, вербальной и невербальной коммуникации и повторяющимся поведением. В публикации 2013 года, озаглавленной «Диагностическое и статистическое руководство по психическим расстройствам (DSM-5)», все аутические расстройства сведены вместе в одном зонтичном диагнозе НАС. Ранее их определяли как различные подтипы, включая аутическое расстройство, дезинтегративное расстройство детского возраста, неуточненное общее расстройство развития (PDD-NOS) и синдром Аспергера. Центры контроля и профилактики заболеваний США диагностируют нарушения аутического спектра приблизительно у 1 из 68 американских детей. Исследования также свидетельствуют о том, что аутизм в четыре-пять раз чаще встречается у мальчиков, чем у девочек. По оценкам, у 1 из 42 мальчиков и у 1 из 189 девочек в Соединенных Штатах диагностирован аутизм. В целом, НАС поражены более 3 миллионов человек в США и десятки миллионов по всему миру (вебсайт Autism Speaks, http://www.autismspeaks.org/). Более того, согласно государственной статистике аутизма предполагается, что скорость распространения заболевания растет. Синдром фрагильной Х-хромосомы (FXS) является наиболее распространенной наследственной причиной умственной недееспособности и наиболее распространенной известной причиной аутизма в мире (Penagarikano et al (2007). The pathophysiology of Fragile X Syndrome. Annu. Rev. Genomics Hum. Genet. 8, 109-129). Такая причинная связь между FXS и аутизмом указывает на то, что ингибитор S6K1, эффективный в лечении FXS, также может быть подходящим для лечения аутизма и НАС.
Синдром Ангельмана
Синдром Ангельмана (СА) представляет собой нейрогенетическое нарушение, обычно диагностируемое у младенцев и характеризующееся задержкой развития, тяжелыми умственными нарушениями, отсутствием речи, поведением с бурным проявлением счастья, нарушением моторных функций и эпилепсией. СА вызван дефицитом экспрессии гена UBE3A, который может быть вызван различными аномалиями хромосомы 15 (Dan, В., Angelman syndrome: Current understanding and research prospects. Epilepsia, 2009. 50(11): p. 2331-2339). Хотя точные цифры неизвестны, распространенность СА среди детей и молодых людей составляет от 1/10000 до 1/20000, в связи с чем СА считается редким заболеванием. При СА были обнаружены мутации в Е3 убиквитин лигазе UBE3A, откуда следует, что при этом заболевании может быть нарушен убиквитин-зависимый белковый обмен, что может привести к повышенным уровням синаптического белка (Jiang and Beaudet, 2004). Кроме того, в мышиной модели синдрома Ангельмана было обнаружено, что ингибирование S6K1 может улучшить синаптическую пластичность гиппокампа и способность к обучению (Cellular and Molecular Life Sciences pp 1-12). Ингибитор киназы S6K1 оказывает воздействие посредством снижения уровней трансляции синаптического белка.
Комплекс туберозного склероза
Комплекс туберозного склероза представляет собой генетическое нарушение, характеризующееся ростом многочисленных нераковых (доброкачественных) опухолей в разных частях тела. Эти опухоли могут возникать в коже, головном мозге, почках и других органах, в некоторых случаях приводя к значительным проблемам со здоровьем. Комплекс туберозного склероза также вызывает трудности в развитии, а признаки и симптомы указанного состояния варьируются от человека к человеку.
Комплекс туберозного склероза часто поражает головной мозг, вызывая судороги, поведенческие проблемы, такие как гиперактивность и агрессия, и умственные нарушения или проблемы в обучении. Некоторые пораженные дети имеют характеристические признаки аутизма, расстройства развития, которое влияет на способность к общению и социальному взаимодействию, как описано выше. Также у людей с комплексом туберозного склероза могут развиваться доброкачественные опухоли головного мозга; такие опухоли могут вызывать серьезные или угрожающие жизни осложнения. Комплекс туберозного склероза поражает приблизительно 1 из 6000 человек (http://ghr.nlm.nih.gov/condition/tuberous-sclerosis-complex).
Мутации гена TSC1 или TSC2 могут вызвать комплекс туберозного склероза. Гены TSC1 и TSC2 обеспечивают образование белков гамартина и туберина, соответственно. Эти белки участвуют в структуре сигнального пути PI3K и действуют как опухолевые супрессоры, ингибируя активацию mTOR посредством Rheb-GTP. В случае, когда TSC1 или TSC2 мутированы, это приводит к потере функции опухолевого супрессора, вызывая избыточную активацию mTOR.
Важно отметить, что было показано, что ингибитор mTORC1 рапамицин эффективно улучшает способность к обучению и восстанавливает недостаток памяти у TSC2 гетерозиготных нокаутных мышей (Ehninger et al., 2008b), откуда следует, что неконтролируемый сигналинг mTORC1 является основным молекулярным механизмом, участвующим в поведенческих аномалиях.
Одним из функциональных эффекторов mTOR является S6K1; поэтому ингибирование функции S6K1 может облегчить течение заболевания.
Синдром дупликации гена МЕСР2
Синдром дупликации гена МЕСР2 представляет собой генетическое состояние, которое наследуется по Х-сцепленному пути и встречается практически только у мужчин. Оно характеризуется умственными нарушениями от умеренной до сильно выраженной степени. Большинство людей, страдающих указанным состоянием, также в младенчестве имеют слабый мышечный тонус, трудности с кормлением, плохую или отсутствующую речь и судороги, которые могут оставаться при лечении мышечной ригидности (спастичности). Люди, страдающие синдромом дупликации гена МЕСР2, отличаются задержкой развития двигательных навыков, таких как способность сидеть и ходить. У многих людей, страдающих синдромом дупликации гена МЕСР2, встречаются рецидивирующие инфекции дыхательных путей. Такие инфекции дыхательных путей являются основной причиной смерти пораженных людей, причем почти половина из них приходится на возраст 25 лет. Распространенность синдрома дупликации гена МЕСР2 неизвестна; в научной литературе сообщалось приблизительно о 120 пораженных людях. Синдром дупликации гена МЕСР2 возникает вследствие дупликации гена МЕСР2, что ведет к избыточному продуцированию белка МеСР2 в головном мозге. МеСР2 является регулятором экспрессии других генов. Хотя МеСР2 играет ключевую роль в обеспечении нормального функционирования головного мозга, его избыток может привести к аномальной регуляции целевых генов (http://ghr.nlm.nih.gov/condition/mecp2-duplication-syndrome). Ингибитор S6K1 может снижать продуцирование белка МеСР2 посредством общего снижения трансляции и может представлять собой подходящее терапевтическое лечение указанного заболевания.
Синдром Дауна
Синдром Дауна (СД) или даунизм, также известный как трисомия 21, представляет собой генетическое нарушение, вызванное наличием третьей копии 21 хромосомы или ее части (Patterson, D (Jul 2009). "Molecular genetic analysis of Down syndrome." Human Genetics 126 (1): 195-214). Обычно он связан с физическими задержками роста, характерными чертами лица и умственными нарушениями от слабой до умеренной степени. СД представляет собой наиболее распространенную хромосомную аномалию у человека, которая встречается приблизительно у одного из 1000 новорожденных детей каждый год (Weijerman, ME; de Winter, JP (Dec 2010). "Clinical practice. The care of children with Down syndrome". European journal of pediatrics 169 (12): 1445-52).
В недавних публикациях было показано, что избыточная активация mTOR участвует в СД на ранних стадиях развития. В контрольном (без СД) гиппокампе фосфорилированный S6 обнаруживался только в предродовой период; однако спустя два месяца после родов перестал обнаруживаться. Напротив, у пациентов, страдающих СД, фосфорилированный S6 и фосфорилированная киназа S6 обнаруживались в предродовой период и сохранялись в течение постнатального развития. Это было связано с повышенной экспрессией фосфорилированного белка S6 (RPS6), фосфорилированной p70S6K, фосфорилированного эукариотической инициации фактор 4Е связывающего белка 1, и фосфорилированного mTOR в гиппокампе с СД по сравнению с контролем (J Neuropathol Exp Neurol. 2014 Jul; 73(7):671-83). Кроме того, было выдвинуто предположение, что ингибиторы mTOR, такие как Рапамицин или другие Рапалоги, могут являться подходящими для лечения когнитивных расстройств, ассоциированных с СД (CNS Neurol Disord Drug Targets. 2014 Feb; 13(1):34-40). Поскольку S6K1 контролирует фосфорилирование и активацию белка S6, ингибитор S6K1 может являться терапевтически подходящим средством для предотвращения избыточной активации сигналинга mTOR у пациентов, страдающих СД.
2. Нейродегенеративные заболевания
Болезнь Альцгеймера
Клинические симптомы болезни Альцгеймера (БА) включают постепенную потерю памяти и последующую деменцию, и нейропатологическое накопление сенильных бляшек и нейрофибриллярных клубков. БА насчитывает от 60% до 70% случаев деменции (Burns, А; Lliffe, S (5 February 2009). "Alzheimer's disease." BMJ (Clinical research ed.) 338: b158). Это изнуряющее и относительно широко распространенное заболевание - по данным за 2010 год, у от 21 до 35 миллионов человек по всему миру была диагностирована БА ("Survival in dementia and predictors of mortality: a review". International journal of geriatric psychiatry 28 (11): 1109-24).
На молекулярном уровне БА ассоциируется с (1) прогрессирующим накоплением β-амилоидных пептидов (Аβ) в форме внеклеточных амилоидных бляшек в головном мозге человека и (2) избыточным фосфорилированием тау-белка. Согласно недавним публикациям сигнальный путь PI3K/mTOR участвует в патогенезе заболевания. Например, было обнаружено, что генетический нокаут белка mTOR в Tg2576 мышах, широко используемой животной модели БА, подавляет накопление амилоида-β и восстанавливает недостаточность памяти у животных (J. Neurosci. 2014 Jun 4; 34(23):7988-98). Кроме того, исследование ткани головного мозга, взятых у пациентов-людей с БА после смерти, показало, что изменение сигналинга mTOR и аутофагия происходит на ранних стадиях БА, что приводит к значительному увеличению уровней Аβ (1-42) и избыточной активации пути PI3K/Akt/mTOR (J. Neurochem. 2015 Jan 27). Уровень экспрессии S6K1, нисходящей мишени mTOR, в этих образцах был повышен, что приводит к заключению, что терапевтическое воздействие ингибитора S6K1 может являться подходящим для контроля синтеза β амилоидного белка и подавления сигналинга mTOR. Кроме того, повышенные уровни фосфорилированного mTOR и S6K1 также обнаруживались в некоторых участках мозга, пораженного БА, например, в коре головного мозга, у двойных APP/PS1 трансгенных мышей, модели БА (Lafay-Chebassier et al., 2005).
Кроме того, в работе Oddo et al (Reducing Ribosomal Protein S6 Kinase 1 Expression Improves Spatial Memory and Synaptic Plasticity in a Mouse Model of Alzheimer's Disease, The Journal of Neuroscience, October 14, 2015, 35(41):14042-14056) опубликованы данные, подтверждающие следующие заключения: (1) активность S6K1 в головном мозге пациентов с БА увеличивается (2) в мышиной модели БА активность S6K1 в головном мозге также повышена по сравнению с контролем (3) Генетическое снижение S6K1 в мышиной модели БА (посредством гаплоидной недостаточности) (1) улучшило синаптическую пластичность и недостаточность пространственной памяти, и (2) уменьшило уровни накопления Амилоида-В (АВ) и фосфо-тау/ общий тау, ключевых неврологических признаков БА. Это подтверждает гипотезу о том, что регуляция активности S6K1 с помощью низкомолекулярного ингибитора S6K1 может представлять собой эффективный терапевтический подход к лечению БА.
Болезнь Хантингтона
Болезнь Хантингтона представляет собой наследственное прогрессирующее нарушение функций головного мозга, вызывающее неконтролируемые движения, эмоциональные нарушения и потерю способности думать (когнитивной деятельности); заболевание существует в двух формах: (1) болезнь Хантингтона, возникающая во взрослом возрасте, наиболее распространенная форма указанного заболевания, которая обычно проявляется после 30 или 40 лет, и (2) болезнь Хантингтона, возникающая в ювенильном периоде, которая менее распространена и начинается в детстве или подростковом возрасте. Обе формы заболевания являются прогрессирующими, и пораженные индивиды обычно живут лишь 10-15 лет после появления признаков и симптомов. Болезнь Хантингтона поражает приблизительно 3-7 человек из 100000 европейской расы.
Болезнь Хантингтона вызвана мутациями гена НТТ, приводящими к продуцированию аномально длинного варианта белка хантингтина (Htt). Удлиненный белок разрезается на более мелкие токсичные фрагменты, которые связываются вместе и накапливаются в нейронах, нарушая нормальные функции указанных клеток. Дисфункция и случайная гибель нейронов в определенных областях головного мозга являются основными признаками и симптомами болезни Хантингтона. В недавних публикациях было показано, что мутантный Htt способствует патогенезу БХ путем усиления активности mTORC1. (Sci. Signal., 28 October 2014, Vol. 7, Issue 349, p. ra103).
Одним из функциональных эффекторов сигналинга mTOR является S6K1; поэтому ингибирование функции S6K1 может ослаблять течение заболевания. Кроме того, ингибирование S6K1 может ограничивать продуцирование белка хантингтина путем общего подавления трансляции белка.
Болезнь Паркинсона
Болезнь Паркинсона (БП) представляет собой прогрессивное нейродегенеративное состояние, вызванное гибелью допамин-содержащих клеток черного вещества. Люди, страдающие БП, обычно проявляют симптомы и признаки, ассоциированные с паркинсонизмом, в частности, брадикинезию, ригидность и тремор в состоянии покоя. БП является распространенным хроническим прогрессирующим неврологическим состоянием, поражающим, по оценкам, 100-180 человек на 100000 населения (от 6 до 11 человек на 6000 общей популяции Великобритании) и ежегодно частота заболеваемости составляет 4-20 человек на 100000. Наблюдается рост заболеваемости с возрастом и более высокая заболеваемость БП у мужчин (https://www.nice.org.uk/guidance/cg035/chapter/introduction).
Несмотря на то, что БП традиционно считается негенетическим заболеванием, в настоящее время известно, что по меньшей мере 5% людей страдают формами указанного заболевания, возникающими вследствие мутации одного или нескольких конкретных генов. Было достоверно показано, что мутации в конкретных генах вызывают БП. Указанные гены кодируют альфа-синуклеин (SNCA), паркин (PRKN), богатую лейцином повторную киназу 2 (LRRK2 или дардарин), PTEN-индуцированную предполагаемую киназу 1 (PINK1), DJ-1 и АТР13А2 (Lesage S, Brice A; Brice (April 2009). "Parkinson's disease: from monogenic forms to genetic susceptibility factors". Hum. Mol. Genet. 18 (R1): R48-59).
Недавние исследования, направленные на изучение механизма нейродегенерации при БП, демонстрируют участие сигнального пути mTORC1 в механизмах выживаемости дофаминергических нейронов. Исследования in vivo и in vitro показывают, что дегенерация, вызванная лечением БП-токсинами, такими как 6-OHDA и МРТР, приводит к повышающей регуляции RTP801, белка, кодируемого геном, чувствительным к стрессу RTP801, который, в свою очередь, снижает активность mTOR-киназы. Было высказано предположение, что молекулярный механизм, связывающий высокие уровни ингибирования RTP801 с mTORC1 и нейродегенерацию, включает TSC2 и Akt (Deyoung et al., 2008; Malagelada et al., 2008). Генетические манипуляции, которые затрагивают TSC2 или увеличивают экспрессию конститутивно активной формы Akt, защищают от токсинов БП, и предотвращают увеличение RTP801 (Malagelada et al., 2008). Однако сообщалось, что рапамицин является нейропротекторным агентом как в культуре клеток, так и в мышиной модели МРТР (мышиная модель БП). Предлагается, что рапамицин способен усиливать активность Akt за счет ингибирования mTORC1-зависимой активации S6K1 и последующего снижения фосфо-IRS-1, который представляет собой каркасный белок, участвующий в активации PI3K и Akt (Shah et al., 2004). Соответственно, возможна ситуация, при которой ингибитор S6K1 (главный эффектор mTOR) расщепляет одну и ту же отрицательную обратную связь IRS-1, что приводит к активации Akt и увеличению выживаемости в нейронах пациентов с БП. Таким образом, ингибитор S6K1 может проявлять терапевтический эффект при дозировании пациенту с БП.
Для лечения всех описанных выше заболеваний было бы целесообразно применять перорально биодоступный ингибитор P70S6K, свойства которого позволяют проникать в головной мозг в концентрации, достаточной для достижения эффективности.
Поэтому представляется целесообразным разработать соединения, способные ингибировать киназу p70S6.
Изобретение
В настоящем изобретении предложен класс новых арилалкиламин-замещенных хиназолинов в качестве ингибиторов киназы p70S6.
Согласно первому варианту реализации (Вариант реализации 1.0) изобретения предложено соединение формулы (1):
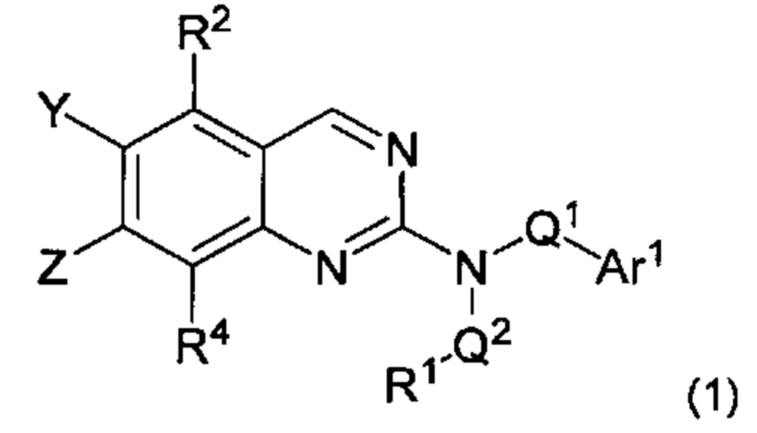
или его соль, таутомер или N-оксид;
где:
один из Y и Z представляет собой R3, а другой представляет собой Ar2;
Q1 представляет собой C1-8 алкиленовую группу, необязательно содержащую один или два заместителя, выбранные из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода; и причем атом углерода C1-8 алкиленовой группы необязательно может быть заменен циклопропан-1,1-диильной или циклобутан-1,1-диильной группой, при условии, что общее количество атомов углерода в алкиленовой группе, содержащей такую замену, не превышает 8;
Q2 представляет собой связь или C1-8 алкиленовую группу, необязательно содержащую один или два заместителя, выбранные из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси- заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода;
R1 выбран из водорода, NRxRy и группы Cy1;
Rx и Ry являются одинаковыми или различными и каждый из них выбран из водорода, С1-4 гидрокарбила или гидрокси-C1-4 гидрокарбила; или NRxRy образует 4-7-членное гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а второй выбран из N, О и S и их окисленных форм, причем указанное гетероциклическое кольцо необязательно содержит один или два заместителя, выбранных из С1-4 гидрокарбила, оксо, амино, моно-С1-4 гидрокарбиламино, ди-С1-4гидрокарбиламино, фтора и гидрокси, при условии что между амино, моно-С1-4 гидрокарбиламино, ди-С1-4 гидрокарбиламино и гидроксильным заместителями в случае наличия и атомом азота в группе NRxRy присутствуют по меньшей мере два атома углерода подряд;
Су1 представляет собой С-связанную 3-7 членную моноциклическую неароматическую карбоцикпическую или гетероциклическую группу, содержащую 0, 1 или 2 гетероатомных члена кольца, выбранных из N, О и S и окисленных форм S, причем указанные карбоциклическая и гетероциклическая группы необязательно содержат один или два заместителя, выбранные из С1-3 гидрокарбила, фтора, оксо и гидрокси;
R2 и R4 являются одинаковыми или различными и каждый из них выбран из водорода, фтора, хлора, С1-2 алкила и С1-2 алкокси, где каждый из С1-2 алкила и С1-2 алкокси необязательно содержит в качестве заместителей два или более атомов фтора;
R3 выбран из водорода, фтора, хлора, С1-2 алкила и С1-2 алкокси, причем каждый C1-2 алкил и С1-2 алкокси необязательно содержит в качестве заместителей один или два атома фтора;
Ar1 представляет собой моноциклическое 5 или 6-членное арильное или гетероарильное кольцо, содержащее 0, 1 или 2 гетероатомных члена кольца, выбранных из О, N и S, причем арил или гетероарил необязательно содержат 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и выбраны из галогена, циано и группы Ra-Rb;
Ra представляет собой связь, О, СО, Х3С(Х4), С(Х4)Х3, Х3С(Х4)Х3, S, SO, SO2, NRc, SO2NRc или NRcSO2;
Rb представляет собой:
• водород;
• карбоциклическую или гетероциклическую группу, содержащую от 3 до 7 членов кольца, 0, 1, 2 или 3 из которых являются гетероатомными членами кольца, выбранными из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; и
• ациклическую C1-8 углеводородную группу, необязательно содержащую один или более заместителей, выбранных из гидрокси; оксо; галогена; циано; карбокси; амино; моно- или ди-С1-4 алкиламино; и карбоциклических или гетероциклических групп, содержащих от 3 до 7 членов кольца, 0, 1, 2 или 3 из которых являются гетероатомными членами кольца, выбранными из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; при этом один или два, но не все атомы углерода ациклической С1-8 углеводородной группы необязательно могут быть заменены О, S, SO, SO2, NRc, Х3С(Х4), С(Х4)Х3 или Х3С(Х4)Х3;
R6 выбран из заместителей R5, за исключением того, что R6 не состоит из карбоциклической или гетероциклической группы или не содержит указанные группы;
X3 представляет собой О, S или NRc; и
X4 представляет собой =O, =S или =NRc; и
Rc представляет собой водород или С1-4 гидрокарбил;
Ar2 представляет собой бициклическую 8-11-членную гетероарильную группу, содержащую 1, 2, 3 или 4 гетероатомных членов кольца, выбранных из О, N и S, и необязательно содержащую 1, 2 или 3 заместителя R7, выбранных из оксо, фтора; хлора; брома; С1-4 гидрокарбила, необязательно содержащего в качестве заместителей один или более атомов фтора; С1-4 гидрокарбилокси, необязательно содержащего в качестве заместителей один или более атомов фтора; гидрокси; циано; N(Rc)2; Rc-C(O)-; Rc-C(O)N(Rc)-; (Rc)2NC(O)-; Rc-SO2NRc-; Rc-NHC(O)NH-; (Rc)2NSO2-; и пяти- и шестичленных моноциклических групп, содержащих от 0 до 3 гетероатомных членов кольца, выбранных из О, N и S, причем указанные пяти- и шестичленные моноциклические группы, не содержащие заместителей или содержащие один или более заместителей R8, выбранных из С1-4 гидрокарбила, С1-4 гидрокарбилокси, циано, гидрокси, оксо, галогена, амино, моно-С1-4 гидрокарбиламино и ди-С1-4 гидрокарбиламино, и при этом гидрокарбильные остатки в случае их присутствия необязательно содержат в качестве заместителей фтор, C1-2 алкокси, гидрокси, амино, моно-ди-С1-2 алкиламино или ди-С1-4 алкиламино;
и при этом в каждом заместителе, состоящем из гидрокарбильной группы или содержащем гидрокарбильную группу, указанная группа выбрана из алкильных, алкенильных, алкинильных и циклоалкильных групп и их комбинаций.
Согласно другому варианту реализации изобретения (Вариант реализации 1.1) предложено соединение формулы (1):

или его соль, таутомер или N-оксид;
где:
один из Y и Z представляет собой R3, а другой представляет собой Ar2;
Q1 представляет собой C1-8 алкиленовую группу, необязательно содержащую один или два заместителя, выбранных из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси- заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода;
Q2 представляет собой связь или C1-8 алкиленовую группу, необязательно содержащую один или два заместителя, выбранных из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода;
R1 выбран из водорода, NRxRy и группы Су1;
Rx и Ry являются одинаковыми или различными и каждый из них выбран из водорода, С1-4 гидрокарбила или гидрокси-С1-4 гидрокарбила; или NRxRy образует 4-7-членное гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а второй выбран из N, О и S и их окисленных форм, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из С1-4 гидрокарбила, оксо, амино, моно-С1-4 гидрокарбиламино, ди-С1-4 гидрокарбиламино, фтора и гидрокси, при условии что между амино, моно-C1-4 гидрокарбиламино, ди-С1-4 гидрокарбиламино и гидрокси-заместителями в случае их наличия и атомом азота в группе NRxRy присутствуют по меньшей мере два атома углерода;
Су1 представляет собой С-связанную 3-7 членную моноциклическую неароматическую карбоциклическую или гетероциклическую группу, содержащую 0, 1 или 2 гетероатомных члена кольца, выбранных из N, О и S и окисленных форм S, причем указанные карбоциклические и гетероциклические группы необязательно содержат один или два заместителя, выбранные из С1-3 гидрокарбила, фтора, оксо и гидрокси;
R2 и R4 являются одинаковыми или различными и каждый из них выбран из водорода, фтора, хлора, С1-2 алкила и С1-2 алкокси, причем каждый С1-2 алкил и С1-2 алкокси необязательно содержит в качестве заместителя один или более атомов фтора;
R3 выбран из водорода, фтора, хлора, С1-2 алкила и С1-2 алкокси, причем каждый C1-2 алкил и С1-2 алкокси необязательно содержит в качестве заместителя один или более атомов фтора;
Ar1 представляет собой моноциклическое 5 или 6-членное арильное или гетероарильное кольцо, содержащее 0, 1 или 2 гетероатомных члена кольца, выбранных из О, N и S, причем арил или гетероарил необязательно содержат 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и выбраны из галогена, циано и группы Ra-Rb;
Ra представляет собой связь, О, СО, Х3С(Х4), С(Х4)Х3, Х3С(Х4)Х3, S, SO, SO2, NRc, SO2NRc или NRcSO2;
Rb представляет собой:
• водород;
• карбоциклическую или гетероциклическую группу, содержащую от 3 до 7 членов кольца, 0, 1, 2 или 3 из которых являются гетероатомными членами кольца, выбранными из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; и
• ациклическую C1-8 углеводородную группу, необязательно содержащую один или более заместителей, выбранных из гидрокси; оксо; галогена; циано; карбокси; амино; моно- или ди-C1-4 алкиламино; и карбоциклических или гетероциклических групп, содержащих от 3 до 7 членов кольца, 0, 1, 2 или 3 из которых являются гетероатомными членами кольца, выбранными из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; при этом один или два, но не все атомы углерода ациклической С1-8 углеводородной группы необязательно могут быть заменены О, S, SO, SO2, NRc, Х3С(Х4), С(Х4)Х3 или Х3С(Х4)Х3;
R6 выбран из заместителей R5, за исключением того, что R6 не состоит из карбоциклической или гетероциклической группы или не содержит указанные группы;
X3 представляет собой О, S или NRc; и
X4 представляет собой =O, =S или =NRc; и
Rc представляет собой водород или С1-4 гидрокарбил;
Ar2 представляет собой бициклическую 8-11-членную гетероарильную группу, содержащую 1, 2, 3 или 4 гетероатомных членов кольца, выбранных из О, N и S, и необязательно содержащую 1, 2 или 3 заместителя R7, выбранных из оксо, фтора; хлора; брома; С1-4 гидрокарбила, необязательно содержащего в качестве заместителей один или более атомов фтора; С1-4 гидрокарбилокси, необязательно содержащего в качестве заместителей один или более атомов фтора; гидрокси; циано; N(Rc)2; Rc-C(O)-; Rc-C(O)N(Rc)-; (Rc)2NC(O)-; Rc-SO2NRc-; Rc-NHC(O)NH-; (Rc)2NSO2-; и пяти- и шестичленных моноциклических групп, содержащих от 0 до 3 гетероатомных членов кольца, выбранных из О, N и S, причем указанные пяти- и шестичленные моноциклические группы, не содержащие заместителей или содержащие один или более заместителей R8, выбранных из С1-4 гидрокарбила, С1-4 гидрокарбилокси, циано, гидрокси, оксо, галогена, амино, моно-С1-4 гидрокарбиламино и ди-С1-4 гидрокарбиламино, и при этом гидрокарбильные остатки в случае их присутствия необязательно содержат в качестве заместителей фтор, С1-2 алкокси, гидрокси, амино, моно-ди-С1-2алкиламино или ди-С1-4 алкиламино;
и при этом в каждом заместителе, состоящем из гидрокарбильной группы или содержащем гидрокарбильную группу, указанная группа выбрана из алкильных, алкенильных, алкинильных и циклоалкильных групп и их комбинаций.
Конкретные и предпочтительные соединения формулы (1) являются такими как определено в Вариантах реализации 1.2-1.92 ниже.
1.2 Соединение согласно Варианту реализации 1.0 или 1.1, где Y представляет собой Ar2 и Z представляет собой R3.
1.3 Соединение согласно Варианту реализации 1.0 или 1.1, где Y представляет собой R3 и Z представляет собой Ar2.
1.4 Соединение согласно Варианту реализации 1.0 или 1.1, имеющее формулу (2):
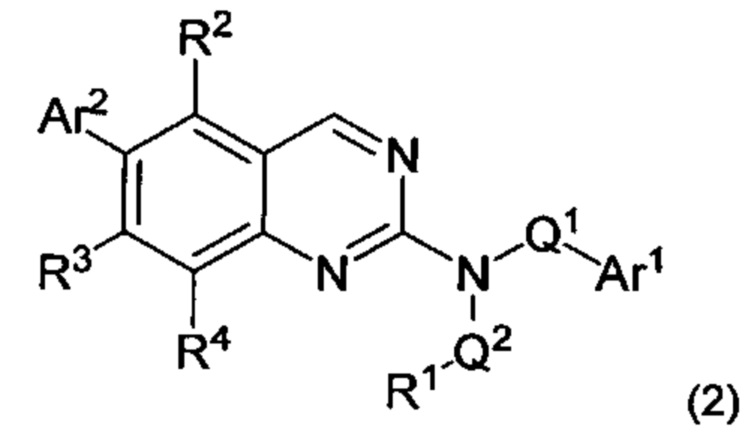
или его соль, таутомер или N-оксид;
причем R1, R2, R3, R4, Ar1, Ar2, Q1 и Q2 все являются такими, как определено в Варианте реализации 1.0 или 1.1.
1.4А Соединение согласно Варианту реализации 1.0, где Q1 представляет собой С1-6 алкиленовую группу, необязательно содержащую один или два заместителя, выбранных из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода; и при этом атом углерода С1-6 алкиленовой группы может быть необязательно заменен цикпопропан-1,1-диильной или циклобутан-1,1-диильной группой при условии, что общее число атомов углерода в алкиленовой группе, содержащей такую замену, не превышает 6.
1.4В Соединение согласно Варианту реализации 1.4А, где Q1 представляет собой С1-5 алкиленовую группу, необязательно содержащую один или два заместителя, выбранные из гидрокси и С1-4 гидрокарбилокси при условии, что в присутствии гидрокси-заместителя между гидрокси- заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода; и при этом атом углерода С1-5 алкиленовой группы может быть необязательно заменен циклопропан-1,1-диильной или циклобутан-1,1-диильной группой при условии, что общее число атомов углерода в алкиленовой группе, содержащей такую замену, не превышает 5.
1.4С Соединение согласно Варианту реализации 1.4В, где Q1 представляет собой С1-4 алкиленовую группу, необязательно содержащую один или два заместителя, выбранные из гидрокси и С1-4 гидрокарбилокси при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода; и при этом атом углерода С1-4 алкиленовой группы может быть необязательно заменен циклопропан-1,1-диильной или циклобутан-1,1-диильной группой при условии, что общее число атомов углерода в алкиленовой группе, содержащей такую замену, не превышает 4.
1.4D Соединение согласно Варианту реализации 1.4С, где Q1 представляет собой циклопропан-1,1-диил.
1.4Е Соединение согласно Варианту реализации 1.4С, где Q1 представляет собой циклобутан-1,1-диил.
1.5 Соединение согласно любому из Вариантов реализации 1.0-1.4, где Q1 представляет собой С1-6 алкилен, необязательно содержащий один или два заместителя, выбранные из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода.
1.6 Соединение согласно Варианту реализации 1.5, где Q1 представляет собой С1-4 алкилен, необязательно содержащий один или два заместителя, выбранные из гидрокси и С1-4 гидрокарбилокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода.
1.7 Соединение согласно Варианту реализации 1.6, где С1-4 алкилен необязательно содержит один гидроксильный заместитель, при условии, что между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода.
1.8 Соединение согласно любому из Вариантов реализации 1.0-1.4, где Q1 представляет собой группу формулы -(CPfRg)р-, где р равен от 1 до 8, каждый Rf независимо выбран из водорода и метила, а каждый Rg независимо выбран из водорода, С1-4 алкила и гидроксил-С1-4 алкила, при условии, что не более, чем один Rg может быть больше метила и при условии, что общее число атомов углерода в -(CRfRg)p- не превышает 8.
1.9 Соединение согласно Варианту реализации 1.8, где общее число атомов углерода в -(CRfRg)p- находится в диапазоне от 1 до 6.
1.10 Соединение согласно Варианту реализации 1.9, где общее число атомов углерода в -(CRfRg)p- находится в диапазоне от 1 до 4.
1.11 Соединение согласно любому из Вариантов реализации 1.8-1.10, где р равен 1 или 2.
1.12 Соединение согласно Варианту реализации 1.11, где p равен 1.
1.13 Соединение согласно любому из Вариантов реализации 1.8-1.12, где Rf представляет собой водород и Rg выбран из водорода, метила, этила, изопропила и гидроксиметила.
1.13А Соединение согласно любому из Вариантов реализации 1.8-1.12, где Rf представляет собой водород и Rg выбран из водорода, метила, этила, изопропила, гидроксиметила и гидроксиэтила.
1.14 Соединение согласно Варианту реализации 1.13, где Q1 выбран из СН2, СН(СН3) и СН(СН2ОН).
1.14А Соединение согласно Варианту реализации 1.13А, где Q1 выбран из СН2, СН(СН3), СН(СН2ОН) и СН(СН2СН2ОН).
1.15 Соединение согласно Варианту реализации 1.13, где Q1 представляет собой СН(СН3).
1.16 Соединение согласно любому из Вариантов реализации 1.0-1.4, где указанная группа -N(Q2-R1)-Q1-Ar1 имеет формулу:
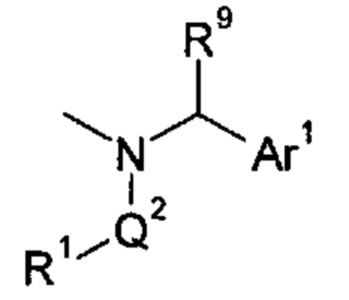
где R9 представляет собой водород или C1-4 алкил, необязательно содержащий гидроксильный заместитель.
1.17 Соединение согласно любому из Вариантов реализации 1.0-1.4, где группа -(Q2-R1)N-Q1-Ar1 имеет формулу:
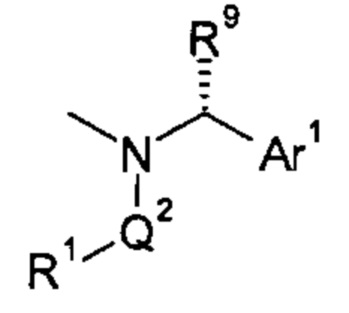
где R9 представляет собой водород или C1-4 алкил, необязательно содержащий гидроксильный заместитель.
1.18 Соединение согласно Варианту реализации 1.16 или 1.17, где R9 представляет собой водород, метил, этил, изопропил или гидроксиметил.
1.18А Соединение согласно Варианту реализации 1.16 или 1.17, где R9 представляет собой водород, метил, этил, изопропил, гидроксиметил или гидроксиэтил.
1.19 Соединение согласно Варианту реализации 1.18, где R9 представляет собой водород, метил или гидроксиметил.
1.19А Соединение согласно Варианту реализации 1.18А, где R9 представляет собой водород, метил, гидроксиметил или гидроксиэтил.
1.20 Соединение согласно Варианту реализации 1.18, где R9 представляет собой водород.
1.21 Соединение согласно Варианту реализации 1.18, где R9 представляет собой метил.
1.22 Соединение согласно Варианту реализации 1.18, где R9 представляет собой гидроксиметил.
1.22А Соединение согласно Варианту реализации 1.18, где R9 представляет собой гидроксиэтил.
1.23 Соединение согласно любому из Вариантов реализации 1.0-1.22, где Q2 представляет собой связь или C1-6алкилен.
1.24 Соединение согласно Варианту реализации 1.23, где Q2 представляет собой связь или C1-3 алкилен.
1.25 Соединение согласно Варианту реализации 1.24, где Q2 выбран из связи, СН2, СН2СН2 и СН2СН2СН2.
1.26 Соединение согласно Варианту реализации 1.25, где Q2 представляет собой связь, СН2, или СН2СН2.
1.27 Соединение согласно Варианту реализации 1.26, где Q2 представляет собой связь.
1.28 Соединение согласно Варианту реализации 1.26, где Q2 представляет собой СН2.
1.29 Соединение согласно любому из Вариантов реализации 1.0-1.28, где R1 выбран из водорода и группы Су1.
1.30 Соединение согласно Варианту реализации 1.29, где R1 представляет собой водород.
1.31 Соединение согласно Варианту реализации 1.29, где R1 представляет собой группу Су1.
1.32 Соединение согласно любому из Вариантов реализации 1.0-1.29 и 1.31, где Су1 выбран из С3-7 циклоалкила и С-связанных 4-7 членных неароматических гетероциклических групп, содержащих 1 или 2 гетероатомных члена кольца, выбранных из N, О и S, причем указанные циклоалкильные и гетероциклические группы необязательно содержат один или два заместителя, выбранных из С1-3 гидрокарбила, фтора, оксо и гидрокси.
1.33 Соединение согласно Варианту реализации 1.32, где Су1 выбран из С3-6 циклоалкила и С-связанных 4-6 членных неароматических гетероциклических групп, содержащих 1 или 2 гетероатомных члена кольца, выбранных из О и S, причем указанные циклоалкильные и гетероциклические группы необязательно содержат один или два заместителя, выбранных из С1-3 гидрокарбила, фтора, оксо и гидрокси.
1.34 Соединение согласно Варианту реализации 1.33, где Су1 выбран из С3-6 циклоалкила и С-связанных 4-6 членных насыщенных неароматических гетероциклических групп, содержащих 1 гетероатомный член кольца, выбранный из О и S, причем указанные циклоалкильные и гетероциклические группы необязательно содержат один или два заместителя, выбранных из насыщенного С1-3 гидрокарбила, фтора, оксо и гидрокси.
1.35 Соединение согласно Варианту реализации 1.33, где Су1 выбран из С3-6 циклоалкила и С-связанных 4-6 членных насыщенных неароматических гетероциклических групп, содержащих 1 гетероатомный член кольца, выбранный из О, причем указанные циклоалкильные и гетероциклические группы необязательно содержат один или два заместителя, выбранных из С1-3 алкила, фтора, оксо и гидрокси.
1.36 Соединение согласно Варианту реализации 1.33, где Су1 выбран из С3-6 циклоалкила и С-связанных 4-6 членных насыщенных неароматических гетероциклических групп, содержащих 1 гетероатомный член кольца, выбранный из О, причем указанные циклоалкильные и гетероциклические группы являются незамещенными или содержат один или два заместителя, выбранных из метила, фтора, оксо и гидрокси.
1.37 Соединение согласно Варианту реализации 1.33, где Су1 выбран из циклопропила, циклобутила, циклопентила, циклогексила, тетрагидропирана и тетрагидрофурана.
1.38 Соединение согласно Варианту реализации 1.33, где Су1 представляет собой тетрагидропиран.
1.39 Соединение согласно Варианту реализации 1.32, где Су1 выбран из С-связанных 4-7 членных неароматических гетероциклических групп, содержащих первый член кольца, представляющий собой азот, и необязательно второй член кольца, выбранный из N, О и S, причем указанные гетероциклические группы необязательно содержат один или два заместителя, выбранных из C1-3 гидрокарбила, фтора, оксо и гидрокси.
1.40 Соединение согласно Варианту реализации 1.39, где Су1 выбран из С-связанных 4-7 членных насыщенных гетероциклических групп, содержащих первый член кольца, представляющий собой азот, и необязательно второй член кольца, выбранный из N, О и S, причем указанные гетероциклические группы необязательно содержат один или два заместителя, выбранных из насыщенного C1-3 гидрокарбила, фтора, оксо и гидрокси.
1.41 Соединение согласно Варианту реализации 1.40, где Су1 выбран из С-связанных 4-7 членных насыщенных гетероциклических групп, содержащих первый член кольца, представляющий собой азот, и необязательно второй член кольца, выбранный из N, О и S, причем указанные гетероциклические группы необязательно содержат один или два заместителя, выбранные из C1-3 алкила, циклопропила, фтора и гидрокси.
1.42 Соединение согласно Варианту реализации 1.41, где Су1 выбран из С-связанного азетидин, пирролидин, пиперидин, пиперазин, морфолин, гомоморфолин и тиоморфолин, каждый из которых необязательно содержит один или два заместителя, выбранных из C1-3 алкила, циклопропила, фтора и гидрокси.
1.43 Соединение согласно Варианту реализации 1.41, где Су1 представляет собой С-связанный морфолин.
1.44 Соединение согласно любому из Вариантов реализации 1.0-1.28, где R1 представляет собой NRxRy.
1.45 Соединение согласно любому из Вариантов реализации 1.0 to 1.28 и 1.44, где Rx и Ry являются одинаковыми или различными, и каждый из них выбран из водорода, С1-4 гидрокарбила или гидрокси-С1-4 гидрокарбила.
1.46 Соединение согласно Варианту реализации 1.45, где Rx и Ry являются одинаковыми или различными, и каждый из них выбран из водорода, насыщенного С1-4 гидрокарбила или насыщенного гидрокси-С1-4 гидрокарбила.
1.47 Соединение согласно Варианту реализации 1.46, где Rx и Ry являются одинаковыми или различными, и каждый из них выбран из водорода, С1-4 алкила, циклопропила, метилциклопропила, циклопропилметила и гидрокси-С2-4 алкила.
1.48 Соединение согласно Варианту реализации 1.47, где Rx и Ry являются одинаковыми или различными, и каждый из них выбран из водорода и С1-4 алкила.
1.49 Соединение согласно Варианту реализации 1.48, где Rx и Ry являются одинаковыми или различными, и каждый из них выбран из водорода и С1-3 алкила.
1.50 Соединение согласно Варианту реализации 1.49, где NRxRy выбран из амино, метиламино и диметиламино.
1.51 Соединение согласно Варианту реализации 1.49, где NRxRy представляет собой диметиламино.
1.52 Соединение согласно любому из Вариантов реализации 1.0-1.28 и 1.44, где NRxRy образует 4-7-членное гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а другой выбран из N, О и S и их окисленных форм, причем указанное гетероциклическое кольцо необязательно содержит один или два заместителя, выбранных из С1-4 гидрокарбила, оксо, амино, моно-С1-4 гидрокарбиламино, ди-С1-4гидрокарбиламино, фтора и гидрокси, при условии, что между амино, моно-С1-4 гидрокарбиламино, ди-С1-4гидрокарбиламино и гидрокси заместителями в случае их присутствия и атомом азота группы NRxRy присутствуют по меньшей мере два последовательно расположенных атома углерода.
1.53 Соединение согласно Варианту реализации 1.52, где NRxRy образует 4-7-членное неароматическое гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а другой выбран из N, О и S, причем указанное гетероциклическое кольцо необязательно содержит один или два заместителя, выбранных из С1-4 гидрокарбила, оксо, амино, насыщенного моно-С1-4 гидрокарбиламино, насыщенного ди-С1-4гидрокарбиламино, фтора и гидрокси, при условии, что между амино, насыщенным моно-С1-4 гидрокарбиламино, насыщенным ди-C1-4гидрокарбиламино и гидрокси заместителями в случае их присутствия и атомом азота группы NRxRy присутствуют по меньшей мере два последовательно расположенных атома углерода.
1.54 Соединение согласно Варианту реализации 1.53, где NRxRy образует насыщенное 4-7-членное гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а другой выбран из N, О и S, причем указанное гетероциклическое кольцо необязательно содержит один или два заместителя, выбранных из С1-4 алкила, фтора, оксо, амино, моно-С1-4 алкиламино, ди-С1-4алкиламино и гидрокси.
1.55 Соединение согласно Варианту реализации 1.54, где NRxRy образует насыщенное 4-7-членное гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а другой выбран из N, О и S, причем указанное гетероциклическое кольцо необязательно содержит один или два заместителя, выбранных из C1-3 алкила, фтора, оксо, амино, моно-C1-2 алкиламино, ди-C1-2алкиламино и гидрокси.
1.56 Соединение согласно Варианту реализации 1.55, где NRxRy образует насыщенное 4-7-членное гетероциклическое кольцо, содержащее в общей сложности 1 или 2 гетероатомных члена кольца, один из которых представляет собой N, а другой выбран из N, О и S, причем указанное гетероциклическое кольцо необязательно содержит один или два заместителя, выбранных из метила, фтора, оксо, амино, метиламино, диметиламино и гидрокси.
1.57 Соединение согласно Варианту реализации 1.55, где NRxRy образует гетероциклическое кольцо, выбранное из азетидина, пирролидина, пиперидина, пиперазина, морфолина, гомоморфолина и тиоморфолина, каждый из которых необязательно содержит один или два заместителя, выбранные из C1-3 алкила, фтора и гидрокси.
1.57 Соединение согласно Варианту реализации 1.55, где NRxRy образует гетероциклическое кольцо, выбранное из азетидина, пирролидина, пиперидина, пиперазина, морфолина, гомоморфолина и тиоморфолина, каждый из которых необязательно содержит один или два заместителя, выбранные из C1-3 алкила, фтора и гидрокси.
1.57А Соединение согласно любому из Вариантов реализации 1.0-1.28, где R1 выбран из:
• водорода;
• группы Су1, где Су1 выбран из 4-7 членных насыщенных гетероциклических групп, содержащих первый член кольца, представляющий собой азот, и необязательно второй член кольца, выбранный из N, О и S, причем указанные гетероциклические группы необязательно содержат один или два заместителя, выбранные из С1-3 алкила, циклопропила, фтора и гидроксила; и
• NRxRy, где Rx и Ry являются одинаковыми или различными и каждый из них выбран из водорода, С1-4 алкила, циклопропила, метилциклопропила, циклопропилметила, и гидрокси-С2-4 алкила.
1.58 Соединение согласно любому из Вариантов реализации 1.0-1.57А, где R2 выбран из водорода, фтора, хлора, метила, метокси, трифторметила и трифторметокси.
1.59 Соединение согласно Варианту реализации 1.58, где R2 представляет собой водород.
1.60 Соединение согласно любому из Вариантов реализации 1.0-1.59, где R3 выбран из водорода, фтора, хлора, метила, метокси, трифторметила и трифторметокси.
1.61 Соединение согласно Варианту реализации 1.60, где R3 представляет собой водород.
1.62 Соединение согласно любому из Вариантов реализации 1.0-1.61, где R4 выбран из водорода, фтора, хлора, метила, метокси, трифторметила и трифторметокси.
1.63 Соединение согласно Варианту реализации 1.62, где R4 представляет собой водород.
1.64 Соединение согласно любому из Вариантов реализации 1.0-1.63, где Ar1 представляет собой моноциклическое арильное или гетероарильное кольцо, выбранное из фенила, фурила, тиенила, пиридила и пиримидинила, каждый из которых содержит 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и являются такими, как определено в Варианте реализации 1.1.
1.65 Соединение согласно Варианту реализации 1.64, где Ar1 представляет собой моноциклическое арильное или гетероарильное кольцо, выбранное из фенила, фурила, тиенила и пиридила, каждый из которых содержит 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и являются такими, как определено в Варианте реализации 1.1.
1.66 Соединение согласно Варианту реализации 1.65, где Ar1 представляет собой фенильное кольцо, необязательно содержащее 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и являются такими, как определено в Варианте реализации 1.1.
1.67 Соединение согласно любому из Вариантов реализации 1.0-1.66, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и выбраны из фтора, хлора, брома, циано и группы Ra-Rb;
Ra представляет собой связь, О, СО, NRcC(=O), C(=O)NRc, NRcC(=O)NRc, С(=O) О, ОС(=O), S, SO, SO2, NRc, SO2NRc или NRcSO2;
Rb представляет собой:
• водород;
• карбоциклическую или гетероциклическую группу, содержащую от 3 до 7 членов кольца, 0, 1, 2 или 3 из которых представляют собой гетероатомные члены кольца, выбранные из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; и
• ациклическую С1-8 углеводородную группу, необязательно содержащую один или более заместителей, выбранных из гидрокси; оксо; галогена; циано; амино; моно- или ди-С1-4 алкиламино; и карбоциклических и гетероциклических групп, содержащих от 3 до 7 членов кольца, 0, 1, 2 или 3 из которых являются гетероатомными членами кольца, выбранными из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; причем один или два, но не все атомы углерода ациклической C1-8 углеводородной группы могут необязательно быть заменены О, S, SO, SO2 или NRc;
R6 выбран из заместителей R5, за исключением того, что R6 не состоит из карбоциклической или гетероциклической группы или не содержит карбоциклическую или гетероциклическую группу; и
Rc представляет собой водород или С1-4 гидрокарбил.
1.68 Соединение согласно Варианту реализации 1.67, отличающееся тем, что Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и выбраны из фтора, хлора, брома, циано и группы Ra-Rb;
Ra представляет собой связь, О, СО, NRcC(=O), C(=O)NRc, NRcC(=O)NRc, С(=O) О, ОС(=O), S, SO, SO2, NRc, SO2NRc или NRcSO2;
Rb представляет собой:
• водород;
• неароматическую карбоциклическую или гетероциклическую группу, содержащую от 3 до 6 членов в кольце, 0, 1 или 2 из которых являются гетероатомными членами кольца, выбранными из О, N, S и SO2, причем указанная неароматическая карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; и
• ациклическую С1-8 углеводородную группу, необязательно содержащую один или более заместителей, выбранных из гидрокси; оксо; галогена; циано; амино; моно- или ди-С1-4 алкиламино; и неароматических карбоциклических и гетероциклических групп, содержащих от 3 до 6 членов кольца, 0, 1 или 2 из которых являются гетероатомными членами кольца, выбранными из О, N S и SO2, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; причем один или два, но не все атомы углерода ациклической С1-8 углеводородной группы могут необязательно быть заменены О, S, SO, SO2 или NRc;
R6 выбран из заместителей R5 за исключением того, что R6 не состоит из карбоциклической или гетероциклической группы или не содержит указанные группы; и Rc представляет собой водород или С1-4 гидрокарбил.
1.69 Соединение согласно Варианту реализации 1.68, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, которые являются одинаковыми или различными и выбраны из фтора, хлора, брома, циано и группы Ra-Rb;
Ra представляет собой связь, О, СО, NRcC(=O), C(=O)NRc, SO2, NRc, SO2NRc или NRcSO2;
Rb представляет собой:
• водород;
• неароматическую карбоциклическую или гетероциклическую группу, содержащую от 3 до 6 членов кольца, 0, 1 или 2 из которых являются гетероатомными членами кольца, выбранными из О, N и S, причем неароматическая карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; и
• насыщенная ациклическая С1-8 углеводородная группа, необязательно содержащая один или более заместителей, выбранных из гидрокси; оксо; фтора; циано; амино; моно- или ди-С1-2 алкиламино; и неароматических карбоциклических и гетероциклических групп, содержащих от 3 до 6 членов кольца, 0,1 или 2 из которых являются гетероатомными членами кольца, выбранными из О, N и S и окисленных форм S, причем карбоциклическая или гетероциклическая группа необязательно содержит один или более заместителей R6; причем один или два, но не все атомы углерода ациклической С1-8 углеводородной группы могут необязательно быть заменены О или NRc;
R6 выбран из заместителей R5, за исключением того, что R6 не состоит из карбоциклической или гетероциклической группы или не содержит указанные группы; и
Rc представляет собой водород, С1-4 алкил, циклопропил или циклопропилметил.
1.70 Соединение согласно любому из Вариантов реализации 1.0-1.66, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из группы L1-Су2; фтора; хлора; брома; C1-3 алкила; C1-3 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-3 алкиламино; ди-С1-3 алкиламино; С1-3 алканоила; С1-3 алканоиламино; карбамоила; моно-C1-3 алкил карбамоила; ди-С1-3 алкил карбамоила; группы O-(CH2)k-OR10; и группы Om-(CH2)n-NR11R12; R10 представляет собой водород или С1-3 алкил; R11 представляет собой водород или С1-3 алкил; R12 представляет собой водород или С1-3 алкил; k равен 2, 3 или 4; m равен 0 или 1; и n равен 1, 2, 3 или 4, при условии, что когда m равен 1, то n равен 2, 3 или 4; L1 представляет собой связь или линкерную группу, выбранную из С1-4 алкилена, -(CH2)p-NH-(CH2)q-, -(CH2)p-N(CH3)-(CH2)q-, -(CH2)p-C(=O)-(CH2)q-, -(CH2)p-C(=O)NH -(CH2)q-, -(CH2)p-C(=O)N(CH3) -(CH2)q-, -(CH2)p-NHC(=O) -(CH2)q- и -(CH2)p-N(CH3)C(=O) -(CH2)q-; каждый из p и q независимо равен 0, 1, 2 или 3 при условии, что общее число р и q не превышает 4; и Су2 представляет собой неароматическое карбоциклическое или гетероциклическое кольцо из от трех до семи членов кольца, содержащее 0, 1 или 2 гетероатомных члена кольца, выбранные из О, N и S, и необязательно содержащее один, два или три заместителя, выбранных из гидрокси, С1-4 гидрокарбила, С1-4 гидрокарбил-С(=O), оксо, амино, моно-С1-4 гидрокарбиламино, ди-С1-4 гидрокарбиламино и фтора.
1.70А Соединение согласно любому из Вариантов реализации 1.0-1.66, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранных из группы L1-Су2; фтора; хлора; брома; С1-3 алкила; С1-3 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-3 алкиламино; ди-С1-3 алкиламино; С1-3 алканоила; С1-3 алкилсульфониламино; С1-3 алканоиламино; карбамоил; моно-C1-3 алкил карбамоила; ди-С1-3 алкил карбамоила; группы O-(CH2)k-OR10; и группы Om-(CH2)n-NR11R12; R10 представляет собой водород или С1-3 алкил; R11 представляет собой водород или С1-3 алкил; R12 представляет собой водород или С1-3 алкил; k равен 2, 3 или 4; m равен 0 или 1; и n равен 1, 2, 3 или 4 при условии, что когда m равен 1, то n равен 2, 3 или 4; L1 представляет собой связь или линкерную группу, выбранную из С1-4 алкилена, -(CH2)p-NH-(CH2)q-, -(CH2)p-N(CH3)-(CH2)q-, -(CH2)p-C(=O)-(CH2)q-, -(CH2)p-C(=O)NH -(CH2)q-, -(CH2)p-C(=O)N(CH3) -(CH2)q-, -(CH2)p-NHC(=O) -(CH2)q- и -(CH2)p-N(CH3)C(=O) -(CH2)q-; каждый из p и q независимо равен 0, 1, 2 или 3 при условии, что общее число р и q не превышает 4; и Су2 представляет собой неароматическое карбоциклическое или гетероциклическое кольцо из от трех до семи членов кольца, содержащее 0, 1 или 2 гетероатомных члена кольца, выбранные из О, N и S, и необязательно содержащее один, два или три заместителя, выбранные из гидрокси, С1-4 гидрокарбила, С1-4 гидрокарбил-С(=O), оксо, амино, моно-С1-4 гидрокарбиламино, ди-С1-4 гидрокарбиламино и фтора.
1.71 Соединение согласно Варианту реализации 1.70, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранных из группы L1-Су2; фтора; хлора; брома; С1-3 алкила; С1-3 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-3 алкиламино; ди-С1-3 алкиламино; С1-3 алканоила; С1-3 алканоиламино; карбамоила; моно-C1-3 алкил карбамоила; ди-С1-3 алкил карбамоила; группы O-(CH2)k-OR10; и группы Om-(CH2)n-NR11R12; R10 представляет собой водород, метил или этил; R11 представляет собой водород, метил или этил; R12 представляет собой водород, метил; или этил; k равен 2 или 3; m равен 0 или 1; и n равен 1, 2 или 3 при условии, что когда m равен 1, то n равен 2 или 3; L1 представляет собой связь или линкерную группу, выбранную из С1-4 алкилена, -(CH2)p-NH-(CH2)q-, -(CH2)p-N(CH3)-(CH2)q-, -(CH2)p-C(=O)-(CH2)q-, -(CH2)p-C(=O)NH-(CH2)q-, -(CH2)p-C(=O)N(CH3) -(CH2)q-, -(CH2)p-NHC(=O) -(CH2)q- и -(CH2)p-N(CH3)C(=O) -(CH2)q-; каждый из p и q независимо равен 0, 1 или 2; и Су2 представляет собой неароматическое карбоциклическое кольцо из от трех до шести членов кольца или гетероциклическое кольцо из от пяти до шести членов кольца, содержащее 1 или 2 гетероатомных члена кольца, выбранные из О, N и S, причем каждое из карбоциклического и гетероциклического колец необязательно содержит один, два или три заместителя, выбранные из гидрокси, С1-4 алкила, циклопропила, циклопропилметила, С1-4 алканоила, циклопропилкарбонила, оксо и фтора.
1.71 А Соединение согласно Варианту реализации 1.70А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранных из группы L1-Су2; фтора; хлора; брома; С1-3 алкила; С1-3 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-3 алкиламино; ди-С1-3 алкиламино; С1-3 алканоила; C1-3 алкилсульфониламино; С1-3 алканоиламино; карбамоила; моно-C1-3 алкил карбамоила; ди-С1-3 алкил карбамоила; группы O-(CH2)k-OR10; и группы Om-(CH2)n-NR11R12; R10 представляет собой водород, метил или этил; R11 представляет собой водород, метил или этил; R12 представляет собой водород, метил, или этил; k равен 2 или 3; m равен 0 или 1; и n равен 1, 2 или 3, при условии, что когда m равен 1, то n равен 2 или 3; L1 представляет собой связь или линкерную группу, выбранную из С1-4 алкилена, -(CH2)p-NH-(CH2)q-, -(CH2)p-N(CH3HCH2)q-, -(CH2)p-C(=O)-(CH2)q-, -(CH2)p-C(=O)NH -(CH2)q-, -(CH2)p-C(=O)N(CH3) -(CH2)q-, -(CH2)p-NHC(=O) -(CH2)q- и -(CH2)p-N(CH3)C(=O) -(CH2)q-; каждый из p и q независимо равен 0, 1 или 2; и Су2 представляет собой неароматическое карбоциклическое кольцо, содержащее от трех до шести членов кольца, или гетероциклическое кольцо, содержащее пять или шесть членов кольца, содержащее 1 или 2 гетероатомных члена кольца, выбранных из О, N и S, причем каждое из карбоциклического и гетероциклического колец необязательно содержит один, два или три заместителя, выбранных из гидрокси, C1-4 алкила, циклопропила, циклопропилметила, С1-4 алканоила, циклопропилкарбонила, оксо и фтора.
1.72 Соединение согласно Варианту реализации 1.71, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранных из группы L1-Су2; фтора; хлора; брома; C1-3 алкила; C1-3 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-2 алкиламино; ди-С1-2 алкиламино; С1-3 алканоила; С2-3 алканоиламино; карбамоила; моно-C1-3 алкил карбамоила; ди-С1-3 алкил карбамоила; группы O-(CH2)k-OR10; и группы Om-(CH2)n-NR11R12; R10 представляет собой водород, метил или этил; R11 представляет собой водород, метил или этил; R12 представляет собой водород, метил; или этил; k равен 2 или 3; m равен 0 или 1; и n равен 1, 2 или 3, при условии, что когда m равен 1, то n равен 2 или 3; L1 представляет собой связь или линкерную группу, выбранную из С1-4 алкилена, -(CH2)p-NH-(CH2)q-, -(CH2)p-N(CH3)-(CH2)q-, -(CH2)p-C(=O)-(CH2)q-, -(CH2)p-C(=O)NH -(CH2)q-, -(CH2)p-C(=O)N(CH3) -(CH2)q-, -(CH2)p-NHC(=O) -(CH2)q- и -(CH2)p-N(CH3)C(=O) -(CH2)q-; каждый из p и q необязательно равен 0 или 1; и Су2 представляет собой неароматическое гетероциклическое кольцо, выбранное из азетидина, пирролидина, пиперидина, пиперазина, морфолина, тиоморфолина, тетрагидрофурана и тетрагидропирана, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из гидрокси, С1-4 алкила, циклопропила, циклопропилметила, С1-4 алканоила, циклопропилкарбонила, оксо и фтора.
1.72А Соединение согласно Варианту реализации 1.71 А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из группы L1-Су2; фтора; хлора; брома; С1-3 алкила; С1-3 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-2 алкиламино; ди-С1-2 алкиламино; С1-3 алканоила; C1-2 алкилсульфониламино; С2-3 алканоиламино; карбамоила; моно-C1-3 алкил карбамоила; ди-С1-3 алкил карбамоила; группы O-(CH2)k-OR10; и группы Om-(CH2)n-NR11R12; R10 представляет собой водород, метил или этил; R11 представляет собой водород, метил или этил; R12 представляет собой водород, метил; или этил; k равен 2 или 3; m равен 0 или 1; и n равен 1, 2 или 3, при условии, что когда m равен 1, то n равен 2 или 3; L1 представляет собой связь или линкерную группу, выбранную из С1-4 алкилена, -(CH2)p-NH-(CH2)q-, -(CH2)p-N(CH3)-(CH2)q-, -(CH2)p-C(=O)-(CH2)q-, -(CH2)p-C(=O)NH -(CH2)q-, -(CH2)p-C(=O)N(CH3) -(CH2)q-, -(CH2)p-NHC(=O) -(CH2)q- и -(CH2)p-N(CH3)C(=O) -(CH2)q-; каждый из p и q независимо равен 0 или 1; и Су2 представляет собой неароматическое гетероциклическое кольцо, выбранное из азетидина, пирролидина, пиперидина, пиперазина, морфолина, тиоморфолина, тетрагидрофурана и тетрагидропирана, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из гидрокси, С1-4 алкила, циклопропила, циклопропилметила, С1-4 алканоила, циклопропилкарбонила, оксо и фтора.
1.73 Соединение согласно Варианту реализации 1.72, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из группы L1-Су2; фтора; хлора; брома; C1-2 алкила; С1-2 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; амино; моно-C1-2 алкиламино; ди-С1-2 алкиламино; ацетила; ацетиламино; карбамоила; моно-C1-2 алкил карбамоила; ди-С1-2 алкил карбамоила; диметиламиноэтокси; где L1 выбран из связи, О, NH, N(СН3), NHC(=O), C(=O)NH, N(СН3)С(=O) и C(=O)N(CH3); и Су2 выбран из пиперидина, пиперазина, морфолина и тетрагидропирана, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из гидрокси, метила и оксо.
1.73А Соединение согласно Варианту реализации 1.72А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из группы L1-Cy2; фтора; хлора; брома; С1-2 алкила; С1-2 алкокси; трифторметила; дифторметила; гидрокси; циано; трифторметокси; дифторметокси; С1-2 алкилсульфониламино; амино; моно-C1-2 алкиламино; ди-С1-2 алкиламино; ацетила; ацетиламино; карбамоила; моно-C1-2 алкил карбамоила; ди-С1-2 алкил карбамоила; диметиламиноэтокси; где L1 выбран из связи, О, NH, N(CH3), NHC(=O), C(=O)NH, N(CH3)C(=O) и C(=O)N(CH3); и Су2 выбран из пиперидина, пиперазина, морфолина и тетрагидропирана, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из гидрокси, метила и оксо.
1.74 Соединение согласно Варианту реализации 1.73, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, брома, метила, гидрокси, метокси, трифторметила, дифторметила, циано, трифторметокси, дифторметокси, морфолинила, пиперазинила, N-метилпиперазинила и диметиламиноэтокси.
1.74А Соединение согласно Варианту реализации 1.73А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, брома, метила, гидрокси, метокси, трифторметила, дифторметила, циано, трифторметокси, дифторметокси, метилсульфониламино, морфолинила, пиперазинила, N-метилпиперазинила и диметиламиноэтокси.
1.75 Соединение согласно любому из Вариантов реализации 1.0-1.66, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из L1-Cy2; фтора, хлора, метила, гидрокси, метокси, трифторметила, дифторметила, трифторметокси и диметиламиноэтокси; где L1 выбран из связи, О, NH, NHC(=O), C(=O)NH и С(=O)N(СН3); и Су2 выбран из пиперидина, пиперазина, морфолина и тетрагидропирана, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из метила и оксо.
1.75А Соединение согласно любому из Вариантов реализации 1.0-1.66, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из L1-Су2; фтора, хлора, метила, гидрокси, метокси, трифторметила, дифторметила, трифторметокси, метилсульфониламино и диметиламиноэтокси; где L1 выбран из связи, О, NH, NHC(=O), C(=O)NH и С(=O)N(СН3); и Су2 выбран из пиперидина, пиперазина, морфолина и тетрагидропирана, причем гетероциклическое кольцо необязательно содержит один или два заместителя, выбранные из метила и оксо.
1.76 Соединение согласно Варианту реализации 1.75, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, брома, метила, метокси, трифторметила, дифторметила, циано, трифторметокси и дифторметокси.
1.76А Соединение согласно Варианту реализации 1.75А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, брома, метила, гидроксила, метокси, метилсульфониламино, трифторметила, дифторметила, циано, трифторметокси и дифторметокси.
1.77 Соединение согласно Варианту реализации 1.76, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, метила, метокси, трифторметила и трифторметокси.
1.77А Соединение согласно Варианту реализации 1.76А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, метила, гидроксила, метокси, метилсульфониламино, трифторметила и трифторметокси.
1.78 Соединение согласно Варианту реализации 1.77, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, метила и метокси.
1.78А Соединение согласно Варианту реализации 1.77А, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора, метилсульфониламино, метила и метокси.
1.79 Соединение согласно Варианту реализации 1.78, где Ar1 является незамещенным или содержит 1, 2 или 3 заместителя R5, выбранные из фтора, хлора и метокси.
1.80 Соединение согласно любому из Вариантов реализации 1.0-1.79, где Ar1 является незамещенным или содержит один или два заместителя R5.
1.81 Соединение согласно Варианту реализации 1.80, где Ar1 не содержит заместителей или содержит один заместитель R5.
1.82 Соединение согласно Варианту реализации 1.81, где Ar1 не содержит заместителей, причем Ar1 не содержит заместителей.
1.83 Соединение согласно Варианту реализации 1.81, где Ar1 содержит один заместитель R5.
1.84 Соединение согласно Варианту реализации 1.80, где Ar1 представляет собой фенильное кольцо, которое является незамещенным или содержит один или два заместителя R5, где по меньшей мере один заместитель R5 расположен в мета- или пара-положении фенильного кольца.
1.85 Соединение согласно Варианту реализации 1.84, где Ar1 представляет собой фенильное кольцо, содержащее один заместитель R5, расположенный в мета-положении фенильного кольца.
1.86 Соединение согласно Варианту реализации 1.84, где Ar1 представляет собой фенильное кольцо, содержащее один заместитель R5, расположенный в пара-положении фенильного кольца.
1.87 Соединение согласно Варианту реализации 1.84, где Ar1 представляет собой фенильное кольцо, которое является незамещенным или содержит один заместитель R5, выбранный из 3-хлор-, 4-хлор-, 3-фтор-, 4-фтор-, 3-метокси-, 4-метокси-, 3-метил- и 4-метил-.
1.88 Соединение согласно Варианту реализации 1.87, где Ar1 представляет собой фенильное кольцо, которое является незамещенным или содержит один заместитель R5, выбранный из 3-хлор-, 3-фтор-, 4-фтор- и 3-метокси.
1.89 Соединение согласно Варианту реализации 1.84, где Ar1 представляет собой фенильное кольцо, которое содержит два заместителя R5.
1.90 Соединение согласно Варианту реализации 1.89, где Ar1 представляет собой фенильное кольцо, которое содержит два заместителя R5, причем один заместитель расположен в пара-положении фенильного кольца, а второй расположен в мета-положении фенильного кольца.
1.91 Соединение согласно Варианту реализации 1.90, отличающееся тем, что Ar1 представляет собой 3,4-дифторфенил.
1.91А Соединение согласно Варианту реализации 1.82, отличающееся тем, что Ar1 представляет собой незамещенный фенил.
1.91В Соединение согласно Варианту реализации 1.66, где Ar1 представляет собой незамещенный фенил или фенил, содержащий один или два заместителя, выбранные из фтора, хлора, метокси, метилсульфониламино и гидроксила.
1.91С Соединение согласно Варианту реализации 1.66, где Ar1 представляет собой незамещенный фенил или фенил, содержащий один или два заместителя, выбранные из фтора, хлора, метокси и метилсульфониламино.
1.91D Соединение согласно Варианту реализации 1.66, где Ar1 выбран из незамещенного фенила, 3-хлорфенила, 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 2,6-дифторфенила, 3,4-дифторфенила, 3-метоксифенила, 3-метилсульфониламинофенила, 2-гидроксифенила, 3-гидроксифенила и 4-гидроксифенила.
1.91Е Соединение согласно Варианту реализации 1.66, где Ar1 выбран из незамещенного фенила, 3-хлорфенила, 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 2,6-дифторфенила, 3,4-дифторфенила, 3-метоксифенила, 3-метилсульфониламинофенила, 2-гидроксифенила, 3-гидроксифенила и 4-гидроксифенила.
1.92 Соединение согласно любому из Вариантов реализации 1.0-1.91, где Ar2 выбран из 5.6 конденсированных гетероароматических колец и 6.6 конденсированных гетероароматических колец, каждое из которых содержит 1, 2, 3 или 4 гетероатомных члена кольца, выбранных из N, О и S, и необязательно содержит 1, 2, или 3 заместителя R7, определенных в Варианте реализации 1.1.
1.93 Соединение согласно Варианту реализации 1.92, где каждое из 5.6 сконденсированных гетероароматических колец и 6.6 сконденсированных гетероароматических колец содержит 1, 2, 3 или 4 атома азота в качестве гетероатомных членов кольца.
1.94 Соединение согласно Варианту реализации 1.92 или 1.93, где Ar2 выбран из 5.6 сконденсированных гетероароматических колец, каждое из которых необязательно содержит 1, 2 или 3 заместителя R7, определенных в Варианте реализации 1.1.
1.95 Соединение согласно Варианту реализации 1.94, где Ar2 выбран из пиримидо-имидазольных, пиридо-имидазольных, пиримидо-пиррольных, пиридо-пиррольных, бензо-имидазольных, бензо-пиррольных, пиримидо-пиразольных, пиридо-пиразольных и бензо-пиразольных групп, каждая из которых необязательно содержит 1, 2 или 3 заместителя R7, определенных в Варианте реализации 1.1.
1.95А Соединение согласно Варианту реализации 1.95, где Ar2 выбран из пиримидо-пиррольных, пиридо-пиррольных, пиримидо-пиразольных, пиридо-пиразольных групп и пиримидо-имидазольных групп, каждая из которых необязательно содержит 1, 2 или 3 заместителя R7, определенных в Варианте реализации 1.1.
1.96 Соединение согласно Варианту реализации 1.95, где Ar2 выбран из пиримидо-пиррольных, пиридо-пиррольных, пиримидо-пиразольных и пиридо-пиразольных групп, каждая из которых необязательно содержит 1, 2 или 3 заместителя R7, определенных в Варианте реализации 1.1.
1.97 Соединение согласно Варианту реализации 1.96, где Ar2 представляет собой пиримидо-пиразольную группу, которая необязательно содержит 1, 2 или 3 заместителя R7, определенных в Варианте реализации 1.1.
1.98 Соединение, определенное согласно любому из Вариантов реализации 1.0-1.97, где Ar2 является незамещенным или содержит один или два заместителя R7, выбранные из оксо, фтора; хлора; брома; С1-4 гидрокарбила, необязательно содержащего в качестве заместителей один или более атомов фтора; С1-4 гидрокарбилокси, необязательно содержащего в качестве заместителей один или более атомов фтора; гидрокси; циано; и пяти- или шести-членных моноциклических групп, содержащих от 0 до 3 гетероатомных членов кольца, выбранных из О, N и S, причем указанные пяти- и шести-членные моноциклические группы являются незамещенными или содержат один или более заместителей R8, выбранных из С1-4 гидрокарбила, С1-4 гидрокарбилокси, циано, гидрокси, оксо, галогена, амино, моно-С1-4 гидрокарбиламино и ди-С1-4гидрокарбиламино.
1.99 Соединение, определенное согласно Варианту реализации 1.98, где Ar2 является незамещенным или содержит один или два заместителя R7, выбранных из оксо, фтора; хлора; брома; С1-4 алкила, необязательно содержащего в качестве заместителей один или более атомов фтора; С1-4 алкокси, необязательно содержащего в качестве заместителей один или более атомов фтора; гидрокси; циано; и пяти- или шести-членных моноциклических групп, выбранных из фенила, пиридила, пиримидинила, пиридазинила, пиразинила, пиррола, пиразола, имидазола, тиофена, фурана, оксазола и изоксазола, причем каждая из указанных пяти- или шести-членных моноциклических групп является незамещенной или содержит один или более заместителей R8, выбранных из С1-4 алкила, С1-4 алкокси, циано, гидрокси, фтора, хлора, амино, метиламино и диметиламино.
1.100 Соединение, определенное согласно Варианту реализации 1.99, где Ar2 является незамещенным или содержит один или два заместителя R7, выбранных из оксо, фтора; хлора; брома; С1-4 алкила, необязательно содержащего в качестве заместителей один или более атомов фтора; С1-4 алкокси, необязательно содержащего в качестве заместителей один или более атомов фтора; гидрокси и циано.
1.101 Соединение, определенное согласно Варианту реализации 1.100, где Ar2 является незамещенным или содержит один или два заместителя R7, выбранных из оксо, метила, дифторметила, трифторметила, амино, гидрокси и циано.
1.102 Соединение, определенное согласно любому из Вариантов реализации 1.0-1.101, где Ar2 является незамещенным или монозамещенным.
1.103 Соединение, определенное согласно Варианту реализации 1.102, где Ar2 является незамещенным или монозамещенным одним заместителем R7, выбранным из метила и амино.
1.103А Соединение, определенное согласно Варианту реализации 1.102, где Ar2 является незамещенным или монозамещенным одним заместителем R7, выбранным из метила и циано.
1.104 Соединение, определенное согласно Варианту реализации 1.103, где Ar2 является незамещенным или содержит один заместитель R7, который представляет собой метил.
1.105 Соединение, определенное согласно Варианту реализации 1.104, где Ar2 является незамещенным.
1.106 Соединение, определенное согласно любому из Вариантов реализации 1.0-1.105, где Ar2 представляет собой:
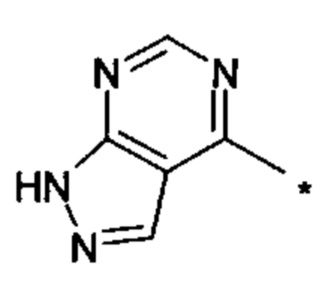
где * обозначает точку присоединения к хиназолиновому кольцу.
1.107 Соединение формулы (3):
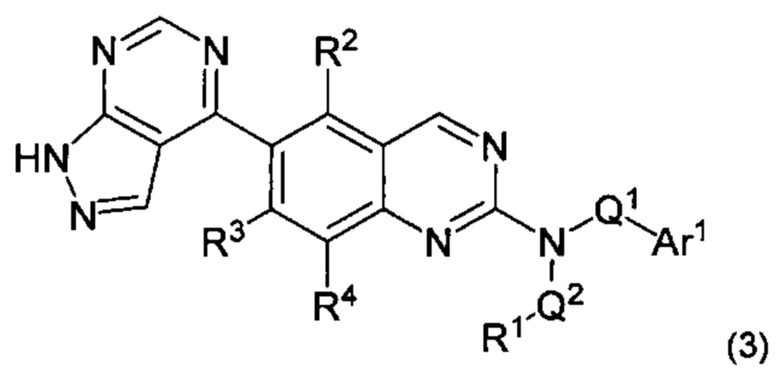
или его соль, таутомер или N-оксид, где R1, R2, R3, R4, Q1, Q2 и Ar1 являются такими как определено в Вариантах реализации 1.0-1.106.
1.108 Соединение формулы (4):
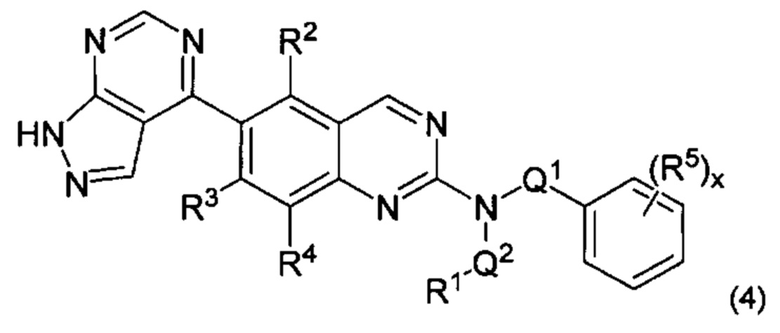
или его соль, таутомер или N-оксид, где R1, R2, R3, R4, R5, Q1 и Q2 являются такими как определено в Вариантах реализации 1.0-1.106; и х равен 0, 1 или 2.
1.109 Соединение согласно Варианту реализации 1.108, где Q1 представляет собой СН2 или СН(СН3), Q2 представляет собой связь или СН2 и R1 представляет собой водород.
1.110 Соединение согласно Варианту реализации 1.108 или Варианту реализации 1.109, где х равен 0 или 1.
1.111 Соединение, выбранное из титульных соединений согласно Примерам 1-43 в настоящей заявке.
1.112 Соединение согласно любому из Вариантов реализации 1.0-1.111, представленное в форме соли.
1.113 Соединение согласно Варианту реализации 1.112, где соль представляет собой соль присоединения кислоты.
1.114 Соединение согласно Варианту реализации 1.112 или Варианту реализации 1.113, где указанная соль представляет собой фармацевтически приемлемую соль.
1.115 Соединение согласно любому из Вариантов реализации 1.0-1.114, представленное в форме сольвата.
1.116 Соединение согласно Варианту реализации 1.115, где сольват представляет собой гидрат.
Определения
Упоминание «карбоциклических» и «гетероциклических» групп, используемое в настоящем изобретении, включает, если в контексте явным образом не указано иное, как ароматические, так и неароматические кольцевые системы. Так, например, термин «карбоциклические и гетероциклические группы» в рамках своего объема включает ароматические, неароматические, ненасыщенные, частично насыщенные и полностью насыщенные карбоциклические и гетероциклические кольцевые системы.
Карбоциклические и гетероциклические группы могут представлять собой арильные или гетероарильные группы. Арильные или гетероарильные группы могут представлять собой моноциклические или бициклические группы, определенные в настоящем изобретении. Термин «арил», используемый в настоящем изобретении, относится к карбоциклической группе с признаками ароматичности, а термин «гетероарил» в настоящем изобретении обозначает гетероциклическую группу с признаками ароматичности. В случаях, когда допускается контекстом, термины «арил» и «гетероарил» могут включать бициклические кольцевые системы, в которых оба кольца являются ароматическими или одно кольцо является неароматическим, а другое является ароматическим. В таких бициклических системах, содержащих одну ароматическую и одну неароматическую группу, указанная группа может быть присоединена через ароматическое кольцо или через неароматическое кольцо.
Термин «С-связанный» (например, «С-связанная 4-5 членная моноциклическая неароматическая или карбоциклическая или гетероциклическая группа»") относится к группе, описанной в настоящем изобретении, где присоединение осуществляется через атом углерода.
В формуле (1), Ar1 представляет собой моноциклическое 5 или 6-членное арильное или гетероарильное кольцо, содержащее 0, 1 или 2 гетероатомных члена кольца, выбранные из О, N и S. Примеры таких колец включают, но не ограничиваются пиррольной, фурановой, тиофеновой, имидазольной, оксазольной, изоксазольной, тиазольной, изотиазольной, пиразольной, пиридиновой, пиразиновой, пиридазиновой, пиримидиновой и фенильной группами.
Арильное или гетероарильное кольцо Ar1 может содержать заместитель, который состоит из 3-7-членной карбоциклической или гетероциклической групп или содержит указанные группы. Указанные карбоциклические или гетероциклические группы могут представлять собой арильные или гетероарильные группы, как определено выше, или они могут представлять собой неароматические группы.
Термин «неароматическая группа» относится к ненасыщенным кольцевым системам без признаков ароматичности, частично насыщенным и полностью насыщенным карбоциклическим и гетероциклическим кольцевым системам. Термины «ненасыщенный» и «частично насыщенный» относятся к кольцам, в которых структура(ы) кольца содержит атомы с более одной общей валентной связью, например, кольцо содержит по меньшей мере одну кратную связь, например, С=С N=C связь. Термин «насыщенный» относится к кольцам, в которых между атомами нет кратных связей. Насыщенные карбоциклические группы включают циклоалкильные группы циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Частично насыщенные карбоциклические группы включают циклоалкенильные группы циклопентенил, циклогексенил, циклогептенил и циклооктенил. Неароматические гетероциклические группы включают азетидин, пирролидин, пиперидин, азепан, пиперазин, морфолин, тиоморфолин, тиоморфолин S-оксид и S,S-диосид, пиран (2Н-пиран или 4Н-пиран), дигидротиофен, дигидропиран, дигидрофуран, дигидротиазол, тетрагидрофуран, тетрагидротиофен, диоксан, тетрагидропиран, имидазолин, имидазолидинон, оксазолин, тиазолин, пиразолин, пиразолидин.
В формуле (1), Ar2 представляет собой бициклическую 8-11-членную гетероарильную группу, содержащую 1, 2, 3 или 4 гетероатомных члена кольца, выбранные из О, N и S. Бициклическая гетероарильная группа может представлять собой, например, группу, выбранную из бензольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце; пиридинового кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце; пиримидинового кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; пиррольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце; пиразольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; имидазольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; оксазольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; изоксазольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; тиазольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; изотиазольного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1 или 2 гетероатома в кольце; тиофенового кольца, сконденсированного с 5-или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце; фуранового кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце; циклогексильного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце; и циклопентильного кольца, сконденсированного с 5- или 6-членным кольцом, содержащим 1, 2 или 3 гетероатома в кольце.
Конкретные примеры бициклических гетероарильных групп, содержащих пятичленное кольцо, сконденсированное с другим пятичленным кольцом, включают, но не ограничиваются указанными, имидазотиазол (например, имидазо[2,1-b]тиазол) и имидазоимидазол (например, имидазо[1,2-а]имидазол). Конкретные примеры бициклических гетероарильных групп, содержащих шестичленное кольцо, сконденсированное с пятичленным кольцом, включают, но не ограничиваются указанными, бензофурановую, бензотиофеновую, бензимидазольную, бензоксазольную, изобензоксазольную, бензизоксазольную, бензотиазольную, бензизотиазольную, изобензофурановую, индольную, изоиндольную, индолизиновую, индолиновую, изоиндолиновую, пуриновую (например, аденин, гуанин), индазольную, пиразолопиримидиновую (например, пиразоло[1,5-а]пиримидин), триазолопиримидиновую (например, [1,2,4]триазоло[1,5-а]пиримидин), бензодиоксольную и пиразолопиридиновую (например, пиразоло[1,5-а]пиридин) группы. Конкретные примеры бициклических гетероарильных групп, содержащих два сконденсированных шестичленных кольца, включают, но не ограничиваются указанными: хинолиновую, изохинолиновую, хромановую, тиохромановую, хроменовую, изохроменовую, хромановую, изохромановую, бензодиоксановую, хинолизиновую, бензоксазиновую, бензодиазиновую, пиридопиридиновую, хиноксалиновую, хиназолиновую, циннолиновую, фталазиновую, нафтиридиновую и птеридиновую группы.
Термин «гидрокарбил», используемый в настоящей заявке, относится к алифатическим, алициклическим и ароматическим группам, содержащим цельноуглеродный остов и состоящим из атомов углерода и водорода, за исключением случаев, когда указано иначе. Примеры гидрокарбильных групп включают алкильную, циклоалкильную, циклоалкенильную, карбоциклическую арильную, алкенильную, алкинильную, циклоалкилалкильную, циклоалкенилклкильную, и карбоциклическую аралкильную, аралкенильную и аралкинильную группы. Такие группы могут быть незамещенными или, где указано, содержать один или более заместителей, определенных в настоящей заявке. В определенных случаях, как описано в настоящей заявке, один или более, но не все атомы углерода углеводородной группы могут быть заменены другим атомом или группой атомов.
Термин «алкилен» (например, С1-4 алкилен с прямой или разветвленной цепью), используемый в настоящей заявке, относится к алкандиильной группе, т.е. дивалентной насыщенной ациклической углеводородной группе с прямой или разветвленной цепью. Примеры алкиленовых групп с прямой цепью включают метилен (СН2), этилен (СН2СН2) и пропилен ((СН2СН2СН2). Примеры алкиленовых групп с разветвленной цепью включают СН(СН3), СН2СН(СН3)СН2 и СН2(СН3)СН2СН2.
Соли
Соединения согласно настоящему изобретению, определенные в Вариантах реализации 1.0-1.111, могут быть представлены в форме солей.
Соли, упомянутые выше (а также определенные в вариантах реализации 1.112, 1.113 и 1.114) обычно являются солями присоединения кислоты.
Соли могут быть синтезированы из исходного соединения традиционными химическими способами, такими как способы, описанные в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Обычно такие соли могут быть получены путем проведения реакции соединения в форме свободного основания с раствором кислоты в воде или органическом растворителе, или в их смеси; обычно применяют неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Соли присоединения кислоты (как определено в Варианте реализации 1.113) могут быть образованы широким разнообразием кислот, как неорганических, так и органических. Примеры солей присоединения кислот включают соли, образованные кислотами, выбранными из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+) камфарной, камфарносульфоновой, (+) -(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламиновой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, бромистоводородной, хлористоводородной, йодистоводородной, изэтиновой, (+) - L-молочной, (±) -DL-молочной, лактобионовой, малеиновой, яблочной, (-) - L-оксиянтарной, малоновой, (±) -DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памоевой, фосфорной, пропионовой, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+) - L-винной, тиоциановой, п-толуолсульфоновой, ундециленовой и валериановой кислот, а также ацилированных аминокислот и катионообменных смол.
Формы солей соединений согласно настоящему изобретению обычно представляют собой фармацевтически приемлемые соли, и примеры фармацевтически приемлемых солей описаны в Berge et el., 1977, "Pharmaceutically Acceptable Salts," J. Pharm. Sci., Vol. 66, pp. 1-19. Однако, соли, которые не являются фармацевтически приемлемыми, могут быть также получены в качестве промежуточных форм, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие формы не фармацевтически приемлемых солей, которые могут быть подходящими, например, для очистки или выделения соединений согласно настоящему изобретению, также являются частью настоящего изобретения.
N-оксиды
Многие соединения согласно Вариантам реализации 1.0-1.116 могут образовывать N-оксиды. В случае, когда соединение содержит несколько аминных функциональных групп, один или более одного атома азота могут быть окислены с образованием N-оксида. Конкретные примеры N-оксидов представляют собой N-оксиды третичного амина или атом азота азот-содержащего гетероцикла.
N-оксиды могут быть получены обработкой соответствующего амина окислителем, таким как перекись водорода или надкислота (например, пероксикарбоновая кислота), см., например, Advanced Organic Chemistry, by Jerry March, 4th Edition, Wiley Interscience, pages. Более конкретно, N-оксиды могут быть получены способом L. W. Deady (Syn. Comm. 1977, 7, 509-514), согласно которому аминные соединения приводят в реакцию с м-хлорпероксибензойной кислотой (МСРВА), например, в инертном растворителе, таком как дихлорметан.
Дополнительные примеры условий образования N-оксидов описаны в более ранней публикации авторов настоящего изобретения WO 2008/139152.
Геометрические изомеры и таутомеры
Соединения согласно настоящему изобретению могут существовать в различных геометрических изомерных и таутомерных формах и отсылки к формуле (1), как определено в Вариантах реализации 1.0-1.116, включают все такие формы. Во избежание неясности в случае, когда соединение может существовать в одной из нескольких геометрических изомерных и таутомерных форм, и только одна форма конкретно описана или показана, все остальные формы в любом случае включены в формулу (1) или ее подгруппы, подмножества, предпочтительные варианты и примеры.
Оптические изомеры
Если соединение содержит один или более хиральных центров и может существовать в форме двух или более оптических изомеров, упоминание соединения включает все его оптически-изомерные формы (например, энантиомеры, эпимеры и диастереомеры), как в виде отдельных оптических изомеров, так и в форме смесей (например, рацемических смесей) или двух или более оптических изомеров, если контекст не требует иного.
Оптические изомеры могут быть охарактеризованы и идентифицированы по их оптической активности (т.е. как + и - изомеры, или d и I изомеры), или они могут быть охарактеризованы с точки зрения абсолютной стереохимии с использованием обозначений "R и S", разработанной Каном, Ингольдом и Прелогом, см Advanced Organic Chemistry by Jerry March, 4th Edition, John Wiley & Sons, New York, 1992, pages 109-114, and see also Cahn, Ingold & Prelog, Angew. Chem. Int. Ed. Engl., 1966, 5, 385-415.
Оптические изомеры можно разделить с использованием ряда методик, включая хиральную хроматографию (хроматографию на хиральном носителе), такие методики хорошо известны специалисту в области техники.
В качестве альтернативы хиральной хроматографии оптические изомеры могут быть разделены путем образования диастереомерных солей с хиральными кислотами, такими как (+)-винная кислота, (-)-проглутаминовая кислота, (-)-ди-толуолил-L-винная кислота, (+)-миндальная кислота, разделения диастереомеров путем фракционной кристаллизации с последующей диссоциацией солей с получением отдельных энантиомеров в форме свободных оснований.
Если соединения согласно настоящему изобретению существуют в виде двух или более оптических изомерных форм, один энантиомер в паре энантиомеров может демонстрировать преимущества относительно другого энантиомера, например, с точки зрения биологической активности. Соответственно, в некоторых ситуациях может быть желательно использовать в качестве терапевтического агента только один из пары энантиомеров и только один из множества диастереоизомеров. Соответственно, в настоящем изобретении предложены композиции, содержащие соединение, содержащее один или более хиральных центров, где по меньшей мере 55% (например, по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%) соединения формулы (1) присутствует в виде единственного оптического изомера (например, энантиомера или диастереоизомера). В общем варианте реализации 99% или более (например, по существу все) общего количества соединения формулы (1) может быть представлено в виде единственного оптического изомера (например, энантиомера или диастереоизомера).
Изотопы
Соединения согласно настоящему изобретению, определенные в любом из Вариантов реализации 1.0-1.116, могут содержать они или более заместителей - изотопов, и указание какого-либо конкретного элемента включает все изотопы этого элемента. Например, термин «водород» включает 1Н, 2Н (D) и 3Н (Т). Аналогично, объем обозначений углерод и кислород включает 12С, 13С, 14С и 16O и 18О.
Изотопы могут быть радиоактивными и нерадиоактивными. В одном варианте реализации изобретения соединения не содержат радиоактивных изотопов. Такие соединения предпочтительны для терапевтического применения. В другом варианте реализации, однако соединение может содержать один или более радиоизотопов. Соединения, содержащие такие радиоизотопы, могут являться подходящими в контексте диагностики.
Сольваты
Соединения формулы (1), определенные в любом из Вариантов реализации 1.0-1.116, могут образовывать сольваты.
Предпочтительные сольваты представляют собой сольваты, образованные в результате включения в структуру в твердом состоянии (например, кристаллическую структуру) соединений согласно настоящему изобретению молекул нетоксичного фармацевтически приемлемого растворителя (называемого далее сольватирующим растворителем). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем рекристаллизации соединений согласно настоящему изобретению с использованием растворителя или смеси растворителей, содержащих сольватирующий растворитель. Образовался ли сольват в любом конкретном случае, можно определить посредством подвергания кристаллов соединения анализу, используя хорошо известные и стандартные методики, такие как термогравиметрический анализ (TGE), дифференциальная сканирующая калориметрия (DSC) и рентгеновская кристаллография.
Сольваты могут представлять собой стехиометрические или нестехиометрические сольваты.
Особенно предпочтительными сольватами являются гидраты, и примеры гидратов включают полугидраты, моногидраты и дигидраты.
Более подробно сольваты и способы, используемые для их получения и определения их характеристик, рассмотрены в Bryn et al., Solid-State Chemistry of Drugs, Second Edition, published by SSCl, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
Пролекарства
Соединения формулы (1), определенные в любом из Вариантов реализации с 1.0 по 1.116, могут быть представлены в форме пролекарства.
Под «пролекарствами» понимают, например, любое соединение, которое в условиях in vivo превращается в биологически активное соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116.
Например, некоторые пролекарства представляют собой сложные эфиры активного соединения (например, физиологически приемлемые, чувствительные к физиологическим условиям эфиры). В процессе метаболизма эфирная группа (-C(=O)OR) расщепляется с образованием активного лекарственного средства. Такие эфиры могут быть образованы путем этерификации, например, любой гидроксильной группы, присутствующей в исходном соединении, в соответствующих случаях с предварительной защитой всех других реакционноспособных групп, присутствующих в исходном соединении, с последующим удалением защиты при необходимости.
Также некоторые пролекарства активируются под действием ферментов с образованием активного соединения или соединения, из которого в результате химической реакции образуется активное соединение (например, как в концепциях ADEPT, GDEPT, LIDEPT и т.д.). Например, пролекарство может быть производным сахара или другим конъюгатом гликозида, или может быть эфирным производным аминокислоты.
Комплексы и клатраты
Также в формулу (1) или ее подгруппы, подмножества, предпочтительные варианты и примеры включены комплексы (например, комплексы включения или клатраты с такими соединениями как циклодекстрины, или комплексы с металлами) соединений.
Биологическая активность
Соединения формулы (1), определенные в любом из вариантов реализации 1.0-1.116, проявляют активность в качестве ингибиторов киназы p70S6. Как таковые, они могут являться подходящими для предотвращения или лечения заболеваний и состояний, в которых киназа p70S6 или ее мутантные формы активно участвуют.
Например, предполагается, что соединения согласно Вариантам реализации 1.0-1.116 будут являться подходящими в лечении ряда пролиферативных заболеваний, таких как раковые заболевания.
Соответственно, в дополнительных вариантах реализации (Вариантах реализации 2.1-2.9) изобретения предложено:
2.1 Соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116 для применения в медицине или терапии.
2.2 Соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116 для применения в предотвращении или лечении заболеваний и состояний, опосредуемых киназой p70S6 или ее мутантными формами.
2.3 Соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116 для применения в предотвращении или лечении заболеваний и состояний, характеризующихся аномальной экспрессией киназы p70S6 (например, избыточной экспрессии или экспрессии мутантной формы киназы p70S6).
2.4 Соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116 для применения в качестве противоракового агента.
2.5 Применение соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116 для получения лекарственного средства для лечения рака.
2.6 Способ лечения рака, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, необязательно совместно с другим противораковым агентом или лучевой терапией.
2.7 Соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116 для применения для усиления терапевтического эффекта лучевой терапии или химиотерапии в лечении пролиферативного заболевания, такого как рак.
2.8 Применение соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116 для приготовления лекарственного средства для усиления терапевтического эффекта лучевой терапии или химиотерапии в лечении пролиферативного заболевания, такого как рак.
2.9 Способ профилактики или лечения пролиферативного заболевания, такого как рак, включающий введение пациенту в комбинации с лучевой терапией или химиотерапией соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116.
Примеры пролиферативных заболеваний (например, раковых заболеваний), определенных в Вариантах реализации 2.4-2.9, включают, но не ограничиваются следующими: карциномы, например карциномы мочевого пузыря, молочной железы, толстой кишки, почек, эпидермиса, печени, легких, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы, желудочно-кишечного тракта или кожи, гематопоэтических опухолей, таких как лейкемия, В-клеточная лимфома, Т-клеточная лимфома, лимфома Ходжкина, неходжкинская лимфома, волосатоклеточный лейкоз или лимфома Беркетта; гематопоэтические опухоли миелоидной линии, например острая и хроническая миелогенная лейкемия, миелодиспластический синдром или промиелоцитарный лейкоз; рак щитовидной железы; опухоли мезенхимного происхождения, например фибросаркома или рабдомиосаркома; опухоли центральной или периферической нервной системы, например астроцитома, нейробластома, глиома или шваннома; меланома; семинома; тератокарцинома; остеосаркома; пигментная ксеродерма; роговая кератома; фолликулярный рак щитовидной железы; или саркома Капоши.
Одним конкретным подмножеством раковых заболеваний, против которых соединения согласно Вариантам реализации 1.0-1.116 будут проявлять особенную активность, являются раковые заболевания, характеризующиеся избыточной экспрессией P70S6 или повышенной экспрессией P70S6 или присутствием мутантных форм P70S6 или повышенными уровнями активированной (фосфорилированной) p70S6K.
Способность соединений согласно настоящему изобретению ингибировать киназу P70S6 может быть определена согласно протоколам, представленным в разделе Примеров ниже.
Дополнительными конкретными примерами раковых заболеваний, против которых соединения согласно Вариантам реализации 1.0-1.116 будут проявлять особенную активность, являются:
- Рак молочной железы (повышенная экспрессия связана с неблагоприятными прогнозами и метастазированием (см. Mol. Can. Ther, 2010, 9, 1180), в частности, трижды негативный рак молочной железы
- Диффузная В-крупноклеточная лимфома: (см. Expert Opin Ther Targets. 2009 Sep; 13(9): 1085-93.)
- Мультиформная глиобластома (ассоциирована с повышенными уровнями P70S6K (см J Clin Oncol. 2005 Aug 10; 23(23):5294-304))
- Колоректальный рак (в котором путь mtor и p70S6K в значительной степени активированы (см Ann. Surg. Oncol. 2009 Sep; 16(9):2617-28. Epub 2009 Jun 11))
Другое подмножество раковых заболеваний, против которых соединения согласно Вариантам реализации 1.0-1.116 будут проявлять особенную активность, включает:
- Рак молочной железы
- Мультиформная глиобластома;
- Аденокарцинома толстой кишки;
- Немелкоклеточный рак легких;
- Мелкоклеточный рак легких;
- Цисплатин-устойчивый мелкоклеточный рак легких;
- Рак яичников;
- Лейкоз;
- Рак поджелудочной железы;
- Рак предстательной железы;
- Карцинома молочной железы;
- Светлоклеточный рак;
- Множественная миелома;
- Саркома Капоши;
- Лимфома Ходжкина;
- Лимфангиолейомиоматоз; и
- Неходжкинская лимфома или саркома
Дополнительная подгруппа раковых заболеваний, против которых соединения согласно Вариантам реализации 1.0-1.116 будут проявлять особенную активность, включают раковые заболевания головного мозга, такие как:
- Метастазы в головном мозге, возникающие из-за трижды негативного рака молочной железы;и
- Глиомы и глиобластомы.
Трижды негативный рак молочной железы
Большинство случаев рака молочной железы представляют собой гормонально-положительные виды рака молочной железы, в которых рост раковых клеток стимулируется воздействием эстрогена и/или прогестерона. Пациентов, страдающих такими раковыми заболеваниями, обычно лечат терапевтическими агентами, которые предотвращают или снижают образование эстрогена в организме или предотвращают связывание эстрогена с клеткой и стимулирование роста. Примеры таких терапевтических агентов включают селективные модуляторы рецепторов эстрогена (SERM), такие как тамоксифен и торемифен; ингибиторы ароматазы, такие как анастрозол, эксеместан и летрозол; ингибиторы рецепторов эстрогена (ERD) такие как фулвестрант; и гормоны, стимулирующие высвобождение лютеинизирующего гормона (LHRH), такие как гозерелин, лейпролид), и трипторелин. Стимуляция прогестерона на гормонально-положительных раковых клетках зависит от активности рецепторов эстрогена; таким образом, если воздействие эстрогена снижается, это также влияет и на чувствительность прогестерона.
Приблизительно четверть всех видов рака молочной железы представляет собой HER2-положительные виды рака молочной железы, которые характеризуются избыточной экспрессией рецептора эпидермального фактора роста человека 2 (HER2). HER2-положительные раковые заболевания обычно лечат терапевтическими агентами (например, Герцептином), которые нацеливаются на рецептор с целью снижения роста и репликации.
Однако, существуют некоторые виды рака молочной железы, которые не являются эстроген- или прогестерон-положительными и не демонстрируют избыточную экспрессию HER2 до уровня, при котором они могли бы считаться HER2-положительными. Такие формы рака молочной железы обычно называют трижды негативным раком молочной железы. Для пациентов, страдающих трижды негативным раком молочной железы, существует меньше вариантов лечения, чем для пациентов, страдающих гормонально-положительным или HER2-положительным раком, и поэтому обычно их лечение более затруднительно, чем лечение эстроген-положительного, прогестерон-положительного и HER2-положительного раковых заболеваний. Также общепризнанным является тот факт, что трижды негативный рак молочной железы с большей вероятностью распространяется (метастазирует) в головной мозг. Как правило, считается, что пациенты, имеющие метастазы в головном мозге, неизлечимы с помощью стандартных подходов лечения.
Соединения формулы (1), определенные в Вариантах реализации 1.0-1.116 в настоящем изобретении, могут применяться в лечении трижды негативного рака молочной железы и лечении метастазов головного мозга, возникающих вследствие трижды негативного рака молочной железы. Соединения также можно применять в лечении метастазов в головном мозге, возникающих вследствие других раковых заболеваний.
Дополнительно, соединения формулы (1), определенные в Вариантах реализации Embodiments 1.0-1.116 согласно настоящем изобретении, могут применяться для предотвращения или лечения метастазов в целом, например, для предотвращения или лечения метастазов в головном мозге, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре.
Соответственно, согласно дополнительным Вариантам реализации 2.10-2.18, в изобретении предложены:
2.10 Соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения в лечении трижды негативного рака молочной железы.
2.11 Соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения в предотвращении или лечении метастазов, например, метастазов в головном мозге, костях, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре, например, метастазов в головном мозге, возникающих вследствие трижды негативного рака молочной железы.
2.12 Соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения в лечении метастазов в головном мозге, возникающих вследствие раковых заболеваний, отличных от рака головного мозга.
2.13 Применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для лечения трижды негативного рака молочной железы.
2.14 Применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для предотвращения или лечения метастазов, например, метастазов в головном мозге, костях, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре, например, метастазов в головном мозге, возникающих вследствие трижды негативного рака молочной железы.
2.15 Применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для лечения метастазов в головном мозге, возникающих вследствие раковых заболеваний, отличных от рака головного мозга.
2.16 Способ лечения трижды негативного рака молочной железы у субъекта (например, представляющего собой человека), нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения, определенного в любом из Вариантов реализации 1.0 to 1.116.
2.17 Способ предотвращения или лечения метастазов, например, метастазов в головном мозге, костях, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре (например, метастазов в головном мозге, возникающих вследствие трижды негативного рака молочной железы) у субъекта (например, представляющего собой человека), нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения, определенного в любом из Вариантов реализации 1.0 - 1.116.
2.18 Способ лечения метастазов в головном мозге, возникающих вследствие раковых заболеваний, отличных от рака головного мозга, у субъекта (например, представляющего собой человека), нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения, определенного в любом из Вариантов реализации 1.0 - 1.116.
Глиомы
Предполагается, что соединения формулы (1), определенные в Вариантах реализации 1.0-1.116 согласно настоящей заявке, будут подходящими для лечения глиом вследствие их эффективности в качестве ингибиторов S6K1 (которые, как известно, играют роль в глиальной трансформации) и их способности достигать места приложения действия, т.е., головного мозга.
Глиомы представляют собой распространенный тип первичных опухолей головного мозга, зарождающихся в глиальных клетках головного мозга, и насчитывают приблизительно 30% всех первичных опухолей головного мозга и центральной нервной системы, и приблизительно 80% всех злокачественных опухолей головного мозга. Глиомы обычно возникают из трех различных типов клеток, которые в здоровом состоянии присутствуют в головном мозге, а именно: астроцитов, олигодендроцитов и эпендимальных клеток. Основные типы глиом включают эпендимомы (ассоциированные с эпендимальными клетками), астроцитомы (ассоциированные с астроцитами), олигодендроглиомы (ассоциированные с олигодендроцитами), глиому ствола головного мозга (развивающуюся в стволе головного мозга), глиому зрительного нерва (которая развивается вили вокруг оптического нерва) и смешанных глиом (которые содержат клетки различных типов глиом).
Эпендимома представляет собой тип глиомы, который развивается из эпендимальных клеток, как правило, выстилающих желудочки головного мозга или спинной мозг. У детей они чаще всего обнаруживаются рядом с мозжечком. Эпендимомы встречаются редко, насчитывают лишь приблизительно 2-3% первичных опухолей головного мозга. Однако, они насчитывают приблизительно 8-10% опухолей головного мозга у детей и чаще всего встречаются у детей младше десятилетнего возраста.
Астроцитомы зарождаются в звездчатых глиальных клетках (астроцитах) в головном мозге. Астроцитомы редко распространяются за пределы головного и спинного мозга и редко поражают другие органы, однако они представляют собой наиболее распространенную глиому и могут встречаться в большинстве участков головного мозга и изредка в спинном мозге. Общепринятым является деление астроцитом на два широких класса, в частности, астроцитомы с узкими зонами инфильтрации (в основном, инвазивные опухоли; например, пилоцитарная астроцитома, субэпендимальная гигантоклеточная астроцитома, плеоморфная ксантоастроцитома), которые часто ясно очерчены на диагностических снимках; и астроцитомы с диффузными зонами инфильтрации (например, низкодифференцируемая астроцитома, анапластическая астроцитома, глиобластома). Мультиформная глиобластома представляет собой злокачественную астроцитому и является наиболее распространенной первичной опухолью головного мозга среди взрослых людей.
Олигодендроглиомы представляют собой тип глиом, развивающийся из олигодендроцитов, которые представляют собой поддерживающие клетки ткани мозга, и обычно обнаруживаются в головном мозге. Около 4% первичных опухолей головного мозга представляют собой олигодендроглиомы, и они наиболее распространены у молодых людей и людей среднего возраста. Очень частым симптомом таких глиом являются пароксизмальные судороги, а также головная боль, слабость или изменения в поведении или сонливости.
Глиомы ствола головного мозга, как становится понятно из названия, представляют собой опухоли, обнаруживаемые в стволе головного мозга. Большую часть опухолей ствола головного мозга нельзя удалить хирургически из-за удаленного расположения и тонкой и сложной функции, контролируемой этой областью. Опухоли ствола головного мозга встречаются почти исключительно у детей, обычно у детей школьного возраста.
Смешанная глиома представляет собой злокачественную глиому, образованную более чем одним типом глиальных клеток. Указанный тип глиомы может также называться олигоастроцитома. Смешанные глиомы часто обнаруживаются в большом мозге (основной части головного мозга), однако могут метастазировать в другие части мозга. Только приблизительно 1% первичных опухолей головного мозга приходится на смешанные глиомы и наиболее часто они обнаруживаются у взрослых людей.
Глиома зрительного нерва представляет собой тип злокачественной глиомы (опухоли головного мозга), обнаруживаемый в перекресте зрительных нервов. Глиомы зрительного нерва часто окружают зрительные нервы, и часто встречаются у людей, страдающих нейрофиброматозом. Человек, страдающий глиомой зрительного нерва, обычно испытывает потерю зрения, а также может страдать от гормональных нарушений, поскольку опухоли чаще всего обнаруживаются у основания головного мозга, где расположены структуры, отвечающие за гормональную регуляцию. Глиомы зрительного нерва обычно трудно поддаются лечению из-за чувствительности окружающих структур мозга.
В дополнение к классификации по типу глиальных клеток, из которых они происходят, или области головного мозга, в которой они развиваются, глиомы можно классифицировать согласно их «степени злокачественности», что является мерой способности к росту и агрессивности опухоли.
Таким образом, наиболее часто глиомы классифицируют как «низкой степени злокачественности» и «высокой степени злокачественности», степень определяется путем оценки патологии опухоли. Опухоли дополнительно могут быть дифференцированы согласно системе оценки Всемирной Организации Здравоохранения (ВОЗ), согласно которой опухоли классифицируются от I (наименьшая степень прогрессии заболевания - наилучшие прогнозы) до IV (наиболее запущенная стадия заболевания - худшие прогнозы).
Также глиомы могут быть классифицированы по расположению: находятся они выше или ниже мембраны, называемой тенториум, которая разделяет область большого мозга (выше) от мозжечка (ниже). Супратенториальные глиомы (т.е., опухоли расположены над тенториумом в большом мозге), в большинстве случаев обнаруживаются у взрослых людей (70%), тогда как инфратенториаотные глиомы (опухоли расположены ниже тенториума в большом мозге) в большинстве случаев обнаруживаются у детей (70%).
Дополнительный класс глиом состоит из опухолей, обнаруживаемых в варолиевом мосте ствола головного мозга. Ствол головного мозга состоит из трех частей (мост, средний мозг и медулла); варолиев мост контролирует важнейшие функции, такие как дыхание, поэтому проводить хирургические операции глиом моста головного мозга чрезвычайно опасно.
Соответственно, согласно дополнительным вариантам реализации 2.19-2.34 в настоящем изобретении предложены:
2.19 Соединение, определенное в любом из вариантов реализации 1.0-1.116, для применения в лечении глиом и глиобластом.
2.20 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой эпендимому.
2.21 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой астроцитому.
2.22 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой глиобластому.
2.23 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой мультиформную глиобластому.
2.24 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой олигодерндроглиому.
2.25 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой глиому ствола головного мозга.
2.26 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой глиому зрительного нерва.
2.27 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой смешанную глиому.
2.28 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой глиому низкой степени злокачественности.
2.29 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой глиому высокой степени злокачественности.
2.30 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой супратенториальную глиому.
2.31 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой инфратенториальную глиому.
2.32 Соединение для применения согласно варианту реализации 2.19, где указанная глиома представляет собой понтинную глиому.
2.33 Применение соединения, определенного в любом из Вариантов реализации 1.0-1.116 для приготовления лекарственного средства для лечения глиомы, как определено в любом из Вариантов реализации 2.19-2.32.
2.34 Способ лечения глиомы, определенный в любом из Вариантов реализации 2.19-2.32 у субъекта, нуждающегося в таком лечении, причем указанный способ включает введение указанному субъекту эффективного терапевтического количества соединения, как определено в любом из Вариантов реализации 2.19-2.32.
Расстройства нервно-психического развития и нейродегенеративные заболевания
Как описано выше, P70S6K также играет важнейшую роль в патологии ряда расстройств нервно-психического развития и нейродегенеративных нарушений и заболеваний, и предполагается, что ингибирование P70S6K обеспечит средства для лечения множества таких заболеваний. Соответственно, согласно дополнительным вариантам реализации 2.35-2.48, в изобретении предложены:
2.35 Соединение, определенное в любом из вариантов реализации 1.0-1.116 для применения в лечении расстройства нервно-психического развития.
2.36 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой синдром фрагильной X-хромосомы.
2.37 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой аутизм или нарушение аутического спектра.
2.38 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой тремор/атаксию, ассоциированную с фрагильной Х-хромосомой (FXTAS).
2.39 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой синдром Ангельмана.
2.40 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой Комплекс туберозного склероза.
2.41 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой синдром дупликации гена МЕСР2.
2.42 Соединение для применения согласно Варианту реализации 2.35, где указанное расстройство нервно-психического развития представляет собой Синдром Дауна.
2.43 Соединение, определенное в любом из Вариантов реализации 1.0-1.116 для применения в лечении нейродегенеративного заболевания.
2.44 Соединение для применения согласно Варианту реализации 2.43, где указанное нейродегенеративное заболевание представляет собой болезнь Альцгеймера.
2.45 Соединение для применения согласно Варианту реализации 2.43, где указанное нейродегенеративное заболевание представляет собой болезнь Хантингтона.
2.46 Соединение для применения согласно Варианту реализации 2.43, где указанное нейродегенеративное заболевание представляет собой болезнь Паркинсона.
2.47 Применение соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116 для получения лекарственного средства для лечения расстройств головного мозга, например, расстройств головного мозга, определены в любом из Вариантов реализации 2.35-2.46.
2.48 Способ лечения расстройств головного мозга (например, расстройств головного мозга, определены в любом из Вариантов реализации 2.35-2.46) у субъекта (например, субъекта-млекопитающего, такого как человек), включающий введение указанному субъекту терапевтически эффективного количества соединения формулы (1), (2), (3) или (4), определенных в любом из Вариантов реализации 1.0-1.116.
Синдром фрагильной Х-хромосомы обычно впервые проявляется в детстве. Задержка речи является распространенным и часто первым симптомом, из-за которого следует обращаться за медицинской помощью (в возрасте около двух или трех лет). Соответственно, согласно дополнительным вариантам реализации в изобретении предложены:
2.48A Соединение согласно любому из Вариантов реализации 1.0-1.116 для применения в лечении синдрома фрагильной Х-хромосомы у пациента в возрасте до 20 лет, например, в возрасте до 15 лет или в возрасте до 12 лет, или в возрасте 10 лет, предпочтительно в возрасте до 8 лет и еще более предпочтительно в возрасте до 5 лет.
2.48B Способ лечения синдрома фрагильной Х-хромосомы, включающий введение пациенту, нуждающемуся в таком лечении, соединения, определенного в любом из Вариантов реализации 1.0-1.116, причем указанный пациент не достиг возраста 20 лет, например, не достиг возраста 15 лет, или не достиг возраста 12 лет, или не достиг возраста 10 лет, предпочтительно не достиг возраста 8 лет, и еще более предпочтительно, не достиг возраста 5 лет.
2.48C Применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для получения лекарственного средства для лечения или профилактики синдрома фрагильной Х-хромосомы у пациента, не достигшего возраста 20 лет, например, не достигшего возраста 15 лет, или не достигшего возраста 12 лет, или не достигшего возраста 10 лет, предпочтительно не достигшего возраста 8 лет, и еще более предпочтительно, не достигшего возраста 5 лет.
Другие заболевания и состояния, в которых может участвовать S6K1
Как обсуждалось выше, ингибиторы P70S6K также могут являться подходящими для лечения синдрома PTEN-гамартомы, нейрофиброматоза 1 типа и лимфангиолейомиоматоза (ЛАМ). Соответственно, дополнительные варианты реализации (Варианты реализации 2.49-2.52), в изобретении предложено:
2.49 Соединение, определенное в любом из Вариантов реализации 1.0-1.116, для лечения состояния, которое представляет собой синдром PTEN-гамартомы.
2.50 Соединение, определенное в любом из Вариантов реализации 1.0-1.116, для лечения состояния, которое представляет собой нейрофиброматоз 1 типа.
2.51 Соединение, определенное в любом из Вариантов реализации 1.0-1.116, для лечения состояния, которое представляет собой лимфангиолейомиоматоз.
2.52 Применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для лечения состояния, определенного в любом из Вариантов реализации 2.49-2.51.
2.53 Способ лечения состояния, определенного в любом из Вариантов реализации 2.49-2.51, у субъекта (например, представляющего собой млекопитающее, такое как человек), включающий введение указанному субъекту терапевтически эффективного количества соединения формулы (1), (2), (3) или (4), определенных в любом из Вариантов реализации 1.0-1.116.
Определение биологических свойств
Способность соединений согласно Вариантам реализации 1.0-1.116 ингибировать пролиферацию клеток, также может быть определена с использованием протоколов, приведенных ниже в разделе Примеры.
Преимущество соединений согласно Вариантам реализации 1.0-1.116 заключается в том, что они являются селективными ингибиторами киназ.
Предпочтительными соединениями согласно Вариантам реализации 1.0-1.116 являются соединения, характеризующиеся IC50 в отношении киназы p70S6, составляющей менее 5 мкМ, или менее 1 мкМ и предпочтительно менее 0,1 мкМ.
Например, соединения согласно Вариантам реализации 1.0-1.116 являются селективными ингибиторами киназы p70S6 по сравнению с их активностью в отношении киназы Akt2. Предпочтительные соединения согласно Вариантам реализации 1.0-1.116 проявляют по меньшей мере в 5 раз большую активность в отношении киназы p70S6, чем в отношении киназы Akt2, и более предпочтительно соединения согласно Вариантам реализации 1.0-1.116 проявляют по меньшей мере в 10 раз или по меньшей мере в 20 раз большую активность в отношении киназы p70S6, чем в отношении киназы Akt2. Особенно предпочтительные соединения согласно Вариантам реализации 1.0-1.116 проявляют по меньшей мере в 100 раз большую активность в отношении киназы p70S6, чем в отношении киназы Akt2.
Кроме того, соединения согласно Вариантам реализации 1.0-1.116 представляют собой селективные ингибиторы киназы p70S6 по сравнению с активностью против киназы Aurora. Предпочтительные соединения согласно Вариантам реализации 1.0-1.116 проявляют по меньшей мере в 5 раз большую активность против киназы p70S6, чем против киназы Aurora А и/или В, и более предпочтительно соединения согласно Вариантам реализации 1.0-1.116 проявляют по меньшей мере в 10 раз большую активность против киназы p70S6, чем против киназы Aurora А и/или В.
Предполагается, что соединения согласно Вариантам реализации 1.0-1.116, характеризующиеся большей селективностью в отношении киназы p70S6 по сравнению с киназой Aurora А и/или Aurora В и/или киназой Akt, будут проявлять улучшенные профили побочных эффектов по отношению к побочным эффектам, возникающим вследствие ингибирования киназы Aurora и киназы Akt. Например, в случае ингибирования киназ Aurora, хорошо известным в клинической практике побочным эффектом является нейтропения.
Соответственно, согласно дополнительным вариантам реализации (Варианты реализации 2.54-2.57), в настоящем изобретении предложены:
2.54 Соединение согласно любому из Вариантов реализации 1.0-1.116, характеризующееся IC50 в отношении киназы p70S6, составляющим менее 5 мкМ.
2.55 Соединение согласно любому из Вариантов реализации 1.0-1.116, характеризующееся IC50 в отношении киназы p70S6, составляющим 1 мкМ или менее.
2.56 Соединение согласно любому из Вариантов реализации 1.0-1.116, характеризующееся IC50 в отношении киназы p70S6, составляющим менее 0,1 мкМ.
2.57 Соединение согласно любому из Вариантов реализации 2.54-2.56 для применения в лекарственном средстве, лечении, способе или применении согласно любому из Вариантов реализации 2.1-2.52.
Многие соединения, определенные в Вариантах реализации 1.0-1.116, отличаются хорошей способностью к пенетрации в головной мозг, и поэтому являются подходящими для лечения нарушений головного мозга, при которых терапевтически эффективным является ингибирование киназы p70S6.
Способность соединений согласно Вариантам реализации 1.0-1.116 проникать в головной мозг может быть определена с помощью in vivo кассетной модели на мышах, которая является стандартным в промышленности способом для оценки пенетрации малых молекул в головной мозг (см., например, "In vitro permeability analysis, pharmacokinetic and brain distribution study in mice of imperatorin, isoimperatorin and cnidilin in Radix Angelicae Dahuricae", Fitoterapia, Volume 85, March 2013, Pages 144-153).
Способы получения соединений согласно настоящему изобретению
В настоящем изобретении также предложены способы получения соединения формулы (1).
Соответственно, согласно другому варианту реализации (Вариант реализации 3.1) в настоящем изобретении предложен способ получения соединения, определенного в любом из Вариантов реализации 1.0-1.116, где Y представляет собой R3 и Z представляет собой Ar2, причем указанный способ включает:
(а) проведение реакции между соединением формулы (8):
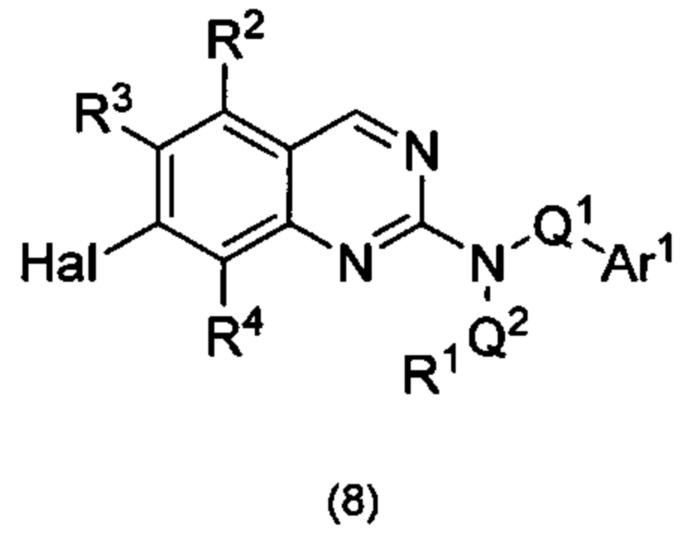
или его защищенной формой, где Hal представляет собой галоген, такой как бром, с бороновой кислотой или боронатным реагентом формулы Ar2-Bor, где Bor представляет собой остаток бороната или бороновой кислоты, в присутствии палладиевого катализатора; и затем необязательно удаление любой присутствующей защитной группы; или
(b) проведение реакции между соединением формулы (9):
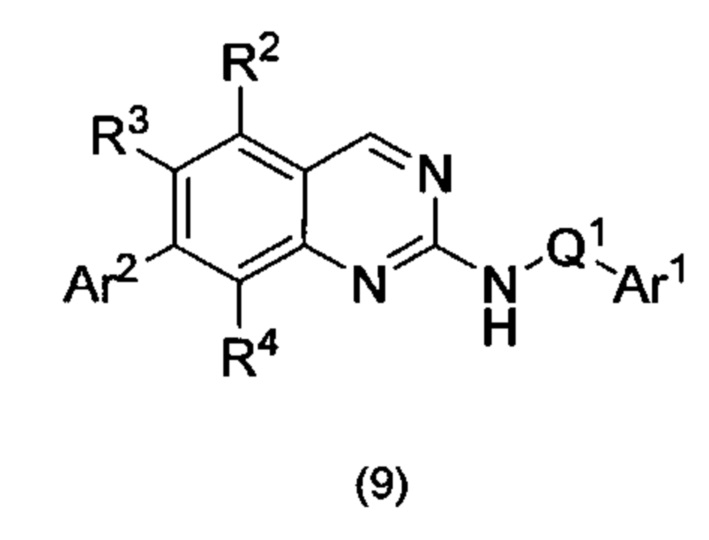
или его защищенной формой, с соединением формулы LG-Q2-R1, где LG представляет собой уходящую группу, такую как галоген; и затем необязательно удаление любой присутствующей защитной группы.
Согласно другому варианту реализации (Вариант реализации 3.2), в изобретении предложен способ получения соединения, определенного в любом из Вариантов реализации 1.0-1.116, где Y представляет собой Ar2 и Z представляет собой R3, причем указанный способ включает:
(а) проведение реакции между соединением формулы (10):
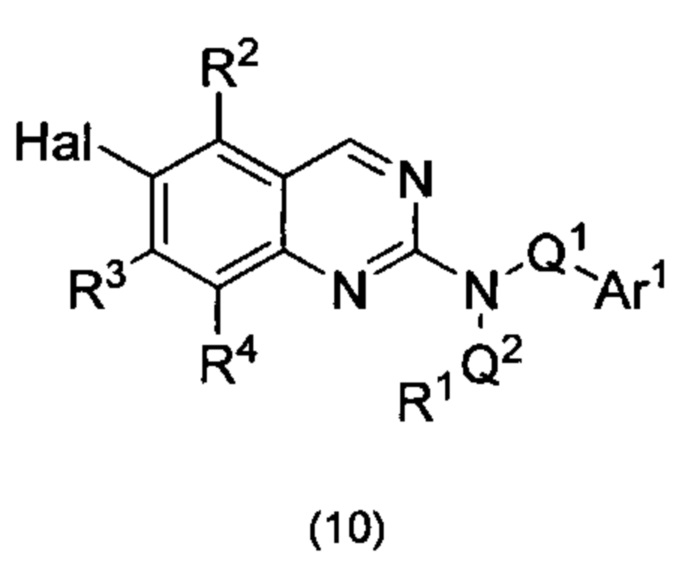
или его защищенной формой, где Hal представляет собой галоген, такой как бром, с бороновой кислотой или боронатным реагентом формулы Ar2-Bor, где Bor представляет собой остаток бороната или бороновой кислоты, в присутствии палладиевого катализатора; и затем необязательно удаление любой присутствующей защитной группы; или
(b) проведение реакции между соединением формулы (11):
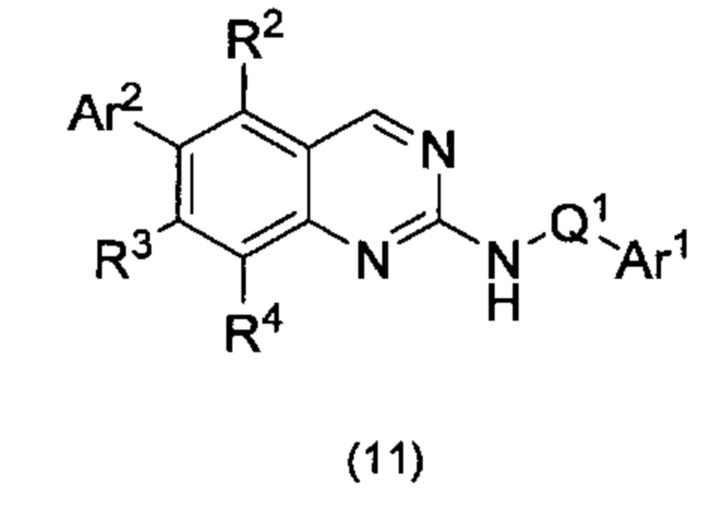
или его защищенной формой, с соединением формулы LG-Q2-R1, где LG представляет собой уходящую группу, такую как галоген; и затем необязательно удаление любой присутствующей защитной группы.
Реакция (а) в Вариантах реализации 3.1 и 3.2, представленная выше, может быть проведена в условиях реакции сочетания Сузуки в присутствии платинового катализатора, такого как бис(три-т-бутилфосфин)палладий (0), и основания (например, карбонат, такой как карбонат калия или карбонат цезия). Реакция может быть проведена в полярном растворителе, таком как диметилформамид (DMF) или диоксан, и как правило, реакционную смесь подвергают нагреванию, например, до температуры свыше 100°C.
Реакция (b) в Вариантах реализации 3.1 и 3.2, представленная выше, обычно проводится при комнатной температуре в полярном растворителе, таком как диметилсульфоксид или диметилформамид, в присутствии ненуклеофильного основания, такого как гидрид щелочного металла (например, гидрид натрия).
Иллюстративные схемы реакций для получения соединений формулы (1) представлены ниже.
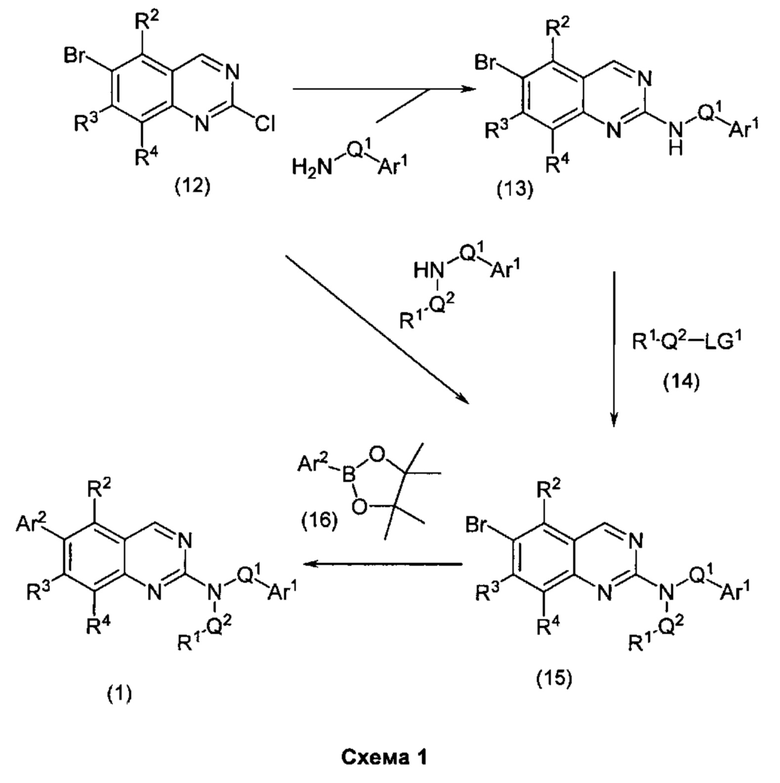
Согласно Схеме 1, когда R2, R3 и R4 представляют собой водород, исходное соединение представляет собой 6-бром-2-хлорхиназолин (12), которое является коммерчески доступным. 6-бром-2-хлорхиназолин (12) вступает в реакцию с арил- или гетероарилалкиламином H2N-Q1-Ar1 в полярном растворителе, таком как диметилсульфоксид, как правило, при комнатной температуре или необязательно при умеренном нагревании, с получением промежуточного соединения (13). Соединение (13) вступает в реакцию с гидридом натрия в диметилформамиде при пониженной температуре около 0°C, и затем соединение формулы (14), где LG1 представляет собой подходящую уходящую группу (например, галоген, метансульфонат или тозилат), добавляют к полученной реакционной смеси. Затем реакционную смесь могут нагревать до умеренных температур около 50°C с получением бром-промежуточного соединения (15).
В качестве альтернативы, бром-промежуточное соединение (15) может быть синтезировано непосредственно из 6-бром-2-хлорхиназолина в качестве исходного соединения (12) путем проведения реакции с амином NH(Q1Ar1)(Q2R1) в полярном растворителе, таком как диметилсульфоксид, как правило, при комнатной температуре или, необязательно, при умеренном нагревании.
Бром-промежуточное соединение (15) затем вступает в реакцию с гетероарил боронатом (16) в полярном растворителе, таком как диоксан, в присутствии бис(три-трет-бутилфосфин)палладия (0) и карбоната цезия или карбоната калия, и необязательно в присутствии йодида калия в условиях реакции Сузуки с получением соединения формулы (1) или его защищенного производного. Гетероарильная группа Ar2 может присутствовать в боронатном соединении (16) в защищенной форме. Например, когда Ar2 содержит группу NH, к атому азота может быть присоединена защитная группа, такая как группа Boc (трет-бутоксикарбонил), замещая водородный атом. После реакции боронатного соединения (16) и промежуточного соединения (15), может потребоваться стадия снятия защиты с целью получения соединения формулы (1). В случае защитной группы Boc она может быть удалена путем обработки кислотой, такой как соляная кислота.
Боронаты и бороновые кислоты формулы Ar2-Bor коммерчески широко доступны или могут быть получены, например, как описано в обзорной статье N. Miyaura and A. Suzuki, Chem. Rev. 1995, 95, 2457. Таким образом, боронаты могут быть получены путем проведения реакции соответствующего бром-содержащего соединения с алкиллитием, таким как бутиллитий, и затем проведение реакции со сложным эфиром борной кислоты. Полученное производное сложного эфира борной кислоты в случае необходимости может быть гидролизовано с получением соответствующей бороновой кислоты.
Соединения формулы (1), где Q2 представляет собой связь, a R1 представляет собой циклическую группу Су1, могут быть получены с помощью последовательности реакций, представленных на Схеме 2.
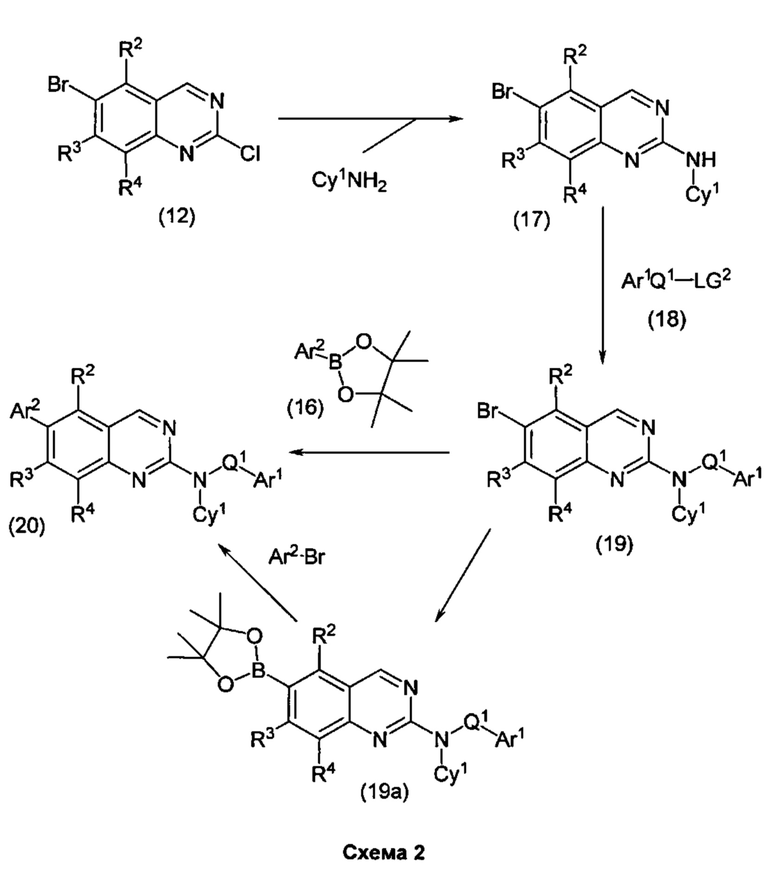
Согласно Схеме 2, 6-бром-2-хлорхиназолин (12) вступает в реакцию с карбоциклическим или гетероциклическим амином Cy1-NH2 в полярном растворителе, таком как диметилсульфоксид, как правило, при комнатной температуре или необязательно при умеренном нагревании, с получением промежуточного соединения (17), которое затем обрабатывают соединением (18), где LG1 является подходящей уходящей группой (например, галогеном, метансульфонатом или тозилатом), в стандартных условиях алкилирования с получением бром-содержащего соединения (19). Бром-содержащее соединение (19) затем вступает в реакцию с гетероарил боронатом (16) в условиях реакции Сузуки, описанных выше, с получением соединения формулы (20), соответствующего соединению формулы (1), в котором Q2 представляет собой связь и R1 представляет собой циклическую группу Су1. В качестве альтернативы, бром-содержащее соединение (19) вступает в реакцию с бис- боран-пинаколовым эфиром с получением бороната (19а), который затем вступает в реакцию с гетероарилбромидом Ar2-Br в условиях реакции сочетания Сузуки с получением соединения (20).
В случае, когда невозможно выделить подходящий реагент Ar2-Bor для применения в реакции Сузуки, реакционность реакции Сузуки может быть обращена, как показано на Схеме 3 ниже.
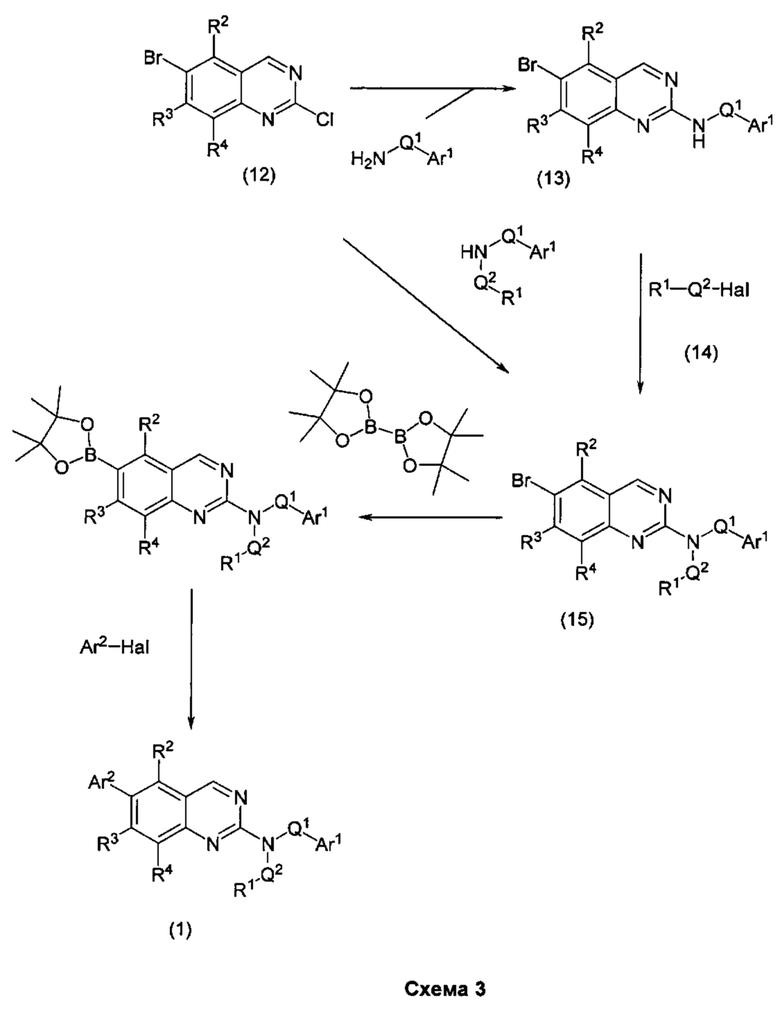
Согласно Схеме 3 бромид (15) превращают в боронатный эфир путем сочетания с бис-пинаколато-дибором в присутствии ацетата калия и с использованием палладиевого катализатора, такого как [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), в комплексе с дихлорметаном. Предпочтительно используют полярный растворитель, такой как DMSO, и реакционную смесь нагревают при 80°C в атмосфере азота или аргона для обеспечения возможности проведения реакции. Полученный боронатный эфир затем вступает в реакцию с галоген-замещенной ароматической или гетероароматической группой в условиях реакции Сузуки, как описано в настоящей заявке.
Соединения формулы (1), где Z представляет собой Ar2 и Y представляет собой R3, могут быть получены, как описано в настоящей заявке, с использованием региоизомера 7-бром-2-хлор-хиназолина в качестве исходного соединения.
После образования соединение формулы (1), или его защищенное производное, может быть превращено в другое соединение формулы (1) с использованием методик, хорошо известных специалисту в области техники. Примеры способов синтеза для превращения одной функциональной группы в другую функциональную группу, приведены в стандартных публикациях, таких как Advanced Organic Chemistry, by Jerry March, 4th edition, 119, Wiley Interscience, New York; Fiesers'Reagents for Organic Synthesis, Volumes 1-17, John Wiley, edited by Mary Fieser (ISBN: 0-471-58283-2); and Organic Syntheses, Volumes 1-8, John Wiley, edited by Jeremiah P. Freeman (ISBN: 0-471-31192-8)).
Во многих описанных выше реакциях может потребоваться защита одной или более групп с целью предотвращения протекания реакции в нежелательном положении молекулы. Примеры защитных групп и способов защиты и снятия защиты функциональных групп можно найти в Protective Groups in Organic Synthesis (Т. Green and P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Соединения, полученные описанными выше способами, могут быть выделены и очищены любым из множества способов, хорошо известных специалистам в области техники, и примеры таких способов включают перекристаллизацию и методы хроматографии, такие как колоночная хроматография (например, флэш-хроматография) и ВЭЖХ.
Лекарственные формы
Хотя активное соединение может быть введено отдельно, предпочтительным является представление указанного активного соединения в виде фармацевтической композиции (например, лекарственной формы).
Соответственно, согласно другому варианту реализации (Вариант реализации 4.1) настоящего изобретения, предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (1), определенное в любом из вариантов реализации 1.0-1.116, совместно с фармацевтически приемлемым вспомогательным веществом.
Фармацевтически приемлемое вспомогательное вещество может представлять собой, например, носитель (например, твердый, жидкий или полутвердый носитель), разбавитель или наполнитель, гранулирующий агент, покрывающий агент, связующий агент, разрыхлитель, смазывающий агент, консервант, антиоксидант, буферный агент, суспендирующий агент, загуститель, вкусоароматический агент, подсластитель, агент, маскирующий вкус, или любое другое вспомогательное вещество, обычно используемое в фармацевтической композиции. Примеры вспомогательных веществ для различных типов фармацевтических композиций более подробно описаны ниже.
Фармацевтические композиции могут быть представлены в любой форме, подходящей для перорального, парентерального, местного, интраназального, офтальмического, ушного, ректального, внутривагинального или трансдермального применения. В случае, когда композиции предназначены для парентерального введения, они могут быть представлены в виде лекарственной формы для внутривенного, внутримышечного, внутрибрюшинного, подкожного введения или для непосредственной доставки в целевой орган или ткань путем инъекции, инфузии или других способов доставки. Доставка может быть осуществлена путем болюсной инъекции, краткосрочной инфузии или долгосрочной инфузии и может осуществляться путем пассивной доставки или путем использования подходящего инфузионного насоса.
Фармацевтические композиции, адаптированные для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики, сорастворители, смеси органических растворителей, циклодекстрин, агенты комплексообразования, эмульгаторы (для формирования и стабилизации эмульсионных форм), компоненты липосом для образования липосом, гелеобразующие полимеры для формирования полимерных гелей, лиопротекторы и комбинации агентов для, среди прочего, стабилизации активного ингредиента в растворимой форме и придания составу изотоничности с кровью предполагаемого реципиента. Фармацевтические составы для парентерального введения также могут иметь форму водных и неводных стерильных суспензий, которые могут включать суспендирующие агенты и загустители (R.G. Strickly, Solubilizing Excipients in oral and injectable formulations, Pharmaceutical Research, Vol 21(2) 2004, p 201-230).
Молекула лекарственного средства, способная к ионизации, может быть растворена до желаемой концентрации путем доведения pH, если pKa лекарственного средства значительно отличается от значения pH лекарственной формы. Приемлемый диапазон pH составляет 2-12 для внутривенного и внутримышечного введения, однако, для подкожного введения диапазон составляет pH 2,7-9,0. pH раствора контролируют либо за счет использования соли лекарственного средства, с помощью сильных кислот/оснований, таких как соляная кислота или гидроксид натрия, или с помощью растворов буферов, которые включают, но не ограничиваются указанными, буферные растворы, образованные из глицина, цитрата, ацетата, малеата, сукцината, гистидина, фосфата, трис(гидроксиметил)аминометана (TRIS), или карбоната.
Комбинацию водного раствора и водорастворимого органического растворителя/поверхностно-активного вещества (т.е., сорастворителя) часто применяют в инъекционных препаратах. Водорастворимые органические растворители и поверхностно-активные вещества, применяемые в инъекционных лекарственных формах, включают, но не ограничиваются указанными, пропиленгликоль, этанол, полиэтиленгликоль 300, полиэтиленгликоль 400, глицерин, диметилацетамид (DMA), N-метил-2-пирролидин (NMP; Pharmasolve), диметилсульфоксид (DMSO), Solutol HS 15, Cremophor EL, Cremophor RH 60, и полисорбат 80. Такие лекарственные формы как правило, но не всегда, могут быть разбавлены перед инъекцией.
Полипропиленгликоль, PEG 300, этанол, Cremophor EL, Cremophor RH 60, и полисорбат 80 являются полностью органическими, смешиваемыми с водой растворителями и поверхностно-активными веществами, используемыми в коммерчески доступных инъецируемых лекарственных формах, и могут применяться в комбинации друг с другом. Полученный органический состав обычно разбавляют в по меньшей мере два раза перед внутривенным введением болюса или внутривенной инфузией.
В качестве альтернативы, повышенная растворимость в воде может быть достигнута за счет образования комплекса молекулы с циклодекстринами.
Составы могут быть представлены в контейнерах, содержащих одну дозу или множество доз, например, в герметичных ампулах и виалах, и могут храниться в лиофилизированном состоянии (высушенные сублимацией), требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед использованием.
Фармацевтический состав может быть получен путем лиофилизации соединения Формулы (1) или его соли присоединения кислоты. Лиофилизацией называется процедура сублимационной сушки композиции. Сублимационная сушка и лиофилизация, соответственно, используются в настоящей заявке как синонимы. Традиционный способ заключается в растворении соединения и доведении полученного состава до прозрачного состояния, стерилизации фильтрованием и асептическом переносе в контейнеры, подходящие для лиофилизации (например, виалы). В случае использования виал они являются частично закрытыми с помощью пробок для лиофилизации. Состав может быть охлажден до замораживания и подвергнут лиофилизации в стандартных условиях, а затем герметично упакован с получением стабильной, сухой лиофилизированной лекарственной формы. Композиция обычно имеет низкое содержание остаточной воды, например, менее чем 5%, например, менее, чем 1% по весу из расчета на вес лиофилизата.
Лиофилизированная лекарственная форма может содержать другие вспомогательные вещества, например, загустители, диспергирующие агенты, буферы, антиоксиданты, консерванты и агенты для регулирования изотоничности. Типичные буферы включают фосфат, ацетат, цитрат и глицин. Примеры антиоксидантов включают аскорбиновую кислоту, бисульфит натрия, метабисульфит натрия, монотиоглицерин, тиомочевину, бутилированный гидрокситолуол, бутилированный гидроксианизол и соли этилендиаминтетрауксусной кислоты. Консерванты могут включать бензойную кислоту и ее соли, сорбиновую кислоту и ее соли, алкиловые сложные эфиры пара-гидроксибензойной кислоты, фенол, хлорбутанол, бензиловый спирт, тимерозал, бензалкония хлорид и цетилпиридиния хлорид. Буферы, упомянутые ранее, также как декстрозу и хлорид натрия, можно применять для регулирования изотоничности раствора при необходимости.
Наполнители обычно используют в методике лиофилизации для облегчения процесса и/или обеспечения объемной и/или механической целостности лиофилизированного остатка. Наполнитель означает легко растворимый в воде твердый разбавитель в виде частиц, который при совместной лиофилизации с соединением или его солью обеспечивает физическую стабильность остатка при лиофилизации, более оптимальный процесс лиофильной сушки и быстрое и полное восстановление. Наполнитель также можно применять для придания раствору изотоничности.
Водорастворимый наполнитель может представлять собой любое фармацевтически приемлемое инертное твердое вещество, обычно используемое для лиофилизации. Такие наполнители включают, например, сахара, такие как глюкоза, мальтоза, сахароза и лактоза; полиатомные спирты, такие как сорбит или маннит; аминокислоты, такие как глицин; полимеры, такие как поливинилпирролидон; и полисахариды, такие как декстран.
Соотношение массы наполнителя к массе активного соединения обычно находится в диапазоне от примерно 1 до примерно 5, например, от примерно 1 до примерно 3, например, в диапазоне примерно от 1 до 2.
В качестве альтернативы, составы могут быть получены в виде раствора, который может быть концентрированным и герметично закупоренным в подходящей виале. Стерилизацию дозированных форм можно проводить путем фильтрования или автоклавирования виал и их содержимого на подходящих стадиях способа получения состава. Полученный состав может потребовать дополнительного разведения или приготовления перед доставкой в организм, например, разведение в подходящих стерильных упаковках для инфузии.
Растворы и суспензии для немедленного введения в виде инъекции могут быть получены из стерильных порошков, гранул и таблеток.
Согласно одному предпочтительному варианту реализации изобретения, фармацевтическая композиция представлена в форме, подходящей для внутривенного введения, например, посредством инъекции или инфузии.
Согласно другому предпочтительному варианту реализации, фармацевтическая композиция представлена в форме, подходящей для подкожного (s.c.) введения.
Фармацевтические дозированные формы, подходящие для перорального применения, включают таблетки, капсулы, каплеты, пилюли, таблетки для рассасывания, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, облатки или пластыри и буккальные пластыри.
Фармацевтические композиции, содержащие соединения формулы (I) могут быть приготовлены в виде лекарственных форм в соответствии с известными методиками, см, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Таким образом, таблетированные композиции могут содержать единицу дозирования активного соединения вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например, лактоза, сахароза, сорбит или маннит; и/или разбавителем, не являющимся производным сахаров, таким как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза или ее производное, такое как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, и крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты в качестве связывающих и гранулирующих агентов, таких как поливинилпирролидон, дезинтегрантов (например, набухающие поперечно-сшитые полимеры, такие как поперечно-сшитая карбоксиметилцеллюлоза), лубриканты (например, стеараты), консерванты (например, парабены), антиоксиданты (например, ВНТ), буферные агенты (например, фосфатный или цитратный буферы) и шипучие агенты, такие как смеси цитрат/бикарбонат. Такие вспомогательные вещества хорошо известны и не нуждаются в подробном описании в рамках настоящей заявки.
Лекарственные формы в виде капсул могут представлять собой твердые желатиновые или мягкие желатиновые варианты, и могут содержать активный компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы могут быть получены из животного желатина или его синтетического или растительного эквивалентов.
Твердые препаративные лекарственные формы (например, таблетки, капсулы и т.д.) могут быть покрыты или не покрыты оболочкой, но обычно имеют покрытие, например, покрытие защитной пленки (например, воск или глазурь) или имеют покрытие для контролируемого высвобождения. Покрытие (например, полимер типа Eudragit ™) может быть разработан для высвобождения активного соединения в желаемом месте желудочно-кишечного тракта. Таким образом, покрытие может быть выбрано для разрушения при определенных условиях pH внутри желудочно-кишечного тракта, таким образом селективно высвобождая соединение в желудке или подвздошной кишке или двенадцатиперстной кишке.
Вместо этого или в дополнение к покрытию, лекарственное средство может быть представлено в твердой матрице, включающей агент контролируемого высвобождения, например, агента замедленного высвобождения, который может быть приспособлен для селективного высвобождения соединения в условиях различной кислотности или основности в желудочно-кишечном тракте. Альтернативно, материал матрицы или покрытия, отсрочивающий высвобождение, может иметь вид эродированного полимера (например, полимер малеинового ангидрида), который по существу непрерывно подвергается эрозии по мере продвижения лекарственной формы по желудочно-кишечному тракту. В качестве дополнительной альтернативы, активное соединение может быть включено в состав системы доставки, которая обеспечивает осмотический контроль за высвобождением соединения. Осмотическое высвобождение и другие препараты с замедленным высвобождением или непрерывным высвобождением могут быть получены в соответствии со способами, хорошо известными специалистам в данной области.
Соединение формулы (1), определенное в любом из Вариантов реализации 1.0-1.116, или его пролекарство, может быть представлено в виде лекарственной формы с носителем и введено в форме наночастиц. Наночастицы обеспечивают возможность непосредственного проникновения в клетку. Системы доставки лекарственных средств с помощью наночастиц описаны в "Nanoparticle Technology for Drug Delivery", edited by Ram В Gupta and Uday B. Kompella, Informa Healthcare, ISBN 9781574448573, published 13th March 2006. Nanoparticles for drug delivery are also described in J. Control. Release, 2003, 91 (1-2), 167-172, and in Sinha et al., Mol. Cancer Ther. August 1, (2006) 5, 1909.
Фармацевтические препараты могут быть представлены пациенту в виде "Упаковки для пациента", содержащей весь курс лечения в одной упаковке, обычно в блистере. Упаковки для пациента имеют преимущество по сравнению с традиционными рецептурными препаратами, когда фармацевт отделяет необходимое пациенту количество лекарства от объемной массы, заключающееся в том, что пациент всегда имеет доступ к инструкции, содержащейся в упаковке для пациента, и, как правило, отсутствующей в рецептах. Было показано, что включение вкладыша в упаковку способствует улучшению соблюдения пациентом инструкций врача.
Композиции для местного применения и назальной доставки препарата включают мази, кремы, спреи, пластыри, гели, жидкие капли и вставки (например, внутриглазные вставки). Такие композиции могут быть приготовлены известными способами.
Композиции для парентерального введения обычно представлены в виде стерильных водных или масляных растворов или тонкодисперсных суспензий, или могут быть представлены в форме мелкодисперсного стерильного порошка для немедленного применения с использованием стерильной воды для инъекций.
Примеры соединений для ректального или внутривагинального введения, включают пессарии и суппозитории, которые могут быть, например, образованы из имеющего определенную форму пластичного или воскообразного материала, содержащего активное соединение.
Композиции для введения путем ингаляции могут иметь форму вдыхаемых порошковых композиций, или жидких или порошковых аэрозолей. Их можно вводить в стандартной форме с помощью порошкового ингалятора или аэрозольных дозаторов. Такие устройства хорошо известны в данной области. Для введения путем ингаляции, в порошкообразные составы обычно включают активное соединение вместе с инертным твердым порошкообразным разбавителем, таким как лактоза.
Соединения согласно Вариантам реализации 1.0-1.116 обычно представлены в дозированной лекарственной форме и, соответственно, будут обычно содержать достаточно соединения для получения желаемого уровня биологической активности. Например, препарат может содержать от 1 нанограмма до 2 грамм активного ингредиента, например, от 1 нанограмма до 2 миллиграмм активного ингредиента. В пределах этих диапазонов, определенные поддиапазоны из содержания соединения - это от 0,1 мг до 2 г активного ингредиента (как правило, от 10 мг до 1 г, например, 50 мг до 500 мг), или от 1 мкг до 20 мг (например, 1 мкг до 10 миллиграмм, или, например, 0,1 мг до 2 миллиграмм активного ингредиента).
Для композиций для перорального введения стандартная лекарственная форма может содержать от 1 мг до 2 г, чаще от 10 мг до 1 г, например, от 50 мг до 1 г, например, от 100 мг до 1 г активного соединения.
Активное соединение может быть введено пациенту, нуждающемуся в этом (например, пациенту-человеку или животному) в количестве, достаточном для достижения желаемого терапевтического эффекта.
Способы лечения
Предполагается, что соединения согласно Вариантам реализации 1.0-1.116 являются подходящими либо в качестве отдельных химиотерапевтических агентов, или, что встречается более часто, в комбинированной терапии с химиотерапевтическими агентами или лучевой терапией для профилактики или ряда пролиферативных заболеваний или состояний. Примеры таких заболеваний и состояний представлены выше.
Конкретные примеры химиотерапевтических агентов, которые могут быть введены совместно с соединениями согласно Вариантам реализации 1.0-1.116, включают:
- Ингибиторы топоизомеразы I
- Антиметаболиты
- Агенты, нацеливающие на тубулин
- Ингибиторы ДНК-связывающих агентов и топоизомеразы II
- Ингибиторы EGFR (например, Гефитиниб- см. Biochemical Pharmacology 78 2009 460-468)
- Ингибиторы mTOR (например, Эверолимус)
- Ингибиторы пути PI3K (например, PI3K, PDK1)
- Ингибиторы Akt
- Алкилирующие агенты (например, темозоломид, циклофосфамид)
- Моноклональные антитела (например, антитела, нацеливающие на CTLA-4, PD-1, PD-L1, CD52 или CD20)
- Антигормоны
- Ингибиторы сигнальной трансдукции
- Ингибиторы протеасомы
- ДНК метилтрансферазы
- Циокины и ретиноиды
- Активированные гипоксией ДНК повреждающие агенты (например, Тирапазамин)
- Ингибиторы ароматазы
- Антитела против Her2, (см, например, http://www.wipo.int/pctdb/en/wo.jsp?wo=2007056118)
- Ингибиторы ангиогенеза
- Ингибиторы HDAC
- Ингибиторы MEK
- Ингибиторы B-Raf
- Ингибиторы ERK
- Низкомолекулярные ингибиторы HER2 (например, лапатиниб)
- Ингибиторы Bcr-Abl-тирозинкиназы (например, иматиниб)
- Ингибитор CDK4/6, например, ибранс
- Ингибиторы VEGFR
- Ингибиторы IGFR-1
- Ингибиторы сигнального пути Hedgehog
Дополнительные примеры химиотерапевтических агентов, которые могут быть введены совместно с соединением, определенным в любом из Вариантов реализации 1.0-1.116, включают:
- Ингибиторы Torc 1
- Ингибиторы пути PI3K (например, PI3K, PDK1)
- Ингибиторы EGFR (например, Гефитиниб - см. Everolimus restores gefitinib sensitivity in resistant non-small cell lung cancer cell lines, Biochemical Pharmacology 78 2009 460-468)
- Таксаны (например, пакситаксел, доцетаксел, кабазитаксел)
- Агенты на основе платины (например, цисплатин, карбоплатин, оксалиплатин)
- Антрациклины (например, доксорубицин)
- Ингибиторы семейства белков Bcl-2, например, АВТ263 (навитоклакс), ингибитор Bcl-2/Bcl-extra large (Bcl-xL)
Одна конкретная комбинация включает соединение согласно любому из Вариантов реализации 1.0-1.116 совместно с ингибитором EGFR, таким как Гефитиниб или Эрлотиниб или с ингибитором mTOR, таким как Эверолимус.
Указанные соединения также могут быть введены в сочетании с лучевой терапией.
Соединения можно вводить в течение длительного периода времени для поддержания полезных терапевтических эффектов или их можно вводить только в течение короткого периода времени. В качестве альтернативы, их можно вводить пульсирующим или непрерывным способом.
Соединения согласно настоящему изобретению вводят в эффективном количестве, т.е., в количестве, эффективном для достижения желаемого терапевтического эффекта. Например, «эффективное количество» может представлять собой количество соединения, которое при введении субъекту, страдающему от рака, замедляет рост опухоли, облегчает симптомы заболевания и/или увеличивает продолжительность жизни.
Количество соединения-ингибитора P70S6 согласно настоящему изобретению, вводимого субъекту, зависит от типа и тяжести заболевания или состояния и характеристик субъекта, таких как общее состояние здоровья, возраст, пол, масса тела и переносимость лекарственных средств. Специалист в области техники способен определить подходящие дозировки, в зависимости от этих и других факторов.
Обычно соединения вводят субъекту, нуждающемуся в таком введении, например, пациенту-человеку или животному, предпочтительно человеку.
Обычная суточная доза соединения согласно любому из Вариантов реализации 1.0-1.116 может находиться в диапазоне от 100 пикограмм до 100 миллиграмм на килограмм массы тела, чаще от 5 нанограмм до 25 миллиграмм на килограмм массы тела, чаще от 10 нанограмм до 15 миллиграмм на килограмм массы тела (например, от 10 нанограмм до 10 миллиграмм, и еще чаще от 1 микрограмма на килограмм до 20 микрограмм на килограмм, например, от 1 микрограмма до 10 миллиграмм на килограмм), хотя в случае необходимости могут быть введены более высокие или низкие дозы. Соединения можно вводить ежедневно или на регулярной основе каждые 2, или 3, или 4, или 5, или 6, или 7, или 10 или 14, или 21, или 28 дней, например.
Согласно одному конкретному режиму дозирования, пациенту вводят инфузию соединения в течение одного часа ежедневно до 10 дней, в частности, до пяти дней в течение одной недели, и лечение повторяют с желаемыми интервалами, такими как от двух до четырех недель, в частности, каждые три недели.
Более конкретно, пациент получает соединение в виде инфузии в течение одного часа ежедневно в течение 5 дней и лечение повторяют каждые три недели.
Согласно другому конкретному режиму дозирования, пациенту вводят инфузию в течение от 30 минут до 1 часа с последующим поддержанием переменной продолжительности инфузий, например, от 1 до 5 часов, например, 3 часа.
Согласно дополнительному конкретному режиму дозирования, пациенту непрерывно вводят инфузию в течение периода от 12 часов до 5 дней, и в частности, непрерывную инфузию в течение от 24 часов до 72 часов.
Однако, в конечном счете, количество вводимого соединения и тип используемой композиции должны соответствовать природе заболевания или физиологического состояния, которое лечат, и определяются на усмотрение лечащего врача.
Способы диагностики
Перед введением соединения согласно любому из Вариантов реализации 1.0-1.116, для пациента может быть проведен скрининг, чтобы определить, будет ли заболевание или состояние, от которого страдает пациент, поддаваться лечению с использованием соединения, обладающего активностью против киназы p70S6.
Например, биологический образец, полученный от пациента, может быть проанализирован для определения того, будет ли состояние или заболевание, такое как рак, от которого страдает или возможно будет страдать пациент, таким, которое характеризуется генетической аномальностью или аномальной экспрессией белка, который приводит к повышающей регуляции киназы p70S6 или к сенсибилизации пути до нормальной активности киназы p70S6 или к избыточной экспрессии киназы p70S6. Термин повышающая регуляция включает повышенную экспрессию или избыточную экспрессию, включая амплификацию гена (т.е. множественные копии гена) и повышенную экспрессию посредством транскрипционного эффекта, и гиперактивность и активацию, включая активацию посредством мутаций. Таким образом, может быть проведено диагностическое исследование пациента с целью обнаружения маркерного признака повышающей регуляции киназы p70S6. Термин «диагноз» включает скрининг. Под маркером в настоящей заявке подразумеваются генетические маркеры, включая, например, измерение композиции ДНК для идентификации мутаций p70S6. Термин «маркер» также включает маркеры, которые являются признаками повышающей регуляции p70S6, включая активность ферментов, концентрацию ферментов и состояние ферментов (например, фосфорилированный фермент или нет) и уровни мРНК вышеуказанных белков.
Опухоли с повышающей регуляцией киназы p70S6 могут быть особенно чувствительными к ингибиторам p70S6. Предпочтительно может быть проведен скрининг опухолей в отношении повышающей регуляции p70S6. Таким образом, может быть проведено диагностическое исследование пациента с целью обнаружения маркерного признака повышающей регуляции киназы p70S6. Диагностические исследования обычно проводят с использованием биологических образцов, выбранных из образцов биопсии опухолей, образцов крови (выделенной и обогащенной рассеянными опухолевыми клетками), биопсии кала, слюны, анализа хромосом, плевральной жидкости, перитонеальной жидкости.
Способы идентификации и анализа мутаций и положительной регуляции белков хорошо известны специалистам в области техники. Способы скрининга могут включать, но не ограниваются указанным, стандартные способы, такие как полимеразно-цепная реакция с обратной транскриптазой ((КТ-ПЦР), или in-situ гибридизация.
При скрининге методом КТ-ПЦР, проводят оценку уровня мРНК в опухоли путем создания кДНК копии мРНК с последующей амплификацией кДНК методом ПЦР. Методы ПЦР-амплификации, выбор праймеров и условий амплификации известны специалисту в данной области техники. Манипулирование нуклеиновыми кислотами и ПЦР проводят с помощью стандартных методов, как описано, например, в Ausubel, F.M. et al., eds. Current Protocols in Molecular Biology, 2004, John Wiley & Sons Inc., или Innis, M.A. et-al., eds. PCR Protocols: a guide to methods and applications, 1990, Academic Press, San Diego. Реакции и манипуляции с нуклеиновыми кислотами также описаны в Sambrook et al., 2001, 3rd Ed, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press. В качестве альтернативы можно использовать коммерчески доступный набор для КТ-ПЦР (например, Roche Molecular Biochemicals) или методики, представленные в патентах США 4,666,828; 4,683,202; 4,801,531; 5,192,659, 5,272,057, 5,882,864, и 6,218,529, включенных в настоящую заявку посредством ссылки.
Примером метода гибридизации in-situ для оценки экспрессии мРНК может являться флуоресцентная in-situ гибридизация (FISH) (см., Angerer, 1987 Meth. Enzymol., 152: 649).
Обычно, гибридизация in situ включает следующие основные стадии: (1) фиксирование ткани для анализа; (2) предгибридизационная обработка образца для повышения доступности целевой нуклеиновой кислоты и для уменьшения неспецифического связывания; (3) гибридизацию смеси нуклеиновых кислот с нуклеиновой кислотой в биологической структуре или ткани; (4) пост- гибридизационную промывку для удаления фрагментов нуклеиновых кислот, не связанных при гибридизации, и (5) обнаружение фрагментов гибридизованной нуклеиновой кислоты. Зонды, используемые для такого применения, обычно являются мечеными, например, радиоизотопами или флуоресцентными репортерами. Предпочтительные зонды являются достаточно длинными, например, приблизительно от 50,100, или 200 нуклеотидов до приблизительно 1000 или более нуклеотидов, с целью обеспечения возможности специфической гибридизации с целевой нуклеиновой кислотой (кислотами) в строгих условиях. Стандартные способы проведения FISH описаны в Ausubel, F.M. et al., eds. Current Protocols in Molecular Biology, 2004, John Wiley & Sons Inc and Fluorescence In Situ Hybridization: Technical Overview by John M. S. Bartlett in Molecular Diagnosis of Cancer, Methods and Protocols, 2nd ed.; ISBN: 1-59259-760-2; March 2004, pps. 077-088; Series: Methods in Molecular Medicine.
Альтернативно, продукты белка, экспрессируемые из мРНК, можно анализировать с помощью иммуногистохимии образцов опухоли, твердофазного иммуноферментного анализа с планшетами для микротитрования, методом вэстерн-блоттинга, 2-мерным электрофорезом на SDS-полиакриламидном геле, ELISA, проточной цитометрией и другими способами, известными в данной области техники для обнаружения определенных белков. Методы обнаружения включают применение сайт-специфических антител. Специалисту в области техники будет понятно, что в данном случае применимы все хорошо известные способы обнаружения повышающей регуляции киназы p70S6.
Соответственно, согласно другому варианту реализации изобретения (Вариант реализации 5.1) предложен способ диагностики и лечения заболевания или состояния, опосредуемого киназой p70S6, включающий (i) проведение скрининга пациента с целью определения, будет ли заболевание или состояние, от которого страдает пациент, поддаваться лечению с применением соединения, обладающего активностью против киназы p70S6; и (ii) если обнаруживается, что заболевание или состояние, от которого страдает пациент, будет поддаваться лечению, введение пациенту соединения, определенного в любом из Вариантов реализации 1.0-1.116.
Согласно другому варианту реализации (Вариант реализации 5.2) предложено применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для получения лекарственного средства для лечения или профилактики заболевания или состояния у пациента, страдающего от или предрасположенного к, как было определено с помощью скрининга, заболевания или состояния, поддающегося лечению с применением соединения, обладающего активностью против киназы p70S6.
Согласно дополнительному варианту реализации (Вариант реализации 5.3), предложено соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения в лечении или профилактике заболевания или состояния у пациента, страдающего от или предрасположенного к, как было определено с помощью скрининга, заболевания или состояния, поддающегося лечению с применением соединения, обладающего активностью против киназы p70S6.
Согласно другому варианту реализации изобретения (Вариант реализации 5.4), предложен способ диагностики и лечения заболевания или состояния, характеризующегося повышающей регуляцией киназы p70S6 или присутствием мутированной формы p70S6, включающий (i) проведение скрининга пациента с целью определения, будет ли заболевание или состояние, от которого страдает пациент, поддаваться лечению с применением соединения, обладающего активностью против киназы p70S6; и (ii) если обнаруживается, что заболевание или состояние, от которого страдает пациент, будет поддаваться лечению, введение пациенту соединения, определенного в любом из Вариантов реализации 1.0-1.116.
Согласно дополнительному варианту реализации (Вариант реализации 5.5) предложен способ лечения заболевания или состояния, характеризующегося повышающей регуляцией киназы p70S6 или присутствием мутированной формы p70S6, включающий введение терапевтически эффективного количества соединения, определенного в любом из Вариантов реализации 1.0-1.116 пациенту, страдающему от или предрасположенному к, как было определено с помощью скрининга, заболевания или состояния, поддающегося лечению с применением соединения, обладающего активностью против киназы p70S6.
Согласно другому варианту реализации изобретения (Вариант реализации 5.6), предложен способ применения соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для лечения или профилактики заболевания, состояния или нарушения, определенного в любом из Вариантов реализации 2.1-2.43 у пациента, страдающего от или предрасположенного к, как было определено с помощью скрининга, заболевания или состояния, поддающегося лечению с применением соединения, обладающего активностью против киназы p70S6.
Согласно дополнительному варианту реализации (Вариант реализации 5.7) предложено соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения в лечении или профилактике заболевания или расстройства мозга у пациента, страдающего от или предрасположенного к заболеванию, состоянию или нарушению, определенному в любом из Вариантов реализации 2.1-2.43, поддающегося лечению с применением соединения, обладающего активностью против киназы p70S6.
Трижды негативный рак молочной железы характеризуется тем, что указанный рак не экспрессирует гены рецептора эстрогена (ER), рецептора прогестерона (PR) или HER2. Присутствие ER и PR можно определить стандартными методами иммуногистохимического окрашивания (см, например, Narod et al, Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence, Clin Cancer Res August 1,2007 13; 4429). В качестве альтернативы можно оценить экспрессию генов этих белков такими методами, как количественная полимеразная цепная реакция в реальном времени (QRT-PCR) с коммерчески доступными методами анализа ПЦР, (см., Jozefczuk et al, Quantitative real-time PCR-based analysis of gene expression, Methods Enzymol. 2011; 500:99-109).
Избыточную экспрессию белка HER2 можно оценить с использованием моноклонального антитела СВ11 в типичных парафиновых срезах каждой опухоли с использованием метода пероксидазы-антипероксидазы для иммуногистохимического анализа. Опухоль определяется как положительная по HER2, когда наблюдается сильное полное окрашивание мембраны по меньшей мере в 10% опухолевых клеток (Narod et al, Clin Cancer Res August 1, 2007 13; 4429).
Соответственно, согласно другому варианту реализации изобретения (Вариант реализации 5.8) предложен способ диагностики и лечения рака, определенного в любом из Вариантов реализации 2.10-2.18, включающий (i) проведение скрининга пациента с целью определения, является ли раковое заболевание, от которого страдает или может страдать пациент, раковым заболеванием, не экспрессирующим рецептор эстрогена, рецептор прогестерона и/или HER2; и (ii) если обнаруживается, что раковое заболевание, от которого страдает пациент, будет поддаваться лечению, введение пациенту соединения, определенного в любом из Вариантов реализации 1.0-1.116.
Согласно другому варианту реализации изобретения (Вариант реализации 5.9) предложено применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для лечения или профилактики рака, определенного в любом из Вариантов реализации 2.10-2.18, у пациента, страдающего или предрасположенного к, как было определено с помощью скрининга, от рака, не экспрессирующего рецептор эстрогена, рецептор прогестерона и/или HER2.
Согласно дополнительному варианту реализации (Вариант реализации 5.10) предложено соединение, определенное в любом из Вариантов реализации 1.0-1.116 для применения в лечении или профилактике рака, как определено в любом из Вариантов реализации 2.10-2.18 у пациента, страдающего или предрасположенного к, как было определено с помощью скрининга, от рака, не экспрессирующего рецептор эстрогена, рецептор прогестерона и/или HER2.
Синдром фрагильной Х-хромосомы возникает в результате мутации гена умственной отсталости, вызванной фрагильной Х-хромосомой (fragile X mental retardation 1) (FMR1). У людей, пораженных FXS, ген FMR1 содержит более 45, чаще более 55 и в некоторых случаях более 200 повторов кодона CGG по сравнению с 5-44 повторами, чаще 29 или 30 повторами у здоровых людей. Эта мутация приводит к нарушению экспрессии белка умственной отсталости, вызванной фрагильной Х-хромосомой (fragile X mental retardation 1) FMRP, в результате чего происходит избыточное продуцирование массива белков, обычно контролируемых FMRP. Поскольку FXS является генетическим заболеванием, можно легко диагностировать FXS путем проведения генетического исследования образца крови или кожи у интересующего пациента. Пациенты с FXS не экспрессируют или характеризуются гораздо меньшими уровнями мРНК FMR1, чем здоровые люди. Уровни мРНК FMR1 можно количественно определить методами ПЦР в реальном времени, которые являются коммерчески доступными. Кроме того, размер повторений CGG можно определить выделением геномной ДНК путем высаливания с последующим проведением ПЦР. См, например, Kumari et al. (HUMAN MUTATION, Vol. 35, No. 12, 1485-1494, 2014) где описаны лабораторные методики получения таких данных. Таким образом, биомаркеры, которые позволяют идентифицировать FXS у пациента, включают уровни мРНК FMR1 и наличие избыточно длинных повторов CGG в геномной ДНК пациента.
Соответственно, согласно другому варианту реализации изобретения (Вариант реализации 5.11) предложен способ диагностики и лечения синдрома фрагильной X-хромосомы, включающий (i) проведение скрининга пациента с целью определения, для обнаружения одного или более биомаркеров, указывающих на синдром фрагильной X-хромосомы; и (ii) если такие маркеры обнаруживаются, введение пациенту соединения, определенного в любом из Вариантов реализации 1.0-1.116.
Согласно другому варианту реализации изобретения (Вариант реализации 5.12), предложено применение соединения, определенного в любом из Вариантов реализации 1.0-1.116, для приготовления лекарственного средства для лечения или профилактики синдрома фрагильной Х-хромосомы у пациента, у которого при скрининге были обнаружены один или более биомаркеров, указывающих на наличие синдрома фрагильной Х-хромосомы.
Согласно дополнительному варианту реализации (Вариант реализации 5.13) предложено соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения для лечения или профилактики синдрома фрагильной Х-хромосомы у пациента, у которого при скрининге были обнаружены один или более биомаркеров, указывающих на наличие синдрома фрагильной Х-хромосомы.
Согласно другому варианту реализации изобретения (Вариант реализации 5.14) предложен способ диагностики и лечения синдрома фрагильной Х-хромосомы, включающий (i) проведение скрининга пациента для определения наличия уровней мРНК FMR1, указывающих на наличие синдрома фрагильной Х-хромосомы; и (ii) в случае, если обнаруживается наличие таких уровней мРНК FMR1, введение пациенту соединения, определенного в любом из Вариантом реализации 1.0-1.116.
Согласно другому варианту реализации (Вариант реализации 5.15), предложено применение соединения, определенного в любом из вариантов реализации с 1.0 по 1.116, для приготовления лекарственного средства для лечения или профилактики синдрома фрагильной Х-хромосомы у пациента, у которого при скрининге были обнаружены уровни мРНК FMR1, указывающие на наличие синдрома фрагильной Х-хромосомы.
В дополнительном варианте реализации (Вариант реализации 5.16) предложено соединение, определенное в любом из Вариантов реализации 1.0-1.116, для применения в лечении или профилактике синдрома фрагильной Х-хромосомы у пациента, у которого при скрининге были обнаружены уровни мРНК FMR1, указывающие на наличие синдрома фрагильной Х-хромосомы.
Краткое описание чертежей
На Фигуре 1 представлены результаты, полученные из in vivo модели трижды негативного рака молочной железы, как описано в Примере 44 настоящей заявки, и в частности, показано процентное соотношение мышей, не имеющих опухоли, в зависимости от количества дней, прошедших после имплантации опухоли, при лечении 100 мг/кг соединения согласно Примеру 1 или носителем.
На Фигуре 2 показан объем опухоли в зависимости от количества дней, прошедших после имплантации опухоли, при лечении 100 мг/кг соединения согласно Примеру 1 или носителем, в in vivo модели трижды негативного рака молочной железы, как описано в Примере 44 настоящей заявки.
На Фигуре 3 показана масса печени субъектов-мышей в Примере 44.
На Фигуре 4 показана масса опухоли субъектов-мышей в Примере 44.
ПРИМЕРЫ
ПРИМЕРЫ 1-43
Соединения согласно Примерам 1-43 в Таблице 1 ниже представлены с целью иллюстрации настоящего изобретения.
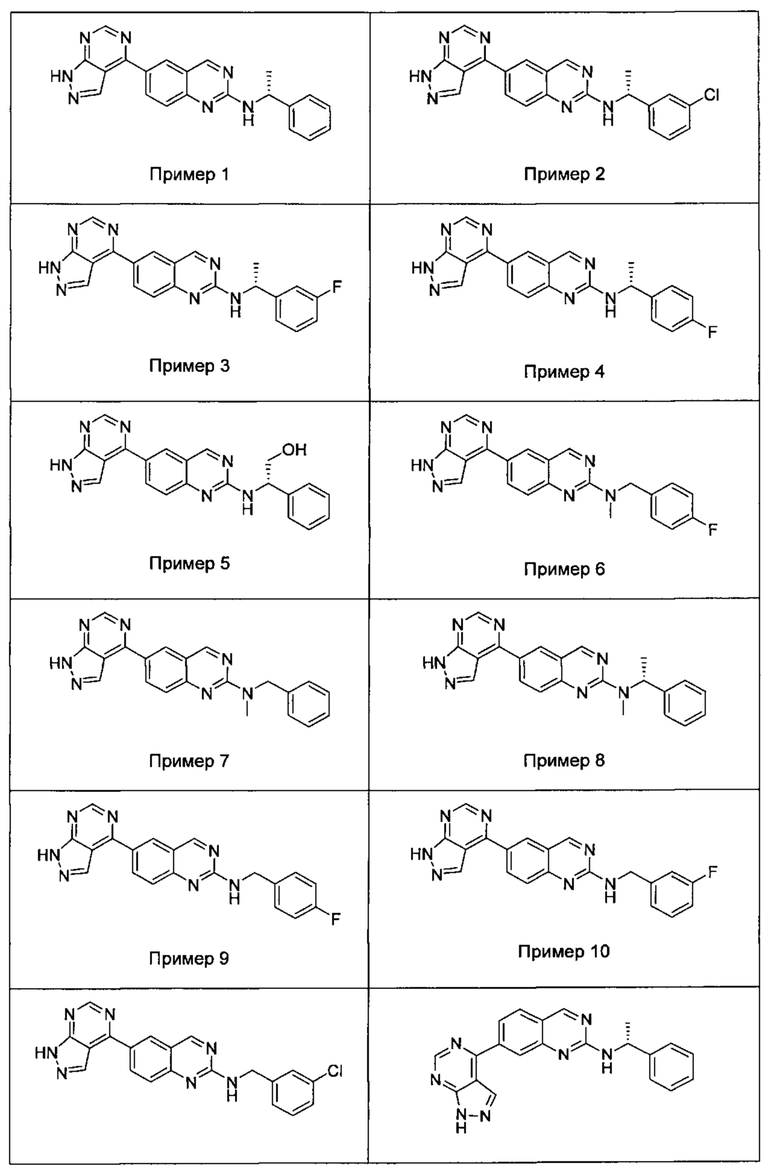
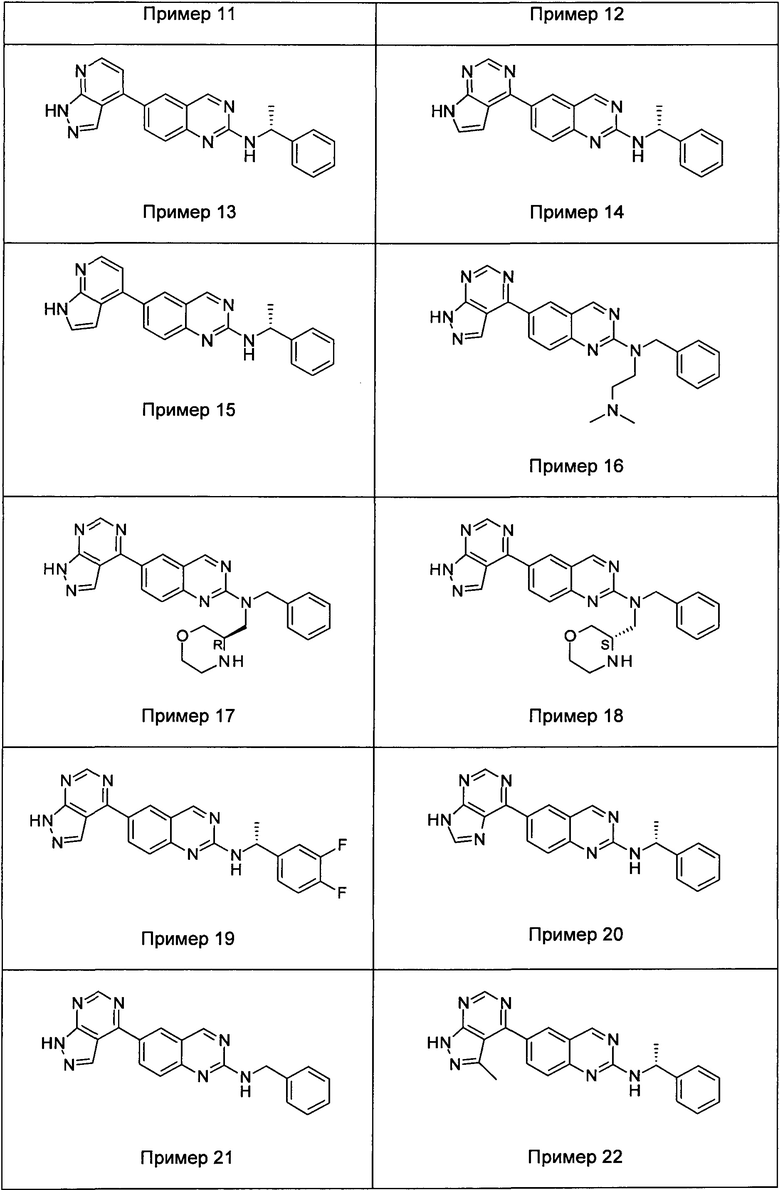
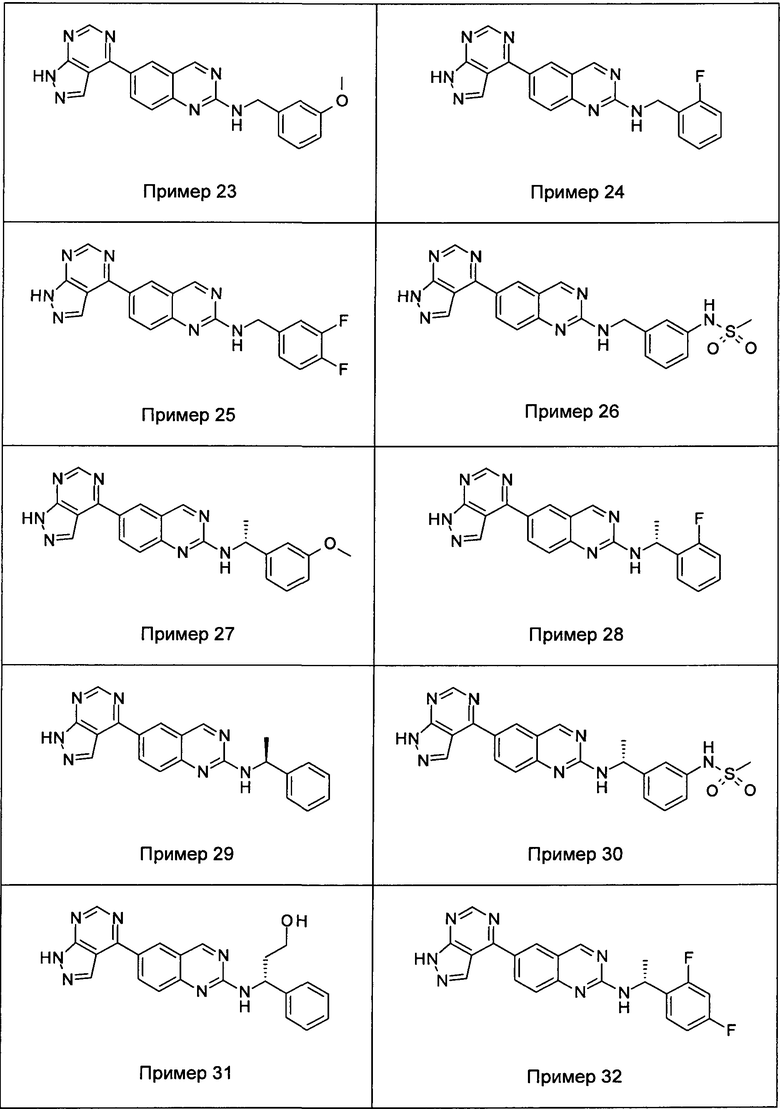
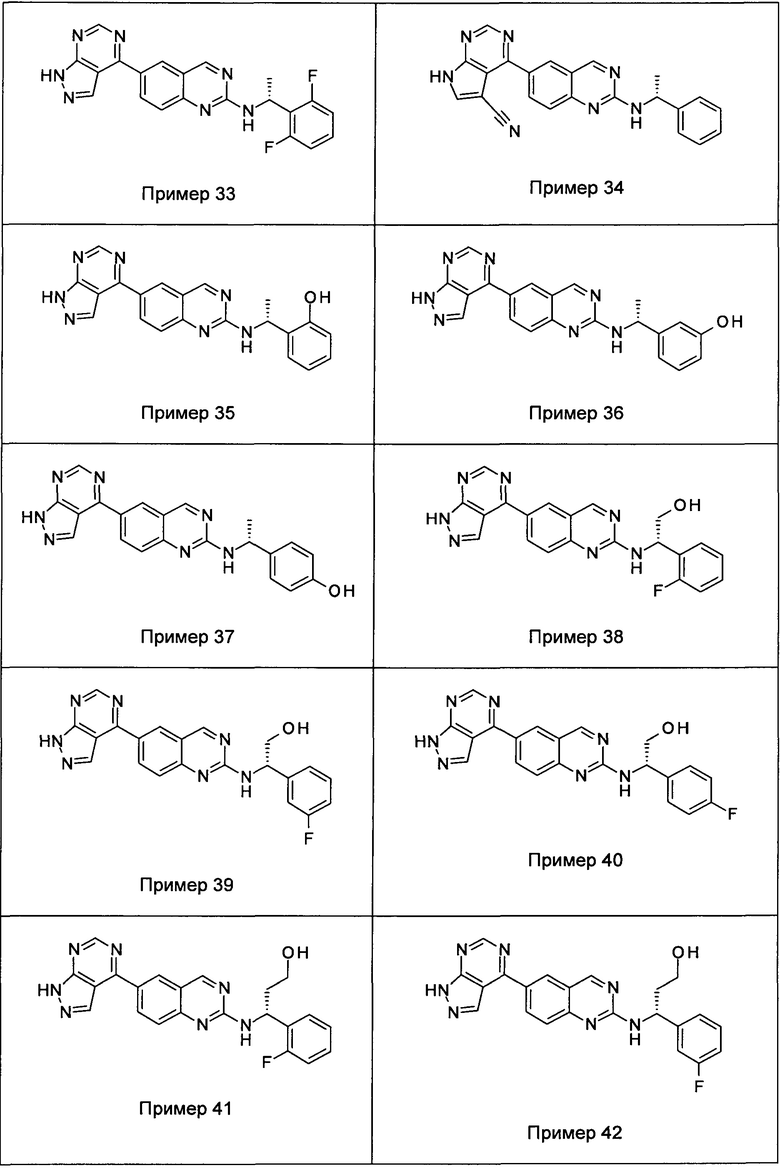
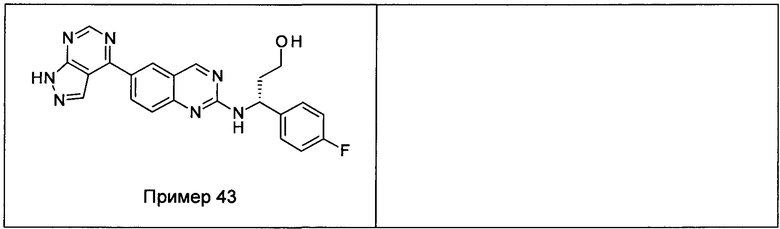
Аналитические данные
1H ЯМР спектры регистрировали на аппарате Bruker 400.
ЖХМС методы:
Анализ ЖХМС проводили с помощью следующего метода(ов):
ЖХМС метод 1
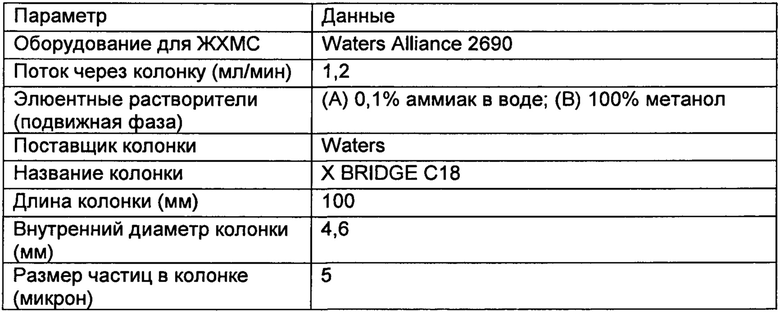
Градиент элюирования:
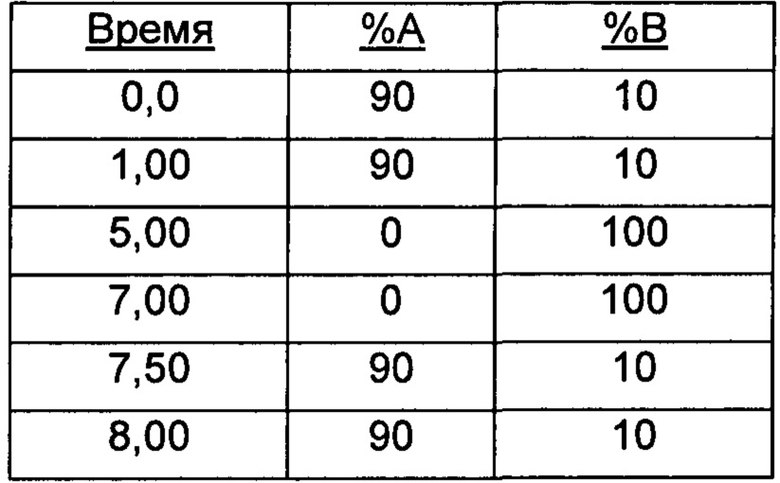
ЖХМС метод 2
ЖХ-МС проводили с использованием устройства Waters Acquity H класса с Qda-Mass детектором с использованием позитивной / негативной ионизации электроспреем.
Использовали следующую колонку: Waters Acquity ВЕН С18 (50×2,1 мм) 1,7 микрон. Скорость потока через колонку: 0,55 мл/мин. Использованная система растворителей: подвижная фаза (А) 5 мм ацетат аммония + 0,1% муравьиная кислота (МК) в воде и (В) 0,1% МК в ацетонитриле в соответствии со следующим градиентом:
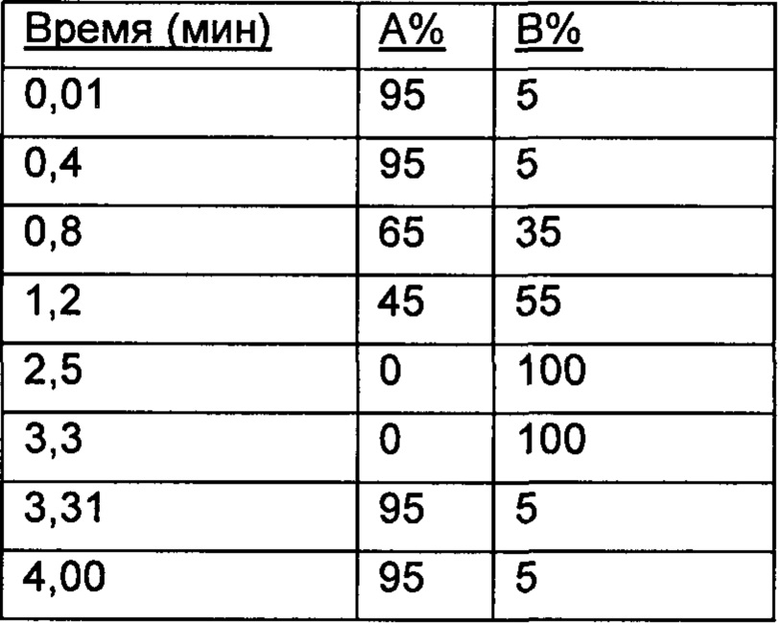
ЖХМС метод 3
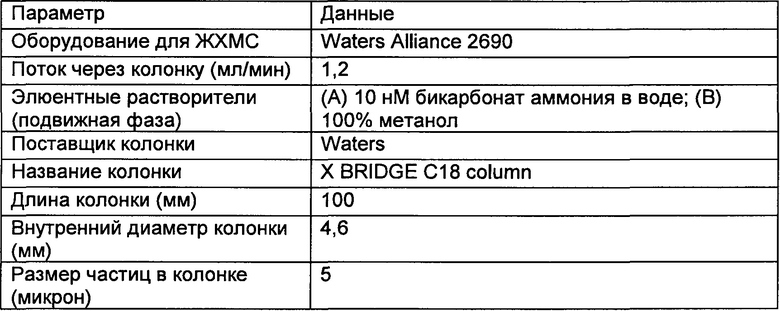
Градиент элюирования:
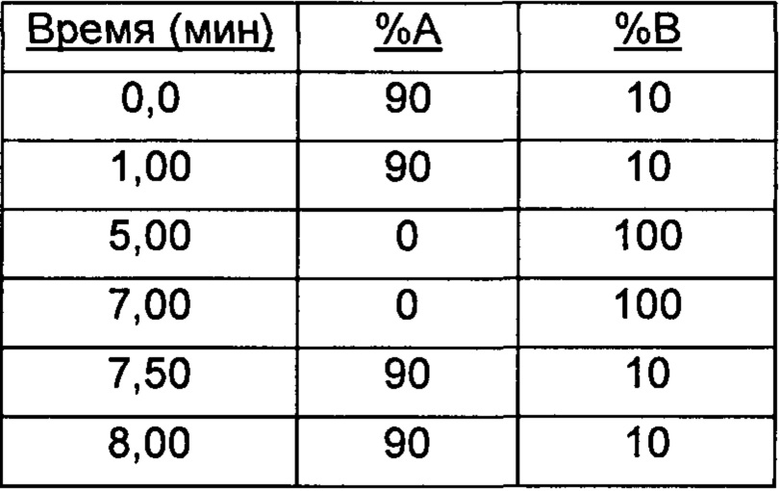
ЖХМС метод 4


Градиент элюирования:
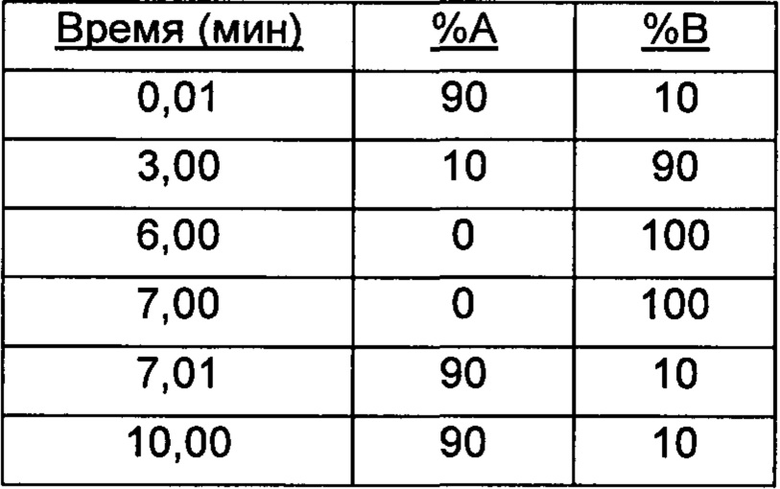
ЖХМС метод 5
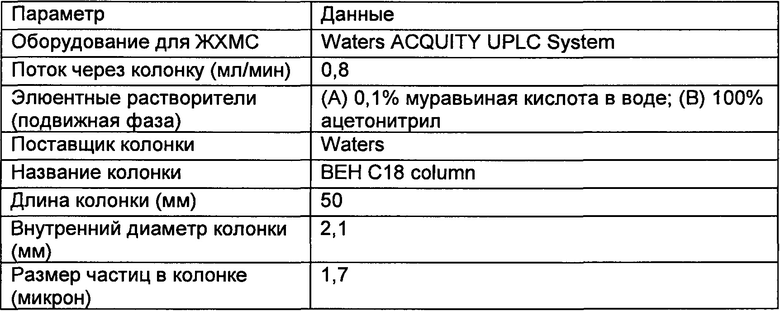
Градиент элюирования:
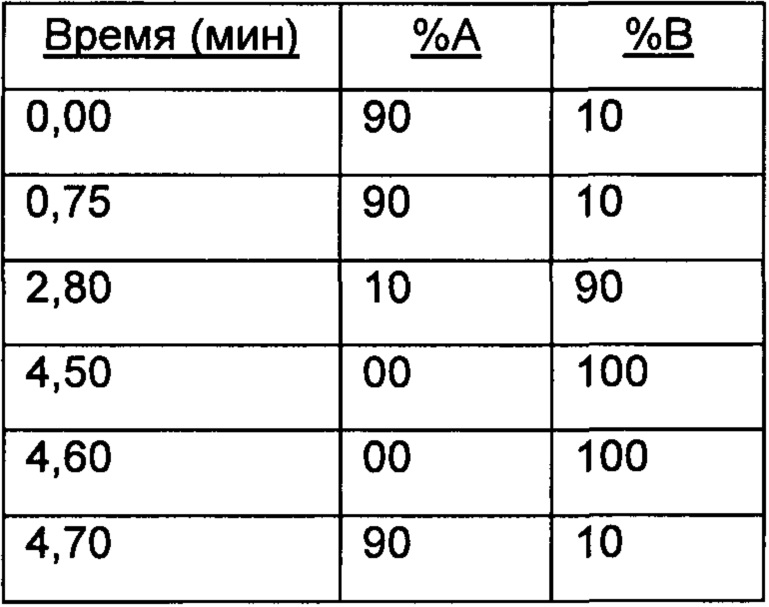
ЖХМС метод 6
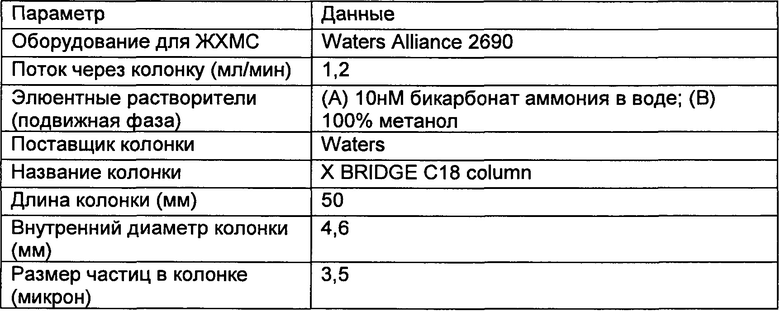
Градиент элюирования:
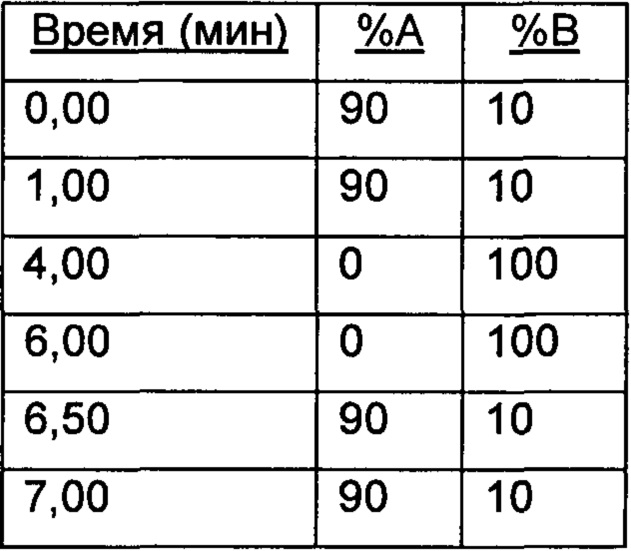
Методы хиральной ВЭЖХ:
Анализ методом хиральной ВЭЖХ проводили с использованием следующих методов:
Хиральная ВЭЖХ метод 1
Хиральную ВЭЖХ проводили с использованием ВЭЖХ аналитического устройства Waters Super-critical Fluid Chromatography (SFC) Investigator с PDA детектором. Скорость потока: 4,0 мл/мин. Объем вводимой пробы: 10 мкл. Используемая аналитическая колонка: Chiralpak IB (250*4,6 мм) с размером частиц 5 микрон. Используемая система растворителей: подвижная фаза (А) диоксид углерода в жидком состоянии (В) 100% метанол согласно следующему градиенту:
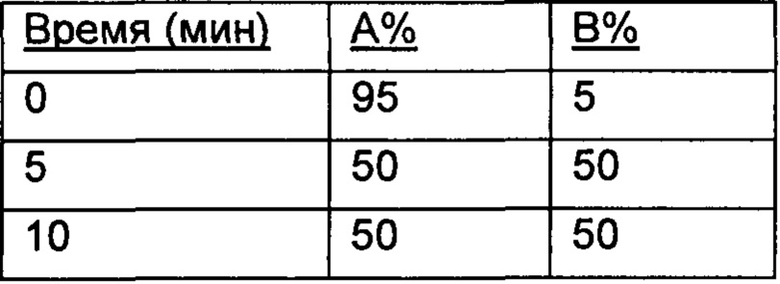
Хиральная ВЭЖХ метод 2
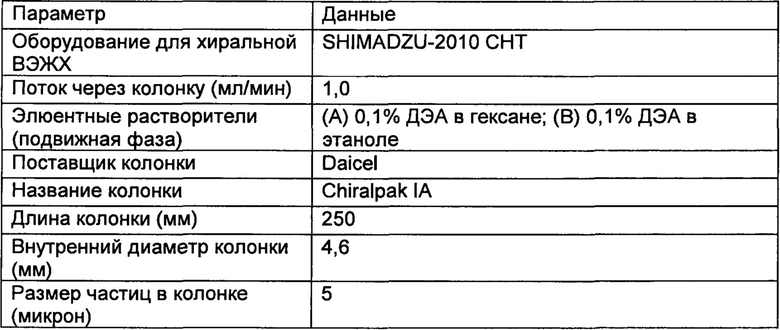
Использовали изократическое элюирование:
Хиральная ВЭЖХ метод 3
Хиральную ВЭЖХ проводили с использованием устройства для ВЭЖХ анализа Waters Super-critical Fluid Chromatography (SFC) Investigator с детектором PDA. Скорость потока: 4,0 мл/ мин. Объем вводимой пробы: 35 мкл. Используемая аналитическая колонка: Chiralpak IB (250*4,6 мм) с размером частиц 5 микрон. Используемая система растворителей: подвижная фаза (А) жидкий диоксид углерода (В) 100% метанол, согласно следующему градиенту:
Хиральная ВЭЖХ метод 4
Хиральную ВЭЖХ проводили с использованием устройства для ВЭЖХ анализа Waters Super-critical Fluid Chromatography (SFC) Investigator с детектором PDA. Скорость потока: 4,0 мл/ мин. Объем вводимой пробы: 20 мкл. Используемая аналитическая колонка: Chiralpak IB (250*4,6 мм) с размером частиц 5 микрон. Используемая система растворителей: подвижная фаза (А) жидкий диоксид углерода (В) 50:50 изопропанол: ацетонитрил, согласно следующему градиенту:
Хиральная ВЭЖХ метод 5
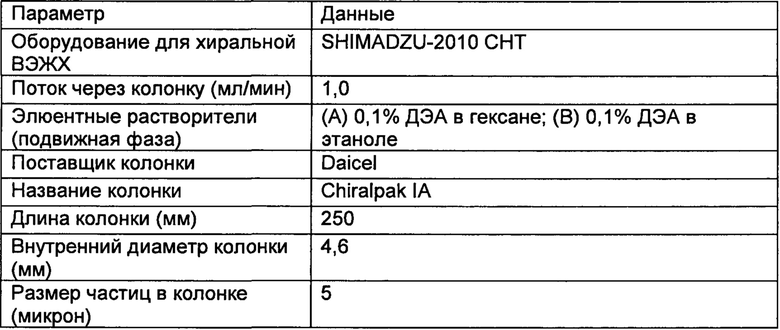
Использовали изократическое элюирование:

Хиральная ВЭЖХ метод 6
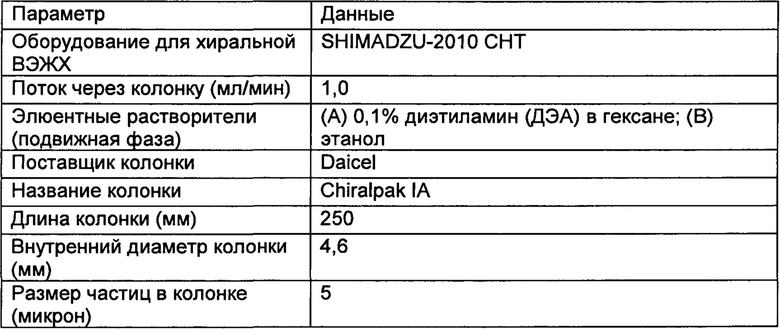
Использовали изократическое элюирование:

Хиральная ВЭЖХ метод 7

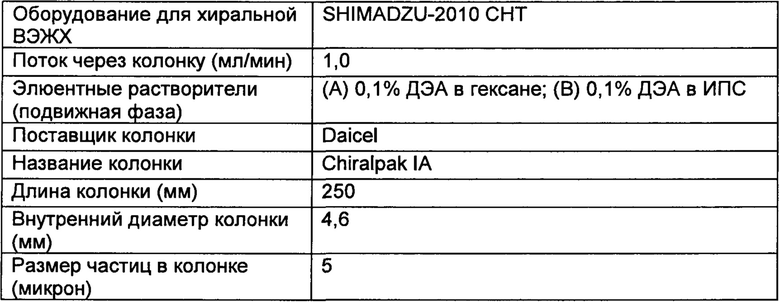
Использовали изократическое элюирование:

Хиральная ВЭЖХ метод 8
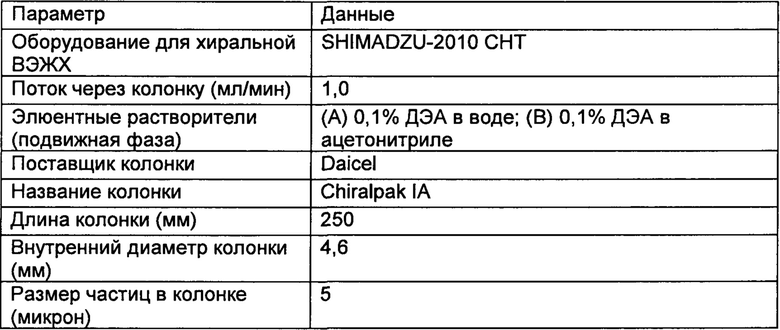
Градиент элюирования:
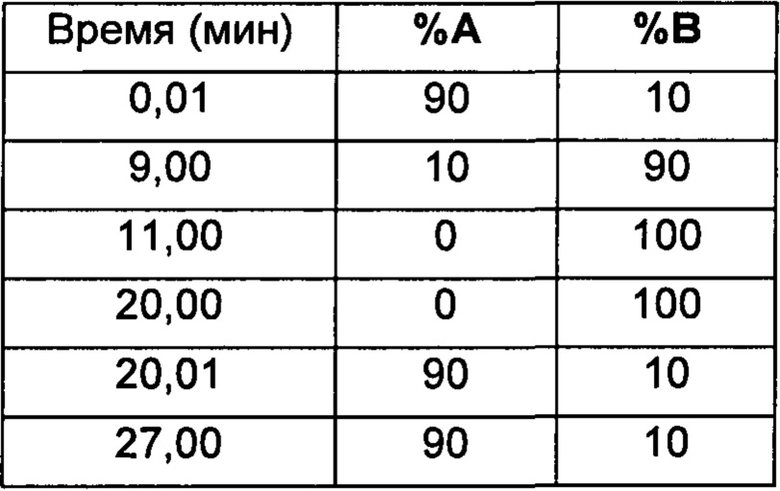
Хиральная ВЭЖХ метод 9

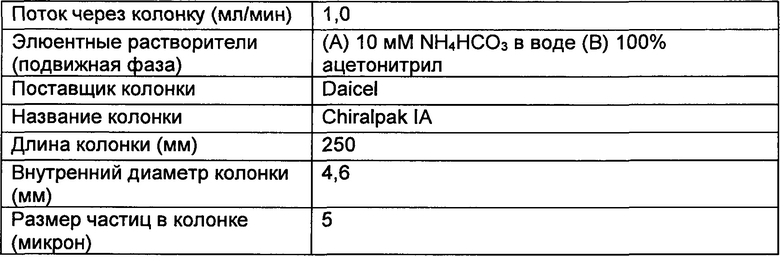
Градиент элюирования:
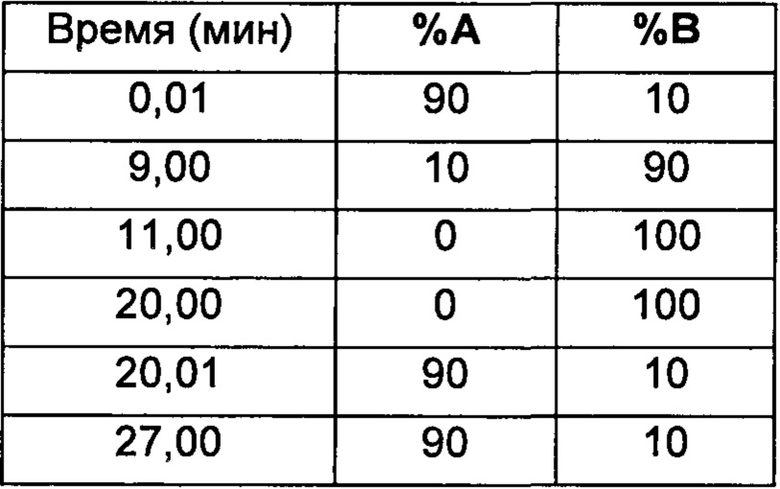
Хиральная ВЭЖХ метод 10
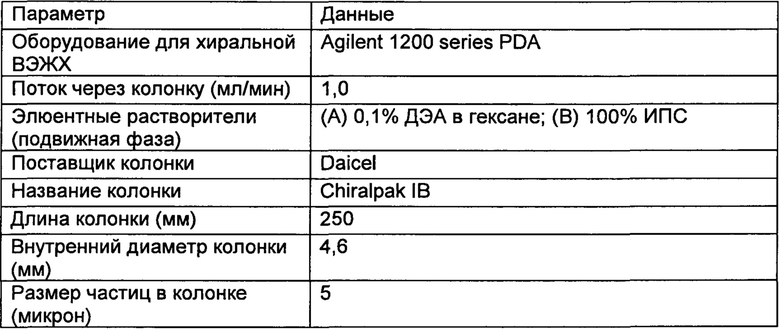
Градиент элюирования:
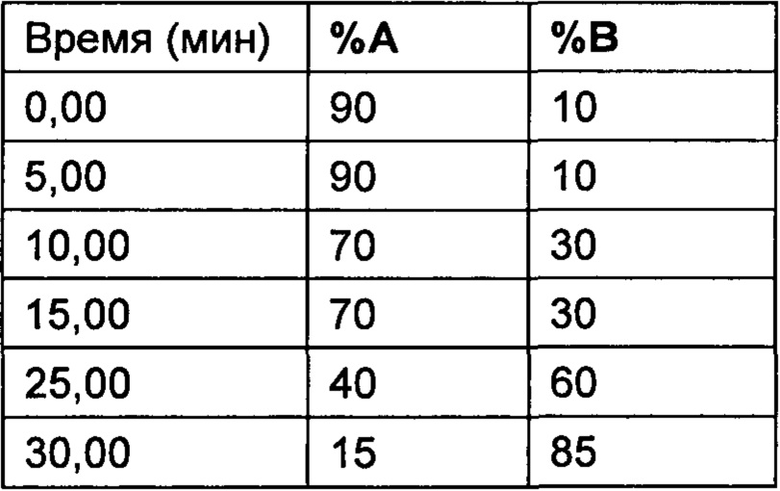

Хиральная ВЭЖХ метод 11
Использовали метод, совпадающий с методом хиральной ВЭЖХ 10, за исключением того, что в качестве растворителя В применяли этанол
Хиральная ВЭЖХ метод 12
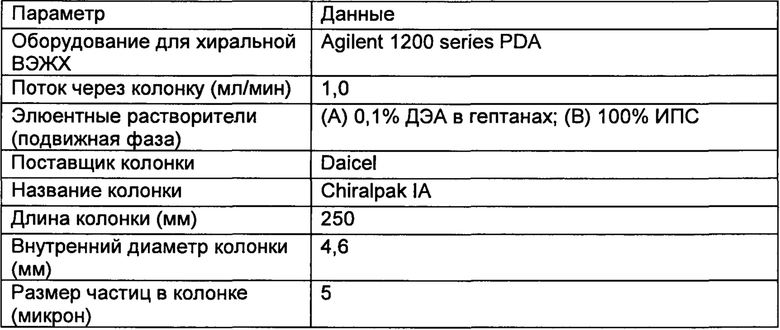
Использовали изократическое элюирование:

Методы препаративной ВЭЖХ:
Препаративная ВЭЖХ метод 1
Очистку проводили с использованием системы препаративной ВЭЖХ Waters PHP-01 с использованием колонки Х Bridge C18 (250 мм длина × 19 мм внутренний объем) с размером частиц 5 микрон и скоростью потока 15 мл/мин. Использованная система растворителей: подвижные фазы (А) 0,1% аммиак в 100% воды и (В) 100% ацетонитрила с использованием следующего градиента:
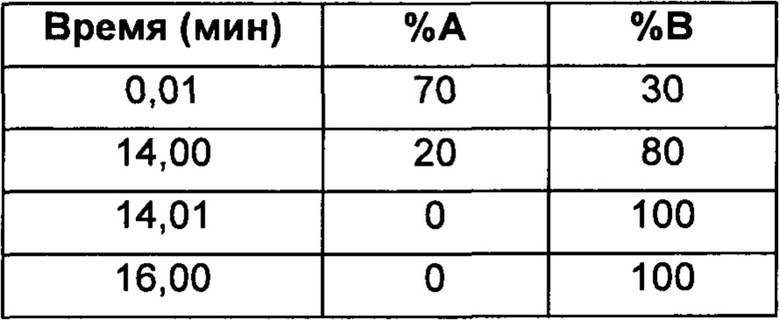

Препаративная ВЭЖХ метод 2
Очистку проводили с использованием системы препаративной ВЭЖХ Waters PHP-01 с использованием колонки Х Bridge C18 (250 мм длина × 30 мм внутренний объем) с размером частиц 5 микрон и скоростью потока 30 мл/мин. Использованная система растворителей: подвижные фазы (А) 0,1% аммиак в 100% воды и (В) 100% ацетонитрила с использованием следующего градиента:
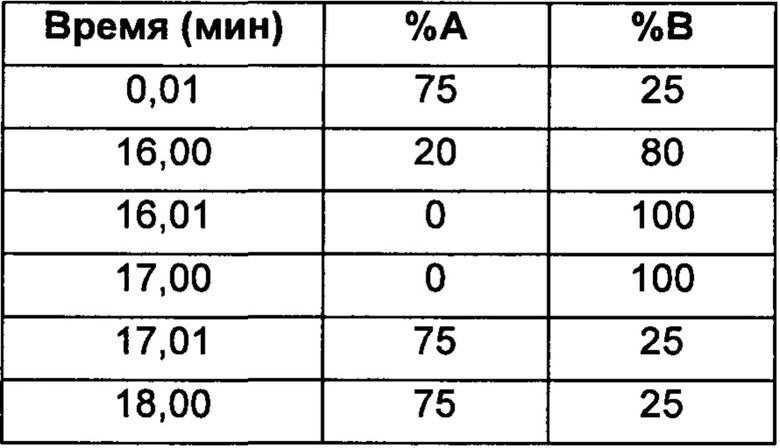
Препаративная ВЭЖХ метод 3

Градиент элюирования:
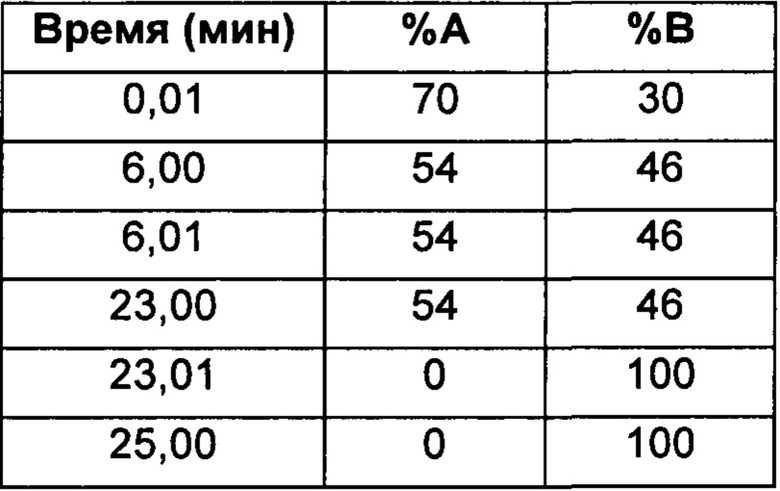

Препаративная ВЭЖХ метод 4
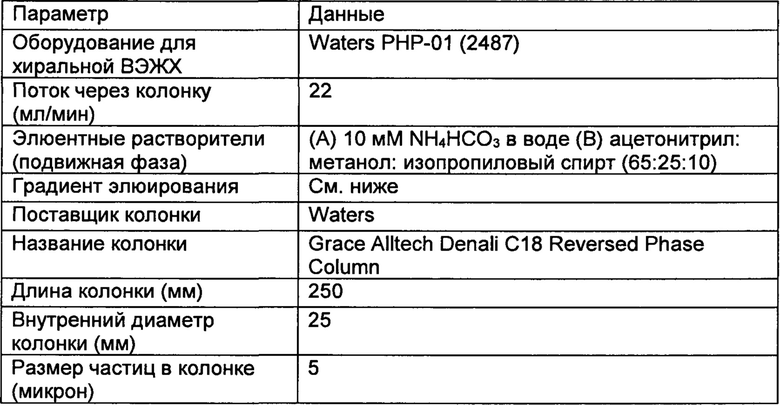
Градиент элюирования:
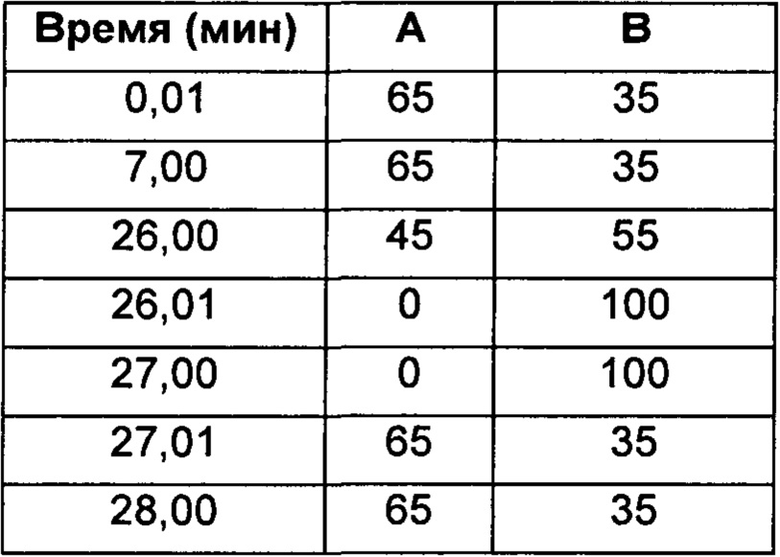
Препаративная ВЭЖХ метод 5


Градиент элюирования:
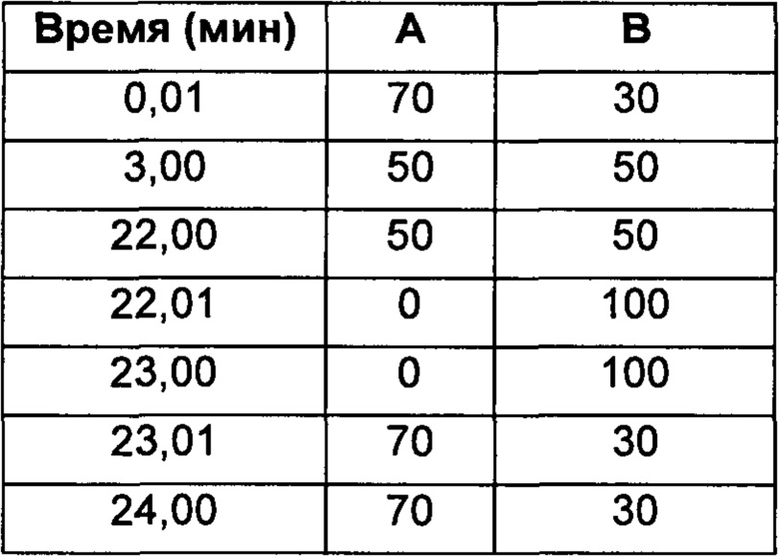
Препаративная ВЭЖХ метод 6
Использовали метод, совпадающий с методом препаративной ВЭЖХ 5, за исключением того, что использовали следующий градиент элюирования:
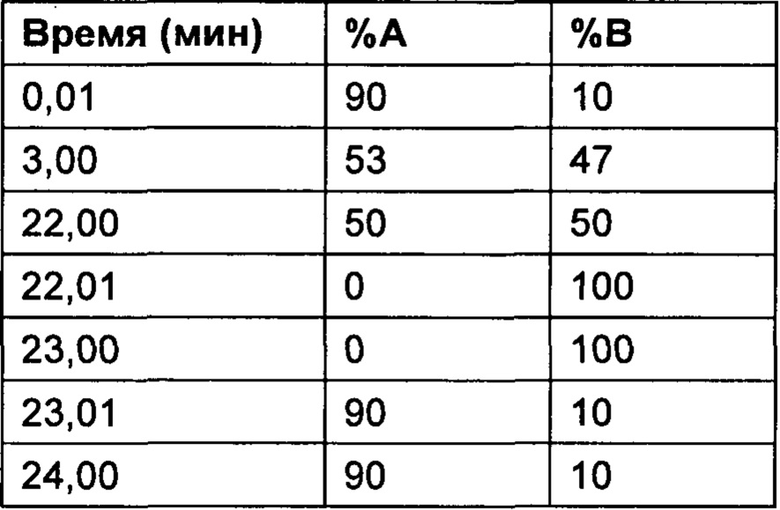
1
Препаративная ВЭЖХ метод 7
Использовали метод, совпадающий с методом препаративной ВЭЖХ 5, за исключением того, что использовали следующий градиент элюирования:
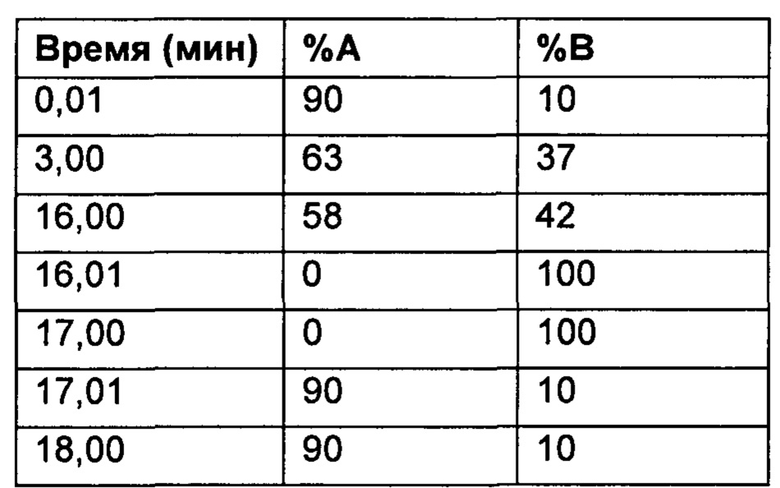
Схема синтеза А
Промежуточное соединение А является коммерчески доступным и также может быть синтезировано с помощью способов, описанных в литературе (Jain, Rama et al, международная публикация PCT WO 2009153313)
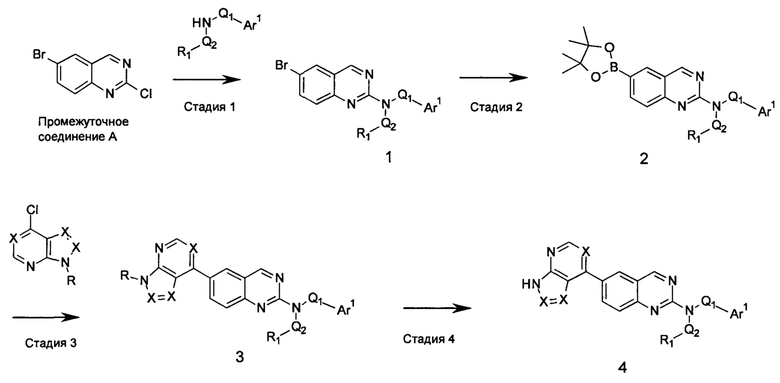
Где X = СН или N и где R = защитная группа, такая как ТНР (тетрагидропиранил) Соединение 4 может быть выделено в форме свободного основания или соли, например, моногидрохлоридной соли, в зависимости от используемого способа выделения.
Схема синтеза В
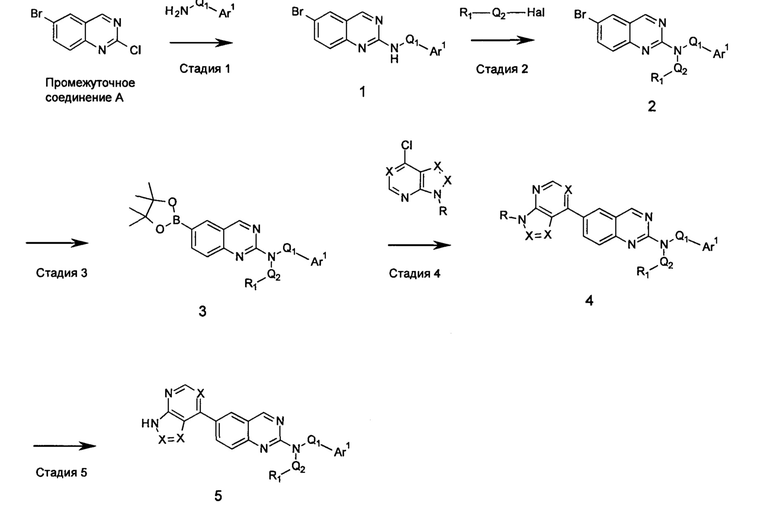
Где X = CH или N и где R = защитная группа, такая как ТНР
Соединение 5 может быть выделено в форме свободного основания или соли, например, моногидрохлоридной соли, в зависимости от используемого способа выделения.
Схема синтеза С
Где X = CH или N и где R = защитная группа, такая как ТНР
Соединение 5 может быть выделено в форме свободного основания или соли, например, моногидрохлоридной соли, в зависимости от используемого способа выделения.
Синтез промежуточных соединений:
Промежуточное соединение А: 6-бром-2-хлоохиназолин
Суспензию 6-бромхиназолин-2-ола (2 г, 8,89 ммоль) в POCl3 (20 мл) перемешивали при 110°С в течение 5 ч. После завершения реакции реакционную смесь концентрировали под вакуумом и остаток медленно выливали в лед, получая в результате твердый осадок. Твердую фазу отфильтровывали, промывали водой, а затем гексанами, после чего высушивали под вакуумом с получением титульного продукта (1,5 г, 69%).
6-бромхиназолин-2-ол является коммерчески доступным и также может быть синтезирован с помощью способов, описанных в литературе (Jain, Rama et al, PCT Int. Appl., 2009153313).
Промежуточное соединение В: 4-хлор-1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин
Стадия 1: 4-хлор-1Н-пиразоло[3,4-d]пиримидин
1Н-пиразоло [3,4-d]пиримидин-4-ол (5,0 г, 36,7 ммоль) растворяли в POCl3 (61,2 мл, 100,67 г, 657 ммоль) при комнатной температуре в атмосфере азота. В реакционную смесь добавляли N,N-диизопропилэтиламин (10,2 мл, 58,5 ммоль) и полученную смесь нагревали с обратным холодильником при 110°С в течение 4 часов. Через 4 часа отгоняли POCl3 под вакуумом при 40°С с получением густой смолы черно-красного цвета. Указанную густую смолу выливали в ледяную воду (25 мл) и экстрагировали DCM (25 мл × 3). Органический слой концентрировали до 25 мл. Полученный продукт использовали в качестве сырья на следующей стадии.
Стадия 2: 4-хлор-1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин
К раствору 4-хлор-1Н-пиразоло[3,4-d]пиримидина (2 г, 12,9 ммоль) и D-10-камфарасульфоновой кислоты (0,324 г, 1,39 ммоль) в безводном этилацетате (20 мл) медленно добавляли 3,4-дигидропиран (6 мл, 5,53 г, 65,8 ммоль) при комнатной температуре. После этого реакционную смесь оставляли для перемешивания при комнатной температуре в течение 24 часов. После завершения реакции, о котором свидетельствовала ТСХ, реакционную смесь концентрировали под вакуумом и очищали методом колоночной флэш-хроматографии на силикагеле с элюированием 10% этилацетатом в гексанах с получением титульного продукта (1,92 г, 62%).
Промежуточное соединение С: N'-бензил-N,N-диметилэтан-1.2-диамин
К раствору бензальдегида (1,0 г, 9,42 ммоль) в метаноле (20 мл) при перемешивании добавляли N,N диметилэтилендиамин (0,83 г, 9,42 ммоль) и ледяную уксусную кислоту (1,13 г, 1,18 ммоль) при КT в атмосфере азота. Полученную смесь перемешивали при КТ в течение 2 часов. К реакционной смеси добавляли триацетоксиборогидрид (5,99 г, 2,83 ммоль) при 0°С и реакционную смесь перемешивали в течение 18 часов при КТ в атмосфере азота. Реакционную смесь концентрировали под вакуумом. Затем к неочищенному продукту добавляли 20 мл 1N HCl и водную фазу экстрагировали этилацетатом (20 мл). Водный слой отделяли, подщелачивали до рН 8 с помощью твердого NaHCO3 и экстрагировали DCM (7×30 мл). Слой DCM отделяли, высушивали над Na2SO4 и концентрировали под вакуумом с получением титульного продукта (1,0 г, 60%).
Промежуточное соединение D: трет-бутиловый эфир (R)-3-аминометилморфолин-4-карбоновой кислоты
Стадия 1: трет-бутиловый эфир (R)-3-(1,3-диоксо-1.3-дигидроизоиндол-2-илметил)-морфолин-4-карбоновой кислоты
К раствору (R)-4-Вос-3-гидроксиметилморфолина (1,5 г, 6,90 ммоль) в ТГФ (7,5 мл) добавляли фталимид (1,21 г, 8,22 ммоль) и трифенилфосфин (5,43 г, 20,70 ммоль) при КТ. К реакционной смеси по каплям добавляли раствор диизопропилазодикарбоксилата (DIAD) (4,18 г, 0,0207 моль) в ТГФ (7,5 мл) и реакционную смесь перемешивали при КT в течение 30 минут. Реакционную смесь выливали в воду и экстрагировали этилацетатом (3×50 мл). Затем органический слой промывали солевым раствором (1×30 мл), а также водой (1×30 мл), высушивали над безводным сульфатом натрия и концентрировали под вакуумом. Затем остаток очищали методом колоночной хроматографии на силикагеле, с элюированием 16% этилацетатом в гексане с получением масла (4,3 г, >100%).
Стадия 2: трет-бутиловый эфир (R)-3-аминометилморфолин-4-карбоновой кислоты 
К раствору трет-бутилового эфира (R)-3-(1,3-диоксо-1,3-дигидроизоиндол-2-илметил)-морфолин-4-карбоновой кислоты (4,3 г, 12,4 ммоль) в этаноле (26 мл) и толуоле (26 мл) добавляли гидразингидрат (99% в воде, 1,19 г, 23,53 ммоль) при КТ. Реакционную смесь нагревали с обратным холодильником в течение 30 минут, концентрировали под вакуумом, и остаток выливали в ледяную воду и экстрагировали этилацетатом (3×100 мл). Органические слои объединяли и промывали солевым раствором, отделяли и высушивали над сульфатом натрия, затем концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт дополнительно очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 4% метанолом в хлороформе с получением титульного продукта (1,63 г, 61%).
Промежуточное соединение Е: трет-бутиловый эфир (S)-3-аминометилморфолин-4-карбоновой кислоты
Промежуточное соединение Е получали из (S)-4-Вос-3-гидроксиметилморфолина тем же способом, как описано для промежуточного соединения D.
Промежуточное соединение F: 6-хлор-9-(тетрагидропиран-2-ил)-9Н-пурин
Промежуточное соединение F является коммерчески доступным (CAS No.: 7306-68-5).
Промежуточное соединение G: 4-хлор-3-метил-1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4d]пиримидин
К суспензии 4-хлор-3-метил-1Н-пиразоло[3,4-d]пиримидина (0,5 г, 2,97 ммоль) в этилацетате (21 мл) последовательно добавляли D-10 камфаросульфоновую кислоту (0,07 г, 0,301 ммоль) и 3,4-дигидропиран (1,24 г, 14,7 ммоль) при КТ. Реакционную смесь перемешивали в течение 24 часов при КТ и затем концентрировали под вакуумом, а затем очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 8% этилацетатом в гексане с получением титульного продукта (0,59 г, 79%).
Промежуточное соединение Н: N-{3-[(6-бромхиназолин-2-иламино)-метил]-фенил}-метансульфонамид
Стадия 1: (6-бромхиназолин-2-ил)-(3-нитробензил)амин
К раствору 6-бром-2-хлорхиназолина (0,6 г, 2,46 ммоль) в DMSO (6 мл) при перемешивании добавляли 3-нитробензиламина гидрохлорид (0,56 г, 2,97 ммоль) и DIPEA (1,28 г, 1,73 мл, 9,89 ммоль) при КТ. Полученную смесь нагревали при 80°С в течение 12 часов. Реакционную смесь выливали в холодную воду и экстрагировали этилацетатом (2×50 мл). Органический слой промывали солевым раствором (20 мл), а затем водой (20 мл), отделяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 18% этилацетатом в гексане с получением титульного продукта (0,97 г, >100%).
Стадия 2: (3-аминобензил)-(6-бромхиназолин-2-ил)-амин
К раствору (6-бромхиназолин-2-ил)-(3-нитробензил)амина (0,97 г, 2,70 ммоль) в метаноле (10 мл) при перемешивании добавляли ледяную уксусную кислоту (0,30 г, 5,0 ммоль) и железный порошок (100 меш) (0,30 г, 5,4 ммоль) при КТ. Полученную смесь нагревали при 60°С в течение 4 часов. Реакционную смесь выливали в холодную воду и экстрагировали этилацетатом (2×50 мл). Органический слой промывали солевым раствором (20 мл), а затем водой (20 мл), отделяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного продукта, непосредственно используемого на следующей стадии.
Стадия 3: N-{3-[(6-бромхиназолин-2-иламино)-метил]-фенил}-метансульфонамид
К раствору (3-аминобензил)-(6-бромхиназолин-2-ил)амина (0,85 г, 2,58 ммоль) в DCM (13 мл) при перемешивании добавляли 2,6-лутидин (0,30 г, 0,33 мл, 2,80 ммоль) и метансульфонилхлорид (0,29 г, 0,196 мл, 2,53 ммоль) при 0°С. Полученную в результате смесь затем перемешивали в течение 6 часов при КТ. Реакционную смесь выливали в холодную воду и экстрагировали этилацетатом (2×50 мл). Органический слой промывали солевым раствором (20 мл), а затем водой (20 мл), отделяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 40% этилацетатом в гексане с получением титульного продукта (0,6 г, 57%).
Промежуточное соединение I: N-{3-[(R)-1-(6-Бром-хиназолин-2-иламино)-этил]-фенил}-метансульфонамид
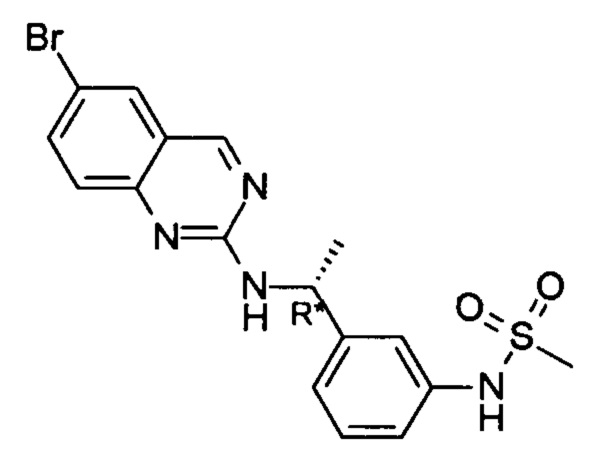
Титульное соединение получали тем же способом, как описано для промежуточного соединения Н, за исключением того, что вместо 3-нитробензиламина гидрохлорида на стадии 1 использовали (R)-1-(3-нитрофенил)этиламина гидрохлорид (0,70 г, 3,45 ммоль). Выход на конечной стадии: 0,7 г (57%).
Представленная схема синтеза А относится к Примеру 1 ниже.
ПРИМЕР 1
((R)-1-фенилэтил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)-хиназолин-2-ил]амин
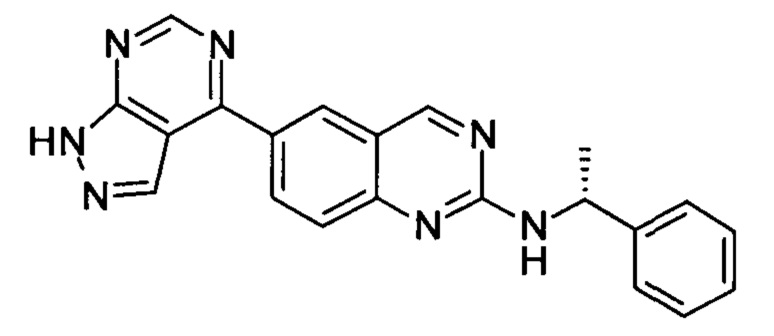
Стадия 1: (6-бромхиназолин-2-ил)-((R)-1-фенилэтил)амин
К раствору 6-бром-2-хлорхиназолина (10 г, 41,07 ммоль) в DMSO (100 мл) добавляли (R)-(+)-1-фенилэтиламин (6,0 г, 49,51 ммоль) и DIPEA (21,4 г, 28,84 мл, 166 ммоль). Полученную в результате смесь нагревали при 80°С в течение 12 часов. Реакционную смесь выливали в холодную воду с получением в результате твердого осадка. Твердую фазу собирали под вакуумом и промывали холодной водой (3×100 мл) с получением титульного продукта, используемого на следующей стадии без очистки (13,2 г, 98%).
Стадия 2: ((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-хиназолин-2-ил]амин
К раствору (6-бромхиназолин-2-ил)-((R)-1-фенилэтил)амина (13,2 г, 40,22 моль) в 1,4-диоксане (149 мл) при перемешивании добавляли бис(пинаколато)дибор (17,4 г, 68,52 ммоль), а затем ацетат калия (11,8 г, 120,2 ммоль) в атмосфере азота при КТ. Газообразный азот барботировали через полученную смесь в течение 30 минут. Затем к реакционной смеси добавляли 1,1-бис(дифенилфосфино)ферроцен палладий(II)дихлорид дихлорметановый комплекс (1,6 г, 1,96 ммоль). Реакционную смесь затем нагревали при 80°С в течение 3 часов. Реакционную смесь выливали в воду и экстрагировали этилацетатом (3×200 мл). Органические слои объединяли и промывали солевым раствором (100 мл), а затем водой (100 мл), затем отделяли и высушивали над безводным сульфатом натрия. Органический слой концентрировали под вакуумом, затем очищали методом колоночной флэш-хроматографии на силикагеле с элюированием 15% этилацетатом в гексанах с получением титульного продукта, который применяли на следующей стадии без дополнительной очистки (19,9 г, >100%).
Стадия 3: ((R)-1-фенилэтил)-{6-[1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил]-хиназолин-2-ил}амин
К раствору ((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-хиназолин-2-ил]амина (19,9 г, 67,89 ммоль) в смеси ТГФ: вода (267 мл, соотношение 4:1) добавляли промежуточное соединение В (19,4 г, 81,28 ммоль), а затем Cs2CO3 (88,3 г, 271,01 ммоль) в атмосфере азота при КТ. Газообразный азот барботировали через полученную смесь в течение 45 минут и затем к реакционной смеси добавляли тетракис(трифенилфосфин)палладий (0) (7,84 г, 6,78 ммоль). Затем реакционную смесь нагревали при 80°С в течение 4 часов в атмосфере азота. Реакционную смесь выливали в воду и экстрагировали этилацетатом (2×300 мл). Органические слои объединяли и промывали водой (100 мл), отделяли, затем высушивали над безводным сульфатом натрия. Органический слой концентрировали под вакуумом, затем очищали методом колоночной флэш-хроматографии на силикагеле с элюированием 45% этилацетатом в гексанах с получением титульного продукта (15 г, 49%).
Стадия 4: ((R)-1-фенилэтил)-[6-(1Н-пиразоло[3,4-о]пиримидин-4-ил)-хиназолин-2-ил]амин
К раствору((R)-1-фенилэтил)-{6-[1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил]-хиназолин-2-ил}амина (15,0 г, 33,2 моль) в смеси метанола (36 мл) и 1,4-диоксана (150 мл) при перемешивании добавляли 4Н HCl в 1,4-диоксане (41,5 мл) в атмосфере азота при КТ. Реакционную смесь дополнительно перемешивали в течение 3 часов при КТ.
Реакционную смесь фильтровали под вакуумом, и затем отфильтрованный осадок промывали этилацетатом (3×50 мл), затем гексаном (3×50 мл) с получением HCl соли. Осадок добавляли к воде (100 мл) и полученную смесь ощелачивали до рН 8,3 с использованием насыщенного водного раствора NaHCO3. Полученную суспензию перемешивали в течение 30 минут при КТ, затем фильтровали под вакуумом. Осадок ресуспендировали в воде на воронке Бюхнера и полученную суспензию перемешивали в течение 10 минут, а затем фильтровали под вакуумом. Процесс суспендирования в воде дополнительно повторяли семь раз. Полученное в результате отфильтрованное твердое вещество высушивали nod вакуумом с получением титульного продукта (9,0 г, 74%).
Необязательно, титульное соединение может оставаться в виде HCl соли, если исключить конечную стадию ощелачивания.
ПРИМЕР 2
((R)-1-(3-хлорфенил)этил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,4 г, 1,64 ммоль) тем же способом, который описан для Примера 1 за исключением следующего: (а) вместо (R)-1-фенил-этиламина на Стадии 1 использовали (R)-1-(3-хлорфенил)этиламин (b) на стадии 4 применяли следующий способ: N-((R)-1-(3-хлорфенил)этил)-6-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-амин (0,12 г, 0,247 ммоль) растворяли в метаноле (1 мл) и при КТ добавляли 4 N HCl в диоксане (3 мл). После перемешивания при КТ в течение 4 часов, полученный таким образом твердый осадок фильтровали и промывали этилацетатом с получением титульного продукта в виде моногидрохлоридной соли (0,050 г, 46%).
ПРИМЕР 3
((R)-1-(3-фторфенил)этил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,4 г, 1,64 ммоль) тем же способом, который описан для Примера 1 за исключением следующего: (а) вместо (R)-1-фенилэтиламина на стадии 1 использовали (R)-1-(3-фторфенил)этиламин (b) на стадии 4 применяли способ, описанный для примера 2, в результате которого получали соединение, выделяемое в виде моногидрохлоридной соли (0,040 г, 68%).
ПРИМЕР 4
((R)-1-(4-фторфенил)этил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,35 г, 1,43 ммоль) тем же способом, который описан для Примера 1 за исключением следующего: (a) (R)-1-(4-фторфенил)этиламин на стадии 1 использовали вместо (R)-1-фенилэтиламина (b) на стадии 4 после концентрирования реакционной смеси под вакуумом, неочищенный продукт очищали методом препаративной ВЭЖХ с получением титульного продукта (0,020 г, 22%).
ПРИМЕР 5
((S)-2-гидрокси-1-фенилэтил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,4 г, 1,64 ммоль) тем же способом, который описан для Примера 1 за исключением следующего: (а) вместо (R)-1-фенилэтиламина на стадии 1 использовали (S)-2-фенилглицинол (b) на стадии 4 после концентрирования реакционной смеси под вакуумом, неочищенный продукт очищали методом препаративной ВЭЖХ с получением свободного основания. К раствору свободного основания в этилацетате (1 мл) при 10°С добавляли 4N HCl в диоксане (0,05 мл), после чего перемешивали при КТ в течение 30 минут. Через 30 минут реакционную смесь концентрировали под вакуумом и тритурировали этилацетатом с получением титульного продукта в виде моногидрохлоридной соли (0,050 г, 37%).
ПРИМЕР 6
(4-фторбензил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)-хиназолин-2-ил]-метиламин
Целевой продукт получали из промежуточного соединения А (0,4 г, 1,64 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) вместо (R)-1-фенил-этиламин на стадии 1 использовали N-(4-фторбензил)-N-метиламин (b) на стадии 4 после концентрирования реакционной смеси под вакуумом продукт тритурировали метанолом и этилацетатом с получением титульного продукта в виде моногидрохлоридной соли (0,080 г, 59%).
ПРИМЕР 7
Бензил-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]метиламин
Целевой продукт получали из промежуточного соединения А (0,6 г, 2,46 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) вместо (R)-1-фенилэтиламина на стадии 1 использовали N-бензил-N-метиламин (b) на стадии 4 применяли способ согласно примеру 6. Целевой продукт выделяли в виде моногидрохлоридной соли (0,080 г, 60%).
ПРИМЕР 8
((R)-1-Фенилэтил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]-метиламин
Целевой продукт получали из промежуточного соединения А согласно следующей схеме В и описанному ниже способу.
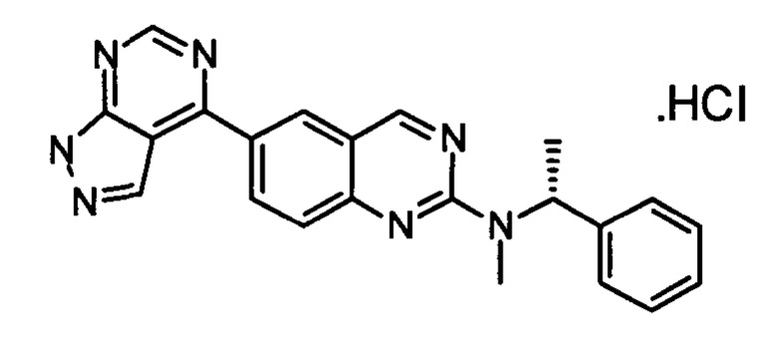
Стадия 1: (6-бромхиназолин-2-ил)-((R)-1-фенилэтил)амин
Как описано в стадии 1 Примера 1
Стадия 2: (6-бромхиназолин-2-ил)метил-((R)-1-фенилэтил)амин
К суспензии NaH (60% в минеральном масле) (0,082 г, 2,05 ммоль) в DMF (5 мл) при 0°С в атмосфере азота добавляли раствор (6-бромхиназолин-2-ил)-((R)-1-фенилэтил)амина (0,45 г, 1,37 ммоль) в DMF (5 мл). Реакционную смесь перемешивали при 0°С в течение 30 минут. Добавляли Mel (0,233 г, 1,64 ммоль) и реакционную смесь перемешивали при КТ в течение 2 часов. Затем реакционную смесь разбавляли водой (30 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои отделяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной флэш-хроматографии на силикагеле с элюированием 5 - 8% этилацетатом в гексанах с получением титульного продукта (0,45 г, 96%).
Стадия 3: Метил-((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)хиназолин-2-ил]-amine
(6-Бром-хиназолин-2-ил)-метил-((R)-1-фенилэтил)амин (0,45 г, 1,31 ммоль) помещали в условия стадии 2, Примера 1 с получением титульного продукта (0,25 г, 49%).
Стадия 4: метил-((R)-1-фенилэтил)-{6-[1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил]хиназолин-2-ил}амин
Метил-((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)хиназолин-2-ил]амин (0,25 г, 0,64 ммоль) помещали в условия стадии 3, Примера 1 с получением титульного продукта (0,1 г, 34%).
Стадия 5: ((R)-1-фенилэтил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]метиламин
К раствору метил-((R)-1-фенилэтил)-{6-[1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил]хиназолин-2-ил}амина (0,1 г, 0,21 ммоль) в метаноле (3 мл) добавляли 4N HCl в диоксане (2 мл) при КТ и реакционную смесь перемешивали при КТ в течение 5 часов. В ходе реакции получали твердый осадок; твердый осадок отфильтровывали под вакуумом и промывали этилацетатом, затем высушивали под вакуумом с получением титульного продукта (0,080 г, 91%).
Соединение выделяли в виде моногидрохлоридной соли.
ПРИМЕР 9
(4-фторбензил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (1,2 г, 4,96 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) вместо (R)-1-фенилэтиламина на стадии 1 использовали 4-фторбензиламин (b) на стадии 4 после обработки 4N HCl в диоксане (6,0 мл) и перемешивания в течение 3 часов реакционную смесь концентрировали под вакуумом, выливали в воду и полученную в результате реакционную смесь ощелачивали до рН 8 с использованием насыщенного (водн) раствора гидрокарбоната. Полученную суспензию фильтровали под вакуумом с получением титульного продукта. Выход на конечной стадии: 0,105 г (48%).
ПРИМЕР 10
(3-фторбензил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (1,0 г, 4,13 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) вместо (R)-1-фенилэтиламина на стадии 1 использовали 3-фторбензиламин (b) на стадии 4 использовали способ выделения, описанный для примера 9, однако, отфильтрованный продукт дополнительно суспендировали в этилацетате (5 мл) в течение 15 минут при КТ, после чего фильтровали лоб вакуумом. Выход на конечной стадии: 0,112 г (34%).
ПРИМЕР 11
(3-хлорбензил)[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,60 г, 2,48 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) вместо (R)-1-фенилэтиламина на стадии 1 использовали 3-хлорбензиламин (b) на стадии 4 использовали способ выделения, описанный для примера 9, однако отфильтрованный продукт дополнительно очищали методом препаративной ВЭЖХ с использованием метода препаративной ВЭЖХ 1. Выход конечной стадии: 0,036 г (17%).
ПРИМЕР 12
((R)-1-фенилэтил)[7-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали с применением способа, описанного в Примере 1, за исключением следующего: (а) вместо 6-бромхиназолин-2-ола в синтезе промежуточного соединения А использовали 7-бромхиназолин-2-ол (1,2 г, 4,96 ммоль) (b) на стадии 4 время реакции составляло 1 час. Использовали способ выделения, описанный для примера 9, однако отфильтрованный осадок дополнительно очищали методом колоночной флэш-хроматографии, eluting with 3% methanol в хлороформе, после чего очищали методом препаративной ВЭЖХ хроматографии с использованием метода препаративной ВЭЖХ 2. Выход конечной стадии: 0,022 г (17%).
7-бромхиназолин-2-ол может быть получен из различных коммерческих источников, наличие указано в базах данных Scifinder, или может быть получен способом, описанным в международной публикации WO 2007117607.
ПРИМЕР 13
((R)-1-фенилэтил)-[6-(1Н-пиразоло[3,4-b]пиридин-4-ил)-хиназолин-2-ил]амин
Целевой продукт получали из ((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)хиназолин-2-ил]амин (0,3 г, 0,799 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) на стадии 3 вместо промежуточного соединения В использовали 4-хлор-1Н-пиразоло[3,4-b]пиридин. Кроме того, на стадии 3 использовали смесь растворителей DMF: вода (4:1, 5 мл) и проводили нагревание при 130°С в микроволновом реакторе. Стадия 4 представляла собой стадию образования HCl соли: к суспензии ((R)-1-фенилэтил)-[6-(1Н-пиразоло[3,4-b]пиридин-4-ил)хиназолин-2-ил]амина (0,080 г, 0,218 ммоль) в этилацетате (0,8 мл) добавляли 4N HCl в диоксане (0,08 мл) при 10°С и реакционную смесь перемешивали при той же температуре в течение 40 минут. Затем реакционную смесь концентрировали под вакуумом и тритурировали этилацетатом с получением титульного продукта в виде моногидрохлоридной соли (0,070 г, 80%)
4-Хлор-1Н-пиразоло[3,4-b]пиридин может быть получен из различных коммерчески доступных источников.
ПРИМЕР 14
((R)-1-фенилэтил)-[6-(7Н-пирроло[2,3-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из ((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-хиназолин-2-ил]амина (0,35 г, 0,933 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) на стадии 3 вместо промежуточного соединения В использовали 4-хлор-7Н-пирроло[2,3-d]пиримидин, (b) была включена стадия образования HCl соли, причем способ для этого описан в примере 13. Соединение выделяли в виде моногидрохлоридной соли (0,055 г, 83%).
4-хлор-7Н-пирроло[2,3-d]пиримидин может быть получен из различных коммерческих источников.
ПРИМЕР 15
((R)-1-фенилэтил[6-(1Н-пирроло[2,3-b]пиридин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из ((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)хиназолин-2-ил]амина (0,20 г, 0,533 ммоль) с применением способа, описанного в Примере 1, за исключением следующего: (а) на стадии 3 вместо промежуточного соединения В использовали 4-бром-1Н-пирроло[2,3-b]пиридин, вместо карбоната цезия использовали карбонат калия, и смесь диоксан: вода (4:1) использовали в качестве растворителя вместо смеси ТГФ: вода (b) была включена стадия образования HCl соли, причем способ для этого описан в примере 13. Соединение выделяли в виде моногидрохлоридной соли (0,070 г, 80%).
4-бром-1Н-пирроло[2,3-b]пиридин может быть получен из различных коммерческих источников.
ПРИМЕР 16
N-бензил-N',N'-диметил-N-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)-хиназолин-2-ил]этан-1,2-диамин
Титульное соединение может быть получено с помощью схемы синтеза А, согласно способу по Примеру 1, с использованием N'-бензил-N,N-диметилэтан-1,2-диамина (промежуточной соединение С) на стадии 1. Стадия 4 проходила согласно следующему способу:
Стадия 4
К раствору N-бензил-N',N'-диметил-N-{6-[1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил]хиназолин-2-ил}-этан-1,2-диамина (0,40 г, 0,79 ммоль) в 1,4-диоксане (4 мл) при перемешивании добавляли 4N HCl в 1,4-диоксане (0,25 мл, 1,0 ммоль) в атмосфере азота при КТ. Реакционную смесь дополнительно перемешивали в течение 4 часов при КТ. Добавляли дополнительную аликвоту 4N HCl в 1,4-диоксане (0,25 мл, 1,0 моль) и реакционную смесь перемешивали в течение 3 часов при КТ. Затем реакционную смесь концентрировали под вакуумом с получением неочищенного продукта, который добавляли к воде. Полученную смесь подщелачивали с помощью насыщенного (водного) раствора NaHCO3 до рН 8. Полученную смесь экстрагировали этилацетатом и затем концентрировали под вакуумом. Остаток очищали методом флэш-хроматографии на силикагеле, с элюированием 2,5% метанолом в хлороформе с получением неочищенного твердого вещества желтого цвета, которое затем тритурировали диэтиловым эфиром. Твердое вещество, восстановленное после тритурации (0,086 г, 0,189 ммоль) растворяли в 1,4-диоксане (0,8 мл) при комнатной температуре и добавляли 4М HCl в 1,4-диоксане (0,06 мл, 0,24 ммоль). Реакционную смесь перемешивали в течение 30 минут при КТ, после чего реакционную смесь концентрировали под вакуумом тритурировали диэтиловым эфиром (1 мл) с получением титульного продукта в виде моногидрохлоридной соли (0,067 г, 19% общий выход).
ПРИМЕР 17
Бензил-(R)-1-морфолин-3-илметил-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)-хиназолин-2-ил]амин
Титульное соединение получали согласно Схеме синтеза С.
Стадия 1: (трет-бутил (R)-3-(((6-бромхиназолин-2-ил)амино)метил)морфолин-4-карбоксилат)
К раствору промежуточного соединения А (1,4 г, 5,75 ммоль) в DMSO (14 мл) при перемешивании добавляли промежуточное соединение D (1,5 г, 6,93 ммоль) и DIPEA (3,0 г, 4,04 мл, 23,21 ммоль) при КТ. Полученную смесь нагревали при 80°С в течение 18 часов. Реакционную смесь выливали в ледяную воду и экстрагировали этилацетатом (3×50 мл). Затем органический слой промывали солевым раствором (50 мл), а также водой (50 мл), отделяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 25% этилацетатом в гексане с получением титульного продукта (1,58 г, 65%).
Стадия 2: (трет-бутил (R)-3-((бензил(6-бромхиназолин-2-ил)амино)метил)морфолин-4-карбоксилат)
К суспензии гидрида натрия (0,74 г, 18,5 ммоль) в сухом DMF (22 мл) добавляли раствор (трет-бутил (R)-3-(((6-бромхиназолин-2-ил)амино)метил)морфолин-4-карбоксилата) (1,58 г, 3,73 ммоль) в DMF (22 мл) при 0°С в атмосфере азота. Реакционную смесь перемешивали в течение 30 минут при 0°С, и затем доводили до КТ перед добавлением бензил бромида (1,92 г, 11,23 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 минут. Реакционную смесь выливали в ледяную воду и экстрагировали этилацетатом (3×50 мл). Органические слои выделяли, объединяли и затем промывали солевым раствором (50 мл), а затем водой (50 мл), высушивали над сульфатом натрия и концентрировали под вакуумом. Неочищенный остаток очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 10% этилацетатом в гексанах с получением титульного продукта (1,8 г, 94%).
Стадия 3: (трет-бутил (R)-3-((бензил(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-2-ил)амино)метил)морфолин-4-карбоксилат)
(трет-бутил (R)-3-((бензил(6-бромхиназолин-2-ил)амино)метил)морфолин-4-карбоксилат) (1,8 г, 3,51 ммоль) помещали в условия, описанные для примера 1, стадии 2 с получением титульного продукта. Выход: 2,3 г (>100%).
Стадия 4: (трет-бутил (3R)-3-((бензил(6-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил)амино)метил)морфолин-4-карбоксилат)
(трет-бутил (R)-3-((бензил(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-2-ил)амино)метил)морфолин-4-карбоксилат) (2,30 г, 4,10 ммоль) помещали в условия, описанные для примера 1, стадии 3 с получением титульного продукта (1,2 г, 46%).
Стадия 5: ((R)-N-бензил-N-(морфолин-3-илметил)-6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-амин)
(трет-бутил (3R)-3-((бензил(6-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил)амино)метил)морфолин-4-карбоксилат) (1,2 г, 1,88 ммоль) помещали в условия, описанные для примера 1, стадии 4 и очищали следующим образом: после завершения реакции реакционную смесь фильтровали под вакуумом, и полученный в результате отфильтрованный осадок промывали этилацетатом (3×50 мл) и гексаном (3×50 мл). Полученное твердое вещество дополнительно тритурировали в ацетоне, затем фильтровали под вакуумом с получением титульного продукта в виде моногидрохлоридной соли (0,48 г, 52%).
ПРИМЕР 18
Бензил-(S)-1-морфолин-3-илметил-[6-(1Н-пиразоло[3.4-d]пиримидин-4-ил)-хиназолин-2-ил]амин
Титульное соединение получали из промежуточного соединения А (1,3 г, 5,34 ммоль) с помощью схемы синтеза С, представленной выше, способом согласно Примеру 17, за исключением следующего: (а) на стадии 1 вместо трет-бутилового эфира (R)-3-аминометилморфолин-4-карбоновой кислоты использовали промежуточное соединение Е, трет-бутиловый эфир (S)-3-аминометилморфолин-4-карбоновой кислоты (полученные способом, аналогичным промежуточному соединению D) (b) на стадии 5 тритурация ацетона была исключена. Соединение выделяли в виде моногидрохлоридной соли. Выход на конечной стадии: 0,63 г (67%).
ПРИМЕР 19
[(R)-1-(3,4-дифторфенил)этил]-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Стадия 1: ((R)-6-бром-N-(1-(3,4-дифторфенил)этил)хиназолин-2-амин)
К раствору Промежуточного соединения А (0,5 г, 2,05 моль) в DMSO (5 мл) при перемешивании добавляли (R)-1-(3,4-дифторфенил)этиламин (0,39 г, 2,48 ммоль) и DIPEA (1,08 г, 1,46 мл, 8,4 ммоль) при комнатной температуре. Полученную смесь нагревали при 80°С в течение 12 часов. Реакционную смесь выливали в ледяную воду с получением в результате осадка. Осадок отфильтровывали лоб вакуумом и промывали холодной водой с получением титульного продукта (0,7 г, 94%).
Стадия 2: ((R)-N-(1-(3,4-дифторфенил)этил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-2-амин)
К раствору((R)-6-бром-N-(1-(3,4-дифторфенил)этил)хиназолин-2-амина) (0,70 г, 1,92 ммоль) в 1,4-диоксане (8 мл) при перемешивании добавляли бис(пинаколато)дибор (0,84 г, 3,31 ммоль) а затем ацетат калия (0,57 г, 5,80 ммоль) в атмосфере азота при КТ. Полученную в результате смесь продували барботированием азота в течение 30 минут. К реакционной смеси добавляли комплекс 1, 1-бис(дифенилфосфино)ферроцен палладий(II)дихлорид дихлорметана (0,08 г, 0,098 ммоль). Затем реакционную смесь нагревали при 80°С в течение 2 часов. Реакционную смесь выливали в воду и экстрагировали этилацетатом (2×40 мл). Объединенные органические слои промывали солевым раствором (20 мл), затем водой (20 мл), отделяли и высушивали над безводным сульфатом натрия, а затем концентрировали под вакуумом. Неочищенный остаток очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 7% этилацетатом в гексане с получением титульного продукта (0,94 г, >100%).
Стадия 3: (N-((R)-1-(3,4-дифторфенил)этил)-6-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-амин)
К раствору((R)-N-(1-(3,4-дифторфенил)этил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-2-амина) (0,94 г, 2,29 ммоль) в смеси ТГФ: вода (12,5 мл, 4:1) при перемешивании добавляли промежуточное вещество В (0,65 г, 2,72 ммоль), а затем Cs2CO3 (2,97 г, 9,12 ммоль) в атмосфере азота при КТ. Смесь продували барботированием азота в течение 45 минут, и к полученной смеси добавляли тетракис(трифенилфосфин)плладий(0) (0,26 г, 0,225 ммоль) при КТ. Затем реакционную смесь нагревали при 80°С в течение 3 часов. Полученную реакционную смесь выливали в воду и продукт экстрагировали с использованием этилацетата (2×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), затем водой (20 мл), отделяли и высушивали над безводным сульфатом натрия, затем концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 45% этилацетатом в гексане с получением титульного продукта (0,6 г, 54%).
Стадия 4: ((R)-N-(1-(3,4-дифторфенил)этил)-6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-амин)
К раствору(N-((R)-1-(3,4-дифторфенил)этил)-6-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-амина) (0,6 г, 1,23 ммоль) в смеси метанола (1,4 мл) и 1,4-диоксана (6 мл) при перемешивании добавляли 4N HCl в 1,4-диоксане (6 мл) в атмосфере азота при КТ. Реакционную смесь дополнительно перемешивали в течение 1 часа при КТ, в результате чего образовывался осадок, который отфильтровывали под вакуумом и промывали этилацетатом (3×30 мл), а затем гексаном (3×30 мл). Затем осадок добавляли к воде (10 мл) и полученную смесь ощелачивали до рН 8 с помощью насыщенного раствора NaHCO3, при перемешивании в течение 30 минут. Реакционную смесь фильтровали под вакуумом, промывали водой с получением титульного продукта (0,27 г, 54%).
ПРИМЕР 20
((R)-1-фенилэтил)-[6-(9Н-пурин-6-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (10 г, 41,1 ммоль) способом, описанным для Примера 19, за исключением следующего: (а) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-1-фенилэтиламин (b) на стадии 3 вместо промежуточного соединения В использовали промежуточное соединение F, 6-хлор-9-(тетрагидропиран-2-ил)-9Н-пурин. Выход на конечной стадии: 0,26 г (46%).
ПРИМЕР 21
Бензил-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали бензиламин. Выход на конечной стадии: 0,2 г (40%).
ПРИМЕР 22
[6-(3-метил-1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]-((R)-1-фенилэтил)амин
Целевой продукт получали из промежуточного соединения А (10 г, 41,1 ммоль) способом, описанным для Примера 19, за исключением следующего: (а) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-1-фенилэтиламин (b) на стадии 3 вместо промежуточного соединения В использовали промежуточное соединение G, 4-хлор-3-метил-1-(тетрагидропиран-2-ил)-1Н-пиразоло[3,4-d]пиримидин (с) на стадии 4 в качестве конечной стадии для очистки титульного продукта использовали препаративную ВЭЖХ (используя метод 3 препаративной ВЭЖХ). Выход на конечной стадии: 0,12 г (24%).
ПРИМЕР 23
(3-метоксибензил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали 3-метокси бензиламин. Выход на конечной стадии: 0,27 г (59%).
ПРИМЕР 24
2-фторбензил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)-хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,4 г, 1,64 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламин использовали 2-фторбензиламин. Выход на конечной стадии: 0,17 г (46%).
ПРИМЕР 25
(3,4-дифторбензил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали 3,4-дифторбензиламин. Выход на конечной стадии: 0,34 г (78%).
ПРИМЕР 26
N-(3-{[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]-метил}-фенил)-метансульфонамид
Промежуточное соединение Н (0,6 г, 1,47 ммоль) помещали в условия стадий 2-4 Примера 19, после чего проводили очистку методом 4 препаративной ВЭЖХ с получением титульного продукта (0,05 г, 11%).
ПРИМЕР 27
[(R)-1-(3-метоксифенил)этил]-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,6 г, 2,46 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-(+)-1-(3-метоксифенил)этиламин. Выход на конечной стадии: 0,4 г (59%).
ПРИМЕР 28
[(R)-1-(2-фторфенил)-этил-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением следующего: (а) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-1-(2-фторфенил)этиламин (b) на стадии 4 применяли конечную стадию тритурирования ацетоном. Выход на конечной стадии: 0,3 г (42%).
ПРИМЕР 29
((S)-1-фенилэтил)-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (2,5 г, 10,3 ммоль) способом, описанным для Примера 19, за исключением следующего: (а) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (S)-(-)-α-метил бензил амин (b) на стадии 4 применяли конечную стадию тритурирования этилацетатом. Выход на конечной стадии: 0,56 г (47%).
ПРИМЕР 30
N-(3-{(R)-1-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}-фенил)метансульфонамид
Промежуточное соединение I (1,0 г, 1,47 ммоль) помещали в условия стадий 2-4 Примера 19 с получением титульного продукта. Выход на конечной стадии: 0,22 г (39%).
ПРИМЕР 31
(R)-3-фенил-3-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]пропан-1-ол
Целевой продукт получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-3-амино-3-фенил-пропан-1-ол. Выход на конечной стадии: 0,14 г (38%).
ПРИМЕР 32
[(R)-1-(2,4-дифторфенил)этил]-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продукт получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-1-(2,4-дифторфенил)-этиламина гидрохлорид. Выход на конечной стадии: 0,205 г (56%).
ПРИМЕР 33
[(R)-1-(2,6-дифторфенил)этил]-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин
Целевой продует- получали из промежуточного соединения А (0,5 г, 2,05 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-1-(2,6-дифторфенил)этанамина гидрохлорид. Выход на конечной стадии: 0,235 г (47%).
ПРИМЕР 34
4-[2-((R)-1-фенилэтиламино)хиназолин-6-ил]-7Н-пирроло[2,3-d]пиримидин-5-карбонитрил
Целевой продукт получали из промежуточного соединения А (1,0 г, 4,11 ммоль) способом, описанным для Примера 19, за исключением следующего: (а) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-1-фенилэтиламин; (b) стадию 3 проводили как описано ниже; и (с) стадию 4 исключали. 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбонитрил является коммерчески доступным. Выход на конечной стадии: 0,142 г (19%).
Стадия 3: 4-[2-((R)-1-фенилэтиламино)хиназолин-6-ил]-7Н-пирроло[2,3-d]пиримидин-5-карбонитрил
К раствору 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбонитрила (0,35 г, 1,96 ммоль) и ((R)-1-фенилэтил)-[6-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)хиназолин-2-ил]амина (0,75 г, 2,00 ммоль) в DMF (7 мл) при перемешивании добавляли 1М водный раствор NaHCO3 (5,9 мл, 5,9 ммоль) в атмосфер азота при КТ. Реакционную смесь продували газообразным азотом в течение 30 минут и затем к реакционной смеси добавляли дихлорбис(трифенилфосфин)палладий(II) (0,07 г, 0,0997 ммоль) при КТ. Затем полученную реакционную смесь нагревали при 80°С в течение 18 часов. Реакционную смесь выливали в воду и экстрагировали этилацетатом (2×20 мл). Органические слои объединяли и промывали солевым раствором (20 мл), затем отделяли, высушивали над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного титульного продукта. Неочищенный продукт очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 60% этилацетатом в гексане с получением титульного продукта, который затем дополнительно очищали методом 5 препаративной ВЭЖХ.
ПРИМЕР 35
2-{(R)-1-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенол
Целевой продукт получали из промежуточного соединения А (0,8 г, 3,29 ммоль) способом, описанным для Примера 19, за исключением следующего: (1) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали 2-((R)-1-аминоэтил)фенол; и (2) на стадии 4 реакционную смесь не фильтровали сразу, а сначала ощелачивали до рН 8 с помощью насыщенного водного раствора NaHCO3. Водную фазу экстрагировали EtOAc (2×20 мл). Органические слои объединяли и концентрировали под вакуумом с получением неочищенного титульного продукта, который очищали методом колоночной флэш-хроматографии на силикагеле, с элюированием 4% метанолом в хлороформе с получением титульного продукта, который затем дополнительно очищали методом 6 препаративной ВЭЖХ. Выход на конечной стадии: 0,032 г (12%).
ПРИМЕР 36
3-{(R)-1-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенол
Целевой продукт получали из промежуточного соединения А (0,8 г, 3,29 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали 3-((R)-1-аминоэтил)фенол. Выход на конечной стадии: 0,126 г (16%).
ПРИМЕР 37
4-{(R)-1-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенол
Целевой продукт получали из промежуточного соединения А (0,8 г, 3,29 ммоль) способом, описанным для Примера 19, за исключением следующего: (1) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали 4-((R)-1-аминоэтил)фенол (2) на стадии 4 реакционную смесь не фильтровали сразу, а сначала ощелачивали до рН 8 с помощью насыщенного водного раствора NaHCO3, и затем фильтровали под вакуумом. Затем выделенный осадок очищали методом 6 препаративной ВЭЖХ с получением титульного продукта. Выход на конечной стадии: 0,1 г (20%).
ПРИМЕР 38
(S)-2-(2-фторфенил)-2-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этанол
Целевой продукт получали из промежуточного соединения А (0,6 г, 2,46 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (5)-2-амино-2-(2-фторфенил)этанол.
ПРИМЕР 39
(S)-2-(3-фторфенил)-2-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этанол
Целевой продукт получали из промежуточного соединения А (0,8 г, 3,29 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (S)-2-амино-2-(3-фторфенил)этанол.
ПРИМЕР 40
(S)-2-(4-фторфенил)-2-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этанол
ТЦелевой продукт получали из промежуточного соединения А (0,8 г, 3,29 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (S)-2-амино-2-(4-фторфенил)этанол.
ПРИМЕР 41
(R)-3-(2-фторфенил)-3-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]пропан-1-ол
Целевой продукт получали из промежуточного соединения А (0,8 г, 3,29 ммоль) способом, описанным для Примера 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-3-амино-3-(2-фторфенил)пропан-1-ол. Выход на конечной стадии: 0,144 г (34%).
ПРИМЕР 42
(R)-3-(3-фторфенил)-3-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)-хиназолин-2-иламино]-пропан-1-ол
Титульное соединение получали из промежуточного соединения А (0,8 г, 3,29 ммоль) с применением способа, описанного в Примере 19, за исключением того, что на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина применяли (R)-3-амино-3-(3-фторфенил)-пропан-1-ол. Выход на последней стадии: 0,1 г (24%).
ПРИМЕР 43
(R)-3-(4-фторфенил)-3-[6-(1Н-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]пропан-1-ол
Целевой продукт получали из промежуточного соединения А (0,6 г, 2,46 ммоль) способом, описанным для Примера 19, за исключением следующего: (1) на стадии 1 вместо (R)-1-(3,4-дифторфенил)этиламина использовали (R)-3-амино-3-(4-фторфенил)пропан-1-ол; и (2) на стадии 4 реакционную смесь концентрировали под вакуумом, добавляли в воду и полученную в результате реакционную смесь ощелачивали до рН 8 с помощью насыщенного водного раствора NaHCO3. Смесь перемешивали в течение 30 минут при КТ и затем фильтровали под вакуумом, промывали водой (3×30 мл), а затем гексаном (2×15 мл). Неочищенное вещество очищали методом 7 препаративной ВЭЖХ. Выход на конечной стадии: 0,203 г (31%).
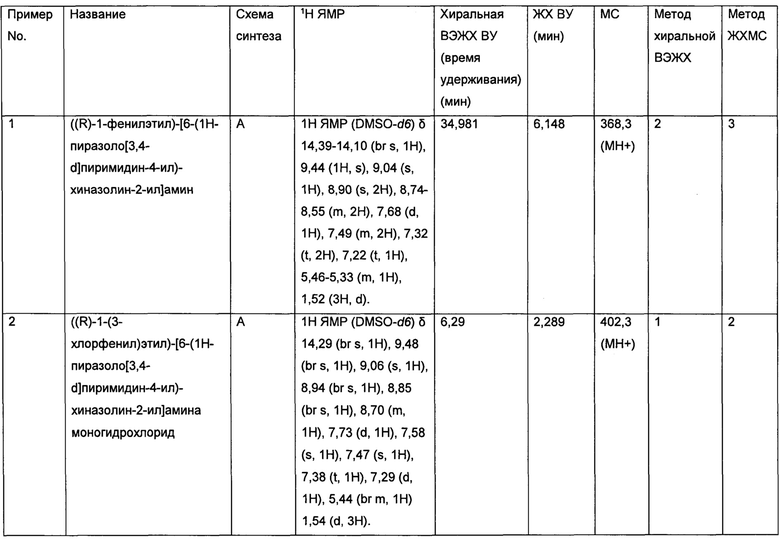
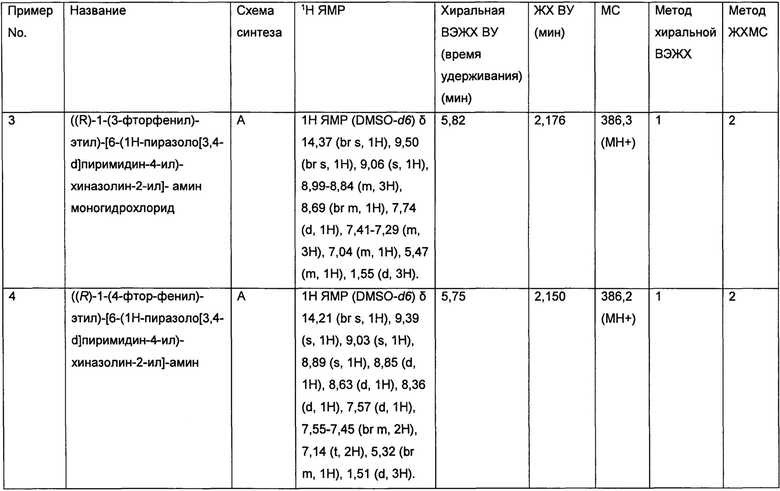
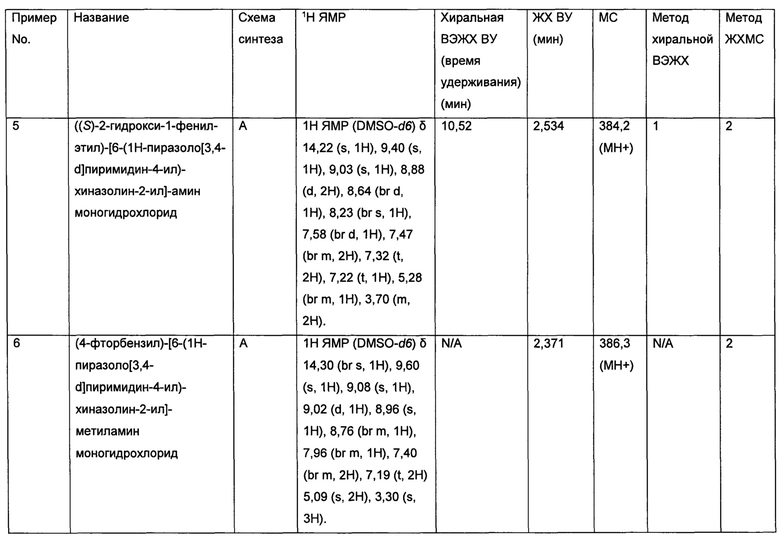
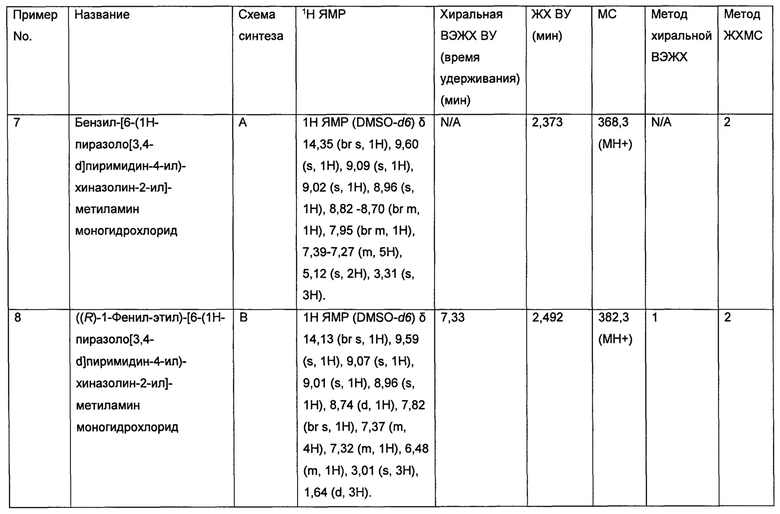
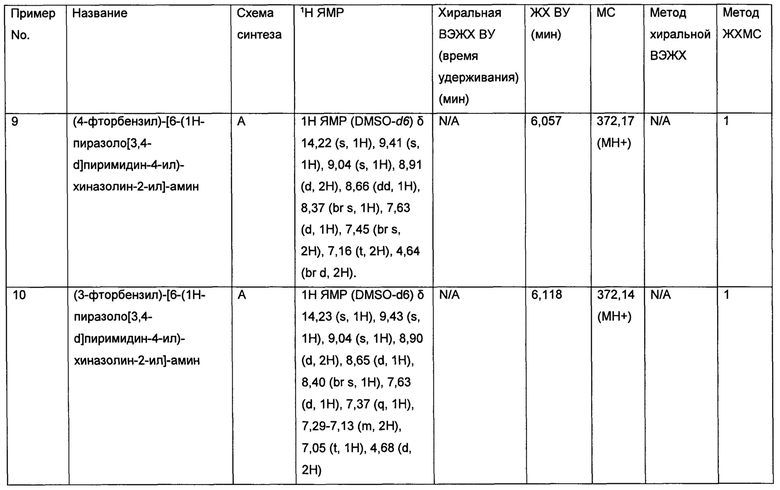
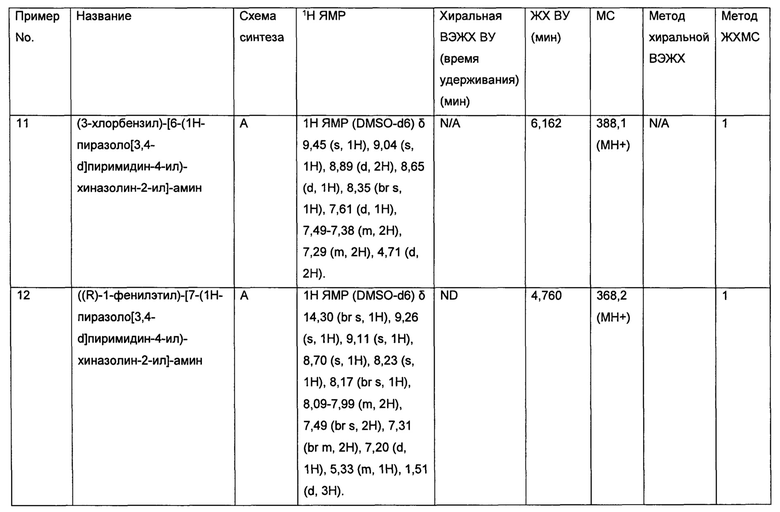
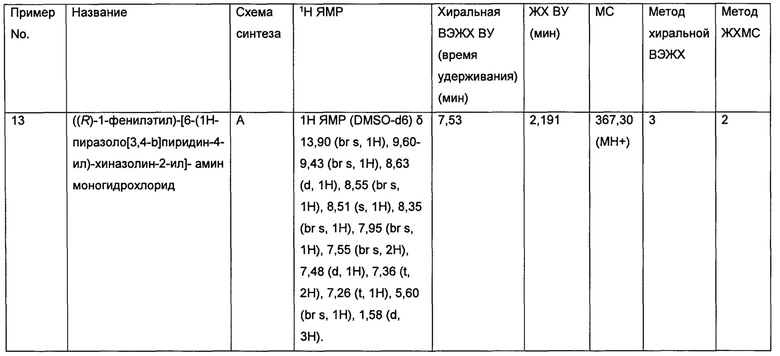
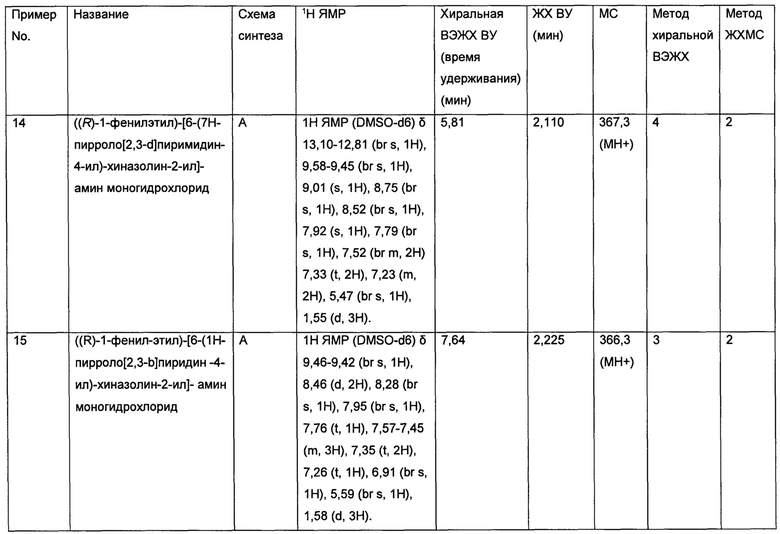
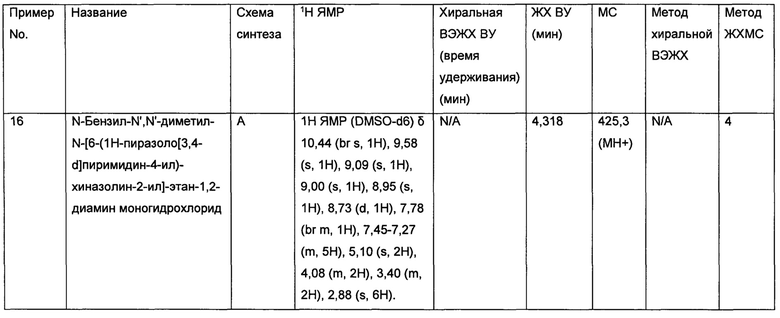
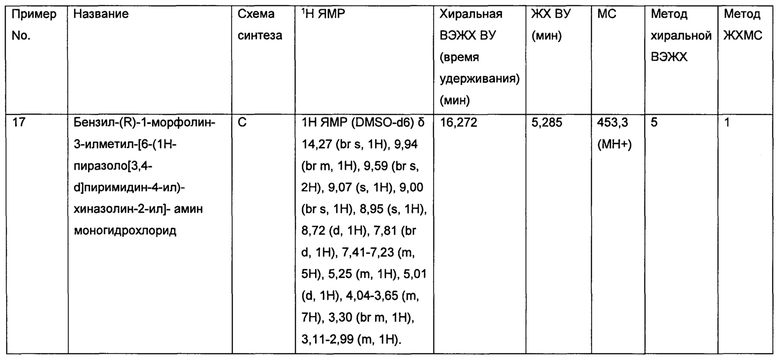
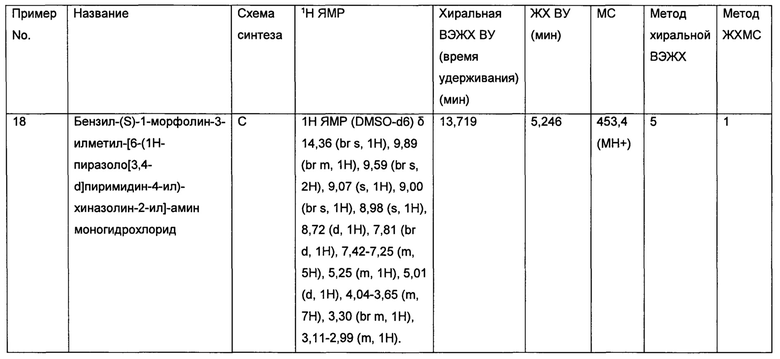
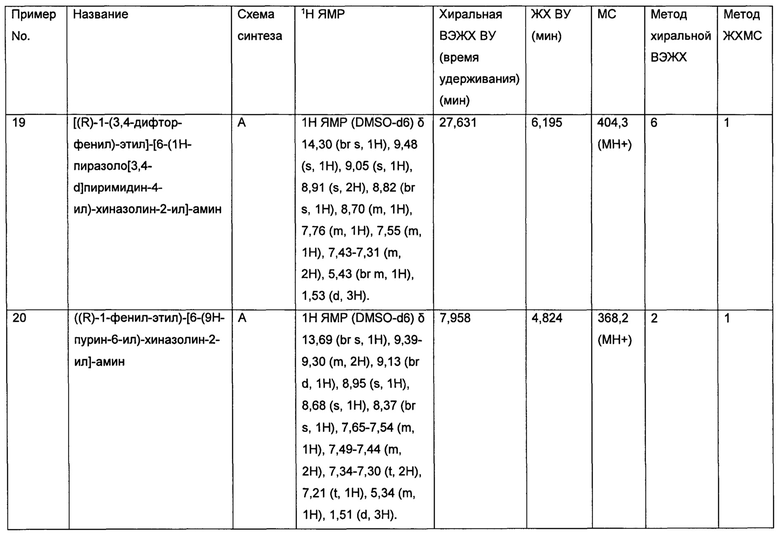
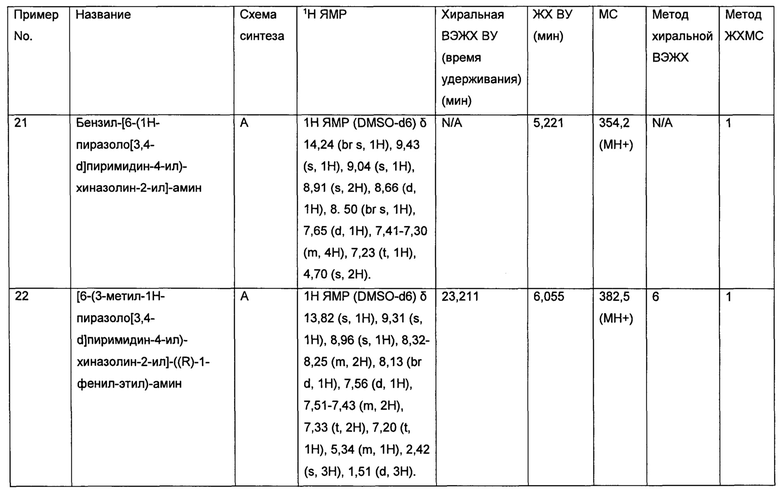
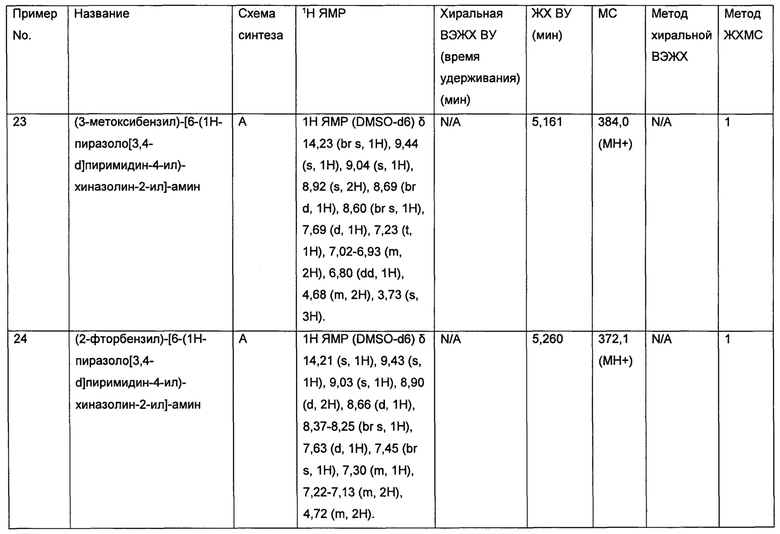
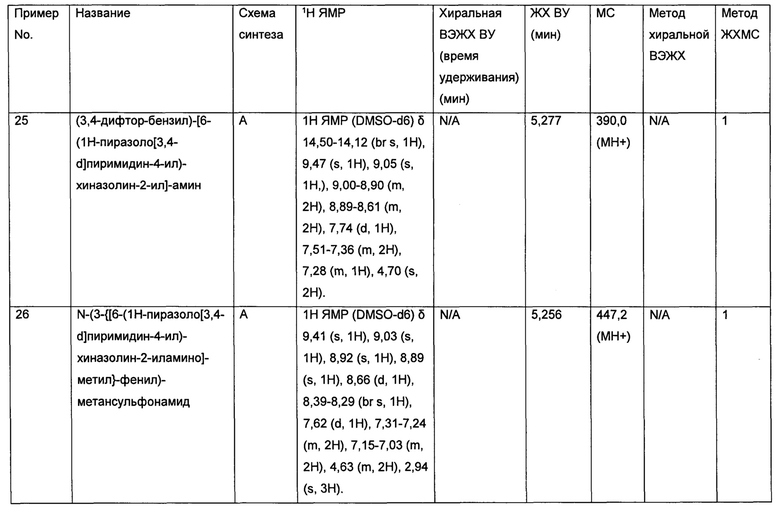
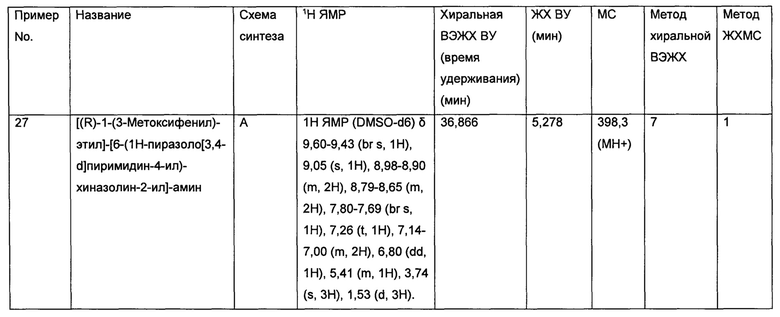
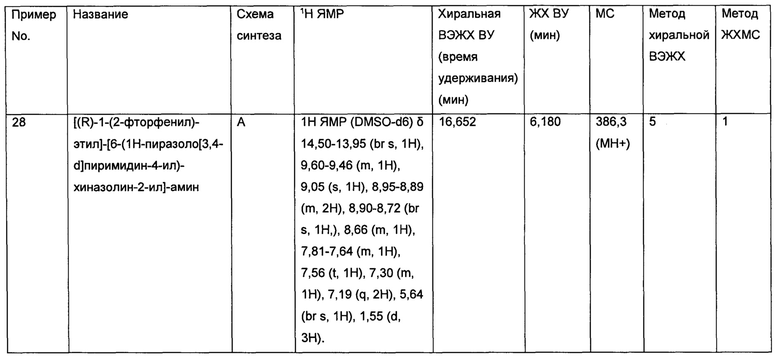
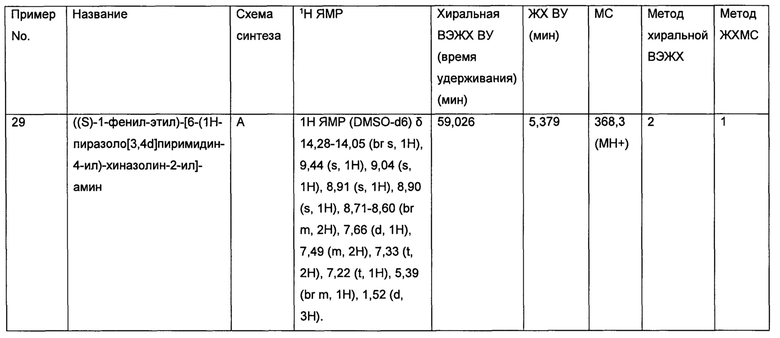
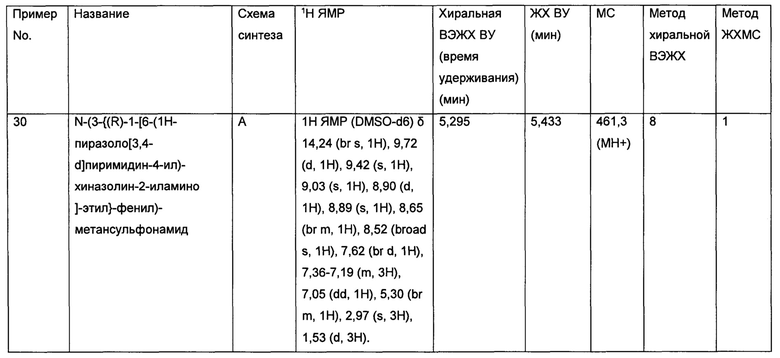
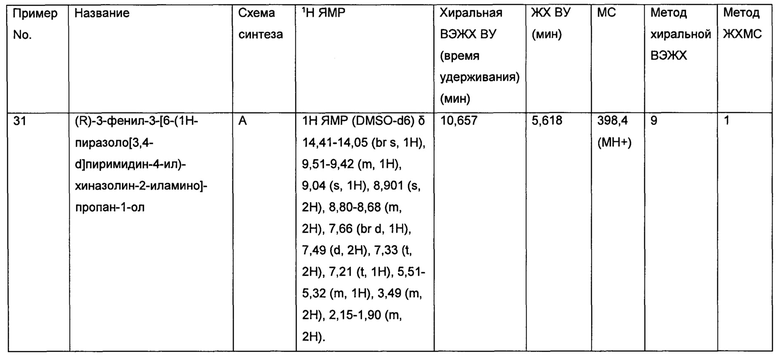
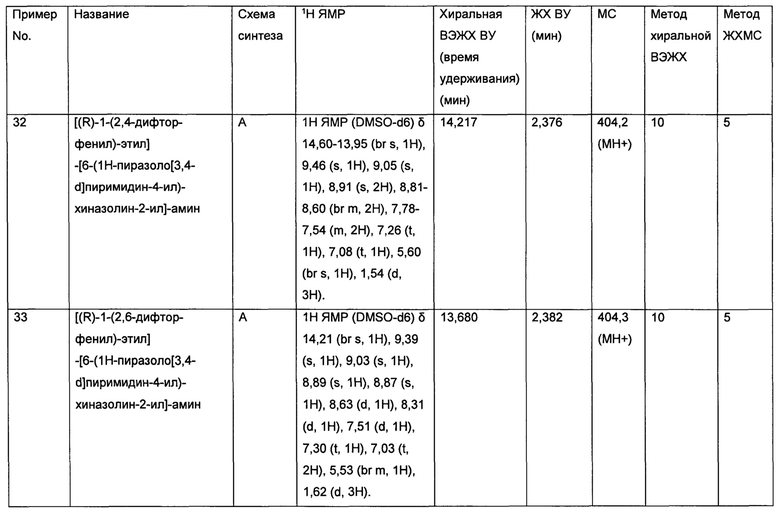
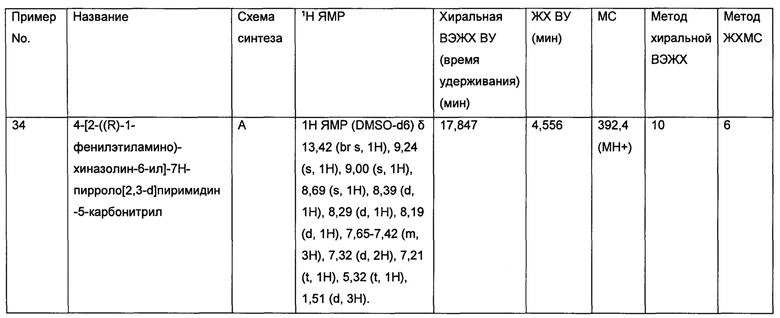
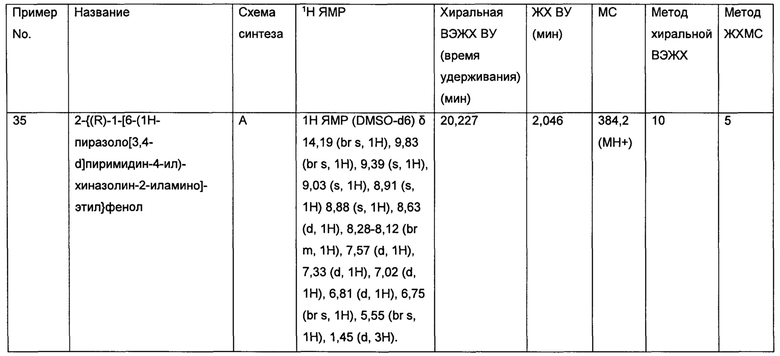
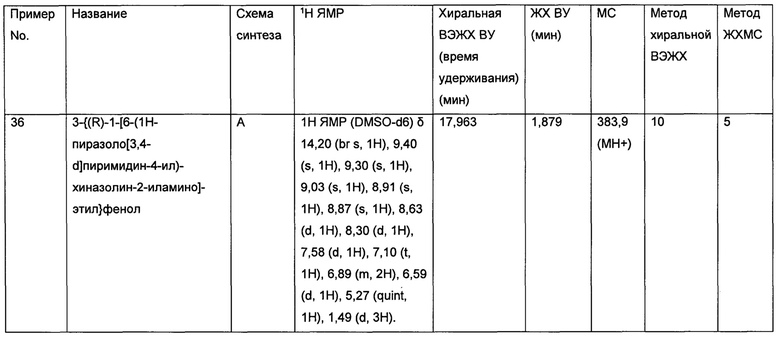
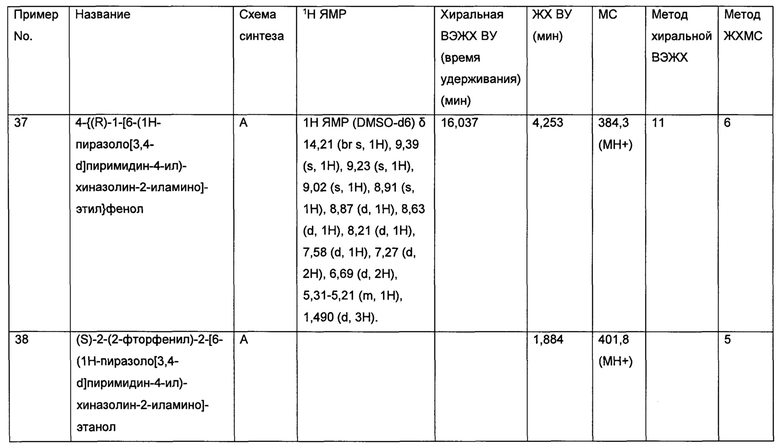
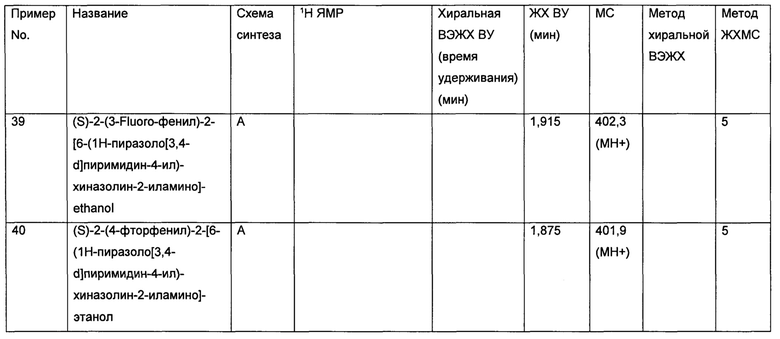
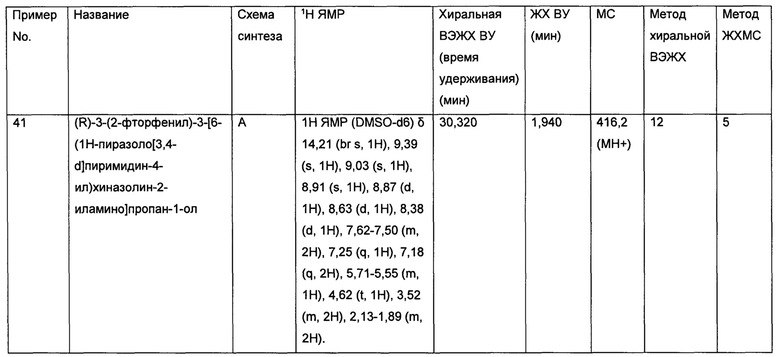
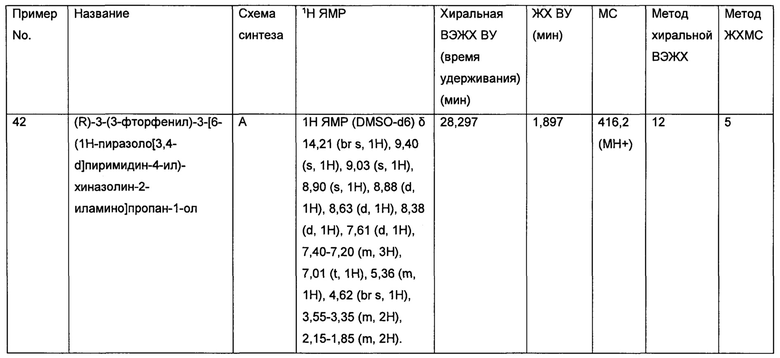
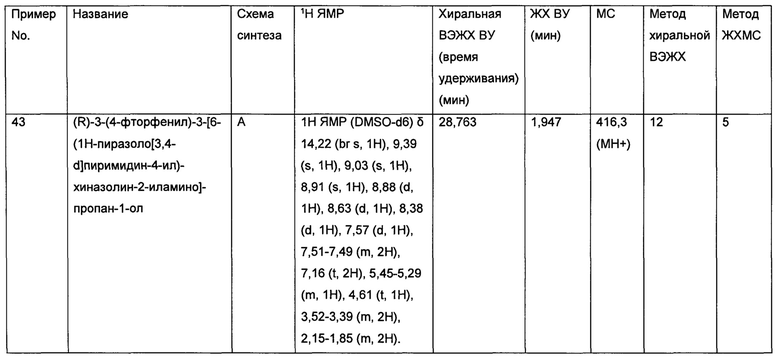
N/A=неприменимо
ПРИМЕР 44
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
(a) Определение ингибиторной активности в отношении p70S6
Способность соединений согласно настоящему изобретению ингибировать киназу P70S6 может быть определена с использованием нижеследующего протокола.
Буферная композиция:
20 мМ MOPS, 1 мМ EDTA, 0,01% Brij-35,5% глицерин, 0,1% b-меркаптоэтанол, 1 мг/мл BSA
Методика:
Исследование киназы p70S6K (h)
В конечном объеме реакции, составляющем 25 мкл, p70S6K (h) (5-10 мЕд) инкубировали с 8 мМ MOPS рН 7,0, 0,2 мМ EDTA, 100 мкМ KKRNRTLTV, 10 мкМ ацетатом Mg и [γ-33Р-АТР] (удельная активность приблизительно 500 cpm/pmol, концентрация по необходимости). Реакцию инициировали добавлением смеси MgATP. После инкубирования в течение 40 минут при комнатной температуре, реакцию останавливали добавлением 5 мкл 3% раствора фосфорной кислоты. 10 мкл реакционной смеси затем наносили на плоский фильтр Р30 и трижды промывали в течение 5 минут в 75 мМ фосфорной кислоте и один раз в метаноле перед высушиванием и сцинтилляционным подсчетом.
(b) Оценка концентрации в головном мозге и плазме крови в in vivo кассетной модели на мышах
Оценку соединений согласно настоящему изобретению для определения концентрации в головном мозге и плазме крови после перорального введения проводили в in vivo кассетной модели на мышах. Это стандартный для промышленности и общепринятый способ оценки пенетрации малых молекул в головной мозг (для ознакомления с недавними литературными статьями см.: in vitro permeability analysis, pharmacokinetic and brain distribution study in mice of imperatorin, isoimperatorin and cnidilin in Radix Angelicae Dahuricae, Fitoterapia, Volume 85, March 2013, Pages 144-153). Также общеизвестно, что более высокие концентрации в головном мозге (и более высокие соотношения концентрации в головном мозге: плазме) приводят к большему воздействию на мозг - что явным образом является целесообразным, если мозг является местом, подвергаемым воздействию.
Экспериментальный метод:
Для отдельного кассетного исследования использовали самцов мышей CD-1 (n=3 в момент времени, три момента времени: 1,0 ч, 3,0 ч и 8,0 ч).
Перорально вводили 5 соединений на кассету (доза 2,5 мг/кг для одного соединения, концентрация доз 0,25 мг/мл, объем доз 10,0 мл/кг).
Для растворения соединений использовали следующий состав: 10% DMSO / 90% гидроксипропил-β-циклодекстрин (20% масс./об. водный)
Конечная стадия представляла собой отбор проб и создание матриц проб плазмы и головного мозга. Для приготовления образцов плазмы белок осаждали с использованием ацетонитрила. Для приготовления образцов головного мозга осуществляли гомогенизацию и осаждение белка с помощью ацетонитрила. Образцы анализировали с использованием ВЭЖХ времяпролетной МС с использованием электрораспылительной ионизации.
Концентрации в головном мозге и плазме и соотношения концентрации в мозге и плазме, определенные для исследуемых соединений, приведены в таблице ниже.
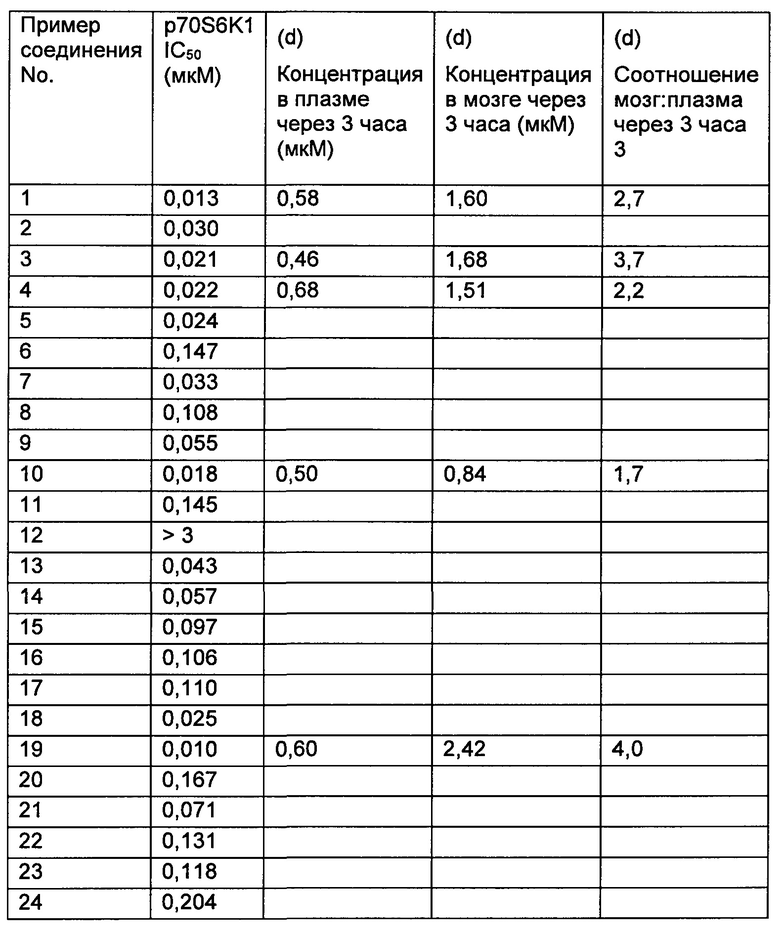
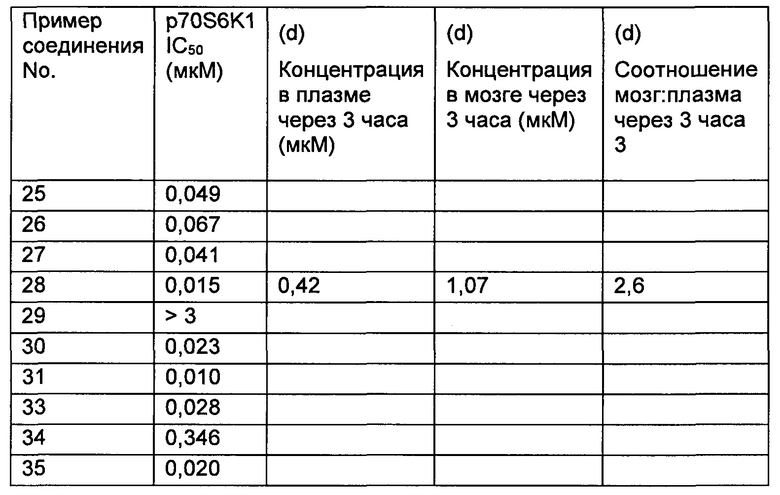
Таким образом, можно заключить, что соединения согласно настоящему изобретению демонстрируют эффективное ингибирование p70S6K1. Исследуемые соединения демонстрируют подходящие концентрации в плазме и головном мозге мышей после перорального приема, при которых концентрации в мозге превышают концентрации в плазме приводя к высоким значениям соотношений концентрация в мозге : в плазме. Обычно считается, что соотношение концентраций в мозге : в плазме, составляющее > 0,5, является благоприятным для лечения заболеваний мозга.
(с) Оценка эффективности соединений для предотвращения образования опухолей и метастазирования в in vivo модели трижды негативного рака молочной железы (TNBC)
Известно, что S6K1 играет важную роль в рецидивах рака TNBC после хирургической операции, в основном из-за активности S6K1 в обеспечении выживаемости раковых клеток в организме хозяина посредством фосфорилирования и активации антиапоптотического белка Bcl2 и Gli1 (Belletti 2014). Кроме того, S6K1 способствует метастазированию клеток TNBC (Akar 2010, Hung 2014).
С целью определения эффективности ингибитора S6K1 в in vivo модели возникновения опухоли и метастазирования, проводили следующий эксперимент:
В общей сложности 22 самки бестимусных голых мышей (Hsd:Athymic Nude-Foxn1nu) приобретали в Harlan (UK) и акклиматизировали в течение 7 дней до начала исследования. Животных помещали в отдельные вентилируемые клетки IVC (5 особей на клетку), где с помощью перфоратора им проделывали отверстия в ушных раковинах с целью определения каждой отдельной особи. Все животные получали свободный доступ к стандартному сертифицированному коммерчески доступному рациону и санированной воде в течение всего исследования. В клетках поддерживали стандартные условия: 20-24°С, 40-70% влажность и 12-часовой цикл день/ночь.
В первый день животных случайным образом распределяли по группам лечения, как указано в таблице ниже, и начинали терапию лекарственными средствами. В день 0, клетки MDA-MB-231 (1×106 в матригеле) имплантировали в жировую ткань молочной железы.
Применяли пероральный путь введения и следующий состав: 10% DMSO / 90% гидроксипропил-β-циклодекстрин (20% масс./об. водный).
Группы лечения:
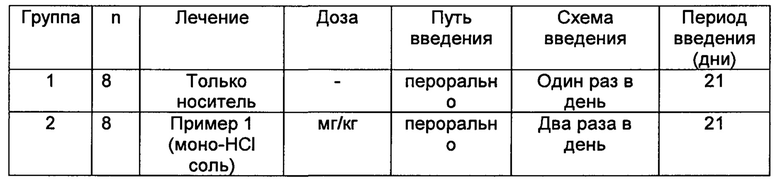
Во время исследования использовали следующие показатели результатов:
(1) Время до пальпации опухоли (латентный период) (2) объем опухоли (3) масса опухоли и органа для определения метастазирования (4) визуальная оценка внешнего вида метастатических узлов в легких
Результаты:
(1) Время до пальпации опухоли
Соединение согласно Примеру 1 отсрочивает время проявления (пальпации) и снижает частоту возникновения (см. Фигуру 1) по сравнению с контролем.
(2) Объем опухоли
Соединение согласно Примеру 1 обеспечивает значительное снижение объема опухолей (Фигура 2) по сравнению с контролем.
Массу печени и опухоли измеряли на 21 день. Лечение соединением согласно Примеру 1 вело к снижению массы печени по сравнению с группой, получавшей носитель, что может указывать на уменьшение метастазов в группе, получавшей лечение (см. Фигуру 3). Кроме того, опухоли животных, получавших лечение, весили значительно меньше (см. Фигуру 4).
(3) Визуальная оценка внешнего вида метастатических узлов в легких
При некроскопии легкие животных, получавших носитель и лечение, исследовали на предмет метастатических узлов:
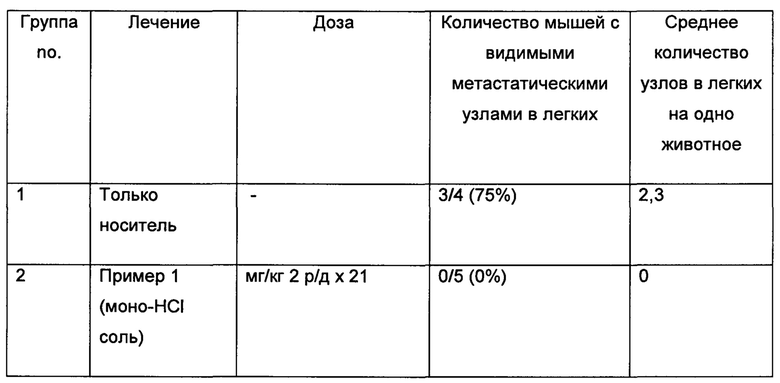
Полученные данные указывают на то, что соединение согласно Примеру 1 эффективно для уменьшения метастазов в легких мышей, возникающего вследствие спонтанного метастазирования первичной TNBC опухоли.
В совокупности это указывает на то, что соединение согласно Примеру 1, ингибитор S6K1, является эффективным для (а) предотвращения возникновения опухолей (b) остановки роста уже существующих опухолей (с) предотвращение образования метастазов в легких, возникающего вследствие первичной опухоли.
(d) Оценка соединений в аудиогенном исследовании пароксизма в in vivo модели фрагильной Х-хромосомы (FXS)
Пароксизмальные судороги возникают в сочетании с FXS и аутизмом примерно у четверти детей, страдающих этими расстройствами (Berry-Kravis Е (2002) Epilepsy in fragile X syndrome. Dev. Med. Child Neurol. 44(11): 724-728). Пароксизмальные судороги, вызванные повышенной восприимчивостью к звукам, называемые аудиогенными судорогами (АГС), является стабильным и подтвержденным клиническим признаков у мышей с FXS (Fmr1 KO), который не встречается у мышей дикого типа. Исследование аудиогенных судорог (АГС) представляет собой хорошо известную модель FXS на мышах (Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome, Epilepsia. 2000 Jan; 41(1): 19-23). В такой модели существует возможность дозирования соединений для наблюдения действия на восприимчивость мышей к аудиогенным судорогам. Для исследования эффективности соединение согласно настоящему изобретению проводили следующий АГС эксперимент:
Использовали следующий план эксперимента:
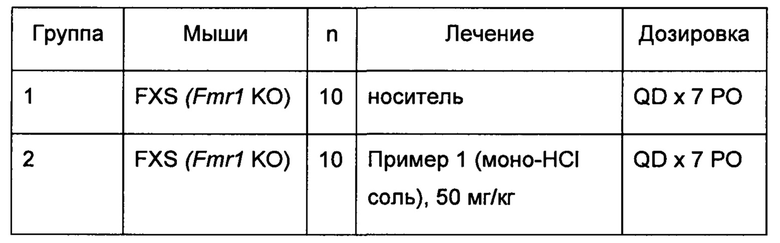
Используемый штамм мыши с FXS: FVB фон
Используемый носитель: 10% DMSO / 90% гидроксипропил-β-циклодекстрин (20% масс./об. водный)
АГС у животных оценивали спустя 7 последовательных дней введения соединений.
Метод:
Экспериментальная камера состояла из пластиковой клетки с размерами 25×25×см, на крыше которой был установлен дверной звонок (Электрический звонок Heath Zenith, модель 172С-А).
Мышей по одной забирали из вивария и переносили в экспериментальную камеру и позволяли изучить новую среду (базовый уровень шума ~65 дБ) в течение 30 секунд, после чего звонили в звонок (124 дБ) в течение 60 секунд.
Полученную в результате двигательную реакцию классифицировали с использованием шкалы, представляющей собой модификацию шкалы, первоначальной описанной у Jobe et al. (1973): нет ответа (NR: остановка или непрекращающееся исследование пространства), неконтролируемые движения (WR), пароксизмальные судороги (S) или остановка дыхания и/или смерть (RA). Оценку двигательного ответа определяли как процентное соотношение животных в группе, реагирующих на стимул.
Результаты:

Полученные данные показывают, что частота аудиогенных судорог, как и степень тяжести ответа, снижались при введении соединения согласно Примеру 1.
ПРИМЕР 45
ФАРМАЦЕВТИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ
(i) Лекарственная форма в виде таблетки
Композиция таблетки, содержащая соединение формулы (1), как описано в любом из Вариантов реализации 1.0-1.116, может быть получена путем смешивания 50 мг соединения со 197 мг лактозы (ВР) в качестве разбавителя, и 3 мг стеарата магния в качестве лубриканта и прессования с получением таблетки известным способом.
(ii) Лекарственная форма в виде капсулы
Препарат в форме капсул получают путем смешивания 100 мг соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, с 100 мг лактозы и необязательно 1% масс. стеарата магния и заполнения полученной смесью стандартных непрозрачных желатиновых капсул.
(iii) Инъецируемая лекарственная форма I
Композиция для парентерального введения путем инъекции может быть приготовления путем растворения соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, в воде, содержащей 10% пропиленгликоля, чтобы получить концентрацию активного вещества 1,5% по массе. Затем раствор стерилизуют фильтрацией, помещают в ампулы и запаивают.
(iv) Инъецируемая лекарственная Форма II
Композицию для парентерального введения путем инъекции получают путем растворения в воде соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, (2 мг/мл) и маннитола (50 мг/мл), стерильной фильтрации раствора и помещения его в запаиваемые ампулы или виалы объемом 1 мл.
v) Инъецируемая лекарственная форма III
Состав для внутривенной доставки путем инъекции или инфузии может быть приготовлен путем растворения соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, (например, в форме соли) в воде в концентрации 20 мг/мл. Затем виалу запечатывают и стерилизуют автоклавированием.
vi) Инъецируемая лекарственная Форма IV
Состав для внутривенной доставки путем инъекции или инфузии может быть приготовлен путем растворения соединения формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, (например, в форме соли) в воде, содержащей буфер (например, 0.2 М ацетат при рН 4.6) в концентрации 20 мг/мл. Затем виалу запечатывают и стерилизуют автоклавированием.
(vii) Лекарственная формула для подкожной инъекции
Композицию для подкожного введения получали путем смешивания соединение формулы (1), определенного в любом из Вариантов реализации 1.0-1.116, с кукурузным маслом фармацевтического класса в концентрации 5 мг/мл. Композицию стерилизовали и помещали в соответствующий контейнер.
viii) Лиофилизированная лекарственная форма
Аликвоты композиции соединения формулы (1), определенные в любом из Вариантов реализации 1.0-1.116, помещали в 50 мл виалы и лиофилизировали. Во время лиофилизации виалы замораживали с использованием протокола замораживания в один этап при (-45°С). Температуру поднимали до -10°С закалки, снижали для замораживания до -45°С, после чего следовала первичная сушка при +25°С в течение приблизительно 3400 минут, после чего следовала вторая сушка с этапами повышения температуры для 50°С. Давление в процессе первичной и вторичной сушки составляло 80 миллитор.
Эквиваленты
Следующие примеры представлены с целью иллюстрации изобретения и их не следует понимать в качестве ограничения объема настоящего изобретения. Должно быть очевидно, что в конкретные варианты реализации изобретения, описанные выше и проиллюстрированные в примерах, могут быть внесены многочисленные модификации и изменения, не отступая от принципов, лежащих в основе настоящего изобретения. Предполагается, что настоящее изобретение охватывает любые такие модификации и изменения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Гетероциклические соединения в качестве ингибиторов киназы RET | 2017 |
|
RU2742115C2 |
| НОВЫЕ ПИРАЗОЛПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2017 |
|
RU2769448C2 |
| ЗАМЕЩЁННЫЕ ПИРАЗОЛОПИРИМИДИНЫ КАК АКТИВАТОРЫ ГЛЮКОЦЕРЕБРОЗИДАЗЫ | 2011 |
|
RU2603637C2 |
| Ингибиторные соединения | 2018 |
|
RU2760669C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| ИНГИБИТОРЫ PI3-КИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2595718C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ МОДУЛИРОВАНИЯ КИНАЗЫ И ПОКАЗАНИЯ К ИХ ПРИМЕНЕНИЮ | 2013 |
|
RU2666146C2 |
| Производные гомопиперазинил- и гомопиперидинил-хиназолин-4(3H)-она, обладающие мультимодальной активностью в отношении боли | 2020 |
|
RU2833258C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2332415C2 |
| КОНДЕНСИРОВАННОЕ АЗА-ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2811975C1 |
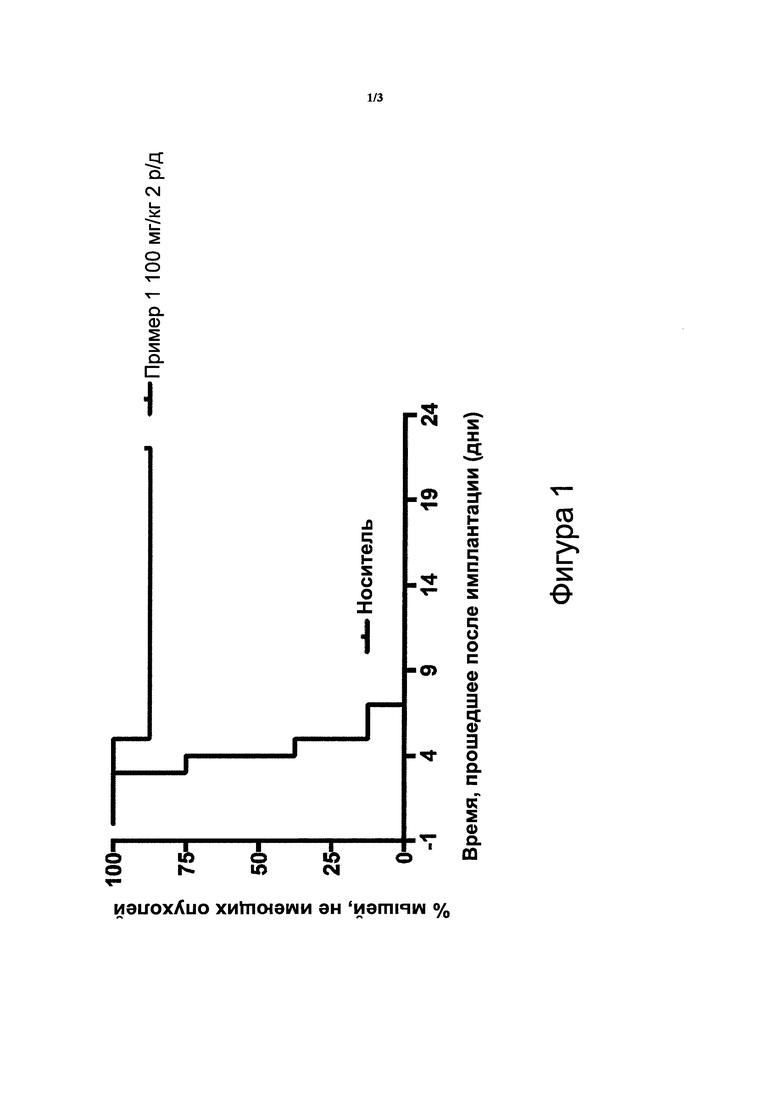
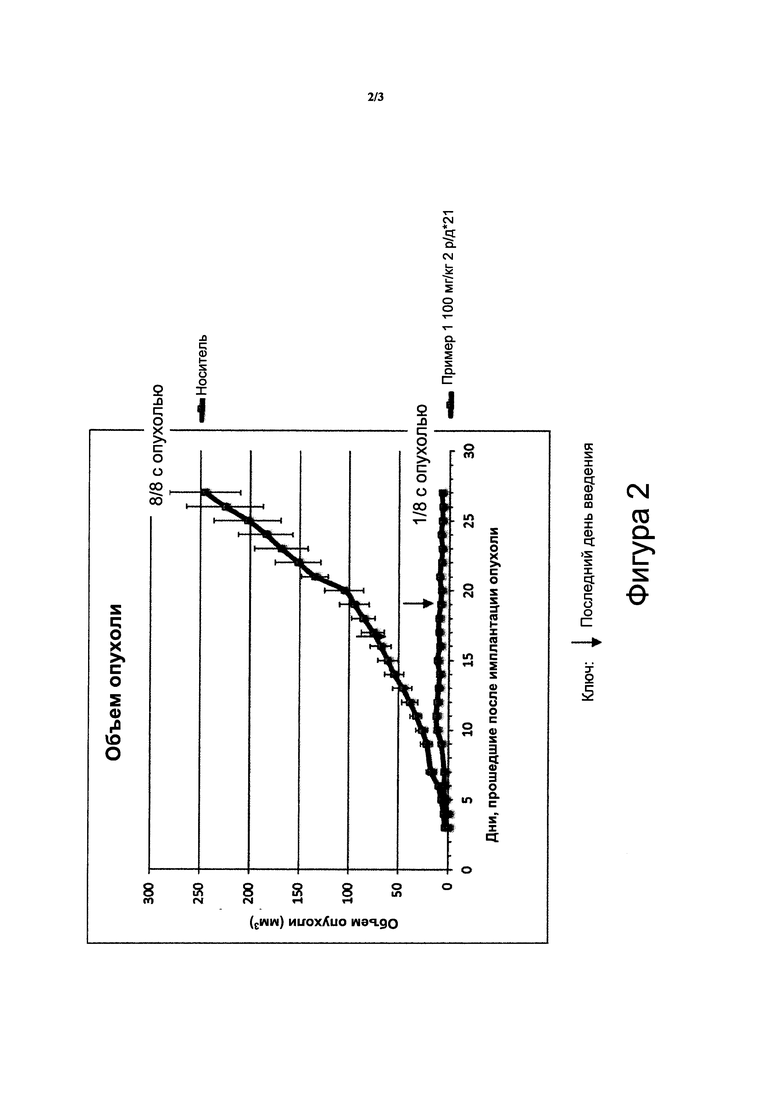
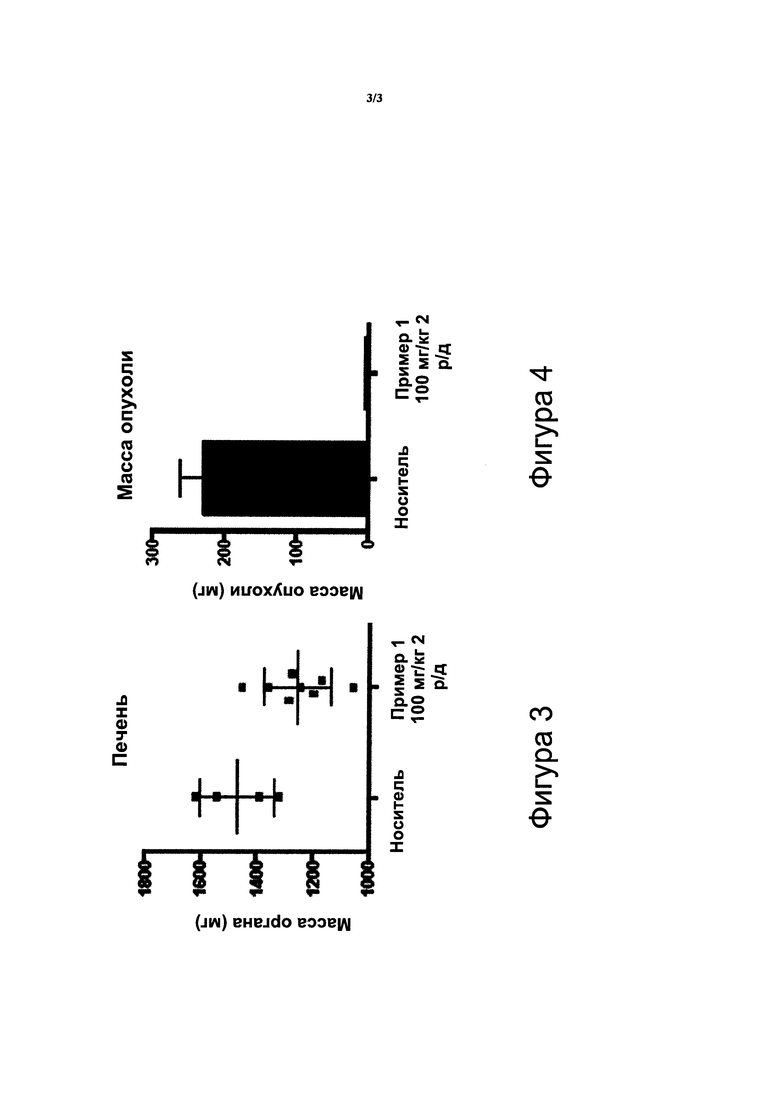
Изобретение относится к области органической химии, а именно к соединению формулы (1) или к его фармацевтически приемлемой соли, где один из Y и Z представляет собой R3, а другой представляет собой Ar2; Q1 представляет собой C1-4 алкиленовую группу, необязательно содержащую один заместитель, выбранный из гидрокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода; Q2 представляет собой связь или C1-3 алкиленовую группу; R1 выбран из водорода, NRxRy и группы Cy1; Rx и Ry являются одинаковыми или различными и каждый из них представляет собой C1-4 алкил; Cy1 представляет собой C-связанный морфолин; все R2, R3 и R4 представляют собой водород; Ar1 представляет собой фенильное кольцо, необязательно содержащее 1 или 2 заместителя R5, которые являются одинаковыми или различными и выбраны из галогена и группы Ra-Rb; Ra представляет собой связь, O или NRcSO2; Rb выбран из водорода, C1-8 алкильной группы; Rc представляет собой водород; и Ar2 представляет собой 5.6 конденсированное гетероароматическое кольцо, содержащее 2, 3 или 4 атома азота в качестве гетероатомных членов кольца, необязательно содержащее 1 заместитель R7, выбранный из C1-4 алкила и циано. Также изобретение относится к конкретным производным соединения формулы (1), фармацевтической композиции на основе соединения формулы (1), способу лечения указанных заболеваний и применению соединения формулы (1). Технический результат: получены новые гетероциклические соединения, обладающие свойствами ингибитора P70S6K. 8 н. и 4 з.п. ф-лы, 4 ил., 44 пр.
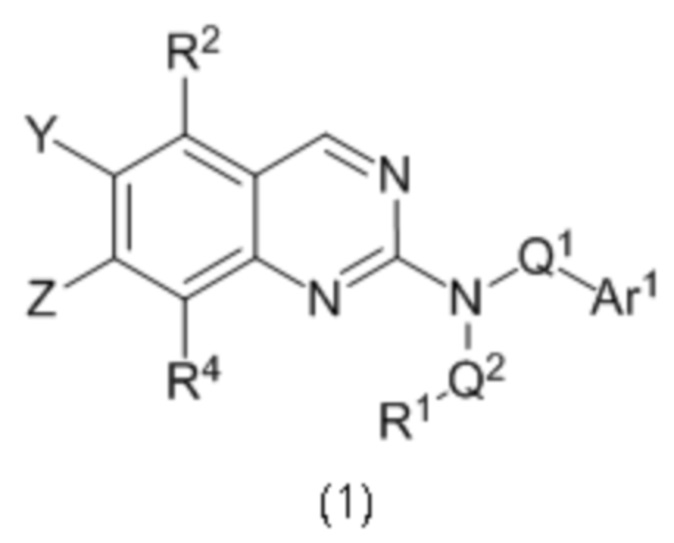
1. Соединение формулы (1)
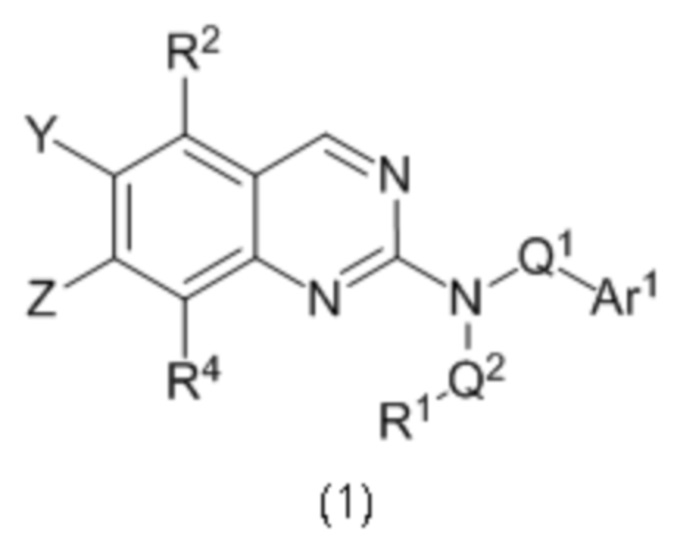
или его фармацевтически приемлемая соль,
где один из Y и Z представляет собой R3, а другой представляет собой Ar2;
Q1 представляет собой C1-4 алкиленовую группу, необязательно содержащую один заместитель, выбранный из гидрокси, при условии, что в присутствии гидрокси-заместителя между гидрокси-заместителем и атомом азота, к которому присоединен Q2, присутствуют по меньшей мере два атома углерода;
Q2 представляет собой связь или C1-3 алкиленовую группу;
R1 выбран из водорода, NRxRy и группы Cy1;
Rx и Ry являются одинаковыми или различными и каждый из них представляет собой C1-4 алкил;
Cy1 представляет собой C-связанный морфолин;
все R2, R3 и R4 представляют собой водород;
Ar1 представляет собой фенильное кольцо, необязательно содержащее 1 или 2 заместителя R5, которые являются одинаковыми или различными и выбраны из галогена и группы Ra-Rb;
Ra представляет собой связь, O или NRcSO2;
Rb выбран из:
• водорода;
• C1-8 алкильной группы;
Rc представляет собой водород; и
Ar2 представляет собой 5.6 конденсированное гетероароматическое кольцо, содержащее 2, 3 или 4 атома азота в качестве гетероатомных членов кольца, необязательно содержащее 1 заместитель R7, выбранный из C1-4 алкила и циано.
2. Соединение по п. 1, отличающееся тем, что Y представляет собой Ar2 и Z представляет собой R3.
3. Соединение по п.1, отличающееся тем, что Q1 выбран из CH2, CH(CH3), CH(CH2OH) и CH(CH2CH2OH).
4. Соединение по п. 1, отличающееся тем, что R1 представляет собой водород.
5. Соединение формулы (3)
или его фармацевтически приемлемая соль или таутомер, где R1, R2, R3, R4, Q1, Q2 и Ar1 являются такими, как определено в любом из пп. 1 - 4.
6. Соединение формулы (4)
или его фармацевтически приемлемая соль или таутомер, где R1, R2, R3, R4, R5, Q1 и Q2 являются такими, как определено в любом из пп. 1 - 4.
7. Соединение по п. 1, выбранное из группы, состоящей из:
((R)-1-фенилэтил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
((R)-1-(3-хлорфенил)этил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
((R)-1-(3-фторфенил)этил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
((R)-1-(4-фторфенил)этил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
((S)-2-гидрокси-1-фенилэтил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
бензил-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]метиламина;
(3-фторбензил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
((R)-1-фенилэтил)-[6-(1H-пиразоло[3,4-b]пиридин-4-ил)хиназолин-2-ил]амина;
бензил-(S)-1-морфолин-3-илметил-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
[(R)-1-(3,4-дифторфенил)этил]-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
бензил-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
(3,4-дифторбензил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
N-(3-{[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]метил}фенил)метансульфонамида;
[(R)-1-(3-метоксифенил)этил]-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
[(R)-1-(2-фторфенил)этил]-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
N-(3-{(R)-1-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенил)метансульфонамида;
(R)-3-фенил-3-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]пропан-1-ола;
[(R)-1-(2,4-дифторфенил)этил]-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
[(R)-1-(2,6-дифторфенил)этил]-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амина;
2-{(R)-1-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенола;
3-{(R)-1-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенола;
4-{(R)-1-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этил}фенола;
(S)-2-(2-фторфенил)-2-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]-этанола;
(S)-2-(3-фторфенил)-2-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]-этанола; и
(S)-2-(4-фторфенил)-2-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-иламино]этанола;
и их фармацевтически приемлемых солей.
8. ((R)-1-фенилэтил)-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин или его фармацевтически приемлемая соль.
9. [(R)-1-(3,4-дифторфенил)этил]-[6-(1H-пиразоло[3,4-d]пиримидин-4-ил)хиназолин-2-ил]амин или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция для
• лечения трижды негативного рака молочной железы;
• предотвращения или лечения метастазов, например метастазов в головном мозге, костях, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре, например метастазов в головном мозге, возникающих при трижды негативном раке молочной железы;
• лечения глиом и глиобластом; или
• лечения нарушения нервно-психического развития (такого как синдром ломкой Х-хромосомы),
содержащая эффективное количество соединения по п. 1, обладающего свойствами ингибитора P70S6K, и фармацевтически приемлемое вспомогательное вещество.
11. Способ
• лечения трижды негативного рака молочной железы;
• предотвращения или лечения метастазов, например метастазов в головном мозге, костях, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре, например метастазов в головном мозге, возникающих при трижды негативном раке молочной железы;
• лечения глиом и глиобластом; или
• лечения нарушения нервно-психического развития (такого как синдром ломкой Х-хромосомы),
включающий введение пациенту эффективного количества соединения по п. 1, обладающего свойствами ингибитора P70S6K.
12. Применение соединения по п. 1, обладающего свойствами ингибитора P70S6K, для:
• лечения трижды негативного рака молочной железы;
• предотвращения или лечения метастазов, например метастазов в головном мозге, костях, легких, печени, поджелудочной железе, почках, мочевом пузыре и желчном пузыре, например метастазов в головном мозге, возникающих при трижды негативном раке молочной железы;
• лечения глиом и глиобластом; или
• лечения нарушения нервно-психического развития (такого как синдром ломкой Х-хромосомы).
| WO 2010056758 А1, 20.05.2010 | |||
| WO 2014015291 A1, 23.01.2014 | |||
| WO 2010136755 A1, 02.12.2010. |

