ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящее изобретение испрашивает приоритет на основании Китайской патентной заявки №202010411386.0, поданной 15 мая 2020 года и озаглавленной «ПИРАЗОЛОПИРИМИДИНОВОЕ АМИДНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ», полное содержание которой включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики и, в частности, относится к соединению конденсированного аза-гетероциклического амида, обладающему ингибирующей активностью в отношении киназ, или таутомеру, стереоизомеру, оптическому изомеру, сольвату, изотопному производному, N-оксиду, пролекарству или фармацевтически приемлемой соли указанного соединения, и фармацевтической композиции, содержащей их, и их применению в качестве лекарственного средства для профилактики и/или лечения ТРК-опосредованных заболеваний.
УРОВЕНЬ ТЕХНИКИ
Тропомиозин-связанная киназа или тропомиозин-рецепторная киназа (TRK) представляет собой тип рецептора фактора роста нервов, семейство которого состоит из трех высоко гомологичных подтипов TRKA, TRKB и TRKC, кодируемых генами рецепторной нейротрофической тирозинкиназы типа 1 (NTRK1), NTRK2 и NTRK3, соответственно. Когда белок рецептора TRK связывается с соответствующим лигандом, различные физиологические функции могут быть достигнуты путем активации нисходящих сигнальных путей, таких как путь RAS/MAPK, путь PLCγ и путь PI3K. Белки семейства TRK обычно в основном экспрессируются в нервных тканях, участвуют в дифференцировке и выживании нервных клеток и формировании аксонов и дендритов, а также играют важную роль в эмбриональном развитии и поддержании нормальных функций нервной системы.
ТРК-киназа активируется при злокачественных новообразованиях с помощью различных механизмов, главным образом структурных перестроек и измененной экспрессии. Например, ген NTRK, кодирующий TRK-киназу, перегруппируется с другими генами с образованием слитого онкогена, который вызывает изменение структуры и экспрессии TRK-киназы и больше не регулируется и не контролируется лигандом фактора роста нервов, а именно конститутивно активируется, и, следовательно, способствует образованию и развитию опухолей. Кроме того, результат секвенирования гена также показывает, что TRK-киназа имеет тесную связь с возникновением, метастазированием и ухудшением различных опухолей и экспрессируется в различных опухолях, таких как немелкоклеточный рак легкого, колоректальный рак, меланома, рак желчного пузыря, рак щитовидной железы, злокачественная глиома и тому подобное.
В настоящее время первое поколение ингибиторов TRK Ларотректиниб (LOXO-101) и Энтректиниб (RXDX-101) было одобрено Управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) в 2018 и 2019 годах соответственно. Ларотректиниб является сильным пероральным и селективным ингибитором тропомиозин-рецепторной киназы, данные об эффективности которого были опубликованы на совещании Американского общества клинической онкологии (ASCO) еще в июне 2017 г. В общей сложности из 55 пациентов, включенных в клинические исследования фазы I и фазы II, 46 оцениваемых пациентов имели общую частоту ответа (ОЧО) 78%. Энтректеиниб является сильным ингибитором белков TRK, ROS1 и ALK и может проходить через гематоэнцефалический барьер, с ОЧО 79% у 24 оцениваемых пациентов в клинических исследованиях фазы I.
Как и другие направленные препараты, ингибиторы ТРК также сталкиваются с проблемами лекарственной устойчивости. Мутация в домене киназы NTRK может вызывать изменение конформации домена протеинкиназы семейства TRK или аффинности связывания с АТФ, тем самым влияя на связывание ингибиторов TRK с мишенями, и типы мутаций представляют собой G595R, G639R, G667C и тому подобное. С целью решения проблемы лекарственной устойчивости ингибиторов ТРК первого поколения были изучены ингибиторы ТРК второго поколения, такие как LOXO-195, ТРХ-005 и тому подобное.


КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Одна из целей настоящего изобретения заключается в обеспечении типа конденсированного аза-гетероциклического амидного соединения с превосходной активностью ингибирования TRK (дикого типа и мутантного типа) и новой структурой, или его таутомера, стереоизомера, сольвата, изотопного производного, N-оксида, пролекарства или фармацевтически приемлемой соли.
Другой целью настоящего изобретения является обеспечение типа конденсированного аза-гетероциклического амидного соединения с лучшей активностью ингибирования ТРК-мутантных опухолевых клеток по сравнению с известным соединением и новой структурой, или его таутомера, стереоизомера, сольвата, изотопного производного, N-оксида, пролекарства или фармацевтически приемлемой соли.
Другой целью настоящего изобретения является обеспечение типа конденсированного аза-гетероциклического амидного соединения с лучшей активностью ингибирования TRK (дикого типа и мутантного типа) и активностью ингибирования ТРК-мутантных опухолевых клеток по сравнению с известным соединением и новой структурой, или его таутомера, стереоизомера, сольвата, изотопного производного, N-оксида, пролекарства или фармацевтически приемлемой соли.
Другой задачей настоящего изобретения является обеспечение типа конденсированного аза-гетероциклического амидного соединения с лучшей ингибирующей активностью в отношении опухолевых клеток с мутацией TRK и противоопухолевой активностью in vivo по сравнению с известным соединением, или его таутомера, стереоизомера, сольвата, изотопного производного, N-оксида, пролекарства или фармацевтически приемлемой соли.
Другой целью настоящего изобретения является обеспечение типа конденсированного аза-гетероциклического амидного соединения с лучшей ингибирующей активностью в отношении опухолевых клеток с мутацией TRK и противоопухолевой активностью in vivo, а также лучшей безопасностью по сравнению с известным соединением, или его таутомера, стереоизомера, сольвата, изотопного производного, N-оксида, пролекарства или фармацевтически приемлемой соли.
Другой целью настоящего изобретения является обеспечение типа конденсированного аза-гетероциклического амидного соединения с лучшей активностью ингибирования TRK (дикого типа и мутантного типа), активностью ингибирования опухолевых клеток с мутацией TRK и противоопухолевой активностью in vivo, а также лучшей безопасностью по сравнению с известным соединением, или его таутомера, стереоизомера, сольвата, изотопного производного, N-оксида, пролекарства или фармацевтически приемлемой соли.
В частности, в настоящем описании предложено соединение формулы (I) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль,
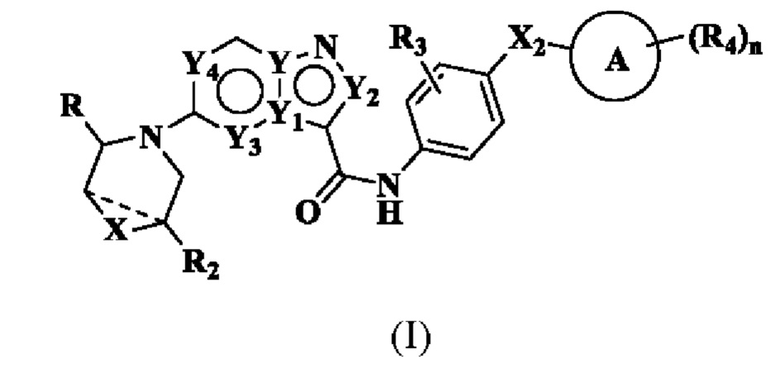
где X выбран из: химической связи, -О-, -S-, -NH- и -CH2-;
Y, Y1, Y2, Y3 и Y4 каждый независимо выбран из: -СН-, N и С;
Х2 выбран из: химической связи, -(СН2)р- и -NH-, где р равен 1, 2, 3 или 4;
 означает, что химическая связь отсутствует или присутствует;
означает, что химическая связь отсутствует или присутствует;
R выбран из: С5-12 арила или гетероарила, где каждый ар ил или гетероарил является незамещенным или замещенным по меньшей мере одним заместителем, выбранным из R1;
Каждый R1, при его наличии, независимо выбран из водорода, галогена, -ОН, амино, -NHC1-6 алкила, -N(C1-6 алкил)2, циано, CI-2 алкила, незамещенного или замещенного по меньшей мере одним R1a, С1-6 алкокси, незамещенного или замещенного по меньшей мере одним R1a, С3-6 циклоалкила, незамещенного или замещенного по меньшей мере одним R1a, С3-6 циклоалкокси, незамещенного или замещенного по меньшей мере одним R1a, и -SC1-6 алкила;
R1a, при его наличии, каждый независимо выбран из C1-6 алкила, С1-6 алкокси, нитро, галогена, -ОН, амино, -NHC1-6 алкила, -N(C1-6 алкил)2 и циано;
R2 выбран из: Н, галогена, гидроксила, амино и замещенного или незамещенного C1-6 алкила, где «замещенный» означает замещенный 1, 2 или 3 заместителями, выбранными из галогена и гидроксила;
R3 выбран из: Н, галогена, -ОН, амино, С1-6 алкила и C1-6 алкокси;
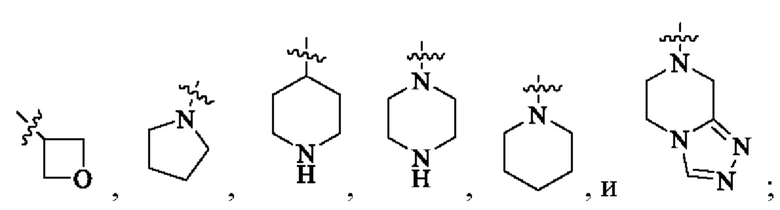
кольцо А выбрано из: циклоалкила, гетероциклоалкила, мостикового циклила, гетеромостикового циклила, конденсированного циклила, гетерокоденсированного циклила, спироциклила и гетероспироциклила, причем гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетероспироциклиле независимо выбраны из О, S и N, и количество гетероатомов выбрано из 1, 2, 3 и 4;
R4 находится в любом замещаемом положении на кольце А и независимо выбран из: -Н, -ОН, галогена, -CN, оксо, замещенного или незамещенного C1-6 алкила, -(СН2)m-ОН, -(СН2)m-СООН, -(CH2)m-CO-NH2, -CO-(CH2)m-NH2, -CO-CR4aR4b-OH и -CO-R4b; где оксо означает, что два атома Н одного и того же сайта замещения замещены одним и тем же атомом О с образованием двойной связи; m выбран из 1, 2, 3 и 4;
R4a выбран из водорода и незамещенного или замещенного cI-4 алкила;
R4b выбран из Н, незамещенного или замещенного C1-6 алкила и незамещенного или замещенного С3-6 циклоалкила, заместитель для замещения независимо выбран из -ОН, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3;
n выбран из 1, 2, 3 и 4.
В одном варианте реализации настоящего изобретения предложено соединение формулы (I) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль, где R выбран из: С5-9 арила и гетероарила, где каждый арил или гетероарил является незамещенным или замещенным по меньшей мере одним заместителем, выбранным из R1; или R выбран из: С5-6 арила и гетероарила, где каждый арил или гетероарил является незамещенным или замещенным по меньшей мере одним заместителем, выбранным из R1.
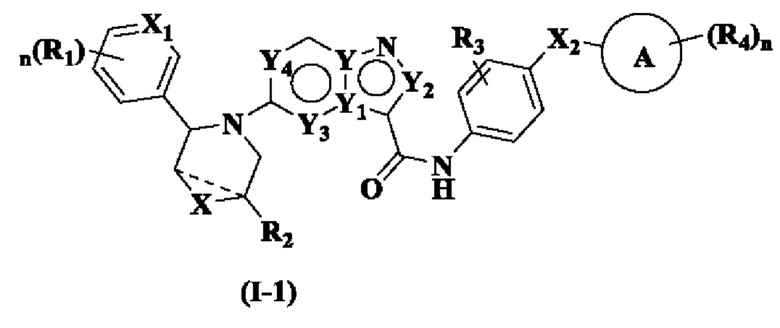
В одном из вариантов реализации указанное соединение имеет структуру формулы (I-1) или формулы (I-2):
 или
или

где R1, R2, R3, R4, X, Х2, Y, Y1, Y2, Y3, Y4, кольцо А и n являются такими, как описано для соединения формулы (I) настоящего изобретения, и X1 выбран из: -СН- и N. В одном варианте реализации настоящего изобретения указанное соединение имеет структуру формулы (I-A):
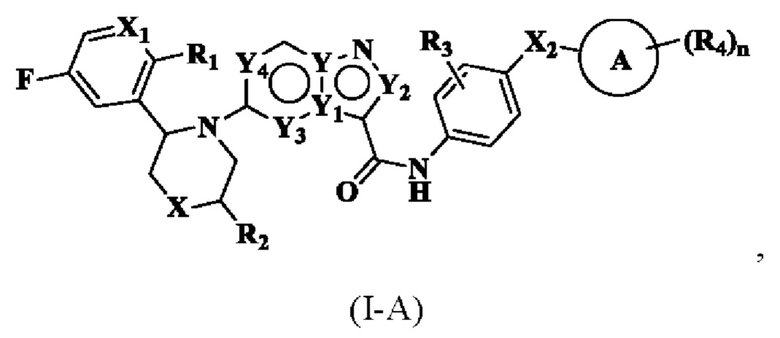
где R1, R2, R3, R4, X, X1, Х2, Y, Y1, Y2, Y3, Y4, кольцо А и n являются такими, как описано для соединения формулы (I-1) или формулы (I-2) настоящего изобретения.
В одном из вариантов реализации, соединение имеет структуру формулы (I-А-1а), формулы (I-A-1b) или формулы (I-А-1с):

где R1, R2, R3, R4, X, X1, Х2, кольцо А и n являются такими, как описано для соединения формулы (I-1) или формулы (I-2) настоящего изобретения.
В одном из вариантов реализации соединение имеет структуру формулы (I-B):
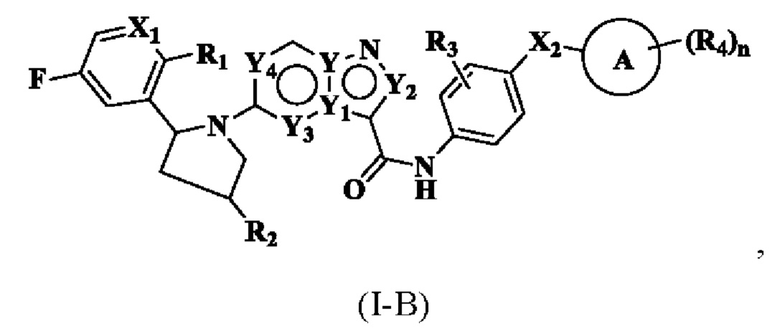
где R1, R2, R3, R4, X1, X2, Y, Y1, Y2, Y3, Y4, кольцо А и n являются такими, как описано для соединения формулы (I-1) или формулы (I-2) настоящего изобретения.
В одном из вариантов реализации, соединение имеет структуру формулы (I-В-1а), формулы (I-B-1b) или формулы (I-В-1с):
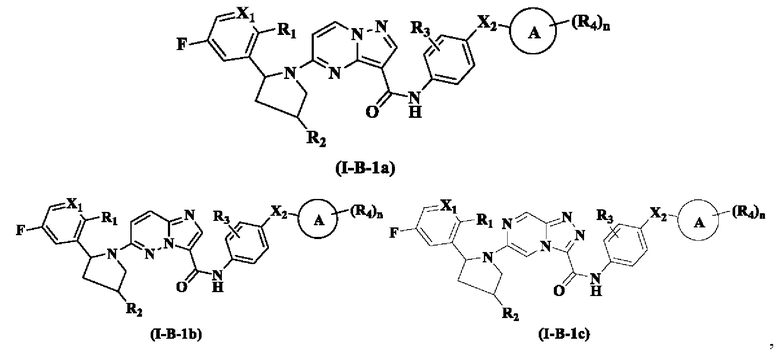
где R1, R2, R3, R4, X1, Х2, кольцо А и n являются такими, как описано для соединения формулы (I-1) или формулы (I-2) настоящего изобретения.
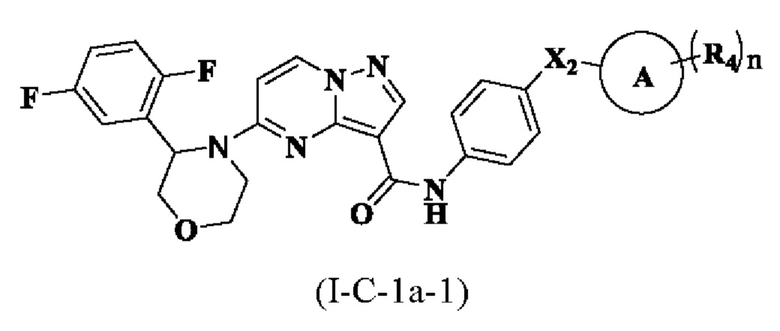
В одном варианте реализации соединение имеет структуру формулы (I-C):
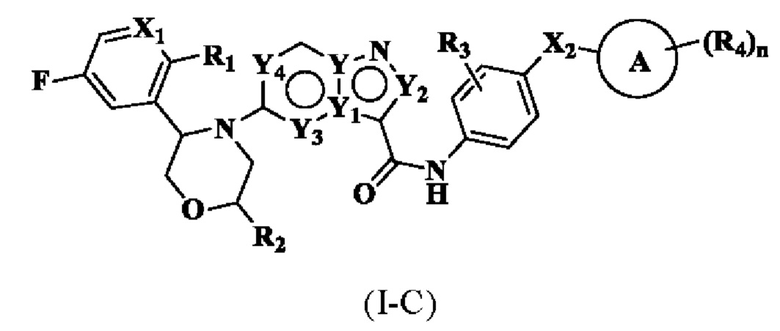
где R1, R2, R3, R4, X1, Х2, Y, Y1, Y2, Y3, Y4, кольцо А и n являются такими, как описано для соединения формулы (I-1) или формулы (I-2) настоящего изобретения. В одном из вариантов реализации, соединение имеет структуру формулы (I-С-1а), формулы (I-C-1b) или формулы (I-С-1с):

где R1, R2, R3, R4, X1, X2, кольцо А и n являются такими, как описано для соединения формулы (I-1) или формулы (I-2) настоящего изобретения.
В одном варианте реализации настоящего изобретения предложено соединение (соединение формулы (I), соединение формулы (I-1), соединение формулы (I-2), соединение формулы (I-A), соединение формулы (I-А-1а), соединение формулы (I-A-1b), соединение формулы (I-А-1 с), соединение формулы (I-B), соединение формулы (I-В-1а), соединение формулы (I-B-1b), соединение формулы (I-В-1с), соединение формулы (I-C), соединение формулы (I-С-1а), соединение формулы (I-C-1b), соединение формулы (I-C-1с)) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль, где каждый R1, при его наличии, независимо выбран из водорода, галогена, -ОН, амино, циано, С1-6 алкила, незамещенного или замещенного по меньшей мере одним R1a, С1-6 алкокси, незамещенного или замещенного по меньшей мере одним R1a, С3-6 циклоалкила, незамещенного или замещенного по меньшей мере одним R1a, и С3-6 циклоалкокси, незамещенного или замещенного по меньшей мере одним R1a; или, каждый R1, при его наличии, независимо выбран из: водорода, галогена, -ОН, амино, циано, С1-6 алкила, незамещенного или замещенного по меньшей мере одним R1a, и C1-6 алкокси, незамещенного или замещенного по меньшей мере одним R1a; или, каждый R1, при его наличии, независимо выбран из: водорода, галогена, -ОН, амино, С1-6 алкила и С1-6 алкокси; или, каждый R1, при его наличии, независимо выбран из: водорода, галогена, -ОН, амино, С1-3 алкила и С1-3 алкокси; или, каждый R1, при его наличии, независимо выбран из: водорода, галогена, -ОН, амино, метила, этила, метокси и этокси; или, каждый R1, при его наличии, независимо выбран из: водорода, F, Cl, -ОН, метила и метокси; или, каждый R1, при его наличии, независимо выбран из: F, -ОН, метила и метокси. В отдельном варианте реализации настоящего изобретения каждый R1a, при его наличии, независимо выбран из С1-3 алкила, С1-3 алкокси, нитро, галогена, -ОН, амино, -NHC1-6 алкила, -N(C1-6 алкил)2 и циано; или R1a выбран из галогена, -ОН, амино, -NHC1-3 алкила, -N(С1-3 алкил)2 и циано; или R1a выбран из галогена, -ОН, амино и циано; или R1a выбран из F, Cl и -ОН.
В одном варианте реализации настоящего изобретения предложено соединение (соединение формулы (I), соединение формулы (I-1), соединение формулы (I-2), соединение формулы (I-A), соединение формулы (I-А-1а), соединение формулы (I-A-1b), соединение формулы (I-А-1с), соединение формулы (I-B), соединение формулы (I-В-1а), соединение формулы (I-B-1b), соединение формулы (I-В-1с), соединение формулы (I-C), соединение формулы (I-С-1а), соединение формулы (I-C-1b), соединение формулы (I-C-1 с)) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль, где R2 выбран из: Н, F, ОН и замещенного или незамещенного C1-6 алкила, где «замещенный» означает замещенный 1, 2 или 3 заместителями, выбранными из галогена и гидроксила; или R2 выбран из Н, F, -ОН и С1-6 алкила; или R2 выбран из Н, F, -ОН и С1-3 алкила; или R2 выбран из Н и F.
В одном варианте реализации настоящего изобретения предложено соединение (соединение формулы (I), соединение формулы (I-1), соединение формулы (I-2), соединение формулы (I-A), соединение формулы (I-А-1а), соединение формулы (I-A-1b), соединение формулы (I-А-1 с), соединение формулы (I-B), соединение формулы (I-В-1а), соединение формулы (I-B-1b), соединение формулы (I-В-1 с), соединение формулы (I-C), соединение формулы (I-С-1а), соединение формулы (I-C-1b), соединение формулы (I-C-1 с)) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, оксид азота, пролекарство или фармацевтически приемлемая соль, где X1 выбран из -СН- и N, и R1 выбран из F и -OCH2.
В одном из вариантов реализации X1 выбран из -СН-, и R1 выбран из F; или X1 выбран из N, и R1 выбран из -ОСН3.
В одном варианте реализации соединение имеет структуру формулы (I-D):
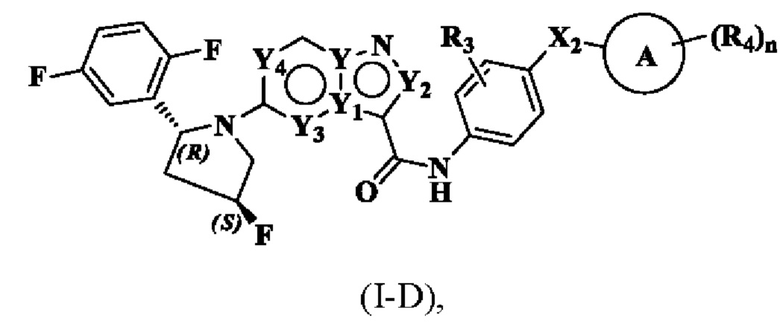
где R3, R4, Х2, Y, Y1, Y2, Y3, Y4, кольцо А и n являются такими, как описано для соединения формулы (I) настоящего изобретения.
В одном из вариантов реализации, соединение имеет структуру формулы (I-D-a), формулы (I-D-b) или формулы (I-D-c):
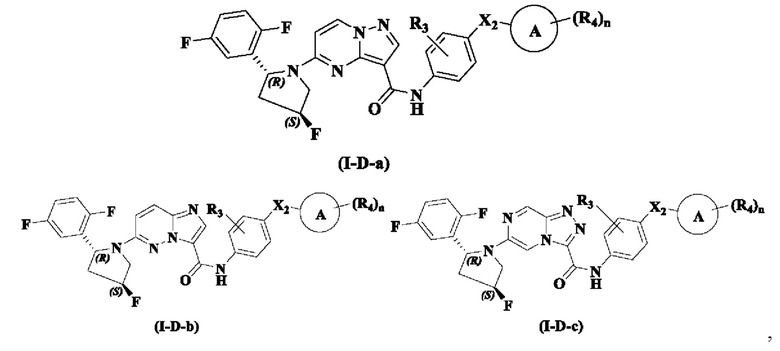
где R3, R4, Х2, кольцо А и n являются такими, как описано для соединения формулы (I) настоящего изобретения.
В одном варианте реализации настоящего изобретения предложено соединение (соединение формулы (I), соединение формулы (I-1), соединение формулы (I-2), соединение формулы (I-A), соединение формулы (I-А-1а), соединение формулы (I-A-1b), соединение формулы (I-А-1 с), соединение формулы (I-B), соединение формулы (I-В-1а), соединение формулы (I-B-1b), соединение формулы (I-В-1 с), соединение формулы (I-C), соединение формулы (I-С-1а), соединение формулы (I-C-1b), соединение формулы (I-C-1 с), соединение формулы (I-D), соединение формулы (I-D-a), соединение формулы (I-D-b), соединение формулы (I-D-c)) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль, где R3 выбран из: Н, галогена, -ОН, амино, С1-3 алкила и С1-3 алкокси; или R3 выбран из: Н, F, Cl, -ОН, метила и метокси; или R3 выбран из: Н и F. В одном из вариантов реализации, соединение имеет структуру формулы (I-D-1a), формулы (I-D-1b) или формулы (I-D-1c):
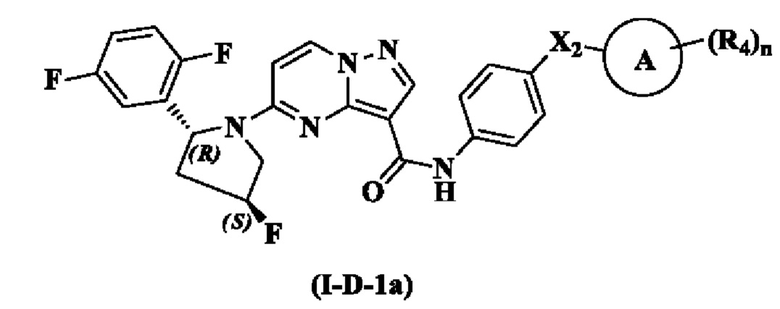

где R4, Х2, кольцо А и n являются такими, как описано для соединения формулы (I) настоящего изобретения.
В одном варианте реализации соединение имеет структуру формулы (I-С-1а-1):
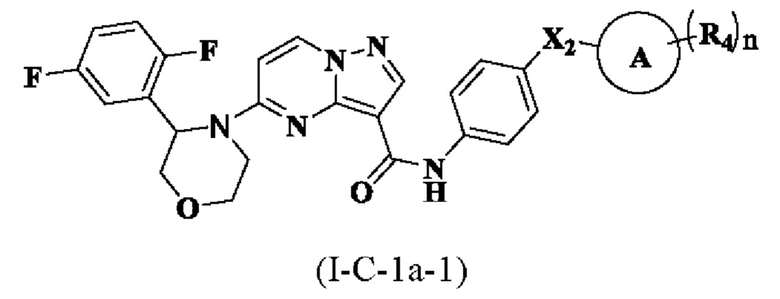
где R4, Х2, кольцо А и n являются такими, как описано для соединения формулы (I) настоящего изобретения.
В одном из вариантов реализации соединение имеет структуру формулы (I-С-1а-2):

где R4, Х2, кольцо А и n являются такими, как описано для соединения формулы (I) настоящего изобретения.
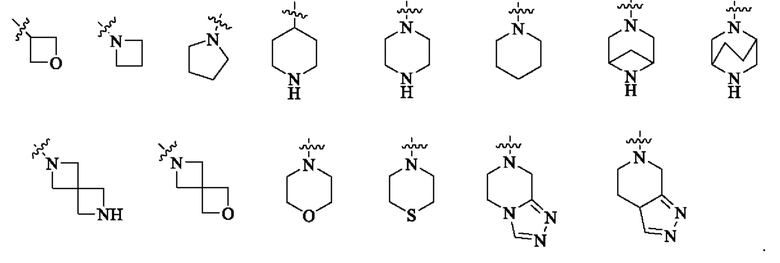
В одном варианте реализации настоящего изобретения предложено соединение (соединение формулы (I), соединение формулы (I-1), соединение формулы (I-2), соединение формулы (I-A), соединение формулы (I-А-1а), соединение формулы (I-A-1b), соединение формулы (I-А-1 с), соединение формулы (I-B), соединение формулы (I-В-1а), соединение формулы (I-B-1b), соединение формулы (I-В-1 с), соединение формулы (I-C), соединение формулы (I-С-1а), соединение формулы (I-C-1b), соединение формулы (I-C-1 с), соединение формулы (I-D), соединение формулы (I-D-a), соединение формулы (I-D-b), соединение формулы (I-D-c), соединение формулы (I-D-la), соединение формулы (I-D-1b), соединение формулы (I-D-1c), соединение формулы (I-С-1а-1), соединение формулы (I-С-1а-2), то же самое ниже) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль, где кольцо А выбрано из: С3-6 циклоалкила, 4-6-членного гетероциклоалкила, C6-8 мостикового циклила, 6-8-членного гетеромостикового циклила, С8-10 конденсированного циклила, 8-10-членного гетероконденсированного циклила, С7-12 моноспироциклила и 7-12-членного гетеромоноспироциклила; или кольцо А выбрано из: 4-6-членного гетероциклоалкила, 6-8-членного гетеромостикового циклила, 8-10-членного гетероконденсированного циклила и 7-12-членного гетеромоноспироциклила; где гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетеромоноспироциклиле независимо выбраны из О, S и N, и количество гетероатомов выбрано из 1, 2, 3 и 4; или кольцо А выбрано из следующих структур:
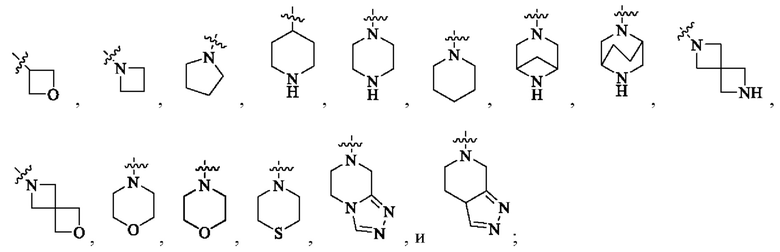
или кольцо А выбрано из следующих структур:
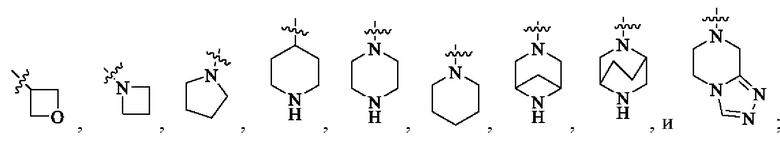
или кольцо А выбрано из следующих структур:
или кольцо А выбрано из следующих структур:

или кольцо А выбрано из следующих структур:

В одном варианте реализации настоящего изобретения предложено соединение (соединение формулы (I), соединение формулы (I-1), соединение формулы (I-2), соединение формулы (I-A), соединение формулы (I-А-1а), соединение формулы (I-A-1b), соединение формулы (I-А-1 с), соединение формулы (I-B), соединение формулы (I-В-1а), соединение формулы (I-B-1b), соединение формулы (I-В-1 с), соединение формулы (I-C), соединение формулы (I-С-1а), соединение формулы (I-C-1b), соединение формулы (I-C-1 с), соединение формулы (I-D), соединение формулы (I-D-a), соединение формулы (I-D-b), соединение формулы (I-D-c), соединение формулы (I-D-1a), соединение формулы (I-D-1b), соединение формулы (I-D-1c), соединение формулы (I-С-1а-1) и соединение формулы (I-С-1а-2)) или его таутомер, стереоизомер, оптический изомер, сольват, изотопное производное, N-оксид, пролекарство или фармацевтически приемлемая соль, где R4 независимо выбран из: -Н, -ОН, галогена, -CN, оксо, замещенного или незамещенного С1-6 алкила, -СООН, -CONH2, -CO-CR4aR4b-OH и -CO-R4b,; или R4 независимо выбран из: -Н, -ОН, галогена, -CN, оксо, замещенного или незамещенного C1-6 алкила, -CO-CR4aR4b-OH и -CO-R4b; или, R4 независимо выбран из: -Н, -ОН, галогена, замещенного или незамещенного С1-6 алкила, -CO-CR4aR4b-OH и -CO-R4b; или, R4 независимо выбран из: -Н, -ОН, галогена, замещенного или незамещенного С1-3 алкила, -CO-CR4aR4b-OH и -CO-R4b; или, R4 независимо выбран из: -Н, -ОН, замещенного или незамещенного С1-3 алкила, -CO-CR4aR4b-OH и -CO-R4b; или, R4 независимо выбран из: -CO-CR4aR4b-OH и -CO-R4b; где оксо означает, что два атома Н одного и того же центра замещения замещены одним и тем же атомом О с образованием двойной связи; R4a выбран из водорода и незамещенного или замещенного С1-4 алкила; R4b выбран из незамещенного или замещенного С1-6 алкила и незамещенного или замещенного С3-6 циклоалкила, заместитель для замещения независимо выбран из -ОН, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3; или заместитель для замещения независимо выбран из -ОН и F, и количество заместителей выбрано из 1, 2 и 3.
В одном из вариантов реализации в настоящем описании предложено соединение или (и) его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, причем указанное соединение имеет структуру формулы (I-А-1а):

где X выбран из: химической связи, -О-, -S-, -NH- и -СН2-;
X1 выбран из: -СН- и N;
Х2 выбран из: химической связи, -(СН2)р- и -NH-, где р равен 1, 2, 3 или 4;
R1 выбран из: Н, галогена, -ОН, амино, C1-6 алкила и C1-6 алкокси;
R2 выбран из: Н, F, ОН и замещенного или незамещенного C1-6 алкила, где «замещенный» означает замещенный 1, 2 или 3 заместителями, выбранными из галогена и гидроксила;
R3 выбран из: Н, галогена, -ОН, амино, C1-6 алкила и C1-6 алкокси; кольцо А выбрано из: циклоалкила, гетероциклоалкила, мостикового циклила, гетеромостикового циклила, конденсированного циклила, гетерокоденсированного циклила, спироциклила и гетероспироциклила, причем гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетероспироциклиле независимо выбраны из О, S и N, и количество гетероатомов выбрано из 1, 2, 3 и 4;
R4 находится в любом замещаемом положении на кольце А и независимо выбран из: -Н, -ОН, галогена, -CN, оксо, замещенного или незамещенного С1-6 алкила, -(СН2)m-ОН, -(СН2)m-СООН, -(CH2)m-CO-NH2, -CO-(CH2)m-NH2, -CO-CR4aR4b-OH и -CO-R4b; где оксо означает, что два атома Н одного и того же центра замещения замещены одним и тем же атомом О с образованием двойной связи; m выбран из 1, 2, 3 и 4; R4a выбран из водорода и незамещенного или замещенного С1-4 алкила; R4b выбран из Н, незамещенного или замещенного C1-6 алкила и незамещенного или замещенного С3-6 циклоалкила, заместитель для замещения независимо выбран из -ОН, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3; n выбран из 1, 2, 3 и 4.
В одном из вариантов реализации в настоящем описании предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом указанное соединение имеет структуру, представленную формулой (I-В-1а):
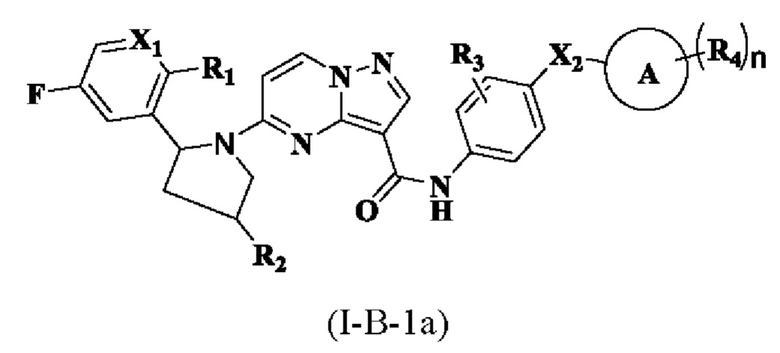
где R1, R2, R3, R4, X1, X2, кольцо А и n являются такими, как описано для соединения формулы (I-А-1а) настоящего изобретения.
В одном из вариантов реализации в настоящем описании предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом указанное соединение имеет структуру, представленную формулой (I-D-1a):
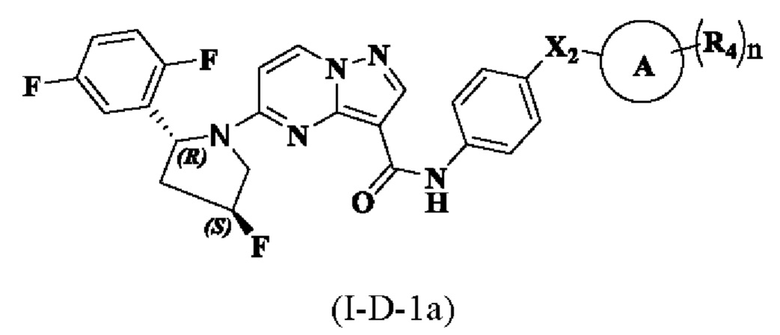
где R1, R3, R4, X1, Х2, кольцо А и n являются такими, как описано для соединения формулы (I-А-1а) настоящего изобретения.
В одном из вариантов реализации в настоящем описании предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом указанное соединение имеет структуру, представленную формулой (I-С-1а):
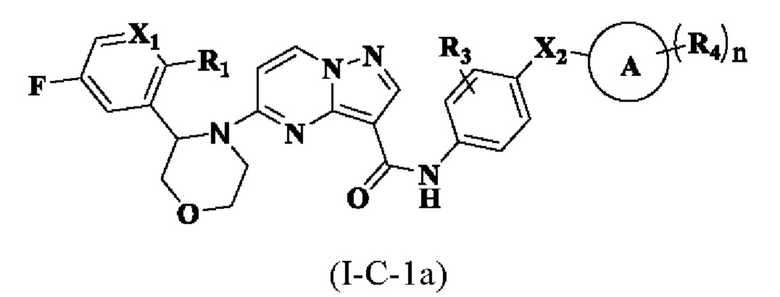
где R1, R3, R4, X1, Х2, кольцо А и n являются такими, как описано для соединения формулы (I-А-1а) настоящего изобретения.
В одном из вариантов реализации в настоящем описании предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом указанное соединение имеет структуру, представленную формулой (I-С-1а-1):
где R4, Х2, кольцо А и n являются такими, как описано для соединения формулы (I-А-1а) настоящего изобретения.
В одном из вариантов реализации в настоящем описании предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом указанное соединение имеет структуру, представленную формулой (I-С-1а-2):

где R4, Х2, кольцо А и n являются такими, как описано для соединения формулы (I-А-1а) настоящего изобретения.
В одном варианте реализации настоящего изобретения предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, где кольцо А выбрано из: С3-6 циклоалкила, 4-6-членного гетероциклоалкила, С6-8 мостикового циклила, 6-8-членного гетеромостикового циклила, С6-10 конденсированного циклила, 8-10-членного гетероконденсированного циклила, С7-12 моноспироциклила и 7-12-членного гетеромоноспироциклила, где гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетеромоноспироциклиле независимо выбраны из О, S и N, и количество гетероатомов выбрано из 1, 2 и 3.
В одном из вариантов реализации в настоящем изобретении предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом кольцо А выбрано из следующих структур:
В одном варианте реализации в настоящем изобретении предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, где R4 независимо выбран из: -Н, -ОН, галогена, -CN, оксо, замещенного или незамещенного С1-6 алкила, -СООН, -CONH2, -CO-CR4aR4b-OH и -СО-R4b, где оксо означает, что два атома Н одного и того же центра замещения замещены одним и тем же атомом О с образованием двойной связи; R4a выбран из водорода и незамещенного или замещенного С1-4 алкила; R4b выбран из незамещенного или замещенного С1-6 алкила и незамещенного или замещенного С3-6 циклоалкила, заместитель для замещения выбран независимо из -ОН, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3.
В одном из вариантов реализации в настоящем изобретении предложено соединение или его таутомер, оптический изомер, сольват, изотопное производное или фармацевтически приемлемая соль, при этом указанное соединение имеет следующие структуры:
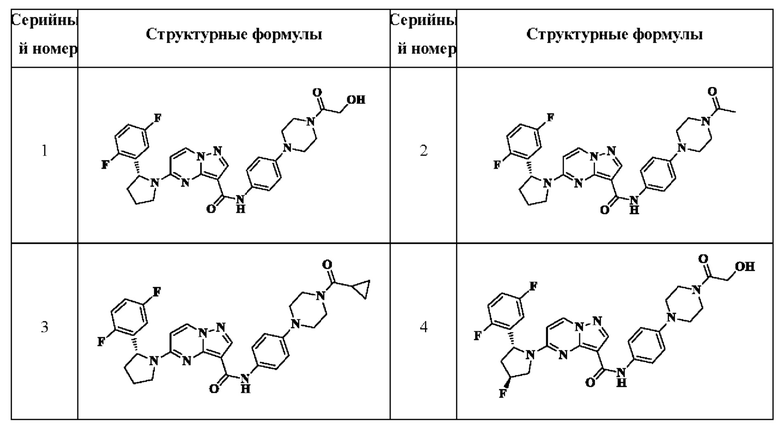





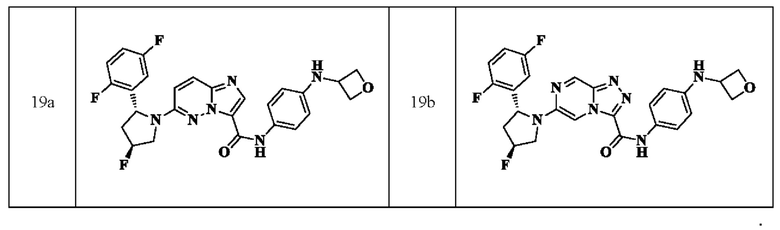
В другом аспекте в настоящем описании дополнительно предложена фармацевтическая композиция, содержащая соединение или его таутомер, стереоизомер, оптический изомер, сольват, N-оксид, пролекарство, изотопное производное или фармацевтически приемлемую соль, раскрытые в настоящем документе.
В другом аспекте в настоящем описании дополнительно предложена фармацевтическая композиция, содержащая соединение или его таутомер, стереоизомер, оптический изомер, сольват, N-оксид, пролекарство, изотопное производное или фармацевтически приемлемую соль, раскрытые в настоящем документе, и фармацевтически приемлемый вспомогательный материал.
В другом аспекте в настоящем описании дополнительно предложено применение соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, раскрытых в настоящем документе, или фармацевтической композиции, раскрытой в настоящем документе, для получения лекарственного средства для предотвращения и/или лечения заболевания, опосредованного TRK.
В одном варианте реализации заболевание выбрано из болевого заболевания, клеточного пролиферативного заболевания, воспалительного заболевания, нейродегенеративного заболевания и инфекционного заболевания.
В одном варианте реализации указанное заболевание опосредовано одним, двумя или тремя из TRKA, TRKB и TRKC.
В одном из вариантов реализации заболевание относится к дисрегуляции гена NTRK, белка TRK или его экспрессии, активности или уровня; предпочтительно, оно относится к слиянию, амплификации, перестройке, мутации или высокой экспрессии гена NTRK; и, кроме того, предпочтительно, оно относится к слиянию или мутации гена NTRK. В другом аспекте в настоящем описании дополнительно предложено соединение или его таутомер, стереоизомер, оптический изомер, сольват, N-оксид, пролекарство, изотопное производное или фармацевтически приемлемая соль, раскрытые в настоящем документе, или фармацевтическая композиция, раскрытая в настоящем документе, для применения для предотвращения и/или лечения заболевания, опосредованного TRK. В другом аспекте в настоящем описании дополнительно предложен способ предотвращения и/или лечения заболевания, опосредованного TRK, включающий: введение пациенту соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе.
В другом аспекте в настоящем описании дополнительно предложено применение соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе, в качестве лекарственного средства.
В одном из вариантов реализации лекарственное средство применяют для лечения заболевания, опосредованного TRK, ALK, ROS1 или их комбинацией. В одном из вариантов реализации лекарственное средство применяют для лечения болевого заболевания, клеточного пролиферативного заболевания, воспалительного заболевания, нейродегенеративного заболевания или инфекционного заболевания. В другом аспекте в настоящем описании дополнительно предложено применение соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе, для получения ингибитора киназы TRK, ингибитора киназы ALK или ингибитора киназы ROS1.
В одном из вариантов реализации ингибитор применяют для лечения заболевания, опосредованного TRK, ALK, ROS 1 или их комбинацией.
В одном варианте реализации ингибитор применяют для лечения болевого заболевания, клеточного пролиферативного заболевания, воспалительного заболевания, нейродегенеративного заболевания или инфекционного заболевания. В другом аспекте в настоящем описании дополнительно предложено применение соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе, для получения лекарственного средства для лечения болевого заболевания, клеточного пролиферативного заболевания, воспалительного заболевания, нейродегенеративного заболевания или инфекционного заболевания.
В одном из вариантов реализации болевое заболевание, клеточное пролиферативное заболевание, воспалительное заболевание, нейродегенеративное заболевание или инфекционное заболевание относится к дисрегуляции гена NTRK, белка TRK или его экспрессии, активности или уровня; предпочтительно, они относятся к слиянию, амплификации, перестройке, мутации или высокой экспрессии гена NTRK; и, кроме того, предпочтительно, они относятся к слиянию или мутации гена NTRK.
В одном из вариантов реализации болевое заболевание, клеточное пролиферативное заболевание, воспалительное заболевание, нейродегенеративное заболевание или инфекционное заболевание относится к дисрегуляции гена ROS1, белка ROS1 или его экспрессии, активности или уровня; предпочтительно, они относятся к слиянию, амплификации, перестройке, мутации или высокой экспрессии гена ROS1; и, кроме того, предпочтительно, они относятся к слиянию или мутации гена ROS1.
В одном из вариантов реализации болевое заболевание, клеточное пролиферативное заболевание, воспалительное заболевание, нейродегенеративное заболевание или инфекционное заболевание относятся к дисрегуляции гена, белка или экспрессии, активности или уровня TRK, ALK, ROS1 или их комбинации; предпочтительно, они относятся к слиянию, амплификации, перестройке, мутации или высокой экспрессии гена NTRK, ALK, ROS 1 или их комбинации; и дополнительно, предпочтительно, они относятся к слиянию или мутации гена NTRK, ALK, ROS 1 или их комбинации.
В одном из вариантов реализации клеточное пролиферативное заболевание представляет собой опухоль или рак.
В одном из вариантов реализации опухоль или рак представляет собой солидную опухоль и гематологическую опухоль, предпочтительно солидную опухоль. В одном варианте реализации изобретения опухоль или рак представляет собой гемобластоз, рак легкого, рак молочной железы, рак яичников, рак предстательной железы, рак поджелудочной железы, глиому головного мозга, колоректальный рак, меланому, рак головы и шеи, рак желчного пузыря, рак щитовидной железы, глиобластому, рак желудка, нейробластому или рак слюнных желез; предпочтительно рак легкого представляет собой немелкоклеточный рак легкого.
В другом аспекте в настоящем описании дополнительно предложено соединение или его таутомер, стереоизомер, оптический изомер, сольват, N-оксид, пролекарство, изотопное производное или фармацевтически приемлемая соль, описанные в настоящем документе, или фармацевтическая композиция, описанная в настоящем документе, для применения для лечения боли, клеточного пролиферативного заболевания, воспаления, нейродегенеративного заболевания или инфекционного заболевания.
В другом аспекте в настоящем описании дополнительно предложен способ лечения заболевания, включающий: введение пациенту соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе, где заболевание представляет собой боль, клеточное пролиферативное заболевание, воспаление, нейродегенеративное заболевание или инфекционное заболевание.
В одном из вариантов реализации указанное соединение или его таутомер, стереоизомер, оптический изомер, сольват, N-оксид, пролекарство, изотопное производное или фармацевтически приемлемую соль, описанные в настоящем документе, или фармацевтическую композицию, описанную в настоящем документе, применяют в комбинации с другим одним, двумя или более лекарственными средствами, обладающими эффектом лечения клеточного пролиферативного заболевания.
В другом аспекте в настоящем описании дополнительно предложено применение соединения или его таутомера, оптического изомера, стереоизомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе, для получения лекарственного средства для предотвращения и/или лечения заболевания, опосредованного TRK, ALK, ROS 1 или их комбинацией.
В одном варианте реализации заболевание выбрано из болевого заболевания, клеточного пролиферативного заболевания, воспалительного заболевания, нейродегенеративного заболевания и инфекционного заболевания.
В одном из вариантов реализации заболевание относится к дисрегуляции гена, белка или экспрессии, активности или уровня TRK, ALK, ROS 1 или их комбинации.
В одном из вариантов реализации заболевание относится к слиянию, амплификации, перестройке, мутации или высокой экспрессии гена NTRK, ALK, ROS1 или их комбинации; предпочтительно, оно относится к слиянию или мутации гена NTRK, ALK, ROS1 или их комбинации.
В другом аспекте в настоящем описании дополнительно предложен способ предотвращения и/или лечения заболевания, опосредованного TRK, ALK, ROS1 или их комбинацией, включающий: введение пациенту соединения или его таутомера, стереоизомера, оптического изомера, сольвата, N-оксида, пролекарства, изотопного производного или фармацевтически приемлемой соли, описанных в настоящем документе, или фармацевтической композиции, описанной в настоящем документе.
Определения
Если не указано иное, следующие термины, упомянутые в настоящем документе, имеют следующие определения.
Термин «алкил» относится к одновалентной насыщенной алифатической углеводородной группе, то есть, линейной или разветвленной группе, содержащей от 1 до 20 атомов углерода, предпочтительно, содержащей от 1 до 10 атомов углерода (то есть, С1-10 алкил), дополнительно, предпочтительно, содержащей от 1 до 8 атомов углерода (C1-8 алкил), и более предпочтительно, содержащей от 1 до 6 атомов углерода (то есть, C1-6 алкил), например, «C1-6 алкил» означает, что группа представляет собой алкил, и количество атомов углерода в углеродной цепи составляет от 1 до 6 (а именно, 1, 2, 3, 4, 5 или 6). Примеры алкила включают, без ограничения, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, неопентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, н-гептил, н-октил и тому подобное.
Термин «карбоциклил» или «карбоцикл» относится к одновалентной или многовалентной, насыщенной или частично ненасыщенной моноциклической, бициклической или трициклической системе, содержащей от 3 до 12 атомов углерода, где моноциклическая, бициклическая или трициклическая система не содержит ароматического кольца. Углеродный бициклил включает мостиковый циклил, спироциклил, конденсированный циклил и тому подобное. Мостиковый циклил означает, что любые два кольца имеют два общих атома, которые могут быть или не быть непосредственно связаны.
Термин «циклоалкил» относится к моноциклической насыщенной алифатической углеводородной группе, содержащей определенное количество атомов углерода, предпочтительно содержащей от 3 до 12 атомов углерода (т.е., С3-12 циклоалкил), более предпочтительно, содержащей от 3 до 10 атомов углерода (С3-10 циклоалкил), и дополнительно предпочтительно, содержащей от 3 до 6 атомов углерода (С3-6 циклоалкил), от 4 до 6 атомов углерода (С4-6 циклоалкил) или от 5 до 6 атомов углерода (С5-6 циклоалкил). Примеры циклоалкила включают, без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, метилциклопропил, 2-этил-циклопентил, диметилциклобутил и тому подобное.
Термин «алкокси» относится к -О-алкилу, где алкил определен выше, т.е. алкил содержит от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 8 атомов углерода, и дополнительно более предпочтительно от 1 до 6 атомов углерода (а именно, 1, 2, 3, 4, 5 или 6). Типичные примеры алкокси включают, без ограничения, метокси, этокси, пропокси, изопропокси, бутокси, 1-метилпропокси, 2-метилпропокси, трет-бутокси, пентилокси, 1-метилбутокси, 2-метилбутокси, 3-метилбутокси, 1,1-диметилпропокси, 1,2-диметилпропокси, 2,2-диметилпропокси, 1-этилпропокси и тому подобное. Термин «галоген» или «гало» относится к F, Cl, Br и I.
Термин «галогеналкил» означает, что один, два или более атомов водорода или все атомы водорода в алкиле, определенном выше, замещены галогеном. Типичные примеры галогеналкила включают CCl3, CF3, CHCl2, CH2Cl, CH2Br, CH2I, CH2CF3, CF2CF3 и тому подобное.
Термин «гетероциклил» относится к насыщенному или частично ненасыщенному моноциклическому, бициклическому или полициклическому углеводородному заместителю и имеет неароматическую структуру, содержащую от 3 до 20 кольцевых атомов, где 1, 2, 3 или более кольцевых атомов выбраны из N, О и S, а остальные кольцевые атомы представляют собой С. Гетероциклил предпочтительно содержит от 3 до 12 атомов в кольце (С3-12 гетероциклил), дополнительно предпочтительно от 3 до 10 атомов в кольце (С3-10 гетероциклил), или от 3 до 8 атомов в кольце (С3-8 гетероциклил), или от 3 до 6 атомов в кольце (С3-6 гетероциклил), или от 4 до 6 атомов в кольце (С4-6 гетероциклил), или от 5 до 6 атомов в кольце (С5-6 гетероциклил). Количество гетероатомов предпочтительно составляет I-4, более предпочтительно I-3 (т.е. 1, 2 или 3). Примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, дигидропирролил, пиперидинил, пиперазинил, пиранил и тому подобное. Полициклический гетероциклил включает гетероциклилы, такие как гетероспироциклил, гетероконденсированный циклил, гетеромостиковый циклил и тому подобное.
Термин «гетеро циклоалкил» относится к насыщенному «гетеро цикл илу», определенному выше, содержащему от 3 до 20 кольцевых атомов, где 1, 2, 3 или более кольцевых атомов выбраны из N, О и S, а остальные кольцевые атомы представляют собой С. Гетероциклоалкил предпочтительно содержит от 3 до 12 атомов в кольце (С3-12 гетероциклоалкил), дополнительно предпочтительно от 3 до 10 атомов в кольце (С3-10 гетероциклоалкил), или от 3 до 8 атомов в кольце (С3-8 гетероциклоалкил), или от 3 до 7 атомов в кольце (С3-7 гетероциклоалкил), или от 3 до 6 атомов в кольце (С3-6 гетероциклоалкил), или от 4 до 6 атомов в кольце (С4-6 гетероциклоалкил), или от 5 до 6 атомов в кольце (С5-6 гетероциклоалкил). Количество гетероатомов предпочтительно составляет I-4, более предпочтительно I-3 (т.е. 1, 2 или 3). Примеры гетероциклоалкила включают азациклопропил, оксациклопропил, тиоциклопропил, азациклобутил, оксациклобутил, тиоциклобутил, пирролидинил, тетрагидрофуранил, оксоциклогексан, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, диоксанил, дитиациклогексил, оксазолидинил, тиазолидинил, пиразолидинил, имидазолидинил и тому подобное.
Термин «арил» относится к моноциклическим, бициклическим и трициклическим ароматическим карбоциклическим системам, содержащим от 6 до 16 атомов углерода, или от 6 до 14 атомов углерода, или от 6 до 12 атомов углерода, или от 6 до 10 атомов углерода, предпочтительно от 6 до 10 атомов углерода, и термин «арил» используется взаимозаменяемо с термином «ароматическое кольцо». Примеры арильной группы могут включать, без ограничения, фенил, нафтил, антрил, фенантрил, пиренил или тому подобное.
Термин «гетероарил» относится к ароматической моноциклической или полициклической кольцевой системе, содержащей 5-12-членную структуру, или предпочтительно 5-10-членную структуру, или 5-8-членную структуру, и более предпочтительно 5-6-членную структуру, при этом 1, 2, 3 или более атомов в кольце представляют собой гетероатомы, а остальные атомы представляют собой углерод, гетероатомы независимо выбраны из О, N и S, и количество гетероатомов предпочтительно равно 1, 2 или 3. Примеры гетероарила включают, без ограничения, фуранил, тиенил, оксазолил, тиазолил, изоксазолил, оксадиазолил, тиадиазолил, пирролил, пиразолил, имидазолил, триазолил, тетразолил, пиридил, пиримидинил, пиразинил, пиридазинил, тиадиазолил, триазинил, фталазинил, хинолинил, изохинолинил, птеридинил, пуринил, индолил, изоиндолил, индазолил, бензофуранил, бензотиенил, бензопиридил, бензопиримидинил, бензопиразинил, бензимидазолил, бензофтализинил, пирроло[2,3-6]пиридил, имидазо[1,2-а]пиридил, пиразоло[1,5-а] пиридинил, пиразоло [1,5-а] пиримидинил, имидазо [1,2-b] пиридазинил, [1,2,4]триазоло[4,3-b]пиридазинил, [1,2,4]триазоло[1,5-а] пиримидинил, [1,2,4]триазоло[1,5-а]пиридинил и т.п.
Термин «фармацевтически приемлемая соль» относится к соли, которая в рамках здравого медицинского суждения подходит для применения в контакте с тканями млекопитающих, особенно людей, без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного и соответствует разумному соотношению польза/риск.
Термин «соль» включает соль, полученную из неорганических кислот, а также включает соль, полученную из органических кислот. Если соединение согласно настоящему изобретению является кислотным, фармацевтически приемлемые нетоксичные основные соли включают соль, полученную из неорганических и органических оснований.
Термин «стереоизомер» относится к изомерам, полученным в результате различного пространственного расположения атомов в молекуле, включая конфигурационные изомеры и конформационные изомеры, где конфигурационные изомеры включают геометрические изомеры (или цис-транс-изомеры) и оптические изомеры (включая энантиомеры и диастереоизомеры).
Геометрические изомеры могут присутствовать в соединении, раскрытом в настоящем документе. Соединение согласно настоящему изобретению может содержать углерод-углеродную двойную связь или углерод-азотную двойную связь в конфигурации Е или Z, при этом термин «Е» относится к следующему по очереди заместителю на противоположной стороне углерод-углеродной или углерод-азотной двойной связи, а термин «Z» представляет собой следующий по очереди заместитель на той же стороне углерод-углеродной или углерод-азотной двойной связи (как определено с помощью правил приоритета Cahn-mgold Prelog). Соединение согласно настоящему изобретению также может присутствовать в виде смесей изомеров «Е» и «Z». Заместители вокруг циклоалкила или гетероциклоалкила упоминаются как цис- или т/адноконфигурации. Оптические изомеры относятся к веществам, которые имеют полностью идентичные молекулярные структуры и аналогичные физико-химические свойства, но различное оптическое вращение. Соединение согласно настоящему изобретению может содержать асимметрично замещенные атомы углерода в конфигурации R или S, где термины «R» и «S» являются такими, как определено в Рекомендациях IUPAC 1974 для раздела Е, Фундаментальная стереохимия, Pure Appl. Chem. (1976) 45, 13-10. Соединения с асимметрично замещенными атомами углерода (с равным количеством конфигураций R и S) являются рацемическими по этим атомам углерода. Наличие избытка атомов с одной конфигурацией (относительно другой), обеспечивает возможность присутствия конфигурации в более высоких количествах, предпочтительно в избытке от примерно 85% до 90%, более предпочтительно в избытке от примерно 95% до 99% и еще более предпочтительно в избытке более примерно 99%. Соответственно, настоящее изобретение включает рацемические смеси, относительные и абсолютные оптические изомеры и смеси относительных и абсолютных оптических изомеров.
Термин «N-оксид» относится к N-оксиду, образованному окислением одного или более атомов азота, когда соединение содержит несколько аминофункциональных групп. Конкретные примеры N-оксидов представляют собой N-оксиды третичных аминов или N-оксиды атома азота азотсодержащего гетероцикла.
Термин «сольват» относится к ассоциированному соединению, образованному одной или более молекулами растворителя, связывающимися с соединением согласно настоящему изобретению.
Термин «таутомер» относится к структурным изомерам, имеющим различные энергии, которые являются взаимопревращаемыми посредством более низкоэнергетического барьера. Если таутомер возможен (например, в растворе), то химическое равновесие таутомера может быть достигнуто. Например, протонный таутомер (также известный как прототропный таутомер) включает взаимную конверсию посредством миграции протонов, такую как кето-енольная изомеризация и имин-енаминовая изомеризация. Валентный таутомер включает взаимопревращение путем рекомбинации некоторых связывающих электронов. Если не указано иное, все таутомерные формы соединений, описанных в настоящем документе, входят в объем настоящего изобретения.
Термин «изотопное производное» означает, что соединение согласно настоящему изобретению может присутствовать в изотопно-меченой или обогащенной форме, содержащей один или более атомов, атомная масса или массовое число которых отличается от атомного числа наибольшего количества атомов, обнаруженных в природе. Изотоп может представлять собой радиоактивный или нерадиоактивный изотоп. Изотопы атомов, таких как водород, углерод, фосфор, сера, фтор, хлор и иод, включают, без ограничения: 2Н, 3Н, 13С, 14С, 15N, l8O, 32Р, 35S, 18F, 36Cl и 125I. Соединение, содержащее другие изотопы этих и/или других атомов, входит в объем настоящего изобретения.
В другом варианте реализации соединения, меченные изотопами, содержат изотопы дейтерия (2Н), трития (3Н) или 14С. Изотопно-меченые соединения согласно настоящему изобретению могут быть получены с помощью общепринятых способов, хорошо известных специалистам в данной области техники. В этой связи, соответствующие ссылки включают: Lizondo, J et al., Drugs Fut, 21(11), 1116 (1996); Brickner, S J et al., J Med Chem, 39(3), 673 (1996); Mallesham, В et al., Org Lett, 5(7), 963 (2003).
Изотоп-содержащие соединения применяли в фармацевтических исследованиях для изучения метаболической судьбы соединений in vivo путем оценки механизма действия и метаболического пути не изотопно меченого исходного соединения (Blake et al., J. Pharm. Sci. 64, 3, 367-391 (1975)). Такие метаболические исследования важны для разработки безопасных и эффективных терапевтических препаратов либо потому, что активное соединение, вводимое пациенту in vivo, либо потому, что метаболиты, полученные из исходного соединения, оказываются токсичными или канцерогенными (Kushner et al., Can. J. Physiol. Pharmacol, 77, 79-88(1999); Foster et al., Advances in Drug Research Vol.14, pp.2-36, Academic press, London, 1985; Kato et al., J. Labelled Comp. Radiopharmaceut, 36(10):927-932 (1995); Kushner et al., Can. J. Physiol. Pharmacol, 11, 19-88 (1999)).
Кроме того, лекарственные средства, содержащие нерадиоактивный активный изотоп, такие как дейтерированные лекарственные средства, называемые «тяжелыми лекарственными средствами», можно применять для лечения связанных заболеваний и состояний. Увеличение количества изотопа, присутствующего в указанном выше соединении, сверх его естественного содержания называется обогащением. Примеры количества обогащения включают от примерно 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 21, 25, 29, 33, 37, 42, 46, 50, 54, 58, 63, 67, 71, 75, 79, 84, 88, 92, 96 до примерно 100% мольн. Любой возможный центр в молекулярной структуре может быть замещен изотопом для получения изотопного производного. Например, любой возможный центр в молекуле может быть замещен дейтерием (2Н) с получением производного в дейтерированной форме.
При применении стабильных изотопно-меченых лекарственных средств физико-химические свойства лекарственных средств, такие как рКа и растворимость в липидах, могут быть изменены. Если изотопное замещение влияет на области, участвующие во взаимодействиях лиганд-рецептор, то эти эффекты и изменения могут влиять на фармакодинамический ответ молекул лекарственного средства. Хотя некоторые физические свойства стабильных изотопно-меченых молекул отличаются от свойств изотопно-немеченых молекул, химические и биологические свойства одинаковы, важным отличием является то, что любая связь между тяжелым изотопом и другим атомом сильнее, чем та же связь между легким изотопом и этим атомом из-за повышенной массы тяжелого изотопа. Соответственно, включение изотопов в центре метаболического или ферментативного превращения может потенциально замедлить реакцию и может изменить фармакокинетические свойства или эффекты по сравнению с неизотопными соединениями.
Термин «пролекарство» представляет собой производное активного лекарственного средства, которое предназначено для улучшения некоторых определенных нежелательных физических или биологических свойств. Физические свойства часто связаны с растворимостью (слишком высокая или недостаточная растворимость в липидах или воде) или стабильностью, в то время как проблемные биологические свойства включают слишком быстрый метаболизм или плохую биодоступность, которая сама по себе может быть связана с физико-химическими свойствами.
Пролекарства, как правило, получают следующим образом: а) образование сложных эфиров, полуэфиров, карбонатов, нитратов, амидов, гидроксамовых кислот, карбаматов, иминов, оснований Манниха, фосфатов, фосфатных эфиров и енаминов активных лекарственных средств, b) функционализация лекарственных средств с помощью азо, гликозида, пептида и простых эфирных функциональных групп, с) применение аминальной, полуаминальной, полимерной, солевой, комплексной, фосфорамидной, ацетальной, полуацетальной и кетальной форм лекарственных средств. См., например, Andrejus Korolkovas, Essentials of Medicinal Chemistry, John Wiley-Interscience Pulications, John Wiley and Sons, New York (1988), pp.97-118, содержание которого полностью включено в настоящую заявку посредством ссылки. Сложные эфиры могут быть получены из субстратов, содержащих гидроксил или карбоксил, при помощи обычных способов, известных специалистам в данной области техники. Типичной реакцией этих соединений является замена одного гетероатома другим атомом. Амиды могут быть получены аналогичным образом из субстратов, содержащих амино или карбоксил. Сложные эфиры также могут вступать в реакцию с аминами или аммиаком с образованием амидов. /Другой способ получения амида заключается в нагревании карбоновой кислоты и амина вместе.
Термин «фармацевтически приемлемый вспомогательный материал» или «фармацевтически приемлемый носитель» включает, без ограничения, любой адъювант, носитель, вспомогательное вещество, глидант, подсластитель, разбавитель, консервант, краситель, красящее вещество, усилитель вкуса, поверхностно-активное вещество, увлажняющий агент, диспергатор, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, который одобрен Управлением по контролю за продуктами и лекарствами США, Национальным управлением по медицинским изделиям и тому подобное для приемлемого применения у людей или домашних животных.
Термин «опухоль» включает доброкачественные опухоли, злокачественные опухоли и пограничные опухоли, причем злокачественные опухоли также коллективно называют раком.
В настоящем документе термин «профилактика» относится к соединению или лекарственному средству, которое при применении для лечения заболевания или состояния (например, рака) может уменьшать частоту или задерживать появление симптомов медицинского состояния у субъекта по сравнению с субъектом, которому не вводили соединение или лекарственное средство (например, комбинированный продукт, как заявлено в настоящем документе).
Термин «лечение», используемый в настоящем документе, относится к облегчению, ослаблению или устранению симптома заболевания или состояния, облегчению основного метаболического симптома и замедлению заболевания или симптома, например, прекращению развития заболевания или состояния, облегчению заболевания или состояния, вызывающему регрессию заболевания или состояния, облегчению нарушения, вызванного заболеванием или состоянием, или прекращению симптома заболевания или состояния.
Термин «клеточное пролиферативное заболевание» в настоящей заявке относится к состоянию, при котором скорость роста популяции клеток ниже или выше, чем ожидаемая скорость для данного физиологического состояния.
В настоящем изобретении используются следующие термины:
ДХМ: дихлорметан; ДИПЭА: диизопропилэтиламин; ДМФА: N,N-диметилформамид; ЭА: этилацетат; NBS: N-бромсукцинимид; ПЭ: петролейный эфир; ДМСО: диметилсульфоксид; ТБТУ: O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат; БОП: бензотриазол-1-илокситрис-(диметиламино)-фосфония гексафторфосфат; АТФ: аденозина 5'-трифосфат; ДТТ: 1,4-дитиотреит; МТТ: 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-2Н-тетразол-3-бромид натрия.
Основываясь на знаниях медицинской химии, N-оксид, изотопное производное, стереоизомер, оптический изомер, сольват, пролекарство и тому подобное соединения согласно настоящему изобретению также могут проявлять эффект in vitro и in vivo, аналогичный эффекту соединения согласно настоящему изобретению.
Положительный эффект настоящего изобретения:
Настоящее изобретение относится к соединению с новой структурой, и тест ингибирования активности киназы in vitro показывает, что: соединение согласно настоящему изобретению проявляет превосходную ингибирующую активность в отношении различных киназ (например, TRK, ALK, ROS1) и их мутантов, в частности, в отношении TRK и их мутантных форм; тест цитостатической активности in vitro показывает, что: соединение согласно настоящему изобретению обладает более сильным ингибирующим действием в отношении различных мутантных клеток TRK и имеет IC50 менее 10 нМ, предпочтительно менее 5 нМ и более предпочтительно менее 1 нМ в отношении ингибирующей активности 6 видов клеток; результаты теста ингибирования опухоли in vivo показывают, что: по сравнению с контрольным соединением, соединение согласно настоящему изобретению обладает лучшим in vivo противоопухолевым действием, лучшей переносимостью и более высокой лекарственной способностью; исследовательский тест механизма действия in vivo показывает, что: соединение согласно настоящему изобретению может ингибировать TRK в опухолевых тканях, дополнительно эффективно ингибировать фосфорилирование PLCγ и АКТ и ингибировать рост опухолевых тканей.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой вестерн-блоттинг, демонстрирующий зависимость от времени ингибирующего действия соединения №4 согласно настоящему изобретению на фосфорилирование нижерасположенных целевых белков в опухолевых тканях мышей с мутантной устойчивостью к опухолям TRKA-G595R.
ПОДРОБНОЕ ОПИСАНИЕ
Исходные материалы, реакционные реагенты, катализаторы или растворители, используемые в следующих конкретных вариантах реализации изобретения, являются коммерчески доступными или могут быть получены обычными способами, известными в данной области техники.
Настоящее изобретение будет дополнительно проиллюстрировано со ссылкой на следующие конкретные примеры. Следует понимать, что эти примеры предназначены только для иллюстрации настоящего изобретения, а не для ограничения объема настоящего изобретения. Процедуры испытаний без определенных условий в следующих примерах, как правило, проводятся в соответствии с обычными условиями или в соответствии с условиями, рекомендованными изготовителем. Если не приведено других определений, все технические и научные термины, используемые в настоящем документе, имеют значение, которое обычно понимается специалистами в данной области техники. Кроме того, в способах согласно настоящему изобретению можно применять любые способы и материалы, аналогичные или эквивалентные описанным в настоящем документе. Предпочтительные варианты реализации и материалы, описанные в настоящем документе, предназначены исключительно для иллюстративных целей.
Получение Примера А: Получение этил-6-хлоримидазо[1,2-А]пиридазин-3-карбоксилата
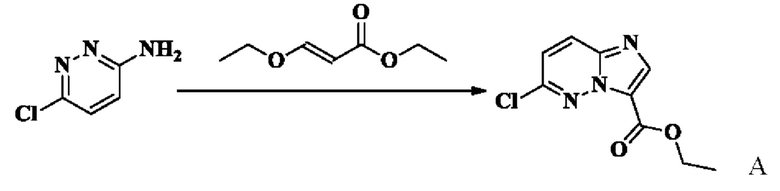
Раствор этил-(E)-3-этоксиакрилата (5,00 г, 34,7 ммоль, 1 экв.) в 1,4-диоксане (50 мл) и воде (50 мл) охлаждали до -10°С, порциями добавляли NBS (6,80 г, 38,15 ммоль, 1,1 экв.) и нагревали до комнатной температуры, и оставляли реагировать в течение 2 часов. В реакционную систему добавляли 6-хлор-3-аминопиридазин (4,5 г, 34,7 ммоль, 1 экв.), нагревали до 80°С и оставляли реагировать в течение 1,5 ч, и охлаждали до комнатной температуры. Затем реакционную систему концентрировали при пониженном давлении с получением остатка. К остатку добавляли воду (100 мл) и ЭА (100 мл), перемешивали и разделяли. Водную фазу экстрагировали этилацетатом (20 мл × 2). Органические фазы объединяли, промывали Н2О (50 мл) и насыщенным раствором соли (50 мл), концентрировали при пониженном давлении для удаления органического растворителя. Остаток разделяли с помощью колоночной хроматографии на силикагеле (элюент: н-гексан:ЭА = от 5:1 до 1:1, об./об.), в результате чего получали этил-6-хлоримидазо[1,2-b]пиридазин-3-карбоксилат (5,2 г, выход 59,75%). (ЭР, m/z): 225,91 [М+Н]+.
Пример получения промежуточного соединения 1: (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновая кислота (промежуточное соединение А1)
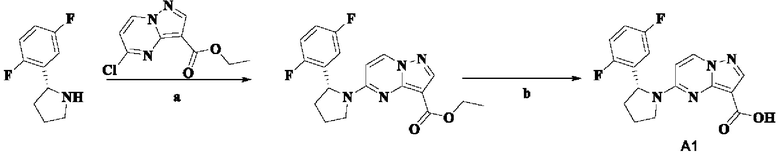
Стадия а: смешанный раствор (R)-2-(2,5-дифторфенил)-пирролидина (5,0 г, 27,292 ммоль), этил-5-хлорпиразоло[1,5-а]пиримидин-3-карбоксилата (6,14 г, 27,292 ммоль), н-бутанола (70 мл) и диизопропиламина (6,9 г, 68,230 ммоль) кипятили с обратным холодильником и перемешивали при 100°С в течение 4 ч, концентрировали при пониженном давлении с получением оранжевого вязкого твердого вещества. К смеси добавляли безводный эфир и перемешивали для осаждения большого количества твердого вещества. Проводили фильтрование с отсасыванием с получением неочищенного этил-(R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло [1,5-а]пиримидин-3-карбоксилата (6,224 г). Полученный неочищенный продукт непосредственно применяли на следующей стадии без очистки. (ЭР, m/z): 373,02 [М+Н]+.
Стадия b: неочищенный этил-(R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксилат (6,224 г, 16,714 ммоль) растворяли в абсолютном этаноле (40 мл) и перемешивали при 75°С до тех пор, пока смесь не становилась прозрачной. Затем в реакционную систему добавляли водный раствор (40 мл) LiOH (2,805 г, 66,856 ммоль) и перемешивали при 75°С в течение 3 ч. После охлаждения до комнатной температуры реакционную систему концентрировали при пониженном давлении для удаления абсолютного этанола. В реакционную систему медленно по каплям добавляли 1 Н водный раствор HCl для доведения рН до 3-4. В осадок выпадало большое количество твердого вещества белого цвета. Реакционную систему перемешивали при комнатной температуре в течение 30 мин, а затем проводили фильтрование с отсасыванием. Остаток на фильтре промывали небольшим количеством очищенной воды. Отфильтрованный осадок собирали, сушили на воздухе и взвешивали с получением (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (5,64 г, 98%), (ЭР, m/z): 345,02 [М+Н]+.
Пример получения промежуточного соединения 2: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]-пиримидин-3-карбоновая кислота (промежуточное соединение А2)
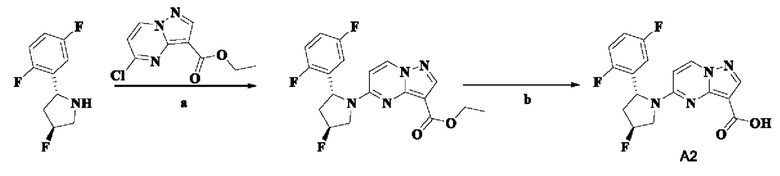
Стадия а: смешанный раствор (2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидина (5,826 г, 28,958 ммоль), этил-5-хлорпиразоло[1,5-а]пиримидин-3-карбоксилата (6,534 г, 28,958 ммоль), н-бутанола (50 мл) и диизопропиламина (8,790 г, 86,874 ммоль) оставляли реагировать при 100°С в течение 4 ч. Реакционную систему концентрировали при пониженном давлении с получением неочищенного этил-5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксилата.
Неочищенный продукт непосредственно применяли на следующей стадии без очистки. (ЭР, m/z): 391,05 [М+Н]+.
Стадия b: неочищенный этил-5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксилат растворяли в абсолютном этаноле (50 мл). Реакционную систему перемешивали при 75°С до тех пор, пока система не становилась прозрачной. Затем в реакционную систему добавляли водный раствор (50 мл) LiOH (4,86 г, 115,832 ммоль) и перемешивали при 75°С в течение 5 часов. После охлаждения до комнатной температуры реакционную систему концентрировали при пониженном давлении для удаления абсолютного этанола. В реакционную систему медленно по каплям добавляли 1 Н водный раствор HCl для доведения рН до 3-4. В осадок выпадало большое количество твердого вещества белого цвета. Реакционную систему перемешивали при комнатной температуре в течение 30 мин, а затем проводили фильтрование с отсасыванием. Остаток на фильтре промывали небольшим количеством очищенной воды. Отфильтрованный осадок собирали, сушили и взвешивали с получением белого порошкообразного твердого вещества 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (9,9 г). Фильтрат экстрагировали этилацетатом (ЭА) (2 ×50 мл). Органические фазы объединяли, промывали водой (2 ×50 мл) и насыщенным водным раствором NaCl (50 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученную смесь очищали с помощью колоночной хроматографии (ПЭ:ЭА = от 4:1 до 2:1, об./об.) с получением белого порошкообразного твердого вещества 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (386 мг). В совокупности получали чистый продукт 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновую кислоту (10,286 г, 98%). (ЭР, m/z): 363,04 [М+Н]+.
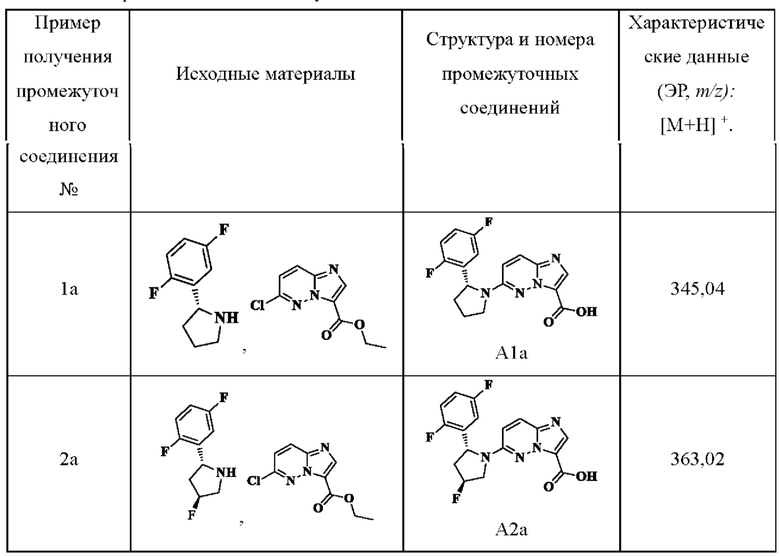
Примеры получения промежуточных соединений 1а-2а:
Ссылаясь на стадии процесса примеров получения промежуточных соединений 1 и 2, промежуточные соединения А1а-А2а получали путем применения следующих соответствующих исходных материалов и способов получения:
Пример получения промежуточного соединения 3: 5-(3-(2,5-дифторфенил)-морфолинил)-пиразоло[1,5-а]пиримидин-3-карбоновая кислота (промежуточное соединение A3)

Стадия а: в атмосфере азота по каплям добавляли триметилсилилцианид (39,66 г, 0,4 моль) в раствор (30 мл) 2,5-дифторбензальдегида (28,4 г, 0,2 моль) в 7 моль/л NH3/CH3OH при 0°С. Реакционную систему перемешивали при комнатной температуре в течение ночи после завершения добавления по каплям. Реакционную жидкость концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии (CH2Cl2:СН3ОН=50:1, об./об.), в результате чего получали 2-амино-2-(2,5-дифторфенил)-ацетонитрил (21,92 г, 65,2%). (ЭР, m/z): 169[М+Н]+.
Стадия b: 2-амино-2-(2,5-дифторфенил)-ацетонитрил (21,92 г, 130 ммоль) добавляли к 2 моль/л водному раствору NaOH (130 мл). Реакционную систему кипятили с обратным холодильником в течение 6 часов и затем охлаждали до 0°С. рН смеси доводили до 3 концентрированной соляной кислотой и затем концентрировали при пониженном давлении. Остаток растворяли в тетрагидрофуране (200 мл), перемешивали в течение 30 мин и фильтровали. Фильтрат сушили над безводным сульфатом натрия в течение ночи, фильтровали и концентрировали при пониженном давлении, в результате чего получали неочищенную 2-амино-2-(2,5-дифторфенил)-уксусную кислоту (20,78 г), которую непосредственно использовали на следующей стадии без очистки. (ЭР, m/z): 186,02[М-Н]-.
Стадия с: в атмосфере азота раствор 2-амино-2-(2,5-дифторфенил)-уксусной кислоты (20,78 г, 111 ммоль) в тетрагидрофуране (600 мл) охлаждали до -10°С - -5°С, добавляли порциями алюмогидрид лития (10,53 г, 278 ммоль) и оставляли реагировать при комнатной температуре в течение ночи после завершения добавления. В реакционную жидкость добавляли насыщенный раствор хлорида аммония (500 мл) для гашения реакции и фильтровали через слой целита. Фильтрат отделяли. Водную фазу экстрагировали этилацетатом (2 ×100 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали; и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (CH2Cl2:СН3ОН=10:1, об./об.), в результате чего получали 2-амино-2-(2,5-дифторфенил)-этанол (7,177 г, 32% двухстадийный выход). (ЭР, m/z): 174,02[М+Н]+.
Стадия d: в атмосфере азота хлорацетилхлорид (5,62 г, 49,736 ммоль) добавляли к раствору 2-амино-2-(2,5-дифторфенил)-этанола (7,177 г, 41,447 ммоль) и триэтиламина (8,388 г, 82,894 ммоль) в тетрагидрофуране (200 мл) при 0°С. Поддерживая указанную температуру, перемешивали реакционную систему в течение 30 мин. В реакционную систему порциями добавляли 60% NaH (4,974 г, 124,341 ммоль). Реакционную смесь оставляли реагировать при комнатной температуре в течение 2 ч после завершения добавления, и гасили насыщенным раствором хлорида аммония (100 мл). Водную фазу экстрагировали этилацетатом (2 × 50 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (CH2Cl2:СН3ОН = от 80:1 до 20:1, об./об.), в результате чего получали 5-(2,5-дифторфенил)-морфолин-3-он (4,532 г, 51,3%). (ЭР, m/z): 214,01 [М+Н]+.
Стадия е: в атмосфере азота алюмогидрид лития (3,229 г, 85,849 ммоль) порциями добавляли в раствор 5-(2,5-дифторфенил)-морфолин-3-она (4,532 г, 21,271 ммоль) в тетрагидрофуране (100 мл) при 0°С. Реакционную систему оставляли реагировать в течение 2 ч при 50°С после завершения добавления, а затем охлаждали до 0°С. Смесь гасили насыщенным хлоридом аммония (90 мл) и концентрировали при пониженном давлении. Водную фазу экстрагировали этилацетатом (ЭА) (3 × 50 мл). Органические фазы объединяли, промывали водой (50 мл) и раствором соли (50 мл), затем сушили над безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (CH2Cl2:СН3ОН=от 50:1 до 25:1, об./об.), в результате чего получали 3-(2,5-дифторфенил)-морфолин (3,40 г, 80,3%). (ЭР, m/z): 200,03[М+Н]+.
Стадия f: смешанный раствор 3-(2,5-дифторфенил)-морфолина (3,40 г, 17,068 ммоль), этил 5-хлорпиразоло[1,5-а]пиримидин-3-карбоксилата (3,851 г, 17,068 ммоль), н-бутанола (50 мл) и диизопропиламина (5,181 г, 51,204 ммоль) оставляли реагировать при 100°С в течение ночи. Реакционную систему концентрировали при пониженном давлении. К остатку добавляли ЭА (100 мл) и воду (100 мл), и смесь перемешивали. Водную фазу экстрагировали этилацетатом (2 × 50 мл). Органические фазы объединяли, промывали водой (100 мл) и раствором соли (50 мл), затем сушили над безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (п-гексан: этил ацетат = от 50:1 до 10:1, об./об.), в результате чего получали этил-5-(3-(2,5-дифторфенил)-морфолинил)-пиразоло[1,5-a]пиримидин-3-карбоксилат (6,066 г, 91,5%). (ЭР, m/z): 389,05[М+Н]+.
Стадия g: к раствору этил-5-(3-(2,5-дифторфенил)-морфолинил)-пиразоло[1,5-а]пиримидин-3-карбоксилата (6,066 г, 15,623 ммоль) в этаноле (60 мл) добавляли водный раствор (60 мл) LiOH (2,622 г, 62,492 ммоль), и смесь нагревали до 75°С для протекания реакции в течение ночи. Реакционную систему охлаждали до комнатной температуры и концентрировали при пониженном давлении. Водную фазу доводили до рН=2-3 1 Н водным раствором соляной кислоты и экстрагировали этилацетатом (ЭА) (3 × 60 мл). Органические фазы объединяли, промывали водой (100 мл) и раствором соли (50 мл), затем сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (ПЭ:ЭА = от 4:1 до 1:1, об./об.) с получением 5-(3-(2,5-дифторфенил)-морфолинил)-пиразоло[1,5-a]пиримидин-3-карбоновой кислоты (5,324 г, 94,6%). (ЭР, m/z): 361,09[М+Н]+.
Пример получения промежуточного соединения 4: 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолино)-пиразоло[1,5-а]пиримидин-3-карбоновая кислота (промежуточное соединение А4)
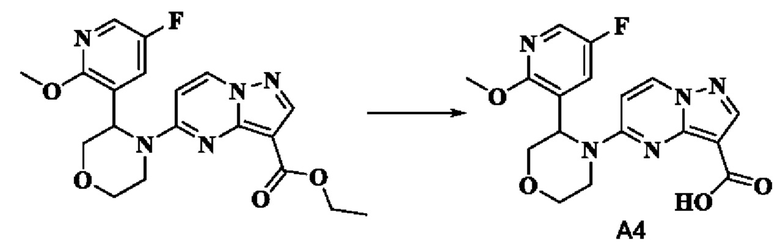
В соответствии со стадиями a-f получения промежуточного продукта, пример 3, этил-5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолинил)-пиразоло[1,5-а]пиримидин-3-карбоксилат получали из 5-фтор-2-метокси-3-пиридинкарбальдегида.
К раствору этил-5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолинил)-пиразоло[1,5-а]пиримидин-3-карбоксилата (5,00 г, 12,46 ммоль) в абсолютном этаноле (50 мл) добавляли водный раствор (50 мл) LiOH (2,091 г, 49,827 ммоль) и оставляли смесь реагировать при 75°С в течение ночи. Реакционную систему охлаждали до комнатной температуры и концентрировали при пониженном давлении. Водную фазу доводили до рН=2-3 водным раствором соляной кислоты 1 Н и экстрагировали этилацетатом (ЭА) (3 × 50 мл). Органические фазы объединяли, промывали водой (50 мл) и раствором соли (50 мл), затем сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (ПЭ:ЭА = от 4:1 до 1:1, об./об.), в результате чего получали 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолино)-пиразоло[1,5-а]пиримидин-3-карбоновую кислоту (4,277 г, 92%). (ЭР, m/z): 374,02[М+Н]+.
Пример получения промежуточного соединения 5: (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (промежуточное соединение А5)

Стадия а: 1-Вос-4-(4-аминофенил)-пиперазин (850 мг, 3,067 ммоль) добавляли к раствору (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло[1,5-a]пиримидин-3-карбоновой кислоты (880 мг, 2,556 ммоль) и ТБТУ (985 мг, 3,067 ммоль) в безводном ДМФА (10 мл) с последующим добавлением по каплям ДИПЭА (991 мг, 7,668 ммоль) при 0°С. Реакционную систему оставляли реагировать при комнатной температуре в течение ночи. К реакционной жидкости добавляли воду (50 мл) и перемешивали. В осадок выпадало твердое вещество. Смесь фильтровали при пониженном давлении с получением осадка на фильтре, который сушили в вакуумной печи, в результате чего получали (R)-трет-бутил-4-(4-(5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенил)-пиперазин-1-карбоксилат (1,450 г, 94%). (ЭР, m/z): 604,52[М+Н]+.
Стадия b: к (R)-трет-бутил-4-(4-(5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенил)-пиперазин-1-карбоксилату (1,45 г, 2,402 ммоль) добавляли ДХМ и CF3COOH (12 мл, 3/1, об./об.), и реакционную систему перемешивали при комнатной температуре в течение 2,5 ч и концентрировали при пониженном давлении. К остатку добавляли воду (12 мл) и ЭА (6 мл) с последующим добавлением водного аммиака для доведения уровня рН до 9. Твердое вещество осаждали при перемешивании. Смесь фильтровали при пониженном давлении с получением осадка на фильтре, который промывали небольшим количеством воды, сушили и взвешивали с получением (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (999 мг). (ЭР, m/z): 504,13 [М+Н]+.
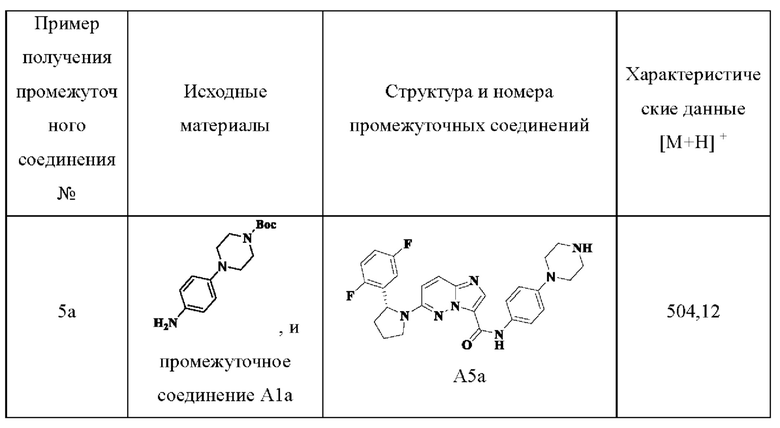
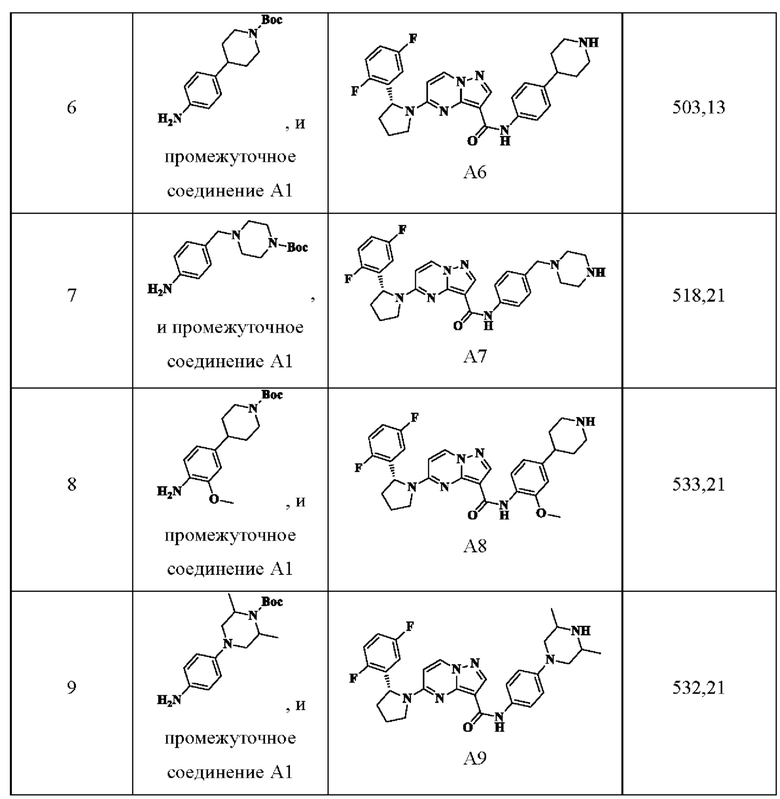
Примеры получения промежуточных соединений 5а и 6-9:
Ссылаясь на стадии процесса в примере получения промежуточного соединения 5, промежуточные соединения А5а и А6-А9 получали путем принятия следующих соответствующих исходных материалов и способов получения:
Пример получения промежуточного соединения 10: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (промежуточное соединение А10)
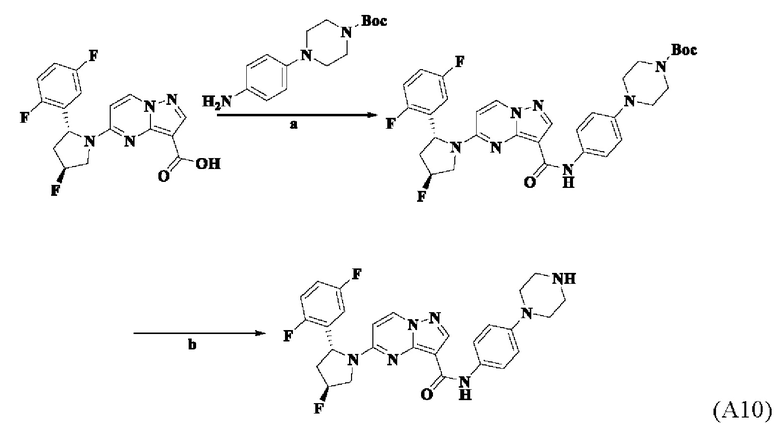
Стадия а: 1-Вос-4-(4-аминофенил)-пиперазин (918 мг, 3,312 ммоль) добавляли к раствору 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (промежуточное соединение А2, 1000 мг, 2,76 ммоль) и ТБТУ (1063 мг, 3,312 ммоль) в безводном ДМ ФА (10 мл) с последующим добавлением по каплям ДИПЭА (1284 мг, 9,936 ммоль) при 0°С; и реакционную систему оставляли реагировать при комнатной температуре в течение ночи. К реакционной жидкости добавляли воду (50 мл) и перемешивали. В осадок выпадало твердое вещество. Смесь фильтровали при пониженном давлении с получением осадка на фильтре, который сушили в вакуумной печи с получением трет-бутил-4-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенилпиперазин-1-карбоксилата (1320 мг, 77%). (ЭР, m/z): 622,09[М+Н]+.
Стадия b: к трет-бутил-4-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенилпиперазин-1-карбоксилату (1,320 г, 2,125 ммоль) добавляли ДХМ и CF3COOH (12 мл, 3/1, об./об.); и реакционную систему перемешивали при комнатной температуре в течение 4 часов и концентрировали при пониженном давлении. К остатку добавляли воду (80 мл) и этилацетат (10 мл) с последующим добавлением водного аммиака для установления рН=9. Твердое вещество осаждали при перемешивании. Смесь фильтровали при пониженном давлении, в результате чего получали осадок на фильтре, который промывали небольшим количеством воды, сушили и взвешивали, в результате чего получали 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (907 мг, 82%), (ЭР, m/z): 522,09 [М+Н]+.
Пример получения промежуточного соединения 10 а:
Ссылаясь на стадии процесса примера получения промежуточного соединения 10, промежуточное соединение А10а получали, применяя следующие соответствующие исходные материалы и способы получения:

Пример получения промежуточного соединения 11: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпир ролидин-1-ил)-N-(4-(пиперидин-4-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (промежуточное соединение A11)

Стадия а: I-Вос-4-(4-аминофенил)-пиперидин (458 мг, 1,656 ммоль) добавляли в раствор 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (промежуточное соединение А2, 500 мг, 1,380 ммоль) и ТБТУ (532 мг, 1,656 ммоль) в безводном ДМФА (5 мл), затем по каплям добавляли ДИПЭА (535 мг, 4,140 ммоль) при 0°С; и реакционную систему оставляли реагировать при комнатной температуре в течение 18 часов в течение ночи. К реакционной жидкости добавляли воду (50 мл) и перемешивали. В осадок выпадало твердое вещество. Смесь фильтровали при пониженном давлении с получением осадка на фильтре, который сушили в вакуумной печи с получением трет-бутил-4-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенилпиперидин-1-карбоксилата (627 мг, 73%). (ЭР, m/z): 621,15 [М+Н]+.
Стадия b: к трет-бутил-4-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенилпиперидин-1-карбоксилату (627 мг, 1,101 ммоль) добавляли ДХМ и CF3COOH (8 мл, 3/1, об./об.), и реакционную жидкость перемешивали при комнатной температуре в течение 4 ч, и концентрировали при пониженном давлении. К остатку добавляли воду (80 мл) и ЭА (50 мл) с последующим добавлением водного аммиака для установления рН=9. Водную фазу экстрагировали ЭА (55 мл × 2), а объединенную экстрагированную фазу промывали H2O (20 мл) и раствором соли (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, в результате чего получали остаток, и очищали полученный остаток с помощью колоночной хроматографии на силикагеле. В частности, элюент собирали с помощью 10% (об./об.) МеОН-ДХМ после того, как менее полярные примеси вымывали 3% (об./об.) МеОН-ДХМ, и концентрировали элюент для получения 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперидин-4-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (437 мг, 76%). (ЭР, m/z): 521,14 [М+Н]+.
Примеры получения промежуточных соединений 11А и 12:
Ссылаясь на стадии процесса в примере промежуточного получения 11, промежуточные соединения A11a и А12 получали путем принятия следующих соответствующих исходных материалов и способов получения:

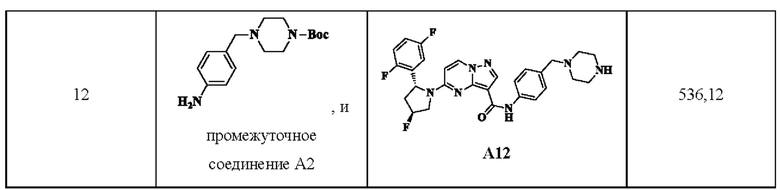
Пример получения промежуточного соединения 13: 5-(3-(2,5-дифторфенил)-морфолинил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (промежуточное соединение А13)

Стадия а: в атмосфере азота добавляли 1-Вос-4-(4-аминофенил)-пиперазин (3,812 г, 13,756 ммоль) в раствор 5-(3-(2,5-дифторфенил)-морфолино)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (4,127 г, 11,46 ммоль) и ТБТУ (4,417 г, 13,756 ммоль) в безводном ДМФА (20 мл), затем по каплям добавляли ДИПЭА (4,441 г, 34,362 ммоль) при 0°С, и реакционную систему оставляли реагировать при комнатной температуре в течение ночи. К реакционной жидкости добавляли ЭА (100 мл) и воду (100 мл), перемешивали и разделяли. Водную фазу экстрагировали этилацетатом (2 × 50 мл). Органические фазы объединяли, промывали водой (2 × 50 мл) и раствором соли (50 мл), затем сушили над безводным Na2SO4 и фильтровали. Растворитель концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии (ПЭ:ЭА = от 80:1 до 60:1, об./об.), в результате чего получали трет-бутил-4-(4-(5-(3-(2,5-дифторфенил)-морфолино)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенил-трет-бутилпиперазин-1-карбоксилат (3,924 г, 55,3%). (ЭР, m/z): 620,49 [М+Н]+.
Стадия b: к раствору трет-бутил-(4-(5-(3-(2,5-дифторфенил)-морфолино)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенил-трет-бутилпиперазин-1-карбоксилата (3,924 г, 6,332 ммоль) в ДХМ (30 мл) добавляли CF3COOH (10 мл), и реакционную систему перемешивали при комнатной температуре в течение 4 ч, и концентрировали при пониженном давлении. К остатку добавляли воду (100 мл) и ЭА (100 мл), с последующим добавлением водного аммиака для установления рН=9, и разделяли. Водную фазу экстрагировали этилацетатом (2 × 50 мл). Органические фазы объединяли, промывали водой (100 мл) и раствором соли (50 мл), затем сушили над безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (CH2Cl2:СН3ОН=от 50:1 до 10:1, об./об.), получая 5-(3-(2,5-дифторфенил)-морфолинил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (2,978 г, 90,5%). (ЭР, m/z): 520,11 [М+Н]+.
Пример получения промежуточного соединения 14:
Ссылаясь на стадии процесса примера получения промежуточного соединения 13, промежуточное соединение А14 получали, применяя следующие соответствующие исходные материалы и способы получения:
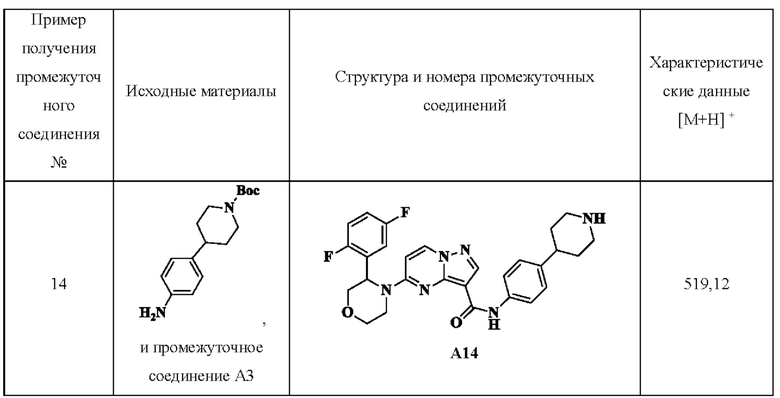
Пример получения промежуточного соединения 15: 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолинил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (промежуточное соединение А15)

Стадия а: в атмосфере азота добавляли 1-Вос-4-(4-аминофенил)-пиперазин (3,813 г, 13,747 ммоль) в раствор 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолино)-пиразоло[1,5-a]пиримидин-3-карбоновой кислоты (4,277 г, 11,46 ммоль) и ТБТУ (4,414 г, 13,747 ммоль) в безводном ДМ ФА (20 мл), затем по каплям добавляли ДИПЭА (4,441 г, 34,368 ммоль) при 0°С, и реакционную систему оставлли реагировать при комнатной температуре в течение ночи. К реакционной жидкости добавляли ЭА (100 мл) и воду (100 мл), перемешивали и разделяли. Водную фазу экстрагировали этилацетатом (2 × 50 мл). Органические фазы объединяли, промывали водой (2 × 50 мл) и раствором соли (50 мл), затем сушили над безводным Na2S04, фильтровали, концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (ПЭ:ЭА = от 100:1 до 50:1, об./об.), в результате чего получали трет-бутил-4-(4-(5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолино)-пиразоло[1,5-а]пиримидин-3-карбоксамидо)-фенил)-пиперазин-1-карбоксилат (4,736 г, 65,3%). (ЭР, m/z): 633,09[М+Н]+.
Стадия b: к раствору трет-бутил-4-(4-(5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолино)-пиразоло [1,5-а] пиримидин-3-карбоксамидо)-фенил)-пиперазин-1-карбоксилата (4,736 г, 7,485 ммоль) в ДХМ (30 мл) добавляли CF3COOH (10 мл), и реакционную систему перемешивали при комнатной температуре в течение 4 ч и концентрировали при пониженном давлении. К остатку добавляли воду (100 мл) и ЭА (100 мл), с последующим добавлением водного аммиака для установления рН=9, и разделяли. Водную фазу экстрагировали этилацетатом (2 × 50 мл). Органические фазы объединяли, промывали водой (100 мл) и раствором соли (50 мл), затем сушили над безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (CH2Cl2:СН3ОН=50:1 до 10:1, об./об.) для получения 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолинил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло [1,5-а] пиримидин-3-карбоксамида (3,685 г, 92,5%). (ЭР, m/z): 533,10 [М+Н]+.
Пример получения промежуточного соединения 16:
Ссылаясь на стадии процесса примера промежуточного получения 15, промежуточное соединение А16 получали, применяя следующие соответствующие исходные материалы и способы получения:

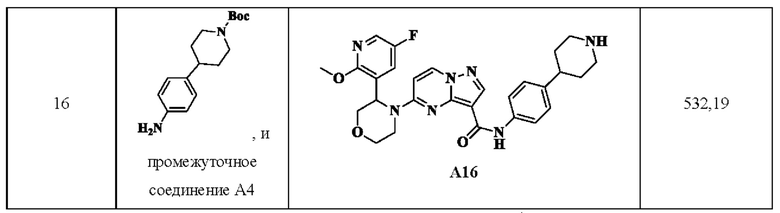
Пример получения промежуточного соединения 17: N1-(оксетан-3-ил)-бензол-1,4-диамин (промежуточное соединение В1)

Стадия а: 3-оксетамин (124 мг, 1,701 ммоль) добавляли в раствор п-фториитробеизола (200 мг, 1,417 ммоль) в ДМСО (3 мл) с последующим добавлением ДИПЭА (366 мг, 2,834 ммоль). Реакционную систему перемешивали при 120°С в течение 5 часов, добавляли воду (20 мл) и перемешивали, осаждая большое количество твердого вещества, которое затем фильтровали при пониженном давлении, в результате чего получали осадок на фильтре N-(4-нитрофенил)-оксетан-3-амин (268 мг, 97%). (ЭР, m/z): 195,01 [М+Н]+. Стадия b: в реакционную колбу добавляли осадок на фильтре М-(4-нитрофенил)-оксетан-3-амина (268 мг, 1,380 ммоль), полученный на предыдущей стадии, и метанол (15 мл). К реакционной смесь добавляли 10% Pd/C, заменяли воздух на H2 и перемешивали в течение 4 ч. Pd/C удаляли фильтрованием с отсасыванием, и фильтрат концентрировали при пониженном давлении, в результате чего получали N1-(оксетан-3-ил)-бензол-1,4-диамин в виде твердого вещества (110 мг). (ЭР, m/z): 164,92 [М+Н]+.
Примеры получения промежуточных соединений с 18 по 24:
Ссылаясь на стадии процесса в примере получения промежуточного соединения 17, промежуточные соединения В2-В8 получали путем принятия следующих соответствующих исходных материалов, п-фторнитробензола и способов получения:

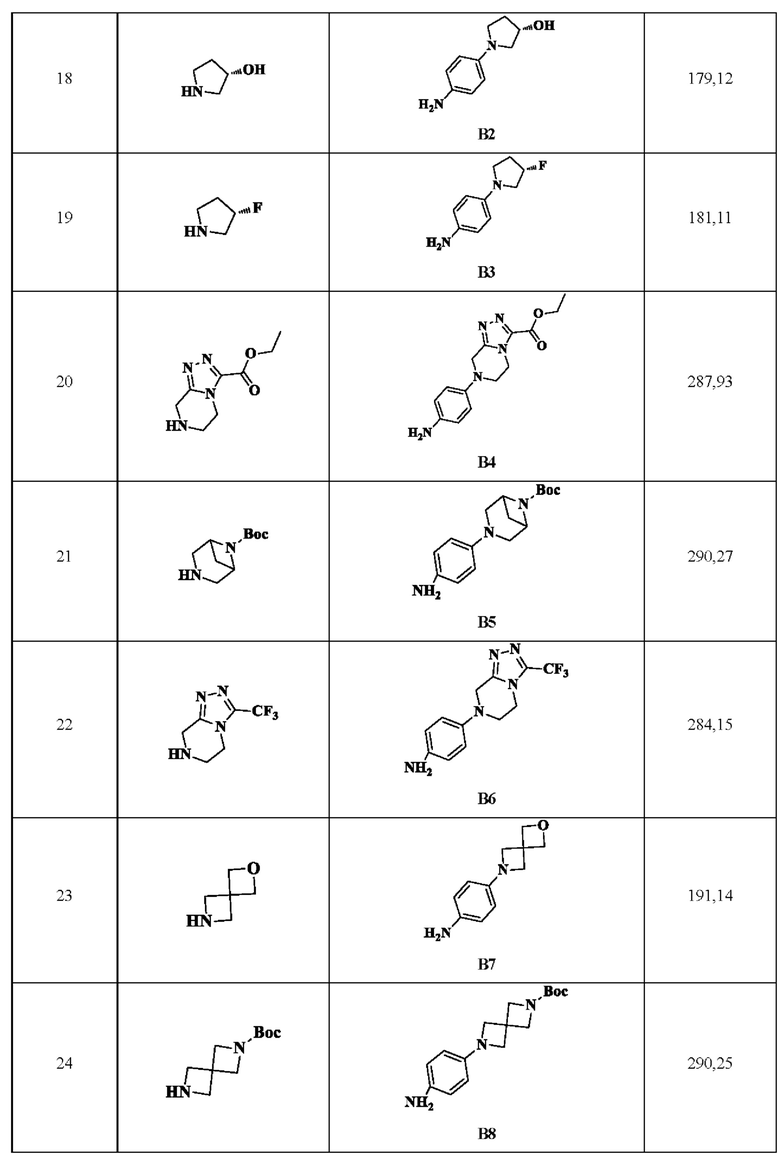
Пример получения промежуточного соединения 25:(R)-6-(2-(2,5-дифторфенил)-пирролидин-1-ил)-[1,2,4]триазоло[4,3-a]пиразин-3-карбоновая кислота (промежуточное соединение С1)
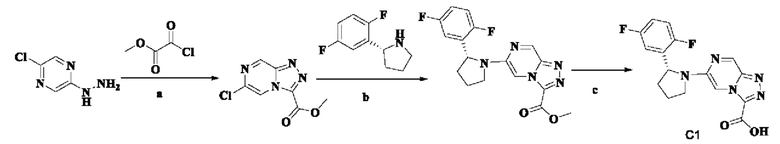
Стадии с а по с: (R)-6-(2-(2,5-дифторфенил)-пирролидин-1-ил)-[1,2,4]триазоло[4,3-а]пиразин-3-карбоновую кислоту получали согласно стадиям 1-3 Примера 1 в патенте CN108794484 В. (ЭР, m/z):346,02 [М+Н]+.
Пример получения промежуточного соединения 26: 6-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-[1,2,4]триазоло[4,3-а]пиразин-3-карбоновая кислота (промежуточное соединение С2)

Стадии с а по с: 6-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-[1,2,4]триазоло[4,3-a]пиразин-3-карбоновую кислоту получали со ссылкой на стадии примера получения промежуточного соединения 25. (ЭР, m/z): 364,03 [М+Н]+.
Примеры получения промежуточных соединений с 27 по 30:
Ссылаясь на стадии процесса в примере получения промежуточного соединения 5, промежуточные соединения С3-С6 получали путем принятия следующих соответствующих исходных материалов и способов получения:

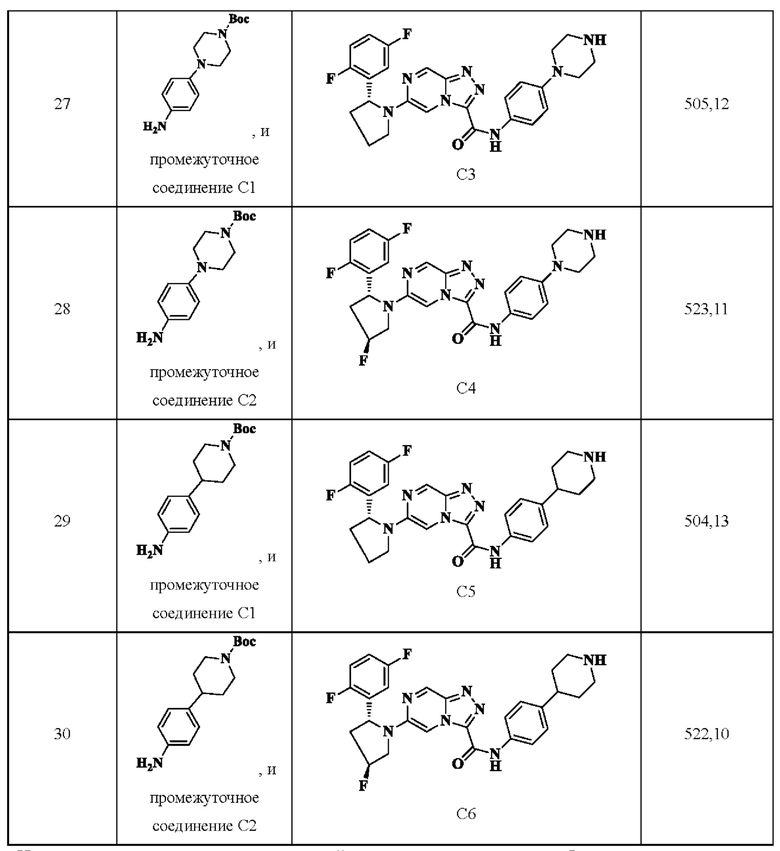
Ниже приведены примеры соединений согласно настоящему изобретению.
Пример 1: (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-R-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 1)

Гидроксиуксусную кислоту (32 мг, 0,417 ммоль) добавляли к раствору (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточное соединение А5, 70 мг, 0,139 ммоль) и БОП (74 мг, 0,167 ммоль) в безводном ДМ ФА (3 мл) с последующим добавлением по каплям ДИПЭА (54 мг, 0,417 ммоль) при 0°С, и реакционную систему перемешивали при комнатной температуре в течение 4 часов. К реакционной жидкости добавляли воду (20 мл) для смешивания и смесь экстрагировали этилацетатом (ЭА) (15 мл × 2). Объединенные органические фазы промывали H2O (20 мл) и раствором соли (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле. В частности, остаток элюировали 1% (об./об.) МеОН-ДХМ, а затем 2% (об./об.) МеОН-ДХМ, и элюент, содержащий целевой продукт, собирали и концентрировали с получением (R)-5-(2-(2,5-дифторфенил)-пирролидин-1-ил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло [1,5-а] пиримидин-3-карбоксамида (44 мг, 56%). (ЭР, m/z):562,13 [М+Н]+.
Примеры 1a, 1b и 2-3:
Ссылаясь на способы получения и операции примера 1, соединения 1a, 1b и 2-3 получали путем принятия следующих материалов и промежуточного соединения А5, А5а или С3 в качестве исходных материалов.
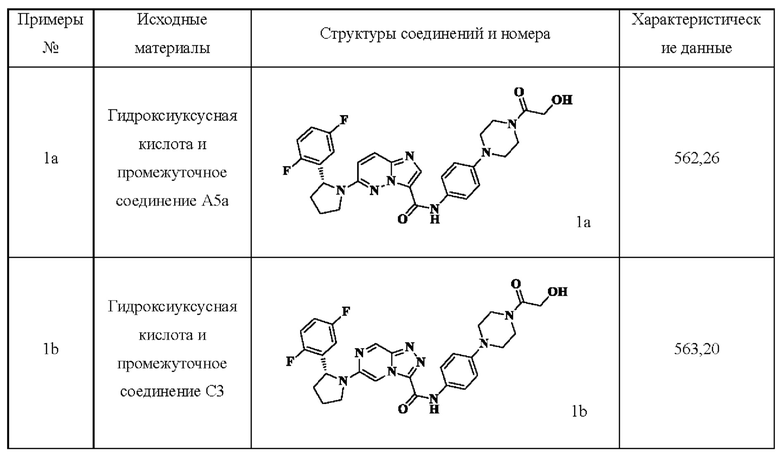
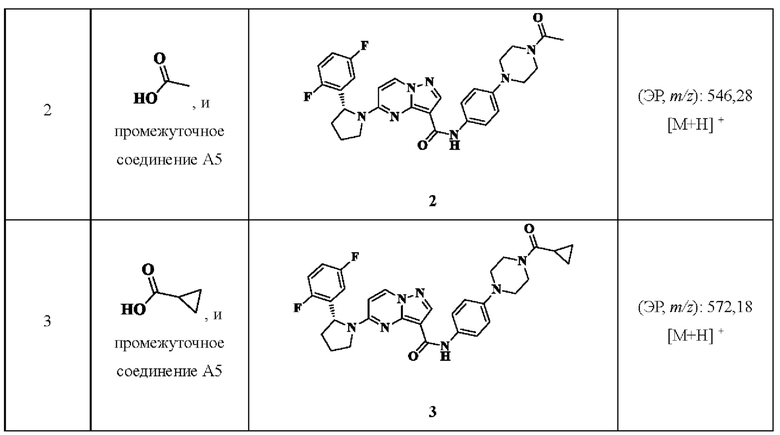
Пример 4: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 4)

Гидроксиуксусную кислоту (306 мг, 4,026 ммоль) добавляли в раствор 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперазин-4-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточное соединение А10, 700 мг, 1,342 ммоль) и БОП (712 мг, 1,610 ммоль) в безводном ДМФА (10 мл) с последующим добавлением по каплям ДИПЭА (520 мг, 4,026 ммоль) при 0°С, и реакционную систему перемешивали при комнатной температуре в течение 4 часов. К реакционной жидкости добавляли воду (80 мл) для смешивания, и смесь экстрагировали ЭА (55 мл × 2). Объединенные органические фазы промывали H2O (80 мл) и раствором соли (80 мл), соответственно. Затем органические фазы высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле. В частности, остаток элюировали 1% (об./об.) МеОН-ДХМ, а затем 2% (об./об.) МеОН-ДХМ, а элюент, содержащий целевой продукт, собирали и концентрировали с получением 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (666 мг, 86%). (ЭР, m/z): 580,14 [М+Н]+. 1Н ЯМР (600 МГц, ДМСО-d6) δ 9,810 (с, 1H), 8,906-8,723 (м, 1H), 8,283-8,229 (м, 1H), 7,623 (с, 1H), 7,343 (с, 1H), 7,210 (с, 2Н), 7,06I-6,842 (м, 4Н), 5,711-5,495 (м, 2Н), 4,631 (т, J=5,4 Гц, 1Н), 4,556-4,548 (м, 1Н), 4,318-4,225 (м, 1Н), 4,150 (д, J=5,4 Гц, 2Н), 3,637 (с, 2Н), 3,513 (с, 2Н), 3,124-3,106 (м, 4Н), 2,957-2,912 (м, 1H).
Примеры 4a-4b, 5а, 7а-7b и 5-9:
В соответствии со способами получения и операциями согласно примеру 4, соединения 4а-4b, 5а, 7а-7b и 5-9 получали путем принятия промежуточного соединения А10, А10а или С4 и следующих материалов в качестве исходных материалов.

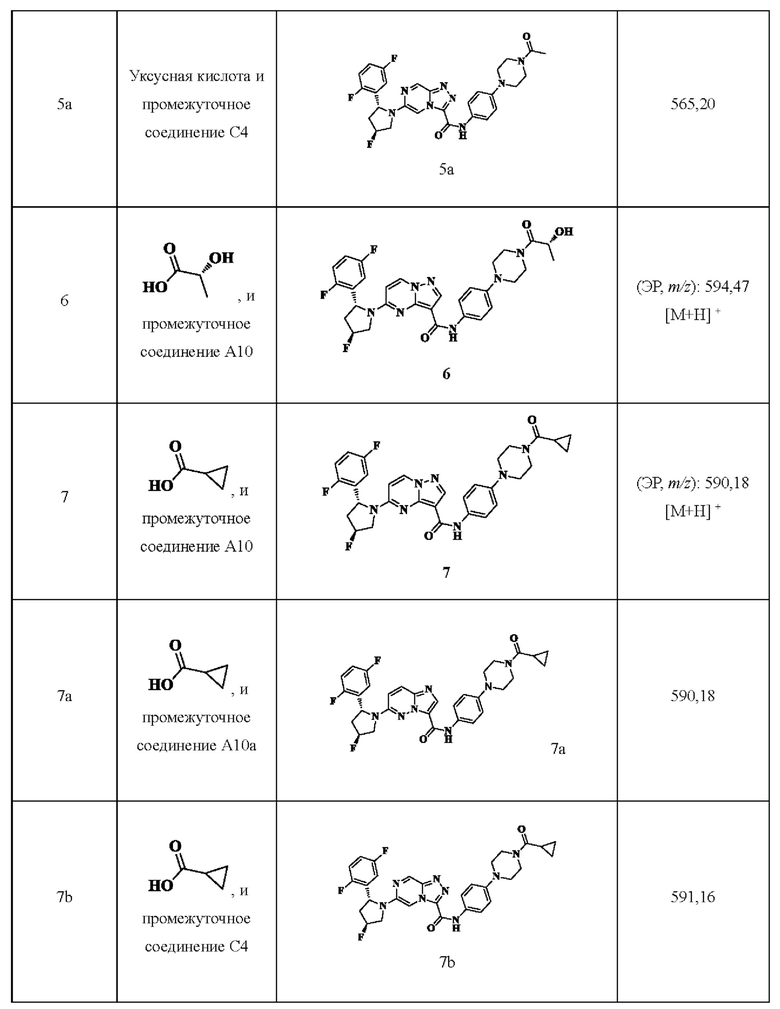
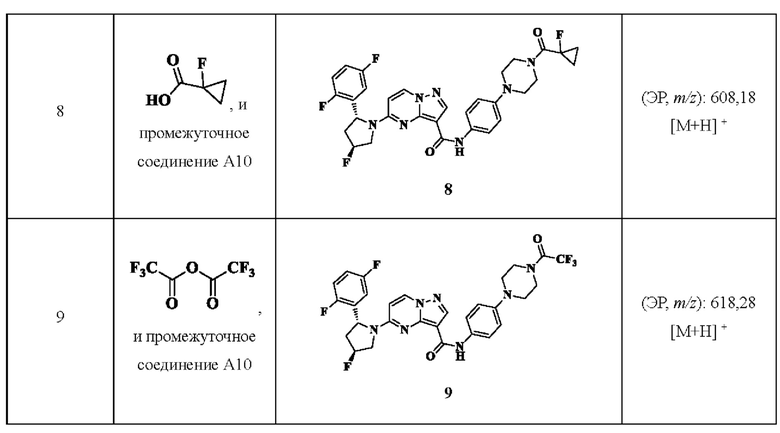
Пример 10: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-l-ил)-N-(4-(1-(2-гидроксиацетил)-пиперидин-4-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 10)
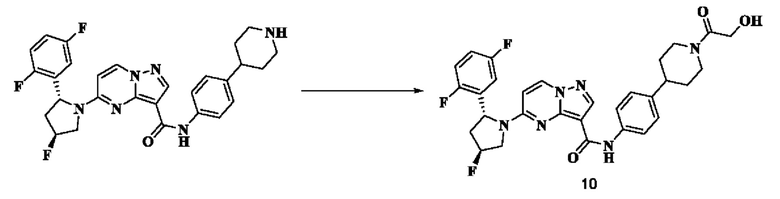
Гидроксиуксусную кислоту (438 мг, 5,763 ммоль) добавляли к раствору 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперидин-4-ил)-фенил)-пиразоло [1,5-а]пиримидин-3-карбоксамида (промежуточное соединение А11, 1000 мг, 1,921 ммоль) и БОП (1274 мг, 2,882 ммоль) в безводном ДМФА (10 мл) с последующим добавлением по каплям ДИПЭА (745 мг, 5,763 ммоль) при 0°С, и реакционную систему перемешивали при комнатной температуре в течение ночи. К реакционной жидкости добавляли воду (100 мл) для смешивания. Смесь экстрагировали ЭА (80 мл × 2). Объединенные органические фазы промывали H2O (100 мл) и раствором соли (110 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле. В частности, остаток элюировали 1% (об./об.) МеОН-ДХМ, а затем 2% (об./об.) МеОН-ДХМ. Элюент, содержащий целевой продукт, собирали и концентрировали с получением 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(1-(2-гидроксиацетил)-пиперидин-4-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (820 мг, 74%). (ЭР, m/z): 579,14 [М+Н]+. 1Н ЯМР (600 МГц, ДМСО-d6) δ 9,952 (с, 1H), 9,022-8,753 (м, 1H), 8,355-8,152 (м, 2Н), 7,663 (с, 1H), 7,343-7,042 (м, 6Н), 5,773-5,552 (м, 2Н), 4,529-4,481 (м, 2Н), 4,358-4,115 (м, 3Н), 3,803-3,779 (м, 1Н), 3,640-3,603 (м, 1H), 3,090-3,049 (м, 1Н), 2,968-2,957 (м, 1H), 2,792-2,752 (м, 1H), 2,722-2,682 (м, 1H), 2,241-2,212 (м, 1H), 1,808 (с, 2Н), 1,608-1,589 (м, 1H), 1,492-1,476 (м, 1H). Примеры 10А-10 В, 11А-11 В и 11-17:
В соответствии со способами получения и операциями Примера 10, соединения 10 А-10 В, 11А-11 В и 11-17 получали путем принятия промежуточного соединения A11, A11a или С6 и следующих материалов в качестве исходных материалов.

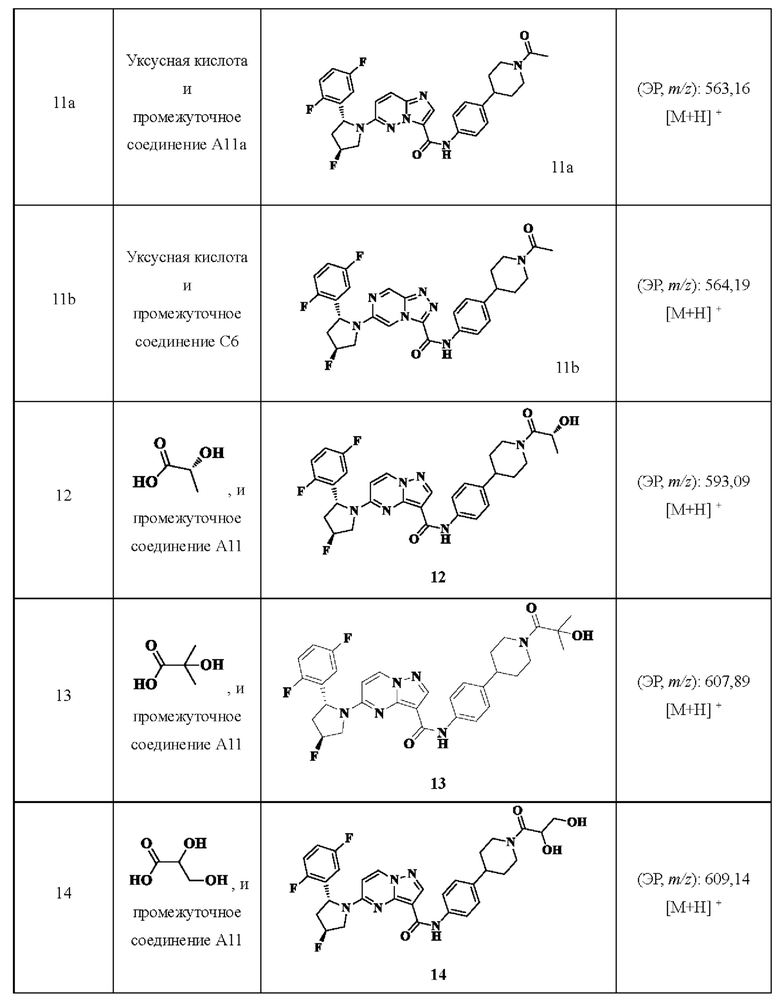

Пример 18: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(4-(2-(2-гидроксиацетил)-пиперазин-1-ил)-метил)-фенил)-пиразоло[1,5-a]пиримидин-3-карбоксамид (соединение 18)

Гидроксиуксусную кислоту (51 г, 0,672 ммоль) добавляли в раствор 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперазин-1-илметил)-фенил)-пиразоло[1,5-a]пиримидин-3-карбоксамида (промежуточное соединение А12, 120 г, 0,224 ммоль) и БОП (149 г, 0,336 ммоль) в безводном ДМ ФА (12 мл) с последующим добавлением по каплям ДИПЭА (87 г, 0,672 ммоль) при 0°С, и реакционную систему перемешивали при комнатной температуре в течение 4 часов. В реакционную жидкость добавляли воду (20 мл) для смешивания и смесь экстрагировали этилацетатом (ЭА) (10 мл × 2). Объединенные органические фазы промывали Н2О (10 мл) и раствором соли (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, и, в частности, остаток элюировали 1% (об./об.) МеОН-ДХМ, а затем 2% (об./об.) МеОН-ДХМ, в результате чего получали 5-((2R,45)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(4-(2-(2-гидроксиацетил)-пиперазин-1-ил)-метил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (30 мг, 22%). (ЭР, m/z): 594,12 [М+Н]+.
Пример 19: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(оксетан-3-иламино)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 19)
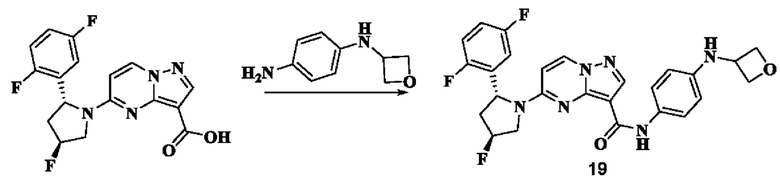
N1-(оксетан-3-ил)-бензол-1,4-диамин (105 мг, 0,640 ммоль) добавляли в раствор 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(1,2,3,4-тетрагидроизохинолин-7-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточное соединение А2, 193 мг, 0,534 ммоль) и ТБТУ (205 мг, 0,640 ммоль) в безводном ДМ ФА (5 мл), затем по каплям добавляли ДИПЭА (205 мг, 1,602 ммоль) при 0°С, и реакционную систему перемешивали в течение ночи при комнатной температуре в течение 14 часов. К реакционной жидкости добавляли воду (25 мл) для смешивания и смесь экстрагировали ЭА (15 мл × 2). Объединенные органические фазы промывали Н2О (20 мл) и раствором соли (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, в результате чего получали остаток, который разделяли с помощью препаративной жидкостной хроматографии (X-Bridge С18 19*150 мм 5 мкм, система элюирования: 5-25% водный раствор ацетонитрила для градиентного элюирования, 0,05% NH4HCO3 для модификации), в результате чего получали 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(оксетан-3-иламино)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (60 мг, 22,1%). (ЭР, m/z): 509,12 [М+Н]+.
Примеры 19А-19 В и 20-24:
Согласно способам получения и операциям Примера 19, соединения 19А-19 В и 20-24 получали путем принятия следующих промежуточных продуктов в качестве исходных материалов:

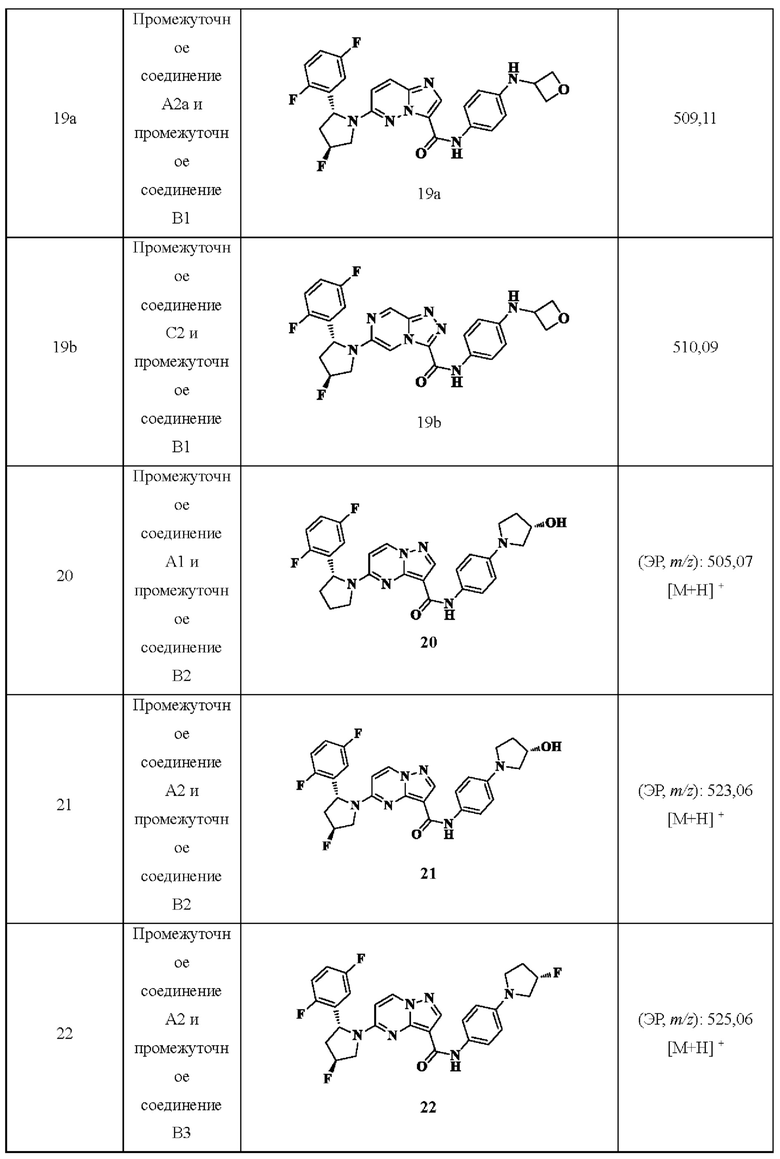
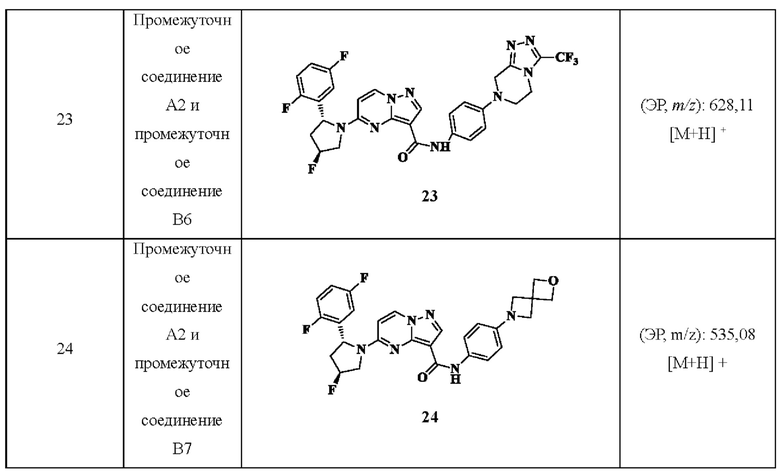
Пример 25: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(6-(2-гидроксиацетил)-3,6-диазабицикло [3.1.1]гепт-3-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 25)
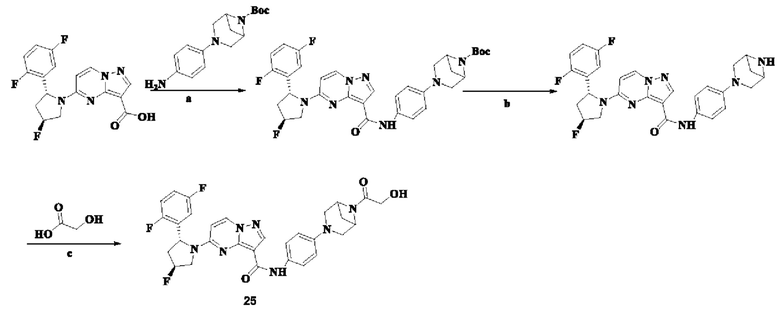
Стадия а: трет-бутил-3-(4-аминофенил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат (442 мг, 1,528 ммоль) (промежуточное соединение В5) добавляли к раствору 5-((2R,45)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-a]пиримидин-3-карбоновой кислоты (промежуточное соединение А2, 460 мг, 1,270 ммоль) и ТБТУ (491 мг, 1,528 ммоль) в безводном ДМФА (5 мл), затем по каплям добавляли ДИПЭА (494 мг, 3,819 ммоль) при 0°С, и реакционную систему перемешивали в течение ночи при комнатной температуре в течение 14 часов. К реакционной жидкости добавляли воду (100 мл) и перемешивали. Большое количество твердого вещества осаждали, фильтровали при пониженном давлении и сушили с получением неочищенного трет-бутил-3-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-формамидо)-фенил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилата.
Стадия b: полученный выше неочищенный трет-бутил-3-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-формамидо)-фенил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилат растворяли в ДХМ (25 мл). К раствору добавляли CF3COOH (5 мл) и перемешивали при комнатной температуре в течение 3 ч. Реакционную жидкость концентрировали путем роторного испарения и добавляли воду для разбавления с последующим добавлением водного аммиака для регулирования рН. Твердое вещество осаждали, фильтровали с отсасыванием и сушили с получением N-(4-(3,6-диазабицикло[3.1.1]гептил-3-ил)-фенил)-5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамида в виде твердого вещества (450 мг).
Стадия с: гидроксиуксусную кислоту (35 мг, 0,450 ммоль) добавляли к раствору N-(4-(3,6-диазабицикло [3.1.1]гептил-3-ил)-фенил)-5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (200 мг, 0,375 ммоль) и ТБТУ (145 мг, 0,450 ммоль) в безводном ДМФА (5 мл), затем по каплям добавляли ДИПЭА (146 мг, 1,125 ммоль) при 0°С, и реакционную систему перемешивали при комнатной температуре в течение 4 часов. К реакционной жидкости добавляли воду (30 мл) для смешивания. Смесь экстрагировали ЭА (20 мл × 2). Объединенные органические фазы промывали H2O (30 мл) и раствором соли (40 мл), сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, в частности, остаток элюировали 1% МеОН-ДХМ, а затем 2% МеОН-ДХМ. Элюент, содержащий целевой продукт, собирали и концентрировали с получением 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(6-(2-гидроксиацетил)-3,6-диазабицикло[3.1.1]гепт-3-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (180 мг, 81%). (ЭР, m/z): 592,02 [М+Н]+.
Пример 26: 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(6-(2-гидроксиацетил)-2,6-диазаспиро[3.3]гептан-2-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 26)
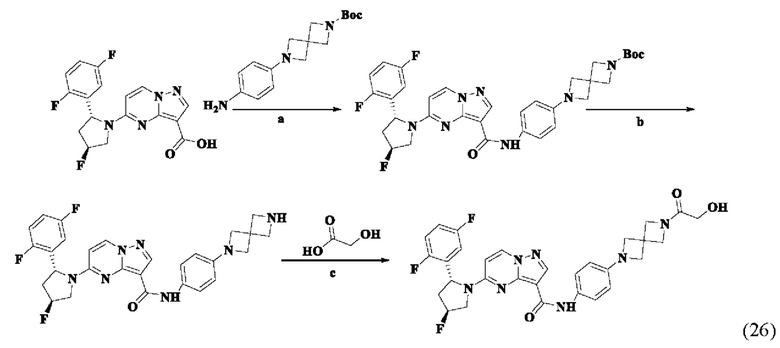
Стадия а: трет-бутил-6-(4-аминофенил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат (178 мг, 0,607 ммоль) (промежуточное соединение В8) добавляли в раствор 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (промежуточное соединение А2, 200 мг, 0,552 ммоль) и ТБТУ (217 мг, 0,607 ммоль) в безводном ДМ ФА (5 мл), затем по каплям добавляли ДИПЭА (217 мг, 1,656 ммоль) при 0°С, и реакционную систему перемешивали в течение ночи при комнатной температуре в течение 14 часов. К реакционной жидкости добавляли воду (30 мл) и перемешивали. Большое количество твердого вещества осаждали, а затем фильтровали при пониженном давлении и сушили с получением неочищенного трет-бутил-6-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-формамидо)-фенил)-2,6-диазаспиро[3.3]гептан-2-карбоксилата в виде твердого вещества. (ЭР, m/z): 534,12 [М+Н]+.
Стадия b: полученный выше неочищенный трет-бутил-6-(4-(5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло [1,5-а] пиримидин-3-формамидо)-фенил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат растворяли в ДХМ (6 мл). К раствору добавляли CF3COOH (2 мл) и перемешивали при комнатной температуре в течение 3 ч. Реакционную жидкость концентрировали путем роторного испарения и добавляли воду для разбавления с последующим добавлением водного аммиака для регулирования рН. Твердое вещество осаждали, фильтровали с отсасыванием и сушили с получением N-(4-(2,6-диазаспиро[3.3]гептил-2-ил)-фенил)-5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксамида в виде твердого вещества (218 мг).
Стадия с: гидроксиуксусную кислоту (37 мг, 0,490 ммоль) добавляли в раствор N-(4-(2,6-диазаспиро[3.3]гептил-2-ил)-фенил)-5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-пиразоло[1,5-a]пиримидин-3-карбоксамида (218 мг, 0,409 ммоль) и ТБТУ (157 мг, 0,490 ммоль) в безводном ДМФА (10 мл) с последующим добавлением по каплям ДИПЭА (158 мг, 1,226 ммоль) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. К реакционной жидкости добавляли воду (30 мл) для смешивания и смесь экстрагировали этилацетатом (20 мл × 2). Объединенные органические фазы промывали H2O (30 мл) и раствором соли (40 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который очищали с помощью препаративной пластины (ДХМ:МеОН=15:1) с получением 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(6-(2-гидроксиацетил)-2,6-диазаспиро[3.3]гептан-2-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (51 мг, 94%). (ЭР, m/z): 592,12 [М+Н]+.
Пример 27: 5-(3-(2,5-дифторфенил)-морфолинил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 27)
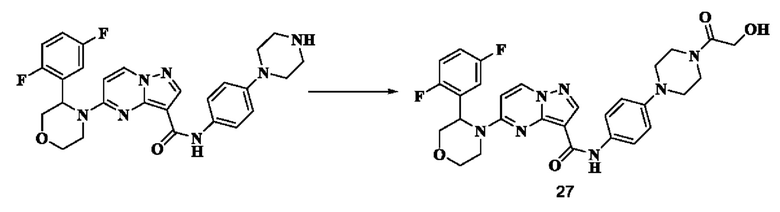
Гидроксиуксусную кислоту (584 мг, 7,676 ммоль) добавляли к раствору 5-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)-N-(4-(пиперидин-4-ил)-фенил)-пиразоло [1,5-а]пиримидин-3-карбоксамида (промежуточное соединение А13, 997 мг, 1,919 ммоль) и БОП (1274 мг, 2,878 ммоль) в безводном ДМФА (10 мл), затем по каплям добавляли ДИПЭА (744 мг, 5,756 ммоль) при 0°С, и реакционную смесь перемешивали при комнатной температуре. К реакционной жидкости добавляли воду (100 мл) для смешивания, и смесь экстрагировали этилацетатом (80 мл × 2). Органические фазы объединяли. Органическую фазу промывали ШО (100 мл) и раствором соли (110 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: ДХМ:СН3ОН = от 100:1 до 50:1, об./об.). Элюент, содержащий целевой продукт, собирали и концентрировали с получением 5-(3-(2,5-дифторфенил)-морфолинил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (609,8 мг, 55%). (ЭР, m/z): 578,12 [М+Н]+.
Пример 28:
В соответствии со способами получения и операциями примера 27, соединение 28 получали путем применения промежуточного соединения А14 и гидроксиуксусной кислоты в качестве исходных материалов.
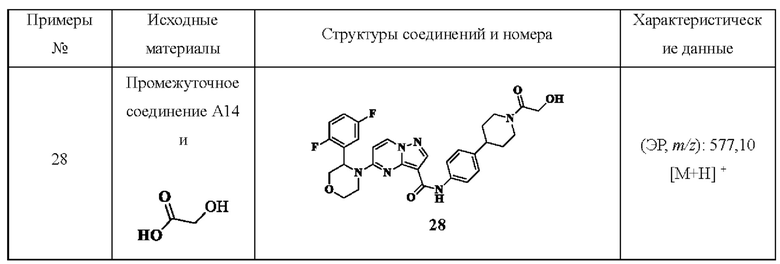
Пример 29: 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолинил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамид (соединение 29)
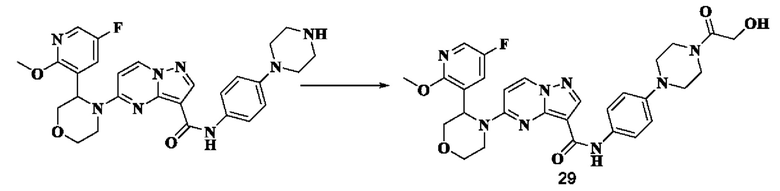
Гидроксиуксусную кислоту (511 мг, 6,723 ммоль) добавляли к раствору 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолино)-N-(4-(пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточное соединение А15, 1,022 г, 1,92 ммоль) и БОП (1274 мг, 2,878 ммоль) в безводном ДМФА (10 мл), затем по каплям добавляли ДИПЭА (744 мг, 5,763 ммоль) при 0°С, и реакционную систему перемешивали при комнатной температуре в течение ночи. К реакционной жидкости добавляли воду (100 мл) для смешивания, и смесь экстрагировали этилацетатом (80 мл × 2). Органические фазы объединяли. Органическую фазу промывали H2O (100 мл) и раствором соли (110 мл), сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: ДХМ:CH2OH = от 100:1 до 50:1). Элюент, содержащий целевой продукт, собирали и концентрировали с получением 5-(3-(5-фтор-2-метоксипиридин-3-ил)-морфолинил)-N-(4-(4-(2-гидроксиацетил)-пиперазин-1-ил)-фенил)-пиразоло[1,5-а]пиримидин-3-карбоксамида (737 мг, 65,02%). (ЭР, m/z): 591,20 [М+Н]+.
Пример 30:
В соответствии со способами получения и операциями Примера 29, соединение 30 получали путем применения промежуточного соединения А16 и гидроксиуксусной кислоты в качестве исходных материалов.
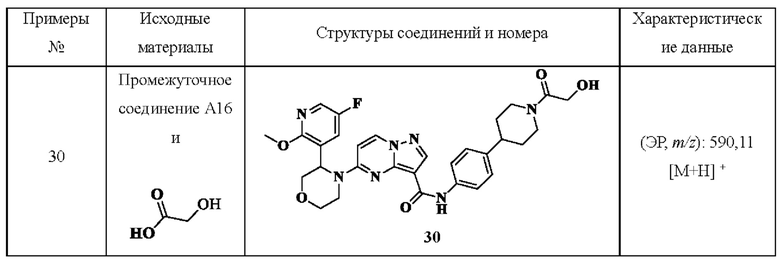
Примеры 31-47:
Ссылаясь на способы получения и операции, описанные в примерах получения промежуточных соединений 1-30 и примерах 1-30, и способ, описанный в CN111936500A, соединения 31-47 получали с применением (2R)-2-(2,5-дифторфенил)-3-азабицикло[3.1.0]гексана и каждого из указанных промежуточных соединений в качестве исходных материалов.
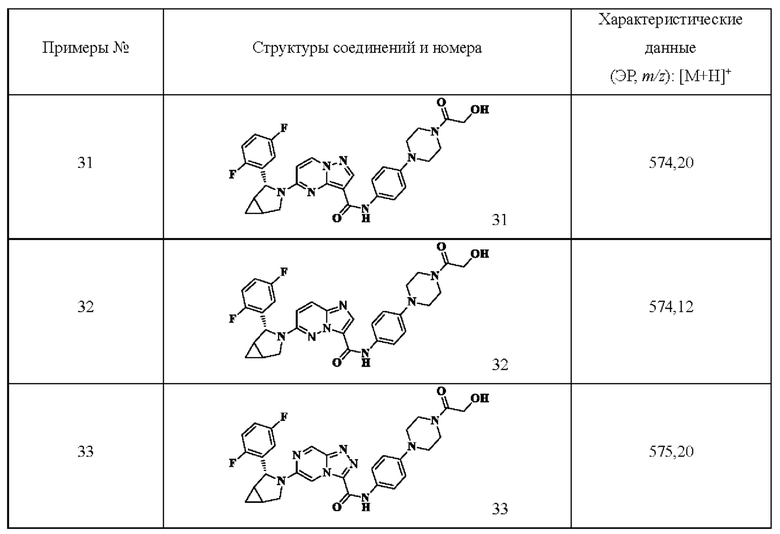
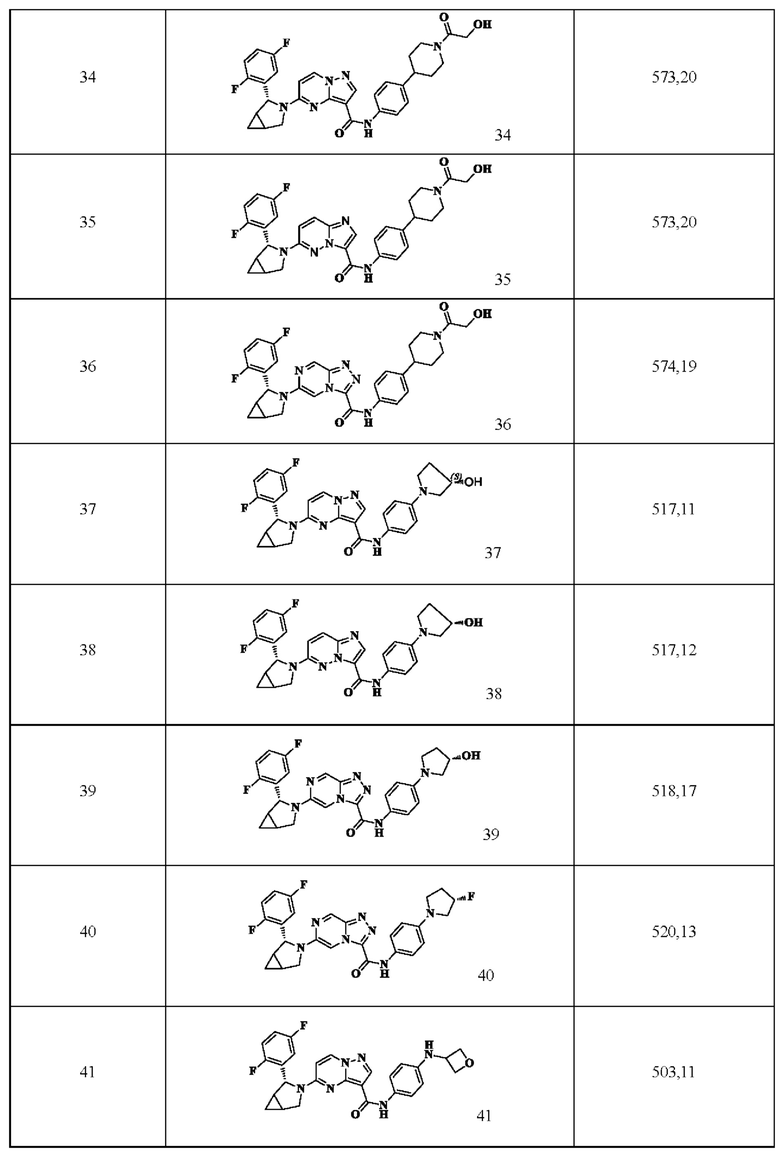

Примеры получения соединений сравнения I-5:
Соединения D1-D5 получали со ссылкой на способы получения и операции в патентных документах WO 2019029629A1 и WO 2012034095А1.

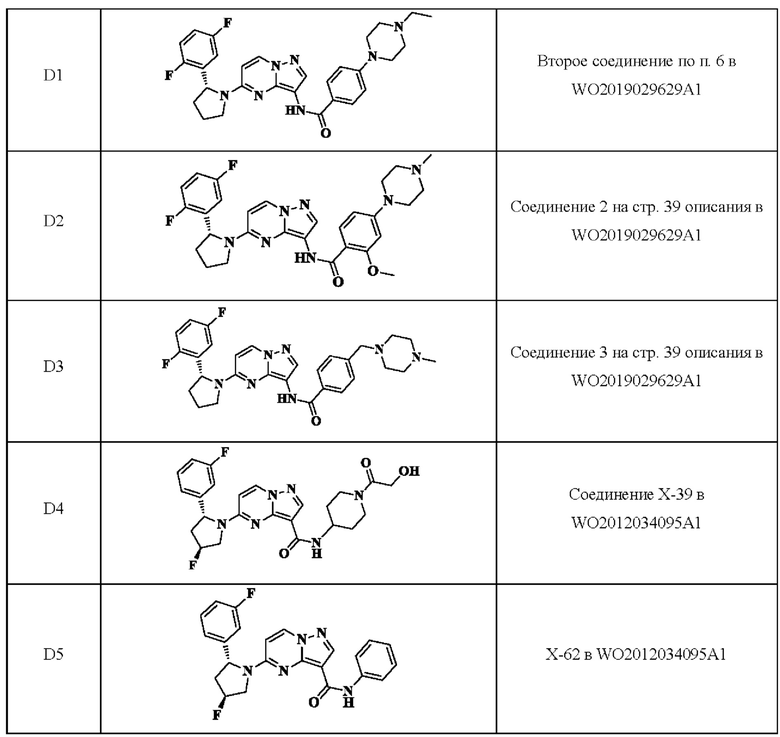
Пример испытания 1. Ингибирование соединений по отношению к TRK
1. Процедуры:
1.1 Реакция киназы:
Соединение, подлежащее испытанию с определенным градиентом концентрации, и раствор фермента последовательно добавляли в планшет (киназный буфер (1× киназный буфер (Cisbio, кат. №62EZBFDD), рН 7,5; 5 мМ MgCl2, 1 мМ DTT) добавляли в лунки отрицательного контроля), и планшете соединением центрифугировали при 1000 об/мин в течение 30 секунд. Планшет герметично закрывали и инкубировали в инкубаторе при постоянной температуре 25°С в течение 30 мин. Готовили раствор субстрата, содержащий TK-Sub-биотин (Cisbio, кат. №61TKOBL) и АТФ (Sigma, кат. №R0441), добавляли раствор субстрата в 384-луночный планшет и центрифугировали при 1000 об/мин в течение 30 секунд. Планшет герметично закрывали и инкубировали в инкубаторе при постоянной температуре 25°С в течение 60 мин.
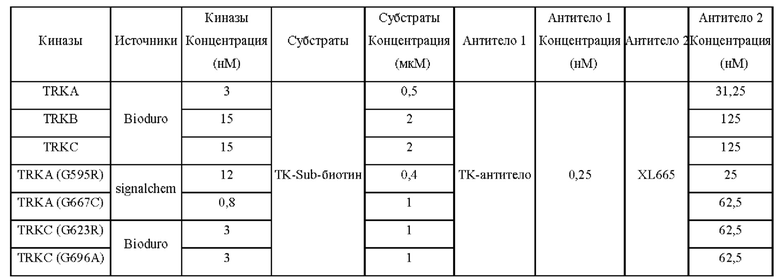
1.2 Анализ киназы:
Антитело ТК и XL665 разбавляли, перемешивали и добавляли в аналитический планшет, и планшет центрифугировали при 1000 об/мин в течение 30 секунд. Планшет герметично закрывали и инкубировали в инкубаторе при постоянной температуре 25°С в течение 60 мин. Планшет для анализа помещали на считыватель Envision для считывания, (отношение HTRF 665/615: значение сигнала при 665 нм /значение сигнала при 615 нм) Ингибирование = (отношениелунка отрицательный контроль - отношение лунка соединение) / (отношениелунка отрицательный контроль - отношениелунка контроль без фермента) × 100%
1.3 Анализ данных и подбор кривой
Обработку данных проводили в надстройке XLFit excel версии 5.4.0.8 для получения значений IC50.
1.4 Параметры контроля качества
Эталонное соединение содержалось в каждом планшете с IC50 в пределах 3-кратного значения каждый раз.
2. Результаты испытаний: как показано в таблице 1
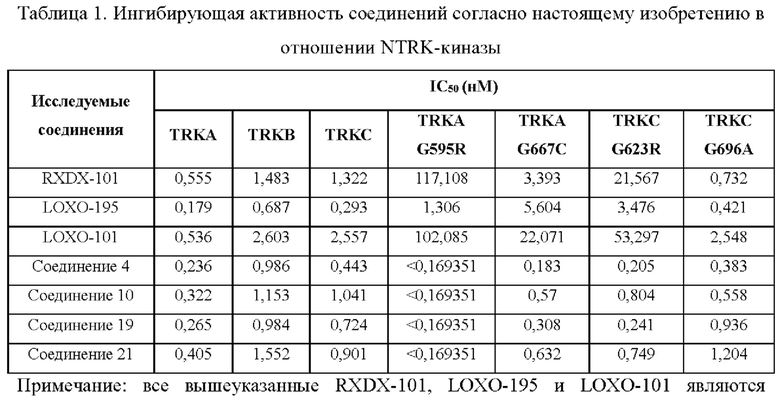

Результаты показывают, что: соединения согласно настоящему изобретению проявляют более высокую ингибирующую активность в отношении различных киназ. Ингибирующая активность соединений согласно настоящему изобретению в отношении TRKA, TRKB, TRKC и TRKC-G696A превосходит или эквивалентна активности соединений RXDX-101, LOXO-195 и LOXO-101, в то время как ингибирующая активность соединений согласно настоящему изобретению в отношении различных мутантных лекарственно-устойчивых киназ (G595R, G667C и G623R) значительно превосходит активность соединений RXDX-101, LOXO-195 и LOXO-101.
Пример испытания 2. Ингибирование соединений в отношении ALK и ROS1
1. Процедуры:
1.1 Реакция киназы:
Соединения разбавляли до определенной концентрации в ДМСО с 4-кратными градиентами. Соединение с определенной концентрацией, раствор фермента и ДМСО добавляли в 384-луночный планшет, и планшет инкубировали при комнатной температуре в течение 10 мин. В планшет добавляли флуоресцеин-меченый пептид и АТФ (sigma, кат. №: A7699-1G, № партии: 987-65-5), инкубировали при температуре 28°С в течение определенного времени и затем добавляли раствор для прекращения реакции. Планшет подвергали считыванию.
Скорость ингибирования для одной концентрации: скорость ингибирования=(ОПлунка отрицательный контроль - ОПлунка соединение) / (ОПлунка отрицательный контроль - ОПлунка контроль без фермента) × 100%
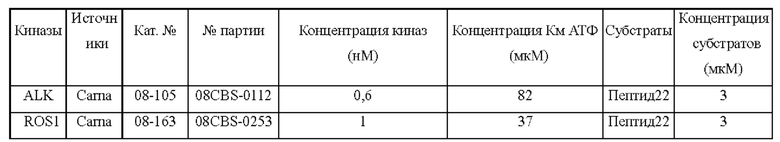
1.2 Анализ данных и подбор кривой
Обработку данных проводили в надстройке XLFit excel версии 4.3.1 для получения значений IC50, и результаты приведены в таблице 2.
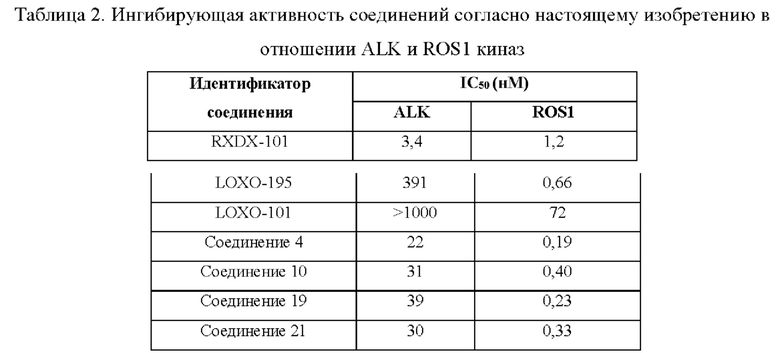
Результаты показывают, что: множество соединений согласно настоящему описанию проявляют более сильную ингибирующую активность в отношении киназы ROS1, которая значительно превосходит активность RXDX-101 и LOXO-101 и превосходит активность LOXO-195. соединения обладают хорошей ингибирующей активностью в отношении ALK-киназы, которая превосходит активность LOXO-101 и LOXO-195.
Пример испытания 3. In vitro активность соединений по ингибированию пролиферации клеток
1. Клеточные линии
Источник 6 видов клеточных линий для испытания: KYinno Biotechnology (Beijing) Co., Ltd.
Тип клеток: мышиные В-клетки
Культуральная среда: RPMI-1640+10%ФБС
2. Метод испытания
Клетки в логарифмической фазе роста собирали и подсчитывали с использованием счетчика тромбоцитов. Суспензию клеток с определенной плотностью равномерно распределяли пипеткой и высевали в 96-луночный планшет с каждой лункой объемом 100 мкл, и встряхивали, чтобы клетки равномерно распределялись по лункам. В каждую лунку добавляли по 100 мкл раствора соединения с определенным градиентом концентрации и устанавливали по 3 повторяющиеся лунки для каждой концентрации. Планшет культивировали в инкубаторе в атмосфере СО2 при 37°С в течение 72 часов. В каждую лунку добавляли 20 мкл рабочего раствора МТТ (5 мг/мл), и планшет помещали при 37°С на 4 ч и центрифугировали при 1000 об/мин в течение 5 мин с помощью центрифугирования планшета. После того, как 180 мкл культуральной среды отбрасывали, в планшет добавляли 150 мкл ДМСО и хорошо перемешивали с помощью шейкера для микропланшетов, дно планшета протирали, и определяли оптическую плотность (ОП) при 550 нм с помощью считывателя для микропланшетов.
3. Анализ данных
Скорость ингибирования - (ОПконтрольная лунка - ОПисследуемая лунка) / (ОПконтрольная лунка - ОПпустая лунка) × 100%, и значения половинной максимальной ингибирующей концентрации IC50 рассчитывали для каждой скорости ингибирования концентрации с использованием программного обеспечения SPSS.
4. Результаты испытаний: как показано в таблице 3:
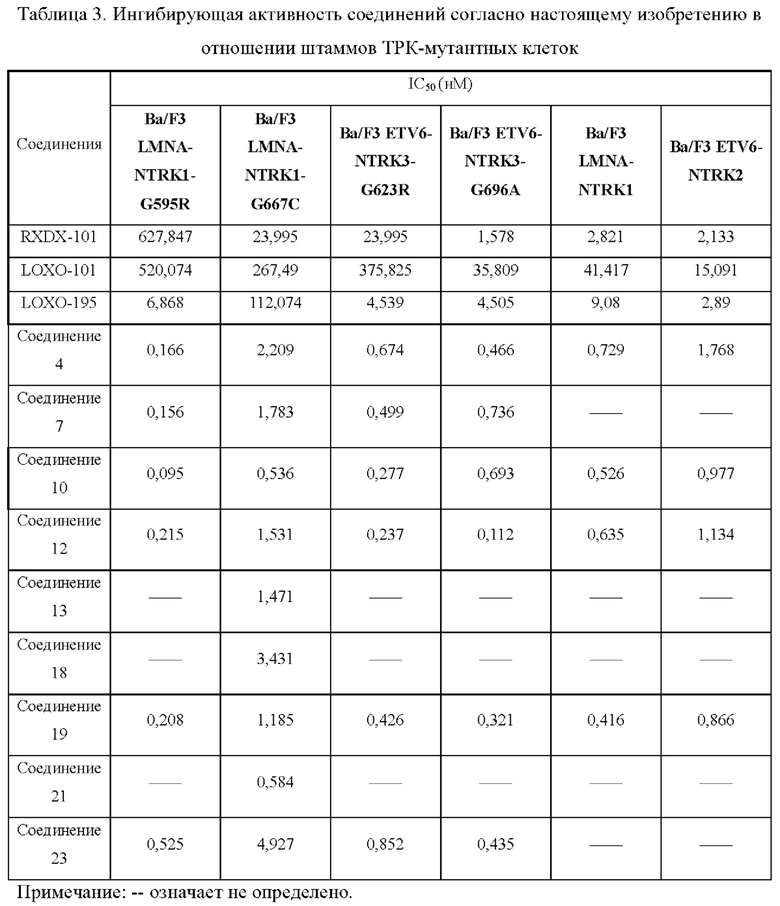
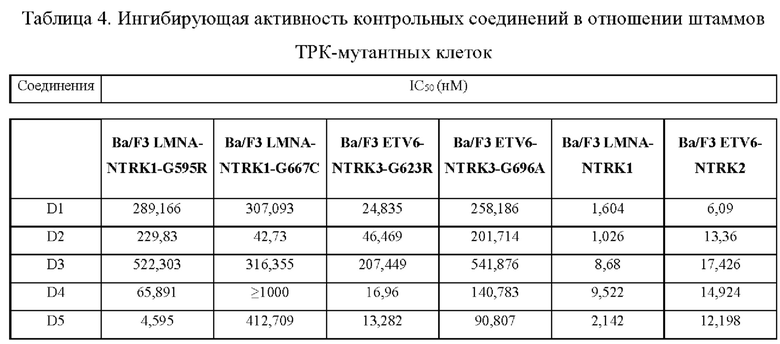
Результаты показывают, что: множество соединений согласно настоящему изобретению проявляют лучшую ингибирующую активность in vitro в отношении клеток различных клеточных штаммов дикого типа и мутантных лекарственно-устойчивых штаммов, и значительно превосходят RXDX-101, LOXO-195, LOXO-101 и соединения DI-D5 согласно предшествующему уровню техники.
Пример испытания 4: Исследование in vivo механизма соединений
1. Метод испытания
1.1 Подготовка модели:
Мутантные лекарственно-устойчивые клетки Ba/F3 LMNA-NTRKI-G595R в логарифмической фазе роста собирали и повторно суспендировали в бессывороточной культуральной среде для достижения концентрации клеток 6×107-10×107 клеток/мл. К клеточной суспензии добавляли равный объем матригеля для получения конечной концентрации клеток при 3×107-5×107 клеток/мл. Модель на животных получали путем подкожной инокуляции 0,1 мл суспензии опухолевых клеток в подмышечной впадине передней конечности мышей NuNu (самки, 4-6 недель, Beijing Vital River Laboratory Animal Technology Co., Ltd.) в количестве 3×106-5×106 клеток/мышь.
1.2 Группировка:
Максимальный и минимальный диаметры опухоли ксенотрансплантатов у голых мышей измеряли с помощью штангенциркуля, а объем опухоли (ОО) рассчитывали по формуле: V=1/2 × а × b2, где а и b представляют собой максимальный и минимальный диаметры опухолевой массы, соответственно. Отбирали голых мышей с соответствующим объемом опухоли, и равномерно разделяли мышей на 7 групп по 3 мыши (200-300 мм3) по объему опухоли с использованием метода случайных чисел.
1.3 Введение
Введение через желудочный зонд осуществляли в соответствии с массой животного при объеме введения 10 мл/кг, и соединение №4 готовили в виде состава с требуемой концентрацией введения с использованием «3% ДМСО+96% HP-β-CD (0,5 г/мл)+1% HCl».
Было отобрано в общей сложности 3 мыши в контрольной группе, и опухолевые ткани извлекали и криоконсервировали через 4 часа после введения носителя. Соединение №4 согласно настоящему изобретению вводили другим группам в дозе 100 мг/кг, а опухолевые ткани извлекали для криоконсервации через 0,25 ч, 1 ч, 4 ч, 8 ч, 12 ч и 24 ч.
1.4 Экстракция белка и количественное определение
Отбирали определенную массу опухолевой ткани и добавляли в соответствующий объем белкового лизата (RTPA лизат (Thermo Fisher, кат. №89900): ингибитор протеазы (cOmplete, Mini, EDTA-free, EASYpack; Roche, кат. №04693159001): ингибитор фосфатазы (PhosStop, EASY pack; Roche, кат. №04906837001)=8:1:1), гомогенизировали и лизировали на ледяной бане в течение 30 мин. Смесь подвергали высокоскоростному центрифугированию при низкой температуре, и затем отбирали супернатант для количественного определения белка ВСА (процедура в соответствии с набором для количественного определения белка ВСА (Qiagen, кат. №РА115-01)). Наконец, после того, как концентрацию белка доводили до однородности с помощью лизата, добавляли загрузочный буфер, и полученную смесь кипятили при 100°С в течение 10 минут.
1.5 Вестерн-блоттинг
Принимали 4-20% 10-луночный предварительно изготовленный гель; количество загрузки образца составляло 100 мкг; 140 В электрофорез проводили в течение I-1,5 ч; влажный перенос 300 мА проводили в течение 1,5-2 ч; мембрану блокировали 5% БСА в течение 2-3 ч; инкубировали с первичным антителом в течение ночи при 4°С (Trk 1:5000, p-Trk, PLCγ1, p-PLCγ1, АКТ, р-АКТ, актин 1:1000) и промывали мембрану 0,1% TBST в течение 5 мин 4 раза; инкубировали с вторичным антителом при комнатной температуре в течение 2 ч (1:5000), облучали ECL и экспонировали.

2. Результаты испытаний: как показано на фиг.1
Из результатов анализа видно, что: с течением времени экспрессия TRK, p-TRK, p-PLCγ1 и р-АКТ на Фиг. 1 значительно снижается, что доказывает, что соединение №4 согласно настоящему изобретению может значительно снижать уровень белка TRK и p-TRK и дополнительно эффективно ингибировать фосфорилирование p-PLCγ1/PLCγ1 и р-АКТ/АКТ, чтобы регулировать рост и пролиферацию клеток.
Пример испытания 5: Фармакодинамический тест in vivo соединений на модели ксенотрансплантата, устойчивой к мутации NTRK
Метод испытаний
1.1 Подготовка модели
Клетки в логарифмической фазе роста собирали и повторно суспендировали в бессывороточной культуральной среде для достижения концентрации клеток 6×107-10×107 клеток/мл. К клеточной суспензии добавляли равный объем матригеля для получения конечной концентрации клеток при 3×107-5×107 клеток/мл. Модель на животных получали путем подкожной инокуляции 0,1 мл суспензии опухолевых клеток в подмышечной впадине передней конечности мышей NuNu (самки, 4-6 недель, Beijing Vital River Laboratory Animal Technology Co., Ltd.) в количестве 3×106-5×106 клеток/мышь.
1.2 Группировка
Максимальный и минимальный диаметры опухоли ксенотрансплантатов у голых мышей измеряли с помощью штангенциркуля, а объем опухоли (ОО) рассчитывали по формуле: V=1/2 × а × b2, где а и b представляют собой максимальный и минимальный диаметры опухолевой массы, соответственно. Отбирали бестимусных мышей с соответствующим объемом опухоли, и равномерно разделяли мышей на 7 групп по 6 мышей (100-200 мм3) по объему опухоли с использованием метода случайных чисел.
1.3 Наблюдение за индексами
Введение через желудочный зонд осуществляли в день группировки в соответствии с массой животного при объеме введения 10 мл/кг, LOXO-195 готовили в виде состава с 0,5% CMC-Na для получения требуемого раствора для введения, а соединения №4 и №10 готовили в виде состава с «3% ДМСО+96% HP-β-CD (0,5 г/мл)+1% HCl» для получения требуемого раствора для введения. Диаметры опухоли измеряли два раза в неделю и рассчитывали объем опухоли. Конкретные индексы описаны далее: Масса животного: животных взвешивали перед введением дозы каждое утро, и потеря массы более 20% определялась как токсический ответ на препарат (наблюдаемый на следующий день после последнего введения);
Объем опухоли (ОО)=V=1/2 × а × b2, где а и b представляют собой максимальный и минимальный диаметры опухолевой массы, соответственно (наблюдаемый на следующий день после последнего введения);
Относительная скорость пролиферации опухоли Т/С (%): Т/С (%)=TRTV / CRTV × 100% (TRTV: группа введения RTV, CRTV: контрольная группа RTV); Скорость ингибирования роста опухоли (TGI)=[1 - (Ti - Т0) / (Vi - V0)] × 100%. (где Ti представляет средний объем опухоли определенной группы введения в определенный день; Т0 представляет средний объем опухоли группы введения в начале введения, Vi представляет средний объем опухоли контрольной группы носителя в определенный день (в тот же день, что и Ti) и V0 представляет средний объем опухоли контрольной группы носителя в начале введения);
Скорость ингибирования опухоли: в конце испытания животных умерщвляли путем удаления шейных позвонков. Опухолевые массы отслаивали, взвешивали и фотографировали, скорость ингибирования опухоли рассчитывали по формуле: скорость ингибирования опухоли=(средняя масса опухоли контрольной группы - средняя масса опухоли группы введения) / средняя масса опухоли контрольной группы × 100%.
Результаты испытаний
2.1 Модель Ba/F3 LMNA-NTRKI-G667C
2.1.1 Влияние лекарственных препаратов на массу мышей с опухолями
Масса мышей в каждой группе дозирования каждого соединения имеет тенденцию к увеличению, и тенденция к увеличению является более значимой, чем у контрольной группы. Масса мышей в каждой группе дозирования каждого соединения значительно увеличивается, что, возможно, связано с соединением, или обусловлено тем, что соединение может ингибировать рост опухоли, так что мыши имеют лучшее состояние и значительное увеличение массы. Результаты представлены в Таблице 5.
2.1.2 Влияние препаратов на массу опухоли и скорость ингибирования опухоли у мышей с опухолями
Результаты данных показывают, что по сравнению с LOXO-195 соединение №4 и соединение №10 согласно настоящему изобретению демонстрируют более значительное ингибирование роста опухоли при той же дозе введения (100 мг/кг); кроме того, соединение №4 и соединение №10 (100 мг/кг) согласно настоящему изобретению также демонстрируют лучший эффект ингибирования опухоли, чем группа LOXO-195 (200 мг/кг) в более высокой дозе. Результаты представлены в Таблице 5.

2.2 Модель Ba/F3 LMNA-NTRK1 -G595R
2.2.1 Влияние лекарственных препаратов на массу мышей с опухолями
Масса мышей в каждой группе дозирования каждого соединения имеет тенденцию к увеличению, и тенденция к увеличению является более значимой, чем у контрольной группы. Масса мышей в каждой группе дозирования каждого соединения значительно увеличивается, что, возможно, связано с соединением, или обусловлено тем, что соединение может ингибировать рост опухоли, так что мыши имеют лучшее состояние и значительное увеличение массы. Результаты представлены в Таблице 6.
2.2.2 Влияние препаратов на массу опухоли и скорость ингибирования опухоли у мышей с опухолями
Результаты данных показывают, что по сравнению с LOXO-195 (100 мг/кг) соединение №4 и соединение №10 согласно настоящему изобретению могут достичь значительного ингибирования массы опухолевых тканей при более низкой дозировке (50 мг/кг) со скоростью ингибирования массы опухоли более 90%. Результаты представлены в Таблице 6.
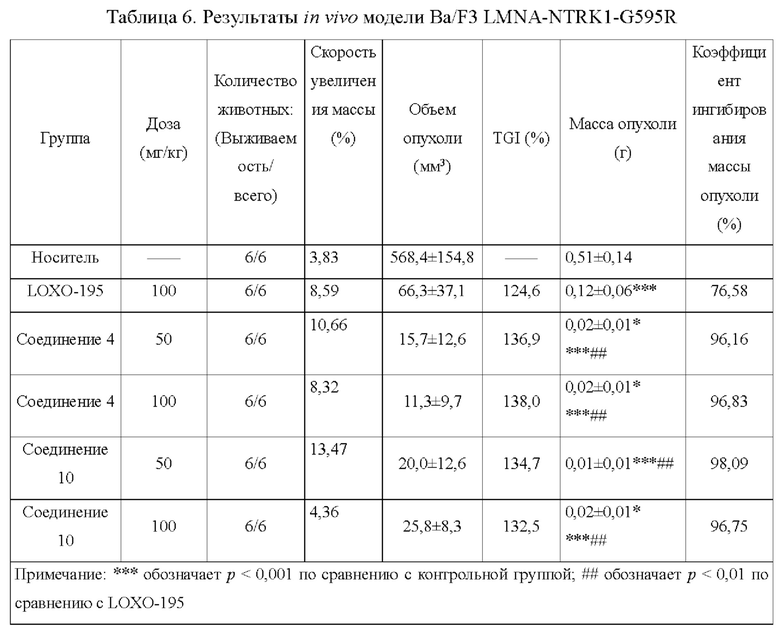
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ TRK | 2019 |
|
RU2803817C2 |
| СОЕДИНЕНИЕ АМИНОПИРАЗОЛОПИРИМИДИНА, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ТИРОЗИНКИНАЗНОГО РЕЦЕПТОРА НЕЙРОТРОФИЧЕСКОГО ФАКТОРА | 2017 |
|
RU2764523C2 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-a]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2009 |
|
RU2523544C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-а]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2014 |
|
RU2666367C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| МАКРОЦИКЛ, СОДЕРЖАЩИЙ АМИНОПИРАЗОЛ И ПИРИМИДИН, И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2779497C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRK | 2010 |
|
RU2584157C2 |
| ЗАМЕЩЕННЫЕ (2-АЗАБИЦИКЛО[3.1.0]ГЕКСАН-2-ИЛ)ПИРАЗОЛО[1.5-a]ПИРИМИДИНОВЫЕ И ИМИДАЗО[1.2-b]ПИРИДАЗИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗ TRK | 2019 |
|
RU2781618C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗ TRK | 2019 |
|
RU2778294C2 |
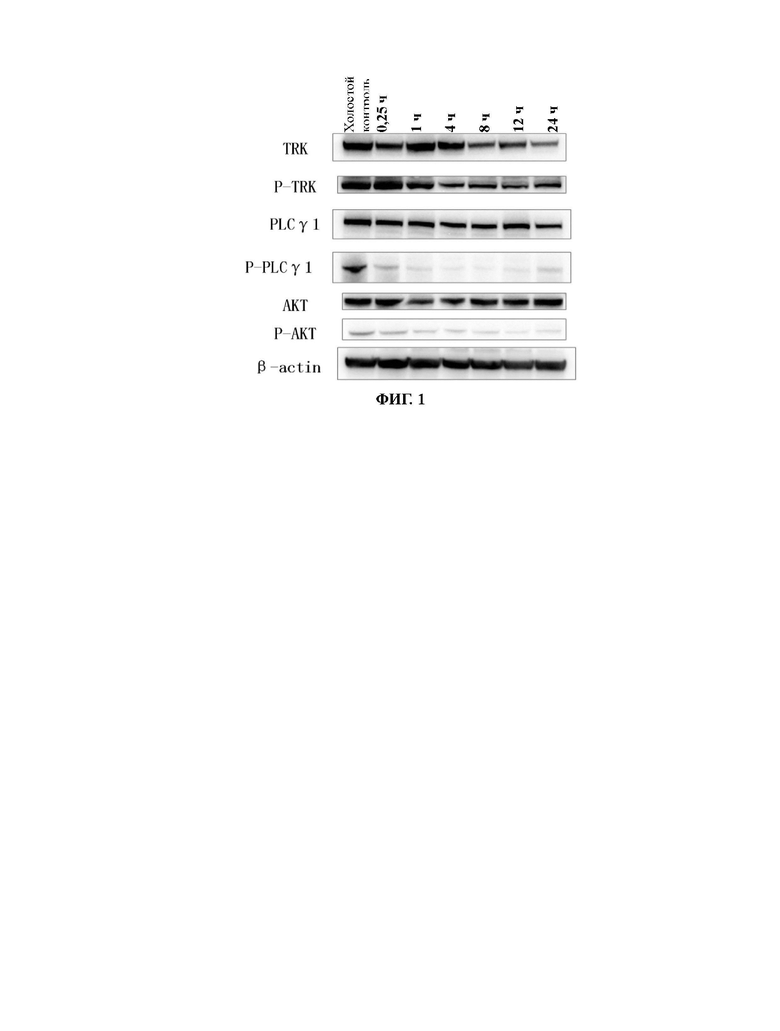
Изобретение относится к соединению формулы (I) или его таутомеру, стереоизомеру, оптическому изомеру или фармацевтически приемлемой соли, где X выбран из химической связи, -O- и -CH2-; Y представляет собой N, Y1 представляет собой C, Y2 представляет собой -CH-, Y3 представляет собой N, и Y4 представляет собой -CH-; X2 выбран из химической связи, -(CH2)p- и -NH-, где p равен 1;  означает, что химическая связь отсутствует или присутствует; R выбран из фенила или пиридинила, где каждый фенил или пиридинил является незамещенным или замещенным по меньшей мере одним заместителем, выбранным из R1; каждый R1, при его наличии, независимо выбран из водорода, галогена, -OH, амино, -NHC1-6 алкила, -N(C1-6 алкил)2, циано, C1-6 алкила, C1-6 алкокси и -SC1-6 алкила; R2 выбран из H, галогена, гидроксила, амино; R3 представляет собой H; кольцо A выбрано из гетероциклоалкила, содержащего от 4 до 6 кольцевых атомов, 6-8-членного гетеромостикового циклила, 8-10-членного гетероконденсированного циклила или 7-10-членного гетероспироциклила, причем гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетероспироциклиле независимо выбраны из O и N, и количество гетероатомов выбрано из 1, 2, 3 или 4; R4 находится в любом замещаемом положении на кольце A и независимо выбран из -H, -OH, галогена, -CN, замещенного или незамещенного C1-6 алкила, -(CH2)m-OH, -(CH2)m-COOH, -(CH2)m-CO-NH2, -CO-(CH2)m-NH2, -CO-CR 4aR4b-OH и -CO-R4b; R4a выбран из водорода и незамещенного или замещенного C1-4 алкила; R4b выбран из H, незамещенного или замещенного C1-6 алкила и незамещенного или замещенного C3-6 циклоалкила, заместитель для замещения независимо выбран из -OH, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3; n выбран из 1, 2, 3 и 4. Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении TRK, на основе указанных соединений. Технический результат – получены новые соединения, которые оказывают сильное ингибирующее действие на различные клетки, содержащие мутации TRK и опухоли, обладают хорошей безопасностью и могут найти применение в медицине в качестве лекарственных средств для лечения гиперпролиферативного заболевания. 4 н. и 18 з.п. ф-лы, 1 ил., 6 табл., 47 пр.
означает, что химическая связь отсутствует или присутствует; R выбран из фенила или пиридинила, где каждый фенил или пиридинил является незамещенным или замещенным по меньшей мере одним заместителем, выбранным из R1; каждый R1, при его наличии, независимо выбран из водорода, галогена, -OH, амино, -NHC1-6 алкила, -N(C1-6 алкил)2, циано, C1-6 алкила, C1-6 алкокси и -SC1-6 алкила; R2 выбран из H, галогена, гидроксила, амино; R3 представляет собой H; кольцо A выбрано из гетероциклоалкила, содержащего от 4 до 6 кольцевых атомов, 6-8-членного гетеромостикового циклила, 8-10-членного гетероконденсированного циклила или 7-10-членного гетероспироциклила, причем гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетероспироциклиле независимо выбраны из O и N, и количество гетероатомов выбрано из 1, 2, 3 или 4; R4 находится в любом замещаемом положении на кольце A и независимо выбран из -H, -OH, галогена, -CN, замещенного или незамещенного C1-6 алкила, -(CH2)m-OH, -(CH2)m-COOH, -(CH2)m-CO-NH2, -CO-(CH2)m-NH2, -CO-CR 4aR4b-OH и -CO-R4b; R4a выбран из водорода и незамещенного или замещенного C1-4 алкила; R4b выбран из H, незамещенного или замещенного C1-6 алкила и незамещенного или замещенного C3-6 циклоалкила, заместитель для замещения независимо выбран из -OH, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3; n выбран из 1, 2, 3 и 4. Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении TRK, на основе указанных соединений. Технический результат – получены новые соединения, которые оказывают сильное ингибирующее действие на различные клетки, содержащие мутации TRK и опухоли, обладают хорошей безопасностью и могут найти применение в медицине в качестве лекарственных средств для лечения гиперпролиферативного заболевания. 4 н. и 18 з.п. ф-лы, 1 ил., 6 табл., 47 пр.
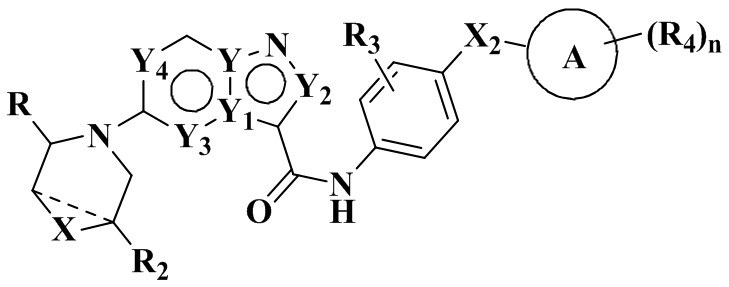 I
I
1. Соединение формулы (I) или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль
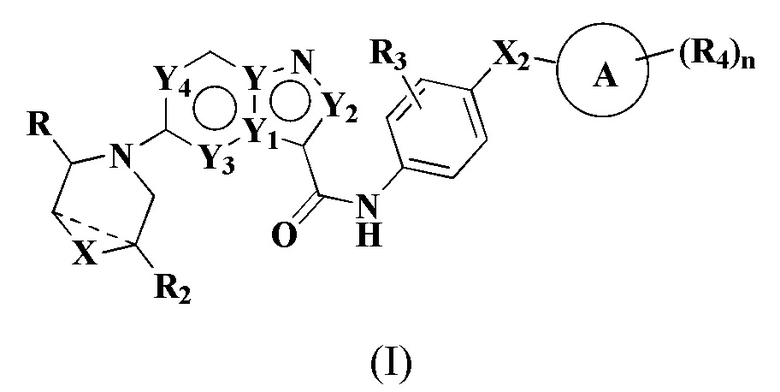 ,
,
где X выбран из химической связи, -O- и -CH2-;
Y представляет собой N, Y1 представляет собой C, Y2 представляет собой -CH-, Y3 представляет собой N, и Y4 представляет собой -CH-;
X2 выбран из химической связи, -(CH2)p- и -NH-, где p равен 1;
 означает, что химическая связь отсутствует или присутствует;
означает, что химическая связь отсутствует или присутствует;
R выбран из фенила или пиридинила, где каждый фенил или пиридинил является незамещенным или замещенным по меньшей мере одним заместителем, выбранным из R1;
каждый R1, при его наличии, независимо выбран из водорода, галогена, -OH, амино, -NHC1-6 алкила, -N(C1-6 алкил)2, циано, C1-6 алкила, C1-6 алкокси и -SC1-6 алкила; R2 выбран из: H, галогена, гидроксила, амино;
R3 представляет собой H;
кольцо A выбрано из гетероциклоалкила, содержащего от 4 до 6 кольцевых атомов, 6-8-членного гетеромостикового циклила, 8-10-членного гетероконденсированного циклила или 7-10-членного гетероспироциклила, причем гетероатомы в гетероциклоалкиле, гетеромостиковом циклиле, гетероконденсированном циклиле и гетероспироциклиле независимо выбраны из O и N, и количество гетероатомов выбрано из 1, 2, 3 или 4;
R4 находится в любом замещаемом положении на кольце A и независимо выбран из -H, -OH, галогена, -CN, замещенного или незамещенного C1-6 алкила, -(CH2)m-OH, -(CH2)m-COOH, -(CH2)m-CO-NH2, -CO-(CH2)m-NH2, -CO-CR 4aR4b-OH и -CO-R4b; R4a выбран из водорода и незамещенного или замещенного C1-4 алкила; R4b выбран из H, незамещенного или замещенного C1-6 алкила и незамещенного или замещенного C3-6 циклоалкила, заместитель для замещения независимо выбран из -OH, -NH2 и галогена, и количество заместителей выбрано из 1, 2 и 3;
n выбран из 1, 2, 3 и 4.
2. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по п. 1, отличающееся тем, что указанное соединение имеет структуру, представленную формулой (I-1) или формулой (I-2):
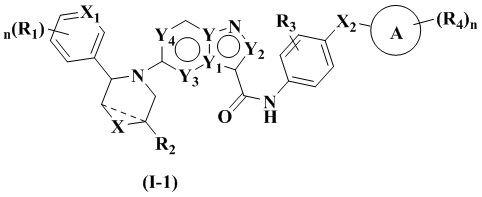 или
или 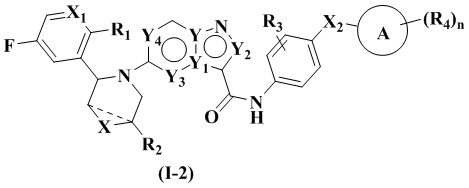 ,
,
где X1 выбран из -CH- и N.
3. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по п. 1 или 2, отличающееся тем, что указанное соединение имеет структуру, представленную формулой (I-A):
 .
.
4. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что указанное соединение имеет структуру формулы (I-B-1a) или формулы (I-C-1a):
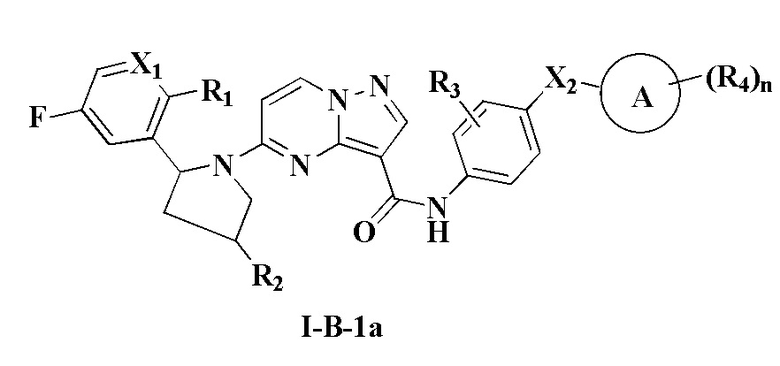 или
или 
5. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-4, где R1, при его наличии, каждый независимо выбран из водорода, галогена, -OH, амино, C1-3 алкила и C1-3 алкокси.
6. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-5, отличающееся тем, что R1, при его наличии, каждый независимо выбран из водорода, F, Cl, -OH, метила, этила, метокси и этокси.
7. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-6, отличающееся тем, что R2 выбран из H, F, OH.
8. Соединение или его таутомер, стереоизомер, оптический изомер или их фармацевтически приемлемая соль по любому из пп. 1-3, отличающееся тем, что указанное соединение имеет структуру, представленную формулой (I-D-1a), формулой (I-C-1a-1) или формулой (I-C-1a-2):
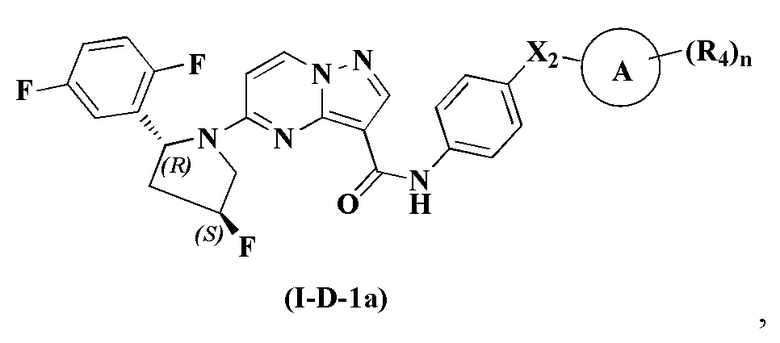
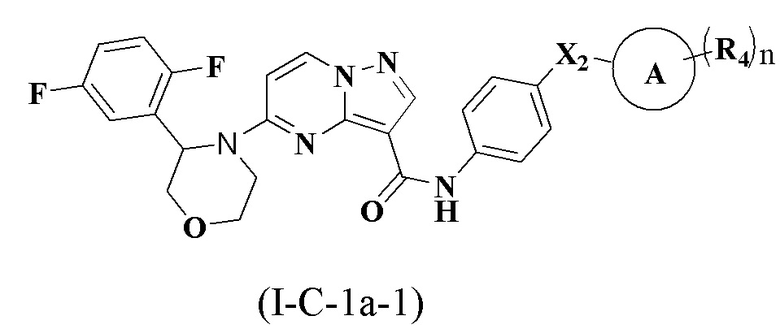 или
или
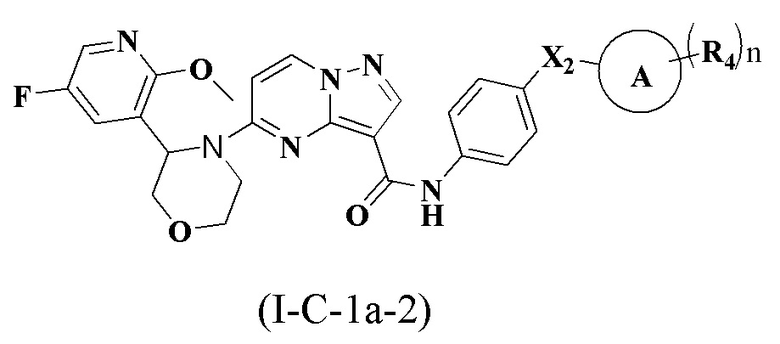 .
.
9. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-8, отличающееся тем, что кольцо A выбрано из следующих структур:
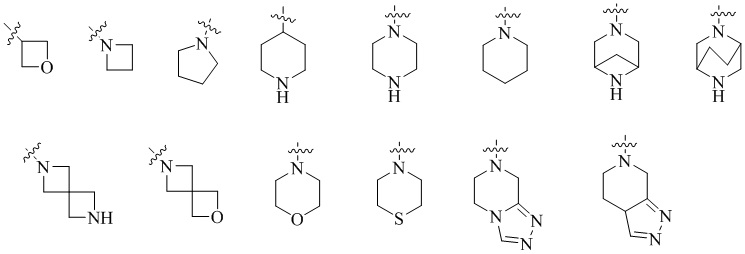 , , , , , , , , , , , , и .
, , , , , , , , , , , , и .
10. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-9, отличающееся тем, что кольцо A выбрано из следующих структур: и .
11. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-10, отличающееся тем, что R4 независимо выбран из -H, -OH, замещенного или незамещенного C1-3 алкила, -CO-CR4aR4b-OH и -CO-R4b.
12. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-11, отличающееся тем, что R4 независимо выбран из -CO-CR4aR4b-OH и -CO-R4b.
13. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-12, отличающееся тем, что R4a выбран из водорода и незамещенного или замещенного C1-4 алкила; R4b выбран из незамещенного или замещенного C1-6 алкила или незамещенного или замещенного C3-6 циклоалкила; заместитель для замещения независимо выбран из -OH и F, и число заместителей выбрано из 1, 2 и 3.
14. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-13, отличающееся тем, что X2 представляет собой связь.
15. Соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемая соль п. 1, отличающееся тем, что указанное соединение имеет следующие структуры:
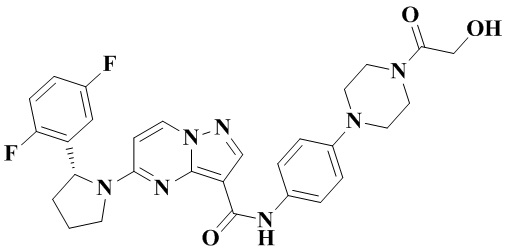
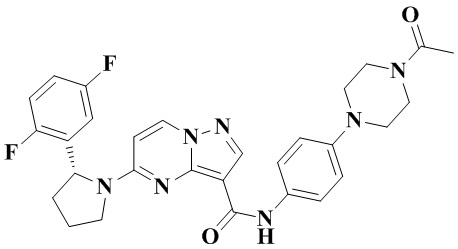
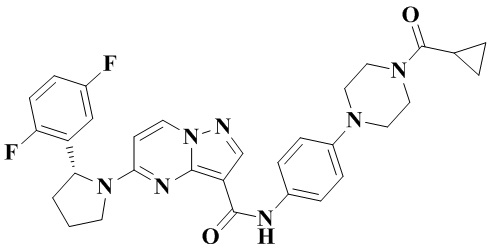
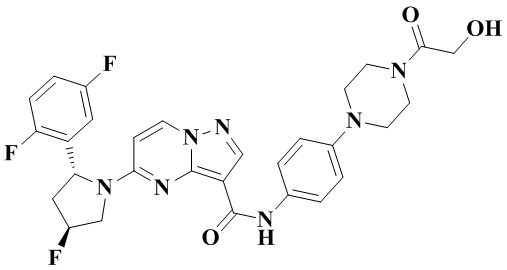

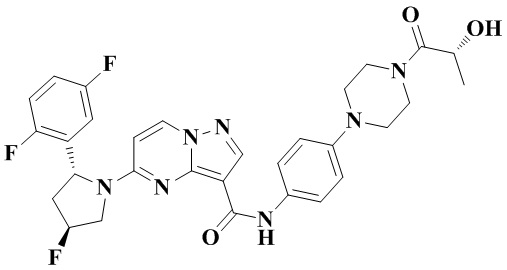
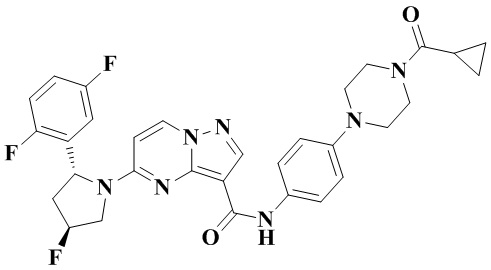
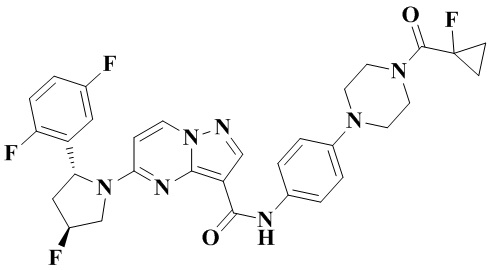
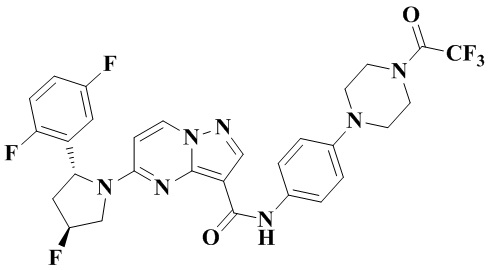
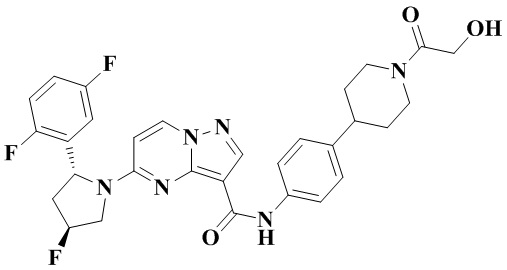
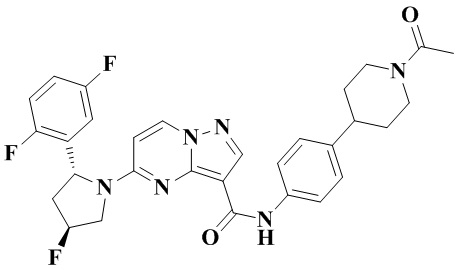
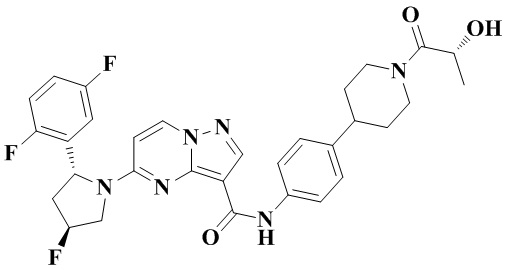
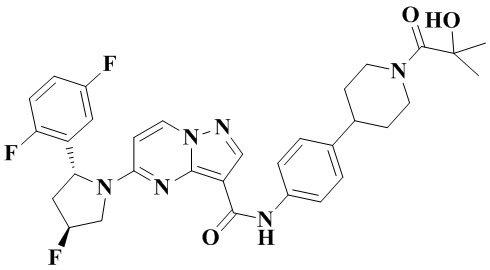
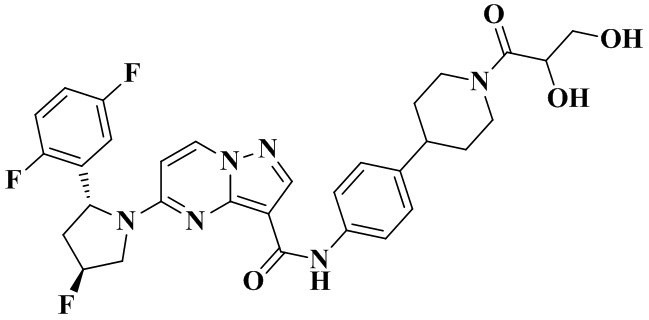

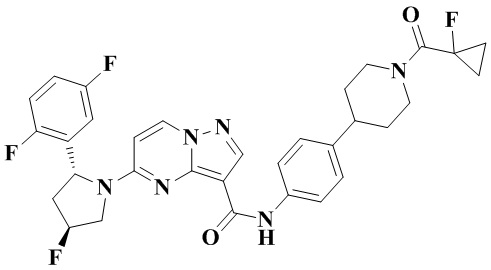
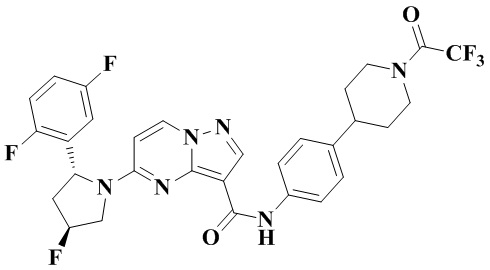
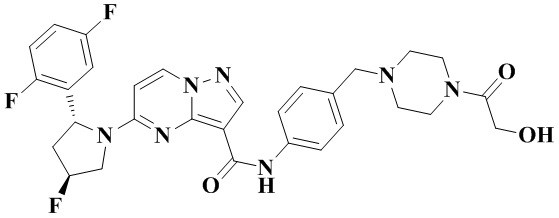
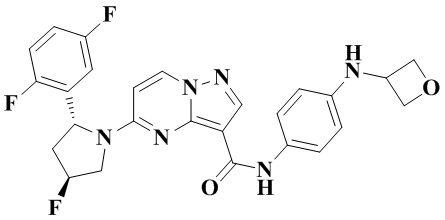
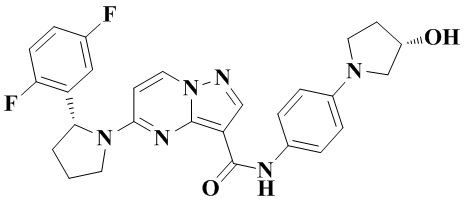
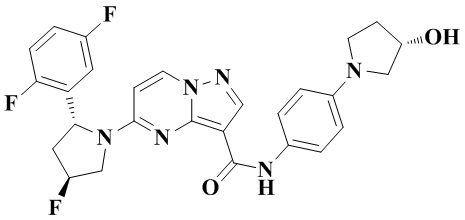
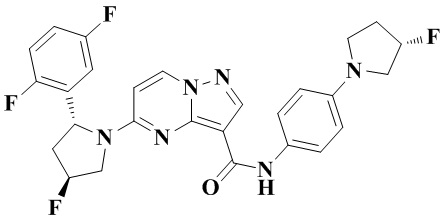
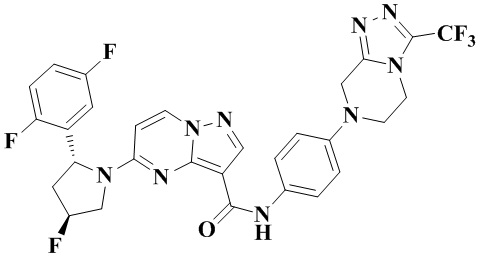
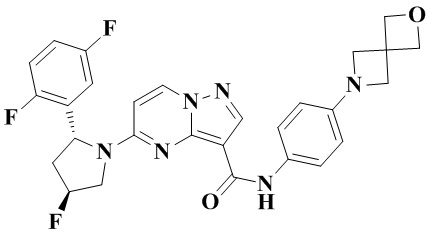
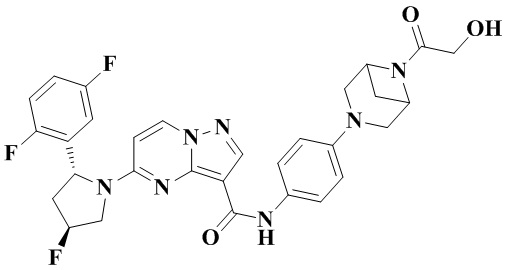
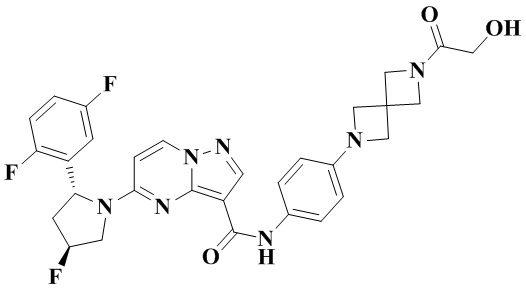
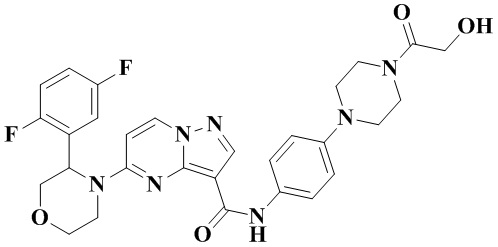
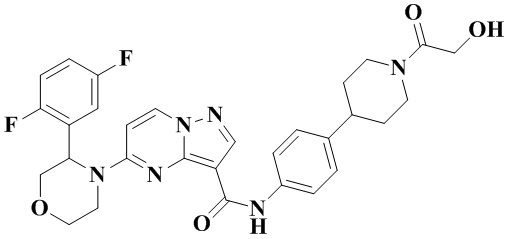
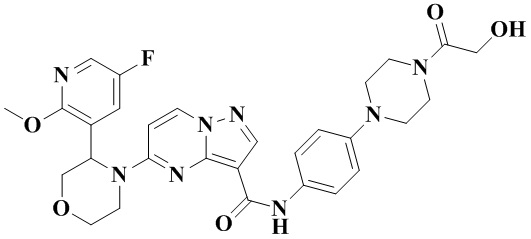
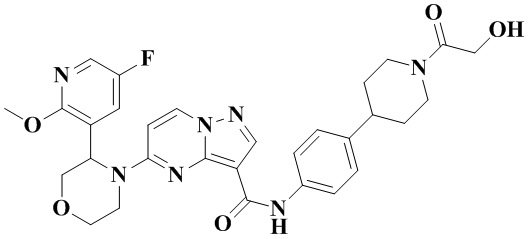
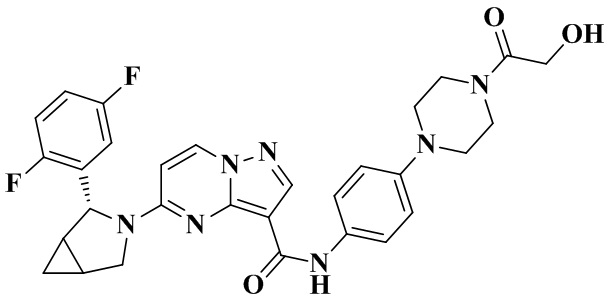
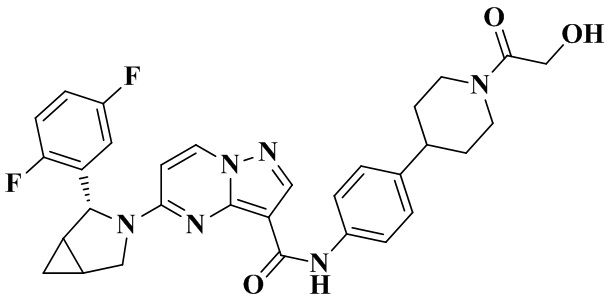
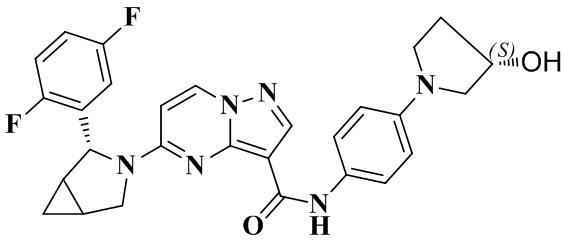

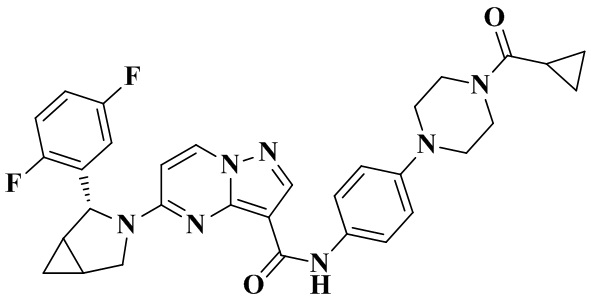
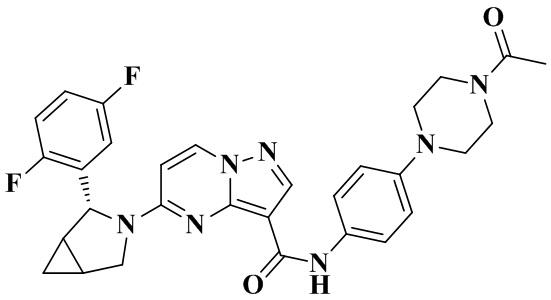
16. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении TRK, содержащая соединение или его таутомер, стереоизомер, оптический изомер или фармацевтически приемлемую соль по любому из пп. 1-15 в качестве единственного активного соединения и фармацевтически приемлемый вспомогательный материал.
17. Применение соединения или его таутомера, стереоизомера, оптического изомера или фармацевтически приемлемой соли по любому из пп. 1-15 или фармацевтической композиции по п. 16 для получения лекарственного средства для предотвращения и/или лечения заболевания, опосредованного TRK, ALK, ROS1 или их комбинацией.
18. Применение соединения или его таутомера, стереоизомера, оптического изомера или фармацевтически приемлемой соли по любому из пп. 1-15 или фармацевтической композиции по п. 16 для получения лекарственного средства для лечения гиперпролиферативного заболевания, где гиперпролиферативное заболевание представляет собой опухоль.
19. Применение по п. 18, отличающееся тем, что опухоль выбрана из солидной опухоли и гематологической опухоли.
20. Применение по п. 18, отличающееся тем, что опухоль выбрана из гемобластоза, рака легкого, рака молочной железы, рака яичников, рака предстательной железы, рака поджелудочной железы, глиомы головного мозга, колоректального рака, меланомы, рака головы и шеи, рака желчного пузыря, рака щитовидной железы, глиобластомы, рака желудка, нейробластомы или рака слюнных желез.
21. Применение по п. 20, отличающееся тем, что рак легкого представляет собой немелкоклеточный рак легкого.
22. Применение по любому из пп. 18-21, отличающееся тем, что указанная опухоль относится к слиянию, амплификации, перестройке, мутации или высокой экспрессии гена NTRK, ALK, ROS1 или их комбинации.
| WO 2013088257 A1, 20.06.2013 | |||
| CN 111039946 A, 21.04.2020 | |||
| WO 2012034095 A1, 15.03.2012 | |||
| WO 2019029629 A1, 14.02.2019 | |||
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-a]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2009 |
|
RU2523544C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRK | 2010 |
|
RU2584157C2 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| Искусственная приманка | 1930 |
|
SU25352A1 |
| ПРОИЗВОДНЫЕ ТИПА АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2640046C2 |

