Область техники, к которой относится изобретение
Изобретение относится к области химико-фармацевтической промышленности и касается нового способа получения софосбувира и новых фосфорамидатов. Предложенный способ получения характеризуется высоким выходом, химической и оптической чистотой полученной субстанции, а также простотой осуществления для реализации в промышленных масштабах. Полученный данными способами софосбувир может использоваться в качестве противовирусного средства в терапии вирусного гепатита С.
Уровень техники
Гепатит С представляет собой заболевание печени, вызываемое вирусом гепатита С (HCV): вирус может приводить к развитию как острого, так и хронического гепатита с разной степенью тяжести - от легкой болезни, длящейся несколько недель, до серьезной пожизненной болезни. В РФ, по разным оценкам, доля людей, которые встречались с вирусом гепатита С, составляет от 2 до 4 процентов от населения РФ, а хроническим гепатитом С больны около 3,5 миллиона человек. Примерно у 30% инфицированных вирус исчезает самопроизвольно без какого-либо лечения в течение шести месяцев после заражения. У остальных 70% инфицированных развивается хроническая инфекция, среди пациентов с хронической инфекцией HCV риск развития цирроза печени в течение следующих 20 лет составляет от 15% до 30%.
Заболевание гепатитом С вызывает РНК-содержащий вирус из семейства Flaviviridae, обнаруженный учеными в 1989 году. Вирус размножается в основном в клетках печени (гепатоцитах). Проникая в них, он использует внутриклеточный механизм репликации (самокопирования генома): каждая вирусная частица производит в день до 50 реплик, которые впоследствии выходят за пределы клетки-хозяина. Основная цель лечения гепатита С - эрадикация вируса (удаление вируса из организма). При невозможности эрадикации целями лечения могут быть: прекращение или замедление воспалительных процессов в печени, предотвращение перехода заболевания в цирроз или рак. Выбор лечения зависит от многих факторов: пола, возраста, вирусной нагрузки, состояния печени, характера течения заболевания (острый, хронический). Если риск развития цирроза высокий, то лечение должно быть назначено как можно скорее. Основным методом лечения гепатита С в настоящее время является противовирусная терапия (ПВТ) с помощью препаратов прямого действия (ПППД), основными мишенями которых являются вирусные белки, необходимые для его размножения (эффективность более 90-95%). ВОЗ рекомендует использовать пангенотипные комбинации ПППД для лечения лиц с хронической HCV-инфекцией в возрасте 18 лет и старше, в частности комбинации софосбувира с велпатасвиром и даклатасвиром в течение 12 недель, для лечения подростков от 12 до 17 лет рекомендуются комбинации софосбувира с ледипасвиром или рибавирином в течение 12 или 24 недель в зависимости от генотипа вируса. Таким образом, применение различных лекарственных комбинаций софосбувира является основой современного подхода к лечению вирусного гепатита С у взрослых и детей старше 12 лет.
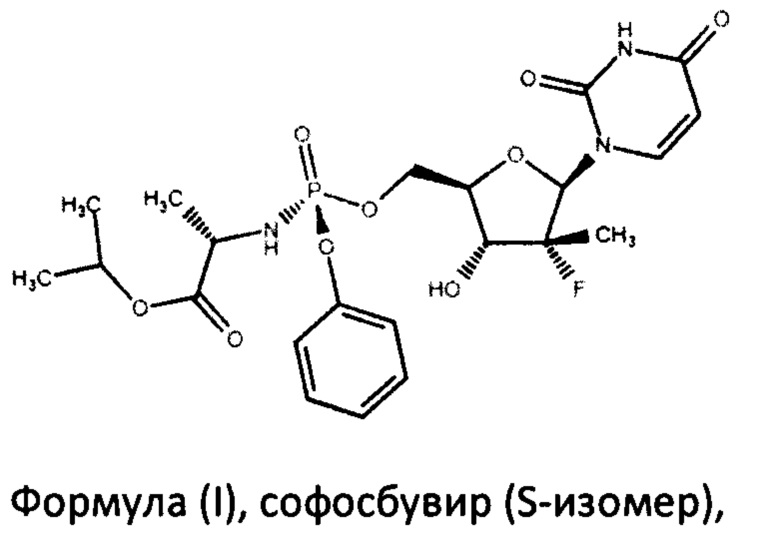
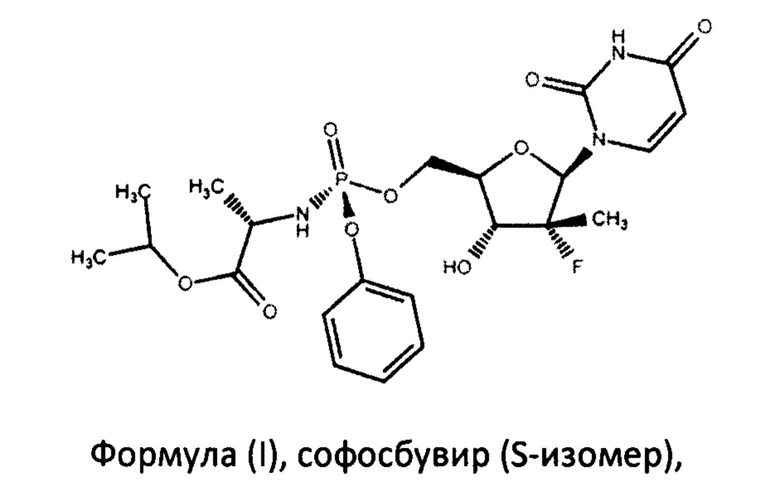
Софосбувир (изопропил-(2S)-2-[(S)-[[(2R,3R,4R,5R)-5-(2,4-диоксипиримидин-1-ил)-4-фторо-3-гидрокси-4-метил-тетрагидрофуран-2-ил]-метокси-фенокси-фосфорил]-амино]пропаноат) (см. WO 2008/121634, RU 2478104) является пангенотипическим ингибитором РНК-зависимой РНК-полимеразы NS5B HCV, необходимой для репликации вируса. Это нуклеотидное пролекарство, которое подвергается внутриклеточному метаболизму, в процессе которого формируется фармакологически активный аналог - 2'-дезокси-2'-α-фтор-β-С-метилуридин-5'-трифосфат (GS-461203). С помощью NS5B полимеразы GS-461203 может встраиваться в строящуюся цепочку РНK HCV и действовать как обрыватель цепи (Fung A., et. al., July 2014, Antimicrobial Agents and Chemotherapy. 58 (7): 3636-45).
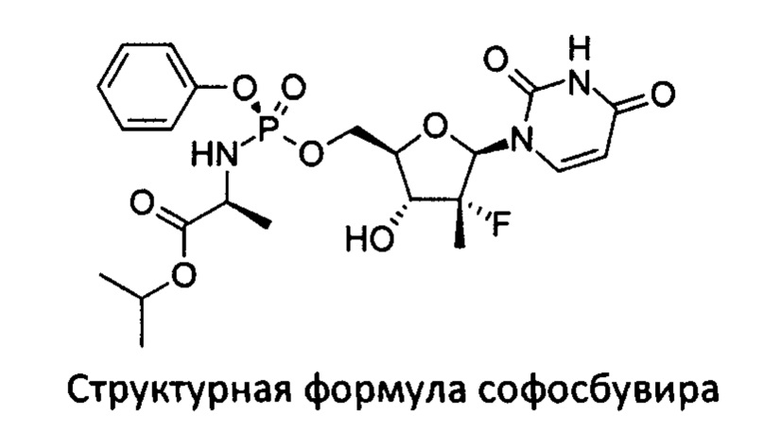
Единственный монопрепарат с МНН софосбувир зарегистрирован в России в марте 2016 года под торговым названием «Совальди» (номер регистрационного удостоверения ЛП-003527; дата выдачи 25.03.2016 года). Ввиду своей высокой эффективности в отношении многих генотипов HCV, сопряженной с низкой частотой возникновения побочных эффектов, препарат пользуется высоким спросом среди конечных потребителей, несмотря на высокую стоимость. Высокая стоимость софосбувира обусловлена, в частности, сложностью его получения. Таким образом, существует острая необходимость в разработке и выводе в гражданский оборот более дешевых воспроизведенных препаратов софосбувира, не уступающих оригинальному препарату по фармакологическим и технологическим свойствам, но обладающие дешевизной за счет более дешевого способа получения софосбувира.
Известно, что атом фосфора в софосбувире является центром хиральности, молекула вещества может существовать в виде R- и S-диастереомера.
I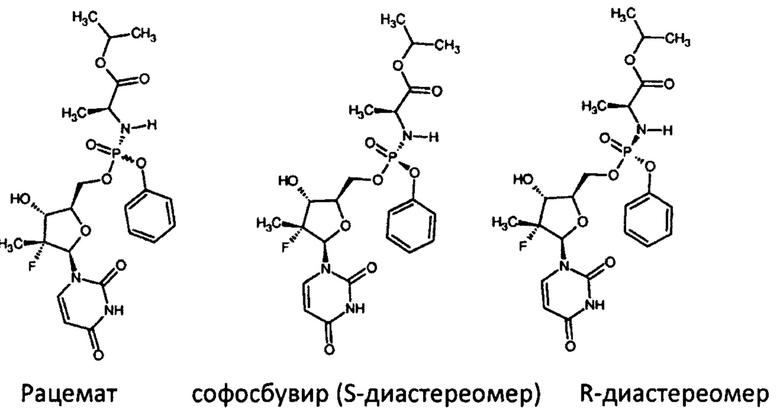
Химический синтез хирального соединения обычно дает рацемическую смесь соединения, в котором R- и S-стереоизомеры присутствуют в равных количествах. Многие биологически активные системы, однако, включают конкретные энантиомеры или диастереоизомеры хиральных соединений. Такие хиральные биологические системы могут реагировать по-разному с различными энантиомерами или диастереоизомерами фармацевтического хирального соединения. Введение пациенту рацемической смеси хирального фармацевтического соединения может означать, что только один стереоизомер соединения может принимать участие в желаемой терапевтической реакции. Синтез хирального фармацевтического соединения может предусматривать дополнительные и дорогостоящие стадии, выполняемые с рацемической смесью для обогащения готового продукта желаемым стереоизомером. Такие стадии предусматривают, например, хиральную хроматографию. Таким образом, неизбежны расходы или вследствие получения рацемической смеси, только часть которой является эффективно фармацевтически активной, или вследствие дополнительных стадий способа, проводимых для удаления, по меньшей мере, некоторой части нежелательного стереоизомера из рацемической смеси перед введением соединения пациенту, нуждающемуся в нем. Так как в случае софосбувира терапевтической эффективностью обладает S-диастереомер, существует потребность в обеспечении более экономически эффективного способа получения хирального соединения для терапевтического использования, где соединение содержит, по меньшей мере, обогащенную часть желаемого стереоизомера.
Впервые софосбувир и способ его получения фосфорилированием нуклеозида (В) с помощью хлорида изопропилаланилфосфорамидата формулы (А) в присутствии N-метилимидазола с получением рацемической смеси R- и S-диастереомеров и последующим хроматографическим разделением диастереомеров на хиральной колонке при помощи ВЭЖХ раскрывается в статье Michael J. Sofia et al., J. Med. Chem. 2010, 53, 7202-7218.
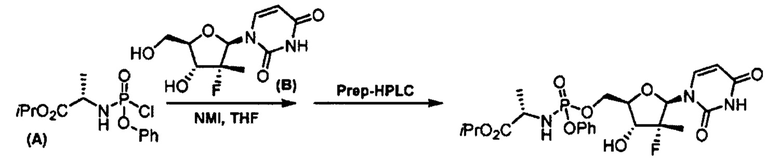
Метод является нестереоселективным, так как реагент (А) не является стабильным и может быть введен в реакцию только в виде смеси диастереомеров. Единственным способом выделить чистый S-диастереомер является хиральная хроматография, которая требует дорогостоящего оборудования, расходных материалов и больших временных затрат, таким образом вышеуказанный способ синтеза не применим для масштабного промышленного получения S-диастереомера софосбувира.
Другой вариант получения стереохимически чистого софосбувира раскрываются в международной заявке WO 2010/135569 (по заявке выданы патенты ЕА 28709, ЕА 28742, действующие на территории РФ). Патенты ЕА 28709 и ЕА 28742 раскрывают способ получения софосбувира с использованием ниже представленных фосфорамидатов:
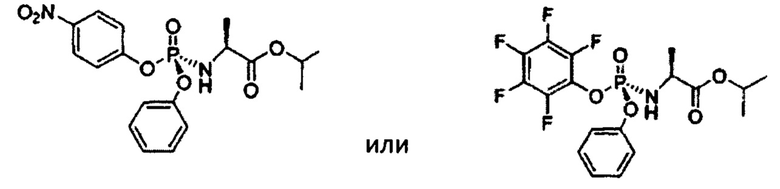
В начале получают смесь диастереомеров, далее S-диастереомер для промежуточных фосфорамидатов выделяют методом перекристаллизации, хроматографии или экстракции. Описанный в патентах подход обладает более высокой стереоселективностью в отличие от описанного в статье Michael J. Sofia et al., J. Med. Chem. 2010, 53, 7202-7218, однако, предполагает использование дорогостоящих и токсичных реактивов (в частности пентафторбензокси производного фосфорамидата). Также использование нитрозамещенных фосфорамидатов раскрывается в международной заявке WO 2012/012465 (по заявке выдан патент ЕА 25311, действующий на территории РФ). Выход кристаллического изопропилаланилфосфорамидата по способу, описанному в патентном документе, составляет 35%, содержание S-диастереомера в кристаллическом материале составляет 95%.
Способ синтеза вещества, содержащего по большей части S-диастереомер реакцией хлорида изопропилаланилфосфорамидата формулы А с нуклеозидом формулы М в присутствии солей металлов, выбранных из группы, включающей медь, железо, лантан и иттербий, раскрывает международная заявка WO 2014/076490 (по заявке выдан патент ЕА 28871, действующий на территории РФ). Способ, описанный в WO 2014/076490, обеспечивает выход 20-45% с содержанием S-диастереомера 98,5%. Однако использование дорогостоящих катализаторов делает данный способ не подходящим для промышленного применения.
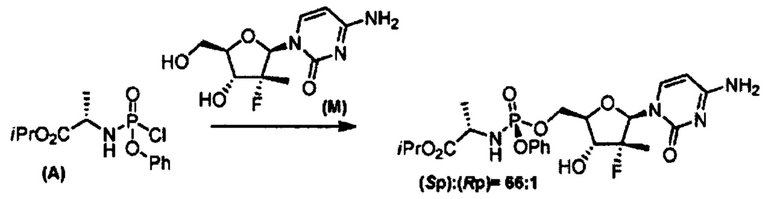
Международная заявка WO 2015/188782 раскрывает способ синтеза софосбувира, обладающий повышенной стереоселективностью, включающий стадии:
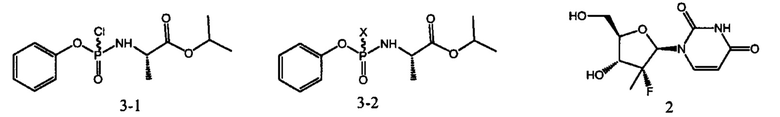
а) реакцию соединения 3-1 с нуклеофилом (азид натрия/калия, цианид натрия/калия, тиоцианат натрия/калия) в присутствии катализатора фазового переноса (тетра-бутиламмония бромида/иодида, 18-краунэфира) с образованием продукта 3-2, в котором X=SCN, CN, N3;
б) реакцию интермедиата 3-2 с нуклеозидом 2 в присутствии иона металла и органического основания с образованием софосбувира.
Согласно описанной в заявке WO 2015/188782 методике, теоретический выход софосбувира составляет от 34 до 40% в зависимости от значения X, количество образующегося S-диастереомера 87,5% требует дополнительной доочистки. Однако данный способ включает сложную для промышленного применения стадию образования интермедиата 3-2, требующую использования дорогостоящих реактивов.
Способ получения софосбувира с более высокой стереоселективностью по отношению к описанным в предыдущих документах, заключающийся в использовании производных изопропилаланилфосфорамидата, в которых уходящая группа представлена 1,3,5-триазин-1-илоксигруппой (1, 2 и 3), раскрывает международная заявка WO 2015/158317 (по заявке выдан патент EA29775, действующий на территории РФ). 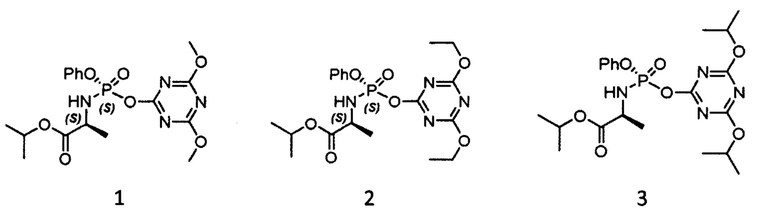
Выход софосбувира, полученного с использованием вышеуказанных фосфорамидатов, составляет около 60% (чистота софосбувира 99,1%, количество S-диастереомера более 99,9%). Однако, способ, раскрытый в заявке WO 2015/158317, является дорогостоящим и трудоемким, так как требует осуществления дополнительных стадий и использования специфических реактивов.
Международная заявка WO 2016/016447 раскрывает способ получения софосбувира, заключающийся в реакции замещенного изопропилаланилфосфорамидата формулы IIA 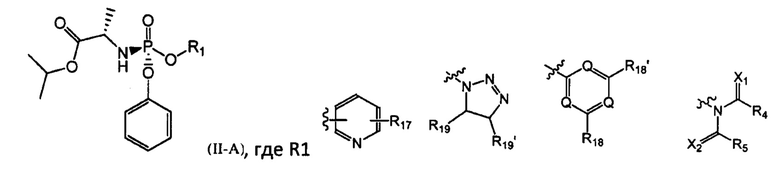
с нуклеозидом в присутствии одной или более кислот Льюиса. Согласно описанной в заявке WO 2016/016447 методике выход софосбувира составляет от 60 до 88% с содержанием S-диастереомера от 92 до 99,7%. Однако методика требует использования дорогостоящих реактивов (например, N-гидроксисукцинимид, 2-бензоксозолинон, 2-хлор-4,6-диметокси-1,3,5-триазин), что делает ее непригодной для промышленного применения.
Способ получения софосбувира через стадию получения интермедиата А раскрывается в международной заявке WO 2016/016865. Далее интермедиат А вступает в реакцию с нуклеозидом с образованием софосбувира.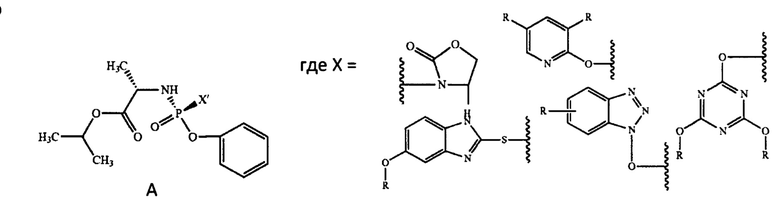
Методика, описанная в заявке WO 2016/016865, также не подходит для масштабного промышленного применения, так как требует применения реактивов с высокой стоимостью, например, таких как 5-4-фенил-2-оксазолидинон или 2-гидрокси-3-нитро-5-(трифторметил)пиридин.
Международная заявка WO 2016/151542 раскрывает способ получения софосбувира конденсацией замещенного изопропилаланилфосфорамидата (1) с нуклеозидом (2) в присутствии литийорганического, магнийорганического соединения, их смеси или амида натрия, калия, лития или магния с последующей перекристаллизацией полученной смеси диастериомеров из алкилацетата или хлорсодержащего растворителя или смеси одного из них с ацетонитрилом или алифатическими углеводородами согласно схеме:
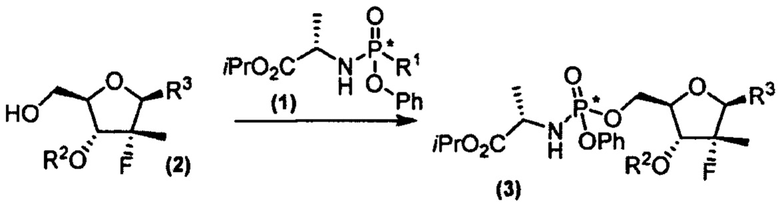
где R1 представляет собой хлор, бром, или сульфонат формулы -OSO2R8 (R8=С1-С4 алкил, арил, замещенный С1-С4 алкилом, галогеном или нитрогруппой арил) или камфоросульфонат, R2 - защитная группа, R3 - урацил. Выход софосбувира согласно данной методике составляет 97%, содержание S-диастереомера 92-93%. Способ, описанный в заявке WO 2016/151542, подразумевает осуществление дополнительных стадий синтеза, таких как введение и снятие защитной группы в молекуле нуклеозида, а также обогащение S-изомером конечной смеси продукта, что в итоге приводит к увеличению стоимости получаемого по данной методике софосбувира и снижению заявленного высокого выхода софосбувира.
Международная заявка WO 2016/196735 раскрывает способ получения софосбувира с повышенной стереоселективностью, включающий использование в качестве исходного соединения интермедиата 2, полученного следующим образом:
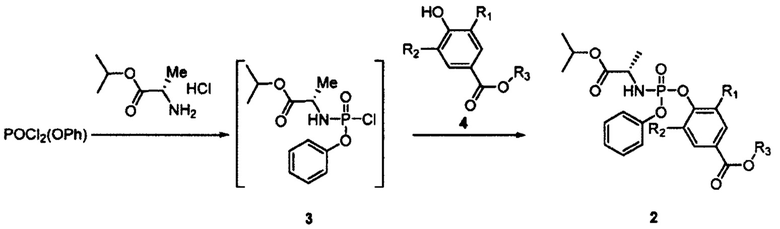
где R1 и R2 независимо друг то друга представляют собой водород или атом галогена, R3 представляет собой метил, этил, С1-С3 алкил, замещенный группой -NR4R5. К недостаткам описанного в WO 2016/196735 способа можно отнести сложную дополнительную стадию получения интермедиата 2, а также наличие двух длительных по времени стадий кристаллизации для получения софосбувира с содержанием S-диастереомера 99,85%.
Стереоселективный способ получения софосбувира, заключающийся в использовании в качестве исходных соединений изопропилаланилфосфорамидатов ниже представленных формул, раскрывает международная заявка WO 2017/029397.
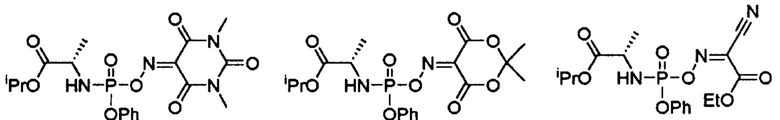
Выше представленная методика требует использования дорогостоящих реагентов (в частности Oxyma-В) для приготовления промежуточных соединений и растворителей (в частности, N-метил имидазол), а также, для того чтобы получить соотношение S-диастереомера к R-диастереомеру более 15:1 требуется использование дорогостоящего основания (turbo-Grignard). Вышеперечисленные недостатки делают методику, раскрытую в заявке WO 2017/029397, не применимой для промышленного применения.
Способ получения стереохимически чистого софосбувира, заключающийся в нагревании исходного замещенного изопропилаланилфосфорамидата в растворителе, в котором ни исходное вещество, ни конечный продукт не растворяются (парафиновое масло) и последующей реакций выделенного чистого S-изопропилаланилфосфорамидата с 2'-дезокси-2'-фтор-2'-С-метилуридином, раскрывает международная заявка WO 2018/025195. В качестве промежуточного продукта для синтеза используется фосформадат, содержащий пентафтороксид в качестве уходящего заместителя, данный способ защищен патентами ЕА 28709 и ЕА 28742 на территории РФ. Кроме того, использование парафинового масла может быть затруднительно с технологической точки зрения, так как оно представляет собой вязкую жидкость, что потребует наличия специфического оборудования.
Способ получения S-диастереомера изопропилаланилфосфорамидата, содержащего пентафторфенокси группу в качестве уходящей из R-изомера в присутствии нуклеофильного основания, выбранного из группы, включающей натрия метоксид, калия/натрия трет-бутоксид, натрия/калия ацетат, натрия/калия пропионат или натрия пентафторфенолат, раскрывает международная заявка WO 2018/033139. Процесс разделения диастереомеров в указанной публикации происходит из рацемической смеси состава 1:1 с использованием дополнительных реактивов, что делает его более дорогостоящим по сравнению с теми, где изомеры разделяются, например, перекристаллизацией.
Несмотря на большой объем опубликованной информации, касающейся способов получения софосбувира, на практике нашли свое применение только способы получения софосбувира с использованием ниже представленных фосфорамидатов (ЕА 28709 и ЕА 28742):
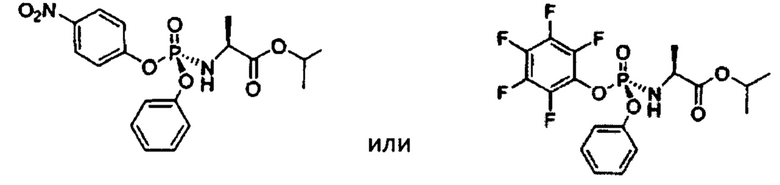
Огромным экономическим преимуществом вышеуказанных фосфорамидатов заключается в том, что S-изомер фосфорамидатов, необходимый для получения софосбувира, достаточно легко выкристаллизовывается из рацемической смеси изомеров фосфорамидатов.
Таким образом, перед авторами настоящего изобретения стояла задача в расширении арсенала способов получения софосбувира с использованием фосфорамидатов за счет получения новых фосфорамидатов, для которых из рацемической смеси изомеров фосфорамидатов выкристаллизовывается S-изомер, необходимый для получения софосбувира. Желательно, чтобы новые фосфорамидаты обеспечивали более высокие выходы софосбувира, большую экономическую выгоду и большую экологичность способа получения софосбувира.
Раскрытие изобретения
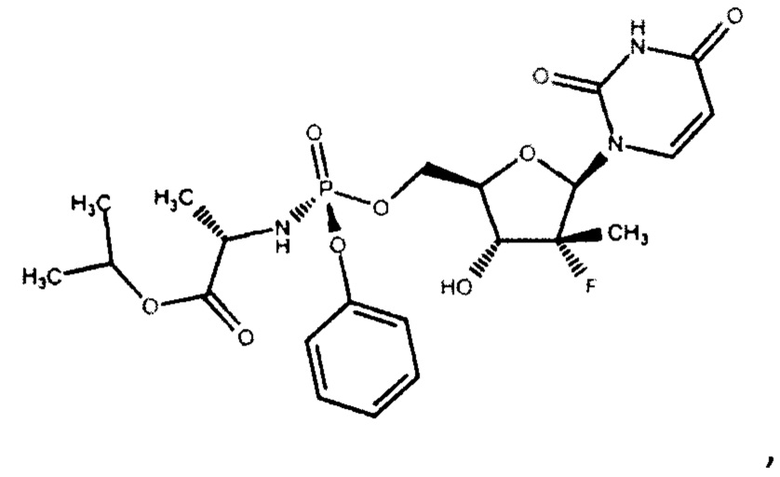
Поставленная задача решается тем, что авторами разработан способ синтеза софосбувира, по существу чистого от R-диастереомера софосбувира, формулы (I)
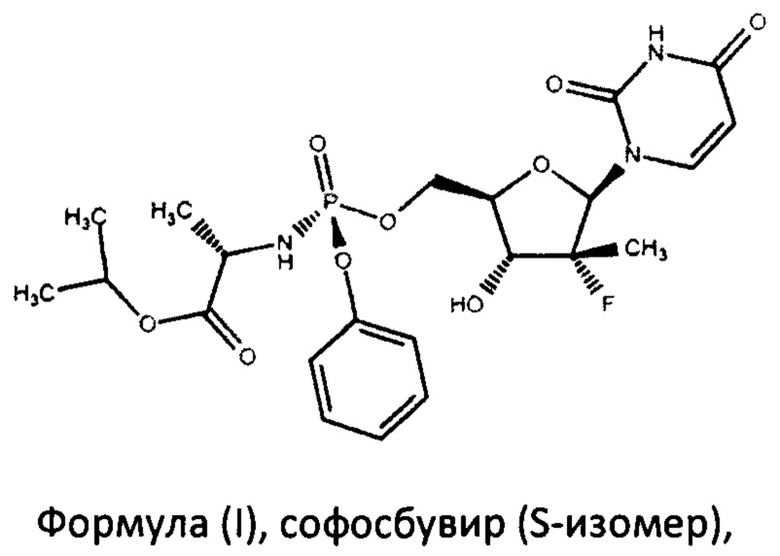
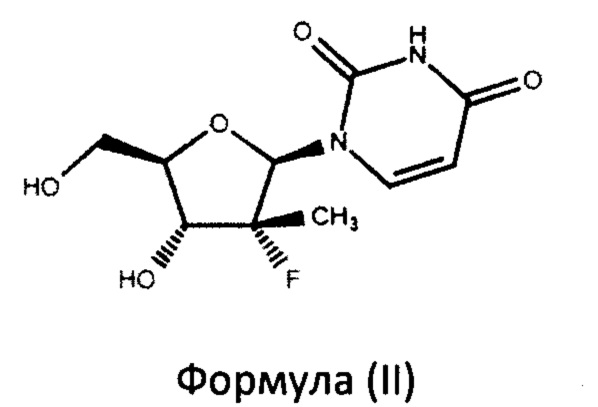
включающий взаимодействие в присутствии основания нуклеозида формулы (II) 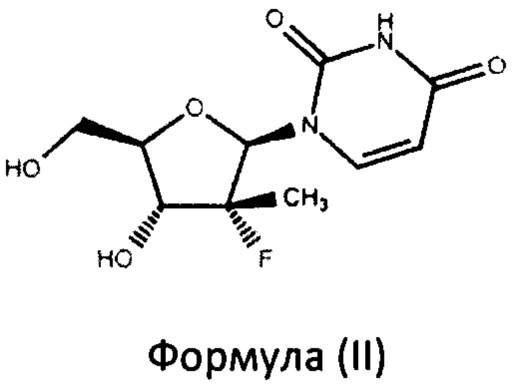
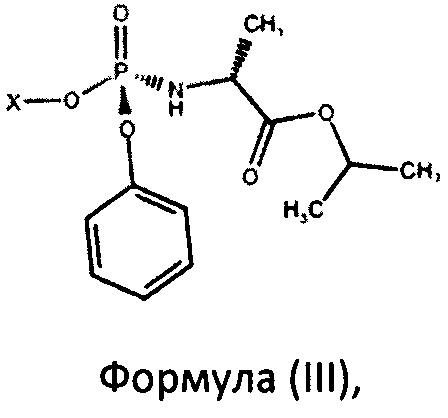
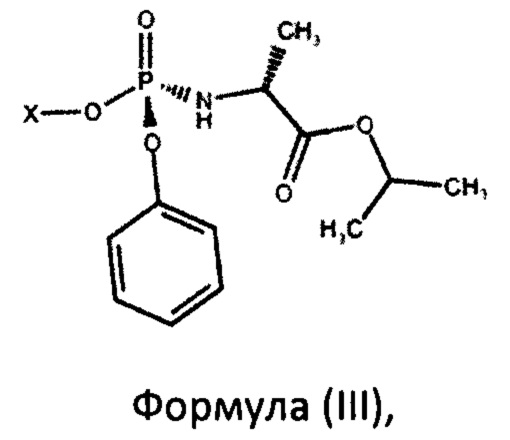
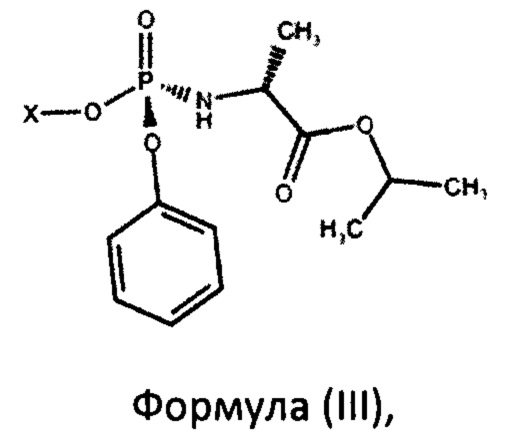
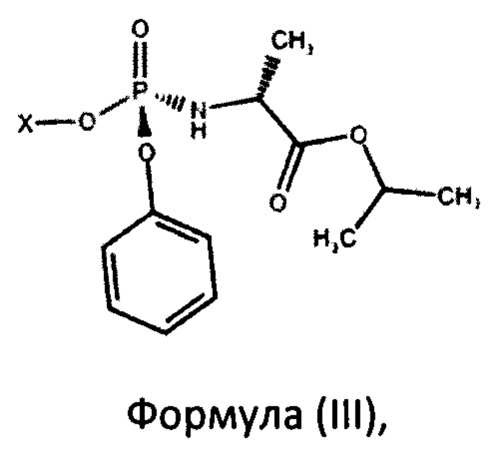
с фосфорамидатом формулы (III)
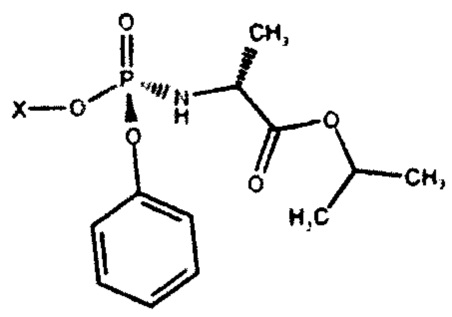
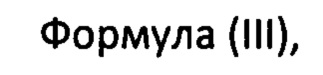
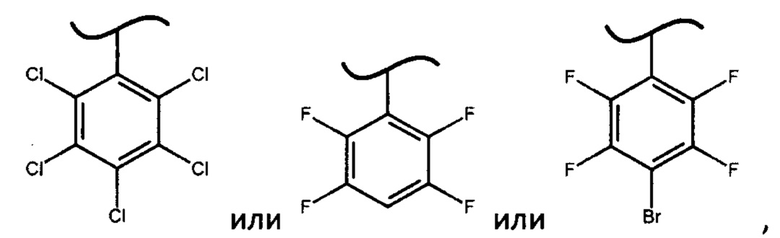
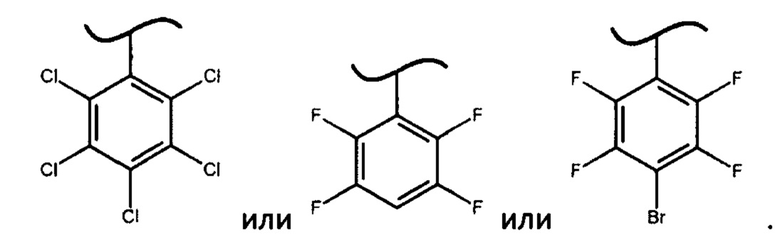
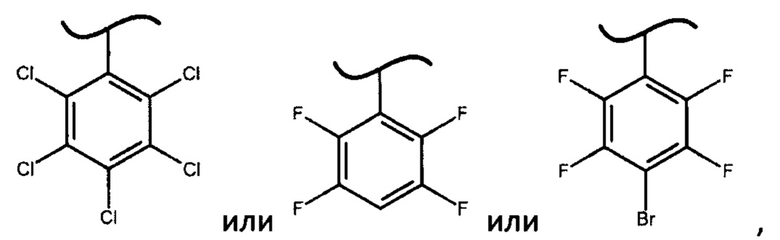
где X представляет собой
с получением софосбувира.
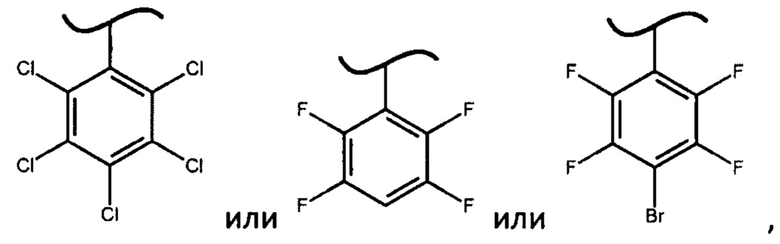
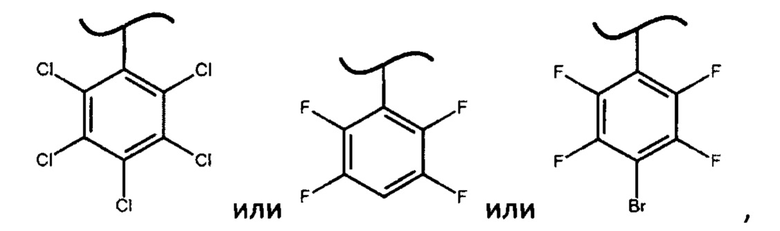
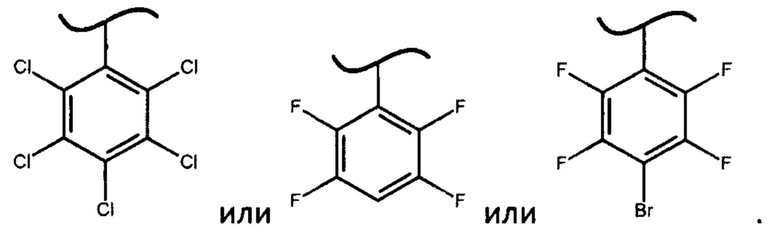
В предпочтительном варианте осуществления изобретения X представляет собой:
В более предпочтительном варианте осуществления изобретения основание выбирают из трет-бутилмагния хлорида, N-метилимидазола, гидрида натрия, гексаметилдисилазана натрия, гексаметилдисилазана лития или диизопропиламида лития.
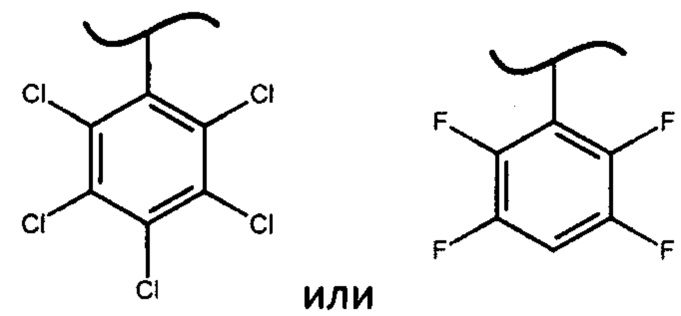
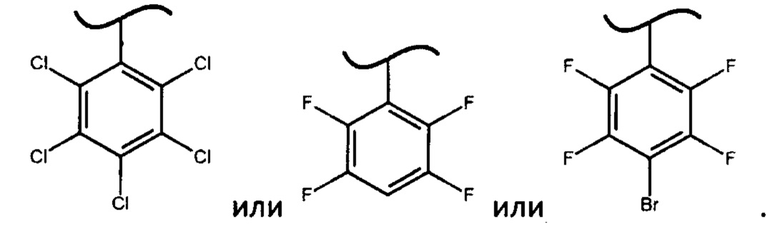
В наиболее предпочтительном варианте изобретения фосфорамидат формулы (III) может быть получен взаимодействием изопропилового эфира L-аланина или его фармацевтически приемлемой соли с дихлорфенилфосфатом и фенолом формулы Х-ОН, где X представляет собой
с получением фосфорамидата формулы (IV) в виде смеси диастереомеров:
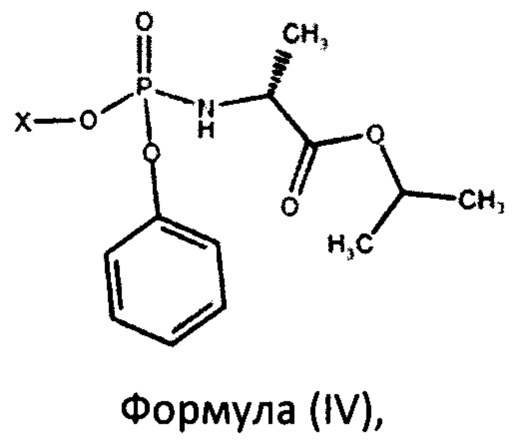
с последующей перекристаллизацией из подходящего растворителя с получением фосфорамидата формулы (III), который представляет собой S-изомер.
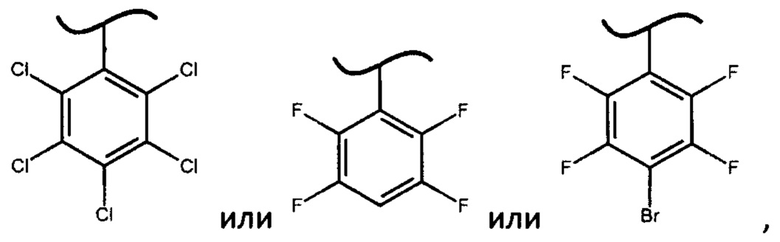
Еще одним вариантом изобретения являются фосфорамидаты формулы (III)
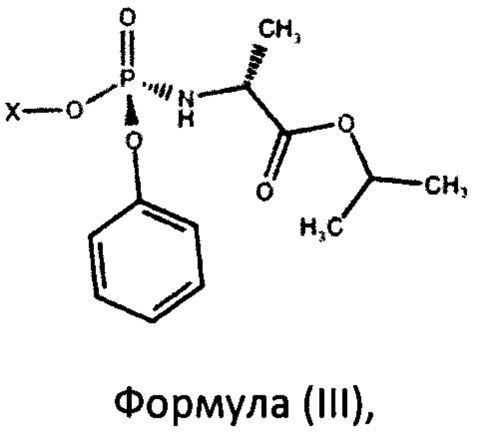
где X представляет собой 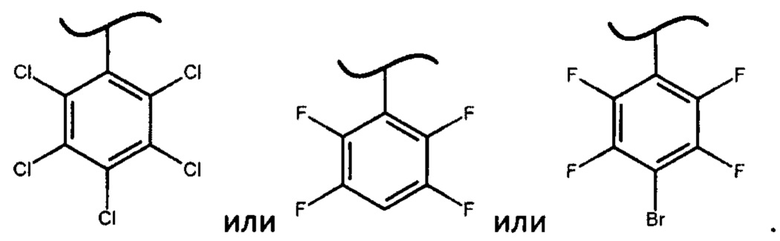
Было неожиданно обнаружено, что указанные фосфорамидаты могут быть легко получены при перекристаллизации из диастереомерной смеси по атому фосфору фосфорамидатов.
Осуществление изобретения
В настоящем изобретении предложен способ получения софосбувира, по существу чистого от R-диастереомера софосбувира, формулы (I)
включающий взаимодействие в присутствии основания нуклеозида формулы (II) 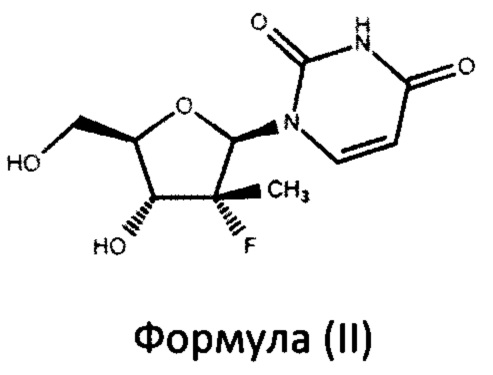
с фосфорамидатом формулы (III)
где X представляет собой
с получением софосбувира.
Реакцию взаимодействия нуклеозида формулы (II) с фосфорамидатом формулы (III) в присутствии основания проводят по известным методикам, например, раскрытым в ЕА 28709, ЕА 29742, WO 2016/016447, WO 2015/158317, которые включены в объем настоящего изобретения.
При этом используют в реакции взаимодействия сильные основания, предпочтительно выбранные из трет-бутилмагния хлорида, N-метилимидазола, гидрида натрия, гексаметилдисилазана натрия, гексаметилдисилазана лития или диизопропиламида лития.
В данном изобретении для ассиметрических хиральных центров используется R/S-система номенклатуры энантиомеров. Так для хиральный центр, относящийся к атому фосфора С или Р, маркируют R или S согласно системе, в которой каждый заместитель на атоме Р определяется старшинством на основе атомного номера согласно правилам старшинства Кана-Ингольда-Прелога (КИП) (J. March, "Advanced Organic Chemistry", John Wiley & Sons (2007)). Правила КИП определяют наименьшее старшинство для конкретного заместителя на хиральном центре С или Р, имеющего наименьший атомный номер.
Например, в случае софосбувира или фосфорамидата для Р хирального центра этот заместитель представляет собой N. Р-центр затем ориентируется так, что N-заместитель направлен от наблюдателя. Атомы или следующие ближайшие атомы, если есть, к трем атомам О, непосредственно связанным с Р, затем рассматриваются согласно правилам КИП. Если атомный номер этих атомов уменьшается по часовой стрелке, энантиомер маркируется как R. Если атомный номер этих атомов уменьшается в направлении против часовой стрелки, энантиомер маркируется как S.
Термин «по существу чистого от R-диастереомера софосбувира» обозначает, что софосбувир, имеющий формулу
не содержит R-диастереоизомер по атому фосфора (Р), либо его содержание не превышает 0,5% по данным ВЭЖХ. Столь низкий процент содержания R-диастереоизомера связан с тем, что в реакцию с нуклеозидом формулы (II) вводят полностью чистый S-изомер фосфорамидата формулы (III).
В рамках настоящего изобретения авторами было неожиданно установлено, что фосфорамидаты формулы (III) в виде S-изомера
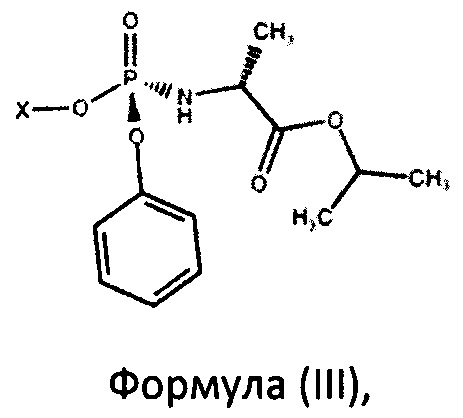
где X представляет собой
могут быть легко получены посредством перекристаллизации из смеси диастереоизомеров фосфорамидатов формулы (IV):
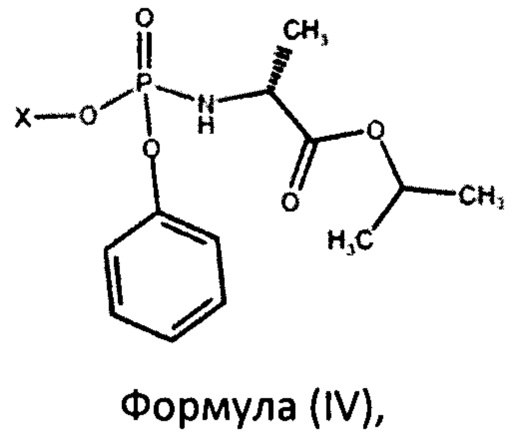
где X такое, как определено выше.
Перекристаллизацию смеси диастереоизомеров фосфорамидатов формулы (IV) можно проводить в любых подходящих растворителях. Предпочтительно использовать в качестве растворителя для перекристаллизации углеводороды и эфиры (также известны как простые эфиры), в которых исходное соединение (IV) растворимо или умеренно растворимо. В качестве углеводородов могут использоваться любые жидкие алифатические и ароматические углеводороды, предпочтительно выбранные из пентана, гексана, гептана, октана, петролейного эфира, бензола, толуола, этилбензола, кумол, ксилолы (о-, м-, п-) или их изомеров (например, 2-метилпентан, 2,2-диметилгексан и другие) и смесей. Наиболее предпочтительно использовать алифатические углеводороды, выбранные из пентана, гексана, гептана, октана, петролейного эфира или их изомеров (например, 2-метилпентан, 2,2-диметилгексан и другие) и смесей. В качестве эфиров могут использоваться любые жидкие эфиры, предпочтительно, выбранные из диэтилового эфира, диизопропилового эфира, метилтретбутилового эфира, этилацетата.
При этом смесь диастереоизомеров фосфорамидатов формулы (IV) по изобретению можно получить взаимодействием изопропилового эфира L-аланина или его фармацевтически приемлемой соли с дихлорфенилфосфатом и фенолом формулы Х-ОН, где X представляет собой
Помимо используемых в настоящем изобретении фосфорамидатов формулы (IV), в которых X представляет собой
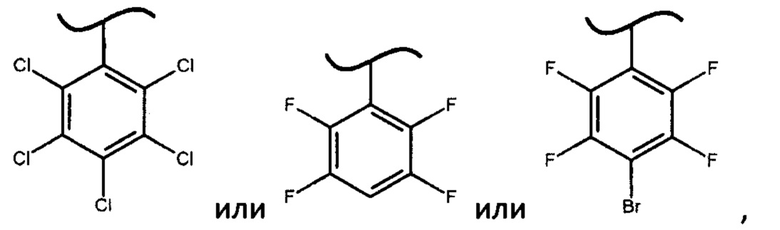
авторами настоящего изобретения были предприняты попытки получить дополнительно более 30 фосфорамидатов формулы (IV), где X представляет собой различные фенилы и дифенилы с акцепторными заместителями, отличными от вышеупомянутых значений X, однако они либо не образуют фосфорамидаты формулы (IV) (например, если X представляет собой 2,6-динитро-4-трифторметилфенил или 2,3,5,6-тетрафтор-2*,3*,4*,5*,6*-пентафтордифенил), либо не позволяют выкристаллизовать фосфорамидат формулы (III) в виде S-изомера (например, если X представляет собой 2,4,6-трибромфенил, 2,4-дибромфенил или 4-цианофенил), либо выкристаллизовывался R-изомер фосфорамидата (например, если X представляет собой 2,4,6-трихлорфенил или 3,5-дифтор-2,6-дихлорфенил).
Неожиданный факт того, что авторам удалось получить диастереомерные фосфорамидаты формулы (IV) и из них при помощи простой перекристаллизации S-изомеры фосфорамидатов формулы (III) только когда X представляет собой
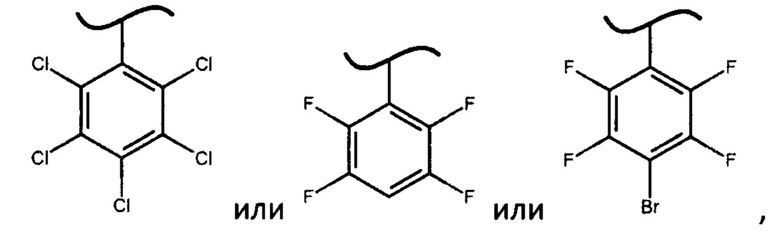
определяет новизну и изобретательский уровень настоящего изобретения. Более того, использование фосфорамидатов формулы (III), в которых X представляет собой
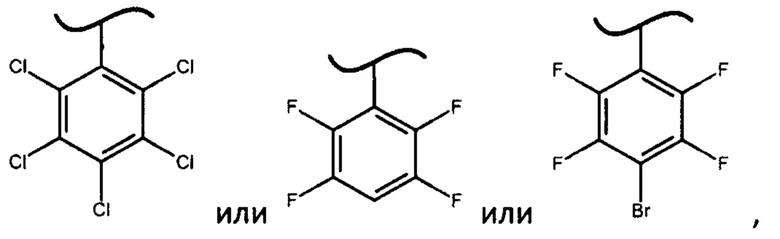
позволило реализовать способ получения софосбувира с выходами и оптической чистотой не меньшими, чем для известных в уровне техники способов (ЕА 28709 и ЕА 28742).
Полученный согласно заявленному способу софосбувир выделяют общепринятыми методами, например, отделяют от жидкой фазы при помощи фильтрования, при необходимости, сушат, например, в сушильном или вакуумном шкафу, предпочтительно, под вакуумом. Выделенный софосбувир при необходимости измельчают и используют для получения готовой лекарственной формы (ГЛФ), например, в виде таблетки или капсулы.
При получении ГЛФ в виде таблеток или желатиновых капсул софосбувир, полученный по настоящему изобретению, смешивают с фармацевтическими носителями, такими как желатины, крахмалы, лактоза, микрокристаллическая целлюлоза, производные винилпирролидона, стеариновая кислота и стеараты, тальк, кремния диоксид и другими. Кроме того, ГЛФ могут иметь пролонгированное или замедленное действие и, таким образом, непрерывно высвобождать определенное количество активного ингредиента.
Софосбувир, полученный по настоящему изобретению, а также содержащие его ГЛФ, могут применяться в способах лечения пациентов, инфицированных вирусом гепатита С генотипа 1 (включая 1а и 1b), генотипа 2, генотипа 3, генотипа 4, генотипа 5 и генотипа 6, предпочтительно в комбинации с другими средствами, включающими, но не ограниченными следующими: интерфероны, рибавирин, ингибиторы NS5A, ингибиторы NS5B, ингибиторы протеазы, ингибиторы альфа-глюкозидазы, гепатопротекторы, агонисты TLR-7, ингибиторы циклофиллина, ингибиторы IRES HCV и другие агенты против HCV.
Еще одним вариантом изобретения являются фосфорамидаты формулы (III)
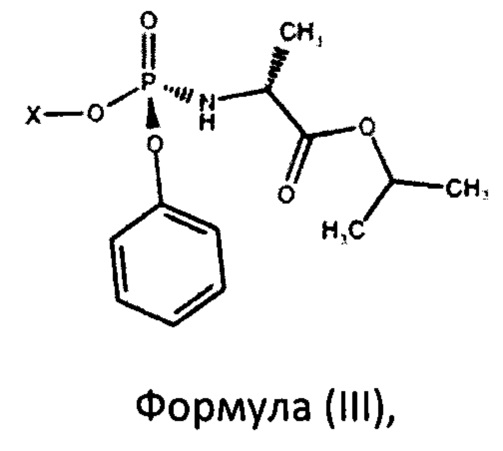
где X представляет собой
ПРИМЕРЫ
Представленные ниже примеры иллюстрируют (без ограничения объема притязаний) наиболее предпочтительные варианты осуществления изобретения, а также подтверждают возможность получения софосбувира и фосфорамидатов и достижения для них указанных технических результатов.
Пример 1. Получение диастереомерного фосфорамидата формулы (IV), в котором X представляет собой пентахлорфенил.
К раствору гидрохлорида изопропилового эфира L-аланина (6,705 г) в хлористом метилене (80.0 мл) при охлаждении (от -15 до -10°С) добавляли фенилдихлорфосфат (6,0 мл) и медленно прикапывали раствор N-метилимидазола (24,6 мл) в хлористом метилене (80,0 мл). Реакционную массу перемешивали (на магнитной мешалке, скорость 300 об/мин) при -15°С в течение 1,0 часа и прибавляли пентахлорфенол (8,2 г). Перемешивали 3 часа при охлаждении (от -15 до -10°С) и 16 часов при 23-25°С. Контролировали по ТСХ. Элюент CHCl3/СН3ОН=10/1.
После завершения реакции реакционную массу фильтровали на воронке Шотта, промывали водой (2×50 мл), IN HCl (50 мл), раствором NaHCO3 (2×50 мл) и рассолом (50 мл) и сушили над Na2SO4.
Получили 18,0 г (86,0%) белой кристаллической массы.
MS: 537 (М+1).
Пример 2. Получение диастереомерного фосфорамидата формулы (IV), в котором X представляет собой 2,3,5,6-тетрафторфенил.
К раствору гидрохлорида изопропилового эфира L-аланина (6,705 г) в хлористом метилене (80,0 мл) при охлаждении (от -15 до -10°С) добавляли фенилдихлорфосфат (6,0 мл) и медленно прикапывали раствор N-метилимидазола (24,6 мл) в хлористом метилене (80,0 мл). Реакционную массу перемешивали (на магнитной мешалке, скорость 300 об/мин) при -15°С в течение 1,0 часа и прибавляли 2,3,5,6-тетрафторфенол (7,9 г). Перемешивали 3 часа при охлаждении (от -15 до -10°С) и 16,0 часов при 23-25°С. Контролировали по ТСХ. Элюент CHCl3/СН3ОН=10/1.
После завершения реакции реакционную массу упаривали на ротационном испарителе. Остаток растворяли в этилацетате (50 мл), промывали водой (2×50 мл), IN HCl (50 мл), раствором NaHCO3 (2×50 мл) и рассолом (50 мл). Органический слой сушили над Na2SO4. Упаривали на ротационном испарителе.
Получили 13,56 г (81,0%) светлого масла.
MS: 436 (М+1).
Пример 3. Получение диастереомерного фосфорамидата формулы (IV), в котором X представляет собой 4-бром-2,3,5,6-тетрафторфенил.
К раствору гидрохлорида изопропилового эфира L-аланина (6,705 г) в хлористом метилене (80,0 мл) при охлаждении (от -15 до -10°С) добавляли фенилдихлорфосфат (6,0 мл) и медленно прикапывали раствор N-метилимидазола (24,6 мл) в хлористом метилене (80,0 мл). Реакционную массу перемешивали (на магнитной мешалке, скорость 300 об/мин) при -15°С в течение 1,0 часа и прибавляли 2,3,5,6-тетрафтор-4-бромфенол (8,6 г). Перемешивали 3 часа при охлаждении (от -15 до -10°С) и 16,0 часов при 23-25°С. Контролировали по ТСХ. Элюент CHCl3/СН3ОН=10/1.
После завершения реакции реакционную массу упаривали на ротационном испарителе. Остаток растворяли в этилацетате (50 мл), промывали водой (2×50 мл), IN HCl (50 мл), раствором NaHCO3 (2x50 мл) и рассолом (50 мл). Органический слой сушили над Na2SO4. Упаривали на ротационном испарителе.
Получили 13,3 г (78,0%) светлого масла.
Пример 4. Кристаллизация S-изомера фосфорамидата формулы (III), в котором X представляет собой пентахлорфенил.
1,0 г диастереомерной смеси пентахлорфенилзамещенного фосфорамидата формулы (IV), полученной в примере 1, растворяли при постоянном перемешивании в 12 мл смеси этилацетата и гексана в соотношении 2:10 в течение 12 часов. В результате выпало 0,46 г бежевых кристаллов S-изомера фосфорамидата формулы (III) (выход 46%).
Соотношение S/R=99,8/0,2 (ВЭЖХ).
1Н NMR (400 MHz, CDCl3, 323К) δ 7.40-7.27 (m, 3Н), 7.26-7.15 (m, 2Н), 5.02 (hept, J=6.2 Hz, 1Н), 4.27-4.09 (m, 1H), 4.06-3.88 (m, 1H), 1.40 (d, J=7.0 Hz, 3H), 1.26 (d, J=6.2 Hz, 3H), 1.24 (d, J=6.2 Hz, 3H);
13C NMR (101 MHz, CDCl3, 323K) δ 172.53, 172.43, 150.47, 150.40, 132.13, 132.11, 129.67,127.37, 127.33, 125.34, 125.33, 120.18, 120.13, 69.46, 50.74, 50.71, 21.57, 21.49, 21.13, 21.09;
LCMS (m/z) вычислено для C18H17Cl5NO5P 534.93, 536.93; найдено 534.67, 536.72.
Пример 5. Кристаллизация S-изомера фосфорамидата формулы (III), в котором X представляет собой 2,3,5,6-тетрафторфенил.
500 мг диастереомерной смеси 2,3,5,6-тетрафторфенилзамещенного фосфорамидата формулы (IV), полученной в примере 2, поместили в 5,5 мл смеси этилацетата и петролейного эфира в соотношении 0,5:5 и перемешивали в течение 12 часов. В результате выпало 296 мг бесцветных игольчатых кристаллов S-изомера фосфорамидата формулы (III) (выход 59%).
Соотношение S/R=99,5/0,5 (ВЭЖХ).
1Н NMR (400 MHz, CDCl3, 323K) δ 7.35-7.26 (m, 4Н), 7.24-7.16 (m, 1Н), 6.97-6.84 (m, 1Н), 5.05 (hept, J=6.3 Hz, 1Н), 4.21-4.13 (m, 1H), 3.95-3.89 (m, 1H), 1.47 (d, J=7.0 Hz, 3H), 1.28-1.25 (m, 6H).
13C NMR (101 MHz, CDCl3, 323K) δ 172.40, 172.31, 150.35, 150.28, 142.15, 142.00, 139.65, 139.50, 129.67, 125.40, 125.39, 120.05, 120.00, 102.06, 102.04, 69.40, 50.62, 50.60, 21.49, 21.46, 20.82, 20.77.
LCMS (m/z) вычислено для C18H18F4NO5P 436.09, 434.08; найдено 436.67, 434.59.
Пример 6. Кристаллизация S-изомера фосфорамидата формулы (III), в котором X представляет собой 4-бром-2,3,5,6-тетрафторфенил.
4,8 г диастереомерной смеси 4-бром-2,3,5,6-тетрафторфенилзамещенного фосфорамидата формулы (IV), полученной в примере 3, растворяли в 33 мл смеси этилацетата и гексана в соотношении 3:30 при перемешивании в течение 4 часов. В результате выпало 2,3 г бесцветных игольчатых кристаллов S-изомера фосфорамидата формулы (III) (выход 48%).
Соотношение S/R=99,5/0,5 (ВЭЖХ).
1Н NMR (400 MHz, CDCl3, 50°С) δ 7.40-7.32 (m, 2Н), 7.32-7.16 (m, 3Н), 5.05 (hept, J=6.3 Hz, 1Н), 4.22-4.08 (m, 1H), 3.98-3.88 (m, 1H), 1.47 (d, J=7.0 Hz, 3H), 1.31-1.18 (m, 6H).
LCMS (m/z) вычислено для C18H17BrF4NO5P 516.00, 514.99; найдено 516.80, 514.74.
Пример 7. Получение софосбувира из нуклеозида формулы (II) и фосфорамидата формулы (III), в котором X представляет собой пентахлорфенил.
К раствору исходного нуклеозида II (7.322 г, 0.028 моль) в сухом тетрагидрофуране (123.0 мл) добавляли трет-бутилмагния хлорид (20 масс % раствор в ТГФ (35.1 мл, 0.060 моль, 2.12 экв.)). Перемешивали (на магнитной мешалке 300 об/мин) 30 минут при температуре 23-25°С и прикапывали раствор S-изомера пентахлорзамещенного фосфорамидата формулы (III), полученного в примере 4 (18.155 г, 0.034 моль, 1.205 экв.) в ТГФ (308 мл). Перемешивали 96 часов.
Контроль протекания реакции осуществляли при помощи ТСХ. Элюент CHCl3/CH3OH=5/1.
По окончании реакции добавили насыщенный раствор хлорида аммония (100 мл), удалили растворитель на роторном испарителе. Добавили этилацетат (700 мл). Промывали органический слой водой (2×150 мл), насыщенным раствором NaHCO3 (5×150 мл) и рассолом (2×75 мл). Сушили над безводным сульфатом натрия. Затем осушитель отфильтровали, промыли этилацетатом (30 мл). Органический растворитель из фильтрата удалили на роторном испарителе. Полученный остаток представляет собой затвердевшее буроватое масло. Прибавили к нему смесь 60 мл дихлорметана и 30 мл диизопропилового эфира. Перемешивали при комнатной температуре в течение 2 часов. Растворители декантировали, добавили к остатку 250 мл н-гексана, нагревали до температуры 55°С, охлаждали до температуры 23-25°С, перемешивали при температуре 23-25°С в течение 2 часов, после чего добавляли 0,05 г затравки софосбувира формы VIII. Реакционную массу перемешивали при температуре 23-25°С в течение 1 часа, осадок отфильтровывали и промывали 35 мл н-гексана. Полученный осадок сушили в вакуумном сушильном шкафу при температуре 30°С.
Получили 8,79 г софосбувира формы VIII. (Выход 59%), содержание S-диастереомера составило 99,6% (ВЭЖХ).
1H NMR (400 MHz, DMSO-d6, 323К) δ 11.37 (s, 1Н), 7.53 (d, J=8.1 Hz, 1H), 7.36-7.32 (m, 2H), 7.21-7.13 (m, 3H), 5.99 (d, J=20.2 Hz, 1H), 5.87 (dd, J=12.6, 9.9 Hz, 1H), 5.51 (d, J=8.1 Hz, 1H), 4.84 (hept, J=6.3 Hz, 1H), 4.38-4.33 (m, 1H), 4.25-4.20 (m, 1H), 4.01-3.98 (m, 1H), 3.82-3.75 (m, 2H), 1.27-1.22 (m, 6H), 1.14 (d, J=6.2 Hz, 6H).
Пример 8. Получение софосбувира из нуклеозида формулы (II) и фосфорамидата формулы (III), в котором X представляет собой 2,3,5,6-тетрафторфенил.
К раствору исходного нуклеозида II (7.322 г, 0.028 моль) в сухом тетрагидрофуране (123.0 мл) добавляли трет-бутилмагния хлорид (20 масс % раствор в ТГФ (35.1 мл, 0.060 моль, 2.12 экв.)). Перемешивали (на магнитной мешалке 300 об/мин) 30 минут при температуре 23-25°С и прикапывали раствор S-изомера 2,3,5,6-тетрафторфенилзамещенного фосфорамидата формулы (III), полученного в примере 5 (14.781 г, 0.034 моль, 1.205 экв.) в ТГФ (308 мл). Перемешивали 96 часов.
Контроль протекания реакции осуществляли при помощи ТСХ. Элюент CHCl3/CH3OH=5/1.
По окончании реакции добавили насыщенный раствор хлорида аммония (100 мл), удалили растворитель на роторном испарителе. Добавили этилацетат (700 мл). Промывали органический слой водой (2×150 мл), насыщенным раствором NaHCO3 (5×150 мл) и рассолом (2×75 мл). Сушили над безводным сульфатом натрия. Затем осушитель отфильтровали, промыли этилацетатом (30 мл). Органический растворитель из фильтрата удалили на роторном испарителе. Полученный остаток представляет собой затвердевшее буроватое масло. Прибавили к нему смесь 60 мл дихлорметана и 30 мл диизопропилового эфира. Перемешивали при комнатной температуре в течение 2 часов. Растворители декантировали, добавили к остатку 250 мл н-гексана, нагревали до температуры 55°С, охлаждали до температуры 23-25°С, перемешивали при температуре 23-25°С в течение 2 часов, после чего добавляли 0,05 г затравки софосбувира формы VIII. Реакционную массу перемешивали при температуре 23-25°С в течение 1 часа, осадок отфильтровывали и промывали 35 мл н-гексана. Полученный осадок сушили в вакуумном сушильном шкафу при температуре 30°С.
Получили 7,9 г софосбувира формы VIII. (Выход 53,0%), содержание S-диастереомера составило 99,5% (ВЭЖХ).
1Н NMR (400 MHz, DMSO-d6, 323К) δ 11.37 (s, 1Н), 7.52 (d, J=8.1 Hz, 1Н), 7.36 - 7.32 (m, 2H), 7.21-7.13 (m, 3H), 5.98 (d, J=20.2 Hz, 1H), 5.86 (dd, J=12.6, 9.9 Hz, 1H), 5.74-5.69 (m, 1H), 5.51 (d, J=8.1 Hz, 1H), 4.84 (hept, J=6.3 Hz, 1H), 4.38-4.33 (m, 1H), 4.25-4.19 (m, 1H), 4.01-3.98 (m, 1H), 3.87-3.74 (m, 2H), 1.27 -1.22 (m, 6H), 1.14 (d, J=6.3 Hz, 6H).
Пример 9. Получение софосбувира из нуклеозида формулы (II) и фосфорамидата формулы (III), в котором X представляет собой 4-бром-2,3,5,6-тетрафторфенил.
Реакцию получения осуществляли по аналогии с примерами 7-8, выход составил 51%, содержание S-диастереомера составило 99,5% (ВЭЖХ).
Изобретение может использоваться в фармацевтической промышленности и относится к способу получения противовирусного соединения софосбувира формулы (I):
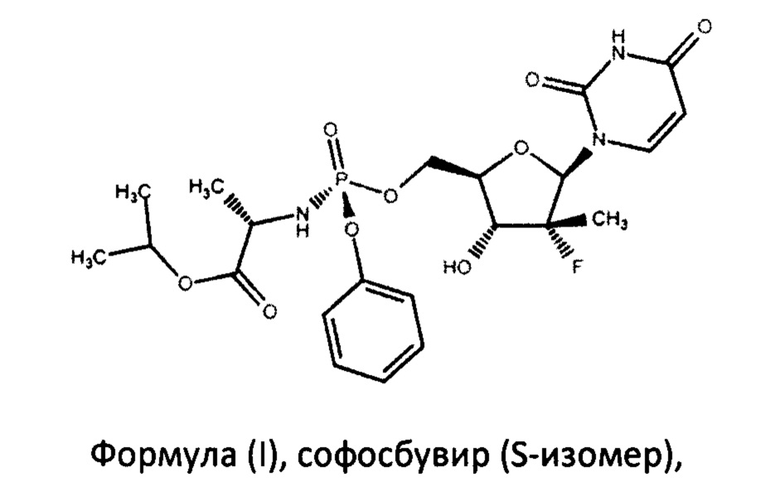
формула (I),
а также промежуточным соединениям для его осуществления. Предложенный способ включает взаимодействие в присутствии основания нуклеозида формулы (II) с фосфорамидатом формулы (III)
где X представляет собой
 .
.
Предложены новые фосфорамидаты и новый эффективный способ получения софосбувира с их использованием. 2 н. и 3 з.п. ф-лы, 9 пр.
1. Способ получения софосбувира формулы (I)
включающий взаимодействие в присутствии основания нуклеозида формулы (II) 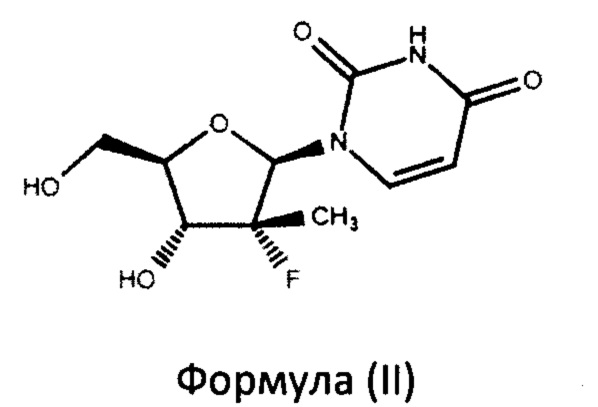
с фосфорамидатом формулы (III)
где X представляет собой
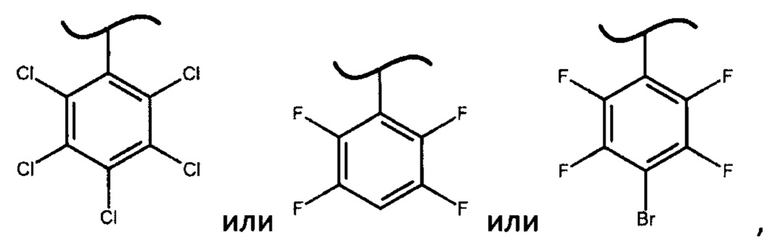
с получением софосбувира.
2. Способ по п. 1, в котором X представляет собой:
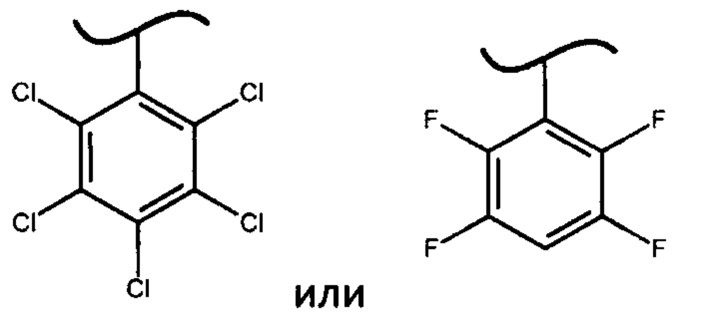
3. Способ по п. 1, в котором основание выбирают из группы, включающей трет-бутилмагния хлорид, N-метилимидазол, гидрид натрия, гексаметилдисилазан натрия, гексаметилдисилазан лития или диизопропиламид лития.
4. Способ по п. 1, в котором фосфорамидат формулы (III) получают взаимодействием изопропилового эфира L-аланина или его фармацевтически приемлемой соли с дихлорфенилфосфатом и фенолом формулы Х-ОН, где X представляет собой
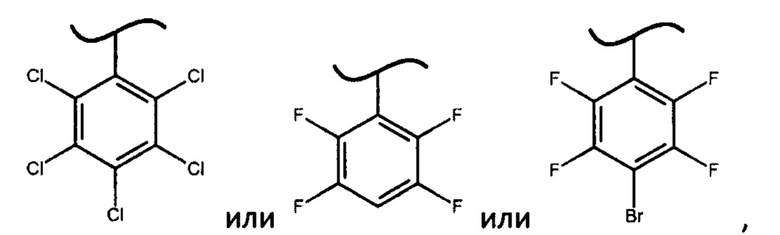
с получением фосфорамидата формулы (IV) в виде смеси диастереомеров:
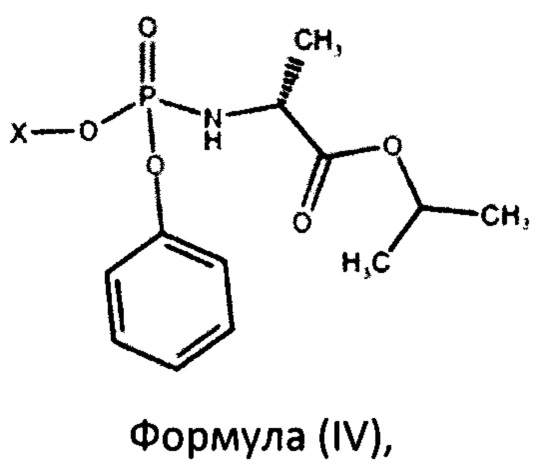
с последующей перекристаллизацией из подходящего растворителя с получением фосфорамидата формулы (III).
5. Фосфорамидаты формулы (III)
где X представляет собой
| Приспособление для смазки холостых шкивов | 1931 |
|
SU28742A1 |
| Папиросная коробка | 1927 |
|
SU28871A1 |
| КРУТКОМЕР | 1931 |
|
SU29775A1 |

