В этой заявке испрашивается приоритет предварительной заявки США №60/068170, поданной 19 декабря 1997 года, на содержание которой ниже включено в ссылки.
Предпосылки создания изобретения
Область изобретения
Настоящее изобретение относится к новому способу получения энантиомерно-обогащенных тетрагидробензотиепиноксидов.
Описание уровня техники.
Известно, что агенты, которые ингибируют передвижение желчных кислот по подвздошной кишке, могут вызвать также понижение уровня холестерина в сыворотке крови. Stedronski в работе "Взаимодействие желчных кислот и холестерина с несистемными агентами, имеющими гипохолистеринэмические свойства" Biochimica et Biophysica Acta, 1210 (1994), 255-287, обсуждает биохимию, физиологию и воздействие известных активных агентов на желчные кислоты и холестерин.
Тетрагидробензотиепин-1,1-диоксиды (THBDO - соединения) представляют собой класс идеальных ингибирующих передвижение желчных кислот соединений, которые, как недавно было обнаружено, пригодны для воздействия на уровень холестерина в сыворотке крови (патентная заявка США №08/816.065=WO 96/08484).
Некоторые классы соединений проявляют усиленное действие в качестве фармакологических препаратов после того, как они были энантиомерно-обогащены (см., например, Richard B.Silverman, The Organic Chemistry of Drug Design and Drug Action, Academic Press, 1992, pp. 76-82). Поэтому THBDO-соединения, которые были энантиомерно обогащены, представляют особенный интерес.
Классом химических соединений, пригодным в качестве промежуточных соединений при получении рацемических THBDO-соединений, является класс тетрагидробензотиепин-1-оксидов (ТНВО-соединения). THBDO-соединения и ТНВО-соединения имеют химические структуры, в которых фенильное кольцо конденсировано с семичленным циклом. Способ получения энантиомерно-обогащенных образцов другой конденсированной циклической системы фенил/семичленное кольцо, бензотиазепинов, описано Хигашикавой (Higashikawa) (JP 59144777), где рацемические производные бензотиазепина разделяют на оптические изомеры на хроматографической колонке, содержащей хиральные краун-эфиры в качестве стационарной фазы. Хотя эффект разделения оптических изомеров и достигается, способ Хигашикавы ограничивается получением только очень малых количеств энантиомерно-обогащенных производных бензотиазепина.
Джордано (Giordano) (СА 2068231) сообщает о циклизации (2S,3S)-аминофенилтиопропионатов в присутствии фосфоновой кислоты с получением (2S, 3S)-бензотиазепин-4-онов. Однако такое получение ограничено необходимостью использования предпочтительней энантиомерно-обогащенных, чем рацемических, исходных материалов. Вдобавок, способ Джордано регулирует стереохимию семичленного кольца бензотиазепин-4-она только в отношении позиций 2 и 3. Позиции 4 и 5 семичленного кольца бензотиазепин-4-она не являются асимметрическими центрами и, следовательно, стереохимию относительно этих позиций нельзя регулировать посредством способа Джордано.
Описанный Ямадой с сотр. (Yamada, et al., J.Org.Chem., 1996, 61 (24), 8586-8590) способ, посредством которого были получены соединения энантиомерно-обогащенного бензотиазепин-3-гидрокси-4(5Н)-она, состоит в асимметрическом восстановлении соединений 1,5-бензотиазепин-3,4 (2Н,5Н)-дионов. Этот продукт получают обработкой рацемического 1,5-бензотиазепин-3,4 (2Н,5Н)-диона продуктом взаимодействия оптически активной альфа-аминокислоты и восстановителя, например боргидрида натрия. Несмотря на то, что получается продукт с высокой оптической чистотой, этот способ ограничен в применении из-за использования относительно дорогой стадии химического восстановления.
Патель и др. (Patel et al., US Patent 5559017) сообщают о микробном восстановлении соединений рацемического 1,5-бензотиазепин-3,4 (2Н,5Н)-диона с получением энантиомерно-обогащенных соединений 1,5-бензотиазепин-3-гидрокси-4(5Н)-она. Этот способ ограничивается проблемами поддержания жизнеспособной и чистой бактериальной культуры подходящего типа и разновидности. Вдобавок, этот способ ограничен в масштабах, производя только микрограммные количества желаемого продукта.
До настоящего времени не было известно о каких-либо способах получения энантиомерно-обогащенных THBDO-соединений или энантиомерно-обогащенных ТНВО-соединений. Более того, не было описано никаких способов регулирования стереохимии в 4 и 5 позициях семичленного кольца THBDO-соединений и ТНВО-соединений.
Сущность изобретения
Удобный и рентабельный способ получения энантиомерно-обогащенных THBDO-соединений и промежуточных соединений для их синтеза представляет большую важность и полезность. В ответ на необходимость этого, авторы настоящего изобретения разработали способ получения энантиомерно-обогащенных тетрагидробензотиепин-1-оксидов или энантиомерно-обогащенных тетрагидробензотиепин-1,1-диоксидов с хиральными центрами в 4 и 5 позициях семичленного кольца.
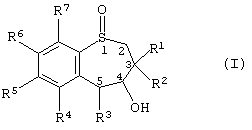
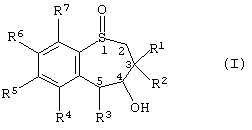
Таким образом, настоящее изобретение предоставляет способ получения энантиомерно-обогащенного тетрагидробензотиепин-1-оксида, имеющего формулу (I):
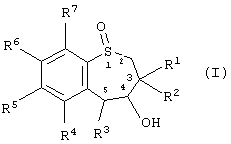
где R1 и R2 независимо друг от друга выбраны из Н, алкила, алкенила, алкинила, циклоалкила, арила и гетероарила;
R3 выбран из группы, включающей Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероцикл, четвертичный гетероцикл, OR24, SR15, S(O)R15, SO2R15 и SO3R15,
где алкил, алкенил, алкинил, арил, циклоалкил, гетероцикл, четвертичный гетероцикл и четвертичный гетероарил могут иметь один или несколько заместителей, независимо выбранных из группы, включающей алкил, алкенил, алкинил, полиалкил, простой полиэфир, арил, галоалкил, циклоалкил, гетероцикл, арилалкил, четвертичный гетероцикл, четвертичный гетероарил, галоген, оксо, OR19, NR19R20, SR19. S(O)R19, SO2R19, SO3R19, NR19OR20, NR19, NR20R21, NO2, CO2R19, CN, OM, SO2OM, SO2NR19R20, C(O) NR19R20, C(O)OM, COR19, P(O) R19R20, P+ R19R20R21A-, P(OR19)OR20, S+ R19R20A- и N+R15R17R18 A-,
где А- представляет собой фармацевтически приемлемый анион и М представляет собой фармацевтически приемлемый катион;
вышеупомянутые алкил, алкенил, алкинил, полиалкил, простой полиэфир, арил, галоалкил, циклоалкил и гетероцикл могут в свою очередь иметь один или несколько заместителей, выбранных из группы, включающей OR13, NR13R14, SR13, S(O)R13, SO2R13, SO3R13, CO2R13, CN, оксо, CONR13R14, N+R13 R14R15A-, алкил, алкенил, алкинил, арил, циклоалкил, гетероцикл, арилалкил, четвертичный гетероцикл, четвертичный гетероарил, P(O)R13R14, P+R13R14R15A- и P(O)(OR13)OR14, и в которых указанные алкильные, алкенильные, алкинильные, полиалкильные, простые полиэфирные, арильные, галоалкильные, циклоалкильные и гетероциклические группы могут иметь один или несколько углеродных атомов, замещенных на О, NR13, N+R13R14A-, S, SO, SO2, S+R13A-, PR13, P(O)R13, P+R13R14A-или фенилен;
R19, R20 и R21 независимо друг от друга выбраны из группы, состоящей из водорода, алкила, алкенила, алкинила, полиалкила, арила, арилалкила, циклоалкила, гетероцикла, гетероарила, четвертичного гетероцикла, четвертичного гетероарила, полиэфира, алкиларилалкила, алкилгетероарилалкила, алкилгетероциклоалкила, гетероциклоалкила, гетероарилалкила, четвертичного гетероциклоалкила, алкиламмонийалкила, карбоксиалкиламинокарбонилалкила и четвертичного гетероарилалкила,
где алкил, алкенил, алкинил, арилалкил, гетероцикл и полиалкил могут иметь один или несколько углеродных атомов, замещенных на О, NR15, N+R15R16A-, S, SO, SO2, S+R15A-, PR15, P+R15R16A-, P(O)R15, фенилен, углевод, аминокислоту, пептид или полипептид,
R19, R20 и R21 могут иметь один или несколько заместителей, выбранных из группы, состоящей из таких групп, как гидрокси, амино, сульфо, карбокси, сульфалкил, карбоксиалкил, алкил, гетерицикл, гетероарил, четвертичный гетероциклоалкил, четвертичный гетероарилалкил, гуанидинил, четвертичный гетероцикл, четвертичный гетероарил, OR15, NR15R16, N+R15R16R17R18A-, SR15, S(O)R15, SO2R15, оксо, CO2R15, CN, галоген, CONR15R16, SO2OM, SO2NR15R16, PO(OR22)OR23, P+R15R16R17A-, S+R15R16A- и C(O)OM,
где R22 и R23 независимо выбраны из заместителей, составляющих R15 и М или
R20 и R21 вместе с атомом азота, с которым они связаны, образуют кольцо;
R24 выбран из группы, содержащей алкил, алкенил, алкинил, циклоалкил, арил, ацил, гетероцикл, аммонийалкил, алкиламмонийалкил и арилалкил;
R13 и R14 независимо выбраны из группы, состоящей из водорода и алкила;
R15 и R16 независимо выбраны из группы, состоящей из водорода, алкила, алкенила, алкинила, циклоалкила, арила, ацила, гетероцикла, аммонийалкила, арилалкила, карбоксиалкила, карбоксигетероарила, карбоксигетероцикла, карбоалкоксиалкила, карбоалкиламиногруппы, гетероарилалкила, гетероциклоалкила и алкиламмонийалкила;
R17 и R18 независимо выбраны из группы, состоящей из Н, алкила, алкенила, алкинила, арила, арилалкила, алкенилалкила, алкинилалкила, гетероцикла, карбоксиалкила, карбалкоксиалкила, циклоалкила, цианалкила, OR15, NR15R16, SR15, S(O)R15, SO2R15, SO3R15, CO3R15, CN, галогена, оксо и CONR15R16, где R15 и R16 такие, как определено выше, или
R17 и R18 вместе с атомом азота или углерода, с которым они связаны, образуют цикл;
R4, R5, R6 и R7 независимо друг от друга выбраны из группы, содержащей водород, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, галоген, алкокси, арилоксигруппу, - NO2, -NR9R10;
R9 и R10 независимо друг от друга выбраны из группы, содержащей Н, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, бутоксикарбонил и карбобензилокси;
R3 и гидроксил в положении 4 энантиомерно-обогащенного тетрагидробензотиепин-1-оксида находятся в син-конформации по отношению друг к другу;
причем алкил, алкенил, алкинил, циклоалкил, арил и гетероарил могут быть замещены одним или несколькими радикалами, выбранными из группы, включающей алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, алкокси, арилокси, -NO2 и галоген;
атом серы в положении 1 семичленного кольца и атомы углерода в положениях 4 и 5 семичленного кольца являются хиральными центрами.
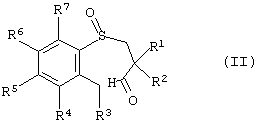
Заявленный способ включает циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида, имеющего формулу (II):

в которой R1, R2, R3, R4, R5, R6 и R7 описаны выше и где атом серы является хиральным центром с образованием энантиомерно-обогащенного тетрагидробензотиепин-1-оксида.
В другой реализации настоящее изобретение также представляет способ получения энантиомерно-обогащенного тетрагидробензотиепин-1-оксида, имеющего формулу (I), причем этот способ включает окисление арил-3-гидроксипропилсульфида, имеющего формулу (IV):

в которой R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, и где окисление проводят в условиях энантиоселективного окисления с получением энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида, имеющего формулу (III):

где R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, а атом серы является хиральным центром; окисление 3-гидроксильной группы энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида с получением эпантиомерно-обогащенного арил-3-пропанальсульфоксида, имеющего формулу (II); и циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида с образованием энантиомерно-обогащенного тетрагидробензотиепин-1 -оксида.
Еще в одном воплощении настоящее изобретение также дает способ получения энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида, имеющего формулу (VII):

в которой R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, R3 и гидроксил в положении 4 энантиомерно-обогащенного тетрагидробензотиепин-1-оксида находятся в син-конформации по отношению друг к другу, а углеродные атомы в положениях 4 и 5 семичленного кольца являются хиральными центрами. Этот способ включает окисление арил-3-гидроксипропилсульфида, имеющего формулу (IV) в условиях энантиоселективного окисления с получением энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида, имеющего формулу (III) и имеющего хиральный центр при атоме серы; окисление энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида с получением энантиомерно-обогащенного арил-3-пропанальсульфоксида, имеющего формулу (II); циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида с образованием энантиомерно-обогащенного тетрагидробензотиепин-1-оксида, имеющего формулу (I) и имеющего хиральные центры на атоме серы в 1-положении семичленного кольца и при атомах углерода в положениях 4 и 5 семичленного кольца; окисление энантиомерно-обогащенного тетрагидротиепин-1-оксида до энантиомерно-обогащенного тетрагидротиепин-1,1-диоксида.
Объем применения настоящего изобретения станет очевидным из детального описания, приведенного ниже. Однако надо иметь ввиду, что последующее детальное описание и примеры, отражающие предпочтительные воплощения данного изобретения, приводятся только в качестве иллюстрации. Различные варианты и модификации в духе и в пределах данного изобретения будут очевидны специалистам из этого детального описания.
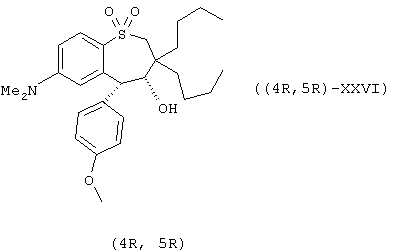
Еще в одном воплощении настоящее изобретение предоставляет идеальное ингибирующее транспорт желчных кислот вещество (IBAT ингибиторы), полезное для профилактики или лечения сердечно-сосудистых заболеваний, включая гиперхолестеролэмию и атеросклероз, при этом указанный IBAT ингибитор имеет структуру формулы ((4 R, 5 R) - XXVII):

И еще в одном воплощении настоящее изобретение предоставляет идеальное ингибирующее транспорт желчных кислот вещество (IBAT ингибиторы), пригодное для профилактики или лечения сердечно-сосудистых заболеваний, включая гиперхолестеролэмию и атеросклероз, при этом указанное IBAT ингибиторное соединение имеет структуру формулы ((4S, 5S) - XXVII):

Краткое описание чертежей
Фиг.1 представляет собой схему реакции, поясняющую получение энантиомерно-обогащенного тетрагидробензотиепин-1,1 -диоксида.
Фиг.2 представляет собой схему реакции, поясняющую получение циклического сульфата.
Детальное описание изобретения
Следующее детальное описание дано, чтобы помочь специалистам применять на практике настоящее изобретение. При этом его не следует истолковывать как ограничивающее объем данного изобретения, поскольку модификации и вариации изложенных воплощений в духе и объеме настоящего изобретения могут выполняться людьми обычной квалификации.
Содержание каждой из ссылок, цитируемых в этой работе, включая содержание ссылок, цитируемых в пределах этих первичных ссылок, полностью здесь включено посредством ссылки.
Определения
Следующие определения даны, чтобы помочь читателю в понимании последующего детального описания:
"Алкил", "алкенил" и "алкинил", если не указано иначе, являются каждый углеводородным радикалом с нормальной цепью, разветвленной цепью или циклическим, или замещенным, имеющим от одного до примерно 20 атомов углерода для алкила, или от двух до примерно 20 атомов углерода для алкенила, или от двух до примерно 20 атомов углерода для алкинила. Алкил может, следовательно, означать, например, метил, этил, пропил, бутил, пентил, гексил, метилциклопентил и их изомеры. Алкенил может означать, например, этенил, пропенил, бутенил, пентенил, или гексенил, или их изомеры. Алкинил может означать, например, этин, пропин, бутин, пентин или гексин, или их изомеры. Указанные алкил-, алкенил- или алкинильные группы могут иметь один или более водородных атомов, содержащих заместители. Такие заместители могут включать, например, циклоалкил, арил, гетероарил, галоген, алкокси, гидрокси, арилокси, -NO2, амино- или алкиламиногруппу.
"Арил" означает ненасыщенный моно- или многоядерный карбоциклический радикал, включающий, в том числе, замещенный или незамещенный фенил, нафтил или антраценил.
"Гетероарил" означает ненасыщенный гетероциклический радикал. "Гетероцикл" означает насыщенный или ненасыщенный моно- или многоядерный карбоциклический радикал, в котором один или более атомов углерода могут быть замещены на N, S, Р или О. Он включает, например, следующие структуры:

где Z, Z', Z" или Z'" представляют собой С, S, Р, О или N, при условии, что один из Z, Z', Z" или Z'" является иным, чем углерод, но не является О или S, когда присоединен к другому Z-атому двойной связью или когда присоединен к другому атому О или S. Более того, возможные заместители присоединены к Z, Z', Z" или Z'" только тогда, когда каждый представляет собой С. Как в "гетероцикле", так и в "гетероариле" точки присоединения к интересующей молекуле могут находиться на гетероатоме или в любом месте данного кольца.
"Галоген" означает фтор, хлор, бром или иод.
"Циклоалкил" означает моно- или многоядерный карбоциклический радикал, в котором каждое кольцо содержит от 3 до 10 атомов углерода и в котором каждое кольцо может содержать одну или больше двойных или тройных связей, и не является полностью ненасыщенным.
"Алкокси" означает алкильную группу, связанную одинарной связью с кислородом. Она включает, например, метокси, алкокси, пропокси, бутокси и их изомеры.
"Арилокси" означает арильную группу, связанную одинарной сязью с кислородом и является, например, фенокси.
"Карбалкил" означает алкильную группу, связанную одинарной связью с карбонильной группой и является, например, -СОСН3, -СОСН2СН3, -СОСН2СН2СН3 и их изомеры.
"Хиральный" (сален) металлический комплекс" означает оптически активный тетрадентатный комплекс Шиффова основания и металла. Это включает, например, (S,S)-(+)-N,N'-бис(3.5-ди-тpeт-бутилсалициклиден)-1,2-циклогександиаминмарганец (III) хлорид или (R,R)-(+)-N,N'-бис(3,5-ди-трет-бутилсалициклиден)-1,2-дифенилэтилендиаминмарганец (III) хлорид.
Если употреблены в сочетании, например, "алкиларил" или "арилалкил", то эти отдельные термины имеют значения, указанные выше.
"Условия энантиоселективного окисления" означает условия, способствующие получению в результате окисления преимущественно одного энантиомера или набора диастереомеров.
"Хиральный" означает невозможность молекулы быть наложенной на свое зеркальное отражение. Хиральный центр в молекуле представляет собой атом, который имеет тетраэдрическую, необращаемую геометрию и для которого каждая вершина тетраэдра отличается от других вершин. Хиральные центры включают, например, атомы углерода, имеющие четыре различных связанных с ним заместителя. Другим примером хирального центра является атом серы в сульфоксидной группе, которая связывает с атомом серы кислород и два других разных заместителя.
"Энантиомерно-обогащенный" означает, что один энантиомер или набор диастереомеров преобладает над комплементарным энантиомером или набором диастереомеров. Энантиомерное обогащение соединения обычно определяют разделением смеси первого энантиомера и второго энантиомера, например, с помощью хиральной хроматографии, интегрируя площади пиков этих двух энантиомеров, суммируя эти площади, деля отдельную площадь каждого энантиомерного пика на суммарную площадь двух пиков и выражая доли как проценты в общей смеси двух энантиомеров. Если первый энантиомер преобладает над вторым энантиомером, процент второго энантиомера вычитают из процента первого энантиомера, а получающуюся в результате разницу выражают как процент энантиомерного обогащения (% э.о.) первым энантиомером. Энантиомерное обогащение может быть от примерно 1 до примерно 100% э.о., предпочтительно от примерно 10 до примерно 100% э.о., более предпочтительно от примерно 20 до примерно 100% э.о., а еще более предпочтительно от примерно 50 до примерно 100% э.о.
"Асимметричный окислитель" означает окислитель, который индуцирует хиральный центр в месте окисления с получением, например, хирального сульфоксида.
"ЯМР" - ядерная магниторезонансная спектроскопия.
"ГХ" - газовая хроматография.
"ВЭЖХ" - высокоэффективная жидкостная хроматография.
"МС" - массспектроскопия.
м-ХНБК - мега-хлор-надбензойная кислота.
Обзор препаративных методов
Среди своих различных воплощений данное настоящее изобретение обеспечивает способ получения энантиомерно-обогащенного тетрагидробензотиепин-1-оксида, имеющего формулу (I):

в которой R1 и R2 независимо выбраны из группы Н, алкил, алкенил, алкинил, циклоалкил, арил и гетероарил;
R3 выбран из группы, состоящей из Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероцикл, четвертичный гетероцикл, OR24, SR15, S(O)R15, SO2R15 и SO3R15,
где алкил, алкенил, алкинил, арил, циклоалкил, гетероцикл, четвертичный гетероцикл и четвертичный гетероарил могут быть замещены одним или более заместителями, независимо выбранными из группы, включающей алкил, алкенил, алкинил, полиалкил, простой полиэфир, арил, галоалкил, циклоалкил, гетероцикл, арилалкил, четвертичный гетероцикл, четвертичный гетероарил, галоген, оксо, OR19, NR19R20, SR19, S(O)R19, SO2R19, SO3R19, NR19OR20, NR19NR20R21, NO2, CO2R19, CN, OM, SO2OM, SO2NR19R20, C(O) NR19R20, C(O)OM, COR19, P(O) R19R20, P+ R19R20R21A-, P(OR19)OR20, S+ R19R20A- и N+R15R17R18 A-,
где А- представляет собой фармацевтически приемлемый анион и М представляет собой фармацевтически приемлемый катион;
указанные алкил, алкенил, алкинил, полилкил, полиэфир, арил, галоалкил, циклоалкил и гетероцикл могут быть дополнительно замещенными одним или более заместителями, выбранными из следующей группы: OR13, NR13R14, SR13, S(O)R13, SO2R13, SO3R13, CO2R13, CN, оксо, CONR13R14, N+R13R14R15A-, алкил,алкенил, алкинил, арил, циклоалкил, гетероцикл, арилалкил, четвертичный гетероцикл, четвертичный гетероарил, P(O)R13R14, P+R13R14R15A- и P(O)(OR13)OR14, и где указанные алкил, алкенил, алкинил, полиалкил, простой полиэфир, арил, галоалкил, циклоалкил и гетероцикл могут иметь один или более атомов углерода, замещенными на О, NR13, N+R13R14A-, S, SO, SO2, S+R13A-, PR13, P(O)R13, P+R13R14A- или фенилен;
R19, R20 и R21 независимо выбраны из группы, включающей водород, алкил, алкенил, алкинил, полиалкил, арил, арилалкил, циклоалкил, гетероцикл, гетероарил, четвертичный гетероцикл, четвертичный гетероарил, простой полиэфир, алкиларилалкил, алкилгетероарилалкил, алкилгетероциклоалкил, гетероциклоалкил, гетероарилалкил, четвертичный гетероциклоалкил, алкиламмонийалкил, карбоксиалкиламинокарбонилалкил и четвертичный гетероарилалкил,
где алкил, алкенил, алкинил, гетероцикл и полиалкил могут иметь один или несколько углеродных атомов, замещенных на О, NR15, N+R15R16A-, S, SO, SO2, S+R15A-, PR15, P+R15R16A-, P(O)R15, фенилен, углевод, аминокислоту, пептид или полипептид, и
R19, R20 и R21 по желанию имеют один или несколько заместителей, выбранных из следующей группы: гидрокси, амино, сульфо, карбокси, сульфалкил, карбоксиалкил, сульфалкил, алкил, гетероцикл, гетероарил, четвертичный гетероциклоалкил, четвертичный гетероарилалкил, гуанидинил, четвертичный гетероцикл, четвертичный гетероарил, OR15, NR15R16, N+R15R17R18A-, SR15, S (O) R15, SO2R15, SO3R15, оксо, СО2R15, CN, галоген, CONR15R16, SO2OM, SO2 NR15R16, PO (OR22) OR23, P+R15R16R17A-, S+R15R16A- и С(O)ОМ,
где R22 и R23 независимо выбраны из заместителей, составляющих R15 и М, или
R20 и R21 вместе с атомом азота, с которым они связаны, образуют цикл;
R24 выбран из группы, включающей алкил, алкенил, алкинил, циклоалкил, арил, ацил, гетероцикл, аммонийалкил, алкиламмонийалкил и арилалкил;
R13 и R14 независимо выбраны из группы, состоящей из водорода и алкила;
R15 и R16 независимо выбраны из группы, включающей Н, алкил, алкенил, алкинил, циклоалкил, арил, ацил, гетероцикл, аммонийалкил, арилалкил, карбоксигетероцикл, карбалкоксиалкил, карбалкиламино, гетероарилалкил, гетероциклоалкил, алкиламмонийалкил; и
R17 и R18 независимо друг от друга выбраны из группы, состоящей из таких радикалов, как Н, алкил, алкенил, алкинил, арил, арилалкил, алкенилалкил, алкинилалкил, гетероцикл, карбоксиалкил, карбалкоксиалкил, циклоалкил, цианалкил, OR15, NR15R16, SR15, S (О) R15, SO2R15, SO3R15, CO3R15, CN, галоген, оксо и CONR15R16, где R15 и R16 такие, как определено выше, или
R17 и R18 вместе с атомами азота или углерода, с которыми они связаны, образуют кольцо; и
R4, R5, R6 и R7 независимо друг от друга выбраны из таких, как Н, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, галоген, алкокси, -NО2 и -NR9R10;
R9 и R10 независимо друг от друга выбраны из таких, как Н, алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, бутоксикарбонил и карбобензилокси;
R3 и гидроксил в положении 4 энантиомерно-обогащенного тетрагидробензотиепин-1-оксида находятся в син-конформации по отношению друг к другу;
где алкил, алкенил, алкинил, циклоалкил, арил и гетероарил могут иметь один или несколько заместителей, выбранных из группы, содержащей алкил алкенил, алкинил, циклоалкил, арил, гетероарил, алкокси, -NO2, галоген; и
атом серы в положении 1 семичленного кольца и атомы углерода в положении 4 и положении 5 семичленного кольца являются хиральными центрами.
Предпочтительней энантиомерно-обогащенный тетрагидробезотиепин-1-оксид формлы (I) имеет структуру, показанную в формулах ((4R, 5R)-XXVII), ((4S, 5S)-XXVII), ((4R, 5R)-XXVI) или ((4S, 5S)-XXVI):



Предпочтительней R1 и R2 представляют собой алкильные группы. Более предпочтительно R1 и R2 представляют собой бутил. В другом предпочтительном воплощении один из R1 и R2 представляет собой этил, а другой представляет собой бутил. Предпочтительней R4, R5, R6 и R7 независимо друг от друга выбраны из Н, -NO2 и -NR9R10. Более предпочтительно, каждый из R4, R6 и R7 является H, a R5 представляет собой -NO2 или -NR9R10. Еще более предпочтительно каждый из R4, R6 и R7 представляет собой Н, а R5 представляет собой диметиламиногруппу. Способ включает циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида, имеющего формулу (II)

в которой R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, и атом серы является хиральным центром с образованием энантиомерно-обогщенного тетрагидробензотиепин-1-оксида.
Предпочтительней R3 имеет формулу (VI):

в которой R11 и R12 независимо выбраны из группы, включающей алкил, простой полиэфир, фторид, хлорид, бромид, иодид, NR20(O)R19, NR19R20 и OR19, в которых указанные алкил и простой полиэфир могут в свою очередь иметь заместители SO3R15, N+R15R17R18A- и четвертичный гетероарил;
R19 выбран из группы, включающей водород, алкил, алкенил, алкинил, полиалкил, арил, арилалкил, циклоалкил, гетероцикл, гетероарил, четвертичный гетероцикл, четвертичный гетероарил, простой полиэфир, алкиларилалкил, алкилгетероарилалкил, алкилгетероциклоалкил, гетероциклоалкил, гетероарилалкил, четвертичный гетероциклоалкил, алкиламмонийалкил, карбоксиалкиламинокарбонилалкил и четвертичный гетероарилалкил;
в вышеуказанном R1 алкил, алкенил, алкинил, арилалкил, гетероцикл и нолиалкил могут иметь один или несколько атомов углерода, замещенных на О, NR15, N+R15R16A-, S, SO, SO2, S+R15A-, PR15, P+R15R16A-, P(O)R15, фенилен, углевод, аминокислоту, пептид или полипептид;
R19 имеет по желанию один или несколько заместителей, выбранных из группы, включающей гидроксил, аминогруппу, сульфогруппу, карбоксил, сульфалкил, карбоксиалкил, сульфалкил, алкил, гетероцикл, гетероарил, четвертичный гетероциклоалкил, четвертичный гетероарилалкил, гуанидинил, четвертичный гетероцикл, четвертичный гетероарил, OR15, NR15R16, N+R15R17R18A-, SR15, S(O)R15, SO2R15, SО3R15, оксо, СO2R15, CN, галоген, CONR15R16, SO2OM, SO2 NR15R16, PO(OR22) OR23, P+R15R16R17A-, S+R15R16A- и С(O)ОМ,
где А- представляет собой фармацевтически приемлемый анион, а М -фармацевтически приемлемый катион,
R15 и R16 независимо друг от друга выбраны из группы, включающей Н, алкил, алкенил, алкинил, циклоалкил, арил, ацил, гетероцикл, аммонийалкил, арилалкил, карбоксиалкил, карбоксигетероарил, карбоксигетероцикл, карбоалкоксиалкил, карбалкиламино, гетероарилалкил, гетероциклоалкил и алкиламмонийалкил;
R17 и R18 независимо выбраны из группы, включающей Н, алкил, алкенил, алкинил, арил, арилалкил, алкенилалкил, алкинилалкил, гетероцикл, карбоксиалкил, карбалкоксиалкил, циклоалкил, цианалкил, OR15, NR15R16, SR15, S(O)R15, SO3R15, CN, галоген, оксо и CONR15R16, где R15 и R16 такие, как определено выше, или
R17 и R18 вместе с атомами азота или углерода, с которыми они связаны, образуют кольцо; и
R22 и R23 независимо друг от друга выбраны из заместителей, составляющих R15 и М, и
R13 и R14 представляют собой водород.
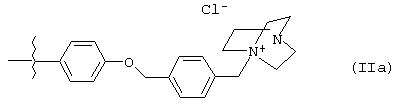
Предпочтительней, когда R3 представляет собой 4-метоксифенил или группу, имеющую структуру формулы (IIа), в которой структура формулы (IIа) находится в положении 4. Более предпочтительно, когда R3 представляет собой группу, имеющую структуру формулы (IIа)

Удивительно, что реакция циклизации энантиомерно-обогащенного арил-3-пропапальсульфоксида (II) протекает с высокой степенью стереоселективности в 4 и 5 положениях семичленного кольца тетрагидробензотиепин-1-оксида. Например, условие реакции можно выбрать так, чтобы получать преимущественно (4R, 5R)-тетрагидробензотиепин-1-оксид или (4S, 5S)-тетрагидробензотиепин-1-оксид.
В одном воплощении настоящего изобретения циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида (III) осуществляют в присутствии основания, например алкоголята. Особенно пригодным основанием для такой циклизации является трет-бутилат калия.
Энантиомерно-обогащенный арил-3-пропанальсульфоксид (II) можно получить, например, окислением энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида, имеющего формулу (III)
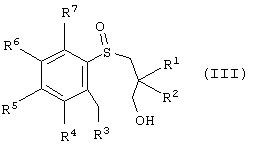
где R1, R2, R3, R4, R5, R6 и R7 такие, как описано ранее, с образованием энантиомерно-обогащенного арил-3-пропанальсульфоксида.
В одном из воплощений настоящего изобретения окисление энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида (III) осуществляют, например, в присутствии комплекса триоксида серы, такого как пиридиновый комплекс триоксида серы. В другом воплощении настоящего изобретения окисление энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида (III) может быть проведено в присутствии хром-пиридиниевого комплекса.
Еще в одном воплощении настоящего изобретения энантиомерно-обогащенный арил-3-гидроксипропилсульфоксид можно получать окислением в условиях энантиоселективного окисления арил-3-гидроксипропилсульфоксида, имеющего формулу (IV):
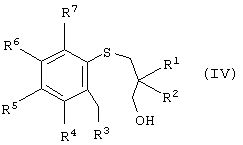
где R1, R2, R3, R4, R5, R6 и R7 описаны ранее с образованием энантиомерно-обогщенного арил-3-гидроксипропилсульфоксида.
Условия энантиоселективного окисления могут включать алкоголят титана (IV) и диалкилтартрат. Предпочтительно, когда алкоголят титана (IV) представляет собой изопропилат титана (IV), а диалкилтартрат является диэтил-D-тартратом. По желанию условия энантиоселективного окисления могут еще включать гидроперекисное соединение, имеющее формулу (V)
R8-O-O-H (V)
где R8 является радикалом, выбранным из группы, содержащей Н, алкил, карбалкил, бензил, бензоил и кумил. Предпочтительней R8 представляет собой алкил или кумил, более предпочтительно - трет-бутил или кумил.
Дополнительные примеры гидроперекисных соединений, пригодных для энантиоселектисиого окисления в настоящем избретении, можно найти в работе Zhao и др., Tetrahedron, 1987, 43 (21), 5135-5144.
В другом воплощении настоящего изобретения условия энантиоселективного окисления для превращения арил-3-гидроксипропилсульфида (IV) в арил-3-гидроксипропилсульфоксид (III) могут включать хиральный (сален) металлический комплекс и окислитель. Окислителем может быть, например, перекись водорода, диалкилпероксид или иодсиларин, такой как диацетат иодбензола. Предпочтительно окислитель представляет собой диацетат иодбензола. (Сален) металлический комплекс предпочтительно представляет собой (S,S)-(+)-N,N'-бис(3,5-ди-трет-бутилсалициклиден-1,2-циклогександиамино-марганец (III) хлорид. Дополнительные примеры (сален) металлических комплексов, пригодных в настоящем изобретении, можно найти в работах: Palucki и др. Tetrahedron Letters, 1992, 33 (47), 7111-7114; Saski и др., Bull. Chem. Soc. Jpn., 1991, 64, 1318-1324.
Еще в одном воплощении настоящего изобретения условия энантиоселективного окисления для превращения арил-3-гидроксипропилсульфида (IV) в арил-3-гидроксипропилсульфоксид (III) могут включать хиральный оксазиридин. Предпочтительно хиральный оксазиридин представляет собой (1R)-(-)-(8,8-дихлор-10-камфор-сульфонил)оксазиридин или (1S)-(+)-(8,8-дихлор-10-камфор-сульфонил)оксазиридин. Дополнительные примеры хиральных оксазиридинов, пригодных для настоящего изобретения, можно найти в работе Dаvis и др. J. Am. Chem. Soc. 1992, 114 (4), 1428-1437.
Способ настоящего изобретения может быть также использован для получения энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида, имеющего формулу (VII):

где R1, R2, R3, R4, R5, R6 и R7 такие, как описано ранее, и где способ включает окисление энантиомерно-обогащенного тетрагидробензотиепин-1-оксида (I) в энантиомерно-обогащенный тетрагидробензотиепин-1,1-диоксид (VII). Окисление может быть осуществлено в присутствии органических надкислот, предпочтительно м-хлорнадбензойной кислоты. Энантиомерно-обогащенный тетрагидробензотиепин-1-оксид (I) можно получить любым из способов, описанных выше. Если желательно энантиомерно-обогащенный тетрагидробензотиепин-1,1-диоксид (VII) может быть использован в дальнейших реакциях.
И еще в одном воплощении настоящее изобретение предоставляет идеальное ингибирующее транспорт желчных кислот соединение (IBAT ингибиторы), полезное для профилактики или лечения сердечно-сосудистых заболеваний, включая гиперхолестеролэмию и атеросклероз, причем вышеуказанные IBAT ингибиторные соединения показаны в таблице.

Биологические испытания
В следующих испытаниях продемонстрирована полезность соединений данного изобретения. Эти испытания проведены in vitro с использованием известной методики, чтобы показать полезность настоящего изобретения.
Испытания in vitro соединений, которые ингибируют осуществляемое через посредство IBAT поглощение [14С] Таурохолата (ТХ) в Н14-клетках
Клетки почки детеныша хомяка (КПХ), трансформированные посредством кДНК человеческого ШАТ (H14 - клетки), посеяны по 60000 клеток на лунку на Топ-каунт чашки с 96 лунками для испытаний, проводимых через 24 часа после посева, по 30000 клеток на лунку для испытаний, проводимых через 48 часов и по 10000 клеток на лунку для испытаний, проводимых через 72 часа.
В день испытаний монослой клеток один раз осторожно промывают порцией в 100 мкл буферного раствора для испытаний (среда Игла, модифицированная по Далбекко с 4,5 г/л глюкозы + 0,2% (вес./об.) свободного от жирных кислот бычьего сывороточного альбумина - (СЖК) БСА). В каждую лунку добавляют по 50 мкл двойного концентрата испытуемого соединения в буферном растворе для испытаний вместе с 50 мкл 6 мкМ [14С] - таурохолата в буферном растворе для испытаний (конечная концентрация 3 мкМ [С] - таурохолата). Чашки с культурой клеток инкубируют в течение 2 часов при 37°С, затем осторожно каждую лунку дважды промывают при 4°С фосфат-буферным физиологическим раствором Далбекко (ФБД), содержащим 0,2% (вес/об) (СЖК) БСА. Затем лунки осторожно промывают один раз при 4°С 100 мкл ФБД без (СЖК) БСА. На каждые 200 мкл раствора добавляют жидкость для сцинтиляционного счетчика, чашки заплавляют и встряхивают 30 мин при комнатной температуре, прежде чем измерять радиоактивность в каждой лунке с помощью счетчика Packard Top-Count.
Испытания in vitro соединений, которые ингибируют поглощение [14С] -аланина
Испытание поглощения аланина проводят так же, как испытание с таурохолатом, с тем исключением того, что таурохолат заменяют аланином.
Описание препаративных способов
Исходные материалы, используемые в способах получения настоящего изобретения, изоестны или могут быть получены обычными способами, известными специалистам, или аналогичными описанным в литературе.
Вообще препаративные способы настоящего изобретения можно представить следующим образом.
Как показано на фиг.1 взаимодействие фенола (VIII) с гидридом натрия (или другим основанием, которое будет отрывать водород от бензольного кольца фенола (VIII)) с последующим добавлением хлорметильного соединения (IX) приводит к получению 2-гидроксибензильного соединения (X). Обработка 2-гидроксибензильного соединения (X) гидридом натрия (или другим основанием, которое будет отрывать водород от соединения (X)), а затем диметилтиокарбамоилхлоридом приводит к образованию тиокарбамата (XI). Тиокарбамат (XI) кипятят с обратным холодильником в растворителе, таком как дифениловый эфир, в течение времени, достаточного для проведения перегруппировки тиокарбамата (XI) (например, в течение ночи), отделяют фильтрованием, а затем обрабатывают основанием, таким как гидроксид натрия, по желанию в растворителе, таком как тетрагидрофуран с метанолом, и получают тиофенол (XII). Обработка 2,2-дизамещенного-1,3-пропандиола (XIII, фиг.2) амином, таким как триэтиламин, с последующим добавлением тионилхлорида приводит к получению циклического сульфита (XIV). Образование циклического сульфита (XIV) может по желанию быть проведено в растворителе, таком как безводный метиленхлорид. Реагирование циклического сульфита (XIV) в условиях, способствующих окислению, дает циклический сульфат (XV). Например, условия, способствующие окислению, могут включать реагирование циклического сульфита (XIV) в присутствии хлорида рутения (III) и периодата натрия в течение ночи с получением циклического сульфата (XV). Обработка тиофенола (XII) основанием, способным удалить водород из сульфгидрильной группы тиофенола (XII) (например, гидридом натрия), с последующим добавлением циклического сульфата (XV) дает арил-3-гидроксипропилсульфид (IV). Окисление арил-3-гидроксипропилсульфида (IV) асимметричным окислителем, например (IR)-(-)-(8,9-дихлор-10-камфорсульфонил)оксазиридином, приводит к получению арил-3-гидроксипропилсульфоксида (III). Реагирование арил-3-гидроксипропилсульфоксида (III) в условиях, способствующих окислению (например, в присутствии комплекса пиридина с триоксидом серы) приводит к образованию арил-3-прпанальсульфоксида (II). Циклизация арил-3-пропанальсульфоксида (II) с основанием (например, основным алкоголятом, таким как трет-бутилат калия), приводит к образованию тетрагидробензотиепин-1 -оксида (I). Окисление тетрагидробензотиепин-1-оксида (I) окислителем, например м-хлорпероксибензойной кислотой, приводит к тетрагидробензотиепин-1,1-диоксиду (VII).
Сокращения, используемые в последующем описании и в примерах, имеют следующие значения:
NaH - гидрид натрия
ClC(S)NMe2 - диметилтиокарбамоилхлорид
Ph2O - дифениловый эфир
SO3Ру - пиридиновый комплекс триоксида серы
трет-ВиОК - трет-бутилат калия
м-СРВА - м-хлорнадбензойная кислота
ТГФ - тетрагидрофуран
ЛАГ - литийалюминийгидрид
КТ - комнатная температура
В реакции, показанной на фиг.1 и 2,
R1 и R2 могут быть независимо выбраны из замещенных и незамещенных радикалов, таких как алкил, содержащий от 1 до 20 атомов С, алкенил, содержащий от 2 до 20 атомов С, арил и гетероарил,
R3 может быть выбран из замещенных и незамещенных радикалов арил и гетероарил,
R4, R5, R6 и R7 могут быть независимо выбраны из замещенных и незамещенных радикалов: алкил, содержащий от 1 до 20 атомов С, алкенил, содержащий от 2 до 20 атомов С, арил, гетероарил, алкокси, арилокси, а также водород, -NO2 и -NR9R10,
R9 и R10 могут быть независимо выбраны из замещенных и незамещенных радикалов: алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, а также водород, бутоксикарбонил, карбобензилокси и другие известные аминзащищающие группы. Заместитель (заместители) у различных групп могут быть выбраны из таких радикалов, как алкил, алкенил, алкинил, циклоалкил, арил, гетероарил, алкокси, арилокси, -NO2 и галоген.
Далее приведены примеры некоторых специфических соединений и условий реакции, которые могут быть использованы в способе данного изобретения. Следующие примеры служат для иллюстрации различных аспектов настоящего изобретения, однако не ограничивают объем охраны изобретения.
Пример 1. Получение 4-фтор-2-((4-метоксифенил)метил)-фенола (XVI)

К раствору 23,66 г 95%-ного гидрида натрия (0,94 моль) в 600 мл сухого толуола при 0°С при перемешивании добавляют 100,0 г 4-фторфенола (0,89 моль). Смесь перемешивают при 50°С в течение 1 часа пока не прекратится газовыделение. Смесь охлаждают до комнатной температуры и добавляют раствор 139,71 г 3-метоксибензилхлорида (0,89 моль) в 400 мл сухого толуола. После кипячения с обратным холодильником в течение 24 ч смесь охлаждают до комнатной температуры и гасят водой (500 мл). Органический слой отделяют, сушат над MgSO4 и концентрируют в высоком вакууме. Остающиеся исходные материалы удаляют отгонкой. Сырое темно-красное масло фильтруют через слой силикагеля (1 л) с помощью чистого гексана и получают 53,00 г (25,6%) продукта в виде розового твердого вещества: 1Н ЯМР (CDCl3) d 3.79 (s, 3Н), 3.90 (s, 2H), 4.58 (s, 1H), 6.70-6.74 (m, 1H), 6.79-6.88 (m, 4H), 7.11-7.16 (т, 2H).
Пример 2. Получение 4-фтор-2-((4-метрксифепил)метил)-тиофенола (XVII)

Стадия 2 а: Получение тиокарбамата (XVIII)

К раствору 50,00 г (215,30 моль) 4-фтор-2-((4-метоксифенил) метил)фенола (XVI) в 500 мл сухого ДМФ при перемешивании добавляют при 2°С 11,20 г 60%-ной дисперсии гидрида натрия в минеральном масле (279,90 ммоль). Смеси дают нагреться до комнатной температуры и добавляют 26,61 г диметилтиокарбамилхлорида (215,30 ммоль). Реакционную смесь при комнатной температуре перемешивают в течение ночи. Реакционную смесь гасят на ледяной бане водой (100 мл). Раствор экстрагируют диэтиловым эфиром (500 мл). Эфирный раствор промывают водой (500 мл) и солевым раствором (500 мл). Эфирный раствор сушат над MgSO4 и эфир отгоняют досуха. Сырой продукт фильтруют через слой из 500 мл силикагеля, используя смесь 5% этилацетат/гексан с получением 48,00 г (69,8%) продукта в виде тусклого белого твердого вещества: 1Н ЯМР(СОСl3) d 3.21 (s, 3Н), 3.46 (s,
3Н), 3.80 (s, 3Н), 3.82 (s, 2H), 6.78-6.86 (m, 3H), 6.90-7.00 (m, 2Н), 7.09 (d, J=8.7 Hz, 2H).
Стадия 2 b: Перегруппировка и гидролиз тиокарбамата (XVIII в 4-фтор-2-((4-метоксифенил)метил)-тиофенол (XVII)
Перемешиваемый раствор 48,00 г (150,29 ммоль) тиокарбамата (XVIII) в 200 мл дифенилового эфира кипятят с обратным холодильником в течение ночи. Раствор охлаждают до комнатной температуры и фильтруют через 1 л силикагеля, используя 2 л гексана, для удаления фенилового эфира. Продукт перегруппировки промывают смесью 5% этилацетат/гексан с получением 45,00 г (95,8%) продукта в виде бледно-желтого твердого вещества: 1Н ЯМР (СDСl3) d 3.02 (s, 3Н), 3.10 (s, 3Н), 3.80 (s, ЗН), 4.07 (s, 2H), 6.82-6.86 (m, 3Н), 6.93 (dt, J=8.4 Hz, 2.7 Hz, 1H), 7.08 (d, J=8.7 Hz, 2H), 7.49 (dd, J=5.0 Hz, 8.7 Hz, 1H).
К раствору 46,00 г (144,02 ммоль) продукта перегруппировки (см. выше) в 200 мл метанола и 200 мл ТГФ добавяют 17,28 г NaOH (432,06 ммоль). Смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Растворитель выпаривают и добавяют 200 мл воды. Водный раствор промывают дважды диэтиловым эфиром по 200 мл и помещают в баню со льдом. Водную смесь подкисляют до рН 6 концентрированным раствором НСl. Раствор экстрагируют дважды диэтиловым эфиром (по 300 мл). Эфирные фазы объединяют, сушат над MgSO4 и выпаривают досуха с получением 27,00 г (75,5%) продукта в виде коричневого масла: 1H ЯМР (CDCl3) d 3.24 (s, 1H), 3.80 (s, 3Н), 3.99 (s, 2H), 6.81-6.87 (m, 4Н), 7.09 (d, J=8.7 Hz, 2H), 7.27-7.33 (m, 1H).
Пример 3. Получение дибутилциклического сульфата (XIX)

Стадия 3а: Получение 2.2-дибутил-1.3-пропандиола
К раствору дибутилдиэтилмалоната (фирмы Aldrich) (150 г, 0,55 моль) в сухом ТГФ (700 мл), помещенном в баню со смесью ацетон/сухой лед, при перемешивании добавляют по каплям 662 мл (1,2 экв., 0,66 моль) ЛАГа (1 М ТГФ), поддерживая температуру в пределах от -20 до 0°С. Реакционную смесь перемешивают в течение ночи при КТ. Затем смесь охлаждают до -20°С и добавляют по каплям 40 мл соды и 80 мл 40% NaOH, и еще 80 мл воды. Образовавшуюся суспензию фильтруют. Фильтрат сушат над сульфатом натрия и концентрируют в вакууме с получением диола в количестве 98,4 г (выход 95%) в виде масла. МС спектры, протонные и углеродные ЯМР спектры сообразны с данным продуктом.
Стадия 3 а: Получение дибутилциклического сульфита (XX)

Раствор 2,2-дибутил-1,3-пропандиола (103 г, 0,548 моль) и триэтиламина (221 г, 2,19 моль) в безводном метиленхлориде (500 мл) перемешивают при 0°С в атмосфере азота. К этой смеси добавяют по каплям тионилхлорид (97,8 г, 0,82 моль) и в течение 5 мин раствор становится желтым, а затем черным, когда добавление проводят в пределах получаса. Реакционную смесь перемешивают в течение 3 ч при 0°С. ГХ показывает отсутствие каких-либо исходных материалов. Смесь промывают дважды ледяной водой, затем дважды солевым раствором. Органическую фазу сушат над сульфатом магния и концентрируют в вакууме с получением 128 г (100%) дибутилциклического сульфита (XX) в виде темного масла. Масс-спектры (МС) сообразуются с данным продуктом.
Стадия 3 b: Окисление дибутилциклического сульфита (XX) до дибутилциклического сульфата (XIX)
К раствору дибутилциклического сульфита (XX) (127,5 г, 0,54 моль) в 600 мл ацетонитрила и 500 мл воды, охлажденному на ледяной бане в атмосфере азота, добавяют хлорид рутения (III) (1 г) и периодат натрия (233 г, 1,08 моль). Реакционную смесь перемешивают в течение ночи, цвет раствора становится черным. ГХ показывает отсутствие исходных материалов. Смесь экстрагируют эфиром (300 мл), и эфирный экстракт трижды промывают раствором. Органическую фазу сушат над MgSO4 и пропускают через целит. Фильтрат концентрируют в вакууме и получают 133 г (97,8%) дибутилциклического сульфата (XIX) в виде масла. Данные протонного и углеродного ЯМР и МС сообразуются с данным продуктом.
Пример 4. Получение арил-3-гидроксипропилсульфида (XXI)
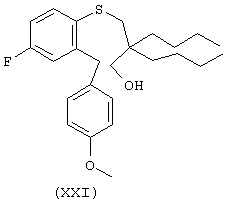
К раствору 27,00 г (108,73 моль) 4-фтор-2-((метоксифенил)метил)тиофенола (XVII) в 270 мл диглима при перемешивании добавляют при 0°С 4,35 г 60% дисперсии гидрида натрия в минеральном масле (108,73 ммоль). После окончания газовыделения добавляют при 0°С 29,94 г (119,60 ммоль) дибутилциклического сульфата (XIX) и перемешивали в течение 10 мин. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Растворитель выпаривают и добавяют 200 мл воды. Раствор промывают диэтиловым эфиром (200 мл) и добавяют 25 мл концентрированной серной кислоты для получения 2,0 М раствора, который кипятят с обратным холодильником в течение ночи. Раствор экстрагируют этилацетатом, органическую часть сушат над MgSO4 и концентрируют в вакууме. Сырой этил-3-гидроксипропилсульфид (XXI) очищают хроматографически на силикагеле (Waters Prep 500), используя смесь 8% этилацетат/гексан, с получением 33,00 г (72,5%) продукта в виде светло-коричнового масла: 1Н ЯМР (CDCl3) d 0.90 (t, J=7.1 Hz, 611), 1.14-1.34 (m, 12K), 2.82 (s, 2H), 3.48 (s, 2H), 3.79 (s, 3Н), 4.10 (s, 2H), 6.77-6.92 (m, 4Н), 7.09 (d, J=8.7 Hz, 2H), 7.41 (dd, J=8.7 Hz. 5.7 Hz, 1Н).
Пример 5. Получение энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида (ХХII)
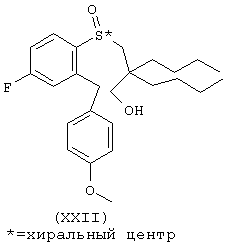
К раствору 20,00 г (47,78 ммоль) арил-3-гидроксипропилсульфида (XXI) в 1 л метиленхлорида при 2°С, при перемешивании добавяют 31,5 г 96%-ного (1R)-(-)-8,8-дихлор-10-камфорсульфонил)оксазиридина (100,34 ммоль, Aldrich). После растворения всего оксазиридина смесь помещают при -30°С в холодильник на 72 ч. Растворитель выпариают и сырое твердое вещество промывают гексаном (1 л). Белое твердое вещество фильтруют, а гексановый раствор концентрируют в вакууме. Сырое масло очищают хроматографически на колонке с силикагелем (Water Prep 500), используя в качестве элюента смесь 15% этилацетат/гексак, и получают 19,00 г (95%) энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида (ХХII) в виде бесцветного масла: Н ЯМР (CDCl3) d 0.82-0.98 (m, 6H), 1.16-1.32 (m, 12Н), 2.29 (d, J=13.8 Hz, 1H), 2.77 (d, J=13.5 Hz, 1H), 3.45 (d, J=12.3 Hz, 1H), 3.69 (d, J=12.3 Hz, 1H), 3.79 (s, 3H), 4.02 (q, J=15.6 Hz, 1H), 6.83-6.93 (m, 3H), 7.00 (d, J=8.1 Hz, 2H), 7.18-7.23 (m, 1H), 7.99-8.04 (т, 1Н).
Энантиомерное обогащение было определено посредством хиральной ВЭЖХ на колонке (R,R)-Whelk-О при использовании смеси 5% этанол/гексан в качестве элюента. Оказалось, что оно составляет 78% э.о. с первым пиком элюции в качестве основного продукта.
Пример 6. Получение энантиомерно-обогащенного арил-3-пропанальсульфоксида (ХХIII)

К раствору 13,87 г триэтиламина (131,16 ммол, фирма Aldrich) в 200 мл диметилсульфоксида при комнатной температуре добавили при перемешивании 19,00 г (43,72 ммоль) энантиомерно-обогащенного арил-3-гидрокси пропилсульфоксида (ХХII) и 20,96 г (131,16 ммол, Aldrich) комплекса пиридина с триоксидом серы. После того как смесь перемешивали при комнатной температуре в течение 48 ч, добавили 500 мл воды и смесь энергично перемешивали. Затем смесь экстрагировали дважды этилацетатом (по 500 мл). Этилацетатный слой отделяли, сушили над MgSO4 и концентрировали в вакууме. Сырое масло фильтровали через 500 мл силикагеля, используя смесь 15% этилацетата/гексана и получили 17,30 г (91%) энантиомерно-обогащенного арил-3-пропанальсульфоксида (XXIII) в виде светло-оранжевою масла: 1Н ЯМР (CDCl3) d 0.85-0.95 (m, 6H), 1.11-1.17 (m, 4H), 1.21-1.39 (m, 4H), 1.59-1.76 (m, 4H), 1.89-1.99 (m, 1H), 2.57 (d, J=14.1 Hz, 1H), 2.91 (d, J=13.8 Hz, 1H), 3.79 (s, 3H), 3.97 (d, J=15.9 Hz, 1H), 4,12 (d, J=15.9 Hz, 1H), 6.84-6.89 (m, 3H), 7.03 (d, J=8.4 Hz, 2H), 7.19 (dt, J,=8.4 Hz, 2.4 Hz, 1H), 8.02 (dd, J=8.7 Hz, 5.7 Hz, 1H), 9.49 (s, 1H).
Пример 7. Получение энантиомерно-обогащенного тетрагидробензотиепин-1-оксида ((4R, 5R)-ХХIV)
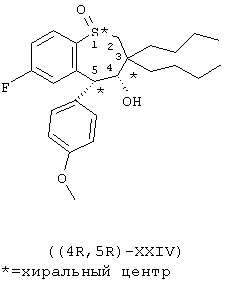
К раствору 17,30 г (39,99 ммоль) энантиомерно-обогащенного арил-3-пропанальсульфоксида (XXIII) в 300 мл сухого ТГФ при -15°С в атмосфере азота при перемешивании добавляют 48 мл 1 М раствора трет-бутилата калия в ТГФ (1,2 эквивалента). Раствор перемешивают при -15°С в течение 4 ч. Затем раствор гасят водой (100 мл) и нейтрализуют концентрированной HСl (4 мл) при 0°С. Слой ТГФ отделяют, сушат над MgSO4 и концентрируют в вакууме. Энантиомерно-обогащенный тетрагидробензотиепин-1-оксид ((4R, 5R)-XXIV) очищают хроматографически на силикагеле (Waters Prep 500), используя смесь 15% этилацетат/гексан, с получением 13,44 г (77,7%) продукта в виде белого твердого вещества: 1Н ЯМР (CDCl3) d 0.87-0.97 (m, 6H), 1.16-1.32 (m, 4H), 1.34-1.48 (m, 4H), 1.50-1.69 (m, 4H), 1.86-1.96 (m, 1H), 2.88 (d, J=13.0 Hz, 1H), 3.00 (d, J=13.0 Hz, 1H), 3.85 (s, 3Н), 4.00 (s, 1H), 4.48 (s, 1H), 6.52 (dd, J=9.9 Hz, 2.4 Hz, 1H), 6.94 (d, J=9 Hz, 2H), 7.13 (dt, J=8.4 Hz, 2.4 Hz, 1H), 7.38 (d, J=8.7 Hz, 2H), 7.82 (dd, J=8.7 Hz, 5.7 Hz, 1H).
Пример 8. Получение энантиомерно-обогащениого тетрагидробензотиепин-1,1-диоксида ((4R. 5R)-ХХV)

К раствору 13,44 (31,07 ммоль) энантиомерно-обогащенного тетрагидробензотиепин-1-оксида ((4R, 5R)-XXIV) в 150 мл метиленхлорида при 0°С при перемешивании добавяют 9,46 г 68%-ной м-хлорнадбензойной кислоты (37,28 ммоль, фирма Sigma). После перемешивания при 0°С в течение 2 ч смеси дают нагреться до комнатной температуры и перемешивают еще в течение 4 ч. В смесь добавляют 50 мл насыщенного раствора Na2SO3 и перемешивают в течение 30 мин. Затем раствор нейтрализуют насыщенным раствором NаНСО3 (50 мл). Мстиленхлоридный слой отделяют, сушат над MgSO4 и концентрируют в вакууме с получением 13,00 г (97,5%) энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида ((4R, 5R)-XXV) в виде светло-желтого твердого вещества: 1Н ЯМР (CDCl3) d 0.89-0.95 (m, 6H), 1.09-1.42 (m, 12Н), 2.16-2.26 (m, 1H), 3.14 (q, J,=15.6 Hz, 1H), 3.8.7 (s, 3H), 4.18 (s, 1H), 5.48 (s, 1H), 6.54 (dd, J=10.2 Hz, 2.4 Hz, 1H), 6.96-7.07 (m, 3H), 7.40 (d, J=8.1 Hz, 2H), 8.11 (dd, J=8.6 Hz, 5.9 Hz, 1H).
Пример 9. Получение энантиомерно-обогащенного 7-(диметиламино) тетрагидробензотиепин-1,1-диоксида ((4R, 5R)-XXVI)

К раствору 13,00 г (28,98 ммоль) энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида ((4R,5R)-XXV) в 73 мл раствора диметиламина (20 М в ТГФ, 146 ммоль) в реакторе Парра добавляют около 20 мл чистого днметиламина. Смесь запаивают и перемешивают при 110°С в течение ночи и охлаждают до комнатной температуры. Избыток диметиламина выпаривают. Сырое масло растворяют в 200 мл этилацетата и промывают водой (100 мл), сушат над MgSO4 и концентрируют в вакууме. Очистка на колонке с силикагелем (Waters Prep 500) с применением смеси 20% этилацетат/гексан дает 12,43 г (90,5%) энантиомерно-обогащенпого 7-(диметиламино)- тетрагидробензотиепин-1,1-диоксида ((4R, 5R)-XXVI) в виде бесцветного твердого нсщества: 1H ЯМР (CDCl3) d 0.87-0.93 (m, 6H), 1.10-1.68 (m, 12H), 2.17-2.25 (m, 1Н), 2.81 (s, 6H), 2.99 (d, J=15.3 Hz, 1H), 3.15 (d, J=15.3 Hz, 1H), 3.34 (s, 3H), 4.11 (d, J=7.5 Hz, 1H), 5.49 (s, 1H), 5.99 (d, J.=2.4 Hz, 1H), 6.51 (dd, J=8.7 Hz, 2.4 Hz, 1H), 6.94 (d, J=8.7 Hz, 2H), 7.42 (d, J=8.4 Hz, 2H), 7.90 (d, J=8.7 Hz, 1H).
С помощью хиральной ВЭЖХ на колонке Chiralpak AD с использованием в качестве элюента 5% этанол/гексан было определено, что данный продукт имеет 78% э.о. Перекристаллизация этого твердого вещества из смеси этилацетат/гексан дает 1,70 г рацемического продукта. Оставшийся раствор концентрируют и перекристаллизовывают с получением 9,8 г бесцветного твердого вещества. Энантиомерное обогащение этого твердого вещества было определено хиральной хроматографией на колонке Chiralpak AD с использованием смеси 5% этанол/гексан в качестве элюента. Оказалось, что оно составляет 95% э.о. с первым пиком элюции в качестве основного продукта.
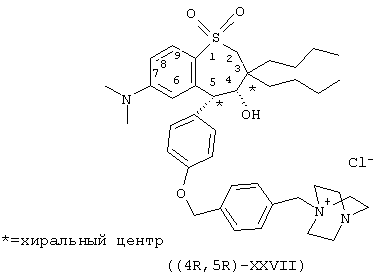
Пример 10. Получение энантиомерно-обогащенного (4R, 5R)-1-[[4-[[4-[3,3-дибутил-7-(диметиламино)-2,3,4,5-тетрагидро-4-гидрокси-1,1-диоксидо-1-бензотиепин-5-ил]фенокси]метил]фенил]метил]-4-аза-1-азониабицикло[2,2,2]октанхлорида ((4R, 5R)-ХХVII)
Стадия 1: Получение 4-фтор-2-((4-метоксифенил)метил)-фенола
К раствору 23,66 г 95% гидрида натрия (0,94 моль) в 600 мл сухого толуола при перемешивании добавляют 100,0 г 4-фторфенола (0,89 моль) при 0°С. Смесь перемешивают при 90°С в течение 1 ч пока до прекращения газовыделения. Затем смеси дают остыть до комнатной температуры и добавляют раствор 139,71 г 3-метоксибензилхлорида (0,89 моль) в 400 мл сухого толуола. После кипячения с обратным холодильником в течение 24 ч смесь охлаждают до комнатной температуры и гасят водой (500 мл). Отделяют органический слой, сушат его над MgSO4 и концентрируют в высоком вакууме. Остающиеся исходные материалы удаляют отгонкой. Сырое темно-красное масло фильтруют через слой 1 л силикагеля с помощью чистого гексана с получением 53,00 г (25,6%) продукта в виде розового твердого вещества: 1Н ЯМР (СDСl3) d 3.79 (s, 3H), 3.90 (s, 2H), 4.58 (s, 1H), 6.70-6.74 (m, 1H), 6.79-6.88 (m, 4Н), 7.11-7.16 (m, 2H).
Стадия 2: Получение 4-фтор-2-((4-метоксифенил)метил)-тиофенола
Стадия 2а. Получение тиокарбамата
К раствору 50,00 г (215,30 ммоль) 4-фтор-2-((4-метоксифенил)метил)-фенола в 500 мл сухого ДМФ при перемешивании добавяют при 2°С 11,20 г 60%-ной дисперсии гидрида натрия в минеральном масле (279,90 ммоль). Смеси дают нагреться до комнатной температуры и добавляют 6,61 г (215,30 ммоль) диметилтиокарбамилхлорида. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь гасят на ледяной бане водой (100 мл). Раствор экстрагируют диэтиловым эфиром (500 мл). Эфирный раствор промывают водой (500 мл) и солевым раствором (500 мл). Эфирный раствор сушат над MgSO4 и выпаривают досуха. Сырой продукт фильтруют через слой из 500 мл силикагеля, используя смесь 5% этилацетат/гексан, с получением 48,00 г (69,8%) продукта в виде тускло-белого твердого вещества: 1H ЯМР (CDCl3) d 3.21 (s, 3H), 3.46 (s, 3Н), 3.80 (s, 3H), 3.82 (s, 2H), 6.78-6.86 (m, 3H), 6.90-7.00 (m, 2H), 7.09 (d, J=8.7 Hz, 2H).
Стадия 2 b: Перегруппировка и гидролиз тиокарбамата до 4-фтор-2-((4-метоксифенил)метил)-тиофенола
Раствор 48,00 г (150,29 ммоль) тиокарбамата (полученного на стадии 2 а) в 200 мл дифенилового эфира при перемешивании кипятят с обратным холодильником при 270°С в течение ночи. Раствор охлаждают до комнатной температуры и фильтруют через 1 л силикагеля с 2 л гексана, чтобы удалить простой фениловый эфир. Продукт перегруппировки промывают смесью 5% этилацетат/гексан с получением 46,00 г (95,8%) продукта в виде бледно-желтого твердого вещества: 1Н ЯМР (CDCl3) d 3.02 (s, 3H), 3.10 (s, 3H), 3.80 (s, 3H), 4.07 (s, 2H), 6.82-6.86 (m, 3H), 6.93 (dt, J=8.4 Hz, 2.7 Hz, 1H), 7.08 (d, J=8.7 Hz, 2H), 7.49 (dd, J=6.0 Hz, 8.7 Hz, 1H).
К раствору 46,00 г (144,02 моль) продукта перегруппировки (указанного выше) в 200 мл метанола и 200 мл ТГФ добавляют 17,28 г NaOH (432,06 ммоль). Смесь в атмосфере азота кипятят с обратным холодильником в течение ночи. Отгоняют растворители и добавляют 200 мл води. Водный раствор дважды промывают диэтиловым эфиром (200 мл) и помещают в баню со льдом. Водную смесь подкисляют до рН 6 концентрированным раствором НСl. Раствор дважды экстрагируют диэтиловым эфиром (по 300 мл). Эфирные фазы объединяют, сушат над MgSO4 и выпаривают досуха с получением 37,00 г (75,5%) продукта в виде коричневого масла: 1Н ЯМР (CDCl3) d 3.24 (s, 1H), 3.80 (s, 3H), 3.99 (s, 2H), 6.81-6.87 (m, 4H), 7.09 (d,.J=8.7 Hz, 2H), 7.27-7.33 (m, 1H).
Стадия 3: Получение дибутилциклического сульфата
Стадия 3 а. Получение 2.2-дибутил-1.3-пропандиола
К раствору дибутилдиэтилмалоната (Aldrich) (150 г, 0,55 моль) в сухом ТГФ (700 мл) на бане ацетон/сухой лед при перемешивании по каплям добавяют ЛАГ (1 М ТГФ) 662 мл (1,2 экв., 0,66 моль), поддерживая температуру между -20 и 0°С. Реакционную смесь перемешивают при КТ в течение ночи. Смесь охлаждают до -20°С и добавляют по каплям 40 мл воды, 80 мл 10% раствора NaOH и еще 80 мл воды. Образовавшуюся суспензию фильтруют. Фильтрат сушат над сульфатом натрия и концентрируют в вакууме с получением диола 98,4 г (выход 95%) в виде масла. МС спектры, протонные и углеродные ЯМР спектры сообразны с данным продуктом.
Стадия 3 b: Получение дибутилциклического сульфита
Раствор 2,2-дибутил-1,3-пропандиола (103 г, 0,548 моль, полученного на стадии 3 а) и триэтиламина (221 г, 2,19 моль) в безводном метиленхлориде (500 мл) перемешивают при 0°С в атмосфере азота. К этой смеси по каплям добавляют тионилхлорид (97,8 г, 0,82 моль) и в течение 5 мин раствор становится желтым, затем черным в течение получаса после завершения добавления. Реакционную смесь перемешивают при 0°С в течение 3 ч. ГХ показывает отсутствие каких-либо исходных материалов. Смесь промывают дважды ледяной водой, затем дважды солевым раствором. Органическую фазу сушат над сульфатом магния и концентрируют в вакууме, получая 128 г (100%) дибутилциклического сульфата в виде темного масла. Масс-спектры (МС) соотносятся с данным продуктом.
Стадия 3 с. Окисление дибутилциклического сульфита до дибутилциклического сульфата
К раствору дибутилциклического сульфита (127,5 г, 0,54 мол, полученного на стадии 3 b) в 600 мл ацетонитрила и 500 мл воды, охлажденному на ледяной бане в атмосфре азота, добавляют хлорид рутения (III) (1 г) и периодат натрия (233 г, 1,08 моль). Реакционную смесь перемешивают в течение ночи и цвет раствора становится черным. ГХ показывает отсутствие каких-либо исходных материалов. Смесь экстрагируют эфиром (300 мл), и этот экстракт промывают трижды солевым раствором. Органическую фазу сушат над сульфатом магния и пропускают через целит. Фильтрат концентрируют в вакууме и получают 133 г (97,8%) дибутилциклического сульфата в виде масла. Спектры протонного и углеродного ЯМР и МС соотносятся с данным продуктом.
Стадия 4. Получение арил-3-гидроксипропилсульфида
К раствору 27,00 г (108,73 ммоль) 4-фтор-2-((4-метоксифенил)метил)тиофенола (полученного на стадии 2) в 270 мл диглима при перемешивании добавляют при 0°С 4,35 г 60% дисперсии гидрида натрия в минеральном масле (108,73 ммоль). После окончания газовыделения при 0°С и при перемешивании в течение 10 мин добавляют 29,94 г (119,60 ммоль) дибутилциклического сульфата (полученного на стадии 3 с). Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Растворитель упаривают и добавляют 200 мл воды. Этот раствор промывают диэтиловым эфиром (200 мл) и добавляют 25 мл концентрированной серной кислоты, чтобы получить 2,0 М раствор, который кипятят с обратным кипятильником в течение ночи. Этот раствор экстрагируют этилацетатом и органический слой сушат над MgSO4 и концентрируют в накууме. Сырой арил-3-гидроксипропилсульфид очищают хроматографически на силикагеле (Waters Prep 500) с использованием элюента 5% этилацетат/гексан с получением 33,00 г (72,5%) продукта в виде светло-коричневого масла: 1H ЯМР (СDСl3) d 0.90 (t, J=7.1 Hz, 6H), 1.14-1.34 (m, 12H), 2.82 (s, 2H), 3.48 (s, 2H), 3.79 (s, 3Н), 4.10 (s, 2H), 6.77-6.92 (m, 4Н), 7.09 (d, J=8.7 Hz, 2H), 7.41 (dd, J.=8.7 Hz, 5.7 Hz, 1H).
Стадия 5: Получение энатиомерно-обогащенного арил-3-гидроксипропилсульфоксида
К раствору 20,00 г (47,78 ммоль) арил-3-гидроксипропилсульфида (полученного на стадии 4) в 1 л метиленхлорида при 0°С добавляют при перемешивании 31,50 г 96% (1R-(-)-(8,8-дихлор-10-камфорсульфонил)-оксазиридина (100,34 ммол, Aldrich). После растворения всего оксазиридина смесь помещают в холодильник при -30°С на 72 ч. Растворитель выпаривают и сырое твердое вещество промывают гексаном (1 л). Белое твердое вещество фильтруют, а гексановый раствор концентрируют в вакууме. Сырое масло очищают на колонке с силикагелем (Waters Prep 500), используя смесь 15% этилацетат/гексан с получением 15,00 г (95%) энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида в виде бесцветного масла: 1H ЯМР (CDCl3) d 0.82-0.98 (m, 6H), 1.16-1.32 (m, 12Н), 2.29 (d, J,=13.8 Hz, 1H), 2.77 (d, J.=13.5 Hz, 1H), 3.45 (d, J=12.3 Hz, 1H), 3.69 (d, J.=12.3 Hz, 1H), 3.79 (s, 3H), 4.02 (q, J=15.6 Hz, 1H), 6.83-6.93 (m, ЗН), 7.00 (d, J.=8.1 Hz, 2H), 7.18-7.23 (m, 1H), 7.99-8.04 (m, 1H).
Энантиомсрное обогащение определяют хиральной ВЭЖХ на колонке (R,R)-Whelk-O с использованием смеси 5% этанол/гексан в качестве элюента. Оказалось, что оно составляет 78% э.о. с первым пиком элюции в качестве основного продукта.
Стадия 6: Получение эпантиомерно-обогащенного арил-3-пронанальсульфоксида
К раствору 13,27 г триэтиламина (131,16 ммол, Aldrich) в 200 мл диметилсульфоксида при перемешивания добавляют при комнатной температуре 19,00 г (43,72 ммоль) энатиомерно-обогащенного арил-3-гидроксипропилсульфоксида (полученного на стадии 5) и 20,96 г пиридинтриоксидсеры (131,16 ммол, Aldrich). После перемешивания смеси при комнатной температуре в течение 48 ч, к ней добавляют 500 мл воды и энергично перемешивают. Затем смесь экстрагируют дважды этилацетатом (500 мл). Этилацетатный слой отделяют, сушат над MgSO4 и концентрируют в вакууме. Сырое масло фильтруют через 500 мл спликагеля, используя 15% этилацетат/гексан в качестве элюента. и получают 17,30 г (91%) энантиомерно-обогащенного арил-3-пропанальсульфоксида в виде светло-оранжевого масла: 1Н ЯМР (CDCl3) d 0.85-0.95 (m, 6H), 1.11-1.17 (m, 4H), 1.21-1.39 (m, 4H), 1.59-1.76 (m, 4H), 1.89-1.99 (m, 1H), 2.57 (d, J=14.1 Hz, 1H), 2.91 (d, J=13.8 Hz, 1H), 3.79 (s, 3H), 3.97 (d, J=15.9 Hz, 1H), 4,12 (d, J=15.9 Hz, 1H), 6.84-6.89 (m, 3H), 7.03 (d, J=8.4 Hz, 2H), 7.19 (dt, J=8.4 Hz, 2.4 Hz, 1H), 8.02 (dd, J:=8.7 Hz, 5.7 Hz, 1H), 9.49 (s, 1H).
Стадия 7. Получение энантиомерно-обогащенного тетрагидробензотиепин-1-оксида (4R, 5R)
К раствору 17,30 г (39,99 моль) энантиомерно-обогащенного арил-3-пропанальсульфоксида (полученного на стадии 6) в 300 мл сухого ТГФ при перемешивании, при -15°С в атмосфере азота добавляют 48 мл 1,0 М трет-бутилата калия в ТГФ (1,2 экв). Раствор перемешивают при -15°С в течение 4 ч. Затем раствор гасят водой (100 мл) и нейтрализуют концентрированной НСl (4 мл) при 0°С. Слой ТГФ отделяют, сушат над MgSO4 и концентрируют в вакууме. Энантиомерно-обогащенный тетрагидробензотиепин-1 -оксид (4R, 5R) очищают хроматографически на силикагеле (колонка Waters Prep 500), используя как элюент 15% этилацетат/гексан, и получают 13,44 г (77,7%) продукта в виде белого твердого вещества: 1Н ЯМР (CDCl3) d 0.87-0.97 (m, 6H), 1.16-1.32 (m, 4H), 1.34-1.48 (m, 4Н), 1.50-1.69 (m, 4Н), 1.86-1.96 (m, 1Н), 2.88 (d, J,=13.0 Hz, 1H), 3.00 (d, J=13.0 Hz, 1H), 3.85 (s, 3H), 4.00 (s, 1H), 4.48 (s, 1H), 6.52 (dd, J=9.9 Hz, 2.4 Hz, 1H), 6.94 (d, J=9 Hz, 2H), 7.13 (dt, J,=8.4 Hz, 2.4 Hz, 1H), 7.38 (d, J=8.7 Hz, 2H), 7.82 (dd, J=8.7 Hz, 5.7 Hz, 1H).
Стадия 8: Получение энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида (4 R, 5R)
К раствору 13,44 г (31,07 ммоль) энантиомерно-обогащенного тетрагидробензотиепин-1-оксида (полученного на стадии 7) в 150 мл метиленхлорида при 0°С при перемешивании добавляют 9,46 г 68%-ной м-хлорнадбензойной кислоты (37,28 ммол, фирма Sigma). После перемешивания при 0°С в течение 2 ч смеси дают нагреться до комнатной температуры и перемешивают еще в течение 4 ч. Затем в смесь добавляют 50 мл насыщенного раствора Nа2SО3 и перемешивют еще в течение 30 мин. Затем этот раствор нейтрализуют насыщенным раствором NaHCO3 (50 мл). Метиленхлоридный слой отделяют, сушат над MgSO4 и концентрировали в вакууме с получением 13,00 г (97,5%) энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида (4R, 5R) в виде светло-желтого твердого вещества: 1Н ЯМР (CDCl3) d 0.89-0.95 (m, 6H), 1.09-1.42 (m, 12H), 2.16-2.26 (m, 1H), 3.14 (q, J=15.6 Hz, 1H), 3.87 (s, 3H), 4.18 (s, 1H), 5.48 (s, 1H), 6.54 (dd, J=10.2 Hz, 2.4 Hz, 1H), 6.96-7.07 (m, 3H), 7.40 (d, J=8.1 Hz, 2H), 8.11 (dd, J=8.6 Hz, 5.9 Hz, 1H).
Стадия 9: Получение энантиомерно-обогащенного 7-(диметиламино)-тетрагидробензотиепин-1,1-диоксида (4R, 5R)
К раствору 13,00 г (28,98 ммоль) энантиомерно-обогащенного тетрагидробензотиепин-1,1-диоксида (полученного на стадии 8) в 73 мл раствора диметиламина (2,0 М в ТГФ, 146 ммоль) в реакторе Парра добавляют около 20 мл чистого диметиламина. Смесь запаивают и перемешивают при 110°С в течение ночи и охлаждают до комнатной температуры. Избыток диметиламнна выпаривают. Сырое масло растворяют в 200 мл этилацетата и промывают водой (100 мл), сушат над MgSO4 и концентрируют в вакууме. Очистка на колонке с силикагелем (Waters Prep 500) с использованием смеси 20% этилацетат/гексан дает 12,43 г (90,5%) энантномерно-обогащенного 7-(диметиламино) тетрагидробензотиепин-1,1-диоксида (4R, 5R) в виде бесцветного твердого вещества: 1Н ЯМР (CDCl3) d 0.87-0.93 (m, 6H), 1.10-1.68 (m, 12Н), 2.17-2.25 (m, 1H), 2.81 (s. 6H), 2.99 (d, J=15.3 Hz, 1H), 3.15 (d, J=15.3 Hz, 1H), 3.84 (s, 3H), 4.11 (d, J=7.5 Hz, 1H), 5.49 (s, 1H), 5.99 (d, J=2.4 Hz, 1H), 6.51 (dd, J=8.7 Hz, 2.4 Hz, 1H), 6.94 (d, J=8.7 Hz, 2H), 7.42 (d, J=8.4 Hz, 2H), 7.90 (d, J=8.7 Hz, 1H).
Было определено посредством хиральной ВЭЖХ на колонке Chiralpak AD при использовании в качестве элюента смеси 5% этанол/гексан, что данный продукт имеет 78% э.о. Перекристаллизация этого твердого вещества из смеси этилацетат/гексан дает 1,70 г рацемического продукта. Оставшийся раствор концентрируют и перекристаллизовают с получением 9,8 г бесцветного твердого вещества. Энантиомерное обогащение этого твердого вещества было определено с помощью хиральной ВЭЖХ на колонке Chiralpak AD с использованием смеси 5% этанол/гексан в качестве элюента. Оказалось, что оно составляет 96% э.о. с первым пиком элюции в качестве основного продукта.
Стадия 10: Деметилирование 5-(4'-метоксифенил)-7-(диметиламино) тетрагидробензотиепин-1,1-диоксида (4R, 5R)
К раствору 47 г (99 ммоль) энантиомерно-обогащенного (диметиламино)-тетрагидробензотиепин-1,1-диоксида (полученного на стадии 9) в 500 мл метиленхлорида при -10°С добавляют по каплям раствор трибромида бора (297 мл, 1 М в метиленхлоридс, 297 ммоль); полученный таким образом раствор перемешивают на холоде (от -5°С до 0°С) в течение 4 ч или пока реакция не завершится. Реакционную смесь охлаждают на бане сухой лед - ацетон при -10°С и медленно гасят водой (300 мл). Затем смесь нагревают до 10°С и далее разбавляют насыщенным раствором бикарбоната нагрня (300 мл) до нейтрализации смеси. Водный слой отделяют и экстрагируют метпленхлоридом (300 мл), а объединенные экстракты промывают водой (200 мл), солевым раствором, сушат над MgSO4 и концентрируют в вакууме. Остаток рчстворяют в 500 мл этилацетата и перемешивают с 50 мл ледяной уксусной кислоты в течение 30 мин при комнатной температуре. Смесь дважды промывают водой (по 200 мл), солевым раствором (200 мл), сушат над MgSO4 и концентрируют в вакууме с получением сырого 4-гидроксифенильного промежуточного соединения. Твердый остаток перекристаллизовывают из метиленхлорида и получают 37,5 г (82%) желаемого (4R,5R)-5-(4'-гидроксифенил)-7-(диметиламино)тетрагидробензотиепин-1,1-диоксида в виде белого твердого вещества: 1Н ЯМР (CDCl3) d 0.84-0.97 (m, 6H), 1.1-1.5 (m, 10Н), 1.57-1.72 (m, 1H), 2.14-2.28 (m, 1H), 2.83 (s, 6H), 3.00 (d, J=15.3 Hz, 1H), 3.16 (d, J=15.3 Hz, 1H), 4.11 (s, 2H), 5.48 (s, 1H), 6.02 (d, J=2.4 Hz, 1H), 6.55 (dd, J=9, 2.4 Hz, 1H), 6.88 (d, 8,7 Hz, 2H), 7.38 (d, J=8.7 Hz, 2H), 7.91 (d, J=9 Hz, 2H).
Стадия 11: Получение энантиомерно-обогащенного хлорбензильного промежуточного соединения
Раствор энантиомерно-обогащенного (4R,5R)-5-(4'-гидроксифенил)-7-(диметиламино)-тетрагидробензотиепин-1,1-диоксида (5,0 г, 10,9 ммоль, полученный на стадии 10) в ацетоне (100 мл) при 25°С в атмосфере азота обрабатывают порошкообразным К2СО3 (2,3 г, 16,3 ммол, 1,5 экв.) и а,а'-дихлор-п-ксилолом (5,7 г, 38,1 ммол, 3,5 экв.). Перемешивают образовавшийся раствор при 65°С в течение приблизительно 48 ч. Смесь охлаждают до 25°С и концентрируют до 1/5 начального объема. Растворяют осадок в этилацетате (150 мл) и промывают водой (2×150 мл). Водный слой экстрагируют этилацегатом (2×150 мл) и объединенные органические экстракты промыиают насыщенным водным раствором NaCl (2×150 мл). Объединенные экстракты сушат над MgSO4 и концентрируют в вакууме, получая сырой продукт. Очистка флэш-хроматографией (5,4×45 см силикагеля, 25-40% этилацетат/гексан) дает энантиомерно-обогащенное хлорбензильное промежуточное соединение.
Стадия 12: Получение энантиомерно-обогащенного (4R, 5R)-1-[[4-[[4-[3.3-дибутил-7-(диметиламино)-2,3,4,5-тетрагидро-4-гидрокси-1,1-диоксид-1-бензотиепин-5-ил]фенокси]метил]фенил]метил]-4-aзa-1-aзониабицикло[2,2,2]-октанхлорида (XXVII)
Раствор энантиомерио-обогащенного хлорбензильного промежуточного соединения (4,6 г, 7,7 ммоль, полученного на упомянутой выше стадии 11) в ацетонитриле (100 мл) при 25°С в атмосфере азота обрабатывают диазабицикло-[2,2,2]октаном (ДАБЦО, 0,95 г, 8,5 ммоль, 1,1 экв.) и перемешивают при 35°С в течение 2 ч. Собирают выпавший осадок и промывают его акрилонитрилом, Перекристаллизация из смеси акрилонитрила и этилового эфира дает желаемое соединение (XXVII).
Приведенные здесь примеры можно с тем же успехом повторить, заменяя условия проведения или реагенты, использованные в предыдущих примерах, на описанные в общем виде или конкретно реагенты и/или условия этого изобретения.
Хотя данное изобретение описано таким образом очевидно, что возможны варианты его реализации различными путями. Такие варианты нельзя рассматривать как отход от духа и границ настоящего изобретения, а все такие модификации и эквиваленты, как это должно быть очевидно специалисту, предназначены быть включенными в объем следующей формулы изобретения.

Настоящее изобретение относится к способу получения энантиомерно-обогащенного тетрагидробензотиепин-1-оксида формулы (I)
циклизацией энантиообогащенного арил-3-пропанальсульфоксида формулы (II), в котором атом серы является хиральным центром
описан также способ получения промежуточного соединения формулы II. Соединения формулы I представляют собой класс идеальных ингибирующих передвижение желчных кислот соединений, которые могут быть использованы для воздействия на уровень холестерина в сыворотке крови. Настоящий способ является удобным и рентабельным. 4 н. и 59 з.п. ф-лы, 1 табл., 2 ил.


где R1 и R2 независимо друг от друга выбраны из Н или алкила;
R3 выбран из арила, возможно замещенного одной или более группой OR19, где R19- алкил, или группы формулы (IIа)

R4, R5, R6 и R7 независимо друг от друга выбраны из группы, включающей водород, галоген и -NR9R10, где R9 и R10 независимо друг от друга выбраны из Н или алкила; причем
R3 и гидроксил в положении 4 энантиомерно-обогащенного тетрагидробензотиепин-1-оксида находятся в син-конформации по отношению друг к другу; и атом серы в положении 1 семичленного кольца и атомы углерода в положениях 4 и 5 семичленного кольца являются хиральными центрами;
путем циклизации энантиомерно-обогащенного арил-3-пропанальсульфоксида формулы (II)

в которой R1, R2, R3, R4, R5, R6 и R7 описаны выше и где атом серы является энантиомерно-обогащенным хиральным центром с образованием энантиомерно-обогащенного тетрагидробензотиепин-1-оксида формулы (I).

в которой R1, R2, R3, R4, R5, R6 и R7 такие, как описано в п.1, и где атом серы является энантиомерно-обогащенным хиральным центром с образованием энантиомерно-обогащенного арил-3-пропанальсульфоксида формулы (II).

в которой R1, R2, R3, R4, R5, R6 и R7 такие, как описано в п.2, с образованием энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида формулы (III).
R8-O-O-H, (V)
где R8 представляет собой радикал, выбранный из группы, состоящей из Н, алкила, карбалкила, бензила, бензоила и кумила.
R8-O-O-H, (V)
в которой R8 представляет собой радикал, выбранный из группы, состоящей из Н, алкила, карбалкила, бензила, бензоила и кумила.

в которой
R11 и R12 независимо представляют собой группу OR19, где R19- алкил.

в которой
R1 и R2 независимо друг от друга выбраны из Н и алкила;
R3 выбран из арила, возможно замещенного одной или более группой OR19, где R19- алкил, или группы формулы (IIа)
R4, R5, R6 и R7 независимо друг от друга выбраны из группы, включающей водород, галоген и -NR9R10, где R9 и R10 независимо друг от друга выбраны из Н или алкила; причем
R3 и гидроксил в 4-положении энантиомерно-обогащенного тетрагидро-бензотиепин-1-оксида находятся в син-конформации по отношению друг к другу; атом серы в положении 1 семичленного кольца и атомы углерода в положениях 4 и 5 семичленного кольца являются хиральными центрами;
включающий:
а) окисление арил-3-гидроксипропилсульфида формулы (IV)
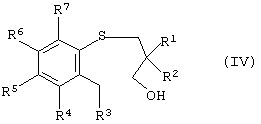
где R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, которое осуществляют в условиях энантиоселективного окисления с получением энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида формулы (III)

где R1, R2, R3, R4 , R5, R6 и R7 такие, как описано выше, а атом серы является энантиомерно-обогащенным хиральным центром;
b) окисление 3-гидроксильной группы энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида с получением энантиомерно-обогащенного арил-3-пропанальсульфоксида формулы (II)

где R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, а атом серы является энантиомерно-обогащенным хиральным центром, и
c) циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида с образованием энантиомерно-обогащенного тетрагидробензотиепин-1-оксида формулы (I).
R8-O-O-H (V)
где R8 представляет собой радикал, выбранный из группы, включающей Н, алкил, карбалкил, бензил, бензоил и кумил.
R8-O-O-H, (V)
в которой R8 представляет собой радикал, выбранный из группы, состоящей из Н, алкила, карбалкила, бензила, бензоила и кумила.

в которой R11 и R12 независимо друг от друга выбраны из Н и -OR10, где R10 - алкил.



где R1 и R2 независимо друг от друга выбраны из Н и алкила;
R3 выбран из арила, возможно замещенного одной или более группой OR19, где R19- алкил, или группы формулы (IIа)

R4, R5, R6 и R7 независимо друг от друга выбраны из группы, включающей водород, галоген и -NR9R10, где R9 и R10 независимо друг от друга выбраны из Н или алкила; причем R3 и гидроксил в 4-положении энантиомерно-обогащенного тетрагидро-бензотиепин-1-оксида находятся в син-конформации по отношению друг к другу и атомы углерода в положениях 4 и 5 семичленного кольца являются хиральными центрами;
включающий:
а) окисление арил-3-гидроксипропилсульфида формулы (IV)
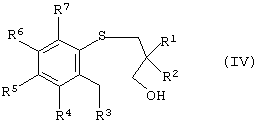
где R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, которое осуществляют в условиях энантиоселективного окисления с получением энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида, имеющего формулу (III)
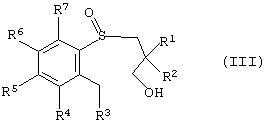
где R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, а атом серы является энантиомерно-обогащенным хиральным центром;
(b) окисление 3-гидроксильной группы энантиомерно-обогащенного арил-3-гидроксипропилсульфоксида с получением энантиомерно-обогащенного арил-3-пропанальсульфоксида формулы (II)

где R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, а атом серы является энантиомерно-обогащенным хиральным центром;
c) циклизацию энантиомерно-обогащенного арил-3-пропанальсульфоксида с образованием энантиомерно-обогащенного тетрагидробензотиепин-1-оксида формулы (I)

в которой R1, R2, R3, R4, R5, R6 и R7 такие, как описано выше, а R3 и гидроксильная группа в 4-положении энантиомерно-обогащенного тетрагидробензотиепин-1-оксида находятся в син-конформации по отношению друг к другу, и атом серы в 1-положении семичленного кольца и атомы углерода в 4-положении и 5-положении семичленного кольца являются энантиомерно-обогащенными хиральными центрами; и
d) окисление энантиомерно-обогащенного тетрагидробензотиепин-1-оксида в энантиомерно-обогащенный тетрагидробензотиепин-1,1-диоксид формулы (VII).


| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2095361C1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

