Изобретение относится к диастереоселективным способам получения оптически активных цис-нуклеозидов и аналогов и производных нуклеозидов. Новые способы в соответствии с настоящим изобретением обеспечивают возможность проведения стереорегулируемого синтеза заданного энантиомера требуемого цис-нуклеозида или аналога или производного нуклеозида с высокой оптической чистотой. Настоящее изобретение касается также интермедиатов, пригодных для использования в способах по настоящему изобретению.
Нуклеотиды и их аналоги и производные являются важным классом лечебных веществ. Например, ряд нуклеозидов проявил антивирусную активность против ретровирусов, таких как вирус иммунодефицита человек (ВИЧ), вирус гепатита B (ВГВ) и T - лимфотропный вирус человека (ТЛВЧ) /PCT публикация W O 89/04662 и публикация европейского патента 0349242 A2 /. К нуклеозидам, которые проявили антивирусную активность, относятся 3'-азидо-3'-дезокситимидин /АЗТ/ и 2'3'-дидезоксицитидин (ДДЦ).
Большинство нуклеозидов и аналогов и производных нуклеозидов содержат, по крайней мере, два хиральных центра (в формуле /A/ они обозначены звездочками) и существуют в форме двух пар оптических изомеров (т.е. два в цис-форме и два в транс-форме). Но обычно полезную биологическую активность проявляют только цис-изомеры.
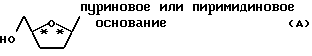
Однако разные энантиомерные формы одного и того же цис-нуклеозида могут весьма различающиеся антивирусные активности. См. M. M. Mansuri et al., "Preparation of the Geometric Isomers of DDC, DDA, DUC and D4T As Potential Anti-HIV Agents", Bioorg. Med. Chem. Lett.,
1/1/, стр. 65 - 68 /1991/. Поэтому важной задачей является общий и экономически привлекательный стереоселективный синтез энантиомеров биологически активных цис-нуклеозидов.
В соответствии с известными способами получения оптически активных нуклеозидов и их аналогов и производных модифицируют встречающиеся в природе /т. е. оптически активные/ нуклеозиды путем изменения основания или путем изменения сахара методами восстановления, такими как деоксигенация или радикально-инициированное восстановление. См. C.K.Chu et al., "General Synthesis Of 2', 3'-Dideoxynucleosides And 2',3'-Didehidro-2',3'- -Dideoxynucleosides", J.Org.Chem., 54, стр. 2217 - 2225 /1989/. Эти превращения включают много стадий, в том числе введение и удаление защитных групп, и обычно дают низкие выходы, кроме того, исходный нуклеозид оптически активен вначале и сохраняет свою оптическую активность в дальнейшем. Поэтому нуклеозиды, полученные указанными способами, ограничиваются специфическими аналогами энантиомерной формы природного нуклеозида. К тому же указанные способы требуют наличия встречающегося в природе нуклеозида, часто являющегося дорогим исходным материалом.
Другие известные способы получения оптически активных нуклеозидов основаны на традиционных процессах гликозилирования для присоединения сахара к основанию. Указанные процессы неизменно дают аномерные смеси цис- и транс-изомеров, что требует трудоемкого разделения и приводит к пониженным выходам целевого биологически активного цис-нуклеозида. Усовершенствованные способы гликозилирования, предназначенные для получения только цис-нуклеозида, требуют присоединения к сахару 2'- или 3'-заместителя. Поскольку 2'- или 3'-заместитель является единственно пригодным для регулирования синтеза цис-нуклеозида в одной конфигурации (когда 2' - или 3' - заместитель занимает транс-положение относительно 4'-заместителя), то требуется много стадий для введения этого заместителя в надлежащей конфигурации. 2'- или 3'-заместитель должен быть затем удален после гликозилирования, что требует дополнительных стадий. См. L. Wilson and D. Liotta, "A General Metbod For Controlling Stereochemistry In Rhe Synthesis Of 2'-Deoxyribose Nucleosides", Tetrahedron Lett.,
31, стр. 1815 - 1818 /1990/. Кроме того, для получения оптически чистого нуклеозида исходный сахар должен быть оптически чистым. Это также требует серии требующих больших затрат времени стадий синтеза и очистки.
Настоящее изобретение устраняет трудности и недостатки известного уровня техники и дает способу получения оптически активных цис-нуклеозидов и аналогов и производных нуклеозидов формулы I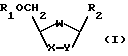
где W - O, S, S = O, SO2, NZ или CH2;
X - O, S, S = O, SO2, NZ, CH2, CHF, CH, CHN3 или CHOH;
Y - O, S, CH2, CH, CHF или CHOH;
Z - водород, гидроксил, алкил или ацил;
R1 - водород или ацил; и
R2 - пуриновое или пиримидиновое основание или его аналог или производное;
при условии, что, когда Y - CH2 и X - O, S, S = O или SO2, то W - не O, S, S = O, или SO2.
Способы в соответствии с настоящим изобретением включают в себя стадию гликозилирования требуемого пуринового или пиримидинового основания или его аналога или производного одиночным энантиомером соединения формулы II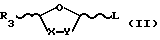
где R3 - замещенный карбонил или замещенное карбонильное производное и L - уходящая группа. Гликозилирование осуществляют, используя кислоту Льюиса формулы III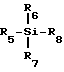
где R5, R6, R7 и R8 определены ниже, и полученный интермедиат восстанавливают и получают в результате нуклеозид или его аналог или производное формулы I.
Преимуществом способов в соответствии с настоящим изобретением является то, что они обеспечивают возможность получения нуклеозида формулы I (или его аналогов или производных) без использования дорогих исходных материалов, обременительных стадий введения и удаления защитных групп или присоединения и удаления 2'- или 3'-заместителей. Способы в соответствии с настоящим изобретением обеспечивают высокие выходы нуклеозидов с высокой чистотой и высокой оптической специфичностью. Другое преимущество способов в соответствии с настоящим изобретением заключается в создании нуклеозидов, стереоизомерную конфигурацию которых можно легко регулировать просто путем выбора подходящих исходных материалов.
В способах получения оптически активных соединений в соответствии с настоящим изобретением в конфигурационно- и диастереоселективной манере используют следующие определения:
R2 - пуриновое или пиримидиновое основание или его аналог или производное.
Пуриновым или пиримидиновым основанием является пуриновое или пиримидиновое основание, найденное в природных нуклеозидах. Его аналогом является основание, которое повторяет такие природные основания в том, что имеет аналогичную с ними структуру /виды атомов и их расположение/, но может иметь дополнительные функциональные свойства или может иметь некоторых из функциональных свойств природных оснований. К таким аналогам относятся те, которые являются результатом замены фрагмента CH атомом азота, например 5-азапиримидины, такие как 5-азацитозин, или наоборот /например, 7-деазапурины, такие, как 7-деазаденин или 7-деазагуанин /либо и те, и другие /например, 7-деаза, 8-азапурины/. Под производными таких оснований или аналогов понимаются те основания, в которых заместители в кольце введены, удалены или модифицированы традиционными заместителями, известными в данной области техники /например, галоген, гидроксил, амино, C1-6 алкил/. Такие пуриновые или пиримидиновые основания, аналоги и производные хорошо известны специалистам в данной области техники.
Аналог или производное нуклеозида - это нуклеозид, который был модифицирован любым из следующих способов или их сочетаниями; модифицирование основания такое, как присоединение заместителя /например, 5-фторцитозин/ или замена одной группы изостерной группой /например, 7-деазааденин/; модифицирование сахара такое, как замещение C-2 и C-3 гидроксильных групп любым заместителем, включающим водород /например, 2',3'-дидеоксинуклеозиды/, замена гетероатомом любой группы CH в кольце или кислорода в кольце; изменение места присоединения сахара к основанию /например, пиримидиновые основания, обычно присоединяемые к сахару в положении N-I, могут быть, например, присоединены в N-3 или C-6, а пурины, обычно присоединяемые в положении N-9, могут быть, например, присоединены в положении N-7/; изменение места присоединения основания к сахару /например, основание может быть присоединено к сахару в C-2, как у изо-DDA/; или изменение формы связи сахара с основанием /например, цис- или транс-формы/.
R3-карбонил, замещенный водородом, гидроксилом, триалкилсилилом, триалкилокси, C1-30 алкилом, C7-30 аралкилом, C1-30 алкокси, C1-30 амином /первичным, вторичным или третичным/, C1-30 тиолом; C6-20 арил; C1-20 алкенил; C1-20 алкинил; 1,2-дикарбонил, такой как 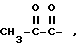 , замещенный C1-6 алкилом или C6-20 арилом; ангидриды, такие как
, замещенный C1-6 алкилом или C6-20 арилом; ангидриды, такие как 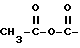 , замещенный C1-6 алкилом или C6-20 арилом; азометин, замещенный у атома азота водородом, C1-20 алкилом или C1-10 алкокси или C1-10 диалкиламино и у атома углерода водородом, C1-2 алкилом или C1-20 алкокси; тиокарбонил /C=S/, замещенный гидроксилом, C1-20 тиолом; гомолог карбонила, например
, замещенный C1-6 алкилом или C6-20 арилом; азометин, замещенный у атома азота водородом, C1-20 алкилом или C1-10 алкокси или C1-10 диалкиламино и у атома углерода водородом, C1-2 алкилом или C1-20 алкокси; тиокарбонил /C=S/, замещенный гидроксилом, C1-20 тиолом; гомолог карбонила, например 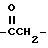 , гомолог тиокарбонила, например
, гомолог тиокарбонила, например 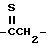 ; или гомолог азометина, такой как
; или гомолог азометина, такой как 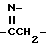 .
.
Предпочтительными замещенными карбонилами или карбонильными производными являются алкоксикарбонилы, такие как метил, этил, изопропил, трет-бутил и ментил; карбоксилы; диэтилкарбоксамид; пирролидинамид; метилкетон и фенилкетон. Более предпочтительными замещенными карбонилами или карбонильными производными являются сложные эфиры и карбоксилы, а наиболее предпочтительны сложные эфиры.
R4 - хиральный вспомогательный реагент. Термин "хиральный вспомогательный реагент" означает асимметрические молекулы, используемые для осуществления химического разделения рацемической смеси. Такой хиральный вспомогательный реагент может иметь один хиральный центр /например, метиблензиламин/ или несколько хиральных центров /например, ментол/. Назначение хирального вспомогательного реагента, когда он введен в состав исходного материала, состоит в обеспечении возможности простого разделения полученной смеси диастереомеров. См., например, J.Jacgues et al., Enantiomers, Racemates And Resolutions, стр. 251-369, John Wiley and Sons, Нью-Йорк /1981/.
R5, R6 и R7 независимо выбирают из группы, состоящей из водорода, C1-20 алкила /например, метила, этила, третбутила/, необязательно замещенного галогенами /F, Cl, Br, I/, C1-6 алкокси /например, метокси/ или C6-20 арилокси /например, фенокси/; C7-20 аралкила /например, бензила/, необязательно замещенного галогеном, C1-20 алкилом или C1-20 алкокси /например, пара-метоксибензил/; C6-20 арила /например, фенила/, необязательно замещенного галогенами, C1-20 алкилом или C1-20 алкокси; триалкилсилила; галогенов /F, Cl, Br, I/.
R8 выбирают из группы, состоящей из галогена /F,Cl, Br, I/; сложных эфиров C1-20 сульфокислоты, необязательно замещенных галогенами /например, трифторметансульфонат/; сложных C1-20 алкиловых эфиров, необязательно замещенных галогеном /например, трифторацетат/; многовалентных галогенидов /например, трииодид/; трехзамещенных силильных групп общей формулы (R5) (R6) (R7)Si, /где R5, R6 и R7 - такие, как определены выше/; насыщенного или ненасыщенного селененил C6-20 арила; замещенного или незамещенного C6-20 арилсульфенила; замещенного или незамещенного C1-20 алкоксиалкила; и триалкилсилокси.
L - "уходящая группа", т.е. атом или группа, которые могут быть замещены при реакции с подходящим пуриновым или пиримидиновым основанием в присутствии или без присутствия кислоты Льюиса. Подходящие уходящие группы включают в себя ацилоксигруппы, алкоксигруппы, например алкоксикарбонильные группы, такие как этоксикарбонил; галогены, такие как иод, бром, хлор или фтор; амидо; азидо; изоцианато; замещенные или незамещенные, насыщенные или ненасыщенные тиолаты, такие как тиометил или тиофенил; замещенные или незамещенные, насыщенные или ненасыщенные селеновые, селениниловые или селенониловые соединения, такие как фенилселенид или алкилселенид.
Подходящей уходящей группой может быть также группа -OR, где R - замещенная или незамещенная, насыщенная или ненасыщенная алкильная группа, например C1-6, алкильная или алкенильная группа; замещенная или незамещенная, алифатическая или ароматическая ацильная группа, например c1-6 алифатическая ацильная группа, такая как ацетил, и замещенная или незамещенная ароматическая ацильная группа, такая как бензоил; замещенная или незамещенная, насыщенная или ненасыщенная алкокси или арилоксикарбонильная группа, такая как метилкарбонат и фенилкарбонат; замещенный или незамещенный сульфонилимидазолид; замещенная или незамещенная, алифатическая или ароматическая аминокарбонильная группа, такая как фенилкарбамат; замещенная или незамещенная алкилимидиатная группа, такая как трихлорацетамидат; замещенный или незамещенный, насыщенный или ненасыщенный фосфонат, такой как диэтилфосфонат; замещенная или незамещенная, алифатическая или ароматическая сульфинильная или сульфонильная группа, такая как тозилат; или водород.
Используемый в данной заявке термин "алкил" представляет замещенный /галогеном, гидроксилом или C6-20 арилом/ или незамещенный неразветвленный, разветвленный или циклический углеводородный фрагмент, имеющий от 1 до 30 атомов углерода, а предпочтительно от 1 до 6 атомов углерода.
Термины "алкенил" и "алкинил" представляют замещенные /галоненом, гидроксилом или C6-20 арилом/ или незамещенные неразветвленные, разветвленные или циклические углеводородные цепи, имеющие от 1 до 20, а предпочтительно от 1 до 5 атомов углерода, и содержащие, по крайней мере, одну ненасыщенную группу /например, аллил/.
Термин "алкокси" представляет замещенную или незамещенную алкильную группу, содержащую от 1 до 30, а предпочтительно от 1 до 6 атомов углерода, причем алкильная группа ковалентно связана с соседним элементом через атом кислорода /например, метокси и этокси/.
Термин "амин" представляет алкильные, арильные, алкенильные, алкинильные или аралалкильные группы, содержащие от 1 до 30, а предпочтительно от 1 до 12 атомов углерода, ковалентно связанные с соседним элементом через атом азота /например, пирролидин/. Они включают в себя первичные, вторичные и третичные амины и соли четвертичного аммония.
Термин "тиол" представляет алкильные, арильные, аралкильные, алкенильные или алкинильные группы, содержащие от 1 до 30, а предпочтительно от 1 до 6 атомов углерода, ковалентно связанных с соседним элементом через атом серы /например, тиометил/.
Термин "арил" представляет карбоциклический фрагмент, который может быть замещен, по крайней мере, одним гетероатомом /например, N, O или S/, содержит, по крайней мере, одно бензольное кольцо и предпочтительно имеет от 6 до 15 углерода /например, фенил или нафтил/.
Термин "аралкил" представляет арильную группу, присоединенную к соседнему атому посредством алкила /например, бензил/.
Термин "алкоксиалкил" представляет алкоксигруппу, присоединенную к соседней группе посредством алкильной группы /например, метоксиметил/.
Термин "арилокси" представляет замещенный /галогеном, трифторметилом или C1-5 алкокси/ или незамещенный арильный фрагмент, ковалентно связанный через атом кислорода /например, фенокси/.
Термин "ацил" относится к радикалу, полученному от карбоновой кислоты, замещенной (галогеном (F, Cl, Br, I), C6-20 арилом или C1-6 алкилом) или незамещенной, путем замены группы -OH. Подобно кислоте, к которой от имеет отношение, ацильный радикал может быть алифатическим или ароматическим, замещенным /галогеном, C1-5 алкоксиалкилом, нитро или O2/ или незамещенным, причем при любой структуре остатка молекулы свойства функциональной группы остаются, по существу, теми же самыми /например, ацетил, пропионил, изобутаноил, пивалоил, гексаноил, трифторацетил, хлорацетил и циклогексаноил/.
Основной особенностью способов в соответствии с настоящим изобретением является использование в качестве R3 замещенного карбонила или карбонильного производного, а незащищенной гидроксиметильной группы, как было описано ранее в данной области техники. Неожиданно оказалось, что замещенный карбонил или карбонильное производное не расщепляется под воздействием кислоты Льюиса, как могли бы ожидать специалисты в данной области техники, при добавлении кислоты Льюиса формулы III к смеси силилированного / с введенной силильной группой/ пуринового или пиримидинового основания и соединения в виде сахара формулы II. Вместо этого замещенный карбонил или карбонильное производное в интермедиате формулы VI заставляет пуриновое или пиридиновое основание (R2) присоединяться к цис-конфигурации относительно замещенной карбонильной или производной карбонильной группы. Без замещенного карбонила или карбонильного производного, присоединенного к C4' /например, когда вместо него используют гидроксиметильную группу/, процедуры соединения /сочетания/ на описанной ниже стадии 4 дают в результате смесь цис- и транс-изомеров.
Другая основная особенность способов в соответствии с настоящим изобретением заключается в выборе кислоты Льюиса. Кислоты Льюиса, используемые для получения соединение формулы I, имеют общую формулу III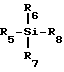 ,
,
где R5, R6, R7 и R8 - такие, как определены ранее. Эти кислоты Льюиса могут быть получены на месте или приготовлены с использованием любого метода, известного в данной области техники /например, A.H.Schmidt, Bromotrimethylsilane and Iodotrimethylsilane-Versatile Reagents for Organis, Synthesis", Aldrichimica Acta, 14, стр. 31 - 38, 1981/. Предпочтительными кислотами Льюиса в соответствии с настоящим изобретением являются иодтриметилсилан и триметилсилилтрифлат. Предпочтительными группами R5, R6 и R7 являются метил или иод. Наиболее предпочтительной группой R5, R6 и R7 является метил. Предпочтительными группами R8 являются иод, хлор, бром или эфиры сульфокислоты. Наиболее предпочтительными группами R8 являются иод и трифторметансульфонат.
В предпочтительном способе в соответствии с настоящим изобретением цис- и транс-изомеры сахара формулы II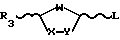 ,
,
разделяют путем дробной кристаллизации и выбирают изомер требуемой конфигурации. Выбранный цис- или транс-изомер может быть затем выделен химически с использованием хирального вспомогательного реагента. Чистый диастереометр хиральный вспомогательный реагент-сахар затем связывают с силилированным пуриновым или пиримидиновым основанием в присутствии кислоты Льюиса, что дает оптически активный нуклеозид цис-конфигурации, который затем восстанавливают, получая в результате нуклеозид формулы I.
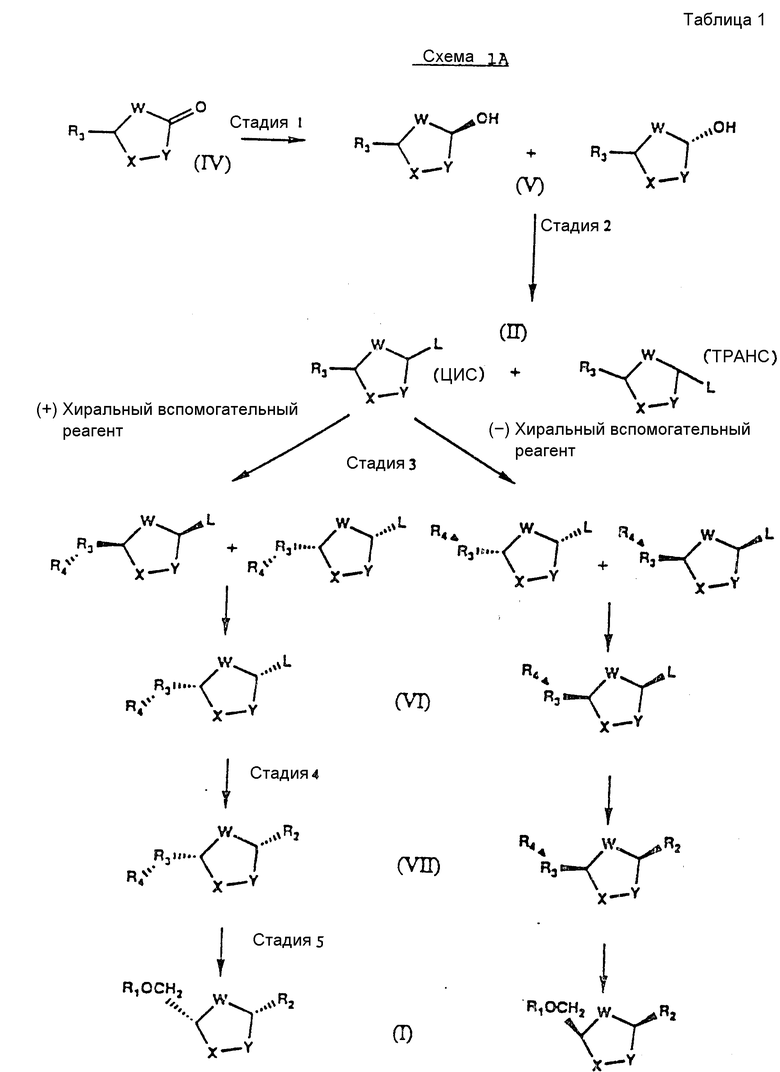
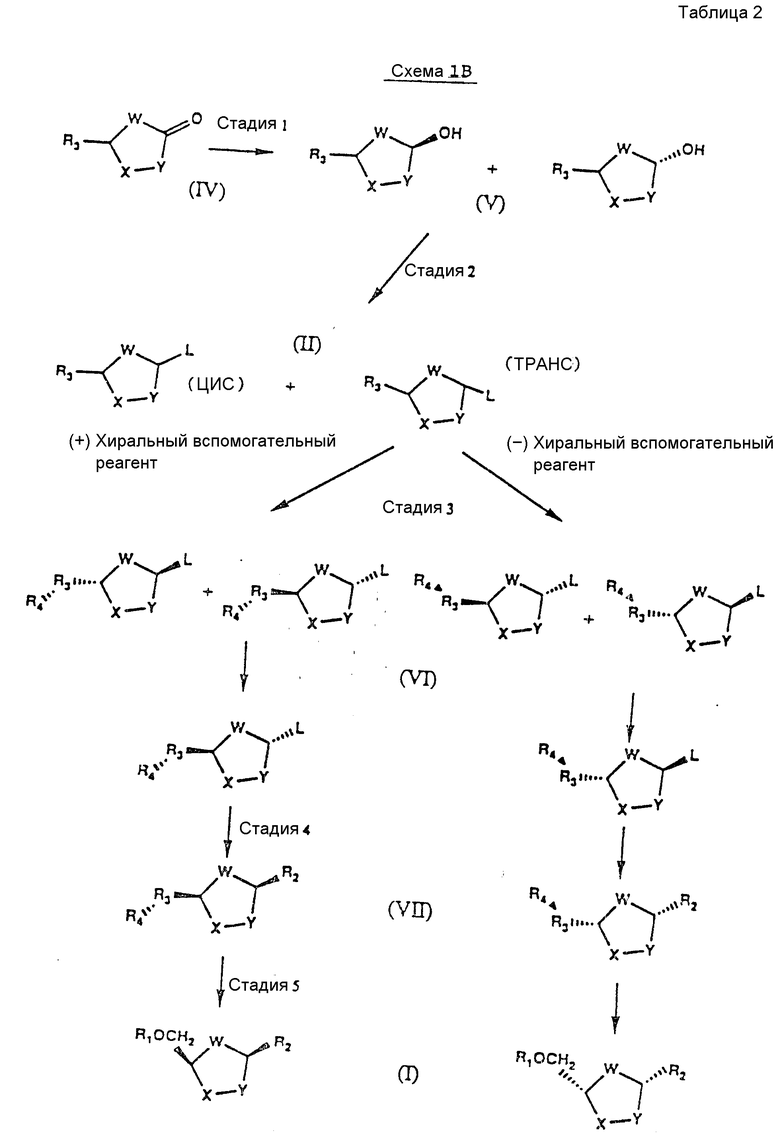
На схемах 1А и 1В показан этот предпочтительный способ в применении к получению любого нуклеозида формулы I (см. табл. 1 и 2).
Различные стадии, показанные на схемах IA и IB, можно вкратце описать следующим образом.
Стадия 1: Исходный карбонил-сахар формулы IV может быть приготовлен любым известным в данной области техники способом. См., например, Farina and Benigni "A New Sinthesis of 2',3' - Dideoxy-nucleosides For Aids Chemotherapy", Tetrahedron Letters, 29, стр. 1239-1242 (1988) and M.Okabe et al., "Synthesis of the Dideoxynucle-osides ddc and CNT From Glutamic Acid, Ribonolactone and Pyrimidine Bases", J.Org.Chem., 53, стр.4780-4786 /1988/. Карбонильную группу этого исходного соединения восстанавливают хемоселективно подходящим восстанавливающим агентом, таким как дисиамилборан, и получают в результате цис- и транс-изомеры формулы V. Обычно цис-изомеров получают меньше, чем транс-изомеров.
Стадия 2: Гидроксильную группу в интермедиате формулы V легко превращают в уходящую группу любым способом, известным в данной области техники /см., например, T.W.Greene Protective Group In Organic Synthesis, стр.50-72, John Weley and Sons, Нью-Йорк /1981//, и получают в результате новые интермедиаты формулы II.
Эту аномерную смесь затем разделяют путем дробной кристаллизации на два пространственных изомера. Чтобы выбрать цис- или транс-изомер, можно регулировать растворитель. См.D.J.Pasto and C.R.Johnson, Organic Structure Determination, стр.7-10, Prentice-Hall, Inc, Нью-Джерси /1969/.
Стадия 3: Цис-/схема IA/ или транс-изомер /схема IB/ формулы II отделяют химически с использованием хирального вспомогательного реагента /R4/. Подходящим хиральным вспомогательным реагентом является реагент высокой оптической чистоты с легко получаемым зеркальным изображением, такой как d- и l- ментол. Полученные диастереомеры формулы VI легко разделяют путем дробной кристаллизации. В соответствии с другим вариантом цис- и транс-изомер может быть отделен ферментативно или другими способами, известными в данной области техники. См. J.Jacgues et al., Enantiomers, Racemates and Resolutions, стр.251-369, John Wiley and Sons, Нью-Йорк /1981/.
Оптическая чистота диастереомера /VI; VII и I/ может быть определена хиральными HP C методами, измерениями удельного вращения и ЯМР методами. Если требуется противоположный энантиомер, он может быть получен путем использования зеркального изображения первоначально использованного хирального вспомогательного реагента. Например, если хиральный вспомогательный реагент d- ментол дает /+/ - энантиомер нуклеозида, то его зеркальное изображение - l-ментол - даст /-/- энантиомер.
Стадия 4: Ранее силилированное /или силилированное на месте/ пуриновое или пиримидиновое основание затем гликозилирует полученным чистым диастереомером в присутствии кислоты Льюиса формулы III, такой как иодтриметилсилан /TMSI/ или триметилсилилтрифлат /TMSOTf/, и получают в результате нуклеозид цис-конфигурации формулы VII. Этот нуклеозид оптически активен и, по существу, свободен от соответствующего транс-изомера /т.е. содержит менее 20%, предпочтительно не более 10% и более предпочтительно не более 5% транс-изомера/. Присоединение интермедиата формулы VI к пуриновому или пиримидиновому основанию на этой стадии обеспечивает более высокий выход цис-изомера.
Предпочтительным силилирующим агентом для пиримидиновых оснований является трет-бутилдиметилсилилтрифлат. Думается, что объемистая трет-бутильная группа увеличивает выход вследствие ослабления взаимодействия между кислотой Льюиса и силилированным пиримидиновым основанием.
Предпочтительный способ смещения реагентов на стадии 4 состоит в том, что сначала добавляют хиральный вспомогательный реагент-сахар /формулы VI/ к силилированному пуриновому или пиримидиновому основанию. Затем к смеси добавляют кислоту Льюиса формулы III.
Стадия 4: Полученный на стадии 4 цис-нуклеозид может быть затем восстановлен подходящим восстанавливающим агентом для удаления хирального вспомогательного реагента и получения конкретного стереоизомера формулы I. Абсолютная конфигурация этого стереоизомера соответствует абсолютной конфигурации интермедиата формулы VII. Как показано на схеме 1, цис- /схема IA/ или транс-изомеры /схема IB/, полученный на стадии 2, дают конечный цис-продукт.
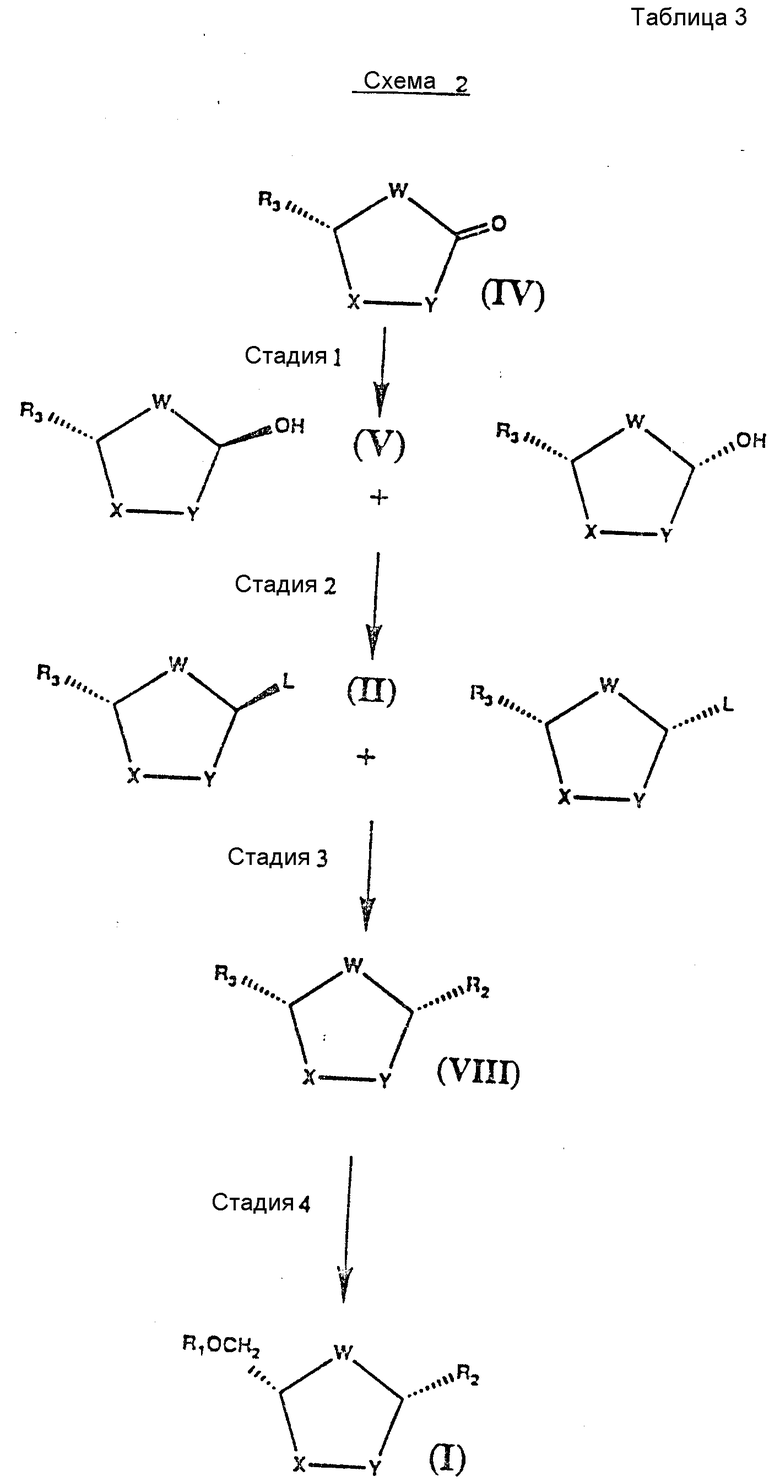
Второй способ диастереоселективного синтеза соединений формулы I проиллюстрирован схемой 2. Способ по схеме 2 пригоден для использования тогда, когда оптически чистый исходный материал может быть легко куплен или легко приготовлен известными способами.
Оптически активный исходный материал хемоселективно восстанавливают и полученную гидроксильную группу превращают в уходящую группу. Диастереомерная смесь может быть далее доведена до соединений формулы /1/ аналогично тому, как это описано в связи со схемой 1. Хотя и не обязательно, диастереомерная смесь может быть разделена дробной кристаллизацией и каждый отдельный оптически активный диастереомер может быть превращен в соединения формулы I.
На схеме 2 показан второй способ в соответствии с настоящим изобретением в применении к любому нуклеозиду /см.табл.3/.
Различные стадии синтеза нуклеозидов формулы I, представленные на схеме 2, можно вкратце описать следующим образом.
Стадия 1: Исходный материал формулы IV может быть куплен в оптически чистом виде или приготовлен в соответствии с процедурами, описанными Farina and Benigni, "A New Synthesis of 2', 3'-Dideoxynucleoside For Aids Chemotherapy," Tetrahedrom Letters, 29, стр. 1239-1242, (1988) and M.Okade et al., Synthesis of The Dideoxynucleosides ddc and CNI From Glutamic Acid, Ribonolactone and Pyrimidine Bases", J.Org Chem., 53, стр. 4780-4786 /1988/. Одиночный изомер формулы IV хемоселективно восстанавливают подходящим восстановителем, таким как дисиамилборан, и в результате получают смесь двух диастереомеров формулы V.
Стадия 2: Гидроксильные группы двух диастереомеров формулы V превращают в уходящие группы любым способом, известным в данной области техники, и получают в результате смесь двух диастереомеров формы II.
Стадия 3: Диастереомерную смесь формулы II вводят в химическое взаимодействие с заранее силированным /или силилированным на месте/ пуриновым или пиримидиновым основанием или налогом или производным. Затем путем добавления кислоты Льюиса формулы III, такой как иодтриметилсилан /TMSI/ или триметилсилилтрифлат /TMSOTf/, получают нуклеозид цис-конфигурации формулы VIII. Этот нуклеозид, по существу, свободен от соответствующего транс-изомера.
Стадия 4: Оптически активный цис-нуклеозид формулы VIII восстанавливают стереоспецифически восстановителем, предпочтительно триэтилборогидридом лития или алюмогидридом лития, а более предпочтительно борогидридом натрия в подходящем растворителе, таком как тетрагидрофуран или простой диэтиловый эфир, и получают соединение формулы I.
В соответствии с другим вариантом в конце стадии 2 либо цис-, либо транс-изомер может быть выделен из диастереомерной смеси формулы II путем дробной кристаллизации или хроматографического разделения. Для выбора цис- или транс-изомера можно регулировать растворитель. Отдельный диастереометр формулы II затем направляют далее на стадии 3 и 4, чтобы получить в результате соединение формулы I.
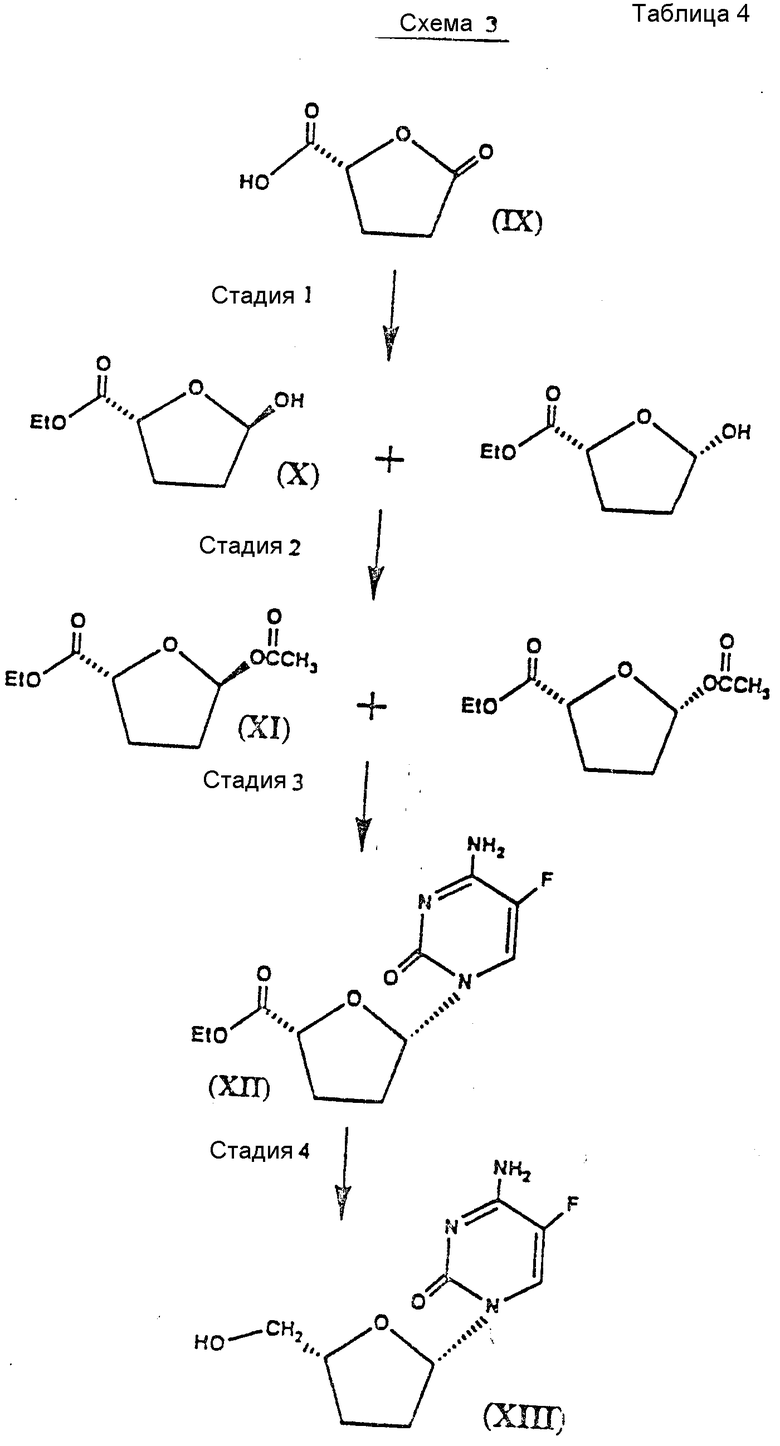
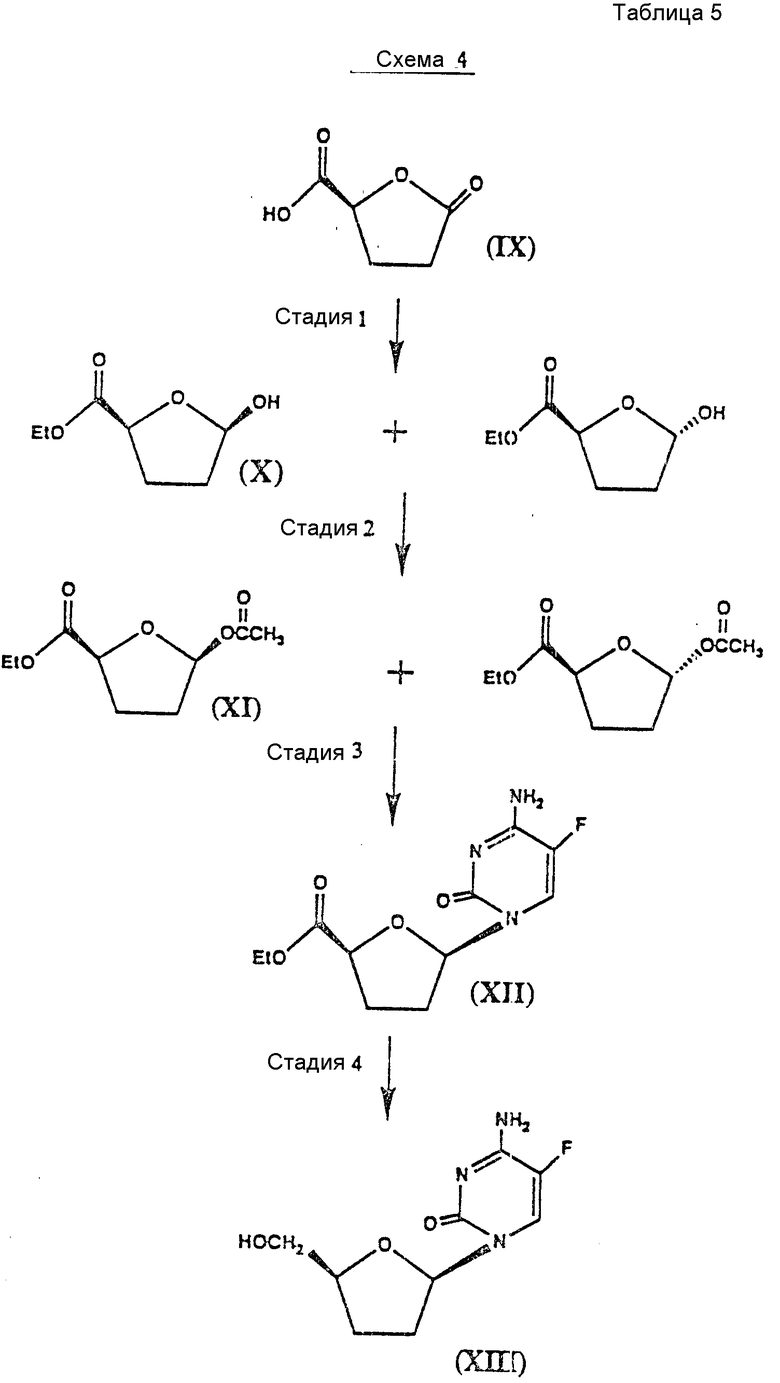
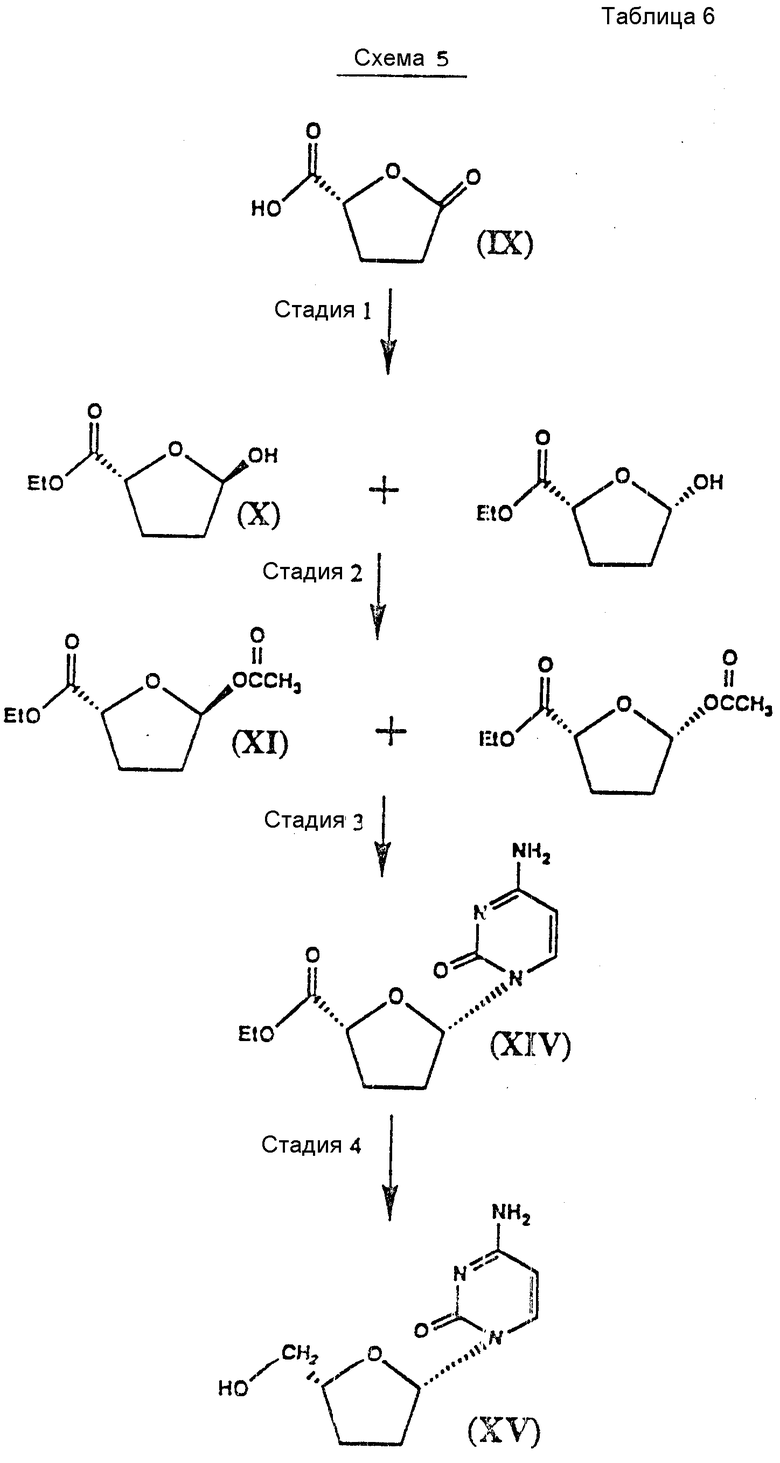
Схемы 3, 4 и 5 иллюстрируют применение способа, показанного на схеме 2, к синтезу энантиомеров аналогов цис-дидеоксинуклеозидов (см. табл. 4-6).
Способ проиллюстрирован с использованием конкретных реагентов и исходных материалов, но специалистам в данной области техники будет понятно, что для получения аналогичных соединений могут быть использованы аналогичные реагенты и исходные материалы.
Различные стадии, показанные на схеме 3, можно вкратце описать следующим образом.
Стадия 1: Исходный материал /2R/-5-оксо-2-тетрагидрофуранкарбоновую кислоту /IX/ покупают или получают путем синтеза из D -глутаминовой кислоты /см. M. okabe et al., "Synthesis of The Dideoxynucleosides ddc and CNT From Glutamic Acid, Ribonolactone and Purimidine Bases", J.Org.Chem., 53, стр. 4780-4786 /1988//. Исходный материал этерифицируют спиртом, таким как этанол, в присутствии ацилирующего агента, такого как оксалилхлорид, и катализатора этерификации, такого как 4-диметиламинопирнимидин, и основания, такого как пиридин, в совместимом растворителе, таком как дихлорметан, этерифицированное соединение восстанавливают подходящим восстановителем, таким как дисиамилборан, в совместимом органическом растворителе, таком как тетрагидрофуран /см. A.Pelter et al., "Borane Reagents", Academic Press,
стр. 426 /1988//, и получают соединения формулы X.
Стадия 2: Соединения формулы X вводят в химическое взаимодействие с хлорангидридом кислоты или ангидридом кислоты, таким как уксусный ангидрид, в присутствии пиридина и катализатора ацилирования, такого как 4-диметиламинопиридин, и получают соединения формулы IX.
Стадия 3: Смесь цис- и транс-ацетоксисоединений формулы IX вводят во взаимодействие с 5-фторцитозином или другим пиримидиновым основанием или его аналогом. Пуриновое или пиримидиновое основание или аналог предпочтительно силилируют гексаметилдисидазаном или, что более предпочтительно, силилируют на месте /in situ/ трет-бутилдиметилсилилтрифлатом в совместимом органическом растворителе, таком как дихлорметан, содержащем затрудненное основание, предпочтительно 2,4,6-коллидин.
Затем добавляют кислоту Льюиса, предпочтительно взятую из соединений формулы III, а более предпочтительно иодтриметилсилан или триметилсилилтрифлат, и получают в результате цис- соединение формулы XII в высоко диастереоселективной манере.
Стадия 5: Оптически активный цис-нуклеозид /с некоторым количеством транс-изомера/ формулы XII восстанавливают стереоспецифически с помощью восстановителя, предпочтительно борогидрида натрия, в соответствующем растворителе, таком как этанол, и получают в результате /после очистки/ соединение формулы XIII.
Специалистам в данной области техники понятно, что для получения энантиомера формулы XIII нужно использовать в качестве материала формулы IX /2S/-5-оксо-2-тетрагидрофуранкарбоновую кислоту /схема 4/ в соответствии со способом, описанным в связи со схемой 3.
Различные стадии, показанные на схеме 5, можно вкратце описать следующим образом.
Стадия 1: Исходный материал /2R/-5-оксо-2-тетрагидрофуранкарбоновую кислоту /IX/ этерифицируют спиртом, например этанолом, в присутствии ацилирующего агента, например оксалилхлорида, и катализатора этерификации, такого как 4-диметиламинопиримидин, и основания, такого как пиридин, в совместимом растворителе, таком как дихлорметан. Этерифицированное соединение восстанавливают подходящим восстановителем, например дисиамилбораном, в совместимом органическом растворителе, таком как тетрагидрофуран, и получают в результате соединения формулы X.
Стадия 2: Соединения формулы X вводят в химическое взаимодействие с хлорангидридом кислоты или ангидридом кислоты, таким как уксусный ангидрид, в присутствии пиридина и катализатора ацилирования, такого как 4-диметиламинопиридин, и получают соединения формулы IX.
Стадия 3: Смесь цис - и транс-ацетоксисоединений формулы IX вводят во взаимодействие с N-ацетилцитозином или другим пиримидиновым соединением или его аналогом. Пуриновое или пиримидиновое основание или аналог предпочтительно силилируют гексаметилдисилазаном или, более предпочтительно, силилируют на месте триметилсилилтрифлатом в совместимом органическом растворителе, таком как дихлорметан, содержащем затрудненное основание, предпочтительно 2,4,6 -коллидина.
Затем добавляют кислоту Льюиса, предпочтительно взятую из соединений формулы III, а более предпочтительно иодтриметилсилан, и в результате получают в высоко диастереоселективной манере цис-нуклеозид. Путем порошкования /растирания в порошок/ с соответствующим растворителем, таким как этилацетат и гексаны, получают чистый цис-нуклеозид.
N-ацетильную группу гидролизуют предпочтительно в кислых условиях и более предпочтительно трифторуксусной кислотой в совместимом органическом растворителе, таком как изопропанол, предпочтительно при нагреве с обратным холодильником и получают деацилированные соединения формулы XIV.
Стадия 4: Оптически активный цис-нуклеозид формулы XIV восстанавливают стереспецифически восстановителем, предпочтительно борогидратом натрия, в подходящем растворителе, таком как этанол, и получают в результате соединение формулы XV.
В диастереоселективных способах в соответствии с настоящим изобретением особенно важны следующие интермедиаты: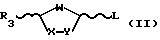
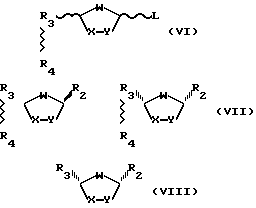
где R3, R4 и L - такие, как те, что определены выше;
цис- и транс-2R-карбоэтокси-5-гидрокситетрагидрофуран;
цис- и транс-2S-карбоэтокси-5-гидрокситетрагидрофуран;
цис- и транс-2R-карбоэтокси-5-ацетокситетрагидрофуран;
цис- и транс-2S-карбоэтокси-5-ацетокситетрагидрофуран;
1'S-/N-4-ацетилцитозин-1-ил/-4'R-карбоэтокситетрагидрофуран;
1'S-/цитозин-1-ил/-4'R-карбоэтокситетрагидрофуран;
1'R-/5-фторцитозин-1-ил/-4'S -карбоэтокситетрагидрофуран и 1'S -/5-фторцитозин-1-ил/-4'S -карбоэтокситетрагидрофуран; и
1'S -/5-фторцитозин-1-ил/-4'R -карбоэтокситетрагидрофуран и 1'R-/5-фторцитозин-1-ил/-4'R-карбоэтокситетрагидрофуран.
Следующие ниже примеры иллюстрируют настоящее изобретение в отношении способа его осуществления, но как таковые не должны рассматриваться как ограничивающие общий объем способов в соответствии с настоящим изобретением. Кроме тех, что специально указаны, все измерения [ [α]D ] были записаны при температуре окружающей среды.
Пример 1. 2R-Карбоэтокси-5-оксо-тетрагидрофуран.
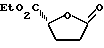
К холодному /0oC/ перемешанному раствору 5-оксо-2R-тетрагидрофуранкарбоновой кислоты /3 г, 23 ммоль/, 4-диметиламинопиридина /141 мг, 0,05 эквивалента/ и пиридина /3,92 мл, 2,1 эквивалента/ в дихлорметане /15 мл/ в среде аргона добавляли оксалилхлорид /2,11 мл, 1,05 эквивалента/ в течение 30 минут. Охлаждающую ванну удаляли и реакционную смесь перемешивали в течение 10 минут. Вводили этанол /2,0 мл, 1,5 эквивалента/ и продолжали перемешивание еще 1 час 40 минут. Реакционную смесь разбавляли водой и дихлорметаном, после чего перемешивали 10 минут. Полученную смесь переносили в делительную воронку. Удаляли водную фазу, и органический слой промывали 1 M HCl, насыщенным NaHCO3, рассолом и затем высушивали /Na2SO4/. Выпаривали растворитель при пониженном давлении, и полученный в результате неочищенный продукт подвергали колоночной хромотографии /1: 1 этилацетат-гексан/, что дало 3,23 г целевого продукта в виде сиропа.
1Н ЯМР/CDCl3/: δ 1,28 /т.,3H, J = 7,1 Гц/, 2,20 - 2,40 /м.,1H/, 4,23 /д. квартетов, 2H, J = 0,9 и 7,1 Гц/, 4,86 - 4,96 /м.,1H/.
Пример 2. Цис- и транс-2R-карбоэтокси-5-гидрокситетрагидрофуран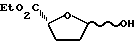
Приготавливали раствор дисиамилборана путем смешивания 35 мл BH3 ТГФ/1 М в ТГФ/ и 35 мл 2-метил-2-бутена /2 М в ТГФ/ при 0oC с последующим перемешиванием при 0oC в течение 75 минут. В этот раствор вводили 2R-карбоэтокси-5-оксотетрагидрофуран, растворенный в ТГФ /6 мл/. Полученной смеси позволяли медленно нагреваться до комнатной температуры в течение 2,5 часов, после чего перемешивали еще 15 часов. Добавляли насыщенный раствор аммонийхлорида и затем разбавляли смесь этилацетатом. Полученную смесь перемешивали 10 минут, после чего переносили в делительную воронку. Органическую фазу промывали последовательно насыщенным раствором аммонийхлорида и рассолом, после чего высушивали сульфатом натрия. Удаляли растворитель на роторном испарителе, и полученный неочищенный продукт очищали путем колоночной хроматографии /40% эталацетат-гексаны/. Целевые продукты выделяли с 70%-ным выходом /2,05 г/ в виде смеси /2: 3/ изомеров, эпимерных у C5. Было также обнаружено /1H ЯМР/ незначительное количество /следы/ изомера открытой формы. Указанные в заголовке соединения показали следующие спектральные характеристики:
1H ЯМР/ CDCl3/: δ 1,28 /т. 2H, J = 7,1 Гц/, 1,30 /т.1H, J = 7,1 Гц/, 1,85 - 2,70 /м, 4H/, 2,59 /д, 0,33H, J = 5,5 Гц/, 2,88 /д, 0,67H, J = 3,1 Гц/, 4,15 - 4,65 /м, 2H/, 4,57 /дд, 0,33H, J = 6,4 и 8,3 Гц/, 4,70 /дд, 0,67H, J = 4,1 и 8,7 Гц/, 5,59 /м, 0,33H/, 5,74 /м, 0,67H/.
Пример 3. Цис- и транс-2R-карбоэтокси-5-ацетокситетрагидрофуран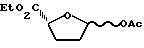
К холодному /-78oC/ перемешанному раствору смеси /2:3/ цис- и транс-2R-карбоэтокси-5-гидрокситетрагидрофурана /2,04 г, 12,75 ммоль/, пиридина /1,24 мл, 1,2 эквивалента/ и 4-диметиламинопиридина /16 мг, 0,01 эквивалента/ в дихлорметане /20 мл/ добавляли ацетилхлорид /1,09 мл, 1,2 эквивалента/ в течение 5 минут. Полученную смесь перемешивали 10 минут. Затем ванну охлаждали до -78oC, заменяли ванной с ледяной водой. Продолжали перемешивание в течение 4,5 часов, позволяя температуре ванны медленно снизиться до комнатной температуры. Реакционную смесь разбавляли дихлорметаном и затем переносили в делительную воронку. Органический слой промывали последовательно водой, IM HCl, насыщенным раствором NaHCO3 и рассолом и затем высушивали /Na2SO4/. Удаляли растворитель на роторном испарителе, и полученный неочищенный продукт очищали путем колоночной хроматографии /40% этилацетат-гексан/, получив в результате 1,757 г указанных в заголовке соединений /смесь с отношением 5:4/ в виде густого масла.
IH ЯМР /CDCl3/: δ 1,28 /т, 1,68H, J=7,1 Гц/, 1,29 /т, 1,32H, J=7,1 Гц/, 1,90 - 2,30 /м, 3H/, 2,30 - 2,50 /м,1H/, 4,10-4,30 /м, 2H/, 4,59 /т, 0,44H, J= 8,0 Гц/, 4,70/ дд, 0,56H, J=3,2 и 8,9 Гц/, 6,33 /дд, 0,44H, J=1,1 и 3,9 Гц/, 6,46/д, 0,56H, J=4,5 Гц/.
Пример 4. 1'S-/N-4-Ацетилцитозин-1-ил/-4'R-карбоэтокситетрагидрофуран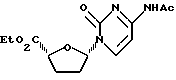
К перемешанной суспензии N-4-ацетилцитозина /50 мг, 0,298 ммоль/ в дихлорметане /0,75 мл/, содержавшей 2,6-лутидин /35 мкл, 0,298 ммоль/, в среде аргона добавляли триметилсилилтрифторметансульфонат /58 млк, 0,298 ммоль/. Полученную смесь перемешивали 15 минут и получили светлую суспензию. В эту суспензию последовательно вводили раствор смеси /5:4/ цис- и транс-2P-карбоэтокси-5-ацетокситетрагидрофурана /50 мг, 0,248 ммоль/ в дифхлометане /1 мл/ и иодтриметилсилан /35 мкл, 0,248 ммоль/, получив в результате гомогенный раствор. Реакции позволяли продолжаться в течение 1 часа 40 минут, после чего резко прекращали полунасыщенным раствором Na2S2O3. Полученную смесь перемешивали 5 минут и затем переносили в делительную воронку с добавлением дихлорметана. Удаляли водную фазу, а органический слой промывали насыщенным раствором Na2S2O3, водой и рассолом и затем высушивали /Na2SO4/. Объединенные промывные воды вновь экстрагировали дихлорметаном. Органические экстракты объединяли и концентрировали при пониженном давлении, получив 83 мг неочищенного продукта. IH ЯМР-анализ неочищенного продукта показал, что была получена смесь /4:1/ цис- и транс- ожидаемых нуклеозидов. Неочищенный продукт растворяли в минимальном количестве хлороформа. Добавление смеси /3: 7/ этилацетата и гексанов в этот раствор дало белый осадок, который улавливали путем вакуумной фильтрации. После высушивания этого твердого вещества получили 25 мг /32%/ указанного в заголовке соединения.
1H ЯМР/CDCl3/: δ 1,33 /т, 3H, J=7,1 Гц/, 1,90-2,08 /м, 1H/, 2,08-2,30 /м, 1H/, 2,23 /с, 3H/, 4,20-4,40 /м, 2H/, 4,64 /т, 1H, J=7,2 Гц/, 6,15 /дд, 1H, J= 4,0 и 5,9 Гц/, 7,46 /д, 1H, J=7,5 Гц/, 8,34 /шир.с,1H/, 8,82 /д, 1H, J=7,5 Гц/.
Промывные воды концентрировали и получили 58 мг цис- и транс- смеси /5: 2/ указанного в заголовке соединения и его 1'-изомера.
Пример 5.
β-L-2',3'-Дидезоксицитидин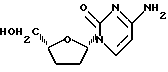
Смесь 1'S-/N-4-ацетилцитозин-1-ил/-4'R-карбоэтокситетрагидрофурана /49 мг, 0,158 ммоль, содержал приблизительно 4% соответствующего 1'R-изомера/ и трифторуксусной кислоты /24 мкл, 2 эквивалента/ в этаноле /1 мл/ нагревали с обратным холодильником в среде аргона в течение 2 часов 40 минут. Полученную смесь, состоявшую из 1'S-/цитозин-1-ил/-4'R-карбоэтокситетрагидрофурана и его 1'-эпимера, охлаждали до комнатной температуры и затем разбавляли этанолом /0,5 мл/. Затем вводили натрия борогидрид /18 мг, 3 эквивалента/, и реакционную смесь перемешивали 1,5 часа. Добавляли еще восстановителя /6 мг/ и продолжали перемешивать еще 1 час 20 минут. Реацию резко прекращали путем добавления 2 капель концентрированного аммонийгидроксида с последующим интенсивным перемешиванием в течение 15 минут. Выпаривали при пониженном давлении растворитель, и полученный неочищенный продукт подвергали колоночной хроматографии /30% метанол-этилацетат/, получив в результате 28 мг /84%/ указанного в заголовке соединения. 1H ЯМР- спектр этого вещества показал наличие приблизительно 3% соответствующего 1'R-изомера. Этот материал растворяли в минимальном количестве метанола. Добавление к этому раствору диэтилового эфира дало 20 мг /60%/ указанного в заголовке соединения в виде кристаллического белого осадка, свободного от 1'R-изомера /1H ЯМР/. Указанное в заголовке соединение показало следующие спектральные характеристики: 1H ЯМР /CD3OD/: δ 1,60-2,00 /м, 3H/, 2,25 - 2,43 /м, 1H/, 3,59 /дд, 1H, J= 4,1 и 12:2 Гц/, 3,78 /дд, IH, J=3,1 и 12,2 Гц/, 4,00-4,12 /м, 1H/, 5,78 /д, 1H, J= 7,4 Гц/, 5,92 /дд, 1H, J=3,1 и 6,7 Гц/, 8,02 /д, 1H, J=7,5 Гц/.
Пример 6. 1'R-/5-фторцитозин-1-ил/-4'S-карбоэтокситетрагидрофуран и 1'S-/5-фторцитозин-1-ил/-4'S-карбоэтокситетрагидрофуран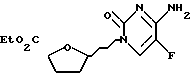
К перемешанной суспензии 5-фторцитозина /192 мг, 1,49 ммоль/ в дихлорметане /2 мл/, содержавшей 2,6-лутидин /346 мкл, 2,98 ммоль/ среде аргона добавляли трет-бутилдиметилсилилтрифторметансульфонат /678 мкл, 2,98 ммоль/. Полученную смесь перемешивали 15 минут и получили гомогенный раствор. В этот раствор последовательно вводили раствор смеси /2:1/ 2S-карбоэтокси-5R-ацетокситетрагидрофурана и 2S-карбоэтокси-5S-ацетокситетрагидрофурана /250 мг, 1,24 ммоль/ в дихлорметане /2 мл/ и иодтриметилсилан /176 мкл, 1,24 ммоль/. Реакции позволяли протекать при комнатной температуре в течение 1 часа 30 минут, после чего ее резко прекращали путем добавления полунасыщенного раствора Na2S2O3. Полученную смесь перемешивали 5 минут и затем переносили в делительную воронку. Удаляли водную фазу, а органический слой промывали насыщенным раствором Na2S2O3, водой и рассолом и затем высушивали /Na2SO4/. Удаляли при пониженном давлении растворитель, получив в результате неочищенный продукт, который подвергали колоночной хроматографии /15% метанол-этилацетат/, что дало 199 мг /59%/ указанных в заголовке соединений в виде смеси [/1'R, 4'S/ : /1'S, 4'S/ = 7:1] согласно 1H ЯМР. Продукт показал следующие спектральные характеристики: 1H ЯМР /CDCL3/ : δ 1,15-1,40 /2 перекрывающихся т. , 3H/, 1,90-2,15 /м, 2H/, 2,25 - 2,55 /м, 2H/, 4,15 - 4,35 /м,:2H/, 4,54 /м, 0,87 H/, 4,82 /дд, 0,13H, J=4,4 и 8,0 Гц/, 5,70-6,80 /нерасщепленный м., 1H/, 6,09 /м, 1H/, 7,40 /д, 0,13H, J=6,7 Гц/, 7,90 - 8,60 / нерасщепленный м., 1H/, 8,48 /д, 0,87H, J=6,7 Гц/.
Пример 7. 1'S-/5-фторцитозин-1-ил/-4'R-карбоэтокситетрагидрофуран и 1'R-/5-фторцитозин 1-ил/-4'R-карбоэтокситетрагидрофуран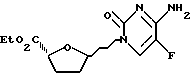
К перемешанной суспензии 5-фторцитозина /38 мг, 0,297 ммоль/ в дихлорметане /1 мл/, содержавшей 2,6-лутидин /69 мкл, 0,594 ммоль/, в среде аргона добавляли трет-бутилдиметилсилилтрифторметансульфонат /137 мкл, 0,594 ммоль/. Полученную смесь перемешивали 15 минут, что дало гомогенный раствор. В этот раствор последовательно вводили раствор смеси /5:4/ 2R-карбоэтокси-5S-ацетокситетрагидрофурана и 2'-карбоэтокси-5R-ацетокситетрагидрофурана /50 мг, 0,248 ммоль/ в дихлорметане /1 мл/ и иодтриметилсилан /35 мкл, 0,248 ммоль/. Реакции позволяли протекать при комнатной температуре 1 час 45 минут, после чего ее резко прекращали полунасыщенным раствором тиосульфата натрия /Na2S2O3/. Полученную смесь перемешивали 5 минут и затем переносили в делительную воронку. Водную фазу удаляли, а органический слой промывали насыщенным раствором тиосульфата натрия, водой и рассолом и затем высушивали /Na2So4/. Удаляли при пониженном давлении растворитель, получив неочищенный продукт, который подвергали колоночной хроматографии /15 % метанол-этилацетат/, что дало 52 мг /78%/ указанных в заготовке соединений в виде смеси с соотношением /1'R,4'R/:/1'S,4'R/ = 11:2 /1Н ЯМР/. Продукт показал следующие спектральные характеристики: 1Н ЯМР /CDCL3/: δ] 1,15-1,40 /2 перекрывающихся т. , 3H/, 1,90-2,10 /м, 2H/, 2,25 - 2,60 /м, 2H/, 4,15 - 4,35 /м, 2H/, 4,57 /м, 0,85H/, 4,84 /дд, 0,15H, J=4,2 и 7,8 Гц/, 5,50 - 6,30 / нерасщепленный м., 1H/, 6,09 /м, 1H/, 7,43 /д, 0,15H, J=6,7 Гц/, 7,50 -9,00 /нерасщепленный м., 1H/, 8,56 /д, 0,85H, J=6,7 Гц/.
Пример 8. β -L-/5-фтор /-2',3'-дидезоксицитидин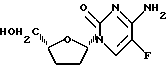
К холодной /0oC/ перемешанной суспензии 1'R-/5-фторцитозин-1-ил/-4'R-карбоэтокситетрагидрофурана и 1'S-/5-фторцитозин-1-ил/-4'R-карбоэтокситетрагидрофурана 307 мг, 1,133 ммоль, отношение изомеров в смеси [/1'R,4'R/: /1'S, 4'R/= 4: 1] в 4 мл этанола добавляли борогидрид натрия /86 мг, 2 эквивалента/. Полученную смесь перемешивали 5 минут, и ванну охлаждения убирали. Продолжали перемешивание 75 минут при комнатной температуре. Реакцию резко прекращали путем добавления 4 капель концентрированного аммонийгидроксида. После перемешивания смеси в течение 15 минут удаляли растворитель при понижении давлении, и полученный неочищенный продукт подвергали колоночной хроматографии /25% метанолэтилацетат/, получив в результате 197 мг /76%/ ожидаемых 4'-гидроксиметиловых продуктов в виде смеси /4:1/. Как было обнаружено /1H ЯМР/, одна из уловленных фракций содержала указанное в заголовке соединение с 97%-ной чистотой. Эту фракцию концентрировали и получили 14 мг пены светло-бежевого цвета.
УФ / λмакс / : 282,7, 236,4 206,7 нм /метанол/;
[α]D - 81o /с 0,7 метанол/;
1H ЯМР /CD3OD/ : δ 1,77 - 1,90 /м, 2H/, 1,90 - 2,03 /м, 1H/, 2,25 - 2,42 /м, 1H/, 3,61 /дд, 1H, J=3,3 и 12,3 Гц/, 3,82 /дд, 1H, J=2,8 и 12,3 Гц/, 4,06 /м, 1H/, 5,87 /м, 1H/, 8,32 /д. 1H, J=7,0 Гц/.
Пример 9. β -D-/5-фтор/-2',3'-дидезоксицитидин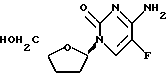
К холодной /0oC/ перемешанной суспензии 1'R-/5-фторцитозин-1-ил/-4'S-карбоэтокситетрагидрофурана и 1'S-/5-фторцитозин-1-ил/-4'S- карбоэтокситетрагидрофурана [199 мг, 0,734 ммоль, отношение изомеров в смеси /1'R,4'S/: /1'S, 4'S/=7:1] в 3 мл этанола добавляли борогидрид натрия /56 мг, 2 эквивалента/. Полученную смесь перемешивали 5 минут, и ванну охлаждения убирали. Продолжали перемешивание всю ночь /примерно 16 часов/ при комнатной температуре. Потом быстро прекращали путем добавления 4 капель концентрированного аммонийгидроксида. После перемешивания смеси в течение 15 минут удаляли растворитель при пониженном давлении, и полученный неочищенный продукт подвергали колоночной хроматографии /20% метанолэтилацетат/, что дало 112 мг /67%/ ожидаемых 4'-гидроксиметиловых продуктов в виде смеси с отношением /1'R, 4'S/:/1'S,4'S/ = 7:1 /1H ЯМР/. Было обнаружено /1H ЯМР/, что одна из уловленных фракций содержала только указанное в заголовке соединение. Эту фракцию концентрировали в вакууме и получили в результате 27 мг белой пены.
УФ / λмакс / : 283,6, 238,2, 202,4 нм /MeOH/;
[α]D +96o /с 0,7, MeOH/;
1H ЯМР /CD3OD/: δ 1,77-1,90 /м, 2H/, 1,90 - 2,03 /м, 1H/, 2,25 - 2,42 /м, 1H/, 3,61 /дд, 1H, J=3,3 и 12,3 Гц/, 3,82 /дд, 1H, J=2,8 и 12,3 Гц/, 4,06 /м, 1H/, 5,87 /м, 1H/, 8,32 /д, 1H, J=7,0 Гц/.
Был представлен ряд вариантов осуществления настоящего изобретения, но специалистам в данной области техники очевидны многочисленные альтернативы и модификации этих вариантов. Следовательно, понятно, что объем настоящего изобретения определяется не представленными выше конкретными примерами, а прилагаемой формулой изобретения.
Использование: в химии нуклеозидов для проведения стереорегулируемого синтеза заданного энантиомера требуемого цис-нуклеозида или аналога, или производного нуклеозида с высокой оптической чистотой. Сущность изобретения: способ диастереоселективного получения оптически активных цис-нуклеозидов, аналогов нуклеозидов или производных формулы I, путем гликозилирования пиримидинового основания или его производного энантиомером соединения формулы II в присутствии кислоты Льюиса формулы III с последующим восстановлением гликозилированного пиримидинового основания или его производного. Предложены также промежуточные соединения формулы II, где W - кислород, X = Y = CH2, R3 - низший алкоксикарбонил, L - группа OR, где R - H или C1 - C6 алифатическая ацильная группа, и формулы VIII, используемые в этом способе. 3 с. и 6 з.п. ф-лы, 6 табл.
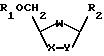
где R1 водород;
R2 пиримидиновое основание или его производное;
W кислород;
Х группа CH2;
Y группа CH2,
отличающийся тем, что включает в себя стадию гликозилирования пиримидинового основания или его производного одиночным энантиомером соединения формулы II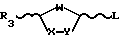
где W, X и Y имеют указанные значения;
R3 низший алкоксикарбонил;
L группа OR, где R атом водорода или С1 С6- алифатическая ацильная группа,
с использованием кислоты Льюиса формулы III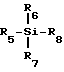
где R5, R6 и R7 независимо выбраны из группы, состоящей из водорода; С1 - 2 0-алкила, необязательно замещенного фтором, бромом, хлором, йодом, С1 - 6-алкокси или С6 - 2 0-арилокси; С6 - 2 0-аралкила, необязательно замещенного галогеном, С1 - 2 0-алкилом или С1 - 2 0-алкокси; С6 - 2 0-арила, необязательно замещенного фтором, бромом, хлором, йодом, С1 - 2 0- алкилом или С1 - 2 0-алкокси, триалкилсилила, фтора, брома, хлора или йода;
R8 выбран из группы, состоящей из фтора, брома, хлора, йода, сложных эфиров С1 C20-сульфокислоты, необязательно замещенных фтором, бромом, хлором или йодом, сложных С1 C20-алкиловых эфиров, необязательно замещенных фтором, бромом, хлором или йодом, поливалентных галогенидов, трехзамещенных силильных групп общей формулы (R5) (R6) (R7)Si -, где R5, R6 и R7 имеют вышеуказанные значения, насыщенного или ненасыщенного селененил-С6 C2-арила, замещенного или незамещенного С6 C20-арилсульфененила, замещенного или незамещенного С6 - C20-алкоксиалкила, и триалкилсилилокси, с последующим восстановлением R3 гликозилированного пиримидинового основания или его производного для получения оптически активных цис-нуклеозидов, аналогов нуклеозидов или производных формулы I.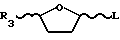
где R3 низший алкоксикарбонил;
L группа OR, где R атом водорода или С1 С6- алифатическая ацильная группа,
в качестве промежуточного соединения для получения оптически активных цис-нуклеозидов, аналогов нуклеозидов или производных по п.1.
цис- и транс-2R-карбоэтокси-5-гидрокситетрагидрофуран;
цис- и транс-2S карбоэтокси-5- гидрокситетрагидрофуран;
цис- и транс-2R-карбоэтокси-5- ацетокситетрагидрофуран;
цис- и транс-2S-карбоэтокси-5-ацетокситетрагидрофуран.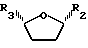
где R2 пиримидиновое основание или его производное;
R3 низший алкоксикарбонил,
в качестве промежуточного соединения для получения оптически активных цис-нуклеозидов, аналогов нуклеозидов или производных по п.1.
1'S-(N-4-ацетилцитозин-1-ил)-4'R- карбоэтокситетрагидрофуран;
1'S-(цитозин-1-ил)-4'R- карбоэтокситетрагидрофуран;
1'R-(5-фторцитозин-1-ил)- 4'S-карбоэтокситетрагидрофуран;
1'S-(5-фторцитозин-1-ил)-4'S- карбоэтокситетрагидрофуран;
1'S-(5-фторцитозин-1-ил)-4'R- карбоэтокситетрагидрофуран и
1'R-(5-фторцитозин-1-ил)-4'R- карбоэтокситетрагидрофуран.
| С.К.Chu et.al | |||
| Journal of Organic Chemistry, 1989, v | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Приспособление для замыкания крышек сосудов | 1917 |
|
SU2217A1 |
| Teirahelron Letters, 1990, v.31, N 13, р | |||
| Электрический конденсатор переменной емкости | 1925 |
|
SU1815A1 |

