Предпосылки создания изобретения
Кинурениновый путь (КР) ответственен за >95% деградации триптофана, представляющего собой незаменимую аминокислоту. Кинурениновый путь для метаболизма триптофана приводит к синтезу незаменимого пиридиннуклеотида NAD+ и множества нейроактивных метаболитов, включая кинуренин (KYN), кинуреновую кислоту (KYNA), нейротоксичный генератор свободных радикалов 3-гидроксикинуренин (3-HK), антраниловую кислоту, 3-НАА, пиколиновую кислоту (PIC), и агонист и нейротоксин возбуждающего рецептора N-метил-D-аспартата (NMDA), хинолиновую кислоту (QUIN). Оставшиеся 5% триптофана метаболизируются триптофан гидроксилазой до 5-гидрокситриптофана и затем далее до 5-гидрокситриптамина (серотонина) и мелатонина.
Как исчерпание триптофана, так и аккумуляция иммуносупрессивных катаболитов триптофана подавляют ответы антиген-специфических Т-клеток и естественных клеток-киллеров, и вызывают формирование регуляторных Т-клеток. Поскольку катаболизм триптофана индуцируется воспалительными медиаторами, особенно IFN-γ, он, как полагают, представляет эндогенный механизм, который ограничивает чрезмерные иммунные ответы, таким образом, предотвращая иммунопатологию. Однако существуют свидетельства, что при болезненных состояниях эта обратная связь может не быть полезной (рассматривается в работе Munn and Mellor, 2013).
Первая стадия катаболизма триптофана катализируется либо триптофан-2,3-диоксигеназой (TDO), либо индоламин-2,3-диоксигеназой (IDO). Оба фермента катализируют окислительное расщепление 2,3 двойной связи в индольном кольце, преобразуя триптофан в N-формилкинуренин. Это соединение является фактором, ограничивающим скорость катаболизма триптофана по кинурениновому пути (Grohmann et al., 2003; Stone and Darlington, 2002). TDO представляет собой гомотетрамер, каждый мономер которого имеет молекулярную массу 48 кДа, тогда как IDO имеет молекулярную массу 45 кДа и мономерную структуру (Sugimoto et al., 2006; Thackray et al., 2008; Zhang et al., 2007). Несмотря на то, что они опосредуют одну и ту же реакцию, TDO и IDO структурно различимы, имея только 10% гомологии, главным образом, в пределах активного центра (Thackray et al., 2008).
Триптофан-2,3-диоксигеназа (TDO) экспрессируется на высоких уровнях в печени и отвечает за регуляцию системных уровней триптофана. Триптофан-2,3-диоксигеназа (TDO) не индуцируется или регулируется сигналами от иммунной системы, однако экспрессия TDO может индуцироваться триптофаном или кортикостероидами (Miller et al., 2004; Salter and Pogson, 1985). Совсем недавно было обнаружено, что TDO экспрессируется в головном мозге, где он регулирует продукцию нейроактивных метаболитов триптофана, таких как кинуреновая кислота и хинолиновая кислота (Kanai et al., 2009).
Индоламин-2,3-диоксигеназа (IDO) является преобладающим триптофан-катаболизирующим ферментом, действующим вне печени, и обнаруживается в многочисленных клетках, включая макрофаги, микроглию, нейроны и астроциты (Guillemin et al., 2007; Guillemin et al., 2001; Guillemin et al., 2003; Guillemin et al., 2005). Транскрипция IDO строго контролируется, отвечая на специфические воспалительные медиаторы. Промоторы мышиных и человеческих генов IDO содержат множественные элементы последовательности, которые придают реактивность к интерферонам типа I (IFN-α/β) и, более мощно, типа II (IFN-γ) (Chang et al., 2011; Dai and Gupta, 1990; Hassanain et al., 1993; Mellor et al., 2003). Различные типы клеток, включая некоторые клетки миелоидного происхождения (макрофаги моноцитарного происхождения и DC), фибробласты, эндотелиальные клетки и некоторые линии опухолевых клеток, экспрессируют IDO после подвергания воздействию IFN-γ (Burke et al., 1995; Hwu et al., 2000; Mellor et al., 2003; Munn et al., 1999; Varga et al., 1996). Однако контроль транскрипции IDO является сложным и специфическим к типу клеток. Активность IDO конститутивно обнаружена в трансплацентарном барьере, экспрессируется вневорсинчатыми тропобластными клетками человека (Kudo and Boyd, 2000). За пределами плаценты функциональная экспрессия IDO у мышей, как сообщалось, была самой высокой в эпидидимисе, пищеварительном тракте (дистальная подвздошная кишка и толстая кишка), лимфатических узлах, селезенке, тимусе и легких (Takikawa et al., 1986).
Также, недавно было показано, что другой вариант фермента IDO катализирует ту же самую ферментативную стадию: индоламин-2,3-диоксигеназа 2 (IDO2). Однако его физиологическая релевантность остается неясной вследствие его очень низкой активности, наличия общих полиморфизмов, которые инактивируют его ферментативную активность у приблизительно половины всех белых людей и азиатов, и наличия множества вариантов сплайсинга (Lob et al., 2008; Meininger et al., 2011; Metz et al., 2007).
IDO-дефицитные мыши являются нормальными на грубом фенотипическом уровне (Mellor et al., 2003), однако они немного более подвержены индукции аутоиммунитета и стимуляции врожденной иммунной системы. Мыши с IDO -/-нокаутом также демонстрируют увеличенный опосредуемый воспалением канцерогенез толстой кишки и резистентность к опосредуемому воспалением раку легкого и кожи (Chang et al., 2011; Yan et al., 2010).
Мыши с TDO -/- нокаутом кажутся фенотипически нормальными. Однако у мышей с нокаутом TDO имеет место 9-кратное увеличение плазменной концентрации L-Trp, в то время как мыши с IDO -/- нокаутом имели уровни L-Trp дикого типа, и это позволяет предположить, что TDO, а не IDO регулирует системный Trp. Абляция TDO увеличивает Trp в головном мозге, а также серотонин (5-НТ), и является поэтому модулятором тревожного поведения (Kanai et al., 2009). TDO также играет роль в поддержании морфологии головного мозга у взрослых мышей, тогда как TDO -/- мыши демонстрируют усиленный нейрогенез в гиппокампе и поджелудочковой зоне в течение взрослой жизни (Funakoshi et al., 2011).
Иммунорегуляция метаболизмом триптофана модулирует иммунную систему путем истощения субстрата TDO/IDO (триптофана) в микросреде и аккумуляции таких продуктов как кинуренин.
Эффекторные Т-клетки особенно чувствительны к низким концентрациям триптофана, поэтому исчерпание незаменимой аминокислоты триптофана из местной микросреды приводит к анергии и апоптозу эффекторных Т-клеток. Деплеция триптофана детектируется путем общего контроля не-дерепрессируемой киназы-2 (GCN2) (Munn et al., 2005). Активация GCN2 запускает программу реакции стресса, которая приводит к остановке клеточного цикла, дифференцировке, адаптации или апоптозу. Т-клетки, не содержащие GCN2 у мышей, не склонны к IDO-опосредуемой анергии миелоидными клетками, включая дендритные клетки в дренирующих опухоль лимфатических узлах (Munn et al., 2005).
Метаболиты триптофана, такие как кинуренин, кинуреновая кислота, 3-гидрокси кинуренин, и 3-гидрокси-антраниловая кислота, подавляют функцию Т-клеток и способны стимулировать апоптоз Т-клеток. Недавние исследования показали, что арил-гидрокарбоновый рецептор (AHR) является прямой мишенью кинуренина (Mezrich et al., 2010; Nguyen et al., 2010; Opitz et al., 2011). AHR является основным транскрипционным фактором семейства спираль-петля-спираль Per-Arnt-Sim (PAS). Поскольку кинуренин аккумулируется в опухоли, KYN связывается с AHR, перемещается в ядро и активирует транскрипцию целевых генов, регулируемых чувствительными к диоксину элементами (DRE). В Т-хелперных клетках кинуренин приводит к генерации регуляторных Т-клеток (Treg).
Фармакологические ингибиторы IDO и/или TDO имеют полезность в широком диапазоне показаний, включая инфекционные болезни, рак, неврологические состояния и множество других заболеваний.
Сущность изобретения
В настоящем документе раскрыты новые соединения формулы (I), которые являются ингибиторами ферментов IDO и/или TDO. Также, в настоящем документе раскрыто применение этих соединений в потенциальном лечении или предупреждении IDO- и/или TDO-ассоциированного заболевания или нарушения. Также, в настоящем документе раскрыты композиции, содержащие одно или несколько этих соединений. Кроме того, в настоящем документе раскрыто применение этих композиций в потенциальном предупреждении или лечении IDO- и/или TDO-ассоциированного заболевания или нарушения.
Подробное описание изобретения
Соединения, раскрытые в настоящем документе, представляют собой ингибиторы IDO и/или TDO. В одном варианте осуществления в настоящем документе раскрыто соединение формулы (I) или его фармацевтически приемлемая соль:
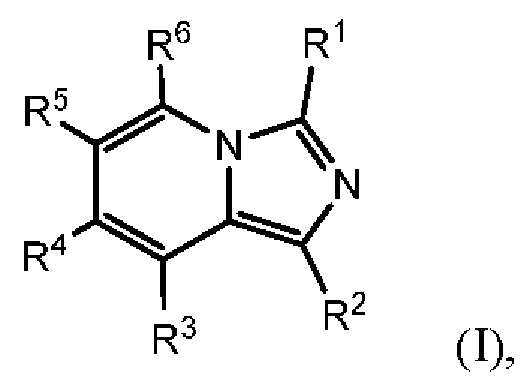
где:
каждый из R1 и R2 независимо выбран из группы, состоящей из (1) Н и (2) NH2;
один из R3 и R6 представляет собой Н, а другой представляет собой Y1;
каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) C1-6-алкила, необязательно замещенного одним-тремя атомами галогена, (4) С3-6-циклоалкила, (5) C1-6-алкокси, необязательно замещенного одним-тремя атомами галогена, и (6) CN, и (7) -NRgRg', где каждый из Rg и R g' независимо выбран из группы, состоящей из Н, C1-6-алкила, -СОН и -COC1-6-алкила;
Y1 представляет собой группу, имеющую следующую формулу:
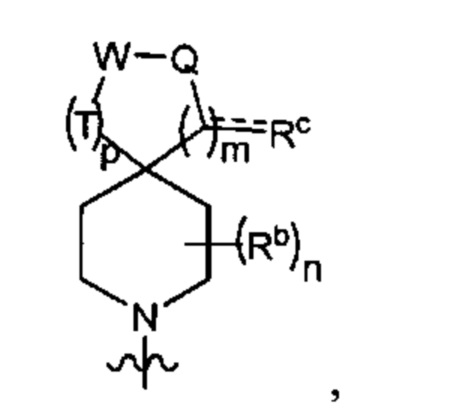
пунктирная линия 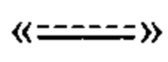 представляет необязательную двойную связь;
представляет необязательную двойную связь;
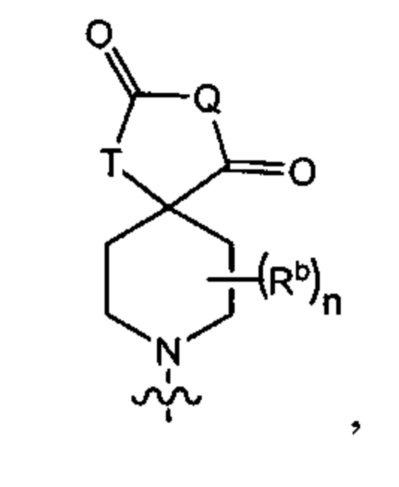
Q представляет собой -C(Ra)(Ra')-, -N(Ra)- или -О-;
Т представляет собой -C(Ra)(Ra')-, -N(Ra)- или -О-;
W представляет собой -S(O)2- или 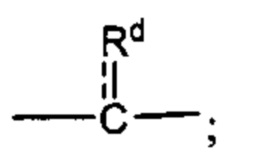
Ra выбран из группы, состоящей из (1) Н, (2) С1-10-алкила, (3) арила, (4) -C(O)-Re, (5) -SO2-NH2 и (6) -SO2-С1-4-алкила; при этом каждый из алкила и арила является необязательно замещенным одним-тремя заместителями, независимо выбранными из галогена и гетероциклила;
Ra' выбран из группы, состоящей из (1) Н и (2) C1-6-алкила;
Rb представляет собой C1-6-алкил или галоген;
каждый из Rc и Rd независимо выбран из группы, состоящей из (1) Н, (2) С1-6-алкила, (3) -С(O)-С1-6-алкила и (4) оксо;
Re выбран из группы, состоящей из (1) C1-6-алкила, (2) арила и (3) гетероарила;
m равен 0, 1 или 2;
n равен 0, 1 или 2; и
p равен 0, 1 или 2.
В одном варианте осуществления формулы (I) каждый из R1 и R2 представляет собой Н.
В одном варианте осуществления формулы (I) m равен 1.
В одном варианте осуществления формулы (I) каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) С1-4-алкила, необязательно замещенного одним-тремя атомами галогена; и m равен 1.
В одном варианте осуществления формулы (I) каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) C1-4-алкила, необязательно замещенного одним-тремя атомами галогена.
В одном варианте осуществления формулы (I) Y1 представляет собой:
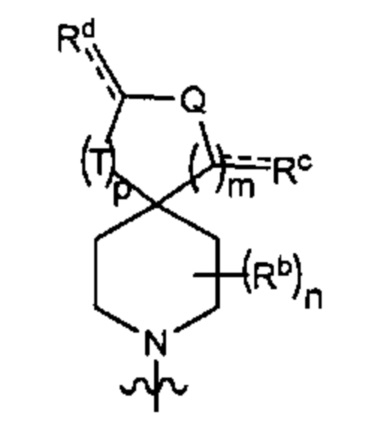
В одном варианте осуществления формулы (I) R3 представляет собой Н и R6 представляет собой Y2, имеющий следующую формулу:
где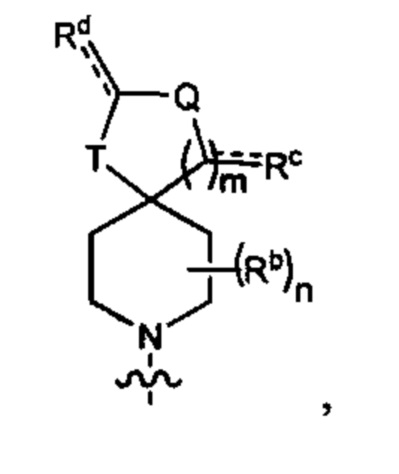
пунктирная линия  представляет необязательную двойную связь;
представляет необязательную двойную связь;
Q представляет собой -CH(Ra)- или -N(Ra)-;
Т представляет собой -CH2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-6-алкила и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним-тремя заместителями, независимо выбранными из галогена и 5- или 6-членной гетеро моноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода, серы и азота;
Rb представляет собой С1-4-алкил;
каждый из Rc и Rd независимо представляет собой Н или оксо; и
m равен 1.
В одном варианте осуществления формулы (I) R3 представляет собой Н, и R6 представляет собой Y3, имеющий следующую формулу:
где:
Q представляет собой -N(Ra)-;
Т представляет собой -CH2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) С1-4-алкила, необязательно замещенного одним-тремя заместителями, независимо выбранными из галогена и 5- или 6-членной гетеро моноциклической группы, содержащей один кольцевой атом кислорода, и (3) фенила;
Rb представляет собой метил или этил; и
n равен 0, 1 или 2.
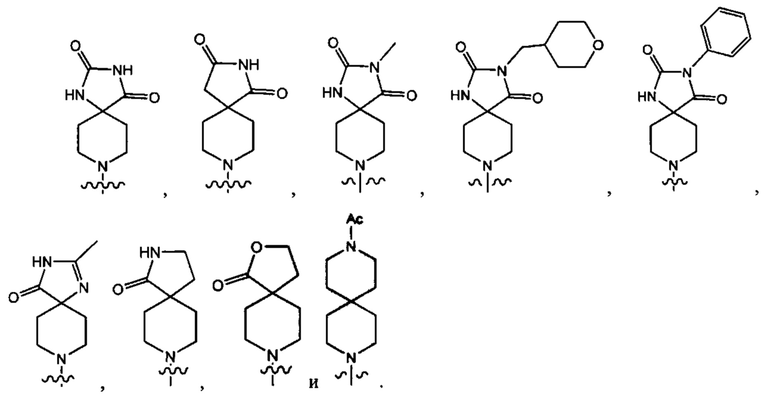
В одном варианте осуществления формулы (I) Y1 выбран из группы, состоящей из:
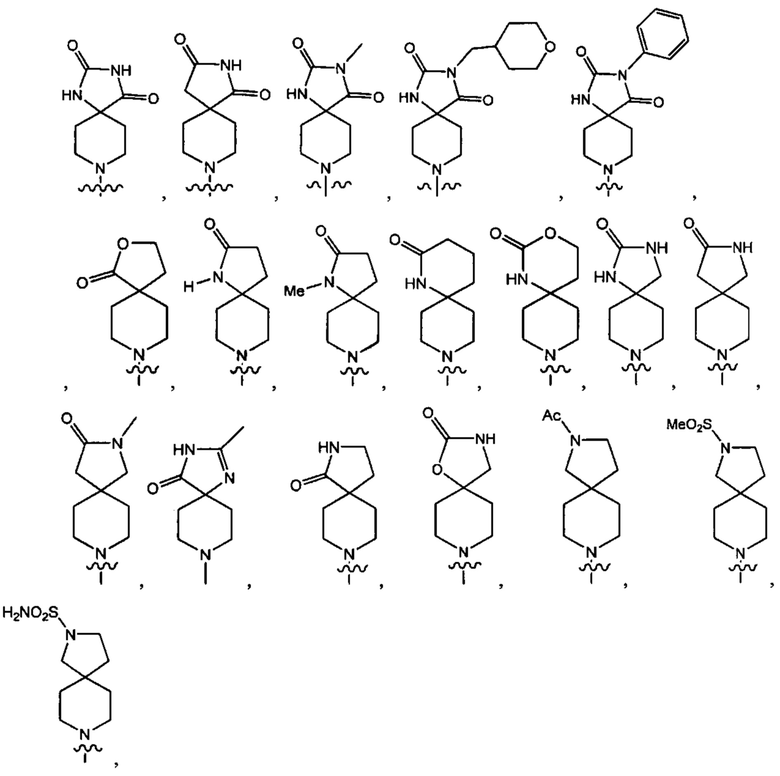
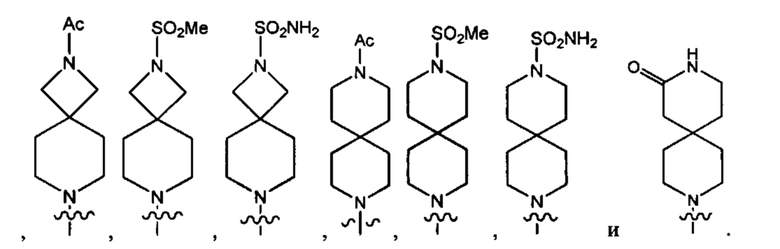
В одном варианте осуществления формулы (I) Y1 выбран из группы, состоящей из:
В одном варианте осуществления формулы (I) соединение представляет собой соединение формулы I(a):
где: 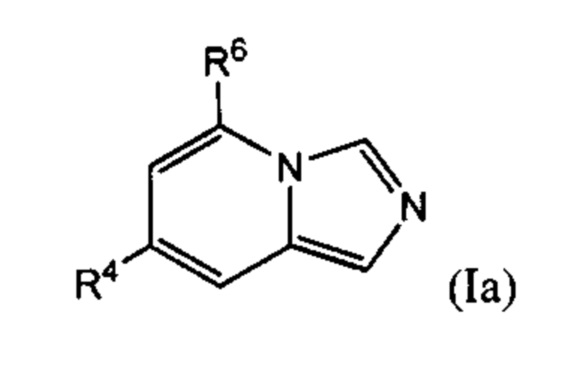
R4 выбран из группы, состоящей из (1) галогена и (2) С1-4-алкила, необязательно замещенного одним-тремя атомами галогена;
R6 представляет собой Y2, имеющий следующую формулу:
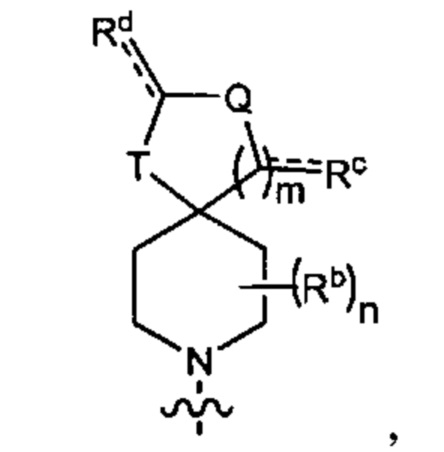
где
пунктирная линия  представляет необязательную двойную связь;
представляет необязательную двойную связь;
Q представляет собой -CH(Ra)- или -N(Ra)-;
Т представляет собой -CH2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-4-алкила и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним-тремя заместителями, независимо выбранными из галогена и гетероциклила;
Rb представляет собой С1-4-алкил;
каждый из Rc и Rd независимо представляет собой Н или оксо;
m равен 1; и
n равен 0, 1 или 2.
В одном варианте осуществления формулы (Ia) R6 представляет собой Y3, имеющий следующую формулу:
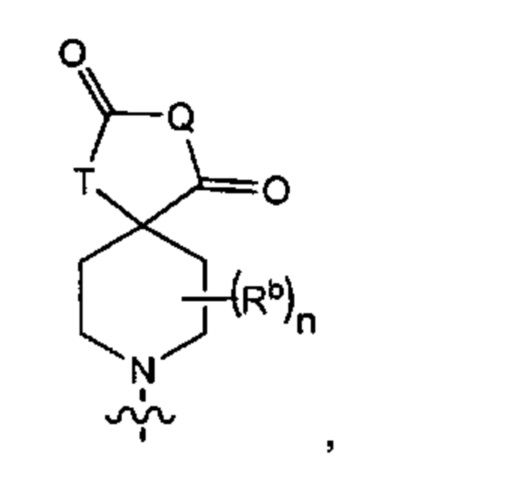
где
Q представляет собой -N(Ra)-;
Т представляет собой -CH2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) С1-4-алкила, необязательно замещенного одним-тремя заместителями, независимо выбранными из галогена и 5- или 6-членной гетеро моноциклической группы, содержащей один кольцевой атом кислорода, и (3) фенила; и
Rb представляет собой метил.
В одном варианте осуществления формулы (Ia) Ra выбран из группы, состоящей из (1) Н, (2) С1-4-алкила, необязательно замещенного одним-тремя заместителями, независимо выбранными из галогена и тетрагидропиранила, и (3) фенила.
В одном варианте осуществления формулы (Ia) Y3 выбран из группы, состоящей из:
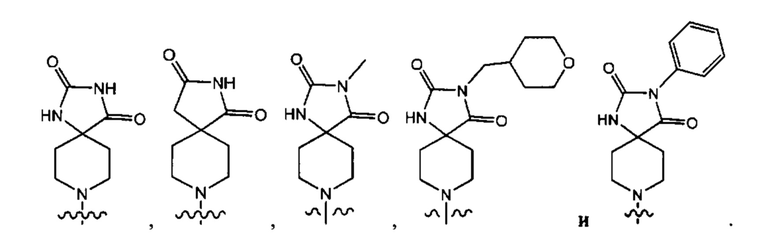
В одном варианте осуществления формулы (I) соединение представляет собой соединение формулы I(b):
где 
R5 выбран из группы, состоящей из (1) галогена и (2) С1-4-алкила, необязательно замещенного одним-тремя атомами галогена;
R3 представляет собой Y2, имеющий следующую формулу:
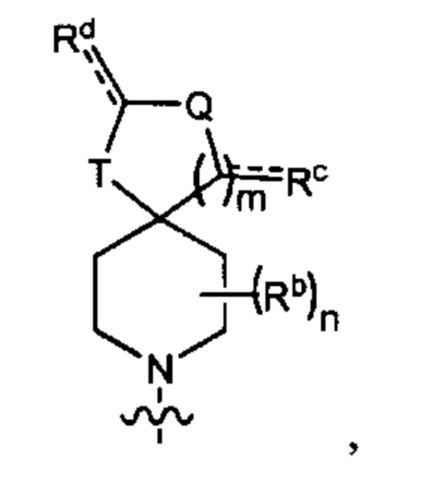
где
пунктирная линия  представляет необязательную двойную связь;
представляет необязательную двойную связь;
Q представляет собой -CH(Ra)- или -N(Ra)-;
Т представляет собой -СН2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-6-алкила и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним-тремя заместителями, независимо выбранными из галогена и гетероциклила;
Rb представляет собой С1-4-алкил;
каждый из Rc и Rd независимо представляет собой Н или оксо;
m равен 1; и
n равен 0, 1 или 2.
В одном варианте осуществления формулы (Ib) R3 представляет собой Y3, имеющий следующую формулу:
где 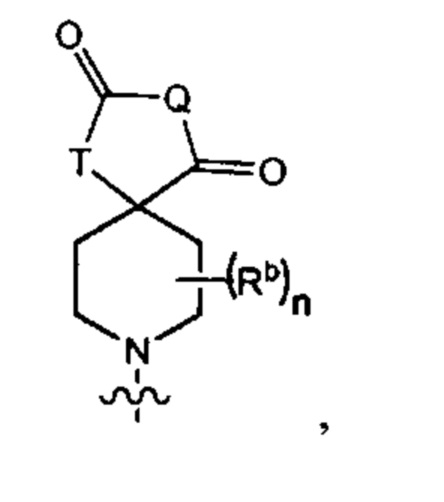
Q представляет собой -N(Ra)-;
Т представляет собой -CH2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) С1-4-алкила, необязательно замещенного одним-тремя заместителями, независимо выбранными из галогена и 5- или 6-членной гетеро моноциклической группы, содержащей один кольцевой атом кислорода, и (3) фенила; и
Rb представляет собой метил.
В одном варианте осуществления формулы (Ib) Ra выбран из группы, состоящей из (1) Н, (2) С1-4-алкила, необязательно замещенного одним-тремя заместителями, независимо выбранными из галогена и тетрагидропиранила, и (3) фенила.
В одном варианте осуществления формулы (Ib) Y3 выбран из группы, состоящей из:
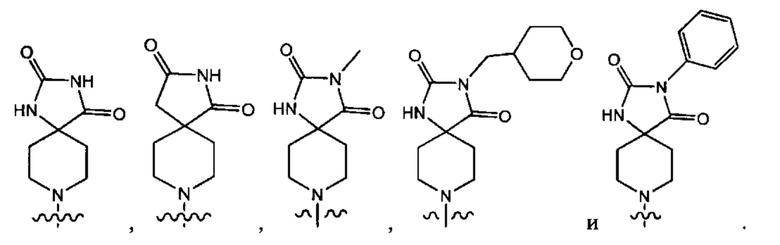
В одном варианте осуществления соединение, раскрытое в настоящем документе, выбрано из группы, состоящей из следующих соединений:
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(6-(трифторметил)имидазо[1,5-а]пиридин-8-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
3-метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
3-((тетрагидро-2Н-пиран-4-ил)метил)-8-(7-(трифторметил)имидазо[1,5-а]-пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
3-фенил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
3,6,6-триметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
6,6-диметил-8-(6-(трифторметил)имидазо[1,5-а]пиридин-8-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
6,6-диметил-3-((тетрагидро-2Н-пиран-4-ил)метил)-8-(7-(трифторметил)имидазо-[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
6,6-диметил-3-фенил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2-окса-8-азаспиро[4,5]декан-1-он,
2-метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4.5]дек-1-ен-4-он,
1-(9-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-3,9-диазаспиро[5.5]-ундекан-3-ил)этан-1-он,
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4.5]декан-1-он,
6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-2-тиа-1,3,8-триазаспиро[4,5]декан-4-он-2,2-диоксид,
(R)- или (S)-8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-2,8-диазаспиро-[4,5]декан-1,3-дион,
(S)- или (R)-6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
(R)- или (S)-8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-2,8-диазаспиро-[4,5]декан-1,3-дион,
(R)- или (S)-4-метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
6,6-дифтор-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро-[4,5]декан-1,3-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
8-(7-метоксиимидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион и
8-(7,8-михлоримидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион;
или их фармацевтически приемлемой соли.
Также, в настоящем документе раскрыта композиция, содержащая соединение формулы I, (Ia) или (Ib) и по меньшей мере один фармацевтически приемлемый носитель.
Также, в настоящем документе раскрыт способ ингибирования активности фермента IDO, включающий приведение в контакт IDO с соединением формулы I, (Ia) или (Ib), или его фармацевтически приемлемой солью.
Также, в настоящем документе раскрыт способ ингибирования активности фермента TDO, включающий приведение в контакт TDO с соединением формулы I, (Ia) или (Ib), или его фармацевтически приемлемой солью.
Также, в настоящем документе раскрыт способ ингибирования активности обоих ферментов, IDO и TDO, включающий приведение в контакт IDO и TDO с соединением формулы I, (Ia) или (Ib), или его фармацевтически приемлемой солью.
Также, в настоящем документе раскрыт способ ингибирования иммуносупрессии, ассоциированной с активностями IDO и/или TDO у пациента, включающий введение указанному пациенту эффективного количества соединения формулы I, (Ia) или (Ib), или его фармацевтически приемлемой соли.
Также, в настоящем документе раскрыт потенциальный способ лечения рака, вирусной инфекции, депрессии, нейродегенеративного нарушения, травмы, возрастной катаракты, болезни трансплантат против хозяина или аутоиммунного заболевания у пациента, включающий введение указанному пациенту эффективного количества соединения формулы I, (1а) или (lb), или его фармацевтически приемлемой соли.
Также, в настоящем документе раскрыт потенциальный способ лечения меланомы у пациента, включающий введение указанному пациенту эффективного количества соединения формулы I, (Ia) или (Ib), или его фармацевтически приемлемой соли.
Кроме того, в настоящем документе раскрыто соединение формулы I, (Ia) или (Ib), или его фармацевтически приемлемая соль для применения в терапии. В одном варианте осуществления в настоящем документе раскрыто применение соединения формулы I, (Ia) или (Ib), или его фармацевтически приемлемой соли для изготовления лекарственного средства для применения в терапии.
Используемый в настоящем документе «алкил» относится к насыщенным алифатическим углеводородным группам как с разветвленной, так и с прямой цепью, содержащим 1-18 атомов углерода или, в частности, 1-12 атомов углерода. Примеры таких групп включают, но без ограничения, метил (Me), этил (Et), н-пропил (Pr), н-бутил (Bu), н-пентил, н-гексил и их изомеры, такие как изопропил (i-Pr), изобутил (i-Bu), втор-бутил (s-Bu), трет-бутил (t-Bu), изопентил и изогексил. Алкильные группы могут быть необязательно замещенными одним или несколькими заместителями, как определено в настоящем документе. «C1-6-алкил» относится к алкильной группе, как определено в настоящем документе, имеющей от 1 до 6 атомов углерода.
«Арил» относится к ароматическому моноциклическому или мультициклическому кольцевому фрагменту, содержащему от 6 до 14 кольцевых атомов углерода, в частности, от 6 до 10 кольцевых атомов углерода. Моноциклические арильные кольца включают, но без ограничения, фенил. Мультициклические кольца включают, но без ограничения, нафтил и бициклические кольца, при этом фенил конденсирован с С5-7-циклоалкильным или С5-7-Циклоалкенильным кольцом. Арильные группы могут быть необязательно замещенными одним или несколькими заместителями, как определено в настоящем документе. Связывание может происходить через любой из атомов углерода любого кольца.
«Циклоалкил» относится к моноциклическому насыщенному карбоциклическому кольцу, имеющему определенное число атомов углерода. Например, С3-7-циклоалкил относится к циклоалкильной группе, как определено в настоящем документе, имеющей от 3 до 7 атомов углерода. Примеры циклоалкила включают, но без ограничения, циклопропил, циклобутил, циклопентил, циклогексил и циклогептанил. Циклоалкильные группы могут быть необязательно замещенными одним или несколькими заместителями, как определено в настоящем документе.
«Галогено» или «галоген» относится к заместителю фтор, хлор, бром или йод, если не указано иначе.
«Гетероцикл» или «гетероциклил» относится к насыщенному, частично ненасыщенному или ароматическому кольцевому фрагменту, имеющему по меньшей мере один кольцевой гетероатом и по меньшей мере один кольцевой атом углерода. В одном варианте осуществления гетероатом представляет собой кислород, серу или азот. Гетероцикл, содержащий более чем один гетероатом, может содержать различные гетероатомы. Гетероциклильные фрагменты включают как моноциклические, так и мультициклические (например, бициклические) кольцевые фрагменты. Бициклические кольцевые фрагменты включают конденсированные, спироциклические и мостиковые бициклические кольца и могут содержать один или несколько гетероатомов в любом из колец. Кольцо, присоединенное к остальной части молекулы, может содержать или не содержать гетероатом. Любое кольцо бициклического гетероцикла может быть насыщенным, частично ненасыщенным или ароматическим. Гетероцикл может быть присоединен к остальной части молекулы через кольцевой атом углерода, кольцевой атом кислорода или кольцевой атом азота. Неограничивающие примеры гетероциклов описаны ниже.
В одном варианте осуществления частично ненасыщенные и ароматические 4-7-членные моноциклические гетероциклильные фрагменты включают, но без ограничения, 2,3-дигидро-1,4-диоксинил, дигидропиранил, дигидропиразинил, дигидропиридазинил, дигидропиридинил, дигидропиримидинил, фуранил, имидазолил, изотиазолил, изоксазолил, оксадиазолил, оксазолил, пиранил, пиразинил, пиразолил, пиридазинил, пиридинил, пиримидинил, пирролил, тетрагидропиразинил, тетрагидропиридазинил, тетрагидропиридинил, тетрагидропиримидинил, тетразолил, тиадиазолил, тиазолил, тиенил, тиофенил и триазолил.
В одном варианте осуществления насыщенные 4-7-членные моноциклические гетероциклильные фрагменты включают, но без ограничения, азетидинил, 1,4-диоксанил, гексагидроазепинил, морфолинил, 1,4-оксазепанил, оксазолидинил, оксетанил, пиперазинил, пиперидинил, пиридин-2-онил, пирролидинил, тетрагидрофуранил, тетрагидропиранил, тиоморфолинил, тетрагидротиенил и тетрагидротиофенил. В одном варианте осуществления насыщенный 4-7-членный моноциклический гетероциклил представляет собой азетидинил.
Гетероциклические группы могут быть необязательно замещенными одним или несколькими заместителями, как определено в настоящем документе.
«Необязательно замещенный» относится к «незамещенному или замещенному», и поэтому описанные здесь общие структурные формулы охватывают соединения, содержащие указанный необязательный заместитель(и), а также соединения, которые не содержат необязательный заместитель(и). Каждый заместитель независимо определяется каждый раз, когда он встречается в определениях общей структурной формулы.
Полиморфизм
Соединение формулы (I), (Ia) или (Ib), включая его соль или сольват, может существовать в кристаллической форме, некристаллической форме или их смеси. Соединение или его соль или сольват могут также проявлять полиморфизм, то есть способность возникать в различных кристаллических формах. Эти различные кристаллические формы обычно известны как «полиморфы». Полиморфы имеют один и тот же химический состав, но отличаются упаковкой, геометрическим расположением и другими описательными свойствами кристаллического твердого состояния. Следовательно, полиморфы могут иметь разные физические свойства, такие как форма, плотность, твердость, деформируемость, стабильность, и свойства растворения. Полиморфы обычно имеют различные температуры плавления, ИК-спектры и рентгеновские порошковые дифрактограммы, все из которых могут быть использованы для идентификации. Специалисту в данной области техники будет понятно, что могут быть получены различные полиморфы, например, путем изменения или регулирования условий, используемых для кристаллизации/перекристаллизации соединения формулы (I), (Ia) или (Ib).
Оптические изомеры - диастереомеры - геометрические изомеры - таутомеры
В настоящий документ включены различные изомеры соединений формулы (I), (Ia) или (Ib). Термин «изомеры» относится к соединениям, которые имеют одинаковый состав и молекулярную массу, но различаются по физическим и/или химическим свойствам. Структурное различие может быть в конституции (геометрические изомеры) или в способности вращать плоскость поляризованного света (стереоизомеры).
Что касается стереоизомеров, соединение формулы (I), (Ia) или (Ib) может содержать один или более асимметричных атомов углерода и может существовать в виде рацемической смеси или в виде отдельных энантиомеров или диастереомеров. Все такие изомерные формы включены в настоящий документ, включая их смеси. Если соединение формулы (I), (Ia) или (Ib) содержит двойную связь, заместитель может находиться в конфигурации Е или Z. Если соединение формулы (I), (Ia) или (Ib) содержит дизамещенный циклоалкил, то циклоалкильный заместитель может иметь цис- или трансконфигурацию.
Любой асимметричный атом (например, углерод) соединения формулы (I), (Ia) или (Ib) может присутствовать в рацемической смеси или энантиомерно обогащенной, например, (R)-, (S)- или (R, S)-конфигурации. В некоторых вариантах осуществления каждый асимметричный атом имеет по меньшей мере 50% энантиомерного избытка, по меньшей мере 60% энантиомерного избытка, по меньшей мере 70% энантиомерного избытка, по меньшей мере 80% энантиомерного избытка, по меньшей мере 90% энантиомерного избытка, по меньшей мере 95% энантиомерного избытка, или по меньшей мере 99% энантиомерного избытка в (R)- или (S)-конфигурации. Заместители на атомах с ненасыщенными двойными связями могут, по возможности, присутствовать в цис- (Z)-или транс- (Е)- форме.
Соединение формулы (I), (Ia) или (Ib) может быть представлено в форме одного из возможных изомеров, ротамеров, атропизомеров, таутомеров или их смесей, например, в виде по существу чистых геометрических (цис или транс) изомеров, диастереомеров, оптических изомеров (антиподы), рацематов или их смесей.
Любые полученные смеси изомеров могут быть разделены на основе физико-химических различий составляющих на чистые или по существу чистые геометрические или оптические изомеры, диастереомеры, рацематы, например, путем хроматографии и/или фракционной кристаллизации.
Любые полученные рацематы конечных соединений примеров или интермедиатов могут быть разделены на оптические антиподы известными способами, например, путем разделения диастереомерных солей, полученных с помощью оптически активной кислоты или основания, и выделения оптически активного кислотного или основного соединения. В частности, таким образом можно использовать основный фрагмент для расщепления соединений согласно настоящему изобретению на их оптические антиподы, например, путем фракционной кристаллизации соли, образованной с помощью оптически активной кислоты, например, винной кислоты, дибензоилвинной кислоты, диацетилвинной кислоты, ди-О, О'-п-толуоилвинной кислоты, миндальной кислоты, яблочной кислоты или камфор-10-сульфоновой кислоты. Рацемические соединения также могут быть разделены методом хиральной хроматографии, например, высокоэффективной жидкостной хроматографии (HPLC) с использованием хирального адсорбента.
Некоторые из соединений, описанных в настоящем документе, могут существовать с различными точками присоединения водорода, и их называют таутомерами. Например, соединения, включающие карбонильные СН2С(O)- группы (кетоформы), могут подвергаться таутомерии с образованием гидроксильных -СН=С(ОН)- групп (енольные формы). Как кето, так и енольные формы, индивидуально, а также их смеси включены в объем настоящего изобретения. Изотопные вариации Соединения формулы (I), (Ia) или (Ib) включают немеченые формы, а также изотопно-меченные формы. Изотопно-меченные соединения имеют структуры, изображенные формулами, приведенными в настоящем документе, за исключением того, что один или более атомов замещены атомом, имеющим выбранную атомную массу или массовое число. Примеры изотопов, которые могут быть введены в соединения, описанные в настоящем документе, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как 2Н (т.е. дейтерий или «D»), 3Н, 11С, 13С, 14С, 13N, 15N, 15O, 17O, 18O, 32Р, 35S, 18F, 123I, 125I и 36CI. Изобретение включает различные изотопно-меченные соединения, как определено в настоящем документе, например, те, в которых присутствуют радиоактивные изотопы, такие как 3Н и 14С, или те, в которых присутствуют нерадиоактивные изотопы, такие как 2Н и 13С. Такие изотопно-меченные соединения являются полезными в метаболических исследованиях (с 14С), кинетических исследованиях реакций (например, с 2Н или 3Н), методах детекции или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая анализы распределения лекарственного средства в ткани или субстрате, или при радиоактивном лечении пациентов. В частности, замещение испускающими позитрон изотопами, такими как 11С, 18F, 15O и 13N, может быть особенно желательным для исследований PET или SPECT.
Изотопно-меченные соединения формулы (I), (Ia) или (Ib) обычно могут быть получены обычными способами, известными специалистам в данной области. Кроме того, замещение более тяжелыми изотопами, в частности, дейтерием (т.е. 2Н или D) может дать определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличенного периода полувыведения in vivo или сниженной дозировки или улучшение терапевтического индекса.
Фармацевтически приемлемые соли
Термин «фармацевтически приемлемая соль» относится к соли, полученной из фармацевтически приемлемого нетоксичного основания или кислоты, включая неорганическое или органическое основание и неорганическую или органическую кислоту. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Конкретные варианты осуществления включают соли аммония, кальция, магния, калия и натрия. Соли в твердой форме могут существовать в более чем одной кристаллической структуре и могут также находиться в форме гидратов. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичного, вторичного и третичного аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Когда соединение формулы (I), (Ia) или (Ib) является основным, соль может быть получена из фармацевтически приемлемой нетоксичной кислоты, включая неорганическую и органическую кислоту. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту, трифторуксусную кислоту (TFA) и т.п. Конкретные варианты осуществления включают лимонную, бромистоводородную, хлористоводородную, малеиновую, фосфорную, серную, фумаровую, винную и трифторуксусную кислоты. Понятно, что, как используется здесь, ссылки на соединения, раскрытые здесь, также должны включать их фармацевтически приемлемые соли.
Способы применения
Эти соединения, раскрытые в настоящем документе, могут быть полезными для потенциального лечения или предупреждения развития заболеваний, связанных с IDO и/или TDO. В одном варианте осуществления эти соединения могут потенциально ингибировать активность фермента IDO, фермента TDO или обоих ферментов, IDO и TDO.
Например, раскрытые в настоящем документе соединения потенциально можно применять для ингибирования активности IDO и/или TDO в клетках или у индивидуума, нуждающегося в модулировании фермента, путем введения эффективного количества соединения. Кроме того, в настоящем документе раскрыты способы ингибирования деградации триптофана в системе, содержащей клетки, экспрессирующие IDO и/или TDO, такой как ткань, живой организм или клеточная культура. В некоторых вариантах осуществления настоящее изобретение обеспечивает способы изменения (например, повышения) внеклеточных уровней триптофана у млекопитающего посредством введения эффективного количества соединения или композиции, обеспеченной в настоящем документе. Способы измерения уровней триптофана и деградации триптофана известны в данной области.
Также, в настоящем документе раскрыты способы ингибирования подавления иммунитета, такого как IDO- и/или TDO-опосредованное подавление иммунитета, у пациента посредством введения пациенту эффективного количества соединения или композиции, представленной в настоящем документе. IDO- и/или TDO-опосредованное подавление иммунитета может быть связано, например, с раком, ростом опухоли, метастазом, вирусной инфекцией, вирусной репликацией и т.д.
Также, в настоящем документе раскрыты способы потенциального лечения заболеваний, связанных с активностью или экспрессией, включая патологическую активность и/или сверхэкспрессию, IDO и/или TDO у индивидуума (например, пациента), путем введения индивидууму, нуждающемуся в таком лечении, эффективного количества или дозы описанного здесь соединения или его фармацевтической композиции. Примеры заболеваний включают любое заболевание, нарушение или патологическое состояние, которое может быть прямо или косвенно связано с экспрессией или активностью фермента IDO и/или TDO, например, сверхэкспрессией или патологической активностью. Заболевание, связанное с IDO и/или TDO, может также включать любое заболевание, нарушение или патологическое состояние, которое можно предупредить, ослабить или излечить путем модулирования активности фермента. Примеры заболеваний, связанных с IDO и/или TDO, включают рак, вирусную инфекцию, такую как HIV и HCV, депрессию, нейродегенеративные нарушения, такие как болезнь Альцгеймера и болезнь Хантингтона, травму, возрастные катаракты, трансплантацию органа (например, отторжение трансплантата органа) и аутоиммунные заболевания, включая астму, ревматоидный артрит, рассеянный склероз, аллергическое воспаление, воспалительное заболевание кишечника, псориаз и системную красную волчанку. Примеры раковых заболеваний, которые поддаются лечению с помощью описанных в настоящем документе способов, включают рак толстой кишки, поджелудочной железы, молочной железы, предстательной железы, легких, головного мозга, яичников, шейки матки, яичек, почек, головы и шеи, лимфому, лейкоз, меланому и т.п. Соединения согласно изобретению можно также применять при лечении ожирения и ишемии. Используемый в настоящем документе термин «клетка» означает клетку, которая находится in vitro, ex vivo или in vivo. В некоторых вариантах осуществления ex vivo клетка может представлять собой часть образца ткани, извлеченной из организма, такого как млекопитающее. В некоторых вариантах осуществления in vitro клетка может представлять собой клетку в клеточной культуре. В некоторых вариантах осуществления in vivo клетка представляет собой клетку, живущую в организме, таком как млекопитающее.
Используемый в настоящем документе термин «приведение в контакт» относится к объединению указанных фрагментов в системе in vitro или в системе in vivo. Например, «приведение в контакт» фермента IDO с соединением, раскрытым в настоящем документе, включает введение соединения согласно настоящему изобретению индивидууму или пациенту, такому как человек, имеющему IDO и/или TDO, а также, например, введение соединения согласно изобретению в образец, содержащий клеточный или очищенный препарат, содержащий фермент IDO и/или TDO.
Субъект, которому вводят раскрытое здесь соединение или его фармацевтически приемлемую соль, обычно представляет собой млекопитающее, такое как человек, мужчину или женщину. Субъект также относится к коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам и птицам. В одном варианте осуществления субъектом является человек.
Используемые в настоящем документе термины «лечение» и «лечить» относятся ко всем процессам, при которых может наблюдаться замедление, прерывание, приостановка, контроль или остановка развития заболевания или нарушения, которое может быть связано с ферментативной активностью IDO и/или TDO. Термины не обязательно указывают на полное устранение всех симптомов заболевания или нарушения. Термины также включают потенциальную профилактическую терапию указанных состояний, особенно у субъекта, предрасположенного к такому заболеванию или нарушению.
Термины «введение» и/или «назначение» соединения следует понимать как доставку описанного в настоящем документе соединения или его фармацевтически приемлемой соли, и композиций вышеуказанного субъекту.
Количество соединения, введенного субъекту, представляет собой количество, достаточное для ингибирования активности ферментов IDO и/или TDO у субъекта. В одном варианте осуществления количество соединения может представлять собой «эффективное количество», при этом указанное соединение вводят в количестве, которое будет вызывать биологический или медицинский отклик в ткани, системе, животном или человеке, необходимый исследователю, ветеринару, врачу или другому клиницисту. Эффективное количество не обязательно включает соображения токсичности и безопасности, связанные с введением соединения. Признано, что специалист в данной области может повлиять на физиологические нарушения, связанные с активностью ферментов IDO и/или TDO, путем лечения субъекта, имеющего нарушения, или путем профилактического лечения субъекта, у которого могут развиться нарушения, с помощью эффективного количества соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли.
Эффективное количество соединения будет варьироваться в зависимости от конкретного выбранного соединения (например, с учетом мощности, эффективности и/или периода полувыведения соединения); выбранного пути введения; патологического состояния, подлежащего лечению; тяжести состояния, подлежащего лечению; возраста, размера, веса и физического состояния субъекта, подлежащего лечению; истории болезни субъекта, подлежащего лечению; продолжительности лечения; природы сопутствующей терапии; желаемого терапевтического эффекта; и подобных факторов, и может быть определено обычным способом квалифицированным специалистом.
Соединения, раскрытые в настоящем документе, могут быть введены любым подходящим путем, включая пероральное и парентеральное введение. Парентеральное введение обычно осуществляют путем инъекции или инфузии и включает внутривенную, внутримышечную и подкожную инъекцию или инфузию.
Соединения, раскрытые в настоящем документе, могут быть введены один раз или в соответствии с режимом дозирования, где ряд доз вводят через различные интервалы времени в течение заданного периода времени. Например, дозы могут быть введены один, два, три или четыре раза в день. Дозы можно вводить до тех пор, пока не будет достигнут желаемый терапевтический эффект, или в течение неопределенного времени, чтобы поддерживать нужный терапевтический эффект. Подходящие режимы дозирования для соединения, раскрытого в настоящем документе, зависят от фармакокинетических свойств этого соединения, таких как абсорбция, распределение и период полувыведения, которые могут быть определены специалистом в данной области. Кроме того, подходящие режимы дозирования, включая продолжительность таких режимов введения для соединения, раскрытого в настоящем документе, зависят от заболевания или патологического состояния, подлежащего лечению, тяжести заболевания или патологического состояния, возраста и физического состояния субъекта, подлежащего лечению, медицинской истории болезни субъекта, подлежащего лечению, характера сопутствующей терапии, желаемого терапевтического эффекта и подобных факторов в рамках знаний и опыта специалиста в данной области. Специалистам в данной области будет также понятно, что подходящие режимы дозирования могут потребовать корректировки с учетом индивидуальной реакции субъекта на режим дозирования или с течением времени, если отдельный субъект нуждается в таких изменениях. Типичные суточные дозы могут варьироваться в зависимости от конкретного выбранного пути введения. Типичные суточные дозы для перорального введения человеку, имеющему вес приблизительно 70 кг, будут находиться в диапазоне от примерно 0,1 мг до примерно 2 г или более конкретно от 0,1 мг до 500 мг или еще более конкретно от 0,2 мг до 100 мг соединения формулы (I), (Ia) или (Ib).
В одном варианте осуществления настоящего изобретения предлагается способ потенциального лечения заболевания или нарушения, связанного с активностью фермента IDO и/или TDO, включающий введение эффективного количества соединения формулы (I), (Ia) или (Ib) субъекту, нуждающемуся в таком лечении. В одном варианте осуществления заболевание или нарушение, связанное с ферментом IDO и/или TDO, представляет собой нарушение клеточной пролиферации.
В одном варианте осуществления в настоящем документе раскрыто применение соединения формулы (I), (Ia) или (Ib) в терапии. Соединение может быть полезным в способе ингибирования активности фермента IDO и/или TDO у субъекта, такого как млекопитающее, нуждающееся в таком ингибировании, включающем введение эффективного количества соединения субъекту.
В одном варианте осуществления в настоящем документе раскрыта фармацевтическая композиция, содержащая соединение формулы (I), (Ia) или (Ib), или его фармацевтически приемлемую соль, для применения в потенциальном лечении нарушения или заболевания, связанного с активностью фермента IDO и/или TDO. Композиции
Используемый в настоящем документе термин «композиция» охватывает дозированную форму, содержащую указанное соединение в определенном количестве, а также любую дозированную форму, которая получена, прямо или косвенно, из комбинации указанного соединения в определенном количестве. Такой термин охватывает дозированную форму, содержащую соединение формулы (I) или (Ia), или его фармацевтически приемлемую соль, и один или несколько фармацевтически приемлемых носителей или вспомогательных веществ. Соответственно, композиции согласно настоящему изобретению охватывают любую композицию, полученную путем смешивания соединения согласно настоящему изобретению и одного или нескольких фармацевтически приемлемых носителей или вспомогательных веществ. Под «фармацевтически приемлемым» подразумевается, что носители или вспомогательные вещества являются совместимыми с соединением, описанным в настоящем документе, и с другими ингредиентами композиции.
В одном варианте осуществления в настоящем документе раскрыта композиция, содержащая соединение формулы (I) или (Ia), или его фармацевтически приемлемую соль, и один или несколько фармацевтически приемлемых носителей или вспомогательных веществ. Композиция может быть изготовлена и упакована в объемной форме, при этом эффективное количество соединения согласно изобретению может быть извлечено, а затем дано субъекту, например, с порошками или сиропами. В альтернативном случае композиция может быть изготовлена и упакована в виде единичной дозированной формы, при этом каждая физически дискретная единица содержит эффективное количество соединения формулы (I), (Ia) или (Ib). В случае изготовления в виде единичной лекарственной формы композиция согласно изобретению обычно содержит от примерно 0,1 мг до 2 г или более конкретно от 0,1 мг до 500 мг или еще более конкретно от 0,2 мг до 100 мг соединения формулы (I), (Ia) или (Ib), или его фармацевтически приемлемой соли.
Соединение, раскрытое в настоящем документе, и фармацевтически приемлемый носитель или вспомогательное вещество(вещества) обычно составляют в дозированную форму, адаптированную для введения субъекту желаемым путем введения. Например, лекарственные формы включают такие лекарственные формы, которые адаптированы для (1) перорального введения, такие как таблетки, капсулы, каплеты, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше и крахмалы; и (2) парентерального введения, такие как стерильные растворы, суспензии и порошки для восстановления. Подходящие фармацевтически приемлемые носители или вспомогательные вещества будут варьироваться в зависимости от конкретной выбранной дозированной формы. Кроме того, подходящие фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны для определенной функции, которую они могут выполнять в композиции. Например, некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны исходя из их способности облегчать изготовление однородных дозированных форм. Некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны исходя из их способности облегчать изготовление стабильных дозированных форм. Некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны исходя из их способности облегчать перенос или транспортировку соединения, раскрытого в настоящем документе, после введения субъекту из одного органа или части тела в другой орган или другую часть тела. Некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны исходя из их способности улучшать соблюдение пациентом режима и схемы лечения.
Подходящие фармацевтически приемлемые вспомогательные вещества включают следующие типы вспомогательных веществ: разбавители, смазывающие вещества, связывающие вещества, разрыхлители, наполнители, скользящие вещества, гранулирующие агенты, агенты для нанесения покрытия, смачивающие агенты, растворители, сорастворители, суспендирующие агенты, эмульгаторы, подсластители, ароматизаторы, маскирующие вкус и запах агенты, окрашивающие агенты, антислеживающие агенты, увлажняющие агенты, хелатирующие агенты, пластификаторы, повышающие вязкость агенты, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные агенты.
Специалист в данной области обладает знаниями и опытом в данной области, чтобы осуществить выбор подходящих фармацевтически приемлемых носителей и вспомогательных веществ в соответствующих количествах для применения в изобретении. Кроме того, существует ряд ресурсов, доступных для квалифицированного специалиста, в которых описаны фармацевтически приемлемые носители и вспомогательные вещества, и которые могут быть полезны при выборе подходящих фармацевтически приемлемых носителей и вспомогательных веществ. Примеры включают Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
Композиции согласно изобретению изготовлены с использованием методик и способов, известных квалифицированным специалистам в данной области. Некоторые способы, обычно используемые в данной области, описаны в Remington's Pharmaceutical Sciences (Mack Publishing Company).
В одном варианте осуществления изобретение относится к твердой пероральной дозированной форме, такой как таблетка или капсула, содержащей эффективное количество соединения согласно изобретению и разбавитель или наполнитель. Подходящие разбавители и наполнители включают лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и прежелатинизированный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и двухосновный фосфат кальция. Пероральная твердая лекарственная форма может дополнительно содержать связывающее вещество. Подходящие связывающие вещества включают крахмал (например, кукурузный крахмал, картофельный крахмал и прежелатинизированный крахмал), желатин, аравийскую камедь, альгинат натрия, альгиновую кислоту, трагакант, гуаровую камедь, повидон, а также целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Пероральная твердая лекарственная форма может, кроме того, содержать дезинтегрирующий агент. Подходящие дезинтегрирующие агенты включают кросповидон, натрия крахмал гликолят, кроскармеллозу, альгиновую кислоту и натрий карбоксиметилцеллюлозу. Пероральная твердая дозированная форма может дополнительно содержать смазывающее вещество. Подходящие смазывающие вещества включают стеариновую кислоту, стеарат магния, стеарат кальция и тальк.
Там, где это необходимо, составы в виде дозированных единиц для перорального введения могут быть инкапсулированы в микрокапсулы. Композиция также может быть изготовлена таким образом, чтобы пролонгировать или замедлить высвобождение, например, путем применения покрытия или заключения вещества в виде частиц в полимеры, воски или т.п.
Соединения, раскрытые в настоящем документе, также могут быть связаны с растворимыми полимерами в качестве нацеливающих носителей лекарственного средства. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатком пальмитоила. Кроме того, соединения согласно изобретению можно сопрягать с классом биоразлагаемых полимеров, пригодных для достижения контролируемого высвобождения лекарственного средства, например, с полимолочной кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианакрилатами и поперечно-связанными или амфипатическими блок-сополимерами гидрогелей.
В одном варианте осуществления изобретение относится к жидкой дозированной форме для перорального введения. Жидкости для перорального введения, такие как растворы, сиропы и эликсиры, могут быть изготовлены в виде единичной дозированной формы таким образом, что заданное количество содержит заранее определенное количество соединения, раскрытого в настоящем документе. Сиропы могут быть изготовлены путем растворения соединенния согласно изобретению в подходящим образом корригированном водном растворе; в то время как эликсиры изготавливают посредством использования нетоксичного спиртового наполнителя. Суспензии могут быть изготовлены путем диспергирования соединения, раскрытого в настоящем документе, в нетоксичном наполнителе. Также, могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые эфиры полиоксиэтиленсорбита, консерванты, корригирующие добавки, такие как масло перечной мяты или другие природные подсластители, сахарин или другие искусственные подсластители и т.п.
В одном варианте осуществления изобретение относится к композициям для парентерального введения. Композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, которые приводят композицию в состояние устойчивой изотоничности с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Композиции могут быть представлены в упаковках с единичной дозой или несколькими дозами, например, в герметично закрытых ампулах и флаконах, и могут храниться в лиофилизированном виде, требующем лишь добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед использованием. Экстемпоральные инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Комбинации
Соединение, раскрытое в настоящем документе, можно применять в комбинации с одним или несколькими другими активными агентами, включая, но без ограничения, другие противораковые агенты, которые применяют для предупреждения, лечения, контроля, ослабления или снижения риска возникновения конкретного заболевания или патологического состояния (например, нарушений клеточной пролиферации). В одном варианте осуществления соединение, раскрытое в настоящем документе, комбинировано с одним или несколькими другими противораковыми агентами, применяющимися для предупреждения, лечения, контроля, ослабления или снижения риска возникновения конкретного заболевания или состояния, для которого соединения, раскрытые в настоящем документе, являются полезными. Такие другие активные агенты могут быть введены способом и в количестве, которые обычно используют, одновременно или последовательно с соединением согласно настоящему изобретению.
Когда соединение, раскрытое в настоящем документе, применяют одновременно с одним или несколькими другими активными агентами, рассматривается композиция, содержащая такие другие активные агенты дополнительно к соединению, раскрытому в настоящем документе. Соответственно, композиции согласно настоящему изобретению включают такие композиции, которые также содержат один или несколько других активных ингредиентов дополнительно к соединению, раскрытому в настоящем документе. Соединение, раскрытое в настоящем документе, можно вводить либо одновременно, либо до или после одного или нескольких других терапевтических агентов. Соединение, раскрытое в настоящем документе, можно вводить отдельно таким же или другим путем введения, или вместе в той же фармацевтической композиции, что и другой агент(ы).
Продукты, представленные в виде комбинированного препарата, включают композицию, содержащую соединение формулы (I), (Ia) или (Ib) и один или несколько других активных агентов вместе в одной и той же фармацевтической композиции, или соединение формулы (I), (Ia) или (Ib) и один или несколько других терапевтических агентов в отдельной форме, например, в форме набора.
Массовое соотношение соединения, раскрытого в настоящем документе, ко второму активному агенту может варьироваться и зависит от эффективной дозы каждого агента. Как правило, будет использоваться эффективная доза каждого из них. Так, например, когда соединение, раскрытое в настоящем документе, комбинировано с другим агентом, массовое соотношение соединения, раскрытого в настоящем документе, к другому агенту обычно будет находиться в диапазоне примерно от 1000:1 до 1:1000, например, примерно от 200:1 до 1:200. Комбинации соединения, описанного в настоящем документе, и других активных агентов, как правило, также будут находиться в пределах вышеуказанного диапазона, но в каждом случае следует использовать эффективную дозу каждого активного агента. В таких комбинациях соединение, раскрытое в настоящем документе, и другие активные агенты можно вводить отдельно или в сочетании. Кроме того, введение одного элемента можно осуществлять до, одновременно или после введения другого агента (агентов).
В одном варианте осуществления в изобретении предлагается композиция, содержащая соединение формулы (I), (Ia) или (Ib) и по меньшей мере еще один терапевтический агент в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. В одном варианте осуществления терапия представляет собой лечение заболевания или нарушения, связанного с активностью фермента IDO и/или TDO.
В одном варианте осуществления в изобретении предлагается набор, содержащий две или более отдельных фармацевтических композиции, по меньшей мере одна из которых содержит соединение формулы (I), (Ia) или (Ib). В одном варианте осуществления набор содержит средства для отдельного содержания указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, обычно используемая для упаковки таблеток, капсул и т.п.
Набор, раскрытый в настоящем документе, может быть использован для введения различных дозированных форм, например, перорального и парентерального, для введения отдельных композиций с различными интервалами дозирования, или для титрования отдельных композиций относительно друг друга. Для содействия облегчению соблюдения пациентом режима и схемы лечения набор согласно изобретению обычно содержит инструкции по введению.
В настоящем документе раскрыто применение соединения формулы (I), (Ia) или (Ib) для лечения заболевания или нарушения, связанного с активностью фермента IDO и/или TDO, при этом лекарственное средство изготовлено для введения другим активным агентом. В изобретении также предлагается применение другого активного агента для лечения заболевания или нарушения, связанного с ферментом IDO и/или TDO, при этом лекарственное средство вводят с соединением формулы (I), (Ia) или (Ib).
В изобретении также предлагается применение соединения формулы (I), (Ia) или (Ib) для лечения заболевания или нарушения, связанного с активностью фермента IDO и/или TDO, при этом пациент ранее (например, в течение 24 часов) подвергался лечению другим активным агентом. В изобретении также предлагается использование другого терапевтического агента для лечения заболевания или нарушения, связанного с активностью фермента IDO и/или TDO, при этом пациент ранее (например, в течение 24 часов) подвергался лечению соединением формулы (I), (Ia) или (Ib). Второй агент можно применять через неделю, несколько недель, месяц или несколько месяцев после введения соединения, раскрытого в настоящем документе.
В одном варианте осуществления другой активный агент выбран из группы, состоящей из ингибиторов рецептора фактора роста эндотелия сосудов (VEGF), ингибиторов топоизомеразы II, ингибиторов белка Smoothen, алкилирующих агентов, противоопухолевых антибиотиков, антиметаболитов, ретиноидов, иммуномодулирующих агентов, включая, но без ограничения, противораковые вакцины, антагонисты CTLA-4, LAG-3 и PD-1.
Примеры ингибиторов рецептора фактора роста эндотелия сосудов (VEGF) включают, но без ограничения, бевацизумаб (продаваемый под торговой маркой AVASTIN фирмы Genentech/Roche), акситиниб (N-метил-2-[[3-[(Е)-2-пиридин-2-илэтенил]-1Н-индазол-6-ил]сульфанил]бензамид, также известный как AG013736 и описанный в публикации РСТ № WO 01/002369), бриваниб аланинат ((S)-((R)-1-(4-(4-фтор-2-метил-1Н-индол-5-илокси)-5-метилпирроло[2,1-f][1,2,4]триазин-6-илокси)-пропан-2-ил)-2-аминопропаноат, также известный как BMS-582664), мотезаниб (N1-(2,3-дигидро-3,3-диметил-1Н-индол-6-ил)-2-[(4-пиридинилметил)амино]-3-пиридинкарбоксамид и описанный в публикации РСТ WO 02/068470), пасиреотид (также известный как SO 230 и описанный в публикации РСТ № WO 02/010192), и сорафениб (продаваемый под торговой маркой NEXAVAR).
Примеры ингибиторов топоизомеразы II включают, но без ограничения, этопозид (также известный как VP-16 и этопозид фосфат, продаваемый под торговыми марками TOPOSAR, VEPESID и ETOPOPHOS), и тенипозид (также известный как VM-26, продаваемый под торговой маркой VUMON).
Примеры алкилирующих агентов включают, но без ограничения, 5-азацитидин (продается под торговой маркой VIDAZA), децитабин (продается под торговой маркой DECOGEN), темозоломид (продается под торговой маркой TEMODAR и TEMODAL фирмы Schering-Plough/Merck), дактиномицин (также известный как актиномицин-D и продаваемый под торговой маркой COSMEGEN), мелфалан (также известный как L-РАМ, L-сарколизин и фенилаланин мустард, продаваемый под торговой маркой ALKERAN), альтретамин (также известный как гексаметилмеламин (НММ), продаваемый под торговой маркой HEXALEN), кармустин (продаваемый под торговой маркой BCNU), бендамустин (продаваемый под торговой маркой TREANDA), бусульфан (продаваемый под торговыми марками BUSULFEX и MYLERAN), карбоплатин (продаваемый под торговой маркой PARAPLATIN), ломустин (также известный как CCNU, продаваемый под торговой маркой CeeNU), цисплатин (также известный как CDDP, продаваемый под торговыми марками PLATINOL и PLATINOL-AQ), хлорамбуцил (продаваемый под торговой маркой LEUKERAN), циклофосфамид (продаваемый под торговыми марками CYTOXAN и NEOSAR), дакарбазин (также известный как DTIC, DIC и имидазол карбоксамид, продаваемый под торговой маркой DTIC-DOME), альтретамин (также известный как гексаметилмеламин (НММ), продаваемый под торговой маркой HEXALEN), ифосфамид (продаваемый под торговой маркой IFEX), прокарбазин (продаваемый под торговой маркой MATULANE), мехлорэтамин (также известный как азотистый иприт, мустина и мехлорэтамина гидрохлорид, продаваемый под торговой маркой MUSTARGEN), стрептозоцин (продаваемый под торговой маркой ZANOSAR), тиотепа (также известный как тиофосфоамид, TESPA и TSPA, и продаваемый под торговой маркой THIOPLEX).
Примеры противоопухолевых антибиотиков включают, но без ограничения, доксорубицин (продаваемый под торговыми марками ADRIAMYCIN и RUBEX), блеомицин (продаваемый под торговой маркой LENOXANE), даунорубицин (также известный как гидрохлорид дауорубицин, дауномицин и рубидомицина гидрохлорид, продаваемый под торговой маркой CERUBIDINE), липосомальная форма даунорубицина (липосомальная форма даунорубицина цитрата, продаваемая под торговой маркой DAUNOXOME), митоксантрон (также известный как DHAD, продаваемый под торговой маркой NOVANTRONE), эпирубицин (продаваемый под торговой маркой ELLENCE), идарубицин (продаваемый под торговыми марками IDAMYCIN, IDAMYCIN PFS) и митомицин С (продаваемый под торговой маркой MUTAMYCIN).
Примеры антиметаболитов включают, но без ограничения, кладрибин (2-хлордеоксиаденозин, продаваемый под торговой маркой LEUSTATIN), 5-фторурацил (продаваемый под торговой маркой ADRUCIL), 6-тиогуанин (продаваемый под торговой маркой PURINETHOL), пеметрекссд (продаваемый под торговой маркой ALIMTA), цитарабин (также известный как цитозина арабинозид (Ara-С), продаваемый под торговой маркой CYTOSAR-U), липосомальный цитарабин (также известный как Liposomal Ara-С, продаваемый под торговой маркой DEPOCYT), децитабин (продаваемый под торговой маркой DACOGEN), гидроксимочевина (продаваемая под торговыми марками HYDREA, DROXIA и MYLOCEL), флударабин (продаваемый под торговой маркой FLUDARA), флоксуридин (продаваемый под торговой маркой FUDR), кладрибин (также известный как 2-хлородеоксиаденозин (2-CdA), продаваемый под торговой маркой LEUSTATIN), метотрексат (также известный как аметоптерин, метотрексат натрия (МТХ), продаваемый под торговыми марками RHEUMATREX и TREXALL) и пентостатин (продаваемый под торговой маркой NIPENT).
Примеры ретиноидов включают, но без ограничения, алитретиноин (продаваемый под торговой маркой PANRETIN), третиноин (полностью транс-ретиноевая кислота, также известный как ATRA, продаваемый под торговой маркой VESANOID), изотретиноин (13-с/s-ретиноевая кислота, продаваемый под торговыми марками ACCUTANE, AMNESTEEM, CLARA VIS, CLARUS, DECUTAN, ISOTANE, IZOTECH, ORATANE, ISOTRET и SOTRET), и бексаротен (продаваемый под торговой маркой TARGRETIN).
«Антагонист PD-1» означает любое химическое соединение или биологическую молекулу, которая блокирует связывание PD-L1, экс премирующегося в раковой клетке, с PD-1, экспрессирующимся на поверхности иммунной клетки (Т-клетки, В-клетки или NKT-клетки) и предпочтительно также блокирует связывание PD-L2, экспрессирующегося на поверхности раковой клетки, с PD-1, экспрессирующимся на поверхности иммунной клетки. Альтернативные названия или синонимы для PD-1 и его лигандов включают: PDCD1, PD1, CD279 и SLEB2 для PD-1; PDCD1L1, PDL1, В7Н1, В7-4, CD274 и В7-Н для PD-L1; и PDCD1L2, PDL2, B7-DC, Btdc и CD273 для PD-L2. В любом из способа лечения, лекарственных средств и областей применения настоящего изобретения, в которых проводят лечение человека, антагонист PD-1 блокирует связывание PD-L1 человека с PD-1 человека и предпочтительно блокирует связывание обоих, PD-L1 и PD-L2 человека, с PD-1 человека. Аминокислотные последовательности PD-1 человека можно найти в NCBI Locus No.: NP_005009. Аминокислотные последовательности PD-L1 и PD-L2 человека можно найти в NCBI Locus No.: NP_054862 и NP_079515, соответственно.
Антагонисты PD-1, используемые в любом из способа лечения, лекарственных средствах и областях применения настоящего изобретения, включают моноклональное антитело (mAb) или его антигенсвязывающий фрагмент, который специфически связывается с PD-1 или PD-L1 и предпочтительно специфически связывается с PD-1 человека или PD-L1 человека. Моноклональное антитело (mAb) может представлять собой человеческое антитело, гуманизированное антитело или химерное антитело и может включать человеческую константную область. В некоторых вариантах осуществления человеческая константная область выбрана из группы, состоящей из константных областей IgG1, IgG2, IgG3 и IgG4, и в предпочтительных вариантах осуществления человеческая константная область представляет собой константную область IgG1 или IgG4. В некоторых вариантах осуществления антигенсвязывающий фрагмент выбран из группы, состоящей из фрагментов Fab, Fab'-SH, F(ab')2, scFv и Fv. Примеры антагонистов PD-1 включают, но без ограничения, пембролизумаб (продаваемый под торговой маркой KEYTRUDA) и ниволумаб (продаваемый под торговой маркой OPDIVO).
Примеры моноклональных антител (mAb), которые связываются с PD-1 человека и могут применяться в способе лечения, лекарственных средствах и областях применения настоящего изобретения, описаны в US 7488802, US 7521051, US 8008449, US 8354509, US 8168757, WO 2004/004771, WO 2004/072286, WO 2004/056875 и US 2011/0271358.
Примеры моноклональных антител (mAb), которые связываются с PD-L1 человека и могут применяться в способе лечения, лекарственных средствах и областях применения настоящего изобретения, описаны в WO 2013/019906, W 02010/077634 А1 и US 8383796. Конкретные моноклональные антитела (mAb) против PD-L1 человека, используемые в качестве антагониста PD-1 в способе лечения, лекарственных средствах и областях применения настоящего изобретения, включают MPDL3280A, BMS-936559, MEDI4736, MSB0010718C и антитело, которое содержит вариабельные области тяжелой цепи и легкой цепи, представленные в SEQ ID NO: 24 и SEQ ID NO: 21, соответственно, как описано в WO 0201/019906.
Другие антагонисты PD-1, которые можно применять в любом из способа лечения, лекарственных средствах и областях применениях настоящего изобретения, включают иммуноадгезин, который специфически связывается с PD-1 или PD-L1 и предпочтительно специфически связывается с PD-1 человека или PD-L1 человека, например, белок слияния, содержащий внеклеточную или связывающую PD-1 часть PD-L1 или PD-L2, слитую с константной областью, такой как Fc-область молекулы иммуноглобулина. Примеры молекул иммуноадгезии, которые специфически связываются с PD-1, описаны в WO 02010/027827 и WO 210166634. Конкретные белки слияния, используемые в качестве антагониста PD-1 в способе лечения, лекарственных средствах и областях применения настоящего изобретения, включают АМР-224 (также известный как B7-DCIg), который представляет собой белок слияния PD-L2-FC и связывается с PD-1 человека.
Примеры других цитотоксических агентов включают, но без ограничения, триоксид мышьяка (продаваемый под торговой маркой TRISENOX), аспарагиназу (также известную как L-аспарагиназа и Erwinia L-аспарагиназа, продаваемые под торговыми марками ELSPAR и KIDROLASE). Экспериментальная часть
Следующие примеры предназначены только для иллюстрации, а не для ограничения каким-либо образом. Используемые сокращения представляют собой сокращения, общепринятые в данной области или представленные далее.
ACN ацетонитрил
°С градусов Цельсия
DCM дихлорметан
DEA диэтиламин
DIBAL-H диизобутилалюминия гидрид
DIPEA ди-изопропилэтиламин
DMEA диметилэтиламин
DMF N,N-диметилформамид
DMSO диметилсульфоксид
EtOAc этилацетат
EtOH этанол
g грамм(ы)
h час(ы)
HPLC высокоэффективная жидкостная хроматография
kg килограмм
L литр(ы)
LC жидкостная хроматография
LCMS жидкостная хроматография и масс-спектрометрия
МеОН метанол
MS масс-спектрометрия
МТВЕ метил трет-бутиловый эфир
min минут(ы)
mL миллилитр(ы)
m/z отношение массы к заряду
nm нанометр
nM наномолярный
N нормальный
NMP N-метил-2-пирролидон
NMR ядерный магнитный резонанс
PPTS пиридиния п-толуолсульфонат
RT комнатная температура
sat. насыщенный
SFC сверхкритическая жидкостная хроматография
TEA триэтиламин
TFA трифторуксусная кислота
TLC тонкослойная хроматография.
Соединения формулы (I), (Ia) или (Ib) могут быть получены способами, известными в области органического синтеза, которые частично описаны с помощью следующих далее схем синтеза, а также методик и условий синтеза для иллюстративных интермедиатов и примеров.
В схемах, описанных ниже, хорошо понятно, что защитные группы для чувствительных или реакционноспособных групп используются там, где это необходимо, в соответствии с общими принципами или химией. Защитными группами манипулируют в соответствии со стандартными способами органического синтеза (Т. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). Эти группы удаляют на удобной стадии синтеза соединений с использованием способов, которые являются очевидными для специалистов в данной области.
Соединения, описанные в настоящем документе, могут быть получены из коммерчески доступных исходных материалов или синтезированы с использованием известных органических, неорганических и/или ферментативных процессов.
Спектры 1Н-ЯМР получали на спектрометре Bruker AVANCE 300 при 300 МГц или на спектрометре Bruker AVANCE 400 при 400 МГц с использованием тетраметилсилана в качестве внутреннего стандарта. Тонкослойную хроматографию (TLC) выполняли с использованием пластин Whatman No. 4500-101 (Diamond No. MK6F silica-gel 60A) Визуализацию пластин TLC выполняли с использованием УФ-излучения (254 нм). Масс-спектры получали на спектрометре Finnigan LCQ-DUO с использованием электрораспылительной ионизации. Анализ HPLC выполняли на приборе Agilent 1100 Series. Содержание примесей представлено в виде %AUC по данным анализа методом HPLC и являются не подтвержденными.
ПРИМЕРЫ
Пример 1: 8-(7-(Трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4.5]декан-1.3-дион
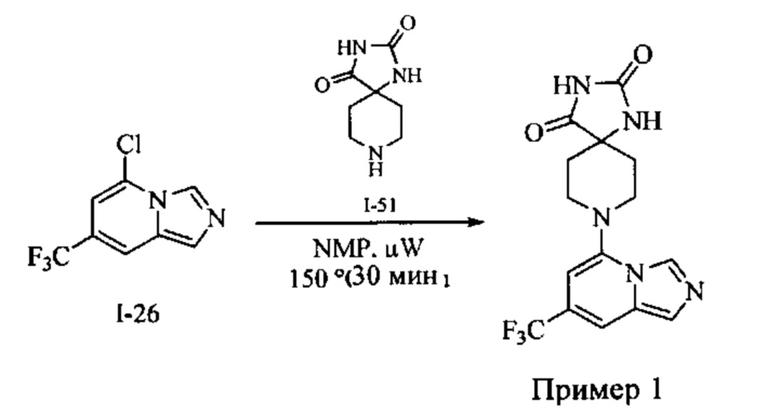
Получение Соединения по примеру 1: Раствор Соединения I-26 (80,0 мг, 0,36 ммоль) и Соединения I-51 (60,0 мг, 0,36 ммоль) в NMP (1,0 мл) подвергали действию микроволнового излучения при 150°С в течение 30 мин. Реакционную смесь охлаждали до RT, разбавляли EtOAc (20 мл) и промывали рассолом (3×20 мл). Органический слой собирали, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Сырое соединение очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, 100% EtOAc) с получением Соединения по примеру 1 в виде твердого вещества. MS (MM) m/z 354.1 [М+Н]+.
1H NMR (300 MHz, DMSO-d6): δ 10.77 (s, 1Н), 8.58 (s, 1H), 8.47 (s, 1H), 7.89 (s, 1H), 7.72 (s, 1H), 6.25 (s, 1H), 3.40-3.24 (m, 2H), 3.12 (t, J=10.8 Hz, 2H), 2.21-2.13 (m, 2H), 1.77 (d, J=13.5 Hz, 2H).
Пример 2: 8-(7-Бромимидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
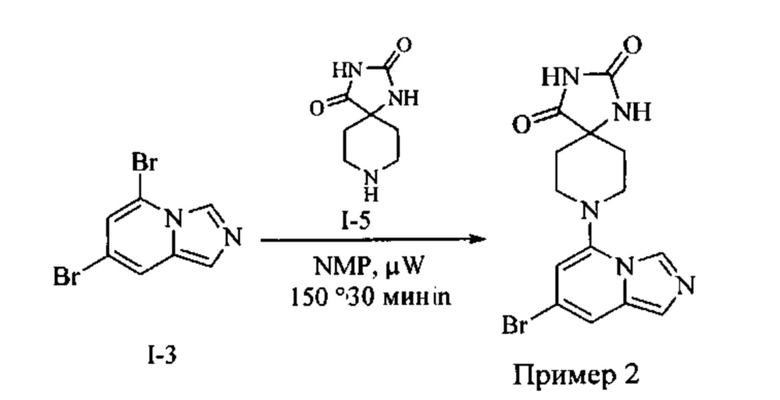
Раствор Соединения I-3 (150 мг, 0,54 ммоль) и Соединения I-5 (68,0 мг, 0,4 ммоль) в NMP (1,0 мл) подвергали действию микроволнового излучения в течение 30 мин при 150 °С. Реакционную смесь охлаждали до RT, наносили на колонку системы Combiflash и очищали с использованием колонки Redisep® (12 г, CH2Cl2/CH3OH, 9:1) с получением указанного соединения в виде твердого вещества. MS (MM) m/z 364.0 [М+Н]+
1Н NMR (300 MHz, DMSO-d6): δ 10.77 (s, 1Н), 8.58 (s, 1H), 8.29 (s, 1H), 7.64 (s, 1H), 7.39 (s, 1H), 6.22 (s, 1H), 3.41-3.32 (m, 2H), 3.08 (t, J=11.4 Hz, 2H), 2.19-2.11 (m, 2H), 1.75 (d, J=13.5 Hz, 2H).
Пример 3: 8-(7-Хлоримидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
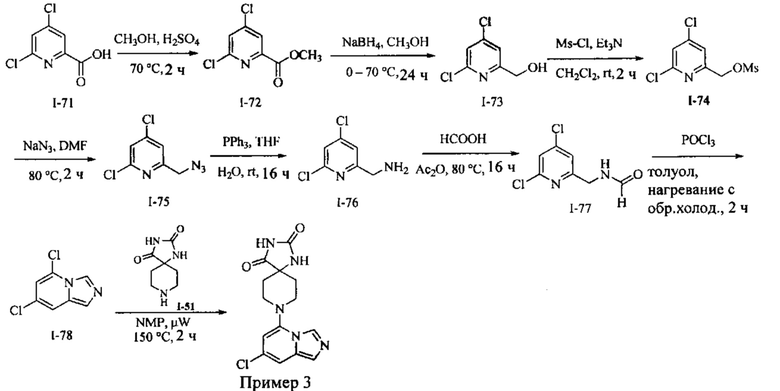
Получение Соединения I-72: В перемешанный раствор Соединения I-71 (20,0 г, 104,1 ммоль) в СН3ОН (200 мл) вносили конц. H2SO4 (1,0 мл) при RT. Реакционную смесь нагревали при 70°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, разбавляли EtOAc (100 мл) и выливали в насыщенный раствор NaHCO3 (80 мл). Слои разделяли, и водный слой экстрагировали EtOAc (2×150 мл). Объединенный органический слой промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения 1-72 в виде жидкости. MS (MM) m/z 207.1 [М+Н]+.
Получение Соединения I-73: В перемешанный раствор Соединения I-72 (20,0 г, 97 ммоль) в СН3ОН (80 мл) порциями вносили NaBH4 (14,35 г, 388 ммоль) в течение 15 мин при 0°С. Реакционную смесь перемешивали при 70°С в течение 24 ч. Реакционную смесь охлаждали до RT, концентрировали при пониженном давлении и разделяли на воду (200 мл) и EtOAc (3×300 мл). Объединенный органический слой промывали водой (100 мл) и солевым раствором (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения по примеру I-73 в виде твердого вещества.
MS (MM) m/z 179.1 [М+Н]+.
Получение Соединения I-74: В раствор Соединения I-73 (18,5 г, 103,9 ммоль) в CH2Cl2 (80 мл) при 0°С вносили Et3N (28 мл, 207,8 ммоль) с последующим MsCl (12 мл, 155,8 ммоль). Реакционную смесь перемешивали при RT в течение 2 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (3×200 мл). Органическую фазу отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения I-74 [20,0 г (сырой)] в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS (MM) m/z 256.1 [М+Н]+.
Получение Соединения I-75: В раствор Соединения I-74 (20,0 г, 78 ммоль) в DMF (80 мл) вносили NaN3 (15,2 г, 235 ммоль) при RT. Реакционную смесь нагревали при 80°С в течение 2 ч. Реакционную смесь разбавляли холодной водой (100 мл) и экстрагировали МТВЕ (3×200 мл). Органическую фазу отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения I-75 в виде жидкости.
Получение Соединения I-76: В перемешанный раствор Соединения I-75 (11,0 г, 54,4 ммоль) в THF (90 мл) и воде (9,0 мл) порциями вносили PPh3 (17,0 г, 65,3 ммоль) при RT в течение 5 мин. Реакционную смесь перемешивали при RT в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли водой (80 мл) и экстрагировали CH2Cl2 (2×50 мл). Водный слой отделяли, подкисляли HCl (2 N, 20 мл) и концентрировали в вакууме с получением соли НС1 Соединения 1-76 в виде твердого вещества. MS (MM) m/z 177.1 [М+Н]+.
Получение Соединения I-77: В перемешанный раствор Соединения I-76 (6,00 г, 34 ммоль) в НСО2Н (100 мл) вносили Ас2О (20 мл) при RT. Реакционную смесь перемешивали при 80°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и совместно выпаривали с толуолом (2×30 мл) с получением Соединения I-77 в виде твердого вещества. MS (MM) m/z 205.1 [М+Н]+.
Получение Соединения I-78: В перемешанный раствор Соединения I-77 (1,20 г, 5,8 ммоль) в толуоле (10 мл) вносили POCl3 (1,2 мл) при 0°С. Реакционную смесь нагревали при 100°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, разбавляли водой (50 мл), подщелачивали водным раствором NaOH (6 N, 20 мл) и экстрагировали EtOAc (3×100 мл). Органическую фазу отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, гексаны/EtOAc, 8:2) с получением Соединения 1-78 в виде твердого вещества.
Получение Соединения по примеру 3: Раствор Соединения I-78 (60,0 мг, 0,32 ммоль) и Соединения 1-51 (548 мг, 3,2 ммоль) в NMP (1,0 мл) подвергали действию микроволнового излучения при 150°С в течение 2 ч. Реакционную смесь разбавляли холодной водой (3,0 мл) и экстрагировали EtOAc (3×8,0 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (4 г, EtOAc/гексаны, 9:1) с получением Соединения по примеру 3 в виде твердого вещества. MS (MM) m/z 320.1 [М+Н]+.
1Н NMR (400 MHz, DMSO-d6): δ 10.77 (s, 1Н), 8.59 (s, 1H), 8.28 (s, 1H), 7.48 (s, 1H), 7.39 (s, 1H), 6.14 (s, 1H), 3.08 (t, J=12.0 Hz, 4H), 2.15 (t, J=11.6 Hz, 2H), 1.76 (d, J=13.2 Hz, 2H).
Пример 4: 8-(6-(Трифторметил)имидазо[1,5-а]пиридин-8-ил)-1,3,8-триазаспиро[4.5]-декан-2,4-дион
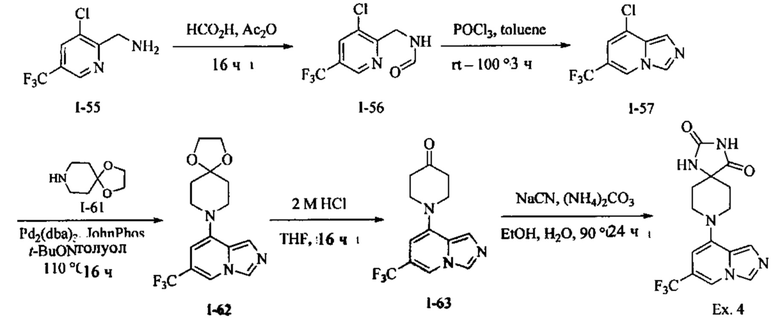
Получение Соединения I-56: В перемешанный раствор Соединения I-55 (5,00 г, 23,91 ммоль) в НСО2Н (50 мл) вносили Ас2О (50 мл) при RT. Реакционную смесь перемешивали при RT в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и совместно выпаривали с толуолом (2×100 мл) с получением Соединения I-56 в виде твердого вещества. MS (MM) m/z 239.1 [М+Н]+.
Получение Соединения I-57: В перемешанный раствор Соединения I-56 (5,00 г, 20,99 ммоль) в толуоле (25 мл) вносили POCl3 (2,5 мл) при RT. Реакционную смесь нагревали при 100°С в течение 3 ч. Реакционную смесь охлаждали, разбавляли EtOAc (500 мл) и выливали в водный раствор NaOH (6 N, 10 мл). Слои разделяли, и водный слой экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали водой (200 мл) и рассолом (200 мл). Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения 1-57 в виде твердого вещества. MS (MM) m/z 221.1 [М+Н]+.
Получение Соединения I-62: В перемешанный раствор Соединения I-57 (1,00 г, 4,5 ммоль) в толуоле (10 мл) вносили Соединение I-61 (700 мг, 4,9 ммоль) и t-BuONa (864 мг, 9,0 ммоль) при RT. Реакционную смесь продували аргоном в течение 20 мин. В реакционную смесь добавляли Pd2(dba)3 (823 мг, 0,89 ммоль) и JohnPhos (40,0 мг, 0,13 ммоль) и нагревали с обратным холодильником до 110°С в течение 16 ч. Реакционную смесь охлаждали до RT и концентрировали при пониженном давлении. Полученную суспензию разбавляли EtOAc (100 мл) и промывали рассолом (2×75 мл). Органическую фазу отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, EtOAc/гексаны, 4:1) с получением Соединения I-62 в виде твердого вещества. MS (MM) m/z 328.1 [М+Н]+.
Получение Соединения I-63: В перемешанный раствор Соединения I-62 (180 г, 5,5 ммоль) в THF (10 мл) вносили HCl (2 М, 5,0 мл) при RT. Реакционную смесь перемешивали в течение 16 ч при этой же температуре. Реакционную смесь разбавляли водой (10 мл) и экстрагировали EtOAc (2×30 мл). Водный слой отделяли, подщелачивали водным раствором NaOH (6 N, 10 мл) и экстрагировали EtOAc (2×30 мл). Органическую фазу отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением Соединения 1-63 в виде твердого вещества. MS (MM) m/z 284.1 [М+Н]+
Получение Соединения по примеру 4: В перемешанный раствор Соединения I-63 (110 мг, 0,38 ммоль) в смеси EtOH (5,0 мл) и H2O (3,0 мл) вносили NaCN (75,0 мг, 1,57 ммоль) и (NH4)2СО3 (754 мг, 7,67 ммоль) при RT. Реакционную смесь нагревали при 90°С в течение 24 ч в герметичной пробирке. Реакционную смесь охлаждали до RT, разбавляли водой (20 мл) и экстрагировали EtOAc (3×30 мл). Органическую фазу отделяли, промывали рассолом (5×20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Сырое соединение перемешивали с МТВЕ (20 мл) и фильтровали с получением Соединения по примеру 4 в виде твердого вещества.
MS (MM) m/z 354.1 [М+Н]+.
1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H), 8.63 (s, 1H), 8.56 (s, 1H), 8.48(s, 1H), 7.56 (s, 1H), 6.13 (s, 1H), 3.66 (d, J=12.4 Hz, 2H), 3.16 (t, J=8.1 Hz, 2H), 2.09-2.03 (m, 2H), 1.72 (d, J=10.2 Hz, 2H).
Пример 5: 6,6-Диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
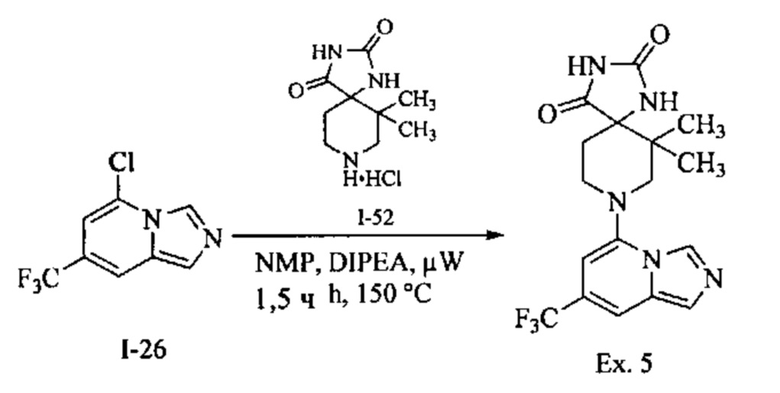
Получение Соединения по примеру 5: Раствор Соединения I-26 (150 мг, 0,90 ммоль), Соединения I-52 (166 мг, 0,99 ммоль) и DIPEA (340 мг, 2,7 ммоль) в NMP (1,0 мл) подвергали действию микроволнового излучения при 150°С в течение 1,5 ч. Реакционную смесь разбавляли EtOAc (30 мл). Органический слой промывали рассолом (3×50 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта. Остаток затем очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (4 г, CH2Cl2/МеОН, 9:1) с получением рацемического 6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-диона в виде твердого вещества. MS (MM) m/z 382.1 [М+Н]+.
1Н NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1Н), 8.46 (s, 1H), 8.27 (s, 1H), 7.89 (s, 1H), 7.73 (s, 1H), 6.28 (s, 1H), 3.28-3.24 (m, 1H), 3.03 (d, J=11.6 Hz, 2H), 2.17-2.06 (m, 2H), 1.07 (s, 6H).
Рацемический 6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион затем разделяли методом хиральной SFC.
Пример 6: 3-Метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
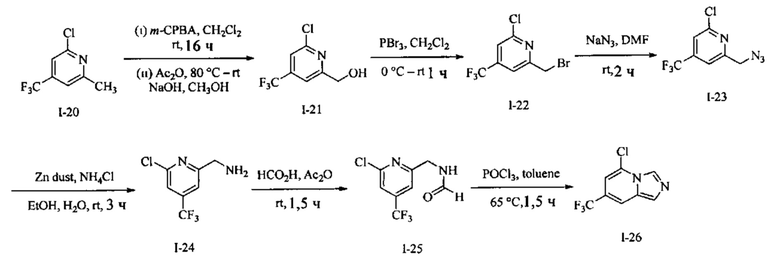
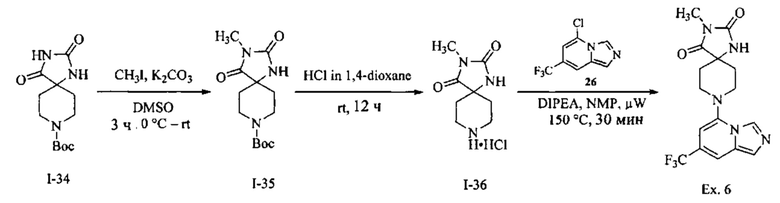
Получение Соединения I-21: В перемешанный раствор Соединения I-20 (5,00 г, 2,5 ммоль) в CH2Cl2 (100 мл) вносили м-хлорпербензойную кислоту (25,0 г, 12,5 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 16 ч. Реакционную смесь разбавляли CH2Cl2 (50 мл) и выливали в насыщенный раствор NaHCO3 (250 мл). Слои разделяли, и водный слой экстрагировали CH2Cl2 (3×150 мл). Объединенный органический слой промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении при 25°С. Полученный сырой остаток добавляли к Ас2О (50 мл) и нагревали с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали до RT, затем концентрировали с получением остатка.
Остаток растворяли в СН3ОН (50 мл), обрабатывали водным раствором NaOH (6 N, 50 мл) и перемешивали в течение 1 ч при RT. Реакционную смесь разбавляли EtOAc (50 мл) и слои разделяли. Водный слой экстрагировали EtOAc (3×150 мл). Объединенные органические слои промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения I-21 в виде смолы. MS (MM) m/z 212.1 [М+Н]+.
Получение Соединения I-22: В раствор Соединения I-21 (1,30 г, 5,8 ммоль) в CH2Cl2 (25 мл) при 0°С вносили PBr3 (1,40 г, 6,5 ммоль) в течение 10 мин. Реакционную смесь перемешивали при RT в течение 1 ч. Реакционную смесь разбавляли CH2Cl2 (25 мл) и значение рН раствора доводили до рН=8 насыщенным раствором NaHCO3 (100 мл). Слои разделяли, и водный слой экстрагировали CH2Cl2 (3×50 мл). Объединенные органические слои промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения I-22 в виде смолы. MS (MM) m/z 215.1 [М+Н]+.
Получение Соединения I-23: В раствор Соединения I-22 (1,25 г, 4,5 ммоль) в DMF (20 мл) вносили NaN3 (2,90 г, 4,5 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 2 ч. Реакционную смесь разбавляли EtOAc (25 мл), а затем водой (100 мл). Слои разделяли, и водный слой экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения I-23 в виде смолы. MS (MM) m/z 237.1 [М+Н]+.
Получение Соединения I-24: В раствор Соединения I-23 (1,05 г, 4,6 ммоль) в EtOH (20 мл) и Н2О (20 мл) вносили порошок Zn (3,00 г, 4,6 ммоль) с последующим NH4Cl (2,47 г, 4.6 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 3 ч. Реакционную смесь фильтровали через слой Celite® и промывали CH2Cl2 (100 мл). Слои разделяли, и водный слой экстрагировали CH2Cl2 (3×50 мл). Объединенные органические слои промывали водой (100 мл) и рассолом (100 мл). Органические слои сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения I-24 в виде твердого вещества. MS (MM) m/z 211.1 [М+Н]+.
Получение Соединения I-25: В перемешанный раствор Соединения I-24 (900 мг, 4,2 ммоль) в НСО2Н (25 мл) вносили Ас2О (5,0 мл) при RT. Реакционную смесь перемешивали при RT в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении и затем совместно выпаривали с толуолом (2×50 мл) с получением Соединения I-25 в виде твердого вещества. MS (MM) m/z 239.1 [М+Н]+.
Получение Соединения I-26: В перемешанный раствор Соединения I-25 (1,00 г, 4,2 ммоль) в толуоле (10 мл) вносили POCl3 (0,5 мл) при RT. Реакционную смесь нагревали при 65°С в течение 1,5 ч. Реакционную смесь охлаждали, разбавляли EtOAc (50 мл) и выливали в водный раствор NaOH (1 N, 50 мл). Слои разделяли, и водный слой экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения I-26 в виде твердого вещества. MS (MM) m/z 221.1 [М+Н]+.
Получение Соединения I-35: В перемешанный раствор Соединения I-34 (500 мг, 1,85 ммоль) в DMSO (10 мл) при 0°С вносили метилйодид (0,29 мг, 2,04 ммоль) и K2CO3 (765 мг, 5,55 ммоль). Реакционную смесь нагревали до RT и перемешивали в течение 3 ч. В реакционную смесь добавляли воду (20 мл) и экстрагировали EtOAc (3×20 мл). Органическую фазу собирали, промывали рассолом (3×20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный сырой продукт перемешивали с МТВЕ (10 мл) и фильтровали с получением Соединения I-35 в виде твердого вещества. MS (MM) m/z 284.1 [М+Н]+.
Получение Соединения I-36: Смесь Соединения I-35 (170 мг, 6,2 ммоль) и HCl в 1,4-диоксане (4 М, 5,0 мл) перемешивали при RT в течение 12 ч. Растворитель выпаривали при пониженном давлении с получением Соединения I-36 в виде твердого вещества.
Получение Соединения по примеру 6: Раствор Соединения I-36 (100 мг, 0,45 ммоль), Соединения I-26 (82,0 мг, 0,45 ммоль) и DIPEA (250 мг, 0,9 ммоль) в NMP (1,0 мл) подвергали действию микроволнового излучения при 150°С в течение 30 мин. Реакционную смесь охлаждали до RT, разбавляли EtOAc (30 мл) и промывали рассолом (3×50 мл). Органический слой собирали, сушили над безводным Na2SO4, концентрировали и сушили при пониженном давлении. Полученное сырое соединение очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (4 г, 100% EtOAc) с получением Соединения по примеру 6 в виде твердого вещества. MS (MM) m/z 368.1 [М+Н]+
1Н NMR (400 MHz, DMSO-d6): δ 8.86 (s, 1Н), 8.75 (s, 1H), 7.96 (s, 1H), 7.86 (s, 1H), 6.36 (s, 1H), 3.44 (d, J= 12.4 Hz, 2H), 3.15 (t, J= 11.6 Hz, 2H), 2.22 (t, J= 10.8 Hz, 1H), 1.77 (d, J= 13.2 Hz, 2H).
Пример 7: 3-((Тетрагидро-2Н-пиран-4-ил)метил)-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.51декан-2,4-дион
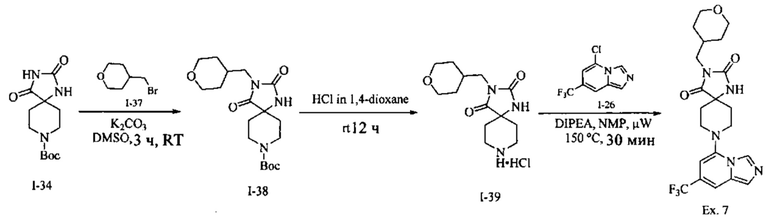
Получение Соединения I-38: В перемешанный раствор Соединения I-34 (500 мг, 1,85 ммоль) в DMSO (10 мл) вносили Соединение I-37 (0,29 мг, 2,04 ммоль) и K2CO3 (765 мг, 5,55 ммоль) при 0°С. Реакционную смесь перемешивали в течение 3 ч при RT. Реакционную смесь обрабатывали водой (20 мл) и экстрагировали EtOAc (3×20 мл). Органическую фазу собирали, промывали рассолом (3×20 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный сырой остаток перемешивали с МТВЕ (10 мл) и фильтровали с получением Соединения I-38 в виде твердого вещества. MS (MM) m/z 368.1 [М+Н]+.
Получение Соединения I-39: Смесь Соединения I-38 (170 мг, 6,2 ммоль) и HCl в 1,4-диоксане (4 М, 5,0 мл) перемешивали при RT в течение 12 ч. Растворитель выпаривали при пониженном давлении с получением Соединения I-39 в виде твердого вещества.
Получение Соединения по примеру 7: Раствор Соединения I-26 (100 мг, 0,45 ммоль), Соединения I-39 (130 мг, 0,45 ммоль) и DIPEA (250 мг, 0,9 ммоль) в NMP (1,0 мл) подвергали действию микроволнового излучения при 150°С в течение 30 мин. Реакционную смесь охлаждали до RT, разбавляли EtOAc (30 мл) и промывали рассолом (3×50 мл). Органический слой собирали, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученное сырое соединение перемешивали с МТВЕ (20 мл) и фильтровали с получением Соединения по примеру 7 в виде твердого вещества. MS (MM) m/z 452.1 [М+Н]+.
1Н NMR (300 MHz, DMSO-d6): δ 8.91 (s, 1Н), 8.50 (s, 1H), 7.90 (s, 1H), 7.73 (s, 1H), 6.27 (s, 1H), 3.83 (d, J= 9.0 Hz, 2H), 3.45 (t, J= 12.3 Hz, 2H), 3.30 - 3.11 (m, 6H), 2.23 (t, J = 14.7 Hz, 2H), 1.92-1.80 (m, 1H), 1.78 (d, J= 13.2 Hz, 2H), 1.49 (d, J = 11.1 Hz, 2H), 1.24-1.16 (m, 2H).
Пример 8: 8-(7-Хлоримидазо[1,5-а]пиридин-5-ил)-6.6-диметил-1,3,8-триазаспиро[4.5]декан-2,4-дион
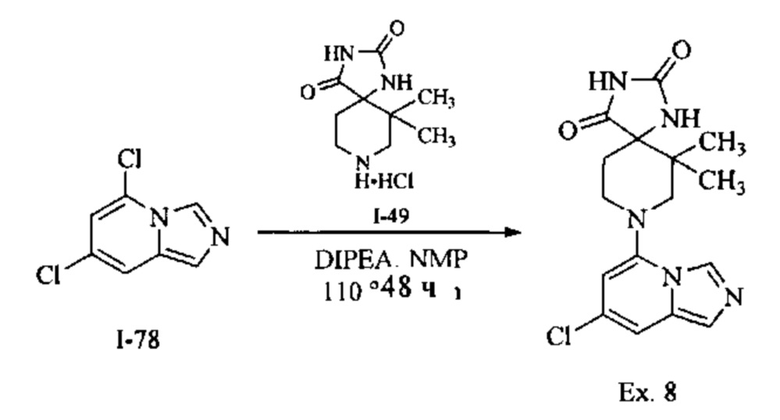
Получение Соединения по примеру 8: Раствор Соединения I-78 (150 мг, 0,80 ммоль), Соединения I-49 (238 мг, 1,2 ммоль) и DIPEA (412 мг, 2,4 ммоль) в NMP (0,8 мл) нагревали при 110°С в течение 48 ч. Реакционную смесь охлаждали до RT, разбавляли холодной водой (3,0 мл) и экстрагировали EtOAc (3×10 мл). Органическую фазу разделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток затем очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (4 г, EtOAc/гексаны, 7:3) с получением рацемического 8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро-[4.5]декан-2,4-диона в виде твердого вещества. MS (MM) m/z 346.1 [М-Н]-.
1Н NMR (300 MHz, DMSO-d6): δ 8.17 (s, 1Н), 7.30 (s, 1H), 7.28 (s, 1H), 6.17 (s, 1H), 3.38-3.30 (m, 3H), 2.87 (d, J= 12.3 Hz, 1H), 2.16-2.05 (m, 2H), 1.14 (s, 3H), 1.02 (s, 3H).
Рацемический 8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4.5]декан-2,4-дион затем очищали методом хиральной SFC (колонка: AS-H; модификатор: 20% МеОН + 0,25% DMEA; скорость потока: 70 мл/мин; температура: 15 °С) с получением Соединения по примеру 8 (пик 1, элюирование через 4,4 мин).
Пример 9: 3-Фенил-8-(7-(трифторметил)имидазо[1.5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
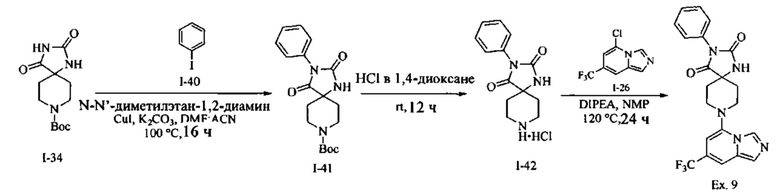
Получение Соединения I-41: В перемешанный раствор Соединения I-34 (500 мг, 1,85 ммоль) в смеси ацетонитрила (20 мл) и DMF (20 мл) вносили Соединение I-40 (483 мг, 0,33 ммоль), K2CO3 (765 мг, 2,2 ммоль), N,N-диметилэтан-1,2-диамин (93,0 мг, 0,55 ммоль) и CuI (200 мг, 0,55 ммоль) при RT. Реакционную смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь разбавляли водой (50 мл) и экстрагировали EtOAc (5×20 мл). Органическую фазу собирали и промывали рассолом (5×20 мл). Органическую фазу сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, 100% EtOAc) с получением Соединения I-41 в виде твердого вещества. MS (MM) m/z 368.1 [М+Н]+.
Получение Соединения I-42: Смесь Соединения I-41 (170 мг, 6,2 ммоль) и HCl в 1,4-диоксане (4 М, 5,0 мл) перемешивали при RT в течение 12 ч. Растворитель выпаривали при пониженном давлении с получением Соединения I-42 в виде твердого вещества. MS (MM) m/z 346.1 [М+Н]+.
Получение Соединения по примеру 9: Раствор Соединения I-26 (100 мг, 0,45 ммоль), Соединения I-42 (130 мг, 0,45 ммоль) и DIPEA (250 мг, 0,9 ммоль) в NMP (1,0 мл) нагревали при 120°С в течение 24 ч. Реакционную смесь охлаждали до RT, разбавляли EtOAc (30 мл) и промывали рассолом (3×50 мл). Реакционную смесь сушили над безводным Na2SO4, концентрировали и сушили при пониженном давлении. Полученный остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (4 г, CH2Cl2/МеОН, 9:1) с получением Соединения по примеру 9 в виде твердого вещества. MS (MM) m/z 430.1 [М+Н]+.
1H NMR (400 MHz, DMSO-d6): δ 9.15 (s, 1H), 8.53 (s, 1H), 7.91 (s, 1H), 7.74 (s, 1H), 7.52-7.48 (m, 2H), 7.43-7.39 (m, 3H), 6.30 (s, 1H), 3.51-3.50 (m, 2H), 3.21 (d, J= 11.6 Hz, 2H), 2.35-2.28 (m, 2H), 1.99 (m, 2H).
Пример 10: 3,6,6-Триметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
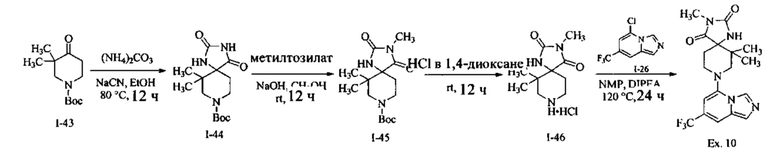
Получение Соединения I-44: В перемешанный раствор Соединения I-43 (7,00 г, 30,8 ммоль) в EtOH (140 мл) и H2O (40 мл) вносили NaCN (3,00 г, 61,6 ммоль) и карбонат аммония (59,1 г, 616 ммоль) при RT. Реакционную смесь перемешивали при 80°С в течение 12 ч в герметично закрытой пробирке. Реакционную смесь разбавляли водой (200 мл) и экстрагировали EtOAc (3×150 мл). Органическую фазу промывали рассолом (5×20 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении с получением Соединения I-44 в виде твердого вещества.
Получение Соединения I-45: В перемешанный раствор Соединения I-44 (1,00 г, 3,3 ммоль) в СН3ОН (140 мл) вносили метилтозилат (1,37 г, 7,4 ммоль) и NaOH (230 мг, 5,94 ммоль) при RT. Реакционную смесь перемешивали в течение 12 ч при RT. Реакционную смесь обрабатывали водой (30 мл) и экстрагировали CH2Cl2 (3×30 мл). Объединенный органический слой сушили над безводным Na2SO4 и выпаривали с получением Соединения I-45 в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Получение Соединения I-46: Смесь Соединения I-45 (350 мг, 6,2 ммоль) и HCl в 1,4-диоксане (4 М, 10 мл) перемешивали при RT в течение 12 ч. Растворитель выпаривали при пониженном давлении с получением Соединения I-46 в виде твердого вещества.
Получение Соединения по примеру 10: Раствор Соединения I-26 (200 мг, 0,45 ммоль), Соединения I-46 (178 мг, 0,49 ммоль) и DIPEA (348 мг, 1,35 ммоль) в NMP (1,0 мл) нагревали при 120°С в течение 24 ч. Реакционную смесь охлаждали до RT, разбавляли EtOAc (30 мл) и промывали рассолом (3×50 мл). Органический слой собирали, сушили над безводным Na2SO4, концентрировали и сушили при пониженном давлении. Полученный остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, CH2Cl2/МеОН, 9:1) с получением Соединения по примеру 10 в виде твердого вещества. MS (MM) m/z 396.1 [М+Н]+.
1Н NMR (300 MHz, DMSO-d6): δ 8.57 (s, 1Н), 8.48 (s, 1H), 7.89 (s, 1H), 7.73 (s, 1H), 6.29 (s, 1H), 3.40-3.25 (m, 3H), 3.05 (d, J=11.7 Hz, 1H), 2.85 (s, 3H), 2.11-2.07 (m, 2H), 1.05 (s, 6H).
Пример 11: 8-(7-Хлоримидазо[1.5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4.5]декан-2,4-дион
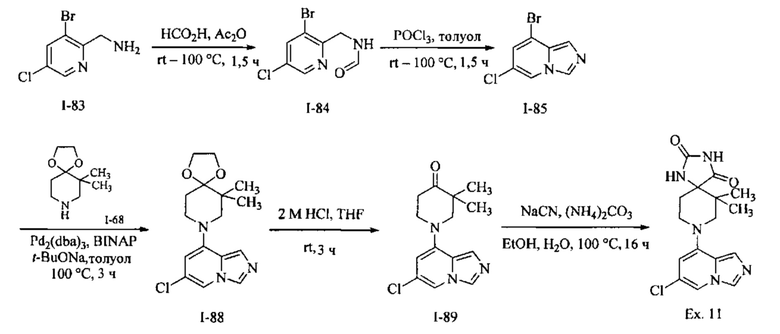
Получение Соединения I-84: В перемешанный раствор Соединения I-83 (15,0 г, 71,0 ммоль) в НСО2Н (370 мл) вносили Ас2О (750 мл) при RT. Реакционную смесь перемешивали при 100°С в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении и совместно выпаривали с толуолом (2×100 мл) с получением Соединения I-84 в виде твердого вещества. MS (MM) m/z 249.1 [М+Н]+.
Получение Соединения I-85: В перемешанный раствор Соединения I-84 (13,0 г, 54,0 ммоль) в толуоле (100 мл) вносили POCl3 (6,0 мл) при RT. Реакционную смесь нагревали при 100°С в течение 1,5 ч. Реакционную смесь охлаждали, разбавляли EtOAc (300 мл) и выливали в водный раствор NaOH (1 N, 300 мл). Слои разделяли, и водный слой экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали водой (2×200 мл) и рассолом (200 мл). Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения I-85 в виде твердого вещества. MS (MM) m/z 230.4 [М+Н]+.
Получение Соединения I-88: В перемешанный раствор Соединения I-85 (800 мг, 3,45 ммоль) в толуоле (20 мл) вносили Соединение I-68 (590 мг, 3,45 ммоль) и порошкообразный t-BuONa (662 мг, 6,9 ммоль) при RT. Реакционную смесь продували аргоном в течение 20 мин. К смеси добавляли Pd2(dba)3 (315 мг, 0,345 ммоль) и BINAP (214 мг, 0,345 ммоль) и нагревали с обратным холодильником при 100°С в течение 3 ч.
Реакционную смесь охлаждали до RT и концентрировали при пониженном давлении. Полученную суспензию разбавляли EtOAc (100 мл) и промывали рассолом (2×75 мл). Органическую фазу разделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (24 г, гексаны/EtOAc, 1:1) с получением Соединения I-88 в виде твердого вещества. MS (ММ) m/z 322.0 [М+Н]+.
Получение Соединения I-89: В перемешанный раствор Соединения I-88 (600 мг, 1,86 ммоль) в THF (3,0 мл) вносили HCl (2 М, 3,0 мл) при RT. Реакционную смесь перемешивали в течение 3 ч. Реакционную смесь подщелачивали насыщенным раствором NaHCO3 (20 мл) и экстрагировали EtOAc (3×50 мл). Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения I-89 в виде твердого вещества.
Получение Соединения по примеру 11: Раствор Соединения I-89 (380 мг, 1,3 ммоль) в EtOH (8,0 мл) и воде (4,0 мл) помещали в герметично закрытую пробирку и вносили NaCN (134 мг, 2,73 ммоль) и карбонат аммония (2,10 г, 27,2 ммоль) при RT. Реакционную смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь выливали в воду (50 мл) и экстрагировали EtOAc (2×100 мл). Органические экстракты промывали водой (2×50 мл) и рассолом (50 мл). Органическую фазу разделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением Соединения по примеру 11 в виде твердого вещества. MS (MM) m/z 348.1 [М+Н]+.
1Н NMR (300 MHz, DMSO-d6): δ 10.70 (s, 1Н), 8.30 (s, 1H), 8.22 (s, 1H), 8.20 (s, 1H), 7.44 (s, 1H), 6.06 (s, 1H), 3.45-3.42 (m, 1H), 3.39-3.30 (m, 2H), 3.17 (d, J=5.1 Hz, 1H), 2.01-1.95 (m, 2H).
Пример 12: 6,6-Диметил-8-(6-(трифторметил)имидазо[1.5-а]пиридин-8-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
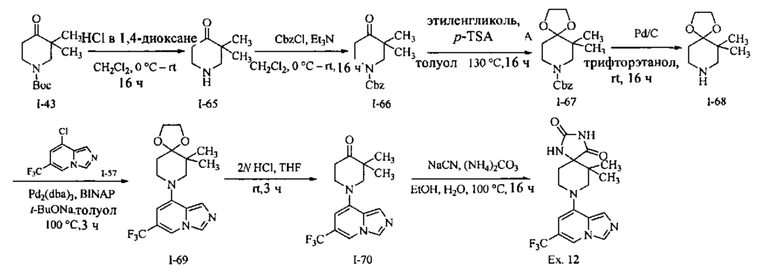
Получение Соединения I-65: В раствор Соединения I-43 (5,00 г, 22,02 ммоль) в CH2Cl2 (20 мл) вносили HCl в 1,4-диоксане (4 М, 20 мл) в течение 15 мин при 0°С. Реакционную смесь перемешивали при RT в течение 16 ч. Растворитель концентрировали при пониженном давлении, совместно перегоняли с МТВЕ (3×200 мл) для удаления избыточного количества HCl и сушили в вакууме с получением Соединения I-65 в виде соли HCl. MS (MM) m/z 128.1 [М+Н]+.
Получение Соединения I-66: В раствор Соединения I-65 (4,20 г, 33,07 ммоль) в CH2Cl2 (80 мл) вносили бензилхлорформиат (6,70 г, 39,68 ммоль) с последующим триэтиламином (5,00 г, 49 ммоль) в течение 20 мин при 0°С. Реакционную смесь перемешивали при RT в течение 16 ч. Органический слой промывали насыщенным раствором NaHCO3 (50 мл), водой (100 мл) и рассолом (50 мл). Органический слой концентрировали при пониженном давлении с получением Соединения I-66 в виде жидкости. MS (MM) m/z 262.1 [М+Н]+.
Получение Соединения I-67: В перемешанный раствор Соединения I-66 (7,00 г, 26,7 ммоль) в толуоле (70 мл) вносили этиленгликоль (1,98 г, 32 ммоль) с последующим р-TSA (28,0 мг, 1 ммоль) при RT и смесь нагревали с обратным холодильником до 130°С в течение 16 ч. Реакционную смесь охлаждали до RT и концентрировали при пониженном давлении. Полученную суспензию разбавляли EtOAc (100 мл) и промывали рассолом (2×500 мл). Органическую фазу разделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (80 г, гексаны/EtOAc, 8:2) с получением Соединения I-67 в виде жидкости. MS (MM) m/z 172.0 [М+Н]+.
Получение Соединения I-68: В перемешанный раствор Соединения I-67 (6,00 г, 305 ммоль) в 2,2,2-трифторэтаноле (60 мл) вносили Pd(OH)2 (600 мг, 10 масс. %) в атмосфере аргона при RT. Атмосферу водорода создавали с помощью баллона и реакционную смесь перемешивали в течение 16 ч. Реакционную смесь фильтровали через слой Celite®, промывали СН3ОН (50 мл) и концентрировали при пониженном давлении с получением Соединения I-68 в виде масла. MS (MM) m/z 172.0 [М+Н]+.
Получение Соединения I-69: В перемешанный раствор Соединения I-57 (1,00 г, 4,54 ммоль) в толуоле (20 мл) вносили Соединение I-68 (261 мг, 4,54 ммоль) и порошкообразный f-BuONa (872 мг, 9,0 ммоль) при RT. Реакционную смесь продували аргоном в течение 20 мин. К реакционной смеси добавляли Pd2(dba)3 (415 мг, 0,45 ммоль) и BINAP (282 мг, 0,45 ммоль) и нагревали с обратным холодильником при 110°С в течение 3 ч. Реакционную смесь охлаждали до RT и концентрировали при пониженном давлении. Полученную суспензию разбавляли EtOAc (100 мл) и промывали рассолом (2×75 мл). Органическую фазу разделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток затем очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (24 г, гексаны/EtOAc, 1:1) с получением Соединения I-69 в виде твердого вещества. MS (ММ)m/z 356.1 [М+Н]+.
Получение Соединения I-70: В перемешанный раствор Соединения I-69 (500 мг, 1,40 ммоль) в THF (3,0 мл) вносили HCl (2 N, 3,0 мл) при RT. Реакционную смесь перемешивали в течение 3 ч. Реакционную смесь подщелачивали насыщенным раствором NaHCO3 (20 мл) и экстрагировали EtOAc (3×50 мл). Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением Соединения I-70 в виде твердого вещества. MS (MM) m/z 312.1 [М+Н]+.
Получение Соединения по примеру 12: В раствор Соединения I-70 (100 мг, 0,32 ммоль) в смеси EtOH:Н2О (2:1, 12 мл) вносили карбонат аммония (500 мг, 6,42 ммоль) с последующим NaCN (31,0 мг, 0,642 ммоль) при комнатной температуре. Реакционную смесь перемешивали в герметично закрытой пробирке при 100°С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (50 мл). Органический слой промывали водой (2×30 мл) и рассолом (20 мл). Органическую фазу разделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением темно-коричневого остатка. Остаток затем очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, DCM/MeOH, 9.7:0.3) с получением Соединения по примеру 12 в виде твердого вещества. MS (MM) m/z 382.1 [М+Н]+.
1H NMR (400 MHz, DMSO-d6): δ 10.72 (s, 1Н), 8.63 (s, 1H), 8.49 (s, 1H), 8.21 (s, 1H), 7.53 (s, 1H), 6.12 (s, 1H), 3.50 - 3.45 (m, 1H), 3.39-3.30 (m, 2H), 3.17 (d, J=12.4, 1H), 2.08-1.97 (m, 2H), 1.05 (d, J=1.2 Hz, 6H)
Пример 13: 8-(7-(Трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2.4-дион
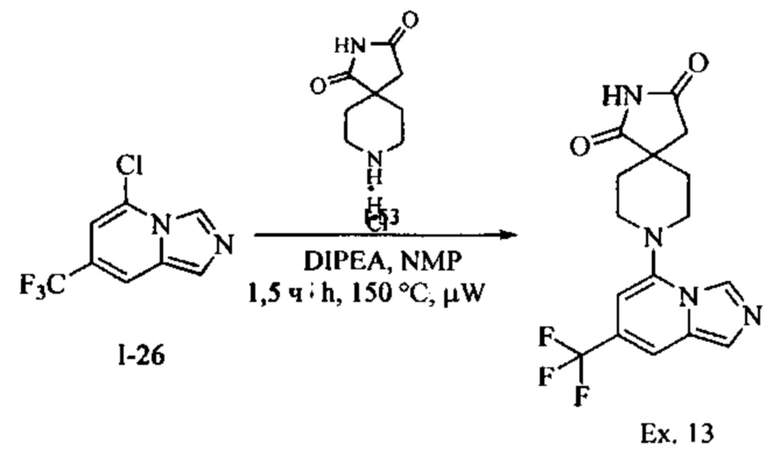
Получение Соединения по примеру 13: Раствор Соединения I-26 (100 мг, 0,45 ммоль), Соединения I-53 (139 мг, 0,68 ммоль) и DIPEA (175 мг, 1,36 ммоль) в NMP (2,0 мл) подвергали действию микроволнового излучения при 150°С в течение 1,5 ч. Реакционную смесь разбавляли холодной водой (10 мл) и экстрагировали EtOAc (2×20 мл). Объединенный органический слой промывали водой (20 мл), рассолом (20 мл) и концентрировали при пониженном давлении. Остаток затем очищали препаративной TLC путем использования смеси (CH2Cl2/CH3OH, 98:2) с получением Соединения по примеру 13 в виде твердого вещества. MS (MM) m/z 353[М+Н]+.
1Н NMR (400 MHz, DMSO-d6): δ 8.44 (s, 1H), 7.88 (s, 1H), 7.72 (s, 1H), 6.30 (s, 1H), 3.42 (d, J=12.4 Hz, 2H), 2.91 (t, J=11.2 Hz, 2H), 2.69 (s, 2H), 2.16-2.08 (m, 2H), 1.79 (d, J=13.2 Hz, 2H).
Пример 14: 6,6-Диметил-3-((тетрагидро-2Н-пиран-4-ил)метил)-8-(7-(трифторметил)имидазо[1.5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
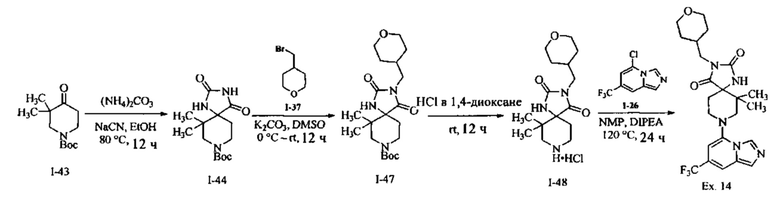
Получение Соединения I-44: В перемешанный раствор Соединения I-43 (7,00 г, 30,8 ммоль) в EtOH (140 мл) и Н2О (40 мл) вносили NaCN (3,00 г, 61,6 ммоль) и карбонат аммония (59,1, 616 ммоль) при RT. Реакционную смесь перемешивали при 80°С в течение 12 ч в герметично закрытой пробирке. Реакционную смесь охлаждали до RT, разбавляли водой (200 мл) и экстрагировали EtOAc (3×150 мл). Объединенный органический слой промывали рассолом (5×20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения I-44 в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS (MM) m/z 368.1 [М+Н]+.
Получение Соединения I-47: В перемешанный раствор Соединения I-44 (1,00 г, 3,35 ммоль) в DMSO (10 мл) вносили Соединение I-37 (660 мг, 2,04 ммоль) и K2CO3 (1,38 г, 10,05 ммоль) при 0°С. Реакционную смесь нагревали до RT и перемешивали в течение 12 ч. К реакционной смеси добавляли воду (20 мл) и экстрагировали EtOAc (3×20 мл). Объединенную органическую фазу промывали рассолом (3×20 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный остаток перемешивали с МТВЕ (10 мл) и фильтровали с получением Соединения I-47 в виде твердого вещества.
Получение Соединения I-48: Смесь Соединения I-47 (1,20 г, 3,03 ммоль) и HCl в 1,4-диоксане (4 М, 20 мл) перемешивали при RT в течение 12 ч. Растворитель выпаривали при пониженном давлении с получением Соединения I-48 в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Получение Соединения по примеру 14: Раствор Соединения I-26 (150 мг, 0,68 ммоль), Соединения I-48 (240 мг, 0,81 ммоль) и DIPEA (260 мг, 2,04 ммоль) в NMP (1,0 мл) нагревали при 120°С в течение 24 ч. Реакционную смесь охлаждали до RT, разбавляли EtOAc (30 мл) и промывали рассолом (3×50 мл). Органический слой собирали, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Сырой остаток очищали препаративной HPLC с получением Соединения по примеру 14 (12,0 мг, 4%) в виде твердого вещества. MS (MM) m/z 480.1 [М+Н]+.
1H NMR (300 MHz, DMSO-d6): δ 8.66 (s, 1Н), 8.54 (s, 1H), 7.95 (s, 1H), 7.79 (s, 1H), 6.36 (s, 1H), 3.89 (br d, J=9.3 Hz, 2H), 3.34-3.10 (m, 7H), 3.12 (d, J=11.7 Hz, 1H), 2.22-2.14 (m, 2H), 1.97-1.92 (m, 1H), 1.54 (d, J=11.7 Hz, 1H), 1.29-1.23 (m, 3H), 1.13 (s, 6H).
Пример 15: 6,6-Диметил-3-фенил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
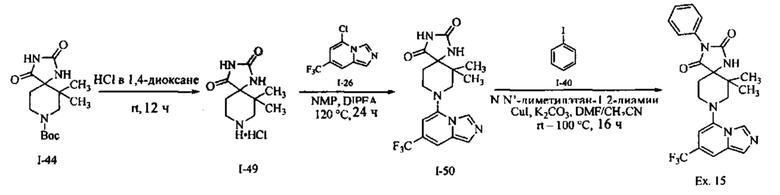
Получение Соединения I-49: Смесь Соединения I-44 (1,00 г, 3,3 ммоль) и HCl в 1,4-диоксане (4 М, 15 мл) перемешивали при RT в течение 12 ч. Растворитель выпаривали при пониженном давлении с получением Соединения I-49 в виде твердого вещества.
Получение Соединения I-50: Раствор Соединения I-26 (200 мг, 0,90 ммоль), Соединения I-49 (166 мг, 0,99 ммоль) и DIPEA (340 мг, 2,7 ммоль) в NMP (1,0 мл) нагревали при 120°С в течение 24 ч. Реакционную смесь охлаждали до RT, разбавляли EtOAc (30 мл) и промывали рассолом (3×50 мл). Органический слой собирали, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением Соединения I-50 в виде твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Получение Соединения по примеру 15: В перемешанный раствор Соединения I-50 (100 мг, 0,26 ммоль) в смеси ацетонитрила (20 мл) и DMF (20 мл) вносили Соединение I-40 (69,0 мг, 0,33 ммоль), K2CO3 (115 мг, 0,84 ммоль), N,N'-диметилэтан-1,2-диамин (7,00 мг, 0,08 ммоль) и CuI (15,0 мг, 0,08 ммоль) при RT. Реакционную смесь нагревали при 100°С в течение 16 ч. Реакционную смесь обрабатывали водой (15 мл) и экстрагировали EtOAc (3×20 мл). Объединенный органический слой промывали рассолом (5×20 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Сырое соединение очищали препаративной TLC (100% EtOAc) с получением Соединения по примеру 15 в виде твердого вещества. MS (MM) m/z 458.2 [М+Н]+.
1Н NMR (400 MHz, DMSO-d6): δ 8.81 (s, 1Н), 8.45 (s, 1H), 7.83 (s, 1H), 7.67 (s, 1H), 7.44-7.41 (m, 2H), 7.36-7.28 (m, 2H), 6.24 (s, 1H), 3.35-3.32 (m, 1H), 3.20-3.10 (m, 2H), 3.05 (d,J=12.0 Hz, 1H), 2.28-2.25 (m, 1H), 2.20-2.10 (m, 1H), 1.10 (s, 6H).
Пример 16: 8-(7-Бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4.5]декан-2,4-дион
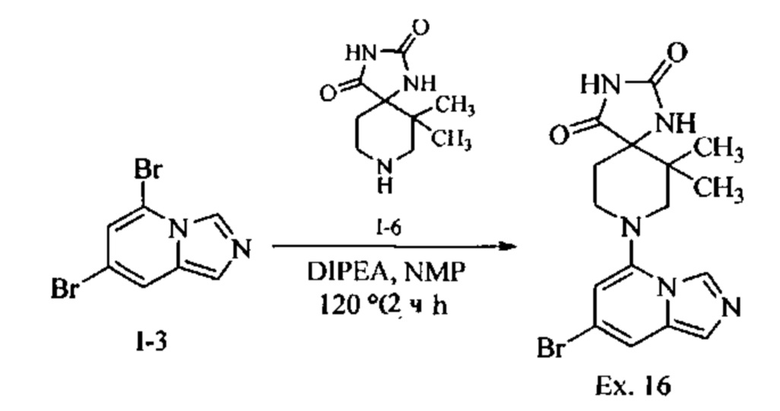
Получение Соединения по примеру 16: Раствор Соединения I-3 (300 мг, 0,365 ммоль), Соединения I-6 (85,0 мг, 0,434 ммоль) и DIPEA (94,0 мг, 0,730 ммоль) в NMP (3,0 мл) подвергали действию микроволнового излучения при 120°С в течение 2 ч. Реакционную смесь охлаждали до RT, разбавляли EtOAc (50 мл), промывали водой (2×50 мл) и рассолом (20 мл). Органический слой отделяли, сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (12 г, гексаны/EtOAc, 1:1) с получением указанного соединения в виде твердого вещества. MS (MM) m/z 392.0 [М+Н]+.
1Н NMR (300 MHz, DMSO-d6): δ 10.72 (s, 1Н), 8.28 (s, 1H), 8.24 (s, 1H), 7.64 (s, 1H), 7.40 (s, 1H), 6.24 (s, 1H), 3.32 (s, 1H), 3.19-3.15 (m, 2H), 2.98 (d, J=10.8 Hz, 1H), 2.08-2.06 (m, 2H), 1.07 (s, 6H).
Рацемический 8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4.5]декан-2,4-дион затем очищали методом хиральной SFC (колонка: ChiralPak AD-3; модификатор: 5-40% EtOH + 0,25% DEA; скорость потока: 2,5 мл/мин; температура: 40°С) с получением Соединения по примеру 16 (пик 2, элюирование через 8,2 мин).
Химические структуры, названия и молекулярная масса соединений в примерах 1-16 представлены в обобщенном виде в следующей далее таблице.
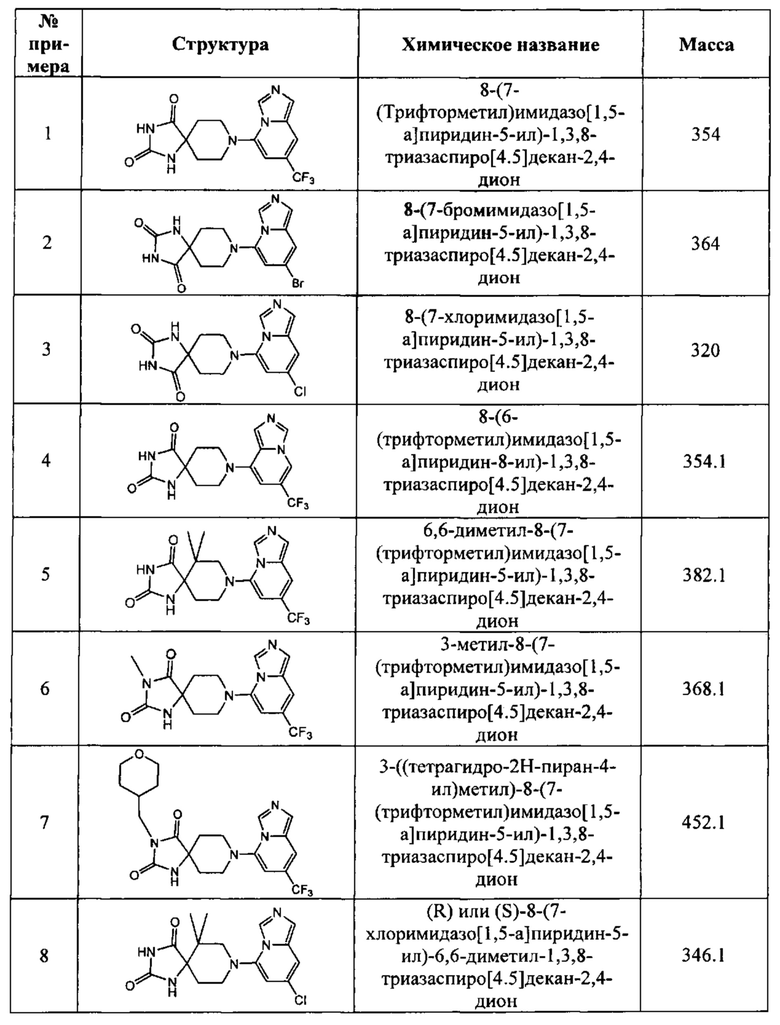
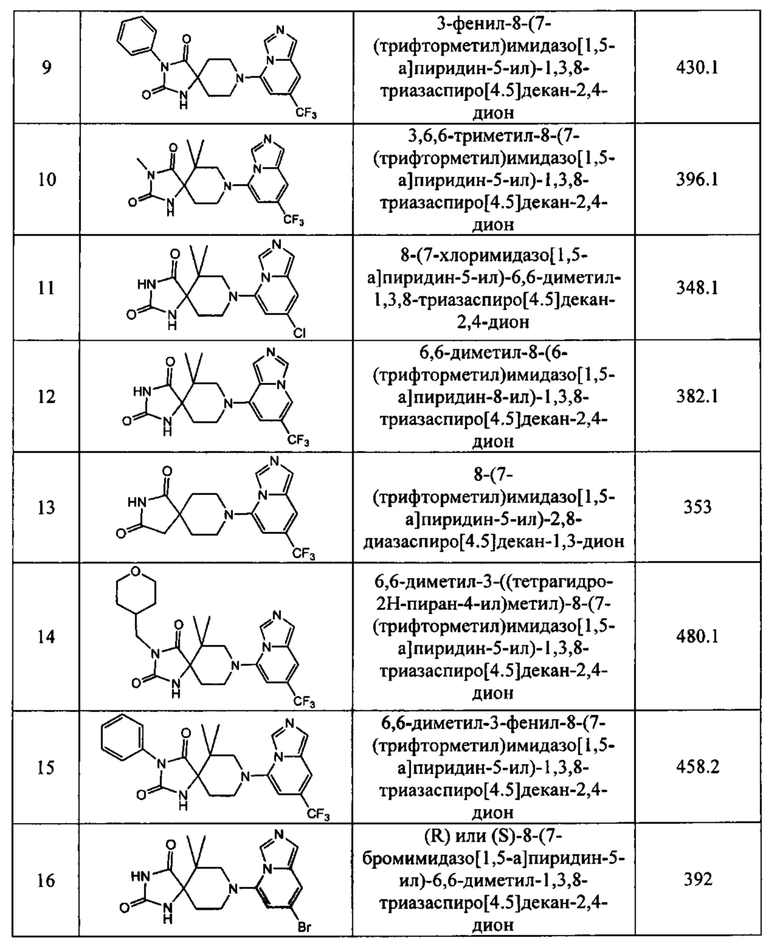
Пример 17: 8-(7-(Трифторметил)имидазо[1,5-а]пиридин-5-ил)-2-окса-8-азаспиро[4.5]декан-1-он
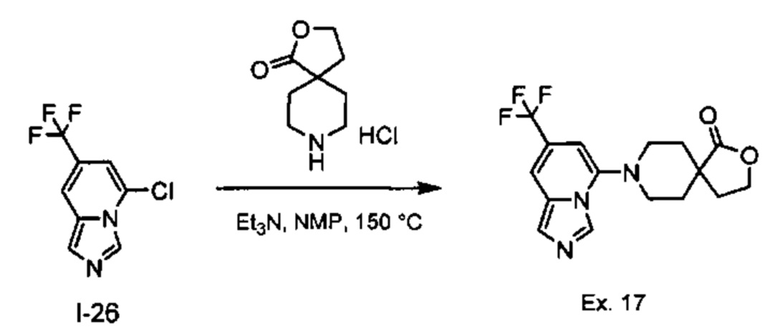
В сосуд добавляли 5-хлор-7-(трифторметил)имидазо[1,5-а]пиридин (I-26) (22 мг, 0,10 ммоль), 2-окса-8-азаспиро[4.5]декан-1-он гидрохлорид (коммерчески доступный от фирмы Enamine Building Blocks) (38 мг, 0,20 ммоль), NMP (300 мкл) и Et3N (100 мкл, 0,72 ммоль). Полученную смесь перемешивали при 150°С в течение 20 ч. Смесь фильтровали и очищали обращенно-фазовой препаративной HPLC (от 15% до 70% ACN в воде с 0,1 % NH4OH) с получением 8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2-окса-8-азаспиро[4.5]декан-1-она (пример 17). [М+Н]+ 340. 1Н NMR (500 MHz, DMSO-d6) δ 8.44 (s, 1Н), 7.88 (s, 1Н), 7.72 (s, 1Н), 6.32 (s, 1Н), 4.40-4.30 (m, 2Н), 3.50-3.25 (m, 2Н), 2.99 (t, J=10 Hz, 1H), 2.60-2.40 (m, 1H), 2.35-2.25 (m, 2H), 2.10-1.95 (m, 2H), 1.85-1.75 (m, 2H); MS (EI) вычислено для C16H17F3N3O2
Пример 18: это соединение синтезировали способом, аналогичным способу получения Соединения по примеру 17, за исключением того, что вместо 2-окса-8-азаспиро[4.5]декан-1-она гидрохлорида использовали 2-метил-1,3,8-триазаспиро[4.5]дек-1-ен-4-он (коммерчески доступный от фирмы Ark Pharm, Inc.).
Пример 19: это соединение синтезировали способом, аналогичным способу получения Соединения по примеру 17, за исключением того, что вместо 2-окса-8-азаспиро[4.5]декан-1-она гидрохлорида использовали 2,8-диазаспиро[4.5]декан-1-она гидрохлорид (коммерчески доступный от фирмы Ark Pharm, Inc.).
Пример 20: 1-(9-(7-(Трифторметил)имидазо[1,5-а]пиридин-5-ил)-3,9-диазаспиро[5.5]ундекан-3-ил)этан-1-он
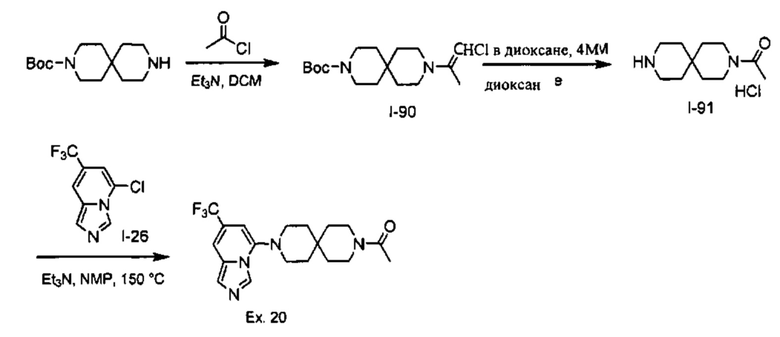
Трет-бутил 3,9-диазаспиро[5.5]ундекан-3-карбоксилат (коммерчески доступный от фирмы Ark Pharm, Inc.) (100 мг, 0,40 ммоль) растворяли в DCM (1 мл) и добавляли TEA (200 мкл, 1,4 ммоль). Раствор охлаждали до 0°С, после чего по каплям добавляли ацетилхлорид (31 мг, 0,4 ммоль) в виде раствора в DCM (0,1 мл). Полученную смесь оставляли нагреваться до RT и перемешивали в течение 18 ч. Затем растворитель удаляли в вакууме с получением трет-бутил 9-ацетил-3,9-диазаспиро[5.5]ундекан-3-карбоксилата (I-90), который использовали без дополнительной очистки. Затем трет-бутил 9-ацетил-3,9-диазаспиро[5.5]ундекан-3-карбоксилат (I-90) растворяли в диоксане (1 мл) и добавляли HCl в диоксане (4М, 1 мл, 4,0 ммоль). Смесь оставляли перемешиваться при RT в течение 18 ч, после чего растворитель удаляли в вакууме с получением 1-(3,9-диазаспиро[5.5]ундекан-3-ил)этан-1-она гидрохлорида (I-91), который использовали без дополнительной очистки. Затем 1-(3,9-диазаспиро[5.5]ундекан-3-ил)этан-1-она гидрохлорид (I-91) растворяли в NMP (1 мл), после чего добавляли 5-хлор-7-(трифторметил)имидазо[1,5-а]пиридин (I-26) (25 мг, 0,11 ммоль) и Et3N (800 мкл, 5,7 ммоль). Смесь нагревали до 150°С и перемешивали в течение 20 ч. Затем смесь фильтровали и очищали непосредственно обращенно-фазовой HPLC масс-спектрометрией (10% - 100% ACN в воде с 0,1% TFA) с получением 1-(9-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-3,9-диазаспиро-[5.5]ундекан-3-ил)этан-1-она (пример 20). MS (EI) вычислено для C19H24F3N4O [М+Н]+ 381.
1H NMR (500 MHz, DMSO-d6) δ 8.59 (s, 1 Н), 7.91 (s, 1Н), 7.83 (s, 1Н), 6.41 (s, 1Н), 3.30-3.00 (m, 4Н), 2.65-2.30 (m, 4Н), 2.01 (s, 3Н), 1.85-1.65 (m, 4Н), 1.65-1.40 (m, 4Н)
Пример 21: это соединение синтезировали способом, аналогичным способу получения Соединения по примеру 17, за исключением того, что вместо Соединения I-26 использовали 2-окса-8-азаспиро[4.5]декан-1-она гидрохлорид и Соединение I-78. Также, карбонат цезия использовали вместо Et3N.
Химические структуры, названия и молекулярная масса соединений в примерах 17-21 представлены в обобщенном виде в следующей далее таблице.
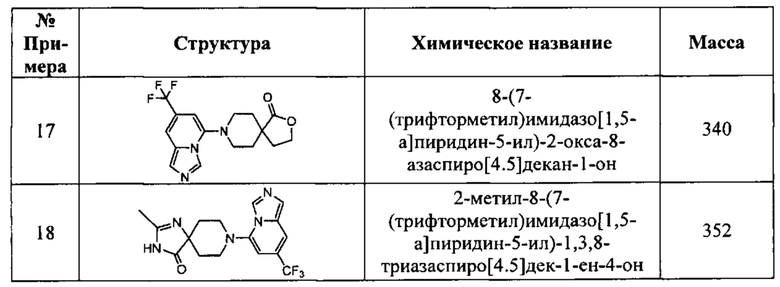
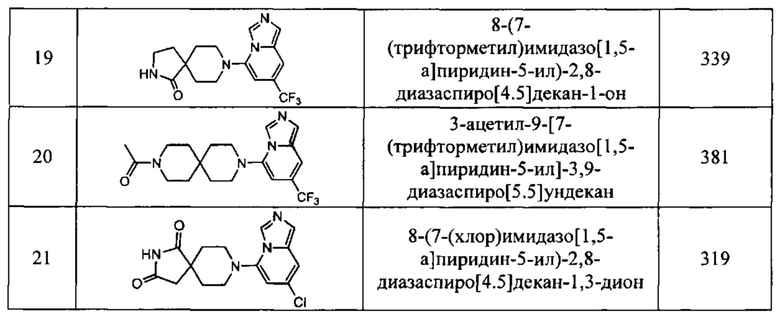
Пример 22: 6,6-Диметил-8-(7-(трифторметил)имидазо[1.5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
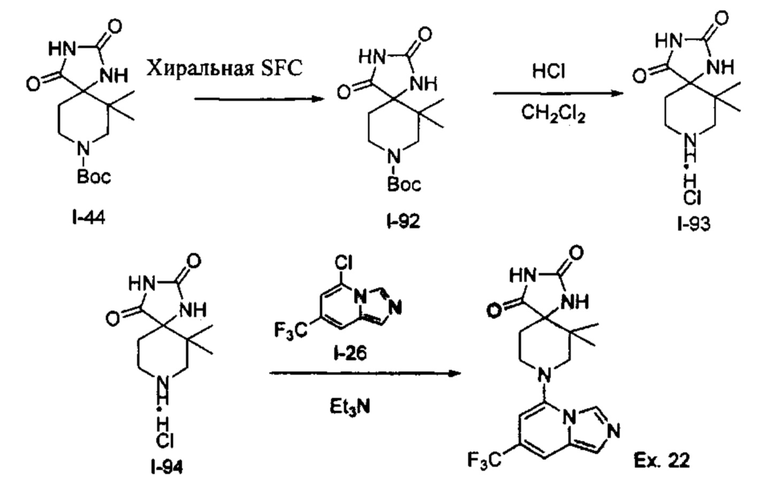
Получение Соединения I-92: Соединение I-44 разделяли методом хиральной SFC (колонка: LUX-2; модификатор: 20% МеОН + 0,25% DMEA; скорость потока: 70 мл/мин; температура: 15°С) с получением Соединения I-92 (пик 1, элюирование через 3,3 мин). MS (EI) [M+H-tBu]+ 242.
Получение Соединения I-93: Соединение I-92 (424 мг, 1,426 ммоль) растворяли в CH2Cl2 (3,5 мл) и охлаждали до 0°С. Затем добавляли HCl (6,8 мл, 27,2 ммоль) в течение 10 мин и реакционную смесь оставляли нагреваться до RT и перемешивали в течение ночи. Затем растворитель удаляли в вакууме и твердое вещество ресуспедировали в эфире. Растворитель снова удаляли с получением Соединения I-94, которое использовали без дополнительной очистки. MS (EI) [М+Н]+ 198.
Получение Соединения по примеру 22: В колбу добавляли Соединение I-26 (50,8 мг, 0,23 ммоль), Соединение I-94 (48,9 мг, 0,209 ммоль), NMP (600 мкл) и Et3N (200 мкл, 1,435 ммоль). Смесь перемешивали при 150°С в течение 18 ч. Затем смесь фильтровали и очищали непосредственного обращенно-фазовой HPLC масс-спектрометрией (10% -100% ACN в воде с 0,1% NH4OH) с получением Соединения по примеру 22. 1Н NMR (600 MHz, DMSO-d6) δ 10.70 (s, 1Н), 8.43 (s, 1H), 8.23 (s, 1H), 7.85 (s, 1H), 7.69 (s, 1H), 6.25 (s, 1H), 3.43-3.07 (m, 3H), 3.00 (d, J=11.6 Hz, 1H), 2.24-1.86 (m, 2H), 1.04 (s, 6H). MS (EI) Вычислено для C17H19F3N5O2 [M+H]+ 382, найдено 382.
Пример 23: 8-(7-Бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4.5]декан-2,4-дион
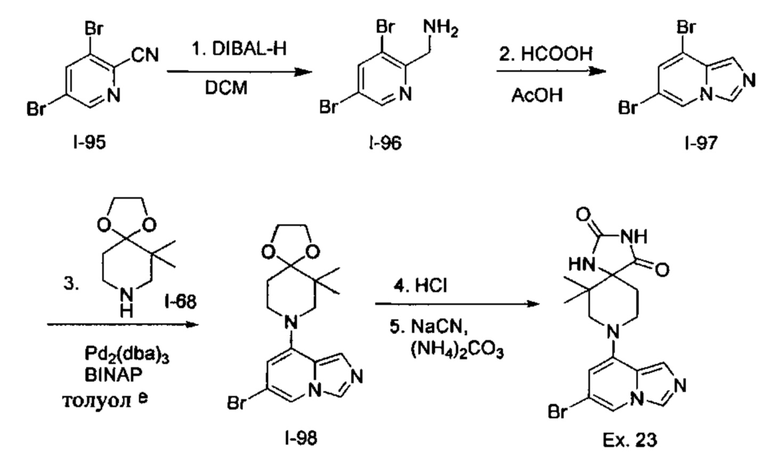
Получение Соединения I-96: К перемешанному раствору Соединения I-95 (5,0 г, 19,2 ммоль) в DCM (50 мл) при 0°С медленно добавляли DIBAL-H (5,4 г, 38,5 ммоль), раствор оставляли нагреваться до RT и перемешивали в течение 2 ч. Реакционную смесь затем гасили холодной водой и экстрагировали DCM (2×500 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток обрабатывали 2М HCl (20 мл) и выпаривали при пониженном давлении с получением Соединения I-96.
Получение Соединения I-97: Раствор Соединения I-96 (2,0 г, 7,32 ммоль) в муравьиной кислоте (50 мл) и уксусной кислоте (50 мл) перемешивали при 100°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали флэш-хроматографией, используя в качестве элюента смесь 30% этилацетат/гексан, с получением Соединения I-97.
Получение Соединения I-98: В перемешанный раствор Соединения I-97 (1,2 г, 4,3 ммоль) в толуоле (10 мл) вносили Соединение I-68 (743 мг, 4,34 ммоль). Реакционную смесь продували аргоном в течение 10 мин, после чего в смесь добавляли Pd2(dba)3 (397 мг, 0,43 ммоль), Cs2CO3 (2,7 г, 8,6 ммоль) и BINAP (270 мг, 0,434 ммоль) и нагревали при 100°С в течение 16 ч. Реакционную смесь охлаждали до RT и разбавляли этилацетатом, промывали водой, органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (гексаны/EtOAc, 1:1) с получением Соединения I-98.
Получение Соединения по примеру 23: В перемешанный раствор Соединения I-98 (290 мг, 0,79 ммоль) в THF (1,5 мл) вносили HCl (2 М, 1,5 мл) при RT. Реакционную смесь перемешивали в течение 16 ч, затем концентрировали при пониженном давлении и экстрагировали этилацетатом. Органический слой сушили над безводным Na2SO4, затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (гексаны/EtOAc, 70:30) с получением 1-(6-бромимидазо[1,5-а]пиридин-8-ил)пиперидин-4-она.
Раствор 1-(6-бромимидазо[1,5-а]пиридин-8-ил)пиперидин-4-она (140 мг, 0,436 ммоль) в EtOH (6,0 мл) и воде (1,5 мл) помещали в герметично закрытую пробирку и вносили NaCN (42 мг, 0,87 ммоль) и карбонат аммония (837 мг, 8,72 ммоль) при RT. Реакционную смесь затем перемешивали при 100°С в течение 16 ч. Реакционную смесь выливали в воду и экстрагировали EtOAc. Органические экстракты затем промывали водой (2×) и рассолом. Органическую фазу разделяли, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на системе Combiflash с использованием колонки Redisep® (MeOH/DCM, 6%), затем полупрепаративной HPLC с получением Соединения по примеру 23. 1Н NMR (300 MHz, DMSO-d6): δ 10.70 (s, 1Н), 8.30 (s, 1H), 8.28 (s, 1H), 8.20 (s, 1H), 7.44 (s, 1H), 6.12 (s, 1H), 3.45-3.42 (m, 1H), 3.39-3.30 (m, 2H), 3.15 (d, J=5.1 Hz, 1H), 2.10-1.90 (m, 2H). MS (MM) m/z 392 [M+H]+.
Пример 24: 8-(7-Хлоримидазо[1,5-а]пиридин-5-ил)-2-тиа-1,3,8-триазаспиро[4.5]декан-4-он 2,2-диоксид
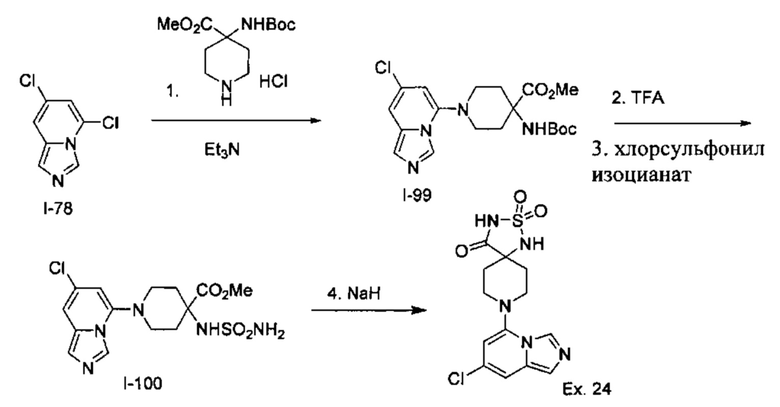
Получение Соединения I-99: В сосуд добавляли Соединение I-78 (300 мг, 1,60 ммоль), метил 4-((трет-бутоксикарбонил)амино)пиперидин-4-карбоксилат (402 мг, 1,56 ммоль), DMSO (3,1 мл) и Et3N (1,1 мл). Смесь перемешивали и нагревали при 120°С в течение 16 ч. Реакционную смесь затем охлаждали, разбавляли насыщенным раствором NaHCO3 и экстрагировали EtOAc. Органический слой отделяли, промывали рассолом, сушили над MgSO4 и концентрировали. Остаток очищали флэш-хроматографией (силикагель, от 0 до 100% 3:1 EtOAc:EtOH в гексанах) с получением метил 4-((трет-бутоксикарбонил)амино)-1-(7-хлоримидазо[1,5-а]пиридин-5-ил)пиперидин-4-карбоксилата (Соединение I-99). Вычислено для C19H26ClN4O4 [М+Н]+, 409 найдено 409.
Получение Соединения I-100: К раствору Соединения I-99 (243 мг, 0,59 ммоль) в DCM (1 мл) при RT добавляли TFA (0,5 мл). Смесь перемешивали в течение 3 ч и затем концентрировали. Сырой остаток, содержащий метил 4-амино-1-(7-хлоримидазо[1,5-а]пиридин-5-ил)пиперидин-4-карбоксилат, использовали на следующей стадии без дополнительной очистки. Вычислено для C14H18ClN4O2 [М+Н]+, 309 найдено 309.
Для разделения реакционной смеси в сосуд, содержащий хлорсульфонил изоцианат (202 мкл, 2,33 ммоль), при 0°С по каплям добавляли муравьиную кислоту (89 мкл, 2,3 ммоль). После добавления реакционную смесь энергично перемешивали при RT в течение 30 мин. Затем смесь метил 4-амино-1-(7-хлоримидазо[1,5-а]пиридин-5-ил)пиперидин-4-карбоксилата из предыдущей стадии растворяли в Et3N (325 мкл, 2,33 ммоль) и добавляли THF (1,2 мл). Реакционную смесь перемешивали при RT в течение 2 ч. Затем смесь концентрировали и остаток очищали обращенно-фазовой HPLC (смесь H2O/CH3CN, содержащая 0,1% TFA) с получением Соединения I-100. C14H19ClN5O4S [М+Н]+, 388 найдено 388.
Получение соединения по примеру 24: Раствор Соединения I-100 (50 мг, 0,129 ммоль) в THF (645 мкл) охлаждали до 0°С и обрабатывали NaH (10,31 мг, 0,258 ммоль) в течение 3 ч. Реакционную смесь затем гасили несколькими каплями воды и концентрировали, затем смесь фильтровали и очищали обращенно-фазовой HPLC масс-спектрометрией (10% - 100% ACN в воде с 0,1% TFA) с получением соединения по примеру 24. 1Н NMR (500 MHz, DMSO-d6) δ 8.67 (s, 1Н), 7.61 (s, 1Н), 7.83 (s, 1Н), δ 7.61 (d, J=12 Hz, 2H), 6.38 (s, 1H), 3.03 (t, J= 12 Hz, 4H), 2.31 (t, J=12 Hz, 2H), 1.97 (t, J=12 Hz, 2H), MS (EI) Вычислено для C13H15ClN5O3S [M+H]+, 356; найдено 356.
Пример 25: (R) или (S)-8-(7-Бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-2,8-диазаспиро[4.5]декан-1,3-дион
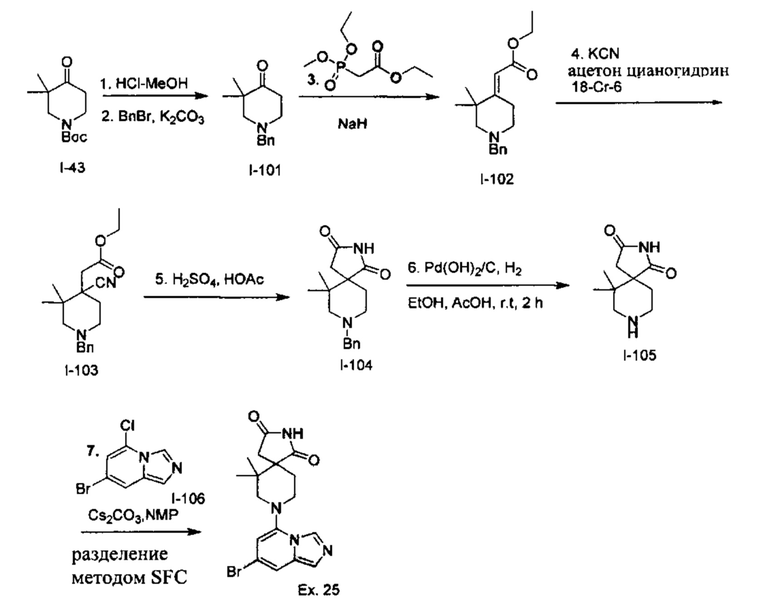
Получение Соединения I-101: Смесь Соединения I-43 (30,0 г, 132 ммоль) и смеси HCl/диоксан (100 мл, 4М) перемешивали при RT в течение 1,5 ч. Реакционную смесь затем концентрировали при пониженном давлении и остаток растворяли в DMF (200 мл). Добавляли K2CO3 (62,1, 449 ммоль) и смесь затем перемешивали в течение 30 мин при RT. Затем в реакционную смесь по каплям добавляли BnBr (26,8 г, 157 ммоль), после чего реакционную смесь нагревали до 65°С и перемешивали в течение 24 ч. Основную часть DMF удаляли путем концентрирования в вакууме, и остаток затем разбавляли водой (150 мл) и экстрагировали EtOAc (200 мл ×2). Объединенные органические слои промывали рассолом (100 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат = 150:1-100:1) с получением Соединения I-101. MS (ESI) m/z: 218 (М+Н+).
Получение Соединения I-102: К раствору этил 2-(диэтоксифосфорил)ацетата (17,0 г, 75,9 ммоль) в THF (150 мл) добавляли NaH (3,04 г, 75,93 ммоль, 60%) при 0°С, после чего полученную смесь перемешивали при 0°С в течение 1,5 ч. Затем по каплям добавляли Соединение I-101 (11 г, 51 ммоль) в THF (50 мл) в течение 15 мин при 0°С, после чего реакционную смесь перемешивали при 80°С в атмосфере азота (1 атм) в течение 20 ч. Реакционную смесь затем охлаждали до RT и большую часть растворителя удаляли путем концентрирования в вакууме. Затем смесь разбавляли водой (150 мл) и экстрагировали EtOAc (200 мл ×3). Объединенные органические слои концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (силикагель, элюирование 100% петролейного эфира) с получением Соединения I-102. MS (ESI) m/z: 288 (М+Н+).
Получение Соединения I-103: К раствору Соединения I-102 (9,0 г, 31 ммоль) в MeCN (90 мл) добавляли KCN (4,08 г, 62,7 ммоль) и 18-краун-6 (9,94 г, 37,6 ммоль) с последующим 2-гидрокси-2-метилпропаннитрилом (5,33 г, 62,6 ммоль) при RT. Полученную смесь нагревали до 75°С в течение 110 ч, после чего реакционную смесь охлаждали до RT и концентрировали при пониженном давлении. Затем добавляли воду (150 мл) и смесь экстрагировали EtOAc (120 мл ×3). Объединенные органические слои промывали рассолом (150 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат = 1:0 -100:1 - 10:1) с получением Соединения I-103. MS (ESI) m/z: 315.1 (М+Н+).
Получение Соединения I-104: Перемешанный раствор Соединения I-103 (500 мг, 1,59 ммоль) в уксусной кислоте (1 мл, 1,59 ммоль) охлаждали до 0°С и по каплям добавляли концентрированную серную кислоту (0,1 мл, 1,59 ммоль). Затем смесь нагревали при 100°С в течение 21 ч, после чего основную часть растворителя удаляли в вакууме. Остаток затем разбавляли водой (40 мл), подщелачивали 2М NaOH до рН ~10 и экстрагировали DCM:MeOH (10:1, 20 мл ×5). Затем органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали обращенно-фазовой HPLC (Phenomenex Synergi С18 (250*21,2 мм*4 мкм) с использованием в качестве элюентов воду (0,1% TFA) и ацетонитрил) с получением Соединения I-104. MS(ESI) m/z: 287 (М+Н+).
Получение Соединения I-105: К перемешанному раствору Соединения I-104 (300 мг, 1,05 ммоль) в EtOH (10 мл) добавляли АсОН (63 мг, 1,1 ммоль) и в реакционный сосуд вносили Pd(OH)2/C (90 мг, 50% масс.) в атмосфере азота. Атмосферу азота заменяли водородом (15 Psi) и смесь перемешивали в течение 2,5 ч при RT в атмосфере водорода (1 атм). Реакционную смесь затем фильтровали через слой целита и промывали EtOH (10 мл ×2). Фильтрат концентрировали при пониженном давлении с получением Соединения I-105, которое использовали на следующей стадии без дополнительной очистки.
Получение Соединения по примеру 25: К раствору 7-бром-5-хлоримидазо[1,5-а]пиридина (Соединение I-106) (200 мг, 0,86 ммоль, коммерчески доступный от фирмы Ellanova Laboratories) и Соединения I-105 (339 мг, 1,73 ммоль) в NMP (6 мл) добавляли карбонат цезия (1,13 г, 3,46 ммоль) при перемешивании при 140°С в течение 14 ч. Затем реакционную смесь охлаждали до RT и разбавляли водой (20 мл), экстрагировали этилацетатом (20 мл ×2), и объединенные органические слои промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой HPLC (Phenomenex Synergi С18 (250*21,2 мм*4 мкм), с использованием в качестве элюентов воду (0,2% муравьиная кислота) и ацетонитрил). Затем рацемический продукт очищали методом SFC (колонка: ChiralPak AS-3; модификатор: 5-40% EtOH + 0,05% DEA; скорость потока: 2,8 мл/мин; температуре: 40°С) с получением Соединения по примеру 25 (пик 2, элюирование через 5,0 мин). 1Н NMR (400 MHz, CD3OD) δ 8.29 (s, 1Н),7.55 (s, 1Н), 7.40 (s, 1Н), 6.34 (d, J=1.5 Hz, 1Н), 3.47 (br s, 1H),3.21 (br s, 1H), 3.00 (d, J=18.5 Hz, 1H), 2.89 (d, J=11.5 Hz, 1H), 2.48 (br d, J=18.3 Hz, 1H), 2.17 (br s, 2H), 1.21 (br s, 3H), 1.14 (br s, 3H); (ESI) m/z: 391 (M+H+).
Пример 26: (S) или (R)-6,6-Диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4.5]декан-1,3-дион
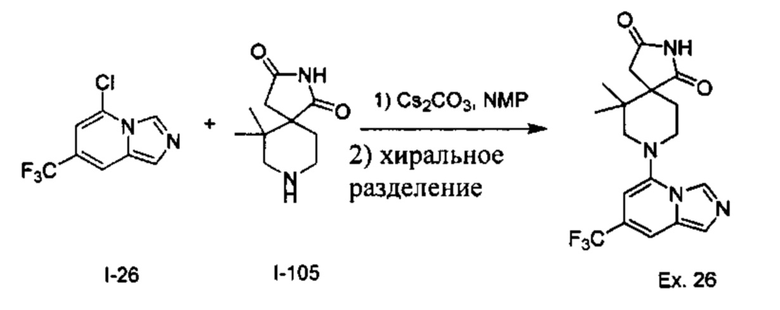
К раствору Соединения I-26 (500 мг, 2,27 ммоль) и Соединения I-105 (534 мг, 2,72 ммоль) в NMP (2,5 ml) добавляли Cs2CO3 (1,48 г, 4,53 ммоль) при перемешивании при RT. После завершения добавления реакционную смесь перемешивали при 150°С в течение 6 ч. Затем реакционную смесь охлаждали до RT и остаток очищали обращенно-фазовой HPLC (YMC-Actus Pro С18 150*30 5u, используя в качестве элюентов поду (0,1% TFA)-CH3CN, с получением рацемического 6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4.5]декан-1,3-диона. Затем рацемический продукт разделяли методом хиральной SFC (колонка: Lux Cellulose-2; модификатор: 5-40% МеОН + 0,05% DEA; скорость потока: 2,5 мл/мин; температруе: 40°С) с получением Соединения по примеру 26 (пик 1, элюирование через 7,5 мин). NMR (400 MHz, CD3OD) δ 9.27 (br s, 1Н), 8.19 (br s, 1H), 7.98 (s, 1H), 6.71 (s, 1H), 3.62 (br s, 1H), 3.39-3.50 (m, 1H), 3.16-3.26 (m, 1H), 2.93-3.07 (m, 2H), 2.49 (br d, J=18.4 Hz, 1H), 2.12-2.35 (m, 2H), 1.22 (s, 3H), 1.16 (br s, 3H); (ESI) m/z: 381 (M+H+).
Соединение по примеру 27 получали способом, аналогичным способу получения Соединения по примеру 26, за исключением того, что Соединение I-78 использовали вместо Соединения I-26. Рацемический продукт разделяли методом хиральной SFC (колонка: Chiralpak AS-3; модификатор: 5-40% EtOH + 0,05% DEA; скорость потока: 2,5 мл/мин; температура: 40°С) с получением Соединения по примеру 27 (пик 2, элюирование через 7,3 мин).
Соединение по примеру 28 получали способом, аналогичным способу получения Соединения по примеру 26, за исключением того, что этил 2-(диэтоксифосфорил)пропаноат использовали вместо этил 2-(диэтоксифосфорил)ацетата, и Соединение I-78 использовали вместо Соединения I-26. Рацемический продукт разделяли методом хиральной SFC (колонка: Lux Cellulose-2; модификатор: 5-40% МеОН + 0,05% DEA; скорость потока: 2,5 мл/мин; температура: 40°С) с получением Соединения по примеру 27 (пик 1, элюирование через 6,6 мин).
Соединение по примеру 29 получали способом, аналогичным способу получения Соединения по примеру 26, за исключением того, что 1-бензил-3,3-дифторпиперидин-4-он (коммерчески доступный от фирмы Merck KgaA) использовали вместо Соединения I-101, и Соединение I-78 использовали вместо Соединения I-26.
Соединение по примеру 30 получали способом, аналогичным способу получения Соединения по примеру 17, за исключением того, что свободное основание Соединения I-53 использовали вместо 2-окса-8-азаспиро[4.5]декан-1-она гидрохлорида, и Соединение I-106 использовали вместо Соединения I-26. Карбонат цезия также использовали вместо Et3N.
Химические структуры, названия и молекулярная масса соединений по примерам 27-30 представлены в обобщенном виде в следующей далее таблице.

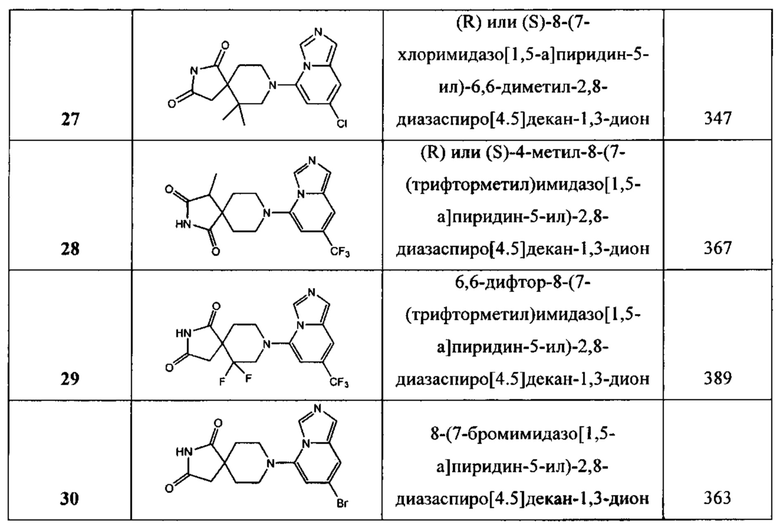
Пример 31: 8-(7-Метоксиимидазо[1.5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
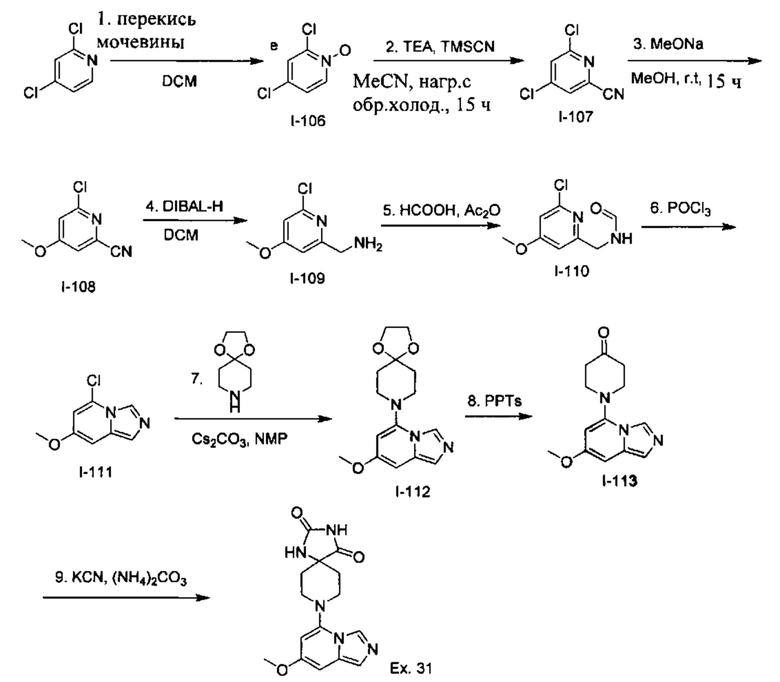
Получение Соединения I-106: К перемешанному раствору 2,4-дихлорпиридина (25 г, 169 ммоль) в DCM (500 мл) медленно добавляли перекись мочевины (31,8 г, 338 ммоль) с последующим трифторуксусным ангидридом (71,0 г, 338 ммоль) при RT. Затем смесь перемешивали при 30°С в течение 17 ч, после чего раствор гасили путем добавления раствора Na2S2O3 (20 г) в Н2О (200 мл). Погашенную смесь затем перемешивали при 30°С в течение 1 ч, после чего органический слой отделяли и водный слой экстрагировали DCM (100×3). Объединенные органические слои промывали рассолом (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюируя смесью петролейный эфир/EtOAc = 3:1 - 0:1) с получением Соединения I-106. MS (ESI) m/z: 164 (М+Н+).
Получение Соединения I-107: К раствору Соединения I-106 (26 г, 159 ммоль) в ацетонитриле (340 мл) добавляли TMSCN (62,9 г, 634 ммоль) и TEA (32,1 г, 317 ммоль), затем смесь перемешивали при 90°С в течение 18 ч. Затем реакционную смесь охлаждали до RT и растворитель удаляли в вакууме, Остаток очищали колоночной хроматографией (силикагель, элюируя смесью петролейный эфир/этилацетат = 10:1) с получением Соединения I-107.
Получение Соединения I-108: NaH (0,983 г, 24,6 ммоль, 60% в масле) добавляли к МеОН (20 мл) при 0°С и затем смесь перемешивали при RT в течение 20 мин. Полученный раствор добавляли к раствору 4,6-дихлорпиколинонитрила (4,25 г, 24,6 ммоль) в МеОН (30 мл) и смесь перемешивали при RT в течение 17 ч. Большую часть растворителя удаляли путем концентрирования в вакууме, после чего к остатку добавляли воду (50 мл). Затем полученную смесь экстрагировали EtOAc (80 мл ×2), и объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (силикагель, элюируя смесью петролейный эфир/этилацетат =20:1 - 10:1) с получением Соединения I-108. MS (ESI) m/z: 169 (М+Н+).
Получение Соединения I-109: К раствору Соединения I-108 (2,83 г, 16,79 ммоль) в DCM (30 мл) при -78°С по каплям добавляли DIBAL-H (84 мл, 84 ммоль) (1М в толуоле) в течение 30 мин в атмосфере азота. Полученную смесь перемешивали при -78°С в течение 2 ч, после чего реакционную смесь гасили водным раствором NH4Cl (30 мл), разбавляли насыщенным раствором тартрата натрия-калия (75 мл) и оставляли для доведения температуры до комнатной, и перемешивали в течение 15 ч. Затем органический слой отделяли и водную фазу экстрагировали EtOAc (80 мл ×2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование DCM/MeOH=50:1) с получением Соединения I-109. MS (ESI) m/z: 172 (М+Н+).
Получение Соединения I-110: К раствору Соединения I-109 (1,06 г, 6,14 ммоль) в муравьиной кислоте (8 мл, 210 ммоль) добавляли Ас2О (1,8 мл, 19,1 ммоль). Смесь перемешивали при RT в течение 16 ч. Затем растворитель удаляли путем концентрирования в вакууме с получением Соединения I-110, которое использовали на следующей стадии без дополнительной очистки. (ESI) m/z: 201.0 (М+Н+).
Получение Соединения I-111: К раствору Соединения I-110 (3,6 г, 18,0 ммоль) в толуоле (40 мл) добавляли POCl3 (8,13 г, 53,0 ммоль) при RT. После завершения добавления смесь нагревали и перемешивали при 100°С в течение 2 ч, после чего раствор охлаждали до RT и медленно выливали в воду (150 мл). Полученную смесь затем подщелачивали насыщенным водным раствором Na2CO3 для доведения значения рН до рН=8. Затем смесь экстрагировали EtOAc (80 мл ×3), объединенные органические слои промывали рассолом (70 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат = 4:1 -3:1) с получением I-111. MS (ESI) m/z: 183 (M+H+).
Получение Соединения I-112: К раствору Соединения I-111 (500 мг, 2,74 ммоль) в NMP (8 мл) добавляли 1,4-диокса-8-азаспиро[4.5]декан (1,18 г, 8,21 ммоль) и Cs2CO3 (2,68 г, 8,21 ммоль). Затем смесь перемешивали при 140°С в течение 16 ч, после чего смесь охлаждали до RT, выливали в воду (50 мл) и экстрагировали EtOAc (70 мл ×4). Объединенные органические слои промывали рассолом (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат =3:1 - 1:1) с получением Соединения I-112. MS (ESI) m/z: 290 (М+Н+).
Получение Соединения I-113: Смесь Соединения I-112 (180 мг, 0,622 ммоль) и HCl (4,0 мл, 12 ммоль) (3 М в воде) в THF (4 мл) перемешивали при RT в течение 15 ч, после чего реакционную смесь подщелачивали насыщенным водным раствором NaHCO3 для доведения значения рН до рН=8. Затем смесь экстрагировали EtOAc (50 мл ×3), и объединенные органические слои промывали рассолом (30 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением Соединения I-113, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z: 246 (М+Н+).
Получение Соединения по примеру 31: К раствору Соединения I-113 (120 мг, 0,49 ммоль) в EtOH (3 мл) и воде (2 мл) добавляли KCN (48 мг, 0,74 ммоль) и (NH4)2CO3 (235 мг, 2,45 ммоль) одной порцией при RT. После завершения добавления смесь нагревали и перемешивали при 80°С в течение 3,5 ч. Реакционную смесь затем охлаждали до RT, выливали в воду (20 мл) и экстрагировали EtOAc (30 мл ×4). Объединенные органические слои промывали рассолом (20 мл), сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали обращенно-фазовой HPLC (Xtimate С18 150*25 мм*5 мкм, элюирование водой (10 мМ NH4HCO3)/CH3CN в качестве элюентов) с получением Соединения по примеру 31. 1Н NMR (400 MHz, DMSO-d6) δ 10.75 (br s, 1H), 8.61 (s, 1H), 8.06 (s, 1H), 7.15 (s, 1H), 6.64 (s, 1H), 5.87 (s, 1H), 3.76 (s, 3H), 3.03 (br t, J=11.2 Hz, 2H), 2.58 - 2.62 (m, 2H), 2.13 (br t, J=10.8 Hz, 2H), 1.75 (br d, J=13.6 Hz, 2H). MS (ESI) m/z: 316 (M+H+).
Пример 32: 8-(7,8-Михлоримидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4.5]декан-2,4-дион
1. перекись мочевины
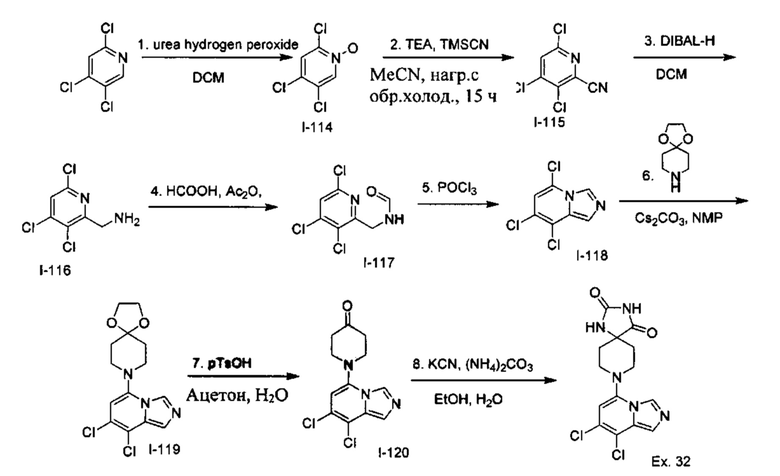
Получение Соединения I-114: К перемешанному раствору 2,4,5-трихлорпиридина (5,0 г, 27 ммоль) в DCM (100 мл) добавляли перекись мочевины (5,16 г, 54,8 ммоль) при RT, после чего медленно добавляли ангидрид трифторуксусной кислоты (11,5 г, 54,8 ммоль). Затем смесь нагревали и перемешивали при 30°С в течение 17 ч, после чего реакционную смесь гасили путем добавления насыщенного водного раствора Na2S2O3 (200 мл). Погашенный раствор затем перемешивали при 30°С в течение 1 ч, после чего органический слой отделяли, и водный слой затем экстрагировали DCM (100×3). Объединенные органические слои промывали рассолом (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/EtOAc = от 3:1 до 0:1) с получением Соединения I-114. MS (ESI) m/z: 198 (М+Н+).
Получение Соединения I-115: К раствору Соединения I-114 (4,3 г, 21,7 ммоль) в ацетонитриле (70 мл) добавляли TMSCN (8,6 г, 87 ммоль) и TEA (4,39 г, 43,3 ммоль), после чего смесь нагревали и перемешивали при 90°С в течение 18 ч. После охлаждения до RT растворитель удаляли в вакууме и остаток очищали колоночной хроматографией (силикагель, элюирование с использованием в качестве элюента смеси петролейный эфир:этилацетат, 10:1) с получением Соединения I-115. MS (ESI) m/z: 209 (М+Н+).
Получение Соединения I-116: К раствору Соединения I-115 (1,9 г, 9,2 ммоль) в DCM (40 мл) по каплям добавляли DIBAL-H (45,8 мл, 45,8 ммоль) (1М в толуоле) в атмосфере азота при -78°C. Полученную смесь перемешивали при -78°С в течение 1,5 ч, после чего реакционную смесь гасили водным раствором NH4Cl (50 мл). Затем температуру реакционной смеси медленно доводили до 0°С, и раствор разбавляли насыщенным раствором тартрата натрия-калия (300 мл). Затем температуру реакционной смеси доводили до RT и перемешивали в течение 15 ч. Затем органический слой отделяли и водную фазу экстрагировали DCM (200 мл ×2). Объединенные органические слои промывали рассолом (200 мл), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток затем очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат =3:1) с получением Соединения I-116. MS (ESI) m/z: 213 (М+Н+).
Получение Соединения I-117: К перемешанному раствору Соединения I-116 (1,0 г, 4,4 ммоль) в муравьиной кислоте (8,71 г, 189 ммоль) добавляли уксусный ангидрид (1,45 г, 14,2 ммоль). Затем смесь перемешивали при RT (30°С) в течение 17 ч. Затем растворитель удаляли в вакууме с получением Соединения I-117, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z: 239 (М+Н+).
Получение Соединения I-118: К раствору Соединения I-117 (1,0 г, 4,2 ммоль) в толуоле (10 мл) добавляли POCl3 (1,92 г, 12,5 ммоль) при перемешивании при RT (30°С). После завершения добавления реакционную смесь нагревали и перемешивали при 100°С в течение 2 ч. Реакционную смесь затем охлаждали до RT и медленно выливали в воду (30 мл). Затем добавляли Na2CO3 для доведения значения рН до рН=8. Затем смесь экстрагировали EtOAc (50 мл ×3), и объединенные органические слои промывали рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат =5:1) с получением Соединения I-118. MS (ESI) m/z: 223 (М+Н+).
Получение Соединения I-119: К раствору Соединения I-118 (300 мг, 1,36 ммоль) в NMP (5 мл) добавляли 1,4-диокса-8-азаспиро[4.5]декана гидрохлорид (487 мг, 2,71 ммоль) и карбонат цезия (1,77 г, 5,42 ммоль). Затем смесь нагревали и перемешивали при 140°С в течение 17 ч, после чего смесь охлаждали до RT. Затем реакционную смесь разбавляли водой (20 мл) и экстрагировали EtOAc (50 мл ×5). Объединенные органические слои промывали водой (50 мл ×4), рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, элюирование смесью петролейный эфир/этилацетат = от 15:1 до 10:1) с получением Соединения I-119. MS (ESI) m/z: 328 (М+Н+).
Получение Соединения I-120: Смесь Соединения I-119 (200 мг, 0,61 ммоль) и р-TosOH (14 мг, 0,075 ммоль) в ацетоне (2 мл) и воде (1 мл) перемешивали при 100°С в течение 2,5 ч под действием микроволнового излучения. Затем реакционную смесь охлаждали до RT, выливали в насыщенный водный раствор NaHCO3 (15 мл) и экстрагировали EtOAc (40 мл ×2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением Соединения I-120, которое использовали на следующей стадии без дополнительной очистки. MS (ESI) m/z: 284 (М+Н+).
Получение Соединения по примеру 32: К раствору Соединения I-120 (140 мг, 0,49 ммоль) в этаноле (3 мл) и воде (2 мл) добавляли KCN (48 мг, 0,74 ммоль) и (NHO2CO3 (474 мг, 5,0 ммоль). Затем реакционную смесь нагревали до 90°С в течение 6 ч, после чего охлаждали до RT. Реакционную смесь затем выливали в воду (20 мл). Осажденное твердое вещество собирали путем фильтрации и промывали водой (10 мл ×2). Твердое вещество затем очищали обращенно-фазовой HPLC (Xtimate С18, 150×25 мм ×5 мкм, элюирование водой (10 мМ NH4HCO3)CH3CN) с получением Соединения по примеру Е32. 1Н NMR (400 MHz, DMSO-d6) δ 10.75 (br s, 1H), 8.59 (s, 1H), 8.43 (s, 1H), 7.51 (s, 1H), 6.31 (s, 1H), 3.41 (br d, J=12.5 Hz, 2H,) 3.10 (br t, J=11.2 Hz, 2H), 2.10-2.22 (m, 2H), 1.76 (br d, J=13.7 Hz, 2 H). MS (ESI) m/z: 354 (M+H+).
Биологические анализы
Иллюстративные соединения, раскрытые в настоящем документе, получали и тестировали для определения их эффекта в качестве ингибиторов IDO и/или TDO. Использовали два разных анализа: 1. клеточный анализ для детекции эффекта тестируемых соединений на продукцию кинуренина в двух разных типах раковых клеток. Данный анализ проводили на раковых клетках, которые экспрессировали TDO или IDO, и его использовали как таковой в качестве средства тестирования активности соединений в отношении этих двух ферментов в клеточном контексте; 2. биохимический сопряженный анализ TDO и IDO, в котором использовали полученные рекомбинантным способом и очищенные ферменты TDO и IDO в комбинации с ферментом формамидазой. Эта сопряженная ферментная система обеспечивала превращение N-формилкинуренина, продуцированного в результате активности TDO или IDO, в кинуренин, который затем определяли количественно посредством флуоресценции после добавления реактива Эрлиха. Протоколы этих анализов изложены ниже.
Анализы на основе клеток А172 и SKOV3 для детекции кинуренина. продуцируемого TDO и/или IDO
Клетки А172 (глиобластома человека) и SKOV3 (аденокарцинома яичников человека) высевали в 96-луночном планшете при плотности 30000 или 40000 клеток на лунку, соответственно, в среде RPMI, не содержащей феноловый красный, дополненной 10% FCS, 2 мМ L-глутамина и 500 мкМ L-триптофана. Экспрессию IDO индуцировали в клетках SKOV3 путем добавления 500 нг/мл IFN-γ. Клетки инкубировали при 37°С с тестируемым соединением или без добавления тестируемого соединения. Через 48 часов клетки удаляли путем центрифугирования и к супернатанту добавляли реактив Эрлиха. Реактив Эрлиха инкубировали в течение 5 минут перед считыванием поглощения при 490 нМ.
Биохимический сопряженный анализ TDO и IDO
Рекомбинантный человеческий IDO или TDO инкубировали в 50 мМ KPO4 (рН 7,0), 0,5 мМ EGTA, 0,5 мМ EDTA, 0,05 % Triton™ Х100, 20 мМ аскорбата, 10 мкМ метиленового синего, 500 Ед/мл каталазы, 50 мкг/мл KynB (кинуренин формамидаза). Анализы TDO проводили в присутствии 330 мкМ L-триптофана, тогда как анализ IDO проводили в присутствии 45 мкМ L-триптофана. После инкубации в течение 17 минут при комнатной температуре реакции останавливали добавлением реактива Эрлиха и инкубировали при комнатной температуре в течение 5 минут перед считыванием флуоресценции.
Значения pIC50 для различных тестируемых соединений показаны в следующей таблице.
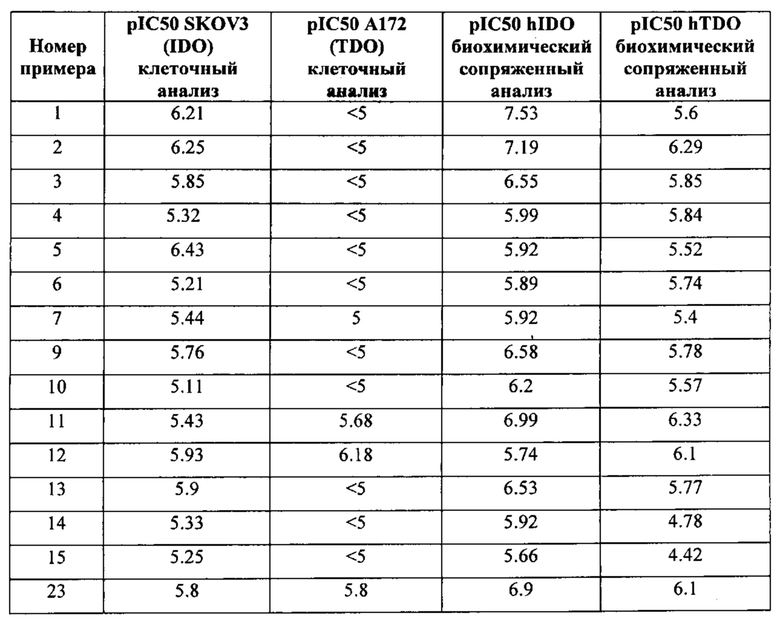
Дополнительно к описанным выше анализам определяли активности некоторых иллюстративных соединений с использованием последующего ферментного анализа IDO1 и клеточного анализа IDO1.
Ферментный анализ IDO1
Соединения, подлежащие тестированию, серийно разбавляли полулогарифмическими шагами с помощью DMSO, начиная с 10 мМ исходных растворов соединений в DMSO. Затем разведения соединений или только DMSO переносили из планшета для разведения в черный 384-луночный аналитический планшет Greiner (номер по каталогу 781086) с использованием устройства Echo 555 Acoustic Liquid Handler (Labcyte).
HIS-меченый белок IDO1 рекомбинантно экспрессировали в клетках Escherichia coli с использованием среды аутоиндукции ZYP5052, дополненной 500 мкМ дельта-аминолевулиновой кислоты, в течение 48 часов при Белок IDO1 очищали аффинной хроматографией на смоле с заряженными ионами Ni2+ и эксклюзионной хроматографией. Очищенный белок затем разводили в аналитическом буфере (50 мМ Tris, рН 7,0, 1% глицерин, 20 мкМ метиленового синего, 0,05% Tween-20, 20 мМ аскорбата натрия, 100 ед/мл каталазы) для получения конечной концентрации IDO1, равной 40 нМ. Раствор IDO1 (30 мкМ) или только буфер (30 мкМ) вносили в лунки аналитического планшета с использованием дозатора для жидкостей BioRAPTR (Beckman Coulter). Аналитические планшеты, содержащие соединение и фермент IDO1, инкубировали при комнатной температуре в течение 30 минут. После этого в каждую лунку аналитического планшета вносили 10 мкл 400 мкМ триптофана с использованием дозатора для жидкостей BioRAPTR. Планшеты инкубировали при комнатной температуре в течение 60 минут и реакции гасили путем добавления 10 мкл 0,5М метил изонипекотата в диметилсульфоксиде. Планшеты герметично закрывали и инкубировали при 37°С в течение 4 часов или при 50°С в течение 2 часов. Планшеты оставляли охлаждаться, а затем центрифугировали в течение 1 минуты при 1000×g. Конечную флуоресценцию измеряли на планшетном ридере Envision (Perkin Elmer) с использованием фильтра возбуждения 400/25 нм и фильтра излучения 510/20 нм.
Интенсивность флуоресценции каждой лунки корректировали на фоновое значение, наблюдаемое в лунках, в которых отсутствовал IDO1, и выражали в виде доли интенсивности, наблюдаемой в лунках, в которых присутствовал фермент IDO1 и только DMSO. Активности рассчитывали путем линейной подгонки методом наименьших квадратов четырехпараметрическому логистическому уравнению IC50.
Анализ IDO1 в клетках HEK293
Соединения, подлежащие тестированию, серийно разбавляли полулогарифмическими шагами с помощью DMSO, начиная с 10 мМ исходных растворов соединений в DMSO. Затем разведения соединений или только DMSO переносили из планшета для разведения в черный 384-луночный аналитический планшет Greiner (номер по каталогу 781086) с использованием устройства Echo 555 Acoustic Liquid Handler (Labcyte).
Клеточные осадки HEK293 ресуспендировали до 5×105 клеток/мл в полной культуральной среде для клеток HEK293 (89% DMEM, 10% FBS, 1% пенициллин/стрептомицин). Суспендированные клетки (2 мл) распределяли в каждую лунку 6-луночного планшета фирмы Corning (номер по каталогу 3516). Клетки оставляли прикрепляться и инкубировали в течение 20 часов при 37°С в инкубаторе, содержащем 5% CO2. Вектор Flag-IDO1 (Genscript True ORF Gold, 2 мкг) в 150 мкл среды Opti-MEM добавляли в каждую лунку 24-луночного планшета фирмы Corning (номер по каталогу 3527) и инкубировали в течение 5 минут при комнатной температуре. В каждую лунку 24-луночного планшета добавляли 150 мкл липофектамина 2000 (Gibco) и планшет инкубировали при комнатной температуре в течение 20-30 минут. В каждую лунку с прикрепленными клетками в 6-луночном планшете осторожно добавляли 250 мкл смеси для трансфекции из 24-луночного планшета и белок IDO1 оставляли экспрессироваться на 24-30 часов при 37°С в инкубаторе, содержащем 5% СО2.
Среду удаляли из клеток, которые затем промывали 2 мл физиологического раствора, забуференного фосфатом Дульбекко (DPBS). После удаления DPBS добавляли 0,5 мл TrypLE (Gibco) и инкубировали в течение 5 минут, пока клетки не поднимутся с поверхности лунок. В каждую лунку добавляли полную культуральную среду HEK293 (4 мл) и клетки собирали и помещали в коническую пробирку. Клетки осаждали центрифугированием при 200×g в течение 5 мин и ресуспендировали в равном объеме полной среды DMEM. Клетки разбавляли до плотности 4×105 клеток на мл в полной среде HEK293. Добавляли L-триптофан с получением конечной концентрации 200 мкМ. Разбавленные трансфицированные клетки (50 мкл) или нетрансфицированные клетки (50 мкл) распределяли в лунки черных 384-луночных аналитических планшетов Greiner (номер по каталогу 781086), содержащих ранее разведенные соединения. Планшет перемешивали в течение короткого периода времени и центрифугировали при 200×g в течение 10 секунд для сбора клеток на дне планшета. Планшеты закрывали и инкубировали в течение 20-24 часов при 37°С в инкубаторе, содержащем 5% СО2. После этого в каждую лунку добавляли 10 мкл 0,5М метил изонипекотата в диметилсульфоксиде, перемешивали, герметично закрывали и центрифугировали при 500 об/мин в течение 10 секунд. Планшеты инкубировали при 37°С в инкубаторе, содержащем 5% СО2, в течение ночи для развития флуоресценции. Планшеты оставляли охлаждаться, а затем центрифугировали в течение 1 минуты при 1000×g. Полученную флуоресценцию измеряли на планшетном ридере Envision (Perkin Elmer) с использованием фильтра возбуждения 400/25 нм и фильтра излучения 510/20 нм.
Интенсивность флуоресценции каждой лунки корректировали на фоновое значение, наблюдаемое в лунках, в которых отсутствовал IDO1, и выражали в виде доли интенсивности, наблюдаемой в лунках, в которых присутствовал фермент IDO1 и только DMSO. Активности рассчитывали путем линейной подгонки методом наименьших квадратов четырехпараметрическому логистическому уравнению IC50.
Анализ IDO1 в клетках Hela, стимулированных IFNγ
Клетки Hela культивировали в полной культуральной среде Hela (90% ЕМЕМ, 10% инактивированная нагреванием фетальная бычья сыворотка) и размножали до около 1×109 клеток. Затем клетки собирали и замораживали при 10×106 клеток/флакон в 1 мл замороженной среды (90% полная культуральная среда Hela, 10% DMSO)
Соединения, подлежащие тестированию, серийно разбавляли полулогарифмическими шагами с помощью DMSO, начиная с 10 мМ исходных растворов соединений в DMSO в планшете(ах) Echo low volume. Разведения соединений или только DMSO затем переносили из планшета(ов) для разведения в черный 384-луночный аналитический планшет(ы) Greiner (номер по каталогу 781086, 50 нл/лунка) с использованием устройства Echo 555 Acoustic Liquid Handler (Labcyte).
Замороженные клетки Hela оттаивали и переносили в аналитическую среду Hela (99% полная культуральная среда Hela, 1% пенициллин/стрептомицин) с 20 мл среды/флакон клеток. Клетки центрифугировали при 250 g в настольной центрифуге в течение 5 мин и суспендировали в таком же объеме аналитической среды Hela. Затем клетки подсчитывали и доводили до плотности 2×105 клеток/мл в аналитической среде Hela. В клетки добавляли L-триптофан, при этом конечная концентрация L-триптофана составила 300 мкМ. Маленькую аликвоту (2 мл/планшет) клеток Hela отбирали и не обрабатывали IFNγ для того, чтобы служить в качестве контроля Мах-Е. В остальные клетки Hela добавляли стерильный IFNγ (номер по каталогу 285-IF), системы R&D при конечной концентрации 100 нг/мл.
Клетки Hela с добавленным IFNγ и без него распределяли в соответствующие лунки 384-луночных аналитических планшетов, содержащих соединения. Планшеты инкубировали в течение около 48 часов при 37°С, в инкубаторе, содержащем 5% СО2. После этого в каждую лунку добавляли 12 мкл 0,5М метил изонипекотата в диметилсульфоксиде и планшеты герметично закрывали и инкубировали при 37°С без CO2 в течение ночи. Планшеты центрифугировали в течение 1 мин при 200×g. Полученную флуоресценцию измеряли на планшетном ридере Spectramax (Molecular Devices) с использованием фильтра возбуждения при 400 нм и фильтра излучения при 510 нм.
Интенсивность флуоресценции каждой лунки корректировали на фоновое значение, наблюдаемое в лунках, не содержащих клетки, обработанные IFNγ, и выражали в виде доли интенсивности, наблюдаемой в лунках, содержащих клетки, обработанные IFNγ, и только DMSO. Активности рассчитывали путем линейной подгонки методом наименьших квадратов четырехпараметрическому логистическому уравнению IC50.
Клеточный анализ TDO в замороженных клетках SW48
Клетки SW48 культивировали в полной культуральной среде RPMI (90% RPMI, 10% инактивированная нагреванием фетальная бычья сыворотка). После достижения состояния, близкого к состоянию конфлюэнтности, клетки собирали и замораживали при плотности 20×106 клеток/флакон в 1 мл замороженной среды (90% полная культуральная среда RPMI, 10% DMSO). Клетки А2780 (с минимальной активностью TDO) культивировали в полной среде RPMI и также аналогично замораживали при 5×106/флакон, чтобы использовать в качестве контроля Мах-Е.
Соединения, подлежащие тестированию, серийно разбавляли полулогарифмическими шагами с помощью DMSO, начиная с 10 мМ исходных растворов соединений в DMSO в планшете(ах) Echo low volume. Разведения соединений или только DMSO затем переносили из планшета(ов) для разведения в черный 384-луночный аналитический планшет(ы) Greiner (номер по каталогу 781086, 50 нл/лунка) с использованием устройства Echo 555 Acoustic Liquid Handler (Labcyte).
Замороженные клетки SW48 и А2780 оттаивали и переносили в полную аналитическую среду RPMI (99% полная культуральная среда RPMI, 1% пенициллин/стрептомицин) с 20 мл среды/флакон с клетками. Клетки центрифугировали при 350 g в настольной центрифуге в течение 5 минут и суспендировали в таком же объеме аналитической среды RPMI. Клетки подсчитывали и доводили до плотности 2×105 клеток/мл в аналитической среде RPMI. К клеткам добавляли стерильный L-триптофан (Sigma, номер по каталогу Т0254) при конечной концентрации 300 мкМ.
Клетки SW48 и А2780 распределяли в соответствующие лунки 384-луночных аналитических планшетов, содержащих соединения. Планшеты инкубировали в течение около 48 часов при 37°С в инкубаторе, содержащем 5% СО2. После этого в каждую лунку добавляли 12 мкл 0,5 М этил изонипекотата (Sigma Aldrich, номер по каталогу Е33505) в диметилсульфоксиде, и планшеты герметично закрывали и инкубировали при 37°С без СО2 в течение ночи. Планшеты центрифугировали в течение 1 минуты при 200×g. Полученную флуоресценцию измеряли на планшетном ридере Spectramax (Molecular Devices) с использованием фильтра возбуждения при 400 нм и фильтра излучения при 510 нм.
Интенсивность флуоресценции каждой лунки корректировали на фоновое значение, наблюдаемое в лунках с клетками А2780, и выражали в виде доли интенсивности, наблюдаемой в лунках, содержащих клетки SW48 и только DMSO. Активности рассчитывали путем линейной подгонки методом наименьших квадратов логистическому уравнению IC50 с четырьмя параметрами.
Значения pIC50 для различных тестируемых соединений показаны в следующей таблице.
Как видно из указанных выше данных по активности, соединения, раскрытые в настоящем документе, являются ингибиторами ферментов IDO и/или TDO.
Анализ IDO1 в цельной крови человека
Соединения, подлежащие тестированию, серийно разбавляли полулогарифмическими шагами с помощью DMSO, начиная с 10 мМ. Три мл разведений соединений или только DMSO затем переносили из планшета для разведения в полипропиленовый 96-луночный аналитический планшет, содержащий 97 мкл среды RPMI с использованием устройства Echo 555 Acoustic Liquid Handler (Labcyte). LPS и IFNγ готовили в RPMI до 10X конечной концентрации (1000 нг/мл), при этом конечная концентрация составила 100 нг/мл.
Цельную кровь человека собирали в покрытые гепарином натрия пробирки у здоровых внутренних доноров. В каждую из лунок 96-луночного планшета с V-образным дном вносили 240 мкл крови. Из промежуточного планшета для разведения переносили 30 мкл соединения и инкубировали в течение 15 мин. Затем 30 мкл от стимуляторов переносили в кровь и тщательно перемешивали. Планшет закрывали воздухопроницаемой мембраной и инкубировали при 37°С в течение ночи (18 ч).
На день 2 изотопно-меченный стандарт кинуренин и триптофан готовили в воде при 10× концентрации и 30 мкл добавляли к крови при конечной концентрации 3 мкМ. Аналитические планшеты центрифугировали при 300×g в течение 10 мин без остановки для отделения плазмы от эритроцитов. Шестьдесят мкл образцов плазмы удаляли без затрагивания эритроцитов. Плазму разбавляли RPMI в соотношении 1:1 и белки осаждали двумя объемами ацетонитрила. Планшеты центрифугировали при 4000×G в течение 60 мин. Двадцать мкл супернатанта осторожно переносили в 384-луночный планшет, содержащий 40 мкл 0,1% муравьиной кислоты в воде, и анализировали методом LC/MS/MS.
Анализы методом LC/MS/MS (жидкостная хроматография/масс-спектрометрия) выполняли с использованием системы Thermo Fisher's LX4-TSQ Quantum Ultra. Эта система состоит из четырех бинарных насосов Agilent для высокоэффективной жидкостной хроматографии (HPLC) и тройного квадрупольного анализатора MS/MS TSQ Quantum Ultra. Для каждого образца 5 мкл вводили на колонку Atlantis Т3 (2,1 мм × 150 мм, размер частиц 3 мкм) фирмы Waters. Градиент подвижной фазы, подаваемой со скоростью 0,8 мл/мин, использовали для элюирования аналитов из колонки при 25°С. Элюирование начинали при 0% В, увеличивали линейно до 25% В до 6,5 мин, удерживали при 25% в течение 1 мин, повторно уравновешивали до 10 мин. Подвижная фаза А состояла из 0,1% раствора муравьиной кислоты в воде. Подвижная фаза В состояла из 0,1% раствора муравьиной кислоты в ацетонитриле. Данные записывали в режиме регистрации положительно заряженных ионов с использованием интерфейса HESI (электрораспылительной ионизации с нагреваемым потоком). Рабочими параметрами для прибора TSQ Quantum Ultra являлись напряжение при распылении 4000 В, температура капилляров 380°С, температура испарителя 400°С, экранирующий газ 60 условных единиц, Aux (вспомогательный) газ 20 условных единиц, линзы трубки 85 и газ для соударений 1,2 mTorr. SRM-хроматограммы кинуренина (Q1:209.2 > Q3:94.0) и внутреннего стандарта (Q1:215.3 > Q3:98.2) записывали в течение 90 сек. Площадь пика интегрировали с помощью программного обеспечения Xcalibur Quan. Соотношения между кинуренином, образовавшимся в ходе реакции, и 2D6-кинуренином, добавленным внутренним стандартом, использовали для получения процентного ингибирования и значений IC50. Соединения титровали, и значения IC50 рассчитывали с помощью формулы подгонки сигмоидальной кривой с 4 параметрами.
Данные по биологической активности выбранных соединений с использованием анализа IDO1 в цельной крови человека, описанного выше, показаны в обобщенном виде в таблице ниже.

Несмотря на то, что изобретение описано и проиллюстрировано с обращением на некоторые конкретные варианты его осуществления, специалистам в данной области будет понятно, что различные адаптации, изменения, модификации, замещения, делеции или добавления к процедурам и протоколам могут быть сделаны без отступления от сущности и объема изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| АЗАСПИРОПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ TRPM8 | 2015 |
|
RU2683309C2 |
| ПРОИЗВОДНОЕ ГИДАНТОИНА | 2013 |
|
RU2678984C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ МЕТАЛЛОФЕРМЕНТЫ | 2017 |
|
RU2764666C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ N-ГИДРОКСИКАРБАМИМИДОИЛ-1,2,5-ОКСАДИАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2765371C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОИЗВОДНЫЕ ГИДАНТОИНА | 2014 |
|
RU2701168C2 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
Изобретение относится к соединения имидазопиридина формулы (I),
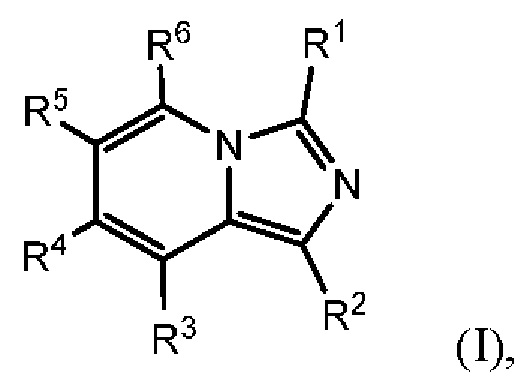
где: каждый из R1 и R2 представляет собой Н; один из R3 и R6 представляет собой Н, а другой представляет собой Y1; каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) C1-6-алкила, необязательно замещенного одним-тремя атомами галогена, и (4) C1-6-алкокси, необязательно замещенного одним-тремя атомами галогена; Y1 представляет собой группу, имеющую следующую формулу:
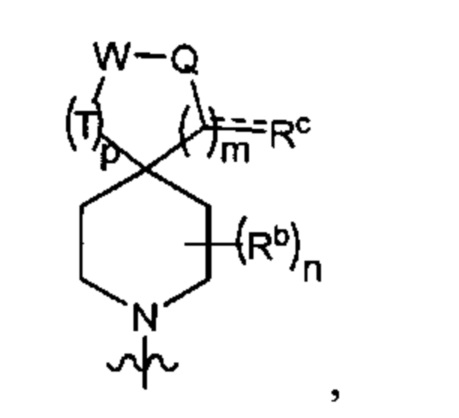
пунктирная линия  представляет собой необязательную двойную связь; Q представляет собой -C(Ra)(Ra')-, -N(Ra)- или -О-; Т представляет собой -C(Ra)(Ra')- или -N(Ra)-; Ra выбран из группы, состоящей из (1) Н, (2) C1-6-алкила, и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода; Ra' выбран из группы, состоящей из (1) Н и (2) C1-6-алкила; Rb представляет собой C1-6-алкил; каждый из Rc и Rd независимо выбран из группы, состоящей из (1) Н, (2) С1-6-алкила, и (3) оксо; m равен 0, 1 или 2; n равен 0, 1 или 2; и р равен 0 или 1, которые являются ингибиторами ферментов индоламина 2,3-диоксигеназы (IDO) и/или триптофан-2,3-диоксигеназы (TDO); также раскрыто применение соединений в потенциальном лечении или предупреждении развития IDO- и/или TDO-ассоциированного заболевания или нарушения, композиции, содержащие эти соединения, и применение соединений в потенциальном лечении или предупреждении развития IDO- и/или TDO-ассоциированного заболевания или нарушения. 6 н. и 11 з.п. ф-лы, 32 пр., 6 табл.
представляет собой необязательную двойную связь; Q представляет собой -C(Ra)(Ra')-, -N(Ra)- или -О-; Т представляет собой -C(Ra)(Ra')- или -N(Ra)-; Ra выбран из группы, состоящей из (1) Н, (2) C1-6-алкила, и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода; Ra' выбран из группы, состоящей из (1) Н и (2) C1-6-алкила; Rb представляет собой C1-6-алкил; каждый из Rc и Rd независимо выбран из группы, состоящей из (1) Н, (2) С1-6-алкила, и (3) оксо; m равен 0, 1 или 2; n равен 0, 1 или 2; и р равен 0 или 1, которые являются ингибиторами ферментов индоламина 2,3-диоксигеназы (IDO) и/или триптофан-2,3-диоксигеназы (TDO); также раскрыто применение соединений в потенциальном лечении или предупреждении развития IDO- и/или TDO-ассоциированного заболевания или нарушения, композиции, содержащие эти соединения, и применение соединений в потенциальном лечении или предупреждении развития IDO- и/или TDO-ассоциированного заболевания или нарушения. 6 н. и 11 з.п. ф-лы, 32 пр., 6 табл.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
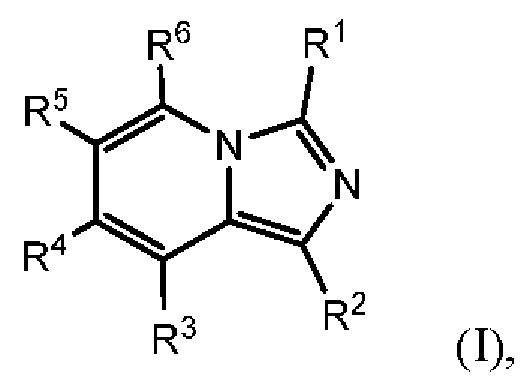
где:
каждый из R1 и R2 представляет собой Н;
один из R3 и R6 представляет собой Н, а другой представляет собой Y1;
каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) C1-6-алкила, необязательно замещенного одним-тремя атомами галогена, и (4) C1-6-алкокси, необязательно замещенного одним-тремя атомами галогена;
Y1 представляет собой группу, имеющую следующую формулу:
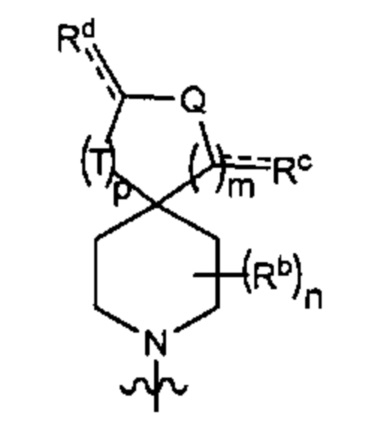
пунктирная линия  представляет собой необязательную двойную связь;
представляет собой необязательную двойную связь;
Q представляет собой -C(Ra)(Ra')-, -N(Ra)- или -О-;
Т представляет собой -C(Ra)(Ra')- или -N(Ra)-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-6-алкила, и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода;
Ra' выбран из группы, состоящей из (1) Н и (2) C1-6-алкила;
Rb представляет собой C1-6-алкил;
каждый из Rc и Rd независимо выбран из группы, состоящей из (1) Н, (2) С1-6-алкила, и (3) оксо;
m равен 0, 1 или 2;
n равен 0, 1 или 2; и
р равен 0 или 1.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) C1-4-алкила, необязательно замещенного одним-тремя атомами галогена.
3. Соединение по любому из пп. 1, 2 или его фармацевтически приемлемая соль, отличающееся тем, что каждый из R4 и R5 независимо выбран из группы, состоящей из (1) Н, (2) галогена, (3) -CF3; и m равен 1.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, отличающееся тем, что
R3 представляет собой Н;
R6 представляет собой Y2, имеющий следующую формулу:
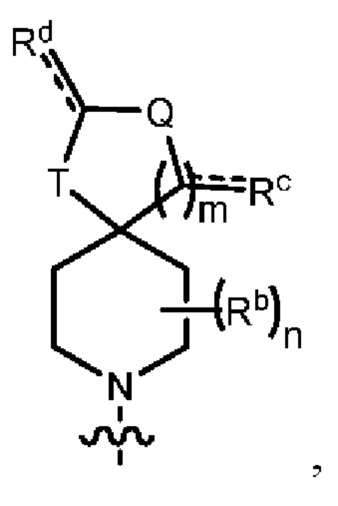
где
пунктирная линия  представляет необязательную двойную связь;
представляет необязательную двойную связь;
Q представляет собой -CH(Ra)- или -N(Ra)-;
Т представляет собой -СН2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) С1-6-алкила и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода;
Rb представляет собой C1-4-алкил;
каждый из Rc и Rd независимо представляет собой Н или оксо; и
m равен 1.
5. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль, отличающееся тем, что:
R3 представляет собой Н;
R6 представляет собой Y3, имеющий следующую формулу:
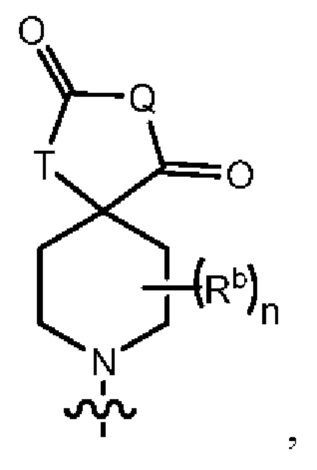
где
Q представляет собой -N(Ra)-;
Т представляет собой -СН2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-6-алкила, необязательно замещенного одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой атом кислорода, и (3) фенила;
Rb представляет собой метил или этил; и
n равен 0, 1 или 2.
6. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, отличающееся тем, что Y1 выбран из группы, состоящей из:
7. Соединение по п. 1, имеющее формулу (Ia), или его фармацевтически приемлемая соль:
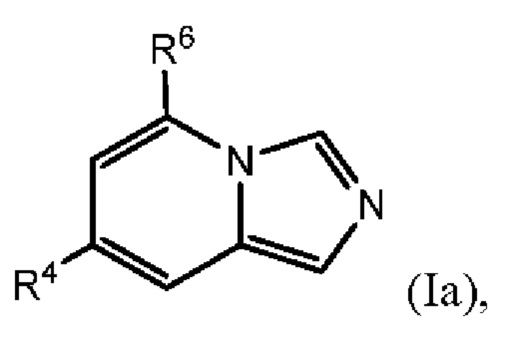
где:
R4 выбран из группы, состоящей из (1) галогена и (2) C1-4-алкила, необязательно замещенного одним-тремя атомами галогена;
R6 представляет собой Y2, имеющий следующую формулу:
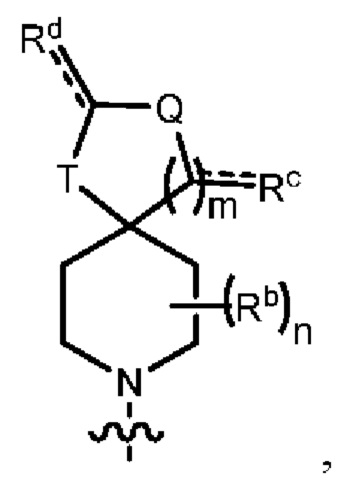
где
пунктирная линия  представляет необязательную двойную связь;
представляет необязательную двойную связь;
Q представляет собой -CH(Ra)- или -N(Ra)-;
Т представляет собой -СН2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-4-алкила и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода;
Rb представляет собой C1-4-алкил;
каждый из Rc и Rd независимо представляет собой Н или оксо;
m равен 1; и
n равен 0, 1 или 2.
8. Соединение по п. 7 или его фармацевтически приемлемая соль, отличающееся тем, что R6 представляет собой Y3, имеющий следующую формулу:
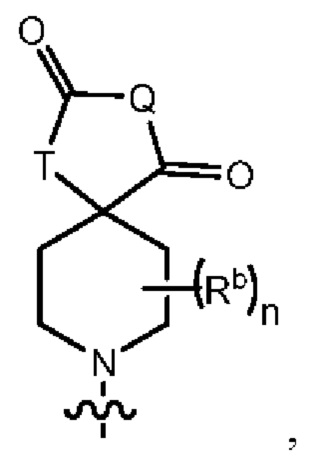
где
Q представляет собой -N(Ra)-;
Т представляет собой -СН2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) C1-4-алкила, необязательно замещенного одним заместителем, независимо выбранным из галогена и тетрагидропиранила, и (3) фенила; и
Rb представляет собой метил.
9. Соединение по п. 1, имеющее формулу (Ib), или его фармацевтически приемлемая соль:
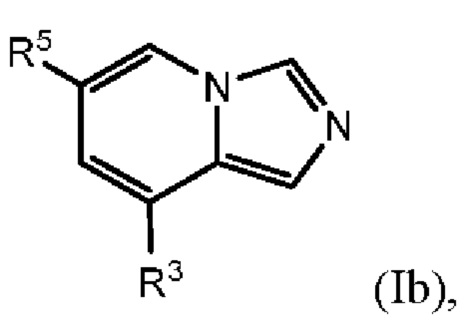
R5 выбран из группы, состоящей из (1) галогена и (2) C1-4-алкила, необязательно замещенного одним-тремя атомами галогена;
R3 представляет собой Y2, имеющий следующую формулу:
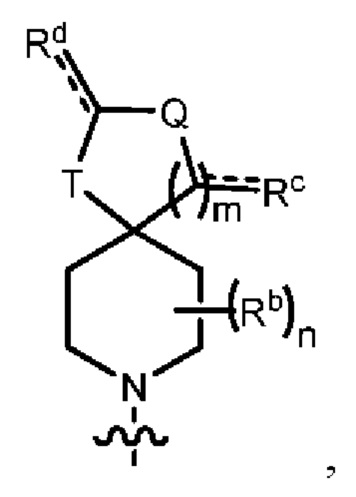
где
пунктирная линия  представляет необязательную двойную связь;
представляет необязательную двойную связь;
Q представляет собой -CH(Ra)- или -N(Ra)-;
Т представляет собой -СН2- или -NH-;
Ra выбран из группы, состоящей из (1) Н, (2) С1-6-алкила и (3) фенила; при этом каждый из алкила и фенила является необязательно замещенным одним заместителем, независимо выбранным из галогена и 6-членной гетеромоноциклической группы, содержащей один кольцевой гетероатом, выбранный из кислорода;
Rb представляет собой C1-4-алкил;
каждый из Rc и Rd независимо представляет собой Н или оксо;
m равен 1; и
n равен 0, 1 или 2.
10. Соединение по п. 9 или его фармацевтически приемлемая соль, отличающееся тем, что R3 представляет собой Y3, имеющий следующую формулу:
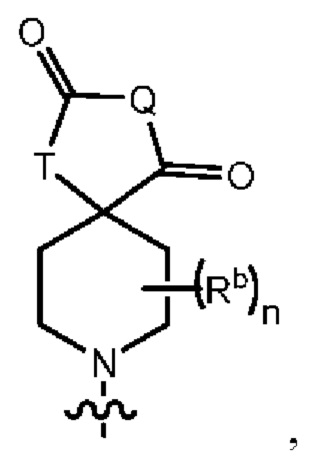
где
Q представляет собой -N(Ra)-;
Т представляет собой -CH2- или NH;
Ra выбран из группы, состоящей из (1) Н, (2) C1-4-алкила, необязательно замещенного одним заместителем, независимо выбранным из галогена и тетрагидропиранила, и (3) фенила; и
Rb представляет собой метил.
11. Соединение, выбранное из группы, состоящей из следующих соединений:
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(6-(трифторметил)имидазо[1,5-а]пиридин-8-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
3-метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
3-((тетрагидро-2Н-пиран-4-ил)метил)-8-(7-(трифторметил)имидазо[1,5-а]-пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
3-фенил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
3,6,6-триметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро [4,5] декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
6,6-диметил-8-(6-(трифторметил)имидазо[1,5-а]пиридин-8-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
6,6-диметил-3-((тетрагидро-2Н-пиран-4-ил)метил)-8-(7-(трифторметил)имидазо-[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
6,6-диметил-3-фенил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]-декан-2,4-дион,
8-(7-(хлор)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2-окса-8-азаспиро[4,5]декан-1-он,
2-метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4.5]дек-1-ен-4-он,
1-(9-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-3,9-диазаспиро[5.5]-ундекан-3-ил)этан-1-он,
8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4.5]декан-1-он,
6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро-[4,5]декан-2,4-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-1,3,8-триазаспиро[4,5]- декан-2,4-дион,
8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-2-тиа-1,3,8-триазаспиро[4,5]декан-4-он-2,2-диоксид,
(R)- или (S)-8-(7-бромимидазо[1,5-а]пиридин-5-ил)-6,6-диметил-2,8-диазаспиро-[4,5]декан-1,3-дион,
(S)- или (R)-6,6-диметил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
(R)- или (S)-8-(7-хлоримидазо[1,5-а]пиридин-5-ил)-6,6-диметил-2,8-диазаспиро-[4,5]декан-1,3-дион,
(R)- или (S)-4-метил-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
6,6-дифтор-8-(7-(трифторметил)имидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро-[4,5]декан-1,3-дион,
8-(7-бромимидазо[1,5-а]пиридин-5-ил)-2,8-диазаспиро[4,5]декан-1,3-дион,
8-(7-метоксиимидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион и
8-(7,8-михлоримидазо[1,5-а]пиридин-5-ил)-1,3,8-триазаспиро[4,5]декан-2,4-дион;
или их фармацевтически приемлемая соль.
12. Фармацевтическая композиция ингибирующая фермент(ы) IDO и/или TDO, содержащая эффективное количество соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
13. Применение соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предупреждения развития IDO- и/или TDO-ассоциированного заболевания или нарушения.
14. Способ лечения или предупреждения IDO- и/или TDO-ассоциированного заболевания или нарушения у субъекта-млекопитающего, который включает введение субъекту эффективного количества соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли.
15. Способ по п. 14, отличающийся тем, что IDO- и/или TDO-ассоциированное заболевание или нарушение представляет собой рак, вирусную инфекцию, HCV-инфекцию, депрессию, нейродегенеративные нарушения, травму, возрастные катаракты, трансплантацию органа и аутоиммунные заболевания.
16. Способ по п. 15, отличающийся тем, что IDO- и/или TDO-ассоциированное заболевание или нарушение представляет собой рак, выбранный из рака толстой кишки, рака поджелудочной железы, рака молочной железы, рака предстательной железы, рака легких, рака головного мозга, рака яичника, рака шейки матки, рака яичка, рака почек, рака головы и шеи, лимфомы, лейкоза и меланомы.
17. Применение соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли в качестве ингибитора фермента(ов) IDO и/или TDO.
| WO 2014159248 A1 02.10.2014 | |||
| WO 2015181532 A1 03.12.2015 | |||
| US 20080234313 A1 25.09.2008 | |||
| Устройство для подогрева питательной воды | 1930 |
|
SU22669A1 |
| Червячные винты для раскладочных, ленточных и т.п. машин | 1932 |
|
SU28424A1 |

