Описание
Перекрестная ссылка на родственные заявки
Настоящее изобретение заявляет приоритет согласно 35 U.S.C. § 119(e) по предварительной заявке на патент США № 62/440,181, поданный 29 декабря 2016. Которая включена в настоящую заявку в полном объеме посредством ссылки.
Уровень техники
Альдостерон представляет собой стероидный гормон, выделяемый из надпочечников, который связывает и активирует минералокортикоидный рецептор (MR). В первичных клетках дистальных канальцев и собирающих протоках почки активация МР приводит к задержке натрия и воды с выделением калия, что приводит к увеличению объема плазмы, приводящему к повышению артериального давления (АД). Избыток альдостерона, измеренный в кровотоке, называется первичным альдостеронизмом (PA) и возникает, когда выработка альдостерона не регулируется системой ренин-ангиотензин-альдостерон (RAAS). PA изначально был выявлен у пациентов с аденомой надпочечников, при этом последние данные свидетельствуют о повышении распространенности, связанной с ожирением. PA является частой причиной вторичной гипертонии с распространенностью PA в диапазоне от 14 до 21% у пациентов с резистентной артериальной гипертонией (RHTN), состояние, определенное как АД, остающееся выше целевого уровня, несмотря на одновременное использование 3 антигипертензивных препаратов различных классов, включая мочегонное средство. Недавние исследования показали связь между избытком альдостерона, RHTN и синдромом остановки дыхания во сне (OSA), который усугубляется опосредованной альдостероном задержкой жидкости.
Локальная сверхпродукция альдостерона была отмечена при нескольких тяжелых болезненных состояниях, даже когда не наблюдается значительного повышения в плазме. У пациентов с хронической сердечной недостаточностью с застойными явлениями (ХСН) уровень альдостерона в поврежденной сердечной ткани выше, чем в периферической плазме. На животных моделях заболеваний почек постулируется локальная продукция альдостерона в корковом веществе почки, способствуя развитию заболевания. В обоих этих состояниях локальные повышенные уровни альдостерона способствуют вредным эффектам посредством как MR-зависимых, так и MR-независимых механизмов, включая генерацию реакционноспсособных соединений кислорода и эндотелиальную дисфункцию, ведущую к воспалению и стимуляции роста и пролиферации клеток, при этом повышающее регулирование отложения коллагена приводит к фиброзу.
Антагонисты MR, включая спиронолактон и эплеренон, широко используются для блокирования эффектов связывания альдостерона с MR. Значительное снижение заболеваемости и смертности у пациентов с сердечной недостаточностью или инфарктом миокарда было продемонстрировано этими препаратами в сочетании с ингибиторами ангиотензин-превращающего фермента (ACE) и диуретиками (исследования RALES & EPHESUS). Побочные эффекты, включая гиперкалиемию, наблюдаются у обоих агентов с неселективным спиронолактоном, также вызывающим гинекомастию через неселективную модуляцию рецепторов прогестерона и андрогена. Кроме того, повышение ренина и альдостерона является результатом антагонизма MR и, следовательно, MR-независимые (не геномные) эффекты альдостерона усугубляются.
В отличие от антагонистов MR, ингибирование CYP11B2 (альдостеронсинтазы), ключевого фермента в биосинтезе альдостерона, должно давать положительные эффекты антагонизма MR без вредного накопления альдостерона, приводящего к активации MR-независимых воспалительных и фиброзных состояний. CYP11B2 представляет собой фермент митохондриального цитохрома P450, который превращает 11-деоксикортикостерон в альдостерон. Избирательное ингибирование CYP11B2 представляет собой перспективное лечение заболеваний, связанных с альдостероном.
Высоко гомологичный металлофермент CYP11B1 (11-b-стероид-гидроксилаза) катализирует образование первичного глюкокортикоидного кортизола из 11-деоксикортизола. Учитывая высокую степень гомологии между CYP11B2 и CYP11B1 (93%), разработка селективных ингибиторов CYP11B2 была серьезной проблемой. Ингибитор Osilodrostat (LCI-699) был разработан как ингибитор CYP11B2 для лечения гипертонии, но был отменен из-за его сильного ингибирования CYP11B1. Селективные соединения, которые блокируют продукцию альдостерона через CYP11B2, без ингибирования продукции кортизола через CYP11B1, раскрываются в настоящей заявке.
Живые организмы имеют развитые глубоко регулируемые процессы, которые специфически импортируют металлы, переносят их во внутриклеточные сайты хранения и, в результате, переносят их в сайты применения. Одной из наиболее полезной функций металлов, таких как цинк и железо, в биологических системах является способность активировать металлоферменты. Металлоферментами являются ферменты, которые включают металлические ионы в активном сайте фермента и используют металл как часть каталитического процесса. Более трети всех охарактеризованных ферментов составляют металлоферменты.
Функция металлоферментов сильно зависит от присутствия иона металла в активном сайте фермента. Хорошо известно, что агенты, которые связываются с металлическим ионом активного сайта и инактивируют его, критическим образом понижают активность фермента. В природе действует та же самая стратегия понижения активности определенных металлоферментов в ходе периодов, когда ферментативная активность нежелательна. Например, белок TIMP (тканевый ингибитор металлопротеаз) связывается с ионом цинка в активном сайте различных матриксных ферментов металлопротеаз и, таким образом, затормаживает ферментативную активность. В фармацевтической промышленности применялась та же самая стратегия при создании терапевтических агентов. Например, азольные противогрибковые агенты флуконазол и вориконазол содержат 1-(1,2,4-триазольную) группу, которая связывается с гем-железом, присутствующим в активном сайте целевого фермента ланостерол деметилазы и, таким образом, инактивирует фермент. Другим примером является связывающая цинк группа гидроксаминовой кислоты, которая была включена в наиболее известные ингибиторы матриксных металлопротеиназ и гистондеацетилаз. Другим примером является связывающая цинк группа карбоновой кислоты, которая была включена в наиболее известные ингибиторы ангиотензин-превращающих ферментов.
При создании клинически безопасных и эффективных ингибиторов металлоферментов желательно применение подходящих металл-связывающей группы для конкретной цели и клинической индикации. Если применяется слабо связывающая металл-связывающая группа, эффективность может быть субоптимальной. С другой стороны, если применяется очень сильно связывающая металл-связывающая группа, селективность для целевого фермента по отношению к родственным металлоферментам может быть субоптимальной. Отсутствие оптимальной селективности может быть причиной клинической токсичности из-за непреднамеренного ингибирования этих нецелевых металлоферментов. Одним примером такой клинической токсичности является непреднамеренное ингибирование ферментов человека, метабилизирующих лекарственные средства, таких как P450 2C9 (CYP2C9), CYP2C19 и CYP3A4, доступными в настоящее время азольным противогрибковыми средствами, такими как флуконазол и вориконазол. Предполагают, что такое нецелевое ингибирование вызвано, прежде всего, неселективным связыванием применяемого в настоящее время 1-(1,2,4-триазола) с железом в активном сайте CYP2C9, CYP2C19 и CYP3A4. Другим примером этого является суставная боль, которая наблюдалась при многих клинических испытаниях ингибиторов матриксных металлопротеиназ. Эта токсичность рассматривается как связанная с ингибированием нецелевых металлоферментов из-за неселективного связывания группы гидроксаминовой кислоты с цинком в нецелевых активных сайтах.
Поэтому поиск металл-связывающих групп, которые могут достигнуть более хорошего баланса между эффективностью и селективностью, остается важной целью и был бы существенным для создания терапевтических средств и способов, для того чтобы удовлетворить неудовлетворенные в настоящее время потребности в лечении и профилактики заболеваний, нарушений и их симптомов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на соединения (например, любое из приведенных в настоящей заявке; любой из формул, приведенных в настоящей заявке), способы модуляции активности металлоферментов и способы лечения заболеваний, нарушений или их симптомов. Способы могут основываться на соединениях, приведенных в описании настоящего изобретения.
Очевидно, что варианты выполнения настоящего изобретения, раскрытые далее в отношении предпочтительных выборов переменных, могут рассматриваться сами по себе или в комбинации с одним или более вариантами выполнения настоящего изобретения, или в отношении предпочтительных выборов переменных, как если бы каждая комбинация была явно указана в настоящей заявке.
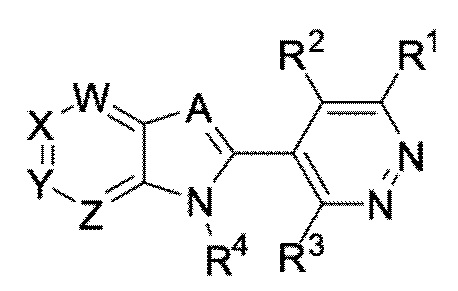
Одним объектом настоящего изобретения являются соединения формулы I:

I,
или фармацевтически приемлемые соли, сокристаллы, таутомеры, стереоизомеры, сольваты, гидраты, полиморфы, изотопически обогащенные производные или их пролекарства, где:
A представляет собой N или CR5;
W представляет собой N или CR6;
X представляет собой N или CR6;
Y представляет собой N или CR6;
Z представляет собой N или CR6;
при условии, что не более чем два из W, X, Y, и Z представляют собой N;
R1 представляет собой водород, галоген, циано, ацил, алкил, алкенил, алкинил, галоалкил, алкокси, галоалкокси, циклоалкил, циклоалкокси, арил, арилалкил, гетероарил, гетероарилалкил, гетероциклоалкил, гетероциклоалкилалкил, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, S(O)Rd, S(O)2Rd, CH2ORf, или CReRfOH, где любой R1 может быть необязательно замещен 1-3 независимыми заместителями R7;
R2 представляет собой водород, галоген, циано, алкил, или галоалкил;
или R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное, гетероциклоалкильное или циклоалкильное кольцо;
R3 представляет собой водород, галоген, циано, ацил, алкил, алкенил, алкинил, галоалкил, алкокси, галоалкокси, циклоалкил, циклоалкокси, арил, арилалкил, гетероарил, гетероарилалкил, гетероциклоалкил, гетероциклоалкилалкил, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH;
R4 представляет собой алкил, циклоалкил, галоалкил, или гетероалкил;
R5 представляет собой водород, алкил, галоалкил, гетероалкил, или циклоалкил;
в каждом случае R6 независимо представляет собой водород, галоген, циано, галоалкил, алкил, циклоалкил, алкокси, галоалкил, или карбоксил;
в каждом случае R7 независимо представляет собой галоген, алкил, алкокси, галоалкил, карбоксил, арил, арил, замещенный 1-3 независимыми заместителями галоген, -(CH2)nC(O)NRgRh, -S(O)2Ri, -CO2Rj, или NRgRh;
каждый n независимо представляет собой 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10;
в каждом случае Ra, Rb, Rc, Rd, Re, и Rf независимо представляет собой водород, ацил, алкоксиалкил, алкил, алкенил, алкинил, гетероалкил, галоалкил, циклоалкил, гетероциклоалкил, арил, гетероарил, C(O)OC1-6 алкил, C(O)C1-6 алкил, защитную группу азота, которая присоединена к атому азота, или защитную группу кислорода, которая присоединена к атому кислорода; или Ra и Rb вместе с атомами, к которым они присоединены, образуют гетероциклоалкильное кольцо; или Re и Rf вместе с атомами, к которым они присоединены, образуют циклоалкильное кольцо; и
в каждом случае Rg, Rh, Ri, и Rj независимо представляет собой водород, ацил, алкил, алкенил, алкинил, гетероалкил, галоалкил, циклоалкил, гетероциклоалкил, арил, гетероарил, C(O)OC1-6 алкил, C(O)C1-6 алкил, или Rg и Rh вместе с атомами, к которым они присоединены, образуют гетероциклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой арил, гетероарил, или гетероциклоалкил, где любой R1 может быть необязательно замещен 1-3 независимыми заместителями R7.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой арил, гетероарил, или гетероциклоалкил, где любой R1 может быть необязательно замещен 1-3 независимыми заместителями R7, и каждый R7 независимо представляет собой галоген, алкил, алкокси, галоалкил, карбоксил, -(CH2)nC(O)NRgRh, -S(O)2Ri, -CO2Rj, или NRgRh, и каждый Rg и Rh независимо представляет собой водород, алкил, C(O)C1-6 алкил, или Rg и вместе с атомами, к которым они присоединены, образуют гетероциклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород, галоген, циано, алкил, алкенил, алкинил, галоалкил, алкокси, галоалкокси, циклоалкил, циклоалкокси, арил, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CORf, или CReRfOH; или R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород, галоген, алкил, алкенил, галоалкил, алкокси, галоалкокси, циклоалкокси, арил, NRaRb, CH2NHSO2Rd, или CReRfOH; или R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород, галоген, алкил, галоалкил, CH2NHSO2Rd, или NRaRb; и Ra, Rb, и Rd независимо представляют собой водород, алкил, или галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой алкил или галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-6 алкил или C1-6 галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-6 галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой фторалкил. В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-6 фторалкил. В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-3 фторалкил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой дифторметил или трифторметил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой дифторметил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой трифторметил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород или алкил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород или C1-6 алкил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород или C1-3 алкил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой алкил. В определенных вариантах выполнения настоящего изобретения, R2 независимо представляет собой C1-6 алкил. В определенных вариантах выполнения настоящего изобретения, R2 независимо представляет собой C1-3 алкил. В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород и R2 представляет собой алкил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород и R2 независимо представляет собой C1-6 алкил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород и R2 независимо представляет собой C1-3 алкил.
В определенных вариантах выполнения настоящего изобретения, R4 представляет собой алкил или циклоалкил.
В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил.
В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой C1-4 алкил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой C3-5 циклоалкил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой циклопентил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой циклобутил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой циклопропил.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, галоген, циано, алкокси, галоалкил, или карбоксил.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, галоген, или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, хлор, фтор, или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, фтор, или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, или галоген.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, хлор, или фтор.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород или фтор.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой галоген. В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой хлор или фтор. В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой фтор. В определенных вариантах выполнения настоящего изобретения, каждый R6 представляет собой циано.
В определенных вариантах выполнения настоящего изобретения, A независимо представляет собой CR5 и R5 представляет собой водород, C1-4 алкил, или C3-5 циклоалкил.
В определенных вариантах выполнения настоящего изобретения, A представляет собой N.
В определенных вариантах выполнения настоящего изобретения, не более чем один из W, X, Y, и Z представляет собой N.
В определенных вариантах выполнения настоящего изобретения, W, X, Y, и Z каждый представляет собой CR6.
В определенных вариантах выполнения настоящего изобретения, Z представляет собой N.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-a:
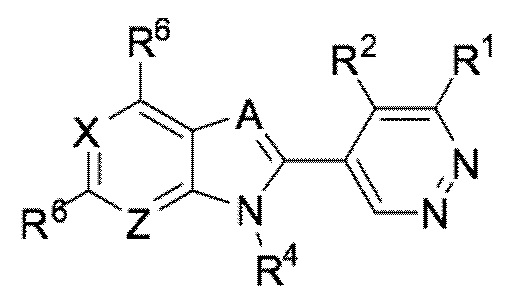
I-a,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, R6, A, X, и Z имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-b:
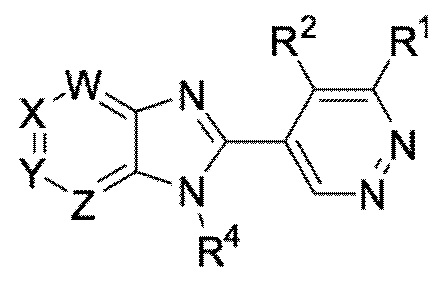
I-b,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, W, X, Y, и Z имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-c:
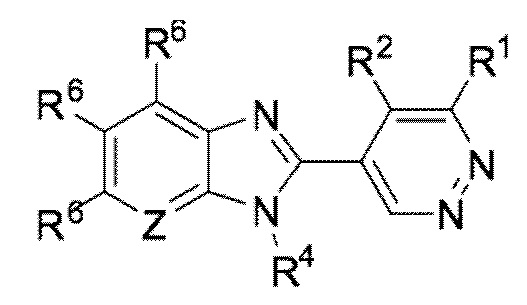
I-c,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, R6, и Z имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-d:
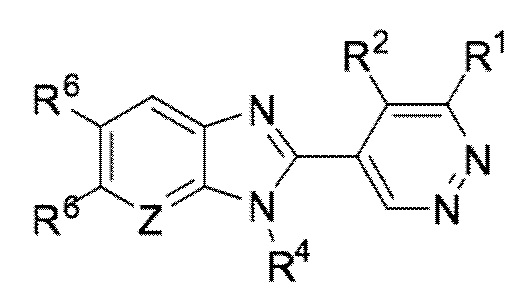
I-d,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, R6, и Z имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-e:

I-e,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R4, R6, и Z имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-f:
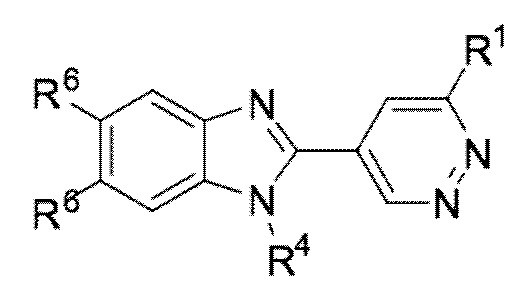
I-f,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R4, и R6 имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой водород, галоген, циано, ацил, алкил, галоалкил, алкокси, галоалкокси, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил, или необязательно замещенный гетероциклоалкил.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 представляет собой алкил или циклоалкил; и R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 представляет собой алкил или циклоалкил; и R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-g:
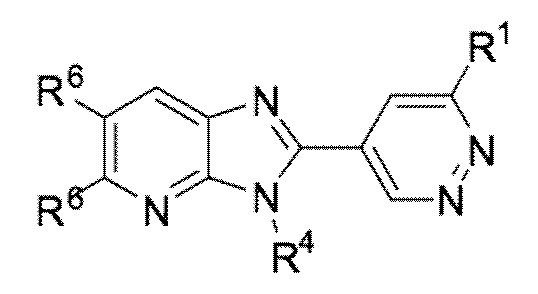
I-g,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R4, и R6 имеют значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой водород, галоген, циано, ацил, алкил, галоалкил, алкокси, галоалкокси, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; R6 представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил, или необязательно замещенный гетероциклоалкил.
Другим объектом настоящего изобретения являются соединения формулы I:
I,
или фармацевтически приемлемые соли, сокристаллы, таутомеры, стереоизомеры, сольваты, гидраты, полиморфы, изотопически обогащенные производные или их пролекарства, где:
A представляет собой N или CR5;
W представляет собой N или CR6;
X представляет собой N или CR6;
Y представляет собой N или CR6;
Z представляет собой N или CR6;
при условии, что не более чем два из W, X, Y, и Z представляют собой N;
R1 представляет собой водород, галоген, циано, ацил, алкил, алкенил, алкинил, галоалкил, алкокси, галоалкокси, циклоалкил, циклоалкокси, арил, арилалкил, гетероарил, гетероарилалкил, гетероциклоалкил, гетероциклоалкилалкил, NRaRb, NHSO2Rc, CH2NRaRb, CReRfNHSO2Rd, ORf, SRf, CO2Re, CORf, S(O)Rd, S(O)2Rd, CH2ORf, или CReRfOH, где любой R1 может быть необязательно замещен 1-3 независимыми заместителями R7;
R2 представляет собой водород, галоген, циано, алкил, или галоалкил;
или R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное, гетероциклоалкильное или циклоалкильное кольцо;
R3 представляет собой водород, галоген, циано, ацил, алкил, алкенил, алкинил, галоалкил, алкокси, галоалкокси, циклоалкил, циклоалкокси, арил, арилалкил, гетероарил, гетероарилалкил, гетероциклоалкил, гетероциклоалкилалкил, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH;
R4 представляет собой алкил, циклоалкил, галоалкил, или гетероалкил;
R5 представляет собой водород, циано, алкил, галоалкил, гетероалкил, или циклоалкил;
в каждом случае R6 независимо представляет собой водород, галоген, циано, галоалкил, алкил, циклоалкил, алкокси, галоалкил, ORf, или карбоксил;
в каждом случае R7 независимо представляет собой галоген, алкил, алкокси, галоалкил, карбоксил, циклоалкил, арил, арил, замещенный 1-3 независимыми заместителями галоген, -(CH2)nC(O)NRgRh, -S(O)2Ri, -CO2Rj, или NRgRh;
каждый n независимо представляет собой 1, 2, 3, 4, 5, 6, 7, 8, 9, или 10;
в каждом случае Ra, Rb, Rc, Rd, Re, и Rf независимо представляет собой водород, ацил, алкоксиалкил, алкил, алкенил, алкинил, гетероалкил, галоалкил, циклоалкил, гетероциклоалкил, арил, гетероарил, C(O)OC1-6 алкил, C(O)C1-6 алкил, защитную группу азота, которая присоединена к атому азота, или защитную группу кислорода, которая присоединена к атому кислорода; или Ra и Rb вместе с атомами, к которым они присоединены, образуют гетероциклоалкильное кольцо; или Re и Rf вместе с атомами, к которым они присоединены, образуют циклоалкильное кольцо; и
в каждом случае Rg, Rh, Ri, и Rj независимо представляет собой водород, ацил, алкил, алкенил, алкинил, гетероалкил, галоалкил, циклоалкил, гетероциклоалкил, арил, гетероарил, C(O)OC1-6 алкил, C(O)C1-6 алкил, или Rg и Rh вместе с атомами, к которым они присоединены, образуют гетероциклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой арил, гетероарил, или гетероциклоалкил, где любой R1 может быть необязательно замещен 1-3 независимыми заместителями R7.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой арил, гетероарил, или гетероциклоалкил, где любой R1 может быть необязательно замещен 1-3 независимыми заместителями R7, и каждый R7 независимо представляет собой галоген, алкил, алкокси, галоалкил, карбоксил, -(CH2)nC(O)NRgRh, -S(O)2Ri, -CO2Rj, или NRgRh, и каждый Rg и Rh независимо представляет собой водород, алкил, C(O)C1-6 алкил, или Rg и Rh вместе с атомами, к которым они присоединены, образуют гетероциклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород, галоген, циано, алкил, алкенил, алкинил, галоалкил, алкокси, галоалкокси, циклоалкил, циклоалкокси, арил, NRaRb, NHSO2Rc, CH2NRaRb, CReRfNHSO2Rd, SRf, CORf, или CReRfOH; или R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное или циклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород, галоген, алкил, алкенил, галоалкил, алкокси, галоалкокси, циклоалкокси, арил, NRaRb, CReRfNHSO2Rd, SRf, или CReRfOH; или R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное или циклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород, галоген, алкил, галоалкил, CReRfNHSO2Rd, SRf, или NRaRb; и Ra, Rb, Rd, Re, и Rf независимо представляют собой водород, алкил, или галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой SRf. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой SRf; и Rf представляет собой водород, алкил, или галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой алкил или галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-6 алкил или C1-6 галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-6 галоалкил.
В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой фторалкил. В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-6 фторалкил. В определенных вариантах выполнения настоящего изобретения, R1 независимо представляет собой C1-3 фторалкил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой дифторметил или трифторметил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой дифторметил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой трифторметил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород или алкил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород или C1-6 алкил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород или C1-3 алкил.
В определенных вариантах выполнения настоящего изобретения, R2 представляет собой алкил. В определенных вариантах выполнения настоящего изобретения, R2 независимо представляет собой C1-6 алкил. В определенных вариантах выполнения настоящего изобретения, R2 независимо представляет собой C1-3 алкил. В определенных вариантах выполнения настоящего изобретения, R2 представляет собой водород.
В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород и R2 представляет собой алкил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород и R2 независимо представляет собой C1-6 алкил. В определенных вариантах выполнения настоящего изобретения, R1 представляет собой водород и R2 независимо представляет собой C1-3 алкил.
В определенных вариантах выполнения настоящего изобретения, R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное или циклоалкильное кольцо. В определенных вариантах выполнения настоящего изобретения, R1 и R2 вместе с атомами, к которым они присоединены, образуют арильное кольцо. В определенных вариантах выполнения настоящего изобретения, R1 и R2 вместе с атомами, к которым они присоединены, образуют циклоалкильное кольцо.
В определенных вариантах выполнения настоящего изобретения, R4 представляет собой алкил или циклоалкил.
В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил.
В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой C1-4 алкил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой C3-5 циклоалкил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой циклопентил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой циклобутил. В определенных вариантах выполнения настоящего изобретения, R4 независимо представляет собой циклопропил.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, галоген, циано, алкокси, галоалкил, или карбоксил.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, галоген, или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, хлор, фтор, или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, фтор, или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, или галоген.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород, хлор, или фтор.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород или фтор.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой водород или циано.
В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой галоген. В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой Хлор или фтор. В определенных вариантах выполнения настоящего изобретения, каждый R6 независимо представляет собой фтор. В определенных вариантах выполнения настоящего изобретения, каждый R6 представляет собой циано.
В определенных вариантах выполнения настоящего изобретения, A независимо представляет собой CR5 и R5 представляет собой водород, циано, C1-4 алкил, или C3-5 циклоалкил.
В определенных вариантах выполнения настоящего изобретения, A представляет собой N.
В определенных вариантах выполнения настоящего изобретения, не более чем один из W, X, Y, и Z представляет собой N.
В определенных вариантах выполнения настоящего изобретения, W, X, Y, и Z каждый независимо представляет собой CR6.
В определенных вариантах выполнения настоящего изобретения, Z представляет собой N.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-a:
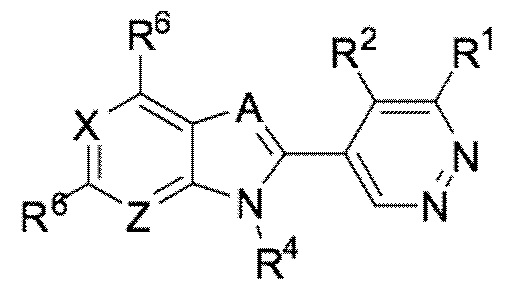
I-a,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, R6, A, X, и Z каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-b:
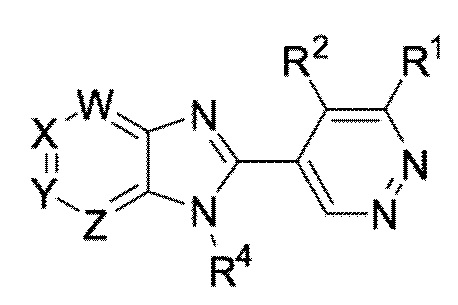
I-b,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, W, X, Y, и Z каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-c:
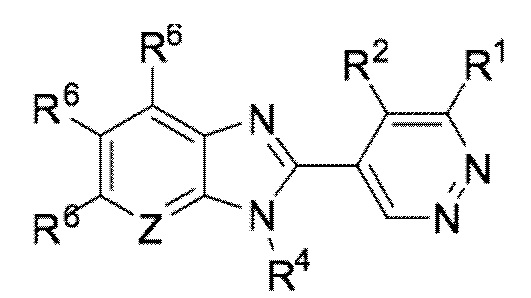
I-c,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, R6, и Z каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-d:
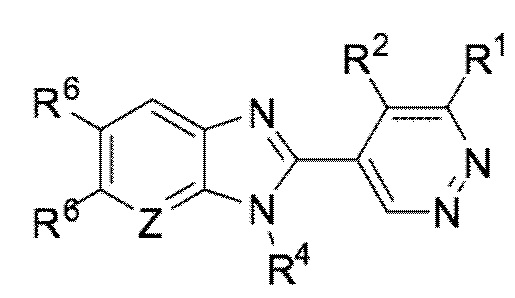
I-d,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R2, R4, R6, и Z каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-e:

I-e,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R4, R6, и Z каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-f:
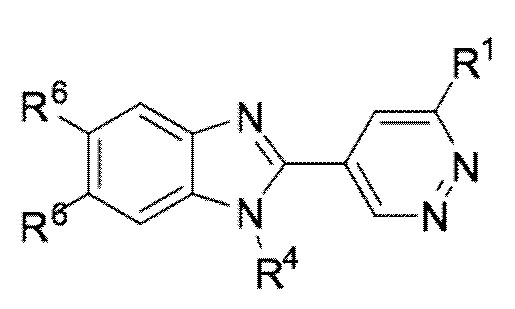
I-f,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R4, и R6 каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых: R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; каждый R6 независимо представляет собой водород, циано, или гало; и R1 представляет собой водород, галоген, циано, ацил, алкил, галоалкил, алкокси, галоалкокси, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой водород, галоген, циано, ацил, алкил, галоалкил, алкокси, галоалкокси, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил, или необязательно замещенный гетероциклоалкил.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение формулы I-g:
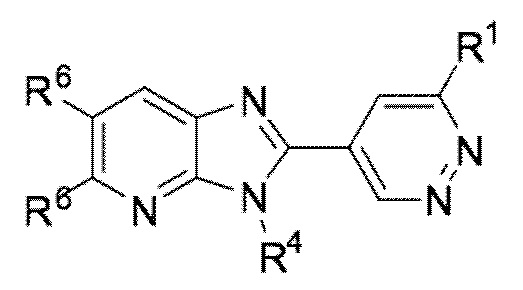
I-g,
или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство, где R1, R4, и R6 каждый независимо имеет значения, как определено в настоящей заявке.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой водород или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой водород или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; каждый R6 независимо представляет собой водород, циано, или гало; и R1 представляет собой водород, галоген, циано, ацил, алкил, галоалкил, алкокси, галоалкокси, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой водород, галоген, циано, ацил, алкил, галоалкил, алкокси, галоалкокси, NRaRb, NHSO2Rc, CH2NRaRb, CH2NHSO2Rd, CO2Re, CORf, CH2ORf, или CReRfOH.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-g являются соединения, в которых:
R4 представляет собой алкил или циклоалкил; каждый R6 независимо представляет собой водород или гало, где по меньшей мере один R6 представляет собой гало; и R1 представляет собой необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил, или необязательно замещенный гетероциклоалкил.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 представляет собой алкил или галоалкил; R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой алкил или циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C1-4 алкил или C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой водород, гало, или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой водород или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой водород или циано.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 представляет собой циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, вышеуказанными соединениями формулы I-f являются соединения, в которых:
R1 независимо представляет собой галоалкил; R4 независимо представляет собой C3-5 циклоалкил; и каждый R6 независимо представляет собой гало.
В определенных вариантах выполнения настоящего изобретения, соединение формулы I представляет собой соединение, выбранное из группы, состоящей из:
1-циклопропил-6-фтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазола (1);
5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбонитрила (2);
2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола (3);
1-циклопропил-6-фтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (4);
метил 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилата (5);
1-циклопропил-2-(6-этилпиридазин-4-ил)-6-фтор-1H-бензо[d]имидазола (6);
1-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифтопентан-1-ола (7);
N-((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)пропионамида (8);
этил ((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)карбамата (9);
4-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)морфолина (10);
1-циклопропил-6-фтор-2-(6-(4-(4-фторфенил)пиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазола (11);
2-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пропан-2-ола (12);
5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)-N-(тетрагидро-2H-пиран-4-ил)пиридазин-3-амина (13);
4-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)циннолина (14);
1-циклопропил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола (15);
N-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этансульфонамида (16);
2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила (17);
2-(4-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)-N-изопропилацетамида (18);
1-циклопропил-5,6-дифтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (19);
2-(4-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)-1-(пирролидин-1-ил)этан-1-она (20);
5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-метилпиридазин-3-амина (21);
2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола (22);
1-циклопропил-5,6-дифтор-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазола (23);
1-циклопропил-5,6-дифтор-2-(6-изопропоксипиридазин-4-ил)-1H-бензо[d]имидазола (24);
5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N,N-диметилпиридазин-3-амина (25);
1-этил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-индола (26);
1-циклопропил-5,6-дифтор-2-(6-(2,2,2-трифторэтокси)пиридазин-4-ил)-1H-бензо[d]имидазола (27);
5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-(2-метоксиэтил)пиридазин-3-амина (28);
1-циклопропил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (29);
1-циклопропил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-индол (30);
N-((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)этансульфонамида (31);
5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-(2,2,2-трифторэтил)пиридазин-3-амина (32);
1-циклопропил-2-(6-этилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (33);
2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила (34);
1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (35);
1-циклопропил-5,6-дифтор-2-(6-(4-метилпиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазола (36);
1-(4-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)этан-1-она (37);
1-циклопропил-5,6-дифтор-2-(6-(4-(метилсульфонил)пиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазола (38);
трет-бутил 4-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-карбоксилата (39);
1-этил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (40);
1-этил-2-(6-этилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (41);
1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (42);
2-(6-циклобутоксипиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола (43);
1-циклопропил-2-(6-(4,4-дифторпиперидин-1-ил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (44);
5-хлор-3-циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридина (45);
1-циклопропил-2-(6-(3,3-дифторпирролидин-1-ил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (46);
1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифтопентан-1-ола (47);
1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-N,N-диметилпирролидин-3-амина (48);
1-циклопропил-5,6-дифтор-2-(6-(4-фторфенил)пиридазин-4-ил)-1H-бензо[d]имидазола (49);
1-циклопропил-5,6-дифтор-2-(6-((4-фторфенил)этинил)пиридазин-4-ил)-1H-бензо[d]имидазола (50);
1-циклопропил-5,6-дифтор-2-(6-изопропилпиридазин-4-ил)-1H-бензо[d]имидазола (51);
N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил) этансульфонамида (52);
1-циклопропил-5,6-дифтор-2-(6-((1,1,1-трифторпропан-2-ил)окси)пиридазин-4-ил)-1H-бензо[d]имидазола (53);
3-циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрила (54);
1-циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазола (55);
1-циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-4-(трифторметил)-1H-бензо[d]имидазола (56);
1-циклопропил-2-(6-(2,2-дифторпропокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (57);
1-циклопропил-5,6-дифтор-2-(5-изопропилпиридазин-4-ил)-1H-бензо[d]имидазола (58);
1-циклопропил-2-(6-циклопропилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (59);
2-(6-хлорпиридазин-4-ил)-3-циклопропил-5-метокси-3H-имидазо[4,5-b]пиридина (60);
1-циклопропил-5,6-дифтор-2-(6-(4-фтор-2-метилфенил)пиридазин-4-ил)-1H-бензо[d]имидазола (61);
1-циклопропил-2-(6-(2,4-дифторфенил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (62);
1-циклопропил-2-(6-(3,4-дифторфенил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (63);
1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-она (64);
2-(6-хлорпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола (65);
1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (66);
2-(6-(дифторметил)пиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола (67);
2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила (68);
2-(6-(дифторметил)пиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила (69);
1-циклопропил-2-(6-(1,1-дифторэтил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (70);
1-циклопропил-5,6-дифтор-2-(6-(2,2,2-трифторэтил)пиридазин-4-ил)-1H-бензо[d]имидазола (71);
2-(6-Циклопропилпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола (72);
5-Хлор-3-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридина (73);
1-Циклопропил-5,6-дифтор-2-(5-метилпиридазин-4-ил)-1H-бензо[d]имидазола (74);
1-Циклопропил-2-(6-циклопропилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (75);
3-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрила (76);
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)метан сульфонамида (77);
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)-N-метилметансульфонамида (78);
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)пропан-2-сульфонамида (79);
1-Циклопропил-5,6-дифтор-2-(6-(метилсульфонил)пиридазин-4-ил)-1H-бензо[d]имидазола (80);
1-Циклопропил-2-(6-(этилсульфонил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (81);
1-Циклопропил-5,6-дифтор-2-(6-(метилсульфинил)пиридазин-4-ил)-1H-бензо[d]имидазола (82);
1-циклопропил-2-(6-(этилсульфинил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d] имидазола (83);
5-Хлор-3-циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридина (84);
1-Циклопропил-5,6-дифтор-2-(6-(трифторметил)пиридазин-4-ил)-1H-бензо[d]имидазола (85);
1-Циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (86);
3-Циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрила (87);
2-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)изотиазолидин 1,1-диоксида (88);
3-Циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрила (89);
1-Циклопропил-2-(6-метилпиридазин-4-ил)-6-(трифторметил)-1H-бензо[d]имидазола (90);
N-(1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этил) а (91);
1-Циклопропил-5,6-дифтор-2-(6-(фторметил)пиридазин-4-ил)-1H-бензо[d]имидазола (92);
1-Циклопропил-2-(5,6-диметилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (93);
(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанола (94);
5-Хлор-3-циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридина (95);
N-(1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этил) этансульфонамида (96);
2-(6-Бутилпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола (97);
1-Циклопропил-6-метил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола (98);
1-Циклопропил-5,6-дифтор-2-(3-метилпиридазин-4-ил)-1H-бензо[d]имидазола (99);
1-Циклопропил-2-(3,6-диметилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (100);
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-6,7-дифтор-1H-бензо[d]имидазола (101);
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиола (102);
2-(6-(Дифторметил)пиридазин-4-ил)-5,6-дифтор-1-метил-1H-бензо[d]имидазола (104);
2-(6-(Дифторметил)пиридазин-4-ил)-5,6-дифтор-1-пропил-1H-бензо[d]имидазола (105);
4-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-5,6,7,8-тетрагидроциннолина (106);
1-Циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол-5-карбонитрила (Пример 107);
1-Циклобутил-5,6-дифтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола (108);
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-ола (109);
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоновой кислоты (110);
1-Циклопропил-7-фтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола (111);
5-Хлор-1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола (112);
4-Хлор-1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола (113);
1-Циклопропил-5,6-дифтор-2-(6-(метоксиметил)пиридазин-4-ил)-1H-бензо[d]имидазол (114);
Метил 5-(6-циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилата (115);
5-(6-Циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоновой кислоты (116);
1-Циклопропил-2-(6-(циклопропилметокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (117);
1-Циклопропил-2-(6-(2,2,2-трифторэтокси)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (118);
6-Хлор-1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазола (119);
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазол-6-карбонитрила (120);
4-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-6,7-дигидро-5H-циклопента[c]пиридазина (121);
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-6-фтор-1H-бензо[d]имидазола (122);
1-Циклопропил-5,6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазола (123);
1-Циклопропил-5,6-дифтор-2-(6-(изопропилтио)пиридазин-4-ил)-1H-бензо[d]имидазола (124);
1-Циклопропил-2-(6-((дифторметил)тио)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (125);
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ола (126);
1-Циклопропил-2-(6-(дифторметокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (127);
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-ола (128);
1-Циклопропил-5,6-дифтор-2-(6-(1-фторэтил)пиридазин-4-ил)-1H-бензо[d]имидазола (129);
1-Циклопропил-5,6-дифтор-2-(6-((трифторметил)тио)пиридазин-4-ил)-1H-бензо[d]имидазола (130)
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазола (131);
2-(6-(Дифторметил)пиридазин-4-ил)-1-пропил-1H-бензо[d]имидазола (132);
1-Циклопропил-2-(6-(дифторметокси)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (133);
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрила (134);
2-(6-(Дифторметил)пиридазин-4-ил)-1-метил-1H-индол-3-карбонитрила (135);
1-Циклопропил-2-(6-(1-фторэтил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (136); их фармацевтически приемлемых солей, сокристаллов, таутомеров, стереоизомеров, сольватов, гидратов, полиморфов, изотопически обогащенных производных и пролекарств.
Другим объектом настоящего изобретения является 2-(6-(дифторметил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (103), или его фармацевтически приемлемая соль, сокристалл, таутомер, стереоизомер, сольват, гидрат, полиморф, изотопически обогащенная производная или пролекарство.
Другим объектом настоящего изобретения является соединение формулы I, которое ингибирует (или идентифицировано как ингибирующее) альдостеронсинтазу (CYP11B2).
Соединения согласно настоящему изобретению включают те, которые идентифицируется как достигающее аффинности, по меньшей мере частично, с металлоферментом, путем образования одной или более из следующих типов химических взаимодействий или связей с металлом: сигма-связи, ковалентные связи, ковалентно-координационные связи, ионные связи, пи-связи, дельта-связи или взаимодействия обратного связывания. Соединения могут также достигать аффинности посредством более слабого взаимодействия с металлом, как например, Ван-дер-Ваальсовы взаимодействия, пи-катионные взаимодействия, пи-анионные взаимодействия, диполь-дипольные взаимодействия, ион-дипольные взаимодействия. В одном варианте выполнения настоящего изобретения соединение идентифицируется как способное к взаимодействию связыванием с пиридазиновой составляющей.
Способы оценки металл-лиганд связывающих взаимодействий известны в данной области техник, как приведено в ссылках, включающих, например, “Principles of Bioinorganic Chemistry” by Lippard и Berg, University Science Books, (1994); “Mechanisms of Inorganic Reactions” by Basolo и Pearson John Wiley & Sons Inc; 2nd edition (September 1967); “Biological Inorganic Chemistry” by Ivano Bertini, Harry Gray, Ed Stiefel, Joan Valentine, University Science Books (2007); Xue et al. “Nature Chemical Biology”, vol. 4, no. 2, 107-109 (2008).
Другим объектом настоящего изобретения являются фармацевтические композиции, содержащие соединение формулы I и фармацевтически приемлемый носитель.
Другим объектом настоящего изобретения являются способы модуляции активности металлофермента у субъекта, включающие контакт субъекта с соединением формулы I, в количестве и при условиях, достаточных для модуляции активности металлофермента.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного нарушению или заболеванию, где субъект был идентифицирован как нуждающийся в лечении нарушения или заболевания, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтической композиции, так что указанный субъект лечится от указанного заболевания.
В другом варианте выполнения настоящего изобретения субъектом является млекопитающее, отличное от человека.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного вызванному металлоферментом нарушению или заболеванию, включающие введение субъекту эффективного количества соединения формулы I или его фармацевтической композиции.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного вызванному металлоферментом нарушению или заболеванию, где субъект был идентифицирован как нуждающийся в лечении вызванного металлоферментом нарушения или заболевания, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтической композиции, так что указанный субъект лечится от указанного заболевания.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного вызванному металлоферментом нарушению или заболеванию, где субъект был идентифицирован как нуждающийся в лечении вызванного металлоферментом нарушения или заболевания, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтической композиции, так что активность металлофермента у указанного субъекта модулируется (например, понижающе регулируется, ингибируется).
Способы согласно настоящему изобретению включают те, в которых заболевание или нарушение вызвано любым из ароматазы (CYP19), члена семейства циклооксигеназ, ланостеролдеметилазы (CYP51), члена семейства синтаз оксида азота, тромбоксансинтазы (CYP5a), тироидной пероксидазы, 17-альфагидроксилазы/17,20-лиазы (CYP17), цитохрома P450 2A6 (CYP2A6), гемоксигеназы, индоламин 2,3-диоксигеназы, гидроксилазы ретиноевой кислоты (CYP26), гидроксилазы витамина D (CYP24), стирол 27- гидроксилазы (CYP27), цитохрома P450 3A5 (CYP3A5), холестерин 24- гидроксилазы (CYP46), цитохрома P450 4F2 (CYP4F2), миелопероксидазы, или 11-бета- гидроксилазы (CYP11B1).
Способы согласно настоящему изобретению включают те, в которых заболеванием или нарушением является рак, сердечнососудистое заболевание, воспалительное заболевание, инфекционное заболевание, нарушение обмена веществ, офтальмологическое заболевание, заболевание центральной нервной системы (ЦНС), урологическое заболевание или желудочно-кишечное заболевание.
Способы согласно настоящему изобретению включают те, в которых заболеванием или нарушением является артериальная гипертония, резистентная артериальная гипертония, смертность, связанная с первичным или вторичным гиперальдостеронизмом или гиперплазией надпочечников, легочно-артериальная гипертония, порок сердца, диастолическая дисфункция, диастолическая дисфункция левого желудочка, диастолический порок сердца, систолическая дисфункция, систолический порок сердца, гипокалиемия, почечная недостаточность, хроническая почечная недостаточность, рестеноз, нефропатия, пост-миокардиальный инфаркт, ишемическая болезнь сердца, фиброз, заболевания, харатеризующиеся повышенным образованием коллагена, фиброз и матриксное ремоделирование после артериальной гипертонии, и матриксное ремоделирование после дисфункции эндотелиальных клеток, сердечнососудистые заболевания, такие как атеросклероз, фибрилляция предсердий, почечная дисфункция, заболевания печени, неалкогольный стеатогепатит, сосудистые заболевания, ретинопатия, заболевание нервной системы, инсулинопатия, эндотелиальная дисфункция, миокардиальный фиброз, сосудистый фиброз, миокардиальные некротические повреждения, сосудистое повреждение, инфаркт миокарда, гипертрофия левого желудочка, гипертрофия стенок сосудов, эндотелиальное сгущение, фибриноидный некроз артерий, заболевания почек, диабетическая нефропатия, гломерулосклероз, гломерулонефрит, нефритический синдром, поликистозная болезнь почек, сахарный диабет, метаболический синдром, резистентность к инсулину, синдром апноэ, обструктивный синдром апноэ, мышечная дистрофия, цирроз печени, неалкогольный стеатогепатоз, нарушение функции почек, диабетическое нарушение функции почек, или инсульт.
Способы, описанные в настоящей заявке, включают те, в которых субъект идентифицируется как нуждающийся в конкретно установленном лечении. Идентификация субъекта как нуждающегося в таком лечении может являться оценкой субъекта или профессионального работника в области здравоохранения и может быть субъективной (например, мнение) или объективной (например, определение с помощь способа тестирования или диагностики).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Для того чтобы можно было более легко понять настоящее изобретение, прежде всего для удобства приводятся определения некоторых терминов.
Как применяется в описании настоящего изобретения термин "лечение" заболевания охватывает профилактику, улучшение, смягчение и/или координацию нарушения и/или состояний, которые могут вызывать нарушение. Термины "лечение" и "терапия" относятся к способу облегчения или ослабления заболевания и/или сопровождающих его симптомов. В соответствии с настоящим изобретением термин "лечение" включает профилактику, блокирование, ингибирование, ослабление, защиту, модуляцию, реверсирование эффектов, вызванных нарушением, и уменьшение наличия, например, вредных эффектов, вызванных нарушением.
Как применяется в описании настоящего изобретения, термин «ингибирование» охватывает предупреждение, уменьшение и остановку развития. Необходимо отметить, что термин “ингибирование фермента” (например, ингибирование металлофермента) уточняется и описывается ниже.
Как применяется в описании настоящего изобретения, термин "модулировать" относится к повышению или уменьшению активности фермента в ответ на воздействие соединения по изобретению.
Термины "выделенный", "очищенный" или "биологически чистый" относятся к материалу, который является по существу или существенно свободным от компонентов, которые, как правило, сопутствуют ему в его природном состоянии. Чистота и гомогенность, как правило, определяются с применением аналитических химических методик, таких как электрофорез на полиакриламидном геле и высокоэффективная жидкостная хроматография. В частности, в вариантах выполнения настоящего изобретения соединение имеет чистоту по меньшей мере 85%, более предпочтительно по меньшей мере 90%, более предпочтительно по меньшей мере 95% и наиболее предпочтительно по меньшей мере 99%.
Термин "ввод" или “введение” включает маршруты введения соединения(ий) субъекту для осуществления предназначенной им функции. Примеры маршрутов введения, которые могут применяться, включают инъекцию (подкожную, внутривенное, парентеральное, интраперитонеальное, интратекальное введение), местное введение, пероральное введение, ингаляцию, ректальное введение и трансдермальное введение.
Термин “эффективное количество” включает количество эффективное, при дозах и в течение необходимых периодов времени, для достижения желательного результата. Эффективное количество соединения может варьироваться в соответствии с факторами, такими как болезненное состояние, возраст и масса субъекта, и способность соединения вызывать желательный ответ у субъекта. Режимы введения доз могут регулироваться для обеспечения оптимального терапевтического эффекта. Эффективным количество также является количество, при котором терапевтические благоприятные эффекты соединения-ингибитора преобладают над токсическими или вредными эффектами (например, побочные эффекты) этого соединения.
Фразы "системное введение," "введенный системно", "периферийное введение" и "введенный периферийно", как применяется в описании настоящего изобретения, означают введение соединения(ий), лекарственного средства или другого материала, таким образом, что оно включается в систему пациента и, таким образом, подвергается метаболизму и другим подобным процессам.
Термин "терапевтически эффективное количество" относится к количеству соединения, подлежащего введению, достаточному для предупреждения развития или улучшению до некоторой степени одного или более симптомов состояния или нарушения, подлежащих лечению.
Терапевтически эффективное количество соединения (то есть эффективная доза) может находиться в интервале от около 0.005 мг/кг до около 200 мг/кг, предпочтительно от около 0.01 мг/кг до около 200 мг/кг, более предпочтительно от около 0.015 мг/кг до около 30 мг/кг массы тела. В других вариантах выполнения настоящего изобретения терапевтически эффективное количество может находиться в интервале от около 1.0 пМ до около10 мкМ, от около 1.0 пМ до около 50 мкМ и от около 1.0 пМ до около 100 мкМ. Специалисту в данной области техники будет очевидно, что определенные факторы могут влиять на дозу, необходимую для эффективного лечения субъекта, включая, но без ограничения к этому, серьезность заболевания или нарушения, предшествующее лечение, общее состояние здоровья и/или возраст субъекта и другие имеющиеся заболевания. Более того, лечение субъекта с применением терапевтически эффективного количества соединения может включать однократное лечение или предпочтительно может включать серию лечений. В одном примере субъекта лечат с применением соединения в количестве в интервале от около 0.005 мкг/кг до около 200 мг/кг массы тела, один раз в день в течение периода времени от 1 до 10 недель, предпочтительно от 2 до 8 недель, более предпочтительно от около 3 до 7 недель и даже более предпочтительно в течение около 4, 5 или 6 недель. В другом примере субъект может подвергаться лечению ежедневно в течение нескольких лет в случае хронического состояния или заболевания. Должно быть оценено по достоинству, что эффективная доза соединения, применяемого для лечения, может повышаться или понижаться в ходе конкретного лечения.
Термин "хиральная" относится к молекулам, которые обладаю свойством несовпадения при наложении с партнером зеркального отражения, тогда как термин "ахиральная" относится к молекулам, которые обладаю свойством совпадения с их зеркальным отражением.
Термин "диастереомеры" относится к стереоизомерам с двумя или более центрами асимметрии, молекулы которых не являются зеркальным отражением друг друга.
Термин "энантиомера" относится к двум стереоизомерам соединения, которые не являются зеркальным отражением друг друга. Эквимолярная смесь двух энантиомеров называется "рацемической смесью" или "рацематом".
Термин "изомеры" или "стереоизомеры" относится к соединениям, которые имеют идентичный химический состав, но различаются с точки зрения расположения атомов или групп в пространстве.
Термин “пролекарство” включает соединения с составляющими, которые могут подвергаться метаболизму in vivo. В общем, пролекарства метаболизируются in vivo эстеразами или посредством других механизмов до активных лекарственных средств. Примеры пролекарств и их применения хорошо известны в данной области техники (смотрите, например, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19). Пролекарства могут быть получены in situ в ходе конечного выделения и очищения соединений или путем отдельной реакции очищенного соединения в его свободной кислотной форме или в форме гидроксила с подходящим эстерифицирующим агентом. Гидроксильные группы могут быть превращены в сложные эфиры путем обработки карбоновой кислотой. Примеры пролекарственных составляющих включают составляющие на основе сложных эфиров замещенных и незамещенных разветвленных или неразветвленных низших алкилов (например, сложные эфиры пропионовой кислоты), низший алкенил-сложные эфиры, ди-низший алкил-амино-низший алкил-сложные эфиры (например, диметиламиноэтиловый сложный эфир), ациламино-низший алкил-сложные эфиры (например, ацетилоксиметиловый сложный эфир), ацилокси-низший алкил-сложные эфиры (например, пивалоилоксиметиловый сложный эфир), ариловые сложные эфиры (фениловый сложный эфир), арил-низший алкил-сложные эфиры (например, бензиловый сложный эфир), замещенный (например, метил, галоген или метокси заместителями) арил и арил-низший алкил-сложные эфиры, амиды, низший алкил-амиды, ди-низший алкил-амиды и гидрокси амиды. Предпочтительными пролекарственными составляющими являются сложные эфиры пропионовой кислоты и ациловые сложные эфиры. Пролекарства, которые превращаются в активные формы через другие механизмы in vivo, также включены в настоящее изобретение. В вариантах выполнения настоящего изобретения соединения по настоящему изобретению представляют собой пролекарства любой из формул, приведенных в настоящем документе.
Термин "субъект" относится к животным, таким как млекопитающие, включая, но без ограничения к этому, приматов (например, людей), коров, овец, козлов, лошадей, собак, кошек, кроликов, крыс, мышей и тому подобное. В определенных вариантах выполнения настоящего изобретения субъектом является человек. Ветеринарные использования или применения относятся к применению, в котором субъектом является млекопитающее, отличное от человека.
Формы единственного числа относятся к “одному или более”, при применении в описании настоящего изобретения, включая формулу изобретения. Таким образом, например, ссылка на “образец” включает множество образцов, если из контекста ясным образом не следует иное (например, множество образцов), и так далее.
В описании изобретения и в формуле изобретения слова “содержат,” “содержит” и “содержание” применяются в смысле, обозначающем отсутствие исключения, если из контекста не следует иное.
Как применяется в описании настоящего изобретения термин “около,” применяемый в отношении значения, означает и охватывает его вариации, в некоторых вариантах выполнения изобретения ± 20%, в некоторых вариантах выполнения изобретения ± 10%, в некоторых вариантах выполнения изобретения ± 5%, в некоторых вариантах выполнения изобретения ± 1%, в некоторых вариантах выполнения изобретения ± 0.5% и в некоторых вариантах выполнения изобретения ± 0.1% от конкретного количества, так как такие вариации подходят для осуществления заявленных способов или применения заявленных композиций.
Термин “ингибитор” в описании настоящего изобретения означает молекулу, которая проявляет ингибирующую активность в отношении металлофермента. Термин “ингибировать”, при применении в описании настоящего изобретения, означает понижение активности металлофермента по сравнению с активностью металлофермента в отсутствии ингибитора. В некоторых вариантах выполнения настоящего изобретения термин “ингибирование” означает понижение активности металлофермента на по меньшей мере около 5%, по меньшей мере около 10%, по меньшей мере около 20%, по меньшей мере около 25%, по меньшей мере около 50%, по меньшей мере около 60%, по меньшей мере около 70%, по меньшей мере около 80%, по меньшей мере около 90% или по меньшей мере около 95%. В других вариантах выполнения настоящего изобретения ингибирование означает понижение активности металлофермента на от около 5% до около 25%, от около 25% до около 50%, от около 50% до около 75% или от около 75% до 100%. В некоторых вариантах выполнения настоящего изобретения ингибирование означает понижение активности металлофермента на около 95% - 100%, например, понижение активности фермента на 95%, 96%, 97%, 98%, 99% или 100%. Такие понижения могут быть измерены с применением множества методик, которые известны специалистам в данной области техники. Конкретный анализ измерения индивидуальной активности описывается ниже.
Кроме того, соединения по настоящему изобретению включают олефины, имеющие одну из двух геометрий: “Z” относится к геометрии, которая обозначается как “цис” (та же сторона) конфигурация, тогда как “E” относится к геометрии, которая обозначается как “транс” (противоположная сторона) конфигурация. В отношении номенклатуры хирального центра применяются термины "d" и "l" конфигурация, как определено согласно рекомендациям ИЮПАК. Термины диастереомер, рацемат, эпимер и энантиомер применяются в описании настоящего изобретения в их обычном контексте для описания стереохимии препаратов.
Как применяется в описании настоящего изобретения термин “алкил” относится к неразветвленной или разветвленной углеводородной группе, содержащей от 1 до 12 атомов углерода. Термин “низший алкил” относится к C1-C6 алкильной цепи. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, трет-бутил и н-пентил. Алкильные группы могут быть при необходимости замещены одним или более заместителями.
Термин “галоалкил” относится к алкильному радикалу, который замещен одним или более гало заместителями. Примеры галоалкильных групп включают фторметилдифторметил, трифторметил, бромметил, хлорметил и 2,2,2-трифторэтил.
Термин “алкенил” относится к ненасыщенной углеводородной цепи, которая может быть неразветвленной цепью или разветвленной цепью, содержащей от 2 до 12 атомов углерода и по меньшей мере одну углерод-углеродную двойную связь. Алкенильные группы могут быть необязательно замещены одним или более заместителями.
Термин “арилалкенил” относится к ненасыщенной углеводородной цепи, которая может быть неразветвленной цепью или разветвленной цепью, содержащей от 2 до 12 атомов углерода и по меньшей мере одну углерод-углеродную двойную связь, где один или более sp2 гибридизованные атомы углерода алкенильной составляющей присоединяются к арильной составляющей. Алкенильные группы могут быть необязательно замещены одним или более заместителями.
Термин “алкинил” относится к ненасыщенной углеводородной цепи, которая может быть неразветвленной цепью или разветвленной цепью, содержащей от 2 до 12 атомов углерода и по меньшей мере одну углерод-углеродную тройную связь. Алкинильные группы могут быть необязательно замещены одним или более заместителями.
Термин “арилалкинил” относится к ненасыщенной углеводородной цепи, которая может быть неразветвленной цепью или разветвленной цепью, содержащей от 2 до 12 атомов углерода и по меньшей мере одну углерод-углеродную тройную связь где один или более sp гибридизованные атомы углерода алкинильной составляющей присоединяются к арильной составляющей. Алкинильные группы могут быть необязательно замещены одним или более заместителями.
sp2 или sp углероды алкенильной группы и алкинильной группы, соответственно, могут при необходимости быть точкой присоединения алкенильной или алкинильной групп.
Термин “алкокси” относится к -O-алкильному радикалу.
Как применяется в описании настоящего изобретения термин "галоген", "гал" или “гало” означает -F, -Cl, -Br или -I.
Термин “алкилтио” относится к -S- алкильному заместителю.
Термин “алкоксиалкил” относится к -алкил-О- алкильному заместителю.
Термин “галоалкокси” относится к -OR заместителю, где R полностью или частично замещен Cl, F, I, или Br или любой их комбинацией. Примеры галоалкокси групп включают трифторметокси и 2,2,2-трифторэтокси.
Термин “галоалкоксиалкил” относится к -алкил-О-алкилу, где алкил замещен одним или более заместителями гало.
Термин “галоалкиламинокарбонил” относится к -С(О)-амино-алкилу, где алкил замещен одним или более заместителями гало.
Термин “галоалкилтио” относится к -S-алкилу, где алкил замещен одним или более заместителями гало. Примеры галоалкилтиогрупп включают трифторметилтио и 2,2,2-трифторэтилито.
Термин “галоалкилкарбонил” относится к -С(О)-алкилу, который замещен одним или более заместителями гало. Примером галоалкилкарбонильной группы является трифторацетил.
Термин “циклоалкил” относится к углеводородной 3-8 членной моноциклической или 7-14 членной бициклической кольцевой системе, содержащей по меньшей мере одно насыщенное кольцо или содержащей по меньшей мере одно неароматическое кольцо, где неароматическое кольцо может иметь некоторую степень ненасыщенности. Циклоалкильные группы могут быть необязательно замещенными одним или более заместителями. В одном варианте выполнения настоящего изобретения 0, 1, 2, 3 или 4 атома каждого кольца циклоалкильной группы могут быть замещены заместителем. Характерные примеры циклоалкильной группы включают циклопропил, циклопентил, циклогексил, циклобутил, циклогептил, циклопентенил, циклопентадиенил, циклогексенил, циклогексадиенил и тому подобное.
Термин “циклоалкокси” относится к -O-циклоалкильному заместителю.
Термин “циклоалкоксиалкил” относится к -алкил-О-циклоалкильному заместителю.
Термин “циклоалкилалкокси” относится к -О-алкил-циклоалкильному заместителю.
Термин “циклоалкиламинокарбонил” относится к -С(О)-NH-циклоалкильному заместителю.
Термин “арил” относится к углеводородной моноциклической, бициклической или трициклической ароматической кольцевой системе. Арильные группы могут быть необязательно замещены одним или более заместителями. В одном варианте выполнения настоящего изобретения 0, 1, 2, 3, 4, 5 или 6 атомов каждого кольца арильной группы могут быть замещены заместителем. Примеры арильных групп включают фенил, нафтил, антраценил, фторенил, инденил, азуленил и тому подобное.
Термин “арилокси” относится к -O-арильному заместителю.
Термин “арилалкокси” относится к -О-алкил-арильному заместителю.
Термин “арилалкилтио” относится к -S-алкил-арильному заместителю.
Термин “арилтиоалкил” относится к -алкил-S-арильному заместителю.
Термин “арилалкиламинокарбонил” относится к -С(О)амино-алкил-арильному заместителю.
Термин “арилалкилсульфонил” относится к -S(O)2-алкил-арильному заместителю.
Термин “арилалкилсульфинил” относится к -S(O)-алкил-арильному заместителю.
Термин “арилоксиалкил” относится к -алкил-O-арильному заместителю.
Термин “алкиларил” относится к -арил-алкильному заместителю.
Термин “арилалкил” относится к -алкил-арильному заместителю.
Термин “гетероарил” относится к ароматической 5-8 членной моноциклической, 8-12 членной бициклической или 11-14 членной трициклической кольцевой системе, имеющей 1-4 кольцевых гетероатомов в случае моноциклической, 1-6 гетероатомов в случае бициклической или 1-9 гетероатомов в случае трициклической, причем указанные гетероатомы выбираются из O, N или S, а оставшимися кольцевыми атомами являются атомы углерода (с соответствующим количеством атомов водорода, если иное не указано). Гетероарильные группы могут быть необязательно замещены одним или более заместителями. В одном варианте выполнения настоящего изобретения 0, 1, 2, 3 или 4 атома каждого кольца гетероарильной группы могут быть замещены заместителем. Примеры гетероарильных групп включают пиридил, фуранил, тиенил, пирролил, оксазолил, оксадиазолил, имидазолил, тиазолил, изоксазолил, хинолинил, пиразолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, изохинолинил, индазолил и тому подобное.
Термин “гетероарилалкил” относится к -алкил-гетероарильному заместителю.
Термин “гетероарилокси” относится к -О-гетероарильному заместителю.
Термин “гетероарилалкокси” относится к -О-алкил-гетероарильному заместителю.
Термин “гетероарилоксиалкил” относится к -алкил-О-гетероарильному заместителю.
Термин “азотсодержащий гетероарил” относится к гетероарильной группе, имеющей 1-4 кольцевых гетероатомов азота в случае моноцикла, 1-6 кольцевых гетероатомов азота в случае бицикла, или 1-9 кольцевых гетероатомов азота в случае трицикла.
Термин "гетероциклоалкил" относится к неароматической 3-8 членной моноциклической, 7-12 членной бициклической или 10-14 членной трициклической кольцевой системе, содержащей 1-3 гетероатома в случае моноцикла, 1-6 гетероатома в случае бицикла или 1-9 гетероатомов в случае трицикла, причем указанные гетероатомы выбираются из O, N, S, B, P или Si, где неароматическая кольцевая система является полностью насыщенной. Гетероциклоалкильные группы могут быть необязательно замещены одним или более заместителями. В одном варианте выполнения настоящего изобретения 0, 1, 2, 3 или 4 атома каждого кольца гетероциклоалкильной группы могут быть замещены заместителем. Примеры гетероциклоалкильных групп включают пиперидинил, пиперазинил, тетрагидропиранил, морфолинил, тиоморфолинил, 1,3-диоксолан, тетрагидрофуранил, тетрагидротиенил, тииренил, и тому подобное.
Термин "гетероциклоалкилалкил” относится к -алкил-гетероциклоалкильному заместителю.
Термин “алкиламино” относится к аминозаместителю, который далее замещается одним или двумя алкильными группами. Термин “аминоалкил” относится к алкильному заместителю, который далее замещается одной или более аминогруппами. Термин “гидроксиалкил” или “гидроксилалкил” относится к алкильному заместителю, который далее замещается одной или более гидроксильными группами. Алкильная или арильная часть алкиламино, аминоалкила, меркаптоалкила, гидроксиалкила, меркаптоалкокси, сульфонилалкила, сульфониларила, алкилкарбонила и алкилкарбонилалкила может быть необязательно замещена одним или более заместителями.
Кислоты и основания, полезные в способах согласно настоящему изобретению, известны в данной области техники. Кислотными катализаторами являются любые известные кислотные соединения, которые могут быть неорганическими (например, соляная, серная, азотная кислоты, трихлорид алюминия) или органическими (например, камфорсульфоновая кислота, п-толуолсульфоновая кислота, уксусная кислота, трифлат иттербия) по своей природе. Кислоты полезны либо в каталитическом, либо в стехиометрическом количествах для содействия химическим реакциям. Основаниями являются любые основные химические соединения, которые могут быть неорганическими (например, бикарбонат натрия, гидроксид калия) или органическими (например, триэтиламин, пиридин) по своей природе. Основания полезны либо в каталитическом, либо в стехиометрическом количествах для содействия химическим реакциям.
Алкилирующие агенты представляют собой любой реагент, который способен эффективно алкилировать рассматриваемую функциональную группу (например, атом кислорода спирта, атом азота аминогруппы). Алкилирующие агенты известны в данной области техники, включая приведенные ссылочные источники, и включают алкилгалогениды (например, метил иодид, бензилбромид или хлорид), алкилсульфаты (например, метилсульфат) или другие комбинации алкильная группа-уходящая группа, известные в данной области техники. Уходящими группами являются любые стабильные группы, которые могут быть отделены от молекулы в ходе реакции (например, реакция элиминирования, реакция замещения) и известны в данной области техники, включая приведенные в описании настоящего изобретения ссылочные источники, и включают галогениды (например, I-, Cl-, Br-, F-), гидрокси, алкокси (например, -OMe, -O-t-Bu), ацилокси анионы (например, -OAc, -OC(O)CF3), сульфонаты (например, мезил, тозил), ацетамиды (например, -NHC(O)Me), карбаматы (например, N(Me)C(O)Ot-Bu), фосфонаты (например, -OP(O)(OEt)2), воду или спирты (протонсодержащие условия) и тому подобное.
В определенных вариантах выполнения настоящего изобретения заместители при любой группе (как например, алкил, алкенил, алкинил, арил, аралкил, гетероарил, гетероаралкил, циклоалкил, гетероциклоалкил) могут находиться при любом атоме этой группы, где любая группа, которая может быть замещена (как например, алкил, алкенил, алкинил, арил, аралкил, гетероарил, гетероаралкил, циклоалкил, гетероциклоалкил) может быть необязательно замещена одним или более заместителями (которые могут быть одинаковыми или различными), причем каждый замещает атом водорода. Примеры подходящих заместителей включают, но без ограничения к этому, алкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, арил, гетероарил, галоген, галоалкил, циано, нитро, алкокси, арилокси, гидроксил, гидроксилалкил, оксо (то есть, карбонил), карбоксил, формил, алкилкарбонил, алкилкарбонилалкил, алкоксикарбонил, алкилкарбонилокси, арилоксикарбонил, гетероарилокси, гетероарилоксикарбонил, тио, меркапто, меркаптоалкил, арилсульфонил, амино, аминоалкил, диалкиламино, алкилкарбониламино, алкиламинокарбонил, алкоксикарбониламино, алкиламино, ариламино, диариламино, алкилкарбонил или ариламино-замещенный арил; арилалкиламино, аралкиламинокарбонил, амидо, алкиламиносульфонил, ариламиносульфонил, диалкиламиносульфонил, алкилсульфониламино, арилсульфониламино, имино, карбамидо, карбамил, тиоуреидо, тиоцианато, сульфоамидо, сульфонилалкил, сульфониларил, меркаптоалкокси, N-гидроксиамидил или N’-арил, N’’-гидроксиамидинил.
Соединения согласно настоящему изобретению могут быть получены способами органического синтеза, известными в данной области техники. Способы оптимизации условий реакции, если необходимо уменьшить конкурирующие побочные продуты, известны в данной области техники. При оптимизации реакции и масштабировании могут предпочтительно применяться оборудование для высокоскоростного параллельного синтеза и микрореакторы, контролируемые компьютером (например, Design and Optimization in Organic Synthesis, 2nd Edition, Carlson R, Ed, 2005; Elsevier Science Ltd.;  et al, Angew. Chem. Int. Ed. Engl. 2004 43: 406; и ссылки приведенные там). Дополнительные реакционные схемы и протоколы могут быть определены специалистами в данной области техники, путем применения коммерчески доступного программного обеспечения для поиска структуры по базам данных, например, SciFinder® (CAS division of the American Chemical Society) и CrossFire Beilstein® (Elsevier MDL), или путем соответствующего поиска по ключевому слову с применением средства для поиска в интернете, такого как Google®, или баз данных, допускающих поиск по ключевому слову, как например база данных текстов патентного ведомства США.
et al, Angew. Chem. Int. Ed. Engl. 2004 43: 406; и ссылки приведенные там). Дополнительные реакционные схемы и протоколы могут быть определены специалистами в данной области техники, путем применения коммерчески доступного программного обеспечения для поиска структуры по базам данных, например, SciFinder® (CAS division of the American Chemical Society) и CrossFire Beilstein® (Elsevier MDL), или путем соответствующего поиска по ключевому слову с применением средства для поиска в интернете, такого как Google®, или баз данных, допускающих поиск по ключевому слову, как например база данных текстов патентного ведомства США.
Как может быть очевидно специалисту в данной области техники способы синтеза соединений приведенных в настоящем изобретении формул будут очевидны специалистам в данной области техники, включая схемы и примеры, приведенные в настоящей заявке. Кроме того, различные стадии синтеза могут осуществляться в альтернативной последовательности или порядке с получением целевых соединений. Кроме того, растворители, температуры, продолжительность реакций и т.д. приводятся в настоящей заявке только в целях иллюстрации, и специалист в данной области техники легко определит, что изменение условий реакции может обеспечить целевые соединения согласно настоящему изобретению.
Соединения согласно настоящему изобретению могут также содержать связи (например, углерод-углеродные связи), где вращение связи ограничено около этой конкретной связи, например, ограничение в результате присутствия кольца или двойной связи. Соответственно, все цис/транс и E/Z изомеры определенным образом включены в объем настоящего изобретения. Соединения согласно настоящему изобретению могут также быть представлены во множественных таутомерных формах, в таком случае настоящее изобретение определенным образом охватывает все таутомерные формы соединений, описанных в настоящем изобретении, даже если только одна таутомерная форма может быть представлена. Все такие изомерные формы таких соединений согласно настоящему изобретению определенным образом включены в объем настоящего изобретения. Все кристаллические формы и полиморфы соединений, описанных в описании настоящего изобретения, определенным образом включены в объем настоящего изобретения. Также вариантами выполнения настоящего изобретения являются экстракты и фракции, содержащие соединения по настоящему изобретению. Термин изомеры, как подразумевается, включает диастереоизомеры, энантиомеры, региоизомеры, структурные изомеры, ротационные изомеры, таутомеры и тому подобное. Для соединений, которые содержат один или более стереогенных центров, например, хиральных соединений, способы согласно настоящему изобретению могут выполняться с энантиомерно обогащенным соединением, рацематом или смесью диастереомеров.
Предпочтительные энанатиомерно обогащенные соединения имеют энантиомерный избыток 50% или более, более предпочтительно соединение имеет энантиомерный избыток 60%, 70%, 80%, 90%, 95%, 98% или 99% или более. В предпочтительных вариантах выполнения настоящего изобретения только один энантиомер или диастереомер хирального соединения согласно настоящему изобретению вводится в клетки или субъекту.
Способы лечения
Одним объектом настоящего изобретения является способ лечения субъекта, страдающего от нарушения или заболевания, или подверженного ему, содержащий введение субъекту эффективного количества соединения или фармацевтической композиции формулы I.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного заболеванию или нарушению, где субъект был идентифицирован как нуждающийся в лечении вызванного металлоферментом нарушения или заболевания, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтической композиции, так что указанный субъект лечится от указанного заболевания.
Другим объектом настоящего изобретения являются способы модуляции металлоферментной активности клетки у субъекта, включающие контакт субъекта с соединением формулы I, в количестве и при условиях, достаточных для модуляции активности металлофермента.
В одном варианте выполнения настоящего изобретения, модуляцией является ингибирование.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного вызванному металлоферментом нарушению или заболеванию, включающие введение субъекту эффективного количества соединения формулы I или его фармацевтической композиции.
Другим объектом настоящего изобретения являются способы лечения субъекта, страдающего от или подверженного вызванному металлоферментом нарушению или заболеванию, где субъект был идентифицирован как нуждающийся в лечении вызванного металлоферментом нарушения или заболевания, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы I или его фармацевтической композиции, так что указанный субъект лечится от указанного заболевания.
В определенных вариантах выполнения настоящего изобретения, обеспечиваются способы лечения заболевания, нарушения или их симптома, где нарушение независимо представляет собой рак, сердечнососудистое заболевание, воспалительное заболевание, инфекционное заболевание, нарушение обмена веществ, офтальмологическое заболевание, заболевание центральной нервной системы (ЦНС), урологическое заболевание или желудочно-кишечное заболевание. В определенных вариантах выполнения настоящего изобретения заболеванием является артериальная гипертония, резистентная артериальная гипертония, смертность, связанная с первичным или вторичным гиперальдостеронизмом или гиперплазией надпочечников, легочно-артериальная гипертония, порок сердца, диастолическая дисфункция, диастолическая дисфункция левого желудочка, диастолический порок сердца, систолическая дисфункция, систолический порок сердца, гипокалиемия, почечная недостаточность, хроническая почечная недостаточность, рестеноз, нефропатия, пост-миокардиальный инфаркт, ишемическая болезнь сердца, фиброз, заболевания, харатеризующиеся повышенным образованием коллагена, фиброз и матриксное ремоделирование после артериальной гипертонии, и матриксное ремоделирование после дисфункции эндотелиальных клеток, сердечнососудистые заболевания, как например атеросклероз, фибрилляция предсердий, почечная дисфункция, заболевания печени, неалкогольный стеатогепатит, сосудистые заболевания, ретинопатия, заболевание нервной системы, инсулинопатия, эндотелиальная дисфункция, миокардиальный фиброз, сосудистый фиброз, миокардиальные некротические повреждения, сосудистое повреждение, инфаркт миокарда, гипертрофия левого желудочка, гипертрофия стенок сосудов, эндотелиальное сгущение, фибриноидный некроз артерий, заболевания почек, диабетическая нефропатия, гломерулосклероз, гломерулонефрит, нефритический синдром, поликистозная болезнь почек, сахарный диабет, метаболический синдром, резистентность к инсулину, синдром апноэ, обструктивный синдром апноэ, мышечная дистрофия, цирроз печени, неалкогольный стеатогепатоз, нарушение функции почек, диабетическое нарушение функции почек, или инсульт.
В определенных вариантах выполнения настоящего изобретения, субъектом является млекопитающее, предпочтительно примат или человек.
В другом варианте выполнения настоящего изобретения, обеспечиваются способы, как описано выше, где эффективное количество соединения формулы I является таким, как описано выше.
В другом варианте выполнения настоящего изобретения, обеспечиваются способы, как описано выше, где соединение формулы I вводят внутривенно, внутримышечно, подкожно, интрацеребровентрикулярно, перорально или местно.
В другом варианте выполнения настоящего изобретения, обеспечиваются способы, как описано в настоящей заявке, где соединение формулы I демонстрирует селективность для диапазона активности против целевого фермента (например, альдостеронсинтазы (CYP11B2) IC50 <1.0 мкМ).
В другом варианте выполнения настоящего изобретения, обеспечиваются способы, как описано выше, где соединение формулы I вводят само по себе или в комбинации с одним или более другими терапевтическими средствами. В другом варианте выполнения настоящего изобретения, дополнительным терапевтическим средством является противораковое средство, противогрибковое средство, сердечнососудистое средство, противовоспалительное средство, химиотерапевтическое средство, средство для развития кровеносных сосудов, цитотоксическое средство, средство против пролиферации, средство против нарушения обмена веществ, средство против офтальмологического заболевания, средство против заболевания центральной нервной системы (ЦНС), средство против урологического заболевания, или средство против желудочно-кишечного заболевания.
В определенных вариантах выполнения настоящего изобретения, дополнительным терапевтическим средством является средство для лечения артериальной гипертонии, средство для лечения первичного гиперальдостеронизма, средство для лечения заболевания почек, средство для лечения конгестивного порока сердца, средство для лечения атеросклеротических заболеваний, средство для лечения диабета, средство для лечения ожирения, или средство для лечения нарушения обмена веществ.
Примерные дополнительные терапевтические средства включают, но без ограничения к этому, ингибиторы ренина, ингибиторы ангиотензин-превращающего фермента (ACE), двойные ингибиторы ангиотензин-превращающего фермента (ACE) и нейтральной эндопептидазы (NEP), блокаторы рецептора ангиотензина II (ARB), антагонисты рецептора минералокортикоида (MRA), ингибиторы нейтральной эндопептидазы (NEP), ингибиторы неприлизина, блокаторы кальциевых каналов, альфа-адренергические блокаторы, бета-адренергические блокаторы, диуретики (включая петлевые диуретики), активаторы калиевых каналов, антагонисты рецепторов эндотелина, агонисты рецептора эндотелина 1, стимуляторы растворимой гуанилатциклазы, сосудорасширяющие средства, ингибиторы HMG-CoA редуктазы, ниацин и агонисты рецептора ниацина, ингибиторы болезни Ниманна-Пика тип C1 (NPC1L1), инсулин или аналоги инсулина, бигуаниды (например, метформин), сульфонилмочевины, агонисты альфа-рецепторов, активируемых пролифераторами пероксисом (PPAR), и частичные агонисты, включая PPARγ агонисты и другие PPAR лиганды, ингибиторы дипептидил пептидазы-4 (DPP4), глюкагон-подобный пептид 1 (GLP-1), агонисты рецептора GLP-1, и ингибиторы натрия-глюкозы совместного транспортера 2 (SGLT2).
Другим объектом настоящего изобретения является применение соединения, как описано в настоящей заявке, (например, соединения формулы I) при получении лекарственного средства при применении в лечении вызванного металлоферментом заболевания или нарушения. Другим объектом настоящего изобретения является применение соединения, как описано в настоящей заявке, (например, соединения формулы I) для применения в лечении вызванного металлоферментом заболевания или нарушения. Другим объектом настоящего изобретения является применение соединения, как описано в настоящей заявке, (например, соединения формулы I) при получении сельскохозяйственной композиции для применения в лечении или профилактики вызванного металлоферментом заболевания или нарушения в сельскохозяйственных или сельских посадок.
Фармацевтические композиции
Другим объектом настоящего изобретения являются фармацевтические композиции, содержащие соединение формулы I и фармацевтически приемлемый носитель.
В другом варианте выполнения настоящего изобретения, обеспечиваются фармацевтические композиции дополнительно содержащие дополнительное терапевтическое средство. В другом варианте выполнения настоящего изобретения дополнительным терапевтическим средством является противораковое средство, противогрибковое средство, сердечнососудистое средство, противовоспалительное средство, химиотерапевтическое средство, антиангиогенное средство, цитотоксическое средство, антипролиферативное средство, средство для лечения нарушения обмена веществ, средство для лечения офтальмологических заболеваний, средство для лечения заболеваний центральной нервной системы (ЦНС), средство для лечения урологических заболеваний или средство для лечения желудочно-кишечных заболеваний.
В определенных вариантах выполнения настоящего изобретения, дополнительным терапевтическим средством является средство для лечения артериальной гипертонии, средство для лечения первичного гиперальдостеронизма, средство для лечения заболевания почек, средство для лечения конгестивного порока сердца, средство для лечения атеросклеротических заболеваний, средство для лечения диабета, средство для лечения ожирения, или средство для лечения нарушения обмена веществ.
Примерные дополнительные терапевтические средства включают, но без ограничения к этому, ингибиторы ренина, ингибиторы ангиотензин-превращающего фермента (ACE), двойные ингибиторы ангиотензин-превращающего фермента (ACE) и нейтральной эндопептидазы (NEP), блокаторы рецептора ангиотензина II (ARB), антагонисты рецептора минералокортикоида (MRA), ингибиторы нейтральной эндопептидазы (NEP), ингибиторы неприлизина, блокаторы кальциевых каналов, альфа-адренергические блокаторы, бета-адренергические блокаторы, диуретики (включая петлевые диуретики), активаторы калиевых каналов, антагонисты рецепторов эндотелина, агонисты рецептора эндотелина 1, стимуляторы растворимой гуанилатциклазы, сосудорасширяющие средства, Ингибиторы HMG-CoA редуктазы, ниацин и агонисты рецептора ниацина, Ингибиторы болезни Ниманна-Пика тип C1 (NPC1L1), инсулин или аналоги инсулина, бигуаниды (например, метформин), сульфонилмочевины, агонисты альфа-рецепторов, активируемых пролифераторами пероксисом (PPAR), и частичные агонисты, включая PPARγ агонисты и другие PPAR лиганды, ингибиторы дипептидил пептидазы-4 (DPP4), глюкагон-подобный пептид 1 (GLP-1), агонисты рецептора GLP-1, и ингибиторы натрия-глюкозы совместного транспортера 2 (SGLT2).
Другим объектом настоящего изобретения являются наборы, содержащие эффективное количество соединения формулы I, в форме однократной дозы, вместе с инструкциями по введению соединения субъекту, страдающему от или подверженному вызванному металлоферментом заболеванию или нарушению, включая рак, сердечнососудистое заболевание, воспалительное заболевание, инфекционное заболевание, нарушение обмена веществ, офтальмологическое заболевание, заболевание центральной нервной системы (ЦНС), урологическое заболевание или желудочно-кишечное заболевание. В других вариантах выполнения настоящего изобретения, заболеванием, нарушением или его симптомом является артериальная гипертония, резистентная артериальная гипертония, смертность, связанная с первичным или вторичным гиперальдостеронизмом или гиперплазией надпочечников, легочно-артериальная гипертония, порок сердца, диастолическая дисфункция, диастолическая дисфункция левого желудочка, диастолический порок сердца, систолическая дисфункция, систолический порок сердца, гипокалиемия, почечная недостаточность, хроническая почечная недостаточность, рестеноз, нефропатия, пост-миокардиальный инфаркт, ишемическая болезнь сердца, фиброз, заболевания, характеризующиеся повышенным образованием коллагена, фиброз и матриксное ремоделирование после артериальной гипертонии, и матриксное ремоделирование после дисфункции эндотелиальных клеток, сердечнососудистые заболевания, как например атеросклероз, фибрилляция предсердий, почечная дисфункция, заболевания печени, неалкогольный стеатогепатит, сосудистые заболевания, ретинопатия, заболевание нервной системы, инсулинопатия, эндотелиальная дисфункция, миокардиальный фиброз, сосудистый фиброз, миокардиальные некротические повреждения, сосудистое повреждение, инфаркт миокарда, гипертрофия левого желудочка, гипертрофия стенок сосудов, эндотелиальное сгущение, фибриноидный некроз артерий, заболевания почек, диабетическая нефропатия, гломерулосклероз, гломерулонефрит, нефритический синдром, поликистозная болезнь почек, сахарный диабет, метаболический синдром, резистентность к инсулину, синдром апноэ, обструктивный синдром апноэ, мышечная дистрофия, цирроз печени, неалкогольный стеатогепатоз, нарушение функции почек, диабетическое нарушение функции почек, или инсульт.
Термин "фармацевтически приемлемые соли" или "фармацевтически приемлемый носитель", согласно его значению, включает соли активных соединений, которые получают с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей, обнаруживаемых в соединениях, описанных в описании настоящего изобретения. Если соединения согласно настоящему изобретению содержат относительно кислотные функциональные группы, основно-аддитивные соли могут быть получены путем контакте нейтральной формы таких соединений с достаточным количеством желательного основания, либо в чистом виде, либо в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей основного добавления включают соль натрия, калия, кальция, аммония, органическую аминосоль или соль магния, или подобную соль. Если соединения согласно настоящему изобретению содержат относительно основные функциональные группы, кислотно-аддитивные соли могут быть получены путем контакта нейтральной формы таких соединений с достаточным количеством желательной кислоты, либо в чистом виде, либо в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей кислотного добавления включают соли, полученные из неорганических кислот, таких как соляная кислота, бромисто-водородная кислота, азотная кислота, угольная кислота, моногидрокарбоновая кислота, фосфорная кислота, моногидрофосфорная кислота, дигидрофосфорная кислота, серная кислота, моногидросерная кислота, иодистоводородная кислота или фосфорная кислота, и тому подобное, а также соли, полученные из относительно нетоксичных органических кислот, как например уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, пробковая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, бензолсульфоновая кислота, п-толилсульфоновая кислота, лимонная кислота, винная кислота, метансульфоновая кислота и тому подобное. Сюда также включены соли аминокислот, такие как аргинат и тому подобное, и соли органических кислот, как например глюкуроновой или галактуроновая кислот, и тому подобное (смотрите, например, Berge et al., Journal of Pharmaceutical Science 66:1-19 (1977)). Определенные специфические соединения согласно настоящему изобретению содержат как основную, так и кислотную функциональные группы, что позволяет соединениям превращаться либо в соли основного добавления, либо в соли кислотного добавления. Другие фармацевтически приемлемые носители, известные специалистам в данной области техники, подходят согласно настоящему изобретению.
Нейтральные формы соединений могут регенерироваться контактированием соли с основанием или кислотой и выделением исходного соединения удобным образом. Исходная форма соединения отличается от различных солевых форм определенными физическими свойствами, такими как растворимость в полярных растворителях, но что касается целей настоящего изобретения, то в остальном соли эквивалентны исходной форме соединений согласно настоящему изобретению.
Помимо солевых форм, настоящее изобретение предоставляет соединения, которые являются пролекарственной формой. Пролекарства активных соединений, описанных в настоящем изобретении, являются неактивными соединениями, которые легко подвергаются химическим превращениям в физиологических условиях с получением активных соединений согласно настоящему изобретению. Кроме того, пролекарства могут подвергаться превращению в активные соединения согласно настоящему изобретению химическими или биохимическими методами ex vivo. Например, пролекарства могут подвергаться медленному превращению в активные соединения согласно настоящему изобретению при размещении их в резервуаре трансдермальной заплаты с подходящим ферментом или химическим реагентом.
Определенные соединения согласно настоящему изобретению могут существовать как в несольватированных формах, так и в сольватированных формах, включая гидратные формы. Как правило, сольватированные формы эквиваленты несольватированным формам, и подразумевается, что они включены в объем настоящего изобретения. Некоторые соединения согласно настоящему изобретению могут существовать во множестве кристаллических или аморфных форм. Обычно все физические формы эквивалентны для областей применения, предусмотренных настоящим изобретением, и подразумевается, что они включены в объем настоящего изобретения.
Настоящее изобретение также обеспечивает фармацевтическую композицию, содержащую эффективное количество соединения согласно настоящему изобретению и фармацевтически приемлемый носитель. В варианте выполнения настоящего изобретения соединение формулы I вводится субъекту с применением фармацевтически приемлемого состава, например, фармацевтически приемлемого состава, который обеспечивает пролонгированную доставку соединения субъекту в течение по меньшей мере 12 часов, 24 часов, 36 часов, 48 часов, одной недели, двух недель, трех недель или четырех недель после введения субъекту фармацевтически приемлемого состава.
Точные уровни доз и временной порядок введения активных ингредиентов в фармацевтических композициях согласно настоящему изобретению может варьироваться таким образом, чтобы получить количество активного ингредиента, которое эффективно для достижения желательного терапевтического ответа для конкретного пациента, композиции и способа введения, без возникновения токсичных эффектов (или неприемлемых токсичных эффектов) в организме пациента.
При применении по меньшей мере одно соединение согласно настоящему изобретению вводится в фармацевтически эффективном количестве субъекту, нуждающемуся в этом, в фармацевтическом носителе, путем внутривенной, внутримышечной, подкожной или интрацеребровентрикулярной инъекции, или путем перорального введения, или путем местного введения. В соответствии с настоящим изобретением соединение согласно настоящему изобретению может вводиться само по себе или в комбинации со вторым отличным терапевтическим средством. Выражение "в комбинации с" означает совместное введение, по существу одновременное введение или последовательное введение. В одном варианте выполнения настоящего изобретения соединение согласно настоящему изобретению вводится пронзительно. Соединение согласно настоящему изобретению может, таким образом, вводиться в течение короткого курса лечения, такого как от около 1 дня до около 1 недели. В другом варианте выполнения настоящего изобретения соединение согласно настоящему изобретению может вводиться в течение более длительного периода лечения для улучшения хронических нарушений, такого как, например, в течение от около одной недели до нескольких месяцев, в зависимости от состояния, которое подлежит лечению.
Термин "фармацевтически эффективное количество", как применяется в описании настоящего изобретения, означает количество соединения согласно изобретению, достаточно высокого для существенной положительной модификации состояния, которое подлежит лечению, но достаточно низкого, чтобы избежать серьезных побочных эффектов (при целесообразном соотношении благоприятный эффект/риск), по результатам тщательной медицинской клинической оценки. Фармацевтически эффективное количество соединения согласно настоящему изобретению будет варьироваться в зависимости от конкретной цели, которая должна быть достигнута, возраста и физического состояния пациента, подлежащего лечению, серьезности излечиваемого заболевания, продолжительности лечения, природы сопутствующей терапии и конкретного применяемого соединения. Например, терапевтически эффективное количество, вводимое ребенку или новорожденному, будет пропорционально уменьшено в соответствии с тщательной медицинской клинической оценки. Эффективным количеством соединения согласно настоящему изобретению будет, таким образом, минимальное количество, которое будет обеспечивать желательный эффект.
Несомненным преимуществом настоящего изобретения при практическом осуществлении является то, что соединение может вводиться обычным образом, например с помощью таких путей введения как внутривенный, внутримышечный, подкожный, пероральный или интрацеребровентрикулярная инъекция, или путем местного введения, например, в виде кремов или гелей. В зависимости от маршрута пути, активные ингредиенты, которые содержат соединение согласно настоящему изобретению, могут требовать покрытия материалом, для защиты соединения от действия ферментов, кислот и других естественных условий, которые могут инактивировать соединение. Для того чтобы ввести соединение согласно изобретению путем введения, отличного от парентерального, соединение может быть покрыто материалом для предотвращения инактивации или вводиться вместе с ним.
Соединение может вводиться парентерально или интраперитонеально. Дисперсии могут также быть получены, например, в глицерине, жидких полиэтиленгликолях и их смесях, или в маслах.
Некоторыми примерами веществ, которые могут служить в качестве фармацевтических носителей, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетаты целлюлозы; порошкообразная трагакантовая камедь; солод; желатин; тальк; стеариновые кислоты; стеарат магния; сульфат кальция; растительные масла, такие как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло, кукурузное масло и какао-масло; полиолы, такие как пропиленгликоль, глицерин, сорбит, маннит и полиэтиленгликоль; агар; альгиновые кислоты; апирогенная вода; изотонический солевой раствор; и раствор фосфатного буфера; порошок из сухого молока; а также другие нетоксичные совместимые вещества, применяемые в фармацевтических композициях, такие как витамин C, эстроген и эхинацея, например. Также могут присутствовать смачивающие средства и смазывающие средства, такие как лаурилсульфат натрия, а также окрашивающие агенты, ароматизирующие вещества, лубриканты, эксципиенты, таблетирующие агенты, стабилизаторы, антиоксиданты и консерванты. Солюбилизирующие агенты, включая, например, кремофор и бета-циклодекстрины, также могут применяться в фармацевтических композициях согласно настоящему изобретению.
Фармацевтические композиции, содержащие активные соединения согласно раскрываемому объекту изобретения (или их пролекарства) могут быть получены посредством обычных процессов смешивания, растворения, грануляции, растирания в порошок с получением драже, эмульгирования, инкапсуляции, включения или лиофилизации. Композиции могут быть получены обычным образом, с применением одного или более фармацевтически приемлемых носителей, разбавителей, эксципиентов или вспомогательных веществ, которые способствуют превращению активных соединений в препараты, которые могут применяться фармацевтическим путем.
Фармацевтические композиции, согласно раскрываемому объекту изобретения, могут иметь форму, подходящую практически для любого пути введения, включая, например, местное введение, окулярное введение, пероральное введение, буккальное введение, системное введение, назальное введение, инъекцию, трансдермальное введение, ректальное введение, вагинальное введение и тому подобное, или форму, подходящую для введения путем ингаляции или вдувания.
Для местного введения активное соединение (активные соединения) или пролекарство(а) могут быть получены в виде растворов, гелей, мазей, кремов, суспензий и тому подобного.
Системные составы включают составы, разработанные для введения путем инъекции, например, подкожной, внутривенной, внутримышечной, интратекальной или интраперитонеальной инъекции, а также составы, разработанные для трансдермального, трансмукозального, перорального или пульмонального введения.
Применяемые инъецируемые препараты включают стерильные суспензии, растворы или эмульсии активного соединения (активных соединений) в водной или масляной среде Композиции могут также содержать вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Составы для инъекции могут быть представлены в стандартной лекарственной форме (например, в ампулах или в контейнерах, содержащих множество доз) и могут содержать добавленные консерванты.
Альтернативным образом, инъецируемый состав может быть получен в форме порошка для восстановления подходящей средой, включая, но без ограничения к этому, стерильную апирогенную воду, буфер, раствор декстрозы и тому подобное, перед применением. С этой целью активное соединение (активные соединения) может быть высушено (могут быть высушены) любим известным в данной области техники методом, таким как лиофилизация, и восстановлены перед применением.
Для трансмукозального введения в составе применяются пенетранты, подходящие для барьера, через который необходимо проникнуть. Такие пенетранты известны в данной области техники.
Для перорального введения фармацевтические композиции могут иметь форму, например, лепешек, таблеток или капсул, полученных обычными средствами с фармацевтически приемлемыми эксципиентами, такими как связывающие агенты (например, предварительно желатинизированный крахмал маиса, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); смазывающие вещества (например, стеарат магния, тальк или диоксид кремния); дезинтегрирующие средства (например, картофельный крахмал или натрия крахмал гликолят); или смачивающие агенты (например, натрия лаурилсульфат). Таблетки могу быть покрыты способами, известными в данной области техники, например, сахарами или энтеросолюбильными покрытиями.
Жидкие препараты для перорального введения могут иметь форму, например, эликсиров, растворов, сиропов или суспензий, или они могут присутствовать в виде сухого продукта для разведения водой или другим подходящим носителем перед применением. Такие жидкие препараты могут быть получены с помощью обычных средств с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, производные целлюлозы или гидрогенизированные пищевые жиры); эмульгирующие агенты (например, лецитин или акация); неводными средами (например, миндальное масло, масляные сложные эфиры, этиловый спирт или фракционированные растительные масла); и консерванты (например, метил или пропил п-гидроксибензоаты или сорбиновая кислота). Препараты также могут содержать буферные соли, консерванты, ароматизирующие вещества, окрашивающие и подслащивающие агенты, при необходимости.
Препараты для перорального введения могут быть подходящим образом получены с обеспечением контролируемого высвобождения активного соединения или пролекарства, как хорошо известно.
Для буккального введения композиции могут иметь форму таблеток или лепешек, полученных обычным образом.
Для ректального и вагинального путей введения активное соединение(ия) могут быть приготовлены в виде растворов (для удерживающих клизм), суппозиториев или мазей, содержащих традиционные основы суппозитория, такие как какао-масло или другие глицериды.
Для назального введения или введения путем ингаляции или вдувания активное соединение (активные соединения) или пролекарство(а) могут, как правило, доставляться в форме аэрозольного спрея из контейнера, находящегося под давлением, или распылителя, с применением подходящего газа-вытеснителя, как например, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, фторуглероды, диоксид углерода или другого подходящего газа. В случае сжатого аэрозоля, единица дозы может быть определена с помощью обеспечения клапана для доставки отмеренного количества. Капсулы и картриджи для применения в ингаляторах или вдувателях (например, капсулы и картриджи, состоящие из желатина) могут быть получены с содержанием порошкообразной смеси соединения и подходящей порошкообразной основы, такой как лактоза или крахмал.
Конкретный пример состава в виде водной суспензии, подходящего для назального введения с применением доступных для приобретения коммерческим путем устройств для распыления в нос, включает следующие ингредиенты: активное соединение или пролекарство (0.5-20 мг/мл); бензалкония хлорид (0.1-0.2 мг/мл); полисорбат 80 (TWEEN® 80; 0.5-5 мг/мл); натрий-карбоксиметилцеллюлозу или микрокристаллическую целлюлозу (1-15 мг/мл); фенилэтанол (1-4 мг/мл); и декстрозу (20-50 мг/мл). Значение pH конечной суспензии может быть доведено до интервала от около 5 до 7, причем pH около 5.5 является типичным.
Для окулярного введения активное(ые) соединение(ия) или пролекарство(а) получают в виде раствора, эмульсии, суспензии или тому подобного, подходящих для введения в глаза. В данной области техники известно многообразии сред, подходящих для введения соединений в глаза. Конкретные неограничивающие примеры приводятся в Патенте США № 6,261,547; Патенте США № 6,197,934; Патенте США № 6,056,950; Патенте США № 5,800,807; Патенте США № 5,776,445; Патенте США № 5,698,219; Патенте США № 5,521,222; Патенте США № 5,403,841; Патенте США № 5,077,033; Патенте США № 4,882,150; и Патенте США № 4,738,851, каждый из которых включен в настоящий документ в полном объеме путем ссылки.
Для пролонгированной доставки активное(ые) соединение(ия) или пролекарство(а) могут быть получены в виде препарата-депо для введения путем имплантации или внутримышечной инъекции. Активный ингредиент может входить в состав вместе с подходящими полимерными или гидрофобными материалами (например, как эмульсия в приемлемом масле) или ион-обменными смолами, или в виде труднорастворимых производных, например, в виде труднорастворимой соли. Альтернативным образом, могут применяться трансдермальные системы доставки получают в виде трансдермального диска или пластыря, который медленно высвобождает активное(ые) соединение(ия) для подкожной абсорбции. С этой целью могут применяться усилители проникновения для облегчения трансдермального проникновения активного(ых) соединения(ий). Подходящие трансдермальные пластыри описываются, например, в Патенте США № 5,407,713; Патенте США № 5,352,456; Патенте США № 5,332,213; Патенте США № 5,336,168; Патенте США № 5,290,561; Патенте США № 5,254,346; Патенте США № 5,164,189; Патенте США № 5,163,899; Патенте США № 5,088,977; Патенте США № 5,087,240; Патенте США № 5,008,110; и Патенте США № 4,921,475, каждый из которых включен в настоящий документ в полном объеме путем ссылки.
Альтернативным образом, могут применяться другие фармацевтические системы доставки. Липосомы и эмульсии являются хорошо известными примерами носителей для доставки, которые могут применяться для доставки активного(ых) соединения(ий) или пролекарства (пролекарств). Также могут применяться определенные органические растворители, такие как диметилсульфоксид (DMSO).
Если желательно, фармацевтические композиции могут присутствовать в виде набора или раздаточного устройства, которые могут содержать одну или более стандартных лекарственных форм, содержащих активное соединение (активные соединения). Набор может, например, содержать металлическую или пластиковую фольгу, как например блистерная упаковка. Набор или блистерная упаковка могут сопровождаться инструкциями по применению.
Активное соединение (активные соединения) или пролекарство(а) согласно раскрываемому объекту изобретения или их композиции, как правило, будут применятся в количестве, эффективном для достижения желательного результата, например, в количестве, эффективном для лечения или профилактики конкретного заболевания, подлежащего лечению. Соединение(ия) может (могут) вводиться терапевтически для достижения терапевтической пользы или профилактически для достижения профилактической пользы. Под терапевтической пользой понимается ликвидация или уменьшение рассматриваемого нарушения, подлежащего лечению, и/или ликвидация или уменьшение одного или более симптомов, связанных с рассматриваемым нарушением, так что улучшается самочувствие или состояние пациента, несмотря на то, что пациент может все еще страдать от рассматриваемого нарушения. Например, введение соединения пациенту, страдающему от аллергии, обеспечивает терапевтическую пользу, но только если рассматриваемая аллергическая реакция исключается или уменьшается, а также если у пациента наблюдается уменьшение серьезности или продолжительности симптомов, связанных с аллергией, после воздействия аллергена. Другой пример, терапевтическая польза в контексте астмы включает улучшение дыхания после начала приступа астмы, или уменьшение частоты или серьезности астматических приступов. Терапевтическая польза также включает прекращение или замедление развития заболевания, независимо от того происходит ли улучшение.
В отношении профилактического введения необходимо отметить, что соединение может вводиться пациент в случае риска развития одного из ранее описанных заболеваний. Пациент, в случае риска развития заболевания, может быть пациентом, обладающим характеристиками, приводящими к помещению пациента в обозначенную группу риска, как определено соответствующим профессионалом в области медицины или группой. Пациентом с риском также может быть пациент, которые обычно или регулярно находится в среде, где происходит развитие рассматриваемого заболевания, которое можно лечить путем введения ингибитора металлоферментов согласно настоящему изобретению. Другими словами, пациентом с риском является пациент, который обычно или регулярно подвергается воздействию условий, вызывающий болезнь или расстройство, или может быть подвержен в высокой степени в течение ограниченного периода времени. Альтернативным образом, профилактическое введение может применяться, чтобы избежать возникновения у пациентов симптомов, позволяющих диагностировать рассматриваемое заболевание.
Количество вводимого соединения будет завесить от множества факторов, включая, например, конкретное показание, подлежащее лечению, путь введения желательно ли достичь профилактики либо лечения, серьезность показания, подлежащего лечению, и возраст и вес пациента, биодоступность конкретного активного соединения и тому подобное. Определение эффективной дозы находится в компетенции специалиста в данной области техники.
Эффективные дозы могут быть оценены первоначально на основе анализов в vitro. Например, начальная доза для применения на животных может быть получена для достижения концентрации активного соединения в циркулирующей крови или сыворотке, которая равно или больше IC50 конкретного соединения, как измерено в анализе in vitro, как например in vitro грибковая MIC или MFC и других in vitro анализах, описанных в разделе Примеры. Расчет доз для достижения таких концентраций в циркулирующей крови или сыворотке учитывает биодоступность конкретного соединения, что входит в компетенцию специалиста в данной области техники. Для руководства смотрите Fingl & Woodbury, “General Principles,” In: Goodman and Gilman’s The Pharmaceutical Basis of Therapeutics, Chapter 1, pp. 1-46, latest edition, Pagamonon Presc, и ссылки, приведенные там, которые включены в настоящий документ посредством ссылки.
Первоначальные дозы также могут быть оценены на основании in vivo данных, как например животные модели. Животные модели, полезные для тестирования эффективности соединений для лечения или профилактики различных заболеваний, описанных выше, хорошо известны в данной области техники.
Количества доз будут, как правило, находится в интервале от около 0.0001 или 0.001 или 0.01 мг/кг/день до около 100 мг/кг/день, но могут быть выше или ниже, в зависимости от, среди других факторов, активности соединения, его биодоступности, пути введения и различных факторов, обсуждаемых выше. Количество доз и интервал введения может регулироваться индивидуально для обеспечения уровней в плазме соединения(ий), которые достаточны для поддержания терапевтического или профилактического эффекта. В случаях локального введения или селективного поглощения, как например локальное местное введение, эффективная локальная концентрация активного соединения (активных соединений) не может быть связана с концентрацией в плазме. Специалисты в данной области техники смогут оптимизировать эффективные локальные дозы без дополнительных экспериментов.
Соединение (соединения) может (могут) вводиться один раз в день, немного или несколько раз в день, или даже множество раз в день, в зависимости от, среди прочего, показания, подлежащего лечению, и предписания лечащего врача.
Предпочтительно соединение(ия) будет (будут) обеспечивать терапевтическую или профилактическую пользу, не вызывая существенной токсичности. Токсичность соединения(ий) может быть определена с применением стандартных фармацевтических методик. Отношение доз между токсическим и терапевтическим (или профилактическим) эффектами представляет собой терапевтический индекс. Предпочтительным (предпочтительными) является (являются) соединение (соединения), которое (которые) проявляет (проявляют) высокий терапевтический индекс.
Перечисление списка химических групп в любом определении переменной в описании настоящего изобретения включает определения этой переменной как любой одиночной группы или комбинации перечисленных групп. Приведение варианта выполнения изобретения для переменной, указанной в настоящем изобретении, включает этот вариант в виде любого одиночного варианта выполнения или в комбинации с любыми другими вариантами выполнения или их частями. Приведение варианта выполнения изобретения в настоящей заявке включает этот вариант в виде любого одиночного варианта выполнения или в комбинации с любыми другими вариантами выполнения или их частями.
Примеры
Для того чтобы изобретение, описанное в настоящей заявке, могло быть понято более полно, изложены следующие примеры. Примеры, описанные в настоящей заявке, предлагаются для иллюстрации соединений, фармацевтических композиций и способов, обеспеченных в настоящей заявке, и сами по себе не должны рассматриваться как ограничивающие их объем настоящего изобретения.
Общие экспериментальные методики
Определения переменных в структурах на схемах, приведенных в настоящей заявке, соответствуют определениям соответствующих положений в формулах, приведенных в настоящей заявке. Примеры соединений, перечисленных в таблице 2, были охарактеризованы методами ВЭЖХ и LCMS, описанными в таблице 1.
Таблица 1. ВЭЖХ и LCMS способы
Общие аббревиатуры:
ACN ацетонитрил
br широкий
d дуплет
dd дуплет дуплетов
dba дибензилиденацетон
DIPEA диизопропилэтиламин
dppf 1,1'-ферроцендиил-бис(дифенилфосфин)
ч час(часы)
HRMS масс-спектрометрия высокого разрешения
ВЭЖХ высокоэффективная жидкостная хроматография
LCMS жидкостная хроматография и масс-спектрометрия
MS масс-спектрометрия
MW микроволны
m мультиплет
мин минуты
мл миллилитр(ы)
m/z соотношение массы и заряда
ЯМР ядерный магнитный резонанс
ppm миллионные доли
кт или RT комнатная температура
s синглет
t триплет
TLC тонкослойная хроматография
Получение Int-1
Схема:
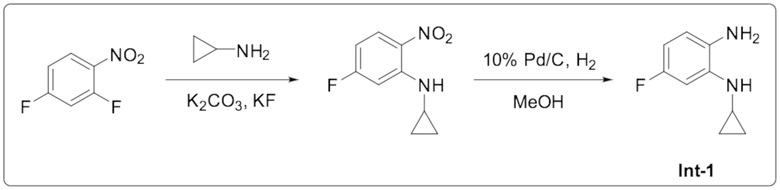
N-Циклопропил-5-фтор-2-нитроанилин
К перемешиваемому раствору 2,4-дифтор-1-нитробензола (25 г, 157.23 ммоль) во фториде калия (9.12 г, 157.23 ммоль) в инертной атмосфере добавили карбонат калия (21.7 г, 157.23 ммоль), а затем циклопропанамин (10.75 г, 188.68 ммоль) по каплям при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После определения израсходования исходного вещества посредством тонкослойной хроматографии (TLC), реакционную смесь разбавили водой (200 мл) и экстрагировали с помощью этилацетата (EtOAc) (2 x 200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% EtOAc/гексан) с получением N-циклопропил-5-фтор-2-нитроанилина (26 г, 132.65 ммоль, 84%) в виде твердого вещества желтого цвета.
1H ЯМР (500МГц, CDCl3): δ 8.20 (dd, J = 9.3, 6.1 Гц, 1H), 6.95 (dd, J = 11.4, 2.7 Гц, 1H), 6.45-6.39 (m, 1H), 2.59-2.53 (m, 1H), 0.97-0.92 (m, 2H), 0.71-0.65 (m, 2H).
N 1-Циклопропил-5-фторбензол-1, 2-диамин (Int-1)
К перемешиваемому раствору N-циклопропил-5-фтор-2-нитроанилина (24 г, 122.45 ммоль) в метаноле (MeOH) (300 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 2.4 г) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли метанолом (100 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклопропил-5-фторбензол-1, 2-диамина Int-1 (18 г, 108.43 ммоль, 88%) в виде сиропа коричневого цвета.
1H ЯМР (400МГц, DMSO-d6): δ 6.52 (dd, J = 11.6, 2.8 Гц, 1H), 6.45 (dd, J = 8.4, 6.0 Гц, 1H), 6.18 (td, J = 8.5, 2.9 Гц, 1H), 5.28 (s, 1H), 4.30 (br s, 2H), 2.36-2.28 (m, 1H), 0.74-0.68 (m, 2H), 0.42-0.37 (m, 2H).
LC-MS: m/z 166.8 [M+H]+ при 1.64 RT (72.46% чистота).
Получение Int-2
Схема:
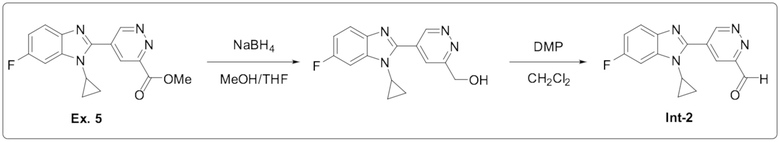
(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанол
К перемешиваемому раствору метил 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбоксилата (250 мг, 0.8 ммоль) в смеси метанол/тетрагидрофурана (THF) (2:1, 15 мл) в инертной атмосфере добавили боргидрид натрия (152 мг, 4.01 ммоль) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь концентрировали при пониженном давлении. Остаток разбавили водой (25 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением (5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метанола (130 мг, 0.46 ммоль, 57%) в виде твердого вещества коричневого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 284.9 [M+H]+ при 1.94 RT (72.20% чистота).
5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегид (Int-2)
К перемешиваемому раствору (5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метанола (130 мг, 0.46 ммоль) в CH2Cl2 (10 мл) в инертной атмосфере добавили периодинан Десса-Мартина (DMP) (291 мг, 0.69 ммоль) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-2 (50 мг, 0.18 ммоль, 39%) в виде твердого вещества коричневого цвета.
1H ЯМР (500МГц, CDCl3): δ 10.50 (s, 1H), 10.06 (d, J = 2.0 Гц, 1H), 8.64 (d, J = 2.3 Гц, 1H), 7.80 (dd, J = 8.8, 4.8 Гц, 1H), 7.34 (dd, J = 8.4, 2.3 Гц, 1H), 7.13 (td, J = 9.3, 2.3 Гц, 1H), 3.73-3.69 (m, 1H), 1.37-1.31 (m, 2H), 0.90-0.85 (m, 2H).
LC-MS: m/z 282.9 [M+H]+ при 1.85 RT (90.42% чистота).
Получение Int-3
Схема:
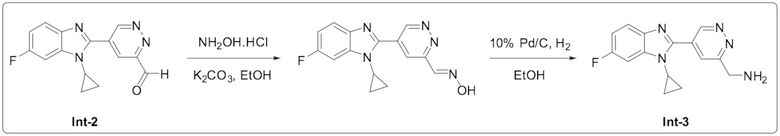
(E)-5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегидоксим
К перемешиваемому раствору 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-2 (100 мг, 0.35 ммоль) в этаноле (EtOH) (2 мл) в инертной атмосфере добавили гидроксиламин гидрохлорид (49 мг, 0.71 ммоль) и карбонат калия (98 мг, 0.71 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь концентрировали при пониженном давлении. Остаток разбавили водой (40 мл) и перемешивали в течение 10 мин. Осажденное твердое вещество отфильтровали, промыли водой (10 мл) и высушили в вакууме с получением (E)-5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегидоксима (70 мг, 0.23 ммоль, 67%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 12.25 (s, 1H), 9.81 (s, 1H), 8.56 (s, 1H), 8.48 (s, 1H), 7.80 (dd, J = 8.7, 4.9 Гц, 1H), 7.55 (dd, J = 9.0, 1.7 Гц, 1H), 7.20 (td, J = 9.8, 2.3 Гц, 1H), 3.95-3.89 (m, 1H), 1.20-1.16 (m, 2H), 0.84-0.79 (m, 2H).
LC-MS: m/z 297.9 [M+H]+ при 1.95 RT (99.11% чистота).
(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанамин (Int-3)
К перемешиваемому раствору (E)-5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегидоксима (70 мг, 0.23 ммоль) в этаноле (5 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 25 мг) при комнатной температуре. Реакционную смесь вакуумировали и перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью MeOH/CH2Cl2 (10:1, 20 мл). Фильтрат концентрировали при пониженном давлении с получением (5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метанамина Int-3 (50 мг, неочищенный) в виде твердого вещества грязновато-белого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 283.9 [M+H]+ при 1.62 RT (64.40% чистота).
Получение Int-4
Схема:
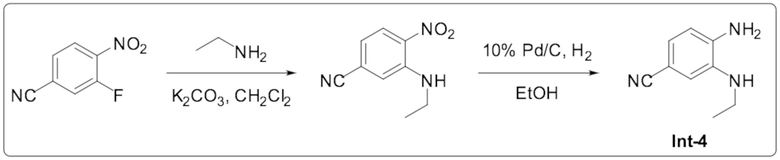
3-(Этиламино)-4-нитробензонитрил
К перемешиваемому раствору 3-фтор-4-нитробензонитрила (2 г, 12.05 ммоль) в CH2Cl2 (250 мл) в инертной атмосфере добавили карбонат калия (3.32 г, 24.09 ммоль) и этиламин (Aq. 70%, 2.17 г, 48.19 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили водой (60 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3-(этиламино)-4-нитробензонитрила (1.9 г, неочищенный) в виде твердого вещества желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
1H ЯМР (500МГц, DMSO-d6): δ 8.22-8.10 (m, 2H), 7.58 (br s, 1H), 7.00 (br d, J = 8.7 Гц, 1H), 3.48-3.38 (m, 2H), 1.21 (br t, J = 6.9 Гц, 3H).
LC-MS: m/z 192.1 [M+H]+ при 4.10 RT (98.96% чистота).
4-Амино-3-(этиламино) бензонитрил (Int-4)
К перемешиваемому раствору 3-(этиламино)-4-нитробензонитрила (1.9 г, неочищенный) в этаноле (20 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 190 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли метанолом (50 мл) и EtOAc (30 мл). Фильтрат концентрировали при пониженном давлении с получением 4-амино-3-(этиламино) бензонитрила Int-4 (1.5 г, неочищенный) в виде твердого вещества грязновато-белого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 161.9 [M+H]+ при 2.11 RT (60.88% чистота).
Получение Int-5
Схема:
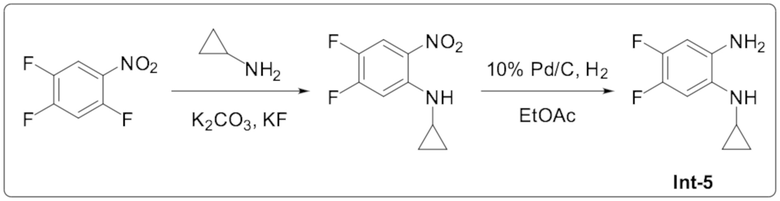
N-Циклопропил-4, 5-дифтор-2-нитроанилин
К 1, 2, 4-трифтор-5-нитробензолу (500 мг, 2.82 ммоль) во фториде калия (164 мг, 2.82 ммоль) в инертной атмосфере добавили карбонат калия (390 мг, 2.82 ммоль) и циклопропанамин (0.23 мл, 3.39 ммоль) по каплям при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-циклопропил-4, 5-дифтор-2-нитроанилина 2 (320 мг, неочищенный) в виде твердого вещества бледно-желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 215.4 [M+H]+ при 4.43 RT (69.03% чистота).
N 1-Циклопропил-4, 5-дифторбензол-1, 2-диамин (Int-5)
К перемешиваемому раствору N-циклопропил-4, 5-дифтор-2-нитроанилина (300 мг, 1.4 ммоль) в EtOAc (10 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 30 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли метанолом (15 мл) и EtOAc (10 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклопропил-4, 5-дифторбензол-1,2-диамина Int-5 (100 мг, неочищенный) в виде вязкого сиропа коричневого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 184.9 [M+H]+ при 2.12 RT (83.53% чистота).
Получение Int-6 и Int-7
Схема:
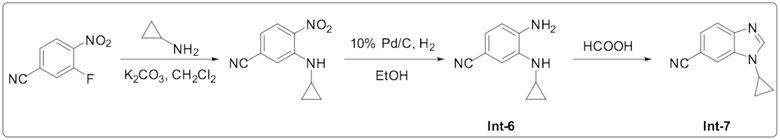
3-(Циклопропиламино)-4-нитробензонитрил
К перемешиваемому раствору 3-фтор-4-нитробензонитрила (1 г, 6.02 ммоль) в CH2Cl2 (5 мл) в инертной атмосфере добавили карбонат калия (1.66 г, 12.05 ммоль) и циклопропанамин (3.33 мл, 48.19 ммоль) по каплям при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3-(циклопропиламино)-4-нитробензонитрил (900 мг, неочищенный) в виде твердого вещества желтого цвета применяемого на следующей стадии без дальнейшей очистки.
1H ЯМР (400МГц, CDCl3): δ 8.24 (d, J = 8.7 Гц, 1H), 8.07 (br s, 1H), 7.64 (d, J = 1.7 Гц, 1H), 6.93 (dd, J = 8.7, 1.7 Гц, 1H), 2.62-2.57 (m, 1H), 1.03-0.97 (m, 2H), 0.72-0.67 (m, 2H).
LC-MS: m/z 201.9 [M-H]+ при 3.25 RT (99.61% чистота).
4-Амино-3-(циклопропиламино) бензонитрил (Int-6)
К перемешиваемому раствору 3-(циклопропиламино)-4-нитробензонитрила (900 мг, 4.43 ммоль) в этаноле (10 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 500 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью EtOAc (30 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 4-амино-3-(циклопропиламино) бензонитрила Int-6 (500 мг, 2.89 ммоль, 65%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400МГц, DMSO-d6): δ 6.93 (d, J = 1.9 Гц, 1H), 6.87 (dd, J = 8.0, 1.9 Гц, 1H), 6.55 (d, J = 8.0 Гц, 1H), 5.49 (s, 2H), 5.43 (s, 1H), 2.40-2.34 (m, 1H), 0.77-0.71 (m, 2H), 0.42-0.37 (m, 2H).
1-Циклопропил-1H-бензо[d]имидазол-6-карбонитрил (Int-7)
Раствор 4-амино-3-(циклопропиламино) бензонитрила Int-6 (500 мг, 2.89 ммоль) в муравьиной кислоте (5 мл) в инертной атмосфере нагрели до температуры возврата флегмы и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток разбавили водой (10 мл), нейтрализовали насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением 1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила Int-7 (250 мг, 1.37 ммоль, 56%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 8.50 (s, 1H), 8.21 (d, J = 0.9 Гц, 1H), 7.82 (d, J = 8.1 Гц, 1H), 7.62 (dd, J = 8.4, 1.4 Гц, 1H), 3.59-3.54 (m, 1H), 1.15-1.05 (m, 4H).
LC-MS: m/z 184.0 [M+H]+ при 2.46 RT (87.33% чистота).
Получение Int-8
Схема:
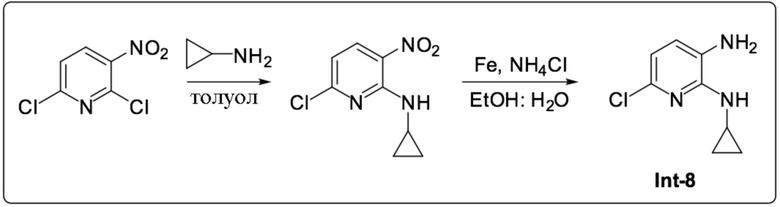
6-Хлор-N-циклопропил-3-нитропиридин-2-амин
К перемешиваемому раствору 2, 6-дихлор-3-нитропиридина (5 г, 26.04 ммоль) в толуоле (25 мл) в инертной атмосфере добавили циклопропил амин (3.7 мл, 52.08 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/Гексан) с получением 6-хлор-N-циклопропил-3-нитропиридин-2-амина (4 г, 18.77 ммоль, 72%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 8.34 (d, J = 8.7 Гц, 2H), 6.66 (d, J = 8.7 Гц, 1H), 3.10-3.05 (m, 1H), 0.97-0.93 (m, 2H), 0.67-0.64 (m, 2H)
6-Хлор-N2-циклопропилпиридин-2, 3-диамин (Int-8)
К перемешиваемому раствору 6-хлор-N-циклопропил-3-нитропиридин-2-амина (1 г, 4.69 ммоль) в EtOH: вода (1: 1, 10 мл) в инертной атмосфере добавили железо (1.3 г, 23.47 ммоль) и хлорид аммония (1.2 г, 23.47 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6-хлор-N2-циклопропилпиридин-2,3-диамина Int-8 (700 мг, неочищенный) в виде твердого вещества бледно-желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 183.9 [M+H]+ при 2.07 RT (58.13% чистота).
Получение Int-9
Схема:
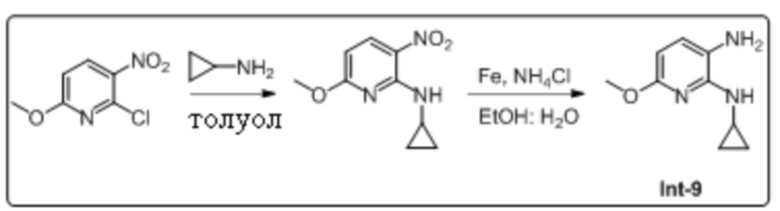
N-Циклопропил-6-метокси-3-нитропиридин-2-амин
К перемешиваемому раствору 2-хлор-6-метокси-3-нитропиридина (5 г, 26.5 ммоль) в толуоле (50 мл) в инертной атмосфере добавили циклопропил амин (3.69 мл, 53 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и нагрели до комнатной температуры и перемешивали в течение 6 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный материал перетерли с н-пентаном с получением N-циклопропил-6-метокси-3-нитропиридин-2-амина (800 мг, 3.82 ммоль, 83%) в виде твердого вещества желтого цвета.
N 2-Циклопропил-6-метоксипиридин-2,3-диамин (Int-9)
К перемешиваемому раствору of N-циклопропил-6-метокси-3-нитропиридин-2-амина (1 г, 4.78 ммоль) в этаноле/воде (1: 1, 10 мл) добавили порошок железа (1.3 г, 24 ммоль) и хлорид аммония (1.29 г, 23.9 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (50 мл). Фильтрат концентрировали при пониженном давлении с получением N2-циклопропил-6-метоксипиридин-2,3-диамина Int-9 (150 мг) в виде жидкости черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, DMSO-d6): δ 6.71 (d, J = 7.9 Гц, 1H), 5.78 (d, J = 7.9 Гц, 1H), 4.09-4.06 (m, 2H), 3.84-3.81 (m, 1H), 3.70 (s, 3H), 2.77-2.68 (m, 1H), 0.69-0.61 (m, 2H), 0.43-0.37 (m, 2H)
LC-MS: m/z 180.1 [M+H]+ при 2.11 RT (75.36% чистота)
Получение Int-10
Схема:
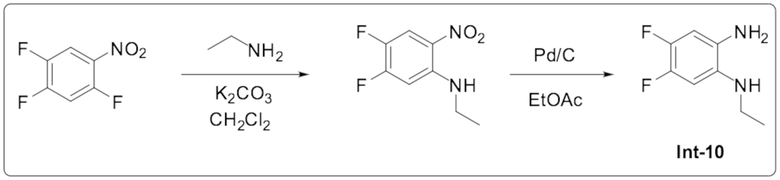
N-Этил-4,5-дифтор-2-нитроанилин
К перемешиваемому раствору 1,2,4-трифтор-5-нитробензола (2 г, 11.29 ммоль) в CH2Cl2 (10 мл) добавили карбонат калия (3.1 г, 22.59 ммоль) и этанамин (559 мг, 12.42 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (100 мл) и экстрагировали с помощью CH2Cl2 (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/гексан) с получением N-этил-4,5-дифтор-2-нитроанилина (800 мг, 3.96 ммоль, 35%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 8.19 (brs, 1H), 8.13 (dd, J = 11.0, 8.7 Гц, 1H), 7.14 (dd, J = 13.9, 7.0 Гц, 1H), 3.40-3.33 (m, 2H), 1.20 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 203.1 [M+H]+ при 3.53 RT (98.40% чистота)
N 1-Этил-4,5-дифторбензол-1,2-диамин (Int-10)
К перемешиваемому раствору N-этил-4,5-дифтор-2-нитроанилина (800 мг, 3.96 ммоль) в этилацетате (5 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 500 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью EtOAc (50 мл). Фильтрат концентрировали при пониженном давлении с получением N1-этил-4,5-дифторбензол-1,2-диамина Int-10 (500 мг, 2.90 ммоль, 73%) в виде жидкости черного цвета, которую применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.47 (dd, J = 12.8, 8.1 Гц, 1H), 6.30 (dd, J = 13.3, 8.1 Гц, 1H), 4.64 (br s, 2H), 4.44 (t, J = 5.2 Гц, 1H), 3.00-2.94 (m, 2H), 1.17 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 173.2 [M+H]+ при 3.16 RT (64.65% чистота)
Получение Int-11
Схема:
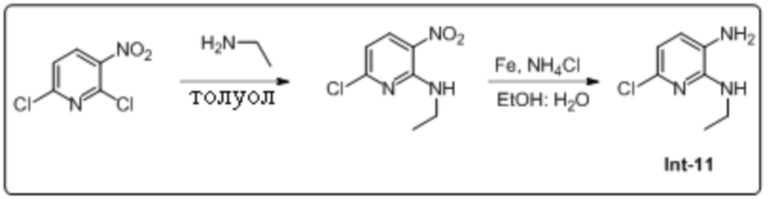
6-Хлор-N-этил-3-нитропиридин-2-амин
К перемешиваемому раствору 2,6-дихлор-3-нитропиридина (2 г, 10.40 ммоль) в толуоле (8.5 мл) в инертной атмосфере добавили этиламин (1.35 мл) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли и высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% EtOAc/гексан) с получением 6-хлор-N-этил-3-нитропиридин-2-амина (800 мг, 3.98 ммоль, 38%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 8.70 (brs, 1H), 8.40 (d, J = 7.5 Гц, 1H), 6.75 (d, J = 8.1 Гц, 1H), 3.55-3.50 (m, 2H), 1.18 (t, J = 7.0 Гц, 3H)
LC-MS: m/z 202 [M+H]+ при 2.63 RT (99.85% чистота)
6-Хлор-N2-этилпиридин-2,3-диамин (Int-11)
К перемешиваемому раствору 6-хлор-N-этил-3-нитропиридин-2-амина (550 мг, 2.73 ммоль) в этаноле/воде (1: 1, 20 мл) добавили порошок железа (763.4 г, 13.68 ммоль) и хлорид аммония (738.7 мг, 13.68 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли соляным раствором (30 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6-хлор-N2-этилпиридин-2,3-диамина Int-11 (452 мг) в виде твердого вещества черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.67 (d, J = 7.5 Гц, 1H), 6.33 (d, J = 8.1 Гц, 1H), 5.85 (brs, 1H), 4.88 (br s, 2H), 3.37-3.25 (m, 2H), 1.15 (t, J = 7.0 Гц, 3H)
LC-MS: m/z 172 [M+H]+ при 1.72 RT (85.88% чистота)
Получение Int-12
Схема:
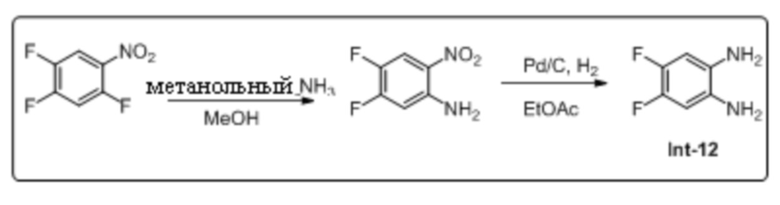
4,5-Дифтор-2-нитроанилин
К перемешиваемому раствору 1,2,4-трифтор-5-нитробензола (5 г, 28.24 ммоль) в метаноле (5 мл) в инертной атмосфере добавили метанольный аммиак (15 мл) при 0°C. Реакционную смесь нагрели до 90°C и перемешивали в течение 2 часов в запаянной трубке. После израсходования исходного вещества (посредством TLC), реакционную смесь концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% EtOAc/гексан) с получением 4,5-дифтор-2-нитроанилина (800 мг, 4.59 ммоль, 16%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 8.00 (dd, J = 10.4, 8.7 Гц, 1H), 6.60 (dd, J = 11.0, 6.4 Гц, 1H), 6.09 (brs, 2H)
LC-MS: m/z 172.8 [M-H]- при 2.49 RT (88.25% чистота)
4,5-Дифторбензол-1,2-диамин (Int-12)
К перемешиваемому раствору 4,5-дифтор-2-нитроанилина (800 мг, 4.59 ммоль) в метаноле (15 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 200 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли метанолом (15 мл) и CH2Cl2 (10 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество промыли н-гексаном (15 мл) с получением 4,5-дифторбензол-1,2-диамина Int-12 (500 мг, 3.47 ммоль, 80%) в виде твердого вещества черного цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 6.47-6.41 (m, 2H), 4.55 (brs, 4H)
LC-MS: m/z 145 [M+H]+ при 1.59 RT (75.16% чистота)
Получение Int-13
Схема:

5-Хлор-N-циклопропил-3-нитропиридин-2-амин
К перемешиваемому раствору  2,5-дихлор-3-нитропиридина (1 г, 5.23 ммоль) в толуоле (10 мл) в инертной атмосфере добавили циклопропиламин (0.73 мл, 10.47 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (40 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5-хлор-N-циклопропил-3-нитропиридин-2-амина (700 мг, 3.28 ммоль, 63%) в виде твердого вещества оранжевого цвета.
2,5-дихлор-3-нитропиридина (1 г, 5.23 ммоль) в толуоле (10 мл) в инертной атмосфере добавили циклопропиламин (0.73 мл, 10.47 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (40 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5-хлор-N-циклопропил-3-нитропиридин-2-амина (700 мг, 3.28 ммоль, 63%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, CDCl3): δ 8.45 (d, J = 2.5 Гц, 1H), 8.41 (d, J = 2.4 Гц, 1H), 8.18 (br s, 1H), 3.05-2.97 (m, 1H), 0.98-0.91 (m, 2H), 0.68-0.62 (m, 2H)
LC-MS: m/z 214 [M+H]+ при 3.03 RT (98.66% чистота)
5-Хлор-N2-циклопропилпиридин-2,3-диамина (Int-13)
К перемешиваемому раствору 5-хлор-N-циклопропил-3-нитропиридин-2-амина (200 мг, 0.938 ммоль) в этаноле/воде (1: 1, 20 мл) добавили порошок железа (262 мг, 4.69 ммоль) и хлорид аммония (253 мг, 4.69 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью EtOAc. Фильтрат разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5-хлор-N2-циклопропилпиридин-2,3-диамина Int-13 (120 мг) в виде твердого вещества черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 7.35 (d, J = 1.7 Гц, 1H), 6.67 (d, J = 2.3 Гц, 1H), 6.00 (br s, 1H), 5.02 (br s, 2H), 2.72-2.63 (m, 1H), 0.68-0.63 (m, 2H), 0.41-0.37 (m, 2H)
LC-MS: m/z 183.9 [M+H]+ при 1.87 RT (88.05% чистота)
Получение Int-14
Схема:

N-Циклопропил-2-фтор-6-нитроанилин
К перемешиваемому раствору 1,2-дифтор-3-нитробензола (200 мг, 1.26 ммоль) в этаноле (2 мл) добавили циклопропанамин (0.13 мл, 1.89 ммоль) при комнатной температуре в инертной атмосфере и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% EtOAc/гексан) с получением N-циклопропил-2-фтор-6-нитроанилина (200 мг, 1.02 ммоль, 81%) в виде вязкого сиропа желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 7.93 (dt, J = 8.8, 1.5 Гц, 1H), 7.82 (brs, 1H), 7.24-7.19 (m, 1H), 6.63-6.58 (m, 1H), 3.12-3.07 (m, 1H), 0.87-0.79 (m, 2H), 0.67-0.61 (m, 2H)
LC-MS: m/z 197.0 [M+H]+ при 3.28 RT (99.89% чистота)
N 1-Циклопропил-6-фторбензол-1,2-диамин (Int-14)
К перемешиваемому раствору N-циклопропил-2-фтор-6-нитроанилина (200 мг, 1.02 ммоль) в этилацетате (5 мл) добавили 10% Pd/C (50% влажность, 30 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью EtOAc (15 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклопропил-6-фторбензол-1,2-диамина Int-14 (160 мг) в виде бесцветного вязкого сиропа. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.61-6.51 (m, 1H), 6.40-6.24 (m, 3H), 4.89 (br s, 2H), 2.62-2.58 (m, 1H), 0.49-0.38 (m, 4H)
Получение Int-15
Схема:
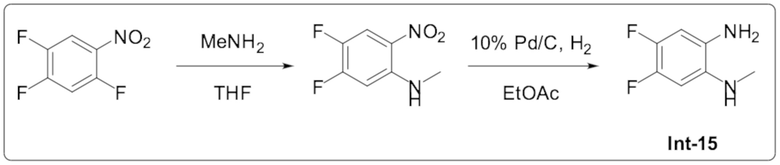
4,5-Дифтор-N-метил-2-нитроанилин
К перемешиваемому раствору 1,2,4-трифтор-5-нитробензола (5 г, 28.25 ммоль) в THF (50 мл) добавили метанамин (2 M в THF, 28.25 мл, 56.5 ммоль) по каплям при -20°C в инертной атмосфере и обеспечили перемешивание при той же температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили соляным раствором (50 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3% EtOAc/гексан) с получением 4,5-дифтор-N-метил-2-нитроанилина (1.1 г, 5.85 ммоль, 20%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 8.06 (dd, J = 10.7, 8.4 Гц, 2H), 6.61 (dd, J = 12.5, 6.7 Гц, 1H), 3.01 (d, J = 5.1 Гц, 3H)
4,5-Дифтор-N1-метилбензол-1,2-диамин (Int-15)
К перемешиваемому раствору 4,5-дифтор-N-метил-2-нитроанилина (1 г, 5.32 ммоль) в этилацетате (15 мл) добавили 10% Pd/C (50% влажность, 250 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целлита, и слой промыли метанолом (30 мл). Фильтрат концентрировали при пониженном давлении с получением 4,5-дифтор-N1-метилбензол-1,2-диамина Int-15 (700 мг) в виде сиропа коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.48 (dd, J = 12.5, 8.4 Гц, 1H), 6.28 (dd, J = 13.3, 8.1 Гц, 1H), 4.71 (s, 1H), 4.59 (br s, 2H), 2.66 (d, J = 5.2 Гц, 3H)
LC-MS: m/z 158.8 [M+H]+ при 2.20 RT (99.55% чистота)
Получение Int-16
Схема:
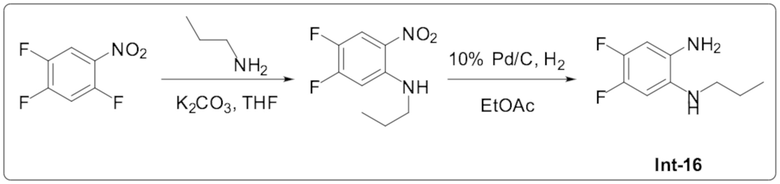
4,5-Дифтор-2-нитро-N-пропиланилин
К перемешиваемому раствору 1,2,4-трифтор-5-нитробензола (5 г, 28.25 ммоль) в THF (150 мл) добавили карбонат калия (5.07 г, 36.72 ммоль) и пропан-1-амин (3.48 мл, 42.37 ммоль) по каплям при комнатной температуре в инертной атмосфере и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3% EtOAc/гексан) с получением 4,5-дифтор-2-нитро-N-пропиланилина (600 мг, 2.77 ммоль, 10%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 8.25 (br s, 1H), 8.14 (dd, J = 11.3, 8.7 Гц, 1H), 7.17 (dd, J = 13.7, 7.2 Гц, 1H), 3.35-3.26 (m, 2H), 1.68-1.56 (m, 2H), 0.93 (t, J = 7.4 Гц, 3H)
4,5-Дифтор-N1-пропилбензол-1,2-диамин (Int-16)
К перемешиваемому раствору 4,5-дифтор-2-нитро-N-пропиланилина (500 мг, 2.31 ммоль) в этилацетате (10 мл) добавили 10% Pd/C (50% влажность, 150 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли метанолом (20 мл). Фильтрат концентрировали при пониженном давлении с получением 4,5-дифтор-N1-пропилбензол-1,2-диамина Int-16 (350 мг) в виде сиропа коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.48 (dd, J = 12.8, 8.1 Гц, 1H), 6.30 (dd, J = 13.3, 8.1 Гц, 1H), 4.66 (s, 2H), 4.47 (br t, J = 4.6 Гц, 1H), 2.96-2.88 (m, 2H), 1.62-1.54 (m, 2H), 0.95 (t, J = 7.5 Гц, 3H)
LC-MS: m/z 186.9 [M+H]+ при 2.88 RT (89.94% чистота)
Получение Int-17
Схема:
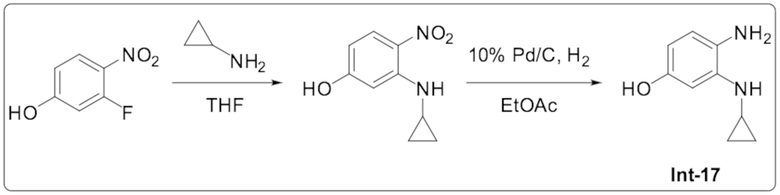
3-(Циклопропиламино)-4-нитрофенол
К перемешиваемому раствору 3-фтор-4-нитрофенола (1 г, 6.37 ммоль) в THF (20 мл) добавили циклопропанамин (726 мг, 12.74 ммоль) в запаянной трубке при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 80°C и перемешивали в течение 6 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в ледяную воду (30 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 3-(циклопропиламино)-4-нитрофенола (1.2 г, 6.18 ммоль, 75%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.86 (br s, 1H), 8.15 (br s, 1H), 7.97 (d, J = 9.3 Гц, 1H), 6.61 (d, J = 1.7 Гц, 1H), 6.22 (dd, J = 9.3, 2.3 Гц, 1H), 2.57-2.55 (m, 1H), 0.91-0.82 (m, 2H), 0.67-0.57 (m, 2H)
LC-MS: m/z 194.9 [M+H]+ при 2.51 RT (99.35% чистота)
4-Амино-3-(циклопропиламино)фенол (Int-17)
К перемешиваемому раствору 3-(циклопропиламино)-4-нитрофенола (1 г, 5.15 ммоль) в этилацетате (10 мл) добавили 10% Pd/C (50% влажность, 250 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли метанолом (30 мл). Фильтрат концентрировали при пониженном давлении с получением 4-амино-3-(циклопропиламино)фенола Int-17 (600 мг) в виде сиропа коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 8.18 (brs, 1H), 6.37-6.29 (m, 2H), 5.86 (dd, J = 8.1, 2.3 Гц, 1H), 4.92 (s, 1H), 3.81 (brs, 2H), 2.27-2.25 (m, 1H), 0.69-0.62 (m, 2H), 0.41-0.35 (m, 2H)
LC-MS: m/z 164.8 [M+H]+ при 1.09 RT (74.22% чистота)
Получение Int-18
Схема:
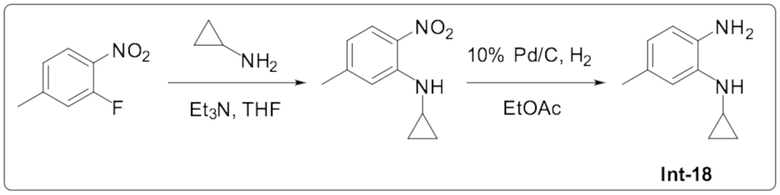
N-Циклопропил-5-метил-2-нитроанилин
К перемешиваемому раствору 2-фтор-4-метил-1-нитробензола (500 мг, 3.22 ммоль) в THF (5 мл) добавили триэтиламин (1.35 мл, 9.68 ммоль), а затем циклопропанамин (919 мг, 16.13 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 60°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% EtOAc/гексан) с получением N-циклопропил-5-метил-2-нитроанилина (500 мг, 2.6 ммоль, 80%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 8.10 (brs, 1H), 8.04 (d, J = 8.8 Гц, 1H), 7.07 (s, 1H), 6.50 (d, J = 8.8 Гц, 1H), 2.58-2.55 (m, 1H), 2.37 (s, 3H), 0.94-0.88 (m, 2H), 0.67-0.62 (m, 2H).
LC-MS: m/z 192.9 [M+H]+ при 3.35 RT (99.53% чистота).
N 1-Циклопропил-5-метилбензол-1,2-диамин (Int-18)
К перемешиваемому раствору N-циклопропил-5-метил-2-нитроанилин (150 мг, 0.78 ммоль) в этилацетате (1 мл) добавили 10% Pd/C (50% влажность, 50 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью этилацетата (15 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклопропил-5-метилбензол-1,2-диамина Int-18 (120 мг) в виде твердого вещества черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.60 (s, 1H), 6.41 (d, J = 8.1 Гц, 1H), 6.24 (br d, J = 7.5 Гц, 1H), 4.91 (s, 1H), 4.21 (br s, 2H), 2.32-2.30 (m, 1H), 2.14 (s, 3H), 0.71-0.64 (m, 2H), 0.41-0.35 (m, 2H)
LC-MS: m/z 162.9 [M+H]+ при 2.49 RT (81.92% чистота)
Получение Int-19
Схема:
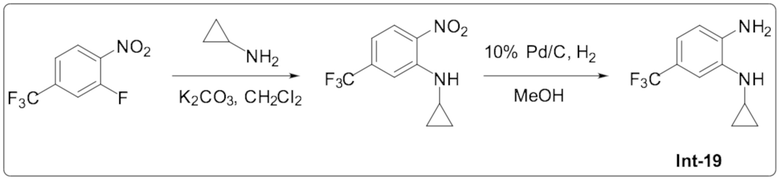
N-Циклопропил-2-нитро-5-(трифторметил)анилин
К перемешиваемому раствору 2-фтор-1-нитро-4-(трифторметил)бензола (1 г, 4.78 ммоль) в CH2Cl2 (40 мл) добавили карбонат калия (1.32 г, 9.57 ммоль) и циклопропанамин (1.09 г, 19.14 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (30 мл) и экстрагировали с помощью CH2Cl2 (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% EtOAc/гексан) с получением N-циклопропил-2-нитро-5-(трифторметил)анилина (1 г, 4.06 ммоль, 85%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 8.25 (d, J = 8.9 Гц, 1H), 8.13 (brs, 1H), 7.62 (d, J = 1.0 Гц, 1H), 7.04 (dd, J = 8.8, 1.8 Гц, 1H), 2.76-2.69 (m, 1H), 0.95-0.87 (m, 2H), 0.69-0.63 (m, 2H)
LC-MS: m/z 245.0 [M-H]- при 4.33 RT (78.90% чистота)
N 1-Циклопропил-5-(трифторметил)бензол-1,2-диамин (Int-19)
К перемешиваемому раствору N-циклопропил-2-нитро-5-(трифторметил)анилина (1 г, 4.06 ммоль) в метаноле (20 мл) добавили 10% Pd/C (50% влажность, 100 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли метанолом (20 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклопропил-5-(трифторметил)бензол-1,2-диамина Int-19 (700 мг) в виде сиропа коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
Получение Int-20
Схема:
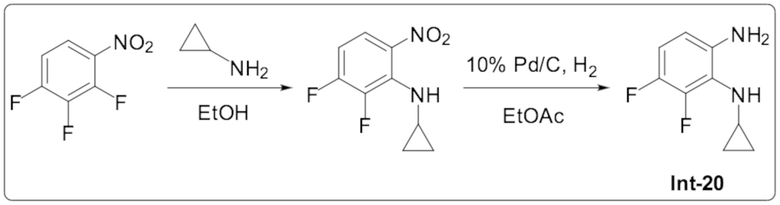
N-Циклопропил-2,3-дифтор-6-нитроанилин
К перемешиваемому раствору 1,2,3-трифтор-4-нитробензола (1 г, 28.25 ммоль) в этаноле (100 мл) добавили циклопропанамин (1.61 г, 28.25 ммоль) при комнатной температуре в инертной атмосфере и реакционную смесь перемешивали в течение 48 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (50 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% EtOAc/гексан) с получением N-циклопропил-2,3-дифтор-6-нитроанилина (3 г, 14.0 ммоль, 50%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 7.95-7.91 (m, 1H), 7.75 (brs, 1H), 6.85-6.80 (m, 1H), 3.02-2.98 (m, 1H), 0.79-0.72 (m, 2H), 0.67-0.65 (m, 2H)
LC-MS: m/z 213.4 [M-H]+ при 4.06 RT (99.81% чистота)
N 1-Циклопропил-5,6-дифторбензол-1,2-диамин (Int-20)
К перемешиваемому раствору N-циклопропил-2,3-дифтор-6-нитроанилина (1 г, 4.67 ммоль) в этилацетате (10 мл) добавили 10% Pd/C (50% влажность, 100 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью этилацетата (20 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклопропил-5,6-дифторбензол-1,2-диамина Int-20 (700 мг) в виде твердого вещества черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.55-6.47 (m, 1H), 6.33-6.27 (m, 1H), 4.70 (br s, 2H), 4.54 (br s, 1H), 2.74-2.66 (m, 1H), 0.59-0.52 (m, 2H), 0.48-0.43 (m, 2H)
LC-MS: m/z 185.0 [M+H]+ при 2.60 RT (67.33% чистота)
Получение Int-21
Схема:
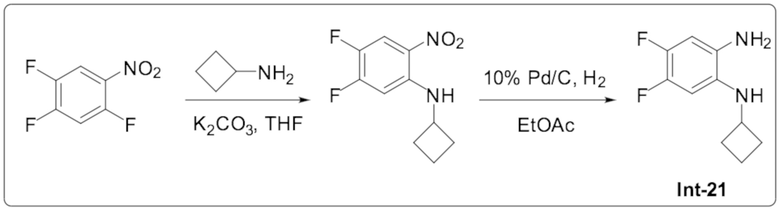
N-Циклобутил-4,5-дифтор-2-нитроанилин
К перемешиваемому раствору 1,2,4-трифтор-5-нитробензола (1 г, 5.65 ммоль) в THF (20 мл) добавили карбонат калия (1.17 г, 8.47 ммоль), а затем циклобутанамин (0.58 мл, 6.78 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 6 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (50 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% EtOAc/гексан) с получением N-циклобутил-4,5-дифтор-2-нитроанилина (450 мг, 1.97 ммоль, 35%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 8.18-8.08 (m, 2H), 7.00 (dd, J = 13.4, 7.0 Гц, 1H), 4.16-4.07 (m, 1H), 2.48-2.39 (m, 2H), 2.06-1.94 (m, 2H), 1.83-1.69 (m, 2H)
LC-MS: m/z 229.0 [M+H]+ при 2.90 RT (97.77% чистота)
N 1-Циклобутил-4,5-дифторбензол-1,2-диамин (Int-21)
К перемешиваемому раствору N-циклобутил-4,5-дифтор-2-нитроанилина (450 мг, 1.97 ммоль) в этилацетате (10 мл) добавили 10% Pd/C (50% влажность, 45 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью этилацетата (15 мл). Фильтрат концентрировали при пониженном давлении с получением N1-циклобутил-4,5-дифторбензол-1,2-диамина Int-21 (350 мг) в виде твердого вещества черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, CDCl3): δ 6.52 (dd, J = 11.3, 8.0 Гц, 1H), 6.33 (dd, J = 12.1, 7.1 Гц, 1H), 3.86-3.76 (m, 1H), 3.40-3.18 (m, 3H), 2.52-2.39 (m, 2H), 1.91-1.78 (m, 4H)
LC-MS: m/z 199.0 [M+H]+ при 2.92 RT (83.44% чистота)
Получение Int-22
Схема:
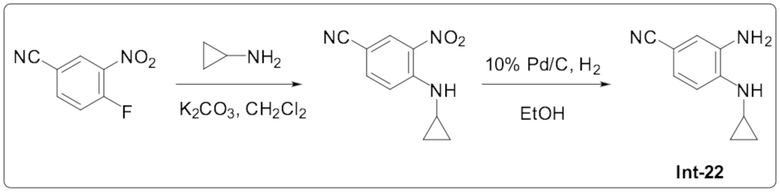
4-(Циклопропиламино)-3-нитробензонитрил
К перемешиваемому раствору 4-фтор-3-нитробензонитрила (1 г, 6.02 ммоль) в CH2Cl2 (5 мл) добавили карбонат калия (1.66 г, 12.05 ммоль), а затем циклопропанамин (3.33 мл, 48.19 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в воду (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4-(циклопропиламино)-3-нитробензонитрила (1.1 г) в виде твердого вещества желтого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, CDCl3): δ 8.49 (d, J = 1.9 Гц, 1H), 8.41 (brs, 1H), 7.66-7.63 (m, 1H), 7.40 (d, J = 9.0 Гц, 1H), 2.68-2.61 (m, 1H), 1.04-0.98 (m, 2H), 0.75-0.70 (m, 2H)
LC-MS: m/z 202.0 [M-H]- при 2.88 RT (99.48% чистота)
3-Амино-4-(циклопропиламино)бензонитрил (Int-22)
К перемешиваемому раствору 4-(циклопропиламино)-3-нитробензонитрила (1.1 г, неочищенный) в этаноле (40 мл) добавили 10% Pd/C (50% влажность, 350 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью этилацетата (20 мл). Фильтрат концентрировали при пониженном давлении с получением 3-амино-4-(циклопропиламино) бензонитрила Int-22 (900 мг) в виде твердого вещества желтого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, CDCl3): δ 7.18 (dd, J = 8.2, 1.8 Гц, 1H), 7.00 (d, J = 8.3 Гц, 1H), 6.92 (d, J = 1.9 Гц, 1H), 4.41 (br s, 1H), 3.24 (br s, 2H), 2.51-2.43 (m, 1H), 0.85-0.78 (m, 2H), 0.59-0.53 (m, 2H)
LC-MS: m/z 173.9 [M+H]+ при 2.40 RT (84.82% чистота)
Получение Int-23
Схема:
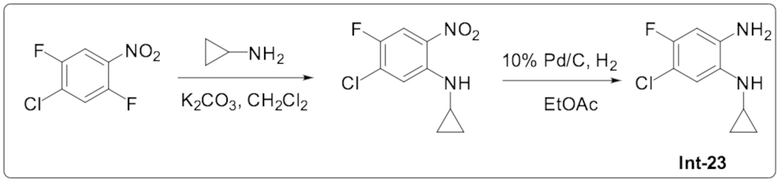
1-Хлор-2,5-дифтор-4-нитробензол
К перемешиваемому раствору 1-хлор-2,5-дифтор-4-нитробензола (500 мг, 2.59 ммоль) в CH2Cl2 (10 мл) в инертной атмосфере добавили карбонат калия (715 мг, 5.18 ммоль) и циклопропанамин (305 мг, 5.18 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-хлор-2,5-дифтор-4-нитробензола (400 мг, 1.73 ммоль, 67%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 7.97 (d, J = 9.3 Гц, 2H), 7.37 (d, J = 6.4 Гц, 1H), 2.57-2.54 (m, 1H), 1.00-0.91 (m, 2H), 0.71-0.59 (m, 2H)
5-Хлор-N1-циклопропил-4-фторбензол-1,2-диамин (Int-23)
К перемешиваемому раствору 1-хлор-2,5-дифтор-4-нитробензола (400 мг, 1.74 ммоль) в EtOAc (10 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 100 мг) при комнатной температуре. Реакционную смесь вакуумировали и перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью EtOAc (40 мл). Фильтрат концентрировали при пониженном давлении с получением 5-хлор-N1-циклопропил-4-фторбензол-1,2-диамина Int-23 (280 мг, 1.40 ммоль, 81%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 6.68 (d, J = 7.5 Гц, 1H), 6.47 (d, J = 11.6 Гц, 1H), 5.16 (br s, 1H), 4.96 (br s, 2H), 2.34-2.27 (m, 1H), 0.74-0.67 (m, 2H), 0.42-0.28 (m, 2H)
Получение Int-24
Схема:
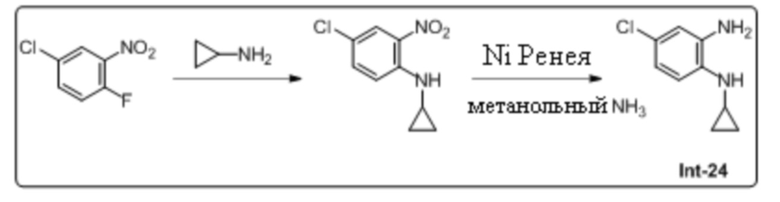
4-Хлор-N-циклопропил-2-нитроанилин
К 4-хлор-1-фтор-2-нитробензолу (2 г, 11.43 ммоль) добавили циклопропанамин (2.6 г, 45.71 ммоль) по каплям при 10°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3-5% EtOAc/гексан) с получением 4-хлор-N-циклопропил-2-нитроанилина (2.08 г, 0.98 ммоль, 83%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 8.16 (d, J = 2.2 Гц, 1H), 8.04 (br s, 1H), 7.42 (dd, J = 8.8, 2.2 Гц, 1H), 7.29 (d, J = 9.3 Гц, 1H), 2.61-2.55 (m, 1H), 0.96-0.90 (m, 2H), 0.69-0.63 (m, 2H)
LC-MS: m/z 213.1 [M+H]+ при 2.85 RT (99.81% чистота)
4-Хлор-N1-циклопропилбензол-1,2-диамин (Int-24)
К перемешиваемому раствору 4-хлор-N-циклопропил-2-нитроанилина (1 г, 4.72 ммоль) в метанольный аммиак (2 M, 50 мл) добавили никель Ренея (500 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью этилацетата (30 мл). Фильтрат концентрировали при пониженном давлении с получением 4-хлор-N1-циклопропилбензол-1,2-диамина Int-24 (700 мг) в виде жидкости коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 6.70 (d, J = 8.3 Гц, 1H), 6.55-6.45 (m, 2H), 5.10 (s, 1H), 4.77 (br s, 2H), 2.31-2.29 (m, 1H), 0.72-0.65 (m, 2H), 0.41-0.34 (m, 2H)
LC-MS: m/z 183.1 [M+H]+ при 2.15 RT (87.51% чистота)
Получение Int-25
Схема:
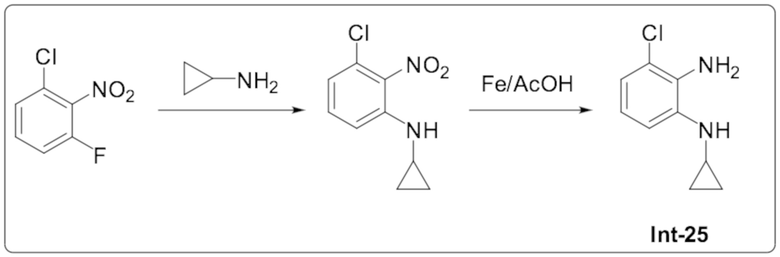
3-Хлор-N-циклопропил-2-нитроанилин
К 1-хлор-3-фтор-2-нитробензолу (500 мг, 2.85 ммоль) добавили циклопропанамин (0.78 мл, 11.4 ммоль) по каплям при комнатной температуре в инертной атмосфере и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3-хлор-N-циклопропил-2-нитроанилина (500 мг) в виде твердого вещества желтого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, CDCl3): δ 7.28-7.22 (m, 1H), 7.15 (dd, J = 8.5, 1.3 Гц, 1H), 6.80 (dd, J = 7.8, 1.3 Гц, 1H), 6.01 (br s, 1H), 2.54-2.46 (m, 1H), 0.88-0.82 (m, 2H), 0.60-0.55 (m, 2H).
LC-MS: m/z 213.1 [M+H]+ при 2.74 RT (98.57% чистота)
3-Хлор-N1-циклопропилбензол-1,2-диамин (Int-25)
К перемешиваемому раствору 3-хлор-N-циклопропил-2-нитроанилина (200 мг, неочищенный) в уксусной кислоте (2 мл) добавили порошок железа (157 мг, 2.83 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 80°C и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором NaHCO3 (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3-хлор-N1-циклопропилбензол-1,2-диамина Int-25 (200 мг) в виде сиропа коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 183.0 [M+H]+ при 2.47 RT (43.71% чистота)
Получение Int-26
Схема:
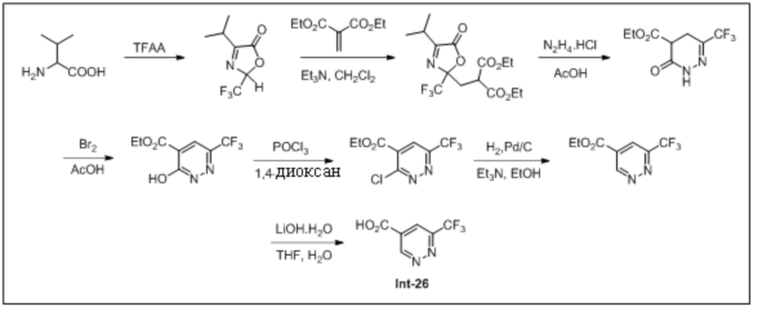
4-Изопропил-2-(трифторметил)оксазол-5(2H)-он
К DL-валину (30 г, 256.41 ммоль) добавили ангидрид трифторуксусной кислоты (72 мл) по каплям при комнатной температуре. Реакционную смесь нагрели до возврата флегмы и перемешивали в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (100 мл) и экстрагировали с помощью CH2Cl2 (250 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4-изопропил-2-(трифторметил)оксазол-5(2H)-она 2 (45 г) в виде жидкости бледно-желтого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, CDCl3): δ 6.11-6.05 (m, 1H), 3.12-3.02 (m, 1H), 1.34 (d, J = 3.0 Гц, 3H), 1.32 (d, J = 3.0 Гц, 3H)
LC-MS: m/z 193.9 [M-H]- при 2.94 RT (75.69% чистота)
Диэтил 2-((4-изопропил-5-оксо-2-(трифторметил)-2,5-дигидрооксазол-2-ил)метил) малонат
К перемешиваемому раствору 4-изопропил-2-(трифторметил)оксазола-5 (2H)-он (45 г, неочищенный) в CH2Cl2 (450 мл) в инертной атмосфере добавили диэтил 2-метиленмалонат (47.63 г, неочищенный) и триэтиламин (48.2 мл, 346.14 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (150 мл) и экстрагировали с помощью CH2Cl2 (2 x 250 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением диэтил 2-((4-изопропил-5-оксо-2-(трифторметил)-2,5-дигидрооксазол-2-ил)метил) малоната (50 г, неочищенный) в виде твердого вещества бледно-желтого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 368.1 [M+H]+ при 3.70 RT (82.98% чистота)
Этил 3-оксо-6-(трифторметил)-2,3,4,5-тетрагидропиридазин-4-карбоксилат
К перемешиваемому раствору диэтил 2-((4-изопропил-5-оксо-2-(трифторметил)-2,5-дигидрооксазол-2-ил)метил)малоната (25 г, неочищенный) в уксусной кислоте (200 мл) в инертной атмосфере добавили гидрохлорид гидразина (23.16 г, 340.59 ммоль) при комнатной температуре. Реакционную смесь нагрели до возврата флегмы и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили насыщенным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (2 x 250 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/гексан) с получением этил 3-оксо-6-(трифторметил)-2,3,4,5-тетрагидропиридазин-4-карбоксилата (15 г, 63.55 ммоль) в виде жидкости бледно-желтого цвета.
LC-MS: m/z 237.0 [M-H]- при 2.19 RT (87.20% чистота)
Этил 3-гидрокси-6-(трифторметил)пиридазин-4-карбоксилат
К перемешиваемому раствору этил 3-оксо-6-(трифторметил)-2,3,4,5-тетрагидропиридазин-4-карбоксилата (7.5 г, 31.51 ммоль) в уксусной кислоте (40 мл) в инертной атмосфере добавили раствор бромина (5.03 г, 31.51 ммоль) в уксусной кислоте (35 мл) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили насыщенным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (2 x 200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/гексан) с получением этил 3-гидрокси-6-(трифторметил)пиридазин-4-карбоксилата (2.5 г, 10.59 ммоль, 34%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 14.16 (br s, 1H), 8.12 (s, 1H), 4.30 (q, J = 7.1 Гц, 2H), 1.29 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 235.0 [M-H]- при 2.03 RT (98.67% чистота)
Этил 3-хлор-6-(трифторметил)пиридазин-4-карбоксилат
К перемешиваемому раствору этил 3-гидрокси-6-(трифторметил)пиридазин-4-карбоксилата (5.0 г, 21.19 ммоль) в 1, 4-диоксане (50 мл) в инертной атмосфере добавили фосфорилтрихлорид (19.6 мл, 211.86 ммоль) при 0°C. Реакционную смесь нагрели до 100°C и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили раствором бикарбоната натрия (100 мл) и экстрагировали с помощью EtOAc (2 x 200 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 15-20% EtOAc/гексан) с получением этил 3-хлор-6-(трифторметил)пиридазин-4-карбоксилата (3 г, 11.81 ммоль, 55%) в виде жидкости бледно-желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 8.15 (s, 1H), 4.53 (q, J = 7.0 Гц, 2H), 1.47 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 255.4 [M+H]+ при 3.61 RT (98.51% чистота)
Этил 6-(трифторметил)пиридазин-4-карбоксилат
К перемешиваемому раствору этил 3-хлор-6-(трифторметил)пиридазин-4-карбоксилата (500 мг, 1.96 ммоль) в этаноле (10 мл) в инертной атмосфере добавили триэтиламин (0.5 мл) и 10% Pd/C (50% влажность, 100 мг) при комнатной температуре. Свободное реакционное пространство быстро поместили под вакуум и быстро обогатили водородом. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью EtOAc (50 мл). Фильтрат концентрировали при пониженном давлении. Полученное неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением этил 6-(трифторметил)пиридазин-4-карбоксилата (300 мг, 1.36 ммоль, 69%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.85 (d, J = 1.8 Гц, 1H), 8.32 (d, J = 1.9 Гц, 1H), 4.53 (q, J = 7.2 Гц, 2H), 1.47 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 221.1 [M+H]+ при 3.09 RT (92.69% чистота)
6-(Трифторметил)пиридазин-4-карбоновая кислота (Int-26)
К перемешиваемому раствору этил 6-(трифторметил)пиридазин-4-карбоксилата (300 мг, 1.36 ммоль) в смеси THF: воды (4:1, 5 мл) добавили гидроксид лития (171.6 г, 4.09 ммоль) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Затем остаток подкислили с применением концентрированной HCl (pH 3-4) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученное твердое вещество промыли с помощью н-пентана (10 мл) и высушили в вакууме с получением 6-(трифторметил)пиридазин-4-карбоновой кислоты Int-26 (210 мг) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, DMSO-d6): δ 14.08 (brs, 1H), 9.83 (brs, 1H), 8.43 (brs, 1H)
LC-MS: m/z 191.0 [M-H]- при 3.79 RT (94.51% чистота)
Получение Int-27
Схема:
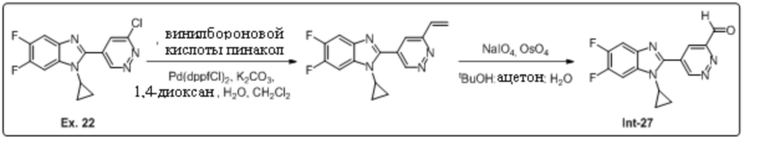
1-Циклопропил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (150 мг, 0.50 ммоль) в 1,4-диоксане (3 мл) и воде (0.5 мл) в инертной атмосфере добавили винилбороновой кислоты сложный пинаколовый эфир (76 мг, 0.50 ммоль) и карбонат калия (203 мг, 1.47 ммоль) при комнатной температуре. Реакционную смесь дегазировали аргоном в течение 10 мин. Pd(dppf)Cl2 (4 мг, 0.005 ммоль) добавили при комнатной температуре и смесь дегазировали аргоном в течение 10 мин. Реакционную смесь нагрели до 80°C и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (70 мг, 0.23 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.69 (s, 1H), 8.45 (s, 1H), 7.90-7.83 (m, 2H), 7.18-7.11 (m, 1H), 6.53 (d, J = 17.7 Гц, 1H), 5.82 (d, J = 11.4 Гц, 1H), 3.99-3.97 (m, 1H), 1.22-1.13 (m, 2H), 0.82-0.60 (m, 2H)
5-(1-Циклопропил-5, 6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегид (Int-27)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (300 мг, 1.00 ммоль) в ацетоне: tBuOH: воде (1:1:1, 18 мл) в инертной атмосфере добавили периодат натрия (430 мг, 2.01 ммоль) и осмия тетроксид (1 M раствор, 6 мл) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (100 мл). Органический слой промыли водой (100 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегида Int-27 (200 мг) в виде твердого вещества черного цвета. Неочищенное вещество применяли без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 10.36 (s, 1H), 10.03 (s, 1H), 8.58 (s, 1H), 7.93-7.81 (m, 2H), 4.00-3.94 (m, 1H), 1.21-1.13 (m, 2H), 0.81-0.72 (m, 2H)
Получение Int-28
Схема:
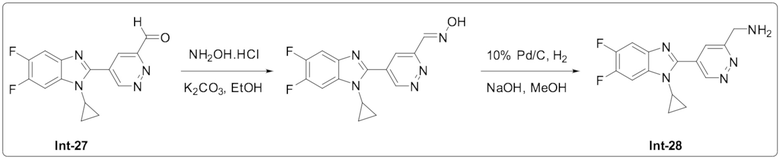
(E)-5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегид оксим
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегида Int-27 (600 мг, 2 ммоль) в этаноле (10 мл) в инертной атмосфере добавили гидроксиламин гидрохлорид (276 мг, 4 ммоль) и карбонат калия (552 мг, 4 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь концентрировали при пониженном давлении. Остаток разбавили водой (70 мл) и экстрагировали с помощью CH2Cl2 (100 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли н-гексаном (20 мл) и высушили в вакууме с получением (E)-5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегид оксима (500 мг, 1.58 ммоль, 79%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 12.26 (s, 1H), 9.80 (d, J = 1.7 Гц, 1H), 8.56 (d, J = 2.3 Гц, 1H), 8.48 (s, 1H), 7.91-7.80 (m, 2H), 3.97-3.88 (m, 1H), 1.22-1.14 (m, 2H), 0.85-0.78 (m, 2H)
LC-MS: m/z 315.9 [M+H]+ при 2.33 RT (97.99% чистота)
(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанамин (Int-28)
К перемешиваемому раствору (E)-5-(1-циклопропил-56-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегидоксима (500 мг, 1.58 ммоль) в этаноле (10 мл) добавили гидроксид натрия (190 мг, 4.76 ммоль) и 10% Pd/C (50% влажность, 150 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита. Фильтрат концентрировали при пониженном давлении. Остаток разбавили с помощью CH2Cl2 (80 мл) и промыли водой (50 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил метанамина Int-28 (400 мг, 1.32 ммоль, 84%) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 9.68 (d, J = 1.7 Гц, 1H), 8.35 (d, J = 1.7 Гц, 1H), 7.89-7.81 (m, 2H), 4.12 (s, 2H), 3.95-3.89 (m, 1H), 2.13 (brs, 2H), 1.27-1.14 (m, 2H), 0.79-0.66 (m, 2H)
LC-MS: m/z 301.9 [M+H]+ при 1.91 RT (95.51% чистота)
Получение Int-29
Схема:
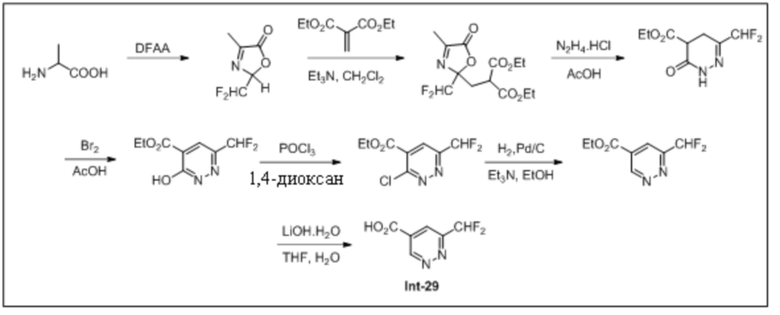
6-(Дифторметил)пиридазин-4-карбоновая кислота (Int-29) получена аналогично получению соединения Int-26 исходя из аланина и ангидрида дифторуксусной кислоты (DFAA).
Получение Int-30
Схема:
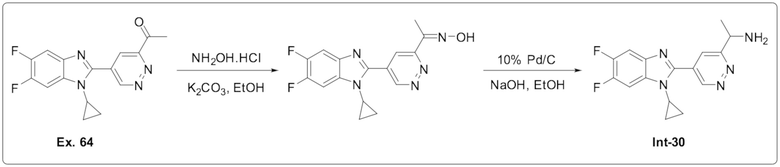
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-он оксим
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-она Пример 64 (170 мг, 0.54 ммоль) в EtOH (10 мл) в инертной атмосфере добавили и гидроксиламин гидрохлорид (75 мг, 1.08 ммоль) и карбонат калия (149 мг, 1.08 ммоль) при 0°C. Реакционную смесь нагрели до 80°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Остаток разбавили водой (30 мл) и экстрагировали с помощью CH2Cl2 (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-он оксима (100 мг) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 12.07 (s, 1H), 9.81 (d, J = 1.7 Гц, 1H), 8.63 (d, J = 1.7 Гц, 1H), 7.91-7.80 (m, 2H), 3.94-3.87 (m, 1H), 2.41 (s, 3H), 1.21-1.14 (m, 2H), 0.85-0.76 (m, 2H)
LC-MS: m/z 329.9 [M+H]+ при 2.52 RT (86.46% чистота)
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-амин (Int-30)
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил)этан-1-он оксима (100 мг, неочищенный) в этаноле (10 мл) в инертной атмосфере добавили гидроксид натрия (37 мг, 0.91 ммоль) и 10% Pd/C (50% влажность, 80 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью MeOH (20 мл). Фильтрат концентрировали при пониженном давлении. Остаток разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-амина Int-30 (80 мг) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли без дальнейшей очистки.
LC-MS: m/z 315.9 [M+H]+ при 1.71 RT (85.70% чистота)
Пример 1
Схема:
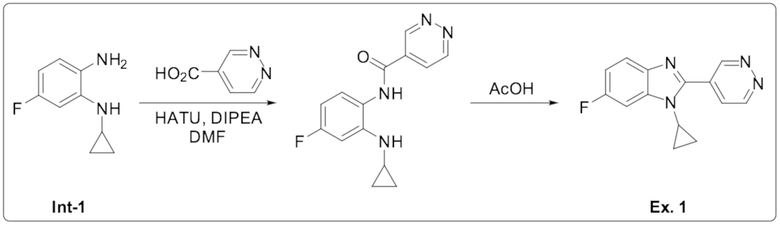
N-(2-(Циклопропиламино)-4-фторфенил) пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5-фторбензол-1, 2-диамина Int-1 (300 мг, 1.81 ммоль) в N,N-диметилформамиде (DMF) (3 мл) в инертной атмосфере добавили пиридазин-4-карбоновую кислоту (224 мг, 1.81 ммоль), N-[(диметиламино)-1H-1,2,3-триазоло-[4,5-b]пиридин-1-илметилен]-N-метилметанаминия гексафторфосфат N-оксид (HATU) (824 мг, 2.17 ммоль) и этилдиизопропиламин (1.26 мл, 7.23 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4-фторфенил) пиридазин-4-карбоксамида (220 мг, 0.81 ммоль, 44%) в виде твердого вещества коричневого цвета.
1H ЯМР (400МГц, DMSO-d6): δ 9.89 (s, 1H), 9.66 (dd, J = 2.2, 1.3 Гц, 1H), 9.47 (dd, J = 5.2, 1.1 Гц, 1H), 8.12 (dd, J = 5.3, 2.3 Гц, 1H), 7.13 (dd, J = 8.5, 6.4 Гц, 1H), 6.75 (dd, J = 11.9, 2.9 Гц, 1H), 6.43 (td, J = 8.5, 2.9 Гц, 1H), 6.12 (s, 1H), 2.38-2.32 (m, 1H), 0.77-0.71 (m, 2H), 0.47-0.40 (m, 2H).
LC-MS: m/z 272.9 [M+H]+ при 2.66 RT (89.14% чистота).
1-Циклопропил-6-фтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазол (Ex 1)
К N-(2-(циклопропиламино)-4-фторфенил) пиридазин-4-карбоксамиду (150 мг, 0.55 ммоль) в уксусной кислоте (3 мл) в инертной атмосфере нагрели до 100°C и перемешивали в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь нейтрализовали насыщенным раствором бикарбоната натрия (50 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% MeOH/CH2Cl2) с получением 1-циклопропил-6-фтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазола Ex 1 (68 мг, 0.27 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400МГц, DMSO-d6): δ 9.82 (dd, J = 2.2, 1.3 Гц, 1H), 9.45 (dd, J = 5.4, 1.1 Гц, 1H), 8.29 (dd, J = 5.4, 2.4 Гц, 1H), 7.79 (dd, J = 8.9, 4.9 Гц, 1H), 7.55 (dd, J = 9.1, 2.4 Гц, 1H), 7.22-7.15 (m, 1H), 3.94-3.87 (m, 1H), 1.22-1.16 (m, 2H), 0.78-0.73 (m, 2H).
LC-MS: m/z 255.0 [M+H]+ при 2.98 RT (97.13% чистота).
ВЭЖХ: 98.24%.
Примеры 2 и 3
Схема:
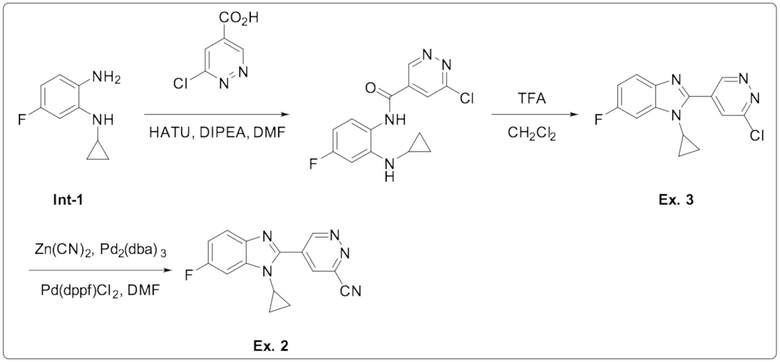
6-Хлор-N-(2-(циклопропиламино)-4-фторфенил) пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5-фторбензол-1, 2-диамина Int-1 (500 мг, 3.01 ммоль) в DMF (3 мл) в инертной атмосфере добавили 6-хлорпиридазин-4-карбоновую кислоту (475 мг, 3.01 ммоль), HATU (1.37 г, 3.61 ммоль) и этилдиизопропиламин (2.2 мл, 12.04 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/Гексан) с получением 6-хлор-N-(2-(циклопропиламино)-4-фторфенил) пиридазин-4-карбоксамида (550 мг, 1.79 ммоль, 60%) в виде твердого вещества коричневого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 9.96 (s, 1H), 9.63 (d, J = 1.4 Гц, 1H), 8.36 (d, J = 1.2 Гц, 1H), 7.13 (dd, J = 8.4, 6.7 Гц, 1H), 6.75 (dd, J = 11.7, 2.7 Гц, 1H), 6.43 (td, J = 8.5, 2.7 Гц, 1H), 6.14 (s, 1H), 2.37-2.33 (m, 1H), 0.77-0.73 (m, 2H), 0.46-0.42 (m, 2H).
LC-MS: m/z 305.0 [M-H]+ при 3.00 RT (83.53% чистота).
2-(6-Хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазол (Пример 3)
К перемешиваемому раствору 6-хлор-N-(2-(циклопропиламино)-4-фторфенил) пиридазин-4-карбоксамида (550 мг, 1.79 ммоль) в CH2Cl2 (10 мл) в инертной атмосфере добавили трифторуксусную кислоту (0.6 мл) по каплям при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток разбавили с помощью EtOAc (50 мл) и промыли с помощью насыщенного раствора бикарбоната натрия (20 мл). Органический слой отделили, высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (200 мг, 0.69 ммоль, 38%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 9.82 (s, 1H), 8.46 (s, 1H), 7.81 (dd, J = 9.0, 4.9 Гц, 1H), 7.58 (dd, J = 9.0, 2.0 Гц, 1H), 7.21 (td, J = 9.3, 2.2 Гц, 1H), 3.97-3.93 (m, 1H), 1.22-1.17 (m, 2H), 0.83-0.79 (m, 2H).
LC-MS: m/z 288.9 [M+H]+ при 2.59 RT (98.16% чистота).
ВЭЖХ: 98.76%.
5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбонитрил (Пример 2)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 2 (100 мг, 0.35 ммоль) в DMF (1 мл) в инертной атмосфере добавили Zn(CN)2 (24 мг, 0.21 ммоль) при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 10 мин. Pd2(dba)3 (16 мг, 0.02 ммоль) и Pd(dppf)Cl2 (13 мг, 0.02 ммоль) добавили при комнатной температуре и реакционную смесь дегазировали под аргоном в течение 5 мин. Реакционную смесь нагрели до 100°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью EtOAc (40 мл), отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (15 мл). Органический слой промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбонитрила Пример 2 (30 мг, 0.11 ммоль, 31%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 10.06 (d, J = 1.7 Гц, 1H), 8.92 (d, J = 1.7 Гц, 1H), 7.83 (dd, J = 8.7, 4.9 Гц, 1H), 7.59 (dd, J = 9.0, 2.0 Гц, 1H), 7.25-7.20 (m, 1H), 3.98-3.93 (m, 1H), 1.24-1.19 (m, 2H), 0.82-0.78 (m, 2H).
LC-MS: m/z 279.8 [M+H]+ при 2.92 RT (99.44% чистота).
ВЭЖХ: 99.21%.
Пример 4
Схема:
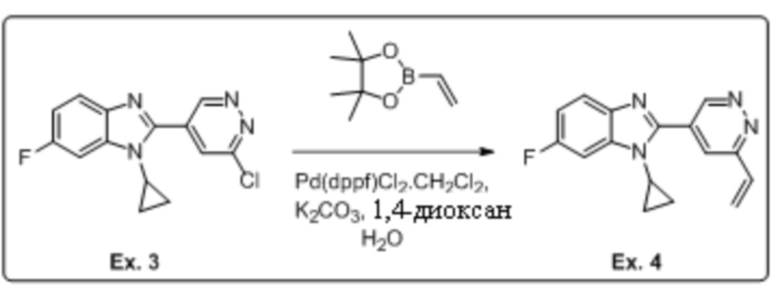
1-Циклопропил-6-фтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 4)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (300 мг, 1.04 ммоль) в смеси 1,4-диоксана (15 мл) и воды (2 мл) в инертной атмосфере добавили карбонат калия (431 мг, 3.12 ммоль) и 4, 4, 5, 5-тетраметил-2-винил-1,3,2-диоксаборолан (160 мг, 1.04 ммоль) при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 15 мин. К этому добавили Pd(dppf)Cl2.CH2Cl2 (8.5 мг, 0.01 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью EtOAc (60 мл), отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (15 мл). Органический слой промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением 1-циклопропил-6-фтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 4 (150 мг, 0.53 ммоль, 51%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 9.68 (d, J = 1.7 Гц, 1H), 8.44 (d, J = 1.7 Гц, 1H), 7.78 (dd, J = 9.0, 4.9 Гц, 1H), 7.55 (dd, J = 9.0, 2.3 Гц, 1H), 7.21-7.09 (m, 2H), 6.52 (d, J = 17.6 Гц, 1H), 5.80 (d, J = 11.0 Гц, 1H), 3.97-3.93 (m, 1H), 1.20-1.14 (m, 2H), 0.77-0.72 (m, 2H).
LC-MS: m/z 280.9 [M+H]+ при 2.43 RT (93.90% чистота).
ВЭЖХ: 95.00%.
Пример 5
Схема:
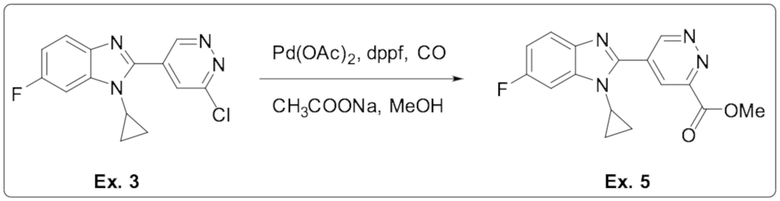
Метил 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилат (Пример 5)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (200 мг, 0.69 ммоль) в MeOH (3 мл) добавили ацетат натрия (171 мг, 2.08 ммоль), 1,1′-Фероценил-бис(дифенилфосфин) (19 мг, 0.03 ммоль) и палладия(II) ацетат (7.8 мг, 0.03 ммоль) при комнатной температуре в стальном сосуде. Стальной сосуд заполнили газом СО (давление 15 бар). Полученную реакционную смесь перемешивали при 50°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целлита, и слой целита промыли метанолом (30 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением метил 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбоксилата Пример 5 (70 мг, 0.22 ммоль, 32%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 10.04 (d, J = 2.0 Гц, 1H), 8.74 (d, J = 2.0 Гц, 1H), 7.82 (dd, J = 9.0, 4.9 Гц, 1H), 7.57 (dd, J = 9.1, 2.5 Гц, 1H), 7.21 (td, J = 9.3, 2.3 Гц, 1H), 4.03 (s, 3H), 3.99-3.95 (m, 1H), 1.23-1.18 (m, 2H), 0.82-0.78 (m, 2H).
LC-MS: m/z 312.9 [M+H]+ при 2.36 RT (95.19% чистота).
ВЭЖХ: 99.01%.
Пример 6
Схема:
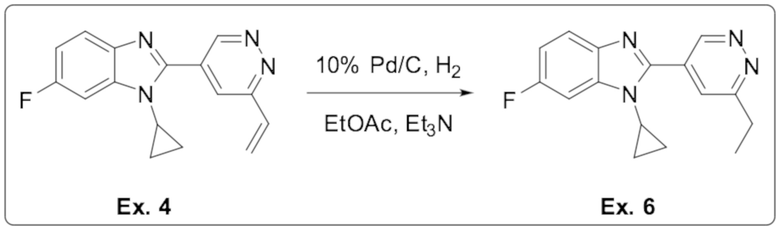
1-Циклопропил-2-(6-этилпиридазин-4-ил)-6-фтор-1H-бензо[d]имидазол (Пример 6)
К перемешиваемому раствору 1-циклопропил-6-фтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 4 (70 мг, 0.25 ммоль) в этилацетате (8 мл) в инертной атмосфере добавили триэтиламин (кат.) и 10% Pd/C (50% влажность, 20 мг) при комнатной температуре. Реакционную смесь вакуумировали и перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (40 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-этилпиридазин-4-ил)-6-фтор-1H-бензо[d]имидазола Пример 6 (50 мг, 0.18 ммоль, 67%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400МГц, DMSO-d6): δ 9.66 (d, J = 2.1 Гц, 1H), 8.18 (d, J = 2.0 Гц, 1H), 7.78 (dd, J = 8.8, 4.9 Гц, 1H), 7.55 (dd, J = 9.0, 2.4 Гц, 1H), 7.22-7.14 (m, 1H), 3.95-3.90 (m, 1H), 3.07 (q, J = 7.6 Гц, 2H), 1.37 (t, J = 7.7 Гц, 3H), 1.21-1.15 (m, 2H), 0.77-0.71 (m, 2H).
LC-MS: m/z 282.9 [M+H]+ при 2.37 RT (98.84% чистота).
ВЭЖХ: 98.29%.
Пример 7
Схема:
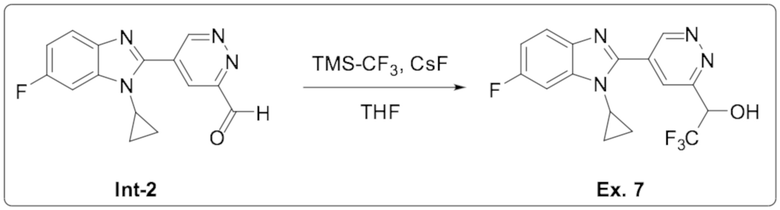
1-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифтопентан-1-ол (Пример 7)
К перемешиваемому раствору 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-2 (50 мг, 0.18 ммоль) в THF (3 мл) в инертной атмосфере добавили триметил (трифторметил) силан (0.05 мл, 0.35 ммоль) при 0°C. Фторид цезия (81 мг, 0.53 ммоль) добавили, и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорид аммония (15 мл) и экстрагировали с помощью EtOAc (2 x 15 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% MeOH/CH2Cl2) с получением 1-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил)-2, 2, 2-трифтопентан-1-ола Пример 7 (15 мг, 0.04 ммоль, 24%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400МГц, CD3OD): δ 9.83 (d, J = 2.1 Гц, 1H), 8.57 (d, J = 2.1 Гц, 1H), 7.76 (dd, J = 8.9, 4.8 Гц, 1H), 7.52 (dd, J = 8.8, 2.3 Гц, 1H), 7.20-7.13 (m, 1H), 5.54 (q, J = 6.9 Гц, 1H), 3.90-3.85 (m, 1H), 1.33-1.20 (m, 2H), 0.87-0.79 (m, 2H).
LC-MS: m/z 352.9 [M+H]+ при 2.59 RT (96.11% чистота).
ВЭЖХ: 93.29%.
Пример 8
Схема:
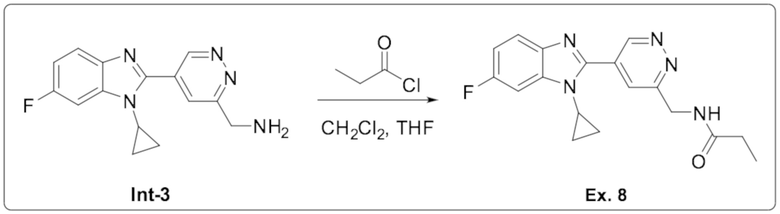
N-((5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил) пропионамид (Пример 8)
К перемешиваемому раствору (5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метанамина Int-3 (140 мг, 0.49 ммоль) в CH2Cl2 (5 мл) в инертной атмосфере добавили пропионил хлорид (0.05 мл, 0.59 ммоль) и триэтиламин (0.14 мл, 0.1 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) с получением N-((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метил) пропионамида Пример 8 (40 мг, 0.12 ммоль, 24%) в виде твердого вещества белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 9.72 (d, J = 1.4 Гц, 1H), 8.60 (br t, J = 5.1 Гц, 1H), 8.13 (d, J = 1.4 Гц, 1H), 7.79 (dd, J = 8.7, 4.9 Гц, 1H), 7.54 (dd, J = 9.0, 2.0 Гц, 1H), 7.18 (td, J = 9.8, 2.3 Гц, 1H), 4.67 (d, J = 6.1 Гц, 2H), 3.89-3.84 (m, 1H), 2.22 (q, J = 7.5 Гц, 2H), 1.25-1.16 (m, 2H), 1.04 (t, J = 7.7 Гц, 3H), 0.77-0.71 (m, 2H).
LC-MS: m/z 340.1 [M+H]+ при 2.07 RT (99.22% чистота).
ВЭЖХ: 99.08%.
Пример 9
Схема:

Этил((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил) карбамат (Пример 9)
К перемешиваемому раствору (5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метанамина Int-3 (125 мг, 0.44 ммоль) в THF (6 мл) в инертной атмосфере добавили этилхлорформиат (0.05 мл, 0.53 ммоль) и триэтиламин (0.12 мл, 0.88 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора хлорида аммония (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением этил ((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метил) карбамата Пример 9 (40 мг, 0.11 ммоль, 26%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400МГц, DMSO-d6): δ 9.75 (d, J = 2.1 Гц, 1H), 8.18 (d, J = 2.0 Гц, 1H), 7.93 (br t, J = 6.0 Гц, 1H), 7.79 (dd, J = 8.9, 4.9 Гц, 1H), 7.55 (dd, J = 9.1, 2.4 Гц, 1H), 7.22-7.15 (m, 1H), 4.60 (d, J = 6.1 Гц, 2H), 4.03 (q, J = 7.1 Гц, 2H), 3.91-3.84 (m, 1H), 1.22-1.16 (m, 5H), 0.78-0.73 (m, 2H).
LC-MS: m/z 356.1 [M+H]+ при 2.33 RT (98.62% чистота).
ВЭЖХ: 99.15%.
Пример 10
Схема:
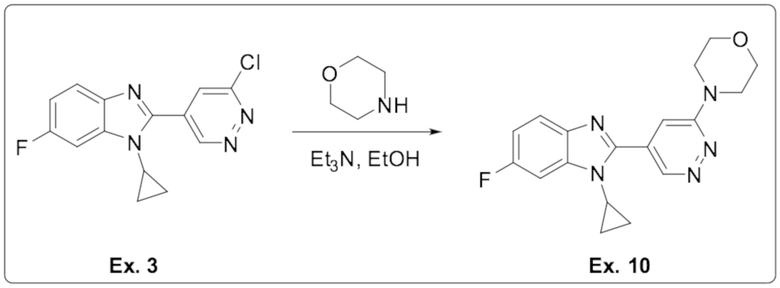
4-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)морфолин (Пример 10)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (150 мг, 0.52 ммоль) в этаноле (0.9 мл) в инертной атмосфере добавили триэтиламин (0.11 мл, 0.78 ммоль) и морфолин (0.07 мл, 0.78 ммоль) при комнатной температуре. Реакционную смесь нагрели до 90°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорид аммония (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 100% EtOAc) с получением 4-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) морфолина Пример 10 (40 мг, 0.12 ммоль, 23%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 9.14 (s, 1H), 7.81-7.71 (m, 2H), 7.53 (br d, J = 7.8 Гц, 1H), 7.16 (br t, J = 7.1 Гц, 1H), 3.93-3.91 (m, 1H), 3.83-3.61 (m, 8H), 1.18-1.16 (m, 2H), 0.75-0.73 (m, 2H).
LC-MS: m/z 340.0 [M+H]+ при 2.33 RT (97.11% чистота).
ВЭЖХ: 97.09%.
Пример 11
Схема:
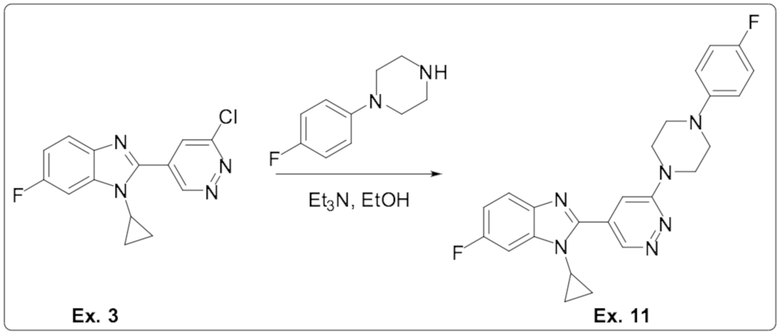
1-Циклопропил-6-фтор-2-(6-(4-(4-фторфенил)пиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 11)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (50 мг, 0.17 ммоль) в EtOH (0.3 мл) в инертной атмосфере добавили триэтиламин (0.036 мл, 0.26 ммоль) и 1-(4-фторфенил) пиперазин (47 мг, 0.26 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 90°C в течение 7 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 90% EtOAc/гексан) с получением 1-циклопропил-6-фтор-2-(6-(4-(4-фторфенил)пиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 11 (40 мг, 0.09 ммоль, 53%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.13 (d, J = 1.6 Гц, 1H), 7.80 (d, J = 1.5 Гц, 1H), 7.77 (dd, J = 8.9, 4.9 Гц, 1H), 7.54 (dd, J = 9.1, 2.4 Гц, 1H), 7.20-7.14 (m, 1H), 7.12-7.02 (m, 4H), 3.96-3.92 (m, 1H), 3.89-3.83 (m, 4H), 3.28-3.24 (m, 4H), 1.21-1.15 (m, 2H), 0.78-0.72 (m, 2H)
LC-MS: m/z 433.1 [M+H]+ при 2.38 RT (99.63% чистота).
ВЭЖХ: 96.82%.
Пример 12
Схема:
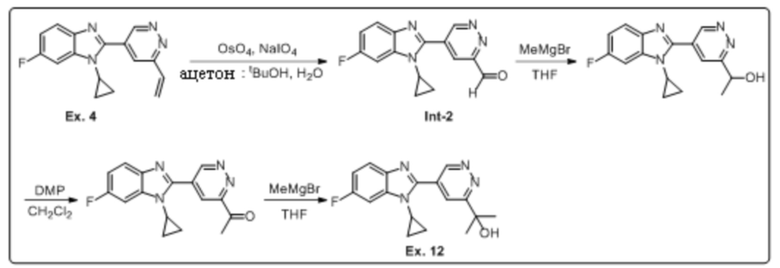
5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегид (Int-2)
К перемешиваемому раствору 1-циклопропил-6-фтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 4 (600 мг, 2.14 ммоль) в ацетоне: трет-бутаноле (tBuOH): воде (1:1:1, 30 мл) в инертной атмосфере добавили периодат натрия (912 мг, 4.28 ммоль) и осмия тетроксид (2.5 мас.% в толуоле, 3 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали, и фильтрат промыли с помощью EtOAc (2 x 30 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 90% EtOAc/гексан) с получением 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-2 (400 мг, 1.41 ммоль, 66%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, CDCl3): δ 10.52 (s, 1H), 10.08 (s, 1H), 8.65 (s, 1H), 7.81 (dd, J = 9.0, 4.9 Гц, 1H), 7.35 (dd, J = 8.4, 2.3 Гц, 1H), 7.17-7.12 (m, 1H), 3.78-3.67 (m, 1H), 1.39-1.30 (m, 2H), 0.94-0.82 (m, 2H)
1-[5-(1-Циклопропил-6-фтор-1H-1,3-бензодиазол-2-ил)пиридазин-3-ил]этан-1-ол
К перемешиваемому раствору 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-2 (200 мг, 0.70 ммоль) в THF (6 мл) в инертной атмосфере добавили метилмагния бромид (2 M в диэтиловом простом эфире, 0.35 мл, 0.70 ммоль) по каплям при -78°C. Реакционную смесь перемешивали при -78°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорид аммония (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2-3% MeOH/CH2Cl2) с получением 1-[5-(1-циклопропил-6-фтор-1H-1,3-бензодиазол-2-ил)пиридазин-3-ил]этан-1-ола (150 мг, 0.50 ммоль, 71%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.72 (s, 1H), 8.34 (s, 1H), 7.79 (dd, J = 9.0, 4.9 Гц, 1H), 7.55 (dd, J = 9.0, 2.0 Гц, 1H), 7.23-7.14 (m, 1H), 5.78 (d, J = 4.6 Гц, 1H), 5.16-5.05 (m, 1H), 3.95-3.90 (m, 1H), 1.52 (d, J = 6.7 Гц, 3H), 1.25-1.15 (m, 2H), 0.77-0.75 (m, 2H)
1-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-он
К перемешиваемому раствору 1-[5-(1-циклопропил-6-фтор-1H-1,3-бензодиазол-2-ил)пиридазин-3-ил]этан-1-ола (100 мг, 0.33 ммоль) в CH2Cl2 (10 мл) в инертной атмосфере добавили периодинан Десса-Мартина (213 мг, 0.51 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 50 мл). Органический слой промыли насыщенным раствором бикарбоната натрия (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2-3% MeOH/CH2Cl2) с получением 1-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) этан-1-она (80 мг, 0.27 ммоль, 81%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.04 (s, 1H), 8.64 (s, 1H), 7.82 (dd, J = 9.0, 4.9 Гц, 1H), 7.57 (dd, J = 9.0, 2.0 Гц, 1H), 7.23-7.11 (m, 1H), 4.02-3.91 (m, 1H), 2.87 (s, 3H), 1.20-1.15 (m, 2H), 0.79-0.76 (m, 2H)
2-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) пропан-2-ол (Пример 12)
К перемешиваемому раствору 1-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) этан-1-она (60 мг, 0.20 ммоль) в THF (2 мл) в инертной атмосфере добавили метилмагния бромид (2 M в диэтиловом простом эфире, 0.1 мл, 0.20 ммоль) по каплям при -78°C. Реакционную смесь перемешивали при -78°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорид аммония (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 2-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) пропан-2-ола Пример 12 (15 мг, 0.05 ммоль, 24%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.65 (s, 1H), 8.57 (s, 1H), 7.73 (dd, J = 8.9, 4.8 Гц, 1H), 7.50 (dd, J = 8.8, 2.4 Гц, 1H), 7.21-7.08 (m, 1H), 3.91-3.76 (m, 1H), 1.71 (s, 6H), 1.30-1.18 (m, 2H), 0.83-0.76 (m, 2H)
LC-MS: m/z 313 [M+H]+ при 2.19 RT (95.83% чистота).
ВЭЖХ: 95.47%.
Пример 13
Схема:
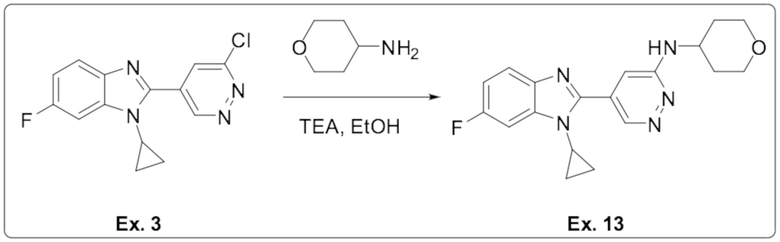
5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)-N-(тетрагидро-2H-пиран-4-ил) пиридазин-3-амин (Пример 13)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (150 мг, 0.52 ммоль) в EtOH (1.5 мл) в инертной атмосфере добавили триэтиламин (0.2 мл, 1.56 ммоль) и тетрагидро-2H-пиран-4-амин (105 мг, 1.04 ммоль) при комнатной температуре. Реакционную смесь перемешивали при возврате флегмы в течение 16 часов. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: EtOAc) с получением 5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)-N-(тетрагидро-2H-пиран-4-ил)пиридазин-3-амина Пример 13 (40 мг, 0.11 ммоль, 22%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.01 (s, 1H), 7.74 (dd, J = 8.7, 4.9 Гц, 1H), 7.51 (dd, J = 9.1, 2.5 Гц, 1H), 7.41 (s, 1H), 7.18-7.11 (m, 1H), 7.07 (d, J = 7.2 Гц, 1H), 4.17-4.07 (m, 1H), 3.93-3.86 (m, 2H), 3.80-3.75 (m, 1H), 3.49-3.44 (m, 2H), 2.05-1.95 (m, 2H), 1.59-1.42 (m, 2H), 1.30-1.02 (m, 2H), 0.87-0.74 (m, 2H)
LC-MS: m/z 354 [M+H]+ при 1.74 RT (97.97% чистота).
ВЭЖХ: 97.95%.
Пример 14
Схема:
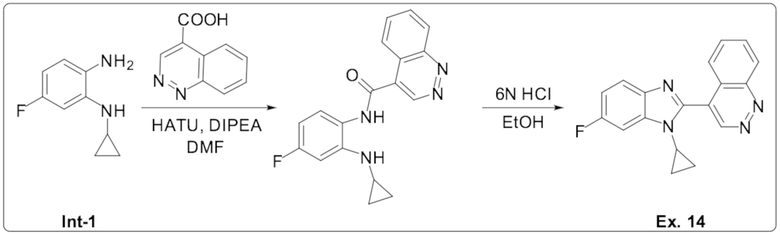
N-(2-(Циклопропиламино)-4-фторфенил) циннолин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5-фторбензол-1, 2-диамина Int-1 (500 мг, 3.01 ммоль) в DMF (5 мл) в инертной атмосфере добавили циннолин-4-карбоновую кислоту (524 мг, 3.01 ммоль), HATU (1.71 г, 4.51 ммоль) и диизопропилэтиламин (1 мл, 6.02 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(2-(циклопропиламино)-4-фторфенил) циннолин-4-карбоксамида (500 мг, неочищенный) в виде твердого вещества коричневого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 322.9 [M+H]+ при 2.71 RT (38.30% чистота).
4-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)циннолин (Пример 14)
К перемешиваемому раствору N-(2-(циклопропиламино)-4-фторфенил) циннолин-4-карбоксамида (400 мг, 1.24 ммоль) в EtOH (5 мл) в инертной атмосфере добавили 6N HCl (4 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 4-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) циннолина Пример 14 (160 мг, 0.52 ммоль, 43%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.69 (s, 1H), 8.65 (d, J = 8.2 Гц, 1H), 8.23 (d, J = 8.5 Гц, 1H), 8.12-8.07 (m, 1H), 8.01-7.97 (m, 1H), 7.82-7.79 (m, 1H), 7.58 (dd, J = 8.8, 2.3 Гц, 1H), 7.25-7.18 (m, 1H), 3.76-3.71 (m, 1H), 0.98-0.93 (m, 2H), 0.65-0.54 (m, 2H)
LC-MS: m/z 304.9 [M+H]+ при 2.59 RT (99.51% чистота).
ВЭЖХ: 99.30%.Пример 15
Схема:
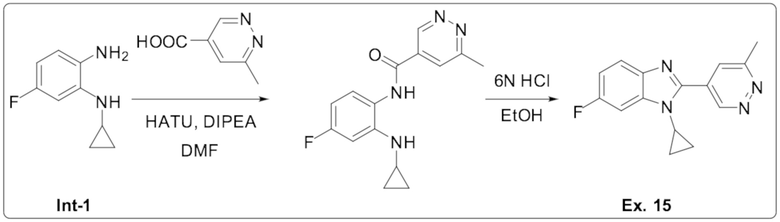
N-(2-(Циклопропиламино)-4-фторфенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору 6-метилпиридазин-4-карбоновой кислоты (174 мг, 1.0 ммоль) в DMF (3 мл) в инертной атмосфере добавили N1-циклопропил-5-фторбензол-1, 2-диамин Int-1 (166 мг, 1.0 ммоль), HATU (570 мг, 1.5 ммоль) и диизопропилэтиламин (1.38 мл, 4 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью хлорид аммония (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(2-(циклопропиламино)-4-фторфенил)-6-метилпиридазин-4-карбоксамида (250 мг, неочищенный) в виде твердого вещества коричневого цвета.
LC-MS: m/z 287.2 [M+H]+ при 3.53 RT (54.60% чистота).
1-Циклопропил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 15)
К перемешиваемому раствору N-(2-(циклопропиламино)-4-фторфенил)-6-метилпиридазин-4-карбоксамида (250 г, 0.87 ммоль) в EtOH (1.5 мл) в инертной атмосфере добавили 6Н HCl (3.5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 4-5% MeOH/CH2Cl2) с получением 1-циклопропил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 15 (80 мг, 0.29 ммоль, 34%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.64 (s, 1H), 8.21 (s, 1H), 7.74 (dd, J = 9.0, 4.6 Гц, 1H), 7.51 (dd, J = 8.8, 2.1 Гц, 1H), 7.19-7.13 (m, 1H), 3.89-3.83 (m, 1H), 2.84 (s, 3H), 1.31-1.23 (m, 2H), 0.86-0.79 (m, 2H)
LC-MS: m/z 268.9 [M+H]+ при 2.19 RT (95.85% чистота).
ВЭЖХ: 97.59%.
Пример 16
Схема:
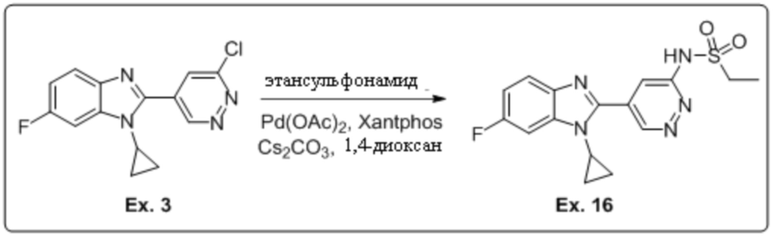
N-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этансульфонамид (Пример 16)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (100 мг, 0.34 ммоль) в 1,4-диоксане (4 мл) добавили этансульфонамид (57 мг, 0.52 ммоль) и карбонат цезия (283 мг, 0.86 ммоль) и смесь продули аргоном в течение 5 мин. Pd(OAc)2 (7.8 мг, 0.004 ммоль) и Xanthphos (30 мг, 0.05 ммоль) затем добавили к реакционной смеси. Реакционную смесь нагрели до 120°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью 10% MeOH: CH2Cl2 (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2-3% MeOH/CH2Cl2) с получением N-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этансульфонамида Пример 16 (30 мг, 0.08 ммоль, 24%) в виде бесцветного сиропа.
1H ЯМР (400 МГц, CD3OD): δ 8.96 (brs, 1H), 8.45 (brs, 1H), 7.76-7.73 (m, 1H), 7.50 (dd, J = 8.8, 2.3 Гц, 1H), 7.25-7.12 (m, 1H), 3.79-3.73 (m, 1H), 3.30-3.21 (m, 2H), 1.46-1.37 (m, 5H), 1.03-0.82 (m, 2H)
LC-MS: m/z 362 [M+H]+ при 2.04 RT (95.41% чистота).
ВЭЖХ: 94.11%.
Пример 17
Схема:
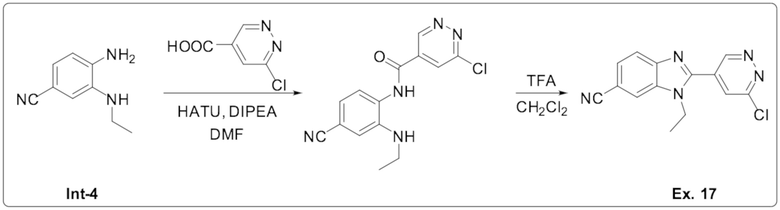
6-Хлор-N-(4-циано-2-(этиламино)фенил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4-амино-3-(этиламино) бензонитрила Int-4 (322 мг, 2 ммоль) в DMF (6 мл) в инертной атмосфере добавили 6-хлорпиридазин-4-карбоновую кислоту (316 мг, 2 ммоль), HATU (1.14 г, 3 ммоль) и этилдиизопропиламин (1.38 мл, 8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6-хлор-N-(4-циано-2-(этиламино)фенил) пиридазин-4-карбоксамида (350 мг, неочищенный) в виде твердого вещества грязновато-белого цвета.
LC-MS: m/z 300.1 [M-H]- при 2.07 RT (18.03% чистота).
2-(6-Хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрил (Пример 17)
К перемешиваемому раствору 6-хлор-N-(4-циано-2-(этиламино) фенил) пиридазин-4-карбоксамида (350 мг, 1.16 ммоль) в CH2Cl2 (3 мл) в инертной атмосфере добавили трифторуксусную кислоту (TFA) (1 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли водой (20 мл), соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила Пример 17 (25 мг, 0.08 ммоль, 7.5%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.63 (d, J = 1.9 Гц, 1H), 8.27 (d, J = 1.8 Гц, 2H), 7.93 (dd, J = 8.4, 0.6 Гц, 1H), 7.69 (dd, J = 8.5, 1.4 Гц, 1H), 4.56-4.51 (m, 2H), 1.51 (t, J = 7.3 Гц, 3H)
LC-MS: m/z 284.2 [M+H]+ при 3.50 RT (99.67% чистота).
ВЭЖХ: 99.48%.
Пример 18
Схема:
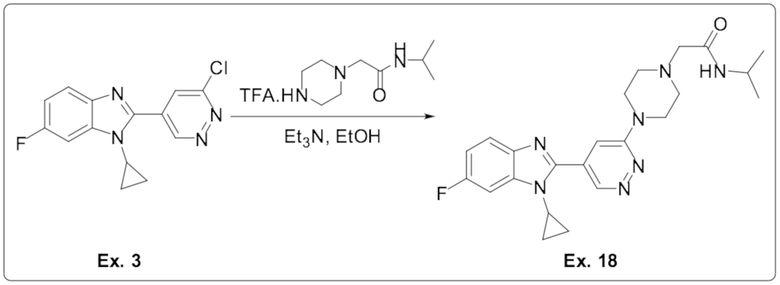
2-(4-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)-N-изопропилацетамид (Пример 18)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (56 мг, 0.19 ммоль) в EtOH (2 мл) в инертной атмосфере добавили триэтиламин (0.04 мл, 0.28 ммоль) и N-изопропил-2-(4-(2, 2, 2-трифторацетил)-4l5-пиперазин-1-ил) ацетамид (60 мг, 0.27 ммоль) при комнатной температуре. Реакционную смесь перемешивали при возврате флегмы в течение 16 часов. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 2-(4-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)-N-изопропилацетамида Пример 18 (12 мг, 0.02 ммоль, 14%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.08 (s, 1H), 7.76 (s, 1H), 7.72 (dd, J = 8.9, 4.8 Гц, 1H), 7.50 (dd, J = 8.8, 2.5 Гц, 1H), 7.15 (dt, J = 9.3, 2.4 Гц, 1H), 4.13-4.02 (m, 1H), 3.87-3.81 (m, 4H), 3.34-3.31 (m, 1H), 3.10 (s, 2H), 2.73-2.69 (m, 4H), 1.28-1.23 (m, 2H), 1.20 (d, J = 6.7 Гц, 6H), 0.86-0.80 (m, 2H)
LC-MS: m/z 438.4 [M+H]+ при 3.76 RT (94.38% чистота).
ВЭЖХ: 94.23%.
Пример 19
Схема:
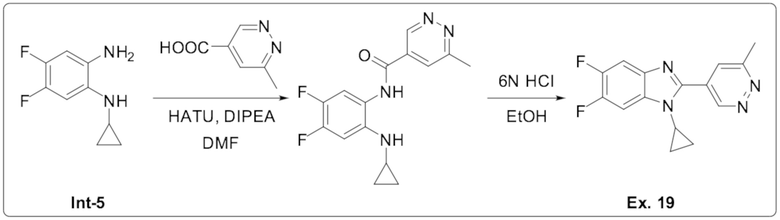
N-(2-(Циклопропиламино)-4,5-дифторфенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4, 5-дифторбензол-1, 2-диамина Int-5 (184 мг, 1.0 ммоль) в DMF (3 мл) в инертной атмосфере добавили 6-метилпиридазин-4-карбоновую кислоту (174 мг, 1.0 ммоль), HATU (570 мг, 1.5 ммоль) и диизопропилэтиламин (1.38 мл, 4.0 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили раствором хлорида аммония (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-6-метилпиридазин-4-карбоксамида (220 мг, неочищенный) в виде сиропа коричневого цвета.
LC-MS: m/z 305.2 [M+H]+ при 3.78 RT (44.76% чистота).
1-Циклопропил-5,6-дифтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 19)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-6-метилпиридазин-4-карбоксамида (220 мг, 0.72 ммоль) в EtOH (1.5 мл) в инертной атмосфере добавили 6Н HCl (3.5 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (100 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3-5% MeOH/CH2Cl2) с получением 1-циклопропил-5,6-дифтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 19 (60 мг, 0.20 ммоль, 29%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.63 (d, J = 1.9 Гц, 1H), 8.20 (d, J = 2.1 Гц, 1H), 7.72 (dd, J = 10.1, 7.1 Гц, 1H), 7.62 (dd, J = 10.4, 7.3 Гц, 1H), 3.90-3.83 (m, 1H), 2.84 (s, 3H), 1.31-1.24 (m, 2H), 0.85-0.80 (m, 2H)
LC-MS: m/z 286.9 [M+H]+ при 2.32 RT (99.57% чистота).
ВЭЖХ: 99.70%.
Пример 20
Схема:
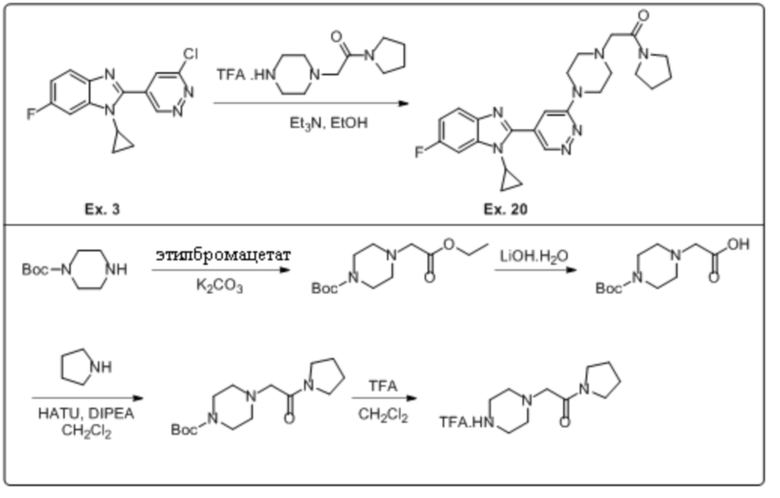
трет-Бутил 4-(2-этокси-2-охоэтил) пиперазин-1-карбоксилат
К перемешиваемому раствору трет-бутилпиперазин-1-карбоксилата (2 г, 10.7 ммоль) в DMF (14 мл) в инертной атмосфере добавили карбонат калия (3.71 г, 26.8 ммоль) и этилбромацетат (1.2 мл, 10.8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением трет-бутил 4-(2-этокси-2-охоэтил) пиперазин-1-карбоксилата (2.8 г, 10.29 ммоль, 95%) в виде масла желтого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 4.12-4.00 (m, 2H), 3.30 (s, 4H), 3.23 (s, 2H), 2.45 (t, J = 4.6 Гц, 4H), 1.39 (s, 9H), 1.18 (t, J = 7.1 Гц, 3H)
2-(4-трет-бутоксикарбонил) пиперазин-1-ил)уксусная кислота
К перемешиваемому раствору трет-бутил 4-(2-этокси-2-охоэтил) пиперазин-1-карбоксилата (2.8 г, 10.29 ммоль) в смеси THF: MeOH: вода (3:1:1, 30 мл) добавили гидроксид лития (1.29 г, 30.8 ммоль) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Затем остаток подкислили до pH 4-5 с помощью раствора 5% лимонной кислоты и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2-(4-(трет-бутоксикарбонил) пиперазин-1-ил) уксусной кислоты (200 мг, неочищенный) в виде масла желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 245.1 [M-H]+ при 1.06 RT (71.81% чистота).
трет-бутил 4-(2-оксо-2-(пирролидин-1-ил)этил)пиперазин-1-карбоксилат
К перемешиваемому раствору 2-(4-(трет-бутоксикарбонил) пиперазин-1-ил) уксусной кислоты (500 мг, 2.04 ммоль) в CH2Cl2 (15 мл) в инертной атмосфере добавили пирролидине (0.2 мл, 2.45 ммоль), HATU (1.1 г, 3.07 ммоль) и этилдиизопропиламин (1.5 мл, 8.19 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением трет-бутил 4-(2-оксо-2-(пирролидин-1-ил) этил) пиперазин-1-карбоксилата (100 мг, 0.33 ммоль, 16%) в виде твердого вещества коричневого цвета.
1H ЯМР (500МГц, DMSO-d6): δ 3.43 (t, J = 6.7 Гц, 2H), 3.31 (s, 2H), 3.29-3.25 (m, 4H), 2.43 (brs, 4H), 1.87-1.82 (m, 2H), 1.78-1.71 (m, 2H), 1.39 (s, 9H), 1.29-1.23 (m, 2H)
4-(2-оксо-2-(пирролидин-1-ил)этил)пиперазин-1-ил 2,2,2-трифторацетатная соль
К перемешиваемому раствору трет-бутил 4-(2-оксо-2-(пирролидин-1-ил) этил) пиперазин-1-карбоксилата (100 мг, 0.33 ммоль) в CH2Cl2 (1 мл) в инертной атмосфере добавили трифторуксусная кислота (0.5 мл) по каплям при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Неочищенное вещество промыли простым эфиром (2 x 5 мл) с получением 4-(2-оксо-2-(пирролидин-1-ил) этил) пиперазин-1-ил 2, 2, 2-трифторацетатной соли (120 мг в виде TFA соли) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 3.72 (brs, 1H), 3.40-3.37 (m, 4H), 3.33-3.30 (m, 4H), 3.24 (s, 2H), 3.08 (brs, 4H), 1.92-1.85 (m, 2H), 1.81-1.76 (m, 2H)
2-(4-(5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)-1-(пирролидин-1-ил)этан-1-он (Пример 20)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-6-фтор-1H-бензо[d]имидазола Пример 3 (100 мг, 0.34 ммоль) в EtOH (2 мл) в инертной атмосфере добавили триэтиламин (0.14 мл, 1.04 ммоль) и 4-(2-оксо-2-(пирролидин-1-ил)этил)пиперазин-1-ил 2,2,2-трифторацетатную соль (34 мг, 0.17 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 32 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 2-(4-(5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) пиперазин-1-ил)-1-(пирролидин-1-ил) этан-1-она Пример 20 (20 мг, 0.041 ммоль, 13%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.16 (s, 1H), 7.75-7.71 (m, 1H), 7.47 (s, 1H), 7.30 (dd, J = 8.6, 2.4 Гц, 1H), 7.11-7.06 (m, 1H), 3.87-3.85 (m, 4H), 3.63-3.57 (m, 1H), 3.53-3.43 (m, 4H), 3.28 (s, 2H), 2.83 (brs, 4H), 2.03-1.93 (m, 2H), 1.89-1.84 (m, 2H), 1.30-1.22 (m, 2H), 0.88-0.80 (m, 2H)
LC-MS: m/z 450.2 [M+H]+ при 1.74 RT (97.66% чистота).
ВЭЖХ: 97.81%.
Примеры 21 и 22
Схема:
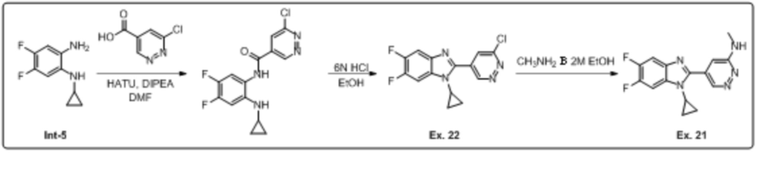
6-Хлор-N-(2-(циклопропиламино)-4,5-дифторфенил)пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4, 5-дифторбензол-1, 2-диамина Int-5 (1 г, 5.43 ммоль) в DMF (10 мл) в инертной атмосфере добавили 6-хлорпиридазин-4-карбоновую кислоты (1.03 г, 6.52 ммоль), HATU (2.48 г, 6.52 ммоль) и диизопропилэтиламин (3.9 мл, 21.72 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 0-2% MeOH/CH2Cl2) с получением 6-хлор-N-(2-(циклопропиламино)-4, 5-дифторфенил) пиридазин-4-карбоксамида (800 мг, 2.46 ммоль, 45%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.50 (s, 1H), 7.95 (d, J = 1.6 Гц, 1H), 7.74 (brs, 1H), 7.37 (dd, J = 10.5, 8.5 Гц, 1H), 7.06 (dd, J = 12.4, 7.7 Гц, 1H), 4.16-4.03 (m, 1H), 2.51-2.40 (m, 1H), 0.83-0.75 (m, 2H), 0.56-0.47 (m, 2H)
2-(6-Хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол (Пример 22)
К перемешиваемому раствору 6-хлор-N-(2-(циклопропиламино)-4, 5-дифторфенил) пиридазин-4-карбоксамида (700 мг, 2.16 ммоль) в EtOH (7 мл) в инертной атмосфере добавили 6N HCl (10.5 мл) при комнатной температуре. Реакционную смесь перемешивали при 70°C в течение 30 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20-30% EtOAc/Гексан) с получением 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (280 мг, 0.91 ммоль, 42%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.78 (d, J = 1.9 Гц, 1H), 8.15 (d, J = 1.9 Гц, 1H), 7.60 (dd, J = 10.0, 7.2 Гц, 1H), 7.44 (dd, J = 9.5, 6.9 Гц, 1H), 3.70-3.62 (m, 1H), 1.41-1.33 (m, 2H), 0.93-0.84 (m, 2H)
LC-MS: m/z 306.9 [M+1]+ при 2.52 RT (99.86% чистота).
ВЭЖХ: 99.51%.
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-метилпиридазин-3-амин (Пример 21)
2-(6-Хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол Пример 22 (600 мг, 1.96 ммоль) растворили в 2 M растворе метиламина (2 M в диэтиловом простом эфире, 3 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2-3% MeOH/CH2Cl2) с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-метилпиридазин-3-амина Пример 21 (50 мг, 0.16 ммоль, 8%) в виде твердого вещества бледно-зеленого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.13 (d, J = 1.8 Гц, 1H), 7.57 (dd, J = 10.2, 7.3 Гц, 1H), 7.41 (dd, J = 9.7, 7.0 Гц, 1H), 7.20 (d, J = 1.8 Гц, 1H), 5.03 (d, J = 4.5 Гц, 1H), 3.59 (tt, J = 7.0, 3.6 Гц, 1H), 3.12 (d, J = 5.1 Гц, 3H), 1.32-1.24 (m, 2H), 0.87-0.80 (m, 2H)
LC-MS: m/z 301.9 [M+1]+ при 1.76 RT (98.78% чистота).
ВЭЖХ: 99.48%.
Пример 23
Схема:
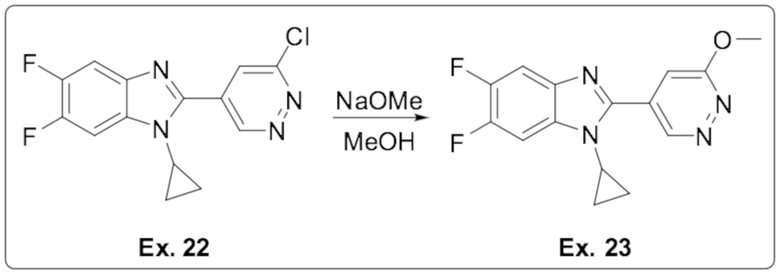
1-Циклопропил-5,6-дифтор-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол (Пример 23)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.32 ммоль) в MeOH (5 мл) в инертной атмосфере добавили метоксид натрия (53 мг, 0.98 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 70°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток разбавили водой (10 мл) и экстрагировали с помощью CH2Cl2 (2 x 10 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/Гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазола Пример 23 (40 мг, 0.13 ммоль, 41%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.45 (s, 1H), 7.89-7.80 (m, 2H), 7.77 (s, 1H), 4.13 (s, 3H), 3.93-3.89 (m, 1H), 1.23-1.12 (m, 2H), 0.82-0.70 (m, 2H)
LC-MS: m/z 302.9 [M+H]+ при 2.30 RT (99.80% чистота).
ВЭЖХ: 99.68%.
Пример 24
Схема:
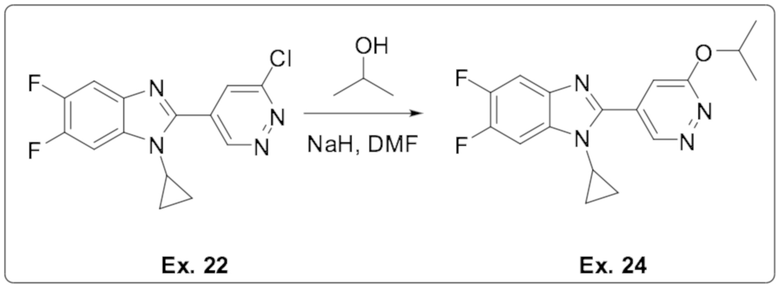
1-Циклопропил-5,6-дифтор-2-(6-изопропоксипиридазин-4-ил)-1H-бензо[d]имидазол (Пример 24)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (70 мг, 0.23 ммоль) в DMF (0.5 мл) в инертной атмосфере добавили гидрид натрия (55% в минеральном масле, 20 мг, 0.46 ммоль) по частям при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 мин. К этой реакционной смеси добавили пропан-2-ол (27.5 мг, 0.46 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (10 мл), соляным раствором (10 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/Гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-изопропоксипиридазин-4-ил)-1H-бензо[d]имидазола Пример 24 (30 мг, 0.09 ммоль, 40%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.39 (s, 1H), 7.89-7.80 (m, 2H), 7.67 (s, 1H), 5.57-5.51 (m, 1H), 3.93-3.89 (m, 1H), 1.42 (d, J = 6.1 Гц, 6H), 1.20-1.11 (m, 2H), 0.86-0.68 (m, 2H)
LC-MS: m/z 330.9 [M+H]+ при 2.88 RT (93.72% чистота).
ВЭЖХ: 95.25%.
Пример 25
Схема:
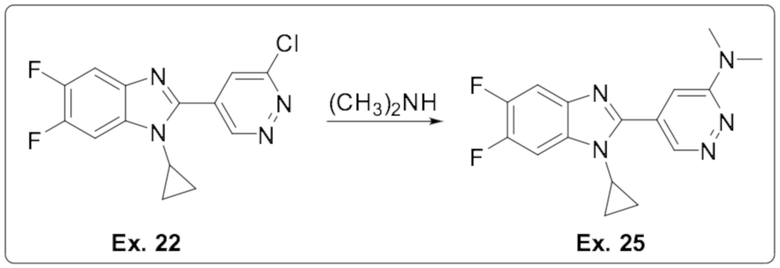
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N,N-диметилпиридазин-3-амин (Пример 25)
2-(6-Хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол Пример 22 (60 мг, 0.19 ммоль) растворили в 2 M растворе диметиламина (2 M в THF, 3 мл) и реакционную смесь перемешивали в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2-3% MeOH/CH2Cl2) с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N, N-диметилпиридазин-3-амина Пример 25 (16 мг, 0.05 ммоль, 26%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.08 (s, 1H), 7.58 (dd, J = 10.2, 7.3 Гц, 1H), 7.41 (dd, J = 9.7, 7.0 Гц, 1H), 7.32 (s, 1H), 3.62-3.57 (m, 1H), 3.28 (s, 6H), 1.29-1.23 (m, 2H), 0.87-0.77 (m, 2H)
LC-MS: m/z 316.1 [M+1]+ при 1.81 RT (98.32% чистота).
ВЭЖХ: 95.98%.
Пример 26
Схема:
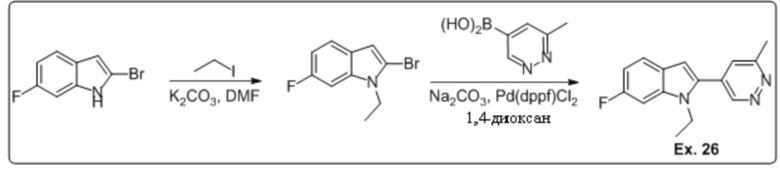
2-Бром-1-этил-6-фтор-1H-индол
К перемешиваемому раствору 2-бром-6-фтор-1H-индола (250 мг, 1.16 ммоль) в DMF (2 мл) в инертной атмосфере добавили карбонат калия (476 мг, 3.50 ммоль), а затем иодэтан(364 мг, 2.33 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2-бром-1-этил-6-фтор-1H-индола (280 мг, неочищенный) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 7.43 (dd, J = 8.6, 5.3 Гц, 1H), 6.98 (dd, J = 9.7, 2.3 Гц, 1H), 6.86-6.84 (m, 1H), 6.54 (d, J = 0.8 Гц, 1H), 4.21-4.16 (m, 2H), 1.34 (t, J = 7.2 Гц, 3H)
1-Этил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-индол (Пример 26)
К перемешиваемому раствору 2-бром-1-этил-6-фтор-1H-индола (200 мг, 0.83 ммоль) в 1, 4-диоксане (2 мл) добавили (6-метилпиридазин-4-ил) бороновую кислоту (214 мг, 1.24 ммоль), раствор карбоната натрия (2M, 0.5 мл) и продули аргоном в течение 10 мин. Затем Pd(dppf)Cl2 (60 мг, 0.08 ммоль) добавили в реакционную смесь, и реакционную смесь перемешивали при 80°C в течение 16 часов в запаянной трубке. Развитие реакции контролировали посредством TLC; реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-этил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-индола Пример 26 (50 мг, 0.19 ммоль, 20%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.25 (d, J = 2.0 Гц, 1H), 7.76 (d, J = 2.1 Гц, 1H), 7.61 (dd, J = 8.7, 5.3 Гц, 1H), 7.28 (dd, J = 10.1, 2.2 Гц, 1H), 6.96-6.90 (m, 2H), 4.35-4.30 (m, 2H), 2.77 (s, 3H), 1.29 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 256 [M+H]+ при 2.73 RT (98.24% чистота)
ВЭЖХ: 97.70%
Пример 27
Схема:
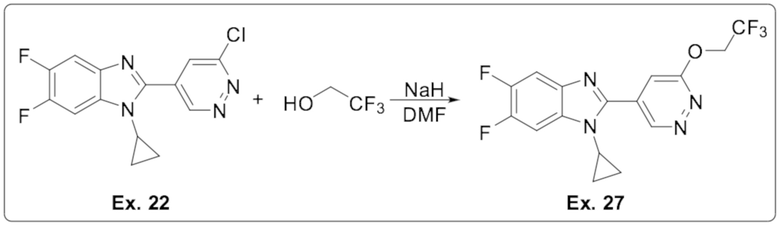
1-Циклопропил-5,6-дифтор-2-(6-(2,2,2-трифторэтокси)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 27)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (70 мг, 0.23 ммоль) в DMF (0.5 мл) в инертной атмосфере добавили гидрид натрия (55% в минеральном масле, 20 мг, 0.46 ммоль) по частям при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 мин. К этой реакционной смеси добавили 2, 2, 2-трифтопентан-1-ол (46 мг, 0.46 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (10 мл), соляным раствором (10 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/Гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(2, 2, 2-трифторэтокси) пиридазин-4-ил)-1H-бензо[d]имидазола Пример 27 (50 мг, 0.13 ммоль, 62%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.55 (s, 1H), 7.97 (s, 1H), 7.89-7.85 (m, 2H), 5.39-5.27 (m, 2H), 3.96-3.93 (m, 1H), 1.21-1.13 (m, 2H), 0.82-0.69 (m, 2H)
LC-MS: m/z 370.9 [M+1]+ при 3.18 RT (99.73% чистота).
ВЭЖХ: 98.32%.
Пример 28
Схема:
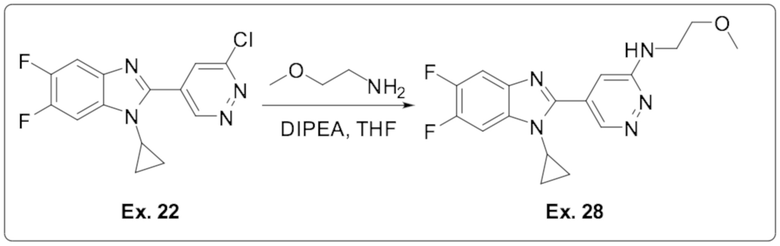
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-(2-метоксиэтил)пиридазин-3-амин (Пример 28)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (50 мг, 0.16 ммоль) в THF (0.5 мл) в инертной атмосфере добавили 2-метоксиэтан-1-амин (0.5 мл, 75.11 ммоль) и этилдиизопропиламин (0.5 мл) при комнатной температуре. Реакционную смесь перемешивали при 110°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью CH2Cl2: Гексана (1: 9, 10 мл) с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-(2-метоксиэтил) пиридазин-3-амина Пример 28 (40 мг, 0.13 ммоль, 92%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.12 (s, 1H), 7.56 (dd, J = 10.1, 7.2 Гц, 1H), 7.41 (dd, J = 9.7, 6.9 Гц, 1H), 7.22 (s, 1H), 5.21 (brs, 1H), 3.76-3.73 (m, 2H), 3.69-3.64 (m, 2H), 3.61-3.59 (m, 1H), 3.41 (s, 3H), 1.31-1.25 (m, 2H), 0.87-0.81 (m, 2H)
LC-MS: m/z 346 [M+H]+ при 1.80 RT (97.27% чистота).
ВЭЖХ: 97.05%.
Пример 29
Схема:
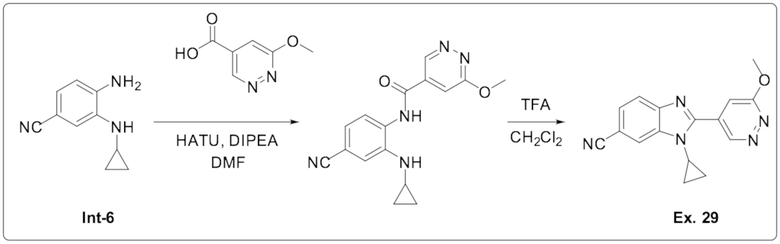
N-(4-Циано-2-(циклопропиламино) фенил)-6-метоксипиридазин-4-карбоксамид
К перемешиваемому раствору 4-амино-3-(циклопропиламино) бензонитрила Int-6 (300 мг, 1.72 ммоль) в DMF (5 мл) в инертной атмосфере добавили 6-метоксипиридазин-4-карбоновую кислоту (281 мг, 1.72 ммоль), HATU (720 мг, 1.89 ммоль) и этилдиизопропиламин (0.8 мл, 5.17 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(4-циано-2-(циклопропиламино) фенил)-6-метоксипиридазин-4-карбоксамида (300 мг, неочищенный) в виде жидкости желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
1-Циклопропил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 29)
К перемешиваемому раствору N-(4-циано-2-(циклопропиламино) фенил)-6-метоксипиридазин-4-карбоксамида (300 мг, неочищенный) в CH2Cl2 (10 мл) в инертной атмосфере добавили трифторуксусную кислоту (0.5 мл) по каплям при 0°C. Реакционную смесь перемешивали при 0°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток разбавили водой (5 мл), подщелочили с помощью насыщенного раствора карбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 29 (40 мг, 0.13 ммоль, 14% (за две стадии)) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.48 (s, 1H), 8.32 (s, 1H), 7.94 (d, J = 8.4 Гц, 1H), 7.82 (s, 1H), 7.71 (dd, J = 8.4, 1.5 Гц, 1H), 4.14 (s, 3H), 4.01-3.85 (m, 1H), 1.23-1.14 (m, 2H), 0.87-0.77 (m, 2H)
LC-MS: m/z 291.9 [M+H]+ при 2.25 RT (95.84% чистота).
ВЭЖХ: 97.16%.
Пример 30
Схема:
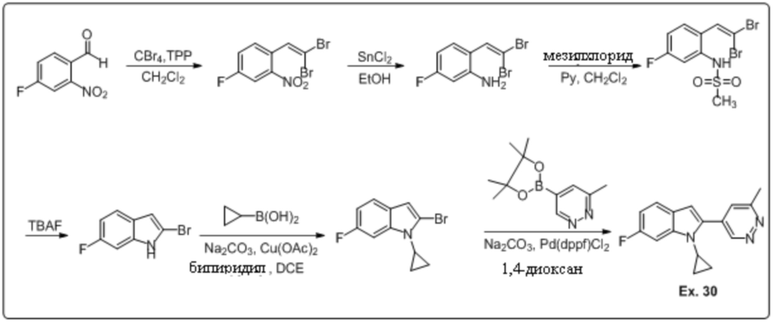
1-(2, 2-Дибромвинил)-4-фтор-2-нитробензол
К перемешиваемому раствору 4-фтор-2-нитробензальдегида (2 г, 11.83 ммоль) в CH2Cl2 (100 мл) добавили тетрабромид углерода (5.8 г, 17.75 ммоль) и трифенилфосфин (9.3 г, 35.50 ммоль) при 0°C в инертной атмосфере. Реакционную смесь перемешивали при 5°C в течение 2 часов. Развитие реакции контролировали посредством TLC; реакционную смесь отфильтровали, фильтрат концентрировали при пониженном давлении с получением 1-(2, 2-дибромвинил)-4-фтор-2-нитробензола (3.2 г, неочищенный) в виде твердого вещества коричневого цвета, которое применяли на следующей стадии без дальнейшей очистки.
2-(2, 2-Дибромвинил)-5-фторанилин
К перемешиваемому раствору 1-(2, 2-дибромвинил)-4-фтор-2-нитробензола (3.2 г, 9.87 ммоль) в EtOH (20 мл) добавили SnCl2. H2O (11.1 г, 46.29 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при возврате флегмы в течение 2 часов. Развитие реакции контролировали посредством TLC; летучие вещества концентрировали при пониженном давлении. Полученный остаток подщелочили с помощью раствора карбоната калия до pH~10 и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/Гексан) с получением 2-(2, 2-дибромвинил)-5-фторанилина (2.1 г, 7.14 ммоль, 72%) в виде твердого вещества коричневого цвета.
LC-MS: m/z 295.6 [M+2H]+ при 3.34 RT (96.98% чистота)
N-(2-(2, 2-Дибромвинил)-5-фторфенил) метансульфонамид
К перемешиваемому раствору 2-(2, 2-дибромвинил)-5-фторанилина (1 г, 3.40 ммоль) в CH2Cl2 (10 мл) добавили пиридин (0.54 мл, 6.80 ммоль) и мезилхлорид (0.38 мл, 5.10 ммоль) при 0°C в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Развитие реакции контролировали посредством TLC; реакционную смесь погасили раствором NaHSO4 и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/Гексан) с получением N-(2-(2, 2-дибромвинил)-5-фторфенил) метансульфонамида (1.1 г, 2.94 ммоль, 87%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.57 (s, 1H), 7.69 (s, 1H), 7.59 (dd, J = 8.7, 6.4 Гц, 1H), 7.23 (dd, J = 10.4, 2.3 Гц, 1H), 7.14 (dt, J = 8.4, 2.3 Гц, 1H), 3.06 (s, 3H)
2-Бром-6-фтор-1H-индол
К перемешиваемому раствору N-(2-(2, 2-дибромвинил)-5-фторфенил) метансульфонамида (200 мг, 0.53 ммоль) в THF (2 мл) добавили TBAF 1M в THF (1 мл) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при 100°C в течение 5 мин под микроволнами. Развитие реакции контролировали посредством TLC; реакционную смесь разбавили водой (10 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2-бром-6-фтор-1H-индола 5 (100 мг, неочищенный) в виде твердого вещества коричневого цвета, которое применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 295.6 [M+2H]+ при 3.34 RT (96.98% чистота)
2-Бром-1-циклопропил-6-фтор-1H-индол
К перемешиваемому раствору 2-бром-6-фтор-1H-индола 5 (400 мг, 1.86 ммоль) в 1, 2-дихлорэтане (5 мл) добавили циклопропилбороновую кислоту (321 мг, 3.73 ммоль), карбонат натрия (571 мг, 5.60 ммоль), ацетат меди (371 мг, 1.86 ммоль) и бипиридил (291 мг, 1.86 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при 80°C в течение 16 часов в запаянной трубке. Развитие реакции контролировали посредством TLC; реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/Гексан) с получением 2-бром-1-циклопропил-6-фтор-1H-индола (300 мг, 1.19 ммоль, 63%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 7.39 (dd, J = 8.6, 5.4 Гц, 1H), 7.23 (dd, J = 10.1, 2.3 Гц, 1H), 6.88-6.83 (m, 1H), 6.51 (s, 1H), 3.14-3.09 (m, 1H), 1.27-1.19 (m, 2H), 1.13-1.08 (m, 2H)
1-Циклопропил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-индол (Пример 30)
К перемешиваемому раствору 2-бром-1-циклопропил-6-фтор-1H-индола (200 мг, 0.79 ммоль) в 1, 4-диоксане (5 мл) добавили 3-метил-5-(4, 4, 5, 5-тетраметил-1, 3, 2-диоксаборолан-2-ил) пиридазин (261 мг, 1.19 ммоль), раствор карбоната натрия (2M, 0.5 мл) и продули аргоном в течение 10 мин. Затем Pd(dppf)Cl2 (58 мг, 0.08 ммоль) добавили в реакционную смесь, и реакционную смесь перемешивали при 80°C в течение 16 часов в запаянной трубке. Развитие реакции контролировали посредством TLC; реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-6-фтор-2-(6-метилпиридазин-4-ил)-1H-индола Пример 30 (36 мг, 0.13 ммоль, 17%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.42 (d, J = 2.1 Гц, 1H), 7.97 (d, J = 2.1 Гц, 1H), 7.60 (dd, J = 8.7, 5.3 Гц, 1H), 7.37 (dd, J = 10.0, 2.4 Гц, 1H), 6.98 (s, 1H), 6.96-6.91 (m, 1H), 3.83-3.68 (m, 1H), 2.80-2.79 (m, 3H), 1.25-1.09 (m, 2H), 0.78-0.63 (m, 2H)
LC-MS: m/z 267.9 [M+H]+ при 2.78 RT (98.96% чистота)
ВЭЖХ: 97.98%
Пример 31
Схема:

N-((5-(1-Циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)этансульфонамид (Пример 31)
К перемешиваемому раствору (5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил) метанамина Int-3 (30 мг, 0.10 ммоль) в THF (2 мл) в инертной атмосфере добавили этансульфонил хлорид (16 мг, 0.12 ммоль) и триэтиламин (0.03 мл, 0.21 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением N-((5-(1-циклопропил-6-фтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метил) этансульфонамида Пример 31 (15 мг, 0.04 ммоль, 38%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.73 (s, 1H), 8.52 (s, 1H), 7.78-7.74 (m, 1H), 7.53 (dd, J = 8.9, 2.4 Гц, 1H), 7.20-7.14 (m, 1H), 4.71 (s, 2H), 3.87-3.82 (m, 1H), 3.24-3.19 (m, 2H), 1.47-1.31 (m, 5H), 0.89-0.80 (m, 2H)
LC-MS: m/z 376 [M+H]+ при 2.21 RT (99.12% чистота).
ВЭЖХ: 98.34%.
Пример 32
Схема:
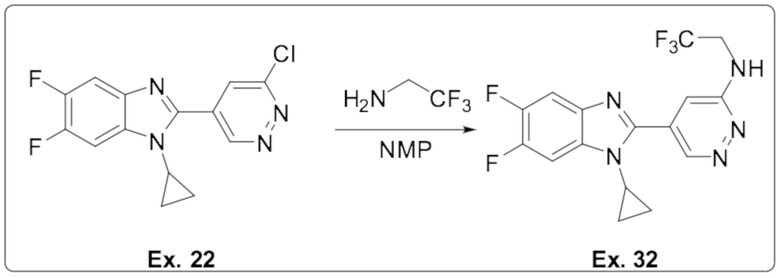
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-(2,2,2-трифторэтил)пиридазин-3-амин (Пример 32)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (80 мг, 0.26 ммоль) в NMP (1.2 мл) в инертной атмосфере добавили 2,2,2-трифторэтан-1-амин (1.2 мл) при комнатной температуре. Реакционную смесь перемешивали при 130°C в течение 6 часов под микроволнами. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3%MeOH/CH2Cl2) с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-N-(2, 2, 2-трифторэтил) пиридазин-3-амина Пример 32 (15 мг, 0.04 ммоль, 15%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.28 (s, 1H), 7.59 (dd, J = 10.1, 7.2 Гц, 1H), 7.44 (dd, J = 9.6, 7.0 Гц, 1H), 7.37 (s, 1H), 5.09 (t, J = 6.0 Гц, 1H), 4.43-4.30 (m, 2H), 3.66-3.60 (m, 1H), 1.36-1.29 (m, 2H), 0.91-0.84 (m, 2H)
LC-MS: m/z 370 [M+H]+ при 2.21 RT (97.23% чистота).
ВЭЖХ: 97.15%.
Пример 33
Схема:
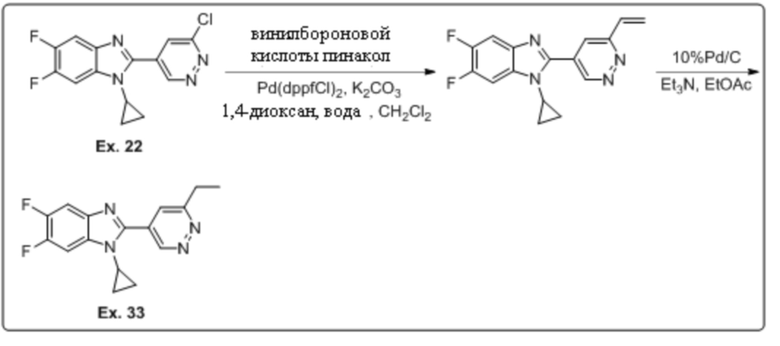
1-Циклопропил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (150 мг, 0.50 ммоль) в 1,4-диоксане (3 мл) и воде (0.5 мл) в инертной атмосфере добавили винилбороновой кислоты сложный пинаколовый эфир(76 мг, 0.50 ммоль) и карбонат калия (203 мг, 1.47 ммоль) при комнатной температуре. Реакционную смесь дегазировали аргоном в течение 10 мин, Pd(dppf)Cl2 (4 мг, 0.005 ммоль) добавили, и реакционную смесь дегазировали аргоном в течение еще 10 мин. Реакционную смесь нагрели до 80°C и перемешивали в течение 5 часов. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/ Гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (70 мг, 0.23 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.69 (s, 1H), 8.45 (s, 1H), 7.90-7.83 (m, 2H), 7.18-7.11 (m, 1H), 6.53 (d, J = 17.7 Гц, 1H), 5.82 (d, J = 11.4 Гц, 1H), 3.99-3.97 (m, 1H), 1.22-1.13 (m, 2H), 0.82-0.60 (m, 2H)
1-Циклопропил-2-(6-этилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 33)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (70 мг, 0.23 ммоль) в EtOAc (3 мл) в инертной атмосфере добавили 10% Pd/C (20 мг) и триэтиламин (каталитическое количество) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли, и слой промыли метанолом (30 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-этилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 33 (35 мг, 0.11 ммоль, 50%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.65 (s, 1H), 8.17 (s, 1H), 7.89-7.81 (m, 2H), 3.95-3.91 (m, 1H), 3.10-3.04 (m, 2H), 1.36 (t, J = 7.6 Гц, 3H), 1.24-1.12 (m, 2H), 0.79-0.64 (m, 2H)
LC-MS: m/z 300.9 [M+H]+ при 2.51 RT (98.98% чистота).
ВЭЖХ: 97.01%.
Пример 34
Схема:
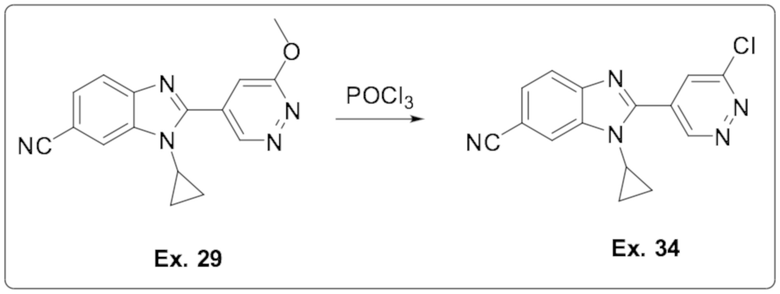
2-(6-Хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрил (Пример 34)
К перемешиваемому раствору 1-циклопропил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 29 (60 мг, 0.20 ммоль) в фосфорилхлориде (POCl3) (0.38 мл, 4.13 ммоль) в инертной атмосфере при комнатной температуре. Реакционную смесь нагрели до 100°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с помощью насыщенного раствора карбоната натрия (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила Пример 34 (10 мг, 0.03 ммоль, 16%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.82 (s, 1H), 8.47 (s, 1H), 8.26 (s, 1H), 7.91 (d, J = 8.4 Гц, 1H), 7.68 (dd, J = 8.4, 1.5 Гц, 1H), 3.95-3.90 (m, 1H), 1.36-1.29 (m, 2H), 0.94-0.84 (m, 2H)
LC-MS: m/z 296.2 [M+H]+ при 3.28 RT (96.40% чистота).
ВЭЖХ: 96.31%.
Пример 35
Схема:
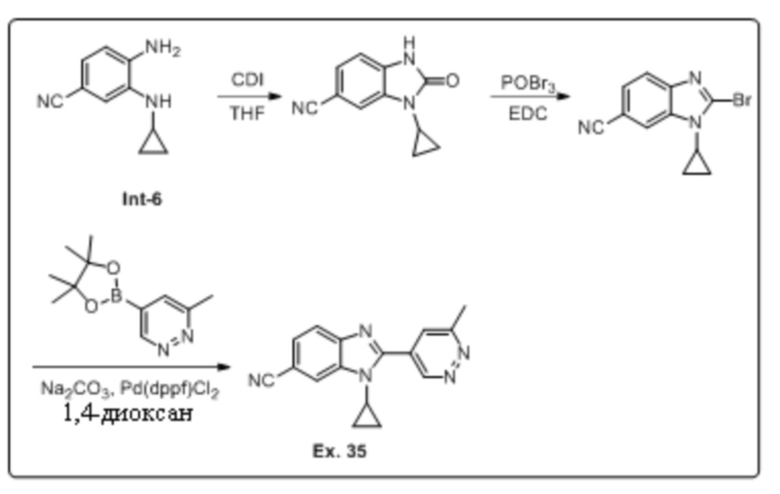
3-Циклопропил-2-оксо-2, 3-дигидро-1H-бензо[d]имидазол-5-карбонитрил
К перемешиваемому раствору 4-амино-3-(циклопропиламино) бензонитрила Int-6 (500 мг, 2.89 ммоль) в THF (10 мл) в инертной атмосфере добавили 1,1’-карбонилдиимидазол (CDI) (702 мг, 4.33 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/Гексан) с получением 3-циклопропил-2-оксо-2, 3-дигидро-1H-бензо[d]имидазол-5-карбонитрил (200 мг, 1.00 ммоль, 35%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 11.29 (brs, 1H), 7.55 (s, 1H), 7.45 (dd, J = 8.0, 1.3 Гц, 1H), 7.09 (d, J = 8.1 Гц, 1H), 2.91-2.85 (m, 1H), 1.05-1.00 (m, 2H), 0.92-0.83 (m, 2H)
2-Бром-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 2-бром-3-циклопропил-2,3-дигидро-1H-бензо[d]имидазол-5-карбонитрила (200 мг, 1.0 ммоль) в 1,2-дихлорэтане (DCE) (4 мл) в инертной атмосфере добавили фосфорилбромид (1.2 г, 4.02 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл), подщелочили с помощью насыщенного раствора карбоната натрия (30 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2-бром-3-циклопропил-2,3-дигидро-1H-бензо[d]имидазол-5-карбонитрила (50 мг, 0.20 ммоль, 19%) в виде твердого вещества белого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 261.8 [M+]+ при 2.52 RT (96.21% чистота).
1-Циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 35)
К перемешиваемому раствору 2-бром-3-циклопропил-2,3-дигидро-1H-бензо[d]имидазол-5-карбонитрила (100 мг, 0.38 ммоль) в 1,4-диоксане (3 мл) в инертной атмосфере добавили 3-метил-5-(4, 4, 5, 5-тетраметил-1, 3, 2-диоксаборолан-2-ил) пиридазин (126.9 мг, 0.57 ммоль) и 2M водный раствор карбоната натрия (0.3 мл, 0.64 ммоль) при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 10 мин, Pd(dppf)Cl2 (23 мг, 0.03 ммоль) добавили, и смесь дегазировали под аргоном в течение еще 10 мин. Реакционную смесь нагрели до 90°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/ Гексан) с получением 1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 35 (20 мг, 0.07 ммоль, 12%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.68 (d, J = 1.8 Гц, 1H), 8.26 (s, 2H), 7.91 (d, J = 8.5 Гц, 1H), 7.68 (dd, J = 8.4, 1.4 Гц, 1H), 3.94-3.90 (m, 1H), 2.86 (s, 3H), 1.36-1.25 (m, 2H), 0.90-0.81 (m, 2H)
LC-MS: m/z 275.9 [M+H]+ при 2.02 RT (99.65% чистота).
ВЭЖХ: 99.26%.
Пример 36
Схема:
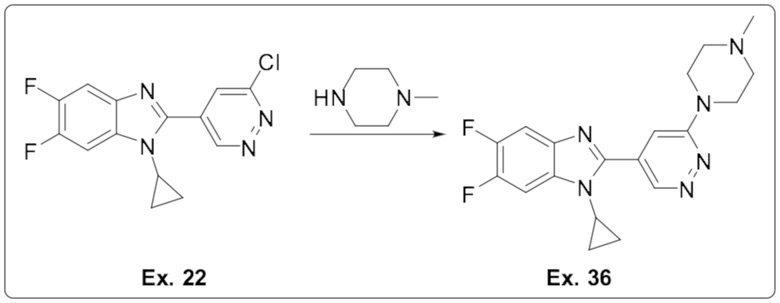
1-Циклопропил-5,6-дифтор-2-(6-(4-метилпиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 36)
Раствор 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (50 мг, 0.16 ммоль) в 1-метилпиперазине (0.5 мл) в инертной атмосфере перемешивали при 130°C в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5%MeOH/CH2Cl2) с получением 1-циклопропил-5,6-дифтор-2-(6-(4-метилпиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 36 (25 мг, 0.06 ммоль, 41%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.13 (s, 1H), 7.57 (dd, J = 10.2, 7.3 Гц, 1H), 7.46 (s, 1H), 7.41 (dd, J = 9.6, 7.0 Гц, 1H), 3.86-3.74 (m, 4H), 3.63-3.57 (m, 1H), 2.64-2.52 (m, 4H), 2.37 (s, 3H), 1.30-1.25 (m, 2H), 0.85-0.81 (m, 2H)
LC-MS: m/z 371 [M+H]+ при 1.71 RT (98.46% чистота).
ВЭЖХ: 97.35%.
Пример 37
Схема:
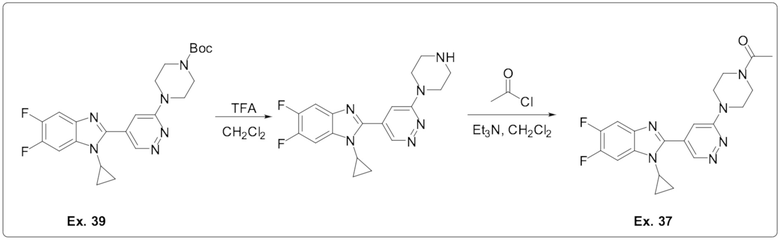
1-Циклопропил-5,6-дифтор-2-(6-(пиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору трет-бутил 4-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) пиперазин-1-карбоксилата Пример 39 (200 мг, 0.43 ммоль) в CH2Cl2 (2 мл) в инертной атмосфере добавили трифторуксуснаяую кислоту (0.6 мл) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1ч. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток нейтрализовали насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли простым эфиром: н-пентаном (1: 1, 2 x 2 мл) с получением 1-циклопропил-5,6-дифтор-2-(6-(пиперазин-1-ил) пиридазин-4-ил)-1H-бензо[d]имидазола (90 мг, неочищенный) в виде твердого вещества коричневого цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 357 [M+H]+ при 1.78 RT (98.01% чистота).
1-(4-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил) этан-1-он (Пример 37)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(6-(пиперазин-1-ил) пиридазин-4-ил)-1H-бензо[d]имидазола (50 мг, 0.14 ммоль) в CH2Cl2 (1 мл) в инертной атмосфере добавили триэтиламин (0.05 мл, 0.42 ммоль) и ацетилхлорид (0.01 мл, 0.14 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% MeOH/CH2Cl2) с получением 1-(4-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-ил)этан-1-она Пример 37 (14 мг, 0.03 ммоль, 25%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.20 (s, 1H), 7.57 (dd, J = 10.1, 7.2 Гц, 1H), 7.49 (s, 1H), 7.42 (dd, J = 9.6, 6.9 Гц, 1H), 3.94-3.89 (m, 2H), 3.84-3.78 (m, 2H), 3.75-3.70 (m, 2H), 3.69-3.64 (m, 2H), 3.64-3.59 (m, 1H), 2.18 (s, 3H), 1.32-1.26 (m, 2H), 0.86-0.81 (m, 2H)
LC-MS: m/z 399.1 [M+H]+ при 1.57 RT (93.31% чистота).
ВЭЖХ: 92.89%.
Пример 38
Схема:
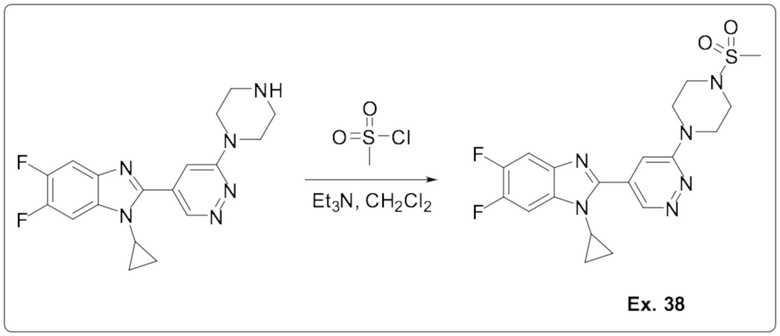
1-Циклопропил-5,6-дифтор-2-(6-(4-(метилсульфонил)пиперазин-1-ил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 38)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(6-(пиперазин-1-ил) пиридазин-4-ил)-1H-бензо[d]имидазола (50 мг, 0.14) в CH2Cl2 (1 мл) в инертной атмосфере добавили триэтиламин (0.05 мл, 0.42 ммоль) и мезилхлорид (0.01 мл, 0.14 ммоль) при 0°C. Реакционную смесь перемешивали в течение 10 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% MeOH/CH2Cl2) с получением 1-циклопропил-5,6-дифтор-2-(6-(4-(метилсульфонил) пиперазин-1-ил) пиридазин-4-ил)-1H-бензо[d]имидазола Пример 38 (14 мг, 0.03 ммоль, 25%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.22 (s, 1H), 7.57 (dd, J = 10.1, 7.2 Гц, 1H), 7.51 (s, 1H), 7.42 (dd, J = 9.6, 6.9 Гц, 1H), 3.96-3.91 (m, 4H), 3.65-3.60 (m, 1H), 3.43-3.38 (m, 4H), 2.84 (s, 3H), 1.34-1.24 (m, 2H), 0.91-0.80 (m, 2H)
LC-MS: m/z 435.1 [M+H]+ при 2.09 RT (98.86% чистота).
ВЭЖХ: 97.10%.
Пример 39
Схема:
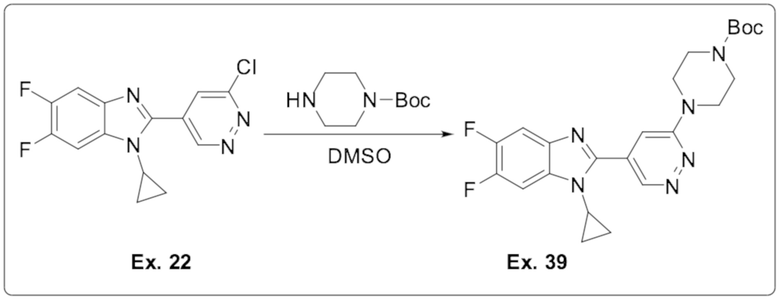
трет-бутил 4-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)пиперазин-1-карбоксилат (Пример 39)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (200 мг, 0.65 ммоль) в диметилсульфоксиде (DMSO) (4 мл) в инертной атмосфере добавили трет-бутилпиперазин-1-карбоксилат (729 мг, 3.97 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 130°C в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли водой (20 мл), солевым раствором (20 мл) высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5%MeOH/CH2Cl2) с получением Пример 39 (230 мг, 0.50 ммоль, 77%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.17 (s, 1H), 7.57 (dd, J = 10.0, 7.2 Гц, 1H), 7.47 (s, 1H), 7.41 (dd, J = 9.7, 6.9 Гц, 1H), 3.78-3.75 (m, 4H), 3.64-3.58 (m, 5H), 1.50 (s, 9H), 1.31-1.26 (m, 2H), 0.86-0.81 (m, 2H)
LC-MS: m/z 457.1 [M+H]+ при 2.52 RT (97.87% чистота).
ВЭЖХ: 95.78%.
Примеры 40 и 41
Схема:
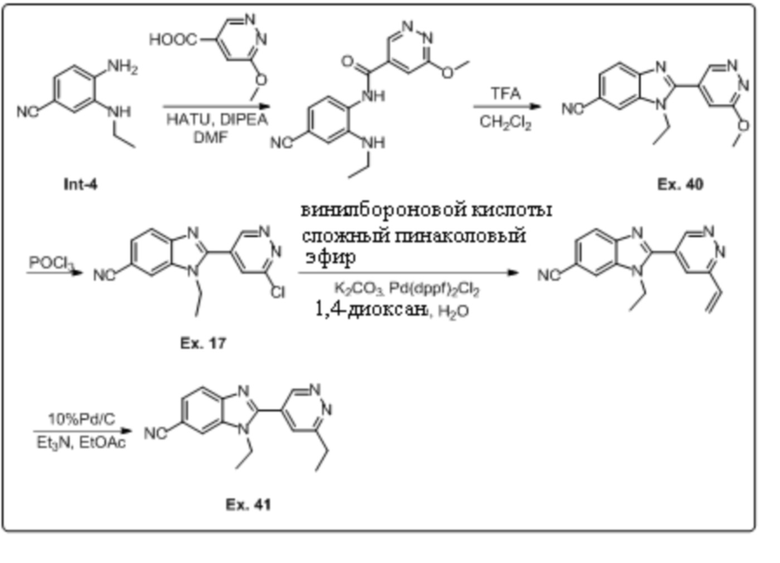
6-Хлор-N-(4-циано-2-(этиламино) фенил) пиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (100 мг, 0.64 ммоль) в DMF (2 мл) в инертной атмосфере добавили 4-амино-3-(этиламино) бензонитрил Int-4 (104 мг, 0.64 ммоль), HATU (369.9 мг, 0.97 ммоль) и этилдиизопропиламин (0.45 мл, 2.59 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью водного раствора хлорида аммония (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6-хлор-N-(4-циано-2-(этиламино) фенил) пиридазин-4-карбоксамида (90 мг, неочищенный) в виде твердого вещества желтого цвета.
LC-MS: m/z 300.9 [M]+ при 2.07 RT (36.25% чистота).
1-Этил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 40)
К перемешиваемому раствору 6-хлор-N-(4-циано-2-(этиламино) фенил) пиридазин-4-карбоксамида (100 мг, 0.33 ммоль) в CH2Cl2 (1.6 мл) в инертной атмосфере добавили трифторуксусную кислоту (0.4 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 6 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли водой (20 мл), солевым раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество перетерли с простым эфиром (2 x 10 мл) и н-пентаном (2 x 10 мл) с получением 1-этил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 40 (25 мг, 0.09 ммоль, 26%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.27 (d, J = 1.9 Гц, 1H), 8.24 (s, 1H), 7.91 (dd, J = 8.4, 0.6 Гц, 1H), 7.68 (dd, J = 8.5, 1.4 Гц, 1H), 7.59 (s, 1H), 4.53-4.49 (m, 2H), 4.22 (s, 3H), 1.48 (t, J = 7.3 Гц, 3H)
LC-MS: m/z 279.8 [M+H]+ при 2.15 RT (98.27% чистота).
ВЭЖХ: 98.65%.
2-(6-Хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрил (Пример 17)
К перемешиваемому раствору 1-этил-2-(6-метоксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 40 (85 мг, 0.30 ммоль) в фосфорилхлориде (0.57 мл, 6.09 ммоль) в инертной атмосфере при комнатной температуре. Реакционную смесь нагрели до 100°C в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с помощью водного раствор карбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (10 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество перетерли с простым эфиром (2 x 5 мл) и н-пентаном (2 x5 мл) с получением 2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрил Пример 17 (100 мг, неочищенный) в виде сиропа бледно-желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
1-Этил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила Пример 17 (130 мг, 0.46 ммоль) в 1, 4-диоксане (3 мл) и воде (0.5 мл) в инертной атмосфере добавили винилбороновой кислоты пинакол (70 мг, 0.46 ммоль) и карбонат калия (190 мг, 1.37 ммоль) при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 10 мин. К этому добавили Pd(dppf)Cl2 (3.7 мг, 0.005 ммоль) при комнатной температуре и смесь дегазировали под аргоном в течение дополнительных 10 мин. Реакционную смесь нагрели до 80°C и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали. Фильтрат концентрировали при пониженном давлении с получением 1-этил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (110 мг, неочищенный) в виде сиропа коричневого цвета.
LC-MS: m/z 275.9 [M+H]+ при 2.18 RT (78.16% чистота).
1-Этил-2-(6-этилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 41)
К перемешиваемому раствору 1-этил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (110 мг, 0.40 ммоль) в EtOAc (5 мл) в инертной атмосфере добавили 10% Pd/C (50% влажность, 20 мг) и триэтиламин (0.005 мл, 0.04 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью EtOAc (20 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 1-этил-2-(6-этилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 41 (4 мг, 14.44 ммоль, 4%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.51 (d, J = 2.0 Гц, 1H), 8.26 (s, 1H), 8.06 (d, J = 2.1 Гц, 1H), 7.92 (d, J = 8.4 Гц, 1H), 7.69 (dd, J = 8.5, 1.4 Гц, 1H), 4.55-4.50 (m, 2H), 3.21-3.15 (m, 2H), 1.53-1.45 (m, 6H)
LC-MS: m/z 277.9 [M+H]+ при 2.12 RT (96.23% чистота).
ВЭЖХ: 96.66%.
Пример 42
Схема:
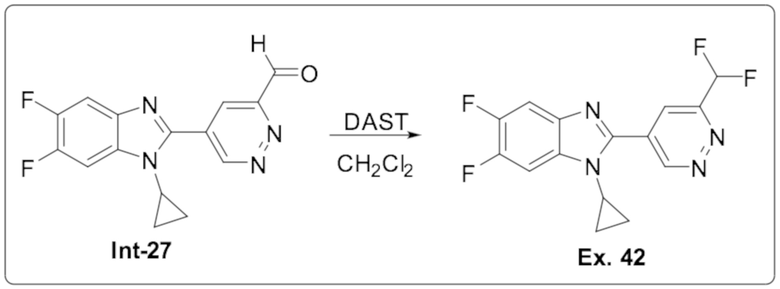
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 42)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-27 (100 мг, 0.33 ммоль) в CH2Cl2 (5 мл) в инертной атмосфере добавили (диэтиламино)серы трифторид (DAST) (0.09 мл, 0.66 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/Гексан) с получением 1-циклопропил-2-(6-(дифторметил) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 42 (40 мг, 0.12 ммоль, 37%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.99 (s, 1H), 8.55 (s, 1H), 7.93-7.85 (m, 2H), 7.42 (t, J = 54.5 Гц, 1H), 4.01-3.97 (m, 1H), 1.25-1.13 (m, 2H), 0.84-0.72 (m, 2H)
LC-MS: m/z 323.3 [M+H]+ при 3.53 RT (98.91% чистота).
ВЭЖХ: 99.33%.
Пример 43
Схема:
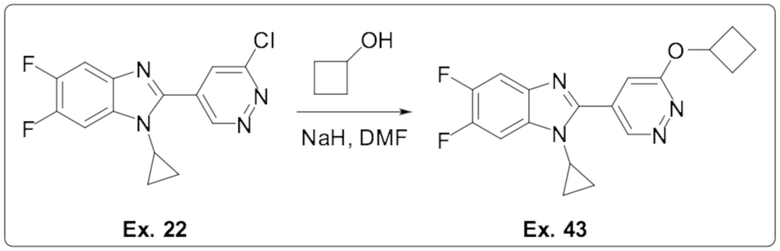
2-(6-Циклобутоксипиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол (Пример 43)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (50 мг, 0.16 ммоль) в DMF (2 мл) в инертной атмосфере добавили гидрид натрия (60% в минеральном масле, 16.3 мг, 0.41 ммоль) по частям при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 мин. Затем циклобутанол (141 мг, 0.20 ммоль) добавили в реакционную смесь при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3% MeOH/CH2Cl2) с получением 2-(6-циклобутоксипиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 43 (45 мг, 0.13 ммоль, 57%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.42 (s, 1H), 7.59 (dd, J = 10.1, 7.2 Гц, 1H), 7.47 (s, 1H), 7.44-7.40 (m, 1H), 5.53-5.48 (m, 1H), 3.62-3.57 (m, 1H), 2.67-2.50 (m, 2H), 2.31-2.08 (m, 2H), 1.99-1.83 (m, 1H), 1.78-1.73 (m, 1H), 1.37-1.24 (m, 2H), 0.89-0.77 (m, 2H)
LC-MS: m/z 343 [M+H]+ при 2.61 RT (95.30% чистота).
ВЭЖХ: 95.28%.
Пример 44
Схема:
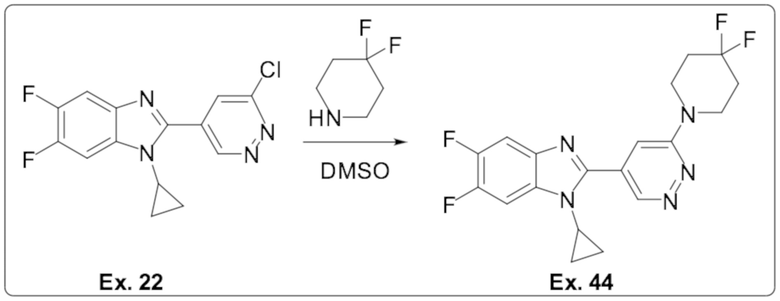
1-Циклопропил-2-(6-(4,4-дифторпиперидин-1-ил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 44)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.32 ммоль) в DMSO (2 мл) в инертной атмосфере добавили 4, 4-дифторпиперидин (59 мг, 0.49 ммоль) при комнатной температуре. Реакционную смесь нагрели до 120-130°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50-60% EtOAc/гексан) с получением 1-циклопропил-2-(6-(4, 4-дифторпиперидин-1-ил) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 44 (40 мг, 0.10 ммоль, 32%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.09 (s, 1H), 7.82 (s, 1H), 7.72-7.68 (m, 1H), 7.62-7.57 (m, 1H), 3.99-3.92 (m, 4H), 3.86-3.83 (m, 1H), 2.19-2.00 (m, 4H), 1.27-1.19 (m, 2H), 0.87-0.76 (m, 2H)
LC-MS: m/z 392.1 [M+H]+ при 2.98 RT (93.89% чистота).
ВЭЖХ: 92.05%.
Пример 45
Схема:
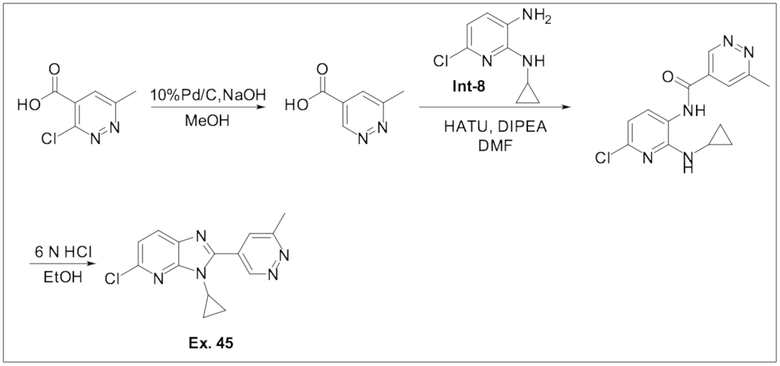
6-Метилпиридазин-4-карбоновая кислота
К перемешиваемому раствору 3-хлор-6-метилпиридазин-4-карбоновой кислоты 1 (500 мг, 2.90 ммоль) в MeOH (50 мл) в инертной атмосфере добавили гидроксид натрия (395 мг, 9.80 ммоль) и 10% Pd/C (50% влажность, 150 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита. Фильтрат концентрировали при пониженном давлении и 6Н HCl добавили до pH~6 и концентрировали при пониженном давлении с получением 6-метилпиридазин-4-карбоновой кислоты (410 мг, неочищенный) в виде жидкости желтого цвета, применяемого на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 9.24 (s, 1H), 7.69 (s, 1H), 2.62 (s, 3H)
N-(6-Хлор-2-(циклопропиламино) пиридин-3-ил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлор-N2-циклопропилпиридин-2, 3-диамина Int-8 (400 мг, 2.18 ммоль) в DMF (5 мл) в инертной атмосфере добавили соединение 6-метилпиридазин-4-карбоновую кислоту (362 мг, 2.62 ммоль), HATU (996 мг, 2.62 ммоль) и этилдиизопропиламин (1.6 мл, 8.75 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(6-хлор-2-(циклопропиламино) пиридин-3-ил)-6-метилпиридазин-4-карбоксамида (200 мг, 0.66 ммоль, 30%) в виде твердого вещества черного цвета, применяемого на следующей стадии без дальнейшей очистки.
LC-MS: m/z 303.9 [M+H]+ при комнатной температуре 2.06 (93.73% чистота).
5-Хлор-3-циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридин (Пример 45)
К перемешиваемому раствору N-(6-хлор-2-(циклопропиламино) пиридин-3-ил)-6-метилпиридазин-4-карбоксамида (100 мг, 0.33 ммоль) в EtOH (2 мл) в инертной атмосфере добавили 6Н HCl (3 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 5-хлор-3-циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридина Пример 45 (12 мг, 0.04 ммоль, 10%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.67 (s, 1H), 8.26 (s, 1H), 8.12 (d, J = 8.4 Гц, 1H), 7.42 (d, J = 8.4 Гц, 1H), 3.83-3.78 (m, 1H), 2.84 (s, 3H), 1.29-1.25 (m, 2H), 0.94-0.92 (m, 2H)
LC-MS: m/z 285.9 [M+H]+ при 2.03 RT (98.73% чистота).
ВЭЖХ: 95.65%.
Пример 46
Схема:
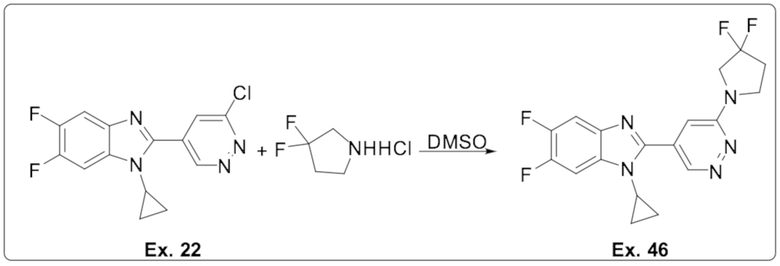
1-Циклопропил-2-(6-(3,3-дифторпирролидин-1-ил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 46)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.32 ммоль) в DMSO (2 мл) в инертной атмосфере добавили 3, 3-дифторпирролидина гидрохлорид (70 мг, 0.50 ммоль) и триэтиламин (0.06 мл, 0.49 ммоль) при комнатной температуре. Реакционную смесь нагрели до 130°C и перемешивали в течение 48 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(3,3-дифторпирролидин-1-ил) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 46 (15 мг, 0.04 ммоль, 12%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.10 (s, 1H), 7.72-7.68 (m, 1H), 7.62-7.57 (m, 1H), 7.49 (d, J = 1.8 Гц, 1H), 4.03 (t, J = 12.9 Гц, 2H), 3.90-3.77 (m, 3H), 2.69-2.54 (m, 2H), 1.30-1.22 (m, 2H), 0.86-0.80 (m, 2H)
LC-MS: m/z 378.1 [M+H]+ при 2.80 RT (97.60% чистота).
ВЭЖХ: 99.55%.
Пример 47
Схема:
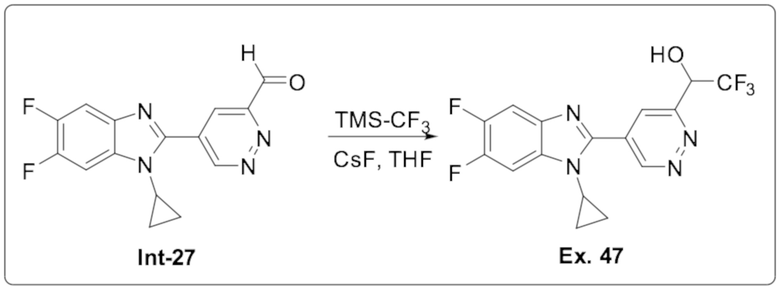
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифтопентан-1-ол (Пример 47)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-27 (50 мг, 0.16 ммоль) в THF (1 мл) в инертной атмосфере добавили триметил (трифторметил) силан (47 мг, 0.33 ммоль) при 0°C. Фторид цезия (76 мг, 0.50 ммоль) добавили в реакционную смесь при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с помощью 1Н раствора HCl (5 мл) при 0°C и экстрагировали с помощью EtOAc (2 x 15 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/Гексан) с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2, 2, 2-трифтопентан-1-ола Пример 47 (10 мг, 0.03 ммоль, 16%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.83 (s, 1H), 8.58 (s, 1H), 7.75-7.71 (m, 1H), 7.67-7.63 (m, 1H), 5.60-5.52 (m, 1H), 3.94-3.81 (m, 1H), 1.35-1.21 (m, 2H), 0.93-0.78 (m, 2H)
LC-MS: m/z 371 [M+H]+ при 2.69 RT (95.24% чистота).
ВЭЖХ: 95.69%.
Пример 48
Схема:
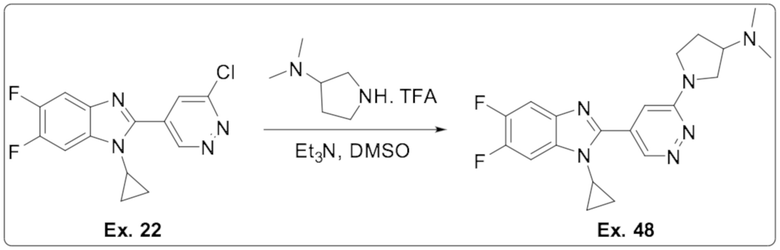
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-N,N-диметилпирролидин-3-амин (Пример 48)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (115 мг, 0.37 ммоль) в DMSO (2 мл) в инертной атмосфере добавили триэтиламин (0.116 мл, 0.84 ммоль) и 1-(3-(диметиламино)-1l5-пирролидин-1-ил)-2, 2, 2-трифторэтан-1-он (120 мг, 0.56 ммоль) при 0°C. Реакционную смесь перемешивали при 90°C в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (10 мл) и экстрагировали с помощью EtOAc (2 x 10 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5-10% MeOH/CH2Cl2) с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил)-N, N-диметилпирролидин-3-амина Пример 48 (10 мг, 0.02 ммоль, 7%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.01 (s, 1H), 7.72-7.67 (m, 1H), 7.61-7.57 (m, 1H), 7.44 (s, 1H), 3.96-3.93 (m, 1H), 3.86-3.81 (m, 2H), 3.62-3.55 (m, 1H), 3.45-3.37 (m, 1H), 3.09-2.99 (m, 1H), 2.38 (s, 6H), 2.08-1.95 (m, 1H), 1.32-1.20 (m, 3H), 0.87-0.78 (m, 2H)
LC-MS: m/z 385.1 [M+H]+ при 2.06 RT (97.17% чистота).
ВЭЖХ: 98.58%.
Пример 49
Схема:
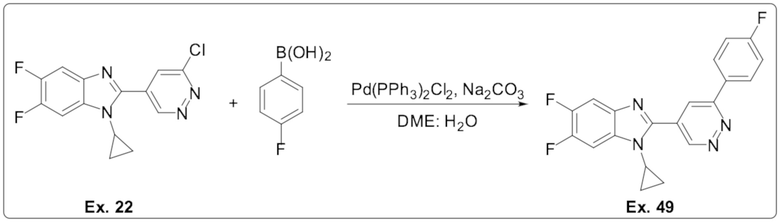
1-Циклопропил-5,6-дифтор-2-(6-(4-фторфенил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 49)
Pd(PPh3)2Cl2 (11.5 мг, 0.01 ммоль) и карбонат натрия (86.4 мг, 0.81 ммоль) в 1,4-диметоксиэтане (DME): воде (4: 1, 1.25 мл) при комнатной температуре продували аргоном в течение 5 мин. 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол Пример 22 (50 мг, 0.16 ммоль) и (4-фторфенил) бороновую кислоту (25.1 мг, 0.17 ммоль) добавили в реакционную смесь при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 12 часов в запаянной трубке. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-5,6-дифтор-2-(6-(4-фторфенил) пиридазин-4-ил)-1H-бензо[d]имидазола Пример 49 (40 мг, 0.10 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.75 (s, 1H), 8.68 (s, 1H), 8.32-8.18 (m, 2H), 7.76-7.72 (m, 1H), 7.67-7.62 (m, 1H), 7.35 (t, J = 8.8 Гц, 2H), 3.98-3.93 (m, 1H), 1.32-1.25 (m, 2H), 0.90-0.84 (m, 2H)
LC-MS: m/z 367 [M+H]+ при 2.58 RT (98.75% чистота).
ВЭЖХ: 98.72%.
Пример 50
Схема:
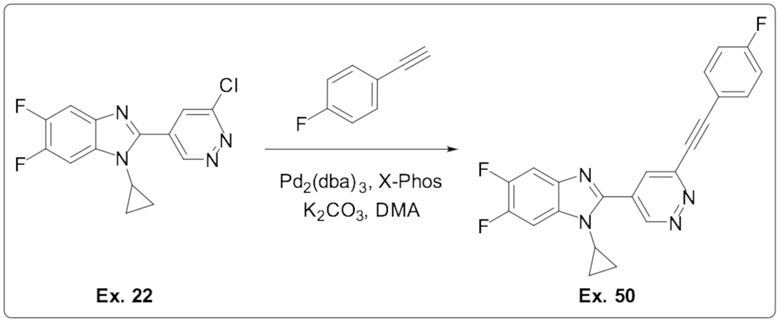
1-Циклопропил-5,6-дифтор-2-(6-((4-фторфенил)этинил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 50)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.32 ммоль) в N,N-диметилацетамиде (DMA) (3 мл) в инертной атмосфере добавили 1-этинил-4-фторбензол (40 мг, 0.32 ммоль) и карбонат калия (90 мг, 0.65 ммоль) при комнатной температуре. Реакционную смесь дегазировали аргоном в течение 10 мин. Pd2(dba)3 (14.8 мг, 0.01 ммоль) и X-phos (8 мг, 0.01 ммоль) добавили при комнатной температуре и смесь дегазировали аргоном в течение еще 5 мин. Реакционную смесь нагрели до 80°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-((4-фторфенил)этинил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 50 (50 мг, 0.13 ммоль, 39%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.81 (s, 1H), 8.49 (s, 1H), 7.91-7.84 (m, 2H), 7.84-7.76 (m, 2H), 7.38 (t, J = 9.0 Гц, 2H), 4.01-3.93 (m, 1H), 1.24-1.16 (m, 2H), 0.85-0.71 (m, 2H)
LC-MS: m/z 391.3 [M+H]+ при 4.58 RT (97.94% чистота).
ВЭЖХ: 96.14%.
Пример 51
Схема:
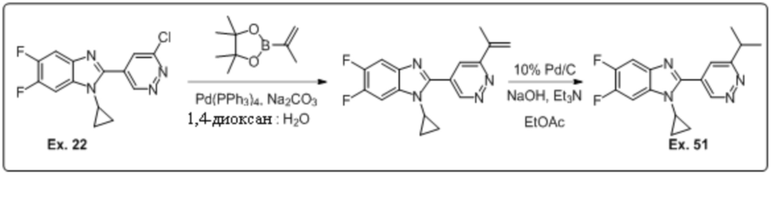
1-Циклопропил-5,6-дифтор-2-(6-(проп-1-ен-2-ил)пиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (120 мг, 0.40 ммоль) в 1,4-диоксане (3.2 мл) и воде (0.8 мл) добавили 4, 4, 5, 5-тетраметил-2-(проп-1-ен-2-ил)-1, 3, 2-диоксаборолан (219 мг, 1.17 ммоль) и карбонат натрия (164.6 мг, 1.56 ммоль) и смесь продули аргоном в течение 10 мин при комнатной температуре. Pd(PPh3)4 (23 мг, 0.02 ммоль) добавили в реакционную смесь. Реакционную смесь нагрели при 110°C в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный материал перетерли с н-пентаном (2 x 5 мл) с получением 1-циклопропил-5,6-дифтор-2-(6-(проп-1-ен-2-ил) пиридазин-4-ил)-1H-бензо[d]имидазола (100 мг, неочищенный) в виде твердого вещества грязновато-белого цвета, которое применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 313.1 [M+H]+ при 2.38 RT (94.08% чистота).
1-Циклопропил-5,6-дифтор-2-(6-изопропилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 51)
К перемешиваемому раствору соединения 1-циклопропил-5,6-дифтор-2-(6-(проп-1-ен-2-ил) пиридазин-4-ил)-1H-бензо[d]имидазола (100 мг, 0.32 ммоль) в этилацетате (5 мл) в инертной атмосфере добавили триэтиламин (0.04 мл), гидроксид натрия (25 мг, 0.64 ммоль) и 10% Pd/C (50% влажность, 30 мг) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (20 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью н-пентана (2 x 2 мл) и простого эфира (2 x 2 мл) с получением 1-циклопропил-5,6-дифтор-2-(6-изопропилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 51 (55 мг, 0.17 ммоль, 55%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.63 (s, 1H), 8.22 (s, 1H), 7.73-7.69 (m, 1H), 7.64-7.60 (m, 1H), 3.91-3.86 (m, 1H), 3.49-3.31 (m, 1H), 1.47 (d, J = 7.0 Гц, 6H), 1.31-1.20 (m, 2H), 0.86-0.73 (m, 2H)
LC-MS: m/z 315.1 [M+H]+ при 2.31 RT (97.95% чистота).
ВЭЖХ: 99.16%.
Пример 52
Схема:
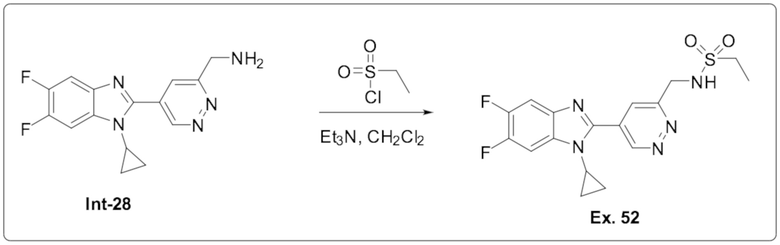
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)этансульфонамид (Пример 52)
К перемешиваемому раствору (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил) метанамина (140 мг, 0.46 ммоль) в CH2Cl2 (4 мл) в инертной атмосфере добавили триэтиламин (0.19 мл, 0.79 ммоль) и этансульфонил хлорид (65.7 мг, 0.51 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил) этансульфонамида Пример 52 (18 мг, 0.04 ммоль, 10%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.72 (s, 1H), 8.52 (s, 1H), 7.76-7.70 (m, 1H), 7.66-7.61 (m, 1H), 4.71 (s, 2H), 3.88-3.82 (m, 1H), 3.24-3.19 (m, 2H), 1.43-1.33 (m, 5H), 0.86-0.79 (m, 2H)
LC-MS: m/z 394 [M+H]+ при 2.32 RT (99.29% чистота).
ВЭЖХ: 98.78%.
Пример 53
Схема:
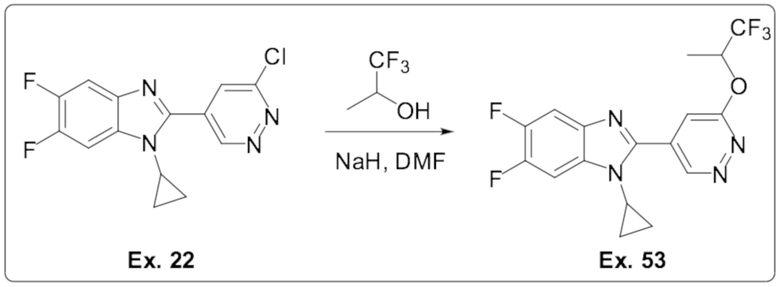
1-Циклопропил-5,6-дифтор-2-(6-((1,1,1-трифторпропан-2-ил)окси)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 53)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (75 мг, 0.24 ммоль) в DMF (2.25 мл) в инертной атмосфере добавили гидрид натрия (60% в минеральном масле, 24.5 мг, 0.61 ммоль) по частям при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 мин. Затем 1, 1, 1-трифторпропан-2-ол (34 мг, 0.30 ммоль) добавили в реакционную смесь при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3% MeOH/CH2Cl2) с получением 1-циклопропил-5,6-дифтор-2-(6-((1, 1, 1-трифторпропан-2-ил) окси) пиридазин-4-ил)-1H-бензо[d]имидазола Пример 53 (60 мг, 0.15 ммоль, 84%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.54 (s, 1H), 7.63 (s, 1H), 7.62-7.58 (m, 1H), 7.45-7.41 (m, 1H), 6.15-6.04 (m, 1H), 3.69-3.42 (m, 1H), 1.64 (d, J = 6.3 Гц, 3H), 1.36-1.24 (m, 2H), 0.92-0.81 (m, 2H)
LC-MS: m/z 385.1 [M+H]+ при 2.76 RT (97.88% чистота).
ВЭЖХ: 97.83%.
Пример 54
Схема:
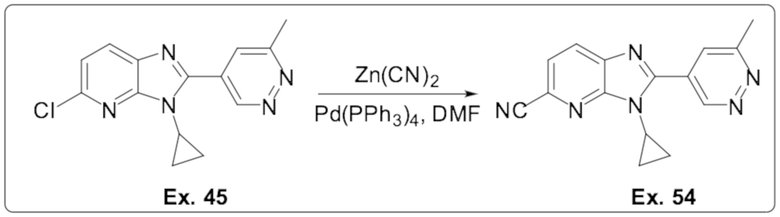
3-Циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрил (Пример 54)
К перемешиваемому раствору 5-хлор-3-циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо [4, 5-b] пиридина Пример 45 (75 мг, 0.26 ммоль) в DMF (0.8 мл) при комнатной температуре добавили цианид цинка (61.5 мг, 0.52 ммоль) и Pd(PPh3)4 (30.3 мг, 0.02 ммоль). Смесь продули аргоном в течение 10 мин и затем нагрели до 170°C в течение 4.5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 3-циклопропил-2-(6-метилпиридазин-4-ил)-3H-имидазо [4, 5-b] пиридин-5-карбонитрила Пример 54 (25 мг, 0.09 ммоль, 35%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.75 (s, 1H), 8.19 (d, J = 8.2 Гц, 1H), 8.01 (s, 1H), 7.72 (d, J = 8.2 Гц, 1H), 3.70-3.64 (m, 1H), 2.90 (s, 3H), 1.41-1.36 (m, 2H), 1.06-0.81 (m, 2H)
LC-MS: m/z 276.9 [M+H]+ при 1.88 RT (98.98% чистота).
ВЭЖХ: 95.03%.
Пример 55
Схема:

N-(2-(Циклопропиламино)-4,5-дифторфенил)пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4, 5-дифторбензол-1, 2-диамина Int-5 (300 мг, 1.63 ммоль) в DMF (3 мл) в инертной атмосфере добавили пиридазин-4-карбоновую кислоту (202 мг, 1.63 ммоль), HATU (743 мг, 1.95 ммоль) и этилдиизопропиламин (1.1 мл, 6.52 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением N-(2-(циклопропиламино)-4, 5-дифторфенил) пиридазин-4-карбоксамида (250 мг, неочищенный) в виде твердого вещества грязновато-белого цвета, которое применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 291[M+H]+ при 2.34 RT (96.97% чистота).
1-Циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 55)
К перемешиваемому раствору N-(2-(циклопропиламино)-4, 5-дифторфенил) пиридазин-4-карбоксамида (200 мг, 0.68 ммоль) в EtOH (4 мл) в инертной атмосфере добавили 6N HCl (2 мл) при комнатной температуре. Реакционную смесь перемешивали при 70°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью н-пентана (2 x 5 мл) с получением 1-циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазола Пример 55 (120 мг, 0.44 ммоль, 64%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.81 (s, 1H), 9.41 (dd, J = 5.4, 1.3 Гц, 1H), 8.33 (dd, J = 5.5, 2.3 Гц, 1H), 7.4-7.69 (m, 1H), 7.65-7.61 (m, 1H), 3.87-3.84 (m, 1H), 1.30-1.24 (m, 2H), 0.88-0.73 (m, 2H)
LC-MS: m/z 272.9 [M+H]+ при 2.21 RT (99.23% чистота).
ВЭЖХ: 99.54%.
Пример 56
Схема:
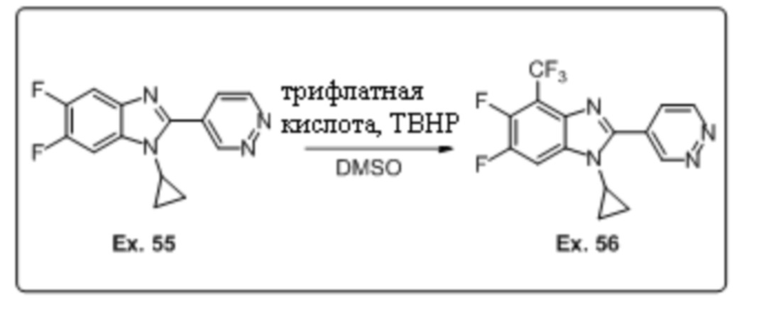
1-Циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-4-(трифторметил)-1H-бензо[d]имидазол (Пример 56)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазола Пример 55 (100 мг, 0.36 ммоль) в DMSO (1.5 мл) в инертной атмосфере добавили трифторметансульфинат цинка (243 мг, 0.73 ммоль) при комнатной температуре. К этому добавили TBHP (141 мг, 1.10 ммоль) при 0°C. Реакционную смесь перемешивали при 50°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-4-(трифторметил)-1H-бензо[d]имидазола Пример 56 (19 мг, 0.05 ммоль, 15%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, CD3OD): δ 9.85 (s, 1H), 9.44 (dd, J = 5.2, 1.2 Гц, 1H), 8.38 (dd, J = 5.2, 2.3 Гц, 1H), 8.06 (dd, J = 9.3, 7.0 Гц, 1H), 3.92-3.86 (m, 1H), 1.38-1.20 (m, 2H), 0.89-0.83 (m, 2H)
LC-MS: m/z 340.9 [M+H]+ при 2.75 RT (91.67% чистота)
ВЭЖХ: 91.79%
Пример 57
Схема:

1-Циклопропил-2-(6-(2,2-дифторпропокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d] имидазол (Пример 57)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (50 мг, 0.16 ммоль) в DMF (1.5 мл) добавили гидрид натрия (60% в минеральном масле, 16.2 мг, 0.40 ммоль) по частям при 0°C в инертной атмосфере и смесь перемешивали в течение 5 мин. 2,2-Дифторпропан-1-ол (18.8 мг, 0.19 ммоль) добавили в реакционную смесь и перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью CH2Cl2: н-пентана (5: 95, 10 мл) с получением 1-циклопропил-2-(6-(2,2-дифторпропокси) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 57 (45 мг, 0.12 ммоль, 82%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.53 (s, 1H), 7.65 (d, 1H), 7.60 (dd, J = 10.0, 7.3 Гц, 1H), 7.43 (dd, J = 9.7, 6.9 Гц, 1H), 4.80 (t, J = 12.1 Гц, 2H), 3.67-3.58 (m, 1H), 1.80 (t, J = 18.6 Гц, 3H), 1.39-1.27 (m, 2H), 0.92-0.76 (m, 2H)
LC-MS: m/z 367 [M+H]+ при 3.08 RT (96.40% чистота)
ВЭЖХ: 94.40%
Пример 58
Схема:

1-Циклопропил-5,6-дифтор-2-(5-изопропилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 58)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(пиридазин-4-ил)-1H-бензо[d]имидазола Пример 55 (100 мг, 0.37 ммоль) в DMSO (1 мл) добавили цинка изопропилсульфинат (205 мг, 0.73 ммоль) при комнатной температуре в инертной атмосфере. Затем трет-бутилгидропероксид (70% в воде, 142 мг, 1.1 ммоль) добавили при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры, и затем нагрели до 50°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с помощью насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 70% EtOAc/гексан), а затем растерли с н-пентаном (2 x 4 мл) и высушили в вакууме с получением 1-циклопропил-5,6-дифтор-2-(5-изопропилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 58 (15 мг, 0.05 ммоль, 13%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.48 (d, J = 0.8 Гц, 1H), 9.31 (d, J = 1.0 Гц, 1H), 7.71 (dd, J = 10.1, 7.1 Гц, 1H), 7.61 (dd, J = 10.5, 7.3 Гц, 1H), 3.62-3.56 (m, 1H), 3.27-3.20 (m, 1H), 1.33 (d, J = 7.0 Гц, 6H), 1.08-1.03 (m, 2H), 0.73-0.68 (m, 2H)
LC-MS: m/z 315.0 [M+H]+ при 2.70 RT (95.37% чистота)
ВЭЖХ: 92.84%
Пример 59
Схема:
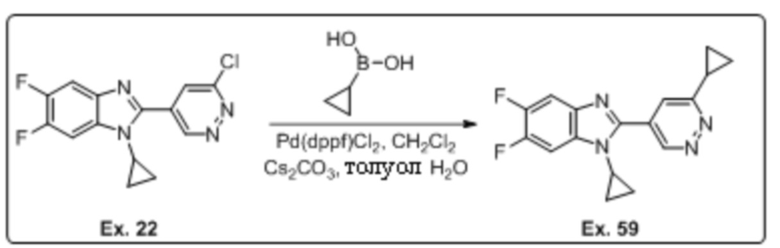
1-Циклопропил-2-(6-циклопропилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 59)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (70 мг, 0.22 ммоль) в толуоле/воде (3: 1, 2 мл) добавили циклопропилбороновую кислоту (24 мг, 0.27 ммоль) и карбонат цезия (186 мг, 0.57 ммоль) в запаянной трубке и смесь продули аргоном в течение 10 мин. Pd(dppf)Cl2 (18.6 мг, 0.02 ммоль) добавили в реакционную смесь с дальнейшим дегазированием в течение 5 мин. Реакционную смесь нагрели до 110°C в течение 16 часов и затем охладили. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-2-(6-циклопропилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 59 (8 мг, 0.02 ммоль, 11%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.57 (s, 1H), 7.81 (s, 1H), 7.59 (dd, J = 10.2, 7.3 Гц, 1H), 7.43 (dd, J = 9.7, 7.0 Гц, 1H), 3.67-3.60 (m, 1H), 2.32-2.24 (m, 1H), 1.35-1.27 (m, 4H), 1.25-1.19 (m, 2H), 0.86-0.80 (m, 2H)
LC-MS: m/z 313.1 [M+H]+ при 2.22 RT (93.72% чистота)
ВЭЖХ: 96.07%
Пример 60
Схема:
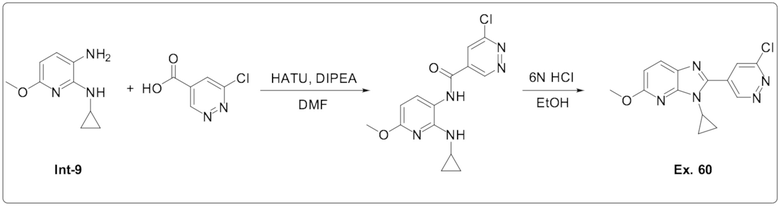
6-Хлор-N-(2-(циклопропиламино)-6-метоксипиридин-3-ил)пиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (500 мг, 3.16 ммоль) в DMF (10 мл) добавили N2-циклопропил-6-метоксипиридин-2,3-диамин Int-9 (566 мг, 3.16 ммоль), HATU (1.8 г, 4.74 ммоль) и диизопропилэтиламин (2.19 мл, 12.64 ммоль) при 0°C в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), солевым раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/гексан) с получением 6-хлор-N-(2-(циклопропиламино)-6-метоксипиридин-3-ил) пиридазин-4-карбоксамида (225 мг, 0.70 ммоль, 22%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.91 (s, 1H), 9.62 (s, 1H), 8.35 (s, 1H), 7.33 (d, J = 8.2 Гц, 1H), 6.50 (d, J = 2.4 Гц, 1H), 6.00 (d, J = 8.0 Гц, 1H), 3.82 (s, 3H), 2.73-2.70 (m, 1H), 0.73-0.64 (m, 2H), 0.52-0.40 (m, 2H)
2-(6-Хлорпиридазин-4-ил)-3-циклопропил-5-метокси-3H-имидазо[4,5-b]пиридин (Пример 60)
К перемешиваемому раствору 6-хлор-N-(2-(циклопропиламино)-6-метоксипиридин-3-ил) пиридазин-4-карбоксамида (150 мг, 0.47 ммоль) в EtOH (3.5 мл) добавили 6 Н HCl (2 мл) при 0°C в инертной атмосфере. Реакционную смесь перемешивали при 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 60-70% EtOAc/гексан) с получением 2-(6-хлорпиридазин-4-ил)-3-циклопропил-5-метокси-3H-имидазо[4,5-b]пиридина Пример 60 (35 мг, 0.11 ммоль, 22%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CD3OD): δ 8.28 (s, 1H), 7.42 (s, 1H), 7.33 (d, J = 8.3 Гц, 1H), 6.03 (d, J = 8.3 Гц, 1H), 3.90 (s, 3H), 2.79-2.74 (m, 1H), 0.77-0.68 (m, 2H), 0.53-0.47 (m, 2H)
LC-MS: m/z 302.1 [M+H]+ при 1.85 RT (95.67% чистота)
ВЭЖХ: 94.24%
Пример 61
Схема:

1-Циклопропил-5,6-дифтор-2-(6-(4-фтор-2-метилфенил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 61)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (70 мг, 0.22 ммоль) в DME/ EtOH (2: 1, 0.9 мл) добавили (4-фтор-2-метилфенил) бороновую кислоту (42 мг, 0.27 ммоль) и карбонат натрия (72.5 мг, 0.68 ммоль) в запаянной трубке и смесь продули аргоном в течение 10 мин. Транс-дихлорбис-(трифенилфосфин)-II (8 мг, 0.01 ммоль) добавили в реакционную смесь, и реакционную смесь перемешивали при 80°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали и фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(4-фтор-2-метилфенил) пиридазин-4-ил)-1H-бензо[d]имидазола Пример 61 (45 мг, 0.11 ммоль, 51%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.81 (s, 1H), 8.14 (s, 1H), 7.60 (dd, J = 10.1, 7.2 Гц, 1H), 7.49 (dd, J = 8.34, 5.83 Гц, 1H), 7.44 (dd, J = 9.6, 6.9 Гц, 1H), 7.11-7.03 (m, 2H), 3.71-3.63 (m, 1H), 2.46 (s, 3H), 1.37-1.30 (m, 2H), 0.93-0.87 (m, 2H)
LC-MS: m/z 381.1 [M+H]+ при 2.60 RT (97.81% чистота)
ВЭЖХ: 96.11%
Пример 62
Схема:
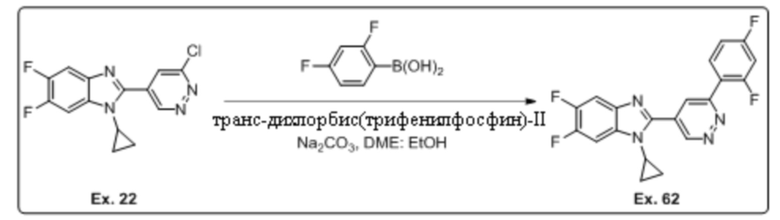
1-Циклопропил-2-(6-(2,4-дифторфенил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 62)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (70 мг, 0.22 ммоль) в DME: EtOH (2: 1, 0.9 мл) добавили (2,4-дифторфенил) бороновую кислоту (43 мг, 0.27 ммоль) и карбонат натрия (75.5 мг, 0.68 ммоль) и смесь продули аргоном в течение 10 мин. Транс-дихлор бис-(трифенил фосфин)-II (8 мг, 0.01 ммоль) добавили в реакционную смесь, и реакционную смесь перемешивали при 80°C в течение 16 часов в запаянной трубке. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали и фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 1-циклопропил-2-(6-(2,4-дифторфенил)пиридазин-4-ил)-5,6-дифтор-1H-бенз[d]имидазола Пример 62 (45 мг, 0.11 ммоль, 51%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.79 (s, 1H), 8.51 (s, 1H), 8.36-8.29 (m, 1H), 7.62 (dd, J = 10.2, 7.3 Гц, 1H), 7.44 (dd, J = 9.6, 7.0 Гц, 1H), 7.16-7.10 (m, 1H), 7.03-6.97 (m, 1H), 3.71-3.64 (m, 1H), 1.38-1.32 (m, 2H), 0.92-0.87 (m, 2H)
LC-MS: m/z 385.1 [M+H]+ при 2.61 RT (96.45% чистота)
ВЭЖХ: 96.95%
Пример 63
Схема:
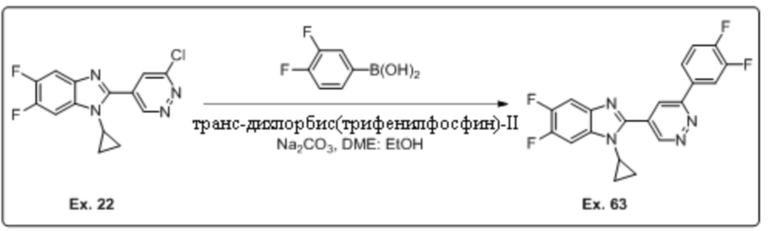
1-Циклопропил-2-(6-(3,4-дифторфенил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 63)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (70 мг, 0.22 ммоль) в DME/EtOH (2: 1, 0.9 мл) добавили (3,4-дифторфенил) бороновую кислоту (43 мг, 0.27 ммоль) и карбонат натрия (75.5 мг, 0.68 ммоль) в запаянной трубке и смесь продули аргоном в течение 10 мин. Транс-дихлор бис-(трифенилфосфин)-II (8 мг, 0.01 ммоль) добавили в реакционную смесь, и реакционную смесь перемешивали при 80°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали и фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 1-циклопропил-2-(6-(3,4-дифторфенил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 63 (45 мг, 0.11 ммоль, 51%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.79 (s, 1H), 8.43 (s, 1H), 8.14-8.09 (m, 1H), 7.96-7.90 (m, 1H), 7.62 (dd, J = 10.0, 7.3 Гц, 1H), 7.46 (dd, J = 9.5, 6.9 Гц, 1H), 7.36 (dd, J = 17.9, 8.7 Гц, 1H), 3.79-3.66 (m, 1H), 1.39-1.29 (m, 2H), 0.93-0.82 (m, 2H)
LC-MS: m/z 385 [M+H]+ при 2.62 RT (98.32% чистота)
ВЭЖХ: 98.68%
Пример 64 и Пример 70
Схема:
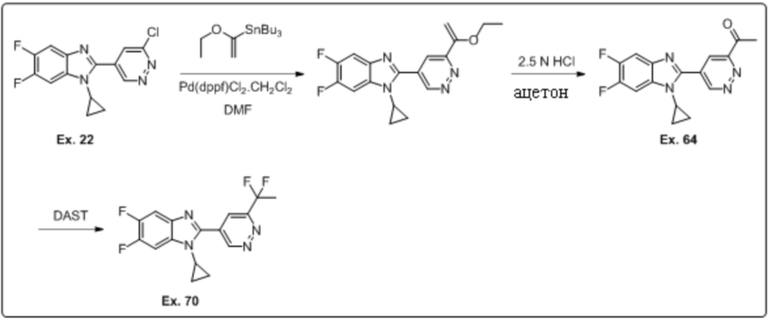
1-Циклопропил-2-(6-(1-этоксивинил) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (200 мг, 0.65 ммоль) в DMF (4 мл) добавили трибутил(1-этоксивинил)станнан (0.26 мл, 0.78 ммоль) и Pd(dppf)Cl2. CH2Cl2 (53 мг, 0.06 ммоль) при комнатной температуре и смесь дегазировали под аргоном в течение 10 мин. Реакционную смесь перемешивали при 100°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (40 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-2-(6-(1-этоксивинил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (180 мг, 0.52 ммоль, 81%) в виде полутвердого вещества бледно-коричневого цвета.
LC-MS: m/z 343.1 [M+H]+ при 3.10 RT (81.31% чистота)
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-он (Пример 64)
К перемешиваемому раствору 1-циклопропил-2-(6-(1-этоксивинил) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (180 мг, 0.52 ммоль) в ацетоне (3 мл) добавили 2.5 N HCl (2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток разбавили с помощью CH2Cl2 (40 мл) и промыли с помощью насыщенного раствора бикарбоната натрия (30 мл) м солевого раствора (30 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-она Пример 64 (110 мг, 0.35 ммоль, 69%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.01 (d, J = 2.3 Гц, 1H), 8.62 (d, J = 2.3 Гц, 1H), 7.91-7.82 (m, 2H), 3.99-3.94 (m, 1H), 2.85 (s, 3H), 1.19-1.14 (m, 2H), 0.79-0.75 (m, 2H)
LC-MS: m/z 315.0 [M+H]+ при 2.65 RT (95.59% чистота)
ВЭЖХ: 93.77%
1-Циклопропил-2-(6-(1,1-дифторэтил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 70)
Раствор 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил) этан-1-она Пример 70 (110 мг, 0.35 ммоль) в DAST (3 мл) в инертной атмосфере перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали с помощью EtOAc (40 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан), которое далее очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(1,1-дифторэтил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 70 (45 мг, 0.13 ммоль, 41%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.98 (d, J = 2.1 Гц, 1H), 8.53 (d, J = 2.0 Гц, 1H), 7.94-7.85 (m, 2H), 4.02-3.97 (m, 1H), 2.22 (t, J = 19.4 Гц, 3H), 1.21-1.14 (m, 2H), 0.83-0.77 (m, 2H)
LC-MS: m/z 337.0 [M+H]+ при 2.99 RT (99.31% чистота)
ВЭЖХ: 99.50%
Пример 65
Схема:

6-Хлор-N-(2-(этиламино)-4,5-дифторфенил)пиридазин-4-карбоксамид
К перемешиваемому раствору N1-этил-4,5-дифторбензол-1,2-диамина Int-10 (500 мг, 2.9 ммоль) в EtOAc (15 мл) в инертной атмосфере добавили 6-хлорпиридазин-4-карбоновую кислоту (505 мг, 3.19 ммоль), триэтиламин (0.8 мл, 5.81 ммоль) и пропилфосфониевый ангидрид (50% в EtOAc, 4.5 мл, 7.26 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (50 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты промыли водой (70 мл), солевым раствором (70 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 15% EtOAc/гексан) с получением 6-хлор-N-(2-(этиламино)-4,5-дифторфенил)пиридазин-4-карбоксамида (500 мг, 1.60 ммоль, 55%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 10.1 (brs, 1H), 9.65 (s, 1H), 8.39 (s, 1H), 7.3 (dd, J = 11.7, 8.1 Гц, 1H), 6.69 (dd, J = 12.1, 7.9 Гц, 1H), 5.5 (brs, 1H), 3.12-3.02 (m, 2H), 1.17 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 312.9 [M+H]+ при 2.69 RT (96.47% чистота)
2-(6-Хлорпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазол (Пример 65)
К перемешиваемому раствору 6-хлор-N-(2-(этиламино)-4,5-дифторфенил)пиридазин-4-карбоксамида (400 мг, 1.28 ммоль) в этаноле (4 мл) в инертной атмосфере добавили 6 Н HCl (6 мл) при комнатной температуре. Реакционную смесь перемешивали при 50°C в течение 15 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в насыщенный раствор бикарбоната натрия (40 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество (100 мг) очистили посредством препаративной ВЭЖХ с получением 2-(6-хлорпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола Пример 65 (30 мг, 0.10 ммоль, 29%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.59 (d, J = 1.9 Гц, 1H), 8.20 (d, J = 1.9 Гц, 1H), 7.73 (dd, J = 10.2, 7.0 Гц, 1H), 7.65 (dd, J = 10.4, 7.3 Гц, 1H), 4.46 (q, J = 7.3 Гц, 2H), 1.48 (t, J = 7.3 Гц, 3H)
LC-MS: m/z 294.9 [M+H]+ при 2.58 RT (98.60% чистота)
ВЭЖХ: 99.85%
Пример 66
Схема:
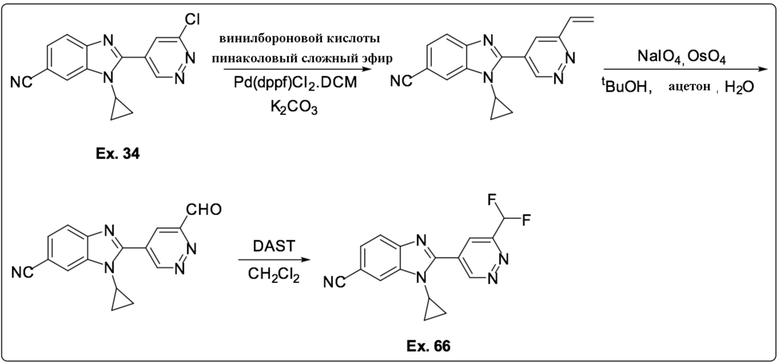
1-Циклопропил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила Пример 34 (200 мг, 0.67 ммоль) в 1, 4-диоксане (5 мл) и оде (1.5 мл) добавили винилбороновой кислоты сложный пинаколовый эфир (313 мг, 2.03 ммоль) и карбонат калия (374 мг, 2.71 ммоль) в запаянной трубке при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 10 мин. К этому добавили Pd(dppf)Cl2. CH2Cl2 (5.3 мг, 0.006 ммоль) при комнатной температуре и смесь далее дегазировали под аргоном в течение 10 мин. Реакционную смесь нагрели до 100°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), солевым раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-циклопропил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (200 мг) в виде густого сиропа коричневого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 288 [M+H]+ при 2.27 RT (73.7% чистота)
1-Циклопропил-2-(6-формилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 1-циклопропил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (200 мг, неочищенный) в смеси трет-бутанола (0.5 мл), ацетона (0.5 мл) и воды (0.5 мл) добавили периодат натрия (29.68 мг, 0.13 ммоль), а затем тетраоксид осмия (0.1 M в толуоле, 4 мл) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (10 мл) и экстрагировали с помощью EtOAc (2 x 10 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-циклопропил-2-(6-формилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (200 мг, неочищенный) в виде твердого вещества оранжевого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 308.1 [M+H2O]+ при 1.78 RT (8.32% чистота)
1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 66)
К перемешиваемому раствору 1-циклопропил-2-(6-формилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (200 мг, неочищенный) в CH2Cl2 (10 мл) в инертной атмосфере добавили DAST (223 мг, 1.38 ммоль) при 0°C в инертной атмосфере. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью насыщенного раствора карбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан), которое далее очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 66 (15 мг, 0.05 ммоль, 7%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.99 (s, 1H), 8.64 (s, 1H), 8.26 (s, 1H), 7.93 (dd, J = 8.4, 0.6 Гц, 1H), 7.68 (dd, J = 8.4, 1.5 Гц, 1H), 7.19 (t, J = 51.1 Гц, 1H), 4.00-3.94 (m, 1H), 1.35-1.30 (m, 2H), 0.91-0.86 (m, 2H)
LC-MS: m/z 312 [M+H]+ при 2.52 RT (98.91% чистота)
ВЭЖХ: 99.83%
Пример 67
Схема:
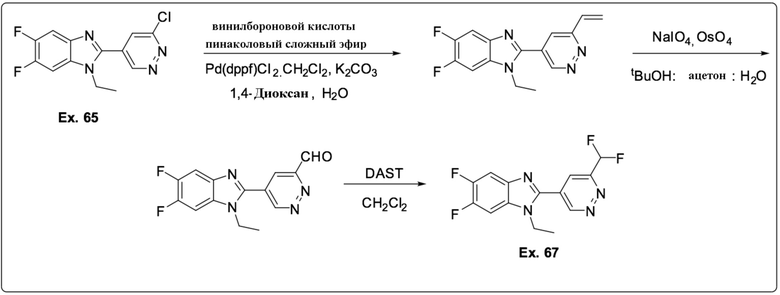
1-Этил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола Пример 65 (400 мг, неочищенный) в 1, 4-диоксане (12 мл) и воде (4 мл) в инертной атмосфере добавили винилбороновой кислоты сложный пинаколовый эфир(419 мг, 2.72 ммоль) и карбонат калия (563 мг, 4.08 ммоль) в запаянной трубке при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 20 мин. К этому добавили Pd(dppf)Cl2.CH2Cl2 (11 мг, 0.013 ммоль) при комнатной температуре и смесь далее дегазировали под аргоном в течение 10 мин. Реакционную смесь нагрели до 80°C и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-этил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (100 мг, неочищенный) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.52 (d, J = 1.7 Гц, 1H), 8.27 (d, J = 1.7 Гц, 1H), 8.02 (dd, J = 10.4, 7.5 Гц, 1H), 7.87 (dd, J = 11.0, 7.5 Гц, 1H), 7.14 (dd, J =, 18.0, 11.0 Гц, 1H), 6.54 (d, J = 17.4 Гц, 1H), 5.82 (d, J = 11.6 Гц, 1H), 4.45 (q, J = 7.5 Гц, 2H), 1.34 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 287 [M+H]+ при 2.47 RT (96.16% чистота)
5-(1-Этил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегид
К перемешиваемому раствору 1-этил-5,6-дифтор-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазола (100 мг, неочищенный) в ацетоне: tBuOH: воде (1:1:1, 6 мл) добавили периодат натрия (149 мг, 0.69 ммоль) и осмия тетроксид (1% в толуоле, 2 мл) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (30 мл). Фильтрат концентрировали при пониженном давлении. Остаток разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5-(1-этил-5, 6-дифтор-1H-бензо[d]имидазол-2-ил пиридазин-3-карбальдегида (100 мг, неочищенный) в виде твердого вещества коричневого цвета. Это неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 10.37 (s, 1H), 9.88 (d, J = 2.3 Гц, 1H), 8.39 (d, J = 2.3 Гц, 1H), 8.04 (dd, J = 10.7, 7.2 Гц, 1H), 7.90 (dd, J = 10.4, 7.5 Гц, 1H), 4.46 (q, J = 7.3 Гц, 2H), 1.37 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 289 [M+H]+ при 1.87 RT (70.81% чистота)
2-(6-(Дифторметил)пиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазол (Пример 67)
К перемешиваемому раствору 5-(1-этил-5,6-дифтор-1H-бензо[d]имидазол-2-ил пиридазин-3-карбальдегида (100 мг, неочищенный) в CH2Cl2 (8 мл) в инертной атмосфере добавили DAST (0.09 мл, 0.69 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты промыли насыщенным раствором бикарбоната натрия (30 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола Пример 67 (30 мг, 0.09 ммоль, 28%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.83 (d, J = 2.1 Гц, 1H), 8.35 (d, J = 2.1 Гц, 1H), 8.04 (dd, J = 10.8, 7.3 Гц, 1H), 7.89 (dd, J = 10.9, 7.5 Гц, 1H), 7.56-7.24 (m, 1H), 4.46 (q, J = 7.3 Гц, 2H), 1.36 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 311.0 [M+H]+ при 2.69 RT (98.69% чистота)
ВЭЖХ: 98.72%
Пример 68 и Пример 69
Схема:
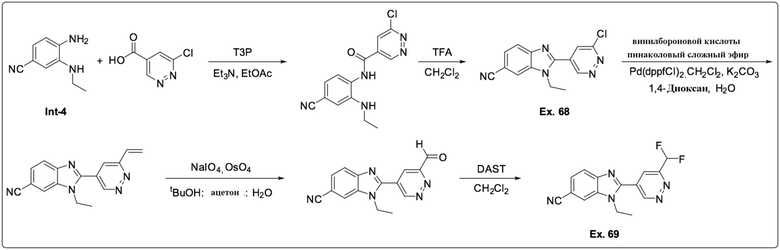
6-Хлор-N-(4-циано-2-(этиламино)фенил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4-амино-3-(этиламино)бензонитрила Int-4 (200 мг, 1.24 ммоль) в EtOAc (10 мл) в инертной атмосфере добавили 6-хлорпиридазин-4-карбоновую кислоту (210 мг, 1.32 ммоль), триэтиламин (0.34 мл, 2.48 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.97 мл, 3.10 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли водой (40 мл), солевым раствором (40 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 6-хлор-N-(4-циано-2-(этиламино)фенил)пиридазин-4-карбоксамида (250 мг, 0.83 ммоль, 67%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.23 (brs, 1H), 9.65 (d, J = 1.2 Гц, 1H), 8.39 (s, 1H), 7.40 (d, J = 8.1 Гц, 1H), 7.06-6.99 (m, 2H), 5.77 (br s, 1H), 3.20-3.15 (m, 2H), 1.17 (t, J = 7.0 Гц, 3H)
2-(6-Хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрил (Пример 68)
К перемешиваемому раствору 6-хлор-N-(4-циано-2-(этиламино)фенил)пиридазин-4-карбоксамида (50 мг, 0.16 ммоль) в CH2Cl2 (2 мл) в инертной атмосфере добавили трифторуксусную кислоту (0.2 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток погасили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила Пример 68 (35 мг, 0.12 ммоль, 74%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.70 (d, J = 1.9 Гц, 1H), 8.50 (s, 1H), 8.35 (d, J = 1.9 Гц, 1H), 7.96 (dd, J = 8.4, 0.5 Гц, 1H), 7.72 (dd, J = 8.5, 1.4 Гц, 1H), 4.52 (q, J = 7.3 Гц, 2H), 1.37 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 284 [M+H]+ при 2.34 RT (97.52% чистота)
ВЭЖХ: 97.70%
1-Этил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила Пример 68 (170 мг, 0.60 ммоль) в смеси 1, 4-диоксана (6 мл) и воды (2 мл) в инертной атмосфере добавили винилбороновой кислоты сложный пинаколовый эфир(185 мг, 1.20 ммоль) и карбонат калия (248.69 мг, 1.80 ммоль). Реакционную смесь дегазировали под аргоном в течение 20 мин. К этому добавили Pd(dppf)Cl2.CH2Cl2 (4.9 мг, 0.06 ммоль) при комнатной температуре. Реакционную смесь нагрели до 80°C и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 1-2% MeOH/CH2Cl2) с получением 1-этил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (100 мг, 0.36 ммоль, 61%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.56 (d, J = 2.1 Гц, 1H), 8.50-8.48 (m, 1H), 8.32 (d, J = 2.1 Гц, 1H), 7.95 (d, J = 8.4 Гц, 1H), 7.72 (dd, J = 8.4, 1.5 Гц, 1H), 7.16 (dd, J = 17.8, 11.0 Гц, 1H), 6.56 (dd, J = 17.7, 0.6 Гц, 1H), 5.84 (d, J = 11.7 Гц, 1H), 4.51 (q, J = 7.2 Гц, 2H), 1.37 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 276.1 [M+H]+ при 2.23 RT (95.18% чистота)
1-Этил-2-(6-формилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 1-этил-2-(6-винилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (100 мг, 0.36 ммоль) в ацетоне: tBuOH: воде (1:1:1, 6 мл) в инертной атмосфере добавили периодат натрия (154.9 мг, 0.72 ммоль) и осмия тетроксид (1% в толуоле, 2 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой целита промыли с помощью EtOAc (20 мл). Фильтрат концентрировали при пониженном давлении. Остаток разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-этил-2-(6-формилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (100 мг, неочищенный) в виде густого сиропа. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 278.1 [M+H]+ при 1.65 RT (43.82% чистота)
2-(6-(Дифторметил)пиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрил (Пример 69)
К перемешиваемому раствору 1-этил-2-(6-формилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (100 мг, неочищенный) в CH2Cl2 (4 мл) в инертной атмосфере добавили DAST (0.09 мл, 0.72 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в ледяную воду (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-1-этил-1H-бензо[d]имидазол-6-карбонитрила Пример 69 (30 мг, 0.10 ммоль) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.81 (d, J = 2.0 Гц, 1H), 8.42 (d, J = 2.0 Гц, 1H), 8.28 (dd, J = 1.4, 0.8 Гц, 1H), 7.94 (dd, J = 8.4, 0.6 Гц, 1H), 7.69 (dd, J = 8.4, 1.5 Гц, 1H), 7.13 (t, J = 54.6 Гц, 1H), 4.54 (q, J = 7.3 Гц, 2H), 1.52 (t, J = 7.3 Гц, 3H)
LC-MS: m/z 300.0 [M+H]+ при 2.41 RT (99.41% чистота)
ВЭЖХ: 99.68%
Пример 71
Схема:
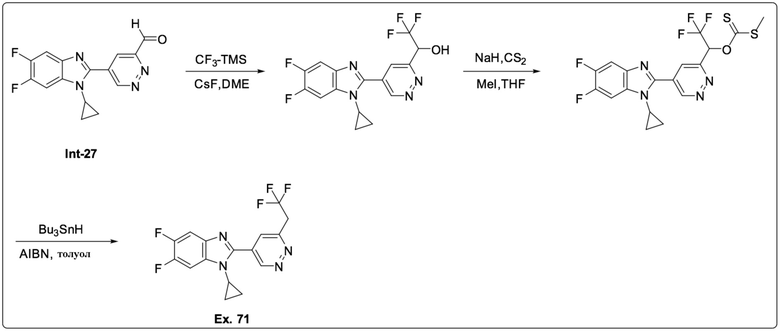
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифтопентан-1-ол
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбальдегида Int-27 (400 мг, 1.33 ммоль) в DME (5 мл) в инертной атмосфере добавили фторид цезия (202.6 мг, 1.33 ммоль) при 0°C и смесь перемешивали в течение 15 мин. TMS-CF3 (284 мг, 1.99 ммоль) добавили в реакционную смесь при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с помощью 1 Н раствора HCl (20 мл), подщелочили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифтопентан-1-ола (160 мг, 0.43 ммоль, 32%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, CD3OD): δ 9.84 (d, J = 2.3 Гц, 1H), 8.58 (d, J = 1.7 Гц, 1H), 7.73 (dd, J = 9.9, 7.0 Гц, 1H), 7.65 (dd, J = 10.4, 7.5 Гц, 1H), 5.55 (q, J = 6.8 Гц, 1H), 4.57 (s, 1H), 3.92-3.86 (m, 1H), 1.29-1.20 (m, 2H), 0.88-0.78 (m, 2H)
LC-MS: m/z 371.1 [M+H]+ при 2.76 RT (75.32% чистота)
O-(1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2,2,2-трифторэтил) S-метил карбондитиоат
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил)-2,2,2-трифтопентан-1-ола (100 мг, 0.27 ммоль) в THF (5 мл) в инертной атмосфере добавили гидрид натрия (60% в минеральном масле, 21.6 мг, 0.54 ммоль) по частям при 0°C и смесь перемешивали в течение 30 мин. Дисульфид углерода (41 мг, 0.54 ммоль) добавили в реакционную смесь при 0°C и перемешивали в течение 30 мин, а затем добавили метилиодид (76 мг, 0.54 ммоль) при 0°C и перемешивание продолжали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорид аммония (15 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением O-(1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-2, 2, 2-трифторэтил) S-метил карбондитиоата (100 мг) в виде густого сиропа черного цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 9.88 (d, J = 2.3 Гц, 1H), 8.56 (d, J = 1.7 Гц, 1H), 7.92 (dd, J = 11.0, 7.5 Гц, 1H), 7.86 (dd, J = 10.4, 7.5 Гц, 1H), 7.52 (q, J = 6.8 Гц, 1H), 3.95-3.88 (m, 1H), 2.67 (s, 3H), 1.20-1.15 (m, 2H), 0.79-0.73 (m, 2H)
LC-MS: m/z 461.1 [M+H]+ при 3.63 RT (77.86% чистота)
1-Циклопропил-5,6-дифтор-2-(6-(2,2,2-трифторэтил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 71)
К перемешиваемому раствору O-(1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил)-2,2,2-трифторэтил) S-метил карбондитиоата (100 мг, 0.21 ммоль) в толуоле (5 мл) в инертной атмосфере добавили AIBN (5.3 мг, 0.032 ммоль) и трибутилстаннан (95 мг, 0.32 ммоль) при комнатной температуре. Реакционную смесь нагрели до 60°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорид аммония (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан), которое далее очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-5,6-дифтор-2-(6-(2,2,2-трифторэтил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 71 (15 мг, 0.04 ммоль, 19%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.84 (d, J = 2.0 Гц, 1H), 8.16 (d, J = 1.6 Гц, 1H), 7.61 (dd, J = 10.0, 7.3 Гц, 1H), 7.44 (dd, J = 9.5, 6.9 Гц, 1H), 3.99 (q, J = 10.5 Гц, 2H), 3.68-3.62 (m, 1H), 1.36-1.29 (m, 2H), 0.88-0.83 (m, 2H)
LC-MS: m/z 355.1 [M+H]+ при 2.86 RT (98.85% чистота)
ВЭЖХ: 99.79%
Пример 72
Схема:
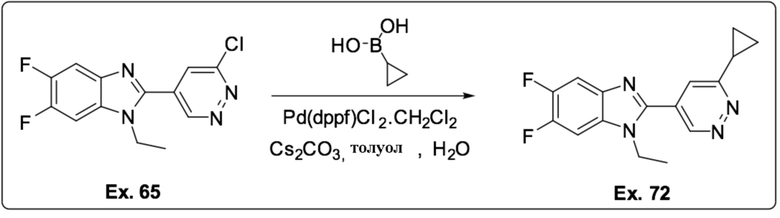
2-(6-Циклопропилпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазол (Пример 72)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола Пример 65 (150 мг, 0.51 ммоль) в толуоле/воде (3: 1, 12 мл) добавили циклопропилбороновую кислоту (52.6 мг, 0.61 ммоль) и карбонат цезия (416 мг, 1.27 ммоль) в запаянной трубке и продули аргоном в течение 20 мин. Pd(dppf)Cl2.CH2Cl2 (4 мг, 0.005 ммоль) добавили в реакционную смесь, и снова продули аргоном в течение 5 мин при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 2-(6-циклопропилпиридазин-4-ил)-1-этил-5,6-дифтор-1H-бензо[d]имидазола Пример 72 (40 мг, 0.133 ммоль, 26%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.35 (d, J = 2.0 Гц, 1H), 7.85 (d, J = 2.1 Гц, 1H), 7.71 (dd, J = 10.3, 7.0 Гц, 1H), 7.63 (dd, J = 10.4, 7.3 Гц, 1H), 4.43 (q, J = 7.3 Гц, 2H), 2.44-2.36 (m, 1H), 1.46 (t, J = 7.2 Гц, 3H), 1.30-1.24 (m, 4H)
LC-MS: m/z 301.0 [M+H]+ при 2.58 RT (99.14% чистота)
ВЭЖХ: 99.64%
Пример 73 и Пример 76
Схема:

Метил 6-хлорпиридазин-4-карбоксилат
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (1.5 г, 9.49 ммоль) в CH2Cl2 (30 мл) в атмосфере аргона добавили диазометан в диэтиловом простом эфире (свеже полученный путем добавления N-нитрозометилмочевины (3 г) к смеси 50% KOH раствора (25 мл) и диэтилового простого эфира (50 мл) при 0°C; нагревания до комнатной температуры и перемешивания в течение 30 мин). После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью CH2Cl2 (30 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением метил 6-хлорпиридазин-4-карбоксилата (1.4 г, 8.13 ммоль, 86%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.58 (d, J = 1.6 Гц, 1H), 8.03 (d, J = 1.8 Гц, 1H), 4.04 (s, 3H)
Метил 6-винилпиридазин-4-карбоксилат
К перемешиваемому раствору метил 6-хлорпиридазин-4-карбоксилата (1.4 г, 8.13 ммоль) в 1, 2-диметоксиэтане (20 мл) в инертной атмосфере добавили карбонат калия (3.36 г, 24.39 ммоль) и винилбороновой кислоты сложный пинаколовый эфир (2.56 мл, 16.2 ммоль) в запаянной трубке при комнатной температуре. Реакционную смесь дегазировали под аргоном в течение 5-10 мин. Pd(dppf)Cl2.CH2Cl2 (66 мг, 0.08 ммоль) добавили в реакционную смесь при комнатной температуре и дегазировали под аргоном в течение 5-10 мин. Реакционную смесь нагрели до 80-90°C в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью EtOAc (50 мл), отфильтровали через слой целита, и слой промыли с помощью EtOAc (50 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/гексан) с получением метил 6-винилпиридазин-4-карбоксилата (1.3 г, 7.92 ммоль, 97%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.51 (d, J = 2.0 Гц, 1H), 8.08 (d, J = 2.0 Гц, 1H), 7.14 (dd, J = 17.8, 11.0 Гц, 1H), 6.38 (d, J = 17.7 Гц, 1H), 5.80 (d, J = 11.0 Гц, 1H), 4.03 (s, 3H)
6-Винилпиридазин-4-карбоновая кислота
К перемешиваемому раствору метил 6-винилпиридазин-4-карбоксилата (1.4 г, 8.53 ммоль) в смеси THF: воды (1:2, 12.75 мл) добавили гидроксид лития (716 мг, 17.07 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1.5 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Остаток подкислили (pH ~1-2), применяя концентрированную HCl, и перемешивали в течение 30 мин при комнатной температуре. Осажденное твердое вещество отфильтровали и высушили в вакууме с получением 6-винилпиридазин-4-карбоновой кислоты (500 мг, 3.33 ммоль, 39%) в виде твердого вещества грязновато-белого цвета, которое применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 14.10 (br s, 1H), 9.42 (d, J = 1.7 Гц, 1H), 8.24 (d, J = 2.3 Гц, 1H), 7.12 (dd, J = 18.0, 11.0 Гц, 1H), 6.53 (d, J = 17.4 Гц, 1H), 5.79 (d, J = 11.0 Гц, 1H)
N-(6-Хлор-2-(циклопропиламино) пиридин-3-ил)-6-винилпиридазин-4-карбоксамид
К перемешиваемому раствору 6-винилпиридазин-4-карбоновой кислоты (40 мг, 0.27 ммоль) в CH2Cl2 (3 мл) в инертной атмосфере добавили 6-хлор-N2-циклопропилпиридин-2,3-диамин Int-8 (50 мг, 0.27 ммоль), бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфат (170 мг, 0.32 ммоль) и диизопропилэтиламин (0.18 мл, 1.08 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили водным раствором хлорида аммония (30 мл) и экстрагировали с помощью EtOAc (40 мл). Органический слой промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/Гексан) с получением N-(6-хлор-2-(циклопропиламино)пиридин-3-ил)-6-винилпиридазин-4-карбоксамида (140 мг, неочищенный) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.01 (s, 1H), 9.51 (d, J = 2.1 Гц, 1H), 8.36 (d, J = 2.0 Гц, 1H), 7.50 (d, J = 7.9 Гц, 1H), 6.86 (d, J = 2.5 Гц, 1H), 6.70-6.65 (m, 1H), 6.47 (d, J = 17.7 Гц, 1H), 5.83 (d, J = 11.4 Гц, 2H), 2.73-2.69 (m, 1H), 0.73-0.68 (m, 2H), 0.49-0.45 (m, 2H)
LC-MS: m/z 316.1 [M+H]+ при 2.36 RT (70.07% чистота)
5-Хлор-3-циклопропил-2-(6-винилпиридазин-4-ил)-3H-имидазо[4, 5-b]пиридин
К перемешиваемому раствору N-(6-хлор-2-(циклопропиламино) пиридин-3-ил)-6-винилпиридазин-4-карбоксамида (140 мг, 0.44 ммоль) в трет-бутаноле (10 мл) в инертной атмосфере добавили трикалия фосфат (222 мг, 1.1 ммоль) при комнатной температуре. Реакционную смесь нагрели до 90°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток разбавили водой (30 мл) и экстрагировали с помощью EtOAc (40 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 60-70% EtOAc/гексан) с получением 5-хлор-3-циклопропил-2-(6-винилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридина (125 мг, неочищенный) в виде сиропа коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.01 (s, 1H), 9.51 (d, J = 1.7 Гц, 1H), 8.36 (d, J = 1.7 Гц, 1H), 7.50 (d, J = 8.1 Гц, 1H), 6.67 (d, J = 8.1 Гц, 1H), 6.48 (d, J = 17.4 Гц, 1H), 5.83 (d, J = 11.0 Гц, 1H), 2.72-2.68 (m, 1H), 0.72-0.68 (m, 2H), 0.49-0.44 (m, 2H)
LC-MS: m/z 297.9 [M+H]+ при 2.35 RT (77.28% чистота)
5-(5-Хлор-3-циклопропил-3H-имидазо[4,5-b]пиридин-2-ил)пиридазин-3-карбальдегид
К перемешиваемому раствору 5-хлор-3-циклопропил-2-(6-винилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридина (125 мг, 0.42 ммоль) в ацетоне: tBuOH: воде (1:1:1, 7.5 мл) в инертной атмосфере добавили периодат натрия (179 мг, 0.84 ммоль) и осмия тетроксид (1% в толуоле, 2.5 мл) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью CH2Cl2 (30 мл). Органический слой промыли водой (20 мл) высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5-(5-хлор-3-циклопропил-3H-имидазо[4,5-b]пиридин-2-ил)пиридазин-3-карбальдегида 8 (130 мг) в виде твердого вещества коричневого цвета, которое применяли на следующей стадии без дальнейшей очистки.
5-Хлор-3-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин (Пример 73)
К перемешиваемому раствору 5-(5-хлор-3-циклопропил-3H-имидазо[4,5-b]пиридин-2-ил) пиридазин-3-карбальдегида (130 мг, неочищенный) в CH2Cl2 (10 мл) в инертной атмосфере добавили DAST (0.11 мл, 0.86 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и подщелочили с помощью водного раствора карбоната натрия (10 мл) и экстрагировали с помощью CH2Cl2 (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/Гексан) с получением 5-хлор-3-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридина Пример 73 (7.2 мг, 0.022 ммоль, 5%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 10.00 (d, J = 2.0 Гц, 1H), 8.64 (d, J = 2.0 Гц, 1H), 8.15 (d, J = 8.4 Гц, 1H), 7.43 (d, J = 8.4 Гц, 1H), 7.33-7.05 (m, 1H), 3.90-3.82 (m, 1H), 1.28-1.25 (m, 2H), 0.99-0.93 (m, 2H)
LC-MS: m/z 321.9 [M+H]+ при 2.56 RT (94.15% чистота)
ВЭЖХ: 92.45%
3-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрил (Пример 76)
К перемешиваемому раствору 5-хлор-3-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридина Пример 73 (50 мг, 0.15 ммоль) в DMF (2 мл) добавили цианид цинка (36 мг, 0.31 ммоль) при комнатной температуре и смесь продули аргоном в течение 5 мин. Pd(PPh3)4 (17.94 мг, 0.01 ммоль) добавили и смесь продули аргоном в течение 5 мин. Реакционную смесь нагрели до 150°C в течение 1.5 часов под микроволнами. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (30 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/гексан) с получением 3-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрила Пример 76 (27 мг, 0.086 ммоль, 56%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 10.05 (d, J = 2.0 Гц, 1H), 8.70 (d, J = 2.1 Гц, 1H), 8.33 (d, J = 8.3 Гц, 1H), 7.88 (d, J = 8.2 Гц, 1H), 7.36-7.08 (m, 1H), 3.94-3.89 (m, 1H), 1.35-1.29 (m, 2H), 1.03-0.97 (m, 2H)
LC-MS: m/z 312.9 [M+H]+ при 2.11 RT (99.81% чистота)
ВЭЖХ: 99.53%
Пример 74
Схема:
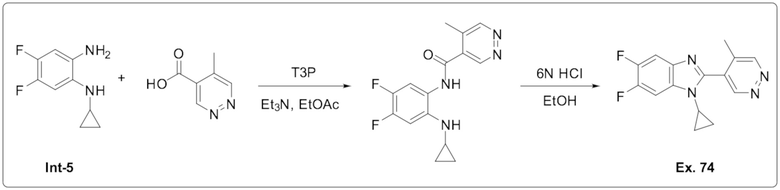
N-(2-(Циклопропиламино)-4, 5-дифторфенил)-5-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4,5-дифторбензол-1,2-диамина Int-5 (250 мг, 1.35 ммоль) в EtOAc (8 мл) в инертной атмосфере добавили 5-метилпиридазин-4-карбоновую кислоту (206 мг, 1.49 ммоль), триэтиламин (0.37 мл, 2.71 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 2.1 мл, 3.39 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью EtOAc (30 мл) и промыли водой (20 мл) и солевым раствором (20 мл). Органический слой высушили над безводным Na2SO4 и концентрировали под давлением. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50-70% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-5-метилпиридазин-4-карбоксамида (210 мг, 0.69 ммоль, 51%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.78 (s, 1H), 9.36 (s, 1H), 9.26 (s, 1H), 7.47 (dd, J = 12.1, 8.7 Гц, 1H), 6.94 (dd, J = 13.4, 8.1 Гц, 1H), 5.85 (s, 1H), 2.43 (s, 3H), 2.12-2.03 (m, 1H), 0.78-0.71 (m, 2H), 0.46-0.39 (m, 2H)
LC-MS: m/z 305.1 [M+H]+ при 2.47 RT (72.24% чистота)
1-Циклопропил-5,6-дифтор-2-(5-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 74)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-5-метилпиридазин-4-карбоксамида (150 мг, 0.49 ммоль) в этаноле (3 мл) добавили 6 Н HCl (3 мл) при комнатной температуре. Реакционную смесь перемешивали при 50°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток подщелочили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали с помощью CH2Cl2 (40 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью диэтилового простого эфира (5 мл) с получением 1-циклопропил-5,6-дифтор-2-(5-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 74 (65 мг, 0.21 ммоль, 46%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.47 (s, 1H), 9.36 (s, 1H), 7.87-7.81 (m, 2H), 3.75-3.69 (m, 1H), 2.44 (s, 3H), 0.99-0.94 (m, 2H), 0.63-0.58 (m, 2H)
LC-MS: m/z 287.0 [M+H]+ при 2.26 RT (98.32% чистота)
ВЭЖХ: 99.05%
Пример 75
Схема:
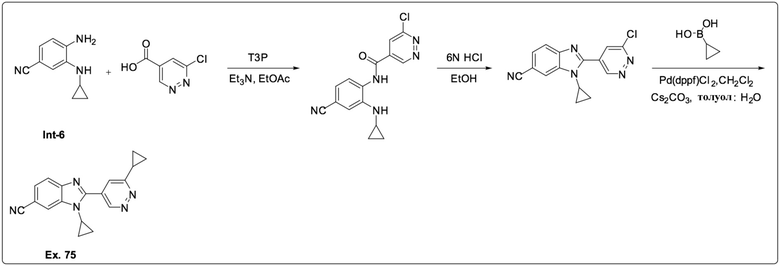
6-Хлор-N-(4-циано-2-(циклопропиламино)фенил)пиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (458 мг, 2.89 ммоль) в EtOAc (30 мл) в инертной атмосфере добавили 4-амино-3-(циклопропиламино)бензонитрил Int-6 (500 мг, 2.89 ммоль), триэтиламин (0.8 мл, 5.78 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 4.6 мл, 7.22 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в насыщенный раствор бикарбоната натрия (70 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты промыли водой (120 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/гексан) с получением 6-хлор-N-(4-циано-2-(циклопропиламино)фенил)пиридазин-4-карбоксамида (650 мг, 2.07 ммоль, 72%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.17 (s, 1H), 9.63 (d, J = 1.8 Гц, 1H), 8.38 (d, J = 1.8 Гц, 1H), 7.42 (d, J = 8.0 Гц, 1H), 7.31 (d, J = 1.8 Гц, 1H), 7.10 (dd, J = 8.0, 1.9 Гц, 1H), 6.31 (s, 1H), 2.45-2.38 (m, 1H), 0.82-0.77 (m, 2H), 0.48-0.43 (m, 2H)
LC-MS: m/z 312.1 [M-H]- при 2.57 RT (80.47% чистота)
2-(6-Хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 6-хлор-N-(4-циано-2-(циклопропиламино)фенил)пиридазин-4-карбоксамида (600 мг, 1.91 ммоль) в этаноле (6 мл) в инертной атмосфере добавили 6 Н HCl (9 мл) при комнатной температуре. Реакционную смесь перемешивали при 55 - 60°C в течение 10 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в насыщенный раствор бикарбоната натрия (100 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 1% MeOH/CH2Cl2) с получением 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила (450 мг, 1.52 ммоль, 80%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.82 (d, J = 1.9 Гц, 1H), 8.20 (d, J = 1.8 Гц, 1H), 8.02 (dd, J = 1.4, 0.7 Гц, 1H), 7.91 (dd, J = 8.5, 0.7 Гц, 1H), 7.63 (dd, J = 8.4, 1.5 Гц, 1H), 3.75-3.69 (m, 1H), 1.45-1.39 (m, 2H), 0.94-0.89 (m, 2H)
LC-MS: m/z 295.9 [M+H]+ при 2.40 RT (95.15% чистота)
1-Циклопропил-2-(6-циклопропилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 75)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила (100 мг, 0.33 ммоль) в толуоле: воде (3: 1, 3.2 мл) в инертной атмосфере добавили циклопропилбороновую кислоту (35 мг, 0.40 ммоль) и карбонат цезия (275.5 мг, 0.84 ммоль) при комнатной температуре и смесь продули аргоном в течение 5 мин. Pd(dppf)Cl2.CH2Cl2 (27.6 мг, 0.03 ммоль) добавили в реакционную смесь при комнатной температуре и смесь дегазировали под аргоном в течение 5 мин. Реакционную смесь перемешивали при 100°C в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в воду (40 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-циклопропилпиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 75 (40 мг, 0.13 ммоль, 39%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.61 (d, J = 2.1 Гц, 1H), 8.00 (s, 1H), 7.89 (dd, J = 8.4, 0.5 Гц, 1H), 7.86 (d, J = 2.0 Гц, 1H), 7.61 (dd, J = 8.3, 1.4 Гц, 1H), 3.72-3.66 (m, 1H), 2.32-2.26 (m, 1H), 1.37-1.32 (m, 4H), 1.27-1.21 (m, 2H), 0.89-0.85 (m, 2H)
LC-MS: m/z 302 [M+H]+ при 2.06 RT (96.07% чистота)
ВЭЖХ:95.17%
Пример 77
Схема:
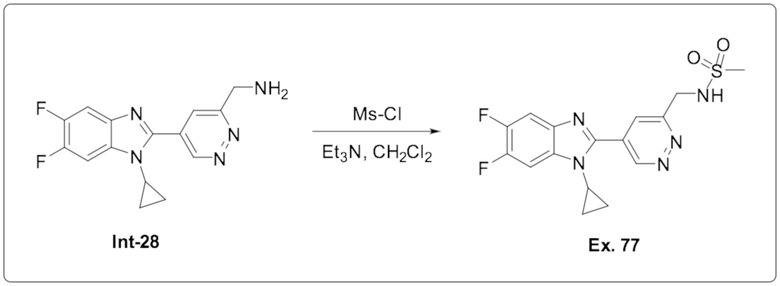
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)метан сульфонамид (Пример 77)
К перемешиваемому раствору (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанамина Int-28 (200 мг, неочищенный) в CH2Cl2 (13 мл) в инертной атмосфере добавили триэтиламин (0.18 мл, 1.32 ммоль) и мезилхлорид (0.08 мл, 0.74 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью CH2Cl2 (40 мл) и промыли водой (30 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)метан сульфонамида Пример 77 (11 мг, 0.029 ммоль, 4% общий выход) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.72 (d, J = 2.0 Гц, 1H), 8.50 (d, J = 2.0 Гц, 1H), 7.73 (dd, J = 10.1, 7.1 Гц, 1H), 7.64 (dd, J = 10.4, 7.3 Гц, 1H), 4.73 (s, 2H), 3.88-3.81 (m, 1H), 3.09 (s, 3H), 1.37-1.32 (m, 2H), 0.86-0.81 (m, 2H)
LC-MS: m/z 380.1 [M+H]+ при 1.99 RT (96.14% чистота)
ВЭЖХ: 98.32%
Пример 78
Схема:
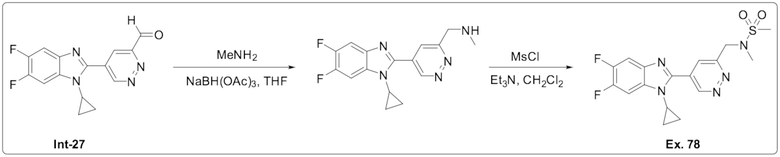
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-N-метилметанамин
К перемешиваемому раствору 5-(1-циклопропил-5, 6-дифтор-1H-бензо [d] имидазол-2-ил) пиридазин-3-карбальдегида Int-27 (200 мг, 0.66 ммоль) в THF (10 мл) в инертной атмосфере добавили раствор метиламина (2 M в THF, 0.66 мл, 1.33 ммоль) по каплям при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 15 мин, затем повторно охладили 0°C. Натрия триацетоксиборгидрид добавили, и реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток разбавили с помощью CH2Cl2 (40 мл) и промыли с помощью насыщенного раствора бикарбоната натрия (30 мл) и воды (30 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-N-метилметанамина (180 мг) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, DMSO-d6): δ 9.69 (d, J = 2.1 Гц, 1H), 8.30 (d, J = 2.1 Гц, 1H), 7.90-7.80 (m, 2H), 4.07 (s, 2H), 3.95-3.87 (m, 1H), 2.90 (br s, 1H), 2.34 (s, 3H), 1.22-1.16 (m, 2H), 0.78-0.73 (m, 2H)
LC-MS: m/z 315.9 [M+H]+ при 1.65 RT (90.51% чистота)
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)-N-метилметансульфонамид (Пример 78)
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)-N-метилметанамина (200 мг, неочищенный) в CH2Cl2 (13 мл) в инертной атмосфере добавили триэтиламин (0.17 мл, 1.26 ммоль) и мезилхлорид (0.08 мл, 0.95 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (по LCMS), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью CH2Cl2 (50 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 80% EtOAc/гексан) с получением N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)-N-метилметансульфонамида Пример 78 (35 мг, 0.089 ммоль, 14%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.80 (d, J = 2.1 Гц, 1H), 8.27 (d, J = 2.0 Гц, 1H), 7.90 (dd, J = 10.9, 7.5 Гц, 1H), 7.84 (dd, J = 10.4, 7.3, Гц, 1H), 4.73 (s, 2H), 3.93-3.87 (m, 1H), 3.08 (s, 3H), 2.87 (s, 3H), 1.23-1.18 (m, 2H), 0.82-0.77 (m, 2H)
LC-MS: m/z 394.1 [M+H]+ при 2.43 RT (99.69% чистота)
ВЭЖХ: 99.74%
Пример 79
Схема:
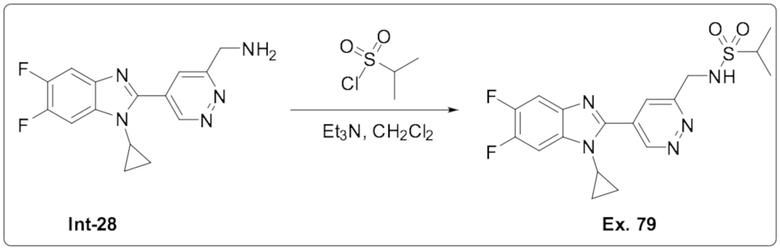
N-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)пропан-2-сульфонамид (Пример 79)
К перемешиваемому раствору (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил) метанамина Int-28 (150 мг, 0.49) в CH2Cl2 (7 мл) в инертной атмосфере добавили триэтиламин (0.13 мл, 0.99 ммоль) и пропан-2-сульфонил хлорид (106 мг, 0.74 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью CH2Cl2 (30 мл) и промыли водой (20 мл) и солевым раствором (20 мл). Органический слой высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2), которое далее очистили посредством препаративной ВЭЖХ с получением N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)пропан-2-сульфонамида Пример 79 (40 мг, 0.09 ммоль, 20%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.78 (d, J = 1.7 Гц, 1H), 8.34 (d, J = 1.7 Гц, 1H), 7.96-7.81 (m, 3H), 4.62 (d, J = 5.8 Гц, 2H), 3.91-3.85 (m, 1H), 3.30-3.27 (m, 1H), 1.28-1.21 (m, 8H), 0.79-0.74 (m, 2H)
LC-MS: m/z 408.0 [M+H]+ при 2.49 RT (98.74% чистота)
ВЭЖХ: 95.26%
Пример 80
Схема:
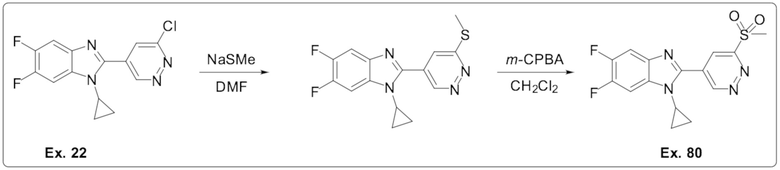
1-Циклопропил-5,6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.32) в DMF (1 мл) в инертной атмосфере добавили натрия метантиолат (15% в воде, 23 мг, 0.32 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC и LC-MS), реакционную смесь разбавили с помощью солевым раствором (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли водой (60 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью н-пентана (5 мл) и высушили в вакууме с получением 1-циклопропил-5,6-дифтор-2-(6-(метилтио) пиридазин-4-ил)-1H-бензо[d]имидазола (80 мг, 0.25 ммоль, 77%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.79-9.49 (m, 1H), 8.15-7.88 (m, 1H), 7.59 (dd, J = 10.1, 7.3 Гц, 1H), 7.42 (dd, J = 9.7, 6.9 Гц, 1H), 3.65-3.58 (m, 1H), 2.80 (s, 3H), 1.33-1.28 (m, 2H), 0.87-0.82 (m, 2H)
LC-MS: m/z 318.9 [M+H]+ при 2.32 RT (97.39% чистота)
1-Циклопропил-5,6-дифтор-2-(6-(метилсульфонил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 80)
К перемешиваемому раствору 1-циклопропил-5, 6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазола (60 мг, 0.18 ммоль) в CH2Cl2 (6 мл) в инертной атмосфере добавили м-хлорпероксибензойную кислоту (81 мг, 0.47 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC и LC-MS), реакционную смесь погасили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли соляным раствором (60 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(метилсульфонил) пиридазин-4-ил)-1H-бензо[d]имидазола Пример 80 (30 мг, 0.085 ммоль, 45%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 10.09 (d, J = 2.0 Гц, 1H), 8.79 (d, J = 2.1 Гц, 1H), 7.64 (dd, J = 9.9, 7.3 Гц, 1H), 7.46 (dd, J = 9.5, 6.9 Гц, 1H), 3.74-3.68 (m, 1H), 3.52 (s, 3H), 1.44-1.37 (m, 2H), 0.94-0.87 (m, 2H)
LC-MS: m/z 351.0 [M+H]+ при 2.64 RT (95.45% чистота)
ВЭЖХ: 96.64%
Пример 81 и Пример 83
Схема:
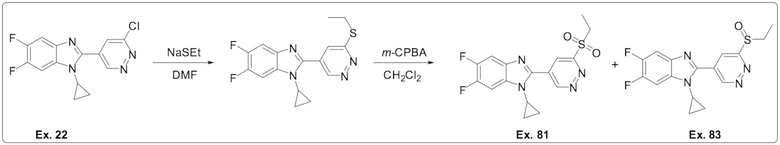
1-Циклопропил-2-(6-(этилтио)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (300 мг, 0.98) в DMF (7 мл) в инертной атмосфере добавили этантиолат натрия (15% в воде, 90 мг, 1.07 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли соляным раствором (40 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью н-пентана (5 мл) и высушили в вакууме с получением 1-циклопропил-2-(6-(этилтио)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (240 мг, 0.72 ммоль, 74%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.49 (d, J = 2.0 Гц, 1H), 7.86 (d, J = 2.0 Гц, 1H), 7.58 (dd, J = 10.2, 7.3 Гц, 1H), 7.42 (dd, J = 9.7, 6.9 Гц, 1H), 3.65-3.57 (m, 1H), 3.44 (q, J = 7.4 Гц, 2H), 1.49 (t, J = 7.3 Гц, 3H), 1.33-1.29 (m, 2H), 0.88-0.83 (m, 2H)
1-Циклопропил-2-(6-(этилсульфонил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 81) и 1-циклопропил-2-(6-(этилсульфинил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d] имидазол (Пример 83)
К перемешиваемому раствору 1-циклопропил-2-(6-(этилтио)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола (260 мг, 0.78) в CH2Cl2 (8 мл) в инертной атмосфере добавили м-хлорпероксибензойную кислоту (296 мг, 1.56 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли водой (60 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50-70% EtOAc/гексан) с получением 1-циклопропил-2-(6-(этилсульфонил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 81 (22 мг, 0.060 ммоль) в виде твердого вещества грязновато-белого цвета и 1-циклопропил-2-(6-(этилсульфинил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 83 (60 мг, 0.17 ммоль) в виде твердого вещества грязновато-белого цвета.
Аналитические данные для примера 81:
1H ЯМР (400 МГц, CD3OD): δ 10.08 (d, J = 2.0 Гц, 1H), 8.89 (d, J = 2.0 Гц, 1H), 7.74 (dd, J =10.1, 7.1 Гц, 1H), 7.67 (dd, J = 10.3, 7.3 Гц, 1H), 3.96-3.89 (m, 1H), 3.69 (q, J = 7.4 Гц, 2H), 1.39 (t, J = 7.4 Гц, 3H), 1.33-1.27 (m, 2H), 0.93-0.86 (m, 2H)
LC-MS: m/z 365.0 [M+H]+ при 2.72 RT (99.37% чистота)
ВЭЖХ: 95.47%
Аналитические данные для примера 83:
1H ЯМР (400 МГц, CD3OD): δ 9.93 (d, J = 2.1 Гц, 1H), 8.76 (d, J = 2.1 Гц, 1H), 7.75 (dd, J = 10.1, 7.1 Гц, 1H), 7.67 (dd, J = 10.4, 7.3 Гц, 1H), 3.94-3.89 (m, 1H), 3.49-3.42 (m, 1H), 3.27-3.19 (m, 1H), 1.35-1.31 (m, 2H), 1.29 (t, J = 7.3 Гц, 3H), 0.92-0.88 (m, 2H).
LC-MS: m/z 348.9 [M+H]+ при 2.09 RT (95.88% чистота)
ВЭЖХ: 95.47%
Пример 82
Схема:
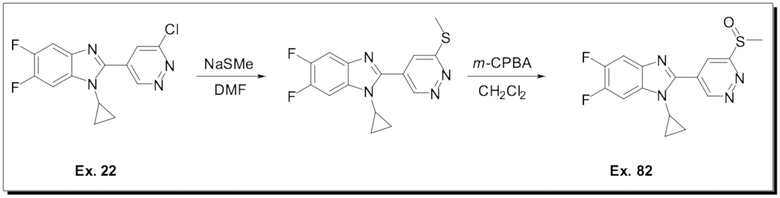
1-Циклопропил-5,6-дифтор-2-(6-(метилтио пиридазин-4-ил)-1H-бензо[d]имидазол
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.32 ммоль) в DMF (1 мл) в инертной атмосфере добавили натрия метантиолат (15% в воде, 23 мг, 0.32 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC и LC-MS), реакционную смесь разбавили с помощью солевым раствором (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли водой (60 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью н-пентана (5 мл), высушили в вакууме с получением 1-циклопропил-5,6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазола (80 мг, 0.25 ммоль, 77%) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.79-9.49 (m, 1H), 8.15-7.88 (m, 1H), 7.59 (dd, J = 10.1, 7.3 Гц, 1H), 7.42 (dd, J = 9.7, 6.9 Гц, 1H), 3.65-3.58 (m, 1H), 2.80 (s, 3H), 1.33-1.28 (m, 2H), 0.87-0.82 (m, 2H)
LC-MS: m/z 318.9 [M+H]+ при 2.32 RT (97.39% чистота)
1-Циклопропил-5,6-дифтор-2-(6-(метилсульфинил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 82)
К перемешиваемому раствору 1-циклопропил-5,6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазола (100 мг, 0.31 ммоль) в CH2Cl2 (6 мл) в инертной атмосфере добавили m-хлорпероксибензойную кислоту (59 мг, 0.34 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты промыли водой (60 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 60-70% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(метилсульфинил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 82 (15 мг, 0.04 ммоль, 14%) в виде твердого вещества грязновато-белого цвета
1H ЯМР (400 МГц, CDCl3): δ 9.95 (d, J = 2.1 Гц, 1H), 8.78 (d, J = 2.1 Гц, 1H), 7.63 (dd, J = 10.0, 7.3 Гц, 1H), 7.45 (dd, J = 9.5, 6.9 Гц, 1H), 3.76-3.68 (m, 1H), 3.08 (s, 3H), 1.39-1.34 (m, 2H), 0.92-0.87 (m, 2H)
LC-MS: m/z 335 [M+H]+ при 2.23 RT (95.79% чистота)
ВЭЖХ: 96.90%
Пример 84
Схема:
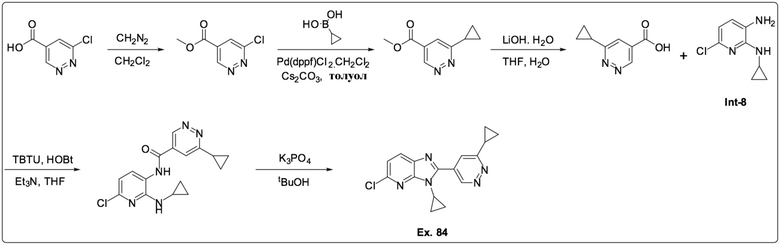
Метил 6-хлорпиридазин-4-карбоксилат
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (1.5 г, 9.49 ммоль) в CH2Cl2 (30 мл) в атмосфере аргона добавили диазометан в диэтиловом простом эфире (свеже полученный путем добавления N-нитрозометилмочевины (3 г) к смеси 50% KOH раствора (25 мл) и диэтилового простого эфира (50 мл) при 0°C) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 30 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и промыли с помощью CH2Cl2 (30 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20-30% EtOAc/гексан) с получением метил 6-хлорпиридазин-4-карбоксилата (1.4 г, 8.13 ммоль, 86%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.58 (d, J = 1.6 Гц, 1H), 8.03 (d, J = 1.8 Гц, 1H), 4.04 (s, 3H)
Метил 6-циклопропилпиридазин-4-карбоксилат
К перемешиваемому раствору метил 6-хлорпиридазин-4-карбоксилата (1 г, 5.81 ммоль) в толуоле (60 мл) добавили циклопропилбороновую кислоту (874 мг, 10.17 ммоль) и карбонат цезия (2.84 г, 8.72 ммоль) в запаянной трубке и смесь продули аргоном в течение 20 мин. Pd(dppf)Cl2.CH2Cl2 (237 мг, 0.290 ммоль) добавили и смесь продули аргоном в течение 5 мин при комнатной температуре. Реакционную смесь нагрели до 100°C и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью EtOAc (40 мл), отфильтровали через слой целита, и слой промыли с помощью EtOAc (25 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением метил 6-циклопропилпиридазин-4-карбоксилата (600 мг, 3.37 ммоль, 58%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.42 (d, J = 2.0 Гц, 1H), 7.72 (d, J = 2.0 Гц, 1H), 4.00 (s, 3H), 2.30-2.22 (m, 1H), 1.30-1.24 (m, 2H), 1.24-1.18 (m, 2H)
LC-MS: m/z 178.9 [M+H]+ при 1.70 RT (96.83% чистота)
6-Циклопропилпиридазин-4-карбоновая кислота
К перемешиваемому раствору метил 6-циклопропилпиридазин-4-карбоксилата (400 мг, 2.24 ммоль) в THF (0.6 мл) добавили раствор гидроксида лития (283 г, 6.74 ммоль) в воде (1 M, 6.7 мл) при комнатной температуре и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Затем остаток подкислили с применением концентрированной HCl (pH 3-4), разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6-циклопропилпиридазин-4-карбоновой кислоты (300 мг, неочищенный) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 14.06 (br s, 1H), 9.31 (d, J = 1.7 Гц, 1H), 7.88 (d, J = 2.3 Гц, 1H), 2.43-2.37 (m, 1H), 1.16-1.11 (m, 4H)
LC-MS: m/z 165.2 [M+H]+ при 2.83 RT (95.00% чистота)
N-(6-Хлор-2-(циклопропиламино)пиридин-3-ил)-6-циклопропилпиридазин-4-карбоксамид
К перемешиваемому раствору 6-циклопропилпиридазин-4-карбоновой кислоты (300 мг, неочищенный) в THF (15 мл) в инертной атмосфере добавили TBTU (1.03 г, 2.74 ммоль) и HOBt (259 мг, 1.92 ммоль) при комнатной температуре, и реакционную смесь перемешивали в течение 10 мин. 6-Хлор-N2-циклопропилпиридин-2, 3-диамин Int-8 (334 мг, 1.82 ммоль) и триэтиламин (0.38 мл, 2.74 ммоль) добавили в реакционную смесь при комнатной температуре и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением N-(6-хлор-2-(циклопропиламино)пиридин-3-ил)-6-циклопропилпиридазин-4-карбоксамида (400 мг, 1.21 ммоль, 54%, за две стадии) в виде густого раствора.
1H ЯМР (500 МГц, DMSO-d6): δ 9.93 (s, 1H), 9.40 (d, J = 1.7 Гц, 1H), 7.89 (d, J = 1.7 Гц, 1H), 7.49 (d, J = 8.1 Гц, 1H), 6.83 (d, J = 2.3 Гц, 1H), 6.66 (d, J = 8.1 Гц, 1H), 2.64-2.62 (m, 1H), 2.41-2.33 (m, 1H), 1.20-1.14 (m, 2H), 1.14-1.09 (m, 2H), 0.73-0.67 (m, 2H), 0.49-0.44 (m, 2H)
LC-MS: m/z 329.9 [M+H]+ при 2.54 RT (89.91% чистота)
5-Хлор-3-циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридин (Пример 84)
К перемешиваемому раствору N-(6-хлор-2-(циклопропиламино)пиридин-3-ил)-6-циклопропилпиридазин-4-карбоксамида (350 мг, 1.06 ммоль) в tBuOH (4 мл) в инертной атмосфере добавили фосфат калия (676 мг, 3.19 ммоль) в запаянной трубке при комнатной температуре. Реакционную смесь нагрели до 110°C и перемешивали в течение 36 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Остаток разбавили водой (50 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 90% EtOAc/Гексан) с получением 5-хлор-3-циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридина Пример 84 (65 мг, 0.20 ммоль, 19%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.59 (d, J = 2.0 Гц, 1H), 8.13-8.08 (m, 2H), 7.42 (d, J = 8.4 Гц, 1H), 3.85-3.77 (m, 1H), 2.46-2.37 (m, 1H), 1.30-1.22 (m, 6H), 0.96-0.89 (m, 2H).
LC-MS: m/z 311.9 [M+H]+ при 2.44 RT (98.03% чистота)
ВЭЖХ: 99.62%
Пример 85
Схема:
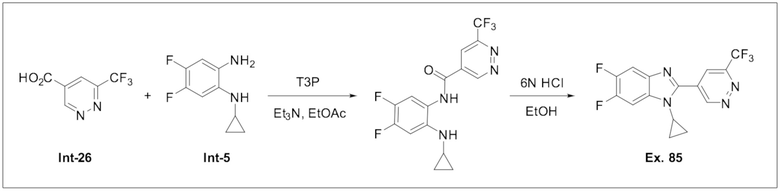
N-(2-(Циклопропиламино)-4,5-дифторфенил)-6-(трифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4,5-дифторбензол-1,2-диамина Int-5 (200 мг, 1.08 ммоль) в EtOAc (12 мл) в инертной атмосфере добавили 6-(трифторметил)пиридазин-4-карбоновую кислоту Int-26 (208 мг, 1.08 ммоль), триэтиламин (0.3 мл, 2.17 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.72 г, 2.71 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-6-(трифторметил)пиридазин-4-карбоксамида (300 мг, 0.83 ммоль, 77%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.18 (s, 1H), 9.91 (d, J = 1.7 Гц, 1H), 8.67 (d, J = 1.7 Гц, 1H), 7.30 (dd, J = 11.0, 8.7 Гц, 1H), 6.93 (dd, J = 13.6, 7.8 Гц, 1H), 6.07 (s, 1H), 2.37-2.34 (m, 1H), 0.78-0.72 (m, 2H), 0.45-0.39 (m, 2H)
LC-MS: m/z 357.0 [M-H]- при 3.17 RT (95.02% чистота)
1-Циклопропил-5,6-дифтор-2-(6-(трифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 85)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-6-(трифторметил)пиридазин-4-карбоксамида (200 мг, 0.55 ммоль) в этаноле (2 мл) в инертной атмосфере добавили 6Н HCl (2.3 мл) при комнатной температуре. Реакционную смесь нагрели до 70°C и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили до pH 8, применяя насыщенного раствора бикарбоната натрия и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20-30% EtOAc/гексан), затем промыли н-пентаном (10 мл) и высушили в вакууме с получением 1-циклопропил-5,6-дифтор-2-(6-(трифторметил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 85 (100 мг, 0.29 ммоль, 53%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 10.07 (d, J = 2.0 Гц, 1H), 8.69 (d, J = 2.0 Гц, 1H), 7.74 (dd, J = 10.1, 7.1 Гц, 1H), 7.66 (dd, J = 10.4, 7.3 Гц, 1H), 3.94-3.89 (m, 1H), 1.32-1.26 (m, 2H), 0.90-0.84 (m, 2H)
LC-MS: m/z 340.9 [M+H]+ при 3.12 RT (97.31% чистота)
ВЭЖХ: 96.06%
Пример 86
Схема:
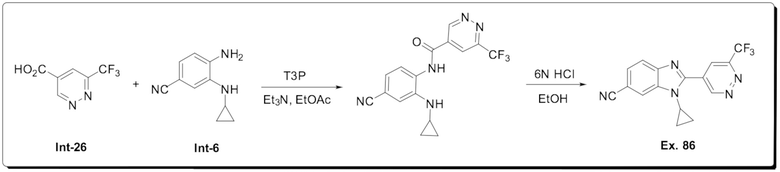
N-(4-Циано-2-(циклопропиламино)фенил)-6-(трифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4-амино-3-(циклопропиламино)бензонитрила Int-6 (200 мг, 1.15 ммоль) в EtOAc (12 мл) в инертной атмосфере добавили 6-(трифторметил) пиридазин-4-карбоновую кислоту Int-26 (221 мг, 1.15 ммоль) и триэтиламин (0.3 мл, 2.31 ммоль) при комнатной температуре. Смесь охладили до 0°C и пропилфосфоновой кислоты ангидрид (T3P, 50% в EtOAc, 1.8 мл, 2.89 ммоль) добавили. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора бикарбоната натрия и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением N-(4-циано-2-(циклопропиламино) фенил)-6-(трифторметил)пиридазин-4-карбоксамида (200 мг) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 346.1 [M-H]- при 2.94 RT (67.16% чистота)
1-Циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 86)
К перемешиваемому раствору N-(4-циано-2-(циклопропиламино)фенил)-6-(трифторметил)пиридазин-4-карбоксамида (200 мг, неочищенный) в EtOH (2 мл) в инертной атмосфере добавили 6 Н HCl (2 мл) при комнатной температуре. Реакционную смесь нагрели до 70°C и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до комнатной температуры, подщелочили насыщенным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением остатка, который далее очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 86 (40 мг, 0.066 ммоль, 21%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 10.10 (d, J = 2.0 Гц, 1H), 8.52 (d, J = 2.0 Гц, 1H), 8.04 (s, 1H), 7.94 (d, J = 8.4 Гц, 1H), 7.64 (dd, J = 8.5, 1.3 Гц, 1H), 3.81-3.74 (m, 1H), 1.46-1.40 (m, 2H), 0.95-0.89 (m, 2H)
LC-MS: m/z 329.9 [M+H]+ при 2.80 RT (99.16% чистота)
ВЭЖХ: 99.42%
Пример 87
Схема:
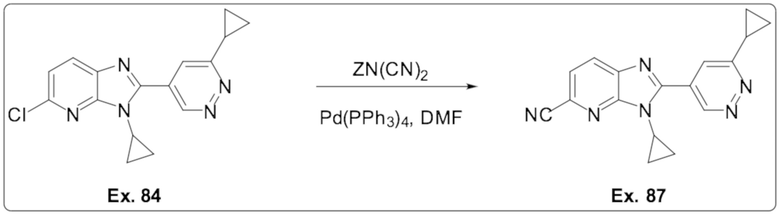
3-Циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрил (Пример 87)
К перемешиваемому раствору 5-хлор-3-циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4, 5-b]пиридина Пример 84 (50 мг, 0.16 ммоль) в DMF (1 мл) добавили цианид цинка (37 мг, 0.32 ммоль) и Pd(PPh3)4 (18.5 мг, 0.01 ммоль) при комнатной температуре в запаянной трубке и смесь продули аргоном в течение 20 мин. Реакционную смесь нагрели до 170°C и перемешивали в течение 9 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 3-циклопропил-2-(6-циклопропилпиридазин-4-ил)-3H-имидазо[4, 5-b] пиридин-5-карбонитрила Пример 87 (40 мг, 0.13 ммоль, 82%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.63 (d, J = 2.0 Гц, 1H), 8.29 (d, J = 8.3 Гц, 1H), 8.16 (d, J = 2.0 Гц, 1H), 7.86 (d, J = 8.3 Гц, 1H), 3.90-3.83 (m, 1H), 2.48-2.41 (m, 1H), 1.32-1.25 (m, 6H), 1.00 - 0.94 (m, 2H)
LC-MS: m/z 303 [M+H]+ при 2.27 RT (93.10% чистота)
ВЭЖХ: 96.03%
Пример 88
Схема:
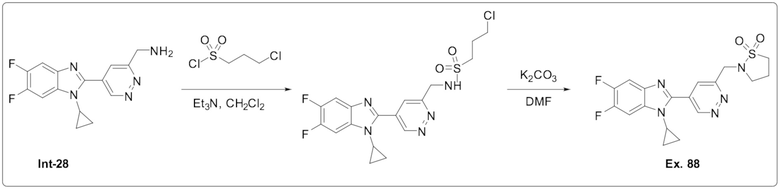
3-Хлор-N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил) метил)пропан-1-сульфонамид
К перемешиваемому раствору (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанамина Int-28 (300 мг, 0.99 ммоль) в CH2Cl2 (10 мл) в инертной атмосфере добавили триэтиламин (0.2 мл, 1.49 ммоль) и 3-хлорпропан-1-сульфонил хлорид (170 мг, 0.99 ммоль) при 0 до 5°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили с помощью CH2Cl2 (60 мл) и промыли водой (40 мл) и солевым раствором (40 мл). Органический слой отделили высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 80% EtOAc/гексан) с получением 3-хлор-N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)пропан-1-сульфонамида (280 мг) в виде бесцветного вязкого сиропа. Неочищенное вещество применяли на следующей стадии без очистки.
LC-MS: m/z 442.1 [M+H]+ при 2.68 RT (90.03% чистота)
2-((5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил) изотиазолидин 1,1-диоксид (Пример 88)
К перемешиваемому раствору 3-хлор-N-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)пропан-1-сульфонамида (200 мг, неочищенный) в DMF (10 мл) в инертной атмосфере добавили карбонат калия (125 мг, 0.90 ммоль) при комнатной температуре и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением 2-((5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метил)изотиазолидин 1,1-диоксида Пример 88 (80 мг, 0.20 ммоль, 44%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.77 (d, J = 1.7 Гц, 1H), 8.28 (d, J = 1.7 Гц, 1H), 7.92-7.82 (m, 2H), 4.59 (s, 2H), 3.92-3.86 (m, 1H), 3.35-3.31 (m, 4H), 2.29 (m, 2H), 1.23-1.17 (m, 2H), 0.79-0.74 (m, 2H)
LC-MS: m/z 406.0 [M+H]+ при 2.39 RT (99.55% чистота)
ВЭЖХ: 97.80%
Пример 90
Схема:
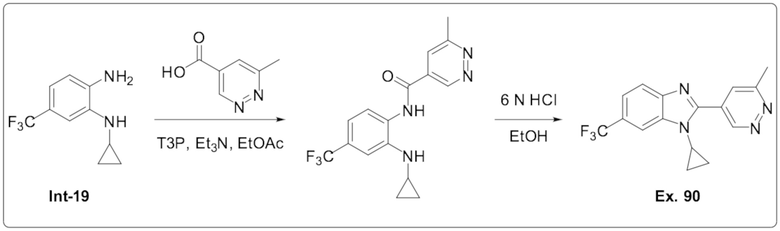
N-(2-(Циклопропиламино)-4-(трифторметил)фенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5-(трифторметил)бензол-1,2-диамина Int-19 (300 мг, неочищенный) и 6-метилпиридазин-4-карбоновой кислоты (266 мг, 1.53 ммоль) в этилацетате (6 мл) добавили триэтиламин (0.39 мл, 2.78 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 2.21 мл, 3.47 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 70% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4-(трифторметил)фенил)-6-метилпиридазин-4-карбоксамида (200 мг, 0.59 ммоль, 42%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.03 (s, 1H), 9.51 (s, 1H), 8.02 (s, 1H), 7.41 (d, J = 8.1 Гц, 1H), 7.23 (s, 1H), 6.98 (br d, J = 8.1 Гц, 1H), 6.24 (s, 1H), 2.74 (s, 3H), 2.43-2.41 (m, 1H), 0.81-0.74 (m, 2H), 0.47-0.45 (m, 2H)
LC-MS: m/z 337.1 [M+H]+ при 2.36 RT (96.19% чистота)
1-Циклопропил-2-(6-метилпиридазин-4-ил)-6-(трифторметил)-1H-бензо[d]имидазол (Пример 90)
К перемешиваемому раствору N-(2-(циклопропиламино)-4-(трифторметил)фенил)-6-метилпиридазин-4-карбоксамида (100 мг, 0.3 ммоль) в этаноле (1 мл) добавили 6 Н HCl (1.5 мл) по каплям при комнатной температуре в инертной атмосфере. Смесь нагрели до 60°C в течение 15 мин на предварительно нагретой масляной бане. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Неочищенное вещество промыли с помощью н-пентана (2 x 5 мл) и высушили в вакууме с получением 1-циклопропил-2-(6-метилпиридазин-4-ил)-6-(трифторметил)-1H-бензо[d]имидазола Пример 90 (40 мг, 0.12 ммоль, 42%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.69 (d, J = 1.9 Гц, 1H), 8.21 (d, J = 2.0 Гц, 1H), 8.06 (s, 1H), 7.97 (d, J = 8.4 Гц, 1H), 7.64 (dd, J = 8.5, 1.4 Гц, 1H), 4.01-3.96 (m, 1H), 2.78 (s, 3H), 1.27-1.21 (m, 2H), 0.81-0.75 (m, 2H)
LC-MS: m/z 319.0 [M+H]+ при 2.61 RT (97.98% чистота)
ВЭЖХ: 97.37%
Пример 91
Схема:
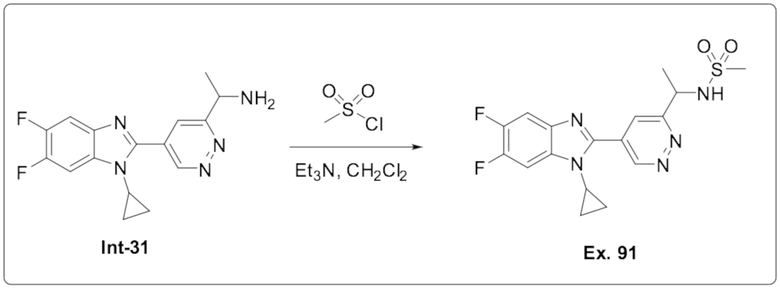
N-(1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этил) метансульфонамид (Пример 91)
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-ил)этан-1-амина Int-30 (70 мг, неочищенный) в CH2Cl2 (3 мл) в инертной атмосфере добавили триэтиламин (0.07 мл, 0.55 ммоль) и метансульфонил хлорид (0.02 мл, 0.33 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 6 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (15 мл) и экстрагировали с помощью CH2Cl2 (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этил)метансульфонамида Пример 91 (36 мг, 0.09 ммоль, 17%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.74 (d, J = 2.1 Гц, 1H), 8.38 (d, J = 2.0 Гц, 1H), 8.02 (d, J = 7.5 Гц, 1H), 7.93-7.82 (m, 2H), 5.00-4.95 (m, 1H), 3.94-3.86 (m, 1H), 2.92 (s, 3H), 1.57 (d, J = 7.0 Гц, 3H), 1.25-1.20 (m, 2H), 0.78-0.75 (m, 2H)
LC-MS: m/z 394.0 [M+H]+ при 2.33 RT (92.93% чистота)
ВЭЖХ: 97.92%
Пример 92 и Пример 94
Схема:
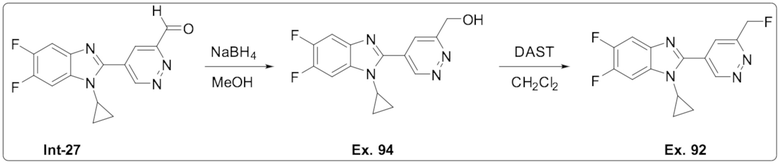
(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанол (Пример 94)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегида Int-27 (100 мг, 0.33 ммоль) в метаноле (5 мл) в инертной атмосфере добавили боргидрид натрия (6.3 мг, 0.16 ммоль) при 0°C. Реакционную смесь перемешивали при той же температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили соляным раствором (10 мл). Летучие соединения удалили при пониженном давлении. Остаток разбавили водой (15 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 70% EtOAc/гексан) с получением (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанола Пример 94 (30 мг, 0.099 ммоль, 30%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.71 (d, J = 2.3 Гц, 1H), 8.28 (d, J = 1.7 Гц, 1H), 7.88-7.79 (m, 2H), 5.77 (t, J = 5.8 Гц, 1H), 4.88 (d, J = 5.8 Гц, 2H), 3.93-3.88 (m, 1H), 1.21-1.15 (m, 2H), 0.77-0.73 (m, 2H)
LC-MS: m/z 302.9 [M+H]+ при 2.39 RT (95.96% чистота)
ВЭЖХ: 96.82%
1-Циклопропил-5,6-дифтор-2-(6-(фторметил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 92)
К перемешиваемому раствору (5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанола Пример 94 (130 мг, 0.43 ммоль) в CH2Cl2 (3 мл) в инертной атмосфере добавили DAST (0.16 мл, 210 мг, 1.29 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором раствор карбоната натрия (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан), которое далее очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-5,6-дифтор-2-(6-(фторметил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 92 (10 мг, 0.03 ммоль, 8%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.80 (d, J = 1.9 Гц, 1H), 8.44 (d, J = 2.1 Гц, 1H), 7.73 (dd, J = 10.1, 7.1 Гц, 1H), 7.64 (dd, J = 10.4, 7.3 Гц, 1H), 5.89 (s, 1H), 5.78 (s, 1H), 3.90-3.85 (m, 1H), 1.31-1.24 (m, 2H), 0.87-0.81 (m, 2H)
LC-MS: m/z 304.9 [M+H]+ при 2.53 RT (99.54% чистота)
ВЭЖХ: 99.73%.
Пример 93
Схема:
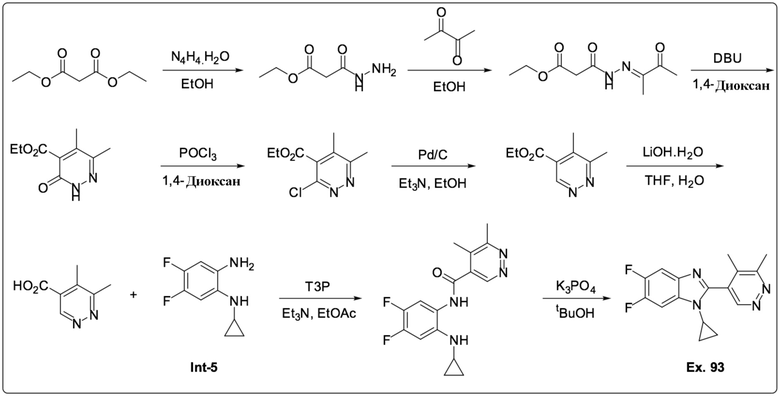
Этил 3-гидразинил-3-оксопропаноат
К перемешиваемому раствору диэтилмалоната (50 г, 312.5 ммоль) в этаноле (32 мл) в инертной атмосфере добавили гидрат гидарзина (5 мл, 103.12 ммоль) при комнатной температуре и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 1% MeOH/CH2Cl2) с получением этил 3-гидразинил-3-оксопропаноата (6 г, 41.09 ммоль, 13%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.16 (brs, 1 H), 4.28 (brs, 2 H), 4.07 (q, J = 7.1 Гц, 2 H), 3.40 (s, 2H), 1.20-1.16 (m, 3H)
LC-MS: m/z 147.2 [M+H]+ при 3.53 RT (89.08% чистота)
Этил (E)-3-оксо-3-(2-(3-оксобутан-2-илиден)гидразинил)пропаноат
К перемешиваемому раствору этил 3-гидразинил-3-оксопропаноата (6 г, 41.09 ммоль) в этаноле (120 мл) в инертной атмосфере добавили диацетил (3.5 г, 41.09 ммоль) при комнатной температуре и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением этил (E)-3-оксо-3-(2-(3-оксобутан-2-илиден) гидразинил) пропаноата (6 г, 28.03 ммоль, 69%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 11.18 (brs, 1H), 4.10 (q, J = 7.1 Гц, 2H), 3.71 (s, 2H), 2.27 (s, 3H), 1.90 (s, 3H), 1.16 (t, J = 7.1 Гц, 3H)
LC-MS: m/z 215 [M+H]+ при 1.69 RT (93.82% чистота)
Этил 5,6-диметил-3-оксо-2,3-дигидропиридазин-4-карбоксилат
К перемешиваемому раствору этил (E)-3-оксо-3-(2-(3-оксобутан-2-илиден) гидразинил) пропаноата (3 г, 14.01 ммоль) в 1,4-диоксане (30 мл) в инертной атмосфере добавили 1,8-диазабицикло(5.4.0)ундец-7-ен (DBU) (2.08 мл, 14.01 ммоль) при комнатной температуре. Реакционную смесь нагрели до 100°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором хлорида аммония (80 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением этил 5,6-диметил-3-оксо-2,3-дигидропиридазин-4-карбоксилата (1.5 г, неочищенный) в виде бесцветного вязкого сиропа. Неочищенное вещество применяли на следующей стадии без очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 12.97 (brs, 1H), 4.30 (q, J = 7.1 Гц, 2H), 2.23 (s, 3H), 2.09 (s, 3H), 1.27 (t, J = 7.0 Гц, 3H)
LC-MS: m/z 196.9 [M+H]+ при 1.50 RT (46.75% чистота)
Этил 3-хлор-5,6-диметилпиридазин-4-карбоксилат
К перемешиваемому раствору этил 5,6-диметил-3-оксо-2,3-дигидропиридазин-4-карбоксилата (1.5 г, неочищенный) в 1,4-диоксане (25 мл) в инертной атмосфере добавили фосфорилтрихлорид (7.3 мл, 76.53 ммоль) при 0°C в запаянной трубке. Реакционную смесь нагрели до 100°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (50 мл) и экстрагировали с помощью EtOAc (2 x 70 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением этил 3-хлор-5,6-диметилпиридазин-4-карбоксилата (250 мг, 1.16 ммоль, 8%, за две стадии) в виде бесцветного вязкого сиропа.
1H ЯМР (400 МГц, DMSO-d6): δ 4.46 (q, J = 7.2 Гц, 2H), 2.63 (s, 3H), 2.28 (s, 3H), 1.34 (t, J = 7.1 Гц, 3H)
LC-MS: m/z 214.9 [M+H]+ при 2.19 RT (97.49% чистота)
Этил 5,6-диметилпиридазин-4-карбоксилат
К перемешиваемому раствору этил 3-хлор-5,6-диметилпиридазин-4-карбоксилата (250 мг, 1.16 ммоль) в этаноле (12 мл) в инертной атмосфере добавили триэтиламин (каталитический) и 10% Pd/C (50% влажность, 120 мг) при комнатной температуре. Реакцию вакуумировалии установили атмосферу водорода (давление баллон), и реакционную смесь перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью этанола (20 мл). Фильтрат концентрировали при пониженном давлении. Полученное неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением этил 5,6-диметилпиридазин-4-карбоксилата (100 мг, 0.55 ммоль, 48%) в виде бесцветного сиропа.
1H ЯМР (500 МГц, DMSO-d6): δ 9.14 (s, 1H), 4.36 (q, J = 7.1 Гц, 2H), 2.66 (s, 3H), 2.44 (s, 3H), 1.33 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 180.9 [M+H]+ при 1.76 RT (98.17% чистота
5,6-Диметилпиридазин-4-карбоновая кислота
К перемешиваемому раствору этил 5,6-диметилпиридазин-4-карбоксилата (100 мг, 0.55 ммоль) в смеси THF: воды (2:1, 2 мл) добавили гидроксид лития моногидрат (47 мг, 1.11 ммоль) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), летучие вещества концентрировали при пониженном давлении. Затем остаток разбавили водой (5 мл) и подкислил с применением концентрированного раствора HCl (~ pH 3-4). Водный слой лиофилизировали с получением 5,6-диметилпиридазин-4-карбоновой кислоты (110 мг, соль) в виде твердого вещества белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, DMSO-d6): δ 9.17 (s, 1H), 2.69 (s, 3H), 2.48 (s, 3H)
LC-MS: m/z 153.2 [M+H]+ при 1.79 RT (99.78% чистота)
N-(2-(Циклопропиламино)-4,5-дифторфенил)-5,6-диметилпиридазин-4-карбоксамид
К перемешиваемому раствору 5,6-диметилпиридазин-4-карбоновой кислоты (67 мг, неочищенный) в EtOAc (5 мл) в инертной атмосфере добавили N1-циклопропил-4, 5-дифторбензол-1,2-диамин Int-5 (80 мг, 0.43 ммоль), триэтиламин (0.24 мл, 1.75 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 0.7 мл, 1.09 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 60% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-5,6-диметилпиридазин-4-карбоксамида (40 мг, 0.12 ммоль, 29%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.76 (s, 1H), 9.18 (s, 1H), 7.51 (dd, J = 11.9, 8.4 Гц, 1H), 6.67 (dd, J = 10.7, 7.2 Гц, 1H), 5.78 (s, 1H), 2.66 (s, 3H), 2.60-2.55 (m, 1H), 2.34 (s, 3H), 1.43-1.34 (m, 2H), 0.44-0.40 (m, 2H)
LC-MS: m/z 318.9 [M+H]+ при 2.90 RT (49.24% чистота).
1-Циклопропил-2-(5,6-диметилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 93)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-5,6-диметилпиридазин-4-карбоксамида (40 мг, 0.125 ммоль) в tBuOH (0.5 мл) в инертной атмосфере добавили трикалия фосфат (66 мг, 0.31 ммоль) при комнатной температуре в запаянной трубке. Реакционную смесь нагрели до 120°C и перемешивали в течение 6 ч. После израсходования исходного вещества (посредством TLC), летучие вещества выпарили при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-2-(5,6-диметилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 93 (18 мг, 0.06 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.18 (s, 1H), 7.71 (dd, J = 10.1, 7.1 Гц, 1H), 7.61 (dd, J = 10.5, 7.2 Гц, 1H), 3.63-3.55 (m, 1H), 2.80 (s, 3H), 2.39 (s, 3H), 1.06-1.00 (m, 2H), 0.72-0.66 (m, 2H)
LC-MS: m/z 300.9 [M+H]+ при 2.34 RT (99.49% чистота)
ВЭЖХ: 99.17%
Пример 95 и Пример 89
Схема:
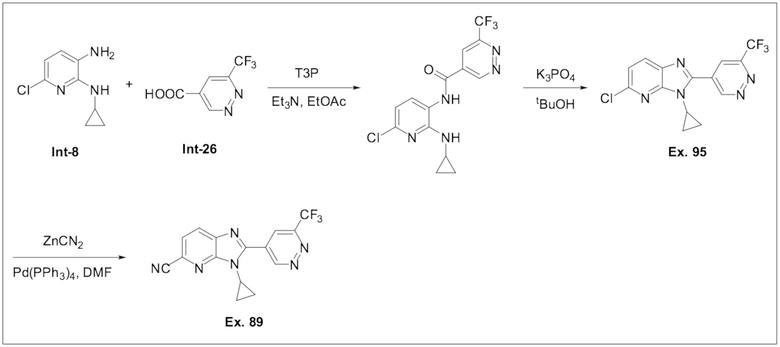
N-(6-Хлор-2-(циклопропиламино)пиридин-3-ил)-6-(трифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлор-N2-циклопропилпиридин-2,3-диамина Int-8 (300 мг, 1.63 ммоль) в EtOAc (10 мл) в инертной атмосфере добавили 6-(трифторметил)пиридазин-4-карбоновую кислоту Int-26 (314 мг, 1.63 ммоль), триэтиламин (0.43 мл, 3.27 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 2.6 мл, 4.09 ммоль) при 0 - 5°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением N-(6-хлор-2-(циклопропиламино)пиридин-3-ил)-6-(трифторметил)пиридазин-4-карбоксамида (250 мг, 0.70 ммоль, 43%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.24 (s, 1H), 9.92 (s, 1H), 8.67 (d, J = 1.7 Гц, 1H), 7.49 (d, J = 8.1 Гц, 1H), 6.87 (d, J = 1.7 Гц, 1H), 6.68 (d, J = 8.1 Гц, 1H), 2.72-2.67 (m, 1H), 0.74-0.69 (m, 2H), 0.49-0.44 (m, 2H)
LC-MS: m/z 357.9 [M+H]+ при 2.87 RT (96.94% чистота)
5-Хлор-3-циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин (Пример 95)
К перемешиваемому раствору N-(6-хлор-2-(циклопропиламино)пиридин-3-ил)-6-(трифторметил)пиридазин-4-карбоксамида (200 мг, 0.56 ммоль) в tBuOH (6 мл) в инертной атмосфере добавили трикалия фосфат (400 мг) при комнатной температуре в запаянной трубке. Реакционную смесь перемешивали при 130°C в течение 24 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 25% EtOAc/гексан) с получением 5-хлор-3-циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридина Пример 95 (150 мг, 0.44 ммоль, 79%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 10.13 (d, J = 2.0 Гц, 1H), 8.76 (d, J = 2.0 Гц, 1H), 8.16 (d, J = 8.4 Гц, 1H), 7.44 (d, J = 8.5 Гц, 1H), 3.91-3.84 (m, 1H), 1.32-1.26 (m, 2H), 1.00-0.95 (m, 2H)
LC-MS: m/z 339.9 [M+H]+ при 2.86 RT (97.81% чистота)
ВЭЖХ: 98.06%
3-Циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрил (Пример 89)
К перемешиваемому раствору 5-хлор-3-циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридина Пример 95 (90 мг, 0.26 ммоль) в DMF (1.5 мл) добавили цианид цинка (62 мг, 0.53 ммоль) и Pd(PPh3)4 (30 мг, 0.02 ммоль) при комнатной температуре в запаянной трубке и смесь продули аргоном в течение 10 мин. Реакционную смесь нагрели до 170°C в течение 8 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью диэтилового простого эфира (2 x 10 мл) и высушили в вакууме. Полученное твердое вещество далее очистили посредством препаративной ВЭЖХ с получением 3-циклопропил-2-(6-(трифторметил)пиридазин-4-ил)-3H-имидазо[4,5-b]пиридин-5-карбонитрила Пример 89 (13 мг, 0.039 ммоль, 15%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 10.16 (d, J = 2.0 Гц, 1H), 8.81 (d, J = 2.0 Гц, 1H), 8.34 (d, J = 8.3 Гц, 1H), 7.87 (d, J = 8.2 Гц, 1H), 3.96-3.88 (m, 1H), 1.35-1.28 (m, 2H), 1.03-0.97 (m, 2H)
LC-MS: m/z 331.2 [M+H]+ при 3.41 RT (99.80% чистота)
ВЭЖХ: 99.55%
Пример 96
Схема:
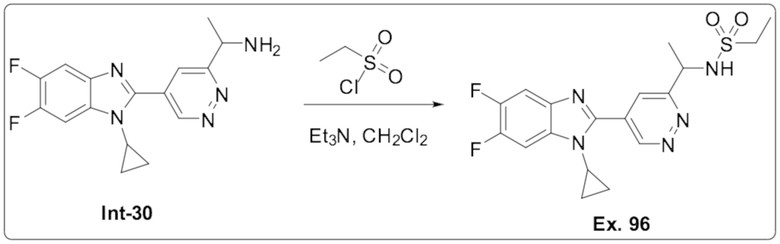
N-(1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этил) этансульфонамид (Пример 96)
К перемешиваемому раствору 1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-амина Int-30 (70 мг, 0.22 ммоль) в CH2Cl2 (5 мл) в инертной атмосфере добавили и триэтиламин (0.07 мл, 0.55 ммоль) и этансульфонил хлорид (43 мг, 0.33 ммоль) при 0°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством препаративной ВЭЖХ с получением N-(1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этил)этансульфонамида (Пример 96) (40 мг, 0.09 ммоль, 44%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.74 (d, J = 2.0 Гц, 1H), 8.39 (d, J = 2.1 Гц, 1H), 8.03 (d, J = 8.0 Гц, 1H), 7.92-7.81 (m, 2H), 4.92 (p, J = 7.3 Гц, 1H), 3.93-3.86 (m, 1H), 3.11-2.90 (m, 2H), 1.57 (d, J = 7.0 Гц, 3H), 1.27-1.20 (m, 2H), 1.16 (t, J = 7.3 Гц, 3H), 0.80-0.72 (m, 2H)
LC-MS: m/z 408.0 [M+H]+ при 2.42 RT (98.97% чистота)
ВЭЖХ: 99.61%
Пример 97
Схема:
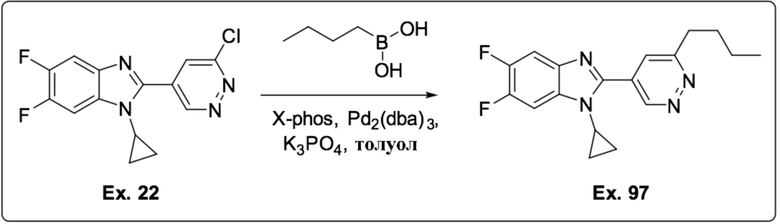
2-(6-Бутилпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол (Пример 97)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (300 мг, 0.98 ммоль) в толуоле (5 мл) добавили X-phos (47 мг, 0.1 ммоль), трикалия фосфат (623 мг, 2.94 ммоль) и бутилбороновую кислоту (148 мг, 1.47 ммоль) в запаянной трубке при комнатной температуре в инертной атмосфере. Реакционную смесь продули аргоном в течение 15 мин. Pd2(dba)3 (90 мг, 0.1 ммоль) добавили при комнатной температуре, и реакционную смесь нагрели до 100°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением остатка, который очистили посредством нормальнофазовой препаративной ВЭЖХ с получением 2-(6-бутилпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 97 (20 мг, 0.06 ммоль, 6%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.63 (d, J = 2.1 Гц, 1H), 8.20 (d, J = 2.1 Гц, 1H), 7.71 (dd, J = 10.1, 7.1 Гц, 1H), 7.62 (dd, J = 10.4, 7.3 Гц, 1H), 3.90-3.84 (m, 1H), 3.15-3.09 (m, 2H), 1.90-1.81 (m, 2H), 1.52-1.43 (m, 2H), 1.29-1.23 (m, 2H), 1.01 (t, J = 7.3 Гц, 3H), 0.85-0.79 (m, 2H)
LC-MS: m/z 329.0 [M+H]+ при 2.99 RT (99.92% чистота)
ВЭЖХ: 99.82%
Пример 98
Схема:
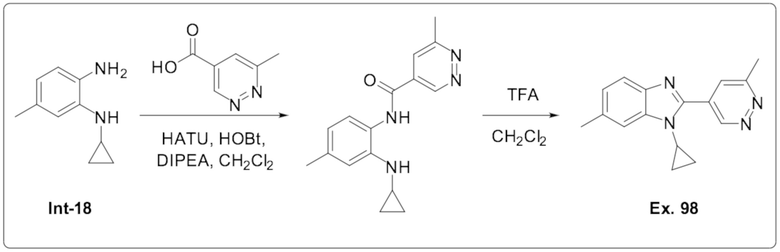
N-(2-(Циклопропиламино)-4-метилфенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5-метилбензол-1,2-диамина Int-18 (120 мг, неочищенный) и 6-метилпиридазин-4-карбоновой кислоты (129 мг, 0.74 ммоль) в CH2Cl2 (5 мл) добавили HATU (310 мг, 0.81 ммоль) и HOBt (110 мг, 0.81 ммоль), а затем этилдиизопропиламин (0.52 мл, 2.96 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(2-(циклопропиламино)-4-метилфенил)-6-метилпиридазин-4-карбоксамида (120 мг, 0.42 ммоль, 57%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.80 (s, 1H), 9.48 (s, 1H), 7.99 (s, 1H), 7.02 (br d, J = 7.7 Гц, 1H), 6.86 (s, 1H), 6.47 (d, J = 7.7 Гц, 1H), 5.63 (brs, 1H), 2.72 (s, 3H), 2.36-2.32 (m, 1H), 2.28 (s, 3H), 0.72-0.69 (m, 2H), 0.43-0.39 (brs, 2H)
LC-MS: m/z 282.9 [M+H]+ при 2.36 RT (94.69% чистота)
1-Циклопропил-6-метил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 98)
К перемешиваемому раствору N-(2-(циклопропиламино)-4-метилфенил)-6-метилпиридазин-4-карбоксамида (120 мг, 0.42 ммоль) в CH2Cl2 (5 мл) добавили трифторуксусную кислоту (0.2 мл) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (5 мл), подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2), а затем препаративной ВЭЖХ с получением 1-циклопропил-6-метил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 98 (25 мг, 0.09 ммоль, 22%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.68 (d, J = 1.9 Гц, 1H), 7.97 (d, J = 1.9 Гц, 1H), 7.70 (d, J = 8.3 Гц, 1H), 7.44-7.41 (m, 1H), 7.18 (dd, J = 8.3, 1.1 Гц, 1H), 3.66-3.61 (m, 1H), 2.84 (s, 3H), 2.56 (s, 3H), 1.31-1.25 (m, 2H), 0.86-0.81 (m, 2H)
LC-MS: m/z 264.9 [M+H]+ при 2.26 RT (99.85% чистота)
ВЭЖХ: 99.84%
Пример 99
Схема:
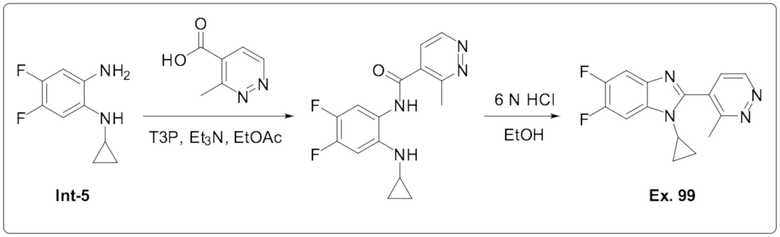
N-(2-(Циклопропиламино)-4,5-дифторфенил)-3-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4,5-дифторбензол-1,2-диамина Int-5 (100 мг, 0.72 ммоль) в этилацетате (6 мл) добавили 3-метилпиридазин-4-карбоновую кислоту (132 мг, 0.72 ммоль) и триэтиламин (0.2 мл, 1.45 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.15 мл, 1.81 ммоль) по каплям при 0°C в инертной атмосфере, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-3-метилпиридазин-4-карбоксамида (100 мг, 0.33 ммоль, 45%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.74 (s, 1H), 9.28 (d, J = 5.0 Гц, 1H), 7.86 (d, J = 5.0 Гц, 1H), 7.48 (dd, J = 11.9, 8.7 Гц, 1H), 6.94 (dd, J = 13.6, 8.0 Гц, 1H), 5.83 (s, 1H), 2.72 (s, 3H), 2.41-2.33 (m, 1H), 0.80-0.71 (m, 2H), 0.46-0.37 (m, 2H)
LC-MS: m/z 305.1[M+H]+ при 2.43 RT (96.65% чистота)
1-Циклопропил-5,6-дифтор-2-(3-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 99)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-3-метилпиридазин-4-карбоксамида (80 мг, 0.26 ммоль) в этаноле (0.8 мл) добавили 6 Н HCl (0.4 мл) по каплям при комнатной температуре в инертной атмосфере. Затем реакционную смесь нагрели до 60°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с помощью насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклопропил-5,6-дифтор-2-(3-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 99 (40 мг, 0.14 ммоль, 53%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.26 (d, J = 5.1 Гц, 1H), 7.63-7.55 (m, 2H), 7.42 (dd, J = 9.6, 7.0 Гц, 1H), 3.41-3.36 (m, 1H), 2.82 (s, 3H), 1.10-1.01 (m, 2H), 0.72-0.62 (m, 2H).
LC-MS: m/z 287.2 [M+H]+ при 3.10 RT (98.57% чистота)
ВЭЖХ: 98.53%
Пример 100
Схема:
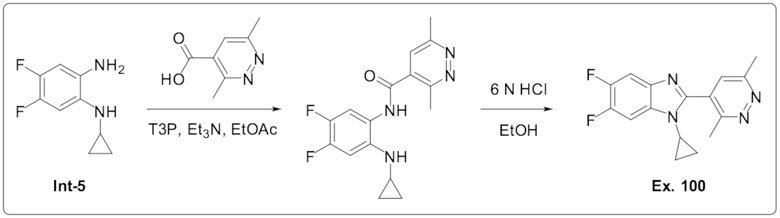
N-(2-(Циклопропиламино)-4,5-дифторфенил)-3,6-диметилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4,5-дифторбензол-1,2-диамина Int-5 (200 мг, 1.09 ммоль) в этилацетате (10 мл) добавили 3,6-диметилпиридазин-4-карбоновую кислоту (165 мг, 1.09 ммоль) и триэтиламин (0.3 мл, 2.17 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.73 мл, 2.71 ммоль) по каплям при 0-5°C в инертной атмосфере, и смесь перемешивали при комнатной температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Вещество объединили с другой партией (100 мг, неочищенный) и очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/EtOAc) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-3,6-диметилпиридазин-4-карбоксамида (260 мг, 0.82 ммоль, 51%) в виде твердого вещества коричневого цвета.
LC-MS: m/z 319.1[M+H]+ при 2.51 RT (98.54% чистота)
1-Циклопропил-2-(3,6-диметилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 100)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-3,6-диметилпиридазин-4-карбоксамида (200 мг, 0.63 ммоль) в этаноле (3 мл) добавили 6 Н HCl (3 мл) по каплям при комнатной температуре в инертной атмосфере. Затем реакционную смесь нагрели до 60°C и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с помощью насыщенного NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Вещество объединили с другой партией (50 мг, неочищенный) и очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением твердого вещества, которое перетерли с EtOAc (5 мл) и высушили в вакууме с получением 1-циклопропил-2-(3,6-диметилпиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 100 (110 мг, 0.37 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 7.87 (s, 1H), 7.71 (dd, J = 10.1, 7.1 Гц, 1H), 7.60 (dd, J = 10.4, 7.3 Гц, 1H), 3.63-3.57 (m, 1H), 2.77 (s, 3H), 2.69 (s, 3H), 1.09-1.02 (m, 2H), 0.73-0.67 (m, 2H)
LC-MS: m/z 300.9 [M+H]+ при 2.35 RT (98.70% чистота)
ВЭЖХ: 99.50%
Пример 101
Схема:
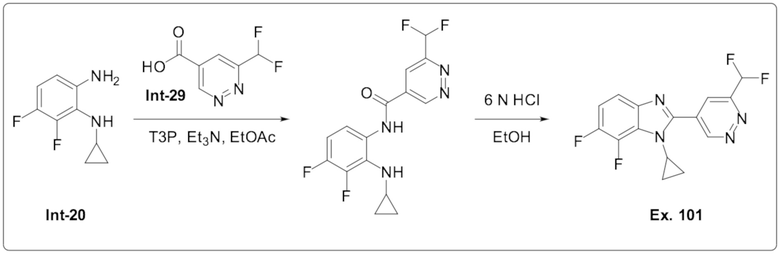
N-(2-(Циклопропиламино)-3,4-дифторфенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5,6-дифторбензол-1,2-диамина Int-20 (250 мг, неочищенный) и 6-(дифторметил)пиридазин-4-карбоновой кислоты Int-29 (260 мг, 1.49 ммоль) в этилацетате (5 мл) добавили триэтиламин (0.38 мл, 2.72 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 2.16 мл, 3.4 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(2-(циклопропиламино)-3,4-дифторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (300 мг, 0.88 ммоль, 65%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.24 (s, 1H), 9.79 (s, 1H), 8.45 (d, J = 1.2 Гц, 1H), 7.56-7.28 (m, 1H), 6.95 (t, J = 6.7 Гц, 1H), 6.68 (q, J = 8.7 Гц, 1H), 5.60 (br s, 1H), 2.82-2.75 (m, 1H), 0.63-0.48 (m, 4H)
LC-MS: m/z 340.9 [M+H]+ при 2.79 RT (92.05% чистота)
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-6,7-дифтор-1H-бензо[d]имидазол (Пример 101)
К перемешиваемому раствору N-(2-(циклопропиламино)-3,4-дифторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (150 мг, 0.44 ммоль) в этаноле (1.5 мл) добавили 6 Н HCl (2.25 мл) по каплям при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали на предварительно нагретой масляной бане при 60°C в течение 15 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 70% EtOAc/гексан) с получением 1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-6,7-дифтор-1H-бензо[d]имидазола Пример 101 (70 мг, 0.22 ммоль, 49%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.97 (d, J = 2.0 Гц, 1H), 8.53 (d, J = 2.0 Гц, 1H), 7.65-7.62 (m, 1H), 7.57-7.29 (m, 2H), 4.20-4.15 (m, 1H), 1.16-1.09 (m, 2H), 0.88-0.84 (m, 2H)
LC-MS: m/z 323.0 [M+H]+ при 2.83 RT (98.57% чистота)
ВЭЖХ: 98.76%
Пример 102
Схема:
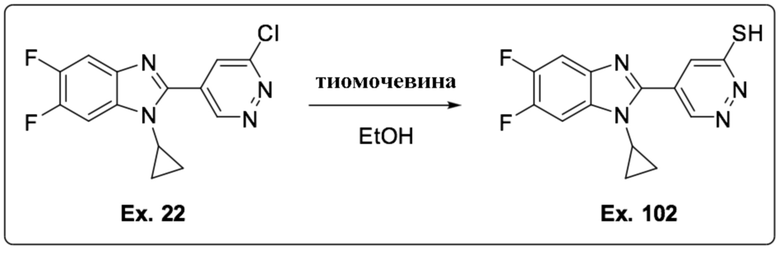
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиол (Пример 102)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (100 мг, 0.33 ммоль) в этаноле (5 мл) добавили тиомочевину (35 мг, 0.49 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до температуры возврата флегмы в течение 4 часов и охладили. Насыщенный раствор гидроксида натрия (2 мл) добавили при комнатной температуре и нагрели до температуры возврата флегмы в течение 1 часа. Реакционную смесь охладили и нейтрализовали до pH ~7 с применением 6 Н HCl. Осажденное твердое вещество отфильтровали, и твердое вещество снова промыли последовательно водой (5 мл), MeOH (5 мл), CH2Cl2 (5 мл), EtOAc (5 мл), Et2O (5 мл) и высушили в вакууме с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиола Пример 102 (50 мг, 0.16 ммоль, 50%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 14.94 (br s, 1H), 8.79 (d, J = 1.8 Гц, 1H), 8.19 (s, 1H), 7.89-7.80 (m, 2H), 3.93-3.83 (m, 1H), 1.22-1.19 (m, 2H), 0.91-0.89 (m, 2H)
LC-MS: m/z 304.9 [M+H]+ при 2.56 RT (94.69% чистота)
ВЭЖХ: 93.13%
Пример 103
Схема:
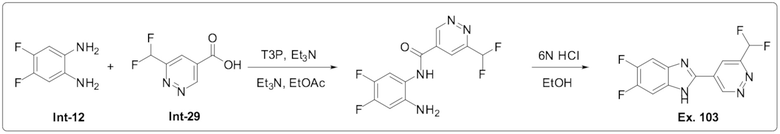
N-(2-Амино-4,5-дифторфенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4, 5-дифторбензол-1,2-диамина Int-12 (199 мг, 1.37 ммоль) в EtOAc (12 мл) в инертной атмосфере добавили 6-(дифторметил)пиридазин-4-карбоновую кислоту Int-29 (200 мг, 1.14 ммоль), триэтиламин (0.32 мл, 2.29 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.83 мл, 2.87 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC и LC-MS), реакционную смесь погасили насыщенным раствором бикарбоната натрия (50 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты промыли водой (70 мл) и солевым раствором (70 мл), высушили над безводным Na2SO4, и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением N-(2-амино-4,5-дифторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (70 мг, 0.23 ммоль, 13%) в виде твердого вещества желтого цвета.
LC-MS: m/z 300.9 [M+H]+ при 2.2 RT (85.12% чистота)
2-(6-(Дифторметил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 103)
К перемешиваемому раствору N-(2-амино-4,5-дифторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (50 мг, 0.16 ммоль) в EtOH (0.75 мл) в инертной атмосфере добавили 6 Н HCl (0.5 мл) при комнатной температуре. Реакционную смесь нагрели до 90°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили раствором бикарбоната натрия (40 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 103 (15 мг, 0.05 ммоль, 23%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 13.88 (brs, 1H), 10.03 (d, J = 2.0 Гц, 1H), 8.58 (d, J = 2.1 Гц, 1H), 7.87-7.81 (m, 2H), 7.55-7.25 (m, 1H)
LC-MS: m/z 283.1 [M+H]+ при 2.12 RT (97.58% чистота)
ВЭЖХ: 98.71%
Пример 104
Схема:
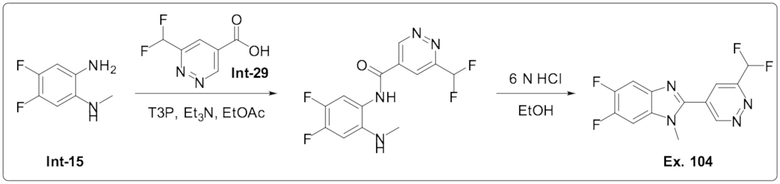
N-(4,5-Дифтор-2-(метиламино)фенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4,5-дифтор-N1-метилбензол-1,2-диамина Int-15 (200 мг, неочищенный) и 6-(дифторметил)пиридазин-4-карбоновой кислоты Int-29 (220 мг, 1.26 ммоль) в этилацетате (20 мл) добавили триэтиламин (0.36 мл, 2.53 ммоль) и пропилфосфоновой кислоты ангидрид (50% in EtOAc, 2 мл, 3.16 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением N-(4,5-дифтор-2-(метиламино)фенил)-6-(дифторметил) пиридазин-4-карбоксамида (210 мг, 0.67 ммоль, 53%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.23 (s, 1H), 9.81 (d, J = 1.7 Гц, 1H), 8.48 (d, J = 1.7 Гц, 1H), 7.55-7.25 (m, 2H), 6.60 (dd, J = 13.6, 7.8 Гц, 1H), 5.68 (br s, 1H), 2.69 (d, J = 4.6 Гц, 3H)
LC-MS: m/z 314.9 [M+H]+ при 2.53 RT (93.02% чистота)
2-(6-(Дифторметил)пиридазин-4-ил)-5,6-дифтор-1-метил-1H-бензо[d]имидазол (Пример 104)
К перемешиваемому раствору N-(4,5-дифтор-2-(метиламино)фенил)-6-(дифторметил) пиридазин-4-карбоксамида (150 мг, 0.48 ммоль) в этаноле (1.5 мл) добавили 6 Н HCl (1.5 мл) при 0°C в инертной атмосфере. Затем реакционную смесь перемешивали на предварительно нагретой масляной бане при 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-5 6-дифтор-1-метил-1H-бензо[d]имидазола Пример 104 (98 мг, 0.33 ммоль, 69%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.89 (s, 1H), 8.44 (d, J = 1.7 Гц, 1H), 7.99 (dd, J = 10.4, 7.5 Гц, 1H), 7.89 (dd, J = 11.0, 7.5 Гц, 1H), 7.54-7.29 (m, 1H), 4.04 (s, 3H)
LC-MS: m/z 296.9 [M+H]+ при 2.46 RT (97.61% чистота)
ВЭЖХ: 97.08%
Пример 105
Схема:
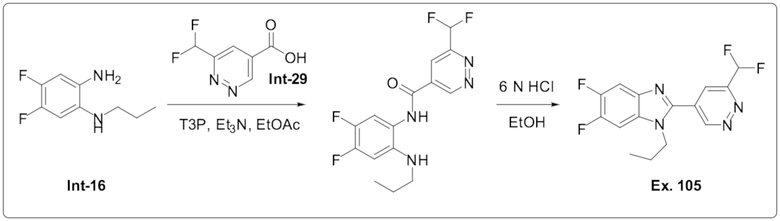
N-(4,5-Дифтор-2-(пропиламино)фенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4,5-дифтор-N1-пропилбензол-1,2-диамина Int-16 (200 мг, неочищенный) и 6-(дифторметил)пиридазин-4-карбоновой кислоты Int-29 (187 мг, 1.07 ммоль) в этилацетате (20 мл) добавили триэтиламин (0.3 мл, 2.15 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.71 мл, 2.69 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением N-(4,5-дифтор-2-(пропиламино)фенил)-6-(дифторметил)пиридазин-4-карбоксамида (200 мг, 0.58 ммоль, 55%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.19 (brs, 1H), 9.82 (d, J = 1.2 Гц, 1H), 8.47 (d, J = 1.7 Гц, 1H), 7.55-7.23 (m, 2H), 6.66 (dd, J = 13.9, 7.5 Гц, 1H), 5.52 (brs, 1H), 3.02 (q, J = 6.6 Гц, 2H), 1.60-1.50 (m, 2H), 0.91 (t, J = 7.5 Гц, 3H)
LC-MS: m/z 342.9 [M+H]+ при 3.00 RT (95.46% чистота)
2-(6-(Дифторметил)пиридазин-4-ил)-5,6-дифтор-1-пропил-1H-бензо[d]имидазол (Пример 105)
К перемешиваемому раствору N-(4,5-дифтор-2-(пропиламино)фенил)-6-(дифторметил)пиридазин-4-карбоксамида (150 мг, 0.44 ммоль) в этаноле (1.5 мл) добавили 6 Н HCl (1.5 мл) при 0°C в инертной атмосфере. Затем реакционную смесь перемешивали на предварительно нагретой масляной бане при 80°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-5,6-дифтор-1-пропил-1H-бензо[d]имидазола Пример 105 (110 мг, 0.34 ммоль, 77%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.85 (d, J = 1.7 Гц, 1H), 8.38 (d, J = 1.7 Гц, 1H), 8.07 (dd, J = 10.7, 7.2 Гц, 1H), 7.89 (dd, J = 11.0, 7.5 Гц, 1H), 7.53-7.28 (m, 1H), 4.42 (t, J = 7.2 Гц, 2H), 1.75-1.68 (m, 2H), 0.77 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 324.9 [M+H]+ при 2.90 RT (97.45% чистота)
ВЭЖХ: 96.79%
Пример 106
Схема:

Диэтил 2-гидрокси-2-(2-оксоциклогексил)малонат
Раствор диэтил 2-охомалоната (500 мг, 2.87 ммоль) и циклогексанона (282 мг, 2.87 ммоль) в запаянной трубке нагрели до 100°C и перемешивали в течение 16 ч и охладили. Реакционную смесь (сироп коричневого цвета), содержащий диэтил 2-гидрокси-2-(2-оксоциклогексил) малонат (500 мг) применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 273.3 [M+H]+ при 2.83 RT (82.91% чистота)
Этил 3-гидрокси-5,6,7,8-тетрагидроциннолин-4-карбоксилат
К перемешиваемому раствору диэтил 2-гидрокси-2-(2-оксоциклогексил) малоната (5 г, неочищенный) в уксусной кислоте (35 мл) добавили гидразина моногидрохлорид (6.25 г, 91.91 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 100°C и перемешивали в течение 3 ч. Развитие реакции контролировали посредством TLC, реакционную смесь погасили с применением насыщенного раствора NaHCO3 (100 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением этил 3-гидрокси-5,6,7,8-тетрагидроциннолин-4-карбоксилата (4 г, 18.0 ммоль, 98%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, CDCl3): δ 4.48 (q, J = 7.1 Гц, 2H), 3.18 (t, J = 6.4 Гц, 2H), 2.81 (t, J = 6.4 Гц, 2H), 1.99-1.92 (m, 2H), 1.89-1.82 (m, 2H), 1.44 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 223.0 [M+H]+ при 1.87 RT (83.82% чистота)
Этил 3-хлор-5,6,7,8-тетрагидроциннолин-4-карбоксилат
К перемешиваемому раствору этил 3-гидрокси-5,6,7,8-тетрагидроциннолин-4-карбоксилата (4 г, 18.0 ммоль) в 1,4-диоксане (40 мл) добавили фосфорилхлорид (16.79 мл, 179.98 ммоль) в запаянной трубке при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 100°C и перемешивали в течение 2 часов. Развитие реакции контролировали посредством TLC, реакционную смесь погасили с применением насыщенного раствора NaHCO3 (100 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением этил 3-хлор-5,6,7,8-тетрагидроциннолин-4-карбоксилата (3.5 г, 14.54 ммоль, 81%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, CDCl3): δ 4.48 (q, J = 7.0 Гц, 2H), 3.17 (t, J = 6.4 Гц, 2H), 2.80 (t, J = 6.7 Гц, 2H), 1.98-1.92 (m, 2H), 1.88-1.82 (m, 2H), 1.43 (t, J = 7.0 Гц, 3H)
LC-MS: m/z 241.0 [M+H]+ при 2.57 RT (99.53% чистота)
Этил 5,6,7,8-тетрагидроциннолин-4-карбоксилат
К перемешиваемому раствору этил 3-хлор-5,6,7,8-тетрагидроциннолин-4-карбоксилата (500 мг, 2.08 ммоль) в этилацетате (10 мл) добавили 10% Pd/C (50% влажность, 150 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 1 часа. Развитие реакции контролировали посредством TLC, реакционную смесь отфильтровали через слой целита, и слой промыли с помощью EtOAc (15 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением этил 5,6,7,8-тетрагидроциннолин-4-карбоксилата (100 мг, 0.48 ммоль, 23%) в виде сиропа коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.26 (s, 1H), 4.41 (q, J = 7.2 Гц, 2H), 3.22 (t, J = 6.5 Гц, 2H), 3.15 (t, J = 6.5 Гц, 2H), 1.98-1.89 (m, 2H), 1.89-1.80 (m, 2H), 1.41 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 206.9 [M+H]+ при 2.12 RT (98.29% чистота)
5,6,7,8-Тетрагидроциннолин-4-карбоновая кислота
К перемешиваемому раствору этил 5,6,7,8-тетрагидроциннолин-4-карбоксилата (20 мг, 0.1 ммоль) в смеси THF/воды (4:1, 1 мл) добавили гидроксид лития моногидрат (12 мг, 0.29 ммоль) при комнатной температуре и перемешивали в течение 2 часов. Затем реакционную смесь лиофилизовали с получением 5,6,7,8-тетрагидроциннолин-4-карбоновой кислоты (25 мг, 0.14 ммоль) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, DMSO-d6): δ 9.24 (s, 1H), 3.14 (t, J = 6.4 Гц, 2H), 3.09 (t, J = 6.4 Гц, 2H), 1.89-1.81 (m, 2H), 1.80-1.72 (m, 2H)
LC-MS: m/z 179.0 [M+H]+ при 2.73 RT (70.16% чистота)
N-(2-(Циклопропиламино)-4,5-дифторфенил)-5,6,7,8-тетрагидроциннолин-4-карбоксамид
К перемешиваемому раствору 5,6,7,8-тетрагидроциннолин-4-карбоновой кислоты (86 мг, неочищенный) в этилацетате (5 мл) добавили N1-циклопропил-4,5-дифторбензол-1,2-диамин Int-5 (88 мг, 0.48 ммоль), триэтиламин (0.27 мл, 1.93 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 0.77 мл, 1.21 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. Развитие реакции контролировали посредством TLC, реакционную смесь погасили с применением насыщенного раствора NaHCO3 (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-5,6,7,8-тетрагидроциннолин-4-карбоксамида (50 мг, 0.14 ммоль, 30%) в виде твердого вещества коричневого цвета.
LC-MS: m/z 343.1 [M-H]- при 2.74 RT (66.07% чистота)
4-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-5,6,7,8-тетрагидроциннолин (Пример 106)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-5,6,7,8-тетрагидроциннолин-4-карбоксамида (50 мг, 0.14 ммоль) в этаноле (0.5 мл) добавили 6 NH HCl (0.2 мл) по каплям при комнатной температуре в инертной атмосфере. Затем реакционную смесь нагрели до 60°C и перемешивали в течение 2 часов. Реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 4-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-5,6,7,8-тетрагидроциннолина Пример 106 (20 мг, 0.06 ммоль, 42%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.20 (s, 1H), 7.71 (dd, J = 10.1, 7.1 Гц, 1H), 7.60 (dd, J = 10.4, 7.3 Гц, 1H), 3.65-3.60 (m, 1H), 3.24 (t, J = 6.6 Гц, 2H), 2.90 (t, J = 6.4 Гц, 2H), 2.07-1.99 (m, 2H), 1.90-1.82 (m, 2H), 1.08-1.02 (m, 2H), 0.73-0.67 (m, 2H)
LC-MS: m/z 327.2 [M+H]+ при 2.22 RT (95.90% чистота)
ВЭЖХ: 96.28%
Пример 107
Схема:
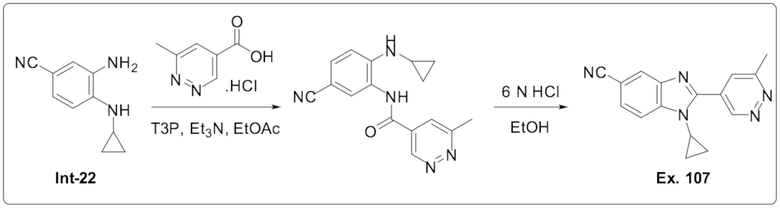
N-(5-Циано-2-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору 3-амино-4-(циклопропиламино)бензонитрила Int-22 (198 мг, неочищенный) и 6-метилпиридазин-4-карбоновой кислоты гидрохлорида (200 мг, 1.15 ммоль) в этилацетате (12 мл) добавили триэтиламин (0.32 мл, 2.29 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.82 мл, 2.86 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 60% EtOAc/гексан) с получением N-(5-циано-2-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамида (400 мг, 1.34 ммоль, 79%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.12 (s, 1H), 9.60 (d, J = 2.0 Гц, 1H), 8.11 (d, J = 2.0 Гц, 1H), 7.71-7.63 (m, 2H), 7.19 (d, J = 8.5 Гц, 1H), 6.83 (s, 1H), 2.84 (s, 3H), 2.57-2.50 (m, 1H), 0.92-0.85 (m, 2H), 0.60-0.55 (m, 2H)
LC-MS: m/z 293.9 [M+H]+ при 2.14 RT (88.98% чистота)
1-Циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол-5-карбонитрил (Пример 107)
К перемешиваемому раствору N-(5-циано-2-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамида (200 мг, 0.68 ммоль) в этаноле (2 мл) добавили 6 Н HCl (3 мл) по каплям при комнатной температуре в инертной атмосфере. Затем реакционную смесь перемешивали на предварительно нагретой масляной бане при 70°C в течение 25 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 до pH ~8 и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 1-2% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол-5-карбонитрила Пример 107 (120 мг, 0.43 ммоль, 64%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.69 (s, 1H), 8.16 (dd, J = 1.4, 0.6 Гц, 1H), 7.98 (d, J = 1.9 Гц, 1H), 7.75 (dd, J = 8.4, 0.6 Гц, 1H), 7.65 (dd, J = 8.4, 1.5 Гц, 1H), 3.74-3.69 (m, 1H), 2.88 (s, 3H), 1.38-1.32 (m, 2H), 0.89-0.83 (m, 2H)
LC-MS: m/z 275.9 [M+H]+ при 2.07 RT (99.17% чистота)
ВЭЖХ: 99.01%
Пример 108
Схема:
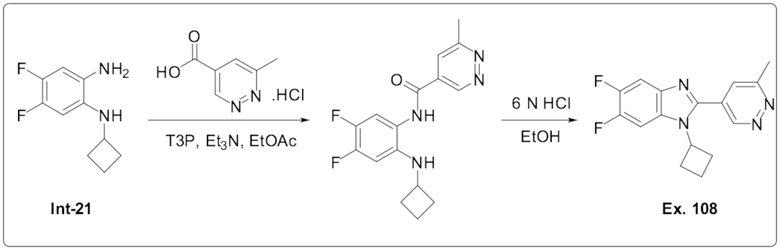
N-(2-(Циклобутиламино)-4,5-дифторфенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклобутил-4,5-дифторбензол-1,2-диамина Int-21 (200 мг, неочищенный) и 6-метилпиридазин-4-карбоновой кислоты гидрохлорида (176 мг, 1.01 ммоль) в этилацетате (10 мл) добавили триэтиламин (0.56 мл, 4.04ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.61 мл, 2.52 ммоль) по каплям при 0°C в инертной атмосфере. Реакцию нагрели до комнатной температуры и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(2-(циклобутиламино)-4,5-дифторфенил)-6-метилпиридазин-4-карбоксамида (250 мг, 0.78 ммоль, 78%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.95 (s, 1H), 9.51 (s, 1H), 8.01 (s, 1H), 7.31 (dd, J = 11.5, 8.8 Гц, 1H), 6.54 (dd, J = 13.7, 7.7 Гц, 1H), 5.58 (d, J = 6.6 Гц, 1H), 3.91-3.79 (m, 1H), 2.74 (s, 3H), 2.40-2.31 (m, 2H), 1.93-1.83 (m, 2H), 1.76-1.66 (m, 2H)
LC-MS: m/z 318.9 [M+H]+ при 2.64 RT (97.50% чистота)
1-Циклобутил-5,6-дифтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 108)
К перемешиваемому раствору N-(2-(циклобутиламино)-4,5-дифторфенил)-6-метилпиридазин-4-карбоксамида (150 мг, 0.47 ммоль) в этаноле (1.5 мл) добавили 6 Н HCl (2.2 мл) по каплям при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 60°C на предварительно нагретой масляной бане в течение 15 минут и охладили. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением 1-циклобутил-5,6-дифтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 108 (100 мг, 0.33 ммоль, 70%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.36 (d, J = 1.1 Гц, 1H), 7.98 (dd, J = 11.0, 7.7 Гц, 1H), 7.88-7.80 (m, 2H), 5.19-5.12 (m, 1H), 2.74 (s, 3H), 2.44-2.42 (m, 4H), 1.89-1.70 (m, 2H).
LC-MS: m/z 300.9 [M+H]+ при 2.51 RT (98.06% чистота)
ВЭЖХ: 98.42%
Пример 109
Схема:

N-(2-(Циклопропиламино)-4-гидроксифенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 4-амино-3-(циклопропиламино)фенола Int-17 (50 мг, неочищенный) и 6-(дифторметил)пиридазин-4-карбоновой кислоты Int-29 (53 мг, 0.3 ммоль) в этилацетате (10 мл) добавили триэтиламин (0.08 мл, 0.61 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 0.48 мл, 0.76 ммоль) по каплям при 0°C в инертной атмосфере. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 до pH ~8 и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4-гидроксифенил)-6-(дифторметил)пиридазин-4-карбоксамида (50 мг, 0.16 ммоль, 51%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.88 (s, 1H), 9.80 (s, 1H), 9.16 (br s, 1H), 8.44 (s, 1H), 7.55-7.26 (m, 1H), 6.89 (d, J = 8.1 Гц, 1H), 6.48 (d, J = 1.7 Гц, 1H), 6.07 (dd, J = 8.4, 2.0 Гц, 1H), 5.71 (s, 1H), 2.30-2.28 (m, 1H), 0.71-0.68 (m, 2H), 0.43-0.39 (m, 2H)
LC-MS: m/z 321.2 [M+H]+ при 1.84 RT (99.05% чистота)
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-ол (Пример 109)
К перемешиваемому раствору N-(2-(циклопропиламино)-4-гидроксифенил)-6-(дифторметил)пиридазин-4-карбоксамида (50 мг, 0.16 ммоль) в этаноле (1 мл) добавили 6 Н HCl (1 мл) по каплям при 0°C в инертной атмосфере. Реакционную смесь нагрели до 70°C на предварительно нагретой масляной бане в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-ола Пример 109 (40 мг, 0.13 ммоль, 85%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.93 (d, J = 2.1 Гц, 1H), 8.54 (d, J = 2.1 Гц, 1H), 7.57 (d, J = 8.8 Гц, 1H), 7.29-7.00 (m, 2H), 6.90 (dd, J = 8.8, 2.3 Гц, 1H), 3.88-3.82 (m, 1H), 1.31-1.22 (m, 2H), 0.86-0.79 (m, 2H)
LC-MS: m/z 302.9 [M+H]+ при 2.01 RT (99.11% чистота)
ВЭЖХ: 98.91%
Пример 110
Схема:

Метил 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилат
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазола Пример 22 (1 г, 2.9 ммоль) в метаноле (50 мл) добавили ацетат натрия (713 мг, 8.69 ммоль) и dppf (80 мг, 0.14 ммоль), а затем Pd(OAc)2 (97 мг, 0.14 ммоль) в стальном сосуде при комнатной температуре. Стальной сосуд заполнили газом СО (200 пси), и реакционную смесь нагрели до 100°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли метанолом (30 мл). Фильтрат концентрировали при пониженном давлении. Остаток разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением метил 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилата (200 мг, 0.6 ммоль, 21%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.03 (d, J = 2.1 Гц, 1H), 8.73 (d, J = 2.1 Гц, 1H), 7.93-7.84 (m, 2H), 4.03 (s, 3H), 4.00-3.96 (m, 1H), 1.23-1.16 (m, 2H), 0.84-0.78 (m, 2H)
LC-MS: m/z 330.9 [M+H]+ при 2.49 RT (94.38% чистота)
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоновая кислота (Пример 110)
К перемешиваемому раствору метил 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил) пиридазин-3-карбоксилата (100 мг, 0.3 ммоль) в смеси THF/воды (3:1, 3 мл) добавили гидроксид лития моногидрат (38 мг, 0.91 ммоль) при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь промыли простым эфиром (10 мл) и органический слой отделили. Водный слой подкислили с применением концентрированной HCl (pH ~2), что привело к образованию осадка. Осажденное твердое вещество отфильтровали, промыли простым диэтиловым эфиром (2 x 5 мл) и высушили в вакууме с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоновой кислоты Пример 110 (40 мг, 0.13 ммоль, 42%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 14.15 (brs, 1H), 10.00 (d, J = 2.1 Гц, 1H), 8.72 (d, J = 2.1 Гц, 1H), 7.93-7.84 (m, 2H), 4.00-3.95 (m, 1H), 1.22-1.16 (m, 2H), 0.83-0.78 (m, 2H)
LC-MS: m/z 316.9 [M+H]+ при 1.55 RT (99.76% чистота)
ВЭЖХ: 99.88%
Пример 111
Схема:
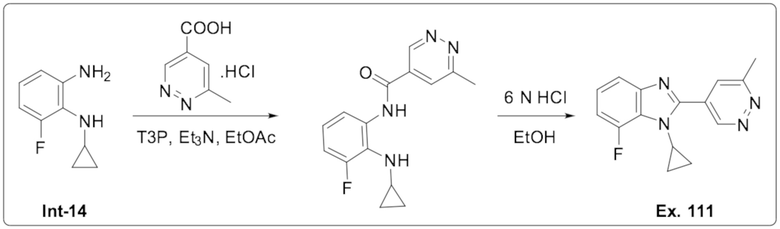
N-(2-(Циклопропиламино)-3-фторфенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-6-фторбензол-1,2-диамина Int-14 (100 мг, неочищенный) и 6-метилпиридазин-4-карбоновой кислоты гидрохлорида (83 мг, 0.6 ммоль) в этилацетате (4 мл) добавили триэтиламин (0.34 мл, 2.41 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 0.95 мл, 1.51 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с применением насыщенного раствора NaHCO3 (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением N-(2-(циклопропиламино)-3-фторфенил)-6-метилпиридазин-4-карбоксамида (200 мг, 0.7 ммоль, 72%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.06 (br s, 1H), 9.47 (s, 1H), 7.98 (s, 1H), 7.05-6.97 (m, 2H), 6.73-6.66 (m, 1H), 5.09 (br s, 1H), 2.72-2.70 (m, 4H), 0.55-0.42 (m, 4H)
LC-MS: m/z 286.9 [M+H]+ при 2.15 RT (91.92% чистота)
1-Циклопропил-7-фтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 111)
К перемешиваемому раствору N-(2-(циклопропиламино)-3-фторфенил)-6-метилпиридазин-4-карбоксамида (200 мг, 0.7 ммоль) в этаноле (3 мл) добавили 6 Н HCl (2 мл) при комнатной температуре в инертной атмосфере. Затем реакционную смесь перемешивали на предварительно нагретой масляной бане при 65°C в течение 15 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором NaHCO3 (30 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3% MeOH/CH2Cl2), а затем промыли с помощью н-пентана (2 x 5 мл) и высушили в вакууме с получением 1-циклопропил-7-фтор-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 111 (110 мг, 0.41 ммоль, 59%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.64 (d, J = 1.7 Гц, 1H), 8.14 (d, J = 2.0 Гц, 1H), 7.58 (d, J = 7.8 Гц, 1H), 7.30-7.17 (m, 2H), 4.11-4.07 (m, 1H), 2.77 (s, 3H), 1.15-1.10 (m, 2H), 0.77-0.75 (m, 2H)
LC-MS: m/z 269.2 [M+H]+ при 2.05 RT (98.96% чистота)
ВЭЖХ: 98.50%
Пример 112
Схема:

N-(5-Хлор-2-(циклопропиламино) фенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору 4-хлор-N1-циклопропилбензол-1,2-диамина Int-24 (200 мг, неочищенный) в этилацетате (8 мл) добавили 6-метилпиридазин-4-карбоновой кислоты гидрохлорид (191 мг, 1.1 ммоль) и триэтиламин (0.61 мл, 4.37 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.74 мл, 2.73 ммоль) по каплям при 0°C в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Эту партию объединили с другой партией (90 мг, неочищенный) и очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением N-(5-хлор-2-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамида (200 мг, 0.66 ммоль, 40%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.94 (s, 1H), 9.49 (d, J = 1.7 Гц, 1H), 8.00 (d, J = 1.9 Гц, 1H), 7.26 (d, J = 2.3 Гц, 1H), 7.19 (dd, J = 8.7, 2.5 Гц, 1H), 7.03 (d, J = 8.7 Гц, 1H), 5.93 (s, 1H), 2.73 (s, 3H), 2.38-2.32 (m, 1H), 0.75-0.70 (m, 2H), 0.45-0.40 (m, 2H)
LC-MS: m/z 302.9 [M+H]+ при 2.55 RT (88.67% чистота)
5-Хлор-1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 112)
К перемешиваемому раствору N-(5-хлор-2-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамида (100 мг, 0.33 ммоль) в этаноле (1 мл) добавили 6 Н HCl (1 мл) по каплям при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 50°C на предварительно нагретой масляной бане в течение 1 часа, охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Эту партию объединили с другой партией (90 мг, неочищенный) и очистили посредством колоночной хроматографии на силикагеле (элюент: 70-80% EtOAc/гексан) с получением 5-хлор-1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 112 (100 мг, 0.35 ммоль, 53%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.66 (d, J = 2.0 Гц, 1H), 8.17 (d, J = 1.9 Гц, 1H), 7.84 (d, J = 1.9 Гц, 1H), 7.75 (d, J = 8.7 Гц, 1H), 7.42 (dd, J = 8.6, 2.0 Гц, 1H), 3.96-3.92 (m, 1H), 2.77 (s, 3H), 1.22-1.17 (m, 2H), 0.77-0.72 (m, 2H)
LC-MS: m/z 284.9 [M+H]+ при 2.42 RT (96.67% чистота)
ВЭЖХ: 96.19%
Пример 113
Схема:
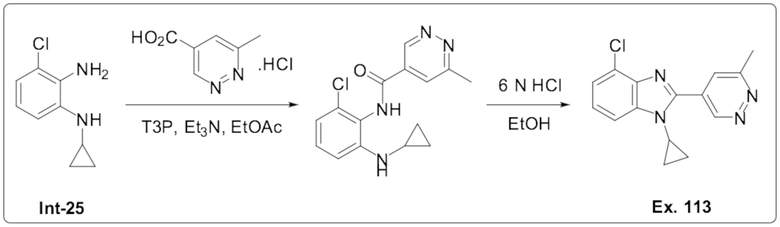
N-(2-Хлор-6-(циклопропиламино) фенил)-6-метилпиридазин-4-карбоксамид
К перемешиваемому раствору 3-хлор-N1-циклопропилбензол-1,2-диамина Int-25 (200 мг, неочищенный) в этилацетате (10 мл) добавили 6-метилпиридазин-4-карбоновой кислоты гидрохлорид (191 мг, 1.1 ммоль) и триэтиламин (0.31 мл, 2.2 ммоль), а затем пропилфосфоновой кислоты ангидрид (50% в EtOAc, 1.75 мл, 2.75 ммоль) по каплям при 0°C в инертной атмосфере, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением N-(2-хлор-6-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамида (50 мг, 0.16 ммоль, 15%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.98 (s, 1H), 9.51 (d, J = 1.9 Гц, 1H), 8.01 (d, J = 2.0 Гц, 1H), 7.19 (t, J = 8.1 Гц, 1H), 6.97 (dd, J = 8.3, 0.9 Гц, 1H), 6.75 (dd, J = 8.0, 1.0 Гц, 1H), 6.16 (s, 1H), 2.74 (s, 3H), 2.35-2.31 (m, 1H), 0.75-0.70 (m, 2H), 0.44-0.40 (m, 2H)
LC-MS: m/z 302.9 [M+H]+ при 2.38 RT (88.61% чистота)
4-Хлор-1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазол (Пример 113)
К перемешиваемому раствору N-(2-хлор-6-(циклопропиламино)фенил)-6-метилпиридазин-4-карбоксамида (50 мг, 0.16 ммоль) в этаноле (0.7 мл) добавили 6 Н HCl (0.3 мл) по каплям при комнатной температуре в инертной атмосфере. Затем реакционную смесь нагрели до 60°C и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с помощью насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 70-80% EtOAc/гексан) с получением 4-хлор-1-циклопропил-2-(6-метилпиридазин-4-ил)-1H-бензо[d]имидазола Пример 113 (30 мг, 0.1 ммоль, 64%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.67 (d, J = 2.0 Гц, 1H), 8.24 (d, J = 2.1 Гц, 1H), 7.75-7.71 (m, 1H), 7.39 (dd, J = 4.6, 0.8 Гц, 2H), 3.91-3.86 (m, 1H), 2.84 (s, 3H), 1.29-1.23 (m, 2H), 0.84-0.79 (m, 2H)
LC-MS: m/z 285.1 [M+H]+ при 2.07 RT (95.04% чистота)
ВЭЖХ: 95.53%
Пример 114
Схема:
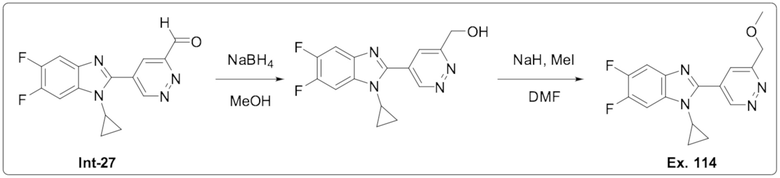
(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанол
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбальдегида Int-27 (400 мг, неочищенный) в метаноле (20 мл) добавили боргидрид натрия (25 мг, 0.67 ммоль) при 0°C в инертной атмосфере, и реакцию перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли с помощью 5% CH2Cl2/н-пентана (10 мл) с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил) метанола (300 мг, 1.0 ммоль, 75%) в виде твердого вещества грязновато-белого цвета.
LC-MS: m/z 302.9 [M+H]+ при 2.10 RT (90.22% чистота)
1-Циклопропил-5,6-дифтор-2-(6-(метоксиметил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 114)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)метанола (150 мг, 0.5 ммоль) в DMF (5 мл) добавили гидрид натрия (55% в минеральном масле, 32 мг, 0.74 ммоль) при 0°C в инертной атмосфере, и смесь перемешивали в течение 15 мин. Иодметан (0.05 мл, 0.74 ммоль) добавили при 0°C, и реакцию перемешивали при комнатной температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 70% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(метоксиметил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 114 (70 мг, 0.22 ммоль, 49%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.77 (d, J = 1.7 Гц, 1H), 8.25 (d, J = 1.7 Гц, 1H), 7.91-7.81 (m, 2H), 4.84 (s, 2H), 3.96-3.92 (m, 1H), 3.44 (s, 3H), 1.21-1.16 (m, 2H), 0.80-0.75 (m, 2H)
LC-MS: m/z 316.9 [M+H]+ при 2.41 RT (96.55% чистота)
ВЭЖХ: 97.19%
Пример 115
Схема:
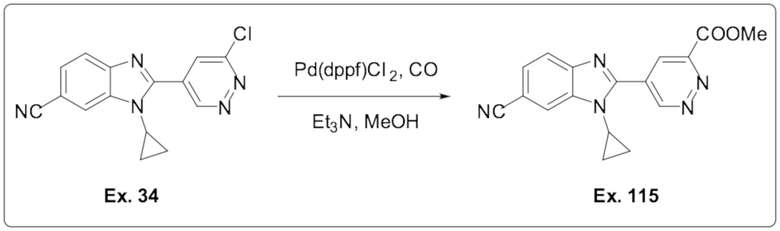
Метил 5-(6-циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилат (Пример 115)
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила Пример 34 (200 мг, 0.68 ммоль) в метаноле (10 мл) добавили триэтиламин (0.19 мл, 1.35 ммоль) и Pd(dppf)Cl2 (99 мг, 0.13 ммоль) в стальном сосуде при комнатной температуре. Стальной сосуд заполнили газом СО (50 пси) и реакционную смесь нагрели до 60°C и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли метанолом (10 мл). Фильтрат концентрировали при пониженном давлении с получением неочищенного вещества, которое очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2), а затем перетерли с CH2Cl2 (1 мл), Et2O (1 мл), и н-пентаном (2 x 5 мл) и наконец высушили в вакууме с получением метил 5-(6-циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилата Пример 115 (140 мг, 0.44 ммоль, 65%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.07 (d, J = 2.1 Гц, 1H), 8.78 (d, J = 2.1 Гц, 1H), 8.36-8.34 (m, 1H), 7.97 (d, J = 8.4 Гц, 1H), 7.73 (dd, J = 8.4, 1.5 Гц, 1H), 4.05-3.99 (m, 4H), 1.24-1.20 (m, 2H), 0.87-0.82 (m, 2H)
LC-MS: m/z 320.2 [M+H]+ при 1.98 RT (95.55% чистота)
ВЭЖХ: 95.69%
Пример 116
Схема:
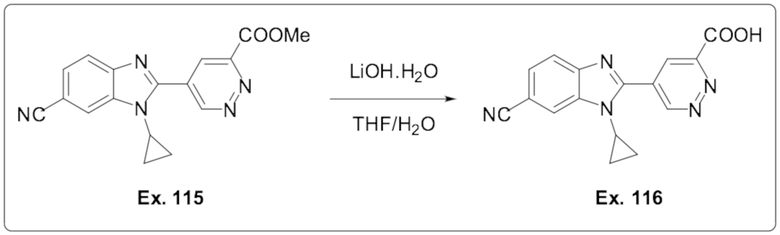
5-(6-Циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоновая кислота (Пример 116)
К перемешиваемому раствору метил 5-(6-циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоксилата Пример 115 (80 мг, 0.25 ммоль) в смеси THF/воды (3:1, 3 мл) добавили гидроксида лития моногидрат (31 мг, 0.75 ммоль) при 0°C. Реакционная смесь превратилась в прозрачный раствор. Через 15-20 мин., реакционная смесь помутнела, и перемешивали при 0°C в течение 1 часа. Реакционную смесь промыли простым эфиром (10 мл) и органический слой отделили. Водный слой подкислили с помощью 6 Н HCl (pH ~2) при 0°C, что дало осаждение твердого вещества, которое отфильтровали, последовательно промыли диэтиловым простым эфиром (2 x 5 мл) и н-пентаном (2 x 5 мл) и высушили в вакууме с получением 5-(6-циано-1-циклопропил-1H-бензо[d]имидазол-2-ил)пиридазин-3-карбоновой кислоты Пример 116 (29 мг, 0.09 ммоль, 38%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 14.18 (br s, 1H), 10.04 (d, J = 2.1 Гц, 1H), 8.77 (d, J = 2.1 Гц, 1H), 8.35-8.33 (m, 1H), 7.97 (d, J = 8.4 Гц, 1H), 7.73 (dd, J = 8.4, 1.5 Гц, 1H), 4.05-3.98 (m, 1H), 1.25-1.18 (m, 2H), 0.87-0.82 (m, 2H)
LC-MS: m/z 306.2 [M+H]+ при 1.75 RT (99.32% чистота)
ВЭЖХ: 98.53%
Пример 117
Схема:

1-Циклопропил-2-(6-(циклопропилметокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 117)
К перемешиваемому раствору циклопропилметанола (0.11 мл, 0.98 ммоль) в DMF (3 мл) добавили гидрид натрия (60% в минеральном масле, 59 мг, 1.47 ммоль) при 0°C в инертной атмосфере и смесь перемешивали в течение 30 мин. 2-(6-Хлорпиридазин-4-ил)-1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол Пример 22 (300 мг, 0.98 ммоль) добавили при 0°C, и реакционную смесь перемешивали при той же температуре в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан), а затем перетерли с н-пентаном (2 x 10 мл) и высушили в вакууме с получением 1-циклопропил-2-(6-(циклопропилметокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 117 (200 мг, 0.58 ммоль, 60%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.41 (d, J = 1.7 Гц, 1H), 7.89-7.81 (m, 2H), 7.77 (d, J = 1.8 Гц, 1H), 4.37 (d, J = 7.2 Гц, 2H), 3.95-3.90 (m, 1H), 1.42-1.30 (m, 1H), 1.21-1.13 (m, 2H), 0.80-0.73 (m, 2H), 0.65-0.59 (m, 2H), 0.44-0.39 (m, 2H)
LC-MS: m/z 343.0 [M+H]+ при 3.07 RT (96.81% чистота)
ВЭЖХ: 97.26%
Пример 118
Схема:
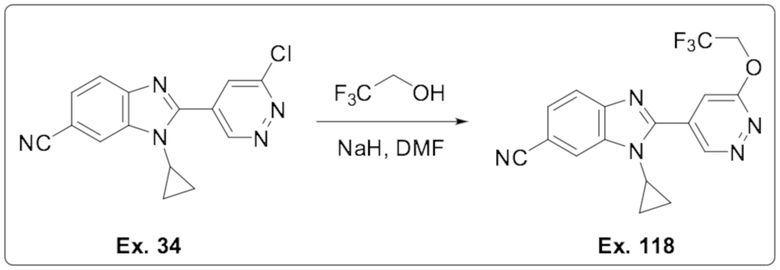
1-Циклопропил-2-(6-(2,2,2-трифторэтокси)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 118)
К перемешиваемому раствору 2,2,2-трифтопентан-1-ола (66 мг, 0.68 ммоль) в DMF (2 мл) добавили гидрид натрия (60% в минеральном масле, 41 мг, 1.02 ммоль) при 0°C в инертной атмосфере и смесь перемешивали при той же температуре в течение 20 мин. Раствор 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила Пример 34 (200 мг, 0.68 ммоль) в DMF (1 мл) добавили при 0°C, и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-2-(6-(2,2,2-трифторэтокси)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 118 (180 мг, 0.5 ммоль, 74%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.58 (d, J = 1.7 Гц, 1H), 8.34 (s, 1H), 8.02 (d, J = 1.7 Гц, 1H), 7.96 (d, J = 8.4 Гц, 1H), 7.72 (dd, J = 8.4, 1.4 Гц, 1H), 5.31 (q, J = 9.0 Гц, 2H), 4.01-3.97 (m, 1H), 1.22-1.17 (m, 2H), 0.85-0.81 (m, 2H)
LC-MS: m/z 360.0 [M+H]+ при 2.86 RT (95.88% чистота)
ВЭЖХ: 96.79%
Пример 119
Схема:
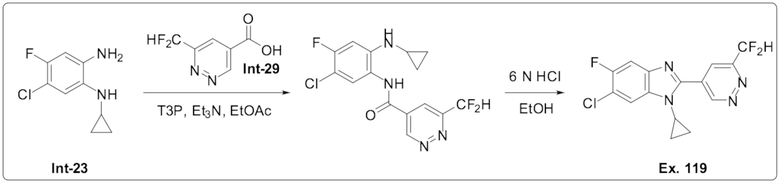
N-(5-Хлор-2-(циклопропиламино)-4-фторфенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 5-хлор-N1-циклопропил-4-фторбензол-1,2-диамин Int-23 (400 мг, 2.0 ммоль) в этилацетате (10 мл) добавили 6-(дифторметил)пиридазин-4-карбоновую кислоту Int-29 (350 мг, 2.0 ммоль), триэтиламин (1.11 мл, 8.0 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 3.18 мл, 5.0 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с применением насыщенного раствора NaHCO3 (20 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Эту партию объединили с другой партией (100 мг, неочищенный) и очистили посредством колоночной хроматографии на силикагеле (элюент: 70% EtOAc/гексан) с получением N-(5-хлор-2-(циклопропиламино)-4-фторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (300 мг, 0.84 ммоль, 34%) в виде твердого вещества желтого цвета.
LC-MS: m/z 357.0 [M+H]+ при 3.08 RT (60.71% чистота)
6-Хлор-1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазол (Пример 119)
К перемешиваемому раствору N-(5-хлор-2-(циклопропиламино)-4-фторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (200 мг, 0.56 ммоль) в этаноле (2 мл) добавили 6 Н HCl (2 мл) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали на предварительно нагретой масляной бане при 50°C в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с применением насыщенного раствора NaHCO3 (30 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 3% MeOH/CH2Cl2) с получением 6-хлор-1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазола Пример 119 (100 мг, 0.29 ммоль, 53%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.00 (d, J = 2.0 Гц, 1H), 8.56 (d, J = 2.1 Гц, 1H), 8.00 (d, J = 6.7 Гц, 1H), 7.89 (d, J = 9.8 Гц, 1H), 7.58-7.28 (m, 1H), 4.02-3.96 (m, 1H), 1.23-1.16 (m, 2H), 0.84-0.78 (m, 2H)
LC-MS: m/z 339.2 [M+H]+ при 3.72 RT (94.81% чистота)
ВЭЖХ: 95.18%Пример 120
Схема:

1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазол-6-карбонитрил (Пример 120)
К перемешиваемому раствору 6-хлор-1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазола Пример 119 (150 мг, 0.44 ммоль) в DMF (1 мл) добавили цианид цинка (104 мг, 0.89 ммоль) в сосуде для обработки микроволнами при комнатной температуре и смесь продули аргоном в течение 20 мин. Pd2(dba)3 (41 мг, 0.04 ммоль) и Pd(dppf)Cl2 (33 мг, 0.04 ммоль) добавили и смесь продули аргоном в течение 5 мин. Реакционную смесь нагрели до 150°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью EtOAc (30 мл). Фильтрат концентрировали при пониженном давлении. Остаток разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-5-фтор-1H-бензо[d]имидазол-6-карбонитрила Пример 120 (50 мг, 0.15 ммоль, 34%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.02 (d, J = 2.0 Гц, 1H), 8.59 (d, J = 2.0 Гц, 1H), 8.47 (d, J = 5.6 Гц, 1H), 7.98 (d, J = 10.0 Гц, 1H), 7.60-7.31 (m, 1H), 4.05-4.00 (m, 1H), 1.23-1.16 (m, 2H), 0.87-0.82 (m, 2H)
LC-MS: m/z 329.9 [M+H]+ при 2.67 RT (99.00% чистота)
ВЭЖХ: 98.49%
Пример 121
Схема:
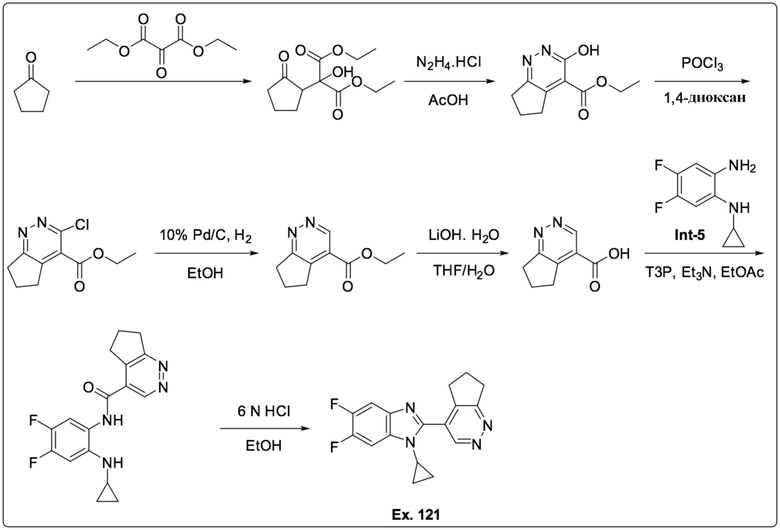
Диэтил 2-гидрокси-2-(2-оксоциклопентил)малонат
Раствор диэтил 2-охомалоната (5 г, 28.73 ммоль) и циклопентанона (2.54 мл, 28.73 ммоль) в запаянной трубке нагрели до 100°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь (сироп коричневого цвета), содержащую диэтил 2-гидрокси-2-(2-оксоциклопентил) малонат (5 г) применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 259.0 [M+H]+ при 2.05 RT (47.02% чистота)
Этил 3-гидрокси-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилат
К перемешиваемому раствору диэтил 2-гидрокси-2-(2-оксоциклопентил) малоната (5 г, неочищенный) в уксусной кислоте (35 мл) добавили гидразина моногидрохлорид (6.59 г, 96.9 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 100°C и перемешивали в течение 3 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с применением насыщенного раствора NaHCO3 (100 мл) и экстрагировали с помощью EtOAc (2 x 60 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением этил 3-гидрокси-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилата (800 мг, 3.84 ммоль, 20%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 12.85 (brs, 1H), 4.26 (q, J = 7.1 Гц, 2H), 2.86 (t, J = 7.4 Гц, 2H), 2.72 (t, J = 7.5 Гц, 2H), 2.05-1.96 (m, 2H), 1.26 (t, J = 7.1 Гц, 3H)
LC-MS: m/z 208.9 [M+H]+ при 1.58 RT (39.74% чистота)
Этил 3-хлор-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилат
К перемешиваемому раствору этил 3-гидрокси-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилата (800 мг, 3.85 ммоль) в 1,4-диоксане (8 мл) добавили фосфорилхлорид (3.59 мл, 38.46 ммоль) в запаянной трубке при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 100°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили с применением насыщенного раствора NaHCO3 (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением этил 3-хлор-6, 7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилата (300 мг, 1.32 ммоль, 34%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3): δ 4.46 (q, J = 7.1 Гц, 2H), 3.25 (t, J = 7.8 Гц, 2H), 3.10 (t, J = 7.6 Гц, 2H), 2.28-2.20 (m, 2H), 1.42 (t, J = 7.2 Гц, 3H)
LC-MS: m/z 227.0 [M+H]+ при 2.25 RT (93.52% чистота)
Этил 6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилат
К перемешиваемому раствору этил 3-хлор-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилата (300 мг, 1.33 ммоль) в этилацетате (8 мл) добавили 10% Pd/C (50% влажность, 120 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и слой целита промыли метанолом (15 мл). Фильтрат концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% MeOH/CH2Cl2) с получением этил 6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилата (100 мг, 0.52 ммоль, 39%) в виде сиропа коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.21 (s, 1H), 4.37 (q, J = 7.0 Гц, 2H), 3.27 (t, J = 7.5 Гц, 2H), 3.18 (t, J = 7.8 Гц, 2H), 2.13-2.07 (m, 2H), 1.35 (t, J = 7.1 Гц, 3H)
LC-MS: m/z 192.9 [M+H]+ при 1.83 RT (92.05% чистота)
6,7-Дигидро-5H-циклопента[c]пиридазин-4-карбоновая кислота
К перемешиваемому раствору этил 6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксилата (100 мг, 0.52 ммоль) в смеси THF/воды (4:1, 2 мл) добавили гидроксида лития моногидрат (66 мг, 1.56 ммоль) при комнатной температуре, и смесь перемешивали в течение 4 ч. Реакционную смесь лиофилизовали с получением 6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоновой кислоты (120 мг, 0.73 ммоль) в виде твердого вещества грязновато-белого цвета. Неочищенное вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (400 МГц, DMSO-d6): δ 9.22 (s, 1H), 3.28 (t, J = 7.6 Гц, 2H), 3.19 (t, J = 7.8 Гц, 2H), 2.14-2.06 (m, 2H)
LC-MS: m/z 165.0 [M+H]+ при 2.03 RT (89.57% чистота)
N-(2-(Циклопропиламино)-4,5-дифторфенил)-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-4,5-дифторбензол-1,2-диамина Int-5 (111 мг, 0.61 ммоль) в этилацетате (6 мл) добавили 6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоновую кислоту (100 мг, неочищенный), триэтиламин (0.34 мл, 2.44 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 0.97 мл, 1.52 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили насыщенным раствором NaHCO3 (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением N-(2-(циклопропиламино)-4,5-дифторфенил)-6,7-дигидро-5H-циклопента[c пиридазин-4-карбоксамида (100 мг, 0.3 ммоль, 50%) в виде твердого вещества коричневого цвета.
LC-MS: m/z 330.9 [M+H]+ при 2.59 RT (38.96% чистота)
4-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-6,7-дигидро-5H-циклопента[c]пиридазин (Пример 121)
К перемешиваемому раствору N-(2-(циклопропиламино)-4,5-дифторфенил)-6,7-дигидро-5H-циклопента[c]пиридазин-4-карбоксамида (75 мг, 0.23 ммоль) в этаноле (0.75 мл) добавили 6 Н HCl (0.75 мл) по каплям при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 50°C и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Эту партию объединили с другой партией (20 мг, неочищенный) и очистили посредством препаративной ВЭЖХ с получением 4-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)-6,7-дигидро-5H-циклопента[c]пиридазина Пример 121 (20 мг, 0.06 ммоль, 28%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.36 (s, 1H), 7.70 (dd, J = 10.0, 7.2 Гц, 1H), 7.61 (dd, J = 10.4, 7.3 Гц, 1H), 3.80-3.75 (m, 1H), 3.36-3.32 (m, 2H), 3.30-3.27 (m, 2H), 2.28-2.21 (m, 2H), 1.18-1.12 (m, 2H), 0.75-0.69 (m, 2H)
LC-MS: m/z 313.0 [M+H]+ при 2.47 RT (99.66% чистота)
ВЭЖХ: 99.76%
Пример 122
Схема:

N-(2-(Циклопропиламино)-4-фторфенил)-6-(дифторметил) пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропил-5-фторбензол-1,2-диамина Int-1 (300 мг, неочищенный) и 6-(дифторметил)пиридазин-4-карбоновой кислоты Int-29 (314 мг, 1.81 ммоль) в этилацетате (10 мл) добавили триэтиламин (0.51 мл, 3.61 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 2.87 мл, 4.52 ммоль) по каплям при 0°C в инертной атмосфере, и реакционную смесь перемешивали при той же температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество промыли н-гексанами (2 x 20 мл) и высушили в вакууме с получением N-(2-(циклопропиламино)-4-фторфенил)-6-(дифторметил)пиридазин-4-карбоксамида (320 мг, 1.0 ммоль, 55%) в виде твердого вещества желтого цвета. Вещество применяли на следующей стадии без дальнейшей очистки.
1H ЯМР (500 МГц, DMSO-d6): δ 10.05 (br s, 1H), 9.79 (d, J = 1.7 Гц, 1H), 8.45 (d, J = 1.7 Гц, 1H), 7.52-7.28 (m, 1H), 7.12 (dd, J = 8.4, 6.4 Гц, 1H), 6.74 (dd, J = 11.9, 2.6 Гц, 1H), 6.41 (td, J = 8.4, 2.9 Гц, 1H), 6.15 (br s, 1H), 2.37-2.31 (m, 1H), 0.76-0.70 (m, 2H), 0.44-0.40 (m, 2H)
LC-MS: m/z 322.9 [M+H]+ при 2.80 RT (99.46% чистота)
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-6-фтор-1H-бензо[d]имидазол (Пример 122)
К перемешиваемому раствору N-(2-(циклопропиламино)-4-фторфенил)-6-(дифторметил) пиридазин-4-карбоксамида 2 (320 мг, 1.0 ммоль) в этаноле (4 мл) добавили 6 Н HCl (4 мл) при 0°C в инертной атмосфере. Реакционную смесь перемешивали на предварительно нагретой масляной бане при 70°C в течение 30 мин. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C; подщелочили с применением насыщенного раствора NaHCO3 (pH ~8) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением 1-циклопропил-2-(6-(дифторметил) пиридазин-4-ил)-6-фтор-1H-бензо[d]имидазола B-680 (250 мг, 0.82 ммоль, 82%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.00 (d, J = 2.1 Гц, 1H), 8.56 (d, J = 2.0 Гц, 1H), 7.82 (dd, J = 8.9, 4.9 Гц, 1H), 7.60-7.28 (m, 2H), 7.24-7.19 (m, 1H), 4.01-3.96 (m, 1H), 1.23-1.15 (m, 2H), 0.83-0.77 (m, 2H)
LC-MS: m/z 304.9 [M+H]+ при 2.73 RT (99.60% чистота)
ВЭЖХ: 99.62%
Пример 123
Схема:
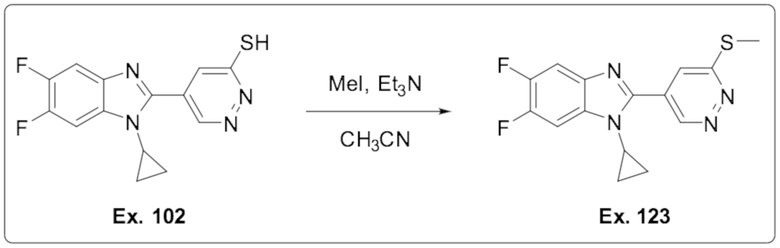
1-Циклопропил-5,6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 123)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиола Пример 102 (100 мг, 0.33 ммоль) в ацетонитриле (3 мл) добавили триэтиламин (0.07 мл, 0.49 ммоль), а затем иодметан (0.02 мл, 0.39 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 24 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(метилтио)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 123 (50 мг, 0.16 ммоль, 48%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.52 (d, J = 1.9 Гц, 1H), 8.17 (d, J = 1.9 Гц, 1H), 7.89-7.82 (m, 2H), 3.95-3.90 (m, 1H), 2.72 (s, 3H), 1.21-1.14 (m, 2H), 0.80-0.74 (m, 2H)
LC-MS: m/z 318.9 [M+H]+ при 2.75 RT (98.20% чистота)
ВЭЖХ: 98.82%
Пример 124
Схема:
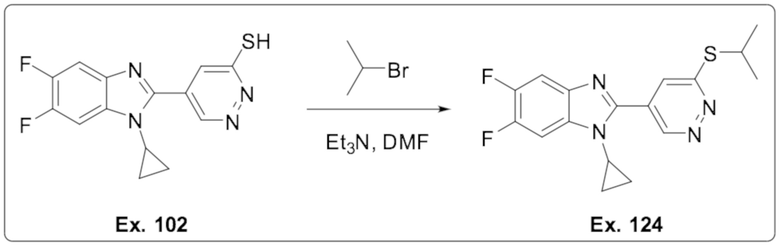
1-Циклопропил-5,6-дифтор-2-(6-(изопропилтио)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 124)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиола Пример 102 (100 мг, 0.33 ммоль) в DMF (3 мл) добавили триэтиламин (0.14 мл, 0.98 ммоль) и 2-бромпропан (61 мг, 0.49 ммоль) при комнатной температуре в инертной атмосфере, и реакционную смесь перемешивали в течение 24 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан), а затем препаративной ВЭЖХ с получением 1-циклопропил-5,6-дифтор-2-(6-(изопропилтио)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 124 (40 мг, 0.11 ммоль, 35%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.52 (d, J = 2.0 Гц, 1H), 8.10 (d, J = 2.0 Гц, 1H), 7.89-7.81 (m, 2H), 4.23-4.16 (m, 1H), 3.94-3.89 (m, 1H), 1.45 (d, J = 6.8 Гц, 6H), 1.19-1.14 (m, 2H), 0.81-0.75 (m, 2H)
LC-MS: m/z 346.9 [M+H]+ при 3.24 RT (98.03% чистота)
ВЭЖХ: 99.28%
Пример 125
Схема:
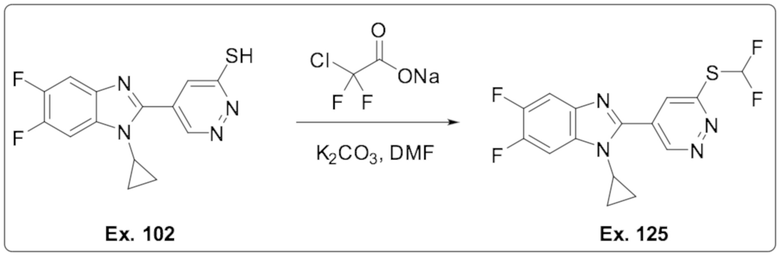
1-Циклопропил-2-(6-((дифторметил)тио)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 125)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиола Пример 102 (100 мг, 0.33 ммоль) в DMF (3 мл) добавили натрия 2-хлор-2,2-дифторацетат (130 мг, 0.66 ммоль) и карбонат калия (68 мг, 0.49 ммоль) в запаянной трубке при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 80°C и перемешивали в течение 18 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили ледяной водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 1-циклопропил-2-(6-((дифторметил)тио)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 125 (40 мг, 0.11 ммоль, 34%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.74 (d, J = 1.8 Гц, 1H), 8.43 (d, J = 1.8 Гц, 1H), 8.32-8.02 (m, 1H), 7.91-7.83 (m, 2H), 3.96-3.88 (m, 1H), 1.23-1.15 (m, 2H), 0.84-0.77 (m, 2H)
LC-MS: m/z 354.9 [M+H]+ при 3.06 RT (98.26% чистота)
ВЭЖХ: 99.31%
Пример 126
Схема:

6-Хлор-N-(2-(циклопропиламино)-4,5-дифторфенил)пиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (200 мг, 1.08 ммоль) в DMF (4 мл) добавили N1-циклопропил-4,5-дифторбензол-1,2-диамин Int-5 (172 мг, 1.08 ммоль), HATU (495 мг, 1.30 ммоль) и диизопропилэтиламин (0.78 мл, 4.35 ммоль) при 0°C в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты промыли водой (20 мл), солевым раствором (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 6-хлор-N-(2-(циклопропиламино)-4,5-дифторфенил)пиридазин-4-карбоксамида (100 мг, 0.30 ммоль, 28%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.50 (s, 1H), 7.95 (d, J = 1.6 Гц, 1H), 7.74 (brs, 1H), 7.37 (dd, J = 10.5, 8.5 Гц, 1H), 7.06 (dd, J = 12.4, 7.7 Гц, 1H), 4.16-4.03 (m, 1H), 2.51-2.40 (m, 1H), 0.83-0.75 (m, 2H), 0.56-0.47 (m, 2H)
5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ол (Пример 126)
К перемешиваемому раствору 6-хлор-N-(2-(циклопропиламино)-4,5-дифторфенил)пиридазин-4-карбоксамида (500 мг, 1.54 ммоль) в EtOH (5 мл) добавили 6 Н HCl (7.5 мл) при 0°C в инертной атмосфере. Реакционную смесь перемешивали при 100°C в течение 3 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ола Пример 126 (300 мг, 1.04 ммоль, 67%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, CD3COOD): δ 8.70 (s, 1H), 7.94 (s, 1H), 7.80 (dd, J = 10.1, 7.2 Гц, 1H), 7.65 (dd, J = 9.9, 7.0 Гц, 1H), 3.82-3.72 (m, 1H), 1.41-1.32 (m, 2H), 1.05-0.90 (m, 2H)
LC-MS: m/z 289.9 [M+H]+ при 2.15 RT (91.69% чистота)
ВЭЖХ: 91.55%
Пример 127
Схема:
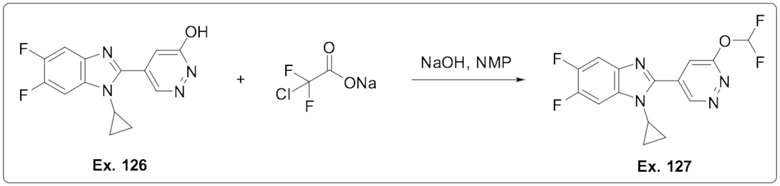
1-Циклопропил-2-(6-(дифторметокси)пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазол (Пример 127)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ола Пример 126 (1 г, 3.47 ммоль) в N-метил-2-пирролидоне (15 мл) добавили гидроксид натрия (972 мг, 24.3 ммоль), а затем натрия 2-хлор-2,2-дифторацетат (1.06 г, 6.94 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 120°C и перемешивали в течение 24 ч. После израсходования исходного вещества (посредством TLC и LCMS), смесь охладили и объединили со второй партией (200 мг). Объединенные реакционные смеси разбавили водой (100 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30% EtOAc/гексан) с получением 1-циклопропил-2-(6-(дифторметокси) пиридазин-4-ил)-5,6-дифтор-1H-бензо[d]имидазола Пример 127 (80 мг, 0.24 ммоль, 6% для двух партий) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 9.69 (d, J = 1.4 Гц, 1H), 8.16-8.00 (m, 2H), 7.93-7.84 (m, 2H), 3.97-3.92 (m, 1H), 1.22-1.17 (m, 2H), 0.83-0.78 (m, 2H)
LC-MS: m/z 338.9 [M+H]+ при 3.00 RT (96.39% чистота)
ВЭЖХ: 96.85%
Пример 128
Схема:
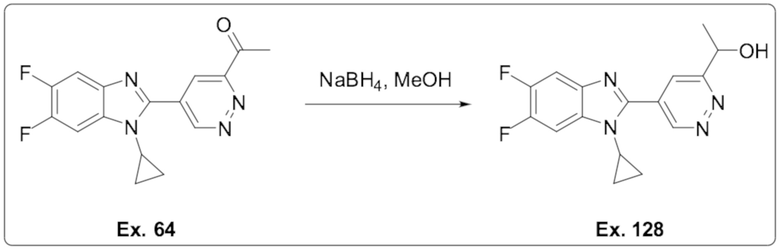
1-(5-(1-Циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-ол (Пример 128)
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-она Пример 64 (300 мг, 0.95 ммоль) в метаноле (12 мл) добавили боргидрид натрия (36 мг, 0.95 ммоль) при 0°C в инертной атмосфере и смесь перемешивали при той же температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток разбавили водой (20 мл) и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40% EtOAc/гексан) с получением 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-ола Пример 128 (150 мг, 0.47 ммоль, 50%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.71 (d, J = 2.3 Гц, 1H), 8.34 (d, J = 2.1 Гц, 1H), 7.90-7.81 (m, 2H), 5.78 (d, J = 4.9 Гц, 1H), 5.15-5.07 (m, 1H), 3.96-3.91 (m, 1H), 1.52 (d, J = 6.5 Гц, 3H), 1.24-1.16 (m, 2H), 0.79-0.73 (m, 2H)
LC-MS: m/z 317.0 [M+H]+ при 2.17 RT (99.24% чистота)
ВЭЖХ: 98.70%
Пример 129
Схема:
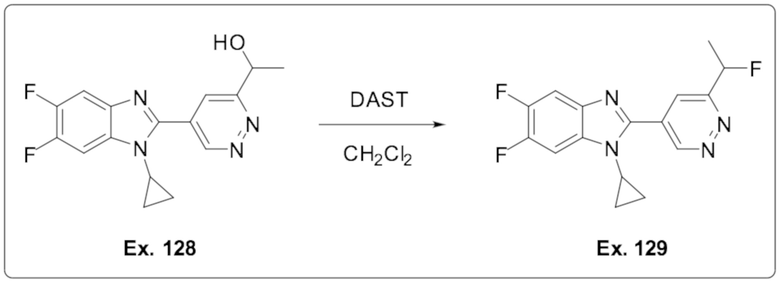
1-Циклопропил-5,6-дифтор-2-(6-(1-фторэтил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 129)
К перемешиваемому раствору 1-(5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-ил)этан-1-ола Пример 128 (100 мг, 0.32 ммоль) в CH2Cl2 (3 мл) добавили диэтиламиносеры трифторид (0.06 мл, 0.47 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (20 мл), нейтрализовали с применением насыщенного раствора карбоната натрия и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Неочищенное вещество объединили с другой партией неочищенного вещества (25 мг), и объединенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-(1-фторэтил)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 129 (40 мг, 0.12 ммоль, 32% для двух партий) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 9.82 (d, J = 2.1 Гц, 1H), 8.37 (d, J = 2.1 Гц, 1H), 7.91-7.83 (m, 2H), 6.22-6.02 (m, 1H), 4.00-3.94 (m, 1H), 1.84-1.75 (m, 3H), 1.21-1.15 (m, 2H), 0.80-0.75 (m, 2H)
LC-MS: m/z 318.9 [M+H]+ при 2.69 RT (98.49% чистота)
ВЭЖХ: 97.92%
Пример 130
Схема:
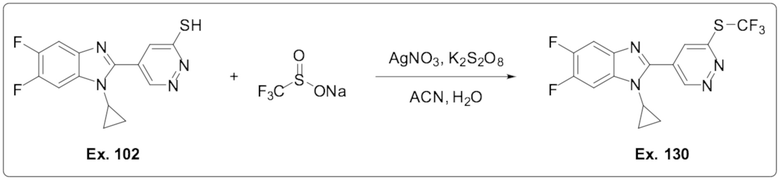
1-Циклопропил-5,6-дифтор-2-(6-((трифторметил)тио)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 130)
К перемешиваемому раствору 5-(1-циклопропил-5,6-дифтор-1H-бензо[d]имидазол-2-ил)пиридазин-3-тиола Пример 102 (200 мг, 0.66 ммоль) в ацетонитриле/воде (1:1, 3 мл) добавили натрия трифторметансульфинат (205 мг, 1.31 ммоль), калия моноперсульфат (355 мг, 1.31 ммоль), и нитрат серебра (11 мг, 0.06 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 80°C и перемешивали в течение 18 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в воду (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 15% EtOAc/гексан) с получением 1-циклопропил-5,6-дифтор-2-(6-((трифторметил)тио)пиридазин-4-ил)-1H-бензо[d]имидазола Пример 130 (75 мг, 0.2 ммоль, 31%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.85 (d, J = 2.0 Гц, 1H), 8.34 (d, J = 1.9 Гц, 1H), 7.62 (dd, J = 10.0, 7.3 Гц, 1H), 7.44 (dd, J = 9.5, 6.9 Гц, 1H), 3.69-3.63 (m, 1H), 1.40-1.33 (m, 2H), 0.92-0.86 (m, 2H)
LC-MS: m/z 373.0 [M+H]+ при 3.21 RT (98.19% чистота)
ВЭЖХ: 98.55%
Пример 131 и Пример 132
Схема:
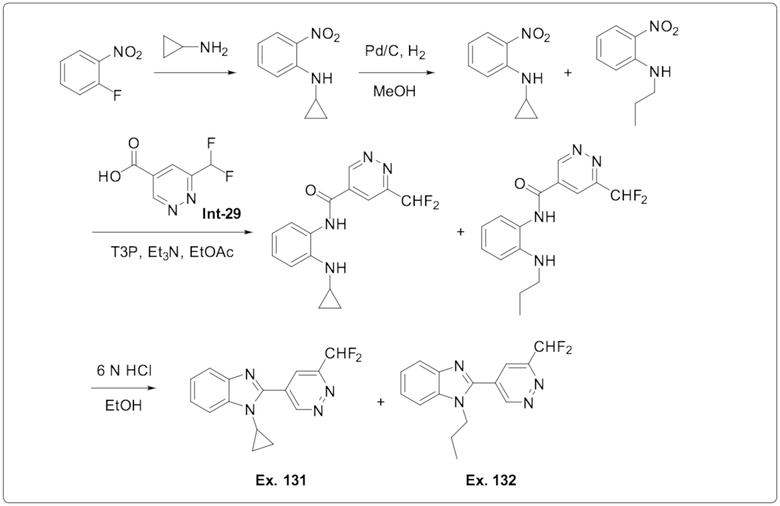

N-Циклопропил-2-нитроанилин
К 1-фтор-2-нитробензолу (1 г, 7.09 ммоль) добавили циклопропанамин (1.47 мл, 21.28 ммоль) по каплям при комнатной температуре в инертной атмосфере, и смесь перемешивали в течение 24 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь разбавили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 40 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2% EtOAc/гексан) с получением N-циклопропил-2-нитроанилина (720 мг, 4.04 ммоль, 60%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 8.15 (dd, J = 8.6, 1.4 Гц, 1H), 8.07 (brs, 1H), 7.50-7.44 (m, 1H), 7.32 (dd, J = 8.6, 1.2 Гц, 1H), 6.72-6.67 (m, 1H), 2.62-2.55 (m, 1H), 0.94-0.89 (m, 2H), 0.69-0.64 (m, 2H)
LC-MS: m/z 179.0 [M+H]+ при 3.11 RT (99.64% чистота)
N 1-Циклопропилбензол-1,2-диамин и N1-пропилбензол-1,2-диамин
К перемешиваемому раствору N-циклопропил-2-нитроанилина (700 мг, 3.93 ммоль) в метаноле (10 мл) добавили 10% Pd/C (50% влажность, 300 мг) при комнатной температуре в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (давление баллон) в течение 5 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита и слой целита промыли метанолом (15 мл). Фильтрат концентрировали при пониженном давлении с получением смеси N1-циклопропилбензол-1,2-диамина и N1-пропилбензол-1,2-диамина (400 мг) в виде вязкого сиропа коричневого цвета. Смесь применяли на следующей стадии без дальнейшей очистки.
N-(2-(Циклопропиламино)фенил)-6-(дифторметил)пиридазин-4-карбоксамид и 6-(дифторметил)-N-(2-(пропиламино)фенил)пиридазин-4-карбоксамид
К перемешиваемому раствору N1-циклопропилбензол-1,2-диамина и N1-пропилбензол-1,2-диамина (400 мг, смесь) в этилацетате (10 мл) добавили 6-(дифторметил) пиридазин-4-карбоновую кислоту Int-29 (470 мг, 2.7 ммоль) и триэтиламин (0.75 мл, 5.4 ммоль), а затем по каплям добавили пропилфосфоновой кислоты ангидрид (50% в EtOAc, 3.44 мл, 5.4 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 5 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с помощью насыщенного раствора NaHCO3 до pH ~8 и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 40-50% EtOAc/гексан) с получением смеси N-(2-(циклопропиламино) фенил)-6-(дифторметил) пиридазин-4-карбоксамида 5 и 6-(дифторметил)-N-(2-(пропиламино) фенил) пиридазин-4-карбоксамида 6 (300 мг, 37%) в виде твердого вещества бледно-желтого цвета. Смесь применяли на следующей стадии без дальнейшей очистки.
LC-MS: m/z 305.0 [M+H]+ при 2.63 RT (51.82% чистота) и m/z 306.9 [M+H]+ при 2.74 RT (41.52% чистота)
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазол (Пример 131) и 2-(6-(дифторметил)пиридазин-4-ил)-1-пропил-1H-бензо[d]имидазол (Пример 132)
К перемешиваемому раствору смеси N-(2-(циклопропиламино)фенил)-6-(дифторметил)пиридазин-4-карбоксамида и 6-(дифторметил)-N-(2-(пропиламино)фенил)пиридазин-4-карбоксамида (300 мг, смесь) в этаноле (10 мл) добавили 6 Н HCl (6 мл) по каплям при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 50°C и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь охладили до 0°C, подщелочили с помощью насыщенного раствора NaHCO3 до pH ~8 и экстрагировали с помощью EtOAc (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества. Неочищенное вещество очистили посредствам нормальнофазовой препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-бензо[d]имидазола (Пример 131) (60 мг, 0.21 ммоль) и 2-(6-(дифторметил)пиридазин-4-ил)-1-пропил-1H-бензо[d]имидазола (Пример 132) (40 мг, 0.14 ммоль) в виде твердых веществ бледно-желтого цвета, соответственно.
Аналитические данные для примера 131:
1H ЯМР (400 МГц, DMSO-d6): δ 10.03 (d, J = 2.0 Гц, 1H), 8.57 (d, J = 2.0 Гц, 1H), 7.82-7.74 (m, 2H), 7.45-7.28 (m, 3H), 4.04-3.99 (m, 1H), 1.24-1.17 (m, 2H), 0.82-0.77 (m, 2H).
LC-MS: m/z 286.9 [M+H]+ при 2.52 RT (98.41% чистота)
ВЭЖХ: 99.92%
Аналитические данные для примера 132:
1H ЯМР (400 МГц, DMSO-d6): δ 9.88 (d, J = 2.1 Гц, 1H), 8.39 (d, J = 2.1 Гц, 1H), 7.82-7.77 (m, 2H), 7.43-7.27 (m, 3H), 4.44 (t, J = 7.5 Гц, 2H), 1.81-1.70 (m, 2H), 0.78 (t, J = 7.4 Гц, 3H)
LC-MS: m/z 288.9 [M+H]+ при 2.61 RT (99.03% чистота)
ВЭЖХ: 99.86%
Пример 133
Схема:
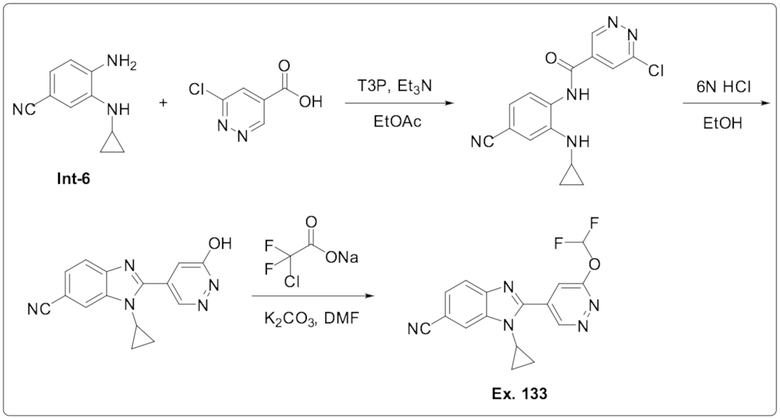
6-Хлор-N-(4-циано-2-(циклопропиламино)фенил)пиридазин-4-карбоксамид
К перемешиваемому раствору 6-хлорпиридазин-4-карбоновой кислоты (458 мг, 2.89 ммоль) в этилацетате (30 мл) добавили 4-амино-3-(циклопропиламино)бензонитрил Int-6 (500 мг, 2.89 ммоль), триэтиламин (0.8 мл, 5.78 ммоль) и пропилфосфоновой кислоты ангидрид (50% в EtOAc, 4.6 мл, 7.22 ммоль) при 0°C в инертной атмосфере. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь перелили в насыщенный раствор бикарбоната натрия (70 мл) и экстрагировали с помощью EtOAc (2 x 100 мл). Объединенные органические экстракты промыли водой (120 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 10% EtOAc/гексан) с получением 6-хлор-N-(4-циано-2-(циклопропиламино)фенил)пиридазин-4-карбоксамида (650 мг, 2.07 ммоль, 72%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 10.17 (s, 1H), 9.63 (d, J = 1.8 Гц, 1H), 8.38 (d, J = 1.8 Гц, 1H), 7.42 (d, J = 8.0 Гц, 1H), 7.31 (d, J = 1.8 Гц, 1H), 7.10 (dd, J = 8.0, 1.9 Гц, 1H), 6.31 (s, 1H), 2.45-2.38 (m, 1H), 0.82-0.77 (m, 2H), 0.48-0.43 (m, 2H)
LC-MS: m/z 312.1 [M-H]- при 2.57 RT (80.47% чистота)
1-Циклопропил-2-(6-гидроксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 6-хлор-N-(4-циано-2-(циклопропиламино)фенил)пиридазин-4-карбоксамида (1 г, 3.19 ммоль) в этаноле (15 мл) добавили 6 Н HCl (22.5 мл) при комнатной температуре в инертной атмосфере. Реакционную смесь нагрели до 60°C и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили до pH ~8 с применением ледяного насыщенного раствора бикарбоната натрия и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 2-5% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-гидроксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (500 мг, 1.8 ммоль, 56%) в виде твердого вещества коричневого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 13.36 (br s, 1H), 8.41 (d, J = 2.3 Гц, 1H), 8.29 (s, 1H), 7.92 (d, J = 8.7 Гц, 1H), 7.72-7.67 (m, 1H), 7.54 (s, 1H), 3.91-3.84 (m, 1H), 1.26-1.19 (m, 2H), 0.94-0.87 (m, 2H)
LC-MS: m/z 277.9 [M+H]+ при 2.10 RT (91.16% чистота)
1-Циклопропил-2-(6-(дифторметокси)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 133)
К перемешиваемому раствору 1-циклопропил-2-(6-гидроксипиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (350 мг, 1.26 ммоль) в DMF (3.5 мл) добавили натрия 2-хлор-2,2-дифторацетат (290 мг, 1.89 ммоль) и карбонат калия (349 мг, 2.53 ммоль) в запаянной трубке при комнатной температуре в инертной атмосфере. Сосуд запаяли, и реакционную смесь нагрели до 90°C и перемешивали в течение 32 ч. Реакционную смесь охладили, погасили водой (30 мл) и экстрагировали с помощью EtOAc (2 x 30 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 30-40% EtOAc/гексан), а затем препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(дифторметокси)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 133 (5 мг, 0.01 ммоль, 1%) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.72 (d, J = 1.8 Гц, 1H), 8.03-8.01 (m, 1H), 7.98-7.77 (m, 3H), 7.64-7.61 (m, 1H), 3.73-3.68 (m, 1H), 1.45-1.38 (m, 2H), 0.95-0.89 (m, 2H)
LC-MS: m/z 328.2 [M+H]+ при 2.33 RT (99.76% чистота)
ВЭЖХ: 99.44%
Пример 134 и Пример 135
Схема:
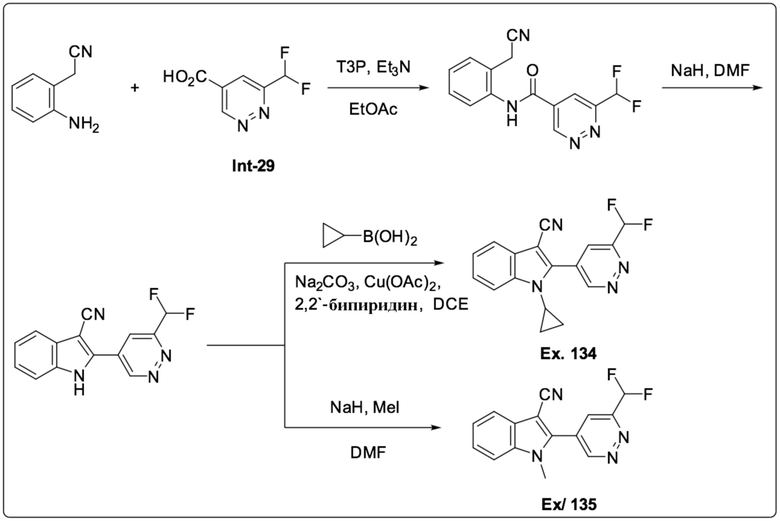
N-(2-(Цианометил)фенил)-6-(дифторметил)пиридазин-4-карбоксамид
К перемешиваемому раствору 6-(дифторметил)пиридазин-4-карбоновой кислоты Int-29 (1 г, 5.75 ммоль) в этилацетате (60 мл) добавили 2-(2-аминофенил)ацетонитрил (759 мг, 5.75 ммоль) и триэтиламин (1.6 мл, 11.49 ммоль) при комнатной температуре в инертной атмосфере. К этому добавили пропилфосфоновой кислоты ангидрид (50% в EtOAc, 9.14 мл, 14.37 ммоль) по каплям при 0°C. Реакционную смесь постепенно нагрели до комнатной температуры и перемешивали в течение 2 часов. После израсходования исходного вещества (посредством TLC), реакционную смесь подщелочили с применением насыщенного раствора бикарбоната натрия до pH ~8 и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный материал перетерли с н-пентаном (2 x 25 мл) и высушили в вакууме с получением N-(2-(цианометил)фенил)-6-(дифторметил)пиридазин-4-карбоксамида (1.4 г, 4.86 ммоль, 85%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, DMSO-d6): δ 10.77 (s, 1H), 9.80 (s, 1H), 8.45 (s, 1H), 7.54-7.49 (m, 1H), 7.45-7.30 (m, 4H), 4.02 (s, 2H)
LC-MS: m/z 288.9 [M+H]+ при 2.25 RT (98.33% чистота)
2-(6-(Дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрил
К перемешиваемому раствору N-(2-(цианометил)фенил)-6-(дифторметил)пиридазин-4-карбоксамида (1 г, 3.47 ммоль) в DMF (20 мл) добавили гидрид натрия (60% в минеральном масле, 139 мг, 3.47 ммоль) при 0°C в инертной атмосфере. Реакционную смесь нагрели до 120°C и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (30 мл) и экстрагировали с помощью EtOAc (2 x 50 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрила (80 мг, 0.3 ммоль, 8%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, DMSO-d6): δ 13.21 (brs, 1H), 9.96 (d, J = 2.3 Гц, 1H), 8.48 (d, J = 2.3 Гц, 1H), 7.80-7.74 (m, 1H), 7.68 (d, J = 8.3 Гц, 1H), 7.50-7.32 (m, 3H)
LC-MS: m/z 270.9 [M+H]+ при 2.71 RT (90.28% чистота)
1-Циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрил (Пример 134)
К перемешиваемому раствору 2-(6-(дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрила (100 мг, 0.37 ммоль) и 1,2-дихлорэтана (15 мл) в запаянной трубке добавили циклопропилбороновую кислоту (64 мг, 0.74 ммоль) и карбонат натрия (78 мг, 0.74 ммоль) при комнатной температуре в инертной атмосфере. Смесь продули аргоном в течение 5 мин. В отдельной запаянной пробирке меди(II) ацетат (67 мг, 0.37 ммоль) и 2,2`-бипиридин (58 мг, 0.37 ммоль) растворили в 1,2-дихлорэтане (10 мл) при комнатной температуре в инертной атмосфере и продули аргоном в течение 5 мин. Сосуд запаяли, и реакционную смесь нагрели до 80°C и перемешивали в течение 3 мин. Полученный раствор синего цвета добавили к предварительно смешанной ранее бороновой кислоте, и реакционную смесь продули аргоном в течение 5 мин. Сосуд запаяли, и реакционную смесь нагрели до 80°C и перемешивали в течение 4 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь подкислили с применением 1 Н раствора HCl (25 мл) и экстрагировали с помощью CH2Cl2 (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 15-20% EtOAc/гексан) с получением 1-циклопропил-2-(6-(дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрила Пример 134 (11 мг, 0.03 ммоль, 9%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.71 (d, J = 2.0 Гц, 1H), 8.17 (d, J = 2.1 Гц, 1H), 7.82 (d, J = 7.9 Гц, 1H), 7.73 (d, J = 8.4 Гц, 1H), 7.49 (td, J = 7.7, 1.0 Гц, 1H), 7.43-7.37 (m, 1H), 7.21-6.91 (m, 1H), 3.67-3.59 (m, 1H), 1.25-1.17 (m, 2H), 0.75-0.68 (m, 2H)
LC-MS: m/z 310.9 [M+H]+ при 3.08 RT (98.05% чистота)
ВЭЖХ: 97.84%
2-(6-(Дифторметил)пиридазин-4-ил)-1-метил-1H-индол-3-карбонитрил (Пример 135)
К перемешиваемому раствору 2-(6-(дифторметил)пиридазин-4-ил)-1H-индол-3-карбонитрила (35 мг, 0.13 ммоль) в DMF (0.8 мл) добавили гидрид натрия (60% в минеральном масле, 6 мг, 0.15 ммоль) при 0°C в инертной атмосфере, и смесь перемешивали в течение 5 мин. Иодметан (0.01 мл, 0.15 ммоль) добавили при 0°C и смесь перемешивали при комнатной температуре в течение 1 часа. После израсходования исходного вещества (посредством TLC), реакционную смесь погасили ледяной водой (10 мл) и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 20% EtOAc/гексан) с получением 2-(6-(дифторметил)пиридазин-4-ил)-1-метил-1H-индол-3-карбонитрила Пример 135 (10 мг, 0.03 ммоль, 28%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3): δ 9.57 (d, J = 2.1 Гц, 1H), 8.05 (d, J = 2.1 Гц, 1H), 7.85 (dt, J = 7.9, 0.9 Гц, 1H), 7.53-7.50 (m, 2H), 7.45-7.39 (m, 1H), 7.23-6.92 (m, 1H), 3.88 (s, 3H)
LC-MS: m/z 284.9 [M+H]+ при 2.81 RT (98.36% чистота)
ВЭЖХ: 99.44%
Пример 136
Схема:
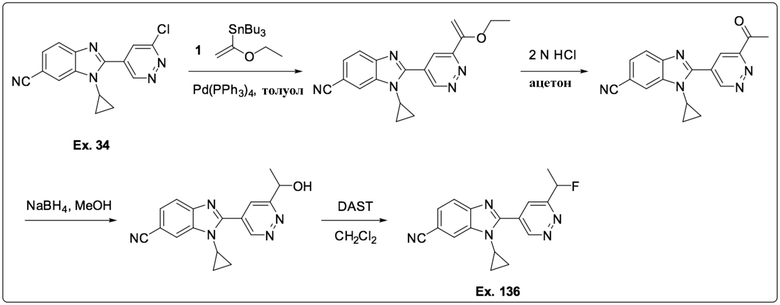
1-Циклопропил-2-(6-(1-этоксивинил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 2-(6-хлорпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила Пример 34 (750 мг, 2.54 ммоль) в толуоле (7.5 мл) добавили трибутил(1-этоксивинил) станнан (0.86 мл, 2.54 ммоль) и Pd(PPh3)4 (294 мг, 0.25 ммоль) при комнатной температуре в инертной атмосфере. Реакционную смесь продули аргоном в течение 5 мин и нагрели до температуры возврата флегмы и перемешивали в течение 16 ч. После израсходования исходного вещества (посредством TLC), реакционную смесь отфильтровали через слой целита, и слой промыли с помощью EtOAc (20 мл). Фильтрат промыли водой (20 мл), высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 50-60% EtOAc/гексан) с получением 1-циклопропил-2-(6-(1-этоксивинил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (425 мг, 1.28 ммоль, 50%) в виде твердого вещества бледно-желтого цвета.
LC-MS: m/z 331.9 [M+H]+ при 2.80 RT (56.84% чистота)
2-(6-Ацетилпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 1-циклопропил-2-(6-(1-этоксивинил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (425 мг, 1.28 ммоль) в ацетоне (2.2 мл) добавили 2.5 N HCl (0.44 мл) при комнатной температуре в инертной атмосфере, и смесь перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток разбавили с помощью CH2Cl2 (50 мл) и промыли с помощью насыщенного раствора бикарбоната натрия (30 мл) и воды (30 мл). Органический слой отделили, высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный материал перетерли с н-пентаном (2 x 10 мл) с получением 2-(6-ацетилпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила (380 мг, 1.25 ммоль, 97%) в виде твердого вещества грязновато-белого цвета.
LC-MS: m/z 304.0 [M+H]+ при 2.35 RT (53.18% чистота).
1-Циклопропил-2-(6-(1-гидроксиэтил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил
К перемешиваемому раствору 2-(6-ацетилпиридазин-4-ил)-1-циклопропил-1H-бензо[d]имидазол-6-карбонитрила (380 мг, 1.25 ммоль) в метаноле (4 мл) добавили боргидрид натрия (48 мг, 1.25 ммоль) при 0°C в инертной атмосфере, и смесь перемешивали в течение 1 ч. После израсходования исходного вещества (посредством TLC), летучие соединения удалили при пониженном давлении. Остаток разбавили водой (20 мл) и экстрагировали с помощью CH2Cl2 (2 x 25 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 4-5% MeOH/CH2Cl2) с получением 1-циклопропил-2-(6-(1-гидроксиэтил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (300 мг, 0.98 ммоль, 78%) в виде твердого вещества грязновато-белого цвета.
LC-MS: m/z 305.9 [M+H]+ при 1.92 RT (56.55% чистота)
1-Циклопропил-2-(6-(1-фторэтил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрил (Пример 136)
К перемешиваемому раствору 1-циклопропил-2-(6-(1-гидроксиэтил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила (300 мг, 0.98 ммоль) в CH2Cl2 (3 мл) добавили диэтиламиносеры трифторид (0.19 мл, 1.47 ммоль) при 0°C в инертной атмосфере, и реакционную смесь перемешивали в течение 30 минут. После израсходования исходного вещества (посредством TLC), реакционную смесь нейтрализовали с применением насыщенного раствора карбоната натрия и экстрагировали с помощью EtOAc (2 x 20 мл). Объединенные органические экстракты высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (элюент: 5% MeOH/CH2Cl2) с получением желаемого соединения (35 мг). Этот материал объединили с другой партией (10 мг) и очистили посредством препаративной ВЭЖХ с получением 1-циклопропил-2-(6-(1-фторэтил)пиридазин-4-ил)-1H-бензо[d]имидазол-6-карбонитрила Пример 136 (21 мг, 0.07 ммоль, 6% для двух партий) в виде твердого вещества грязновато-белого цвета.
1H ЯМР (400 МГц, CD3OD): δ 9.83 (d, J = 2.0 Гц, 1H), 8.50 (d, J = 2.1 Гц, 1H), 8.28-8.26 (m, 1H), 7.93 (dd, J = 8.4, 0.6 Гц, 1H), 7.70 (dd, J = 8.4, 1.5 Гц, 1H), 6.19-6.01 (m, 1H), 3.99-3.94 (m, 1H), 1.93-1.81 (m, 3H), 1.37-1.28 (m, 2H), 0.93-0.86 (m, 2H)
LC-MS: m/z 308.0 [M+H]+ при 2.55 RT (99.50% чистота)
ВЭЖХ: 99.43%
Пример 137: Активность металлофермента
A. Клетки V79-4, экспрессирующие рекомбинантные андренодоксин и андренодоксинредуктазу с либо рекомбинантным человеческим CYP11B2, либо CYP11B1, получали в соответствии с методами, описанными ранее (LaSala et al 2009 Anal Bioch 394: 56-61). Микросомальную фракцию, обогащенную ферментом, получали из клеточных лизатов и затем использовали в качестве источника фермента для определения значений IC50 ингибиторов. Значения Km для субстрата были определены экспериментально для 11-деоксикортикостерона (субстрат CYP11B2) и 11-деоксикортизола (субстрат CYP11B1). Ферментные анализы для скрининга ингибиторов использовали микросомы, обогащенные ферментами CYP11B2 и CYP11B1, и проводились при Km соответствующих субстратов. Продукты ферментативных реакций, альдостерон для CYP11B2 или кортизол для CYP11B1, измеряли посредством LC-MS. Анализы проводили в условиях менее 20% обмена субстрата. Значения IC50 ингибитора получали путем определения образования продукта в отсутствие или в присутствии ингибитора при различных концентрациях. В отсутствие тестируемого соединения образовавшийся продукт (Pt) в каждом наборе данных определяли как 100% -ную активность. В отсутствие фермента образовавшийся продукт (Pb) в каждом наборе данных определяли как активность 0%. Процент активности в присутствии каждого ингибитора вычисляли по следующему уравнению: % активности = (P-Pb)/(Pt-Pb), где P = продукт, образовавшийся в присутствии ингибитора. Значение IC50 определяли как концентрацию ингибитора, вызывающую снижение активности на 50% относительно контрольной реакции без ингибитора.
Таблица 2. Результаты: CYP11B2 активность


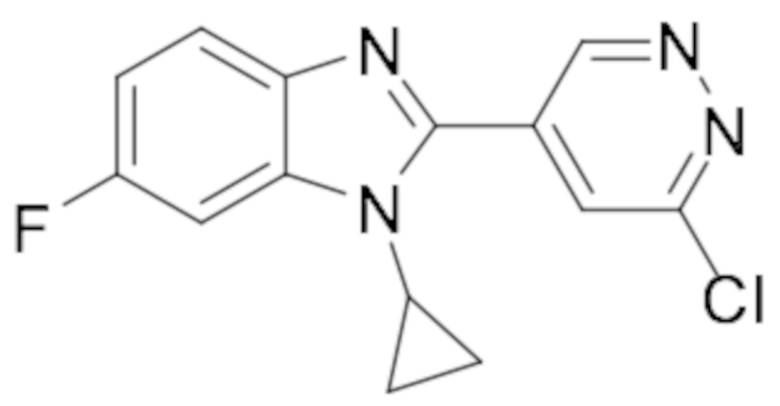
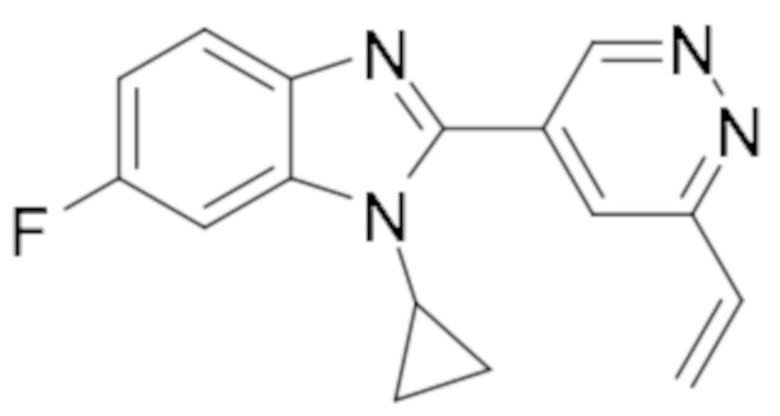

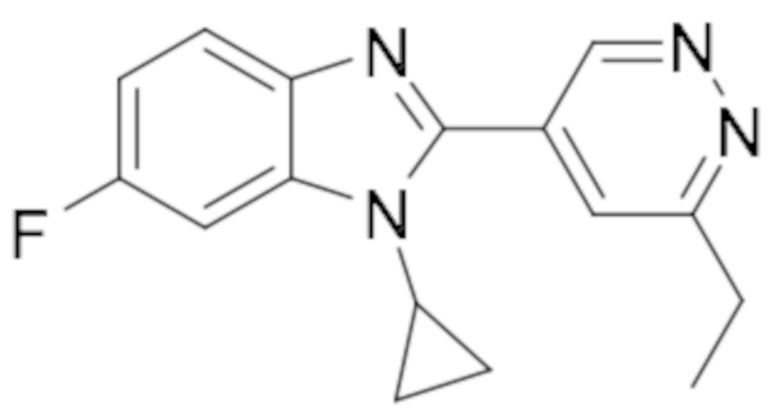
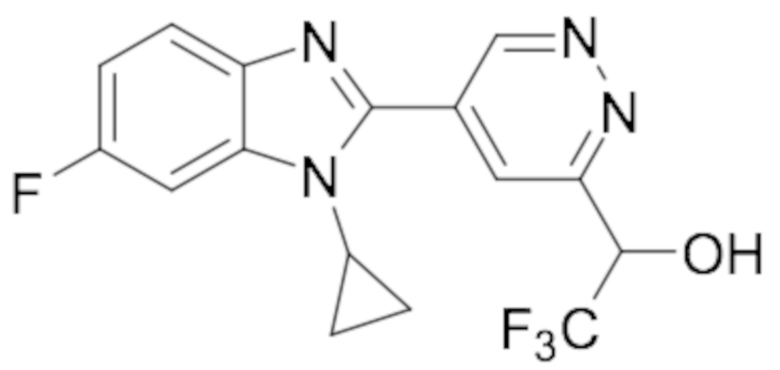

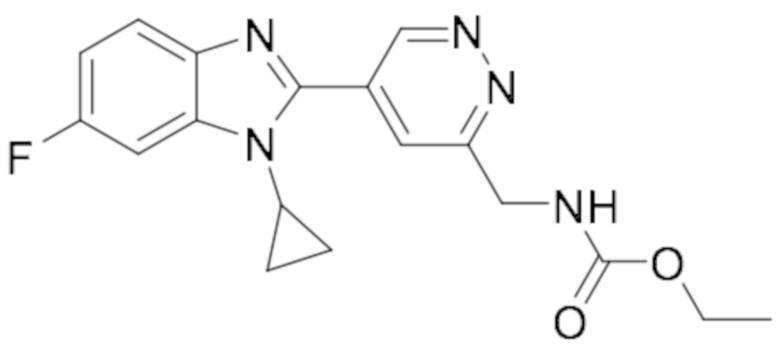
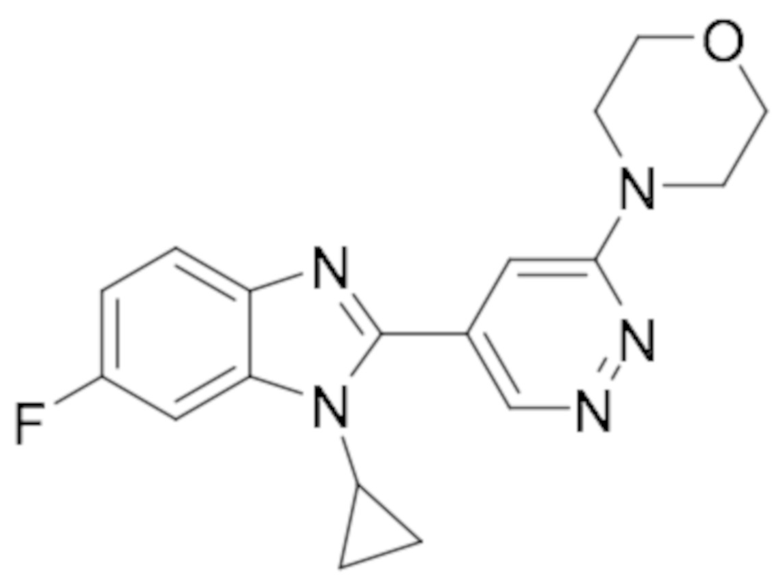
[M+H]C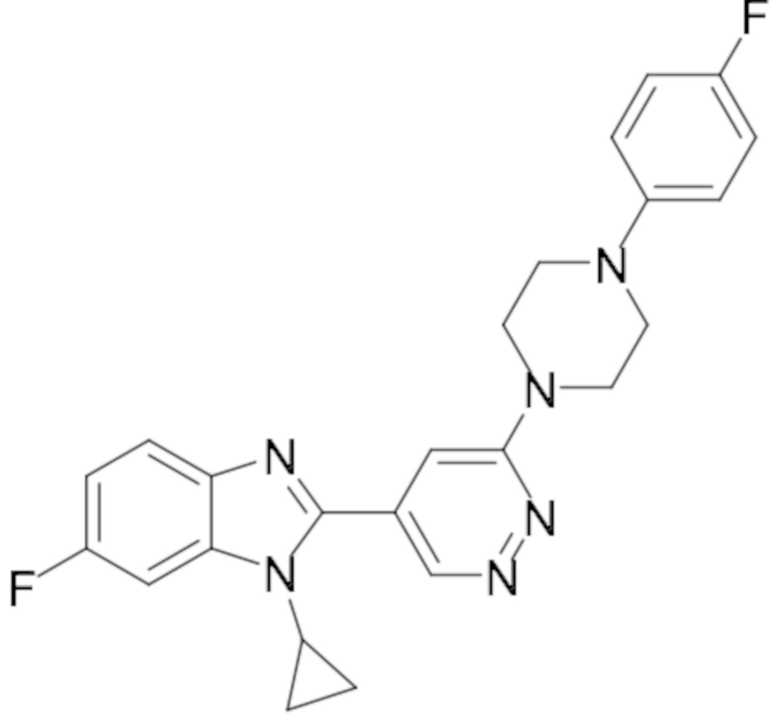
[M+H]b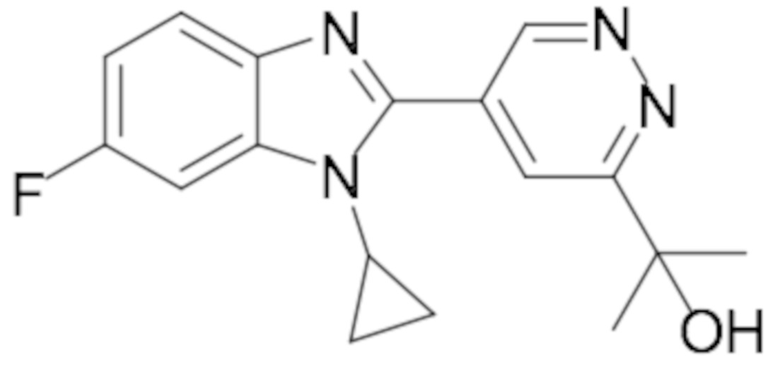
[M+H]C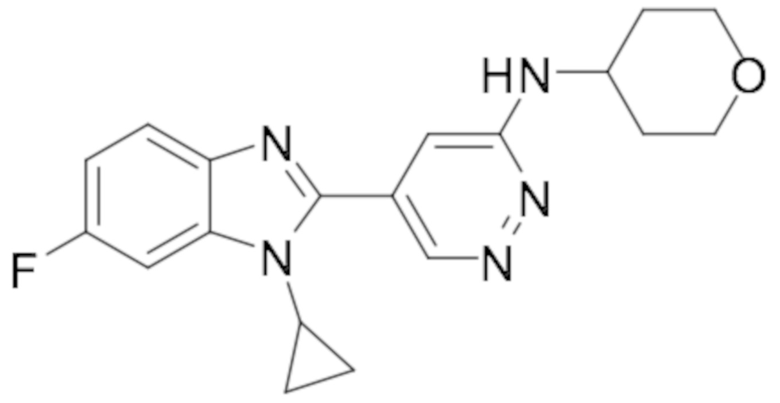
[M+H]b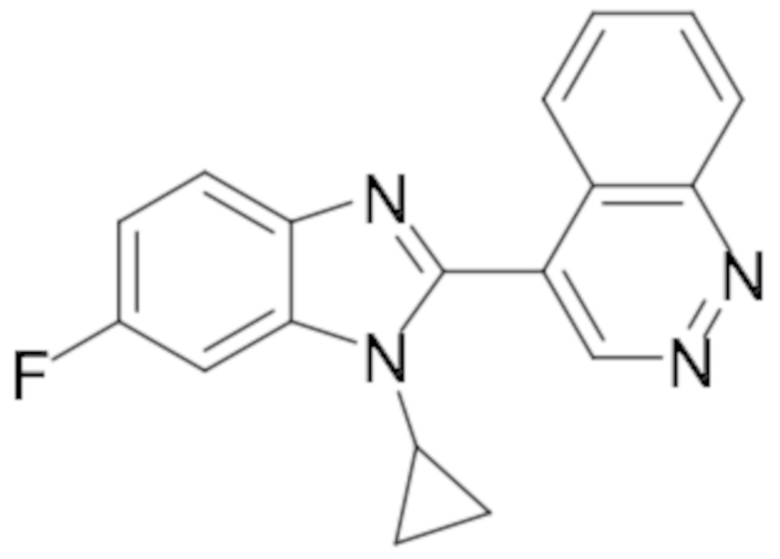
[M+H]b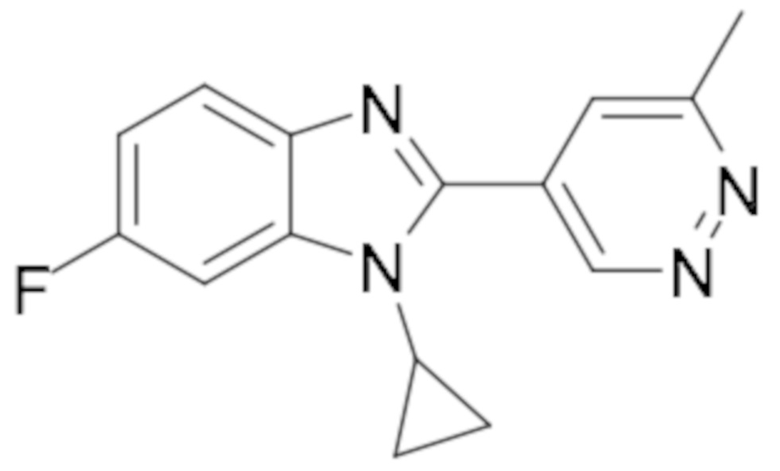
[M+H]c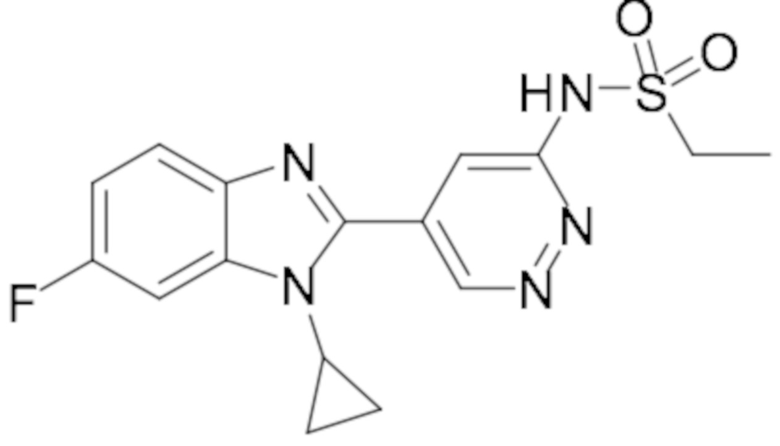
[M+H]d
[M+H]d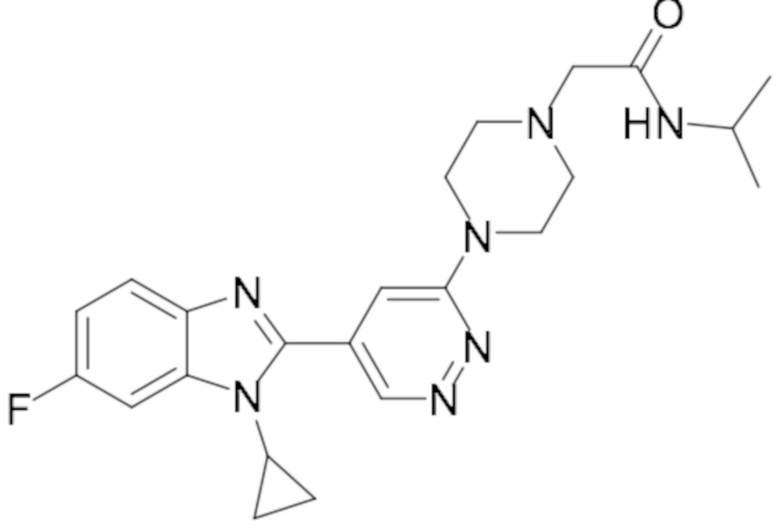
[M+H]b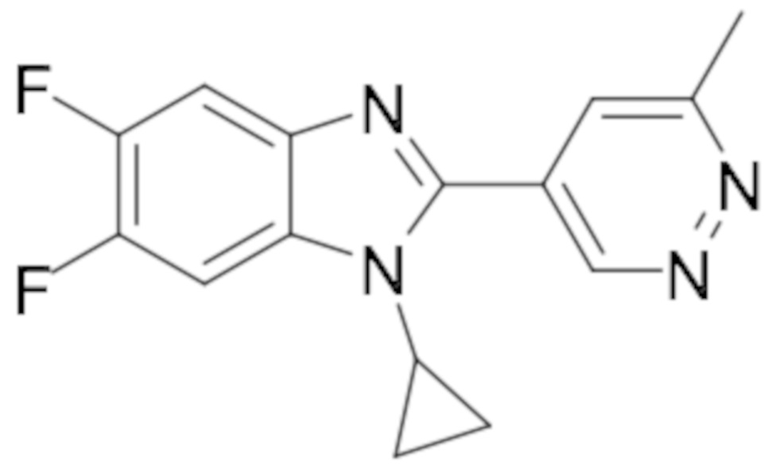
[M+H]C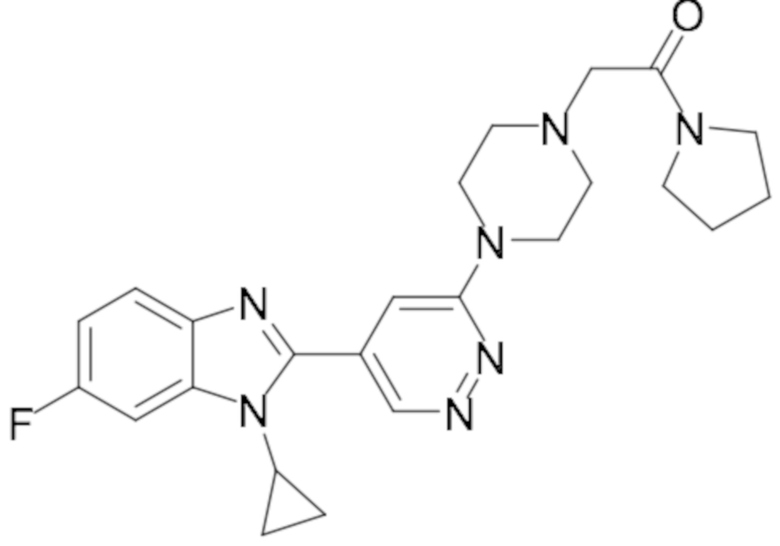
[M+H]C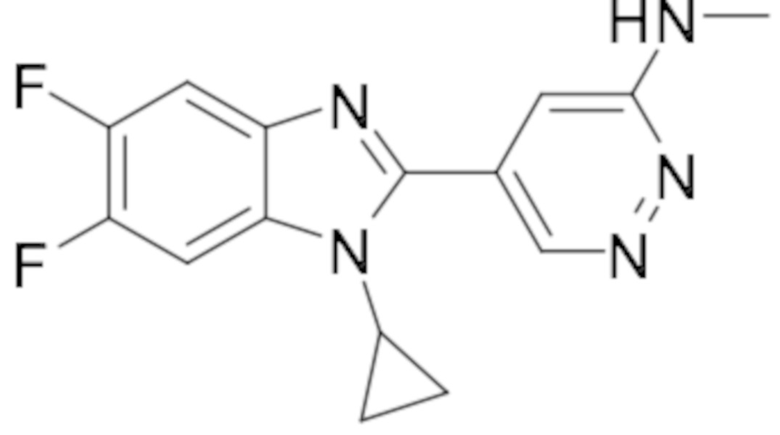
[M+H]C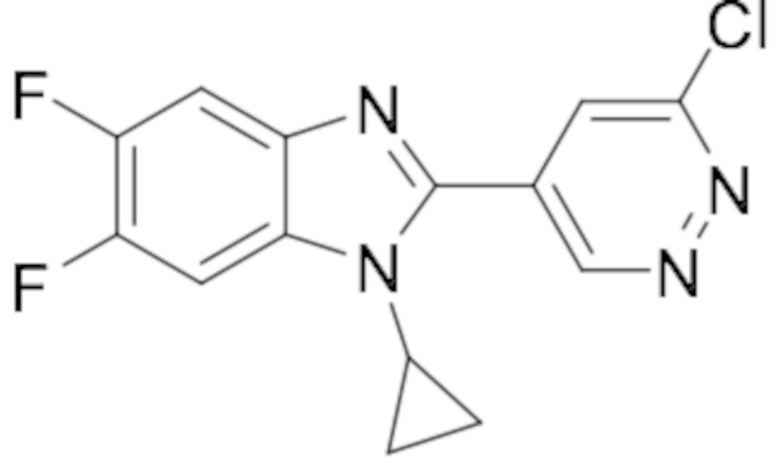
[M+H]C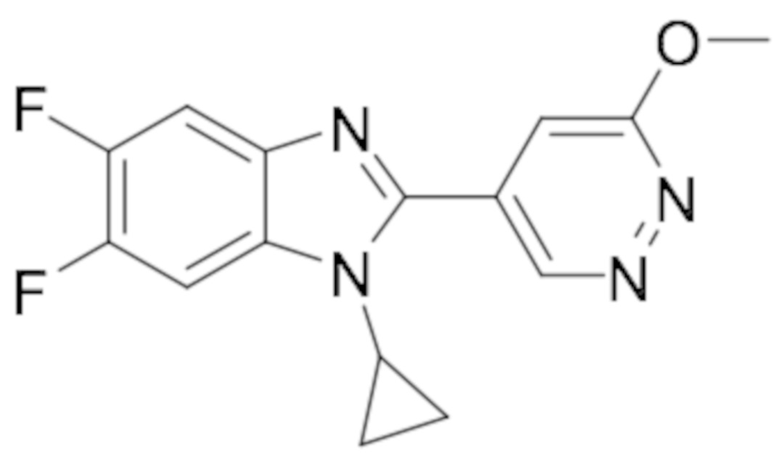
[M+H]b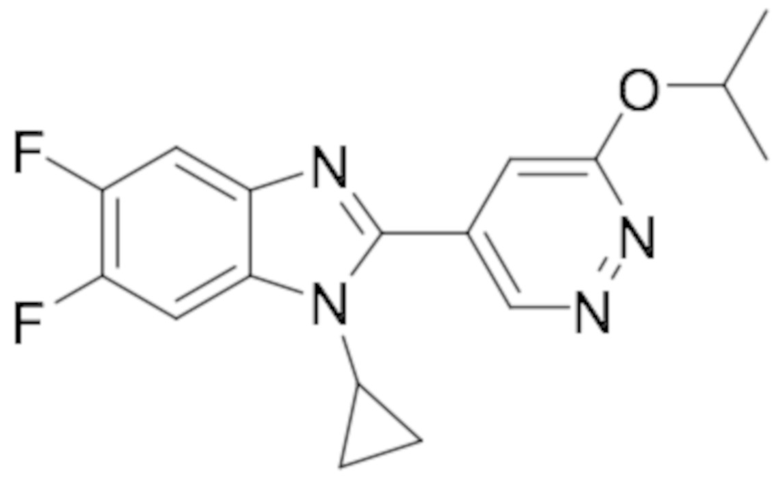
[M+H]C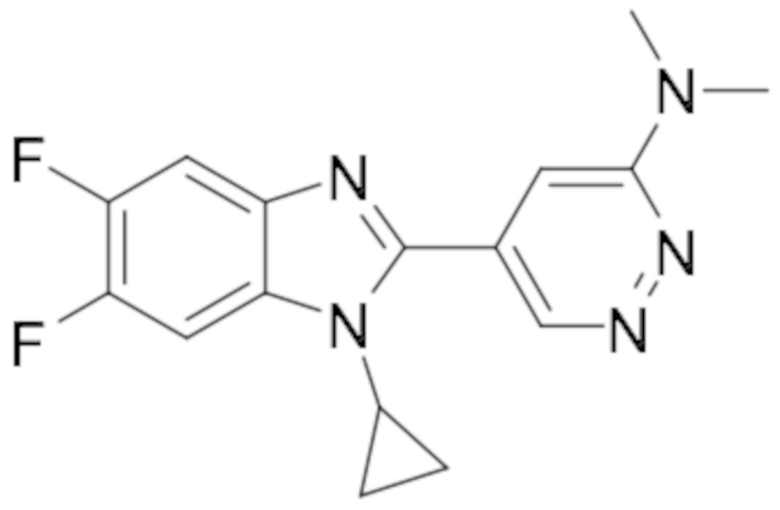
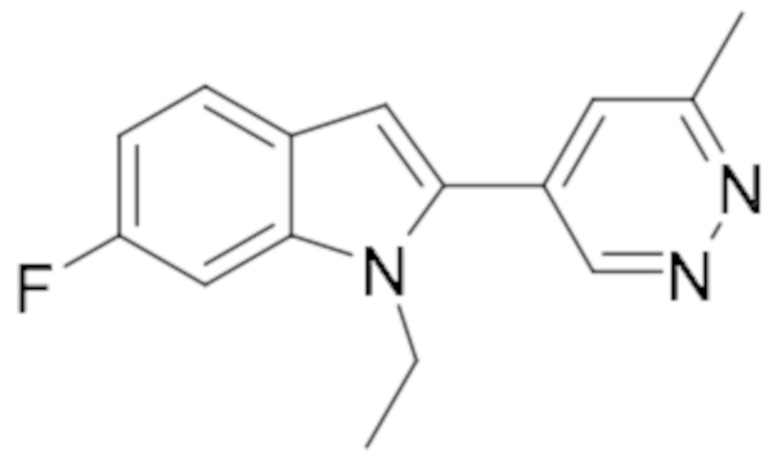
[M+H]b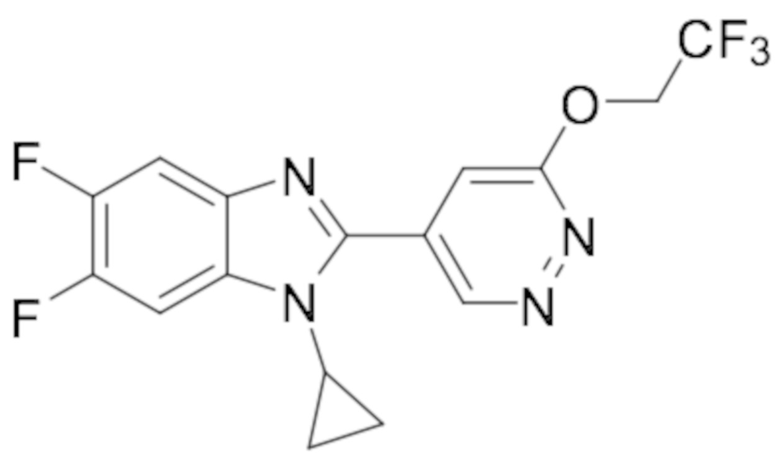
[M+H]C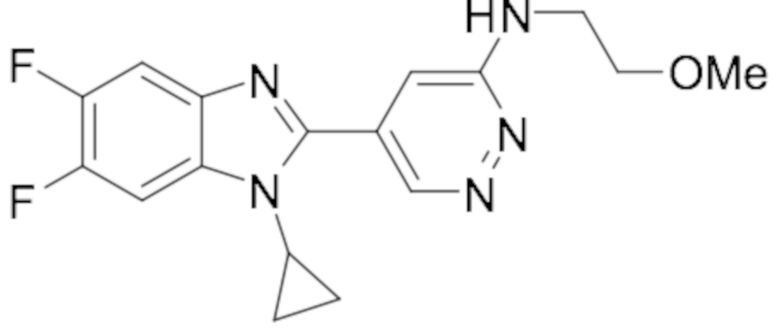
[M+H]b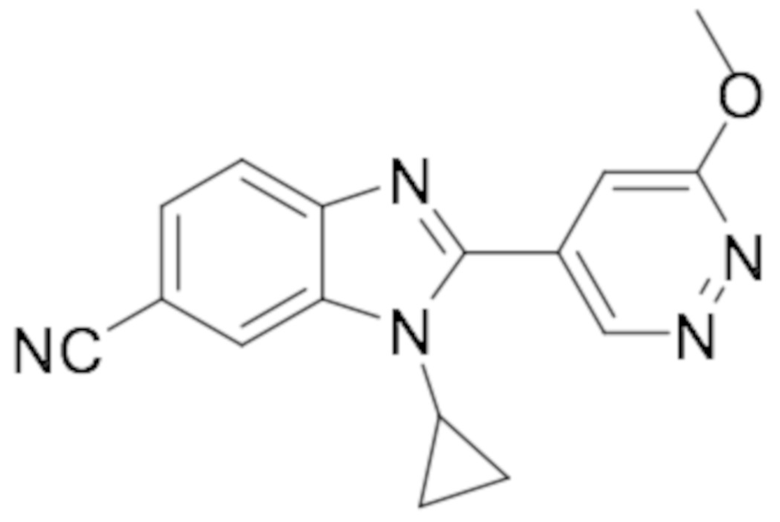
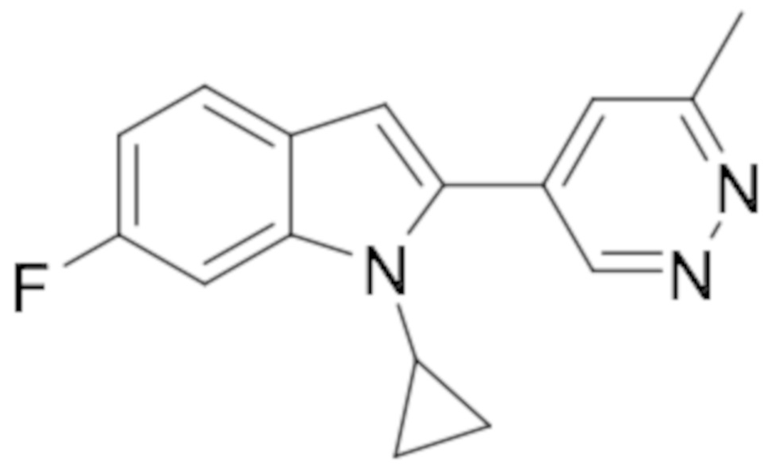
[M+H]b
[M+H]C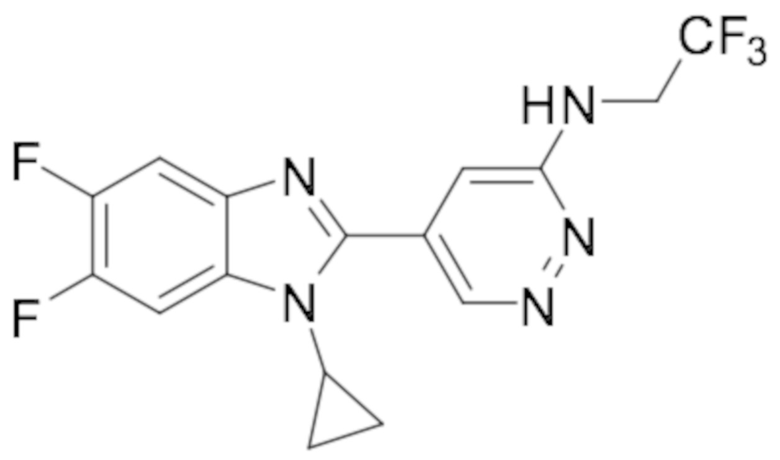
[M+H]b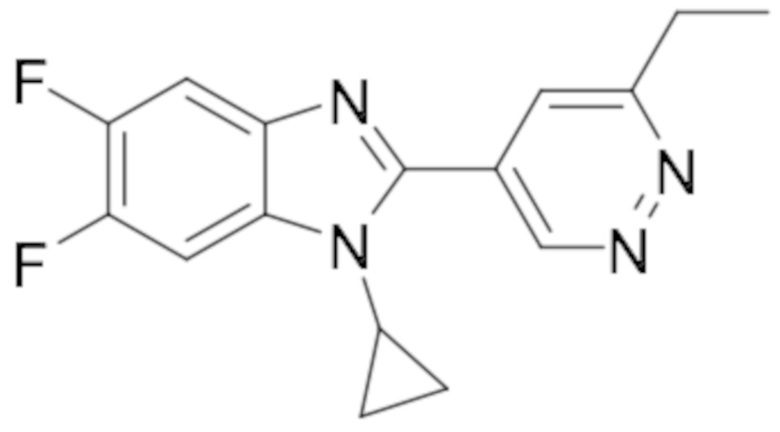
[M+H]e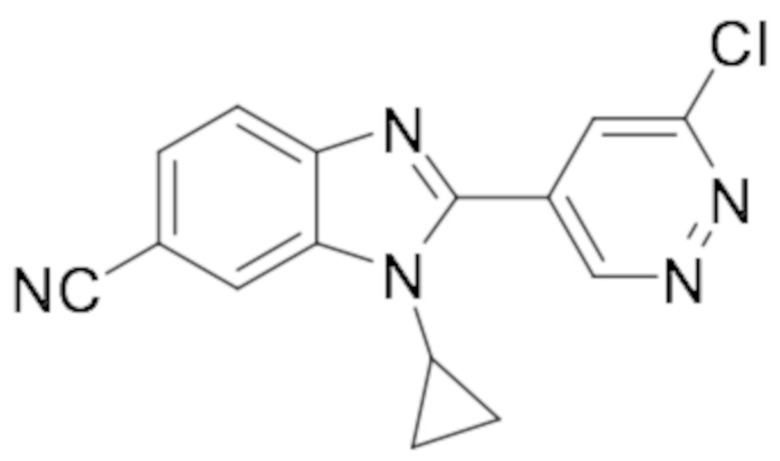
[M+H]b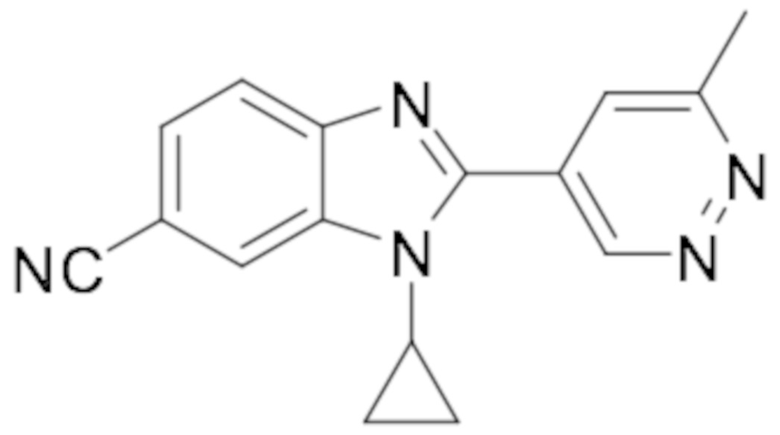
[M+H]C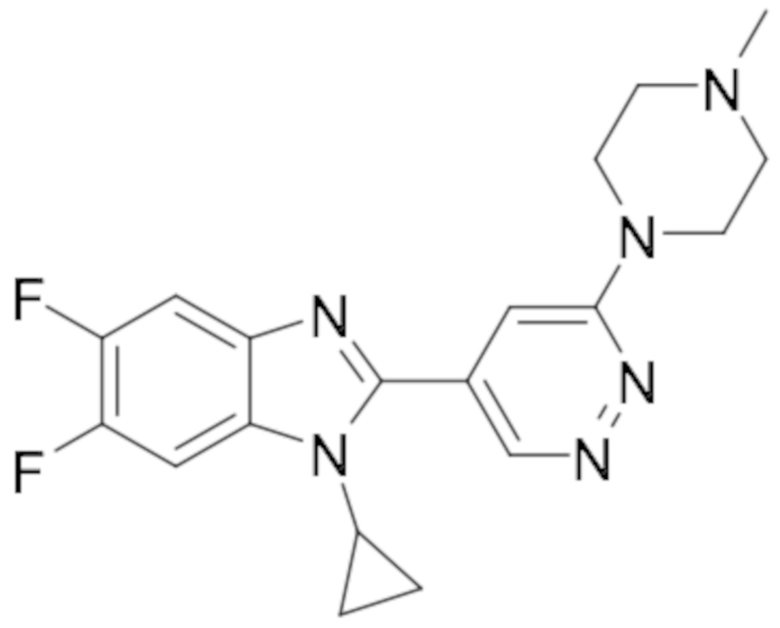
[M+H]C
[M+H]C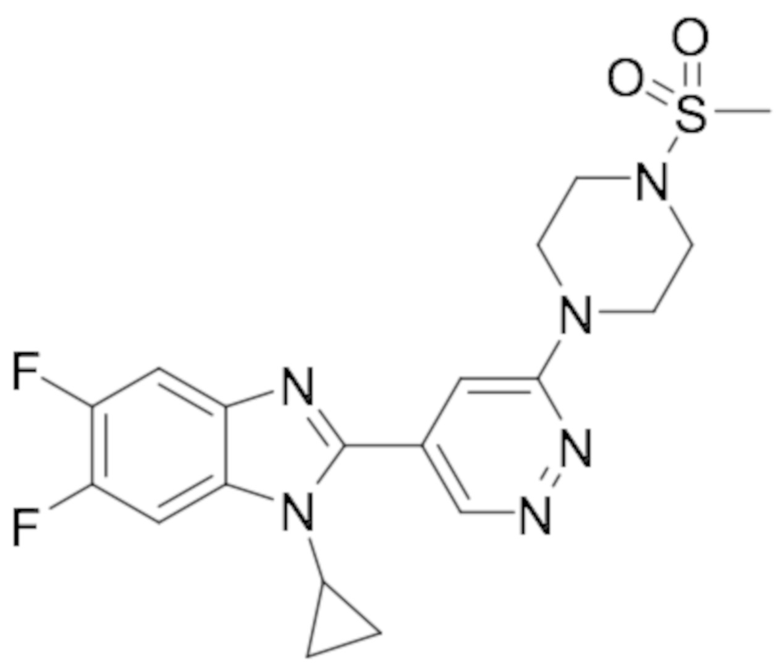
[M+H]C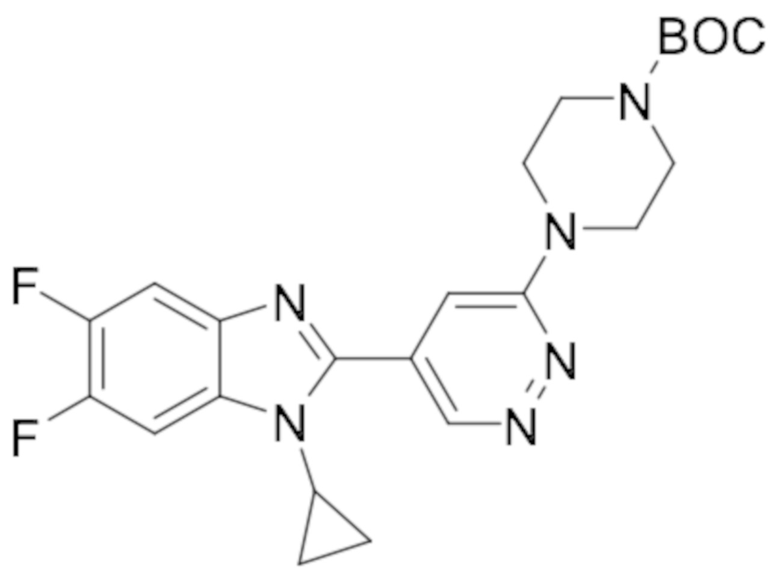
[M+H]b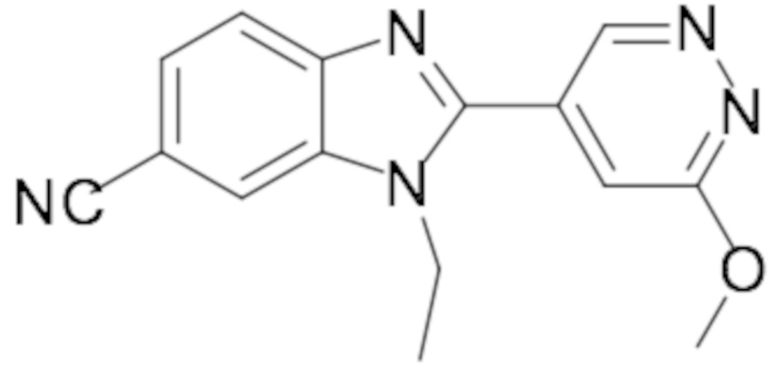
[M+H]b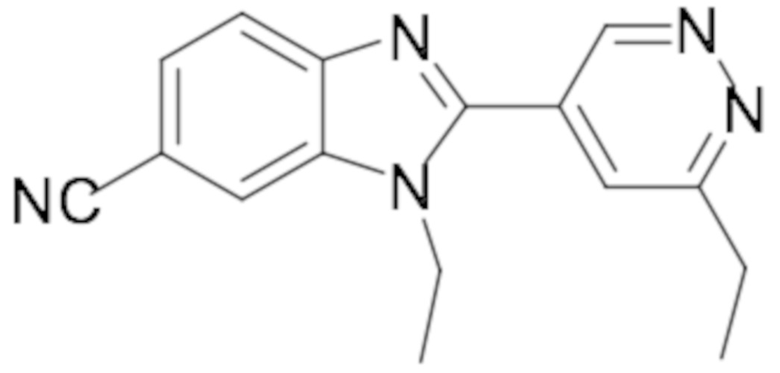
[M+H]e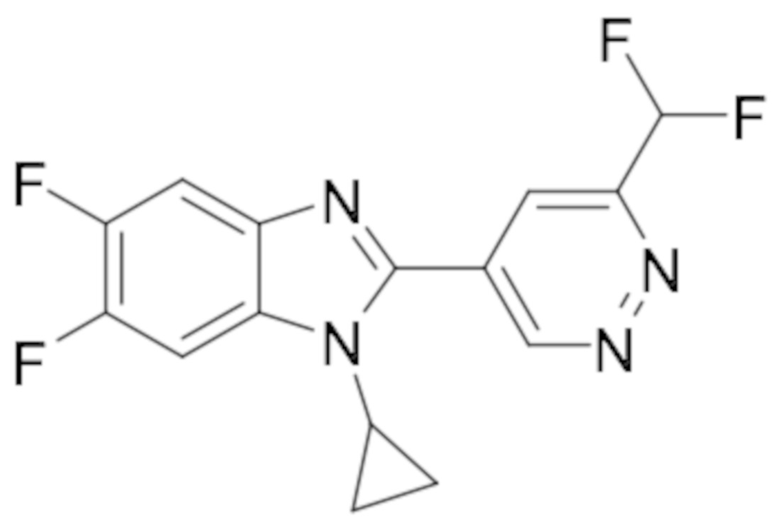
[M+H]C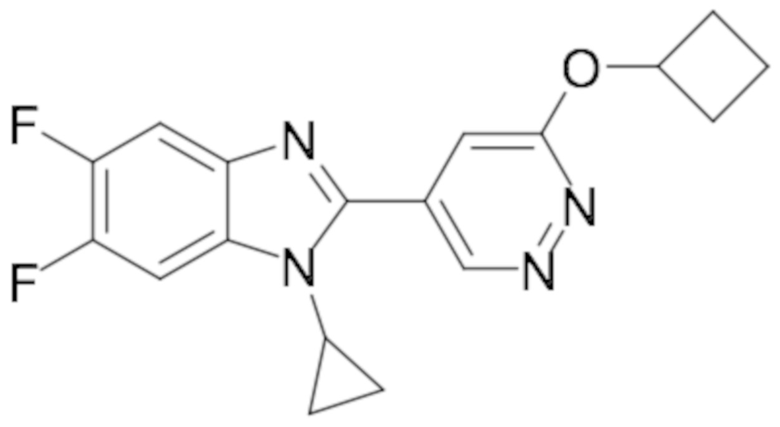
[M+H]b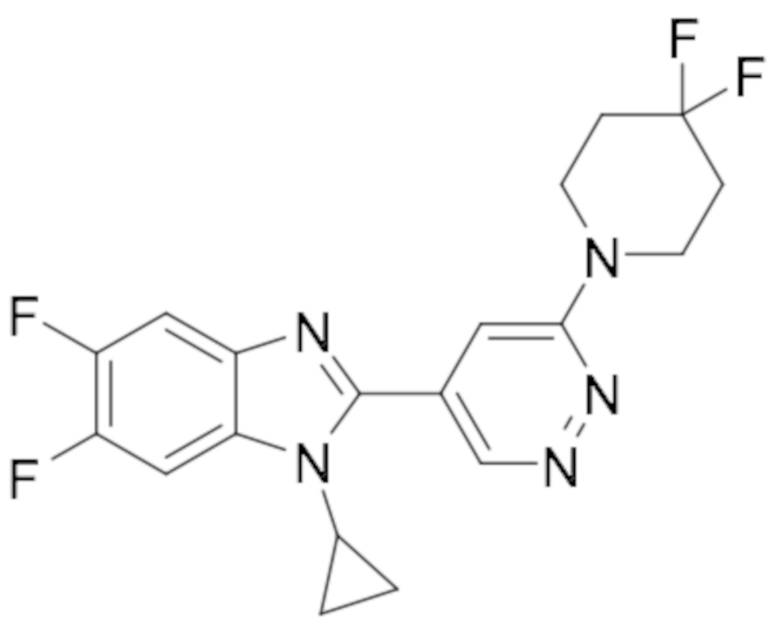
[M+H]b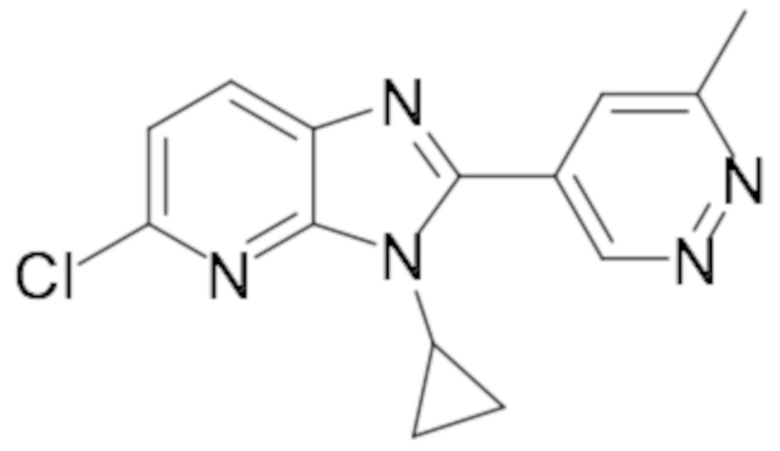
[M+H]b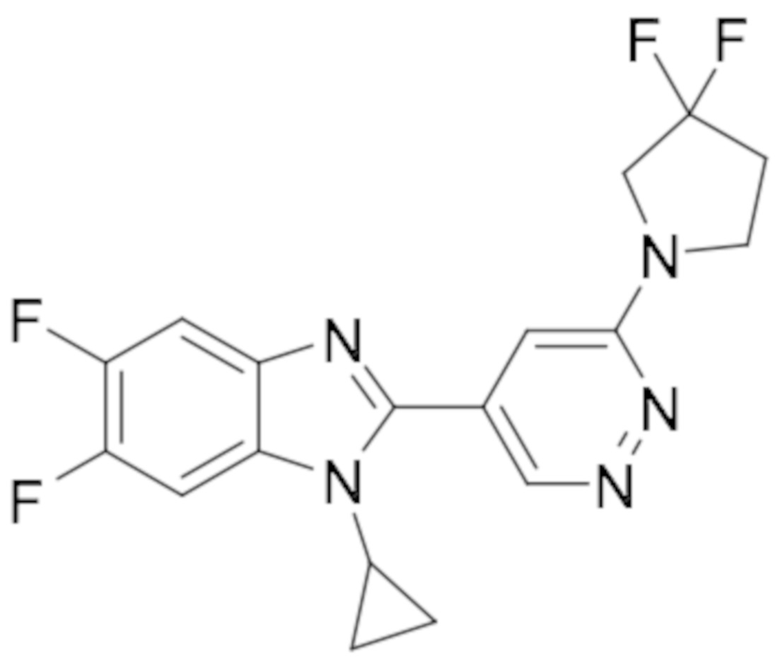
[M+H]b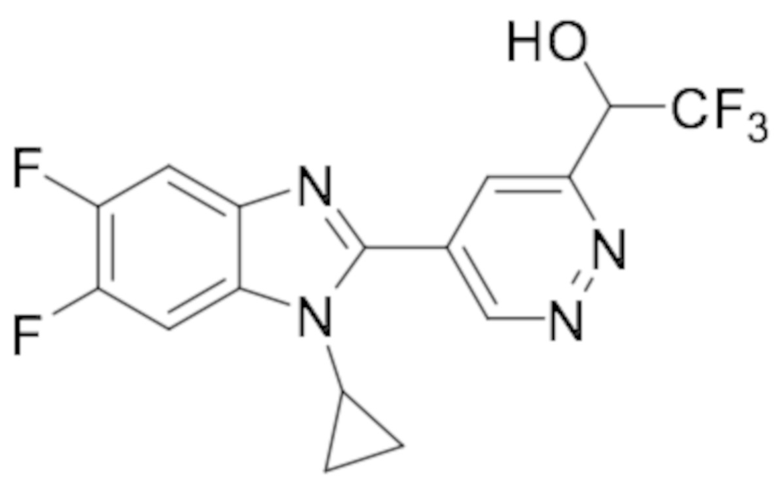
[M+H]b
[M+H]c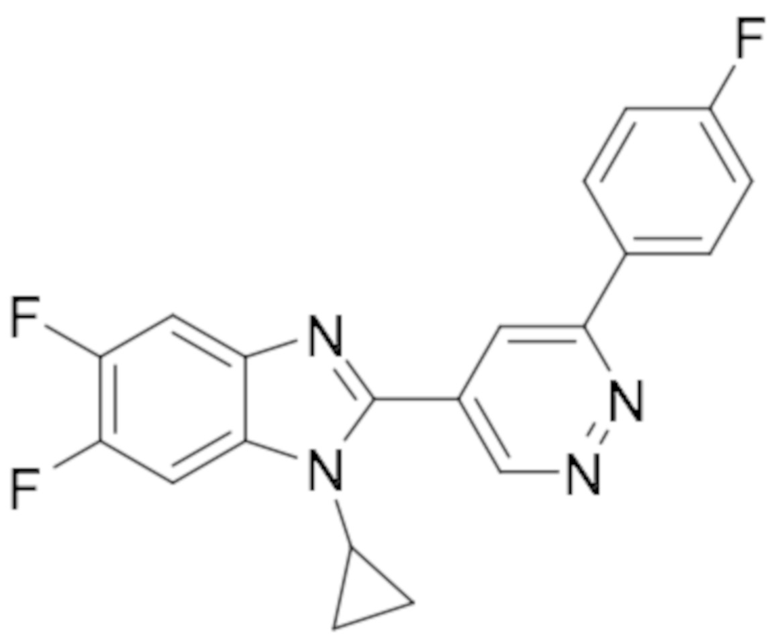
[M+H]e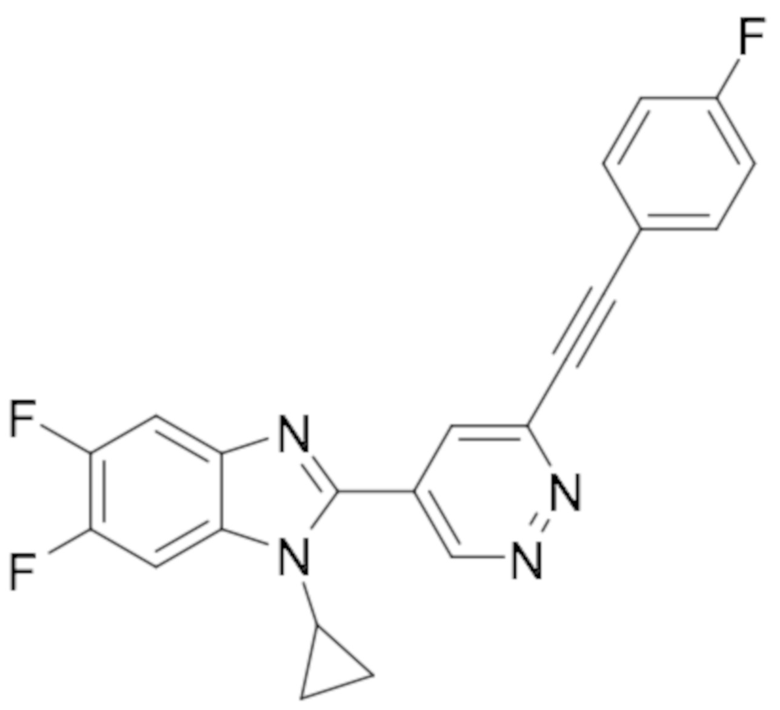
[M+H]C
[M+H]b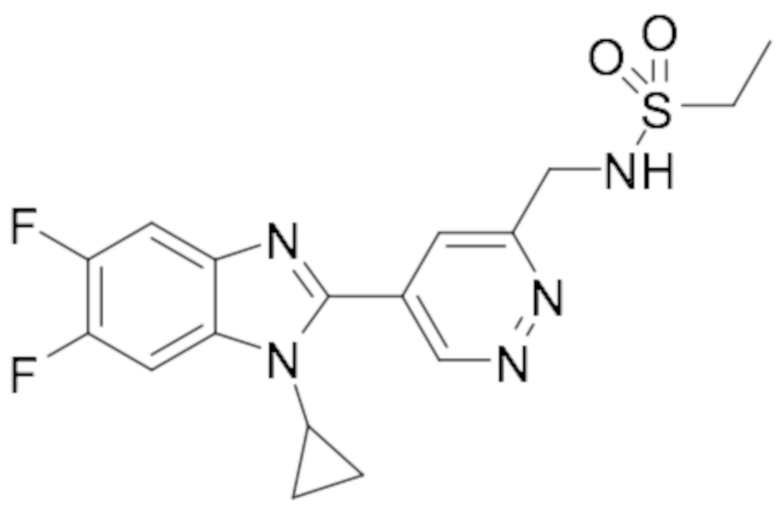
[M+H]C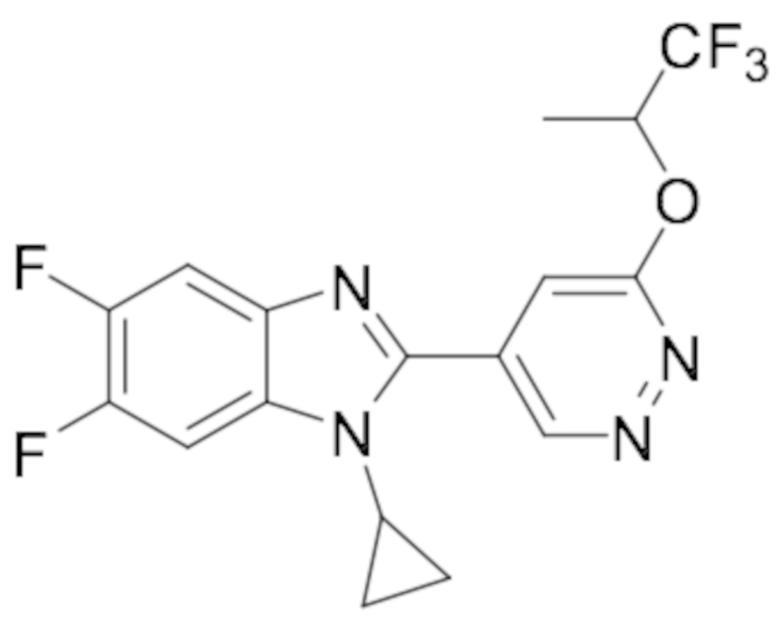
[M+H]b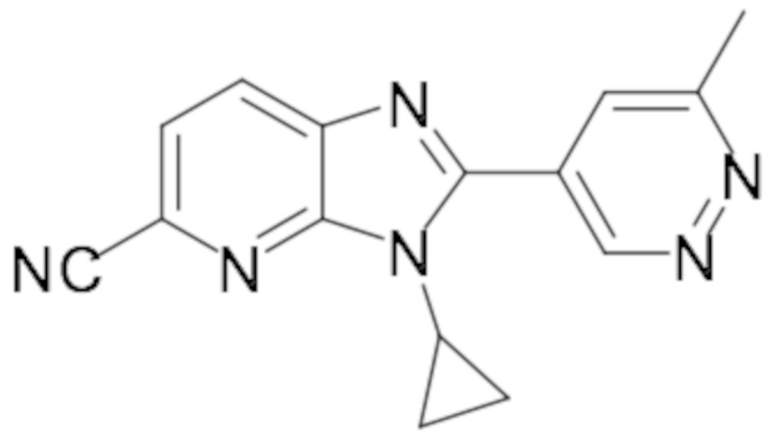
[M+H]b
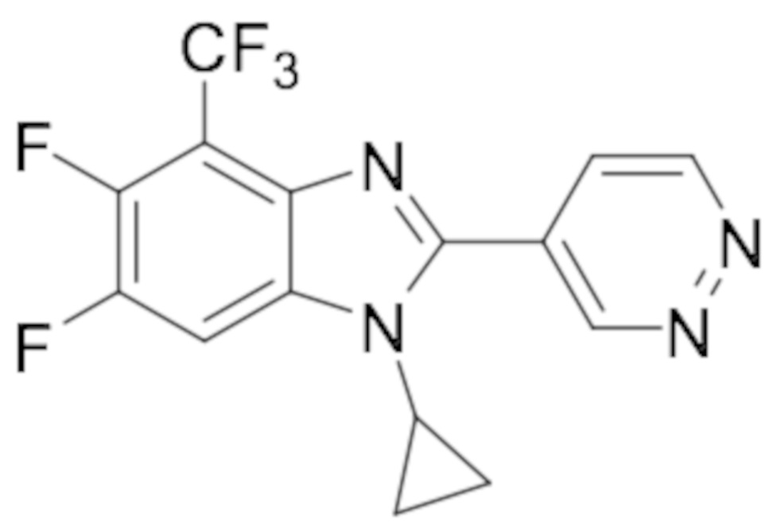
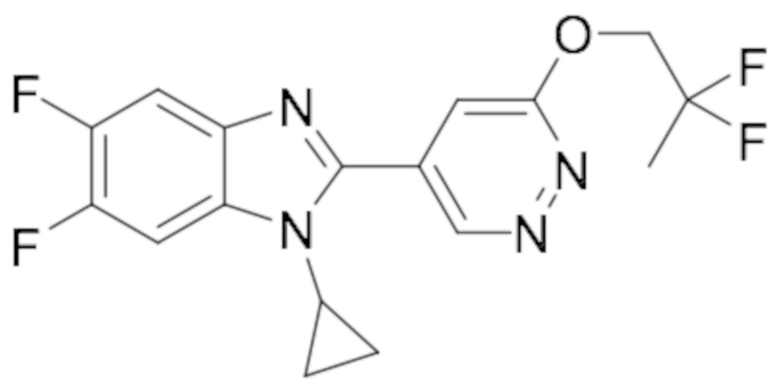
[M+H]b

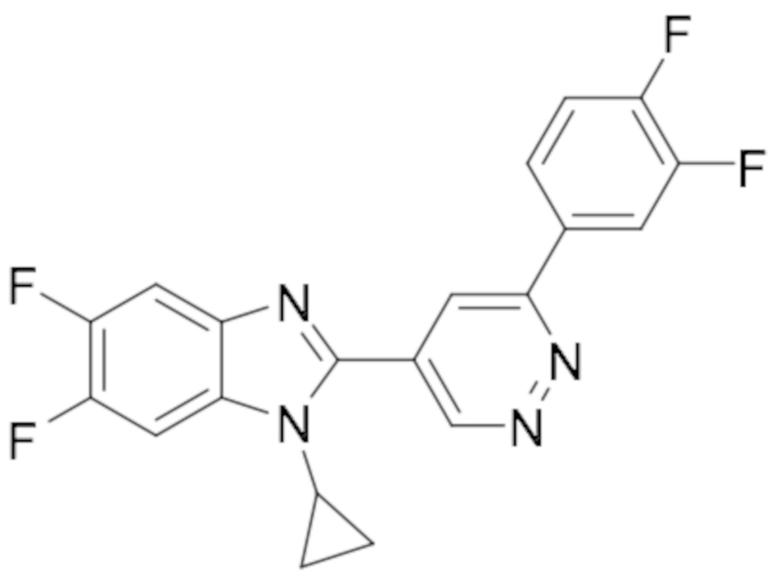
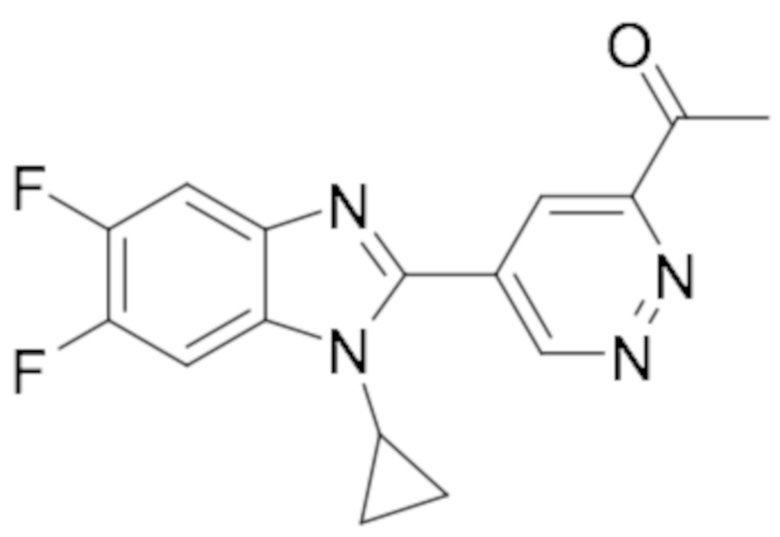
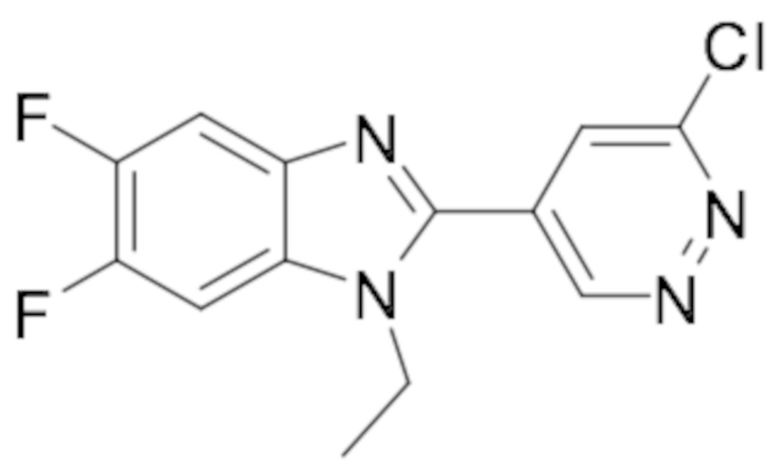
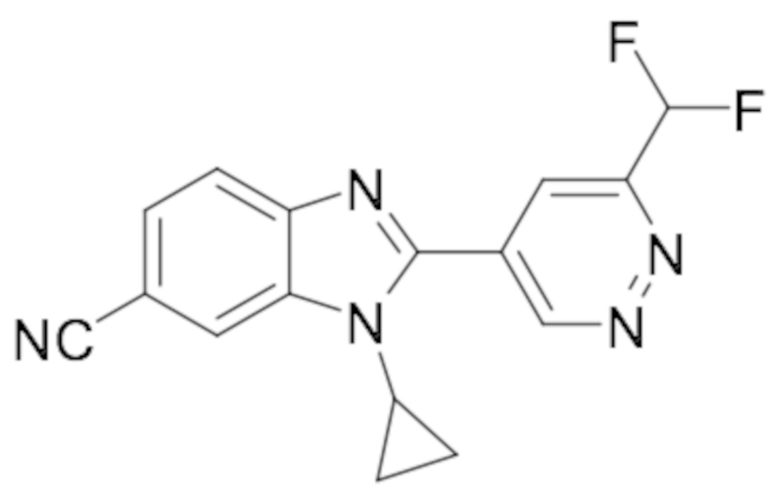
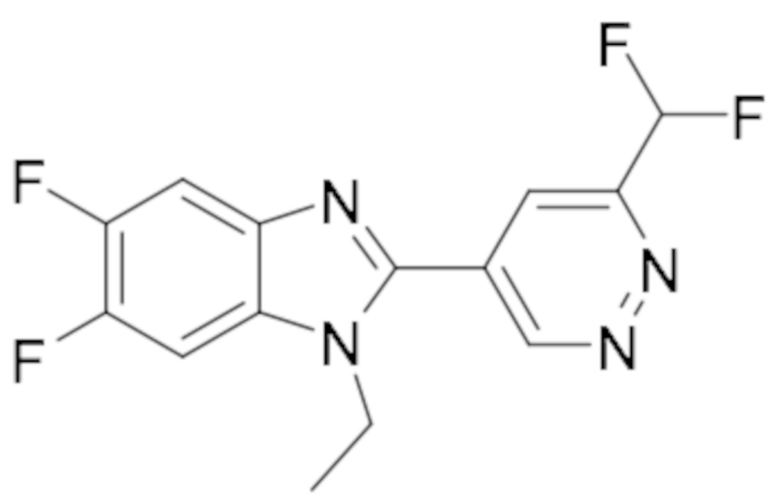
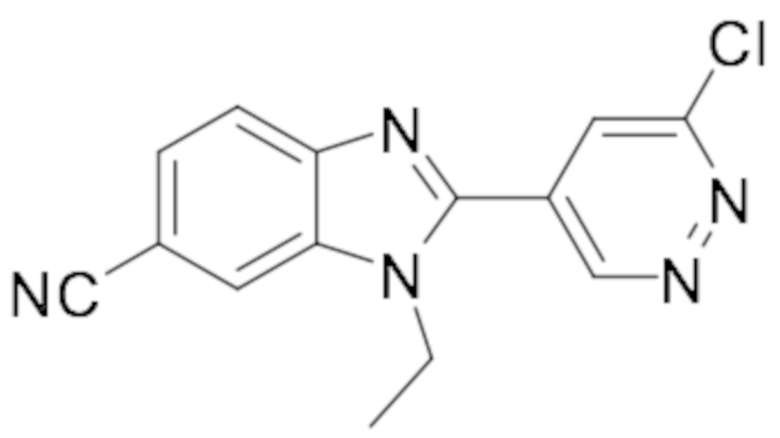
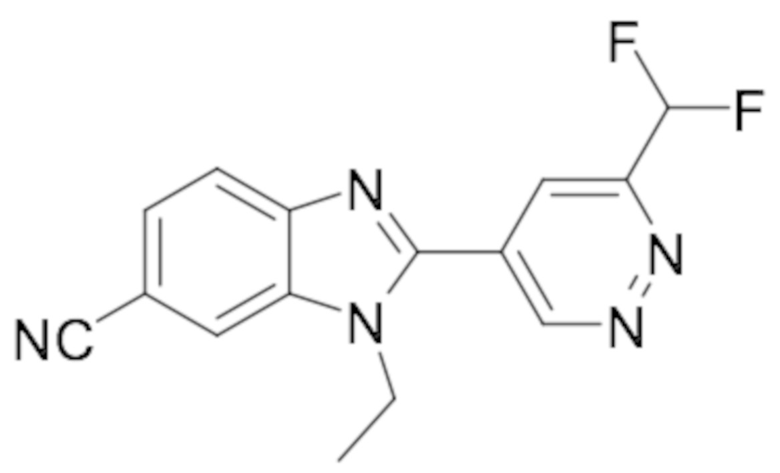
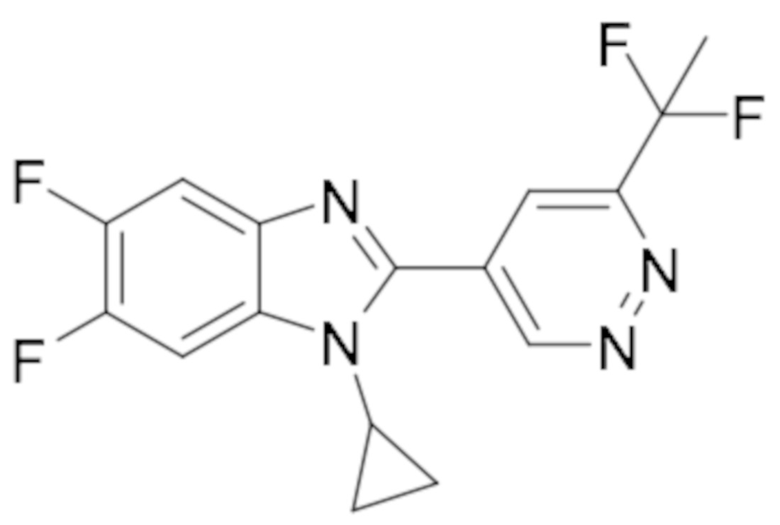
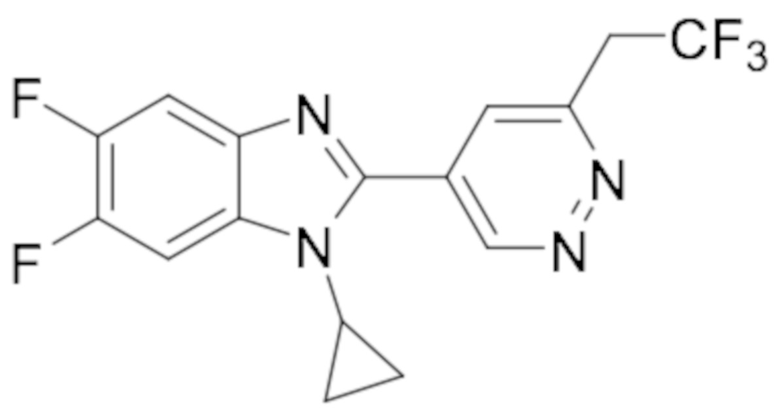
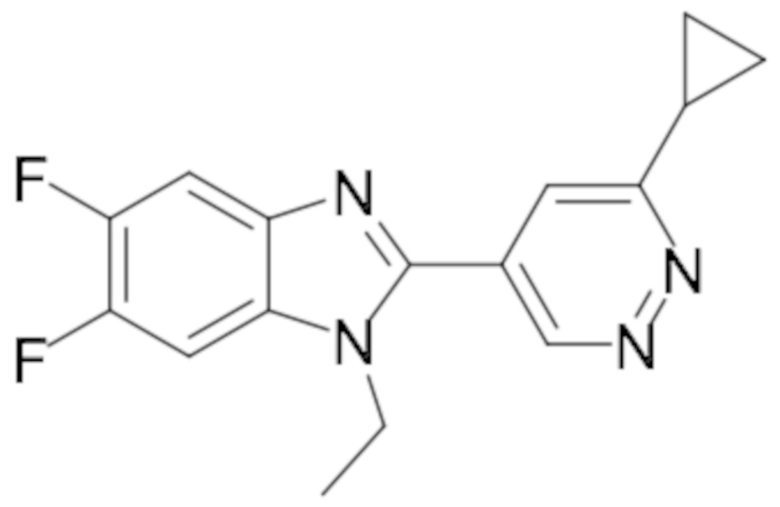
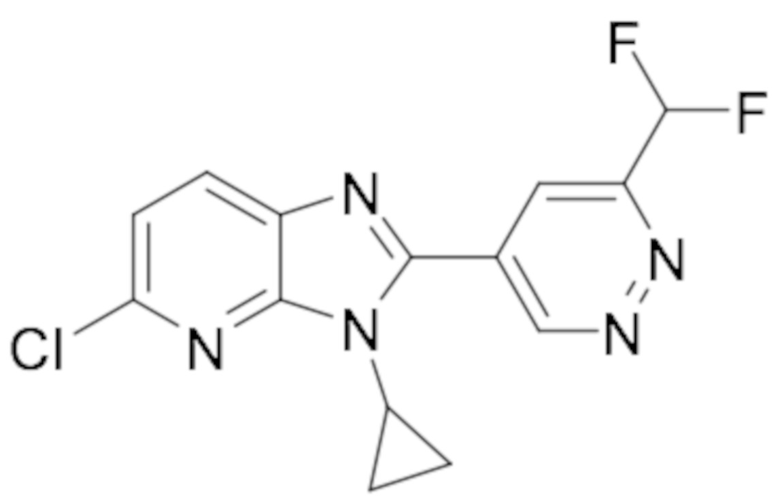
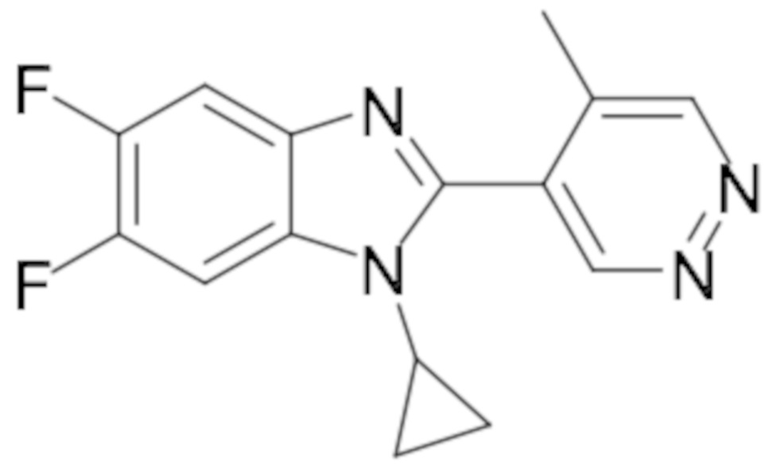
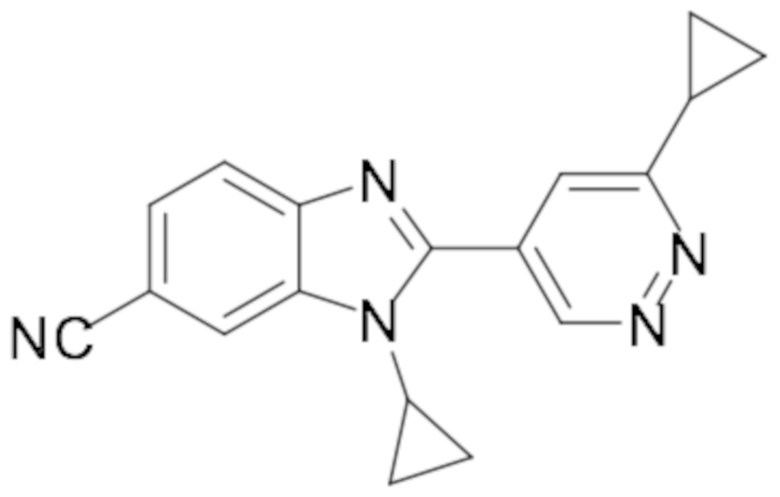
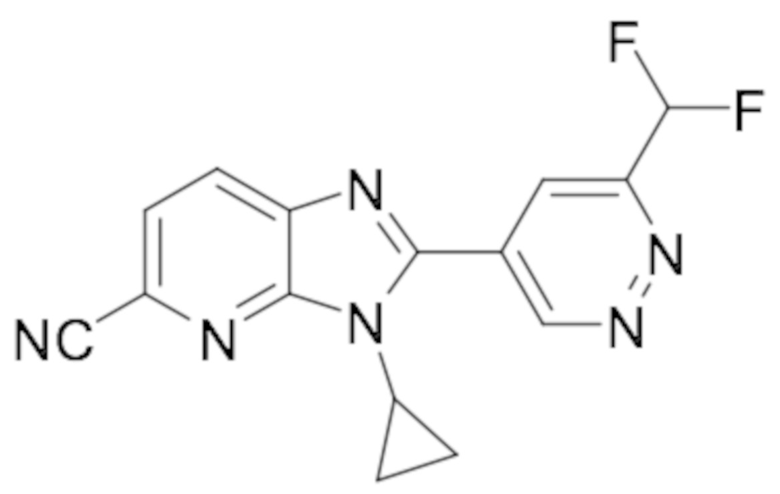
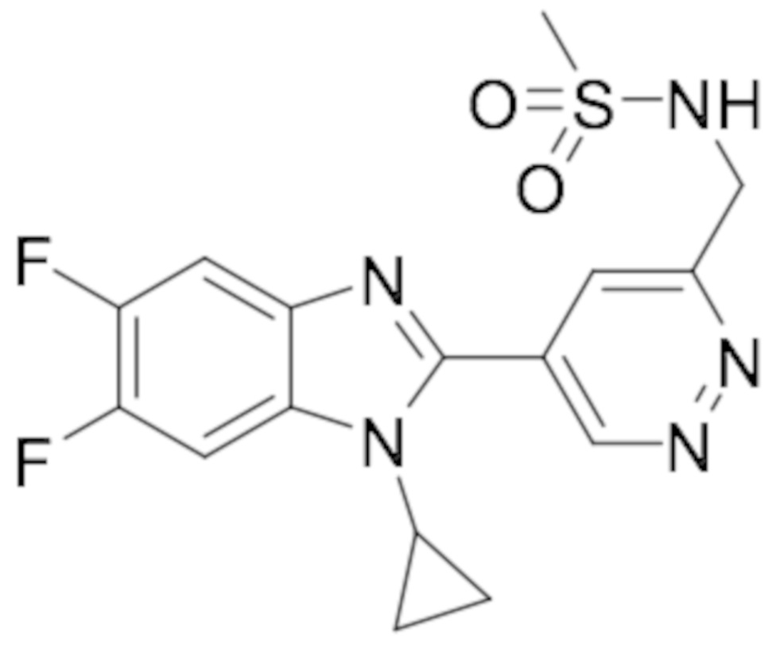
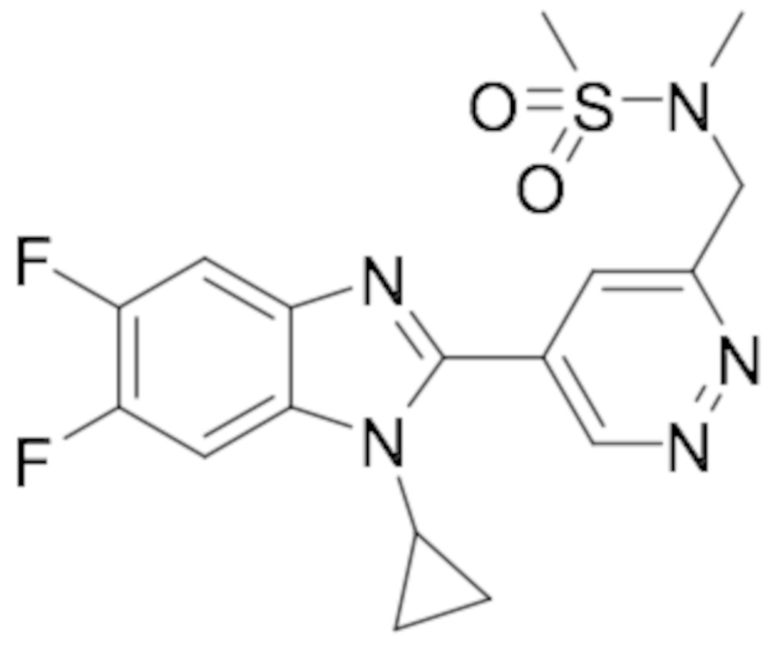
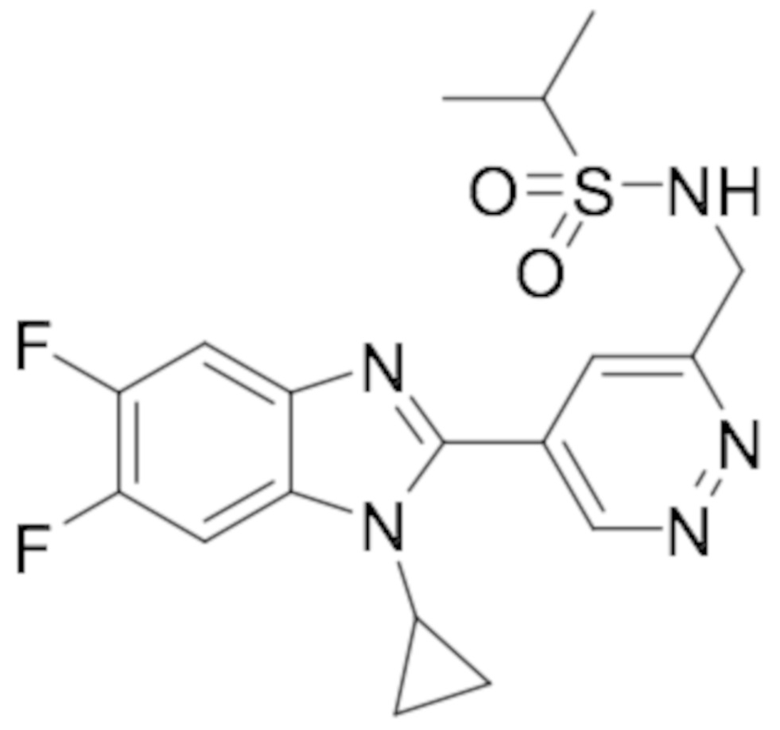
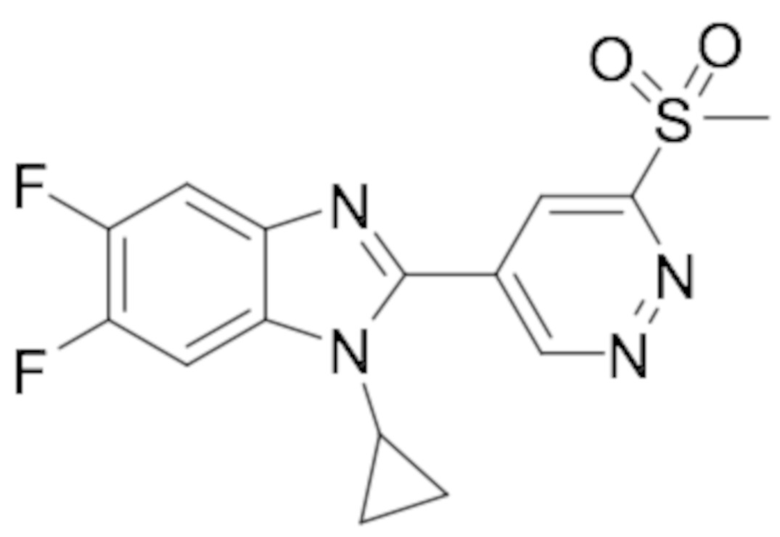

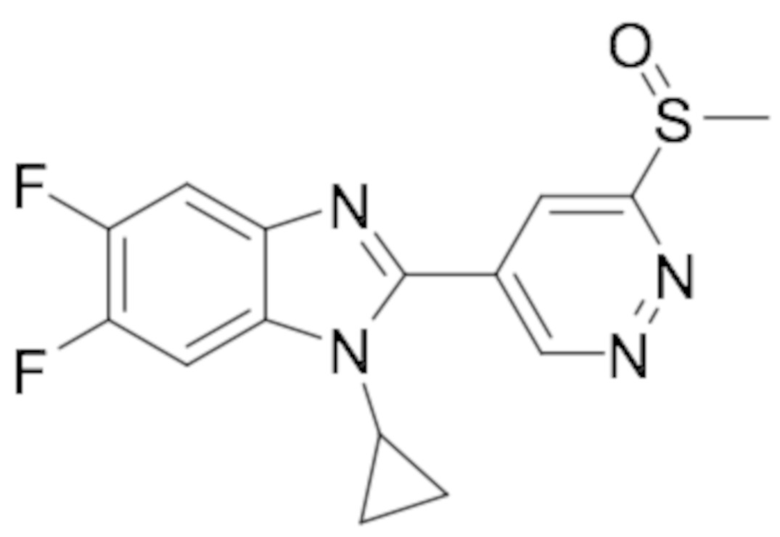

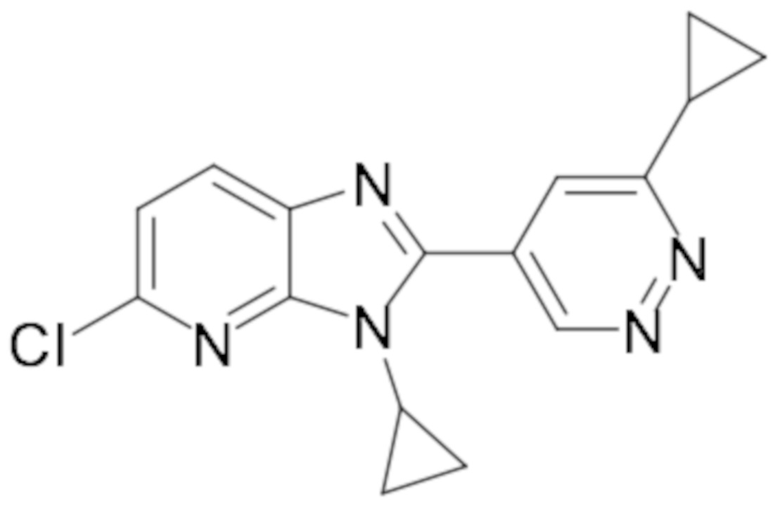
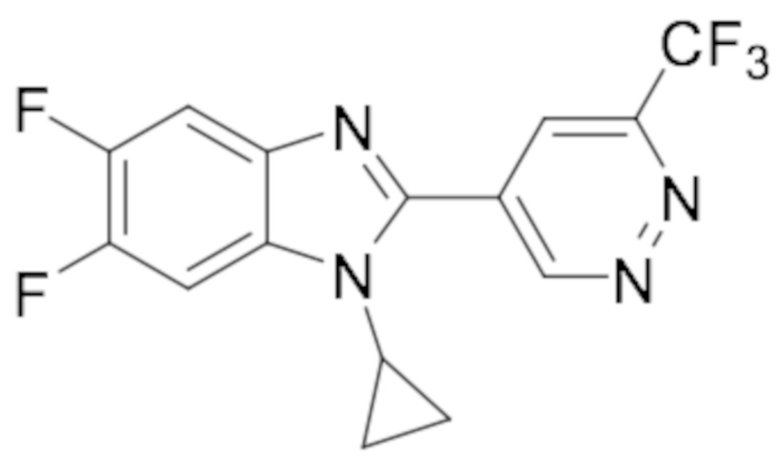
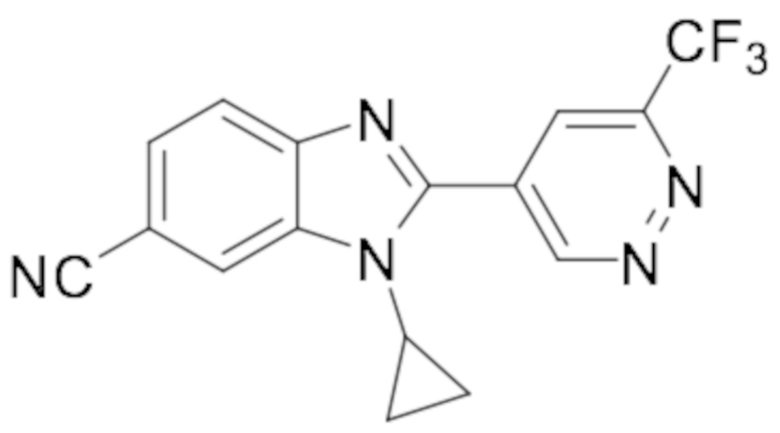
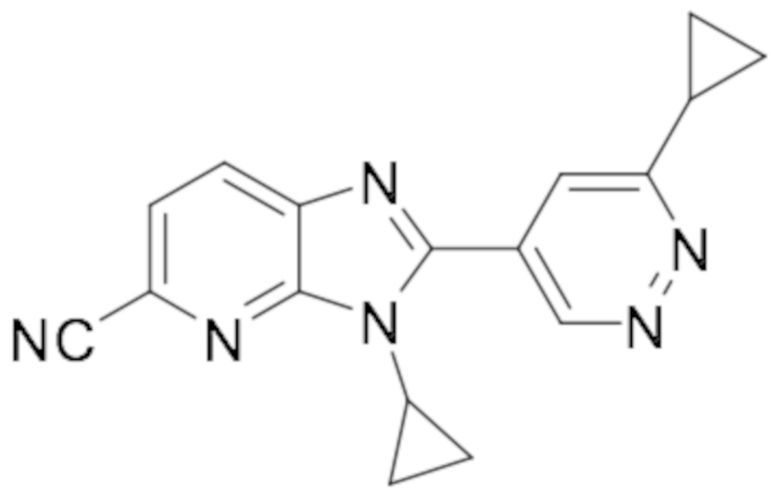
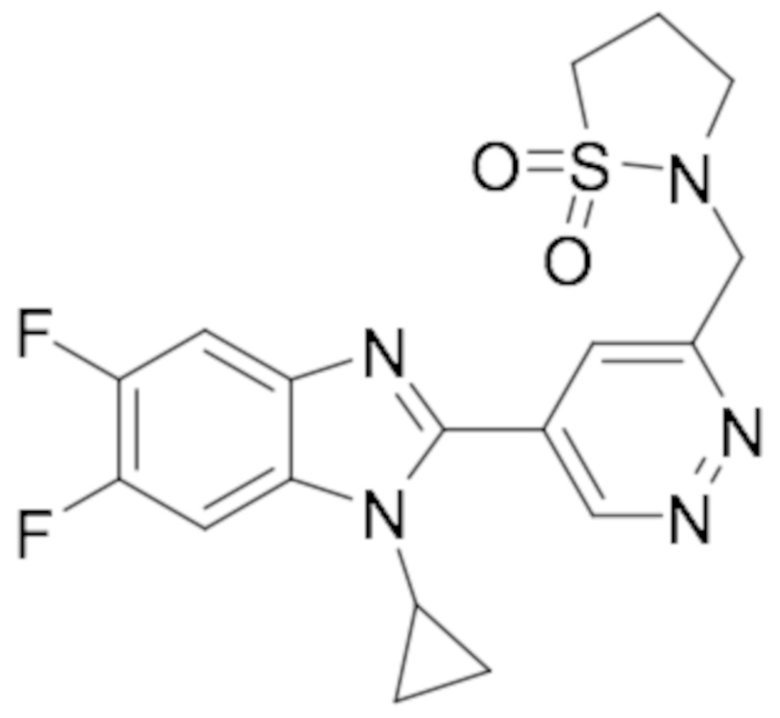
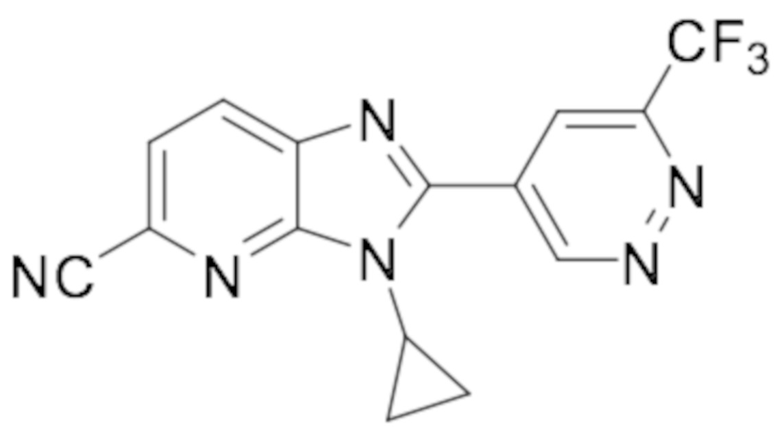
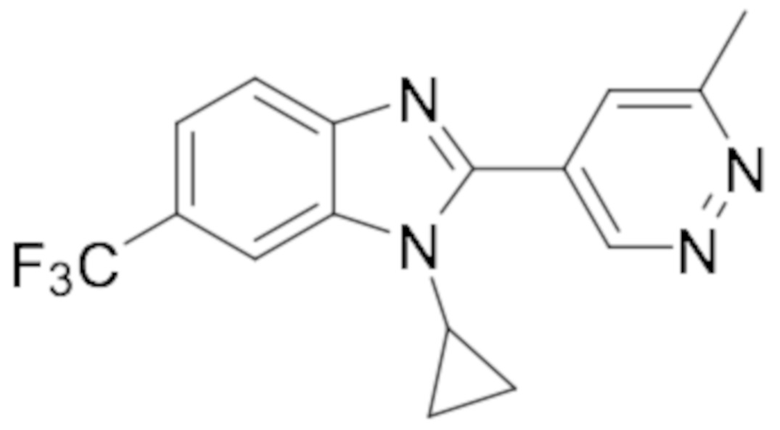



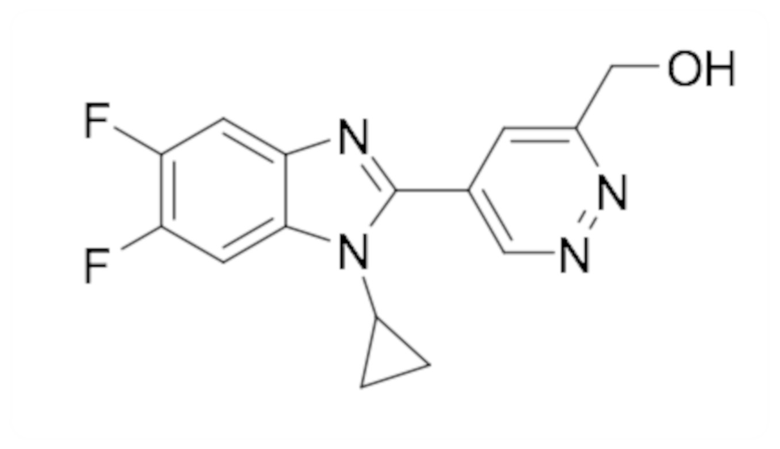
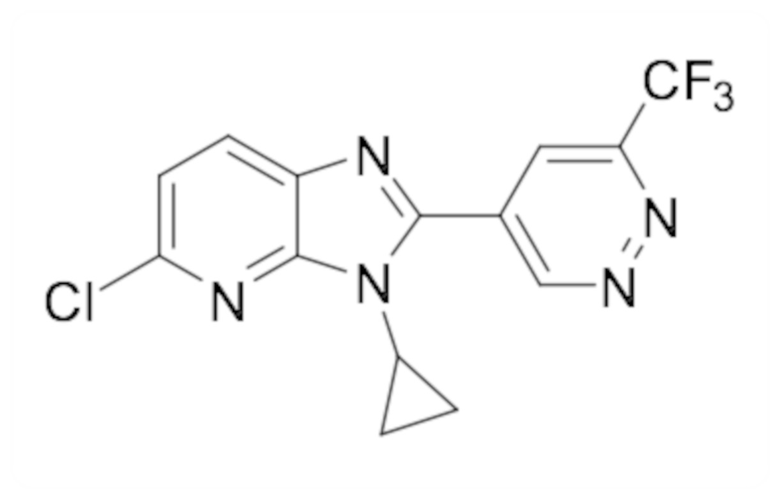

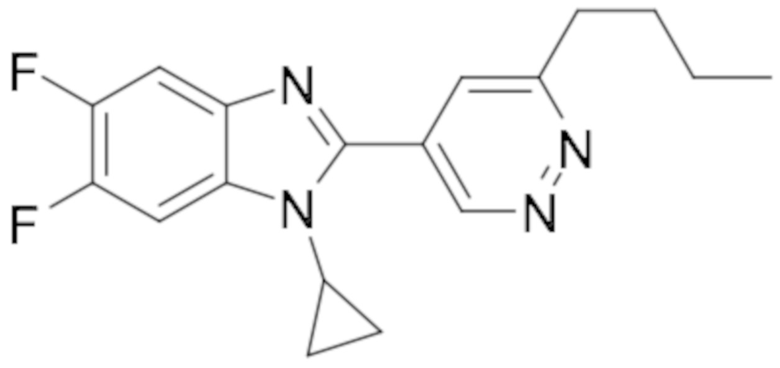
[M+H]e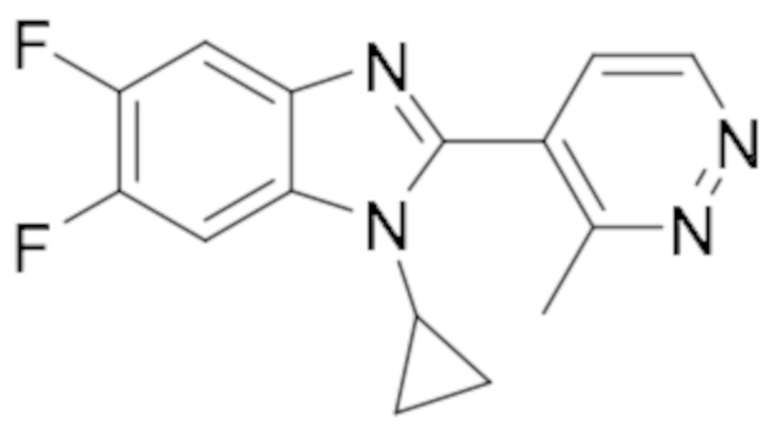
[M+H]b
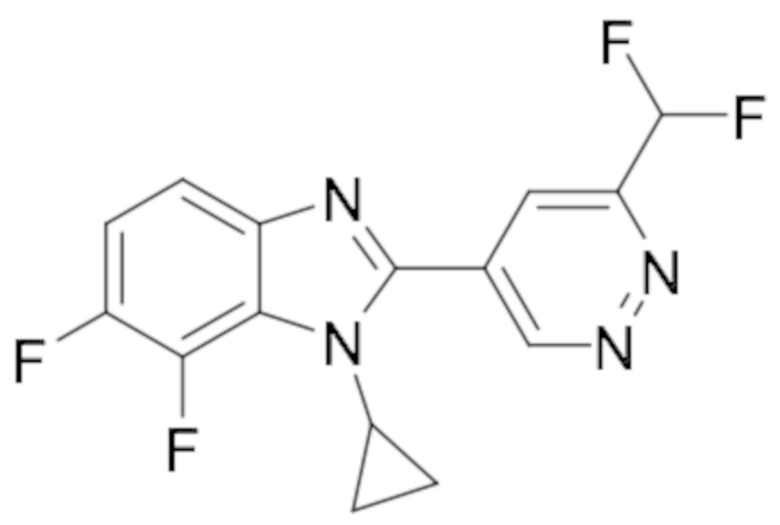
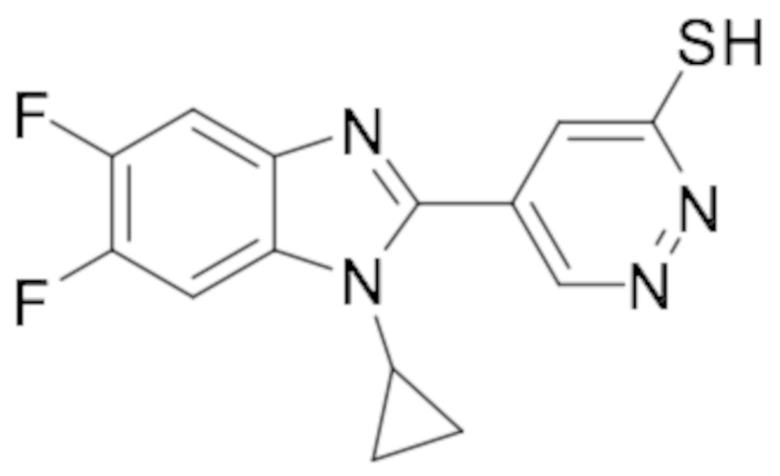
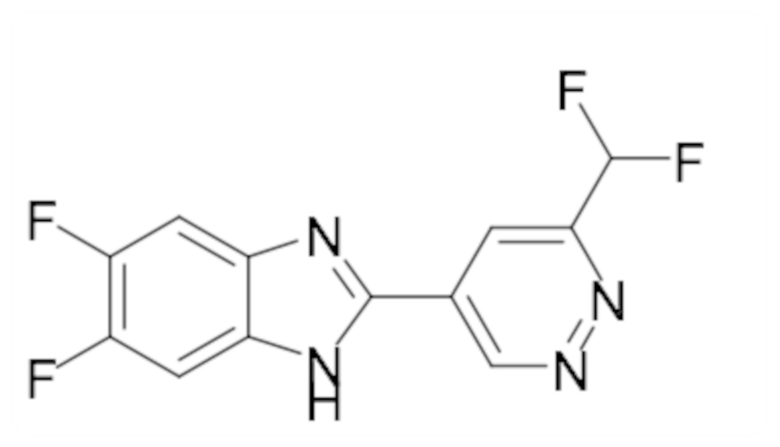
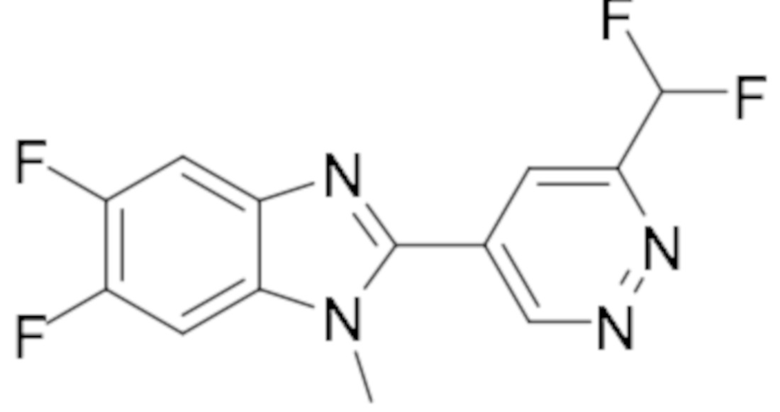
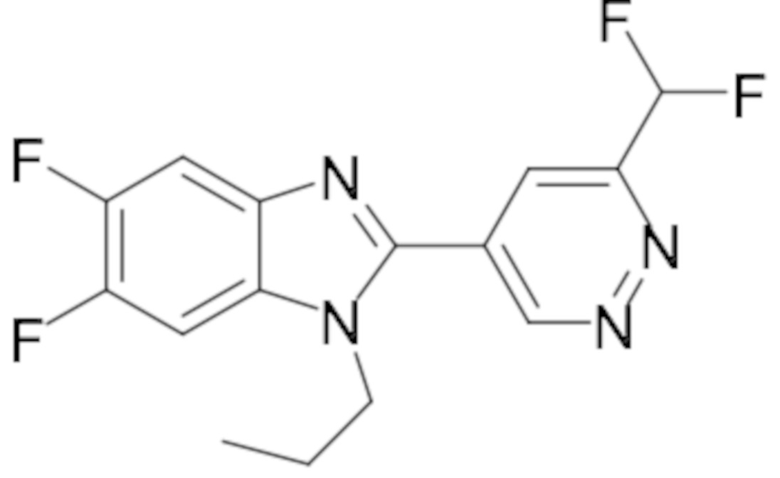
[M+H]c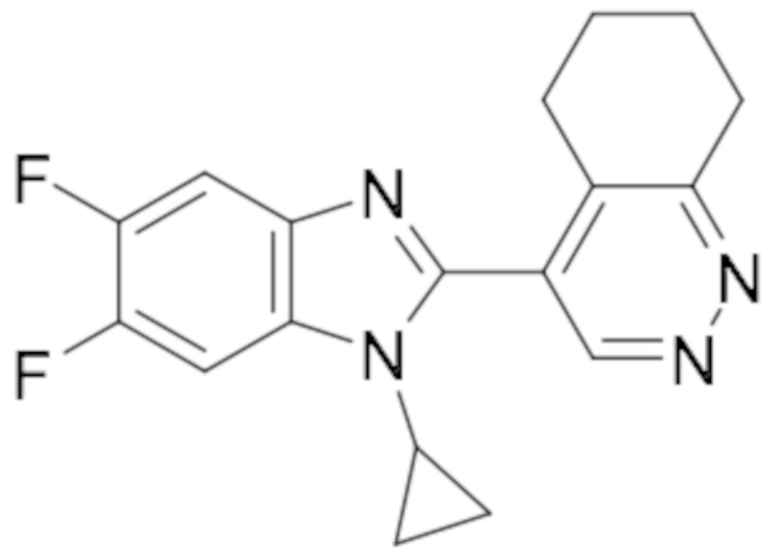
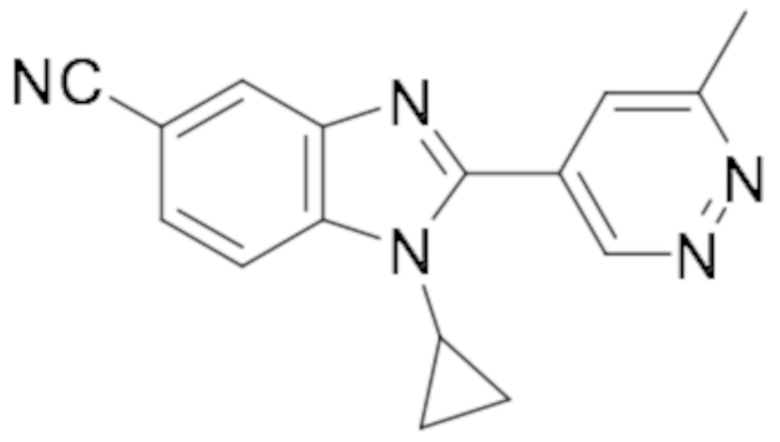
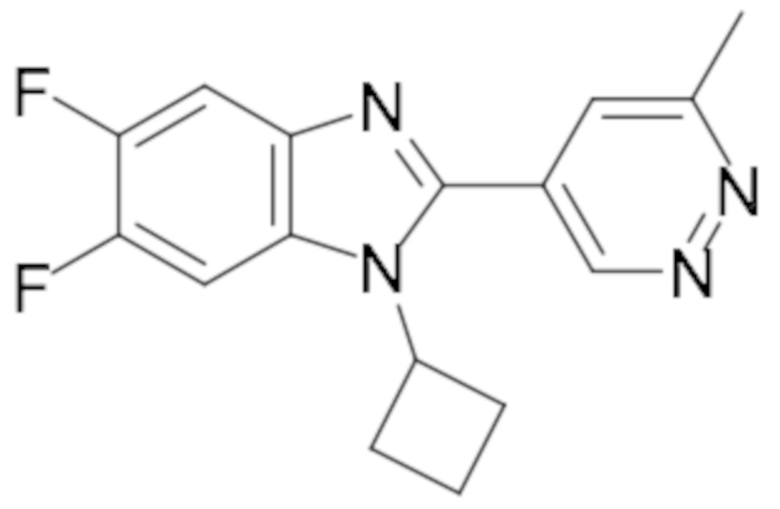

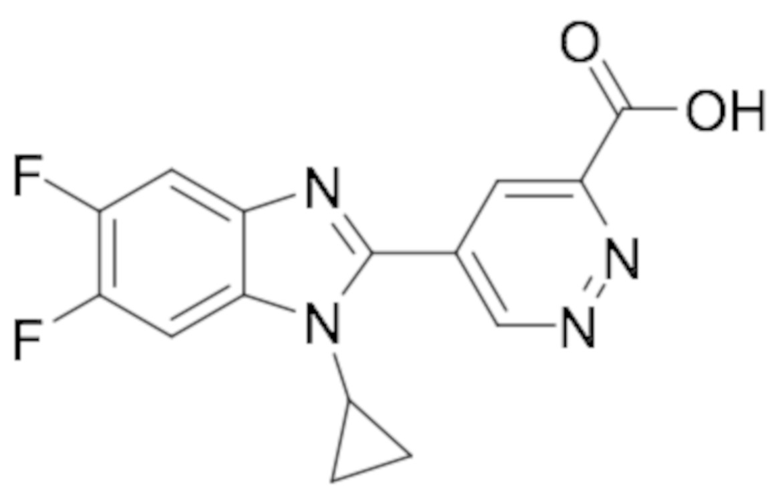
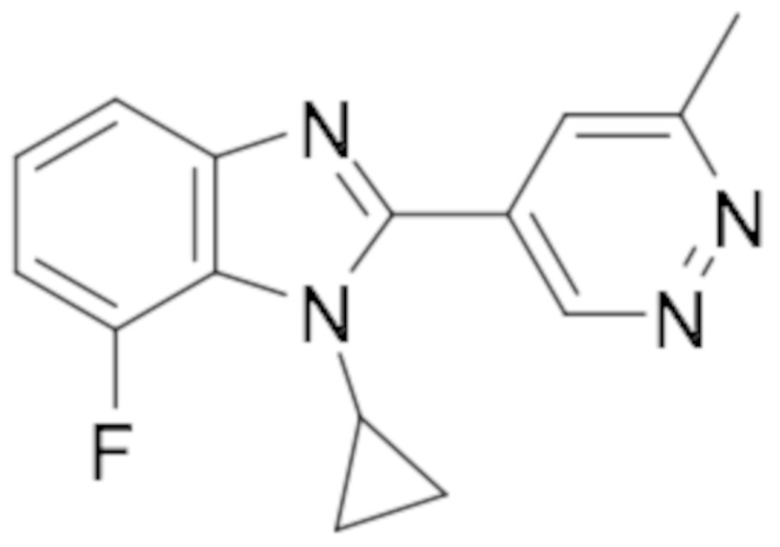
[M+H]b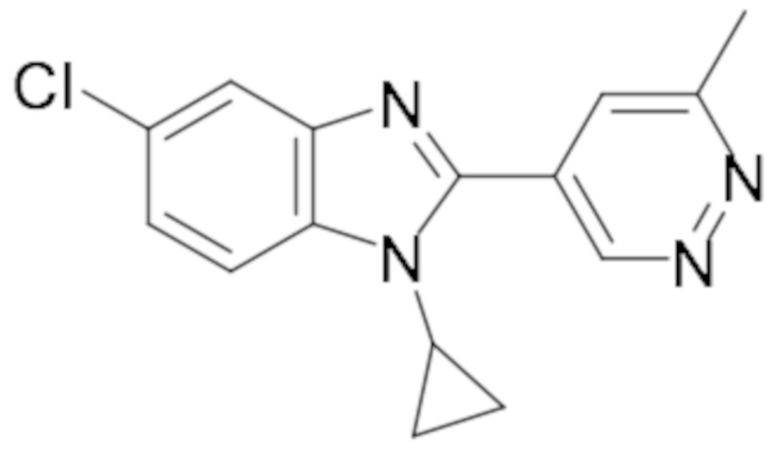
[M+H]c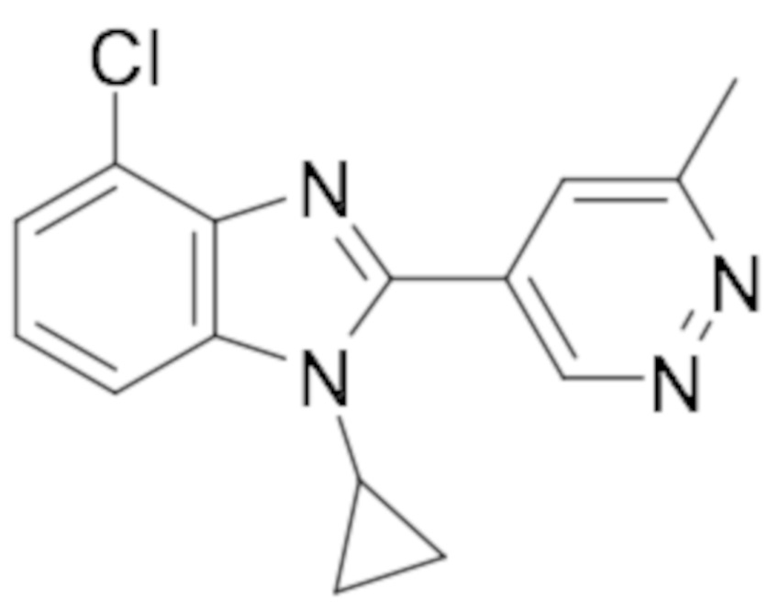

[M+H]c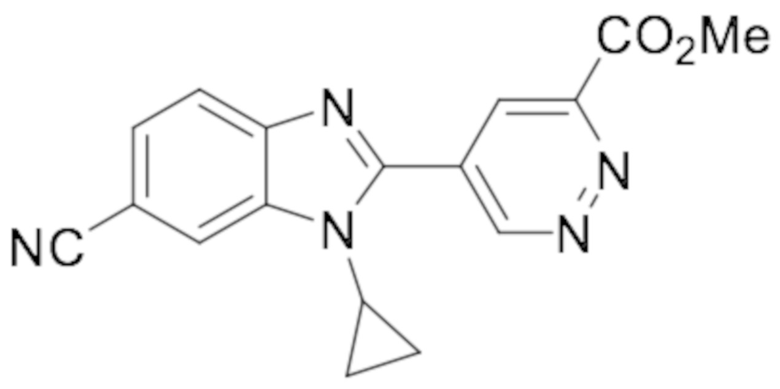
[M+H]c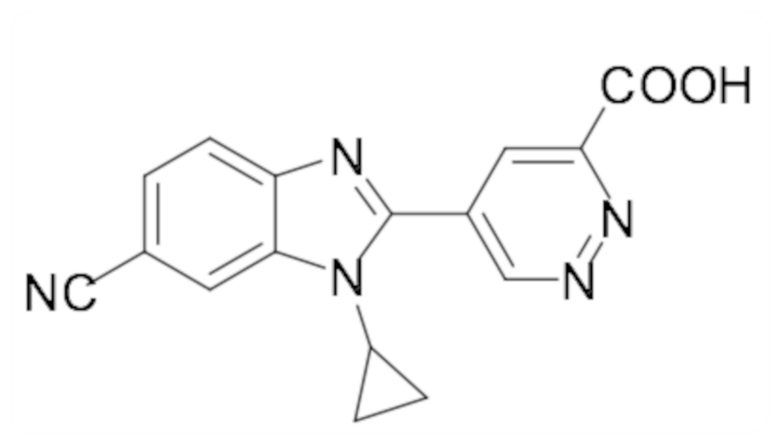

[M+H]e
[M+H]b
[M+H]b
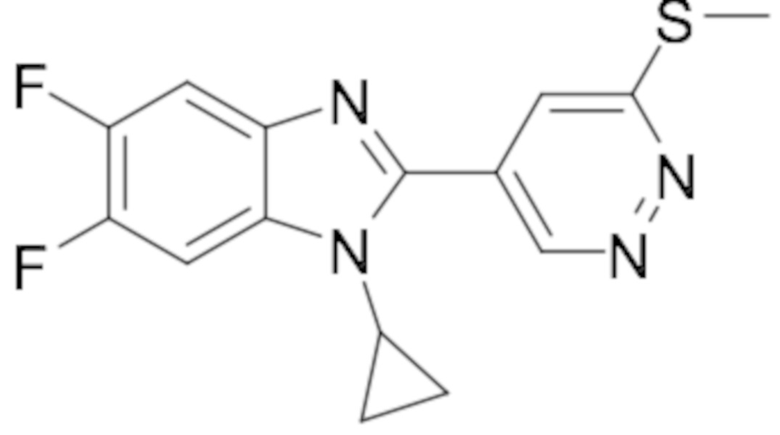
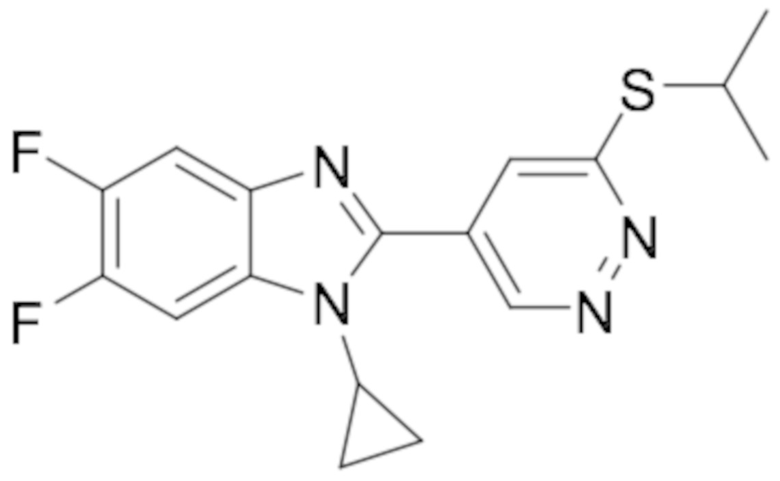
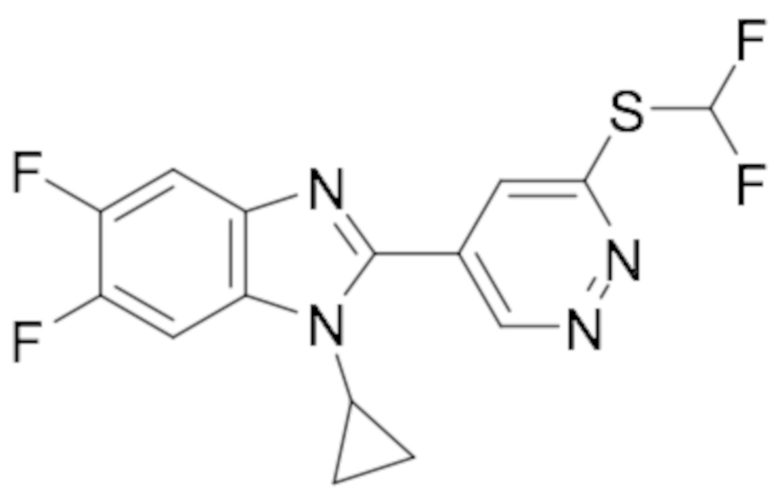
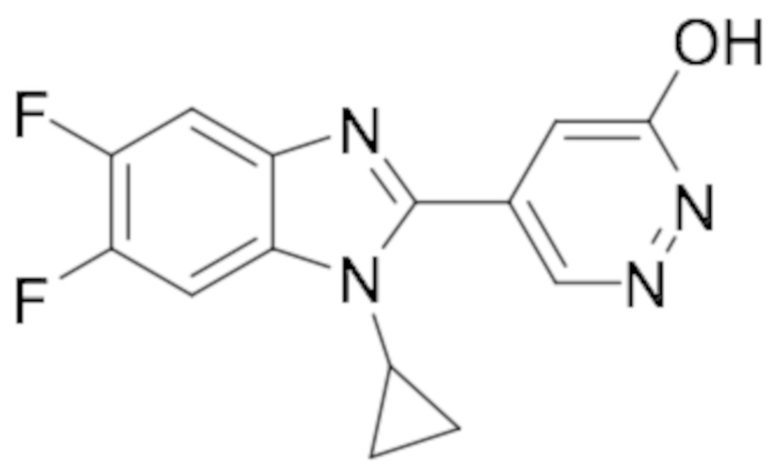
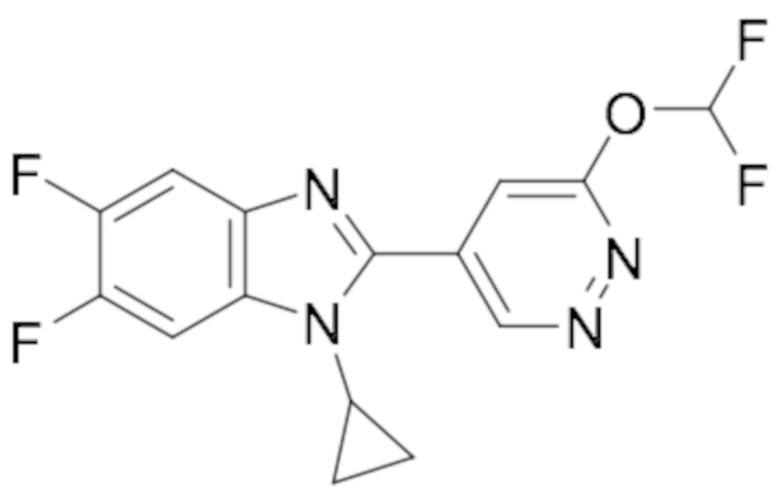
[M+H]b
[M+H]b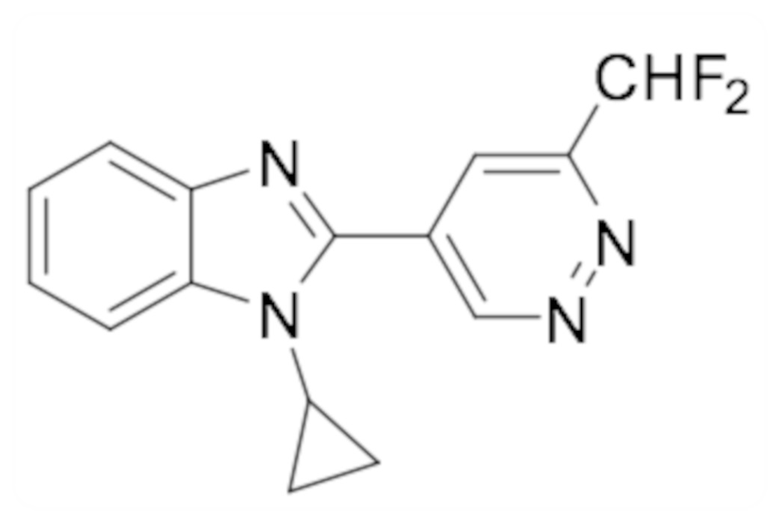
[M+H]b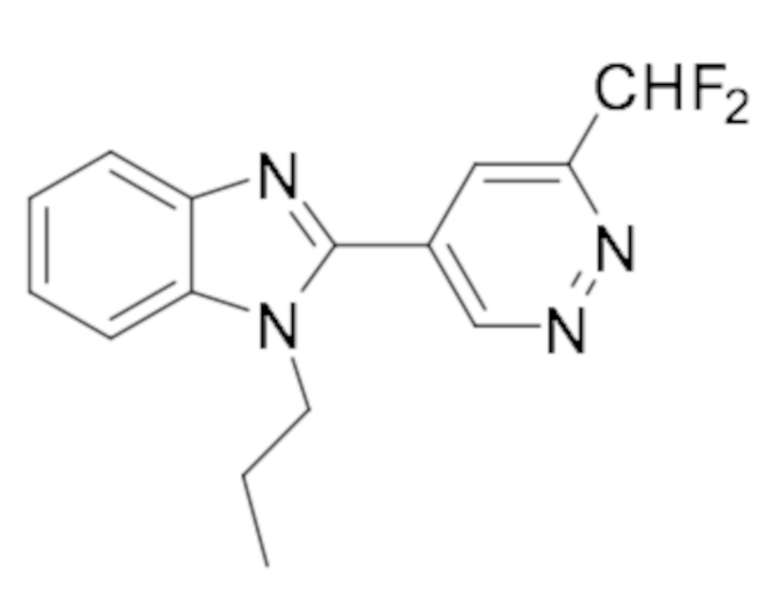
[M+H]c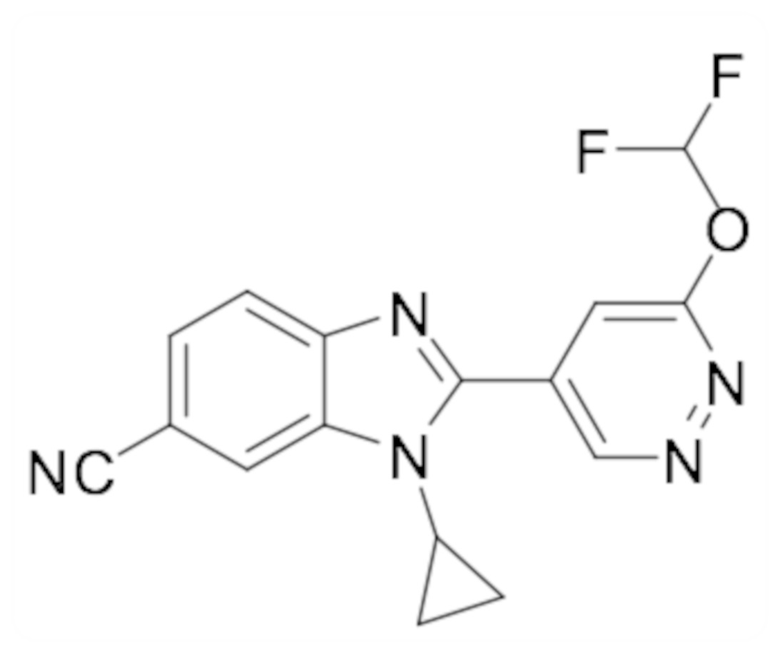
[M+H]b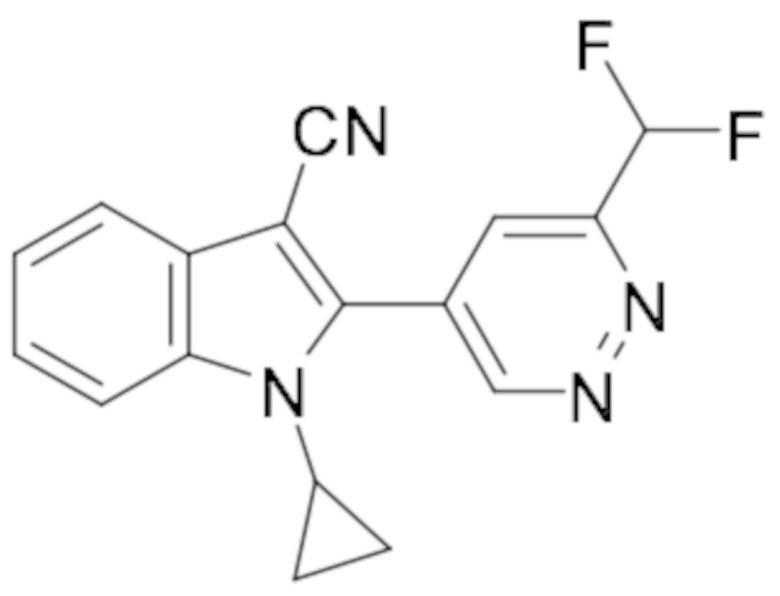
[M+H]b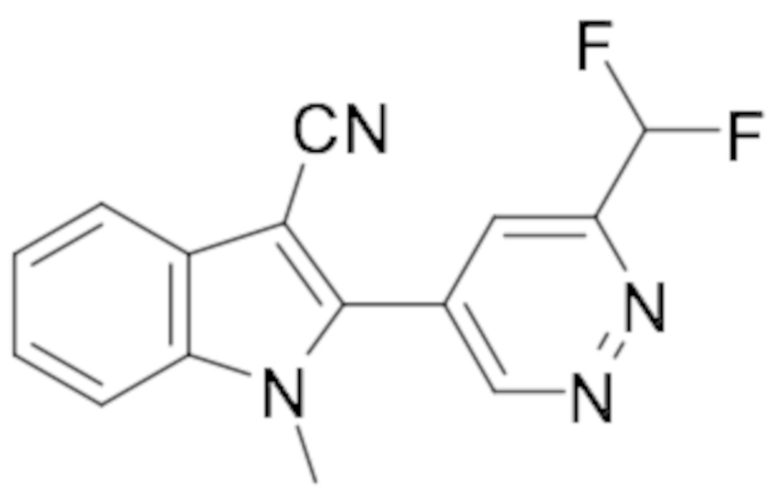
[M+H]b
f Смотрите таблицу 1 для ВЭЖХ и LCMS способов g IC50 диапазоны: + > 10 мкМ; ++ 1-10 мкМ; +++ 0.1-1 мкМ; ++++ 0.001-0.1 мкМ
Результаты в таблице 2 демонстрируют, что соединения формулы I обладают сильной активностью в отношении CYP11B2, и многие соединения формулы I обладают значительной селективностью в отношении ингибирования CYP11B2 по сравнению с CYP11B1.
Примеры сравнения были также получены. Таблицы 3 и 4 показывают, что замена пиридазина в соединениях формулы I пиридином приводит к соединениям (например, примеры 137-140), которые показывают повышенное ингибирование CYP11B1 (увеличение в 7-54 раза) для каждого из сравнений. Эти структурные изменения также приводят к соединениям, которые менее селективны в отношении CYP11B2 по сравнению с CYP11B1. Обе эти особенности нежелательны для ингибитора CYP11B2, поскольку, как описано выше, активность CYP11B1 является неотъемлемой частью продукции кортизола. Кроме того, примеры 14 и 42 продемонстрировали пониженное ингибирование CYP1A2 и CYP19 по сравнению с их соответствующими примерами сравнения. Примеры 42 и 19 также показали гораздо большую метаболическую стабильность при воздействии на микросомы печени яванских макак и крыс, чем их пиридиновые аналоги, примеры 139 и 140. Аналогично, в Таблице 5 показано, что известные ингибиторы CYP11B2, такие как Примеры 141-143, также обладают нежелательной активностью в отношении CYP11B1, ограничивая их эффективность в качестве терапевтических средств. Однако аналогичные примеры 31 и 52 являются гораздо менее эффективными ингибиторами CYP11B1. Примеры 31 и 52 также обладают большей метаболической стабильностью, чем пример 141. Взятые вместе, эти результаты демонстрируют, что пиридазиновые соединения формулы I потенциально имеют большую терапевтическую полезность для людей, чем известные соединения, отчасти из-за их пониженного ингибирования CYP11B1, повышенной селективности в отношении ингибирования CYP11B2 по сравнению с другими родственными металлоферментами и их метаболической стабильности.
Таблица 3. Результаты сравнения


a pKa (N2 пиридазинового атома или пиридина N), LogP (Chemaxon Method), и общая полярная площадь поверхности (tPSA), вычисленная с MarvinSketch 15.5.4.0
b Селективность = CYP11B1 IC50/CYP11B2 IC50
c Процент соединения, остающийся при 65 мин в микросомах печени крыс или яванских макак d ACS Med. Chem. Lett. 2015, 6, 573.
e WO 2012/012478
Таблица 4. Результаты сравнения
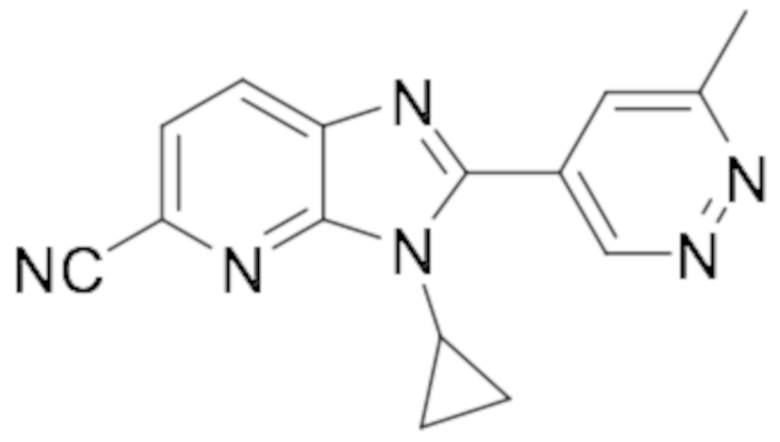
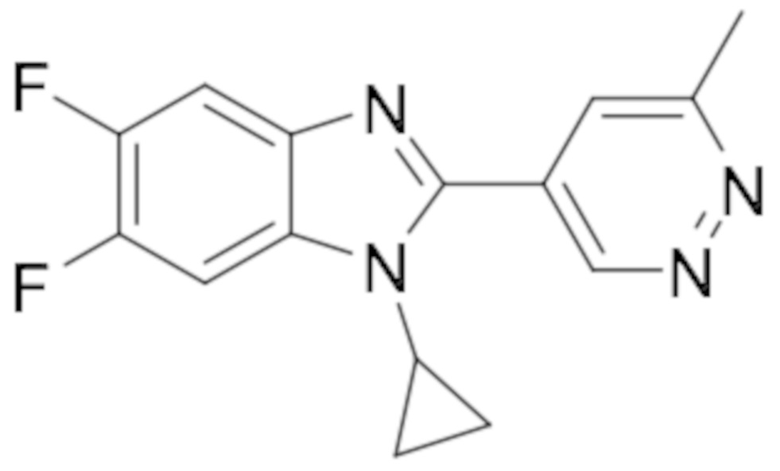
a pKa (N2 пиридазинового атома или пиридина N), LogP (Chemaxon Method), общая полярная площадь поверхности (tPSA), вычисленная с MarvinSketch 15.5.4.0
b Селективность = CYP11B1 IC50/CYP11B2 IC50
c Процент соединения, остающийся при 65 мин в микросомах печени крыс или яванских макак
Таблица 5. Результаты сравнения


(мкм)
(мкм)
(мкм)
(мкм)
Стабильно-стьc
Стабильно-стьc
a pKa (N2 пиридазинового атома или пиридина N), вычисленная с помощью MarvinSketch 15.5.4.0
b селективность = CYP11B1 IC50/CYP11B2 IC50
c Процент соединения, остающийся при 65 мин в микросомах печени крыс или яванских макак
d J. Med. Chem. 2015, 58, 9382.
e ACS Med. Chem. Lett. 2013, 4, 1203.
Включение посредством ссылки
Содержания всех ссылочных источников (включая литературные источники, выданные патенты, опубликованные заявки на выдачу патента и совместно рассматриваемые патентные заявки), процитированные в настоящей заявке, таким образом, определенно включены в настоящую заявку в их полном объеме посредством ссылки.
Эквиваленты
Специалистам в данной области техники очевидны или они могут установить, без проведения дополнительных рутинных экспериментов, множество эквивалентов конкретных вариантов выполнения описанного в настоящей заявке изобретения. Такие эквиваленты, как подразумевается, охватываются приложенной формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
| МОДУЛЯТОРЫ ПИРУВАТКИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2797518C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2019 |
|
RU2812128C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ KDM5, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2840256C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
Изобретение относится к соединению, выбранному из:
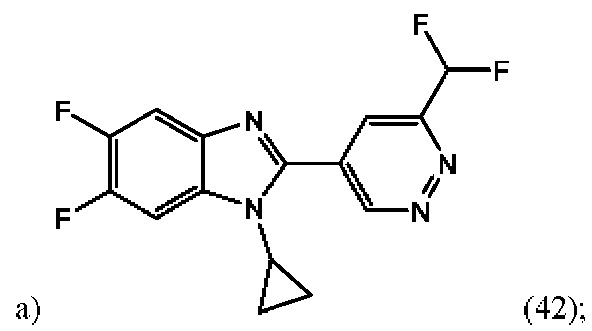
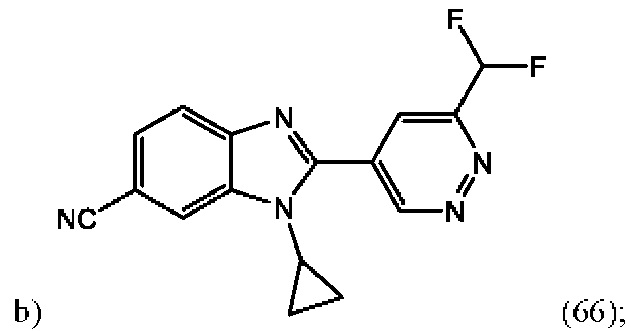
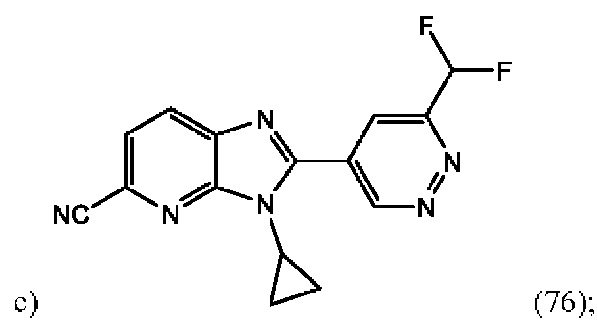
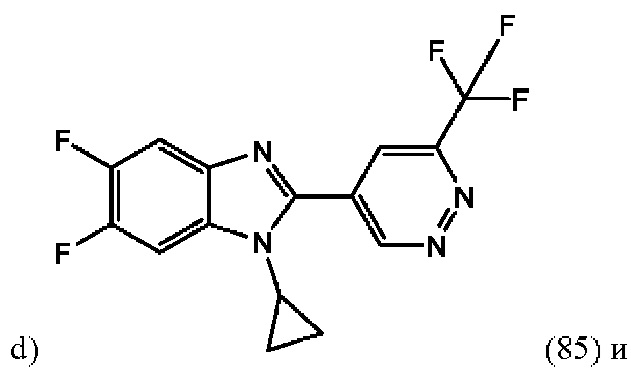
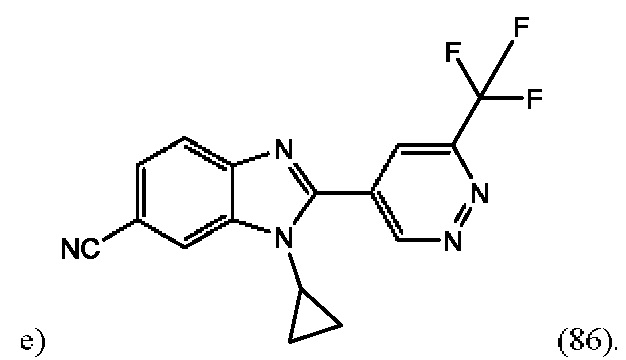
Также предложены способ ингибирования и способ модуляции активности металлофермента альдостеронсинтазы (CYP11B2), способы лечения заболеваний или нарушений, вызванных таким металлоферментом, и фармацевтическая композиция. Предложенные соединения способны ингибировать активность металлофермента альдостеронсинтазы (CYP11B2) и могут быть использованы для лечения заболеваний или нарушений, вызванных таким металлоферментом. 7 н. и 8 з.п. ф-лы, 5 табл., 137 пр.
1. Соединение, выбранное из:
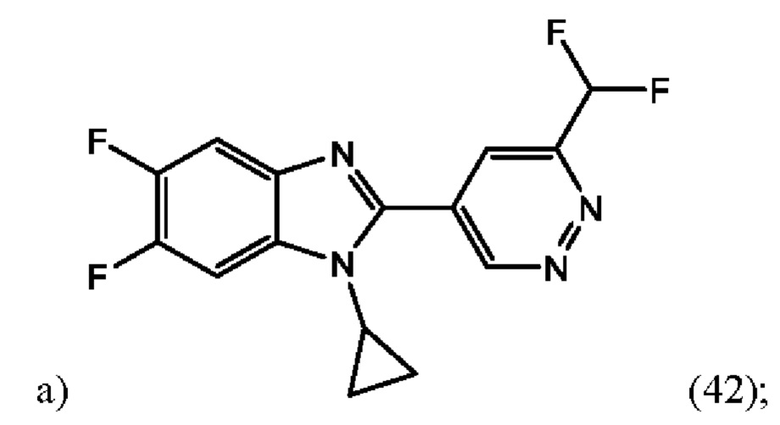

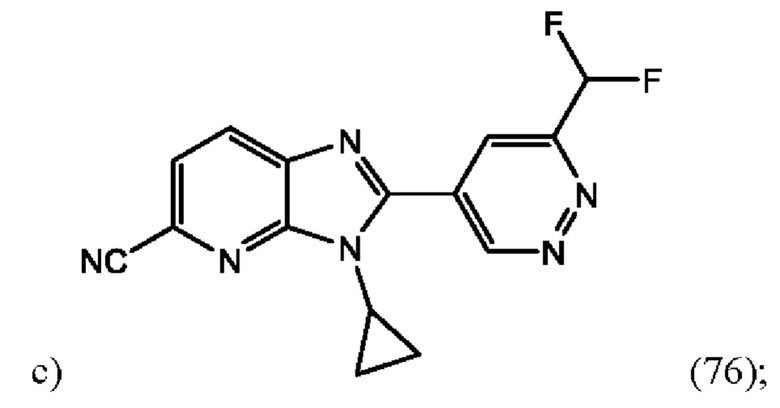
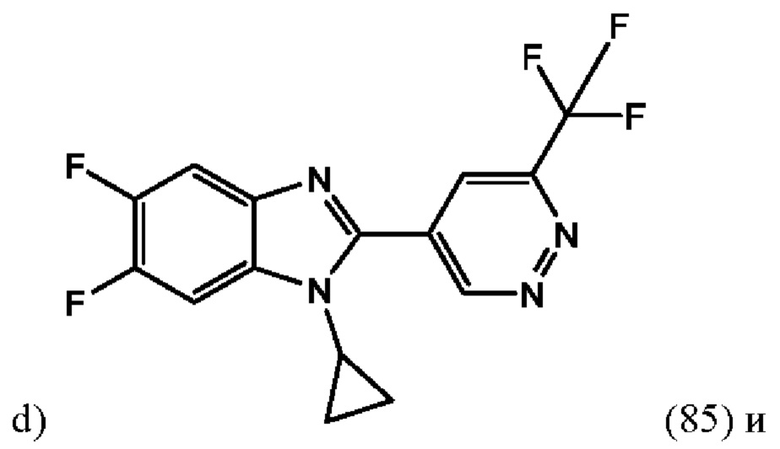
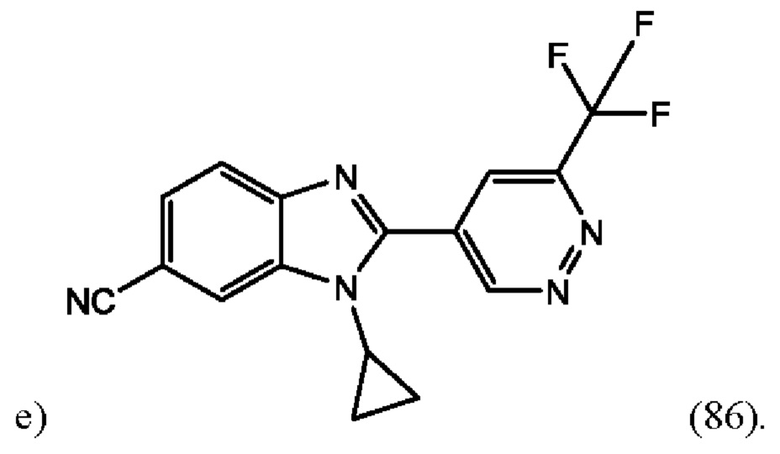
2. Способ ингибирования активности металлофермента альдостеронсинтазы (CYP11B2), включающий контакт соединения по п. 1 с металлоферментом.
3. Способ по п. 2, где контакт осуществляется in vivo.
4. Способ по п. 2, где контакт осуществляется in vitro.
5. Способ по п. 2, дополнительно включающий введение соединения субъекту-человеку.
6. Способ модуляции активности металлофермента альдостеронсинтазы (CYP11B2) у субъекта, включающий контакт субъекта с соединением по п. 1 в количестве и при условиях, достаточных для понижающей регуляции или ингибирования активности металлофермента.
7. Способ лечения субъекта, страдающего от или подверженного вызванному металлоферментом альдостеронсинтазой (CYP11B2) нарушению или заболеванию, включающий введение субъекту эффективного количества соединения по п. 1.
8. Способ лечения субъекта, страдающего от или подверженного вызванному металлоферментом альдостеронсинтазой (CYP11B2) нарушению или заболеванию, где субъект был идентифицирован как нуждающийся в лечении вызванного металлоферментом нарушения или заболевания, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по п. 1, так что указанный субъект лечится от указанного заболевания или нарушения.
9. Способ лечения субъекта, страдающего от или подверженного вызванному металлоферментом альдостеронсинтазой (CYP11B2) нарушению или заболеванию, где субъект был идентифицирован как нуждающийся в лечении вызванного металлоферментом нарушения или заболевания, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по п. 1, так что активность металлофермента у указанного субъекта понижающе регулируется или ингибируется.
10. Способ по любому из пп. 7-9, где нарушение или заболевание независимо представляет собой рак, сердечно-сосудистое заболевание, воспалительное заболевание, инфекционное заболевание, нарушение обмена веществ, офтальмологическое заболевание, заболевание центральной нервной системы (ЦНС), урологическое заболевание или желудочно-кишечное заболевание.
11. Способ по любому из пп. 7-10, где нарушением или заболеванием является артериальная гипертония, резистентная артериальная гипертония, смертность, связанная с первичным или вторичным гиперальдостеронизмом или гиперплазией надпочечников, легочно-артериальная гипертония, порок сердца, диастолическая дисфункция, диастолическая дисфункция левого желудочка, диастолический порок сердца, систолическая дисфункция, систолический порок сердца, гипокалиемия, почечная недостаточность, хроническая почечная недостаточность, рестеноз, нефропатия, постмиокардиальный инфаркт, ишемическая болезнь сердца, фиброз, заболевания, характеризующиеся повышенным образованием коллагена, фиброз и матриксное ремоделирование после артериальной гипертонии, и матриксное ремоделирование после дисфункции эндотелиальных клеток, сердечно-сосудистые заболевания, как например атеросклероз, фибрилляция предсердий, почечная дисфункция, заболевания печени, неалкогольный стеатогепатит, сосудистые заболевания, ретинопатия, заболевание нервной системы, инсулинопатия, эндотелиальная дисфункция, миокардиальный фиброз, сосудистый фиброз, миокардиальные некротические повреждения, сосудистое повреждение, инфаркт миокарда, гипертрофия левого желудочка, гипертрофия стенок сосудов, эндотелиальное сгущение, фибриноидный некроз артерий, заболевания почек, диабетическая нефропатия, гломерулосклероз, гломерулонефрит, нефритический синдром, поликистозная болезнь почек, сахарный диабет, метаболический синдром, резистентность к инсулину, синдром апноэ, обструктивный синдром апноэ, мышечная дистрофия, цирроз печени, неалкогольный стеатогепатоз, нарушение функции почек, диабетическое нарушение функции почек или инсульт.
12. Фармацевтическая композиция для ингибирования активности металлофермента альдостеронсинтазы (CYP11B2) у субъекта, содержащая соединение по п. 1 и фармацевтически приемлемый носитель.
13. Фармацевтическая композиция по п. 12, дополнительно содержащая дополнительное терапевтическое средство.
14. Фармацевтическая композиция по п. 12, дополнительно содержащая, дополнительное терапевтическое средство, которое представляет собой противораковое средство, противогрибковое средство, сердечно-сосудистое средство, противовоспалительное средство, химиотерапевтическое средство, средство для развития кровеносных сосудов, цитотоксическое средство, средство против пролиферации, средство против нарушения обмена веществ, средство против офтальмологического заболевания, средство против заболевания центральной нервной системы (ЦНС), средство против урологического заболевания или средство против желудочно-кишечного заболевания.
15. Способ по любому из пп. 7-11, где субъектом является млекопитающее, отличное от человека.
| US 2008255085 A1, 16.10.2008 | |||
| HOYT S | |||
| B | |||
| et al., Discovery of Benzimidazole CYP11B2 Inhibitors with in Vivo Activity in Rhesus Monkeys, ACS Medicinal Chemistry Letters, 2015, vol | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Котел для водяного отопления с внутренним перегревателем воды для побуждения циркуляции в сети и с регулятором наружной температуры котла | 1924 |
|
SU573A1 |
| [PUBCHEM] PubChem SID 45792664, введено 21.06.2010 | |||
| ДЕАЗАПУРИНЫ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2380368C2 |

