УРОВЕНЬ ТЕХНИКИ
Триптофан (Trp) является незаменимой аминокислотой, необходимой для биосинтеза белков, ниацина и нейромедиатора 5-гидрокситриптамина (серотонина). Фермент индоламин-2,3-диоксигеназа (IDO) катализирует первую и скорость-лимитирующую стадию деградации L-триптофана до N-формилкинуренина. В клетках человека истощение Trp, являющееся результатом активности IDO, является важным индуцируемым интерфероном гамма (ИФНγ) противомикробным эффекторным механизмом. Стимуляция ИФНγ индуцирует активацию IDO, что приводит к истощению Trp, таким образом, приводя к прекращению роста Trp-зависимых внутриклеточных патогенов, таких как Toxoplasma gondii и Chlamydia trachomatis. Активность IDO также обладает антипролиферативным эффектом в отношении многих опухолевых клеток, и индукцию IDO наблюдают in vivo во время отторжения аллогенных опухолей, что свидетельствует о возможной роли этого фермента в процессе отторжения опухоли (Daubener, et al, 1999, Adv. Exp. Med. Biol, 467: 517-24; Taylor, et al, 1991, FASEB J., 5: 2516-22). Показано, что клетки HeLa, сокультивируемые с лимфоцитами периферической крови (PBL), приобретают иммуноингибиторный фенотип посредством положительной регуляции активности IDO. Считают, что снижение пролиферации PBL после обработки интерлейкином-2 (ИЛ-2) возникает из-за IDO, высвобождаемой опухолевыми клетками в ответ на секрецию ИФНγ PBL. Этот эффект реверсируют обработкой 1-метилтриптофаном (IMT), специфическим ингибитором IDO. Предполагают, что активность IDO в опухолевых клетках может нарушать противоопухолевые ответы (Logan, et al, 2002, Immunology, 105: 478-87).
Несколько линий доказательств позволяют предполагать, что IDO участвует в индукции иммунологической толерантности. Исследования беременности у млекопитающих, опухолевой резистентности, хронических инфекций и аутоиммунных заболеваний показали, что клетки, экспрессирующие IDO, могут супрессировать T-клеточные ответы и способствовать толерантности. Ускоренный катаболизм Trp наблюдают при заболеваниях и нарушениях, ассоциированных с клеточной активацией иммунитета, таких как инфекции, злокачественные новообразования, аутоиммунные заболевания и СПИД, а также во время беременности. Например, при аутоиммунных заболеваниях определяют повышенные уровни ИФН и повышенные уровни метаболитов Trp в моче; предполагают, что системное или локальное истощение Trp, возникающее при аутоиммунных заболеваниях, может быть связано с деградацией и симптомами истощения при этих заболеваниях. В поддержку этой гипотезы, высокие уровни IDO определяли в клетках, выделенных из синовиальной жидкости суставов при артрите. ИФН также повышены у пациентов с вирусом иммунодефицита человека (ВИЧ), и повышенные уровни ИФН ассоциированы с ухудшением прогноза. Таким образом, предполагают, что IDO хронически индуцирована при инфекции ВИЧ и дополнительно повышается при оппортунистических инфекциях, и что хроническая потеря Trp запускает механизмы, отвечающие за кахексию, деменцию и диарею и, возможно, иммуносупрессию у пациентов со СПИД (Brown, et al., 1991, Adv. Exp. Med. Biol, 294: 425-35). В связи с этим, недавно показано, что ингибирование IDO может повышать уровни вирус-специфических T-клеток и одновременно снижать количество инфицированных вирусом макрофагов в модели ВИЧ на мышах (Portula et al., 2005, Blood, 106: 2382-90).
Считают, что IDO играет роль в иммуносупрессорных процессах, предотвращающих отторжение плода in utero. Более 40 лет назад было показано, что во время беременности генетически отличающийся концептус млекопитающего выживал, несмотря на то, что можно было бы предположить, исходя из иммунологии трансплантации тканей (Medawar, 1953, Symp. Soc. Exp. Biol. 7: 320-38).
Анатомическое разделение матери и плода и антигенная незрелость плода не могут полностью объяснить выживание аллотрансплантата плода. В последнее время внимание сфокусировано на иммунологической толерантности матери. Т.к. IDO экспрессируется синцитиотрофобластными клетками человека, и системная концентрация триптофана падает во время нормальной беременности, предполагали, что экспрессия IDO на трансплацентарном барьере необходима для предотвращения иммунологического отторжения аллотрансплантатов плода. Для проверки этой гипотезы беременных мышей (несущих сингенные или аллогенные плоды) подвергали IMT и наблюдали быстрое, индуцируемое T-клетками отторжение всех аллогенных концептусов. Таким образом, катаболизируя триптофан, концептус млекопитающего, по-видимому, супрессирует активность T-клеток и защищает себя от отторжения, и блокирование катаболизма триптофана во время беременности мыши позволяет материнским T-клеткам провоцировать отторжение аллотрансплантата плода (Moan, et al., 1998, Science, 281: 1191-3).
Дополнительное доказательство того, что механизм опухолевой иммунологической резистентности основан на деградации триптофана IDO, получено при наблюдении того, что большинство опухолей человека конститутивно экспрессируют IDO, и что экспрессия IDO иммуногенными опухолевыми клетками мыши предотвращает их отторжение предварительно иммунизированными мышами. Этому эффекту сопутствует отсутствие накопления специфических T-клеток в очаге опухоли, и он может быть частично реверсирован посредством системного введения мышам ингибитора IDO в отсутствие значительной токсичности. Таким образом, предполагают, что эффективность терапевтической вакцинации пациентов со злокачественными новообразованиями можно улучшать посредством одновременного введения ингибитора IDO (Uyttenhove et al., 2003, Nature Med., 9: 1269-74). Также показано, что ингибитор IDO 1-MT может действовать синергически с химиотерапевтическими средствами, снижая рост опухоли у мышей, что позволяет предполагать, что ингибирование IDO также может повышать противоопухолевую активность общепринятой цитотоксической терапии (Muller et al, 2005, Nature Med., 11: 312-9).
Одним из механизмов, вносящих вклад в иммунологическую неотвечаемость в отношении опухолей, может являться презентирование опухолевых антигенов толерогенными APC организма-хозяина. Описана субпопуляция IDO-экспрессирующих антигенпрезентирующих клеток (APC) человека, коэкспрессирующих CD123 (IL3RA) и CCR6 и ингибирующих пролиферацию T-клеток. Зрелые и незрелые CD123-положительные дендритные клетки супрессировали активность T-клеток, и эту супрессорную активность IDO блокировали с помощью 1MT (Munn, et al, 2002, Science, 297: 1867-70). Также показано, что дренирующие опухоль лимфоузлы (TDLN) мыши содержат субпопуляцию плазмоцитоидных дендритных клеток (pDC), конститутивно экспрессирующих иммуносупрессорные уровни IDO. Несмотря на то, что они составляют лишь 0,5% клеток лимфоузла, in vitro эти pDC мощно супрессируют T-клеточные ответы на антигены, презентируемые самими pDC, а также с доминантном режиме супрессируют T-клеточные ответы на сторонние антигены, презентируемые несупрессорными APC. В популяции pDC основная часть функциональной IDO-опосредованной супрессорной активности разделена с новой субпопуляцией pDC, коэкспрессирующих B-лимфоцитарный маркер CD19. Таким образом, предполагают, что IDO-опосредованная супрессия под действием pDC в TDLN создает локальное микроокружение, мощно супрессирующее противоопухолевые T-клеточные ответы организма-хозяина (Munn, et al., 2004, J. Clin. Invest, 114(2): 280-90).
IDO вызывает деградацию индолового остатка триптофана, серотонина и мелатонина и инициирует продукцию нейроактивных и иммунорегуляторных метаболитов, в совокупности известных как кинуренины. С помощью локального истощения триптофана и повышения проапоптотических кинуренинов IDO, экспрессируемая дендритными клетками (DC), может в значительной степени влиять на пролиферацию и выживание T-клеток. Индукция IDO в DC может являться общим механизмом делеционной толерантности, регулируемой регуляторными T-клетками. Т.к. можно ожидать, что такие толерогенные ответы будут возникать при разных патофизиологических состояниях, метаболизм триптофана и продукция кинуренина могут представлять собой ключевую область контакта между иммунной и нервной системами (Grohmann, et al, 2003, Trends Immunol, 24: 242-8). В состоянии непрерывной иммунной активации доступность свободного Trp в сыворотке уменьшается, и вследствие сниженной продукции серотонина влиянию также могут подвергаться серотонинергические функции (Wirleitner, et al., 2003, Curr. Med. Chem., 10: 1581-91).
В свете потенциальной роли IDO в иммуносупрессии, резистентности и/или отторжении опухоли, хронических инфекциях, инфекции ВИЧ, СПИД (включая такие его проявления, как кахексия, деменция и диарея), аутоиммунных заболеваниях или нарушениях (таких как ревматоидный артрит), иммунологической толерантности и предотвращении отторжения плода in utero, желательными являются терапевтические средства, направленные на супрессию деградации триптофана посредством ингибирования активности IDO. Ингибиторы IDO можно использовать для активации T-клеток и, таким образом, повышения активации T-клеток, когда T-клетки супрессированы беременностью, злокачественным новообразованием или вирусом, таким как ВИЧ. Ингибирование IDO также может являться важной стратегией лечения в случае пациентов с неврологическими или психоневрологическими заболеваниями или нарушениями, такими как депрессия. Соединения, представленные в настоящем описании, можно использовать в возможном лечении или профилактике связанных с IDO заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям формулы (I), являющимся ингибиторами ферментов IDO. Настоящее изобретение также относится к применению этих соединений в возможном лечении или профилактике IDO-ассоциированного заболевания или нарушения. Настоящее изобретение также относится к композициям, содержащим одно или более из соединений. Настоящее изобретение дополнительно относится к применению этих композиций в возможной профилактике или лечении IDO-ассоциированного заболевания или нарушения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли:
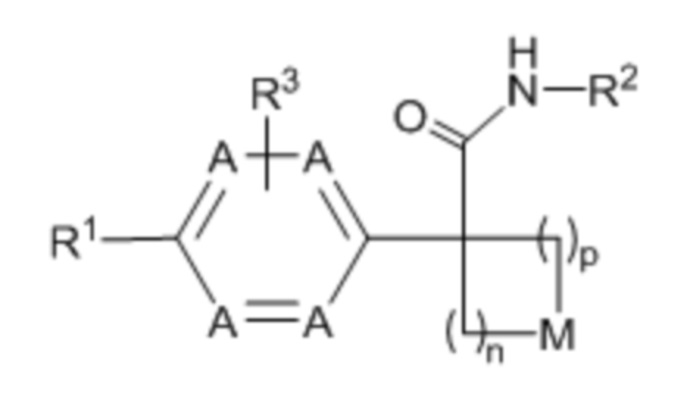 (I),
(I),
где:
n выбрано из 1, 2 и 3;
p выбрано из 0, 1 и 2;
в каждом случае A независимо выбрано из -CH= и -N=, при условии, что по меньшей мере одно A представляет собой -CH=;
M выбрано из -O-, -S- и -CRaRb-, каждый из Ra и Rb независимо выбран из H, галогена, -OH и -C1-8-алкила; или, альтернативно, Ra и Rb вместе с атомом углерода, к которому они присоединены, образуют C3-4-карбоциклическое кольцо, необязательно, замещенное 1-2 заместителями, независимо выбранными из галогена и C1-4-алкила;
R1 выбран из:
(1) арила и
(2) гетероциклила;
где арил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-8-алкила, необязательно, замещенного 1-5 атомами галогена,
(f) -O-C3-8-циклоалкила,
(g) -C1-8-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH, -NH2, NHC(O)Rc и -S(O)2-C1-8-алкила, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(h) -NH-S(O)2-Rc, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(i) -C(O)-Re, где Re выбран из -OH и -C1-8-алкила,
(j) арила, необязательно, замещенного 1-3 атомами галогена, и
(k) гетероциклила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена и -C1-8-алкила; и
где гетероциклил (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-8-алкила, необязательно, замещенного 1-5 атомами галогена,
(f) -O-C3-8-циклоалкила,
(g) -C1-8-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH, -NH2, NHC(O)Rc и -S(O)2-C1-8-алкила, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(h) -NH-S(O)2-Rc, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(i) -C(O)-Rf, где Rf выбран из -OH, -NH2 и -NH-C1-8-алкила,
(j) арила, необязательно, замещенного 1-3 атомами галогена, и
(k) гетероциклила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена и -C1-8-алкила;
R2 выбран из:
(1) C1-8-алкила,
(2) C3-8-карбоциклила,
(3) арила и
(4) гетероциклила;
где C1-8-алкил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила,
(c) -O-C1-8-алкила и
(d) гетероциклила; и
где каждый из C3-8-карбоциклила (2), арила (3) и гетероциклила (4), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила,
(c) -CN,
(d) -O-C1-8-алкила, необязательно, замещенного 1-3 атомами галогена, и
(e) -C1-8-алкила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена, -OH и -NH2; и
R3 выбран из H, галогена и -C1-8-алкила, необязательно, замещенного -OH.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n выбрано из 1 и 2;
p выбрано из 0 и 1;
M выбрано из -O- и -CRaRb-, каждый из Ra и Rb независимо выбран из H и галогена; или, альтернативно, Ra и Rb вместе с атомом углерода, к которому они присоединены, образуют C3-4-циклоалкильное кольцо;
R1 выбран из:
(1) арила и
(2) гетероциклила;
где арил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(e) -O-C3-6-циклоалкила,
(f) -C1-6-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(g) -C(O)-Re, где Re выбран из -OH и -C1-6-алкила;
где гетероциклил (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(f) -O-C3-6-циклоалкила,
(g) -C1-6-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH и -NH2,
(h) -C(O)-Rf, где Rf выбран из -OH, -NH2 и -NH-C1-6-алкила,
(i) фенила, необязательно, замещенного 1-3 атомами галогена;
R2 выбран из:
(1) C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(2) C3-6-циклоалкила,
(3) арила и
(4) 4-7-членного моноциклического гетероциклила;
где каждый из C3-6-циклоалкила (2), арила (3) и гетероциклила (4), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила,
(c) -CN,
(d) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена, и
(e) -C1-6-алкила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена и -OH; и
R3 выбран из H, галогена и -C1-6-алкила, необязательно, замещенного -OH.
В одном из вариантов осуществления соединения формулы (I) его или фармацевтически приемлемой соли
n представляет собой 1;
p представляет собой 1;
каждая группа A представляет собой -CH=;
или, альтернативно, одна группа A представляет собой -N=, и каждая из остальных групп A представляет собой -CH=;
или, альтернативно, каждая из двух групп A представляет собой -N=, и каждая из двух остальных групп A представляет собой -CH=; и
M выбрано из -O-, -CH2-, -CHF, -CF2- и  .
.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли R3 выбран из H, галогена и -CH2-OH.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила,
(2) 4-7-членного моноциклического гетероциклила, выбранного из насыщенного, частично ненасыщенного и ароматического кольца, содержащего 1-4 гетероатома, независимо выбранных из N, O и S, и
(3) 7-10-членного конденсированного бициклического гетероциклила, содержащего 1-3 гетероатома, независимо выбранных из N, O и S, в любом из колец;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила, необязательно замещенного -OH,
(c) -CN,
(d) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(e) -O-C3-6-циклоалкила,
(f) -C1-6-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(g) -C(O)-Re, где Re выбран из -OH и -C1-6-алкила; и
где каждый из моноциклического гетероциклила (2) и конденсированного бициклического гетероциклила (3), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила, необязательно замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(f) -O-C3-6-циклоалкила,
(g) -C1-6-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH и -NH2,
(h) -C(O)-Rf, где Rf выбран из -OH, -NH2 и -NH-C1-6-алкила,
(i) фенила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила;
(2) моноциклического гетероциклила, выбранного из имидазолила, оксазолила, пиперидинила, пиразолила, пиридинила, пиримидинила, тиазолила, тетразолила и 1,2,4-оксадиазолила; и
(3) конденсированного бициклического гетероциклила, выбранного из 3a,4,5,6,7,7a-гексагидро-1H-бензо[d]имидазолила, имидазол[4,5-b]пиридинила, имидазол[4,5-c]пиридинила, индолила, изоиндолинила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного-OH,
(c) циклобутила, необязательно, замещенного-OH,
(d) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(e) -O-циклопропила,
(f) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(g) -C(O)-C1-4-алкила; и
где каждый из моноциклического гетероциклила (2) и конденсированного бициклического гетероциклила (3) необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -CN,
(e) оксогруппы,
(f) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(g) -O-циклопропила,
(h) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(i) фенила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R2 выбран из:
(1) C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(2) C3-6-циклоалкила,
(3) фенила, и
(4) 5-6-членного моноциклического гетероциклила;
где каждый из C3-6-циклоалкила (2), фенила (3) и гетероциклила (4), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила,
(c) -CN,
(d) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена, и
(e) -C1-6-алкила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена и -OH.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R2 выбран из:
(1) C1-4-алкила,
(2) C3-6-циклоалкила,
(3) фенила, и
(4) 5-6-членного моноциклического гетероциклила, выбранного из оксазолила, пиридинила и тиазолила;
где каждый из C3-6-циклоалкила (2), фенила (3) и гетероциклила (4), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена, и
(e) -C1-4-алкила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n представляет собой 1;
p представляет собой 1;
каждая группа A представляет собой -CH=;
или, альтернативно, одна группа A представляет собой -N=, и каждая из остальных групп A представляет собой -CH=;
или, альтернативно, две группы A представляют собой -N=, и каждая из двух остальных групп A представляет собой -CH=;
M выбрано из -O-, -CH2-, -CHF, -CF2- и ;
R1 выбран из:
(1) фенила;
(2) моноциклического гетероциклила, выбранного из имидазолила, оксазолила, пиперидинила, пиразолила, пиридинила, пиримидинила, тиазолила, тетразолила и 1,2,4-оксадиазолила; и
(3) конденсированного бициклического гетероциклила, выбранного из 3a,4,5,6,7,7a-гексагидро-1H-бензо[d]имидазолила, имидазол[4,5-b]пиридинила, имидазол[4,5-c]пиридинила, индолила, изоиндолинила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(e) -O-циклопропила,
(f) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(g) -C(O)-C1-4-алкила; и
где каждый из моноциклического гетероциклила (2) и конденсированного бициклического гетероциклила (3), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -CN,
(e) оксогруппы,
(f) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(g) -O-циклопропила,
(h) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(i) фенила, необязательно, замещенного 1-3 атомами галогена;
R2 выбран из:
(1) C1-4-алкила,
(2) C3-6-циклоалкила,
(3) фенила, и
(4) 5-6-членного моноциклического гетероциклила, выбранного из оксазолила, пиридинила и тиазолила;
где каждый из C3-6-циклоалкила (2), фенила (3) и гетероциклила (4), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена, и
(e) -C1-4-алкила, необязательно, замещенного 1-3 атомами галогена; и
R3 выбран из H, галогена и -CH2-OH.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
каждая группа A представляет собой -CH=;
M представляет собой -O-;
R1 выбран из:
(1) фенила; и
(2) моноциклического гетероциклила, выбранного из имидазолила, оксазолила, пиперидинила, пиразолила, пиридинила, пиримидинила, тиазолила, тетразолила и 1,2,4-оксадиазолила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(d) -O-циклопропила, и
(e) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
где моноциклический гетероциклил (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(d) -O-циклопропила,
(e) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH;
R2 выбран из:
(1) фенила, и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 атомами галогена; и
R3 представляет собой H.
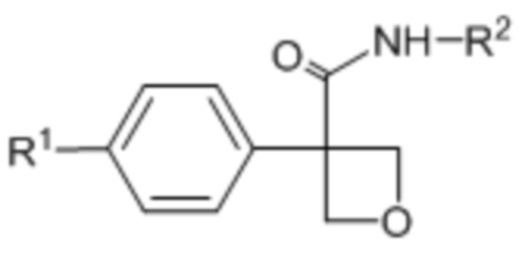
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу (Ia):
 (Ia),
(Ia),
где:
R1 выбран из:
(1) фенила; и
(2) моноциклического гетероциклила, выбранного из имидазолила, оксазолила, пиперидинила, пиразолила, пиридинила, пиримидинила, тиазолила, тетразолила и 1,2,4-оксадиазолила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(e) -O-циклопропила, и
(f) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
(g) -C(O)-C1-4-алкила; и
где моноциклический гетероциклил (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(c) -O-циклопропила и
(d) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH; и
R2 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (Ia), или его фармацевтически приемлемой соли:
R1 представляет собой пиридинил, необязательно, замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CH3,
(c) -CHF2,
(d) -CF3 и
(e) -C(CH3)2OH; и
R2 представляет собой фенил, необязательно, замещенный 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу, выбранную из (Ii), (Ij) и (Ik):
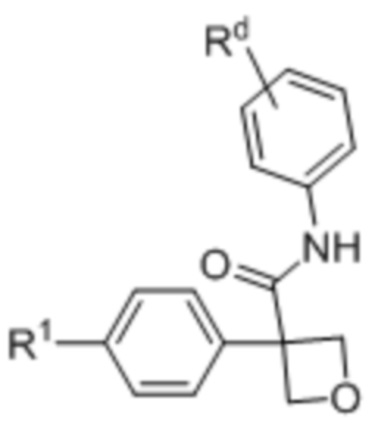 (Ii),
(Ii), 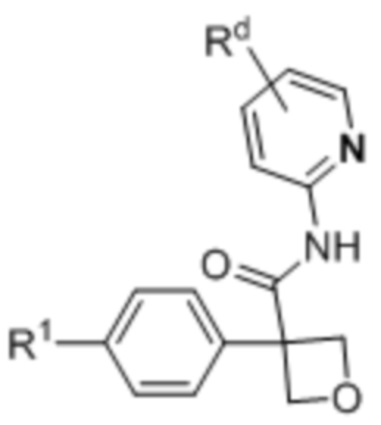 (Ij) и
(Ij) и 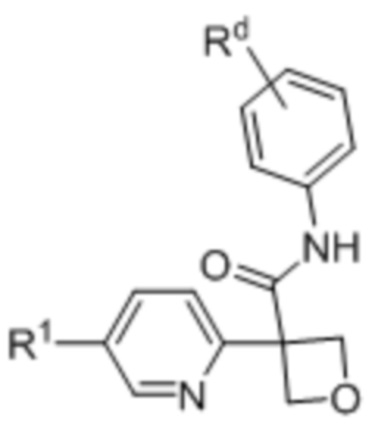 (Ik);
(Ik);
где:
R1 выбран из:
(1) фенила и
(2) моноциклического гетероциклила, выбранного из имидазолила, оксазолила, пиперидинила, пиразолила, пиридинила, пиримидинила, тиазолила, тетразолила и 1,2,4-оксадиазолила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(d) -O-циклопропила и
(e) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH, и
где моноциклический гетероциклил (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена,
(d) -O-циклопропила,
(e) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена и -OH; и
Rd выбран из:
(a) галогена,
(b) -CN,
(c) -O-C1-3 -алкила, необязательно, замещенного 1-3 атомами галогена, и
(d) C1-3 -алкила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (Ii), (Ij) или (Ik) или его фармацевтически приемлемой соли:
R1 представляет собой пиридинил, необязательно, замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CH3,
(c) -CHF2,
(d) -CF3 и
(e) -C(CH3)2OH; и
Rd выбран из:
(a) галогена,
(b) -CN,
(c) -CH3 и
(d) -CF3.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n выбрано из 1, 2 и 3;
p выбрано из 0, 1 и 2;
M выбрано из -O-, -S- и -CRaRb-, каждый из Ra и Rb независимо выбран из H, галогена, -OH и -C1-6-алкила; или, альтернативно, Ra и Rb вместе с атомом углерода, к которому они присоединены, образуют C3-4-циклоалкильное кольцо, необязательно, замещенное 1-2 заместителями, независимо выбранными из галогена и C1-4-алкила;
R1 выбран из:
(1) арила и
(2) гетероциклила;
где арил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-8-алкила, необязательно, замещенного 1-5 атомами галогена,
(f) -O-C3-8-циклоалкила,
(g) -C1-8-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH, -NH2, NHC(O)Rc и -S(O)2-C1-8-алкила, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(h) -NH-S(O)2-Rc, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(i) -C(O)-OH,
(j) арила, необязательно, замещенного 1-3 атомами галогена, и
(k) гетероциклила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена и -C1-8-алкила;
где гетероциклил (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-8-алкила, замещенного 1-5 атомами галогена,
(f) -O-C3-8-циклоалкила,
(g) -C1-8-алкила, замещенного 1-4 заместителями, независимо выбранными из галогена, -NH2, NHC(O)Rc и -S(O)2-C1-8-алкила, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(h) -NH-S(O)2-Rc, где Rc выбран из -C1-8-алкила и -C3-8-циклоалкила,
(i) -C(O)-OH,
(j) арила, необязательно, замещенного 1-3 атомами галогена, и
(k) гетероциклила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена и -C1-8-алкила;
R2 выбран из:
(1) C1-8-алкила,
(2) C3-8-карбоциклила,
(3) арила и
(4) гетероциклила;
где C1-8-алкил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила,
(c) -O-C1-8-алкила и
(d) гетероциклила; и
где каждый из C3-8-карбоциклила (2), арила (3) и гетероциклила (4), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила,
(c) -CN,
(d) -O-C1-8-алкила, необязательно, замещенного 1-3 атомами галогена, и
(e) -C1-8-алкила, необязательно, замещенного 1-3 заместителями, независимо выбранными из галогена, -OH и -NH2; и
R3 выбран из H, галогена и -C1-8-алкила.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила,
(2) моноциклического гетероциклила, выбранного из изоксазолила, изотиазолила, оксадиазолила, оксазолила, пиразинила, пиразолила, пиридазинила, пиридинила, пиримидинила, тетрагидрофуранила, тетрагидропиранила, тетразолила и 1,2,3-тиадиазолила, и
(3) бициклического гетероциклила, выбранного из 3a,4,5,6,7,7a-гексагидро-1H-бензо[d]имидазолила, индолила, 1,2,3-тиадиазолила, 1H-бензо[d]имидазолила, 3H-имидазо[4,5-c]пиридинила, 3H-имидазо[4,5-b]пиридинила, имидазо-[1,2-a]пиридинила и имидазо-[1,2-b]пиридазинила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) C3-4-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-4-алкила, необязательно, замещенного 1-5 атомами галогена,
(f) -O-циклопропила,
(g) -C1-4-алкила, необязательно, замещенного 1-4 заместители, независимо выбранными из галогена, -OH, -NH2, NHC(O)C1-3-алкила и -S(O)2-C1-4-алкила,
(h) -NH-S(O)2-Rc, где Rc выбран из метила, этила, пропила и циклопропила,
(i) -C(O)-OH,
(j) фенила, необязательно, замещенного 1-3 атомами галогена, и
(k) оксадиазолила, необязательно, замещенного метилом или этилом; и
где моноциклический гетероциклил (2) и бициклический гетероциклил (3) замещены 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) C3-4-циклоалкила, необязательно, замещенного -OH,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-4-алкила, замещенного 1-5 атомами галогена,
(f) -O-циклопропила,
(g) -C1-4-алкила, замещенного 1-4 заместителями, независимо выбранными из галогена, -NH2, NHC(O)C1-3-алкила и -S(O)2-C1-4-алкила,
(h) -NH-S(O)2-Rc, где Rc выбран из метила, этила, пропила и циклопропила,
(i) -C(O)-OH,
(j) фенила, необязательно, замещенного 1-3 атомами галогена, и
(k) оксадиазолила, необязательно, замещенного метилом или этилом.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила и
(2) моноциклического гетероциклила, выбранного из изоксазолила, изотиазолила, оксадиазолила, оксазолила, пиразинила, пиразолила, пиридазинила, пиридинила, пиримидинила, тетрагидрофуранила, тетрагидропиранила, тетразолила и 1,2,3-тиадиазолила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-3-алкила, необязательно, замещенного 1-5 атомами галогена,
(e) -O-циклопропила и
(f) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH и -NH2; и
где моноциклический гетероциклил (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-3-алкила, замещенного 1-5 атомами галогена,
(e) -O-циклопропила и
(f) -C1-4-алкила, замещенного 1-4 заместители, независимо выбранными из галогена и -NH2.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R2 выбран из:
(1) фенила,
(2) пиридинила и
(3) пиримидинила;
где каждый из фенила (1), пиридинила (2) и пиримидинила (3), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно, замещенного 1-3 атомами галогена, и
(d) C1-4-алкила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
R2 выбран из:
(1) фенила,
(2) пиридинила, и
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CH3,
(f) -CH2F,
(g) -CHF2 и
(h) -CF3.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n представляет собой 1;
p представляет собой 1;
каждая группа A представляет собой -CH=; или, альтернативно, одна группа A представляет собой -N=, и каждая из трех остальных групп A представляет собой -CH=;
M выбрано из -O-, -CH2-, -CF2- и ;
R1 выбран из:
(1) фенила,
(2) пиридинила и
(3) пиримидинила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-CH3,
(e) -O-CH2CH3,
(f) -O-CF3,
(g) -O-CHF2,
(h) -O-CF2CF3,
(i) -CH3,
(j) -CH2F,
(k) -CHF2,
(l) -CF3,
(m) -CH2CF3,
(n) -CH2OH,
(o) -CH2CH3,
(p) -CH(CH3)OH,
(q) -CH2CH2OH,
(r) -CH(CHF2)OH,
(s) -C(CH3)2OH,
(t) -C(CF3)2OH,
(u) -O-циклопропила и
(v) -O-циклобутила;
где каждый из пиридинила (2) и пиримидинила (3) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-CF3,
(e) -O-CHF2,
(f) -O-CF2CF3,
(g) -CH2F,
(h) -CHF2,
(i) -CF3,
(j) -CH2CF3,
(k) -CH(CHF2)OH,
(l) -C(CF3)2OH,
(m) -O-циклопропила и
(n) -O-циклобутила;
R2 выбран из:
(1)фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CH3,
(f) -CH2F,
(g) -CHF2 и
(h) -CF3; и
R3 представляет собой H.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n представляет собой 1;
p представляет собой 1;
каждая группа A представляет собой -CH=; или, альтернативно, одна группа A представляет собой -N=, и каждая из трех остальных групп A представляет собой -CH=;
M выбрано из -O-, -CH2-, -CF2- и ;
R1 выбран из:
(1) фенила и
(2) пиридинила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -CHF2,
(g) -CF3,
(h) -CH2CF3,
(i) -CH2OH,
(j) -CH2CH3,
(k) -CH(CH3)OH,
(l) -CH2CH2OH,
(m) -CH(CHF2)OH,
(n) -C(CH3)2OH,
(o) -C(CF3)2OH и
(p) -O-циклопропила;
где пиридинил (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CF3,
(d) -O-CHF2,
(e) -CHF2,
(f) -CF3,
(g) -CH2CF3,
(h) -CH(CHF2)OH,
(i) -C(CF3)2OH и
(j) -O-циклопропила;
R2 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CHF2 и
(f) -CF3; и
R3 представляет собой H.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n представляет собой 1;
p представляет собой 1;
каждая группа A представляет собой -CH=; или, альтернативно, одна группа A представляет собой -N=, и каждая из трех остальных групп A представляет собой -CH=;
M выбрано из -O-, -CH2-, -CF2- и ;
R1 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -CHF2,
(g) -CF3,
(h) -CH2CF3,
(i) -CH2OH,
(j) -CH2CH3,
(k) -CH(CH3)OH,
(l) -CH2CH2OH,
(m) -CH(CHF2)OH,
(n) -C(CH3)2OH,
(o) -C(CF3)2OH и
(p) -O-циклопропила; и
R2 представляет собой фенил, необязательно, замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CH3,
(f) -CH2F,
(g) -CHF2 и
(h) -CF3; и
R3 представляет собой H.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли:
n представляет собой 1;
p представляет собой 1;
каждая группа A представляет собой -CH=; или, альтернативно, одна группа A представляет собой -N=, и каждая из трех остальных групп A представляет собой -CH=;
M выбрано из -O-, -CH2-, -CF2- и ;
R1 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -CHF2,
(g) -CF3,
(h) -CH2CF3,
(i) -CH2OH,
(j) -CH(CH3)OH,
(k) -CH2CH2OH,
(l) -CH(CHF2)OH,
(m) -C(CH3)2OH и
(n) -C(CF3)2OH; и
R2 представляет собой фенил, необязательно, замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CHF2 и
(f) -CF3 и
R3 представляет собой H.
В одном из вариантов осуществления соединения формулы (I), описанного выше, или его фармацевтически приемлемой соли n представляет собой 1; p представляет собой 1; и M представляет собой -O-.
В одном из вариантов осуществления соединения формулы (I), описанного выше, или его фармацевтически приемлемой соли:
каждая группа A представляет собой -CH=;
R1 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -CHF2,
(g) -CF3,
(h) -CH2CF3,
(i) -CH2OH,
(j) -CH(CH3)OH,
(k) -CH2CH2OH,
(l) -CH(CHF2)OH,
(m) -C(CH3)2OH и
(n) -C(CF3)2OH; и
R2 представляет собой фенил, необязательно, замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CHF2 и
(f) -CF3; и
R3 представляет собой H.
В одном из вариантов осуществления соединения формулы (I), описанного выше, или его фармацевтически приемлемой соли:
одна группа A представляет собой -N=, и каждая из трех остальных групп A представляет собой -CH=;
R1 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -CHF2,
(g) -CF3,
(h) -CH2CF3,
(i) -CH2OH,
(j) -CH(CH3)OH,
(k) -CH2CH2OH,
(l) -CH(CHF2)OH,
(m) -C(CH3)2OH и
(n) -C(CF3)2OH; и
R2 представляет собой фенил, необязательно, замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CHF2 и
(f) -CF3; и
R3 представляет собой H.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу (Ia) или (Ib):
(Ia) 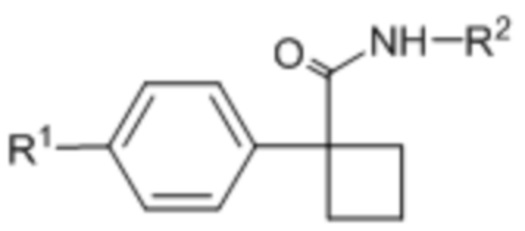 (Ib).
(Ib).
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу (Ic) или (Id):
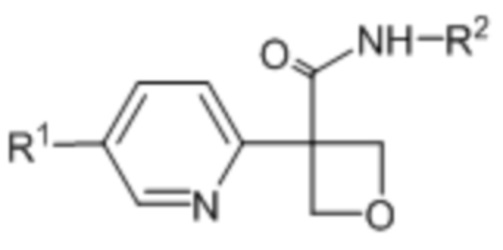 (Ic),
(Ic), 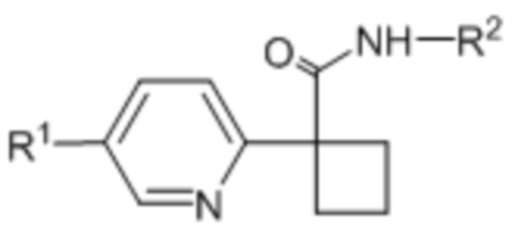 (Id).
(Id).
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу (Ie) или (If):
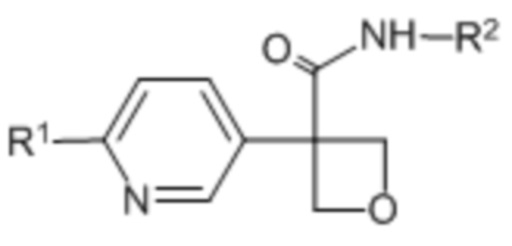 (Ie),
(Ie), 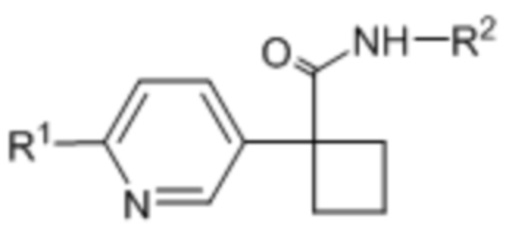 (If).
(If).
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу (Ig) или (Ih):
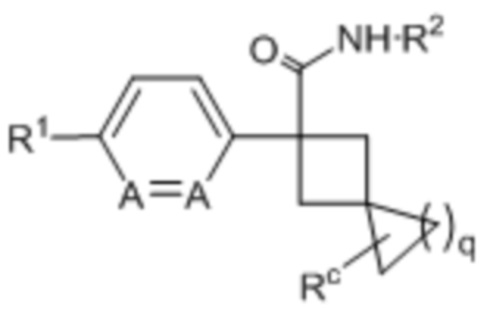 (Ig) или
(Ig) или 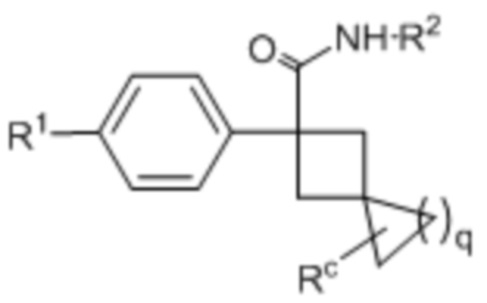 (Ih);
(Ih);
где q представляет собой 1 или 2; каждое A независимо представляет собой -CH= или -N=; и Rc представляет собой H, галоген или C1-3-алкил.
В одном из вариантов осуществления соединения формулы (I) или его фармацевтически приемлемой соли соединение имеет формулу (Ii), (Ij) или (Ik):
(Ii), (Ij) или (Ik);
где:
R1 выбран из:
(1) фенила и
(2) моноциклического гетероциклила, выбранного из изоксазолила, изотиазолила, оксадиазолила, оксазолила, пиразинила, пиразолила, пиридазинила, пиридинила, пиримидинила, тетрагидрофуранила, тетрагидропиранила, тетразолила и 1,2,3-тиадиазолила;
(3) 6-12-членного конденсированного бициклического гетероциклила, содержащего 1-3 гетероатома, независимо выбранных из N, O и S, в любом из колец;
где каждый из фенила (1), моноциклического гетероциклила (2) и конденсированного бициклического гетероциклила (3) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-3-алкила, необязательно, замещенного 1-5 атомами галогена,
(e) -O-циклопропила и
(f) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH и -NH2; и
Rd выбран из:
(a) галогена,
(b) -CN,
(c) -O-C1-3-алкила, необязательно, замещенного 1-3 атомами галогена, и
(d) C1-3-алкила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (Ii), (Ij) или (Ik) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -CHF2,
(g) -CF3,
(h) -CH2CF3,
(i) -CH2OH,
(j) -CH2CH3,
(k) -CH(CH3)OH,
(l) -CH2CH2OH,
(m) -CH(CHF2)OH,
(n) -C(CH3)2OH,
(o) -C(CF3)2OH и
(p) -O-циклопропила; и
Rd выбран из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CH3,
(f) -CH2F,
(g) -CHF2 и
(h) -CF3.
В одном из вариантов осуществления соединения формулы (Ia), (Ib), (Ic), (Id), (Ie), (If), (Ig), (Ih), (Ii), (Ij) или (Ik) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила;
(2) моноциклического гетероциклила, выбранного из насыщенного, частично ненасыщенного и ароматического 4-7-членного кольца, содержащего 1-4 гетероатома, независимо выбранных из N, O и S; и
(3) 6-12-членного конденсированного бициклического гетероциклила, содержащего 1-3 гетероатома, независимо выбранных из N, O и S, в любом из колец;
где фенил (1) необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(f) -O-C3-6-циклоалкила,
(g) -C1-6-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH, -NH2 и -S(O)2-C1-6-алкила,
(h) -NH-S(O)2-Rc, где Rc выбран из -C1-6-алкила и -C3-6-циклоалкила,
(i) -C(O)-OH,
(j) фенила, необязательно, замещенного 1-3 атомами галогена, и
(k) ароматического 4-7-членного моноциклического кольца, содержащего 1-3 гетероатома, независимо выбранных из N, O и S, необязательно, замещенного -C1-6-алкила;
где моноциклический гетероциклил (2) и конденсированный бициклический гетероциклил (3) замещены 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-6-циклоалкила,
(c) -CN,
(d) оксогруппы,
(e) -O-C1-6-алкила, замещенного 1-3 атомами галогена,
(f) -O-C3-6-циклоалкила,
(g) -C1-6-алкила, замещенного 1-4 заместителями, независимо выбранными из галогена, -NH2 и -S(O)2-C1-6-алкила,
(h) -NH-S(O)2-Rc, где Rc выбран из -C1-6-алкила и -C3-6-циклоалкила,
(i) -C(O)-OH,
(j) фенила, необязательно, замещенного 1-3 атомами галогена, и
(k) ароматического 4-7-членного моноциклического кольца, содержащего 1-3 гетероатома, независимо выбранных из N, O и S, необязательно, замещенного -C1-6-алкила; и
R2, если он есть, выбран из:
(1) C1-6-алкила, необязательно, замещенного 1-3 атомами галогена,
(2) C3-6-циклоалкила,
(3) C4-6-мостикового бициклического насыщенного карбоциклила,
(4) фенила и
(5) моноциклического гетероциклила, выбранного из насыщенного, частично ненасыщенного и ароматического 4-7-членного кольца, содержащего 1-4 гетероатома, независимо выбранных из N, O и S;
где каждый из C3-6-циклоалкила (2), C4-6-мостикового бициклического насыщенного карбоциклила (3), фенила (4) и гетероциклила (5), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-6-алкила, необязательно, замещенного 1-3 атомами галогена, и
(d) -C1-6-алкила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (Ia), (Ib), (Ic), (Id), (Ie), (If), (Ig), (Ih), (Ii), (Ij) или (Ik) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила; и
(2) моноциклического гетероциклила, выбранного из изоксазолила, изотиазолила, оксадиазолила, оксазолила, пиразинила, пиразолила, пиридазинила, пиридинила, пиримидинила, тетрагидрофуранила, тетрагидропиранила, тетразолила и 1,2,3-тиадиазолила;
где фенил (1), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-3-алкила, необязательно, замещенного 1-5 атомами галогена,
(e) -O-циклопропила и
(f) -C1-4-алкила, необязательно, замещенного 1-4 заместителями, независимо выбранными из галогена, -OH и -NH2;
где моноциклический гетероциклил (2) замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) циклобутила, необязательно, замещенного -OH,
(d) -O-C1-3-алкила, замещенного 1-5 атомами галогена,
(e) -O-циклопропила и
(f) -C1-4-алкила, замещенного 1-4 заместителями, независимо выбранными из галогена и -NH2; и
R2, если он есть, выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-3-алкила, необязательно, замещенного 1-3 атомами галогена, и
(d) C1-3-алкила, необязательно, замещенного 1-3 атомами галогена.
В одном из вариантов осуществления соединения формулы (Ia), (Ib), (Ic), (Id), (Ie), (If), (Ig), (Ih), (Ii), (Ij) или (Ik) или его фармацевтически приемлемой соли:
R1 выбран из:
(1) фенила и
(2) пиридинила;
где каждый из фенила (1) и пиридинила (2), необязательно, замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно, замещенного -OH,
(c) -O-CH3,
(d) -O-CF3,
(e) -O-CHF2,
(f) -O-CF2CF3,
(g) -CH3,
(h) -CH2F,
(i) -CHF2,
(j) -CF3,
(k) -CH2CF3,
(l) -CH2OH,
(m) -CH(CH3)OH,
(n) -CH2CH2OH,
(o) -CH(CHF2)OH,
(p) -C(CH3)2OH,
(q) -C(CF3)2OH, и
R2, если он есть, представляет собой фенил, необязательно замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-CHF2,
(d) -O-CF3,
(e) -CH3,
(f) -CH2F,
(g) -CHF2 и
(h) -CF3.
В одном из вариантов осуществления соединение, представленное в настоящем описании, выбрано из группы, состоящей из соединений, примеры которых приведены в примерах 1-131; или их фармацевтически приемлемых солей, сольватов или гидратов.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, представленное в настоящем описании, и по меньшей мере один фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способу ингибирования активности индоламин-2,3-диоксигеназы (IDO), включающему приведение IDO в контакт с соединением, представленным в настоящем описании, или его фармацевтически приемлемой солью, сольватом или гидратом.
Настоящее изобретение также относится к способу ингибирования иммуносупрессии у пациента, включающему введение указанному пациенту эффективного количества соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата.
Настоящее изобретение также относится к способу лечения злокачественного новообразования, вирусной инфекции, депрессии, нейродегенеративного нарушения, травмы, возрастной катаракты, отторжения трансплантата органа или аутоиммунного заболевания у пациента, включающему введение указанному пациенту эффективного количества соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата.
Настоящее изобретение также относится к способу лечения меланомы у пациента, включающему введение указанному пациенту эффективного количества соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата.
Настоящее изобретение дополнительно относится к соединению, представленному в настоящем описании, или его фармацевтически приемлемой соли для применения в терапии. В одном из вариантов осуществления настоящее изобретение относится к применению соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата для получения лекарственного средства для применения в терапии.
Термин "алкил" относится к разветвленным и неразветвленным насыщенным алифатическим углеводородным группам из 1-18 атомов углерода, или более конкретно - 1-12 атомов углерода. Неограничивающие примеры таких групп включают метил (Me), этил (Et), n-пропил (Pr), n-бутил (Bu), n-пентил, n-гексил и их изомеры, такие как изопропил (i-Pr), изобутил (i-Bu), втор-бутил (s-Bu), трет-бутил (t-Bu), изопентил и изогексил. Алкильные группы, необязательно, могут быть замещенными одним или более заместителями, как представлено в настоящем описании. Термин "C1-6-алкил" относится к алкильной группе, как представлено в настоящем описании, содержащей 1-6 атомов углерода.
Термин "арил" относится к ароматическому моноциклическому или полициклическому кольцу, содержащему 6-14 кольцевых атомов углерода, или более конкретно - 6-10 кольцевых атомов углерода. Моноциклические арильные кольца включают, в качестве неограничивающих примеров, фенил. Полициклические кольца включают, в качестве неограничивающих примеров, нафтил и бициклические кольца, где фенил конденсирован с C4-7-циклоалкильным или C4-7-циклоалкенильным кольцом. Арильные группы, необязательно могут быть замещены одним или более заместителями, как представлено в настоящем описании. Связывание можно осуществлять через любой из атомов углерода любого кольца. В одном из вариантов осуществления арил представляет собой фенил.
Термин "карбоциклил" относится к неароматическому (т.е. насыщенному или частично ненасыщенному) моноциклическому карбоциклическому радикалу или конденсированному бициклическому, мостиковому бициклическому или спироциклическому карбоциклическому радикалу, содержащему определенные кольцевые атомы углерода. Например, термин "C3-8-карбоциклил" относится к неароматическому 3-8-членному моноциклическому карбоциклическому радикалу или неароматическому 6-8-членному конденсированному бициклическому, мостиковому бициклическому или спироциклическому карбоциклическому радикалу. Карбоцикл можно присоединять по любому атому цикла, что приводит к получению стабильной структуры. Неограничивающие примеры 3-8-членных моноциклических карбоциклов включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептанил и циклогептенил. Неограничивающие примеры 6-8-членных конденсированных бициклических карбоциклических радикалов включают бицикло[3.3.0]октан. Неограничивающие примеры 5-8-членных мостиковых бициклических карбоциклических радикалов включают бицикло[1.1.1]пентанил, бицикло[2.2.2]гептанил, бицикло[2.2.2]октанил и бицикло[3.2.1]октанил. Неограничивающие примеры 6-8-членных спироциклических карбоциклических радикалов включают спиро[3.3]гептанил и спиро[3.4]октанил.
Термин "циклоалкил" относится к моноциклическому насыщенному карбоциклическому кольцу, содержащему определенное количество кольцевых атомом углерода. Например, термин "C3-8-циклоалкил" относится к циклоалкильной группе, как представлено в настоящем описании, содержащей 3-8 атомов углерода. Неограничивающие примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептанил. Циклоалкильные группы, необязательно, могут быть замещены одним или более заместителями, как представлено в настоящем описании.
Термин "галоген", если не указано иначе, относится к фтору, хлору, брому или йоду.
Термин "гетероцикл" или "гетероциклил" относится к насыщенному, частично ненасыщенному или ароматическому кольцевому остатку, содержащему по меньшей мере один кольцевой гетероатом и по меньшей мере один кольцевой атом углерода. В одном из вариантов осуществления гетероатом представляет собой кислород, серу или азот. Гетероцикл, содержащий несколько гетероатомов, может содержать разные гетероатомы. Остатки гетероциклила включают моноциклические и полициклические (например, бициклические) кольцевые остатки. Бициклические кольцевые остатки включают конденсированные, спироциклические и мостиковые бициклические кольца и могут содержать один или более гетероатомов в любом из колец. Кольцо, присоединенное к остальной части молекулы, может содержать или не содержать гетероатом. Любое кольцо в бициклическом гетероцикле может являться насыщенным, частично ненасыщенным или ароматическим. Гетероцикл можно присоединять к остальной части молекулы через кольцевой атом углерода, кольцевой атом кислорода или кольцевой атом азота. Неограничивающие примеры гетероциклов описаны ниже.
В одном из вариантов осуществления гетероциклил представляет собой 4-7-членный моноциклический гетероциклил, выбранный из насыщенного, частично ненасыщенного и ароматического кольца, содержащего 1-4 гетероатома, независимо выбранных из N, O и S.
В одном из вариантов осуществления гетероциклил представляет собой 6-12-членный конденсированный бициклический гетероциклил, содержащий 1-3 гетероатома, независимо выбранных из N, O и S, в любом из колец
В одном из вариантов осуществления гетероциклил представляет собой моноциклический гетероциклил, выбранный из имидазолила, оксазолила, пиперидинила, пиразолила, пиридинила, пиримидинила, тиазолила, тетразолила и 1,2,4-оксадиазолила.
В одном из вариантов осуществления гетероциклил представляет собой моноциклический гетероциклил, выбранный из оксазолила, пиридинила и тиазолила.
В одном из вариантов осуществления гетероциклил представляет собой пиридинил.
В одном из вариантов осуществления гетероциклил представляет собой конденсированный бициклический гетероциклил, выбранный из 3a,4,5,6,7,7a-гексагидро-1H-бензо[d]имидазолила, имидазол[4,5-b]пиридинила, имидазол[4,5-c]пиридинила, индолила, изоиндолинила.
Гетероциклические группы, необязательно, могут быть замещены одним или более заместителями, как представлено в настоящем описании.
Термин "необязательно замещенный" относится к "незамещенному или замещенному", и, таким образом, общие структурные формулы, представленные в настоящем описании, включают соединения, содержащие определенные необязательные заместители, а также соединения, несодержащие необязательные заместители. Каждый заместитель независимо определяют каждый раз, когда он встречается в определениях общих структурных формул.
Полиморфизм
Соединение, представленное в настоящем описании, включая его соль, сольват или гидрат, может существовать в кристаллической форме, некристаллической форме или их смеси. Соединение или его соль или сольват также могут проявлять полиморфизм, т.е. способность существовать в разных кристаллических формах. Эти разные кристаллические формы, как правило, известны как "полиморфные формы". Полиморфные формы имеют одинаковую химическую композицию, но отличаются упаковкой, геометрическим расположением и другими описательными свойствами твердого кристаллического состояния. Таким образом, полиморфные формы могут иметь разные физические свойства, такие как форма, плотность, твердость, деформируемость, стабильность и растворение. Полиморфные формы, как правило, демонстрируют разную температуру плавления, ИК-спектры и профили порошковой рентгеновской дифрактограммы, все из которых можно использовать для идентификации. Специалисту в этой области будет понятно, что можно получать разные полиморфные формы, например, изменяя или корректируя условия, используемые в кристаллизации/рекристаллизации соединения, представленного в настоящем описании.
Оптические изомеры - Диастереомеры - Геометрические изомеры - Таутомеры
В настоящее описание включены различные изомеры соединений, представленных в настоящем описании. Термин "изомеры" относится к соединениям, имеющим одинаковую композицию и молекулярную массу, но отличающимся физическими и/или химическими свойствами. Структурное различие может состоять в строении (геометрические изомеры) или способности вращать плоскость поляризованного света (стереоизомеры).
Что касается стереоизомеров, соединение, представленное в настоящем описании, может содержать один или более асимметричных атомов углерода и может встречаться в виде смесей (таких как рацемическая смесь) или в качестве отдельных энантиомеров или диастереомеров. Все такие изомерные формы включены в настоящее описание, включая их смеси. Если соединение, представленное в настоящем описании, содержит двойную связь, заместитель может находиться в E- или Z-конфигурации. Если соединение, представленное в настоящем описании, содержит дизамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию. Все таутомерные формы также предназначены для включения в настоящее описание.
Любой асимметричный атом (например, атом углерода) в соединении, представленном в настоящем описании, может находиться в рацемической смеси или быть энантиомерно обогащенным, например, в (R)-, (S)- или (R, S)-конфигурации. В определенных вариантах осуществления каждый асимметричным атом имеет по меньшей мере 50% энантиомерного избытка, по меньшей мере 60% энантиомерного избытка, по меньшей мере 70% энантиомерного избытка, по меньшей мере 80% энантиомерного избытка, по меньшей мере 90% энантиомерного избытка, по меньшей мере 95% энантиомерного избытка или по меньшей мере 99% энантиомерного избытка в (R)- или (S)-конфигурации. Заместители в атомах с ненасыщенными двойными связями, по возможности, могут находиться в цис- (Z)- или транс- (E)-форме.
Соединение, представленное в настоящем описании, может находиться в форме одного из возможных изомеров, ротамеров, атропизомеров, таутомеров или их смесей, например, в виде, по существу, чистых геометрических (цис или транс) изомеров, диастереомеров, оптических изомеров (антиподов), рацематов или их смесей.
Любые получаемые смеси изомеров можно разделять с учетом физико-химических различий составных частей на чистые или, по существу, чистых геометрических или оптических изомеров, диастереомеров, рацематов, например, посредством хроматографии и/или фракционной кристаллизации.
Любые получаемые рацематы конечных соединений примеров или промежуточные соединения можно разрешать на оптические антиподы известными способами, например, посредством разделения их диастереомерных солей, полученных с использованием оптически активной кислоты или основания и выделения оптически активного кислого или основного соединения. В частности, таким образом, можно использовать основный остаток для разрешения соединений по настоящему изобретению на их оптические антиподы, например, посредством фракционной кристаллизации соли, образованной с оптически активной кислотой, например, винной кислотой, дибензоилвинной кислотой, диацетилвинной кислотой, ди-O, O'-p-толуоилвинной кислотой, миндальной кислотой, яблочной кислотой или камфор-10-сульфоновой кислотой. Рацемические соединения также можно разрешать посредством хиральной хроматографии, например, высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хирального адсорбента.
Некоторые из соединений, представленных в настоящем описании, могут существовать с разными точками присоединения водорода, их обозначают как таутомеры. Например, соединения, включающие карбонильные -CH2C(O)- группы (кето-формы) могут подвергаться таутомерии с образованием гидроксильных -CH=C(OH)- групп (енольные формы). Кето-формы и енольные формы в отдельности, а также в их смесях, включены в объем настоящего изобретения.
Изотопные варианты
Соединения, представленные в настоящем описании, включают немеченые формы, а также формы, меченые изотопами. Меченые изотопами соединения имеют структуры, представленные формулами, приведенными в настоящем описании, за исключением того, что один или более атомов замещают атомам, имеющим выбранную атомную массу или массовое число. Примеры изотопов, которые можно включать в соединения, представленные в настоящем описании, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как 2H (т.е. дейтерий или "D"), 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 32P, 35S, 18F, 123I, 125I и 36Cl. настоящее изобретение включает различные меченые изотопами соединения, представленные в настоящем описании, например, соединения, в которых присутствуют радиоактивные изотопы, такие как 3H и 14C, или соединения, в которых присутствуют нерадиоактивные изотопы, такие как 2H и 13C. Такие меченые изотопами соединения можно использовать в метаболических исследованиях (14C), исследованиях кинетики реакций (например, 2H или 3H), способах детекции или визуализации, таких как позитронно-эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография (SPECT), включая анализы распределения лекарственных средств или субстратов в ткани, или в лучевой терапии пациентов. В частности, замена позитронно-активными изотопами, такими как 11C, 18F, 15O и 13N, может быть особенно желательной для исследований с использованием ПЭТ или SPECT.
Меченые изотопами соединения, представленные в настоящем описании, как правило, можно получать общепринятыми способами, известными специалистам в этой области. Кроме того, замена более тяжелыми изотопами, в частности, дейтерием (т.е. 2H или D), может привести к некоторым терапевтическим преимуществам, являющимся результатом более высокой метаболической стабильности, например повышенного времени полужизни in vivo, или снижения необходимых доз, или улучшения терапевтического индекса.
Фармацевтически приемлемые соли
Термин "фармацевтически приемлемая соль" относится к соли, полученной из фармацевтически приемлемого нетоксичного основания или кислоты, включая неорганическое или органическое основание и неорганическую или органическую кислоту. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца (VI), марганца (II), калия, натрия, цинка и т.п. Конкретные варианты осуществления включают соли аммония, кальция, магния, калия и натрия. Соли в твердой форме могут существовать в виде нескольких кристаллических структур, а также могут находиться в форме гидратов. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, таких как аргинин, бетаин, кофеин, холин, N, N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Если соединение, представленное в настоящем описании, является основным, соль можно получать из фармацевтически приемлемой нетоксичной кислоты, включая неорганическую и органическую кислоту. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, соляную, изэтиновую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, p-толуолсульфоновую кислоту, трифторуксусную кислоту (TFA) и т.п. Конкретные варианты осуществления включают лимонную, бромистоводородную, соляную, малеиновую, фосфорную, серную, фумаровую, винную и трифторуксусную кислоты.
Способы применения
Соединения, представленные в настоящем описании, могут ингибировать активность фермента индоламин-2,3-диоксигеназы (IDO). Например, соединения, представленные в настоящем описании, потенциально можно использовать для ингибирования активности IDO в клетке или у индивидуума, нуждающегося в модуляции фермента, посредством введения эффективного количества соединения. Настоящее изобретение дополнительно относится к способам ингибирования деградации триптофана в системе, содержащей клетки, экспрессирующей IDO, такой как ткань, живой организм или культура клеток. В некоторых вариантах осуществления настоящее изобретение относится к способам изменения (например, повышения) уровней внеклеточного триптофана у млекопитающего посредством введения эффективного количества соединения или композиции, представленных в настоящем описании. Способы измерения уровней и деградации триптофана общеизвестны в этой области.
Настоящее изобретение также относится к способам ингибирования иммуносупрессии, такой как IDO-опосредованная иммуносупрессия, у пациента посредством введения пациенту эффективного количества соединения или композиции, представленных в настоящем описании. IDO-опосредованная иммуносупрессия ассоциирована, например, со злокачественными новообразованиями, ростом опухоли, метастазированием, вирусной инфекцией, репликацией вируса и т.д.
Настоящее изобретение также относится к способам лечения заболеваний, ассоциированных с активностью или экспрессией, включая аномальную активность и/или гиперэкспрессию, IDO у индивидуума (например, пациента) посредством введения индивидууму, нуждающемуся в таком лечению, эффективного количества или дозы соединения, представленного в настоящем описании, или его фармацевтической композиции. Пример заболевания может включать любое заболевание, нарушение или состояние, которое может быть прямо или косвенно связано с экспрессией или активностью фермента IDO, такой как гиперэкспрессия или аномальная активность. IDO-ассоциированное заболевание также может включать любое заболевание, нарушение или состояние, которое можно подвергать профилактике, улучшать или излечивать посредством модуляции активности фермента. Примеры IDO-ассоциированных заболеваний включают злокачественные новообразования, вирусную инфекцию, такую как ВИЧ и HCV, депрессию, нейродегенеративные нарушения, такие как болезнь Альцгеймера и болезнь Гентингтона, травму, возрастную катаракту, трансплантацию органа (например, отторжение трансплантата органа) и аутоиммунные заболевания, включая астму, ревматоидный артрит, рассеянный склероз, аллергическое воспаление, воспалительное заболевание кишечника, псориаз и системную красную волчанку. Примеры злокачественных новообразований, которые потенциально можно лечить способами, представленными в настоящем описании, включают рак толстого кишечника, поджелудочной железы, молочной железы, предстательной железы, легких, злокачественное новообразование головного мозга, рак яичника, шейки матки, яичка, почки, головы и шеи, лимфому, лейкоз, меланому и т.п. Соединения по изобретению также можно использовать в лечении ожирения и ишемии. В рамках изобретения термин "клетка" предназначен для обозначения клетки in vitro, ex vivo или in vivo. В некоторых вариантах осуществления клетка ex vivo может являться частью образца ткани, удаленного из организма, например, млекопитающего. В некоторых вариантах осуществления клетка in vitro может являться клеткой в культуре клеток. В некоторых вариантах осуществления клетка in vivo является живой клеткой в организме, например, млекопитающего.
В рамках изобретения термин "приведение в контакт" относится к сближению указанных остатков в системе in vitro или системе in vivo. Например, "приведение в контакт" фермента IDO с соединением, представленным в настоящем описании, включает введение соединения по настоящему изобретению индивидууму или пациенту, такому как человек, а также, например, введение соединения по изобретению в образец, содержащий клеточный или очищенный препарат, содержащий фермент IDO.
Индивидуум, которому вводят соединение, представленное в настоящем описании, или его фармацевтически приемлемую соль, сольват или гидрат, как правило, является млекопитающим, таким как человек, мужского или женского пола. Термин "индивидуум" также относится к коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам и птицам. В одном из вариантов осуществления индивидуум является человеком.
В рамках изобретения термин "лечение" относится ко всем процессам, включающим замедление, препятствование, прекращение, контроль или остановку прогрессирования заболевания или нарушения, которые могут быть ассоциированы с активностью фермента IDO. Термин не обязательно означает полное устранение всех симптомов заболевания или нарушения. Термины также включают возможное профилактическое лечение указанных состояний, в частности, у индивидуума, предрасположенного к такому заболеванию или нарушению.
Термин "введение" соединения следует понимать как включающий предоставление индивидууму соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата и композиций указанных выше соединений.
Количество соединения, вводимого индивидууму, является количеством, достаточным для ингибирования активности фермента IDO у индивидуума. В варианте осуществления количество соединения может являться "эффективным количеством", где соединение по изобретению вводят в количестве, которое будет вызывать биологический или медицинский ответ ткани, системы, животного или человека, которого добивается исследователь, ветеринар, врач или другой клиницист. Эффективное количество не обязательно включает факторы токсичности и безопасности, связанные с введением соединения. Известно, что специалист в этой области может влиять на физиологические нарушения, ассоциированные с активностью фермента IDO, посредством лечения индивидуума, в настоящее время страдающего нарушениями, или посредством профилактического лечения индивидуума, который, вероятно, будет страдать нарушениями, с использованием эффективного количества соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата.
Эффективное количество соединения будет варьироваться в зависимости от конкретного выбранного соединения (например, с учетом активности, эффективности и/или времени полужизни соединения); выбранного пути введения; состояния, подвергаемого лечению; тяжести состояния, подвергаемого лечению; возраста, размера, массы и физического состояния индивидуума, подвергаемого лечению; медицинского анамнеза индивидуума, подвергаемого лечению; природы сопутствующей терапии; желаемого терапевтического эффекта и подобных факторов, и специалисты в этой области могут определять его общепринятыми способами.
Соединения, представленные в настоящем описании, можно вводить любым подходящим путем, включая пероральное и парентеральное введение. Парентеральное введение, как правило, осуществляют посредством инъекции или инфузии, и оно включает внутривенную, внутримышечную и подкожную инъекцию или инфузию.
Соединения, представленные в настоящем описании, можно вводить однократно или по схеме введения, где ряд доз вводят с разными временными интервалами в течение определенного периода времени. Например, дозы можно вводить один, два, три или четыре раза в сутки. Дозы можно вводить до достижения желаемого терапевтического эффекта или неопределенно долго для поддержания желаемого терапевтического эффекта. Подходящие схемы введения соединения, представленного в настоящем описании, зависят от фармакокинетических свойств этого соединения, таких как абсорбция, распределение и время полужизни, которые могут определять специалисты в этой области. Кроме того, подходящие схемы введения, включая длительность использования таких схем введения, соединения, представленного в настоящем описании, зависят от заболевания или состояния, подвергаемых лечению, тяжести заболевания или состояния, возраста и физического состояния индивидуума, подвергаемого лечению, медицинского анамнеза индивидуума, подвергаемого лечению, природы сопутствующей терапии, желаемого терапевтического эффекта и подобных факторов, определение которых входит в пределы знаний и компетенции специалистов в этой области. Специалистам в этой области также следует понимать, что для подходящих схем введения может потребоваться корректировка с учетом индивидуального ответа индивидуума на схему введения или с течением времени, т.к. для отдельного индивидуума могут потребоваться изменения. Типичные суточные дозы могут варьироваться в зависимости от конкретного выбранного пути введения. Типичные суточные дозы для перорального введения человеку, весящему приблизительно 70 кг, будут находиться в диапазоне от приблизительно 0,1 мг до приблизительно 2 г, или более конкретно - от 0,1 мг до 500 мг, или даже более конкретно - от 0,2 мг до 100 мг соединения, представленного в настоящем описании.
Один из вариантов осуществления настоящего изобретения относится к способу лечения заболевания или нарушения, ассоциированного с активностью фермента IDO, включающему введение эффективного количества соединения, представленного в настоящем описании, индивидууму, нуждающемуся в лечении. В одном из вариантов осуществления заболевание или нарушение, ассоциированное с ферментом IDO, представляет собой нарушение пролиферации клеток.
В одном из вариантов осуществления настоящее изобретение относится к применению соединения, представленного в настоящем описании, в терапии. Соединение можно использовать в способе ингибирования активности фермента IDO у индивидуума, такого как млекопитающее, нуждающегося в таком ингибировании, включающему введение индивидууму эффективного количества соединения.
В одном из вариантов осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение, представленное в настоящем описании, или его фармацевтически приемлемую соль, сольват или гидрат, для применения в возможном лечении нарушения или заболевания, связанного с активностью фермента IDO.
Композиции
В рамках изобретения термин "композиция" предназначен для включения лекарственной формы, содержащей определенное соединение в определенном количестве, а также любой лекарственной формы, получаемой прямо или косвенно посредством комбинирования определенного соединения в определенном количестве. Такой термин предназначен для включения лекарственной формы, содержащей соединение, представленное в настоящем описании, или его фармацевтически приемлемую соль, сольват или гидрат, и один или более фармацевтически приемлемых носителей или эксципиентов. Таким образом, композиции по настоящему изобретению включают любую композицию, получаемую посредством смешивания соединения по настоящему изобретению и одного или более фармацевтически приемлемых носителей или эксципиентов. Термин "фармацевтически приемлемый" означает, что носители или эксципиенты совместимы с соединением, представленным в настоящем описании, и с другими ингредиентами композиции.
В одном из вариантов осуществления настоящее изобретение относится к композиции, содержащей соединение, представленное в настоящем описании, или его фармацевтически приемлемую соль, сольват или гидрат и один или более фармацевтически приемлемых носителей или эксципиентов. Композицию можно получать и упаковывать в нерасфасованную форму, где эффективное количество соединения по изобретению можно отделять, а затем вводить индивидууму, например, как в случае порошков или сиропов. Альтернативно, композицию можно получать и упаковывать в стандартную лекарственную форму, где каждая физически дискретная единица содержит эффективное количество соединения, представленного в настоящем описании. При получении в стандартной лекарственной форме композиция по изобретению, как правило, содержит от приблизительно 0,1 мг до 2 г, или более конкретно - от 0,1 мг до 500 мг, или даже более конкретно - от 0,2 мг до 100 мг соединения, представленного в настоящем описании, или его фармацевтически приемлемой соли, сольвата или гидрата.
Соединение, представленное в настоящем описании, и фармацевтически приемлемые носители или эксципиенты, как правило, будут составлять в лекарственной форме, адаптированной для введения индивидууму желаемым путем. Например, лекарственные формы включают формы, адаптированные для (1) перорального введения, такие как таблетки, капсулы, каплеты, пилюли, троше, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше и крахмальные капсулы; и (2) парентерального введения, такие как стерильные растворы, суспензии и порошки для разведения. Подходящие фармацевтически приемлемые носители или эксципиенты будут варьироваться в зависимости от конкретной выбранной лекарственной формы. Кроме того, подходящие фармацевтически приемлемые носители или эксципиенты можно выбирать по конкретной функции, которую они могут выполнять в композиции. Например, некоторые фармацевтически приемлемые носители или эксципиенты можно выбирать по их способности облегчать получение однородных лекарственных форм. Некоторые фармацевтически приемлемые носители или эксципиенты можно выбирать по их способности облегчать получение стабильных лекарственных форм. Некоторые фармацевтически приемлемые носители или эксципиенты можно выбирать по их способности облегчать транспорт соединения, представленного в настоящем описании, после введения индивидууму от одного органа или части организма в другой орган или другую часть организма. Некоторые фармацевтически приемлемые носители или эксципиенты можно выбирать по их способности повышать соблюдение пациентом схемы лечения.
Подходящие фармацевтически приемлемые эксципиенты включают следующие типы эксципиентов: дилюенты, смазочные средства, связывающие средства, разрыхлители, наполнители, глиданты, гранулирующие средства, покрывающие средства, увлажнители, растворители, сорастворители, суспендирующие средства, эмульгаторы, подсластители, ароматизаторы, средства для маскировки вкуса, красители, средства против слеживания, увлажнители, хелатирующие средства, пластификаторы, загустители, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные средства.
Специалисты в этой области обладают знаниями и навыками для выбора подходящих фармацевтически приемлемых носителей и эксципиентов в подходящих количествах для использования в настоящем изобретении. Кроме того, существует ряд ресурсов, доступных специалистам в этой области, где описаны фармацевтически приемлемые носители и эксципиенты, и которые можно использовать в выборе подходящих фармацевтически приемлемых носителей и эксципиентов. Примеры включают Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited) и The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and Pharmaceutical Press).
Композиции по изобретению получают способами, известными специалистам в этой области. Некоторые способы, общеупотребительные в этой области, описывают в Remington's Pharmaceutical Sciences (Mack Publishing Company).
В одном из вариантов осуществления изобретение относится к твердой пероральной лекарственной форме, такой как таблетка или капсула, содержащей эффективное количество соединения по изобретению и дилюент или наполнитель. Подходящие дилюенты и наполнители включают лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и прежелатинизированный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и гидроортофосфат кальция. Пероральная твердая лекарственная форма может дополнительно содержат связывающее средство. Подходящие связывающие средства включают крахмал (например, кукурузный крахмал, картофельный крахмал и прежелатинизированный крахмал), желатин, гуммиарабик, альгинат натрия, альгиновую кислоту, трагакантовую камедь, гуаровую камедь, повидон и целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Пероральная твердая лекарственная форма может дополнительно содержать разрыхлитель. Подходящие разрыхлители включают кросповидон, крахмалгликолят натрия, кроскармеллозу, альгиновую кислоту и натрий-карбоксиметилцеллюлозу. Пероральная твердая лекарственная форма может дополнительно содержать смазочное средство. Подходящие смазочные средства включают стеариновую кислоту, стеарат магния, стеарат кальция и тальк.
При необходимости, стандартные лекарственные формы для перорального введения можно микроинкапсулировать. Композицию также можно получать для пролонгирования или поддержания высвобождения, например, посредством покрывания или заключения дисперсного материала в полимеры, воск или т.п.
Соединения, представленные в настоящем описании, также можно соединять с растворимыми полимерами в качестве направляемых носителей лекарственных средств. Такие полимеры могут включать поливинилпирролидон, пирансополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоиловыми остатками. Кроме того, соединения по изобретению можно соединять с классом биодеградируемых полимеров, которые можно использовать для достижения контролируемого высвобождения лекарственного средства, например, полимолочной кислотой, полиэпсилон капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианакрилатами и перекрестно сшитыми или амфипатическими блок-сополимерами гидрогелей.
В одном из вариантов осуществления изобретение относится к жидкой пероральной лекарственной форме. Пероральные жидкости, такие как раствор, сиропы и эликсиры, можно получать в стандартной лекарственной форме таким образом, что указанное количество включает заранее определенное количество соединения, представленного в настоящем описании. Сиропы можно получать посредством растворения соединения по изобретению в соответствующим образом ароматизированном водном растворе; в то время как эликсиры получают с использованием нетоксичного спиртового носителя. Суспензии можно составлять посредством диспергирования соединения, представленного в настоящем описании, в нетоксичном носителе. Также можно добавлять солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые эфиры полиоксиэтилена и сорбита, консерванты, вкусовые добавки, такие как масло перечной мяты или другие природные подсластители или сахарин или другие искусственные подсластители и т.п.
В одном из вариантов осуществления изобретение относится к композициям для парентерального введения. Композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и солюты, делающие состав изотоническим по отношению к крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие средства и загустители. Композиции могут находиться в контейнерах для однократных доз или многодозовых контейнерах, например, запаянных ампулах и флаконах, и их можно хранить в лиофилизированном состоянии, требующем лишь добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед использованием. Экстемпоральные инъекционные растворы и суспензии можно получать из стерильных порошков, гранул и таблеток.
Комбинации
Соединение, представленное в настоящем описании, можно использовать в комбинации с одним или более другими активными средствами, включая, в качестве неограничивающих примеров, другие противоопухолевые средства, используемые в профилактике, лечении, контроле, улучшении или снижении риска конкретного заболевания или состояния (например, нарушений пролиферации клеток). В одном из вариантов осуществления соединение, представленное в настоящем изобретении, комбинируют с одним или более другими противоопухолевыми средствами для использования в профилактике, лечении, контроле, улучшении или снижении риска конкретного заболевания или состояния, для которого соединения, представленные в настоящем описании, являются полезными. Такие другие активные средства можно вводить путем и в количестве, общеупотребительных для них, одновременно или последовательно с соединением по настоящему изобретению.
Если соединение, представленное в настоящем описании, используют одновременно с одним или более другими активными средствами, предусмотрена композиция, содержащая другие такие активные средства в дополнение к соединению, представленному в настоящем описании. Таким образом, композиции по настоящему изобретению включают композиции, также содержащие один или более других активных ингредиентов в дополнение к соединению, представленному в настоящем описании. Соединение, представленное в настоящем описании, можно вводить одновременно, или до, или после одного или более других терапевтических средств. Соединение, представленное в настоящем описании, можно вводить раздельно одним или разными путями введения или совместно в той же фармацевтической композиции, что и другие средства.
Продукты, предоставляемые в виде комбинированного препарата, включают композицию, содержащую соединение, представленное в настоящем описании, и одно или более других активных средств вместе в одной фармацевтической композиции, или соединение, представленное в настоящем описании, и одно или более других терапевтических средств в отдельных формах, например, в форме набора.
Массовое соотношение соединения, представленного в настоящем описании, и второго активного средства может варьироваться и будет зависеть от эффективной дозы каждого средства. Как правило, будут использовать эффективную дозу каждого средства. Таким образом, например, если соединение, представленное в настоящем описании, комбинируют с другим средством, массовое соотношение соединения, представленного в настоящем описании, и другого средства, как правило, будет находиться в диапазоне от приблизительно 1000:1 до приблизительно 1:1000, например, от приблизительно 200:1 до приблизительно 1:200. Комбинации соединения, представленного в настоящем описании, и других активных средств, как правило, также будут находиться в пределах указанного выше диапазона, но в каждом случае следует использовать эффективную дозу каждого активного средства. В таких комбинациях соединение, представленное в настоящем описании, и другие активные средства можно вводить раздельно или вместе. Кроме того, введение одного элемента можно осуществлять до, одновременно или после введения других средств.
В одном из вариантов осуществления изобретение относится к композиции, содержащей соединение, представленное в настоящем описании, и по меньшей мере одно другое терапевтическое средство в виде комбинированного препарата для одновременного, раздельного или последовательного использования в терапии. В одном из вариантов осуществления терапия представляет собой лечение заболевания или нарушения, ассоциированного с активностью фермента IDO.
В одном из вариантов осуществления изобретение относится к набору, содержащему две или более отдельных фармацевтических композиций, по меньшей мере одна из которых содержит соединение, представленное в настоящем описании. В одном из вариантов осуществления набор содержит средства для раздельного удержания указанных композиций, такие как контейнер, разделенная бутыль или разделенный фольгированный пакет. Примером такого набора является блистерная упаковка, как правило, используемая для упаковывания таблеток, капсул и т.п.
Набор, представленный в настоящем описании, можно использовать для введения разных лекарственных форм, например, пероральных и парентеральных, для введения отдельных композиций с разными интервалами между введениями или для титрования отдельных композиций друг относительно друга. Для облегчения соблюдения пациентом схемы лечения набор по изобретению, как правило, содержит инструкции по введению.
Настоящее изобретение относится к применению соединения, представленного в настоящем описании, для лечения заболевания или нарушения, ассоциированного с активностью фермента IDO, где лекарственное средство получают для введения с другим активным средством. Изобретение также относится к применению другого активного средства для лечения заболевания или нарушения, ассоциированного с ферментом IDO, где лекарственное средство вводят с соединением, представленным в настоящем описании.
Изобретение также относится к применению соединения, представленного в настоящем описании, для лечения заболевания или нарушения, ассоциированного с активностью фермента IDO, где пациента ранее (например, в пределах 24 часов) лечили другим активным средством. Изобретение также относится к применению другого терапевтического средства для лечения заболевания или нарушения, ассоциированного с активностью фермента IDO, где пациента ранее (например, в пределах 24 часов) лечили соединением, представленным в настоящем описании. Второе средство можно использовать через неделю, несколько недель, месяц или несколько месяцев после введения соединения, представленного в настоящем описании.
В одном из вариантов осуществления другое активное средство выбрано из группы, состоящей из ингибиторов рецептора фактора роста эндотелия сосудов (VEGF), ингибиторов топоизомеразы II, ингибиторов Smoothened, алкилирующих средств, противоопухолевых антибиотиков, антиметаболитов, ретиноидов, иммуномодулирующих средств, включая, в качестве неограничивающих примеров, противоопухолевые вакцины, антагонистов CTLA-4, LAG-3 и PD-1.
Неограничивающие примеры ингибиторов рецептора фактора роста эндотелия сосудов (VEGF) включают бевацизумаб (продаваемый Genentech/Roche под торговым названием AVASTIN), акситиниб, (N-метил-2-[[3-[([E])-2-пиридин-2-илэтенил]-1H-индазол-6-ил]сульфанил]бензамид, также известный как AG013736 и описанный в публикации PCT № WO 01/002369), бриваниб аланинат ((S)-((R)-1-(4-(4-фтор-2-метил-1H-индол-5-илокси)-5-метилпирроло[2,1-f][1,2,4]триазин-6-илокси)пропан-2-ил)2-аминопропаноат, также известный как BMS-582664), мотесаниб (N-(2,3-дигидро-3,3-диметил-1H-индол-6-ил)-2-[(4-пиридинилметил)амино]-3-пиридинкарбоксамид, описанный в публикации PCT № WO 02/068470), пасиреотид (также известный как SO 230 и описанный в публикации PCT № WO 02/010192) и сорафениб (продаваемый под торговым названием NEXAVAR).
Неограничивающие примеры ингибиторов топоизомеразы II включают этопозид (также известный как VP-16 и этопозид фосфат, продаваемый под торговыми названиями TOPOSAR, VEPESID и ETOPOPHOS) и тенипозид (также известный как VM-26, продаваемый под торговым названием VUMON).
Неограничивающие примеры алкилирующих средств включают 5-азацитидин (продаваемый под торговым названием VIDAZA), децитабин (продаваемый под торговым названием DECOGEN), темозоломид (продаваемый Schering-Plough/Merck под торговыми названиями TEMODAR и TEMODAL), дактиномицин (также известный как актиномицин-D и продаваемый под торговым названием COSMEGEN), мелфалан (также известный как L-PAM, L-сарколизин и фенилаланин-иприт, продаваемый под торговым названием ALKERAN), алтретамин (также известный как гексаметилмеламин (HMM), продаваемый под торговым названием HEXALEN), кармустин (продаваемый под торговым названием BCNU), бендамустин (продаваемый под торговым названием TREANDA), бусульфан (продаваемый под торговыми названиями BUSULFEX и MYLERAN), карбоплатин (продаваемый под торговым названием PARAPLATIN), ломустин (также известный как CCNU, продаваемый под торговым названием CeeNU), цисплатин (также известный как CDDP, продаваемый под торговыми названиями Platinol и Platinol-AQ), хлорамбуцил (продаваемый под торговым названием Leukeran), циклофосфамид (продаваемый под торговыми названиями Cytoxan и Neosar), дакарбазин (также известный как DTIC, DIC и имидазол-карбоксамид, продаваемый под торговым названием DTIC-Dome), алтретамин (также известный как гексаметилмеламин (HMM), продаваемый под торговым названием Hexalen), ифосфамид (продаваемый под торговым названием Ifex), прокарбазин (продаваемый под торговым названием Matulane), мехлоретамин (также известный как азотистый иприт, мустин и мехлорэтамин гидрохлорид, продаваемый под торговым названием Mustargen), стрептозоцин (продаваемый под торговым названием Zanosar), тиотепа (также известная как тиофосфамид, TESPA и TSPA и продаваемый под торговым названием Thioplex).
Неограничивающие примеры противоопухолевых антибиотиков включают доксорубицин (продаваемый под торговыми названиями ADRIAMYCIN и Rubex), блеомицин (продаваемый под торговым названием lenoxane), даунорубицин (также известный как даунорубицин гидрохлорид, дауномицин и рубидомицин гидрохлорид, продаваемый под торговым названием Cerubidine), липосомный даунорубицин (липосомный даунорубицин цитрат, продаваемый под торговым названием DaunoXome), митоксантрон (также известный как DHAD, продаваемый под торговым названием Novantrone), эпирубицин (продаваемый под торговым названием Ellence), идарубицин (продаваемый под торговыми названиями Idamycin, Idamycin PFS) и митомицин C (продаваемый под торговым названием Mutamycin).
Неограничивающие примеры антиметаболитов включают кладрибин (2-хлордезоксиаденозин, продаваемый под торговым названием LEUSTATIN), 5-фторурацил (продаваемый под торговым названием ADRUCIL), 6-тиогуанин (продаваемый под торговым названием PURINTHOL), пеметрексед (продаваемый под торговым названием ALIMTA), цитарабин (также известный как арабинозилцитозин (Ara-C), продаваемый под торговым названием CYTOSAR-U), липосомный цитарабин (также известный как липосомный Ara-C, продаваемый под торговым названием DepoCyt), децитабин (продаваемый под торговым названием Dacogen), гидроксимочевину (продаваемую под торговыми названиями Hydrea, Droxia и Mylocel), флударабин (продаваемый под торговым названием FLUDARA), флоксуридин (продаваемый под торговым названием FUDR), кладрибин (также известный как 2-хлордезоксиаденозин (2-CdA), продаваемый под торговым названием Leustatin), метотрексат (также известный как аметоптерин, метотрексат натрия (MTX), продаваемый под торговыми названиями Rheumatrex и Trexall) и пентостатин (продаваемый под торговым названием Nipent).
Неограничивающие примеры ретиноидов включают алитретиноин (продаваемый под торговым названием Panretin), третиноин (полностью транс-ретиноевую кислоту, также известную как ATRA, продаваемую под торговым названием Vesanoid), изотретиноин (13-цис-ретиноевую кислоту, продаваемую под торговыми названиями Accutane, Amnesteem, Claravis, Clarus, Decutan, Isotane, Izotech, Oratane, Isotret и Sotret) и бексаротен (продаваемый под торговым названием Targretin).
Термин "антагонист PD-1" означает любое химическое соединение или биологическую молекулу, блокирующую связывание PD-L1, экспрессирующегося на злокачественной клетке, с PD-1, экспрессирующимся на иммунной клетке (T-клетке, B-клетке или NKT-клетке), и, предпочтительно, также блокирующую связывание PD-L2, экспрессирующегося на злокачественной клетке, с PD-1, экспрессирующимся на иммунной клетке. Альтернативные названия или синонимы PD-1 и его лигандов включают: PDCD1, PD1, CD279 и SLEB2 в случае PD-1; PDCD1L1, PDL1, B7H1, B7-4, CD274 и B7-H в случае PD-L1; и PDCD1L2, PDL2, B7-DC, Btdc и CD273 в случае PD-L2. В любом из способов лечения, лекарственных средств и применении по настоящему изобретению, в котором лечению подвергают человека, антагонист PD-1 блокирует связывание PD-L1 человека с PD-1 человека и, предпочтительно, блокирует связывание PD-L1 и PD-L2 человека с PD-1 человека. Аминокислотные последовательности PD-1 человека можно найти в NCBI под регистрационным номером NP_005009. Аминокислотные последовательности PD-L1 и PD-L2 человека можно найти в NCBI под регистрационными номерами NP_054862 и NP_079515, соответственно.
Антагонисты PD-1, которые можно использовать в любом из способов лечения, лекарственных средств и применения по настоящему изобретению, включают моноклональное антитело (mAb), или его антигенсвязывающий фрагмент, специфически связывающийся с PD-1 или PD-L1 и, предпочтительно, специфически связывающийся с PD-1 человека или PD-L1 человека. mAb может являться антителом человека, гуманизированным антителом или химерным антителом и может включать константную область человека. В некоторых вариантах осуществления константная область человека выбрана из группы, состоящей из константных областей IgG1, IgG2, IgG3 и IgG4, и в предпочтительных вариантах осуществления константная область человека является константной областью IgG1 или IgG4. В некоторых вариантах осуществления антигенсвязывающий фрагмент выбран из группы, состоящей из Fab-, Fab'-SH-, F(ab')2-, scFv- и Fv-фрагментов. Неограничивающие примеры антагонистов PD-1 включают пембролизумаб (продаваемый под торговым названием Keytruda) и ниволумаб (продаваемый под торговым названием Opdivo).
Примеры mAb, связывающихся с PD-1 человека, и которые можно использовать в способах лечения, лекарственных средствах и применении по настоящему изобретению, описаны в US7488802, US7521051, US8008449, US8354509, US8168757, WO2004/004771, WO2004/072286, WO2004/056875 и US2011/0271358.
Примеры mAb, связывающихся с PD-L1 человека, и которые можно использовать в способах лечения, лекарственных средствах и применении по настоящему изобретению, описаны в WO2013/019906, W02010/077634 A1 и US8383796. Специфические mAb против PD-L1 человека, которые можно использовать в качестве антагониста PD-1 в способах лечения, лекарственных средствах и применении по настоящему изобретению, включают MPDL3280A, BMS-936559, MEDI4736, MSB0010718C и антитело, содержащее вариабельные области тяжелой цепи и легкой цепи SEQ ID NO: 24 и SEQ ID NO: 21, соответственно, из WO2013/019906.
Другие антагонисты PD-1, которые можно использовать в любом из способов лечения, лекарственных средств и применении по настоящему изобретению, включают иммуноадгезин, специфически связывающийся с PD-1 или PD-L1 и, предпочтительно, специфически связывающийся с PD-1 человека или PD-L1 человека, например, слитый белок, содержащий внеклеточную или PD-1-связывающую часть PD-L1 или PD-L2, слитую с константной областью, такой как Fc-область молекулы иммуноглобулина. Примеры молекул иммуноадгезии, специфически связывающихся с PD-1, описаны в WO2010/027827 и WO2011/066342. Специфические слитые белки, которые можно использовать в качестве антагониста PD-1 в способах лечения, лекарственных средствах и применении по настоящему изобретению, включают AMP-224 (также известный как B7-DCIg), являющийся слитым белком PD-L2-FC и связывающийся с PD-1 человека.
Неограничивающие примеры других цитотоксических средств включают триоксид мышьяка (продаваемый под торговым названием Trisenox), аспарагиназу (также известную как L-аспарагиназа, и L-аспарагиназа Erwinia, продаваемую под торговыми названиями Elspar и Kidrolase).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Следующие схемы синтеза и примеры представлены исключительно в иллюстративных целях, а не для какого-либо ограничения. Используемые сокращения являются общепринятыми в этой области или следующими.
ОБЩИЕ СХЕМЫ СИНТЕЗА
Соединения формулы (I) можно получать известными в этой области способы органического синтеза, частично приведенными в следующих схемах синтеза и способах и условиях синтеза для иллюстративных промежуточных соединений и примеров.
Из описанных ниже схем очевидно, что защитные группы для чувствительных или реакционноспособных групп используют, при необходимости, в соответствии с общими принципами химии. На защитные группы воздействуют стандартными способами органического синтеза (T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). Эти группы удаляют на удобной стадии синтеза соединения способами, очевидными специалистам в этой области.
Схема 1
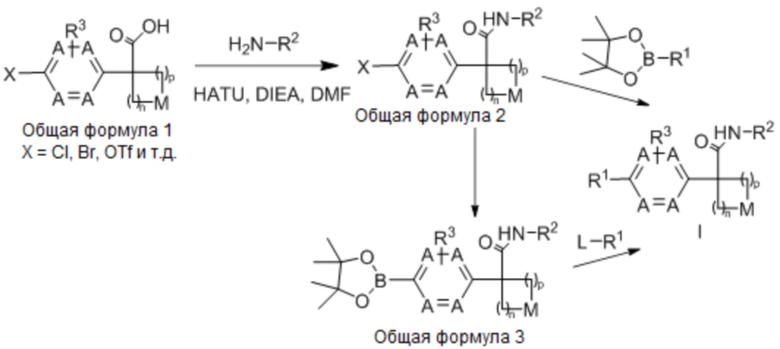
Соединения формулы I можно получать по схеме 1. Подходящую карбоновую кислоту общей формулы 1 подвергают реакции сочетания с подходящим амином R2-NH2 в стандартных условиях сочетания амида для получения промежуточного амидного соединения общей формулы 2. Соединение общей формулы 2 реагирует с подходящей бороновой кислотой, пинаколовым сложным эфиром бороновой кислоты, силикон-содержащим средством, цинкатом, станнаном или подходящим металлическим средством в условиях реакции сочетания Сузуки, реакции Негиши, реакции Стилла или др. с образованием соединений формулы I. Альтернативно, соединения общей формулы 2 превращают в соответствующий пинаколовый сложный эфир бороновой кислоты общей формулы 3 посредством реакции с бис(пинаколато)дибороном (B2pin2) в условиях Pd-катализируемой реакции перекрестного сочетания. Посредством реакции соединения общей формулы 3 с подходящим галогенидом, трифлатом и т.д. в условиях реакции сочетания Сузуки получают соединения формулы I.
Соединения, представленные в настоящем описании, можно получать из коммерчески доступных исходных материалов или синтезировать известными органическими, неорганическими и/или ферментативными способами.
Спектры 1H ЯМР получали с помощью спектрометра Bruker Ultra Shield при 600 МГц или спектрометра Varian 500 при 499 МГц с использованием тетраметилсилана в качестве внутреннего референса. Спектры LC/MS получали с помощью квадрупольных масс-спектрометров Agilent 6120 с использованием ионизации электрораспылением.
ПРИМЕРЫ
Пример 1: 3-(4-(6-циклопропилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
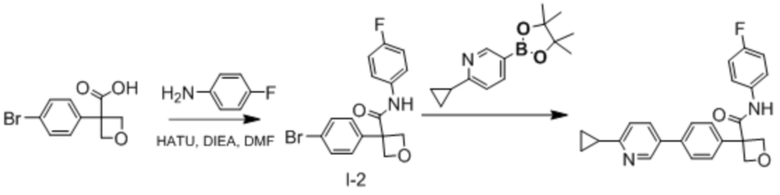
Стадия 1: 3-(4-бромфенил)- N -(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору коммерчески доступной 3-(4-бромфенил)оксетан-3-карбоновой кислоты (4000,0 мг, 15,56 ммоль) в DCM (20,0 мл) добавляли HATU (7099 мг, 18,67 ммоль) и перемешивали суспензию в течение 15 мин при RT. Затем последовательно добавляли 4-фторанилин (1729 мг, 15,56 ммоль) и DIEA (8,15 мл, 46,7 ммоль) и перемешивали реакционную смесь в течение 4 ч. при RT. Реакционную смесь разводили этилацетатом и промывали 1 Н водным раствором HCl (3 раза), водой (2 раза), соляным раствором и насыщенным водным раствором NaHCO3. Затем органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка для флэш-хроматографии на силикагеле 120 г SepaFlash®, 10-100% этилацетат/DCM) для получения титульного соединения (I-2). MS (ESI) m/z 350 [M+H]+.
Далее в настоящем описании условия реакции на стадии 1 обозначают как стандартные условия амидного сочетания.
Стадия 2: 3-(4-(6-циклопропилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемой суспензии 2-циклопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (47,6 мг, 0,194 ммоль), 3-(4-бромфенил)-N-(4-фторфенил)оксетан-3-карбоксамида (68,0 мг, 0,194 ммоль) и дихлор[1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (II) (15,92 мг, 0,017 ммоль) в 1,4-диоксане (1,5 мл) добавляли карбонат натрия (0,194 мл, 0,388 ммоль). Реакционную смесь выкачивали и помещали обратно с азотом три раза и нагревали до 80°C в течение 4 ч., когда посредством LC-MS обнаруживали полное превращение. Реакционную смесь охлаждали до RT и фильтровали через целитовую прокладку. Фильтрат концентрировали под вакуумом. Остаток переносили в CH2Cl2 и промывали соляным раствором. Органический слой сушили над MgSO4, фильтровали и удаляли растворитель под вакуумом. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 389. 1H ЯМР (600 МГц, DMSO-d6) δ 10,00 (с, 1H), 8,82 (с, 1H), 8,21 (с, 1H), 7,79 (д, J=8,1 Гц, 2H), 7,70-7,58 (м, 4H), 7,50 (д, J=8,2 Гц, 1H), 7,15 (т, J=8,7 Гц, 2H), 5,23 (д, J=6,5 Гц, 2H), 4,91 (д, J=6,5 Гц, 2H), 2,29-2,16 (м, 1H), 1,14-1,09 (м, 2H), 1,05 (с, 2H).
Далее в настоящем описании условия реакции на стадии 2 обозначают как стандартные условия реакции перекрестного сочетания Сузуки.
Пример 2: 3-(4-(6-циклопропил-4-фторпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
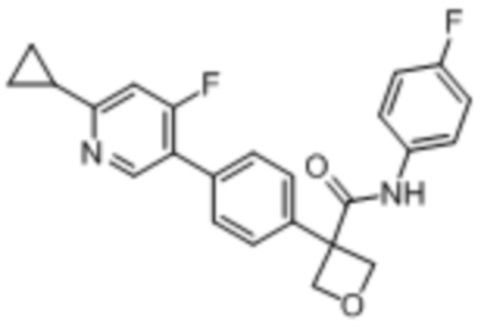
Титульное соединение получали аналогично синтезу примера 1, за исключением того, что использовали 2-циклопропил-4-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин. MS (ESI) [M+H]+: m/z 407, 1H ЯМР (499 МГц, DMSO-d6) δ 10,03 (с, 1H), 8,58 (д, J=10,7 Гц, 1H), 7,72-7,51 (м, 4H), 7,39 (д, J=11,7 Гц, 1H), 7,15 (т, J=8,8 Гц, 2H), 5,22 (д, J=6,5 Гц, 2H), 4,91 (д, J=6,5 Гц, 2H), 2,26-2,12 (м, 1H), 1,05 (т, J=7,7 Гц, 2H), 1,01 (с, 2H).
Пример 3: 3-(4-(6-циклопропил-4-метилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
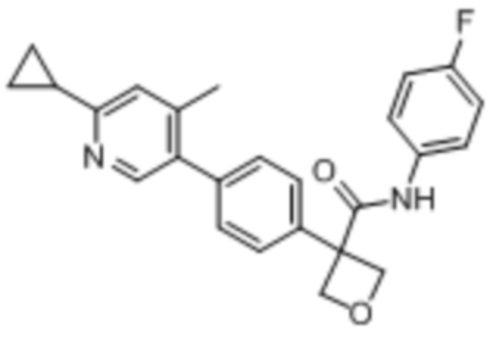
Титульное соединение получали аналогично синтезу примера 1, за исключением того, что использовали 2-циклопропил-4-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин. MS (ESI) [M+H]+: m/z 403. 1H ЯМР (499 МГц, DMSO-d6) δ 10,11 (с, 1H), 8,48 (с, 1H), 7,70-7,60 (м, 4H), 7,58-7,49 (м, 3H), 7,15 (т, J=8,8 Гц, 2H), 5,25 (д, J=6,5 Гц, 2H), 4,92 (д, J=6,5 Гц, 2H), 2,38 (с, 3H), 2,28 (тт, J=8,5, 5,0 Гц, 1H), 1,32-1,22 (м, 2H), 1,20-1,07 (м, 2H).
Пример 4: N-(4-фторфенил)-3-(4-(7-метилимидазо[1,2-a]пиридин-6-ил)фенил)оксетан-3-карбоксамид
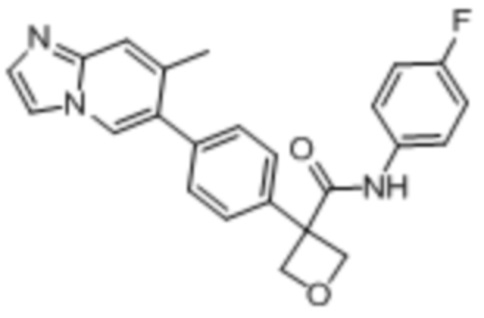
Титульное соединение получали аналогично синтезу примера 1, за исключением того, что использовали (7-метилимидазо[1,2-a]пиридин-6-ил)бороновую кислоту. MS (ESI) [M+H]+: m/z 402, 1H ЯМР (499 МГц, DMSO-d6) δ 10,09 (с, 1H), 8,83 (с, 1H), 8,19 (с, 1H), 8,15 (с, 1H), 7,92 (с, 1H), 7,66 (т, J=7,5 Гц, 4H), 7,56 (д, J=8,1 Гц, 2H), 7,17 (т, J=8,8 Гц, 2H), 5,26 (д, J=6,5 Гц, 2H), 4,93 (д, J=6,5 Гц, 2H), 2,42 (с, 3H).
Пример 5: 3-(4-(4,6-диметилпиримидин-5-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид
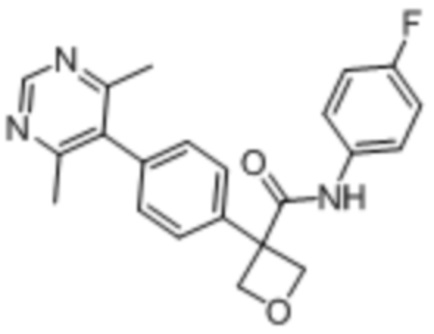
Титульное соединение получали аналогично синтезу примера 1, за исключением того, что использовали (4,6-диметилпиримидин-5-ил)бороновую кислоту. MS (ESI) [M+H]+: m/z 378, 1H ЯМР (499 МГц, DMSO-d6) δ 10,10 (с, 1H), 8,90 (с, 1H), 7,77-7,56 (м, 4H), 7,38 (д, J=8,1 Гц, 2H), 7,17 (т, J=8,8 Гц, 2H), 5,24 (д, J=6,5 Гц, 2H), 4,92 (д, J=6,5 Гц, 2H), 2,20 (с, 6H).
Пример 6: N -(4-фторфенил)-3-(4-(4-метил-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
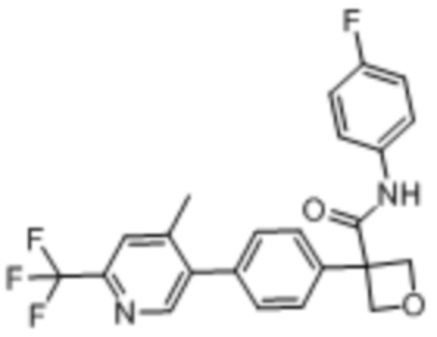
Титульное соединение получали аналогично синтезу примера 1, за исключением того, что в качестве партнера по реакции сочетания использовали 4-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридин. MS (ESI) [M+H]+: m/z 431; обнаружено, 431,1H ЯМР (499 МГц, DMSO-d6) δ 10,07 (с, 1H), 8,57 (с, 1H), 7,92 (с, 1H), 7,72-7,61 (м, 4H), 7,55 (д, J=8,0 Гц, 2H), 7,17 (т, J=8,7 Гц, 2H), 5,24 (д, J=6,5 Гц, 2H), 4,93 (д, J=6,5 Гц, 2H), 2,39 (с, 3H).
Пример 7: N -(4-фторфенил)-3-(4-(6-метокси-2,4-диметилпиридин-3-ил)фенил)оксетан-3-карбоксамид
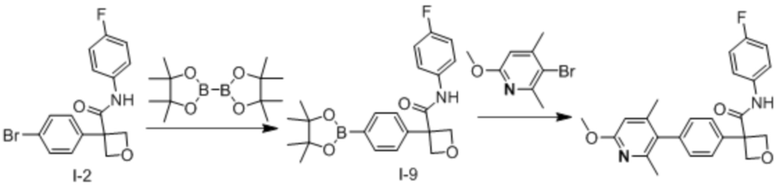
Стадия 1: N -(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид
В сухую круглодонную колбу помещали 3-(4-бромфенил)-N-(4-фторфенил)оксетан-3-карбоксамид (I-2) (200,0 мг, 0,571 ммоль), бис(пинаколато)диборон (377 мг, 1,485 ммоль), ацетат калия (166 мг, 1,691 ммоль) и PdCl2(dppf)-CH2Cl2 (46,6 мг, 0,057 ммоль) в диоксане (5,0 мл). Затем полученную смесь выкачивали и помещали обратно с азотом (3 раза). Смесь нагревали до 80°C в течение 4 ч., затем охлаждали до RT и фильтровали через целитовую прокладку. Фильтрат концентрировали под вакуумом для получения остатка, который растворяли в дихлорметане. После промывки водой (3 раза) и соляным раствором дихлорметановый слой сушили над безводным Na2SO4 и концентрировали под вакуумом для получения неочищенного продукта, который очищали посредством хроматографии на колонках (силикагель, EtOAc/гексан, 12-100%) для получения титульного соединения(I-9). MS (ESI) [M+H]+: m/z 398.
Стадия 2: N -(4-Фторфенил)-3-(4-(6-метокси-2,4-диметилпиридин-3-ил)фенил)оксетан-3-карбоксамид
Раствор 3-бром-6-метокси-2,4-диметилпиридина (16,32 мг, 0,076 ммоль), N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (30,0 мг, 0,076 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (4,92 мг, 7,55 мкмоль) и карбоната натрия (0,076 мл, 0,151 ммоль) в 1,4-диоксане (2,0 мл) выкачивали и помещали обратно с азотом три раза и нагревали смесь в атмосфере азота при 80°C в течение 4 ч. Затем растворители удаляли под вакуумом и получаемый остаток суспендировали в EtOAc/DCM, фильтровали через целитовую прокладку, которую промывали EtOAc/DCM. Объединенные фильтраты концентрировали и очищали неочищенный материал посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 407, 1H ЯМР (499 МГц, DMSO-d6) δ 10,09 (с, 1H), 7,66 (дд, J=8,7, 5,0 Гц, 2H), 7,59 (д, J=8,0 Гц, 2H), 7,26 (д, J=8,0 Гц, 2H), 7,17 (т, J=8,8 Гц, 2H), 6,68 (с, 1H), 5,23 (д, J=6,5 Гц, 2H), 4,90 (д, J=6,5 Гц, 2H), 3,86 (с, 3H), 2,11 (с, 3H), 1,97 (с, 3H).
Примеры 8-33 в таблице ниже получали аналогично примеру 7. Способы синтеза выбранных исходных материалов представлены в настоящем описании.
Примеры общих условий реакции сочетания Сузуки с использованием X-Phos (X-phosG2 или G3) при температурах в диапазоне 45-70°C приведены для препаратов в примере 11 и соединения I-14 ниже.
N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (I-9) (70 мг, 0,141 ммоль), (5-бром-2-метоксипиридин-4-ил)метанол (30,7 мг, 0,141 ммоль) и X-PhosPdG2 (11,09 мг, 0,014 ммоль) добавляли в реакционную пробирку объемом 4 мл, выкачивали и помещали обратно с азотом 3 раза. Добавляли THF (564 мкл) и ортофосфат калия (водный раствор 1 M) (282 мкл, 0,282 ммоль). Реакционную смесь нагревали до 70°C в течение 1 ч. и охлаждали до RT. Реакционную смесь разводили 4 мл DCM, пропускали через фазовый сепаратор, собирали органическую фазу и концентрировали ее под вакуумом. Остаток растворяли в 1 мл DMSO и подвергали обращенно-фазовой очистке (градиент ACN/вода с модификатором 0,1% TFA). Чистые фракции замораживали и лиофилизировали для получения N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-метоксипиридин-3-ил)фенил)оксетан-3-карбоксамида (пример 11) в качестве соли TFA в виде твердого вещества.
Получение 2-(3-хлор-5-(трифторметил)пиридин-2-ил)этан-1-ола (I-11): партнера по реакции сочетания для примера 15
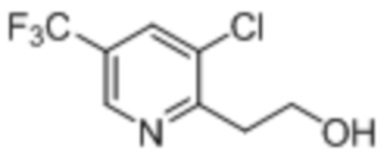 (I-11)
(I-11)
Метил 2-(3-хлор-5-(трифторметил)пиридин-2-ил)ацетат (480 мг, 1,893 ммоль) растворяли в THF (9464 мкл) и охлаждали смесь до -78°C. Затем добавляли DIBAL-H (4732 мкл, 4,73 ммоль) и смеси позволяли нагреться до RT за 1 ч. Реакцию гасили насыщенным водным раствором тартрата калия натрия и перемешивали в течение ночи при RT. Органический слой отделяли и экстрагировали водный слой с помощью 50 мл EtOAc 1 раз. Объединенные органические соединения промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом для получения 2-(3-хлор-5-(трифторметил)пиридин-2-ил)этанола в виде оранжевого масла, которое использовали для получения примера 15 без последующей очистки. MS (ESI) [M+H]+: m/z 226.
Получение 2-(5-бром-2-(трифторметил)пиридин-4-ил)пропан-2-ола (I-12): партнера по реакции сочетания для примера 12
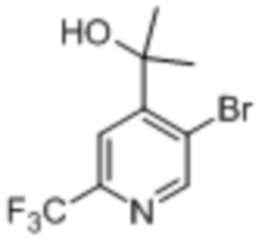 (I-12)
(I-12)
К раствору метил-5-бром-2-(трифторметил)изоникотината (1,5 г, 5,28 ммоль) в THF (21,12 мл) при -78°C добавляли бромид метилмагния (3 M в Et2O) (3,52 мл, 10,56 ммоль) и позволяли смеси нагреться до RT в течение 1 ч. Реакционную смесь гасили насыщенным водным раствором хлорида аммония и перемешивали в течение ночи при RT. Отделяли органические соединения, промывали соляным раствором, сушили над сульфатом натрия и удаляли растворители под вакуумом для получения 2-(5-бром-2-(трифторметил)пиридин-4-ил)пропан-2-ола (I-12) в виде твердого вещества, которое использовали для получения примеров 12 и 20 без последующей очистки. MS (ESI) [M+H]+: m/z 284/286.
Альтернативно, I-12 можно получать следующим способом: в круглодонную колбу в атмосфере азота помещали LDA (2 M, 33,2 мл). В смесь по каплям добавляли раствор 5-бром-2-(трифторметил)пиридина (15 г, 66,4 ммоль) в THF (15 мл) при ≤-65°C в течение 3 ч. и перемешивали смесь при ≤-65°C еще в течение 1 ч. По каплям добавляли ацетон (3,8 г, 66,4 ммоль) в течение 0,5 ч. при ≤-65°C и перемешивали смесь при ≤-65°C в течение 1 ч. Затем смесь разводили этилацетатом (270 мл) и гасили водой (170 мл). Фазы разделяли и водную фазу экстрагировали этилацетатом (70 мл, 2 раза). Объединенные органические экстракты промывали соляным раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали посредством хроматографии на колонках для получения I-12. 1H ЯМР (400 МГц, DMSO-d6) δ 8,87 (с, 1H), 8,19 (с, 1H), 5,82 (с, 1H), 1,62 (с, 6 H).
Получение 1-(5-бром-2-(трифторметил)пиридин-4-ил)этан-1-ола (I-13): партнера по реакции сочетания для примеров 16-17
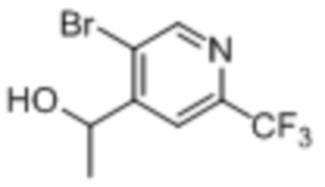 (I-13)
(I-13)
5-бром-2-(трифторметил)изоникотинальдегид (333 мг, 1,311 ммоль) растворяли в THF (6555 мкл) в сосуде объемом 40 мл и раствор дегазировали в атмосфере азота и охлаждали до -78°C. Добавляли бромид метилмагния (3 M в Et2O) (524 мкл, 1,573 ммоль) и получаемую смесь медленно нагревали до RT за 1 ч. Реакционную смесь гасили насыщенным водным раствором хлорида аммония и перемешивали в течение 1 ч. Отделяли органический слой, экстрагировали водный слой с помощью 50 мл EtOAc и объединенные органические соединения промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом для получения 1-(5-бром-2-(трифторметил)пиридин-4-ил)этанола, который использовали для получения примеров 16-17 без последующей очистки. MS (ESI) [M+H]+: m/z 270/272.
Рацемическую смесь, полученную посредством реакции сочетания Сузуки, разделяли посредством SFC с использованием колонки 21×250 мм Lux-2 и 20% MeOH/0,25% DMEA в качестве модификатора при скорости потока 70 мл/мин. Первый элюируемый пик собирали и концентрировали для получения соединения 16. Второй элюируемый пик собирали и концентрировали для получения соединения 17.
Получение 2-(5-бром-2-(дифторметокси)пиридин-4-ил)пропан-2-ола (I-14): партнера по реакции сочетания для примера 18
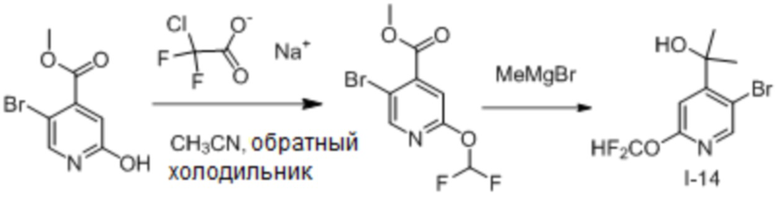
Стадия 1: Метил-5-бром-2-(дифторметокси)изоникотинат
К метил-5-бром-2-гидроксиизоникотинату (1,13 г, 4,87 ммоль) добавляли хлордифторацетат натрия (0,891 г, 5,84 ммоль) и ACN (30 мл) и нагревали смесь до конденсации в течение ночи. Добавляли дополнительный хлордифторацетат натрия (0,891 г, 5,84 ммоль) и нагревали реакционную смесь до конденсации в течение ночи. Реакцию гасили насыщенным раствором хлорида аммония, экстрагировали 2 раза с помощь. 50 мл EtOAc, объединенные органические соединения промывали соляным раствором и сушили над сульфатом натрия. Неочищенный продукт очищали на силикагеле, 0-10% Et2O/гексаны, для получения метил-5-бром-2-(дифторметокси)изоникотината в виде твердого вещества. MS (ESI) [M+H]+: m/z 282/284.
Стадия 2: 2-(5-бром-2-(дифторметокси)пиридин-4-ил)пропан-2-ол (I-14)
Соединение получали так же, как проиллюстрировано выше для примера 16. MS (ESI) [M+H]+: m/z 282/284.
Получение 2-(5-бром-2-(дифторметокси)пиридин-4-ил)пропан-2-ола (I-15): партнера по реакции сочетания для примера 32
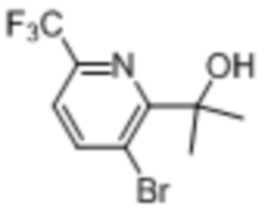 (I-15)
(I-15)
Соединение получали из метил-3-бром-6-(трифторметил)никотината так же, как проиллюстрировано выше для примера 16. MS (ESI) [M+H]+: m/z 284/286.
Получение (5-бром-2-(дифторметокси)пиридин-4-ил)метанола (I-16): партнера по реакции сочетания для примера 13
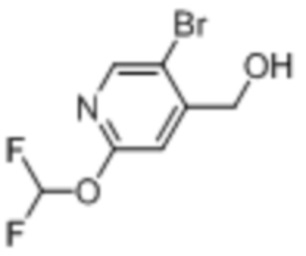 (I-16)
(I-16)
I-16 получали из метил-5-бром-2-(трифторметил)изоникотината и DIBAL-H аналогично получению соединения I-11. MS (ESI) [M+H]+: m/z 254/256.
Синтез (3-бром-6-циклопропилпиридин-2-ил)метанола (I-17) приведен на следующей схеме:
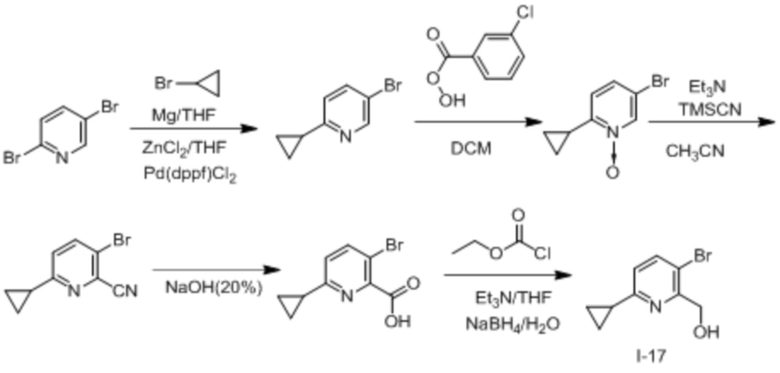
Стадия 1: Синтез 5-бром-2-циклопропилпиридина
В 4-горлую круглодонную колбу объемом 5 л, подвергнутую продуву азотом и поддерживаемую в инертной атмосфере азота, помещали раствор Mg (200 г, 8,33 моль, 10,00 экв.) в тетрагидрофуране (1500 мл), I2 (1 г) и бромциклопропане (400 г, 3,33 моль, 4,00 экв.). Реакционную смесь перемешивали в течение 3 ч. при 65°C, а затем добавляли в раствор ZnCl2 (560 г, 4,12 моль, 5,00 экв.) в тетрагидрофуране (2000 мл) при 10°C. Полученный раствор перемешивали в течение 2 ч. при RT, а затем добавляли Pd(dppf)Cl2 (20 г), 2,5-дибромпиридин (200 г, 843,88 ммоль, 1,00 экв.). Реакционную смесь перемешивали в течение ночи при RT и гасили посредством добавления 500 мл воды. Полученную смесь концентрировали под вакуумом, а затем экстрагировали 3 раза с помощью 5 л этилацетата. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колонки с силикагелем с EtOAc/петролейным эфиром (1/2) для получения 5-бром-2-циклопропилпиридина в виде твердого вещества.
Стадия 2: Синтез 1-оксида 5-бром-2-циклопропилпиридина
В 4-горлую круглодонную колбу объемом 3 л помещали раствор 5-бром-2-циклопропилпиридин (184 г, 924,62 ммоль, 1,00 экв.) в дихлорметане (1500 мл). К раствору частями добавляли 3-хлорбензопероксоевую кислоту (209 г, 1,22 моль, 1,00 экв.) при 0°C в течение 5 мин. Полученный раствор перемешивали в течение ночи при RT. pH раствора доводили до 11 с использованием гидроксида натрия (20%). Полученный раствор экстрагировали 4 раза с помощью 400 мл этилацетата, объединенный органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колонки с силикагелем с этилацетатом для получения 1-оксида 5-бром-2-циклопропилпиридина в виде твердого вещества.
Стадия 3: Синтез 3-бром-6-циклопропилпиколинонитрила
В 4-горлую круглодонную колбу объемом 3 литра помещали раствор 1-оксида 5-бром-2-циклопропилпиридина (100 г, 467,29 ммоль, 1,00 экв.) в ACN (1200 мл), TMSCN (190 г, 1,92 моль, 4,00 экв.) и TEA (192 мл, 3,00 экв.). Полученный раствор перемешивали в течение ночи с обратным холодильником, а затем разводили водой и экстрагировали 4 раза с помощью 500 мл этилацетата. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колонки с силикагелем с этилацетатом/петролейным эфиром (1/50) для получения 3-бром-6-циклопропилпиколинонитрила в виде твердого вещества.
Стадия 4: Синтез 3-бром-6-циклопропилпиколиновой кислоты
В 4-горлую круглодонную колбу объемом 3 литра помещали 3-бром-6-циклопропилпиколинонитрил (74 г, 331,84 ммоль, 1,00 экв.) и гидроксид натрия (20%, 1500 мл). Полученный раствор перемешивали в течение ночи с обратным холодильником, охлаждали до RT, а затем экстрагировали 3 раза с помощью 200 мл простого эфира. pH комбинированного водного слоя доводили до 4 соляной кислотой (3 Н) и полученный раствор экстрагировали 4 раза с помощью 300 мл этилацетата. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колонки с силикагелем с этилацетатом/петролейным эфиром (1/5) для получения 3-бром-6-циклопропилпиколиновой кислоты в виде твердого вещества.
Стадия 5: Синтез (3-бром-6-циклопропилпиридин-2-ил)метанола (I-17)
В 4-горлую круглодонную колбу объемом 3 литра помещали раствор 3-бром-6-циклопропилпиколиновой кислоты (80 г, 330,58 ммоль, 1,00 экв.) в тетрагидрофуране (20 мл). В реакционную смесь по каплям добавляли триэтиламин (72 мл, 1,50 экв.) с перемешиванием при -5-0°C в течение 5 мин, а затем по каплям добавляли этилхлорформиат (41 г, 379,63 ммоль, 1,30 экв.) с перемешиванием при -20°C. Полученный раствор перемешивали в течение 60 мин при -20°C. Твердое вещество отфильтровывали и к фильтрату по каплям добавляли раствор NaBH4 (28 г, 2,00 экв.) в воде (84 г) с перемешиванием при -10°C. Полученный раствор перемешивали еще в течение 20 мин при -10°C. Затем реакцию гасили посредством добавления NH4Cl (насыщенного). Полученный раствор экстрагировали 3 раза с помощью 1000 мл этилацетата, объединенный органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом. Остаток очищали с помощью колонки с силикагелем с петролейным эфиром/этилацетатом (50/1). Таким образом, получали соединение I-17 в виде твердого вещества. MS (ESI) [M+H]+: m/z 229, 1H ЯМР (300 МГц, DMSO-d6) δ 7,85 (1H, d,J=8,1 Гц), 7,16 (1H, т), 4,98 (1H, т,J=5,7 Гц), 4,52 (2H, д,J=5,7 Гц). Этот материал использовали для получения примера 26.
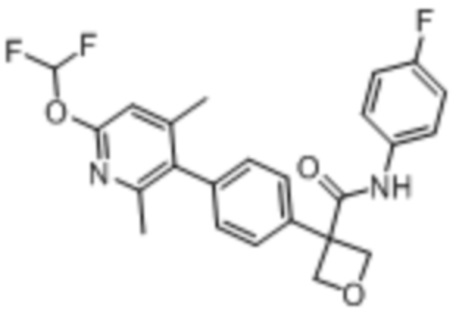
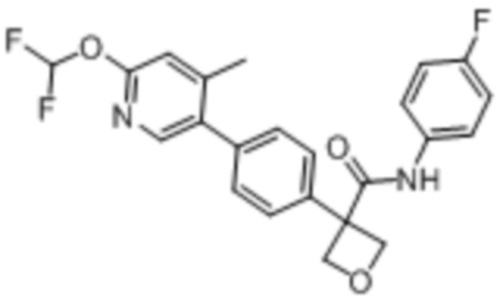
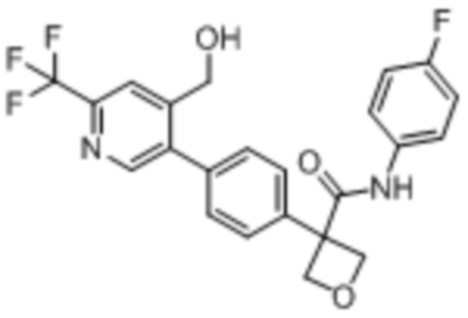
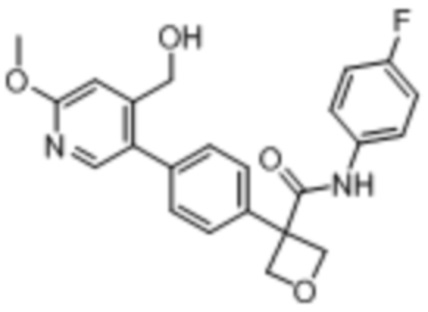
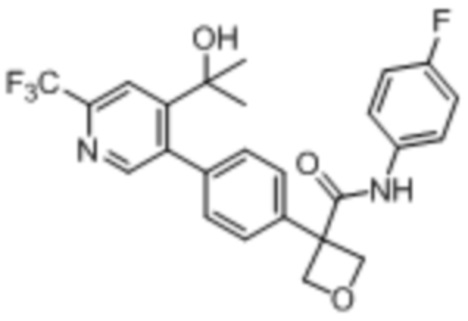
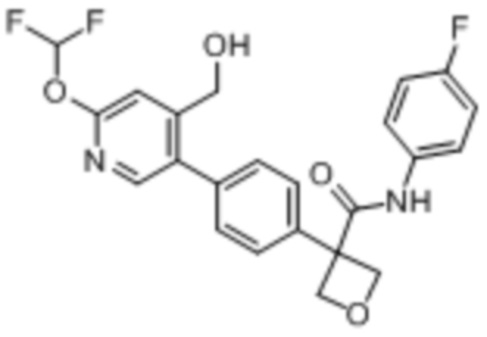
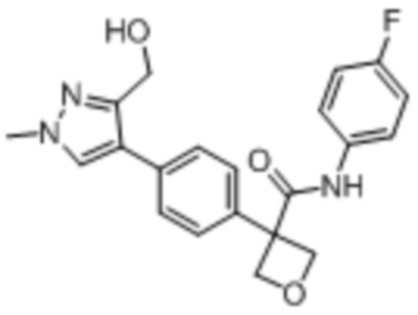
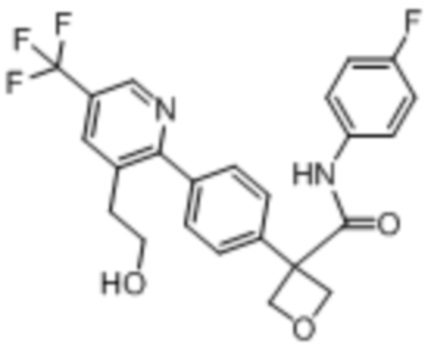
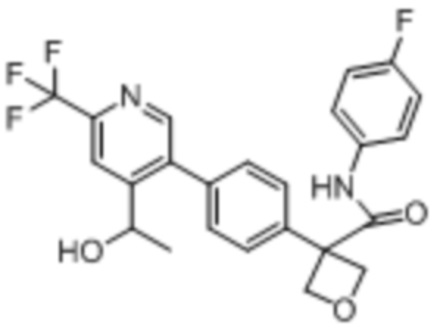
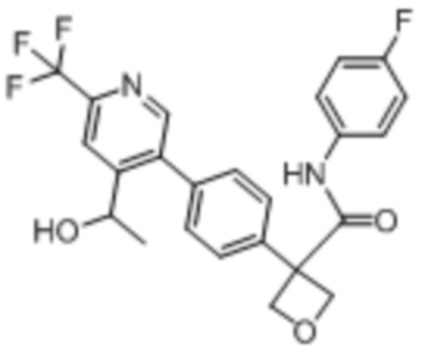
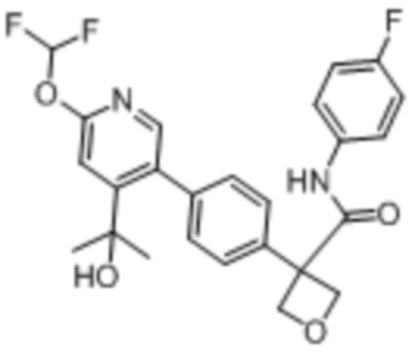
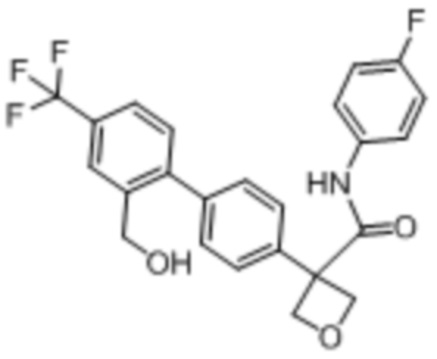
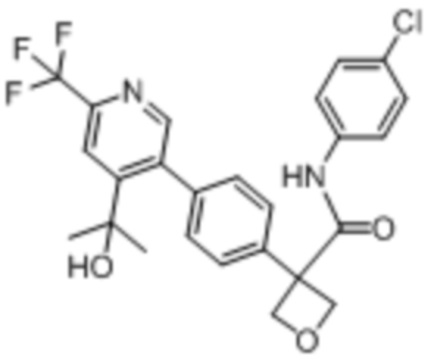
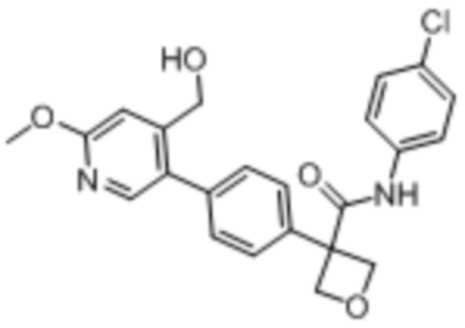
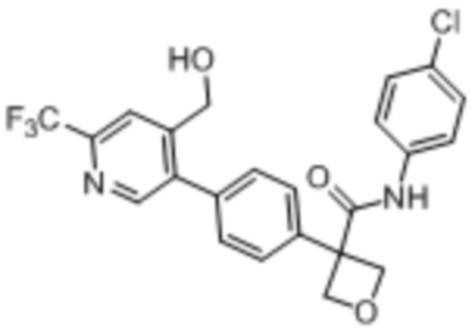
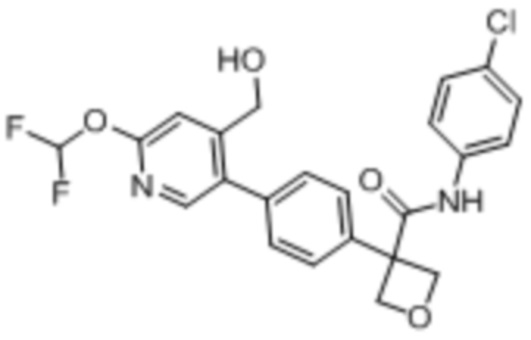
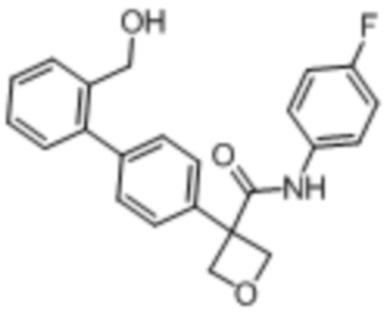
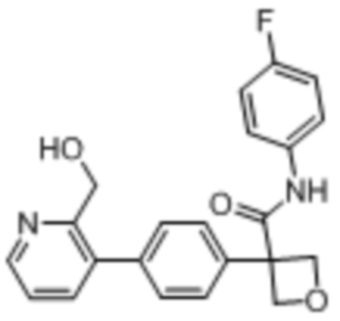
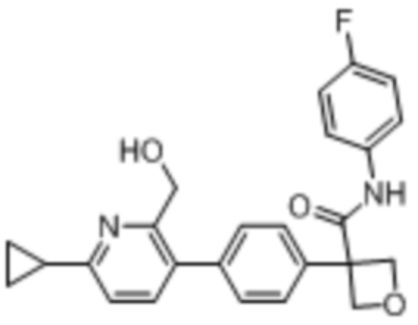
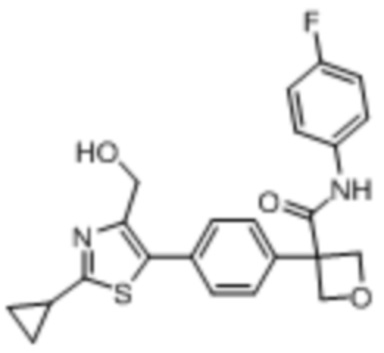
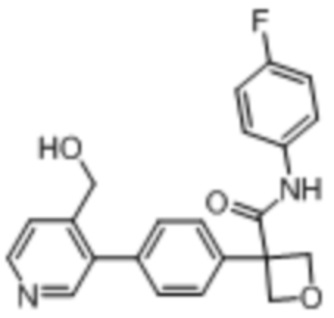
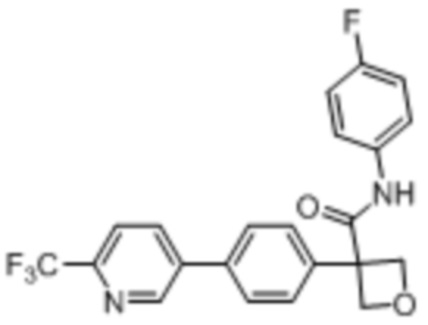
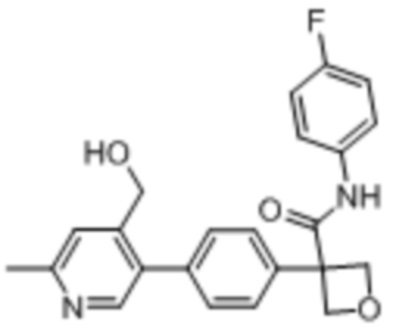
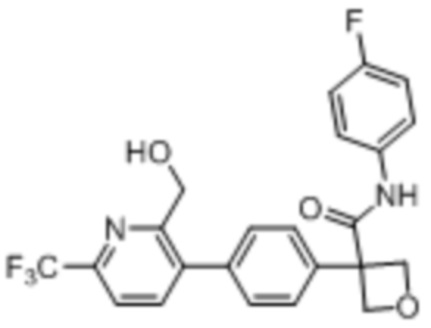
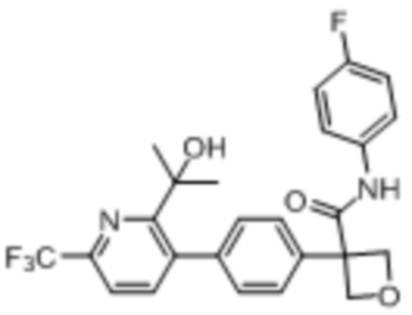
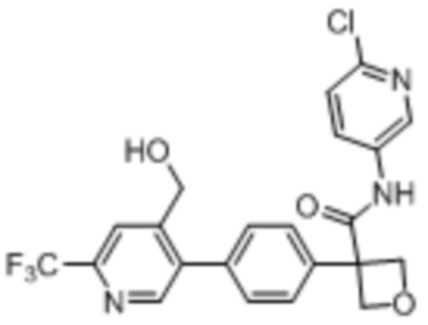
Пример 10 также можно получать следующим способом:
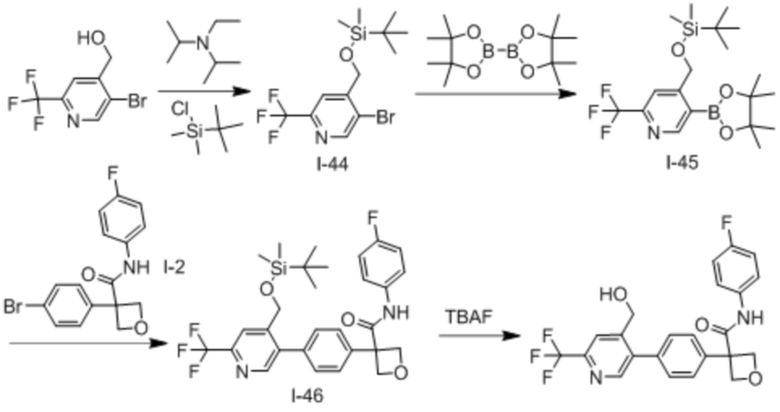
Стадия 1: Синтез 5-бром-4-(((трет-бутилдиметилсилил)окси)метил)-2-(трифторметил)пиридина (I-44)
(5-бром-2-(трифторметил)пиридин-4-ил)метанол (600,0 мг, 2,344 ммоль) растворяли в смеси DCM/DMF (об./об. 4:1, 12 мл) с перемешиванием и полученный таким образом раствор охлаждали до 0°C. Добавляли DIEA (0,573 мл, 3,28 ммоль) и трет-бутилдиметилсилилхлорид (495 мг, 3,28 ммоль) и перемешивали смесь при RT в течение 20 ч. Затем реакционную смесь концентрировали под вакуумом и разделяли с помощью EtOAc (30 мл) и воды (30 мл). Органический слой промывали водой (20 мл) и соляным раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием колонки с силикагелем с золотом 40 г RediSep, 2-20% MeOH/DCM в качестве элюентов) для получения соединения I-44. MS (ESI) [M+H]+: m/z 370.
Стадия 2: Синтез 4-(((трет-бутилдиметилсилил)окси)метил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридина (I-45)
Смесь соединения I-44 (720,0 мг, 1,944 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (593 мг, 2,333 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (127 мг, 0,194 ммоль) и ацетата калия (573 мг, 5,83 ммоль) в 1,4-диоксане (5,0 мл) выкачивали и помещали обратно с азотом 3 раза. Смесь нагревали до 80°C в запаянной трубке в течение 2 ч. После охлаждения до RT реакционную смесь фильтровали через целитовую прокладку. Фильтрат концентрировали под вакуумом для получения остатка, который растворяли в дихлорметане. После промывки водой (3 раза) и соляным раствором дихлорметановый слой сушили над безводным Na2SO4 и концентрировали под вакуумом для получения неочищенного продукта, который использовали непосредственно на следующей стадии без очистки. MS (ESI) [M+H]+: m/z 418.
Стадия 3: Синтез 3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамида (I-46)
Раствор (4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)бороновой кислоты (96 мг, 0,286 ммоль), 3-(4-бромфенил)-N-(4-фторфенил)оксетан-3-карбоксамида (I-2) (100 мг, 0,286 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (18,61 мг, 0,029 ммоль) и карбоната натрия (0,286 мл, 0,571 ммоль) в 1,4-диоксан (2,0 мл) подвергали реакции перекрестного сочетания Сузуки в стандартных условиях и неочищенный продукт очищали посредством хроматографии на колонках с силикагелем (этилацетат/DCM, 10-70%) для получения соединения I-46. MS (ESI) [M+H]+: m/z 561.
Стадия 4: N -(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
К раствору соединения I-46, (143,0 мг, 0,255 ммоль) в THF (5,0 мл) добавляли тетра-N-бутилхлорид аммония в 1 M THF (0,255 мл, 0,255 ммоль) при 0°C и перемешивали реакционную смесь в течение 2 ч. при RT. Растворитель выпаривали под вакуумом и перерастворяли остаток в дихлорметане. После последовательной промывки водой, насыщенным водным раствором бикарбоната натрия и соляным раствором органическую фазу сушили над сульфатом натрия, фильтровали и выпаривали фильтрат под вакуумом. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения.
Пример 12: N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
В 3-горлую круглодонную колбу в атмосфере азота помещали N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (I-9) (5 г, 12,5 ммоль), 2-(5-бром-2-(трифторметил)пиридин-4-ил)пропан-2-ол (I-12) (4 г, 14 ммоль) и PdCl2(dtbpf) (0,53 г, 8,15 ммоль) и колбу подвергали циклу обработки вакуумом/азотом три раза. Затем добавляли этанол (100 мл), а затем водный раствор фосфата калия (40 мл, 1 M). Смесь снова подвергали циклу обработки вакуумом/азотом три раза, а затем нагревали до 60°C в атмосфере азота в течение 4 ч. Смеси позволяли охлаждаться до RT, переносили на делительную воронку и разводили водой (50 мл) и дихлорметаном (100 мл). Собирали органическую фазу и еще раз экстрагировали водную фазу дихлорметаном (100 мл). Объединенные органические экстракты промывали соляным раствором и перемешивали с SiliaMetS Thiol (6,5 г). Полученную суспензию сушили над безводным сульфатом натрия, фильтровали и концентрировали фильтрат досуха при пониженном давлении. Полученный неочищенный продукт очищали посредством хроматографии на силикагеле с использованием градиента этилацетата в гексане для получения неочищенного материала. Этот неочищенный материал ресуспендировали в диэтиловом простом эфире (25 мл) и перемешивали в течение 15 мин. Твердые вещества фильтровали и промывали диэтиловым простым эфиром для получения титульного соединения. MS (ESI+) m/z [M+H]+: 475. 1H ЯМР (600 МГц, DMSO-d6) δ 10,07 (с, 1H), 8,35 (с, 1H), 8,22 (с, 1H), 7,67-7,62 (м, 2H), 7,58 (д, 2H), 7,41 (д, 2H), 7,20-7,14 (м, 2H), 5,49 (с, 1H), 5,24 (д, 2H), 4,91 (д, 2H), 1,25 (с, 6H).
Пример 34: 3-(4-(4-(фторметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
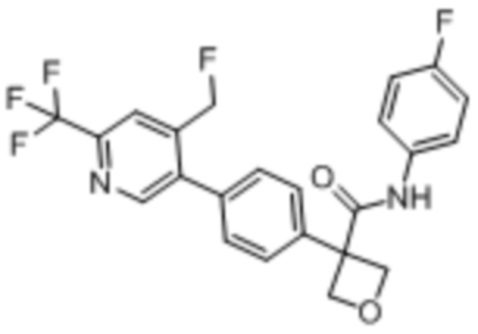
К раствору N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида (I-20) (70,0 мг, 0,157 ммоль) в DCM (3 мл) при 0°C добавляли 1,1,1-трифтор-N, N-бис(2-метоксиэтил)-1,4-сульфонамин. После перемешивания при 0°C в течение 1 ч. реакцию осторожно гасили водным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой концентрировали и очищали неочищенный материал посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 449. 1H ЯМР (499 МГц, DMSO-d6) δ 10,07 (с, 1H), 8,76 (с, 1H), 8,03 (с, 1H), 7,65 (т, J=6,9 Гц, 4H), 7,57 (д, J=8,1 Гц, 2H), 7,17 (т, J=8,8 Гц, 2H), 5,60 (д, J=46,4 Гц, 2H), 5,25 (д, J=6,5 Гц, 2H), 4,93 (д, J=6,5 Гц, 2H).
Пример 35: 3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-(трифторметокси)фенил)оксетан-3-карбоксамид
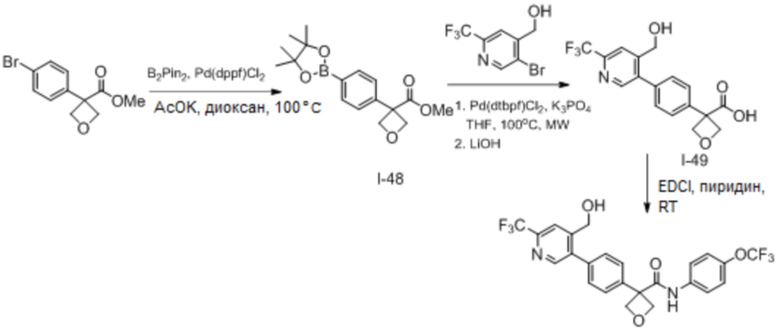
Стадия 1: Метил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксилат (I-48)
К раствору метил-3-(4-бромфенил)оксетан-3-карбоксилата (2,1 г, 7,75 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (2,065 г, 8,13 ммоль) в диоксане (30 мл) добавляли AcOK (2,281 г, 23,24 ммоль) и Pd(dppf)Cl2 (0,567 г, 0,775 ммоль) при перемешивании при RT в атмосфере азота. После завершения добавления реакционную смесь перемешивали при 80°C в течение 14 ч., охлаждали до RT и разводили EtOAc (30 мл). Смесь фильтровали и концентрировали фильтрат. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка для флэш-хроматографии с силикагелем-CS Agela® (12 г), элюция с использованием градиента 0~7% этилацетата/петролейного эфира при скорости 30 мл/мин) для получения соединения I-48 в виде твердого вещества. MS (ESI) m/z 360 [M+ACN+H+].
Стадия 2: Получение 3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоновой кислоты (I-49)
К раствору соединения I-48 (500 мг, 1,571 ммоль) и (5-бром-2-(трифторметил)пиридин-4-ил)метанола (402 мг, 1,571 ммоль) в THF (6 мл) и воде (1 мл) добавляли K3PO4 (1,001 г, 4,71 ммоль) и Pd(dtbpf)Cl2 (102 мг, 0,157 ммоль). Реакционную смесь запаивали, перемешивали при 100°C в атмосфере азота, переносили в микроволновую печь. После перемешивания при 100°C в течение 0,5 ч. реакционную смесь охлаждали до RT и разводили водой (10 мл). Смесь экстрагировали с помощью EtOAc (30 мл, 3 раза). Объединенные органические слои промывали соляным раствором (20 мл) и сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле (ISCO®; колонка для флэш-хроматографии с силикагелем-CS Agela® (12 г), элюция с использованием градиента 0~27% этилацетата/петролейного эфира при скорости 30 мл/мин) для получения метил-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилата в виде твердого вещества. MS (ESI) m/z 368,0 [M+H+].
К раствору метил-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилата, полученного, как описано выше, (520 мг, 1,416 ммоль) в THF (4 мл), MeOH (4 мл) и воде (2 мл) добавляли LiOH (102 мг, 4,25 ммоль) при RT и перемешивании и перемешивали реакционную смесь при RT в течение 14 ч. К смеси добавляли 3 Н HCl при перемешивании до достижения pH ~4. Затем реакционную смесь разводили водой (5 мл) и экстрагировали с помощью EtOAc (10 мл, 5 раз). Объединенный органический слой промывали соляным раствором (10 мл) и сушили над Na2SO4, фильтровали и концентрировали для получения соединения I-49 в виде твердого вещества, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 354 [M+H+].
Стадия 3: 3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-(трифторметокси)фенил)оксетан-3-карбоксамид
К раствору соединения I-49 (30 мг, 0,085 ммоль) и 4-(трифторметокси)анилина (30 мг, 0,169 ммоль) в пиридине (1,0 мл) добавляли EDCI (50 мг, 0,261 ммоль) при перемешивании и RT. После завершения добавления реакционную смесь перемешивали при RT в течение 1 ч. Реакционную смесь концентрировали и очищали остаток посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой aYMC-Actus Pro C18 150×30×5 мкм, с использованием воды (0,1% TFA) и ACN в качестве элюентов. Желаемые фракции концентрировали, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 513,2 [M+H+]. 1H ЯМР (400 МГц, CD3OD) δ 8,54 (с, 1H), 8,07 (с, 1H), 7,61-7,74 (м, 4 H), 7,50 (д, J=8,3 Гц, 2H), 7,24 (д, J=8,3 Гц, 2H), 5,37 (д, J=6,6 Гц, 2H), 5,04 (д, J=6,6 Гц, 2H), 4,65 (с, 2H).
Примеры 36-42 в следующей таблице получали аналогично примеру 35.
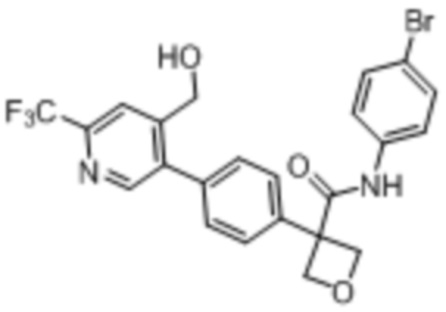
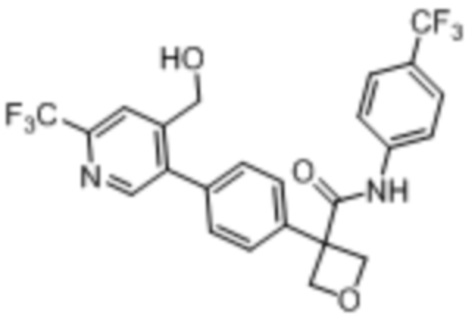
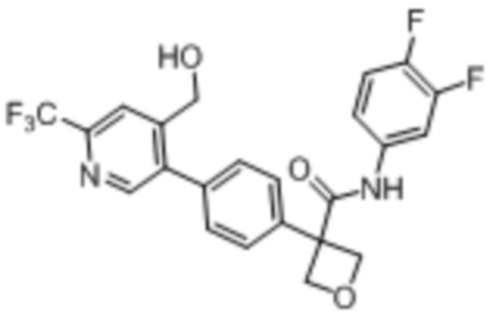
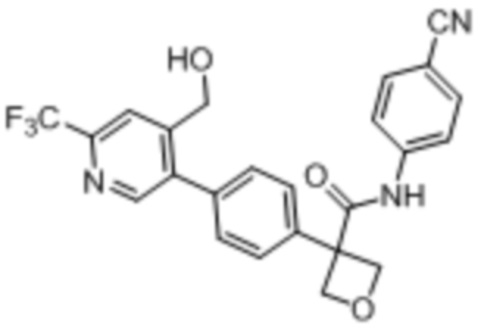
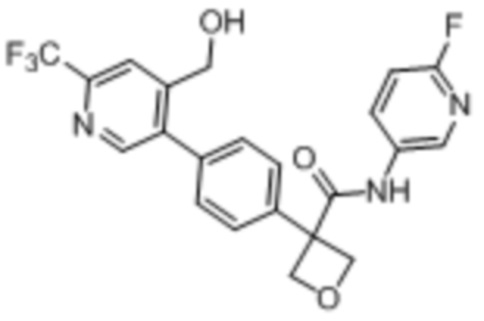
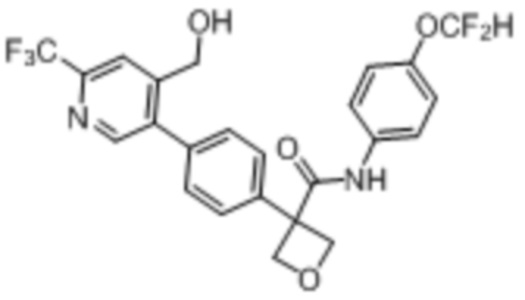
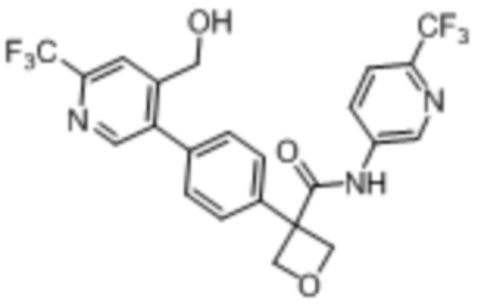
Пример 43: 3-(4-(1H-тетразол-5-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
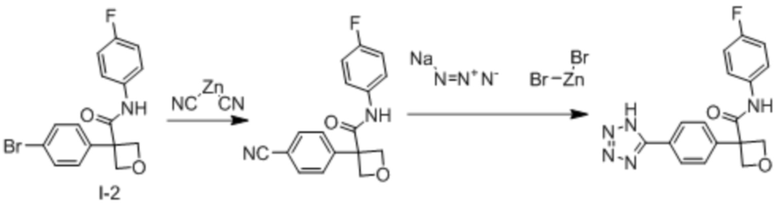
Стадия 1: Синтез N -(4-фторфенил)-3-(4-изоцианофенил)оксетан-3-карбоксамида
В сухую колбу помещали 3-(4-бромфенил)-N-(4-фторфенил)оксетан-3-карбоксамид (I-2) (1000,0 мг, 2,86 ммоль) в безводном DMF (10,0 мл). К нему добавляли цианид цинка (671 мг, 5,71 ммоль), Pd2(dba)3 (105 мг, 0,114 ммоль) и dppf (79 мг, 0,143 ммоль), а затем цинковую пыль (18,67 мг, 0,286 ммоль). Затем реакционную смесь продували азотом в течение 5 мин. Реакционную смесь нагревали при 120°C в течение 4 ч. и охлаждали до RT, фильтровали через целитовую прокладку, которую затем промывали DCM. Фильтрат концентрировали под вакуумом и неочищенный продукт очищали на колонке с силикагелем Biotage (EtOAc/гексан 5-40%) для получения N-(4-фторфенил)-3-(4-изоцианофенил)оксетан-3-карбоксамида. MS (ESI) [M+H]+: m/z 297.
Стадия 2: 3-(4-( 1H -тетразол-5-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид (соединение 43)
N-(4-фторфенил)-3-(4-изоцианофенил)оксетан-3-карбоксамид (41,0 мг, 0,138 ммоль), азид натрия (17,99 мг, 0,277 ммоль) и бромид цинка (31,2 мг, 0,138 ммоль) добавляли к смеси воды (923 мкл) и 2-пропанола (461 мкл). Затем полученную суспензию нагревали до конденсации и перемешивали в течение ночи. Смесь охлаждали до RT, фильтровали через целитовую прокладку и концентрировали. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 340,1H ЯМР (600 МГц, DMSO-d6) δ 10,03 (с, 1H), 8,09 (д, J=8,1 Гц, 2H), 7,71 (д, J=8,1 Гц, 2H), 7,61 (дд, J=8,6, 5,0 Гц, 2H), 7,15 (т, J=8,7 Гц, 2H), 5,23 (д, J=6,5 Гц, 2H), 4,92 (д, J=6,5 Гц, 2H).
Пример 44: N-(4-фторфенил)-3-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)фенил)оксетан-3-карбоксамид
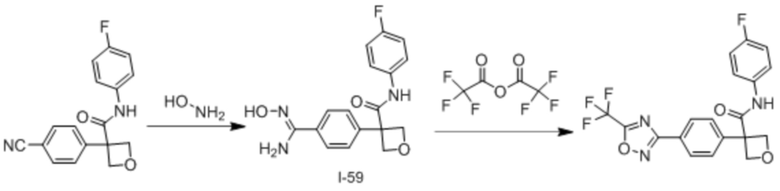
Стадия 1: Синтез (Z)- N -(4-фторфенил)-3-(4-(N'-гидроксикарбамимидоил)фенил)оксетан-3-карбоксамида (I-59)
Нитрил-N-(4-фторфенил)-3-(4-изоцианофенил)оксетан-3-карбоксамид (60,0 мг, 0,203 ммоль) растворяли в MeOH (1,0 мл), а затем добавляли гидроксиламин (50% масс. в воде) (0,137 мл, 2,228 ммоль). Смесь нагревали до 60°C в течение 2 ч. Реакционную смесь разводили соляным раствором и экстрагировали с помощью EtOAc (3 раза). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали фильтрат при пониженном давлении для получения соединения I-59. MS (ESI) [M+H]+: m/z 330. Неочищенный продукт использовали непосредственно на следующей стадии без очистки.
Стадия 2: N -(4-фторфенил)-3-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)фенил)оксетан-3-карбоксамид
TFAA (0,064 мл, 0,455 ммоль) добавляли к смеси соединения I-59 (50,0 мг, 0,152 ммоль) и пиридина (0,037 мл, 0,455 ммоль) в толуоле (2,0 мл) при 10°C. После завершения добавления смесь перемешивали при 110°C в течение 3 ч. После охлаждения до RT смесь концентрировали при пониженном давлении и остаток разделяли с помощью EtOAc и воды. Водный слой экстрагировали с помощью EtOAc (2 раза) и собранные органические слои промывали соляным раствором, сушили над безводным Na2SO4. Смесь фильтровали и концентрировали фильтрат при пониженном давлении. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 408, 1H ЯМР (600 МГц, DMSO-d6) δ 10,07 (с, 1H), 8,13 (д, J=8,2 Гц, 2H), 7,74 (д, J=8,2 Гц, 2H), 7,61 (дд, J=8,5, 5,0 Гц, 2H), 7,16 (т, J=8,7 Гц, 2H), 5,24 (д, J=6,6 Гц, 2H), 4,91 (д, J=6,6 Гц, 2H).
Пример 45: 3-(4-(1H-бензо[d]имидазол-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
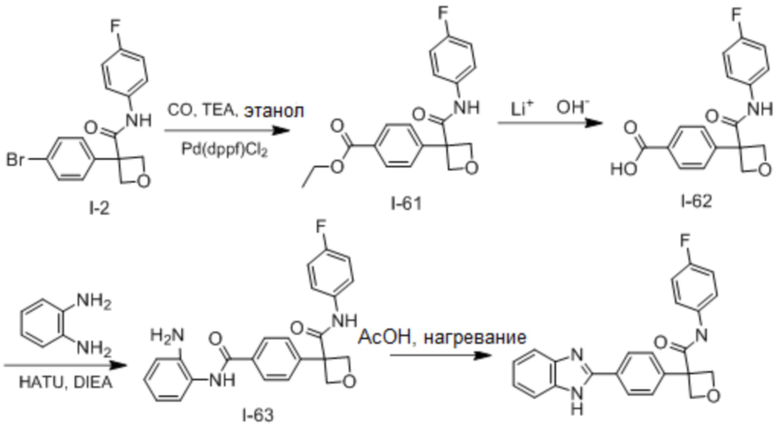
Стадия 1: Синтез этил-4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)бензоата (I-61)
Комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроценпалладия (II) в дихлорметане (152 мг, 0,186 ммоль) добавляли к перемешиваемому раствору, содержащему соединение I-2 (325,0 мг, 0,928 ммоль), TEA (517 мкл, 3,71 ммоль) в EtOH (1084 мкл, 18,56 ммоль) и DMF (4640 мкл). Смесь перемешивали в атмосфере диоксида углерода при 90°C в течение ночи. Реакционную смесь охлаждали до RT, а затем фильтровали через целитовую прокладку. Фильтрат концентрировали, разводили этилацетатом, промывали водой (2 раза) и соляным раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали на колонке Biotage (силикагель) с использованием EtOAc/гексанов (5-40%) в качестве элюентов для получения соединения I-61. MS (ESI) [M+H]+: m/z 344.
Стадия 2: Синтез 4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)бензойной кислоты (I-62)
1 M LiOH (3,0 мл, 3,00 ммоль) добавляли к раствору соединения I-61 (300,0 мг, 0,874 ммоль) в MeOH (5,0 мл). Эту смесь перемешивали при RT в течение 4 ч. и выпаривали при пониженном давлении. Остаток разводили водой и подкисляли водным раствором соляной кислоты (2 Н). Эту водную смесь экстрагировали этилацетатом (2 раза, 50 мл) и объединенные органические экстракты промывали соляным раствором, сушили над MgSO4, фильтровали и выпаривали при пониженном давлении для получения соединения I-62. MS (ESI) [M+H]+: m/z 316.
Стадия 3: 3-(4-(1H-бензо[d]имидазол-2-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид
В сосуд объемом 20 мл помещали якорь магнитной мешалки, соединение I-62 (50,0 мг, 0,159 ммоль) и DMF (1,91 мл). Добавляли HATU (60,3 мг, 0,159 ммоль) при перемешивании и перемешивали смесь в течение нескольких минут. Затем к смеси добавляли бензол-1,2-диамин (17,15 мг, 0,159 ммоль), DIEA (83 мкл, 0,476 ммоль) и перемешивали реакционную смесь в течение 4 ч. при RT. Реакционную смесь разводили 50 мл этилацетата и промывали 1 Н водным раствором HCl, водой, соляным раствором и насыщенным водным раствором NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали фильтрат. Остаток растворяли в DCM и очищали посредством хроматографии на силикагеле (колонка для флэш-хроматографии 12 г, EtOAc в гексане 0-50%, 15 CV) для получения соединения I-63. MS (ESI) [M+H]+: m/z 406.
Соединение I-63 растворяли в MeOH (1910 мкл) и уксусной кислоте (200,0 мкл, 3,49 ммоль), а затем нагревали при 100°C в течение 1 ч. Смесь разводили MeOH, фильтровали и очищали посредством масс-направленной обращенно-фазовой ВЭЖХ (ACN/вода, TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 388, 1H ЯМР (600 МГц, DMSO-d6) δ 10,06 (с, 1H), 8,25 (с, 1H), 8,23 (с, 1H), 7,79 (с, 1H), 7,76 (дд, J=7,4, 4,6 Гц, 3H), 7,62 (дд, J=8,6, 5,0 Гц, 2H), 7,51-7,38 (м, 2H), 7,16 (т, J=8,8 Гц, 2H), 5,25 (д, J=6,6 Гц, 2H), 4,96 (д, J=6,6 Гц, 2H).
Примеры 46-50 получали так же, как и пример 45, с использованием кислоты I-62 и 4-хлорфенилендиамина, пиридин-3,4-диамина, 4-фторбензол-1,2-диамина, 4,5-дифторбензол-1,2-диамина, пиридин-2,3-диамина и 5-бромпиридин-2,3-диамина, соответственно.
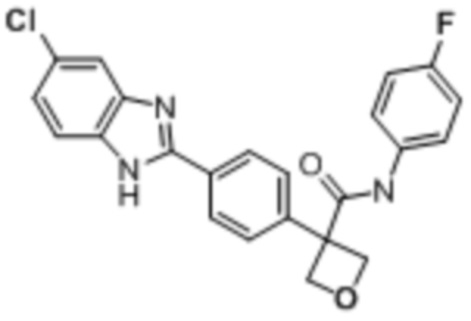
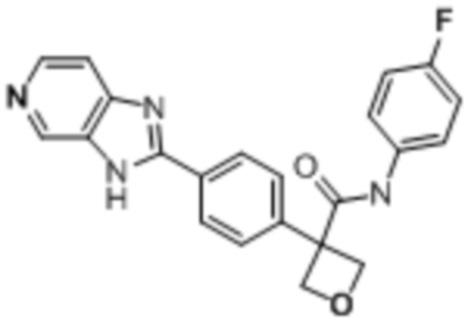
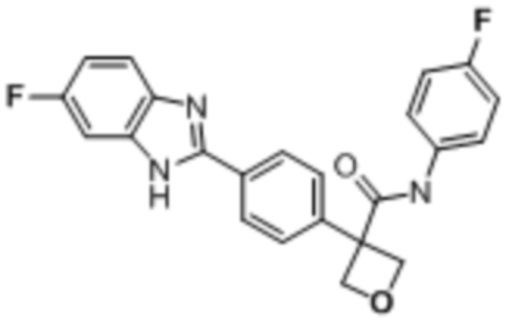
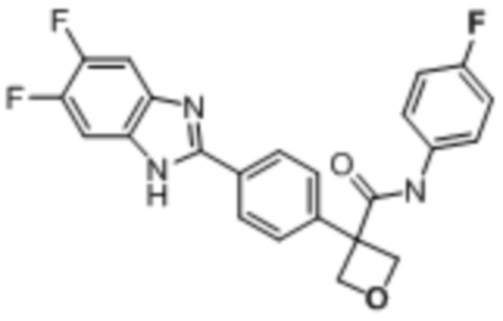
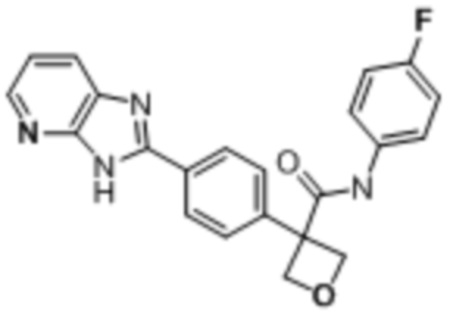
Пример 51: 3-(4-(6-циано-3H-имидазо[4,5-b]пиридин-2-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид
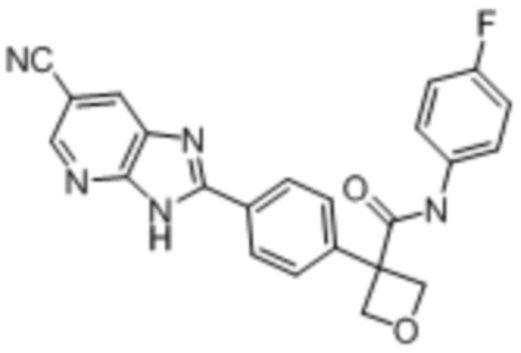
3-(4-(6-бром-3H-имидазо[4,5-b]пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид (полученный способом, описанным для примера 45) (70,0 мг, 0,150 ммоль), цианид цинка (42,2 мг, 0,360 ммоль), цинковую пыль (7,05 мг, 0,108 ммоль), Pd2(dba)3 (21,95 мг, 0,024 ммоль) и DPPF (26,6 мг, 0,048 ммоль) в N, N-диметилацетамиде (2,0 мл) добавляли в реакционный сосуд для микроволновой печи. Затем смесь облучали при 130°C в течение 60 мин. Реакционную смесь охлаждали, а затем фильтровали через целитовую прокладку и концентрировали. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 414, 1H ЯМР (499 МГц, DMSO-d6) δ 10,05 (с, 1H), 8,77 (с, 1H), 8,61 (с, 1H), 8,32 (д, J=8,2 Гц, 2H), 7,71 (д, J=8,2 Гц, 2H), 7,63 (дд, J=8,7, 5,0 Гц, 2H), 7,16 (т, J=8,8 Гц, 2H), 5,24 (д, J=6,5 Гц, 2H), 4,94 (д, J=6,5 Гц, 2H).
Пример 52: 3-(4-(6-циклопропилпиридин-3-ил)фенил)- N -(5-фторпиридин-2-ил)оксетан-3-карбоксамид
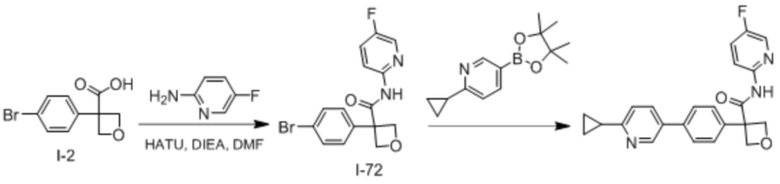
Стадия 1: 3-(4-бромфенил)-N-(5-фторпиридин-2-ил)оксетан-3-карбоксамид
В сосуд объемом 100 мл помещали якорь магнитной мешалки, 3-(4-бромфенил)оксетан-3-карбоновую кислоту (1000,0 мг, 3,89 ммоль) и DMF (10,0 мл). К этому добавляли HATU (1775 мг, 4,67 ммоль) при RT и перемешивании и перемешивали реакционную смесь в течение нескольких минут. Добавляли 5-фторпиридин-2-амин (436 мг, 3,89 ммоль) и DIEA (2,038 мл, 11,67 ммоль) и перемешивали реакционную смесь в течение 4 ч. при RT. Реакционную смесь разводили этилацетатом и промывали 1 Н водным раствором HCl (3 раза), водой, соляным раствором и насыщенным водным раствором NaHCO3. Затем органический раствор сушили над Na2SO4, фильтровали и концентрировали фильтрат. Неочищенный продукт очищали на колонке Biotage с DCM/EtOAc (10-100%) для получения соединения I-72. MS (ESI) [M+H]+: m/z 351.
Стадия 2: 3-(4-(6-циклопропилпиридин-3-ил)фенил)- N -(5-фторпиридин-2-ил)оксетан-3-карбоксамид
К перемешиваемой суспензии соединения I-72, (50,0 мг, 0,142 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (11,68 мг, 0,013 ммоль) в 1,4-диоксане (1,5 мл) добавляли карбонат натрия (0,142 мл, 0,285 ммоль). Реакционную смесь выкачивали и помещали обратно с азотом три раза, а затем нагревали до 80°C в течение 4 ч. Реакционную смесь охлаждали до RT, фильтровали через целитовую прокладку и концентрировали. Продукт очищали посредством масс-направленной обращенно-фазовой хроматография (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 390, 1H ЯМР (499 МГц, DMSO-d6) δ 12,86 (с, 1H), 9,25 (с, 1H), 8,74 (с, 1H), 8,45 (т, J=7,3 Гц, 1H), 8,05 (д, J=7,6 Гц, 1H), 7,75 (д, J=8,3 Гц, 2H), 7,51 (д, J=8,3 Гц, 2H), 7,42 (дд, J=15,3, 6,3 Гц, 2H), 5,57 (д, J=15,0 Гц, 2H), 5,22 (д, J=15,0 Гц, 2H), 2,24-2,11 (м, 1H), 1,10-1,02 (м, 2H), 0,99 (с, 2H).
Пример 53: 3-(4-(3-циклопропил-1,2,4-оксадиазол-5-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид
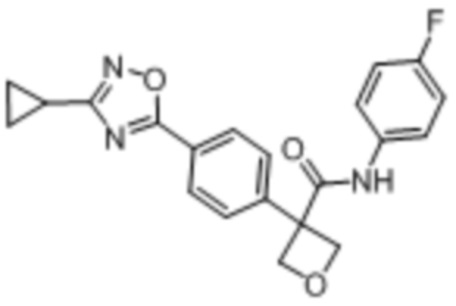
N, N'-карбонилдиимидазол (94 мг, 0,579 ммоль)) добавляли к перемешиваемой смеси 4-(3-(4-фторфенил)карбамоил)оксетан-3-ил)бензойной кислоты (I-62) (112,0 мг, 0,355 ммоль) в DCM (2,0 мл) и перемешивали смесь при RT в течение 2 ч. Затем добавляли N'-гидроксициклопропанкарбоксимидамид (89 мг, 0,888 ммоль) и перемешивали смесь при RT еще в течение 2 ч, концентрировали, выпаривали с толуолом, перерастворяли в толуоле (2 мл) и нагревали 110°C в течение 2 ч. Смесь охлаждали, гасили водой и экстрагировали этилацетатом (3 раза). Объединенные органические фракции промывали соляным раствором, сушили над Na2SO4, фильтровали и выпаривали растворители при пониженном давлении. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматография (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 380. 1H ЯМР (600 МГц, DMSO-d6) δ 10,06 (с, 1H), 8,10 (д, J=8,2 Гц, 2H), 7,71 (д, J=8,2 Гц, 2H), 7,60 (дд, J=8,6, 5,0 Гц, 2H), 7,15 (т, J=8,8 Гц, 2H), 5,23 (д, J=6,6 Гц, 2H), 4,90 (д, J=6,6 Гц, 2H), 2,19 (тт, J=8,4, 4,8 Гц, 1H), 1,20-1,06 (м, 2H), 1,06-0,91 (м, 2H).
Пример 54: 3-(4-(4-циклопропил-6-оксопиримидин-1( 6H )-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид
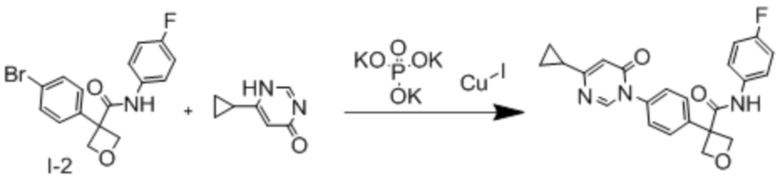
В реакционный сосуд помещали 6-циклопропилпиримидин-4(1H)-он (43,0 мг, 0,316 ммоль), йодид меди (6,01 мг, 0,032 ммоль), 3-(4-бромфенил)-N-(4-фторфенил)оксетан-3-карбоксамид (I-2) (111 мг, 0,316 ммоль) и транс-N, N'-диметилциклогексан-1,2-диамин (8,98 мг, 0,063 ммоль). Затем эту смесь выкачивали и помещали обратно с азотом (3 раза). Затем добавляли сухой, дегазированный 1,4-диоксан (1263 мкл), а затем нагревали смесь при 110°C в течение 24 ч. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 406, 1H ЯМР (499 МГц, DMSO-d6) δ 10,05 (с, 1H), 8,33 (с, 1H), 7,63 (т, J=8,4 Гц, 4H), 7,49 (д, J=8,4 Гц, 2H), 7,15 (т, J=8,8 Гц, 2H), 6,44 (с, 1H), 5,23 (д, J=6,5 Гц, 2H), 4,91 (д, J=6,6 Гц, 2H), 3,36 (с, 2H), 1,95 (п, J=6,9 Гц, 1H), 1,05-0,71 (м, 2H).
Пример 55: 3-(4-(4-циклопропил-6-метил-2-оксопиримидин-1( 2H )-ил)фенил)- N -(4-фторфенил)оксетан-3-карбоксамид
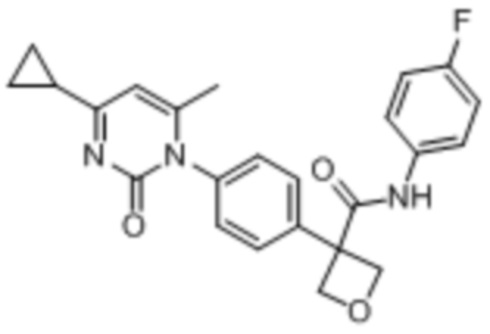
Пример 55 получали аналогично примеру 54. MS (ESI) [M+H]+: m/z 420. 1H ЯМР (499 МГц, DMSO-d6) δ 9,96 (с, 1H), 7,79 (д, J=8,3 Гц, 2H), 7,60 (дд, J=8,8, 5,0 Гц, 2H), 7,29 (д, J=8,3 Гц, 3H), 7,15 (т, J=8,8 Гц, 2H), 5,17 (д, J=6,5 Гц, 2H), 4,82 (д, J=6,5 Гц, 2H), 3,37 (с, 4H), 2,50 (с, 3H), 2,08 (с, 1H).
Пример 56: N -(4-фторфенил)-1-(6-(2-фенилоксазол-4-ил)пиридин-3-ил)циклобутан-1-карбоксамид
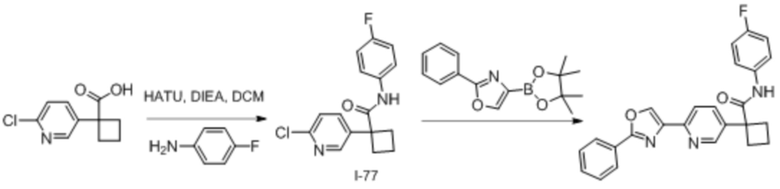
Стадия 1: Синтез 1-(6-хлорпиридин-3-ил)- N -(4-фторфенил)циклобутан-1-карбоксамида (I-77)
В сосуд объемом 100 мл помещали якорь магнитной мешалки, 1-(6-хлорпиридин-3-ил)циклобутанкарбоновую кислоту (715,0 мг, 3,38 ммоль) в DCM (20,0 мл). Добавляли HATU (1541 мг, 4,05 ммоль) при перемешивании и перемешивали реакционную смесь в течение нескольких минут. Добавляли 4-фторанилин (375 мг, 3,38 ммоль) и DIEA (1,770 мл, 10,13 ммоль) и перемешивали реакционную смесь в течение 4 ч. при RT. Реакционную смесь разводили этилацетатом и промывали 1 н. HCl (2 раза), водой, соляным раствором и насыщенным водным раствором NaHCO3. Затем органический слой сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колонки с силикагелем Biotage с использованием DCM/EtOAc (10 до 100%) в качестве элюентов для получения соединения I-77. MS (ESI) [M+H]+: m/z 305.
Стадия 2: N -(4-фторфенил)-1-(6-(2-фенилоксазол-4-ил)пиридин-3-ил)циклобутан-1-карбоксамид
Соединение I-77 (50 мг, 0,164 ммоль), смесь ацетата палладия (II), 1,1'-бис(ди-трет-бутилфосфино)ферроцена и фосфата калий (13,46 мг, 0,015 ммоль) растворяли в 1,4-диоксане (1,5 мл) в круглодонной колбе объемом 20 мл. Добавляли карбонат натрия (0,164 мл, 0,328 ммоль), реакционную смесь выкачивали, помещали обратно с азотом 3 раза и нагревали при 80°C в течение 12 ч. Затем реакционную смесь охлаждали и фильтровали через целитовую прокладку. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 414.
Пример 57-63 в следующей таблице получали аналогично примеру 56 с использованием соответствующих боронатов.
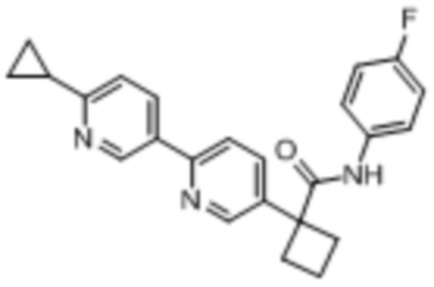
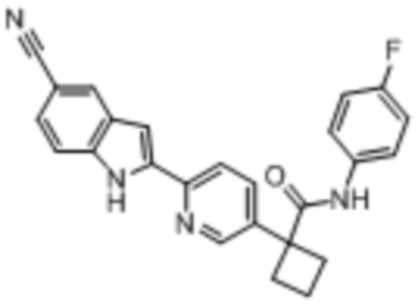
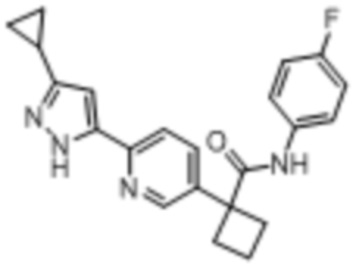
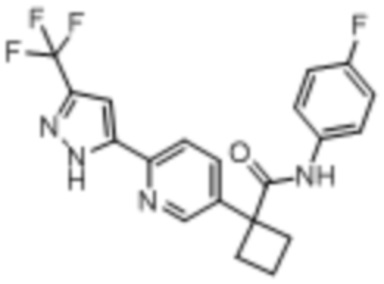
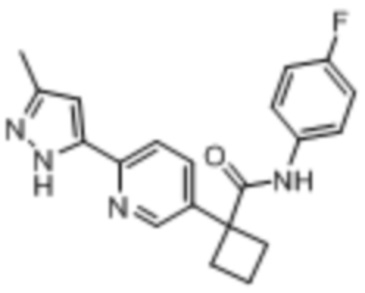
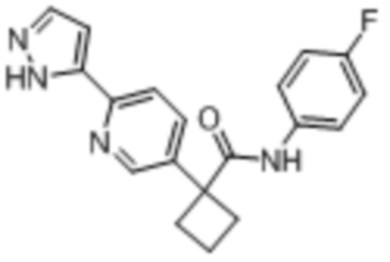
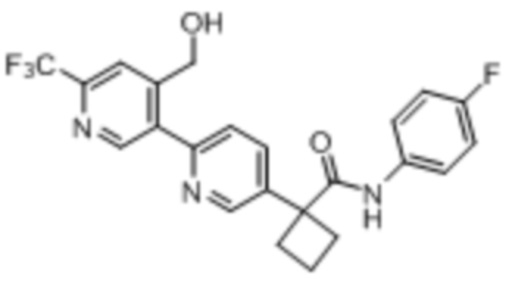
Пример 64: 1-(4-(4-циклопропил-1H-пиразол-1-ил)фенил)-N-пропилциклобутан-1-карбоксамид
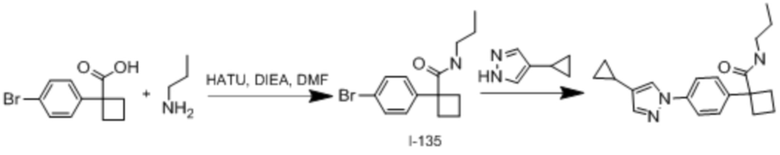
Стадия 1: Синтез 1-(4-бромфенил)-N-пропилциклобутан-1-карбоксамида (I-135)
В сосуд емкостью 20 мл помещали якорь магнитной мешалки, при перемешивании добавляли 1-(4-бромфенил)циклобутанкарбоновую кислоту (800,0 мг, 3,14 ммоль) в DMF (4,0 мл) и HATU (1431 мг, 3,76 ммоль) и перемешивали реакционную смесь в течение нескольких минут. Затем добавляли пропан-1-амин (185 мг, 3,14 ммоль) и DIEA (1,643 мл, 9,41 ммоль) и перемешивали смесь в течение 4 ч. при RT. Реакционную смесь разводили этилацетатом и промывали 1 Н водным раствором HCl (3 раза), водой, соляным раствором и насыщенным водным раствором NaHCO3. Затем раствор сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием колонки с силикагелем с золотом 48 г RediSep, 10-100% EtOAc/гексаны в качестве элюентов) для получения соединения I-135. MS (ESI) [M+H]+: m/z 296.
Стадия 2: Синтез 1-(4-(4-циклопропил-1H-пиразол-1-ил)фенил)-N-пропилциклобутан-1-карбоксамида
В сосуд объемом 20 мл добавляли соединение I-135 (25,0 мг, 0,084 ммоль) и йодид меди (1,607 мг, 8,44 мкмоль), а затем 4-циклопропил-1H-пиразол (9,13 мг, 0,084 ммоль) и транс-N, N'-диметилциклогексан-1,2-диамин (2,401 мг, 0,017 ммоль). Затем эту смесь выкачивали и помещали обратно с азотом (3 раза). Затем добавляли сухой, дегазированный 1,2-диоксан (338 мкл). Смесь нагревали при 110°C в течение 24 ч. и охлаждали до RT. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 324. 1H ЯМР (600 МГц, DMSO-d6) δ 8,22 (с, 1H), 7,70 (д, J=8,4 Гц, 2H), 7,59 (к, J=6,1, 5,5 Гц, 1H), 7,51 (д, J=18,9 Гц, 1H), 7,39 (д, J=8,4 Гц, 2H), 2,96 (п, J=7,0 Гц, 2H), 2,77-2,62 (м, 2H), 2,33 (дк, J=19,2, 10,1, 9,3 Гц, 2H), 1,86-1,68 (м, 3H), 1,32 (п, J=7,1 Гц, 2H), 0,86 (т, J=6,2 Гц, 2H), 0,70 (т, J=7,3 Гц, 3H), 0,58 (д, J=4,0 Гц, 2H).
Пример 65: 1-(4-(2-(4-Фторфенил)оксазол-4-ил)фенил)-N-пропилциклобутан-1-карбоксамид
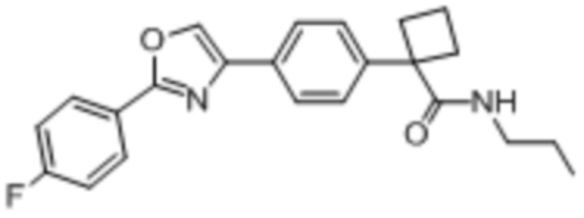
Титульное соединение получали из промежуточного бромосоединения I-98 и (2-(4-фторфенил)оксазол-4-ил)бороновой кислоты посредством реакции сочетания Сузуки в стандартных условиях. MS (ESI) [M+H]+: m/z 279.
Пример 66: 1-(4-(6-циклопропилпиридин-3-ил)фенил)-N-пропилциклобутан-1-карбоксамид
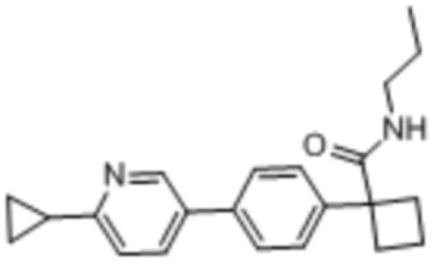
Это соединение получали аналогично синтезу примера 64, за исключением того, что использовали (6-циклопропилпиридин-3-ил)бороновую кислоту. MS (ESI) [M+H]+: m/z 335.
Пример 67: 1-(4-(6-(4-фторфенил)пиридин-3-ил)фенил)-N-пропилциклобутан-1-карбоксамид
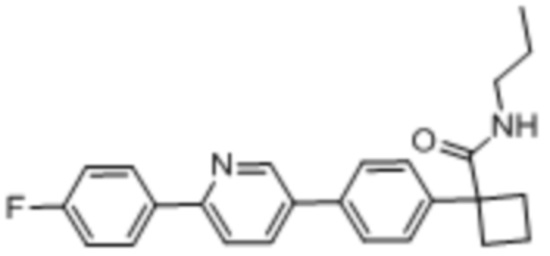
Это соединение получали аналогично синтезу примера 64, за исключением того, что использовали (6-(4-фторфенил)пиридин-3-ил)бороновую кислоту. MS (ESI) [M+H]+: m/z 389.
Пример 68: 1-(4-(6-циклопропилпиридин-3-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
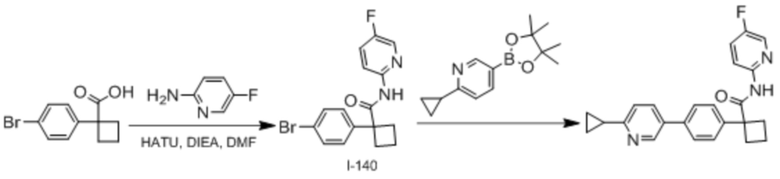
Стадия 1: 1-(4-бромфенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид (I-140)
В сосуд объемом 100 мл помещали якорь магнитной мешалки, при перемешивании добавляли 1-(4-бромфенил)циклобутанкарбоновую кислоту (1000,0 мг, 3,92 ммоль) в DMF (10,0 мл) и HATU (1789 мг, 4,70 ммоль) и перемешивали смесь в течение нескольких минут. Добавляли 5-фторпиридин-2-амин (439 мг, 3,92 ммоль), DIEA (2,054 мл, 11,76 ммоль) и перемешивали реакционную смесь в течение 4 ч. при RT. Реакцию проводили, как описано выше, и очищали неочищенный продукт посредством хроматографии (система Isco CombiFlash, с использованием колонки с силикагелем с золотом 48 г RediSep, 10-100% EtOAc/гексаны в качестве элюентов) для получения соединения I-140. MS (ESI) [M+H]+: m/z 349.
Стадия 2: 1-(4-(6-циклопропилпиридин-3-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
Смесь 2-циклопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (42,1 мг, 0,172 ммоль), соединения I-140 (60,0 мг, 0,172 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (14,09 мг, 0,015 ммоль) и карбоната натрия (0,172 мл, 0,344 ммоль) в 1,4-диоксане (1,5 мл) подвергали реакции сочетания Сузуки в стандартных условиях. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 388, 1H ЯМР (499 МГц, DMSO-d6) δ 10,19 (с, 1H), 8,82 (с, 2H), 8,29 (д, J=2,7 Гц, 1H), 8,08 (дд, J=9,2, 4,0 Гц, 1H), 7,74 (д, J=8,0 Гц, 2H), 7,64 (д, J=8,1 Гц, 2H), 7,52 (д, J=8,3 Гц, 2H), 2,89 (к, J=8,6 Гц, 2H), 2,25 (с, 2H), 1,83 (дт, J=22,2, 8,5 Гц, 3H), 1,24-1,10 (м, 2H), 1,07 (с, 2H).
Пример 69: 1-(4-(6-циклопропил-4-фторпиридин-3-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
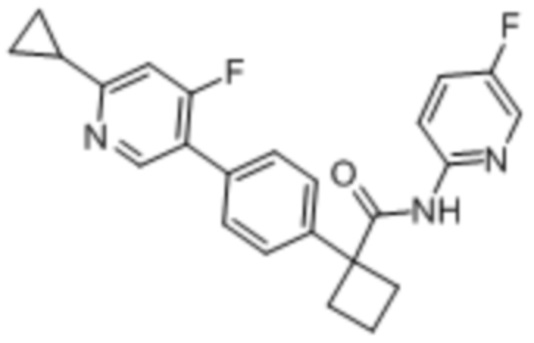
Это соединение получали аналогично синтезу примера 68, за исключением того, что использовали 2-циклопропил-4-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин. MS (ESI) [M+H]+: m/z 406, 1H ЯМР (499 МГц, DMSO-d6) δ 10,20 (с, 1H), 8,59 (д, J=10,5 Гц, 1H), 8,29 (д, J=2,7 Гц, 1H), 8,09 (дд, J=9,2, 4,0 Гц, 1H), 7,76-7,68 (м, 1H), 7,63 (д, J=8,2 Гц, 2H), 7,57 (д, J=7,9 Гц, 2H), 7,40 (д, J=11,7 Гц, 1H), 2,88 (к, J=8,6 Гц, 2H), 2,20 (д, J=4,8 Гц, 2H), 1,84 (дт, J=16,3, 8,4 Гц, 3H), 1,05 (д, J=7,9 Гц, 2H), 1,02 (с, 2H).
Пример 70: 1-(4-(6-циклопропил-4-метилпиридин-3-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
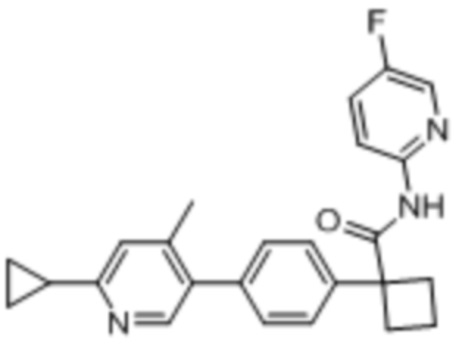
Это соединение получали аналогично синтезу примера 68, за исключением того, что использовали 2-циклопропил-4-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин. MS (ESI) [M+H]+: m/z 402. 1H ЯМР (499 МГц, DMSO-d6) δ 10,25 (с, 1H), 8,45 (с, 1H), 8,30 (д, J=2,8 Гц, 1H), 8,10 (дд, J=9,2, 4,1 Гц, 1H), 7,73 (тд, J=8,8, 2,9 Гц, 1H), 7,64 (д, J=8,1 Гц, 2H), 7,50 (с, 1H), 7,45 (д, J=8,0 Гц, 2H), 2,99-2,72 (м, 3H), 2,37 (с, 3H), 2,28-2,19 (м, 1H), 1,84 (дт, J=18,4, 8,5 Гц, 3H), 1,31-1,18 (м, 2H), 1,11 (с, 2H).
Пример 71: 1-(4-(6,7-дифтор-1H-бензо[d]имидазол-2-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
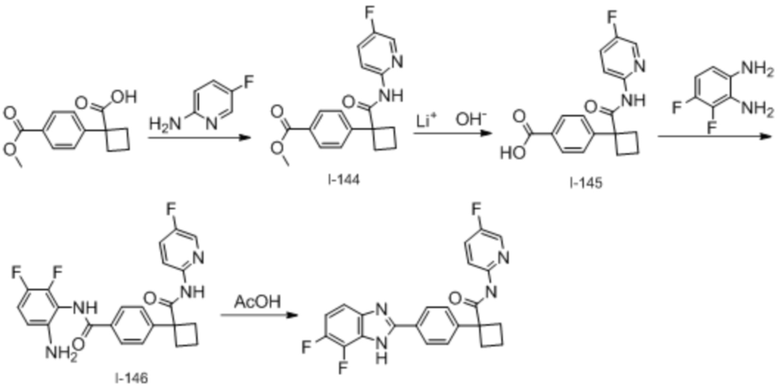
Стадия 1: Метил-4-(1-((5-фторпиридин-2-ил)карбамоил)циклобутил)бензоат (I-144)
1-(4-(метоксикарбонил)фенил)циклобутанкарбоновую кислоту (2000,0 мг, 8,54 ммоль) и оксалилхлорид (0,747 мл, 8,54 ммоль) перемешивали в DCM (10,0 мл), к ним добавляли DMF (0,2 мл). Реакционную смесь перемешивали при RT в течение 4 ч, концентрировали под вакуумом и оставляли на ночь в лиофилизаторе. Затем к неочищенному продукту добавляли смесь 2-амино-5-фторпиридина (957 мг, 8,54 ммоль) в пиридине (10,0 мл) и охлаждали смесь до 0°C. Смесь медленно нагревали до RT, перемешивали в течение ночи, концентрировали под вакуумом и неочищенный продукт очищали с помощью Biotage (SiO2, CH2Cl2/MeOH; 0-10%) для получения соединения I-144. MS (ESI) [M+H]+: m/z 329.
Стадия 2: 4-(1-((5-фторпиридин-2-ил)карбамоил)циклобутил)бензойная кислота (I-145)
В сосуд, содержащий соединение I-144 (1000,0 мг, 3,05 ммоль) в тетрагидрофуране (4,0 мл) и MeOH (1,333 мл), добавляли LiOH в воде (6,09 мл, 12,18 ммоль) и перемешивали смесь при RT в течение 24 ч. Органические растворители выпаривали и подкисляли водный слой до pH~3 посредством добавления HCl (1 Н), а затем экстрагировали DCM 3 раза. Органические фазы объединяли, промывали соляным раствором, сушили и концентрировали для получения соединения I-145. MS (ESI) [M+H]+: m/z 315.
Стадия 3: Синтез N-(6-амино-2,3-дифторфенил)-4-(1-((5-фторпиридин-2-ил)карбамоил)циклобутил)-бензамида (I-146)
В сосуд объемом 20 мл помещали соединение I-145 (30,0 мг, 0,095 ммоль) в DMF (1,0 мл), затем добавляли HATU (43,6 мг, 0,115 ммоль) при перемешивании. Через несколько минут добавляли 1,2-диамино-3,4-дифторбензол (13,76 мг, 0,095 ммоль) и DIEA (0,100 мл, 0,573 ммоль) и перемешивали реакционную смесь в течение 4 ч. при RT. Реакционную смесь разводили этилацетатом и промывали 1 н. HCl (3 раза). Органический слой концентрировали и остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием колонки с силикагелем с золотом 12 г RediSep и 0-20% MeOH/DCM в качестве элюентов) для получения соединения I-146. MS (ESI) [M+H]+: m/z 441.
Стадия 4: 1-(4-(6,7-дифтор-1H-бензо[d]имидазол-2-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
Раствор соединения I-146 (17,0 мг, 0,039 ммоль) в AcOH (1,5 мл) нагревали при 150°C в микроволновой печи в течение 30 мин. Смесь выпаривали при пониженном давлении. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 423. 1H ЯМР (499 МГц, DMSO-d6) δ 10,24 (с, 1H), 8,29 (д, J=2,7 Гц, 1H), 8,18 (д, J=8,2 Гц, 3H), 8,09 (дд, J=9,2, 4,0 Гц, 1H), 7,73 (дд, J=8,6, 2,6 Гц, 1H), 7,69 (д, J=8,3 Гц, 3H), 7,37 (дд, J=8,6, 3,1 Гц, 1H), 7,31-7,17 (м, 1H), 2,90 (дт, J=14,6, 8,6 Гц, 2H), 2,59-2,53 (м, 2H), 1,86 (дт, J=14,8, 7,4 Гц, 2H).
Пример 72: 1-(4-(7-фтор-1H-бензо[d]имидазол-2-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
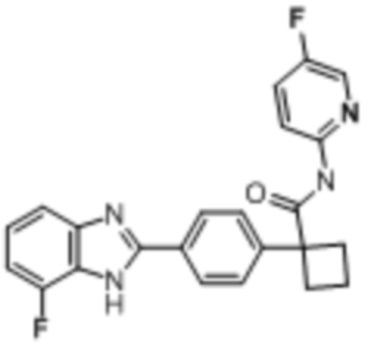
Титульное соединение получали аналогично синтезу примера 71, за исключением того, что использовали 3-фторбензол-1,2-диамин. MS (ESI) [M+H]+: m/z 405, 1H ЯМР (499 МГц, DMSO-d6) δ 10,26 (с, 1H), 8,29 (д, J=2,7 Гц, 1H), 8,16 (д, J=8,3 Гц, 2H), 8,09 (дд, J=9,2, 4,0 Гц, 1H), 7,78-7,66 (м, 4H), 7,54 (д, J=8,9 Гц, 1H), 7,23 (т, J=9,2 Гц, 1H), 2,92 (дт, J=14,6, 8,6 Гц, 2H), 2,60-2,53 (м, 2H), 1,98-1,78 (м, 2H).
Пример 73: 1-(4-(5-циано-1H-бензо[d]имидазол-2-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
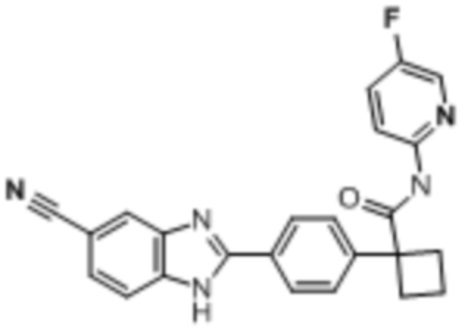
Титульное соединение получали аналогично синтезу примера 71, за исключением того, что использовали 3,4-диаминобензонитрил. MS (ESI) [M+H]+: m/z 412, 1H ЯМР (499 МГц, DMSO-d6) δ 10,25 (с, 1H), 8,29 (д, J=2,7 Гц, 1H), 8,20 (д, J=8,2 Гц, 2H), 8,16 (с, 1H), 8,09 (дд, J=9,2, 4,0 Гц, 1H), 7,76 (д, J=8,3 Гц, 1H), 7,71 (д, J=8,3 Гц, 3H), 7,62 (д, J=8,3 Гц, 1H), 2,91 (дт, J=14,6, 8,6 Гц, 2H), 2,59-2,52 (м, 2H), 1,86 (дт, J=17,4, 8,6 Гц, 2H).
Пример 74: 1-(4-(4,7-дифтор-1H-бензо[d]имидазол-2-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
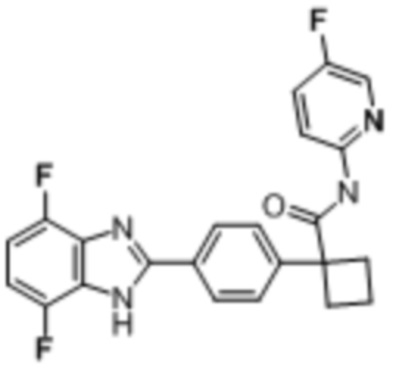
Титульное соединение получали аналогично синтезу примера 71, за исключением того, что использовали 3,6-дифторбензол-1,2-диамин. MS (ESI) [M+H]+: m/z 423. 1H ЯМР (499 МГц, DMSO-d6) δ 10,25 (с, 1H), 8,29 (д, J=2,7 Гц, 1H), 8,24 (д, J=8,2 Гц, 2H), 8,09 (дд, J=9,2, 4,0 Гц, 1H), 7,73 (дд, J=8,6, 2,7 Гц, 1H), 7,69 (д, J=8,3 Гц, 2H), 7,08-7,00 (м, 2H), 2,91 (дт, J=14,7, 8,6 Гц, 2H), 2,59-2,52 (м, 2H), 1,86 (дт, J=17,2, 8,6 Гц, 2H).
Пример 75: 1-(4-(4,4-дифтор-3a,4,5,6,7,7a-гексагидро-1H-бензо[d]имидазол-2-ил)фенил)-N-(5-фторпиридин-2-ил)циклобутан-1-карбоксамид
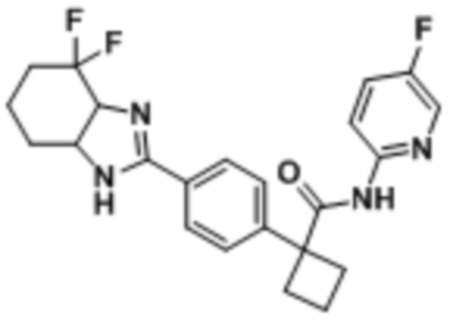
Титульное соединение получали аналогично синтезу примера 71, за исключением того, что использовали (1R,2R)-3,3-дифторциклогексан-1,2-диамин. MS (ESI) [M+H]+: m/z 429, 1H ЯМР (499 МГц, DMSO-d6) δ 11,12 (с, 1H), 11,01 (с, 1H), 10,32 (с, 1H), 8,30 (д, J=2,8 Гц, 1H), 8,11-7,97 (м, 3H), 7,80 (д, J=8,3 Гц, 1H), 7,73 (тд, J=8,8, 2,9 Гц, 1H), 4,71 (с, 2H), 3,19-2,72 (м, 2H), 2,55 (д, J=8,6 Гц, 2H), 2,24-2,07 (м, 1H), 1,97-1,85 (м, 3H), 1,85-1,58 (м, 4H).
Пример 76: N -(4-фторфенил)-3-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
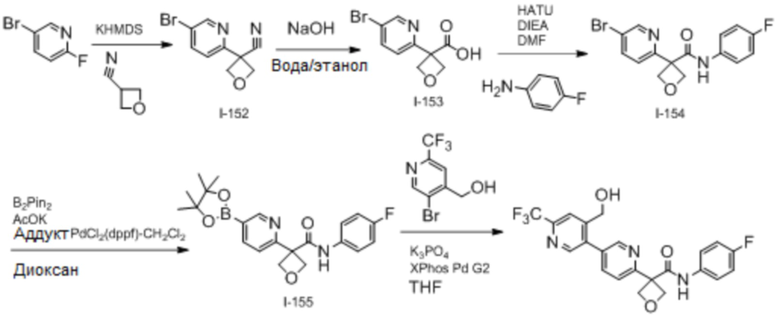
Стадия 1: 3-(5-бромпиридин-2-ил)оксетан-3-карбонитрил (I-152)
В сосуд с якорем магнитной мешалки добавляли 5-бром-2-фторпиридин (58,5 мкл, 0,568 ммоль), оксетан-3-карбонитрил (47,2 мкл, 0,625 ммоль) и толуол (2840 мкл). Реакционную смесь охлаждали до 0°C при перемешивании в атмосфере азота. К перемешиваемой реакционной смеси медленно добавляли 1,0 M KHMDS в THF (682 мкл, 0,682 ммоль). Через 5 мин реакцию гасили MeOH (~5 мл). Неочищенную реакционную смесь фильтровали через целитовую прокладку, которую промывали этилацетатом. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на колонках с силикагелем (0-100% EtOAc/гексаны). Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении для получения соединения I-152. MS (ESI) [M+H]+: m/z 239.
Стадия 2: Получение 3-(5-бромпиридин-2-ил)оксетан-3-карбоновой кислоты (I-153)
В сосуд с якорем магнитной мешалки добавляли соединение I-152 (190 мг, 0,795 ммоль), NaOH (127 мг, 3,18 ммоль), этанол (2660 мкл) и воду (1310 мкл). Сосуд запаивали и нагревали до 80°C в течение 20 ч. Через 20 ч. неочищенную реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в EtOAc и доводили pH до ~2 посредством добавления 1 н. HCl по каплям. Смесь промывали водой. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении для получения соединения I-153. MS (ESI) [M+H]+: m/z 258.
Стадия 3: Получение 3-(5-бромпиридин-2-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (I-154)
В сосуд с якорем магнитной мешалки добавляли соединение I-153 (127 мг, 0,493 ммоль), HATU (281 мг, 0,739 ммоль) и DMF (4930 мкл). Добавляли 4-фторанилин (56,0 мкл, 0,591 ммоль), а затем DIEA (258 мкл, 1,48 ммоль). Реакционную смесь перемешивали при RT в течение 21 ч. Через 21 ч. неочищенную реакционную смесь разводили этилацетатом и промывали насыщенным раствором NaHCO3 и экстрагировали водный слой этилацетатом. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на колонках с силикагелем (0-100% EtOAc/гексаны) для получения соединения I-154. MS (ESI) [M+H]+: m/z 351.
Стадия 4: Получение N-(4-фторфенил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбоксамида (I-155)
В сосуд с якорем магнитной мешалки добавляли соединение I-154 (113 мг, 0,320 ммоль), бис(пинаколато)диборон (203 мг, 0,801 ммоль), ацетат калия (94 мг, 0,961 ммоль) и аддукт PdCl2(dppf)-CH2Cl2 (26,2 мг, 0,032 ммоль) в диоксане (1600 мкл). Сосуд продували азотом в течение 5 мин, а затем запаивали и нагревали до 80°C в течение 23 ч. Неочищенную реакционную смесь фильтровали через целитовую прокладку, которую промывали этилацетатом. Объединенные органические соединения концентрировали при пониженном давлении. Остаток разводили этилацетатом и водой и экстрагировали этилацетатом. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в ACN/воде и сушили в лиофилизаторе в течение ночи для получения соединения I-155. MS (ESI) [M+H]+: m/z вычислено: 399; обнаружено 317 (масса бороновой кислоты).
Стадия 5: N -(4-фторфенил)-3-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
В сосуд с якорем магнитной мешалки добавляли соединение I-155 (17 мг, 0,043 ммоль), (5-бром-2-(трифторметил)пиридин-4-ил)метанол (10,9 мг, 0,043 ммоль), хлор(2-дициклогексилфосфино-2',4',6'-три-I-пропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил) палладий (II) (3,36 мг, 4,27 мкмоль), ортофосфат калия (водный раствор 1 M) (85 мкл, 0,085 ммоль) и THF (427 мкл). Сосуд продували азотом, запаивали и нагревали до 40°C в течение 1 ч. Через 1 ч. неочищенную реакционную смесь фильтровали через целитовую прокладку, которую промывали этилацетатом. Реакционную смесь концентрировали при пониженном давлении, остаток разводили этилацетатом и промывали насыщенным раствором NaCl. Органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на колонках с силикагелем (0-100% EtOAc/гексаны) для получения титульного соединения. MS (ESI) [M+H]+: m/z 448. 1H ЯМР (600 МГц, DMSO-d6) δ 10,15 (с, 1H), 8,80 (с, 1H), 8,72 (с, 1H), 8,12-8,03 (м, 2H), 7,75-7,69 (м, 3H), 7,21 (т, J=8,6 Гц, 2H), 5,76 (т, J=5,2 Гц, 1H), 5,22 (д, J=6,3 Гц, 2H), 5,08 (д, J=6,2 Гц, 2H), 4,62 (д, J=5,2 Гц, 2H).
Пример 77: 3-(6'-(дифторметокси)-4'-(гидроксиметил)-[3,3'-бипиридин]-6-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
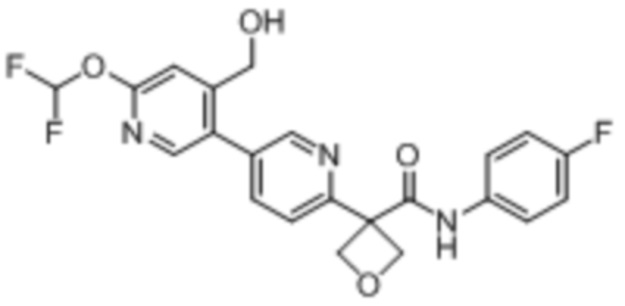
Титульное соединение получали из промежуточного соединения I-155 и (5-бром-2-(дифторметокси)пиридин-4-ил)метанола аналогично синтезу примера 76. MS (ESI)) [M+H]+: m/z 446. 1H ЯМР (600 МГц, DMSO-d6) δ 10,14 (с, 1H), 8,72 (с, 1H), 8,20 (с, 1H), 7,99 (д, J=7,9 Гц, 1H), 7,74-7,69 (м, 2H), 7,67 (д, J=8,4 Гц, 1H), 7,26 (с, 1H), 7,20 (т, J=8,6 Гц, 2H), 5,14 (дд, J=80,5, 6,2 Гц, 4H), 4,52 (с, 2H).
Пример 78: 3-(6'-(дифторметокси)-4'-(2-гидроксипропан-2-ил)-[3,3'-бипиридин]-6-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
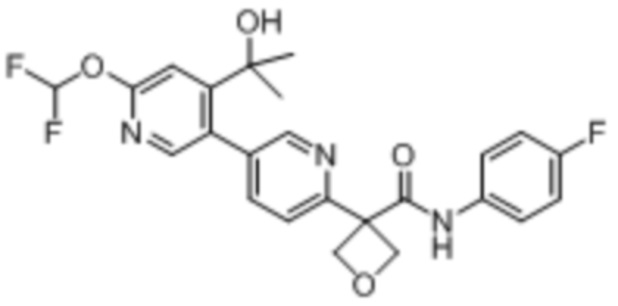
Титульное соединение получали аналогично примеру 76. MS (ESI) [M+H]+: m/z 474. 1H ЯМР (600 МГц, DMSO-d6) δ 10,18 (с, 1H), 8,60 (с, 1H), 7,94 (с, 1H), 7,86 (д, J=8,0 Гц, 1H), 7,78 (т, J=72 Гц, 1H), 7,74-7,70 (м, 2H), 7,60 (д, J=8,1 Гц, 1H), 7,34 (с, 1H), 7,21 (т, J=8,7 Гц, 2H), 5,13 (дд, J=83,1, 6,2 Гц, 4H), 1,32 (с, 6H).
Пример 79: N-(4-фторфенил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
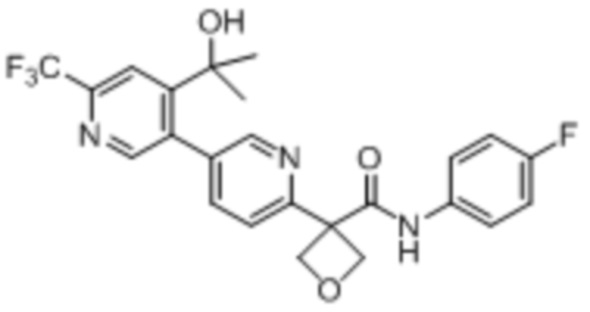
Титульное соединение получали аналогично примеру 76. MS (ESI) [M+H]+: m/z 476.
Пример 80: 3,3-дифтор-N-(4-фторфенил)-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоксамид
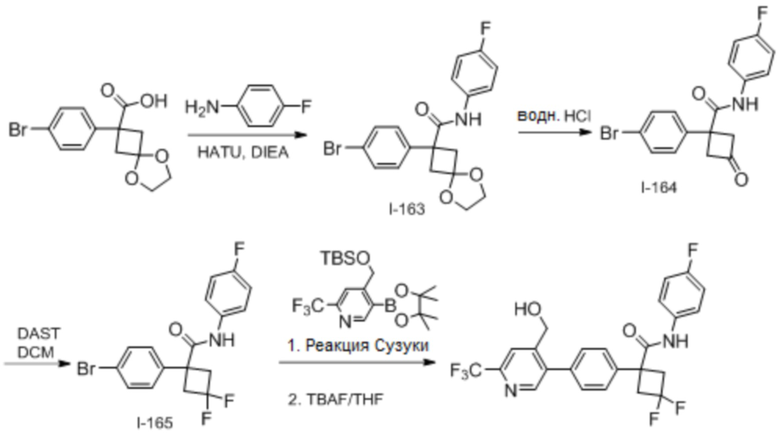
Стадия 1. 2-(4-бромфенил)-N-(4-фторфенил)-5,8-диоксаспиро[3,4]октан-2-карбоксамид (I-163)
К раствору 2-(4-бромфенил)-5,8-диоксаспиро[3,4]октан-2-карбоновой кислоты (1,0 г, 3,2 ммоль) в DMF (6,4 мл) добавляли 4-фторанилин (0,30 мл, 3,2 ммоль) и основание Хунига (1,12 мл, 6,4 ммоль), а затем частями добавляли HATU (1,58 г, 4,2 ммоль). Смесь перемешивали при RT в течение 14 ч. Реакционную смесь разводили водным раствором NaHCO3 и экстрагировали с помощью EtOAc. Органический слой разделяли, промывали водой, соляным раствором, сушили над MgSO4 и концентрировали. Остаток очищали посредством флэш-хроматографии (0-100% EtOAc/гексаны) для получения соединения I-163. MS (ESI) [M+H]+: m/z 406.
Стадия 2: 1-(4-бромфенил)-N-(4-фторфенил)-3-оксоциклобутан-1-карбоксамид (I-164)
В колбу, содержащую соединение I-163 (380 мг, 0,94 ммоль), добавляли HCl (4 Н в диоксане, 2 мл) и H2O (2 мл). Смесь нагревали при 80°C в течение 2 ч. Смесь охлаждали, нейтрализовали насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc. Органический слой промывали соляным раствором, сушили над MgSO4 и концентрировали для получения соединения I-164 в виде твердого вещества. Этот материал использовали непосредственно на следующей стадии. MS (ESI) [M+H]+: m/z 362.
Стадия 3: 1-(4-бромфенил)-3,3-дифтор-N-(4-фторфенил)циклобутан-1-карбоксамид (I-165)
К раствору соединения I-164 (127 мг, 0,35 ммоль) в CH2Cl2 (2,3 мл) при -30°C добавляли DAST (185 мкл, 1,4 ммоль). После добавления реакционную смесь медленно нагревали до RT и продолжали перемешивание в течение 3 ч. В этот момент смесь гасили посредством добавления водного раствора NaHCO3 и экстрагировали с помощью EtOAc. Органический слой отделяли, промывали соляным раствором, сушили над MgSO4 и концентрировали. Остаток очищали посредством флэш-хроматографии (0-100% EtOAc/гексаны) для получения соединения I-165. MS (ESI) [M+H]+: m/z 384.
Стадия 4: 3,3-дифтор-N-(4-фторфенил)-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоксамид
Смесь 4-(((трет-бутилдиметилсилил)окси)метил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридина (98 мг, 0,23 ммоль), соединения I-165 (60 мг, 0,15 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (15 мг, 0,023 ммоль) и карбоната натрия (195 мкл, 0,39 ммоль) в 1,4-диоксане (1,0 мл) выкачивали и помещали обратно с азотом 3 раза и нагревали смесь в атмосфере азота при 90°C в течение 2 ч. Реакционную смесь охлаждали, разводили водой и экстрагировали с помощью EtOAc. Органический слой отделяли, промывали соляным раствором, сушили над MgSO4 и концентрировали. Остаток очищали посредством флэш-хроматографии (0-100% EtOAc/гексаны) для получения 1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-3,3-дифтор-N-(4-фторфенил)циклобутан-1-карбоксамида в виде масла. MS (ESI) [M+H]+: m/z 595.
В колбу, содержащую 1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-3,3-дифтор-N-(4-фторфенил)циклобутанкарбоксамид (83 мг, 0,14 ммоль), при RT добавляли TBAF (1,0 M в THF, 42 мкл, 0,42 ммоль) и THF (0,3 мл). Смесь продолжали перемешивать при RT в течение 1 ч. Смесь разводили насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc. Органический слой промывали соляным раствором, сушили над MgSO4 и концентрировали. Остаток очищали посредством флэш-хроматографии (0-100% EtOAc/гексаны) для получения титульного соединения. MS (ESI) [M+H]+: m/z 480. 1H ЯМР (500 МГц, DMSO-d6): δ 9,92 (с, 1H), 8,61 (с, 1H), 8,02 (с, 1H), 7,75-7,59 (м, 4H), 7,53 (д, J=8,0 Гц, 2H), 7,14 (т, J=8,8 Гц, 2H), 5,66 (т, J=5,3 Гц, 1H), 4,55 (д, J=5,2 Гц, 2H), 3,55 (к, J=13,2 Гц, 2H), 3,21 (к, J=13,2 Гц, 2H).
Пример 81: N-(4-фторфенил)-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоксамид
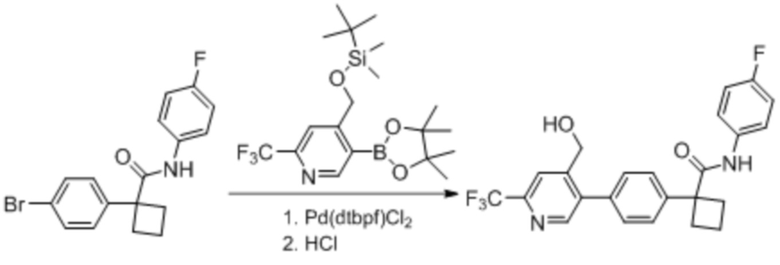
Стадия 1. 1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)циклобутан-1-карбоксамид
Смесь 4-(((трет-бутилдиметилсилил)окси)метил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридина (180 мг, 0,43 ммоль), 1-(4-бромфенил)-N-(4-фторфенил)циклобутанкарбоксамида (100 мг, 0,29 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (28 мг, 0,043 ммоль) и карбоната натрия (287 мкл, 0,57 ммоль) в 1,4-диоксане (1,9 мл) выкачивали и помещали обратно с азотом 3 раза и нагревали смесь в атмосфере азота при 90°C в течение 3 ч. Реакционную смесь охлаждали, разводили водой и экстрагировали с помощью EtOAc. Органический слой отделяли, промывали соляным раствором, сушили над MgSO4 и концентрировали. Остаток очищали посредством флэш-хроматографии (0-100% EtOAc/гексаны) для получения титульного соединения в виде твердого вещества. MS (ESI) [M+H]+: m/z 559.
Стадия 2. N-(4-фторфенил)-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоксамид
В колбу, содержащую 1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)циклобутанкарбоксамид (107 мг, 0,19 ммоль), при RT добавляли TBAF (1,0 М в THF, 575 мкл, 0,575 ммоль) и THF (0,3 мл). Смесь продолжали перемешивать при RT в течение 1 ч. Смесь разводили насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc. Органический слой промывали соляным раствором, сушили над MgSO4 и концентрировали. Остаток очищали посредством флэш-хроматографии (0-100% EtOAc/гексаны) для получения титульного соединения. MS (ESI) [M+H]+: m/z 445,1H ЯМР (500 МГц, DMSO-d6) δ 9,62 (с, 1H), 8,62 (с, 1H), 8,03 (с, 1H), 7,75-7,55 (м, 4H), 7,50 (д, J=7,5 Гц, 2H), 7,14 (т, J=8,2 Гц, 2H), 5,68 (с, 1H), 4,57 (д, J=4,3 Гц, 2H), 2,95-2,75 (м, 2H), 2,40-2,60 (м, 2H), 1,98-1,77 (м, 2H).
Пример 82: N-(4-фторфенил)-5-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)спиро[2,3]гексан-5-карбоксамид
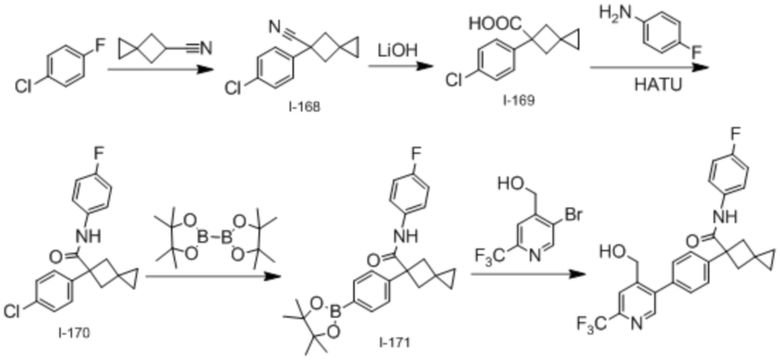
Стадия 1: 5-(4-хлорфенил)спиро[2,3]гексан-5-карбонитрил (I-168)
1-хлор-4-фторбензол (193 мкл, 1,8 ммоль) и спиро[2,3]гексан-5-карбонитрил (179 мкл, 1,5 ммоль) добавляли в колбу в атмосфере азота, а затем добавляли 1,5 мл THF. К этому по каплям добавляли KHMDS (1 M в THF, 1,58 мл, 1,58 ммоль). Смесь перемешивали в течение 15 ч. при RT, выпаривали под вакуумом, а затем проверяли превращение посредством ЯМР. Остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием гексанов и этилацетат в качестве элюентов) для получения соединения I-168, 1H ЯМР (600 МГц, CDCl3) δ 7,46 (д, J=8,2 Гц, 2H), 7,37 (д, J=8,3 Гц, 2H), 2,96 (д, J=12,3 Гц, 2H), 2,68 (д, J=12,4 Гц, 2H), 0,77-0,69 (м, 2H), 0,60-0,53 (м, 2H).
Стадия 2: 5-(4-хлорфенил)спиро[2,3]гексан-5-карбоновая кислота (I-169)
Гидроксид лития (99 мг, 4,1 ммоль) добавляли в колбу, содержащую соединение I-168 (150 мг, 0,69 ммоль). К этому добавляли 1 мл воды и 1 мл этанола. Затем смесь нагревали в запаянной колбе в атмосфере аргона при 65°C в течение 72 ч. По завершении продукт подкисляли 1 M HCl (водным раствором) до достижения pH~3. Затем продукт экстрагировали этилацетатом, сушили над MgSO4, фильтровали через целитовую прокладку, а затем выпаривали под вакуумом для получения соединения I-169, которое использовали непосредственно на следующей стадии. MS (ESI) [M+H]+: m/z 237.
Стадия 3: 5-(4-хлорфенил)-N-(4-фторфенил)спиро[2,3]гексан-5-карбоксамид (I-170)
5-(4-хлорфенил)спиро[2,3]гексан-5-карбоновую кислоту (125 мг, 0,53 ммоль) и HATU (221 мг, 0,58 ммоль) добавляли в сосуд с 1,5 мл DMF. К этому добавляли 4-фторанилин (55 мкл, 0,58 ммоль), а затем DIPEA (231 мкл, 1,3 ммоль). Эту смесь перемешивали в течение 2 ч., после чего ее выпаривали под вакуумом. Неочищенный остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием гексанов и этилацетата в качестве элюентов) для получения соединения I-170. MS (ESI) [M+H]+: m/z 330.
Стадия 4: N-(4-фторфенил)-5-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)спиро[2,3]-гексан-5-карбоксамид (I-171)
В сухую круглодонную колбу помещали соединение I-170 (118 мг, 0,36 ммоль), бис(пинаколато)диборон (236 мг, 0,93 ммоль), ацетат калия (105 мг, 1,1 ммоль) и XPhos G3 (5%, 15 мг). Затем добавляли диоксан (2,4 мл), продували аргоном и нагревали до 80°C в течение 15 ч. Реакционную смесь охлаждали до RT и фильтровали через целитовую прокладку. Фильтрат концентрировали под вакуумом для получения остатка, который очищали посредством хроматографии (система Isco CombiFlash, с использованием гексанов и этилацетата в качестве элюентов) для получения соединения I-171. MS (ESI) [M+H]+: m/z 422.
Стадия 5: N-(4-фторфенил)-5-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)спиро[2,3]гексан-5-карбоксамид
В сухой сосуд с якорем магнитной мешалки помещали Xphos G3 (8,46 мг, 10,00 мкмоль), (5-бром-2-(трифторметил)пиридин-4-ил)метанол (28,2 мг, 0,11 ммоль) и соединение I-171 (42,1 мг, 0,1 ммоль) и держали в атмосфере азота. К этому добавляли THF 0,50 мл и ортофосфат калия (1 M в H2O, 0,200 мл, 0,200 ммоль). Затем реакционную смесь продували аргоном и нагревали при 70°C в течение ночи. Ей позволяли охлаждаться до RT, а затем фильтровали через целитовую прокладку. Этот неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 471, 1H ЯМР (600 МГц, DMSO-d6) δ 9,71 (с, 1H), 8,61 (с, 1H), 8,02 (с, 1H), 7,71-7,66 (м, 2H), 7,63 (д, J=7,6 Гц, 2H), 7,49 (д, J=7,5 Гц, 2H), 7,13 (т, J=8,3 Гц, 2H), 4,57 (с, 2H), 3,04 (д, J=11,9 Гц, 2H), 2,63 (д, J=11,9 Гц, 2H), 0,49 (с, 4H).
Пример 83: 1-(4-(4,7-дифтор-1-оксоизоиндолин-2-ил)фенил)-N-(4-фторфенил)циклобутан-1-карбоксамид
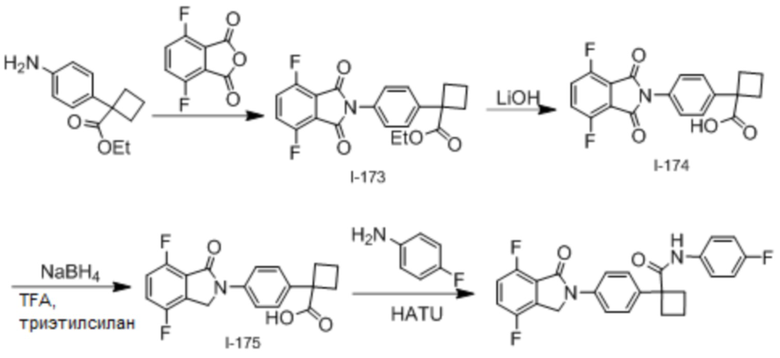
Стадия 1: Этил-1-(4-(4,7-дифтор-1,3-диоксоизоиндолин-2-ил)фенил)циклобутан-1-карбоксилат (I-173)
В сосуд помещали 4,7-дифторизобензофуран-1,3-дион (184 мг, 1 ммоль) и этил-1-(4-аминофенил)циклобутанкарбоксилат (329 мг, 1,500 ммоль). В этот сосуд добавляли 4 мл ледяной уксусной кислоты, а затем сосуд продували аргоном и нагревали при 80°C в течение 15 ч. Затем реакционную смесь концентрировали под вакуумом. Этот неочищенный материал растворяли в DCM (10 мл) и переносили на делительную воронку. Его промывали насыщенным раствором бикарбоната натрия и отделяли органические соединения, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием гексанов и этилацетата в качестве элюентов) для получения соединения I-173. MS (ESI) [M+H]+: m/z 386.
Стадия 2: 1-(4-(4,7-дифтор-1,3-диоксоизоиндолин-2-ил)фенил)циклобутан-1-карбоновая кислота (I-174)
В сосуд помещали соединение I-173 (200 мг, 0,519 ммоль) и к нему добавляли 2 мл диоксана. Затем добавляли гидроксид лития (37,3 мг, 1,557 ммоль), а затем добавляли 2 мл воды. Эту смесь перемешивали при RT в течение 2 ч. Затем органические соединения выпаривали под вакуумом и подкисляли водный слой 1 M HCl до pH ~2. Затем их экстрагировали 3 раза с помощью DCM (5 мл), сушили над сульфатом магния, фильтровали и выпаривали под вакуумом. В этих условиях открывалось лактамное кольцо, поэтому этот неочищенный материал затем растворяли в 2 мл уксусной кислоты в сосуде, сосуд продували аргоном и нагревали до 100°C в течение 15 ч. Затем реакционную смесь концентрировали под вакуумом. Этот неочищенный материал растворяли в DCM (10 мл) и переносили на делительную воронку. Его промывали насыщенным раствором карбоната натрия и отделяли органические соединения, сушили над сульфатом натрия, фильтровали и концентрировали для получения соединения I-174, которое сразу использовали. MS (ESI) [M+H]+: m/z 358.
Стадия 3: 1-(4-(4,7-дифтор-1-оксоизоиндолин-2-ил)фенил)циклобутан-1-карбоновая кислота (I-175)
Соединение I-174 (90 мг, 0,252 ммоль) и NaBH4 (10,48 мг, 0,277 ммоль) добавляли в сосуд в атмосфере азота и держали в атмосфере азота. К этому добавляли 1 мл THF и 1 мл MeOH. Эту смесь перемешивали в течение 5 мин при 0°C, а затем перемешивали при RT в течение 2 ч. По завершении добавляли 2 капли уксусной кислоты и выпаривали под вакуумом. Затем добавляли 5 мл DCM и 5 мл насыщенного раствора бикарбоната натрия, и отделяли органические соединения. Их экстрагировали еще два раза с помощью 5 мл DCM и органические соединения объединяли, сушили над сульфатом магния, фильтровали и выпаривали под вакуумом. Затем этот неочищенный материал растворяли в TFA (1 мл) и добавляли триэтилсилан (0,161 мл, 1,008 ммоль). Эту смесь перемешивали при RT в течение 30 мин и выпаривали под вакуумом. Неочищенный остаток очищали посредством хроматографии (система Isco CombiFlash, с использованием гексанов и этилацетата в качестве элюентов) для получения соединения I-175. MS (ESI) [M+H]+: m/z 344.
Стадия 4: 1-(4-(4,7-дифтор-1-оксоизоиндолин-2-ил)фенил)- N -(4-фторфенил)циклобутан-1-карбоксамид
Соединение I-175 (17 мг, 0,050 ммоль) и HATU (19,77 мг, 0,052 ммоль) добавляли в сосуд с 1,5 мл DMF. К этому добавляли 4-фторанилин (5,17 мкл, 0,054 ммоль), а затем DIPEA (12,97 мкл, 0,074 ммоль). Смесь перемешивали в течение 24 ч. По завершении смесь выпаривали под вакуумом. Неочищенный материал очищали посредством масс-направленной обращенно-фазовой хроматографии (градиент ACN/вода с модификатором 0,1% TFA) для получения титульного соединения. MS (ESI) [M+H]+: m/z 437, 1H ЯМР (600 МГц, DMSO-d6) δ 9,49 (с, 1H), 7,85 (д, J=8,1 Гц, 2H), 7,65-7,59 (м, 2H), 7,59-7,55 (м, 1H), 7,53 (д, J=8,1 Гц, 2H), 7,45-7,39 (м, 1H), 7,10 (т, J=8,3 Гц, 2H), 5,10 (с, 2H), 2,88-2,80 (м, 2H), 2,50-2,44 (м, 2H), 1,89-1,77 (м, 2H).
Пример 84: N-(4-фторфенил)-3-(4'-(гидроксиметил)-6'-(трифторметил)-[2,3'-бипиридин]-5-ил)оксетан-3-карбоксамид
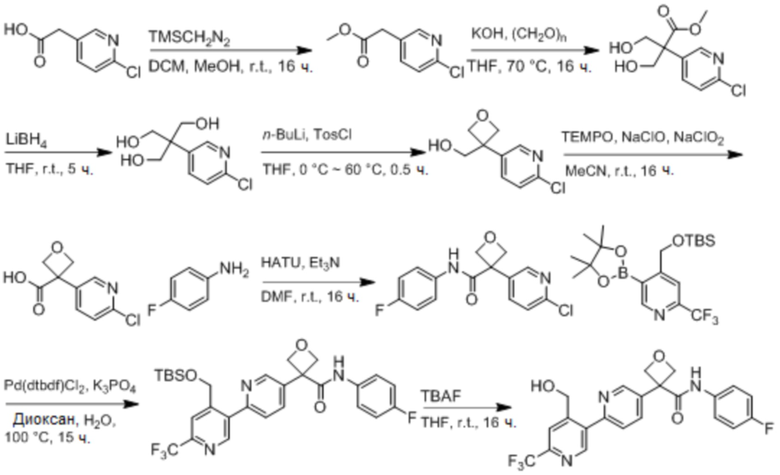
Стадия 1: Метил-2-(6-хлорпиридин-3-ил)ацетат
К перемешиваемому раствору 2-(6-хлорпиридин-3-ил)уксусной кислоты (10 г, 58,3 ммоль) в DCM (100 мл) и MeOH (50 мл) добавляли ((триметилсилил)метил)диазен ( 8,3 мл, 175 ммоль) при 0°C. Реакционную смесь перемешивали при RT в течение 16 ч., а затем растворитель концентрировали при пониженном давлении. Остаток разводили водой (100 мл), экстрагировали с помощью EtOAc (100 мл, 3 раза) и органические слои собирали, промывали соляным раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 185,8 [M+H+].
Стадия 2: 4-метил-2-(6-хлорпиридин-3-ил)-3-гидрокси-2-(гидроксиметил)пропаноат
К перемешиваемому раствору метил-2-(6-хлорпиридин-3-ил)ацетата (7,5 г, 40,4 ммоль) в THF (50 мл) добавляли гидроксид калия (0,227 г, 4,04 ммоль), параформальдегид (4,85 г, 162 ммоль) при RT и перемешивали реакционную смесь при 60°C в течение 16 ч. Смесь фильтровали и концентрировали фильтрат при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 245,9 [M+H+].
Стадия 3: 2-(6-хлорпиридин-3-ил)-2-(гидроксиметил)пропан-1,3-диол
К перемешиваемому раствору метил-2-(6-хлорпиридин-3-ил)-3-гидрокси-2-(гидроксиметил)пропаноата (4,6 г, 18,73 ммоль) в THF (50 мл) добавляли тетрагидроборат лития (1,224 г, 56,2 ммоль) при 0°C и перемешивали реакционную смесь при 0°C в течение 5 ч. Смесь гасили MeOH (50 мл) и концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 217,8 [M+H+].
Стадия 4: (3-(6-хлорпиридин-3-ил)оксетан-3-ил)метанол
К перемешиваемому раствору 2-(6-хлорпиридин-3-ил)-2-(гидроксиметил)пропан-1,3-диола (1,7 г, 7,81 ммоль) в THF (70 мл) по каплям добавляли n-BuLi (4 мл, 10,0 ммоль) (2,5 M) при 0°C и перемешивали смесь при 0°C в течение 30 мин. Затем добавляли раствор TsCl (1,340 г, 7,03 ммоль) в 5 мл THF при 0°C и продолжали перемешивание при 0°C в течение 1 ч. К указанной выше смеси добавляли n-BuLi (3,12 мл, 7,80 ммоль) (2,5 M) при 0°C и перемешивали смесь при 0°C в течение еще 0,5 ч, а затем нагревали до 60°C в течение 0,5 ч. После охлаждения до RT реакцию гасили водным раствором NH4Cl (30 мл), экстрагировали с помощью EtOAc (30 мл, 3 раза) и органические слои собирали, промывали соляным раствором (20 мл), сушили над Na2SO4, фильтровали, фильтрат концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения. MS (ESI) m/z 199,9 [M+H+].
Стадия 5: 3-(6-хлорпиридин-3-ил)оксетан-3-карбоновая кислота
К перемешиваемому раствору (3-(6-хлорпиридин-3-ил)оксетан-3-ил)метанола (270 мг, 1,352 ммоль) в ACN (10 мл) последовательно добавляли TEMPO (42 мг, 0,269 ммоль), хлорит натрия (489 мг, 5,41 ммоль) в 1 мл воды и гипохлорит натрия (1007 мг, 1,352 ммоль) (10% в воде) при RT. Смесь перемешивали при RT в течение 16 ч. Реакционную смесь обрабатывали 2 M NaOH до достижения pH 10, затем добавляли 10% тиосульфата натрия (30 мл). Смесь разделяли с помощью этилацетата (30 мл) и воды (15 мл), подкисляли водную фазу с помощью HCl (2 М в воде) до pH 4 и экстрагировали этилацетатом (30 мл, 3 раза). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 214,0 [M+H+].
Стадия 6: 3-(6-хлорпиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(6-хлорпиридин-3-ил)оксетан-3-карбоновой кислоты (250 мг, 1,170 ммоль) в DMF (10 мл) добавляли HATU (667 мг, 1,755 ммоль), Et3N (0,5 мл, 3,59 ммоль) и 4-фторанилин (195 мг, 1,755 ммоль) при RT и продолжали перемешивание в течение 16 ч. Реакционную смесь обрабатывали и очищали неочищенный продукт посредством препаративной TLC (петролейный эфир/EtOAc=1:1) для получения титульного соединения в виде масла. MS (ESI) m/z 307,1 [M+H+].
Стадия 7: 3-(4'-(((трет-бутилдиметилсилил)окси)метил)-6'-(трифторметил)-[2,3'-бипиридин]-5-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(6-хлорпиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (30 мг, 0,098 ммоль) и 4-(((трет-бутилдиметилсилил)окси)метил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридина (49 мг, 0,117 ммоль) в диоксане (2 мл) и воде (0,4 мл) добавляли K3PO4 (62 мг, 0,292 ммоль) и Pd(dtbpf)Cl2 (7 мг, 10,74 мкмоль) при RT. Смесь нагревали до 100°C при перемешивании в течение 15 ч. и охлаждали до RT. Реакционную смесь разводили этилацетатом (5 мл) и фильтровали. Фильтрат промывали водой (2 мл) и соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc=1:1 в качестве элюентов) для получения титульного соединения в виде масла. MS (ESI) m/z 562,3[M+H+].
Стадия 8: N-(4-фторфенил)-3-(4'-(гидроксиметил)-6'-(трифторметил)-[2,3'-бипиридин]-5-ил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(4'-(((трет-бутилдиметилсилил)окси)метил)-6'-(трифторметил)-[2,3'-бипиридин]-5-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (21 мг, 0,037 ммоль) в THF (2 мл) добавляли TBAF (0,05 мл, 0,050 ммоль) при RT и перемешивали смесь при RT в течение 16 ч. Смесь концентрировали при пониженном давлении и очищали остаток посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi (C18 150*30 мм*4 мкм) с использованием воды (0,225% FA) и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,85 (д, 1H), 8,79 (с, 1H), 8,12 (т, 2H), 7,82 (д, 1H), 7,60-7,56 (м, 2H), 7,07 (т, 2H), 5,38 (д, 2H), 5,06 (д, 2H), 4,82 (с, 2H); MS (ESI) m/z 448,0 [M+H+].
Пример 85: N-(4-фторфенил)-3-(6-(2-(гидроксиметил)-4-(трифторметил)фенил)пиридин-3-ил)оксетан-3-карбоксамид
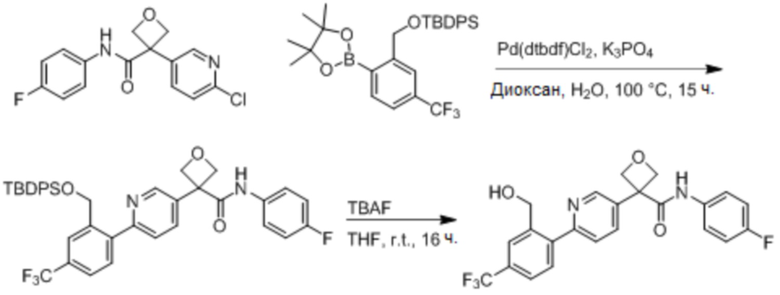
Стадия 1: 3-(6-(2-(((трет-бутилдифенилсилил)окси)метил)-4-(трифторметил)фенил)пиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(6-хлорпиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (42 мг, 0,137 ммоль) и трет-бутилдифенил((2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5-(трифторметил)бензил)окси)силана (89 мг, 0,164 ммоль) в диоксане (1,0 мл) и воде (0,2 мл) добавляли K3PO4 (87 мг, 0,411 ммоль) и Pd(dtbpf)Cl2 (9 мг, 0,014 ммоль) при RT. Смесь нагревали до 100°C при перемешивании в течение 15 ч. Реакционную смесь охлаждали до RT, разводили EtOAc (5 мл) и фильтровали. Фильтрат промывали водой (2 мл) и соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc=1:1 в качестве элюентов) для получения титульного соединения в виде масла. MS (ESI) m/z 685,4[M+H+].
Стадия 2: N-(4-фторфенил)-3-(6-(2-(гидроксиметил)-4-(трифторметил)фенил)пиридин-3-ил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(6-(2-(((трет-бутилдифенилсилил)окси)метил)-4-(трифторметил)фенил)пиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (40 мг, 0,058 ммоль) в THF (2 мл) добавляли TBAF (0,1 мл, 0,100 ммоль) при RT. Смесь перемешивали при RT в течение 16 ч. Смесь концентрировали при пониженном давлении и очищали остаток посредством обращенно-фазовой ВЭЖХ для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,80 (д, 1H), 8,12 (дд, 8,3 Гц, 1H), 7,92 (с, 1H), 7,78 (д, 1H), 7,71 (с, 2H), 7,59 (дд, 9,2 Гц, 2H), 7,10-7,04 (м, 2H), 5,38 (д, 2H), 5,06 (д, 2H), 4,66 (с, 2H); MS (ESI) m/z 447,0 [M+H+].
Пример 86: N-(4-фторфенил)-3-(6-(2-(2-гидроксипропан-2-ил)-4-(трифторметил)фенил)пиридин-3-ил)оксетан-3-карбоксамид
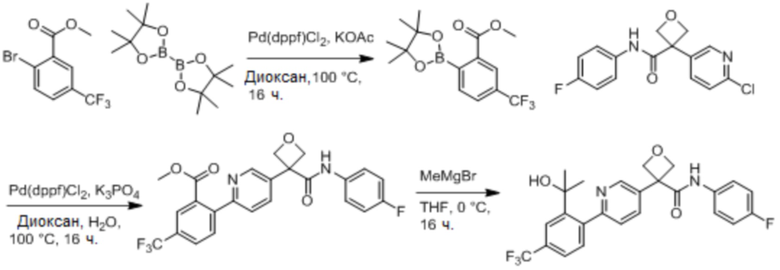
Стадия 1: Метил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5-(трифторметил)бензоат
К перемешиваемому раствору метил-2-бром-5-(трифторметил)бензоата (510 мг, 1,802 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (686 мг, 2,70 ммоль) в диоксане (2 мл) добавляли ацетат калия (531 мг, 5,41 ммоль) и Pd(dppf)Cl2 (132 мг, 0,180 ммоль) при RT. Смесь нагревали до 100°C при перемешивании в течение 16 ч. и концентрировали растворитель при пониженном давлении. К остатку добавляли насыщенный раствор NaHCO3 для доведения pH до >8 и смесь экстрагировали с помощью EtOAc (20 мл, 3 раза). Органические слои собирали, промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и фильтрат концентрировали под вакуумом. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc=5:1) для получения титульного соединения в виде масла. MS (ESI) m/z 331,1 [M+H+].
Стадия 2: Метил-2-(5-(3-((4-фторфенил)карбамоил)оксетан-3-ил)пиридин-2-ил)-5-(трифторметил)бензоат
К перемешиваемому раствору 3-(6-хлорпиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (30 мг, 0,098 ммоль) и (2-(2-(метоксикарбонил)-4-(трифторметил)фенил)-4,5,5-триметил-1,3,2-диоксаборолан-4-ил)метилия (62 мг, 0,188 ммоль) в диоксане (2 мл) и воде (0,4 мл) добавляли K3PO4 (62 мг, 0,292 ммоль) и Pd(dtbpf)Cl2 (7 мг, 10,74 мкмоль) при RT. Смесь нагревали до 100°C в течение 15 ч. Реакционную смесь разводили EtOAc (5 мл) и фильтровали. Фильтрат промывали водой (2 мл) и соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали посредством препаративной TLC (SiO2, петролейный эфир/EtOAc=1:1 в качестве элюентов) для получения титульного соединения в виде масла. MS (ESI) m/z 475,2 [M+H+].
Стадия 3: N-(4-фторфенил)-3-(6-(2-(2-гидроксипропан-2-ил)-4-(трифторметил)фенил)-пиридин-3-ил)оксетан-3-карбоксамид
К перемешиваемому раствору метил-2-(5-(3-((4-фторфенил)карбамоил)оксетан-3-ил)пиридин-2-ил)-5-(трифторметил)бензоата (30 мг, 0,063 ммоль) в THF (2 мл) добавляли 3 M MeMgBr (0,1 мл, 0,300 ммоль) при 0°C. Реакционную смесь перемешивали при RT в атмосфере азота в течение 15 ч. Реакционную смесь медленно гасили насыщенным раствором NH4Cl (10 мл, водный раствор) и экстрагировали с помощью EtOAc (20 мл, 3 раза). Объединенный органический слой промывали соляным раствором (5 мл), сушили над безводным Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281 с колонкой Phenomenex Synergi C18 150×30 мм×4 мкм для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,74 (д, 1H), 8,16 (дд, 8,2 Гц, 1H), 7,96 (с, 1H), 7,70 (д, 1H), 7,66 (ш.д., 1H), 7,57 (тдд, 6,9 Гц, 2H), 7,47 (д, 1H), 7,07 (т, 2H), 5,38 (д, 2H), 5,05 (д, 2H), 1,43 (с, 6H); MS (ESI) m/z 475,2 [M+H+].
Пример 87: N-(4-фторфенил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[2,3'-бипиридин]-5-ил)оксетан-3-карбоксамид
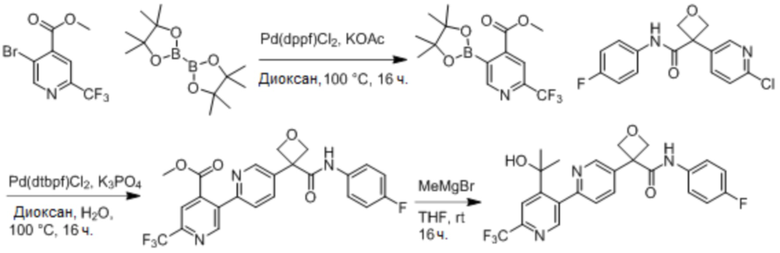
Стадия 1: Метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)изоникотинат
К перемешиваемому раствору метил-5-бром-2-(трифторметил)изоникотината (520 мг, 1,831 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (697 мг, 2,75 ммоль) в диоксане ( 2 мл) добавляли ацетат калия (539 мг, 5,49 ммоль) и Pd(dppf)Cl2 (134 мг, 0,183 ммоль) при RT. Смесь нагревали до 100°C при перемешивании в течение 16 ч. Смесь концентрировали при пониженном давлении и добавляли насыщенный раствор NaHCO3 для доведения pH до >8. Затем смесь экстрагировали с помощью EtOAc (20 мл, 3 раза) и органические слои собирали, промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc =5:1) для получения титульного соединения в виде масла. 1H ЯМР (500 МГц, CD3OD) δ 8,86 (с, 1H), 8,44 (с, 1H), 3,86 (с, 3H), 1,37-1,32 (м, 8H), 1,30 (с, 1H).
Стадия 2: Метил-5-(3-((4-фторфенил)карбамоил)оксетан-3-ил)-6'-(трифторметил)-[2,3'-бипиридин]-4'-карбоксилат
К перемешиваемому раствору 3-(6-хлорпиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (30 мг, 0,098 ммоль) и метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)изоникотината (48 мг, 0,145 ммоль) в диоксане (2 мл) и воде (0,4 мл) добавляли K3PO4 (62 мг, 0,292 ммоль) и Pd(dtbpf)Cl2 (15 мг, 0,023 ммоль) при RT. Реакционную смесь подвергали реакции в тех же условиях, что и на стадии 1, и неочищенный продукт очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 150×30 мм×4 мкм, для получения титульного соединения в виде масла. MS (ESI) m/z 475,9 [M+H+].
Стадия 3: N-(4-фторфенил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[2,3'-бипиридин]-5-ил)оксетан-3-карбоксамид
К перемешиваемому раствору метил-5-(3-((4-фторфенил)карбамоил)оксетан-3-ил)-6'-(трифторметил)-[2,3'-бипиридин]-4'-карбоксилата (10 мг, 0,021 ммоль) в THF (2 мл) по каплям добавляли бромид метилмагния (0,1 мл, 0,300 ммоль) (3 M в простом эфире) при RT. Реакционную смесь перемешивали при RT в течение 16 ч. Реакционную смесь разводили водой (5 мл), а затем фильтровали при пониженном давлении для получения остатка, который очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 150×30 мм×4 мкм, для получения титульного соединения в виде твердого вещества. 1H ЯМР (500 МГц, CD3OD) δ 8,83 (д, 1H), 8,57 (с, 1H), 8,19-8,16 (м, 1H), 8,14 (с, 1H), 7,73 (д, 1H), 7,63-7,60 (м, 2H), 7,60-7,59 (м, 1H), 7,10 (т, 2H), 5,42 (д, 2H), 5,09 (д, 2H), 2,71 (ш.с., 1H), 2,55 (ш.с., 1H), 2,07-2,03 (м, 1H), 1,47 (с, 6H). MS (ESI) m/z 476,2[M+H+]
Пример 88: 3-(6'-циклопропокси-4'-(гидроксиметил)-[2,3'-бипиридин]-5-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
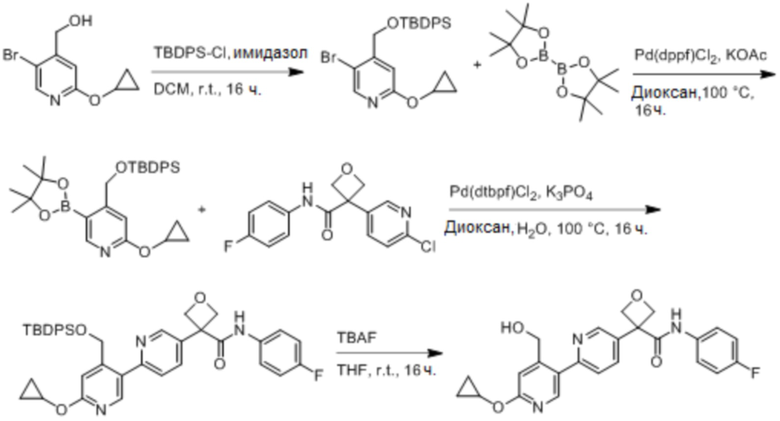
Стадия 1: 5-бром-4-(((трет-бутилдифенилсилил)окси)метил)-2-циклопропоксипиридин
К перемешиваемому раствору (5-бром-2-циклопропоксипиридин-4-ил)метанола (500 мг, 2,048 ммоль) in DCM (10 мл) добавляли трет-бутилхлордифенилсилан (619 мг, 2,253 ммоль) и 1H-имидазол (307 мг, 4,51 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 16 ч. Смесь разводили водой (50 мл), экстрагировали с помощью EtOAc (50 мл, 3 раза) и органические слои собирали, промывали соляным раствором (20 мл) и сушили над Na2SO4. После фильтрации фильтрат концентрировали под вакуумом и остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения. 1H ЯМР (400 МГц, CD3OD) δ 8,12 (с, 1H), 7,70-7,63 (м, 4H), 7,48-7,36 (м, 6H), 7,29 (с, 1H), 4,71 (с, 2H), 4,09 (тт, 1H), 1,11 (с, 9H), 0,82-0,72 (м, 4H)
Стадия 2: 4-(((трет-бутилдифенилсилил)окси)метил)-2-циклопропокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
К перемешиваемому раствору 5-бром-4-(((трет-бутилдифенилсилил)окси)метил)-2-циклопропоксипиридина (940 мг, 1,948 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (742 мг, 2,92 ммоль) в диоксане ( 20 мл) добавляли ацетат калия (574 мг, 5,84 ммоль) и Pd(dppf)Cl2 (143 мг, 0,195 ммоль) при RT. Реакционную смесь перемешивали при 100°C в течение 16 ч. После охлаждения до RT смесь экстрагировали с помощью EtOAc (50 мл, 3 раза), органические слои собирали, промывали соляным раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 530,2 [M+H+].
Стадия 3: 3-(4'-(((трет-бутилдифенилсилил)окси)метил)-6'-циклопропокси-[2,3'-бипиридин]-5-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(6-хлорпиридин-3-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (30 мг, 0,098 ммоль) и 4-(((трет-бутилдифенилсилил)окси)метил)-2-циклопропокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (62 мг, 0,117 ммоль в диоксане (2 мл) и воде (0,4 мл) добавляли K3PO4 (62 мг, 0,292 ммоль) и Pd(dtbpf)Cl2(7 мг, 10,74 мкмоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях как указано выше. Неочищенный продукт очищали посредством препаративной TLC (SiO2, петролейный эфир/EtOAc=1:1 в качестве элюентов) для получения титульного соединения в виде масла. MS (ESI) m/z 674,2[M+H+].
Стадия 4: 3-(6'-циклопропокси-4'-(гидроксиметил)-[2,3'-бипиридин]-5-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(4'-(((трет-бутилдифенилсилил)окси)метил)-6'-циклопропокси-[2,3'-бипиридин]-5-ил)-N-(4-фторфенил)оксетан-3-карбоксамида (25 мг, 0,037 ммоль) в THF (2 мл) добавляли фторид тетрабутиламмония (0,1 мл, 0,100 ммоль) при RT. Смесь перемешивали при RT в течение 16 ч. Растворитель концентрировали при пониженном давлении и остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (150×30 мм×4 мкм) для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,77 (д, 1H), 8,27 (с, 1H), 8,08 (дд, 1H), 7,73 (д, 1H), 7,58 (дд, 2H), 7,22 (с, 1H), 7,07 (т, 2H), 5,37 (д, 2H), 5,04 (д, 2H), 4,68 (с, 2H), 4,23-4,17 (м, 1H), 0,88-0,83 (м, 2H), 0,77 (ш.с., 2H). MS (ESI) m/z 436,1 [M+H+].
Пример 89: N-(5-фтортиазол-2-ил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
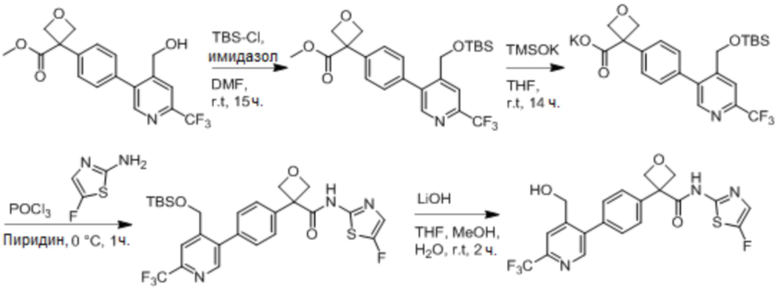
Стадия 1: Метил-3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилат
К раствору метил-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилата (220 мг, 0,599 ммоль) и имидазола (82 мг, 1,198 ммоль) в DMF (2,0 мл) добавляли TBS-Cl (108 мг, 0,719 ммоль) при перемешивании при RT. Реакционную смесь перемешивали при RT в течение 14 ч. Реакционную смесь разводили водой (15 мл) и экстрагировали с помощью EtOAc (15 мл, 3 раза). Объединенный органический слой промывали соляным раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 482,3 [M+H+].
Стадия 2: 3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилат калия
К раствору метил-3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилата (168 мг, 0,349 ммоль) в THF (5,0 мл) добавляли TMSOK (50 мг, 0,390 ммоль) при перемешивании при RT в атмосфере азота. Реакционную смесь перемешивали при RT в течение 14 ч. Растворитель концентрировали для получения титульного соединения в виде твердого вещества, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 468,3 [M+H+].
Стадия 3: 3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(5-фтортиазол-2-ил)оксетан-3-карбоксамид
К раствору 3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксилата калия (15 мг, 0,030 ммоль) и 5-фтортиазол-2-амина (8 мг, 0,068 ммоль) в пиридине (0,5 мл) добавляли POCl3 (0,03 мл, 0,326 ммоль) с перемешиванием при 0°C и перемешивали полученную смесь при 0°C в течение 1 ч. Реакцию гасили посредством добавления насыщенного раствора Na2CO3 (1,0 мл), а затем разводили водой (5 мл), и экстрагировали с помощью EtOAc (5 мл, 3 раза). Объединенный органический слой промывали соляным раствором (5 мл), сушили над Na2SO4, фильтровали и концентрировали для получения неочищенного продукта, который использовали на следующей стадии без последующей очистки. MS (ESI) m/z 568,1 [M+H+].
Стадия4: N-(5-фтортиазол-2-ил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
К раствору 3-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(5-фтортиазол-2-ил)оксетан-3-карбоксамида (16 мг, 0,028 ммоль) в THF (2,0 мл), воды (1,0 мл) и MeOH (2,0 мл) добавляли LiOH (5 мг, 0,209 ммоль) при перемешивании при RT. Смесь перемешивали при RT в течение 1 ч. Реакцию гасили посредством добавления 1 н. HCl до достижения pH~7 и концентрировали. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Xtimate C18 (150×25 мм×5мкм) с использованием воды и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,53 (с, 1H), 8,07 (с, 1H), 7,56-7,65 (м, 2H), 7,50 (д, 2H), 7,09 (д, 1H), 5,34 (д, 2H), 5,06 (д, 2H), 4,64 (с, 2H). MS (ESI) m/z 453,9 [M+H+].
Примеры 90-93 в следующей таблице получали аналогично примеру 89.
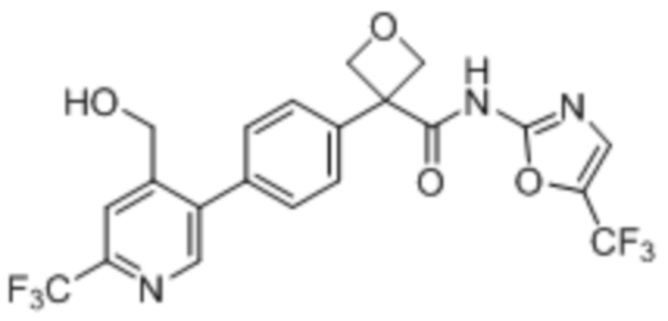
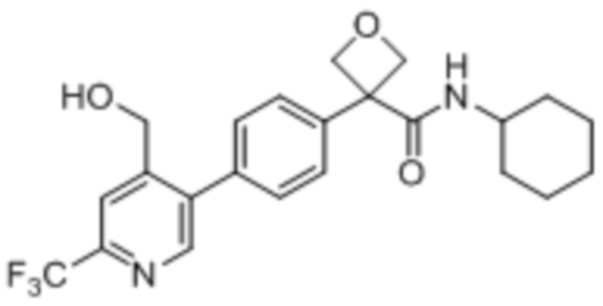
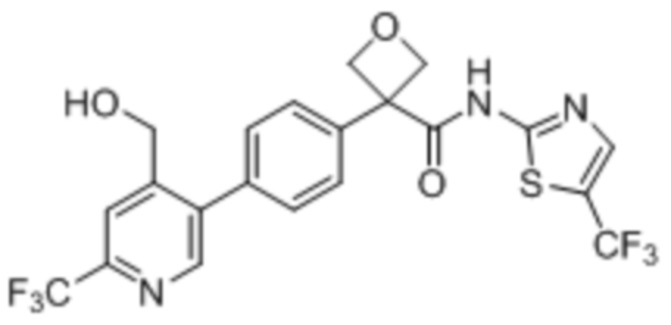
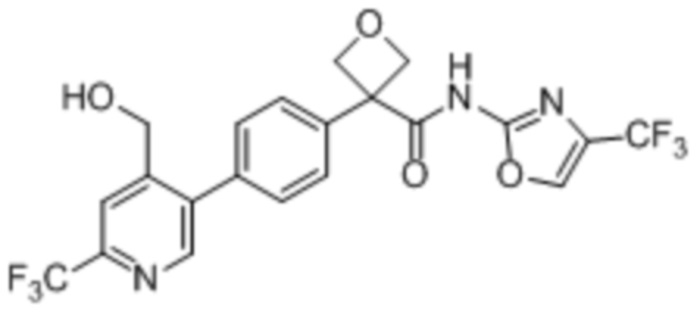
Пример 94: N-(4-фторфенил)-1-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)циклобутанкарбоксамид
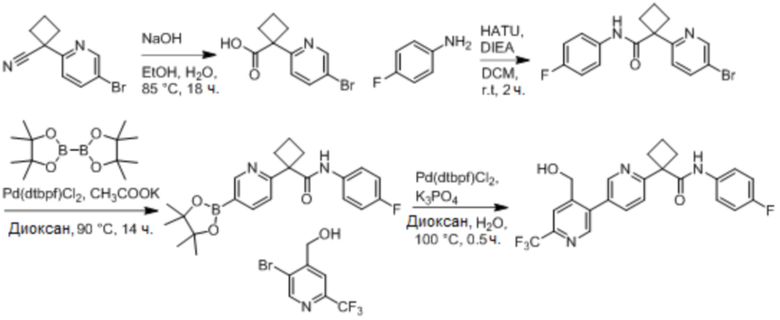
Стадия 1: 1-(5-бромпиридин-2-ил)циклобутанкарбоновая кислота
К раствору 1-(5-бромпиридин-2-ил)циклобутанкарбонитрила (2,0 г, 8,44 ммоль) в воде (2 мл) и EtOH (15 мл) добавляли NaOH (1,687 г, 42,2 ммоль) при перемешивании при RT в атмосфере азота и перемешивали реакционную смесь при 85°C в течение 18 ч. Реакционную смесь охлаждали до RT и концентрировали. Остаток разводили DCM (20 мл) и фильтровали. Отфильтрованный осадок суспендировали в EtOAc (20 мл) и воде (20 мл) при перемешивании, затем добавляли 3 Н HCl до достижения pH~3 и растворяли все твердые вещества. Смесь экстрагировали с помощью EtOAc (20 мл, 2 раза), органические слои объединяли, промывали соляным раствором (60 мл), сушили над Na2SO4, фильтровали и концентрировали для получения титульного соединения в виде масла, которое использовали непосредственно на следующей стадии без последующей очистки. MS (ESI) m/z 257,9 [M+H+].
Стадия 2: 1-(5-бромпиридин-2-ил)-N-(4-фторфенил)циклобутанкарбоксамид
К перемешиваемому раствору 1-(5-бромпиридин-2-ил)циклобутанкарбоновой кислоты (2,16 г, 8,43 ммоль) и DIEA (4,72 мл, 27,0 ммоль) в DCM (20 мл) добавляли HATU (4,11 г, 10,80 ммоль) и 4-фторанилин (1,0 г, 9,00 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 2 ч. Реакционную смесь наливали в воду (100 мл) и экстрагировали с помощью EtOAc (15 мл, 3 раза). Объединенный органический слой промывали соляным раствором (20 мл) и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на колонках с силикагелем для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 350,8[M+H+].
Стадия 3: N-(4-фторфенил)-1-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)циклобутанкарбоксамид
К раствору 1-(5-бромпиридин-2-ил)-N-(4-фторфенил)циклобутанкарбоксамида (500 мг, 1,432 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (545 мг, 2,148 ммоль) в диоксане (20 мл) добавляли KOAc (281 мг, 2,86 ммоль) и Pd(dtbpf)Cl2 (47 мг, 0,072 ммоль) при перемешивании при RT в атмосфере азота. Реакционную смесь перемешивали при 90°C в течение 14 ч. После охлаждения до RT и разведения водой (30 мл) смесь экстрагировали с помощью EtOAc (30 мл, 3 раза). Объединенный органический слой промывали соляным раствором (20 мл) и сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 315,0 (масса соответствующей бороновой кислоты).
Стадия 4: N-(4-фторфенил)-1-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)циклобутанкарбоксамид
К раствору N-(4-фторфенил)-1-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)циклобутанкарбоксамида (50 мг, 0,126 ммоль) в 1,4-диоксане (1 мл) и воде (0,2 мл) добавляли Pd(dtbpf)Cl2 (8 мг, 0,012 ммоль), (5-бром-2-(трифторметил)пиридин-4-ил)метанол (33 мг, 0,129 ммоль) и фосфат калия (80 мг, 0,379 ммоль) при RT. Реакционную смесь перемешивали при 100°C в микроволновой печи в течение 0,5 ч. Удаляли растворитель, остаток разводили водой (5 мл) и экстрагировали с помощью EtOAc (5 мл, 3 раза). Органические слои собирали, промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Реакционную смесь очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Waters XSELECT C18 (150×30 мм×5 мкм) с использованием воды (0,1% TFA) и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,71 (д, 1H), 8,61 (с, 1H), 8,09 (с, 1H), 8,02 (ш.д., 1H), 7,74 (д, 1H), 7,51-7,58 (м, 2H), 7,03 (т, 2H), 4,66 (с, 2H), 2,91-3,09 (м, 2H), 2,71-2,88 (м, 2H), 1,93-2,16 (м, 2H). MS (ESI) m/z 446,0 [M+H+].
Пример 95: N-(4-фторфенил)-1-(4'-(1-гидроксициклобутил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)циклобутанкарбоксамид
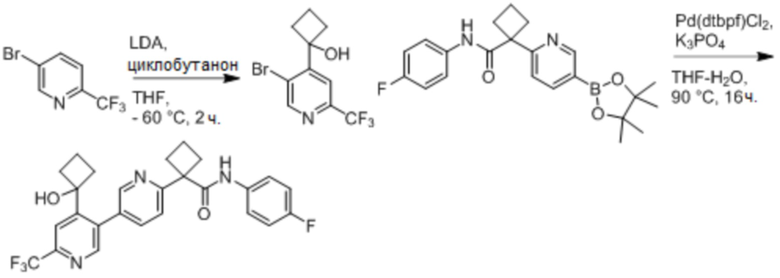
Стадия 1: 1-(5-бром-2-(трифторметил)пиридин-4-ил)циклобутанол
К раствору 5-бром-2-(трифторметил)пиридина (1 г, 4,42 ммоль) в THF (20 мл) по каплям добавляли LDA (2,4 мл, 4,87 ммоль) при -60°C в атмосфере азота. Через 3 ч. по каплям добавляли циклобутанон (0,372 г, 5,31 ммоль) при -60°C, смесь перемешивали при этой температуре в течение 1 ч. и позволяли ей нагреться до RT. Реакцию гасили насыщенным водным раствором NH4Cl (10 мл) и экстрагировали с помощью EtOAc (15 мл, 2 раза). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 295,9, 297,9 [M+H+].
Стадия 2: N-(4-фторфенил)-1-(4'-(1-гидроксициклобутил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)циклобутанкарбоксамид
К раствору N-(4-фторфенил)-1-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)циклобутанкарбоксамида (50 мг, 0,126 ммоль) и 1-(5-бром-2-(трифторметил)пиридин-4-ил)циклобутанола (45 мг, 0,152 ммоль) в THF (2,0 мл) и воде (0,2 мл) добавляли K3PO4 (80 мг, 0,379 ммоль) и Pd(dtbpf)Cl2 (8 мг, 0,012 ммоль) при перемешивании при RT в атмосфере азота. Реакционную смесь перемешивали при 90°C в течение 14 ч. Реакционную смесь охлаждали до RT, разводили водой (5 мл) и экстрагировали смесь с помощью EtOAc (5 мл, 3 раза). Объединенный органический слой промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой YMC-Actus Pro C18 (150×30×5 мкм) с использованием воды (0,1% TFA) и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,79 (с, 1H), 8,59 (с, 1H), 8,14 (дд, 1H), 7,80 (с, 1H), 7,73 (д, 1H), 7,53 (дд, 2H), 7,03 (т, J=8,8 Гц, 2H), 2,93-3,04 (м, 2H), 2,74-2,84 (м, 2H), 2,27-2,39 (м, 2H), 1,91-2,13 (м, 5 H), 1,62 (ш.д., 1H). MS (ESI) m/z 486,3 [M+H+].
Примеры 96-97 в следующей таблице получали аналогично примеру 95.
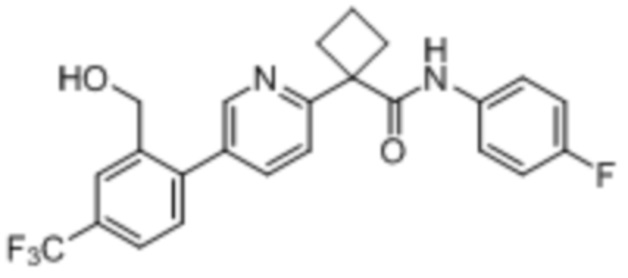
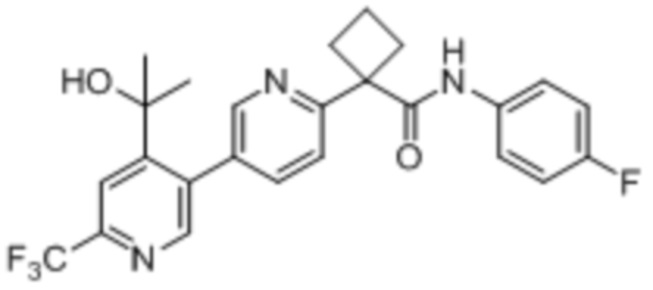
Пример 98: N-(4-фторфенил)-3-(4-(3-(гидроксиметил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамид
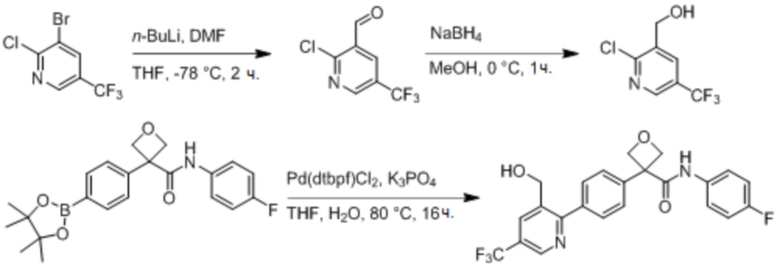
Стадия 1: 2-хлор-5-(трифторметил)никотинальдегид
К перемешиваемому раствору 3-бром-2-хлор-5-(трифторметил)пиридина (3 г, 11,52 ммоль) в толуоле (60 мл) добавляли n-BuLi (6 мл, 15,00 ммоль) (2,5 M гексан) при -78°C и перемешивали реакционную смесь при -78°C в течение 1,5 ч. Затем добавляли DMF (1,2 мл, 15,5 ммоль) при -78°C и полученную смесь перемешивали при -78°C в течение 0,5 ч. Реакцию гасили HCl (1 M) (60 мл), затем экстрагировали с помощью EtOAc (50 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. 1H ЯМР (500 МГц, CDCl3) δ 10,44-10,51 (м, 1H) 8,87 (д, 1H) 8,47 (д, 1H).
Стадия 2: (2-хлор-5-(трифторметил)пиридин-3-ил)метанол
К перемешиваемому раствору 2-хлор-5-(трифторметил)никотинальдегида (780 мг, 3,72 ммоль) в MeOH (5 мл) добавляли NaBH4 (141 мг, 3,72 ммоль) при 0°C и перемешивали реакционную смесь при 0°C в течение 1 ч. Реакционную смесь гасили насыщенным раствором NH4Cl (5 мл), затем экстрагировали с помощью EtOAc (5 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 212,0 [M+H+].
Стадия 3: N-(4-фторфенил)-3-(4-(3-(гидроксиметил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору (2-хлор-5-(трифторметил)пиридин-3-ил)метанола (40 мг, 0,189 ммоль) и N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил-)оксетан-3-карбоксамида (75 мг, 0,189 ммоль) в THF (2 мл) и воде (0,4 мл) добавляли K3PO4 (120 мг, 0,567 ммоль) и Pd(dtbpf)Cl2 (13 мг, 0,020 ммоль) при RT. Реакционную смесь перемешивали при 80°C в течение 16 ч. После охлаждения до RT реакционную смесь разводили водой (2 мл) и экстрагировали с помощью EtOAc (2 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Boston Green ODS (150×30×5 мкм) с использованием воды (0,1% TFA)и CH3CN в качестве элюентов, а затем концентрировали (при температуре ниже 50°C) для получения титульного соединения в виде масла. 1H ЯМР (500 МГц, CD3OD) δ 8,85 (ш.с., 1H) 8,38 (с, 1H) 7,63-7,70 (м, 4H) 7,52-7,61 (м, 2H) 7,00-7,10 (м, 2H) 5,36 (д, 2H), 5,04 (д, 2H) 4,67 (с, 2H). MS (ESI) m/z 447,0[M+H+].
Пример 99: N-(4-фторфенил)-3-(4-(4-(1-гидроксициклобутил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
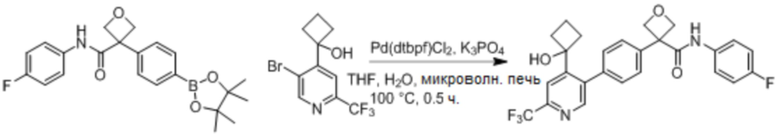
К раствору N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (35 мг, 0,088 ммоль) и 1-(5-бром-2-(трифторметил)пиридин-4-ил)циклобутанола (30 мг, 0,101 ммоль) в THF (2,0 мл) и воде (0,2 мл) добавляли K3PO4 (56 мг, 0,264 ммоль) и Pd(dtbpf)Cl2 (6 мг, 9,21 мкмоль). Реакционную смесь запаивали и перемешивали при 100°C в микроволновой печи в течение 0,5 ч. Реакционную смесь охлаждали до RT и разводили водой (5 мл). Смесь экстрагировали с помощью EtOAc (5 мл, 3 раза), объединенный органический слой промывали соляным раствором (5 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой YMC-Actus Pro C18 (150×30×5 мкм), с использованием воды (0,1% TFA) и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,52 (с, 1H), 7,75 (с, 1H), 7,52-7,64 (м, 6 H), 6,98-7,13 (м, 2H), 5,36 (д, 2H), 5,03 (д, 2H), 2,22-2,32 (м, 2H), 2,02-2,12 (м, 1H), 1,93-2,01 (м, 2H), 1,52-1,62 (м, 1H). MS (ESI) m/z 487,0 [M+H+].
Пример 100: N-(4-фторфенил)-3-(4-(4-(1-гидроксициклопропил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
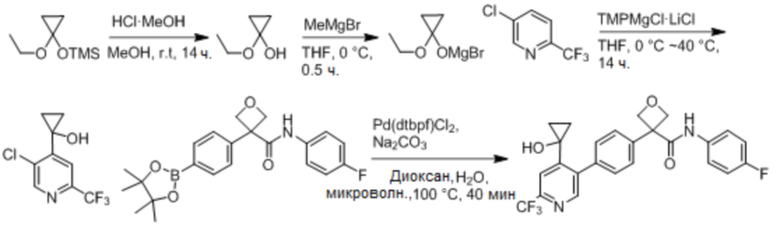
Стадия 1:1-этоксициклопропанол
К раствору (1-этоксициклопропокси)триметилсилана (5,0 г, 28,7 ммоль) в MeOH (35 мл) добавляли 4,0 M HCl (0,07 мл, 0,274 ммоль, в MeOH) при перемешивании при RT. Реакционную смесь перемешивали при RT в течение 14 ч. Удаляли растворитель и остаток очищали посредством дистилляции при пониженном давлении (65°C, 10-12 мбар) для получения титульного соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 3,76 (к, 2H), 3,14 (ш.с., 1H), 1,22 (т, 3 H), 0,91-0,99 (м, 4H).
Стадия 2: 1-(5-хлор-2-(трифторметил)пиридин-4-ил)циклопропан-1-ол
К раствору 5-хлор-2-(трифторметил)пиридина (2,0 г, 11,02 ммоль) в THF (8 мл) по каплям добавляли TMPMgCl·LiCl (12,12 мл, 12,12 ммоль) при 0°C в атмосфере азота. Смесь перемешивали при RT в течение 0,5 ч. Одновременно в отдельную колбу, содержащую раствор 1-этоксициклопропанола (1,238 г, 12,12 ммоль) в THF (12,0 мл), по каплям добавляли бромид метилмагния (4,04 мл, 12,12 ммоль) при 0°C и полученную белую суспензию перемешивали при 0°C в течение 10 мин. Затем указанный раствор литийорганического соединения добавляли в эту суспензию и полученную реакционную смесь перемешивали при температуре окружающей среды в течение 30 мин, а затем перемешивали при 40°C в течение 14 ч. Реакционную смесь охлаждали до 0°C и гасили посредством медленного добавления насыщенного раствора NH4Cl (25 мл). Смесь разводили EtOAc (20 мл) и фильтровали. Фильтрат экстрагировали с помощью EtOAc (15 мл, 3 раза). Объединенный органический слой промывали соляным раствором (15 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии на колонках с силикагелем с использованием петролейного эфира/EtOAc (20:1-10:1) в качестве элюентов для получения неочищенного продукта, который затем очищали посредством препаративной TLC (петролейный эфир/этилацетат 4:1 в качестве элюентов) для получения 1-(5-хлор-2-(трифторметил)пиридин-4-ил)циклопропанола в виде масла. MS (ESI) m/z 238 [M+H+],
Стадия 3: N-(4-фторфенил)-3-(4-(4-(1-гидроксициклопропил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
К раствору 1-(5-хлор-2-(трифторметил)пиридин-4-ил)циклопропанола (10 мг, 0,042 ммоль) и N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (17 мг, 0,043 ммоль) в диоксане (1,0 мл) и воде (0,1 мл) добавляли карбонат натрия (18 мг, 0,170 ммоль) и Pd(dtbpf)Cl2 (3 мг, 4,60 мкмоль) при перемешивании при RT в атмосфере азота. Смесь запаивали и нагревали до 100°C в микроволновой печи в течение 40 мин, а затем охлаждали до RT. Охлажденную смесь разводили водой (2 мл) и экстрагировали с помощью EtOAc (5 мл, 3 раза). Объединенный органический слой концентрировали и очищали остаток посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Xtimate C18 (150×25 мм×5 мкм), с использованием воды (10 мМ NH4HCO3) и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (500 МГц, CDCl3) δ 8,62-8,74 (м, 1H), 7,78 (с, 1H), 7,67-7,72 (м, 1H), 7,71 (д, J=8,2 Гц, 1H), 7,49 (д, J=8,2 Гц, 2H), 7,41 (т, J=6,4 Гц, 2H), 6,97-7,07 (м, 3 H), 5,43 (д, J=6,0 Гц, 2H), 5,12 (д, J=6,0 Гц, 2H), 2,48 (с, 1H), 1,05-1,16 (м, 2H), 0,79-0,89 (м, 2H). MS (ESI) m/z 473,1[M+H+].
Примеры 101-102 в следующей таблице получали аналогично примеру 100.
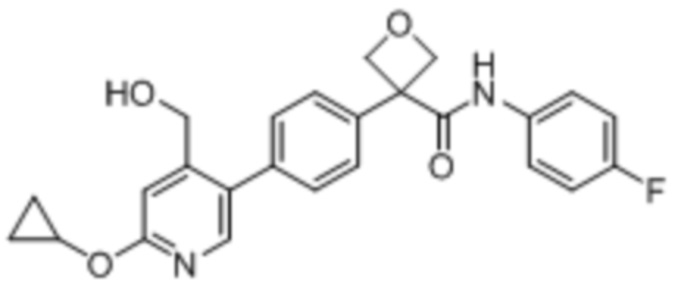
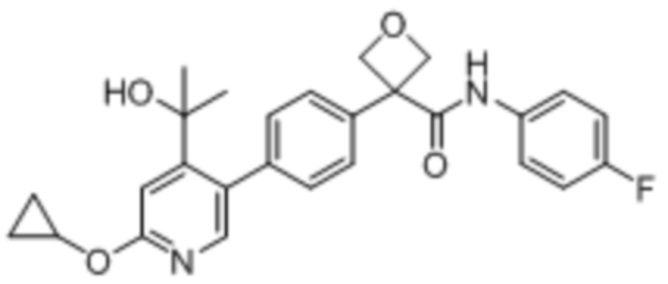
Пример 103: 5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-N-метил-2-(трифторметил)изоникотинамид
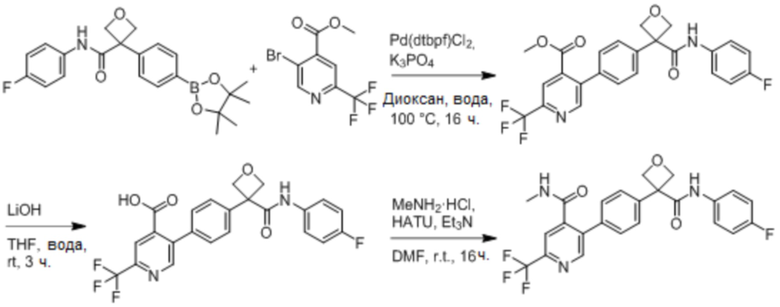
Стадия 1: Метил-5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-2-(трифторметил)изоникотинат
К раствору N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (200 мг, 0,503 ммоль) в диоксане (2,5 мл) и воде (0,5 мл) добавляли Pd(dtbpf)Cl2 (32,8 мг, 0,050 ммоль), метил-5-бром-2-(трифторметил)изоникотинат (143 мг, 0,503 ммоль) и фосфат калия (321 мг, 1,510 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и осуществляли способ для получения неочищенного продукта, который очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 475,2 [M+H+].
Стадия 2: 5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-2-(трифторметил)изоникотиновая кислота
К раствору метил-5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-2-(трифторметил)изоникотината (150 мг, 0,316 ммоль) в THF (5 мл) и воде (2,5 мл) добавляли LiOH (15 мг, 0,626 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 3 ч. Растворитель удаляли и подкисляли остаток до pH 3 с помощью 6 M HCl. Водную смесь экстрагировали с помощью EtOAc (20 мл, 3 раза), во время экстракции к водному слою добавляли соляной раствор. Объединенные органические соединения промывали соляным раствором, сушили над Na2SO4 и концентрировали под вакуумом для получения титульного соединения в виде масла. MS (ESI) m/z 461,2 [M+H+].
Стадия 3: 5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-N-метил-2-(трифторметил)изоникотинамид
К раствору 5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-2-(трифторметил)изоникотиновой кислоты (120 мг, 0,261 ммоль) в DMF (5 мл) добавляли TEA (0,2 мл, 1,435 ммоль), HATU (99 мг, 0,261 ммоль) и гидрохлорид метиламина (18 мг, 0,267 ммоль) при RT. Реакционную смесь перемешивали в течение 16 ч. при RT. Реакционную смесь разводили водой (40 мл) и экстрагировали с помощью EtOAc (30 мл, 3 раза). Объединенный органический слой промывали соляным раствором (20 мл) и сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством обращенно-фазовой ВЭЖХ с использованием колонки Phenomenex Synergi C18 150×30 мм×4мкм и воды (0,1% TFA) и ACN в качестве элюентов для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,80 (с, 1H) 7,86 (с, 1H) 7,60-7,64 (м, 2H) 7,52-7,58 (м, 4H) 7,04 (т, 2H) 5,34 (д, 2H) 5,01 (д, 2H) 2,70-2,74 (м, 3H). MS (ESI) m/z 474,2 [M+H+].
Пример 104 в следующей таблице получали аналогично примеру 103.
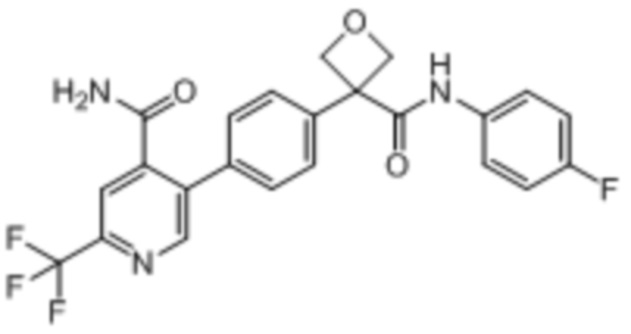
Пример 105: (R)-3-(4-(4-(1-амино-2,2,2-трифторэтил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
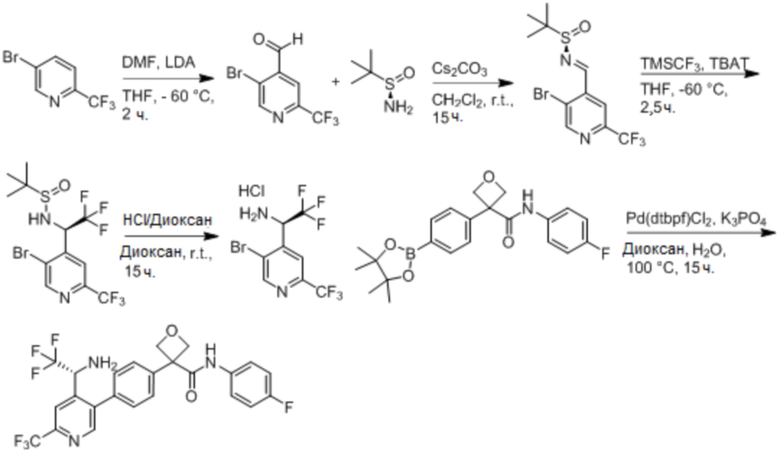
Стадия 1:-5-бром-2-(трифторметил)изоникотинальдегид
К раствору диизопропиламина (1,343 г, 13,27 ммоль) в THF (20 мл) по каплям добавляли бутиллитий (4,60 мл, 11,50 ммоль) при -60°C в атмосфере азота. Через 1 ч. по каплям добавляли 5-бром-2-(трифторметил)пиридин (2,0 г, 8,85 ммоль) в THF (5 мл) при -60°C и перемешивали смесь при этой температуре в течение 1 ч. В указанную выше смесь добавляли DMF (3,43 мл, 44,2 ммоль) и перемешивали раствор при -60°C еще в течение 1 ч. Реакцию гасили насыщенным водным раствором NH4Cl (100 мл) и экстрагировали с помощью EtOAc (20 мл×2). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 10,4 (с, 1H), 9,0 (с, 1H), 8,1 (с, 1H).
Стадия 2: (R, E)-N-((5-бром-2-(трифторметил)пиридин-4-ил)метилен)-2-метилпропан-2-сульфинамид
К перемешиваемому раствору 5-бром-2-(трифторметил)изоникотинальдегида (721 мг, 2,84 ммоль) в CH2Cl2 (20 мл) добавляли (R)-2-метилпропан-2-сульфинамид (344 мг, 2,84 ммоль) и Cs2CO3 (1110 мг, 3,41 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 15 ч. Реакционную смесь концентрировали для удаления CH2Cl2, а затем разводили водой (30 мл) и экстрагировали с помощью EtOAc (30 мл, 2 раза). Органические слои собирали, промывали соляным раствором (20 мл), сушили над Na2SO4 и после фильтрации концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,9-9,0 (м, 2H), 8,2 (с, 1H), 1,3 (с, 9 H).
Стадия 3: (R)-N-(1-(5-бром-2-(трифторметил)пиридин-4-ил)-2,2,2-трифторэтил)-2-метилпропан-2-сульфинамид
К перемешиваемому раствору (R, Z)-N-((5-бром-2-(трифторметил)пиридин-4-ил)метилен)-2-метилпропан-2-сульфинамида (659 мг, 1,845 ммоль) в THF (10 мл) добавляли TBAT (1195 мг, 2,214 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 0,5 ч. Затем смесь охлаждали до -60°C, к ней добавляли триметил(трифторметил)силан (1312 мг, 9,22 ммоль) и продолжали перемешивание при -60°C в течение 2 ч. Смесь гасили водным раствором NH4Cl (15 мл), экстрагировали с помощью EtOAc (30 мл, 2 раза) и органические слои собирали, промывали соляным раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 467,8[M+ACN+H+].
Стадия 4: (R)-1-(5-бром-2-(трифторметил)пиридин-4-ил)-2,2,2-трифторэтанамин гидрохлорид
К перемешиваемому раствору (R)-N-(1-(5-бром-2-(трифторметил)пиридин-4-ил)-2,2,2-трифторэтил)-2-метилпропан-2-сульфинамида (90 мг, 0,211 ммоль) в 1,4-диоксане (5 мл) добавляли 4 M HCl (5 мл, 20,00 ммоль, в диоксане) при RT. Реакционную смесь перемешивали при RT в течение 15 ч. Растворитель концентрировали под вакуумом для получения титульного соединения в виде твердого вещества, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 363,8 [M+ACN+H+].
Стадия 5: (R)-3-(4-(4-(1-амино-2,2,2-трифторэтил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору гидрохлорида (R)-1-(5-бром-2-(трифторметил)пиридин-4-ил)-2,2,2-трифторэтанамина (76 мг, 0,211 ммоль) в 1,4-диоксане (2,5 мл) и воде (0,5 мл) добавляли N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (101 мг, 0,254 ммоль), K3PO4 (135 мг, 0,634 ммоль) и Pd(dtbpf)Cl2 (16 мг, 0,025 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного титульного соединения, которое очищали посредством ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Waters XSELECT C18 150×30 мм×5 мкм, с использованием воды (0,1% TFA) и CH3CN в качестве подвижных фаз для получения титульного соединения в виде твердого вещества. 1H ЯМР (500 МГц, CDCl3) δ 8,7 (с, 1H), 8,0 (с, 1H), 7,4-7,5 (м, 6 H), 7,1 (ш.с., 1H), 7,0 (т, 2H), 5,4 (т, 2H), 5,1 (дд, 2H), 4,6 (к, 1H). MS (ESI) m/z 514,2 [M+H+].
Пример 106: (S)-3-(4-(4-(1-амино-2,2,2-трифторэтил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
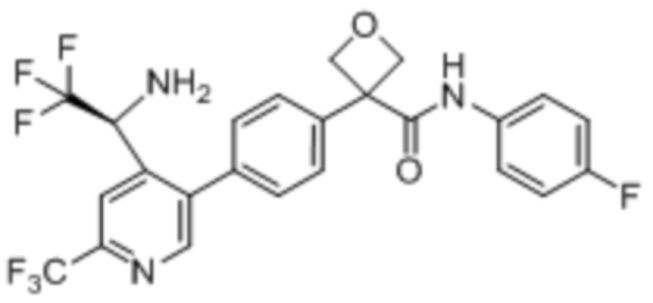
Стадия 1: (с,E)-N-((5-бром-2-(трифторметил)пиридин-4-ил)метилен)-2-метилпропан-2-сульфинамид
Это соединение получали аналогично примеру 105, за исключением того, что использовали (S)-2-метилпропан-2-сульфинамид. MS (ESI) m/z 400,1 [M+ACN+H+].
Стадия 2: N-((S)-1-(5-бром-2-(трифторметил)пиридин-4-ил)-2,2,2-трифторэтил)-2-метилпропан-2-сульфинамид
Это соединение получали аналогично примеру 105. MS (ESI) m/z 470,1 [M+ACN+H+].
Стадия 3: (S)-1-(5-бром-2-(трифторметил)пиридин-4-ил)-2,2,2-трифторэтанамин гидрохлорид
Гидролиз осуществляли аналогично стадии 3 примера 105. MS (ESI) m/z:364,0 [M+ACN+H+].
Стадия 5: (S)-3-(4-(4-(1-амино-2,2,2-трифторэтил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
Титульное соединение получали аналогично примеру 105, 1H ЯМР (400 МГц, CDCl3) δ 8,7 (с, 1H), 8,0 (с, 1H), 7,4-7,5 (м, 6H), 7,1 (ш.с., 1H), 7,0 (ш.т., 2H), 5,4 (т, 2H), 5,1-5,2 (м, 2H), 4,6 (к, 1H). MS (ESI) m/z 514,1 [M+H+].
Пример 107: N-(4-фторфенил)-3-(4-(3-(2-гидроксипропан-2-ил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамид
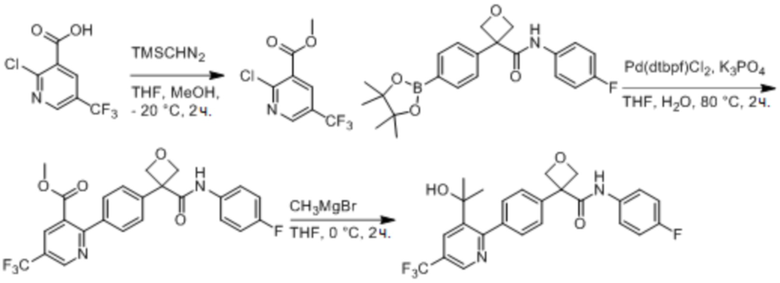
Стадия 1: Метил-2-хлор-5-(трифторметил)никотинат
К перемешиваемому раствору 2-хлор-5-(трифторметил)пиридин-3-карбоновой кислоты (2 г, 8,87 ммоль) в THF (5 мл) и MeOH (5 мл) по каплям добавляли (триметилсилил)диазометан (8,87 мл, 17,73 ммоль) (2 M в гексане) при -20°C и перемешивали реакционную смесь при -20°C в течение 2 ч. Растворитель концентрировали при пониженном давлении и очищали остаток посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 242,2 [M+H+]
Стадия 2: Метил-2-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-5-(трифторметил)никотинат
К перемешиваемому раствору метил-2-хлор-5-(трифторметил)никотината (100 мг, 0,417 ммоль) и N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (166 мг, 0,417 ммоль) в THF (2 мл) добавляли Pd(dtbpf)Cl2 (28 мг, 0,043 ммоль) и фосфат калия (266 мг, 1,252 ммоль) при RT. Смесь перемешивали при 80°C в течение 2 ч. После охлаждения до RT реакционную смесь разводили водой (2 мл), экстрагировали с помощью EtOAc (1 мл, 2 раза) и органические слои собирали, промывали соляным раствором, сушили над Na2SO4. После фильтрации фильтрат концентрировали под вакуумом. Остаток очищали посредством хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 474,9 [M+H+].
Стадия 3: N-(4-фторфенил)-3-(4-(3-(2-гидроксипропан-2-ил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору бромида метилмагния (0,1 мл, 0,300 ммоль) (3 M в гексане) в THF (0,5 мл) добавляли метил-2-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-5-(трифторметил)никотинат (50 мг, 0,105 ммоль) в THF (0,5 мл) при 0°C. Смесь перемешивали при 0°C в течение 2 ч. Реакцию гасили NH4Cl (3 мл), смесь разводили водой (2 мл) и экстрагировали с помощью EtOAc (5 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4 и после фильтрации концентрировали фильтрат под вакуумом. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой YMC-Actus Pro C18 150×30×5 мкм, с использованием воды (0,1% TFA) и CH3CN в качестве элюентов для получения титульного соединения в виде твердого вещества после лиофилизации. 1H ЯМР (500 МГц, CD3OD) δ 8,75-8,80 (м, 1H) 8,68 (д, 1H) 7,60-7,65 (м, 2H) 7,52-7,58 (м, 2H) 7,45-7,51 (м, 2H) 7,01-7,12 (м, 2H) 5,37 (д, 2H) 5,05 (д, 2H) 1,33-1,43 (м, 6H).MS (ESI) m/z 475,2 [M+H+]
Пример 108: 3-(4-(5-циклопропокси-3-(гидроксиметил)пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
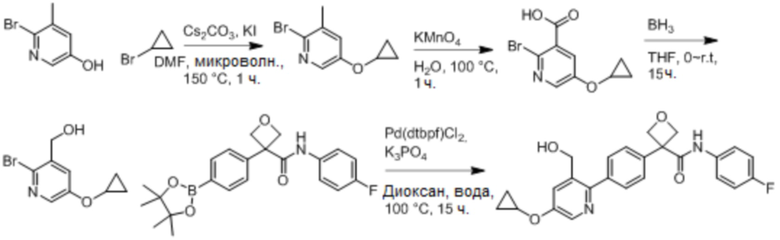
Стадия 1: 2-бром-5-циклопропокси-3-метилпиридин
К раствору 6-бром-5-метилпиридин-3-ола (500 мг, 2,66 ммоль), бромциклопропана (1287 мг, 10,64 ммоль) и йодида калия (50 мг, 0,301 ммоль) в DMF (4,0 мл) добавляли карбонат цезия (1300 мг, 3,99 ммоль) при RT. Реакционный сосуд запаивали и нагревали в микроволновой печи при 150°C в атмосфере азота в течение 1 ч. Реакционную смесь охлаждали до RT, наливали в воду (20,0 мл) и экстрагировали этилацетатом (10,0 мл, 3 раза). Объединенный органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 229,9 [M+H+].
Стадия 2: 2-бром-5-циклопропоксиникотиновая кислота
К перемешиваемой смеси 2-бром-5-циклопропокси-3-метилпиридина (160 мг, 0,701 ммоль) в воде (10,0 мл) частями добавляли KMnO4 (443 мг, 2,81 ммоль) при RT. Реакционную смесь перемешивали при 100°C в течение 3 ч. Затем горячую полученную смесь фильтровали через целитовую прокладку, фильтрат охлаждали и промывали EtOAc (5 мл, 3 раза). Водный слой подкисляли 6 Н HCl до pH=4, концентрировали до небольшого объема (5 мл) и экстрагировали с помощью EtOAc (10 мл, 3 раза). Объединенный органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали под вакуумом для получения неочищенного титульного соединения в виде твердого вещества. MS (ESI) m/z 259,9 [M+H+].
Стадия 3: (2-бром-5-циклопропоксипиридин-3-ил)метанол
К перемешиваемому раствору 2-бром-5-циклопропоксиникотиновой кислоты (30 мг, 0,116 ммоль) в THF (2,0 мл) по каплям добавляли BH3·THF (0,50 мл, 0,500 ммоль) при 0°C и перемешивали реакционную смесь при RT в течение 15 ч. Смесь осторожно гасили MeOH (5 мл), затем концентрировали под вакуумом. Остаток очищали посредством препаративной TLC для получения титульного соединения в виде масла. MS (ESI) m/z 243,9 [M+H+].
Стадия 4: 3-(4-(5-циклопропокси-3-(гидроксиметил)пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (41 мг, 0,103 ммоль) в диоксане (2,0 мл) и воде (0,4 мл) добавляли K3PO4 (65 мг, 0,306 ммоль) и Pd(dtbpf)Cl2 (7 мг, 10,74 мкмоль) при RT. Реакционную смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного продукта, который очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (250×21,2 мм×4 мкм), с использованием воды (0,2% муравьиной кислоты) и ACN в качестве элюентов, а затем лиофилизировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,34 (д, 1H), 8,01 (д, 1H), 7,52-7,68 (м, 6 H), 7,06 (т, 2H), 5,36 (д, 2H), 5,04 (д, 2H), 4,59 (с, 2H), 4,02 (дт, 1H), 0,79-0,96 (м, 4H).MS (ESI) m/z 435,1 [M+H+].
Пример 109: 3-(4-(5-циклопропокси-3-(2-гидроксипропан-2-ил)пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
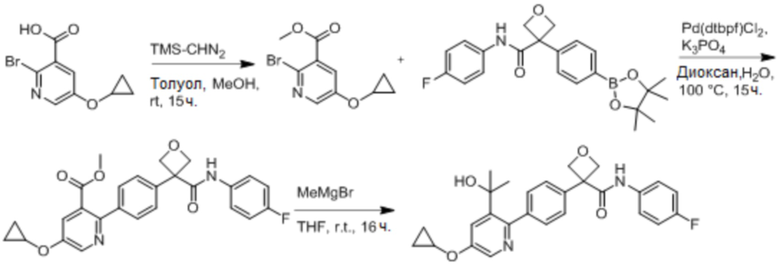
Стадия 1: Метил-2-бром-5-циклопропоксиникотинат
К перемешиваемому раствору 2-бром-5-циклопропоксиникотиновой кислоты (0,11 г, 0,426 ммоль) в DCM (5 мл) и MeOH (2,5 мл) добавляли ((триметилсилил)метил)диазен (0,64 мл, 1,280 ммоль) при 0°C и перемешивали смесь при RT в течение 15 ч. Растворитель концентрировали при пониженном давлении и остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc=2:1) для получения титульного соединения в виде масла. MS (ESI) m/z 271,8 и 273,8 [M+H+].
Стадия 2: Метил-5-циклопропокси-2-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)никотинат
К перемешиваемому раствору N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (52 мг, 0,131 ммоль) и метил-2-бром-5-циклопропоксиникотината (30 мг, 0,110 ммоль) в диоксане (2 мл) и воде (0,4 мл) добавляли K3PO4 (70 мг, 0,330 ммоль) и Pd(dtbpf)Cl2 (7 мг, 10,74 мкмоль) при RT. Смесь нагревали до 100°C при перемешивании в течение 16 ч. После охлаждения до RT смесь экстрагировали с помощью EtOAc (30 мл, 2 раза), органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc=1:1) для получения титульного соединения в виде масла. MS (ESI) m/z 463,0 [M+H+].
Стадия 3: 3-(4-(5-циклопропокси-3-(2-гидроксипропан-2-ил)пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
К перемешиваемому раствору метил-5-циклопропокси-2-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)никотината (13 мг, 0,028 ммоль) в THF(1 мл) добавляли 3 M MeMgBr (0,08 мл, 0,240 ммоль) при RT. Смесь перемешивали при RT в течение 15 ч. Смесь разводили водой (20 мл), экстрагировали с помощью EtOAc (20 мл, 3 раза), органические слои собирали, промывали соляным раствором (10 мл), сушили над Na2SO4 и после фильтрации концентрировали фильтрат под вакуумом. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (150×30 мм×4 мкм), с использованием воды (0,225% FA) и ACN в качестве элюентов для получения титульного соединения в виде твердого вещества после лиофилизации желаемых фракций. 1H ЯМР (400 МГц, CD3OD) δ 8,41 (д, 1H), 8,36 (д, 1H), 7,65 (д, 2H), 7,57-7,52 (м, 4H), 7,06 (т, 2H), 5,37 (д, 2H), 5,04 (д, 2H), 4,08 (ш.с., 1H), 1,38 (с, 6H), 0,94 (ш.д., 2H), 0,85 (ш.с., 2H). MS (ESI) m/z 463,2 [M+H+].
Пример 110: 3-(4'-циклопропокси-2'-(2-гидроксипропан-2-ил)-[1,1'-бифенил]-4-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
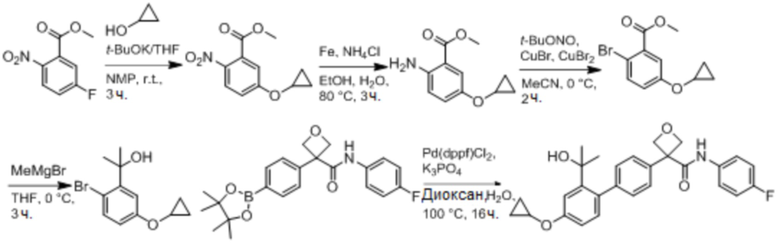
Стадия 1: Метил-5-циклопропокси-2-нитробензоат
К перемешиваемому раствору метил-5-фтор-2-нитробензоата (1 г, 5,02 ммоль) и циклопропанола (0,3 г, 5,17 ммоль) в NMP (20 мл) по каплям добавляли 2-метилпропан-2-олат калия (7,5 мл, 7,50 ммоль) (1 M в THF) при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 мин. Смесь нагревали до RT и перемешивали при RT в течение 3 ч. Реакционную смесь разделяли с помощью EtOAc/петролейного эфира (100 мл, 1/1 об./об.) и воды (80 мл). Органический слой отделяли, промывали соляным раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. 1H ЯМР (400 МГц, CD3OD) δ 8,07 (д, 1H), 7,32 (дд, 2,8 Гц, 1H), 7,26-7,30 (м, 1H), 3,96 (тт, 3,0 Гц, 1H), 3,89 (с, 3H), 0,85-0,91 (м, 2H), 0,76-0,80 (м, 2H).
Стадия 2: Метил-2-амино-5-циклопропоксибензоат
К раствору метил 5-циклопропокси-2-нитробензоата (300 мг, 1,265 ммоль) в EtOH (10 мл) и вода (2 мл) добавляли железо (353 мг, 6,32 ммоль) и хлорид аммония (338 мг, 6,32 ммоль) при RT. Смесь перемешивали при 80°C в течение 3 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток разводили водой (30 мл), экстрагировали с помощью EtOAc (15 мл, 2 раза) и объединенный органический слой промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 208,1 [M+H+].
Стадия 3: Метил-2-бром-5-циклопропоксибензоат
К раствору метил-2-амино-5-циклопропоксибензоата (50 мг, 0,241 ммоль) в CH3CN (5 мл) добавляли бромид меди (I) (70 мг, 0,483 ммоль) и трет-бутилнитрит (50 мг, 0,483 ммоль) при 0°C. После перемешивания при RT в течение 16 ч. реакционную смесь разводили водой (30 мл) и экстрагировали с помощью DCM (30 мл, 3 раза). Объединенный органический слой промывали соляным раствором (20 мл) и сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc=2:1) для получения титульного соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 7,52 (д, 1H), 7,46 (д, 1H), 7,00 (дд, 1H), 3,92 (с, 3 H), 3,71-3,76 (м, 1H), 0,75-0,81 (м, 4H).
Стадия 4: 2-(2-бром-5-циклопропоксифенил)пропан-2-ол
К раствору метил-2-бром-5-циклопропоксибензоата (70 мг, 0,258 ммоль) в THF (5 мл) добавляли бромид метилмагния (0,3 мл, 0,900 ммоль) (3 M в этоксиэтане) при 0°C. После завершения добавления реакционную смесь перемешивали при RT в течение 16 ч., медленно гасили насыщенным раствором NH4Cl (20 мл, водный раствор) и экстрагировали с помощью EtOAc (10 мл, 3 раза). Объединенный органический слой промывали соляным раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки. 1H ЯМР (400 МГц, CD3OD) δ 7,51 (д, J1 H), 7,44 (д, 1H), 6,83 (дд, 1H), 3,75 (тд, 1H), 1,68 (с, 6H), 0,78 (ш.д., 2H), 0,67 (ш.с., 2H).
Стадия 5: 3-(4'-циклопропокси-2'-(2-гидроксипропан-2-ил)-[1,1'-бифенил]-4-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К раствору 2-(2-бром-5-циклопропоксифенил)пропан-2-ола (80 мг, 0,295 ммоль) в диоксане (5 мл) и воде (1 мл) добавляли N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (117 мг, 0,295 ммоль), Pd(dtbpf)Cl2 (19,23 мг, 0,030 ммоль) и K3PO4 (188 мг, 0,885 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного продукта, который очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Xtimate C18 150×25 мм×5 мкм, и желаемые фракции собирали и концентрировали для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,56 (т, 2H), 7,43-7,48 (м, 3H), 7,32 (д, 2H), 7,06 (т, 2H), 6,89-6,94 (м, 2H), 5,34 (д, 2H), 5,02 (д, 2H), 3,80 (тт, 1H), 1,32 (с, 6 H), 0,76-0,83 (м, 2H), 0,67-0,73 (м, 2H). MS (ESI) m/z 484,3 [M+Na+].
Пример 111: 3-(4'-циклопропокси-2'-(гидроксиметил)-[1,1'-бифенил]-4-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
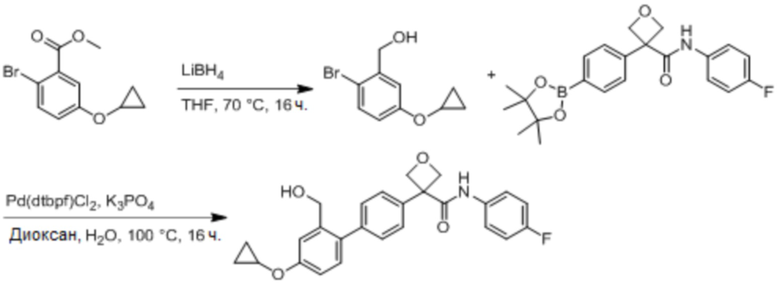
Стадия 1: (2-бром-5-циклопропоксифенил)метанол
К перемешиваемому раствору метил-2-бром-5-циклопропоксибензоата (70 мг, 0,258 ммоль) в THF (5 мл) добавляли тетрагидроборат лития (16,87 мг, 0,775 ммоль) при 0°C и смесь нагревали до 70°C в течение 16 ч. Реакционную смесь охлаждали, фильтровали и концентрировали фильтрат при пониженном давлении. Остаток разводили водой (30 мл), экстрагировали с помощью EtOAc (15 мл, 2 раза) и объединенный органический слой промывали соляным раствором (10 мл) и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки.
Стадия 2: 3-(4'-циклопропокси-2'-(гидроксиметил)-[1,1'-бифенил]-4-ил)-N-(4-фторфенил)оксетан-3-карбоксамид
К раствору (2-бром-5-циклопропоксифенил)метанола (50 мг, 0,206 ммоль) в воде (1 мл) и 1,4-диоксане (5 мл) добавляли N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (82 мг, 0,206 ммоль), Pd(dppf)Cl2 (15,05 мг, 0,021 ммоль) и фосфат калия (131 мг, 0,617 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного продукта, который очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (150×30 мм×4 мкм), для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,46-7,61 (м, 4H), 7,40 (д, 2H), 7,26 (д, 1H), 7,16 (д, 1H), 6,96-7,11 (м, 3H), 5,33 (д, 2H), 5,01 (д, 2H), 4,48 (с, 2H), 3,81 (дт, 1H), 0,75-0,84 (м, 2H), 0,66-0,75 (м, 2H). MS (ESI) m/z 456,1 [M+Na+].
Пример 112: N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-изопропоксипиридин-3-ил)фенил)оксетан-3-карбоксамид
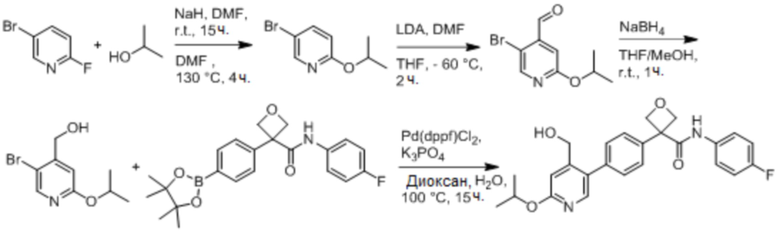
Стадия 1:5-бром-2-изопропоксипиридин
К перемешиваемому раствору пропан-2-ола (2,77 г, 46,0 ммоль) в DMF (30 мл) добавляли гидрид натрия (0,920 г, 23,01 ммоль, 60% в масле) в атмосфере азота при 0°C и перемешивали реакционную смесь при RT в течение 15 ч. Затем добавляли 5-бром-2-фторпиридин (2,7 г, 15,34 ммоль) в DMF (5 мл) и нагревали реакционную смесь до 130°C в течение 4 ч. После охлаждения до RT растворитель концентрировали и остаток гасили водой (20 мл) и экстрагировали с помощью EtOAc (30 мл, 2 раза). Органические слои объединяли, промывали соляным раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 215,9 [M+H+].
Стадия 2: 5-бром-2-изопропоксиизоникотинальдегид
К раствору 5-бром-2-изопропоксипиридина (1,8 г, 8,33 ммоль) в THF (20 мл) добавляли LDA (5,00 мл, 10,00 ммоль) при -60°C и смесь перемешивали при этой температуре в течение 1 ч. Затем добавляли N, N-диметилформамид (0,913 г, 12,50 ммоль) и перемешивали смесь при RT в течение 1 ч. Растворитель концентрировали, остаток гасили водой (20 мл) и экстрагировали с помощью EtOAc (30 мл, 2 раза). Органические слои собирали, промывали соляным раствором (20 мл), сушили над Na2SO4 и после фильтрации концентрировали фильтрат под вакуумом. Остаток очищали посредством хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 243,9 [M+H+].
Стадия 3: (5-бром-2-изопропоксипиридин-4-ил)метанол
К перемешиваемому раствору 5-бром-2-изопропоксиизоникотинальдегида (1,286 г, 5,27 ммоль) в THF (20 мл) и MeOH (20 мл) добавляли NaBH4 (0,299 г, 7,90 ммоль) при 0°C и перемешивали реакционную смесь при RT в течение 1 ч. Реакцию гасили NH4Cl (20 мл) и смесь экстрагировали с помощью EtOAc (30 мл, 2 раза). Органические слои собирали, промывали соляным раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством хроматографии на силикагеле для получения титульного соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 8,14 (с, 1H), 6,89 (с, 1H), 5,23 (спп, 1H), 4,67 (д, 2H), 4,12 (к, 1H), 1,34 (д, 6H).
Стадия 4: N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-изопропоксипиридин-3-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (300 мг, 0,755 ммоль) в 1,4-диоксане (2,5 мл) и воде (0,5 мл) добавляли PdCl2(dppf) (45 мг, 0,062 ммоль) и K3PO4 (388 мг, 1,829 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного титульного соединения, которое очищали посредством ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Waters Xbridge Prep OBD C18 (100×19 мм×5 мкм), используя воду (0,225% муравьиной кислоты)/CH3CN в качестве элюентов для получения титульного соединения в виде твердого вещества после концентрирования желаемых фракций. 1H ЯМР (400 МГц, CDCl3) δ 7,36-7,48 (м, 6 H), 6,94-7,06 (м, 4H), 5,41 (д, 1H), 5,29-5,38 (м, 1H), 5,09 (д, 2H), 4,63 (с, 2H), 1,40 (д, 6H). MS (ESI) m/z 437,2 [M+H+].
Пример 113: N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридазин-3-ил)фенил)оксетан-3-карбоксамид
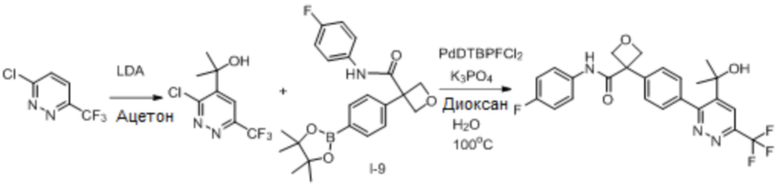
Стадия 1: 2-(3-хлор-6-(трифторметил)пиридазин-4-ил)пропан-2-ол
К раствору 2,5 M BuLi в гексанах (1,3 мл, 3,3 ммоль) по каплям добавляли THF (5 мл) при перемешивании при -78°C в атмосфере азота. Добавляли 2,2,6,6-тетраметилпиперидин (503 мг, 3,56 ммоль) при 0°C. После перемешивания при 0°C в течение 0,5 ч. смесь охлаждали до -70°C. В смесь по каплям добавляли 3-хлор-6-(трифторметил)пиридазин (500 мг, 2,74 ммоль). Реакционную смесь перемешивали при -70°C в течение 1,5 ч. В смесь по каплям добавляли ацетон (3 мл, 40,9 ммоль). Реакционную смесь перемешивали при -70°C в течение 1 ч., гасили насыщенным водным раствором хлорида аммония (5 мл) и экстрагировали с помощью EtOAc (20 мл, 2 раза). Объединенные органические слои промывали соляным раствором, сушили над MgSO4, концентрировали под вакуумом и очищали остаток посредством флэш-хроматографии на силикагеле, а затем посредством препаративной TLC (SiO2, петролейный эфир/EtOAc=3/1) для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 240,9 [M+H+].
Стадия 2: N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридазин-3-ил)фенил)оксетан-3-карбоксамид
К раствору 2-(3-хлор-6-(трифторметил)пиридазин-4-ил)пропан-2-ола (30 мг, 0,125 ммоль) в диоксане (1 мл) и воде (0,2 мл) добавляли N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (I-9) (59 мг, 0,149 ммоль), ортофосфат калия (79 мг, 0,374 ммоль) и 1,1′-бис(ди-трет-бутилфосфино)ферроцен]дихлорпалладий (II) (8 мг, 0,012 ммоль). Реакционную смесь перемешивали при 100°C в течение 16 ч. в атмосфере азота. Смесь концентрировали при пониженном давлении для получения остатка, который очищали посредством препаративной ВЭЖХ (колонка Phenomenex Synergi C18 (150×30 мм×4 мкм)) с использованием воды (0,1% TFA)и ACN в качестве подвижных фаз для получения титульного соединения в виде твердого вещества. 1H ЯМР (400 МГц, MeOD) δ 8,47 (с, 1H), 7,68-7,66 (м, 2H), 7,58-7,55 (м, 4H), 5,38-5,37 (м, 2H), 5,06-5,05 (м, 2H), 3,3 (м, 4H), 1,38 (м, 6H). MS (ESI) m/z 476 [M+H+].
Пример 114: N-(4-хлорфенил)-1-(5-(2-(гидроксиметил)-4-(трифторметил)фенил)пиразин-2-ил)циклобутан-1-карбоксамид
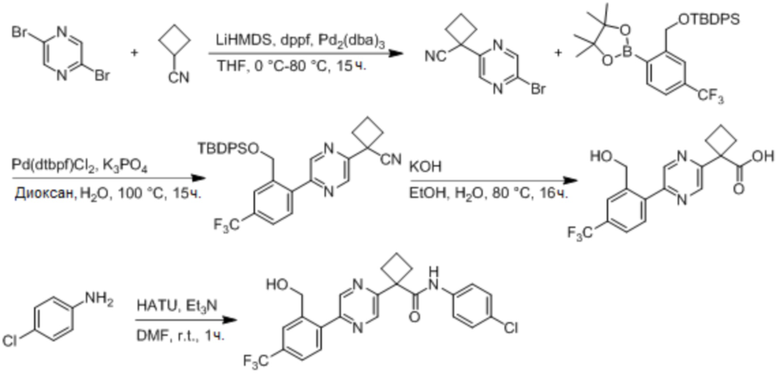
Стадия 1: 1-(5-бромпиразин-2-ил)циклобутан-1-карбонитрил
К раствору 2,5-дибромпиразина (200 мг, 0,841 ммоль) в THF (10,0 мл) добавляли NiXantphos (46 мг, 0,083 ммоль), Pd2(dba)3 (40 мг, 0,044 ммоль) и циклобутанкарбонитрил (75 мг, 0,925 ммоль), затем добавляли LiHMDS (1,6 мл, 1,600 ммоль) (1 M в THF) при 0°C. Полученную смесь нагревали до 80°C в течение 15 ч. и охлаждали до RT, гасили NH4Cl (10,0 мл) и экстрагировали с помощью EtOAc (20 мл, 2 раза). Объединенный органический слой промывали соляным раствором и концентрировали под вакуумом. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонки Phenomenex Synergi C18 (250×21,2 мм×4 мкм), с использованием воды (0,2% муравьиной кислоты) и ACN в качестве элюентов. Желаемые фракции собирали и концентрировали посредством лиофилизации для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 237,9 [M+H+].
Стадия 2: 1-(5-(2-(((трет-бутилдифенилсилил)окси)метил)-4-(трифторметил)фенил)пиразин-2-ил)циклобутан-1-карбонитрил
К перемешиваемому раствору 1-(5-бромпиразин-2-ил)циклобутанкарбонитрила (50 мг, 0,210 ммоль) и трет-бутилдифенил((2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5-(трифторметил)-бензил)окси)силана (125 мг, 0,231 ммоль) в диоксане (1,0 мл) и воде (0,2 мл) добавляли K3PO4 (134 мг, 0,630 ммоль) и Pd(dtbpf)Cl2 (10 мг, 0,015 ммоль) при RT. Реакционную смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного титульного соединения, которое очищали посредством препаративной TLC (петролейный эфир/этилацетат=5:1 в качестве элюентов) для получения титульного соединения в виде масла. MS (ESI) m/z 572,2 [M+H+].
Стадия 3: 1-(5-(2-(гидроксиметил)-4-(трифторметил)фенил)пиразин-2-ил)циклобутан-1-карбоновая кислота
К перемешиваемому раствору 1-(5-(3-(((трет-бутилдифенилсилил)окси)метил)-4-(трифторметил)фенил)пиразин-2-ил)циклобутанкарбонитрила (45 мг, 0,079 ммоль) в этаноле (6,0 мл) и воде (2,0 мл) добавляли KOH (45 мг, 0,802 ммоль) при RT и нагревали реакционную смесь до 80°C в течение 16 ч. Растворитель удаляли посредством концентрирования под вакуумом и разводили остаток водой (2,0 мл) и экстрагировали с помощью EtOAc (3,0 мл). Водный слой подкисляли 3 Н HCl до pH=5 и экстрагировали с помощью EtOAc (3 мл, 3 раза). Объединенный органический слой промывали соляным раствором, сушили над Na2SO4 и концентрировали под вакуумом для получения титульного соединения в виде масла, которое использовали непосредственно на следующей стадии без последующей очистки. MS (ESI) m/z 353,0 [M+H+].
Стадия 4: N-(4-хлорфенил)-1-(5-(2-(гидроксиметил)-4-(трифторметил)фенил)пиразин-2-ил)циклобутан-1-карбоксамид
К перемешиваемому раствору 4-хлоранилина (20 мг, 0,16 ммоль), TEA (40 мг, 0,395 ммоль) и 1-(5-(3-(гидроксиметил)-4-(трифторметил)фенил)пиразин-2-ил)циклобутанкарбоновой кислоты (27 мг, 0,077 ммоль) в DMF (1,0 мл) добавляли HATU (50 мг, 0,131 ммоль) при RT и перемешивали реакционную смесь при RT в течение 1 ч. Остаток очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (250×21,2 мм×4 мкм), с использованием воды (0,2% муравьиной кислоты) и ACN в качестве элюентов. Титульное соединение получали в виде твердого вещества после лиофилизации желаемых фракций. 1H ЯМР (400 МГц, CDCl3) δ 8,95 (д, 1H), 8,78 (д, 1H), 8,17 (с, 1H), 7,84 (с, 1H), 7,71-7,78 (м, 2H), 7,49 (д, 2H), 7,29 (с, 2H), 4,57 (с, 2H), 3,08 (ддд, 2H), 2,73-2,83 (м, 2H), 2,13-2,29 (м, 1H), 1,94-2,11 (м, 1H). MS (ESI) m/z 461,9 [M+H+].
Пример 115: N-(6-фторпиридин-3-ил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
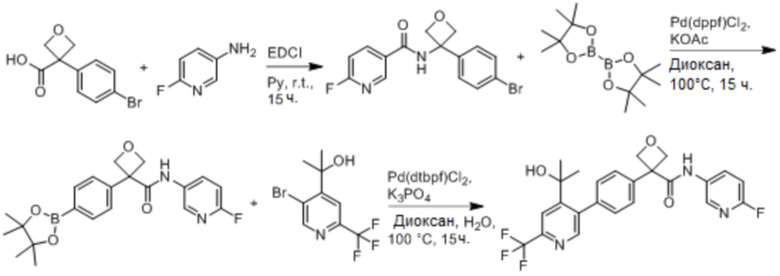
Стадия 1: 3-(4-бромфенил)-N-(6-фторпиридин-3-ил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(4-бромфенил)оксетан-3-карбоновой кислоты (1 г, 3,89 ммоль) в пиридине (20 мл) добавляли 6-фторпиридин-3-амин (0,654 г, 5,83 ммоль) и EDC (2,237 г, 11,67 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 15 ч. Концентрировали растворитель и разводили остаток водой (20 мл) и экстрагировали этилацетатом (30 мл, 2 раза). Органические слои собирали, промывали соляным раствором (20 мл), сушили над Na2SO4 и после фильтрации концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 351,0 [M+H+].
Стадия 2: N-(6-фторпиридин-3-ил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору 3-(4-бромфенил)-N-(6-фторпиридин-3-ил)оксетан-3-карбоксамида (1,3 г, 3,70 ммоль) в 1,4-диоксане (10 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,128 г, 4,44 ммоль), ацетат калия (1,090 г, 11,11 ммоль) и Pd(dppf)Cl2 (0,271 г, 0,370 ммоль) при RT. Реакционную смесь нагревали до 100°C при перемешивании в течение 15 ч. После охлаждения до RT реакцию гасили водой (30 мл) и смесь экстрагировали с помощью EtOAc (30 мл, 2 раза). Органические слои собирали, промывали соляным раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 399,1 [M+H+].
Стадия 3: N-(6-фторпиридин-3-ил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору N-(6-фторпиридин-3-ил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (220 мг, 0,552 ммоль) в 1,4-диоксане (2,5 мл) и воде (0,5 мл) добавляли 2-(5-бром-2-(трифторметил)пиридин-4-ил)пропан-2-ол (189 мг, 0,665 ммоль), K3PO4 (352 мг, 1,657 ммоль) и Pd(dtbpf)Cl2 (36 мг, 0,055 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения неочищенного титульного соединения, которое очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Waters Xbridge Prep OBD C18 (100×19 мм×5 мкм), с использованием воды (0,225% муравьиной кислоты) и CH3CN в качестве элюентов для получения титульного соединения в виде твердого вещества. 1H ЯМР (500 МГц, CD3OD) δ 8,4 (ш.с., 1H), 8,3 (с, 1H), 8,2-8,3 (м, 2H), 7,6 (ш.д., 2H), 7,4 (ш.д., 2H), 7,1 (дд, 1H), 5,4 (ш.д., 2H), 5,1 (ш.д., 2H), 1,4 (с, 6 H). MS (ESI) m/z 476,1 [M+H+].
Примеры 116 и 117 в следующей таблице получали аналогично примеру 115.
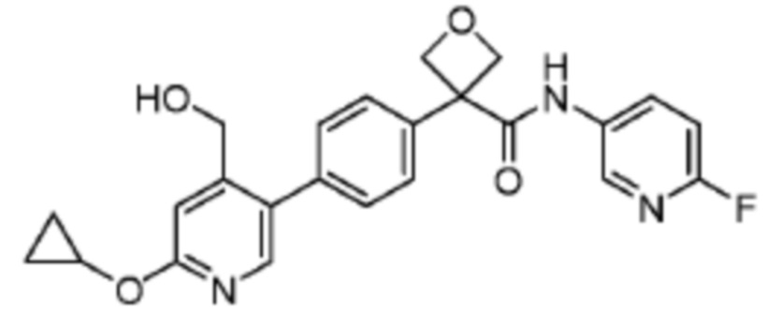
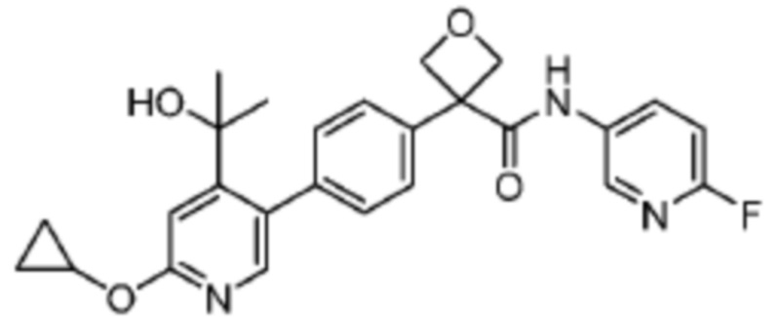
Пример 118: 3-фтор-N-(4-фторфенил)-1-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)циклобутанкарбоксамид
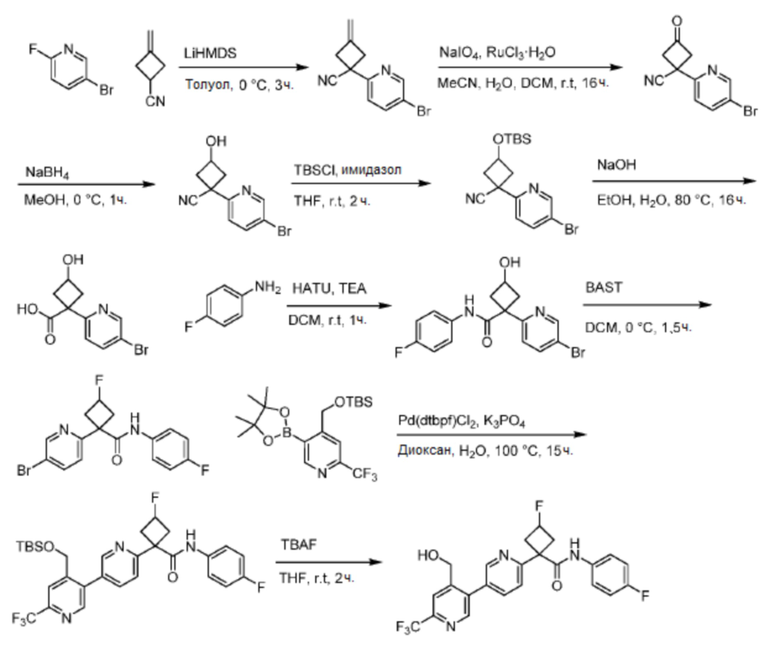
Стадия 1: 1-(5-бромпиридин-2-ил)-3-метиленциклобутанкарбонитрил
К раствору 5-бром-2-фторпиридина (10 г, 56,8 ммоль) и 3-метиленциклобутанкарбонитрила (5,29 г, 56,8 ммоль) в толуоле (50 мл) добавляли LiHMDS (31,3 мл, 62,5 ммоль) при 0°C и перемешивали смесь при 0°C в течение 3 ч. Реакционную смесь разводили водным раствором NH4Cl (200 мл), экстрагировали с помощью EtOAc (100 мл, 2 раза), собирали органические соединения и промывали соляным раствором, сушили над безводным Na2SO4, а затем концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла.
Стадия 2: 1-(5-бромпиридин-2-ил)-3-оксоциклобутанкарбонитрил
К перемешиваемому раствору 1-(5-бромпиридин-2-ил)-3-метиленциклобутанкарбонитрила (500 мг, 2,007 ммоль) в ACN (4 мл), DCM (4 мл) и воде (6 мл) частями добавляли гидрат хлорида рутения (III) (46 мг, 0,204 ммоль) и NaIO4 (2147 мг, 10,04 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 16 ч. Затем реакцию гасили посредством добавления насыщенного раствора Na2S2O3 (50 мл) и экстрагировали получаемую смесь с помощью EtOAc (100 мл, 2 раза). Органические слои собирали, промывали соляным раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 292,0; 294,0 [M+ACN+H+].
Стадия 3: 1-(5-бромпиридин-2-ил)-3-гидроксициклобутанкарбонитрил
К раствору 1-(5-бромпиридин-2-ил)-3-оксоциклобутанкарбонитрила (430 мг, 1,713 ммоль) в MeOH (10 мл) добавляли NaBH4 (130 мг, 3,43 ммоль) при 0°C и перемешивали смесь при 0°C в течение 1 ч. Реакционную смесь осторожно разводили водой (50 мл) и экстрагировали с помощью EtOAc (20 мл, 2 раза). Собирали органические соединения и промывали соляным раствором, сушили над безводным Na2SO4, фильтровали и концентрировали фильтрат при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле для получения соответствующего спирта в виде твердого вещества.
Стадия 4: 1-(5-бромпиридин-2-ил)-3-((трет-бутилдиметилсилил)окси)циклобутанкарбонитрил
К раствору 1-(5-бромпиридин-2-ил)-3-гидроксициклобутанкарбонитрила (300 мг, 1,185 ммоль) в THF (10 мл) добавляли имидазол (161 мг, 2,371 ммоль) и трет-бутилхлордиметилсилан (214 мг, 1,422 ммоль) при RT. Смесь перемешивали при RT в течение 2 ч. Реакционную смесь разводили водой (100 мл) и экстрагировали с помощью EtOAc (50 мл, 2 раза). Собирали органические соединения и промывали соляным раствором (10 мл), сушили над безводным Na2SO4, фильтровали и концентрировали фильтрат при пониженном давлении. Остаток очищали посредством препаративной TLC для получения титульного соединения в виде масла.
Стадия 5: 1-(5-бромпиридин-2-ил)-3-гидроксициклобутанкарбоновая кислота
К перемешиваемому раствору 1-(5-бромпиридин-2-ил)-3-((трет-бутилдиметилсилил)окси)циклобутанкарбонитрила (200 мг, 0,544 ммоль) в EtOH (5 мл) и воде (2,5 мл) добавляли NaOH (87 мг, 2,178 ммоль) при RT. Реакционную смесь нагревали до 80°C при перемешивании в течение 16 ч. Растворитель удаляли посредством концентрирования под вакуумом и остаток растворяли в EtOAc. По каплям добавляли водный раствор 3 Н HCl для доведения pH до ~2 и смесь экстрагировали с помощью EtOAc (20 мл, 3 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4 и после фильтрации концентрировали фильтрат для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 272,0, 274,0 [M+H+].
Стадия 6: 1-(5-бромпиридин-2-ил)-N-(4-фторфенил)-3-гидроксициклобутанкарбоксамид
К перемешиваемому раствору 1-(5-бромпиридин-2-ил)-3-гидроксициклобутанкарбоновой кислоты (70 мг, 0,257 ммоль) и HATU (293 мг, 0,772 ммоль) в DCM (2 мл) добавляли TEA (0,2 мл, 1,435 ммоль) и 4-фторанилин (35 мг, 0,315 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 1 ч. Реакционную смесь разводили водой (50 мл), экстрагировали с помощью EtOAc (20 мл, 3 раза), органические слои собирали, промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством препаративной TLC для получения титульного соединения в виде масла. MS (ESI) m/z 365,1, 367,1 [M+H+].
Стадия 7: 1-(5-бромпиридин-2-ил)-3-фтор-N-(4-фторфенил)циклобутанкарбоксамид
К перемешиваемому раствору 1-(5-бромпиридин-2-ил)-N-(4-фторфенил)-3-гидроксициклобутанкарбоксамида (800 мг, 2,191 ммоль) в DCM (20 мл) добавляли DAST (0,29 мл, 2,195 ммоль) при 0°C и перемешивали реакционную смесь при RT в течение 1,5 ч. Реакцию гасили посредством добавления насыщенного раствора NaHCO3 (20 мл) и экстрагировали с помощью DCM (10 мл, 2 раза). Органические слои собирали, промывали соляным раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством хроматографии на силикагеле для получения титульного соединения в виде твердого вещества. MS (ESI) m/z 366,9[M+H+].
Стадия 8: 1-(4'-(((трет-бутилдиметилсилил)окси)метил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)-3-фтор-N-(4-фторфенил)циклобутанкарбоксамид
К перемешиваемому раствору 4-(((трет-бутилдиметилсилил)окси)метил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридина (273 мг, 0,654 ммоль) в 1,4-диоксане (5 мл) и воде (1 мл) добавляли K3PO4 (347 мг, 1,634 ммоль), Pd(dtbpf)Cl2 (36 мг, 0,055 ммоль) и 1-(5-бромпиридин-2-ил)-3-фтор-N-(4-фторфенил)циклобутанкарбоксамид (200 мг, 0,545 ммоль) при RT. Смесь подвергали реакции сочетания Сузуки в стандартных условиях и обрабатывали для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 578,2 [M+H+].
Стадия 9: 3-фтор-N-(4-фторфенил)-1-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)циклобутанкарбоксамид
К раствору 1-(4'-(((трет-бутилдиметилсилил)окси)метил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)-3-фтор-N-(4-фторфенил)циклобутанкарбоксамида (50 мг, 0,087 ммоль) в THF (2 мл) добавляли TBAF (0,17 мл, 0,170 ммоль) (1 M в THF) при RT. Смесь перемешивали при RT в течение 2 ч. Удаляли растворитель и очищали остаток посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (150×30 мм×4 мкм), с использованием воды (0,1% TFA)/ACN в качестве элюентов. Титульное соединение получали в виде твердого вещества после лиофилизации желаемых фракций. 1H ЯМР (500 МГц, CD3OD) δ 8,71 (д, 1H), 8,63 (с, 1H), 8,11 (с, 1H), 7,97 (дд, 1H), 7,62 (д, 1H), 7,53-7,58 (м, 2H), 7,04-7,09 (м, 2H), 5,09-5,30 (м, 1H), 4,68 (с, 2H), 3,36-3,43 (м, 2H), 2,89-3,01 (м, 2H). MS (ESI) m/z 464,1 [M+H+]
Пример 119: 4-фтор-N-(1-(7-гидрокси-5-(2-метилпиримидин-4-ил)-5,6,7,8-тетрагидро-1,5-нафтиридин-2-ил)циклопропил)бензамид
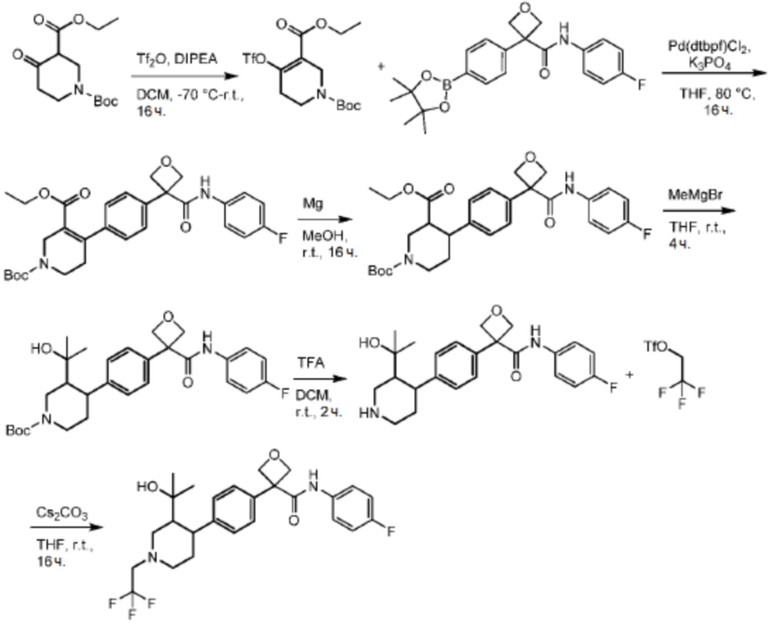
Стадия 1: 1-трет-бутил-3-этил-4-(((трифторметил)сульфонил)окси)-5,6-дигидропиридин-1,3(2H)-дикарбоксилат
К перемешиваемому раствору 1-трет-бутил-3-этил-4-оксопиперидин-1,3-дикарбоксилата (4,0 г, 14,74 ммоль) и DIPEA (4,76 г, 36,9 ммоль) в DCM (60 мл) по каплям добавляли Tf2O (3,2 мл, 18,94 ммоль) при -70°C и перемешивали реакционную смесь при -70°C в атмосфере азота в течение 3 ч. Смесь разводили водой (40 мл) и экстрагировали с помощью DCM (20 мл, 3 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде масла.
Стадия 2: 1-трет-бутил 3-этил 4-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-5,6-дигидропиридин-1,3(2H)-дикарбоксилат
К перемешиваемому раствору 1-трет-бутил-3-этил-4-(((трифторметил)сульфонил)окси)-5,6-дигидропиридин-1,3(2H)-дикарбоксилата (1,218 г, 3,02 ммоль) и N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамида (1,0 г, 2,52 ммоль) в THF (18 мл) добавляли фосфат калия (1,603 г, 7,55 ммоль) и Pd(dtbpf)Cl2(9 мг, 0,014 ммоль) при RT. Реакционную смесь перемешивали при 80°C в атмосфере азота в течение 16 ч. После охлаждения до RT реакционную смесь разводили водой (80 мл) и экстрагировали с помощью EtOAc (100 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4 и после фильтрации концентрировали фильтрат под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения в виде твердого вещества.
Стадия 3: 1-трет-бутил-3-этил-4-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)пиперидин-1,3-дикарбоксилат
К перемешиваемому раствору 1-трет-бутил-3-этил 4-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-5,6-дигидропиридин-1,3(2H)-дикарбоксилата (1,1 г, 2,097 ммоль) в MeOH (20 мл) добавляли магний (0,153 г, 6,29 ммоль) при RT. Смесь перемешивали при RT в течение 16 ч. Реакционную смесь фильтровали и промывали осадок MeOH (10 мл, 2 раза). Фильтраты комбинировали и концентрировали под вакуумом. Остаток разводили EtOAc (30 мл), промывали HCl (20 мл, 2 M) и соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 549,0 [M+Na+].
Стадия 4: Трет-бутил-4-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-3-(2-гидроксипропан-2-ил)пиперидин-1-карбоксилат
К перемешиваемому раствору бромида метилмагния (2,215 мл, 6,65 ммоль, 3 M в Et2O) в THF (5 мл) по каплям добавляли 1-трет-бутил-3-этил-4-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)пиперидин-1,3-дикарбоксилата (1,0 г, 1,899 ммоль) в THF (20 мл) при RT. Реакционную смесь перемешивали при RT в течение 4 ч. Реакционную смесь разводили водой (20 мл) и экстрагировали с помощью EtOAc (10 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом. Остаток очищали посредством хроматографии на силикагеле для получения титульного соединения в виде масла. MS (ESI) m/z 513,1 [M+H+].
Стадия 5: N-(4-фторфенил)-3-(4-(3-(2-гидроксипропан-2-ил)пиперидин-4-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору трет-бутил-4-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-3-(2-гидроксипропан-2-ил)пиперидин-1-карбоксилата (310 мг, 0,605 ммоль) в DCM (5 мл) добавляли TFA (3 мл, 38,9 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 2 ч. Реакционную смесь разводили насыщенным раствором NaHCO3 (40 мл) и экстрагировали с помощью DCM (15 мл, 2 раза). Органические слои собирали, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали фильтрат под вакуумом для получения титульного соединения в виде масла, которое использовали на следующей стадии без последующей очистки. MS (ESI) m/z 413,1 [M+H+].
Стадия 6: N-(4-фторфенил)-3-(4-(3-(2-гидроксипропан-2-ил)-1-(2,2,2-трифторэтил)пиперидин-4-ил)фенил)оксетан-3-карбоксамид
К перемешиваемому раствору N-(4-фторфенил)-3-(4-(3-(2-гидроксипропан-2-ил)пиперидин-4-ил)фенил)оксетан-3-карбоксамида (210 мг, 0,509 ммоль) в CH3CN (10 мл) добавляли 2,2,2-трифторэтил трифторметансульфонат (236 мг, 1,018 ммоль) и Cs2CO3 (332 мг, 1,018 ммоль) при RT. Реакционную смесь перемешивали при RT в течение 2 ч. После фильтрации реакционную смесь очищали посредством обращенно-фазовой ВЭЖХ с помощью устройства GILSON 281, оборудованного колонкой Phenomenex Synergi C18 (150×30 мм×4 мкм), с использованием воды (0,1% TFA) и CH3CN в качестве элюентов для получения пика 1, изомера 1 в виде твердого вещества. При последующей элюции получали пик 2, изомер 2 в виде твердого вещества. MS (ESI) m/z 495,0 [M+H+].
Изомер 1 дополнительно разрешали посредством разделения с помощью SFC с использованием колонки Phenomenex-Amylose-1 (250 мм×30 мм×5 мкм) для получения примера 119a (энантиомер 1, пик 1) и примера 119b (энантиомер 2, пик 2).
Пример 119a: 1H ЯМР (400 МГц, CD3OD) δ 7,50 (дд, 2H), 7,44 (д, 2H), 7,34 (д, 2H), 7,03 (т, 2H), 5,29 (д, 2H), 4,94 (д, 2H), 3,75 (ш.с., 3H), 3,61-3,62 (м, 1H), 3,34-3,40 (м, 1H), 2,62-2,96 (м, 3H), 2,23-2,34 (м, 1H), 1,81-2,04 (м, 2H), 1,06 (с, 3H), 0,70 (с, 3H). MS (ESI) m/z 495,1 [M+H+].
Пример 119b: 1H ЯМР (400 МГц, CD3OD) δ 7,50 (дд, 2H), 7,44 (д, 2H), 7,34 (д, 2H), 7,03 (т, 2H), 5,29 (д, 2H), 4,94 (д, 2H), 3,75 (ш.с., 3H), 3,61-3,62 (м, 1H), 3,34-3,40 (м, 1H), 2,62-2,96 (м, 3H), 2,23-2,34 (м, 1H), 1,81-2,04 (м, 2H), 1,06 (с, 3H), 0,70 (с, 3H). MS (ESI) m/z 495,1 [M+H+].
Изомер 2 аналогично разрешали посредством разделения с помощью SFC с использованием колонки Phenomenex-Amylose-1 (250 мм×30 мм×5мкм) для получения энантиомера 2-1 и энантиомера 2-2.
Пример 119c: 1H ЯМР (400 МГц, CD3OD) δ 7,42-7,56 (м, 6H), 7,04 (т, 2H), 5,30 (дд, 2H), 4,94 (ш.т., 2H), 3,71-4,04 (м, 3H), 3,41-3,61 (м, 2H), 3,35 (ш.с., 1H), 3,17-3,25 (м, 1H), 2,16-2,58 (м, 3H), 1,21 (с, 3H), 0,71 (с, 3H). MS (ESI) m/z 495,1 [M+H+].
Пример 119d: 1H ЯМР (400 МГц, CD3OD) δ 7,42-7,58 (м, 6H), 7,04 (т, 2H), 5,30 (дд, 2H), 4,94 (т, 2H), 3,75-4,10 (м, 3H), 3,53 (ш.с., 2H), 3,34 (ш.с., 1H), 3,20-3,28 (м, 1H), 2,17-2,58 (м, 3H), 1,22 (с, 3H), 0,71 (с, 3H). MS (ESI) m/z 495,1 [M+H+].
Пример 120: N -(4-фторфенил)-3-(4-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
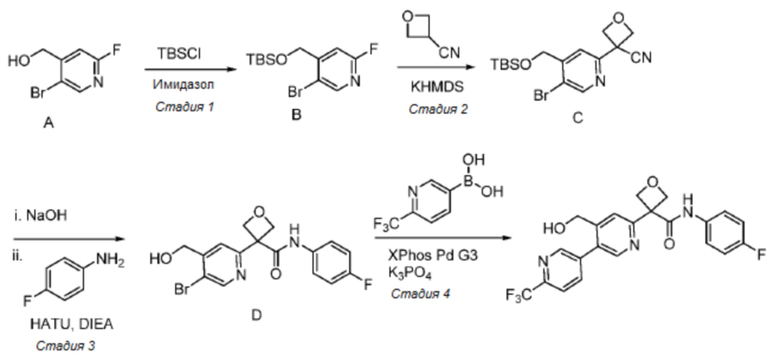
Стадия 1: 5-бром-4-(((трет-бутилдиметилсилил)окси)метил)-2-фторпиридин (B)
В сосуд с якорем магнитной мешалки добавляли (5-бром-2-фторпиридин-4-ил)метанол (A) (500 мг, 2,43 ммоль), DMF (4,9 мл), имидазол (363 мг, 5,34 ммоль) и трет-бутилдиметилсилилхлорид (402 мг, 2,67 ммоль). Смесь перемешивали при RT в течение 63 ч. Затем реакционную смесь разводили насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc. Затем органический слой отделяли, промывали соляным раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии для получения титульного соединения. MS (ESI) m/z 320, 322 [M+H]+.
Стадия 2: 3-(5-бром-4-(((трет-бутилдиметилсилил)окси)метил)пиридин-2-ил)оксетан-3-карбонитрил (C)
В круглодонную колбу объемом 50 мл с якорем магнитной мешалки в атмосфере азота добавляли 5-бром-4-(((трет-бутилдиметилсилил)окси)метил)-2-фторпиридин (B) (712 мг, 2,22 ммоль), оксетан-3-карбонитрил (185 мкл, 2,45 ммоль) и толуол (18 мл). Колбу охлаждали до 0°C и по каплям добавляли KHMDS (2668 мкл, 2,67 ммоль, 1,0 M в THF) при перемешивании. Раствор перемешивали в течение 10 мин при 0°C, после чего реакцию гасили посредством медленного добавления MeOH при перемешивании. Полученную смесь разводили EtOAc и водой, водный слой экстрагировали с помощью EtOAc и объединенные органические соединения промывали соляным раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Затем остаток очищали посредством флэш-хроматографии для получения титульного соединения. MS (ESI): 383, 385 [M+H]+.
Стадия 3: 3-(5-бром-4-(гидроксиметил)пиридин-2-ил)-N-(4-фторфенил)оксетан-3-карбоксамид (D)
В сосуд с якорем магнитной мешалки добавляли 3-(5-бром-4-(((трет-бутилдиметилсилил)окси)метил)пиридин-2-ил)оксетан-3-карбонитрил (D) (410 мг, 1,07 ммоль), NaOH (1,07 мл, 1,07 ммоль, 1 Н в воде) и этанол (2,7 мл). Сосуд запаивали и нагревали до 75°C в течение 16 ч. Затем реакционную смесь охлаждали и добавляли EtOAc (4 мл), затем по каплям добавляли HCl (1 Н в воде) для доведения pH до ~2. Затем реакционную смесь дополнительно разводили водой и экстрагировали с помощью EtOAc. Объединенные органические соединения промывали соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении для получения 3-(5-бром-4-(гидроксиметил)пиридин-2-ил)оксетан-3-карбоновой кислоты (в условиях реакции наблюдали потерю группы TBS).
В сосуд с якорем магнитной мешалки добавляли 3-(5-бром-4-(гидроксиметил)пиридин-2-ил)оксетан-3-карбоновую кислоту (308 мг, 1,069 ммоль, неочищенный материал, полученный на предыдущей стадии), HATU (610 мг, 1,604 ммоль) и DMF (5345 мкл). Затем добавляли 4-фторанилин (304 мкл, 3,21 ммоль), а затем DIEA (560 мкл, 3,21 ммоль). Реакционную смесь перемешивали при RT в течение 48 ч. Затем добавляли воду и EtOAc и экстрагировали смесь с помощью EtOAc. Объединенные органические соединения промывали соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Затем остаток очищали посредством флэш-хроматографии (силикагель, элюировали в градиенте 0-100% EtOAc в гексанах) для получения 3-(5-бром-4-(гидроксиметил)пиридин-2-ил)-N-(4-фторфенил)оксетан-3-карбоксамида. MS (ESI): 381, 383 [M+H]+.
Стадия 4: N-(4-фторфенил)-3-(4-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
В сосуд с якорем магнитной мешалки помещали 3-(5-бром-4-(гидроксиметил)пиридин-2-ил)-N-(4-фторфенил)оксетан-3-карбоксамид (65 мг, 0,17 ммоль), (6-(трифторметил)пиридин-3-ил)бороновую кислоту (48,8 мг, 0,256 ммоль) и Xphos Pd G3 (14,4 мг, 0,017 ммоль). Затем сосуд запаивали и выкачивали и помещали обратно с аргоном (3 раза). Затем добавляли THF (3,4 мл) и ортофосфат калия (512 мкл, 0,512 ммоль, 1 M водный раствор) и нагревали реакционную смесь до 50°C в течение 1 ч., а затем до 80°C в течение 1 ч. Затем реакционную смесь охлаждали до RT и разводили EtOAc и водой. Реакционную смесь экстрагировали с помощью EtOAc и объединенные органические соединения промывали соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в DMSO и очищали посредством препаративной обращенно-фазовой ВЭЖХ (от 5:95 до 95:5 ацетонитрил:вода: 0,1% об./об. модификатора TFA) для получения титульного соединения в виде соли TFA. 1H ЯМР (600 МГц, DMSO-d6) δ 10,11 (с, 1H), 8,87 (д, J=1,9 Гц, 1H), 8,58 (с, 1H), 8,22 (дд, J=8,1, 2,0 Гц, 1H), 8,04 (д, J=8,0 Гц, 1H), 7,81 (с, 1H), 7,67-7,71 (м, 2H), 7,24-7,08 (м, 2H), 5,20 (д, J=6,4 Гц, 2H), 5,05 (д, J=6,4 Гц, 2H), 4,51 (с, 2H). MS (ESI): 448 [M+H]+.
Пример 121: 3-(4-(6-(2,2-дифторэтокси)-4-(гидроксиметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
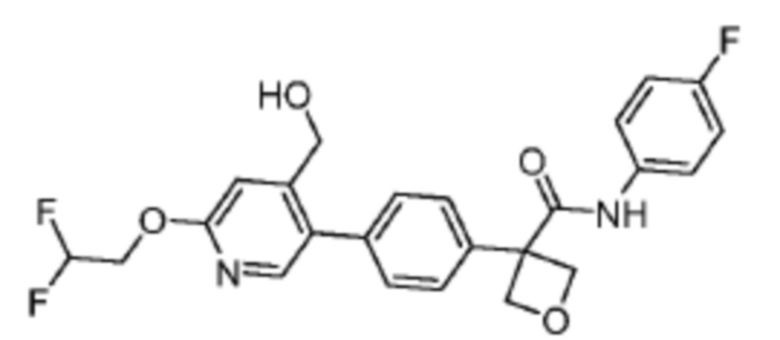
В сосуд добавляли N-(4-фторфенил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-карбоксамид (50 мг, 0,12 ммоль), (5-бром-2-(2,2-дифторэтокси)пиридин-4-ил)метанол (38,5 мг, 0,143 ммоль), XPhos Pd G3 (10 мг, 0,012 ммоль), THF (600 мкл) и K3PO4 (1M) (250 мкл, 0,250 ммоль). Смесь выкачивали и помещали обратно с азотом 4 раза и нагревали при 45°C в течение 2 ч. Смесь фильтровали через целитовую прокладку. Фильтрат разводили водой и EtOAc, переносили на делительную воронку. Органический слой промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения остатка, который очищали посредством хроматографии на колонках с силикагелем для получения титульного соединения. 1H ЯМР (600 МГц, DMSO-d6) δ 10,05 (с, 1H), 8,00 (с, 1H), 7,72-7,61 (м, 2H), 7,57 (д, 2H), 7,43 (д, 2H), 7,16 (т, 2H), 7,06 (с, 1H), 6,41 (т, 1H), 5,23 (д, 2H), 4,91 (д, 2H), 4,70-4,50 (м, 2H), 4,44 (с, 2H). MS (EI) m/z 459 [M+H]+.
Примеры 122-124 в следующей таблице получали аналогично примеру 121.
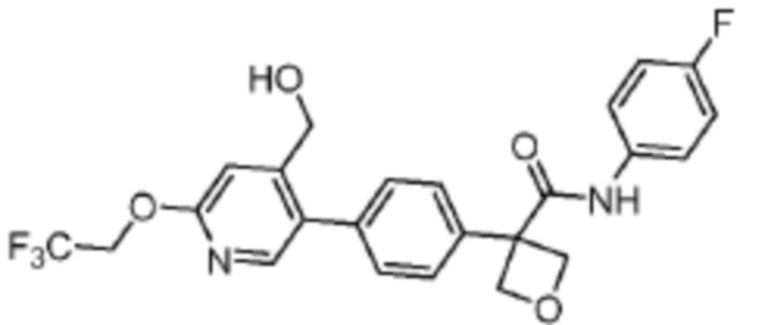
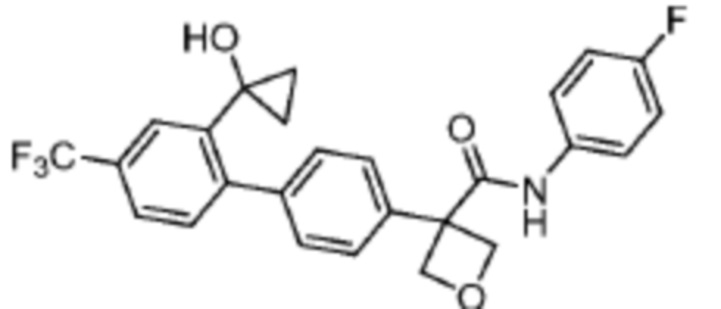
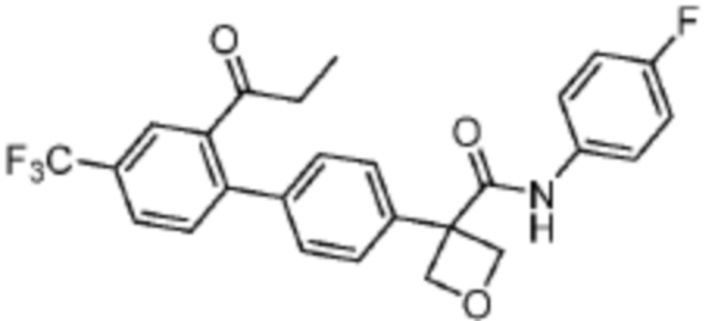
Пример 125: N-(4-фторфенил)-3-(2'-(гидроксиметил)-4'-(трифторметокси)-[1,1'-бифенил]-4-ил)оксетан-3-карбоксамид
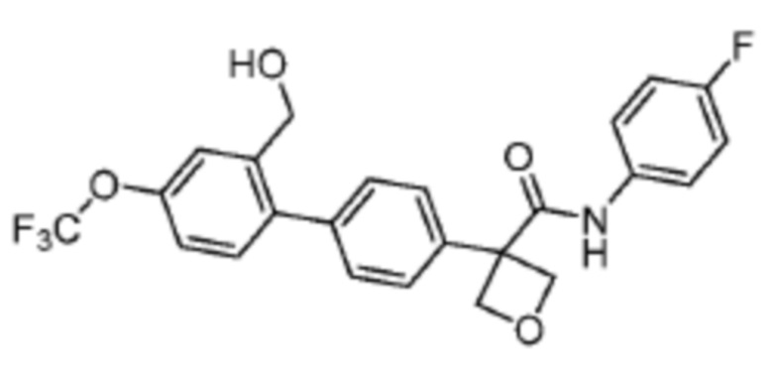
Стадия 1: (2-бром-5-(трифторметокси)фенил)метанол
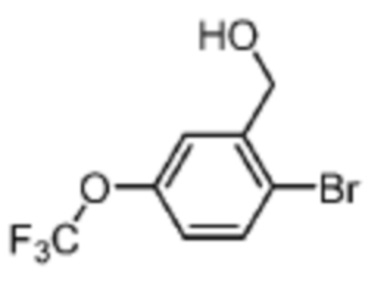
К перемешиваемому раствору метил-2-бром-5-(трифторметокси)бензоата (2,0 г, 6,7 ммоль) в THF (21 мл) и MeOH (7 мл) добавляли NaBH4 (1,26 г, 33,4 ммоль) при 0°C. Смесь перемешивали при RT в течение 1 ч., затем гасили NH4Cl (насыщенным раствором), разводили EtOAc и водой. Органическую фазу отделяли от водной фазы и снова экстрагировали водную фазу с помощью EtOAc. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и концентрировали под вакуумом для получения титульного соединения, которое использовали непосредственно на следующей стадии. 1H ЯМР (600 МГц, DMSO-d6) δ 7,72 (д, 1H), 7,46 (с, 1H), 7,30-7,16 (м, 1H), 5,68 (т, 1H), 4,52 (д, 2H).
Стадия 2: N-(4-фторфенил)-3-(2'-(гидроксиметил)-4'-(трифторметокси)-[1,1'-бифенил]-4-ил)оксетан-3-карбоксамид
Титульное соединение получали аналогично примеру 121. 1H ЯМР (600 МГц, DMSO-d6) δ 10,04 (с, 1H), 7,66 (ддт, 2H), 7,57 (д, 2H), 7,52 (с, 1H), 7,44 (д, 2H), 7,36 (д, 1H), 7,34-7,29 (м, 1H), 7,21-7,13 (м, 2H), 5,23 (д, 2H), 4,91 (д, 2H), 4,43 (с, 2H). MS (EI) m/z 462 [M+H]+.
Пример 126: 3-(3-фтор-4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
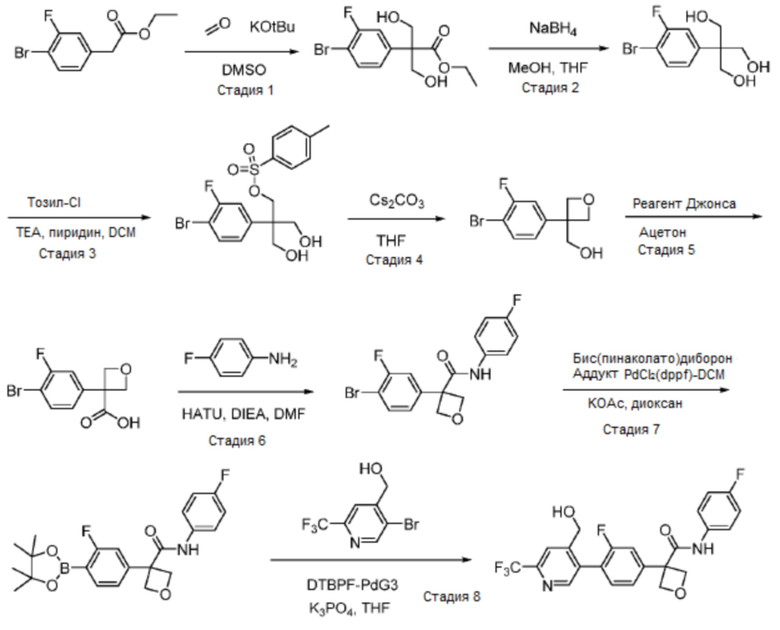
Стадия 1: Этил-2-(4-бром-3-фторфенил)-3-гидрокси-2-(гидроксиметил)пропаноат
В колбу помещали этил-2-(4-бром-3-фторфенил)ацетат (4,01 г, 15,4 ммоль), DMSO (40 мл) и формальдегид (1,84 г, 61,4 ммоль). К этой суспензии добавляли KOtBu (0,34 г, 3,07 ммоль) за один раз. Смесь перемешивали при RT в течение 30 мин, гасили 1 M HCl (водным раствором) и разводили водой. Смесь переносили на делительную воронку и экстрагировали с помощью EtOAc. Объединенный органический слой промывали соляным раствором, сушили над MgSO4, фильтровали и концентрировали под вакуумом для получения неочищенного продукта, который очищали посредством хроматографии на колонках с силикагелем для получения титульного соединения. MS (EI) m/z 321 [M+H]+.
Стадия 2: 2-(4-бром-3-фторфенил)-2-(гидроксиметил)пропан-1,3-диол
В колбу добавляли этил 2-(4-бром-3-фторфенил)-3-гидрокси-2-(гидроксиметил)-пропаноат (456,6 мг, 1,422 ммоль), MeOH (2000 мкл) и THF (6000 мкл). К этой смеси добавляли NaBH4 (300 мг, 7,93 ммоль) за один раз при 0°C. Смесь нагревали при 60°C в течение 2 ч., добавляли другую часть NaBH4 (172 мг, 4,55 ммоль) и продолжали нагревание при 60°C в течение еще 1 ч. Реакцию гасили 5 мл NH4Cl (насыщенным раствором) и 5 мл HCl (1 M) для достижения pH~2, затем экстрагировали с помощью EtOAc. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали, концентрировали под вакуумом для получения неочищенного продукта, который растворяли в 2 мл DMSO и 1 мл воды. Смесь перемешивали при RT в течение ночи, затем концентрировали под вакуумом, растворяли остаток в DCM и промывали NaHCO3, соляным раствором, сушили над MgSO4, фильтровали и концентрировали для получения титульного соединения. 1H ЯМР (600 МГц, DMSO-d6) δ 7,58 (т, 1H), 7,40 (дд, 1H), 7,22 (д, 1H), 4,51 (т, 3H), 3,67 (д, 6H).
Стадия 3: 2-(4-бром-3-фторфенил)-3-гидрокси-2-(гидроксиметил)пропил-4-метилбензолсульфонат
В сосуд, содержащий 2-(4-бром-3-фторфенил)-2-(гидроксиметил)пропан-1,3-диол (118 мг, 0,423 ммоль), добавляли DCM (3500 мкл) и TEA (200 мкл, 1,43 ммоль). К этому раствору добавляли раствор тозил-Cl (81 мг, 0,42 ммоль) в DCM (1000 мкл) при 0°C. Затем смесь перемешивали при RT в течение 24 ч. При LCMS определяли низкую степень превращения. К этой смеси добавляли пиридин (150 мкл, 1,86 ммоль) и другую партию тозил-Cl (20 мг, 0,11 ммоль). Смесь перемешивали при RT еще в течение 4 ч., разводили DCM, промывали NaHCO3 (насыщенным раствором), соляным раствором, сушили над MgSO4, фильтровали, концентрировали под вакуумом для получения титульного соединения, которое использовали непосредственно на следующей стадии. MS (EI) m/z 455 [M+Na]+.
Стадия 4: (3-(4-бром-3-фторфенил)оксетан-3-ил)метанол
В сосуд добавляли 2-(4-бром-3-фторфенил)-3-гидрокси-2-(гидроксиметил)пропил-4-метилбензолсульфонат (47,8 мг, 0,110 ммоль), THF (1000 мкл) и Cs2CO3 (108 мг, 0,331 ммоль). Смесь нагревали при 65°C в течение 18 ч., затем при 70°C в течение 3 ч. Смесь разводили водой и EtOAc. Водный слой экстрагировали с помощью EtOAc 3 раза. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения титульного соединения, которое использовали непосредственно на следующей стадии. MS (EI) m/z 261 [M+H]+.
Стадия 5: 3-(4-бром-3-фторфенил)оксетан-3-карбоновая кислота
В сосуд, содержащий (3-(4-бром-3-фторфенил)оксетан-3-ил)метанол (26,2 мг, 0,100 ммоль), добавляли ацетон (500 мкл) и реагент Джонса (110 мкл, 0,72 ммоль). Смесь перемешивали при RT в течение 5 ч., затем разводили EtOAc и водой. Органическую фазу промывали водой, соляным раствором, сушили над MgSO4, фильтровали и концентрировали под вакуумом для получения титульного соединения, которое использовали непосредственно на следующей стадии. MS (EI) m/z 275 [M+H]+.
Стадия 6: 3-(4-бром-3-фторфенил)-N-(4-фторфенил)оксетан-3-карбоксамид
В сосуд добавляли 3-(4-бром-3-фторфенил)оксетан-3-карбоновую кислоту (19,6 мг, 0,0710 ммоль), HATU (54,2 мг, 0,143 ммоль), DMF (800 мкл), 4-фторанилин (30 мг, 0,27 ммоль) и DIEA (100 мкл, 0,573 ммоль). Смесь перемешивали при RT в течение 18 ч, разводили EtOAc и промывали 1 M HCl, NaHCO3 (насыщенным раствором), соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения титульного соединения, которое использовали непосредственно на следующей стадии. MS (EI) m/z 368 [M+H]+.
Стадия 7: 3-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
В сосуд добавляли 3-(4-бром-3-фторфенил)-N-(4-фторфенил)оксетан-3-карбоксамид (26,2 мг, 0,0710 ммоль), бис(пинаколато)диборон (45,2 мг, 0,178 ммоль), ацетат калия (20,9 мг, 0,213 ммоль), аддукт PdCl2(dppf)-CH2Cl2 (5,81 мг, 7,12 мкмоль) и диоксан (700 мкл). Затем смесь выкачивали и помещали обратно с N2 3 раза. Смесь нагревали до 80°C в течение 20 ч., охлаждали до RT и фильтровали через целитовую прокладку. Фильтрат концентрировали под вакуумом для получения остатка, который растворяли в DCM, промывали водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения титульного соединения, которое использовали непосредственно на следующей стадии. MS (EI) m/z 416 [M+H]+.
Стадия 8: 3-(3-фтор-4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид
В сосуд добавляли 3-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамид (29,5 мг, 0,0710 ммоль), (5-бром-2-(трифторметил)пиридин-4-ил)метанол (35 мг, 0,14 ммоль), DTBPF-Pd G3 (8,8 мг, 10 мкмоль), THF (700 мкл) и K3PO4 (1 M, 200 мкл, 0,200 ммоль). Смесь выкачивали и помещали обратно с азотом 4 раза и нагревали при 50°C в течение 1,5 ч. Смесь фильтровали через целитовую прокладку, разводили водой и EtOAc и переносили на делительную воронку. Органический слой промывали водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения остатка, который очищали посредством хроматографии на колонках с силикагелем для получения титульного соединения.1H ЯМР (600 МГц, DMSO-d6) δ 10,08 (с, 1H), 9,97 (с, 1H), 8,63 (с, 1H), 8,05 (с, 1H), 7,65 (дд, 1H), 7,61-7,51 (м, 2H), 7,43 (д, 1H), 7,26-7,04 (м, 2H), 5,68 (т, 1H), 5,24 (д, 2H), 4,95 (д, 2H), 4,46 (д, 2H). MS (EI) m/z 465 [M+H]+.
Пример 127: N-(6-хлорпиридин-3-ил)-3-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
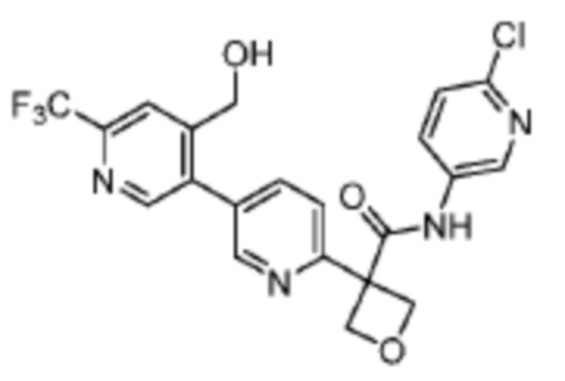
Стадия 1: 3-(5-бромпиридин-2-ил)-N-(6-хлорпиридин-3-ил)оксетан-3-карбоксамид
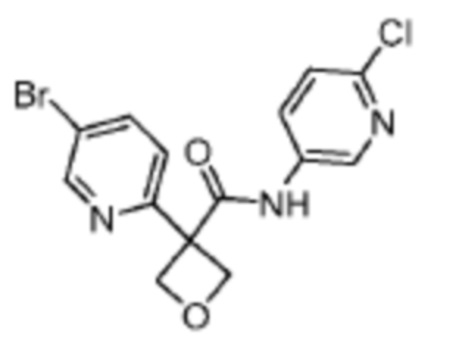
В сосуд с якорем магнитной мешалки добавляли 3-(5-бромпиридин-2-ил)оксетан-3-карбоновую кислоту (1023 мг, 3,96 ммоль), HATU (2261 мг, 5,95 ммоль) и DMF (9910 мкл). К этому добавляли 6-хлорпиридин-3-амин (612 мг, 4,76 ммоль), а затем DIEA (2077 мкл, 11,89 ммоль). Смесь перемешивали при 45°C в течение 2 ч., разводили EtOAc, и промывали насыщенным раствором NaHCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле для получения титульного соединения. MS (ESI) [M+H]+ m/z 368.
Стадия 2: Получение N-(6-хлорпиридин-3-ил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбоксамида
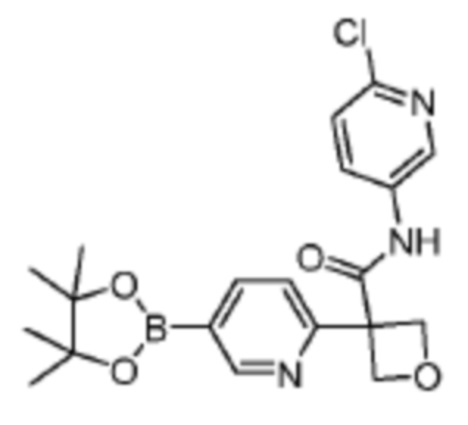
В сосуд с якорем магнитной мешалки добавляли 3-(5-бромпиридин-2-ил)-N-(6-хлорпиридин-3-ил)оксетан-3-карбоксамид (952 мг, 2,58 ммоль), бис(пинаколато)диборон (1640 мг, 6,46 ммоль), ацетат калия (761 мг, 7,75 ммоль) и аддукт PdCl2(dppf)-CH2Cl2 (211 мг, 0,26 ммоль) в диоксане (8612 мкл). Сосуд продували азотом в течение 5 мин, запаивали и нагревали до 80°C в течение 23 ч. После охлаждения до RT, смесь разводили EtOAc и промывали насыщенным раствором NaHCO3. Органические соединения сушили над MgSO4, фильтровали и сухими вводили в силикагель, который вводили в колонку с 80 г силикагеля. Колонку элюировали 100% DCM к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении для получения титульного соединения. MS (ESI): вычислено для C20H23BClN3O4 [M+H]+, 334; обнаружено, 334 (определена масса бороновой кислоты).
Стадия 3: Получение N-(6-хлорпиридин-3-ил)-3-(4'-(гидроксиметил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамида
В сосуд с якорем магнитной мешалки добавляли N-(6-хлорпиридин-3-ил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбоксамид (212 мг, 0,51 ммоль), (5-бром-2-(трифторметил)пиридин-4-ил)метанол (131 мг, 0,51 ммоль), ортофосфат калия (водный раствор, 1 M) (1020 мкл, 1,02 ммоль), метансульфонат (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия (II) (43,2 мг, 0,05 ммоль) и THF (2550 мкл). Сосуд продували азотом, запаивали и нагревали до 40°C в течение 1 ч. Через 1 ч. неочищенный продукт фильтровали через целитовую прокладку и промывали метанолом. Объединенные органические соединения концентрировали при пониженном давлении. Материал растворяли в EtOAc и сухим вводили в силикагель. Материал вводили в колонку 40 г с золотом; прогоняли от 100% DCM к 100% EtOAc. Элюировали желаемый продукт; фракции собирали и концентрировали при пониженном давлении. Препаративное разрешение получаемого материала осуществляли с использованием суперкритической жидкостной хроматографии с помощью Sepiatec Prep 100. В качестве неподвижной фазы использовали колонку ES Industries GreenSep с этилпиридином (5 мкм, 20 мм×250 мм, ES Industries, West Berlin, NJ). Смесь соединений растворяли в смеси 1:1:1 N, N-диметилформамида, метанола и ACN. Впрыскивание и сбор осуществляли с использованием следующих условий изократической SFC: 80% диоксида углерода и 20% метанола с 0,25% диметилэтиламина в качестве подвижной фазы, длина волны УФ 245 нм, давление на выходе 100 бар, температура компартментов колонки 40°C, общая скорость потока 70 мл/мин. Время удержания для сбора пиков составляло 2,7 мин. MS (ESI): вычислено для C21H16ClF3N4O3 [M+H]+, 465; обнаружено, 465, 1H ЯМР (600 МГц, DMSO-d6) δ 10,45 (с, 1H), 8,79 (д, J=1,7 Гц, 1H), 8,71 (с, 1H), 8,70 (д, J=2,7 Гц, 1H), 8,19 (дд, J=8,7, 2,8 Гц, 1H), 8,09 (с, 1H), 8,08 (д, J=2,4 Гц, 1H), 7,79-7,73 (м, 1H), 7,53 (д, J=8,7 Гц, 1H), 5,75 (т, J=5,5 Гц, 1H), 5,16 (дд, J=91,1, 6,4 Гц, 4H), 4,61 (д, J=5,4 Гц, 2H).
Пример 128: N-(4-фторфенил)-3-(5-(2-(гидроксиметил)-4-(трифторметил)фенил)пиридин-2-ил)оксетан-3-карбоксамид
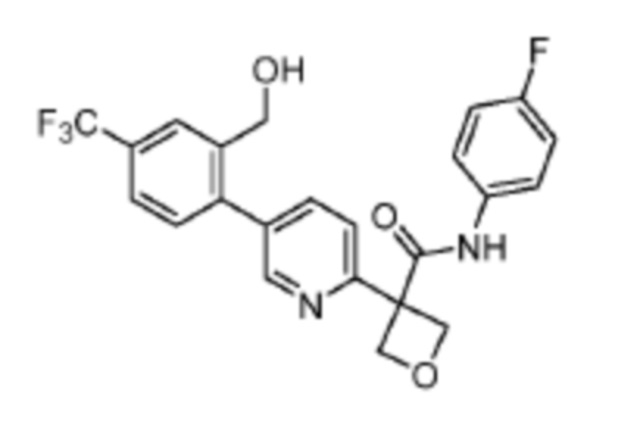
В сосуд с якорем магнитной мешалки добавляли N-(4-фторфенил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбоксамид (I-155; см. получение в примере 76) (500 мг, 1,26 ммоль), (2-бром-5-(трифторметил)фенил)метанол (320 мг, 1,26 ммоль), ортофосфат калия (водный раствор, 1 M) (2511 мкл, 2,51 ммоль), метансульфонат (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия (II) и THF (1,26E+04 мкл). Сосуд продували азотом, запаивали и нагревали до 40°C в течение 1,5 ч. Через 1,5 ч. неочищенный продукт промывали EtOAc и насыщенным раствором NaHCO3. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Материал сухим вводили в колонку 120 г и прогоняли от 100% DCM к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении. Материал добавляли в сосуд и медленно, по каплям добавляли ACN. Практически сразу твердое вещество распадалось, и получали жидкость. По каплям добавляли еще ACN, перемешивали посредством вращения, и твердое вещество не попадало в раствор. Смесь нагревали с помощью фена для получения однородного раствора. Смесь оставляли на 3,5 ч. Через 3,5 ч. образец переносили в холодильную камеру на 12 ч. Через 12 ч. смесь промывали холодным (охлажденным на ледяной бане) ACN и получали титульное соединение в виде твердого вещества. MS (ESI) вычислено для C23H18F4N2O3 [M+H]+, 447; обнаружено, 447. 1H ЯМР (600 МГц, DMSO-d6) δ 10,14 (с, 1H), 8,75 (д, J=1,7 Гц, 1H), 8,01 (дд, J=8,2, 2,4 Гц, 1H), 7,97 (с, 1H), 7,78 (д, J=6,9 Гц, 1H), 7,75-7,70 (м, 2H), 7,63 (дд, J=60,7, 8,0 Гц, 2H), 7,25-7,17 (м, 2H), 5,51 (т, J=5,4 Гц, 1H), 5,15 (дд, J=76,2, 6,4 Гц, 4H), 4,52 (д, J=5,4 Гц, 2H).
Пример 129: N-(6-хлорпиридин-3-ил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
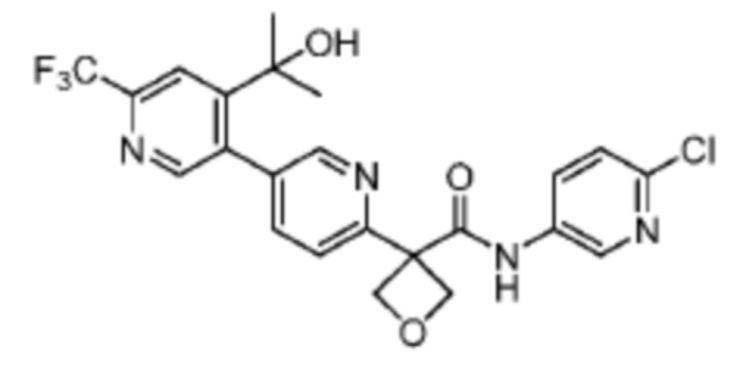
Стадия 1: 3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбонитрил
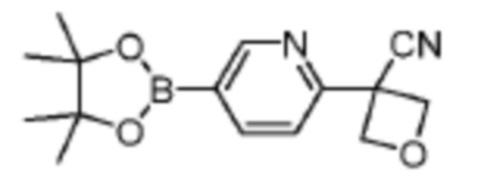
В колбу с якорем магнитной мешалки добавляли 3-(5-бромпиридин-2-ил)оксетан-3-карбонитрил (2 г, 8,37 ммоль), бис(пинаколато)диборон (4,25 г, 16,73 ммоль), ацетат калия (2,46 г, 25,10 ммоль) и аддукт PdCl2(dppf)-CH2Cl2 (0,68 г, 0,84 ммоль). Смесь продували азотом в течение 5 мин. Добавляли диоксан (41,8 мл) и перемешивали смесь. Полученную смесь нагревали до 80°C при перемешивании в атмосфере азота в течение 24 ч. Через 24 ч. неочищенную реакционную смесь фильтровали через целитовую прокладку и промывали EtOAc. Объединенные органические соединения концентрировали при пониженном давлении. Остаток промывали EtOAc и водой. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в DCM и вводили в колонку 120 г с золотом. Колонку прогоняли от 100% DCM к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении для получения титульного соединения. MS (ESI): вычислено для C15H19BN2O3[M+H]+, 205; обнаружено, 205 (определяли массу бороновой кислоты).
Стадия 2: Получение 3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбонитрила
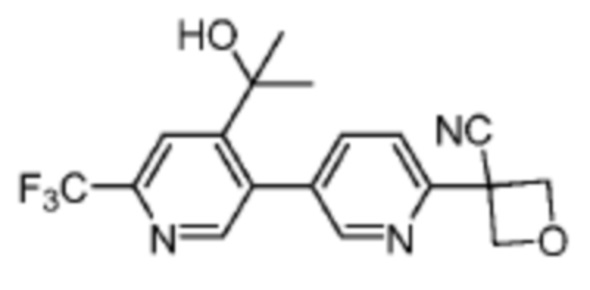
В круглодонную колбу с якорем магнитной мешалки (в атмосфере азота) добавляли 3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбонитрил (1,72 г, 6,01 ммоль), 2-(5-бром-2-(трифторметил)пиридин-4-ил)пропан-2-ол (1,88 г, 6,61 ммоль), ортофосфат калия (водный раствор, 1 M) (12,02 мл, 12,02 ммоль), дихлорид 1,1'-бис(ди-трет-бутилфосфино)ферроцен-палладия (0,39 г, 0,60 ммоль) и диоксан (10,02 мл). Сосуд запаивали с перегородкой и продували азотом при нагревании до 65°C в течение 4 ч. Через 4 ч. реакционную смесь охлаждали до RT. Реакционную смесь фильтровали через целитовую прокладку и промывали EtOAc. Объединенные органические соединения концентрировали при пониженном давлении и промывали EtOAc и водой; объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученную смесь вводили в колонку 80 г с золотом; колонку прогоняли от 100% DCM к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении для получения титульного соединения. MS (ESI): вычислено для C18H16F3N3O2[M+H]+, 364; обнаружено, 364.
Стадия 3: Получение 3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоновой кислоты
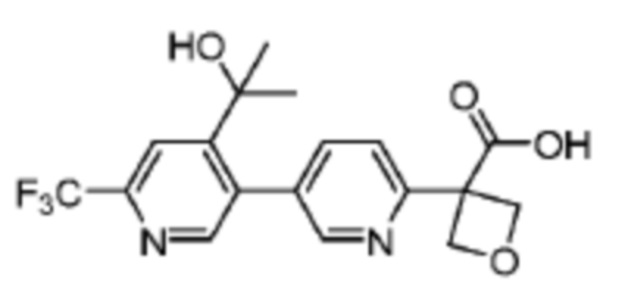
В круглодонную колбу с якорем магнитной мешалки добавляли 3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбонитрил (932 мг, 2,57 ммоль), NaOH (410 мг, 10,26 ммоль), этанол (8550 мкл) и воду (4275 мкл). Колбу запаивали и нагревали до 65°C в течение 2 ч. Через 2 ч. реакционную смесь охлаждали до RT и концентрировали при пониженном давлении. Реакционную смесь растворяли в EtOAc и по каплям добавляли 1 н. HCl для достижения pH ~2. Смесь промывали водой и EtOAc. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении для получения титульного соединения. MS (ESI): вычислено для C18H17F3N2O4[M+H]+, 383; обнаружено, 383.
Стадия 4: Получение N-(6-хлорпиридин-3-ил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамида
В сосуд с якорем магнитной мешалки добавляли 3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоновую кислоту (34,5 мг, 0,09 ммоль), HATU (51,5 мг, 0,14 ммоль) и DMF (902 мкл). Смесь перемешивали в течение 5 мин. Добавляли 6-хлорпиридин-3-амин (58,0 мг, 0,45 ммоль), а затем DIEA (47,3 мкл, 0,27 ммоль). Смесь перемешивали при 45°C в течение 48 ч. Через 48 ч. смесь сухой вводили в колонку 24 г с золотом; колонку прогоняли от 100% DCM к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении. Смесь растворяли в ACN/воде, затем лиофилизировали в течение ночи. Полученную смесь растворяли в 1,5 мл DMSO и сразу подвергали очистке посредством ВЭЖХ (очистка посредством ВЭЖХ, элюирующий градиент ACN/вода с модификатором TFA, линейный градиент) для получения титульного соединения. MS (ESI): вычислено для C23H20ClF3N4O3 [M+H]+, 493; обнаружено, 493. 1H ЯМР (600 МГц, DMSO-d6) δ 10,51 (с, 1H), 8,71 (д, J=2,7 Гц, 1H), 8,66 (д, J=1,8 Гц, 1H), 8,46 (с, 1H), 8,21 (д, J=2,8 Гц, 1H), 8,20 (д, J=2,7 Гц, 2H), 7,94 (дд, J=8,1, 2,3 Гц, 1H), 7,70 (д, J=8,0 Гц, 1H), 7,55 (д, J=8,7 Гц, 1H), 5,16 (дд, J=94,6, 6,4 Гц, 4H), 1,35 (с, 6H).
Примеры 130-131 в следующей таблице получали аналогично примеру 129.
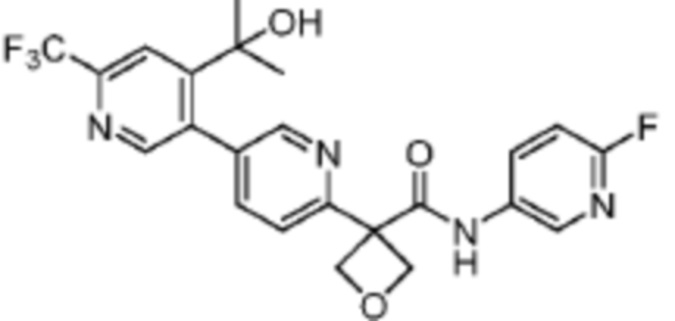
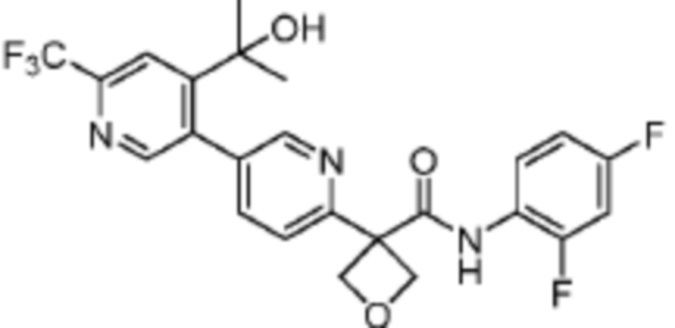
Пример 132: N-(4-хлорфенил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
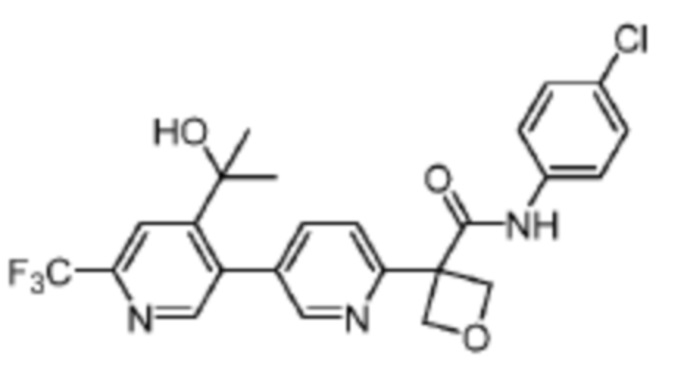
Стадия 1: 3-(5-бромпиридин-2-ил)-N-(4-хлорфенил)оксетан-3-карбоксамид
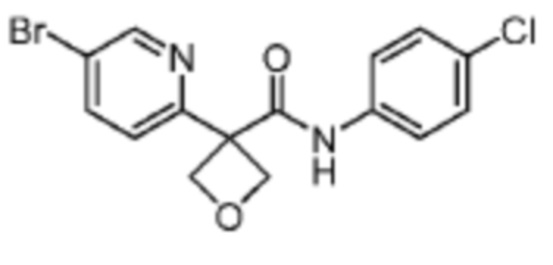
В сосуд с якорем магнитной мешалки добавляли 3-(5-бромпиридин-2-ил)оксетан-3-карбоновую кислоту (500 мг, 1,94 ммоль), HATU (1105 мг, 2,91 ммоль) и DMF (4844 мкл). Добавляли 4-хлоранилин (297 мг, 2,33 ммоль), а затем DIEA (1015 мкл, 5,81 ммоль). Смесь перемешивали при 45°C в течение 4 ч. Через 4 ч. неочищенный продукт промывали EtOAc и насыщенным раствором NaHCO3. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Материал сухим вводили в колонку 120 г с золотом и колонку прогоняли от 100% гексанов к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении для получения титульного соединения. MS (ESI): вычислено для C15H12BrClN2O2[M+H]+, 367; обнаружено, 367.
Стадия 2: N-(4-хлорфенил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбоксамид
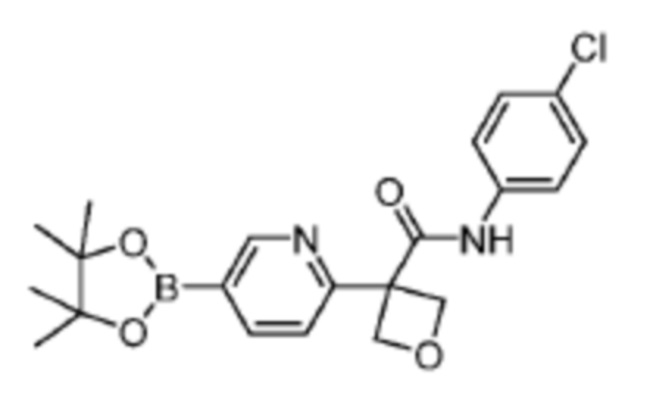
В колбу с якорем магнитной мешалки добавляли 3-(5-бромпиридин-2-ил)-N-(4-хлорфенил)оксетан-3-карбоксамид (200 мг, 0,54 ммоль), бис(пинаколато)диборон (207 мг, 0,82 ммоль), ацетат калия (160 мг, 1,63 ммоль) и аддукт PdCl2(dppf)-CH2Cl2 (44,4 мг, 0,054 ммоль). Смесь продували азотом в течение 5 мин. Добавляли диоксан (2720 мкл) и перемешивали смесь. Сосуд нагревали до 80°C в течение 12 ч. Через 12 ч. неочищенный продукт промывали EtOAc и насыщенным раствором NaHCO3. Объединенные органические соединения сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Материал сухим вводили в колонку 40 г с золотом и колонку прогоняли от 100% DCM к 100% EtOAc. Элюировали желаемый продукт, фракции собирали и концентрировали при пониженном давлении для получения титульного соединения. MS (ESI): вычислено для C21H24BClN2O4[M+H]+, 333; обнаружено, 333 (определяли массу бороновой кислоты).
Стадия 3: N-(4-хлорфенил)-3-(4'-(2-гидроксипропан-2-ил)-6'-(трифторметил)-[3,3'-бипиридин]-6-ил)оксетан-3-карбоксамид
В сосуд с якорем магнитной мешалки добавляли N-(4-хлорфенил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксетан-3-карбоксамид (93,7 мг, 0,23 ммоль), 2-(5-бром-2-(трифторметил)пиридин-4-ил)пропан-2-ол (70,6 мг, 0,25 ммоль), ортофосфат калия (водный раствор, 1 M) (452 мкл, 0,45 ммоль), PdCl2(dtbpf) (29,5 мг, 0,045 ммоль) и диоксан (1130 мкл). Сосуд продували азотом, запаивали и нагревали до 65°C в течение 22 ч. Через 22 ч. добавляли 1 небольшую ложку MgSO4, затем EtOAc, и отфильтровывали твердые вещества. Органические соединения концентрировали при пониженном давлении, растворяли в 1,5 мл DMSO и сразу подвергали очистке посредством ВЭЖХ (очистка посредством ВЭЖХ, элюирующий градиент ACN/вода с модификатором TFA, линейный градиент). Фракции возвращали и лиофилизировали в лиофилизаторе в течение ночи для получения титульного соединения. MS (ESI): вычислено для C24H21ClF3N3O3[M+H]+, 492; обнаружено, 492, 1H ЯМР (600 МГц, DMSO-d6) δ 10,27 (с, 1H), 8,67 (д, J=1,7 Гц, 1H), 8,46 (с, 1H), 8,20 (с, 1H), 7,93 (дд, J=8,1, 2,3 Гц, 1H), 7,79-7,70 (м, 2H), 7,66 (д, J=8,0 Гц, 1H), 7,47-7,36 (м, 2H), 5,15 (дд, J=86,8, 6,4 Гц, 4H), 1,35 (с, 6H).
Примеры 133-139 в следующей таблице получали аналогично примеру 112 с использованием промежуточного соединения I-9 (см. получение в примере 7) и соответствующих спиртов.
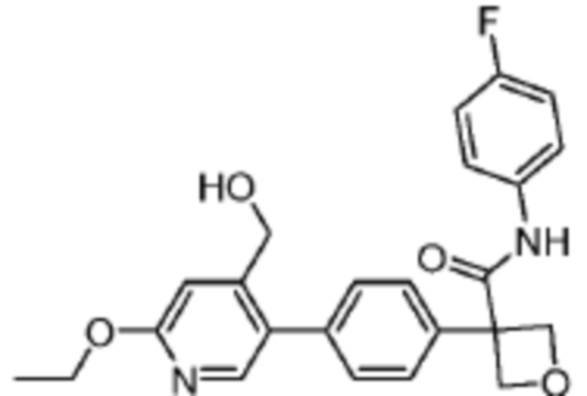
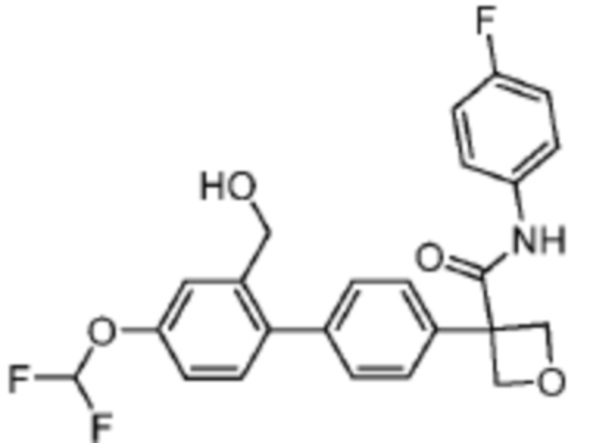
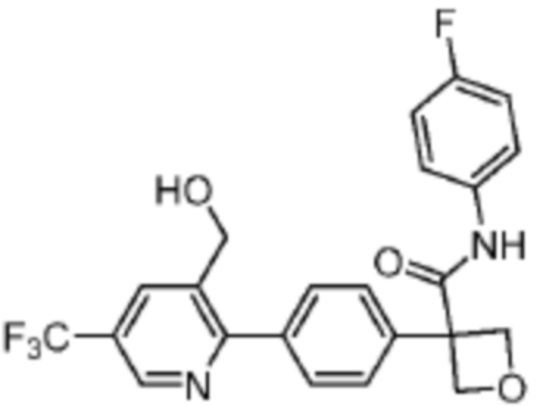
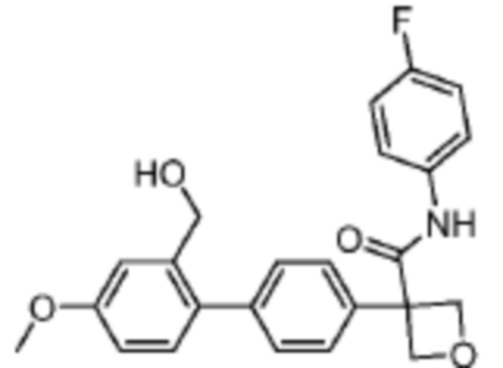
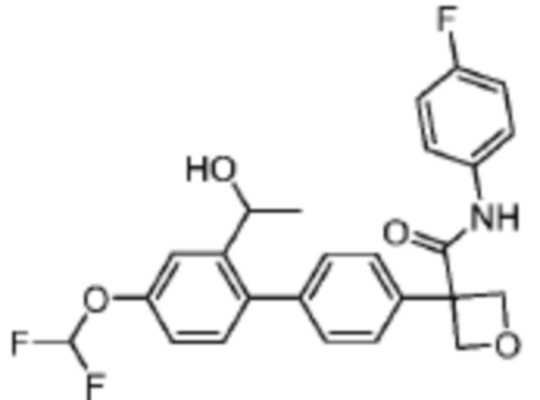
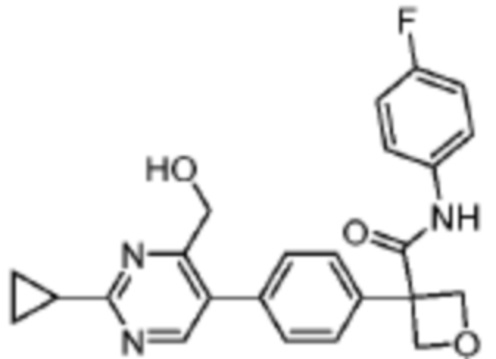
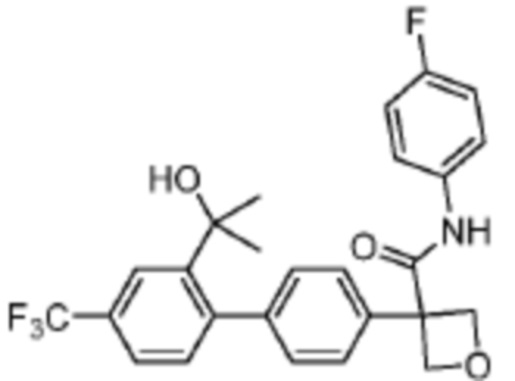
Примеры 140-148 в следующей таблице получали аналогично примеру 76 с использованием промежуточного соединения I-155 (см. получение в примере 76) и соответствующих спиртов-партнеров по сочетанию.
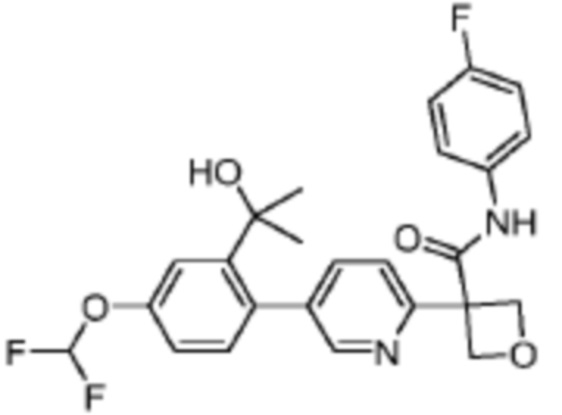
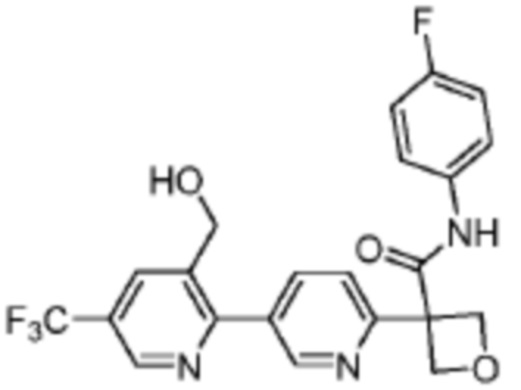
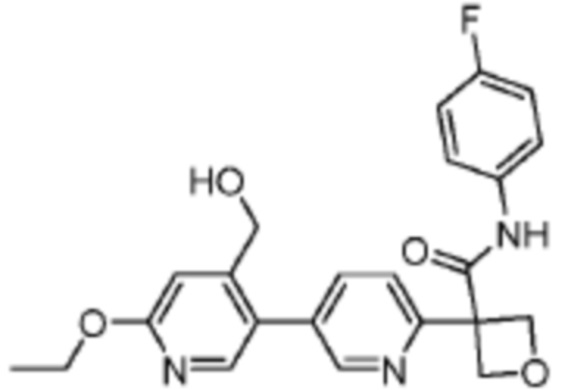
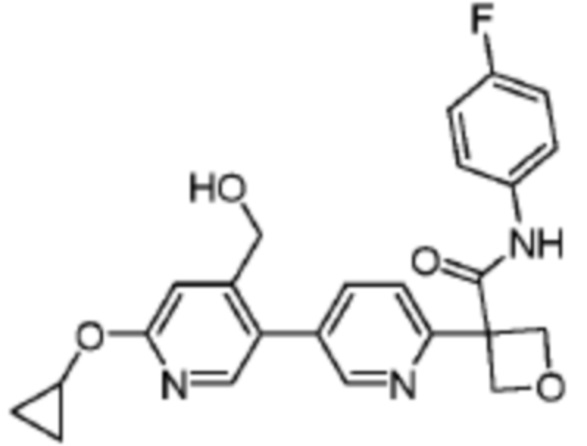
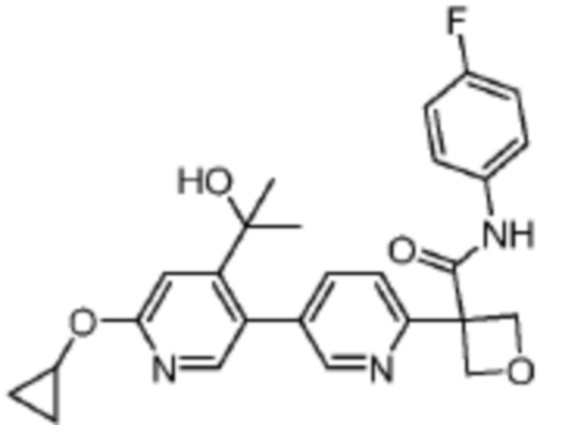
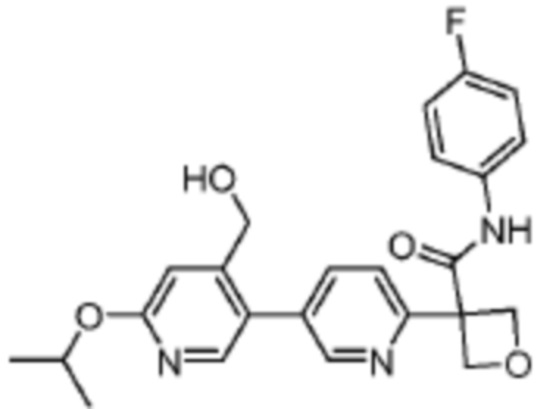
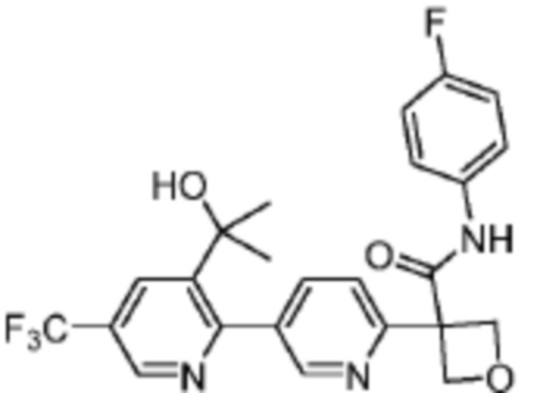
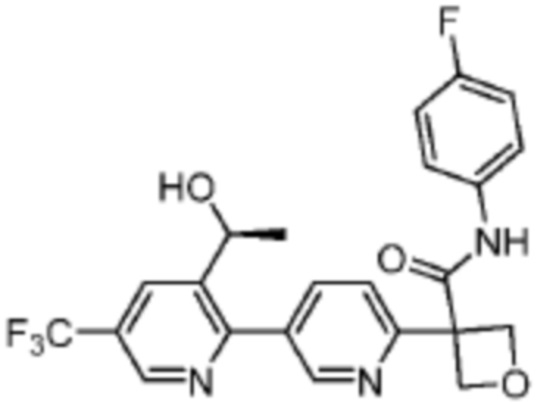
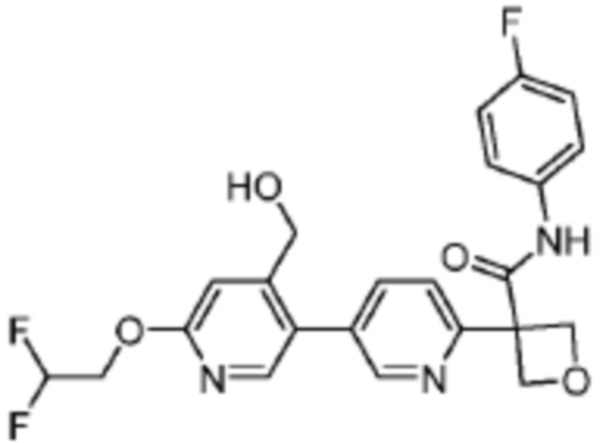
Примеры 149-151 в следующей таблице получали аналогично примеру 81.
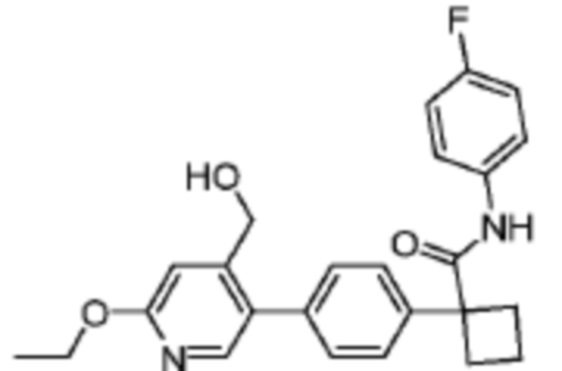
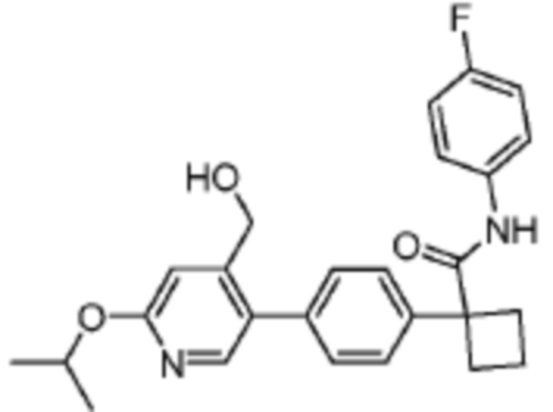
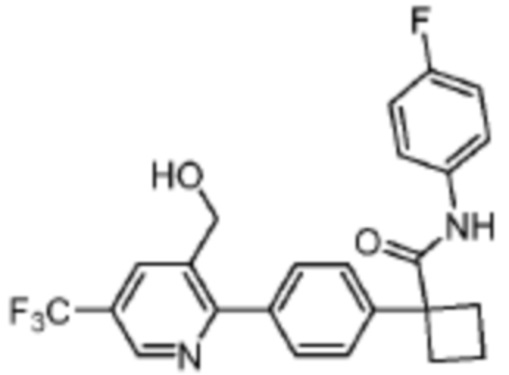
Пример 152: 3-фтор-N-(4-фторфенил)-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоксамид
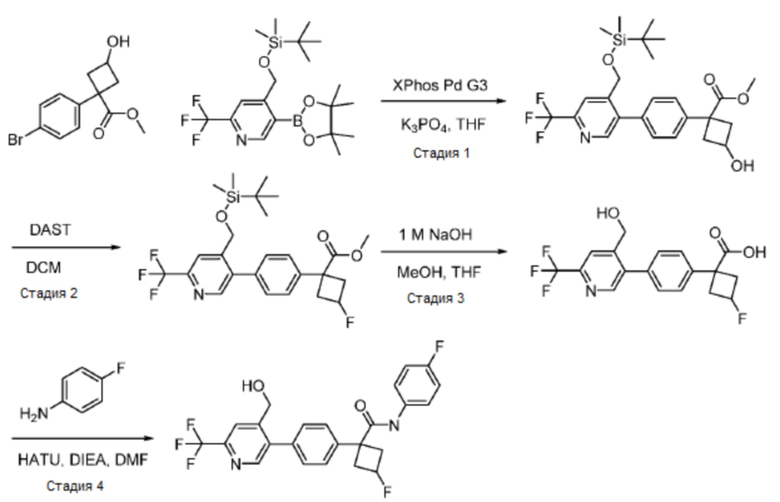
Стадия 1: Метил-1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-3-гидроксициклобутан-1-карбоксилат
В сосуд добавляли метил-1-(4-бромфенил)-3-гидроксициклобутанкарбоксилат (113 мг, 0,396 ммоль), 4-(((трет-бутилдиметилсилил)окси)-метил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридин (248 мг, 0,594 ммоль), XPhos Pd G3 (33 мг, 0,04 ммоль), THF (2000 мкл) и K3PO4 (1000 мкл, 1 ммоль). Смесь выкачивали и помещали обратно с азотом 4 раза и нагревали при 60°C в течение 1 ч. Смесь разводили водой и EtOAc. Водный слой экстрагировали с помощью EtOAc 3 раза. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом для получения остатка, который очищали посредством хроматографии на колонках с силикагелем для получения титульного соединения. MS (EI) m/z 496 [M+H+].
Стадия 2: Метил-1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-3-фторциклобутан-1-карбоксилат
Раствор метил-1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-3-гидроксициклобутанкарбоксилата (147 мг, 0,297 ммоль) в DCM (2500 мкл) обрабатывали DAST (96 мг, 0,59 ммоль) при 0°C. Смесь перемешивали при RT в течение 3 ч., затем гасили NaHCO3 (насыщенным раствором) и разводили DCM. Органическую фазу концентрировали под вакуумом и очищали посредством хроматографии на колонках с силикагелем для получения титульного соединения. MS (EI) m/z 498 [M+H+].
Стадия 3: 3-фтор-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоновая кислота
В сосуд, содержащий метил-1-(4-(4-(((трет-бутилдиметилсилил)окси)метил)-6-(трифторметил)пиридин-3-ил)фенил)-3-фторциклобутанкарбоксилат (21 мг, 0,043 ммоль), добавляли MeOH (100 мкл), THF (300 мкл) и NaOH (1 M) (50 мкл, 0,050 ммоль). Смесь перемешивали при RT в течение 18 ч. Растворитель концентрировали под вакуумом для получения остатка, который растворяли в 0,1 мл воды. Раствор доводили до pH~3 с помощью HCl (1 M), затем концентрировали под вакуумом. Остаток сушили посредством азеотропной осушки два раза с ACN и один раз с толуолом для получения титульного соединения, которое использовали непосредственно на следующей стадии. MS (EI) m/z 370 [M+H+].
Стадия 4: 3-фтор-N-(4-фторфенил)-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутан-1-карбоксамид
В сосуд, содержащий 3-фтор-1-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)циклобутанкарбоновую кислоту (16 мг, 0,043 ммоль), добавляли DMF (400 мкл), 4-фторанилин (21 мг, 0,19 ммоль), HATU (33 мг, 0,087 ммоль) и DIEA (30 мкл, 0,17 ммоль). Смесь перемешивали при RT в течение 15 мин. Смесь фильтровали и очищали посредством обращенно-фазовой ВЭЖХ, проводя элюцию с использованием воды (0,1% TFA) и ACN, для получения титульного соединения. 1H ЯМР (600 МГц, DMSO-d6) δ 9,71 (с, 1H), 8,61 (с, 1H), 8,02 (с, 1H), 7,69-7,60 (м, 2H), 7,56 (д, 2H), 7,50 (д, 2H), 7,19-7,08 (м, 2H), 5,65 (т, 1H), 5,17 (дп, 1H), 4,56 (д, 2H), 3,50-3,36 (м, 2H), 2,75-2,58 (м, 2H). MS (EI) m/z 463 [M+H+].
Биологические анализы
Клеточный анализ IDO1 с использованием клеток HeLa, стимулируемых ИФНγ
Клетки HeLa культивировали в полной среде для культивирования HeLa (90% EMEM, 10% термически инактивированной эмбриональной телячьей сыворотки) и выращивали до количества приблизительно 1×109 клеток. Затем клетки собирали и замораживали в количестве 1×107 клеток/флакон в 1 мл среды для замораживания (90% полной среды для культивирования HeLa, 10% DMSO).
Тестируемые соединения серийно разводили в десяти 3-кратных разведениях в DMSO, начиная со стока 10 мМ DMSO, в малообъемных планшетах Echo. Затем разведения соединений или DMSO в отдельности переносили из планшетов для разведения в черные 384-луночные аналитические планшеты Greiner (кат. № 781086, 50 нл/лунку) с использованием акустического жидкостного манипулятора Echo 550 (Labcyte).
Замороженные клетки HeLa размораживали и переносили в среду для анализа HeLa (99% полной среды для культивирования HeLa, 1% пенициллина/стрептомицина) с использованием 20 мл среды/флакон клеток. Клетки центрифугировали при 250×g с помощью настольной центрифуги в течение 5 мин и суспендировали в том же объеме среды для анализа HeLa. Затем клетки подсчитывали и доводили до плотности 2×105 клеток/мл в среде для анализа HeLa. К клеткам добавляли стерильный L-триптофан в конечной концентрации 300 мкМ L-триптофана. Небольшую аликвоту (2 мл/планшет) клеток HeLa откладывали и не обрабатывали ИФНγ, используя ее в качестве контроля Max-E. К остальным клеткам HeLa добавляли стерильный ИФНγ (кат. № 285-IF, R&D systems) в конечной концентрации 100 нг/мл.
Клетки HeLa с ИФНγ и без него переносили в соответствующие лунки 384-луночных аналитических планшетов, содержащих соединения. Планшеты инкубировали в течение приблизительно 48 часов при 37°C в инкубаторе с 5% CO2. Затем в каждую лунку добавляли 12 мкл 0,5 M метилизонипекотата в диметилсульфоксиде, планшеты герметично закрывали и инкубировали при 37°C без CO2 в течение ночи. Планшеты центрифугировали в течение 1 мин при 200×g. Конечную флуоресценцию измеряли с помощью спектрофотометра для чтения планшетов Spectramax (Molecular Devices) с использованием фильтра возбуждения 400 нм и фильтра эмиссии 510 нм.
Интенсивность флуоресценции в каждой лунке корректировали по фоновой флуоресценции, определяемой в лунках с клетками, необработанными ИФНγ, и выражали как долю интенсивности, определяемой в лунках с клетками, обработанными ИФНγ, и только с DMSO. Активности вычисляли нелинейным методом наименьших квадратов в отношении четырех-параметрического логистического уравнения для IC50.
Данные о биологической активности, полученные с использованием клеточного анализа IDO1, описанного выше, приведены в таблице ниже. Соединения, представленные в настоящем описании, как правило, имеют IC50 от приблизительно 0,1 нМ до приблизительно 20000 нМ, или более конкретно - от приблизительно 1 нМ до приблизительно 10000 нМ, или более конкретно - от приблизительно 5 нМ до приблизительно 5000 нМ, или более конкретно - от приблизительно 10 нМ до приблизительно 1000 нМ, или еще более конкретно - от приблизительно 10 нМ до приблизительно 500 нМ. Данные о IC50 специфической активности для примеров соединений, представленных в настоящем описании, приведены в следующей таблице.
Анализ IDO1 человека в цельной крови
Тестируемые соединения серийно разводили в десяти 3-кратных разведениях DMSO, начиная с 10 мМ. Затем 3 мкл разведений соединений или DMSO в отдельности переносили из планшета для разведения в полипропиленовый 96-луночный аналитический планшет, содержащий 97 мкл RPMI, с использованием акустического жидкостного манипулятора Echo 555 (Labcyte). LPS и ИФНγ подготавливали в RPMI в 10-кратной конечной концентрации (1000 нг/мл), конечная концентрация составляет 100 нг/мл.
Цельную кровь человека, получаемую от здоровых доноров, отбирали в пробирки с гепарином натрия. 240 мкл крови переносили в каждую из лунок 96-луночного планшета с V-образным дном. 30 мкл соединения переносили из промежуточного планшета для разведения и инкубировали в течение 15 мин. Затем 30 мкл стимулирующих средств переносили в кровь и тщательно смешивали. Планшет закрывали воздухопроницаемой мембраной и инкубировали при 37°C в течение ночи (18 ч).
В день 2 получали меченый изотопом стандарт кинуренина и триптофана в воде в 10-кратной концентрации и 30 мкл добавляли к крови в конечной концентрации 3 мкМ. Аналитические планшеты центрифугировали при 300×g в течение 10 мин без ограничителя для отделения плазмы от эритроцитов. 60 мкл образцов плазмы удаляли, не трогая эритроциты. Плазму разводили RPMI в соотношении 1:1 и осаждали белки двумя объемами ацетонитрила. Планшеты центрифугировали при 4000×g в течение 60 мин. 20 мкл супернатанта осторожно переносили в 384-луночный планшет, содержащий 40 мкл 0,1% муравьиной кислоты в воде, и анализировали посредством LC/MS/MS.
Анализы LC/MS/MS осуществляли с использованием системы Thermo Fisher's LX4-TSQ Quantum Ultra. Эта система состоит из четырех бинарных насосов для высокоэффективной жидкостной хроматографии (ВЭЖХ) Agilent и тройного квадрупольного масс-спектрометра TSQ Quantum Ultra. В случае каждого образца 5 мкл впрыскивали в колонку Atlantis T3 (2,1 мм×150 мм, размер частиц 3 мкм) от Waters. Градиент подвижной фазы, перекачиваемой со скоростью 0,8 мл/мин, использовали для элюции аналитов из колонки при 25°C. Элюцию начинали с 0% B, линейно повышая до 25% B за 6,5 мин, удерживая на уровне 25% в течение 1 мин, повторно уравновешивая до 10 мин. Подвижная фаза A состояла из 0,1% муравьиной кислоты в воде. Подвижная фаза B состояла из 0,1% муравьиной кислоты в ацетонитриле. Данные получали в режиме детекции положительных ионов с использованием интерфейса HESI. Рабочие параметры устройства TSQ Quantum Ultra представляли собой напряжение при электростатическом напылении 4000 В, температуру капилляров 380°C, температуру испарителя 400°C, защитный газ 60 условных единиц, выходной газ 20 условных единиц, цилиндрические линзы 85 и газ для соударений 1,2 мТорр. Хроматограммы SRM кинуренина (Q1: 209,2>Q3:94,0) и внутреннего стандарта (Q1: 215,3>Q3:98,2) регистрировали в течение 90 сек. Площадь пика интегрировали с помощью программного обеспечения Xcalibur Quan. Соотношения между кинуренином, образовавшимся в ходе реакции, и внутренним стандартом с добавлением 2D6-кинуренина использовали для получения процента ингибирования и значений IC50. Титровали соединения и с помощью 4-параметрической формулы аппроксимации сигмоидальной кривой вычисляли IC50.
Данные о биологической активности селективных соединений при использовании анализа IDO1 человека в цельной крови, описанного выше, приведены в таблице ниже.
Хотя настоящее изобретение описано и проиллюстрировано со ссылкой на некоторые конкретные варианты осуществления, специалистам в этой области будет понятно, что можно осуществлять различные адаптации, изменения, модификации, замены, исключения или включения в способах без отклонения от сущности и объема изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ | 2019 |
|
RU2798336C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ ROR-ГАММА | 2017 |
|
RU2760366C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, которые обладают ингибирующей активностью по отношению к индоламин-2,3-диоксигеназе (IDO). В указанной формуле n представляет собой 1; p представляет собой 1; в каждом случае A представляет собой -CH=; M представляет собой -O-; R1 представляет собой пиридинил; где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из: (a) галогена, (b) -C3-8-циклоалкила, необязательно замещенного -OH, (e) -O-C1-8-алкила, необязательно замещенного 1-5 атомами галогена, (f) -O-C3-8-циклоалкила, (g) -C1-8-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена, -OH, -NH2, (i) -C(O)-Rf, где Rf выбран из -OH, -NH2 и -NH-C1-8-алкила; R2 представляет собой фенил; где фенил необязательно замещен 1-3 заместителями, независимо выбранными из: (a) галогена, (c) -CN, (d) -O-C1-8-алкила, необязательно замещенного 1-3 атомами галогена, и (e) -C1-8-алкила, необязательно замещенного 1-3 заместителями, независимо выбранными из галогена; и R3 выбран из H и галогена. Изобретение также относится к фармацевтической композиции, ингибирующей индоламин-2,3-диоксигеназу (IDO) и содержащей фармацевтически приемлемый носитель и эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, и отдельному соединению N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамиду или его фармацевтически приемлемой соли. 3 н. и 13 з.п. ф-лы, 16 табл., 152 пр.
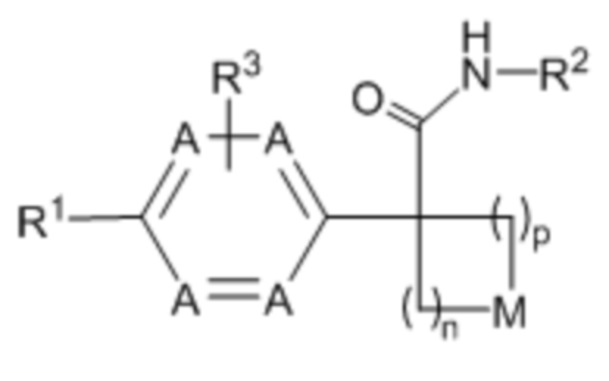 (I)
(I)
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
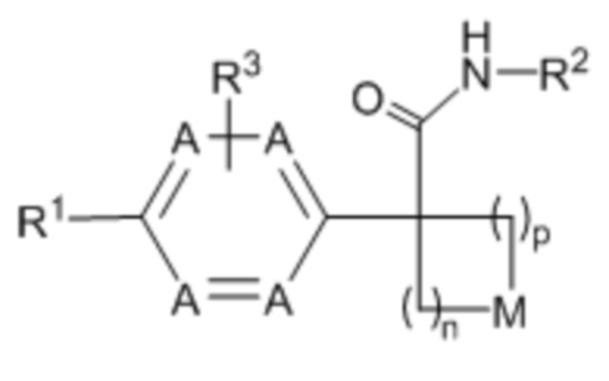 (I),
(I),
где:
n представляет собой 1;
p представляет собой 1;
в каждом случае A представляет собой -CH=;
M представляет собой -O-;
R1 представляет собой пиридинил;
где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -C3-8-циклоалкила, необязательно замещенного -OH,
(e) -O-C1-8-алкила, необязательно замещенного 1-5 атомами галогена,
(f) -O-C3-8-циклоалкила,
(g) -C1-8-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена, -OH, -NH2,
(i) -C(O)-Rf, где Rf выбран из -OH, -NH2 и -NH-C1-8-алкила;
R2 представляет собой фенил;
где фенил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(c) -CN,
(d) -O-C1-8-алкила, необязательно замещенного 1-3 атомами галогена, и
(e) -C1-8-алкила, необязательно замещенного 1-3 заместителями, независимо выбранными из галогена; и
R3 выбран из H и галогена.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
M представляет собой -O-; и
R3 представляет собой H.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
R1 представляет собой пиридинил;
где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно замещенного -OH,
(c) циклобутила, необязательно замещенного -OH,
(f) -O-C1-4-алкила, необязательно замещенного 1-3 атомами галогена,
(g) -O-циклопропила и
(h) -C1-4-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена и -OH.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
R2 представляет собой фенил;
где фенил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(c) -CN,
(d) -O-C1-6-алкила, необязательно замещенного 1-3 атомами галогена, и
(e) -C1-6-алкила, необязательно замещенного 1-3 заместителями, независимо выбранными из галогена.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
R2 представляет собой фенил;
где фенил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно замещенного 1-3 атомами галогена, и
(d) -C1-4-алкила, необязательно замещенного 1-3 атомами галогена.
6. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
M представляет собой -O-;
R1 представляет собой пиридинил;
где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) циклопропила, необязательно замещенного -OH,
(c) -O-C1-4-алкила, необязательно замещенного 1-3 атомами галогена, и
(d) -C1-4-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена и -OH; и
R2 представляет собой фенил;
где фенил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CN,
(c) -O-C1-4-алкила, необязательно замещенного 1-3 атомами галогена, и
(d) -C1-4-алкила, необязательно замещенного 1-3 атомами галогена.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, где:
R1 представляет собой пиридинил;
где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена и
(b) -C1-4-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена и -OH;
R2 представляет собой фенил;
где фенил необязательно замещен 1-3 атомами галогена; и
R3 представляет собой H.
8. Соединение по п. 1 формулы (Ia) или его фармацевтически приемлемая соль:
 (Ia),
(Ia),
где:
R1 представляет собой пиридинил;
где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -O-C1-4-алкила, необязательно замещенного 1-3 атомами галогена,
(c) -O-циклопропила и
(d) -C1-4-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена и -OH; и
R2 представляет собой фенил, необязательно замещенный 1-3 атомами галогена.
9. Соединение по п. 8 или его фармацевтически приемлемая соль, где:
R1 представляет собой пиридинил, необязательно замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CH3,
(c) -CHF2,
(d) -CF3 и
(e) -C(CH3)2OH; и
R2 представляет собой фенил, необязательно замещенный 1-2 атомами галогена.
10. Соединение по п. 1 формулы (Ii) или его фармацевтически приемлемая соль:
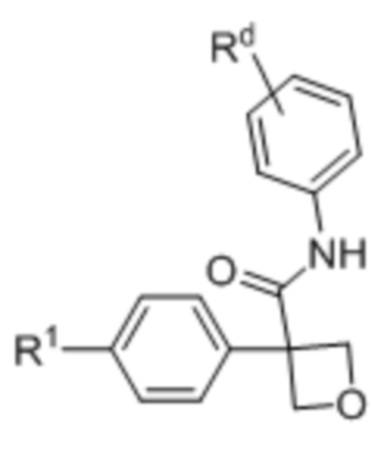 (Ii);
(Ii);
где:
R1 представляет собой пиридинил;
где пиридинил необязательно замещен 1-3 заместителями, независимо выбранными из:
(a) галогена,
(c) -O-C1-4-алкила, необязательно замещенного 1-3 атомами галогена,
(d) -O-циклопропила и
(e) -C1-4-алкила, необязательно замещенного 1-4 заместителями, независимо выбранными из галогена и -OH; и
Rd выбран из:
(a) галогена,
(b) -CN,
(c) -O-C1-3-алкила, необязательно замещенного 1-3 атомами галогена, и
(d) C1-3-алкила, необязательно замещенного 1-3 атомами галогена.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, где:
R1 представляет собой пиридинил, необязательно замещенный 1-3 заместителями, независимо выбранными из:
(a) галогена,
(b) -CH3,
(c) -CHF2,
(d) -CF3,
(e) -CH2OH и
(f) -C(CH3)2OH; и
Rd выбран из:
(a) галогена, выбранного из F, Cl и Br,
(b) -CH3 и
(c) -CF3.
12. Соединение по п. 1 или его фармацевтически приемлемая соль, выбранные из группы, состоящей из:
3-(4-(6-циклопропилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(6-циклопропил-4-фторпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(6-циклопропил-4-метилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-метил-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-(4-(6-(дифторметокси)-2,4-диметилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(6-(дифторметокси)-4-метилпиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-[4-[6-(дифторметокси)-4-(гидроксиметил)-3-пиридил]фенил]-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-[4-[3-(2-гидроксиэтил)-5-(трифторметил)-2-пиридил]фенил]оксетан-3-карбоксамида,
(R или S)-N-(4-фторфенил)-3-(4-(4-(1-гидроксиэтил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-[4-[6-(дифторметокси)-4-(1-гидрокси-1-метил-этил)пиридин-1-ий-3-ил]фенил]-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-хлорфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
N-(4-хлорфенил)-3-[4-[4-(гидроксиметил)-6-(трифторметил)-3-пиридил]фенил]оксетан-3-карбоксамида,
N-(4-хлорфенил)-3-[4-[6-(дифторметокси)-4-(гидроксиметил)-3-пиридил]фенил]оксетан-3-карбоксамида,
3-[4-[6-циклопропил-2-(гидроксиметил)пиридин-1-ий-3-ил]фенил]-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-[4-[6-(трифторметил)пиридин-1-ий-3-ил]фенил]оксетан-3-карбоксамида,
N-(4-фторфенил)-3-[4-[2-(гидроксиметил)-6-(трифторметил)-3-пиридил]фенил]оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(2-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-(4-(4-(фторметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-(трифторметокси)фенил)оксетан-3-карбоксамида,
N-(4-бромфенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-(трифторметил)фенил)оксетан-3-карбоксамида,
N-(3,4-дифторфенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
N-(4-цианофенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
N-(4-(дифторметокси)фенил)-3-(4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(3-(гидроксиметил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-(1-гидроксициклобутил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-(1-гидроксициклопропил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-(4-(6-циклопропокси-4-(гидроксиметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(6-циклопропокси-4-(2-гидроксипропан-2-ил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-N-метил-2-(трифторметил)изоникотинамида,
5-(4-(3-((4-фторфенил)карбамоил)оксетан-3-ил)фенил)-2-(трифторметил)изоникотинамида,
(R)-3-(4-(4-(1-амино-2,2,2-трифторэтил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
(S)-3-(4-(4-(1-амино-2,2,2-трифторэтил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(3-(2-гидроксипропан-2-ил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамида,
3-(4-(5-циклопропокси-3-(гидроксиметил)пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(5-циклопропокси-3-(2-гидроксипропан-2-ил)пиридин-2-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-изопропоксипиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-(4-(6-(2,2-дифторэтокси)-4-(гидроксиметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
N-(4-фторфенил)-3-(4-(4-(гидроксиметил)-6-(2,2,2-трифторэтокси)пиридин-3-ил)фенил)оксетан-3-карбоксамида,
3-(3-фтор-4-(4-(гидроксиметил)-6-(трифторметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида,
3-(4-(6-этокси-4-(гидроксиметил)пиридин-3-ил)фенил)-N-(4-фторфенил)оксетан-3-карбоксамида и
N-(4-фторфенил)-3-(4-(3-(гидроксиметил)-5-(трифторметил)пиридин-2-ил)фенил)оксетан-3-карбоксамида.
13. Фармацевтическая композиция, ингибирующая индоламин-2,3-диоксигеназу (IDO), содержащая фармацевтически приемлемый носитель и эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли.
14. Соединение, представляющее собой N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид или его фармацевтически приемлемую соль.
15. Соединение по п. 14, представляющее собой N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамид.
16. Соединение по п. 14, представляющее собой фармацевтически приемлемую соль N-(4-фторфенил)-3-(4-(4-(2-гидроксипропан-2-ил)-6-(трифторметил)пиридин-3-ил)фенил)оксетан-3-карбоксамида.
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| WO 2017076742 A1, 11.05.2017 | |||
| ИНГИБИТОРЫ ИДО | 2012 |
|
RU2613579C2 |

