ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к фармацевтическим препаратам, которые содержат в качестве активного ингредиента производное гидантоина, которое имеет высокую метаболическую стабильность и проявляет сильные PTH-подобные эффекты, и позволяет создать лекарственные препараты, которые индуцируют анаболизм костной и/или хрящевой ткани для предупреждения, лечения, и обеспечения восстановления и сращения при остеопорозе, предупреждения уменьшения костной массы при периодонтальной болезни, дефекте альвеолярной кости после удаления зуба, при остеоартрите, дефиците суставного хряща, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, переломе кости и т.п.
УРОВЕНЬ ТЕХНИКИ
Паратгормон (PTH) известен как гормон, который действует на клетки-мишени в почках и костях и регулирует гомеостаз кальция (Ca) и фосфора (Pi) (непатентный документ 1). Сывороточная концентрация Ca поддерживается PTH, главным образом, путем прямого и косвенного воздействия на желудочно-кишечный тракт, костную ткань и почки. PTH повышает ресорбцию Ca в почечных канальцах и таким образом подавляет экскрецию Ca из организма во внешнюю среду. PTH также увеличивает синтез фермента, который превращает витамин D в активный витамин D в почках, и тем самым способствует опосредованному активным витамином D всасыванию Са в желудочно-кишечном тракте. Кроме того, РТН увеличивает дифференцировку остеокластов опосредованно, через остеобласты и способствуют высвобождению Ca из костей. Считают, что указанные действия PTH осуществляются, главным образом, посредством увеличения циклического аденозин-3',5'-монофосфата (цАМФ) и/или активации фосфолипазы C (PLC), что происходит при связывании PTH с PTH1R.
У человека препараты PTH [PTH (1-34) и PTH (1-84)] оказывают мощный анаболический эффект на костную ткань и вызывают значительное увеличение минеральной плотности костной ткани (BMD) и прочности костей. В настоящее время большинство лекарственных препаратов против остеопороза предлагаемых для человека, представляют собой ингибиторы резорбции кости, и единственный тип лекарственного средства с анаболическим действием на костную ткань, который, например, активно увеличивает BMD, представляет собой препараты PTH. Таким образом, препарат PTH рассматривается в качестве одного из наиболее эффективных средств для лечения остеопороза (непатентный документ 2); однако поскольку PTH представляет собой пептид, его необходимо вводить инвазивным способом. Таким образом, существует потребность в создании лекарственного средства, которое имеет PTH-подобные эффекты и которое можно вводить неинвазивно.
Остеоартрит представляет собой дегенеративное заболевание, характеризующееся дегенерацией и разрушением хряща в суставах всего организма, таких как колени, тазобедренные суставы, позвоночник, пальцы; синовитом; отвердеванием субхондральной кости; или дисфункцией суставов в результате образования остеофитов и хронической болью. Считается, что сорок или более процентов популяции в возрасте 65 лет и старше поражены остеоартритом, и он стал огромной нагрузкой для медицинской экономики (непатентные документы 3 и 4). Причины остеоартрита включают физически чрезмерную весовую нагрузку на суставной хрящ, воспаление синовиальной мембраны и костного мозга, генетическую предрасположенность компонентов хрящевого матрикса и усиление метаболизма костной ткани субхондральной кости; однако не существует лекарственных препаратов, которые подавляют дегенерацию и разрушение суставного хряща, и медицинская потребность в них остается высокой.
Аггеканазы (ADAMTS-4, ADAMTS-5, и т.д.), матриксные металлопротеиназы (MMP-3, MMP-9, MMP-13, и т.д.; непатентный документ 5), и воспалительные цитокины (IL-1, IL-6, и т.д.; непатентный документ 6), которые участвуют в разрушении хрящевого матрикса, рассматривались в качестве мишеней для терапевтических средств, но такие средства не использовались в практических целях. С другой стороны, были проведены клинические испытания фармацевтических средств, воздействующих на усиление метаболического обновления субхондральной кости (ризедронат, кальцитонин; непатентные документы 7 и 8); однако дегенерация и разрушение суставного хряща не были подавлены. Кроме того, эффекты подавления деструкции суставного хряща в дополнение к указанному механизму были продемонстрированы в ходе клинических испытаний ранелата стронция, который оказывает комбинированное воздействие, способствуя образованию костной ткани, а также способствуя формированию хряща (непатентный документ 9); однако препарат не достиг стадии практического использования.
С другой стороны, в недавних исследованиях описано перерождение суставного хряща из постоянного хряща в кальцинированный хрящ в патогенезе остеоартрита, и его подавление привлекло внимание в качестве мишени для лекарственных средств (непатентный документ 10). Сообщалось, что фармацевтические препараты с несколькими типами подавления конечной дифференцировки хондроцитов суставов, на основе указанного механизма действия подавляли дегенерацию и разрушение суставного хряща в моделях остеоартрита животных, что предполагает возможность введения лекарственных средств на основе указанного механизма в практическое использование (непатентные документы 11 и 12).
Авторы настоящего изобретения представили патентную заявку на основании своего открытия, что соединение, представленное формулой (A):
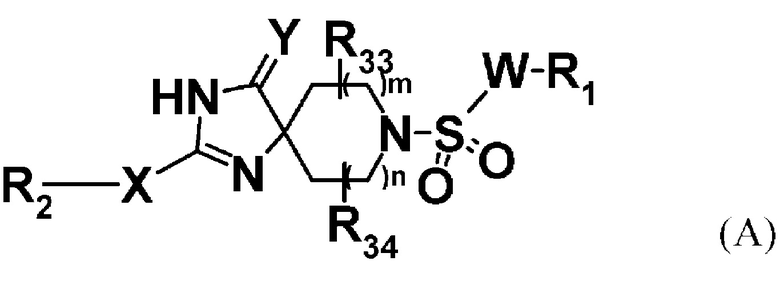
[можно сослаться на патентный документ 1 для W, X, Y, m, n, R1, R2, R33, и R34 в формуле]
и его фармацевтически приемлемые соли можно использовать в качестве соединений, имеющих PTH-подобные эффекты, или более предпочтительно, в качестве агониста PTH1R, и можно использовать для предупреждения и/или лечения остеопороза, переломов, остеомаляции, артрита, тромбоцитопении, гипопаратиреоза, гиперфосфатемии, или опухолевого кальциноза или для мобилизации стволовых клеток (патентный документ 1).
Для создания фармацевтических средств, которые имеют высокую клиническую ценность и могут быть введены неинвазивно, необходимо учитывать кинетику in vivo, такую как абсорбция, распределение, метаболизм и экскрекция, лекарства, помимо его непосредственного действия на мишень. Поэтому желательно иметь фармацевтическое средство, имеющее PTH-подобные эффекты, которое обладает высокой метаболической стабильностью против микросом печени человека и сильной PTH1R-опосредованной способностью продуцировать цАМФ у человека.
[Документы предшествующего уровня техники]
[Патентные документы]
[Патентный документ 1] WO 2010/126030
[Непатентные документы]
[Непатентный документ 1] Kronenberg, H.M., et al., In Handbook of Experimental Pharmacology, Mundy, G.R., and Martin, T.J., (edс), pp.185-201, Springer-Verlag, Heidelberg (1993)
[Непатентный документ 2] Tashjian and Gagel, J. Bone Miner. Res. 21:354-365 (2006)
[Непатентный документ 3] Sem Arth Rheumatism 2013; 43: 303-13
[Непатентный документ 4] CPMP/EWP/784/97 Rev. 1. 2010, European Medicines Agency
[Непатентный документ 5] Osteoarth Cart 2010; 18: 1109-1116
[Непатентный документ 6] Osteoarth Cart 2013; 21: 16-21
[Непатентный документ 7] Arthritis Rheum. 2006;54(11):3494-507
[Непатентный документ 8] J Clin Pharmacol. 2011;51(4):460-71
[Непатентный документ 9] Ann Rheum Dis. 2013 Feb;72(2):179-86
[Непатентный документ 10] Arth Rheum 2006; 54(8): 2462-2470
[Непатентный документ 11] Nat Med 2009; 15(12): 1421-1426
[Непатентный документ 12] Sci Trans Med 2011;3: 101ra93
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[Проблемы, решаемые с помощью изобретения]
Целью настоящего изобретения является предоставление способа предупреждения, лечения и облегчения восстановления и сращения при остеопорозе, уменьшении костной массы при периодонтальной болезни, дефекте альвеолярной кости после удаления зуба, при остеоартрите, дефиците суставного хряща, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, переломе кости и т.п. путем индукции анаболизма костной/хрящевой ткани путем неинвазивного системного воздействия или местного воздействия производных гидантоина, имеющих высокую метаболическую стабильность и проявляющих сильные PTH-подобные эффекты.
[Способы решения проблем]
Авторы настоящего изобретения обнаружили с помощью дополнительных исследований, что вновь обнаруженные производные гидантоина по настоящему изобретению проявляют сильную цАМФ-продуцирующую способность в клетках, экспрессирующих PTH1R человека, и обладать высокой стабильностью в отношении метаболизма в микросомах печени человека. Кроме того, при введении соединений по настоящему изобретению, авторы настоящего изобретения обнаружили, что соединения индуцируют анаболизм хрящевой и/или костной ткани, и их можно использовать в виде фармацевтических композиций для предупреждения, лечения и обеспечения восстановления и срастания при остеопорозе, предупреждения уменьшения костной массы при заболеваниях пародонта, при альвеолярном костном дефекте после удаления зуба, остеоартрите, дефиците суставного хряща, адинамическом заболевании костей, ахондроплазии, гипохондроплазии, остеомаляции, переломе кости и т.п.
Настоящее изобретение относится к указанному ниже.
[1] Фармацевтическая композиция для индукции анаболизма хрящевой и/или костной ткани, которая содержит в качестве активного ингредиента соединение, представленное общей формулой (1), приведенной ниже, или его фармацевтически приемлемую соль:
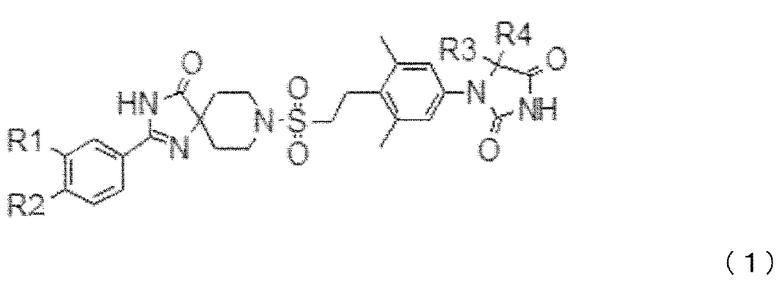
где,
если R1 и R2 оба не являются атомами водорода, R1 и R2 представляют собой независимо:
1) атом водорода;
2) атом галогена;
3) алкильную группу, содержащую один или два атома углерода, которые могут быть замещены от одного до пяти атомами фтора; или
4) алкоксигруппу, содержащую один или два атома углерода, которые могут быть замещены от одного до пяти атомами фтора; или
R1 и R2 связаны друг с другом для создания группы, представленной формулой, приведенной ниже:
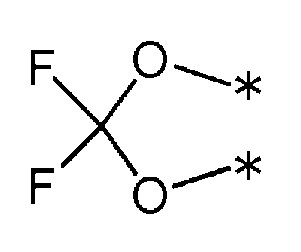
(где каждая * обозначает место связывания с фенильной частью); и
R3 и R4 представляют собой независимо метильную группу, которая может быть замещена от одного до трех атомами фтора; или
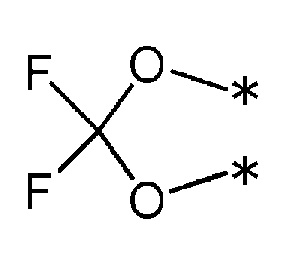
R3 и R4, вместе со связанным атомом углерода, образуют трех-шестичленное карбоциклическое кольцо (в котором один из атомов углерода, образующих кольцо, может быть заменен атомом кислорода, атомом серы, или метил-замещенным или незамещенным атомом азота).
В отношении соединения, содержащегося в качестве активного ингредиента в фармацевтической композиции по настоящему изобретению, соединение, в котором комбинация R1 и R2 представляет собой трифторметильную группу и атом водорода, и в котором R3 и R4 вместе со связанным атомом углерода образуют циклопентильное кольцо, может быть исключено из вышеупомянутых соединений, представленных формулой (1).
[2] Фармацевтическая композиция по [1], в которой R1 и R2 соединения, представленного общей формулой (1) или его фармацевтически приемлемой солью, выбирают из комбинаций, приведенных ниже:
1) R1 представляет собой атом водорода или атом галогена, и R2 представляет собой атом водорода, трифторметильную группу, или трифторметоксильную группу (при условии, что R1 и R2 оба не являются атомами водорода);
2) R1 представляет собой трифторметильную группу или трифторметоксильную группу, и R2 представляет собой атом водорода или атом галогена;
3) R1 и R2 связаны друг с другом для создания группы, представленной формулой, приведенной ниже :
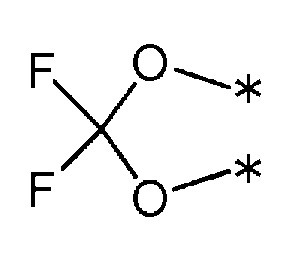
(где каждая * обозначает место связывания с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4, вместе со связанным атомом углерода, образуют кольцо, выбранное из приведенных ниже колец:
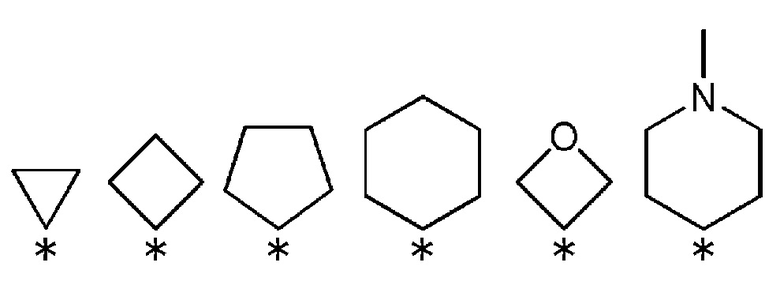
(где * указывает место связывания с частью имидазолидин-2,4-диона).
[3] Фармацевтическая композиция по [1], в которой R1 и R2 соединения, представленного общей формулой (1) или его фармацевтически приемлемую соль выбирают из комбинаций, приведенных ниже:
1) R1 представляет собой трифторметоксильную группу и R2 представляет собой атом фтора;
2) R1 представляет собой атом брома и R2 представляет собой атом водорода;
3) R1 представляет собой трифторметоксильную группу и R2 представляет собой атом фтора;
4) R1 представляет собой атом фтора и R2 представляет собой трифторметоксильную группу;
5) R1 представляет собой трифторметильную группу и R2 представляет собой атом водорода;
6) R1 представляет собой атом водорода и R2 представляет собой трифторметоксильную группу;
7) R1 и R2, связанные друг с другом, для создания группы, представленной формулой, приведенной ниже:
(где каждая * обозначает место связывания с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4, вместе со связанным атомом углерода, образуют кольцо, выбранное из представленных ниже колец:
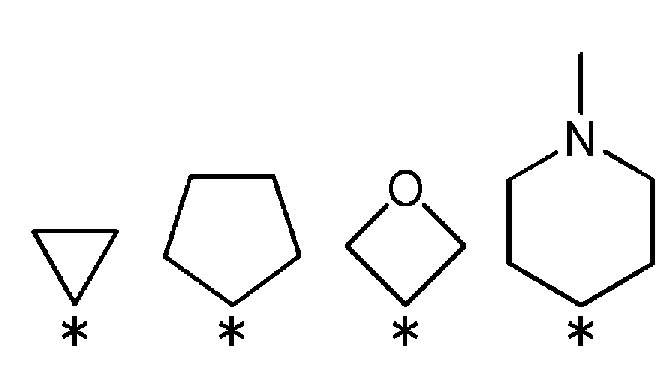
(где * указывает место связывания с частью имидазолидин-2,4-диона).
[4] Фармацевтическая композиция по [1], где R3 и R4 соединения, представленного общей формулой (1) или его фармацевтически приемлемой соли представляют собой метильные группы.
[5] Фармацевтическая композиция по [1], где R3 и R4 соединения, представленного общей формулой (1) или его фармацевтически приемлемой соли, вместе со связанным атомом углерода, образуют кольцо, выбранное из представленных ниже колец:
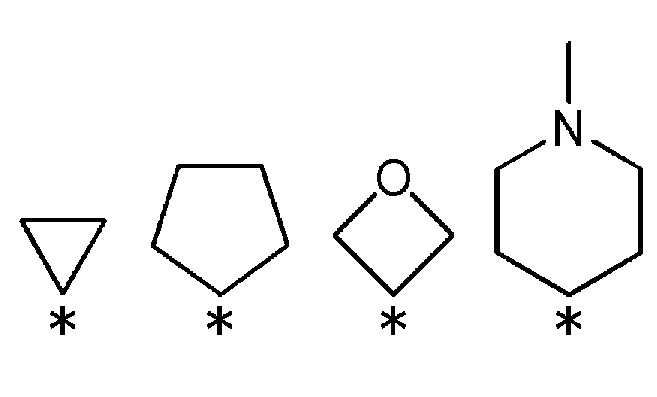
(где * указывает место связывания с частью имидазолидин-2,4-диона).
[6] Фармацевтическая композиция по [1], которая содержит в качестве активного ингредиента соединение или его фармацевтически приемлемую соль, где соединение выбирают из группы, состоящей из:
1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8- триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(3-бромфенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5- диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(4-фтор-3-(трифторметил)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(3-фтор-4-(трифторметокси)фенил)-4-оксо-1,3,8- триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5- диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-диона);
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3- диазаспиро[4.4]нонан-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8- метил-1,3,8-триазаспиро[4.5]декан-2,4-диона;
5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-oxa- 5,7-диазаспиро[3,4]октан-6,8-диона; и
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6- диазаспиро[2,4]гептан-5,7-диона.
[7] Фармацевтическая композиция по [1], в которой соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион или его фармацевтически приемлемую соль.
[8] Фармацевтическая композиция по [1], в которой соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион или его фармацевтически приемлемую соль.
[9] Фармацевтическая композиция по [1], в которой соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион или его фармацевтически приемлемую соль.
[10] Фармацевтическая композиция по [1], предназначенная для предупреждения или лечения остеопороза, предупреждения снижения костной массы при периодонтальном заболевании, обеспечения восстановления дефекта альвеолярной кости после удаления зуба, предупреждения или лечения остеоартрита, обеспечения восстановления дефицита суставного хряща, предупреждения или лечения адинамической болезни кости, предупреждения или лечения ахондроплазии, предупреждения или лечения гипохондроплазии, предупреждения или лечения остеомаляции, или облегчения восстановления после перелома кости;
[11] способ индукции анаболизма костной и/или хрящевой ткани, который включает введение соединения по любому из пунктов с [1] по [9] или его фармацевтически приемлемой соли в фармацевтически эффективном количестве пациенту, нуждающемуся в предупреждении или лечении остеопороза, предупреждении снижения костной массы при периодонтальной болезни, облегчения восстановления дефекта альвеолярной кости после удаления зуба, в предупреждении или лечении остеоартрита, облегчении восстановления дефицита суставного хряща, в предупреждении или лечении адинамической болезни кости, в предупреждении или лечении ахондроплазии, в предупреждении или лечении гипохондроплазии, в предупреждении или лечении остеомаляции, или в облегчении восстановления после перелома кости;
[12] способ по [11], где способ индукции анаболизма костной и/или хрящевой ткани представляет собой способ предупреждения или лечения остеопороза, способ предупреждения е снижения костной массы при периодонтальной болезни, способ облегчения восстановления дефекта альвеолярной кости после удаления зуба, способ предупреждения или лечения остеоартрита, способ облегчения восстановления дефицита суставного хряща, способ предупреждения или лечения адинамической болезни кости, способ предупреждении или лечения ахондроплазии, способ предупреждении или лечения гипохондроплазии, способ профилактики или лечения остеомаляции, или способ обеспечения восстановления после перелома кости;
[13] применение соединения по любому из пунктов с [1] по [9] или его фармацевтически приемлемой соли для изготовления фармацевтической композиции, предназначенной для предупреждения или лечения остеопороза, предупреждения снижения костной массы при периодонтальном заболевании, обеспечения восстановления дефекта альвеолярной кости после удаления зуба, предупреждения или лечения остеоартрита, обеспечения восстановления дефицита суставного хряща, предупреждения или лечения адинамической болезни кости, предупреждения или лечения ахондроплазии, предупреждения или лечения гипохондроплазии, предупреждения или лечения остеомаляции, или обеспечения восстановления после перелома кости;
[14] применение соединения по любому из пунктов с [1] по [9] или его фармацевтически приемлемой соли для создания фармацевтической композиции, предназначенной для индукции анаболизма костной и/или хрящевой ткани; и
[15] соединение по любому из пп. с [1] по [9], или его фармацевтически приемлемая соль, которое применяют для предупреждения или лечения остеопороза, предупреждения снижения костной массы при периодонтальном заболевании, обеспечения восстановления дефекта альвеолярной кости после удаления зуба, предупреждения или лечения остеоартрита, обеспечения восстановления дефицита суставного хряща, предупреждения или лечения адинамической болезни кости, предупреждения или лечения ахондроплазии, предупреждения или лечения гипохондроплазии, предупреждения или лечения остеомаляции, или обеспечения восстановления после перелома кости.
Кроме того, настоящее изобретение относится к способам лечения патологических состояний, которые можно предотвратить, лечить или устранить с помощью анаболизма костной и/или хрящевой ткани путем введения соединения формулы (1) или его фармацевтически приемлемой соли.
[Эффекты изобретения]
Настоящее изобретение способствует предупреждению, лечению, и обеспечению восстановления и/или излечения при остеопорозе, уменьшению костной массы при периодонтальном заболевании, дефекте альвеолярной кости после удаления зуба, при остеоартрите, дефиците суставного хряща, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, и переломе кости путем индукции анаболизма костной и/или хрящевой ткани путем использования производных гидантоина, которые обладают сильными PTH-подобными эффектами и высокой метаболической стабильностью.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 представлены значения минеральной плотности поясничного отдела позвоночника и бедренной кости, полученные у крыс с удаленными яичниками после регулярного введения в течении шести недель. Более конкретно, представлены результаты измерений минеральной плотности костной ткани, полученные для поясничного отдела позвоночника и бедренной кости с использованием двухэнергетического рентгеновского сканера минеральной плотности кости, при регулярном введении носителя, соединения 7, или hPTH (1-34) один раз в день в течение шесть недель крысам с удаленными яичниками.
На фиг. 2 представлены значения минеральной плотности поясничного отдела позвоночника и берцовой кости, полученные у обычных крыс после регулярного введения в течение четырех недель. Более конкретно, представлены результаты измерений минеральной плотности костной ткани, полученные для поясничного отдела позвоночника и костей голени с использованием двухэнергетического рентгеновского сканнера минеральной плотности кости, при регулярном введении носителя, соединения 7, или hPTH (1-34) один раз в день в течение четырех недель обычным крысам.
На фиг. 3 представлены значения минеральной плотности мандибулы (кости нижней челюсти) у обычных крыс после регулярного введения в течение четырех недель. Более конкретно, представлены результаты измерений минеральной плотности кости, полученные для нижней челюсти с использованием двухэнергетического рентгеновского сканнера минеральной плотности кости, при регулярном введении носителя, соединения 7, или hPTH(1-34) один раз в день в течение четырех недель обычным крысам.
На фиг. 4 показано ингибирующее действие соединения 7 в отношении конечной дифференцировки хондроцитов суставов берцовой кости кролика. Более конкретно на фотографиях представлены результаты оценки ингибирующего действия соединения 7 и hPTH(1-34) в отношении конечной дифференцировки хондроцитов суставов берцовой кости кролика с использованием окраски на щелочную фосфатазу (A) и окрашивания ализариновым красным С (B).
На фиг. 5 показано количество протеогликана, синтезрованного хондроцитами человека. Более конкретно, представлены результаты оценки эффекта соединения 7 и hPTH(1-34) в активации синтеза протеогликанов хондроцитами человека.
На фиг. 6 показана доля области повреждения суставного хряща берцовой кости в модели частичного удаления мениска у кроликов. Более конкретно, представлены результаты измерения доли области повреждения суставного хряща берцовой кости, когда носитель или соединение 7 постоянно вводили в коленные суставы кроликов в модели с частичным удалением мениска.
На фиг. 7 представлены визуально наблюдаемые изменения суставного хряща берцовой кости через две недели после операции в модели у кроликов с частично удаленными менисками. Более конкретно, представлены результаты визуального наблюдения изменений суставного хряща берцовой кости через две недели после операции, при постоянном введении носителя или соединения 7 в коленные суставы кроликов в модели с частичным удалением менисков.
На фиг. 8 представлены микрофотографии типичных примеров суставного хряща на дистальном конце бедренной кости обычных кроликов после четырех недель регулярного перорального введения. Более конкретно, представлены результаты гистопатологического исследования, полученные с помощью световой микроскопии типичных примеров суставного хряща на дистальном конце бедренной кости у обычных крыс после четырех недель повторного ежедневного перорального введения носителя или соединения 7.
На фиг. 9 показаны средние значения изменений сывороточной концентрации Ca в пределах 24 часов после перорального введения каждого соединения в дозе 30 мг/кг крысам в модели TPTX.
[Способ осуществления изобретения]
Настоящее изобретение относится к производным гидантоина, которые обладают высокой метаболической стабильностью и проявляют сильные PTH-подобные эффекты, и к их применению. Авторы настоящего изобретения синтезировали соединения, представленные вышеупомянутой формулой (1) или их фармацевтически приемлемые соли, и обнаружили, что эти соединения или их соли вызывают анаболизм костной и/или хрящевой ткани.
В настоящем документе "алкил" относится к одновалентной группе, полученной путем удаления любого одного атома водорода из алифатического углеводорода, и охватывает подмножество структур гидрокарбильных или углеводородных групп, не содержащих гетероатом или ненасыщенную углерод-углеродную связь и содержащих атомы водорода и углерода в основной цепи. Примеры алкильных групп включают группы, имеющие линейную или разветвленную структуру. Алкильная группа предпочтительно представляет собой алкильную группу, содержащую один или два атома углерода. Алкильная группа представляет собой в частности, например, метильную группу или этильную группу, и предпочтительно представляет собой метильную группу.
Термин "алкокси" относится к оксигруппе, с которой связывается упомянутый выше "алкил", и предпочтительно относится к алкоксигруппе, содержащей один или два атома углерода. Конкретные примеры включают метокси- и этокси-группы, и предпочтительным примером является метоксигруппа.
В настоящем документе фраза "B необязательно замещен A" обозначает, что любой атом(ы) водорода в B может быть замещен любым числом атомов A.
В настоящем изобретении, число заместителей не ограничено, если не указано иное. Например, число заместителей может составлять от 1 до 5, от 1 до 4, от 1 до 3, от 1 до 2, или 1.
"Атом галогена" относится к атому фтора, атому хлора, атому брома или атому йода.
Символ "*" в химической формуле обозначает место связывания.
Соединения по настоящему изобретению, представленные формулой (1), обладают сильными PTH-подобными эффектами и высокой метаболической стабильностью.
В настоящем документе "PTH-подобный эффект" относится к активности образования внутриклеточного цАМФ (цАМФ: циклический аденозинмонофосфат) в результате воздействия на рецептор PTH или воздействия на путь передачи сигнала через рецептор PTH.
В настоящем изобретении, имеется ли "сильный PTH-подобный эффект", или является ли "PTH-подобный эффект сильным", может быть подтверждено с помощью измерения активности сигнального пути цАМФ, путем анализа сигнального пути цАМФ, например, согласно способу, описанному в J. Bone. Miner. Res. 14:11-20, 1999. В частности, например, согласно способу, описанному в ссылочном примере тестирования 1, количество цАМФ, произведенное клетками, экспрессирующими PTH1R человека, определяют с помощью коммерчески доступного набора cAMP EIA (например, Biotrack cAMP EIA system, GE health care) для измерения концентрации каждого соединения при 20% активности сигнальной системы цАМФ (EC20) или его концентрации при 50% активности сигнальной системы цАМФ (EC50), при этом активность сигнальной системы цАМФ, полученную после введения 100 нМ PTH (1-34)человека, принимают за 100%. В настоящем изобретении в случае "сильного PTH-подобного эффекта" или когда "PTH-подобный эффект является сильным", например, значение EC20 (мкМ), измеренное упомянутым выше способом, составляет предпочтительно 5,0 или меньше, более предпочтительно 3,0 или меньше, и еще более предпочтительно 2,0 или меньше. Для EC50, значение (мкМ), измеренное упомянутым выше способом, составляет, например, предпочтительно 25,0 или меньше, более предпочтительно 15,0 или меньше, и еще более предпочтительно 10,0 или меньше.
Есть ли "высокая метаболическая стабильность" или является ли "метаболическая стабильность высокой" можно подтвердить с использованием общепринятого способа измерения. Например, клетки печени, клетки тонкого кишечника, микросомы печени, микросомы тонкого кишечника, S9-фракция клеток печени, и т.п. можно использовать для подтверждения. В частности, например, стабильность соединения в микросомах печени можно подтвердить путем проведения измерений в соответствии с описанием T. Kronbach et al. (Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol, 1989, 36(1), 89-96). Более конкретно, стабильность может быть подтверждена с помощью приведенного ниже способа, описанного в ссылочном примере тестирования 3. В настоящем изобретении "высокая метаболическая стабильность" или "метаболическая стабильность является высокой" имеют место, когда величина клиренса (мкл/мин/мг) в тесте метаболической стабильности с использованием микросом печени человека, описанном в упомянутом выше ссылочном примере тестирования, составляет предпочтительно 60 или меньше, более предпочтительно 40 или меньше, и еще более предпочтительно 35 или меньше. В частности, высокая метаболическая стабильность может быть получена в вышеупомянутой формуле (1), кроме тех случаев, когда комбинация R1 и R2 представляет собой трифторметильную группу и атом водорода, и R3 и R4, вместе со связанным атомом углерода, образуют циклопентильное кольцо.
"Индуцируется ли анаболизм костной и/или хрящевой ткани" можно подтвердить с помощью известных способов.
Индукция анаболизма костной ткани может быть подтверждена, например, путем непрерывного введения тестируемого соединения в течение определенного периода времени, и последующего измерения минеральной плотности костной ткани или костной массы с использованием обычного способа измерения, и последующего сравнения полученного результата с контролем. В частности, например, измерения минеральной плотности костной ткани могут быть осуществлены с использованием двухэнергетического рентгеновского сканера минеральной плотности кости [например, DCS-600EX (Aloka)] по способу, описанному в документе Takeda et al. (Bone 2013; 53(1):167-173). Если минеральная плотность костной ткани является высокой по сравнению с контрольной группой, получавшей носитель, можно считать, что анаболизм костной ткани индуцируется. Соединения по настоящему изобретению представляют собой предпочтительно, например, соединения, для которых наблюдаются повышения, равные или большие, чем уровень повышения минеральной плотности костной ткани, наблюдаемый при введении hPTH(1-34) в качестве лекарственного средства при остеопорозе испытуемым в клинически эквивалентной дозе. Более конкретно, например, повышение от 8% до 12% минеральной плотности костной ткани по сравнению с контрольной группой, получающей носитель, является предпочтительным, и увеличение на 12% или больше является более предпочтительным.
Индукцию анаболизма хрящевой ткани можно подтвердить, например, путем культивирования хондроцитов в присутствии соединения по настоящему изобретению, и последующего измерения уровня матриц, продуцируемых хондроцитами (таких как протеогликан). Также индукцию анаболизма можно подтвердить путем определения происходит ли супрессия конечной дифференцировки и кальцификации хондроцитов. В частности, например, продукцию матрикса хрящевой ткани можно измерить с помощью следующих способов, описанных в документах Loester et al. (Atrh Rheum 2003; 48(8): 2188-2196) и Ab-Rahim et al. (Mol Cell Biochem 2013; 376: 11-20). Супрессию конечной дифференцировки можно оценить согласно способу, описанному в документе Okazaki et al. (Osteoarth Cart 2003;11(2):122-32). Если величина продукции матрикса хрящевой ткани увеличивается, и конечная дифференцировка и кальцификация супрессируются по сравнению с контролем, может иметь место индукция анаболизма хрящевой ткани. Соединения по настоящему изобретению представляют собой предпочтительно, например, соединения, которые имеют эффекты, равные или большие, чем эффекты PTH в отношении продукции матрикса хрящевой ткани и супрессии конечной дифференцировки хондроцитов. По сравнению с PTH, соединения по настоящему изобретению имеют высокую метаболическую стабильность, и поэтому они обладают достаточными эффектами в отношении упомянутых выше патологических состояний, и можно выбрать различные пути введения. Кроме того, когда "более-высокий-чем-РТН" эффект может быть получен в отношении величины продукции матрикса хрящевой ткани, становится возможным получение эффектов PTH-превосходящих эффектов против вышеуказанных патологических состояний.
Например, индукция анаболизма хрящевой ткани может быть подтверждена путем получения эндохондральной кости у субъекта, которому постоянно вводили тестируемое вещество в течение определенного периода времени, проведения гистопатологических наблюдений, и экспериментального определения утолщения суставного хряща и пластинки роста. В частности, толщину суставного хряща и хряща пластинки роста можно измерить гистологически. Если толщина хряща увеличена по сравнению с толщиной контроля, тестируемое соединение может индуцировать анаболизм хрящевой ткани. В частности, если увеличение толщины хряща является заметным по сравнению с увеличением толщины хряща под действием PTH, предпочтительно имеются соответствующие эффекты против вышеуказанных патологических состояний, и более предпочтительно, если эффекты получают в результате перорального введения.
Индукция анаболизма хрящевой ткани также может быть подтверждена, например, согласно способу Kikuchi et al. (Osteoarth Cart 1996;4(2):99-110) и согласно способу Sampson et al. (Sci Transl Med 2011; 3: 10193) путем постоянного введения тестируемого соединения в течение определенного периода времени животным (грызунам и не-грызунам) с частично удаленным мениском для дестабилизации коленного сустава и индукции остеоартрита, и последующей визуальной или гистопатологической оценки дегенеративного состояния суставного хряща коленного сустава. Если дегенерация суставного хряща в коленном суставе подавляется аналогичным образом, как в случае применения PTH, можно прийти к заключению, что тестируемое соединение является эффективным благодаря эффектам анаболизма в хрящевой ткани и подавлению конечной дифференцировки. Более предпочтительно, если указанные эффекты получают путем перорального введения тестируемого соединения.
Оценка также может быть осуществлена, например, по способу Wakitani et al. (Bone Joint Surg Br. 1989;71(1):74-80) путем введения тестируемого соединения в течение определенного периода времени субъектам, у которых были повреждены суставной хрящ и субхондральная кость, и анализа состояния регенерации хряща в участке повреждения. Наличие эффекта регенерации хряща, превосходящего эффект, наблюдаемый в контрольной группе, предполагает индукцию эффекта анаболизма хрящевой ткани тестируемым соединением. В частности, резко выраженный эффект регенерации хряща по сравнению с эффектом PTH, является благоприятным, поскольку соответствующие эффекты могут быть получены в отношении вышеуказанных патологических состояний, и более предпочтительно, если тестируемое соединение проявляет указанные эффекты при пероральном введении.
Указанные эффекты также могут быть подтверждены путем измерения PTH-подобного действия. PTHrP, который активирует PTH1R, рецептор PTH, с помощью паракринного действия, является важным фактором, вовлеченным в регуляцию роста и дифференцировки хондроцитов, и известно, что он подавляет конечную дифференцировку хондроцитов и оказывает действие по поддержанию хрящевых тканей (Science 1996; 273: 663-666). Анаболизм хрящевой ткани при активации PTH1R также можно оценить, например, по способу Xie et al. (Human Mol Genet 2012; 21(18): 3941-3955) путем введения тестируемого соединения в течение определенного периода времени обычным субъектам или субъектам с генетическим нарушением роста, и анализа скорости роста хрящевой кости и гистологического анализа утолщения пластинки роста. Если увеличение скорости роста и толщины пластинки роста можно подтвердить путем сравнения с контролем, можно считать, что тестируемое соединение обладает эффектом анаболизма в хрящевой ткани. В частности, эффекты тестируемого соединения предпочтительно превосходят эффекты PTH, и более предпочтительно, если исследуемое соединение проявляет эффекты при пероральном введении.
Соединения согласно настоящему изобретению, как в свободной форме, так и в форме фармакологически приемлемых солей, включены в настоящее изобретение. Примеры таких "солей " включают соли неорганических кислот, соли органических кислот, соли неорганических оснований, соли органических оснований и кислые или основные соли аминокислот.
Предпочтительные примеры солей неорганических кислот включают гидрохлориды, гидробромиды, сульфаты, нитраты и фосфаты. Предпочтительные примеры солей органических кислот включают ацетаты, сукцинаты, фумараты, малеаты, тартраты, цитраты, лактаты, стеараты, бензоаты, метансульфонаты, бензолсульфонаты и п-толуолсульфонаты.
Предпочтительные примеры солей неорганических оснований включают соли щелочных металлов, такие как соли натрия и соли калия, соли щелочноземельных металлов, такие как соли кальция и соли магния, соли алюминия и соли аммония. Предпочтительные примеры солей органических оснований включают соли диэтиламина, соли диэтаноламина, соли меглумина и соли N,N- дибензилэтилендиамина.
Предпочтительные примеры солей кислых аминокислот включают аспартаты и глутаматы. Предпочтительные примеры солей основных аминокислот включают соли аргинина, лизина и орнитина.
Соединения по настоящему изобретению могут адсорбировать воду, содержать адсорбированную воду или образовывать гидраты, когда их оставляют на воздухе. Такие гидраты также включены в соли настоящего изобретения.
Кроме того, соединения по настоящему изобретению могут адсорбировать некоторые другие растворители с образованием сольватов. Такие соли также включены в настоящее изобретение в качестве солей соединений формулы (1).
В настоящем документе, структурная формула соединения может представлять определенный изомер для удобства. Однако соединения по настоящему изобретению включают все изомеры, такие как геометрические изомеры, оптические изомеры, основанные на асимметричных атомов углерода, стереоизомеры и таутомеры, а также смеси указанных изомеров, которые возникают вследствие структуры соединений, без ограничения формулами, описанными для удобства, и могут представлять собой либо один из изомеров, либо их смесь. Таким образом, соединения по настоящему изобретению могут иметь асимметричный атом углерода в молекуле, и могут присутствовать в виде оптически активных форм и рацематов, но настоящее изобретение не ограничивается любой из них и включает в себя обе активные формы.
Настоящее изобретение включает все изотопы соединений, представленных формулой (1). В изотопах соединений по настоящему изобретению по меньшей мере один атом заменен на атом, имеющий тот же атомный номер (число протонов), но имеющий другое массовое число (сумма числа протонов и числа нейтронов). Примеры изотопов, содержащихся в соединениях по настоящему изобретению, включают атом водорода, атом углерода, атом азота, атом кислорода, атом фосфора, атом серы, атом фтора и атом хлора, в том числе 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F и 36Cl, соответственно. В частности, радиоизотопы, которые распадаются с радиоактивным излучением, такие как 3H и 14C, можно использовать в тестах распределения фармацевтических средств или соединений в тканях организма. Стабильные изотопы не распадаются, присутствуют почти в одинаково большом количестве и не являются радиоактивными, и поэтому безопасны при использовании. Изотопы в соединениях по настоящему изобретению могут быть изменены обычными способами путем замены реагента, используемого во время синтеза, реагентом, содержащим соответствующий изотоп.
Соединения согласно настоящему изобретению могут проявлять кристаллический полиморфизм, и они не особенно ограничены каким-либо из них, но они могут находиться в любой из кристаллических форм или существовать в виде смеси двух или более кристаллических форм.
Соединения согласно настоящему изобретению включают их пролекарства. Пролекарства представляют собой производные соединений по настоящему изобретению, которые имеют химически или метаболически разрушаемые группы и превращаются обратно в исходные соединения после введения in vivo с проявлением своей исходной эффективности, и включают комплексы, образованные без ковалентных связей, и соли.
Соединения, представленные приведенной выше формулой, (1) согласно настоящему изобретению являются предпочтительно такими, как указано далее.
В формуле R1 и R2 выбирают из комбинаций, приведенных ниже:
1) R1 представляет собой атом водорода или атом галогена, и R2 представляет собой атом водорода, трифторметильную группу, или трифторметоксильную группу (при условии, что R1 и R2 оба не являются атомами водорода);
2) R1 представляет собой трифторметильную группу или трифторметоксильную группу, и R2 представляет собой атом водорода or атом галогена;
3) R1 и R2 связаны друг с другом с образованием группы, представленной приведенной ниже формулой:
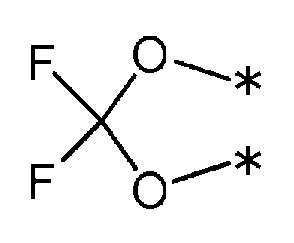
(где каждая * указывает место связывания с фенильной частью); и
R3 и R4 представляют собой метильные группы; или
R3 и R4, вместе со связанным атомом углерода, образуют кольцо, выбранное из представленных ниже колец:
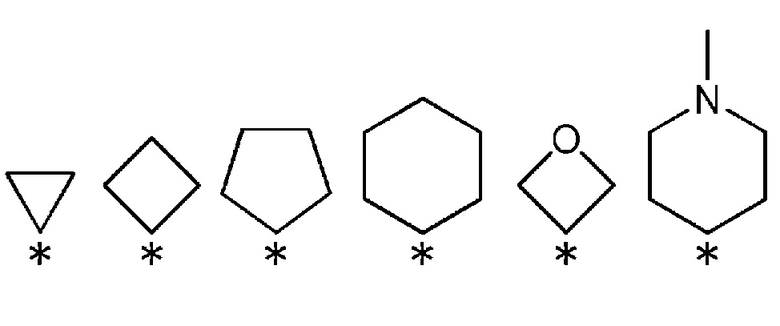
(где * указывает место связывания с частью имидазолидин-2,4-диона).
Соединения, представленные приведенной выше формулой, (1) согласно настоящему изобретению более предпочтительно являются такими, как указано далее.
В формуле, R1 и R2 выбирают из комбинаций, приведенных ниже:
1) R1 представляет собой трифторметоксильную группу и R2 представляет собой атом фтора;
2) R1 представляет собой атом брома и R2 представляет собой атом водорода;
3) R1 представляет собой трифторметоксильную группу и R2 представляет собой атом фтора;
4) R1 представляет собой атом фтора и R2 представляет собой трифторметоксильную группу;
5) R1 представляет собой трифторметильную группу и R2 представляет собой атом водорода;
6) R1 представляет собой атом водорода и R2 представляет собой трифторметоксильную группу;
7) R1 и R2 связаны друг с другом с образованием группы, представленной приведенной ниже формулой:
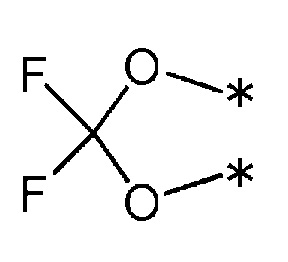
(где каждая * указывает место связывания с фенильной частью); и
R3 и R4 представляют собой метильные группы; or
R3 и R4, вместе со связанным атомом углерода, образуют кольцо, выбранное из представленных ниже колец:
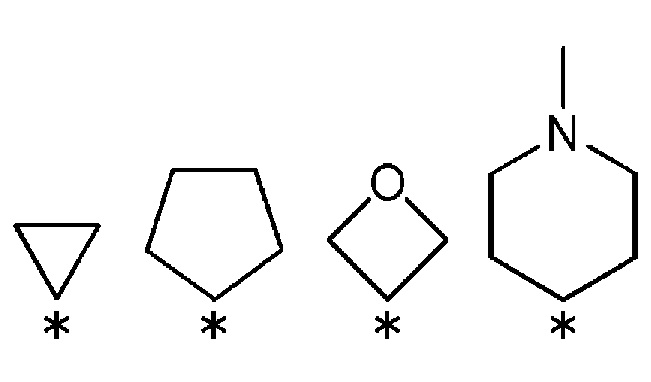
(где * указывает место связывания с частью имидазолидин-2,4-диона).
Соединения, представленные приведенной выше формулой, (1) согласно настоящему изобретению дополнительно представляют собой предпочтительно соединение, выбранное из группы, состоящей из приведенных ниже соединений, или его фармацевтически приемлемую соль.
Соединение 1:
1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8- триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 2:
1-(4-(2-((2-(3-бромфенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 3:
1-(4-(2-((2-(4-фтор-3-(трифторметил)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 4:
1-(4-(2-((2-(3-фтор-4-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 5:
1-(4-(2-((2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион;
Соединение 6:
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-дион;
Соединение 7:
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-дион);
Соединение 8:
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3- диазаспиро[4.4]нонан-2,4-дион;
Соединение 9:
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-дион;
Соединение 10:
5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-oxa-5,7-диазаспиро[3.4]октан-6,8-дион; и
соединение 11:
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6- диазаспиро[2.4]гептан-5,7-дион.
Из соединений с 1 по 11, указанных выше, соединения 6, 7 и 8 являются более предпочтительными.
Такие соединения по настоящему изобретению, представленные вышеупомянутой формулой (1), или их фармацевтически приемлемые соли индуцируют анаболизм костной и/или хрящевой ткани, и благодаря таким эффектам их можно использовать для предупреждения или лечения остеопороза, предупреждения снижения костной массы при периодонтальном заболевании, обеспечения восстановления дефекта альвеолярной кости после удаления зуба дефекта альвеолярной кости после удаления зуба, для предупреждения или лечения остеоартрита, обеспечения восстановления дефицита суставного хряща, для предупреждения или лечения адинамической болезни кости, для предупреждения или лечения ахондроплазии, для предупреждения или лечения гипохондроплазии, или для предупреждения или лечения остеомаляции.
Соединения их соли согласно настоящему изобретению могут быть включены обычными способами в состав таблеток, порошков, мелких гранул, гранул, драже, капсул, сиропов, пастилок, ингаляций, суппозиториев, инъекций, мазей, глазных мазей, офтальмологических препаратов, назальных препаратов, ушных препаратов, горячих компрессов, лосьонов и т.п. Обычно используемые носители, такие как вспомогательные вещества, связывающие вещества, смазывающие вещества, красители, корригенты, и, при необходимости, стабилизаторы, эмульгаторы, стимуляторы абсорбции, сурфактанты, регуляторы pH, консерванты, антиоксиданты и т.п., можно использовать для создания лекарственной формы, и их смешивают с ингредиентами, обычно используемыми в качестве сырья для фармацевтических препаратов и включают в состав общепринятыми способами.
Например, пероральные препараты получают путем добавления к соединению или его фармацевтически приемлемой соли согласно настоящему изобретению, вспомогательного вещества, и при необходимости, связывающего вещества, разрыхлителя, смазывающего вещества, красителя, корригента и т.п., и затем включения их в состав порошка, мелких гранул, гранул, таблеток, таблеток, покрытых оболочкой, капсул и т.п. общепринятым способом.
Примеры таких ингредиентов включают животные жиры и растительные масла, такие как соевое масло, говяжий жир и синтетический глицерид; углеводороды, такие как жидкий парафин, сквалан и твердый парафин; эфирные масла, такие как октилдодецилмиристат и изопропилмиристат; высшие спирты, такие как цетостеариловый спирт и бегениловый спирт; силиконовая смола; силиконовое масло; сурфактанты, такие как полиоксиэтиленовый эфир жирной кислоты, эфир сорбита и жирной кислоты, эфир глицерина и жирной кислоты, полиоксиэтиленовый эфир сорбита и жирной кислоты, полиэтиленгликоля касторовое масло гидрогенизированное и блок-сополимер полиоксиэтилена и полиоксипропилена; водорастворимые полимеры, такие как гидроксиэтилцеллюлоза, полиакриловая кислота, карбоксивиниловый полимер, полиэтиленгликоль, поливинилпирролидон и метилцеллюлоза; низшие спирты, такие как этанол и изопропанол; многоатомные спирты, такие как глицерин, пропиленгликоль, дипропиленгликоль и сорбит; сахара, такие как глюкоза и сахароза; неорганические порошки, такие как кремниевый ангидрид, алюмосиликат магния и алюмосиликат; и очищенную воду.
Примеры вспомогательных веществ включают лактозу, кукурузный крахмал, белый гранулированный или порошкообразный сахар, глюкозу, маннит, сорбит, микрокристаллическую целлюлозу и диоксид кремния.
Примеры связывающих веществ включают поливиниловый спирт, поливиниловый эфир, метилцеллюлозу, этилцеллюлозу, аравийскую камедь, трагакант, желатин, шеллак, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, поливинилпирролидон, блок-сополимер полипропиленгликоля и полиоксиэтилена и меглумин.
Примеры разрыхлителей включают крахмал, агар, желатиновый порошок, микрокристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстрин, пектин и кальций-карбоксиметилцеллюлозу.
Примеры смазывающих веществ включают стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и гидрогенизированное растительное масло.
Используемые красители представляют собой красители, утвержденные в качестве добавок к лекарственным средствам. Используемые корригенты представляют собой порошок какао, ментол, присыпной порошок, мятное масло, борнеол, порошок корицы и т.п.
Очевидно, указанные таблетки и гранулы могут быть покрыты сахаром или в других случаях могут быть покрыты соответствующим образом по назначению. Жидкие препараты, такие как сиропы и инъекционные препараты изготавливаются путем добавления вещества, регулирующего pH, растворителя, вещества, регулирующего тоничность, и т.п., и по необходимости, солюбилизирующего компонента, стабилизатора и т.п. к соединению или его фармацевтически приемлемой соли согласно настоящему изобретению и включению их в состав общепринятым способом.
Способ изготовления препаратов для наружного применения не ограничен, и они могут быть изготовлены обычными способами. В частности, различные исходные вещества, обычно используемые для фармацевтических препаратов, квази-лекарств, косметических средств и т.п., можно использовать в качестве основных веществ для препарата. Конкретные примеры используемых основных веществ включают исходные вещества, такие как животные и растительные масла, минеральные масла, сложноэфирные синтетические масла, воски, высшие спирты, жирные кислоты, силиконовое масло, сурфактанты, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и очищенную воду. Кроме того, регуляторы рН, антиоксиданты, хелаторы, консерванты и фунгициды, красители, корригенты и т.п. могут быть добавлены при необходимости. Основные вещества для препаратов для наружного применения согласно настоящему изобретению не ограничиваются указанными веществами.
Ингредиенты, такие как ингредиенты, имеющие эффект индукции дифференцировки, стимуляторы кровотока, антибактериальные средства, противовоспалительные средства, клеточные активаторы, витамины, аминокислоты, смачивающие средства и кератолитические средства также могут быть смешаны при необходимости. Вышеупомянутые основные вещества добавляют в количестве, соответствующем концентрации, обычно выбираемой для изготовления препаратов для наружного применения.
Способ применения соединений или их солей, или гидратов соединений или их солей согласно настоящему изобретению особенно не ограничен, и они могут быть введены перорально или парентерально обычно используемыми способами. Например, они могут быть включены в состав препаратов, таких как таблетки, порошки, гранулы, капсулы, сиропы, пастилки, ингаляции, суппозитории, инъекции, мази, глазные мази, глазные препараты, назальные препараты, ушные препараты, горячие компрессы и лосьоны, и введены.
Доза и способ введения лекарственного средства согласно настоящему изобретению можно выбрать соответствующим образом в зависимости от тяжести симптомов, возраста, пола, массы тела, способа применения, типа соли, конкретного типа заболевания и т.п.
Хотя доза может существенно изменяться в зависимости от типа заболевания и тяжести симптомов, наблюдаемых у пациента, возраста пациента, половых различий и разницы в чувствительности к лекарствам среди пациентов и т.п., доза обычно составляет приблизительно от 0,03 до 1000 мг, предпочтительно от 0,1 до 500 мг и более предпочтительно от 0,1 до 100 мг в день для взрослых и ее вводят раздельно в виде от одной до нескольких доз в день.
Способ введения выбирают соответствующим образом с учетом типа заболевания и степени выраженности симптомов у пациента, возраста и пола пациента, разницы в чувствительности к лекарственному средству и т.п. Способ введения особенно не ограничен, при условии, что он представляет собой способ, в котором соединение по настоящему изобретению неинвазивно воздействует системно или местно, и достигается эффект индукции анаболизма костной или хрящевой ткани. Примеры таких способов введения включают пероральное введение, внутривенное введение, трансназальное введение, чрескожное введение, транспульмональное введение, и внутрисуставное введение.
При изготовлении соединений по настоящему изобретению, представленных приведенной выше формулой (1), исходные соединения и различные реагенты могут образовывать соли, гидраты или сольваты, которые изменяются в соответствии с используемым исходным веществом, растворителем, и т.п., и они особенно не ограничены, если не ингибируют реакцию.
Используемый растворитель также изменяется в зависимости от исходного вещества, реагента и т.п., и растворитель особенно не ограничен, если он не ингибирует реакцию и растворяет исходное вещество до известной степени, разумеется.
Различные изомеры (например, геометрические изомеры, оптические изомеры, на основе асимметричных атомов углерода, ротамеры, стереоизомеры и таутомеры) можно очистить и выделить с помощью общепринятых способов разделения, например, перекристаллизации, способов с использованием диастереоизомерных солей, методов ферментативного расщепления и различных хроматографических методов (например, тонкослойная хроматография, колоночная хроматография, высокоэффективная жидкостная хроматография и газовая хроматография).
Соединения согласно настоящему изобретению, полученные в виде свободных форм, могут быть преобразованы в соли, которые могут быть образованы с помощью соединений, или в гидраты соединений в соответствии с общепринятыми способами. Соединения согласно настоящему изобретению, полученные в виде солей или гидратов соединений, также могут быть преобразованы в свободные формы соединений в соответствии с общепринятыми способами.
Соединения согласно настоящему изобретению можно выделить и очистить с использованием общепринятых химических процессов, таких как экстракция, концентрирование, упаривание, кристаллизация, фильтрование, перекристаллизация и различные хроматографические методы.
Все документы предшествующего уровня техники, упомянутые в описании, включены в виде ссылки.
ОБЩИЕ СПОСОБЫ СИНТЕЗА
Соединения по настоящему изобретению можно синтезировать различными способами, некоторые из которых будут описаны со ссылкой на приведенные далее схемы. Схемы являются иллюстративными, и настоящее изобретение не ограничивается только химическими реакциями и условиями, указанными в явной форме. Хотя некоторые заместители исключены в приведенных далее схемах для ясности, указанное исключение не предназначено для ограничения раскрытия схем. Типичные соединения по настоящему изобретению можно синтезировать с использованием соответствующих интермедиатов, известных соединений и реагентов. R1, R2, R3 и R4 в формулах приведенных ниже общих способах синтеза являются такими, как определено для R1, R2, R3 и R4 в соединениях, представленных вышеуказанной общей формулой (1) (соединения, представленные формулой 1 в приведенных ниже общих способах синтеза).
Соединения по настоящему изобретению (формула 1) можно синтезировать с помощью способов получения (способы A и B), представленных ниже.
Схема 1 (способ A)
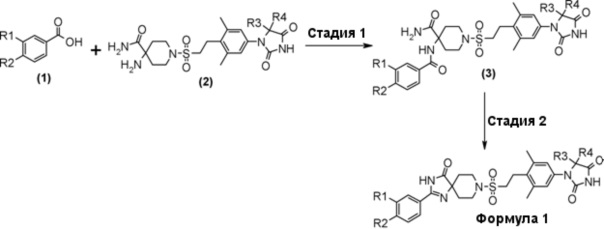
На схеме 1 представлен способ получения производного гидантоина (формула 1) с помощью амидирования производного карбоновой кислоты (1) и аминоамидного производного (2) с получением амидо-амидного производного (3), и затем построения спироимидазолонового кольца путем внутримолекулярной циклизации.
Стадия 1 представляет собой образование амидной связи между производным карбоновой кислоты (1) и аминоамидным производным (2). Примеры конденсирующего реагента включают N,N'-дициклогексилкарбодиимид (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (EDC), O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (HATU) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинхлорид n-гидрат (DMT-MM). Примеры основания включают триэтиламин или N,N-диизопропилэтиламин. При необходимости, 4-(диметиламино)пиридин (DMAP) можно использовать в качестве катализатора. Примеры подходящего растворителя включают дихлорметан или N, N-диметилформамид. Примеры подходящего реакционного растворителя при использовании DMT-MM включают метанол, этанол и ацетонитрил. Температура реакции составляет от 0°C до комнатной температуры, например, и время реакции составляет от 0,5 до 24 часов. Полученное амино-амидопроизводное (3) выделяют стандартным способом и, если необходимо, оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 2 представляет собой способ циклизации амидоамидного производного (3) в присутствии подходящего основания, такого как водный раствор гидроксида натрия или трет-бутоксид калия, в подходящем растворителе, таком как этанол, трет-бутанол, или диметилсульфоксид. Температура, при которой осуществляют реакцию, составляет, например, от комнатной температуры до температуры кипения в течение от одного до 24 часов. Полученное производное гидантоина (формула 1) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Аминоамидное производное (2), указанное на схеме 1, можно синтезировать из производного пиперидина (4). Способ синтеза аминоамидного производного (2) показан на схеме 2.
Схема 2
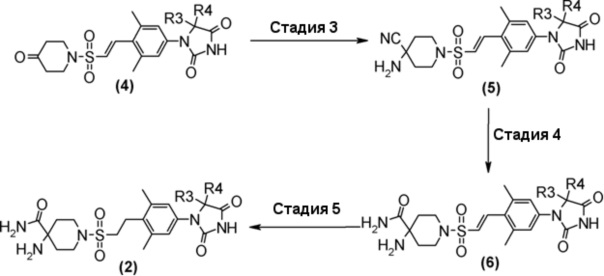
Стадия 3 представляет собой синтез Штреккера для превращения производного пиперединона (4) в производное аминонитрила (5). В частности, синтез представляет собой способ реакции пиперидинона (4) с цианидом натрия или цианидом калия и хлоридом аммония или ацетатом аммония в подходящем растворителе, таком как метанол, этанол или тетрагидрофуран, в присутствии/отсутствии воды. Температура реакции составляет от комнатной температуры до 80°C, например, и время реакции составляет от 2 до 72 часов. Полученное производное аминонитрила (5) выделяют стандартным способом и, если необходимо, оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 4 представляет собой способ превращения нитрильной группы в амидную группу в условиях щелочного гидролиза в присутствии перекиси водорода. Указанная реакция может быть осуществлена со ссылкой на Chemistry-A European Journal (2002), 8(2), 439-450, например.
Стадия 5 представляет собой способ гидрогенизации олефинового соединения (6) в инертном растворителе, таком как метанол, этанол, трифторэтанол, диметилформамид или диметилацетамид в присутствии катализатора, такого как палладированный уголь или гидроксид палладия на угле, соответственно, в атмосфере H2. Температура реакции составляет от комнатной температуры до 80°C, и реакция может быть проведена под давлением. Полученное аминоамидное производное (2) выделяют стандартным способом, и, если необходимо, оно может быть очищено с помощью кристаллизации или хроматографии.
Производное пиперидинона (4), показанное в схеме 2, можно синтезировать из известного кетал-винилсульфонилпроизводного (7) и гидантоин-арилбромидпроизводного (8). Способ синтеза производного пиперидина (4) показан на схеме 3.
Схема 3
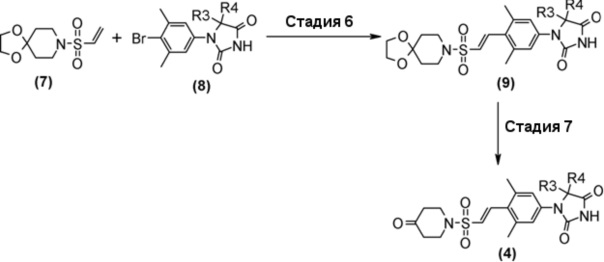
Стадия 6 представляет собой способ синтеза кетальарилвинилсульфонильного производного (9) путем сочетания кетальвинилсульфонильного производного (7) и гидантоинарилбромидного производного (8) в атмосфере N2 в присутствии палладиевого катализатора, такого как трис(дибензилидинацетон)палладий (0) или бис(дибензилидинацетон)палладий, и путем добавления фосфинового лиганда, такого как три-трет-бутилфосфинтетрафторборная кислота, и подходящего основания, такого как метилдициклогексиламин, в подходящем растворителе, таком как N-метил-2-пипридон (NMP). Температура реакции составляет от 90°С до температуры кипения. Полученное кетальарилвинилсульфонильное производное (9) выделяют общепринятыми способами, и при необходимости оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 7 представляет собой способ превращения кеталя кетальарилвинилсульфонильного производного (9) в кетон в подходящем растворителе, таком как водный тетрагидрофуран, в присутствии кислоты, такой как соляная кислота. Температура реакции составляет, например, температуры кипения растворителя, и время реакции составляет приблизительно от 1 до 24 часов. Полученное производное пиперидина (4) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Гидантоинарилбромидное производное (8), представленное в схеме 3, можно синтезировать из 4-бром-3,5-диметиланилина (10) и производного бромуксусной кислоты (11), или из 2-бром-5-иод-1,3- диметилбензола (13) и производного аминокислоты (14). Способ синтеза гидантоинарилбромидного производного (8) показан на схеме 4.
Схема 4
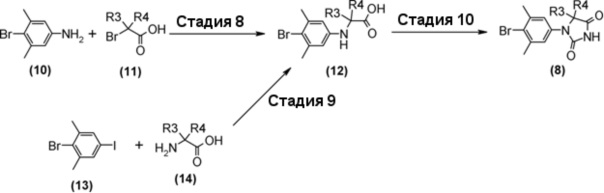
Стадия 8 представляет собой способ алкилирования 4-бром-3,5-диметиланилина (10) с производным бромуксусной кислоты (11) в присутствии подходящего основания, такого как диизопропилэтиламин, и в подходящем растворителе, таком как N-метил-2-пиперидон (NMP). Температура реакции составляет, например, от комнатной температуры до 100°С, и время реакции составляет от 1 до 24 часов. Полученное производное арилбромидаминокислоты (12) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 9 представляет собой способ синтеза производного арилбромидаминокислоты (12) путем соединения 2-бром-5-иод-1,3- диметилбензола (13) и производного аминокислоты (14) в присутствии металлического катализатора, такого как йодид меди (I). Реакцию можно осуществить в присутствии подходящего основания, такого как диазабициклоундецен (DBU) и в подходящем растворителе таком как N,N-диметилацетамид (DMA), при температуре реакции приблизительно от 80°C до 120°C. Полученное производное арилбромидаминокислоты (12) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 10 представляет собой способ синтеза гидантоинарилбромидного производного (8) с помощью реакции производного арилбромидаминокислоты (12) с цианатом натрия в кислой среде. Растворитель представляет собой, например, смешанный растворитель, такой как уксусная кислота - дихлорметан; температура реакции составляет от комнатной температуры до 60°C; и время реакции составляет от 1 до 24 часов. Полученное гидантоин-арилбромидное производное (8) может быть выделено общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Гидантоин-арилбромидное производное (8), представленное на схеме 3, также можно синтезировать из 4-бром-3,5-диметиланилина (10) и производного кетона (15). Способ синтеза гидантоин-арилбромидного производного (8) представлен на схеме 5.
Схема 5
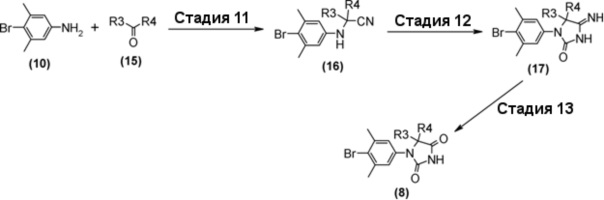
Стадия 11 представляет собой синтез Штреккера, в котором производное кетона (15) превращается в ариламинонитрильное производное (16). Более конкретно, синтез представляет собой способ проведения реакции производного кетона (15) с 4-бром-3,5-диметиланилином (10) и триметилсилилцианидом в подходящем растворителе, таком как уксусная кислота. Реакция может проходить при комнатной температуре, и время реакции составляет приблизительно от одного до трех часов. Полученное ариламинонитрильное производное (16) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 12 представляет собой способ проведения реакции ариламинонитрильного производного (16) с 2,2,2-трихлорацетилизоцианатом в подходящем растворителе, таком как дихлорметан, и затем синтеза иминопроизводного гидантоина (17) путем добавления реагентов, таких как метанол, вода, и триэтиламин и их реагирования в условиях нагревания. Полученное иминопроизводное гидантоина (17) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Стадия 13 представляет собой способ превращения производного иминогидантоина (17) в гидантоинарилбромидное производное (8) в кислой среде. Например, синтез может быть осуществлен в растворителе уксусная кислота-вода с нагреванием приблизительно до 65°C в течение примерно от одного до шести часов. Полученное гидантоинарилбромидное производное (8) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Схема 6 представляет собой способ проведения реакции Хека между производным винилсульфонамида (18) и производным гидантоин-арилбромида (8) в присутствии металлического катализатора, и последующего гидрирования олефинового соединения (19) для получения производного гидантоина (формула 1).
Схема 6 (Способ B)
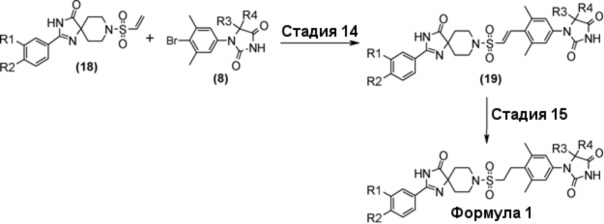
Производное гидантоина (формула 1) можно синтезировать путем проведения реакции стадии 14 согласно методу стадии 6 и реакции стадии 15 согласно методу стадии 5. Полученное производное гидантоина (формула 1) выделяют общепринятыми способами, и при необходимости, оно может быть очищено с помощью кристаллизации или хроматографии.
Производное винилсульфонамида (18), используемое на стадии 14, можно синтезировать, обращаясь к схемам 2, 3, и 12 в WO2010/126030(A1).
Все документы предшествующего уровня техники, цитируемые в описании, включены в виде ссылки.
Далее настоящее изобретение будет дополнительно проиллюстрировано с помощью ссылок на примеры, но изобретение не следует истолковывать, как ограниченное ими.
ПРИМЕРЫ
[Пример 1] Воздействие на минеральную плотность костной ткани в результате шести недель повторного введения крысам после овариэктомии
Самок крыс Crl:CD(SD), приобретенных в Charles River Japan, Inc., акклиматизировали в течение одной недели или больше в стандартных лабораторных условиях: от 20°C до 26°C и от 35% до 75% влажности, и затем использовали в экспериментах. Крысы имели свободный доступ к водопроводной воде и стандартному корму для грызунов (СЕ-2) (Clea Japan Inc.), содержащий 1,1% кальция, 1,0% фосфорной кислоты и 250 МЕ/100 г витамина D3.
Крысам в возрасте двенадцати недель проводили операцию по удалению обоих яичников (OVX) или ложную операцию (Sham). После того, как вес тела определяли на четвертой неделе после операции, крыс распределяли по группам таким образом, чтобы значения среднего веса тела в каждой группе из шести крыс, были бы равны. На следующий день после распределения по группам начинали регулярное введение препарата каждой крысе один раз в день в течение шести недель. Растворитель для перорального введения (носитель) и растворитель для подкожного введения (буфер PC) вводили соответственно перорально и подкожно крысам в контрольной группе Sham. Носитель и буфер PC вводили перорально и подкожно крысам в группе OVX-контроль, соответственно. Крысам в группе OVX-соединение 7, упомянутое выше соединение 7, растворенное в носителе, вводили перорально в дозе 30 мг/кг, и буфер PC вводили подкожно. Крысам в группе OVX-hPTH(1-34), носитель вводили перорально, и hPTH(1-34), растворенный в буфере PC, вводили подкожно в дозе 0,9 нмоль/кг.
Значение AUC (площадь под кривой концентрация в крови - время) было таким же, как при введении человеку 20 мкг Forteo®, терапевтического средства, используемого при остеопорозе, когда вводимая доза у крыс составляла 0,9 нмоль/кг. Вводимые дозы составляли 5 мл/кг для перорального введения и 1 мл/кг для подкожного введения во всех группах. Используемый носитель представлял собой композицию, состоящую из 10% диметилсульфоксида (Wako Pure Chemical Industries, Ltd.), 10% Kolliphor EL® (Sigma-Aldrich Japan), и 10% гидроксипропил-β-циклодекстрин (Nihon Shokuhin Kako Co., Ltd), значение pH которой доводили до 10, используя глицин (Wako Pure Chemical Industries, Ltd.) и гидроксид натрия (Wako Pure Chemical Industries, Ltd.). Используемый буфер PC представлял собой композицию, состоящую из 25 ммоль/л фосфатно-цитратного буфера, 100 ммоль/л NaCl, и 0,05% Tween80, значение pH которой доводили до 5,0. На следующий день после последнего введения крыс подвергали эвтаназии путем забора крови из брюшной аорты, и затем осуществляли аутопсию для получения поясничного отдела позвоночника и бедренной кости. Поясничный отдел позвоночника и бедренную кость хранили в 70% этаноле. Значения минеральной плотности поясничного отдела позвоночника и бедренной кости определяли с использованием двухэнергетического рентгеновского сканера минеральной плотности кости (DCS-600EX, Aloka). Минеральную плотность поясничного отдела позвоночника определяли путем измерения показателя со второго по четвертый поясничных позвонков; и минеральную плотность бедренной кости определяли путем вертикального деления бедренной кости на десять частей, и измерения показателя в трех частях на дистальном конце коленного сустава. Результаты представлены на фиг. 1.
Данные представлены в виде среднее значение + стандартная ошибка (SE). Программное обеспечение SAS preclinical package ver. 5.00 (SAS Institute Japan) использовали для проведения приведенных далее статистических анализов. Устанавливали уровень значимости 5% для обеих сторон. Что касается минеральной плотности поясничного отдела позвоночника и бедренной кости, использовали t-тест для двух групп для сравнения группы Sham-Контроль и группы OVX-Контроль (#P<0,05), для сравнения группы OVX-Контроль и группы OVX-соединение 7 (*P<0,05), и для сравнения группы OVX-Контроль и группы OVX-hPTH(1-34) (ʃP<0,05).
Как показано на фиг. 1, в отношении минеральной плотности бедренной кости, в группе OVX-Контроль наблюдали существенное снижение минеральной плотности по сравнению с группой Sham-Контроль, и в группе OVX-hPTH(1-34), которая является положительным контролем, наблюдали существенное увеличение по сравнению с группой OVX-Контроль. Процентное значение увеличения в группе OVX-hPTH(1-34) по сравнению с группой OVX-Контроль составляло 8%. В группе OVX-соединение 7 наблюдали существенное увеличение по сравнению с группой OVX-Контроль, и процентное значение увеличения составляло 12%. В отношении минеральной плотности поясничного отдела позвоночника, в группе OVX-Контроль наблюдали существенное снижение минеральной плотности относительно группы Sham-Контроль, и в группе OVX-соединение 7 отмечали тенденцию к увеличению по сравнению с группой OVX-Контроль, хотя увеличение было незначительным. В группе OVX-hPTH(1-34) наблюдали существенное увеличение по сравнению с группой OVX-Контроль, и процентное значение увеличения составляло 12%.
Как описано выше, повторное пероральное введение соединения 7 вызвало увеличение минеральной плотности костной ткани у крыс с OVX, патологической моделью остеопороза. Таким образом, соединение 7 может быть эффективным профилактики, лечения, обеспечения и улучшения восстановления при патологических состояниях, которые требуют индукции анаболизма костной ткани, увеличения костной массы, или регенерации кости, таких как остеопороз, снижение костной массы при периодонтальном заболевании, и дефект альвеолярной кости после удаления зуба. Кроме того, подтвердили, что соединения, представленные формулой (1), обладают сильными PTH-подобными эффектами и высокой метаболической стабильностью в ссылочных примерах 1-5, и они могут обеспечивать эффект увеличения минеральной плотности костной ткани путем анаболизма костной ткани с помощью PTH-подобных функций. Таким образом, соединения, представленные формулой (1), могут быть эффективны для профилактики, лечения, обеспечения и улучшения восстановления при патологических состояниях, которые требуют индукции анаболизма костной ткани, увеличения костной массы, или регенерации кости, таких как остеопороз, снижение костной массы при периодонтальном заболевании и дефект альвеолярной кости после удаления зуба.
[Пример 2] Воздействие на минеральную плотность костной ткани в результате четырех недель повторного введения обычным крысам
Самок крыс RccHan:WIST приобретенных в Japan Laboratory Animals Inc. акклиматизировали в течение одной недели или больше в стандартных лабораторных условиях: от 20°C до 26°C и от 35% до 75% влажности, и затем использовали в экспериментах. Крысы имели свободный доступ к водопроводной воде и стандартному корму для грызунов (CR-LPF) (Oriental Yeast Co., Ltd.).
Внутривенный катетер устанавливали крысам в возрасте в восьми недель. Катетер вводили из бедренной вены в паховой области, и выводили кончик в каудальную полую вену для размещения в указанном участке. Вес тела определяли в течение первой недели после операции, и распределяли крыс в группы, по десять животных на группу, так чтобы средние значения веса тела в каждой группе были равны. Через два дня после распределения по группам всем крысам начинали осуществлять повторные внутривенные введения один раз в день в течение четырех недель. Введение осуществляли путем подсоединения инфузионного насоса (MEDFUSION SYRINGE INFUSION PUMP Model 2001) к постоянному катетеру.
Растворитель (носитель) вводили внутривенно в группе Носитель-Контроль. В группах Соединение 7-20 мг/кг, -30 мг/кг, и -50 мг/кг, соединение 7, растворенное в носителе, вводили внутривенно в дозе 20 мг/кг, 30 мг/кг, и 50 мг/кг, соответственно. Во всех группах объем введения составлял 5 мл/кг и скорость введения составляла 5 мл/кг/мин. Состав используемого носителя включал 5% диметилсульфоксид (Wako Pure Chemical Industries, Ltd.), 25% пропиленгликоль (Kanto Chemical Co., Inc.)/20% этанол (Junsei Chemical Co., Ltd.)/15% гидроксипропил-β-циклодекстрин (Nihon Shokuhin Kako Co., Ltd)/300 мМ глицин (Wako Pure Chemical Industries, Ltd.)/192 мМ гидроксид натрия (Wako Pure Chemical Industries, Ltd.)/физиологический раствор (Otsuka Pharmaceutical Factory, Inc.). На следующий день после последнего введения крыс подвергали эвтаназии путем забора крови из брюшной аорты, и затем осуществляли аутопсию для получения поясничного отдела позвоночника, берцовой кости, и нижней челюсти. Поясничный отдел позвоночника, берцовую кость и нижнюю челюсть хранили в 70% этаноле. Значения минеральной плотности поясничного отдела позвоночника (со второго по четвертый поясничные позвонки), берцовой кости, и нижней челюсти определяли с использованием двухэнергетического рентгеновского сканера минеральной плотности кости (DCS-600EX, Aloka). Результаты представлены на фиг. 2 и 3.
Данные представлены в виде среднее значение + стандартная ошибка (SE). Программное обеспечение SAS preclinical package ver. 5.00 (SAS Institute Japan) использовали для проведения приведенных далее статистических анализов. Устанавливали уровень значимости 5% для обеих сторон. Что касается показателей минеральной плотности поясничного отдела позвоночника, берцовой кости и нижней челюсти, осуществляли множественное сравнение с использованием параметрического критерия Даннета (*P<0,05) для трех групп доз соединения 7 с использованием в качестве контроля группы Носитель-Контроль.
Как показано на фиг. 2, описывающей минеральную плотность поясничного отдела позвоночника и берцовой кости, в группах, которым вводили соединение 7, наблюдали существенный дозозависимый эффект увеличения минеральной плотности кости по сравнению с группой Носитель-Контроль. Кроме того, процентное значение увеличения минеральной плотности поясничного отдела позвоночника в трех группах, которым вводили соединение 7: группа 20 мг/кг, группа 30 мг/кг, и группа 50 мг/кг, по сравнению с группой Носитель-Контроль составляло 16%, 21%, и 25%, соответственно; и процентное значение увеличения минеральной плотности берцовой кости составляло 7%, 16% и 19%, соответственно. Как показано на фиг. 3, описывающей минеральную плотность нижней челюсти, в группе Соединение 7-30 мг/кг наблюдали существенный эффект увеличения минеральной плотности кости по сравнению с группой Носитель-Контроль. Процентное значение увеличения по сравнению с группой Носитель-Контроль составляло 7%.
Как описано выше, поскольку повторное пероральное введение соединения 7 крысам OVX в патологической модели остеопороза, вызывало увеличение минеральной плотности бедренной кости, и повторное внутривенное введение соединения 7 вызывало увеличение минеральной плотности поясничного отдела позвоночника, берцовой кости, и нижней челюсти у обычных крыс, системное воздействие соединения 7 может быть эффективным для профилактики, лечения, и обеспечения восстановления от патологических состояний, которые требуют индукции анаболизма костной ткани, увеличения костной массы, или регенерации кости, таких как остеопороз, снижение костной массы при периодонтальном заболевании, и дефект альвеолярной кости после удаления зуба. Кроме того, подтвердили, что соединения, представленные формулой (1), обладают сильными PTH-подобными эффектами и высокой метаболической стабильностью в ссылочных примерах 1-5, и они могут давать эффект увеличения минеральной плотности кости за счет анаболизма костной ткани с помощью PTH-подобного действия. Таким образом, соединения, представленные формулой (1), могут быть эффективны для профилактики, лечения, улучшения и способствования выздоровлению от патологических состояний, которые требуют индукции анаболизма костной ткани, увеличения костной массы, или регенерации кости, таких как остеопороз, уменьшение костной массы при периодонтальном заболевании, и дефект альвеолярной кости после удаления зуба.
[Пример 3] Супрессивные эффекты соединения 7 на конечную дифференцировку хондроцитов суставов кролика
Кроликов NZW (в возрасте 4 недель, Oriental Yeast Co., Ltd.) подвергали эвтаназии, и затем получали суставной хрящ берцовой кости, и переносили в 50 мл пробирку (Nippon Becton Dickinson Company). PBS (Nacalai Tesque, Inc.), содержащий 1% трипсин (Wako Pure Chemical Industries, Ltd.) добавляли в пробирку, и мягкие ткани разрушались при 37°C в течение одного часа. Затем осуществляли центрифугирование при 1200 об/мин в течение пяти мин, удаляли супернатант и PBS(-) добавляли для суспендирования хрящевой ткани. Смесь центрифугировали при 1200 об/мин в течение пяти мин, с последующим удалением супернатанта, и отмывали три раза суспендированием в PBS(-). Смесь центрифугировали при 1200 об/мин в течение пяти мин, и затем клеточный осадок разрушали обработкой DMEM (Life Technologies Japan, Ltd.), содержащим 0,2% коллагеназы II типа (CLS-2, Worthington Biochemical Corp.) при 37°C в течение трех часов. Фетальную бычью сыворотку (Life Technologies Japan, Ltd.) добавляли в количестве 10% об/об для остановки реакции, и смесь энергично пипетировали с использованием 10-мл пипетки (Nippon Becton Dickinson Company) для выделения хондроцитов. Смесь центрифугировали при 1200 об/мин в течение пяти мин с последующим удалением супернатанта и отмывали три раза путем суспендирования в 10% FCS-содержащем DMEM, и хондроциты высевали в количестве 1×104 клеток/лунку на покрытый коллагеном I типа 96-луночный культуральный планшет (AGC TECHNO GLASS CO., LTD.). Среду меняли три раза в неделю; и после того как клетки достигали конфлюентности, их культивировали в среде DMEM, содержащей 100 мкг/мл n-гидрата магниевой соли фосфата L-аскорбиновой кислоты (Wako Pure Chemical Industries, Ltd.), 10 ммоль/л β-глицерофосфата пентагидрат (Wako Pure Chemical Industries, Ltd.) и 10% FCS. Приведенные ниже условия использовали для культивирования клеток и окраски на щелочную фосфатазу (культивировали в течение 14 дней) и окрашивания ализариновым красным (культивировали в течение 21 дней) осуществляли для оценки конечной дифференцировки хондроцитов.
Окраску на щелочную фосфатазу осуществляли путем удаления среды, последующей однократной отмывки хондроцитов 200 ммоль/л буфером Tris-HCl pH 8,2, окрашивания клеток согласно протоколу, прилагаемому к набору для окраски на щелочную фосфатазу (Vector Red Alkaline Phosphatase Substrate Kit I, Vector Laboratories, Inc.), и получали фотографии с помощью инвертированного микроскопа (Nikon Corporation) (объектив 4x).
В результате показали, что BMP-2 увеличивало активность щелочной фосфатазы, и как соединение 7 так и PTH(1-34) подавляли активность щелочной фосфатазы зависимым от концентрации образом (фиг. 4A).
Окрашивание ализариновым красным осуществляли путем удаления среды с последующей двукратной отмывкой хондроцитов PBS, фиксации клеток 100% этанолом (Wako Pure Chemical Industries Ltd.) в течение 15 мин, удаления этанола, и затем окрашивания 1% ализариновым красным S (Wako Pure Chemical Industries Ltd.) в течение 15 мин, с последующей отмывкой дистиллированной водой; и получали фотографии с помощью инвертированного микроскопа. В результате BMP-2 существенно увеличивал способность к окрашиванию ализариновым красным и способствовал кальцификации; и как соединение 7, так и PTH(1-34) подавляли способность к окрашиванию ализариновым красным зависимым от концентрации образом и подавляли кальцификацию (фиг. 4B).
[Пример 4] Эффекты соединения 7 на синтез протеогликанов хондроцитами суставов человека
После приобретения криоконсервированных хондроцитов суставов человека (Lot 2867, Cell Applications Inc.), клетки размораживали на водяной бане при 37°C, 15 мл основной среды, дополненной 10% ростовой добавки (Growth Medium, Cell Applications Inc.) помещали в культуральный флакон T75 (CORNING, Corning Japan K. K.) для культивирования клеток, и на следующий день добавляли 15 мл питательной среды; и культивировали клетки в течение трех дней. Затем питательную среду удаляли и HBSS (Cell Applications Inc.) использовали для отмывки слоя хондроцитов. Добавляли 1 мл раствора трипсин/ЭДТА (Cell Applications Inc.), оставляли флакон при комнатной температуре приблизительно на пять минут и отделяли хондроциты от поверхности флакона. С добавлением 10 мл нейтрализующего раствора (Cell Applications Inc.), использовали гемоцитометр Bulker-Turk для определения числа клеток, и затем клетки переносили в 15 мл пробирку для центрифугирования (1200 об/мин в течение пяти минут, Tomy Seiko Co., Ltd.) для получения осадка хондроцитов. Супернатант удаляли и помещали клетки в 1,2% раствор альгината натрия (25 ммоль/л HEPES/150 ммоль/л раствор хлорида натрия, pH 7,0) в количестве 2×106 клеток/мл. Раствор набирали в 1 мл шприц (Terumo Corporation), снабженный инъекционной иглой размером 22G, добавляли по пять капель раствора в каждую лунку 24-луночного планшета (Corning Japan K.K.), содержащего 2 мл 102 ммоль/л водного раствора CaCl2, и оставляли планшет на пять минут для образования гранул. Затем, планшет отмывали три раза 150 ммоль/л раствором хлорида натрия и инкубировали в течение одного дня в питательной среде, и заменяли среду основной средой, дополненной 1% ростовой добавки (Cell Applications Inc.). В данном способе, указанные ниже факторы добавляли в среду и культивировали клетки в течение 13 дней, причем замену среды осуществляли три раза в неделю.
На 12-й день инкубирования 35S-меченую серную кислоту (PerkinElmer Japan Co., Ltd.) добавляли в количестве 370 кБк/лунку, и собирали среду через 24 часа в пробирку и хранили при 4°C. К гелю альгината добавляли 55 ммоль/л раствор цитрата натрия (Nacalai Tesque, Inc.; 1 мл/лунку) и инкубировали при 37°C в течение десяти минут для превращения геля в золь. Золь собирали в микропробирку (Eppendorf AG) и затем центрифугировали (1200 об/мин, пять мин.) для получения осадка хондроцитов. Осадок суспендировали путем добавления 0,5 мл 1 мг/мл актиназа Е (Kaken Pharmaceutical Co., Ltd.)-содержащего раствора 0,2 моль/л Tris-HCl (Sigma-Aldrich Co. LLC.)/5 ммоль/л CaCl2 (Nacalai Tesque, Inc.), pH 7,8, в микропробирку. Суспензию переносили в 12-луночный планшет (Corning Japan K.K.), герметично закрывали и затем инкубировали в течение ночи в инкубаторе для яиц (ESPEC CORP.), установленном на 50°C. Переносили 0,4 мл указанного раствора продуктов расщепления клеток в пробирку, добавляли 250 мкл 0,1 мг/мл водного раствора хондроитина сульфата (Wako Pure Chemical Industries, Ltd.), и по 2,5 мл каждого из растворов 2 ммоль/л MgSO4 (Wako Pure Chemical Industries, Ltd.), 0,2 моль/л Tris-HCl (Sigma Aldrich)/5 ммоль/л CaCl2 (Nacalai Tesque, Inc.) pH 7,8, 1% цетилпиридиния хлорида (CPC, Wako Pure Chemical Industries, Ltd.)/20 ммоль/л NaCl (Nacalai Tesque, Inc.), и указанную смесь инкубировали при 37°C в течение трех часов. Указанный раствор фильтровали через стеклянный фильтр (GC-50, ADVANTEC) путем отсасывания с помощью вакуумного насоса, и 1% CPC/20 ммоль/л NaCl использовали при отмывке для удаления свободной 35S-меченой серной кислоты. Стеклянный фильтр переносили во флакон для жидкостного сцинтилляционного счетчика, добавляли 5 мл сцинтилляционного коктейля (Hionic-Fluor, PerkinElmer Japan Co., Ltd.) и измеряли радиоактивность с помощью жидкостного сцинтилляционного счетчика (TRI-CARB, PerkinElmer Japan Co., Ltd.).
Оставшийся 0,1 мл раствора продуктов расщепления клеток использовали для количественного анализа ДНК. К 1 мл буфера, включенного в набор для количественного определения ДНК (Cosmo Bio Co., Ltd.), 100 мкл окрашивающего раствора и 450 мкл раствора продуктов расщепления клеток добавляли вместе, и измеряли интенсивность флуоресценции при 458 нм с возбуждением при 356 нм (Infinite M200, Tecan Group Ltd.).
Стандартную кривую для определения концентрации ДНК получали путем приготовления растворов с серийным двукратным разведением стандартного раствора (100 мкг/мл), входящего в набор. Линейное уравнение регрессии получали (Excel, Microsoft) по результатам измерения стандартной кривой, и рассчитывали концентрации ДНК в образцах.
Радиоактивность каждой лунки стандартизировали по содержанию ДНК (кБк/мкг ДНК).
В результате, для положительного контроля, TGF-β1, наблюдали 10-кратное увеличение радиоактивности относительно контрольной группы, получавшей носитель, и повышенное количество синтеза протеогликанов. Для PTH(1-34) наблюдали 6-кратное увеличение радиоактивности относительно контрольной группы, получавшей носитель. Для соединения 7 наблюдали 3-кратное увеличение радиоактивности относительно контрольной группы, получавшей носитель, для дозы 10-6 моль/л, и 10-ратное увеличение радиоактивности относительно контрольной группы, получавшей носитель, для дозы 10-5 моль/л (фиг. 5). Результаты показывают, что соединение 7 обладает эффектом стимуляции синтеза хрящевого матрикса в хондроцитах суставов человека.
[Пример 5] Эффекты соединения 7 в модели с использованием кроликов с частично удаленным мениском
Самцов кроликов NZW в возрасте двенадцати недель (Oriental Yeast Co., Ltd.) акклиматизировали в течение пяти дней, и затем под анестезией изофлураном, рассекали кожу на латеральной поверхности левого коленного сустава и латеральную коллатеральную связку и сесамовидную связку хирургически удаляли для открытия латерального мениска. Центр латерального мениска шириной 3-4 мм иссекали для создания модели остеоартрита (Kikuchi T et al., Osteoarth Cart 1999; 4(2): 99-110). Кончики трех полиэтиленовых трубок (PE60, Nippon Becton Dickinson Company), соединенных с тремя осмотическими насосами (2ML1, Durecm, Road Cupertino, CA, UС), которые внедряли подкожно на левом бедре, помещали в сустав, и раствор препарата непрерывно вводили в коленный сустав. Каждый осмотический насос заполняли одним из перечисленного 1) носитель, в качестве контроля (50% диметилсульфоксид/50% физиологический раствор об./об.); 2) соединение 7 в дозе 3,0 мкг/мл; или 3) соединение 7 в дозе 30 мкг/мл. На 7-й день после операции кроликов вновь подвергали анестезии изофлураном, и первоначально установленный насос заменяли осмотическим насосом, заполненным тем же фармацевтическим агентом, каким был заполнен первоначально установленный насос. Кроликов подвергали эвтаназии через 14 дней, хирургически получали часть мениска. Выделяли бедренную и берцовую кости и фиксировали их выдерживанием в 20% нейтральном забуференном растворе формалина. Затем грубую структуру на поверхности суставного хряща окрашивали тушью (фиг. 7). Изображения структуры поверхности берцовой кости получали с помощью цифрового микроскопа (VHX-2000, Keyence Corporation), определяли площадь участка повреждения, окрашенного тушью, и площадь неповрежденного наружного мыщелка, и рассчитывали долю, которую занимает участок повреждения во всем наружном мыщелоке (фиг. 6). В результате, обнаружили, что соединение 7 уменьшает площадь участка повреждения дозозависимым образом. Фотографии поверхности суставного хряща берцовой кости, полученные во время данного эксперимента, представлены на фиг. 4.
[Пример 6] Эффекты в отношении хряща ростовой пластинки после четырех недель повторного перорального введения обычным крысам.
Самок крыс RccHan:WISТ, полученных из Japan Laboratory Animals Inc., акклиматизировали в течение по меньше мере одной недели в стандартных лабораторных условиях 20°C-26°C и 30%-70% влажности, и затем использовали в экспериментах. Крысы имели свободный доступ к водопроводной воде и стандартному корму для грызунов (CE-2, Clea Japan Inc.), содержащему 1,1% кальция, 1,0% фосфорной кислоты и 250 МЕ/100 г витамина D3.
После измерения веса тела у шестинедельных крыс, их распределяли по группам, так чтобы значения среднего веса тела в каждой группе из десяти крыс были бы одинаковыми. На следующий день после распределения по группам всем крысам вводили препарат или носитель один раз в день в течение четырех недель. Растворитель (носитель) вводили перорально группе Носитель-Контроль. В группе Соединение 7-6 мг/кг, группе Соединение 7-60 мг/кг, и группе Соединение 7-600 мг/кг, соединение 7, суспендированное в носителе, вводили перорально в дозе 6 мг/кг, 60 мг/кг, и 600 мг/кг, соответственно. Для всех групп вводимый объем составлял 2 мл/кг. Пропиленгликоль (специальная марка, Kanto Chemical Co., Inc.) использовали в качестве носителя. Крыс подвергали эвтаназии под анестезией через один день после последнего введения соединения, путем забора крови из брюшной аорта, и затем осуществляли аутопсию для получения бедренной кости. Бедренную кость фиксировали 10% нейтральным буферным раствором формалина, и после деминерализации, готовили образцы срезов тканей, залитых парафином (окрашенных гематоксилин-эозином). Дистальные концы бедренной кости полученных образцов исследовали гистопатологически с помощью светового микроскопа. Результаты представлены в таблице 1, и типичные гистологические изображения представлены на фиг. 8.
Гистологические изменения в хряще ростовой пластинки бедренной кости после четырех недель повторного перорального введения соединения обычным крысам
контроль
Как показано в таблице 1, в группах, получавших соединение 7, наблюдали дозозависимое утолщение хряща ростовой пластинки бедренной кости по сравнению с контрольной группой, получавшей носитель. Когда ширину типичного хряща ростовой пластинки (указанную стрелкой) определяли на основании изображения ткани на фиг. 8 с использованием шкалы, она составляла приблизительно 390 мкм для индивида в контрольной группе, получавшей носитель, и приблизительно 2940 мкм для индивида в группе, получавшей соединение 7-600 мг/кг.
Как описано выше, повторное пероральное введение соединения 7 вызывало утолщение хряща ростовой пластинки бедренной кости крысы. Такие эффекты обусловлены индукцией анаболизма хряща, подавлением конечной дифференцировки хряща или стимуляцией роста хряща с помощью соединения 7, и следовательно пероральное введение соединения 7 может быть эффективным для лечения остеоартрита. Кроме того, подтвердили, что соединения, представленные формулой (1), обладают сильными PTH-подобными эффектами и высокой метаболической стабильностью в ссылочных примерах 1-5, и предполагается, что они будут эффективны для лечения остеоартрита благодаря анаболизму хрящевой ткани за счет PTH-подобного действия.
[Ссылочные примеры]
Содержание настоящего изобретения будет описано более подробно с помощью следующих примеров и примера тестирования; однако настоящее изобретение не ограничивается содержанием примеров и примера тестирования. Все исходные вещества и реагенты получены от коммерческих поставщиков или синтезированы с использованием известных способов. Спектры 1H-ЯМР измеряли с использованием Mercury300 (производства Varian), ECP-400 (производства JEOL) или 400-MR (производства Varian) с использованием или без Me4Si в качестве внутреннего стандарта (s=синглет, d=дуплет, t=триплет, brs=широкий синглет, m=мультиплет). Масс-спектрометрическое измерение проводили с использованием масс-спектрометра, ZQ2000 (производства Waterс), SQD (производства Waters) или 2020 (производства Shimazu).
Ссылочный пример 1
1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-дион (соединение 1)
Реакция (1-1)
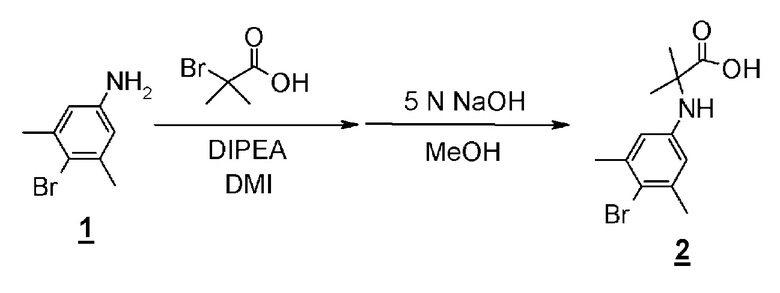
К раствору 4-бром-3,5-диметиланилина (3,47 г, 17,4 ммоль) и диизопропилэтиламина (5,3 мл, 30,4 ммоль) в DMI (13 мл), добавляли 2-бромизомасляную кислоту (3,86 г, 23,1 ммоль) при комнатной температуре. Смесь перемешивали при 100°C в течение одного часа. Затем добавляли 2-бромизобутират (496 мг, 2,97 ммоль) и диизопропилэтиламин (0,8 мл, 4,59 ммоль) и смесь перемешивали при 100°C в течение одного часа.
Метанол (52 мл) и 5 н водный раствор гидроксида натрия (52 мл, 260 ммоль) добавляли к реакционной смеси при комнатной температуре и затем указанную смесь перемешивали при 75°C в течение 1,5 часов. Реакционную смесь охлаждали, с последующим добавлением воды и доведением значения pH до 5 с помощью 1 N водного раствора гидросульфата калия, и затем экстрагировали с использованием этилацетата. Органический слой отмывали водой, затем высушивали над безводным сульфатом магния, и концентрировали с получением 2-((4-бром-3,5-диметилфенил)амино)-2-метилпропановой кислоты в виде неочищенного продукта (5,79 г).
MS(ESI) m/z=286, 288 (M+H)+
(Реакция 1-2)
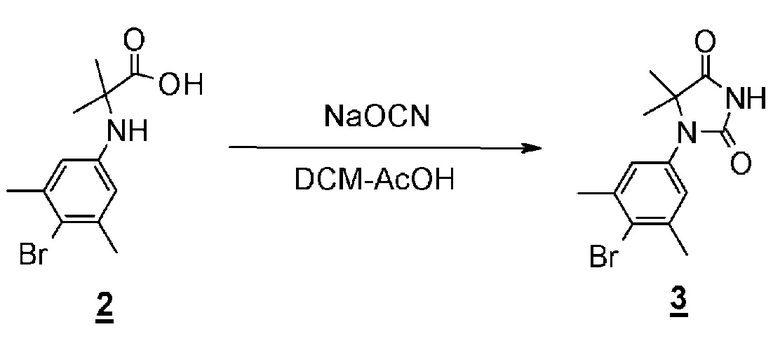
К смеси 2-((4-бром-3,5-диметилфенил)амино)-2- метилпропионовой кислоты (5,79 г соединения, полученного в реакции 1-1) в дихлорметане (62 мл) и уксусной кислоты (62 мл), добавляли цианат натрия (5,03 г, 59,8 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение трех часов. Насыщенный раствор гидрокарбоната натрия (400 мл) добавляли для доведения значения pH до 7-8 с использованием 5 н водного раствора гидроксида натрия, и указанную смесь экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученное твердое вещество промывали последовательно этилацетатом-гексаном и затем дихлорметаном-гексаном с получением 1-(4-бром-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (3,80 г, 66%).
MS(ESI) m/z=311, 313 (M+H)+
(Реакция 1-3)
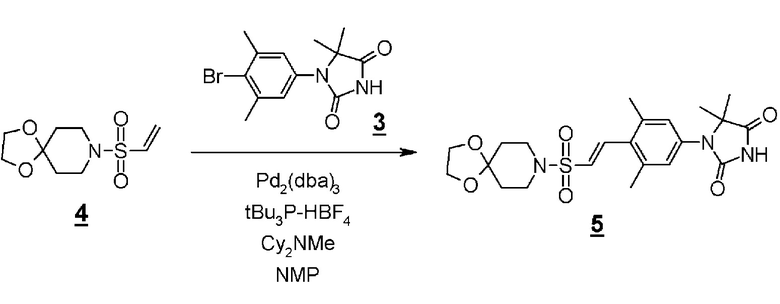
Смесь 8-(винилсульфонил)-1,4-диокса-8-азаспиро[4,5]декана (431 мг, 1,85 ммоль), 1-(4-бром-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (575 мг, 1,85 ммоль), трис(дибензилиденацетон)палладия (0) (508 мг, 0,55 ммоль), три-терт-бутилфосфинтетрафторборной кислоты (165 мг, 0,55 ммоль) и метилциклогексиламина (2,1 мл, 9,25 ммоль) в N-метил-2-пирролидоне (18,5 мл) перемешивали в атмосфере азота при 110°C в течение двух часов. Реакционную смесь охлаждали, гасили водой, и затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Осадок очищали колоночной хроматографии на аминосиликагеле (дихлорметан - метанол) с получением (E)-1-(4-(2-(1,4-диокса-8-азаспиро[4,5]декан-8-ил-сульфонил) винил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (584 мг, 68%).
MS(ESI) m/z=464 (M+H)+
(Реакция 1-4)
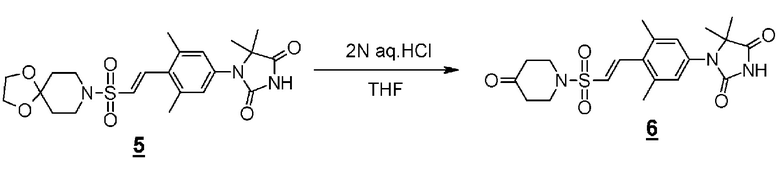
К раствору (E)-1-(4-(2-(1,4-диокса-8-азаспиро[4,5]декан-8-ил-сульфонил) винил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (1,2 г, 2,58 ммоль) в тетрагидрофуране (26 мл), 2 N водный раствор соляной кислоты (26 мл, 52 ммоль) добавляли по каплям в течение десяти минут. Смесь перемешивали при 60°C в течение двух часов. Реакционную смесь охлаждали, с последующим доведением значения pH до 7 с помощью 2 N водного раствора гидроксида натрия, и указанную смесь экстрагировали этилацетатом. Органический слой отмывали солевым раствором, сушили над безводным сульфатом магния, и затем концентрировали при пониженном давлении. Осадок очищали с помощью колоночной хроматографии на силикагеле (дихлорметан - этилацетат) с получением (E)-1-(3,5-диметил-4-(2-((4-оксопиперидин-1-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (998 мг, 92%).
MS(ESI) m/z=420 (M+H)+
(Реакция 1-5)
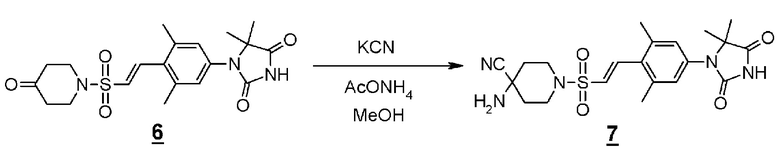
К раствору (E)-1-(3,5-диметил-4-(2-((4-оксопиперидин-1-ил)сульфонил)винил) фенил)-5,5-диметилимидазолидин-2,4-диона (994 мг, 2,37 ммоль) в метаноле (24 мл) добавляли цианид калия (188 мг, 2,84 ммоль) и ацетат аммония (237 мг, 3,08 ммоль) при комнатной температуре. Смесь перемешивали при 60-70°C в течение трех часов. Реакционную смесь охлаждали, концентрировали при пониженном давлении, и затем разводили этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния, и затем концентрировали при пониженном давлении. Осадок очищали с помощью колоночной хроматографии на силикагеле (этилацетат - гексан) с получением (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбонитрила (681 мг, 68%).
1H-ЯМР (300 МГц, ДМСО-d6) δ: 1,3 (6H, с), 1,7 (2H, м), 2,0 (2H, м), 2,3 (6H, с), 2,7 (2H, с), 2,9 (2H, м), 3,4 (2H, м), 6,9 (1H, д, J=15,9 Гц), 7,1 (2H, с), 7,4 (1H, д, J=15,9 Гц), 11,2 (1H, уш.с)
(Реакция 1-6)
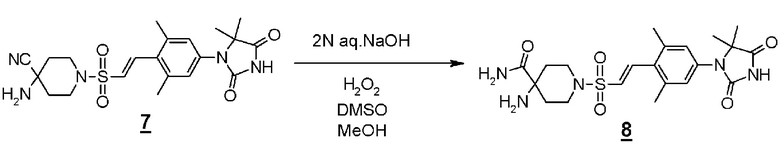
К раствору (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбонитрила (675 мг, 1,50 ммоль) в метаноле (7,5 мл) и диметилсульфоксида (0,195 мл) при комнатной температуре, добавляли 2 н водный раствор гидроксида натрия (1,6 мл, 1,6 ммоль) и затем добавляли медленно по каплям 30% водный раствор перекиси водорода (0,2 мл, 1,95 ммоль). Смесь перемешивали при комнатной температуре в течение одного часа. Этилацетат, гексан, и насыщенный водный раствор хлорида аммония добавляли к реакционной смеси. Твердую фазу собирали фильтрацией, промывали и сушили с получением (E)-4-амино-1-((4-(5,5-диметил-2,4-диoxoимидазолидин-1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4- карбоксамида (498 мг, 72%).
MS(ESI) m/z=464 (M+H)+
(Реакция 1-7)
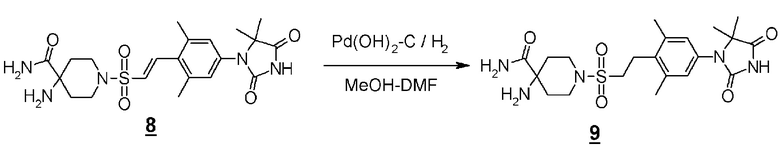
Смесь (E)-4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин -1-ил)-2,6-диметилстирил)сульфонил)пиперидин-4-карбоксамида (1,3 г, 2,8 ммоль) и гидроксида палладия на угле (20% Pd) (смоченного приблизительно 50% воды) (1,3 г) в метаноле (21 мл) и диметилформамида (7 мл) перемешивали в атмосфере водорода при комнатной температуре в течение четырех часов. Реакционную смесь фильтровали и отмывали, и затем фильтрат концентрировали при пониженном давлении с получением 4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)пиперидин-4-карбоксамида (998 мг, 77%).
MS(ESI) m/z=466 (M+H)+
(Реакция 1-8)
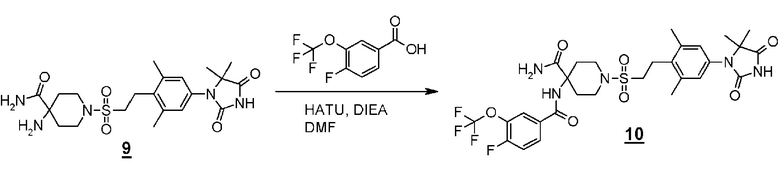
К раствору 4-амино-1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил) пиперидин-4-карбоксамида (120 мг, 0,258 ммоль) добавляли 4-фтор-3-(трифторметокси)бензойную кислоту (69 мг, 0,309 ммоль), и диизопропилэтиламин (0,09 мл, 0,516 ммоль) в диметилформамиде (2,5 мл), O-(7-азабензотриазол-1-ил)-1,1,3,3- тетраметилуронийгексафторфосфат (HATU) (118 мг, 0,309 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь гасили водой и затем экстрагировали дихлорметаном. Органический слой промывали насыщенным солевым раствором, промывали безводным сульфатом натрия, и затем концентрировали при пониженном давлении с получением 1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)-4-(4-фтор-3-(трифторметокси)бензамид)пиперидин-4-карбоксамида (150 мг, 67%).
MS(ESI) m/z=672 (M+H)+
(Реакция 1-9)
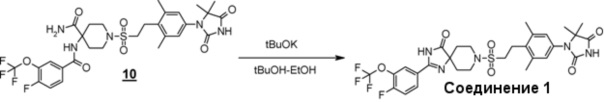
К смешанному раствору 1-((4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-2,6-диметилфенетил)сульфонил)-4-(4-фтор-3-(трифторметокси)бензамид)пиперидин-4- карбоксамида (150 мг, 0,223 ммоль) в трет-бутаноле (2,5 мл) и этаноле (2,5 мл), добавляли трет-бутоксид калия (75 мг, 0,670 ммоль) при 0°C. Смесь перемешивали в атмосфере азота при 50°C в течение 1,5 часов. Реакционную смесь охлаждали, разводили водой, гасили насыщенным водным раствором хлорида аммония и затем экстрагировали дихлорметаном. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный осадок очищали с помощью колоночной хроматографии на силикагеле (дихлорметан - метанол) с получением 1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил) -4-оксо-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона 118 мг, 81%).
MS(ESI) m/z=654 (M+H)+, 1H-ЯМР (400 МГц, CD3OD) δ: 1,40 (6H, с), 1,71-1,80 (2H, м), 2,00-2,08 (2H, м), 2,43 (6H, с), 3,22 (4H, с), 3,47-3,57 (2H, м), 3,80-3,88 (2H, м), 7,01 (2H, с), 7,50-7,57 (1H, м), 7,97-8,04 (1H, м), 8,05-8,12 (1H, м)
Приведенные далее соединения ссылочных примеров синтезировали с помощью действий, аналогичных действиям реакций 1-8 и 1-9 в ссылочном примере 1, с использованием подходящих исходных карбоновых кислот, реагентов и растворителей and solvents.
(соединение 2-5)
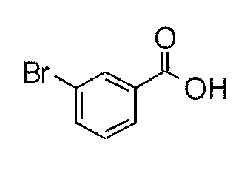
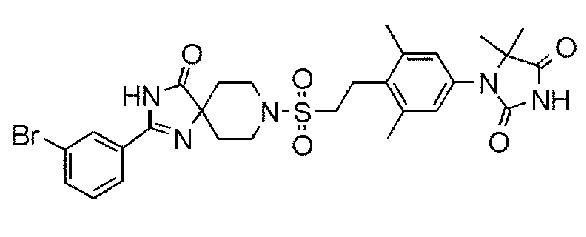
1H-ЯМР (400МГц, ДМСО-d6) δ: 1,30 (6H, с), 1,56-1,63 (2H, м), 1,80-1,90 (2H, м), 2,37 (6H, с), 3,00-3,08 (2H, м), 3,23-3,30 (2H, м), 3,32-3,41 (2H, м), 3,67-3,73 (2H, м), 7,00 (2H, с), 7,50 (1H, дд, J=8,8 Гц), 7,77-7,82 (1H, м), 7,95-8,00 (1H, м), 8,13-8,20 (1H, м), 11,10 (1H, уш.с), 11,70 (1H, уш.с)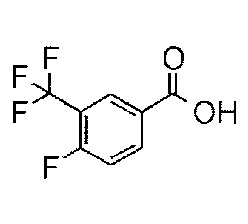
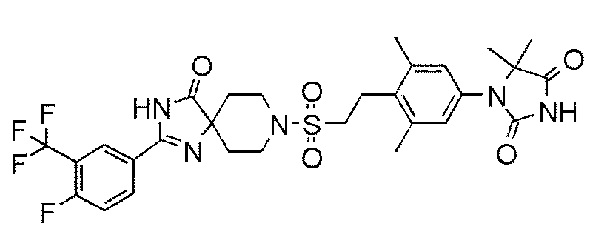
1H-ЯМР (400МГц, CDCI3) δ: 1,47 (6H, с), 1,70-1,78 (2H, м), 2,09-2,18 (2H, м), 2,40 (6H, с), 3,00-3,08 (2H, м), 3,20-3,28 (2H, м), 3,44-3,54 (2H, м), 3,80-3,88 (2H, м), 6,94 (2H, С), 7,34 (1H, т, J=9,6 Гц), 8,02 (1H, уш.с), 8,08-8,13 (1H, м), 8,20-8,24 (1H, м), 10,10 (1H, уш.с)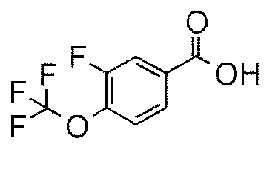
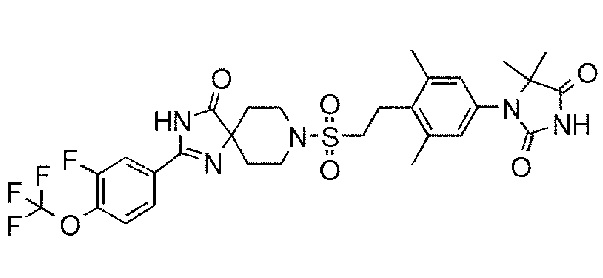
1H-ЯМР (400МГц, ДМСО-d6) δ: 1,30 (6H, с), 1,58-1,64 (2H, м), 1,81-1,91(2H, м), 2,37 (6H, с), 3,00-3,08 (2H, м), 3,22-3,31 (2H, м), 3,32-3,42 (2H, м), 3,68-3,73 (2H, м), 7,00 (2H, с), 7,76-7,82 (1H, м), 7,95 (1H, д, J=9,6Гц), 8,05 (1H, дд, J=9,6, 2 Гц), 11,09 (1H,с),
11,79 (1H, с)
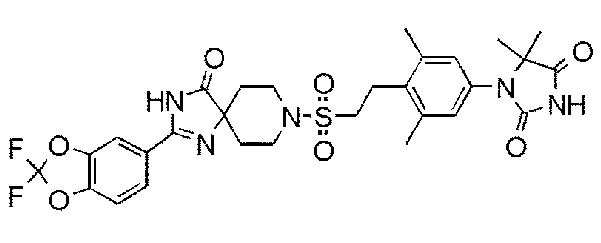
1H-ЯМР (400МГц, CDCI3) δ: 1,47 (6H, с), 1,65-1,73 (2H, м), 2,11-2,20 (2H, м), 2,39 (6H, с), 2,98-3,04 (2H, м), 3,18-3,25 (2H, м), 3,40-3,52 (2H, м), 3,82-3,90 (2H, м), 6,94 (2H, с), 7,17 (1H, д, J=8,4 Гц), 7,63 (1H, д, J=8,4 Гц), 7,75 (1H, с), 8,49 (1H, уш.с), 10,46 (1H, уш.с)
Ссылочный пример 2
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-дион (соединение 6)
(Реакция 2-1)
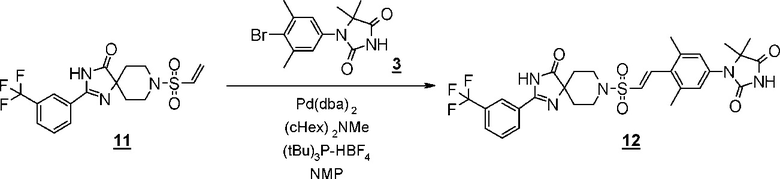
Смесь 2-(3-(трифторметил)фенил)-8-(винилсульфонил) -1,3,8-триазаспиро[4,5]дека-1-ен-4-она (150 мг, 0,387 ммоль), синтезированного по способу, описанному в схемах 2, 3, и 12 WO2010/126030(A1), 1-(4-бром-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона (169 мг, 0,542 ммоль), бис(дибензилиденацетон)палладия (45 мг, 0,077 ммоль), три-трет-бутилфосфинтетрафторборной кислоты (22 мг, 0,077 ммоль), и метилциклогексиламина (0,123 мл, 0,581 ммоль) в N-метил-2-пирролидоне (0,97 мл) перемешивали при 100°C в течение одного часа в атмосфере азота. Реакционную смесь охлаждали, гасили водой и затем экстрагировали этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный осадок очищали с помощью колоночной хроматографии на силикагеле (этилацетат - гексан) с получением (E)-1-(3,5-диметил-4- (2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (197 мг, 82%).
MS(ESI) m/z=618 (M+H)+
(Реакция 2-2)
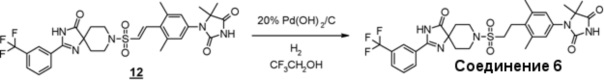
Смесь (E)-1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)винил)фенил)-5,5-диметилимидазолидин-2,4-диона (195 мг, 0,316 ммоль) и гидроксида палладия/угля (20% Pd) (смоченного приблизительно 50% воды) (195 мг, 0,139 ммоль) в 2,2,2-трифторэтаноле (6 мл) перемешивали при комнатной температуре в течение 14 часов в атмосфере водорода. Смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный осадок очищали с помощью колоночной хроматографии на силикагеле (этилацетат - гексан) с получением 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-диона (121 мг, 62%).
MS(ESI) m/z=620 (M+H)+, 1H-ЯМР (400 МГц, CD3OD) δ: 1,40 (6H, с), 1,72-1,81 (2H, м), 2,00-2,10 (2H, м), 2,44 (6H, с), 3,22 (4H, с), 3,50-3,58 (2H, м), 3,80-3,88 (2H, м), 7,01 (2H, с), 7,72-7,79 (1H, м), 7,88-7,94 (1H, м), 8,16-8,23 (1H, м), 8,31 (1H, с).
Ссылочный пример 3
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5- диметилимидазолидин-2,4-дион (соединение 7)
(Реакция 3)
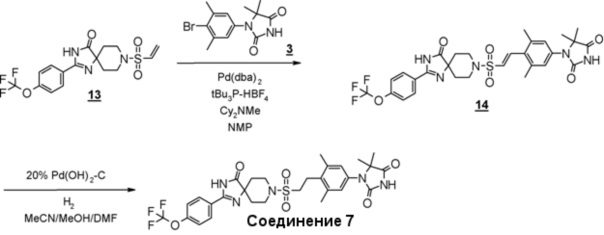
При использовании соответствующих исходных веществ и растворителей 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион (соединение 7) синтезировали с помощью действий, аналогичных действиям, описанным в ссылочном примере 2.
MS(ESI) m/z=636 (M+H)+, 1H-ЯМР (400 МГц, CDCl3) δ: 1,47 (6H, с), 1,70-1,78 (2H, м), 2,10-2,19 (2H, м), 2,40 (6H, с), 3,00-3,07 (2H, м), 3,19-3,25 (2H, м), 3,45-3,53 (2H, м), 3,81-3,88 (2H, м), 6,94 (2H, с), 7,35 (2H, д, J=8,0 Гц), 7,73 (1H, уш.с), 7,93 (2H, д, J=8,0 Гц), 9,37 (1H, уш.с)
Ссылочный пример 4
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3- диазаспиро[4,4]нонан-2,4-дион (соединение 8)
(Реакция 4-1)
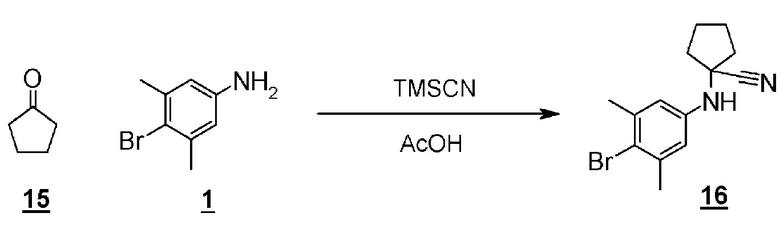
К смеси циклопентанона (42 мг, 0,500 ммоль) и 4-бром-3,5-диметиланилина (100 мг, 0,500 ммоль) в уксусной кислоте (0,5 мл), добавляли триметилсилилцианид (0,063 мл, 0,500 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1,5 часов в атмосфере азота. Реакцию в смеси гасили 28% водным раствором аммиака (1 мл), разводили водой и экстрактировали дихлорметаном. Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением 1-((4-бром-3,5-диметилфенил)амино) циклопентанкарбонитрила в виде неочищенного продукта (152 мг).
1H-ЯМР (400 МГц, CDCl3) δ: 1,83-1,92 (4H, м), 2,07-2,15 (2H, м), 2,33-2,42 (2H, м), 2,37 (6H, м), 3,71 (1H, уш.с), 6,56 (2H, с)
(Реакция 4-2)
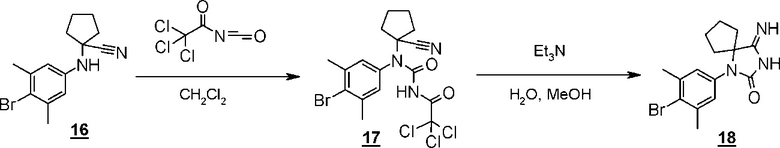
К раствору 1-((4-бром-3,5-диметилфенил)амино) циклопентанкарбонитрила (145 мг, 0,495 ммоль) в дихлорметане (5 мл), 2,2,2-трихлороактилизоцианат (0,070 мл, 0,593 ммоль) добавляли при комнатной температуре. Смесь перемешивали при комнатной температуре в течение одного часа в атмосфере азота.
Добавляли триэтиламин (0,103 мл, 0,742 ммоль), воду (0,045 мл), и метанол (0,10 мл) и смесь нагревали в сосуде с обратным холодильником в течение 1,5 часов в атмосфере азота. Реакционную смесь охлаждали, с последующим разбавлением водой и доведением значения pH до 5 с использованием 1 N водного раствора соляной кислоты, и затем экстрагировали дихлорметаном. Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем концентрировали при пониженном давлении с получением 1-(4-бром-3,5-диметилфенил)-4-имино-1,3-диазаспиро[4,4]нонан-2-она в виде неочищенного продукта.
MS(ESI) m/z=336, 338 (M+H)+
(Реакция 4-3)
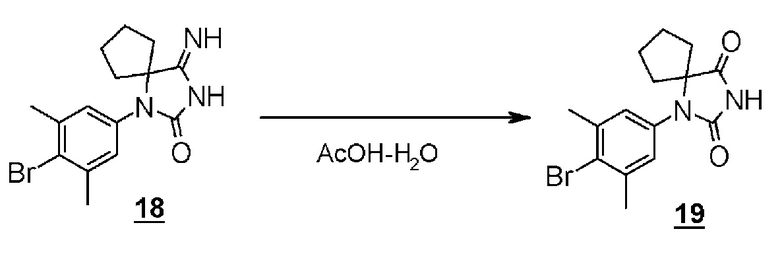
Смесь 1-(4-бром-3,5-диметилфенил)-4-имино-1,3-диазаспиро[4,4]нонан-2- она (неочищенный продукт, полученный в предыдущей реакции) в уксусной кислоте (1,0 мл) и воды (0,25 мл) перемешивали в течение 1,5 часов при 65°C в атмосфере азота. После дополнительного добавления уксусной кислоты (1,0 мл) и воды (0,25 мл), смесь перемешивали в течение 17 часов при 65°C в атмосфере азота. Реакционную смесь охлаждали, с последующим разведением водой и доводили значение pH до 8 с использованием насыщенного водного раствора гидрокарбоната натрия, и экстрагировали этилацетатом. Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем концентрировали при пониженном давлении. Осадок очищали с помощью колоночной хроматографии на силикагеле (этилацетат - гексан) с получением 1-(4-бром-3,5-диметилфенил)-1,3-диазаспиро[4,4]нонан-2,4-диона (121 мг).
MS(ESI) m/z=337, 339 (M+H)+
(Реакция 4-4)
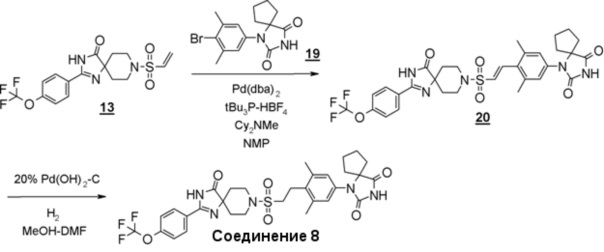
При использовании соответствующих исходных веществ и растворителей, 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4,4]нонан-2,4-дион (соединение 8) получали с помощью действий, аналогичных действиям, описанным в ссылочном примере 2.
MS(ESI) m/z=662 (M+H)+, 1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,36-1,44 (2H, м), 1,60-1,70 (4H, м), 1,82-1,91 (2H, м), 1,91-2,06 (4H, м), 2,38 (6H, с), 3,01-3,09 (2H, м), 3,22-3,30 (2H, м), 3,30-3,42 (2H, м), 3,70-3,77 (2H, м), 7,03 (2H, с), 7,57 (2H, д, J=8,4 Гц), 8,14 (2H, д, J=8,4 Гц)
Ссылочный пример 5
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8-метил-1,3,8-триазаспиро[4,5]декан-2,4-дион (соединение 9)
(Реакция 5-1)
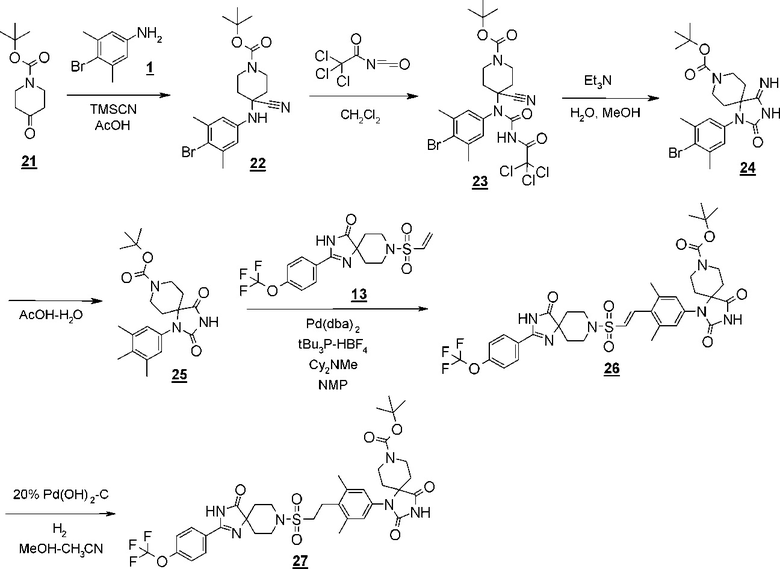
При использовании трет-бутилового эфира 4-оксопипередин-1- карбоновой кислоты в качестве исходного вещества, и соответствующего растворителя, трет-бутиловый эфир 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8- триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2,4-диоксо-1,3,8-триазаспиро[4,5]декан-8-карбоновой кислоты получали с помощью действий, аналогичных действиям, описанным в ссылочном примере 4.
MS(ESI) m/z=777 (M+H)+.
(Реакция 5-2)
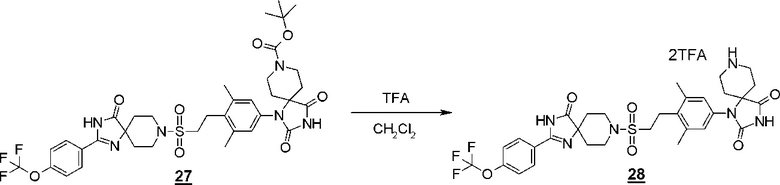
К смешанному раствору трет-бутилового эфира 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8- триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2,4-диоксо-1,3,8-триазаспиро[4,5]декан-8-карбоновой кислоты (11,7 мг, 0,015 ммоль) в дихлорметане (0,13 мл), добавляли трифторуксусную кислоту (0,05 мл, 0,673 ммоль) при комнатной температуре. Смесь помещали под струю азота и перемешивали при комнатной температуре в течение одного часа. Реакционную смесь концентрировали при пониженном давлении для получения соли 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8- триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3,8- триазаспиро[4,5]декан-2,4-дион-2-трифторуксусной кислоты (13,6 мг).
MS(ESI) m/z=677 (M+H)+.
(Реакция 5-3)
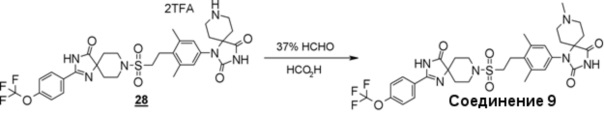
К смеси соли 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3,8-триазаспиро[4,5]декан-2,4-дион-2-трифторуксусной кислоты (21,1 мг, 0,022 ммоль) и муравьиной кислоты (0,033 мл), добавляли 37% водный раствор формальдегида (0,055 мл). Смесь помещали под струю азота, и перемешивали в течение трех часов при нагревании до 80°C. Реакционную смесь концентрировали и полученный остаток разводили этилацетатом. Органический слой отмывали разбавленным водным раствором гидроксида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии (дихлорметан - метанол) для очистки с получением 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил-8-метил-1,3,8-триазаспиро[4,5]декан-2,4-диона (4,5 мг, 30%).
MS(ESI) m/z=691 (M+H)+, 1H-ЯМР (400 МГц, CD3OD) δ: 1,76-1,84 (2H, м), 1,92-2,02 (2H, м), 2,02-2,12 (4H, м), 2,38 (3H, с), 2,46 (6H, с), 2,81-2,88 (2H, м), 2,92-3,02 (2H, м), 3,23 (4H, с), 3,51-3,60 (2H, м), 3,72-3,80 (2H, м), 7,01 (2H, с), 7,48 (2H, д, J=8,0 Гц), 8,10 (2H, д, J=8,0 Гц)
Ссылочный пример 6
5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3,4]октан-6,8-дион (соединение 10)
(Реакция 6)
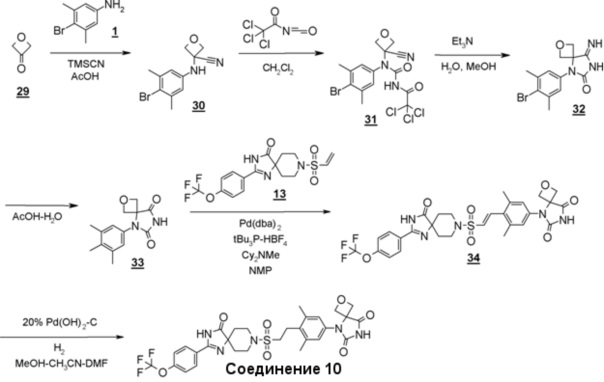
При использовании окситан-3-она, в качестве исходного вещества, и соответствующих растворителей, 5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8- триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3,4]октан-6,8-дион получали с помощью действий, аналогичных действиям ссылочного примера 4.
MS(ESI) m/z=650 (M+H)+, 1H-ЯМР (400 МГц, CDCl3) δ: 1,69-1,77 (2H, м), 2,12-2,22 (2H, м), 2,45 (6H, с), 3,03-3,11 (2H, м), 3,22-3,29 (2H, м), 3,46-3,53 (2H, м), 3,84-3,91 (2H, м), 4,86 (2H, д, J=7,2 Гц), 5,03 (2H, д, J=7,2 Гц), 7,07 (2H, с), 7,35 (2H, д, J=8,4 Гц), 7,98 (2H, д, J=8,4 Гц), 8,56 (1H, с), 10,34 (1H, с)
Ссылочный пример 7
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6- диазаспиро[2,4]гептан-5,7-дион (соединение 11)
(Реакция 7-1)
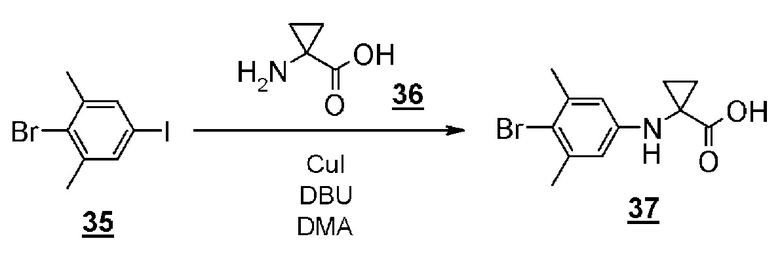
Смесь 2-бром-5-иод-1,3-диметилбензола (300 мг, 0,965 ммоль), 1- аминоциклопропанкарбоновой кислоты (195 мг, 1,93 ммоль), иодида меди (I) (37 мг, 0,194 ммоль), и диазабициклоундецена (0,50 мл, 3,35 ммоль) в диметилацетамиде (2,6 мл) перемешивали при 120°C в течение трех часов в атмосфере азота. Реакционную смесь очищали с помощью колоночной хроматографии с силикагелем (Wakosil C18, ацетонитрил - вода (0,1% муравьиная кислота)) с получением 1-((4-бром-3,5-диметилфенил)амино)циклопропанкарбоновой кислоты (219 мг, 80%).
MS(ESI) m/z=284, 286 (M+H)+.
(Реакция 7-2)
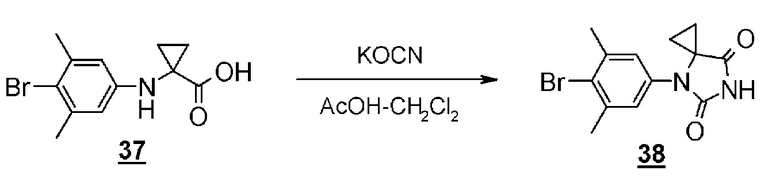
К смеси 1-((4-бромо-3,5-диметилфенил)амино) циклопропанкарбоновой кислоты (198 мг, 0,697 ммоль) в уксусной кислоте (3 мл) и дихлорметана (1,5 мл), добавляли цианат калия (424 мг, 5,23 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение одного часа, и затем перемешивали при 60°C в течение двух часов. Насыщенный водный раствор гидрокарбоната натрия добавляли для доведения значения pH до 8, и указанную смесь экстрагировали этилацетатом. Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии с силикагелем (этилацетат - гексан) с получением 4-(4-бром-3,5-диметилфенил)-4,6-диазаспиро[2,4]гептан-5,7-диона (49 мг, 23%).
MS(ESI) m/z=309, 311 (M+H)+.
(Реакция 7-3)
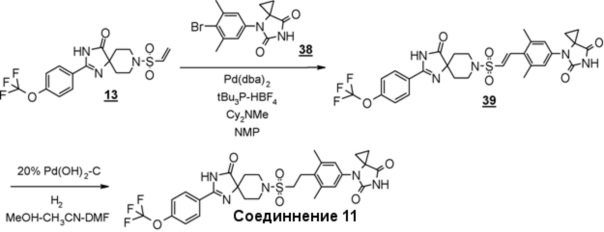
При использовании соответствующих исходных веществ и растворителей, 4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6-диазаспиро[2,4]гептан-5,7-дион (соединение 11) получали с помощью действий, аналогичных действиям ссылочного примера 2.
MS(ESI) m/z=634 (M+H)+, 1H-ЯМР (400 МГц, ДМСО-d6) δ: 0,99-1,03 (2H, м), 1,19-1,27 (4H, м), 1,58-1,64 (2H, м), 1,81-1,90 (2H, м), 2,35 (6H, с), 2,99-3,04 (2H, м), 3,22-3,29 (2H, м), 3,67-3,73 (2H, м), 6,95 (2H, с), 7,56 (2H, д, J=8,4 Гц), 8,12 (2H, д, J=8,4 Гц)
Ссылочный пример 8
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3- диазаспиро[4,4]нонан-2,4-дион (соединение 12)
(Реакция 8)
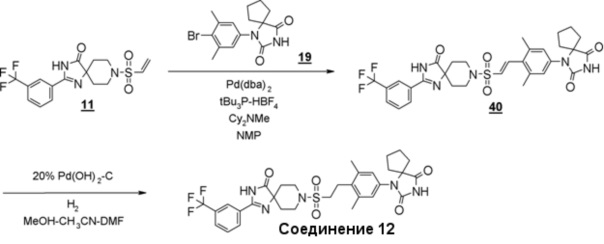
При использовании соответствующих исходных веществ и растворителей, 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4,4]нонан-2,4-дион получали с помощью действий, аналогичных действиям ссылочного примера 2.
MS(ESI) m/z=646 (M+H)+, 1H-ЯМР (400 МГц, ДМСО-d6) δ: 1,40-1,48 (2H, м), 1,62-1,71 (4H, м), 1,88-1,97 (2H, м), 1,97-2,08 (4H, м), 2,41 (6H, с), 3,03-3,10 (2H, м), 2,29-3,34 (2H, м), 3,38-3,47 (2H, м), 3,72-3,79 (2H, м), 7,06 (2H, с), 7,84 (1H, дд, J=7,6, 7,6 Гц), 8,02 (1H, д, J=7,6 Гц), 8,33 (1H, д, J=7,6 Гц), 8,38 (1H, с)
Ссылочные примеры тестирования
В отношении соединений по настоящему изобретению, результаты тестов измерения активности продукции цАМФ с использованием PTH1R человека, активности продукции цАМФ с использованием PTH1R крысы, метаболической стабильности с использованием микросом печени человека, метаболической стабильности с использованием гепатоцитов крысы, и кальцемического действия в модели крыс с TPTX показаны в ссылочных примерах тестирования с 1 по 5 соответственно. Соединения, описанные в WO2010/126030A1, которые представлены в таблице 3, использовали в качестве соединений для сравнения.
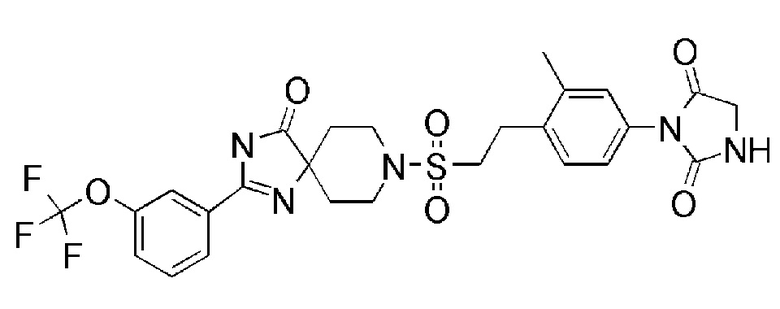
Соединение 799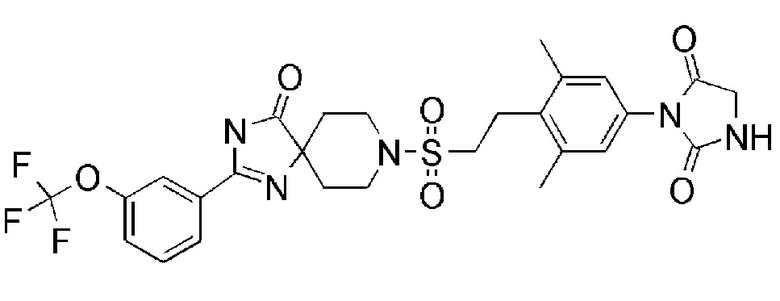
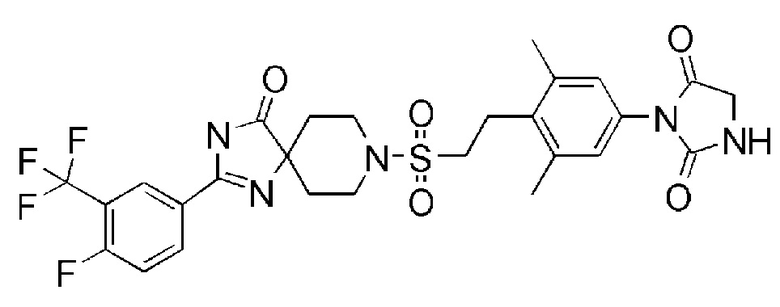
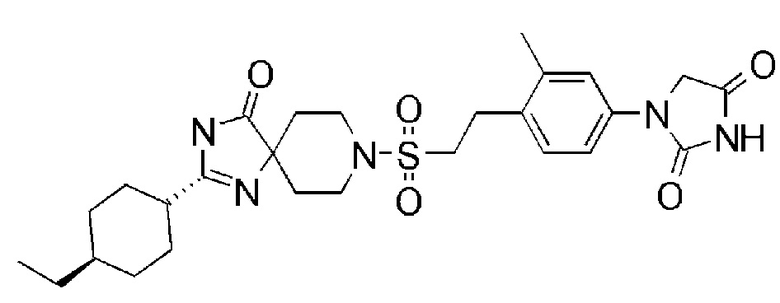
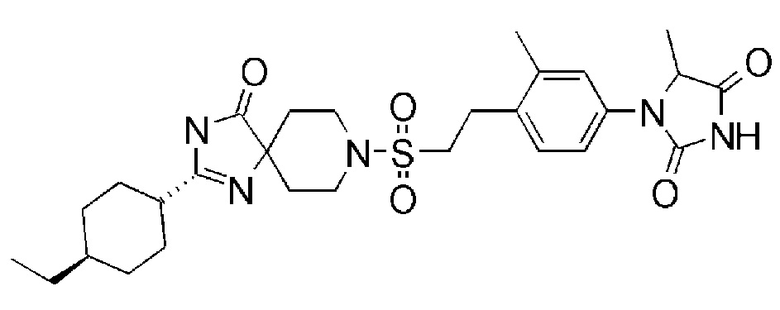
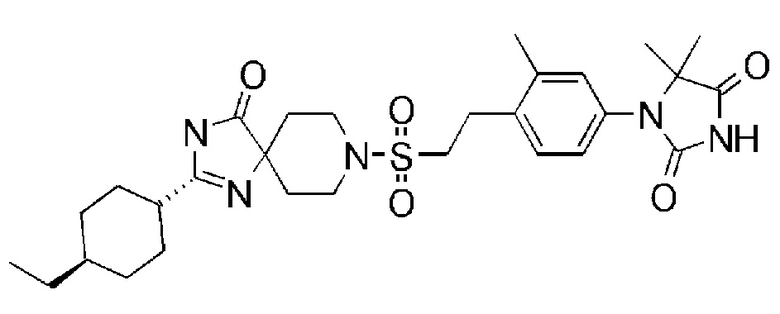
Ссылочный пример тестирования 1. Измерение in vitro активности цАМФ-сигнального пути для соединений с помощью PTH1R человека
(Пептиды)
Человеческий PTH(1-34) и кальцитонин приобретали в Peptide Institute, Inc. (Osaka, Japan), растворяли в 10 мМ растворе уксусной кислоты до 1 мМ и хранили в морозильной камере при -80°C.
(Клеточная культура)
Клетки культивировали в модифицированной по способу Дульбекко среде Игла (DMEМ), дополненной 10% фетальной бычьей сыворотки (Hyclone), 100 ед/мл пенициллина G и 100 мкг/мл сульфата стрептомицина (Invitrogen Corp) при 37°C в увлажненной атмосфере, содержащей 5% CO2.
Анализ сигнальной трансдукции с участием цАМФ, с использованием клеток LLC-PK1, не экспрессирующих PTH1R, и клеток HKRK-B7, которые представляют собой клетки LLC-PK1, сверхэкспрессирующие PTH1R человека в количестве 9,5×105 рецепторов/клетка (Takasu et al., J. Bone. Miner. Res. 14:11-20, 1999).
(Стимуляция цАМФ)
Клетки HKRK-B7 или клетки LLC-PK1 высевали в 96-луночный планшет в количестве 1×105 клеток/лунку и инкубировали в течение ночи. На следующий день добавляли 50 мкл буфера для количественного определения цАМФ (DMEM, 2 мМ IBMX, 0,2 мг/мл бычьего сывороточного альбумина, 35 мМ Hepes-NaOH, pH 7,4) содержащего PTH(1-34) человека или каждое соединение, и помещали планшет в инкубатор при 37°C. Клетки инкубировали в течение 20 мин. После удаления среды, клетки однократно отмывали 100 мкл буфера для количественного определения цАМФ. Планшет помещали на порошок из сухого льда. Клетки лизировали 40 мкл 50 мМ HCl и снова замораживали на сухом льду. Количество произведенного внутриклеточного цАМФ измеряли с использованием коммерчески доступного набора cAMP EIA (Biotrack cAMP EIA system, GE health care).
(Расчет 20% эффективной концентрации (EC20) и 50% эффективной концентрации (EC50) для измерения in vitro цАМФ-индуцирующей способности)
Анализы осуществляли с использованием уравнения, описывающего S-образную кривую доза-ответ с вариабельным градиентом. Активность сигнального пути с участием цАМФ, наблюдаемую для PTH(1-34)человека в количестве 100 нМ, принимали за 100%, и концентрацию, при которой для каждого соединения наблюдали 20% или 50% цАМФ-сигнальную активность рассчитывали, как EC20 или EC50.
Результаты, полученные для клеток HKRK-B7, представлены в таблице 4.
Уровень ответа цАМФ в клетках LLC-PK1 был ниже, чем уровень ответа в клетках HKRK-B7.
(мкМ)
(мкМ)
(мкМ)
(мкМ)
Ссылочный пример тестирования 2. Измерение in vitro цАМФ-сигнальной активности для соединений с использованием PTH1R крысы
Вместо клеток HKRK-B7, клетки LLC-PK46_RATO_PTH1R, сверхэкспрессирующие PTH1R крысы, которые были созданы в Chugai Pharmaceutical, использовали для проведения измерений, аналогичных измерениям ссылочного примера тестирования 1.
Результаты, полученные с использованием клеток LLC-PK46_RATO_PTH1R, представлены в таблице 5.
Значения EC20 in vitro активности сигнального пути с участием цАМФ рецептора PTH1 крысы имели выраженную корреляцию со значениями, полученными для PTH1R человека. Также наблюдали выраженную корреляцию между значениями EC50, полученными для рецепторов крысы и человека.
(мкМ)
(мкМ)
(мкМ)
(мкМ)
Ссылочный пример тестирования 3. Исследование метаболической стабильности с использованием микросом печени человека.
В 0,1 М фосфатном буфере (рН 7,4) инкубировали микросомы печени человека с соединением или сравнительным примером в присутствии NADPH при 37°С в течение определенного периода времени. Концентрацию исходного соединения в каждый момент времени реакции измеряли с помощью метода ЖХ/МС/МС, и собственный клиренс (мкл/мин/мг белка) рассчитывали, исходя из наклона кривой "время реакции и остаточный уровень".
<Условия проведения теста>
Концентрация соединения: 1 мкМ
Микросомы: 0,5 мг/мл
NADPH: 1 мМ
Время реакции: 0, 5, 15, и 30 мин
Результаты представлены в таблице 6. Соединения с 1 по 11 показали высокую метаболическую стабильность против микросом печени человека по сравнению со сравнительными примерами 1-6.
(мкл/мин/мг)
(мкл/мин/мг)
Ссылочный пример тестирования 4. Исследование метаболической стабильности с использованием гепатоцитов крысы
Клетки печени получали из печени крыс (SD, самки) перфузионным методом с использованием коллагеназы. Добавляли соединение ссылочных примеров или сравнительного примера, и указанную смесь инкубировали при 37°C в течение определенного периода времени, с последующим добавлением раствора для остановки реакции. Концентрацию исходного соединения в каждый момент времени реакции измеряли с использованием метода ЖХ/МС/МС, и естественный клиренс (мкл/106 клеток/мин) рассчитывали, исходя из наклона кривой "время реакции и остаточный уровень".
<Условия проведения теста>
Концентрация клеток: 1×106 клеток/мл
Концентрация соединения: 1 мкМ
Среда: среда Williams E
Время реакции: 0, 15, 30, 60, 120, и 240 минут
Раствор для остановки реакции: ацетонитрил/2-пропанол (4/6, об/об).
Результаты представлены в таблице 7. Метаболическая стабильность соединений 2, 4, 5, 6, 7, 8, 9, 10, и 11 в присутствии гепатоцитов крысы повышена по сравнению со сравнительными примерами 1, 2, 3, 5, и 6.
(мкл/106 клеток/мин)
(мкл/106 клеток/мин)
Ссылочный пример тестирования 5. Кальцемическое действие в модели с использованием крыс с TPTX
Самок крыс Crl: CD (SD) в возрасте четырех недель получали из Charles River Japan (Atsugi Breeding Center) и проводили акклиматизацию в стандартных лабораторных условиях 20-26°C и 35-75% влажности в течение одной недели. Крысам давали водопроводную воду и кормили ad libitum стандартным кормом для грызунов (CE-2) (CLEA Japan, Inc.), содержащим 1,1% кальция, 1,0% фосфорной кислоты, и 250 МЕ/100 г витамина D3.
TPTX осуществляли у крыс в возрасте пяти недель. Некоторых крыс подвергали ложной операции (Sham). Индивидов, у которых концентрация Ca в сыворотке была ниже, чем 8 мг/дл через четыре дня после операции, отбирали для использования в качестве крыс TPTX. Через пять дней после операции крыс распределяли в восемь групп TPTX и одну группу Sham (n=5, каждая группа) на основе их массы тела и сывороточной концентрации Ca, измеренной через четыре дня после операции. Только один растворитель вводили перорально в группе Sham и в группе TPTX-носитель в объеме 10 мл/кг. Каждый тестируемый препарат вводили перорально по отдельности в каждой группе TPTX-тестируемый препарат путем растворения препарата в растворителе в дозе 30 мг/10 мл/кг. Состав растворителя включал 10% диметилсульфоксида (Wako Pure Chemical Industries, Ltd.), 10% Кремофора EL (Sigma-Aldrich Japan LLC), 20% гидроксипропил-β-циклодекстрина (Nihon Shokuhin Kako Co., Ltd.), глицин (Wako Pure Chemical Industries, Ltd.); и значение pH доводили до 10. Непосредственно перед введением каждого образца осуществляли забор крови для определения исходного уровня показателя, и забор крови осуществляли через 2, 6, 10, и 24 часов после введения препарата для измерения концентрации Ca в сыворотке. Каждый забор крови осуществляли из яремной вены под ингаляционной анестезией изофлураном.
Измерение Ca в сыворотке. Измерение в сыворотке, полученной центрифугированием из собранной крови, осуществляли с использованием автоматического анализатора TBA-120FR (Toshiba Medical Systems Corporation).
Для статистического анализа исследований, проведенных на животных, данные представлены в виде среднее ± стандартная ошибка (SE). Статистический анализ проводили с помощью непарного теста с использованием пакета программ SAS Preclinical Package (Ver.5.00.010720, SAS Institute Japan, Tokyo, Japan). Р-значение <0,05 считали статистически значимым. Статистическую значимость для каждой группы тестируемого препарата в сравнении с группой TPTX-носитель, группой Сравнительного примера 1, и группой Сравнительного примера 2 показана как #, *, и ∫ соответственно.
Исходное значение концентрации Са в сыворотке составляло 9,9 мг/дл в группе Sham, и 5,3-6,2 мг/дл в каждой из групп TPTX. Концентрации Са в сыворотке для каждого соединения через 24 часа после введения представлены на фиг. 9 в виде среднего значения изменения от исходного значения. Кроме того, для всех соединений, концентрация Са в сыворотке достигала максимального значения в течение шести часов после введения или в течение десяти часов после введения каждого соединения.
Для соединений 6, 7 и 8, которые имеют высокую метаболическую стабильность в присутствии гепатоцитов крысы, наблюдали высокие положительные изменения показателя от исходного значения, и при их пероральном введении наблюдали сильные эффекты на кальцемическое действие. С другой стороны, для соединения 1 и сравнительных примеров 1 и 2, которые имеют низкую метаболическую стабильность в присутствии гепатоцитов крысы, наблюдали меньшие положительные изменения от исходного значения, по сравнению с соединениями 6, 7 и 8. В частности, для соединений 7 и 8 наблюдали статистически значимые изменения, по сравнению со сравнительными примерами 1 и 2.
Кроме того, для соединений 6, 7 и 8, которые имеют высокую метаболическую стабильность в присутствии гепатоцитов крысы, наблюдали индивидуальные максимальные значения от 7,8 до 8,5 мг/дл в течение шести или десяти часов после введения, и терапевтический целевой уровень сывороточной концентрации Ca от 7,6 до 8,8 мг/дл достигался у пациентов с гипопаратиреозом. С другой стороны, указанный терапевтический целевой уровень не может быть достигнут в какой-либо из моментов времени проведения измерения в случае соединения 1 и сравнительных примеров 1 и 2, которые имеют низкую метаболическую стабильность в присутствии гепатоцитов крысы.
На основании вышеуказанных результатов тестов, обнаружили, что соединения 6, 7 и 8, которые обладают сильной цАМФ-сигнальной активностью в клетках, экспрессирующих PTH1R крысы, и высокой стабильностью против метаболического расщепления в гепатоцитах крысы, проявляют сильные эффекты в отношении кальцемического действия у крыс при пероральном введении. Указанные соединения также обладают цАМФ-сигнальной активностью в клетках, экспрессирующих PTH1R человека, и высокой метаболической стабильностью против микросом печени человека, по сравнению со сравнительными соединениями; и предполагается, что они будут иметь высокий терапевтический эффект при пероральном введении пациентам с гипопаратиреозом. Кроме того, предполагается, что соединения, представленные формулой (1), которые имеют цАМФ-сигнальную активность в клетках, экспрессирующих PTH1R человека, и проявляют метаболическую стабильность против микросом печени человека в такой же степени, как соединения 6, 7 и 8, также будут иметь высокий терапевтический эффект у пациентов с гипопаратиреозом.
Промышленная применимость
Настоящее изобретение относится к лекарственным средствам, предназначенным для профилактики, лечения и обеспечения восстановления и устранения остеопороза, уменьшения костной массы при периодонтальном заболевании, дефекта альвеолярной кости после удаления зуба, остеоартрита, дефицита суставного хряща, адинамической болезни кости, ахондроплазии, гипохондроплазии, остеомаляции, перелома кости, и т.п., которые индуцируют анаболизм костной/хрящевой ткани путем неинвазивного системного воздействия или местного воздействия производных гидантоина, которые обладают высокой метаболической стабильностью и проявляют сильные PTH-подобные эффекты.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ГИДАНТОИНА | 2013 |
|
RU2678984C2 |
| Новые замещенные соединения имидазопиридина в качестве ингибиторов индоламин-2,3-диоксигеназы и/или триптофан-2,3-диоксигеназы | 2017 |
|
RU2741911C2 |
| 5-[(ПИПЕРАЗИН-1-ИЛ)-3-ОКСО-ПРОПИЛ]-ИМИДАЗОЛИДИН-2,4-ДИОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ADAMTS ДЛЯ ЛЕЧЕНИЯ ОСТЕОАРТРИТА | 2015 |
|
RU2693459C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2814288C1 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2288228C2 |
| 1-СУЛЬФОНИЛ-1,3-ДИГИДРОИНДОЛ-2-ОНЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ (ВАРИАНТЫ), СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2003 |
|
RU2259999C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СПИРОСОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ MAGL | 2018 |
|
RU2726631C1 |
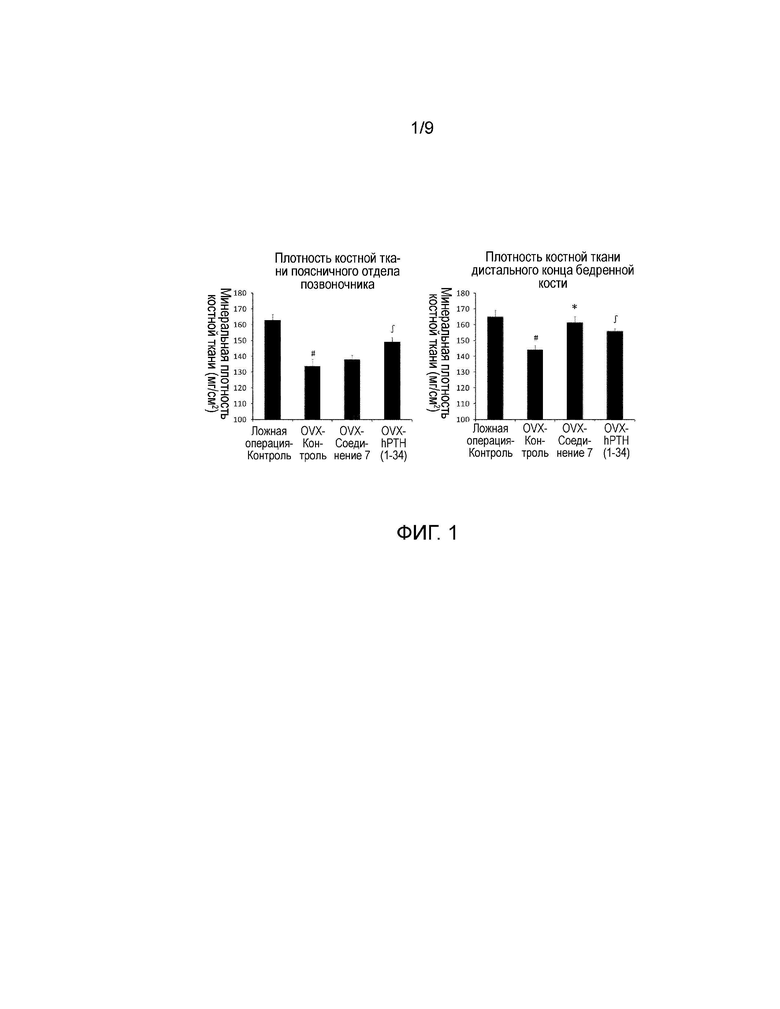
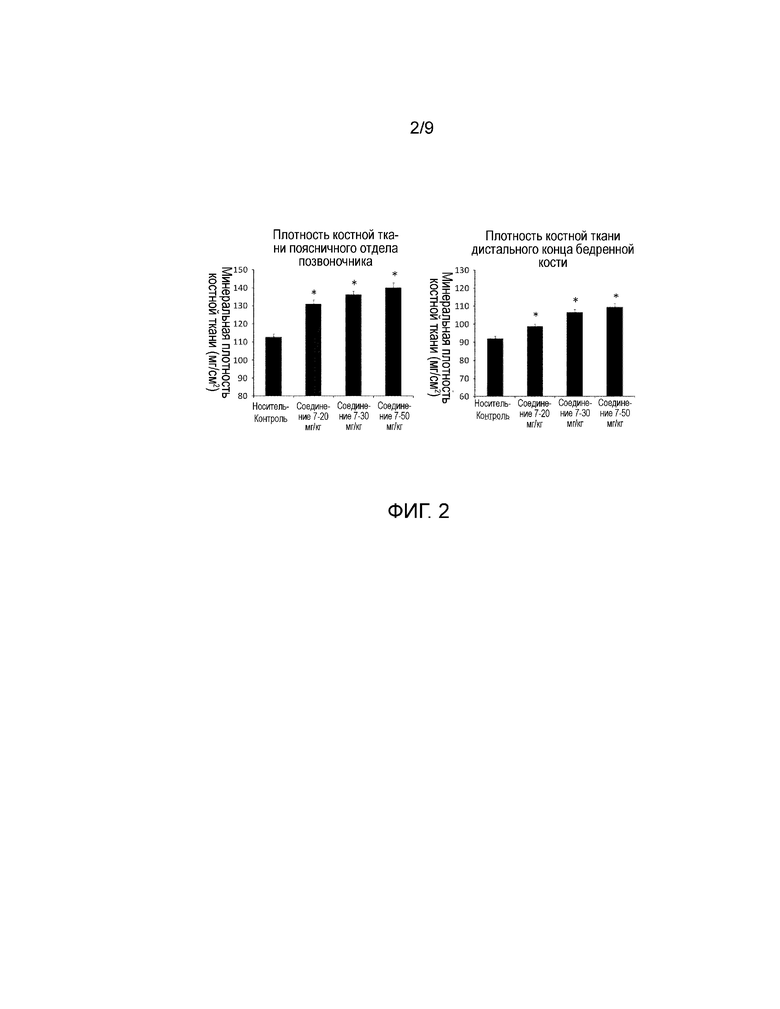
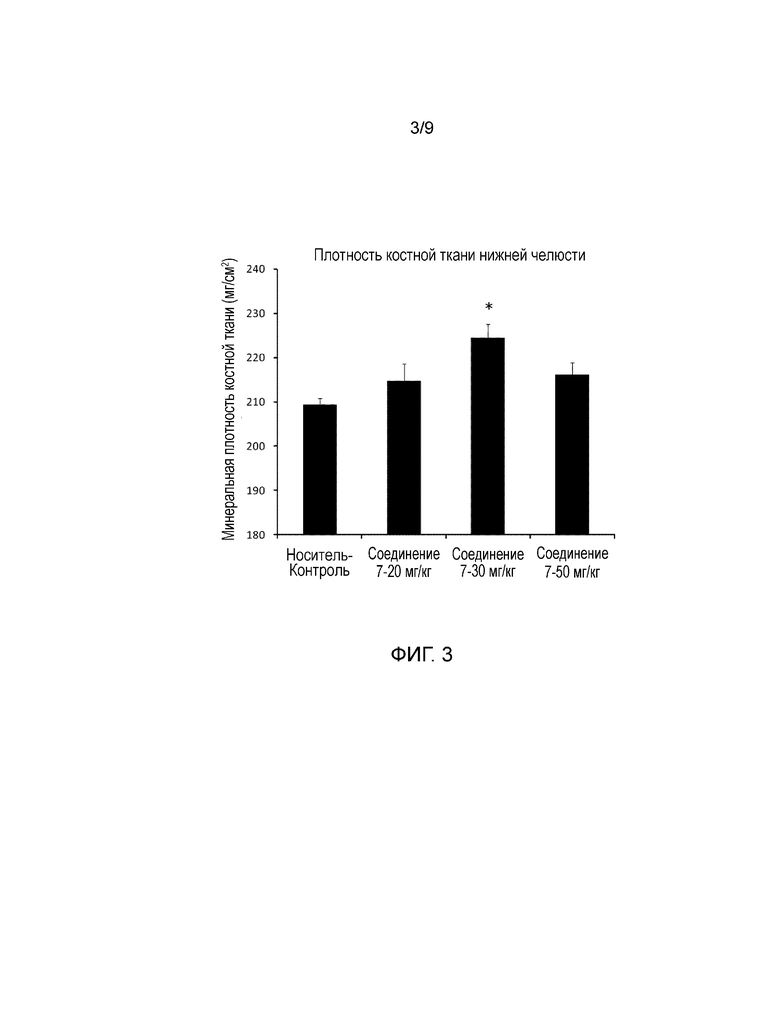

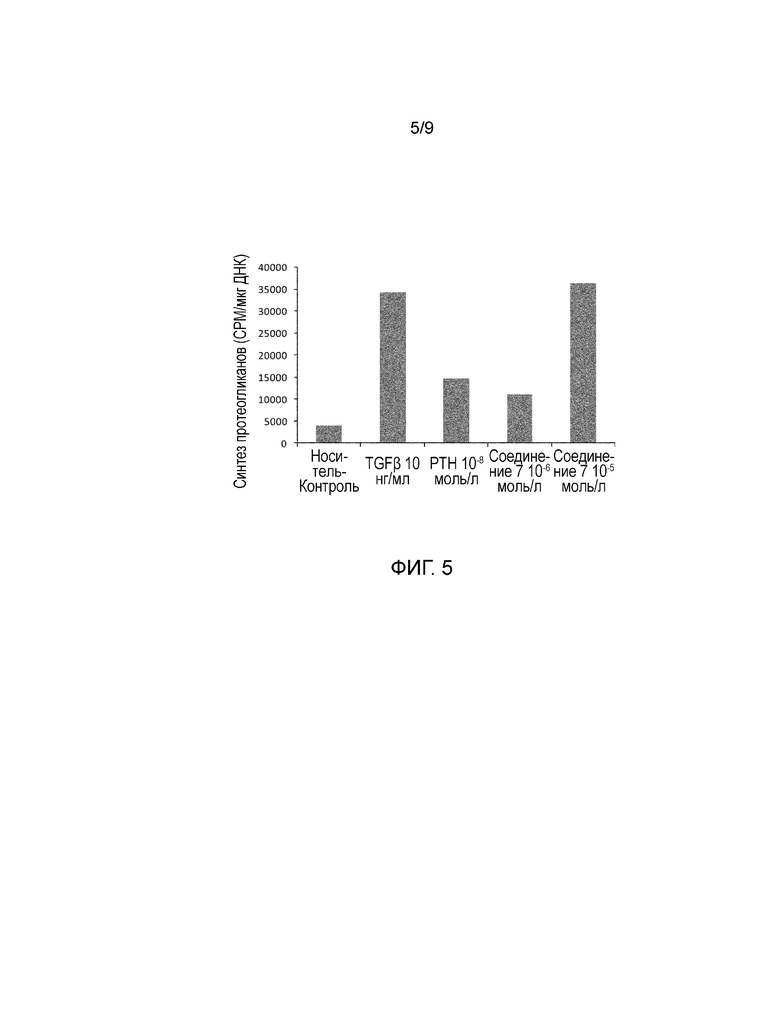
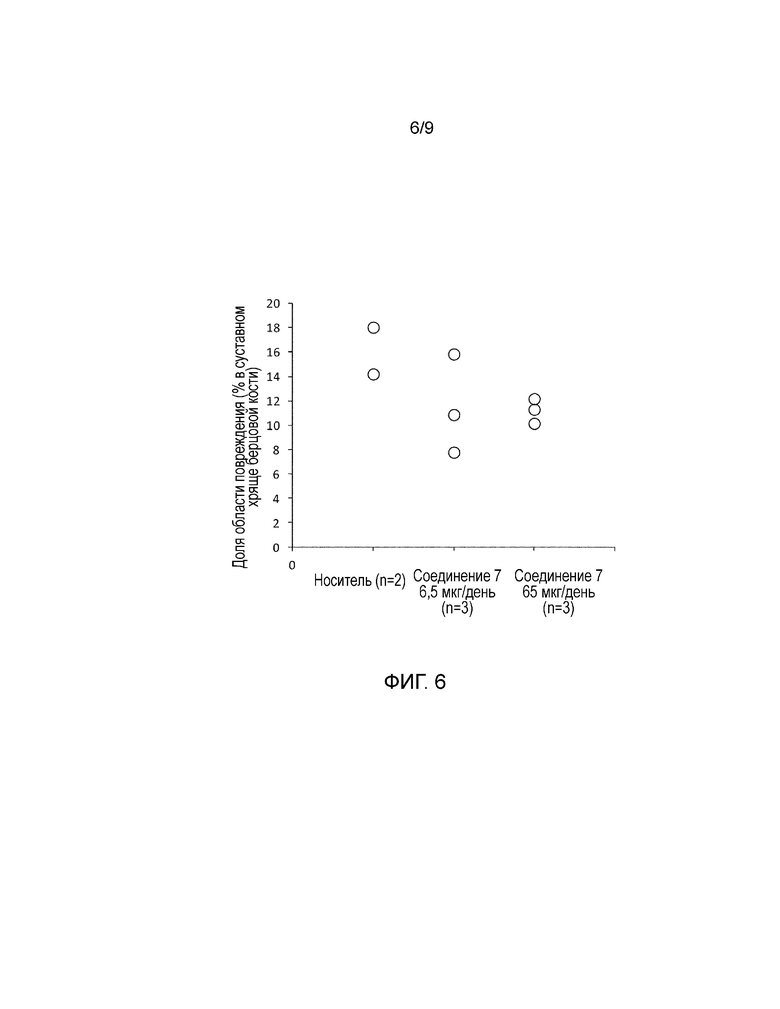

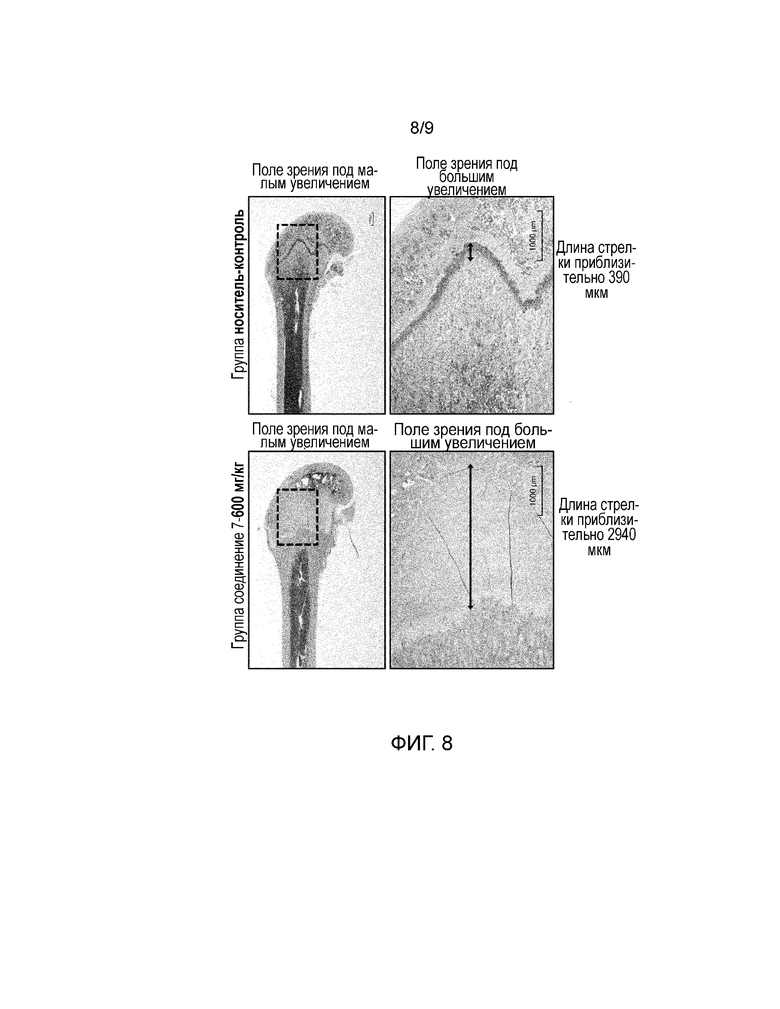
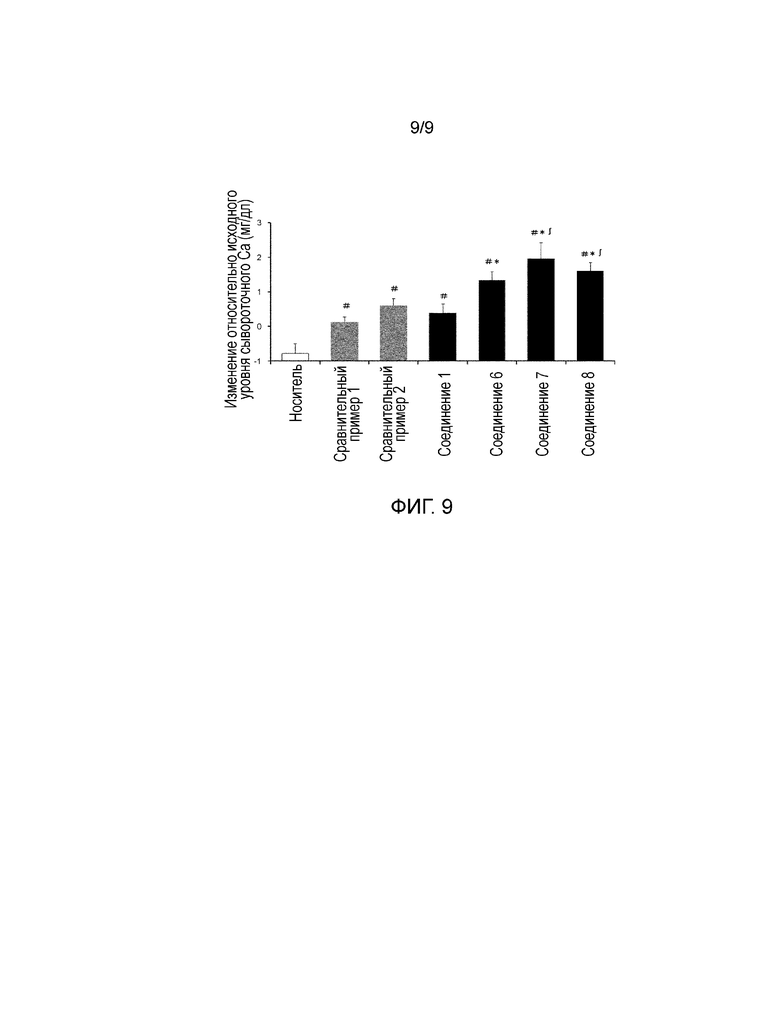
Группа изобретений относится к химико-фармацевтической промышленности и представляет собой фармацевтическую композицию, предназначенную для индукции анаболизма костной и/или хрящевой ткани, содержащую конкретные производные гидантоина. Группа изобретений также включает применение указанной композиции и способ индукции анаболизма костной и/или хрящевой ткани пациенту, нуждающемуся в предупреждении снижения костной массы при периодонтальном заболевании, облегчения восстановления дефекта альвеолярной кости после удаления зуба или облегчения восстановления дефицита суставного хряща. Группа изобретений позволяет применять конкретные производные гидантоина, проявляющие сильную цАМФ-продуцирующую способность в клетках, экспрессирующих РТН1R человека и обладающих высокой стабильностью в отношении метаболизма в микросомах печени человека. 4 н. и 5 з.п. ф-лы, 9 ил., 7 табл., 6 пр.
1. Фармацевтическая композиция, предназначенная для индукции анаболизма костной и/или хрящевой ткани, которая содержит в качестве активного ингредиента соединение или его фармацевтически приемлемую соль, где соединение выбрано из группы, состоящей из:
1-(4-(2-((2-(4-фтор-3-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(3-бромфенил)-4-оксо-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(4-фтор-3-(трифторметил)фенил)-4-оксо-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(3-фтор-4-(трифторметокси)фенил)-4-оксо-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(4-(2-((2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-4-оксо-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)-3,5-диметилфенил)-5,5-диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-диона);
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-диона;
1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4,5]дека-1-ен-8-ил)сульфонил)этил)фенил)-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-диона;
5-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-2-окса-5,7-диазаспиро[3,4]октан-6,8-диона; и
4-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-4,6-диазаспиро[2.4]гептан-5,7-диона,
при условии, что анаболизм костной и/или хрящевой ткани не включает остеопороз, перелом, адинамическую болезнь кости, ахондроплазию, гипохондроплазию, остеомаляцию, остеоартрит, артрит, тромбоцитопению, гипопаратиреоз, гиперфосфатемию, опухолевый кальциноз и мобилизацию стволовых клеток.
2. Фармацевтическая композиция по п. 1, в которой соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(3-(трифторметил)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион или его фармацевтически приемлемую соль.
3. Фармацевтическая композиция по п. 1, в которой соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-5,5-диметилимидазолидин-2,4-дион или его фармацевтически приемлемую соль.
4. Фармацевтическая композиция по п. 1, в которой соединение представляет собой 1-(3,5-диметил-4-(2-((4-оксо-2-(4-(трифторметокси)фенил)-1,3,8-триазаспиро[4.5]дека-1-ен-8-ил)сульфонил)этил)фенил)-1,3-диазаспиро[4.4]нонан-2,4-дион или его фармацевтически приемлемую соль.
5. Фармацевтическая композиция по п. 1 для применения для предупреждения снижения костной массы при периодонтальном заболевании, обеспечения восстановления дефекта альвеолярной кости после удаления зуба или обеспечения восстановления дефицита суставного хряща.
6. Способ индукции анаболизма костной и/или хрящевой ткани, который включает введение фармацевтической композиции по любому из пп. 1-4 пациенту, нуждающемуся в предупреждении снижения костной массы при периодонтальном заболевании, облегчения восстановления дефекта альвеолярной кости после удаления зуба или облегчения восстановления дефицита суставного хряща.
7. Способ по п. 6, где способ индукции анаболизма костной и/или хрящевой ткани представляет собой способ предупреждения снижения костной массы при периодонтальном заболевании, способ облегчения восстановления дефекта альвеолярной кости после удаления зуба или способ стимулирования восстановления дефицита суставного хряща.
8. Применение фармацевтической композиции по любому из пп. 1-4 для изготовления фармацевтической композиции, предназначенной для предупреждения снижения костной массы при периодонтальном заболевании, облегчения восстановления дефекта альвеолярной кости после удаления зуба или облегчения восстановления дефицита суставного хряща.
9. Применение фармацевтической композиции по любому из пп. 1-4 для изготовления фармацевтической композиции, предназначенной для индукции анаболизма костной и/или хрящевой ткани, при условии, что анаболизм костной и/или хрящевой ткани не включает остеопороз, перелом, адинамическую болезнь кости, ахондроплазию, гипохондроплазию, остеомаляцию, остеоартрит, артрит, тромбоцитопению, гипопаратиреоз, гиперфосфатемию, опухолевый кальциноз и мобилизацию стволовых клеток.
| WO 2010126030 A1, 04.11.2010 | |||
| БЕЛИКОВ В.Г., Фармацевтическая химия, М., Высшая школа, 1993, с | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Д.А | |||
| Харкевич, "Фармакология", М., Медицина, 1987, стр | |||
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |
| JP 2008515895 A, 15.05.2008 | |||
| WO 2008148689 A1, 11.12.2008 | |||
| WO 2007149873 A2, 27.12.2007 | |||
| JP 2004523583 A, 05.08.2004 | |||
| Строительный элемент для сооружения стен и т.п. | 1929 |
|
SU17696A1 |

