Настоящее изобретение относится к ингибиторам вируса гепатита В (ВГВ, HBV) в качестве химиотерапевтических средств для лечения ВГВ.
Гепатит В - это инфекционное воспалительное заболевание печени, возникающее вследствие внедрения в организм ВГВ и представляющее серьезную глобальную проблему здравоохранения. Он может вызывать как острые, так и хронические заболевания и подвергать людей высокому риску смерти от цирроза и рака печени.
По оценкам ВОЗ, 257 миллионов человек живут с инфекцией ВГВ. В 2015 году у пациентов, инфицированных ВГВ, было 887000 случаев смерти, в основном из-за осложнений (включая цирроз и гепатоцеллюлярную карциному). Гепатит В является важной профессиональной опасностью для медицинских работников. Тем не менее, это может быть предотвращено в настоящее время доступной, безопасной и эффективной вакциной. Вакцина против гепатита В доступна с 1982 года. Вакцина эффективна на 95% для профилактики инфекции и развития хронических заболеваний и рака печени из-за гепатита В.
Распространенность ВГВ наиболее высока в Регионе Западной части Тихого океана и Африканском регионе ВОЗ, где инфицировано соответственно 6,2% и 6,1% взрослого населения. В Регионе Восточного Средиземноморья ВОЗ, регионе Юго-Восточной Азии ВОЗ и Европейском регионе ВОЗ, соответственно, инфицировано 3,3%, 2,0% и 1,6% населения в целом. В регионе Америки инфицировано 0,7% населения [http://www.who.int/mediacentre/factsheets/fs204/en/]. Число инфицированных в России по разным оценкам достигает от 3 до 6 млн. человек. Заболеваемость хроническими формами гепатита В, в целом по населению, находится на уровне 13-14 на 100 тыс. населения Российской Федерации [http://58.rospotrebnadzor.ru/].
ВГВ уникален среди патогенных вирусов человека, относится к гепаднавирусам - ДНК-содержащим гепатотропным вирусам. ВГВ имеет сферическую форму, диаметр 42 нм, сложную структуру. В центре нуклеокапсида, состоящего из 90 димеров белка кор-антигена (HBcAg), расположен геном вируса, представленный частично двух цепочечной кольцевой ДНК, и фермент ДНК-полимераза. Наружная липопротеиновая оболочка содержит поверхностный антиген HBsAg (австралийский антиген), который имеет 4 подтипа, обладающих антигенными различиями. Существуют и мутантные штаммы вируса по антигенам HBsAg и HbeAg. Процесс репликации вируса сложен, возможно образование полноценных вирусов и дефектных, состоящих из HBsAg-частиц, которые не имеют инфекционных свойств. В последние годы открыты новые антигены ВГВ, однако роль их неясна. HBeAg и HBsAg циркулируют в крови, HBcAg обнаруживается только в ткани печени, антитела образуются ко всем основным антигенам - анти-НВс, анти-НВе и анти-HBs. ВГВ отличается исключительно высокой устойчивостью в окружающей среде. При 100°С погибает через 30 мин, в холодильнике сохраняется до года, в замороженном состоянии - 20 лет, в сухой плазме - 25 лет. Раствор 1-2% хлорамина инактивирует вирус через 2 ч, а 1,5% раствор формалина - через 7 сут. При автоклавировании при 120°С вирус гибнет через 5 мин [http://medbe.ru].
Несмотря на наличие эффективной профилактической вакцины против ВГВ, бремя хронической инфекции ВГВ по-прежнему является серьезной неудовлетворенной мировой медицинской проблемой из-за субоптимальных вариантов лечения и устойчивых темпов новых инфекций в большинстве районов развивающегося мира. Современные методы лечения ограничиваются только двумя классами агентов (интерфероном и нуклеозидными аналогами - ингибиторами вирусной полимеразы). Лекарственная устойчивость, низкая эффективность и проблемы переносимости ограничивают их воздействие. Низкие скорости излечения ВГВ объясняются, по меньшей мере, частично присутствием и стойкостью ковалентно замкнутой кольцевой ДНК (cccDNA) в ядре инфицированных гепатоцитов. Однако стойкое подавление ДНК ВГВ замедляет прогрессирование заболевания печени и помогает предотвратить гепатоцеллюлярную карциному. Текущие цели терапии для ВГВ-инфицированных пациентов направлены на снижение сывороточной ДНК ВГВ до низкого или неопределяемого уровня и, в конечном счете, снижение или предотвращение развития цирроза и гепатоцеллюлярной карциномы.
У большинства людей лечение не излечивает инфекцию ВГВ, а только подавляет репликацию вируса. Поэтому большинство людей, которые начинают лечение ВГВ, должны продолжать его всю жизнь [http://www.who.int/mediacentre/factsheets/fs204/en/].
В этой связи в терапии ВГВ существует потребность в новых терапевтических агентах, которые лечат, улучшают или предотвращают ВГВ-инфекцию. Введение этих терапевтических агентов инфицированному ВГВ пациенту либо в виде монотерапии, либо в сочетании с другими анти-ВГВ препаратами или вспомогательными методами приведет к значительному улучшению прогноза, уменьшению прогрессирования заболевания и его излечения.
Вирусные капсиды ВГВ, состоящие из молекул кор-антигена, играют существенную роль в жизненном цикле ВГВ. Капсиды ВГВ защищают вирусный геном при его передаче между клетками, а также играют центральную роль в процессе вирусной репликации, включая инкапсидацию и репликацию вирусного генома, образование и секрецию вирусных частиц. Вирусные капсиды также реагируют на изменение внешних условий при попадании в клетку, и обеспечивают освобождение вирусного генома от вирусных белков оболочки. Было показано, что правильная сборка вирусных капсид необходима для вирулентности вирусных частиц.
Важные функции кор-антигена накладывают существенные эволюционные ограничения на его первичную структуру, что приводит к низкой вариабельности последней. В соответствии с этим, мутации в гене кор-антигена, нарушающие его самосборку в вирусные капсиды, приводят к полной или частичной потере жизнеспособности вируса. Чем меньше вариабельности наблюдается в молекулярной мишени лекарственного препарата, тем менее вероятно появление в клинике жизнеспособных мутантов вируса, резистентных к препарату. Действительно, спонтанные мутации в гене кор-антигена наблюдаются только в 4 из 183 кодонов. Таким образом, ингибиторы кор-антигена могут обладать меньшей вероятностью развития резистентности по сравнению с применяемыми в настоящее время препаратами. Далее, терапия, направленная на ингибирование кор-антигена возможно будет менее подвержена развитию резистентности по сравнению с традиционной терапией, направленной на ингибирование активных центров ферментов. Описанные ранее антивирусные препараты для ВИЧ, риновирусов и ВГВ, связывающиеся с вирусными капсидами и ингибирующие их функции, являются фармакологическим доказательством того, что вирусный кор-антиген является перспективной молекулярной мишенью для разработки антивирусных препаратов.
Одним из первых достаточно эффективных ингибиторов кор-антигена ВГВ ин витро является гетероарилдигидропиримидин BAY 41-4109. Противовирусная активность BAY 41-4109 на клетках HepG2.2.15, конститутивно экспрессирующие ВГВ, показали IC50 около 202 нМ без клеточной токсичности [N. Brezillon at al. Antiviral activity of Bay 41-4109 on hepatitis В virus in humanized Alb-uPA/SCID mice. PLoS One. 2011; 6(12): e25096. doi:10.1371/journal.pone.0025096. Epub 2011 Dec 5.].

Сравнительно недавно была предложена фармацевтическая композиция [Patent ЕР 3085368, prior. 01.07.2011], включающая по меньшей мере одно соединение общей формулы DVR

где:
R2 представляет собой заместитель выбранный из группы, включающей Н, СН3, CF3, F и О;
R3 представляет собой заместитель выбранный из группы, включающей Н, СН3, F и Cl;
R4 представляет собой заместитель выбранный из группы, включающей Н, СН3, F и Cl;
R5 представляет собой заместитель выбранный из группы, включающей Н и Cl; R7 представляет собой заместитель выбранный из группы, включающей Н, Cl, F и Br;
R9 представляет собой заместитель выбранный из группы, включающей Н, СН3, F и Cl;
Rx представляет собой заместитель выбранный из группы, включающей фрагменты первичных и вторичных аминов, азепан-1-ил, пиперидин-1-ил, морфолин-4-ил и тиоморфолин-4-ил.
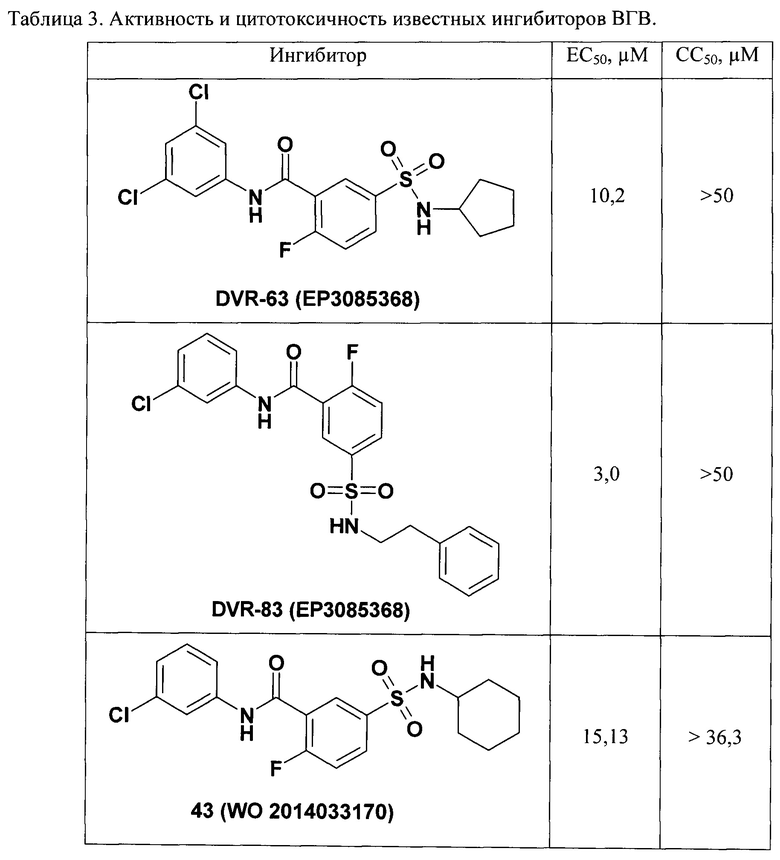
Наиболее активные соединения в ЕР 3085368 являются N-(3,4-дифторфенил)-2-фтор-5-сульфамоил-бензамиды (II) представленные в табл. 7 на страницах 37 и 38 ЕР 3085368, в которых Rx = втор-бутиламино, циклопентиламино, циклогептиламино: ЕС50 = 0.39 мМ, СС50 > 50 мМ в клеточной линии AML 12HBV10.

Недавно был предложен также способ лечения инфекции ВГВ у нуждающихся в ней индивидуумов, включающий введение индивидууму терапевтически эффективного количества сульфамоилбензамида (СБА) общей формулы NVR [US 9066932, prior. 21.12.2011]. Эти СБА являются ингибиторов кор-антигена ВГВ. В патенте представлены многочисленные соединения, соответствующие общей формуле NVR, для которых отсутствуют данные по их активности. В таблицах 1-3 представлены соединения общей формулы NVR, для которых приведена «активность» по отношению к ВГВ более 50% при их концентрации 10 мМ. Это значит, что активность соединения общей формулы NVR имеет значение ЕС50 < 10 мМ (точные значения IC50 для отдельных соединений в патенте не приводится).

где: X и G1 представляют собой галоген, G2 представляют собой С1-С4 алкил или галоген, G4 представляют собой С1-С4 алкил, галоген или ОН.
Опубликована также патентная заявка WO 2014033170 (prior. 28.08.2012) в которой заявляются новые соединения общей формулы А1 и фармацевтические композиции, включающие эти соединения

где В - представляет собой 5-ти или 6-ти членный ароматический цикл, необязательно содержащий один или несколько гетероатомов, каждый из которых независимо выбран из группы, состоящей из О, S и N, и необязательно замещенный одним или несколькими заместителями, каждый из которых независимо выбран из группы, состоящей из водорода, галогена, С1-С3 алкила, CN, CFH2, CF2H и CF3;
R1 представляет собой водород или С1-С3 алкил;
R2 представляет собой С1-С6 алкил, С1-С3 алкил, бензил, С(=O)-R5, CFH2, CF2H, CF3 или 3-7-членное насыщенное кольцо, необязательно содержащее один или несколько гетероатомов, каждый из которых независимо выбран из группы, О, S и N, такое 3-7-членное насыщенное кольцо или С1-алкалкил, необязательно замещенный одним или несколькими заместителями, каждый из которых независимо выбран из группы, состоящей из водорода, галогена, С1-С4-алкилокси, оксо, С(=О)-С1-С3 алкил, С1-С4-алкил, ОН, CN, CFH2, CF2H и CF3;
или R1 и R2 вместе с азотом, к которому они присоединены, образуют фрагмент 1,4-диокса-8-азаспиро[4,5] или 5-7-членное насыщенное кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, каждый из которых независимо выбран из группы, состоящей из О, S и N, такое 5-7-членное насыщенное кольцо необязательно замещено одним или несколькими заместителями, каждый из которых независимо выбран из группы, состоящей из водорода, галогена, С1-С4 алкилокси, оксо, С(=О)-С1-С3-алкила, С1-С4алкила, ОН, CN, CFH2, CF2, Н и CF3.
В этом ряду соединений имеется значительное число ингибиторов с наномолярной активностью ЕС50 < 300 nM в клеточной линии HepG2.2.15. В заявке WO 2014033170 максимальную активность с ЕС50 < 100 nM показали соединения 85 (ЕС50 = 50 nM), 93 (ЕС50 = 70 nM), 99 (ЕС50 = 90 nM), 183 (ЕС50 = 80 nM), 201 (ЕС50 = 60 nM) и 222 (ЕС50 = 50 nM),


Позже был опубликован скрининг библиотеки, состоящей из 26 900 малых молекул, который привел к открытию ряда СБА, соответствующих общей формуле NVR, которые значительно уменьшали количество внутриклеточной ДНК HBV. Наиболее активными СБА оказались соединения DVR-23, DVR-43 и DVR-56, которые проявили суб- или низкомикромолярную активность как в клетках AML12HBV10, так и в HepDES19 (Таблица 1). Все три соединения DVR-23, DVR-43 и DVR-56 имеют общие структурные особенности, включают в определенных положениях три атома фтора и алкилсульфамидный фрагмент. Авторы данной работы считают, что это наилучшая комбинация заместителей для оптимальной активности. [М.R. Campagna, F. Liu, R. Мао, С. Mills, D. Cai, F. Guo, X. Zhao, H. Ye, A. Cuconati, H. Guo, J. Chang, X. Xu, Т.M. Block and J.T. Guo, J. Virol., 2013, 87, 6931-6942.].
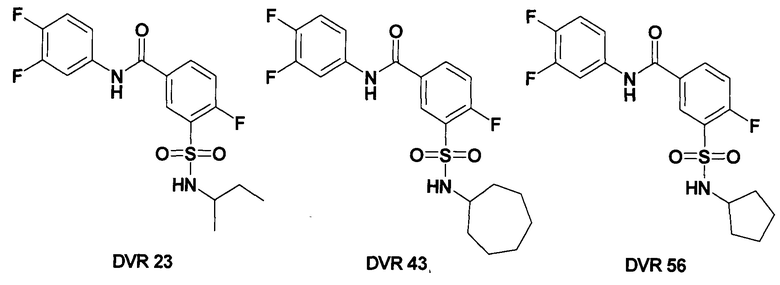

Дальнейшая оптимизация структуры ингибиторов кор-антигена ВГВ в ряду СБА привела к соединению NVR 3-778, структура которого до настоящего времени полностью не раскрыта.

Ингибитор NVR 3-778 имеет ЕС50 = 0,24 мМ в клетках HepG2.2.15 [E.J. Ganel at al. Phase 1a Safety and Pharmacokinetics of NVR 3-778, a Potential First-In-Class HBV Core Inhibitor, https://noviratherapeutics.com/wp-content/uploads/2014/11/Novira_2014_AASLD_poster_LB-19.pdf]. Результаты международного исследовании фазы 1b (64 пациента, ранее не получавших никакого лечения гепатитом В; шесть когорт: 100 мг, 200 мг, 400 мг ежедневно, 600 мг два раза в день и 600 мг два раза в день в сочетании С пегилированным интерфероном) показали, что NVR 3-778 эффективен при лечении ВГВ и хорошо переносится [M-F Yuen et al. Phase lb Efficacy and Safety of NVR 3-778, a First-In-Class HBV Core Inhibitor, in HBeAg-Positive Patients with Chronic HBV Infection, [http://www.natap.org/2016/EASL/EASL_91.htm].
Несмотря на достигнутые результаты по разработке ингибиторов кор-антигена ВГВ, остается актуальным расширение ассортимента ингибиторов ВГВ для терапии ВГВ, в частности, с более высокой активностью. Введение этих терапевтических агентов инфицированному ВГВ пациенту либо в виде монотерапии, либо в сочетании с другими анти-ВГВ препаратами или вспомогательными методами приведет к значительному уменьшению прогрессирования этого заболевания и его излечению.
Предметом данного изобретения являются новые ингибиторы ВГВ для лечения и профилактики инфекций ВГВ человека. Заявляемые ингибиторы обладают высокой антивирусной активностью ин витро, благоприятным метаболическим профилем, распределением по тканям, безопасностью и другими привлекательными фармацевтическими свойствами, и пригодны для применения в клинике.
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если иным не ограничены в конкретных случаях либо по отдельности, либо как часть большей группы.
Термин «алкил», используемый здесь, относится к насыщенным с линейной или разветвленной цепью углеводородным радикалам, содержащим от одного до шести атомов углерода. Примеры алкильных радикалов С1-С6, включают, но не ограничиваются ими, метил, этил, пропил, изопропил, н-бутил и трет-бутил.
Термин «циклоалкил» означает моновалентную насыщенную карбоциклическую группу, которая может быть моноциклической или мультициклической. Репрезентативные циклоалкильные группы включают в себя, в качестве примеров, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и пр.
Термин «необязательно замешенный» означает, что упомянутая группа может быть замещена в одном или более положениях любым одним или любой комбинацией радикалов.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактериального и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «терапевтический коктейль» представляет одновременно администрируемую комбинацию двух и более лекарственных препаратов, обладающих различным механизмом фармакологического действия, и направленных на различные биомишени, участвующие в патогенезе заболевания.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя активное соединение (субстанцию) и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного компонента, одного или в комбинации с другим активным компонентом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному и ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для человеческого использования. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартно фармацевтической практики.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня,, достаточного для предотвращения всплеска инфекции.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, кур, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительно субъектом является человек.
Термин «фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкил аммония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
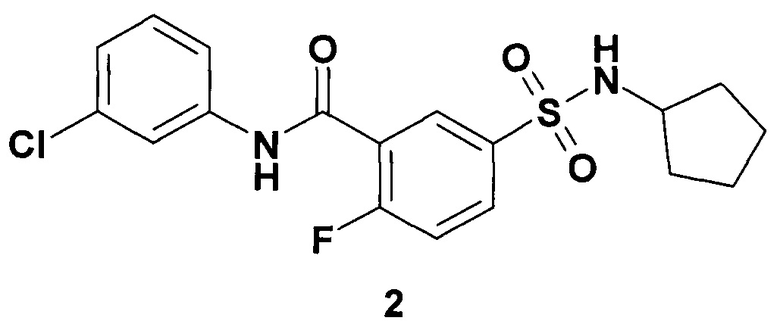
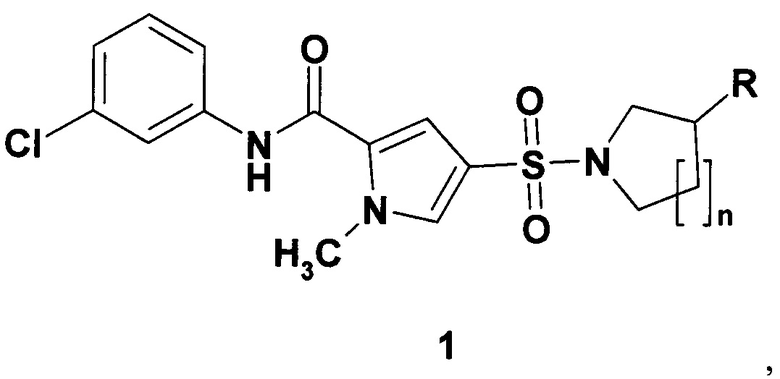
Предметом данного изобретения являются новый ингибитор ВГВ, представляющий собой 1-метил-4-сульфамоил-1Н-пиррол-2-(3-хлорфенил)карбоксамид общей формулы 1 или N-(3-хлорфенил)-5-циклопентилсулфамоил-2-фторобензамид формулы 2, его изотопно-обогащенный аналог, фармацевтически приемлемую соль, гидрат, сольват, кристаллическую или поликристаллическую форму,

где R представляет собой водород или метил; n=1, 2 или 3.
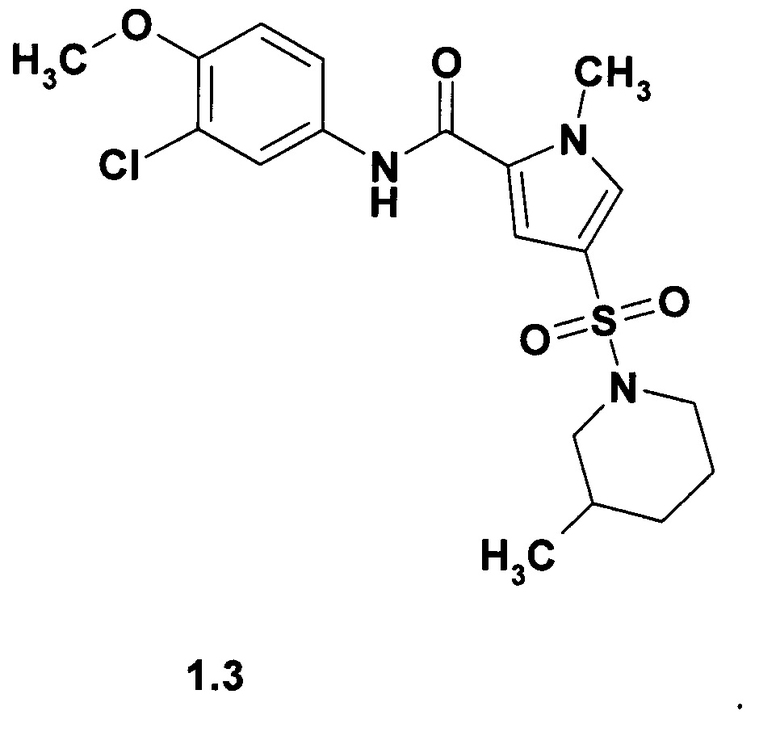
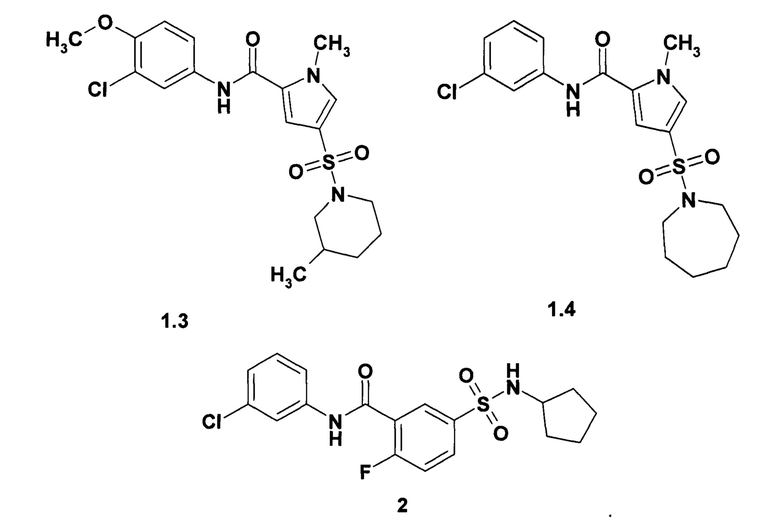
Предпочтительными ингибиторами общей формулы 1 являются 4-(пирролидин-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.1), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.2), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлор-4-метоксифенил)-карбоксамид (1.3) и 4-(азепан-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.4), их изотопно-обогащенные аналоги, фармацевтически приемлемые соли, гидраты, сольваты, кристаллические и поликристаллические формы.

Неожиданно новые ингибиторы общей формулы 1 и формулы 2 имеют сравнимую или более высокую активность, чем известные аналоги (таблица 2). В ряду новых ингибиторов соединение формулы 2 является рекордсменом по активности (ЕС50 = 0,042 цМ) в сравнении с наиболее активными известными ингибиторами ВГВ.

Неожиданный результат состоит также в том, что ближайшие по структуре известные соединения DVR-63 и DVR-83 по патенту ЕР 3085368 и ингибиторы 43 и 62 по патентной заявке WO 2014033170 являются микромолярными ингибиторами (Таблица 3) и уступают по активности новым ингибиторам (Таблица 2) на 1-2 порядка.

Новые ингибиторы общей формулы 1 и формулы 2, оказались также более активными чем ингибиторы DVR-23, DVR-43 и DVR-56 (Таблица 1), имеющие до настоящего изобретения наилучшую комбинацию заместителей (включающую 3,4-дифторфанилиновый фрагмент) для оптимальной активности [М.R. Campagna, F. Liu, R. Мао, С. Mills, D. Cai, F. Guo, X. Zhao, H. Ye, A. Cuconati, H. Guo, J. Chang, X. Xu, Т.M. Block and J.T. Guo, J. Virol., 2013, 87, 6931-6942.], и более активны, чем ингибитор NVR 3-778 [E.J. Ganel at al. Phase la Safety and Pharmacokinetics of NVR 3-778, a Potential First-In-Class HBV Core Inhibitor, https://noviratherapeutics.com/wp-content/uploads/2014/11/ Novira_2014_AASLD_ poster_LB-19.pdf].
Предметом данного изобретения являются фармацевтическая композиция, в форме таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки для комбинированной терапии гепатита В, содержащая в терапевтически эффективном количестве ингибитор общей формулы 1 или формулы 2, в том числе выбранный из ингибиторов ВГВ формулы 1.1, 1.2, 1.3 или 1.4, и необязательно вспомогательные вещества.
Противовирусная комбинаторная терапия, в то числе ВГВ, в последние годы успешно развивается и заключается в использовании комбинации препаратов ВГВ с разным механизмом действия. В частности в случае вируса гепатита В используют или предполагаются к использованию нуклеозидные ингибиторы ДНК-полимеразы ВГВ, сходной с обратной транскриптазой ВИЧ (NRTI).
В качестве NRTI используют Ламивудин формулы 4а [https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Epivir-HBV/pdf/EPIVIR-HBV-PI-PIL.PDF]. Фторзамещенный Ламивудин, известный как Эмтрицитабин (Brand Name: Emtriva) формулы 4b [https://www.ncbi.nlm.nih.gov/pubmed/ 16323102], в настоящее время используется в комбинированной терапии ВИЧ [https://en.wikipedia.org/wiki/Emtricitabine], но еще не одобрен в качестве препарата для лечения ВГВ [https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient]

Недавно были получены новые NRTI, представляющие собой фосфорамидатные производные Ламивудина и Эмтрицибина формулы 4с-l, обладающие противо-ВГВ активностью и представляющие интерес для комбинаторной терапии ВГВ [Pat. Appl. US 15221613 (2016). Пат. заявка RU 2017/106611 (2017)]


4: R=i-Pr (2c,h), CH2=CHCH2 (2d,i), CH3OCH2CH2 (2e,j), циклобутил (2f, k), i-PrOC(O)C(S)H(CH3)(2g,l). C(S) - хиральный атом углерода.
В качестве NRTI используют Тенофовир дизопроксил фумарат (Viread®) формулы 5 [https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022577lbl.pdf] и Тенофовир алафенамид полуфумарат (TAF, Vemlidy®) формулы 6а [https://www.gilead.com/~/media/files/pdfs/medicines/liver-disease/vemlidy/vemlidy_pi.pdf?la=en].

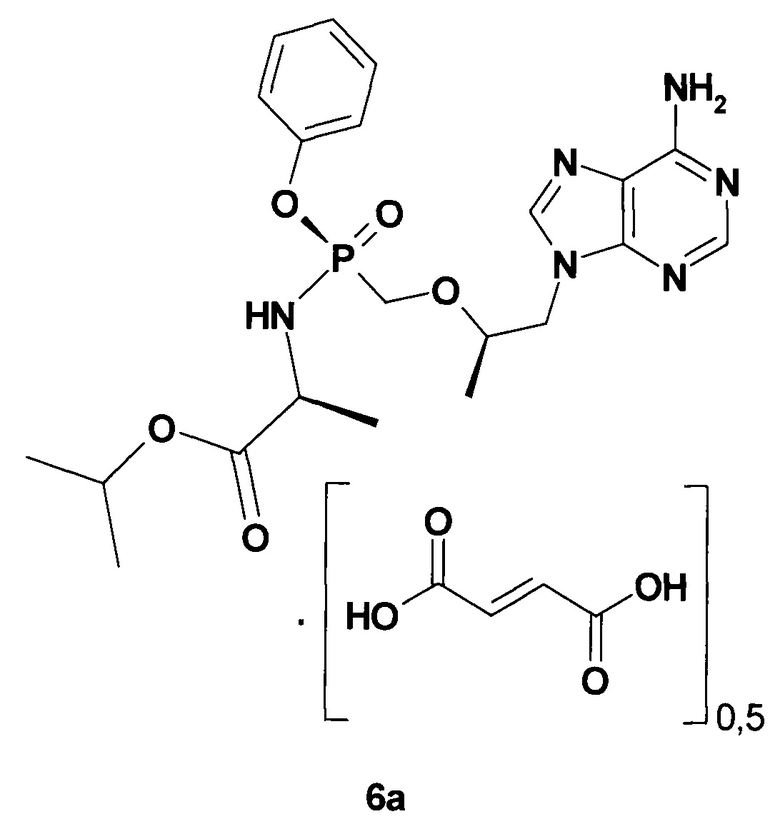
Недавно были получены новые NRTI, представляющие собой фосфорамидатные производные Тенофовира формулы 6b-r, обладающие противо-ВГВ активностью (Таблица 4) и представляющие интерес для комбинаторной терапии ВГВ [Pat. Appl. US 15221613 (2016). Пат. заявки RU 2017/106609 (2017); RU 2017/106610 (2017); RU 2017/106611 (2017); RU 2017/106615 (2017)].

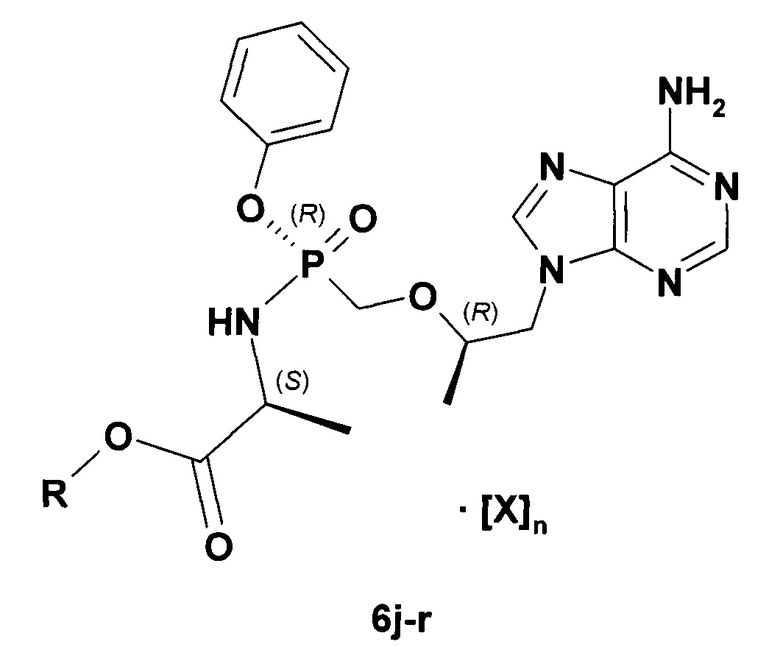
X=HO2CCH=CHCO2H, n=1 : R=СН2=СНСН2 (6b,j), СН3ОСН2СН2 (6с,k), циклобутил (6d,I), i-PrOC(O)C(S)H(CH3)(6e,m).
X=HO2CCH=CHCO2H,; n=0,5 : R=CH2=CHCH2 (6f, n), циклобутил (6g,o).
X=HCl, n=1 : R=i-Pr (6h,p), циклобутил, (6i,r).

Предметом данного изобретения является комбинированный лекарственный препарат в форме таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки для комбинированной терапии гепатита В, содержащий в терапевтически эффективном количестве ингибитор общей формулы 1.1, 1.2, 1.3, 1.4 или формулы 2, NRTI и необязательно вспомогательные вещества.
Лучшие результаты достигаются, если в качестве NRTI используют соединение, выбранное из ряда 4а-l, 5 и 6а-r.
Более предпочтительным NRTI является Ламивудин формулы 4а и его производное формулы 4c-g, более предпочтительным NRTI является замещенный Тенофовир формулы 6j, g-r.
Предметом данного изобретения является способ лечения субъекта, инфицированного ВГВ, включающий введение субъекту терапевтически эффективной дозы фапмацевтической крмпозиции в форме таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащего ингибитор кор-антигена ВГВ общей формулы 1.1, 1.2, 1.3, 1.4 или формулы 2 и необязательно вспомогательные вещества.
Предметом данного изобретения является также способ лечения субъекта, инфицированного ВГВ, включающий введение субъекту терапевтически эффективной дозы комбинированной фармацевтической композиции в форме таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащей ингибитор кор-антигена ВГВ общей формулы 1 или 1.1 или 1.2, в том числе выбранный из ингибиторов кор-антигена ВГВ формулы 1.1, 1.2, 1.3, 1.4 или формулы 2, NRTI и необязательно вспомогательные вещества.
Предпочтительным вариантом является способ с использованием комбинированной фармацевтической композиции, включающей в качестве NRTI соединение выбранное из соединений формул 4а-l 3, 6а-r и необязательно вспомогательные вещества.
Более предпочтительным вариантом является способ с использованием комбинированного лекарственного препарата, включающего в качестве предшественника NRTI Ламивудин формулы 2а или его производное формулы 2c-g, замещенный Тенофовир формулы 4j, g-r и необязательно вспомогательные вещества.
Предметом данного изобретения является способ производства лекарственного препарата для комбинированной терапии гепатита В, смешиванием терапевтически эффективного количества ингибитора ВГВ общей формулы 1 или формулы 2 и вспомогательных веществ с последующей переработкой полученной смеси в таблетки, желатиновые капсулы, пилюли, порошки, гранулы или жевательные резинки.
Предметом данного изобретения является также способ производства комбинированного лекарственного препарата для лечения гепатита В, смешиванием терапевтически эффективного количества ингибитора кор-антигена ВГВ общей формулы 1 или формулы 2, NRTI и вспомогательных веществ с последующей переработкой полученной смеси в таблетки, желатиновые капсулы, пилюли, порошки, гранулы или жевательные резинки.
Предметом данного изобретения является способ ингибирования кор-антигена ВГВ, включающий введение в клетку соединения общей формулы 1 или формулы 2, его изотопно-обогащенного аналога, фармацевтически приемлемой соли, гидрата, сольвата, кристаллической и поликристаллической формы.
Предметом данного изобретения является также способ ингибирования ВГВ, включающий введение в клетку соединения общей формулы 1 или формулы 2, его изотопно-обогащенного аналога, фармацевтически приемлемой соли, гидрата, сольвата, кристаллической и поликристаллической формы, и NRTI.
Новые ингибиторы представляют собой соединения, структурные формулы которых известны. Однако в научной и патентной литературе отсутствуют данные об способе их получения, физико-химических, спектральных свойствах, а также об их биологической активности.
Поэтому предметом данного изобретения является также способ получения соединений общей формулы 1 и формулы 2, их изотопно-обогащенных аналогов, фармацевтически приемлемых солей, гидратов, сольватов, кристаллических и поликристаллических форм.
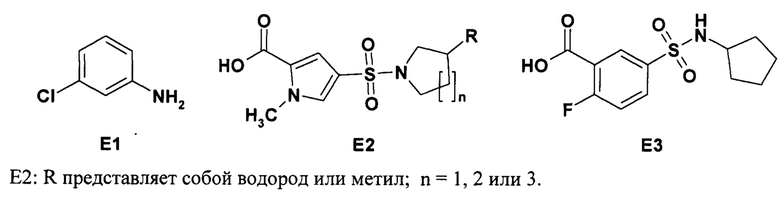
Способ получения соединений общей формулы 1 или формулы 2, включающий ацилирование 3-хлоранилина (Е1) соответственно 1-метил-4-сульфамоил-1Н-пиррол-2-карбоновой кислотой общей формулы Е2 или 3-циклопентилсульфамоил-4-фтор-бензойной кислотой Е3.



Е2: R и n имеют вышеуказанное значение.
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Синтез 4-(пирролидин-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.1), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.2), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлор-4-метоксифенил)-карбоксамид (1.3) и 4-(азепан-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1,4). Синтез сульфамоилпирролкарбоксамидов 1.1, 1.2, 1.3 и 1.4 осуществляли по схеме 1.
Схеме 1. Схема синтеза сульфамоилпирролкарбоксамидов 1.2(1), 1.2(2) и 1.2(5).
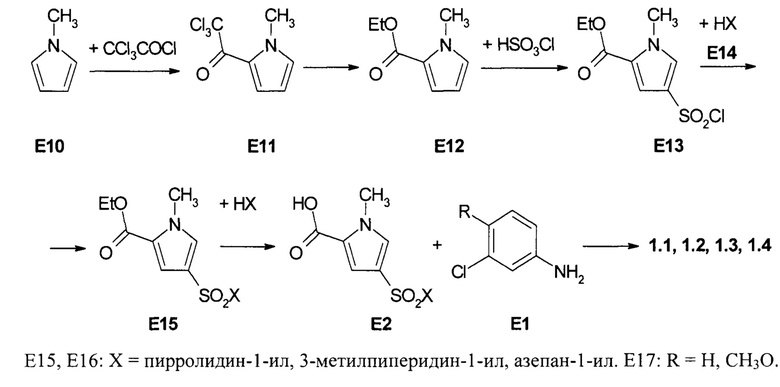
В трехгорлую круглодонную колбу, снабженную герметичной механической мешалкой, капельной воронкой и эффективным обратным холодильником, загружают раствор 1 моль трихлорацетилхлорида в 200 мл безводного эфира. К полученному раствору прибавляют при перемешивании по каплям в течение 3 ч раствор 0,94 моль свежеперегнанного 1-метил-1Н-пиррола (Е10) в 640 мл безводного эфира. Реакционную массу дополнительно перемешивают 1 ч, а затем медленно добавляют раствор 100 г (0,72 моль) карбоната калия в 300 мл воды. Органический слой отделяют и сушат над сульфатом магния, обрабатывают 6 г норита и фильтруют. Растворитель удаляют на паровой бане, а остаток растворяют в 225 мл гексана. Полученный темный раствор кристаллизовали при охлаждении льдом и перекристаллизовывали из гексана. Получали 2,2,2-трихлор-1-(1-метил-1Р-пиррол-2-ил)этанон (E11) с выходом 60%. Растворяют 2,5 г (0,04 моль) натрия в 300 мл безводного этаноле. К полученному раствору постепенно добавляют 0,3 моль соединения E11. Полученный раствор перемешивают в 4 ч, затем упаривают в вакууме досуха. Маслянистый остаток распределяли при перемешивании между 200 мл эфира и 25 мл 3N HCl. Эфирный слой отделяли, а водный промывали 100 мл эфира. Эфирные растворы объединяли, промывали 25 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом магния и концентрировали. Полученный сырой продукт очищали колоночной хроматографией на силикагеле. Получали этил 1-метил-1Н-пиррол-2-карбоксилат (Е12) с выходом 45-50% в виде вязкого масла.
Добавляют в течение 30 мин к 64 мл (0,96 моль) хлорсульфоновой кислоты при интенсивном перемешивании и при 10°С 20 г (0,16 моль) соединения Е12. Реакционную смесь перемешивали при 20°С в течение 4 ч и выливали после охлаждения на 300 г льда. Осадок соединения отфильтровывали, промывали водой, сушили и перекристаллизовывали из бензола. Получали этил 1-метил-4-хлорсульфонил-1Н-пиррол-2-карбоксилат (Е13) с выходом 65%.
К перемешиваемому раствору 0,1 моль амина Е14 и 17 мл (0,12 моля) триэтиламина в 300 мл диоксана добавляют 0,1 моль сульфохлорида Е13 в течение 5 мин при комнатной температуре. Реакционную смесь перемешивают 3 ч при 60°С. Затем смесь выливают в воду. Осадок отфильтровывают, промывают водой и перекристаллизовывают из изопропанола. Получают замещенный этил 1-метил-4-сульфамоил-1Н-пиррол-2-карбоксилат Е15 с выходом 60%.
Смесь 0,15 моль соединения Е15, 200 мл 10%-ного водного раствора NaOH и 7 мл этанола энергично перемешивают и нагревают до 95°С. Затем нерастворимый осадок отфильтровывают, фильтрат охлаждают до комнатной температуры и при перемешивании добавляют 15% HCl до рН<3. Осадок отфильтровывают, промывают водой, сушат при 100°С и перекристаллизовывают из смеси вода-ДМФ. Получают 1-метил-4-сульфамоил-1Н-пиррол-2-карбоновую кислоту (Е16) с выходом 55%.
Суспендируют в 5 мл безводного диоксана 0,0011 моль кислоты Е16 и 0,16 г (0,001 моль) карбодиимидазола. Полученную смесь при перемешивании нагревают до 40-60°С и получают прозрачный раствор, к которому прибавляют 0,0011 моль соответствующего амина Е17. Реакционную смесь кипятят с обратным холодильником в течение 1-2 ч, охлаждают, разбавляли водой, осадок отфильтровывают и перекристаллизовывают из этанола. Получают соответствующие сульфамоилпирролкарбоксамиды 1.1, 1.2, 1.3 и 1.4 с выходом 30-70%. Сульфамоилпирролкарбоксамид 1.1, m/z 377 (М+1), вычислен для молекулярной формулы C16H18ClN3O3S молекулярный вес М = 367.86. Сульфамоилпирролкарбоксамид 1.2, 1Н NMR (400 MHz, DMSO) δ: 10.15 (s, 1Н), 7.90 (t, J=2.0 Hz, 1H), 7.67-7.62 (m, 2H), 7.39-7.33 (m, 2H), 7.14 (d, J=8.1 Hz, 1H), 3.94 (s, 3H), 3.41 (t, J=12.0 Hz, 2H), 2.22 (t, J=11.0 Hz, 1H), 1.90 (t, J=11.0 Hz, 1H), 1.74-1.60 (m, 3H), 1.57-1.46 (m, 1H), 0.87 (d, J=6.5 Hz, 3H), 0.92-0.81 (m, 1H). Сульфамоилпирролкарбоксамид 1.3, 1Н NMR (400 MHz, DMSO) δ: 10.01 (s, 1H), 7.87 (d, J=2.4 Hz, 1H), 7.63-7.57 (m, 2H), 7.30 (d, J=2.0 Hz, 1H), 7.13 (d, J=9.0 Hz, 1H), 3.93 (s, 3H), 3.83 (s, 3H), 3.41 (t, J=12.0 Hz, 2H), 2.21 (t, J=11.0 Hz, 1H), 1.89 (t, J=11.0 Hz, 1H), 1.74-1.59 (m, 3H), 1.57-1.45 (m, 1H), 0.86 (d, J=6.5 Hz, 3H), 0.92-0.81 (m, 1H). Сульфамоилпирролкарбоксамид 1.4, 1H NMR (400 MHz, DMSO) δ: 10.12 (s, 1H), 7.90 (s, 1H), 7.66-7.61 (m, 2H), 7.39-7.32 (m, 2H), 7.13 (d, J=7.9 Hz, 1H), 3.92 (s, 3H), 3.15 (t, J=6.1 Hz, 4H), 1.68-1.61 (m, 4H), 1.55-1.50 (m, 4H).
Пример 2. Синтез N-(3-хлорфенил)-5-циклопентилсулфамоил-2-фторобензамида (2) осуществляют по схеме 2.
Схема 2. Схема синтеза сульфамоилбензамида 2.
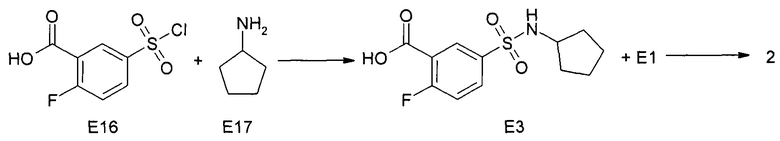
В стакан емкостью 2 л помещают 1 л воды, 1,543 моль КОН, 0,617 моль амина Е17 и 500 г мелкотолченого льда. Далее при интенсивном перемешивании прибавляют порциями 0,617 моль сульфохлорида Е16. Смесь перемешивают 3 часа и оставляют на ночь при 20°С. Далее реакционную массу подкисляют 15% раствором HCl. Выпавший осадок отфильтровывают, тщательно промывают водой и перекристаллизовывают из этанола. Получают бензолсульфамид Е3 с выходом 60-65%.
Смесь 4,1 ммоль бензолсульфамид (Е3), 0,78 г (6,27 ммоль) 3-хлоранилина (Е1) и триэтиламина (1,2 г, (11,9 ммоль) в диоксане (80 мл) охлаждали на ледяной бане и прибавляли 1,0 г (6,47 ммоль) хлорокиси фосфора. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем выливали в холодную воду (500 мл). Продукт экстрагируют этилацетатом (3×100 мл) и очищали на короткой колонке с силикагелем, элюируя эфиром, и получали в виде белого твердого вещества сульфамоилбензамид 2 с выходом 68%. Сульфамоилбензамид 2, 1Н NMR (400 MHz, DMSO) δ: 10.78 (s, 1Н), 8.15-8.04 (m, 1H), 8.05-7.95 (m, 1H), 7.90 (s, 1H), 7.80 (d, J=7.0 Hz, 1H), 7.68-7.52 (m, 2H), 7.41 (t, J=8.1Hz, 1H), 7.21 (d, J=8.0Hz, 1H), 3.55-3.39 (m, 1H), 1.75-1.47 (m, 4H), 1.47-1.21 (m, 4H).
Пример 3. Противовирусная активность ингибиторов ВГВ общей формулы 1 и формулы 2.
Противовирусную активность тестируемых соединений определяли в клеточной линии гепатомы человека АД38, несущую интегрированную ДНК вируса гепатита В (ВГВ) с терминальными повторами [Lander S, et. al, Antimicrobal Agents and Chemotherapy, 1997, pg. 1715-1720]. Данная клеточная линия была предоставлена Dr. C. Seeger, Fox Chase Cancer Center, Philadelphia, PA). Цитотоксичность соединений оценивали в параллельном режиме.
Клетки культивировали в полной среде DMEM/F12 с 2 мМ L-Глютамина (Thermo Scientific, Cat #11320033), 10% фетальной бычьей сыворотки (ThermoFisher Scientific, Cat#), 1% раствора антибиотиков-антимикотиков (ThermoFisher Scientific, Cat# 15240096), и 0.3 мкг/мл тетрациклина (Sigma, Cat # T7660-5G). Клетки высевали в 96-луночные планшеты Corning Biocoat (Corning, Cat # 356407) в 225 мкл полной среды без тетрациклина, 20000 клеток на лунку. Тестируемые вещества сперва растворяли в ДМСО (Sigma cat. D2650), затем в среде DMEM/F12, после чего 9 разведений с 3-кратным шагом добавляли к клеткам в объеме 225 мкл. Конечные концентрации тестируемых соединений составляли от 10 мкМ до 1 нМ. Каждое разведение препарата тестировалось на трех идентичных лунках. В качестве контроля на ингибирование использовали клетки, культивируемые в присутствии Тетрациклина, т.к. Тетрациклин полностью останавливает репликацию ВГВ в данной клеточной линии. Далее клетки инкубировали при 37°С в увлажненной атмосфере 5% CO2 в течение 4 дней.
Выделение секретируемой ДНК ВГВ. Через 4 дня инкубации, вирусную ДНК выделяли из культуральных супернатантов при помощи набора реагентов PureLink® Pro 96 Genomic DNA Purification Kit (ThermoFisher Scientific, Cat # K183104A), используя рекомендации производителя. После элюции очищенную ДНК хранили при -20°С.
Метод количественной полимеразной цепной реакции реального времени (ПЦР, RT-qPCR).
Метод ПЦР применяли с использованием прибора CFX96TM Real-Time System (Bio-Rad, Hercules, CA) и полимеразы AmpliTaq Gold® DNA Polymerase (Applied Biosystems®).
Состав реакционной смеси:
Программа циклов:
50°С - 2 мин
95°С - 10 мин
40 циклов:
95°С - 15 секунд
60°С - 30 секунд
72°С - 30 секунд + 1 секунда на каждый последующий цикл
Считывание флуоресцентного сигнала производили в конце каждого цикла.
Праймеры и флуоресцентные пробы были получены от компании IDT (San Diego, СА):
 ;
;
 ;
;
 .
.
Значения Ct ("пороговый цикл", на котором начинается заметная амплификация ДНК ВГВ), нормализованные к клеточным культурам без тестируемых соединений, определяли по формуле Е=(1/(1+100%))∧(Ct[тестируемый образец]-Ct[K-]), где Е - нормализованный уровень ДНК ВГВ, Ct[K-] и Ct[тестируемый образец] - значения Ct для образцов без- и с тестируемым препаратом соответственно. Значения ЕС50 тестируемых соединений (Таблица 2) рассчитывали при помощи программы Graph Prizm.
Цитотоксичностъ тестируемых соединений определяли параллельно на той-же клеточной линии АД38. Клетки культивировали в черной микроплате с прозрачным дном (96 ячеек, 104 клеток на лунку) в полной среде DMEM/F12 с 2 мМ L-Глютамина (Thermo Scientific, Cat #11320033), 10% фетальной бычьей сыворотки (ThermoFisher Scientific, Cat#), 1% раствора антибиотиков-антимикотиков (ThermoFisher Scientific, Cat# 15240096). Клетки AD38 высевались в 96-луночные планшеты (7.5×103 клеток на лунку в 100 мкл питательной среды), растворы тестируемых соединений в среде ДМЕМ готовились непосредственно перед использованием. Всего готовилось 9 серийных трехкратных разведений. Через 4 часа после высевания клеток, серийные разведения препаратов добавлялись к клеткам (100 мкл на лунку). Конечная концентрация тестируемых соединений составляла от 30 мкМ до 10 нМ, а ДМСО - 0.5%. При необходимости исследовались более высокие концентрации тестируемых веществ. Далее клетки инкубировали в течение трех дней при 37°С/5% в увлажненной атмосфере СО2. Количество живых клеток определяли при помощи набора ATPLite (Perkin Elmer, Бостон, США) в соответствии с инструкциями производителя. Для каждого соединения использовали три независимых повтора. Промывали дважды каждую лунку фосфатно-солевым буфером (0,2 мл/лун) и затем лизировали клетки добавлением клеточного буфера (50 мкл/лун, все указанные реактивы входят в комплект набора ATPLite). Микроплату инкубировали в течение 5 минут на вращающейся платформе при 600 об/мин, после чего добавляли в каждую лунку 50 мкл раствора субстрата (часть набора ATPLite). Инкубировали еще 5 минут на вращающейся платформе при 600 об/мин, выдерживали 10 минут в темноте и затем измеряли люминесценцию на приборе TopCount NXT (Packard, Perkin Elmer). В качестве количественного параметра для оценки цитотоксичности использовали величину СС50, которая соответствует концентрации вещества, при которой погибает 50% клеток.
Расчет параметра СС50: для расчета эффективности ингибирования (% Инг) использовали формулу: %Инг=[(Лпоз-Лэкс)/Лпоз-Лотр)]⋅100%, где Лпоз - положительный контроль, люминесценция в ячейках с клетками без вещества; Лотр - отрицательный контроль, люминесценция в ячейках со средой без клеток; Лэкс - люминесценция в ячейках с веществом в определенной концентрации. Значения СС50 тестируемых соединений (Таблица 2) затем рассчитывали при помощи программы XLfit 4.
Пример 4. Приготовление лекарственного средства в виде таблеток. Смешивают 1750 мг крахмала, 1750 мг размолотой лактозы, 500 мг талька и 1000 мг ингибитора формулы 1.1 или 1.2 или 1.3 или 1.4 или 2 и прессуют в брусок. Полученный брусок измельчают в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетируют в таблетки пригодных форм весом 500 мг каждая.
Пример 5. Получение противовирусной композиции в виде таблетки. Крахмал (500 мг), молотую лактозу (800 мг), тальк (200 мг), и 1500 мг ингибитора формулы 1.1 или 1.2 или 1.3 или 1.4 или 2 смешивали друг с другом и прессовали в бар. Полученный брусок измельчали в гранулы и просеивали через сито, чтобы собрать гранулы размером 14-16 меш. Полученные таким образом гранулы были сформированы в таблетки подходящей формы весом 400 или 800 мг каждая.
Пример 6. Приготовление лекарственного средства в виде капсул. Ингибитор формулы 1.1 или 1.2 или 1.3 или 1.4 или 2 тщательно смешивают с лактозой в соотношении 2:1. Полученную порошкообразную смесь упаковывают по 300 мг в желатиновые капсулы подходящего размера.
Пример 7. Получение противовирусной композиции в виде капсул, ингибитор формулы 1.1 или 1.2 или 1.3 или 1.4 или 2 тщательно смешивали с порошком лактозы в соотношении 1:1. Полученную порошкообразную смесь упаковывали в желатиновые капсулы подходящего размера по 200 мг 400 мг в каждой капсуле.
Пример 8. Общий способ получения комбинированной фармацевтической композиции. Тщательно измельчают и смешивают 30 г ингибитора формулы.1 или 1.2 или 1.3 или 1.4 или 2, 20-30 г Тенофовир дизопроксил фумарата формулы (5) или 1,5-3,5 г Тенофовира формулы 6а-r. Получают комбинированную фармацевтическую композицию (Таблица 5), которую используют для получения известным способом комбинированного лекарственного препарата в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки.
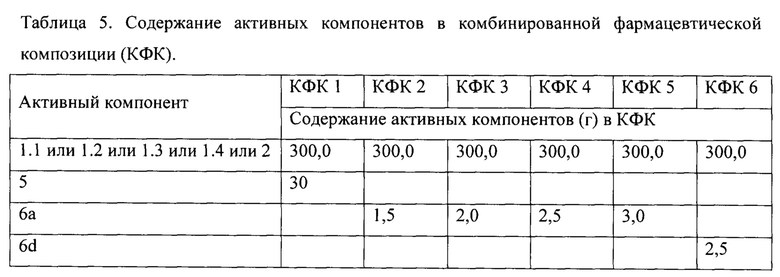
Пример 9. Общий способ получения комбинированной фармацевтической композиции (КФК). Тщательно измельчают и смешивают 30 г ингибитора формулы 1.1 или 1.2 или 1.3 или 1.4 или 2, 30.0 г соединения (4a-i) и/или 15-35 мг тенофовира (6а-r). Получают комбинированную фармацевтическую композицию (Таблица 6), которую используют для получения известным способом комбинированного лекарственного препарата в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки.

| название | год | авторы | номер документа |
|---|---|---|---|
| Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения | 2019 |
|
RU2717101C1 |
| Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноаты, способ их получения и применения | 2017 |
|
RU2647576C1 |
| Ингибитор вируса гепатита В (ВГВ) | 2019 |
|
RU2736975C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АГОНИСТЫ РЕЦЕПТОРОВ ЖЕЛЧНЫХ КИСЛОТ TGR5, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2543485C2 |
| Противо-РНК вирусное, в том числе противокоронавирусное средство - замещенный хиноксалин, фармацевтическая композиция и применения | 2020 |
|
RU2744429C1 |
| Ингибитор вируса гепатита В (ВГВ) | 2019 |
|
RU2726456C1 |
| Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения | 2017 |
|
RU2644156C1 |
| Ингибитор входа вируса гепатита и фармацевтическая композиция для лечения гепатита | 2017 |
|
RU2662161C1 |
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение | 2017 |
|
RU2659388C1 |
Настоящее изобретение относится к новым ингибиторам вируса гепатита В (ВГВ, HBV) в качестве химиотерапевтических средств для лечения ВГВ. Эти соединения связываются с кор-антигеном ВГВ и вызывают формирование аберрантных вирусных капсид, не несущих вирусный геном, что приводит к образованию и секреции дефектных (невирулентных) вирусных частиц. Согласно изобретению ингибитор ВГВ представляет собой соединение общей формулы 1, 1.3 и 2
 ,
,
где R представляет собой водород или метил; n=1, 2 или 3,

Соединение формулы 2 является новым. Способ получения соединений 1, 2 и 1.3 заключается в ацилировании 3-хлоранилина (Е1) или 4-метокси-3-хлоранилина соответственно 1-метил-4-сульфамоил-1Н-пиррол-2-карбоновой кислотой общей формулы Е2 или 3-циклопентилсульфамоил-4-фтор-бензойной кислотой Е3
8 н. и 5 з.п. ф-лы, 6 табл., 8 пр.
1. Применение соединений формул 1, 2 или 1.3 в качестве ингибитора ВГВ
R представляет собой водород или метил; n=1, 2 или 3;


2. Применение по п. 1 соединения 1-метил-4-сульфамоил-1Н-пиррол-2-(3-хлорфенил)карбоксамид общей формулы 1
 ,
,
где R принимает вышеуказанное значение.
3. Соединение N-(3-хлорфенил)-5-циклопентилсулфамоил-2-фторобензамид формулы 2

4. Применение по п. 1 соединения N-(3-хлорфенил)-5-циклопентилсулфамоил-2-фторобензамид формулы 2

5. Применение по п. 1 соединения, выбранного из ряда 4-(пирролидин-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.1), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.2), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлор-4-метоксифенил)-карбоксамид (1.3) и 4-(азепан-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.4)

6. Фармацевтическая композиция, обладающая свойствами ингибитора вируса гепатита В, содержащая в терапевтически эффективном количестве ингибитор ВГВ- соединение общей формулы 1, или формулы 2, или формулы 1.3 и фармацевтически приемлемые и фармацевтически совместимые вспомогательные вещества.
7. Фармацевтическая композиция по п. 6 в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки.
8. Лекарственный препарат в форме таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки для терапии гепатита В, содержащий в терапевтически эффективном количестве ингибитор по п. 1, или 2, или 4 и вспомогательные вещества.
9. Способ лечения субъекта, инфицированного ВГВ, заключающийся во введении субъекту эффективной дозы фармацевтической композиции по п. 6, 7 или лекарственного препарата по п. 8.
10. Способ производства лекарственного препарата по п. 8, включающий смешивание терапевтически эффективного количества ингибитора ВГВ по любому из пп. 1, 2, 3 или 4 со вспомогательным веществом.
11. Способ производства лекарственного препарата, включающий смешивание терапевтически эффективного активного ингибитора ВГВ по п.10 с последующим формированием и/или упаковкой полученной смеси для получения таблетки, гранулы, желатиновой капсулы, пилюли, порошка или жевательной резинки.
12. Способ ингибирования ВГВ, включающий введение в клетку соединения, выбранного из ряда, включающего 4-(пирролидин-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.1), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.2), 1-метил-4-(3-метилпиперидин-1-сульфонил)-1Н-пиррол-2-(3-хлор-4-метоксифенил)-карбоксамид (1.3) и 4-(азепан-1-сульфонил)-1-метил-1Н-пиррол-2-(3-хлорфенил)карбоксамид (1.4), N-(3-хлорфенил)-5-циклопентилсулфамоил-2-фторобензамид (2)

13. Способ получения соединений общей формулы 1, 1.3 и 2, включающий ацилирование 3-хлоранилина (Е1) или 4-метокси-3-хлоранилина соответственно 1-метил-4-сульфамоил-1Н-пиррол-2-карбоновой кислотой общей формулы Е2 или 3-циклопентилсульфамоил-4-фтор-бензойной кислотой Е3

| WO 2014033170 A1, 06.03.2014 | |||
| DATABASE REGISTRY, CHEMICAL ABSTRACTS SERVICE, COLUMBIO OHIO, US: RN 959531-00-1, 26 Dec 2007, Entered from STN | |||
| Способ получения замороженных полуфабрикатов из сыра | 1985 |
|
SU1358892A1 |
| Установка для мойки пакета тарелок сепаратора | 1980 |
|
SU902880A1 |
| WO 2004064845 А1, 05.08.2004 | |||
| В.В.КОМИСАРОВ и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ получения волокон из листьев агав, юккацей и проч. | 1924 |
|
SU578A1 |
| US 0009066932 B2, 30.06.2015. | |||

