Данное изобретение относится к композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе, а также к способу получения упомянутой композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе, исходя из графитового материала и с применением ультразвуковой технологии.
В данном тексте под композицией, содержащей графен и графеновые нанопластинки, диспергированные в растворителе, подразумевают стабильную дисперсию графена и графеновых нанопластинок, в которой не наблюдают образования осадков или выделившихся в растворителе фаз.
Под графитовым материалом подразумевают материал, состоящий по существу из графитизированного углерода, как он определен IUPAC в документе «Recommended Terminology for the Description of Carbon as a Solid» (Рекомендуемая терминология для описания углерода как твердого вещества) (IUPAC Recommendations (Рекомендации IUPAC), 1995).
В качестве исходного материала для получения данной композиции, описанной и заявленной в данной патентной заявке, в дополнение к материалу графита также может присутствовать неграфитированный углерод, такой как, например, сажа.
Целью способа по данному изобретению является получение композиций, содержащих графен и графеновые нанопластинки, обладающие высокой степенью эксфолиации, вплоть до получения моноатомного слоя в случае графена, или ограниченного числа слоев в случае графеновых нанопластинок.
Термин «эксфолиация» (расслоение) относится к способу, посредством которого слои многослойного материала (например, графитового материала, применяемого в данном изобретении) отделяют друг от друга (((Definitions of terms relating to the structure and processing of sols, gels, networks and inorganic-organic hybrid materials)) (Определения терминов, относящихся к структуре и переработке золей, гелей, пространственных структур и неорганически-органических гибридных материалов) - IUPAC Recommendations 2007).
Композиции, содержащие графен и графеновые нанопластинки, диспергированные в растворителе, описанные и заявленные в данной патентной заявке, можно применять для получения следующих материалов:
- полимерных композитов, или полимерных нанокомпозитов, или маточных смесей (последние также известны как концентрированные полимерные композиты), содержащих графен и графеновые нанопластинки; и эти соединения обладают высокими механическими характеристиками, высокой термостойкостью, антистатическими свойствами, свойствами электромагнитной изоляции, или имеют высокую теплопроводность;
- вспениваемых полимерных композитов, или вспениваемых полимерных нанокомпозитов, из которых можно получить изделия и вспененные полимеры, обладающие высокой теплоизолирующей способностью, термостойкостью и высокими механическими характеристиками;
- графена и графеновых нанопластинок в твердой форме, полученных из композиций, описанных, заявленных и полученных с помощью описанного и заявленного способа.
Вышеуказанные продукты применяют в качестве промежуточных продуктов в последующих процессах. В частности, полученные таким образом графен и графеновые нанопластинки, а также относящиеся к ним композиции или дисперсии, обладающие высокой чистотой, можно использовать для применений в электронике.
Полимерный нанокомпозит определяют как композит, в котором по меньшей мере одна из фазовых областей имеет по меньшей мере один размер порядка нанометров.
Заявитель также предполагает защитить права по полимерным композитам, или полимерным нанокомпозитам, также и вспениваемым, у которых полимер представляет собой винилароматический тип; упомянутые композиты можно получить исходя из полимерных композиций, описанных и заявленных в данном тексте.
Упомянутые полимерные композиты или полимерные нанокомпозиты, возможно, вспениваемые, можно применять для получения полимерных гранул, промышленных товаров или изделий из вспениваемых полимеров, расширяющихся полимерных пен, обладающих высокой теплоизолирующей способностью, термостойкостью и высокими механическими характеристиками; и они также являются объектом данного изобретения.
В данном тексте термин "GRS" относится к графену и графеновым нанопластинкам.
В данном тексте термин «графен и графеновые нанопластинки» относится к графену, или к графеновым нанопластинкам, или и к тому, и к другому.
В данном тексте термин «растворитель» относится к текучей фазе, в которой диспергированы графен и графеновые нанопластинки, безотносительно к возможным растворяющим свойствам компонентов.
В данном тексте термин «жидкая фаза» относится также к композиции, включающей жидкую фазу и твердые материалы в виде частиц (например, графеновые нанопластинки), которые в ней диспергированы.
Для целей данного изобретения все рабочие условия, указанные в тексте, следует рассматривать как условия предпочтительные, даже если это не указано конкретно.
Для целей данного изобретения термин «содержать» или «включать» включает также термин «состоять из» или «по существу состоящий из».
Для целей данного изобретения определения диапазонов всегда включают крайние точки, если только конкретно не указано иное.
Графен представляет собой моноатомный слой атомов углерода, sp2-гибридизованных, выстроенных в соответствии с кристаллической структурой, имеющей гексагональные ячейки. Эта базовая структура обладает плоской конформацией и, следовательно, данный моноатомный слой выглядит как двухмерный материал. Он представляет собой базовую структуру всех аллотропных графитных форм углерода: графит, фактически, состоит из листов графена, сложенных в стопку и разделенных расстоянием 3,37 А; фуллерены можно представить как свернутую в кольцо секцию графенового листа, в то время как углеродные нанотрубки можно получить, скручивая в трубку графеновые листы. В течение более чем 60 лет полагали, что этот тип материала не может существовать в изолированном состоянии, так как он является термодинамически нестабильным в отношении образования изогнутых структур, таких как фуллерены или нанотрубки. Впервые он был выделен в 2004 году двумя русскими исследователями A.K. Geim и K.S. Novoselov. Из-за специфической геометрии системы и электронной конфигурации углерода графен имеет очень специфичную структуру полос электронного спектра и обладает исключительными механическими, а также электронными свойствами. Обладая модулем Юнга, равным 1 ТПа, и пределом прочности на разрыв, равным 130 ГПа, графен является наиболее прочным материалом из всех, на которых до сих пор проводили измерения. Кроме того, он обладает теплопроводностью, равной 5000 Вт/мK, и электрическим сопротивлением 4×10-5 Ом⋅см. В дополнение к его замечательным механическим, термическим и электрическим свойствам, графен обладает также исключительными оптическими свойствами: фактически, даже если графеновый лист имеет толщину, равную размеру одного атома, он способен поглощать существенную долю, равную 2,3%, от падающего светового потока. Также было замечено, что поглощение не зависит от длины λ волны (от УФ до почти инфракрасных длин волн) и возрастает с увеличением числа слоев; на практике каждый слой графена добавляет дополнительные 2,3% поглощения (следовательно, 2 слоя графена поглощают 4,6% падающего светового потока).
С момента открытия графена были разработаны различные способы его получения; но самой важной задачей является получение высококачественных образцов с помощью способа, который может быть масштабируемым. Фактически поведение данного материала зависит, в дополнение к общему качеству кристаллической решетки, от количества присутствующих слоев (Honeycomb Carbon: A Review of Graphene (Сотообразный углерод: обзор по графену); Matthew J. Allen, Vincent С. Tung и Richard В. Kaner; Chem. Rev. 2010, 110, 132-145).
Способы получения графена можно разделить на две макро-категории: восходящие способы и нисходящие способы.
К первой категории принадлежат различные способы, такие как CVD (химическое осаждение из паровой фазы), эпитаксиальный рост на SiC, дуговой разряд, разворачивание углеродных нанотрубок. Эти способы позволяют получить однослойный графен, или двухслойный, или с небольшим количеством слоев, высокой чистоты и с большими размерами сторон, но они дороги и позволяют вести получение лишь в небольшом масштабе; таким образом, они не пригодны для производства полимерных нанокомпозитов. Например, патент US 2011/244661 описывает способ получения нанолент (полос, имеющих ширину порядка нм) из графена, посредством разворачивания углеродных нанотрубок. Этот способ заключается в частичном окислении углеродных нанотрубок, диспергировании их в органическом растворителе и осуществлении механического перемешивания раствора, чтобы раскрыть нанотрубки.
Нисходящие способы заключаются в получении графена и модифицированного графена посредством эксфолиации графита или его производных (таких как оксид графита, например). Эти способы являются, конечно, более предпочтительными с экономической точки зрения, чем восходящие способы, так как графит является минералом, широко распространенным в природе и легко добываемым, и, следовательно, дешевым (Graphene/Polymer Nanocomposites (Нанокомпозиты графен/полимер); Hyunwoo Kim, Ahmed A. Abdala и Christopher W. Macosko; Macromolecules 2010, 43, 6515-6530; Graphene based materials: Past, present and future (Материалы на основе графена: прошлое, настоящее и будущее); Virendra Singh, Daeha Joung, Lei Zhai, Soumen Das, Saiful I. Khondaker, Sudipta Seal; Prog. Mat. Sci 56, 2011, 1178-1271).
Другой способ крупномасштабного получения графена основан на эксфолиации и восстановлении оксида графита. Последний получают методами Staudenmaier или Hummers (или с помощью их вариаций) путем окисления графита KMnO4, KClO3 и NaNO3 в присутствии азотной или серной кислоты (Staudenmaier, L., Ber. Dtsch. Chem. Ges. 1898, 31, 1481-87; Hummers, W.S., Jr., Offerman, R.E., J. Am. Chem. Soc. 1958, 80, 1339).
По сравнению с графитом как таковым оксид имеет гидроксильные и эпоксидные группы на sр3-гибридизованных атомах углерода базовой плоскости, в дополнение к карбонильным и карбоксильным группам, расположенным на кромках слоев на sp2-гибридизованных атомах углерода. Таким образом, оксид графита является в высокой степени гидрофильным, и может легко расслаиваться в воде; кроме того, расстояние между слоями больше, чем расстояние между слоями графита (около 0,34 нм у графита по сравнению с 0,6-1 нм у оксида) из-за смещения sp3-aтомов углерода выше и ниже исходной базовой плоскости и присутствия атомов кислорода, связанных ковалентными связями.
Присутствие функциональных групп обеспечивает реакционноспособные активные центры для различных реакций модификации поверхности, чтобы получить функционализованный оксид графита.
Основным недостатком этого способа является то, что присутствующие функциональные группы разрушают сопряженную электронную структуру графена, внося дефекты и разупорядоченности. Это оказывает сильное воздействие на оптические и электрические свойства, которые в сильной степени зависят от пространственного распределения функциональных групп и от дефектов структуры; оксид графита фактически является электроизоляционным и термически нестабильным материалом.
Кроме того, оптические свойства также полностью отличаются от свойств графена. В частности, оксид графита теряет свойство поглощать инфракрасное излучение, независимо от длины волны, и это сводит к нулю возможность его применения для повышения теплоизоляции во вспененных полимерах.
Введенные топологические дефекты можно классифицировать как изолированные (присутствие пентагонов и гептагонов вместо гексагональных ячеек) и экстенсивные (почти аморфные структуры углерода).
Восстановление оксида графита, как химическим, так и термическим способом, может восстановить проводимость (электрическую и оптическую), но только частично, до значений, которые в любом случае на порядки по величине ниже, чем у чистого графена. Восстановление оксида фактически является неполным, и внутри структуры остается значительное содержание кислорода, также в виде дефектов и разупорядоченностей.
После восстановления в окисленных областях восстанавливается структура sp2 атомов углерода, но кристалличность исходно применяемого графита в любом случае утрачивается. В частности, присутствие атомов углерода с sp3-гибридизацией в сильной степени ограничивает электронные возможности и взаимодействие с инфракрасным излучением полученного таким образом графена. Более того, разупорядоченные области, подвергнутые восстановлению, вводят напряжения и деформации как внутри, так и за пределами базовой плоскости.
Химическое восстановление происходит с применением различных восстанавливающих агентов (таких как, например, гидразин, боргидрид натрия), но опасная природа и стоимость этих химических агентов могут ограничивать такие применения.
Другим способом проведения восстановления оксида графита является быстрое нагревание до высоких температур. Материал нагревают до температур порядка 1000°С в течение нескольких секунд, в инертной среде; таким образом получают как восстановление, так и эксфолиацию. При разложении эпоксидных и гидроксильных групп, фактически, выделяется СО2; и когда создаваемое газом давление превышает силы Ван-дер-Ваальса, которые удерживают слои вместе, происходит эксфолиация.
Термическое восстановление имеет то преимущество, что оно не требует применения растворителей, но из-за структурных дефектов, вызванных потерей СО2, слои в сильной степени смяты и завернуты друг на друга.
WO 2012/166001 описывает способ получения графена посредством обработки графита серной кислотой, с последующим окислением перманганатом калия и окончательным восстановлением оксидов спиртами.
Аналогично, CN 103408000 описывает способ получения оксида графита с большими размерами сторон и большой площадью поверхности. В этом случае природный графит также обрабатывают азотной и серной кислотами и далее окисляют с использованием сильного окисляющего агента (пероксида водорода).
Как указано выше, оксид графита, полученный с помощью процессов, описанных в вышеупомянутых патентах, представляет собой материал, отличающийся присутствием дефектов структуры, который, даже в восстановленном состоянии, не возвращается к кристаллической структуре исходного материала графита. Кроме того, заявленные в этих патентах способы предусматривают применение химических агентов, которые часто вызывают коррозию и наносят вред окружающей среде (таких как сильные кислоты и окислители); следовательно, перевод их в промышленное производство будет сложным и дорогостоящим.
Механическая эксфолиация графита представляет собой способ, с помощью которого впервые был выделен однослойный графен. Этот способ, который невозможно применять в промышленном масштабе, заключается в использовании липкой ленты для разделения слоев графита; он позволяет получить высококачественный графен, с большими размерами сторон, но в очень малых количествах.
Прямую эксфолиацию графита можно провести в жидкой фазе, посредством обработки ультразвуком в подходящем растворителе. Этот способ обладает различными преимуществами: потенциально он является легко масштабируемым, следовательно, можно в промышленных количествах получать однослойный графен или графен с небольшим количеством слоев, возможно, функционализованный. Недостатки, обычно присутствующие в этом способе получения, можно охарактеризовать следующим образом: низкий выход и сложность разделения или извлечения графеновых листов или графеновых нанопластинок из раствора. В частности, извлечение путем фильтрования или центрифугирования приводит к некоторой степени штабелирования и повторной сборки графеновых слоев, что снижает эффективность данного способа. Кроме того, слишком интенсивная обработка ультразвуком может привести к чрезмерному уменьшению размеров сторон слоев, что вызывает уменьшение аспектного отношения (отношения между размером стороны и толщиной).
Выбор растворителя является основополагающим вопросом, так как для того, чтобы получить хорошую дисперсию и эксфолиацию графита, поверхностное натяжение на границе раздела растворителя и материала графита должно быть сведено к минимуму ("High-yield production of graphene by liquid-phase exfoliation of graphite" (Получение графена с высоким выходом путем жидкофазной эксфолиации графита), Hernandez Y., Nicolosi V., Lotya M, Blighe F.M., Sun Z., De S., Mc Govern I.T., Holland В., Byrne M., Gun'ko Y.K., Boland J.J., Niraj P., Duesberg G., Krishnamurthy S., Goodhue R., Hutchison J., Scardaci V., Ferrari A.C., Coleman J.N., Nature Nanotechnology 2008, 3, 563-568). Если межфазное поверхностное натяжение является высоким, частицы графита фактически будут склонны слипаться друг с другом и, следовательно, необходимая для разделения двух соседних слоев энергия на единицу поверхности будет более высокой. Идеальными для диспергирования графита являются те растворители, у которых межфазное поверхностное натяжение составляет около 40 мДж/м2, такие как, например, N-метилпирролидон, диметилформамид и орто-дихлорбензол.
В вышеупомянутой статье Hernandez et al. "High-yield production of graphene by liquid-phase exfoliation of graphite" авторы получали дисперсию графена в N-метилпирролидоне (NМП) с концентрациями до 0,01 мг/мл, исходя из графита, с получением неокисленного графена без дефектов, с выходом однослойного графена около 1 мас.%.
Однако применяемый растворитель (NМП) обладает многочисленными недостатками: кроме того, что он является токсичным, вызывающим раздражение и уродства, он обладает также высокой температурой кипения (203°С), что помимо прочего ограничивает манипуляции с ним и его применение в области электроники, где требуется полное удаление растворителя, который иначе влияет на характеристики.
CN 103466612 описывает способ получения графена исходя из графита и применения двух или более компонентов при различных частотах ультразвука в диапазоне от 20 до 1000 кГц. В качестве растворителей используют воду и спирты, но также и растворы неорганических кислот, альдегидов, кетонов, жидкие алканы или их комбинации.
WO 2013/010211 описывает способ эксфолиации слоистого материала (таким образом, не обязательно графена) посредством ультразвуковой обработки в подходящем поверхностно-активном веществе.
CN 103112848 описывает способ получения графена, который включает диспергирование графита в кислом водном растворе хитозана, посредством ультразвуковой обработки в течение 0,5-50 часов.
Затем дисперсию оставляют для декантации, и надосадочную жидкость центрифугируют для извлечения эксфолиированного материала с минимальным количеством слоев.
KR 20110077606 включает обработку графита ультразвуком в пропаноле в течение 10 - 20 минут, с последующим центрифугированием при 4500-5500 об/мин для устранения неэксфолиированных частиц графита.
Для того чтобы провести эксфолиацию графита, можно прибегнуть к подготовительной стадии, заключающейся в интеркаляционной обработке.
Графит обрабатывают соответствующими веществами, которые размещаются между слоями, образуя так называемые интеркаляционные соединения графита (ИСГ). Способ, обычно применяемый для интеркаляции, включает диспергирование графита в азотной кислоте, серной кислоте или смеси обеих этих кислот, но часто также применяют другие вещества, такие как хлорат калия, хромовая кислота, перманганат калия, хлорная кислота. Далее интеркалированный графит подвергают быстрому нагреванию до высоких температур (700-1000°С), в ходе которого частицы расширяются до 80-1000 раз по отношению к их исходному объему, принимая специфичную структуру гармошки в направлении, перпендикулярном кристаллическим слоям графита.
Этот способ описан в различных документах, например, в US 2010/140792 и WO 2011/162727, где для разделения слоев после интеркалирования предусматривают применение процессов электрохимической, термической, акустической природы, сверхвысоких частот и ультразвука.
WO 2008/060703 описывает способ получения наноструктур (нанотрубок, фуллеренов и графеновых нанопластинок) путем интеркалирования графита муравьиной или уксусной кислотой, водой или их комбинацией, с последующим приведением интеркалированного соединения в контакт со сверхкритической текучей средой и проведением термической эксфолиации при температуре по меньшей мере 1450°С, подавая интеркалированный графит в плазму из инертного газа.
Недостатком интеркаляционного способа является применение не безопасных для окружающей среды веществ; кроме того, этот процесс производит различные виды сернокислых и азотнокислых соединений, как в жидкой, так и в газообразной фазе, а это требует обработки по очистке. Дополнительным недостатком интеркаляции является то, что в случае этого процесса трудно контролировать степень эксфолиации, и его нельзя применять для получения графеновых нанопластинок, имеющих лишь небольшое количество слоев.
В статье "High concentration few-layer graphene sheets obtained by liquid phase exfoliation of graphite in ionic liquid" (Графеновые листы высокой концентрации с небольшим количеством слоев, полученные путем жидкофазной эксфолиации графита в ионной жидкости) (Nuvoli D., Valentini L., Alzari V., Scognamillo S., Bittolo Bon S., Piccinini M., Illescas J. и Mariani A.; J. Mater. Chem., 2011, 21, 3428-3431), авторы предлагали ультразвуковую обработку графита в ионной жидкости (гексафторфосфат 1-этил-3-метилимидазола). Даже при получении концентрации 5,33 мг/мл, было подтверждено присутствие частиц, имеющих толщину 2 нм, что демонстрирует низкий выход эксфолиации.
KR 20130068515 также описывает способ получения графена с применением ионных жидкостей, подвергая смесь ионных жидкостей, содержащих фтор, и материала графита действию ультразвука, с последующим воздействием сверхвысоких частот, а затем опять ультразвука.
В статье "Preparation of Graphene by Using an Intense Cavitation Field in a Pressurized Ultrasonic Reactor" (Получение графена с использованием интенсивного кавитационного поля в работающем под давлением ультразвуковом реакторе) (Stengl V., Chem, Eur. J. 2012, 18, 14047-14054), авторы представили способ получения высококачественного и неокисленного графена исходя из природного графита, с использованием кавитации в ультразвуковом реакторе, работающем под давлением. Графит добавляли к смеси воды и этиленгликоля (в отношении 9:1) и подвергали воздействию кавитации в течение 50 минут. Кавитацию проводят в ультразвуковом реакторе под давлением 0,5 МПа (5 бар) и при подаваемой в жидкость интенсивности (ультразвука) свыше 300 Вт/см2.
WO 2011/014347 описывает способ эксфолиации графитового материала с получением графеновых нанопластинок, который заключается в диспергировании материала графита в жидкой среде, дающей при контакте с графитом контактный угол ниже 90°, и в обработке полученной суспензии ультразвуком.
На последующей стадии данный способ включает также добавление к суспензии графеновых нанопластинок в растворителе мономера или полимера, с получением предшественника нанокомпозита, который можно превратить в твердое вещество, удаляя растворитель или полимеризуя мономер.
Во всех случаях эти способы не позволяют получить полимерный композит, содержащий графен или графеновые нанопластинки; или, в любом случае, позволяют получить их, но не за одну стадию. Извлечение графена или нанопластинок из раствора является деликатной операцией, которая легко может вызвать агломерацию этого материала, таким образом снижая эффективность обработки. Кроме того, поскольку это требует применения растворителей, поверхностно-активных веществ и других химических агентов, это может оказывать существенное воздействие на окружающую среду, что делает процесс очень продолжительным, дорогостоящим и сложным.
Другим фактором, который характеризует вышеописанные способы и который отличает их от способа, описанного в данном изобретении, является фактическое время обработки ультразвуком, которое во всех случаях больше, чем время обработки ультразвуком, применяемое в данном изобретении.
В статье "Sonochemical Preparation of Functionalized Graphenes" (Сонохимическое получение функционализованных графенов) (Hangxun Xu, Kenneth S. Suslick; J. Am. Chem. Soc, 2011, 133, 9148-9151), авторы предлагали одностадийный способ для получения графена, функционализованного стиролом, исходя из графита (природного) в форме частиц. В соответствии с этим способом природный графит смешивают со стиролом; и эту смесь облучают ультразвуком высокой интенсивности; ультразвуковую обработку проводят посредством зонда при частоте 20 кГц и интенсивности 50 Вт/см2, при 0°С в течение двух часов, в токе аргона. Этот способ позволяет получить эксфолиацию графита до однослойного графена или графена с малым количеством слоев, обычно меньше 5, и одновременно полимеризацию стирола и функционализацию графена образованными полистирольными цепями.
Следовательно, в результате этого способа получают функционализованный графен, у которого полистирольные цепи абсорбированы на поверхности графена. Данная статья фактически объясняет, что в ходе обработки ультразвуком в указанных условиях стирол полимеризуется, образуя реакционноспособные радикалы, в то время как 3D структура графита разрушается, образуя 2D структуру графена. На этой стадии реакционноспособные радикалы связываются с поверхностью графена, таким образом образуя графен, функционализованный полистиролом. Данная статья также утверждает, что полученные функционализованные графены являются стабильными и растворимыми в обычных органических растворителях, и их можно использовать для получения материалов на основе упомянутых графенов.
В отличие от способа, описанного в статье Suslick et al., способ по данному изобретению позволяет производить полимерный нанокомпозит, полученный эксфолиацией материала графита с одновременной полимеризацией растворителя.
Статья Suslick et al. не упоминает о возможности полимеризации предложенной системы. В любом случае специалист может считать, что при используемых условиях было бы чрезвычайно трудно получить значительное количество полимера и, следовательно, соединения с полимером.
Условия, используемые Suslick et al. отличаются от условий, применяемых в способе, описанном и заявленном в данной патентной заявке, как станет очевидно далее по тексту. В отношении условий, применяемых в статье Suslick et al., фактически способ по данному изобретению проводят при более коротком времени обработки ультразвуком; обработку ультразвуком осуществляют при более высоком давлении, в дегазированной среде и обычно при значительно более высокой температуре. Заявитель хотел бы снова подчеркнуть, что способ по данному изобретению проводят в отсутствие газообразной фазы в ходе обработки ультразвуком, в то время как в соответствии с Suslick et al. обработку ультразвуком проводят в присутствии аргона: чтобы иметь возможность осуществить способ, описанный и заявленный в данной патентной заявке, предпочтительно удалить и то небольшое количество газа, которое обычно растворено в применяемом растворителе.
Для того чтобы лучше понять сущность данного изобретения, описанного и заявленного, заявитель хотел бы обсудить две статьи, которые имеют отношение к полимеризации стирола при обработке его ультразвуком.
В статье "Polymerization of Styrene Initiated by Ultrasonic Cavitation" (Полимеризация стирола, инициированная ультразвуковой кавитацией) (P. Kruus, D. McDonald, T.J. Patraboy; J. Phys. Chem. 1987, 91, 3041-3047), которую далее упоминают как "Kruus et al.", авторы исследуют полимеризацию стирола в ходе обработки ультразвуком, оценивая скорость полимеризации стирола в связи с температурой реакции (температурой в объеме) и реакционной средой. В статье Kruus et al. обработку ультразвуком проводят с мощностью 60 Вт, удельной мощностью 21 Вт/см2 и частотой ультразвука 20 кГц; обработка ультразвуком происходит в присутствии аргона. Присутствие аргона необходимо как для ограничения кавитационного шума, так и для того, чтобы благоприятствовать полимеризации в отношении образования окрашенных соединений.
В статье Kruus et al. авторы сделали заключение, что скорость полимеризации стирола уменьшается со снижением температуры полимеризации. В частности, ниже 48°С скорость полимеризации значительно снижается, и наблюдается получение окрашенных соединений. Добавление жидких углеводородов с высоким давлением пара, таких как, например, н-гексан, н-гептан, толуол или циклогексан, увеличивает скорость полимеризации и подавляет образование окрашенных соединений.
В статье "Polymerization and depolymerization by Ultrasound" (Полимеризация и деполимеризация ультразвуком) (P. Kruus, J.A.G. Lawrie, M.L. O'Neill, Ultrasonics 1988, т. 26, ноябрь 352-355), упоминаемой далее как "Lawrie et al.", авторы исследуют действие присутствия потока аргона на скорость полимеризации стирола и распределение его по молекулярному весу при обработке его ультразвуком.
В статье Lawrie et al. описана обработка стирола ультразвуком, проводимая при мощности 86 Вт, удельной мощности 34 Вт/см2 и частоте ультразвука 20 кГц, в присутствии различных по интенсивности потоков аргона.
Данное исследование показывает, что в присутствии потока аргона полимеризация стирола является явной; если ток аргона прекращается, превращение стирола резко снижается. Кроме того, в период, когда поток аргона прерывают, авторы наблюдают деполимеризацию образованного полимера и образование окрашенных соединений.
На основе концепций вышеупомянутых статей можно сделать заключение, что низкие температуры в значительной степени снижают полимеризацию стирола, если при этом его подвергают воздействию ультразвука. Кроме того, из данных, приведенных Suslick (см. статью и приложенную к ней дополнительную информацию), можно оценить, что 18% полимера, связанного с графеном, составляет порядка десятков млн. ч по отношению к исходному полимеру. Таким образом, можно сделать заключение, что степень превращения, полученная Suslick, является чрезвычайно низкой по сравнению со степенью превращения, полученной способом, описанным и заявленным в данной заявке.
Кроме того, в соответствии с концепциями вышеупомянутых статей, если обработку ультразвуком проводят в отсутствии газообразного потока (например, аргона), полимеризация стирола прерывается и начинается деполимеризация. Способ, описанный и заявленный в данной патентной заявке, напротив, проводят при более высоких температурах и в отсутствии газообразной фазы в ходе обработки ультразвуком.
В WO 2011/055198 описан способ получения графеновых нанопластинок, исходя из графита и проведения функционализации кислородом посредством термического окисления, приводя графит в контакт с кислородом или веществом, способным его выделять (озоном). Затем частично окисленный графит восстанавливают соединениями-восстановителями, такими как гидразин, метилгидразин, водород. Данная патентная заявка описывает также композиции на основе термопластичных полимеров и графеновых нанопластинок, с улучшенными механической прочностью, электропроводностью и термоизолирующей способностью.
В WO 2011/042800 описана композиция на основе вспениваемых термопластичных полимеров, обладающая наилучшими термоизолирующими свойствами, содержащая графеновые нанопластинки в качестве непроницаемых для инфракрасного излучения агентов. Как было показано, полимерные пены, полученные из этих вспениваемых соединений, отличаются более высокими термоизолирующими свойствами по сравнению с полимерными пенами, содержащими другие непроницаемые для инфракрасного излучения агенты, такие как графит, уголь, алюминиевые частицы.
Однако ни одна из вышеупомянутых патентных заявок не использует ультразвук для получения нанопластинок или для получения связанных с ними соединений.
U. Khan et al. "High-Concentration Solvent Exfoliation of Graphene" (Эксфолиация графена в растворителе с получением высоких концентраций), Small, т.6, Выпуск 7, сс.864-871, 2010, описывает способ получения дисперсий графена с высокой концентрацией (до 1,2 мг/мл) посредством обработки ультразвуком в N-метилпирролидоне (NMП). Однако время, необходимое для получения столь высоких концентраций, является чрезвычайно длительным, обработка ультразвуком продолжается до 460 часов.
A. Ciesielski, P. Samori, "Graphene via sonification assisted liquid-phase exfoliation" (Получение графена жидкофазной эксфолиацией под действием ультразвука), Chem. Soc. Rev., 2014, 43, 381-398, описывает различные способы и результаты, полученные при производстве графена посредством обработки ультразвуком в жидкой фазе. Подробно обсуждают время, необходимое для обработки ультразвуком, которое может достигать 1000 часов. Продолжительное время обработки ультразвуком необходимо для получения растворов с высокой концентрацией (см. Фиг. 6, приведенную в статье); необходимо по меньшей мер 100 часов для достижения концентрации, равной 1 мг/мл. Как подтверждает эта же статья, продолжительная обработка ультразвуком вызывает уменьшение размеров сторон полученных нанопластинок, а это нежелательно, поскольку размер стороны является критическим параметром для многочисленных применений.
Краткое описание изобретения
Недостатки и ограничения вышеописанного существующего уровня техники преодолевают с помощью композиции и способа, описанных и заявленных в данном патентном описании.
Предмет данной патентной заявки относится к композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе, при этом указанная композиция отличается тем, что:
a) она содержит по меньшей мере 1 мас.%, по отношению к общей массе растворителя, винилароматического полимера,
b) она содержит массовую концентрацию графена и графеновых нанопластинок (GRS) в диапазоне от 0,001% до 10 мас.% по отношению к общей массе растворителя;
при этом указанный винилароматический полимер получают путем частичной или полной полимеризации соответствующего винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных способных к сополимеризации мономеров; и при условии, что сумма возможного содержания указанных непрореагировавших мономеров и содержания образованного винилароматическог полимера равна по меньшей мере 10 мас.% по отношению к общей массе растворителя.
Данное изобретение относится также к способу получения по меньшей мере частично полимеризованной композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе, исходя из материала графита; при этом указанный способ включает следующие стадии:
а) приведение материала графита в контакт с основным растворителем, содержащим по меньшей мере 10 мас.% по отношению к общей массе основного растворителя, винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных способных к сополимеризации мономеров, образуя исходную композицию;
b) воздействие на указанную исходную композицию ультразвука, отличающегося частотным спектром в диапазоне от 18 кГц до 1000 кГц; и давления не менее 0,2 МПа (2 бар) абс; в контейнере или камере для обработки ультразвуком, где в контакте с указанной композицией не должна присутствовать отдельная текучая фаза в газообразном состоянии;
при этом указанный способ отличается тем, что по меньшей мере 1% винилароматического мономера, присутствующего в основном растворителе, является полимеризованным.
Данное изобретение предпочтительно представляет собой способ, как он описан и заявлен, для получения композиций, описанных и заявленных в данном тексте.
После получения композиций, описанных и заявленных в данном тексте, в ходе последующей дополнительной стадии полимеризации основного растворителя можно получить полимерные композиты, или полимерные нанокомпозиты, или маточные смеси (известные также как концентрированные полимерные композиты), содержащие графен и графеновые нанопластинки.
Или же упомянутые полимерные композиты, или полимерные нанокомпозиты, или маточные смеси можно получить непосредственно с помощью способа, описанного и заявленного в данном тексте, завершая полимеризацию в ходе того же процесса.
Полимерные композиции, полимерные нанокомпозиты или концентраты, которые можно получить из композиций, описанных и заявленных в данном тексте, обладают тем преимуществом, что они содержат графен и графеновые нанопластинки с высокой степенью эксфолиации и высокой степенью химической и кристаллической чистоты.
Способ - объект данного изобретения - выгодно отличается простотой, может быть легко масштабирован для целей крупномасштабных производств и имеет низкую стоимость и малое воздействие на окружающую среду.
В способе по данному изобретению нет необходимости проводить операции «обмена растворителей», то есть замещения основного растворителя, применяемого для эксфолиации, растворителем, необходимым для данного применения.
Фактически в способе по данному изобретению эксфолиацию проводят непосредственно в основном растворителе, содержащем мономер, из которого путем полимеризации получают полимер, присутствующий в полимерном композите или полимерном нанокомпозите.
Операция «обмена растворителей», в дополнение к тому, что она усложняет процесс, что как правило создает проблемы, связанные с окружающей средой, фактически может привести к частичной агломерации слоев эксфолиированного материала графита, таким образом снижая качество полученного полимерного нанокомпозита.
Способ - объект данной патентной заявки - обеспечивает преимущество, состоящее в том, что он не создает проблем, связанных со здоровьем, безопасностью и окружающей средой, а также с обработкой наноматериалов. Фактически при обработке не образуются нанопорошки в свободном состоянии. Нанопорошки всегда находятся в растворе с основным растворителем, или непосредственно заключены в полимерный компонент конечного нанокомпозита.
Описанный и заявленный способ обеспечивает преимущество, состоящее в том, что эффективное время, в течение которого материал графита подвергают действию ультразвука, короче, чем времена, обычно используемые в способах существующего уровня техники.
Способ, описанный и заявленный в данном тексте, обеспечивает преимущество, состоящее в том, что он не зависит от межфазного поверхностного натяжения основного растворителя.
Фактически упомянутый способ позволяет обеспечить эксфолиацию материала графита и образование стабильных во времени растворов и с использованием растворителей, имеющих межфазное поверхностное натяжение, отличное от поверхностного натяжения самого материала графита, или, в любом случае, растворителей, которые не принадлежат к тем, которые считают более приемлемыми, например, таких растворителей как стирол и глицерин, вопреки тому, что описано в литературе. Следует сослаться, например, на дополнительную информацию уже упомянутой статьи Hernandez et al. "High-yield production of graphene by liquid-phase exfoliation of graphite". Таблица S1 указывает наилучший растворитель для эксфолиации и диспергирования графена: по-видимому, ацетон является значительно менее эффективным (примерно на 70% менее) чем бензилбензоат; стирол и глицерин даже не упоминаются.
Статья Hernandez et al. "High-yield production of grapheme by liquid-phase exfoliation of graphite" указывает в качестве подходящих растворителей те, которые имеют поверхностное натяжение в диапазоне от 40 до 50 мДж/м2; кроме того, в случае, когда растворителем является бензоилбензоат, который считают наиболее пригодным, после центрифугирования остается 8,3 мас.% исходного растворителя; в случае NМП после центрифугирования остается 7,6 мас.% исходного растворителя. Данное изобретение, напротив, применяет стирол, имеющий поверхностное натяжение при 120°С 26 мДж/м2, или этилбензол, который при такой же температуре имеет поверхностное натяжение 18 мДж/м2. Таким образом, удивительно, что при использовании этих растворителей и при этих режимах обработки можно получить высокую степень эксфолиации и стабильность растворов во времени.
Что касается глицерина, упомянутый выше WO 2011/014347 анализирует 50 различных растворителей для эксфолиации графита до графеновых нанопластинок. Глицерин рассматривают как пример неэффективного растворителя при получении нанопластинок графена с применением ультразвука высокой мощности: предварительные данные указывают, что растворители, имеющие контактный угол не менее 90°, такие как глицерин, являются неэффективными при получении графеновых нанопластинок, исходя из материала графита, при обработке ультразвуком высокой мощности.
С другой стороны, в способе, описанном и заявленном в данном тексте, неожиданно оказалось возможным получить стабильный во времени раствор графеновых нанопластинок также и с использованием глицерина.
Дополнительные цели и преимущества данного изобретения станут более очевидными из последующего описания и прилагаемых чертежей, обеспеченных только для иллюстративных, а не ограничивающих целей, которые представляют предпочтительные примеры воплощения данного изобретения.
Фиг. 1 изображает способ по данному изобретению в периодическом режиме.
Фиг. 2 изображает способ по данному изобретению в непрерывном режиме, в котором насос (4) расположен ниже камеры (2) обработки ультразвуком по ходу технологического потока.
Фиг. 3 изображает способ по данному изобретению в непрерывном режиме, в котором насос (4) расположен выше камеры (2) обработки ультразвуком по ходу технологического потока.
Фиг. 4 изображает способ по данному изобретению в непрерывном режиме, в котором насос (4) находится выше камеры (2) обработки ультразвуком по ходу технологического потока, а теплообменник (6) находится ниже камеры (2) обработки ультразвуком по ходу технологического потока.
Фиг. 5 изображает конфигурацию, сходную с конфигурацией Фиг. 4, но имеющую также емкость для текучей среды (7), рассчитанной на аварийные ситуации.
Фиг. 6 изображает конфигурацию, сходную с конфигурацией Фиг. 5, но имеющую также напорную емкость (8) и сливной резервуар (9).
Фиг. 7 изображает конфигурацию, сходную с конфигурацией Фиг. 6, но имеющую также бак - отстойник (10).
Фиг. 8 изображает конфигурацию, сходную с конфигурацией Фиг. 7, но включающую еще накопительную емкость (11).
Фиг. 9 изображает конфигурацию, сходную с конфигурацией Фиг. 8, но включающую еще концентратор (12).
На всех чертежах от 2 до 9 конечный продукт собирают в специальном сборном резервуаре (5).
Фиг. 10 представляет собой фотографию продукта 1В (пример 1), полученную с помощью трансмиссионной электронной микроскопии (ТЭМ).
Фиг. 11 представляет собой фотографию продукта 2В (пример 2), полученную с помощью трансмиссионной электронной микроскопии (ТЭМ).
Фиг. 12 представляет собой фотографию продукта 4В (пример 4), полученную с помощью трансмиссионной электронной микроскопии (ТЭМ).
Фиг. 13 представляет собой фотографию продукта 1А (см. Пример 1) в сравнении с такой же композицией примера 1, не обработанной ультразвуком.
Фиг. 14 представляет собой фотографию продукта 1А (см. Пример 1) в сравнении с такой же композицией Примера 1, обработанной ультразвуком при атмосферном давлении (Сравнительный Пример 1).
Подробное описание изобретения
Теперь данное изобретение, объект данной патентной заявки, будет описано подробно.
В первую очередь данное изобретение относится к композиции, содержащей растворитель, графен и графеновые нанопластинки, стабильно диспергированные в упомянутом растворителе. Упомянутая композиция отличается тем, что она содержит по меньшей мере 1% масс, по отношению к общей массе растворителя, винилароматического полимера, а также содержит массовую концентрацию графена и графеновых нанопластинок (GRS) в диапазоне от 0,001% до 10 мас.%, по отношению к общей массе растворителя.
Упомянутый винилароматический полимер получают путем частичной или полной полимеризации соответствующего винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных способных к сополимеризации мономеров, и при условии, что сумма возможного содержания упомянутых непрореагировавших винилароматических мономеров, возможно смешанных с дополнительными непрореагировавшими способными к сополимеризации мономерами; и содержания упомянутого винилароматического полимера равна по меньшей мере 10 мас.% по отношению к общей массе растворителя.
В случае частичной полимеризации мономеров винилароматический мономер составляет предпочтительно по меньшей мере 10 мас.% по отношению к сумме масс всех мономеров и упомянутого винилароматического полимера; еще более предпочтительно винилароматический мономер составляет по меньшей мере 25 мас.% по отношению к сумме масс всех мономеров и упомянутого винилароматического полимера.
Содержание винилароматического полимера предпочтительно составляет по меньшей мере 5 мас.% более предпочтительно по меньшей мере 10 мас.% по отношению к общей массе растворителя. Содержание винилароматического полимера предпочтительно равно или ниже 70 мас.% по отношению к общей массе растворителя.
Содержание винилароматического полимера предпочтительно находится в диапазоне от 5 мас.% до 70 мас.% по отношению к общей массе растворителя, более предпочтительно от 10 мас.% до 70 мас.% по отношению к общей массе растворителя.
Сумма возможного содержания упомянутых непрореагировавших винилароматических мономеров, возможно смешанных с дополнительными непрореагировавшими способными к сополимеризации мономерами, и содержания упомянутого винилароматического полимера предпочтительно равна по меньшей мере 20 мас.%, более предпочтительно по меньшей мере 80 мас.%, по отношению к общей массе растворителя.
Массовая концентрация графена и графеновых нанопластинок (GRS) в описанной и заявленной композиции предпочтительно находится в диапазоне от 0,05% до 5 мас.%, еще более предпочтительно от 0,2% до 2,5 мас.%, по отношению к общей массе растворителя.
GRS предпочтительно могут отличаться средней толщиной равной или менее 50 нм, более предпочтительно равной или менее 20 нм, еще более предпочтительно равной или менее 5 нм, и в любом случае равной или менее толщины одного графенового слоя.
Толщину измеряют на нескольких образцах GRS, нанесенных на кремниевую подложку, покрытую оксидом кремния, с помощью атомно-силовой микроскопии (АСМ; AFM) с режимом периодического контактирования (АСМ с прерывистым контактом).
GRS предпочтительно может иметь мольное отношение углерод/кислород (С/О) равное или выше 10, более предпочтительно равное или выше 30, еще более предпочтительно равное или выше 50, еще более предпочтительно равное или выше 100. Отношение (С/О) получают с помощью рентгеновской фотоэлектронной спектроскопии (XPS) GRS.
В описанных и заявленных композициях графен и графеновые нанопластинки предпочтительно имеют мольное отношение углерод/кислород равное 10 или более и среднюю толщину равную или менее 50 нм, и в любом случае равную толщине одного графенового слоя или более.
Полученная композиция имеет средневзвешенную молекулярную массу (ММ) в диапазоне от 50 до 500 кДа, измеренную с помощью высокопроизводительной гель-проникающей хроматографии по ASTM D5296-11, и имеет показатель текучести расплава (ПТР при 200°С, 5 кг) в диапазоне от 3 до 50. Показатель текучести измеряют в соответствии с ISO-1133, четвертое издание, 2005. Распределение по молекулярной массе измеряют посредством высокопроизводительной гельпроникающей хроматографии по ASTM D5296-11, с использованием рефрактометрического детектора; измерение проводят на полимере после отделения его от GRS и последующего осаждения.
Следующий объект данного изобретения относится к способу получения по меньшей мере частично полимеризованной композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе, исходя из материала графита; где указанный способ включает следующие стадии:
а) приведение материала графита в контакт с основным растворителем, содержащим по меньшей мере 10 мас.%, по отношению к общей массе основного растворителя, винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных способных к сополимеризации мономеров, формируя исходную композицию;
b) воздействие на указанную исходную композицию ультразвуком, который отличается частотным спектром в диапазоне от 18 кГц до 1000 кГц, при давлении, равном не менее 0,2 МПа (2 бар) (абс.), в контейнере или камере для проведения обработки ультразвуком, в которой, в контакте с указанной композицией, не должно присутствовать отдельной фазы текучей среды, находящейся в газообразном состоянии;
при этом указанный способ отличается тем, что по меньшей мере 1% присутствующего в основном растворителе винилароматического мономера полимеризован.
В одном из предпочтительных воплощений данного изобретения предложен способ получения композиций, описанных и заявленных в данной заявке, исходя из материала графита; при этом указанный способ включает следующие стадии:
a) приведение материала графита в контакт с основным растворителем, содержащим по меньшей мере 10 мас.%, по отношению к общей массе основного растворителя, винилароматического мономера, одного или в смеси с до 50 мас.% других способных к сополимеризации мономеров, формируя исходную композицию;
b) воздействие на указанную исходную композицию ультразвуком, который отличается частотным спектром в диапазоне от 18 кГц до 1000 кГц, при давлении, составляющим не менее 0,2 МПа (2 бар) (абс.), в контейнере или камере для проведения обработки ультразвуком, в которой, в контакте с указанной композицией, не должно присутствовать отдельной фазы текучей среды, находящейся в газообразном состоянии;
при этом указанный способ отличается тем, что по меньшей мере 1% присутствующего в основном растворителе винилароматического мономера полимеризован.
В описанном и заявленном способе и во всех предпочтительных его воплощениях стадии (а) и (b) могут происходить одновременно или последовательно.
В описанном и заявленном способе и во всех предпочтительных примерах его воплощения по меньшей мере 1%, предпочтительно по меньшей мере 5%, более предпочтительно от 10% до 80%, и еще более предпочтительно от 15% до 50% мономеров, присутствующих в основном растворителе, полимеризованы.
Способ, описанный и заявленный в данном описании, можно осуществить или в непрерывном проточном режиме, или с непрерывным потоком, или при изготовлении партиями, или в режиме с периодическим включением потока.
В конце описанного и заявленного процесса получают частично или полностью полимеризованную композицию, в которой графен и графеновые нанопластинки стабильно диспергированы в растворителе; предпочтительно композицию, описанную и заявленную в данном тексте.
Упомянутые композиции можно впоследствии подвергнуть, как это более подробно разъясняется далее в тексте, дополнительной стадии полимеризации, завершая полимеризацию и таким образом формируя полимерные композиты, полимерные нанокомпозиты или маточные смеси (также известные как концентрированные полимерные нанокомпозиты).
Стадия или частичной, или полной полимеризации композиций, содержащих графен и графеновые нанопластинки, стабильно диспергированные в растворителе, происходит непосредственно в ходе обработки ультразвуком, путем продолжения полимеризации, или частичной, или полной, используемых способных к полимеризации мономеров, предпочтительно винилароматического мономера, содержащегося в основном растворителе.
После того, как полимеризация полностью завершена, в ходе обработки ультразвуком непосредственно получают полимерные композиты или полимерные нанокомпозиты, содержащие графен и графеновые нанопластинки, отличающиеся высокой дисперсией графена и графеновых нанопластинок.
Данный процесс можно также провести в одну стадию, исходя из материала графита и основного реакционноспособного растворителя, который содержит по меньшей мере 10 мас.% винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных способных к сополимеризаци мономеров. В случае полной полимеризации может быть предпочтительным применять также, в дополнение к реакционноспособному мономеру, инертное вещество (такое как, например, этилбензол), способное ограничивать вязкость раствора при высоком содержании полимера в растворе. Это вещество может уже исходно присутствовать в растворе или его можно добавить в ходе обработки ультразвуком, когда вязкость начинает повышаться из-за увеличения количества полимера (см. «вторичный растворитель», описанный далее). Под термином «одна стадия» Заявитель подразумевает, что в ходе обработки ультразвуком одновременно происходит эксфолиация материала графита, диспергирование в основном растворителе и частичная или полная полимеризация мономеров, присутствующих в основном растворителе.
В ходе частичной или полной полимеризации полимеризуется по меньшей мере 1% мономера, присутствующего в основном растворителе, более предпочтительно по меньшей мере 5%, еще более предпочтительно от 10% до 80%, а еще более предпочтительно от 15% до 50% мономеров, присутствующих в основном растворителе.
Полимеризация мономера проявляется содержанием твердых веществ в продукте, подвергнутом испарению под вакуумом.
Основной растворитель, в дополнение к непосредственному влиянию на эффективность обработки ультразвуком (как описано в примерах), можно с успехом использовать для регулирования вязкости композиции в ходе ультразвуковой обработки, а также желаемой степени полимеризации. Кроме того, основной растворитель можно также использовать для предотвращения запуска неконтролируемых реакций разгона, с использованием, например, этилбензола.
Растворителем предпочтительно является винилароматический мономер, возможно смешанный с дополнительными способными к полимеризации сомономерами и, возможно, с соответствующим полимером, который образуется в ходе полимеризации.
Основной растворитель предпочтительно выбирают из винилароматических мономеров, органических соединений, способных растворять винилароматические полимеры и винилароматические мономеры, пентана и их смесей; более предпочтительно его выбирают из винилароматических мономеров, этилбензола и его смесей; еще более предпочтительными являются стирол, этилбензол и их смеси.
В ходе процесса по данному изобретению можно добавлять дополнительные химические агенты, выбранные из полимеров, зародышеобразователей и инициаторов, чтобы активизировать полимеризацию. Среди полимеров предпочтение отдают полимерам, растворимым в самом основном растворителе. Более предпочтительно полимер может содержать мономерные блоки, соответствующие блокам, присутствующим в способном к полимеризации мономере. Упомянутые дополнительные химические агенты можно добавлять перед обработкой ультразвуком или в ходе нее.
Среди инициаторов предпочтительными являются пероксиды; более предпочтительно выбранные из 1,1-ди(третбутилперокси)циклогексана (Trigonox 22Е50, AkzoNobel), трет-бутилперокси 2-этилгексилкарбоната (Trigonox 117, AkzoNobel), дибензоилпероксида (Perkadox L-W75, AkzoNobel), дикумилпероксида (Perkadox BC-FF, AkzoNobel) или веществ со слабой связью между атомами углерода, таких как 2,3-диметил-2,3-дифенилбутан (Perkadox 30, AkzoNobel). Дополнительные химические агенты и, в частности, инициаторы, можно добавлять в фазе получения исходной композиции или в ходе обработки ультразвуком.
Благодаря способу, описанному и заявленному в данном тексте, графен и графеновые нанопластинки чрезвычайно хорошо диспергированы как в конечной композиции, которая может находиться в жидкой фазе, так и в полимерном композите или полимерном нанокомпозите, полученными путем полимеризации исходя из упомянутой конечной композиции.
Графен и графеновые нанопластинки, присутствующие в конечной композиции, или в полимерном композите, или в полимерном нанокомпозите, описанных и заявленных в данном описании, отличаются средней толщиной равной или менее 50 нм, более предпочтительно равной или менее 20 нм, еще более предпочтительно равной или менее 5 нм, и в любом случае равной или менее толщины одного слоя графена.
Толщину измеряют на нескольких образцах GRS, нанесенных на кремниевую подложку, покрытую оксидом кремния, с помощью атомно-силовой микроскопии (АСМ; AFM) с режимом периодического контактирования (АСМ с прерывистым контактом).
Графен и графеновые нанопластинки, присутствующие в конечной композиции, в полимерном композите или в полимерном нанокомпозите, описанными и заявленными в данном тексте, отличаются высокой кристалличностью и химической чистотой. В частности, мольное отношение углерод/кислород (С/О) равно 10 или более, более предпочтительно равно 30 или более, еще более предпочтительно равно 50 или более, еще более предпочтительно равно 100 или более.
Отношение (С/О) получают с помощью рентгеновской фотоэлектронной спектроскопии (XPS) GRS.
В случае дальнейшего получения вспениваемых композитов полученные полимерные нанокомпозиты дают следующие характеристики: хорошие свойства в отношении электропроводности, хорошие механические характеристики и хорошие термоизолирующие способности.
После того, как получены полимерные композиты, полимерные нанокомпозиты или маточные смеси, их можно переработать с помощью обычных технологий, принятых в области преобразования полимерных материалов, таких как, например, экструзия, литье под давлением и прямое формование, с получением производных полимеров.
Как видно из последующих примеров, образование стабильной дисперсии показывает тот факт, что дисперсии, полученные способом по данному изобретению, являются визуально стабильными, то есть без какого-либо явного образования осадков или выделенных фаз, даже через 30 дней. Кроме того, образование стабильной дисперсии возможно только в том случае, если произошла по меньшей мере частичная эксфолиация материала графита. Фотографии, полученные с помощью трансмиссионного электронного микроскопа (ТЭМ), также демонстрируют образование графеновых нанопластинок, как это проиллюстрировано на Фиг. 9, 10 и 11.
Как уже было указано, материал графита, применяемый для целей данного изобретения, состоит по существу из графитизированного углерода. Графитизированный углерод исходного материала графита предпочтительно может быть выбран из синтетического графита, природного графита, вспученного графита, высокоупорядоченного пиролитического графита (ВУПГ), природного кристаллического графита или интеркалированного графита.
Вместе с материалом графита может присутствовать также неграфитированный углерод, предпочтительно выбранный из кокса или сажи, или графитизированного кокса.
Нет необходимости приводить конкретные методики приготовления исходной композиции, получаемой на стадии а) описанного и заявленного способа. В соответствии с предпочтительной процедурой жидкие и твердые компоненты исходной композиции смешивают друг с другом с помощью подходящего устройства для перемешивания, не обязательно интенсивного, но достаточного для придания смеси исходной однородности. Эта исходная однородность служит в основном для того, чтобы сделать исходную композицию текучей, чтобы избежать, например, проблем на стадии перекачивания (например, возможные насосы могут не работать, или работать плохо, если в них подают исключительно твердый материал).
Для этой цели, например, можно применять любой вид мешалок, или статических смесителей, или динамических смесителей, таких как, например, лопастные насосы или экструдеры. Обычно достаточно уже 30 секунд, но равным образом можно использовать более продолжительное время. Может быть полезно проводить данную операцию при температурах, отличающихся от комнатной температуры, чтобы получить вязкость, совместимую с применяемым смешивающим устройством: например, если применяют винтовой смеситель, температура должна быть такой, чтобы получить вязкость в пределах диапазона 0,2-5000 сПз.
Ультразвуковую обработку проводят в контейнере или камере для обработки ультразвуком, в которой в контакте с упомянутой композицией не должна присутствовать отдельная текучая фаза в газообразном состоянии.
Следовательно, вторая текучая фаза, находящаяся в газообразном состоянии, не должна присутствовать в камере для обработки ультразвуком, в контакте с упомянутой композицией. В частности за пределы объема данного изобретения выходит обработка ультразвуком, проводимая в открытых контейнерах, где имеется прямой контакт с атмосферным воздухом или другими газами, такими как, например, азот или аргон, которые таким образом формируют вторую фазу в газообразном состоянии, находящуюся в контакте с исходной композицией, подвергаемой действию ультразвука.
Исходная композиция, циркулирующая в контуре обработки ультразвуком в ходе обработки, предпочтительно имеет вязкость по меньшей мере 0,1 Па с, которая не превышает 1000 Па с, более предпочтительно составляет не менее 0,1 Па с и не выше 100 Па с. С этой целью может быть полезным применение вторичного растворителя, пригодного для разбавления исходной композиции в ходе обработки ультразвуком. Упомянутым вторичным растворителем может быть, например, этилбензол; или он может быть идентичным основному растворителю. Добавление упомянутого вторичного растворителя является необязательным. Упомянутый вторичный растворитель добавляют в ходе ультразвуковой обработки.
Обработку ультразвуком предпочтительно осуществляют при удельной мощности ультразвука, рассчитанной как электрическая мощность, генерируемая ультразвуковым генератором, по отношению к объему камеры, в которую ультразвук подают, по меньшей мере 60 Вт/см3. Еще более предпочтительно удельная мощность ультразвука составляет по меньшей мере 110 Вт/см3.
Для способа, описанного и заявленного в данной патентной заявке, времена ультразвуковой обработки составляют порядка минут, более предпочтительно секунд; они указаны также как эффективная продолжительность или эффективное время.
Эффективная продолжительность обработки ультразвуком составляет предпочтительно 20 минут или менее и 0,1 секунду или более; еще более предпочтительно она составляет в диапазоне от 0,5 секунд до 5 минут, еще более предпочтительно от 1 секунды до 1 минуты; и еще более предпочтительно она находится в диапазоне от 2 секунд до 30 секунд.
Эффективное время обозначает время, в течение которого композицию эффективно подвергают облучению ультразвуком.
В периодическом режиме, например, упомянутое время соответствует общему времени, в течение которого генерируют ультразвуковое облучение. В непрерывном проточном режиме упомянутое время соответствует (в случае непрерывного генерирования ультразвука) времени пребывания в камере, в которой генерируют ультразвук; обычно его рассчитывают как отношение между занимаемым объемом и объемным расходом. Если ультразвук генерируют циклически, то есть если генерирование ультразвука перемежается периодами, когда генерирование ультразвука по существу отсутствует, то эффективное время рассчитывают, умножая время пребывания на долю времени, когда эффективно генерируют ультразвук.
Данные, полученные по фактической продолжительности обработки ультразвуком по данному изобретению, являются чрезвычайно неожиданными, поскольку традиционно в процессах, описанных в уровне техники, время обработки ультразвуком, необходимое для получения хорошего диспергирования и эксфолиации графита, кроме того с высокой концентрацией, составляет порядка часов или даже дней.
Время обработки ультразвуком порядка минут является не только более эффективным с точки зрения энергии и экономики, но также облегчает масштабирование производства до промышленного объема. Кроме того, при снижении фактического времени обработки ультразвуком уменьшают также модификацию графеновых нанопластинок и графена и, в частности, ограничивают уменьшение размера сторон частиц графеновых нанопластинок и графена, что могло бы привести к уменьшению аспектного отношения (отношения размера стороны и толщины). Уменьшение аспектного отношения обычно является нежелательным, так как это повышает порог электрической и реологической перколяции (просачивания) в полученных полимерных композитах, а это уменьшает способность поглощения инфракрасного излучения и снижает механические характеристики.
В ходе ультразвуковой обработки исходную композицию следует довести до давления равного или ниже 0,2 МПа (2 бар) (абс.), предпочтительно в диапазоне от 0,8 до 5 МПа (от 8 до 50 бар) (абс.), еще более предпочтительно в диапазоне от 1,2 до 2,5 МПа (от 12 до 25 бар) (абс). Предпочтительно давление может быть выше, чем давление пара растворителя, применяемого в данном способе, если растворитель представляет собой одно химическое соединение; или химического соединения, присутствующего в большем количестве по массе, если растворитель образован большим количеством химических соединений.
Повышение давления исходной композиции может происходить перед обработкой ультразвуком или в ходе нее.
Для получения конечных композиций, описанных и заявленных в данной патентной заявке, важно проводить работу при давлении выше атмосферного, а особенно в пределах вышеописанного диапазона. В частности, было показано, что это существенно для получения дисперсии стабильной по меньшей мере 30 дней и для получения эксфолиации и полимеризации на одной стадии, как было описано ранее.
Специалист мог бы, фактически, предположить, что высокое давление может препятствовать кавитации и, следовательно, снижать эффективность обработки ультразвуком, так как увеличение давления вызывает увеличение порога кавитации. Это происходит, поскольку звуковое давление в ходе отрицательной фазы (в течение которой генерируют кавитационные пузырьки) должно преодолеть не только предел прочности на разрыв сред, в которых распространяется ультразвук, но и дополнительное гидростатическое давление.
Следовательно, в соответствии с концепцией существующего уровня техники, увеличение давления может ослабить обработку ультразвуком, поскольку оно ослабляет кавитацию; интенсивность сонотрода (мощность сонотрода) следует увеличить, чтобы способствовать обработке ультразвуком.
В соответствии с концепциями данного изобретения, напротив, высокие значения давления являются существенными (как указано выше) в ходе обработки ультразвуком. Следовательно, в соответствии с тем, что утверждается в уровене техники, для специалистов увеличение значений давления могло бы сделать саму обработку неэффективной.
Повышение давления перед обработкой ультразвуком или в ходе нее можно осуществить в соответствии с любым способом, известным в уровне техники. Его можно осуществить перед обработкой ультразвуком, например, с помощью накачивающего и/или подающего устройства, такого как шестеренчатый насос. Упомянутый насос может быть, например, тем же самым насосом, которым подают исходную композицию в схему, в которой происходит обработка ультразвуком. В соответствии с другим режимом повышение давления можно осуществить с применением расширяющейся текучей среды, которую прямо или косвенно приводят в контакт с текучей средой, обрабатываемой ультразвуком, как ниже по тексту описано более подробно.
В соответствии с другой методикой, после ультразвуковой обработки в противотоке может быть размещено второе перекачивающее устройство (например, с направлением перекачивания, противоположным направлению потока), чтобы поддерживать обрабатываемый ультразвуком поток под давлением.
После того, как давление исходной композиции повышено, может быть полезно поддерживать давление стабильным во времени. Этого можно достичь с помощью любой процедуры, известной в уровне техники; например, с использованием регулирующего клапана, расположенного ниже обработки ультразвуком по ходу технологического потока, или путем регулирования скорости подающего насоса, или с помощью расширительного резервуара (прямого или косвенного действия).
Технологию стабилизации с помощью расширительного резервуара, прямого или косвенного действия, можно использовать или для периодической схемы, или для непрерывной схемы, или в установках, которые могут предусматривать использование как в непрерывном, так и в периодическом режиме; такой вариант является особенно гибким. Расширительный резервуар косвенного действия включает устройство, содержащее подвижную или гибкую перегородку, разделяющую две камеры, одна из которых содержит текучую среду, которая должна быть стабилизирована, в данном контексте исходную композицию, а другая - стабилизирующую текучую среду, предпочтительно инертный газ. Последняя оказывает давление на перегородку, которая, поскольку она является подвижной или гибкой, в свою очередь оказывает давление на текучую среду, которая должна быть стабилизирована. Таким образом две текучие среды поддерживают при одном и том же давлении, не приводя их в какой-либо контакт друг с другом. Сжимаемость газа больше, чем сжимаемость жидкостей, из-за чего изменения объема жидкости, например, из-за термических расширений, введения добавочного количества или отбора проб, или из-за химических реакций, могут привести к сильному изменению давления. С другой стороны, газ способен поглощать эти изменения без существенного изменения давления. Следовательно, применение такого устройства, как вышеописанное, позволяет стабилизировать давление.
Альтернативно, и более предпочтительно, стабилизацию давления осуществляют с помощью расширительного резервуара прямого действия.
В этом режиме обрабатываемую композицию помещают в прямом контакте с газообразной фазой, или экспансионой текучей средой в жидком состоянии, которая, в свою очередь, находится в контакте с газообразной фазой. Газообразная фаза в любом случае имеет достаточно большой объем, чтобы можно было стабилизировать давление в результате, например, изменения объема обрабатываемой композиции после изменения ее температуры. Объем газообразной фазы равен, например, 1/10 или более от объема обрабатываемой композиции.
В случае расширительного резервуара прямого действия при приложении давления к одной из двух фаз обе приобретают одинаковое давление за счет прямого контакта. Этот режим является предпочтительным по сравнению с применением расширительного резервуара косвенного действия, поскольку он позволяет поглотить значительные изменения объема, и на него не влияет неизбежная механическая жесткость перегородки, которая некоторым образом снижает эффективность устройства. Перегородка также должна быть совместимой с исходной композицией в химическом и физическом отношении.
Однако желательно ограничить поглощение газа упомянутой композицией, так как эффективность обработки ультразвуком в значительной степени зависит от присутствия газа в упомянутой композиции. Если, фактически, с одной стороны пузырьки газа действуют как центры кавитации, способствуя ультразвуковому процессу, то с другой стороны они снижают интенсивность ударной волны из-за эффекта поглощения. Непосредственный контакт между жидкой фазой и газообразной фазой приводит к медленному растворению части газообразной фазы в жидкости и со временем - к изменению действия ультразвука.
Заявитель обнаружил, что эту проблему можно решить, уменьшая или устраняя поверхность контакта жидкость-газ.
Предпочтительным путем снижения поверхности контакта жидкость-газ является осуществление контакта жидкость-газ внутри вертикальной трубы, имеющей достаточно малый диаметр (например, диаметр, изменяющийся от 3 до 15 мм, для контура обработки ультразвуком, имеющего объем в диапазоне от 1 до 8 литров); или в эквивалентном устройстве, длина которого является по меньшей мере такой, чтобы соответствующий объем был больше, чем объем жидкости, который должен быть перемещен для компенсации давления; и имеющем отношение длины к диаметру, равное по меньшей мере 10, предпочтительно равное по меньшей мере 20, еще более предпочтительно больше 50. Таким образом контакт можно минимизировать, в то же время допуская большие изменения объема.
Поверхность контакта между обрабатываемой композицией и газом находится за пределами камеры обработки ультразвуком. Например, труба, в которой находится контакт между жидкостью и газом, может быть соединена с контуром обработки ультразвуком посредством бокового патрубка или другого ответвления, как более подробно описано далее. Таким образом, вся жидкость, содержащаяся в упомянутой трубе, также как и соединение с контуром обработки ультразвуком, находится вне области, в которой циркулирует обрабатываемая композиция.
Для того, чтобы дополнительно уменьшить количество газа, который растворяется при контакте между двумя фазами, также предпочтительно можно использовать жидкую текучую среду, также называемую экспансионной текучей средой, помещая ее между обрабатываемой ультразвуком композицией и стабилизирующим газом. Это может происходить или перед применением ультразвука к композиции, или одновременно с этим. Экспансионную текучую среду можно выбрать из самой исходной композиции, предпочтительно без полимерного компонента, или из другой жидкой текучей среды, которая предпочтительно не содержит твердых фаз.
В соответствии с этим режимом обрабатываемую композицию помещают в прямом контакте с экспансионной текучей средой, например в вертикальной трубе или эквивалентном устройстве, имеющем диаметр, который достаточно мал для того, чтобы минимизировать диффузию одной текучей среды по направлению к другой. При этом вертикальная труба или эквивалентное устройство имеет, например, длину, которая является по меньшей мере такой, чтобы соответствующий объем был больше, чем объем жидкости, который должен быть перемещен для компенсации давления; и имеет отношение длина-диаметр, равное по меньшей мере 10, предпочтительно равное по меньшей мере 20, еще более предпочтительно больше 50.
Экспансионная текучая среда, в свою очередь, может быть соединена со стабилизирующим устройством прямого или косвенного действия, как описано ранее. В этом случае стабилизирующее устройство прямого действия не обязательно должно иметь малую поверхность, так как нет необходимости ограничивать поверхность контакта. Экспансионная текучая среда находится фактически между стабилизирующим устройством прямого действия и контактом с технологической текучей средой.
Применяя эти режимы осуществления, как будет более очевидно из приведенных далее примеров, наблюдали более высокую стабильность обработки ультразвуком, совместно с более высокой эффективностью передачи ультразвуковых волн.
Альтернативно или совместно с вышеописанными процедурами, как в режиме периодического действия, так и в непрерывном режиме можно применять также другие устройства для регулирования давления, известные специалистам, такие как, например, предохранительные клапаны, разрывные диски, клапаны сброса давления; и перекачивающие устройства, такие как шестеренчатые насосы, скорость которых можно регулировать для поддержания желаемого противодавления. Ультразвук можно применять в соответствии с прямым режимом или косвенным режимом, как более подробно описано далее. Это справедливо, если способ, объект данного изобретения, осуществляют как в непрерывном, так и в периодическом режиме.
Ультразвук можно генерировать с помощью любого известного устройства, пригодного для данной цели, которое содержит активные материалы, способные расширяться и сжиматься с соответствующим усилием. Эти активные материалы предпочтительно выбирают из пьезоэлектрической керамики, которую подвергают действию соответствующего высоковольтного переменного электрического поля; или материалов, обладающих характеристиками гигантского магнитострикционного эффекта. Периодическое изменение длины этих активных материалов обычно является чрезвычайно малым (например, не более 50 мкм), и, таким образом, может потребовать соответствующего усиления, которое обычно осуществляют в обладающем соответствующей формой металлическом материале, имеющем резонансную частоту, близкую к резонансной частоте электрического/магнитного поля, приложенного к активному материалу.
Упомянутый металлический материал, далее по тексту называемый преобразователем, наносят, в соответствии с прямым режимом, одной стороной на активный материал, а с другой стороны он находится в прямом контакте с исходной обрабатываемой композицией («прямой преобразователь»). С другой стороны, в соответствии с косвенным режимом, преобразователь наносят одной стороной на активный материал, а с другой стороны он находится, посредством склеивания или сварки, в контакте с контейнером, содержащим исходную обрабатываемую композицию («косвенный преобразователь»). В последнем случае звуковые волны проходят сначала через стенки контейнера, в который введена обрабатываемая композиция, а затем в обрабатываемую композицию.
В соответствии с концепцией данного изобретения, можно применять любой режим генерирования ультразвука. Предпочтительным является прямой режим.
Не имеется конкретных ограничений по числу преобразователей, применяемых в контейнере. Количество преобразователей рассчитывают на основе мощности ультразвука, который должен быть приложен к композиции: на малые объемы, например, менее 1 литра, может быть достаточно одного преобразователя, в то время как на более крупные объемы, например, более 1 литра, может быть удобным использовать два или большее количество преобразователей.
Резонансная частота преобразователей не обязательно должна быть одинаковой для всех применяемых преобразователей. Между различными преобразователями может быть образован любой угол. В соответствии с режимом по данному изобретению, если применяют большее число преобразователей, может быть предпочтительным располагать преобразователи таким образом, чтобы они были противоположно направленными, то есть чтобы композицию пропускали в пространстве между концами одних и тех же преобразователей.
Устройством, в котором проводят обработку ультразвуком, может быть любое устройство, в которое можно подавать исходную композицию и создавать в нем ультразвуковое поле, в ранее указанных условиях по температуре, давлению и интенсивности звука.
Как указано ранее, способ - объект данного изобретения - можно осуществлять в непрерывном режиме или в периодическом режиме.
В периодическом режиме исходную композицию, которая должна быть обработана, загружают в устройство, пригодное для проведения ультразвуковой обработки, например, отбирая упомянутую композицию из накопительной емкости с помощью насоса или под действием силы тяжести. Затем повышают давление исходной композиции, которая должна быть обработана, и доводят ее до желаемой температуры; после этого проводят обработку ультразвуком в течение необходимого времени, и, наконец, обработанную композицию выгружают из устройства.
С другой стороны, в непрерывном режиме исходную композицию постоянно загружают в камеру обработки ультразвуком, например, посредством насоса, и затем проводят обработку ультразвуком. Обычно перед тем, как исходная композиция покинет устройство, она многократно циркулирует в камере обработки ультразвуком, посредством циркуляционного насоса. Таким образом, ее многократно подвергают действию ультразвука, и оборудование может работать непрерывно (24 ч/24 ч). Это позволяет также поддерживать постоянными во времени условия по температуре, давлению и расходу, таким образом максимизируя производительность и качественное постоянство полученного продукта.
Контур обработки ультразвуком состоит из комбинации объемов, через которые циркулирует текучая среда, подвергаемая воздействию ультразвука. Эти объемы состоят из объемов, находящихся в контакте с технологической текучей средой циркуляционного насоса, камеры обработки ультразвуком, любых возможных теплообменников, труб, адаптеров, измерительного оборудования, стабилизирующих давление устройств, клапанов и т.д.
Если способ - объект данного изобретения - проводят в непрерывном режиме, устройство, в котором происходит ультразвуковая обработка (камера обработки ультразвуком), имеет объем, занятый исходной композицией, который составляет самое большое 50% общего объема контура обработки ультразвуком, предпочтительно самое большое 15%, еще более предпочтительно самое большое 2%.
Как в случае непрерывной процедуры, так и в случае периодической процедуры ультразвук можно подавать в соответствии с прямым режимом или косвенным режимом, описанными ранее. Предпочтительно применяют прямой режим.
Поверхность, на которой происходит генерирование ультразвука, предпочтительно равна по меньшей мере 10% от поверхности камеры обработки ультразвуком, еще более предпочтительно по меньшей мере 30% от поверхности упомянутой камеры.
В предпочтительном случае ультразвук пропускают через исходную композицию импульсами, перемежая периоды, когда упомянутую композицию подвергают действию ультразвука (также называемые активными периодами), периодами, когда упомянутую композицию не подвергают воздействию какого-либо ультразвукового поля, или подвергают воздействию ультразвукового поля, имеющего мощность по меньшей мере в 10 раз меньшую, чем мощность ультразвука, пропускаемого в течение активного периода. Этот период называют пассивным периодом.
Охарактеризованный выше импульсный режим должен происходить с некоторым отношением между активным периодом и суммой активного и пассивного периодов, которое определяют в данной патентной заявке как «рабочий цикл»; и которое находится в диапазоне от 0,0001 до 1, более предпочтительно от 0,0005 до 0,3, еще более предпочтительно от 0,001 до 0,05.
Фактически, при рассмотрении, например, режима непрерывного потока, и при отслеживании некоторого малого объема текучей среды в контуре обработки ультразвуком, этот объем будут подвергать действию ультразвука в течение периода времени, в который он находится в камере обработки ультразвуком (активный период). При выходе из камеры обработки ультразвуком на упомянутый объем текучей среды больше не действует ультразвуковое поле. В лучшем случае, вблизи впускного или выпускного отверстия камеры обработки ультразвуком упомянутый объем текучей среды может подвергаться воздействию ультразвукового поля, имеющего значительно более низкую мощность (по меньшей мере в 10 раз меньше). Таким образом, время, которое упомянутый объем текучей среды проводит вне камеры обработки ультразвуком, является пассивным периодом, в то время как время, проведенное в камере обработки ультразвуком, является активным периодом.
Генерирование и испускание ультразвука можно поддерживать на постоянном уровне мощности в течение всей продолжительности теста, или его можно модулировать во времени, периодически изменяя мощность испускаемого ультразвука. Обычно эту модуляцию можно проводить, периодически прерывая генерирование ультразвука на заданное время (нулевая мощность), или периодически модулируя электрический ток, подаваемый на преобразователь, например, в соответствии с синусоидальной или пилообразной формой.
Если мощность ультразвука модулируют во времени, активный период может быть дополнительно сокращен, так как данный объем текучей среды может входить в камеру обработки ультразвуком во время периода периодического прерывания ультразвука.
В любом случае, с модуляцией мощности ультразвука или без нее, данный объем текучей среды в контуре обработки ультразвуком подвергают действию ультразвука только в течение некоторой доли времени, так как за активными циклами следуют пассивные циклы и так далее.
В случае технологии с периодическим потоком импульсный режим можно получить, модулируя генерирование звуковых волн во временных циклах, как указано выше.
В режиме непрерывного потока, если ультразвук генерируют без модуляций по времени (например, при постоянной во времени мощности), рабочий цикл соответствует отношению между объемом камеры обработки ультразвуком и объемом контура обработки ультразвуком. Фактически, раствор непрерывно циркулирует в контуре обработки ультразвуком, но его подвергают воздействию ультразвука в течение той доли времени, которую он проводит в камере обработки ультразвуком.
И опять, в режиме непрерывного потока, если генерирование ультразвука модулируют во времени, рабочий цикл задан соотношением между объемом камеры обработки ультразвуком и объемом контура обработки ультразвуком, умноженным на долю времени, в течение которого генерирование ультразвука является активным; или, в более общем случае, умноженным на долю средней мощности, подаваемой на генератор ультразвука.
В «импульсном режиме» количество периодов, за которые исходную композицию подвергают действию ультразвука, предпочтительно равно 10 или более и менее или равно 100000, еще более предпочтительно равно 100 или более и менее или равно 100000.
Предпочтительно исходную композицию подвергают обработке ультразвуком при температуре не менее 20°С, более предпочтительно равной или выше 40°С, еще более предпочтительно равной или выше 60°С, и еще более предпочтительно равной или выше 90°С.
Обработку можно также проводить при температуре равной или менее 110°С. Предпочтительно исходную композицию подвергают обработке ультразвуком при температуре равной или менее 200°С, более предпочтительно равной или менее 170°С, еще более предпочтительно равной или менее 150°С, и еще более предпочтительно равной или менее 130°С.
Исходную композицию предпочтительно подвергают обработке ультразвуком при температуре в диапазоне от 20°С до 200°С, предпочтительно от 40°С до 200°С, еще более предпочтительно от 40°С до 170°С, и еще более предпочтительно от 60°С до 130°С.
В уровне техники известно, что температура, обычно применяемая при обработке ультразвуком, представляет собой комнатную температуру или, в некоторых случаях, температуры ниже комнатной и близкие к 0°С.
Результаты данного изобретения, напротив, предпочтительно и неожиданно получают при температурах выше комнатной температуры, как указано выше.
Фактически, избыточно высокая температура обычно делает процесс обработки ультразвуком менее эффективным. В то же время применяемые температурные условия лежат за пределами диапазона, который применяют в промышленности при полимеризации выбранных мономеров.
С другой стороны, описанный и заявленный способ демонстрирует, что неожиданно можно на одной стадии получить эффективную обработку ультразвуком, с эксфолиацией материала графита до GRS, и одновременно полимеризацию способного к полимеризации мономера.
Температуру можно поддерживать в пределах необходимого диапазона с помощью одного или большего количества теплообменников, или посредством резистивных или термических рубашек, обмоток, электрических сопротивлений, элементов Пельтье, или с использованием того же самого ультразвука в качестве источника тепла. Регулирование температуры можно осуществлять за пределами камеры, в которой композицию подвергают действию ультразвука (камеры обработки ультразвуком), непрерывно или периодически рециркулируя обрабатываемую композицию в теплообменник.
Температура не обязательно является постоянной во времени и пространстве, так как она может изменяться в пределах указанного диапазона.
Например, ультразвук можно подавать на композицию при комнатной температуре, постепенно доводя ее до заданной температуры. Или, опять-таки, температура может изменяться во времени и/или пространстве в соответствии с заданной программой.
При периодическом режиме ультразвук можно пропускать и не в импульсном режиме. В этом режиме композиция постоянно находится в камере обработки ультразвуком, где ультразвук предпочтительно излучают непрерывно (например, без перерывов во времени) на всем протяжении обработки.
Объем, в котором проводят обработку ультразвуком, называемый камерой обработки ультразвуком, представляет собой любое устройство, в которое подают композицию и на которое воздействуют ультразвуковым полем, при ранее указанных условиях по температуре, давлении и интенсивности звука.
В режиме непрерывного потока камера, в которой происходит обработка ультразвуком, предпочтительно имеет занятый смесью объем, составляющий максимально 50% общего объема контура обработки ультразвуком, предпочтительно максимально 15%, еще более предпочтительно максимально 2%. Это делает саму обработку более эффективной. Таким образом, действие ультразвука фактически можно сконцентрировать в уменьшенном объеме текучей среды, что приводит к более короткому, но более интенсивному, а следовательно более эффективному, действию. Повышенная эффективность позволяет также снизить потребление электроэнергии на единицу полученного композита.
Как при непрерывном потоке, так и в периодическом режиме, поверхность, на которой происходит генерирование ультразвука, может составлять по меньшей мере 10% от поверхности камеры, в которой осуществляют обработку ультразвуком, еще более предпочтительно по меньшей мере 30% от поверхности упомянутой камеры.
В предпочтительном воплощении исходную композицию, полученную на стадии (а) описанного и заявленного способа, дегазируют перед обработкой ультразвуком или в ходе нее, чтобы выпустить те количества газа, которые обычно растворены в жидкой фазе. Дегазацию можно проводить путем выпуска газа из исходной композиции, предпочтительно из испытуемых образцов, помещенных в самой высокой точке контура или контейнера, в которых композиция находится (например, для выпуска в атмосферу); или путем помещения исходной композиции в достаточно большой контейнер, чтобы можно было выпустить газ через отверстие, расположенное в верхней части этого контейнера.
Такая обработка позволяет удалить присутствующие газы, в частности газы, присутствующие в объеме устройства перед загрузкой исходной композиции, а также газ, присутствующий в упомянутой композиции и/или полученный в ходе обработки ультразвуком (например, низкокипящие продукты разложения растворителя).
То же самое действие ультразвука облегчает отделение жидкости от растворенного газа; таким образом, в ходе обработки можно успешно провести дегазацию, как в периодическом режиме, так и в режиме непрерывного потока.
Способ, описанный и заявленный в данном тексте, неожиданно позволяет диспергировать и эксфолиировать материал графита до графена и графеновых нанопластинок, по существу не изменяя электронных свойств исходного материала графита и его химической чистоты.
В дополнение к диспергированию и эксфолиации, способ - объект данного изобретения - в то же время позволяет провести частичную или полную полимеризацию способного к полимеризации мономера, проводя таким образом прямой синтез полимерного композита.
Согласно существующему уровню техники, для получения полимерного композита полученные графен и графеновые нанопластинки следует предварительно извлечь, а затем диспергировать в полимере. Это не только приводит к необходимости проведения сложных дополнительных отдельных операций и небезопасных для окружающей среды процессов удаления растворителя, но также снижает эффективность обработки, так как извлечение графена и графеновых нанопластинок неизбежно влечет за собой частичную повторную агрегацию эксфолиированных ранее слоев.
Как указано ранее, растворитель, применяемый в способе - объекте данной патентной заявки - должен содержать по меньшей мере 10 мас.%, винилароматического мономера, одного или в смеси с до 50 мас.% других способных к сополимеризации мономеров.
Термин «винилароматический мономер», как его используют в данном описании и формуле изобретения, относится по существу к продукту, который соответствует следующей общей формуле:
где R представляет собой водород или метальную группу, n равно нулю или целому числу в диапазоне от 1 до 5, a Y представляет собой галоген, выбранный из хлора или брома, или алкильный или алкоксильный радикал, имеющий от 1 до 4 атомов углерода.
Предпочтительные винилароматические мономеры можно выбрать из: стирола, α-метилстирола, метилстирола, этилстирола, бутилстирола, диметилстирола, моно-, ди-, три-, тетра- и пентахлорстирола, бромстирола, метоксистирола, ацетоксистирола и т.д. Предпочтительными винилароматическими мономерами являются стирол и α-метилстирол.
Как уже указывали, винилароматические мономеры, имеющие общую формулу (I), можно применять сами по себе или в смеси с до 50 мас.% дополнительных способных к сополимеризации мономеров. Примерами таких дополнительных способных к сополимеризации мономеров являются (мет)акриловая кислота, С1-С4 алкильные эфиры (мет)акриловой кислоты, такие как метилакрилат, метилметакрилат, этилакрилат, этилметилакрилат, изопропилакрилат, бутилакрилат, амиды и нитрилы (мет)акриловой кислоты, такие как акриламид, метакриламид, акрилонитрил, метакрилонитрил, бутадиен, этилен, дивинилбензол, малеиновый ангидрид и т.д. Предпочтительными способными к сополимеризации мономерами являются акрилонитрил, малеиновый ангидрид, глицидилметакрилат и метилметакрилат.
В конце осуществления способа, описанного и заявленного в данном тексте, полученную композицию можно, после того, как она была предварительно обработана, подвергнуть дополнительной полимеризации, чтобы завершить полимеризацию и таким образом сформировать полимерные композиты, полимерные нанокомпозиты или маточные смеси.
Фактически перед проведением дополнительной полимеризации конечную композицию, содержащую графен и графеновые нанопластинки, диспергированные в растворителе, можно обработать в соответствии с различными методиками.
По первой методике упомянутую конечную композицию можно оставить для декантации, чтобы дать возможность не подвергнутому эксфолиации материалу графита выпасть в осадок, образуя отстой, и таким образом отделить первую жидкую фазу, содержащую графен и графеновые нанопластинки. После стадии декантации отделенную первую жидкую фазу (первую надосадочную жидкость) можно подвергнуть центрифугированию со скоростью, достаточной для дополнительного отделения второй жидкой фазы, содержащей графен и графеновые нанопластинки, от не подвергшегося эксфолиации материала графита.
По второй, альтернативной, методике конечную композицию, содержащую графен и графеновые нанопластинки, диспергированные в растворителе, после обработки ультразвуком непосредственно подвергают центрифугированию, без предварительной декантации, отделяя конечную жидкую фазу, содержащую графен и графеновые нанопластинки, от не подвергшегося эксфолиации материала графита.
В ходе декантации упомянутую конечную композицию оставляют для отстаивания на период времени, равный по меньшей мере 2 минутам, предпочтительно не менее чем 20 минутам, еще более предпочтительно по меньшей мере одному часу, таким образом формируя осадок.
Скорость центрифугирования может изменяться от 500 об/мин до 30000 об/мин; его проводят в течение времени в диапазоне от 2 минут до 60 минут. Центрифугирование можно проводить в периодическом, полупериодическом и непрерывном режиме. В полупериодическом режиме конечную композицию, поступающую с обработки ультразвуком, или первую жидкую фазу, отделенную после декантации, загружают партиями в центрифугу, из которой непрерывно извлекают вторую жидкую фазу (надосадочную жидкость).
С другой стороны, в случае центрифуг непрерывного действия, конечную композицию, поступающую с обработки ультразвуком, или жидкую фазу, отделенную после декантации, которую следует подвергнуть центрифугированию, подают в центрифугу непрерывно, и непрерывно извлекают по меньшей мере вторую жидкую фазу (вторую надосадочную жидкость).
После декантации и/или центрифугирования, первую, вторую жидкую фазу или конечную фазу, содержащую графен и графеновые нанопластинки, стабильно диспергированные в растворителе, отделяют в соответствии с описанными ранее методиками; и, возможно с внесенными дополнительными добавками, доводят до желаемой температуры, чтобы завершить полимеризацию, образуя полимерные композиты, полимерные нанокомпозиты или полимерные маточные смеси, содержащие композиции, описанные и заявленные в данной патентной заявке. Показано, что упомянутые соединения обладают высокими механическими характеристиками, высокой термостойкостью, антистатическими свойствами, свойствами электромагнитной изоляции и высокой теплопроводностью.
Дополнительные добавки выбирают из огнезащитных агентов, не поддерживающих горение агентов, синергетических агентов, зародышеобразователей, смазок, пигментов, нейтрализующих кислоту веществ и поглотителей свободных радикалов.
Вспениваемые полимерные композиты или вспениваемые полимерные нанокомпозиты можно получить из полимерных композитов, полимерных нанокомпозитов или полимерных маточных смесей, содержащих графен и графеновые нанопластинки, путем добавления вспенивающего агента, который предпочтительно выбирают из пентана или смеси изомеров пентана; и упомянутый вспенивающий агент предпочтительно составляет от 3% до 8 мас.% по отношению к массе вспениваемого полимерного нанокомпозита.
Таким образом, дополнительный объект данной патентной заявки относится к вспениваемым полимерным композитам или вспениваемым полимерным нанокомпозитам, содержащим ранее описанные полимерные композиты, или полимерные нанокомпозиты, или маточные смеси, а также по меньшей мере один вспенивающий агент.
Исходя из упомянутых вспениваемых полимерных композитов или вспениваемых полимерных нанокомпозитов, можно получить изделия и пористые полимерные пены, обладающие высокими теплоизолирующими способностями, термостойкостью и высокими механическими характеристиками.
Процесс полимеризации происходит в соответствии с методиками, известными на существующем уровне техники, такими как, например, суспензионная полимеризация, эмульсионная полимеризация, полимеризация в растворе или полимеризация в непрерывной массе.
Как описано в US 4231919 и US 3898300, включенных в данную патентную заявку в виде ссылки, в случае суспензионной полимеризации и эмульсионной полимеризации первую или вторую жидкую фазу, отделенную в соответствии с методикой, описанной ранее в данном тексте, можно добавить к системе, которая включает суспендирующий агент и эмульгирующий агент, например, воду и подходящие суспендирующие и эмульгирующие агенты, образуя продукт уже в форме твердых частиц или гранул, в котором по существу весь мономер прореагировал. Таким образом получают полимерный композит, или полимерный нанокомпозит, возможно, концентрированный (маточная смесь), в гранулах.
В случае полимеризации в непрерывной массе, как описано, например, в US 6348549 и US 2593399, включенных в данную патентную заявку в виде ссылки, первую или вторую жидкую фазу, отделенную в соответствии с методиками, описанными ранее в данном тексте, можно полимеризовать в расплавленном состоянии. Затем полученный полимер, после возможного удаления растворителей и летучих продуктов, экструдируют и нарезают на гранулы (нарезка нитей).
Альтернативно, расплавленный полимер пропускают через пластину фильеры, в которой имеется большое количество отверстий, и гранулируют в головке, например, с помощью грануляторов с водяной завесой, таких как грануляторы, описанные в WO 2007/048536, включенном в данную патентную заявку в виде ссылки, таким образом получая полимерный композит, или полимерный нанокомпозит, возможно концентрированный (маточная смесь), в гранулах.
Полученный продукт можно использовать непосредственно в производстве полуфабрикатов или полупродуктов, для улучшения их механических, электрических, теплоизолирующих свойств. Полимерные нанокомпозиты, например, можно экструдировать, формируя изделие путем литья под давлением или прямого формования. Полученные полимерные нанокомпозиты можно также применять в качестве концентратов (маточных смесей) для последующего производства полуфабрикатов и полупродуктов, упомянутых ранее.
Полученные таким образом полимерные композиты, полимерные нанокомпозиты или полимерные маточные смеси предпочтительно можно использовать для получения пенополистирола (ППС), обладающего высокими теплоизолирующими свойствами, в соответствии с одним из способов, известных на существующем уровне техники, например, суспензионной полимеризацией, как это описано, например, в US 2673194, US 4500692, ЕР 2475709, включенных в данную патентную заявку в виде ссылок, или полимеризацией в непрерывной массе.
В ходе полимеризации в непрерывной массе полимерные нанокомпозиты можно добавлять к другим соединениям, например, полистиролу, огнезащитным добавкам и соответствующим синергетическим агентам, порообразователям, возможным агентам-зародышеобразователям и другим агентам, обеспечивающим непрозрачность для ИК-излучения. Данные компоненты смешивают, например, с помощью статических смесителей, возможно доводят до расчетной температуры (например, 170°С), а затем подают в пластину фильеры, в которой сделаны серии отверстий, через которые экструдируют композицию. Композицию, выходящую из отверстий в пластине фильеры, гранулируют, например, с помощью ряда вращающихся ножей, и подвергают соответствующему охлаждению, например, с помощью воды или другой охлаждающей текучей среды, чтобы иметь возможность нарезать вспененную полимерную композицию на гранулы. Например, упомянутые грануляторы могут принадлежать к типу с охлаждением в воде, или к типу со струями воды, выходящими из многочисленных распылительных сопел, как описано в US 7320585, включенном в данную патентную заявку в виде ссылки. Для более подробного описания можно сделать ссылку на патентные заявки WO 2003/053651, WO 2008/141766 и WO 2008/141767, включенные в данную патентную заявку в виде ссылок.
Способ - объект данного изобретения - можно осуществлять в непрерывном режиме или в периодическом режиме. Фиг. 1-9 иллюстрируют оба эти режима, как предпочтительные воплощения данного изобретения.
Фиг. 1 описывает способ по данному изобретению в режиме периодического потока. Исходную композицию приготовляют в лабораторном стакане или другом контейнере, пригодном для данной цели (не показан на Фиг. 1), в котором материал графита, основной растворитель, содержащий по меньшей мере 10 мас.% винилароматического мономера, и возможно другие дополнительные добавки приводят в контакт и перемешивают с помощью механической или магнитной мешалки.
После того, как исходная композиция приготовлена, ее вводят, посредством потока f1, в соответствующее устройство (1), способное выдержать давление, предусмотренное для обработки ультразвуком. Ультразвук подают непосредственно в исходную композицию, в течение заданного времени, посредством сонотрода (2), соединенного с генератором (3) ультразвука, после чего обработанную композицию удаляют из нижней части устройства (f2).
При периодическом режиме данного способа в устройство (1) загружают исходную композицию, которая должна быть обработана, до тех пор, пока весь объем устройства не будет заполнен; следовательно, газ не будет присутствовать. Повышение давления в устройстве проводят в соответствии с одной из методик, описанных ранее, таких как, например: путем подачи композиции с помощью насоса, который таким образом повышает давление во всем контейнере (1); или путем применения повышающего давление устройства с расширительным резервуаром прямого или косвенного действия, описанного ранее в данном тексте, соединенного с устройством (1), например, посредством отверстия (f3) в крышке контейнера. В частности, камера обработки ультразвуком и расширительный резервуар экспансионной текучей среды, как описано ранее. В этом случае расширительный резервуар предпочтительно может занимать верхнюю часть контейнера (1), в то время как камера обработки ультразвуком занимает нижнюю часть, чтобы облегчить дегазацию композиции.
Фиг. 1 изображает дополнительный поток, называемый «f1 бис», благодаря которому можно осуществить увеличение давления в резервуаре с прямым расширением. Для этой цели систему снабжают сифоном (1 бис). Упомянутый сифон заполняют самой композицией, пропуская ее из контейнера (1) по потоку «f1бис», чтобы удалить находящийся в нем газ. Сифон «1 бис» соединен потоком «f1 бис» с баллоном с аргоном, для повышения давления в контейнере (1), в то же время, предотвращая поступление газа (аргона) в контейнер. Это же соединение не только позволяет осуществить повышение давления, но также дает возможность поддерживать давление постоянным в ходе всего процесса обработки ультразвуком (путем компенсации, например, термических расширений). Таким образом, описанная здесь система с сифоном представляет работу в режиме расширительного резервуара прямого действия, где экспансионной текучей средой является сама композиция.
Регулирование температуры можно осуществить с помощью любого способа, известного на существующем уровне техники. Устройство Фиг. 1, например, можно снабдить рубашкой для регулирования температуры. В рубашке циркулирует теплоноситель (h1, h2), регулируемый внешним блоком контроля температуры.
Исходную композицию, загруженную в устройство (1), подвергают действию непрерывного, или импульсного, ультразвукового поля. Как уже было указано ранее, при описании периодического режима, импульсный режим получают путем модуляции генерирования звуковых волн во временных циклах: преобразователи сонотрода возбуждают электрическим/магнитным полем в течение ограниченного времени, определяемого здесь как «активный» период; после этого следует время, при котором возбуждение активного материала отсутствует, или само возбуждение в сильной степени снижено, которое определяют как «пассивный» период.
Устройство (1) может быть снабжено системой перемешивания (не показанной на Фиг. 1). Упомянутую систему перемешивания предпочтительно получают путем циркуляции исходной композиции через насос, или с помощью мешалки, помещенной внутри самого контейнера.
Устройство (1) предпочтительно может быть снабжено аварийной системой откачивания (не показанной на Фиг. 1), системой введения инертной жидкости для предотвращения реакций разгона, системой отбора проб или, в любом случае, другими системами управления, известными на существующем уровне техники.
Газ, присутствующий в (1) перед введением раствора, можно удалить из отверстия (f3), сделанного в верхней части контейнера. Регулирование давления можно осуществить с помощью подающего насоса и/или путем отбора некоторого количества раствора из верхней (f3) или нижней (f2) части устройства. После завершения обработки ультразвуком обработанную композицию можно сразу же удалить, замещая ее объем равным объемом газа или жидкости, то есть ее можно подвергнуть обработке декантацией в том же устройстве (1). После декантации образуются две фазы: верхняя и нижняя: продукт удаляют из верхней фазы с помощью выпускного отверстия, находящегося на боковой части устройства (не показано на Фиг. 1), или с использованием погружной трубки. Оставшуюся нижнюю фазу, выделившуюся в нижней части устройства, затем размещают в виде отходов или повторно используют в последующей партии.
В дополнение к декантации, или в качестве альтернативы, можно провести обработку концентрированием, которая заключается в испарении части растворителя, последующем устранении полученных таким образом паров и сборе концентрированного продукта. Эту операцию можно провести как в устройстве (1), так и за его пределами, в устройствах, пригодных для этой цели.
Фиг. 2 и 3 описывают способ по данному изобретению, осуществляемый в режиме непрерывного потока. Исходную композицию загружают в устройство (1) посредством потока f1, и перекачивают посредством насоса (4) в устройство (2), в котором происходит ультразвуковая обработка (соединенное с генератором 3 ультразвука), посредством потоков f2 и f3. Затем, ниже ультразвуковой обработки (f4) по ходу технологического потока, композицию возвращают в устройство (1), посредством потока f5, и рециркулируют ее в пределах контура. Таким образом, эта схема образует замкнутый контур сонификации. Насос (4) можно расположить выше (Фиг. 3) или ниже (Фиг. 2) устройства (2), в котором проводят обработку ультразвуком. В конце обработки ультразвуком обработанную композицию собирают в подходящий контейнер (5), посредством потока f6.
Контейнер (1) может быть снабжен системой перемешивания, как показано на Фиг. 2-9. Можно использовать любую систему перемешивания, известную на существующем уровне техники, например, внутренние вращающиеся мешалки, такие как пропеллеры, крыльчатки или анкерные или спирально-лопастные мешалки, приводимые в действие соединенным с ними двигателем; или перемешивающий элемент, приводимый в движение соответствующим внешним магнитным полем, например, с помощью анкера.
Если насос размещен выше устройства для обработки ультразвуком по ходу технологического потока (как на Фиг. 3), ниже последнего можно разместить контрольный клапан или клапан для сброса давления, чтобы гарантировать, что исходная композиция в ходе обработки ультразвуком останется при заданном давлении, которое выше, чем давление в том же устройстве (1).
Это, например, позволяет снизить стоимость, а также упростить конструкцию устройства (1), так как это устройство можно сконструировать при более низком давлении, или даже при атмосферном давлении.
Фиг. 4 описывает способ, снова осуществляемый в непрерывном режиме, подобно способу, приведенному на Фиг. 3, который включает один или большее количество теплообменников (6) ниже устройства (2) обработки ультразвуком по ходу технологического потока, для регулирования температуры обрабатываемой композиции с помощью теплоносителя (h3, h4). Например, первый теплообменник мог бы способствовать нагреванию смеси, доводя ее до желаемой рабочей температуры перед включением ультразвука, в то время как второй теплообменник, содержащий воду или охлаждающую смесь, может способствовать снижению температуры в случае начинающейся неконтролируемой полимеризации (разгона).
Конкретные ограничения по типу используемого теплообменника отсутствуют; однако предпочтительными являются теплообменники, в которых операция очистки является простой, такие как, например, теплообменники типа труба в трубе, или кожухотрубные теплообменники, в которых технологическая текучая среда проходит по трубам. Также и в этом случае, ниже обработки ультразвуком и прохождения через теплообменник, композиция (f5) возвращается в устройство (1) посредством потока f6. В конце ультразвуковой обработки обработанную композицию собирают в подходящий контейнер (5) посредством потока f7.
Дополнительный, приведенный в качестве примера воплощения режим, называемый «с аварийным сбросом», представлен на Фиг. 5. Этот режим предусматривает наличие накопительной емкости (7), содержащей загружаемую посредством потока f8 применяемую при аварийных ситуациях текучую среду, которая предпочтительно представляет собой основной растворитель. В случае возникновения реакций разгона введение этой текучей среды в схему посредством потока f9 позволяет снизить вязкость системы и уменьшить температуру, предотвращая любые возможные закупорки в схеме и насосе. Эти закупорки могут возникнуть в результате того, что при неконтролируемом возрастании температуры образуется полимер. Для использования системы с аварийным сбросом предпочтительно, чтобы раствор можно было быстро выпустить в сборную емкость, называемую сливным резервуаром (9), который можно видеть на Фиг. 6, таким образом, чтобы выпускаемый (из емкости (7)) раствор мог заместить в схеме и соответствующем оборудовании раствор, в котором начались реакции разгона. Для этой цели можно обеспечить управляемые или вручную, или автоматически клапаны, которые позволяют открывать поток раствора в направлении сливного резервуара (9) (см. соединения а, b, с, d и соединение контура обработки ультразвуком со сливным резервуаром (9)), и открывать клапан на аварийной накопительной емкости (7), предварительно, одновременно или последовательно. Аварийную накопительную емкость (7) предпочтительно поддерживают под давлением, которое выше, чем давление в схеме, чтобы способствовать быстрому замещению растворителя также и в отсутствии электрической энергии. В качестве альтернативы, или одновременно, можно обеспечить средства перекачивания, такие как, например, шестеренчатый насос, чтобы облегчить перенос аварийной текучей среды в раствор.
Сливной резервуар (9), в дополнение к тому, что он позволяет удалить содержимое из схемы в замкнутом контуре, также выполняет ту функцию, что он заключает в себе все отверстия для очистки системы, обычно расположенные в самых высоких и самых низких точках схемы, а также выше и ниже циркуляционного насоса по ходу технологического потока, с помощью которых устраняют любые пузырьки газа, возможно присутствующие в исходной композиции; или которые могут быть захвачены в ходе операций загрузки. Предпочтительно в ходе ультразвуковой обработки эти отверстия для очистки можно временно открывать для удаления дополнительной газовой фазы, возможно образованной в ходе процесса. Фактически в ходе ультразвуковой обработки может образоваться газовая фаза, происходящая из газа, исходно растворенного в композиции или генерированного в ходе этой ультразвуковой обработки.
Приведенный в качестве примера воплощения режим, представленный на Фиг. 6, обеспечивает повышение давления в схеме с использованием экспансионной текучей среды, приведенной в контакт с технологической текучей средой (f11) посредством специальной напорной емкости или эквивалентной системы (8), в которой экспансионная текучая среда контактирует с газообразной фазой. Как уже описано, эта напорная емкость или эквивалентная система может состоять из простой трубы, расположенной вертикально. Упомянутая экспансионная текучая среда находится между композицией и стабилизирующим газом (f10), подводимым посредством потока f10 бис, чтобы предотвратить растворение газа в жидкой фазе технологической текучей среды (так как экспансионная текучая среда является неподвижной, диффузия газа, растворенного в экспансионной текучей среде, в направлении технологической текучей среды является чрезвычайно медленной и, фактически, пренебрежимо малой).
Другой пример воплощения, представленный на Фиг. 7, предусматривает наличие второго устройства (10), расположенного ниже по ходу технологического потока основной схемы (контура обработки ультразвуком), которая включает устройство (2), где происходит ультразвуковая обработка; бак (1) для загрузки; насос (4) и теплообменник (6). Упомянутое устройство (10) работает как отстойник.
Обработанную композицию можно подавать в отстойник в конце ультразвуковой обработки, или непрерывно. В последнем случае поток f7 переносит часть текучей среды, выходящей из камеры (2) обработки ультразвуком, в отстойник (10), в то время как оставшаяся часть проходит через теплообменник (6), и ее снова рециркулируют в контур обработки ультразвуком. Обработанная композиция (f7) может оставаться в отстойнике (10), чтобы не подвергшиеся эксфолиации частицы материала графита осаждались на дне, в то время как в наибольшей степени эксфолиированные частицы, которые остаются в суспензии, можно было легко отобрать в верхней части отстойника, предпочтительно с помощью соответствующих перегородок, находящихся внутри устройства (10).
Осажденную часть извлекают из нижней части контейнера (10), и предпочтительно ее можно рециркулировать в бак (1) для загрузки, посредством потока f12, чтобы снова подвергнуть обработке ультразвуком. Обработанную композицию направляют в предназначенный для этой цели контейнер (5), посредством потока f13.
Дополнительный пример воплощения, представленный на Фиг. 8, обеспечивает наличие накопительной емкости (11), имеющей значительно больший объем, чем объем основной схемы (контура обработки ультразвукомв): например, в два раза больше, более предпочтительно в десять раз больше; которая выполняет функцию хранения композиции, которая должна быть обработана, загружаемой в упомянутый бак посредством потока f14. Композицию, которая должна быть обработана, постепенно подают в бак (1) для загрузки посредством потока f1, с помощью подающего устройства, такого как, например, шестеренчатый насос, а затем обрабатывают ультразвуком. Затем часть текучей среды, возможно равную части, перемещенной из емкости (11) в основную схему, чтобы сохранить объем жидкой фазы в ней постоянным, отбирают и направляют в отстойник (10) посредством потока f7.
Из отстойника (10) отбирают обработанную композицию, находящуюся в верхней части устройства отстойника (f13), и собирают в контейнере (5), предназначенном для этой цели. Ее место занимает раствор, содержащий не подвергшуюся эксфолиации часть материала графита, из нижней части устройства (10) отстойника; или, предпочтительно, его рециркулируют обратно в устройство (11), чтобы снова подвергнуть действию ультразвуковой обработки (f12).
Наличие емкости (11) позволяет использовать непрерывный режим в течение более длительного времени; кроме того, конечно, это дает возможность обрабатывать более значительный объем композиции.
Альтернативно накопительной емкости (11) можно использовать любое устройство, способное подавать композицию в основную схему. Например, и преимущественно, приводя материал графита в контакт с растворителем, содержащим по меньшей мере 10 мас.% способного к полимеризации мономера, можно проводить приготовление композиции непосредственно выше контура обработки ультразвуком по ходу технологического потока, также и в непрерывном режиме, с помощью емкостей, снабженных системой смешивания или статическими смесителями. Таким образом, весь процесс является полностью непрерывным.
Дополнительный приведенный в качестве примера воплощения режим, представленный на Фиг. 9, обеспечивает дополнительное наличие контейнера (12), называемого далее по тексту концентратором, в который (f13) поступает надосадочная жидкость из отстойника (10). Часть наиболее летучих растворителей испаряют в концентраторе (12) и удаляют через выпускное отверстие (g), находящееся в его верхней части, так что концентрация GRS в выходящей из концентратора композиции (f15) возрастает.
Концентрирование можно провести в соответствии с любым из способов, известных в уровне техники. Например, концентрирование можно проводить путем инжекции газа, чтобы провести отгонку наиболее летучих растворителей. В качестве альтернативы или в комбинации с этим концентрирование можно провести, доводя композицию в (12) до более высокой температуры; или опять-таки, в качестве альтернативы, или в сочетании с этим, путем снижения давления в (12).
В соответствии с данным изобретением концентрирование предпочтительно проводят путем отгонки легких фракций с применением инертного газа, обычно азота, при снижении давления до более низкого уровня, обычно до 10 кПа (100 мбар).
Концентратор (12) предпочтительно поддерживают при постоянной температуре, например, при такой же температуре, какую имеет композиция в (10), посредством рубашки, в которой циркулирует регулирующий теплоноситель. Извлеченные газы (g) можно сконденсировать и использовать повторно, например, снова направить в контейнер (11) для смешивания исходных материалов. Концентрирование в (12) позволяет увеличить концентрацию GRS в композиции по меньшей мере на 20%, более предпочтительно по меньшей мере на 50%, еще более предпочтительно по меньшей мере на 100%.
Схемы могут также включать дополнительные элементы, которые являются частью существующего уровня техники, такие как, например: фильтрование раствора перед всасыванием в насос, преобразователи давления, температуры, расходомеры, системы наблюдения и контроля, а также все, что специалист может считать полезным ввести.
Далее, для лучшего понимания объема данного изобретения и заявки, приведены несколько примеров, которые никоим образом не ограничивают объем данного изобретения.
Для краткости термин «части» относится далее к массовым частям; «г» или «гр» - к граммам, а «л» или «лт» - к литрам.
Пример 1
В данном примере описан способ приготовления композиции, содержащей графен и графеновые нанопластинки, в соответствии с режимом непрерывного потока, представленным на Фиг. 4.
2 части коммерческого синтетического графита SFG6, полученного от Timcal, диспергируют в 98 частях стирола (Sigma Aldrich) с помощью магнитной анкерной мешалки, при комнатной температуре, в течение 15 минут, с получением однородной композиции графита в жидкости.
Затем перемешивание прекращают, и композицию немедленно загружают в бак (1) для загрузки, при комнатной температуре, с помощью насоса (не показан на чертеже). Одновременно включают циркуляционный насос (4). В ходе загрузки непрерывно отводят поток (f1 ter) газа, исходно содержащегося в контуре обработки ультразвуком, посредством патрубка, размещенного в верхней части контейнера (1), чтобы полностью заполнить контейнер (1) композицией. Загрузку композиции прекращают, когда композиция начинает вытекать из контейнера (1) потоком «f1 ter».
После того, как загрузка композиции завершена, поток «f1 ter» перекрывают и включают подающий насос, чтобы увеличить давление в контейнере (1) до величины 1,3 МПа (13 бар) абс.
Спускной клапан, расположенный в самой высокой точке контура обработки ультразвуком, частично открывают на несколько секунд, чтобы удалить захваченный газ, и включают насос, чтобы снова довести давление до 1,3 МПа (13 бар) абс. Эту операцию повторяют на выпускном клапане, из которого будут проводить отбор проб (5). Затем запускают генератор (3) ультразвука в непрерывном режиме, с помощью сонотрода, при частоте 20 кГц и с удельной энергией около 100 Вт/см3, постоянной во времени (и, следовательно, немодулируемой). Насос (4), который подает композицию в камеру, где проводят обработку (2) ультразвуком, регулируют посредством преобразователя частоты таким образом, чтобы время пребывания в камере обработки ультразвуком, рассчитанное как объем камеры, деленный на объемный расход перерабатываемого раствора, составляло 0,15 секунд. Время пребывания в баке (1) для загрузки и в других устройствах, формирующих контур обработки ультразвуком (в котором постоянно циркулирует композиция), снова рассчитанное как отношение общий объем/объемный расход, равно 15 секунд. Таким образом, рабочий цикл равен примерно 1%. Ниже устройства (2) обработки ультразвуком по ходу технологического потока обеспечивают теплообменник (6) для регулирования температуры. Общее время обработки равно 7200 секунд. Следовательно фактическое время обработки ультразвуком составляет 72 секунды. Тест проводят при постоянном давлении, начиная с комнатной температуры (20°С) и увеличивая температуру примерно на 2 градуса в минуту в течение первых 30 минут, затем примерно на 1 градус в минуту в течение последующих 30 минут, а затем примерно на 0,5 градусов в минуту в течение следующих 60 минут, пока в конце теста не будет достигнута температура 140°С. Увеличение температуры в основном является результатом поглощения технологической текучей средой ультразвуковых волн в камере обработки ультразвуком. В любом случае увеличение температуры было бы более высоким, если бы часть подаваемого тепла не рассеивалась из-за потерь тепла в окружающую среду.
В конце теста смесь охлаждают с помощью теплообменника (6), поддерживая непрерывную работу циркуляционного насоса (4). При достижении температуры 90°С отбирают образец в соответствующий контейнер (5) и оставляют для декантации на 24 часа, чтобы позволить возможному неэксфолиированному материалу графита осесть на дно. Образец, отобранный из надосадочной жидкости образца, оставленный для декантации (примерно 40 мас.%), называют «Продукт 1А».
«Продукт 1А», полученный из предшествующей декантации, подвергают центрифугированию (центрифуга модели Sorvall Evolution RC, с максимальной скоростью вращения 25000 об/мин) при 8000 об/мин в течение 30 минут, чтобы удалить материал графита, который не был полностью эксфолиирован. В конце центрифугирования отбирают надосадочную жидкость, называемую «Продукт 1В»; это конечный продукт процесса обработки ультразвуком, частично полимеризованный.
«Продукт 1 В» подвергают отгонке остаточного стирола с помощью системы вакуумной печи (СВП), чтобы определить процентное содержание твердых веществ в образце, то есть образованного полимера и присутствующих графена и графеновых нанопластинок. СВП по существу представляет собой нагревающее устройство, в которое помещают небольшое количество «Продукта 1В» (около 3 граммов) на 30 минут при температуре 230°С. Давление доводят до 60 кПа (600 мбар) в течение 10 минут, затем до 40 кПа (400 мбар) за дополнительные 10 минут и наконец до 5 кПа (50 мбар) в течение оставшегося времени теста. Полученный таким образом продукт - полимерный нанокомпозит - называют «Продукт 1С». В упомянутом «Продукте 1С» непрореагировавший остаточный мономер удален; описанный способ (СВП) представляет собой возможный режим для получения конечного продукта - полимерного нанокомпозита.
Отношение массы конечного (сухого) образца («Продукт 1С») и исходного образца, подаваемого в СВП («Продукт 1В»), называют процентным содержанием твердого вещества или, что эквивалентно, долей твердого вещества. Доля твердого вещества, которая, как было показано, составляет около 18 мас.%, представляет собой процентное содержание полимера и GRS, присутствующих в надосадочной жидкости.
Для получения процентного содержания GRS Продукт 1С был подвергнут термогравиметрическому анализу (ТГА).
Для этой цели образец нагревали от комнатной температуры до 600°С в атмосфере азота, со скоростью 50°С/мин (в ходе этой стадии устраняют полимерную матрицу), а затем от 600 до 850°С на воздухе, снова со скоростью 50°С/мин; на этой стадии происходит сгорание в воздухе неорганического углерода, то есть GRS, так как это единственный присутствующий материал неорганического углерода. Процентное содержание углерода рассчитывают из потери массы, которая происходит после замены атмосферы азота на воздух, то есть из разницы между массой при 600°С в атмосфере азота и массой при 850°С на воздухе. Показано, что это значение равно 1,2 мас.% Таким образом, процентное содержание GRS в «Продукте 1В» по отношению к общей массе равно 0,22% масс, то есть равно концентрации примерно 2,2 г/л.
«Продукт 1 В» анализировали с помощью трансмиссионной электронной микроскопии (ТЭМ), чтобы проверить полученную степень эксфолиации. Одно из полученных ТЭМ изображений приведено на Фиг. 10. Тот же продукт был также подвергнут атомно-силовой микроскопии (АСМ (AFM)); таким образом подтвердили, что полученные GRS имеют толщину, не превышающую 10 нм.
Оставшийся «Продукт 1А» был оставлен для выдержки в течение 30 дней во втором контейнере, закрытом и прозрачном; при этом не наблюдали какой-либо видимой седиментации. Это предполагает, что и «Продукт 1А» уже представляет собой концентрированную и стабильную суспензию.
Пример 2
Повторяют Пример 1 с использованием 0,05 частей инициатора Pekadox BC-FF (производства AkzoNobel), 2 частей графита SFG6 и 97,95 частей стирола, с получением «Продукта 2А» после декантации, «Продукта 2В» после центрифугирования и «Продукта 2С» после отгонки остаточного стирола, аналогично тому, что описано в Примере 1. «Продукт 2 В» подвергали ТЭМ-анализу: из фотографий (Фиг. 11) можно видеть хорошую степень эксфолиации. Можно также оценить, с помощью АСМ, что GRS имеет толщину не более 10 нм. Концентрацию GRS в «Продукте 2В» определяли с помощью такого же метода, который описан в Примере 1, а также термогравиметрического анализа (ТГА): результат составляет 2,5 г/л. Было показано, что доля твердых веществ равна 22%.
Пример 3
Повторяют Пример 1, но с использованием 0,2 частей инициатора Perkadox BC-FF, 2 частей графита SFG6 и 97,8 частей стирола, с получением «Продукта 3А» после декантации, «Продукта 3В» после центрифугирования и «Продукта 3С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. Как и для Примеров 1 и 2, проводили анализы ТЭМ и АСМ, и концентрацию GRS определяли в соответствии с такими же методиками, примененными для «Продукта 3В». Из АСМ-анализа «Продукта 3 В» можно оценить, что GRS имеет толщину не более 10 нм, в то время как концентрация в «Продукте 3 В», измеренная тем же способом, который описан в Примере 1, а также с помощью термогравиметрического анализа (ТГА), составляет 4 г/л. Показано, что доля твердых веществ равна 25%. И в этом случае «Продукт 3А» был оставлен для выдержки во втором закрытом и прозрачном контейнере. В результате была получена стабильная и концентрированная суспензия, которая через 30 дней не проявляла никаких очевидных признаков седиментации.
Пример 4
Повторяли Пример 1, но с использованием композиции, составленной из 10 частей полистирола Edistir N1782 (М.в.=180000 г/моль; ПТР (200°С, 5 кг)=7,5 г/10 мин), 2 частей коммерческого синтетического графита SFG6 производства Timcal и 88 частей стирола (Sigma Aldrich). «Продукт 4А» был получен после декантации, «Продукт 4В» после центрифугирования и «Продукт 4С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. «Продукт 4В» подвергали ТЭМ-анализу, как можно видеть на Фиг. 12. И в этом случае проводили анализ АСМ, оценивая толщину GRS не более 10 нм. Концентрация GRS в «Продукте 4В» составляет 3 г/л; ее получают с использованием такого же способа, как и описанный в Примере 1, а также термогравиметрического анализа (ТГА).
Пример 5
Повторяют Пример 1, но с использованием 0,05 частей инициатора Perkadox L-W75 (производства AkzoNobel), 2 частей графита SFG6, 30 частей этилбензола и 67,95 частей стирола.
«Продукт 5А» был получен после декантации, «Продукт 5 В» после центрифугирования и «Продукт 5С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. «Продукт 5В» подвергали ТЭМ-анализу. Также посредством АСМ оценили, что толщина GRS составляет не более 10 нм. Концентрация GRS, рассчитанная по «Продукту 5В», полученная с использованием того же способа, который описан в Примере 1, а также термогравиметрического анализа (ТГА), составляет около 1,1 г/л.
Пример 6
Этот пример описывает способ получения полимерного нанокомпозита.
Часть «Продукта 1В» помещают в стеклянный автоклав, снабженный мешалкой. Раствор нагревали до 120°С и поддерживали при этой температуре, под вакуумом, примерно в течение 1 часа. Показано, что процентное содержание твердых продуктов, снова измеренное способом, описанным в Примере 1, составляет 35%.
Полученный таким образом продукт затем помещают в стальной цилиндр, помещаемый в печь и выдерживаемый при температуре 150°С в течение 3 часов, для завершения реакции полимеризации. Полученный продукт затем помещают в вакуум (5 кПа (50 мбар) абс.) на 30 минут при температуре 230°С, чтобы устранить любое возможное количество остаточного стирола. Полученный таким образом материал, называемый «Продукт 1D», охлаждают, извлекают из цилиндра и дробят. Материал подвергают термогравиметрическому анализу (ТГА) в соответствии с методиками, описанными в Примере 1; было получено количество присутствующего GRS, равное 1%.
Данный материал можно впоследствии переработать с помощью обычных технологий, применяемых в области преобразования полимерных материалов, таких как, например, экструзия, литье под давлением, прямое формование.
Пример 7
Повторяли Пример 1, но после загрузки раствора в (1), включения насоса (4) и повышения давления композицию доводили до температуры 80°С с помощью теплообменника (6), выдерживая ее при этой температуре при циркуляции посредством насоса (4) перед включением ультразвука. Получали следующие продукты: 7А после декантации, 7 В после центрифугирования и 7С после отгонки легких фракций, аналогично тому, что описано в Примере 1.
Процентное содержание GRS в «Продукте 7В» по отношению к общему количеству, полученное с использованием такой же методики, как описанная в Примере 1, а также термогравиметрического анализа (ТГА), составляет 0,25 мас.% Показано, что доля твердого вещества равна 24%.
Пример 8
Повторяют Пример 1, но проводя повышение давления с помощью экспансионной текучей среды, находящейся в напорной емкости (8), согласно схеме, изображенной на Фиг. 6.
Для этой цели напорную емкость (8) заполняют стиролом (применяемая экспансионная текучая среда) посредством потока (f10 бис). В ходе загрузки композиции (поток f1) насос (4) включают, и газ, исходно находящийся в контуре обработки ультразвуком, непрерывно отводят из патрубка, расположенного в верхней части контейнера (1), (поток «е»); поддерживая приемник (9) под вакуумом посредством предназначенного для этого насоса (не показан на чертеже). Затем открывают клапан на потоке (f11), чтобы привести экспансионную текучую среду в контакт с композицией. Клапаны на потоках (а), (b) и (d) тоже открывают на короткие периоды времени, чтобы выпустить газ из возможных газовых карманов и полностью заполнить контур сонификации композицией. Затем повышают давление, подавая газообразный аргон (напорный газ) по потоку f10; включают генератор ультразвука и регулируют насос (4), снова как указано в Примере 1. Затем проводили тест с таким же температурным профилем и временем обработки, как в Примере 1. Используемое давление составляло до 1,3 МПа (13 бар) абс.
Также и в этом случае получают стабильную суспензию, без какой-либо видимой седиментации через 30 дней. Затем получали «Продукт 8А» после декантации, «Продукт 8В» после центрифугирования и «Продукт 8С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. Показано, что концентрация GRS в «Продукте 8 В», определенная с помощью той же методики, которая описана в Примере 1, а также термогравиметрического анализа (ТГА), равна 0,25 мас.%
Фотографии ТЭМ показывают хорошую степень эксфолиации, а анализ АСМ показывает присутствие GRS с толщиной, не превышающей 10 нм.
Пример 9
Повторяют Пример 8, но с использованием давления 0,8 МПа (8 бар) абс. Таким образом «Продукт 9А» был получен после декантации, «Продукт 9В» после центрифугирования и «Продукт 9С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. Показано, что концентрация GRS в «Продукте 9В», полученная с использованием такого же способа, как описанный в Примере 1, а также термогравиметрического анализа (ТГА), равна примерно 0,06 мас.%.
Пример 10
Повторяют Пример 8, но с использованием давления 2,1 МПа (21 бар) абс. Затем получали «Продукт 10А», «Продукт 10 В» и «Продукт 10С», с помощью способов, уже описанных в Примере 1. Показано, что концентрация GRS в «Продукте 10 В», полученная с использованием такого же способа, как описанный в предыдущих примерах, равна примерно 0,15 мас.%.
Сравнительный Пример 1
Повторяют Пример 1 без повышения давления и, соответственно, поддерживая атмосферное давление на всем протяжении теста. В конце обработки раствор извлекают в прозрачный контейнер. Было показано, что через 1 час раствор в контейнере полностью разделяется на две фазы, при этом по существу весь материал графита оседает на дне (Фиг. 14, контейнер слева).
Пример 11
Повторяют Пример 1, но с удельной мощностью облучения ультразвуком около 500 Вт/см3. «Продукт 11А» был получен после декантации, «Продукт 11В» после центрифугирования и «Продукт 11С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. Показано, что концентрация GRS в «Продукте 11В», полученная с использованием такого же способа, как описанный в Примере 1, а также термогравиметрического анализа (ТГА), равна 0,35 мас.%
Пример 12
Повторяли Пример 1, но с использованием 2 частей графита SFG6, 10 частей стирола и 88 частей глицерина (Carlo Erba). Продукт, извлеченный после обработки, был помещен в закрытый прозрачный контейнер. Показано, что через 30 дней полученная таким образом суспензия была однородной. Показано, что концентрация GRS в продукте равна 0,4 мас.%
Сравнительный Пример 2
Повторяли Пример 1, но с использованием 2 частей графита SFG6 и 98 частей N-метилпирролидона (Carlo Erba) вместо стирола.
В конце обработки раствор извлекают в прозрачный контейнер. Показано, что через 1 час раствор в контейнере полностью разделился на две фазы, при этом по существу весь материал графита был осажден на дне.
Сравнительный Пример 3
Повторяли Пример 1, но с использованием 2 частей графита SFG6 и 98 частей этилбензола вместо стирола.
В конце обработки раствор извлекают в прозрачный контейнер. Показано, что через 1 час раствор в контейнере полностью разделился на две фазы, при этом по существу весь материал графита был осажден на дне.
Пример 13 (периодический режим)
2 части коммерческого синтетического графита SFG6 производства Timcal, диспергировали в 98 частях стирола (Sigma Aldrich) с помощью магнитной анкерной мешалки при комнатной температуре в течение 15 минут, чтобы получить однородную дисперсию графита в жидкости. Композицию загружают в приемное устройство (1) (Фиг. 1) при комнатной температуре, заполняя весь доступный объем и удаляя газ, который ранее находился в этом контейнере, через патрубок, расположенный на верхнем фланце (f3). Композицию также пропускают, посредством потока (f1 бис), из контейнера (1) в сифон (1 бис), чтобы удалить присутствующий в нем газ. Затем сифон (1 бис) соединяют с баллоном аргона, редуктор которого установлен на 1,3 МПа (13 бар) абс. Таким образом в контейнере (1) повышают давление до такого же значения, в то же время предотвращая поступление аргона в контейнер (1).
В дополнение к повышению давления, это же соединение позволяет также поддерживать давление постоянным в ходе всего процесса ультразвуковой обработки (компенсируя, например, термические расширения).
После повышения давления композицию подвергают действию ультразвука, в периодическом режиме, посредством сонотрода, с частотой 20 кГц и с удельной мощностью около 200 Вт/см3. Эту мощность модулируют во времени, циклами по 10 секунд каждые 60 секунд, чтобы полученный рабочий цикл составлял 17%.
Сонотрод погружают непосредственно в приемное устройство (1), содержащее композицию, которая должна быть обработана. Приемное устройство (1) снабжено рубашкой, внутри которой циркулирует масло для нагревания композиции. Таким образом, в ходе обработки композицию, которая вначале находится при комнатной температуре, нагревают со скоростью 55°С/ч, до тех пор, пока она не достигнет максимальной температуры 130°С.
Обработка продолжается 2 часа; в конце обработки материал отбирают непосредственно из нижней части приемного устройства и оставляют для выдержки, чтобы наблюдать, не произойдет ли седиментации материала графита.
В этом случае также было показано, что отобранная композиция является стабильной и однородной даже через один месяц.
Пример 14
Этот пример описывает получение пенополистирола, содержащего GRS.
48,48 частей полистирола Edistir N1782, 1,8 частей Emerald 3000 (продаваемого фирмой Chemtura; полимерный бромированный огнезащитный агент), 0,22 частей Perkadox 30 и 50 частей «Продукта 1D», полученного в Примере 6, смешивают в экструдере с двойным шнеком. Затем добавляют 5 частей смеси н-пентана (75%) и изопентана (25%). Приготовленную таким образом композицию смешивают посредством серии статических смесителей; шестеренчатый насос увеличивает давление смеси до 20 МПа (200 бар) изб. Впоследствии смесь охлаждают примерно до 170°С с помощью теплообменника.
Затем композицию направляют к нагретой пластине фильеры, где ее экструдируют через множество отверстий, имеющих диаметр 0,5 мм, немедленно охлаждают с помощью водяной струи и нарезают рядом вращающихся ножей (как указано, например, в патенте US 7320585). Относительное давление в грануляционной камере составляет 0,5 МПа (5 бар), а скорость нарезки задают таким образом, чтобы получить гранулы, имеющие средний диаметр 1,2 мм. Гранулы сушат с помощью центробежной сушилки и затем на них наносят покрытие, как описано ниже.
Гранулы подвергают термогравиметрическому анализу с помощью способа, описанного в Примере 1; было получено значение содержания GRS, равное 0,45 мас.%.
Покрытие получают путем добавления к гранулам 3 частей моностеарата глицерила, совместно с 1 частью стеарата цинка и 0,2 частями глицерина, на 1000 частей высушенных гранул. Входящие в состав покрытия добавки смешивают с гранулами посредством шнекового смесителя.
Покрытый таким образом продукт вспенивают до насыпной плотности 17 г/л посредством пара при температуре 100°С, оставляют стареть на 24 часа и, наконец, используют для формовки блоков (1040×1030×550 мм).
Блоки нарезали с получением плоских листов, на которых были проведены измерения теплопроводности. Измеренная теплопроводность была равна 31 мВт/мК.
Некоторые из листов, полученных из одних и тех же блоков, были помещены в печь при 70°С на 2 дня, и затем были получены образцы для проведения испытаний, имеющие размеры 9×19×2 см. Эти образцы были подвергнуты испытанию на огнестойкость в соответствии со стандартом DIN 4102. Образцы прошли этот тест.
Образцы подвергли также испытаниям на механическую прочность на сжатие в соответствии со стандартом EN ISO 844.
Показано, что напряжение при 10% сжатии равно 113 кПа.
Сравнительный Пример 4
Этот пример описывает получение пенополистирола, содержащего графит.
Пена из пенополистирола была получена в соответствии с методиками, описанными в Примере 14, но с заменой 50 частей «Продукта 1D» на 0,5 частей графита SFG6, не подвергнутого какой-либо ультразвуковой обработке, и 49,5 частей Edistir N 1782 (таким образом N 1782 составляет в целом 97,98 частей). Гранулы сушили с помощью центробежной сушилки и затем на них наносили покрытие, как описано в Примере 14. Гранулы подвергали термогравиметрическому анализу с использованием способа, описанного в Примере 1; было получено содержание GRS, равное 0,45 мас.%.
Образцы для проведения испытаний на теплопроводность, прочность на сжатие и огнестойкость были получены, как указано в Примере 14.
Были получены следующие результаты: 33 мВт/мК для теплопроводности и 108 кПа для напряжения при 10% сжатии. Тест на огнестойкость был пройден.
Пример 15
Повторяют Пример 1, но с использованием схемы, приведенной на Фиг. 9. Повышение давления осуществляют посредством экспансионной текучей среды, содержащейся в напорной емкости (8). Давление, применяемое в контуре обработки ультразвуком, составляло 1,2 МПа (12 бар) изб.
Упомянутая экспансионная текучая среда состоит из стирола; в качестве обеспечивающего давление газа используют аргон.
Расход композиции, подаваемой из накопительной емкости (11) в контур обработки ультразвуком, равен 1% от расхода рециркуляционного насоса (4), и его регулируют с использованием дополнительного шестеренчатого насоса (13), расположенного между (11) и (1), скорость которого регулируют с помощью частотного преобразователя.
Небольшую часть текучей среды, находящейся в контуре обработки ультразвуком, периодически удаляют из самой высокой точки контура (точка d на Фиг. 9) и собирают в сливном резервуаре (9).
Часть обработанной ультразвуком текучей среды отбирают из контура обработки ультразвуком и направляют в отстойник (10). Так как давление в контуре обработки ультразвуком поддерживают постоянным при 1,2 МПа (12 бар) изб., то расход текучей среды, направляемой в (10), по существу равен расходу композиции, подаваемой из накопительной емкости (11) в контур обработки ультразвуком.
Полезный объем отстойника (10) является таким, чтобы обеспечить время пребывания композиции в отстойнике, равное 90 минутам. Давление в отстойнике поддерживают на 0,05 МПа (0,5 бар) ниже, чем давление в контуре обработки ультразвуком.
Расход надосадочной жидкости, направляемой из отстойника (10) в концентратор (12), составляет 10% от расхода, подаваемого из (11) в контур обработки ультразвуком. Оставшееся количество удаляют из нижней части отстойника (10) и снова рециркулируют в накопительную емкость (11).
Концентратор (12) поддерживают при давлении 10 кПа (100 мбар) абс. Давление регулируют путем конденсации газов, собранных в (е), с помощью теплообменника и удаления неконденсирующихся газов вакуумным насосом.
Азот вводят со дна концентратора (12), для осуществления отгонки летучего растворителя (не показано на Фиг. 9).
Композицию, выходящую из концентратора (f15), обрабатывали в СВП, получая содержание твердых веществ 40%. Термогравиметрический анализ, проведенный в соответствии с вышеописанным способом на твердом продукте, полученном из СВП, дал величину, равную 3,2%. Это показывает, что концентрация GRS на выходе из концентратора была равна около 1,3%.
Пример 16
Повторяют Пример 1, но перед включением ультразвука теплообменник (6) используют для доведения раствора до 60°С в течение 30 минут. Затем включают ультразвук, как в Примере 1.
Общая продолжительность обработки ультразвуком составляет 90 минут. Скорость нагревания составляет 1,5 градусов в минуту в течение первых 40 минут, затем примерно 1 градус в минуту в течение последующих 30 минут, а затем примерно 0,5 градуса в минуту в течение последних 20 минут, пока в конце теста температура не достигает 160°С.
После первых 50 минут с начала обработки ультразвуком верхний выпускной клапан снова открывают на несколько секунд, чтобы выпустить газ, возможно скопившийся в схеме.
И снова, в соответствии с Примером 1, «Продукт 16А» получали после декантации, «Продукт 16В» после центрифугирования и «Продукт 16С» после отгонки легких фракций. Концентрацию GRS в «Продукте 16В» получали с использованием такого же способа, который описан в Примере 1, а также термогравиметрического анализа (ТГА); результат составляет 5,5 г/л. Показано, что доля твердого вещества равна 30%.
Сравнительный Пример 5
Этот пример описывает обработку ультразвуком в соответствии с косвенным режимом (ультразвуковая ванна), в присутствии газа.
2 части коммерческого синтетического графита SFG6 производства фирмы Timcal диспергируют в 98 частях стирола (Sigma Aldrich) с помощью магнитной анкерной мешалки, при комнатной температуре, в течение 15 минут, чтобы получить однородную дисперсию графита в жидкости. Дисперсию помещают в химический стакан, находящийся в ультразвуковой ванне (модель Branson 8219), а затем в контакт с атмосферным воздухом. После этого включают ультразвук. Через 2 часа обработки отбирают первый образец. Еще через 4 часа обработки отбирают второй образец. Оба образца оставляют для выдержки. Как показано, через день материал графита в обоих образцах полностью оседает на дно.
Сравнительный Пример 6
Повторяют Пример 8, но без загрузки экспансионной текучей среды и без осуществления выведения газа из газовых карманов клапанов на потоках (а), (b) и (d). Таким образом, в ходе обработки ультразвуком насос (4) увлекает газ в камеру обработки ультразвуком, где он вступает в прямой контакт с композицией. После 2 часов обработки отбирают образец и оставляют его выстаиваться в лабораторном стакане. Как показано, через день материал графита, находящийся в стакане, полностью оседает на дно.
Пример 17
Этот пример описывает обработку ультразвуком в соответствии с режимом модуляции испускания ультразвука.
Повторяют Пример 1, но модулируя испускание ультразвука во времени, то есть периодически изменяя мощность испускаемого ультразвука. Более конкретно, периоды длительностью 15 минут, в течение которых испускали ультразвук с постоянной мощностью, перемежают периодами длительностью 5 минут, в течение которых испускание ультразвука прекращают (нулевая мощность). Фактическая продолжительность остается неизменной (72 секунды), и, таким образом, общая продолжительность теста составляет 9600 секунд.
«Продукт 17А» был получен после декантации, «Продукт 17В» после центрифугирования и «Продукт 17С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. «Продукт 17В» был подвергнут ТЭМ анализу; с помощью АСМ было оценено, что GRS имеет толщину, не превышающую 10 нм. Концентрация GRS в «Продукте 17 В» была получена с использованием такого же способа, как описанный в Примере 1, а также термогравиметрического анализа (ТГА); результат составляет 5 г/л.
Пример 18
Повторяют Пример 1, но с использованием более продолжительного общего времени обработки (и, следовательно, более продолжительного фактического времени обработки ультразвуком). Общее время равно 28800 секунд (8 часов); так как рабочий цикл равен 1%, фактическое время обработки ультразвуком равно 288 секунд.
Тест проводят при постоянном давлении, начиная с комнатной температуры (20°С) и увеличивая температуру примерно на 2 градуса в минуту в течение первых 30 минут, затем примерно на 1 градус в минуту в течение последующих 30 минут, а затем примерно на 0,5 градуса в минуту в течение последующих 20 минут, пока, через 80 минут после начала теста, не достигают температуры 120°С. Затем температуру поддерживают постоянной, и регулируют на 120°С в течение остального времени теста. «Продукт 18А» был получен после декантации, «Продукт 18В» после центрифугирования и «Продукт 18С» после отгонки легких фракций, аналогично тому, что описано в Примере 1. «Продукт 18В» подвергали ТЭМ и АСМ анализу; и в этом случае также можно оценить, что толщина GRS не превышает 10 нм. Концентрация GRS в «Продукте 18В» была получена с использованием такого же способа, который описан в Примере 1, а также термогравиметрического анализа (ТГА); результат составляет 0,26 мас.%, то есть примерно 2,6 г/л. Показано, что доля твердого вещества равна 30%.
Анализ образцов, полученных в вышеописанных примерах, в отношении осаждения
Образцы всех продуктов из примеров по данному изобретению и сравнительных примеров помещают после приготовления в закрытый прозрачный контейнер.
Образцы, полученные во всех вышеуказанных примерах по данному изобретению, показывают стабильную дисперсию, то есть даже при подсвечивании образца ярким светом (например, вспышкой Nikon Speedlight SB-80DX, установленной на максимальную интенсивность), невозможно увидеть какого-либо осаждения продукта, как на полученном фотографическом изображении, так и, в еще меньшей степени, визуально. Продукт выглядит однородно непрозрачным и темным. Это свойство сохраняется даже через месяц.
Образцы из сравнительных примеров, напротив, вначале показывают равномерное распределение, но не более чем через один день можно наблюдать сильное осаждение продукта.
Стабильная дисперсия указывает на то, что была получена хорошая эксфолиация материала графита. Таким образом, образцы, полученные в примерах по данному изобретению, показывают хорошую дисперсию и эксфолиацию материала графита в GRS, в то время как образцы из сравнительных примеров показывают плохую дисперсию и эксфолиацию.
В частности, если проводить обработку при атмосферном давлении (Сравнительный Пример 1), то уже через час после отбора пробы можно наблюдать, что материала графита выпал в осадок, что указывает на то, что способ, заявленный в данном изобретении, обычно является неэффективным при атмосферном давлении. Это отличает данное изобретение от большинства процессов, известных на существующем уровне техники, которые, напротив, проводят при атмосферном давлении.
В литературе (см., например, уже цитированную статью Hernandez et al. "High-yield production of graphene by liquid-phase exfoliation of graphite", Nat. Nanotechnol., 2008, 3, 563-568), как уже упоминалось, для эксфолиации графита рекомендуют применять такие растворители, как N-метилпирролидон. Неожиданно, однако, оказалось, что при использовании этого растворителя в способе, раскрытом в данном изобретении (Сравнительный Пример 2) эксфолиация не только не улучшается, но, напротив, данный растворитель явно отрицательно на нее влияет. Фактически лишь через несколько минут после отбора пробы образец показывал явное осаждение подвергнутого ультразвуковой обработке материала графита.
Сравнительный Пример 3 также показывал быстрое осаждение продукта.
Неожиданно оказалось, что полученные суспензии не только являются стабильными во времени, но и содержат GRS в значительно более высокой концентрации, чем продукты, известные в литературе. Например, Hernandez et al. получал концентрацию примерно 0,01 г/л в NMП, при обработке дисперсии ультразвуком в течение 30 минут. Напротив, концентрация GRS, полученного в Примере 1, значительно выше. Более высокие концентрации были получены, но при использовании значительно более высоких времен обработки ультразвуком. Khan et al., например, получал концентрацию около 1,2 г/л после обработки ультразвуком в течение 460 часов. Показано, что для времен обработки ультразвуком менее 100 часов концентрация всегда ниже 1 г/л.
Как уже указывалось, такое высокое время обработки ультразвуком не только невыгодно для внедрения в промышленность, но также легко может привести к дефектам в эксфолиированном материале графита.
Таким образом, из сравнения между примерами по данному изобретению и сравнительными примерами, а также с литературой, можно сделать вывод, что выбор растворителя и давления, при которых проводят обработку ультразвуком, в дополнение к другим рекомендациям, приведенным в данном изобретении, является принципиальным для получения заявленных результатов; и что неожиданные результаты, раскрытые данным изобретением, невозможно было предвидеть в рамках существующего уровня техники.
Сравнительные Примеры 5 и 6 показывают, что без удаления газа из подвергающейся обработке ультразвуком текучей среды, или в любом случае, если обрабатываемая композиция находится в контакте с находящейся над ней отдельной газообразной фазой (атмосферным воздухом), то результаты данного изобретения получить невозможно. В частности, Сравнительный Пример 5 показывает, что такая же композиция, обработанная в традиционной ультразвуковой ванне, не приводит к результатам, полученным с помощью данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНА | 2016 |
|
RU2657504C2 |
| ПРОЦЕСС КАТИОННОЙ ПОЛИМЕРИЗАЦИИ ДЛЯ СИНТЕЗА НАНОСТРУКТУРНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩИХ ГРАФЕН | 2014 |
|
RU2652115C2 |
| ПОЛУПРОВОДНИКОВАЯ ПОЛИОЛЕФИНОВАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭЛЕКТРОПРОВОДНЫЙ НАПОЛНИТЕЛЬ | 2011 |
|
RU2543178C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНОВОГО МАТЕРИАЛА | 2018 |
|
RU2693755C1 |
| ВСПЕНИВАЕМЫЕ ТЕРМОПЛАСТИЧНЫЕ НАНОКОМПОЗИЦИОННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ С УЛУЧШЕННОЙ ТЕПЛОИЗОЛЯЦИОННОЙ СПОСОБНОСТЬЮ | 2010 |
|
RU2537311C9 |
| КУМУЛЕНОВОЕ ВЕЩЕСТВО, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2661876C2 |
| НЕФТЕПРОМЫСЛОВОЕ УСТРОЙСТВО, НЕФТЕПРОМЫСЛОВЫЙ ЭЛЕМЕНТ УКАЗАННОГО УСТРОЙСТВА, СОДЕРЖАЩИЙ ФУНКЦИОНАЛИЗИРОВАННЫЕ ГРАФЕНОВЫЕ ПЛАСТИНКИ, СПОСОБ ОСУЩЕСТВЛЕНИЯ НЕФТЕПРОМЫСЛОВОЙ ОПЕРАЦИИ И СПОСОБ МОДИФИКАЦИИ ФУНКЦИОНАЛИЗИРОВАННЫХ ГРАФЕНОВЫХ ПЛАСТИНОК | 2008 |
|
RU2476457C2 |
| Способ получения дисперсий углеродных наноматериалов | 2016 |
|
RU2618881C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ НАНОРАЗМЕРНЫХ ГРАФЕНОВЫХ ПЛАСТИНОК С ВЫСОКОЙ ДИСПЕРГИРУЕМОСТЬЮ В НИЗКОПОЛЯРНЫХ ПОЛИМЕРНЫХ МАТРИЦАХ И СООТВЕТСТВУЮЩИЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2552477C9 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНОСОДЕРЖАЩИХ МАТЕРИАЛОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2016 |
|
RU2648892C2 |
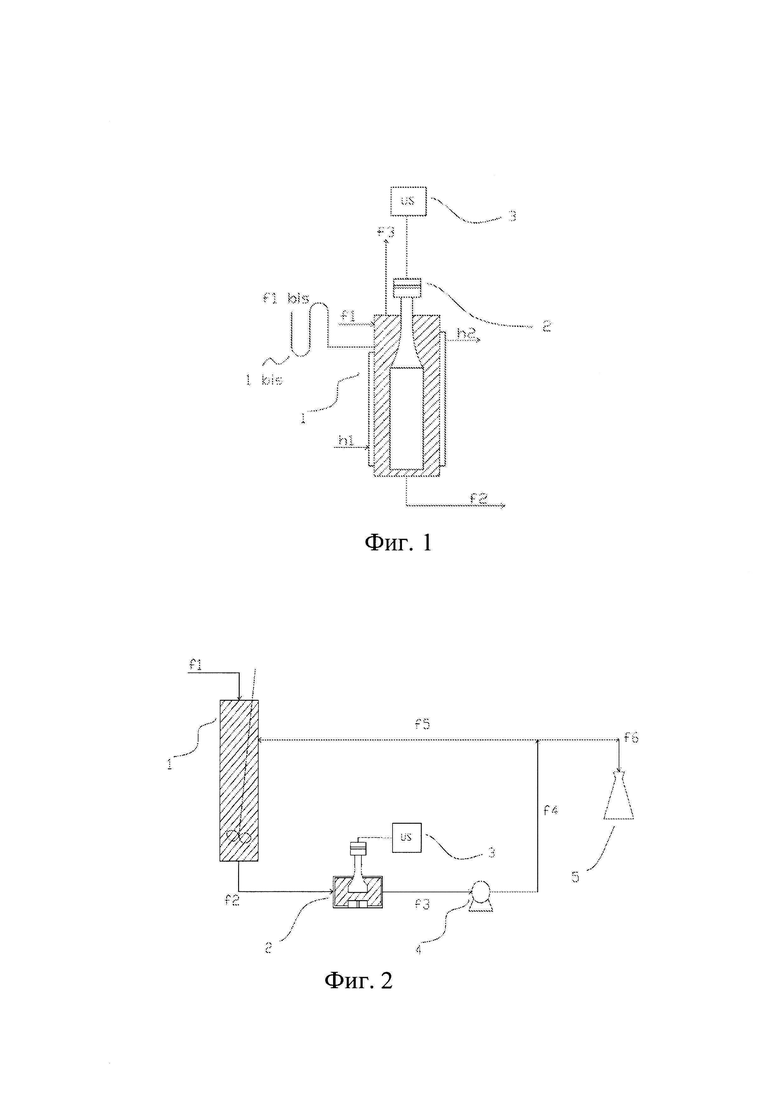
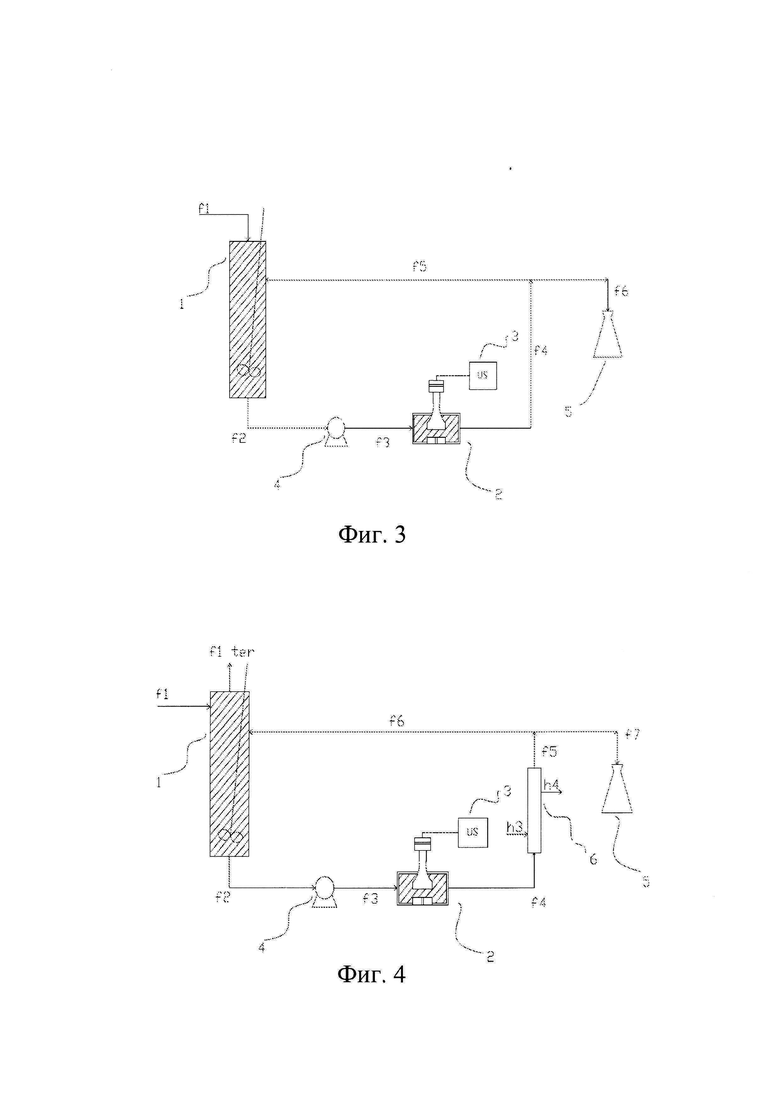
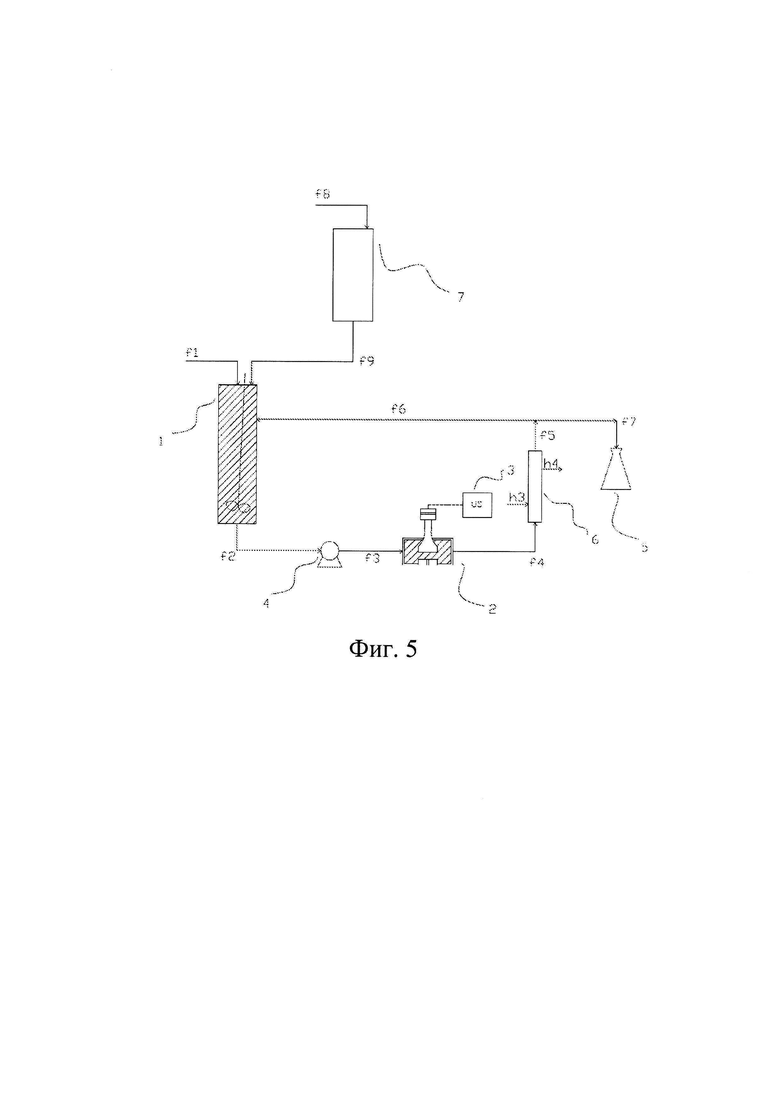

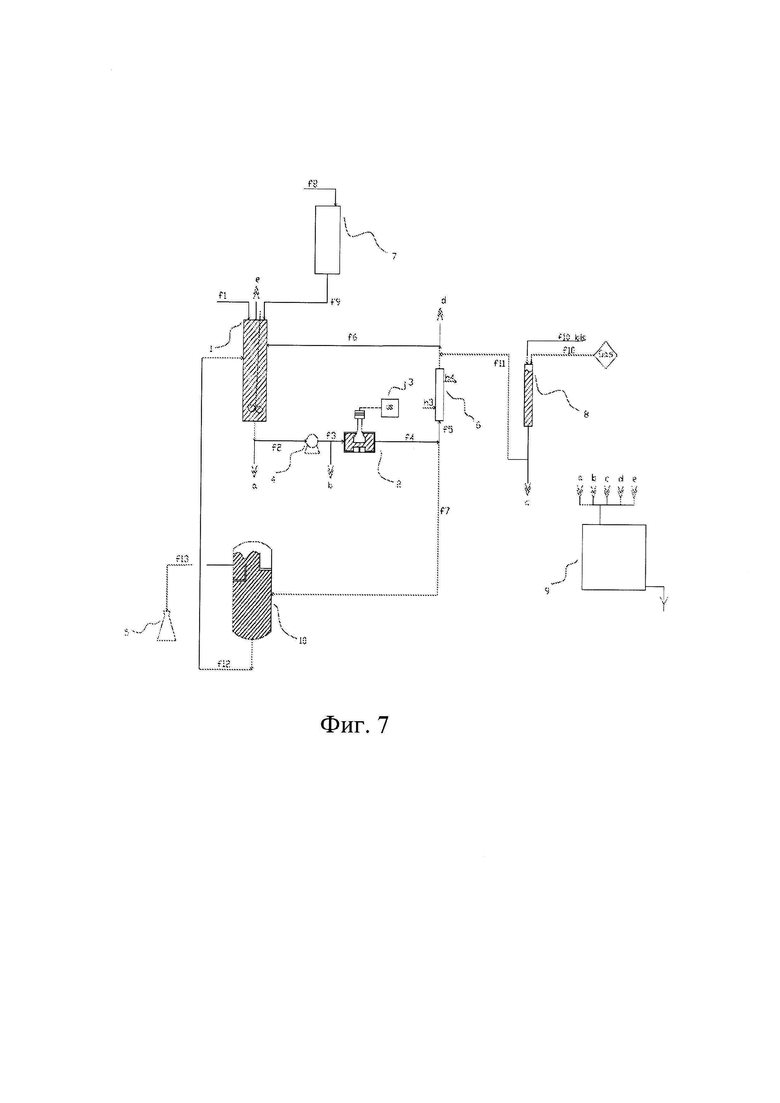





Данное изобретение относится к композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе. Упомянутая композиция отличается тем, что она содержит по меньшей мере 1 мас.%, по отношению к общей массе растворителя, винилароматического полимера, включает массовую концентрацию графена и графеновых нанопластинок (GRS), которая находится в диапазоне от 0,05% до 5 мас.%, по отношению к общей массе растворителя; где указанный винилароматический полимер получен путем частичной или полной полимеризации соответствующего винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных, способных к сополимеризации мономеров, и при условии, что сумма возможного содержания указанных непрореагировавших мономеров и содержания образованного винилароматического полимера равна по меньшей мере 10 мас.% по отношению к общей массе растворителя, и где графен и графеновые нанопластинки имеют среднюю толщину, равную 50 нм или менее. Технический результат - получение композиций, содержащих графен и графеновые нанопластинки, обладающие высокой степенью эксфолиации, создание стабильных дисперсий. 5 н. и 23 з.п. ф-лы, 18 пр., 14 ил.
1. Композиция для получения полимерных композитов, или полимерных нанокомпозитов, или маточных смесей, содержащих графен и графеновые нанопластинки, содержащая графен и графеновые нанопластинки, стабильно диспергированные в растворителе, отличающаяся тем, что она
а) содержит по меньшей мере 1 мас.%, по отношению к общей массе растворителя, винилароматического полимера,
b) включает массовую концентрацию графена и графеновых нанопластинок (GRS), которая находится в диапазоне от 0,05% до 5 мас.%, по отношению к общей массе растворителя;
где указанный винилароматический полимер получен путем частичной или полной полимеризации соответствующего винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных, способных к сополимеризации мономеров, и при условии, что сумма возможного содержания указанных непрореагировавших мономеров и содержания образованного винилароматического полимера равна по меньшей мере 10 мас.% по отношению к общей массе растворителя, и где графен и графеновые нанопластинки имеют среднюю толщину, равную 50 нм или менее, и в любом случае выше или равную толщине одного слоя графена, причем толщину измеряют на нескольких образцах графена и графеновых нанопластинок, нанесенных на кремниевую подложку, покрытую оксидом кремния, с помощью атомно-силовой микроскопии с режимом периодического контактирования, называемой АСМ с прерывистым контактом, и при этом способные к сополимеризации мономеры выбраны из метакриловой кислоты, С1-С4 алкиловых эфиров метакриловой кислоты, амидов и нитрилов метакриловой кислоты, бутадиена, этилена, дивинилбензола, малеинового ангидрида.
2. Композиция по п. 1, в которой содержание винилароматического полимера составляет по меньшей мере 5 мас.% по отношению к общей массе растворителя.
3. Композиция по п. 1 или 2, в которой сумма возможного содержания указанных непрореагировавших мономеров и содержания винилароматического полимера равна по меньшей мере 20 мас.% по отношению к общей массе растворителя.
4. Композиция по любому из пп. 1-3, в которой графен и графеновые нанопластинки имеют мольное отношение углерод/кислород, равное 10 или более.
5. Композиция по п. 4, в которой графен и графеновые нанопластинки имеют среднюю толщину, равную 50 нм или менее, и мольное отношение углерод/кислород, равное 10 или более.
6. Способ получения, исходя из графитового материала, по меньшей мере частично полимеризованной композиции, содержащей графен и графеновые нанопластинки, стабильно диспергированные в растворителе, где указанный способ включает следующие стадии:
а) приведение графитового материала в контакт с основным растворителем, содержащим по меньшей мере 10 мас.%, по отношению к общей массе основного растворителя, винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных, способных к сополимеризации мономеров, образуя исходную композицию;
b) воздействие на указанную исходную композицию ультразвуком, который характеризуется частотным спектром от 18 кГц до 1000 кГц, и давлением, составляющим 0,2 МПа (2 бар) (абс.) или более, в контейнере или камере для проведения обработки ультразвуком, в которой, в контакте с указанной композицией, не должно присутствовать отдельной фазы текучей среды, находящейся в газообразном состоянии;
при этом указанный способ отличается тем, что по меньшей мере 1 % винилароматического мономера, присутствующего в основном растворителе, является полимеризованным.
7. Способ получения, исходя из графитового материала, по меньшей мере частично полимеризованных композиций по любому из пп. 1-6, включающий следующие стадии:
а) приведение графитового материала в контакт с основным растворителем, содержащим по меньшей мере 10 мас.%, по отношению к общей массе основного растворителя, винилароматического мономера, одного или в смеси с до 50 мас.% дополнительных, способных к сополимеризации мономеров, образуя исходную композицию;
b) воздействие на указанную исходную композицию ультразвуком, который характеризуется частотным спектром от 18 кГц до 1000 кГц, при давлении, составляющем 0,2 МПа (2 бар) (абс.) или более, в контейнере или камере для проведения обработки ультразвуком, в которой, в контакте с указанной композицией, не должно присутствовать отдельной фазы текучей среды, находящейся в газообразном состоянии;
при этом указанный способ отличается тем, что по меньшей мере 1 % винилароматического мономера, присутствующего в основном растворителе, является полимеризованным.
8. Способ по п. 6 или 7, в котором стадии (а) и (b) проводят одновременно или последовательно.
9. Способ по любому из пп. 6-8, в котором стадии (а) и (b) проводят в соответствии с непрерывным режимом или в соответствии с периодическим режимом.
10. Способ по любому из пп. 6-9, в котором приготовленную таким образом композицию декантируют, чтобы осадить не подвергшийся эксфолиации графитовый материал, образуя осадок и отделяя первую жидкую фазу, которая содержит графен и эксфолиированные графеновые нанопластинки, а также часть неэксфолиированного графитового материала.
11. Способ по п. 10, в котором после декантации указанную первую жидкую фазу подвергают центрифугированию, чтобы отделить вторую жидкую фазу, содержащую графен и эксфолиированные графеновые нанопластинки, от указанной части неэксфолиированного графитового материала.
12. Способ по любому из пп. 6-9, в котором композицию, содержащую графен и графеновые нанопластинки, стабильно диспергированные в растворителе, непосредственно подвергают центрифугированию, отделяя конечную жидкую фазу, содержащую графен и эксфолиированные графеновые нанопластинки, от неэксфолиированного графитового материала.
13. Способ по п. 10 или 12, в котором после декантации и/или центрифугирования завершают полимеризацию первой или второй жидкой фазы, образуя таким образом полимерные композиты, полимерные нанокомпозиты или полимерную маточную смесь, содержащие графен и графеновые нанопластинки.
14. Способ по любому из пп. 6-9, в котором композиции, содержащие графен и графеновые нанопластинки, стабильно диспергированные в растворителе, частично или полностью полимеризуют непосредственно в ходе обработки ультразвуком (стадия (b)).
15. Способ по любому из пп. 6-14, в котором основной растворитель представляет собой винилароматический мономер, возможно в смеси с дополнительными способными к полимеризации сомономерами и с возможным соответствующим полимером, образованным в ходе полимеризации.
16. Способ по любому из пп. 6-14, в котором основной растворитель выбирают из винилароматических мономеров, органических соединений, способных растворять винилароматические полимеры, пентана и их смесей.
17. Способ по любому из пп. 6-16, в котором помимо графитового материала присутствует также неграфитированный углерод.
18. Способ по любому из пп. 6-17, в котором графитовый материал выбирают из синтетического графита, природного графита, вспученного графита, высокоупорядоченного пиролитического графита, природного кристаллического графита, интеркалированного графита.
19. Способ по п. 17, в котором неграфитированный углерод выбирают из кокса, сажи или графитизированного кокса.
20. Способ по любому из пп. 6-19, в котором в ходе обработки ультразвуком добавляют вторичный растворитель.
21. Способ по любому из пп. 6-20, в котором обработку ультразвуком проводят при удельной мощности ультразвука, измеряемой как электрическая мощность, генерированная ультразвуковым генератором, по отношению к объему камеры, в которую подают ультразвук, составляющей по меньшей мере 60 Вт/см3.
22. Способ по любому из пп. 6-21, в котором эффективное время, в течение которого исходную композицию подвергают воздействию ультразвука, составляет 20 мин или менее и равно 0,1 сек или более.
23. Способ по любому из пп. 6-22, в котором в ходе обработки ультразвуком исходную композицию доводят до давления, составляющего от 0,8 до 5 МПа (от 8 до 50 бар) (абс.).
24. Способ по любому из пп. 6-23, в котором после повышения давления давление исходной композиции поддерживают постоянным посредством расширительного резервуара прямого действия или расширительного резервуара косвенного действия.
25. Способ по любому из пп. 6-24, в котором стадию, на которой исходную композицию подвергают обработке ультразвуком, проводят при температуре, равной 20°С или выше.
26. Способ по любому из пп. 6-25, в котором ультразвук передают в соответствии с прямым режимом или в соответствии с косвенным режимом, или его передают импульсами, чередуя активные периоды, когда исходную композицию подвергают воздействию ультразвука, и пассивные периоды, во время которых указанную композицию не подвергают воздействию ультразвука или ее подвергают воздействию ультразвука с мощностью по меньшей мере в 10 раз ниже, чем мощность воздействия в ходе активного периода, и отношение активного периода к сумме активного и пассивного периодов составляет от 0,0001 до 1.
27. Полимерные композиты, или полимерные нанокомпозиты, или маточные смеси для получения полимерных гранул, промышленных товаров или изделий из вспененных полимеров, содержащие композицию по пп. 1-5.
28. Вспениваемые полимерные композиты или вспениваемые полимерные нанокомпозиты, содержащие композиты, или полимерные нанокомпозиты, или маточные смеси по п. 27 и по меньшей мере вспенивающий агент.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| WO 2008045778 A1, 17.04.2008 | |||
| СПОСОБ ПРИГОТОВЛЕНИЯ НАНОРАЗМЕРНЫХ ГРАФЕНОВЫХ ПЛАСТИНОК С ВЫСОКОЙ ДИСПЕРГИРУЕМОСТЬЮ В НИЗКОПОЛЯРНЫХ ПОЛИМЕРНЫХ МАТРИЦАХ И СООТВЕТСТВУЮЩИЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2552477C9 |
| ВСПЕНИВАЕМЫЕ ТЕРМОПЛАСТИЧНЫЕ НАНОКОМПОЗИЦИОННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ С УЛУЧШЕННОЙ ТЕПЛОИЗОЛЯЦИОННОЙ СПОСОБНОСТЬЮ | 2010 |
|
RU2537311C9 |
| WO 9851735 A1, 19.11.1998 | |||
| КОМПОЗИТНЫЙ МАТЕРИАЛ НА ОСНОВЕ ВИНИЛАРОМАТИЧЕСКИХ ПОЛИМЕРОВ, ИМЕЮЩИХ УЛУЧШЕННЫЕ ТЕПЛОИЗОЛЯЦИОННЫЕ СВОЙСТВА, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2008 |
|
RU2476456C2 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |

