Область техники
Настоящее изобретение относится к области медицины, в частности к лечению митохондриальных заболеваний и, более конкретно, к таким митохондриальным заболеваниям, вызванным делецией/истощением митохондриальной ДНК (мтДНК).
Предшествующий уровень техники
Митохондриальный геном (мтДНК) представляет собой молекулу ДНК (англ. DNA, deoxyribonucleic acid - дезоксирибонуклеиновая кислота), содержащую 16,5 тысяч пар нуклеотидов (англ. kb, kilobase (pairs)), которая обычно присутствует в нескольких копиях в отдельных митохондриях.
Значительная группа менделевских митохондриальных заболеваний вызывается мутациями в ядерных генах, продукты которых участвуют в репликации или поддержании сохранности мтДНК. Эти редкие расстройства также известны как дефекты межгеномной коммуникации, заболевания связанные с истощением мтДНК, множественными делециями мтДНК или заболевания, связанные с истощением и делециями митохондрий (англ. MDDS, mitochondrial depletion and deletions diseases). Такие расстройства имеют специальные орфанные коды: ORPHA35698 - синдром истощения митохондриальной ДНК и ORPHA254807 - синдром множественных делеций митохондриальной ДНК, а также распознаются в базе данных OMIM (англ. Online Mendelian Inheritance in Man - онлайн-каталог фенетических маркеров у человека; менделевское наследование у человека): http://www.om im.org/phenotypicSeries/PS603041 для истощения митохондриальной ДНК и http://www.omim.org/phenotypicSeries/PS157640 для множественных делеций митохондриальной ДНК. Эти заболевания представляют собой комплексную группу генетически и клинически гетерогенных заболеваний, характеризующихся наличием аберраций мтДНК в одной или в комбинации пораженных тканей (например, скелетные мышцы, печень, мозг).
Тяжесть и развитие таких заболеваний очень разнообразны и могут изменяться от легких проявлений (например, прогрессивная внешняя офтальмоплегия) до тяжелых фенотипов, которые могут привести к смерти в младенчестве или раннем детском возрасте, как это наблюдается в классических синдромах истощения мтДНК.
Большинство генов, до настоящего времени соотносимых с дефектами межгеномной коммуникации, либо непосредственно участвуют в репликации мтДНК, либо заняты в метаболизме дезоксирибонуклеозидтрифосфатов (дНТФ), строительных блоков синтеза ДНК. Однако все большее количество мутаций, приводящих к MDDS, идентифицируется в генах, вызывающих нестабильность мтДНК посредством пока еще неизвестного патомеханизма (ОРА1, MPV17, FBXL4 и т.д.). Классически считается, что дефекты в определенных генах специфически приводят к истощению или к множественным делециям мтДНК. Например, дефекты в генах DGUOK обычно приводят к истощению мтДНК, тогда как дефекты в генах ОРА1 обычно вызывают множественные делеции мтДНК. Однако при этом растет число фактов в пользу того, что истощение мтДНК и множественные делеции можно рассматривать как проявления развития тех же патологических процессов, которые влияют на репликацию и восстановление мтДНК. Было обнаружено, что мутации в генах, которые до недавнего времени связывали только с истощением мтДНК с манифестацией в младенческом возрасте (ТК2, DGUOK), также вызывали множественные делеции мтДНК, в некоторых случаях приобретенные.
Поскольку MDDS представляют собой полиорганные расстройства, для оказания поддержки и осуществления симптоматического лечения связанных с этим осложнений необходима многопрофильная бригада, в состав которой входят различные специалисты.
До настоящего времени не разработаны эффективные методы лечения данных комплексных заболеваний. Это связано, главным образом, с отсутствием информации о точных параметрах механизмов, вызывающих такие заболевания, с вариабельностью между одним и другим синдромами и т.д.
Несмотря на изложенное выше, были предприняты попытки поиска подходящих способов лечения MDDS, вызываемых дефектами метаболизма дНТФ, при которых, как считается, нарушена доступность дНТФ. Например, недавние экспериментальные исследования показали, что минуя дефектную стадию биосинтеза дезоксирибонуклеотида, можно преодолеть истощение мтДНК. В частности, сообщалось, что с помощью добавления пуриновых дезокси-нуклеотид (dN) монофосфатов (дНМФ) можно восстановить истощение мтДНК у ТК2-КО мышей [1]. Кроме того, Camara Y. et al. [2] сообщали, что использование дезоксирибонуклеозидов и/или специфических ингибиторов их катаболизма может быть эффективным фармакологическим подходом к лечению различных MDDS, обусловленных дефектами гомеостаза дНТФ.
Несмотря на предпринятые усилия, по-прежнему сохраняется потребность в терапевтических подходах к таким и другим случаям MDDS.
Сущность изобретения
Авторами настоящего изобретения было установлено, что введение дезоксирибонуклеозидов позволяет восстановить уровни мтДНК при MDDS заболеваниях, не обусловленных дефектом метаболизма дНТФ.
До настоящего времени существовало общее мнение о том, что введение нуклеозидов может быть эффективным только при лечении митохондриальных заболеваний, обусловленных дефектом метаболизма дНТФ. Фактически, на предшествующем уровне техники предполагалось, что заболевание, обусловленное дефектом метаболизма дНТФ, можно преодолеть введением "дефектного" нуклеозида [2]. Таким образом, до настоящего момента считалось, что только заболевания, вызванные дефицитом конкретного нуклеотида, можно лечить введением достаточного количества данного «дефектного» нуклеотида/нуклеозида.
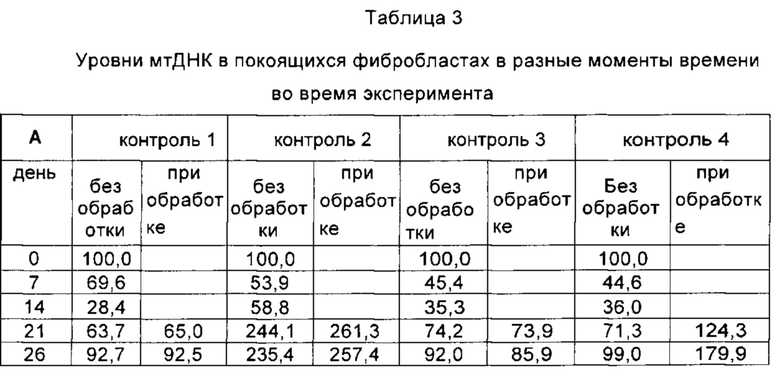
В отличие от способов предшествующего уровня техники авторы настоящего изобретения вводили дезоксирибонуклеозиды в образцы фибробластов пациентов, у которых ранее были диагностированы MDDS, несущие определенные мутации, влияющие на каталитическую субъединицу белка полимеразы гамма 1 (одного из ферментов, вовлеченных в репликативный комплекс мтДНК). Неожиданно было обнаружено, что введение дезоксирибонуклеозидов в эти образцы восстанавливало уровни мтДНК до "нормальных" (здоровых) уровней (см. Таблицу 3, ниже) независимо от мутации, имеющейся у пациента. Невозможно было предсказать, что аномальная функция гена POLG1, кодирующего фермент (которая в конечном итоге отрицательно влияет на правильную работу репликативного комплекса), обусловленная различными мутациями, может быть в равной степени преодолена путем введения дезоксирибонуклеозидов в случае клеток, полученных из организмов всех трех пациентов. Показательно, что восстановление уровней мтДНК путем введения дезоксирибонуклеозидов не зависит от мутации, ответственной за дефект репликативного комплекса мтДНК.
Таким образом, согласно первому аспекту, настоящее изобретение предлагает композицию, содержащую один или более канонический дезоксирибонуклеозид, для применения при лечении синдрома истощения и/или делеции митохондриальной ДНК, при условии, что синдром не обусловлен дефектом метаболизма дезоксирибонуклеозидтрифосфатов (дНТФ). Этот аспект также может быть сформулирован, как применение композиции, содержащей один или более канонический дезоксирибонуклеозид, для приготовления лекарственного средства для лечения синдрома истощения и/или делеции митохондриальной ДНК, при условии, что синдром не обусловлен дефектом метаболизма дезоксирибонуклеозидтрифосфатов (дНТФ). Этот аспект также может быть сформулирован, как способ лечения синдрома истощения и/или делеции митохондриальной ДНК, не обусловленного дефектом метаболизма дезоксирибонуклеозидтрифосфатов (дНТФ), при этом способ включает в себя введение терапевтически эффективного количества одного или более канонического дезоксирибонуклеозида субъекту, нуждающемуся в этом.
Экспериментальные данные были получены в клетках пациентов, страдающих различными клиническими проявлениями, обусловленными дефицитом POLG.
POLG представляет собой ген, кодирующий каталитическую субъединицу митохондриальной ДНК-полимеразы, называемой ДНК-полимеразой гамма. В эукариотических клетках митохондриальные ДНК реплицируются ДНК-полимеразой гамма, тримерным белковым комплексом, состоящим из каталитической субъединицы размером 140 кДа, кодируемой геном POLG, и димерной вспомогательной субъединицы размером 55 кДа, кодируемой геном POLG2. Каталитическая субъединица обладает тремя ферментными активностями: ДНК-полимеразной активностью, 3'-5' экзонуклеазной активностью, корректирующей ошибочно включенные нуклеотиды, и 5'-dRP леазной активностью, необходимой для эксцизионной репарации оснований ДНК. В приведенных ниже примерах пациенты страдали дефицитом POLG вследствие мутаций в экзонуклеазном или полимеразном домене (R309C (в экзонуклеазном домене), и G848S и V1177L (в полимеразном домене)) и в линкерной области (W748S).
Авторы настоящего изобретения неожиданно обнаружили, что введение дезоксирибонуклеозидов "работает" в случае всех исследованных мутаций POLG и, что независимо от того, влияет ли мутация на функцию или структуру POLG, наблюдается заметное улучшение ферментной активности мутированной формы POLG в такой степени, что уровни мтДНК восстанавливаются и находятся в таком же состоянии, как и у здорового субъекта. Другими словами, введение дезоксирибонуклеозидов гиперстимулирует ферментную форму POLG, которая до указанного введения была частично лишена полимеразной активности.
Экспериментальные данные, представленные ниже (обобщенные в приведенной ниже Таблице 3) позволяют заключить, что введение дезоксирибонуклеозидов может быть достаточным для повышения скорости полимеризации мтДНК независимо от мутации в гене POLG. Однако эти данные также свидетельствуют о том, что любое другое MDDS заболевание, которое, как известно, характеризуется снижением мтДНК (либо за счет уменьшения числа копий мтДНК, либо за счет множественных делеций мтДНК), и которое обусловлено мутациями в белках репликативного комплекса, как такового (POLG1, POLG2, который кодирует вспомогательную единицу полимеразы гамма; РЕO1, MGME1, и ДНК2 (англ. DNA2), наряду с прочими), или косвенно участвует в репликации мтДНК (как, например, MPV17), также может быть эффективно подвергнуто лечению путем гиперстимуляции ферментной активности POLG через посредство введения дезоксирибонуклеозидов: усиление активности POLG приводит к существенному повышению уровней мтДНК, которое может "нейтрализовать" потерю мтДНК независимо от причины такой потери (частная мутация в конкретном белке) и восстановить "нормальный" уровень.
Вследствие этого, стратегия лечения, основанная на dNs, могла бы частично или полностью противодействовать истощению мтДНК при любом дефекте, при котором возникают проблемы с репликацией мтДНК, обеспечиваемые за счет частично или полностью действующей полимеразной активности.
На основании представленных ниже экспериментальных данных можно сделать вывод, что восстановление уровней митохондриальной ДНК может быть независимым от тяжести заболевания пациента, что придает изобретению большое терапевтическое значение.
Другими преимуществами, связанными с введением дезоксирибонуклеозидов при лечении объекта с MDDS по настоящему изобретению, являются стоимость (невысокая) и отсутствие специальных требований к их сохранению. Кроме того, канонические дезоксирибонуклеозиды являются природными соединениями, обычно присутствующими во всех живых организмах.
Краткое описание графических материалов
На Фиг. 1 изображена аминокислота белка POLG1 в соответствии с базой данных NCBI (англ. National Center for Biotechnology Information - Национальный центр биотехнологической информации). Подчеркнуты следующие мутации: в позиции 309 - аргинин на цистеин, в позиции 748 - триптофан на серии, в позиции 848 -глицин на серии, в позиции 1143 - глутаминовая кислота на глицин, в позиции 1177 - валин на лейцин.
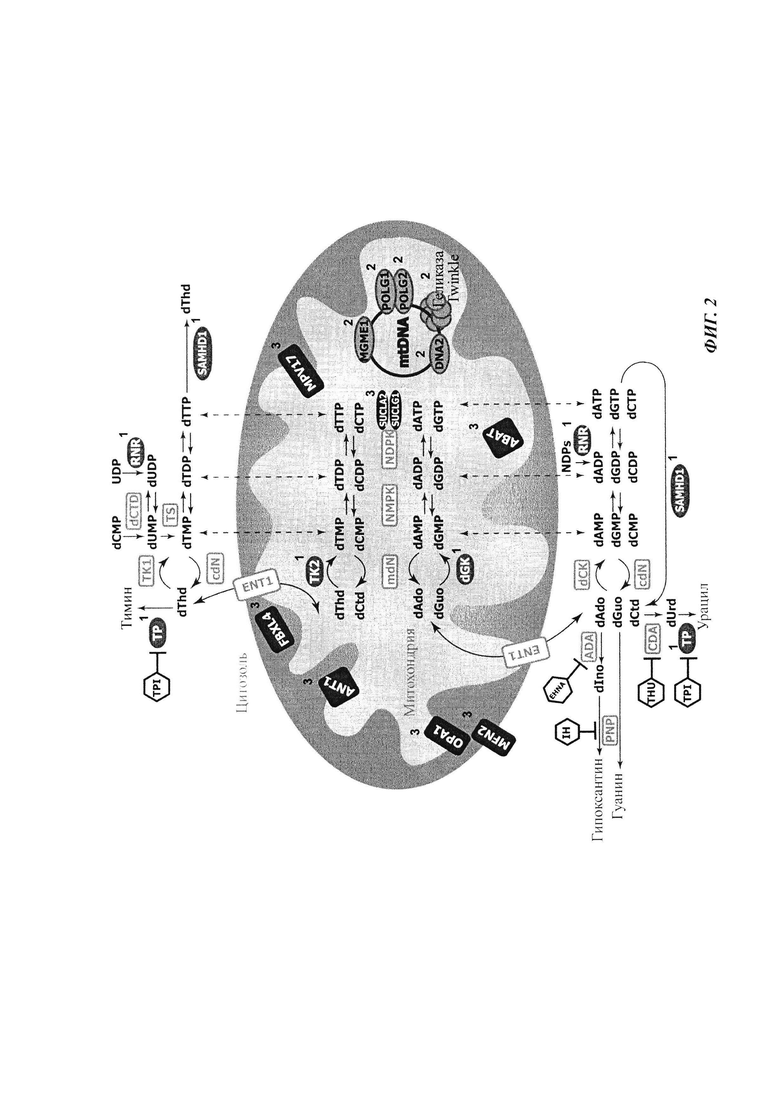
На Фиг. 2 представлены пути метаболизма, задействованные в синдромах истощения и делеции мтДНК (англ. MDDS). Белки, дисфункция которых связана с MDDS, помечены номером 1 (участвовавшие в метаболизме дНТФ), номером 2 (принадлежащие к репликативному комплексу) или номером 3 (связанные с MDDS через посредство неизвестного патомеханизма). Хотя была зарегистрирована связь генов SUCLA2 и SUCLG1 с ферментом нуклеотиддифосфаткиназой, их связь с метаболизмом дНТФ не была четко подтверждена. Изображены также другие белки, участвующие в метаболизме дНТФ, но которые пока еще не соотносили с MDDS, и специфические ингибиторы ферментов катаболизма дезоксирибонуклеозидов: тетрагидроуридин (THU); гидрохлорид 5-хлор-6-[1-(2-иминопирролидинил)метил]урацила (англ. TPI), иммуцилин Н (IH) и эритро-9-(2-гидрокси-3-нонил)аденин (EHNA). Сокращения: АВАТ: 4-аминобутиратаминотрансфераза; ADA: аденозиндеаминаза; ANT1: адениннуклеотидтранслоказа 1; CDA: цитидиндеаминаза; cdN: цитозольная дезоксирибонуклеотидаза; dAdo: дезоксиаденозин; dCK: дезоксицитидинкиназа; dCTD: деаминаза dCMP; dCtd: дезоксицитидин; dGK: дезоксигуанозинкиназа; dGuo: дезоксигуанозин; dlno: дезоксиинозин; ДНК2: ДНК-геликаза 2; dThd: тимидин; dUrd: дезоксиуридин; ENT1: равновесный нуклеозидный транспортер 1; FBXL4: белок 4, содержащий F-бокс и богатые лейцином повторы; mdN: митохондриальная дезоксирибонуклеотидаза; MFN2: митофузин-2; MGME1: экзонуклеаза 1, обеспечивающая сохранность митохондриального генома; MPV17: митохондриальная внутренняя мембрана MPV17; NDPK: нуклеотиддифосфаткиназа; NMPK: нуклеотидмонофосфаткиназа; ОРА1: атрофия 1 зрительного нерва; PNP: пуриннуклеозидфосфорилаза; POLG1: субъединица 1 полимеразы гамма; POLG2: субъединица 2 полимеразы гамма; RNR: рибонуклеотидредуктаза; SAMHD1: SAM-домен и HD-доменсодержащий белок 1; SUCLA2: бета-субъединица, сукцинат-СоА-лигаза; SUCLG1: а-субъединица, сукцинат-СоА-лигаза; ТК1: тимидинкиназа 1; ТК2: тимидинкиназа 2; TP: тимидинфосфорилаза; TS: тимидилатсинтаза; Twinkle: митохондриальная геликаза Twinkle.
Фиг. 3 представляет собой схему экспериментального исследования для определения уровней мтДНК в клетках, собранных в определенные моменты времени (t0, t7, t14, t21 и t26). Стабильность dNs контролировали в клеточных средах после 2-3 дней культивирования (t16-t17).
Подробное описание изобретения
Настоящее изобретение основано на обнаружении того факта, что восстановление уровней мтДНК может быть достигнуто при введении дезоксирибонуклеозидов пациентам, страдающим MDDS, вызванными дефектом, отличным от дефекта метаболизма дНТФ. Нуклеозиды представляют собой гликозиламины, которые можно рассматривать как нуклеотиды без фосфатной группы. Нуклеозид состоит только из нуклеинового основания (также называемого азотистым основанием) и 5-углеродного сахара (рибозы или дезоксирибозы), тогда как нуклеотид состоит из нуклеинового основания, пятиуглеродного сахара и одной или более фосфатной группы. В нуклеозиде основание связано с рибозой или дезоксирибозой посредством бета-гликозидной связи. Примеры нуклеозидов включают цитидин, уридин, аденозин, гуанозин, тимидин и инозин.
Как было пояснено выше, MDDS заболевания представляют собой группу заболеваний, обусловленных дефектами в репликации митохондриальнх ДНК (мтДНК), и включают расстройства, характеризующиеся либо сокращением числа копий мтДНК (синдром истощения мтДНК), либо множественными делециями мтДНК (синдром делеции мтДНК). Они являются официально признанными категориями в каталоге орфанных (редких) заболеваний и в онлайн-каталоге фенетических маркеров у человека (Online Mendelian Inheritance in Man), представляющих собой справочные сайты для специалистов в области такого рода патологий.
Эта группа митохондриальных заболеваний составляет широко известную группу расстройств, обусловленных мутациями в определенной группе генов, а потому хорошо известна специалистам в области митохондриальных расстройств. В некоторых случаях название, под которым известна эта группа заболеваний, может меняться: синдромы истощения и делеций мтДНК [3, 4]; дефекты межгеномной коммуникации (или сигнализации) [5]; дефекты репликации мтДНК [6]; и т.д. Два или более таких названий иногда сосуществуют в одной и той же публикации.
Заболевания, вызываемые дефектами репликации мтДНК, могут быть обусловлены мутациями в:
- А-генах, кодирующих белки, относящиеся к репликативному комплексу мтДНК (обозначены номером "2" на Фиг. 2) [7, 8, 9, 10, 11];
- В-генах, кодирующих белки, принимающие участие в катаболизме или анаболизме нуклеозидов/нуклеотидов (обозначены номером "1" на Фиг. 2) [12, 13, 14, 15];
- С-генах, кодирующих белки с неизвестной функцией, или функция которых не принадлежит к категориям А или В и не может быть биохимически связана с процессом репликации мтДНК (обозначены номером "3" на Фиг. 2) [16, 17, 18, 19, 20, 21, 22, 23, 24].
Эта классификация очевидным образом признана специалистами в данной области техники [3,4,6,25,26,27].
дНТФ необходимы для репликативной вилки в качестве субстратов для синтеза ДНК. Клетки млекопитающих получают предшественники синтеза и восстановления ДНК из двух разных метаболических источников: первичного цитозольного этапа синтеза и «реутилизационного» пути (пути утилизации отходов метаболизма), причем последний основан на двух параллельных наборах ферментов, расположенных в цитоплазме и митохондриальном матриксе. Синтез дНТФ связан с репликацией ядерной ДНК, момент, когда требования клеток к предшественникам ДНК являются наивысшими. Пул дНТФ заметно уменьшается после завершения фазы репликации (S фазы) или в неделящихся клетках. Первичный цитозольный этап синтеза основан на цитозольной активности рибонуклеотидредуктазы (RNR) (за исключением dTMP, для которого недавно была идентифицирована митохондриальная тимидилатсинтаза). RNR катализирует аллостерически сбалансированное восстановление всех четырех рибонуклеозиддифосфатов в соответствующие дезоксирибонуклеозиды (dNs), обеспечивая клетку высокими концентрациями дНТФ во время S-фазы клеточного цикла. RNR представляет собой гетеротетрамер, содержащий две копии большой субъединицы (R1) и две небольшие субъединицы (R2 или p53R2). В то время как R2 подвергается протеасомной деградации в позднем митозе, p53R2 присутствует на протяжении всего клеточного цикла, и его экспрессия сохраняется стабильной также и в неделящихся клетках. В отличие от репликации ядерного генома, синтез мтДНК протекает независимо от клеточного деления. Хотя первичный цитозольный этап синтеза, поддерживаемый p53R2, является значительно более коротким по сравнению с обеспечиваемым с помощью R2 во время S-фазы, было доказано, что он является необходимым для поддержания мтДНК, поскольку мутации гена p53R2 приводят к истощению мтДНК.
Реутилизационный путь основан на последовательном фосфорилировании нуклеозидов-предшественников с образованием дНМФ, dNDP и, в конечном итоге, дНТФ. Первая и лимитирующая стадия этого пути необратимо катализируется дезоксинуклеозидкиназами. Тимидинкиназа 1 (ТК1) и дезоксицитидинкиназа (dCK) действуют в цитозоли, тогда как тимидинкиназа 2 (ТК2) и дезоксигуанозинкиназа (dGK) локализуются внутри митохондрии. Мутации в митохондриальных киназах, участвующих в реутилизационном пути, вызывают тяжелые MDDS, что свидетельствует о зависимости митохондрий от использования отходов метаболизма дНТФ для поддержания сохранности и восстановления ДНК. Дополнительные нуклеотидкиназы завершают фосфорилирование всех четырех дНМФ до соответствующих конечных дНТФ, необходимых для синтеза ДНК. Цитозольный и митохондриальный матрикс являются независимыми компартментами, однако они активно контактируют, двунаправленно обмениваясь компонентами пула через внутреннюю митохондриальную мембрану посредством носителей, которые на сегодняшний день еще плохо охарактеризованы. Считается, что в результате такого перекрестного обмена изменения размеров пула дНТФ протекают параллельно в обоих компартментах. Вследствие этого митохондрии становятся более уязвимыми к дефектам на их собственном пути реутилизации дНТФ в пост-миотических клетках, где цитозольный пул в значительной степени уменьшается.
Размер пула дНТФ зависит от баланса между анаболическими путями, упоминавшимися ранее, скоростью включения в ДНК и катаболическими процессами, ответственными за деградацию дНТФ.
дНТФ требуются в репликативной вилке в качестве субстратов, которые должны быть включены в ДНК с помощью митохондриальной полимеразы гамма (POLG). Хотя точный режим репликации мтДНК в настоящее время обсуждается, основная митохондриальная реплисома, образующаяся с помощью полимеразы гамма (состоящей из каталитической субъединицы, кодируемой геном POLG1, и двух вспомогательных субъединиц, кодируемых геном POLG2), геликазы Twinkle и белка, связывающего одноцепочечную ДНК (SSB-белка), была восстановлена in vitro [28]. Для полной репликации in vivo молекулы мтДНК (например, праймазы, топоизомеразы) и для инициирования и регуляции процесса в ответ на различные раздражающие воздействия и стресс требуются дополнительные полностью неописанные действия. Некоторыми примерами являются гены MGME1 и ДНК2, кодирующие белки, участвующие на определенном уровне в процессе репликации (созревание 7S РНК, активность геликазы, соответственно), мутации которых недавно соотнесли с MDDS.
Согласно одному из вариантов осуществления, лечение синдрома достигается за счет увеличения активности полимеразы гамма.
Согласно одному из вариантов осуществления, один или более дезоксирибонуклеозиды представляют собой канонические дезоксирибонуклеозиды. Предпочтительно, при использовании таких дезоксирибонуклеозидов начало эффекта может быть получено раньше, поскольку не требуется дополнительной обработки метаболита. Кроме того, поскольку канонические дезоксирибонуклеозиды являются по существу идентичными эндогенным дезоксирибонуклеозидам, возможно снижение риска возникновения побочных эффектов, связанных с лечением данного заболевания.
Согласно одному из вариантов осуществления первого аспекта изобретения, синдром обусловлен дефектом репликативного комплекса митохондриальной ДНК.
Согласно другому варианту осуществления первого аспекта изобретения, дефект обусловлен одной или несколькими мутациями в одном или нескольких белках репликативного комплекса митохондриальной ДНК.
Согласно еще одному варианту осуществления, белок выбирают из группы, состоящей из: субъединицы ДНК-полимеразы гамма-1 (POLG1), субъединицы ДНК-полимеразы гамма-2 (POLG2), белка Twinkle (РЕO1), экзонуклеазы 1, поддерживающей сохранность митохондриального генома (MGME1) и белка - ДНК2 геликазы/нуклеазы человека. Согласно другому варианту осуществления, белок представляет собой субъединицу ДНК-полимеразы гамма-1 (POLG1) или белок Twinkle.
Полимераза гамма представляет собой гетеротример, состоящий из одной каталитической субъединицы (кодируемой геном POLG1) и двух вспомогательных субъединиц, действующих в качестве факторов процессивности и модуляторов связывания ДНК (кодируемых геном POLG2). Ее номер доступа в базе данных NCBI - NP_001119603.1 (также представлен как Фиг. 1 и SEQ ID NO: 1).
Расстройства, связанные с POLG, представляют собой целый диапазон широких и перекрывающихся фенотипов, охватывающих возраст от раннего детства до пожилого возраста. Клинические фенотипы расстройств, связанных с POLG, включают в себя аутусомно-рецессивную и аутосомно-доминантную формы приобретенной прогрессирующей наружной офтальмоплегии (РЕО), синдром миоклонической эпилепсии, миопатии, сенсорной атаксии (MEMSA), спектр атаксии-нейропатии, включающий синдром митохондриальной рецессивной атаксии (MIRAS), и синдром сенсорной атаксии, нейропатии, дизартрии, офтальмоплегии (SANDO), а также гепатоцеребральный MOS (синдром Альперса-Гутенлохера). Совсем недавно мутации POLG были идентифицированы у лиц с клиническими признаками MNGIE, но без лейкоэнцефалопатии.
Распространенность синдрома Альперса-Гутенлохера оценивается приблизительно в 1:50000. Это наиболее тяжелый фенотип, связанный с мутациями POLG и характеризующийся прогрессирующей энцефалопатией с инкурабельной эпилепсией и психомоторными нарушениями, нейропатией и печеночной недостаточностью. Данным заболеванием обычно страдают лица в возрасте от 2 до 4 лет, заболевание сопровождается эпилептическими припадками (фокальными, генерализованными, миоклоническими, парциальными непрерывными эпилепсиями или эпилептическими статусами), головными болями, обычно сопровождающимися зрительными ощущениями или зрительными аурами, гипотонией и психомоторной регрессией. На ранних стадиях заболевания присутствуют арефлексия и гипотония, позже присоединяется спастический парапарез, который развивается в течение от нескольких месяцев до нескольких лет, приводя в конечном итоге к психомоторной регрессии. У страдающих этим заболеванием лиц развивается дисфункция печени с повышенными трансаминазами, гипоальбуминемия, коагулопатия, гипогликемия и гипераммониемия. Поражения печени могут развиваться быстро до конечной стадии печеночной недостаточности в течение нескольких месяцев. Белок CSF обычно повышен. Нейровизуализация может показать глиоз и генерализованную атрофию мозга. Гистологические исследования печени могут продемонстрировать макро- и микровезикулярный стеатоз, центрилобулярный некроз, фиброз, цирроз, пролиферацию желчевыводящих путей и митохондриальную пролиферацию. Содержание мтДНК в печени снижено. Прогрессирование болезни варьируется, а ожидаемая продолжительность жизни от начала симптомов колеблется от 3 месяцев до 12 лет.
Согласно одному из вариантов осуществления, синдром истощения и/или делеции митохондриальной ДНК вызван дефектом митохондриального пути репликации, при этом указанный дефект обусловлен одной или более из следующих мутаций в белке POLG1: мутация в позиции 309 с заменой аргинина на цистеин [29], мутация в позиции 748 с заменой остатка триптофана на серии (rs113994097), мутация в позиции 848 с заменой глицина на серии (rs113994098), мутация в позиции 1143 с заменой глутаминовой кислоты на глицин (rs2307441) и мутация в позиции 1177 с заменой валина на лейцин.
В качестве альтернативы, согласно другому варианту осуществления первого аспекта изобретения, дефект обусловлен одной или более мутациями в одном или более белке, выбранном из группы, состоящей из: ANT1, MPV17, SUCLA2, FBXL4, АВАТ, SUCLG1, MFN2 и ОРА1. Согласно другому варианту осуществления первого аспекта, дефект обусловлен одной или более мутациями в белке, выбранном из группы, состоящей из: ОРА1, SUCLA2 и SUCLG1.
Согласно другому варианту осуществления, один или более дезоксирибонуклеозиды представляют собой канонические дезоксирибонуклеозиды. Согласно другому варианту осуществления, один или более дезоксирибонуклеозиды выбраны из группы, состоящей из: дезоксиаденозина, дезоксигуанозина, дезоксицитидина и дезокситимидина. Согласно еще одному варианту осуществления, композиция содержит четыре канонических дезоксирибонуклеозида. Согласно еще одному варианту осуществления, композиция содержит четыре канонических дезоксирибонуклеозида и не содержит каких-либо дополнительных нуклеозидов (это означает, что композиция в качестве "дезоксирибонуклеозидного компонента" содержит только эти четыре дезоксирибонуклеозида, но может также включать в себя наполнители, носители и т.д.) Согласно еще одному варианту осуществления, композиция содержит дезоксиаденозин, дезокситимидин, дезоксицитидин и дезоксигуанозин. Согласно еще одному варианту осуществления, композиция содержит дезоксиаденозин, дезокситимидин, дезоксицитидин и дезоксигуанозин и не содержит каких-либо дополнительных нуклеозидов (это означает, что композиция в качестве "нуклеозидного компонента" содержит только эти четыре нуклеозида, но может также включать в себя наполнители, носители и т.д.).
Специалист в данной области техники способен определить количество каждого из дезоксирибонуклеозидов, необходимое для восстановления уровней мтДНК (то есть терапевтически эффективное количество для улучшения проявлений и симптомов митохондриальных расстройств). Согласно другому варианту осуществления, когда композиция содержит более одного канонического дезоксирибонуклеозида, нуклеозиды присутствуют в эквимолярном соотношении.
Согласно одному из вариантов осуществления, композиция содержит комбинацию, состоящую из дезоксиаденозина (dAdo), дезоксицитидина (dCtd), дезоксигуанозин (dGuo) и дезокситимидина (обычно называемого тимидином, dThd), при этом все нуклеозиды присутствуют в эквимолярном соотношении.
Согласно другому варианту осуществления, композиция также содержит один или более фармацевтически приемлемый ингибитор деградации нуклеозидов.
Ингибиторы деградации нуклеозидов хорошо известны из современного уровня техники. Иллюстративными неограничивающими примерами являются: иммуцилин (Immucilin) Н или фородезин (Forodesine) в качестве ингибиторов деградации dGuo, тетрагидроуридин в качестве ингибитора деградации dCtd, гидрохлорид 5-хлор-6-[1-(2-иминопирролидинил)метил]урацила (TPI) в качестве ингибитора деградации dThd и эритро-9-(2-гидрокси-3- нонил)аденин (EHNA) в качестве ингибитора деградации dAdo. На Фиг. 1 показан механизм действия этих ингибиторов.
Специалист в данной области техники способен определить количество ингибитора (ингибиторов), необходимое для того, чтобы обеспечить биодоступность нуклеозида (нуклеозидов) для восстановления уровней мтДНК.
Согласно одному из вариантов осуществления, композиция также содержит один фармацевтически приемлемый ингибитор деградации нуклеозидов. Согласно другому варианту осуществления, фармацевтически приемлемый ингибитор представляет собой ингибитор деградации дезоксиаденозина. Согласно другому варианту осуществления, ингибитор представляет собой эритро-9-(2-гидрокси-3-нонил)аденин (EHNA).
Активные компоненты, описанные для применения в данном контексте, могут быть объединены с фармацевтически приемлемыми вспомогательными веществами или носителями, выбранными таким образом, чтобы сделать такие композиции пригодными для доставки пероральным, ректальным, парентеральным (например, внутривенно, внутримышечно, внутриартериально, внутрибрюшинно и тому подобное) или ингаляционным способами, посредством осмотического насоса, местно, офтальмологически и т.д.
Мази представляют собой полутвердые препараты, состоящие из активного ингредиента, включенного в жировую, восковую или синтетическую основу.
Примеры подходящих кремов включают, не ограничиваясь перечнем, эмульсии типа "вода в масле" и "масло в воде". Крема типа "вода в масле" могут быть приготовлены при использовании подходящего эмульгатора со свойствами, сходными, но не ограничивающимися свойствами жирных спиртов, таких как цетиловый спирт или цетостеариловый спирт, и свойствами эмульгирующего воска. Крема типа "масло в воде" могут быть приготовлены с использованием эмульгатора, такого как эмульгирующий воск с цетомакроголем. Подходящие свойства включают способность изменять вязкость эмульсии, а также физическую и химическую стабильность в широком диапазоне величин рН. Растворимая в воде или смешивающаяся с водой основа для крема может содержать смесь консервантов, а также может содержать буфер для поддержания приемлемой физиологической величины рН.
Помимо местного способа введения, описанного выше, существуют различные способы введения соединений по настоящему изобретению системно. Одно из таких средств будет включать аэрозольную суспензию вдыхаемых частиц, состоящих из активного соединения, которую субъект вдыхает. Активное соединение будет сорбироваться кровотоком через легкие и контактировать с общим кровообращением в фармацевтически эффективном количестве. Вдыхаемые частицы могут быть жидкими или твердыми, с размером частиц, достаточно малым, чтобы проходить через ротовую полость и гортань при вдыхании.
Другим средством системного введения активных соединений субъекту будет введение жидкости/жидкой суспензии в форме назальных капель жидкого препарата или в виде назального спрея вдыхаемых частиц, которые пациент вдыхает. Жидкие фармацевтические композиции активного соединения для приготовления назального спрея или назальных капель могут быть приготовлены путем объединения активного соединения с подходящим носителем, таким как стерильная апирогенная вода или стерильный физиологический раствор, с помощью способов, известных специалистам в данной области.
Другие способы системного введения активного соединения будут включать пероральное введение, при котором фармацевтические композиции, содержащие соединения Формулы I, имеют форму твердого вещества, раствора, эмульсии, дисперсии, мицеллы, липосомы и тому подобного, где конечный препарат содержит активные соединения, предполагаемые для использования в данном контексте, в смеси с органическим или неорганическим носителем или вспомогательным веществом, подходящим для назальных, энтеральных или парентеральных применений. Активные ингредиенты могут быть смешаны, например, с обычными нетоксичными, фармацевтически или физиологически приемлемыми носителями для таблеток, пеллет, капсул, лепешек, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, суппозиториев, растворов, эмульсий, суспензий, твердых или мягких капсул, капсуловидных таблеток либо сиропов или эликсиров и любых других форм, подходящих для применения. Носители, которые могут быть использованы, включают аравийскую камедь, желатин, маннит, крахмальную пасту, трисиликат магния, тальк, кукурузный крахмал, кератин, коллоидный диоксид кремния, картофельный крахмал, мочевину, среднецепочечные триглицериды, декстраны и другие носители, пригодные для использования при приготовлении препаратов в твердой, полутвердой или жидкой форме. Кроме того, могут использоваться вспомогательные, стабилизирующие, загущающие агенты и красители. Активные соединения, предполагаемые для использования в данном контексте, включены в состав фармацевтического препарата в количестве, достаточном для достижения требуемого эффекта при введении (то есть, в терапевтически эффективном количестве).
Порошок, раствор, суспензия или таблетка содержат активное соединение в физиологически совместимом носителе, который специалисты в области разработки систем пероральной доставки могут выбрать на основании общепринятых критериев. Например, такие препараты могут содержать один или более агент, выбранный из вкусоароматических добавок (таких как мята перечная, винтергреневое масло или вишневое масло), красителей, консервантов и тому подобного, чтобы обеспечить фармацевтически элегантные и вкусные препараты. Таблетки, содержащие активные ингредиенты в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, также могут быть изготовлены известными способами. Используемыми вспомогательными веществами могут быть, например, (1) инертные разбавители, такие как карбонат кальция, лактоза, фосфат кальция, фосфат натрия и тому подобное; (2) гранулирующие средства и вещества для улучшения распадаемости, такие как кукурузный крахмал, картофельный крахмал, альгиновая кислота и тому подобное; (3) связующие агенты, такие как трагакантовая камедь, кукурузный крахмал, желатин, аравийская камедь и тому подобное; и (4) смазывающие вещества, такие как стеарат магния, стеариновая кислота, тальк и тому подобное. Таблетки могут быть непокрытыми, либо они могут иметь покрытие, нанесенное с помощью известных способов, для задержки распада и абсорбции в желудочно-кишечном тракте, обеспечивающее тем самым пролонгированное действие в течение более длительного периода. Например, можно использовать материал с отсроченным высвобождением, такой как глицерилмоностеарат или глицерилдистеарат.
Если препараты для перорального применения имеют форму твердых желатиновых капсул, активные ингредиенты в них могут быть смешаны с твердым инертным разбавителем, например, карбонатом кальция, фосфатом кальция, каолином или тому подобным. Препараты также могут иметь форму мягких желатиновых капсул, в которых активные ингредиенты смешаны с водной или масляной средой, например, арахисовым маслом, жидким парафином, оливковым маслом и тому подобным.
Дополнительные средства для системного введения активного соединения субъекту могут включать суппозиторную форму активного соединения, такую чтобы терапевтически эффективное количество соединения достигало большого круга кровообращения.
В зависимости от растворимости конкретного вводимого препарата, содержащего активное соединение, суточная доза для улучшения проявлений и симптомов митохондриальных расстройств может быть разделена на прием одной или нескольких разовых доз.
В описании и формуле изобретения слово "содержит" и различные формы этого слова не предназначены для исключения других технических особенностей, добавок, компонентов или стадий. Кроме того, слово "содержит" включает в себя также и вариант «состоящий из». Дополнительные объекты, преимущества и признаки изобретения станут очевидными для специалистов в данной области техники по рассмотрению описания или могут быть изучены путем осуществления изобретения на практике. Следующие примеры приведены в качестве иллюстрации и не предназначены для ограничения настоящего изобретения. Кроме того, настоящее изобретение охватывает все возможные комбинации конкретных и предпочтительных вариантов осуществления, описанных в данном контексте.
Описание примеров осуществления изобретения
1. Методики
Пациенты
В исследование были включены клетки, полученные от трех пациентов, страдающих дефицитом POLG. Все субъекты дали информационное согласие в соответствии с внутренним Институциональным наблюдательным советом и Хельсинкской декларацией. У всех 3 пациентов с помощью секвенирования по Сэнгеру были выявлены мутации в POLG (RefSeq NP_001119603.1). Общую ДНК выделяли из фибробластов с помощью QiaAMP Mini (Qiagen), а фрагменты длиной приблизительно 500 bp, включающие исследуемый сайт мутации, амплифицировали с помощью обычной ПЦР (англ. PCR, polymerase chain reaction - полимеразная цепная реакция) со следующими праймерными парами, перечисленными ниже, и rTaq (Takara).
Были использованы следующие праймеры:
Праймеры 1 (R309C, 925 c->t)
Прямой праймер GTCCACACCACCAAGCAGT (SEQ ID NO: 2)
Обратный праймер GGTCCCAAGCACTATGCTCC(SEQ ID NO: 3)
Праймеры 2 (W748S с. 2243G->C)
Прямой праймер CCTTGCTGAA TGCAGGTGCT (SEQ ID NO: 4)
Обратный праймер TGTGCCTGAAATCACACTCTGT (SEQ ID NO:5)
Праймеры 3 (G848S с. 2542G->A)
Прямой праймер ATGGTCTGCTGAGTGGTTGT (SEQ ID NO: 6)
Обратный праймер CCCTCAGAGCCCAGTTTCTAC (SEQ IDNO: 7)
Праймеры 3 (E1143G с. 3428A->G)
Прямой праймер CCCAGTTTATGACCAGCCGT (SEQ ID NO: 8)
Обратный праймер CAAGGAACGCTCACCCAAAG(SEQ ID NO: 9)
Праймеры 4 (V1177L c.3529G->C)
Прямой праймер AGGGGAAGCCCTGCTCTAAG (SEQ ID NO: 10)
Обратный праймер ACAAATGTGTTGTGCTCACCC (SEQ ID NO: 11)
Реакции секвенирования проводили с этими же праймерами и набором для секвенирования BigDye v3.1 (Life Technologies), очищали с помощью набора для очистки BigDye X-Terminator (Life Technologies) и секвенировали в секвенаторе ABI 3130 (Applied Biosystems). Пациент 1 (гомозиготный по мутации P.R309C) страдал тяжелым неврологическим фенотипом (нейропатия, энцефалопатия, MNGIE (англ. mitochondrial neurogastrointestinal encephalopathy syndrome - синдром митохондриальной нейрогастроинтестинальной энцефалопатии)), который приводит к смерти в возрасте 20 лет; пациент 2 (сложная гетерозигота по мутациям p.W748S и p.G848S) страдал менее выраженным фенотипом, но также преимущественно неврологическим (нейропатия, психиатрические симптомы, MNGIE); у пациента 3 (гетерозиготный по двум доминантным аминокислотным заменам в цис р.V1177L и р.Е1143G) был выявлен семейный паттерн доминантного наследования РЕО (прогрессивной внешней офтальмоплегии), психиатрические симптомы и слабость проксимальных мышц. Скелетные мышцы свидетельствовали о накоплении делеций мтДНК, но не показали какого-либо значительного истощения.
Культура клеток
Первичные культивируемые фибробласты были получены из биопсии кожи пациентов 1-3 и 4 здоровых доноров. Все субъекты дали информационное согласие в соответствии с внутренним Институциональным наблюдательным советом и Хельсинкской декларацией.
Клетки высевали в 6-луночные планшеты, 9,5 см2, в среде Игла, модифицированной Дульбекко (DMEM), с добавлением 4,5 г/л глюкозы, дополненной 2 мМ L-глутамина, 100 ед./мл пенициллина и стрептомицина и 10% диализированной фетальной бычьей сыворотки (FBS) (Invitrogen), во влажной камере при температуре 37°С и 5% СO2. После того как было достигнуто плотное слияние, FBS уменьшали до 0,1%, чтобы индуцировать состояние покоя, и начинали одновременную обработку 5 нг/мл EtBr (бромид этидия, Merck) (день 0, t0). Клеточную среду заменяли при помощи указанной выше обработки каждые 2-3 дня в течение двух недель. На 14-й день (t14) EtBr удаляли из клеточной среды и начинали обработку набора всех клеток с помощью 200 мкМ всех четырех дезоксинуклеозидов (dNs): dAdo (дезоксиаденозин, Sigma), dCtd (дезоксицитидин, Sigma), dGuo (дезоксигуанозин, Sigma), dThd (дезокситимидин, Sigma) и 5 мкМ EHNA (Sigma). Ряд клеток оставляли необработанными и параллельно контролировали все их параметры. Клеточную среду заменяли с помощью такой же обработки каждые 2-3 дня в течение еще 12 дней. На 16 или 17 день среды собирали и хранили при температуре -20°С до дальнейшего использования. Для анализа ДНК клетки собирали в 0, 7, 14, 21 и 26 день с помощью трипсинизации, промывали фосфатно-солевым буферным раствором, осаждали центрифугированием и хранили при температуре -20°С до выделения ДНК (Фиг. 2). Общую ДНК выделяли с помощью набора реагентов для выделения ДНК QiaAMP DNA mini kit (Qiagen).
Оценка стабильности дезоксинуклеозидов в среде для куьтивирования клеток dNs и некоторые родственные метаболиты анализировали методом жидкостной хроматографии стандемной масс-спектрометрией (ЖХ/МС/МС, англ. LC-MS/MS) на приборе Acquity UPLC-MS/MS (масс-спектрометр Acquity UPLC-Xevo ТМ TQ, Waters, Милфорд, Массачусетс), как описано ранее [2]. Клеточную среду депротеинизировали ультрафильтрацией (ультрафильтр миллипоровый Amicon Ultra, 3 кДа) при 14000 × g и температуре 4°С в течение 30 минут перед инъекцией в систему ЖХ/МС/МС.
Исследования мтДНК
Число копий мтДНК оценивали с помощью количественной ПЦР, как было описано ранее [2].
Делеции мтДНК исследовали с помощью ПЦР длинных фрагментов в соответствии с условиями ПЦР и праймерами, раскрытыми в работе Nishigaki et al. [30],:
Прямой праймер (F1142-1516):
ACCGCCCGTCACCCTCCTCAAGTATACTTCAAAGG (SEQ ID NO: 12)
Обратный праймер (R1180-1146):
ACCGCCAGGTCCTTTGAGTTTTAAGCTGTGGCTCG (SEQ ID NO: 13) 2.
Результаты
Воздействие EtBr индуцирует большее истощение мтДНК в POLG-дефицитных покоящихся фибробластах
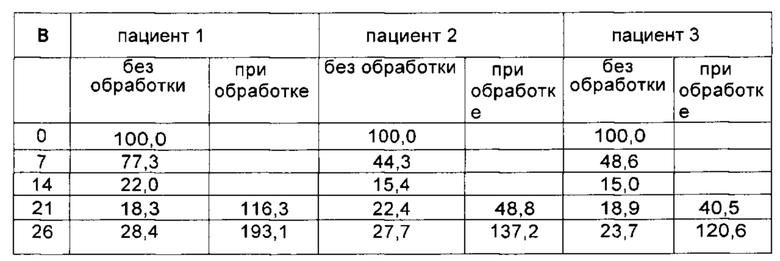
Важным ограничением при тестировании потенциальных методик лечения истощения мтДНК является тот факт, что во многих случаях клетки, полученные у пациента с MDDS, не обнаруживают каких-либо аномалий мтДНК в культуре клеток. По этой причине были разработаны различные модели для демонстрации молекулярных дефектов, затрагивающих репликацию мтДНК. На данном уровне техники для инициирования истощения мтДНК в культивируемых клетках и анализа их репликативной способности восстанавливать нормальные уровни мтДНК периодически используют воздействие EtBr (Pontarin et al. "Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells (Субъединица p53R2 рионуклеотидредуктазы млекопитающих необходима для репликации митохондриальной ДНК и репарации ДНК в покоящихся клетках)", 2012, PNAS, v. 109(33), р. 13302-13307). Истощение мтДНК индуцировали и в здоровых контрольных клетках, и в POLG-дефицитных покоящихся клетках путем воздействия в течение 14 дней 5 нг/мл EtBr. Было отмечено заметное истощение мтДНК во всех клетках. Однако все POLG-дефицитные клетки показали более высокую степень истощения мтДНК по сравнению с наблюдавшейся в линии фибробластов, полученных от контрольной группы здоровых людей (усредненный процент остаточных уровней мтДНК плюс/минус стандартная ошибка среднего (СОС) составил: 17,5 плюс/минус 2,3%; в контрольных клетках: 39,68 плюс/минус 6,6%). Эти данные свидетельствуют о том, что молекулярный дефект, обусловленный мутациями POLG, может усиливать вмешательство EtBr в процесс репликации.
PQLG-дефицитные фибробласты полностью восстанавливают уровни мтДНК после инициированного с помощью EtBr истощения при лечении комбинацией dNs плюс EHNA
После удаления EtBr изучали влияние, оказываемое добавлением среды для культивирования клеток, содержащей все четыре dNs, на восстановление уровней мтДНК. Ранее сообщалось о том, что dAdo особенно чувствителен к внеклеточному и внутриклеточному ферментному расщеплению, главным образом, с помощью ADA (аденозиндеаминазы) [2]. Вследствие этого в среду добавляли эксимолярную концентрацию всех четырех канонических dNs (по 200 мкМ dGuo, dAdo, dThd и dCtd) плюс 5 мкМ EHNA (Sigma), чтобы обеспечить частичное ингибирование катаболизма dAdo и улучшить его стабильность. На 16-17 день измеряли концентрацию добавленных dNs и некоторых их производных метаболитов в 2-3 суточных кондиционированных средах в соответствии с протоколом, раскрытым в работе Camara Y. et al., 2014 [2]. Несмотря на частичное разложение, концентрация всех dNs во всех случаях оставалась выше 70% от первоначально добавленной (Таблица 2). Никаких существенных различий в стабильности dNs между кондиционированными средами из контрольных и полученных у пациента клеток не наблюдалось.
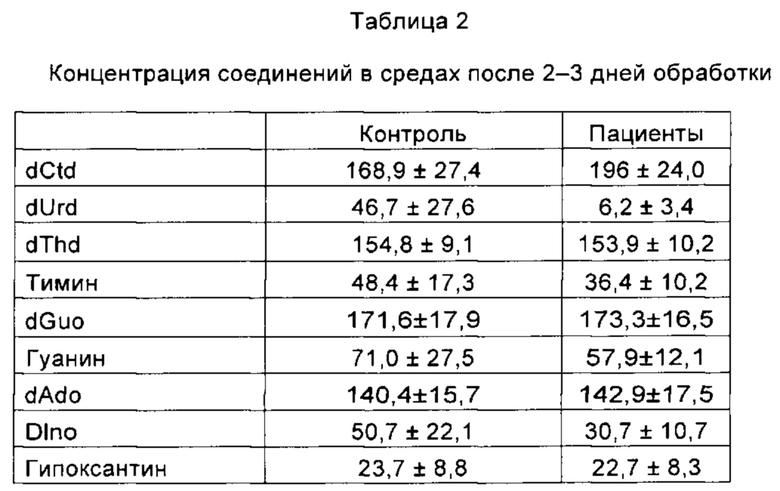
Результаты представляют собой среднее значение плюс/минус SD (англ. standard deviation - среднеквадратическое отклонение) для трех различных POLG-дефицитных и четырех контрольных клеточных линий. dUrd: дезоксиуридин; dlno: дезоксиинозин.
Восстановление мтДНК контролировали до и после удаления EtBr в присутствии или в отсутствие добавок dNs (в соответствии с тем же протоколом, что описан в работе Camara Y. et al., 2014 [2]). Число копий мтДНК от контрольных клеток достигало величин более 100% от начальных уровней через 12 дней после удаления EtBr независимо от обработки dNs (усредненный процент остаточных уровней мтДНК плюс/минус СОС: 129,8 плюс/минус 35,2% без обработки; 153,9 плюс/минус 40,6% при обработке).
Напротив, POLG-дефицитные клетки были не способны восстановить нормальные уровни мтДНК (26.6 плюс/минус 1.5%), не будучи дополненными dNs.
Величины выражены в процентах от числа копий мтДНК по отношению к величине в 0 день. После индуцированного с помощью EtBr истощения клетки обрабатывают или не обрабатывают dNs+EHNA с 14 дня эксперимента.
Результаты, представленные в Таблице 3, позволяют заключить, что введение канонических нуклеозидов позволяет достичь у пациентов с MDDS уровней мтДНК, сравнимых с таковыми у здоровых субъектов (150,3 плюс/минус 21,9%, Таблица 3). Таким образом, полученные данные свидетельствуют о том, что введение нуклеозидов может восстанавливать уровни мтДНК у пациентов, страдающих MDDS, обусловленными дефектом, отличным от дефекта метаболизма дНТФ, до уровней, соответствующих "здоровому" состоянию, что свидетельствует о терапевтическом потенциале комбинации при лечении такого рода заболеваний.
Список документов
1. Garone С, Garcia-Diaz В, Emmanuele V, Lopez LC, Tadesse S, et al. (2014) Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency (Обходная терапия дефицита тимидинкиназы 2 с использованием монофосфата дезоксипиримидина), EMBO Mol. Med., 6:1016-1027.
2. Camara Y, Gonzalez-Vioque E, Scarpelli M, Torres-Torronteras J, Caballero A, et al. (2014) Administration of deoxyribonucleosides or inhibition of their catabolism as a pharmacological approach for mitochondrial DNA depletion syndrome (Введение дезоксирибонуклеозидов или ингибирование их катаболизма в качестве фармакологического подхода к синдрому истощения митохондриальной ДНК), Hum. Mol. Genet., 23: 2459-2467.
3. Camara Y, Gonzalez-Vioque E, Scarpelli M, Torres-Torronteras J, Marti R (2013) Feeding the deoxyribonucleoside salvage pathway to rescue mitochondrial DNA (Питание для пути утилизации отходов метаболизма дезоксирибонуклеозида для восстановления митохондриальной ДНК), Drug Discov. Today, 18:950-957.
4. Suomalainen A, Isohanni P (2010) Mitochondrial DNA depletion syndromes -many genes, common mechanisms (Синдромы истощения митохондриальной ДНК - множество генов, общие механизмы) Neuromuscul Disord., 20:429-437.
5. Spinazzola A, Zeviani М (2005) Disorders of nuclear-mitochondrial intergenomic signaling (Расстройства ядерно-митохондриальной интергеномной сигнальной системы), Gene, 354:162-168.
6. Copeland WC (2012) Defects in mitochondrial DNA replication and human disease (Дефекты репликации митохондриальных ДНК и болезни человека), Grit. Rev. Biochem. Mol. Biol., 47:64-74.
7. Naviaux RK, Nyhan WL, Barshop BA, Poulton J, Markusic D, et al. (1999) Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers' syndrome (Дефицит митохондриальной ДНК-полимеразы гамма и истощение мтДНК у ребенка с синдромом Альперса), Ann. Neurol., 45: 54-58.
8. Hakonen АН, Isohanni Р, Paetau A, Herva R, Suomalainen A, et al. (2007) Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion (Рецессивные мутации Twinkle при раннем начале энцефалопатии с истощением мтДНК), Brain, 130:3032-3040.
9. Kornblum С, Nicholls TJ, Haack ТВ, Scholer S, Peeva V, et al. (2013) Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease (Мутации с потерей функции в MGME1 нарушают репликацию мтДНК и вызывают мультисистемное митохондриальное заболевание), Nat. Genet.
10. Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, et al. (2013) Mutations in DNA2 Link Progressive Myopathy to Mitochondrial DNA Instability (Мутации в ДНК2 связывают прогрессивную миопатию с нестабильностью митохондриальной ДНК), Am. J. Hum. Genet.
11. Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, et al. (2006) Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia (Мутации в POLG2 разрушают субъединицы ДНК-полимеразы гамма и приводят к прогрессивной внешней офтальмоплегии), Am. J. Hum. Genet., 78:1026-1034.
12. Mandel H, Szargel R, Labay V, Elpeleg 0, Saada A, et al. (2001) The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA (Ген дезоксигуанозинкиназы является мутированным у индивидуумов с истощением митохондриальной ДНК гепатоцеребральной формы), Nat. Genet., 29:337-341.
13. Saada A, Shaag A, Mandel Н, Nevo Y, Eriksson S, et al. (2001) Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy (Мутантная митохондриальная тимидинкиназа при миопатии с делециями митохондриальной ДНК), Nat. Genet., 29:342-344.
14. Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, et al. (2007) Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion (Мутация RRM2B, кодирующего р53-контролируемую рибонуклеотидредуктазу (p53R2), вызывает серьезное истощение митохондриальной ДНК), Nat. Genet., 39: 776-780.
15. Nishina I, Spinazzola A, Hirano M (1999) Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder (Мутации гена, кодирующего тимидинфосфорилазу, при MNGIE, митохондриальном расстройстве человека), Science, 283: 689-692.
16. Rouzier С, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, et al. (2012) The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy 'plus' phenotype (Ген MFN2 является ответственным за нестабильность митохондриальной ДНК и атрофию зрительного нерва "плюс" фенотипа), Brain, 135:23-34.
17. Renaldo F, Amati-Bonneau P, Slama A, Romana C, Forin V, et al. (2012) MFN2, a new gene responsible for mitochondrial DNA depletion (MFN2, новый ген, ответственный за истощение митохондриальной ДНК), Brain.
18. Ostergaard Е, Schwartz М, Batbayli М, Christensen Е, Hjalmarson O, et al. (2005) A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria (Новая миссенс-мутация в SUCLG1, связанная с истощением митохондриальной ДНК, энцефаломиопатическая форма, с метилмалоновой ацидурией), Eur. J. Pediatr., 169: 201-205.
19. Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, et al. (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes (Мутации OPA1 индуцируют нестабильность митохондриальной ДНК и атрофию зрительного нерва "плюс" фенотипов), Brain, 131: 338-351.
20. Blakely EL, Butterworth A, Hadden RD, Bodi I, He L, et al. (2012) MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle (MPV17 мутация вызывает нейропатию и лейкоэнцефалопатию с множественными делециями мтДНК в мышцах), Neuromuscul Disord., 22: 587-591.
21. Spinazzola A, Viscomi С, FePHKndez-Vizarra Е, Carrara F, D'Adamo P, et al. (2006) MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion (MPV17 кодирует внутренний белок митохондриальной мембраны и мутирует при истощение митохондриальной ДНК печени ребенка), Nat. Genet., 38: 570-575.
22. Wedding IM, Koht J, Tran GT, Misceo D, Selmer KK, et al. (2014) Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions (Спастическая параплегия 7 типа связана с множественными делециями митохондриальной ДНК), PLoS One 9:е86340.
23. Bonnen РЕ, Yarham JW, Besse A, Wu P, Faqeih EA, et al. (2013) Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance (Мутации в FBXL4 вызывают митохондриальную энцефалопатию и расстройство сохранности митохондриальной ДНК), Am. J. Hum. Genet., 93: 471-481.
24. Gai X, Ghezzi D, Johnson MA, Biagosch CA, Shamseldin HE, et al. (2013) Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy (Мутации в FBXL4, кодирующем митохондриальный белок, вызывают митохондриальную энцефаломиопатию с ранним началом), Am. J. Hum. Genet., 93: 482-495.
25. Nogueira С, Almeida LS, Nesti C, Pezzini I, Videira A, et al. (2014) Syndromes associated with mitochondrial DNA depletion (Синдромы, связанные с истощением митохондриальной ДНК), Ital. J. Pediatr., 40: 34.
26. Copeland WC (2008) Inherited mitochondrial diseases of DNA replication (Наследственные митохондриальные заболевания репликации ДНК), Annu. Rev. Med., 59: 131-146.
27. Copeland WC (2014) Defects of mitochondrial DNA replication (Десректы репликации митохондриальной ДНК), J. Child. Neurol, 29:1216-1224.
28. Korhonen JA, Pham XH, Pellegrini M, Falkenberg M (2004) Reconstitution of a minimal mtDNA replisome in vitro (Восстановление минимальной реплисомы мтДНК in vitro), Embo. J., 23: 2423-2429.
29. Amiot A, Tchikviladze M, Joly F, Slama A, Hatem DC, et al. (2009) Frequency of mitochondrial defects in patients with chronic intestinal pseudo-obstruction (Частота митохондриальных дефектов у пациентов с хронической кишечной псевдообструкцией), Gastroenterology, 137:101-109.
30. Nishigaki Y, Marti R, Hirano M (2004) ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy (ND5 является "горячей точкой" для множественных атипичных делеций митохондриальной ДНК при митохондриальной нейрогастроинтестинальной энцефаломиопатии), Hum. Mol. Genet., 13: 91-101.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕЧЕНИЕ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2834339C1 |
| ДЕЗОКСИНУКЛЕОЗИДНАЯ ТЕРАПИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ НЕСБАЛАНСИРОВАННЫМИ ПУЛАМИ НУКЛЕОТИДОВ, В ТОМ ЧИСЛЕ СИНДРОМОВ ИСТОЩЕНИЯ МИТОХОНДРИАЛЬНОЙ ДНК | 2016 |
|
RU2827432C2 |
| ДЕЗОКСИНУКЛЕОЗИДНАЯ ТЕРАПИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ НЕСБАЛАНСИРОВАННЫМИ ПУЛАМИ НУКЛЕОТИДОВ, В ТОМ ЧИСЛЕ СИНДРОМОВ ИСТОЩЕНИЯ МИТОХОНДРИАЛЬНОЙ ДНК | 2016 |
|
RU2721492C2 |
| Способ определения доли мтДНК с делециями в биологических образцах | 2018 |
|
RU2676897C1 |
| СПОСОБ КОРРЕКЦИИ МИТОХОНДРИАЛЬНОЙ ДИСФУНКЦИИ С ПОМОЩЬЮ ГЕНЕТИЧЕСКОЙ КОНСТРУКЦИИ | 2016 |
|
RU2642972C1 |
| Способ репликации человеческого митохондриального генома в клетках дрожжей Yarrowia lipolytica | 2016 |
|
RU2660715C2 |
| Способ измерения числа копий митохондриальной ДНК человека методом цифровой полимеразной цепной реакции | 2021 |
|
RU2755663C1 |
| Нуклеиновая кислота для аллотопической экспрессии гена MT-ND4 | 2023 |
|
RU2809065C1 |
| Пептид митохондриальной локализации, нуклеиновая кислота для аллотопической экспрессии гена MT-ND4, содержащий ее экспрессионный вектор и его применение | 2023 |
|
RU2817420C1 |
| СПОСОБ ДИАГНОСТИКИ ЗАБОЛЕВАНИЯ, СОПРОВОЖДАЮЩЕГОСЯ ПОВЫШЕННОЙ ГИБЕЛЬЮ КЛЕТОК, И НАБОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2016 |
|
RU2630648C2 |






Изобретение относится к применению композиции, содержащей дезоксиаденозин, дезоксигуанозин, дезоксицитидин, дезокситимидин и один или более фармацевтически приемлемых ингибиторов деградации нуклеозидов, для лечения синдрома истощения и/или делеции митохондриальной ДНК, обусловленного дефектом в субъединице ДНК-полимеразы гамма-1 (POLG-1). Изобретение обеспечивает восстановление уровней митохондриальной ДНК независимо от тяжести заболевания пациента, а следовательно, значительную терапевтическую ценность. 8 з.п. ф-лы, 1 пр., 3 табл., 3 ил.
1. Применение композиции, содержащей дезоксиаденозин, дезоксигуанозин, дезоксицитидин, дезокситимидин и один или более фармацевтически приемлемых ингибиторов деградации нуклеозидов, для лечения синдрома истощения и/или делеции митохондриальной ДНК, обусловленного дефектом в субъединице ДНК-полимеразы гамма-1 (POLG-1).
2. Применение композиции по п.1, где лечение синдрома достигается за счет увеличения активности полимеразы гамма.
3. Применение композиции по любому из пп. 1-2, где указанный дефект обусловлен одной или более из следующих мутаций в POLG1: R309C, W748S, V1177L, G848S и E1143G, указанные позиции относятся к SEQ ID NO: 1.
4. Применение композиции по п.1, где указанная композиция не содержит какого-либо другого нуклеозида.
5. Применение композиции по п.1, где фармацевтически приемлемый ингибитор является ингибитором деградации дезоксиаденозина.
6. Применение композиции по п.5, где ингибитором является эритро-9-(2-гидрокси-3-нонил)аденин (англ. EHNA).
7. Применение композиции по п.1, где дефект находится в экзонуклеазном домене POLG1.
8. Применение композиции по п.1, где дефект находится в полимеразном домене POLG1.
9. Применение композиции по п.1, где дефект находится в линкерной области POLG1.
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ | 1999 |
|
RU2279880C2 |
| US 5246708 A, 21.09.1993 | |||
| US 6417170 B2, 09.07.2002. | |||

