Область техники, к которой относится изобретение
Изобретение относится к катализатору окисления для дизельного двигателя и к выхлопной системе для дизельного двигателя, содержащей катализатор окисления. Изобретение также относится к способам и вариантам использования катализатора окисления для обработки выхлопного газа дизельного двигателя.
Уровень техники, предшествующий изобретению
Дизельные двигатели вырабатывают выхлопные газы, которые, как правило, содержат по меньшей мере четыре класса загрязнителей, которые запрещены в законодательном порядке межправительственными организациями по всему миру: угарный газ (CO), несгоревшие углеводороды (HC), оксиды азота (NOx) и твердые примеси (PM). Нормы выбросов для дизельных двигателей, как стационарных, так и мобильных (например, автотранспортных дизельных двигателей), становятся все более жесткими. Имеется потребность в обеспечении улучшенных катализаторов и выхлопных систем, которые могут соответствовать этим стандартам, и которые являются экономичными.
Для дизельных двигателей катализатор окисления (известный как дизельный катализатор окисления (DOC)), как правило, используется для обработки выхлопного газа, вырабатываемого такими двигателями. Дизельные катализаторы окисления, как правило, катализируют окисление (1) угарного газа (CO) до углекислого газа (CO2) и (2) HC до углекислого газа (CO2) и воды (H2O). Температуры выхлопного газа для дизельных двигателей, особенно для транспортных средств с дизельным двигателем малой мощности, являются относительно низкими (например, приблизительно 400°C) и, таким образом, одна из задач состоит в разработке каталитических составов с большим сроком службы, имеющих низкие «рабочие» температуры.
Активность катализаторов окисления, таких как DOC, часто определяется исходя из его «рабочей» температуры, которая представляет собой температуру, при которой катализатор начинает способствовать протеканию определенной каталитической реакции или способствует протеканию этой реакции в определенной степени. Обычно «рабочие» температуры указываются исходя из определенной степени превращения реагирующего вещества, например превращения угарного газа. Таким образом, температура T50 часто указывается в качестве «рабочей» температуры, потому что она представляет собой самую низкую температуру, при которой катализатор катализирует превращение реагирующего вещества на 50%.
Выхлопные системы для дизельных двигателей могут включать несколько устройств контроля токсичности выхлопных газов. Каждое устройство контроля токсичности выхлопных газов имеет специализированное назначение и отвечает за обработку одного или более классов загрязнителей в выхлопном газе. Эффективность функционирования расположенного выше по потоку устройства контроля токсичности выхлопных газов может влиять на эффективность функционирования расположенного ниже по потоку устройства контроля токсичности выхлопных газов. Это вызвано тем, что выходящий из расположенного выше по потоку устройства контроля токсичности выхлопных газов выхлопной газ передается на впуск расположенного ниже по потоку устройства контроля токсичности выхлопных газов. Взаимное влияние между всеми устройствами контроля токсичности выхлопных газов в выхлопной системе является важным для общей эффективности функционирования системы.
Катализаторы окисления, такие как дизельные катализаторы окисления (DOC), как правило, способствуют окислению угарного газа (CO) и углеводородов (HC) в выхлопном газе, выработанном дизельным двигателем. Дизельные катализаторы окисления могут также способствовать окислению части оксида азота (NO), который присутствует в выхлопном газе, до диоксида азота (NO2). Несмотря на то, что диоксид азота (NO2) сам по себе является загрязнителем, превращение NO в NO2 может быть выгодным. Полученный NO2 может использоваться для восстановления твердых примесей (PM), которые были задержаны, например, расположенным ниже по потоку фильтром твердых частиц выхлопа дизельных двигателей (DPF) или расположенным ниже по потоку каталитическим фильтром сажи (CSF). Как правило, NO2, образованный за счет катализатора окисления, увеличивает отношение NO2:NO в выхлопном газе на выпуске катализатора окисления по сравнению с выхлопным газом на впуске. Такое увеличенное отношение может быть выгодным для выхлопных систем, содержащих расположенный ниже по потоку катализатор селективного каталитического восстановления (SCR) или катализатор селективного каталитического восстановления на фильтрующей подложке(SCRF™). Отношение NO2:NO в выхлопном газе, вырабатываемом непосредственно дизельным двигателем, может быть слишком низким для оптимальной работы катализатора SCR или SCRF.
В то время как в большинстве случаев выгодно включать в выхлопную систему катализатор окисления, такой как DOC, который имеет хорошую активность в отношении образования NO2, использование катализатора окисления таким образом может быть проблематичным при попытках получить оптимальные рабочие характеристики для расположенного ниже по потоку устройства контроля токсичности выхлопных газов (например, катализатора SCR или SCRF™). Среднее количество NO2, которое образуется за счет катализатора окисления при заданной температуре выхлопного газа, может значительно меняться в течение срока его службы. Это может привести к трудностям в регулировании дозирования азотсодержащего восстановителя для осуществления активного SCR.
Сущность изобретения
Катализаторы, которые используются для окисления угарного газа (CO), углеводородов (HC), а также иногда оксидов азота (NOx) в выхлопном газе, выбрасываемом из двигателя с воспламенением от сжатия, как правило, содержат по меньшей мере один металл платиновой группы, такой как платина или палладий. При катализировании окисления CO и HC в выхлопном газе двигателя с воспламенением от сжатия платина является более активной, чем палладий, и включения палладия в такие катализаторы, как правило, избегали из-за его чувствительности к отравлению серой. Однако, использование топлив со сверхнизким содержанием серы, соотношение стоимости палладия и платины, а также улучшения в отношении срока службы катализатора, которые могут быть достигнуты путем включения палладия, обусловили то, что каталитические составы, содержащие палладий, особенно составы, содержащие как палладий, так и платину, становятся предпочтительными.
Даже при том, что обычно стоимость палладия исторически была ниже, чем стоимость платины, и палладий и платина являются дорогими металлами. Востребованы катализаторы окисления, которые показывают улучшенную каталитическую активность без увеличения суммарного количества платины и палладия, или которые показывают каталитическую активность, аналогичную активности существующих катализаторов окисления при более низком количестве платины и палладия.
Авторы изобретения неожиданно обнаружили, что катализатор окисления, имеющий подходящую активность, можно получить при включении в каталитический состав комбинации (i) марганца и (ii) платины. Было обнаружено, что такие катализаторы имеют превосходную низкотемпературную окислительную активность в отношении CO. Катализаторы являются особенно эффективными при конверсии относительно высоких концентраций CO в выхлопном газе, выработанном двигателем с воспламенением от сжатия, в частности при температурах ниже 250°C. Катализаторы могут также показывать хорошую окислительную активность в отношении HC. Окислительная активность катализатора при относительно низкой температуре делает его особенно подходящим для использования в комбинации с другими устройствами контроля токсичности выхлопных газов в выхлопной системе.
Авторы изобретения также обнаружили, что при использовании такой комбинации можно получить превосходную активность в отношении окисления оксида азота (NO) в выхлопном газе дизельного двигателя. Платина является дорогой и часто включается в катализаторы окисления в относительно больших количествах, в том числе, для его окислительной активности в отношении NO. Включение марганца (Mn) в комбинации с платиной (Pt) может привести к улучшению окислительной активности в отношении NO или обеспечить возможность использования уменьшенного количества Pt для достижения заданного уровня окисления NO.
Изобретение предлагает катализатор окисления для обработки выхлопного газа дизельного двигателя, при этом катализатор окисления содержит: первую область пористого покрытия, содержащую платину (Pt), марганец (Mn) и первый материал-носитель; вторую область пористого покрытия, содержащую металл платиновой группы (PGM) и второй материал-носитель; и подложку, имеющую впускной конец и выпускной конец. Вторая область пористого покрытия предпочтительно размещена таким образом, чтобы контактировать с выхлопным газом на выпускном конце подложки и после контактирования выхлопного газа с первой областью пористого покрытия.
Окислительная активность комбинации Pt и Mn в отношении угарного газа (CO) и оксида азота (NO) может быть особенно выгодной, когда в качестве материала-носителя используются некоторые материалы на основе оксида алюминия.
Изобретение дополнительно предлагает выхлопную систему для дизельного двигателя. Выхлопная система содержит катализатор окисления согласно изобретению и устройство контроля токсичности выхлопных газов.
Количество NO2 в выхлопном газе может влиять на эффективность функционирования расположенного ниже по потоку устройства контроля токсичности выхлопных газов для селективного каталитического восстановления оксидов азота (NOx). Для оптимальной эффективности каталитического действия катализаторы селективного каталитического восстановления (SCR) и катализаторы селективного каталитического восстановления на фильтрующей подложке (SCRF™) для обработки NOx (например, NO2+NO) часто нуждаются в том, чтобы отношение NO2 к NO во входящем газе находилось в пределах заданного диапазона. Оптимальная доля NO2 в NOx, как правило, зависит от типа композиции, используемой для катализатора SCR или SCRF™, но отношение NO2 к NO в выхлопном газе непосредственно из дизельного двигателя обычно является слишком низким для оптимальной работы катализатора.
Катализатор окисления согласно изобретению может использоваться для превращения NO в NO2 и, следовательно, увеличения количества NO2 в выхлопном газе, который выходит из катализатора, по сравнению с количеством NO2 в выхлопном газе на впуске катализатора (т.е. отношения NO2:NOx и NO2:NO в выхлопном газе, который выходит из катализатора окисления имеют большие значения, чем соответствующие отношения выхлопного газа на впуске катализатора окисления). Катализатор окисления может регулировать состав NOx в выхлопном газе для оптимальных показателей SCR.
Проблема с использованием катализатора окисления для «повышения» содержания NO2 в выхлопном газе состоит в том, что окислительная активность катализатора окисления в отношении NO обычно изменяется в течение срока его службы. Как правило, по мере того как катализатор «стареет» (т.е. катализатор используется в течение длительного срока), окислительная активность катализатора в отношении NO уменьшается. Тогда как количество NO2 в выхлопном газе, который выходит из «старого» катализатора окисления, может быть достаточным для оптимального функционирования расположенного ниже по потоку устройства контроля токсичности выхлопных газов (например, катализатора SCR), такое изменение в количестве образующегося NO2 является проблематичным для регулирования дозирования азотсодержащего восстановителя для осуществления активного SCR.
Было обнаружено, что катализатор окисления согласно изобретению может демонстрировать относительно устойчивую окислительную активность в отношении NO в течение своего срока службы. Таким образом, различие в окислительной активности катализатора окисления в отношении NO в свежем состоянии (т.е. когда он является «новым» и не подвергался многократному, длительному использованию) и в состоянии после старения, как правило, является небольшим.
Еще один аспект изобретения относится к транспортному средству или устройству (например, стационарному или мобильному устройству). Транспортное средство или устройство содержит дизельный двигатель и либо катализатор окисления, либо выхлопную систему согласно изобретению.
Изобретение также относится к нескольким вариантам использования и способам.
В соответствии с первым аспектом способа согласно изобретению предлагается способ обработки выхлопного газа дизельного двигателя. Способ включает либо контактирование выхлопного газа с катализатором окисления согласно изобретению, либо пропускание выхлопного газа через выхлопную систему согласно изобретению. В данном контексте выражение «обработка выхлопного газа» относится к окислению угарного газа (CO), углеводородов (HC) и оксида азота (NO) в выхлопном газе дизельного двигателя.
В соответствии со вторым аспектом способа предлагается способ регулирования содержания NOx в выхлопном газе дизельного двигателя для устройства контроля токсичности выхлопных газов. Способ включает: (a) регулирование содержания NOx в выхлопном газе путем контактирования выхлопного газа с катализатором окисления согласно изобретению для получения обработанного выхлопного газа; и (b) передачу обработанного выхлопного газа в устройство контроля токсичности выхлопных газов.
Первый аспект использования согласно изобретению относится к использованию катализатора окисления для обработки выхлопного газа дизельного двигателя, необязательно в комбинации с устройством контроля токсичности выхлопных газов. Как правило, катализатор окисления используется для обработки (например, окисления) угарного газа (CO) и углеводородов (HC) в выхлопном газе дизельного двигателя.
Во втором аспекте использования изобретение относится к использованию катализатора окисления для регулирования содержания NOx в выхлопном газе дизельного двигателя для устройства контроля токсичности выхлопных газов (например, расположенного ниже по потоку устройства контроля токсичности выхлопных газов).
Третий аспект использования относится к использованию катализатора окисления для регенерации устройства контроля токсичности выхлопных газов, имеющего фильтрующую подложку (например, расположенного ниже по потоку устройства контроля токсичности выхлопных газов, имеющего фильтрующую подложку).
Четвертый аспект использования относится к использованию марганца (Mn), предпочтительно в комбинации с платиной (Pt), в катализаторе окисления для дизельного двигателя для усиления окисления оксида азота (NO) в выхлопном газе дизельного двигателя.
Пятый аспект использования относится к использованию марганца (Mn), предпочтительно в комбинации с платиной (Pt), в катализаторе окисления для дизельного двигателя для стабилизации окислительной активности катализатора окисления в отношении NO в течение срока его службы.
В аспектах использования с первого по пятый катализатор окисления представляет собой катализатор окисления, соответствующий изобретению.
Краткое описание чертежей
Фигуры с 1 по 10 представляют собой схематические представления катализаторов окисления согласно изобретению. На фигурах левая сторона катализатора окисления представляет собой впускной конец, а правая сторона представляет собой выпускной конец.
На фигуре 1 показан катализатор окисления, содержащий первую область (1) пористого покрытия и вторую область/зону (2) пористого покрытия, размещенные на подложке (3).
На фигуре 2 показан катализатор окисления, содержащий первую область (1) пористого покрытия и вторую область/зону (2) пористого покрытия. Первая область (1) пористого покрытия размещена непосредственно на подложке (3). Вторая область/зона (2) пористого покрытия размещена на первой области пористого покрытия (1).
На фигуре 3 показан катализатор окисления, содержащий первую область (1) пористого покрытия и вторую область/зону (2) пористого покрытия. Имеется наложение между первой областью (1) пористого покрытия и второй областью/зоной (2) пористого покрытия. Часть первой области (1) пористого покрытия размещена на второй области/зоне (2) пористого покрытия. Как первая область (1) пористого покрытия, так и вторая область/зона (2) пористого покрытия размещены на подложке (3).
На фигуре 4 показан катализатор окисления, содержащий первую область (1) пористого покрытия и вторую область/зону (2) пористого покрытия. Имеется наложение между первой областью (1) пористого покрытия и второй областью/зоной (2) пористого покрытия. Часть второй области/зоны (2) пористого покрытия размещена на первой области (1) пористого покрытия. Как первая область (1) пористого покрытия, так и вторая область/зона (2) пористого покрытия размещены на подложке (3).
На фигуре 5 показан катализатор окисления, содержащий первый слой (1) пористого покрытия и второй слой (2) пористого покрытия, размещенные на подложке (3). Второй слой (2) пористого покрытия размещен на первом слое (1) пористого покрытия.
На фигуре 6 показан катализатор окисления, содержащий первую зону (1) пористого покрытия и вторую область/зону (2) пористого покрытия. Как первая область/зона (1) пористого покрытия, так и вторая область/зона (2) пористого покрытия размещены на третьей области/слое (4) пористого покрытия. Третья область/слой (4) пористого покрытия размещена на подложке (3).
На фигуре 7 показан катализатор окисления, содержащий первую зону (1) пористого покрытия, вторую зону (2) пористого покрытия и третью зону пористого покрытия (4). Первая зона (1) пористого покрытия и вторая зона (2) пористого покрытия вместе размещены на подложке (3). Третья зона (4) пористого покрытия размещена на второй зоне (2) пористого покрытия.
На фигуре 8 показан катализатор окисления, содержащий первую зону (1) пористого покрытия, вторую зону (2) пористого покрытия и третью зону (4) пористого покрытия. Первая зона (1) пористого покрытия и третья зона (4) пористого покрытия вместе размещены на подложке (3). Вторая зона (2) пористого покрытия размещена на третьей зоне (4) пористого покрытия.
На фигуре 9 показан катализатор окисления, содержащий первую зону (1) пористого покрытия, вторую зону (2) пористого покрытия и третью зону (4) пористого покрытия. Первая зона (1) пористого покрытия и вторая зона (2) пористого покрытия вместе размещены на подложке (3). Третья зона (4) пористого покрытия размещена на первой зоне (1) пористого покрытия.
На фигуре 10 показан катализатор окисления, содержащий первую зону (1) пористого покрытия, вторую зону (2) пористого покрытия, третью зону (4) пористого покрытия и четвертую зону (5) пористого покрытия. Первая зона (1) пористого покрытия и третья зона (4) пористого покрытия вместе размещены на подложке (3). Вторая зона (4) пористого покрытия размещена на третьей зоне (2) пористого покрытия. Четвертая зона (5) пористого покрытия размещена на первой зоне (1) пористого покрытия.
Подробное описание изобретения
Катализатор окисления согласно изобретению содержит или может состоять из первой области пористого покрытия, второй области пористого покрытия и подложки.
Как правило, вторая область пористого покрытия размещена таким образом, чтобы контактировать с выхлопным газом на выпускном конце подложки и после контактирования выхлопного газа с первой областью пористого покрытия.
Катализатор может демонстрировать подходящую окислительную активность (например, в отношении CO, HC и NO), когда он имеет схему размещения, которая облегчает контактирование выхлопного газа с областью пористого покрытия, содержащей Pt и Mn, непосредственно после поступления выхлопного газа в катализатор и предпочтительно до его контактирования со второй областью пористого покрытия. При такой схеме размещения катализатора окисления, в тот момент, когда выхлопной газ поступает в катализатор, он сначала контактирует с первой областью пористого покрытия для окисления CO и HC. После того как выхлопной газ прошел сквозь или через первую область пористого покрытия, он контактирует со второй областью пористого покрытия, прежде чем он окончательно пройдет через выпуск катализатора окисления.
Как правило, вторая область пористого покрытия размещена или ориентирована таким образом, чтобы контактировать с выхлопным газом после того, как он контактировал с первой областью пористого покрытия. Как правило, первая область пористого покрытия размещена или ориентирована таким образом, чтобы контактировать с выхлопным газом раньше, чем вторая область пористого покрытия. Таким образом, первая область пористого покрытия может размещаться таким образом, чтобы контактировать с выхлопным газом, когда он поступает в катализатор окисления, а вторая область пористого покрытия может размещаться таким образом, чтобы контактировать с выхлопным газом, когда он покидает катализатор окисления. Примеры таких схем размещения описаны в данном документе.
Первая область пористого покрытия предназначена для окисления угарного газа (CO) и углеводородов (HC), а также необязательно для окисления оксида азота (NO).
Первая область пористого покрытия содержит или может состоять в основном из платины (Pt), марганца (Mn) или их оксида, а также первого материала-носителя.
Платина (Pt), как правило, размещается или наносится на первый материал-носитель. Платина может размещаться непосредственно на или быть нанесенной непосредственно на первый материал-носитель (например, нет никакого промежуточного материала-носителя между платиной и первым материалом-носителем). Например, платина может быть распределена на первом материале-носителе.
Первая область пористого покрытия, как правило, имеет суммарное содержание платины от 0,18 до 10,59 г/дм3. Предпочтительно, чтобы первая область пористого покрытия имела суммарное содержание платины от 0,35 до 8,83 г/дм3 (например, от 2,65 до 6,18 г/дм3), более предпочтительно от 0,53 до 7,06 г/дм3 (например, от 1,77 до 5,30 г/дм3), еще более предпочтительно от 0,71 до 5,30 г/дм3.
Первая область пористого покрытия может дополнительно содержать палладий, такой как палладий, размещенный или нанесенный на первый материал-носитель.
Как правило, первая область пористого покрытия имеет в своем составе отношение по суммарной массе платины к палладию≥1:1 (например, Pt:Pd от 1:0 до 1:1 или от 10:1 до 1:1), предпочтительно≥2:1 (например, Pt:Pd от 1:0 до 2:1 или от 10:1 до 2:1), более предпочтительно≥4:1 (например, Pt:Pd от 1:0 до 4:1 или от 10:1 до 4:1) и еще более предпочтительно≥10:1 (например, Pt:Pd от 1:0 до 10:1).
Когда первая область пористого покрытия содержит палладий (Pd), тогда платина (Pt) и палладий (Pd) предпочтительно являются единственными металлами платиновой группы (т.е. в первой области пористого покрытия).
Первая область пористого покрытия может содержать цеолитный катализатор, такой как цеолитный катализатор согласно приведенному ниже в данном документе определению. Когда первая область пористого покрытия содержит цеолитный катализатор, тогда первая область пористого покрытия может иметь в своем составе отношение по суммарной массе платины к палладию, нанесенному на второй материал-носитель, составляющее≥2:1 (например, Pt:Pd от 1:0 до 2:1), более предпочтительно≥4:1 (например, Pt:Pd от 1:0 до 4:1).
Как правило, предпочтительно, чтобы первая область пористого покрытия практически не содержала палладия, в частности практически не содержала палладия (Pd), размещенного или нанесенного на первый материал-носитель. Более предпочтительно первая область пористого покрытия не содержит палладия, в частности палладия, размещенного или нанесенного на первый материал-носитель. Присутствие палладия, особенно в большом количестве, может негативно сказываться на окислительной активности в отношении HC.
Как правило, первая область пористого покрытия в качестве единственного металла платиновой группы содержит платину (Pt). Первая область пористого покрытия предпочтительно не содержит одного или более других металлов платиновой группы, таких как рутений (Ru), родий (Rh), палладий (Pd), осмий (Os) и/или иридий (Ir). Более предпочтительно первая область пористого покрытия не содержит одного или более других металлов платиновой группы, таких как рутений (Ru), родий (Rh), палладий (Pd), осмий (Os) и/или иридий (Ir), нанесенных на первый материал-носитель.
Предполагается, что основное назначение первой области пористого покрытия заключается в окислении угарного газа (CO) и углеводородов (HC). Однако следует принимать во внимание, что в некоторых вариантах осуществления катализатора окисления первая область пористого покрытия может при использовании также способствовать окислению части оксида азота (NO) до диоксида азота (NO2), особенно когда присутствует значительное количество платины.
Первая область пористого покрытия также содержит марганец (Mn). Марганец может присутствовать в элементной форме или в виде оксида. Первая область пористого покрытия, как правило, содержит марганец или его оксид.
Марганец (Mn), как правило, размещается или наносится на первый материал носитель. Марганец (Mn) может размещаться непосредственно на или быть нанесенным непосредственно на первый материал-носитель (например, нет никакого промежуточного материала-носителя между Mn и первым материалом-носителем).
Первая область пористого покрытия, как правило, имеет суммарное содержание марганца (Mn) от 0,18 до 17,66 г/дм3. Предпочтительно, чтобы первая область пористого покрытия имела суммарное содержание марганца (Mn) от 0,35 до 8,83 г/дм3 (например, от 2,65 до 6,18 г/дм3), более предпочтительно от 0,53 до 7,06 г/дм3 (например, от 1,77 до 5,30 г/дм3), еще более предпочтительно от 0,71 до 5,30 г/дм3.
Как правило, первая область пористого покрытия имеет в своем составе отношение Mn:Pt по массе≤5:1, более предпочтительно <5:1.
Как правило, первая область пористого покрытия имеет в своем составе отношение Mn:Pt по массе≥0,2:1 (например, ≥0,5:1), более предпочтительно >0,2:1 (например, >0,5:1).
Первая область пористого покрытия может иметь в своем составе отношение по суммарной массе марганца (Mn) и платины, составляющее от 5:1 до 0,2:1, такое как от 5:1 до 0,5:1 (например, от 5:1 до 2:3 или от 5:1 до 1:2), предпочтительно от 4,5:1 до 1:1 (например, от 4:1 до 1,1:1), более предпочтительно от 4:1 до 1,5:1. Отношение Mn:Pt по массе может быть важным для достижения описанной в данном документе активности.
Как правило, первый материал-носитель содержит или состоит в основном из тугоплавкого оксида металла. Тугоплавкие оксиды металла, подходящие для использования в качестве каталитического компонента катализатора окисления для дизельного двигателя, являются известными в данной области.
Тугоплавкий оксид металла, как правило, выбирают из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, диоксида церия и смешанного или сложного оксида на их основе, такого как смешанный или сложный оксид из двух или более указанных оксидов. Например, тугоплавкий оксид металла можно выбирать из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, диоксида церия, диоксида кремния-оксида алюминия, диоксида титана-оксида алюминия, диоксида циркония-оксида алюминия, диоксида церия-оксида алюминия, диоксида титана-диоксида кремния, диоксида циркония-диоксида кремния, диоксида циркония-диоксида титана, диоксида церия-диоксида циркония и оксида алюминия-оксида магния.
Первый материал-носитель или соответствующий тугоплавкий оксид металла необязательно может быть легирован (например, с помощью легирующей добавки). Легирующую добавку можно выбирать из группы, состоящей из циркония (Zr), титана (Ti), кремния (Si), иттрия (Y), лантана (La), празеодима (Pr), самария (Sm), неодима (Nd) и их оксида.
Включение легирующей добавки может термически стабилизировать тугоплавкий оксид металла или материал-носитель. Следует понимать, что любое упоминание слова «легированный» в данном контексте относится к материалу, в котором основная или исходная кристаллическая решетка тугоплавкого оксида металла является легированной путем замещения или легированной путем внедрения с использованием легирующей добавки. В некоторых случаях небольшие количества легирующей добавки могут присутствовать на поверхности тугоплавкого оксида металла. Однако большая часть легирующей добавки, как правило, будет присутствовать в теле тугоплавкого оксида металла. Присутствие легирующей добавки часто влияет на химические и/или физические свойства тугоплавкого оксида металла.
Когда первый материал-носитель или соответствующий тугоплавкий оксид металла легирован, суммарное количество легирующей добавки составляет от 0,25 до 5% масс., предпочтительно от 0,5 до 3% масс. (например, приблизительно 1% масс.).
Особенно предпочтительно, чтобы первый материал-носитель или соответствующий тугоплавкий оксид металла содержал или состоял в основном из оксида алюминия, легированного легирующей добавкой.
Оксид алюминия может быть легирован легирующей добавкой, содержащей кремний (Si), магний (Mg), барий (Ba), лантан (La), церий (Ce), титан (Ti) или цирконий (Zr), либо комбинацию из двух или более указанных элементов. Легирующая добавка может содержать или состоять в основном из оксида кремния, оксида магния, оксида бария, оксида лантана, оксида церия, оксида титана или оксида циркония. Предпочтительно, легирующая добавка содержит или состоит в основном из кремния, магния, бария, церия или их оксида, особенно из кремния или церия, либо их оксида. Более предпочтительно, легирующая добавка содержит или состоит в основном из кремния, магния, бария или их оксида; особенно из кремния, магния или их оксида; особенно из кремния или его оксида.
Примеры оксида алюминия, легированного легирующей добавкой, включают оксид алюминия, легированный диоксидом кремния, оксид алюминия, легированный оксидом магния, оксид алюминия, легированный барием или оксидом бария, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный диоксидом церия, особенно оксид алюминия, легированный диоксидом кремния, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный диоксидом церия. Предпочтительно, чтобы оксид алюминия, легированный легирующей добавкой, представлял собой оксид алюминия, легированный диоксидом кремния, оксид алюминия, легированный барием или оксидом бария, или оксид алюминия, легированный оксидом магния. Более предпочтительно оксид алюминия, легированный легирующей добавкой, представляет собой оксид алюминия, легированный диоксидом кремния, или оксид алюминия, легированный оксидом магния. Еще более предпочтительно оксид алюминия, легированный легирующей добавкой, представляет собой оксид алюминия, легированный диоксидом кремния. Оксид алюминия, легированный легирующей добавкой, можно получить с использованием известных в данной области способов или, например, с помощью способа, описанного в US 5045519.
Когда оксид алюминия представляет собой оксид алюминия, легированный диоксидом кремния, тогда оксид алюминия легирован диоксидом кремния в суммарном количестве от 0,5 до 45% масс. (т.е. % от массы оксида алюминия), предпочтительно от 1 до 40% масс., более предпочтительно от 1,5 до 30% масс. (например, от 1,5 до 10% масс.), в частности от 2,5 до 25% масс., конкретнее от 3,5 до 20% масс. (например, от 5 до 20% масс.), еще более предпочтительно от 4,5 до 15% масс.
Когда оксид алюминия представляет собой оксид алюминия, легированный оксидом магния, тогда оксид алюминия легирован магнием в указанном выше количестве или количестве, составляющем от 1 до 30% масс. (т.е. % от массы оксида алюминия), предпочтительно от 5 до 25% масс.
Предпочтительно, чтобы первый материал-носитель или соответствующий тугоплавкий оксид металла не был легирован легирующей добавкой, содержащей или состоящей в основном из марганца. Таким образом, первый материал-носитель или соответствующий тугоплавкий оксид металла не промотирован промотором, таким как промотор, выбираемый из группы, состоящей из олова, марганца, индия, металлов VIII группы (например, Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt, особенно Ir) и их комбинаций.
В соответствии с другим вариантом или кроме того, первый материал-носитель или соответствующий тугоплавкий оксид металла может содержать или состоять в основном из алюмината щелочноземельного металла. Термин «алюминат щелочноземельного металла», как правило, относится к соединению с формулой MAl2O4, где «M» обозначает щелочноземельный металл, такой как Mg, Ca, Sr или Ba. Такие соединения, как правило, образуют шпинельную структуру. Такие соединения можно получить с применением обычных известных в данной области способов или с применением способа, описанного в EP 0945165, US 6217837 или US 6517795.
Как правило, алюминат щелочноземельного металла представляет собой алюминат магния (MgAl2O4), алюминат кальция (CaAl2O4), алюминат стронция (SrAl2O4), алюминат бария (BaAl2O4) или смесь из двух или более указанных соединений. Предпочтительно, алюминат щелочноземельного металла представляет собой алюминат магния (MgAl2O4).
Как правило, когда первый материал-носитель или соответствующий тугоплавкий оксид металла содержит или состоит в основном из смешанного или сложного оксида на основе оксида алюминия (например, диоксида кремния-оксида алюминия, оксида алюминия-оксида магния или смеси оксида алюминия и диоксида церия), тогда смешанный или сложный оксид на основе оксида алюминия предпочтительно содержит по меньшей мере от 50 до 99% масс. оксида алюминия, более предпочтительно от 70 до 95% масс. оксида алюминия, еще более предпочтительно от 75 до 90% масс. оксида алюминия.
Когда первый материал-носитель или соответствующий тугоплавкий оксид металла содержит или состоит в основном из диоксида церия-диоксида циркония, тогда диоксид церия-диоксид циркония может преимущественно иметь в своем составе от 20 до 95% масс. диоксида церия и от 5 до 80% масс. диоксида циркония (например, от 50 до 95% масс. диоксид церия и от 5 до 50% масс. диоксида циркония), предпочтительно от 35 до 80% масс. диоксида церия и от 20 до 65% масс. диоксида циркония (например, от 55 до 80% масс. диоксида церия и от 20 до 45% масс. диоксида циркония), еще более предпочтительно от 45 до 75% масс. диоксида церия и от 25 до 55% масс. диоксида циркония.
Первая область пористого покрытия может содержать первый материал-носитель в количестве от 6,25 до 281,25 г/дм3 (например, от 15,63 до 262,50 г/дм3), предпочтительно от 18,75 до 237,50 г/дм3, еще более предпочтительно от 31,25 до 187,50 г/дм3 (от 62,50 до 171,88 г/дм3 или от 46,88 до 93,75 г/дм3) и еще более предпочтительно от 37,50 до 156,25 г/дм3 (например, от 46,88 до 143,75 г/дм3).
Первая область пористого покрытия может дополнительно содержать адсорбирующий углеводороды материал. Адсорбирующий углеводороды материал может представлять собой цеолит.
Предпочтительно цеолит представляет собой среднепористый цеолит (например, цеолит, имеющий максимальный размер кольца, составляющий десять тетраэдрических атомов) или крупнопористый цеолит (например, цеолит, имеющий максимальный размер кольца, составляющий двенадцать тетраэдрических атомов). Может быть предпочтительным, чтобы цеолит не представлял собой мелкопористый цеолит (например, цеолит, имеющий максимальный размер кольца, составляющий восемь тетраэдрических атомов).
Примеры подходящих цеолитов или типы цеолита включают фоязит, клиноптилолит, морденит, силикалит, феррьерит, цеолит X, цеолит Y, ультрастабильный цеолит Y, цеолит типа AEI, цеолит ZSM-5, цеолит ZSM-12, цеолит ZSM-20, цеолит ZSM-34, цеолит CHA, цеолит SSZ-3, цеолит SAPO-5, оффретит, цеолит бета или медьсодержащий цеолит CHA. Цеолит предпочтительно представляет собой ZSM-5, цеолит бета или цеолит Y.
Когда первая область пористого покрытия содержит адсорбент углеводородов, суммарное количество адсорбента углеводородов составляет от 3,13 до 187,50 г/дм3, в частности от 6,25 до 125,00 г/дм3, конкретнее от 12,50 до 62,50 г/дм3. Например, суммарное количество адсорбента углеводородов может составлять от 50,00 до 109,38 г/дм3, например, от 62,50 до 93,75 г/дм3.
Однако может быть предпочтительным, чтобы первая область пористого покрытия не содержала адсорбирующего углеводороды материала.
Первая область пористого покрытия может дополнительно содержать цеолитный катализатор, такой как цеолитный катализатор согласно приведенному ниже в данном документе определению. Предпочтительно, чтобы первая область пористого покрытия содержала цеолитный катализатор, когда вторая область пористого покрытия содержит цеолитный катализатор. Таким образом, и первая область пористого покрытия, и вторая область пористого покрытия содержат цеолитный катализатор.
В соответствии с другим вариантом первая область пористого покрытия практически не содержит цеолитного катализатора, такого как цеолитный катализатор, описанный ниже в данном документе. Таким образом, первая область пористого покрытия может не содержать цеолитного катализатора.
Однако может быть предпочтительным, чтобы первая область пористого покрытия не содержала родия, щелочного металла и/или щелочноземельного металла, в частности щелочного металла и/или щелочноземельного металла, размещенного или нанесенного на материал-носитель (например, первый материал-носитель, материал-носитель для платины и/или материал-носитель для палладия). Таким образом, первая область пористого покрытия может не содержать родия, щелочного металла и/или щелочноземельного металла, в частности щелочного металла и/или щелочноземельного металла, размещенного или нанесенного на материал-носитель.
Первая область пористого покрытия, как правило, не содержит индия и/или иридия. Более предпочтительно, вторая область пористого покрытия не содержит индия, иридия и/или магния.
Может быть предпочтительным, чтобы первая область пористого покрытия не содержала оксида церия или его смешанного или сложного оксида, такого как (i) смешанный или сложный оксид из оксида церия и оксида алюминия и/или (ii) смешанный или сложный оксид из оксида церия и диоксида циркония.
Кроме того или в соответствии с другим вариантом, первая область пористого покрытия может практически не содержать родия, щелочного металла и/или щелочноземельного металла, в частности щелочного металла и/или щелочноземельного металла, размещенного или нанесенного на первый материал-носитель. Таким образом, первое пористое покрытие может не содержать родия, щелочного металла и/или щелочноземельного металла, в частности щелочного металла и/или щелочноземельного металла, размещенного или нанесенного на первый материал-носитель.
Катализатор окисления согласно изобретению содержит вторую область пористого покрытия. Предпочтительно, чтобы вторая область пористого покрытия являлась подходящей для окисления оксида азота (NO) до диоксида азота (NO2).
Вторая область пористого покрытия содержит металл платиновой группы (PGM).
Как правило, PGM выбирают из группы, состоящей из платины, палладия и комбинации платины и палладия. PGM может представлять собой платину. PGM может состоять в основном из платины (например, PGM представляет собой только платину). PGM может представлять собой палладий. PGM может состоять в основном из палладия (например, PGM представляет собой только палладий). PGM может представлять собой комбинацию платины и палладия. PGM может состоять в основном из платины и палладия (например, PGM представляет собой только платину и палладий). Предпочтительно, чтобы PGM выбирали из группы, состоящей из платины и комбинации платины и палладия.
Вторая область пористого покрытия может содержать PGM в качестве единственного(ых) металла(ов) платиновой группы. Таким образом, единственный(е) PGM присутствующий(ие) во второй области пористого покрытия обозначается(ются) как PGM.
Когда PGM представляет собой комбинацию платины и палладия, тогда PGM может находиться в форме сплава, предпочтительно биметаллического сплава. Таким образом, PGM может содержать или состоять в основном из сплава платины и палладия.
Когда PGM представляет собой палладий или комбинацию платины и палладия, тогда вторая область пористого покрытия может дополнительно содержать золото. Вторая область пористого покрытия может содержать сплав палладий-золото (например, палладий первого металла платиновой группы может присутствовать в виде сплава с золотом). Катализаторы, содержащие золото (Au), могут быть получены с использованием способа, описанного в WO 2012/120292.
Когда вторая область пористого покрытия содержит золото, например, сплав палладий-золото, тогда, как правило, вторая область пористого покрытия имеет в своем составе отношение суммарной массы палладия (Pd) к суммарной массе золота (Au), составляющее от 9:1 до 1:9, предпочтительно от 5:1 до 1:5 и более предпочтительно от 2:1 до 1:2.
Вторая область пористого покрытия, как правило, имеет суммарное содержание PGM от 0,18 до 10,59 г/дм3. Предпочтительно, чтобы вторая область пористого покрытия имела суммарное содержание PGM от 0,35 до 8,83 г/дм3 (например, от 2,65 до 6,18 г/дм3), более предпочтительно от 0,53 до 7,06 г/дм3 (например, от 1,77 до 5,30 г/дм3), еще более предпочтительно от 0,71 до 5,30 г/дм3.
Когда PGM представляет собой комбинацию платины и палладия, тогда, как правило, вторая область пористого покрытия имеет в своем составе отношение по массе платины к палладию, составляющее от 20:1 до 1:20 (например, от 15:1 до 1:15), предпочтительно от 10:1 до 1:10 (например, от 7,5:1 до 1:7.5), более предпочтительно от 5:1 до 1:5 (например, от 3:1 до 1:3), и еще более предпочтительно от 2,5:1 до 1:1.
Предпочтительно, когда PGM представляет собой комбинацию платины и палладия, тогда вторая область пористого покрытия содержит суммарную массу платины, которая имеет большее значение или равна суммарной массе палладия (например, отношение Pt:Pd по массе составляет≥1:1). Более предпочтительно, вторая область пористого покрытия имеет в своем составе суммарную массу платины, которая имеет большее значение, чем суммарная масса палладия (например, отношение Pt:Pd по массе составляет >1:1). Подходящая окислительная активность в отношении NO может быть получена, когда вторая область пористого покрытия обогащена платиной.
Как правило, предпочтительно, чтобы вторая область пористого покрытия имела в своем составе отношение по массе платины к палладию, составляющее от 20:1 до 1:1 (например, от 15,1:1 до 1,1:1), более предпочтительно от 10:1 до 1,25:1 (например, от 7,5:1 до 1,5:1) и еще более предпочтительно от 5:1 до 2:1.
Как правило, PGM размещается или наносится на второй материал-носитель. PGM может быть размещен непосредственно на или нанесен непосредственно на второй материал-носитель (например, нет никакого промежуточного материала-носителя между PGM и вторым материалом-носителем). Например, платина и/или палладий могут быть распределены на втором материале-носителе.
Как правило, второй материал-носитель содержит или состоит в основном из тугоплавкого оксида металла. Тугоплавкий оксид металла, как правило, выбирают из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, диоксида церия и смешанного или сложного оксида на их основе, такого как смешанный или сложный оксид из двух или более указанных оксидов. Например, тугоплавкий оксид металла можно выбирать из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, диоксида церия, диоксида кремния-оксида алюминия, диоксида титана-оксида алюминия, диоксида циркония-оксида алюминия, диоксида церия-оксида алюминия, диоксида титана-диоксида кремния, диоксида циркония-диоксида кремния, диоксида циркония-диоксида титана, диоксида церия-диоксида циркония и оксида алюминия-оксида магния.
Второй материал носитель или соответствующий тугоплавкий оксид металла необязательно может быть легированным (например, легирующей добавкой). Легирующую добавку можно выбирать из группы, состоящей из циркония (Zr), титана (Ti), кремния (Si), иттрия (Y), лантана (La), празеодима (Pr), самария (Sm), неодима (Nd) и их оксида.
Когда второй материал-носитель или соответствующий тугоплавкий оксид металла легирован, суммарное количество легирующей добавки составляет от 0,25 до 5% масс., предпочтительно от 0,5 до 3% масс. (например, приблизительно 1% масс.).
Второй материал-носитель или соответствующий тугоплавкий оксид металла может содержать или состоять в основном из оксида алюминия, легированного легирующей добавкой. Особенно предпочтительно, чтобы второй материал носитель или соответствующий тугоплавкий оксид металла содержал или состоял в основном из оксида алюминия, легированного легирующей добавкой. Было обнаружено, что комбинация марганца (Mn) платины (Pt) и легированного оксида алюминия в качестве материала-носителя, особенно материала-носителя на основе оксида алюминия, легированного диоксидом кремния, обеспечивает превосходную окислительную активность в отношении NO и может стабилизировать окислительную активность катализатора окисления в отношении NO в течении его срока службы.
Оксид алюминия может быть легирован легирующей добавкой, содержащей кремний (Si), магний (Mg), барий (Ba), лантан (La), церий (Ce), титан (Ti) или цирконий (Zr) или комбинацию из двух или более указанных элементов. Легирующая добавка может содержать или состоять в основном из оксида кремния, оксида магния, оксида бария, оксида лантана, оксида церия, оксида титана или оксида циркония. Предпочтительно, легирующая добавка содержит или состоит в основном из кремния, магния, бария, церия или их оксида; особенно кремния, церия или их оксида. Более предпочтительно легирующая добавка содержит или состоит в основном из кремния, магния, бария или их оксида; особенно кремния, магния или их оксида; особенно кремния или его оксида.
Как правило, когда второй материал-носитель или соответствующий тугоплавкий оксид металла содержит или состоит в основном из смешанного или сложного оксида на основе оксида алюминия (например, диоксида кремния-оксида алюминия, оксида алюминия-оксида магния или смеси оксида алюминия и диоксида церия), тогда предпочтительно смешанный или сложный оксид на основе оксида алюминия содержит по меньшей мере от 50 до 99% масс. оксида алюминия, более предпочтительно от 70 до 95% масс. оксида алюминия, еще более предпочтительно от 75 до 90% масс. оксида алюминия.
Примеры оксида алюминия, легированного легирующей добавкой, включают оксид алюминия, легированный диоксидом кремния, оксид алюминия, легированный оксидом магния, оксидом алюминия, легированный барием или оксидом бария, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированный диоксидом церия, особенно оксид алюминия, легированный диоксидом кремния, оксид алюминия, легированный оксидом лантана, или оксид алюминия, легированным диоксидом церия. Предпочтительно, чтобы оксид алюминия, легированный легирующей добавкой, представлял собой оксид алюминия, легированный диоксидом кремния, оксид алюминия, легированный барием или оксидом бария, или оксид алюминия, легированный оксидом магния. Более предпочтительно оксид алюминия, легированный легирующей добавкой, представляет собой оксид алюминия, легированный диоксидом кремния или оксид алюминия, легированный оксидом магния. Еще более предпочтительно оксид алюминия, легированный легирующей добавкой, представляет собой оксид алюминия, легированный диоксидом кремния.
Когда оксид алюминия представляет собой оксид алюминия, легированный диоксидом кремния, тогда оксид алюминия легирован диоксидом кремния в суммарном количестве от 0,5 до 45% масс. (т.е. % от массы оксида алюминия), предпочтительно от 1 до 40% масс., более предпочтительно от 1,5 до 30% масс. (например, от 1,5 до 10% масс.), в частности от 2,5 до 25% масс., конкретнее от 3,5 до 20% масс. (например, от 5 до 20% масс.), еще более предпочтительно от 4,5 до 15% масс.
Когда оксид алюминия представляет собой оксид алюминия, легированный оксидом магния, тогда оксид алюминия легирован магнием в указанном выше количестве или количестве, составляющем от 1 до 30% масс. (т.е. % от массы оксида алюминия), предпочтительно от 5 до 25% масс.
Когда второй материал-носитель или соответствующий тугоплавкий оксид металла содержит или состоит в основном из диоксида церия-диоксида циркония, тогда диоксид церия-диоксид циркония может иметь в своем составе преимущественно от 20 до 95% масс. диоксида церия и от 5 до 80% масс. диоксида циркония (например, от 50 до 95% масс. диоксида церия и от 5 до 50% масс. диоксида циркония), предпочтительно от 35 до 80% масс. диоксида церия и от 20 до 65% масс. диоксида циркония (например, от 55 до 80% масс. диоксида церия и от 20 до 45% масс. диоксида циркония), еще более предпочтительно от 45 до 75% масс. диоксида церия и от 25 до 55% масс. диоксид циркония.
В соответствии с другим вариантом или кроме того, второй материал-носитель или соответствующий тугоплавкий оксид металла может содержать или состоять в основном из алюмината щелочноземельного металла, такого как алюминат щелочноземельного металла, имеющий шпинельную структуру.
Как правило, алюминат щелочноземельного металла представляет собой алюминат магния (MgAl2O4), алюминат кальция (CaAl2O4), алюминат стронция (SrAl2O4) или алюминат бария (BaAl2O4), либо смесь из двух или более указанных соединений. Предпочтительно, алюминат щелочноземельного металла представляет собой алюминат магния (MgAl2O4).
Вторая область пористого покрытия, как правило, содержит второй материал-носитель в количестве от 6,25 до 281,25 г/дм3 (например, от 15,63 до 250,00 г/дм3), предпочтительно от 31,25 до 187,50 г/дм3, более предпочтительно от 37,50 до 156,25 г/дм3 (например, от 46,88 до 93,75 г/дм3).
Как правило, первая область пористого покрытия и вторая область пористого покрытия имеют различные составы (т.е. разные компоненты и/или разное количество компонентов).
В первом варианте осуществления второй области пористого покрытия вторая область пористого покрытия содержит марганец (Mn). Марганец может присутствовать в элементной форме или в виде оксида. Вторая область пористого покрытия, как правило, содержит марганец или его оксид.
Марганец (Mn), как правило, размещается или наносится на второй материал-носитель. Марганец (Mn) может быть размещен непосредственно на или нанесен непосредственно на второй материал-носитель (например, отсутствует какой-либо промежуточный материал-носитель между Mn и вторым материалом-носителем).
Вторая область пористого покрытия, как правило, имеет суммарное содержание марганца (Mn) от 0,18 до 17,66 г/дм3. Предпочтительно, чтобы вторая область пористого покрытия имела суммарное содержание марганца (Mn) от 0,35 до 8,83 г/дм3 (например, от 2,65 до 6,18 г/дм3), более предпочтительно от 0,53 до 7,06 г/дм3 (например, от 1,77 до 5,30 г/дм3), еще более предпочтительно от 0,71 до 5,30 г/дм3.
Когда вторая область пористого покрытия содержит марганец (Mn), тогда PGM содержит платину (например, PGM выбирают из группы, состоящей из платины и комбинации платины и палладия). Было обнаружено, что вторая область пористого покрытия, содержащая Mn, может обеспечить хороший показатель окисления NO, особенно в сочетании с первой областью пористого покрытия, которая способствует окислению CO, HC и части NO.
Как правило, вторая область пористого покрытия имеет в своем составе отношение Mn:Pt по массе ≤5:1, более предпочтительно <5:1.
Как правило, вторая область пористого покрытия имеет в своем составе отношение Mn:Pt по массе≥0,2:1 (например, ≥0,5:1), более предпочтительно >0,2:1 (например, >0,5:1).
Вторая область пористого покрытия может иметь в своем составе отношение по суммарной массе марганца (Mn) к платине, составляющее от 5:1 до 0,2:1, например, от 5:1 до 0,5:1 (например, от 5:1 до 2:3 или от 5:1 до 1:2), предпочтительно от 4,5:1 до 1:1 (например, от 4:1 до 1,1:1), более предпочтительно от 4:1 до 1,5:1. Отношение Mn:Pt по массе может быть важным для достижения активности в отношении NO, описанной в данном документе.
Платина (Pt), как правило, размещается или наносится на второй материал-носитель. Платина может размещаться непосредственно на или наноситься непосредственно на второй материал-носитель (например, отсутствует какой-либо промежуточный материал-носитель между платиной и вторым материалом-носителем). Например, платина может быть распределена на втором материале-носителе.
Вторая область пористого покрытия, как правило, имеет суммарное содержание платины от 0,18 до 10,59 г/дм3. Предпочтительно, чтобы вторая область пористого покрытия имела суммарное содержание платины от 0,35 до 8,83 г/дм3 (например, от 2,65 до 6,18 г/дм3), более предпочтительно от 0,53 до 7,06 г/дм3 (например, от 1,77 до 5,30 г/дм3), еще более предпочтительно от 0,71 до 5,30 г/дм3.
Вторая область пористого покрытия может дополнительно содержать палладий (например, PGM представляет собой комбинацию платины и палладия). Палладий может размещаться или наноситься на второй материал носитель.
Когда вторая область пористого покрытия содержит палладий, тогда отношение по суммарной массе платины к палладию, как правило, ≥2:1 (например, Pt:Pd составляет от 1:0 до 2:1), более предпочтительно≥4:1 (например, Pt:Pd составляет от 1:0 до 4:1).
Как правило, предпочтительно, чтобы вторая область пористого покрытия практически не содержала палладия, в частности практически не содержала палладия (Pd), размещенного или нанесенного на второй материал-носитель. Более предпочтительно, вторая область пористого покрытия не содержит палладия, в частности палладия, размещенного или нанесенного на второй материал-носитель. Присутствие палладия, особенно в большом количестве, во второй области пористого покрытия может негативно сказываться на окислительной активности в отношении NO. Окислительная активность палладия в отношении NO, как правило, является низкой при обычных условиях использования для дизельного катализатора окисления. Кроме того, любое количество присутствующего палладия может реагировать с некоторой частью присутствующей платины с образованием сплава. Это также может негативно сказываться на окислительной активности второй области пористого покрытия в отношении NO, поскольку сплавы платины и палладия не так активны в отношении окисления NO, как платина сама по себе.
Как правило, вторая область пористого покрытия содержит платину (Pt) в качестве единственного металла платиновой группы. Вторая область пористого покрытия предпочтительно не содержит одного или более других металлов платиновой группы, таких как рутений (Ru), родий (Rh), палладий (Pd), осмий (Os) и/или иридий (Ir). Более предпочтительно, вторая область пористого покрытия не содержит одного или более других металлов платиновой группы, таких как рутений (Ru), родий (Rh), палладий (Pd), осмий (Os) и/или иридий (Ir), нанесенных на второй материал-носитель.
Во втором варианте осуществления второй области пористого покрытия вторая область пористого покрытия содержит цеолитный катализатор, такой как цеолитный катализатор согласно приведенному ниже в данном документе определению.
Когда вторая область пористого покрытия содержит цеолитный катализатор, тогда вторая область пористого покрытия может иметь в своем составе отношение по суммарной массе платины к палладию, нанесенному на второй материал-носитель, составляющее≥2:1 (например, Pt:Pd составляет от 1:0 до 2:1), более предпочтительно≥4:1 (например, Pt:Pd составляет от 1:0 до 4:1).
Как правило, цеолитный катализатор содержит или состоит в основном из благородного металла и цеолита. Цеолитные катализаторы могут быть получены согласно способу, описанному в WO 2012/166868.
Благородный металл, как правило, выбирают из группы, состоящей из палладия (Pd), платины (Pt), родия (Rh), золота (Au), серебра (Ag), иридия (Ir), рутения (Ru), осмия (Os) и смеси из двух или более указанных металлов. Предпочтительно, благородный металл выбирают из группы, состоящей из палладия (Pd), платины (Pt), родия (Rh), золота (Au), серебра (Ag), иридия (Ir), рутения (Ru) и смеси из двух или более указанных металлов. Более предпочтительно, благородный металл выбирают из группы, состоящей из палладия (Pd), платины (Pt) и родия (Rh). Еще более предпочтительно, благородный металл представляет собой палладий (Pd). Кроме того, предпочтительно, чтобы цеолитный катализатор содержал палладий в качестве единственного благородного металла.
Цеолитный катализатор может дополнительно содержать неблагородный металл. Таким образом, цеолитный катализатор может содержать или состоять в основном из благородного металла, цеолита и необязательно неблагородного металла.
Неблагородный металл можно выбирать из группы, состоящей из железа (Fe), меди (Cu), марганца (Mn), хрома (Cr), кобальта (Co), никеля (Ni), и олова (Sn), а также смеси из двух или более указанных металлов. Предпочтительно, чтобы неблагородный металл выбирали из группы, состоящей из железа, меди и кобальта, более предпочтительно, железа и меди. Еще более предпочтительно неблагородный металл представляет собой железо.
В соответствии с другим вариантом цеолитный катализатор может практически не содержать неблагородного металла. Таким образом, цеолитный катализатор не содержит неблагородного металла.
Как правило предпочтительно, чтобы цеолитный катализатор не содержал неблагородного металла.
Цеолит, как правило, выбирают из алюмосиликатного цеолита и алюмофосфатного цеолита.
Как правило, цеолит может быть металлзамещенным цеолитом (например, металлзамещенным алюмосиликатным цеолитом или металлзамещенный алюмофосфатным цеолитом). Металл в металлзамещенном цеолите может представлять собой благородный металл (например, цеолит представляет собой замещенный благородным металлом цеолит). Когда цеолитный катализатор содержит неблагородный металл, тогда цеолит может представлять собой замещенный благородным и неблагородным металлом цеолит.
Цеолитный катализатор, как правило, содержит по меньшей мере 1% масс. (т.е. от количества благородного металла в цеолитном катализаторе) благородного металла, находящегося в порах цеолита, предпочтительно по меньшей мере, 5% масс., более предпочтительно по меньшей мере 10% масс., например, по меньшей мере 25% масс., еще более предпочтительно по меньшей мере 50% масс.
Цеолит можно выбирать из мелкопористого цеолита (т.е. цеолита, имеющего максимальный размер кольца, составляющий восемь тетраэдрических атомов), среднепористого цеолита (т.е. цеолита, имеющего максимальный размер кольца, составляющий десять тетраэдрических атомов) и крупнопористого цеолита (т.е. цеолита, имеющего максимальный размер кольца, составляющий двенадцать тетраэдрических атомов). Более предпочтительно цеолит выбирают из мелкопористого цеолита и среднепористого цеолита.
Как правило, цеолит состоит из алюминия, кремния и/или фосфора. Цеолит, как правило, имеет трехмерную структуру из SiO4, AlO4 и/или PO4, которые соединены за счет связей с общими атомами кислорода. Цеолит может иметь анионную структуру. Заряд анионной структуры может быть скомпенсирован катионами, такими как катионы щелочных и/или щелочноземельных элементов (например, Na, K, Mg, Ca, Sr и Ba), катионами аммония и/или протонами.
В первом варианте осуществления цеолитного катализатора цеолит представляет собой мелкопористый цеолит. Мелкопористый цеолит предпочтительно имеет тип структуры, выбираемый из группы, состоящей из ACO, AEI, AEN, AFN, AFT, AFX, ANA, APC, APD, ATT, CDO, CHA, DDR, DFT, EAB, EDI, EPI, ERI, GIS, GOO, IHW, ITE, ITW, LEV, KFI, MER, MON, NSI, OWE, PAU, PHI, RHO, RTH, SAT, SAV, SIV, THO, TSC, UEI, UFI, VNI, YUG и ZON, а также смеси или сростка из любых двух или более указанных структур. Сросток предпочтительно выбирают из KFI-SIV, ITE-RTH, AEW-UEI, AEI-CHA и AEI-SAV. Более предпочтительно мелкопористый цеолит имеет тип структуры, который представляет собой AEI, CHA или сросток AEI-CHA. Еще более предпочтительно мелкопористый цеолит имеет тип структуры, который представляет собой AEI или CHA.
Во втором варианте осуществления цеолитного катализатора цеолит имеет тип структуры, выбираемый из группы, состоящей из AEI, MFI, EMT, ERI, MOR, FER, BEA, FAU, CHA, LEV, MWW, CON и EUO, а также смесей из любых двух или более указанных структур.
В третьем варианте осуществления цеолитного катализатора цеолит представляет собой среднепористый цеолит. Среднепористый цеолит предпочтительно имеет тип структуры, выбираемый из группы, состоящей из MFI, FER, MWW и EUO, более предпочтительно MFI.
В четвертом варианте осуществления цеолитного катализатора цеолит представляет собой крупнопористый цеолит. Крупнопористый цеолит предпочтительно имеет тип структуры, выбираемый из группы, состоящей из CON, BEA, FAU, MOR и EMT, более предпочтительно BEA.
Цеолит, как правило, имеет мольное отношение диоксида кремния к оксиду алюминию (SAR), составляющее от 10 до 200 (например, от 10 до 40), более предпочтительно от 15 до 80 (например, от 15 до 30).
Цеолитный катализатор согласно первому, третьему и четвертому варианту осуществления цеолитного катализатора (а также для некоторых типов структуры второго варианта осуществления цеолитного катализатора) может иметь инфракрасный спектр, имеющий характерный пик поглощения в области от 750 см-1 до 1050 см-1 (помимо пиков поглощения цеолита самого по себе). Предпочтительно, характерный пик поглощения находится в области от 800 см-1 до 1000 см-1, более предпочтительно в области от 850 см-1 до 975 см-1.
Во втором варианте осуществления второй области пористого покрытия вторая область пористого покрытия может практически не содержать марганца или его оксида. Вторая область пористого покрытия предпочтительно не содержит марганца или его оксида.
Неожиданно было обнаружено, что область пористого покрытия, содержащая цеолитный катализатор согласно первому варианту осуществления цеолитного катализатора, действует как пассивный адсорбент NOx (PNA). Область пористого покрытия с активностью PNA может использоваться для накапливания NOx, когда значения температуры выхлопного газа являются относительно низкими, такими как значения непосредственно после запуска дизельного двигателя. Накапливание NOx цеолитным катализатором происходит при значениях температуры (например, менее 200°C), которые ниже значения температуры, при котором компоненты на основе платины и марганца первой области пористого покрытия способствуют осуществлению окисления оксида азота (NO) до диоксида азота (NO2) в значительно степени.
Когда дизельный двигатель нагревается, увеличивается температура выхлопного газа, и температура цеолитного катализатора (и области пористого покрытия, действующей как PNA) будет также увеличиваться. При таких более высоких значениях температуры (например, 200°C или выше) адсорбированный NOx будет высвобождаться из цеолитного катализатора. Адсорбированный NOx может высвобождаться из цеолитного катализатора, когда компоненты на основе платины и марганца во второй области пористого покрытия достигают соответствующей им эффективной для того, чтобы способствовать окислению NO температуры, или адсорбированный NOx может высвобождаться из цеолитного катализатора при значении температуры немного ниже указанной эффективной температуры. Неожиданно было обнаружено, что цеолитный катализатор имеет высокую температуру высвобождения адсорбированного NOx. Также неожиданно было обнаружено, что область пористого покрытия, содержащая цеолитный катализатор согласно второму варианту осуществления цеолитного катализатора, действует как катализатор, работающий при запуске холодного двигателя (CSC™). Такое действие может снижать выбросы в период запуска холодного двигателя путем адсорбции NOx и углеводородов (HC) при относительно низких значениях температуры выхлопного газа (например, менее 200°C). Адсорбированные NOx и/или HC могут высвобождаться из области пористого покрытия, когда температура цеолитного катализатора имеет значение близкое или выше эффективной температуры для окисления NO и/или HC, соответствующей другим каталитическим компонентам. Активность катализатора, работающего при холодном запуске двигателя, является особенно выгодной для цеолитного катализатора, когда он присутствует в сочетании с содержащей марганец первой областью пористого покрытия.
Как правило, предпочтительно (включая первый и второй варианты осуществления второй области пористого покрытия), чтобы второй материал-носитель или соответствующий тугоплавкий оксид металла не был легирован легирующей добавкой, содержащей или состоящей в основном из марганца. Таким образом, второй материал-носитель или соответствующий тугоплавкий оксид металла не промотирован промотором, таким как промотор, выбираемый из группы, состоящей из олова, марганца, индия, металлов VIII группы (например, Fe, Co, Ni, Ru, Rh, Pd, Os, Ir и Pt, особенно Ir) и их комбинации.
Для некоторых вариантов применения, как правило, может быть предпочтительным, чтобы вторая область пористого покрытия практически не содержала адсорбирующего углеводороды материала, в частности цеолита. Таким образом, вторая область пористого покрытия может не содержать адсорбирующего углеводороды материала.
Также может быть предпочтительным, чтобы вторая область пористого покрытия практически не содержала цеолитного катализатора, такого как цеолитный катализатор, описанный выше в данном документе. Таким образом, вторая область пористого покрытия может не содержать цеолитного катализатора.
Вторая область пористого покрытия, как правило, не содержит индия и/или иридия. Более предпочтительно, вторая область пористого покрытия не содержит индия, иридия и/или магния.
Может быть предпочтительным, чтобы вторая область пористого покрытия не содержала оксида церия или смешанного или сложного оксида на его основе, такого как (i) смешанный или сложный оксид из оксида церия и оксида алюминия и/или (ii) смешанный или сложный оксид из оксида церия и диоксида циркония.
Кроме того или в соответствии с другим вариантом, вторая область пористого покрытия может практически не содержать родия, щелочного металла и/или щелочноземельного металла, в частности щелочного металла и/или щелочноземельного металла, размещенного или нанесенного на второй материал-носитель. Таким образом, второе пористое покрытие может не содержать родия, щелочного металла и/или щелочноземельного металла, в частности щелочного металла и/или щелочноземельного металла, размещенного или нанесенного на второй материал-носитель.
Как правило, первая область пористого покрытия содержит >25% от суммарной концентрации металла платиновой группы (т.е. в катализаторе окисления). Предпочтительно, чтобы первая область пористого покрытия содержала >30%, более предпочтительно≥40% от суммарной концентрации металла платиновой группы.
Как правило, суммарная концентрация платины в первой области пористого покрытия имеет большее значение, чем суммарная концентрация PGM во второй области пористого покрытия.
Как правило, катализатор окисления содержит суммарное количество материала-носителя (например, первого материала-носителя и второго носителя) от 6,25 до 281,25 г/дм3 (например, от 15,63 до 262,50 г/дм3), предпочтительно от 12,50 до 237,50 г/дм3, например, от 18,75 до 187,50 г/дм3, в частности от 31,25 до 156,25 г/дм3 (например, от 46,88 до 143,75 г/дм3), еще более предпочтительно от 37,50 до 125,00 г/дм3 и еще более предпочтительно от 46,88 до 109,38 г/дм3.
Первая область пористого покрытия и/или вторая область пористого покрытия могут размещаться или наноситься на подложку.
Первая область пористого покрытия может размещаться непосредственно на подложке (т.е. первая область пористого покрытия контактирует с поверхностью подложки; см. фигуры с 1 по 5). Вторая область пористого покрытия может:
(a) размещаться или наноситься на первую область пористого покрытия (например, см. фигуры 2, 4 и 5); и/или
(b) размещаться непосредственно на подложке [т.е. вторая область пористого покрытия контактирует с поверхностью подложки] (например, см. фигуры 1, 3, 4); и/или
(c) контактировать с первой областью пористого покрытия [т.е. вторая область пористого покрытия располагается рядом или прилегает к первой области пористого покрытия].
Когда вторая область пористого покрытия размещена непосредственно на подложке, тогда часть или участок второй области пористого покрытия может контактировать с первой областью пористого покрытия, или первая область пористого покрытия и вторая область пористого покрытия могут быть отделены друг от друга (например, промежутком).
Когда вторая область пористого покрытия размещена или нанесена на первую область пористого покрытия, вся поверхность или часть второй области пористого покрытия предпочтительно размещена непосредственно на первой области пористого покрытия (т.е. вторая область пористого покрытия контактирует с поверхностью первой области пористого покрытия). Вторая область пористого покрытия может представлять собой второй слой пористого покрытия, а первая область пористого покрытия может представлять собой первый слой пористого покрытия.
Вторая область пористого покрытия может размещаться непосредственно на подложке (т.е. вторая область пористого покрытия контактирует с поверхностью подложки; см. фигуры 1, 3 и 4). Первая область пористого покрытия может:
(i) размещаться или наноситься на вторую область пористого покрытия (например, см. фигуры 3 и 4); и/или
(ii) размещаться непосредственно на подложке [т.е. первая область пористого покрытия контактирует с поверхностью подложки] (например, см. фигуры 3 и 4); и/или
(iii) контактировать со второй областью пористого покрытия [т.е. первая область пористого покрытия располагается рядом или прилегает ко второй области пористого покрытия].
Первая область пористого покрытия может размещаться непосредственно на второй области пористого покрытия (т.е. первая область пористого покрытия контактирует с поверхностью второй области пористого покрытия).
Предпочтительно, чтобы только участок или часть первой области пористого покрытия был размещен или нанесен на вторую область пористого покрытия. Таким образом, первая область пористого покрытия не накладывается или не покрывает полностью вторую область пористого покрытия.
Как правило, возможно, чтобы первая область пористого покрытия и вторая область пористого покрытия вместе не размещались непосредственно на подложке (т.е. ни первая область пористого покрытия, ни вторая область пористого покрытия не контактируют с поверхностью подложки).
В катализаторе окисления согласно изобретению вторая область пористого покрытия, как правило, размещена таким образом, чтобы контактировать с выхлопным газом на выпускном конце подложки. Это достигается путем размещения второй области/слоя/зоны пористого покрытия на выпускном конце подложки.
Вторая область пористого покрытия, как правило, размещена или ориентирована таким образом, чтобы контактировать с выхлопным газом после его контактирования с первой областью пористого покрытия. Это может быть достигнуто путем размещения различными способами второй области пористого покрытия на подложке относительно первой области пористого покрытия.
Таким образом, вторая область пористого покрытия размещена или ориентирована таким образом, чтобы контактировать с выхлопным газом после его контактирования с первой областью пористого покрытия, когда:
(a) вторая область пористого покрытия является второй зоной пористого покрытия, размещенной на выпускном конце подложки, и необязательно, первая область пористого покрытия является первой зоной пористого покрытия, размещенной на впускном конце подложки;
(b) первая область пористого покрытия представляет собой первый слой пористого покрытия, а вторая область пористого покрытия представляет собой вторую зону пористого покрытия, где вторая зона пористого покрытия размещена на первом слое пористого покрытия на выпускном конце подложки; или
(c) первая область пористого покрытия представляет собой первый слой пористого покрытия, а вторая область пористого покрытия представляет собой второй слой пористого покрытия, где при этом второй слой пористого покрытия размещен на первом слое пористого покрытия.
Как правило, первая область пористого покрытия размещена или ориентирована таким образом, чтобы контактировать с выхлопным газом раньше, чем вторая область пористого покрытия. Таким образом, первая область пористого покрытия может размещаться таким образом, чтобы контактировать с выхлопным газом, когда он поступает в катализатор окисления, а вторая область пористого покрытия может размещаться таким образом, чтобы контактировать с выхлопным газом, когда он покидает катализатор окисления. Зонная схема размещения первой и второй областей пористого покрытия, показанная на фигурах 1 и 6-10, является особенно подходящей в этом отношении.
Вторая область пористого покрытия размещена таким образом, чтобы контактировать с выхлопным газом на выпускном конце подложки и после контактирования выхлопного газа с первой областью пористого покрытия в любой из описанных ниже в данном документе схем размещения катализатора окисления с первой по третью.
Предпочтительно, чтобы вторая область пористого покрытия представляла собой вторую зону пористого покрытия. Более предпочтительно, вторая зона пористого покрытия размещена или нанесена в области или вблизи выпускного конца подложки.
Вторая зона пористого покрытия, как правило, имеет длину, составляющую от 10 до 90% длины подложки (например, от 10 до 45%), предпочтительно от 15 до 75% длины подложки (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 30 до 65%, например, от 25 до 45%) длины подложки, еще более предпочтительно от 25 до 65% (например, от 35 до 50%).
В первой схеме размещения катализатора окисления первая область пористого покрытия размещена или нанесена выше по потоку относительно второй зоны пористого покрытия. Предпочтительно, первая область пористого покрытия является первой зоной пористого покрытия. Более предпочтительно, первая зона пористого покрытия размещена или нанесена в области или вблизи впускного конца подложки.
Как правило, первая зона пористого покрытия имеет длину, составляющую от 10 до 90% длины подложки (например, от 10 до 45%), предпочтительно от 15 до 75% длины подложки (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 30 до 65%, например, от 25 до 45%) длины подложки, еще более предпочтительно от 25 до 65% (например, от 35 до 50%). Предпочтительно, чтобы длина первой зоны пористого покрытия была больше длины второй зоны пористого покрытия.
Первая зона пористого покрытия может прилегать ко второй зоне пористого покрытия. Предпочтительно, первая зона пористого покрытия контактирует со второй зоной пористого покрытия. Когда первая зона пористого покрытия прилегает ко второй зоне пористого покрытия, или первая зона пористого покрытия контактирует со второй зоной пористого покрытия, тогда первая зона пористого покрытия и вторая зона пористого покрытия могут быть размещены или нанесены на подложку в виде слоя (например, одного слоя). Таким образом, слой (например, один) может быть образован на подложке, когда первая и вторая зоны пористого покрытия прилегают или контактируют друг с другом. Такая схема размещения может помочь избежать проблем с противодавлением.
Первая зона пористого покрытия может быть отделена от второй зоны пористого покрытия. Может присутствовать промежуток (например, пространство) между первой зоной пористого покрытия и второй зоной пористого покрытия.
Первая зона пористого покрытия может накладываться на вторую зону пористого покрытия. Таким образом, концевой участок или часть первой зоны пористого покрытия может быть размещена или нанесена на вторую зону пористого покрытия. Первая зона пористого покрытия может полностью или частично накладываться на вторую зону пористого покрытия. Когда первая зона пористого покрытия накладывается на вторую зону пористого покрытия, предпочтительно, чтобы первая зона пористого покрытия накладывалась на вторую зону пористого покрытия только частично (т.е. верхняя, наиболее удаленная от центра поверхность второй зоны пористого покрытия не полностью покрыта первой зоной пористого покрытия).
В соответствии с другим вариантом вторая зона пористого покрытия может накладываться на первую зону пористого покрытия. Таким образом, концевой участок или часть второй зоны пористого покрытия может быть размещена или нанесена на первую зону пористого покрытия. Вторая зона пористого покрытия, как правило, только частично накладывается на первую зону пористого покрытия.
Предпочтительно, чтобы первая зона пористого покрытия и вторая зона пористого покрытия практически не накладывались друг на друга.
Во второй схеме размещения катализатора окисления первая область пористого покрытия представляет собой первый слой пористого покрытия. Предпочтительно, чтобы первый слой пористого покрытия тянулся по всей длине (т.е. практически по всей длине) подложки, в частности по всей длине каналов монолитной подложки.
Вторая зона пористого покрытия, как правило, размещается или наносится на первый слой пористого покрытия. Предпочтительно, вторая зона пористого покрытия размещается непосредственно на первом слое пористого покрытия (т.е. вторая зона пористого покрытия контактирует с поверхностью первого слоя пористого покрытия).
Когда вторая зона пористого покрытия размещена или нанесена на первый слой пористого покрытия, предпочтительно, чтобы вся длина второй зоны пористого покрытия была размещена или нанесена на первый слой пористого покрытия. Длина второй зоны пористого покрытия меньше длины первого слоя пористого покрытия.
В третьей схеме размещения катализатора окисления первая область пористого покрытия представляет собой первый слой пористого покрытия. Предпочтительно, чтобы первый слой пористого покрытия тянулся по всей длине (т.е. практически по всей длине) подложки, в частности по всей длине каналов монолитной подложки.
Вторая область пористого покрытия представляет собой второй слой пористого покрытия. Предпочтительно, чтобы второй слой пористого покрытия тянулся по всей длине (т.е. практически по всей длине) подложки, в частности по всей длине каналов монолитной подложки.
Второй слой пористого покрытия размещается или наносится на первый слой пористого покрытия. Предпочтительно, второй слой пористого покрытия размещается непосредственно на первом слое пористого покрытия (т.е. второй слой пористого покрытия контактирует с поверхностью первого слоя пористого покрытия).
Когда второй слой пористого покрытия размещается на первом слое пористого покрытия, в частности, когда второй слой пористого покрытия не содержит марганца, Mn-содержащая область пористого покрытия неожиданным образом является устойчивой к отравлению серой из дизельного топлива.
Катализатор окисления может дополнительно содержать третью область пористого покрытия. Таким образом, по меньшей мере одна область из первой области пористого покрытия и второй области пористого покрытия может размещаться или наноситься на третью область пористого покрытия. Третья область пористого покрытия может содержать или может не содержать металла платиновой группы.
Третья область пористого покрытия может размещаться непосредственно на подложке (т.е. третья область пористого покрытия контактирует с поверхностью подложки; см. фигуры 6, 8 и 10). Вторая область пористого покрытия может:
(a) размещаться или наноситься на третью область пористого покрытия (например, см. фигуры 6, 8 и 10); и/или
(b) размещаться непосредственно на подложке [т.е. вторая область пористого покрытия контактирует с поверхностью подложки]; и/или
(c) контактировать с третьей областью пористого покрытия [т.е. вторая область пористого покрытия располагается рядом или прилегает к третьей области пористого покрытия].
Кроме того или в соответствии с другим вариантом, третья область пористого покрытия может размещаться или наноситься на первую область пористого покрытия и/или вторую область пористого покрытия (например, см. фигуры 7 и 9).
Когда третья область пористого покрытия размещается непосредственно на подложке, тогда первая область пористого покрытия может размещаться или наноситься на третью область пористого покрытия (например, см. фигуру 6). Вторая область пористого покрытия может также размещаться или наноситься на третью область пористого покрытия. Третья область пористого покрытия может представлять собой третий слой пористого покрытия, первая область пористого покрытия может представлять собой первую зону пористого покрытия, а вторая область пористого покрытия может представлять собой вторую зону пористого покрытия. Вторая область/зона пористого покрытия может контактировать с первой областью/зоной пористого покрытия [т.е. вторая область/зона пористого покрытия располагается рядом или прилегает к первой области/зоне пористого покрытия]. В соответствии с другим вариантом первая область/зона пористого покрытия и вторая область/зона пористого покрытия могут быть отделены друг от друга (например, промежутком).
Когда третья область пористого покрытия размещена непосредственно на подложке, тогда первая область пористого покрытия может размещаться непосредственно на подложке (например, см. фигуры 8 и 10). Вторая область пористого покрытия может размещаться или наноситься на третью область пористого покрытия и/или первую область пористого покрытия, предпочтительно вторая область пористого покрытия размещается или наносится на третью область пористого покрытия. Третья область пористого покрытия может представлять собой третью зону пористого покрытия, а первая область пористого покрытия может представлять собой первую зону пористого покрытия. Третья область/зона пористого покрытия может контактировать с первой областью/зоной пористого покрытия [т.е. третья область/зона пористого покрытия располагается рядом или прилегает к первой области/зоне пористого покрытия]. В соответствии с другим вариантом первая область/зона пористого покрытия и третья область/зона пористого покрытия могут быть отделены друг от друга (например, промежутком).
Вторая область пористого покрытия может представлять собой второй слой пористого покрытия или вторую зону пористого покрытия, предпочтительно вторую зону пористого покрытия. Когда вторая область пористого покрытия представляет собой вторую зону пористого покрытия, катализатор окисления может дополнительно содержать четвертую область пористого покрытия (например, см. фигуру 10). Четвертая область пористого покрытия может размещаться или наноситься на первую зону пористого покрытия. Четвертая область пористого покрытия может контактировать со второй зоной пористого покрытия [т.е. четвертая область пористого покрытия располагается рядом или прилегает ко второй зоне пористого покрытия]. В соответствии с другим вариантом четвертая область пористого покрытия и вторая зона пористого покрытия могут быть отделены друг от друга (например, промежутком).
Четвертая область пористого покрытия может представлять собой четвертую зону пористого покрытия.
Третья область пористого покрытия может размещаться или наноситься на вторую область пористого покрытия (например, см. фигуру 9). Вторая область пористого покрытия может представлять собой вторую зону пористого покрытия, а первая область пористого покрытия может представлять собой первую зону пористого покрытия. Вторая область/зона пористого покрытия может контактировать с первой областью/зоной пористого покрытия [т.е. вторая область/зона пористого покрытия располагается рядом или прилегает к первой области/зоне пористого покрытия]. В соответствии с другим вариантом первая область/зона пористого покрытия и вторая область/зона пористого покрытия могут быть отделены друг от друга (например, промежутком).
Третья область пористого покрытия может представлять собой третий слой пористого покрытия или третью зону пористого покрытия.
Когда третья область пористого покрытия представляет собой третью зону пористого покрытия, тогда третья зона пористого покрытия может размещаться или наноситься на вторую зону пористого покрытия (например, см. фигуру 7). Первая область пористого покрытия может представлять собой первую зону пористого покрытия. Третья зона пористого покрытия может контактировать с первой зоной пористого покрытия [т.е. третья зона пористого покрытия располагается рядом или прилегает к первой зоне пористого покрытия]. В соответствии с другим вариантом первая зона пористого покрытия и третья зона пористого покрытия могут быть отделены друг от друга (например, промежутком).
Когда третья область пористого покрытия представляет собой третью зону пористого покрытия, тогда третья зона пористого покрытия может размещаться или наноситься на первую зону пористого покрытия (например, см. фигуру 9). Вторая область пористого покрытия может представлять собой вторую зону пористого покрытия. Третья зона пористого покрытия может контактировать со второй зоной пористого покрытия [т.е. третья зона пористого покрытия располагается рядом или прилегает ко второй зоне пористого покрытия]. В соответствии с другим вариантом вторая зона пористого покрытия и третья зона пористого покрытия могут быть отделены друг от друга (например, промежутком) и/или могут не контактировать (например, вторая зона пористого покрытия не находится в физическом контакте с третьей зоной пористого покрытия).
Когда третья область пористого покрытия представляет собой третий слой пористого покрытия, тогда третий слой пористого покрытия может размещаться или наноситься как на первую область/зону пористого покрытия, так и на вторую область/зону пористого покрытия.
Когда третья область пористого покрытия представляет собой третью зону пористого покрытия, тогда третья зона пористого покрытия, как правило, имеет длину, составляющую от 10 до 90% длины подложки (например, от 10 до 45%), предпочтительно от 15 до 75% длины подложки (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 30 до 65%, например, от 25 до 45%) длины подложки, еще более предпочтительно от 25 до 65% (например, от 35 до 50%).
Катализатор окисления согласно изобретению может содержать цеолитный катализатор. Третья область пористого покрытия и/или четвертая область пористого покрытия могут содержать цеолитный катализатор.
Третья область пористого покрытия может содержать или состоять в основном из цеолитного катализатора.
Четвертая область пористого покрытия может содержать или состоять в основном из цеолитного катализатора.
Когда третья область пористого покрытия и четвертая область пористого покрытия вместе содержат цеолитный катализатор, тогда третья область пористого покрытия может содержать или состоять в основном из первого цеолитного катализатора, а четвертая область пористого покрытия может содержать или состоять в основном из второго цеолитного катализатора, где первый цеолитный катализатор отличается (т.е. разный состав) от второго цеолитного катализатора. Первый цеолитный катализатор может представлять собой описанный выше в данном документе цеолитный катализатор. Второй цеолитный катализатор может представлять собой описанный выше в данном документе цеолитный катализатор.
Когда третья область пористого покрытия содержит или состоит в основном из цеолитного катализатора, тогда предпочтительно, первая область пористого покрытия и/или вторая область пористого покрытия не содержат цеолитного катализатора.
Когда третья область пористого покрытия содержит или состоит в основном из цеолитного катализатора, в частности цеолитного катализатора согласно первому или второму варианту осуществления цеолитного катализатора, температура, при которой адсорбированный NOx высвобождается из цеолитного катализатора, может изменяться (т.е. увеличиваться), когда катализатор окисления имеет определенную схему размещения. Подходящее изменение (т.е. увеличение) температуры цеолитного катализатора может быть получено, когда вторая область пористого покрытия размещается или наносится на третью область пористого покрытия (см., например, фигуры 6, 8 и 10).
Когда область пористого покрытия содержит цеолитный катализатор, особенно третья область пористого покрытия и/или четвертая область пористого покрытия, тогда суммарное содержание благородного металла (т.е. в цеолитном катализаторе в каждой области пористого покрытия), как правило, составляет от 0,04 до 8,83 г/дм3 (например, от 0,04 до 2,65 г/дм3), предпочтительно от 0,18 до 5,30 г/дм3 (например, от 0,18 до 1,77 г/дм3), более предпочтительно от 0,35 до 3,53 г/дм3 (например, от 0,35 до 1,41 г/дм3).
Подложки для нанесения катализаторов окисления для обработки выхлопного газа дизельного двигателя являются известными в данной области. Способы создания пористых покрытий и нанесения пористых покрытий на подложку также известны в данной области (см., например, наши публикации WO 99/47260, WO 2007/077462 и WO 2011/080525).
Подложка, как правило, имеет множество каналов (например, для протекания выхлопного газа). Как правило, подложка представляет собой керамический материал или металлический материал.
Предпочтительно, чтобы подложка изготавливалась или состояла из кордиерита (SiO2-Al2O3-MgO), карбида кремния (SiC), сплава Fe-Cr-Al, сплава Ni-Cr-Al или сплава нержавеющей стали.
Как правило, подложка представляет собой монолит (также упоминается в данном документе как монолитная подложка). Такие монолиты являются известными в данной области. Монолитная подложка может представлять собой проточный монолит или фильтрующий монолит.
Проточный монолит, как правило, включает монолит с сотовой структурой (например, металлический или керамический монолит с сотовой структурой), имеющий множество каналов, простирающихся через него, при этом такие каналы являются открытыми с обоих концов. Когда подложка представляет собой проточный монолит, тогда катализатор окисления согласно изобретению, как правило, является дизельным катализатором окисления (DOC) или предназначен для использования в качестве дизельного катализатора окисления (DOC).
Фильтрующий монолит, как правило, содержит множество впускных каналов и множество выпускных каналов, где впускные каналы открыты с расположенного выше по потоку конца (т.е. со стороны впуска выхлопного газа) и перекрыты или закупорены с расположенного ниже по потоку конца (т.е. со стороны выпуска выхлопного газа), выпускные каналы перекрыты или закупорены с расположенного выше по потоку конца и открыты с расположенного ниже по потоку конца, и где каждый впускной канал отделен от выпускного канала пористой структурой. Когда подложка представляет собой фильтрующий монолит, тогда катализатор окисления согласно изобретению, как правило, является каталитическим фильтром сажи (CSF) или предназначен для использования в качестве каталитического фильтра сажи (CSF).
Когда монолит представляет собой фильтрующий монолит, предпочтительно, чтобы фильтрующий монолит представлял собой фильтр с проточными стенками. В фильтре с проточными стенками каждый впускной канал поочередно отделяется от выпускного канала с помощью стенки с пористой структурой и наоборот. Предпочтительно, чтобы впускные каналы и выпускные каналы размещались с образованием сотовой структуры. Когда имеется сотовая структура, предпочтительно, чтобы каналы, являющиеся соседними в вертикальном и боковом направлениях к впускному каналу, были перекрыты с расположенного выше по потоку конца и наоборот (т.е. чтобы каналы, соседние в вертикальном и боковом направлениях к выпускному каналу, были перекрыты с расположенного ниже по потоку конца). При рассмотрении с любого конца, поочередно перекрытые и открытые концы каналов принимают вид шахматной доски.
В принципе, подложка может иметь любую форму или размер. Однако форму и размер подложки обычно выбирают таким образом, чтобы оптимизировать подвергание контактированию каталитически активных материалов в катализаторе с выхлопным газом. Подложка может, например, иметь трубчатую, волокнистую форму, или может состоять из частиц. Примеры подходящих несущих подложек включают подложку в виде монолитного сотового кордиерита, подложку в виде монолитного сотового SiC, подложку типа слоистого волокна или трикотажа, подложку пенистого типа, подложку поперечноточного типа, подложку в виде металлической проволочной сетки, подложку в виде металлического пористого тела и подложку в виде керамических частиц.
Как правило, катализатор окисления согласно изобретению предназначен для использования в качестве дизельного катализатора окисления (DOC) или каталитического фильтра сажи (CSF). На практике, каталитические составы, используемые в DOC и CSF, являются схожими. Как правило, принципиальное различие между DOC и CSF заключается в подложке, на которую наносится покрытие из каталитического состава, а также в суммарном количестве платины, палладия и любых других каталитически активных металлов, которые наносятся на подложку.
Следует понимать, что любое упоминание катализатора окисления согласно изобретению, предназначенного для использования в качестве дизельного катализатора окисления (DOC), может включать действие в качестве пассивного адсорбента NOx (DOC-PNA) или действие в качестве катализатора, работающего при запуске холодного двигателя (DOC-CSC).
Изобретение также предлагает выхлопную систему, содержащую катализатор окисления и устройство контроля токсичности выхлопных газов. Примеры устройства контроля токсичности выхлопных газов включают фильтр твердых частиц выхлопа дизельных двигателей (DPF), ловушку NOx, образующихся при сгорании обедненной смеси (LNT), катализатор восстановления NOx углеводородами (LNC), катализатор селективного каталитического восстановления (SCR), дизельный катализатор окисления (DOC), каталитический фильтр сажи (CSF), катализатор селективного каталитического восстановления на фильтрующей подложке (SCRF™), катализатор окисления проскочившего аммиака (ASC), а также комбинации из двух или более указанных устройств. Все указанные устройства контроля токсичности выхлопных газов являются известными в данной области.
Некоторые из вышеуказанных устройств контроля токсичности выхлопных газов имеют фильтрующие подложки. Устройство контроля токсичности выхлопных газов, имеющее фильтрующую подложку, можно выбирать из группы, состоящей из фильтра твердых частиц выхлопа дизельных двигателей (DPF), каталитического фильтра сажи (CSF) и катализатора селективного каталитического восстановления на фильтрующей подложке (SCRF™).
Предпочтительно, чтобы выхлопная система содержала устройство контроля токсичности выхлопных газов, выбираемое из группы, состоящей из ловушки NOX, образующихся при сгорании обедненной смеси (LNT), катализатора окисления проскочившего аммиака (ASC), фильтра твердых частиц выхлопа дизельных двигателей (DPF), катализатора селективного каталитического восстановления (SCR), каталитического фильтра сажи (CSF), катализатора селективного каталитического восстановления на фильтрующей подложке (SCRF™), а также комбинации из двух или более указанных устройств. Более предпочтительно устройство контроля токсичности выхлопных газов выбирают из группы, состоящей из фильтра твердых частиц выхлопа дизельных двигателей (DPF), катализатора селективного каталитического восстановления (SCR), каталитического фильтра сажи (CSF), катализатора селективного каталитического восстановления на фильтрующей подложке (SCRF™), а также комбинации из двух или более указанных устройств. Еще более предпочтительно устройство контроля токсичности выхлопных газов представляет собой катализатор селективного каталитического восстановления (SCR) или катализатора селективного каталитического восстановления на фильтрующей подложке (SCRF™).
Когда выхлопная система согласно изобретению содержит катализатор SCR или катализатор SCRF™, тогда выхлопная система может дополнительно содержать инжектор для впрыскивания азотсодержащего восстановителя, такого как аммиак или предшественник аммиака, такой как мочевина или формиат аммония, предпочтительно мочевина, в выхлопной газ ниже по потоку относительно катализатора окисления и выше по потоку относительно катализатора SCR или катализатора SCRF™. Такой инжектор может иметь соединение по текучей среде с источником (например, емкостью) предшественника азотсодержащего восстановителя. Управляемое с помощью клапана дозирование предшественника в выхлопной газ можно регулировать с помощью подходящих запрограммированных средств управления двигателем и замкнутой системы обратной связи или разомкнутой системы без обратной связи, снабженных датчиками, осуществляющими контроль состава выхлопного газа. Аммиак может также образовываться при нагревании карбамата аммония (твердое вещество), при этом образовавшийся аммиак можно впрыскивать в выхлопной газ.
В качестве другого варианта или в дополнение к инжектору, аммиак может образовываться непосредственно в системе (например, во время регенерации в условиях обогащенной смеси LNT, размещенной выше по потоку относительно катализатора SCR или катализатора SCRF™). Таким образом, выхлопная система может дополнительно содержать средства управления двигателем для обогащения выхлопного газа углеводородами.
Катализатор SCR или катализатор SCRF™ могут содержать металл, выбираемый из группы, состоящей по меньшей мере из одного металла из Cu, Hf, La, Au, В, V, лантаноидов и переходных металлов VIII группы (например, Fe), в которых металл наносится на тугоплавкий оксид или молекулярные сита. Металл предпочтительно выбирают из Ce, Fe, Cu и комбинации из любых двух или более указанных металлов, более предпочтительно металл представляет собой Fe или Cu.
Тугоплавкий оксид для катализатора SCR или катализатора SCRF™ можно выбирать из группы, состоящей из Al2O3, TiO2, CeO2, SiO2, ZrO2 и смешанных оксидов, содержащих два или более указанных оксидов. Нецеолитный катализатор может также включать оксид вольфрама (например, V2O5/WO3/TiO2, WOx/CeZrO2, WOx/ZrO2 или Fe/WOx/ZrO2).
Особенно предпочтительно, когда катализатор SCR, катализатор SCRF™ или их пористое покрытие содержат по меньшей мере одно молекулярное сито, такое как алюмосиликатный цеолит или SAPO. По меньшей мере одно молекулярное сито может быть мелкопористым, среднепористым или крупнопористым молекулярным ситом. Под «мелкопористым молекулярным ситом» в данном документе мы имеем в виду молекулярные сита, имеющие максимальный размер кольца, равный 8, такие как CHA; под «среднепористым молекулярным ситом» в данном документе мы имеем в виду молекулярное сито, имеющее максимальный размер кольца, равный 10, такое как ZSM-5; а под «крупнопористым молекулярным ситом» в данном документе мы имеем в виду молекулярное сито, имеющее максимальный размер кольца, равный 12, такое как бета. Мелкопористые молекулярные сита являются потенциально выгодными для использования в катализаторах SCR.
В выхлопной системе согласно изобретению предпочтительные молекулярные сита для катализатора SCR или катализатора SCRF™ представляют собой молекулярные сита на основе синтетического алюмосиликатного цеолита, выбираемые из группы, состоящей из AEI, ZSM-5, ZSM-20, ERI, включая ZSM-34, морденита, феррьерита, BEA, включая бета, Y, CHA, LEV, включая Nu-3, MCM-22 и EU-1, предпочтительно AEI или CHA, и имеющие отношение диоксида кремния к оксиду алюминия, составляющее от приблизительно 10 до приблизительно 50, как например, от приблизительно 15 до приблизительно 40.
В первом варианте осуществления выхлопной системы выхлопная система содержит катализатор окисления согласно изобретению, предпочтительно такой как DOC, и каталитический фильтр сажи (CSF). Такую схему размещения можно назвать DOC/CSF. За катализатором окисления, как правило, следует (например, находящийся выше по потоку) каталитический фильтр сажи (CSF). Таким образом, например, выпуск катализатора окисления соединен с впуском каталитического фильтра сажи.
Во втором варианте осуществления выхлопной системы выхлопная система содержит дизельный катализатор окисления и катализатор окисления согласно изобретению, предпочтительно такой как каталитический фильтр сажи (CSF). Такую схему размещения можно также назвать схемой размещения DOC/CSF. Как правило, за дизельным катализатором окисления (DOC) следует (например, находящийся выше по потоку) катализатор окисления согласно изобретению. Таким образом, выпуск дизельного катализатора окисления соединен с впуском катализатора окисления согласно изобретению.
Третий вариант осуществления выхлопной системы относится к выхлопной системе, содержащей катализатор окисления согласно изобретению, предпочтительно такой как DOC, каталитический фильтр сажи (CSF) и катализатор селективного каталитического восстановления (SCR). Такую схему размещения можно назвать DOC/CSF/SCR, и она является предпочтительной выхлопной системой для транспортного средства с дизельным двигателем малой мощности. За катализатором окисления, как правило, следует (например, находящийся выше по потоку) каталитический фильтр сажи (CSF). За каталитическим фильтром сажи, как правило, следует (например, находящийся выше по потоку) катализатор селективного каталитического восстановления (SCR). Инжектор азотсодержащего восстановителя может размещаться между каталитическим фильтром сажи (CSF) и катализатором селективного каталитического восстановления (SCR). Таким образом, за каталитическим фильтром сажи (CSF) может следовать (например, находящийся выше по потоку) инжектор азотсодержащего восстановителя, а за инжектором азотсодержащего восстановителя может следовать (например, находящийся выше по потоку) катализатор селективного каталитического восстановления (SCR).
Четвертый вариант осуществления выхлопной системы относится к выхлопной системе, содержащей дизельный катализатор окисления (DOC), катализатор окисления согласно изобретению, предпочтительно, такой как каталитический фильтр сажи (CSF), и катализатор селективного каталитического восстановления (SCR). Это также представляет собой схему размещения DOC/CSF/SCR. За дизельным катализатором окисления (DOC), как правило, следует (например, находящийся выше по потоку) катализатор окисления согласно изобретению. За катализатором окисления согласно изобретению, как правило, следует (например, находящийся выше по потоку) катализатор селективного каталитического восстановления (SCR). Инжектор азотсодержащего восстановителя может размещаться между катализатором окисления и катализатором селективного каталитического восстановления (SCR). Таким образом, за катализатором окисления может следовать (например, находящийся выше по потоку) инжектор азотсодержащего восстановителя, а за инжектором азотсодержащего восстановителя может следовать (например, находящийся выше по потоку) катализатор селективного каталитического восстановления (SCR).
В пятом варианте осуществления выхлопной системы, выхлопная система содержит катализатор окисления согласно изобретению, предпочтительно, такой как DOC, катализатор селективного каталитического восстановления (SCR) и либо каталитический фильтр сажи (CSF), либо фильтр твердых частиц выхлопа дизельных двигателей (DPF). Схема размещения представляет собой либо DOC/SCR/CSF, либо DOC/SCR/DPF.
В пятом варианте осуществления выхлопной системы за катализатором окисления согласно изобретению, как правило, следует (например, находящийся выше по потоку) катализатор селективного каталитического восстановления (SCR). Инжектор азотсодержащего восстановителя может размещаться между катализатором окисления и катализатором селективного каталитического восстановления (SCR). Таким образом, за катализатором окисления может следовать (например, находящийся выше по потоку) инжектор азотсодержащего восстановителя, а за инжектором азотсодержащего восстановителя может следовать (например, находящийся выше по потоку) катализатор селективного каталитического восстановления (SCR). За катализатором селективного каталитического восстановления (SCR) следует (например, находящийся выше по потоку) каталитический фильтр сажи (CSF) или фильтр твердых частиц выхлопа дизельных двигателей (DPF).
Шестой вариант осуществления выхлопной системы содержит катализатор окисления согласно изобретению, предпочтительно, такой как DOC, и катализатор селективного каталитического восстановления на фильтрующей подложке (SCRF™). Такую схему размещения можно назвать DOC/SCRF™. За катализатором окисления согласно изобретению, как правило, следует (например, находящийся выше по потоку) катализатор селективного каталитического восстановления на фильтрующей подложке (SCRF™). Инжектор азотсодержащего восстановителя может размещаться между катализатором окисления и катализатором селективного каталитического восстановления на фильтрующей подложке (SCRF™). Таким образом, за катализатором окисления может следовать (например, находящийся выше по потоку) инжектор азотсодержащего восстановителя, а за инжектором азотсодержащего восстановителя может следовать (например, находящийся выше по потоку) катализатор селективного каталитического восстановления на фильтрующей подложке (SCRF™).
В любом из описанных выше вариантов осуществления выхлопной системы с третьего по шестой, катализатор ASC может размещаться ниже по потоку относительно катализатора SCR или катализатора SCRF™ (т.е. в виде отдельной монолитной подложки), или более предпочтительно зона на расположенном ниже по потоку или заднем конце монолитной подложки, содержащей катализатор SCR, может использоваться для нанесения ASC.
Другой аспект изобретения относится к транспортному средству или устройству. Транспортное средство или устройство содержат дизельный двигатель. Дизельный двигатель может быть двигателем компрессионного воспламенения однородной смеси (HCCI), двигателем компрессионного воспламенения предварительно перемешанной смеси (PCCI) или двигателем низкотемпературного горения (LTC). Предпочтительно, чтобы дизельный двигатель представлял собой обычный (т.е. традиционный) дизельный двигатель.
Транспортное средство может представлять собой транспортное средство с дизельным двигателем малой мощности (LDV), например, соответствующее определению в американском или европейском законодательстве. Транспортное средство с дизельным двигателем малой мощности, как правило, имеет массу <2840 кг, более предпочтительно массу <2610 кг.
В США транспортное средство с дизельным двигателем малой мощности (LDV) относится к транспортному средству с дизельным двигателем, имеющему полную массу≤3856 кг. В Европе термин транспортное средство с дизельным двигателем малой мощности (LDV) относится к (i) пассажирским транспортным средствам, содержащим не более восьми мест помимо места для водителя, и имеющим максимальную массу не более 5 тонн, а также к (ii) транспортным средствам для грузоперевозок, имеющим максимальную массу не более 12 тонн.
В соответствии с другим вариантом транспортное средство может представлять собой транспортное средство с дизельным двигателем большой мощности (HDV), такое как транспортное средство с дизельным двигателем, имеющее полную массу >3856 фунтов, согласно определению в американском законодательстве.
Изобретение также предлагает способ регулирования содержания NOx в выхлопном газе дизельного двигателя для устройства контроля токсичности выхлопных газов. Стадия (b) поступления обработанного выхлопного газа в устройство контроля токсичности выхлопных газов, как правило, включает прямое поступление обработанного выхлопного газа в устройство контроля токсичности выхлопных газов. Таким образом, выпуск катализатора окисления непосредственно соединен (например, при отсутствии промежуточной стадии) с впуском устройства контроля токсичности выхлопных газов.
Как правило, устройство контроля токсичности выхлопных газов представляет собой катализатор селективного каталитического восстановления (SCR), катализатор селективного каталитического восстановления на фильтрующей подложке (SCRF™), фильтр твердых частиц выхлопа дизельных двигателей (DPF) или каталитический фильтр сажи (CSF). Предпочтительно, чтобы устройство контроля токсичности выхлопных газов представляло собой катализатор селективного каталитического восстановления (SCR) или катализатор селективного каталитического восстановления на фильтрующей подложке (SCRF™).
Любое упоминание «регулирования содержания NOx» в данном контексте, особенно в отношении аспектов способа или использования согласно изобретению, относится к изменению (т.е. регулированию) или поддержанию, предпочтительно изменению, отношения (в ч./млн. или % об., как правило, при температуре и давлении выхлопного газа) NO:NO2 таким образом, чтобы оно находилось в пределах заданного отношения при конкретной температуре выхлопного газа или в конкретном диапазоне значений температуры. Заданное отношение, как правило, составляет менее 17:3, предпочтительно должно составлять от 5:1 до 1:5, более предпочтительно от 2,5:1 до 1:2,5 и еще более предпочтительно от 2:1 до 1:2 (например, от 1,5:1 до 1:1,5 или приблизительно 1:1).
Изобретение также относится к использованию катализатора окисления при регенерации устройства контроля токсичности выхлопных газов, имеющего фильтрующую подложку (например, расположенного ниже по потоку устройства контроля токсичности выхлопных газов, имеющего фильтрующую подложку).
Устройство контроля токсичности выхлопных газов, имеющее фильтрующую подложку, можно выбирать из группы, состоящей из фильтра твердых частиц выхлопа дизельных двигателей (DPF), каталитического фильтра сажи (CSF), катализатора селективного каталитического восстановления на фильтрующей подложке (SCRF™) и комбинации из двух или более указанных устройств.
Когда катализатор окисления согласно изобретению используется при регенерации устройства контроля токсичности выхлопных газов, имеющего фильтрующую подложку, он может использоваться при активной или пассивной регенерации устройства контроля токсичности выхлопных газов, предпочтительно при активной регенерации.
Катализатор окисления может использоваться для регенерации устройства контроля токсичности выхлопных газов, имеющего фильтрующую подложку, при температуре не менее 220°C, предпочтительно не менее 240°C, более предпочтительно не менее 260°C, еще более предпочтительно не менее 280°C, путем окисления оксида азота (NO) до диоксида азота (NO2).
Определения
Термин «пористое покрытие» является известным в данной области и относится к плотно прилегающему покрытию, которое обычно наносится на подложку при получении катализатора.
Термин «область пористого покрытия» в данном контексте относится к участку пористого покрытия на подложке. «Область пористого покрытия» может, например, размещаться или наноситься на подложку в виде «слоя» или «зоны». Площадь или размещение пористого покрытия на подложке, как правило, задают в процессе нанесения пористого покрытия на подложку. «Область пористого покрытия», как правило, имеет четкие границы или края (т.е. можно отличить одну область пористого покрытия от другой области пористого покрытия при использовании обычных аналитических методов).
Как правило, «область пористого покрытия» имеет практически равномерную длину. Упоминание «практически равномерной длины» в данном контексте относится к длине, значение которой не отклоняется (например, разница между максимальным и минимальным значениями длины) более чем на 10%, предпочтительно не отклоняется более чем на 5%, более предпочтительно не отклоняется более чем на 1%, от ее среднего значения.
Предпочтительно, чтобы каждая «область пористого покрытия» имела практически однородный состав (т.е. отсутствуют какие-либо существенные различия в составе пористого покрытия при сравнении одной части области пористого покрытия с другой частью указанной области пористого покрытия). Практически однородный состав в данном контексте относится к материалу (например, области пористого покрытия), в котором различие в составе при сравнении одной части области пористого покрытия с другой частью области пористого покрытия составляет 5% или менее, обычно 2,5% или менее, и наиболее часто 1% или менее.
Термин «зона пористого покрытия» в данном контексте относится к области пористого покрытия, имеющей длину меньше суммарной длины подложки, например, ≤75% от суммарной длины подложки. «Зона пористого покрытия», как правило, имеет длину (т.е. практически равномерную длину) не менее 5% (например, ≥5%) от суммарной длины подложки.
Суммарная длина подложки - это расстояние между ее впускным концом и ее выпускным концом (например, противоположными концами подложки).
Любое упоминание «зоны пористого покрытия, размещенной на впускном конце подложки» в данном контексте относится к зоне пористого покрытия, размещенной или нанесенной на подложку, где зона пористого покрытия находится ближе к впускному концу подложки по сравнению с положением зоны пористого покрытия относительно выпускного конца подложки. Таким образом, середина зоны пористого покрытия (т.е. на половине ее длины) находится ближе к впускному концу подложки по сравнению с положением середины относительно выпускного конца подложки. Аналогичным образом, любое упоминание «зоны пористого покрытия, размещенной на выпускном конце подложки» в данном контексте относится к зоне пористого покрытия, размещенной или нанесенной на подложку, где зона пористого покрытия находится ближе к выпускному концу подложки по сравнению с положением зоны пористого покрытия относительно впускного конца подложки. Таким образом, середина зоны пористого покрытия (т.е. на половине ее длины) находится ближе к выпускному концу подложки по сравнению с положением середины относительно впускного конца подложки.
Когда подложка представляет собой фильтр с проточными стенками, тогда, как правило, любое упоминание «зоны пористого покрытия, размещенной на впускном конце подложки» относится к зоне пористого покрытия, размещенной или нанесенной на подложку, которая:
(a) находится ближе к впускному концу (например, открытому концу) впускного канала подложки по сравнению с положением зоны пористого покрытия относительно закрытого конца (например, заблокированного или перекрытого конца) впускного канала, и/или
(b) находится ближе к закрытому концу (например, заблокированному или перекрытому концу) выпускного канала подложки по сравнению с положением зоны пористого покрытия относительно выпускного конца (например, открытого конца) выпускного канала.
Таким образом, середина зоны пористого покрытия (т.е. на половине ее длины) (a) находится ближе к впускному концу впускного канала подложки по сравнению с положением середины относительно закрытого конца впускного канала и/или (b) находится ближе к закрытому концу выпускного канала подложки по сравнению с положением середины относительно выпускного конца выпускного канала.
Аналогичным образом, любое упоминание «зоны пористого покрытия, размещенной на выпускном конце подложки», когда подложка представляет собой фильтр с проточными стенками, относится к зоне пористого покрытия, размещенной или нанесенной на подложку, которая:
(a) находится ближе к выпускному концу (например, открытому концу) выпускного канала подложки по сравнению с положением зоны пористого покрытия относительно закрытого конца (например, заблокированного или перекрытого) выпускного канала, и/или
(b) находится ближе к закрытому концу (например, заблокированному или перекрытому концу) впускного канала подложки по сравнению с ее положением относительно впускного конца (например, открытого конца) впускного канала.
Таким образом, середина зоны пористого покрытия (т.е. на половине ее длины) (a) находится ближе к выпускному концу выпускного канала подложки по сравнению с положением середины относительно закрытого конца выпускного канала и/или (b) находится ближе к закрытому концу впускного канала подложки по сравнению с положением середины относительно впускного конца впускного канала.
Зона пористого покрытия может соответствовать как (a), так и (b), когда пористое покрытие присутствует в стенке фильтра с проточными стенками (т.е. зона пористого покрытия внутри стенки).
Аббревиатура «PGM» в данном контексте обозначает «металл платиновой группы». Термин «металл платиновой группы», как правило, относится к металлу, выбираемому из группы, состоящей из Ru, Rh, Pd, Os, Ir и Pt, предпочтительно металлу, выбираемому из группы, состоящей из Ru, Rh, Pd, Ir и Pt. Как правило, термин «PGM» предпочтительно относится к металлу, выбираемому из группы, состоящей из Rh, Pt и Pd.
Термин «смешанный оксид» в данном контексте, как правило, относится к однофазной смеси оксидов, что обычно известно в данной области. Термин «сложный оксид» в данном контексте, как правило, относится к композиции из оксидов, имеющей более одной фазы, что обычно известно в данной области.
Любое упоминание зон пористого покрытия, которые «практически не накладываются», в данном контексте относится к наложению (т.е. между концами соседних зон на подложке) менее 10% от длины подложки, предпочтительно менее 7,5% от длины подложки, более предпочтительно менее 5% от длины подложки, особенно менее 2,5% от длины подложки, еще более предпочтительно менее 1% от длины подложки и наиболее предпочтительно наложение отсутствует.
Выражение «состоит в основном» в данном контексте ограничивает объем признака таким образом, чтобы он включал конкретные материалы, а также любые другие материалы или стадии, которые не оказывают существенного влияния на основные характеристики указанного признака, такие как, например, незначительные примеси. Выражение «состоит в основном из» включает в себя выражение «состоящий из».
Выражение «практически не содержит» в данном контексте в отношении материала, как правило, в контексте содержимого области пористого покрытия, слоя пористого покрытия или зоны пористого покрытия, означает, что материал присутствует в незначительном количестве, таком как≤5% масс., предпочтительно≤2% масс., более предпочтительно≤1% масс. Выражение «практически не содержит» включает в себя выражение «не содержит».
Любая ссылка на количество легирующей добавки, в частности на суммарное количество, выраженное в виде % масс., в данном контексте имеет отношение к массе материала-носителя или соответствующего тугоплавкого оксида металла.
Примеры
Теперь изобретение будет проиллюстрировано посредством следующих неограничивающих примеров.
Пример 1
Первая суспензия была получена следующим образом: порошок диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. Добавили растворимую платиновую соль, а затем нитрат марганца. Смесь перемешали для получения однородного состояния.
Суспензию нанесли на выпускном конце кордиеритового проточного монолита, имеющего 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Затем она была высушена и прокалена при 500°C. Содержание Pt на участке составляло 0,53 г/дм3. Содержание марганца на участке составляло 1,77 г/дм3.
Для получения второй суспензии взяли порошок диоксида кремния-оксида алюминия и измельчили до d90<20 мкм. В суспензию добавили ацетат бария, а затем подходящие количества растворимых солей платины и палладия. Добавили цеолит бета, в результате чего суспензия содержала 77% диоксида кремния-оксида алюминия и 23% цеолита по массе. Затем суспензию перемешали для получения однородного состояния. Полученный состав для нанесения пористого покрытия нанесли на впускные каналы проточного монолита с использованием стандартных методов нанесения покрытия. Участок высушили и прокалили при 500°C. Конечный катализатор имел суммарное содержание Pt 1,59 г/дм3 и содержание Pd 0,53 г/дм3.
Пример 2
Первая суспензия была получена и нанесена на кордиеритовый проточный монолит, имеющий 62 ячейки на см2 с использованием таких же способов, как в примере 1. Содержание Pt на участке составляло 0,53 г/дм3. Содержание марганца на участке составляло 1,77 г/дм3.
Для получения второй суспензии взяли порошок диоксида кремния-оксида алюминия и измельчили до d90<20 мкм. Добавили растворимую платиновую соль, а затем нитрат марганца. Добавили цеолит бета, в результате чего суспензия содержала 77% диоксида кремния-оксида алюминия и 23% цеолита по массе. Затем суспензию перемешали для получения однородного состояния. Полученный состав для нанесения пористого покрытия нанесли на впускные каналы проточного монолита с использованием стандартных методов нанесения покрытия. Участок высушили и прокалили при 500°C. Конечный катализатор имел суммарное содержание Pt 1,91 г/дм3 и содержание марганца 3,53 г/дм3.
Результаты эксперимента
Измерение окислительной активности
Для каждого из катализаторов согласно примерам 1 и 2 были взяты образцы сердцевины. Сердцевины были подвергнуты гидротермальному «старению» в сушильном шкафу при 750°C в течение 15 часов с использованием 10% воды. Каталитическую активность определили, применяя лабораторные испытания с использованием синтетического газа. Сердцевины были испытаны в газовом устройстве для модельного испытания активности катализатора (SCAT) при использовании входящей газовой смеси, указанной в таблице 1. В каждом случае остальная часть представляла собой азот. Окислительную активность в отношении CO и HC определяли по рабочей температуре, при которой достигалась конверсия 50% (T50). Окислительную активность в отношении NO определяли как процент конверсии при 300°C.
Таблица 1
Результаты
Результаты испытаний SCAT показаны ниже в таблице 2.
Таблица 2
Результаты в таблице 2 показывают, что примеры 1 и 2 имеют очень близкие значения рабочих температур T50 для CO и HC. Это означает, что примеры 1 и 2 имеют очень близкие показатели окислительной активности в отношении CO и HC. Пример 2 содержит одновременно платину и марганец как в покрытии на впуске, так и в покрытии на выпуске. Пример 1 содержит одновременно платину и марганец только в покрытии на выпуске. Пример 2 имеет более высокую окислительную активность в отношении NO при 300°C, чем пример 1.
Пример 3
Порошок диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. Добавили подходящие количества растворимых солей платины и палладия. Смесь перемешали для получения однородного состояния. Состав для нанесения пористого покрытия нанесли на кордиеритовый проточный монолит, имеющий 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Затем его высушили и прокалили при 500°C. Суммарное содержание PGM для участка составляло 2,12 г/дм3. Отношение Pt:Pd составляло 4:1 по массе.
Пример 4
Порошок диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. Добавили подходящие количества растворимых солей платины и палладия, а затем нитрат марганца. Смесь перемешали для получения однородного состояния. Состав для нанесения пористого покрытия нанесли на кордиеритовый проточный монолит, имеющий 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Затем его высушили и прокалили при 500°C. Суммарное содержание PGM для участка составляло 2,12 г/дм3. Отношение Pt:Pd составляло 4:1 по массе. Содержание марганца составляло 3,53 г/дм3.
Пример 5
Порошок диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. Добавили подходящие количества растворимых солей платины и палладия. Смесь перемешали для получения однородного состояния. Состав для нанесения пористого покрытия нанесли на кордиеритовый проточный монолит, имеющий 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Затем его высушили и прокалили при 500°C. Суммарное содержание PGM для участка составляло 2,12 г/дм3. Отношение Pt:Pd составляло 10:1 по массе.
Пример 6
Порошок диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. Добавили подходящие количества растворимых солей платины и палладия, а затем нитрат марганца. Смесь перемешали для получения однородного состояния. Состав для нанесения пористого покрытия нанесли на кордиеритовый проточный монолит, имеющий 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Затем его высушили и прокалили при 500°C. Суммарное содержание PGM для участка составляло 2,12 г/дм3. Отношение Pt:Pd составляло 10:1 по массе. Содержание марганца составляло 3,53 г/дм3.
Результаты эксперимента
Измерение окислительной активности
Для каждого из катализаторов согласно примерам 3, 4, 5 и 6 были взяты образцы сердцевины. Сердцевины были подвергнуты гидротермальному «старению» в сушильном шкафу при 800°C в течение 16 часов с использованием 10% воды. Каталитическую активность определили, применяя лабораторные испытания с использованием синтетического газа. Сердцевины были испытаны в газовом устройстве для модельного испытания активности катализатора (SCAT) при использовании входящей газовой смеси, указанной в таблице 1. В каждом случае остальная часть представляла собой азот. Окислительную активность в отношении CO и HC определяли по рабочей температуре, при которой достигалась конверсия 50% (T50).
Результаты
Результаты испытаний SCAT показаны ниже в таблице 3.
Таблица 3
Результаты в таблице 3 показывают рабочие температуры T50 для CO и HC для примеров 3, 4, 5 и 6. Примеры 3 и 4 имеют одинаковое содержание благородных металлов и массовое отношение Pt:Pd, равное 4:1. Пример 4, который содержит марганец, имеет более низкое значение рабочей температуры T50 для CO, чем пример 3, который не содержит марганца. Значения рабочей температуры T50 для HC для примеров 3 и 4 являются идентичными. Примеры 5 и 6 имеют одинаковое содержание благородных металлов и массовое отношение Pt:Pd, равное 10:1. Пример 6, который содержит марганец, имеет более низкое значение рабочей температуры катализатора T50 для CO и HC, чем пример 5, который не содержит марганца.
Более значительное улучшение в отношении рабочей температуры для содержащего марганец катализатора наблюдается при массовом отношении Pt:Pd, равном 10:1, чем при 4:1.
Пример 7
Порошок диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. Добавили растворимую платиновую соль и перемешали смесь для получения однородного состояния. Суспензию нанесли на кордиеритовый проточный монолит, имеющий 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Затем ее высушили и прокалили при 500°C. Содержание Pt на участке составляло 1,06 г/дм3.
Пример 8
Повторили способ согласно примеру 1, за исключением того, что добавили растворимую платиновую соль, а затем нитрат марганца, перед тем как перемешать смесь для получения однородного состояния. Содержание Pt на участке составляло 1,06 г/дм3. Содержание марганца на участке составляло 1,41 г/дм3. Массовое отношение Pt к марганцу составляло 3:4.
Пример 9
Использовался способ согласно примеру 2, за исключением того, что вместо порошка диоксида кремния-оксида алюминия суспендировали в воде и измельчили до d90<20 мкм порошок оксида алюминия. Содержание Pt на участке составляло 1,06 г/дм3. Содержание марганца на участке составляло 1,41 г/дм3. Массовое отношение Pt к марганцу составляло 3:4.
Пример 10
Легированный диоксидом кремния порошок оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. В суспензию добавили ацетат бария, а затем подходящие количества растворимых солей платины и палладия. Добавили цеолит бета, в результате чего суспензия содержала 78% легированного диоксидом кремния оксида алюминия и 22% цеолита по массе. Затем суспензию перемешали для получения однородного состояния. Полученный состав для нанесения пористого покрытия нанесли на впускные каналы кордиеритового проточного монолита, имеющего 62 ячейки на см2, с использованием стандартных методов нанесения покрытия, после чего высушили.
При получении второй суспензии брали легированный диоксидом кремния порошок оксида алюминия и измельчали до d90<20 мкм. В суспензию добавили растворимую платиновую соль, а затем раствор нитрата марганца. Суспензию перемешали для получения однородного состояния. Данную вторую суспензию нанесли на выпускные каналы проточного монолита с использованием стандартных методов нанесения покрытия. Участок высушили и затем прокалили при 500°C. Конечный катализатор имел суммарное содержание PGM 2,65 г/дм3 и содержание марганца в покрытии на выпуске 3,53 г/дм3. Массовое отношение платины к марганцу составляло 3:5.
Пример 11
Легированный диоксидом кремния порошок оксида алюминия суспендировали в воде и измельчили до d90<20 мкм. В суспензию добавили ацетат бария, а затем подходящие количества растворимых солей платины и палладия. Добавили цеолит бета, в результате чего суспензия содержала 78% легированного диоксидом кремния оксида алюминия и 22% цеолита по массе. Затем суспензию перемешали для получения однородного состояния. Полученный состав для нанесения пористого покрытия нанесли на впускные каналы кордиеритового проточного монолита, имеющего 62 ячейки на см2, с использованием стандартных методов нанесения покрытия, после чего высушили.
При получении второй суспензии брали легированный диоксидом кремния порошок оксида алюминия и измельчали до d90<20 мкм. В суспензию добавили подходящее количество растворимой платиновой соли, а также ее перемешали для получения однородного состояния. Данную вторую суспензию нанесли на выпускные каналы проточного монолита с использованием стандартных методов нанесения покрытия. Участок высушили и затем прокалили при 500°C. Конечный катализатор имел суммарное содержание PGM 2,65 г/дм3.
Результаты эксперимента
Измерение в отношении окисления NO
Для каждого из катализаторов согласно примерам с 1 по 5 были взяты образцы сердцевины. Все сердцевины были подвергнуты гидротермальному «старению» в сушильном шкафу при 750°C в течение 15 часов. Взяли дополнительные сердцевины для примеров 4 и 5 и оставили их в «свежем» состоянии (т.е. их не подвергали какой-либо тепловой обработке в сушильном шкафу).
Каталитическую активность определили, применяя лабораторные испытания на активность с использованием синтетического газа (SCAT). Находящиеся в свежем состоянии и подвергнутые старению сердцевины были испытаны в газовом устройстве для модельного испытания активности катализатора (SCAT) при использовании входящей газовой смеси, указанной в таблице 1. В каждом случае остальная часть представляла собой азот.
Результаты
Результаты испытаний SCAT показаны ниже в таблицах 4-6.
Таблица 4
Результаты в Таблице 4 показывают окислительную активность в отношении NO для примеров 7, 8 и 9 при 250°C. Пример 7 (не содержит марганца) имеет самую низкую окислительную активность в отношении NO. Пример 8 (содержит марганец и носитель на основе диоксида кремния-оксида алюминия) имеет самую высокую окислительную активность в отношении NO. Пример 9 (содержит марганец и носитель на основе оксида алюминия) имеет промежуточный показатель окислительной активности в отношении NO. Включение марганца дает в результате улучшенную окислительную активность в отношении NO. Результаты для примера 8 показывают, что комбинация марганца с материалом-носителем, содержащим легированный оксид алюминия, такой как легированный диоксидом кремния оксид алюминия, является особенно выгодной.
Результаты для примеров 10 и 11 показаны в таблицах 5 и 6.
Таблица 5
Результаты в таблице 5 показывают, что катализатор согласно примеру 10 (содержит марганец в содержащей Pt зоне) в состоянии после старения имеет большую окислительную активность в отношении NO при 200°C, чем катализатор согласно примеру 11.
Таблица 6
Результаты в таблице 6 показывают, что катализатор согласно примеру 10 в состоянии после старения показывает лучшую окислительную активность в отношении NO при 220°C, чем катализатор согласно примеру 11.
В таблицах 5 и 6 также показано, что различие в окислительной активности в отношении NO между свежим состоянием и состоянием после старения катализатора имеет меньшее значение для примера 10, чем для примера 11. Таким образом, катализатор согласно примеру 10 показывает более стабильную окислительную активность в отношении NO, чем катализатор согласно примеру 11. Стабильная окислительная активность в отношении NO при переходе от свежего состояния к состоянию после старения является выгодной для регулирований дозирования в процессе SCR ниже по потоку.
Пример 12
К суспензии мелкопористого цеолита со структурой CHA добавили нитрат Pd и перемешали. Суспензию нанесли на кордиеритовый проточный монолит, имеющий структуру, содержащую 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Покрытие высушили и прокалили при 500°C. Получили покрытие, содержащее Pd-замещенный цеолит. Содержание Pd в данном покрытии составляло 1,41 г/дм3.
Вторую суспензию получили с использованием порошка диоксида кремния-оксида алюминия, измельченного до d90<20 мкм. Добавили растворимую платиновую соль, при этом смесь перемешали для получения однородного состояния. Суспензию нанесли на выпускной конец проточного монолита с использованием стандартных методов нанесения покрытия. Затем покрытие высушили.
Третью суспензию получили с использованием порошка диоксида кремния-оксида алюминия, измельченного до d90<20 мкм. Добавили подходящие количества растворимых солей платины и палладия, а затем цеолит бета, в результате чего суспензия содержала 75% легированного диоксидом кремния оксида алюминия и 25% цеолита по массе. Затем суспензию перемешали для получения однородного состояния. Полученный состав для нанесения пористого покрытия нанесли на впускные каналы проточного монолита с использованием стандартных методов нанесения покрытия. Затем участок высушили и прокалили при 500°C. Содержание Pt для готового участка составляло 1,80 г/дм3, а содержание Pd составляло 1,68 г/дм3.
Пример 13
К суспензии мелкопористого цеолита со структурой CHA добавили нитрат Pd и перемешали. Суспензию нанесли на кордиеритовый проточный монолит, имеющий структуру, содержащую 62 ячейки на см2, с использованием стандартных методов нанесения покрытия. Покрытие высушили и прокалили при 500°C. Получили покрытие, содержащее Pd-замещенный цеолит. Содержание Pd в данном покрытии составляло 1,41 г/дм3.
Вторую суспензию получили с использованием 5% марганца на носителе из порошка диоксида кремния-оксида алюминия, измельченного до d90<20 мкм. Добавили растворимую платиновую соль, при этом смесь перемешали для получения однородного состояния. Суспензию нанесли на выпускной конец проточного монолита с использованием стандартных методов нанесения покрытия. Затем покрытие высушили.
Третью суспензию получили с использованием порошка диоксида кремния-оксида алюминия, измельченного до d90<20 мкм. Добавили подходящие количества растворимых солей платины и палладия, а затем цеолит бета, в результате чего суспензия содержала 75% легированного диоксидом кремния оксида алюминия и 25% цеолита по массе. Затем суспензию перемешали для получения однородного состояния. Полученный состав для нанесения пористого покрытия нанесли на впускные каналы проточного монолита с использованием стандартных методов нанесения покрытия. Затем участок высушили и прокалили при 500°C. Содержание Pt для готового участка составляло 1,80 г/дм3, а содержание Pd составляло 1,68 г/дм3.
Результаты эксперимента
Измерение в отношении окисления NO
Для катализаторов согласно примерам 12 и 13 были взяты образцы сердцевины. Обе сердцевины были подвергнуты гидротермальному «старению» в сушильном шкафу при 800°C в течение 16 часов при 10% воды.
Каталитическую активность определили, применяя лабораторные испытания на активность с использованием синтетического газа (SCAT). Подвергнутые старению сердцевины были испытаны в газовом устройстве для модельного испытания активности катализатора (SCAT) при использовании входящей газовой смеси, указанной в таблице 1. В каждом случае остальная часть представляла собой азот.
Результаты
Результаты испытаний SCAT показаны ниже в таблице 7.
Результаты в таблице 7 показывают окислительную активность в отношении NO для примеров 12 и 13 при 250°C. Пример 13, который содержит марганец, показывает более высокую степень протекания окисления NO при 250°C, чем пример 12.
Во избежание каких-либо недоразумений, полное содержание любого и всех документов, упомянутых в данном документе, включено в качестве ссылки в настоящую заявку.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИЗЕЛЬНЫЙ ОКИСЛИТЕЛЬНЫЙ КАТАЛИЗАТОР, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ АДСОРБЕРА NOx | 2015 |
|
RU2704801C2 |
| КАТАЛИТИЧЕСКИЙ ФИЛЬТР САЖИ ДЛЯ ОБРАБОТКИ ВЫХЛОПНОГО ГАЗА ДВИГАТЕЛЯ КОМПРЕССИОННОГО ВОСПЛАМЕНЕНИЯ | 2013 |
|
RU2650522C2 |
| КАТАЛИЗАТОР ДЛЯ ХОЛОДНОГО ЗАПУСКА И ЕГО ПРИМЕНЕНИЕ В ВЫХЛОПНЫХ СИСТЕМАХ | 2014 |
|
RU2692809C1 |
| КАТАЛИЗАТОР ДЛЯ ОЧИСТКИ ВЫХЛОПНОГО ГАЗА | 2015 |
|
RU2724261C2 |
| КАТАЛИЗИРОВАННЫЙ БЛОК-НОСИТЕЛЬ | 2012 |
|
RU2618058C2 |
| ДИЗЕЛЬНЫЙ КАТАЛИЗАТОР ОКИСЛЕНИЯ, ИМЕЮЩИЙ ОБЛАСТЬ УЛАВЛИВАНИЯ ДЛЯ ПРИМЕСЕЙ В ВЫХЛОПНЫХ ГАЗАХ | 2016 |
|
RU2750389C2 |
| ВЫХЛОПНАЯ СИСТЕМА, СОДЕРЖАЩАЯ ПАССИВНЫЙ NO-АДСОРБЕР | 2016 |
|
RU2716957C2 |
| ПАССИВНЫЙ NOx-АДСОРБЕР | 2016 |
|
RU2737175C2 |
| КАТАЛИЗАТОР ОКИСЛЕНИЯ ДИЗЕЛЬНОГО ТОПЛИВА, ИМЕЮЩИЙ ОБЛАСТЬ ЗАХВАТА ДЛЯ СЕРОСОДЕРЖАЩИХ ПРИМЕСЕЙ | 2017 |
|
RU2783210C2 |
| ОКИСЛИТЕЛЬНЫЙ КАТАЛИЗАТОР И СИСТЕМА ВЫПУСКА ВЫХЛОПНЫХ ГАЗОВ ДЛЯ ДИЗЕЛЬНОГО ДВИГАТЕЛЯ | 2015 |
|
RU2733407C2 |
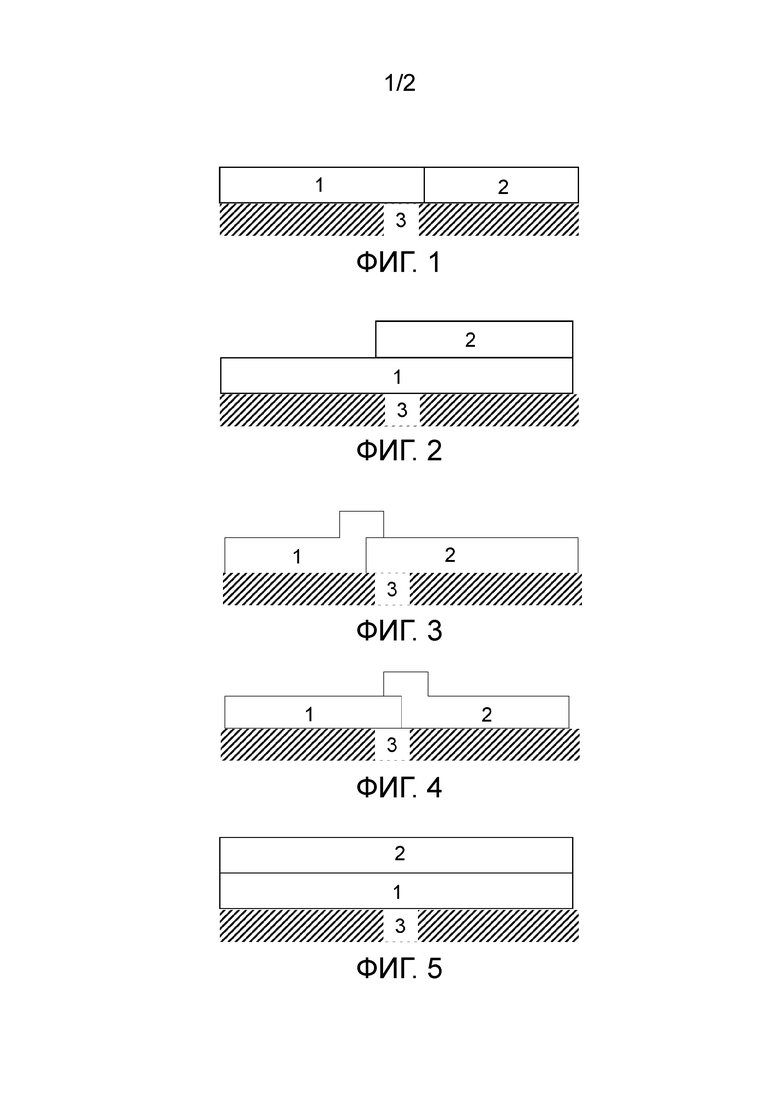
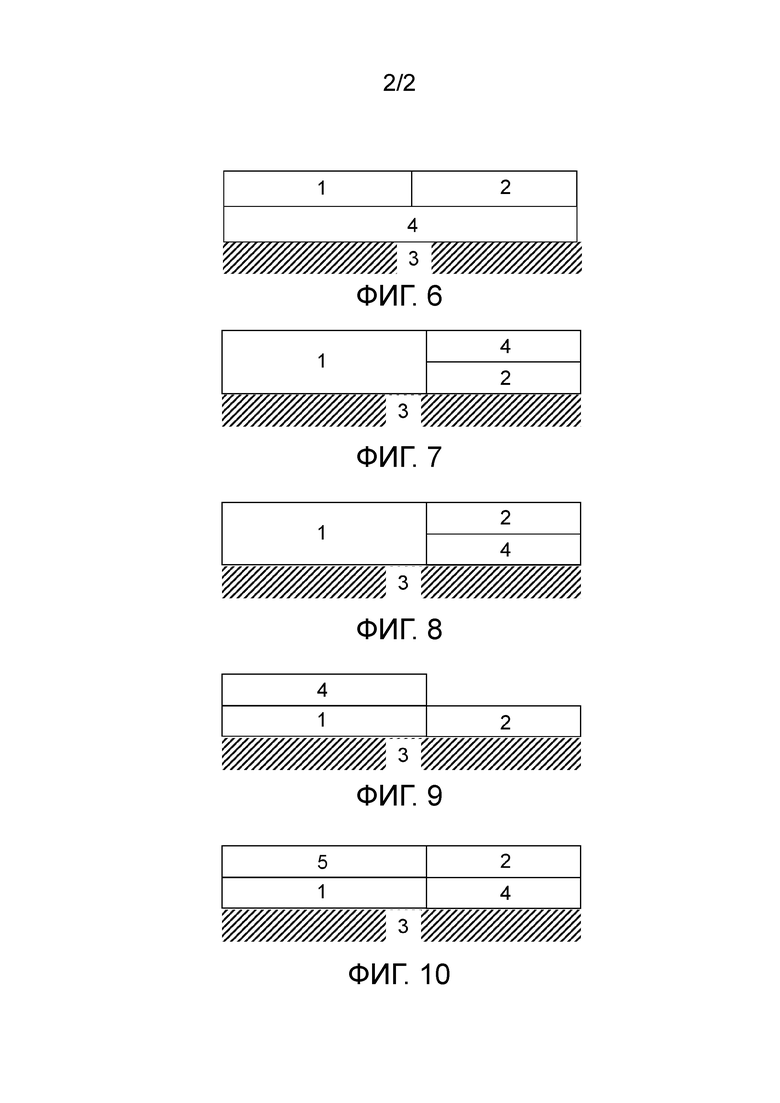
Изобретение относится к выхлопной системе для дизельного двигателя, включающей катализатор окисления для обработки выхлопного газа из дизельного двигателя и устройство контроля токсичности выхлопных газов, в которой катализатор окисления содержит: первую область пористого покрытия, содержащую платину (Pt), марганец (Mn), первый материал-носитель и адсорбирующий углеводороды материал, который представляет собой цеолит, и при этом марганец (Mn) размещен на первом материале-носителе или нанесен на первый материал-носитель; вторую область пористого покрытия, содержащую металл платиновой группы (PGM), марганец (Mn) и второй материал-носитель, содержащий тугоплавкий оксид металла, который представляет собой диоксид кремния-оксид алюминия или оксид алюминия, легированный диоксидом кремния, где указанная вторая область пористого покрытия содержит платину (Pt) в качестве единственного металла платиновой группы (PGM), и где металл платиновой группы (PGM) размещен на втором материале-носителе или нанесен на второй материал-носитель, марганец (Mn) размещен на втором материале-носителе или нанесен на второй материал-носитель, и где второй материал-носитель не содержит адсорбирующего углеводороды материала, который представляет собой цеолит; и подложку, имеющую впускной конец и выпускной конец; в котором первая область пористого покрытия представляет собой первую зону пористого покрытия, размещенную на впускном конце подложки, а вторая область пористого покрытия представляет собой вторую зону пористого покрытия, размещенную на выпускном конце подложки, так что вторая область пористого покрытия размещена таким образом, чтобы контактировать с выхлопным газом на выпускном конце подложки после контактирования выхлопного газа с первой областью пористого покрытия. Изобретение также относится к транспортному средству и способу обработки выхлопного газа дизельного двигателя. Технический результат заключается в разработке каталитических составов с большим сроком службы, имеющих низкие «рабочие» температуры. 3 н. и 14 з.п. ф-лы, 10 ил., 7 табл., 12 пр.
1. Выхлопная система для дизельного двигателя, включающая катализатор окисления для обработки выхлопного газа из дизельного двигателя и устройство контроля токсичности выхлопных газов, в которой катализатор окисления содержит:
первую область пористого покрытия, содержащую платину (Pt), марганец (Mn), первый материал-носитель и адсорбирующий углеводороды материал, который представляет собой цеолит, и при этом марганец (Mn) размещен на первом материале-носителе или нанесен на первый материал-носитель;
вторую область пористого покрытия, содержащую металл платиновой группы (PGM), марганец (Mn) и второй материал-носитель, содержащий тугоплавкий оксид металла, который представляет собой диоксид кремния-оксид алюминия или оксид алюминия, легированный диоксидом кремния, где указанная вторая область пористого покрытия содержит платину (Pt) в качестве единственного металла платиновой группы (PGM), и где металл платиновой группы (PGM) размещен на втором материале-носителе или нанесен на второй материал-носитель, марганец (Mn) размещен на втором материале-носителе или нанесен на второй материал-носитель, и где вторая область пористого покрытия не содержит адсорбирующего углеводороды материала, который представляет собой цеолит; и
подложку, имеющую впускной конец и выпускной конец;
в котором первая область пористого покрытия представляет собой первую зону пористого покрытия, размещенную на впускном конце подложки, а вторая область пористого покрытия представляет собой вторую зону пористого покрытия, размещенную на выпускном конце подложки, так что вторая область пористого покрытия размещена таким образом, чтобы контактировать с выхлопным газом на выпускном конце подложки после контактирования выхлопного газа с первой областью пористого покрытия.
2. Выхлопная система по п. 1, в которой первая зона пористого покрытия и вторая зона пористого покрытия размещаются на подложке в виде одного единого слоя.
3. Выхлопная система по п. 1, в которой первая область пористого покрытия представляет собой первый слой пористого покрытия, и в котором вторая зона пористого покрытия размещается на первом слое пористого покрытия.
4. Выхлопная система по п. 1, в которой первая область пористого покрытия представляет собой первый слой пористого покрытия, а вторая область пористого покрытия представляет собой второй слой пористого покрытия, и в котором второй слой пористого покрытия размещается на первом слое пористого покрытия.
5. Выхлопная система по п. 4, в которой второй слой пористого покрытия распространяется по всей длине подложки.
6. Выхлопная система по любому из пп. 1-5, в которой первая область пористого покрытия дополнительно содержит палладий.
7. Выхлопная система по п. 6, в которой первая область пористого покрытия имеет в своем составе платину и палладий в отношении по суммарной массе платины к палладию ≥ 1:1
8. Выхлопная система по любому из пп. 1-5, в которой первая область пористого покрытия содержит платину (Pt) как единственный металл платиновой группы (PGM).
9. Выхлопная система по любому из пп. 1-5, в которой первый материал-носитель содержит тугоплавкий оксид металла, выбираемый из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, диоксида церия, а также смешанного или сложного оксида на их основе, в котором тугоплавкий оксид металла необязательно легирован легирующей добавкой.
10. Выхлопная система по п. 9, где тугоплавкий оксид металла выбирают из группы, состоящей из оксида алюминия, диоксида кремния, диоксида титана, диоксида циркония, диоксида церия, диоксида кремния-оксида алюминия, диоксида титана-оксида алюминия, диоксида циркония-оксида алюминия, диоксида церия-оксида алюминия, диоксида титана-диоксида кремния, диоксида циркония-диоксида кремния, диоксида циркония-диоксида титана, диоксида церия-диоксида циркония и оксида алюминия-оксида магния.
11. Выхлопная система по п. 10, в которой тугоплавкий оксид металла представляет собой оксид алюминия, легированный диоксидом кремния.
12. Выхлопная система по любому из пп. 1-5, в которой катализатор окисления дополнительно содержит третью область пористого покрытия, где третья область пористого покрытия размещается непосредственно на подложке, и где первая область пористого покрытия нанесена на третью область пористого покрытия и вторая область пористого покрытия нанесена на третью область пористого покрытия.
13. Выхлопная система по п. 12, в которой третья область пористого покрытия содержит цеолитный катализатор, где цеолитный катализатор содержит благородный металл и цеолит, и при этом благородный металл представляет собой палладий.
14. Выхлопная система по любому из пп. 1-5, в которой подложка является проточной подложкой.
15. Выхлопная система по любому из пп. 1-5, в которой устройство контроля токсичности выхлопных газов выбирают из группы, состоящей из ловушки NOx, образующегося при сгорании обедненной смеси (LNT), катализатора окисления проскочившего аммиака (ASC), фильтра твердых частиц выхлопа дизельных двигателей (DPF), катализатора селективного каталитического восстановления (SCR), каталитического фильтра сажи (CSF), катализатора селективного каталитического восстановления на фильтрующей подложке (SCRF™), а также комбинаций из двух или более указанных устройств.
16. Транспортное средство, содержащее дизельный двигатель и выхлопную систему по любому из пп. 1-5.
17. Способ обработки выхлопного газа дизельного двигателя, включающий пропускание выхлопного газа через выхлопную систему по любому из пп. 1-5.
| WO 2006044974 A1, 27.04.2006 | |||
| Бетоноукладчик,например,для подводного бетонирования | 1981 |
|
SU1046423A2 |
| US 8449852 B1, 28.05.2013 | |||
| EP 1264629 B1, 27.10.2004 | |||
| US 6221804 B1, 24.04.2001 | |||
| СПОСОБ СЖИГАНИЯ УГЛЕВОДОРОДНЫХ ТОПЛИВ (ВАРИАНТЫ) И КАТАЛИЗАТОРЫ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2008 |
|
RU2372556C2 |

