Родственная заявка
Настоящая заявка испрашивает приоритет на основании заявки на патент Индии под номером 5080/СНЕ/2015, поданной 23 сентября 2015, и заявки под номером 6013/СНЕ/2015, поданной 06 ноября 2015, которые включены в содержание настоящего документа посредством ссылки.
Область техники
Настоящее изобретение относится к новым кристаллическим формам промежуточного продукта позаконазола и способу получения позаконазола посредством этих форм. В частности, настоящее изобретение относится к новым кристаллическим формам промежуточного продукта позаконазола и способам получения новых кристаллических форм промежуточного продукта позаконазола, представленного следующей структурной формулой:
который представляет собой ключевой промежуточный продукт в производстве позаконазола. Позаконазол, известный под торговым названием NOXAFIL, представляет собой триазольный противогрибковый агент, показанный для профилактики инвазивной инфекции Aspergillus и Candida у пациентов в возрасте 13 лет и старше, которые имеют высокий риск развития этих инфекций по причине серьезного ослабления иммунитета, например у реципиентов ТКСК (трансплантация кроветворных стволовых клеток) с БТПХ (болезнь «трансплантат против хозяина») или у пациентов, имеющих злокачественные заболевания системы крови, с пролонгированной нейтропенией, вызванной химиотерапией. NOXAFIL также показан к применению для лечения орофарингеального кандидоза (ОФК), включая трудно поддающийся лечению ОФК (тОФК) с помощью итраконазола и/или флуконазола.
Предшествующий уровень техники
Позаконазол, известный под торговым названием NOXAFIL, представляет собой триазольный противогрибковый агент, показанный для лечения инвазивных грибковых инфекций.
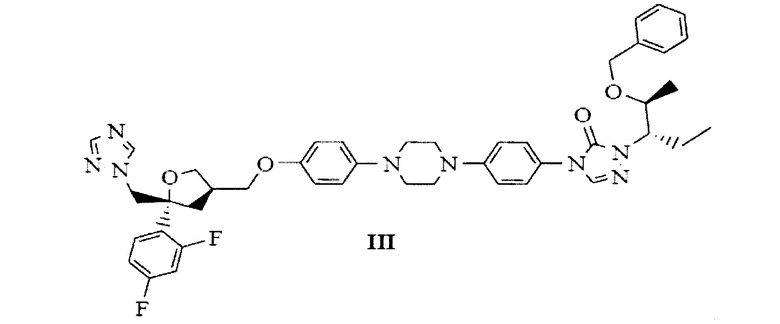
В качестве эффективного и высокопроизводительного способа получения позаконазола предложено следующее: получение промежуточного продукта позаконазола, здесь и далее по тексту обозначаемого как бензилпозаконазол, представленного структурной формулой III:
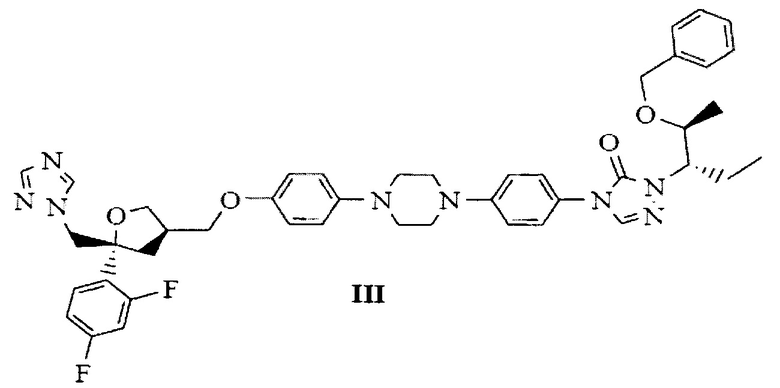
подходит для крупномасштабного производства; данный промежуточный продукт представляет собой кристаллическое вещество, что обуславливает легкую растворимость данного кристаллического промежуточного продукта и в целом влияет на выход активного фармацевтического ингредиента (АФИ) позаконазола.
О кристаллических формах бензилпозаконазола сообщалось ранее. В международной заявке на патент WO 2011158248 (А2) заявлен бензилпозаконазол в форме А, которая характеризуется порошковой рентгеновской дифрактограммой с пиками примерно при значениях 2,04; 6,1; 12,24; 15,06; 15,73; 17,17; 17,37; 18,15; 19,42; 19,97; 24,34; 26,0; а в WO 2013042138 (A3) заявлен кристаллический бензилпозаконазол в форме М, которая характеризуется порошковой рентгеновской дифрактограммой с пиками примерно при значениях 3,90; 5,86; 7,82; 9,82; 11,79; 12,59; 13,38; 13,74; 16,24; 16,95; 17,83; 18,90; 20,09; 21,49; 22,89; 24,93; 26,53; 27,56; 28,73; 29,88 и 34,20. В целях обеспечения способа с более высоким выходом позаконазола в крупномасштабном производстве, необходимо разработать новые кристаллические формы бензилпозаконазола.
Вышеуказанная необходимость обеспечивается настоящим изобретением за счет новых кристаллических полиморфов бензилпозаконазола В-1, В-2 и В-3. Эти новые полиморфы характеризуются порошковой рентгеновской дифрактограммой с указанными ниже пиками.
Пиковые значения 2θ кристаллической формы В-1: 4,14; 6,85; 9,92; 15,28; 15,69; 16,59; 17,29; 17,92; 18,27; 18,91; 20,63; 19,95; 23,58; и 25,12 плюс/минус 0,2 градуса 2θ.
Пиковые значения 2θ кристаллической формы В-2: 4,20; 9,27; 14,22; 15,46; 15,82; 16,45; 17,31; 19,09; 20,47; 21,18; и 24,74 плюс/минус 0,2 градуса 2θ.
Пиковые значения 2θ кристаллической формы В-3: 7,07; 14,49; 15,68; 16,50; 18,05; 20,13; 20,87; 22,85; и 24,42 плюс/минус 0,2 градуса 2θ.
Стандартная точность значений 2-тета кристаллических форм В-1, В-2 и В-3 находится в интервале примерно плюс/минус 0,2 градуса 2θ.
Патентная ссылка 1: международная публикация согласно РСТ WO 2011158248;
Патентная ссылка 2: международная публикация согласно РСТ WO 2013042138.
Цель настоящего документа - предложить новые кристаллические полиморфные формы бензилпозаконазола и способы их получения в промышленном масштабе.
Целью настоящего документа является также предложить эффективный и высокопроизводительный способ получения аморфной формы позаконазола; предложен однореакторный способ с применением новых кристаллических форм бензилпозаконазола В-1, В-2 и В-3.
О способах получения аморфных форм позаконазола сообщалось ранее в данной области техники. В международной патентной публикации WO 2013042138 (А2) раскрыто несколько однореакторных способов получения аморфной формы позаконазола с помощью 5% Pd-C и 5 н HCl в метаноле в качестве растворителя. В публикации WO 2015059716 (A3) раскрыто несколько способов получения аморфной формы позаконазола, включающих растворение позаконазола в дихлорметане, перемешивание реакционной смеси, фильтрацию реакционной смеси, внесение фильтрата в метил-трет-бутиловый эфир (МТБЭ), перемешивание реакционной смеси, фильтрацию твердого вещества и затем высушивание с получением аморфной формы позаконазола. В публикации CN 104370894 раскрыты способы получения аморфного позаконазола: позаконазол растворяют в смешанном растворителе, состоящего из спиртов и сложных эфиров, и затем вносят по каплям к алкану или ароматическому углеводороду с получением аморфного позаконазола.
Патентная ссылка 3: международная публикация согласно РСТ WO 2015059716 (A3);
Патентная ссылка 4: заявка на патент Китая CN 104370894.
Указанные выше описания получения аморфной формы позаконазола имеют различные недостатки в отношении выхода и качества аморфного позаконазола. В некоторых случаях для однореакторных способах использование катализаторов на основе тяжелых металлов с целью дебензилирования с последующим выделением аморфного вещества приводит к более высоким издержкам производства, а в других описаниях включено выделение позаконазола в твердой форме, за которым следует еще одна стадия аморфизации, что опять же приводит к увеличению затратности и длительности производственного процесса.
Тем не менее, цель настоящего изобретения - предложить способ получения аморфного позаконазола, который несложен в осуществлении, подходит для масштабов промышленного производства, затратоэффективен и имеет высокий выход.
Целью настоящего документа является также предложить однореакторный способ для получения аморфного позаконазола с помощью новой кристаллической полиморфной формы бензилпозаконазола В-3.
Краткое описание фигур
Для того чтобы содержание настоящего документа было легче понять и осуществить на практике, ниже даны ссылки на примеры воплощений, представленных со ссылкой на сопутствующие фигуры. Эти фигуры вместе с представленным ниже подробным описанием включены в описание настоящего изобретения и составляют его часть, и служат наглядным, более подробным представлением воплощений и разъяснением различных принципов и преимуществ, в соответствии с содержанием настоящего документа, в том числе:
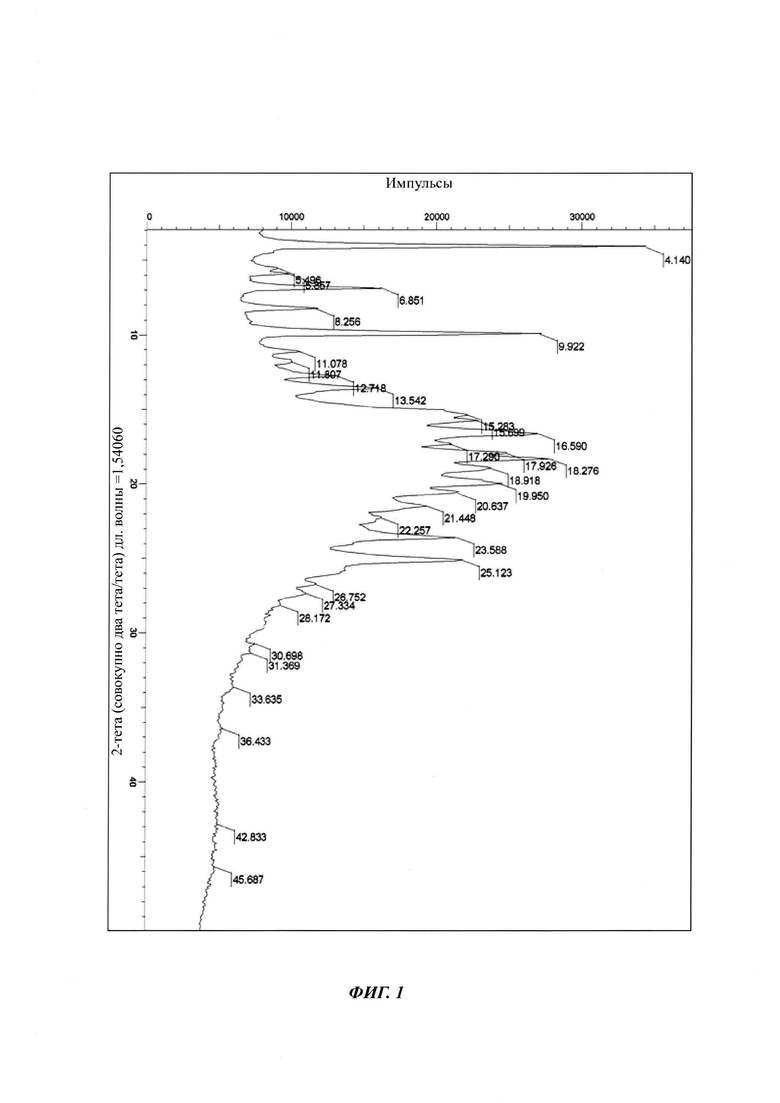
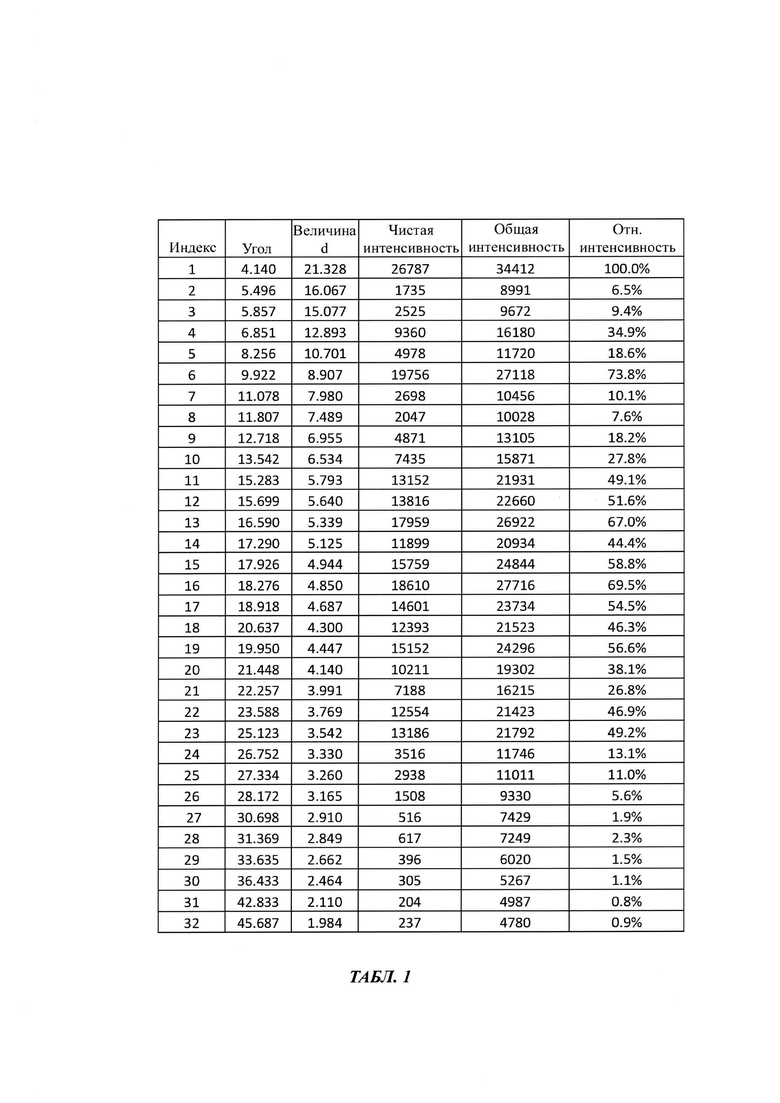
на Фиг. 1 показана рентгеновская дифрактограмма кристаллической формы В-1 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III);
в Таблице 1 представлены данные рентгеновской дифрактограммы кристаллической формы В-1 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III), как показано на фиг. 1. В частности, в Таблице 1 представлены соответствующие значения угла 2 тета кристаллической формы В-1. Где значения угла 2 тета кристаллической формы В-1 характеризуются пиками, выраженными в градусах 2θ плюс/мингус 0,2θ, при значениях около 4,14, 5,49, 5,85, 6,85, 8,25, 9,92, 11,07, 11,80, 12,71, 13,54, 15,28, 15,69, 16,59, 17,29, 17,92, 18,27, 18,92, 19,95, 20,63, 21,44, 22,25, 23,58, 25,12, 26,75, 27,33, 28,17, 30,69, 31,36, 33,63, 36,43, 42,83, 45,68;
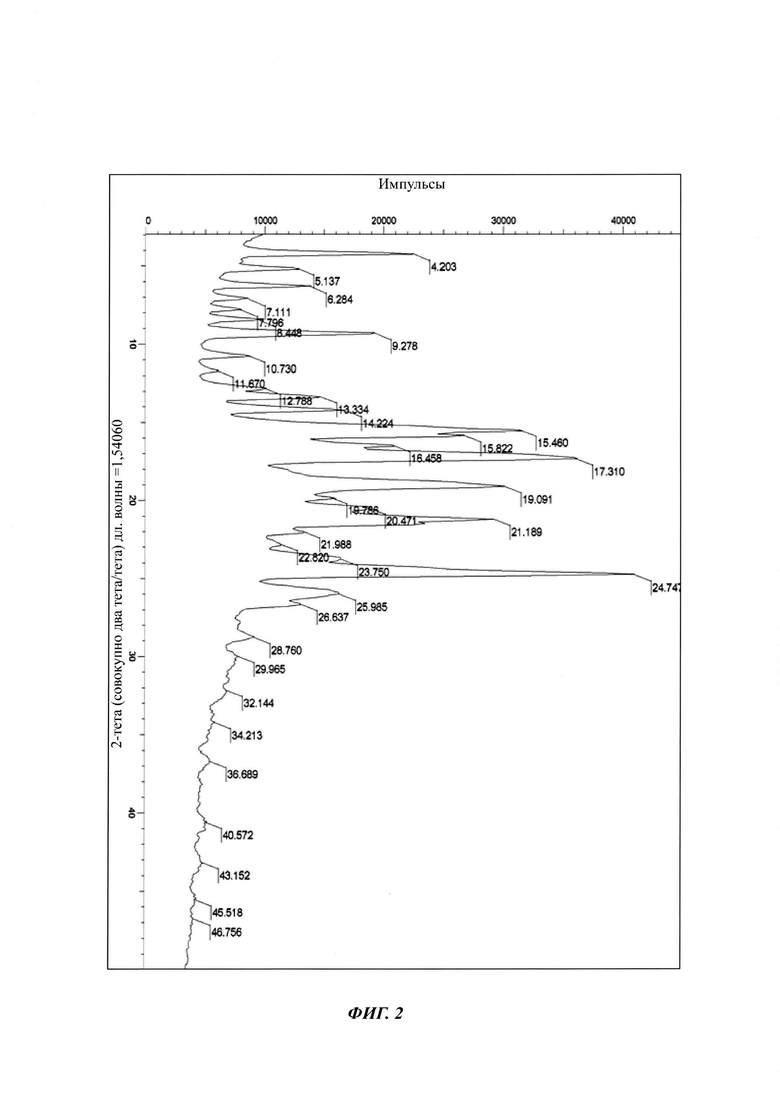
на Фиг. 2 показана рентгеновская дифрактограмма кристаллической формы В-2 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III);
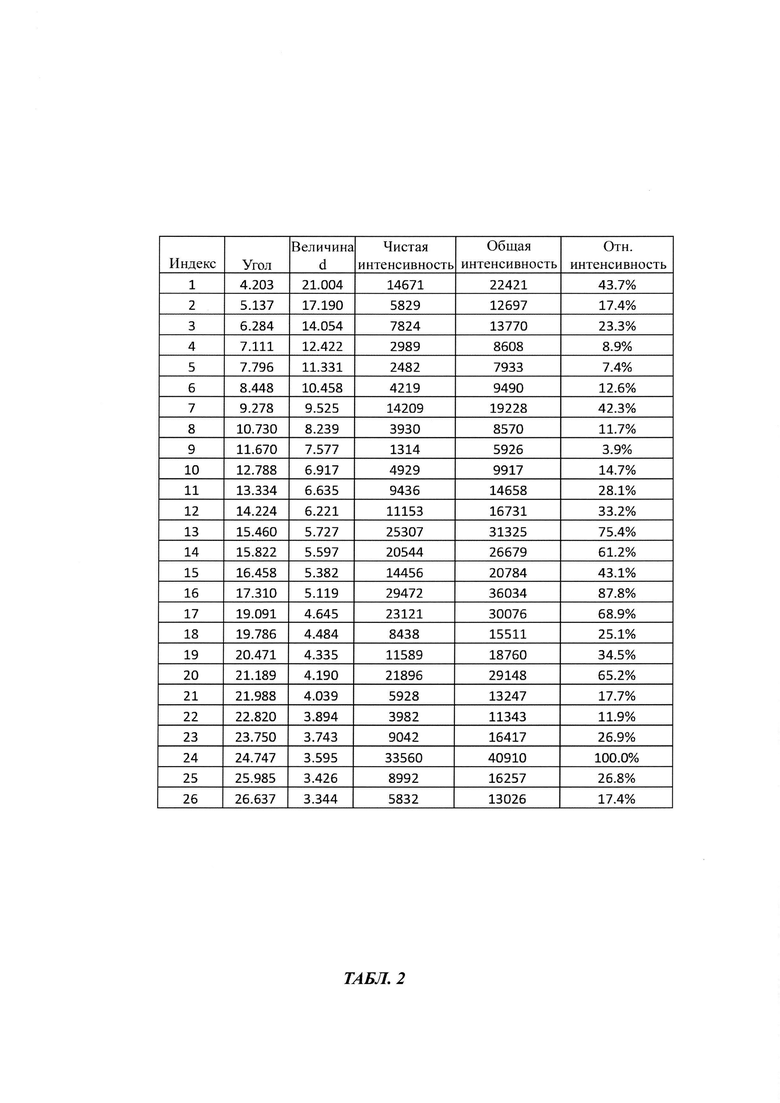
в Таблице 2 представлены данные рентгеновской дифрактограммы кристаллической формы В-2 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III), как показано на фиг. 2. В частности, в Таблице 2 представлены соответствующие значения угла 2 тета кристаллической формы В-2. Где значения угла 2 тета кристаллической формы В-2 характеризуются пиками, выраженными в градусах 2θ плюс/мингус 0,2θ, при значениях около 4,20, 5,13, 6,28, 7,11, 7,79, 8,44, 9,27, 10,73, 11,67, 12,78, 13,33, 14,22, 15,46, 15,82, 16,45, 17,31, 19,09, 19,78, 20,47, 21,18, 21,98, 22,82, 23,75, 24,74, 25,98, 26,63;
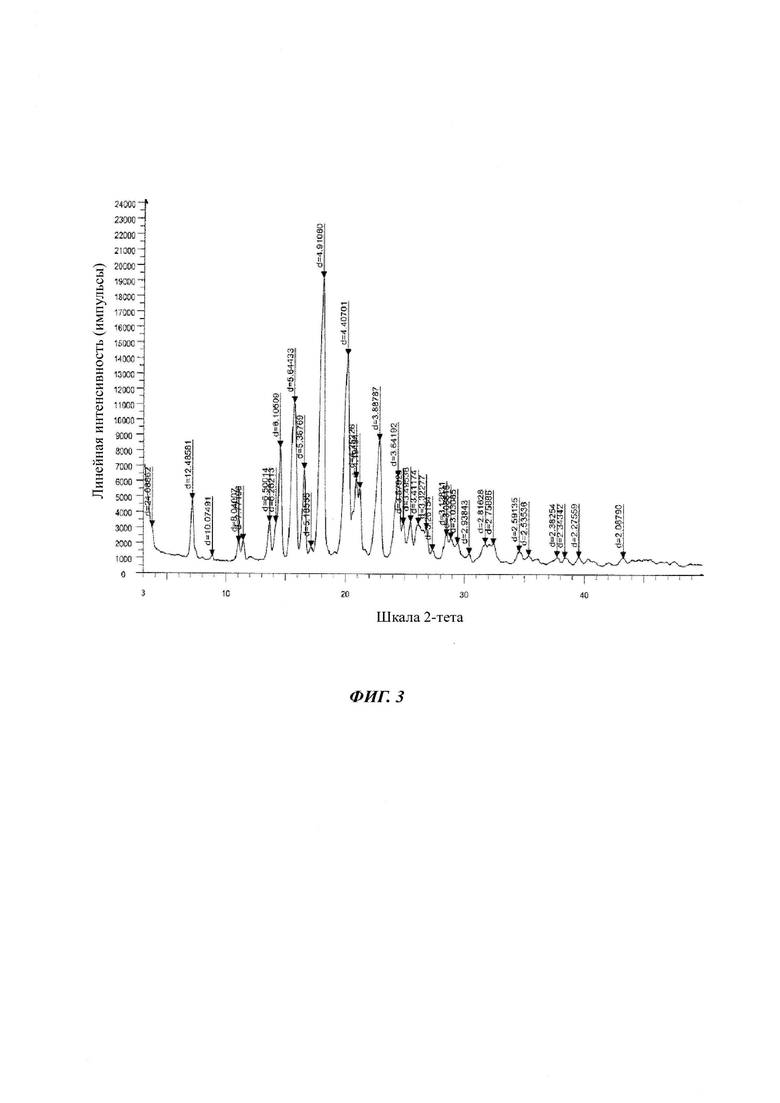
на Фиг. 3 показана рентгеновская дифрактограмма кристаллической формы В-3 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III);
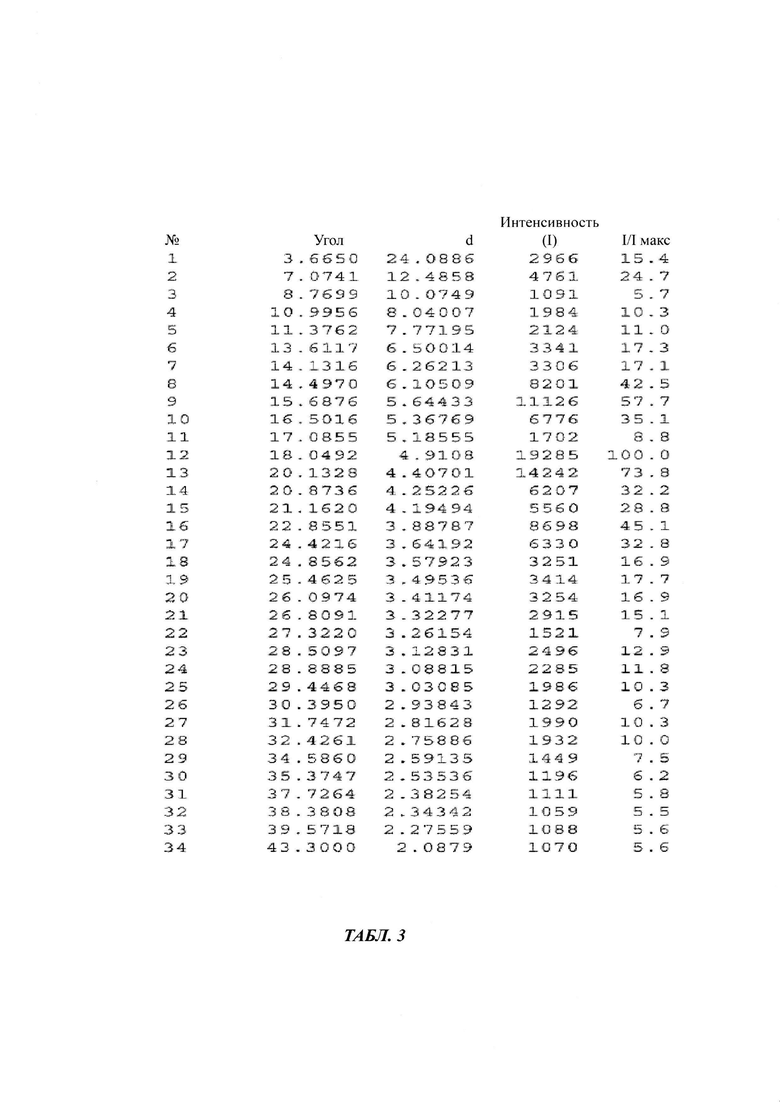
в Таблице 3 представлены данные рентгеновской дифрактограммы кристаллической формы В-3 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III), как показано на фиг. 3. В частности, в Таблице 3 представлены соответствующие значения угла 2 тета кристаллической формы В-3. Где значения угла 2 тета кристаллической формы В-3 характеризуются пиками, выраженными в градусах 2θ плюс/мингус 0,2θ, при значениях около 3,66, 7,07, 8,76, 10,99, 11,37, 13,61, 14,13, 14,49, 15,68, 16,50, 17,08, 18,04, 20,13, 20,87, 21,16, 22,85, 24,42, 24,85, 25,46, 26,09, 26,80, 27,32, 28,50, 28,88, 29,44, 30,39, 31,74, 32,42, 34,58, 35,37, 37,72, 38,38, 39,57, 43,30;
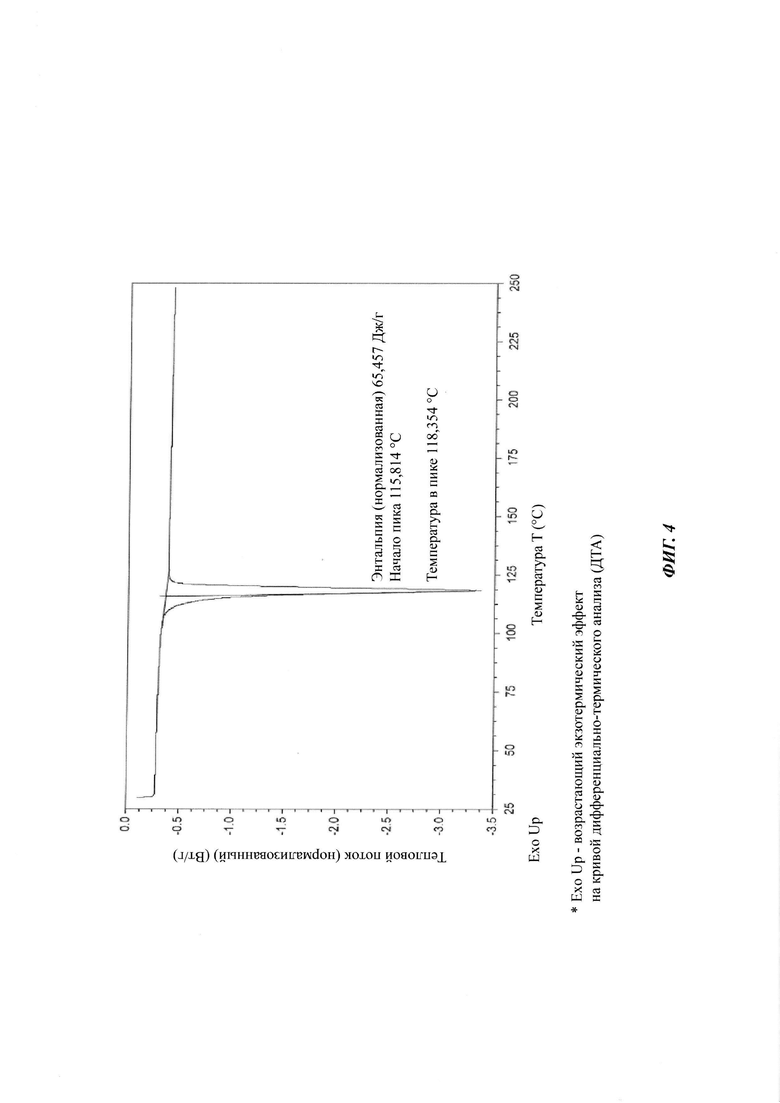
на Фиг. 4 показан спектр ДСК (дифференциальная сканирующая калориметрия) кристаллической формы В-3 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III);
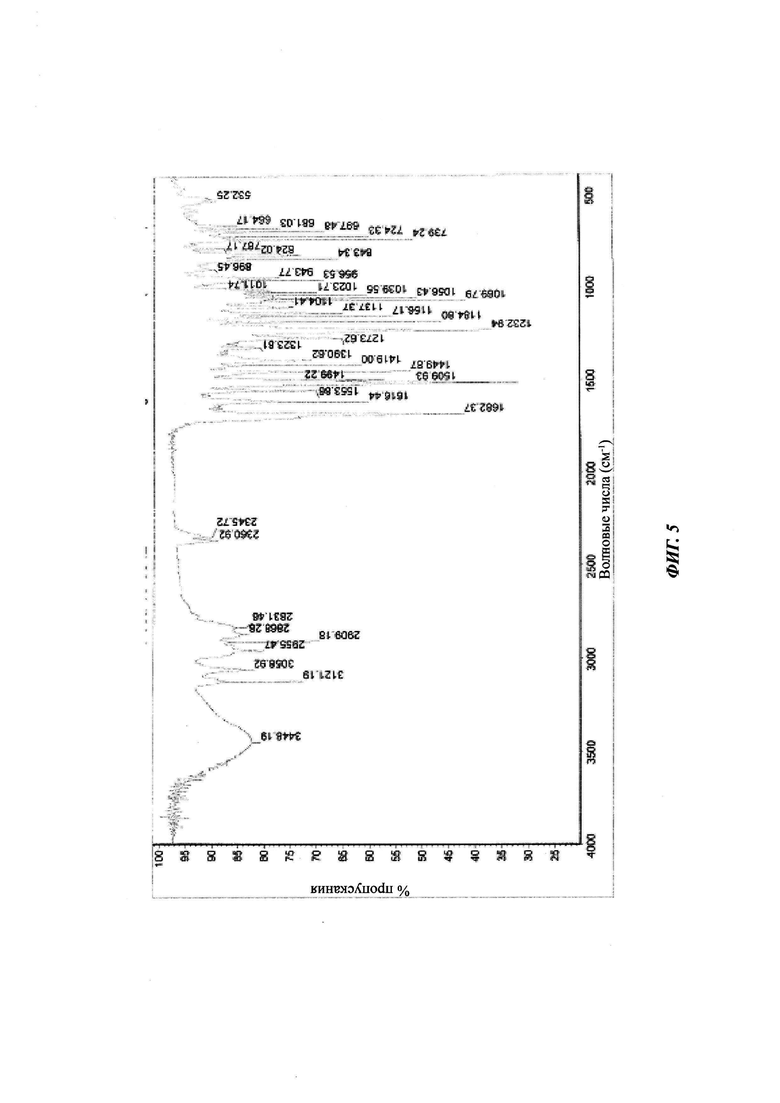
на Фиг. 5 показан ИК-спектр кристаллической формы В-3 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III);
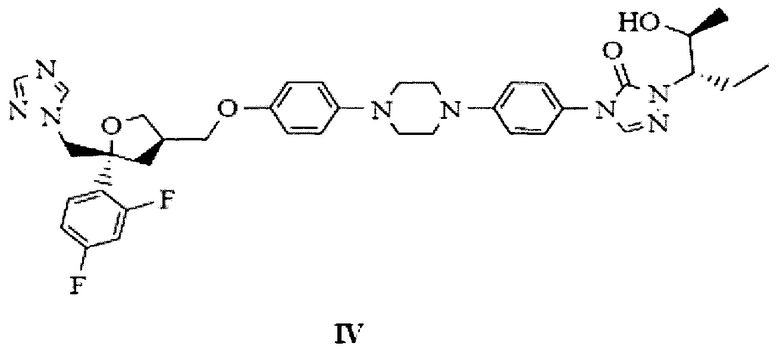
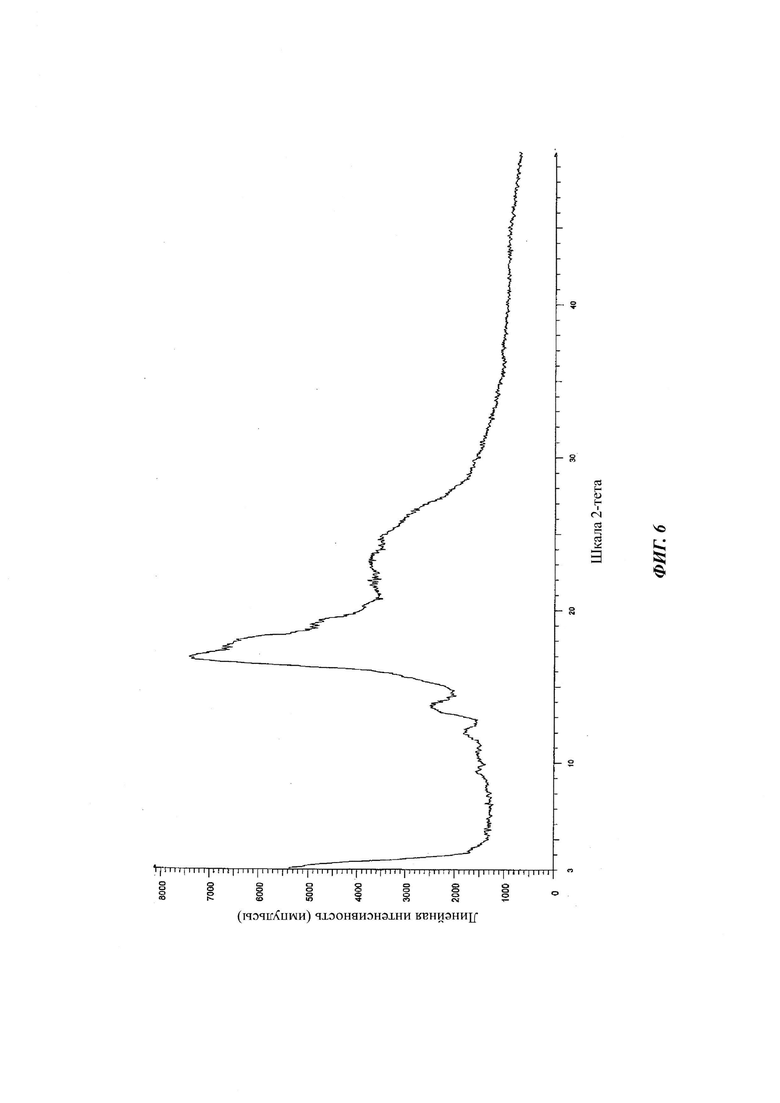
на Фиг. 6 показана рентгеновская дифрактограмма аморфной формы 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV);
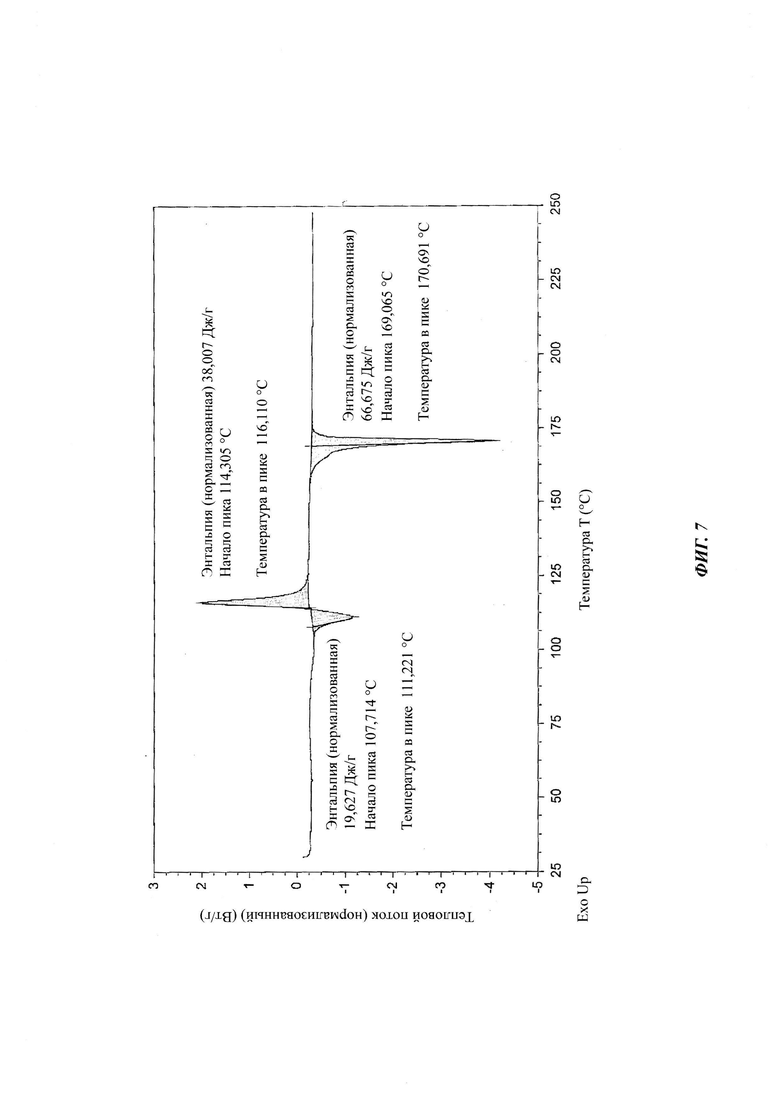
на Фиг. 7 показан спектр ДСК аморфной формы 4-[4-[4-[4-[[(3R,5R)-5- (2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV).
Способы анализа соединений, представленные на фигурах выше, описаны далее.
Порошковая рентгеновская дифракция
Примерно 300 мг порошкообразного образца помещали на держатель для образца, плотно и гомогенно утрамбовывали на держателе для образца с помощью стеклянной пластинки, и записывали спектр порошковой рентгеновской дифракции на дифрактометре Bruker D8 Advance (Bruker-AXS, Карлсруэ, Германия) с помощью рентгеновского излучения Cu-Ka (λ равен 1,5406  ) при 40 кВ и 30 мА. Порошковые рентгеновские дифрактограммы накапливали в интервале 2θ 3-50° при скорости сканирования 1°/мин.
) при 40 кВ и 30 мА. Порошковые рентгеновские дифрактограммы накапливали в интервале 2θ 3-50° при скорости сканирования 1°/мин.
Анализ ДСК (дифференциальная сканирующая калориметрия)
ДСК осуществляли на модуле Mettler Toledo DSC 822е. Образец (4-6 мг) помещали в завальцованные, но имеющие отверстие алюминиевые кюветы для образца. Температурный интервал составлял 30-250°С при 10°С/мин. Образцы продували током азота, который подавали со скоростью 80 мл/мин.
ИК-анализ (инфракрасная спектроскопия)
ИК-спектроскопию осуществляли на приборе Fisher Scientific (NICOLET-iS50-FTIR). Примерно 5 мг образца распределяли на участке для образцов, с использованием метода НПВО (нарушенное полное внутреннее отражение; в качестве кристалла использовали алмаз), и снимали спектр образца в интервале от 4000 см-1 до 400 см-1 до получения спектра подходящей интенсивности (около 60% трансмиссии при 2000 см-1).
Подробное описание изобретения
Настоящее изобретение относится к кристаллическим формам бензилпозаконазола структурной формулы III, полезным при получении позаконазола, представленного следующей структурной формулой IV.
Настоящим изобретением предложено следующее: при конденсации ((3S,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил-4-метилбензолсульфоната, представленного следующей структурной формулой (I)
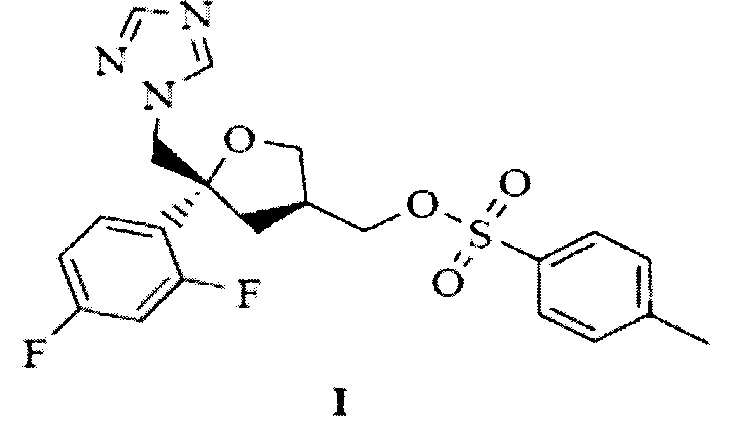
и 1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-она, представленного следующей структурной формулой (II)
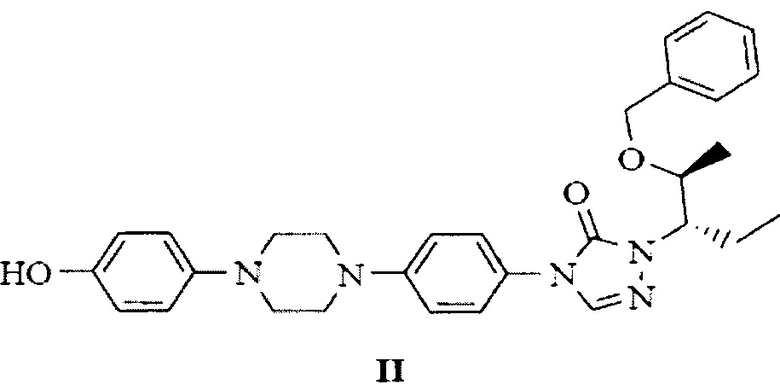
с получением 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она, представленного следующей структурной формулой (III), и с получением при этом новых кристаллических полиморфов В-1, В-2 и В-3 бензилпозаконазола структурной формулы (III).
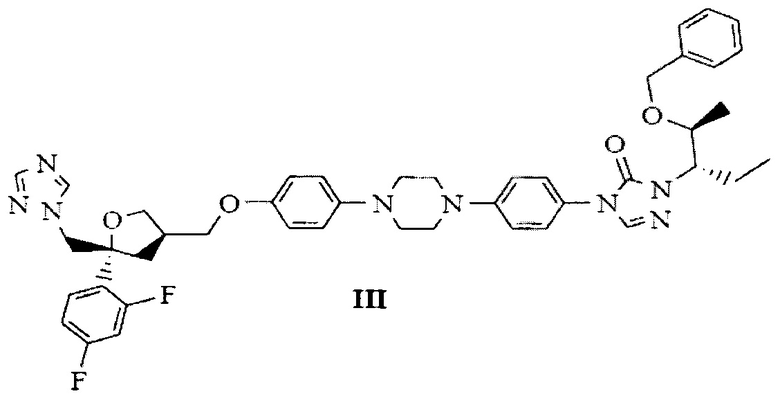
В первом аспекте настоящего изобретения предложена стабильная кристаллическая форма В-1 бензилпозаконазола структурной формулы III. Кристаллическая форма В-1 характеризуется пиковыми значениями 2θ: 4,14; 6,85; 9,92; 15,28; 15,69; 16,59; 17,29; 17,92; 18,27; 18,91; 20,63; 19,95; 23,58; и 25,12 плюс/минус 0,2 градуса 2θ.
Настоящее изобретение предлагает способ получения кристаллической формы В-1 следующим образом.
Раствор 1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (II) в полярном органическом растворителе обрабатывают щелочным раствором при более низкой температуре (около 5-10°С) и перемешивают в течение 10 мин, затем вносят ((3S,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил-4-метилбензолсульфонат структурной формулы (I), реакционную массу медленно нагревают до комнатной температуры и перемешивают в течение 40-45 ч. Реакционную массу разбавляют растворителем - алифатическим сложным эфиром и охлаждают до 10-20°С, затем вносят воду и перемешивают. Слои разделяют; собирают органический слой. Водный слой снова экстрагируют растворителем - алифатическим сложным эфиром. Объединенные органические слои промывают водой и концентрируют. К указанному выше остатку добавляют галогенированный углеводородный растворитель и алифатический углеводородный растворитель и перемешивают при 28 плюс/минус 5°С в течение 2 ч, затем охлаждают до 0-10°С и перемешивают в течение 1 ч. Твердое вещество отфильтровывают; промывают алифатическим углеводородным растворителем. Высушивают в вакуумной лоточной сушилке при 50°С с получением 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) в кристаллической форме В-1.
Вторым аспектом настоящего изобретение предложена стабильная кристаллическая форма В-2 бензилпозаконазола структурной формулы III. Кристаллическая форма В-2 характеризуется пиковыми значениями 2θ: 4,20; 9,27; 14,22; 15,46; 15,82; 16,45; 17,31; 19,09; 20,47; 21,18; и 24,74 плюс/минус 0,2 градусов 2θ. Настоящее изобретение предлагает способ получения кристаллической формы В-2 следующим образом.
Раствор 1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (II) растворяют в полярном органическом растворителе при 25 плюс/минус 5°С. К реакционной смеси добавляют щелочной раствор при 5-10°С и перемешивают в течение 10-30 мин. К реакционной смеси добавляют ((3S,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил-4-метилбензол-сульфонат и продолжают перемешивать в течение 1 ч при 5-10°С. Температуру реакции повышают до 28 плюс/минус 2°С и перемешивают в течение 40-45 ч. К реакционной массе добавляют растворитель - алифатический сложный эфир (5,0 об.) и охлаждают до 15-20°C, затем вносят воду (5,0 об.) и перемешивают. Слои разделяют; собирают органический слой. Водный слой снова экстрагируют растворителем - алифатическим сложным эфиром. Объединенные органические слои промывают водой (3 об.) и частично концентрируют. К частично концентрированному раствору добавляют алифатический углеводородный растворитель и перемешивают при 40 плюс/минус 5°С в течение 30 мин, затем медленно охлаждают до 25 плюс/минус 2°С и продолжают перемешивать при 25 плюс/минус 2°С в течение 2 ч. Указанную выше смесь охлаждают до 0-5°С и перемешивают в течение 1 ч. Твердое вещество отфильтровывают; промывают алифатическим углеводородным растворителем. Высушивают в вакуумной лоточной сушилке при 50°C с получением 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) в кристаллической форме В-2.
Третьим аспектом настоящего изобретения предложена стабильная кристаллическая форма В-3 бензилпозаконазола структурной формулы III. Кристаллическая форма В-3 характеризуется пиковыми значениями 2θ: 7,07; 14,49; 15,68; 16,50; 18,05; 20,13; 20,87; 22,85; и 24,42 плюс/минус 0,2 градуса 2θ.
Настоящее изобретение предлагает способ получения кристаллической формы В-3 следующим образом.
Раствор 1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (II) растворяют в полярном органическом растворителе при 25 плюс/минус 2°С в атмосфере азота и охлаждают до 10-20°С. К реакционной смеси добавляют щелочной раствор и перемешивают в течение 10 мин. К реакционной смеси добавляют ((3S,5S)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил-4-метилбензолсульфонат и продолжают перемешивать в течение 1 ч при 10-20°С. Температуру реакции повышают до 28 плюс/минус 2°С и перемешивают в течение 45-50 ч. К реакционной массе добавляют растворитель - алифатический сложный эфир и охлаждают до 15-20°С, затем вносят воду, реакционную массу медленно нагревают до 25 плюс/минус 2°С и перемешивают. Слои разделяли; собирают органический слой. Водный слой снова экстрагируют растворителем - алифатическим сложным эфиром. Объединенные органические слои промывают водой (3 об.) и органический слой частично концентрируют. Указанный выше частично концентрированный раствор охлаждают до 25 плюс/минус 2°С, добавляют алифатический углеводородный растворитель, перемешивают при 28 плюс/минус 2°С в течение 30 мин и дополнительно разбавляют алифатическим углеводородным растворителем, подогретым до 42 плюс/минус 2°С, перемешивают в течение 30 мин и затем медленно охлаждают до 28 плюс/минус 2°С и продолжают перемешивать при 28 плюс/минус 2°С в течение 2 ч. Указанную выше смесь охлаждают до 0-5°С и перемешивают в течение 1 ч. Твердое вещество отфильтровывают; промывают алифатическим углеводородным растворителем. Высушивают в вакуумной лоточной сушилке при 60 плюс/минус 5°С с получением 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) в кристаллической форме В-3.
Четвертым аспектом настоящего изобретения предложен однореакторный способ получения аморфного позаконазола структурной формулы IV из новой кристаллической формы В-3 промежуточного продукта позаконазола, т.е. бензилпозаконазола структурной формулы III:
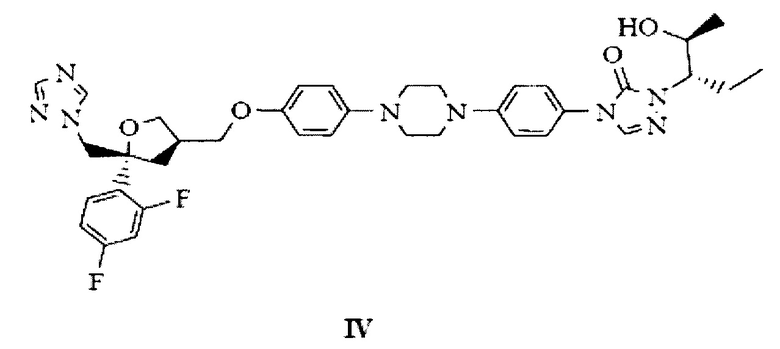
Настоящее изобретение также предлагает дебензилирование бензилпозаконазола концентрированной соляной кислотой при повышенной температуре с последующим внесением органического растворителя, нейтрализацию путем промывки основанием, частичное концентрирование и внесение органического растворителя, с последующим добавлением к углеводородному растворителю при более низкой температуре и медленное доведение смеси до комнатной температуры, фильтрацию с последующей промывкой углеводородным растворителем и высушивание с получением в аморфной форме позаконазола структурной формулы (IV).
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S, 3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) кристаллической формы В-3 вносят в концентрированную соляную кислоту при 25 плюс/минус 2°С. Реакционную смесь медленно нагревают до 63 плюс/минус 2°С и перемешивают при 63 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждают до 25 плюс/минус 2°С и вносят галогенированный углеводородный растворитель, перемешивают и слои разделяют. В водный слой добавляют галогенированный углеводородный растворитель и воду. Содержимое охлаждают до 15 плюс/минус 5°С, рН указанной выше массы доводят до рН 10-12 щелочным раствором. Слои разделяют. Водный слой повторно экстрагируют галогенированным углеводородным растворителем. Объединенные органические слои промывают 10% водным гидроксидом натрия, а затем водой. Органический слой концентрируют в вакууме при 43 плюс/минус 2°С вплоть до 4,0 объемов. К реакционной массе добавляют растворитель - алифатический кетон. Эту реакционную массу по каплям добавляют к углеводородному растворителю при более низкой температуре и непрерывном перемешивании, реакционную массу медленно нагревают до комнатной температуры и продолжают перемешивать при этой же температуре в течение 1 ч. Фильтруют, промывают углеводородным растворителем и высушивают с отсасыванием; высушивают в вакуумной лоточной сушилке в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
Пятым аспектом настоящего изобретения предложен способ получения кристаллической формы I позаконазола структурной формулы IV из аморфной формы позаконазола.
Аморфную форму позаконазола структурной формулы (IV) растворяют в галогенированном органическом растворителе с получением прозрачного раствора. Этот раствор выпаривают с получением отвержденного вещества. Этот твердый продукт высушивали с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в кристаллической форме I.
Упомянутый выше в аспектах изобретения термин "полярный органический растворитель" обозначает диметилсульфоксид, диметилформамид и т.п.; термин "растворитель - алифатический сложный эфир" обозначает этилацетат, метилацетат, изопропилацетат и т.п.; термин "галогенированный углеводородный растворитель" обозначает дихлорметан, хлороформ, дихлорэтан, четыреххлористый углерод и т.п.; термин "щелочной раствор" обозначает водный раствор гидроксида натрия, гидроксида калия, бикарбоната натрия и т.п.; термин "алифатический углеводородный растворитель" обозначает гексан, гептан, циклогексан, пентан и т.п. Термин «более низкая температура» обозначает температуру в интервале 0-25°С.
Эффект изобретения
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III), получаемый как промежуточный продукт в способе получения по настоящему изобретению, хорошо кристаллизуется, его легко отделять от побочных продуктов и легко обрабатывать. Таким образом, кристаллические полиморфы В-1, В-2 и В-3 по настоящему изобретению служат отличными промежуточными продуктами для промышленного производства позаконазола.
4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он структурной формулы (IV) аморфной формы, полученный по настоящему изобретению, стабилен; производственный цикл - короткий, с высоким выходом, и подходит для промышленного производства, с применением стандартных растворителей и недорогих реагентов.
Примеры
Схема:
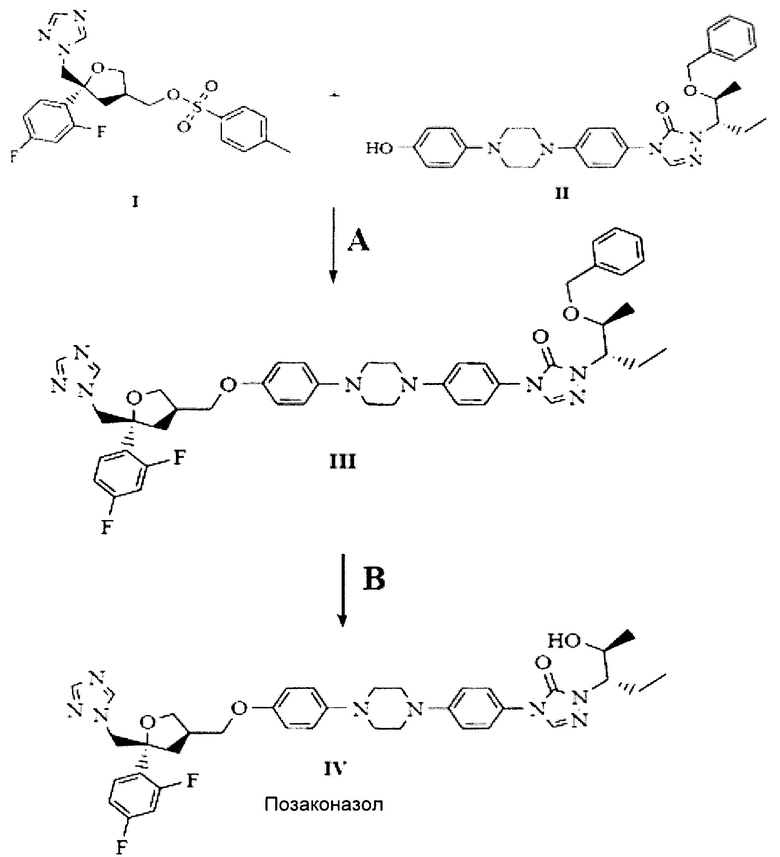
Реагенты: А) гидроксид натрия/ вода/ диметилсульфоксид; В) концентрированная соляной кислота/ дихлорметан/ водный раствор гидроксида натрия/ ацетон/ циклогексан.
Данная схема представлена в ниже следующих примерах. Эти примеры служат лишь иллюстрацией, и их не следует воспринимать как ограничение объема настоящего изобретения.
Пример 1. Получение 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) в кристаллической форме В-1
1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-он (25,0 г) структурной формулы (II) растворяли в диметилсульфоксиде (6,0 об.) при 25 плюс/минус 2°С и охлаждали до 5-10°С. К реакционной смеси добавляли 25% водный раствор гидроксида натрия (1,3 экв.) и перемешивали в течение 10 мин. К реакционной смеси добавляли ((3S,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил 4-метилбензолсульфонат структурной формулы (I) (25,16 г) и продолжали перемешивать в течение 2 ч при 5-10°С. Температуру реакции повышали до 28 плюс/минус 2°С и перемешивали в течение 40-45 ч. К реакционной массе добавляли этилацетат (5,0 об.) и охлаждали до 15-20°С, а затем вносили воду (5,0 об.) и перемешивали. Слои разделяли; этилацетатный слой собирали. Водный слой повторно экстрагировали этилацетатом (3 об.). Объединенные органические слои промывали водой (3 об.) и концентрировали. К полученному остатку добавляли дихлорметан (2,0 об.) и гептан (7,0 об.) и перемешивали при 28 плюс/минус 2°С в течение 2 ч, а затем охлаждали до 0-5°С и перемешивали в течение 1 ч. Твердое вещество отфильтровывали; промывали гептаном (5 об.). Высушивали в вакуумной лоточной сушилке при 50°C с получением 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) кристаллической формы В-1 с выходом 95%.

Пример 2. Получение 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) в кристаллической форме В-2
1-((2S,3S)-2-(Бензилокси)пентан-3-ил)-4(-4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-он (25,0 г) структурной формулы (II) растворяли в диметилсульфоксиде (6,0 об.) при 25 плюс/минус 2°С и охлаждали до 5-10°С. К реакционной смеси добавляли 25% водный раствор гидроксида натрия (1,3 экв.) и перемешивали в течение 10 мин. К реакционной смеси добавляли ((3S,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил-4-метилбензолсульфонат структурной формулы (I) (25,16 г) и продолжали перемешивать в течение 1 ч при 5-10°С.Температуру реакции повышали до 28 плюс/минус 2°С и перемешивали в течение 40-45 ч. К реакционной массе добавляли этилацетат (5,0 об.) и охлаждали до 15-20°С, затем вносили воду (5,0 об.) и перемешивали. Слои разделяли. Собирали этилацетатный слой. Водный слой повторно экстрагировали этилацетатом (3 об.). Объединенные органические слои промывали водой (3 об.) и частично концентрировали до содержания 3,0 об. этилацетата. К этому частично концентрированному раствору добавляли гептан (8,0 об.) и перемешивали при 40 плюс/минус 5°С в течение 30 мин, затем медленно охлаждали до 25 плюс/минус 2°С и продолжали перемешивать при 25 плюс/минус 2°С в течение 2 ч. Указанную выше смесь охлаждали до 0-5°С и перемешивали в течение 1 ч. Твердое вещество отфильтровывали; промывали гептаном (5 об.). Высушивали в вакуумной лоточной сушилке при 50°C с получением 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) кристаллической формы В-2 с выходом 88%.

Пример 3. Получение 4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) кристаллической формы В-3
1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидроксифенил)пиперазин-1-ил)фенил)-1Н-1,2,4-триазол-5(4Н)-он (4,0 кг; 1,0 экв.) структурной формулы (II) растворяли в диметилсульфоксиде (6,0 об.) при 25 плюс/минус 2°С в атмосфере азота и охлаждали до 15-20°С. К реакционной смеси добавляли 25% водный раствор гидроксида натрия (1,3 экв.) и перемешивали в течение 10 мин. К реакционной смеси добавляли ((3S,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метил-4-метилбензолсульфонат структурной формулы (I) (4,02 кг) и продолжали перемешивать в течение 1 ч при 15-20°С. Температуру реакции повышали до 28 плюс/минус 2°С и перемешивали в течение 45-50 ч. К реакционной массе добавляли этилацетат (5,0 об.) и охлаждали до 15-20°С, затем вносили воду (5,0 об.), реакционную массу медленно нагревали до 25 плюс/минус 2°С и перемешивали. Слои разделяли; собирали органический слой. Водный слой повторно экстрагировали этилацетатом (3 об.). Объединенные органические слои промывали водой (3 об.) и органический слой частично концентрировали до содержания 5,0 об. этилацетата. Этот частично концентрированный раствор охлаждали до 25 плюс/минус 2°С и добавляли н-гептан (5,0 об.), перемешивали при 28 плюс/минус 2°С в течение 30 мин, дополнительно разбавляли н-гептаном (2,0 об.), нагретым до 42 плюс/минус 2°С, перемешивали в течение 30 мин, затем медленно охлаждали до 28 плюс/минус 2°С и продолжали перемешивать при 28 плюс/минус 2°С в течение 2 ч. Указанную выше смесь охлаждали до 0-5°С и перемешивали в течение 1 ч. Твердое вещество отфильтровывали; промывали гептаном (5 об.). Высушивали в вакуумной лоточной сушилке при 60 плюс/минус 5°C с получением 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)-пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-она структурной формулы (III) кристаллической формы В-3 с 96% выходом.


Пример 4. Получение 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) (10,0 г) кристаллической формы В-3 вносили в круглодонную колбу, содержащую концентрированную соляную кислоту (5,0 об.), при 25 плюс/минус 2°С. Реакционную смесь медленно нагревали до 62 плюс/минус 2°С и перемешивали при 62 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждали до 25 плюс/минус 2°С и вносили дихлорметан (5,0 об. × 2 раза), перемешивали в течение 30 мин и слои разделяли. В водный слой добавляли дихлорметан (6,0 об.) и воду (5,0 об.). Содержимое охлаждали до 15 плюс/минус 5°С, рН указанной выше массы доводили до рН 9-10 25% водным гидроксидом натрия. Слои разделяли. Водный слой повторно экстрагировали дихлорметаном (5,0 об. × 2 раза). Объединенные дихлорметановые слои промывали 10% водным гидроксидом натрия (5,0 об. × 2 раза), а затем водой (5,0 об.). Дихлорметановый слой высушивали над безводным сульфатом натрия и концентрировали дихлорметановый слой в вакууме при 43 плюс/минус 2°С вплоть до 3,0 объемов. Эту реакционную массу охлаждали до 25°С и добавляли ацетон (4,0 об.). К этой реакционной массе добавляли циклогексан (20,0 об.) при той же температуре, нагревали до 45 плюс/минус 2°С и перемешивали в течение 30 мин. Реакционную массу медленно охлаждали до 25 плюс/минус 2°С. Фильтровали и промывали циклогексаном (5,0 об.) и высушивали с отсасыванием; высушивали в вакуумной лоточной сушилке при 70 плюс/минус 2°С в течение 30-40 ч в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме с 92% выходом.
Пример 5. Получение 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в кристаллической форме I.
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) (10,0 г) кристаллической формы В-3 вносили в круглодонную колбу с концентрированной соляной кислотой (5,0 об.) при 25 плюс/минус 2°С. Реакционную смесь медленно нагревали до 62 плюс/минус 2°С и перемешивали при 62 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждали до 25 плюс/минус 2°С и вносили дихлорметан (5,0 об. × 2 раза), перемешивали в течение 30 мин и слои разделяли. В водный слой добавляли дихлорметан (6,0 об.) и воду (5,0 об.). Содержимое охлаждали до 15 плюс/минус 5°С, рН указанной выше массы доводили до рН 9-10 25% водным гидроксидом натрия. Слои разделяли. Водный слой повторно экстрагировали дихлорметаном (5,0 об. × 2 раза). Объединенные дихлорметановые слои промывали 10% водным гидроксидом натрия (5,0 об. × 2 раза), а затем водой (5,0 об.). Дихлорметановый слой высушивали над безводным сульфатом натрия и концентрировали этот дихлорметановый слой в вакууме при 43 плюс/минус 2°С вплоть до 3,0 объемов. Этот дихлорметановый раствор охлаждали до 25°С и добавляли ацетон (4,0 об.). Реакционную массу вносили по каплям в циклогексан (20,0 об.) при 0-10°С при непрерывном перемешивании, после внесения полученную реакционную массу медленно нагревали до 25°С и продолжали перемешивать при этой же температуре в течение 1 ч. Фильтровали, промывали циклогексаном (5,0 об.) и высушивали с отсасыванием; высушивали в вакуумной лоточной сушилке при 70 плюс/минус 2°С в течение 30-40 ч в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
Указанную выше полученную аморфную форму растворяли в дихлорметане с получением прозрачного раствора. Этот раствор выпаривали при 70°С с получением отвержденного вещества. Этот твердый продукт высушивали при 70°С в течение 7 ч с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в кристаллической форме I.
Пример 6. Получение 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) (10,0 г) в кристаллической форме В-3 вносили в круглодонную колбу с концентрированной соляной кислотой (5,0 об.) при 25 плюс/минус 2°С. Реакционную смесь медленно нагревали до 62 плюс/минус 2°С и перемешивали при 62 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждали до 25 плюс/минус 2°С и вносили дихлорметан (5,0 об. × 2 раза), перемешивали в течение 30 мин и слои разделяли. В водный слой добавляли дихлорметан (6,0 об.) и воду (5,0 об.). Содержимое охлаждали до 15 плюс/минус 5°С, рН указанной выше массы доводили до рН 9-10 25% водным гидроксидом натрия. Слои разделяли. Водный слой повторно экстрагировали дихлорметаном (5,0 об. × 2 раза). Объединенные дихлорметановые слои промывали 10% водным гидроксидом натрия (5,0 об. × 2 раза), а затем водой (5,0 об.). Дихлорметановый слой высушивали над безводным сульфатом натрия и концентрировали дихлорметановый слой в вакууме при 43 плюс/минус 2°С вплоть до 3,0 объемов. К этой массе добавляли ацетон (4,0 об.) и замораживали реакционную массу внесением жидкого азота (-95 плюс/минус 5°С). К указанной выше реакционной массе добавляли циклогексан (20,0 об.) и перемешивали при 2 плюс/минус 2°С в течение 1 ч. Фильтровали и промывали циклогексаном (5,0 об.) и высушивали с отсасыванием; высушивали в вакуумной лоточной сушилке при 70 плюс/минус 2°С в течение 30-40 ч в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме с 75% выходом.
Пример 7. Получение 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) (350,0 г) в кристаллической форме В-3 вносили в круглодонную колбу с концентрированной соляной кислотой (5,0 об.) при 25 плюс/минус 2°С. Реакционную смесь медленно нагревали до 63 плюс/минус 2°С и перемешивали при 63 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждали до 25 плюс/минус 2°С и вносили дихлорметан (5,0 об. × 2 раза), перемешивали и слои разделяли. В водный слой добавляли дихлорметан (6,0 об.) и воду (5,0 об.). Содержимое охлаждали до 15 плюс/минус 5°С, рН указанной выше массы доводили до 10-12 рН 25% водным гидроксидом натрия. Слои разделяли. Водный слой повторно экстрагировали дихлорметаном (5,0 об. × 2 раза). Объединенные дихлорметановые слои промывали 10% водным гидроксидом натрия (3,0 об. × 2 раза), а затем водой (3,0 об.). Дихлорметановый слой концентрировали в вакууме при 43 плюс/минус 2°С вплоть до 4,0 объемов. К реакционной массе добавляли ацетон (4,0 об.). Эту реакционную массу вносили по каплям в циклогексан (20,0 об.) при 10-14°C при непрерывном перемешивании, полученную реакционную массу медленно нагревали до 25°С и продолжали перемешивать при этой же температуре в течение 1 ч. Фильтровали, промывали циклогексаном (5,0 об.) и высушивали с отсасыванием; высушивали в вакуумной лоточной сушилке при 60 плюс/минус 5°С в течение 30-40 ч в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме с 95% выходом.

Пример 8. Получение 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) (10,0 г) в кристаллической форме В-3 вносили в круглодонную колбу с концентрированной соляной кислотой (5,0 об.) при 25 плюс/минус 2°С. Реакционную смесь медленно нагревали до 62 плюс/минус 2°С и перемешивали при 62 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждали до 25 плюс/минус 2°С и вносили дихлорметан (5,0 об. × 2 раза), перемешивали в течение 30 мин и слои разделяли. В водный слой добавляли дихлорметан (6,0 об.) и воду (5,0 об.). Содержимое охлаждали до 15 плюс/минус 5°С, рН указанной выше массы доводили до рН 9-10 25% водным гидроксидом натрия. Слои разделяли. Водный слой повторно экстрагировали дихлорметаном (5,0 об. × 2 раза). Объединенные дихлорметановые слои промывали 10% водным гидроксидом натрия (5,0 об. × 2 раза), а затем водой (5,0 об.). Дихлорметановый слой высушивали над безводным сульфатом натрия и концентрировали дихлорметановый слой в вакууме при 43 плюс/минус 2°С вплоть до 3,0 объемов. К реакционной массе добавляли ацетон (4,0 об.). Эту реакционную массу вносили по каплям в циклогексан (20,0 об.) при 25 плюс/минус 2°С при непрерывном перемешивании и продолжали перемешивать при этой же температуре после внесения в течение 1 ч. Фильтровали и промывали циклогексаном (5,0 об.) и высушивали с отсасыванием; высушивали в вакуумной лоточной сушилке при 70 плюс/минус 2°С в течение 30-40 ч в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме с 90% выходом.
Пример 9. Получение 4-[4-[4-[4-[[(3R,5R)-5- (2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме.
4-(4-(4-(4-(((3R,5R)-5-((1Н-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1Н-1,2,4-триазол-5(4Н)-он структурной формулы (III) (10,0 г) в кристаллической форме В-3 вносили в круглодонную колбу с концентрированной соляной кислотой (5,0 об.) при 25 плюс/минус 2°С. Реакционную смесь медленно нагревали до 62 плюс/минус 2°С и перемешивали при 62 плюс/минус 2°С в течение 2-3 ч. Реакционную массу охлаждали до 25 плюс/минус 2°С и вносили дихлорметан (5,0 об. × 2 раза), перемешивали в течение 30 мин и слои разделяли. В водный слой добавляли дихлорметан (6,0 об.) и воду (5,0 об.). Содержимое охлаждали до 15 плюс/минус 5°С, рН указанной выше массы доводили до рН 9-10 25% водным гидроксидом натрия. Слои разделяли. Водный слой повторно экстрагировали дихлорметаном (5,0 об. × 2 раза). Объединенные дихлорметановые слои промывали 10% водным гидроксидом натрия (5,0 об. × 2 раза), а затем водой (5,0 об.). Дихлорметановый слой высушивали над безводным сульфатом натрия и концентрировали полученный дихлорметановый слой в вакууме при 43 плюс/минус 2°С вплоть до 3,0 объемов. Эту реакционную массу вносили по каплям в циклогексан (20,0 об.) при 23 плюс/минус 2°С при непрерывном перемешивании и продолжали перемешивать при этой же температуре в течение 1 ч. Фильтровали и промывали циклогексаном (5,0 об.) и высушивали с отсасыванием; высушивали в вакуумной лоточной сушилке при 70 плюс/минус 2°С в течение 30-40 ч в вакууме с получением 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1Н-1,2,4-триазол-1-илметил)-3-фуранил]метокси]фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-она структурной формулы (IV) в аморфной форме с 92% выходом.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА ИЛИ ТЕТРАГИДРОПИРАНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ГИДРАТЫ, ИЛИ СОЛЬВАТЫ ЭТИХ СОЕДИНЕНИЙ, ИЛИ ИХ СОЛЕЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2084439C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛОВ, КОНЕЧНЫЕ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ СПОСОБА | 1996 |
|
RU2156760C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА И ЕГО АНАЛОГОВ | 2013 |
|
RU2619928C2 |
| АЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОТИВОГРИБКОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ГРИБКОВОЙ ИНФЕКЦИИ | 1998 |
|
RU2189982C2 |
| N-(1-(1-БЕНЗИЛ-4-ФЕНИЛ-1Н-ИМИДАЗОЛ-2-ИЛ)-2,2-ДИМЕТИЛПРОПИЛ)БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНЕЗИНОВОГО БЕЛКА ВЕРЕТЕНА (KSP) ДЛЯ ЛЕЧЕНИЯ РАКА | 2005 |
|
RU2427572C2 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА 2-{ 3-[2-(1-{ [3,5-БИС(ДИФТОРМЕТИЛ)-1Н-ПИРАЗОЛ-1-ИЛ]АЦЕТИЛ} ПИПЕРИДИН-4-ИЛ)-1,3-ТИАЗОЛ-4-ИЛ]-4,5-ДИГИДРО-1,2-ОКСАЗОЛ-5-ИЛ} -3-ХЛОРФЕНИЛМЕТАНСУЛЬФОНАТА | 2014 |
|
RU2691948C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| СИНТЕТИЧЕСКОЕ ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1-(2-ДЕЗОКСИ-2-ФТОР-4-ТИО-β-D-АРАБИНОФУРАНОЗИЛ)ЦИТОЗИНА, СИНТЕТИЧЕСКОЕ ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ТИОНУКЛЕОЗИДА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2633355C2 |
Изобретение относится к однореакторному способу получения аморфной формы 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1H-1,2,4-триазол-1-илметил)-3-фуранил]метокси]-фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-она формулы (IV) (позаконазол), который включает a) обработку 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она формулы (III) концентрированной соляной кислотой и перемешивание реакционной смеси при температуре 55-70°C, b) охлаждение реакционной смеси и внесение галогенированного углеводородного растворителя, c) разделение слоев, d) внесение галогенированного углеводородного растворителя в водный слой и доведение pH до 8-12, e) частичное концентрирование органического слоя и возможное внесение в реакционную смесь растворителя - алифатического кетона, f) смешивание реакционной смеси стадии e) и алифатического углеводородного растворителя при температуре 10-25°C и g) фильтрацию и возможную промывку алифатическим углеводородным растворителем с получением аморфного соединения формулы IV, где 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-он формулы (III) представляет собой кристаллическую В-3 форму, которая характеризуется рентгеновской дифрактограммой, представленной на фиг.3. Изобретение также относится к кристаллической форме В-3 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она формулы (III), которая является промежуточным продуктом для получения аморфного позаконазола, и способу ее получения. 3 н. и 2 з.п. ф-лы, 3 табл., 7 ил., 9 пр.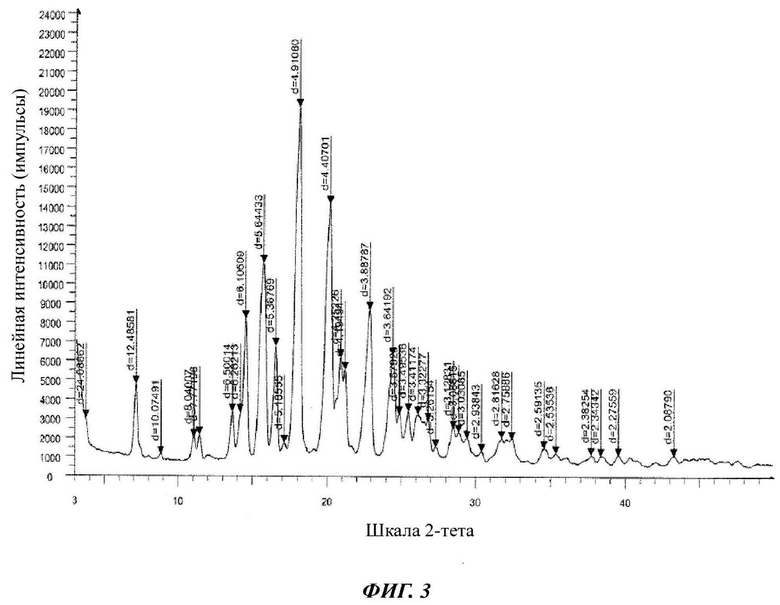
1. Способ получения кристаллической B-3 формы 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)-метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она (III), включающий:
a) обработку раствора 1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4- гидроксифенил)пиперазин-1-ил)фенил)-1H-1,2,4-триазол-5(4H)-она (II) в полярном органическом растворителе в атмосфере азота щелочным раствором при температуре 10-20°C;
b) внесение ((3S,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)-тетрагидрофуран-3-ил)метил-4-метилбензолсульфоната (I) в раствор стадии a);
c) внесение растворителя - алифатического сложного эфира и воды в раствор стадии b);
d) разделение слоев;
e) частичное концентрирование органического слоя и внесение алифатического углеводородного растворителя при температуре 20-30°C;
f) перемешивание реакционной массы стадии e) при температуре 40-50°C; и
g) охлаждение реакционной массы стадии f), фильтрацию и возможную промывку алифатическим углеводородным растворителем с получением соединения III в кристаллической форме B-3, которая характеризуется порошковой рентгеновской дифрактограммой с пиками при значениях около 7,07, 14,49, 15,68, 16,50, 18,05, 20,13, 20,87, 22,85 и 24,42 плюс/минус 0,2 градуса 2θ.
2. Способ получения кристаллической B-3 формы 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)-метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она (III) по п. 1, включающий:
a) обработку раствора 1-((2S,3S)-2-(бензилокси)пентан-3-ил)-4-(4-(4-(4-гидрокси-фенил)пиперазин-1-ил)фенил)-1H-1,2,4-триазол-5(4H)-она (II) в диметилсульфоксиде в атмосфере азота раствором гидроксида натрия при 10-20°C;
b) внесение ((3S,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)-тетрагидрофуран-3-ил)метил-4-метилбензолсульфоната (I) в раствор стадии a);
c) внесение этилацетата и воды в раствор стадии b);
d) разделение слоев;
e) частичное концентрирование органического слоя до 4-7 объемов и внесение н-гептана при 20-30°C;
f) перемешивание реакционной массы стадии e) при 40-50°C; и
g) охлаждение реакционной массы стадии f), фильтрацию и возможную промывку н-гептаном с получением соединения структурной формулы III в кристаллической форме B-3.
3. Кристаллическая B-3 форма 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она (III), которая характеризуется порошковой рентгеновской дифрактограммой с пиками при значениях около 7,07, 14,49, 15,68, 16,50, 18,05, 20,13, 20,87, 22,85 и 24,42 плюс/минус 0,2 градуса 2θ.
4. Кристаллическая B-3 форма 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она (III) по п. 3, которая характеризуется рентгеновской дифрактограммой, представленной на Фиг. 3.
5. Однореакторный способ получения аморфной формы 4-[4-[4-[4-[[(3R,5R)-5-(2,4-дифторфенил)тетрагидро-5(1H-1,2,4-триазол-1-илметил)-3-фуранил]метокси]-фенил]-1-пиперазинил]фенил]-2-[(1S,2S)-1-этил-2-гидроксипропил]-2,4-дигидро-3H-1,2,4-триазол-3-она формулы (IV), включающий:
a) обработку 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-она формулы (III) концентрированной соляной кислотой и перемешивание реакционной смеси при температуре 55-70°C;
b) охлаждение реакционной смеси и внесение галогенированного углеводородного растворителя;
c) разделение слоев;
d) внесение галогенированного углеводородного растворителя в водный слой и доведение pH до 8-12;
e) частичное концентрирование органического слоя и возможное внесение в реакционную смесь растворителя - алифатического кетона;
f) смешивание реакционной смеси стадии e) и алифатического углеводородного растворителя при температуре 10-25°C; и
g) фильтрацию и возможную промывку алифатическим углеводородным растворителем с получением аморфного соединения формулы IV, где 4-(4-(4-(4-(((3R,5R)-5-((1H-1,2,4-триазол-1-ил)метил)-5-(2,4-дифторфенил)тетрагидрофуран-3-ил)метокси)фенил)пиперазин-1-ил)фенил)-1-((2S,3S)-2-(бензилокси)пентан-3-ил)-1H-1,2,4-триазол-5(4H)-он формулы (III) представляет собой кристаллическую В-3 форму по пп. 3, 4.
| WO 2015059716 A2, 30.04.2015 | |||
| WO 2013042138 A2, 28.03.2013 | |||
| WO 2009141837 A2, 26.11.2009 | |||
| WO 2011158248 A2, 22.12.2011 | |||
| RU 2012154807 A, 27.06.2014. |

