Данное изобретение относится к улучшенному способу получения противогрибкового лекарственного вещества вориконазола и его аналогов.
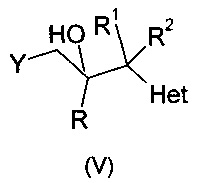
Опубликованная европейская патентная заявка ЕР 0357241 А1 описывает противогрибковые триазолы формулы:
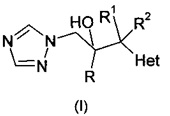
и их фармацевтически приемлемые соли, где R означает фенил, необязательно замещенный 1-3 заместителями, каждый из которых независимо выбран из гало и CF3; R1 означает С1-С4-алкил; R2 означает Н или С1-С4-алкил; и Het, присоединенный к смежному атому углерода кольцевым атомом углерода, выбирают из следующих: пиридинил, пиридазинил, пиримидинил, пиразинил и триазинил, указанный Het необязательно замещен С1-С4-алкилом, С1-С4-алкокси, гало, CF3, CN, NO2, NH2, -NH(C1-С4-алканоилом) или -NHCO2(С1-С4-алкилом). Описано, что соединения могут быть получены путем (а) депротонирования соединения формулы:
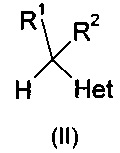
сильным основанием и реакцией с кетоном формулы:
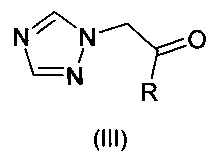
или (b) реакцией эпоксида формулы:
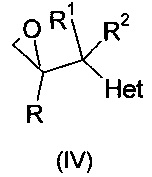
или соединения, содержащего отходящую группу Y формулы:
с триазолом.
Опубликованная европейская патентная заявка ЕР 0440372 А1 описывает группу фунгицидных триазолов формулы
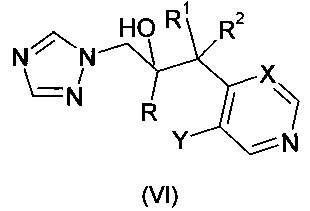
где R означает фенил, замещенный 1-3 заместителями, каждый из которых независимо выбран из гало, -CF3, и -OCF3; R1 означает С1-С4-алкил; R2 означает Н или С1-С4-алкил; X означает СН или N; и Y означает F или Cl. Описано, что соединения могут быть получены теми же путями, что описаны выше по отношению к соединениям формулы (I). предлагается дополнительный путь, где соединение формулы
содержащее одну или две восстанавливаемые группы Z2 и Z3 (например, группы хлора), восстанавливают, например, путем гидрогенолиза.
Одним из соединений, описанных в ЕР 0440372 А1, является (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол формулы:
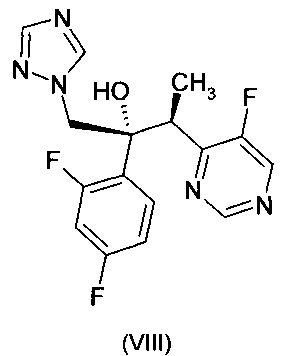
(смотреть Примеры 7-9). Это соединение в общем известно как вориконазол и было коммерциализировано под торговой маркой VFEND® для лечения грибковых заболеваний.
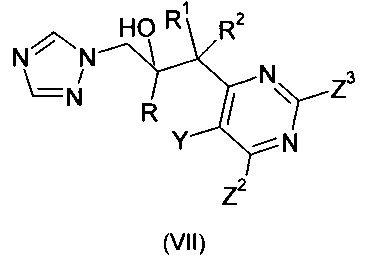
Международная патентная публикация WO-1997/06160 А1 описывает способ получения соединения формулы

где R означает фенил, необязательно замещенный 1-3 заместителями, каждый из которых независимо выбран из гало и трифторметила; R1 означает С1-С6-алкил; и Het означает пиримидинил, необязательно замещенный 1-3 заместителями, независимо выбранными из следующих: С1-С4-алкил, С1-С4-алкокси, гало, оксо, бензил и бензилокси; где соединение формулы (III) (смотреть выше) реагирует с соединением формулы:
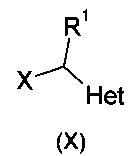
где X означает хлор, бром или йод, в присутствии цинка, йода и/или кислоты Льюиса. Реакция так же может осуществляться с одним или двумя дополнительными атомами хлора или брома на Het, которые впоследствии удаляют путем восстановления (например, гидрогенолиза).
Вориконазол является одиночным (2R,3S) стереоизомером, и поэтому контроль как относительной, так и абсолютной стереохимии является важной целью в любом способе синтеза, разработанном для его получения. Описанные выше способы способны обеспечивать различные степени относительного стехиометрического контроля в конструкции двух смежных стереоцентров, однако ни один способ не способен обеспечить абсолютный стехиометрический контроль любого вида. Таким образом, например, в Примере 7 опубликованной европейской патентной заявки ЕР 0440372 А1 вориконазол получают в виде рацемата, который растворяется с использованием 1R-(-)-10-камфорсульфоновой кислоты с целью получения чистого (2R,3S) энантиомера. Таким образом, желательно обеспечить способ получения вориконазола, контролирующий как относительную, так и абсолютную стереохимию двух хиральных центров и обеспечивающий непосредственно (2R,3S) стереоизомер. Попытки контролировать абсолютную стереохимию реакции Реформатского, описанные в ЕР 0440372 А1, путем добавления хиральных лигандов, однако, были неуспешными.
Так же в литературе было описано, что добавление определенных нуклеофилов на основе меди к кетонам может осуществляться энантиоселективным путем, используя хиральные лиганды. Таким образом, например, медные нуклеофилы были получены из α,β-ненасыщенных кетонов, сложных эфиров и тиоэфиров и энантиоселективно добавлены к альдегидам и кетонам (Tetrahedron Letters, 2012, 53, 4199-4201; Chem. Asian J., 2010, 5, 478; Chem. Commun., 2008, 4309-4311; J. Am. Chem. Soc., 2008, 130 (9), 2747; J. Am. Chem. Soc., 2008, 130, 14378-14379; Org. Biomol. Chem., 2011, 9, 6143-6147; Org. Biomol. Chem., 2012, 10, 5971-5978; Org. Letters, 2006, 8 (26), 6059-6062); Org. Letters, 2006, 8 (26), 5943-5946; Angew. Chem. Int. Ed., 2006, 45, 1292-1297; Synlett., 2009, 8, 1299-1302; Tetrahedron Lett., 2006, 47, 1403-1407). Так было продемонстрировано энантиоселективное добавление к кетонам медных нуклеофилов, полученных из алленов (J. Am. Chem. Soc., 2006, 128, 14440-14441; Tetrahedron Lett., 2006, 47, 1403-1407). Недавно было описано получение медных нуклеофилов из определенных гетероароматических соединений винила и их добавление к определенному диапазону кетонов (J. Am. Chem. Soc., 2012, 134, 8428). Однако ни один из этих документов не описывает получение медного нуклеофила из 4-винилпиримидина и его энантиоселективное добавление к кетону. Так же они не описывают добавление любого медного нуклеофила к фенилметилкетону, содержащему заместитель на метильной группе.
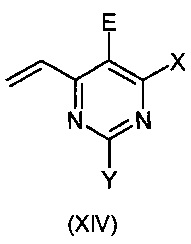
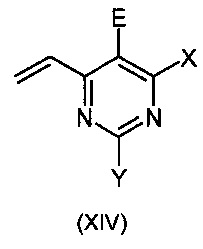
Данное изобретение относится к способу получения соединения формулы:
 или
или 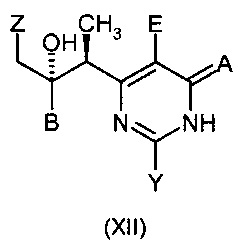
где:
X означает Н, гало, -NH2, -NН(С1-С6-алкил), -N(С1-С6-алкил)(С1-С6-алкил), -Si(R3)3 [где R3 означает, независимо в каждом случае, C1-C6-алкил, арил или арил(С1-С6-алкил)], -O(C1-С6-алкил), -О-арил, -S(C1-С6-алкил), -OSO2(С1-С6-алкил), -NHSO2(С1-С6-алкил) или -S-арил;
Y означает Н, гало, -NH2, -NН(С1-С6-алкил), -N(С1-С6-алкил)(С1-С6-алкил), -Si(R3)3 [где R3 является таким как указано выше], -О(C1-С6-алкил), -О-арил, -S(С1-С6-алкил), -OSO2(C1-С6-алкил), -NHSO2(С1-С6-алкил) или -S-арил;
Z означает необязательно замещенный гетероарил, -Si(R3)3 (где R3 является таким как указано выше), -ОН, защищенной гидроксильной группой, гало, нитро, циано, -SH, защищенной тиогруппой, C1-С6-алкил или C1-С6-алкокси;
А означает О, S или NH;
В означает фенилзамещенный одним или больше атомами галогена; и
Е означает атом галогена;
путем взаимодействия соединения формулы:
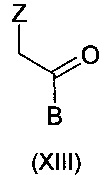
где Z и В являются такими, как указано выше, с соединением формулы:
 или
или 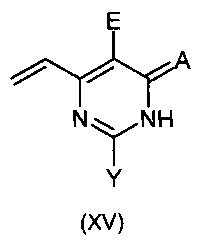
соответственно, где X, Y, А и Е являются такими как указано выше, в присутствии катализатора переходного металла, лиганда, приемлемого для использования с катализатором, и восстанавливающего агента. Такой тип реакции обычно описывают как восстановительная альдольная конденсация. Образование нуклеофила переходного металла из 4-винилпиримидина и его добавление к замещенному ацетофенону является неочевидным.
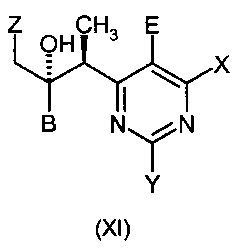
Полученная относительная стереохимия показана на формулах (XI) и (XII). При использовании хирального лиганда продукт будет рацемическим. С другой стороны, при использовании хирального лиганда реакция проходит энантиоселективно.
Когда Z означает гетероарильную группу, она желательно означает или (i) 6-членный ароматический гетероцикл, содержащий 1-3 атома N, или (ii) 5-членный ароматический гетероцикл, содержащий или (а) 1-4 атома N или (b) 1 атом О или S и 0-3 атома N. Гетероарильные группы могут быть присоединены посредством атома углерода кольца (во всех случаях) или атома азота кольца с подходящей валентностью. В случае замещения, заместитель может быть расположен на атоме углерода кольца (во всех случаях) или атоме азота кольца с подходящей валентностью (если заместитель присоединен посредством атома углерода). Определенные примеры гетероарила включают тиенил, фуранил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридил, пиридазинил, пиримидинил и пиразинил. Возможные заместители включают С1-С6-алкил, С3-С8 циклоалкил, гало, -CN, -NO2, O(C1-С6-алкил), -N(С1-С6-алкил)(C1-С6-алкил), -S(C1-С6-алкил), -SO(С1-С6-алкил), -SO2(С1-С6-алкил), -СО(С1-С6-алкил), -ОСО(С1-С6-алкил), -СОО(С1-С6-алкил), -N(C1-С6-алкил)CO(C1-С6-алкил), -CON(C1-С6-алкил)(C1-C6-алкил), -N(С1-С6-алкил)SO2(С1-С6-алкил), -SO2N(С1-С6-алкил)(С1-С6-алкил), -N(С1-С6-алкил)CON(С1-С6-алкил)(С1-С6-алкил), -N(С1-С6-алкил)СОО(С1-С6-алкил) и -N(C1-C6-алкил)SO2N(С1-С6-алкил)(C1-C6-алкил).
В случае, когда Z означает защищенную гидрокси или тиогруппу, приемлемые защитные группы широко известны специалистам в отрасли. Смотреть, например, 'Protective Groups in Organic Chemistry' by Wuts and Greene (Wiley-Blackwell). Желательные защитные группы включают триалкилсилил группы, такие как триметилсилил и трет-бутилдиметилсилил, и арилметил группы, такие как бензил.
Арил означает фенил или нафтил, указанные фенил и нафтил являются необязательно замещенными 1-5 заместителями, каждый из которых независимо выбран из следующих: C1-C6-алкил, С3-С8 циклоалкил, гало, -CN, -NO2, -O(С1-С6-алкил), -N(С1-С6-алкил)(C1-C6-алкил), -S(C1-С6-алкил), -SO(C1-C6-алкил), -SO2(С1-С6-алкил), -СО(С1-С6-алкил), -ОСО(С1-С6-алкил), -СОО(C1-С6-алкил), -N(С1-С6-алкил)СО(С1-С6-алкил), -СОN(С1-С6-алкил)(C1-C6-алкил), -N(С1-С6-алкил)SO2(С1-С6-алкил), -SO2N(C1-C6-алкил)(С1-C6-алкил), -N(С1-C6-алкил)CON(С1-С6-алкил)(C1-C6-алкил), -N(С1-С6-алкил)COO(C1-C6-алкил) и -N(C1-C6-алкил)SO2N(С1-С6-алкил)(C1-C6-алкил).
Термин "алкил", отдельно или в комбинации, означает ацикличный насыщенный углеводород формулы СnН2n+1, который может быть линейным или разветвленным. Примеры таких групп включают метил, этил, н-пропил, изобутил, н-бутил, изобутил, втор.-бутил, трет.-бутил, пентил, изо-амил и гексил. Пока не указано иное, алкил включает 1-6 атомов углерода.
Термин "алкокси" означает алкил, присоединенный посредством атома кислорода, например, метокси (СН3-O-), этокси (СН3СН2-O-).
Содержание атома углерода в алкиле и других углеводородо-содержащих остатков обозначено подстрочным символом, указывающим верхнюю и нижнюю границы количества атомов углерода в остатке, то есть, подстрочный символ Сi-Сj, указывает на остаток с целым числом от "i" до "j" атомов углерода, включительно. Таким образом, например, C1-С6-алкил относиться к алкилу, содержащему от 1 до 6 атомов углерода, включительно.
Термин "гало" означает фтор, хлор, бром или йод.
Желательно, Z означает необязательно замещенный гетероарил или хлор.
Наиболее предпочтительно, Z означает 1,2,4-триазол-1-ил.
Желательно, X означает хлор, a Y означает Н.
Желательно, В означает 2,4-дифторфенил.
Желательно, Е означает фтор.
В одном наиболее предпочтительном варианте осуществления, Z означает 1,2,4-триазол-1-ил; X означает хлор; Y означает Н; В означает 2,4-дифторфенил и Е означает фтор.
Более предпочтительно, изобретение обеспечивает способ получения соединения формулы:
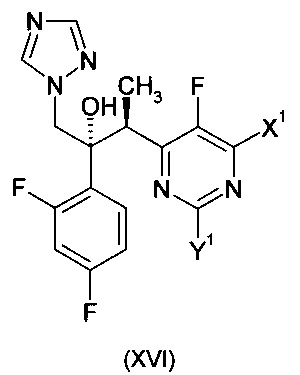
где X1 и Y1 оба означают Н или один из X1 и Y1 означает Н, а другой хлор;
путем взаимодействия соединения формулы:
с соединением формулы:
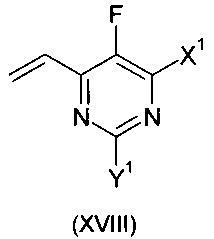
где X1 и Y1 являются такими, как указано выше, в присутствии медного катализатора, хирального фосфинового лиганда и восстанавливающего агента.
Желательно, реакцию осуществляют при температуре от -30°С до +80°С, наиболее предпочтительно при температуре от -12°С до 0°С. Температура на уровне -9°С является оптимальной.
Реакцию осуществляют в присутствии органического растворителя. Спирт является предпочтительным. Наиболее предпочтительным является третичный спирт, оптимальным растворителем является 2-метил-2-бутанол. Примеры приемлемых растворителей включают тетрагидрофуран, метилтетрагидрофуран, диметоксиэтан, диэтоксиэтан, метанол, этанол, 2-пропанол, изобутилацетат, этилацетат, н-бутилацетат, толуол, тетралин, н-бутанол, трет.-бутанол, бензиловый спирт, 1-пентанол, 3-пентанол, 1-гексанол, 2-гексанол, 1-гептанол, 2-гептанол, 4-гептанол, 1-нонанол, 1-метилциклогексанол, 2-метил-1-бутанол, 2-метил-2-бутанол, 3-метил-3-пентанол, 3-этил-3-пентанол, 2,4-диметил-3-пентанол, 1,2-пропандиол, 2,3-диметил-3-пентанол, анизол, н-бутоксиэтанол, дихлорметан, трибутиламин, N-метилпирролидон, ацетонитрил, ацетон, диметилсульфоксид и диоксан.
Необязательно, реакция может осуществляться в присутствии до 10 эквивалентов воды (на основе ограничивающего реагента).
Приемлемые переходные металлы катализаторы хорошо известны специалистам в отрасли (смотреть, например, J. Am. Chem. Soc., 1999, 121 (51), 12202-12203). переходные металлы катализаторы желательно представлены палладиевым катализатором (смотреть, например, Tetrahedron Lett., 1998, 39, 5237-5238), кобальтовым катализатором (смотреть, например, Chem., Lett., 1989, 2005-2008), никилевым катализатором (смотреть, например, Org. Lett., 2007, 9 (3), 537-540), иридиевым катализатором (смотреть, например, Org, Lett., 2001, 12 (3), 1829-1831), индиевым катализатором (смотреть, например, Adv. Synth.Catal., 2002, 344, 283-287; Angew. Chem. Int. Ed., 2004, 43, 711-714), родиевым катализатором (смотреть, например, Eur. J. Org. Chem., 2006, 5594-5600) или медным катализатором, наиболее предпочтительно медным катализатором.
Предпочтительным медным катализатором является стабильная соль меди (I) или меди (II). Если используется соль меди (II), к реакционной смеси нужно добавлять трет.-бутоксид натрия. В целом, соли меди (I) являются предпочтительными, а именно CuF(PPh3)3.MeOH (CuF(PPh3)3.метанол сольват), CuF(PPh3)3.EtOH (CuF(PPh3)3.этанол сольват) и CuOtBu. Примеры приемлемых солей меди (II) включают CuCl2 и Cu(ОСОСН3)2. Сольваты метанола и этанола CuF(PPh3)3 обычно содержат от 1 до 2 молярных эквивалентов растворителя и являются коммерчески доступными. Получение и применение CuOtBu описано в J. Am. Chem. Soc., 1972, 94, 658 и Angew Chemie, 2008, 47, 9961.
Оптимальным медным катализатором является сольват CuF(PPh3)3, такой как CuF(PPh3)3.метанол сольват или CuF(PPh3)3.этанол сольват, а именно CuF(PPh3)3.метанол сольват.
Загрузка катализатора от 0,1 моль % до 2,5 моль % (относительно ограничивающего реагента) является предпочтительной. Загрузка катализатора от 0,1 моль % до 0,5 моль % является наиболее предпочтительной. Оптимальная загрузка составляет 0,1 моль % - 0,2 моль %.
Лиганд, приемлемый для применения с выбранным переходным металлом катализатором, может быть легко выбран специалистом в отрасли на основании общих знаний (смотреть, например, Heterobidentate and Monodentate Phosphine Ligands for Asymmetric Catalysis by Suzanna Christine Milheiro, Yale University, 2011 or Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis edited by Kamer and van Leeuwen, Wiley 2012). Применение ахирального лиганда, такого как BINAP, приведет к синтезу рацемического продукта. Применение хирального лиганда, с другой стороны, приведет к энантиоселективному синтезу. Предпочтительным лигандом является фосфиновый лиганд. Наиболее предпочтительные является хиральный фосфиновый лиганд.
Большинство хиральных фосфиновых лигандов, приемлемых для применения с медным катализатором, дают прекрасные результаты в реакции. Конкретные примеры приемлемых хиральных лигандов включают: (R)-1-[(SP)-2-(дифенилфосфино)ферроценил]этилди-трет.-бутилфосфин; (R)-1-[(SP)-2-(дициклогексилфосфино)ферроценил]этилдициклогексилфосфин; (R)-1-[(SP)-2-(дициклогексилфосфино)ферроценилэтил]дифенилфосфин; (R)-1-[(SP)-2-(дифенилфосфино)ферроценил]этилди(3,5-ксилил)фосфин; (R)-1-{(SP)-2-[бис[3,5-бис(трифторметил)фенил]фосфино]ферроценил}этилдициклогексилфосфин; (R)-1-{(SP)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил}этилдициклогексилфосфин; (R)-1-{(SP)-2-[бис[3,5-бис(трифторметил)фенил]фосфино]ферроценил}этилди(3,5-ксилил)фосфин; (R)-1-[(SP)-2-(дициклогексилфосфино)ферроценил]этилди-трет.-бутилфосфин; (R)-1-{(SP)-2-[бис[4-(трифторметил)фенил]фосфино]ферроценил}этилди-трет.-бутилфосфин; (R)-1-[(SP)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил}этилди-трет.-бутилфосфин; (R)-1-{(SP)-2-[ди(2-фурил)фосфино]ферроценил}этилди(3,5-ксилил)фосфин; (R)-1-{(SP)-2-[ди(2-фурил)фосфино]ферроценил}этилди-трет.-бутилфосфин; (R)-1-{(SP)-2-[ди(1-нафтил)фосфино]ферроценил}этилди-трет.-бутилфосфин; (R)-1-{(SP)-2-[ди(1-нафтил)фосфино]ферроценил}этилди(3,5-ксилил)фосфин; (R)-1-{(SP)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил}-этилди(3,5-ксилил)фосфин; (R)-1-{(SP)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил}-этилбис(2-метилфенил)фосфин; (R)-1-{(SP)-2-[ди(2-фурил)фосфино]ферроценил}этилбис(2-метилфенил)фосфин; (R)-1-[(SP)-2-(ди-трет.-бутилфосфино)ферроценил]этилдифенилфосфин; (R)-1-[(SP)-2-(ди-трет.-бутилфосфино)ферроценил]этилбис(2-метилфенил)фосфин; (R)-(+)-(6,6'-диметоксибифенил-2,2'-диил)бис(дифенилфосфин); (R)-(6,6'-диметоксибифенил-2,2'-диил)бис[бис(3,5-ди-трет.-бутил-4-метоксифенил)фосфин; (SP,S'P)-1,1'-бис[(R)-α-(диметиламино)бензил]-2,2'-бис(дифенилфосфино)ферроцен; (SP,S'P)-1,1'-биc(дициклoгeкcилфocфинo)-2,2'-биc[(R)-α-(димeтиламинo)бeнзил]фeppoцeн; (SP,S'P)-1,1'-биc{биc[3,5-биc(тpифтopмeтил)фeнил]фocфинo}-2,2'-биc[(R)-α-(диметиламино)бензил]ферроцен; (SP,S'P)-1,1'-бис[бис(4-метокси-3,5-димeтилфeнил)фocфинo]-2,2'-биc[(R)-α-(димeтиламинo)бeнзил]фeppoцeн; (SP,S'P)-1,1'-биc[(R)-α-(димeтилaминo)бeнзил]-2,2'-биc[ди(3,5-кcилил)фocфинo]фeppoцeн; (SP,S'P)-1,1'-бис[бис(2-метилфенил)фосфино]-2,2'-бис[(R)-α-(диметиламино)бензил]ферроцен; (RP)-1-[(R)-α-(диметиламино)-2-(дифенилфосфино)бензил]-2-дифенилфосфиноферроцен; (PR)-1-дициклогексилфосфино-2-[(R)-α-(диметиламино)-2-(дициклогексилфосфино)бензил]ферроцен; (R)-1-{(RP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилбис[3,5-бис-(трифторметил)фенил]фосфин; (R)-1-{(RP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилдифенилфосфин; (R)-1-{(RP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилдициклогексилфосфин; (R)-1-{(RP)-2-[2-[бис(4-метокси-3,5-диметилфенил)фосфино]фенил]ферроценил}этилбис[3,5-бис(трифторметил)фенил]фосфин; (R)-1-{(RP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилди(3,5-ксилил)фосфин; (R)-1-{(RP)-2-[2-(дициклогексилфосфино)фенил]ферроценил}этилбис[3,5-бис(трифторметил)фенил]фосфин; (R)-1-{(RP)-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроценил}этилди(3,5-ксилил)фосфин; (R)-1-{(RP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилди(2-норборнил)фосфин; (1R,1'R,2S,2'S)-2,2'-ди-трет.-бутил-2,3,2',3'-тетрагидро-1H,1'Н-(1,1')биизофосфиндолил; (1S,1S',2R,2R')-1,1'-ди-трет.-бутил-(2,2')-дифосфолан; (+)-1,2-бис[(2S,5S)-2,5-диметилфосфолано]бензол; [N-[(1R,2R)-2-(амино-N)-1,2-(2R,3R)-(-)-2,3-бис(дифенилфосфино)-бицикло[2,2,1]гепт-5-ен; (R)-(+)-2,2',6,6'-тетраметокси-4,4'-бис(ди(3,5-ксилил)фосфино)-3,3'-бипиридин; (S)-(+)-(3,5-диокса-4-фосфациклогепта[2,1-а:3,4-а']динафтален-4-ил)пиперидин; (R)-2,2-бинафтоил-(S,S)-ди(1-фенилэтил)аминоилфосфин; (-)-1,2-бис[(2R,5R)-2,5-диметилфосфолано]бензол; (-)-1,2-бис[(2S,5S)-2,5-диметилфосфолано]этан; (R)-(+)-5,5'-дихлор-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенил; (R)-(+)-(1,1'-бинафтален-2,2'-диил)бис(дифенилфосфин).
Предпочтительным хиральным фосфиновым лигандом является (S)-1-{(SP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилбис[3,5-бис-(трифторметил)фенил]фосфин.
Кроме фосфиновых линадов, другие упомянутые лиганды включают N-гетероциклические карбеновые лиганды (особенно для применения с медью; смотреть, например, Org. Lett., 2006, 8 (26), 6059-6062), фосфорамидитовые и фосфонитовые лиганды (особенно для применения с родием; смотреть, например, J. Am. Chem. Soc., 2008, 130, 2746-2747 and Synthesis, 2011, 13, 2011-2013) и бизоксазолиновые (box) и бизоксазолинилпиридиновые (pybox) лиганды (особенно для применения с иридием и родием; смотреть, например, Org. Lett., 2001, 12 (3), 1829-1831 and Tetrahedron, 2008, 64, 9408-9412).
Количество использованного лиганда должно составлять как минимум 1 молярный эквивалент относительно количества использованного переходного металла. Молярное соотношение катализатор : лиганд от 1:1 до 1:6 является предпочтительным, молярное соотношение от 1:2 до 1:3 является особенно предпочтительным.
Восстановительный агент должен быть способен генерировать гидрид переходного металла (например, гидрид меди (I)) in situ. Предпочтительным восстановительным агентом является силан, такой как фенилсилан, диметилфенилсилан, триэтоксисилан, тетраметилдисилоксан, дифенилсилан или полиметилгидросилоксан. Предпочтительным силаном является фенилсилан. Другие восстановительные агенты включают пинаколборан. Желательно, количество использованного восстановительного агента составляет от 0,5 до 3 эквивалентов, на основе ограничивающего реагента.
Реакция является диастереоселектиной во всех случаях, давая преимущественно относительную стереохимию около двух хиральных центров, как показано на формуле (XI) и (XII). Обычно, образуется более чем 86% этого диастереомера (диастереомерный избыток 76% или больше). При использовании хирального лиганда получают продукт в качестве преимущественно одного стереоизомера. Обычно получают энантиомерный избыток более 50%, энантиомерный избыток более 90% тоже не редок.
Когда получают соединение формулы (XVI), где X1 и Y1 оба означают Н, продуктом реакции является вориконазол. Когда один из X1 и Y1 означает хлор, продукт может быть легко преобразован в вориконазол путем восстановления, например, путем гидрогенолиза, описанного в ЕР 0440372 А1. Такое гидрирование желательно проходит при температуре от 20°С до 80°С, наиболее предпочтительно при температуре от 40°С до 70°С, например, при приблизительно 40°С. Предпочтительным катализатором является палладий на угле. Гидрирование может быть осуществлено в неочищенной реакционной смеси, полученной от восстановительной альдольной конденсации, однако, в предпочтительном варианте осуществления указанная неочищенная реакционная смесь разделена между толуолом и слабой водной кислотой (например, водный раствор лимонной кислоты), и водный слой отбрасывают перед гидрированием. Приемлемые растворители для стадии гидрирования включают толуол, этил ацетат, 3-метил-3-пентанол и 2-метил-2-бутанол.
Реакция может осуществляться различными путями с получением продукта, способного преобразовываться в вориконазол путем простого превращения функциональной группы. Например, группа В в соединении формулы (XIII) может означать 2-хлор-4-фторфенил, 2,4-дифторфенил или 2,4-дихлорфенил и атомы хлора или может быть преобразована в атомы фтора путем замещения. Равным образом, группа Е в соединении формулы (XI) или (XII) может означать атом хлора, преобразованный атом фтора путем замещения. Такое замещение проиллюстрировано в способах, описанных в J. Med. Chem., 2011, 54, 8343-8352 и Tet, Lett., 2010, 2652-2654. Альтернативно, В в соединении формулы (XIII) может означать группу 2,4-фторфенил, несущую одну или больше дополнительных групп хлора, которые позже могут быть удалены путем гидролиза.
Неочищенный вориконазол, полученный с использованием этих процедур, может быть очищен путем обработки раствора неочищенного продукта кислотой с целью осаждения соответствующей соли, и последующей нейтрализации соли основанием, таким как ацетат натрия. Применение сульфоновой кислоты является предпочтительным. Особенно предпочтительной является камфорсульфоновая кислота. Приемлемые растворители для стадии осаждения включают толуол, этилацетат, метанол, этанол, 2-пропанол, воду, ацетон, метилэтиловый кетон, метилизобутиловый кетон, тетрагидрофуран и их смеси. Предпочтительным растворителем является смесь толуола, воды и ацетона. При применении хиральной сульфоновой кислоты (например, камфорсульфоновой кислоты) энантиомерный избыток продукта может быть дополнительно улучшен, обычно до уровня 98-100%.
Данное изобретение так же относится к новым промежуточным соединениям формулы:
 и
и 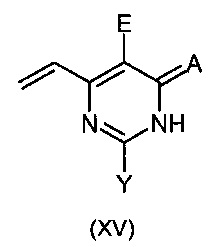
где
X означает Н, гало, -NH2, -NH(С1-С6-алкил), -N(С1-С6-алкил)(С1-С6-алкил), -Si(R3)3 [где R3 независимо в каждом случае означает С1-С6-алкил, арил или арил(C1-С6-алкил)], -O(C1-С6-алкил), -О-арил, -S(С1-алкил), -OSO2(С1-С6-алкил), -NHSO2(С1-С6-алкил) или -S-арил;
Y означает Н, гало, -NH2, -NH(С1-С6-алкил), -N(С1-С6-алкил)(С1-С6-алкил), -Si(R3)3 [где R3 является таким как указано выше], -O(С1-С6-алкил), -О-арил, -OSO2(С1-С6-алкил), -NHSO2(С1-С6-алкил) или -S-арил;
А означает О, S или NH; и
Е означает атом галогена;
при условии, что соединение формулы (XIV) не является 5-бром-4-винилпиримидином.
Следующие Примеры иллюстрируют практическое применение вышеописанных процедур.
Пример 1 - Получение вориконаэола
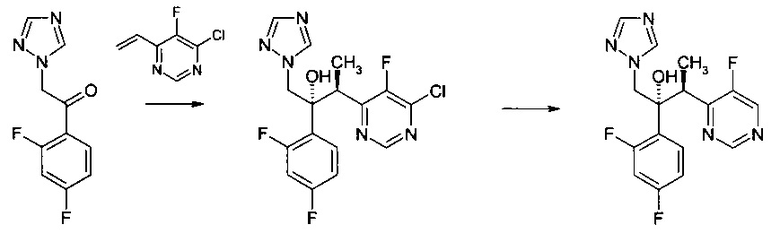
Стадия 1
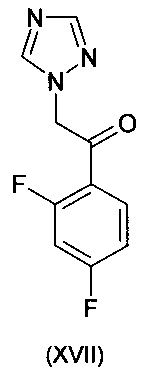
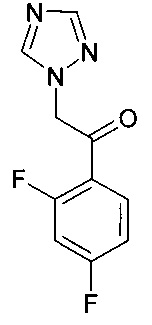
К смеси CuF(PPh3)3 метанол сольвата (0,019 г) и (S)-1-{(SP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилбис[3,5-бис-(трифторметил)фенил]фосфина (0,093 г) добавляли 2-метил-2-бутанол (16 мл). Смесь перемешивали при комнатной температуре, в атмосфере азота, на протяжении 30 мин до растворения всех твердых веществ. К полученному раствору потом добавляли 1-(2,4-дифторфенил)-2-(1Н-1,2,4-триазол-1-ил)этанон (2,69 г) и воду (0,36 г). Потом реакционную суспензию перемешивали при комнатной температуре на протяжении 20 мин перед охлаждением до -9°С.
К реакционной суспензии при -9°С потом добавляли раствор 4-хлор-5-фтор-6-винилпиримидина (1,59 г, ограничивающий реагент) и фенилсилана (1,09 г) в 2-метил-2-бутаноле (4 мл) на протяжении 45 мин. После перемешивания при -9°С на протяжении 270 мин, к этому времени уже не наблюдалось 4-хлор-5-фтор-6-винилпиримидина с помощью ВЭЖХ, реакционную смесь нагревали до комнатной температуры и выдерживали при этой температуре на протяжении 18 часов. Потом добавляли воду (3,98 г) для гашения реакции. Потом добавляли толуол (30 мл) к погашенной реакционной смеси с получением двухфазной смеси, содержащей (2R,3S)-3-(6-хлор-5-фторпиримидин-4-ил)-2-(2,4-дифторфенил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол.
Растворитель удаляли в вакууме при 45-50°С до получения остатка объемом приблизительно 8 мл. К нему добавляли еще толуол (50 мл), после чего 50 мл водного раствора лимонной кислоты (20% м./о.). Двухфазную смесь перемешивали при комнатной температуре на протяжении 30 мин., и потом слои разделялись на протяжении 20 мин. Водный слой отбрасывали и к слою толуола добавляли водный раствор лимонной кислоты (50 мл, 20% м./о.). Двухфазную смесь перемешивали на протяжении 10 мин и слои разделялись на протяжении 20 мин. Снова водный слой отбрасывали. К слою толуола добавляли воду (9,5 мл). Двухфазную смесь перемешивали на протяжении 10 мин., оставляли для расслоения (20 мин) и разделяли.
К сохраненному слою толуола добавляли активированный уголь (0,192 г). Смесь после этого нагревали до 50°С на протяжении 3 часов, перед охлаждением до комнатной температуры. Катализатор палладий на угле (Evonik Е101 NE/W 10% Pd/C, 50% смоченный водой, 0,546 г) добавляли к смеси, после чего ацетат натрия (2,06 г) и воду (5,97 г). Реакционную смесь нагревали до 40°С перед герметизацией реакционной колбы водородом (5 бар). Реакционную смесь перемешивали при 40°С и 5 бар водорода на протяжении 7 часов, перед охлаждением до ~21°С на протяжении последующих 15 часов. Гидрированную реакционную смесь потом разводили насыщенным водным раствором бикарбоната натрия (9,5 мл), перед фильтрованием через Celite®. Фильтр промывали водой (1,59 мл) и толуолом (2×100 мл).
Часть неочищенного продукта очищали с помощью колоночной хроматографии на силикагеле и анализировали с помощью хиральной колоночной хроматографии (колонка Chiralcel OD-RH 150×4,6 мм, 30°С, скорость потока 1 мл/мин, элюент 60:40 гептан : этанол, противодавление ~600 psi). Результаты показали, что соотношении желаемого (2R,3S) энантиомера к нежелательному (2S,3R) энантиомеру составляет 97:3 (энантиомерный избыток 94%).
При таких же условиях, но с использованием лиганда ((RP)-1-[(R)-α-(диметиламино)-2-(дифенилфосфино)бензил]-2-дифенилфосфиноферроцен), наблюдали энантиоселективность на уровне приблизительно 84% (что соответствует приблизительно 70% энантиомерного избытка).
Двухфазную смесь потом разделяли на протяжении 10 мин перед отбрасыванием водного слоя. Слой толуола потом промывали водой (10 мл) перед концентрированием в вакууме до ~45 мл. Раствор толуола (содержащий вориконазол API - (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол) нагревали до 45°С и потом обрабатывали раствором камфорсульфоновой кислоты (CSA) (1,40 г) в ацетоне (40 мл) на протяжении 60 мин. Полученный раствор потом охлаждали до 5°С на протяжении 60 мин перед перемешиванием на протяжении еще 120 мин. Твердые вещества потом удаляли, промывали толуолом (2×20 мл) и высушивали на протяжении 18 часов при пониженном давлении с получением (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол ((1R,4R)-7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфоната (2,52 г, 43% на основе исходного материала пиримидина).
К высушенному (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол ((1R,4R)-7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфонату (2,52 г) добавляли раствор EtOH/ацетон (соотношение 3:1, 22 мл). Суспензию потом нагревали до 50°С на протяжении 30 мин, перед охлаждением до 5°С на протяжении 60 мин. Твердые вещества потом удаляли и высушивали на протяжении 24 часов при 40°С при пониженном давлении с получением (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол ((1R,4R)-7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфоната в виде белого твердого вещества (2,39 г, выход 41,0% на основе исходного материала пиримидина).
1Н ЯМР (400 МГц, CDCl3): δ (млн ч.) = 9,42 (1Н, с, ArH), 9,10 (1Н, с, ArH), 8,67 (1Н, с, ArH), 8,14 (1Н, с, ArH), 7,49 (1Н, м, ArH), 7,53-7,42 (2Н, м, 2×ArH), 4,96 (1Н, д, СН2), 4,48 (1Н, д, СН2), 4,11 (1Н, септ., СН), 3,25 (1Н, д, СН2), 2,81 (1Н, д, СН2), 2,56-2,41 (1Н, м, СН2), 2,38-2,25 (1Н, м, СН2), 2,11-1,95 (2Н, м, СН2), 1,90 (1Н, д, СН2), -1,86-1,73 (1Н, м, СН2), 1,45-1,35 (1Н, м, СН2), 1,15 (3Н, д, СН3), 1,03 (3Н, с, СН3), 0,82 (3Н, с, СН3).
Стадия 2
К (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол ((1R,4R)-7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфонату (2,125 г) добавляли EtOH (3,8 мл) и воду (3,8 мл). Суспензию нагревали до 32,5°С, что приводило к получению практически бесцветного раствора. Этот раствор потом добавляли маленькими аликвотами (1 мл) к охлажденному (0°С) раствору ацетата натрия (0,30 г) в воде (4,1 мл) на протяжении 45 мин. К полученной суспензии потом добавляли раствор EtOH (0,45 мл) и воду (0,45 мл). Смесь потом перемешивали при 2°С на протяжении 30 мин, перед постепенным добавлением воды (4,5 мл) на протяжении 20 мин. Суспензию потом перемешивали при 2°С на протяжении 13,5 часов, перед выделением твердых веществ, и дважды ресуспендировали при 0-5°С в воде (6,4 мл). Выделенные твердые вещества после высушивали при 45°С на протяжении ~48 часов при пониженном давлении с получением (2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ола в виде белого твердого вещества (0,969 г, выход 76%).
1Н ЯМР (400 МГц, CDCl3): δ (млн ч.) = 8,95 (1Н, с, ArH), 8,61 (1Н, с, ArH), 8,10 (1Н, с, ArH), 7,62-7,56 (1Н, м, ArH), 7,25 (1Н, с, ArH), 6,90-6,79 (2Н, м, 2×ArH), 6,50 (1Н, ш с, ОН), 4,76 (1Н, д, СН2), 4,37 (1Н, д, СН2), 4,14 (1Н, септ., СН), 1,11 (3Н, д, СН3).
Пример 2 - Получение вориконазола
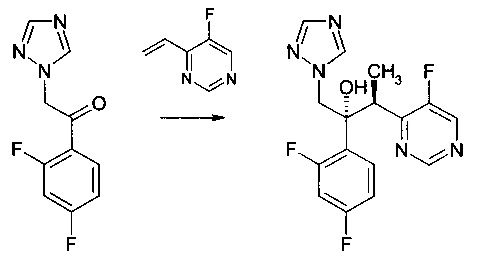
К смеси CuF(PPh3)3 метанол сольвата (0,0038 г) и (S)-1-{(SP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилбис[3,5-бис-(трифторметил)фенил]фосфина (0,0187 г) добавляли 3-метил-3-пентанол (0,5 мл). Смесь перемешивали при комнатной температуре в атмосфере аргона на протяжении 20 мин до растворения всех твердых веществ. Полученный раствор потом охлаждали до 0°С перед добавлением 5-фтор-4-винилпиримидина (0,050 г, ограничивающий реагент) и 1-(2,4-дифторфенил)-2-(1Н-1,2,4-триазол-1-ил)этанона (0,0899 г) в 3-метил-3-пентанол (2 мл). К этой смеси при 0°С потом добавляли раствор фенилсилана (0,0436 г) в 3-метил-3-пентаноле (0,5 мл) на протяжении приблизительно 30 мин.
Реакционную смесь перемешивали при 0°С на протяжении 21 часа до полного поглощения 5-фтор-4-винилпиримидина, что наблюдали с помощью ВЭЖХ. Обнаружили, что анализируемая реакционная смесь содержала желаемый продукт ((2R,3S)-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол) с выходом приблизительно 40% (на основе исходного материала пиримидина).
Пример 3 - Получение вориконазола
Используя подобную процедуру, что описана в Примере 2, с CuF(PPh3)3.MeOH в качестве медного катализатора, (S)-1-{(SР)-2-[2-(дифенилфосфино)фенил]ферроценил}этилбис [3,5-бис-(трифторметил)фенил]фосфином в качестве хирального фосфинового лиганда и 2-метил-2-бутанол в качестве растворителя получали вориконазол с 70% энантиомерным избытком.
Используя похожие условия, но с ((RР)-1-[(R)-α-(диметиламино)-2-(дифенилфосфино)бензил]-2-дифенилфосфиноферроценом) в качестве хирального фосфинового лиганда получали an энантиомерный избыток 50%.
Пример 4 - Получение предшественника вориконазола
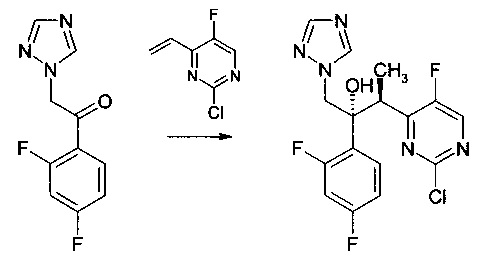
К смеси CuF(PPh3)3 метанол сольвата (0,0029 г) и (RР)-1-[(R)-α-(диметиламино)-2-(дифенилфосфино)бензил]-2-дифенилфосфиноферроцена (0,0108 г) добавляли н-бутил ацетат (1 мл). Смесь перемешивали при комнатной температуре в атмосфере аргона на протяжении 30 мин до растворения всех твердых веществ. Полученный раствор потом охлаждали до 0°С перед добавлением 2-хлор-5-фтор-4-винилпиримидина (0,050 г, ограничивающий реагент) и 1-(2,4-дифторфенил)-2-(1Н-1,2,4-триазол-1-ил)этанона (0,074 г) в н-бутилацетате (1 мл). К этой смеси при 0°С потом добавляли раствор фенилсилана (0,034 г) в н-бутилацетате (0,25 мл) на протяжении приблизительно 5 мин.
Реакционную смесь перемешивали при 0°С на протяжении 24 часов до полного поглощения 2-хлор-5-фтор-4-винилпиримидина. Реакционную смесь анализировали с помощью ВЭЖХ и выяснилось, что она содержит желаемый продукт ((2R,3S)-3-(2-хлор-5-фторпиримидин-4-ил)-2-(2,4-дифторфенил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол) с выходом приблизительно 5%.
Пример 5 - Получение промежуточного продукта вориконазола
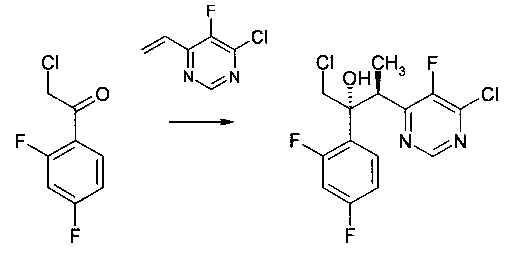
К CuF(PPh3)3 метанол сольвату (0,0094 г) и (RP)-1-[(R)-α-(диметиламино)-2-(дифенилфосфино)бензил]-2-дифенилфосфиноферроцене (0,0068 г) в атмосфере аргона добавляли тетрагидрофуран (1 мл). Смесь перемешивали при 20°С (в атмосфере аргона) на протяжении 30 мин до растворения всех твердых веществ. Раствор потом охлаждали до -20°С и добавляли фенилсилан (0,027 г). Через 10 мин добавляли раствор 4-хлор-5-фтор-6-винилпиримидина (0,180 г) и 2-хлор-1-(2,4-дифторфенил)этанона (0,095 г) в тетрагидрофуране (1,5 мл) на протяжении приблизительно 5 мин. Реакционную смесь перемешивали на протяжении часа при -20°С до полного поглощения винилпиримидина, что наблюдали с помощью ВЭЖХ. Реакцию гасили добавлением водного раствора хлорида аммония (1 М, 3 мл), после чего метилтрет.-бутиловым эфиром (5 мл). Органический слой высушивали до остатка, который потом очищали с помощью колоночной хроматографии (эллюируя 0-20% этилацетатом в циклогексане) с получением (2R,3S)-1-хлор-3-(6-хлор-5-фторпиримидин-4-ил)-2-(2,4-дифторфенил)бутан-2-ола (0,259 г, выход 65%) в виде белого твердого вещества.
1Н ЯМР (500 МГц, CDCl3): δ (млн ч.) = 8,71 (1Н, с, ArH), 7,75 (1Н, м, ArH), 6,90 (1Н, м, ArH), 6,78 (1Н, м, ArH), 5,51 (1Н, S, ОН), 4,00 (1Н, септ., СН), 3,94 (1Н, д, СН2), 3,51 (1Н, д, СН2), 1,08 (3Н, д, СН3).
Хиральная ВЭЖХ (Chiralcel-OJ-H, с 95:5 гексан : IPA, при 1 мл/мин) продукта продемонстрировала энантиоселективность ~95,7% в реакции восстановительной альдольной конденсации (~91% э.и.)
Пример 6 - Получение промежуточного продукта вориконазола

К CuF(PPh3)3 метанол сольвату (0,137 г) и (RР)-1-[(R)-α-(диметиламино)-2-(дифенилфосфино)бензил]-2-дифенилфосфиноферроцену (0,100 г) добавляли тетрагидрофуран (14 мл). Смесь перемешивали при 0-5°С (в атмосфере аргона) на протяжении 30 мин до растворения всех твердых веществ. К этому раствору потом добавляли фенилсилан (0,789 г). Через 10 мин добавляли раствор 5-фтор-4-винилпиримидина (0,903 г) и 2-хлор-1-(2,4-дифторфенил)этанона (1,68 г) в тетрагидрофуране (22 мл) на протяжении 30 мин. Реакционную смесь перемешивали на протяжении часа при 0-5°С, после чего не наблюдалось присутствие 5-фтор-4-винилпиримидина с помощью ВЭЖХ. Реакцию гасили добавлением водного раствора хлорида аммония (1 М, 15 мл), после чего метилтрет.-бутиловым эфиром (30 мл). Органический слой высушивали до остатка, который потом очищали с помощью колоночной хроматографии (эллюируя с 5-40% этилацетатом в циклогексане) с получением (2R,3S)-1-хлор-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)бутан-2-ола в виде белого твердого вещества (1,70 г, выход 74%).
1Н ЯМР (500 МГц, CDCl3): δ (млн ч.) = 8,93 (1Н, с, ArH), 8,57 (1Н, м, ArH), 7,76 (1Н, м, ArH), 6,90 (1Н, м, ArH), 6,78 (1Н, м, ArH), 5,79 (1Н, S, ОН), 3,97 (1Н, септ., СН), 3,93 (1Н, д, СН2), 3,49 (1Н, д, СН2), 1,05 (3Н, д, СН3).
Хиральная ВЭЖХ (колонка Целлюлоза-1, используя элюент в соотношении 60:40 ацетонитрил : вода при 1 мл/мин. & 25°С) (2R,3S)-1-хлор-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)бутан-2-ола продемонстрировала энантиоселективность ~93,2% в реакции восстановительной альдольной конденсации (~86% энантиомерный избыток).
Следующие примеры получения показывают, как можно получить исходные материалы винилгетероарила.
Получение 1: 4-хлор-5-фтор-6-винилпиримидин
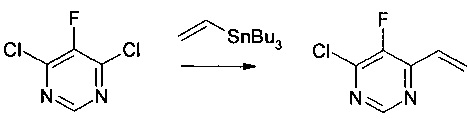
Смесь 4,6-дихлор-5-фторпиримидина (5,0 г, 30,0 ммоль, 1,0 экв.) и трибутил(винил)олова (10,4 г, 33,0 ммоль, 1,1 экв.) в дихлорметане (50 мл) дегазировали потоком азота на протяжении 10 мин. Добавляли бис(трифенилфосфин) палладия(II) хлорид (0,53 г, 0,75 ммоль, 0,025 экв.). Полученную смесь дегазировали потоком азота на протяжении дополнительных 15 мин и кипятили с обратным холодильником на протяжении 72 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водным раствором фторида калия (2 М, 75 мл, 5 экв.). Полученную смесь перемешивали на протяжении 2 часов и фильтровали через Celite®. Фильтрат выливали в разделительную воронку и разделяли. Органический слой промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении при 20°С. Полученный неочищенный продукт очищали на колонке AnaLogix® (SF40-115g). Градиент, использованный для очистки, включал 10 мин изократический пентан, после чего 20 мин линейное изменение до 5% диэтилового эфира в пентане. Чистые фракции объединяли и концентрировали при пониженном давлении при 20°С с получением 4-хлор-5-фтор-6-винилпиримидина (3,0 г, 63% выход).
Масс-спектр (позитивный режим): m/z 158,0 (М+). 1Н ЯМР (300 МГц, CDCl3): δ (млн ч.) 8,71 (с, 1Н), 6,99 (м, 1Н), 6,75 (дд, J=17,4 Гц, 1,8 Гц, 1Н), 5,90 (дд, J=10,5 Гц, 1,5 Гц, 1Н). 19F ЯМР (282 МГц, CDCl3): δ 133,88 (с).
Получение 2: 4-хлор-5-фтор-6-винилпиримидин, альтернативный путь
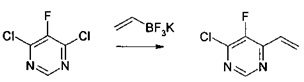
4,6-Дихлор-5-фторпиримидин (6 г, 36,0 ммоль) реагировал с винилтрифторборатом калия (1,60 г, 37,8 ммоль, 1,05 экв.) и карбонатом цезия (17,58 г, 1,5 экв.) в вода/метил тетрагидрофуран. Реакцию обрабатывали Pd(PPh3)2Cl2 (504 мг, 0,02 экв.) и PPh3 (189 мг, 0,02 экв.) в атмосфере аргона. Реакционную смесь кипятили с обратным холодильником и продолжали кипячение на протяжении 20 часов. Реакцию гасили добавлением дополнительного количества воды и трет.-бутилметилового эфира. Органическую фазу атмосферно дистиллировали для удаления растворителей. Остаток очищали с помощью колоночной хроматографии, 12-100% дихлорметан в гексане. Объединенные фракции концентрировали фракционным дистилляцией с получением 4,1 г (71% выход) продукта в виде светло-желтого масла.
Получение 3: 4-хлор-5-фтор-6-винилпиримидин, альтернативный путь

Стадия 1
Метоксид натрия (151,6 г, 2,81 моль) частями добавляли к метанолу (1,75 л), поддерживая температуру ниже 30°С, используя охлаждающую баню лед/метанол. Формамид ацетат (146,12 г, 1,40 моль) добавляли одной порцией. Смесь охлаждали до 5°С и после добавляли этилфторацетоацетат (218.3 г, 1,47 моль) на протяжении 10 мин. охлаждая. Реакционную смесь нагревали до 25°С и перемешивали при этой температуре на протяжении 2 часов. Уксусную кислоту (252,8 г, 4,21 моль) добавляли к смеси на протяжении 5 мин, после чего выпаривали растворители. Потом добавляли толуол (400 мл) и полученную суспензию концентрировали снова путем выпаривания. Этилацетат (808 мл) добавляли к полученной суспензии и смесь нагревали до 40°С на протяжении 15 мин. Нерастворимый материал отфильтровывали и промывали этилацетатом (2×153 мл).
Фильтрат выпаривали и полученная суспензия со временем затвердевала. Добавляли диэтиловый эфир (400 мл) и твердую массу разбивали. Через 4 часа при комнатной температуре твердый продукт отфильтровывали и промывали диэтиловым эфиром (2×150 мл). После высушивания в теплой печке на протяжении ночи получали 4-гидрокси-5-фтор-6-винилпиримидин в виде воскового твердого вещества, 264,6 г (147%). Протон ЯМР показал, что требуемый продукт содержал 0,86 молярных экв. ацетата натрия (выход корректировали по ацетату натрия = 94,9%).
Стадия 2
4-Гидрокси-5-фтор-6-винилпиримидин (954,2 г, неочищенный продукт стадии 1, 4,80 моль) частями добавляли на протяжении 15 мин к оксихлориду фосфора (1622 мл, 986 г, 6,43 моль, 1,7 об.), удерживая температуру на уровне 40°С охлаждением на ледяной бане. Охлаждение убирали и температура возрастала до 50°С. Реакция продолжалась при 50°С на протяжении 15 мин и потом нагревалась при 80°С на протяжении 2 часов. На протяжении этого времени все твердые вещества растворялись с получением коричневого раствора.
Полученный раствор добавляли к воде (7,35 л) по капле на протяжении 90 мин, тщательно перемешивая, поддерживая температуру на уровне 20°С охлаждением лед/метанол и контролируя скорость добавления. Реакционную смесь перемешивали при 20°С на протяжении еще 30 мин. Соль (NaCl) добавляли до насыщения раствора и смесь экстрагировали дихлорметаном (3,63 л, после чего 6×1,8 л). Экстракты дихлорметана промывали насыщенным раствором бикарбоната натрия (363 мл), высушивали над MgSO4 и концентрировали с получением светло-коричневого масла. Масло дистиллировали при давлении 180 мбар. 4-Хлор-5-фтор-6-метилпиримидин дистиллировали в диапазоне 100-106°С при таком давлении.
Стадия 3
К раствору хлорида N-изобутил-N-метиленпропан-2-аминия (13,3 г, 88,7 ммоль) в ацетонитриле (55 мл) добавляли 4-хлор-5-фтор-6-метилпиримидин (10,0 г, 68.2 ммоль). Реакционную колбу промывали аргоном и смесь кипятили с обратным холодильником на протяжении 24 часов, перемешивая, потом охлаждали до комнатной температуры. Воду (130 мл) добавляли и смесь экстрагировали дихлорметаном (140 мл). Органическую фазу промывали 10% водным KHSO4 (400 мл) и высушивали над Na2SO4. Раствор фильтровали и выпаривали в вакууме при давлении 300 мбар и 35°С. Добавляли трет.-бутилкатехол (30 мг, 0,2 мас. %, на основе массы неочищенного материала). Оставшийся растворитель удаляли в вакууме при 50°С и неочищенный продукт потом дистиллировали при давлении 5 мбар и 50°С (масляная баня). Добавляли трет.-бутилкатехол (0,1 мас. %) к дистиллированному 4-хлор-5-фтор-6-винилпиримидину (7,9 г, 73%), полученному в виде светло-желтого масла.
Получение 4: 5-фтор-4-винилпиримидин
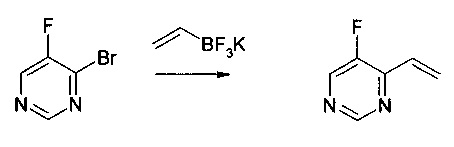
4-Бром-5-фторпиримидин (5 г) реагировал с винилтрифторборатом калия (1,05 эквивалентов) в присутствии (PPh3)2PdCl2 (0,02 экв.), PPh3 (0,02 экв.) и Cs2CO3 (3 эквивалентов) в смеси метилтетрагидрофурана (85 мл) и воды (8,5 мл). Реакционную смесь нагревали при 75°С на протяжении приблизительно 5,5 часов. Реакционную смесь потом разводили метил трет.-бутиловым эфиром (50 мл), после чего экстрагировали водой. Неочищенный продукт очищали дистилляцией при 170 мбар (90-110°С). Продукт получали в виде бесцветного масла (1,44 г, 41% выход).
Масс-спектр (позитивный режим): m/z 124,0 (М+). 1Н ЯМР (300 МГц, CDCl3): δ (млн ч.) 8,98 (с, 1Н), 6,96 (дд, 1Н), 6,70 (м, 1Н), 5,82 (д, 1Н). 19F ЯМР (282 МГц, CDCl3): 138,60 (с).
Получение 5: 2-хлор-5-фтор-4-винилпиримидин
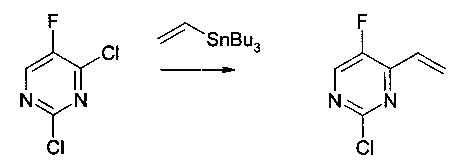
Смесь 2,4-дихлор-5-фторпиримидина (5,0 г, 30,0 ммоль, 1,0 экв.), трибутил(винил)олова (10,4 г, 33,0 ммоль, 1,1 экв.) в дихлорметане (50 мл) дегазировали потоком азота на протяжении 10 мин. Добавляли хлорид бис(трифенилфосфин)палладия (II) (0,53 г, 0,75 ммоль, 0,025 экв.). Полученную смесь дегазировали потоком азота на протяжении дополнительных 15 мин и кипятили с обратным холодильником на протяжении 24 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водным раствором фторида калия (2 М, 75 мл, 5 экв.). Полученную смесь перемешивали на протяжении 2 часов и фильтровали через Celite®. Фильтрат выливали в разделительную воронку и разделяли. Органический слой промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении при 20°С. Полученный неочищенный продукт очищали на колонке AnaLogix® (SF40-115g). Градиент, использованный для очистки, составлял 10 мин изократичный пентан, после чего 20 мин линейное изменение до 5% диэтилового эфира в пентане. Чистые фракции объединяли и концентрировали при пониженном давлении при 20°С с получением 2-хлор-5-фтор-4-винилпиримидина в виде масла (3,65 г, 77% выход).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФУНГИЦИД НА ОСНОВЕ ГЕТЕРОЦИКЛИЛ-ПИРИМИДИНИЛ-АМИНОПРОИЗВОДНЫХ | 2008 |
|
RU2471793C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИАРИЛЗАМЕЩЕННОЙ 4-АМИНОМАСЛЯНОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ИНГИБИТОРОВ НЭП | 2007 |
|
RU2469019C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХПРИ ПОЛУЧЕНИИИНГИБИТОРОВ NEP | 2011 |
|
RU2573824C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2797316C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ, ТИОФЕНКАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ | 2020 |
|
RU2837147C1 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2797513C2 |
| СПОСОБ ГИДРОГЕНИЗАЦИИ ИМИНОВ И СПОСОБ ПОЛУЧЕНИЯ АМИНА | 1995 |
|
RU2150464C1 |
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
Изобретение относится к способу получения соединения формулы:
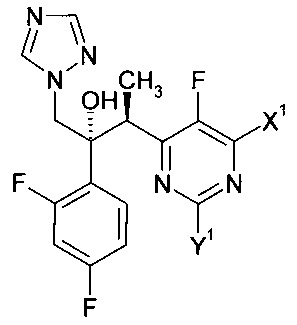
в которой X1 и Y1 означает Н или один из X1 и Y1 означает Н, а другой означает хлор; путем реагирования соединения формулы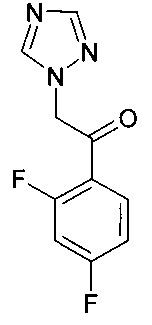 с соединением формулы
с соединением формулы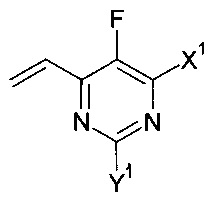 , в присутствии медного катализатора, хирального фосфинового лиганда и восстанавливающего агента, при этом реакцию проводят в присутствии органического растворителя, который является спиртом. Изобретение так же относится к новым соединениям, которые являются промежуточными, в заявленном способе получения. Технический результат: предложен улучшенный способ получения вориконазола и его аналогов, обеспечивающий получение (2R,3S) стереоизомера. 2 н. и 9 з.п. ф-лы, 6 пр.
, в присутствии медного катализатора, хирального фосфинового лиганда и восстанавливающего агента, при этом реакцию проводят в присутствии органического растворителя, который является спиртом. Изобретение так же относится к новым соединениям, которые являются промежуточными, в заявленном способе получения. Технический результат: предложен улучшенный способ получения вориконазола и его аналогов, обеспечивающий получение (2R,3S) стереоизомера. 2 н. и 9 з.п. ф-лы, 6 пр.
1. Способ получения соединения формулы:
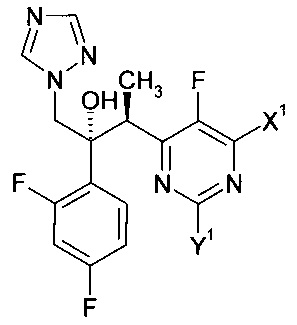
где:
X1 и Y1 означает Н или один из X1 и Y1 означает Н, а другой означает хлор; путем реагирования соединения формулы:
с соединением формулы:
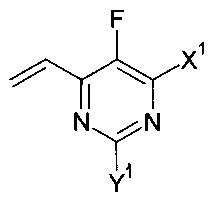
соответственно, где X1 и Y1 являются такими, как указано выше, в присутствии медного катализатора, хирального фосфинового лиганда и восстанавливающего агента, при этом реакцию проводят в присутствии органического растворителя, который является спиртом.
2. Способ по п. 1, который отличается тем, что органическим растворителем является третичный спирт.
3. Способ по п. 2, который отличается тем, что органическим растворителем является 2-метил-2-бутанол.
4. Способ по п. 1, который отличается тем, что X1 означает хлор, a Y1 означает Н.
5. Способ по п. 1, который отличается тем, что катализатором является медь (I).
6. Способ по п. 5, который отличается тем, что катализатор означает CuF(PPh3)3.метанол сольват или CuF(PPh3)3.этанол сольват.
7. Способ по п. 1, который отличается тем, что лиганд означает (S)-1-{(SP)-2-[2-(дифенилфосфино)фенил]ферроценил}этилбис[3,5-бис-(трифторметил)фенил]фосфин.
8. Способ по п. 1, который отличается тем, что восстанавливающий агент означает фенилсилан.
9. Способ по п. 1, который отличается тем, что температура реакции составляет приблизительно -9°С.
10. Способ по п. 1, который отличается тем, что продуктом является вориконазол, или продукт впоследствии обрабатывают на одной или нескольких стадиях с получением вориконазола.
11. Соединение представляет собой:
4-хлор-5-фтор-6-винилпиримидин;
5-фтор-4-винилпиримидин; или
2-хлор-5-фтор-4-винилпиримидин.
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1992 |
|
RU2095358C1 |
| ТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1991 |
|
RU2114838C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛОВ, КОНЕЧНЫЕ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ СПОСОБА | 1996 |
|
RU2156760C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
| WO 2006065726 A2, 22.06.2006. | |||

