Настоящее изобретение относится к новой полиморфной форме соединения, пригодного в качестве фармацевтического средства, к способам получения указанной полиморфной формы, композициям, содержащим указанную полиморфную форму, и к ее применению для лечения и предотвращения аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит, и других воспалительных заболеваний, опосредованных простагландином D2 (PGD2) или другими агонистами, которые действуют через рецептор CRTH2 на клетки, включающие эозинофилы, базофилы и лимфоциты Th2.
PGD2 представляет собой эйкозаноид, который представляет собой класс химических медиаторов, синтезируемый клетками в ответ на локальное повреждение ткани, нормальные или гормональные стимулы или в результате путей активации клеток. Эйкозаноиды связываются со специфическими поверхностными рецепторами клеток в различных тканях всего организма и опосредуют различные эффекты в этих тканях. Известно, что PGD2 продуцируется мастоцитами, макрофагами и лимфоцитами Th2; кроме того, PGD2 был обнаружен в высоких концентрациях в дыхательных путях у пациентов страдающих астмой, которым вводят антиген (Murray et al., (1986), N. Engl. J. Med. 315: 800-804). Инсталляция PGD2 в дыхательные пути может вызвать многие признаки астматического ответа, в том числе бронхоконстрикцию (Hardy et al., (1984) N. Engl. J. Med. 311: 209-213; Sampson et al., (1997) Thorax 52: 513-518) и аккумуляцию эозинофилов (Emery et al., (1989) J. Appl. Physiol. 67: 959-962).
Способность экзогенно применяемого PGD2 вызывать воспалительные реакции была подтверждена путем применения сверхэкспрессирующей PGD2-синтазы человека у трансгенных мышей, которые проявляют повышенное эозинофильное легочное воспаление и продуцирование цитокинов Th2 в качестве ответа на антиген (Fujitani et al., (2002) J. Immunol. 168: 443-449).
Первым обнаруженным рецептором, специфичным для PGD2, был рецептор DP, который связан с повышением внутриклеточных уровней cAMP. Однако предполагают, что PGD2 опосредуют большую часть своей провоспалительной активности посредством взаимодействия с рецептором, сопряженным с G-белком, который называют CRTH2 (молекула, гомологичная хемоаттрактантному рецептору, экспрессируемая на клетках Th2) и который экспрессируют лимфоциты Th2, эозинофилы и базофилы (Hirai и et al., (2001) J. Exp Med. 193: 255-261, EP 0851030, EP-A-1211513 и Bauer et al., EP-A-1170594). Представляется очевидным, что влияние PGD2 на активацию лимфоцитов Th2 и эозинофилов осуществляется посредством CRTH2, поскольку селективные агонисты CRTH2 13,14-дигидро-15-кето-PGD2(DK-PGD2) и 15R-метил-PGD2 могут вызывать этот ответ, а влияние PGD2 f анти-CRTH2 антителом (Hirai et al., 2001; Monneret et al., (2003) J. Pharmacol. Exp. Ther. 304: 349-355). В отличие от этого селективный агонист DP BW245C не способствует миграции лимфоцитов Th2 или эозинофилов (Hirai et al., 2001; Gervais et al., (2001) J. Allergy Clin. Immunol. 108: 982-988). На основании этих данных использование антагонистов PGD2 на рецепторе CRTH2 представляет собой привлекательный подход к лечению воспалительного компонента аллергических заболеваний, зависимых от Th2, таких как астма, аллергический ринит и атопический дерматит.
В ЕР-А-1170594 предполагают, что способ, к которому относится указанный документ, можно применять для идентификации соединений, пригодных для лечения аллергической астмы, атопического дерматита, аллергического ринита, аутоиммунного, реперфузионного нарушения и ряда воспалительных состояний, все из которых опосредованы действием PGD2 или других агонистов на рецептор CRTH2.
С момента публикации документа ЕР-А-1170594 появилось множество публикаций, относящихся к соединениям, имеющим антагонистическую активность по отношению к CRTH2.
В более ранних заявках авторов настоящего изобретения WO-A-2005/044260, WO 2006/095183 и WO 2008/012511 описаны соединения, которые представляют собой антагонисты PGD2 рецептора CRTH2. Указанные соединения представляют собой производные индол-1-уксусной кислоты, замещенные в 3 положении CH2-арильной группой, которая может быть замещена одним или более дополнительными заместителями. Соединения, описанные в указанных документах, представляют собой мощные in vitro антагонисты PGD2 на рецепторе CRTH2.
Более ранняя заявка авторов настоящего изобретения WO 2009/090414 относится к пиридиловым аналогам соединений, описанных в WO 2008/012511. Неожиданно было обнаружено, что конкретные региоизомеры пиридила и его замещенные варианты приводят к оптимальному балансу активности и фармакокинетических свойств. В частности, было обнаружено, что введение фенил-сульфонильного заместителя во 2 положение региоизомера пиридин-3-ила обеспечивает соединения, обладающие хорошей активностью в функциональном анализе in vitro и хорошей фармакокинетикой in vivo.
Соединения, описанные в WO 2009/090414, как было предсказано, пригодны для лечения заболеваний и состояний, опосредованных действием PGD2 на рецептор CRTH2. Одно из указанных соединений, [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусная кислота (соединение 1), является особенно полезным.
Способ синтеза [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты изложен в документе WO 2009/090414, но авторы настоящего изобретения обнаружили, что партии соединения, полученные согласно указанному способу, являются аморфными.
Таким образом, авторами настоящего изобретения была поставлена задача разработать кристаллическую форму соединения 1. Кристаллические формы часто более стабильны по сравнению с аморфными формами, и аморфная форма может со временем спонтанно превращаться в кристаллическую форму. Безусловно, для фармацевтически активных соединений это является недостатком, поскольку различные формы соединения могут иметь различные фармакокинетические свойства.
Авторы настоящего изобретения также стремились получить кристаллическую форму несольватированной формы соединения 1. Несольватированные формы часто являются более подходящими для получения фармацевтических композиций, поскольку многие сольваты являются термодинамически неустойчивыми при температуре окружающей среды, хотя в целом гидраты предпочтительнее, чем другие сольваты.
Авторами настоящего изобретения было обнаружено, что [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусная кислота является энантиотропной и, следовательно, стабильная полиморфная форма указанного соединения зависит от температуры. Авторы настоящего изобретения обнаружили три различные полиморфные формы этого соединения: первую, обозначенную как форма 1, которая является термодинамически стабильной при высоких температурах, хотя определить точную температуру перехода было затруднительно; вторую (обозначенную как форма 2), которая является термодинамически стабильной при температурах до примерно 60-65°C; и третью форму (форма 3), которая является стабильной при температурах между диапазонами стабильности формы 2 и формы 1.
Таким образом, вторая полиморфная форма представляет собой стабильную форму при комнатной температуре и при всех температурах, при которых могут храниться фармацевтические средства, и, следовательно, указанная форма 2 представляет собой особенно предпочтительную форму соединения 1.
Таким образом, в первом аспекте настоящего изобретения предложена полиморфная форма [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1), характеризующаяся КР-спектром с Фурье-преобразованием с характеристическими пиками на 3063±2 см-1, 1578±2 см-1, 1423±2 см-1, 1209±2 см-1, 1187±2 см-1, 1166±2 см-1, 1150±2 см-1, 930±2 см-1, 883±2 см-1, 770±2 см-1, 356±2 см-1, 304±2 см-1, 167±2 см-1, 119±2 см-1.
Полный спектр комбинационного рассеяния с Фурье-преобразованием полиморфной формы 2 соединения 1 характеризуется пиками на 3083±2 см-1, 3063±2 см-1, 2941±2 см-1, 2919±2 см-1, 1629±2 см-1, 1589±2 см-1, 1578±2 см-1, 1571±2 см-1, 1461±2 см-1, 1423±2 см-1, 1385±2 см-1, 1356±2 см-1, 1301±2 см-1, 1209±2 см-1, 1187±2 см-1, 1166±2 см-1, 1150±2 см-1, 1130±2 см-1, 1094±2 см-1, 1056±2 см-1, 1024±2 см-1, 930±2 см-1, 904±2 см-1, 883±2 см-1, 841±2 см-1, 832±2 см-1, 770±2 см-1, 717±2 см-1, 655±2 см-1, 630±2 см-1, 570±2 см-1, 441±2 см-1, 356±2 см-1, 304±2 см-1, 279±2 см-1, 167±2 см-1, 119±2 см-1; где подчеркнуты сигналы, отличающиеся от сигналов других полиморфных форм соединения 1 по меньшей мере на 2 см-1.
Спектр формы 2 был успешно проиндексирован, и было обнаружено, что кристаллическая решетка представляет собой триклинную кристаллическую решетку.
Во втором аспекте настоящего изобретения предложена полиморфная форма [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1), характеризующаяся следующими параметрами кристаллической решетки:


Указанная полиморфная форма (известная как форма 2) является термодинамически устойчивой при комнатной температуре, и поэтому особенно полезна для получения фармацевтических композиций, поскольку данная кристаллическая форма указанного соединения не будет спонтанно превращаться в другую полиморфную форму при любой из тех температур, при которых может храниться фармацевтический состав.
Полиморфная форма 2 имеет пик плавления при 196°C измеренный методом дифференциальной сканирующей калориметрии и устойчива при температурах вплоть до примерно 60-65°C. Выше этой температуры форма 3 является термодинамически более стабильной формой, при этом форма 1 термодинамически стабильна при очень высоких температурах, хотя температуру перехода между формами 2 и 3 определить затруднительно, поскольку измерение обычно проводят в эксперименте по установлению равновесия в суспензии из смеси различных форм соединения 1, а в ходе данного эксперимента, по-видимому, соединение 1 разлагается до достижения температуры перехода.
Предпочтительно полиморфная форма 2 соединения 1 будет чистой или в значительной степени чистой. Следовательно, она обычно будет содержать не более 10% других форм соединения 1, предпочтительно не более 5%, более предпочтительно не более 2% и наиболее предпочтительно не более 1% других форм соединения 1. Другие формы соединения 1 могут представлять собой аморфную форму или формы 1 или 3.
Также предпочтительно полиморфная форма 2 соединения 1 по существу не содержит других примесей, например, следов растворителя. Таким образом, предпочтительно указанная полиморфная форма 2 соединения 1 содержит не более 1% по массе растворителя (например, ацетонитрила, метилэтилкетона или метилизобутилкетона), более предпочтительно не более 0,5% по массе, предпочтительно не более 0,2% и более предпочтительно не более 0,1% по массе.
Способ получения соединения 1 описан в WO 2009/090414. Однако, как уже обсуждалось, описанный способ, по-видимому, приводит к получению аморфной формы соединения. Перекристаллизация этого продукта привела к образованию полиморфной формы 1.
Полиморфная Форма 2 может быть получена из продукта, описанного в WO 2009/090414, или из полиморфных форм 1 или 3 соединения 1 при установлении равновесия между фазами в течение длительного периода времени, обычно от 15 до 30 дней при комнатной температуре в ацетонитриле, смеси ацетонитрила и воды или в кетоновом растворителе, таком как метилэтилкетон, метилизобутилкетон или смеси указанных веществ.
Таким образом, в дополнительном аспекте настоящего изобретения предложен способ получения полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты, как определено выше, включающий:
a. суспендирование [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1) в растворителе, содержащем ацетонитрил, смесь ацетонитрила и воды или кетоновый растворитель, такой как метилэтилкетон или метилизобутилкетон, причем соединение 1 находится в виде аморфной формы, кристаллической формы, отличной от полиморфной формы 2, или в виде смеси формы 2 с одной или более другими полиморфными формами;
b. перемешивание указанной суспензии при температуре от примерно 15 до 25°C в течение от 15 до 30 дней; и
c. выделение и высушивание указанной кристаллической [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты.
После выполнения указанного способа было обнаружено, что продукт представляет собой полиморфную форму 2.
Исходное соединение 1, применяемое в способе согласно настоящему изобретению, может представлять собой аморфный материал, полиморфную форму 1, полиморфную форму 3, смесь любого из указанных веществ или смеси полиморфной формы 2 с одним или более из вышеуказанного. Указанный способ особенно полезен в том случае, если исходный материал находится в аморфной форме, например, материал, полученный согласно способу, описанному в WO 2009/090414. В другом особенно предпочтительном варианте реализации настоящего изобретения исходный материал представляет собой смесь полиморфных форм 2 и 3 соединения 1.
Установление равновесия между фазами обычно проводят в течение примерно от 15 до 20 дней, например, в течение 17 дней.
Как упоминалось выше, установление равновесия между фазами можно проводить в ацетонитриле, в смеси ацетонитрила и воды или в кетоновом растворителе, таком как метилэтилкетон, метилизобутилкетон или в смеси указанных веществ. Однако в некоторых случаях, например, когда исходный материал представляет собой аморфную форму соединения 1 и в качестве растворителя применяют метилэтилкетон, получают полиморфную форму 2 в смеси с другой полиморфной формой, обозначенной как форма 3.
Таким образом, в одном варианте реализации настоящего изобретения исходный материал представляет собой аморфную форму соединения 1 и способ осуществляют в ацетонитриле, необязательно в смеси с водой.
Тем не менее смесь полиморфных форм 2 и 3 представляет собой полезный продукт, поскольку она представляет собой хороший исходный материал для получения полиморфной формы 2 способом установления равновесия между фазами. Таким образом, в другом варианте реализации настоящего изобретения исходный материал для описанного выше способа представляет собой смесь полиморфных форм 2 и 3 соединения 1.
Для получения в большем масштабе по существу чистой полиморфной формы 2 был разработан альтернативный способ, включающий:
получение насыщенного раствора соединения 1 в растворителе, выбранном из ацетонитрила, ацетонитрила и воды или кетона, например, метилэтилкетона или метилизобутилкетона или смеси указанных веществ;
добавление в насыщенный раствор кристаллов полиморфной формы 2 Соединения 1 в качестве затравок; выдерживание с осуществлением кристаллизации; и
выделение кристаллов полиморфной формы 2 Соединения 1.
В одном варианте реализации настоящего изобретения указанный растворитель представляет собой ацетонитрил или смесь ацетонитрила и воды, причем указанный способ особенно успешен в том случае, когда растворитель представляет собой ацетонитрил. Насыщенный раствор можно получить при суспендировании исходного материала в растворителе и последующем нагревании суспензии до повышенной температуры, составляющей примерно от 55 до 80°C, обычно примерно 60°C, с получением раствора соединения 1. Предпочтительно указанный раствор выдерживают при повышенной температуре в течение по меньшей мере 3 минут, например от 3 до 15 минут, обычно примерно от 4 до 10 минут, стандартно примерно 5 минут, после чего раствор можно охладить до температуры, составляющей примерно от 45 до 50°C. Наиболее предпочтительно указанную суспензию нагревают до примерно 60°C и оставляют при этой температуре в течение примерно 5 минут, после чего полученный раствор охлаждают до температуры 48-50°C, обычно примерно 49°C, при этом охлаждение обычно происходит при примерно 10 K/час. Если на стадии охлаждения из раствора осаждается какой-либо твердый осадок, то раствор можно повторно нагреть до повышенной температуры (обычно примерно 60°C) и затем повторно дать ему остыть.
Затравливание может включать добавление затравочных кристаллов непосредственно в раствор или, в качестве альтернативы, можно получить затравочную суспензию, содержащую примерно от 1,5 до 5 мас. %, обычно примерно 2,5 мас. % затравочных кристаллов полиморфной формы 2, в перенасыщенном растворе соединения 1 в ацетонитриле; эту суспензию можно добавлять к раствору соединения 1 и перемешивать при температуре от 45 до 50°C, обычно примерно от 48 до 50°C, предпочтительно примерно при 49°C в течение от 1 до 5 часов.
После затравливания можно снизить температуру суспензии для осуществления процесса кристаллизации. Например, указанная суспензия может достигать конечной температуры, составляющей от 5 до 25°C, более предпочтительно от 10 до 25°C.
Охлаждение можно осуществлять в несколько этапов, например, в одном варианте реализации настоящего изобретения указанную суспензию можно охлаждать до 30-40°C в течение примерно от 4 до 12 часов, например, от 6 до 10 часов, обычно примерно 8 часов. Обычно охлаждение продолжают и суспензию оставляют для достижения комнатной температуры (от 15 до 25°C, предпочтительно около 20°C) в течение дополнительного времени приблизительно от 3 до 7 часов, предпочтительно около 5 часов. Затем можно продолжить перемешивание при комнатной температуре в течение дополнительного времени от 6 до 14 часов, обычно от 8 до 12 часов и наиболее предпочтительно примерно 10 часов.
Также можно способствовать кристаллизации при помощи концентрирования раствора, например до примерно 1:2 мас./об. соединение 1 : растворитель. Это может быть достигнуто при помощи удаления растворителя при пониженном давлении (например, <100 мбар) до тех пор, пока не будет получен требуемый объем растворителя.
Затем можно выделить кристаллы полиморфной формы 2 соединения 1 при помощи фильтрации и сушки при температуре примерно 20-45°C.
Когда в качестве растворителя для любого из указанных выше способов применяют ацетонитрил, то его удаление на стадии сушки может быть затруднено. Таким образом, каждый из способов или оба способа могут дополнительно включать промывку кристаллов полиморфной формы 2 еще одним растворителем и сушку. Кетоновые растворители, такие как метилэтилкетон, метилизобутилкетон и смеси указанных веществ, являются особенно подходящими дополнительными растворители для применения на стадии промывки.
Стадия промывки может включать:
добавление дополнительного растворителя к полиморфной форме 2, полученной способом, описанным выше, для получения суспензии или густой суспензии;
выпаривание растворителя; и
высушивание указанного продукта.
Стадию промывки можно повторять несколько раз, например, 1, 2 или 3 раза.
Когда в качестве дополнительного растворителя на стадии промывки применяют метилэтилкетон, то кристаллы могут быть суспендированы в метилэтилкетоне (МЕК) в концентрации примерно 500-600 г/л. Обычно метилэтилкетон выпаривают из указанной суспензии при комнатной температуре (примерно от 15 до 25°C) и при давлении приблизительно от 80 до 120 мбар, обычно 100 мбар. Сушку можно проводить в вакууме при комнатной температуре.
В альтернативном варианте реализации настоящего изобретения растворитель удаляют из продукта процесса кристаллизации, представляющего собой полиморфную форму 2, с применением способа горячей густой суспензии. В таком способе кристаллический продукт, представляющий собой полиморфную форму 2, можно перемешивать в условиях густой суспензии с дополнительным растворителем при повышенной температуре, например, от 45 до 55°C, обычно при примерно 50°C. В этом варианте реализации настоящего изобретения указанный дополнительный растворитель может быть таким, как описано выше, особенно подходящим является метилизобутилкетон.
Соотношение полиморфной формы 2 к дополнительному растворителю может составлять от примерно 1:2 до 1:20 мас./об., обычно от 1:2 до примерно 1:10 мас./об. Более предпочтительно соотношение полиморфный формы 2 к дополнительному растворителю составляет примерно от 1:3 до 1:5 мас./об.
Как обсуждается в WO 2009/090414, соединение 1 обладает антагонистической активностью по отношению к CRTH2, и, следовательно, пригодно для лечения состояний, которые опосредованы PGD2 или другими агонистами, связывающимися с CRTH2.
Таким образом, в дополнительном аспекте настоящего изобретения предложена полиморфная форма 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты, как описано выше, для применения в медицине, в частности, для лечения или предотвращения аллергических заболеваний, астматических состояний и воспалительных заболеваний, примерами которых являются астма, обострения бронхиальной астмы, хроническая обструктивная болезнь легких, аллергический ринит, конъюнктивит, полипы носовой полости, атопический дерматит, контактная гиперчувствительность (включая контактный дерматит), эозинофильный кашель, эозинофильный бронхит, эозинофильный гастроэнтерит, эозинофильный эзофагит, пищевые аллергии, воспалительное заболевание кишечника, язвенный колит, болезнь Крона, мастоцитоз, крапивница, гиперэозинофильный синдром, синдром гиперпродукции IgE, фиброзные заболевания, синдром Черджа-Стросс и рассеянный склероз.
Указанное соединение также применяют для лечения инфекции.
Термин «астма» включает все виды астмы, например, аллергическую астму, неаллергическую астму, эозинофильную астму, резистентную к стероидам астму, Th2 зависимую астму, не-Th2 зависимую астму и астму, индуцированную аспирином. В одном варианте реализации настоящего изобретения указанная астма представляет собой аллергическую астму и в другом варианте реализации настоящего изобретения указанная астма представляет собой эозинофильную астму.
Термин «обострение астмы» включает обострения, вызванные вирусными инфекциями, особенно заражением респираторно-синцитиальным вирусом (RSV) или риновирусом.
Аллергический ринит включает как круглогодичный аллергический ринит, так и сезонный аллергический ринит.
Термин «конъюнктивит» включает, в частности, аллергический конъюнктивит, весенний кератоконъюнктивит и атопический кератоконъюнктивит.
Термин «инфекция» включает бактериальные, вирусные или грибковые инфекции. Инфекция может возникнуть у пациентов, подверженных атопии или подверженных риску стать атопии и может представлять собой, например, риновирус, грипп или инфекцию, вызванную респираторно-синцитиальным вирусом (RSV), особенно у пациентов, страдающих астмой. В качестве альтернативы указанная инфекция может представлять собой бактериальную инфекцию, например, инфекцию золотистого стафилококка Staphylococcus aureus, особенно у пациентов, страдающих атопическим дерматитом.
Термин «фиброзные заболевания» включает, в частности, фиброзные заболевания, вызываемые/усугубляемые иммунными ответами Th2, например, идиопатический легочный фиброз, склеродермию и гипертрофические рубцы.
Полиморфную форму 2 соединения 1 также можно применять для лечения других PGD2-опосредованных заболеваний. Заболевания, которые могут быть опосредованы PGD2, включают аутоиммунные заболевания, такие как системная красная волчанка, псориаз, акне, отторжение аллотрансплантата, ревматоидный артрит, псориатический артрит и остеоартрит.
Настоящее изобретение дополнительно относится к способу лечения или предотвращения заболевания или состояния, выбранного из перечисленных выше, при этом указанный способ включает введение пациенту, нуждающемуся в таком лечении, эффективного количества полиморфной формы 2 соединения 1, определенной выше.
Пациент будет представлять собой млекопитающего, например, человека.
Также предложено применение полиморфной формы 2 соединения 1, как определено выше, для получения лекарственного средства для лечения или предотвращения заболевания или состояния, выбранного из перечисленных выше.
Полиморфная форма 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты должна быть соответствующим образом приготовлена в виде составов в зависимости от заболеваний или состояний, подлежащих лечению.
Таким образом, в дополнительном аспекте настоящего изобретения предложена фармацевтическая или ветеринарная композиция, содержащая полиморфную форму 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты, как определено выше, вместе с фармацевтически или ветеринарно приемлемым вспомогательным веществом. Также могут присутствовать другие активные вещества, если это может быть сочтено целесообразным или желательным для заболевания или состояния, подлежащего лечению или предотвращению.
Вспомогательное вещество, или, если присутствует больше, чем одно, то каждое из вспомогательных веществ должно быть приемлемым, т.е. совместимым с другими ингредиентами композиции и не оказывать отрицательного влияния на реципиента.
Составы включают такие составы, которые пригодны для перорального (включая вязкие пероральные препараты), ректального, назального, бронхиального (ингаляционного), местного (включая трансбуккальное, подъязычное, а также глазные капли), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения и могут быть получены любыми способами, хорошо известными в области фармацевтики.
Пути введения будут зависеть от состояния, подлежащего лечению, но предпочтительные композиции приготовлены для перорального, назального, бронхиального или местного введения.
Композицию можно приготовить путем объединения полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты со вспомогательным веществом. Обычно указанные составы получают путем равномерного и тщательного объединения указанного активного агента с жидкими носителями или тонко измельченными твердыми носителями или и тем, и другим, а затем, если это необходимо, формованием продукта. Настоящее изобретение относится к способам получения фармацевтической композиции, включающим сочетание или объединение полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты с фармацевтически или ветеринарно приемлемым вспомогательным веществом.
Составы для перорального введения согласно настоящему изобретению могут быть представлены в виде: отдельных единиц, таких как капсулы, порошки для приготовления раствора, принимаемого внутрь (сашеты), таблетки, формованные пастилки и пастилки для рассасывания, каждая из которых содержит заранее определенное количество полиморфной формы 2 соединения 1, в виде порошка или гранул; в виде раствора или суспензии активного агента в водной или неводной жидкости; или в виде жидкой эмульсии «масло в воде» или жидкой эмульсии «вода в масле»; или в виде сиропа или эликсира; или в виде болюса и т.д.
Для композиций для перорального введения (например, таблеток, капсул, составов, содержащих мукоадгезив и т.д.) термин «приемлемый носитель» включает носители, такие как обычные вспомогательные вещества, например, связующие агенты, например, сироп, гуммиарабик, желатин, сорбит, трагакант, поливинилпирролидон (повидон), метилцеллюлозу, этилцеллюлозу, натрий карбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу, сахарозу и крахмал; наполнители и носители, например, кукурузный крахмал, желатин, лактозу, сахарозу, микрокристаллическую целлюлозу, каолин, маннит, дикальцийфосфат, хлорид натрия и альгиновую кислоту; смачивающие агенты/поверхностно-активные вещества, такие как полоксамеры, полисорбаты, докузат натрия и лаурилсульфат натрия; разрыхлители, такие как крахмал или карбоксиметилкрахмал натрия; и смазывающие вещества, такие как стеарат магния, стеарат натрия и стеараты других металлов, стеарат глицерина, стеариновая кислота, силиконовое масло, тальк, воски, масла и коллоидный диоксид кремния. Также можно применять подслащивающие и ароматизирующие агенты, такие как перечная мята, масло грушанки, вишневый ароматизатор и т.п. Может быть желательным добавление красителя, чтобы сделать стандартную лекарственную форму легко идентифицируемой. Таблетки также можно покрывать оболочкой способами, хорошо известными в данной области техники.
Таблетку можно изготовить прессованием или формованием, необязательно с одним или более вспомогательными ингредиентами. Прессованные таблетки можно получить прессованием в подходящем устройстве полиморфной формы 2 соединения 1 в свободно текучей форме, такой как порошок или гранулы, необязательно смешанными со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки можно получить формованием в подходящем устройстве смеси указанного порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки необязательно могут быть покрыты оболочкой или на них может быть нанесена насечка и таблетки можно приготовить таким образом, чтобы обеспечить медленное или контролируемое высвобождение указанного активного агента.
Некоторые составы могут содержать мукоадгезив, например, мукополисахарид, такой как гиалуронат натрия. Такие композиции можно приготовить в виде, например, жидкостей, жидких сиропов, мягких гелей, жидких гелей, текучих гелей или водных суспензий и они могут, в дополнение к активному агенту и мукоадгезиву, также содержать одно или более дополнительных вспомогательных веществ, как указано выше. Жидкие композиции обычно будут содержать жидкий носитель, который может представлять собой растворитель или суспендирующий агент, например, воду или физиологический раствор и может также содержать вещество для повышения вязкости, например, натрий карбоксиметилцеллюлозу, сорбит или декстран.
Другие составы, подходящие для перорального введения, включают пастилки для рассасывания, содержащие указанный активный агент в ароматизированной основе, обычно сахарозу и гуммиарабик или трагакант; пастилки, содержащие полиморфную форму 2 соединения 1 в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и жидкости для полоскания рта, содержащие активный агент в подходящем жидком носителе.
Для местного нанесения на кожу указанную композицию можно приготовить в виде крема, мази, желе, раствора или суспензии и т.п. Составы кремов или мазей, которые можно применять для полиморфной формы 2 соединения 1, представляют собой обычные составы, хорошо известные в данной области техники, например, описанные в обычных учебниках по фармацевтике, таких как British Pharmacopoeia.
Композицию, определенную выше, можно применять для лечения дыхательных путей с помощью назального, бронхиального или трансбуккального введения, например, аэрозолей или спреев, которые могут распылять фармакологически активный ингредиент в виде порошка или в виде капель раствора или суспензии. Фармацевтические композиции, обладающие свойством диспергировать порошок, включают ингаляторы сухого порошка и ингаляторы отмеренных доз. Ингаляторы сухих порошков обычно содержат, в дополнение к полиморфной форме 2 соединения 1, подходящий носитель, такой как лактоза и, при желании, вспомогательные вещества, такие как поверхностно-активные вещества и/или разбавители и/или агенты для повышения текучести и/или смазывающие вещества. Ингаляторы отмеренных доз для диспергирования порошков обычно содержат, в дополнение к полиморфной форме 2 соединения 1, жидкий пропеллент с температурой кипения ниже комнатной температуры и, если требуется, вспомогательные вещества, такие как жидкие или твердые неионогенные или анионные поверхностно-активные вещества и/или разбавители. Фармацевтические композиции для лечения дыхательных путей, в которых фармакологически активный ингредиент находится в растворе (например, раствор для аэрозольного распыления или ингаляторы отмеренных доз), дополнительно содержат подходящий пропеллент и кроме того, при необходимости, дополнительный растворитель и/или стабилизатор. Вместо указанного пропеллента также можно применять сжатый воздух, если это будет производиться в соответствии с требованиями посредством подходящего устройства для сжатия и расширения.
Парентеральные составы обычно должны быть стерильными.
Обычно доза соединения 1 будет составлять примерно от 0,01 до 100 мг/кг, таким образом, чтобы поддерживать концентрацию лекарственного средства в плазме крови в концентрации, эффективной для ингибирования PGD2 на рецепторе CRTH2. Точное количество соединения 1, которое является терапевтически эффективным, и путь введения, при помощи которого такое соединение лучше всего вводить, легко определяется средним специалистом в данной области техники при помощи сравнения уровня агента в крови с концентрацией, необходимой для достижения терапевтического эффекта.
Предпочтительно указанная фармацевтическая композиция приготовлена для введения один раз в день, хотя в некоторых случаях можно применять и более частое дозирование, например, дозирование два, три или четыре раза в день. С другой стороны, иногда дозировать можно реже, чем один раз в день, например, один раз в два дня. В некоторых обстоятельствах можно применять такую схему приема лекарственного средства, при которой указанную композицию вводят в течение первого периода времени, а затем в течение второго периода времени введение прекращают или, в качестве альтернативы, указанную композицию вводят в более низкой дозе. Такая схема приема лекарственного средства описана в WO 2009/063202.
Полиморфную форму 2 соединения 1, как определено выше, можно применять в комбинации с одним или более активными агентами, пригодными для лечения заболеваний и состояний, перечисленных выше, при этом указанные активные вещества не обязательно являются ингибиторами PGD2 на рецепторе CRTH2.
Таким образом, фармацевтическая композиция, описанная выше, может дополнительно содержать один или более из этих активных агентов.
Настоящее изобретение также относится к применению полиморфной формы 2 соединения 1, как определено выше, для получения агента для лечения заболеваний и состояний, опосредованных агонистами рецептора CRTH2, особенно PGD2, при этом указанный агент также содержит дополнительный активный агент, пригодный для лечения тех же заболеваний и состояний.
Эти дополнительные активные агенты могут представлять собой другие антагонисты рецептора CRTH2 или могут иметь совершенно иной принцип действия. Они включают существующие средства для лечения аллергических и других воспалительных заболеваний, в том числе:
суплатаст тозилат и подобные соединения;
агонисты β2 адренорецептора, такие как метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, сальметерол, индакатерол, тербуталин, орципреналин, битолтерола мезилат и пирбутерол или метилксантины, такие как теофиллин и аминофиллин, стабилизаторы мембран тучных клеток, такие как кромогликат натрия или антагонисты мускариновых рецепторов, такие как тиотропий;
антигистамины, например, антагонисты гистаминового рецептора H1, такие как лоратадин, цетиризин, дезлоратадин, левоцетиризин, фексофенадин, астемизол, азеластин и хлорфенирамин или антагонисты рецептора H4;
агонисты адренорецептора α1 и α2, такие как пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид и этилнорепинфрина гидрохлорид;
модуляторы функции рецептора хемокина, например, CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства C-C) или CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства C-X-C) и CX3CR1 для семейства С-Х3-С; антагонисты лейкотриена, такие как монтелукаст, пранлукаст и зафирлукаст;
ингибиторы биосинтеза лейкотриена, такие как ингибиторы 5-липоксигеназы или ингибиторы активирующего белка 5-липоксигеназы (FLAP), такие как зилеутон, АВТ-761, фенлеутон, тепоксалин, Abbott-79175, N-(5-замещенные)тиофен-2-алкилсолфонамиды, 2,6-ди-трет-бутилфенол гидразоны, метокситетрагидропираны, такие как ZD2138, SB-210661, пиридинил-замещенные-2-цианонафталеновые соединения, такие как L-739010, 2-цианохинолиновые соединения, такие как L-746,530, индоловые и хинолиновые соединения, такие как МК-591, МК-886 и BAYx1005;
ингибиторы фосфодиэстеразы, включая ингибиторы PDE4, такие как рофлумиласт;
терапия анти-IgE-антител, такая как омализумаб;
антибактериальные средства, такие как фузидовая кислота (в частности, для лечения атопического дерматита);
противогрибковые средства, такие как клотримазол (в частности, для лечения атопического дерматита);
иммунодепрессанты, такие как такролимус и особенно пимекролимус в случае воспалительного заболевания кожи, или альтернативно FK-506, рапамицин, циклоспорин, азатиоприн или метотрексат;
иммунотерапевтические агенты, включая аллерговакцинацию, такие как Гразакс (Grazax);
кортикостероиды, такие как преднизон, преднизолон, флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, мометазона фуроат и флутиказона фуроат, лекарственные средства, которые способствуют цитокиновому Th1 ответу, такие как интерфероны, ФНО (TNF) или GM-CSF.
Антагонисты рецептора CRTH2 также можно комбинировать со средствами, находящимися в разработке для воспалительных показаний к применению, в том числе:
другие антагонисты PGD2, действующие на другие рецепторы, такие как антагонисты DP;
лекарственные средства, модулирующие производство цитокинов, такие как ингибиторы ФНОα конвертирующего фермента (ТАСЕ), анти-ФНО моноклональные антитела, рецептор ФНО молекул иммуноглобулина, ингибиторы других изоформ ФНО, неселективные ингибиторы СОХ-1/СОХ-2, такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак и апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин; ингибиторы СОХ-2, такие как мелоксикам, целекоксиб, рофекоксиб, вальдекоксиб и эторикоксиб, низкодозированный метотрексат, лефуномид, циклесонид, гидроксихлорохин, d-пеницилламин, ауранофиновое или парентеральное или пероральное золото;
лекарственные средства, модулирующие активность Th2 цитокинов IL-4 и IL-5, такие как блокирующие моноклональные антитела и растворимые рецепторы;
PPAR-γ агонисты, такие как розиглитазон; или с анти-RSV антителами, такими как Синагис (Synagis), (паливизумаб) и агентами, которые могут применяться для лечения риновирусной инфекции в будущем, например, интерферон-альфа, интерферон-бета или другие интерфероны.
Особенно предпочтительны комбинации полиморфной формы 2 соединения 1, как определено выше, с антагонистами лейкотриенов, такими как монтелукаст, пранлукаст и зафирлукаст, особенно в комбинации с монтелукастом.
В другом аспекте настоящего изобретения предложен продукт, содержащий полиморфную форму 2 соединения 1, как определено выше, и один или более агентов, перечисленных выше, в качестве комбинированного состава для одновременного, раздельного или последовательного применения для лечения заболевания или состояния, опосредованного действием PGD2 на рецептор CRTH2.
В другом аспекте настоящего изобретения предложен набор для лечения заболевания или состояния, опосредованного действием PGD2 на указанный рецептор CRTH2, содержащий первый контейнер, содержащий полиморфную форму 2 соединения 1, как определено выше, и второй контейнер, содержащий один или более активных агентов, перечисленных выше.
Настоящее изобретение будет описано более подробно со ссылками на прилагаемые фигуры, на которых:
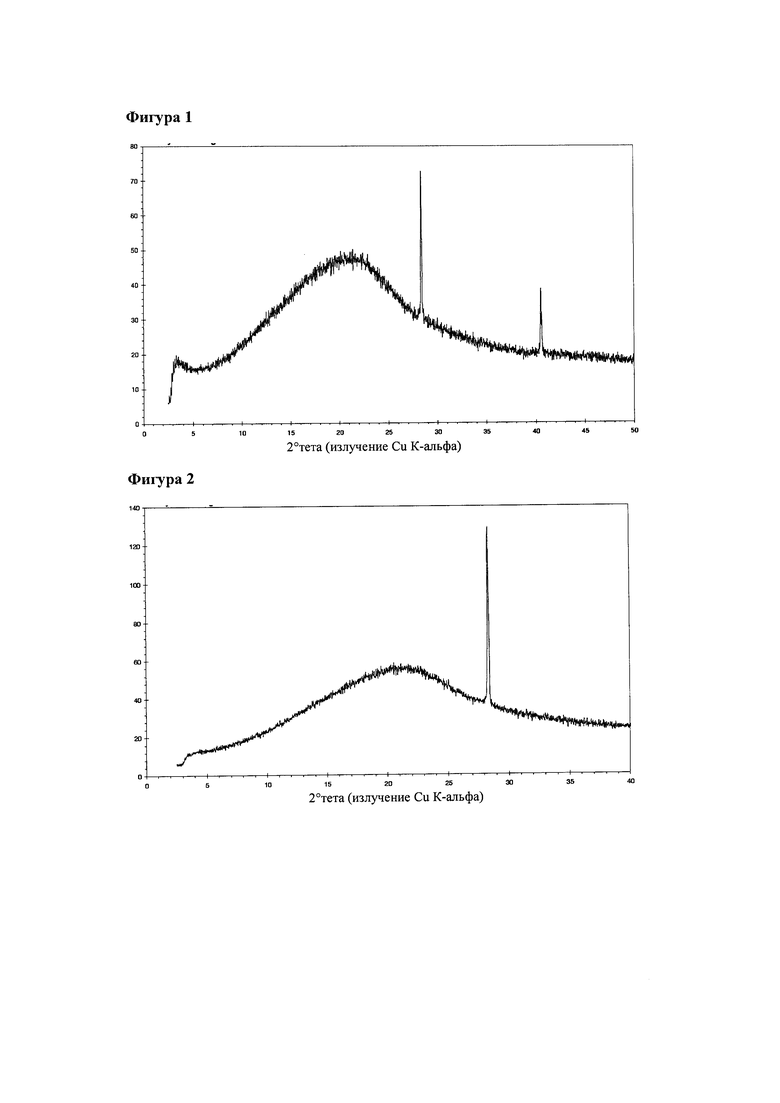
На фигуре 1 представлена рентгенограмма, полученная методом порошковой рентгеновской дифракции (порошковая рентгенограмма) соединения 1, партия 1.
На фигуре 2 представлена порошковая рентгенограмма соединения 1, партия 3.
На фигуре 3 представлен КР-спектр с Фурье-преобразованием соединения 1, партии 3. Указанный спектр применяли в качестве стандарта при предварительном исследовании полиморфизма. На фигуре обозначены наиболее выраженные КР-пики.
На фигуре 4 представлен график, полученный совместным анализом методами термогравиметрии и инфракрасной спектроскопии (ТГ-Фурье-ИК) соединения 1, партии 3 в интервале температур от 25°C до 250°C при скорости нагрева 10°C/мин.
На фигуре 5 представлена кривая дифференциальной сканирующей калориметрии для соединения 1, партии 1.
На фигуре 6 представлена кривая динамической сорбции паров для соединения 1, партии 1, показывающая относительную влажность по образцу и массу образца в процентах в зависимости от времени.
На фигуре 7 представлена дополнительная кривая динамической сорбции паров для соединения 1, партии 1, показывающая массу образца в процентах в зависимости от относительной влажности.
На фигуре 8 представлена порошковая рентгенограмма соединения 1, партия 2.
На фигуре 9 представлена порошковая рентгенограмма продукта, полученного путем перекристаллизации соединения 1, партии 1, из этилацетата и дальнейшей сушки полученного твердого кристаллического вещества (пример 3).
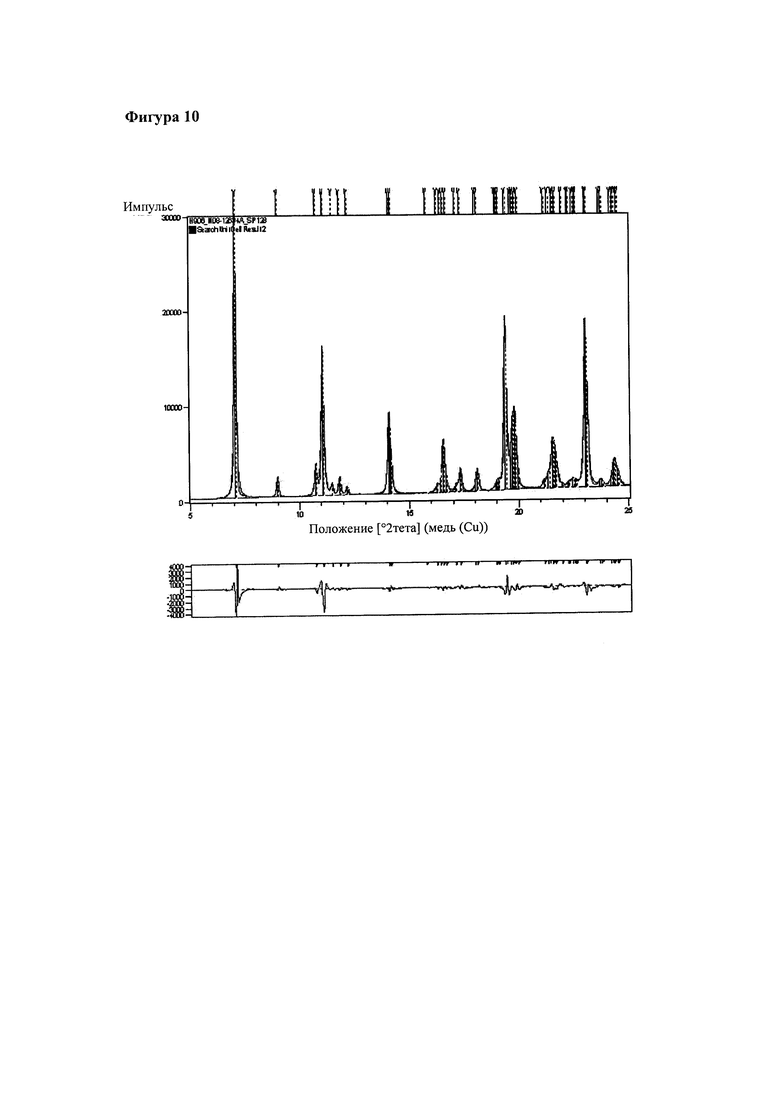
На фигуре 10 представлен фрагмент порошковой рентгенограммы перекристаллизованного и высушенного продукта, полученного в примере 3 (полиморфная форма 1), показано соответствие между полученной рентгенограммой продукта из примера 3 (форма 1, красная линия, файл: Н906) и вычисленными данными на основании анализа Ле Бейля (синяя линия). На той же фигуре внизу представлен график, на котором красным цветом показаны различия.
На фигуре 11 представлен 1H-ЯМР спектр продукта, полученного в эксперименте Р15 в примере 4 (полиморфная форма 1) в ДМСО-d6.
На фигуре 12 представлен КР-спектр с Фурье-преобразованием продукта, полученного в эксперименте Р9 в примере 4 (полиморфная форма 2).
На фигуре 13 представлена порошковая рентгенограмма продукта, полученного в эксперименте Р9 в примере 4 (полиморфная форма 2). Указанный материал представляет собой кристаллическое вещество.
На фигуре 14 представлен 1H-ЯМР спектр продукта, полученного в эксперименте Р9 в примере 4 (полиморфная форма 2) в ДМСО-d6.
На фигуре 15 представлен КР-спектр с Фурье-преобразованием продукта Р6, полученного в примере 4 (форма 3 в смеси с формой 2).
На фигуре 16 представлен фрагмент порошковой рентгенограммы продукта Р6, полученного в примере 4 в сравнении с порошковой рентгенограммой продуктом Р9, полученного в примере 4 (полиморфная форма 2).
На фигуре 17 представлен фрагмент порошковой рентгенограммы продукта Р24, полученного в примере 6 (полиморфная форма 2), показано соответствие между полученной рентгенограммой продукта Р24 (форма 1, красная линия, файл: Н893) и вычисленными данными на основании анализа Ле Бейля (синяя линия). На той же фигуре внизу представлен график, на котором красным цветом показаны различия.
На фигуре 18 представлен спектр, полученный совместным анализом методами термогравиметрии и инфракрасной спектроскопии с преобразованием Фурье (ТГ-Фурье-ИК) продукта Р9 в интервале температур от 25°C до 250°C при скорости нагрева 10°C/мин.
В примерах были использованы следующие условия измерений.
1H-NMR: Спектры 1Н-ЯМР регистрировали при помощи спектрометра Bruker DPX300 с частотой протонов 300,13 МГц, при импульсе возбуждения 30° и задержкой повторного цикла 1 с. Накапливали по 16 сканирований. В качестве растворителя применяли d6-ДМСО.
DSC: Дифференциальную сканирующую калориметрию проводили при помощи прибора Perkin Elmer DSC-7 (закрытая золотая кювета с образцом в атмосфере N2).
Динамическая сорбция паров DVS (SPS): Система измерения сорбции SPS11-100n. Образец помещали в алюминиевый тигель и оставляли для установления равновесия при заданной относительной влажности перед началом установленной программы влажности. Программу, которая была использована для измерений, можно обнаружить на соответствующих фигурах (синяя линия).
КР-спектроскопия с Фурье-преобразованием: КР-спектр с Фурье-преобразованием регистрировали на системе КР-спектроскопии с Фурье-преобразованием Bruker RFS 100, оснащенной лазером с излучением в ближайшей ИК-области спектра Nd:YAG с длиной волны 1064 нм, и германиевым детектором, охлаждаемым жидким азотом. Для каждого образца накапливали как минимум 64 сканирования с разрешением 2 см-1. Применяли лазер с мощностью 300 мВт. Данные КР-спектроскопии с Фурье-преобразованием показаны в области от 3500 до 100 см-1. Ниже 100 см-1 данные не имеют смысла из-за фильтра с ограниченной полосой пропускания.
Порошковая рентгеновская дифракция: Bruker D8; излучение Cu Ка, 40 кВ/40 мА; детектор LynxEye, размер шага 0,02° 2 тета, шаг по времени 37 сек. Подготовка образцов: Обычно образцы измеряли без какой-либо специальной обработки, за исключением применения небольшого давления для получения плоской поверхности. Применяли держатели образцов из монокристалла кремния (глубина 0,1 мм). Во время измерения образцы вращали.
Растворители: Для всех экспериментов применяли растворители аналитической степени чистоты Fluka, Merck или ABCR.
ТГ-Фурье-ИК: Термогравиметрические измерения проводили при помощи прибора Netzsch Thermo-Microbalance TG 209, присоединенного к Фурье-ИК-спектрометру Bruker FTIR Vector 22 или IFS 28 (тигли для образца с отверстием, атмосфера N2, скорость нагрева 10°C/мин, диапазон от 25°C до 250°C).
В примерах использовали следующие сокращения:
Пример 1 - Получение [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1)
Соединение 1 получали согласно способу, изложенному в WO 2009/090414, а именно:
i. 2-(4-фторбензолсульфонил)-пиридин-3-карбоксальдегид

2-хлор-3-пиридинкарбоксальдегид (4,04 г, 2,86 ммоль) и натриевую соль 4-фторбензолсульфиновой кислоты (5,73 г, 3,14 ммоль) растворяли в ДМСО (100 мл) и смесь нагревали при 100°C в течение 72 часов в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (500 мл) и экстрагировали EtOAc (3x). Объединенные органические слои промывали водой, рассолом, сушили (MgSO4) и выпаривали досуха с получением 7,89 г неочищенного продукта. Указанное неочищенное соединение предварительно абсорбировали на силикагеле и очищали с помощью хроматографии на "сухих" колонках при пониженном давлении, элюируя гептаном с применением градиента EtOAc с получением 4,14 г (41%) целевого продукта в виде желтого твердого вещества (пластинки) (Тпл=131-131,3°C; IR=1691 см-1; ВЭЖХ=7,21 мин >99%).
1H ЯМР (400 МГц; CDCl3): 7,23-7,29 (2Н, m) 7,60 (1Н, dd) 8,05-8,10 (2H, m) 8,37 (2H, dd) 8,67 (1H, dd) 11,1 (1H, s).
13C ЯМР (100 МГц, CDCl3): 116,6 (d) 116,8 (d) 127,3 (d) 130,7 (s) 132,6 (d) 134,0 (s) 137,9 (d) 152,5 (s) 159,7 (s) 165,1 (s) 167,7 (s) 188,5 (d).
ii. Этиловый эфир [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты

Раствор этилового эфира (5-фтор-2-метил-индол-1-ил)-уксусной кислоты (1,0 г, 4,4 ммоль) и 2-(4-фторбензолсульфонил)-пиридин-3-карбоксальдегида (1,13 г, 4,3 ммоль) в сухом DCM (50 мл) добавляли в течение 5-10 мин к перемешиваемому раствору TMSOTf в сухом DCM (15 мл) при 0°C. Смесь выдерживали в течение 15 мин перед добавлением чистого триэтилсилана (2,05 мл, 12,8 ммоль) одной порцией. Смесь перемешивали еще в течение 15 часов и затем оставляли нагреваться до температуры окружающей среды. Реакцию гасили при добавлении по каплям насыщенного раствора NaHCO3 (10 мл) и двухфазную смесь экстрагировали DCM (2×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), затем сушили (MgSO4) и выпаривали досуха. Указанную реакцию повторяли в одинаковом масштабе, затем два сырых материала очищали по отдельности. Указанные неочищенные реакционные материалы очищали при помощи колоночной хроматографии с использованием гептана и градиента этилацетата с получением 0,90 г (43%) и 1,50 г (72%) целевого соединения в виде бледно-фиолетового твердого вещества и коричневого твердого вещества соответственно, с различной степенью чистоты (96,0% и 94,5% по данным ВЭЖХ) (Тпл=150,5-151,5°C, IR=1751 см-1; ВЭЖХ=12,24 мин).
1H ЯМР (400 МГц; CDCl3): 1,26 (3H, t) 2,29 (3H, s) 4,22 (2Н, q) 4,62 (2Н, s) 4,80 (2Н, s) 6,79 (1H, dd) 6,86 (1Н, ddd) 7,10 (1H, dd) 7,19 (1Н, dd) 7,23-7,28 (2Н, m) 7,36 (1Н, dd) 8,05-8,11 (2Н, m) 8,29 (1Н, dd).
13C ЯМР (100 МГц, CDCl3): 10,4 (q) 14,2 (q) 25,3 (t) 45,2 (t) 61,9 (t) 103,4 (d) 103,6 (d) 108,0 (s) 108,1 (s) 109,1 (d) 109,2 (d) 109,5 (d) 109,8 (d) 116,2 (d) 116,4 (d) 127,0 (d) 128,5 (s) 128,6 (s) 132,2 (d) 132,3 (d) 133,3 (s) 135,1 (s) 136,4 (s) 136,6 (s) 139,4 (d) 146,2 (d) 156,2 (s) 157,0 (s) 159,4 (s) 164,7 (s) 167,3 (s) 168,6 (s).
iii. [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусная кислота (соединение 1)

Способ А
KOH (0,34 г, 5,94 ммоль) растворяли в воде (7 мл) и добавляли к интенсивно перемешиваемому раствору этилового эфира [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты (0,96 г, 1,98 ммоль) в ТГФ (21 мл) в атмосфере азота при температуре окружающей среды. Реакцию контролировали с помощью ТСХ и ЖХ-МС. Через 2 ч растворитель удаляли в вакууме перед доведением pH до 1,5 с применением 0,1 М раствора HCl. Указанный осадок тщательно перемешивали в течение 15 мин и затем выделяли при помощи фильтрации с отсасыванием. Собранное твердое вещество промывали водой и затем метил-трет-бутиловым эфиром (MTBE), поднимали в воздух и затем сушили в вакууме при 50°C с получением 870 мг (97%) продукта в виде розового твердого вещества (Тпл=125-126°C; IR=1729 см-1; ВЭЖХ=10,80 мин, 99,3%).
1H ЯМР (400 МГц; ДМСО): 2,29 (3H, s) 4,56 (2Н, s) 4,97 (2Н, s) 6,85-6,91 (2Н, m) 7,37-7,7,45 (2Н, m) 7,47 (1Н, dd) 7,51-7,57 (2Н, m) 8,06-8,15 (2Н, m) 8,36 (1H, dd).
13C ЯМР (100 МГц, ДМСО): 10,5 (q) 25,0 (t) 45,5 (t) 102,7 (d) 102,9 (d) 107,7 (s) 107,8 (s) 108,8 (d) 109,1 (d) 110,9 (d) 111,0 (d) 117,1 (d) 117,3 (d) 128,1 (d) 128,2 (d) 128,3 (d) 132,7 (d) 132,8 (d) 133,8 (d) 135,5 (s) 136,8 (s) 138,1 (s) 140,4 (d) 147,0 (d) 155,9 (s) 156,6 (s) 158,9 (s) 164,6 (s) 167,1 (s) 171,1 (s).
Партии 1 и 3 соединения 1, используемые далее, получали описанным выше способом, который идентичен способу, изложенному в WO 2009/090414. Для партии 2 соединения 1 применяли следующую процедуру.
Способ В
KOH (0,514 г, 9,16 ммоль) растворяли в воде (11 мл) и добавляли к интенсивно перемешиваемому раствору этилового эфира [5-фтор-3-({2-[(4-фторбензол)сульфонил]-пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты (1,48 г, 3,05 ммоль) в ТГФ (32 мл) в атмосфере азота при температуре окружающей среды. Реакцию контролировали с помощью ТСХ и ЖХ-МС. Через 2 часа реакционный сосуд содержал основной водный раствор калиевой соли соединения 1. Вместо удаления растворителя, как указано в WO 2009/090414, водный раствор промывали этилацетатом с получением суспензии. Осажденное твердое вещество удаляли при помощи фильтрования и доводили pH водной фазы до 1,5 при помощи 0,1 М раствора HCl, энергично перемешивали в течение 15 минут перед выделением фильтрацией с отсасыванием. Собранное твердое вещество промывали водой и затем метил-трет-бутиловым эфиром (MTBE), поднимали в воздух и затем сушили в вакууме при 50°C с получением 900 мг (64%) продукта в виде рыжевато-коричневого твердого вещества.
Пример 2 - Определение характеристик продукта, полученного в примере 1
Три партии продукта, полученных при помощи способа из примера 1, были охарактеризованы методами КР-спектроскопии с Фурье-преобразованием, порошковой рентгеновской дифракции (PXRD), термогравиметрии в сочетании с инфракрасной спектроскопией с преобразованием Фурье (ТГ-Фурье-ИК), дифференциальной сканирующей калориметрии (ДСК) и динамической сорбции паров (DVS).
На фигуре 1 представлена порошковая рентгенограмма партии 1. Проводили съемку образца в том виде, в каком он был получен. Указанный материал представлял собой аморфное вещество. Сигналы на 28,4 °2 тета и 40,5 °2 тета, наиболее вероятно, можно отнести к KCl.
На фигуре 2 представлена порошковая рентгенограмма партии 3, проводили съемку образца в том виде, в каком он был получен. Указанный материал представляет собой аморфное вещество. Сигнал на 28,3 °2 тета, наиболее вероятно, можно отнести к KCl.
На фигуре 3 представлен КР-спектр с Фурье-преобразованием партии 3. Указанный спектр применяли в качестве стандарта при предварительном исследовании полиморфизма. На фигуре обозначены наиболее выраженные КР-пики.
Партию 3 анализировали при помощи ТГ-Фурье-ИК в диапазоне температур от 25°C до 250°C при скорости нагрева 10°C/мин. Анализ ТГ-Фурье-ИК показал потерю 1,2 мас. % массы (остаточный H2O) от комнатной температуры до 160°C. Выше ~160°C происходило разложение, следовательно, вероятно, что материал представляет собой несольватированную форму. Это показано на фигуре 4.
Партию 1 также анализировали при помощи дифференциальной сканирующей калориметрии, полученная кривая DSC представлена на фигуре 5. При первом сканировании (красная линия) точка стеклования находилась при температуре примерно 79°C (ΔСр: 0,4 Дж/г*°C), сигнал перекристаллизации наблюдали при температуре примерно 155°C. После охлаждения указанный образец все еще был частично аморфным. На второй стадии нагревания (синяя линия) точка стеклования находилась при температуре примерно 73°C с ΔСр 0,2 Дж/г*°C, сигнал плавления наблюдали при температуре примерно 200°C, далее образец разлагался. Сдвиг точки стеклования до более низких температур, вероятно, обусловлен частичным разложением во время первой стадии нагревания.
Для того, чтобы исследовать поведение партии 1 при переменном давлении паров воды, образец анализировали методом динамической сорбции паров (DVS). На фигуре 6 представлена полученная кривая DVS, на которой приведены относительная влажность воздуха над образцом и масса образца в массовых процентах по отношению ко времени. На фигуре 7 представлен график массы образца в массовых процентах по отношению к относительной влажности. Образец кондиционировали при относительной влажности 50% перед началом установленной программы влажности со скоростью сканирования 5% изменения относительной влажности в час. При относительной влажности ниже 50% наблюдали непрерывную потерю массы. На втором этапе (от 0% относительной влажности до 95% относительной влажности) образец демонстрировал непрерывное поглощение воды. При значениях относительной влажности более 84% наблюдали повышенное поглощение воды. При значении относительной влажности 95% не было достигнуто равновесие, что является обычным для аморфного материала. В конце измерения (конечная влажность 50% отн. влажн.) масса образца составляла на 0,6% выше массы исходного материала. Тот же самый образец исследовали методом КР-спектроскопии с Фурье-преобразованием (до и после измерения DVS). После получения кривой DVS указанный образец не демонстрировал какого-либо фазового перехода.
Аналогичные результаты были получены для партии 3.
В отличие от партий 1 и 3, порошковая рентгенограмма партии 2 (фигура 8) показала, что материал являлся кристаллическим. Материал подготавливали с применением альтернативной процедуры, способа В, изложенного выше.
Пример 3 - Рекристаллизация партии 1 соединения 1
Перед скрининговым исследованием соли удаляли примеси KCl при кристаллизации 5 г партии 1 соединения 1 в этилацетате. Продукт был охарактеризован методами КР-спектроскопии с Фурье-преобразованием и ТГ-Фурье-ИК. Измерение методом ТГ-Фурье-ИК показало потерю массы 8,2% этилацетата при температуре 140°C, т.е. выше, чем при температуре кипения. Это показывает, что растворитель является связанным, что типично для сольватов. Образец сушили в вакууме при комнатной температуре и указанный продукт характеризовали методами КР-спектроскопии с Фурье-преобразованием, ТГ-Фурье-ИК и порошковой рентгеновской дифрактометрии. После сушки до несольватированной формы указанный материал изменялся, поскольку этилацетат больше не идентифицировался по данным ТГ-Фурье-ИК. На фигуре 9 представлена порошковая рентгенограмма перекристаллизованного продукта. Указанный материал является кристаллическим.
Порошковая рентгенограмма показывает, что этот перекристаллизованный и высушенный продукт представлял собой ту же кристаллическую форму, что и партия 2 соединения 1, данная кристаллическая форма была обозначена как форма 1.
Пример 4 - Эксперименты по установлению равновесия в суспензии и кристаллизации при охлаждении
Примерно 100 мг соединения партии 3 суспендировали в растворителях и смесях растворителей и перемешивали в течение 17 дней при 22°C. Твердые вещества отфильтровывали и анализировали методом КР-спектроскопии с Фурье-преобразованием (2 измерения: 1. влажный материал, 2. высушенный материал).
Проводили дополнительный эксперимент по установлению равновесия в суспензии (Ехр Р23) в соответствии со способом получения. К 250 мг соединения партии 3 добавляли 62,5 мкл воды и 440 мкл муравьиной кислоты (98%). Наблюдали желтовато-коричневую суспензию. Добавляли 95 мкл толуола. После кратковременной обработки ультразвуком наблюдали осаждение. Перемешивание не было возможно. Дополнительно добавляли 1 мл H2O и 80 мкл толуола. Температуру зацикливали на 25°C в течение одного часа и 50°C в течение двух часов (с временем вывода установки в рабочий режим в два часа) в общей сложности в течении трех дней.
Для эксперимента по кристаллизации при охлаждении растворяли примерно 100 мг соединения партии 3 при повышенной температуре в соответствующих количествах растворителя с получением насыщенного раствора. Температуру в дальнейшем увеличивали на 5°C для получения раствора, не содержащего затравочных кристаллов. Указанные растворы в дальнейшем охлаждали до 5°C. Твердые вещества отфильтровывали и анализировали методом КР-спектроскопии с Фурье-преобразованием (2 измерения: 1. влажный материал, 2. высушенный материал). Если не было получено твердых веществ, то растворы перемешивали или хранили при 5°C, и, если осадка все еще не наблюдалось, то растворы выпаривали в атмосфере азота при комнатной температуре.
Новые формы были дополнительно охарактеризованы методами рентгеновской порошковой дифракции и ТГ-Фурье-ИК.
В таблице 1 приведены результаты экспериментов по установлению равновесия в суспензии и кристаллизации при охлаждении.


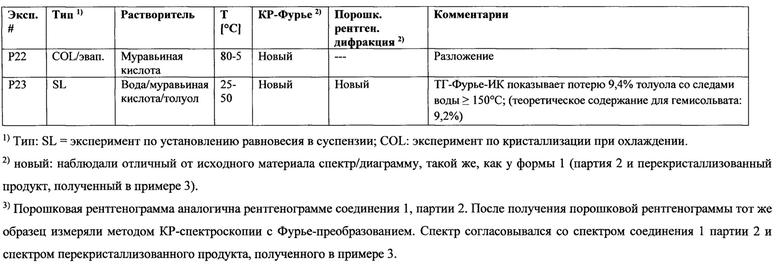
Результаты:
В экспериментах Р2, Р3, Р4, Р5, Р8, Р10, P11, Р16, Р17, Р21 и Р23 происходило образование сольватов (в том числе гидратов) или гемисольватов;
В эксперименте Р18 получен гелеобразный продукт;
В эксперименте Р22 происходило образование продукта, который разложился;
В экспериментах Р7, Р13, Р14, Р15 и Р20 происходило образование кристаллической формы 1 (т.е. той же полиморфной формы, что и партия 2 соединения 1 и продукт, полученный в примере 3);
В экспериментах Р9 и Р12 произошло образование новой кристаллической формы, обозначенной как форма 2; и
В эксперименте Р6 произошло образование полиморфной формы 2 в смеси с третьей полиморфной формой, обозначенной как форма 3.
Пример 5 - Дальнейшее определение характеристик полиморфных форм
i. Полиморфная форма 1
Как подробно описано выше, эксперимент по перекристаллизации соединения из примера 3 привел к образованию продукта, который, вероятно, представлял собой сольват этилацетата. Однако при высушивании он переходил в несольватированную форму, обозначенную как форма 1. После сушки продукты, полученные в экспериментах Р7 (сольват ТГФ), Р13 (сольват ацетона), Р14 (сольват муравьиной кислоты), Р15 (сольват дихлорметана) и Р20 (сольват этанола) приняли кристаллическую форму, которая по данным КР-спектроскопии с Фурье-преобразованием имела характеристики аналогичные характеристикам партии 2 соединения 1 и перекристаллизованного продукта, полученного в примере 3. Не представлялось возможным определить стехиометрию сольватов из-за их нестабильности.
Эта полиморфная форма была обозначена как форма 1 и ее дополнительно характеризовали при помощи порошковой рентгеновской дифракции. Для того, чтобы определить, является ли данный образец чистой твердой фазой можно провести индексирование порошковой рентгенограммы. Порошковая рентгенограмма перекристаллизованного материала (файл: Н906) была успешно проиндексирована и было обнаружено, что кристаллическая решетка представляет собой триклинную кристаллическую решетку. Полученные параметры кристаллической решетки представлены в таблице 2. Соответствие между полученной и расчетной рентгенограммами показано на фигуре 10, при этом низкие значения R (таблица 2) подтверждают хорошее соответствие. Это подтверждает, что форма 1 соответствует именно полиморфной форме, а не смеси форм.

Получен 1H-ЯМР спектр продукта, полученного в эксперименте Р15 (полиморфная форма 1) в ДМСО-d6. Приведенный спектр подтверждает химическую целостность (см. фигуру 11).
ii. Полиморфная форма 2
Форму 2 получали в экспериментах по установлению равновесия между фазами при комнатной температуре в смеси вода / ацетонитрил (1:1) (эксперимент Р9) и ацетонитриле (эксперимент Р12). Эксперимент по установлению равновесия между фазами при комнатной температуре в метилэтилкетоне (эксперимент Р6) также привел к образованию формы 2 в смеси с другой новой формой (формой 3).
КР-спектр с Фурье-преобразованием продукта, полученного в эксперименте Р9 из примера 4 (форма 2) приведен на фигуре 12, а его порошковая рентгенограмма приведена на фигуре 13. Данная рентгенограмма была успешно проиндексирована, что подтверждает, что форма 2 соответствует именно полиморфной форме, а не смеси форм.
Получен 1H-ЯМР спектр продукта, полученного в эксперименте Р9 из примера 4 (полиморфная форма 2) в ДМСО-d6 (см. фигуру 14). Приведенный спектр подтверждает химическую целостность.
iii. Полиморфная форма 3
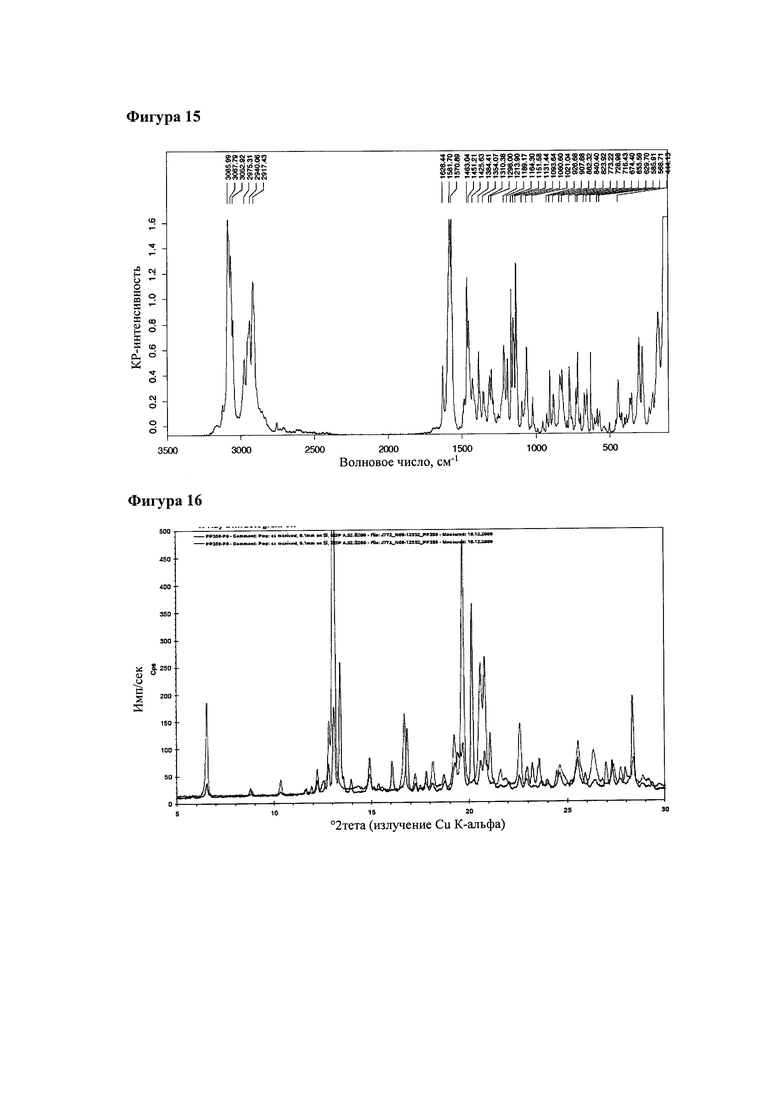
Форму 3 получали из смеси с формой 2 в эксперименте по установлению фазового равновесия при комнатной температуре в метилэтилкетоне (пример 4, эксперимент Р6). На фигуре 15 приведен КР-спектр с Фурье-преобразованием продукта Р6 (форма 3 в смеси с формой 2). На указанной фигуре обозначены наиболее выраженные пики комбинационного рассеяния.
Порошковая рентгенограмма продукта Р6 показала, что указанный материал представлял собой кристаллический материал; на фигуре 16 приведена порошковая рентгенограмма продукта Р6 в сравнении с порошковой рентгенограммой продукта Р9 (форма 2). У продукта Р6 наблюдаются все сигналы, что и у продукта Р9, а также дополнительные сигналы. Это свидетельствует о том, что продукт Р6 представляет собой смесь формы 2 и другой формы. Эта другая форма была обозначена как форма 3.
Пример 6 - Термодинамическая стабильность полиморфных форм
Смесь с одинаковыми соотношениями продуктов, полученных в примере 4 в эксперименте 15 (форма 1), в примере 4 в эксперименте 9 (форма 2+х) и в примере 4 в эксперименте 6 (форма 2+форма 3) суспендировали в ацетонитриле и встряхивали в течение 13 дней при температуре 22°C с получением продукта, обозначенного как продукт Р24. Твердое вещество извлекали при фильтрующем центрифугировании и охарактеризовывали при помощи порошковой рентгеновской дифрактометрии. На фигуре 16 представлена порошковая рентгенограмма продукта Р24 в сравнении с порошковой рентгенограммой продукта, полученного в эксперименте Р9 в примере 4. Две рентгенограммы по существу одинаковы, хотя в рентгенограмме продукта Р9 присутствует несколько дополнительных сигналов. Эти сигналы могут быть (i) отнесены к другой кристаллическую форме или (ii) эти сигналы не были обнаружены для продукта Р24, поскольку рентгенограмма имеет более низкую интенсивность.
Таким образом, порошковая рентгенограмма продукта после 13 дней при 22°C порошковая рентгенограмма соответствует порошковой рентгенограмме формы 2. На основании этого результата можно сделать вывод о том, что форма 2 представляет собой наиболее стабильную при комнатной температуре форму.
Для того, чтобы определить, является ли данный образец чистой твердой фазой, можно провести индексирование порошковой рентгенограммы. Порошковая рентгенограмма продукта Р24 (файл: J893) была успешно проиндексирована и было обнаружено, что кристаллическая решетка представляет собой триклинную кристаллическую решетку. Полученные параметры кристаллической решетки представлены в таблице 3. Соответствие между полученной и расчетной рентгенограммами показано на фигуре 17, при этом низкие значения R (таблица 3) подтверждают хорошее соответствие. Это подтверждает, что форма 2 соответствует именно полиморфной форме, а не смеси форм.
Рентгенограмма продукта Р9, полученного в примере 4, также была индексирована в той же пространственной группе с аналогичными параметрами кристаллической решетки. Однако несколько сигналов продукта Р9 проиндексированы не были. Эти сигналы можно отнести к другой кристаллической форме (например, возможны: полиморфная форма, примесь или разложение).

Пример 7 - Получение полиморфной формы 2 из смеси полиморфных форм 2 и 3
В примере 4 в эксперименте Р6 получена смесь полиморфных форм 2 и 3. Смесь полиморфных форм 2 и 3 можно преобразовать в чистую полиморфную форму 2 с применением следующего способа.
Соединение 1 (12,79 г; смесь полиморфных форм 2 и 3) суспендировали в 150 мл ацетонитрила при 25°C (скорость перемешивания: 400 оборотов в минуту): Суспензию нагревали до 60°C со скоростью нагрева 30 К/час. При 54°C получили раствор желтого цвета. Температуру выдерживали при 60°C в течение 5 мин. После этого раствор охлаждали с 60°C до 49°C при 10 К/ч (при 49°C наблюдали суспензию; нагревали до 56°C при 30 К/ч, наблюдали раствор, снова охлаждали до 49°C при 10 К/ч; при 49°C наблюдали раствор). 320,1 мг соединения 1 формы 2 суспендировали в 1 мл перенасыщенного раствора. Указанную суспензию добавляли к указанному раствору для кристаллизации при 49°C. Стеклянный сосуд промывали 0,2 мл перенасыщенного раствора. Наблюдали суспензию желтого цвета. Указанную суспензию перемешивали в течение 3 ч при 49°C и затем охлаждали до температуры 35°C при скорости охлаждения 1,8 К/ч и с 35°C до 20°C при 3 К/ч. Указанную суспензию перемешивали в течение 10 ч при температуре 20°C. Сохраняли дополнительный образец 1 мл. Твердое вещество извлекали при помощи вакуумной фильтрации (стеклообразная фритта Р04; 59,4 мг влажного материала). Указанное твердое вещество охарактеризовывали методом порошковой рентгеновской дифракции. Образец соответствовал форме 2. Растворитель суспензии частично выпаривали на роторном испарителе при температуре 23°C и давлении 107-110 мбар. Оставшуюся суспензию, имеющую объем ~25 мл, фильтровали при помощи вакуумной фильтрации (стеклообразная фритта Р04, выход: 12,32 г). Все образцы из предыдущих экспериментов кристаллизации содержали примерно 0,4-0,5 мас. % ацетонитрила. В целях уменьшения содержания ацетонитрила проводили дальнейшие исследования.
Пример 8 - удаление ацетонитрила из полиморфной формы 2 (способ 1)
Отфильтрованный твердый продукт, полученный в примере 7, суспендировали в 21 мл метилэтилкетона. Затем метилэтилкетон выпаривали в роторном испарителе при комнатной температуре и ~100 мбар. Трижды выполняли этап промывки. Твердое вещество сушили в вакууме (50 мбар) при комнатной температуре в течение ночи. Полученный материал охарактеризовывали методами 1H-ЯМР и порошковой рентгеновской дифракции. Высушенное твердое вещество (10,68 г) снова промывали 18 мл метилэтилкетона; метилэтилкетон выпаривали в вакууме (110 мбар) при комнатной температуре. Трижды выполняли этап промывки. Твердое вещество сушили в роторном испарителе в вакууме (20 мбар) при комнатной температуре в течение ночи. Оставшийся материал охарактеризовывали методами 1H-ЯМР и порошковой рентгеновской дифракции. Порошковые рентгенограммы образцов соответствовали форме 2. Порошковая рентгенограмма образца, отобранного после второй стадии промывки, содержит небольшой дополнительный сигнал. 1H-ЯМР спектры всех образцов показали наличие меньшего количества ацетонитрила по сравнению с предыдущими образцами (вероятно <0,1 мас. %).
Пример 9 - удаление ацетонитрила из полиморфной формы 2 (способ 2)
Густую суспензию готовили из полиморфной формы 2 соединения 1, полученной в соответствии со способом из примера 7 (5 г) с метилизобутилкетоном (3 объема, 15 мл). Густую суспензию перемешивали при температуре 50°C в течение ночи, после чего ее охлаждали до комнатной температуры, фильтровали и промывали метилизобутилкетоном с получением 4,1 г (82%) порошка почти белого цвета, который по данным порошковой рентгеновской дифракции оказался полиморфной формой 2. Данные 1ЯМР показали отсутствие ацетонитрила и только следы метилизобутилкетона.








Настоящее изобретение относится к способу получения полиморфной формы [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислот. Способ получения полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1), которая характеризуется КР-спектром с Фурье-преобразованием с характеристическими пиками при 3063 ± 2 см-1, 1578 ± 2 см-1, 1423 ± 2 см-1, 1209 ± 2 см-1, 1187 ± 2 см-1, 1166 ± 2 см-1, 1150 ± 2 см-1, 930 ± 2 см-1, 883 ± 2 см-1, 770 ± 2 см-1, 356 ± 2 см-1, 304 ± 2 см-1, 167 ± 2 см-1, 119 ± 2 см-1 или следующими параметрами кристаллической решетки:
осуществляют путем (а) суспендирования соединения 1 в растворителе, содержащем ацетонитрил, смесь ацетонитрила и воды или кетоновый растворитель, выбранный из метилизобутилкетона, метилэтилкетона и их смесей, причем соединение 1 находится в аморфной форме, в кристаллической форме, отличной от полиморфной формы 2, или в виде смеси полиморфной формы 2 с одной или более другими полиморфными формами; (b) перемешивания суспензии при температуре примерно от 15 до 25°С в течение от 15 до 30 дней; и (c) выделения и высушивания твердой [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты. Также изобретение относится к способу получения полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1), путем получения насыщенного раствора соединения 1 в растворителе, выбранном из ацетонитрила, ацетонитрила и воды или кетонового растворителя, выбранного из метилизобутилкетона, метилэтилкетона или их смеси; затравливания указанного насыщенного раствора кристаллами полиморфной формы 2 соединения 1; выдерживания с осуществлением кристаллизации; и выделения кристаллов полиморфной формы 2 соединения 1. Технический результат – получение термодинамически стабильной полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты. 2 н. и 4 з.п. ф-лы, 18 ил., 3 табл., 9 пр.
1. Способ получения полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1), которая характеризуется КР-спектром с Фурье-преобразованием с характеристическими пиками при 3063 ± 2 см-1, 1578 ± 2 см-1, 1423 ± 2 см-1, 1209 ± 2 см-1, 1187 ± 2 см-1, 1166 ± 2 см-1, 1150 ± 2 см-1, 930 ± 2 см-1, 883 ± 2 см-1, 770 ± 2 см-1, 356 ± 2 см-1, 304 ± 2 см-1, 167 ± 2 см-1, 119 ± 2 см-1 или следующими параметрами кристаллической решетки:
при этом способ включает:
а. суспендирование соединения 1 в растворителе, содержащем ацетонитрил, смесь ацетонитрила и воды или кетоновый растворитель, выбранный из метилизобутилкетона, метилэтилкетона и их смесей, причем соединение 1 находится в аморфной форме, в кристаллической форме, отличной от полиморфной формы 2, или в виде смеси полиморфной формы 2 с одной или более другими полиморфными формами;
b. перемешивание суспензии при температуре примерно от 15 до 25°С в течение от 15 до 30 дней; и
c. выделение и высушивание твердой [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты.
2. Способ по п. 1, отличающийся тем, что на стадии (а) растворитель представляет собой ацетонитрил или смесь ацетонитрила и воды.
3. Способ получения полиморфной формы 2 [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]уксусной кислоты (соединение 1), которая характеризуется КР-спектром с Фурье-преобразованием с характеристическими пиками при 3063 ± 2 см-1, 1578 ± 2 см-1, 1423 ± 2 см-1, 1209 ± 2 см-1, 1187 ± 2 см-1, 1166 ± 2 см-1, 1150 ± 2 см-1, 930 ± 2 см-1, 883 ± 2 см-1, 770 ± 2 см-1, 356 ± 2 см-1, 304 ± 2 см-1, 167 ± 2 см-1, 119 ± 2 см-1 или следующими параметрами кристаллической решетки:
при этом способ включает:
получение насыщенного раствора соединения 1 в растворителе, выбранном из ацетонитрила, ацетонитрила и воды или кетонового растворителя, выбранного из метилизобутилкетона, метилэтилкетона или их смеси;
затравливание указанного насыщенного раствора кристаллами полиморфной формы 2 соединения 1;
выдерживание с осуществлением кристаллизации; и
выделение кристаллов полиморфной формы 2 соединения 1.
4. Способ по п. 3, отличающийся тем, что растворитель представляет собой ацетонитрил.
5. Способ по п. 3 или 4, дополнительно включающий промывку кристаллов полиморфной формы 2 дополнительным растворителем и высушивание.
6. Способ по п. 5, отличающийся тем, что указанный дополнительный растворитель представляет собой метилэтилкетон, метилизобутилкетон или их смесь.
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТОВ CRTH2 | 2009 |
|
RU2503672C2 |
| Mino R | |||
| Caira, "Crystalline Polymorphism of Organic Compounds", Topics in Current Chemistry, Vol | |||
| Складная решетчатая мачта | 1919 |
|
SU198A1 |
| WO 2006095183 A1, 14.09.2006 | |||
| Прибор для исследования электрических свойств почв, грунтов и тому подобных материалов | 1935 |
|
SU54417A1 |

