Настоящее изобретение относится к соединениям, которые полезны в качестве лекарственных средств, к способам получения указанных соединений, композициям, которые содержат указанные соединения, и к их использованию при лечении и профилактике аллергических заболеваний, таких как астма, аллергический ринит, атопический дерматит и других воспалительных заболеваний, опосредованных простагландином D2(PGD2) или другими агонистами, действующими на CRTH2 рецептор, расположенный на клетках, включающих эозинофилы, базофилы и Th2 лимфоциты.
PGD2 относится к эйкозаноидам, классу химических медиаторов, синтезируемых клетками в ответ на локальное повреждение ткани, в ответ на нормальные стимулы или гормональные стимулы или через клеточные пути активации. Эйкозаноиды связываются со специфическими рецепторами клеточной поверхности в разнообразных тканях всего организма и опосредуют различные эффекты в этих тканях. Известно, что PGD2 продуцируется тучными клетками, макрофагами и Th2 лимфоцитами, он обнаружен в высоких концентрациях в дыхательных путях пациентов, страдающих бронхиальной астмой, после провокации антигеном (Murray et al, (1986), N. Engl. J. Med. 315: 800-804). Инсталляция PGD2 в дыхательные пути может провоцировать многие проявления астматического ответа, включая бронхоспазм (Hardy el al., (1984) N.Engl. J.Med. 311: 209-213; Sampson et al, (1997) Thorax 52: 513-518) и накопление эозинофилов (Emery el al., (1989) J.Appl. Physiol. 67: 959-962).
Способность экзогенно вводимого PGD2 вызывать воспалительную реакцию подтверждена с использованием трансгенных мышей со сверэкспрессией PGD2 - синтазы человека, у которых проявляются повышенная эозинофильная воспалительная реакция в легких и повышенная Th2 продукция цитокинов в ответ на антиген (Fujitani et al, (2002). / Immunol. 168: 443-449).
Первым обнаруженным специфичным для PGD2 рецептором был DP рецептор, который связан с повышением внутриклеточных уровней цАМФ. Тем не менее, предполагают, что PGD2 опосредует большую часть своей провоспалительной активности через рецептор, сопряженный с G-белком, называемый CRTH2 (хемоаттрактантная рецепторноподобная молекула (chemoattxactant receptor-homologous molecule) экспрессируемая на Th2 клетках), которая экспрессируется Th2 лимфоцитами, эозинофилами и базофилами (Hirai et al, (2001) J. Exp. Med. 193: 255-261, and EP 0851030 and EP-A-1211513 and Bauer et al, EP-A-1170594). Представляется понятным, что действие PGD2 в отношении активации Th2 лимфоцитов и эозинофилов опосредуется CRTH2, так как данную реакцию могут вызывать селективные агонисты CRTH2 13,14-дигидро-15-кето-PGD2 и 15R-МЕТИЛ-PGD2, и указанное действие PGD2 блокируются антителами против CRTH2 (Hirai et al, 2001; Monneret et al, (2003) J. Pharmacol. Exp. Ther. 304: 349-355). В отличие от этого, селективный агонист DP - BW245C не способствует миграции Th2 лимфоцитов или эозинофилов (Hirai et al, 2001; Gervais et al, (2001) J. Allergy Clin. Immunol. 108: 982-988). На основании приведенных выше данных, антагонистическое воздействие на взаимодействие PGD2 с CRTH2 рецептором является привлекательным подходом для лечения воспалительного компонента Th2-зависимых аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит.
В ЕР-А-1170594 предложен способ, который согласно данному документу можно использовать для идентификации соединений, которые полезны при лечении аллергической астмы, атопического дерматита, аллергического ринита, аутоиммунных состояний, реперфузионных повреждений и ряда воспалительных состояний, все из которых опосредованы действием PGD2 или других агонистов на CRTH2 рецептор.
Соединения, которые связываются с CRTH2 рецептором, рассмотрены в WO-A-03066046 и WO-A-03066047. Данные соединения не являются новыми, их свойства были впервые раскрыты, наряду со сходными соединениями, в GB 1356834, GB 1407658 и GB 1460348, в качестве соединений обладающих противовоспалительной, анальгетической и жаропонижающей активностью. В WO-A-03066046 и WO-A-03066047 сообщают, что раскрытые в данных документах соединения являются модуляторами активности CRTH2 рецептора и поэтому полезны для лечения или профилактики обструктивных заболеваний дыхательных путей, таких как астма, хроническое обструктивное заболевание легких (ХОЗЛ) и ряда других заболеваний, включая различные патологические состояния костей и суставов, кожи и глаз, ЖКТ, центральной и периферической нервной системы и других тканей, а также реакции отторжения трансплантата. Все данные соединения являются производными индола, замещенными уксусной кислотой в положении 3 индольного кольца.
PL 65781 и JP 43024418 также относятся к производным индол-3-уксусной кислоты, которые сходны по структуре с индометацином и показано, что, подобно индометацину, они обладают противовоспалительным и жаропонижающим действием. Таким образом, хотя это нельзя было установить на момент публикации данных документов, описанные в них соединения представляют собой ингибиторы ЦОГ (циклооксигеназы), т.е. проявляют активность, которая совершенно отлична от активности соединений согласно настоящему изобретению. Действительно, ингибиторы ЦОГ противопоказаны при лечении многих заболеваний и состояний, например астмы и воспалительных заболеваний кишечника, для которых могут быть полезны соединения согласно настоящему изобретению, хотя они могут иногда быть использованы для лечения артритов.
Существует другие источники в уровне техники, которые относятся к соединениям индол-1-уксусной кислоты, хотя указанные соединения и не раскрыты в них в качестве CRTH2 антагонистов. Например, WO-A-9950268, WO-A-0032180, WO-A-01518489 и WO-А-0164205 относятся к соединениям, которые представляют собой производные индол-1-уксусной кислоты, но указано, что эти соединения представляют собой ингибиторы альдозы, которые полезны для лечения сахарного диабета (WO-A-9950268, WO-A-0032180 и WO-A-0164205) или гипоуремические агенты (WO-A-0151849). Ни один из указанных документов не содержит указаний на то, что данные соединения могут быть полезны при лечении заболеваний и состояний, опосредованных PGD2 или другими агонистами CRTH2 рецептора.
US 4,363,912 относится к производным индол-1-алкил карбоновых кислот (включая аналоги индол-1-уксусной кислоты), которые указаны в качестве ингибиторов тромбоксан-синтетазы, полезных для лечения таких состояний, как тромбоз, ишемическая болезнь сердца и инсульт. Данные соединения имеют незамещенные 3-пиридил или 4-пиридил заместители в положении, эквивалентном положению пиридильной группы в общей формуле (I). Сравнение ближайшего аналога (5-фтор-2-метил-3-(пиридин-3-илметил)-индол-1-ил)-уксусной кислоты, который рассматривается в заявке US 4,363,912, и соединений согласно настоящему изобретению показывает, что указанный аналог значительно менее активен в качестве антагониста CRTH2 по сравнению с соединениями согласно настоящему изобретению. В отличие от соединений согласно настоящему изобретению (все из которых являются производными индол-1-уксусной кислоты) предпочтительные соединения US 4,363,912 представляют собой производные 3-(индол-1-ил)-пропионовой кислоты.
WO-A-9603376 относится к соединениям, которые указаны в качестве ингибиторов SPLA2, которые полезны для лечения бронхиальной астмы и аллергического ринита. Данные соединения имеют амидные или гидразидные заместители вместо производных карбоновой кислоты у соединений согласно настоящему изобретению.
JP 2001247570 относится к способу получения 3-бензотиазолил-индол-уксусной кислоты, которую рассматривают в качестве ингибитора альдозы.
US 4,859,692 относится к соединениям, которые рассматривают в качестве 5 антагонистов лейкотриена полезными для лечения патологических состояний, таких как астма, сенная лихорадка и аллергический ринит, а также некоторых воспалительных состояний, таких как бронхит, атоническая и эктопическая экзема. Некоторые из соединений, рассматриваемых в данном документе, являются индол-1-уксусными кислотами, но те же авторы, в J. Med. Chem., 33, 1781-1790 (1990), указывают, что соединения с уксуснокислой группой при азоте индола не имеют значимой пептидолейкотриеновой активности.
US 4,273,782 относится к производным индол-1-уксуксной кислоты, которые считают полезными для лечения таких патологических состояний, как тромбоз, ишемическая болезнь сердца, инсульт, транзиторный ишемический приступ и сосудистые осложнения диабета. В данном документе нет упоминания о состояниях, опосредованных PGD2 или другими агонистами CRTH2 рецептора.
US 3,557,142 относится к 3-замещенным-1-индол карбоновым кислотам и эфирам, которые порлезны для лечения воспалительной патологии.
WO-A-03/097598 относится к соединениям, которые представляют собой антагонисты рецептора CRTH2. Указанные соединения не имеют ароматического заместителя в положении 3 индола.
Публикация Cross et al., J. Med. Chem. 29, 342-346 (1986) относится к способу получения производных индол-1-уксусной кислоты из соответствующих эфиров. Данные соединения рассматривают в качестве ингибиторов тромбоксан-синтетазы.
ЕР-А-0539117 относится к производным индол-1-уксусной кислоты, которые представляют собой антагонисты лейкотриена.
US 2003/0153751 относится к производным индол-1-уксусной кислоты, которые являются ингибиторами sPLA2. Тем не менее, все представленные соединения содержат объемные заместители в положениях 2 и 5 позициях индольной системы и поэтому существенным образом отличаются от соединений согласно настоящему изобретению.
US 2004/0116488 относится к производным индол-1-уксусной кислоты, которые представляют собой ингибиторы PAI-1. Отсутствуют указания на то, что данные соединения могли бы проявлять активность антагонистов CRTH2.
WO 2004/058164 относится к соединениям, которые рассматривают в качестве модуляторов астмы и аллергического воспаления. Единственные соединения, для которых показана активность, полностью отличаются по структуре от производных индол-1-уксусной кислоты согласно настоящему изобретению.
Соединения, которые связываются с CRTH2 рецептором, раскрыты в WO-A-03/097042 и WO-A-03/097598. Эти соединения представляют собой индолуксусные кислоты, но в WO-A-03/097042 индольная система соединена в положениях 2-3 с 5-7-членным карбоциклическим кольцом. В WO-A-03/097598 в положении 3 индола присутствует пирролидиновая группа.
WO-A-03/101981, WO-A-03/101961 и WO-A-2004/007451 относятся к производным индол-1-уксусной кислоты, которые рассматривают в качестве антагонистов CRTH2, но которые по структуре отличаются от соединений общей формулы (I) из-за отсутствия спейсера или -S- или -SO2- группы, присоединенной к индолу в положении 3 на месте группы СН2 соединений согласно настоящему изобретению, как описано ниже.
В WO-A-2005/019171 также описаны производные индол-1-уксусной кислоты, которые рассматривают в качестве антагонистов CRTH2 и которые считают полезными для лечения различных респираторных заболеваний. Все указанные соединения имеют заместитель, который присоединен по положению 3 индола через кислородный спейсер.
В WO-A-2005/094816 также описывает соединения индол-1-уксусной кислоты, но которые имеют алифатический заместитель в положении 3 индольного кольца. Данные соединения рассматривают в качестве антагонистов CRTH2.
WO-A-2006/034419 относится к антагонистам CRTH2 индольного ряда, которые имеют гетероциклический или гетероароматический заместитель, напрямую присоединенный по положению 3 системы индольного кольца.
В более ранней заявке авторов настоящего изобретения, WO-A-2005/044260, были описаны соединения, которые представляют собой антагонисты PGD2 в отношении CRTH2 рецептору. Данные соединения представляют собой производные индол-1-уксусной кислоты, замещенные в положении 3 группой CR8R9, где R9 представляет собой водород или алкил, a R8 представляет собой арильную группу, которая может быть замещена одним или более заместителем. Соединения, описанные в указанном документе, являются эффективными антагонистами PGD2 в отношении рецептора CRTH2 in vitro. Тем не менее, авторы настоящего изобретения обнаружили, что при тестировании in vivo, фармакокинетический профиль некоторых соединения не является оптимальным и их эффективность в тесте изменения формы эозинофилов цельной крови, который дает представление об активности соединений in vivo, часто ниже, чем можно было бы ожидать на основании результатов связывания in vitro.
В другой более ранней заявке авторов настоящего изобретения, WO 2006/095183. производные индол-1 уксусной кислоты замещены по положению 3 1-бензолсульфонил-1Н-пиррол-2-ил-метильной группой, причем фенильная группа молекулы бензолсульфонила может быть в свою очередь замещена. Данные соединения являются высокоактивными антагонистами CRTH2, но они ускоренно метаболизируются, что определяли посредством инкубации с препаратами микросом человека.
Заявка авторов настоящего изобретения, WO 2008/012511, также относится к антагонистам CRTH2, которые в данном случае представляют собой производные индол-1-уксусной кислоты, замещенные 2-фенилсульфонилбензильной группой в положении 3.
Было обнаружено, что положение группы фенилсульфонила оказывало значительное влияние, как на активность соединений, так и на их фармакокинетический профиль.
Настоящее изобретение относится к пиридильным аналогам соединений согласно WO 2008/012511. Данные соединения лишены недостатков в отношении метаболической нестабильности, которыми обладают соединения согласно WO 2006/095183 и, неожиданно, было обнаружено, что специфические позиционные изомеры пиридила и их замещенные формы приводят к оптимальному балансу активности и фармакокинетических свойств. В частности, было обнаружено, что введение фенилсульфонильного заместителя в положение 2 позиционного изомера пиридин-3-ила дает соединения с высокой активностью в функциональном тесте in vitro и хорошей фармакокинетикой in vivo. То, что данная комбинация должна в итоге давать выигрышное сочетание свойств не является очевидным, и на это нет указаний в литературе и патентных заявках, относящихся к антагонистам CRTH2. Особенно неожиданным оказалось то, что соединения 2-бензолсульфонил-пиридин-3-ила являются эффективными и специфичными антагонистами CRTH2 рецептора, как по данным теста связывания с рецептором, так и по данным функционального теста in vitro, поскольку авторы настоящего изобретения обнаружили, что аналогичное соединение - 3-бензолсульфонил-пиридин-2-ил значительно менее активно и, что 3-бензолсульфонил-пиридин-4-ил проявляет более низкую активность в функциональном тесте in vitro, чем можно было ожидать, учитывая степень его сродства к рецептору. Таким образом оказалось, что положение азота пиридила является особенно значимым в соединениях согласно настоящему изобретению.
Поэтому настоящее изобретение относится к новым соединениям, которые связываются с CRTH2 рецептором и которые, поэтому, полезны для лечения заболеваний и патологических состояний, опосредованных действием PGD2 на CRTH2 рецептор.
Настоящее изобретение обеспечивает соединение общей формулы (I)
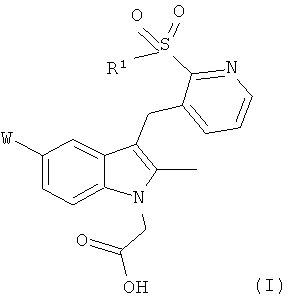
где
W представляет собой хлор или фтор;
R1 представляет собой фенил, возможно замещенный одним или более заместителями, выбранными из галогена, -CN, -C1-С6 алкила, -SOR3, -SO2R3, -SO2N(R2)2, -N(R2)2, -NR2C(O)R3, -CO2R2, -CONR2R3, -NO2, -OR2, -SR2, -O(CH2)pOR2, и -O(CH2)pO(CH2)qOR2 где
каждый R2 независимо представляет собой водород, -С1-С6 алкил, -С3-C8 циклоалкил,
арил или гетероарил;
каждый R3 независимо представляет собой -C1-С6 алкил, -С3-C8 циклоалкил, арил или гетероарил;
р и q независимо представляют собой целое число от 1 до 3;
или фармацевтически приемлемую соль, гидрат, сольват, комплекс или пролекарство указанного соединения.
Соединения общей формулы (I) представляют собой антагонисты CRTH2 рецептора и полезны для лечения состояний, которые опосредованы PGD2 или другими агонистами, которые связываются с CRTH2. Указанные состояния включают: аллергические заболевания, астматические состояния и воспалительные заболевания, примерами которых могут служить астма, включая аллергическую астму, бронхиальная астма, обострения астмы и родственные аллергические заболевания, вызванные вирусной инфекцией, в особенности, обострения которых вызваны риновирусом и респираторным синцитиальным вирусом, астма физического напряжения, астма, вызванная лекарственными средствами, пылью; лечение кашля, включая хронический кашель, связанный с воспалительными патологическими состояниями и нарушениями секреторной функции дыхательных путей, а также ятрогенный кашель; острые и хронические риниты, включая медикаментозный ринит, вазомоторный ринит, постоянный аллергический ринит, сезонный аллергический ринит, назальный полипоз; острые вирусные инфекции, включая обычную простуду, инфицирование респираторным синцитиальным вирусом, вирусом гриппа, коронавирусом и аденовирусом; атопический дерматит, повышенную контактную чувствительность (включая контактный дерматит), экзематозный дерматит, фитодерматит, фотодерматит, себорейный дерматит, герпетический дерматит, плоский красный лишай, склерозирующий и атрофический лишай, гангренозную пиодермию, саркоид кожи, красную дискоидную волчанку, пузырчатку, пемфигоид, буллезный крапивочный эпидермолиз, ангиневротический отек, васкулит, токсическую эритему, кожную эозинофилию, очаговую алопецию, мужскую форму облысения, синдром Свита, синдром Вебер-Кристиана, мультиформную эритему, целлюлит, панникулит, кожную лимфому, немеланомный рак кожи и другие очаги дисплазии; блефарит, конъюктивит, особенно аллергический конъюктивит, передний и задний увеит, хориоидит, аутоиммунное, дегенеративные или воспалительные расстройства, поражающие сетчатку, офтальмит; бронхит, включая инфекционный и эозинофилический бронхит; эмфизему, бронхоэктазы, легкое "фермера", пневмонит сверхчувствительность, идиопатическую интерстициальную пневмонию, осложнения при трансплантации легкого, сосудистые и тромботические нарушения легочного сосудистого русла, легочную гипертензию, пищевую аллергию, гингивит, глоссит, периодонтит, эзофагит, включая рефлюкс-эзофагит, эозинофилический гастроэнтерит, проктит, pruris ani, целиакию, аллергию, связанную с пищей, воспалительное заболевание кишечника, язвенный колит и болезнь Крона, мастоцитоз; а также другие, опосредованные CRTH2 заболевания, например аутоиммунные заболевания, такие как синдром повышенного иммуноглобулина Е, тиреоидит Хашимото, болезнь Грэвиса, болезнь Аддисона, сахарный диабет, идиопатическая тромбоцитопеническая пурпура, эозинофилический фасцит, антифосфолипидный синдром и системная красная волчанка; СПИД, лепра, синдром Сезари, паранеопластический синдром; смешанные или недифференцированные заболевания соединительной ткани, воспалительные миопатии, включая дерматомиозит и полимиозит; ревматическую полимиалгию, ювенильный артрит, ревматическую лихорадку, васкулит, включая гигантоклеточный артериит, артериит Такаясу, синдром Черга-Страуса, узловой полиартрит, микроскопические полиартерииты, артериит височной артерии, миастению, острую и хроническую боль, синдромы невропатической боли, осложнения опухолевых, инфекционных и аутоиммунных процессов центральной и периферической нервной системы, поясничные боли, наследственную средиземноморскую лихорадку, синдром Макла-Уэльса, наследственную ирландскую лихорадку, болезнь Кикучи, псориаз, акнэ, рассеянный склероз, реакцию отторжения трансплантанта, реперфузионное повреждение, хроническое обструктивное заболевание легких; а также ревматоидный артрит, болезнь Стила, анкилозирующий спондилит, реактивный артрит, недифференцированную спондартропатию, псориатический артрит, септический артрит и другие связанные с инфекцией артропатии; костные нарушения и остеоартрит; острый и хронический индуцированный кристаллами синовиит, включая подагру, болезни отложения кристаллов пирофосфата кальция, кальцевый паптит, связанный с ним сухожильный синдром и синовиит, болезнь Бехчета, первичный и вторичный синдром Съегрен; системный склероз и ограниченную склеродерму; гепатит, цирроз печени, холецистит, панкреатит, нефрит, нефритический синдром, цистит и язву Ханнера, острый и хронический уретрит, простатит, эпидимит, овариит, сальпингит, вульво-вагинит, болезнь Пейрони, эректильную дисфункцию, болезнь Альцгеймера и другие деменции; перикардит, миокардит, воспалительную и аутоиммунную кардиомиопатию, включая кардиальный саркоид, ишемические реперфузионные повреждения, эндокардит, вальвулит, аортит, флебит, тромбоз, лечение онкологических Заболеваний и фибротических патологических состояний, таких как идиопатический легочный фиброз, включая криптогенный фиброзирующий альвсолит, келоиды, чрезмерные постхирургические фиброзные шрамы и сращения, фиброз печени, включая фиброз, ассоциированный с гепатитом В и С, фибромы матки, саркоидоз, включая невральный саркоидоз, склеродерма, связанный с диабетом фиброз почек, фиброз ассоциированный, с ревматоидным артритом; атеросклероз, включая церебральный атеросклероз, васкулит, миокардиальный фиброз в результате инфаркта миокарда, фиброз мочевого пузыря, рестеноз, системный склероз, болезнь Дюшоитрена, фиброз, как осложнение противоопухолевой терапией и хронической инфекцией, включая туберкулез и аспергиллез и другие грибковые инфекции, фиброз ЦНС, вследствие инсульта, или для предотвращения образования рубцов в процессе заживления.
Улучшенные показатели активности в тесте изменения формы эозинофилов цельной крови и фармакокинетического профиля соединений общей формулы (I) особенно неожиданны, поскольку некоторые соединения WO-A-2005/044260, которые по структуре являются ближайшими аналогами соединений общей формулы (I) не обладают указанными благоприятными свойствами. В частности, соединение Примера 17, приведенное в WO-A-2005/044260, сходно с соединениями согласно настоящему изобретению и у него следовало бы ожидать наличие похожих свойств. Тем не менее, в экспериментах in vivo, проведенных на собаках, замена 4-метилсульфонилбензильной группы в соединении Примера 17 WO-A-2005/044260 на 2-(бензолсульфонил)пиридин-ил-метильную группу в соединениях формулы (I) оказывало значительное влияние на фармакокинетический профиль данных соединений, поскольку, когда Соединение 17 вводили перорально, его фармакокинетический профиль in vivo уступал профилю для соединений общей формулы (I).
Кроме того, авторы настоящего изобретения обнаружили в отношении многих соединений WO-A-2005/044260, что их in vitro активность в тесте изменения формы эозинофилов цельной крови часто была ниже, чем можно было бы ожидать, учитывая их активность in vitro, которую измеряли в экспериментах по связыванию радиолиганда с CRTH2 рецептором.
Более того, улучшение показателей активности является очень специфичным для группы соединений общей формулы (I). Соединения, которые представляют собой даже более близкие аналоги соединений приведенных в WO-A-2005/044260, не обладают такими благоприятными свойствами. Например, аналоги соединений общей формулы (1), у которых группа SO2R находится в положении пиридинового кольца отличном от положения, смежного к метиленовому спейсеру, присоединенному в положении 3 индолильного остова, менее активны в in vitro в тесте изменения формы эозинофилов цельной крови.
В настоящем описании "C1-C6алкил" относится к неразветвленной или разветвленной углеводородной цепи, содержащей 1-6 атомов углерода и, как вариант, замещенной одним или более заместителем - галогеном и/или одной или более C3-C8 циклоалкильной группой. Примеры включают в себя метил, этил, н-пропил, изопропил, трет-бутил, 2-хлорэтил, метиленциклопропил, метиленциклобутил и метиленциклопентил.
Термин "C1-C18алкил" имеет сходное с указанным выше значение, с той разницей, что он относится к неразветвленной или разветвленной углеводородной цепи, содержащей 1-18 атомов углерода.
В настоящем описании "C3-C8циклоалкил" относится к насыщенной карбоциклической группе, содержащей от 3-х до 8-и атомов в кольце, которая может иметь один или более заместитель. Примеры включают в себя циклопропил, циклопентил, циклогексил и 4-фтор-циклогексил.
В настоящем описании "галоген" относится к фтору, хлору, брому, или йоду
Термин "арил" в контексте настоящего описания относится к кольцевой системе ароматического характера, имеющей от 5 до 14 атомов углерода и содержащей до 3-х колец. В случае, когда арильная группа содержит более одного кольца, полностью ароматическими могут быть не все кольца. Примерами ароматических структур являются бензол, нафталин, индан и инден.
Термин "гетероарил" в контексте настоящего описания относится к кольцевой системе ароматического характера, имеющей от 5 до 14 атомов кольца, по меньшей мере один из которых представляет собой гетероатом, выбранный из N, О и S, и содержащей до 3-х колец. В случае, когда гетероарильная группа содержит более одного кольца, полностью ароматическими могут быть не все кольца. Примеры гетероарильных групп включают в себя пиридин, пиримидин, индол, бензофуран, бензимидазол и индолен.
Подходящие, приемлемые в фармации и ветеринарии соли соединений общей формулы (I) включают в себя соли, полученные с помощью добавления оснований, такие как соли натрия, калия, кальция, алюминия, цинка, магния и других солей металлов, а также холина, диэтаноламина, этаноламина, этилдиамина, мегульмина и других хорошо известных солей, как это подробно изложено у Paulekuhn et al., (2007) J. Med. Chem. 50: 6665-6672 и/или известно специалистам в данной области.
Соли, которые не приемлемы в фармации и ветеринарии могут все же представлять ценность как промежуточные соединения.
Пролекарства представляют собой любые ковалентно-связанные соединения, которые высвобождают in vivo активное лекарственное средство, соответствующее общей формуле (I). Примеры пролекарств включают в себя алкиловые эфиры соединений общей формулы (I), например, эфиры общей формулы (II), приведенной ниже.
В особенно перспективных соединениях общей формулы (I), W представляет собой фтор, а фенильная группа R1 остается незамещенной, или она замещена единственным заместителем галогеном, обычно фтором или хлором, которые находятся, как правило, в положении 4 фенильной группы R1.
Особенно активны следующие соединения согласно настоящему изобретению: (3-{[2-(бензолсульфонил)пиридин-3-ил]метил}-5-фтор-2-метилиндол-1-ил)-уксусная кислота;
[5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусная кислота;
[3-({2-[(4-хлорбензол)сульфонил]пиридин-3-ил}метил)-5-фтор-2-метилиндол-1-ил]-уксусная кислота;
или C1-C6алкил, арил, (CH2)mOC(=O)C1-C6алкил,
((CH2)mO)nCH2CH2X,
(CH2)mN(R5)2 или CH((CH2)mO(C=O)R6)2 эфир указанных соединений;
m равно 1 или 2;
n равно от 1 до 4;
X представляет собой OR или N(R5)2;
R5 представляет собой водород или метил;
R6 представляет собой C1-C18алкил.
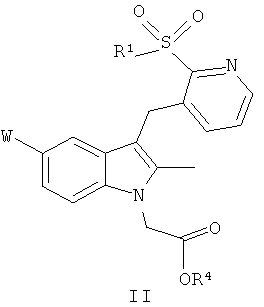
В следующем аспекте настоящего изобретения представлено соединение общей формулы (II):
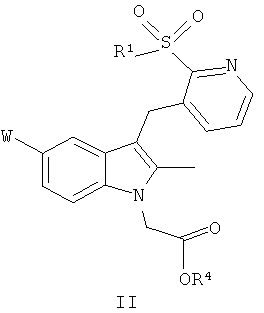
где W и R1 определены также, как и в общей формуле (I); а
R4 представляет собой C1-C6алкил, C1-C6алкил, замещенный арилом, арил, (CH2)mOC(=O)C1-C6алкил, ((CH2)mO)nCH2CH2X, (CH2)mN(R5)2 или CH((CH2)mO(C=O)R6)2;
m равно 1 или 2;
n равно от 1 до 4;
X представляет собой OR или N(R5)2;
R5 представляет собой водород или метил;
R6 представляет собой C1-C18алкил.
или фармацевтически приемлемую соль, гидрат, сольват, комплекс, или пролекарство указанного соединения.
Соединения общей формулы (II) являются новыми и могут быть использованы в качестве пролекарств для соединений общей формулы (1). В случае, если соединение общей формулы (II) действует как пролекарство, оно впоследствии превращается в лекарство под действием эстеразы в крови или тканях пациента.
Примеры особенно подходящих R4 групп, когда соединение общей формулы (II) используют, в качестве пролекарства включают: метил, этил, пропил, фенил,
→ -O(CH2)2O(CH2)2OR5, -O(CH2)2O(CH2)2O(CH2)2OR5, -O(CH2)2O(CH2)2NR5 2, -O(CH2)2O(CH2)2O(CH2)2NR5 2, -CH2OC(=O)tBu, -CH2CH2N(Me)2, -CH2CH2NH2 или -CH(CH2O(C=O)R6)2, где R5 и R6, такие же, как определено выше.
В дополнение к их использованию в качестве пролекарств, соединения формулы (II), в которых R4 представляет собой C1-C6алкил или бензил, можно использовать в способе получения соединения общей формулы (I), указанный способ включает в себя осуществление взаимодействие соединения общей формулы (II) с основанием, таким как гидроксид натрия или гидроксид лития. Такая реакция может проходить в водном или органическом растворителе или в их смеси. Типичный растворитель, используемый для данной реакции представляет собой смесь тетрагидрофурана и воды.
Соединения общей формулы (II) могут быть получены из соединений общей формулы (III):
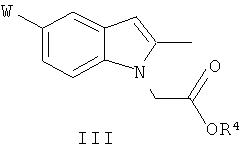
где W идентичен определенному для общей формулы (1), a R4 идентичен определенному для общей формулы (II); путем взаимодействия с альдегидом общей формулы (IV):
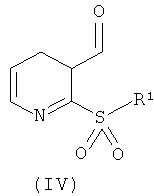
где R1 идентичен определенному для общей формуле (I). Данную реакцию можно проводить в присутствии триметилсилилтрифлата (TMSOTf) в неполярном органическом растворителе при пониженной температуре, например от -5 до 10°C, как правило, при 0°C. Промежуточный продукт затем восстанавливают, например, с помощью триалкилсилана, такого как триэтилсилан.
Процедуры получения соединений общей формулы (III) известны специалистам в данной области и, в общем случае, включают в себя алкилирование 5-галоген-индольного производного в положении производным 1 альфа-бромацетата или родственным алкилирующим агентом. Производные 5-галоген-индола коммерчески доступны или могут быть приготовлены известными методами.
Соединения общей формулы (IV) могут быть получены посредством осуществления взаимодействия соединения общей формулы (V):
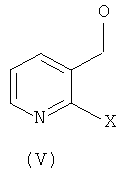
где X - уходящая группа, такая как галоген, в частности, фтор или хлор;
с соединением общей формулы (VI):
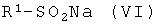
где R1 идентичен определенному для общей формулы (I).
Данная реакция может быть проведена в полярном органическом растворителе, таком как ДМСО при повышенной температуре, как правило, температуре рефлюкса в течение продолжительного периода, например от 48 до 120 часов.
Соединения общих формул (V) и (VI) коммерчески доступны.
Соединения общей формулы (I) представляют собой антагонисты CRTH2 рецептора, а соединения общей формулы (II) представляют собой пролекарства соединений общей формулы (I). Поэтому соединения общих формул (I) и (II) полезны для лечения заболеваний и состояний, опосредованных PGD2 или других агонистов CRTH2 рецептора, данный способ включает в себя введение пациенту, нуждающемуся в указанном лечении, подходящего количества соединения общей формулы (I) или (II).
В третьем своем аспекте настоящее изобретение относится к соединению общей формулы (I) или (II) для применения в медицине, в частности, применения для лечения и профилактики заболеваний и состояний, опосредованных агонистами CRTH2 рецептора, особенно PGD2
Кроме тога, также обеспечено применение соединения общей формулы (I) или (II) для приготовления агента для лечения или профилактики заболеваний и состояний, опосредованных агонистами CRTH2 рецептора, в особенности PGD2.
Как указано выше, такие заболевания и патологические состояния включают в себя аллергические заболевания, астматические состояния и воспалительные заболевания, примерами, которых являются: астма, включая аллергическую астму, бронхиальная астма, эндогенная, экзогенная, астма физического напряжения, астма, индуцированная лекарственными средствами, пылью, лечение кашля, включая хронический кашель, связанный с воспалительными патологическими состояниями и нарушениями секреторной функции дыхательных путей, а также ятрогенный кашель, острые и хронические риниты, включая медикаментозный ринит, вазомоторный ринит, постоянный аллергический ринит, сезонный аллергический ринит, назальный полипоз, острые вирусные инфекции, включая обычную простуду, инфекцию респираторным синцитиальным вирусом, вирусом гриппа, коронавирусом и аденовирусом; атопический дерматит, повышенная контактная чувствительность (включая контактный дерматит), экзематозный дерматит, фитодерматит, фотодерматит, себорейный дерматит, герпетический дерматит, плоский красный лишай, склерозирующий и атрофический лишай, гангренозная пиодермия, саркоид кожи, красная дискоидная волчанка, пузырчатка, пемфигоид, буллезный крапивочный эпидермолиз, ангиневротический отек, васкулит, токсическая эритема, кожная эозинофилия, очаговая алопеция, мужская форма облысения, синдром Свита, синдром Вебер-Кристиана, мультиформная эритема, целлюлит, панникулит, кожная лимфома, немеланомный рак кожи и другие очаги дисплазии; блефарит, конъюктивит, особенно аллергический конъюктивит, передний и задний увеит, хориоидит, аутоиммунные, дегенеративные или воспалительные расстройства, поражающие сетчатку, офтальмит; бронхит, включая инфекционный и эозинофилический бронхит, эмфизема, бронхоэктазы, легкое "фермера", пневмонит сверхчувствительность, идиопатическая иитерстициальная пневмония, осложнения при трансплантации легкого, сосудистые и тромботические нарушения легочного сосудистого русла, легочная гипертензия, пищевая аллергия, гингивит, глоссит, периодонтита, эзофагит, включая рефлюкс-эзофагит, эозинофилический гастроэнтерит, проктит, pruris ani, целиакия, аллергия, связанная с пищей, воспалительное заболевание кишечника, язвенный колит и болезнь Крона, мастоцитоз, а также другие, опосредованные CRTH2 заболевания, например аутоиммунные заболевания, такие как синдром повышенного иммуноглобулина Е, тиреоидит Хашимото, болезнь Грэвиса, болезнь Аддисона, сахарный диабет, идиопатическая тромбоцитопеническая пурпура, эозинофилический фасцит, антифосфолипидный синдром и системная красная волчанка, СПИД, лепра, синдром Сезари, паранеопластический синдром, смешанные или недифференцированные заболевания соединительной ткани, воспалительные миопатии, включая дерматомиозит и полимиозит, ревматическая полимиалгия, ювенильный артрит, ревматическая лихорадка, васкулит, включая гигантоклеточный артериит, артериит Такаясу, синдром Черга-Страуса, узловой полиартрит, микроскопический полиартериит, артериит височной артерии, миастения, острая и хроническая боль, синдромы невропатической боли, осложнения опухолевых, инфекционных и аутоиммунных процессов центральной и периферической нервной системы, поясничные боли, наследственная средиземноморская лихорадка, синдром Макла-Уэльса, наследственная ирландская лихорадка, болезнь Кикучи, псориаз, акнэ, рассеянный склероз, реакция отторжения трансплантанта, реперфузионное повреждение, хроническое обструктивное заболевание легких, а также ревматоидный артрите, болезнь Стила, анкилозирующий спондилит, реактивный артрит, недифференцированная спондартропатия, псориатический артрит, септический артритеи другие связанные с инфекцией артропатии, костные нарушения и остеоартрит; острый и хронический индуцированный кристаллами синовиит, включая подагру, болезни отложения кристаллов пирофосфата кальция, кальцевый паптит, связанный с ним сухожильный синдром и синовиит, болезнь Бехчета, первичный и вторичный синдром Съегрен, системный склероз и ограниченная склеродерма; гепатит, цирроз печени, холецистит, панкреатит, нефрит, нефритический синдром, цистит и язва Ханнера, острый и хронический уретрит, простатит, эпидимит, овариит, сальпингит, вульво-вагинит, болезнь Пейрони, эректильная дисфункция, болезнь Альцгеймера и другие деменции; перикардит, миокардит, воспалительная и аутоиммунная кардиомиопатия, включая кардиальный саркоид, ишемическое реперфузионное повреждение, эндокардит, вальвулит, аортит, флебит, тромбоз, лечение онкологических заболеваний и фибротических патологических состояний, таких как: идиопатический легочный фиброз, включая криптогенный фиброзирующий альвеолит, келоиды, чрезмерные постхирургические фиброзные шрамы и сращения, фиброз печени, включая фиброз, ассоциированный с гепатитом B и C, фибромы матки, саркоидоз, включая невральный саркоидоз, склеродерма, фиброз почек вследствие диабета, фиброз, ассоциированный с ревматоидным артритом, атеросклероз, включая церебральный атеросклероз, васкулит, миокардиальный фиброз вследствие инфаркта миокарда, фиброз мочевого пузыря, рестеноз, системный склероз, болезнь Дюпюитрена, фиброз, как осложнения противоопухолевой терапии и хронической инфекции, включая туберкулез и аспергиллез и другие грибковые инфекции, фиброз ЦНС вследствие инсульта. Данные соединения также полезны для предотвращения образования фиброзных рубцов во время лечения.
Данные соединения особенно эффективны, когда их применяют для лечения или профилактики аллергической астмы, круглогодичного аллергического ринита, сезонного аллергического, ринита, атопического дерматита, повышенной контактной чувствительности (включая контактный дерматит), конъюктивита, особенно аллергического конъюктивита, ювенильного кератоконъюктивита и атопического кератоконъюктивита, эозинофильного бронхита, пищевой аллергии, эозинофильного гастроэнтерита, воспалительного заболевания кишечника, язвенного колита и болезни Крона, мастоцитоза, а также других PGD2-опосредованных заболеваний, например аутоиммунных заболеваний, таких как синдром гиперпродукции иммуноглобулина Е и системная красная волчанка, псориаз, акне, рассеянный склероз, реакция отторжения трансплантанта, реперфузионное повреждение, хроническое обструктивное заболевание легких, а также ревматоидный артрит, псориатический артрит, остеоартрит и фиброзные заболевания, вызванные или обострившиеся под влиянием Th2 иммунного ответа, например идиопатический легочный фиброз и гипертрофические рубцы.
Соединения общей формулы (I) или (II) должны быть приготовлены подходящим способом в зависимости от заболевания или состояния, для лечения которого они необходимы.
Поэтому в следующем аспекте настоящего изобретения предложена фармацевтическая композиция, которая содержит соединение общей формулы (I) или (II) вместе с фармацевтическим вспомогательным веществом (эксципиентом) или наполнителем. Другие активные материалы также могут присутствовать, поскольку могут считаться подходящими или желательными для лечения или профилактики указанного заболевания или состояния,.
Наполнитель или каждый из наполнителей, если их присутствует более одного, должен быть приемлем в плане совместимости с другими ингредиентами лекарственной формы и быть безвредным для пациента.
Лекарственные формы включают в себя те, которые подходят для введения перорально, ректального, назального, бронхиального (ингаляция), наружного (включая глазные капли, буккальное и сублингвальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения и могут быть приготовлены любыми хорошо известными в области фармации способами.
Путь введения будет зависеть от состояния, подлежащего лечению, но предпочтительные композиции разрабатывают для перорального, назального, бронхиального или местного введения.
Композиция может быть приготовлена путем объединения определенного выше активного агента с наполнителем. В целом, лекарственные формы готовят посредством единообразного и тщательного объединения указанного активного агента с жидким наполнителем или тонко размолотым твердым наполнителем или с обоими, и после этого, если необходимо придают продукту определенную форму. Настоящее изобретение распространяется, и на способ приготовления фармацевтической композиции, который включает объединение или ассоциацию соединения общей формулы (I) или (II) с фармацевтически или ветеринарно приемлемым наполнителем.
Лекарственные композиции для приема перорально согласно настоящему изобретению могут быть представлены как: отдельные единицы дозировки, такие как капсулы, пакетики или таблетки, каждая из которых содержит строго определенное количество активного агенты; в виде порошка или гранул; в виде растворов или суспензий указанного активного агента в воде или неводной жидкости; или в виде масляно-водных жидких эмульсий или водно-масляных жидких эмульсий; или в виде болюсов и т.д.
Для композиций для приема перорально (например таблеток и капсул), термин "приемлемый наполнитель" включает в себя носители, такие, как общепринятые эксципиенты, например связывающие агенты, например патоку, гуммиарабик, желатин, сорбит, трагант, поливинилпирролидон (Povidone), метилцеллюлозу, этилцеллюлозу, натрийкарбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу, сахарозу и крахмал; наполнители, например, кукурузный крахмал, желатин, лактозу, сахарозу, микрокристиллическую целлюлозу, каолин, маннит, фосфат кальция, хлорид натрия и альгиновую кислоту; и любриканты, такие как стеарат магния, стеарат натрия и другие стеараты металлов, стеарат глицерина, стеариновую кислоту, жидкий силикон, тальк, воски, масла и коллоидную кремниевую кислоту. Можно также использовать ароматизаторы, такие как перечная мята, масло гаультерии, вишневый ароматизатор и тому подобные. Может быть желательным добавить краситель, для того чтобы сделать лекарственную форму легко идентифицируемой. На таблетки может быть также нанесено покрытие способами хорошо известными в данной области техники.
Таблетка может быть изготовлена путем прессования или формовки, возможно с одним или более, вспомогательным ингредиентом. Прессованные таблетки могут быть приготовлены прессованием в подходящем устройстве активного агента в свободной сыпучей форме, такой как порошок или гранулы, возможно смешанной со связующим веществом, лубрикантом, инертным разбавителем, консервантом, ПАВ или эмульгатором. Формованные таблетки могут быть изготовлены формовкой в подходящем устройстве смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Таблетки, возможно, могут быть покрыты оболочкой или иметь риски, и могут быть получены таким образом, что бы обеспечивать медленное или контролируемое высвобождение активного агента.
Другие лекарственные формы, подходящие для приема перорально, включают в себя пастилки, содержащие активный агент в ароматизированной основе, обычно сахарозе и патоке или траганте; пастилы, содержащие активный агент в инертной основе, такой как желатин и глицерин, или сахароза и патока; и средства для полоскания, содержащие активный агент и подходящий жидкий наполнитель.
Для местного применения, соединения общей формулы (I) или (II) могут быть приготовлены в виде крема, мази, геля, раствора или суспензии и т.д. Лекарственные формы в виде крема или мази, которые могут быть использованы для данного лекарственного средства, являются обычными лекарственными формами хорошо известными в данной области, например, как описано в стандартных пособиях по фармации, таких как Британская Фармакопея.
Соединения общей формулы (I) или (II) можно применять для лечения дыхательной системы посредством назального, бронхиального или буккального введения, например, аэрозолей или спреев, которые могут распространять фармакологически активный ингредиент в форме порошка или в форме капель раствора или суспензии. Фармацевтические композиции со свойствами порошкового распыления обычно содержат, в дополнение к активному ингредиенту, жидкий пропеллент с температурой кипения ниже комнатной и, если необходимо, добавки, такие как жидкие или твердые неионные или анионные сурфактанты и/или разбавители. Фармацевтические композиции, в которых фармакологически активный ингредиент находится в растворе, содержат, в дополнение к нему, подходящий пропеллент, и даже более того, если необходимо дополнительный растворитель и/или стабилизатор. Вместо пропеллента, может быть также использован сжатый воздух, при необходимости его можно получать с помощью подходящего устройства сжатия/расширения.
Парентеральные лекарственные формы должны быть, как правило, стерильны.
Обычно, доза соединения согласно настоящему изобретению должна составлять примерно от 0,01 до 100 мг/кг; так чтобы поддерживать концентрацию лекарственного средства в плазме на уровне достаточном для ингибирования воздействия PGD2 на CRTH2 рецептор. Точное количество соединения общей формулы (I) или (II), которое терапевтически эффективно, и способ введения наилучший для такого соединения, легко определяется с помощью обычного для данной области способа, а именно, посредством сравнения уровня содержания указного соединения в крови с концентрацией необходимой для достижения терапевтического эффекта.
Соединения общей формулы (I) или (II) можно использовать в комбинации с одним или более активными агентами, которые пригодны для лечения заболеваний и патологических состояний, перечисленных выше, хотя указанные активные агенты не обязательно являются ингибиторами PGD2 в отношении CRTH2 рецептора.
Таким образом, фармацевтическая композиция, описанная выше, может дополнительно содержать один или более из указанных активных агентов.
Также предложено применение соединения общей формулы (I) или (II) в приготовлении агента для лечения заболеваний и патологических состояний, опосредованных агонистами CRTH2 рецептора, в особенности PGD2, отличающееся тем, что указанный агент включает в себя дополнительное активное вещество полезное для лечения того же заболевания или состояния.
Указанные дополнительные активные вещества могут быть другими антагонистами CRTH2 рецептора или могут иметь полностью отличный механизм действия. К ним относятся существующие средства для лечения аллергических и других воспалительных заболеваний, которые включают:
суплатаста тозилат и сходные соединения;
агонисты β2 адренорецепторов, такие как метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, салметерол, индакатерол, тербуталин, орципреналин, битолтерола мезилат и пирбутерол или метилксантины, такие как теофиллин и аминофиллин, стабилизаторы тучных клеток, такие как кромогликат натрия или антагонисты мускариновых рецепторов, такие как тиотропиум;
антигистаминовые средства, например антагонисты H1 рецепторов гистамина, такие как лоратидин, цетиризин, деслоратидин, левоцетиризин, фексофенадин, астемизол, азеластин и хлорфенирамин или антагонисты H4 рецепторов;
агонисты α1 и α2 адренорецепторов, такие как пропилгекседрин фенилэфрин, фенилпропаноламин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид и этилнорадреналина гидрохлорид;
модуляторы функции хемокиновых рецепторов, например CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С) или CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства C-X-C) и CX3CR1 для семейства C-X3-C;
антагонисты лейкотриена, такие как монтелукаст и зафирлукаст;
ингибиторы биосинтеза лейкотриена, такие как ингибиторы 5-липооксигеназы или ингибиторы белка, активирующего 5-липооксигеназу (FLAP), такие как цилеутон, АВТ-761, фенлеутон, тепоксалин, Abbott-79175, N-(5-замещенные)-тиофен-2-алкилсульфонамиды, гидразоны 2,6-ди-терт-бутилфенола, метокситетрагидропираны, такие как ZD2138, SB-210661, соединения пиридинил-замещенного-2-цианонафталена, такие как L-739010, соединения 2-цианохинолина, такие как L-746,530, соединения индола и хинолина, такие как МК-591, МК-886 и BAY×1005;
ингибиторы фосфодиэстеразы, включая ингибиторы PDE4, такие как рофлумиласт;
лекарственные средства на основе антител к иммуноглобулину Е, такие как омализумаб;
противомикробные средства, такие как фузидиевая кислота (особенно для лечения атопического дерматита);
противогрибковые средства, такие как клотримазол (особенно для лечения атопического дерматита);
иммунодепрессанты, такие как такролимус и особенно пимекролимус в случае воспалительных заболеваний кожи или, в качестве альтернативы, FK-506, рапамицин, циклоспорин, азатиоприн или метотрексат;
средства для иммунотерапии, включая иммунотерапию с использованием аллергенов, такие как Grazax;
кортикостероиды, такие как преднизон, преднизолон, флунизолид, триамцинолон, ацетонид, беклометазона дипропионат, будезонид, флутиказона пропионат, мометазона фуроат и флутиказона фуорат; лекарственные средства, которые способствуют Th1 цитокиновому ответу, такие как интерферон, TNF (фактор некроза опухоли) или GM-CSF (гранулоцит/макрофаг - колониестимулирующий фактор).
Антагонисты CRTH2 можно также комбинировать со средствами, которые находятся в состоянии разработки и показаны при воспалительных состояниях:
другие антагонисты PGD2, действующие на другие рецепторы, такие как антагонисты DP;
лекарственные средства, которые модулируют продукцию цитокинов, такие как ингибиторы TNFα конвертирующего фермента (ТАСЕ), моноклональные антитела к TNF, молекулы иммуноглобулина к TNF рецептору, ингибиторы других изоформ TNF, неселективные ЦОГ1/ЦОГ2 ингибиторы, такие как пироксикам, диклофенак, производные пропионовой кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак и апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин; ЦОГ-2 ингибиторы, такие как мелоксикам, целекоксиб, фофекоксиб, валдекоксиб и эторикоксиб, низкие дозы метотрексата, лефуномида, цикленозида, гидроксихлорохина, d-пеницилламина, ауранофина или золота, вводимого парентерально или перорально;
лекарственные средства, которые модулируют активность Th2 цитокинов IL-4 и IL-5, такие как блокирующие моноклональные антитела и растворимые рецепторы;
PPAR-γ агонисты, такие как росиглитазон; или с
антителами к RSV, такими как Synagis (palivizumab) и агентами, которые могут быть использованы в будущем для лечения риновирусной инфекции, например, интерферон-альфа, интерферон-бета или другие интерфероны.
В следующем своем аспекте настоящее изобретение относится к продукту, который содержит соединение общей формулы (I) или (II) и одно или более вещество, перечисленное выше, в виде комбинированного препарата для одновременного, раздельного или последовательного применения для лечения заболевания или состояния, опосредованного действием PGD2 на CRTH2 рецептор.
В следующем своем аспекте настоящее изобретение относится к набору для лечения заболевания или состояния, опосредованного действием PGD2 на CRTH2 рецептор, включающему первый контейнер, который содержит соединение общей формулы (I) или (II) и второй контейнер, который содержит одно или более активных веществ, перечисленных выше.
Далее настоящее изобретение будет описано более подробно на основе следующих неограничивающих примеров.
В Примере 1, были получены 1H ЯМР спектры, с помощью спектрометра Bruker Advance II, работающего на частоте 300 МГц. Все сигналы были соотнесены сигналу остаточного протонного растворителя.
В Примерах 2 и 3, были записаны ЯМР спектры на спектрометре Jeol JNM-GSX, работающего на частоте 400 МГц для получения 1H ЯМР данных и 100 МГц для получения 13C ЯМР данных.
В Примере, 1, был проведен ВЭЖХ-КАД-МС анализ на хроматографе Gilson 321 с детекцией, выполненной посредством ESA Corona CAD и масс-спектрометра Finnigan AQA, работающем в режиме позитивной электроспрей ионизации. Для высокоэффективной жидкостной хроматографии использовали колонку Phenomenex Gemini C18 50×4.6 мм 3 мкм, разделение проводили в режиме градиента подвижной фазы, меняя ее состав со 100% 0.1% муравьиной кислоты в воде до 100% 0.1% муравьиной кислоты в ацетонитриле за 2.5 минуты, общее время анализа 6.5 минут. В ряде случаев только данные масс-спектрометриии были получены, используя прибор, описанный выше.
В Примерах 2 и 3, хроматографический анализ проводили на Agilent 1050, детекцию осуществляли спектрофотометрически на 220 нм. Использовали колонку YMC-Pack, ODS-А 150×4.6 мм 5 мкм, разделение проводили в режиме градиента, меняя состав подвижной фазы со 100%-0.01% трифторуксусной кислоты в воде до 100%-0.01% трифторуксусной кислоты в ацетонитриле за 16 минут, общее время анализа 21 минута.
Пример 1: Получение (3-{[2-(бензолсульфонил)пиридин-3-ил]метил}-5-фтор-2-метилиндол-1-ил)-уксусной кислоты (Соединение 1)
2-(бензолсульфонил)изоникотинальдегид
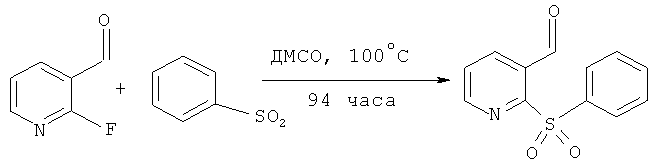
К перемешиваемой суспезии натриевой соли бензолсульфоновой кислоты (9.36 г, 0.057 моль) в ДМСО (45 мл), добавляли 2-фтор-3-пиридинкарбоксальдегид (5.20 мл, 0.052 моль). Полученную смесь перемешивали при 100°C 94 часа. После охлаждения до комнатной температуры, реакционная смесь была распределена между этилацетатом и водой. Органический слой отделяли, а водный далее экстрагировали этилацетатом (3×150 мл). Объединенные органические экстракты промывали водой (100 мл) и солевым раствором (100 мл), высушивали над безводным MgSO4, фильтровали и выпаривали. Полученный сухой остаток очищали колоночной хроматографией, колонку уравновешивали гексаном, элюировали смесью этилацетат: гексан 60:40 об./об., выход компонента, указанного в заглавии, (ВЭЖХ-МС время удерживания =5.63 мин, MH+ 248) составил 6.56 г (51%).
1H ЯМР (ДМСО): 10.89 (1Н, d, J0.68 Гц), 8.82 (1Н, dd, J 1.7, 4.7 Гц), 8.32 (1Н, dd, J 1.7, 7.9 Hz), 8.08-8.02 (2Н, m), 7.85 (1H, dd, J 7.9, 0.7 Гц), 7.81 (1H, dt, J 1.3, 7.5 Гц), 7.73-7.65 (2H, m).
этиловый эфир [3-(2-бензолсульфонил-пиридин-3-илметил)-5-фтор-2-метил-индол-1-ил1-уксусной кислоты
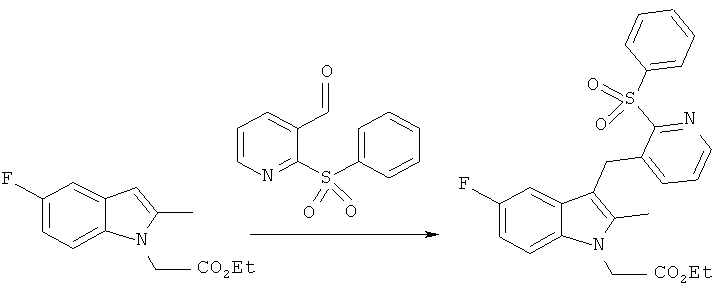
Раствор этилового эфира 5-фтор-2-метил-индол-1-ил-уксусной кислоты (0.95 г, 4.04 мМ), приготовленного как описано в Примере 1 WO/2006/092579, и 2-(бензолсульфонил)изоникотинальдегид (1.0 г, 4.04 мМ) в безводном дихлорметане (45 мл) медленно добавляли в течение 5 минут к перемешиваемому раствору TMSOTf (1.46 мл, 8.08 мМ) в безводном дихлорметане (12.5 мл), охлажденном до 0°C в атмосфере N2. Эту смесь перемешивали 15 минут и одномоментно добавляли триэтилсилан (1.94 мл, 12.12 мМ). Реакционную смесь перемешивали 2 часа 30 минут, подогревали до комнатной температуры, и реакцию прекращали медленным добавлением насыщенного водного раствора NaHCO3 (10 мл). Полученную в результате двухфазную смесь экстрагировали дихлорметаном. Объединенные органические слои промывали солевым раствором, высушивали над безводным MgSO4, фильтровали и выпаривали. Сухой остаток очищали колоночной хроматографией колонку уравновешивали гексаном, элюировали смесью этилацетат. тексан 60:40 об/об, выход компонента, указанного в заглавии, (ВЭЖХ-МС время удерживания +6.63 мин, МН+ 466.8) составил 1.21 г (64%).
1Н ЯМР (CDCI3): 8.38 (1H, dd, J 1.6, 4.5 Гц), 8.14-8.07 (2Н, m), 7.67 (3Н, ddt, J 1.3, 27.7, 7.4 Гц), 7.40-7.34 (1Н, m), 7.22 (1Н, dd, J 4.6, 7.9 Гц), 7.12 (1H dd, J 4.2, 8.9 Гц), 6.90 (1Н, dt, J 2.5, 9.0 Гц), 6.72 (1Н, dd, J2.4, 9.5 Гц), 4.82 (2Н, s), 4.62 (2Н, s), 4.24 (2Н, q, J 7.2 Гц), 2.30 (3Н, s), 1.29 (3Н, t, J 7.2 Гц).
(3-{[2-бензолсульфонил)пиридин-3-ил]метил}-5-фтор-2-мегил-индол-1-ил)-уксусная кислота (Соединение 1)
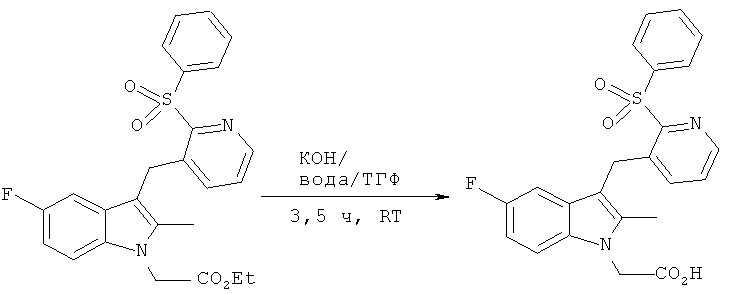
К перемешиваемому раствору этилового эфира [3-(2-бензолсульфонил-пиридин-3-илметил)-5-фтор-2-метил-индол-1-ил]-уксусной кислоты (1.20 г, 2.56 мМ) в ТГФ (26 мл) добавляли раствор гидроксида калия (0.43 г, 7.68 мМ) в воде (9 мл). Полученный раствор перемешивали при комнатной температуре 3.5 часов. ТГФ удаляли in vacuo и оставшийся водный слой подкисляли водным раствором HCl (0.1 М, 25 мл). Продукт собирали фильтрацией, промывали водой и высушивали in vacuo, выход компонента, указанного в заглавии, (ВЭЖХ-МС время удерживания =4.58 мин, М+ -Н 437.2) составил 1.12 г (100%).
1H ЯМР (ДМСО): 8.41-8.27 (1H, m), 8.06-7.91 (2Н, m), 7.84-7.62 (3H, m), 7.50-7.31 (3H, m), 6.93-6.78 (2H, m), 4.99 (2H, s), 4.55 (2H, s), 2.27 (3H, s).
Пример 2 - Получение [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты (Соединение 2)
2-(4-фторбензолсульфонил)-пиридин-3-карбоксальдегид
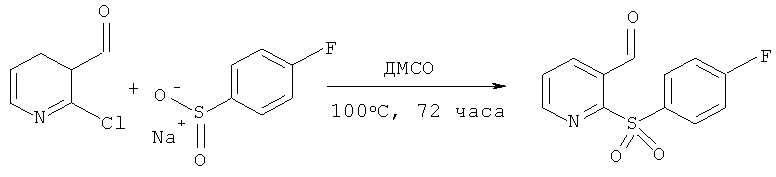
2-хлор-3-пиридинкарбоксальдегид (4.04 г, 2.86 мМ) и натриевую соль 4-фторбензолсульфоновой кислоты растворяли в ДМСО (100 мл) и эту смесь нагревали при 100°C 72 часа в атмосфере азота. После охлаждения до температуры окружающей среды смесь разбавляли водой (500 мл) и экстрагировали этилацетатом (3х). Объеденные органические фракции промывали водой, солевым раствором, высушивали (MgSO4) и выпаривали, получая 7.89 г сухого остатка. Полученное соединение адсорбировали на силикагеле и очищали колоночной хроматографией DPS (dry pad suction), элюируя гептаном с использованием градиента этилацетата, с получением 4.14 г (41%) целевого продукта в виде желтого твердого остатка (пластинки) (Температура плавления = 131-131.3°C; Инфракрасный спектр=1691 cm-1; ВЭЖХ время удерживания =7.21 min>99%).
1H ЯМР (400 МГц; CDCl3): 7.23-7.29 (2H, m) 7.60 (1H, dd) 8.05-8.10 (2H, m) 8.37 (2H, dd)8.67 (1H, dd) 11.1 (1H, s).
13C ЯМР (100 МГц, CDCb): 116.6 (d) 116.8 (d) 127.3 (d) 130.7 (s) 132.6 (d) 134.0 (s) 137.9 (d) 152.5 (s) 159.7 (s) 165.1 (s) 167.7 (s) 188.5 (d).
этиловый эфир [5-фтор-3-((2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты
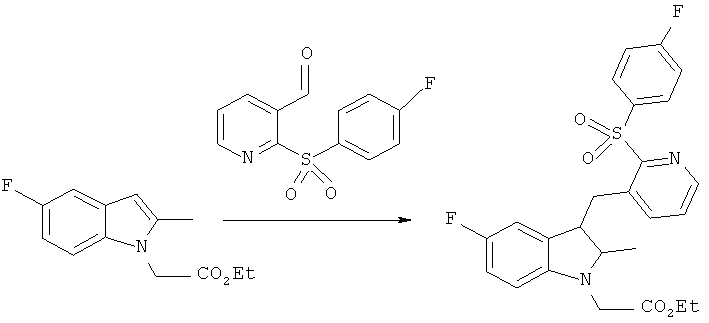
Раствор этилового эфира (5-фтор-2-метил-индол-1-ил)-уксусной кислоты (1.0 г, 4.4 мМ) и 2-(4-фторбензолсульфонил)-пиридин-3-карбоксальдегида (1.13 г, 4.3 мМ) в безводном ДХМ (дихлорметан) (50 мл) добавляли в течение 5-10 мин к перемешиваемому раствору TMSOTf в безводном ДХМ (15 мл) при 0°C. Смесь оставляли на 15 мин перед одномоментным добавлением чистого триэтилсилана (2.05 мл, 12.8 мМ). Далее смесь перемешивали еще 15 часов и прекращали охлаждать, она нагревалась до температуры окружающей среды. Реакцию прекращали капельным добавлением насыщенного раствора NaHCO3 (10 мл) и двухфазную смесь экстрагировали ДХМ (2×50 мл). Объединенные вытяжки промывали солевым раствором (50 мл), затем высушивали (MgSO4) и выпаривали до сухого остатка. Эту реакцию повторяли в том же масштабе и два полученных продукта очищали раздельно. Полученные продукты реакции очищали колоночной хроматографией, используя гептан и градиент этилацетата, выход целевых компонентов составил 0.90 г (43%) и 1.50 г (72%) в виде, соответственно, бледно пурпурного твердого остатка и коричневого твердого остатка разной степени чистоты (96.0% и 94.5% по данным ВЭЖХ) (Температура плавления = 150.5-151.5°C, Инфракрасный спектр =1751 cm-1; ВЭЖХ время удерживания =12.24 min).
1H ЯМР (400 МГц; CDCl3): 1.26 (3H, t) 2.29 (3H, s) 4.22 (2H, q) 4.62 (2H, s) 4.80 (2H, s) 6.79 (1H, dd) 6.86 (1H, ddd) 7.10 (1H, dd) 7.19 (1H, dd) 7.23-7.28 (2H, m) 7.36 (1H, dd) 8.05-8.11 (2H, m) 8.29 (1H, dd).
13C ЯМР (100 МГц, CDCl3): 10.4 (q) 14.2 (q) 25.3 (t) 45.2 (t) 61.9 (t) 103.4 (d) 103.6 (d) 108.0 (s) 108.1 (s) 109.1 (d) 109.2 (d) 109.5 (d) 109.8 (d) 116.2 (d) 116.4 (d) 127.0 (d) 128.5 (s) 128.6 (s) 132.2 (d) 132.3 (d) 133.3 (s) 135.1 (s) 136.4 (s) 136.6 (s) 139.4 (d) 146.2 (d) 156.2 (s) 157.0 (s) 159.4 (s) 164.7 (s) 167.3 (s) 168.6 (s).
[5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил)метил)-2-метилиндол-1-ил1-уксусная кислота (Соединение 2) 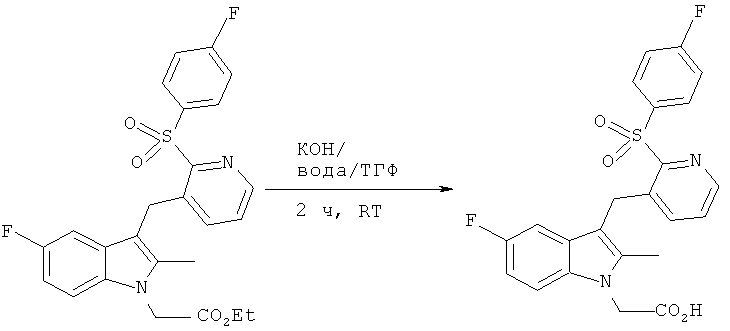
КОН (0.34 г, 5.94 мМ) растворяли в воде (7 мл) и добавляли к интенсивно перемешиваемому раствору этилового эфира [5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты (0.96 г, 1.98 мМ) в ТГФ (21 мл) в атмосфере азота при комнатной температуре. За ходом реакции следили с помощью тонкослойной хроматографии и ВЭЖХ-МС. Спустя 2 ч растворитель удаляли in vacuo, после чего pH доводили до 1.5, используя 0.1М раствор HCl. Преципитат интенсивно перемешивали в течение 15 мин до того как выделить его вакуум-фильтрацией. Собранный твердый остаток промывали водой, а затем МТВЕ, оставляли на воздухе и затем сушили in vacuo при 50°C, что давало в итоге 870 мг (97%) продукта в виде розового твердого остатка (Температура плавления = 125-126°C; Инфракрасный спектр=1729 cm-1 ВЭЖХ время удерживания = 10.80 min 99.3%).
1H ЯМР (400 МГц; ДМСО): 2.29 (3H, s) 4.56 (2H, s) 4.97 (2H, s) 6.85-6.91 (2H, m) 7.37-7.7.45 (2H, m) 7.47 (1H, dd) 7.51-7.57 (2H, m) 8.06-8.15 (2H, m) 8.36 (1H, dd).
13C ЯМР (100 МГц; ДМСО): 10.5 (q) 25.0 (t) 45.5 (t) 102.7 (d) 102.9 (d) 107.7 (s) 107.8 (s) 108.8 (d).109.1 (d) 110.9 (d) 111.0 (d) 117.1 (d) 117.3 (d) 128.1 (d) 128.2 (d) 128.3 (d) 132.7 (d) 132.8 (d) 133.8 (d) 135.5 (s) 136.8 (s) 138.1 (s) 140.4 (d) 147.0 (d) 155.9 (s) 156.6 (s) 158.9 (s) 164.6 (s) 167.1 (s) 171.1 (s).
Пример 3 - Получение [3-({2-[(4-хлорбензол)сульфонил]пиридин-3-ил}метил)-5-фтор-2-метилиндол-1-ил]-уксусной кислоты (Соединение 3)
2-(4-хлорбензолсульфонил)-пиридин-3-карбоксальдегид

2-хлор-3-пиридинкарбоксальдегид (5.0 г, 35.0 мМ) и натриевую соль 4-хлорбензосульфоновой кислоты (7.75 г, 38.8 мМ) растворяли в ДМСО (120 мл) и эту смесь нагревали при 100°C 72 ч в атмосфере азота. После охлаждения до комнатной температуры смесь разбавляли, водой (500 мл) и экстрагировали этилацетатом (3х). Объединенные вытяжки промывали водой, солевым раствором, затем высушивали (MgSO4) и выпаривали до сухого остатка, получая в итоге 8.1 г исходного материала. Полученное соединение адсорбировали на силикагеле и очищали колоночной хроматографией DPS (dry pad suction), элюируя гептаном с использованием градиента этилацетата, с получением 4.62 г (61%) целевого компонента в виде белого порошкообразного продукта (Температура плавления =100.5-101°C; Инфракрасный спектр =1698 cm-1; ВЭЖХ время удерживания =8.00 min>99%).
1H ЯМР (400 МГц; CDCl3): 7.56 (1H, dd) 7.60 (1H, dd) 7.99 (1H, dd) 8.38 (1H, dd) 8.67 (1Н, dd) 11.1 (1Н, s).
13C ЯМР (100 МГц, CDCl3): 127.3 (d) 129.6 (d) 130.8 (s) 131.1 (d) 136.5 (s) 138.0 (d) 141.4 (s) 152.5 (d) 159.5 (s) 188.4 (d).
этиловый эфир 3-((2-[(4-хлорбензол)сульфонил]пиридин-3-ил}метил)-5-фтор-2-метилиндол-1-ил]-уксусной кислоты
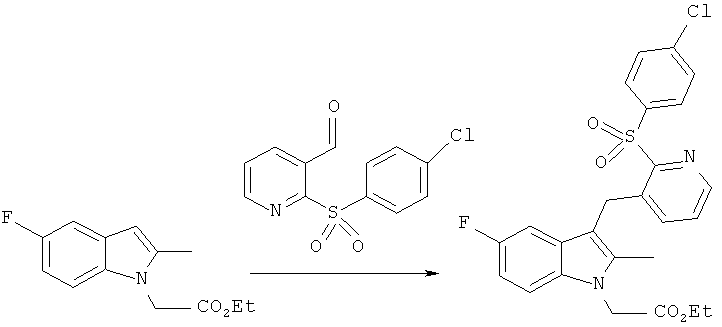
Раствор этилового эфира (5-фтор-2-метил-индол-1-ил)-уксусной кислоты (1.0 г, 4.25 мМ) и 2-(4-хлорбензолсульфонил)-пиридин-3-карбоксальдегида в безводном DCM (50 мл) добавляли в течение 5-10 мин к перемешиваемому раствору TMSOTf в безводном ДХМ (15 мл) при 0°C. Смесь оставляли на 15 мин перед одномоментным добавлением чистого триэтилсилана (2.05 мл, 12.7 мМ). Далее смесь перемешивали еще 15 часов, прекращали охлаждать, и она нагревалась до температуры окружающей среды. Реакцию останавливали капельным добавлением насыщенного раствора NaHCO3 (10 мл) и двухфазную смесь экстрагировали ДХМ (2×50 мл). Объединенные вытяжки промывали солевым раствором (50 мл), затем высушивали (MgSO4) и выпаривали до сухого остатка. Эту реакцию повторяли в том же масштабе и два полученных продукта объединяли. Полученный материал очищали колоночной хроматографией, используя гептан и градиент этилацетата, выход целевого компонента составил 1.80 г (42%) в виде бледно оранжевого твердого остатка (Температура плавления =124.6-124.9°C; ИК-спектр =1741 cm-1; ВЭЖХ время удерживания = 12.75 min 97.3%).
1H ЯМР (400 МГц; CDCl3): 1.26 (3h, t) 2.29 (3H, s) 4.20 (2H, q) 4.62 (2H, s) 4.80 (2H, s) 6.80 (1H, dd) 6.87 (1H, ddd) 7.10 (1H, dd) 7.19 (1H, dd) 7.37 (1H, dd) 7.54 (2Н, dd) 8.00 (2Н, dd)8.28(1H, dd).
13С ЯМР (100 МГц, CDCl3): 10.4 (q) 14.3 (q) 25.3 (t) 45.2 (t) 61.9 (t) 103.4 (d) 103.6 (d) 108.0 (s) 108.1 (s) 109.2 (d) 109.2 (d) 109.5 (d) 109.8 (d) 127.0 (d) 128.5 (s) 128.6 (s) 129.3 (d) 130.8 (d) 133.3 (s) 136.4 (s) 136.6 (s) 137.6 (s) 139.4 (d) 140.5 (s) 146.2 (d) 156.1 (s) 157.0 (s) 159.4 (s) 168.6 (s).
3-({2-[(4-хлорбензол)сульфонил]пиридин-3-ил)метил)-5-фтор-2-метилиндол-1-ил]-уксусная кислота (Соединение 3)
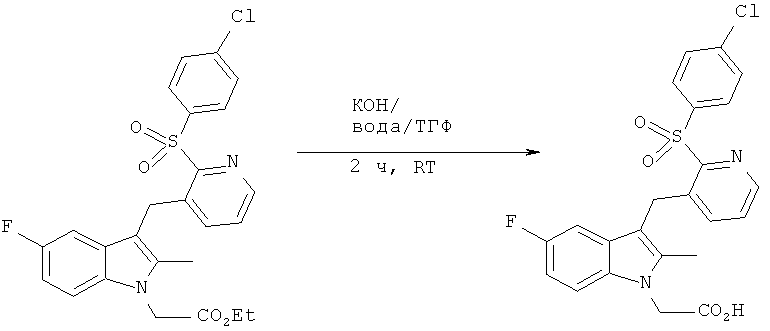
КОН (0.60. г, 10.7 мМ), растворенный в воде (14 мл), добавляли к интенсивно перемешиваемому раствору этилового эфира 3-({2-[(4-хлорбензол)сульфонил]пиридин-3-ил}метил)-5-фтор-2-метилиндол-1-ил]-уксусной кислоты (1.17 г, 3.49 мМ) в ТГФ (21 мл) в атмосфере азота при комнатной температуре. За ходом реакции следили с помощью тонкослойной хроматографии и ВЭЖХ-МС. Спустя 2 ч растворитель удалили in vacuo, после чего pH доводили до 1.5, используя 0.1М раствор HCl. Преципитат интенсивно перемешивали в течение 15 мин до того как выделить его вакуум-фильтрацией. Собранный твердый остаток промывали водой, а затем МТВЕ, оставляли на воздухе и затем сушили in vacuo при 50°C, что давало в итоге 1.31 г (78%) продукта, указанного в заглавии, в виде розового твердого остатка (Температура плавления = 125.2-126°C; Инфракрасный спектр = 1729 cm-1; ВЭЖХ время удерживания = 11.37 мин >99%).
1H ЯМР (400 МГц; ДМСО): 2.29 (3Н, s) 4.56 (2Н, s) 4.96 (2Н, s) 6.85-6.91 (2Н, m) 7.39 (1Н, dd) 7.44 (1Н, dd) 7.49 (1Н, dd) 7.76-7.79 (2Н, m) 8.00-8.8.03 (2Н, m) 8.36 (1Н, dd).
13C ЯМР (100 МГц, ДМСО): 10.5 (q) 25.0 (t) 45.6 (t) 102.7 (d) 102.9 (d) 107.6 (s) 107.7 (s) 108.8 (d) 109.0 (d) 110.9 (d) 111.0 (d) 128.1 (d) 130.0 (d) 131.4 (d) 133.9 (d) 136.9 (s) 138.1 (d) 139.8 (s) 140.5 (s) 147.1 (d) 155.7 (s) 156.5 (s) 158.9 (s) 171.1 (s).
В следующих примерах, Соединения 1-3 были протестированы по отношению к следующим соединениям сравнения:
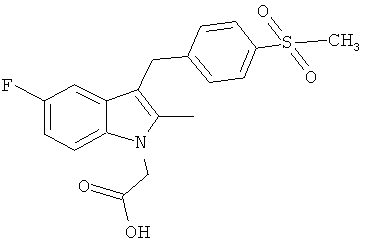
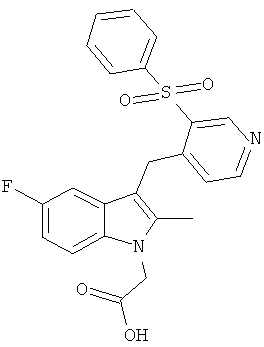
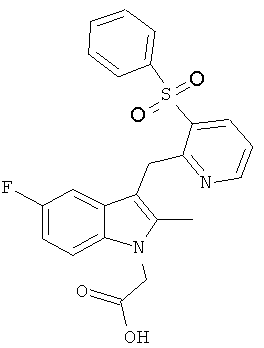
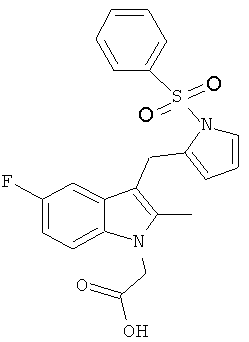
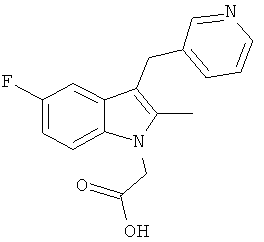
Соединения B, C и E были приготовлены с использованием способа аналогичному способу получения Соединения 1. Соединение A является Соединением 17 в WO 2005/044260 и способ получения этого соединения изложен в Примере 1 указанного документа. Соединение D является Соединением 1 в WO 2006/095183 и способ его получения изложен в Примере 1 указанного документа.
Пример 4 - Измерение активности CRTH2 антагонистов
Материалы и методы
Материалы
Моно-поли разрешащая среда (M-PRM) была получена от Dainippon Pharmaceuticals (Osaka, Japan). MACS анти-CD16 микрочастицы от Miltenyi biotec (Bisley, Surrey). Планшеты ChemoTx были закуплены у Neuroprobe (Gaithersburg, MD). 96-луночные планшеты, покрытые поли-D-лизином были получены от Greiner (Gloucestershire, UK). [3H]PGD2 от Amersham Biosciences (Buckinghamshire, UK). [3H]SQ29548 был закуплен у Perkin Elmer Life Sciences (Buckinghamshire, UK). Все другие реагенты были от Sigma- Aldrich (Dorset, UK), если не указано другое.
Методы
Клеточные культуры
Яйцеклетки китайского хомячка трансфецировали CRTH2 или DP рецепторами (CHO/CRTH2 и CHO/DP) и выращивали в культуре в увлажненной атмосфере при 37°C (5% CO2) в минимальной необходимой среде (MEM) с добавкой 10% эмбриональной бычьей сыворотки, 2 мМ глутамина и 1 мг/мл активного G418. Клетки пересевали каждые 2-3 дня. Для анализа связывания меченого изотопом лиганда клетки были приготовлены в трехслойных колбах или в 175 см2 квадратных флаконах (для приготовления мембран).
Приготовление клеточных мембран
Мембраны получали или из клеток CHO/CRTH2 и CHO/DP, или из тромбоцитов (как источника TP рецепторов). Выросшие до состояния конфлюэнтности клетки СНО промывали PBS и отделяли от подложки, используя раствор Versene (15 мл на колбу). Когда клетки выращивали в 175 см2 плоских колбах, их собирали скрэппингом в PBS. Клеточные суспезии центрифугировали (1700 об/мин, 10 мин, 4°C) и ресуспендировали в 15 мл буфера (1×HBSS, с добавкой 10 мМ HEPES, pH 7.3). Клеточные суспензии затем гомогенизировали с помощью Ultra Turrax в режиме 4-6 в течение 20 секунд. Гомогенизат центрифугировали на 1700 об/мин 10 мин, супернатант собирали и центрифугировали на 20000 об/мин 1 час при 4°C. Полученный осадок ресуспедировали в буфере и хранили на - 80°C в аликвотах по 200-500 мкл. Концентрация белка определяли по Bradford (1976), используя бычий сывороточный альбумин в качестве стандарта. Осадок промывали центрифугированием на 600xg в течение 10 мин, ресуспедировали в ледяном аналитическом буфере (10 мМ Tris-HCl, рН 7.4, 5 мМ глюкозы, 120 мМ NaCl, 10 мкМ индометацина) и сразу же центрифугировали на 20000 об/мин 30 мин при 4°C. Полученный осадок обрабатывали, как описано выше.
Радиолигандный анализ
Эксперименты по связыванию [3H]PGD2 (160 С//мМ) проводили на мембранах, приготовленных, как описано выше. Анализ проводили в конечном объеме буфера 100 мкл (1XHBSS/HEPES 10 мМ, рН 7.3). Клеточные мембраны (15 мкг) предварительно инкубировали при комнатной температуре с различными концентрациями конкурирующего лиганда в течение 15 минут. Затем добавляли [3H]PGD2 и инкубацию продолжали еще час при комнатной температуре. Реакцию прекращали добавлением в каждую лунку ледяного аналитического буфера, зачем следовала быстрая фильтрация через стеклянные волокнистые фильтры Whatman GF/B с помощью Unifilter Cell harvester (PerkinElmer Life Sciences) и шесть промывок по 300 мкл ледяного буфера. Планшеты Unifilter высушивали при комнатной температуре по меньшей мере в течение 1 ч и остаточную радиоактивность на фильтрах определяли счетчиком Beta Trilux (PerkinElmer Life Sciences) после добавления 40 мкл жидкого сцинтиллятора Optiphase Hi-Safe 3 (Wallac). Неспецифическое связывание определяли в присутствие 10 мкл немеченого PGD2. Анализы проводили в параллелях.
Результаты экспериментов по связыванию радиолиганда с CRTH2 приведены в Таблице 1.
Соединения C и E связывались с CRTH2 рецептором очень слабо и поэтому в дальнейшем не тестировались.
Пример 5 - Тест изменения формы эозинофилов цельной крови человека
Тестировали действие соединений 1-3 на индуцированное PGD2 изменение формы эозинофилов и сравнивали их действие с соединениями сравнения A, B и D.
Методы
Тест изменения формы в цельной крови
Соединения (1 мкл, 200 × конечная концентрация) добавляли непосредственно к 200 мкл цельной крови, тщательно смешивали и инкубировали в течение 15 мин, 37°C, 5% CO2. По прошествии указанного времени, форму клеток фиксировали добавлением 300 мкл буфера Cytofix™ (BD Biosciences), 15 мин на льду. К зафиксированным клеткам добавляли 10 мл лизирующего буфера RBC, инкубировали 5 мин при комнатной температуре и центрифугировали, 300×g в течение 5 мин. Удаляли супернатант (содержащий лизированные эритроциты) и проводили лизис повторно. Лейкоциты ресуспензировали в 250 мкл RPMI/10% FCS и анализировали изменение формы с помощью флуоресцентно-активированной клеточной сортировки (FACS). Эозинофилы выделяли на основании их автофлуоресценции и считывали сигналы от 2000 эозинофилов в каждой пробе. Данные анализировали трижды.
Результаты теста изменения формы эозинофилов приведены в Таблице 2.
Наиболее подходящие для применения в качестве фармацевтических субстанций соединения имеют в тесте изменения формы эозинофилов значения IC50 около 1 и 10 нМ. Поэтому, хотя Соединение В специфически связывается с рецептором CRTH2 (Таблица 1), оно не является особенно эффективным антагонистом CRTH2 при физиологических условиях.
Особо следует отметить, что соединения сравнения, являющиеся наиболее близкими по структуре Соединению 1, это Соединения B и C. Из них Соединение C не связывается с рецептором CRTH2 специфичным образом, а Соединение В намного менее активно, чем Соединение 1.
Пример 6 - микросомальная стабильность
Микросомальную стабильность тестируемых соединений определяли следующим способом.
Тестируемое соединение в концентрации 1 мкМ инкубировали с микросомами из печени человека (общая концентрация белка 0.3 мг/мл) в течение 60 мин. Измеряли процентное содержание тестируемого соединения, оставшегося в пробе, по прошествии 1 часа для определения скорости метаболизма тестируемого соединения. Результаты приведены в Таблице 3, в которой представлены результаты двух экспериментов и полученное среднее значение.
Результаты, представленные в Таблице 3 демонстрируют, что после 120 минут, 96% Соединения 1 и 98% Соединения 2 не подвергались метаболическим превращениям в микросомах печени человека. Эти данные сравниваются со значениями, соответственно, 69% и 90% для Соединений B и C, которые являются региональными изомерами Соединения 1 и значением всего лишь 24% для Соединения D.
Таким образом, описанные в данных примерах эксперименты в целом демонстрируют, что Соединения C и E сильно не связываются с рецептором CRTH2, Соединение В намного менее активно, чем Соединение 1 в тесте изменения формы эозинофилов цельной крови, а Соединение D обладает низкой стабильностью в микросомах человека, что ограничивает его применимость в качестве фармацевтической субстанции. Соединения 1-3 проявляют неожиданно высокую активность в качестве антагонистов CRTH2 и неожиданную стабильность по сравнению с соединениями, с которыми они структурно наиболее тесно связаны. Более того, соединения согласно настоящему изобретению обладают значительно улучшенными фармакокинетическими профилями in vivo у собак по сравнению с соединением А. Периоды полувыведения в плазме для Соединений 1 и 2 составили, соответственно, 3 и 5 часов, тогда как Соединение А имело период полувыведения всего лишь 1 час.
Изобретение относится к соединению общей формулы (I) и соединению общей формулы (II)
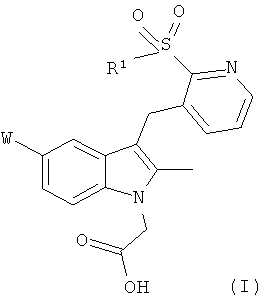
где W представляет собой хлор или фтор, R1 представляет собой фенил, возможно замещенный одним или более галогеновыми заместителями; и R4 представляет собой C1-C6алкил, или фармацевтически приемлемые соли указанных соединений. Кроме того, изобретение относится к способу получения соединения формулы II, к применению соединений I и II для получения препарата для лечения или профилактики таких заболеваний, как астма, аллергический ринит и атопический дерматит. Также описан способ лечения указанных заболеваний, а кроме того, фармацевтическая композиция, применяемая для лечения аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит. Технический результат: получены и описаны новые соединения, которые могут найти свое применение в лечении аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит. 7 н. и 8 з.п. ф-лы, 6 пр., 3 табл.
1. Соединение общей формулы (I)
где W представляет собой хлор или фтор,
R1 представляет собой фенил, возможно замещенный одним или более галогеновыми заместителями;
или фармацевтически приемлемая соль указанного соединения.
2. Соединение общей формулы (II):
где W и R1 определены так же, как и в п.1; а
R4 представляет собой C1-C6алкил,
или фармацевтически приемлемая соль указанного соединения.
3. Соединение по п.1 или 2, отличающееся тем, что W представляет собой фтор.
4. Соединение по п.1 или 2, отличающееся тем, что фенильная группа R1 является незамещенной или содержит один заместитель, который представляет собой галоген.
5. Соединение по п.4, отличающееся тем, что указанный галоген представляет собой фтор или хлор.
6. Соединение по п.5, отличающееся тем, что указанный заместитель фтор или хлор находится в положении 4 фенильной группы R1.
7. Соединение по п.1, выбранное из:
(3-{[2-(бензолсульфонил)пиридин-3-ил]метил}-5-фтор-2-метилиндол-1-ил)-уксусной кислоты;
[5-фтор-3-({2-[(4-фторбензол)сульфонил]пиридин-3-ил}метил)-2-метилиндол-1-ил]-уксусной кислоты;
[3-({2-[(4-хлорбензол)сульфонил]пиридин-3-ил}метил)-5-фтор-2-метилиндол-1-ил]-уксусной кислоты;
или фармацевтически приемлемая соль указанного соединения.
8. Соединение по любому из пп.1-2, 5-7 для применения для лечения и профилактики аллергической астмы, круглогодичного аллергического ринита, сезонного аллергического ринита, атопического дерматита, повышенной контактной чувствительности (включая контактный дерматит), конъюктивита, в частности аллергического конъюктивита, весеннего кератоконъюктивита и атопического кератоконъюктивита, эозинофилического бронхита, пищевой аллергии, эозинофилического гастроэнтерита, воспалительного заболевания кишечника, язвенного колита и болезни Крона, мастоцитоза, синдрома повышенного иммуноглобулина Е, рассеянного склероза, хронического обструктивного заболевания легких, фиброза, включая идиопатический легочный фиброз, назального полипоза, крапивницы, эзофагита, мастоцитоза и синдрома Черга-Страусса.
9. Применение соединения по любому из пп.1-8 для получения препарата для лечения или профилактики аллергической астмы, круглогодичного аллергического ринита, сезонного аллергического ринита, атопического дерматита, повышенной контактной чувствительности (включая контактный дерматит), конъюктивита, в частности аллергического конъюктивита, весеннего кератоконъюктивита и атопического кератоконъюктивита, эозинофилического бронхита, пищевой аллергии, эозинофилического гастроэнтерита, воспалительного заболевания кишечника, язвенного колита и болезни Крона, мастоцитоза, синдрома повышенного иммуноглобулина Е, рассеянного склероза, хронического обструктивного заболевания легких, фиброза, включая идиопатический легочный фиброз, назального полипоза, крапивницы, эзофагита, мастоцитоза и синдрома Черга-Страусса.
10. Способ получения соединения общей формулы (I) по любому из пп.1 или 3-8, включающий осуществление реакции соединения общей формулы (II):
где W и R1 такие, как определено в п.1; и
R4 представляет собой C1-C6алкил;
с основанием.
11. Способ лечения или профилактики заболевания или состояния, опосредованного PGD2 или другими агонистами CRTH2 рецептора, включающий введение в организм пациента, нуждающегося в подобном лечении, подходящего количества соединения согласно любому из пп.1-8.
12. Способ по п.11, отличающийся тем, что указанное заболевание или патологическое состояние выбрано из группы, состоящей из: аллергической астмы, круглогодичного аллергического ринита, сезонного аллергического ринита, атопического дерматита, повышенной контактной чувствительности (включая контактный дерматит), конъюктивита, в частности аллергического конъюктивита, весеннего кератоконъюктивита и атопического кератоконъюктивита, эозинофилического бронхита, пищевой аллергии, эозинофилического гастроэнтерита, воспалительного заболевания кишечника, язвенного колита и болезни Крона, мастоцитоза, синдрома повышенного иммуноглобулина Е, рассеянного склероза, хронического обструктивного заболевания легких, фиброза, включая идиопатический легочный фиброз, назального полипоза, крапивницы, эзофагита, мастоцитоза и синдрома Черга-Страусса.
13. Фармацевтическая композиция для лечения или профилактики аллергической астмы, круглогодичного аллергического ринита, сезонного аллергического ринита, атопического дерматита, повышенной контактной чувствительности, включая контактный дерматит, конъюктивита, в частности аллергического конъюктивита, весеннего кератоконъюктивита и атопического кератоконъюктивита, эозинофилического бронхита, пищевой аллергии, эозинофилического гастроэнтерита, воспалительного заболевания кишечника, язвенного колита и болезни Крона, мастоцитоза, синдрома повышенного иммуноглобулина Е, рассеянного склероза, хронического обструктивного заболевания легких, фиброза, включая идиопатический легочный фиброз, назального полипоза, крапивницы, эзофагита, мастоцитоза и синдрома Черга-Страусса, содержащая соединение по любому из пп.1-8 совместно с фармацевтическим эксципиентом или наполнителем.
14. Фармацевтическая композиция по п.13, приготовленная в форме для перорального, ректального, назального, бронхиального (ингаляция), местного (включая глазные капли, буккальное и сублингвальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения.
15. Способ получения фармацевтической композиции по любому из пп.13 и 14, который включает приведение соединения по любому из пп.1-8 в сочетание или в ассоциацию с фармацевтически или ветеринарно приемлемым наполнителем или носителем.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Прибор для исследования электрических свойств почв, грунтов и тому подобных материалов | 1935 |
|
SU54417A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МСР-1 | 2000 |
|
RU2235090C2 |

