ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное раскрытие относится к винилсульфоновому ангидриду, способу его получения и к способу получения винилсульфонилфторида. Более конкретно, данное раскрытие относится к винилсульфоновому ангидриду, который применяют в качестве синтетического промежуточного соединения для синтеза фторированного мономера, который может служить в качестве исходного материала для полимерных электролитов на основе фтора, таких как мембраны для топливных батарей, связывающие полимеры катализа для топливных батарей и мембраны для хлорщелочного электролиза; к способу получения такого винилсульфонового ангидрида; и к способу получения фторированного мономера с применением винилсульфонового ангидрида.
УРОВЕНЬ ТЕХНИКИ
Полимерные электролиты на основе фтора, представленные следующей общей формулой (5), обычно применяют в качестве основных компонентов мембран для топливных батарей, связывающих полимеров катализа для топливных батарей и мембран для хлорщелочного электролиза, и подобных:

(где p является целым числом от 0 до 6, и q является целым числом от 1 до 6).
Хорошо известно, что полимерный электролит на основе фтора, представленный общей формулой (5), может быть получен подверганием сополимера фторированного мономера, представленного следующей общей формулой (6), и тетрафторэтилена (ТФЭ) омылению и обработке кислотой:
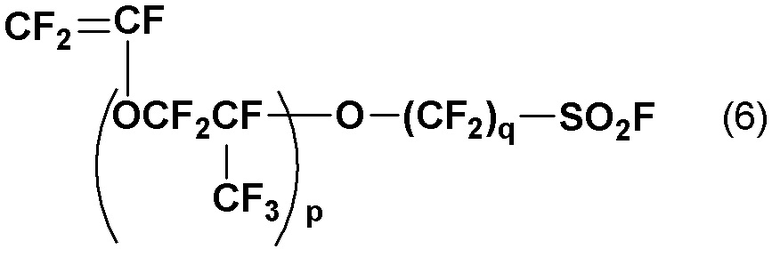
(где p и q такие, как определены в общей формуле (5)).
Среди фторированных мономеров, представленных общей формулой (6), широко применяются полимерные электролиты на основе фтора, полученные из мономеров, где p равно 1 и q равно от 2 до 4. Хорошо известно, что мономеры, где p равно 1 и q равно от 2 до 4 могут быть получены, например, следующим путем:

В это же время, хорошо известно, что полимерные электролиты на основе фтора, где p равно 0 в общей формуле (5), имеют более короткие спейсерные части между основной цепью и группами сульфоновой кислоты, чем в полимерах, где p равно 1 или более, и, таким образом, имеют более высокие температуры стеклования и более высокую механическую прочность, чем полимеры, где p равно 1 или более.
Однако синтез фторированных мономеров, представленных общей формулой (6), которые служат сырьем для таких полимерных электролитов на основе фтора, затруднен, если p равно 0, что является проблемой. Более конкретно, когда CF3CF(COF)O(CF2)qSO2F подвергают декарбоксилированию и винилированию так же, как в указанной выше реакции, где p равно 1 или более, в попытке синтезировать фторированный мономер, где p равно 0 в общей формуле (6), циклизация становится доминирующей. В результате, известно, что выход представляющего интерес фторированного мономера, имеющего короткую структуру цепи, где p равно 0, становится крайне низким. Например, если q равно 2, проходит только циклизация, что затрудняет получение фторированного мономера (см. НПЛ 1, например).
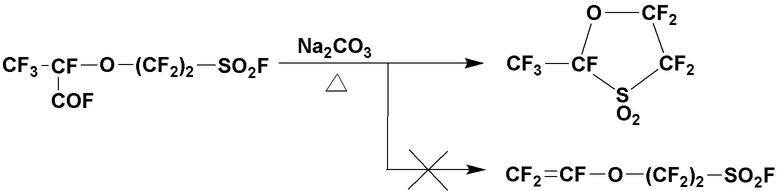
В качестве другого способа синтеза фторированного мономера, где p равно 0 в общей формуле (6), раскрыт способ синтеза с применением фторэпоксида, содержащего атом хлора (см. ПТЛ 1, например). Однако этот способ далек от практического применения, поскольку для него требуется специальный фторэпоксид, содержащий атом хлора, который не является широкодоступным и синтез которого является трудоемким.
Раскрыт еще один способ синтеза фторированного мономера, где p равно 0 в общей формуле (6) (см. ПТЛ 2, например). Более конкретно, 5-членное циклическое соединение получают декарбоксилированием CF3CF(COF)O(CF2)2SO2F при нагревании с карбонатом натрия. 5-членное циклическое соединение затем подвергают реакции с метоксидом натрия (NaOCH3) с получением CF2=CFO(CF2)2SO3Na, который затем подвергают реакции с пентахлоридом фосфора (PCl5) с получением CF2=CFO(CF2)2SO2Cl, с последующей реакцией с фторидом натрия (NaF) с получением фторированного мономера (CF2=CFO(CF2)2SO2F) имеющего p равное 0 и q равное 2 в общей формуле (5).

СПИСОК ЦИТАТ
Патентная литература
ПТЛ 1: JPS57-28024A
ПТЛ 2: US3560568B
Не патентная литература
НПЛ 1: The 155th Committee on Fluorine Chemistry, Japan Society for the Promotion of Science, "Introduction to Fluorine Chemistry 2010: The Frontiers of Basics and Applications," April 2010, pp. 353-355.
НПЛ 2: Gronwald, Oliver, et al. "Synthesis of difluoroethyl perfluorosulfonate monomer and its application." Journal of Fluorine Chemistry 129 (2008) 535-540.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
(Техническая проблема)
Когда CF2=CFO(CF2)2SO3Na смешивают с пентахлоридом фосфора и нагревают, однако, побочные продукты оксихлорида фосфора (POCl3) и хлорида натрия (NaCl), образуются вместе с CF2=CFO(CF2)2SO2Cl в качестве целевого продукта:
CF2=CFO(CF2)2SO3Na+PCl5 → CF2=CFO(CF2)2SO2Cl+POCl3+NaCl.
В ПТЛ 2 описан способ получения фторированного мономера (CF2=CFO(CF2)2SO2F) получением смеси CF2=CFO(CF2)2SO2Cl и оксихлорида фосфора в виде дистиллированной фракции, которую затем повергают реакции с фторидом натрия. Однако это способ далек от того, чтобы модернизироваться до промышленных масштабов по следующим причинам. Когда фторид натрия и CF2=CFO(CF2)2SO2Cl подвергают реакции в присутствии оксихлорида фосфора, образование фторированного мономера может ингибироваться влиянием высокореакционноспособного оксихлорида фосфора. Далее, оксихлорид фосфора может ограничивать условия реакции, такие как растворитель, применяемый во время реакции, и температура реакции. Таким образом, трудоемкие методы требуются для отделения и удаления оксихлорида фосфора из смеси. На этом фоне существует потребность в разработке нового процесса, который позволяет эффективно получать фторированные мономеры без использования пентахлорида фосфора.
(Решение проблемы)
После интенсивного изучения решений вышеупомянутых проблем мы обнаружили, что фторированный мономер и соединение винилсульфоновой кислоты были получены путем взаимодействия винилсульфонового ангидрида, полученного из соединения винилсульфоновой кислоты, и агента ангидридизации и их смешивания с фторирующим агентом, чтобы таким образом обнаружить, что винилсульфоновый ангидрид служит ключевым синтетическим промежуточным соединением, из которого получается фторированный мономер. Кроме того, мы также обнаружили, что соединение винилсульфоновой кислоты, выделенное из реакционной смеси фторированного мономера и соединения винилсульфоновой кислоты, может быть преобразовано агентом ангидридизации обратно в винилсульфоновый ангидрид, который может быть повторно использован в качестве синтетического промежуточного соединения для получения фторированного мономера, тем самым завершая настоящее раскрытие.
Более конкретно, в настоящем раскрытии представлено следующее.
[1] Винилсульфоновый ангидрид, представленный следующей общей формулой (1):

(где m является целым числом от 0 до 3, и n является целым числом от 1 до 6).
[2] Способ получения винилсульфонового ангидрида, представленного следующей общей формулой (1):
(где m является целым числом от 0 до 3, и n является целым числом от 1 до 6),
где способ включает:
стадию (a) получения соединения винилсульфоновой кислоты, представленного следующей общей формулой (2), контактом и смешиванием с агентом ангидридизации:

где m является целым числом от 0 до 3, n является целым числом от 1 до 6 и M является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью).
[3] Способ получения винилсульфонилфторида, включающий:
стадию (b) получения винилсульфонового ангидрида, представленного следующей общей формулой (1), контактом и смешиванием с фторирующим агентом:
(где m является целым числом от 0 до 3 и n является целым числом от 1 до 6)
с получением реакционной смеси, содержащей винилсульфонилфторид, представленный следующей общей формулой (3):
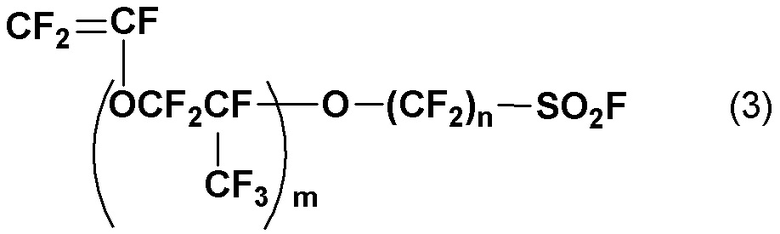
(где m является целым числом от 0 до 3 и n является целым числом от 1 до 6); и
соединение винилсульфоновой кислоты, представленное следующей общей формулой (2):
(где m является целым числом от 0 до 3, n является целым числом от 1 до 6 и M является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью).
[4] Способ получения винилсульфонилфторида по [3], где винилсульфоновым ангидридом является винилсульфоновый ангидрид, полученный способом получения винилсульфонового ангидрида по [2].
[5] Способ получения винилсульфонилфторида по [3] или [4], дополнительно содержащий стадию (c) отделения винилсульфонилфторида, представленного общей формулой (3) и соединения винилсульфоновой кислоты, представленного общей формулой (2), из реакционной смеси, полученной на стадии (b).
[6] Способ получения винилсульфонового ангидрида по [2], где стадия (a) включает контакт и смешивание соединения винилсульфоновой кислоты, представленного общей формулой (2), полученного на стадии (c) по [5] с агентом ангидридизации.
[7] Способ получения винилсульфонового ангидрида по [2] или [6], дополнительно включающий стадию (d) получения соединения винилсульфоновой кислоты, в котором M в общей формуле (2) является щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью, контактом и смешиванием с кислым веществом для превращения в винилсульфоновую кислоту, представленную следующей общей формулой (4):
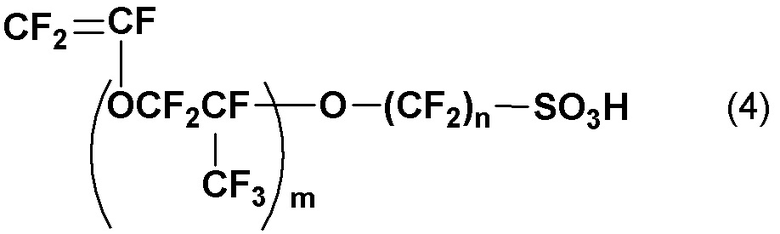
(где m и n такие, как определены в общей формуле (2)).
[8] Способ получения винилсульфонилфторида по любому из [3]-[5], где фторирующим агентом является один или более, выбранные из группы, состоящей из фторводорода, фторида металла, фторида четвертичного аммония и фторида четвертичного фосфония.
[9] Способ получения винилсульфонового ангидрида по любому из [2], [6] и [7], где агентом ангидридизации является один или более, выбранных из группы, состоящей из пентоксида фосфора, уксусного ангидрида, трифторуксусного ангидрида, метансульфонового ангидрида, трифторметансульфонового ангидрида, п-толуолсульфонового ангидрида, тионилхлорида, дициклогексилкарбодиимида, цианурхлорида, тетрахлорида титана и бензолсульфонилхлорида.
[10] Способ получения винилсульфонового ангидрид по [2], где стадия (a) включает стадию отделения винилсульфонового ангидрида, представленного общей формулой (1).
[11] Способ получения винилсульфонового ангидрида по [10], дополнительно включающий стадию, после отделения винилсульфонового ангидрида, представленного общей формулой (1), сбора соединения винилсульфоновой кислоты, представленного общей формулой (2), из полученного остатка.
(Полезный эффект)
В соответствии с настоящим раскрытием может быть предоставлен винилсульфоновый ангидрид, который может использоваться в качестве синтетического промежуточного соединения для синтеза фторированного мономера, который может служить в качестве сырья для полимерного электролита на основе фтора, используемого как мембраны для топливных батарей, связывающие полимеры катализа для топливных батарей и мембраны для хлорщелочного электролиза и подобных. Кроме того, может быть достигнуто эффективное производство такого винилсульфонового ангидрида, и этот винилсульфоновый ангидрид может быть использован для эффективного производства фторированного мономера.
ПОДРОБНОЕ ОПИСАНИЕ
Далее будет подробно описан вариант осуществления настоящего раскрытия (в дальнейшем называемый просто «настоящий вариант осуществления»).
Винилсульфоновым ангидридом данного раскрытия является винилсульфоновый ангидрид, представленный следующей общей формулой (1) (в дальнейшем также может называться "соединение (1)"):
(где m является целым числом от 0 до 3 и n является целым числом от 1 до 6).
Соединение (1) является полезным синтетическим промежуточным соединением, способным давать соединение винилсульфоновой кислоты, представленное указанной ниже общей формулой (2), а также винилсульфонилфторид, представленный указанной ниже общей формулой (3).
В соединении (1), m является целым числом от 0 до 3 и n является целым числом от 1 до 6. С точки зрения эффективности как сырья для полимерных электролитов на основе фтора, таких как мембраны для топливных батарей, связывающие полимеры катализа для топливных батарей и мембраны для хлорщелочного электролиза, полученного из сополимера винилсульфонилфторида, представленного следующей общей формулой (3) (фторированного мономера) и ТФЭ, и простоты синтеза винилсульфонилфторида, представленного следующей общей формулой (3), m равное целому числу от 0 до 2 и n равное целому числу от 2 до 6 является предпочтительным, m равное целому числу от 0 до 1 и n равное целому числу от 2 до 6 является более предпочтительным, и m равное целому числу от 0 до 1 и n равное целому числу от 2 до 4 является наиболее предпочтительным.
Способ получения винилсульфонового ангидрид данного раскрытия включает стадию (a) получения соединения винилсульфоновой кислоты, представленного следующей общей формулой (2) (в дальнейшем также может называться "соединение (2)") контактом и смешиванием с агентом ангидридизации:
(где m является целым числом от 0 до 3, n является целым числом от 1 до 6 и M является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью).
m и n в соединении (2) являются такими же, как m и n в соединении (1), которое получают контактом и смешиванием соединения (2) с агентом ангидридизации.
Способ получения винилсульфонилфторида данного раскрытия включает стадию (b) получения соединения (1) контактом и смешиванием с фторирующим агентом с получением реакционной смеси, включающей винилсульфонилфторид, представленный следующей общей формулой (3) (в дальнейшем также может называться "соединение (3)"):
(где m и n такие, как определены в общей формуле (1)); и
соединение (2).
В настоящем раскрытии, стадия (c) включает отделение каждого из соединения (3) и соединения (2) от реакционной смеси, содержащей соединение (3) и соединение (2), полученной на стадии (b). Сополимер, полученный из соединения (3), полученного на стадии (c) и ТФЭ может применяться для получения полимерных электролитов на основе фтора, таких как мембраны для топливных батарей, связывающие полимеры катализа для топливных батарей и мембраны для хлорщелочного электролиза. Далее, соединение (2), полученное на стадии (c), подвергают контакту и смешиванию с агентом ангидридизации с получением соединения (1), которое может быть повторно использовано в качестве синтетического промежуточного соединения для получения фторированного мономера.
Кроме того, в настоящем раскрытии, в случае, где M в соединении (2) является щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью, включена стадия (d) получения соединения (2) контактом и смешиванием с кислым веществом для превращения в винилсульфоновую кислоту, представленную следующей общей формулой (4):
(где m и n такие, как определены в общей формуле (2)).
Согласно способу получения винилсульфонилфторида настоящего варианта осуществления, стадии (a)-(d) позволяют применять соединение (2) в качестве сырья для получения соединения (3), которое применяют в качестве мономера на основе фтора, в виде соединения (1) в качестве полезного синтетического промежуточного соединения настоящего варианта осуществления, а также позволяет собирать и повторно использовать соединение (2).

<Стадия (a)>
Стадия (a) является стадий получения соединения (1) из соединения (2) и агента ангидридизации.
Способ получения винилсульфонового ангидрида данного раскрытия включает, по меньшей мере, стадию (a).
Методика получения соединения (2) особенно не ограничена, и соединение винилсульфоновой кислоты, где m равно 0 в соединении (2), например, может быть получено реакцией CF3CF(COF)O(CF2)nSO2F и гидроксида натрия с последующим нагреванием и декарбоксилированием с получением соединения (2) (см НПЛ 2, например). Мы также обнаружили другой способ получения соединения (2) термическим разложением карбоксилата щелочного металла, полученного из CF3CF(COF)O(CF2)nSO2F, и карбоната натрия с последующей реакцией с соединением силанола. Кроме того, как будет описано ниже, соединение (2) также может быть получено реакцией соединения (1) с фторирующим агентом (стадия (b)) и отделением каждого из полученных соединения (3) и соединения (2).
Примеры указанных выше агентов ангидридизации включают пентоксид фосфора, уксусный ангидрид, трифторуксусный ангидрид, метансульфоновый ангидрид, трифторметансульфоновый ангидрид, п-толуолсульфоновый ангидрид, тионилхлорид, дициклогексилкарбодиимид, цианурхлорид, тетрахлорид титана и бензолсульфонилхлорид. С точки зрения доступности и высокой управляемости реакции предпочтительны пентоксид фосфора, трифторуксусный ангидрид, трифторметансульфоновый ангидрид, тионилхлорид, цианурхлорид и тетрахлорид титана; пентоксид фосфора, трифторуксусный ангидрид, трифторметансульфоновый ангидрид и тионилхлорид являются более предпочтительными; и пентоксид фосфора или тионилхлорид являются наиболее предпочтительными. Вышеупомянутый агент ангидридизации можно использовать отдельно или в сочетании двух или более.
Агент ангидридизации предпочтительно применяют на стадии (a) в количестве 1 молярного эквивалента или более к 1 молю соединения (2). Для достижения эффективного применения агента ангидридизации и для улучшения производственного выхода соединения (1), агент ангидридизации применяют в количестве, предпочтительно, 1-20 молярных эквивалентов, более предпочтительно, 1-10 молярных эквивалентов, и особенно предпочтительно, 1-5 молярных эквивалентов по отношению к 1 молю соединения (2).
На стадии (a) реакция может проводиться в присутствии растворителя или в условиях без растворителя.
В случае, где стадию (a) проводят в присутствии растворителя, предпочтительными растворителями являются растворители, инертные к сырью и продуктам реакции. Среди них надлежащим образом применяют растворитель на основе нитрила и растворитель на основе фтора.
Любой из обычно используемых растворителей на основе нитрила может быть использован без каких-либо конкретных ограничений, и примеры растворителя на основе нитрила включают алкилнитрилы, имеющие, по меньшей мере, одну нитрильную группу в качестве заместителя в насыщенном углеводородном скелете, такие как ацетонитрил, пропионитрил, бутиронитрил и адипонитрил. Эти соединения можно использовать по отдельности или в комбинации двух или более. Среди растворителей на основе нитрила предпочтительными являются ацетонитрил и пропионитрил, а более предпочтительным является ацетонитрил. Это связано с тем, что их более низкие температуры кипения выгодны для повышения рентабельности стадии, когда отгонка растворителя требуется на любой стадии или на двух или более стадиях до, во время и после реакции. Кроме того, в случае, когда время реакции может быть сокращено, чтобы тем самым сделать стадию более рентабельной, для повышения температуры реакции предпочтительно используется растворитель на основе нитрила, имеющий высокую температуру кипения, и более предпочтителен адипонитрил.
Любой из обычно используемых растворителей на основе фтора может использоваться без каких-либо конкретных ограничений, и примеры растворителей на основе фтора включают фторалкилсульфоновые ангидриды (CmF2m+1SO2)2O (m=1-10), сложные эфиры фторалкилсульфоновой кислоты CmF2m+1SO2OCnF2n+1 (m=1-10 и n=1-10), перфторалканы CmF2m+2 (m=4-20), перфторалкиламины (CmF2m+1)3N (m=2-10) и перфторполиэфиры. С точки зрения легкости сбора винилсульфонового ангидрида после реакции особенно предпочтительны фторалкилсульфоновый ангидрид, перфторалканы и перфторполиэфиры. Можно использовать один растворитель или можно использовать два или более растворителя в комбинации.
В случае, где стадию (a) проводят в присутствии растворителя, температура реакции обычно составляет от -40°C до 250°C. С точки зрения тепловой стабильности соединения (1) и соединения (2) и объема выпуска соединения (1), температура реакции предпочтительно составляет от -20°C до 230°C, более предпочтительно, от 0°C до 200°C, и особенно предпочтительно, от 10°C до 150°C.
В случае, где стадию (a) проводят в присутствии растворителя, время реакции обычно составляет от 0,01 до 100 часов и, предпочтительно, от 0,1 до 80 часов.
В случае, где стадию (a) проводят в условиях без растворителя, температура реакции обычно составляет от -40°C до 300°C. С точки зрения тепловой стабильности соединения (1) и соединения (2) и объема выпуска соединения (1), температура реакции предпочтительно составляет от -20°C до 280°C, более предпочтительно, от 0°C до 250°C и особенно предпочтительно, от 10°C до 200°C.
В случае, где стадию (a) проводят в условиях без растворителя, время реакции обычно составляет от 0,01 до 200 часов и, предпочтительно, от 0,1 до 180 часов.
Атмосферой реакции на стадии (a) может быть любая из широко используемых атмосфер без каких-либо конкретных ограничений, независимо от того, проводится ли реакция в условиях без растворителя или в присутствии растворителя. Обычно используются воздушная атмосфера, атмосфера азота, атмосфера аргона и подобные. Среди них предпочтительны атмосфера азота и атмосфера аргона, поскольку, в некоторых случаях можно подавить образование побочных продуктов окислением. Кроме того, более предпочтительна атмосфера азота, поскольку она имеет тенденцию быть превосходной по рентабельности.
Реакционное давление на стадии (а) особенно не ограничено независимо от того, проводится ли реакция в условиях без растворителя или в присутствии растворителя, и реакция проводится при атмосферном давлении, повышенном давлении или пониженном давлении. В случае, когда присутствует соединение, которое может быть летучим, и желательно предотвращение улетучивания, повышенное давление выше атмосферного давления является эффективным. Наоборот, в случае, когда желательно удаление летучего компонента из смеси, эффективным является пониженное давление ниже атмосферного.
На стадии (а) можно использовать любую из обычно используемых методик контакта и смешивания без каких-либо конкретных ограничений, независимо от того, проводится ли реакция в условиях без растворителя или в присутствии растворителя. Типовые методики включают методики с помощью перемешивающей лопасти (например, вентилятора, пропеллера, крестовины, бабочки, складывающейся крыльчатки, турбины, дисковой турбины, диспергатора, лопасти, наклонной лопасти или винтовой лопасти); методики с помощью шлифовальной машины (например, щековой дробилки, гирационной дробилки, ударной дробилки, конусной дробилки, валковой дробилки, фрезерной мельницы, штамповой мельницы, кольцевой мельницы, валковой мельницы, роторной мельницы, вибрационной мельницы, планетарной мельницы, молотковой мельницы, бисерной мельницы, дезинтегратора или штифтовой мельницы); и методики с помощью резонанса (например, резонансного акустического смесителя). Эти методики можно использовать по отдельности или в сочетании двух или более. Кроме того, может использоваться измельчающая среда, обычно используемая в шаровой мельнице или подобных, такая как дробь из оксида алюминия, стеклянная дробь, дробь из диоксида циркония, дробь из оксида циркония и стальная дробь, поскольку они могут повысить эффективность контакта и смешивания (и дробь может быть шариками). Эту измельчающую среду можно использовать по отдельности или в комбинации из двух или более.
Любая из методик отделения и сбора соединения (1) из полученной реакционной смеси на стадии (а) может использоваться без каких-либо конкретных ограничений, независимо от того, проводится ли реакция в условиях без растворителя или в присутствии растворителя. Примеры методик включают извлечение соединения (1) из реакционной смеси с использованием растворителя; и отделение соединения (1) от смеси во время реакции и/или после реакции дистилляцией.
В качестве добавляемого растворителя предпочтительны растворители, инертные к соединению (1). Среди них подходящими являются растворитель на основе нитрила и растворитель на основе фтора. Любой из обычно используемых растворителей на основе нитрила может быть использован без каких-либо конкретных ограничений, и примеры растворителя на основе нитрила включают алкилнитрилы, имеющие, по меньшей мере, одну нитрильную группу в качестве заместителя в насыщенном углеводородном скелете, такие как ацетонитрил, пропионитрил, бутиронитрил. и адипонитрил. Любой из обычно используемых растворителей на основе фтора может быть использован без каких-либо конкретных ограничений, и примеры растворителя на основе фтора включают перфторалканы CmF2m+2 (m=4-20), перфторбензол, перфтортолуол, перфтор(2-бутилтетрагидрофуран), 2H,3H-перфторпентан, перфторалкиламины (CmF2m+1)3N (m=2-10) и перфторполиэфиры. Можно использовать один растворитель или можно использовать два или более растворителя в комбинации. Эти растворители можно использовать по отдельности или в комбинации двух или более.
В случае, когда реакцию проводят на стадии (а) в условиях без растворителя, например, соединение (1) можно отогнать дистилляцией путем нагревания полученной реакционной смеси при одновременном снижении давления внутри реакторной системы.
В случае, когда реакцию проводят с использованием растворителя на стадии (а), например, соединение (1) можно отделить от растворителя путем удаления любых нерастворимых компонентов в полученной реакционной смеси путем фильтрации или подобного с последующим нагреванием собранного фильтрата для отгонки растворителя и соединения (1) дистилляцией. Отделенный растворитель можно повторно использовать на стадии (а).
После отделения и сбора соединения (1) на стадии (а), соединение (2) можно собрать из полученного остатка. Любая методика сбора может использоваться без каких-либо особых ограничений. Типовые методики включают контакт остатка, полученного после разделения и сбора соединения (1) с растворителем для экстракции соединения (2), тем самым собирая соединение (2); отделение и сбор соединения (1) дистилляцией и сбор соединения (2) путем повышения температуры и/или снижения давления; и контакт остатка, полученного после разделения и сбора соединения (1) с растворителем, способным растворять остаток, с получением раствора, который смешивают с растворителем, имеющим более низкую совместимость с раствором для экстракции соединения (2), тем самым собирая соединение (2), например.
Растворителем, способным растворять остаток, полученный после разделения и сбора соединения (1), является, но не ограничен ими, предпочтительно, растворитель, имеющий гидроксильную группу и, более предпочтительно, спирт, такой как метанол, этанол или пропанол, или вода, и даже более предпочтительно, вода. Эти растворители можно использовать по отдельности или в комбинации двух или более. Кроме того, могут быть добавлены соединения, растворимые в этих растворителях. Примеры растворимого соединения включают, но не ограничиваются ими, сульфат натрия, гидросульфат натрия, фосфат натрия, гидрофосфат динатрия, дигидрофосфат натрия, гидрокарбонат натрия, карбонат натрия, хлорид натрия, хлорид калия, нитрат натрия и нитрат калия. Эти соединения можно использовать по отдельности или в комбинации двух или более.
Примеры растворителя, имеющего более низкую совместимость с растворителем, способным растворять остаток, полученный после разделения и сбора соединения (1), включают, но не особо ограничиваются ими, алифатические углеводороды, такие как пентан, гексан и гептан; ароматические углеводороды, такие как бензол, толуол, ксилол, гексафторбензол и бензотрифторид; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, тетрагидрофуран, метилтетрагидрофуран, диметоксиэтан, циклопентилметиловый эфир и 4-метилтетрагидропиран; и сложные эфиры, такие как этилацетат и бутилацетат. Среди них предпочтительными являются простые эфиры, и более предпочтительны диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, циклопентилметиловый эфир и 4-метилтетрагидропиран. Эти растворители можно использовать по отдельности или в комбинации двух или более.
<Стадия (b)>
Стадия (b) является стадией контакта и смешивания соединения (1) с фторирующим агентом с получением реакционной смеси, содержащей соединение (2) и соединение (3).
Способ получения винилсульфонилфторида настоящего варианта осуществления включает, по меньшей мере, стадию (b).
Соединением (1) на стадии (b) предпочтительно является соединение (1), полученное указанным выше способом получения винилсульфонового ангидрида этого варианта осуществления, и соединение, полученное способом получения винилсульфонового ангидрид может непрерывно использоваться на этой стадии (b) или может использоваться на стадии (b) после хранения.
На стадии (b), дополнительное соединение может быть включено в пределах интервала, не ингибирующего реакцию. Например, может применяться смесь соединения (1) и соединения (2), полученная на стадии (a).
Примеры вышеупомянутого фторирующего агента включают фторводород, фторид металла, фторид четвертичного аммония и фторид четвертичного фосфония и, по меньшей мере, может быть выбран один из фторводорода, фторида металла, фторида четвертичного аммония и фторида четвертичного фосфония.
В случае, когда фторводород используется в качестве фторирующего агента, фторводород может быть подвергнут реакции один или в присутствии органического основания. В случае использования органического основания можно использовать следующие:
- первичные амины, такие как метиламин, этиламин, изопропиламин и бутиламин;
- вторичные амины, такие как диметиламин, диэтиламин, диизопропиламин и морфолин;
- третичные амины, такие как триметиламин, триэтиламин и диизопропилэтиламин; и
- азотсодержащие ароматические гетероциклические соединения, такие как пиридин, 2,6-лутидин, имидазол и хинолин.
Эти органические основания можно использовать по отдельности или в сочетании двух или более органических оснований.
В случае, когда в качестве фторирующего агента используется фторид металла, примеры металла включают щелочные металлы (Li, Na, K, Rb и Cs), щелочноземельные металлы (Mg и Ca) и Ag. Конкретные примеры фторида металла включают LiF, NaF, KF, RbF, CsF, MgF2, CaF2 и AgF. С точки зрения реакционной способности с соединением (1), фторирующим агентом является, более предпочтительно, по меньшей мере, один или несколько фторидов металлов, выбранных из группы, состоящей из LiF, NaF, KF, RbF, CsF и AgF, и NaF и KF особенно предпочтительны.
В случае, когда фторид четвертичного аммония используется в качестве фторирующего агента, его конкретные примеры включают фторид тетраметиламмония, фторид тетраэтиламмония и фторид тетрабутиламмония.
В случае, когда фторид четвертичного фосфония используется в качестве фторирующего агента, его конкретные примеры включают фторид тетраметилфосфония, фторид тетраэтилфосфония и фторид тетрабутилфосфония.
Вышеупомянутые фторирующие агенты могут использоваться по отдельности или в виде смеси двух или более.
Фторирующий агент применяют на стадии (b) в количестве обычно от 0,95 до 20 молей по отношению к 1 молю соединения (1). тем не менее, если любой неиспользованный фторирующий агент остается после стадии (b), неиспользованный фторирующий агент будет оставаться вместе с соединением (2) после отделения соединения (3). Таким образом, желательно, чтобы неиспользованный фторирующий агент оставался в как можно меньшем количестве после стадии (b). По этой причине фторирующий агент используют в количестве, предпочтительно, от 0,98 до 10 молей, более предпочтительно, от 1 до 3 молей, и особенно предпочтительно, от 1 до 2 молей, по отношению к 1 молю соединения (1).
Стадия (b) может быть проведена в условиях отсутствия растворителя или в присутствии растворителя.
В случае, где стадию (b) проводят в присутствии растворителя, предпочтительными растворителями являются растворители, инертные по отношению к сырью и продуктам реакции. Примеры растворителя включают алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; хлорированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, трет-бутилметиловый эфир, циклопентилметиловый эфир, 4-метилтетрагидропиран, моноглим, диглим, триглим и тетраглим; кетоны, такие как ацетон и метилэтилкетон; сложные эфиры, такие как этилацетат и бутилацетат; амиды, такие как N, N-диметилформамид, N, N-диметилацетамид и N-метилпирролидон; нитрилы, такие как ацетонитрил, пропионитрил, бутиронитрил, изобутиронитрил и 2-метилбутиронитрил; диметилсульфоксид; и воду. Среди них предпочтительны гептан, толуол, хлороформ, моноглим, диглим, триглим, тетраглим, ацетонитрил, пропионитрил, бутиронитрил, изобутиронитрил, 2-метилбутиронитрил и вода; ацетонитрил, пропионитрил, бутиронитрил, изобутиронитрил, 2-метилбутиронитрил и вода являются более предпочтительными; и особенно предпочтителен ацетонитрил. Можно использовать один растворитель или можно использовать два или более растворителя в комбинации.
В случае, где стадию (b) проводят в присутствии растворителя, температура реакции обычно составляет от -40°C до 250°C. С точки зрения тепловой стабильности соединения (1), соединения (2) и соединения (3) и объемов выпуска соединения (2) и соединения (3), температура реакции предпочтительно составляет от -20°C до 200°C, более предпочтительно, от 0°C до 180°C и особенно предпочтительно, от 10°C до 150°C.
В случае, где стадию (b) проводят в присутствии растворителя, время реакции обычно составляет от 0,01 до 50 часов и предпочтительно составляет от 0,1 до 30 часов.
В случае, где стадию (b) проводят в условиях без растворителя, температура реакции обычно составляет от -40°C до 300°C. С точки зрения тепловой стабильности соединения (1), соединения (2) и соединения (3) и объемов выпуска соединения (2) и соединения (3), температура реакции предпочтительно составляет от -20°C до 280°C, более предпочтительно, от 0°C до 250°C и особенно предпочтительно, от 10°C до 200°C.
В случае, где стадию (b) проводят в условиях без растворителя, время реакции обычно составляет от 0,01 до 40 часов и предпочтительно составляет от 0,1 до 20 часов.
M в соединении (2), полученном на стадии (b) является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью, и M может быть соответствующим образом модифицирован в соответствии с описанным выше фторирующим агентом. Например, в случае, когда фторид металла используется в качестве фторирующего агента, M в соединении (2) может быть таким же, как атом металла в этом фториде металла.
<Стадия (c)>
Стадия (c) является стадией отделения соединения (3) от реакционной смеси, содержащей соединение (3) и соединение (2), полученной на вышеупомянутой стадии (b). Любые методики могут применяться для отделения соединения (3) от реакционной смеси.
Способ получения винилсульфонилфторида настоящего варианта осуществления включает стадию (b) и, предпочтительно, также включает стадию (c).
В случае, когда реакцию проводят в присутствии растворителя на стадии (b), например, типовые методики отделения соединения (3) от реакционной смеси включают следующие:
- добавление воды к полученной реакционной смеси и отделение соединения (3) экстракционного растворителя; и
- нагревание полученной реакционной смеси для отделения растворителя и соединения (3) друг от друга дистилляцией.
В случае использования экстракционного растворителя, предпочтительными растворителями являются растворители, которые нерастворимы в воде, инертны по отношению к соединению (3) и способны растворять соединение (3). Примеры растворителя включают алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; хлорированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, трет-бутилметиловый эфир, циклопентилметиловый эфир и 4-метилтетрагидропиран; сложные эфиры, такие как этилацетат и бутилацетат; и растворители на основе фтора, такие как HFC-4310mee, фторалкилсульфоновые ангидриды (CmF2m+1SO2)2O (m=1-10), сложные эфиры фторалкилсульфоновой кислоты CmF2m+1SO2OCnF2n+1 (m=1 до 10 и n=1-10), перфторалканы CmF2m+2 (m=4-20), перфторалкиламины (CmF2m+1)3N (m=2-10) и перфторполиэфиры. Растворители для экстракции можно использовать по отдельности или в сочетании двух или более растворителей.
В случае если реакцию проводят в условиях без растворителя на стадии (b), методика отделения соединения (3) от реакционной смеси может заключаться, например, в удалении соединения (3) из смеси во время и/или после реакции на стадии (b) дистилляцией с отделением соединения (3).
Соединение (3), отделенное на вышеупомянутой стадии, может быть использовано в качестве фторированного мономера как есть для сополимеризации с ТФЭ или другими веществами. Альтернативно, соединение (3) может быть использовано в качестве фторированного мономера для сополимеризации с ТФЭ или другими веществами после того, как соединение (3) промывают водой и, необязательно, очищают дистилляцией для повышения чистоты фторированного мономера.
Соединение (2), отделенное на вышеупомянутой стадии, можно превратить в соединение (1) на стадии (а) или можно превратить в соединение (1) на стадии (d), описанной ниже, и далее через стадию (а).
<Стадия (d)>
Стадия (d) является стадией получения соединения винилсульфоновой кислоты, в котором M в общей формуле (2) является щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью, контактом и смешиванием с кислым веществом для превращения в винилсульфоновую кислоту, представленную следующей общей формулой (4):
(где m и n такие, как определены в общей формуле (2)).
Соединение винилсульфоновой кислоты общей формулы (2), в котором M является щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью, может быть подвергнуто стадии (d), и соединение (4), полученное на стадии (d), можно использовать для получения соединения (1) на стадии (а).
В качестве кислого вещества неорганическая кислота, такая как хлористоводородная кислота, серная кислота, азотная кислота, дымящая серная кислота, фосфорная кислота или бромводород; или органическая кислота, такая как метансульфоновая кислота, бензолсульфоновая кислота, паратолуолсульфоновая кислота, трифторметансульфоновая кислота или перфторбутансульфоновая кислота; сильнокислая ионообменная смола; или подобные, могут контактировать и смешиваться с соединением (2). Эти кислые вещества можно использовать по отдельности или в комбинации двух или более.
Количество используемого кислого вещества зависит от валентности используемой кислоты, и кислое вещество может использоваться в количестве 1 моль или более по отношению к 1 молю соединения (2).
На стадии (d) соединение (2) может быть превращено в соединение (4) в условиях без растворителя, или соединение (2) может быть превращено в соединение (4) в присутствии растворителя.
В случае, когда соединение (2) превращается в соединение (4) в присутствии растворителя на стадии (d), предпочтительными растворителями являются растворители, инертные по отношению к кислому веществу. Предпочтительные растворители включают алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; хлорированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, трет-бутилметиловый эфир, циклопентилметиловый эфир, 4-метилтетрагидропиран, моноглим, диглим, триглим и тетраглим; сложные эфиры, такие как этилацетат и бутилацетат; нитрилы, такие как ацетонитрил, пропионитрил, бутиронитрил, изобутиронитрил и 2-метилбутиронитрил; и воду. Можно использовать один растворитель или можно использовать два или более растворителя в комбинации.
В случае, когда стадию (d) проводят в присутствии растворителя, и тот же растворитель используют на последующей стадии (а), оставшийся растворитель можно использовать без удаления. В случае, когда используется другой растворитель или реакция проводится в условиях без растворителя на последующей стадии (а), растворитель предпочтительно отгоняют. Соединение (4) с высокой чистотой можно получить, подвергая полученное соединение (4) дистилляции или подобному.
В случае, когда соединение (2) превращают в соединение (4) в условиях без растворителя на стадии (d), соединение (4) высокой чистоты можно получить, подвергая полученное соединение (4) дистилляции или подобному.
Как описано выше, синтез с применением винилсульфонового ангидрида, полученного из соединения винилсульфоновой кислоты и агента ангидридизации, может позволить эффективное производство фторированного мономера, который может служить сырьем для полимерных электролитов на основе фтора, обладающих высокой термостойкостью, таких как мембраны для топливных батарей, связывающие полимеры катализа для топливных батарей и мембраны для хлорщелочного электролиза.
ПРИМЕРЫ
Хотя настоящее раскрытие теперь будет описано более подробно со ссылкой на примеры и сравнительные примеры, понятно, что настоящее раскрытие не ограничивается этими примерами. В примерах и сравнительных примерах используются следующие аналитические и оценочные методы:
- Анализ ядерного магнитного резонанса (ЯМР): анализ молекулярной структуры с применением 19F-ЯМР.
Измерительный аппарат: Avance 500 (доступный от Bruker Corporation)
Измерительная пробирка: двухслойная пробирка для ЯМР (внешняя трубка содержит дейтерохлороформ (ссылочный материал: CFCl3 (0 ч./млн.)) и внутренняя пробирка содержит образец (внутренний стандарт: гексафторбензол))
Индекс накопления: 16 раз
Газовая хроматография (ГХ)
Аппарат для измерения: GC-2010 Plus (доступный от Shimadzu Corporation)
Колонка: капиллярная колонка Rtx-200, доступная от Restek Corporation, USA (имеющая внутренний диаметр 0,25 мм, длину 60 м и толщину пленки 1 мкм)
Газ-носитель: гелий
Скорость потока газа-носителя: 30 мл/мин
Объем впрыска: 1 мкл
Коэффициент деления потока: 30
Температура испарительной камеры: 200°C
Программа для температуры колонки: температуру выдерживают 40°C в течение 10 мин, повышают со скоростью 20°C/мин и затем выдерживают 280°C в течение 10 мин
Определение: ПИД при 280°C
- Газовая хроматография - масс спектр (ГХ-МС)
Аппарат для измерения: GCMS-QP2020 (доступный от Shimadzu Corporation)
Колонка: капиллярная колонка Rtx-200, доступная от Restek Corporation, USA (имеющая внутренний диаметр 0,25 мм, длину 60 м и толщину пленки 1 мкм)
Газ-носитель: гелий
Скорость потока газа-носителя: 30 мл/мин
Объем впрыска: 1 мкл
Коэффициент деления потока: 30
Температура испарительной камеры: 200°C
Программа для температуры колонки: температуру выдерживают 40°C в течение 10 мин, повышают со скоростью 20°C/мин и затем выдерживают 280°C в течение 10 мин
Определение: ПИД при 280°C
Источник ионов: ЭИ
(Пример 1)
- Синтез CF2=CFOCF2CF2SO3Na (соединение (2)) из CF3CF(COF)OCF2CF2SO2F с применением циклического соединения
3-литровую круглодонную колбу, оборудованную механической мешалкой, капельной воронкой и конденсатором с обратным холодильником загружают карбонатом натрия (328,6 г, 3,10 моль), который сушат при 180°C в течение 6 часов, и тетраглимом (1000 мл). Пока температуру внутри реактора поддерживают 30°C или ниже, CF3CF(COF)OCF2CF2SO2F (934,2 г, 2,70 моль) добавляют по каплям в течение более 3 часов. После завершения добавления по каплям, реакционную смесь дополнительно перемешивают при 40°C в течение 1 часа с получением натриевой соли карбоновой кислоты (CF3CF(CO2Na)OCF2CF2SO2F). Полученную реакционную смесь нагревают при 160°C под нормальным давлением, чтобы вызвать декарбоксилирование. Летучий компонент отгоняют, собирают в охлажденный на льду сосуд. Эту бесцветную жидкость идентифицируют как следующее циклическое соединение (741,9 г, 2,65 моль; выход: 98%) по 19F-ЯМР.
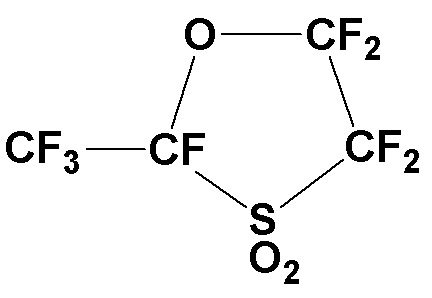
19F-ЯМР: δ (ч./млн.) -124,7 (1F), -120,6 (1F), -115,4 (1F), -90,1 (1F), -80,5 (3F), -78,0 (1F)
В 500 мл 4-горлую колбу в атмосфере азота загружают циклическое соединение (20,39 г, 0,073 моль), полученное на вышеупомянутой стадии, и затем охлаждают до 0°C. Затем раствор триметилсиланолата натрия (доступный от Sigma Aldrich Co., LLC; 16,03 г, 0,143 моль), растворенный в 4-метилтетрагидропиране (120,85 г) добавляют по каплям в эту колбу в течение 1 часа, затем дополнительно перемешивают при комнатной температуре в течение 2 часов. Вакуумная концентрация полученной реакционной смеси дает твердый остаток (28,7 г). Этот твердый остаток идентифицируют, как содержащий 70,0% массовых (выход: 92%) CF2=CFOCF2CF2SO3Na по 19F-ЯМР (внутренний стандарт: трифторэтанол).
CF2=CFOCF2CF2SO3Na
19F-ЯМР: δ (ч./млн.) -136,3 (1F), -123,3 (1F), -118,8 (2F), -115,4 (1F), -85,4 (2F)
- Синтез CF2=CFOCF2CF2SO3H (соединение (4)) из CF2=CFOCF2CF2SO3Na (соединение (2)) (стадия (d))
В 300 мл четырехгорлую колбу загружают CF2=CFOCF2CF2SO3Na (70,02 г, 0,233 моль) и серную кислоту (79,31 г, 0,79 моль) при комнатной температуре, и смесь перемешивают при 70°C, сохраняя давление внутри колбы 30 кПа. Через некоторое время получают однородное растворение. Затем, после постепенного нагревания колбы до температуры 145°C, в то время как давление внутри колбы снижается до 0,33 кПа, жидкость отгоняют (полученное количество: 59,91 г). Эту жидкость идентифицируют как CF2=CFOCF2CF2SO3H (0,216 моль, выход: 93%) по 19F-ЯМР.
- Синтез (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3H (соединение (4)) (стадия (a))
В 300 мл четырехгорлую колбу загружают пентоксид фосфора (P2O5) (80,8 г, 0,569 моль) в перчаточной камере в атмосфере сухого воздуха. Конденсатор Лейбига и капельную воронку присоединяют к колбе, и смесь нагревают при 50°C в потоке азота. Затем, после добавления по каплям винилсульфоновой кислоты (CF2=CFOCF2CF2SO3H) (79,13 г, 0,285 моль) из капельной воронки, колбу нагревают до 140°C, и давление внутри колбы меняют от нормального давления до 60 кПа. После дальнейшего нагревания колбы до 160°C и снижения давления внутри колбы до 20 кПа, жидкость отгоняют (63,53 г). Эту жидкость идентифицируют как содержащую 90% массовых (0,106 моль, выход: 74%) (CF2=CFOCF2CF2SO2)2O и 10% массовых (0,023 моль) CF2=CFOCF2CF2SO3H по 19F-ЯМР.
(CF2=CFOCF2CF2SO2)2O
19F-ЯМР: δ (ч./млн.) -138,6 (2F), -123,3 (2F), -116,0 (2F), -111,9 (4F), -84,6 (4F)
ЭИ-МС: m/z 261, 194, 169, 147, 131, 119, 100, 97, 81, 69, 50, 47, 31
- Синтез CF2=CFOCF2CF2SO2F (соединение (3)) и CF2=CFOCF2CF2SO3Na (соединение (2)) из (CF2=CFOCF2CF2SO2)2O (соединение (1)) и NaF (стадии (b) и (c))
NaF (5,26 г, 0,125 моль) взвешивают в 100 мл трехгорлой колбе и затем сушат в вакууме при 150°C в течение 1 часа, и колбу возвращают в условия нормального давления и комнатной температуры. Конденсатор Лейбига и капельную воронку присоединяют к колбе. Смесь (44,92 г) (CF2=CFOCF2CF2SO2)2O и CF2=CFOCF2CF2SO3H (смесь 96% массовых (0,080 моль) (CF2=CFOCF2CF2SO2)2O и 4% массовых (0,0065 моль) CF2=CFOCF2CF2SO3H) добавляют по каплям из капельной воронки при комнатной температуре в потоке азота. Затем, после нагревания колбы до 190°C при пониженном давлении (95 кПа), жидкость отгоняют (17,56 г). Эту жидкость идентифицируют как CF2=CFOCF2CF2SO2F (0,063 моль) по 19F-ЯМР. Затем остаток (31,17 г) в перегонном кубе идентифицируют как содержащий 94,56% массовых (0,098 моль) CF2=CFOCF2CF2SO3Na, 0,27% массовых (0,00015 моль) (CF2=CFOCF2CF2SO2)2O и 5,17% массовых (0,038 моль) NaF по 19F-ЯМР, что осуществляют добавлением ацетонитрила и гексафторбензола (внутренний стандарт) к остатку.
- Синтез CF2=CFOCF2CF2SO3H (соединение (4)) из CF2=CFOCF2CF2SO3Na (соединение (2)), полученного на вышеупомянутой стадии (c) (стадия (d))
Сильнокислотную катионообменную смолу IR120B (доступную от ORGANO CORPORATION) (93 мл, 0,184 экв.) промывают 1-N водным раствором серной кислоты (1000 мл) и затем дополнительно промывают деионизированной водой (1000 мл). Деионизированную воду (265,5 г) добавляют к остатку (29,50 г) в дистилляционном кубе, полученному на вышеупомянутой стадии (который содержит 94,56% массовых (0,093 моль) CF2=CFOCF2CF2SO3Na, 0,27% массовых (0,00015 моль) (CF2=CFOCF2CF2SO2)2O и 5,17% массовых (0,036 моль) NaF) для получения водного раствора, который затем пропускают через сильнокислотную катионообменную смолу. Затем, после того, как деионизированную воду (214 г) дополнительно пропускают через катионообменную смолу, собранный водный раствор (509 г) концентрируют при пониженном давлении с получением 31,50 г жидкости. Эту жидкость идентифицируют как содержащую 78% массовых (0,088 моль, выход: 95%) CF2=CFOCF2CF2SO3H по 19F-ЯМР. Далее, дистилляция жидкости при пониженном давлении (0,33 кПа) дает 23,20 г CF2=CFOCF2CF2SO3H.
- Синтез (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3H (соединение (4)), полученного на вышеупомянутой стадии (d) (стадия (a))
В 200 мл четырехгорлую колбу загружают пентоксид фосфора (P2O5) (24,97 г, 0,176 моль) в перчаточной камере в атмосфере сухого воздуха. Конденсатор Лейбига и капельную воронку присоединяют к колбе, и смесь нагревают при 50°C в потоке азота. Затем, после добавления по каплям CF2=CFOCF2CF2SO3H (16,42 г, 0,059 моль), полученного на вышеупомянутой стадии (d) из капельной воронки, колбу нагревают до 140°C, и давление внутри колбы меняют от нормального давления до 60 кПа. После дальнейшего нагревания колбы до 160°C и снижения давления внутри колбы до 20 кПа, жидкость отгоняют (11,35 г). Эту жидкость идентифицируют как содержащую 95% массовых (0,020 моль, выход: 68%) (CF2=CFOCF2CF2SO2)2O и 5% массовых (0,0021 моль) CF2=CFOCF2CF2SO3H по 19F-ЯМР.
(Пример 2)
Синтез (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3Na (соединение (2)) с применением CF2=CFOCF2CF2SO3H (соединение (4)) (стадии (d) и (a))
Сильнокислотную катионообменную смолу IR120B (доступную от ORGANO CORPORATION) (357 мл, 0,714 экв.) промывают 1-N водным раствором серной кислоты (3570 мл) и затем дополнительно промывают деионизированной водой (3570 мл). Водный раствор 10% массовых CF2=CFOCF2CF2SO3Na (1500 г, 0,50 моль) пропускают через катионообменную смолу и затем через нее пропускают деионизированную воду (524 г). Концентрация собранного водного раствора дает жидкость (143 г). Эту жидкость идентифицируют как содержащую 93,8% массовых (0,48 моль, выход: 96%) CF2=CFOCF2CF2SO3H по 19F-ЯМР (внутренний стандарт: бензотрифторид).
В 1000 мл четырехгорлую колбу загружают пентоксид фосфора (P2O5) (217,3 г, 1,531 моль) в перчаточной камере в атмосфере сухого воздуха. Конденсатор Лейбига и капельную воронку присоединяют к колбе, и смесь нагревают при 50°C в потоке азота. Затем, после добавления по каплям вышеупомянутого концентрата (143 г) из капельной воронки, колбу нагревают до 140°C, и давление внутри колбы меняют от нормального давления до 60 кПа. После дальнейшего нагревания колбы до 160°C и снижения давления внутри колбы до 20 кПа, жидкость отгоняют (104,7 г). Эту жидкость идентифицируют как содержащую 90% массовых (0,18 моль, выход: 70%) (CF2=CFOCF2CF2SO2)2O и 9% массовых (0,034 моль) CF2=CFOCF2CF2SO3H по 19F-ЯМР.
(Пример 3)
Синтез (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3Na (соединение (2)) с применением CF2=CFOCF2CF2SO3H (соединение (4)) (стадии (d) и (a))
К CF2=CFOCF2CF2SO3Na (300 г, 1 моль) добавляют 30% массовых водный раствор серной кислоты (490 г), и смесь затем перемешивают при комнатной температуре. Затем добавляют циклопентилметиловый эфир (ЦПМЭ) (1500 г), с последующим перемешиванием при комнатной температуре в течение 1 часа. После выдерживания смеси в покое после остановки перемешивания, смесь разделяется на два слоя. После отделения органического слоя и последующей концентрации при пониженном давлении, получают жидкость (291,0 г). Эту жидкость идентифицируют как содержащую 93,6% массовых (0,98 моль, выход: 98%) CF2=CFOCF2CF2SO3H по 19F-ЯМР (внутренний стандарт: бензотрифторид).
В 2000 мл четырехгорлую колбу загружают пентоксид фосфора (P2O5) (442,4 г, 3,117 моль) в перчаточной камере в атмосфере сухого воздуха. Конденсатор Лейбига и капельную воронку присоединяют к колбе, и смесь нагревают при 50°C в потоке азота. Затем, после добавления по каплям вышеупомянутого концентрата (291,0 г) из капельной воронки, колбу нагревают до 140°C, и давление внутри колбы меняют от нормального давления до 60 кПа. После дальнейшего нагревания колбы до 160°C и снижения давления внутри колбы до 20 кПа, жидкость отгоняют (213,2 г). Эту жидкость идентифицируют как содержащую 90% массовых (0,36 моль, выход: 71%) (CF2=CFOCF2CF2SO2)2O и 9% массовых (0,07 моль) CF2=CFOCF2CF2SO3H по 19F-ЯМР.
(Пример 4)
Синтез CF2=CFOCF2CF2SO2F (соединение (3)) и CF2=CFOCF2CF2SO3K (соединение (2)) из (CF2=CFOCF2CF2SO2)2O (соединение (1)) и KF (стадии (b) и (c))
KF (2,60 г, 0,045 моль) взвешивают в 100 мл трехгорлой колбе и затем сушат в вакууме при 150°C в течение 1 часа, и колбу возвращают в условия нормального давления и комнатной температуры. Конденсатор Лейбига и капельную воронку присоединяют к колбе. Смесь (14,25 г) (CF2=CFOCF2CF2SO2)2O и CF2=CFOCF2CF2SO3H (смесь 88% массовых (0,023 моль) (CF2=CFOCF2CF2SO2)2O и 12% массовых (0,006 моль) CF2=CFOCF2CF2SO3H) добавляют по каплям из капельной воронки при комнатной температуре в потоке азота. Затем, после нагревания колбы до 190°C при пониженном давлении (90 кПа), жидкость отгоняют (6,30 г). Эту жидкость идентифицируют как содержащую 92% массовых (0,017 моль) CF2=CFOCF2CF2SO2F и 0,4% массовых (0,000045 моль) (CF2=CFOCF2CF2SO2)2O по 19F-ЯМР. Затем остаток (10,80 г) в перегонном кубе идентифицируют как содержащий 87% массовых (0,030 моль) CF2=CFOCF2CF2SO3K по 19F-ЯМР, что осуществляют добавлением ацетонитрила и гексафторбензола (внутренний стандарт) к остатку.
(Пример 5)
Синтез CF2=CFOCF2CF2SO2F (соединение (3)) и CF2=CFOCF2CF2SO3Na (соединение (2)) из (CF2=CFOCF2CF2SO2)2O (соединение (1)) и NaF (стадия (b))
CF2=CFOCF2CF2SO2)2O (0,66 г, 0,00123 моль), NaF (0,083 г, 0,0020 моль и ацетонитрил (2,8 г) помещают в 50 мл тестовую пробирку с винтовым верхом и оставляют взаимодействовать при 40°C в течение 1 часа. Полученную реакционную смесь идентифицируют как содержащую CF2=CFOCF2CF2SO2F (0,00074 моль) и CF2=CFOCF2CF2SO3Na (0,0016 моль) по 19F-ЯМР.
(Пример 6)
Синтез CF2=CFOCF2CF2SO2F (соединение (3)) и CF2=CFOCF2CF2SO3K (соединение (2)) из (CF2=CFOCF2CF2SO2)2O (соединение (1)) и KF (стадия (b))
(CF2=CFOCF2CF2SO2)2O (0,69 г, 0,00128 моль), KF (0,11 г, 0,0019 моль) и ацетонитрил (2,9 г) помещают в 50 мл тестовую пробирку с винтовым верхом и оставляют взаимодействовать при 40°C в течение 1 часа. Полученную реакционную смесь идентифицируют как содержащую CF2=CFOCF2CF2SO2F (0,00082 моль) и CF2=CFOCF2CF2SO3K (0,0017 моль) по 19F-ЯМР.
(Пример 7)
В синтезе (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3H (соединение (4)) в примере 1, после отгонки жидкости оставшийся остаток (10 г) добавляют к раствору, содержащему карбонат натрия (12,7 г) и воду (57,9 г) с последующим перемешиванием. Затем добавляют 4-метилтетрагидропиран (40,3 г) с последующим дальнейшим перемешиванием. После выдерживания смеси в покое после остановки перемешивания, смесь разделяется на два слоя. После удаления слоя 4-метилтетрагидропирана и концентрации при пониженном давлении получают твердое вещество (0,792 г). Это твердое вещество идентифицируют как содержащее 98% массовых (0,00259 моль) CF2=CFOCF2CF2SO3Na по 19F-ЯМР (внутренний стандарт: бензотрифторид).
(Пример 8)
Синтез (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3Na (соединение (2)) и пентоксида фосфора (P2O5) (стадия (a))
Пентоксид фосфора (P2O5) (94,6 г, 0,666 моль) и соль винилсульфоновой кислоты (CF2=CFOCF2CF2SO3Na) (100 г, 0,333 моль) помещают в мини скоростную мельницу (MS-05, доступную от Labonect Co. Ltd.) и перемешивают в течение 30 минут с получением смеси. Полученную смесь помещают в 500 мл четырехгорлую колбу. Конденсатор Лейбига присоединяют к колбе, и давление снижают до 0,3 кПа. После постепенного нагревания колбы до 200°C жидкость отгоняют. Колбы выдерживают при 200°C в течение 45 минут. Полученную жидкость взвешивают 17,9 г. Эту жидкость идентифицируют как содержащую 95% массовых (0,0317 моль) (CF2=CFOCF2CF2SO2)2O по 19F-ЯМР.
(Пример 9)
Синтез (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3Na (соединение (2)) и пентоксида фосфора (P2O5) (стадия (a))
Пентоксид фосфора (P2O5) (9,72 г, 0,0685 моль) и соль винилсульфоновой кислоты (CF2=CFOCF2CF2SO3Na) (10,3 г, 0,0342 моль) и циркониевую дробь, имеющую диаметр 1 мм (200 г, YTZ-1, доступную от Nikkato Corporation) помещают в Тефлоновый (зарегистрированный товарный знак) контейнер. Контейнер помещают в низкочастотный резонансный акустический смеситель (LabRAMII, доступный от Resodyn Acoustic Mixers) и обрабатывают при ускорении 100 G в течение 25 минут с получением обработанного продукта. Обработанный продукт фильтруют через сито 140 меш с получением смеси пентоксида фосфора (P2O5) и соли винилсульфоновой кислоты (CF2=CFOCF2CF2SO3Na). Эту методику повторяют множество раз с получением 50 г смеси. Полученную смесь помещают в 200 мл трехгорлую колбу. Конденсатор Лейбига присоединяют к колбе, и давление снижают до 0,3 кПа. После постепенного нагревания колбы до 200°C жидкость отгоняют. Колбы выдерживают при 200°C в течение 45 минут. Полученную жидкость взвешивают 9,68 г. Эту жидкость идентифицируют как содержащую 95% массовых (0,0171 моль) (CF2=CFOCF2CF2SO2)2O по 19F-ЯМР.
(Пример 10)
В синтезе (CF2=CFOCF2CF2SO2)2O (соединение (1)) из CF2=CFOCF2CF2SO3Na (соединение (2)) и пентоксида фосфора (P2O5) в примере 9, после отгонки жидкости, оставшийся остаток (10 г) добавляют к раствору, содержащему карбонат натрия (4,50 г) и воду (20,5 г) с последующим перемешиванием. Затем добавляют 4-метилтетрагидропиран (17,5 г) с последующим дальнейшим перемешиванием. После выдерживания смеси в покое после остановки перемешивания, смесь разделяется на два слоя. После удаления слоя 4-метилтетрагидропирана и концентрации при пониженном давлении получают твердое вещество (2,59 г). Это твердое вещество идентифицируют как содержащее 98% массовых (0,00845 моль) CF2=CFOCF2CF2SO3Na по 19F-ЯМР (внутренний стандарт: бензотрифторид).
ПРОМЫШЛЕНННАЯ ПРИМЕНИМОСТЬ
Согласно настоящему раскрытию можно получить фторированный мономер с хорошим выходом, который является сырьем для полимерных электролитов на основе фтора, обладающих высокой термостойкостью, полезных для различных применений, таких как мембраны для топливных батарей, связывающие полимеры катализа для топливных батарей и мембраны для хлорщелочного электролиза.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ФТОРСУЛЬФОНИЛЬНУЮ ГРУППУ, МОНОМЕР, СОДЕРЖАЩИЙ ФТОРСУЛЬФОНИЛЬНУЮ ГРУППУ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2766150C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННОГО СУЛЬФОНИЛФТОРИДА | 2004 |
|
RU2379285C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННОГО СОЕДИНЕНИЯ | 2014 |
|
RU2675377C2 |
| СПОСОБ ПОЛУЧЕНИЯ VIC-ДИХЛОРФТОРАНГИДРИДА | 2000 |
|
RU2252210C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРПРОПИЛЕНКАРБОНАТА | 2009 |
|
RU2470019C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩЕГО СОЕДИНЕНИЯ | 2001 |
|
RU2248346C2 |
| ФТОРИРОВАННЫЙ ДИЕН, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ПОЛИМЕР НА ЕГО ОСНОВЕ, ОПТИЧЕСКОЕ ПЕРЕДАЮЩЕЕ УСТРОЙСТВО И ОПТИЧЕСКОЕ ПЛАСТМАССОВОЕ ВОЛОКНО | 2001 |
|
RU2272020C2 |
| СОЛЬ ПЕРФТОРКАРБОНОВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2453529C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОМЕРА, СОДЕРЖАЩЕГО СУЛЬФОГРУППУ | 2019 |
|
RU2751162C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ГАЛОГЕНИРОВАННЫХ ФТОРИРОВАННЫХ ЭФИРСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2015 |
|
RU2671967C1 |
Настоящее раскрытие направлено на предоставление винилсульфонового ангидрида, который может быть использован в качестве синтетического промежуточного соединения для синтеза фторированного мономера. Оно также направлено на эффективное производство винилсульфонового ангидрида. Оно также направлено на эффективное производство фторированного мономера с использованием винилсульфонового ангидрида. Винилсульфоновый ангидрид данного раскрытия выражается общей формулой (1) (где m равен 0 и n является целым числом от 1 до 6). Кроме того, способ получения винилсульфонового ангидрида данного раскрытия включает контакт и смешивание соединения винилсульфоновой кислоты, представленного общей формулой (2) (где m равен 0, n является целым числом от 1 до 6 и M является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью), с агентом ангидридизации. Кроме того, способ получения винилсульфонилфторида данного раскрытия включает стадию (b) контакта и смешивания винилсульфонового ангидрида, представленного общей формулой (1), с фторирующим агентом для приготовления реакционной смеси, включающей винилсульфонилфторид, представленный общей формулой (3) (где m равен 0 и n является целым числом от 1 до 6), и соединение винилсульфоновой кислоты, представленное общей формулой (2). 3 н. и 8 з.п. ф-лы, 10 пр.

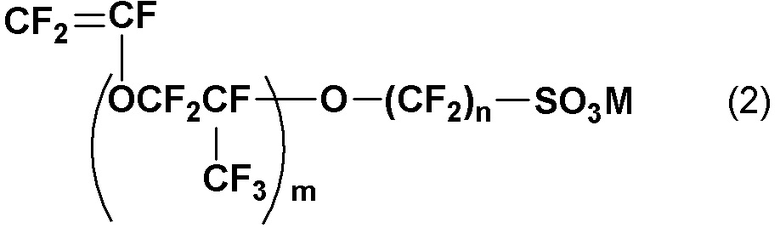

1. Винилсульфоновый ангидрид, представленный следующей общей формулой (1):

(где m равен 0 и n является целым числом от 1 до 6).
2. Способ получения винилсульфонового ангидрида, представленного следующей общей формулой (1):
(где m равен 0 и n является целым числом от 1 до 6),
где способ включает:
стадию (a) получения соединения винилсульфоновой кислоты, представленного следующей общей формулой (2), контактом и смешиванием с агентом ангидридизации:

(где m равен 0, n является целым числом от 1 до 6 и M является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью).
3. Способ получения винилсульфонилфторида, включающий:
стадию (b) получения винилсульфонового ангидрида, представленного следующей общей формулой (1), контактом и смешиванием с фторирующим агентом:
(где m равен 0 и n является целым числом от 1 до 6)
с получением реакционной смеси, содержащей винилсульфонилфторид, представленный следующей общей формулой (3):

(где m равен 0, и n является целым числом от 1 до 6); и
соединение винилсульфоновой кислоты, представленное следующей общей формулой (2):
(где m равен 0, n является целым числом от 1 до 6 и M является атомом водорода, щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью).
4. Способ получения винилсульфонилфторида по п. 3, где винилсульфоновым ангидридом является винилсульфоновый ангидрид, полученный способом получения винилсульфонового ангидрида по п. 2.
5. Способ получения винилсульфонилфторида по п. 3 или 4, дополнительно содержащий стадию (c) отделения винилсульфонилфторида, представленного общей формулой (3), и соединения винилсульфоновой кислоты, представленного общей формулой (2), из реакционной смеси, полученной на стадии (b).
6. Способ получения винилсульфонового ангидрида по п. 5, где стадия (a) включает контакт и смешивание соединения винилсульфоновой кислоты, представленного общей формулой (2), полученного на стадии (c) по п. 5, с агентом ангидридизации.
7. Способ получения винилсульфонового ангидрида по п. 2 или 6, дополнительно включающий стадию (d) получения соединения винилсульфоновой кислоты, в котором M в общей формуле (2) является щелочным металлом, щелочноземельным металлом, Ag, четвертичной аммониевой солью или четвертичной фосфониевой солью, контактом и смешиванием с кислым веществом для превращения в винилсульфоновую кислоту, представленную следующей общей формулой (4):
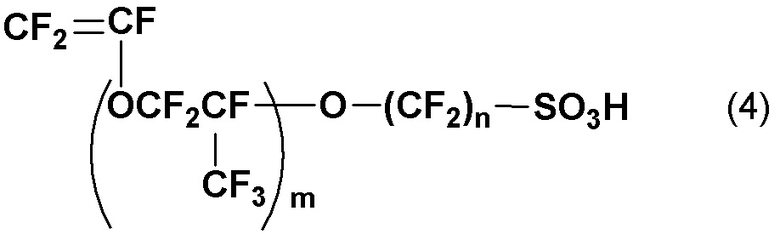
(где m и n такие, как определены в общей формуле (2)).
8. Способ получения винилсульфонилфторида по любому из пп. 3-5, где фторирующим агентом является один или более, выбранные из группы, состоящей из фторводорода, фторида металла, фторида четвертичного аммония и фторида четвертичного фосфония.
9. Способ получения винилсульфонового ангидрида по любому из пп. 2, 6 и 7, где агентом ангидридизации является один или более, выбранных из группы, состоящей из пентоксида фосфора, уксусного ангидрида, трифторуксусного ангидрида, метансульфонового ангидрида, трифторметансульфонового ангидрида, п-толуолсульфонового ангидрида, тионилхлорида, дициклогексилкарбодиимида, цианурхлорида, тетрахлорида титана и бензолсульфонилхлорида.
10. Способ получения винилсульфонового ангидрида по п. 2, где стадия (a) включает стадию отделения винилсульфонового ангидрида, представленного общей формулой (1).
11. Способ получения винилсульфонового ангидрида по п. 10, дополнительно включающий стадию, после отделения винилсульфонового ангидрида, представленного общей формулой (1), сбора соединения винилсульфоновой кислоты, представленного общей формулой (2), из полученного остатка.
| JP 2010218982 А, 30.09.2010 | |||
| JP 2015224218 A, 14.12.2015 | |||
| JP S5728024 A, 15.02.1982 | |||
| ФТОРСОДЕРЖАЩИЕ ПОЛИМЕРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2131888C1 |

