Перекрестные ссылки на родственные заявки
По настоящей заявке испрашивается приоритет согласно предварительной заявке на патент США No. 62/378455, поданной 23 августа 2016 г. Настоящая заявка включена в настоящее описание в качестве ссылки в полном объеме.
Уровень техники
Рак печени является второй по значимости причиной смертности от любого типа рака и 16-й наиболее распространенной причиной смерти в мире (Llovet JM, et al., 2015 ʺAdvances in targeted therapies for hepatocellular carcinoma in the genomic era.ʺ Nat. Rev. Clin Oncology 12, 408-424). Гепатоцеллюлярная карцинома (HCC) составляет до 90% всех первичных раковых заболеваний печени (Llovet JM et al. 2015).
Различные сигнальные пути вовлечены в HCC, включая факторы роста фибробластов (FGF) (в частности, FGF19/FGFR4), эпидермальный фактор роста (EGF), фактор роста эндотелия сосудов (VEGF), ERK/MAPK и механистическую мишень рапамицина (mTOR), среди прочих (Llovet JM et al., 2015). FGF19 сверхэкспрессируется примерно в трети всех HCC, и эта сверхэкспрессия предположительно гиперактивирует FGFR4 и его нисходящий сигнальный путь, приводящий к усиленному росту опухоли (Xie MH et al., 1999 ʺFGF-19, a novel fibroblast growth factor with unique specificity for FGFR4.ʺ Cytokine. 1999 Oct;11(10):729-35; Sawey, et al., 2011 ʺIdentification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening.ʺ Cancer Cell. 2011 Mar 8;19(3):347-58.). Подобно пути FGF19/FGFR4, активация пути CDK4/6 также участвует в патогенезе HCC (Rivadeneira et al., 2010 ʺProliferative Suppression by CDK4/6 Inhibition: Complex Function of the Retinoblastoma Pathway in Liver Tissue and Hepatoma Cells.ʺ Gastroenterology 138:1920-1930).
Соединение 1 представляет собой селективный перорально биодоступный низкомолекулярный ингибитор FGFR4 со структурой, показанной в формуле I, и химическим названием N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид:
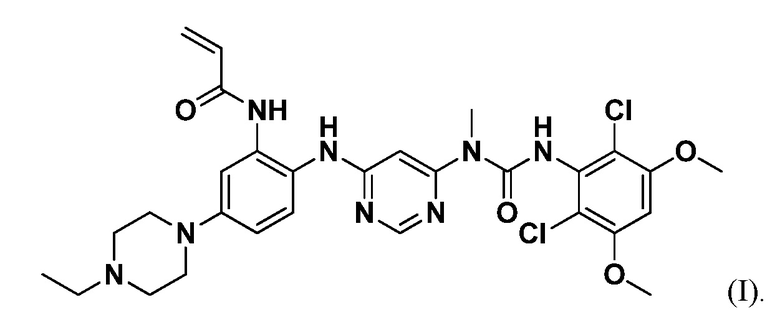
Соединение 1 и его синтез описаны в публикации международной заявки РСТ No. WO2015/057938, опубликованной 23 апреля 2015 г. Этот документ включен в настоящее описание в качестве ссылки.
Палбоциклиб (6-ацетил-8-циклопентил-5-метил-2-{[5-(пиперазин-1-ил)пиридин-2-ил]амино}пиридо[2,3-d]пиримидин-7(8H)-он) является одобренным FDA ингибитором циклин-зависимой киназы (CDK) 4 и 6. Палбоциклиб имеет следующую структуру:
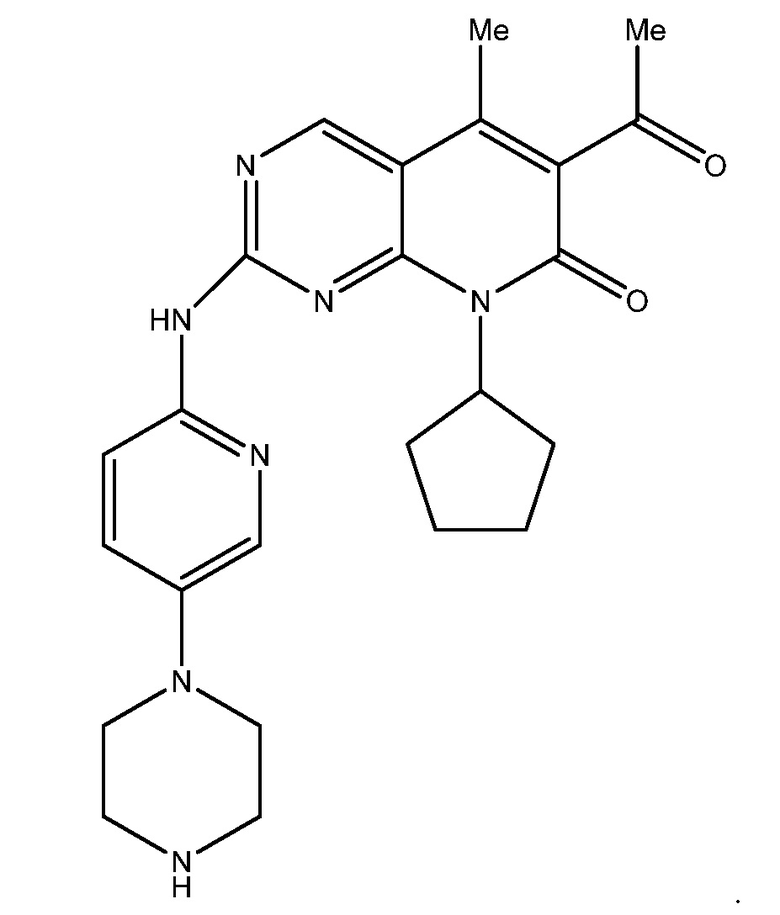
См. патент США No. 6936612; 7208489 и 7456168, который включен в настоящее описание в качестве ссылки.
Рибоциклиб (диметиламид 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты) является одобренным FDA ингибитором циклин-зависимой киназы (CDK) 4 и 6. Рибоциклиб имеет следующую структуру:
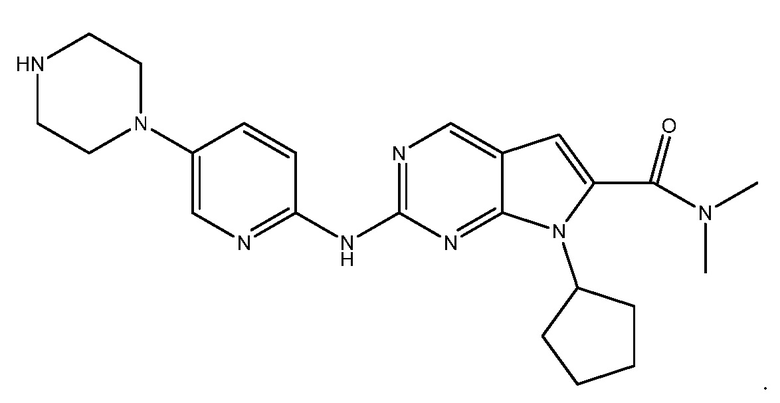
См. опубликованную патентную заявку США US20120115878, публикацию РСТ No WO2007140222, публикацию РСТ No WO2012061156; публикацию РСТ No WO2011130232; публикацию РСТ No WO2011101417; и публикацию РСТ No WO2010020675, все из которых включены в настоящее описание в качестве ссылки.
Абемациклиб представляет собой ингибитор CDK 4/6 с названием N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиримидин-2-амин. Абемациклиб имеет следующую структуру:
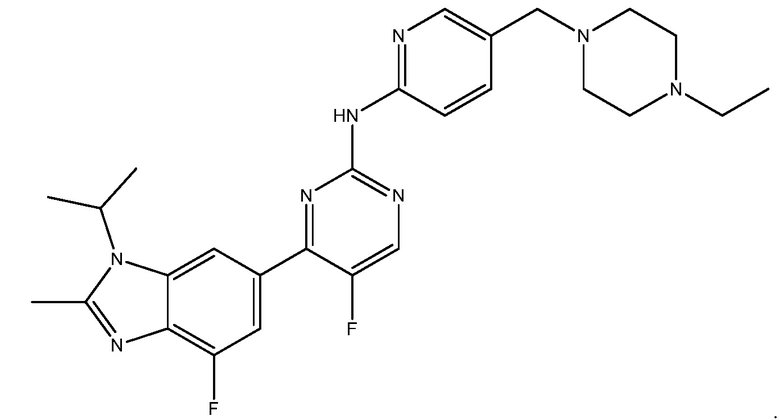
См. O'Leary, et al., ʺTreating Cancer with Selective CDK 4/6 Inhibitorsʺ Nat. Rev. (Published Online March. 31, 2016); публикацию РСТ No WO2016110224, опубликованную патентную заявку США No 20100160340; и публикацию РСТ No WO2016025650, все из которых включены в настоящее описание в качестве ссылки.
G1T-38 (также называемый как GZ-38-1 или G1T38-1) является зарегистрированным ингибитором CDK 4/6. G1T-38, который изучается G1 Therapeutics, Inc., of Research Triangle Park, North Carolina, is reported in Abstract #2824 of the 2016 AACR Annual Meeting, held April 16-20 in New Orleans, Louisiana, entitled ʺG1T38, A Novel, Oral, Potent and Selective CDK 4/6 Inhibitor for the Treatment of RB Competent Tumors,ʺ by J. Sorrentino, J. Bisi, P. Roberts, and J. Strum. Этот документ включен в настоящее описание в качестве ссылки. G1T38 имеет химическое название 2'-((5-(4-изопропилпиперазин-1-ил)пиридин-2-ил)амино)-7',8'дигидро-6'H-спиро[циклогексан1,9'пиразино[1',2':1,5]пирроло[2,3-d]пиримидин]-6'-он ди-гидрохлорид, и структуру, показанную ниже:
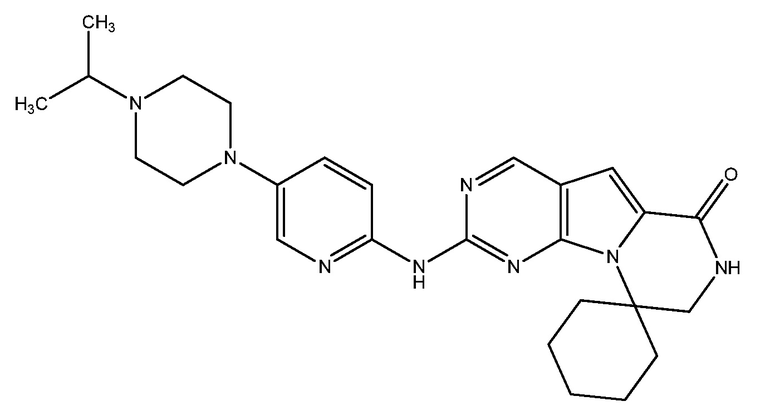
См. Bisi, et al., ʺPreclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors,ʺ Oncotarget, Advance Publications 2017 (March 15, 2017), включенную в настоящее описание ссылкой.
G1T-28 представляет собой ингибитор CDK 4/6 с названием 2'-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7',8'-дигидро-6'H-спиро[циклогексан-1,9'-пиразино[1',2':1,5]пирроло[2,3-d]пиримидин]-6'-он. G1T-28 имеет следующую структуру:
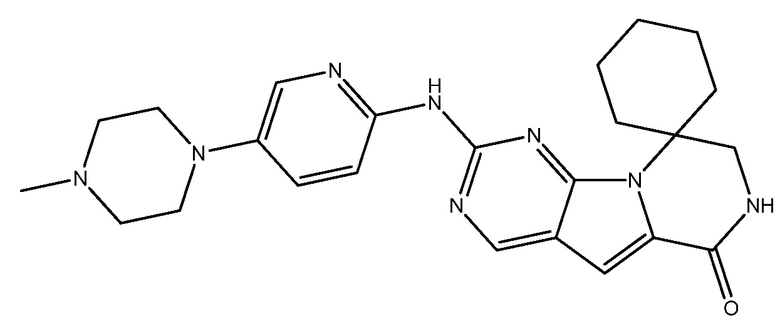 .
.
См., например, Bisi, et al., ʺPreclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-induced Myelosuppressionʺ Mol. Cancer Ther.; 15(5) 783-93, May 2016; опубликованную патентную заявку США No US20160220569; публикации международной заявки на патент PCT No WO2014144326; WO2014144847; и WO2016040848, все из которых включены в настоящее описание в качестве ссылки.
AT-7519 представляет собой ингибитор CDK 4/6 с названием N-(4-пиперидинил)-4-(2,6-дихлорбензоиламино)-1H-пиразол-3 карбоксамид. AT-7519 имеет следующую структуру:
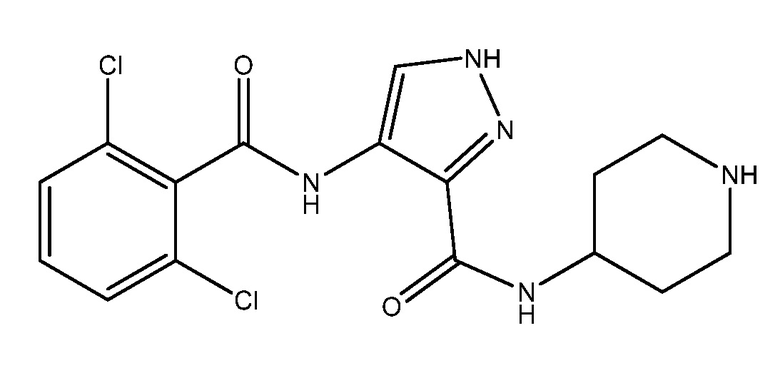 .
.
См., например, публикацию международной заявки на патент PCT No WO 2005012256; WO 2006077424; WO 2006077426; WO 2008001101; WO 2006077425; WO 2006077428; WO 2008007113; WO 2008007122; и WO 2008009954, которые включены в настоящее описание в качестве ссылки.
FLX-925 (также известный как AMG-925) представляет собой ингибитор CDK 4/6 с названием 2-гидрокси-1-[2-[[9-(транс-4-метилциклогексил)-9H-пиридо[4',3':4,5]пирроло[2,3-d]пиримидин-2-ил]амино]-7,8-дигидро-1,6-нафтиридин-6(5H)-ил]этанон. FLX-925 имеет следующую структуру:
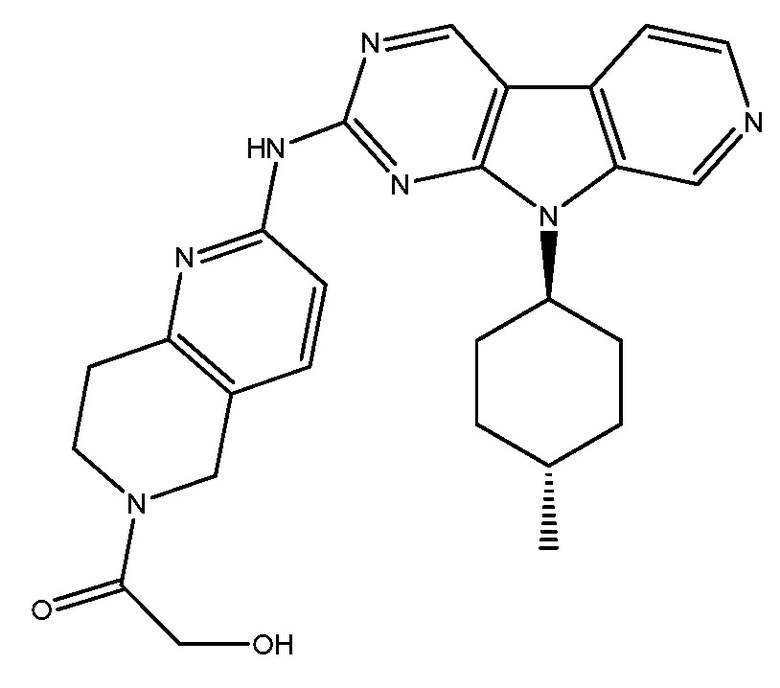 .
.
См., например, опубликованную патентную заявку США No. 2014163052 и публикацию международной заявки на патент PCT No WO 2012129344, обе из которых ключены в настоящее описание в качестве ссылки.
Альвоцидиб представляет собой ингибитор CDK 4/6 с названием 2-(2-хлорфенил)-5,7-дигидрокси-8-((3S,4R)-3-гидрокси-1-метилпиперидин-4-ил)-4H-хромен-4-он. Альвоцидиб имеет следующую структуру:
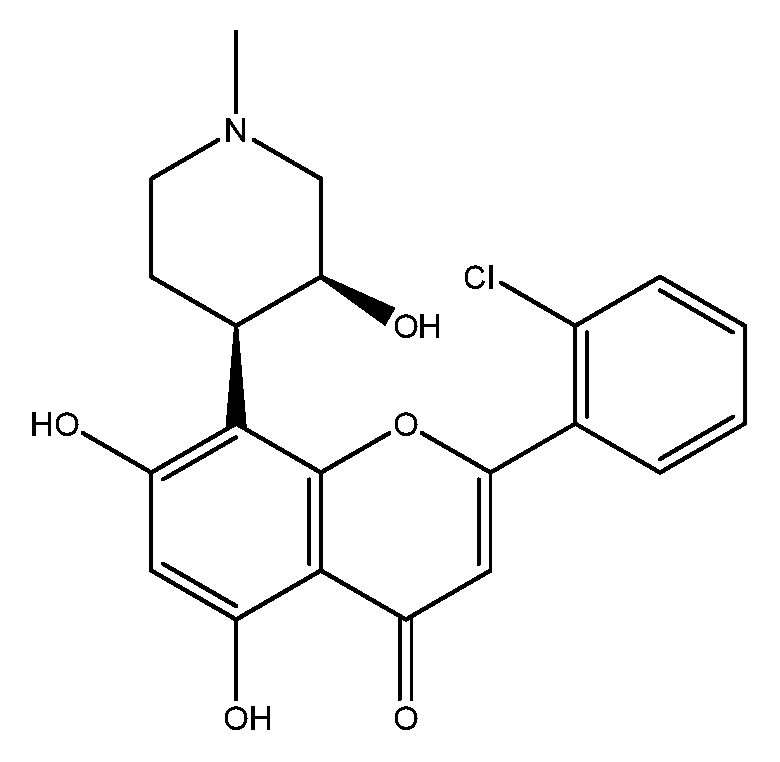 .
.
См., например, опубликованную патентную заявку США No US2011189175 и US2011189175; публикацию международной заявки на патент PCT No WO 2000044362; WO 2001041747; WO 2001053293; WO 2001053294; WO 2002022133; WO 2007010946, все из которых включены в настоящее описание в качестве ссылки.
Несмотря на достижения в лечении HCC, существует необходимость в улучшении лечения HCC. Несмотря на достижения в лечении IHCC, существует необходимость в улучшении лечения IHCC.
Сущность изобретения
Варианты осуществления предоставляют комбинированную терапию, включающую эффективное количество соединения 1 и эффективное количество ингибитора CDK4/6. В определенных вариантах осуществления ингибитор CDK 4/6 представляет собой палбоциклиб. В других вариантах осуществления ингибитор CDK 4/6 представляет собой рибоциклиб. В других вариантах осуществления ингибитор CDK 4/6 представляет собой абемациклиб. В других вариантах осуществления ингибитор CDK 4/6 представляет собой G1T38. В других вариантах осуществления ингибитор CDK 4/6 представляет собой G1T28. Комбинированная терапия, представленная в настоящем документе, может привести к усилению снижения жизнеспособности клеток HCC и может привести к ингибированию роста опухоли HCC у пациентов, нуждающихся в лечении. Комбинированная терапия, представленная в настоящем документе, также может быть эффективной при лечении холангиокарциномы печени, включая, например, внутрипеченочную холангиокарциному (IHCC).
Варианты осуществления могут обеспечивать способ лечения гепатоцеллюлярной карциномы у пациента, нуждающегося в этом, включая введение пациенту комбинации N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида или его фармацевтически приемлемой соли и ингибитора CDK 4/6 или его фармацевтически приемлемой соли. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль вводят в суточной дозе от 50 мг до 600 мг. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль вводят в суточной дозе от 200 мг до 400 мг. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль вводят в суточной дозе 300 мг.
В некоторых вариантах осуществления ингибитор CDK 4/6 выбран, например, из 6-ацетил-8-циклопентил-5-метил-2-{[5-(пиперазин-1-ил)пиридин-2ил]амино}пиридо[2,3-d]пиримидин-7(8H)-она (палбоциклиб); диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (рибоциклиб); и N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиримидин-2-амина (абемациклиб).
В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой палбоциклиб. Палбоциклиб можно вводить, например, в дозе 75, 100 или 125 мг/день. Обычно дозу вводят перорально в виде одной капсулы в течение 21 последовательного дня, после чего следует 7 дневный период без лечения.
В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой рибоциклиб. Рибоциклиб можно вводить, например, в дозе 200, 400 или 600 мг/день. Обычно рибоциклиб вводят перорально в виде капсул или таблеток по 200 мг, в течение 21 последовательного дня, после чего следует 7 дневный период без лечения.
В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой абемациклиб. Абемациклиб можно вводить, например, в дозе 200, 300 или 400 мг/день. Обычно абемациклиб вводят два раза в день в дозах 100, 150 или 200 мг/доза. Абемациклиб обычно вводят в течение 21 последовательного дня или 28 последовательных дней, после чего следует 7 дневный период без лечения.
В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой G1T-38. G1T-38 можно вводить, например, в дозе 10, 50 или 100 мг/кг. В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой G1T-28. G1T-28 можно вводить, например, в дозе от 190 до 200 мг/м2.
В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой AT-7519. AT-7519 можно вводить, например, в дозе 14,4-32,4 мг/м2. AT-7519 можно вводить каждые три недели, с введением лекарства в дни 1, 4, 8 и 11. В одном варианте осуществления доза составляет 27 мг/м2, с вышеуказанной кратностью введения.
В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой FLX-925. В некоторых вариантах осуществления ингибитор CDK 4/6 представляет собой альвоцидиб. Альвоцидиб можно вводить, например, в количестве от 8 до 122 мг/м2. Альвоцидиб можно вводить в виде инфузии в течение 72 часов. Максимальные переносимые дозы альвоцидиба были отмечены как 40, 50 или 78 мг/м2.
В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят в виде отдельных композиций. Обычно время между приемами каждого препарата не превышает 12 часов. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят в виде одной композиции. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят последовательно с другими лекарственными средствами. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят одновременно.
В некоторых вариантах осуществления форма N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, которая вводится, является формой свободного основания. В некоторых вариантах осуществления форма N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, которая вводится, представляет собой форму гидрохлоридной соли.
Дополнительные варианты осуществления могут обеспечивать фармацевтическую композицию, включающую N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид представляет собой форму свободного основания. В некоторых вариантах осуществления N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид представляет собой форму гидрохлоридной соли.
Дополнительные варианты осуществления могут обеспечивать использование комбинации N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида или его фармацевтически приемлемой соли и ингибитора CDK 4/6 при лечении гепатоцеллюлярной карциномы. Дополнительные варианты осуществления могут обеспечивать использование комбинации N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида или его фармацевтически приемлемой соли и ингибитора CDK 4/6 в получении лекарственного средства для лечения гепатоцеллюлярной карциномы.
Краткое описание фигур
На фиг.1A - фиг.1C показаны противоопухолевые эффекты соединения 1 и ингибитора CDK 4/6 рибоциклиба на ксенотрансплантатной модели JHH7 гепатоцеллюлярной карциномы. Соединение 1 и рибоциклиб, каждое в виде свободного основания, давали перорально (PO) один раз в день (QD) в течение 8 дней. Данные представляют собой среднее±SEM для объема опухоли.
На фиг. 1A показан рибоциклиб в качестве единственного средства (****P ≤ 0,0001 по сравнению с носителем в качестве контролей с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг. 1В показаны два уровня дозы рибоциклиба в комбинации с 300 мг/кг соединения 1 (**** P≤0,0001 по сравнению с группой, получавшей 300 мг/кг соединения 1 в качестве единственного средства, с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг. 1C показаны два уровня дозы рибоциклиба в комбинации с 500 мг/кг соединения 1 (****P≤0,0001 для группы, получавшей 500 мг/кг соединения 1 в качестве единственного средства, по сравнению с носителем в качестве контролей и **** P≤0,0001 для комбинаций по сравнению с группой, получавшей 500 мг/кг соединения 1 в качестве единственного средства, с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг.2A - фиг.2C показаны противоопухолевые эффекты соединения 1 и ингибитора CDK 4/6 палбоциклиба на ксенотрансплантатной модели JHH гепатоцеллюлярной карциномы. Соединение 1 и палбоциклиб, каждое в виде свободного основания, давали перорально (PO) один раз в день (QD) в течение 8 дней. Данные представляют собой среднее±SEM для объема опухоли.
На фиг. 2A показан палбоциклиб в качестве единственного средства (*P≤0,05 по сравнению с носителем в качестве контролей с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг. 2B показаны два уровня дозы палбоциклиба в комбинации с 300 мг/кг соединения 1 (**P≤0,01 по сравнению с контролем носителем и ****P≤0,0001 по сравнению с группой, получавшей 300 мг/кг соединения 1 в качестве единственного средства, с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг. 2C показаны два уровня дозы палбоциклиба в комбинации с 500 мг/кг соединения 1 (****P≤0,0001 для группы, получавшей 500 мг/кг соединения 1 в качестве единственного средства, по сравнению с носителем в качестве контролей и ****P≤0,0001 для комбинаций по сравнению с группой, получавшей 500 мг/кг соединения 1 в качестве единственного средства, с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг. 3A и фиг. 3B показаны противоопухолевые эффекты соединения 1 и ингибитора CDK 4/6 палбоциклиба на ксенотрансплантатной модели гепатоцеллюлярной карциномы, полученной у пациента LIX066. Соединение 1 и палбоциклиб давали перорально (PO) один раз в день (QD) в течение 8 дней. Данные представляют собой среднее ± SEM для объема опухоли.
На фиг. 3A показано 300 мг/кг соединения 1 или в качестве единственного средства или в комбинации с 100 мг/кг палбоциклиба (***P≤0,001 для палбоциклиба 100 мг/кг в качестве единственного средства по сравнению с носителем в качестве контролей, ****P≤0,0001 для группы, получавшей 300 мг/кг соединения 1 в качестве единственного средства, по сравнению с носителем в качестве контролей и ****P≤0,0001 для комбинаций по сравнению с группой, получавшей 300 мг/кг соединения 1 в качестве единственного средства, с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
На фиг. 3В показано 500 мг/кг соединения 1 или в качестве единственного средства или в комбинации с 100 мг/кг палбоциклиба (***P≤0,001 для палбоциклиба 100 мг/кг в качестве единственного средства по сравнению с носителем в качестве контролей, ****P≤0,0001 для группы, получавшей 500 мг/кг соединения 1 в качестве единственного средства, по сравнению с носителем в качестве контролей и ****P≤0,0001 для комбинаций по сравнению с группой, получавшей 500 мг/кг соединения 1 в качестве единственного средства, с использованием двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak).
Подробное описание вариантов осуществления
В настоящем документе предоставлены комбинированные терапии, полезные в лечении гепатоцеллюлярной карциномы (HCC) и внутрипеченочной холангиокарциномы (IHCC). В некоторых вариантах осуществления, комбинированная терапия включает введение соединения 1 в комбинации с ингибитором CDK 4/6. В определенных вариантах осуществления ингибитор CDK 4/6 представляет собой палбоциклиб. В других вариантах осуществления, ингибитор CDK 4/6 представляет собой рибоциклиб. В еще других вариантах осуществления ингибитор CDK 4/6 представляет собой абемациклиб.
В настоящем документе предоставлены комбинации терапевтических средств и способы введения комбинации средств для лечения гепатоцеллюлярной карциномы. Как используется в настоящем документе термин «комбинация терапевтических средств» и подобные термины относятся к комбинации двух типов терапевтических средств: (1) соединение 1 и/или его фармакологически активные соли и (2) ингибитор CDK 4/6 и/или их фармакологически активные соли. «Комбинация», как используется в настоящем документе (включая термин «комбинация терапевтических средств»), относится к этим типам терапевтических средств, комбинированных в одной лекарственной форме, отдельно составленных и совместно вводимых, или отдельно составленных и последовательно вводимых.
Соединение 1 представляет собой селективный перорально биодоступный низкомолекулярный ингибитор FGFR4 со структурой, показанной в формуле I:
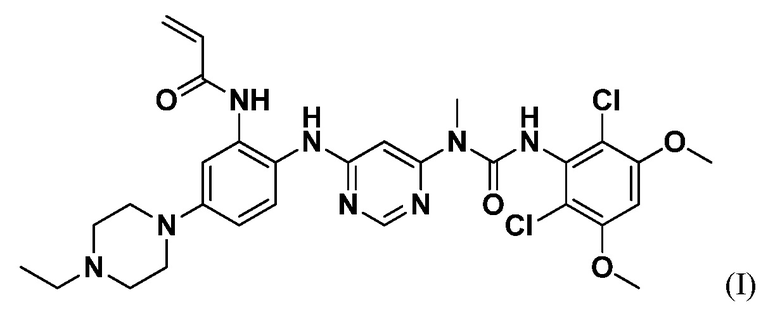 .
.
Соединение 1 и его синтез описаны в публикации международной заявки РСТ No. WO2015/057938, опубликованной 23 апреля 2015 г. Этот документ включен в настоящее описание в качестве ссылки. Соединение 1 также можно использовать отдельно или в комбинациях, описанных в настоящем документе, для лечения HCC или холангиокарциномы, включая внутрипеченочную холангиокарциному (IHCC). При использовании отдельно или в комбинациях, как описано в настоящем документе, соединение 1 можно вводить пациентам в любой из следующих суточных доз: 150 мг, 300 мг, 600 мг, 1000 мг, 1500 мг или 2000 мг. Величина суточной дозы может составлять от 50 мг до 3000 мг, от 50 мг до 600 мг или от 200 мг до 400 мг. Суточная доза может быть частью циклического режима, продолжительностью 14 дней или 21 день. Количество суточной дозы можно вводить в виде одной дозы или в виде нескольких доз.
Ингибиторы CDK 4/6, подходящие для использования в настоящем документе, могут включать, например, рибоциклиб, палбоциклиб и абемациклиб, и их фармацевтически приемлемые соли и гидраты.
Введение комбинации терапевтических средств включает введение отдельных терапевтических средств в комбинации в одной композиции или стандартной дозированной форме, введение отдельных терапевтических средств комбинации одновременно, но отдельно, или введение отдельных средств комбинации последовательно любым подходящим путем. Дозировка отдельных терапевтических средств комбинации может потребовать более частого введения одного из средств по сравнению с другим средством в комбинации. Следовательно, для обеспечения надлежащей дозировки упакованные фармацевтические продукты могут включать одну или несколько лекарственных форм, которые включают комбинацию средств, и одну или несколько лекарственных форм, которые включают одну из комбинаций средств, но не другое средство(а) комбинации.
Комбинации, как описано в настоящем документе, могут включать варианты осуществления, в которых одно или несколько из соединения 1 и ингибитора CDK 4/6 вводят в виде фармацевтически приемлемой соли или в виде свободного основания. Нет требования, чтобы оба соединения вводили в виде одной и той же фармацевтически приемлемой соли, но они могут быть. В конкретных вариантах осуществления комбинации включают форму свободного основания соединения 1 и форму свободного основания ингибитора CDK 4/6. В других вариантах осуществления комбинации включают HCl-форму соединения 1 и ингибитор CDK 4/6. В некоторых вариантах осуществления ингибитор CDK 4/6 может быть свободным основанием. В некоторых вариантах осуществления ингибитор CDK4/6 может быть фармацевтически приемлемой солью. В некоторых вариантах осуществления ингибитор CDK4/6 может быть гидратом.
«Фармацевтически приемлемая соль», как используется в настоящем документе, относится к кислотно-аддитивным солям или основно-аддитивным солям соединений по настоящему изобретению. Фармацевтически приемлемая соль представляет собой любую соль, которая сохраняет активность исходного соединения и не оказывает какого-либо чрезмерно вредного или нежелательного эффекта на субъекта, которому ее вводят, и в ситуации, в которой ее вводят. Фармацевтически приемлемые соли включают, но не ограничиваются ими, комплексы металлов и соли как неорганических, так и карбоновых кислот. Фармацевтически приемлемые соли также включают соли металлов, таких как алюминий, кальций, железо, магний, марганец и комплексные соли. Кроме того, фармацевтически приемлемые соли включают, но не ограничиваются ими, соли кислот, таких как уксусная, аспарагиновая, алкилсульфоновая, арилсульфоновая, аксетиловая, бензолсульфоновая, бензойная, дикарбоновая, дисерная, дивинная, масляная, кальция эдетат, камсиловая, угольная, хлорбензойная, лимонная, эдетовая, этандисульфоновая, лаурилсерная, этансульфоновая, этансульфиновая, муравьиная, фумаровая, глюкогептоновая, глюконовая, глутаминовая, гликолевая, гликолиларсаниловая, гексамовая, гексилрезорциновая, гидрабаминовая, бромистоводородная, хлористоводородная, иодистоводородная, гидроксинафтойная, изэтионовая, молочная, лактобионновая, малеиновая, яблочная, малоновая, миндальная, метансульфоновая, метилазотная, метилсерная, слизевая, муконовая, напсиловая, азотная, щавелевая, п-нитрометансульфоновая, памовая, пантотеновая, фосфорная, моноводородфосфорная, диводородфосфорная, фталевая, полигалактуроновая, пропионовая, салициловая, стеариновая, янтарная, сульфаминовая, сульфаниловая, сульфоновая, серная, дубильная, винная, теоклиновая, толуолсульфоновая и тому подобное.
Варианты осуществления могут быть гидрохлоридными солями. Фармацевтически приемлемые соли могут быть получены из аминокислот, включая, но не ограничиваясь этим, цистеин. Способы получения соединений в виде солей известны специалистам в данной области (см., например, Stahl et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Acta, Zurich, 2002; Berge et al., J. Pharm. Sci. 66: 1, 1977).
«Эффективное количество» комбинации терапевтических средств (например, соединение 1 и ингибитор CDK 4/6) представляет собой количество, достаточное для обеспечения наблюдаемого терапевтического эффекта по сравнению с HCC или IHCC, оставленными без лечения, у субъекта или пациента.
Активные средства, описанные в настоящем документе, могут быть объединены с фармацевтически приемлемым носителем для получения их фармацевтических композиций. Конкретный выбор носителя и состава будет зависеть от конкретного пути введения, для которого предназначена композиция.
«Фармацевтически приемлемый носитель», как используется к настоящем описании, относится к нетоксичному носителю, адъюванту или наполнителю, который не разрушает фармакологическую активность соединения, с которым оно составлено. Фармацевтически приемлемые носители, адъюванты или наполнители, которые можно использовать в композициях по настоящему изобретению, включают, но не ограничиваются ими, сорбиновую кислоту, сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, динатрийгидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий карбоксиметилцеллюлоза, полиакрилаты, воски, полиэтиленгликоль и ланолин.
Композиции по настоящему изобретению могут быть введены парентерально, перорально, ингаляцией в виде спрея, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар и тому подобное. В некоторых вариантах осуществления, композиция включает ингредиенты, которые получены из природных или неприродных источников. В некоторых вариантах осуществления, композиция или носитель могут быть представлены в стерильной форме. Неограничивающие примеры стерильного носителя включают воду, не содержащую эндотоксинов, или воду, не содержащую пирогенов.
Термин «парентеральный», как используется в настоящем описании, включает методы подкожной, внутривенной, внутримышечной, внутрисуставной, внутрисиновиальной, интрастернальный, интратекальной, внутрипеченочной, внутриочаговой и внутричерепной инъекций или инфузий. В конкретных вариантах осуществления соединения вводят внутривенно, перорально, подкожно или посредством внутримышечного введения. Стерильные инъекционные формы композиций по настоящему изобретению могут представлять собой водную или масляную суспензию. Эти суспензии могут быть получены в соответствии с методиками, известными в данной области, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе. Среди приемлемых носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные жирные масла обычно используются в качестве растворителя или суспендирующей среды.
Для этой цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты и их глицеридные производные пригодны для получения инъекционных препаратов, так же как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных видах. Эти масляные растворы или суспензии могут также содержать спиртовой разбавитель или диспергатор с длинной цепью, такой как карбоксиметилцеллюлоза или подобные диспергирующие агенты, которые обычно используются в составе фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Для целей получения также могут быть использованы другие обычно используемые поверхностно-активные вещества, такие как Tweens, Spans и другие эмульгирующие агенты, которые обычно используются при изготовлении фармацевтически приемлемых твердых, жидких или других лекарственных форм.
Для перорального введения соединение или соль могут быть предоставлены в приемлемой пероральной лекарственной форме, включая, но не ограничиваясь этим, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Также могут быть добавлены лубриканты, такие как стеарат магния. Для перорального введения в форме капсул полезные разбавители включают лактозу и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, активный ингредиент можно комбинировать с эмульгирующими и суспендирующими агентами. При желании также могут быть добавлены определенные подсластители, ароматизаторы или красители. Кроме того, могут быть добавлены консерванты. Подходящие примеры фармацевтически приемлемых консервантов включают, но не ограничиваются ими, различные антибактериальные и противогрибковые агенты, такие как растворители, например, этанол, пропиленгликоль, бензиловый спирт, хлорбутанол, соли четвертичного аммония и парабены (такие как метилпарабен, этилпарабен, пропилпарабен и тому подобное).
Под «немедленным высвобождением» подразумевается обычное высвобождение, при котором высвобождение лекарственного средства начинается сразу после введения. Как используется в настоящем описании, термин «немедленное высвобождение» включает лекарственные формы, которые позволяют лекарственному средству растворяться в желудочно-кишечном содержимом без намерения отсрочки или пролонгации растворения или абсорбции лекарственного средства. Задача состоит в том, чтобы лекарственное средство быстро высвобождалось после введения, например, чтобы было возможно высвободить по меньшей мере 80% лекарственного средства в течение приблизительно 30 минут после начала растворения в тесте на растворение.
Термин «замедленное высвобождение» или «пролонгированное высвобождение» включает лекарственные формы, характеристики высвобождения лекарственного средства которых по времени и/или месту выбраны для достижения терапевтических целей или удобства, не предлагаемых обычными лекарственными формами, такими как раствор или лекарственная форма с немедленным высвобождением.
Термин «устойчивое состояние» означает, что концентрация в плазме данного активного агента или комбинации активных агентов была достигнута и которая поддерживается последующими дозами активного агента(ов) на уровне, который находится на уровне или выше минимального эффективный терапевтической концентрации и ниже минимальной токсической концентрации в плазме для данного активного агента(ов).
Термин «единая композиция», как используется в настоящем документе, относится к единому носителю или наполнителю, приготовленному для доставки эффективных количеств обоих терапевтических средств пациенту. Единый носитель предназначен для доставки эффективного количества каждого из агентов вместе с любыми фармацевтически приемлемыми наполнителями или эксципиентами. В некоторых вариантах осуществления, носитель представляет собой таблетку, капсулу, пилюлю или пластырь.
Термин «стандартная доза» используется в настоящем документе для обозначения одновременного введения обоих агентов вместе в одной лекарственной форме пациенту, подлежащего лечению. В некоторых вариантах осуществления, стандартная доза представляет собой единую композицию. В определенных вариантах осуществления, стандартная доза включает один или несколько носителей, так что каждый носитель включает эффективное количество по меньшей мере одного из агентов (соединение 1 или ингибитор CDK 4/6) вместе с фармацевтически приемлемыми наполнителями и эксципиентами. В некоторых вариантах осуществления, стандартная доза представляет собой одну или несколько таблеток, капсул, пилюль или пластырей, вводимых пациенту одновременно.
Термин «диапазон доз», как используется в настоящем документе, относится к верхнему и нижнему пределу допустимого изменения количества указанного агента. Обычно дозу агента в любом количестве в указанном диапазоне можно вводить пациентам, подвергающимся лечению.
Термин «лечить» используется в настоящем документе для обозначения облегчения, уменьшения или ослабления по меньшей мере одного симптома заболевания у субъекта. Например, в отношении HCC термин «лечить» может означать остановку, отсрочку наступления (то есть период до клинического проявления заболевания или симптома заболевания) и/или снижение риска развития или ухудшения состояния симптома заболевания. Термин «защищать» используется в настоящем документе для обозначения предотвращения задержки или лечения, или всех, в случае необходимости, развития или продолжения или обострения симптомов заболевания у субъекта.
Термин «субъект» или «пациент» предназначен для обозначения животных, которые могут страдать от HCC или IHCC или страдают от них. Примеры субъектов или пациентов включают млекопитающих, например, людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных животных, отличных от человека. В определенных вариантах осуществления, субъектом является человек, например, человек, страдающий от, подверженный риску поражения или потенциально поддающийся поражению HCC или IHCC.
Термин «примерно» или «приблизительно» обычно означает в пределах 20%, более предпочтительно в пределах 10% и наиболее предпочтительно еще в пределах 5% от заданного значения или диапазона. Альтернативно, особенно в биологических системах, термин «примерно» означает в пределах приблизительно log (то есть на порядка) от заданного значения, в том числе оно может отличаться от него в два раза.
Использование обозначений единственного числа и аналогичных ссылок в контексте описания изобретения (особенно в контексте следующей формулы изобретения) должно истолковываться как охватывающее как единственное, так и множественное число, если иное не указано в настоящем документе или явно не противоречит контексту. Термины «содержащий», «имеющий», «включающий» и «состоящий» следует истолковывать как неограничивающие термины (то есть означающие «включающий, но не ограничивающийся ими»), если не указано иное. Перечисление диапазонов значений в настоящем документе просто предназначено для того, чтобы служить кратким способом индивидуальной ссылки на каждое отдельное значение, попадающее в этот диапазон, если в настоящем документе не указано иное, и каждое отдельное значение включено в описание, как если бы оно было отдельно указано в настоящем документе.
Термин «усиленный эффект», как используется в настоящем документе, относится к действию двух агентов, которые вводят вместе, обеспечивают больший или улучшенный результат, чем когда отдельные агенты вводятся отдельно без совместного введения другого агента. Введение агентов вместе может обеспечить усиленный эффект, когда они вводятся одновременно или последовательно. Последовательное введение агентов включает введения, разделенные несколькими секундами, минутами, часами или днями. Совместное введение агентов может обеспечить усиленный эффект, когда агенты вводят или в виде части одной композиции, или при введении в отдельных композициях. Примеры агентов, которые можно вводить вместе, включают соединение 1 и ингибиторы CDK4/6. Дополнительные примеры агентов, которые можно вводить вместе, включают i) соединение 1 и рибоциклиб; ii) соединение 1 и палбоциклиб; и iii) соединение 1 и абемациклиб.
Более высокий или улучшенный результат усиленного эффекта может включать, например, одно или несколько из следующего: i) улучшенное качество ответа опухоли, ii) улучшенная скорость ответа опухоли и iii) ответ опухоли, который больше, чем аддитивность ответа, который мог бы быть достигнут, если бы отдельные агенты вводились раздельно. Примеры улучшенного качества ответа опухоли могут включать полную регрессию (CR) вместо частичной регрессии (PR), стабильного заболевания (SD) или прогрессирования заболевания (PD). Другой пример улучшенного качества ответа опухоли может включать частичную регрессию (PR) вместо стабильного заболевания (SD) или прогрессирующего заболевания (PD). Другой пример улучшенного качества ответа опухоли может включать стабильное заболевание (SD) вместо прогрессирования заболевания (PD). Могут быть проведены контролируемые исследования для определения, приводило ли введение совместно агентов к усиленному эффекту ответа опухоли, больше, чем аддитивность соответствующих ответов, достигаемых, когда отдельные агенты соответственно вводятся отдельно, например, на мышах, крысах, собаках, обезьянах или других животных. Такие контролируемые исследования могут оценивать, например, результирующий объем опухоли или метастатический или другой статус. Аналогичным образом, контролируемые исследования могут быть использованы для определения усиленного эффекта, приводящего к более быстрому ответу опухоли.
В некоторых вариантах осуществления, лечение предоставляется субъекту, имеющему гепатоцеллюлярную карциному с измененным статусом FGFR4 и/или FGF19 (фактор роста фибробластов 19).
В некоторых вариантах осуществления, лечение может включать или проводиться в комбинации с анализом статуса FGFR4 и/или FGF19 в биологическом образце, содержащем клетки указанной гепатоцеллюлярной карциномы, и если указанная гепатоцеллюлярная карцинома демонстрирует изменение FGFR4 и/или FGF19, лечение субъекта эффективным для лечения количеством терапевтической комбинации, как описано в настоящем документе.
Способы лечения
В настоящем описании представлена комбинированная терапия, полезная для лечения HCC или IHCC. Как будет показано ниже, комбинации, представленные в настоящем документе, могут иметь ряд преимуществ.
Одним из преимуществ комбинации, раскрытой в настоящем документе, является неожиданный усиленный эффект комбинации соединения 1 и ингибитора CDK 4/6 на лечение ингибирования роста опухоли и лечение НСС или IHCC.
В некоторых вариантах осуществления, в настоящем документе представлена единая фармацевтическая композиция, содержащая комбинацию соединения 1 и ингибитора CDK 4/6. Преимущество, представленное в настоящем документе, представляет собой усиленный эффект, который приводит к лечению HCC по сравнению с лечением однократной дозой любого лекарственного средства. Когда лекарственные средства предоставляются в виде единичной стандартной дозы или единой композиции, «количество таблеток» у пациента, страдающего HCC, не увеличивается.
В соответствии с вышеизложенным, в одном аспекте в настоящем документе предложена комбинация лекарственных средств, полезная для лечения, предотвращения, остановки, задержки начала и/или снижения риска развития или реверсии HCC у млекопитающего, включающая введение указанному млекопитающему комбинированной терапии, включающей эффективное количество соединения 1 и эффективное количество ингибитора CDK 4/6.
В некоторых вариантах осуществления, определяют, что подлежащий лечению субъект (например, пациент) не отвечает или устойчив к одной или нескольким терапиям HCC, например, к соединению 1. В других вариантах осуществления, человек, подлежащий лечению, реагирует на терапию соединением 1, но терапия была улучшена с введением ингибитора CDK 4/6. Например, пациенту вводят соединение 1 (например, от 50 мг до 600 мг в день, от 200 мг до 400 мг в день или 300 мг в день в течение некоторого периода времени, например, более одного дня, более двух дней, более трех дней, более одной недели, в течение 21 дня, более одного месяца и т.д. После этого времени этому пациенту можно вводить ингибитор CDK 4/6 в комбинации с соединением 1.
Количество ингибитора CDK 4/6 может варьироваться в зависимости от используемого ингибитора CDK 4/6. Например, палбоциклиб можно вводить, например, в дозе 75, 100 или 125 мг/день; рибоциклиб можно вводить, например, в дозе 200, 400 или 600 мг/день. Обычно доза вводится перорально в виде одной капсулы в течение 21 последовательного дня, после чего следует 7 дневный период без лечения.
Суточная доза может быть частью циклического режима, продолжительностью 14-21 дня или дольше. Количество суточной дозы можно вводить в виде одной дозы или в виде нескольких доз.
Специалистам в данной области понятно, что эффективная доза активного лекарственного средства может быть ниже, чем вводимое фактическое количество. Таким образом, в настоящем описании представлены дозы, необходимые для достижения терапевтической дозы.
В различных вариантах осуществления в настоящем документе предоставлены способы лечения HCC путем введения эффективного количества соединения 1 и ингибитора CDK 4/6 индивидууму, имеющему HCC. Количество комбинации агентов является эффективным для лечения HCC. В одном варианте осуществления комбинация агентов имеет усиленный эффект. В одном варианте осуществления, даже если один или несколько агентов, вводимых отдельно в определенной дозировке, могут быть эффективными, при введении в комбинации при той же дозировке каждого агента, лечение является более эффективным. Например, в одном варианте осуществления комбинация соединения 1 и палбоциклиба является более эффективной, чем введение каждого агента отдельно. В другом варианте осуществления комбинация соединения 1 и рибоциклиба является более эффективной, чем введение каждого агента отдельно.
Дозы
Оптимальная доза комбинации агентов для лечения HCC может быть определена эмпирически для каждого индивидуума с использованием известных способов и будет зависеть от множества факторов, включая активности агентов; возраст, массу тела, общее состояние здоровья, пол и питание индивидуума; время и путь введения; и другие лекарственные средства, которые принимает индивидуум. Оптимальные дозы могут быть установлены с использованием рутинных испытаний и процедур, которые хорошо известны в данной области.
Для комбинированной терапии по настоящему изобретению суточная доза соединения 1 находится в диапазоне от 50 мг до 600 мг. В некоторых вариантах осуществления, суточная доза соединения 1 составляет до 600 мг. В определенных вариантах осуществления, суточная доза соединения 1 составляет до 400 мг. В различных вариантах осуществления, суточная доза соединения 1 составляет до 300 мг. В определенных вариантах осуществления, суточная доза соединения 1 составляет от 200 мг до 400 мг. В одном варианте осуществления, суточная доза составляет 300 мг.
Время введения может быть выбрано таким, чтобы оба лекарственных средства вводятся одновременно, по отдельности или последовательно, или утром, или ночью. Кроме того, одно лекарственное средство можно вводить утром, а другое ночью. В определенных вариантах осуществления, оба лекарственных средства можно вводить в виде единого состава таблетки, капсулы, пилюли, пластыря или желе, один раз в день, утром или ночью.
Количество комбинации агентов, которые можно комбинировать с материалами-носителями для получения единичной лекарственной формы, будет варьироваться в зависимости от индивидуума, получающего лечение, и конкретного способа введения. В некоторых вариантах осуществления стандартные лекарственные формы, содержащие комбинацию агентов, как описано в настоящем документе, будут содержать количества каждого агента комбинации, которые обычно вводят, когда агенты вводят по отдельности.
Фармацевтические композиции и пути введения
В настоящем документе предоставлены фармацевтические композиции, включающие комбинацию агентов для лечения HCC. Фармацевтические композиции могут дополнительно включать носитель или наполнитель, стабилизатор, ароматизатор и/или краситель.
Комбинация агентов может быть введена с использованием различных путей введения, известных специалистам в данной области. Пути введения включают пероральное введение. В определенных вариантах осуществления, фармацевтическая композиция, включающая комбинацию агентов, может приниматься перорально в форме жидкости, сиропа, таблетки, капсулы, порошка, капель, жевательных таблеток или растворимого диска. Альтернативно, фармацевтические композиции по настоящему изобретению можно вводить внутривенно или трансдермально. Дополнительные способы введения известны специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, Gennaro A. R., Ed., 20.sup.th Edition, Mack Publishing Co., Easton, Pa.).
В некоторых вариантах осуществления, соединение 1 и ингибитор CDK 4/6 получают в виде пасты, желе или суспензии. Например, лекарственные средства растворяют, инкапсулируют или суспендируют в форме частиц лекарственного средства, микрокапсулированных частиц или частиц лекарство-полимер в желатиновом растворе или полутвердом состоянии. Преимущество перорального желеобразного состава состоит в том, что лекарственные средства легче вводить пациентам, которым трудно глотать таблетки, капсулы или пилюли. В определенных вариантах осуществления, оба агента тщательно перемешивают и суспендируют в подходящей среде для образования пасты или геля. Дополнительные агенты необязательно могут быть смешаны для придания вкуса при пероральном введении. Арахисовое масло или альгинат, ароматизированные малиной и подсластителем, являются примерами многих подходящих средств, маскирующих вкус. В различных вариантах осуществления, паста или желе также могут быть сформулированы с подходящими связующими веществами или эксципиентами, известными в данной области для местного применения.
Способы получения лекарственных форм с замедленным высвобождением в форме таблеток, капсул или пилюль известны в данной области. В некоторых вариантах осуществления, лекарственную форму с замедленным высвобождением получают путем покрытия активного ингредиента лекарственного средства полимером, предпочтительно нерастворимым в воде полимером. Например, нерастворимый в воде полимер, используемый в фармацевтической области в качестве покрывающего агента с замедленным высвобождением, кишечнорастворимый покрывающий агент или желудочнорастворимый покрывающий агент. Нерастворимый в воде полимер может включать, например, этилцеллюлозу, очищенный шеллак, белый шеллак, аминоалкилметакрилатный сополимер RS, гидроксипропилметилцеллюлоза фталат, гидроксипропилметилцеллюлозы ацетата сукцинат, карбоксиметилэтилцеллюлозу, ацетатфталат целлюлозы, метакриловой кислоты сополимер L, метакриловой кислоты сополимер LD, метакриловой кислоты сополимер S, аминоалкилметакрилатный сополимер E или поливинилацетил диэтиламиноацетат.
Тип, степень замещения и молекулярная масса нерастворимых в воде полимеров могут зависеть от растворимости активного ингредиента в воде или спирте, желаемого уровня замедленного высвобождения и тому подобного. Нерастворимые в воде полимеры можно использовать отдельно или в комбинации. Кроме того, могут быть включены гидрогенизированное масло, стеариновая кислота или цетанол в качестве вспомогательного покрывающего агента, и триглицерид со средней цепью, триацетин, триэтилцитрат или цетанол в качестве желатинизатора.
В некоторых вариантах осуществления, лекарственная форма с замедленным высвобождением представляет собой матричную таблетку или гранулу. Активный ингредиент может быть покрыт до 3 различных типов полимеров. Эти три различных типа полимеров могут включать: 1) нерастворимый в воде полимер, такой как этилцеллюлоза; 2) рН-независимый гелеобразующий полимер, такой как гидроксипропилметилцеллюлоза; и 3) рН-зависимый гелеобразующий полимер, такой как альгинат натрия. Эти три различных типа полимеров можно использовать вместе для ослабления скорости высвобождения лекарственного средства.
Лекарственные формы: свойства высвобождения
Составы с замедленным высвобождением могут достигать определенной степени устойчивого эффекта. Однако степень воздействия и/или биодоступность активного ингредиента может варьироваться в зависимости от множества факторов, таких как, например, окно абсорбции, носители или эксципиенты, используемые в препарате, способ доставки препарата и/или время прохождения активного ингредиента через желудочно-кишечный тракт пациента.
Комбинированная терапия может включать по меньшей мере одну часть с замедленным высвобождением для выполнения функции замедленного высвобождения и одну часть с немедленным высвобождением для выполнения функции немедленного высвобождения. В определенных вариантах осуществления, когда комбинированная терапия находится в одной лекарственной форме, она может быть в форме таблеток, образованных из смеси гранул с замедленным высвобождением, составляющих часть с замедленным высвобождением, и гранул с немедленным высвобождением, составляющих часть с немедленным высвобождением, капсульного препарата, полученного путем наполнения капсулы гранулами длительного высвобождения и гранулами немедленного высвобождения или таблетками, покрытыми прессованием, в которых внешний слой, составляющий часть немедленного высвобождения, формируют на внутреннем ядре, составляющем часть замедленного высвобождения. Однако нет никаких ограничений для вышеуказанных вариантов осуществления.
Кроме того, нет особых ограничений в отношении состояния содержания каждого лекарственного средства в композиции или в части с немедленным высвобождением или в части с замедленным высвобождением; соединение 1 может быть равномерно диспергировано в композиции, части с немедленным высвобождением или части с замедленным высвобождением или может содержаться только в одной части композиции, части с немедленным высвобождением или части с замедленным высвобождением, или может содержаться так, что существует градиент концентрации.
Часть с замедленным высвобождением в композиции по настоящему изобретению может содержать, по меньшей мере, одно независимое от рН полимерное вещество или рН-зависимое полимерное вещество для контролирования высвобождения лекарственного средства.
Используемое здесь независимое от рН полимерное вещество может содержать полимерное вещество, состояние заряда которого практически не изменяется в условиях рН, обычно встречающихся в желудочно-кишечном тракте, в частности, от рН 1 до рН 8. Это означает, например, полимерное вещество, которое не имеет функциональные группы, состояние заряда которых изменяется в зависимости от pH, такие как основные функциональные группы, такие как аминогруппы, или кислотные функциональные группы, такие как группы карбоновых кислот. Следует отметить, что независимое от рН полимерное вещество может быть включено для придания композиции в соответствии с настоящим изобретением функции замедленного высвобождения, но также может быть включено для другой цели. Кроме того, независимое от рН полимерное вещество, используемое в настоящем изобретении, может быть нерастворимым в воде или может разбухать в воде или растворяться в воде с образованием геля.
Примеры нерастворимых в воде независимых от рН полимерных веществ включают, но не ограничиваются ими, простые эфиры целлюлозы, сложные эфиры целлюлозы и сополимеры метакриловой кислоты и акриловой кислоты (торговое название Eudragit, производство Rohm GmbH & Co. KG, Darmstadt, Germany). Примеры включают, но не ограничиваются ими, алкиловые эфиры целлюлозы, такие как этилцеллюлоза (торговое название Ethocel, производство Dow Chemical Company, USA), этилметилцеллюлоза, этилпропилцеллюлоза или изопропилцеллюлоза, и бутилцеллюлоза, аралкиловые эфиры целлюлозы, такие как бензилцеллюлоза, цианоалкиловые эфиры целлюлозы, такие как цианоэтилцеллюлоза, сложные эфиры целлюлозы и органических кислот, такие как ацетобутират целлюлозы, ацетат целлюлозы, пропионат целлюлозы или бутират целлюлозы, и ацетопропионат целлюлозы, сополимеры этилакрилата и метилметакрилата (торговое название Eudragit NE, производство Rohm GmbH & Co. KG, Darmstadt, Germany), и аминоалкилметакрилатный сополимер RS (торговые названия Eudragit RL, Eudragit RS). Не существует особых ограничений в отношении среднего диаметра частиц нерастворимого в воде полимера, используемого в настоящем изобретении, но обычно чем ниже этот средний диаметр частиц, тем лучше характеристики, причем средний диаметр частиц предпочтительно составляет от 0,1 до 100 мкм, более предпочтительно от 1 до 50 мкм, особенно предпочтительно от 3 до 15 мкм, наиболее предпочтительно от 5 до 15 мкм. Кроме того, примеры водорастворимых или разбухающих в воде независимых от рН полимерных веществ включают, но не ограничиваются ими, полиэтиленоксид (торговое название Polyox, производство Dow Chemical Company, молекулярная масса 100000-7000000), гидроксипропилцеллюлозу с низкой степенью замещения (торговое название L-HPC, производство Shin-Etsu Chemical, Japan), гидроксипропилцеллюлозу (торговое название HPC, производство Nippon Soda, Co., Ltd, Japan), гидроксипропилметилцеллюлозу (торговые названия Metolose 60SH, 65SH, 90SH, производство Shin-Etsu Chemical, Japan), и метилцеллюлозу (торговое название Metolose SM, производство Shin-Etsu Chemical, Japan).
В некоторых вариантах осуществления в композиции может содержаться одно независимое от pH полимерное вещество, или может содержаться множество независимых от pH полимерных веществ. Независимое от рН полимерное вещество, если оно используется в описанных в настоящем описании вариантах осуществления, может представлять собой нерастворимое в воде полимерное вещество, более предпочтительно этилцеллюлозу, сополимер этилакрилата и метилметакрилата (торговое название Eudragit NE), или аминоалкилметакрилатный сополимер RS (торговое название Eudragit RL, Eudragit RS). Особенно предпочтительным является, по меньшей мере, одно из этилцеллюлозы и аминоалкилметакрилатного сополимера RS. Наиболее предпочтительной является этилцеллюлоза. Не существует особых ограничений на количество независимого от рН полимерного вещества, содержащегося в композиции; это количество может быть соответствующим образом скорректировано в соответствии с такой целью, как контроль за замедленным высвобождением лекарственного средства.
Зависимое от рН полимерное вещество, которое можно использовать в описанных в настоящем документе вариантах осуществления, может представлять собой полимерное вещество, состояние заряда которого изменяется в условиях pH, обычно встречающихся в желудочно-кишечном тракте, в частности, от pH 1 до pH 8. Это означает, например, полимерное вещество, имеющее функциональные группы, состояние заряда которых изменяется в зависимости от рН, такие как, основные функциональные группы, такие как аминогруппы, или кислотные функциональные группы, такие как группы карбоновых кислот. Зависимые от рН функциональные группы рН-зависимого полимерного вещества предпочтительно представляют собой кислотные функциональные группы, причем рН-зависимое полимерное вещество наиболее предпочтительно имеет группы карбоновых кислот.
Зависимое от рН полимерное вещество, используемое в настоящем изобретении, может быть нерастворимым в воде или может разбухать в воде или растворяться в воде с образованием геля. Примеры рН-зависимых полимерных веществ, используемых в настоящем изобретении, включают, но не ограничиваются ими, энтеросолюбильные полимерные вещества. Примеры энтеросолюбильных полимерных веществ включают, но не ограничиваются ими, сополимеры метакриловой кислоты и метилметакрилата (Eudragit L100, Eudragit S100, производство Rohm GmbH & Co. KG, Darmstadt, Germany), сополимеры метакриловой кислоты и этилакрилата (Eudragit L100-55, Eudragit L30D-55, производство Rohm GmbH & Co. KG, Darmstadt, Germany), гидроксипропилметилцеллюлоза фталат (HP-55, HP-50, производство Shin-Etsu Chemical, Japan), гидроксипропилметилцеллюлозы ацетата сукцинат (AQOAT, производство Shin-Etsu Chemical, Japan), карбоксиметил этилцеллюлозу (CMEC, производство Freund Corporation, Japan), и ацетатфталат целлюлозы.
Примеры рН-зависимых полимерных веществ, которые разбухают в воде или растворяются в воде с образованием геля, включают, но не ограничиваются ими, альгиновую кислоту, пектин, карбоксивиниловый полимер и карбоксиметилцеллюлозу. В настоящем изобретении в композиции может содержаться одно зависимое от рН полимерное вещество или может содержаться множество зависимых от рН полимерных веществ. Зависимое от рН полимерное вещество, используемое в настоящем изобретении, предпочтительно представляет собой энтеросолюбильное полимерное вещество, более предпочтительно, сополимер метакриловой кислоты и этилакрилата, сополимер метакриловой кислоты и метилметакрилата, гидроксипропилметилцеллюлоза фталат или гидроксипропилметилцеллюлозы ацетата сукцинат, особенно предпочтительно сополимер метакриловой кислоты и этилакрилата.
При использовании рН-зависимого полимерного вещества в процессе изготовления композиции в соответствии с настоящим изобретением коммерчески доступный продукт порошкового типа или гранулированного типа, или в виде суспензии, в котором рН-зависимое полимерное вещество заранее диспергировано в растворителе, может быть использован как есть, или такой коммерчески доступный продукт может быть использован диспергированным в воде или органическом растворителе. Чем ниже диаметр частиц pH-зависимого полимерного вещества, тем лучше характеристики, причем pH-зависимое полимерное вещество предпочтительно относится к порошковому типу. В случае сополимера метакриловой кислоты и этилакрилата примером является Eudragit L100-55. Не существует особых ограничений в отношении среднего диаметра частиц pH-зависимого полимерного вещества, используемого в настоящем изобретении, но средний диаметр частиц предпочтительно составляет от 0,05 до 100 мкм, более предпочтительно от 0,05 до 70 мкм, наиболее предпочтительно от 0,05 до 50 мкм. Более того, нет особых ограничений на количество pH-зависимого полимерного вещества, например, в случае энтеросолюбильного полимерного вещества, количество обычно составляет от 0,1 до 90 мас.ч., предпочтительно от 1 до 70 масс.ч. более предпочтительно от 5 до 60 мас.ч., особенно предпочтительно от 10 до 50 масс.ч. в расчете на 100 масс.ч. композиции.
Комбинированный препарат в соответствии с вариантами осуществления, описанными в настоящем документе, может дополнительно включать любую из различных добавок, таких как любой из различных фармакологически приемлемых носителей, таких как разбавители, лубриканты, связующие вещества и разрыхлители, а также консерванты, красители, подсластители, пластификаторы, пленкообразующие агенты и т.д. по мере необходимости. Примеры разбавителей включают, но не ограничиваются ими, лактозу, маннит, двухосновный фосфат кальция, крахмал, прежелатинизированный крахмал, кристаллическую целлюлозу, легкий кремниевый ангидрид, синтетический силикат алюминия, метасиликат алюмината магния или тому подобное. Примеры лубрикантов включают, но не ограничиваются ими, стеарат магния, стеарат кальция, тальк, стеарилфумарат натрия или тому подобное. Примеры связующих веществ включают, но не ограничиваются ими, гидроксипропилцеллюлозу, метилцеллюлозу, натрийкарбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон или тому подобное. Примеры разрыхлителей включают, но не ограничиваются ими, карбоксиметилцеллюлозу, кальций карбоксиметилцеллюлозу, кроскармеллозу натрия, натрий карбоксиметилкрахмал, гидроксипропилцеллюлозу с низкой степенью замещения или тому подобное. Примеры консервантов включают, но не ограничиваются ими, сложные эфиры параоксибензойной кислоты, хлорбутанол, бензиловый спирт, фенетиловый спирт, дегидроуксусную кислоту, сорбиновую кислоту или тому подобное. Предпочтительные примеры красителей включают, но не ограничиваются ими, нерастворимые в воде красочные пигменты, природные пигменты (например, β-каротин, хлорофилл, красный оксид железа), желтый оксид железа, красный оксид железа, черный оксид железа или тому подобное. Предпочтительные примеры подсластителей включают, но не ограничиваются ими, сахарин натрия, дикалийглицирризат, аспартам, стевию или тому подобное. Примеры пластификаторов включают, но не ограничиваются ими, сложные эфиры глицерина и жирных кислот, триэтилцитрат, пропиленгликоль, полиэтиленгликоль или тому подобное. Примеры агентов для нанесения пленочных покрытий включают, но не ограничиваются ими, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу или тому подобное.
Методы изготовления
Для изготовления вариантов осуществления, описанных в настоящем документе, может быть использован один традиционный способ или комбинация традиционных способов. Например, при изготовлении гранул, содержащих лекарственное средство, в виде части с замедленным высвобождением или части с немедленным высвобождением, гранулирование является основной операцией, но это может сочетаться с другими операциями, такими как смешивание, сушка, просеивание и классификация. В качестве способа гранулирования, например, способ влажного гранулирования, в котором связующее и растворитель добавляют к порошку и проводят грануляцию, метод сухого гранулирования, при котором порошок прессуют и проводят грануляцию, способ гранулирования расплава, в котором добавляют связующее, которое плавится при нагревании, и проводят нагревание и гранулирование или тому подобное.
Кроме того, в соответствии со способом гранулирования, можно использовать способ получения, такой как способ гранулирования смешиванием с использованием планетарного миксера, шнекового смесителя или тому подобного, способ гранулирования с интенсивным смешиванием с использованием миксера Хеншель, супер-миксера или тому подобного, способ гранулирование способом экструзии с использованием цилиндрического гранулятора, роторного гранулятора, шнекового экструдера-гранулятора, гранулятор типа пресс-гранулятора или тому подобное, способ влажной грануляция с большим усилием сдвига, способ грануляции в псевдоожиженном слое, способ гранулирования под давлением, способ гранулирования измельчением или способ гранулирования распылением. После гранулирования можно проводить сушку с использованием сушилки, псевдоожиженного слоя или тому подобного, дробление и просеивание для получения гранул или мелких гранул для использования. Кроме того, может быть использован растворитель для грануляции при приготовлении композиции по настоящему изобретению. Не существует особых ограничений в отношении такого растворителя для грануляции, которым может быть вода или любой из различных органических растворителей, например, вода, низший спирт, такой как метанол или этанол, кетон, такой как ацетон или метилэтилкетон, метиленхлорид или их смесь.
Для гранул замедленного высвобождения, содержащихся в вариантах осуществления, по меньшей мере одно лекарственное средство и по меньшей мере одно, выбранное из независимых от рН полимерных веществ и рН-зависимых полимерных веществ, смешивают вместе, разбавитель и связующее добавляют по мере необходимости, и проводят гранулирование для получения гранулированных веществ. Полученное гранулированное вещество сушат с использованием полочной сушилки, сушилки с псевдоожиженным слоем или тому подобного, и просеивание проводят с использованием мельницы или вибратора, в результате чего могут быть получены гранулы с замедленным высвобождением. Альтернативно, в качестве способа изготовления гранул замедленного высвобождения в настоящем изобретении, можно добавить, по меньшей мере, одно лекарственное средство, по меньшей мере, одно, выбранное из независимых от рН полимерных веществ и рН-зависимых полимерных веществ, и, при необходимости, разбавитель и связующее вещество с использованием сухого пресса, такого как роликовый пресс или поршневая таблетировочная машина, и выполняют формование прессованием при перемешивании, а затем проводят гранулирование путем измельчения до подходящего размера. Гранулированное вещество, полученное с использованием такого гранулятора, может быть использовано как есть, в виде гранул или мелких гранул согласно настоящему изобретению или может быть дополнительно подвергнуто крекингу с использованием силовой мельницы, валкового гранулятора, роторной мельницы и т.п. просеивали для получения гранул замедленного высвобождения. Следует отметить, что гранулы с немедленным высвобождением могут быть изготовлены так же, как и гранулы с замедленным высвобождением.
Полученный прямым прессованием продукт может быть изготовлен в виде содержащей лекарственное средство части с замедленным высвобождением или части с немедленным высвобождением, или в виде композиции, описанной в настоящем документе, с использованием одного общепринятого способа или комбинации общепринятых способов. Например, используют по меньшей мере одно лекарственное средство, по меньшей мере, одно, выбранное из независимых от pH полимерных веществ и pH-зависимых полимерных веществ, разбавителя, такого как маннит или лактоза, связующего, такого как поливинилпирролидон или кристаллическая целлюлоза, дезинтегранта, такого как кармеллоза натрия или кросповидон, и лубриканта, такого как стеарат магния или тальк, и таблетирование осуществляют с использованием обычного метода, посредством которого может быть получен продукт, формованный прессованием. В этом случае таблетирование является основной операцией в способе изготовления продукта, формованного прессованием, но это может сочетаться с другими операциями, такими как смешивание, сушка, образование сахарного покрытия и нанесение покрытия.
Примеры способа таблетирования включают, но не ограничиваются этим, формирование прямым прессованием, при котором по меньшей мере одно лекарственное средство и фармакологически приемлемые добавки смешивают вместе, а затем смесь непосредственно прессуют в таблетки с использованием таблетировочной машины, и сжатие сухих гранул или сжатие влажных гранул, при котором гранулы с замедленным высвобождением или гранулы с немедленным высвобождением согласно настоящему изобретению подвергают формованию под давлением после добавления лубриканта или разрыхлителя по мере необходимости. Нет никаких особых ограничений на таблетировочную машину, используемую при формовании прессованием; например, можно использовать однопуансонную таблетировочную машину, роторную таблетировочную машину или таблетировочную машину для прессованных покрытий.
Содержащие лекарственное средство гранулы с замедленным высвобождением или гранулы с немедленным высвобождением, или прессованный под давлением продукт в соответствии с вариантами осуществления, приведенными в настоящем документе, могут быть использованы как есть в форме гранул или таблетки в качестве препарата, но могут также быть подвергнуты последующей обработке для получения препарата. Например, на прессованный под давлением продукт или гранулы можно нанести пленочное покрытие, с использованием пленочного основного материала, такого как этилцеллюлоза, казеин, метилцеллюлоза, гидроксипропилметилцеллюлоза, сополимер метакриловой кислоты L, ацетатфталат целлюлозы, шеллак или тому подобное, или нанести сахарное покрытие с использованием жидкости для сахарного покрытия, содержащей сахарозу, сахарный спирт, порошок гуммиарабика, тальк или тому подобное, в результате чего получают таблетки, покрытые пленочной оболочкой, или таблетки, покрытые сахарной оболочкой. Одним растворителем в этой технологии нанесения покрытия может быть очищенная вода, но также можно использовать органический растворитель, такой как спирт, кетон, простой эфир или хлорированный углеводород, или их смесь. Например, этанол, ацетон, метиленхлорид или тому подобное можно использовать в качестве органического растворителя. Кроме того, в качестве устройства для нанесения покрытия можно использовать устройство, обычно используемое в технологиях нанесения покрытия для изготовления лекарственных средств, с примерами, включающими устройство для нанесения покрытия распылением, в котором нанесение покрытия осуществляют путем распыления покрывающей жидкости или тому подобного, и роторный гранулятор для нанесения слоя в псевдоожиженном слое.
В случае изготовления препаратов в виде капсул, препараты в виде капсул могут быть изготовлены путем наполнения гранулами замедленного высвобождения или гранулами немедленного высвобождения, как указано выше, или мини-таблеткам твердых желатиновых капсул или капсул HPMC с использованием автоматической машины для наполнения капсул. Альтернативно, в случае препаратов для введения через трубку или сухого сиропа, который используют смешанным с водой или тому подобным, когда требуется, гранулы замедленного высвобождения или гранулы немедленного высвобождения, как указано выше, могут быть смешаны с загустителем или диспергатором так, чтобы диспергировать эти гранулы, в этом случае смесь получают в виде гранул или таблеток. Кроме того, жидкость или желе могут быть приготовлены с использованием воды и веществ, выбранных из диспергаторов, эмульгаторов, загустителей, консервантов, регуляторов pH, подсластителей, вкусовых добавок, ароматизаторов и так далее. Тем не менее, в отношении других способов изготовления, нет никаких ограничений на вышеизложенное.
Для более полного понимания описанных в настоящем документе вариантов осуществления приводятся следующие примеры. Следует понимать, что эти примеры приведены только в иллюстративных целях и не должны толковаться как ограничивающие.
Примеры
Материалы и методы
Исследованные клеточные линии
Используемая клеточная линия, JHH7, была получена из Japanese Collection of Research Bioresources Cell Bank (JCRB), проверена на отсутствие микобактерий и подтверждена на идентичность с помощью анализа короткого тандемного повтора 9 маркеров.
Условия поддержания и изучения клеточной линии
Клетки JHH7 содержали в среде E William's (Thermo Fisher Scientific, 12551-032) с 10% фетальной бычьей сывороткой. Клетки поддерживали до и во время экспериментов при 37°С, 5% СО2 и при 95% относительной влажности. Количество пассажей клеток было ограничено от 12 до 20.
Получение ксенотрансплантата, дозирование и измерение противоопухолевой активности
Клеточную линию гепатоцеллюлярного рака человека JHH7 культивировали в среде E Уильяма (Thermo Fisher Scientific, 12551-032), содержащей 10% эмбриональной бычьей сыворотки при 37°C в атмосфере 5% CO2 и поддерживали в экспоненциальной фазе роста. Для сбора клетки промывали забуференным фосфатом солевым раствором, инкубировали с 0,25% трипсин-EDTA и суспендировали в смеси 1:1 среды E Уильяма и матригеля (Corning) в конечной концентрации 5×107клеток/мл. Для получения ксенотрансплантатов 0,1 мл инокулята инъецировали подкожно в правую боковую область NU/NU иммунокомпрометированных самок мышей 6-8 недель, получая конечную концентрацию 5×106 клеток/мышь. Когда средний объем опухоли (TV) достиг приблизительно 170 мм3 (через 10 дней после имплантации), 144 мыши были отобраны на основе их TV и были разделены на 18 групп лечения по 8 животных на группу. Пероральное (PO) лечение соединением 1 (300 и 500 мг/кг) отдельно или в комбинации с носителем (контроль) или палбоциклибом (50 и 100 мг/кг) или рибоциклибом (75 и 150 мг/кг), вводимым один раз в день (QD), продолжалось 8 дней. Объем введения (0,1 мл/10 г массы тела) рассчитывали из массы тела отдельной мыши (BW) перед введением. Массу тела измеряли ежедневно и измерения опухоли проводили два раза в неделю.
Модель первичной гепатоклеточной карциномы человека LIX066 модель от ChemPartner (shangpharma.com) имплантировали самкам мышей с тяжелым комбинированным иммунодефицитом (SCID). Когда опухоли развивались у мышей, мышей умерщвляли, а опухоли резецировали и имплантировали самкам голых мышей для сохранения опухоли, диагностики гистопатологии и исследования эффективности in vivo. Ткани солидной опухоли были лишены некротических компонентов, разрезаны на кусочки по 10-15 мг и смешаны. Три-пять кусочков смешивали с 15-30 мл матригеля и имплантировали в бок 6-8-недельных NU/NU иммунокомпрометированных самок мышей весом 18-20 г голых мышей. 30 мышей со средним объемом опухоли 160 мм3 отбирали и рандомизировали на 6 групп по 5 мышей в каждой. Все первичные опухоли человека, использованные в этом исследовании, перенесли 3-5 пассажей in vivo, и гистология опухоли каждого из них сохранялась в процессе серийной трансплантации. Пероральное (PO) лечение Соединением 1 (300 и 500 мг/кг) отдельно или в комбинации с носителем (контроль) или палбоциклибом (100 мг/кг) вводимым один раз в день (QD) продолжалось в течение 17 дней. Объем введения (0,1 мл/10 г массы тела) рассчитывали из массы тела отдельной мыши (BW) перед введением. Массу тела измеряли ежедневно и измерения опухоли проводили два раза в неделю.
TV в мм3 рассчитывали по следующей формуле: TV=длина×ширина2×0,5 длина: наибольший диаметр опухоли (мм) ширина: диаметр перпендикулярно длине (мм) % ингибирования роста опухоли (TGI) рассчитывали по следующей формуле:
((Средний контрольный TV День Х - Лечение TV День Х)/ Средний контрольный TV День Х)×100
где День X представляет собой любой день измерения
Противоопухолевые эффекты лечения, частичной (PR) и полной регрессии (CR), стабильного (SD) и прогрессирующего (PD) заболевания были определены критериями ответа модели ксенотрансплантата (см. ниже). Мышей с потерей массы тела> 20% по сравнению с их массой тела в день 1 или опухолями с большим диаметром > 2000 мм немедленно подвергли эвтаназии, чтобы предотвратить любую боль или страдания у животного в соответствии с рекомендациями IACUC.
Статистический анализ
Данные выражали в виде среднего ± SEM для объема опухоли (TV). Различия в ТV между группами анализировали с помощью двухфакторного дисперсионного анализа, затем апостериорного критерия Sidak. Статистический анализ проводили с использованием версии GraphPad Prism version 6 (GraphPad Software, La Jolla, CA).
Критерий ответа модели ксенотрансплантата
Прогрессирование заболевания (PD): 3 последовательных измерения >120% от начального объема или 3 последовательных увеличивающихся измерения от наилучшего ответа, стабилизация заболевания (SD): 3 последовательных измерения >50% и <120% от начального объема, частичная регрессия (PR): 3 последовательных измерения <50% от начального объема, полная регрессия (CR): 3 последовательных измерения <30 мм3.
Получение ингибиторов CDK 4/6
В приведенных ниже примерах, рибоциклиб и палбоциклиб были сформулированы следующим образом. Этот тип состава является примерным и не требуется в конкретных вариантах осуществления изобретения. В этих примерах оба рибоциклиб и палбоциклиб были представлены в качестве свободных оснований.
Палбоциклиб формулировали в 25 мМ бикарбонате натрия, 15 мМ растворе молочной кислоты с 2% cremaphor. Сначала добавляли cremaphor и обрабатывали ультразвуком до образования равномерной суспензии. Это соединение должно быть свежим каждый раз.
Рибоциклиб формулировали в 0,5% метилцеллюлоза в дистиллированной воде с 1% cremaphor. Сначала добавляли cremaphor и обрабатывали ультразвуком до образования равномерной суспензии. Это соединение должно быть свежим каждый раз
Пример 1 - Соединение 1 и рибоциклиб
Клеточную линию JHH7 выращивали в виде ксенотрансплантата у иммунокомпрометированных самок голых мышей и мыши-опухоленосители, перорально получали ежедневно в течение 8 дней 300 или 500 мг/кг соединения 1 в качестве единственного средства или в комбинации с 75 и 150 мг/кг рибоциклиба. Рибоциклиб, в качестве единственного средства в дозе 75 мг/кг значительно не ингибирует рост опухоли с 13% TGI, тогда как 150 мг/кг значительно ингибирует рост опухоли (P ≤ 0,0001) с 23% TGI по сравнению с носителем в качестве контролей. Соединение 1, в качестве единственного средства, в дозах 300 и 500 мг/кг приводило к значительному ингибированию роста опухоли (P ≤ 0,0001) с TGI 14% и 35%, соответственно, по сравнению с носителем в качестве контролей. Комбинация 300 мг/кг соединения 1 и 75 мг/кг рибоциклиба привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению с 300 мг/кг соединения 1, в качестве единственного средства, с TGI 44%. Комбинация 300 мг/кг соединения 1 и 150 мг/кг рибоциклиба также привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 300 мг/кг соединения 1 с TGI 64%. Комбинация 500 мг/кг соединения 1 и 75 мг/кг рибоциклиба привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001 по сравнению только с 500 мг/кг соединения 1 с TGI 61%. Комбинация 500 мг/кг соединения 1 и 150 мг/кг рибоциклиба также привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 500 мг/кг соединения 1 с TGI 73%.
Эти данные демонстрируют, что рибоциклиб может значительно усиливать противоопухолевые эффекты соединения 1. Лечение соединением 1 в качестве единственного средства или в комбинации с рибоциклибом не вызывало никакое CR, PR или SD, и все группы имели PD. Все группы комбинированного дозирования были хорошо переносимы на основании измерений массы тела и рутинного клинического наблюдения.
Результаты показаны на фиг. 1А-фиг. 1С.
Пример 2 - Соединение 1 и палбоциклиб
Клеточную линию JHH7 выращивали в виде ксенотрансплантата у иммунокомпрометированных самок голых мышей и мыши-опухоленосители перорально получали ежедневно в течение 8 дней 300 или 500 мг/кг соединения 1 в качестве единственного средства или в комбинации с 50 и 100 мг/кг палбоциклиба. Палбоциклиб, в качестве единственного средства, в дозе 50 мг/кг значительно не ингибирует рост опухоли с 11% TGI, тогда как 100 мг/кг значительно ингибирует рост опухоли (P ≤ 0,05) с 26% TGI по сравнению с носителем в качестве контролей. Соединение 1, в качестве единственного средства, при 300 мг/кг приводило к значительному ингибированию роста опухоли (P ≤ 0,01) с TGI 12% и 500 мг/кг также приводило к значительному ингибированию роста опухоли (P ≤ 0,0001) с 32% по сравнению с носителем в качестве контролей. Комбинация 300 мг/кг соединения 1 и 50 мг/кг палбоциклиба привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению с 300 мг/кг соединения 1, в качестве единственного средства, с TGI 59%.
Комбинация 300 мг/кг соединения 1 и 100 мг/кг палбоциклиба также привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 300 мг/кг соединения 1 с TGI 77%. Комбинация 500 мг/кг соединения 1 и 50 мг/кг палбоциклиба привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 500 мг/кг соединения 1 с TGI 62%. Комбинация 500 мг/кг соединения 1 и 100 мг/кг палбоциклиба также привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 500 мг/кг соединения 1 с TGI 77%.
Эти данные демонстрируют, что палбоциклиб может значительно усиливать противоопухолевые эффекты соединения 1. Лечение соединением 1 в качестве единственного средства или в комбинации с палбоциклибом не вызывало никакое CR, PR или SD, и все группы имели PD. Все группы комбинированного дозирования были хорошо переносимы на основании измерений массы тела и рутинного клинического наблюдения.
Результаты показаны на фиг. 2А-фиг. 2С.
Пример 3 - Соединение 1 и палбоциклиб
От полученной от пациента ксенотрансплантатной модели (PDX) LIX066 PDX фрагменты инокулировали самкам иммунокомпрометированных голых мышей, и мыши-опухоленосители перорально получали ежедневно в течение 17 дней 300 или 500 мг/кг соединения 1 в виде отдельного средства или в комбинации с 100 мг/кг палбоциклиба. Палбоциклиб, в качестве отдельного средства, в дозе 100 мг/кг значительно ингибировал рост опухоли (P ≤ 0,001), по сравнению с носителем в качестве контролей, с 62% TGI. Все зарегистрированные животные в этой группе показали PD. Соединение 1, как единственное средство, при 300 мг/кг и 500 мг/кг значительно ингибировало рост опухоли (P ≤ 0,0001) по сравнению с носителем в качестве контролей, с 59% TGI и 70% TGI, соответственно. Все животные в группе соединения 1 при дозе 300 мг/кг и 500 мг/кг показали PD.
Комбинация 300 мг/кг соединения 1 и 100 мг/кг палбоциклиба привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 300 мг/кг соединения 1 с TGI 96%. Эта комбинация приводила к SD у 3/5 животных и PR у 2/5 животных. Комбинация 500 мг/кг соединения 1 и 100 мг/кг палбоциклиба также привела к значительному усилению ингибирования роста опухоли (P ≤ 0,0001) по сравнению только с 300 мг/кг соединения 1 с TGI 97%. Эта комбинация приводила к SD у 1/5 животных и PR у 4/5 животных. Все группы комбинированного дозирования были хорошо переносимы на основании измерений массы тела и рутинного клинического наблюдения.
Результаты показаны на фиг.3А – фиг.3В. В соответствии с открытием, что палбоциклиб может усиливать противоопухолевые эффекты соединения 1 в моделях HCC, дополнительное тестирование еще в 7 моделях PDX показало аналогичное улучшение в 3 из 7 моделей.
| название | год | авторы | номер документа |
|---|---|---|---|
| Комбинированные терапии для лечения гепатоцеллюлярной карциномы | 2019 |
|
RU2787993C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2018 |
|
RU2764724C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| КОМБИНАЦИИ ИНГИБИТОРОВ TGFβ И ИНГИБИТОРОВ CDK ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2019 |
|
RU2784852C2 |
| ИНГИБИТОРЫ FGFR4 | 2014 |
|
RU2715708C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2747228C2 |
| СПОСОБЫ ЛЕЧЕНИЯ УСТОЙЧИВОГО К ИНГИБИТОРАМ CDK4/6 РАКА | 2019 |
|
RU2820478C2 |
| СПОСОБЫ ЛЕЧЕНИЯ AR+ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2017 |
|
RU2769527C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ, СОДЕРЖАЩАЯ TNO155 И РИБОЦИКЛИБ | 2020 |
|
RU2813111C2 |
| КОМПЛЕКСНАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2810487C2 |
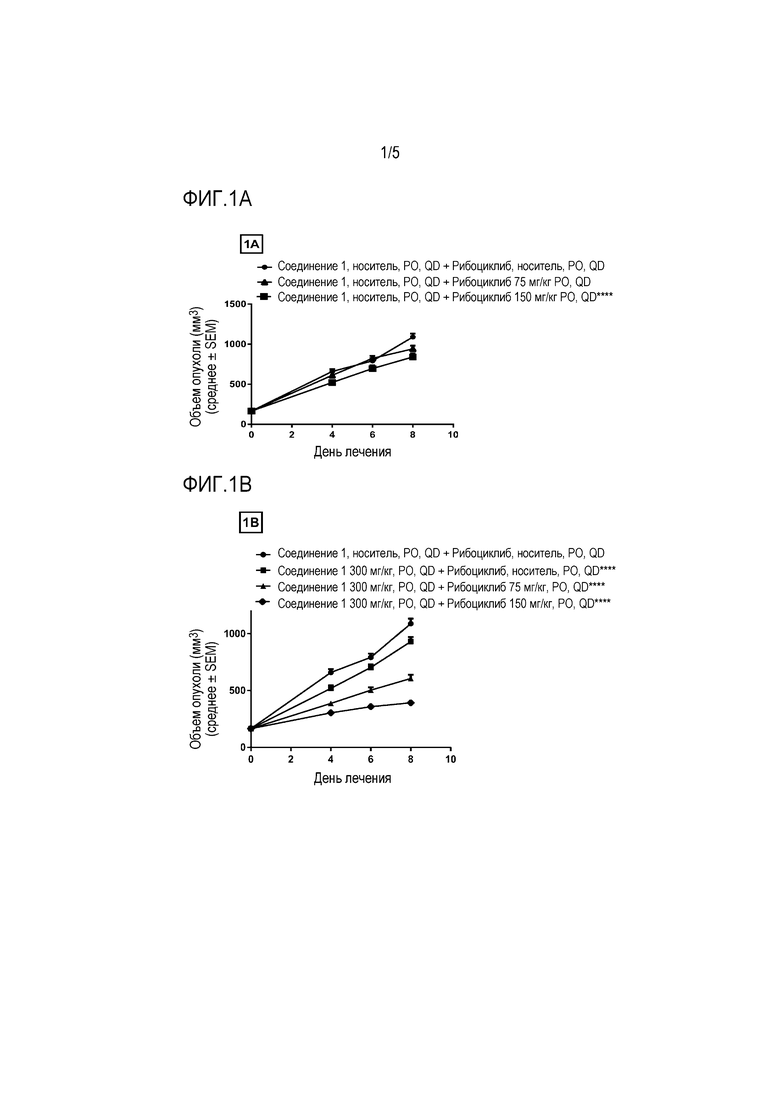
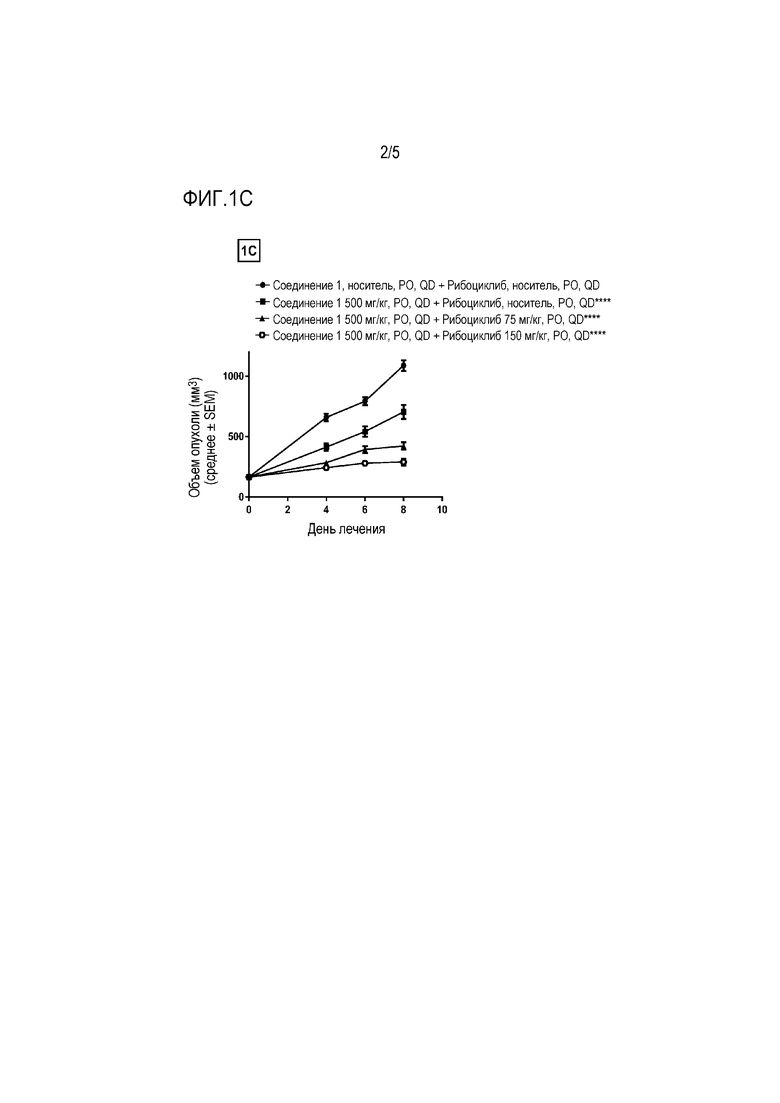
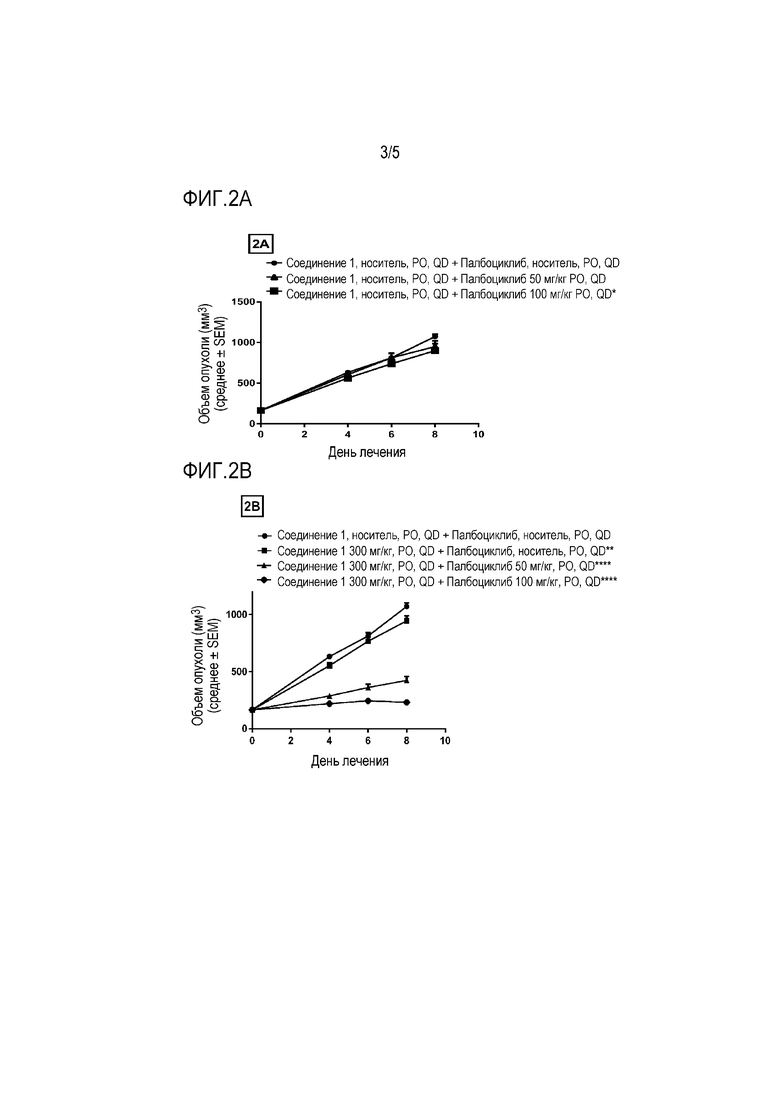
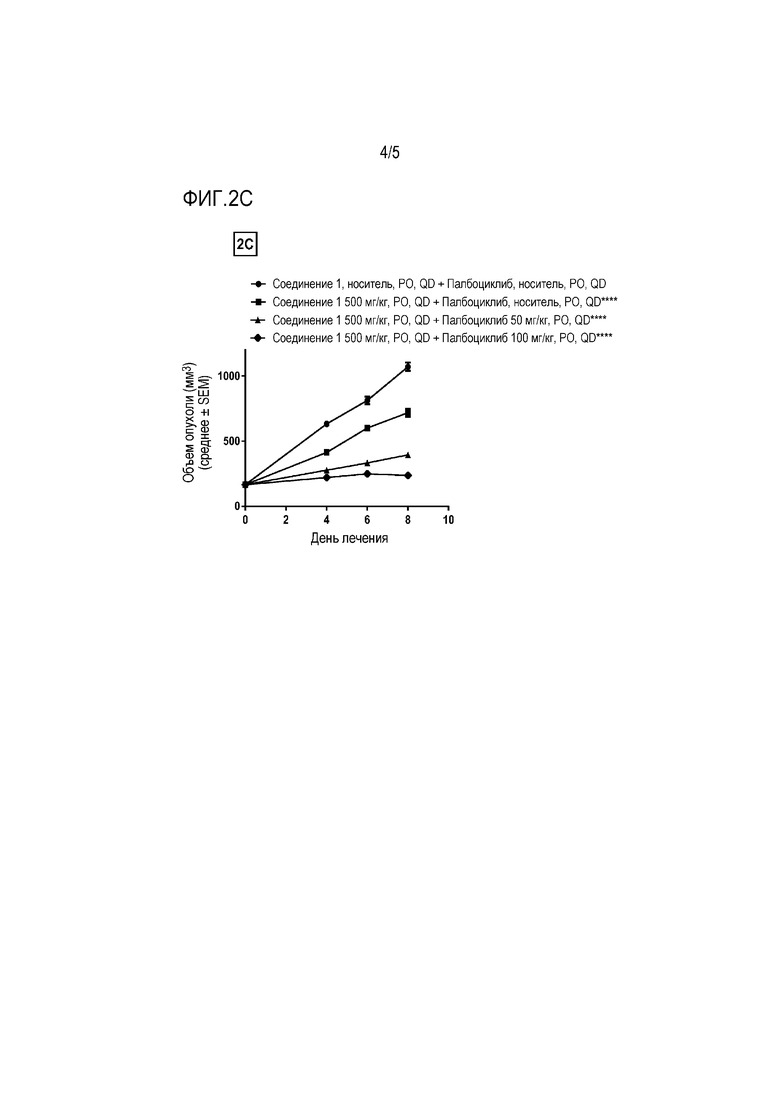
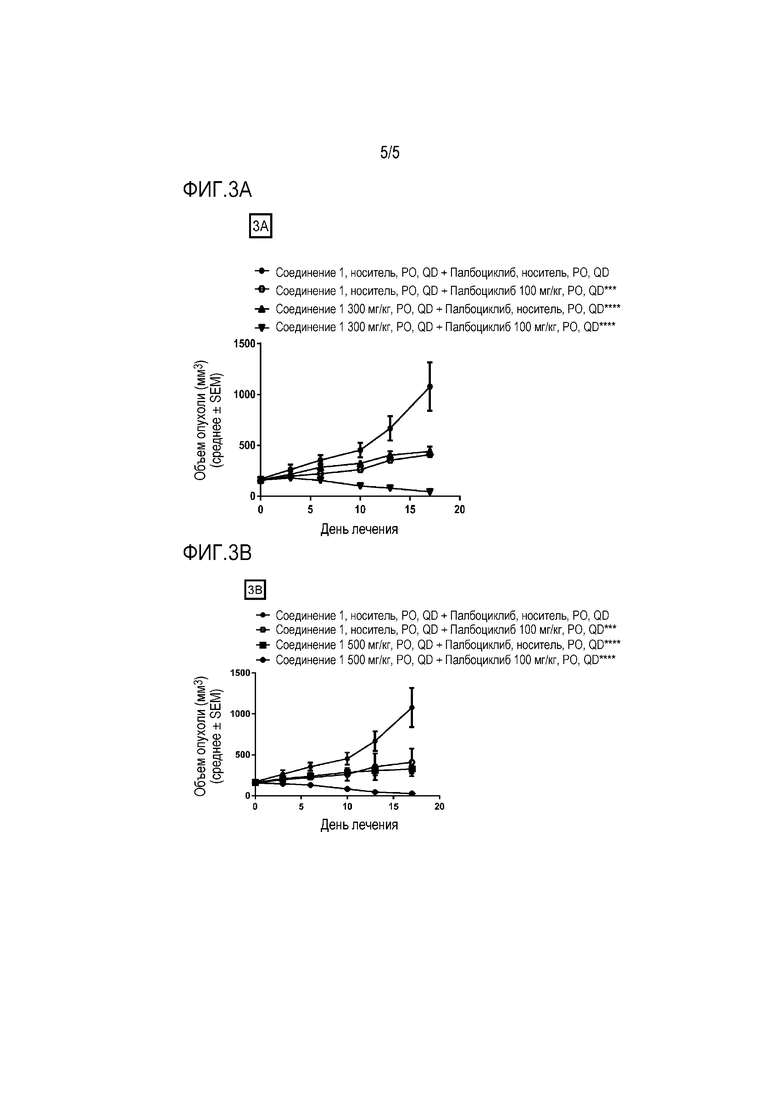
В данном настоящем описании представлена комбинированная терапия, полезная для лечения гепатоцеллюлярной карциномы. Комбинация включает ингибитор FGFR4 и ингибитор CDK 4/6. Данное сочетание обеспечивает усиленный противоопухолевый эффект. 4 н. и 19 з.п. ф-лы, 3 пр., 8 ил.
1. Способ лечения гепатоцеллюлярной карциномы у пациента, нуждающегося в этом, включающий введение пациенту комбинации N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида или его фармацевтически приемлемой соли и ингибитора CDK 4/6 или его фармацевтически приемлемой соли, где ингибитор CDK 4/6 выбран из группы, состоящей из 6-ацетил-8-циклопентил-5-метил-2-{[5-(пиперазин-1-ил)пиридин-2ил]амино}пиридо[2,3-d]пиримидин-7(8H)-она (палбоциклиб) и его фармацевтически приемлемых солей и диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (рибоциклиб) и его фармацевтически приемлемых солей.
2. Способ по п. 1, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль вводят в суточной дозе от 50 мг до 600 мг.
3. Способ по п. 2, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль вводят в суточной дозе от 200 мг до 400 мг.
4. Способ по п. 3, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль вводят в суточной дозе 300 мг.
5. Способ по п. 1, где ингибитор CDK 4/6 представляет собой палбоциклиб.
6. Способ по п. 5, где палбоциклиб вводят в суточной дозе 75 мг.
7. Способ по п. 5, где палбоциклиб вводят в суточной дозе 100 мг.
8. Способ по п. 5, где палбоциклиб вводят в суточной дозе 125 мг.
9. Способ по п. 1, где ингибитор CDK 4/6 представляет собой рибоциклиб.
10. Способ по п. 9, где рибоциклиб вводят в суточной дозе 200 мг/день.
11. Способ по п. 9, где рибоциклиб вводят в суточной дозе 400 мг/день.
12. Способ по п. 9, где рибоциклиб вводят в суточной дозе 600 мг/день.
13. Способ по любому из пп. 1-12, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят в виде отдельных композиций.
14. Способ по любому из пп. 1-12, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят в виде одной композиции.
15. Способ по любому из пп. 1-12, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят последовательно.
16. Способ по любому из пп. 1-12, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль вводят одновременно.
17. Способ по любому из пп. 1-16, где N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид представляет собой форму свободного основания N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида.
18. Способ по любому из пп. 1-16, где фармацевтически приемлемая соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида представляет собой форму гидрохлоридной соли N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида.
19. Фармацевтическая композиция для лечения гепатоцеллюлярной карциномы, содержащая N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид или его фармацевтически приемлемую соль и ингибитор CDK 4/6 или его фармацевтически приемлемую соль, где ингибитор CDK 4/6 выбран из группы, состоящей из 6-ацетил-8-циклопентил-5-метил-2-{[5-(пиперазин-1-ил)пиридин-2ил]амино}пиридо[2,3-d]пиримидин-7(8H)-она (палбоциклиб) и его фармацевтически приемлемых солей и диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (рибоциклиб) и его фармацевтически приемлемых солей.
20. Фармацевтическая композиция по п. 19, включающая форму свободного основания N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида.
21. Фармацевтическая композиция по п. 19, где фармацевтически приемлемая соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида представляет собой форму гидрохлоридной соли N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида.
22. Применение комбинации N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида или его фармацевтически приемлемой соли и ингибитора CDK 4/6 или его фармацевтически приемлемой соли при лечении гепатоцеллюлярной карциномы, где ингибитор CDK 4/6 выбран из группы, состоящей из 6-ацетил-8-циклопентил-5-метил-2-{[5-(пиперазин-1-ил)пиридин-2ил]амино}пиридо[2,3-d]пиримидин-7(8H)-она (палбоциклиб) и его фармацевтически приемлемых солей и диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (рибоциклиб) и его фармацевтически приемлемых солей.
23. Применение комбинации N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида или его фармацевтически приемлемой соли и ингибитора CDK 4/6 или его фармацевтически приемлемой соли в получении лекарственного средства для лечения гепатоцеллюлярной карциномы, где ингибитор CDK 4/6 выбран из группы, состоящей из 6-ацетил-8-циклопентил-5-метил-2-{[5-(пиперазин-1-ил)пиридин-2ил]амино}пиридо[2,3-d]пиримидин-7(8H)-она (палбоциклиб) и его фармацевтически приемлемых солей и диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (рибоциклиб) и его фармацевтически приемлемых солей.
| WO 2015057938 A1, 2015.04.23 | |||
| WO 2016025650 A1, 2016.02.18 | |||
| WO 2011130232 A1, 2011.10.20 | |||
| WO 2013006368 A1, 2013.01.10 | |||
| Jessica A | |||
| Sorrentino et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

