ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка претендует на приоритет предварительной заявки на патент США № 62/147313, поданной 14 апреля 2015 г. Эта заявка включена в данное описание в виде ссылки.
УРОВЕНЬ ТЕХНИКИ
Факторы роста фибробластов (FGF) представляют собой семейство более чем из 20 структурно родственных белков с разнообразными биологическими активностями. Их основные рецепторы, такие как рецепторы фактора роста фибробластов (FGFR1, FGFR2, FGFR3 и FGFR4) являются семейством рецепторных тирозинкиназ, которые связываются с FGF и которые вовлечены в процессы пролиферации и дифференцировки клеток. Дерегуляция сигнальных сетей FGFR вовлечена в ряд патофизиологических состояний, включая многие типы злокачественных опухолей человека.
Известно, что "рецептор фактора роста фибробластов 4" или "FGFR4" регулирует пролиферацию и антиапоптоз, и он экспрессируется или сверхэкспрессируется при многих видах рака. См. например, Dieci et al. 2013, Cancer Discovery, 0F1-0F16. Исследования показали, что экспрессия FGFR4 является прогностическим явлением для наиболее агрессивного фенотипа рака, а нокдаун или снижение экспрессии FGFR4 связано с уменьшением пролиферации и стимулированием апоптоза. См. например, Wesche et al. 11, Biochem J 437: 199-213.
Например, экспрессия или сверхэкспрессия FGFR4 связана с агрессивностью рака при раке желудка (Ye et al. 2011, Сancer, 5304-5313), при раке простаты (Xu et al. 2011, ВМС Сancer, 11; 84), при саркоме, такой как рабдомиосаркома (Taylor VI et al. 2009, J Clin Invest, 119, (11):3395-3407), при раке кожи, таком как меланома (Streit et al. 2006, British J cancer, 94:1879-1886), при раке печени, таком как холангиокарцинома (Sia et al. 2013, Gastroenterology, 144:829-840) и гепатоцеллюлярная карцинома (French et al. 2012, PLoS ONE 7(5): e367313; Miura et al. 2012, BMC Cancer 12: 56; Chiang et al. 2008, Cancer Res. 68 (16): 6779-6788; Sawey et al. 2011, Cancer cell 19:347-358), при раке поджелудочной железы, таком как интраэпителиальная неоплазия поджелудочной железы и аденокарцинома поджелудочной железы (Motoda et al. Int'l J Oncol. 38:133-143), при раке легких, таком как немелкоклеточный рак легкого (Fawdar et al. 2013, PNAS 110(30):12426-12431), при колоректальном раке (Pelaez-Garcia et al. 2013, PLoS ONE 8(5): e63695; Barderas et al. 2012, J Proteomics 75:4647-4655) и при раке яичника (Zaid et al. 2013, Clin Cancer Res 19:809-820).
Клиническое исследование нескольких ингибиторов FGFR подтвердило возможность их применения в качестве противоопухолевых средств. См. Dieci et al. 2013, Cancer Discovery, 0F1-0F16. Однако необходимы новые агенты, которые пригодны для целевого воздействия на FGFR, и, в частности, FGFR4.
Кроме того, при изготовлении фармацевтических продуктов соединение должно находиться в форме, которую можно легко проводить манипуляции и перерабатывать. В этом отношении химическая стабильность и физическая стабильность активного вещества являются важными факторами. Предпочтительно, когда соединение и содержащие его фармацевтические композиции способны эффективно храниться в течение длительных периодов времени без существенного изменения физико-химических характеристик.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Варианты осуществления изобретения обеспечивают кристаллическую форму соединения:
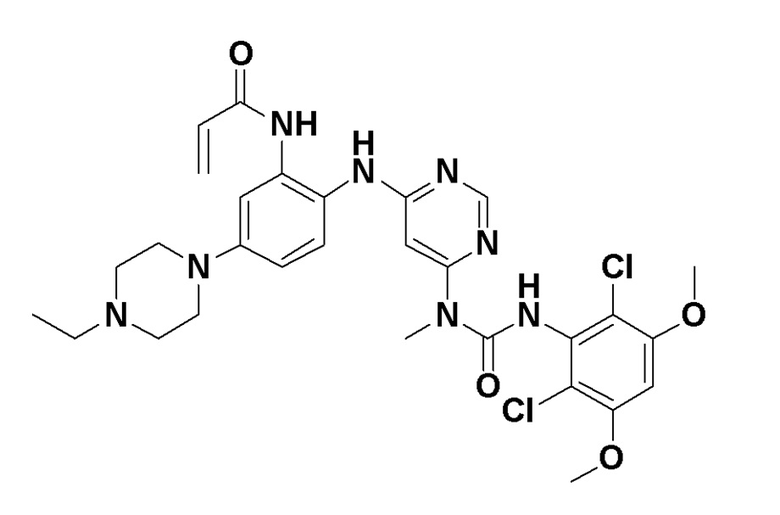
N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид.
В некоторых вариантах осуществления изобретения кристаллическая форма может быть представлена, например, в форме свободного основания, в форме гидрохлоридной соли, в форме моногидрохлоридной соли, в форме дигидрохлоридной соли или в форме этансульфонатной соли.
Варианты осуществления изобретения могут предоставить кристаллическую форму в виде свободного основания N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (Соединение 108):

N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил) акриламид (Соединение 108).
В некоторых вариантах осуществления изобретения кристаллическая форма соединения в виде свободного основания имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме (анализ PXRD) по меньшей мере с одним, двумя, тремя, четырьмя, пятью или шестью следующими значениями 2Θ°: 7,8-8,2; 10,1-10,5; 14,6-15,0; 16,3-16,7; 16,9-17,3; и 21,6-22,0. Например, соединение может иметь по меньшей мере, одно, два, три, четыре, пять или шесть значений 2Θ° (±0,2°), выбранных из группы, состоящей из: 8,0, 10,3, 14,8, 16,5, 17,1 и 21,8. В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания характеризуется по существу спектром PXRD, показанным на фигуре 5 (Фиг. 5). В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 10,3, 8,0 и 9,5; по меньшей мере 10,3, 8,0, 9,5, 14,8, 16,5 и 17,1; или по меньшей мере 10,3, 8,0, 9,5, 14,8, 16,5, 17,1, 19,3, 21,8,23,7 и 24,5.
В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания характеризуется кривой дифференциальной сканирующей калориметрии (DSC), по существу такой, как показано на фигуре 7 (Фиг. 7).
В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания характеризуется спектром 13C-ЯМР, по существу таким, как показано на фигуре 10 (Фиг. 10).
Некоторые варианты осуществления изобретения обеспечивают соединение, которое представляет собой кристаллическую моногидрохлоридную соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. В некоторых вариантах осуществления изобретения кристаллическая моногидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 26,6, 19,9 и 11,3; по меньшей мере 26,6, 19,9, 11,3, 9,0, 23,5 и 25,4; или по меньшей мере 26,6, 19,9, 11,3, 9,0, 23,5, 25,4, 27,6, 23,0, 18,1 и 29,0. В некоторых вариантах осуществления изобретения моногидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется спектром PXRD по существу таким, как представлено на фигуре 13 (Фиг. 13).
В некоторых вариантах осуществления изобретения моногидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется спектром 13C-ЯМР, по существу таким, как показано на фигуре 11 (Фиг. 11).
Некоторые варианты осуществления изобретения обеспечивают соединение, которое представляет собой кристаллическую дигидрохлоридную соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. В некоторых вариантах осуществления изобретения кристаллическая дигидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 26,6, 22,8 и 21,3; по меньшей мере 26,6, 22,8, 21,3, 8,0, 26,2 и 13,5; или по меньшей мере 26,6, 22,8,21,3, 8,0, 26,2, 13,5, 12,4, 16,1, 28,0 и 18,7. В некоторых вариантах осуществления изобретения дигидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется спектром PXRD по существу таким, как представлено на фигуре 16 (Фиг. 16).
В некоторых вариантах осуществления изобретения дигидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется спектром 13C-ЯМР, по существу таким, как показано на фигуре 14 (Фиг. 14).
Некоторые варианты осуществления изобретения обеспечивают соединение, которое представляет собой кристаллическую этансульфонатую соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. В некоторых вариантах осуществления изобретения кристаллическая этансульфонатая соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 19,2, 15,1 и 23,3; по меньшей мере 19,2, 15,1, 23,3, 20,3, 11,2 и 21,8; или по меньшей мере 19,2, 15,1, 23,3, 20,3, 11,2, 21,8, 9,4, 22,4, 23,6 и 24,0. В некоторых вариантах осуществления изобретения кристаллическая этансульфонатая соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется спектром PXRD по существу таким, как представлено на фигуре 19 (Фиг. 19).
В некоторых вариантах осуществления изобретения этансульфонатая соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется спектром 13C-ЯМР, по существу таким, как показано на фигуре 17 (Фиг. 17).
Другой целью изобретения является фармацевтическая композиция, содержащая кристаллическую форму соединения, описанного здесь, и фармацевтически приемлемый носитель. В некоторых вариантах осуществления изобретения композиция представлена в форме для перорального или парентерального введения.
Другой целью изобретения является способ получения кристаллического соединения:
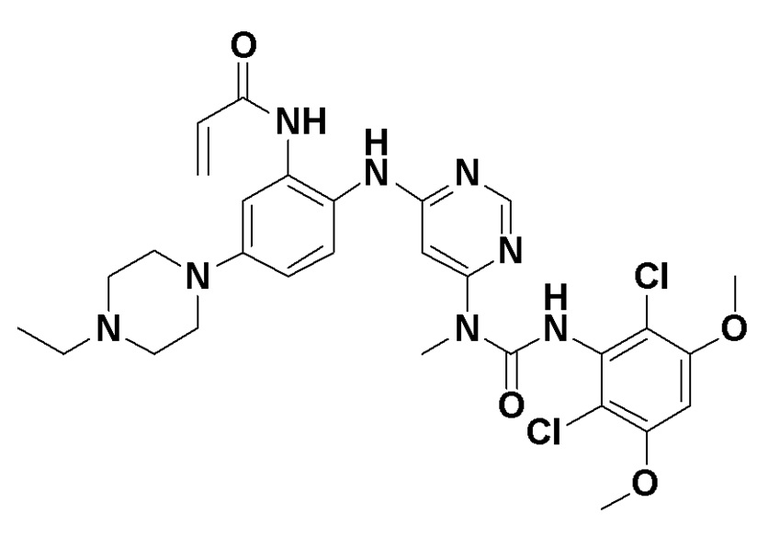
N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида в форме свободного основания.
Такие способы могут включать одну или несколько стадий из числа следующих:
a) предоставление композиции, содержащей соединение:

где Х представляет собой (2-(триметилсилил)этокси)метил,
в растворителе;
b) добавление кислоты к указанной композиции с такой скоростью, чтобы поддерживать температуру композиции при температуре≤50°С (например, при≤30, 20 или 15°С);
c) поддержание композиции, полученной на стадии b), в нагретом состоянии (например, при комнатной температуре); и затем
d) добавление композиции к насыщенному раствору гидроксида аммония (например, к охлажденному, при температуре 0-5°C) с такой скоростью, чтобы поддерживать температуру композиции при температуре≤25°С; и затем
e) экстрагирование композиции смесью несмешивающихся растворителей (например, дихлорметан/метанол) с образованием органической фазы; и
f) добавление подходящего растворителя к органической фазе,
с получением, в итоге, кристаллического соединения в форме свободного основания.
Другой целью изобретения является способ лечения гепатоцеллюлярной карциномы у субъекта, нуждающегося в этом, включающий введение указанному субъекту эффективного количества кристаллической формы соединения, описанного здесь. В некоторых вариантах осуществления изобретения гепатоцеллюлярная карцинома имеет измененный статус FGFR4 и/или FGF19 (например, повышенная экспрессия FGFR4 и/или FGF19)
Другой целью изобретения является способ лечения гепатоцеллюлярной карциномы у субъекта, нуждающегося в этом, включающий обнаружение измененного статуса FGFR4 и/или FGF19 (например, повышенной экспрессия FGFR4 и/или FGF19) в биологическом образце, содержащем клетки указанной гепатоцеллюлярной карциномы, и, если указанная гепатоцеллюлярная карцинома имеет указанный измененный статус FGFR4 и/или FGF19, то способ включает введение кристаллической формы соединения, описанного здесь, указанному субъекту в количестве, эффективном для лечения.
Другой целью является применение кристаллической формы соединения, описанного здесь, в способе лечения гепатоцеллюлярной карциномы.
Другой целью изобретения является применение кристаллической формы соединения, описанного здесь, для получения лекарственного средства для лечения гепатоцеллюлярной карциномы.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фиг. 1 представлены результаты оценки эффективности in vivo в модели гепатоцеллюлярной карциномы с использованием клеток HUH7. Соединение 108 (25 мг/кг или 37,5 мг/кг) или контрольный носитель вводили путем интраперитонеальной инъекции, и объем опухоли измеряли дважды в неделю в течение 15 дней.
На Фиг. 2 представлены результаты исследования эффективности in vivo в модели гепатоцеллюлярной карциномы с использованием клеток HEP3B. Соединение 108 (12,5 мг/кг, 25 мг/кг или 37,5 мг/кг) или контрольный носитель вводили путем внутрибрюшинной инъекции, и объем опухоли измеряли дважды в неделю в течение 15 дней.
На Фиг. 3 представлены результаты оценки эффективности in vivo в модели гепатоцеллюлярной карциномы с использованием клеток JHH7. Соединение 108 (12,5 мг/кг, 25 мг/кг или 37,5 мг/кг) или контрольный носитель вводили путем внутрибрюшинной инъекции, и объем опухоли измеряли дважды в неделю в течение 15 дней.
На Фиг. 4 представлены результаты сравнительной оценки эффективности in vivo в модели гепатоцеллюлярной карциномы с использованием клеток HEP3B. Соединение 108 (25 мг/кг, 37,5 мг/кг или 50 мг/кг) вводили дважды в день путем внутрибрюшинной инъекции, или соединение BGJ398 (30 мг/кг или 60 мг/кг) вводили перорально дважды в день.
На Фиг. 5 представлен спектр PXRD, полученный для кристаллического соединения в форме свободного основания
На Фиг. 6 представлены спектры PXRD, полученные для трех партий кристаллического соединения 108 в форме свободного основания.
На Фиг. 7 представлена кривая DSC для кристаллического соединения 108 в форме свободного основания.
На Фиг. 8 представлены кривые DSC, полученные для трех партий кристаллического соединения 108 в форме свободного основания.
На Фиг. 9А-9D представлены спектры 1Н-ЯМР, подтверждающие структуру соединения 108.
На Фиг. 10 представлен спектр 13C-ЯМР кристаллического соединения 108 в форме свободного основания.
На Фиг. 11 представлен 13C-ЯМР спектр кристаллического моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (то есть спектр кристаллической моногидрохлоридной соли соединения (108)), полученного в Примере 10.
На Фиг. 12 представлен 1Н-ЯМР спектр моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученного в Примере 10.
На Фиг. 13 представлен спектр PXRD, полученный для кристаллического моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученного в Примере 10.
На Фиг. 14 представлен 13C-ЯМР спектр кристаллического дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (то есть спектр кристаллической дигидрохлоридной соли соединения (108)), полученного в Примере 13.
На Фиг. 15 представлен 1Н-ЯМР спектр дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученного в Примере 13.
На Фиг. 16 представлен спектр PXRD, полученный для кристаллического дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученного в Примере 13.
На Фиг. 17 представлен 13C-ЯМР спектр кристаллического этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (то есть спектр кристаллической этансульфонатной соли соединения (108)), полученного в Примере 16.
На Фиг. 18 представлен 1Н-ЯМР спектр этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученного в Примере 16.
На Фиг. 19 представлен спектр PXRD, полученный для кристаллического этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученного в Примере 16
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Представлены кристаллические формы соединения:

N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид, который используется в качестве селективного ингибитора FGFR4.
В некоторых вариантах осуществления изобретения кристаллическое соединение представляет собой свободное основание N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. Вариант кристаллической формы свободного основания имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме (анализ PXRD) по меньшей мере с одним, двумя, тремя, четырьмя, пятью или шестью следующими значениями 2Θ°: 7,8-8,2; 10,1-10,5; 14,6-15,0; 16,3-16,7; 16,9-17,3; и 21,6-22,0. Например, кристаллическая форма свободного основания может иметь по меньшей мере одно, два, три, четыре, пять или шесть значений 2Θ° (±0,2°), выбранных из группы, состоящей из: 8,0, 10,3, 14,8, 16,5, 17,1 и 21,8. В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания характеризуется по существу спектром PXRD, показанным на Фиг. 5. В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при следующих значениях 2Θ° (±0,2): равных по меньшей мере 10,3, 8,0 и 9,5; по меньшей мере 10,3, 8,0, 9,5, 14,8, 16,5 и 17,1; или по меньшей мере 10,3, 8,0, 9,5, 14,8, 16,5, 17,1, 19,3, 21,8,23,7 и 24,5.
В некоторых вариантах осуществления изобретения кристаллическое соединение в форме свободного основания характеризуется кривой дифференциальной сканирующей калориметрии (DSC), по существу такой, как показано на фигуре 7.
Некоторые варианты осуществления изобретения обеспечивают соединение, которое представляет собой кристаллическую моногидрохлоридную соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. В некоторых вариантах осуществления изобретения кристаллическая моногидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 26,6, 19,9 и 11,3; по меньшей мере 26,6, 19,9, 11,3, 9,0, 23,5 и 25,4; или по меньшей мере 26,6, 19,9, 11,3, 9,0, 23,5, 25,4, 27,6, 23,0, 18,1 и 29,0. В таком варианте осуществления изобретения соединение характеризуется спектром PXRD по существу таким, как представлено на фигуре 13.
Некоторые варианты осуществления изобретения обеспечивают соединение, которое представляет собой кристаллическую дигидрохлоридную соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. В некоторых вариантах осуществления изобретения кристаллическая дигидрохлоридная соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 26,6, 22,8 и 21,3; по меньшей мере 26,6, 22,8, 21,3, 8,0, 26,2 и 13,5; или по меньшей мере 26,6, 22,8,21,3, 8,0, 26,2, 13,5, 12,4, 16,1, 28,0 и 18,7. В таком варианте осуществления изобретения соединение характеризуется спектром PXRD по существу таким, как представлено на 16.
Некоторые варианты осуществления изобретения обеспечивают соединение, которое представляет собой кристаллическую этансульфонатую соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида. В некоторых вариантах осуществления изобретения кристаллическая этансульфонатая соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида имеет спектральные пики на порошковой рентгеноструктурной дифрактограмме при значениях 2Θ° (±0,2), равных по меньшей мере 19,2, 15,1 и 23,3; по меньшей мере 19,2, 15,1, 23,3, 20,3, 11,2 и 21,8; или по меньшей мере 19,2, 15,1, 23,3, 20,3, 11,2, 21,8, 9,4, 22,4, 23,6 и 24,0. В таком варианте осуществления изобретения соединение характеризуется спектром PXRD по существу таким, как представлено на фигуре 19.
Как используется в настоящем документе, выражение "по существу как показано" или "по существу как представлено", со ссылкой, например, на спектр, означает, что квалифицированный специалист, при сравнении таких спектров, полученных с использованием одних и тех же методов получения результатов, например, как показано на фигуре 6, сделает вывод о том, что спектры являются достаточно схожими для того, чтобы указывать на одно и то же кристаллическое соединение в форме свободного основания, как описано здесь.
В изобретении также предлагаются способы синтеза и кристаллизации соединений, как описано здесь. Для кристаллического соединения в форме свободного основания способы могут включать одну или несколько стадий из числа следующих:
a) предоставление композиции, содержащей соединение:
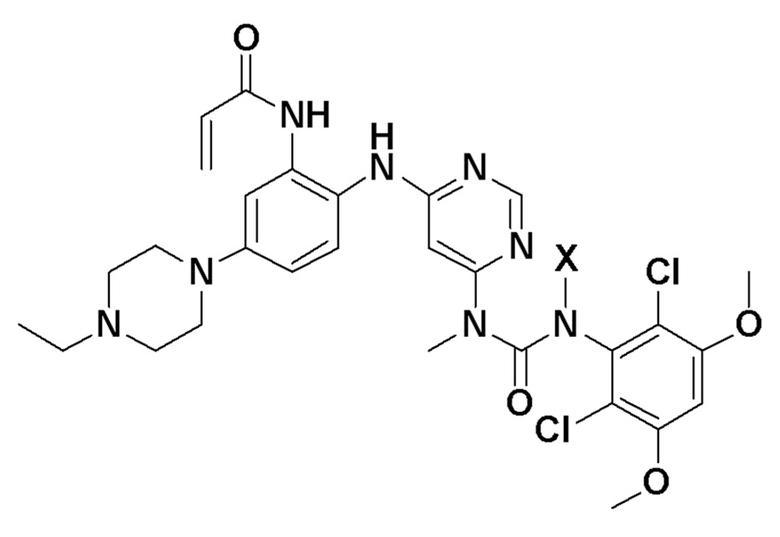
где Х представляет собой (2-(триметилсилил)этокси)метил,
в растворителе;
b) добавление кислоты к указанной композиции с такой скоростью, чтобы поддерживать температуру композиции при температуре≤50°С (например, при≤30, 20 или 15°С);
c) поддержание композиции, полученной на стадии b), в нагретом состоянии (например, при комнатной температуре); и затем
d) добавление композиции к насыщенному раствору гидроксида аммония (например, к охлажденному, при температуре 0-5°C) с такой скоростью, чтобы поддерживать температуру композиции при температуре≤25°С; и затем
e) экстрагирование композиции смесью несмешивающихся растворителей (например, дихлорметан/метанол) с образованием органической фазы; и
f) добавление подходящего растворителя к органической фазе,
с получением, в итоге, кристаллического соединения в форме свободного основания.
Кристаллическое соединение, описанное здесь, может быть объединено с фармацевтически приемлемым носителем для получения фармацевтических композиций. Конкретный выбор носителя и формы композиции зависит от конкретного пути введения, для использования в котором предназначена композиция. В некоторых вариантах осуществления носитель выбирают таким образом, чтобы перед введением сохранялась кристаллическая форма соединения.
Термин "фармацевтически приемлемый носитель", как он используется здесь, относится к нетоксичному носителю, вспомогательному веществу или средству для доставки, которое не разрушает фармакологической активности соединения, когда оно представлено в составе композиции. В некоторых вариантах осуществления изобретения фармацевтически приемлемый носитель выбирают таким образом, чтобы он поддерживал кристаллическую форму свободного основания соединения. Фармацевтически приемлемые носители, вспомогательные вещества или средства для доставки могут включать, но без ограничения, сорбиновую кислоту, сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, динатрийгидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воски, полиэтиленгликоль и ланолин.
Композиции настоящего изобретения предназначены для парентерального, перорального, распылительно-ингаляционного, местного, ректального, назального, трансбуккального, вагинального или резервуарно-имплантационного и т.п. введения. В некоторых вариантах осуществления изобретения, вводимый препарат содержит ингредиенты, которые получены из природных или неприродных источников. В некоторых вариантах осуществления изобретения композиция или носитель могут быть представлены в стерильной форме. Неограничивающие примеры стерильного носителя включают свободную от эндотоксина воду или апирогенную воду.
Термин "парентеральный", как он используется здесь, включает подкожные, внутривенные, внутримышечные, внутрибрюшинные, интрасиновиальные, интрастернальные, интратекальные, внутрипеченочные, внутричерепные и интракраниальные инъекции или инфузии. В конкретных вариантах осуществления изобретения соединения вводят внутривенно, перорально, подкожно или путем внутримышечного введения. Стерильные инъекционные формы композиций по изобретению могут представлять собой водную или масляную суспензию. Эти суспензии могут быть приготовлены в соответствии с методиками, известными в данной области, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный препарат для инъекций может также представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе. Среди приемлемых носителей и растворителей могут быть использованы вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла.
Для этой цели может быть использовано любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты и их глицеридные производные пригодны для приготовления препаратов для инъекций, так же как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно их полиоксиэтилированные варианты. Эти масляные растворы или суспензии могут также содержать длинноцепочечный спиртовой разбавитель или диспергатор, такой как карбоксиметилцеллюлоза или аналогичные диспергирующие агенты, которые обычно используются в составе фармацевтически приемлемых дозированных лекарственных форм, включая эмульсии и суспензии. Другие обычно используемые поверхностно-активные вещества, такие как Tween, Span и другие эмульгирующие агенты, которые обычно используются в производстве фармацевтически приемлемых твердых, жидких или других дозированных лекарственных форм, могут также использоваться с целью получения лекарственных препаратов.
Для перорального введения соединение или соль может быть предоставлено в виде приемлемой пероральной дозированной лекарственной форме, включая, без ограничения, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Также могут быть добавлены смазывающие агенты, например, стеарат магния. Для перорального введения препарата в форме капсул полезные разбавители включают лактозу и высушенный кукурузный крахмал. Когда для перорального применения предусматривается использование водной суспензии, то активный ингредиент может быть объединен с эмульгирующим и суспендирующим агентами. Если желательно, то могут быть добавлены некоторые подсластители, флаворанты или красящие агенты. Кроме того, могут быть добавлены консерванты. Подходящие примеры фармацевтически приемлемых консервантов включают, без ограничения, различные антибактериальные и противогрибковые агенты, такие как растворители, например, этанол, пропиленгликоль, бензиловый спирт, хлорбутанол, четвертичные аммонийные соли и парабены (такие как метилпарабен, этилпарабен, пропилпарабен и т.п.).
Субъекты и способы применения
Кристаллическая форма соединения, описанная здесь, может быть использована для лечения гепатоцеллюлярной карциномы.
Термины "лечение" и "лечить" относятся к улучшению состояния, облегчению, замедлению начала развития или ослаблению иным образом заболевания или нарушения, как описано здесь. В некоторых вариантах осуществления изобретения лечение может быть начато после развития одного или нескольких симптомов. В других вариантах осуществления изобретения лечение может быть осуществлено при отсутствии симптомов. Например, лечение восприимчивого индивидуума может быть начато до начала проявления симптомов (например, в свете истории симптомов и/или в свете генетических или других факторов восприимчивости).
Лечение также может продолжаться и после устранения симптомов, например, для предотвращения или задержки их рецидивов.
"Пациент" или "субъект", как используется здесь, представляет собой животное, предпочтительно млекопитающее, и особенно человека (включая как мужчин, так и женщин), и включает неонатальные, детские, ювенильные, подростковые, взрослые и гериатрические субъекты. Субъекты могут также включать и других млекопитающих (например, собаки, кошки, лошади, коровы, овцы, козы, обезьяны, птицы и т.п.), которые используются в лабораторных целях или в области ветеринарии.
В некоторых вариантах осуществления изобретения лечение проводят в отношении субъекта, имеющего гепатоцеллюлярную карциному с измененным статусом FGFR4 и/или FGF19 (фактор роста фибробластов 19).
В некоторых вариантах осуществления изобретения лечение может включать или выполняться в сочетании с анализом (например, измерением или определением) статуса FGFR4 и/или FGF19 в биологическом образце, содержащем клетки указанной гепатоцеллюлярной карциномы, и если указанная гепатоцеллюлярная карцинома характеризуется изменением FGFR4 и/или FGF19, то осуществляют лечение субъекта эффективным количеством активного агента, как описано здесь.
Термин "измененный статус", используемый здесь в отношении FGFR4 и/или FGF19, включает повышенную экспрессию (например, повышенные уровни мРНК или повышенные уровни белка), увеличенное количество копий в геноме и/или повышенную активность кодируемого белка в результате мутации, по сравнению с соответствующей нераковой тканью. В некоторых вариантах осуществления изобретения измененный статус FGFR4 и/или FGF19 включает мутации гена и/или кодируемого белка, которые приводят к увеличению активности гепатоцеллюлярной карциномы или которые иным образом связаны с более агрессивной формой гепатоцеллюлярной карциномы.
Термин "экспрессия" FGFR4 и/или FGF19 означает, что ген, кодирующий эти рецепторы, транскрибируется и предпочтительно транслируется. Как правило, экспрессия кодирующей области приводит к продуцированию кодируемого полипептида.
Белки FGFR4 и FGF19 известны, и их измененный статус и/или экспрессию можно измерить с использованием известных в данной области техники методик, например, для анализа геномных мутаций или аберрации числа копий, таких как амплификация нуклеиновой кислоты, секвенирование и/или методы на основе гибридизации, анализ экспрессии РНК, такой как нозерн-блоттинг или количественная ПЦР с детекцией в реальном времени (qRT-PCR), вестерн-блоттинг или другой иммуноблоттинг или иммунологический анализ, сортировка флюоресцентно-активированных клеток (FACS) и т.п.
Для того чтобы описанное здесь изобретение можно было более ясно понять, ниже приведены следующие примеры. Следует понимать, что эти примеры предназначены только для иллюстративных целей, и они не должны рассматриваться как ограничивающие.
ПРИМЕРЫ
Пример 1: Процедуры синтеза соединения

N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид (Соединение 108)
Общие указания:
Микроволновое нагревание проводили с использованием прибора Biotage Emrys Liberator или Biotage Initiator. Колоночную хроматографию проводили с использованием установки Isco Rf200d. Удаление растворителя осуществляли с использованием роторного испарителя Büchi rotary evaporator или центробежного испарителя Genevac centrifugal evaporator. Препаративную LC/MS проводили с использованием системы очистки Waters autopurifier и колонки 19×100 мм XTerra 5 мкм MS C18 с использованием кислой подвижной фазы. Спектры ЯМР регистрировали с использованием спектрометра Varian 400 MHz. Аналитические масс-спектры (MS) были получены с использованием установки Waters Acquity UPLC с одиночным квадрупольным детектором (Waters SQD).
Условия препаративной ВЭЖХ, используемой для очистки
Условия осуществления хроматографии:
Прибор: Waters 2767-SQD Mass trigger Prep System
Колонка: Waters Xbridge C18 150 мм * 19 мм * 5 мкм
Детектор: VWD SQD
Скорость потока: 15 мл/мин
Градиент во времени:
Типовая подвижная фаза:
1) Подвижная фаза А: 0,1% TFA в воде
Подвижная фаза В: ACN
2) Подвижная фаза А: 0,1% NH4HCO3 в воде
Подвижная фаза В: ACN
3) Подвижная фаза A: 0,1% NH4OAc в воде
Подвижная фаза В: ACN
4) Подвижная фаза A: 0,1% NH4OH в воде
Подвижная фаза В: ACN
Определения
Следующие обозначения и сокращения имеют указанные значения:
ACN: ацетонитрил
Boc2O: ди-трет-бутилдикарбонат
Brettphos: 2-(дициклогексилФосфино)-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил
tBuONa: трет-бутоксид натрия
CH3I: иодметан
Cs2CO3: карбонат цезия
DCC: N,N'-дициклогексилкарбодиимид
DCM: дихлорметан
DIEA: N,N-диизопропилэтиламин
DIPEA: N,N-диизопропилэтиламин
DMAP: 4-(диметиламино)пиридин
DME: диметиловый эфир
DMF: диметилФормамид
DMSO: диметилсульфоксид
EGTA: этиленгликольтетрауксусная кислота
ESI-MS: электрораспылительная ионизационная масс-спектрометрия
EtOH: этанол
HATU: гексафторфосфат 1-[бис(диметиламино)метилен]-1H-1,2,3-тризоло[4,5-b]пиридиний-3-оксида
H2SO4: серная кислота
iPrOH: изопропанол
К2СО3: карбонат калия
KHMDS: бис(триметилсилил)амид калия
КОН: гидроксид калия
LCMS: жидкостная хроматография-масс-спектрометрия
MeOH: метанол
MsCl: метансульфонилхлорид
NaBH3CN: цианоборгидрид натрия
NaBH(OAc)3: триацетоксиборгидрид натрия
NH4Cl: хлорид аммония
NH4HCO3: бикарбонат аммония
NaI: иодид натрия
NaNO3: нитрат натрия
NaOAc: ацетат натрия
MTBE: метил/трет-бутиловый эфир
nBuOH: н-бутанол
prep-HPLC: препаративная высокоэффективная жидкостная хроматография (ВЭЖХ)
prep- TLC: препаративная тонкослойная хроматография (TCX)
TBAF: фторид тетрабутиламмония
TBDMS-CL: трет-бутилдиметилсилилхлорид
TBSCl: трет-бутилдиметилсилилхлорид
TBSOTf: трет-бутилдиметилсилил-трифторметансульфонат
TEA: триэтиламин
TESCl: хлортриэтилсилан
TFA: трифторуксусная кислота
THF: тетрагидрофуран
Ti(OiPr)4: изопропоксид титана
TLC: Тонкослойная хроматография
PPTS: п-толуолсульфонат пиридиния
PE: петролейный эфир
PEG: поли(этиленгликоль)
PtO2: диоксид платины
EtOAc: этилацетат
Pd/C: палладий (0) на угле
Pd2(dba)3: трис(дибензилиденацетон)дипалладий (0)
Pd(dppf)2Cl2: [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II)
Ruphos: 2-дициклогексилФосфино-2',6'-диизопропоксибифенил
Xantphos: 4,5-бис(дифенилфосфино)-9,9-диметилксантен
Как использовано в описании и формуле изобретения, формы единственного числа включают в себя множество ссылок, если только содержание не диктует иное. Так, например, указание "соль HCl" или "гидрохлоридная соль" относится к моногидрохлоридным солям, дигидрохлоридным солям, 1,5-гидрохлоридным солям и другие стехиометрическим и нестехиометрическим гидрохлоридным солям.

N-[2-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино}-5-(4-этил-пиперазин-1-ил)-фенил]-акриламидметан
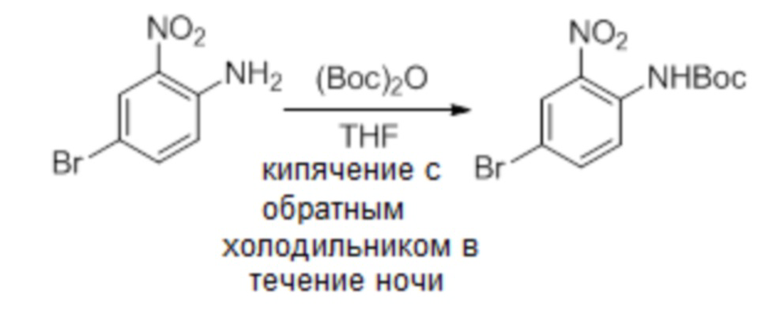
а. трет-бутил-4-бром-2-нитрофенилкарбамат
Смесь 4-бром-2-нитроанилина (4 г, 18,4 ммоль), (Boc)2O (4,4 г, 20,24 ммоль) в THF (50 мл) кипятили с обратным холодильником в течение ночи. Смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния элюированием смесью PE:EtOAc=20:1, получая указанное в заголовке соединение (5,4 г, выход: 93%) MS (ESI): 317, 319 [M+H]+.

b. трет-бутил-4-(4-этилпиперазин-1-ил)-2-нитрофенилкарбамат
Дегазированную смесь трет-бутил-4-бром-2-нитрофенилкарбамата (5,4 г, 17 ммоль), 1-этилпиперазина (2,91 г, 25,5 ммоль), Pd2(dba)3 (2,1 г, 3,4 ммоль), xantphos (ксантфос) (3,92 г, 6,8 ммоль) и Cs2CO3 (11,1 г, 34 ммоль) в толуоле (85 мл) нагревают при 100°С в течение 4 часов. Реакционную смесь концентрировали, и остаток очищали флэш-хроматографией на диоксиде кремния элюированием смесью MeOH: DCM=1:50 ~ 1:20, получая указанное в заголовке соединение (3,3 г, выход: 55%) MS (ESI): 351 [M+H]+.
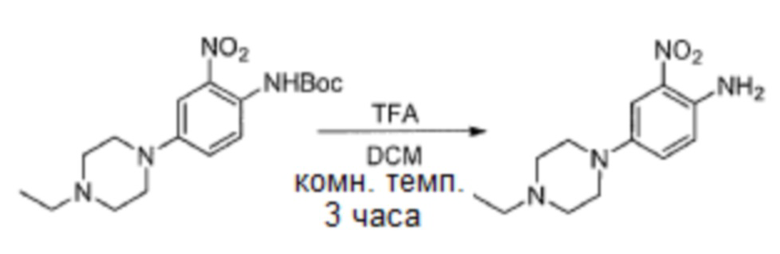
c. 4-(4-этилпиперазин-1-ил)-2-нитроанилин
К раствору трет-бутил-4-(4-этилпиперазин-1-ил)-2-нитрофенилкарбамата (3,3 г, 9,43 ммоль) в DCM (50 мл) добавляли TFA (20 мл) при 0°С, и полученную смесь перемешивали в течение 3 часов при комнатной температуре. После удаления всех летучих веществ в вакууме остаток повторно растворяли в DCM, нейтрализовали насыщенным водным раствором карбоната калия и экстрагировали DCM. Объединенные экстракты концентрировали, получая указанное в заголовке соединение (2,1 г, выход: 90%), которое использовали непосредственно на следующей стадии. 1H-ЯМР (400 Мгц, DMSO-d6) δ 1,02 (т, 3H), 2,36 (кв, 2H), 2,47-2,49 (м, 4H) 2,97-3,00 (м, 4H), 6,97 (д, 1H), 7,20 (с, 2H), 7,25 (с, 1H), 7,34 (дд, 1H); MS (ESI): 251 [M+H]+.

d. N4-(4-(4-этилпиперазин-1-ил)2-нитрофенил)-N6-метилпиримидин-4,6-диамин
Дегазированную смесь 4-(4-этилпиперазин-1-ил)-2-нитроанилина (2,1 г, 8,4 ммоль) 6-хлор-N-метилпиримидин-4-амина (см. Процедура 2А, стадия е; 1,2 г, 8,4 ммоль), Pd2(dba)3 (1,54 г, 1,68 ммоль), xantphos (1,94 г, 3,36 ммоль) и Cs2CO3 (5,48 г, 16,8 ммоль) в толуоле (45 мл) нагревали при 100°С в течение 1 часа. Реакционную смесь концентрировали, и остаток очищали флэш-хроматографией на диоксиде кремния элюированием смесью MeOH:DCM=1:40 ~ 1:20, получая указанное в заголовке соединение (870 мг, выход: 29%) МС (ESI): 358 [M+H]+.
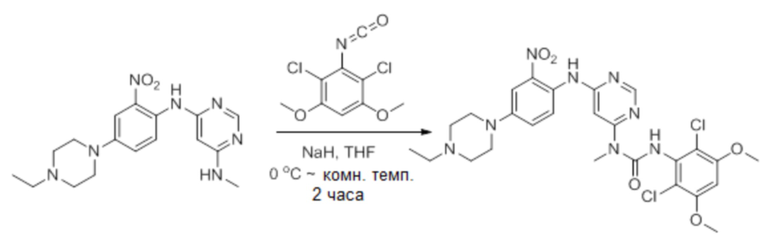
е. 3-(2,6-дихлор-3,5-диметоксифенил)-1-(6-(4-(4-этилпиперазин-1-ил)-2-нитрофениламино)пиримидин-4-ил)-1-метилмочевина
К раствору N4-(4-(4-этилпиперазин-1-ил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамина (870 мг, 2,44 ммоль) в THF (15 мл) добавляли NaH (60%, 200 мг, 5 ммол) при 0°С и смесь перемешивали в течение 30 минут при комнатной температуре. Раствор 2,4-дихлор-3-изоцианато-1,5-диметоксибензола (см. Процедура 2А, стадии a-d; 908 мг, 3,66 ммоль) в THF добавляли по каплям при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщенный водный раствор NH4Cl (2 мл) для гашения реакции. Смесь концентрировали и экстрагировали с помощью DCM. Объединенные экстракты промывали насыщенным солевым раствором, сушили над безводным Na2SO4, и концентрировали, получая неочищенный продукт, который затем очищали флэш-хроматографией на диоксиде кремния, получая указанное в заголовке соединение (330 мг, выход: 21%) в виде красного масла. 1H-ЯМР (400 Мгц, CDCl3) δ 1,44 (т, 3H), 3,01 (т, 2H), 3,21 (кв, 2H), 3,41-3,49 (м, 5H), 3,73-3,80 (м, 4H), 3,92 (с, 6H), 6,27 (с, 1Н), 6,55 (с, 1Н), 7,25 (д, 1Н), 7,69 (с, 1Н), 8,32 (д, 1Н), 8,52 (с, 1Н), 10,28 (уш.с, 1H), 12,05 (уш.с, 1H); MS (ESI): 605 [M+H]+.
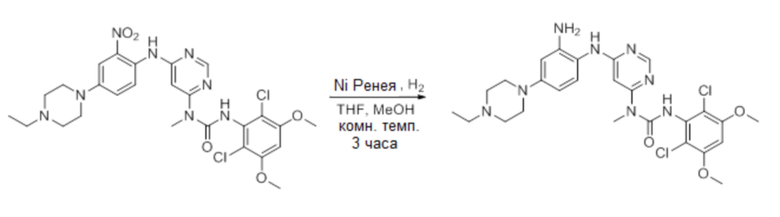
f. 1-(6-(2-амино-4-(4-этилпиперидин-1-ил)-(фениламино)пиримидин-4-ил)-3--(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевина
К раствору 3-(2,6-дихлор-3,5-диметоксифенил)-1-(6-(4-(4-этилпиперазин-1-ил)-2-(нитрофениламино)пиримидин-4-ил)-1-метилмочевины (330 мг, 0,546 ммоль) в THF (20 мл) и МеОН (20 мл) добавляли никель Ренея (суспензия в воде) при комнатной температуре, и полученную смесь перемешивали в течение 3 часов в атмосфере водорода (1 атм). Реакционную смесь фильтровали и концентрировали. Остаток дважды промывали с помощью MeOH, получая указанное в заголовке соединение (280 мг, чистота: 90%), которое использовали непосредственно на следующей стадии. MS (ESI): 575 [M+H]+.

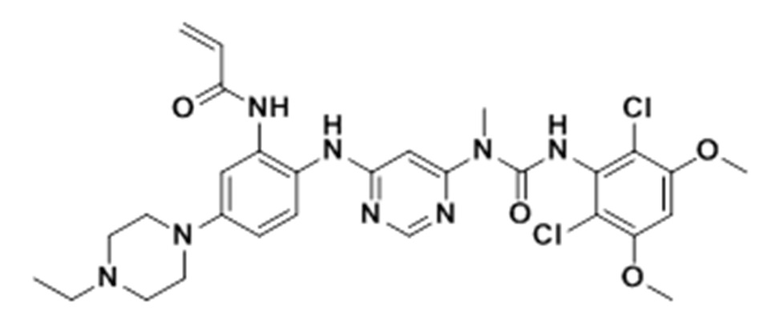
g. N-(2-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
К раствору 1-(6-(2-амино-4-(4-этилпиперазин-1-ил)фениламино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (280 мг, чистота: 90%, 0,44 ммоль) в THF (30 мл) добавляли раствор акрилоилхлорида в THF (20 мг/мл), 2 мл, 0,44 ммоль) при -10°С, и полученную смесь перемешивали в течение 1 часа при этой температуре. Для гашения реакции добавляли МеОН (1 мл). Смесь концентрировали, и остаток очищали препаративной ВЭЖХ и препаративной ТСХ, получая указанное в заголовке соединение 108 (20 мг, выход: 7%).
1H-ЯМР (400 Мгц, CDCl3) δ 1,31 (т, 3H), 2,65 (кв, 2H), 2,62-2,68 (м, 2H) (м, 4H), 3,27 (с, 3H), 3,36-3,38 (м, 4H), 3,91 (с, 6H), 5,76 (д, 1H), 5,90 (с, 2H), 6,24 (дд, 1H), 6,41 (д, 1H), 6,52 (с, 1H), 6,74 (дд, 1H), 7,07 (уш.с, 1H), 7,23 (д, 1H), 7,72 (уш.с, 1H), 7,98 (уш.с, 1Н), 8,37 (с, 1Н), 12,52 (с, 1H); MS (ESI): 629 [M+H]+.
Пример 2. Анализы биологической активности
Анализ связывания с FGFR4 . Очищенный рекомбинантный FGFR4 предварительно инкубировали с 10 мкМ соединения в течение ночи при 4°С или в течение 1 часа при комнатной температуре. После предварительной инкубации FGFR4 концентрировали и заменяли на буфер на белок-концентрирующей и обессоливающей колонке OPTI-TRAP C4 (Optimize Technologies). Белки элюировали ацетонитрилом, содержащем 0,1% муравьиной кислоты, и вводили прямой инъекцией в прибор Thermo Scientific Q ExactiveTM LCMS для идентификации модифицированного интактного FGFR4.
Результаты, представленные ниже в Таблице 1, подтверждают образование ковалентных аддуктов испытуемого соединения с пептидами по ожидаемой массе аддукта "пептид-лиганд" с наблюдающейся массой.
Таблица 1
IC50 профили ингибирования активности киназы . Соединения оценивали по профилю ингибирования активности FGFR в фирме Reaction Biology Corporation (Malvern, Pennsylvania) с использованием анализа Kinase HotSpotSM. См. Anansiadis et al. 2011, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039-1045.
Рекомбинантный FGFR1 (2,5 нМ), FGFR2 (1 нМ), FGFR3 (5 нМ) или FGFR4 (12 нМ) (Invitrogen™) получали в виде смеси с субстратом KKKSPGEYVNIEFG (SEQ ID NO: 1) (20 мкМ, субстрат для FGFR1); и Poly[Е,Y] 4:1 (0,2 мг/мл, субстрат для FGFR2,3,4) в киназном реакционном буфере (20 мМ HEPES-HCl, рН 7,5, 10 мМ MgCl2, 2 мМ MnCl2, 1 мМ EGTA, 0,02% Brij 35, 0,1 мМ Na3VO4, 0,02 мг/мл BSA, 2 мМ DTT и 1% DMSO). Соединение добавляли к смеси фермент/субстрат с использованием акустической обработки (Labcyte® Echo 550, Sunnyvale, California) (см. Olechno et al. 2006,, Improving IC50 results with acoustic droplet ejection. JALA 11, 240-246), и предварительно инкубировали в течение 0, 15 или 60 минут при комнатной температуре. После предварительной инкубации добавляли смесь АТФ (Sigma-Aldrich) и 33P-γ-АТФ (Perkin Elmer) до конечной концентрации 10 мкМ для инициирования киназных реакций. Реакционные смеси инкубировали в течение 120 минут при комнатной температуре, а затем наносили на ионообменную фильтровальную бумагу Whatman™ P81. Несвязанный фосфат удаляли с помощью интенсивной промывки фильтров в 0,75%-ой фосфорной кислоте. См. Anansiadis et al. 2011,, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039-1045.
Результаты, полученные для FGFR4 и FGFR1, показаны в Таблице 2. Соединение 108 показало селективное ингибирование FGFR4 с более высоким значением IC50 для FGFR1.
Таблица 2
Не вдаваясь в теорию, следует отметить, что активность по отношению к FGFR1, выраженная через IC50, как правило, отражает активности по отношению к FGFR1, FGFR2 и FGFR3. См. также Dieci et al. 2013, Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discovery, F1-F16.
Для подтверждения результатов соединение также тестировали в отношении ингибирования FGFR2 и FGFR3. Эти результаты, представленные ниже в Таблице 3, согласуются с IC50 для FGFR1, которая, как правило, отражает активность в отношении FGFR1, FGFR2 и FGFR3, и дополнительно демонстрирует селективность этого ингибитора FGFR4.
Таблица 3
Эффективность на моделях опухолей in vivo . Соединение 108 оценивали по его способности ингибировать рост опухоли у голых мышей с опухолевыми ксенотрансплантатами из трех различных линий опухолевых клеток гепатоцеллюлярной карциномы человека. Эти клеточные линии представляют собой злокачественные опухоли, имеющие измененный статус FGFR4 и/или FGF19. См. Sawey et al. Cancer cell 19(3): 347-358 (2011).
Животные: Голых мышей в возрасте 6-8 недель и весящих приблизительно 19-25 г, приобретали в фирме Taconic (Taconic, Hudson, New York) Все эксперименты на животных проводили в соответствии с протоколами, одобренными Комитетом по содержанию и использованию лабораторных животных (IACUC).
Опухолевые ксенотрансплантаты и лечение: 7,5 × 106 клеток HUH7 (HSRRB; Cat. no. JCRB0403) 5 × 106 клеток Hep3B (ATCC; Cat. no. HB8064), или 2,5 × 106 клеток JHH7 (HSRRB; Cat. no. JCRB1031), каждые из которых в общем объеме 100 мкл, 1:1 Matrigel (Corning Inc. Corning, NY) вводили подкожно (s.c.) в правый бок животных. При достижении размера опухолей 150-200 мм3, мышей случайным образом распределяли в группы лечения по 5-10 животных на группу. Дозирование проводили дважды в день путем интраперитонеальной инъекции в указанных дозах в течение 15 дней с использованием соединения 108, приготовленного в виде препарата в носителе, состоящим из 5% DMSO (Alfa Aesar, Ward Hill, MA), 10% PEG300 (Sigma, St Louis, МО) 8% TWEEN® 80 (Sigma, St Louis, МО), 77% солевого раствора по USP, в желаемой концентрации. Объемы опухолей регистрировали два раза в неделю, используя формулу объем=(длина * ширина2)/2. Массу тела животных также регистрировали два раза в неделю. Все животные наблюдались и получали уход в соответствии с Руководством по уходу и использованию лабораторных животных (The Guide for Care and Use of Laboratory Animals, 8th edition (National Academies Press, Washington D.C.).
Статистические методы: Статистические сравнения проводили в конце эксперимента с использованием дисперсионного анализа повторных измерений с дополнительным тестом Бонферрони для сравнения групп, получавших лечение, используя программное обеспечение GraphPad Prism 5. Использовали следующие критерии для определения тяжести состояния: Прогрессия заболевания, Стабилизация заболевания, Частичная регрессия и Полная регрессия. Прогрессию заболевания определяли по трем последовательным измерениям, в которых регистрировали увеличение объема опухоли по сравнению с наилучшим ответом или увеличение объема опухоли на >120% от начального. Стабилизацию заболевания определяли по трем последовательным измерениям, в которых регистрировали объем опухоли, составляющий <120% и >50% от начального объема опухоли. Три последовательных измерения, в которых регистрировали объем опухоли как <50% от исходного объема, квалифицировали как частичную регрессию. Полная регрессия квалифицировалась в случаях, когда объем опухоли составил <30 мм3 в трех последовательных измерениях. Критерий хи-квадрат использовали для сравнения ответов между группами, получавшими лечение (программное обеспечение - Microsoft Excel).
Результаты, полученные при оценке животных с опухолями из раковых клеток HUH7, HEP3B и JHH7, показаны на Фиг. 1-3, соответственно, и также представлены в Таблице 4.
Таблица 4. Ингибирование роста опухоли в ксенотрансплантатах HCC с повышенным уровнем FGF19
HUH7 (n=10 в группе)
HEP3B (n=5 в группе)
JHH7 (n=10 в группе)
Эти данные показывают, что соединение 108 эффективно во всех моделях. Среди трех моделей клетки HEP3B являются наиболее чувствительными, JHH7 менее чувствительными, а HUH7 показали промежуточную чувствительность к соединению 109. Хотя на Фиг. 3 показан ответ в зависимости от дозы для клеток JHH7, во всех испытанных уровни прогрессия заболевания зависели от дозы.
Сравнительные исследования соединения 108 и BGT398 . Сравнительные исследования проводили с соединением 108 и с соединением BJG398, известным ингибитором FGFR.
Протокол биохимического анализа киназы для получения IC50: Рекомбинантный FGFR1 (2,5 нМ) или FGFR4 (12 нМ) получали в виде смеси с субстратом KKKSPGEYVNIEFG (SEQ ID NO: 1) (20 мкМ, субстрат для FGFR1); Poly[Е,Y] 4:1 (0,2 мг/мл, субстрат для FGFR 2,3,4) в киназном реакционном буфере (20 мМ HEPES-HCl, рН 7,5), 10 мМ MgCl2, 2 мМ MnCl2, 1 мМ EGTA, 0,02% Brij35, 0,1 мМ Na3VO4, 0,02 мг/мл BSA, 2 мМ DTT и 1% DMSO). Соединение добавляли к смеси фермент/субстрат с использованием акустической обработки и предварительно инкубировали в течение 0, 15 или 60 минут при комнатной температуре. После предварительной инкубации соединения добавляли 33P-γ-АТФ при конечной концентрации 10 мкМ для инициирования киназных реакций. Реакционные смеси инкубировали в течение 120 минут при комнатной температуре. Фосфорилирование субстрата контролировали с помощью анализа на фильтре, как описано выше. Результаты показаны в Таблице 5. Результаты показывают, что соединение 108 является более сильным ингибитором FGFR4, в то время как BGJ398 является более сильным ингибитором FGFR1.
Таблица 5. Сравнительные испытания соединения 108 и соединения BGJ398 с помощью биохимического анализа киназы
IC50 (нМ)
IC50 (нМ)
Протокол анализа жизнеспособности клеток для определения GI50: Линии клеток культивировали при 37°С в атмосфере 5%CO2 и при влажности 95%. Культуральные среды приобретали в GIBCO®. Для анализа жизнеспособности клеток в 96-луночные планшеты высевали 2000 клеток на лунку, и инкубировали в течение 24 часов перед обработкой соединением. После добавления соединения планшеты инкубировали в течение 72 часов при 37°С в атмосфере 5% CO2, а затем анализировали с помощью анализа CTG (CellTiter-Glo® Luminescent Cell Viability Assay, Cat. No.: G7572, Promega). Результаты анализа показаны в Таблице 6. Данные, приведенные в Таблице, показывают, что соединение 108 является более активным, чем соединение BGJ398 в линии клеток Hep3B с повышенным уровнем FGF19. Активности соединения 108 и соединения BGJ398 в отношении двух других линий клеток HUH7 и JHH7, с повышенным уровнем FGF19, являются сравнимыми. Клеточные линии HepG2 (ATCC; cat. no. HB-8065), SNU398 (ATCC; cat. no. CRL-2233) и SNU449 (ATCC; cat. no. CRL-2234) представляют собой линии клеток без повышенных уровней FGF19, и их использовали в качестве контролей.
GI50 представляет собой концентрацию испытуемого лекарственного вещества, при которой 100 × (T-T0)/(C-T0)=50. См. например, Monks et al., Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines, J Natl Cancer Inst (1991) 83(11):757-766; Boyd et al., Data Display and Analysis Strategies for the NCI Disease-oriented In Vitro Antitumor Drug Screen, in Cytotoxic Anticancer Drugs: Models and Concepts for Drug Discovery and Development, Valeriote et al., eds. (1990), pp. 11-34. Люминесценция тест-лунки после 72-часового периода воздействия тестируемого препарата соответствует T, люминесценция в момент времени 0 соответствует T0, а контрольная люминесценция соответствует C. Величина GI50 характеризует мощность ингибирования роста у испытуемого соединения.
Таблица 6. Сравнительные испытания соединений 108 и BGJ398 в анализах жизнеспособности клеток
GI50 (нМ)
GI50 (нМ)
Сравнение эффективности in vivo: Для этих экспериментов использовали голых мышей, как описано выше. 5,0 × 106 клеток Hep3B в общем объеме 100 мкМ, 1:1 Matrigel (Corning Inc. Corning, New York), вводили подкожно в правый бок животных. При достижении размера опухолей 150-200 мм3, мышей случайным образом распределяли в группы лечения по 5-10 животных на группу. Затем начинали лечение с использованием соединения 108, приготовленного в виде препарата в носителе, состоящим из 5% DMSO (Alfa Aesar, Ward Hill, MA), 10% PEG300 (Sigma, St Louis, МО), 8% TWEEN® 80 (Sigma, St Louis, МО), 77% солевого раствора по USP, в желаемой концентрации. Оба лекарства дозировали в течение 18 дней, за исключением одной группы лечения (см. ниже). Объемы опухолей регистрировали два раза в неделю, используя формулу объем=(длина * ширина2)/2. Массу тела животных также регистрировали два раза в неделю. Все животные наблюдались и получали уход в соответствии с Руководством по уходу и использованию лабораторных животных (The Guide for Care and Use of Laboratory Animals, 8th edition (National Academies Press, Washington D.C.). Результаты этого сравнительного исследования in vivo показаны на Фиг. 4.
Полученные результаты показывают, что соединение 108 является более эффективным, чем соединение BGJ398, при переносимых уровнях дозировки. Хотя соединение BGJ398 при дозе 60 мг/кг показало эффективность, сравнимую с активностью соединения 108, введение этой группы соединения BGJ398 в дозе 60 мг/кг было прекращено на 11-й день из-за плохого состояния животных. Это различие в токсичности не связано с путями введения, поскольку группа животных, которым перорально вводили соединение BGJ398 в дозе 30 мг/кг, не показала ухудшения состояния здоровья.
Пример 3: Альтернативные способы синтеза и кристаллизация Соединения 108.
Синтез бис(Boc)-4-бром-2-нитроанилина

В трехгорлую 5-литровую круглодонную колбу загружали 4-бром-3-нитроанилин (200 г, 922 ммоль), Boc-ангидрид (412 г, 1889 ммоль, 2,05 экв.) и THF (3 л). К перемешиваемой смеси добавляли DMAP (1,26 г, 92,2 ммоль, 0,10 экв.), смесь нагревали до 65°С и перемешивали при этой температуре пока завершение реакции не подтверждали методом ВЭЖХ (оставшееся количество 4-бром-3-нитроанилина≤2%, примерно 3,5 часа), а затем охлаждали до комнатной температуры. Смесь переносили в 12-литровый сосуд для дальнейшей обработки, добавляли этилацетат (3 л), и смесь последовательно промывали 1н HCl (1 л), насыщенным водным раствором NaHCO3 (1 л) и 10%-ным водным раствором NaCl (1 л). Органический слой концентрировали до минимально перемешиваемого объема и добавляли этилацетат (1 л). Раствор дважды промывали гептаном (1,5 л), смесь концентрировали до общего объема 1,5 л после второй заключительной промывки. Полученную суспензию фильтровали, промывали гептаном (3 × 200 мл) и сушили в вакууме, получая указанное в заголовке соединение (348,5 г, 90% выход) в виде беловатого твердого вещества.
Трет-бутил-(4-(4-этилпиперазин-1-ил)-2-нитрофенил)карбамат

В 5-литровую трехгорлую круглодонную колбу загружали бис(Boc)-4-бром-2-нитроанилин (250 г, 599 ммоль), карбонат цезия (234г, 719 ммоль, 1,2 экв.) и катализатор Бухвальда Ru-Phos (CAS# 1375325-68-00, 20 г, 27,4 ммоль, 0,046 экв.). К твердым частицам добавляли предварительно дегазированный толуол (1 л) и N-этилпиперазин (156 мл, 1228 ммоль, 2,05 экв.). Полученную смесь барботировали азотом в течение 30 минут, затем смесь нагревали до 95-105°С и перемешивали при этой температуре до тех пор, пока анализ ВЭЖХ не показал завершение реакции (примерно 6 часов). Смесь охлаждали до комнатной температуры и фильтровали через целит Celite® 545 (125 г), осадок промывали толуолом (3 × 60 мл) и полученный раствор концентрировали, получая указанное в заголовке соединение (210 г, 100%-ный выход), которое использовали далее без дальнейшей очистки.
4-(4-этилпиперазин-1-ил)-2-нитроанилин

К суспензии трет-бутил-4-(4-этилпиперазин-1-ил)-2-нитрофенилкарбамата (210 г, 599 ммоль) в минимальном количестве толуола загружали 4н водный раствор HCl (2,1 л, 14 экв.) при механическом перемешивании с поддержанием внутренней температуры≤40°С. Полученную суспензию перемешивали при комнатной температуре с продувкой азотом для удаления любого оставшегося органического растворителя, пока анализ ВЭЖХ не показал завершение реакцию. К суспензии добавляли MTBE (1 л). Смесь перемешивали в течение 30 минут и разделяли слои. Нижний водный слой охлаждали до 0-5°С и добавляли 12н водный раствор NaOH до рН 9-10 (примерно потребовалось 480 мл). Полученное твердое вещество отфильтровывали и промывали холодной (0-5°C) водой (3 × 400 мл). Влажный осадок отфильтровали в вакууме и/или продували азотом при 30°С, получая указанное в заголовке соединение (133,3 г, выход 91%) в виде красного твердого вещества.
Трет-бутил-4-бром-2-нитрофенилкарбамат - альтернативная методика

Смесь 4-бром-2-нитроанилина (4 г, 18,4 ммоль) и (Boc)2О (4,4 г, 20,24 ммоль) в THF (50 мл) кипятили с обратным холодильником в течение ночи. Смесь концентрировали, и остаток очищали флэш-хроматографией на диоксиде кремния элюированием смесью PE:EtOAc=20:1, получая указанное в заголовке соединение (5,4 г, выход: 93%). MS (ESI): 317, 319 [M+H]+.
Трет-бутил-4-(4-этилпиперазин-1-ил)-2-нитрофенилкарбамат - альтернативная методика
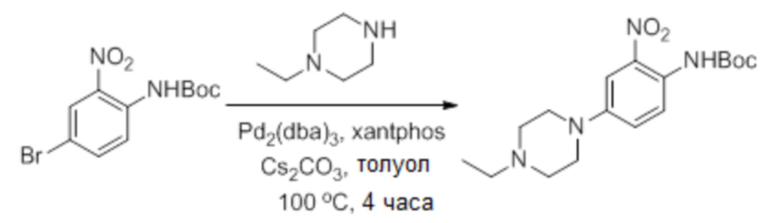
Дегазированную смесь трет-бутил-4-бром-2-нитрофенилкарбамата (5,4 г, 17 ммоль), 1-этилпиперазина (2,91 г, 25,5 ммоль), Pd2(dba)3 (2,1 г, 3,4 ммоль), xantphos (3,92 г, 6,8 ммоль) и Cs2CO3 (1,1 г, 34 ммоль) в толуоле (85 мл) нагревали при 100°С в течение 4 часов. Реакционную смесь концентрировали, и остаток очищали флэш-хроматографией на диоксиде кремния элюированием смесью MeOH:DCM=1:50 ~ 1:20, получая указанное в заголовке соединение (3,3 г, выход: 55%) MS (ESI): 351 [M H]+.
a. 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевино

В трехлитровую трехгорлую круглодонную колбу загружали 2,6-дихлор-3,5-диметоксианилин (9,65 г, 899 ммоль), трифосген (93г, 315 ммоль, 0,35 экв) и 1,4-диоксан (1,9 л). Смесь нагревали до 100°С и перемешивали при этой температуре в течение 3,5 часов. Затем смесь охлаждали до 20-25°С и фильтровали. Твердый остаток промывали 1,4-диоксаном (200 мл), фильтрат концентрировали до минимального перемешиваемого объема и 3 раза промывали диоксаном (700 мл для каждой промывки). Раствор после последней промывки концентрировали до минимального перемешиваемого объема, а затем повторно растворяли в 1,4-диоксане (730 мл). В эту суспензию добавляли 6-хлор-N-метилпиримидин-4-амин (129 г, 899 ммоль, 1 экв.). Полученную смесь нагревали до 80°С и перемешивали при этой температуре в течение 60 часов, и за это время образовывалось значительное количество осадка. Смесь охлаждали до 20-22°С и фильтровали. Твердый осадок промывали диоксаном (2 × 90 мл) и сушили в вакууме при комнатной температуре в атмосфере азота, получая указанное в заголовке соединение (191 г, выход 54%) в виде твердого вещества.
b. 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевина
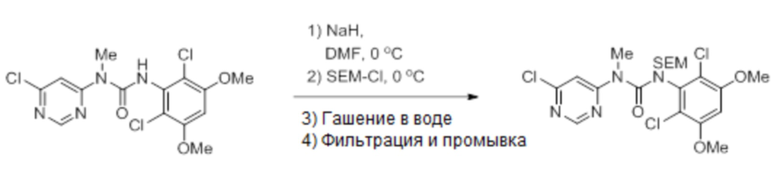
В 5-литровую трехгорлую круглодонную колбу загружали 3-(2,6-дихлор-3,5-диметоксифенил)-1-(6-(4-(4-этилпиперазин-1-ил)-2-нитрофениламино)пиримидин-4-ил)-1-метилмочевину (135 г, 345 ммоль), иодид калия (6,87 г, 41,4 ммоль, 0,12 экв.) и DMF (400 мл). Смесь охлаждали до 0-5°С и добавляли по порциям NaH (60%-ая дисперсия в минеральном масле, 14,9 г, 448 ммоль, 1,3 экв.), поддерживая внутреннюю температуру≤5°С. Смесь перемешивали в течение 1 часа при 0-5°С, после чего добавляли по каплям SEM-Cl (73,2 мл, 414 ммоль, 1,2 экв.), поддерживая внутреннюю температуру≤5°С. Реакционную смесь перемешивали при 0-5°С, пока ВЭЖХ анализ не показал завершение реакции (примерно 1 час). Полученный продукт переносили во вторую 5-литровую трехгорлую круглодонную колбу, содержащую холодную (0-5°C) воду (4 л). Полученную суспензию перемешивали в течение 15 минут, затем фильтровали. Осадок промывали водой (3 × 300 мл) и сушили в вакууме, получая указанное в заголовке соединение (184 г, 102%) в виде твердого вещества.
c. 1-(2,6-дихлор-3,5-диметоксифенил)-3-(6-((4-(4-этилпиперазин-1-ил)-2-нитрофенил)амино)пиримидин-4-ил)-3-метил-1-((2-(триметилсилил)этокси)метил)мочевина

В трехгорлую круглодонную колбу объемом 3 л загружали 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-(триметилсилил)этокси)метил)мочевину (186 г, 349 ммоль), 4-(4-этилпиперазин-1-ил)-2-нитроанилин (87 г, 349 ммоль, 1 экв.), прекатализатор BrettPhos (CAS# 1470372-59-8), 15,6 г, 17,2 ммоль, 0,05 экв.) и карбонат цезия (136 г, 418 ммоль, 1,2 экв.). Систему продували азотом и добавляли предварительно дегазированный DMF (910 мл). Раствор барботировали азотом в течение 30 минут, затем оставляли перемешиваться при 22-25°С, пока анализ ВЭЖХ не показал завершение реакции (3-4 часа). Смесь загружали в 5-литровую трехгорлую круглодонную колбу, содержащую воду (2,7 л) с такой скоростью, чтобы поддерживать внутреннюю температуру≤35°С. Полученную суспензию охлаждали до 22-25°С, а затем фильтровали. Осадок промывали водой (4 × 150 мл) и сушили в вакууме, получая указанное в заголовке соединение (257 г, 100%-ный выход) в виде твердого вещества.
d. Синтез 1-(2,6-дихлор-3,5-диметоксифенил)-3-(6-((4-(4-этилпиперазин-1-ил)-2-нитрофенил)амино)пиримидин-4-ил)-3-метил-1-((2-(триметилсилил)этокси)метил)мочевины - альтернативная методика
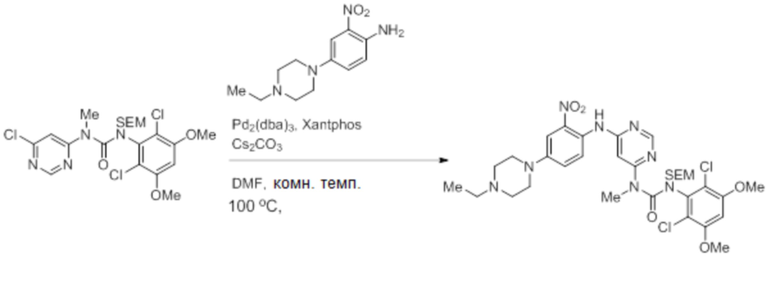
В трехгорлую круглодонную колбу объемом 3 л, снабженную обратным холодильником, магнитной мешалкой и системой продувки азотом, добавляли 4-(4-этилпиперазин-1-ил)-2-нитроанилин (50 г, 0,199 моль), 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевину (125,1 г, 0,239 моль, 1,2 экв.), DMF (500 мл) и карбонат цезия (130 г, 0,399 моль, 2,0 экв.). Через полученную смесь барботировали азот в течение 5-10 мин при комнатной температуре. К смеси добавляли Pd2(dba)3 (18,29 г, 0,0199 моль, 0,10 экв.), Xantphos (1,5 г, 0,0199 моль, 0,10 экв.) и продолжали продувать азотом еще 10 минут. Затем реакционную смесь нагревали до 100°C, пока анализ ТСХ не показал завершение реакции (примерно 2 часа). Смесь фильтровали через слой целита и обрабатывали водой, желаемый продукт экстрагировали EtOAc (3 × 500 мл), объединенные органические фазы промывали солевым раствором (2 × 500 мл), сушили над Na2SO4, и концентрировали, получая указанное в заголовке соединение в виде красновато-коричневого твердого вещества (142 г), которое далее использовали в полученном виде.
е. 1-(6-(2-амино-4-(4-этилпиперазин-1-ил)фениламино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевина

К раствору 3-(2,6-дихлор-3,5-диметоксифенил)-1-(6-(4-(4-этилпиперазин-1-ил)-2-нитрофениламино)пиримидин-4-ил)-1-метилмочевины (330 мг, 0,546 ммоль) в THF (20 мл) и МеОН (20 мл) добавляли никель Ренея (суспензия в воде) при комнатной температуре, и полученную смесь перемешивали в течение 3 часов в атмосфере водорода (1 атм). Реакционную смесь фильтровали и концентрировали. Остаток дважды промывали MeOH, получая указанное в заголовке соединение (280 мг, чистота: 90%), которое использовали непосредственно на следующей стадии. MS (ESI): 575 [M+H]+.
f. Получение N-(2-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(4-этилпиперазин-1-ил)фенил)акриламида

В 5-литровую трехгорлую круглодонную колбу загружали дихлорметан (1 л), акриловую кислоту (14,6 мл, 213 ммоль, 1,5 экв.) и основание Хюнига (39,5 мл, 227 ммоль, 1,6 экв.). Раствор охлаждали до 0-5°C и добавляли метилхлорформиат (15,4 мл, 198 ммоль, 1,4 экв.) со скоростью, чтобы поддерживать внутреннюю температуру≤7°С. Смесь нагревали до 20-25°С и перемешивали при этой температуре в течение 1 часа, а затем охлаждали до 0-5°С. В отдельной 500-миллилитровой круглодонной колбе растворяли 1-(6-(2-амино-4-(4-этилпиперазин-1-ил)фенил)амино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевину (100 г, 142 ммоль, 1 экв.) в дихлорметане (150 мл). Раствор субстрата переносили в раствор смешанного ангидрида, промывали дихлорметаном (200 мл), и переносили эту промывочную жидкость в раствор ангидрида. К полученной смеси добавляли пиридин (27 5 мл, 340 ммоль, 2,4 экв.), и полученную смесь нагревали до 20-25°С. Реакционную смесь перемешивали при этой температуре, пока анализ ВЭЖХ не показал завершение реакции (примерно 1 час). Затем смесь охлаждали до 0-5°С и добавляли насыщенный водный раствор NaHCO3 (1 л). Реакционную смесь перемешивали в течение 20 минут, затем выдерживали для отстаивания и разделения фаз. Нижнюю органическую фазу промывали второй раз раствором NaHCO3 (600 мл). Затем нижнюю органическую фазу промывали 10%-ным водным раствором NaCl (600 мл). Эту нижнюю органическую фазу сушили над твердым Na2SO4 (250 г), фильтровали и концентрировали, получая указанное в заголовке соединение (108 г, 100%), которое использовали на следующей стадии без очистки.
g. Синтез и кристаллизация свободного основания соединения 108
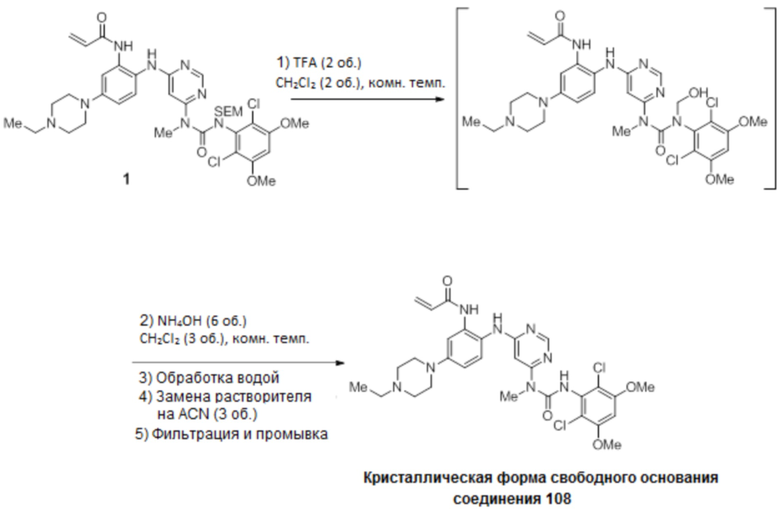
Перемешиваемый раствор N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (1,51 г, 67,12 моль) в дихлорметане (102 мл) охлаждали до 0-5°С. К этому раствору добавляли трифторуксусную кислоту (102 мл, 1585 моль, 23,6 экв.) со скоростью, чтобы поддерживать внутреннюю температуру≤15°С. После завершения добавления, смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов, и в этот момент анализ ВЭЖХ показал завершение реакции. Смесь вносили во вторую колбу, содержащую холодный (0-5°C) насыщенный раствор гидроксида аммония (550 мл), со скоростью, чтобы поддерживать внутреннюю температуру≤25°С. Смесь перемешивали в течение 5-10 минут, и анализ ВЭЖХ показал завершение реакцию. Смесь переносили в делительную воронку и дважды экстрагировали смесью дихлорметан/метанол (5:1, 600 мл) и один раз смесью дихлорметан/метанол (5:1, 300 мл). Объединенные органические фазы концентрировали до минимального перемешиваемого объема, и добавляли ацетонитрил (150 мл). Полученное твердое вещество суспендировали в течение 15 минут, а затем фильтровали. Твердый осадок промывали ацетонитрилом (4 × 30 мл) и сушили на фильтре в вакууме с продувкой азотом, получая N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид (соединение 108, 29,56 г, выход 70%) в виде твердого вещества. 1Н-ЯМР (d6-DMSO): δ 1,04 (т, J=7,15 Гц, 3Н); 2,37 (м, 2H); 3,13 (м, 4H); 3,22 (м, 4H); 3,31 (с, 3H); 3,93 (с, 6H); 5,72 (дд, 1H, J=10,27, 1,83 Гц); 6,23 (дд, 1Н, J=17,06, 1,83 Гц; уш.с, 1H); 6,48 (дд, 1Н, J=16,87, 10,27 Гц); 6,81 (дд, 1H, J=8,99, 2,75 Гц); 6,89 (с, 1Н); 7,29 (уш.д, J=9,17 Гц, 2H); 8,32 (с, 1H); 8,71 (с, 1Н); 9,58 (с, 1Н) (Фиг. 9А-9С). MS: [M+H]+=629,2. Спектр получен на установке Varian Inova при 500 мГц.
Перекристаллизация свободного основания соединения 108
N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид (соединение 108, 42 г) растворяли в смеси дихлорметан/метанол (4:1, 500 мл). К этому раствору добавляли ацетонитрил (150 мл) и смесь концентрировали при пониженном давлении до общего объема 150 мл. К полученной смеси добавляли дополнительное количество ацетонитрила (150 мл) и смесь снова концентрировали при пониженном давлении до общего объема 150 мл. Полученную суспензию фильтровали. Твердый осадок промывали ацетонитрилом (4 × 30 мл) и сушили на фильтре в вакууме с продувкой азотом, получая перекристаллизованное свободное основание N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-(1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (соединение 108, 36 г, выход после перекристаллизации 86%) в виде твердого вещества.
Пример 4. Характеризация кристаллической формы соединения 108 в виде свободного основания с помощью метода рентгеноструктурной дифракции на порошке (PXRD)
В этом примере результаты PXRD для образцов получали на приборе Rigaku MultiFlex Instrument (мишень: Cu; напряжение трубки 40 кВ; ток трубки: 30 мА, диапазон 3,0-45,0=42,0°). Образцы готовили путем внесения порошков в стандартные круглые алюминиевые держатели образцов, и поверхность образцов выравнивали с помощью стеклянного предметного стекла.
Кристаллическая форма соединения 108 в виде свободного основания характеризуется дифрактограммой PXRD, показанной на Фиг. 5, ограниченной для показа диапазон 3,0-35,0=32,0°. Наложение дифрактограмм PXRD для трех партий кристаллической соединения 108 в виде свободного основания (см. Фиг. 16), демонстрируют воспроизводимость результатов.
Как представлено в Таблице 7, кристаллическая форма соединения 108 в виде свободного основания имеет дифрактограмму PXRD с шестью характеристическими пиками.
Таблица 7: Характеристические пики кристаллической формы соединения 108 в виде свободного основания на дифрактограмме PXRD
Пример 5. Характеризация кристаллической формы соединения 108 в виде свободного основания с помощью метода дифференциальной сканирующей калориметрии (DSC)
В этом примере результаты DSC для образцов получали на приборе Mettler-Toledo DSC 1/700 (условия измерения: начальная температура 35°С, конечная температура 325°С, скорость нагрева 10° С/мин).
Приблизительно от 4 до 8 мг кристаллической формы соединения 108 в виде свободного основания вносили в алюминиевые стаканчики емкостью 70 мкл, которые закрывали, обжимали и прокалывали для образования одного отверстия. Образец и пустой стаканчик, подготовленные указанным образом, закрепляли на поверхности термопары, и прибор уравновешивали при начальной температуре. Камеру нагревали до 325°С со скоростью 10°С/мин и получали дифференциальную термограмму.
Дифференциальную термограмму кристаллической формы соединения 108 в виде свободного основания получали с использованием прибора для дифференциальной сканирующей калориметрии (DSC) (см. Фиг. 7, где представлена термограмма, усеченная после температуры приблизительно равной 254°С). Кристаллическая форма соединения 108 в виде свободного основания характеризуется наличием единственного эндотермического пика с началом при температуре 213,6°С (±1°С). Сравнение начала температур для пиков, полученных у трех различных партий кристаллической формы соединения 108 в виде свободного основания, показано в Таблице 8 и на Фиг 8 (термограмма ограничена до температуры, приблизительно равной 246°С).
Таблица 8. Сравнение начала температур пиков для трех партий
кристаллической формы соединения 108 в виде свободного основания
Пример 6. Растворимость кристаллической формы соединения 108 в виде свободного основания
Растворимость кристаллической формы соединения 108 в виде свободного основания в 0,1 Н HCl составляет 5,2 мг/мл.
ПРИМЕР 7
В этом примере представлены дополнительные данные, полученные для отдельной партии кристаллической формы соединения 108 в виде свободного основания, по сравнению с материалом, в отношении которого были получены результаты, указанные выше.
13С-ЯМР (100 Мгц, твердое состояние) δ (мд): 11,9, 31,1, 52,2, 54,8, 56,5, 88,6, 95,7, 106,2, 110,8, 112,3, 114,5, 123,0, 129,0, 130,3, 132,3, 132,9, 133,8, 149,9, 153,2, 154,8, 155,4, 160,0, 162,1, 164,4. Спектр показан на Фиг. 10
Спектр 1Н-ЯМР (DMSO-d6) δ (мд): 1,03 (3H, т, J=7,2 Гц), 2,37 (2H, кв, J=7,2 Гц), 2,46-2,51 (4H, м), 3,12 (4Н, т, J=5,0 Гц), 3,21 (3H, с), 3,92 (6Н, с), 5,71 (1H, дд, J=10,3, 1,7 Гц), 6,13-6,26 (1Н, м), 6,22 (1Н, дд, J=17,1, 1,8 Гц), 6,48 (1Н, дд, J=17,0, 10,3 Гц), 6,80 (1Н, дд, J=8,9, 2,6 Гц), 6,88 (1Н, с), 7,23-7,33 (1Н, м), 7,28 (1Н, д, J=8,9 Гц), 8,30 (1H, с), 8,69 (1H, уш.с), 9,57 (1H, уш.с), 12,05 (1H, с). Спектр получен с использованием прибора Avance 600 Мгц (Bruker). Спектр показан на Фиг. 9D.
Пример 8. Получение N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Раствор (4 мл) ацетона, содержащий 10% (об./об.) DMSO с добавкой раствора соляной кислоты (2,76 мкл, 1 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (20,25 мг), обрабатывали ультразвуком и перемешивали при комнатной температуре в течение 7 дней. Полученные кристаллы отфильтровывали и промывали ацетоном, получая моногидрохлоридную соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида.
Пример 9. Получение кристаллов моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Раствор (2,5 мл) ацетона, содержащий 10% (об./об.) DMSO с добавкой раствора соляной кислоты (7,25 мкл, 1 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (53,12 мг), обрабатывали ультразвуком и перемешивали при комнатной температуре в течение дня. Кристаллическую форму, полученную в Примере 8, добавляли к реакционной смеси и дополнительно перемешивали в течение 5 дней. Полученные кристаллы отфильтровывали и промывали этанолом (1 мл), получая кристаллы моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (50,60 мг).
Пример 10: Получение кристаллов моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Раствор (25 мл) ацетона, содержащий 10% (об./об.) DMSO с добавкой раствора соляной кислоты (68,51 мкл, 1 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (502,1 мг), и обрабатывали ультразвуком. К реакционной смеси добавляли кристаллическую форму, полученную в Примере 9, и перемешивали при комнатной температуре в течение 7 дней. Полученные кристаллы отфильтровывали, промывали этилацетатом (1,5 мл) и сушили при пониженном давлении в течение 3 дней, получая кристаллы моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (543,0 мг).
13С-ЯМР (100 Мгц, твердое вещество) δ (мд): 10,9, 32,1, 34,1, 45,4, 49,2, 51,9, 53,7, 56,0, 91,3, 95,7, 112,5, 121,2, 124,1, 128,2, 130,7, 134,3, 144,7, 153,6, 154,8, 160,3, 166,9. Спектр показан на Фиг. 11.
Спектр 1Н-ЯМР (DMSO-d6) δ (мд): 1,26 (3Н, м), 2,98-3,21 (6H, м), 3,23 (3H, с), 3,55 (2H, уш.с), 3,77 (2H, уш.с), 3,92 (6Н, с), 5,72 (1Н, дд, J=10,3, 1,3 Гц), 6,23 (1Н, дд, J=17,0, 1,6 Гц), 6,26 (1H, уш.с), 6,52 (1Н, дд, J=17,0, 10,2), 6,87 (1H, дд, J=9,0, 2,0 Гц), 6,89 (1H, с), 7,37 (1Н, д, J=8,8 Гц), 7,41 (1Н, уш.с), 8,32 (1Н, с), 8,88 (1Н, с), 9,67 (1Н, с), 10,17 (1H, уш.с), 12,02 (1H, с). Спектр показан на Фиг. 12.
ПРИМЕР 11
В этом примере результаты PXRD для образцов были получены на приборе Rigaku RINT TTR-III Instrument (мишень: Cu; напряжение трубки: 50 кВ; ток трубки: 300 мА, диапазон 5,0-35,0=30°).
Кристаллический моногидрохлорид N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученный в Примере 10, характеризуется дифрактограммой PXRD, показанной на Фиг. 13.
Как представлено в Таблице 8, кристаллическая форма моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется дифрактограммой PXRD, имеющей по меньшей мере десять характеристических пиков.
Таблица 8: Характеристические пики кристаллической формы моногидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида на дифрактограмме PXRD
Пример 12. Получение кристаллов дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Ацетон (3 мл), с добавкой раствора соляной кислоты (12,36 мкл, 3 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (30,19 мг), обрабатывали ультразвуком и перемешивали при комнатной температуре в течение 4 дней. Полученные кристаллы отфильтровывали, получая кристаллы дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида.
Пример 13. Получение кристаллов дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Раствор (10 мл) ацетона, содержащий 10% (об./об.) DMSO, с добавкой раствора соляной кислоты (137,7 мкл, 2 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (504,4 мг), и обрабатывали ультразвуком. К реакционной смеси добавляли кристаллическую форму, полученную в Примере 12, и перемешивали при комнатной температуре в течение 7 дней. Полученные кристаллы отфильтровывали, промывали этилацетатом (1,5 мл) и сушили при пониженном давлении в течение 3 дней, получая кристаллы дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (565,4 мг).
13С-ЯМР (100 Мгц, твердое состояние) δ мд): 9,9, 30,4, 35,0, 39,8, 41,5, 45,5, 46,7, 51,3, 53,8, 56,1, 57,6, 93,2, 96,1, 109,9, 111,5, 112,5, 115,6, 119,0, 128,2, 130,0, 131,6, 134,0, 148,6, 154,6, 161,4, 163,7. Спектр показан на Фиг. 14.
Спектр 1H-ЯМР (DMSO-d6) δ (мд): 1,27 (3H, т, J=7,3 Гц), 3,03-3,21 (6H, м), 3,25 (3H, с), 3,52 -3,59 (2H, м), 3,72-3,83 (2H, м), 3,92 (6H, с), 5,72 (1H, дд, J=10,3, 1,5 Гц), 6,23 (1H, дд, J=17,1, 1,6 Гц), 6,32 (1Н, уш.с), 6,53 (1H, дд, J=17,0, 10,4), 6,88 (1H, дд, J=8,9, 2,5 Гц), 6,89 (1H, с), 7,37 (1H, д, J=8,8 Гц), 7,42 (1H, уш.с), 8,34 (1H, с), 9,03 (1H, уш.с), 9,73 (1H, с), 10,43 (1H, уш.с), 11,88 (1Н, уш.с). Спектр показан на Фиг. 15.
ПРИМЕР 14
В этом примере результаты PXRD для образцов были получены на приборе Rigaku RINT TTR-III Instrument (мишень: Cu; напряжение трубки: 50 кВ; ток трубки: 300 мА, диапазон 5,0-35,0=30°).
Кристаллический дигидрохлорид N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученный в Примере 13, характеризуется дифрактограммой PXRD, показанной на Фиг. 16.
Как представлено в Таблице 9, кристаллическая форма дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется дифрактограммой PXRD, имеющей по меньшей мере десять характеристических пиков.
Таблица 9: Характеристические пики кристаллической формы дигидрохлорида N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида на дифрактограмме PXRD
Пример 15. Получение этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Ацетон (5 мл) с добавкой раствора этансульфоновой кислоты (13,51 мкл, 1 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (100,6 мг), обрабатывали ультразвуком, и перемешивали при комнатной температуре в течение 7 дней. Полученные кристаллы отфильтровывали, получая кристаллы этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (103,7 мг).
Пример 16. Получение этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида
Ацетон (20 мл) с добавкой раствора этансульфоновой кислоты (67,22 мкл, 1 экв.), добавляли к N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламиду (500,6 мг), и обрабатывали ультразвуком. К реакционной смеси добавляли кристаллическую форму, полученную в Примере 15, и перемешивали при комнатной температуре в течение 3 дней. Полученные кристаллы отфильтровывали, промывали этилацетатом (1,5 мл) и сушили при пониженном давлении в течение 3 дней, получая кристаллы этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида (562,0 мг).
13С-ЯМР (100 Мгц, твердое состояние) δ (мд): 10,3, 33,4, 44,0, 44,3, 45,0, 45,9, 47,0, 50,0, 53,0, 55,9, 87,6, 95,5, 107,4, 111,0, 124,8, 129,5, 131,5, 134,5, 144,2, 154,6, 159,2, 159,9, 164,1. Спектр показан на Фиг. 17.
Спектр 1Н-ЯМР (DMSO-d6) δ (мд): 1,05 (3H, т, J=7,5 Гц), 1,25 (3Н, т, J=7,3 Гц), 2,37 (2H, кв, J=7,4 Гц), 2,98 (2H, т, J=11,7 Гц), 3,13 (2H, м), 3,21 (2H, м), 3,24 (3H, с), 3,58 (2H, уш.д, J=11,6 Гц), 3,80 (2H, уш.д, J=12,9 Гц), 3,92 (6H, с), 5,72 (1Н, дд, J=10,3, 1,5 Гц), 6,22 (1H, дд, J=17,0, 1,7 Гц), 6,25 (1H, уш.с), 6,50 (1H, дд, J=17,0, 10,3), 6,84-6,92 (2H, м ), 7,36 (1Н, д, J=8,9 Гц), 7,41 (1H, уш.с), 8,32 (1H, с), 8,81 (1H, с), 9,34 (1H, уш.с), 9,58 (1H, уш.с) 11,99 (1H, с). Спектр показан на Фиг. 18.
ПРИМЕР 17
В этом примере результаты PXRD для образцов были получены на приборе Rigaku RINT TTR-III Instrument (мишень: Cu; напряжение трубки: 50 кВ; ток трубки: 300 мА, диапазон 5,0-35,0=30°).
Кристаллический этансульфонат N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, полученный в Примере 16, характеризуется дифрактограммой PXRD, показанной на Фиг. 19.
Как представлено в Таблице 10, кристаллическая форма этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида характеризуется дифрактограммой PXRD, имеющей по меньшей мере десять характеристических пиков.
Таблица 10: Характеристические пики кристаллической формы этансульфоната N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида на дифрактограмме PXRD
Вышеприведенные экспериментальные материалы является иллюстративными для настоящего изобретения, и их не следует истолковывать как ограничивающие настоящее изобретение. Изобретение определяется следующей формулой изобретения с эквивалентами, которые включены в нее.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ FGFR4 | 2014 |
|
RU2715708C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ ГЕПАТОЦЕЛЛЮЛЯРНОЙ КАРЦИНОМЫ | 2017 |
|
RU2769251C2 |
| Комбинированные терапии для лечения гепатоцеллюлярной карциномы | 2019 |
|
RU2787993C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
| ИНГИБИТОР FGFR И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2745035C1 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2014 |
|
RU2704112C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ FGFR4, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СОСТАВЫ, СОДЕРЖАЩИЕ ИХ, И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2827699C1 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНОМОЧЕВИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2005 |
|
RU2430093C2 |

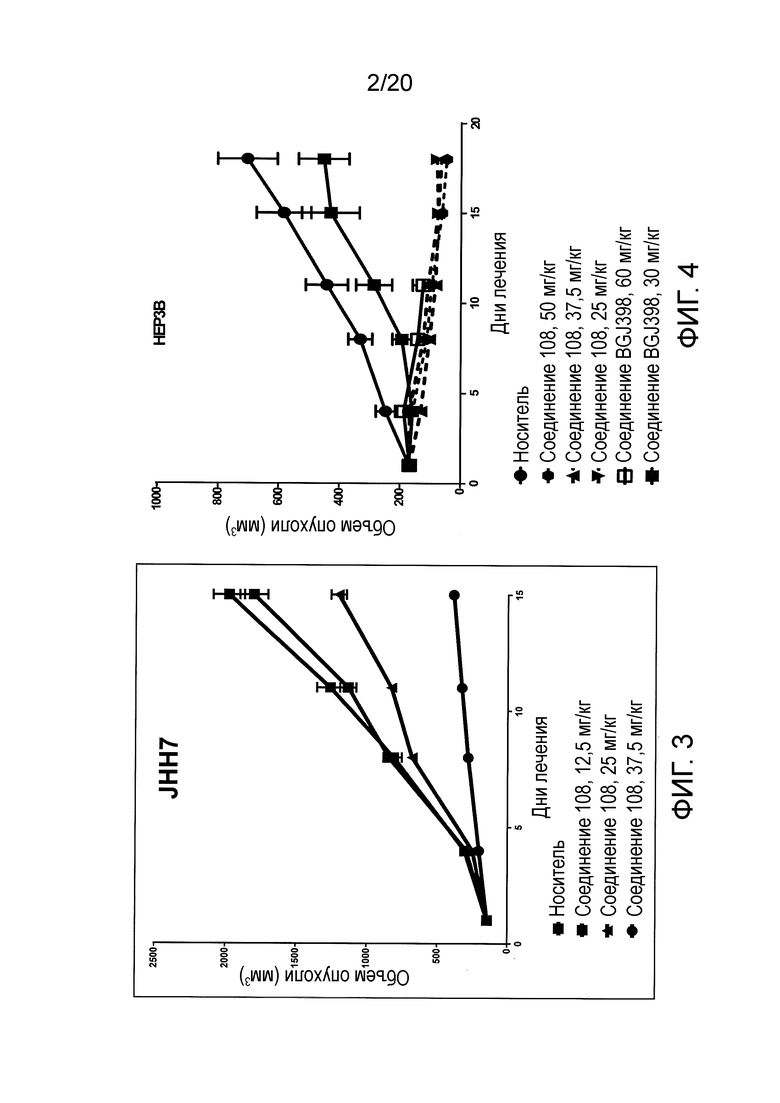
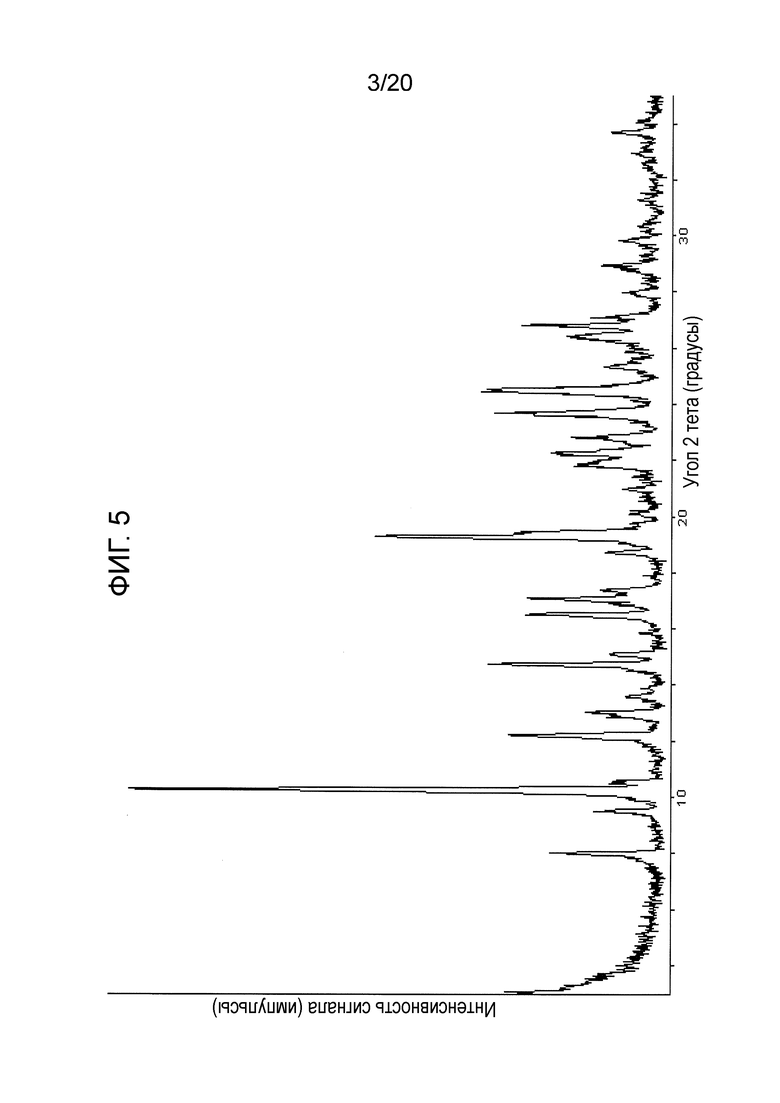

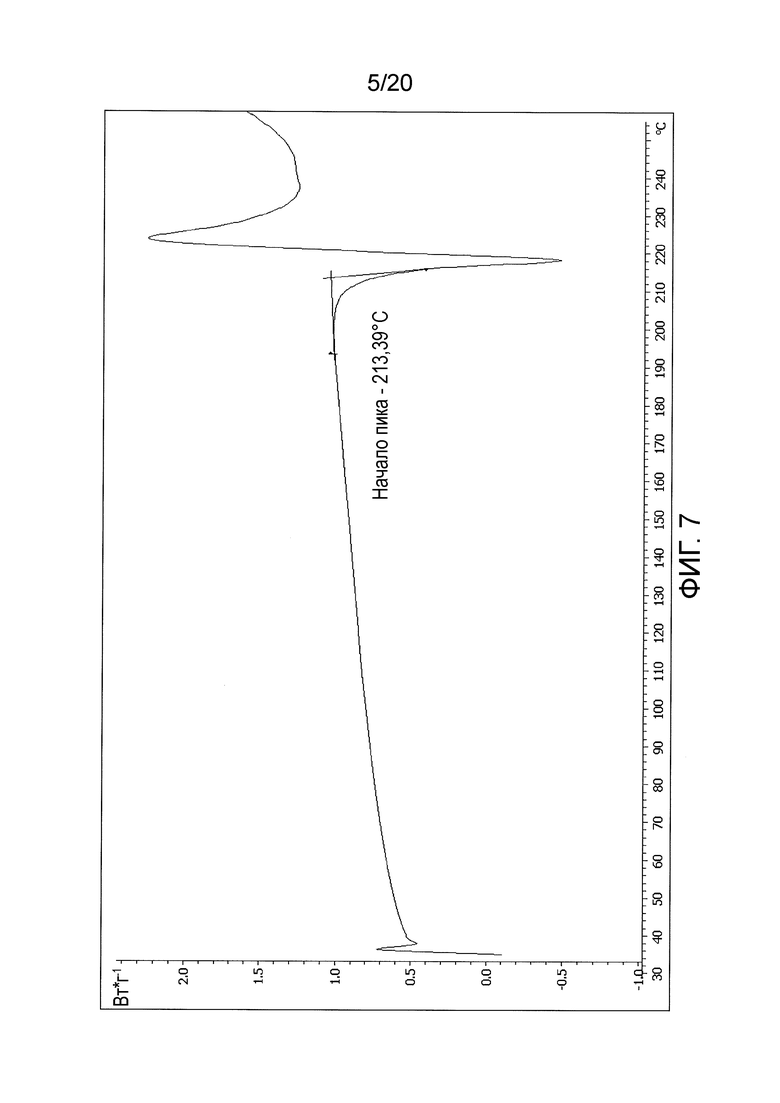
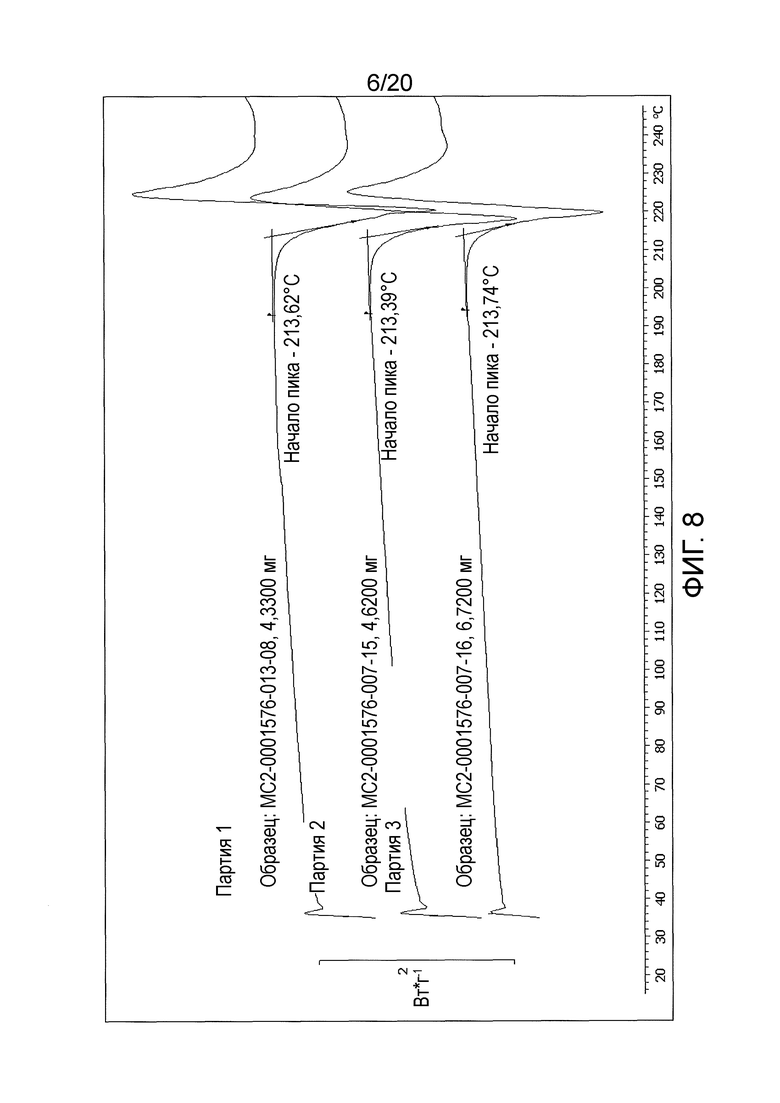

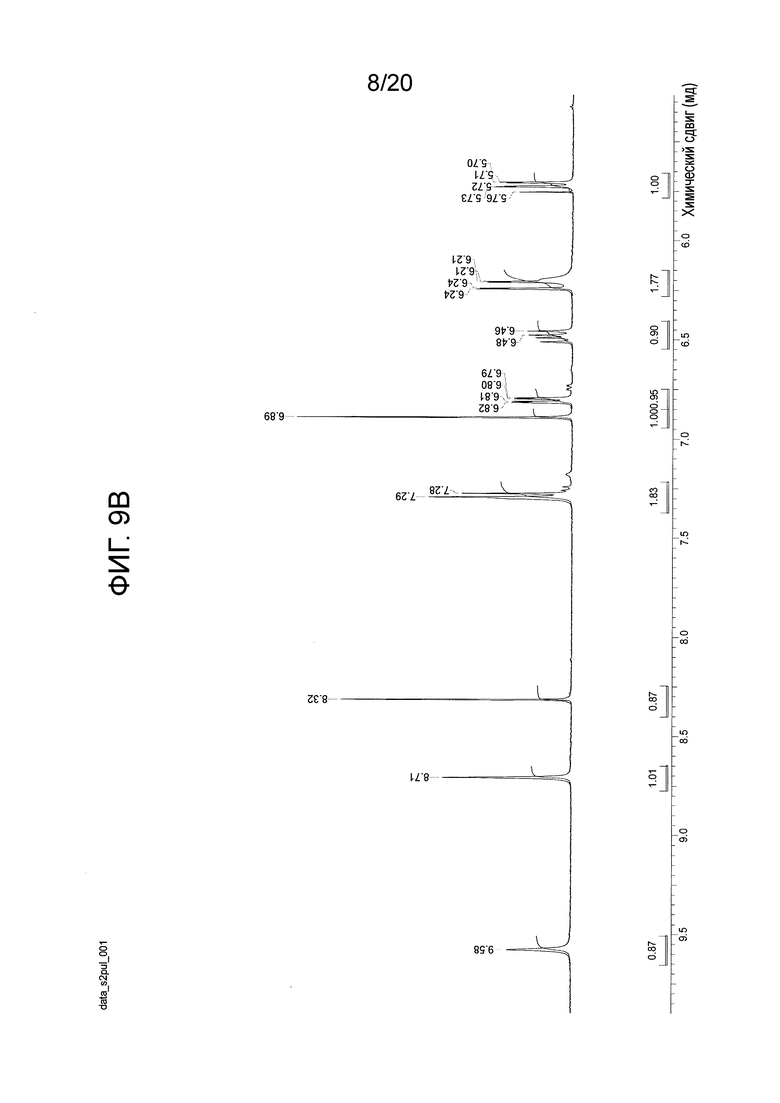
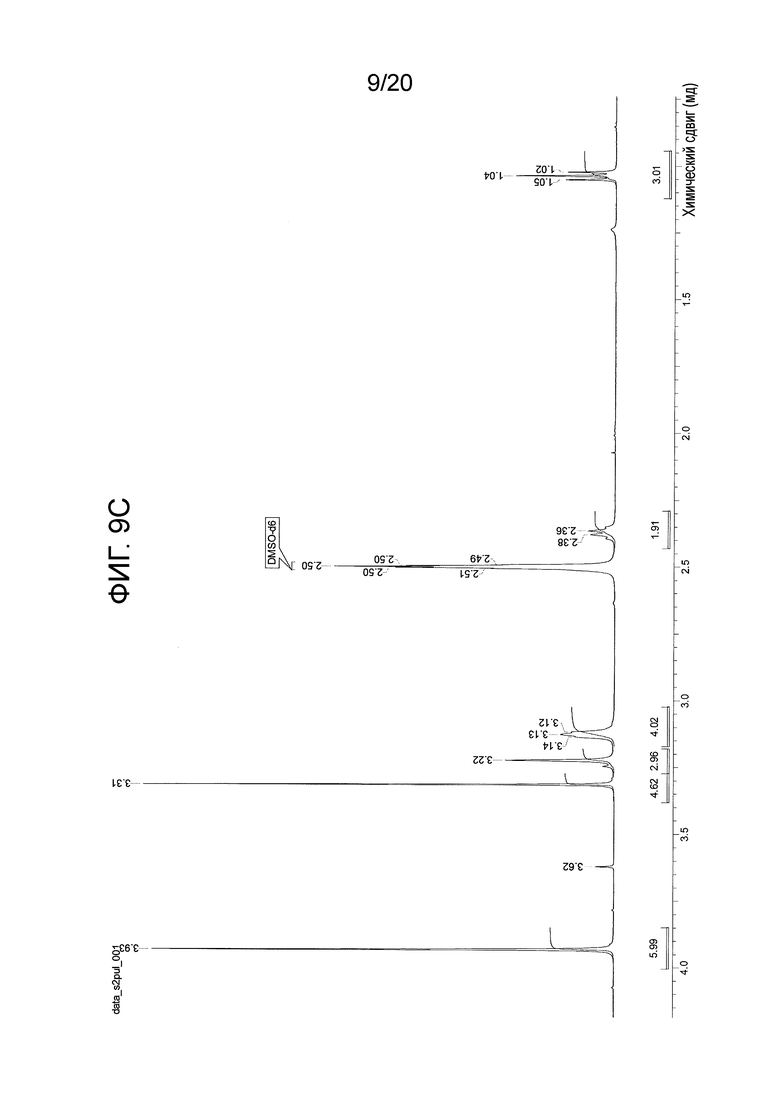


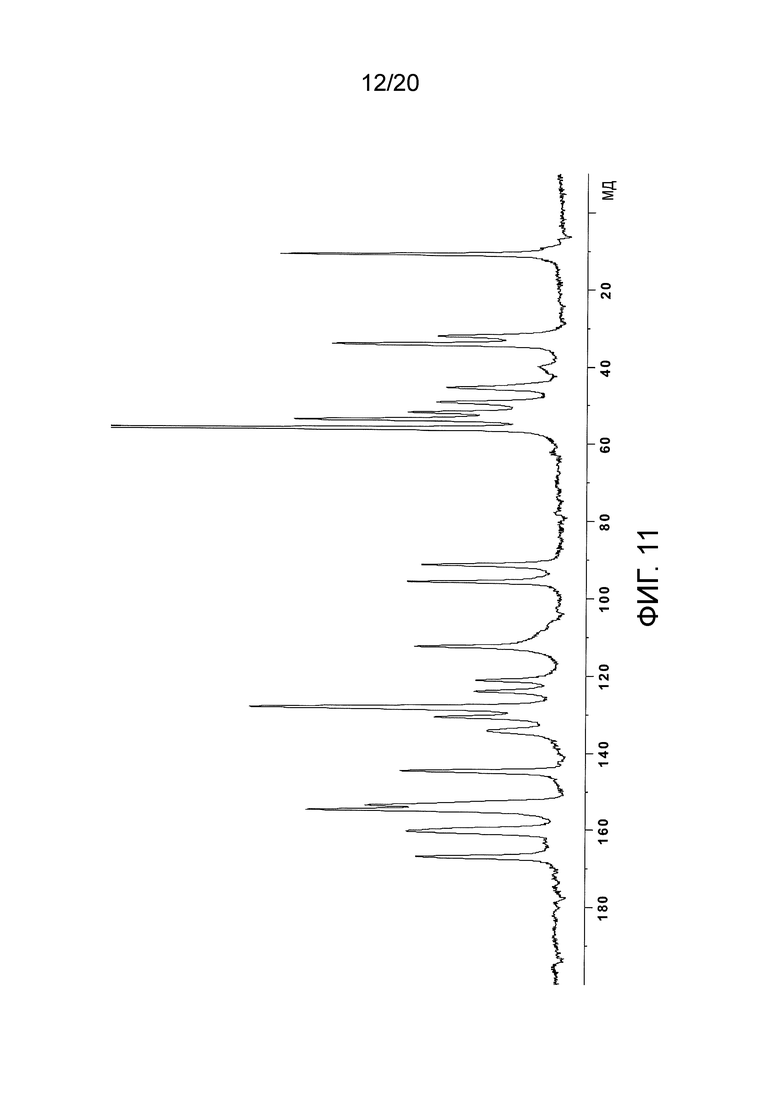
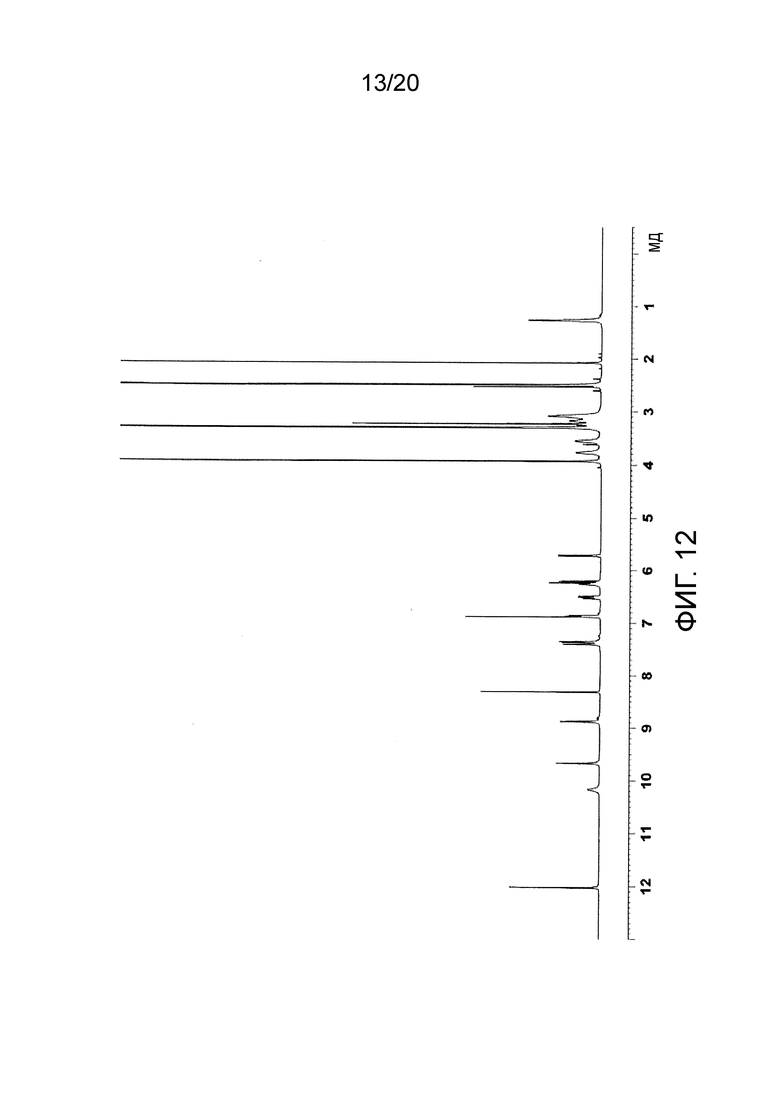



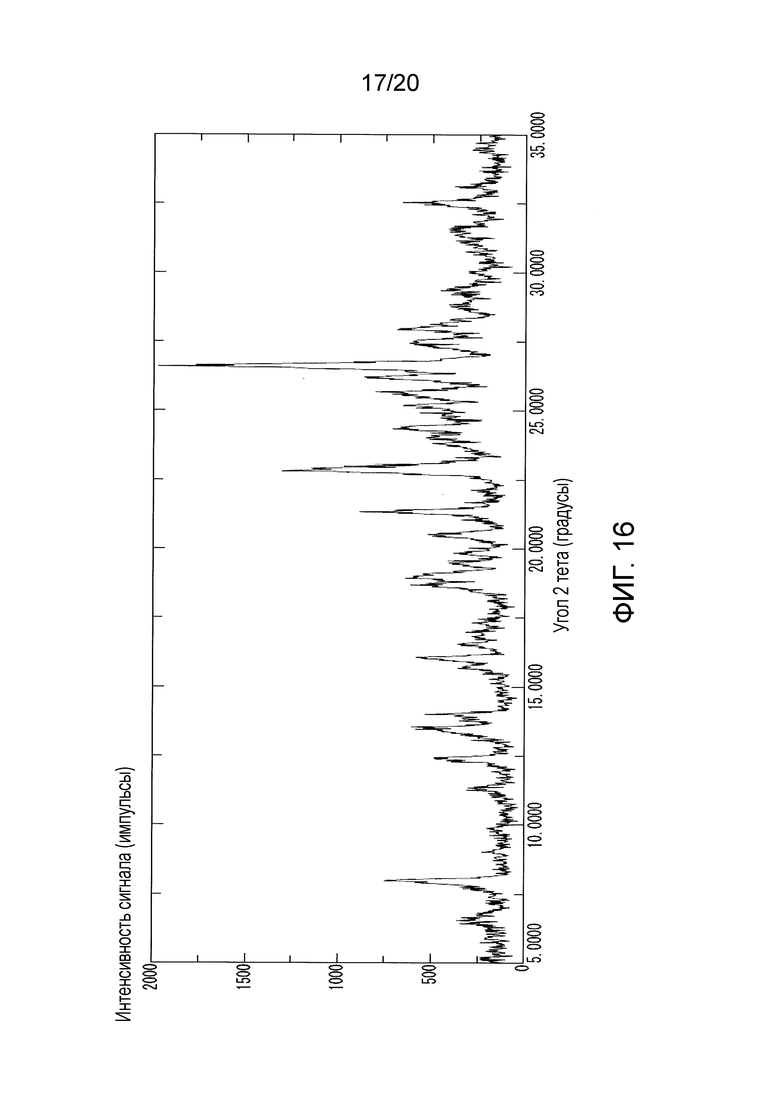
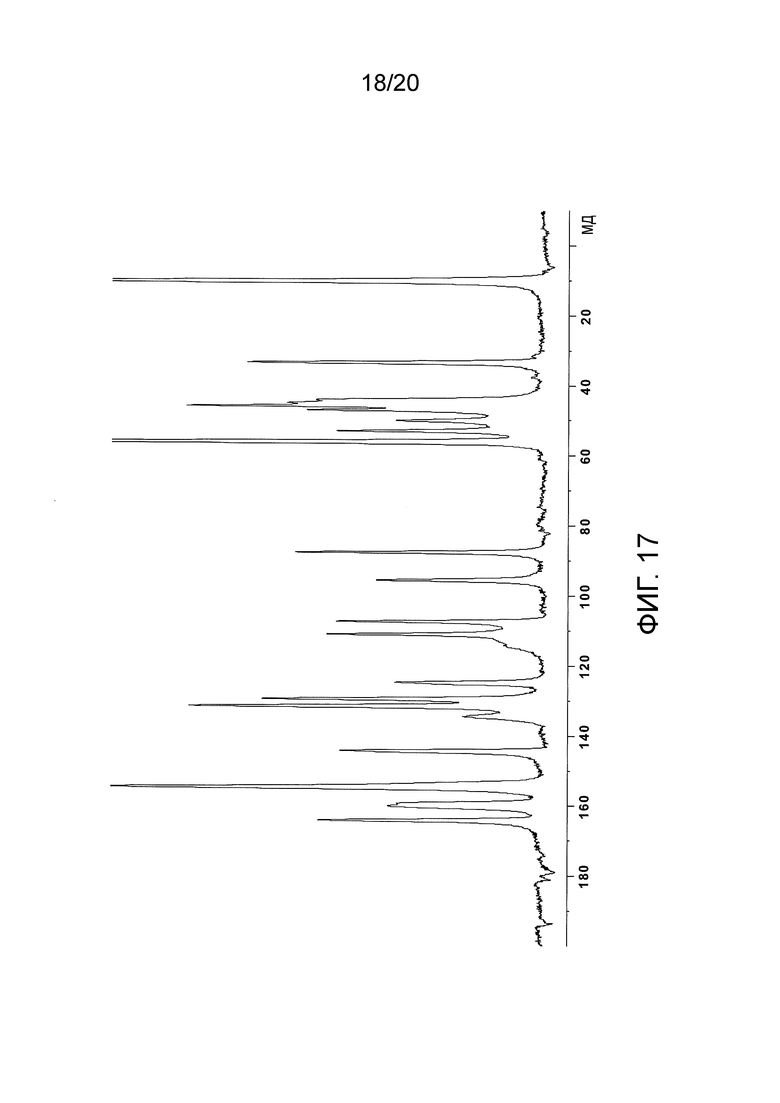
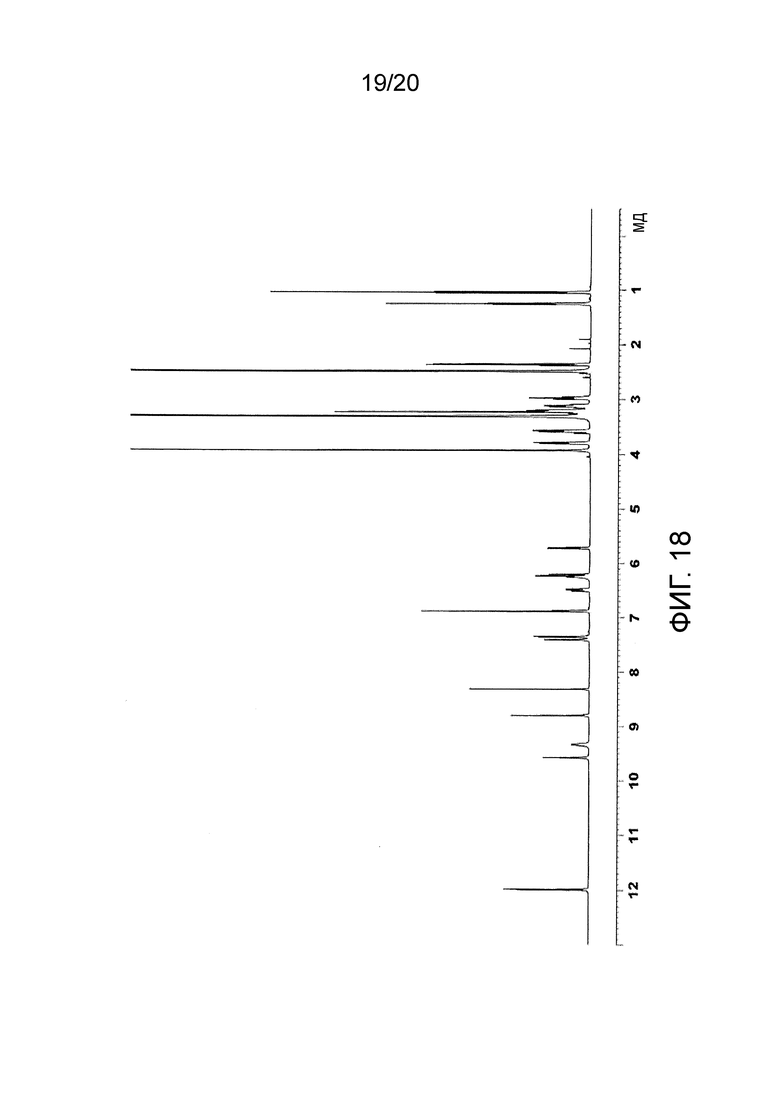

Изобретение относится к ингибиторам FGFR и их применению в качестве противоопухолевых средств. Предложена кристаллическая форма N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида в виде свободного основания. Также предложены способы получения указанной кристаллической формы и ее применение для лечения гепатоцеллюлярной карциномы. Технический результат – предложенная кристаллическая форма стабильна при хранении и хорошо растворима. 7 н. и 15 з.п. ф-лы, 19 ил., 10 табл., 17 пр.
1. Кристаллическая форма соединения
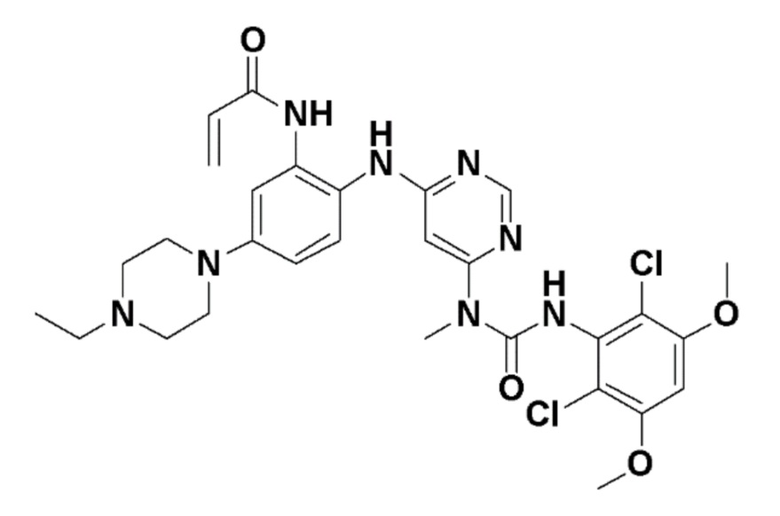
N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид,
где указанное соединение представляет собой кристаллическую форму свободного основания N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламида, которая имеет по меньшей мере следующие спектральные пики на порошковой рентгеноструктурной дифрактограмме 2Θ° (±0,2): 10,3, 8,0, 14,8, 16,5, 17,1 и 21,8.
2. Кристаллическое соединение в виде свободного основания по п.1, где кристаллическая форма указанного соединения характеризуется кривой дифференциальной сканирующей калориметрии (DSC), имеющей единственный эндотермический пик с началом при температуре 213,6°С (±1°С).
3. Кристаллическое соединение в виде свободного основания по п.2, которая характеризуется DSC, показанной на Фиг.7.
4. Кристаллическое соединение в виде свободного основания по п.1, где кристаллическое соединение в виде свободного основания имеет по меньшей мере следующие спектральные пики на порошковой рентгеноструктурной дифрактограмме 2Θ° (±0,2): 10,3, 8,0, 9,5, 14,8, 16,5, 17,1, 19,3, 21,8, 23,7 и 24,5.
5. Кристаллическое соединение в виде свободного основания по п.1, где кристаллическая форма соединения в виде свободного основания характеризуется по существу спектром PXRD, показанным на Фиг. 5.
6. Кристаллическое соединение в виде свободного основания по п.1, где кристаллическое соединение в виде свободного основания характеризуется 13С-ЯМР (100 МГц, твердое состояние) δ (мд): 11,9, 31,1, 52,2, 54,8, 56,5, 88,6, 95,7, 106,2, 110,8, 112,3, 114,5, 123,0, 129,0, 130,3, 132,3, 132,9, 133,8, 149,9, 153,2, 154,8, 155,4, 160,0, 162,1, 164,4.
7. Кристаллическое соединение в виде свободного основания по п.1, которое характеризуется 13С-ЯМР спектром, показанным на Фиг. 10.
8. Кристаллическое соединение в виде свободного основания по п.1, где растворимость кристаллического соединения в виде свободного основания в 0,1 Н HCl составляет 5,2 мг/мл.
9. Фармацевтическая композиция для лечения гепатоцеллюлярной карциномы у субъекта, нуждающегося в этом, содержащая эффективное количество кристаллического соединения по любому из пп.1-8 и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п.9, где указанная композиция представлена в форме для перорального, внутривенного или подкожного введения.
11. Способ получения кристаллической формы соединения по п.1, которая представляет собой кристаллическую форму соединения в виде свободного основания, включающий:
a) предоставление композиции, содержащей соединение
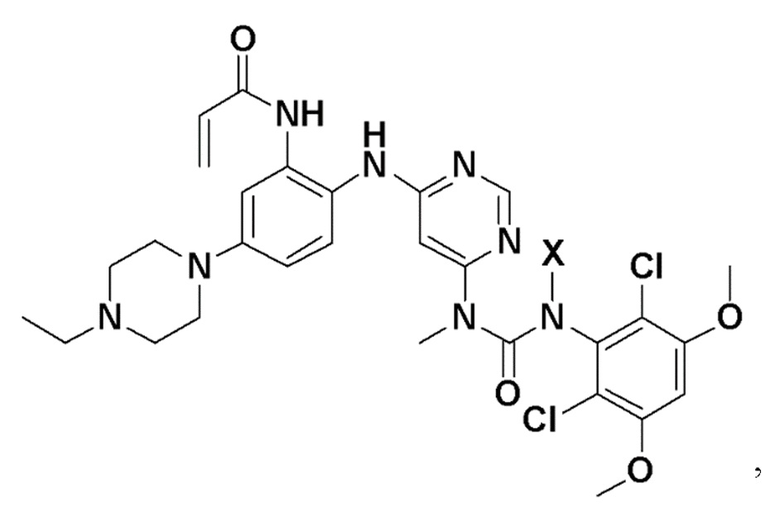
где Х представляет собой (2-(триметилсилил)этокси)метил,
в растворителе, где растворитель представляет собой дихлорметан;
b) добавление трифторуксусной кислоты к указанной композиции с поддержанием температуры композиции ≤50°С; и затем
c) добавление к композиции насыщенного раствора гидроксида аммония; и затем
e) экстрагирование композиции смесью несмешивающихся растворителей с образованием органической фазы, где смесь несмешивающихся растворителей представляет собой смесь дихлорметана/метанола (5:1); и
f) добавление подходящего растворителя к органической фазе, где подходящий растворитель представляет собой ацетонитрил,
с получением, в итоге, кристаллического соединения в форме свободного основания по п.1.
12. Способ лечения гепатоцеллюлярной карциномы у субъекта, нуждающегося в этом, включающий введение указанному субъекту эффективного для лечения количества соединения по любому из пп.1-8 или фармацевтической композиции по п.9 или 10.
13. Способ по п.12, где указанная гепатоцеллюлярная карцинома имеет измененный статус FGFR4 и/или FGF19.
14. Способ по п.13, где указанный измененный статус FGFR4 и/или FGF19 включает повышенную экспрессию FGFR4 и/или FGF19.
15. Способ лечения гепатоцеллюлярной карциномы у субъекта, нуждающегося в этом, включающий:
обнаружение измененного статуса FGFR4 и/или FGF19 в биологическом образце, содержащем клетки указанной гепатоцеллюлярной карциномы, и, в случае если указанная гепатоцеллюлярная карцинома имеет указанный измененный статус FGFR4 и/или FGF19,
введение кристаллической формы соединения по любому из пп.1-8 или фармацевтической композиции по п.9 или 10 указанному субъекту в эффективном для лечения количестве.
16. Способ по п.15, где указанный измененный статус FGFR4 и/или FGF19 включает повышенную экспрессию FGFR4 и/или FGF19.
17. Применение кристаллической формы соединения по любому из пп.1-8 или фармацевтической композиции по п.9 или 10 в способе лечения гепатоцеллюлярной карциномы.
18. Применение по п.17, в котором указанная гепатоцеллюлярная карцинома имеет измененный статус FGFR4 и/или FGF19.
19. Применение по п.18, где указанный измененный статус FGFR4 и/или FGF19 включает повышенную экспрессию FGFR4 и/или FGF19.
20. Применение кристаллической формы соединения по любому из пп.1-8 для получения лекарственного средства для лечения гепатоцеллюлярной карциномы.
21. Применение по п.20, где указанная гепатоцеллюлярная карцинома имеет измененный статус FGFR4 и/или FGF19.
22. Применение по п.21, где указанный измененный статус FGFR4 и/или FGF19 включает повышенную экспрессию FGFR4 и/или FGF19.
| WO 2006000420 A1, 05.01.2006 | |||
| Vito Guagnano et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Journal of Medicinal Chemistry, 2011, | |||

