ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Описание в целом относится к С5-анилинохиназолиновым соединениям и их фармацевтически приемлемым солям. Данные соединения и их фармацевтически приемлемые соли селективно модулируют KIT, в том числе KIT дикого типа и KIT с первичными и вторичными мутациями, и, следовательно, настоящее описание также относится к применению таких соединений и их солей для лечения или предупреждения опосредованного KIT заболевания, в том числе рака. Настоящее описание дополнительно относится к кристаллическим формам С5-анилинохиназолиновых соединений и их фармацевтически приемлемых солей; фармацевтическим композициям, содержащим такие соединения и соли; наборам, содержащим такие соединения и соли; способам изготовления таких соединений и солей, а также к способам лечения опосредованного KIT заболевания, в том числе рака, с применением таких соединений и солей.
УРОВЕНЬ ТЕХНИКИ
Рецепторные тирозинкиназы (RTK) могут быть онкогенными факторами при раке вследствие генетических нарушений, таких как амплификация, мутации или события слияния, или за счет сверхэкспрессии (М.A. Lemmon, K.М. Ferguson, Cell 130, 213 (2007)). Большинство нарушений в RTK приводят к лиганд-независимой активации рецептора и активации нисходящих путей передачи сигнала, стимулирующих рост и пролиферацию клеток, а также увеличение выживаемости. RTK класса III, в том числе KIT, рецепторы альфа и бета тромбоцитарного фактора роста (PDGFR), рецептор колониестимулирующего фактора-1 (CSF1R) и рецептор Fms-подобной тирозинкиназы-3 (FLT3) причастны к целому ряду форм рака человека (K. Verstraete, S.N. Savvides, Nat. Rev. Cancer 12, 753 (2012)).
Ген, кодирующий KIT, расположен на хромосоме 4 и содержит 21 экзон (J. Lennartsson, L. Ronnstrand, Physiol Rev. 92, 1619 (2012)). 976 аминокислот белка KIT разделяются на основные домены: внеклеточный домен, трансмембранный домен, околомембранный домен (JM) и киназный домен, разделенный посередине киназным вставочным доменом (KID). После N-гликозилирования зрелый белок имеет массу ~145 кДа и экспрессируется на клеточной поверхности. После связывания фактора стволовых клеток (SCF) димеризация усиливает собственную активность киназы с фосфорилированием тирозиновых остатков в домене JM (Y547, Y553, Y568 и Y570) с последующим фосфорилированием в KID (Y703, Y721, Y729/730) и наконец в петле активации (Y823) (J.P. DiNitto et al., J. Biochem. 147, 601 (2010)). Некоторые сайты фосфорилирования на KIT представляют собой ключевые сайты стыковки адаптеров и нижележащих эффекторов, передающих сигнал активации. PI3K, Src и MAPK представляют собой ключевые пути передачи сигнала, активируемые ниже по каскаду относительно KIT. Регуляция передачи сигнала KIT предусматривает интернализацию и последующее разрушение рецептора, фосфорилирование Ser 741 и 746 и дефосфорилирование тирозиновых остатков под действием фосфатаз, таких как SHP1.
Обусловленная KIT передача сигнала играет ключевую роль в специфических типах клеток, в том числе в интерстициальных клетках Кахаля (ICC), меланоцитах, тучных клетках, половых клетках и некоторых гемопоэтических стволовых клетках (J. Lennartsson, L. Ronnstrand, Physiol Rev. 92, 1619 (2012)). Нарушения в KIT наблюдаются у злокачественных новообразований, происходящих из данных типов клеток. Например, сообщалось о мутациях KIT в стромальных опухолях желудочно-кишечного тракта (происходящих из ICC), при мастоцитозе и в меланомах.
Мутации в KIT при раке затрагивают несколько экзонов, при этом горячие точки мутаций наблюдаются в домене JM и киназном домене (J. Lennartsson, L. Ronnstrand, Physiol Rev. 92, 1619 (2012)). Считается, что мутации в домене JM устраняют автоингибирующее взаимодействие домена JM с киназным доменом (J.P. DiNitto et al., J. Biochem. 147, 601 (2010)). С более низкой частотой мутации возникают в экзоне 9 (внеклеточный домен-5 Ig) и 13 (АТФ-связывающий карман и "гейткипер"). Мутации в домене JM встречаются при GIST, тогда как мутации, затрагивающие киназный домен, в частности А-петлю, часто встречаются при мастоцитозе. Аналогичным образом, мутации PDGFR при GIST затрагивают как домен JM, так и киназный домен (С. Bahlawane et al., Cell Commun. Signal. 13, 21 (2015)).
Стромальные опухоли желудочно-кишечного тракта (GIST) представляют собой наиболее распространенные мезенхимальные опухоли желудочно-кишечного тракта (С.М. Barnett, С.L. Corless, М.С.Heinrich, Hematol. Oncol. Clin. North Am. 27, 871 (2013)). Чаще всего GIST обнаруживаются в желудке и тонком кишечнике. Неопластическая GIST происходит из тех же клеток-предшественников, что и ICC, и подавляющее большинство GIST экспрессируют белок KIT, который изначально назывался CD117. Мутации KIT, затрагивающие экзон 11, впервые были идентифицированы при GIST в 1998 году (S. Hirota et al., Science 279, 577 (1998)). В той же публикации также сообщалось об онкогенности мутаций KIT, экспрессируемой эктопически в клетках Ва/F3, и вызываемой ими конститутивной активации киназы. В 75-80% случаев GIST характеризуются мутациями KIT и ~10% случаев мутациями PDGFR (J.A. Fletcher, Cancer Res. 76, 6140 (2016)). Редкие нарушения в BRAF, NF1 и SDH соответствуют тому, что обозначается как KIT WT (С.М. Barnett, С.L. Corless, М.С. Heinrich, Hematol. Oncol. Clin. North Am. 27, 871 (2013)).
Иматиниб был первым ингибитором KIT, протестированным в отношении GIST, который демонстрирует значительную активность у пациентов с GIST на поздней стадии (G.D. Demetri et al., N. Engl. J. Med. 347, 472 (2002), J. Verweij et al., Lancet 364, 1127 (2004), C.D. Blanke et al., J. Clin. Oncol. 26, 626 (2008)). При метаанализе 2 крупных клинических исследований сделали вывод, что в случае пациентов с мутациями KIT в экзоне 9 или другими мутациями прогноз был хуже, чем в случае пациентов с мутациями в экзоне 11 (Metagist, J. Clin. Oncol. 28, 1247 (2010)). Кроме того, высокая доза иматиниба (800 мг) не улучшала показатель выживаемости без прогрессирования у пациентов с мутациями в экзоне 9 по сравнению со стандартной дозой (400 мг). О клинических случаях устойчивости к иматинибу впервые сообщалось в 2005 году (С.R. Antonescu et al., Clin. Cancer Res. 11, 4182 (2005)), однако более крупное исследование, отслеживающее пациентов, которых лечили иматинибом в качестве части фазы II исследования В2222, продемонстрировало реактивацию KIT и передачи сигнала с участием KIT, когда у пациентов, которые изначально получили пользу от иматиниба, происходил рецидив (М.С. Heinrich et al., J. Clin. Oncol. 24, 4764 (2006)). Вторичные мутации, приводящие к устойчивости, отмечались в ключевых остатках: V654A в АТФ-связывающем кармане, T670I в остатках "гейткипера" и А-петли (D816X, D820X, N822K, Y823D). Кроме того, так называемая "первичная устойчивость" к иматинибу наблюдалась, главным образом, у пациентов с мутациями в экзоне 9. В целом у 50% пациентов в течение 2 лет развилась устойчивость (С.D. Blanke et al., J. Clin. Oncol 26, 626 (2008)).
Сунитиниб представляет собой мультикиназный ингибитор, ингибирующий в том числе KIT и PDGFR. Сунитиниб продемонстрировал клиническую активность на пациентах с GIST, у которых наблюдалось прогрессирование при приеме иматиниба (G.D. Demetri et al., Lancet 368, 1329 (2006)). Клиническую пользу при приеме сунитиниба наблюдали у пациентов с первичными мутациями в экзоне 9. Кроме того, пациенты с вторичными мутациями, затрагивающими экзон 13 и 14, характеризовались более продолжительными выживаемостью без прогрессирования и общей выживаемостью по сравнению с пациентами с вторичными мутациями, затрагивающими А-петлю (М.С. Heinrich et al., J. Clin. Oncol. 26, 5352 (2008)). Клинические случаи прогрессирования при приеме сунитиниба наблюдали в пределах 1 года лечения. Эктопическая экспрессия KIT с первичными и вторичными мутациями в клетках СНО показала, что сунитиниб снижал фосфорилирование KIT преимущественно в тех случаях, когда нарушения KIT затрагивали АТФ-связывающий карман или "гейткипер".
Регорафениб, другой мультикиназный ингибитор, продемонстрировал клиническую активность у пациентов с GIST после рецидива на фоне приема иматиниба и сунитиниба (G.D. Demetri et al., Lancet 381, 295 (2013)). По данным исследования фазы III медианная PFS составляла 4,8 месяца.
Соответственно, существует потребность в ингибиторах KIT, которые ингибируют KIT с вторичными мутациями и, кроме того, являются селективными в отношении KDR, в частности ввиду того, что существующие средства лечения являются неэффективными в отношении таких вторичных мутаций. Также существует потребность в ингибиторах KIT, которые ингибируют KIT с первичными мутациями и KIT дикого типа.
КРАТКОЕ ОПИСАНИЕ
Было обнаружено, что соединения по настоящему изобретению обладают сильной противоопухолевой активностью и применимы в ингибировании целого ряда KIT с вторичными мутациями, включающими V654A, D816H и T670I, а также KIT с первичными мутациями и дикого типа, и, кроме того, являются селективными в отношении KDR. Соединения по настоящему изобретению характеризуются необходимыми фармацевтическими свойствами, например хорошими свойствами PK.
Вкратце, настоящее описание, помимо прочего, относится к соединению формулы (I):
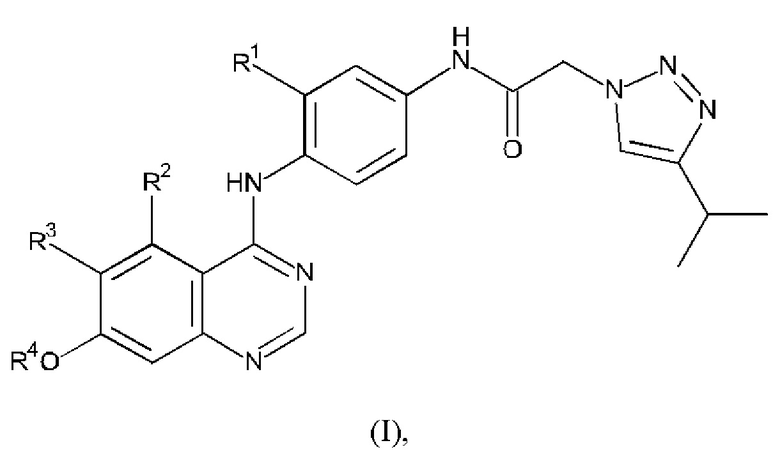
или его фармацевтически приемлемой соли, где:
R1 выбран из водорода и фтора;
R2 выбран из фтора и С1-2алкокси;
R3 выбран из водорода и метокси; и
R4 представляет собой С1-3алкил, необязательно замещенный группой, выбранной из C1-3алкокси и NR5R6, где каждый из R5 и R6 независимо представляет собой водород или метил; или 4-6-членное гетероциклильное кольцо, содержащее один атом кислорода.
В настоящем описании, помимо прочего, также описана фармацевтическая композиция, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый разбавитель или носитель.
В настоящем описании, помимо прочего, также описано соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
В настоящем описании, помимо прочего, также описано соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака.
В настоящем описании, помимо прочего, также описано соединение формулы (I) или его фармацевтически приемлемая соль для применения для изготовления лекарственного препарата для лечения рака.
В настоящем описании, в частности, также описан способ лечения рака у теплокровного животного, нуждающегося в таком лечении, который предусматривает введение теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре А показана XRPD для формы А N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (соединение X, пример 12).
На фигуре В показана DSC для формы А N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (соединение X, пример 12).
На фигуре С показана XRPD для формы В N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (соединение X, пример 12).
На фигуре D показана DSC для формы В N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (соединение X, пример 12).
На фигуре Е показана XRPD для формы А тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (тозилатная соль Y, пример 12А).
На фигуре F показана DSC для формы А тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (тозилатная соль Y, пример 12А).
На фигуре G показана XRPD для формы В тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (тозилатная соль Y, пример 12А).
На фигуре Н показана DSC для формы В тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (тозилатная соль Y, пример 12А).
На фигуре I показана XRPD для формы D тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (тозилатная соль Y, пример 12А).
На фигуре J показана DSC для формы D тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (тозилатная соль Y, пример 12А).
ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Многие варианты осуществления настоящего изобретения подробно представлены на протяжении всего настоящего описания и будут понятны читателю, который является специалистом в данной области техники. Настоящее изобретение не должно интерпретироваться как ограниченное каким(-и)-либо определенным(-и) вариантом(-ами) его осуществления.
В первом варианте осуществления предусмотрено соединение формулы (I):
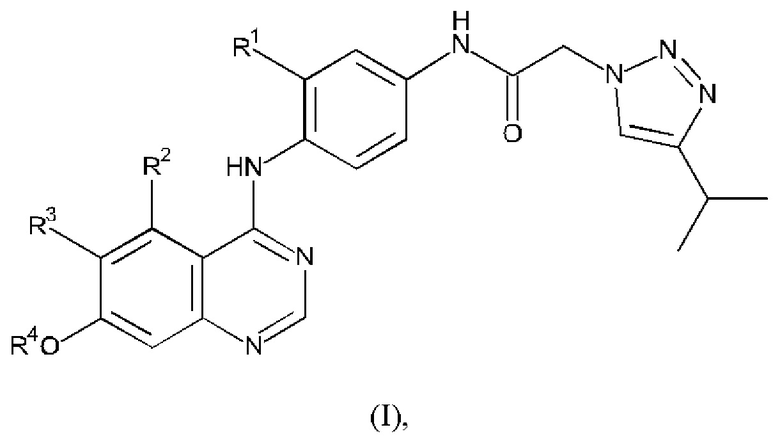
или его фармацевтически приемлемая соль, где:
R1 выбран из водорода и фтора;
R2 выбран из фтора и С1-2алкокси;
R3 выбран из водорода или метокси; и
R4 представляет собой С1-3алкил, необязательно замещенный группой, выбранной из С1-3алкокси и NR5R6, где каждый из R5 и R6 независимо представляет собой водород или метил; или 4-6-членное гетероциклильное кольцо, содержащее один атом кислорода.
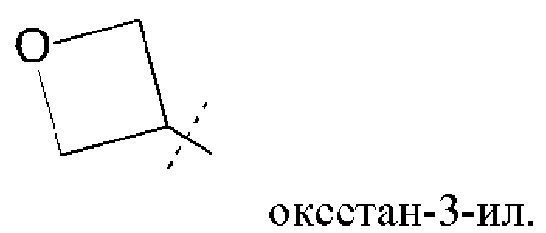
Подходящие 4-6-членные гетероциклильные кольца, содержащие один атом кислорода, включают оксетанильное кольцо, тетрагидрофуранильное кольцо и оксанильное кольцо.
Термин "оксетанильное" кольцо включает оксетан-3-ил, структура которого показана ниже:
Термин "тетрагидрофуранил" включает тетрагидрофуран-3-ил, структура которого показана ниже:
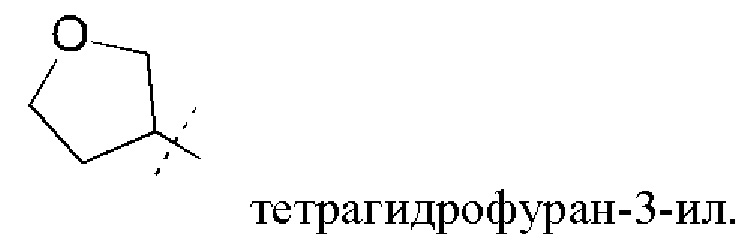
Термин "оксанильное" кольцо включает оксан-3-ильную и океан-4-ильную группы, структуры которых показаны ниже:

В приведенных выше структурах пунктирная линия обозначает положение образования связи у соответствующей группы.
Оксанильное кольцо также может обозначаться как тетрагидропиранильное кольцо. Аналогичным образом, оксан-4-ильное кольцо может обозначаться как тетрагидропиран-4-ильное кольцо, а оксан-3-ильное кольцо может обозначаться как тетрагидропиран-3-ильное кольцо.
Префикс Ср-q в Ср-qалкиле и других терминах (где р и q составляют целые числа) указывает на диапазон числа атомов углерода, которые присутствуют в группе, например С1-3алкил включает С1алкил (метил), С2алкил (этил) и С3алкил (пропил, такой как н-пропил и изопропил).
Термин Ср-qалкокси предусматривает -O-Ср-qалкильные группы.
При применении термина "необязательно" подразумевается, что последующий признак может существовать или может не существовать. Соответственно, применение термина "необязательно" включает случаи, когда признак присутствует, а также случаи, когда признак не присутствует. Например, группа, "необязательно замещенная одной метоксигруппой", включает группы с метокси-заместителем и без него.
Термин "замещенный" означает, что один или несколько атомов водорода (например, один или два атома водорода или в качестве альтернативы один атом водорода) в обозначенной группе замещен указанным заместителем(-ями) (например, одним или двумя заместителями или в качестве альтернативы одним заместителем) при условии, что любой атом(-ы), несущий(-ие) заместитель, сохраняет(-ют) допустимую валентность. Комбинации заместителей охватывают только стабильные соединения и стабильные промежуточные соединения в ходе синтеза. "Стабильный" означает, что соответствующее соединение или промежуточное соединение является достаточно устойчивым для выделения и применимо либо в качестве промежуточного соединения в ходе синтеза, либо в качестве средства, характеризующегося потенциальной терапевтической применимостью. Если группа не описана как "замещенная" или "необязательно замещенная", ее следует рассматривать как незамещенную (т.е. ни один из атомов водорода в обозначенной группе не был заменен).
Термин "фармацевтически приемлемый" применяется для указания того, что объект (например, соль, лекарственная форма, разбавитель или носитель) является подходящим для применения у пациентов. Иллюстративный перечень фармацевтически приемлемых солей можно найти в Handbook of Pharmaceutical Salts: Properties, Selection and Use, P.H. Stahl and C.G. Wermuth, editors,  2002. Подходящей фармацевтически приемлемой солью соединения формулы (I) является, например, соль присоединения кислоты. Соль присоединения кислоты соединения формулы (I) может быть образована посредством приведения соединения в контакт с подходящей неорганической или органической кислотой в условиях, известных специалисту в данной области технике. Например, соль присоединения кислоты может быть образована с применением неорганической кислоты, выбранной из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты и фосфорной кислоты. Соль присоединения кислоты также может быть образована с применением органической кислоты, выбранной из группы, состоящей из трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, щавелевой кислоты, уксусной кислоты, муравьиной кислоты, бензойной кислоты, фумаровой кислоты, янтарной кислоты, винной кислоты, молочной кислоты, пировиноградной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты и пара-толуолсульфоновой кислоты.
2002. Подходящей фармацевтически приемлемой солью соединения формулы (I) является, например, соль присоединения кислоты. Соль присоединения кислоты соединения формулы (I) может быть образована посредством приведения соединения в контакт с подходящей неорганической или органической кислотой в условиях, известных специалисту в данной области технике. Например, соль присоединения кислоты может быть образована с применением неорганической кислоты, выбранной из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты и фосфорной кислоты. Соль присоединения кислоты также может быть образована с применением органической кислоты, выбранной из группы, состоящей из трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, щавелевой кислоты, уксусной кислоты, муравьиной кислоты, бензойной кислоты, фумаровой кислоты, янтарной кислоты, винной кислоты, молочной кислоты, пировиноградной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты и пара-толуолсульфоновой кислоты.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где фармацевтически приемлемая соль представляет собой тозилатную, мезилатную или безилатную соль. В случае данных солей продемонстрировано улучшение технологических свойств при получении составов, содержащих соединения формулы (I). В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где фармацевтически приемлемая соль представляет собой тозилатную соль. В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где фармацевтически приемлемая соль представляет собой моно-тозилатную соль, т.е. стехиометрическое соотношение соединения формулы (I) и тозилата составляет 1:1.
В дополнительном варианте осуществления предусмотрен любой из вариантов осуществления, определенных в данном документе (например, вариант осуществления по пункту 1), при условии, что один или несколько конкретных примеров (например, один, два или три конкретных примера), выбранных из группы, состоящей из примеров 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 12А, 13, 14, 15, 16, 17, 18, 19, 20, 21 и 22, по отдельности исключены.
В дополнительном варианте осуществления предусмотрен любой из вариантов осуществления, определенных в данном документе (например, вариант осуществления по пункту 1), при условии, что один или несколько конкретных примеров (например, один, два или три конкретных примера), выбранных из группы, состоящей из примеров 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 и 22, по отдельности исключены.
В одном варианте осуществления R1 представляет собой водород. В одном варианте осуществления R1 представляет собой фтор.
В одном варианте осуществления R2 выбран из фтора, метокси и этокси. В одном варианте осуществления R2 представляет собой фтор. В одном варианте осуществления R2 представляет собой метокси. В одном варианте осуществления R2 представляет собой этокси.
В одном варианте осуществления R3 представляет собой водород. В одном варианте осуществления R3 представляет собой метокси.
В одном варианте осуществления R4 выбран из С1-3алкила, необязательно замещенного группой, выбранной из метокси и NR5R6, где каждый из R5 и R6 независимо представляет собой водород или метил; и оксетанильного, тетрагидрофуранильного и оксанильного кольца.
В одном варианте осуществления R4 выбран из С1-3алкила, необязательно замещенного группой, выбранной из метокси и NR5R6, где каждый из R5 и R6 представляет собой метил; и оксетанильного, тетрагидрофуранильного и оксанильного кольца.
В одном варианте осуществления R4 выбран из метила, этила, изопропила, 2-(диметиламино)этила, 2-метоксиэтила, оксетан-3-ила, тетрагидрофуран-3-ила и оксан-4-ила.
В одном варианте осуществления R4 представляет собой метил. В одном варианте осуществления R4 представляет собой этил. В одном варианте осуществления R4 представляет собой изопропил.
В одном варианте осуществления R4 представляет собой 2-диметиламиноэтил. В одном варианте осуществления R4 представляет собой 2-метоксиэтил. В одном варианте осуществления R4 представляет собой оксетан-3-ил. В одном варианте осуществления R4 представляет собой тетрагидрофуран-3-ил. В одном варианте осуществления R4 представляет собой оксан-4-ил.
В одном варианте осуществления R1 представляет собой водород, R2 представляет собой метокси, R3 представляет собой водород, и R4 представляет собой метил.
В одном варианте осуществления R1 представляет собой водород, R2 представляет собой фтор, R3 представляет собой водород, и R4 представляет собой 2-метоксиэтил.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из:
N-{4-[(5,7-диметоксихиназолин-4-ил)амино]-3-фторфенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
N-{4-[(5-фтор-6,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
(R)-N-(4-{[5-этокси-7-(тетрагидрофуран-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
(S)-N-(4-{[5-этокси-7-(тетрагидрофуран-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
N-(4-((5-этокси-7-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида;
N-(4-((7-(2-(диметиламино)этокси)-5-этоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида;
N-(4-((5-этокси-7-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида;
N-(4-((5-этокси-7-(2-метоксиэтокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида;
N-(4-((5-этокси-7-(оксетан-3-илокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида;
N-(4-{[5-метокси-7-(тетрагидро-2H-пиран-4-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]-N-{4-[(5,6,7-триметоксихиназолин-4-ил)амино]фенил}ацетамида;
N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
(R)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамида;
(S)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамида;
N-(4-{[5-метокси-7-(пропан-2-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида;
N-(4-{[5-метокси-7-(оксетан-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида;
N-[4-({7-[2-(диметиламино)этокси]-5-метоксихиназолин-4-ил}амино)фенил]-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида;
N-{4-[(7-этокси-5-метоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида;
N-{4-[(5,7-диэтоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида;
N-(4-{[5-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида;
N-{4-[(5-фтор-7-метоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида и
N-{4-[(5,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из:
N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида и
N-{4-[(5,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение представляет собой N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид.
В одном варианте осуществления предусмотрено соединение формулы (I), где соединение представляет собой N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамид в форме свободного основания (также обозначаемый как соединение X).
В одном варианте осуществления предусмотрено соединение формулы (I), где соединение представляет собой тозилатную соль N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (также обозначаемую как тозилатная соль Y).
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, где соединение представляет собой N-{4-[(5,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид.
Соединения и соли, описываемые в настоящем описании, могут существовать в сольватированных формах и несольватированных формах. Например, сольватированная форма может представлять собой гидратированную форму, такую как полугидрат, моногидрат, дигидрат, тригидрат или другая степень гидратации. Настоящее изобретение охватывает все такие сольватированные и несольватированные формы соединений формулы (I), в частности, при условии, что такие формы обладают ингибирующей KIT активностью, например, измеренной с применением описываемых в данном документе испытаний.
Атомы соединений и солей, описываемых в настоящем описании, могут существовать в виде своих изотопов. Настоящее изобретение охватывает все соединения формулы (I), в которых атом замещен одним или несколькими своими изотопами (например, соединение формулы (I), в котором один или несколько атомов углерода представляют собой изотоп углерода 11С или 13С, или в котором один или несколько атомов водорода представляют собой изотоп 2Н или 3Н, или в котором один или несколько атомов азота представляют собой изотоп 15N, или в котором один или несколько атомов кислорода представляют собой изотоп 17О или 18О).
Соединения и соли, описанные в настоящем описании, могут существовать в виде оптически активных или рацемических форм вследствие наличия одного или нескольких асимметрических атомов углерода. Настоящее изобретение включает любую оптически активную или рацемическую форму соединения формулы (I), которая обладает ингибирующей KIT активностью, например, измеренной с применением описываемых в данном документе испытаний. Синтез оптически активных форм можно осуществлять с помощью стандартных методик органической
химии, хорошо известных из уровня техники, например, с помощью синтеза с применением оптических активных исходных веществ или за счет разделения рацемической формы.
Следовательно, в одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль, которые представляют собой отдельный оптический изомер, находящийся в энантиомерном избытке (% э.и.), составляющем ≥95%, ≥98% или ≥99%. В одном варианте осуществления отдельный
оптический изомер присутствует в энантиомерном избытке (% э.и.), составляющем ≥99%.
Некоторые из соединений формулы (I) могут быть кристаллическими и могут характеризоваться несколькими кристаллическими формами. Следует понимать, что настоящее изобретение охватывает любую кристаллическую или аморфную форму или их смеси, которые обладают свойствами, применимыми для обеспечения ингибирующей KIT активности. Хорошо известно как определить эффективность кристаллической или аморфной формы с помощью стандартных испытаний, описанных далее в данном документе.
В целом известно, что кристаллические материалы можно анализировать с применением традиционных методик, таких как, например, анализ на основе порошковой рентгеновской дифрактометрии (далее в данном документе XRPD) и дифференциальная сканирующая калориметрия (далее в данном документе DSC).
В качестве примера, соединение из примера 12, соединение X, проявляет кристалличность и характеризуется двумя кристаллическими формами, формой А и формой В, которые были идентифицированы и были получены с применением условий, описанных далее в данном документе в экспериментальном разделе
Полиморфная форма А соединения X
Соответственно, дополнительный аспект настоящего изобретения представляет собой форму А соединения X (пример 12).
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=6,7° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=18,7° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при приблизительно 2-тета=6,7° и 18,7° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD, имеющей специфические пики при приблизительно 2-тета=3,4, 6,7, 9,9, 16,2, 18,7, 22,1, 23,3 25,1, 26,6, 28,9° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А, соединения X, которая характеризуется рентгенограммой XRPD, фактически идентичной рентгенограмме XRPD, показанной на фигуре А, согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А, соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=6,7° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=18,7° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при 2-тета=6,7° и 18,7°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А соединения X, которая характеризуется рентгенограммой XRPD со специфическими пиками при 2-тета=3,4, 6,7, 9,9, 16,2, 18,7, 22,1, 23,3, 25,1, 26,6, 28,9°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
DSC-анализ формы А соединения X демонстрирует эндотерму плавления с началом при 235,7°С и пиком при 237,6°С (фигура В).
Таким образом, DSC-анализ демонстрирует, что форма А соединения X представляет собой тугоплавкое твердое вещество с началом плавления при приблизительно 235,7°С и пиком при приблизительно 237,6°С.
Полиморфная форма В соединения X
Дополнительный аспект настоящего изобретения представляет собой форму В соединения X.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=4,2° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=7,7° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при приблизительно 2-тета=4,2° и 7,7° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD со специфическими пиками при приблизительно 2-тета=4,2, 6,6, 7,7, 9,8, 13,1, 13,7, 18,6, 20,0, 26,5, 28,8° согласно измерениям с применением CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В, соединения X, которая характеризуется рентгенограммой XRPD, фактически идентичной XRPD, показанной на фигуре С, согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В, соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=4,2° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=7,7° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при 2-тета=4,2° и 7,7°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В соединения X, которая характеризуется рентгенограммой XRPD со специфическими пиками при 2-тета=4,2, 6,6, 7,7, 9,8, 13,1, 13,7, 18,6, 20,0, 26,5, 28,8°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
DSC-анализ формы В соединения X показан на фигуре D.
В качестве дополнительного примера соединение из примера 12А, представляющее собой тозилатную соль Y, проявляет кристалличность и характеризуется четырьмя кристаллическими формами, формой А, формой В, формой С и формой D, которые были идентифицированы и были получены с применением условий, описанных далее в данном документе в экспериментальном разделе.
Полиморфная форма А тозилатной соли Y
Соответственно, дополнительный аспект настоящего изобретения представляет собой форму А тозилатной соли Y (пример 12А).
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=13,4° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=14,3° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при приблизительно 2-тета=13,4° и 14,3° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD со специфическими пиками при приблизительно 2-тета=11,7, 12,2, 13,4, 14,3, 17,3, 20,2, 21,4, 23,6, 23,7, 24,4° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, фактически идентичной рентгенограмме XRPD, показанной на фигуре Е.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=13,4° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=14,3° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при 2-тета=13,4° и 14,3°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма А тозилатной соли Y, которая характеризуется рентгенограммой XRPD со специфическими пиками при 2-тета=11,7, 12,2, 13,4, 14,3, 17,3, 20,2, 21,4, 23,6, 23,7, 24,4°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
DSC-анализ формы А тозилатной соли Y демонстрирует эндотерму плавления с началом при приблизительно 165,7°С и пиком при приблизительно 170,8°С (фигура F).
Таким образом, DSC-анализ демонстрирует, что форма А тозилатной соли Y представляет собой тугоплавкое твердое вещество с началом плавления при приблизительно 165,7°С и пиком при приблизительно 170,8°С.
Полиморфная форма В тозилатной соли Y
Дополнительный аспект настоящего изобретения представляет собой форму В тозилатной соли Y.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=7,1° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=9,2° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при приблизительно 2-тета=7,1° и 9,2° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD со специфическими пиками при приблизительно 2-тета=7,1, 9,2, 11,8, 14,2, 19,4, 20,4, 20,9, 22,5, 23,9, 25,2° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, фактически идентичной рентгенограмме XRPD, показанной на фигуре G.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=7,1° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=9,2° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при 2-тета=7,1° и 9,2°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма В тозилатной соли Y, которая характеризуется рентгенограммой XRPD со специфическими пиками при 2-тета=7,1, 9,2, 11,8, 14,2, 19,4, 20,4, 20,9, 22,5, 23,9, 25,2°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
DSC-анализ формы В тозилатной соли Y демонстрирует эндотерму плавления с началом при приблизительно 140,0°С и пиком при приблизительно 146,2°С (фигура Н).
Таким образом, DSC-анализ демонстрирует, что форма В тозилатной соли Y представляет собой твердое вещество с началом плавления при приблизительно 140,0°С и пиком при приблизительно 146,2°С.
Полиморфная форма D тозилатной соли Y
Дополнительный аспект настоящего изобретения представляет собой форму D тозилатной соли Y.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=4,4° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при приблизительно 2-тета=5,6° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при приблизительно 2-тета=4,4° и 5,6° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD со специфическими пиками при приблизительно 2-тета=4,4, 5,6, 8,8, 16,8, 19,1, 19,7, 21,9, 22,3, 24,8, 26,9° согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, фактически идентичной рентгенограмме XRPD, показанной на фигуре I.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=4,4° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере один специфический пик при 2-тета=5,6° плюс или минус 0,2° 2-тета согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD, имеющей по меньшей мере два специфических пика при 2-тета=4,4° и 5,6°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
В соответствии с настоящим изобретением предусматривается кристаллическая форма, форма D тозилатной соли Y, которая характеризуется рентгенограммой XRPD со специфическими пиками при 2-тета=4,4, 5,6, 8,8, 16,8, 19,1, 19,7, 21,9, 22,3, 24,8, 26,9°, где указанные значения могут отклоняться на плюс или минус 0,2° 2-тета, согласно измерениям с применением излучения CuKα.
DSC-анализ формы D тозилатной соли Y демонстрирует эндотерму плавления с началом при приблизительно 116,5°С и пиком при приблизительно 122,3°С (фигура J).
Таким образом, DSC-анализ демонстрирует, что форма D тозилатной соли Y представляет собой твердое вещество с началом плавления при приблизительно 116,5°С и пиком при приблизительно 122,3°С.
Когда определено, что настоящее изобретение относится к кристаллической форме, представляющей собой форму А или форму В соединения X или форму А, форму В или форму D тозилатной соли Y, степень кристалличности преимущественно составляет более приблизительно 60%, более преимущественно более приблизительно 80%, предпочтительно более приблизительно 90% и более предпочтительно более приблизительно 95%. Наиболее предпочтительно степень кристалличности превышает приблизительно 98%.
Будет понятно, что значения угла 2-тета на рентгенограмме порошковой рентгеновской дифрактометрии могут слегка варьироваться от одного аппарата к другому или от одного образца к другому, и поэтому приведенные значения не следует рассматривать как абсолютные.
Известно, что может быть получена рентгенограмма порошковой рентгеновской дифрактометрии, которая характеризуется одной или несколькими ошибками измерения в зависимости от условий измерения (таких как применяемые оборудование или аппарат). В частности, общеизвестно, что значения интенсивности на рентгенограмме порошковой рентгеновской дифрактометрии могут колебаться в зависимости от условий измерения. Следовательно, должно быть понятно, что форма А и форма В соединения X и форма А, форма В и форма D тозилатной соли Y по настоящему изобретению не ограничиваются кристаллами, которые дают рентгенограммы порошковой рентгеновской дифрактометрии, идентичные рентгенограмме порошковой рентгеновской дифрактометрии, показанной на фигурах А, С, Е, G и I, и любые кристаллы, дающие рентгенограммы порошковой рентгеновской дифрактометрии, показанные на фигурах А, С, Е, G и I, попадают в объем настоящего изобретения. Специалист в области порошковой рентгеновской дифрактометрии сможет сделать вывод о существенной степени идентичности рентгенограмм порошковой рентгеновской дифрактометрии.
Специалистам в области порошковой рентгеновской дифрактометрии будет понятно, что на относительную интенсивность пиков могут воздействовать, например, зерна с размером более 30 микрон и неунитарными соотношениями сторон, которые могут воздействовать на анализ образцов. Специалисту в данной области техники также будет понятно, что на положение отражений могут воздействовать точная высота, на которой находится образец в дифрактометре, и калибровка нуля дифрактометра. Незначительное влияние также может оказывать плоскостность поверхности образца. Следовательно, представленные данные рентгенограмм дифрактометрии не следует принимать как абсолютные значения. (Jenkins, R & Snyder, R.L. 'Introduction to X-Ray Powder Diffractometry' John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H.P. & Alexander, L.E. (1974), X-Ray Diffraction Procedures).
Как правило, ошибка измерения угла дифракции на рентгеновской порошковой дифрактограмме составляет примерно плюс или минус 0,2° 2-тета, и такая степень ошибки измерения должна быть принята во внимание при рассмотрении рентгенограммы порошковой рентгеновской дифрактометрии на фигурах А, С, Е, G и I и при чтении таблиц А-Е (см. примеры 12 и 12А). Кроме того, следует понимать, что значения интенсивности могут колебаться в зависимости от экспериментальных условий и способа получения образца (предпочтительной ориентации).
Соединения формулы (I) включают один или несколько хиральных центров. В тех случаях, когда структура или химическое название в настоящем описании не указывают на хиральность, предполагается, что структура или название охватывают любой отдельный стереоизомер (т.е. любой отдельный хиральный изомер), соответствующий таким структуре или названию, а также любую смесь стереоизомеров (например, рацемат). Из уровня техники хорошо известно как можно получать такие оптически активные формы. Например, отдельный стереоизомер можно получать за счет выделения его из смесей изомеров (например, рацемата) с применением, например, хирального хроматографического разделения. В других вариантах осуществления отдельный стереоизомер получают путем прямого синтеза, например, из хирального исходного материала.
Соединения формулы (I), где R2 представляет собой фтор, можно получать, например, путем проведения реакции соединения формулы (II) или его соли:
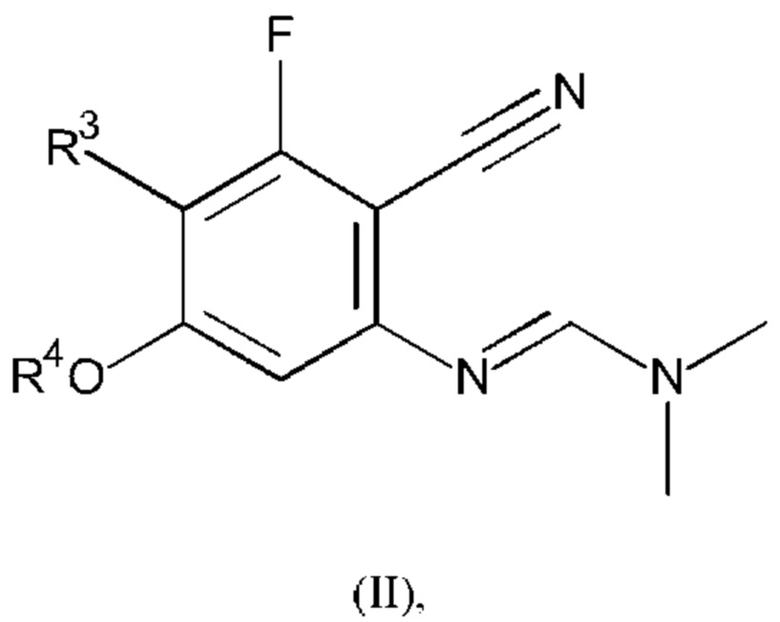
где R3 и R4 являются такими, как определено в любом из вариантов осуществления в данном документе, с соединением формулы (III):
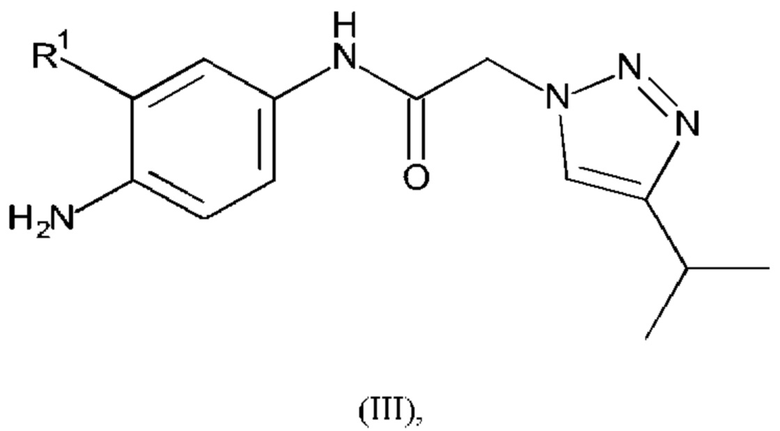
или его солью, где R1 является таким, как определено в любом из вариантов осуществления в данном документе. Преимущественно реакцию проводят в подходящем растворителе (например, уксусной кислоте) при подходящей температуре (например, температуре, составляющей приблизительно 25-100°С).
Следовательно, соединения формулы (II) и (III) и их соли применимы в качестве промежуточных соединений при получении соединений формулы (I), где R2 представляет собой фтор, и обеспечивают дополнительный вариант осуществления.
В случае любого из вариантов осуществления, в котором упоминается соединение формулы (II) или (III) или его соль, следует понимать, что такие соли не обязательно должны быть фармацевтически приемлемыми солями.
Соединение формулы (II) можно получать, например, путем проведения реакции соединения формулы (IV):
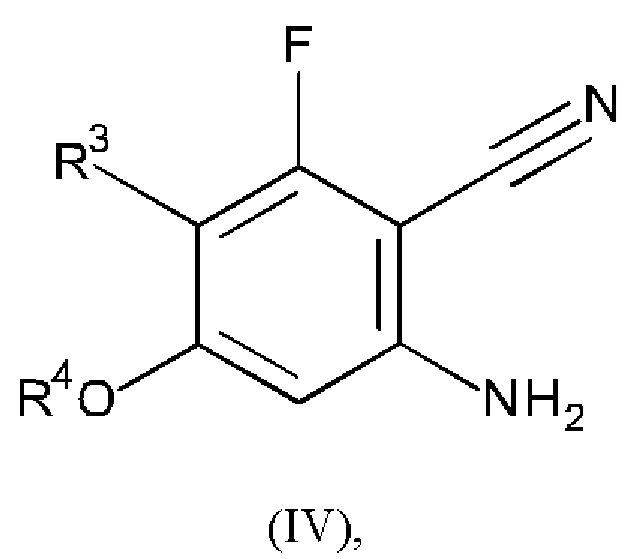
или его соли, где R3 и R4 являются такими, как определено в любом из вариантов осуществления в данном документе, с диалкилацеталем DMF (например, 1,1-диметокси-N,N-диметилметанамином).
Соединение формулы (IV) можно получать, например, путем проведения реакции соединения формулы (V):
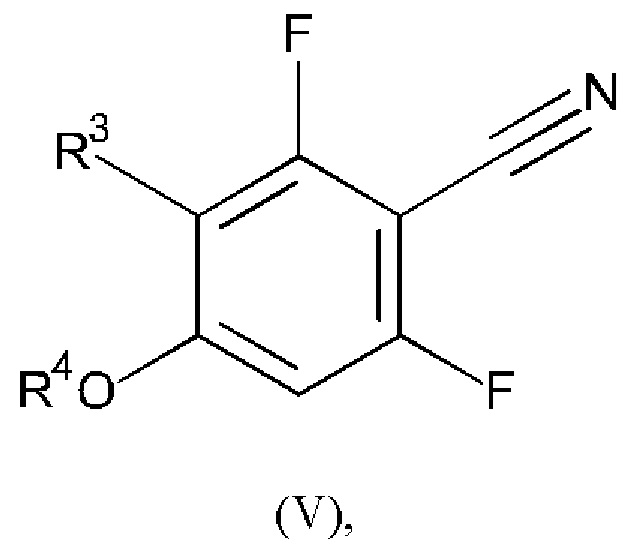
где R3 и R4 являются такими, как определено в любом из вариантов осуществления в данном документе, со слабым основанием (например, водным аммиаком) в условиях высоких температуры и давления (например, при температуре приблизительно 80-100°С и давлении приблизительно 3-15 бар).
Соединение формулы (V) можно получать, например, путем проведения реакции соединения формулы (VI):
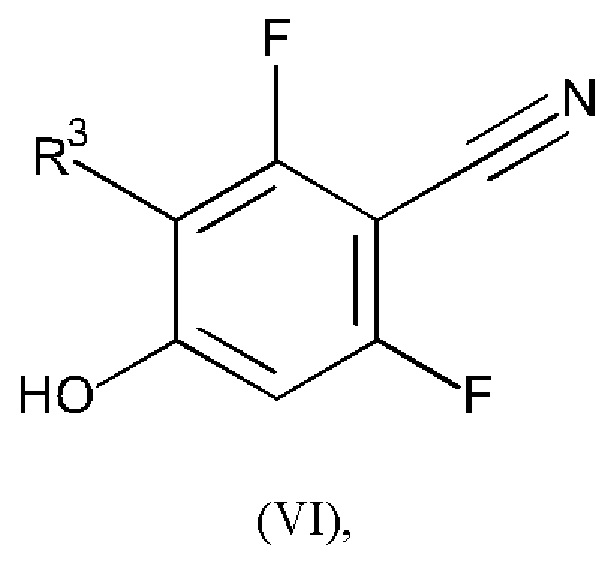
или его соли, где R3 является таким, как определено в любом из вариантов осуществления в данном документе, с R4-Br, где R4 является таким, как определено в любом из вариантов осуществления в данном документе, в присутствии подходящего основания (например, карбоната калия) и растворителя (например, диметилформамида) и при подходящей температуре (например, приблизительно 80°С).
Соединение формулы (III) можно получать, например, путем проведения реакции соединения формулы (VII):
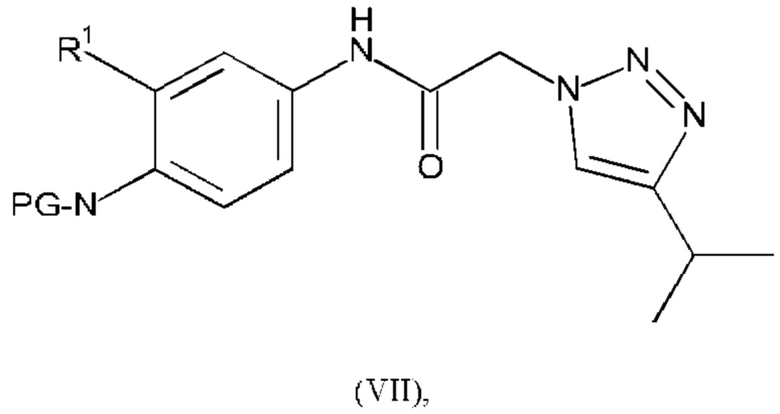
где R1 является таким, как определено в любом из вариантов осуществления в данном документе, a PG представляет собой подходящую защитную группу (например, трет-бутилоксикарбонильную (ВОС)), с подходящим средством для удаления защитной группы (например, HCl в диоксане) и в подходящем растворителе (например, DCM).
Соединение формулы (VII) можно получать, например, путем проведения реакции соединения формулы (VIII):
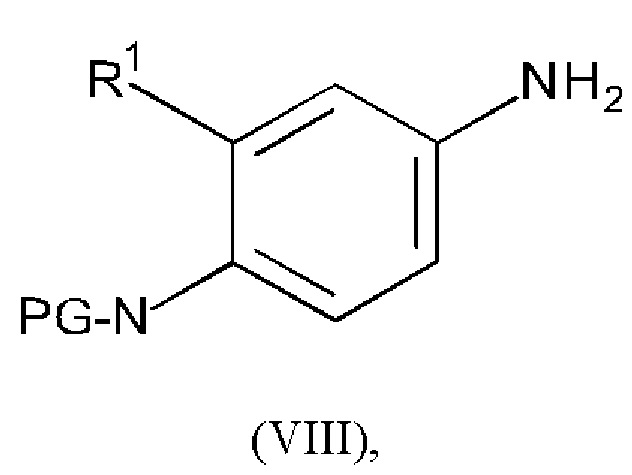
или его соли, с соединением формулы (IX):
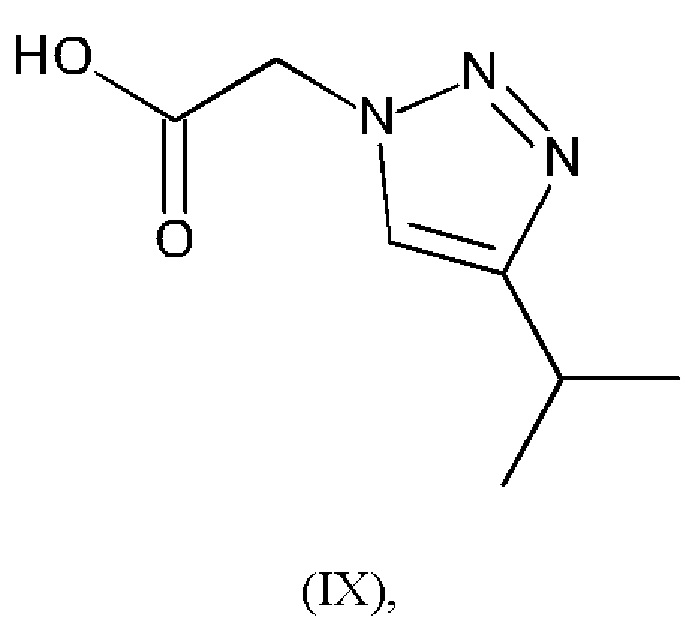
или его солью, где R1 является таким, как определено в любом из вариантов осуществления в данном документе, a PG представляет собой подходящую защитную группу (например, трет-бутилоксикарбонильную (ВОС)), в присутствии подходящего реагента и основания для образования пептидной связи (например, HATU и DIPEA, соответственно) и подходящего растворителя (например, DMF).
Соединения формулы (I), где R2 представляет собой С1-2алкокси, можно получать, например, путем проведения реакции соединения формулы (X):
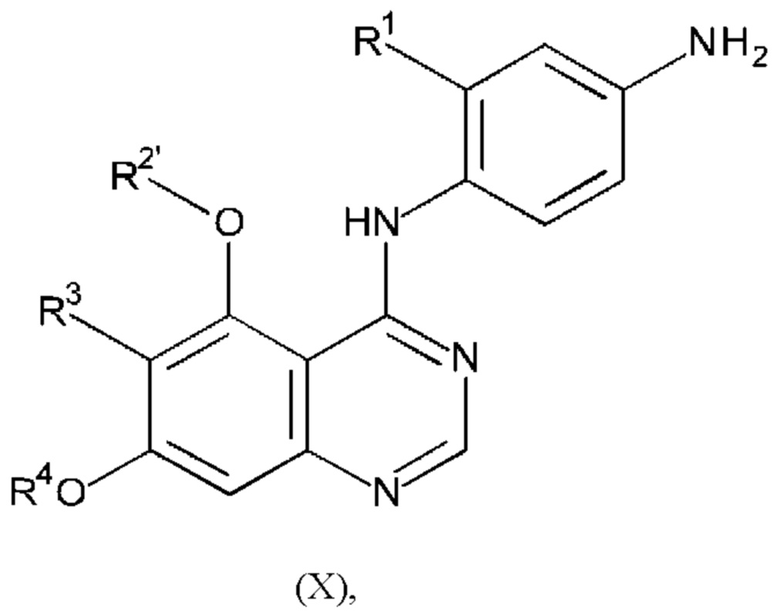
или его соли, где R2' представляет собой С1-2алкил, a R1, R3 и R4 являются такими, как определено в любом из вариантов осуществления в данном документе, с соединением формулы (XI):
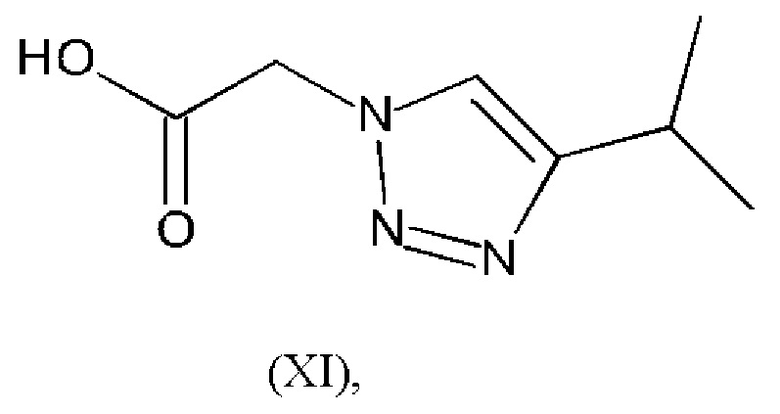
или его солью в присутствии подходящего реагента и основания для образования пептидной связи (например, HATU и DIPEA, соответственно) и подходящего растворителя (например, DMF) и при подходящей температуре (например, комнатной температуре). В одном варианте осуществления R2' представляет собой метил. В одном варианте осуществления R2' представляет собой этил.
Следовательно, соединения формулы (X) и (XI) и их соли применимы в качестве промежуточных соединений при получении соединений формулы (I), где R2 представляет собой С1-2алкокси, и обеспечивают дополнительный вариант осуществления.
В случае любого из вариантов осуществления, где упоминается соединение формулы (X) или (XI) или его соль, следует понимать, что такие соли не обязательно должны быть фармацевтически приемлемыми солями.
Соединение формулы (X) можно получать, например, путем проведения реакции соединения формулы (XII):
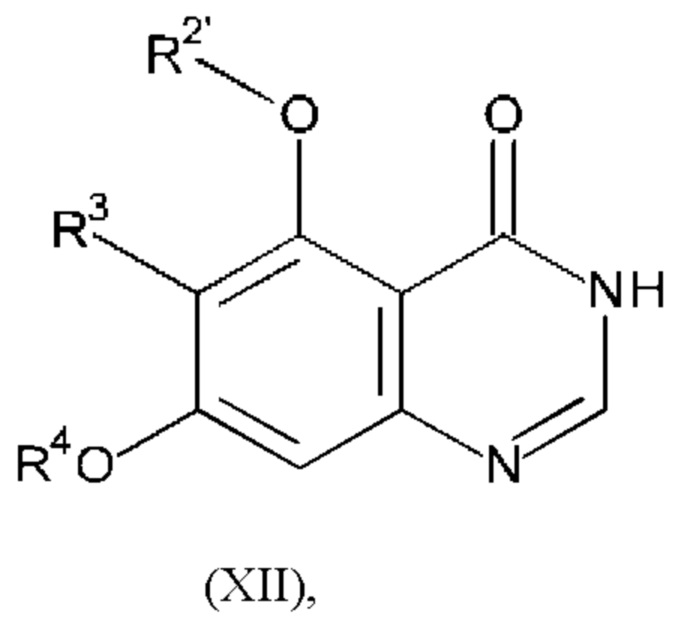
или его соли, где R2', R3 и R4 являются такими, как определено в любом из вариантов осуществления в данном документе, с соединением формулы (XIII):
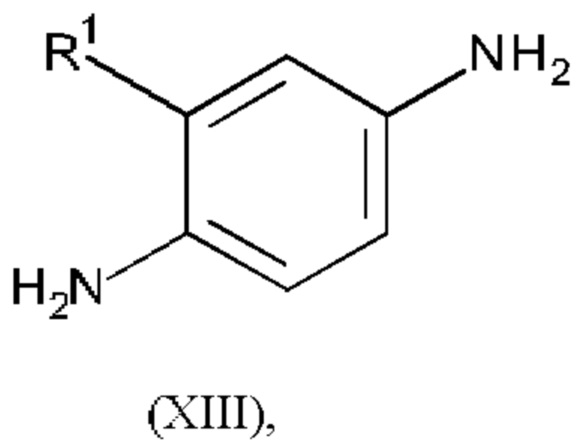
или его солью, где R1 является таким, как определено в любом из вариантов осуществления в данном документе, в присутствии подходящего реагента для образования пептидной связи (например, РуВОР), сильного органического основания (например, DBU) и подходящего растворителя (например, MeCN).
Соединение формулы (XII) можно получать, например, путем проведения реакции соединения формулы (XIV):
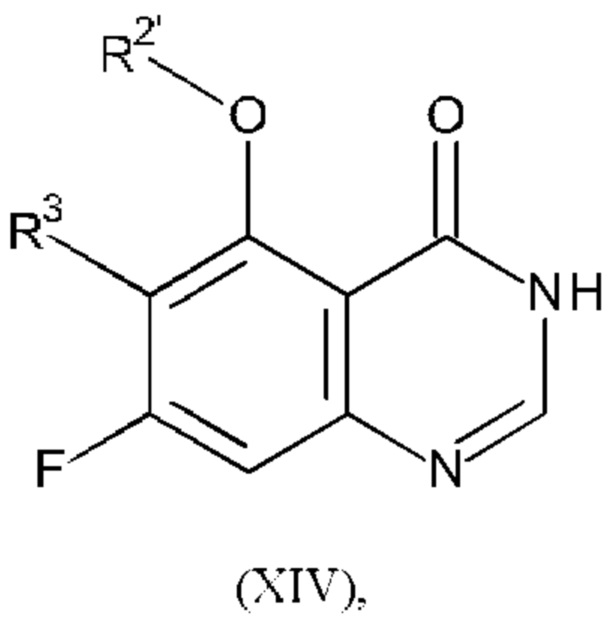
где R2' и R3 являются такими, как определено в любом из вариантов осуществления в данном документе, с KOR4 или другим подходящим алкоксидом щелочного металла при подходящей температуре (например, приблизительно 60-100°С), где R4 является таким, как определено в любом из вариантов осуществления в данном документе.
Соединение формулы (XIV) можно получать, например, путем проведения реакции соединения формулы (XV):
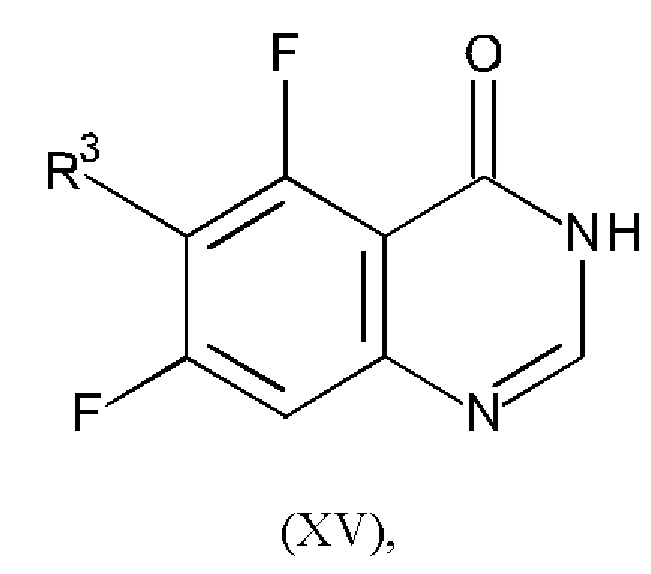
где R3 является таким, как определено в любом из вариантов осуществления в данном документе, с подходящим алкоксидом щелочного металла (например, NaOR2') при подходящей температуре (например, комнатной температуре), где R2' является таким, как определено в любом из вариантов осуществления в данном документе.
Будет понятно, что определенные из различных заместителей кольца в соединениях по настоящему изобретению можно вводить за счет стандартных реакций замещения в ароматическом ядре или получать за счет традиционных модификаций функциональных групп либо до, либо непосредственно после упомянутых выше способов, и, в связи с этим, они включены в относящийся к способу аспект настоящего изобретения. Например, соединения формулы (I) можно превращать в дополнительные соединения формулы (I) за счет стандартных реакций замещения в ароматическом ядре или за счет традиционных модификаций функциональных групп.Такие реакции и модификации включают, например, введение заместителя посредством реакции замещения в ароматическом ядре, восстановление заместителей, алкилирование заместителей и окисление заместителей. Реагенты и условия реакции для таких процедур хорошо известны в области химии.
Также будет понятно, что в некоторых из реакций, упомянутых в данном документе, может быть необходима/требоваться защита каких-либо чувствительных групп в соединениях. Случаи, когда защита является необходимой или требуется и подходящие способы защиты известны специалистам в данной области техники. Традиционные защитные группы можно применять в соответствии со стандартной практикой (для иллюстрации см. T.W. Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). В силу этого, если реагенты содержат группы, такие как амино, карбокси или гидрокси, в некоторых из реакций, упомянутых в данном документе, может требоваться защита данной группы.
Соединения формулы (I), (II) и (III) и любые промежуточные соединения, применяемые для их получения, можно получать посредством способов, аналогичных способам, показанным в разделе Примеры.
Биологические анализы
Для измерения эффектов соединений по настоящему изобретению применяли следующий анализ.
кДНК для 3 различных KIT, кодирующих делецию в экзоне 11 (557-558) и вторичную мутацию (V654A, T670I, D816H), которые были получены от Genescript, встраивали в вектор pLVX-IRES Puro (Clontech). Лентивирусные частицы получали с применением набора для упаковки Trans-lentiviral ORF (TLP 5918) от Thermo Scientific (Уолтем, Массачусетс) в клетках HEK293-Т/17 согласно инструкциям производителя.
Tel-KDR myc клонировали в pBCS2004, ретровирусный вектор, в котором KDR (K790-V1356) слит с С-концом Tel. Ретровирусные частицы получали в клетках HEK293T. Плазмиду Tel-KDR трансфицировали совместно с вирусами-помощниками (gag-Pol и VSV-G) в клетки HEK293T с применением фосфата кальция и вирус собирали через 72 ч после трансфекции.
Клетки Ba/F3 в экспоненциальной фазе роста (1,5×106 клеток в 2 мл среды) инфицировали с помощью 2 мл вирусной суспензии в 6-луночном планшете в присутствии mIL-3 (10 нг/мл) и полибрена (4 мкг/мл) (Sigma Aldrich, Сент-Луис, Миссури) и инкубировали в течение 24 ч. Через 24 ч клетки центрифугировали и вирусный супернатант удаляли. Затем клетки ресуспендировали в свежей среде и им обеспечивали возможность восстановления в течение еще одного дня. На следующий день клетки высевали в полную среду без IL-3 мыши. Через одну или две недели, когда клетки начинали разрастаться, проводили отбор путем постепенного повышения концентрации пуромицина до 0,5 мкг/мл. Как только клетки достигали экспоненциальной фазы роста в присутствии пуромицина, партии клеток замораживали для создания банка.
Влияние ингибиторов KIT на жизнеспособность Ba/F3, экспрессирующих KIT с мутациями, определяли с применением анализа MTS, который представляет собой колориметрическое чувствительное количественное определение жизнеспособных клеток в анализе пролиферации и цитотоксичности. В анализе MTS применяли 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид (МТТ) в присутствии феназин-метосульфата (PMS). Митохондриальная редуктаза образует формазан, который характеризуется поглощением при 490 нм. Клетки в экспоненциальной фазе роста добавляли в 384-луночные планшеты, содержащий предварительно отмеренные соединения (самая высокая концентрация 10 мкМ, кривая по 10 точкам). Клетки инкубировали в течение 72 ч при 37°С и 5% CO2. Через 72 ч в планшеты добавляли реагент MTS и инкубировали в течение дополнительных 2 ч при 37°С перед проведением измерения поглощения при 490 нм на микропланшетном ридере Tecan с применением программного обеспечения Magellan (Tecan Trading AG, Швейцария).
Значения поглощения нормализовали следующим образом: (считанное значение - значение контроля для дня 0)/(значение контроля для дня 3 - значение контроля для дня 0)*100. Значения GI50 получали с применением программного обеспечения Genedata Screener (Genedata; Лексингтон, Массачусетс). Нелинейную регрессию с ограничениями для верхнего и нижнего пределов в диапазоне от 100 до -100 и без ограничений для коэффициента Хилла применяли для получения значений GI50. Значения GI50, приведенные ниже, представляют собой рассчитанный средних результат по меньшей мере 3 биологических повторностей для всех протестированных клеточных линий.
Следующие данные были получены для примеров (данные ниже могут представлять собой результат одного эксперимента или среднее значение нескольких повторяемых экспериментов).
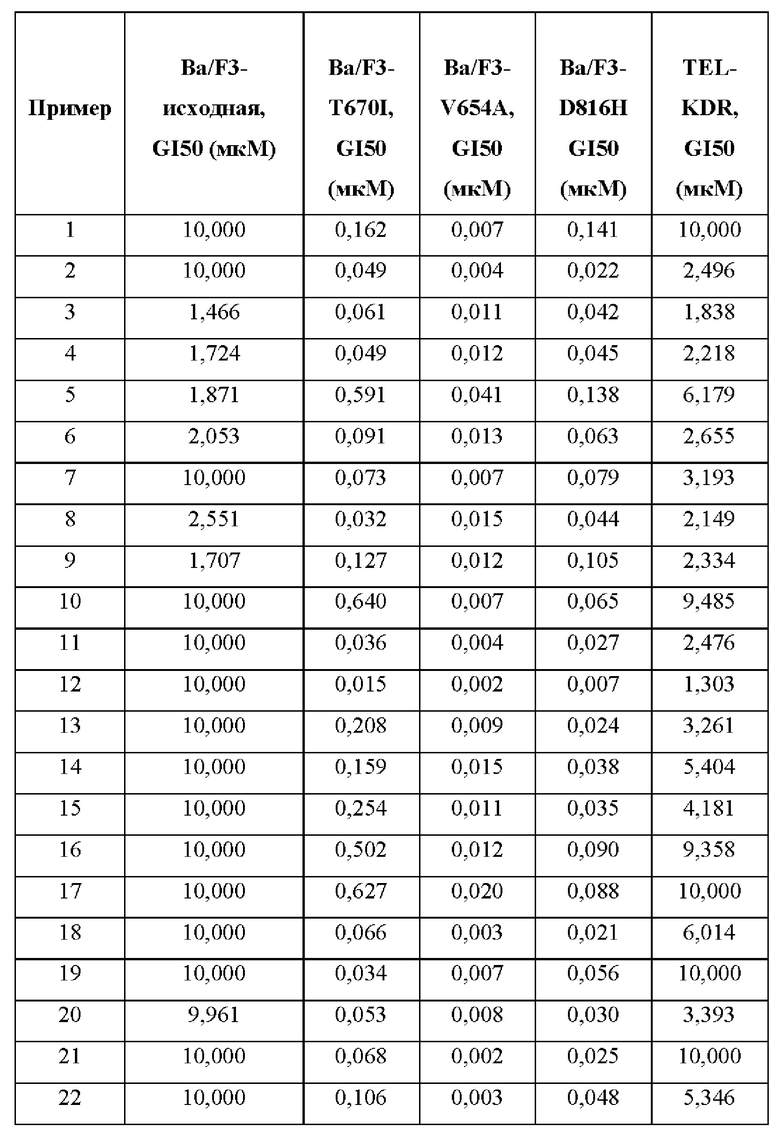
Данные показывают, что соединения по настоящему изобретению ингибируют KIT, одновременно несущую как первичные, так и вторичные мутации KIT, и, кроме того, являются селективными в отношении KDR. В некоторых вариантах осуществления фармацевтически приемлемые соли соединения формулы (I) превращаются в форму свободного основания in vivo. Например, тозилатная соль из примера 12 (также обозначаемая в данном документе как пример 12А и тозилатная соль Y) превращается в свободное основание in vivo, и именно свободное основание, а не тозилатная соль, проходит через клеточную мембрану. Следовательно, введение тозилатной соли будет приводить к активности свободного основания, проиллюстрированной выше для примера 12.
Соединения можно дополнительно отбирать на основе дополнительных биологических или физических свойств, которые можно измерять с помощью методик, известных из уровня техники и которые можно применять при оценке или отборе соединений для терапевтического или профилактического применения.
Ожидается, что ввиду их ингибирующей KIT активности соединения формулы (I) и их фармацевтически приемлемые соли будут применимы в терапии.
Авторы настоящего изобретения обнаружили, что соединения формулы (I) обладают сильной противоопухолевой активностью, которая, как полагают, достигается путем ингибирования как KIT дикого типа, так и мутантных KIT. Авторы настоящего изобретения также обнаружили, что соединения формулы (I), в частности, могут также выступать в качестве иммуноонкологического лекарственного средства.
Подразумевается, что термин "терапия" имеет свое обычное значение, обозначающее борьбу с заболеванием, чтобы полностью или частично ослабить один, несколько или все его симптомов или исправить или компенсировать лежащую в его основе патологию. Термин "терапия" также включает "профилактику", если нет конкретных указаний об обратном. Термины "терапевтический" и "терапевтически" должны интерпретироваться соответствующим образом.
Подразумевается, что термин "профилактика" имеет свое обычное значение и включает первичную профилактику для предупреждения развития заболевания и вторичную профилактику, при которой заболевание уже развилось, и пациента временно или постоянно защищают от обострения или усугубления заболевания или развития новых симптомов, ассоциированных с заболеванием.
Термин "лечение" применяют как синоним "терапии". Аналогичным образом термин "лечить" можно рассматривать как "применять терапию", где "терапия" определена в данном документе.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
В одном варианте осуществления предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного препарата.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении заболевания, опосредованного KIT. В одном варианте осуществления заболевание, опосредованное KIT, представляет собой рак. В одном варианте осуществления рак выбран из группы, состоящей из стромальной опухоли желудочно-кишечного тракта (GIST), меланомы, форм рака легкого, глиобластомы, лейкозов, форм карциномы яичка и форм рака головы и шеи. Формы рака легкого включают мелкоклеточный рак легкого (SCLC), формы аденокарциномы и формы плоскоклеточной карциномы легкого. Лейкозы включают острый миелоидный лейкоз (AML) и тучноклеточные лейкозы.
В одном варианте осуществления рак представляет собой стромальную опухоль желудочно-кишечного тракта. GIST представляет собой тип опухоли, которая возникает в желудочно-кишечном тракте, наиболее часто в желудке или тонком кишечнике.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака.
В одном варианте осуществления предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли для изготовлении лекарственного препарата для лечения заболевания, опосредованного KIT. В одном варианте осуществления заболевание, опосредованное KIT, представляет собой рак. В одном варианте осуществления рак выбран из группы, состоящей из стромальных опухолей желудочно-кишечного тракта (GIST), меланомы, форм рака легкого, глиобластомы, лейкозов, форм карциномы яичка и форм рака головы и шеи. Формы рака легкого включают мелкоклеточный рак легкого (SCLC), формы аденокарциномы и формы плоскоклеточной карциномы легкого. Лейкозы включают острый миелоидный лейкоз (AML) и тучноклеточные лейкозы.
В одном варианте осуществления предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного препарата для лечения рака.
В одном варианте осуществления предусмотрен способ лечения заболевания, при котором благоприятным является ингибирование KIT, у теплокровного животного, нуждающегося в таком лечении, который предусматривает введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном варианте осуществления заболевание представляет собой рак. В одном варианте осуществления рак выбран из группы, состоящей из стромальных опухолей желудочно-кишечного тракта (GIST), меланом, форм рака легкого, глиобластомы, лейкозов, форм карциномы яичка и форм рака головы и шеи. Формы рака легкого включают мелкоклеточный рак легкого (SCLC), формы аденокарциномы и формы плоскоклеточной карциномы легкого. Лейкозы включают острый миелоидный лейкоз (AML) и тучноклеточные лейкозы.
В одном варианте осуществления рак представляет собой стромальную опухоль желудочно-кишечного тракта.
Термин "терапевтически эффективное количество" относится к количеству соединения формулы (I), описываемого в любом из вариантов осуществления в данном документе, которое является эффективным для обеспечения "терапии" у субъекта или для "лечения" заболевания или нарушения у субъекта. В случае рака терапевтически эффективное количество может вызывать любое из изменений, которые можно наблюдать или
измерять у субъекта, как описано выше в определении "терапии", "лечения" и "профилактики". Например, эффективное количество может снижать число раковых или опухолевых клеток; снижать общий размер опухоли; подавлять или останавливать инфильтрацию опухолевых клеток в периферические органы, включая, например, мягкую ткань и кость; подавлять и останавливать метастазирование опухолей;
подавлять и останавливать рост опухолей; в некоторой степени ослаблять один или несколько симптомов, ассоциированных с раком; снижать тяжесть заболевания и смертность; улучшать качество жизни или обеспечивать комбинацию таких эффектов. Эффективное количество может представлять собой количество, достаточное для снижения интенсивности симптомов заболевания, чувствительного к ингибированию активности KIT. В случае терапии рака in-vivo эффективность можно измерять, например, с помощью оценки продолжительности выживания, периода времени до прогрессирования заболевания (ТТР), частоты случаев ответа (RR), продолжительности ответа и/или качества жизни. Как признается специалистами в данной области техники, эффективные количества могут варьироваться в зависимости от пути введения, применения вспомогательного вещества и совместного применения с другими средствами. Например, если применяют комбинированную терапию, количество соединения формулы (I) или его фармацевтически приемлемой соли, описываемых в настоящем описании, и количество другого(-их) фармацевтически активного(-ых) средства(средств), в случае их комбинирования, в совокупности является эффективным для лечения целевого нарушения у пациента-животного. В данном контексте объединенные количества представляют собой "терапевтически эффективное количество", если, в случае их комбинирования, они являются достаточными для уменьшения интенсивности симптомов заболевания, чувствительного к ингибированию активности KIT, как описано выше. Как правило, такие количества может определять специалист в данной области техники, например, начиная с диапазона доз, описанного в настоящем описании для соединения формулы (I) или его фармацевтически приемлемой соли, и утвержденного(-ых) или иным образом опубликованного(-ых) диапазона(-ов) доз другого(-их) фармацевтически активного(-ых) соединения(-й).
"Теплокровные животные" включают, например, людей.
В одном варианте осуществления предусмотрен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, который предусматривает введение указанному теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном варианте осуществления указанный рак выбран из группы, состоящей из стромальных опухолей желудочно-кишечного тракта (GIST), меланомы, форм рака легкого, глиобластомы, лейкозов, форм карциномы яичка и форм рака головы и шеи. Формы рака легкого включают мелкоклеточный рак легкого (SCLC), формы аденокарциномы и формы плоскоклеточной карциномы легкого. Лейкозы включают острый миелоидный лейкоз (AML) и тучноклеточные лейкозы.
В одном варианте осуществления рак представляет собой стромальную опухоль желудочно-кишечного тракта.
Противораковое лечение, описанное в настоящем описании, может быть пригодным в виде монотерапии или в дополнение к введению соединения формулы (I) или его фармацевтически приемлемой соли может предусматривать традиционные хирургическое вмешательство, лучевую терапию или химиотерапию или комбинацию таких дополнительных видов терапии. Такое традиционное хирургическое вмешательство, лучевая терапия или химиотерапия могут применяться одновременно, последовательно или раздельно с лечением с помощью соединения формулы (I) или его фармацевтически приемлемой соли.
Если средства комбинированной терапии вводятся "одновременно", это предусматривает лечение пациента с применением единичной лекарственной формы (например, таблетки), содержащей как соединение формулы (I) или его фармацевтически приемлемую соль, так и дополнительное противораковое вещество; а также одновременное введение дозы отдельных лекарственных форм, каждая из которых по отдельности содержит один из соответствующих партнеров по комбинации.
Если средства комбинированной терапии вводятся "последовательно" или "раздельно", это предусматривает лечение пациента с помощью первой лекарственной формы (например, таблетки), содержащей соединение формулы (I) или его фармацевтически приемлемую соль, с последующим лечением того же пациента с помощью второй лекарственной формы, содержащей дополнительное противораковое вещество; или лечение пациента с помощью единичной лекарственной формы (например, таблетки), содержащей конкретное противораковое вещество, с последующим лечением того же пациента с помощью второй лекарственной формы, содержащей соединение формулы (I) или его фармацевтически приемлемую соль. Интервал между последовательными или раздельными дозами может быть определен практикующим специалистом с учетом информации, изложенной в настоящем описании.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят до хирургического вмешательства.
Введение соединения формулы (I) или его фармацевтически приемлемой соли до хирургического вмешательства для полного или частичного устранения рака может обозначаться как "неоадъювантная терапия". В такой ситуации целью введения соединения формулы (I) или его фармацевтически приемлемой соли обычно является уменьшение размера целевой опухоли с тем, чтобы повысить шансы успешной резекции. Соответственно, продолжительность введения дозы соединения формулы (I) или его фармацевтически приемлемой соли до хирургического вмешательства может определять практикующий специалистом с учетом информации, изложенной в настоящем описании.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят после хирургического вмешательства.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с по меньшей мере одним дополнительным противораковым веществом.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, последовательно или раздельно с по меньшей мере одним дополнительным противораковым веществом.
Противораковое лечение, определенное в данном документе, может применяться в качестве монотерапии или в дополнение к применению соединений по настоящему описанию может включать традиционное хирургическое вмешательство, или лучевую терапию, или химиотерапию. Такая химиотерапия может включать одно или несколько из следующих категорий противоопухолевых средств:
(i) ингибиторы функции факторов роста и их нисходящих путей передачи сигнала: предусматриваются Ab-модуляторы любых мишеней, представляющих собой факторы роста или рецепторы факторов роста, рассмотренные в обзоре Stern et al. Critical Reviews in Oncology/Haematology, 2005, 54, pp 11-29); также предусматриваются низкомолекулярные ингибиторы таких мишеней, например ингибиторы киназ - примеры включают трастузумаб, представляющий собой антитело к erbB2 [Herceptin™], панитумумаб, представляющий собой антитело к EGFR, цетуксимаб, представляющий собой антитело к EGFR [Erbitux, С225], и ингибиторы тирозинкиназ, включая ингибиторы рецепторов семейства erbB, такие как ингибиторы тирозинкиназ семейства рецепторов эпидермального фактора роста (EGFR/erbB1), такие как гефитиниб или эрлотиниб, ингибиторы тирозинкиназы erbB2, такие как лапатиниб, и смешанные ингибиторы erb1/2, такие как афатаниб; аналогичные стратегии доступны для других классов факторов роста и их рецепторов, например, ингибиторы семейства факторов роста гепатоцитов или их рецепторов, включая c-met и ron; ингибиторы инсулина и семейства инсулиновых факторов роста или их рецепторов (IGFR, IR), ингибиторы семейства факторов роста тромбоцитов или их рецепторов (PDGFR) и ингибиторы передачи сигнала, опосредованной другими рецепторными тирозинкиназами, такими как c-kit, AnLK и CSF-1R; также предусматриваются модуляторы, которых нацеливаются на сигнальные белки в пути передачи сигнала PI3-киназы, например, ингибиторы изоформ PI3-киназы, таких как PI3K-α/β/γ, и ser/thr-киназ, таких как AKT, mTOR (такой как AZD2014), PDK, SGK, PI4K или PIP5K; также предусматриваются ингибиторы не перечисленных выше серин-треониновых киназ, например, ингибиторы raf, такие как вемурафениб, ингибиторы MEK, такие как селуметиниб (AZD6244), ингибиторы Abl такие как иматиниб или нилотиниб, ингибиторы Btk, такие как ибрутиниб, ингибиторы Syk, такие как фостаматиниб, ингибиторы аврора-киназы (например, AZD1152), ингибиторы других ser/thr-киназ, таких как разновидности JAK, разновидности STAT и IRAK4, и ингибиторы циклинзависимых киназ, например ингибиторы CDK1, CDK7, CDK9 и CDK4/6, такие как палбоциклиб;
(ii) модуляторы путей апоптоза и клеточной гибели, такие как модуляторы семейства Bcl (например, АВТ-263/навитоклаке, АВТ-199);
(iii) иммунотерапевтические подходы, в том числе, например, ex vivo и in vivo подходы к повышению иммуногенности опухолевых клеток пациента, такие как трансфекция с применением цитокинов, таких как интерлейкин-2, интерлейкин-4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, подходы для уменьшения толерантности Т-клеток, подходы с применением трансфицированных иммунных клеток, таких как трансфицированные цитокинами дендритные клетки, подходы с применением трансфицированных цитокинами линий опухолевых клеток и подходы с применением антиидиотипических антител. Конкретные примеры включают моноклональные антитела, нацеливающиеся на PD-1 (например, BMS-936558) или CTLA4 (например, ипилимумаб и тремелимумаб).
Следовательно, в одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное противоопухолевое вещество для применения в лечении рака. В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с дополнительным противоопухолевым веществом. В одном варианте осуществления применяют одно дополнительное противоопухолевое вещество. В одном варианте осуществления применяют два дополнительных противоопухолевых вещества. В одном варианте осуществления применяют три или более дополнительных противоопухолевых вещества.
В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное противоопухолевое вещество для применения в одновременном, раздельном или последовательном лечении рака. В одном варианте осуществления предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении рака, где соединение формулы (I) или его фармацевтически приемлемую соль вводят одновременно, раздельно или последовательно с дополнительным противоопухолевым веществом.
В одном варианте осуществления предусмотрен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, который предусматривает введение указанному теплокровному животному соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного дополнительного противоопухолевого вещества, где количества соединения формулы (I) или его фармацевтически приемлемой соли и дополнительного противоопухолевого вещества в совокупности являются эффективными для осуществления противоракового эффекта.
В одном варианте осуществления предусмотрен способ лечения рака у теплокровного животного, нуждающегося в таком лечении, который предусматривает введение указанному теплокровному животному соединения формулы (I) или его фармацевтически приемлемой соли и одновременное, раздельное или последовательное введение по меньшей мере одного дополнительного противоопухолевого вещества указанному теплокровному животному, где количества соединения формулы (I) или его фармацевтически приемлемой соли и дополнительного противоопухолевого вещества в совокупности являются эффективными для осуществления противоракового эффекта.
В любом варианте осуществления дополнительное противоопухолевое вещество выбрано из группы, состоящей из одного или нескольких противоопухолевых веществ, перечисленных в пунктах (i)-(iii) выше.
В одном варианте осуществления предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) и по меньшей мере одно дополнительное противоопухолевое вещество, для применения в лечении рака. В одном варианте осуществления фармацевтическая композиция также содержит по меньшей мере один фармацевтически приемлемый разбавитель или носитель. В одном варианте осуществления противоопухолевое вещество представляет собой антинеопластическое средство.
В соответствии с дополнительным вариантом осуществления предусмотрен набор, содержащий:
a) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
b) еще одно дополнительное противоопухолевое вещество в дополнительной стандартной лекарственной форме;
c) средства-контейнеры для размещения указанных первой и дополнительной стандартных лекарственных форм и необязательно
d) инструкции по применению.
Соединения формулы (I) и их фармацевтически приемлемые соли можно вводить в виде фармацевтических композиций, содержащих один или несколько фармацевтически приемлемых разбавителей или носителей.
Следовательно, в одном варианте осуществления предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый разбавитель или носитель. Композиции могут находиться в форме, подходящей для перорального применения (например, в виде таблеток, пастилок, твердых или мягких желатиновых капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или настоек), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкодисперсного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде тонкодисперсного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного или внутримышечного введения дозы) или в виде суппозитория для ректального введения дозы. Композиции можно получать с помощью традиционных процедур с применением традиционных фармацевтических вспомогательных веществ, хорошо известных из уровня техники. Таким образом, композиции, предназначенные для перорального применения, могут содержать, например, один или несколько красителей, подсластителей, ароматизаторов и/или консервантов.
В одном варианте осуществления предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый разбавитель или носитель, для применения в терапии.
В одном варианте осуществления предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый разбавитель или носитель, для применения в лечении рака. В одном варианте осуществления указанный рак выбран из группы, состоящей из стромальных опухолей желудочно-кишечного тракта (GIST), меланомы, форм рака легкого, глиобластомы, лейкозов, форм карциномы яичка и форм рака головы и шеи. Формы рака легкого включают мелкоклеточный рак легкого (SCLC), формы аденокарциномы и формы плоскоклеточной карциномы легкого. Лейкозы включают острый миелоидный лейкоз (AML) и тучноклеточные лейкозы.
Обычно соединение формулы (I) будут вводить теплокровному животному в однократной дозе в диапазоне 2,5-5000 мг/м2 площади поверхности тела животного или примерно 0,05-100 мг/кг, и это обычно обеспечивает терапевтически эффективную дозу. Стандартная лекарственная форма, такая как таблетка или капсула, будет обычно содержать, например, 0,1-500 мг активного ингредиента. Суточная доза обязательно будет варьироваться в зависимости от получающего лечение субъекта, конкретного пути введения, каких-либо вводимых совместно терапевтических средств и тяжести заболевания, подлежащего лечению. Следовательно, практикующий врач, который лечит какого-либо конкретного пациента, может определить оптимальную дозу.
ПРИМЕРЫ
Аспекты настоящего изобретения могут быть дополнительно определены посредством ссылки на следующие неограничивающие примеры, в которых подробно описано получение определенных соединений и промежуточных соединений по настоящему изобретению и способы применения соединений по настоящему изобретению. Специалистам в данной области техники будет очевидно, что на практике могут быть осуществлены множество модификаций как материалов, так и способов без отступления от объема настоящего изобретения.
Если не определено иное:
(i) все синтезы проводили при температуре окружающей среды, т.е. в диапазоне 17-25°С, и в атмосфере инертного газа, такого как азот, если не определено иное;
(ii) выпаривание проводили путем ротационного выпаривания или с использованием оборудования Genevac или испарителя Biotage v10 in vacuo, а процедуры выделения продукта проводили после удаления остаточных твердых веществ путем фильтрации;
(iii) колоночную флэш-хроматографию осуществляли на диоксиде кремния Merck Kieselgel (арт. 9385), или на диоксиде кремния для обращенно-фазовой хроматографии (силикагель Fluka 90 С18), или на картриджах Silicycle (диоксид кремния размером 40-63 мкм, масса 4-330 г), или на картриджах Grace Resolv (4-120 г), или на колонках для флэш-хроматографии RediSep Rf 1.5, или на колонках для высокоэффективной флэш-хроматографии RediSep Rf Gold (масса 150-415 г), или на колонках для обращенно-фазовой хроматографии RediSep Rf Gold С18 (диоксид кремния 20-40 мкм), либо вручную, либо автоматически с применением системы Isco CombiFlash Companion или аналогичной системы;
(iv) препаративную обращенно-фазовую HPLC осуществляли на устройстве Waters (600/2700 или 2525), оснащенном масс-спектрометрами ZMD или ZQ ESCi и колонкой для обращенно-фазовой хроматографии Waters X-Terra, или Waters X-Bridge, или Waters SunFire (С-18, диоксид кремния размером 5 микрон, диаметр 19 мм или 50 мм, длина 100 мм, расход 40 мл/минута), с применением смесей с уменьшающейся полярностью из воды (содержащей 1% аммиака) и ацетонитрила или смесей с уменьшающейся полярностью из воды (содержащей 0,1% муравьиной кислоты) и ацетонитрила, в качестве элюентов;
(v) способы хиральной HPLC проводили с применением Gilson GX-281 HPLC и Daicel CHIRALPAK IC (2×25 см, 5 мкм) или Daicel CHIRALPAK IF (2×25 см, 5 мкм); обычно расход составлял 10-350 мл/минута, а обнаружение осуществляли с помощью УФ-поглощения при типичной длине волны 254 нм. Применяли образец с концентрацией приблизительно 1-100 мг/мл в подходящей смеси растворителей с объемом вводимой пробы 0,5-10 мл, и временем прогона 10-150 минут, и типичной температурой в печи, составляющей 25-35°С; способы аналитической хиральной HPLC проводили с применением Shimadzu UFLC и Daicel CHIRALPAK IC-3 (50×4,6 мм, 3 мкм) или Daicel CHIRALPAK IF-3 (50×4,6 мм, 3 мкм); обычно расход составлял 1 мл/минута и обнаружение осуществляли с помощью УФ-поглощения при типичной длине волны 254 нм. Применяли образец с концентрацией приблизительно 1 мг/мл в подходящем растворителе, таком как EtOH, с объемом вводимой пробы приблизительно 10 мкл, и временем прогона 10-60 минут, и типичной температурой в печи 25-35°С;
(vi) значения выхода, если они присутствуют, не обязательно представляют собой максимально достижимые;
(vii) обычно структуры конечных продуктов соединений формулы (I) подтверждали с помощью спектроскопии ядерного магнитного резонанса (ЯМР); значения химического сдвига при ЯМР измеряли по дельта-шкале [спектры протонного магнитного резонанса определяли с применением устройства Bruker Avance 500 (500 МГц), Bruker Avance 400 (400 МГц), Bruker Avance 300 (300 МГц) или Bruker DRX (300 МГц)]; измерения проводили при температуре окружающей среды, если не указано иное; были использованы следующие сокращения: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; dd, дублет дублетов; ddd, дублет дублетов дублетов; dt, дублет триплетов; bs, широкий сигнал;
(viii) обычно конечные продукты формулы (I) также характеризовали с помощью масс-спектроскопии после жидкостной хроматографии (LCMS или UPLC); обычно применяли диоксид кремния С18 для обращено-фазовой хроматографии при расходе 1 мл/минуту и обнаружение осуществляли с помощью масс-спектрометрии с электрораспылением и с помощью записи УФ-поглощения при длине волны в диапазоне 220-320 нм. Аналитическую UPLC осуществляли на диоксиде кремния CSH С18 для обращено-фазовой хроматографии с применением колонки Waters XSelect CSH C18 с размерами 2,1×50 мм и размером частиц 1,7 микрона. Использовали градиентный анализ с применением в качестве элюента смесей с уменьшающейся полярностью, например смесей с уменьшающейся полярностью из воды (содержащей 0,1% муравьиной кислоты или 0,1% аммиака) в качестве растворителя А и ацетонитрила в качестве растворителя В. При типичном способе аналитической UPLC продолжительностью 2 минуты будет использоваться градиент растворителя в течение 1,3 минуты, при расходе примерно 1 мл в минуту, от смеси 97:3 растворителей А и В до смеси 3:97 растворителей А и В, соответственно. Описываемый молекулярный ион соответствует [М+Н]+, если не указано иное; для молекул с множественными изотопами (Br, Cl и т.д.) приведенное значение является значением, полученным для изотопа с наименьшей массой, если не указано иное;
(ix) ионообменную очистку обычно осуществляли с применением картриджа SCX-2 (Biotage);
(x) если в описании реакции ссылаются на применение микроволновой обработки, применяли один из следующих реакторов для микроволновой обработки: Biotage Initiator, Personal Chemistry Emrys Optimizer, Personal Chemistry Smithcreator или СЕМ Explorer.
(xi) чистоту промежуточного соединения оценивали с помощью тонкослойной хроматографии-масс-спектроскопии, LCMS, UPLC/MS, HPLC и/или ЯМР-анализа;
(xii) применяли следующие сокращения:
ВЕН - гибрид с этиленовыми мостиками
ВОР - (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат
DBU - 1,8-диазабицикло(5.4.0)ундец-7-ен
DCM - дихлорметан
DEA - диэтиламин
DIPEA - диизопропилэтиламин
DMF - N,N-диметилформамид
DMF-DMA - N,N-диметилформамида диметилацеталь
DMSO - диметилсульфоксид
э.и. - энантиомерный избыток
HATU - (1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксидгексафторфосфат)
HCl - хлористоводородная кислота
HPLC - высокоэффективная жидкостная хроматография
MS - масс-спектрометрия
NMR - ядерный магнитный резонанс
PAT - технология анализа процессов
РуАОР - (гексафторфосфат(7-азабензотриазол-1-илокси)трипирролидинофосфония)
РуВОР - (гексафторфосфат бензотриазол-1-ил-окситрипирролидинофосфония)
ТВМЕ/МТВЕ - трет-бутилметиловый эфир
TEA - триэтиламин
TFA - трифторуксусная кислота
THF - тетрагидрофуран
tR - время удерживания
PTSA - пара-толуолсульфоновая кислота
UPLC - ультравысокоэффективная жидкостная хроматография
(xiii) XRPD: аналитическое устройство: Bruker D4
Рентгенограмму порошковой рентгеновской дифрактометрии получали путем помещения образца кристаллического материала на пластинчатый держатель Bruker (Bruker D4) из монокристалла кремния (SSC) и распределения образца в виде тонкого слоя с помощью предметного стекла. Образец вращали со скоростью 30 оборотов в минуту (для улучшения статистики подсчета) и облучали рентгеновским излучением, генерируемым медной длинной острофокусной трубкой, эксплуатируемой при 40 кВ и 40 мА и длине волны 1,5418 ангстрема. Коллимированное рентгеновское излучение пропускали через автоматическую переменную щель расходимости, установленную на V20, а отраженное излучение направляли через антирассеивающую щель шириной 5,89 мм и щель детектора шириной 9,55 мм. Образцы измеряли в отношении геометрии отражения в конфигурации θ-2θ в диапазоне сканирования от 2° до 40° 2θ с номинальной выдержкой 0,12 секунды на шаг 0,02°. Устройство было оборудовано детектором, определяющим изменение положения (Lynxeye). Специалистам в области порошковой рентгеновской дифрактометрии будет понятно, что на относительную интенсивность пиков могут воздействовать, например, зерна с размером более 30 микрон и неунитарными соотношениями сторон, которые могут воздействовать на анализ образцов. Специалисту в данной области техники также будет понятно, что на положение отражений могут воздействовать точная высота, на которой находится образец в дифрактометре, и калибровка нуля дифрактометра. Незначительное влияние также может оказывать плоскостность поверхности образца. Следовательно, представленные данные рентгенограмм дифрактометрии не следует принимать как абсолютные значения;
(xiv) дифференциальная сканирующая калориметрия: аналитическое устройство: Q2000 DSC от ТА Instruments
Как правило, менее 3 мг материала, содержащегося в стандартном алюминиевом тигле, оснащенном крышкой, нагревали в диапазоне температур от 25°С до 300°С при постоянной скорости нагрева, составляющей 10°С в минуту. Для продувки газом применяли азот при расходе 50 мл в минуту. Температурные данные анализировали с применением стандартного программного обеспечения, например Universal v.4.5А от ТА INSTRUMENTS®.
(xv) для термогравиметрического анализа (TGA) применяли устройство Q5000 TGA от ТА Instruments
Как правило, менее 5 мг помещали в алюминиевый тигель для образцов и переносили в печь для TGA. Устройство продували азотом со скоростью 50 мг/мин и данные собирали в диапазоне от 25°С до температуры, непосредственно ниже точки плавления соединения, с применением постоянной скорости нагрева, составляющей 10°С/минута. Температурные данные анализировали с применением стандартного программного обеспечения, например Universal v.4.5А от ТА INSTRUMENTS®.
Пример 1
N-{4-[(5,7-диметоксихиназолин-4-ил)амино]-3-фторфенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
4-Хлор-5,7-диметоксихиназолин (78 мг, 0,4 ммоль) добавляли к N-(4-амино-3-фторфенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (80 мг, 0,3 ммоль) в изопропаноле (2,5 мл) в атмосфере азота. Полученную смесь перемешивали при 80°С в течение 4 часов. Реакционную смесь разбавляли водой. Осадок собирали фильтрацией, промывали водой (10 мл) и сушили под вакуумом с получением неочищенного продукта в виде пурпурного твердого вещества. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде бежевого твердого вещества (80 мг, 58%). 1Н ЯМР (400 МГц, DMSO-d6) δ 1,25 (6Н, d), 2,95-3,06 (1H, m), 3,91 (3Н, s), 4,06 (3Н, s), 5,29 (2Н, s), 6,72 (1H, d), 6,80 (1H, d), 7,28-7,36 (1H, m), 7,67-7,75 (1Н, m), 7,88 (1H, s), 8,19 (1H, t), 8,41 (1Н, s), 9,81 (1Н, s), 10,71 (1Н, s); масса/заряд (ES+), [М+Н]+=466; кислота, HPLC Rt=1,53 мин.
Промежуточные соединения, применяемые в примере 1, получали следующим образом.
Получение 2-фторбензол-1,4-диамина
Порошок цинка (4,2 г, 64,1 ммоль) добавляли к хлориду аммония (3,4 г, 64,1 ммоль), 3-фтор-4-нитроанилину (500 мг, 3,2 ммоль) и воде (4 мл) в этаноле (15 мл) в атмосфере азота. Полученную смесь перемешивали при 25°С в течение 2 часов. Смесь фильтровали и фильтрат выпаривали до сухого состояния с получением неочищенного остатка, который очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 1 до 10% метанола в DCM (0,1% DIPEA). Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде черного масла (405 мг, 100%). 1H ЯМР (DMSO-d6, 300 МГц) δ 6,27 (1H, dd), 6,39 (1H, dd), 6,58 (1H, dd), 9,74 (2Н, s); масса/заряд (ES+), [М+Н]+=127; кислота, HPLC Rt=0,227 мин.
Получение этил-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетата
30% раствор этил-2-азидоацетата в DCM (19,5 г, 45,4 ммоль) добавляли в виде раствора в ацетонитриле (27 мл) в течение 5 минут к суспензии йодида меди(I) (0,17 г, 0,9 ммоль), 3-метилбут-1-ина (5,1 мл, 49,9 ммоль) и триэтиламина (0,13 мл, 0,9 ммоль) в ацетонитриле (27 мл) при комнатной температуре. Смесь перемешивали в течение 3 дней при комнатной температуре. Смесь концентрировали и остаток разделяли между водой (150 мл) и этилацетатом (150 мл). Водный слой экстрагировали с помощью этилацетата (100 мл) и экстракты объединяли с органическим слоем. Объединенные экстракты сушили и выпаривали до сухого состояния. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 30 до 50% этилацетата в гептане. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (8,06 г, 90%). 1H ЯМР (500 МГц, DMSO, 27°С) δ 1,21 (3Н, t), 1,22 (6Н, d), 2,98 (1H, гептет), 4,16 (2Н, q), 5,30 (2Н, s), 7,82 (1H, d); масса/заряд: ES+ [М+Н]+ 198.
Получение 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоты
Гидрат гидроксида лития (10,2 г, 242,5 ммоль) добавляли в виде раствора в воде (540 мл) к этил-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетату (15,9 г, 80,8 ммоль) в THF (180 мл). Смесь перемешивали в течение 90 минут, затем концентрировали. Полученный водный раствор подкисляли до рН 5 с помощью 2 М HCl и экстрагировали с помощью этилацетата (200 мл). Водный слой выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества, содержащего LiCl (28,2 г, 100%, концентрация 48%), которое применяли без дополнительной очистки. 1H ЯМР (500 МГц, DMSO, 27°С) δ 1,20 (6Н, d), 2,92 (1H, гептет), 4,59 (2Н, s), 7,62 (1H, d); масса/заряд: ES+ [М+Н]+ 170.
Получение N-(4-амино-3-фторфенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида
HATU (678 мг, 1,8 ммоль) добавляли к 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоте (201 мг, 1,2 ммоль), 2-фторбензол-1,4-диамину (150 мг, 1,2 ммоль) и DIPEA (0,3 мл, 1,8 ммоль) в DMF (7 мл) в атмосфере азота. Полученную смесь перемешивали при 25°С в течение 6 часов, затем выпаривали до сухого состояния. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде черного твердого вещества (200 мг, 61%). 1Н ЯМР (DMSO-d6, 400 МГц) δ 1,24 (6Н, d), 2,99 (1H, dt), 4,98 (2Н, s), 5,18 (2Н, s), 6,72 (1H, dd), 6,99 (1H, dd), 7,38 (1H, dd), 7,85 (1H, s), 10,25 (1H, s); масса/заряд (ES+), [М+Н]+ = 278; кислота, HPLC Rt = 0,979 мин.
Пример 2
N-{4-[(5-фтор-6,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
HATU (84 мг, 0,2 ммоль) порциями добавляли к N1-(5-фтор-6,7-диметоксихиназолин-4-ил)бензол-1,4-диамину (100 мг, 0,2 ммоль), 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоте (52 мг, 0,2 ммоль) и DIPEA (0,06 мл, 0,4 ммоль) в DMF (1 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали и разбавляли с помощью DCM (100 мл) и последовательно промывали с помощью 0,1 М HCl (20 мл), воды (10 мл) и насыщенного раствора Na2CO3 (20 мл). Органический слой сушили, фильтровали и выпаривали до сухого состояния. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (60 мг, 70%). 1Н ЯМР (400 МГц, DMSO-d6) δ 1,26 (6Н, d), 2,94-3,07 (1Н, m), 3,91 (3Н, s), 4,00 (3Н, s), 5,27 (2Н, s), 7,15 (1H, d), 7,59 (2Н, d), 7,69 (2Н, d), 7,88 (1H, d), 8,45 (1H, s), 8,94 (1H, d), 10,48 (1H, s); масса/заряд (ES+), [М+Н]+ = 466; кислота, HPLC Rt = 1,38 мин.
Промежуточные соединения, применяемые в примере 2, получали следующим образом.
Получение 2-фтор-3,4-диметоксибензальдегида
Хлорид титана(IV) (8 г, 42,3 ммоль) в DCM (12 мл) по каплям добавляли к 1-фтор-2,3-диметоксибензолу (4 г, 25,6 ммоль) в DCM (40 мл) при 0°С на протяжении 15 минут в атмосфере азота. За этим следовало добавление по каплям дихлор(метокси)метана (3,2 г, 28,2 ммоль) в безводном DCM (8 мл) в течение 12 минут. Полученный раствор перемешивали при 0°С в течение 30 минут, затем перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь гасили водой (200 мл), экстрагировали с помощью DCM (2×100 мл), органический слой сушили, фильтровали и выпаривали до сухого состояния. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 2% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения (4,6 г, 98%) в виде желтого твердого вещества. 1H ЯМР (DMSO-d6, 300 МГц) δ 3,84 (3Н, d), 3,95 (3Н, s), 7,11 (1H, р), 7,61 (1H, р), 10,06 (1H, s); масса/заряд (ES+), [М+Н]+ = 185; кислота, HPLC Rt = 1,434 мин.
Получение 2-фтор-3,4-диметокси-6-нитробензальдегида
Нитрат калия (2,8 г, 27,4 ммоль) порциями добавляли к 2-фтор-3,4-диметоксибензальдегиду (4,2 г, 22,8 ммоль) и концентрированной серной кислоте (30 мл, 562,9 ммоль) при 0°С. Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь выливали в ледяную воду. Осадок собирали фильтрацией, промывали ледяной водой (75 мл) и сушили под вакуумом с получением указанного в заголовке соединения в виде коричневого твердого вещества (3,2 г, 61%). 1H ЯМР (DMSO-d6, 300 МГц) δ 3,95 (3Н, d), 4,02 (3Н, s), 7,71 (1H, d), 10,08 (1Н, s); масса/заряд (ES+), [М+Н]+ = 230; кислота, HPLC Rt = 1,206 мин.
Получение 2-фтор-3,4-диметокси-6-нитробензойной кислоты
Перборат натрия (4,3 г, 27,9 ммоль) порциями добавляли к 2-фтор-3,4-диметокси-6-нитробензальдегиду (3,2 г, 14 ммоль) в уксусной кислоте (45 мл) на протяжении 2 минут. Полученную смесь перемешивали при 50°С в течение 3 дней. Реакционную смесь выпаривали до сухого состояния и растворяли в DCM (200 мл) и последовательно промывали водой (2×100 мл). Водный слой разделяли, замораживали и лиофилизировали с получением указанного в заголовке соединения в виде желтого твердого вещества (2,9 г, 85%). 1H ЯМР (DMSO-d6, 300 МГц) δ 3,89 (6Н, d), 7,41 (1H, d); масса/заряд (ES+), [М+Н]+ = не обнаружили; кислота, HPLC Rt = 1,041 мин.
Получение 2-фтор-3,4-диметокси-6-нитробензамида
SOCl2 (100 мл, 1370 ммоль) порциями добавляли к 2-фтор-3,4-диметокси-6-нитробензойной кислоте (2,9 г, 11,8 ммоль). Полученную смесь перемешивали при 90°С в течение 2 часов. Растворитель удаляли при пониженном давлении и полученный остаток растворяли в THF (30 мл). Растворитель охлаждали до 0°С и медленно добавляли аммиак (0,5 М в THF) (47,3 мл, 23,7 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь выпаривали до сухого состояния, и растворяли в DCM (200 мл), и последовательно промывали насыщенным раствором NaHCO3 (2×50 мл) и водой (2×50 мл). Органический слой сушили, фильтровали и выпаривали до сухого состояния. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 6% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде коричневого твердого вещества (0,6 г, 21%). 1H ЯМР (DMSO-d6, 300 МГц) δ 3,98 (6Н, d), 7,69 (1H, d).
Получение 6-амино-2-фтор-3,4-диметоксибензамида
Железо (412 мг, 7,4 ммоль) порциями добавляли к 2-фтор-3,4-диметокси-6-нитробензамиду (600 мг, 2,5 ммоль) в уксусной кислоте (3 мл). Полученную смесь перемешивали при 105°С в течение 15 минут. Реакционную смесь выпаривали до сухого состояния и растворяли в DCM (100 мл) и последовательно промывали водой (2×50 мл). Органический слой сушили, фильтровали и выпаривали с получением указанного в заголовке соединения в виде желтого масла (500 мг, 95%), которое применяли без дополнительной очистки. Масса/заряд (ES+), [М+Н]+ = 215; кислота, HPLC tR = 0,964 мин.
Получение 5-фтор-6,7-диметоксихиназолин-4(3H)-она
PTSA (35,5 мг, 0,2 ммоль) добавляли к 6-амино-2-фтор-3,4-диметоксибензамиду (200 мг, 0,9 ммоль) и триметоксиметану (5 мл) при 25°С в атмосфере азота. Полученную суспензию перемешивали при 100°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры. Осадок собирали фильтрацией, промывали этилацетатом (5 мл) и выпаривали до сухого состояния с получением указанного в заголовке соединения в виде бежевого твердого вещества (140 мг, 67%), которое применяли без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 3,83 (3Н, s), 3,95 (3Н, s), 7,01-7,08 (1H, m), 8,00 (1H, d), 12,12 (1H, s); масса/заряд (ES+), [М+Н]+ = 225; кислота, HPLC Rt = 0,87 мин.
Получение N1-(5-фтор-6,7-диметоксихиназолин-4-ил)бензол-1,4-диамина
РуАОР (393 мг, 0,8 ммоль) добавляли к 5-фтор-6,7-диметоксихиназолин-4(3H)-ону (130 мг, 0,6 ммоль) и DBU (0,22 мл, 1,5 ммоль) в ацетонитриле (4 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 10 минут. К нему добавляли бензол-1,4-диамин (94 мг, 0,9 ммоль) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили водой (20 мл), экстрагировали с помощью DCM (2×25 мл), органический слой сушили, фильтровали и выпаривали с получением темного масла, которое затвердевало при отстаивании. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 2% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде желтого твердого вещества (220 мг, >100%). 1H ЯМР (400 МГц, DMSO-d6) δ 3,89 (3Н, s), 3,98 (3Н, s), 5,05 (2Н, s), 6,57 (2Н, d), 7,09 (1H, s), 7,26 (2Н, d), 8,34 (1H, s), 8,65 (1Н, d); масса/заряд (ES+), [М+Н]+ = 315; кислота, HPLC Rt = 0,87 мин.
Пример 3 и пример 4
(R)-N-(4-{[5-этокси-7-(тетрагидрофуран-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамид и (S)-N-(4-{[5-этокси-7-(тетрагидрофуран-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
Трет-бутоксид калия (150 мг, 1,3 ммоль) добавляли к тетрагидрофуран-3-олу (78 мг, 0,9 ммоль) и N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (200 мг, 0,4 ммоль) в DMF (4 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при 80°С в течение 5 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением рацемической формы указанного в заголовке соединения в виде белого твердого вещества (160 мг, 70%); масса/заряд (ES+), [М+Н]+ = 518; TFA, HPLC tR = 1,443 мин. Данное соединение очищали с помощью препаративной хиральной HPLC на колонке Chiralpak IA при изократическом элюировании с помощью 30% этанола в МТВЕ (0,1% DEA) в качестве элюента. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением одного энантиомера N-(4-((5-этокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида в виде белого твердого вещества (68 мг, 43%). 1H ЯМР (400 МГц, DMSO, 20°С) δ 1,25 (6Н, d), 1,56 (3Н, t), 1,99-2,06 (1Н, m), 2,25-2,35 (1H, m), 2,95-3,02 (1H, m), 3,75-3,95 (4Н, m), 4,32 (2Н, t), 5,22 (1H, t), 5,27 (2Н, s), 6,68 (1H, s), 6,76 (1H, s), 7,60 (2Н, d), 7,78 (2Н, d), 7,88 (1H, s), 8,46 (1H, s), 9,98 (1H, s), 10,50 (1H, s); масса/заряд (ES+), [М+Н]+ = 518; TFA, HPLC Rt = 1,463 мин. Затем за ним следовал другой энантиомер N-(4-((5-этокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида в виде белого твердого вещества (62 мг, 39%). 1Н ЯМР (400 МГц, DMSO, 20°С) δ 1,25 (6Н, d), 1,56 (3Н, t), 1,98-2,05 (1H, m), 2,25-2,35 (1Н, m), 2,95-3,02 (1H, m), 3,75-3,95 (4Н, m), 4,32 (2Н, t), 5,22 (1H, t), 5,27 (2Н, s), 6,68 (1H, s), 6,76 (1Н, s), 7,60 (2Н, d), 7,78 (2Н, d), 7,88 (1H, s), 8,46 (1H, s), 9,99 (1H, s), 10,50 (1H, s); масса/заряд (ES+), [М+Н]+ = 518; TFA, HPLC Rt = 1,470 мин.
Промежуточные соединения, применяемые в примере 3 и примере 4, получали следующим образом.
Получение 5-этокси-7-фторхиназолин-4(3H)-она
Этанолят натрия (7,5 г, 109,8 ммоль) добавляли к 5,7-дифторхиназолин-4(3H)-ону (4 г, 22 ммоль) в DMSO (20 мл), охлажденному при 0°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой (200 мл) и доводили до рН 7 с помощью 2 М HCl. Полученное твердое вещество выделяли фильтрацией с получением указанного в заголовке соединения в виде желтого твердого вещества (4,4 г, 96%). 1H ЯМР (300 МГц, DMSO, 21°С) δ 1,37 (3H, t), 4,09-4,16 (2Н, m), 6,88-6,94 (2Н, m), 7,98 (1H, s), 11,98 (1Н, s); масса/заряд (ES+), [M+H]+ = 209; TFA, HPLC Rt = 0,988 мин.
Получение N1-(5-этокси-7-фторхиназолин-4-ил)бензол-1,4-диамина
ВОР (8,9 г, 20,2 ммоль) добавляли к 5-этокси-7-фторхиназолин-4(3H)-ону (3 г, 14,4 ммоль) и DBU (4,3 мл, 28,8 ммоль) в ацетонитриле (20 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при 60°С в течение 3 часов. К нему добавляли бензол-1,4-диамин (2,2 г, 20,2 ммоль) при 25°С в атмосфере азота. Полученный раствор перемешивали при 60°С в течение 3 часов. Растворитель удаляли при пониженном давлении. Реакционную смесь гасили водой (10 мл), экстрагировали с помощью DCM (3×25 мл), органический слой сушили, фильтровали и выпаривали с получением темного масла. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 4% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде желтого твердого вещества (2,6 г, 59%). 1H ЯМР (300 МГц, DMSO, 23°С) δ 1,68 (3Н, t), 4,27-4,29 (2Н, m), 6,65 (1Н, s), 6,74-6,77 (2Н, m), 7,06 (1H, d), 7,45-7,48 (2Н, m), 8,54 (1H, d), 9,69 (1H, s); масса/заряд (ES+), [М+Н]+ = 299; TFA, HPLC Rt = 0,871 мин.
Получение N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида
HATU (4,7 г, 12,3 ммоль) добавляли к N1-(5-этокси-7-фторхиназолин-4-ил)бензол-1,4-диамину (2,5 г, 8,2 ммоль), 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоте (2,1 г, 12,3 ммоль) и DIPEA (4,3 мл, 24,6 ммоль) в DMF (10 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли этилацетатом (150 мл) и последовательно промывали водой (2×150 мл) и насыщенным солевым раствором (75 мл). Органический слой сушили, фильтровали и выпаривали до сухого состояния. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 4% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде желтого твердого вещества (1,5 г, 41%). 1Н ЯМР (300 МГц, DMSO, 23°С) δ 1,25 (6Н, d), 1,58 (3Н, t), 2,95-3,05 (1H, m), 4,35-4,42 (2Н, m), 5,26 (2Н, s), 7,05-7,12 (2Н, m), 7,60-7,63 (2Н, m), 7,77-7,80 (2Н, m), 7,87 (1H, s), 8,50 (1H, s), 10,03 (1Н, s), 10,51 (1H, s); масса/заряд (ES+), [М+Н]+ = 450; TFA, HPLC Rt = 1,048 мин.
Пример 5
N-(4-((5-этокси-7-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамид
Трет-бутоксид калия (112 мг, 1 ммоль) добавляли к N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (150 мг, 0,3 ммоль) и тетрагидро-4H-пиран-4-олу (102 мг, 1 ммоль) в DMF (2 мл) при комнатной температуре. Полученный раствор перемешивали при 80°С в течение 7 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения (75 мг, 42%) в виде белого твердого вещества. 1Н ЯМР (DMSO-d6, 400 МГц) δ 1,26 (6Н, d), 1,58 (3Н, t), 1,65 (2Н, t), 2,01-2,11 (2Н, m), 2,95-3,07 (1H, m), 3,52-3,58 (2Н, m), 3,86-3,91 (2Н, m), 4,35 (2Н, q), 4,80-4,86 (1H, m), 5,27 (2Н, s), 6,70 (1H, d), 6,88 (1H, d), 7,57-7,65 (2Н, m), 7,75-7,84 (2Н, m), 7,89 (1H, d), 8,44 (1H, s), 9,98 (1H, s), 10,50 (1H, s); масса/заряд (ES+), [М+Н]+ = 532; кислота, HPLC Rt = 1,485 мин.
Пример 6
N-(4-((7-(2-(диметиламино)этокси)-5-этоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамид
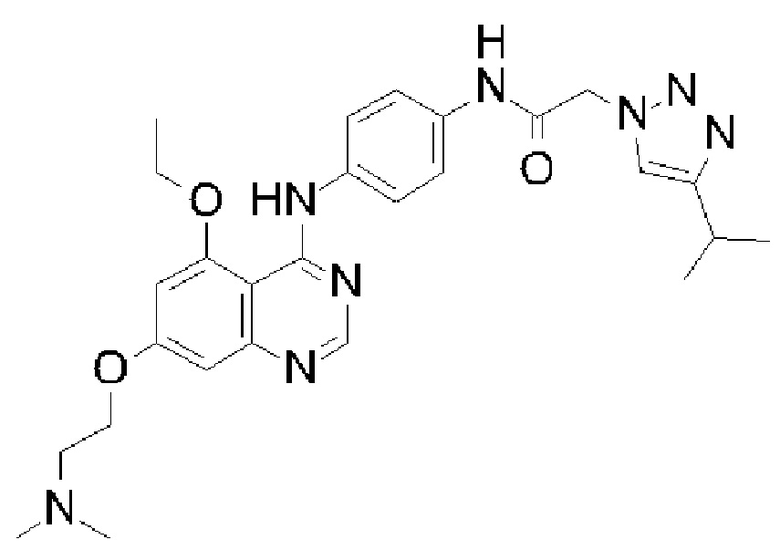
Трет-бутоксид калия (75 мг, 0,7 ммоль) добавляли к 2-(диметиламино)этан-1-олу (39,7 мг, 0,4 ммоль) и N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (100 мг, 0,2 ммоль) в DMF (2 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при 80°С в течение 15 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (51 мг, 44%). 1Н ЯМР (400 МГц, DMSO, 20°С) δ 1,26 (6Н, d), 1,58 (3Н, t), 2,25 (6Н, s), 2,65-2,69 (2Н, t), 2,95-3,05 (1H, m), 4,19 (2H, t), 4,31-4,36 (2Н, m), 5,27 (2Н, s), 6,90 (1H, s), 6,80 (1H, s), 7,61 (2Н, d), 7,78 (2Н, d), 7,88 (1H, s), 8,45 (1H, s), 9,97 (1H, s), 10,50 (1H, s); масса/заряд (ES+), [М+Н]+ = 519; TFA, HPLC tR = 1,193 мин.
Пример 7
N-(4-((5-этокси-7-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамид
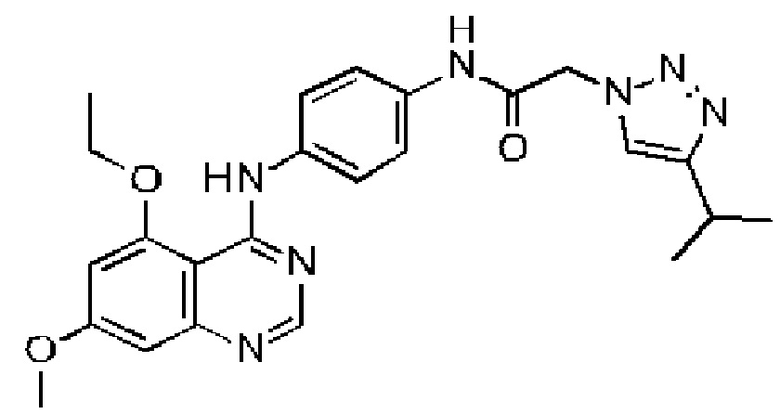
Метоксид натрия (24 мг, 0,4 ммоль) добавляли к метанолу (43 мг, 1,3 ммоль) и N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (100 мг, 0,2 ммоль) в DMF (2 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при комнатной температуре в течение 5 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (70 мг, 68%). 1Н ЯМР (400 МГц, DMSO, 20°С) δ 1,26 (6Н, d), 1,58 (3Н, t), 2,95-3,05 (1H, m), 3,90 (3Н, s), 4,31-4,36 (2Н, m), 5,27 (2Н, s), 6,70 (1H, s), 6,79 (1Н, s), 7,61 (2H, d), 7,79 (2H, d), 7,88 (1H, s), 8,46 (1H, s), 9,97 (1H, s), 10,50 (1H, s).
Масса/заряд (ES+), [M+H]+ = 462; TFA, HPLC tR = 1,445 мин.
Пример 8
N-(4-((5-этокси-7-(2-метоксиэтокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамид
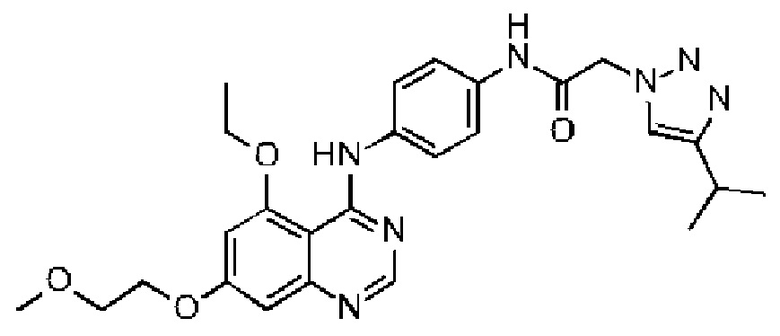
2-Метоксиэтан-1-ол (34 мг, 0,4 ммоль) добавляли к N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (100 мг, 0,2 ммоль) и трет-бутоксиду калия (75 мг, 0,7 ммоль) в DMF (2 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при 80°С в течение 5 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (63 мг, 56%). 1Н ЯМР (400 МГц, DMSO, 20°С) δ 1,26 (6Н, d), 1,58 (3Н, t), 2,95-3,05 (1H, m), 3,34 (3Н, s), 3,71 (2Н, t), 4,25 (2Н, t), 4,31-4,36 (2Н, m), 5,27 (2Н, s), 6,72 (1H, s), 6,79 (1Н, s), 7,61 (2Н, d), 7,78 (2Н, d), 7,88 (1H, s), 8,45 (1H, s), 9,97 (1H, s), 10,50 (1Н, s); масса/заряд (ES+), [М+Н]+ = 506; TFA, HPLC Rt = 1,445 мин.
Пример 9
N-(4-((5-этокси-7-(оксетан-3-илокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамид
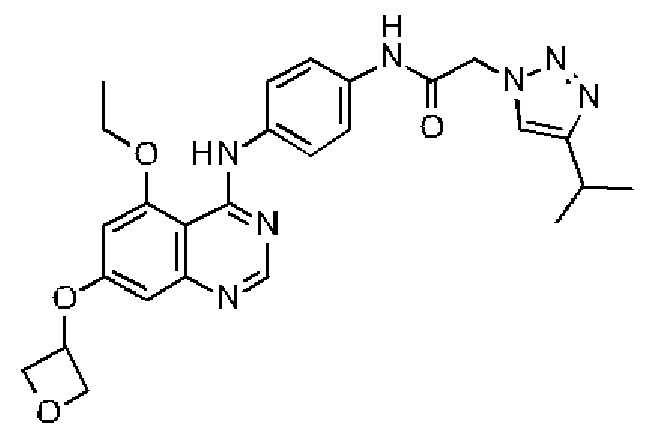
Трет-бутоксид калия (50 мг, 0,4 ммоль) добавляли к оксетан-3-олу (33 мг, 0,4 ммоль) и N-(4-((5-этокси-7-фторхиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (100 мг, 0,2 ммоль) в DMF (2 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при 80°С в течение 5 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (68 мг, 61%). 1Н ЯМР (400 МГц, DMSO, 20°С) δ 1,26 (6Н, d), 1,58 (3Н, t), 2,98-3,05 (1H, m), 4,33-4,38 (2Н, m), 4,60 (2Н, t), 5,00 (2Н, t), 5,27 (2Н, s), 5,45-5,50 (1H, m), 6,46 (1H, s), 6,72 (1H, s), 7,61 (2Н, d), 7,78 (2Н, d), 7,88 (1H, s), 8,45 (1H, s), 9,97 (1H, s), 10,50 (1H, s); масса/заряд (ES+), [М+Н]+ = 504; TFA, HPLC Rt = 1,883 мин.
Пример 10
N-(4-{[5-метокси-7-(тетрагидро-2H-пиран-4-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
Трет-бутоксид калия (85 мг, 0,8 ммоль) добавляли к N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (110 мг, 0,3 ммоль) и тетрагидро-4H-пиран-4-олу (77 мг, 0,8 ммоль) в DMF (3 мл) при комнатной температуре. Полученный раствор перемешивали при 80°С в течение 7 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (38 мг, 29%). 1Н ЯМР (DMSO-d6, 400 МГц) δ 1,26 (6Н, d), 1,60-1,69 (2Н, m), 2,05 (2Н, d), 2,95-3,10 (1H, m), 3,56 (2Н, t), 3,89 (2Н, t), 4,09 (3Н, s), 4,81-4,85 (1H, m), 5,27 (2Н, s), 6,69 (1H, d), 6,87 (1H, d), 7,57-7,66 (2Н, m), 7,74-7,84 (2Н, m), 7,89 (1H, d), 8,40 (1H, s), 9,78 (1H, s), 10,52 (1H, d); масса/заряд (ES+), [М+Н]+ = 518; TFA, HPLC Rt = 8,32 мин.
Промежуточные соединения, применяемые в примере 10, получали следующим образом.
Получение 7-фтор-5-метоксихиназолин-4(3H)-она
Метоксид натрия (в метаноле) (9,9 г, 54,9 ммоль) добавляли к 5,7-дифторхиназолин-4(3H)-ону (2 г, 11 ммоль) в DMSO (10 мл), охлажденному до 0°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой, затем нейтрализовали с помощью 2 М HCl. Полученный осадок собирали фильтрацией, промывали водой (20 мл) и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (1,9 г, 89%), которое применяли без дополнительной очистки. 1H ЯМР (300 МГц, DMSO-d6) δ 3,87 (3Н, s), 6,87 - 7,03 (2Н, m), 8,00 (1H, s), 12,04 (1Н, s); масса/заряд (ES+), [М+Н]+ = 195; кислота, HPLC Rt = 0,90 мин.
Получение N1-(7-фтор-5-метоксихиназолин-4-ил)бензол-1,4-диамина
ВОР (3,2 г, 7,2 ммоль) добавляли к 7-фтор-5-метоксихиназолин-4(3H)-ону (1 г, 5,2 ммоль) и DBU (1,6 мл, 10,3 ммоль) в ацетонитриле (20 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при 60°С в течение 15 минут. К нему добавляли бензол-1,4-диамин (0,8 г, 7,2 ммоль) при 25°С в атмосфере азота. Полученный раствор перемешивали при 60°С в течение 1 часа. Растворитель удаляли при пониженном давлении. Реакционную смесь гасили водой (10 мл), экстрагировали с помощью DCM (3×25 мл), органический слой сушили, фильтровали и выпаривали с получением темного масла. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 4% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде желтого твердого вещества (2,1 г, >100%). 1H ЯМР (400 МГц, DMSO-d6) δ 4,10 (3Н, s), 5,12 (2Н, s), 6,59 (2Н, d), 6,97 - 7,07 (2Н, m), 7,31 (2Н, d), 8,34 (1H, s), 9,59 (1H, s); масса/заряд (ES+), [М+Н]+ = 285; кислота, HPLC Rt = 0,81 мин.
Получение N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида
HATU (1,9 г, 4,9 ммоль) порциями добавляли к N1-(7-фтор-5-метоксихиназолин-4-ил)бензол-1,4-диамину (2,1 г, 4,4 ммоль), 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоте (1,2 г, 4,9 ммоль) и DIPEA (1,70 мл, 9,8 ммоль) в ацетонитриле (100 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении. Реакционную смесь разбавляли водой. Осадок собирали фильтрацией, промывали смесью вода/ацетонитрил (50 мл, 5:1) и сушили под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,2 г, 62%), которое применяли без дополнительной очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ 1,25 (6Н, d), 2,95-3,05 (1H, m), 4,13 (3Н, s), 5,27 (2Н, s), 7,01-7,14 (2Н, m), 7,61 (2Н, d), 7,75 (2Н, d), 7,88 (1H, d), 8,46 (1H, s), 9,87 (1H, s), 10,51 (1H, s); масса/заряд (ES+), [М+Н]+ = 436; основание, HPLC Rt = 0,82 мин.
Пример 11
2-[4-(Пропан-2-ил)-1H-1,2,3-триазол-1-ил]-N-{4-[(5,6,7-триметоксихиназолин-4-ил)амино]фенил}ацетамид
HATU (64 мг, 0,2 ммоль) добавляли к суспензии 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоты (42 мг, 0,1 ммоль), N1-(5,6,7-триметоксихиназолин-4-ил)бензол-1,4-диамина (44 мг, 0,1 ммоль) и N-этил-N-изопропилпропан-2-амина (0,05 мл, 0,3 ммоль) в DMF (5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов. Реакционную смесь гасили водой (10 мл), разбавляли этилацетатом (10 мл) и слои разделяли. Водный слой промывали этилацетатом (3×10 мл) и органические слои объединяли, промывали солевым раствором (10 мл), сушили, фильтровали и концентрировали in vacuo. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (41 мг, 76%). 1Н ЯМР (500 МГц, DMSO, 27°С) δ 1,24 (3Н, s), 1,25 (3Н, s), 2,99 (1H, pd), 3,86 (3Н, s), 3,95 (3Н, s), 4,12 (3Н, s), 5,25 (2Н, s), 7,06 (1H, s), 7,59 (2Н, d), 7,82 (2Н, d), 7,86 (1Н, d), 8,43 (1Н, s), 9,88 (1Н, s), 10,45 (1Н, s); масса/заряд: ES+ [М+Н]+ 478.
Промежуточные соединения, применяемые в примере 11, получали следующим образом.
Получение 5,6,7-триметоксихиназолин-4(3H)-она
Раствор гидрохлорида этил-6-амино-2,3,4-триметоксибензоата (200 мг, 0,7 ммоль) и ацетата формимидамида (214 мг, 2,1 ммоль) нагревали в 2-метоксиэтаноле (5 мл) при 125°С в течение 2 часов. Смесь охлаждали до температуры окружающей среды и выпаривали. Остаток обрабатывали водой (10 мл). Полученный осадок собирали фильтрацией, промывали водой и сушили под вакуумом с получением указанного в заголовке соединения в виде коричневого твердого вещества (164 мг, 100%), которое применяли без дополнительной очистки. 1H ЯМР (500 МГц, CDCl3, 27°С) δ 3,95 (3Н, s), 3,99 (2Н, s), 4,02 (3Н, s), 7,01 (1H, s), 7,99 (1H, s), 11,02 (1Н, s). масса/заряд: ES+ [М+Н]+ 237.
Получение N1-(5,6,7-триметоксихиназолин-4-ил)бензол-1,4-диамина
DBU (0,2 мл, 1,4 ммоль) добавляли к смеси 5,6,7-триметоксихиназолин-4(3H)-она (130 мг, 0,6 ммоль) и РуВОР (372 мг, 0,7 ммоль) в ацетонитриле (10 мл) и нагревали при 60°С в течение 1 часа. Добавляли бензол-1,4-диамин (119 мг, 1,1 ммоль) и перемешивание при 60°С продолжали в течение дополнительных 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали in vacuo. Неочищенную смесь растворяли в ацетоне и добавляли 2 мл 2 М HCl в диэтиловом эфире с образованием осадка. Осадок фильтровали и промывали ацетоном, затем растворяли в насыщенном водном растворе гидрокарбоната натрия (20 мл) и этилацетата (20 мл). Слои разделяли и водный слой экстрагировали этилацетатом (3×20 мл). Объединенные органические слои сушили, фильтровали и концентрировали in vacuo с получением указанного в заголовке соединения в виде бежевого твердого вещества (73 мг, 41%), которое применяли без дополнительной очистки. Масса/заряд: ES+ [М+Н]+ 327.
Пример 12
N-{4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
Смесь N-(4-аминофенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида (5,4 г, 20,7 ммоль) и (E)-N'-(2-циано-3-фтор-5-(2-метоксиэтокси)фенил)-N,N-диметилформимидамида (5,2 г, 19,7 ммоль) в уксусной кислоте (12 мл) перемешивали при 60°С в течение 35 минут. Смесь выливали в воду (150 мл), смесь перемешивали и подвергали воздействию ультразвука. Полученный осадок собирали фильтрацией, промывали водой и сушили. Твердое вещество растворяли в DCM/метаноле (12:1, 600 мл) и раствор промывали с помощью 0,2 М раствора NaHCO3 (600 мл). Водный слой экстрагировали с помощью DCM/метанола (12:1, 2×200 мл) и экстракты объединяли с органическим слоем. Объединенные органические экстракты сушили, фильтровали и выпаривали с получением бежевого твердого вещества. Неочищенный продукт кристаллизовали из горячего этанола (700 мл). После охлаждения до температуры окружающей среды и перемешивания в течение 2 часов кристаллическое твердое вещество собирали фильтрацией, промывали холодным этанолом и сушили под высоким вакуумом при 50°C с получением 7,4 г неочищенного продукта. Неочищенный продукт дополнительно очищали с помощью перекристаллизации в горячем этаноле (800 мл). После охлаждения до температуры окружающей среды и перемешивания в течение 20 часов кристаллическое твердое вещество собирали фильтрацией, твердые вещества собирали и сушили под вакуумом при 50°С в течение 72 часов с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (6,2 г, 58%). 1Н ЯМР (500 МГц, DMSO, 27°С) δ 1,24 (6Н, d), 2,99 (1H, pd), 3,32 (3Н, s), 3,67-3,73 (2Н, m), 4,24-4,3 (2Н, m), 5,26 (2Н, s), 7,04 (1Н, d), 7,13 (1H, dd), 7,53-7,61 (2Н, m), 7,63-7,69 (2Н, m), 7,86 (1H, d), 8,44 (1H, s), 8,95 (1H, d), 10,47 (1Н, s); масса/заряд: ES+ [М+Н]+ 480.
Промежуточные соединения, применяемые в примере 12, получали следующим образом.
Получение трет-бутил-(4-(2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамидо)фенил)карбамата
HATU (18,4 г, 48,3 ммоль) добавляли к раствору трет-бутил-(4-аминофенил)карбамата (8,4 г, 40,3 ммоль), 2-(4-изопропил-1Н-1,2,3-триазол-1-ил)уксусной кислоты (19,4 г, 44,3 ммоль) и DIPEA (10,5 мл, 60,4 ммоль) при температуре окружающей среды. Смесь перемешивали при температуре окружающей среды в течение 16 часов. Смесь концентрировали до объема 75 мл, разбавляли водой (700 мл) и экстрагировали с помощью этилацетата (3×300 мл). Объединенные этилацетатные экстракты промывали с помощью 0,5 М раствора лимонной кислоты (300 мл), воды (4×300 мл), 0,5 М раствора NaHCO3 (200 мл), воды (200 мл), солевого раствора (200 мл) и сушили. Раствор выпаривали до сухого состояния и остаток перекристаллизовывали из ацетонитрила с получением указанного в заголовке соединения в виде белого твердого вещества (8,2 г, 57%). 1H ЯМР (500 МГц, DMSO, 27°С) δ 1,23 (6Н, d), 1,45 (9Н, s), 2,98 (1H, гептет), 5,20 (2Н, s), 7,38 (2Н, d), 7,44 (2Н, d), 7,83 (1H, d), 9,27 (1H, s), 10,31 (1H, s), масса/заряд: ES+ [М+Н]+ 360.
Получение N-(4-аминофенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида
4 М хлористого водорода в диоксане (15,3 мл, 61,2 ммоль) добавляли к смеси трет-бутил-(4-(2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамидо)фенил)карбамата (2,2 г, 6,1 ммоль) в DCM (20 мл) и метаноле (20 мл). Смесь перемешивали при температуре окружающей среды в течение 3 часов, при этом в течение данного времени добавляли дополнительную порцию 4 М хлористого водорода в диоксане (8,0 мл, 24 ммоль). Смесь выпаривали до сухого состояния и остаток растворяли в воде (70 мл). Данный водный раствор медленно добавляли к перемешиваемому 1 М раствору карбоната калия (150 мл), что вызывало осаждение белого твердого вещества. Смесь перемешивали в течение 10 минут при температуре окружающей среды. Осадок собирали фильтрацией, промывали водой и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (1,4 г, 89%), которое применяли без очистки. 1Н ЯМР (500 МГц, DMSO, 27°С) δ 1,23 (6Н, d), 2,98 (1H, гептет), 4,90 (2Н, s), 5,14 (2Н, s), 6,50 (2Н, d), 7,20 (2Н, d), 7,82 (1Н, s), 9,99 (1H, s); масса/заряд: ES+ [М+Н]+ 260.
Получение 2,6-дифтор-4-(2-метоксиэтокси)бензонитрила
1-Бром-2-метоксиэтан (8,4 мл, 89 ммоль) добавляли к перемешиваемой суспензии 2,6-дифтор-4-гидроксибензонитрила (11,5 г, 74,1 ммоль) и карбоната калия (30,7 г, 222,4 ммоль) в DMF (175 мл). Смесь нагревали до 85°С в течение 5 часов. Смесь охлаждали до температуры окружающей среды и выливали в воду (1250 мл). Смесь экстрагировали этилацетатом (2×400 мл). Объединенные экстракты промывали водой (4×400 мл), насыщенным солевым раствором (200 мл), сушили и выпаривали до сухого состояния с получением оранжевого масла. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 20 до 45% этилацетата в гептане. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (16,1 г, 93%). 1H ЯМР (500 МГц, DMSO, 27°С) δ 3,28 (3Н, s), 3,62-3,68 (2Н, m), 4,21-4,27 (2Н, m), 7,05-7,14 (2Н, m); масса/заряд: ES+ [М+Н]+ 214.
Получение 2-амино-6-фтор-4-(2-метоксиэтокси)бензонитрила 2,6-Дифтор-4-(2-метоксиэтокси)бензонитрил (23 г, 107,9 ммоль) разделяли на 14 флаконов для микроволновой обработки, каждый из которых содержал субстрат (1,64 г, 7,7 ммоль). Каждую партию суспендировали в изопропаноле (3 мл) и добавляли концентрированный водный раствор аммиака (8 мл, 3237 ммоль). Каждый флакон герметически закрывали и нагревали до 100°С в реакторах для микроволновой обработки в течение 13 часов. Все партии объединяли; твердое вещество, которое кристаллизовалось из раствора, собирали фильтрацией, промывали водой и сушили с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (19,6 г, 87%). 1Н ЯМР (500 МГц, DMSO, 27°С) δ 3,28 (3Н, s), 3,57-3,64 (2Н, m), 4,02-4,07 (2Н, m), 6,10 (1H, dd), 6,17 (1H, dd), 6,35 (2Н, s); масса/заряд: ES- [М-Н]- 209.
Получение (E)-N'-(2-циано-3-фтор-5-(2-метоксиэтокси)фенил)-N,N-диметилформимидамида
1,1-Диметокси-N,N-диметилметанамин (62,6 мл, 471 ммоль) добавляли к 2-амино-6-фтор-4-(2-метоксиэтокси)бензонитрилу (11 г, 52,3 ммоль) при 25°С. Полученный раствор перемешивали при 80°С в течение 2 часов, затем охлаждали до комнатной температуры. Смесь выливали в перемешиваемую воду (200 мл) (экзотермическая реакция, применяли охлаждение холодной водой) и реакционную смесь перемешивали в течение 1 часа. Смесь экстрагировали этилацетатом (2×150 мл). Объединенные экстракты промывали водой (3×150 мл), насыщенным солевым раствором (100 мл), сушили и выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (13,9 г, 100%). 1H ЯМР (500 МГц, DMSO, 27°С) δ 2,98 (3Н, s), 3,07 (3Н, s), 3,29 (3Н, s), 3,61-3,66 (2Н, m), 4,14-4,17 (2Н, m), 6,55-6,6 (2Н, m), 8,03 (1H, s); масса/заряд: ES+ [М+Н]+ 266.
Форма А соединения X
Конечный продукт, N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамид, анализировали с помощью XRPD и DSC и обнаружили, что он является кристаллическим. XRPD образца материала дала рентгенограмму дифрактометрии, показанную на фигуре А. Форма А N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида характеризовалась по меньшей мере одним пиком при значении 2θ 6,7° и 18,7° согласно измерениям с применением излучения CuKα. Десять наиболее выраженных пиков XRPD показаны в таблице А.

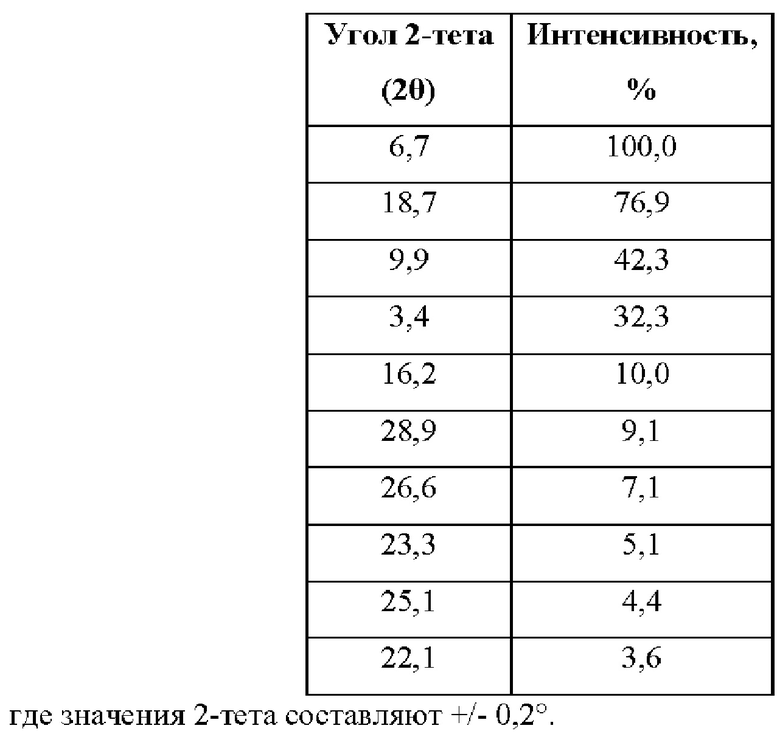
DSC-анализ формы А N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида продемонстрировали эндотерму плавления с эндотермой плавления с началом при 235,7°С и пиком при 237,6°С. Кривая DSC-анализа показана на фигуре В.
Форма В соединения X
Материал формы В получали путем образования взвеси формы А N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида в воде при комнатной температуре. Примерно 10 мг исходного материала помещали в 1,5-мл стеклянный флакон с магнитным мешателем и добавляли примерно 0,5 мл воды, затем флакон герметично укупоривали колпачком и оставляли перемешиваться на платформе магнитной мешалки. Через примерно 4 дней образец убирали с платформы, колпачок снимали и взвесь оставляли высушиваться в условиях окружающей среды перед анализом с помощью XRPD, DSC и TGA. С помощью XRPD определили, что полученный материал (форма В) является кристаллическим. DSC- и TGA-анализ продемонстрировал, что данный материал соответствует моногидрату. При нагревании до 220°С наблюдали потерю массы на 4,1%. Кривая для N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида показана на фигуре D.
XRPD образца материала дала рентгенограмму дифрактометрии, показанную на фигуре С. Форма В N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида характеризовалась по меньшей мере одним пиком при значении 2θ 4,2° и 7,7° согласно измерениям с применением излучения CuKα. Десять наиболее выраженных пиков XRPD показаны в таблице В.

Пример 12.1
Увеличение масштаба получения N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида
1ая группа сходящегося пути синтеза
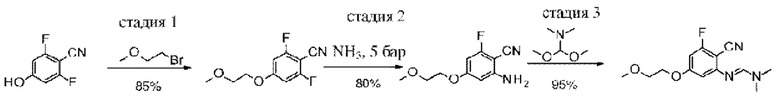
Стадия 1
1-Бром-2-метоксиэтан (65,4 мл, 696,31 ммоль) добавляли одной порцией к 2,6-дифтор-4-гидроксибензонитрилу (90 г, 580,26 ммоль) и карбонату калия (241 г, 1740,77 ммоль) в DMF (1200 мл). Полученный раствор перемешивали при 85°С в течение 5 часов.
Реакционную смесь выливали в воду (400 мл), экстрагировали с помощью EtOAc (2×200 мл), органический слой сушили над Na2SO4, фильтровали и выпаривали с получением неочищенного продукта.
Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 20 до 30% EtOAc в петролейном эфире. Чистые фракции выпаривали до сухого состояния с получением 2,6-дифтор-4-(2-метоксиэтокси)бензонитрила (115 г, 93%) в виде желтого масла. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,17-7,08 (m, 2Н), 4,29-4,22 (m, 2Н), 3,70-3,61 (m, 2Н), 3,30 (s, 3Н).
Стадия 2
Водный раствор аммиака (360 мл, 16,64 моль) добавляли к 2,6-дифтор-4-(2-метоксиэтокси)бензонитрилу (56 г, 0,26 моль) в iPrOH (120 мл) при комнатной температуре. Полученный раствор перемешивали при 95°С в течение 12 часов.
Твердое вещество кристаллизовали из раствора, твердое вещество собирали фильтрацией. Твердое вещество промывали с помощью 15% изопропанола в воде, затем промывали водой и сушили. В результате получали 2-амино-6-фтор-4-(2-метоксиэтокси)бензонитрил (50,8 г, 92%) в виде белого твердого вещества. 1Н ЯМР (DMSO-d6, 400 МГц) δ 3,29 (3Н, s), 3,59-3,66 (2Н, m), 4,03-4,10 (2Н, m), 6,09-6,23 (2Н, m), 6,38 (2Н, s).
Стадия 3
DMF-DMA (500 мл, 3734,35 ммоль) добавляли к 2-амино-6-фтор-4-(2-метоксиэтокси)бензонитрилу (90 г, 428,15 ммоль). Полученный раствор перемешивали при 80°С в течение 2 часов.
Реакционную смесь выливали в ледяную воду (1,5 л), экстрагировали с помощью EtOAc (3×500 мл), органический слой сушили над Na2SO4, фильтровали и выпаривали с получением (Е)-N'-(2-циано-3-фтор-5-(2-метоксиэтокси)фенил)-N,N-диметилформимидамида (110 г, 97%) в виде бесцветного твердого вещества. 1H ЯМР (DMSO-d6, 400 МГц) δ 3,04 (6Н, d), 3,61-3,69 (2Н, m), 4,13-4,21 (2Н, m), 6,55-6,63 (2Н, m), 8,05 (1H, s).
2ая группа сходящегося пути синтеза
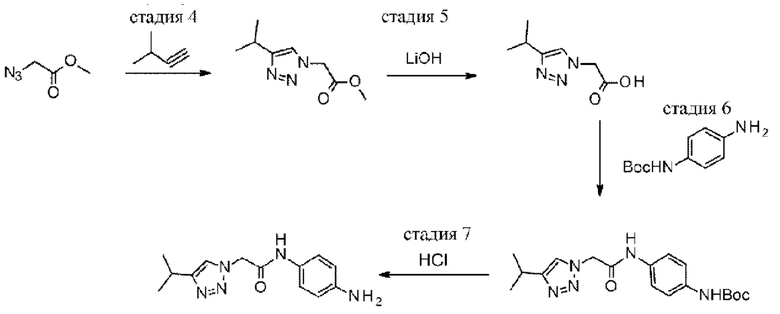
Стадия 4
Данную стадию осуществляли параллельно. 30% раствор этил-2-азидоацетата в DCM (60 г, 464,69 ммоль) смешивали с ацетонитрилом (500 мл) и затем по каплям добавляли к перемешиваемому раствору 3-метилбут-1-ина (36,4 г, 534,39 ммоль), йодида меди(1) (1,770 г, 9,29 ммоль) и TEA (1,295 мл, 9,29 ммоль) в ацетонитриле (500 мл) при 22°С на протяжении 5 минут в атмосфере азота. Полученный раствор перемешивали при 22°С в течение 12 часов.
Затем полученные растворы объединяли и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 40% EtOAc в петролейном эфире. Чистые фракции выпаривали до сухого состояния с получением этил-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетата (160 г, 175%) в виде бесцветного масла. 1Н ЯМР (Хлороформ-d, 300 МГц) δ 1,06-1,18 (9Н, m), 2,88-2,97 (1H, m), 4,07 (2Н, q), 4,99 (2Н, s), 7,35 (1H, d).
Стадия 5
Раствор LiOH (38,9 г, 1622,41 ммоль) в воде (200 мл) по каплям добавляли к перемешиваемому раствору этил-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетата (160 г, 811,20 ммоль) в THF (800 мл) при 22°С на протяжении 5 минут в атмосфере азота. Полученный раствор перемешивали при 22°С в течение 12 часов.
Растворитель удаляли при пониженном давлении. Реакционную смесь подкисляли с помощью 2 М HCl до рН 6. Твердое вещество собирали и сушили под вакуумом при 40°C с получением 2-(4-изопропил-1Н-1,2,3-триазол-1-ил)уксусной кислоты (120 г, 87%) в виде белого твердого вещества. 1H ЯМР (Метанол-d4, 300 МГц) δ 1,33 (6Н, d), 3,04-3,09 (1H, m), 4,93 (2Н, s), 7,69 (1H, d).
Стадия 6
HATU (147 г, 386,88 ммоль) добавляли к трет-бутил-(4-аминофенил)карбамату (67,1 г, 322,40 ммоль), 2-(4-изопропил-1Н-1,2,3-триазол-1-ил)уксусной кислоте (60 г, 354,64 ммоль) и DIPEA (84 мл, 483,60 ммоль) в DMF (200 мл). Полученный раствор перемешивали при комнатной температуре в течение 12 часов.
Реакционную смесь выливали в воду (600 мл). Полученный твердый осадок собирали фильтрацией и сушили с получением светло-пурпурного твердого вещества.
Твердое вещество перекристаллизовывали из ацетонитрила с получением трет-бутил-(4-(2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамидо)фенил)карбамата (105 г, 91%) в виде белого твердого вещества. 1H ЯМР (DMSO-d6, 400 МГц) δ 1,25 (6Н, d), 1,47 (9Н, s), 2,94 - 3,04 (1H, m), 5,22 (2Н, s), 7,36 - 7,50 (4Н, m), 7,85 (1H, d), 9,30 (1H, s), 10,33 (1H, s).
Стадия 7
HCl (4 M в диоксане) (598 мл, 2392,68 ммоль) добавляли к трет-бутил-(4-(2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамидо)фенил)карбамату (86 г, 239,27 ммоль) в DCM (400 мл) и МеОН (400 мл). Полученный раствор перемешивали при 25°С в течение 16 часов. Растворитель удаляли при пониженном давлении. Реакционную смесь разбавляли с помощью DCM. Реакционную смесь доводили до рН 10 с помощью насыщенного раствора Na2CO3. Осадок собирали фильтрацией, промывали водой (300 мл) и сушили под вакуумом с получением N-(4-аминофенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида (51,0 г, 82%) в виде белого твердого вещества, которое применяли без дополнительной очистки. 1H ЯМР (400 Гц, DMSO) δ: 1,24 (6Н, d), 2,96-3,02 (1H, m), 5,11-5,16 (4Н, m), 6,53 (2Н, d), 7,23 (2Н, d), 7,83 (1Н, s), 10,04 (1H, s).
Схождение
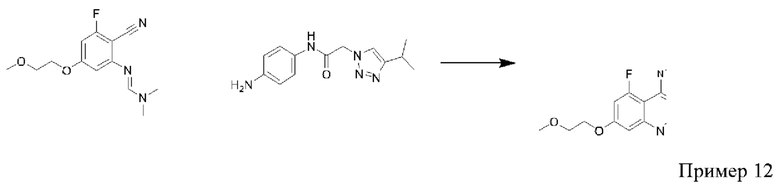
Пример 12
(Е)-N'-(2-циано-3-фтор-5-(2-метоксиэтокси)фенил)-N,N-диметилформимидамид (37,6 г, 141,91 ммоль) добавляли к N-(4-аминофенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамиду (46 г, 177,39 ммоль) в уксусной кислоте (300 мл). Полученный раствор перемешивали при 60°С в течение 40 минут.
Для выделения и очистки данную реакционную смесь объединяли с двумя другими партиями, которые синтезировали в соответствии со способом, описанным в абзаце выше.
Реакционную смесь выливали в ледяную воду. Осадок собирали фильтрацией, промывали водой (200 мл) и сушили под вакуумом с получением неочищенного продукта.
Неочищенный продукт перекристаллизовывали с помощью EtOH и сушили с получением N-(4-((5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида (45,0 г, 66,1%) в виде белого твердого вещества. 1H ЯМР (DMSO-d6, 400 МГц) δ 1,26 (6Н, d), 2,93-3,07 (1H, m), 3,33 (3Н, s), 3,68-3,75 (2Н, m), 4,24-4,32 (2Н, m), 5,29 (2Н, s), 7,05 (1H, d), 7,14 (1H, q), 7,57-7,64 (2Н, m), 7,63-7,72 (2Н, m), 7,89 (1H, d), 8,46 (1H, s), 8,98 (1H, d), 10,50 (1Н, s).
Пример 12А
Увеличение масштаба получения тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида
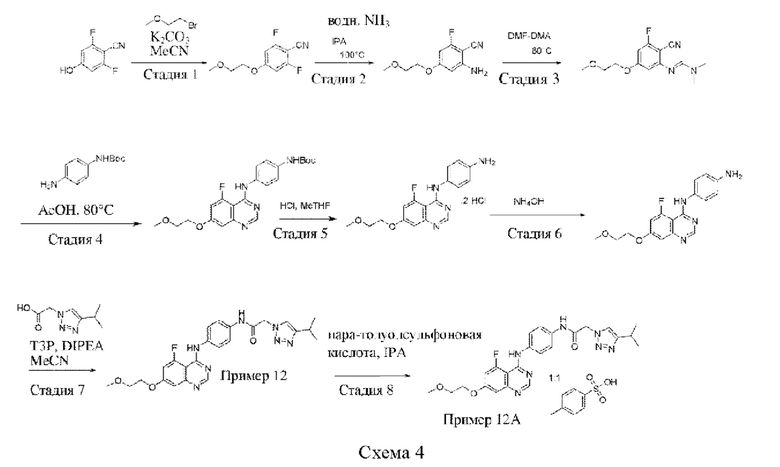
Стадия 1
1-Бром-2-метоксиэтан (9,6 кг, 69,06 моль) добавляли одной порцией к 2,6-дифтор-4-гидроксибензонитрилу (8,6 кг, 55,45 моль) и карбонату калия (23 кг, 166,43 моль) в CH3CN (70,9 кг). Полученный раствор перемешивали при 80-85°С в течение 8-10 часов.
Реакционную смесь охлаждали до 40-45°С и фильтровали. Осадок на фильтре промывали с помощью CH3CN (13,4 кг). Объединенный фильтрат концентрировали под вакуумом до 34-43 л. Добавляли производственную воду (92,0 кг) и смесь концентрировали под вакуумом до 77-85 л. Суспензию фильтровали с получением 2,6-дифтор-4-(2-метоксиэтокси)бензонитрила (13,70 кг, 100%) в виде влажного твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 3,29 (3Н, s), 3,65-3,67 (2Н, t), 4,24-4,26 (2Н, t), 7,11 (1H, s), 7,14 (1H, s)
Стадия 2
25% водный раствор аммиака (187,8 кг, 1341,43 моль) добавляли к 2,6-дифтор-4-(2-метоксиэтокси)бензонитрилу (13,7 кг, 56,68 моль, чистота: 88,2%) в iPrOH (42,6 кг) при комнатной температуре. Полученный раствор перемешивали при 100-110°С в течение 16-20 часов в автоклаве.
Твердое вещество кристаллизовали из раствора и собирали фильтрацией. Твердое вещество промывали 15% изопропанолом в воде и сушили. В результате получили 2-амино-6-фтор-4-(2-метоксиэтокси)бензонитрил (7,4 кг, 62%) в виде белого твердого вещества. 1H ЯМР (DMSO-d6, 400 МГц) δ 3,29 (3Н, s), 3,61-3,63 (2Н, t), 4,04-4,07 (2Н, t), 6,11 (1H, s), 6,16-6,19 (1H, d), 6,37 (2Н, s)
Стадия 3
DMF-DMA (16,8 кг, 140,99 моль) добавляли к 2-амино-6-фтор-4-(2-метоксиэтокси)бензонитрилу (7,4 кг, 35,20 моль) в МТВЕ (46,8 кг). Полученный раствор перемешивали при 55-60°С в течение 6-8 часов.
Реакционную смесь охлаждали до 20-30°С и промывали солевым раствором. Для удаления воды к органической фазе добавляли молекулярные сита (11,0 кг). Органический раствор концентрировали под вакуумом до 37-44 л, а затем добавляли МТВЕ с получением раствора (Е)-N'-(2-циано-3-фтор-5-(2-метоксиэтокси)фенил)-N,N-диметилформимидамида (53,25 кг, чистота: 15,9%, 91%). 1Н ЯМР (DMSO-d6, 400 МГц) δ 2,99 (3Н, s), 3,08 (3Н, s), 3,30 (3Н, s), 3,64-3,67 (2Н, t), 4,15-4,18 (2Н, t), 6,57-6,60 (2Н, m), 8,04 (1H, s)
Стадия 71
Трет-бутил-(4-аминофенил)карбамат (8,4 кг, 40,33 моль) добавляли к раствору (Е)-N'-(2-циано-3-фтор-5-(2-метоксиэтокси)фенил)-N,N-диметилформимидамида (9,0 кг, 33,92 моль) в МТВЕ (30 кг). Затем добавляли АсОН (31,6 кг). Полученный раствор перемешивали при 50-60°С в течение 3-4 часов.
Реакционную смесь охлаждали до 20-30°С и фильтровали. Для промывки осадка на фильтре применяли МТВЕ (26 кг). Твердое вещество сушили с получением трет-бутил-(4-((5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил)амино)фенил)карбамата (13,10 кг, чистота: 87,5%, 79%). 1Н ЯМР (DMSO-d6, 400 МГц) δ 1,49 (9Н, s), 3,3 (3Н, s), 3,70-3,72 (2Н, t), 4,27-4,29 (2Н, t), 7,04 (1Н, s), 7,11-7,15 (1H, dd), 7,43-7,56 (4Н, m), 8,42 (1H, s), 8,90-8,93 (1H, d), 9,35 (1H, s)
Стадия 5
Газообразный HCl (85 кг, 2328,77 моль) добавляли к трет-бутил-(4-((5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил)амино)фенил)карбамату (10,2 кг, 23,80 моль) в 2-MeTHF (160 кг) и воде (10 кг). Полученный раствор перемешивали при 20-25°С в течение 4-6 часов.
Реакционную смесь фильтровали и промывали с помощью 2-MeTHF (40 кг). Твердое вещество сушили с получением дигидрохлорида N1-(5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил)бензол-1,4-диамина (9,10 кг, 95%). 1H ЯМР (DMSO-d6, 400 МГц) δ 3,33 (3Н, s), 3,72-3,74 (2Н, t), 4,30-4,35 (2Н, t), 7,29-7,62 (6Н, m), 8,79 (1H, s), 10,57 (2Н, brs)
Стадия 6
25% водный раствор аммиака (27 кг, 192,86 моль) добавляли к дигидрохлориду N1-(5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил)бензол-1,4-диамина (9,10 кг, 22,68 моль) в воде (150 кг). Полученный раствор перемешивали при 20-25°С в течение 2-3 часов.
Реакционную смесь фильтровали и промывали водой (60 кг). Твердое вещество сушили с получением N1-(5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил)бензол-1,4-диамина (6,15 кг, 83%). 1H ЯМР (DMSO-d6, 400 МГц) δ 3,3 (3Н, s), 3,70-3,72 (2Н, t), 4,25-4,29 (2Н, t), 5,07 (2Н, s), 6,56-6,59 (2Н, d), 7,00 (1Н, s), 7,09-7,10 (1H, d), 7,23-7,25 (2Н, d), 8,35 (1H, s), 8,69-8,72 (1H, d)
Стадия 7
N-[5-фтор-7-(2-метоксиэтокси)-4-хиназолинил]-1,4-бензолдиамин (4,184 кг, 12,5 моль) загружали с [4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]уксусной кислотой (2,41 кг, 13,8 моль) и ацетонитрилом (62 л) и нагревали до 65°С. N-этилдиизопропиламин (4,36 л, 25,0 моль) загружали с 50% раствором 1-пропанфосфонового ангидрида в ацетонитриле (9,96 кг, 16,3 моль) при <80°С. Смесь перемешивали при 65°С и затем охлаждали до 10-20°С и добавляли 2-пропанол (20,5 л). Затем смесь охлаждали до 0-10°С перед проведением фильтрации, промывки ацетонитрилом (2×10 л) и затем высушивания с получением N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (5,530 кг, 91,1%) в виде грязно-белого твердого вещества. 1H ЯМР (500 МГц, DMSO-d6) δ 1,2 (d, 6Н) 3,0 (spt, 1Н) 3,3 (s, 3Н) 3,7-3,7 (m, 2Н) 4,2-4,3 (m, 2Н) 5,3 (s, 2Н) 7,0 (d, 1Н) 7,1 (dd, 1Н) 7,5-7,6 (m, 2Н) 7,6-7,7 (m, 2Н) 7,9 (s, 1Н) 8,4 (s, 1Н) 9,0 (d, 1Н) 10,5 (s, 1Н)
Стадия 8
N-(4-{[5-Фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамид (2 кг, 4,13 моль) и пара-толуолсульфоновую кислоту (0,842 кг, 4,34 моль) нагревали при встряхивании в смеси пропан-2-ола (26 л, 340 моль) и диметилсульфоксида (6,5 л, 92 моль), пока не образовывался раствор. Полученный раствор фильтровали горячем состоянии и фильтрат охлаждали и вводили затравку из тозилата N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида, при этом затравку получали в соответствии со способом, описанным ниже под заголовком "форма А тозилатной соли Y". Полученный тозилат N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида фильтровали при пониженном давлении и осадок на фильтре промывали смесью пропан-2-ола (0,4 л, 5 моль) и диметилсульфоксида (0,1 л, 1 моль). После удаления жидкости из осадка на фильтре и конечной промывки осадка на фильтре с помощью пропан-2-ола твердые вещества сушили в вакуумной печи с получением тозилата N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (2,458 кг, 91,2%) в виде ярко-желтого твердого вещества. 1H ЯМР (500 МГц, DMSO-d6) δ 1,2 (d, 6Н) 2,3 (s, 3Н) 3,0 (spt, 1Н) 3,3 (s, 3Н) 3,7-3,8 (m, 2Н) 4,3-4,4 (m, 2Н) 5,3 (s, 2Н) 7,0-7,1 (m, 3Н) 7,4-7,6 (m, 5Н) 7,6-7,7 (m, 2Н) 7,9 (s, 1Н) 8,8 (s, 1Н) 10,5 (br d, 1Н) 10,6 (s, 1Н)
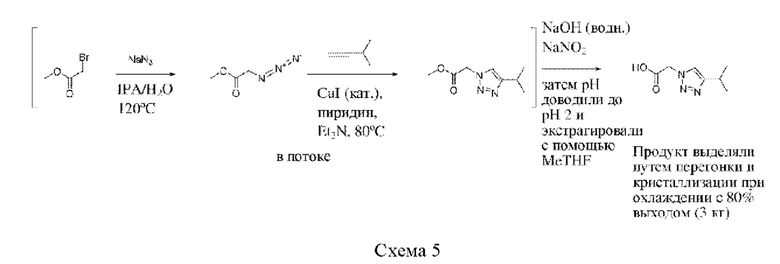
Схема 5
[4-(Пропан-2-ил)-1Н-1,2,3-триазол-1-ил]уксусную кислоту, используемую на стадии 7 схемы 4, получали с помощью способа, изображенного на схеме 5. Проводили реакцию раствора метил-2-бромацетата (5,4 кг, 36 моль) в 2-пропаноле (19 л) и азида натрия (2,3 кг, 35 моль) в воде (23 л) в проточном реакторе Alfa Laval при 120°С со временем удержания 40 секунд, при этом реакцию отслеживали с помощью PAT (IR и RAMAN). Затем проводили дальнейшую реакцию выходящего материала с подаваемым раствором, содержащим 3-метилбут-1-ин (3,1 кг, 44 моль) с йодидом меди (0,21 кг) и триметиламином (1,1 кг) в пиридине (8,1 л), при 80°С со временем удерживания 152 секунды. Выходящий продукт (отслеживали с помощью PAT IR) собирали во взбалтываемый сосуд вместе с подаваемым раствором, содержащим эдетат динатрия (2,9 кг), нитрит натрия (0,14 кг) и гидроксид натрия (5 кг, 58 моль) в воде (7,95 л). Полученную суспензию фильтровали при пониженном давлении. К фильтрату добавляли трет-бутилметиловый эфир (45 л) и смесь взбалтывали в течение 30 минут. Взбалтывание прекращали, а слоям давали разделиться и водный слой собирали. Водный слой подкисляли путем медленного добавления водного раствора серной кислоты (3,5 М) со взбалтыванием, поддерживая температуру при <26°С, пока не будет достигнут рН 2,5. Затем добавляли 2-метилтетрагидрофуран (32,5 л) и смесь взбалтывали в течение дополнительных 10 минут. Взбалтывание прекращали, а слоям давали разделиться. Собирали органический слой. К полученному водному слою добавляли 2-метилтетрагидрофуран (33 л) и смесь взбалтывали в течение 10 минут. Взбалтывание прекращали, а слоям давали разделиться и органический слой собирали. Полученный водный слой доводили до рН 2,5 с помощью 3,5 М HCl со взбалтыванием и добавляли 2-метилтетрагидрофуран (16 л) и смесь взбалтывали в течение 10 минут. Взбалтывание прекращали, а слоям давали разделиться. Органический слой собирали и объединяли с ранее собранными 2-метилтетрагидрофурановыми органическими экстрактами. Объединенные органические экстракты перегоняли при атмосферном давлении с удалением 15 относительных объемов (48,6 л) органических веществ. К оставшимся органическим веществам дополнительно добавляли 2-метилтетрагидрофуран (33 л) и смесь дистиллировали при атмосферном давлении с удалением дополнительных 12 относительных объемов (40 л) органических веществ. Оставшиеся органические вещества охлаждали до 70°С со взбалтыванием, затем охлаждали с линейным изменение температуры до 0°С на протяжении 4 часов. Трет-бутилметиловый эфир (13 л) добавляли в течение 20 минут и смесь взбалтывали при 0°С в течение 3 часов. Полученную суспензию фильтровали при пониженном давлении. Осадок на фильтре промывали предварительно охлажденным (0°С) трет-бутилметиловым эфиром (13 л) и осадок на фильтре собирали для сушки. Полученные твердые вещества сушили в вакуумной печи (40°С) с получением [4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]уксусной кислоты в виде белого кристаллического твердого вещества (6,2 кг, 40%). 1H ЯМР (DSMO-d6, 500 МГц): 13,3 (1H, br s, ОН) 7,82 (1Н, s, 5-Н), 5,21 (2Н, s, CH2CO2H), 2,99 (1H, гептет, J=5 Гц, СНМе2), 1,24 (6Н, d, J=5 Гц, 2 х Me). 13С ЯМР (DMSO-d6, 125 МГц): 168,9 (С=O), 153,1 (С, С-4), 121,9 (СН, С-5), 50,45 (СН2, CH2CO2H), 23,32 (СН, СНМе2), 22,51 (СН3, 2 х Me). UPLC MS (BEH/MeCN/TFA): Rt = 0,61 мин (λ макс. = 221,1). Масс-спектр: 170,06 (М+Н), 154,05 (М - СН3), 125,9 (М - CO2), 112,0 (М - CH2CO2H).
Форма А тозилатной соли Y
Чистое свободное основание N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида и моногидрат пара-толуолсульфоновой кислоты растворяли в 16 относительных объемах 20% диметилсульфоксида:пропан-2-ола при 20°С. В сосуд добавляли затравку формы А тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (0,02%) и содержимое сосуда нагревали до 75°С в течение минимум 60 минут. Полученный раствор процеживали в кристаллизатор при 75°С через встроенный фильтр. Полученный раствор охлаждали до 40°С в течение 20 минут и в реакционный сосуд по каплям добавляли пропан-2-ол (6,5 относительного объема) в качестве антирастворителя с доведением композиции растворителя до 15% диметилсульфоксида:пропан-2-ола. В кристаллизатор снова вводили затравку из формы А тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида (0,06%), при этом взвесь поддерживали при 40°С. Взвесь продолжали перемешивать при 40°С в течение 3 дней. Для проверки полиморфной формы проводили XRPD-анализ. Полученную взвесь фильтровали, промывали 15% диметилсульфоксидом:пропан-2-олом и твердое вещество сушили в вакуумной печи при 40°С, пока не получали постоянную массу. Из полученного порошка отбирали образцы для проведения XRPD и HPLC для проверки полиморфной формы и чистоты, соответственно, и также определяли потерю жидкости.
При некоторых обстоятельствах может потребоваться дополнительная стадия, которая предусматривает суспендирование материала в изопропилацетате, введение затравки из формы А тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида и образование взвеси при 80°С в течение 24 часов. Полученный материал анализируют с помощью XRPD для подтверждения образования формы А, которую выделяют и сушат, как описано выше. Форма А характеризуется по меньшей мере одним пиком при значении 20, составляющем 13,4° и 14,3° согласно измерениям с применением излучения CuKα. Десять наиболее выраженных пиков XRPD показаны в таблице С.
Затравка из формы А тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида
Затравку, применяемую в способе, описанном выше для формы А тозилатной соли Y, получали следующим образом. Чистое свободное основание N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида вносили в бутыль Duran и в бутыль добавляли при перемешивании 20 относительных объемов этилацетата для получения взвеси. Раствор моногидрата пара-толуолсульфоновой кислоты (1,1 эквивалента) получали путем его полного растворения в 40 относительных объемах этилацетата. Раствор пара-толуолсульфоновой кислоты по каплям добавляли к раствору API при энергичном перемешивании. Полученный желтый материал подвергали образованию взвеси в условиях окружающей среды в течение 5 дней в закрытой бутыли Duran. Полученный материал проверяли с помощью XRPD для демонстрации образования высококристаллического материала, который соответствует 1:1 тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида согласно анализу с помощью помощи ЯМР, DSC и идентификации кристаллической структуры посредством рентгенографии. Твердое вещество выделяли с помощью вакуумной фильтрации и сушили в вакуумной печи.

Форма В тозилатной соли Y
Форма В тозилатной соли Y представляет собой гидратированную форму, полученную в экспериментах по увеличению масштаба получения соли в этаноле при добавлении затравки из формы С тозилатной соли Y.
Форму В получали, воздействуя на форму С тозилатной соли Y высокими уровнями влажности, как, например, в эксперименте DVS. В качестве альтернативы форму В получали во время эксперимента увеличению масштаба получения соли в этаноле. Чистое свободное основание N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида и моногидрат пара-толуолсульфоновой кислоты добавляли в 1-литровый стеклянный реактор, оснащенный мешалкой с верхним приводом. В реактор добавляли десять относительных объемов этанола при 20°С. Содержимое сосуда нагревали до 75°С в течение 2 часов с получением прозрачного желтого раствора. Содержимое реактора охлаждали до 60°С в течение 20 минут. 1% смеси форм С и В, полученной в способе получения формы С, описанного в разделе ниже, добавляли в реактор при перемешивании. Содержимое выдерживали при 60°С в течение 5 часов с последующим охлаждением до 5°С в течение 10 часов. Твердое вещество выделяли при 5°С посредством вакуумной фильтрации, промывали охлажденным этанолом и сушили в вакуумной печи при 20°С на протяжении ночи с последующим периодом высушивания в течение 25 минут при 40°С. Полученное твердое вещество анализировали с помощью XRPD и получали кристаллическую форму, называемую формой В. Форма В характеризуется по меньшей мере одним пиком при значении 20, составляющим 4,2° и 7,7° согласно измерениям с применением излучения CuKα. Десять наиболее выраженных пиков XRPD показаны в таблице D.

Форма С тозилатной соли Y
Форма С тозилатной соли Y представляет собой форму этанольного сольвата, полученную в экспериментах по измерению растворимости в этаноле и ацетонитриле.
Форму С получали во время экспериментов по измерению растворимости на основе формы А тозилатной соли Y. Кривые растворимости получали в этаноле и ацетонитриле. Во флаконы вносили образцы формы А различной массы и перемешивали в этаноле и ацетонитриле. Устанавливали нагревание с линейным изменением температуры от 25°С до 75°С при скорости 0,03°С/минуту. Все материалы растворялись при повышенных температурах. Растворы вновь охлаждали до 25°С при скорости 0,03°С/минута и твердое вещество перекристаллизовывалось во флаконах в различные моменты цикла охлаждения. Полученные твердые вещества анализировали с помощью XRPD в виде взвесей, и они обеспечивали кристаллическую форму, называемую формой С. Форма С не является стабильной формой и будет превращаться в форму В как во время вакуумной сушки, так и в условиях окружающей среды. Следовательно, обычно выделяют смесь форм С и В, которая в конечном счете превратится в форму В вследствие испарения этанола из кристаллической структуры. Выделенную смесь форм С и В можно использовать в качестве затравки в способе для получения формы В, описанном в разделе выше.
Форма D тозилатной соли Y
Форма D тозилатной соли Y представляет собой форму диметилсульфоксидного сольвата, полученную в экспериментах по увеличению масштаба получения соли в 20% диметилсульфоксиде:пропан-2-оле.
Форму D получали путем кристаллизации тозилатной соли N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида из насыщенного раствора в 20% диметилсульфоксиде:пропан-2-оле. Исходно система растворителей, применяемая для получения формы А, представляла собой 20% диметилсульфоксид:пропан-2-ол. Позже стало понятно, что перекристаллизация из исходного раствора после изначального выделения формы А в приведенной выше системе растворителей приводила к получению формы D. Поэтому количество диметилсульфоксида в смеси растворителей для получения формы А уменьшали с 20% до 15%, чтобы гарантировать получение формы А. Форму А получали при температуре 40°С, в то время как форму D получали при температурах ниже 25°С. Форма D характеризуется по меньшей мере одним пиком при значении 20, составляющем 4,4° и 5,6° согласно измерениям с применением излучения CuKα. Десять наиболее выраженных пиков XRPD показаны в таблице Е.

Пример 13 и пример 14
(R)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамид и (S)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамид
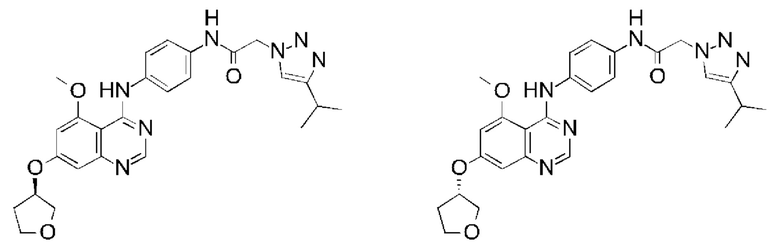
Трет-бутоксид калия (150 мг, 1,4 ммоль) добавляли к тетрагидрофуран-3-олу (121 мг, 1,4 ммоль) и N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (300 мг, 0,7 ммоль) в DMF (3 мл) при 25°С в атмосфере азота. Полученную смесь перемешивали при 80°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали с получением неочищенного остатка. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 10% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением рацемической формы указанного в заголовке соединения в виде бледно-желтого твердого вещества (150 мг, 43%). Рацемический продукт очищали с помощью препаративной хиральной HPLC на колонке Chiralpak IB при изократическом элюировании с помощью 30% изопропанола в ТВМЕ (модифицированном с помощью 0,1% DEA) в качестве элюента. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением одного энантиомера 2-(4-изопропил-1H-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамида в виде бледно-желтого твердого вещества (52 мг, 15%, >99,9% э.и.). 1H ЯМР (300 МГц, DMSO-d6) δ 1,25 (6Н, d), 1,97-2,11 (1H, m), 2,27-2,41 (1H, m), 2,94-3,06 (1H, m), 3,75-4,00 (4Н, m), 4,11 (3Н, s), 5,22-5,28 (1H, m), 5,29 (2Н, s), 6,80-6,84 (2Н, m), 7,61 (2Н, d), 7,67 (2Н, d), 7,88 (1H, s), 8,62 (1Н, s), 10,43 (1H, s), 10,65 (1H, s); масса/заряд (ES+), [М+Н]+ = 504; кислота, HPLC Rt = 1,35 мин. За ним следовал другой энантиомер 2-(4-изопропил-1H-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамида в виде бледно-желтого твердого вещества (51 мг, 15%, 98,7% э.и.). 1Н ЯМР (300 МГц, DMSO-d6) δ 1,25 (6Н, d), 1,96-2,10 (1H, m), 2,25-2,39 (1H, m), 2,94-3,06 (1H, m), 3,74-3,98 (4Н, m), 4,08 (3Н, s), 5,20-5,26 (1H, m), 5,27 (2Н, s), 6,67 (1Н, d), 6,75 (1H, d), 7,60 (2Н, d), 7,77 (2Н, d), 7,88 (1Н, s), 8,41 (1H, s), 9,80 (1H, s), 10,49 (1H, s); масса/заряд (ES+), [М+Н]+ = 504; кислота, HPLC Rt = 1,35 мин.
Пример 15
N-(4-{[5-метокси-7-(пропан-2-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид

Гидрид натрия (41 мг, 1 ммоль) добавляли к N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (150 мг, 0,3 ммоль) и изопропанолу (62,1 мг, 1 ммоль) в DMF (1,5 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Полученный раствор перемешивали при 80°С в течение 7 часов. Реакционную смесь фильтровали. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (72 мг, 44%). 1Н ЯМР (300 МГц, DMSO-d6) δ 1,26 (6Н, d), 1,35 (6Н, d), 2,94-3,07 (1H, m), 4,08 (3Н, s), 4,77-4,90 (1H, m), 5,27 (2Н, s), 6,64 (1H, d), 6,77 (1Н, d), 7,60 (2Н, d), 7,78 (2Н, d), 7,88 (1Н, s), 8,40 (1H, s), 9,77 (1H, s), 10,49 (1H, s)
масса/заряд (ES+), [М+Н]+ = 476; кислота, HPLC Rt = 6,90 мин.
Пример 16
N-(4-{[5-метокси-7-(оксетан-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
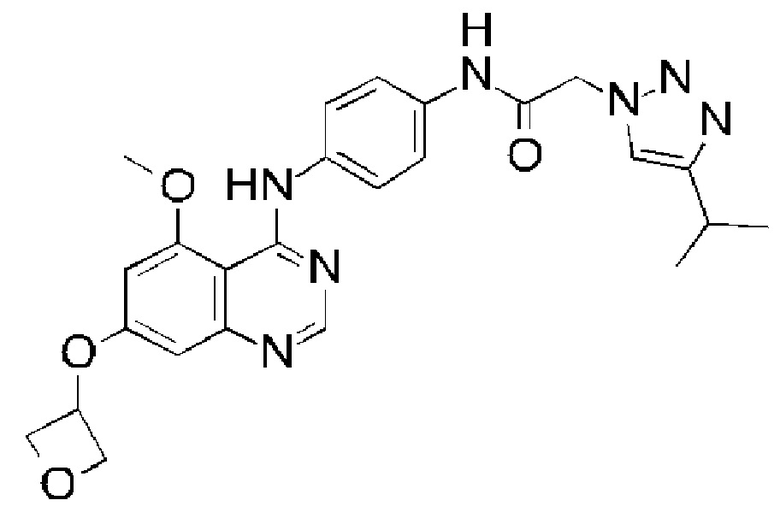
Трет-бутоксид калия (52 мг, 0,5 ммоль) добавляли к оксетан-3-олу (34 мг, 0,5 ммоль) и N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (100 мг, 0,2 ммоль) в DMF (0,5 мл) при 25°С в атмосфере воздуха. Полученную смесь перемешивали при 80°С в течение 5 часов. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения (42 мг, 37%) в виде грязно-белого твердого вещества. 1H ЯМР (300 МГц, DMSO, 23°С) δ 1,25 (6Н, d), 2,94-3,06 (1H, m), 4,10 (3Н, s), 4,59 (2Н, t), 5,00 (2Н, t), 5,26 (2Н, s), 5,42-5,50 (1H, m), 6,45 (1H, s), 6,72 (1H, s), 7,58-7,60 (2Н, m), 7,75-7,77 (2Н, m), 7,87 (1H, s), 8,40 (1Н, s), 9,78 (1H, s), 10,48 (1Н, s). масса/заряд (ES+), [М+Н]+ = 490; TFA, HPLC Rt = 2,319 мин.
Пример 17
N-[4-({7-[2-(диметиламино)этокси]-5-метоксихиназолин-4-ил}амино)фенил]-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамид
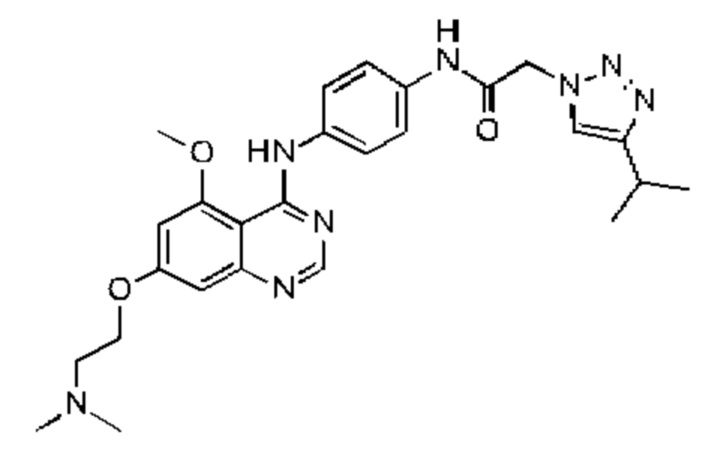
Трет-бутоксид калия (52 мг, 0,5 ммоль) добавляли к N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (100 мг, 0,2 ммоль) и 2-(диметиламино)этан-1-олу (31 мг, 0,3 ммоль) в DMF (1 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при 100°С в течение 3 часов. Реакционную смесь очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 10% метанола (1 М NH3) в DCM. Чистые фракции выпаривали до сухого состояния с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (36 мг, 31%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,25 (6Н, d), 2,24 (6Н, s), 2,67 (2Н, t), 2,92-3,06 (1H, m), 4,08 (3Н, s), 4,19 (2Н, t), 5,27 (2Н, s), 6,68 (1Н, d), 6,79 (1H, d), 7,59 (2Н, d), 7,78 (2Н, d), 7,88 (1H, d), 8,40 (1H, s), 9,78 (1H, s), 10,48 (1Н, s); масса/заряд (ES+), [М+Н]+ = 505; кислота, HPLC Rt = 1,12 мин.
Пример 18
N-{4-[(7-этокси-5-метоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
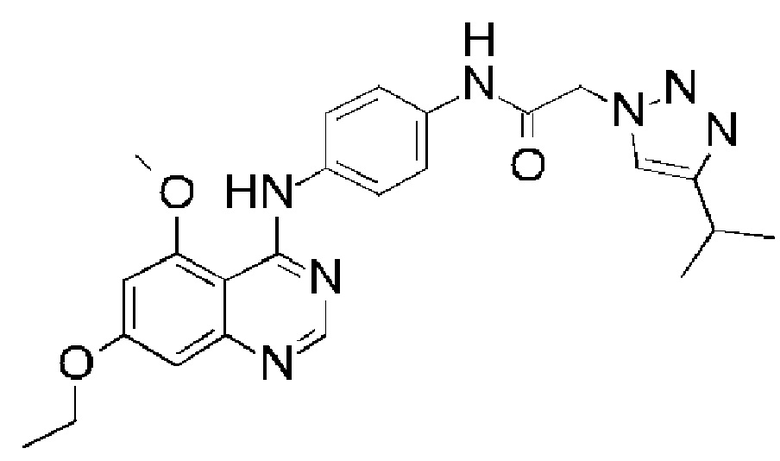
Этоксид натрия (в этаноле) (66 мг, 1 ммоль) добавляли к N-(4-((7-фтор-5-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамиду (85 мг, 0,2 ммоль) в DMF (5 мл) при комнатной температуре. Полученный раствор перемешивали при 80°С в течение 3 часов. Реакционную смесь выливали в насыщенный водный раствор NH4Cl (100 мл). Смесь фильтровали и неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (30 мг, 33%). 1H ЯМР (DMSO-d6, 400 МГц) δ 1,26 (6Н, d), 1,40 (3Н, t), 2,93-3,08 (1H, m), 4,08 (3Н, s), 4,18 (2Н, d), 5,27 (2Н, s), 6,73 (2Н, dd), 7,60 (2Н, d), 7,78 (2Н, d), 7,88 (1H, s), 8,41 (1H, s), 9,78 (1H, s), 10,49 (1H, s); масса/заряд (ES+), [М+Н]+ = 462; кислота, HPLC Rt = 1,406 мин.
Пример 19
N-{4-[(5,7-диэтоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамид
HATU (140 мг, 0,4 ммоль) добавляли к 4-((5,7-диэтоксихиназолин-4-ил)окси)анилину (260 мг, 0,3 ммоль), 2-(4-изопропил-1Н-1,2,3-триазол-1-ил)уксусной кислоте (79 мг, 0,3 ммоль) и DIPEA (0,12 мл, 0,7 ммоль) в DMF (3 мл) при 23°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 8% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением неочищенного продукта в виде желтого твердого вещества. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (97 мг, 61%). 1Н ЯМР (400 МГц, DMSO-d6) δ 1,25 (6Н, d), 1,39 (3Н, t), 1,57 (3Н, t), 2,95-3,06 (1H, m), 4,11-4,24 (2Н, m), 4,26-4,40 (2Н, m), 5,27 (2Н, s), 6,68 (1H, d), 6,76 (1H, d), 7,61 (2Н, d), 7,79 (2Н, d), 7,88 (1H, s), 8,44 (1H, s), 9,97 (1H, s), 10,51 (1H, s); масса/заряд (ES+), [М+Н]+ = 476; кислота, HPLC Rt = 1,48 мин.
Промежуточные соединения, применяемые в примере 19, получали следующим образом.
Получение 5,7-диэтоксихиназолин-4(3H)-она
Этоксид натрия (2,6 г, 7,9 ммоль) добавляли к 7-фтор-5-метоксихиназолин-4(3H)-ону (1,4 г, 7,2 ммоль) в DMF (15 мл) в атмосфере азота. Полученный раствор перемешивали при 80°С в течение 5 часов. Реакционную смесь выливали в ледяную воду. Полученный осадок собирали фильтрацией, промывали холодной водой (100 мл), эфиром (50 мл) и сушили под вакуумом с получением твердого вещества. Неочищенный продукт очищали с помощью препаративной HPLC. Фракции, содержащие необходимое соединение, выпаривали до сухого состояния с получением вначале 7-этокси-5-метоксихиназолин-4(3H)-она (0,5 г, 29%), 1Н ЯМР (DMSO-d6, 300 МГц) δ 1,38 (3Н, t), 3,83 (3Н, s), 4,16 (2Н, q), 6,53 (1H, d), 6,65 (1H, d), 7,92 (1H, s), 11,78 (1H, s), а затем 5,7-диэтоксихиназолин-4(3H)-она в виде белого твердого вещества (0,2 г, 9%). 1H ЯМР (DMSO-d6, 300 МГц) δ 1,35 (6Н, t), 4,10 (4Н, dq), 6,49 (1H, d), 6,61 (1Н, d), 7,88 (1Н, s), 11,71 (1H, s).
Получение N1-(5,7-диэтоксихиназолин-4-ил)бензол-1,4-диамина
DBU (0,27 мл, 1,8 ммоль) добавляли к 5,7-диэтоксихиназолин-4(3H)-ону (160 мг, 0,7 ммоль) и ВОР (393 мг, 0,9 ммоль) в ацетонитриле (5 мл) при комнатной температуре в атмосфере азота. Полученный раствор перемешивали при 60°С в течение 1 часа. Добавляли пара-фенилендиамин (148 мг, 1,4 ммоль). Полученный раствор перемешивали при 60°С в течение 5 часов. Реакционную смесь концентрировали и разбавляли с помощью DCM (100 мл) и последовательно промывали насыщенным водным раствором NH4Cl (10 мл), водой (10 мл) и насыщенным раствором NaHCO3 (10 мл). Органический слой сушили, фильтровали и выпаривали с получением неочищенного продукта. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 4% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде желтого твердого вещества (280 мг, >100%). 1H ЯМР (400 МГц, DMSO-d6) δ 1,38 (3Н, t), 1,54 (3Н, t), 4,12-4,18 (2Н, m), 4,24-4,37 (2Н, m), 5,60 (2Н, s), 6,58 (1H, s), 6,61 (2Н, d), 6,70 (1H, s), 7,39 (2Н, d), 8,33 (1H, s), 9,68 (1H, s); масса/заряд (ES+), [М+Н]+ = 325; основание, HPLC Rt = 0,83 мин.
Пример 20
N-(4-{[5-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
HATU (10 мг, 0,03 ммоль) добавляли к АТ-(5-метокси-7-(2-метоксиэтокси)хиназолин-4-ил)бензол-1,4-диамину (1,5 г, 4,4 ммоль), 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоте (1,1 г, 4,9 ммоль) и DIPEA (1,1 г, 8,8 ммоль) в DMF (30 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой. Полученный осадок собирали фильтрацией, промывали ацетонитрилом/H2O (1:10, 50 мл) и сушили под вакуумом. Неочищенный продукт очищали посредством кристаллизации из ацетонитрила с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,5 г, 70%). 1H ЯМР (400 МГц, DMSO-d6) δ 1,25 (6Н, d), 2,93-3,07 (1H, m), 3,33 (3Н, s), 3,65-3,77 (2Н, m), 4,09 (3Н, s), 4,20-4,30 (2Н, m), 5,26 (2Н, s), 6,72 (1H, d), 6,78 (1Н, d), 7,59 (2Н, d), 7,77 (2Н, d), 7,87 (1H, s), 8,41 (1Н, s), 9,79 (1H, s), 10,48 (1H, s); масса/заряд (ES+), [М+Н]+ = 492; кислота, HPLC Rt = 1,44 мин.
Промежуточные соединения, применяемые в примере 20, получали следующим образом.
Получение 5-метокси-7-(2-метоксиэтокси)хиназолин-4(3H)-она
Трет-бутоксид калия (4,3 г, 38,6 ммоль) добавляли к 7-фтор-5-метоксихиназолин-4(3H)-ону (3 г, 15,5 ммоль) и 2-метоксиэтан-1-олу (1,76 г, 23,2 ммоль) в DMSO (30 мл) в атмосфере азота. Полученный раствор перемешивали при 80°С в течение 15 часов. Реакционную смесь нейтрализовали с помощью 2 М HCl. Объединенный водный слой сушили с помощью лиофилизации. Реакционную смесь разбавляли с помощью DCM (30 мл) и ТВМЕ (70 мл). Полученный осадок собирали фильтрацией, промывали с помощью DCM (10 мл) и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (2,2 г, 57%), которое применяли без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 3,66-3,77 (2Н, m), 3,88 (3Н, s), 4,19-4,32 (2Н, m), 6,71 (1H, d), 6,81 (1Н, d), 8,74 (1Н, s); масса/заряд (ES+), [М+Н]+ = 251; Основание, HPLC Rt = 0,46 мин.
Получение гидрохлорида N1-(5-метокси-7-(2-метоксиэтокси)хиназолин-4-ил)бензол-1,4-диамина
РуАОР (5,5 г, 10,6 ммоль) добавляли к 5-метокси-7-(2-метоксиэтокси)хиназолин-4(3H)-ону (2,2 г, 8,8 ммоль) и DBU (2,7 мл, 17,6 ммоль) в ацетонитриле (100 мл) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 5 минут. К нему добавляли бензол-1,4-диамин (1,5 г, 14,1 ммоль) при 25°С в атмосфере азота. Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали и разбавляли с помощью DCM (500 мл) и последовательно промывали насыщенным раствором NaHCO3 (100 мл) и насыщенным водным раствором NH4Cl (100 мл). Органический слой сушили, фильтровали и выпаривали с получением неочищенного продукта. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 4% метанола в DCM. Чистые фракции выпаривали до сухого состояния с получением неочищенного продукта в виде бледно-желтого масла. Реакционную смесь разбавляли с помощью HCl (4Mb диоксане, 2 мл) и ацетона (10 мл). Осадок собирали фильтрацией, промывали ацетоном (10 мл) и сушили под вакуумом с получением указанного в заголовке соединения в виде коричневого твердого вещества. Твердое вещество растворяли в DCM (300 мл) и последовательно промывали насыщенным раствором NaHCO3 (50 мл). Органический слой сушили, фильтровали и выпаривали с получением указанного в заголовке соединения в виде желтого твердого вещества (1,2 г, 40%). 1H ЯМР (400 МГц, DMSO-d6) δ 3,33 (3Н, s), 3,67-3,74 (2Н, m), 4,05 (3Н, s), 4,20-4,26 (2Н, m), 4,98 (2Н, s), 6,58 (2Н, d), 6,65 (1H, d), 6,72 (1H, d), 7,33 (2Н, d), 8,28 (1Н, s), 9,47 (1H, s); масса/заряд (ES+), [М+Н]+ = 341; Основание, HPLC Rt = 0,7 мин.
Пример 21
N-{4-[(5-фтор-7-метоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
HATU (0,3 г, 0,8 ммоль) добавляли к раствору 1H-(5-фтор-7-метоксихиназолин-4-ил)бензол-1,4-диамина (0,2 г, 0,7 ммоль) и DIPEA (0,5 мл, 2,7 ммоль) в DMF (5 мл). Смесь перемешивали при температуре окружающей среды в течение 6 часов. Реакционную смесь выливали в воду (40 мл) и перемешивали в течение 5 минут. Добавляли этилацетат (30 мл) и органический слой отделяли. Водный слой экстрагировали с помощью этилацетата (30 мл) и экстракты объединяли и промывали насыщенным солевым раствором (2×30 мл) и выпаривали до сухого состояния. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния при элюировании с помощью градиента 0-10% (10:1 этилацетат : метанол с 1% NH3) в этилацетате. Подходящие фракции выпаривали до сухого состояния с получением указанного в заголовке соединения в виде белого твердого вещества (70 мг, 25%). 1Н ЯМР (500 МГц, DMSO, 27°С) δ 1,24 (3Н, s), 1,25 (3Н, s), 2,99 (1H, гептет), 3,92 (3Н, s), 5,26 (2Н, s), 7,04 (1H, d), 7,11 (1H, dd), 7,55-7,62 (2Н, m), 7,63-7,7 (2Н, m), 7,86 (1Н, d), 8,45 (1H, s), 8,95 (1H, d), 10,48 (1Н, s); масса/заряд: ES+ [М+Н]+ 436.
Промежуточные соединения, применяемые в примере 21, получали следующим образом.
Получение 2-амино-6-фтор-4-метоксибензонитрила
Гидроксид аммония (5,2 мл, 41,4 ммоль) вносили во флакон для микроволновой обработки, содержащий 2,6-дифтор-4-метоксибензонитрил (1 г, 5,9 ммоль) в изопропаноле (1 мл). Полученный раствор герметизировали и перемешивали при 80°С в течение 16 часов. Реакционную смесь концентрировали и добавляли этилацетат (75 мл). Органический слой отделяли и промывали насыщенным солевым раствором (10 мл), затем выпаривали с получением указанного в заголовке соединения (0,8 г, 86%), которое применяли без дополнительной очистки. 1H ЯМР (500 МГц, DMSO, 27°С) δ 3,72 (3Н, s), 6,11 (1H, dd), 6,15 (1H, dd), 6,36 (2Н, s); масса/заряд: ES- [М-Н]- 165.
Получение (E)-N'-(2-циано-3-фтор-5-метоксифенил)-N,N-диметилформимидамида
1,1-Диметокси-N,N-диметилметанамин (6,9 мл, 51,8 ммоль) добавляли к 2-амино-6-фтор-4-метоксибензонитрилу (0,9 г, 5,2 ммоль) при 25°С. Полученную взвесь перемешивали при 80°С в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли воду (10 мл) и реакционную смесь перемешивали в течение 1 часа. Образовавшийся осадок собирали фильтрацией с получением указанного в заголовке соединения в виде розового твердого вещества (0,8 г, 72%), которое применяли без дополнительной очистки. 1H ЯМР (500 МГц, DMSO, 27°С) δ 2,98 (3Н, s), 3,07 (3Н, s), 3,81 (3Н, s), 6,53 - 6,59 (2Н, m), 8,01 (1H, s); масса/заряд: ES+ [М+Н]+ 222.
Получение 5-фтор-7-метокси-N-(4-нитрофенил)хиназолин-4-амина
4-Нитроанилин (259 мг, 1,9 ммоль) добавляли к (E)-N'-(2-циано-3-фтор-5-метоксифенил)-N,N-диметилформимидамиду (277 мг, 1,3 ммоль) в уксусной кислоте (7 мл). Полученный раствор перемешивали при 120°С в течение 2 часов. При охлаждении реакционная смесь твердела. Добавляли эфир (15 мл) и обеспечивали получение взвеси твердого вещества в течение 10 минут. Полученное желтое твердое вещество собирали фильтрацией, промывали дополнительным количеством эфира и сушили под вакуумом с получением указанного в заголовке соединения в виде рыжевато-коричневого твердого вещества (210 мг, 53%), которое применяли без дополнительной очистки. 1Н ЯМР (500 МГц, DMSO, 27°С) δ 3,95 (3Н, s), 7,14 (1Н, d), 7,22 (1H, dd), 8,08 (2Н, d), 8,21-8,29 (2Н, m), 8,66 (1H, s), 9,52 (1H, s); масса/заряд: ES+ [М+Н]+ 315.
Получение N4-(5-фтор-7-метокси-хиназолин-4-ил)бензол-1,4-диамина
5-Фтор-7-метокси-N-(4-нитрофенил)хиназолин-4-амин (0,2 г, 0,7 ммоль) в DMF (8 мл) добавляли к 10% палладию на угле (0,07 г, 0,1 ммоль) в атмосфере азота. Обеспечивали перемешивание реакционной смеси в течение 24 часов в атмосфере водорода. Реакционную смесь фильтровали через целит, промывали этилацетатом (30 мл) и полученные жидкости концентрировали с получением указанного в заголовке соединения в виде желтой камеди (190 мг, 100%). 1Н ЯМР (500 МГц, DMSO, 27°С) δ 3,90 (3Н, s), 5,01 (2Н, s), 6,53-6,60 (2Н, m), 6,99 (1H, d), 7,05 (1H, dd), 7,20-7,27 (2Н, m), 8,34 (1H, s), 8,67 (1H, d); масса/заряд: ES+ [М+Н]+ 285.
Пример 22
N-{4-[(5,7-Диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамид
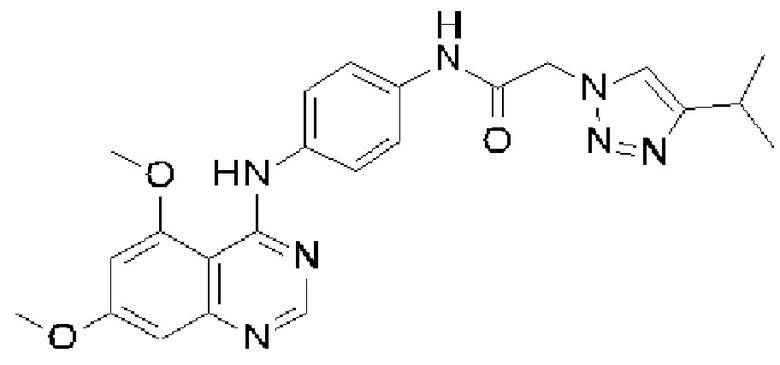
HATU (15,6 г, 40,9 ммоль) добавляли к смеси N1-(5,7-диметоксихиназолин-4-ил)бензол-1,4-диамина (8,1 г, 27,3 ммоль), 2-(4-изопропил-1H-1,2,3-триазол-1-ил)уксусной кислоты (11,9 г, 34,1 ммоль) и DIPEA (14,3 мл, 81,9 ммоль) в DMF (250 мл). Смесь перемешивали при температуре окружающей среды в течение 16 часов. Смесь гасили добавлением воды (20 мл). Полученный раствор концентрировали до объема 100 мл и добавляли в быстро перемешиваемую смесь этилацетата (250 мл) и воды (1300 мл). Смесь перемешивали в течение 1 часа. Полученный осадок собирали фильтрацией и промывали водой (400 мл) с получением бежевого твердого вещества. Твердое вещество обрабатывали с помощью 0,1 М раствора NaHCO3 (400 мл) и полученную суспензию подвергали воздействию ультразвука и перемешивали в течение 1 часа. Осадок собирали фильтрацией и промывали водой. Твердое вещество обрабатывали водой (400 мл) и полученную суспензию подвергали воздействию ультразвука и перемешивали в течение 30 минут. Осадок собирали фильтрацией, промывали водой и сушили с получением бежевого твердого вещества (11,2 г). Твердое вещество перекристаллизовывали из ацетонитрила (300 мл) с получением указанного в заголовке соединения в виде грязно-белого полукристаллического твердого вещества (8,8 г, 72%). 1H ЯМР (500 МГц, DMSO, 27°С) δ 1,24 (6Н, d), 2,99 (1H, гептет), 3,89 (3Н, s), 4,07 (3Н, s), 5,25 (2Н, s), 6,68 (1H, d), 6,77 (1Н, d), 7,58 (2Н, d), 7,76 (2Н, d), 7,86 (1H, d), 8,40 (1H, s), 9,76 (1H, s), 10,45 (1H, s); масса/заряд: ES+ [М+Н]+ 448.
Промежуточные соединения, применяемые в примере 22, получали следующим образом.
Получение N1-(5,7-диметоксихиназолин-4-ил)бензол-1,4-диамина
DBU (18,8 мл, 126 ммоль) добавляли к смеси 5,7-диметоксихиназолин-4(3H)-она (10 г, 48,5 ммоль) и РуВОР (32,8 г, 63 ммоль) в ацетонитриле (500 мл). Образовавшийся бесцветный раствор нагревали при 60°С в течение 1 часа. Добавляли бензол-1,4-диамин (10,5 г, 96,9 ммоль) и перемешивание при 60°С продолжали в течение дополнительных 2 часов. Смесь выпаривали и остаток разделяли между DCM (700 мл) и насыщенным раствором хлорида аммония (600 мл). Органический слой промывали насыщенным раствором хлорида аммония (300 мл), водой (600 мл), насыщенным раствором NaHCO3 (600 мл) и солевым раствором (300 мл), сушили и выпаривали до сухого состояния. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния при градиенте элюирования от 0 до 6% (10:1 метанол/конц. NH3 (водн.)) в этилацетате. Фракции выпаривали с получением неочищенного продукта (26 г) в виде коричневого полутвердого вещества. Данное твердое вещество растворяли в ацетоне (400 мл) и добавляли HCl в диэтиловом эфире (2 М, 25 мл). Полученное твердое вещество собирали фильтрацией и промывали ацетоном с получением неочищенного продукта, который разделяли между насыщенным раствором NaHCO3 (300 мл) и DCM (300 мл). Водный слой экстрагировали с помощью DCM (200 мл) и экстракты объединяли с органическим слоем. Объединенные органические экстракты фильтровали через бумагу для разделения фаз и выпаривали до сухого состояния. Остаток растирали в порошок с диэтиловым эфиром с получением указанного в заголовке соединения в виде оранжевого твердого вещества (8 г, 55%). 1H ЯМР (500 МГц, DMSO, 27°С) δ 3,87 (3Н, s), 4,03 (3Н, s), 4,97 (2Н, s), 6,57 (2Н, d), 6,63 (1Н, d), 6,71 (1Н, d), 7,31 (2Н, d), 8,28 (1H, s), 9,45 (1Н, s); масса/заряд: ES+ [М+Н]+ 297.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНДАЗОЛИЛИЗОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ТАКИХ КАК ЗЛОКАЧЕСТВЕННОЕ НОВООБРАЗОВАНИЕ | 2020 |
|
RU2836662C2 |
| 4-(ИМИДАЗО[1,2-а]ПИРИДИН-3-ИЛ)-ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2822388C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Ха | 2004 |
|
RU2346944C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ C-KIT | 2016 |
|
RU2754858C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2002 |
|
RU2302244C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2013 |
|
RU2650107C2 |
| ПРОИЗВОДНЫЕ АЗОЛОВ | 2012 |
|
RU2622639C2 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
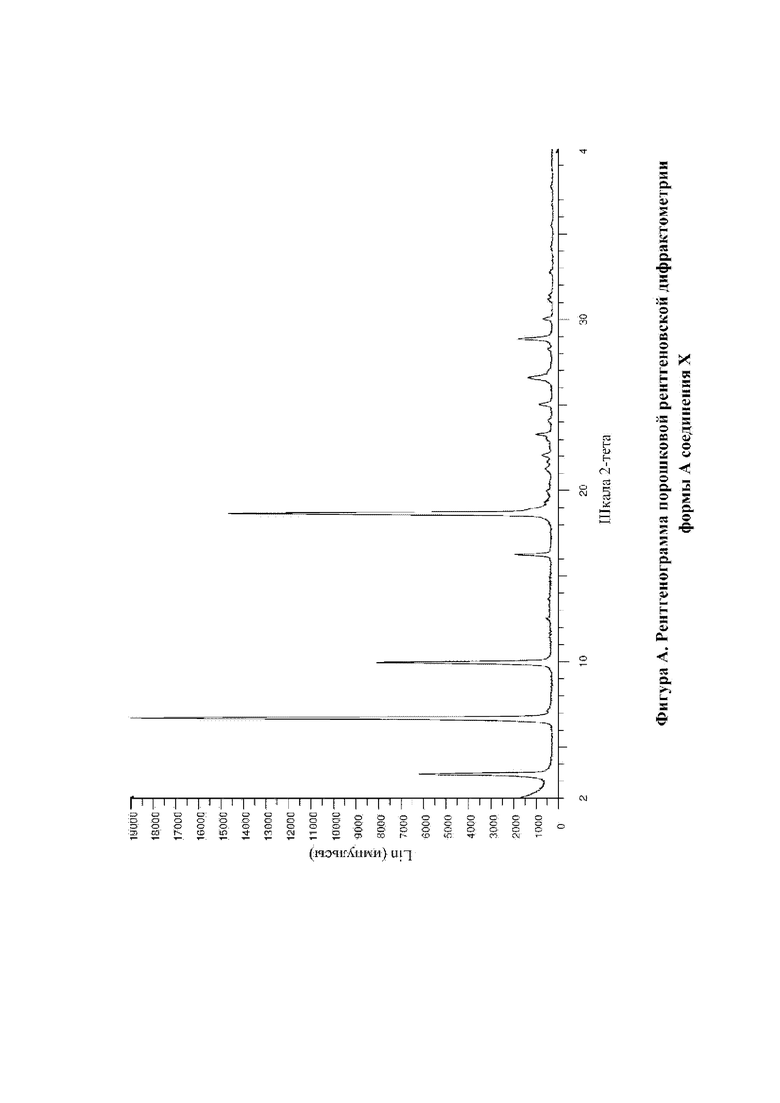
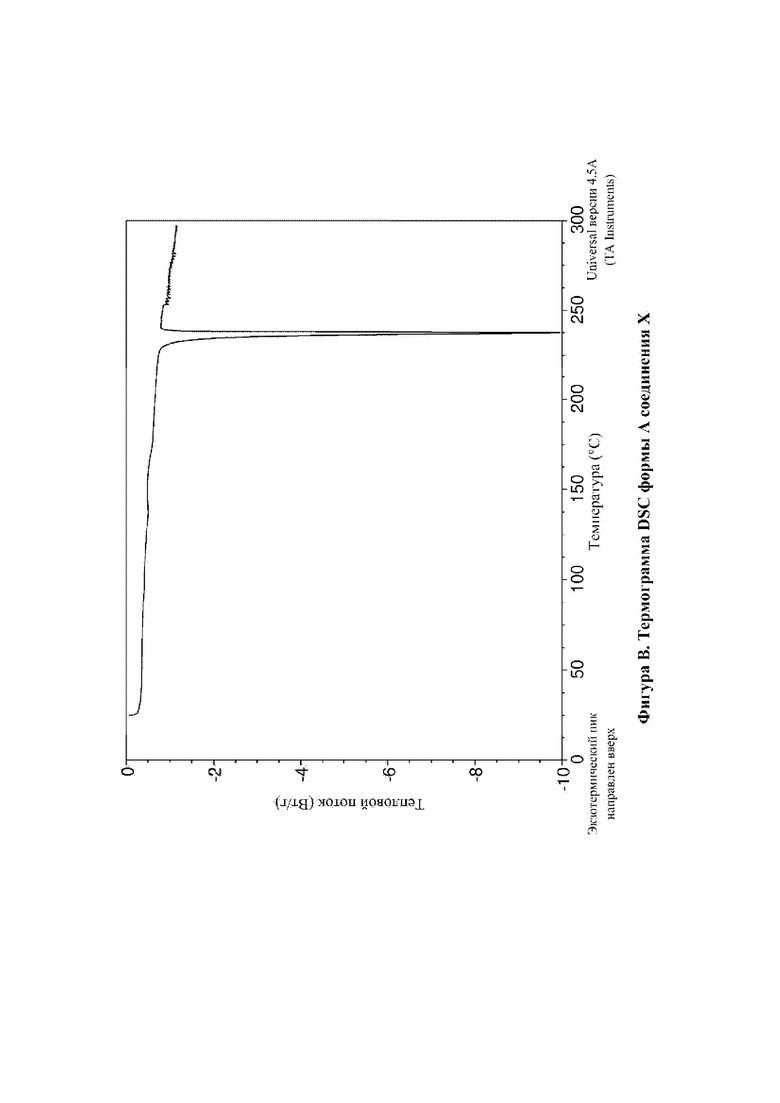
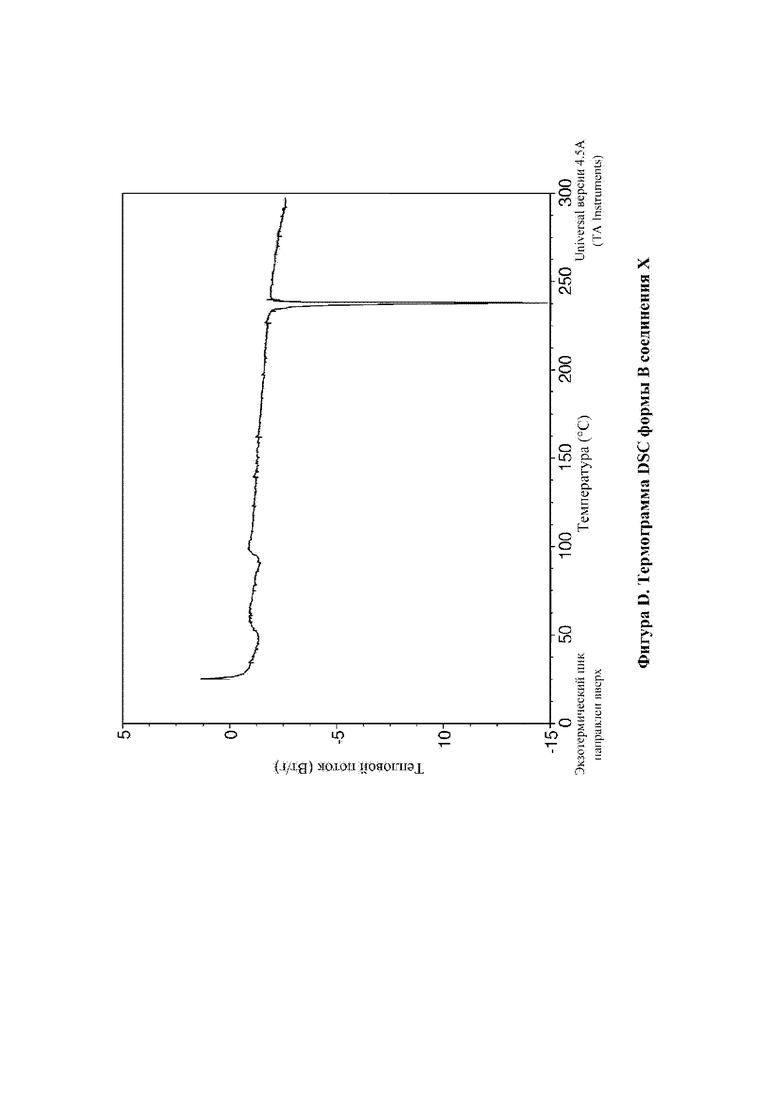
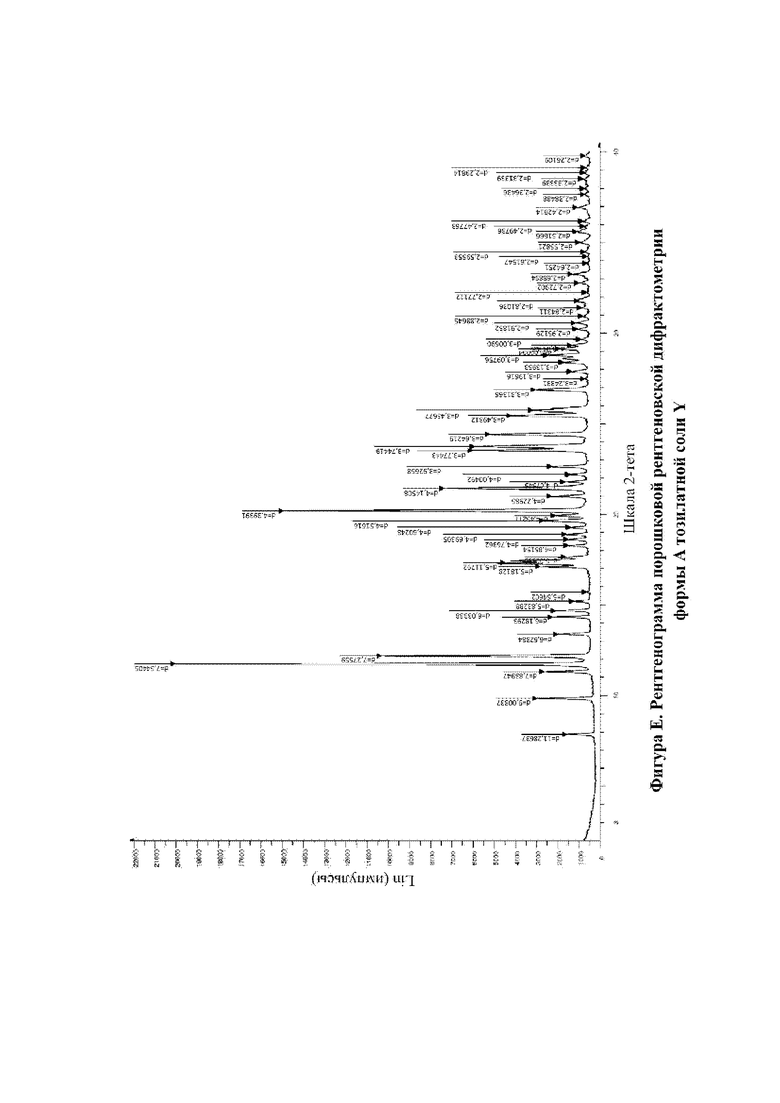
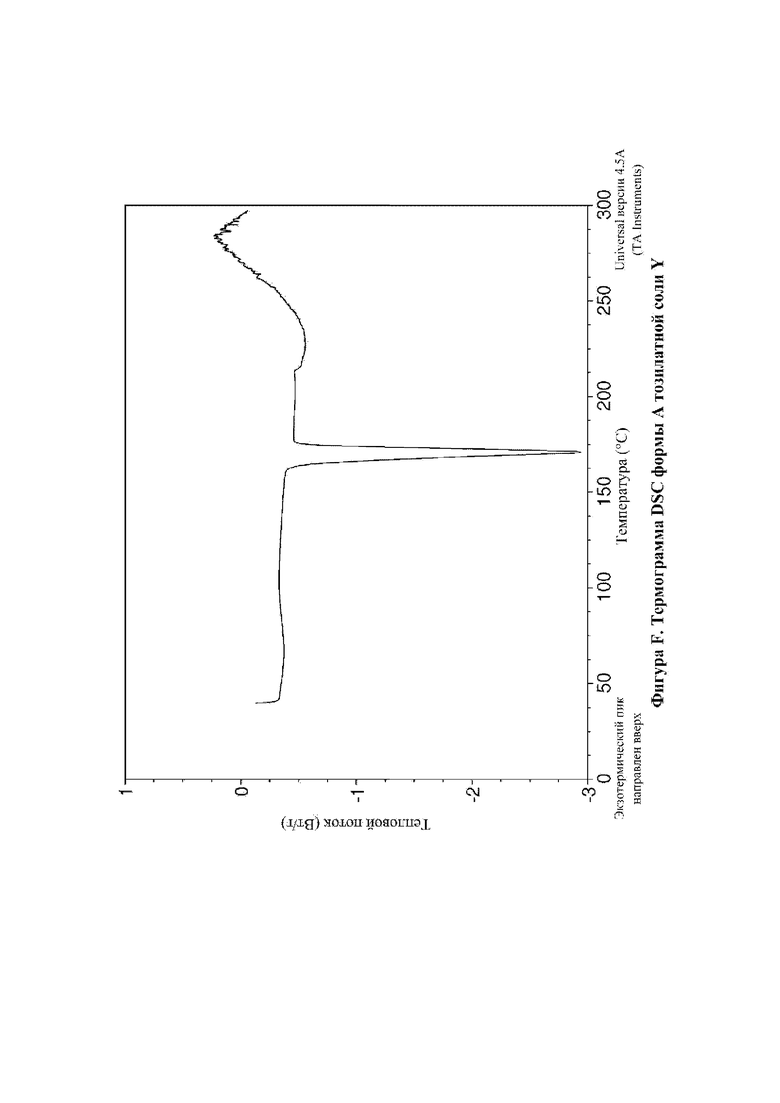
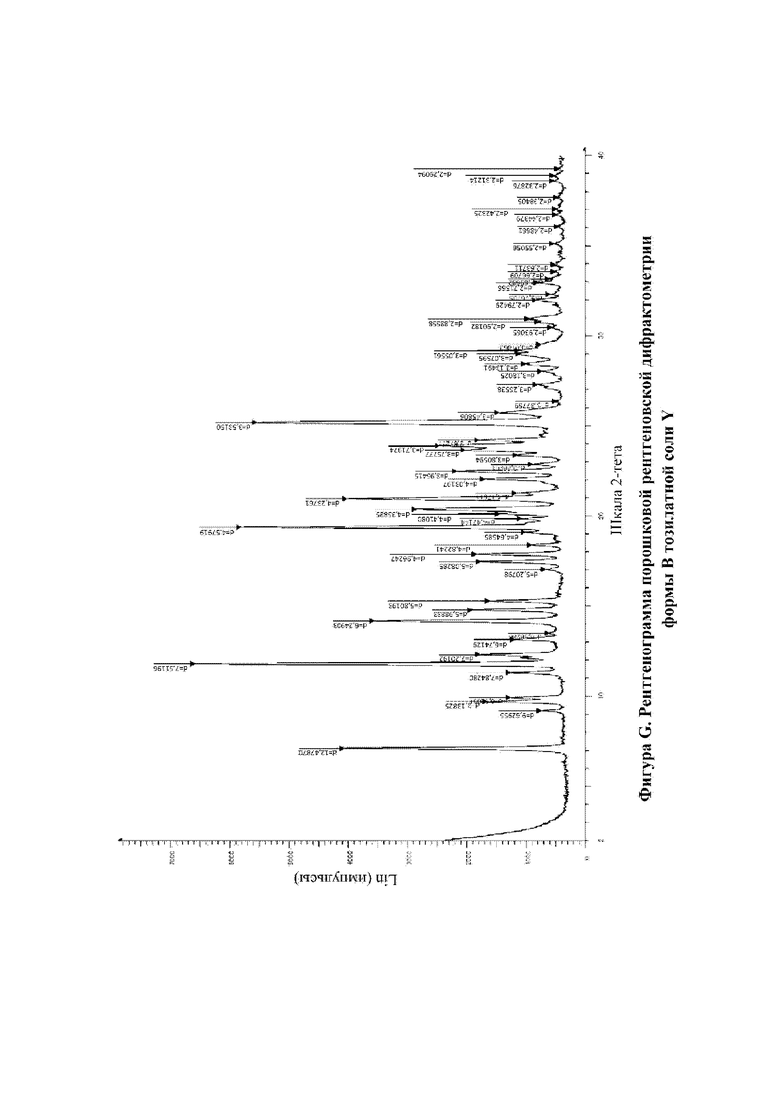
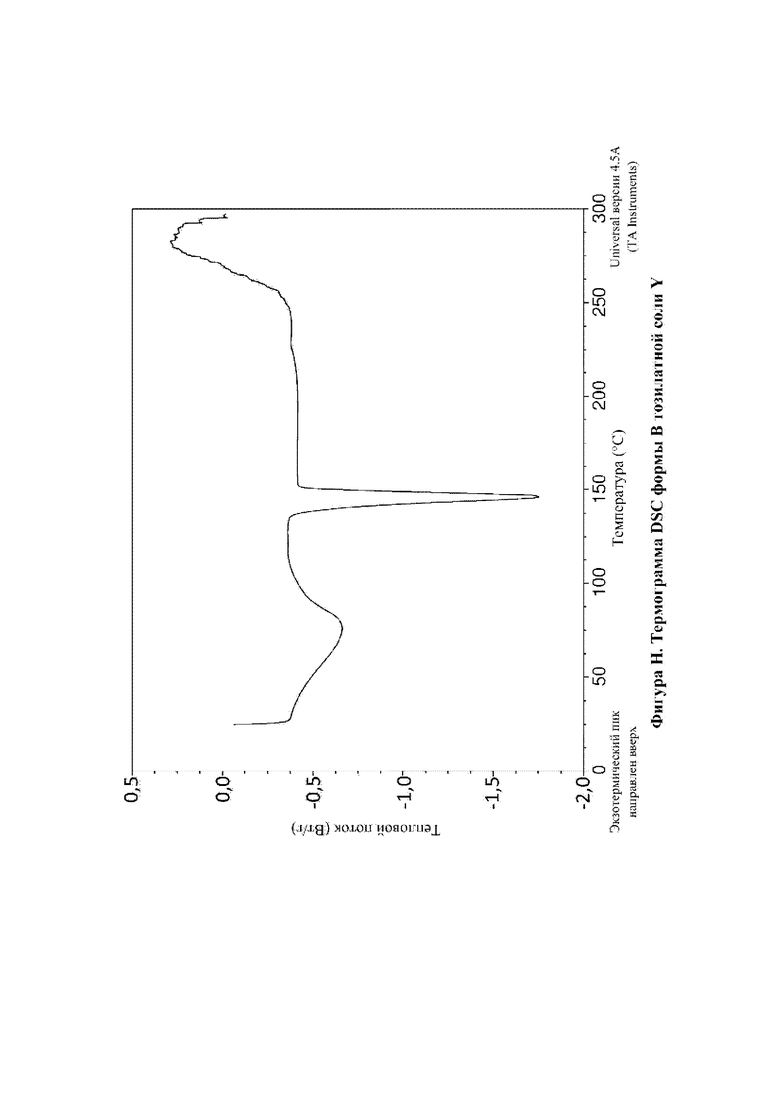


Изобретение относится к гетероциклическому соединению формулы (I), его изомеру или фармацевтически приемлемой соли, R1 выбран из водорода и фтора; R2 выбран из фтора и С1-2алкокси; R3 выбран из водорода и метокси и R4 представляет собой C1-3алкил, необязательно замещенный группой, выбранной из C1-3алкокси и NR5R6, где каждый из R5 и R6 независимо представляет собой метил; или 4-6-членное гетероциклильное кольцо, содержащее один атом кислорода. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), его изомера или фармацевтически приемлемой соли, их применению и способу лечения KIT-опосредованного рака. Технический результат: получены новые соединения, полезные для лечения KIT-опосредованного рака. 4 н. и 16 з.п. ф-лы, 10 ил., 5 табл., 22 пр.
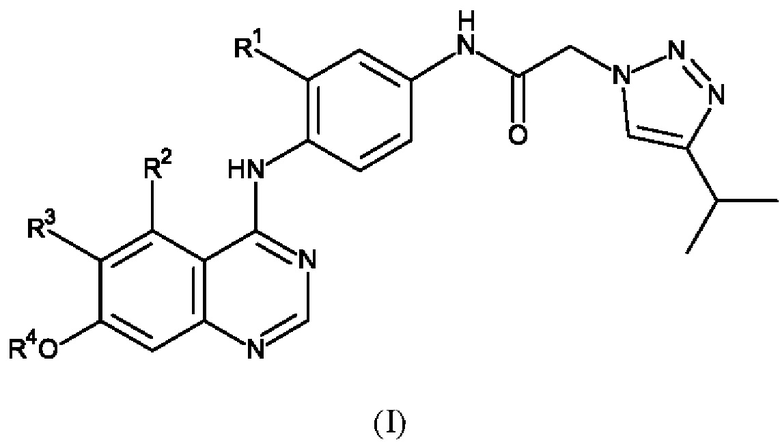
1. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль
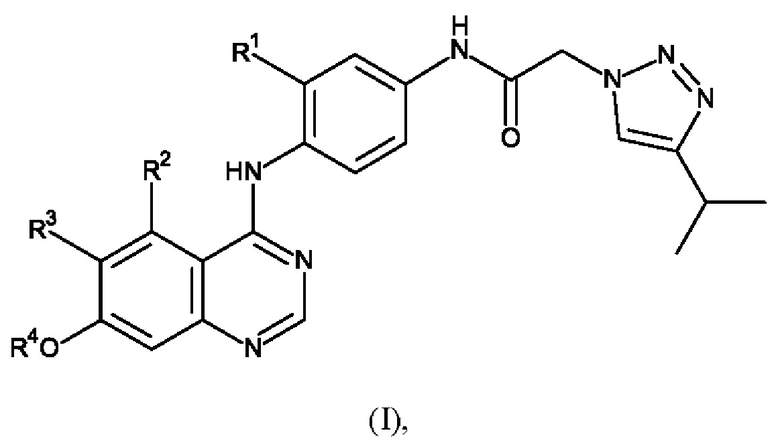
где R1 выбран из водорода и фтора;
R2 выбран из фтора и С1-2алкокси;
R3 выбран из водорода и метокси и
R4 представляет собой C1-3алкил, необязательно замещенный группой, выбранной из C1-3алкокси и NR5R6, где каждый из R5 и R6 независимо представляет собой метил; или 4-6-членное гетероциклильное кольцо, содержащее один атом кислорода.
2. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой водород.
3. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по любому из предыдущих пунктов, где R2 представляет собой фтор.
4. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по любому из предыдущих пунктов, где R3 представляет собой водород.
5. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по любому из предыдущих пунктов, где R4 выбран из метила, этила, изопропила, оксетанила, тетрагидрофуранила, оксанила, 2-диметиламиноэтила и 2-метоксиэтила.
6. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по п. 5, где R4 представляет собой 2-метоксиэтил.
7. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из группы, состоящей из:
N-{4-[(5,7-диметоксихиназолин-4-ил)амино]-3-фторфенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-{4-[(5-фтор-6,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
(R)-N-(4-{[5-этокси-7-(тетрагидрофуран-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
(S)-N-(4-{[5-этокси-7-(тетрагидрофуран-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-(4-((5-этокси-7-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида,
N-(4-((7-(2-(диметиламино)этокси)-5-этоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида,
N-(4-((5-этокси-7-метоксихиназолин-4-ил)амино)фенил)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)ацетамида,
N-(4-((5-этокси-7-(2-метоксиэтокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида,
N-(4-((5-этокси-7-(оксетан-3-илокси)хиназолин-4-ил)амино)фенил)-2-(4-изопропил-1H-1,2,3-триазол-1-ил)ацетамида,
N-(4-{[5-метокси-7-(тетрагидро-2H-пиран-4-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил] ацетамида,
2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]-N-{4-[(5,6,7-триметоксихиназолин-4-ил)амино]фенил}ацетамида,
N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
(R)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамида,
(S)-2-(4-изопропил-1Н-1,2,3-триазол-1-ил)-N-(4-((5-метокси-7-((тетрагидрофуран-3-ил)окси)хиназолин-4-ил)амино)фенил)ацетамида,
N-(4-{[5-метокси-7-(пропан-2-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-(4-{[5-метокси-7-(оксетан-3-илокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-[4-({7-[2-(диметиламино)этокси]-5-метоксихиназолин-4-ил}амино)фенил]-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-{4-[(7-этокси-5-метоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-{4-[(5,7-диэтоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-(4-{[5-метокси-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида,
N-{4-[(5-фтор-7-метоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1-ил]ацетамида,
N-{4-[(5,7-диметоксихиназолин-4-ил)амино]фенил}-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида.
8. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по п. 1, где соединение представляет собой N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1H-1,2,3-триазол-1 ил]ацетамид.
9. Соединение формулы (I) или его изомеры по любому из пп. 1-8, где соединение находится в форме свободного основания.
10. Соединение формулы (I) по п. 8 в кристаллической форме, которая характеризуется рентгенограммой XRPD, имеющей специфические пики при значениях 2-тета, равных 3,4°±0,2°, 6,7°±0,2°, 9,9°±0,2°, 16,2°±0,2°, 18,7°±0,2°, 22,1°±0,2°, 23,3°±0,2°, 25,1°±0,2°, 26,6°±0,2°, 28,9°±0,2°, согласно измерениям с применением излучения CuKα.
11. Соединение формулы (I) по п. 8 в кристаллической форме, которая характеризуется рентгенограммой XRPD, имеющей специфические пики при значениях 2-тета, равных 4,2°±0,2°, 6,6°±0,2°, 7,7°±0,2°, 9,8°±0,2°, 13,1°±0,2°, 13,7°±0,2°, 18,6°±0,2°, 20,0°±0,2°, 26,5°±0,2°, 28,8°±0,2°, согласно измерениям с применением излучения CuKα.
12. Соединение формулы (I) или его изомеры по п. 1, где соединение представляет собой тозилатную соль N-(4-{[5-фтор-7-(2-метоксиэтокси)хиназолин-4-ил]амино}фенил)-2-[4-(пропан-2-ил)-1Н-1,2,3-триазол-1-ил]ацетамида.
13. Соединение формулы (I) по п. 12 в кристаллической форме, которая характеризуется рентгенограммой XRPD, имеющей специфические пики при значениях 2-тета, равных 11,7°±0,2°, 12,2°±0,2°, 13,4°±0,2°, 14,3°±0,2°, 17,3°±0,2°, 20,2°±0,2°, 21,4°±0,2°, 23,6°±0,2°, 23,7°±0,2°, 24,4°±0,2°, согласно измерениям с применением излучения CuKα.
14. Соединение формулы (I) по п. 12 в кристаллической форме, которая характеризуется рентгенограммой XRPD, имеющей специфические пики при значениях 2-тета, равных 7,1°±0,2°, 9,2°±0,2°, 11,8°±0,2°, 14,2°±0,2°, 19,4°±0,2°, 20,4°±0,2°, 20,9°±0,2°, 22,5°±0,2°, 23,9°±0,2°, 25,2°±0,2°, согласно измерениям с применением излучения CuKα.
15. Соединение формулы (I) по п. 12 в кристаллической форме, которая характеризуется рентгенограммой XRPD, имеющей специфические пики при значениях 2-тета, равных 4,4°±0,2°, 5,6°±0,2°, 8,8°±0,2°, 16,8°±0,2°, 19,1°±0,2°, 19,7°±0,2°, 21,9°±0,2°, 22,3°±0,2°, 24,8°±0,2°, 26,9°±0,2°, согласно измерениям с применением излучения CuKα.
16. Фармацевтическая композиция для лечения KIT-опосредованного рака, которая содержит терапевтически эффективное количество соединения формулы (I), его изомеров или его фармацевтически приемлемой соли по любому из пп. 1-15 и по меньшей мере один фармацевтически приемлемый разбавитель или носитель.
17. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по любому из пп. 1-15 для применения в терапии KIT-опосредованного рака.
18. Соединение формулы (I), его изомеры или его фармацевтически приемлемая соль по любому из пп. 1-15 для применения в лечении KIT-опосредованного рака.
19. Применение соединения формулы (I), его изомеров или его фармацевтически приемлемой соли по любому из пп. 1-15 для изготовления лекарственного препарата для лечения KIT-опосредованного рака.
20. Способ лечения KIT-опосредованного рака у теплокровного животного, нуждающегося в таком лечении, который предусматривает введение теплокровному животному терапевтически эффективного количества соединения формулы (I), его изомеров или его фармацевтически приемлемой соли по любому из пп. 1-15.
| US 2006178382 A1, 10.08.2006 | |||
| US 7709479 B1, 04.05.2010 | |||
| Rao, G.-W | |||
| и др.: Synthesis, Antitumor Evaluation and Docking Study of Novel 4-Anilinoquinazoline Derivatives as Potential Epidermal Growth Factor Receptor (EGFR) Inhibitors, ChemMedChem, 2013, 8(6), с.928-933 | |||
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |

