Изобретение относится к области медицины, а именно к клинической фармакологии и может быть использовано для одновременного количественного определения противоэпилептических препаратов, таких как карбамазепин, ламотриджин, леветирацетам, зонисамид, окскарбазепин (по метаболиту 10,11-дигидро-10-гидроксикарбазепину) в рамках одного хроматографического анализа у пациентов получающих политерапию антиконвульсантами при различных видах эпилепсии в условиях стационара.
Эпилепсия является одним из наиболее распространенных нервно-психических заболеваний. По данным ВОЗ, число больных во всем мире составляет около 50 миллионов человек. Заболеваемость эпилепсией в мире составляет 50-70 на 100000 человек, а распространенность - 5-10 на 1000 (0,5-1%). По оценкам, доля общего населения с активной формой эпилепсии составляет 4-10 на 1000 человек, и эта доля увеличивается в странах с низким и средним уровнем дохода. Средняя заболеваемость по России составляет 44 на 100000 человек, причем диапазон значений меняется от региона к региону: так, в Москве эта цифра составляет 11,78 (данные за 2004 год), а в Якутии - 114 (2008 год). Эпилепсия является социально и экономически значимым заболеванием. Она составляет 0,5% глобального бремени болезней (это показатель на определенный момент времени, объединяющий годы жизни, прожитые в состояниях ниже уровня полноценного здоровья и утраченные вследствие преждевременной смерти) и имеет ощутимые экономические последствия с точки зрения удовлетворения потребностей в медико-санитарной помощи, преждевременной смертности и утраченной производительности труда. Довольно распространенной является ситуация развития фармакорезистентности, при которой врачи неврологи вынужденно назначают больным комбинации из трех и более антиконвульснтов, с целью достижения стойкой ремиссии. Вынужденная полипрагмазия нередко ведет к малопредсказуемым последствиям, в том числе - к нежелательными лекарственным реакциям (НЛР), связанным в том числе и с межлекарственными взаимодейстиями. Согласно определению ВОЗ, к нежелательным (неблагоприятным) лекарственным реакциям относят любую реакцию на лекарственное вещество (ЛВ), вредную и нежелательную для организма, возникающую при его назначении для лечения, диагностики и профилактики заболеваний. Считают, что нежелательные реакции развиваются у 4-29% больных, принимающих различные лекарственные препараты. Частота возникновения нежелательных лекарственных реакций в первую очередь зависит от индивидуальных особенностей, пола, возраста больного, тяжести основного и сопутствующих заболеваний, фармакодинамики и фармакокинетики лекарственного вещества, дозы, длительности приема, путей введения препарата, его взаимодействий. Снижение числа и уменьшение выраженности нежелательных лекарственных реакций является приоритетной задачей, стоящей перед современным здравоохранением, и магистральным путем преодоления этих проблем является широкое внедрение технологий терапевтического лекарственного мониторинга и прочих элементов доказательной медицины. Терапевтический лекарственный мониторинг - это лабораторное измерение определенных параметров, которые, при адекватной интерпретации, смогут непосредственно повлиять на режим дозирования лекарственных препаратов. Для многих (но не всех) лекарственных средств основным фактором, определяющим выраженность клинической реакции, является концентрация, которая может быть достигнута в месте действия (в месте связывания с клеточным рецептором или в локусе инфекции) (Patsalos, P.N., Berry, D.J., Bourgeois, B.F., Cloyd, J.С, Glauser, Т А., Johannessen, S.I., Leppik, I.E., Tomson, T. & Perucca, E. Antiepileptic drugs-best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies Epilepsia, 2008, 49, 1239-1276). Востребованность данного типа анализа возрастает с каждым годом, однако большинство пациентов получающих политерапию вынуждены ограничиваться анализом одного или, реже, двух препаратов из списка принимаемых, поскольку содержание каждого антиконвульсанта зачастую анализируется либо отдельными иммуноферментными наборами, либо посредством спектрофотометрического детектирования с низкой избирательностью в отношении исследуемых соединений. Каждый анализ оплачивается пациентом отдельно и это создает высокую финансовую нагрузку на пациентов, что нередко приводит к отказу от необходимых анализов и обедняет информацию, поступающую лечащему врачу.
На сегодняшний день известен способ одновременного определения окскарбазепина и его основных метаболитов методом высокоэффективной жидкостной хроматографии со спектрофотометрическим детектированием на длине волны 237 нм (V. Kimiskidis, М. Spanakis, I. Niopas. Development and validation of a high performance liquid chromatographic method for the determination of oxcarbazepine and its main metabolites in human plasma and cerebrospinal fluid and its application to pharmacokinetic study Journal of Pharmaceutical and Biomedical Analysis 43 (2007) 763-768). Недостатками данного метода является то, что спектрофотометричеекое детектирование имеет низкую избирательность в отношении исследуемых соединений, ввиду чего нередко происходит интерференция хроматографических пиков коэкстрактивных веществ биологической матрицы с аналитами, в том числе и с образованием так называемых «критических пар», что делает данный метод недостаточно селективным и ограничивает сферу его применения.
Известен способ количественного определения ликарбазепина путем газовой хроматографии. При этом проводят жидкостно-жидкостную экстракцию из плазмы крови, дериватизацию продуктов экстракции с последующей газовой хромато-масс-спектрометрией продуктов дериватизации и расчетом концентрации дериватизированного ликарбазепина (G.E. von Unruht and W D. Paar Gas Chromatsgraphic/Mass Spectrometric Assays for Oxcarbazepine and Its Main Metabolites, 10-Hydroxy-Carbazepine and Carbazepine-10,11-trans-diol BIOMEDICAL AND ENVIRONMENTAL MASS SPECTROMETRY, 1986, Vol. 13, 651-656). Недостатками способа является применение сложной дериватизации с использованием силилирующего агента MSTFA. Наличие данной стадии дериватизации негативным образом сказывается на робастности метода, поскольку дериватизированное производное ликарбазепина стабильно при комнатной температуре только в течение несколько часов. Кроме того, сами авторы метода указывают, что если образец, дериватизированный 30 мкл MSTFA, вводится неоднократно, то раствор с образцом может частично затвердевать в хроматографической виале, особенно если влажность в помещении высока, что также негативно сказывается на рабочих характеристиках указанной методики.
Наиболее близким техническим решением к заявленному изобретению является способ количественного определения ликарбазепина в плазме крови путем проведения жидкостно-жидкостной экстракции ликарбазепина из плазмы крови с последующей газовой хромато-масс-спектрометрией продуктов экстракции и расчетом концентрации ликарбазепина, который включает экстракцию анализируемого соединения и внутреннего стандарта - нордазепама, газовую хромато-масс-спектрометрию раствора продуктов экстракции, включающего анализируемое соединение и внутренний стандарт, с последующим расчетом концентрации ликарбазепина по формуле: Y=6,710⋅X+0,7567, где Y - концентрация ликарбазепина (мкг/мл), X - отношение площади хроматографического пика ликарбазепина к площади пика внутреннего стандарта - нордазепама. Недостатками способа является невозможность одновременного количественного определения группы противоэпилептических препаратов, в рамках одного хроматографического анализа у пациентов получающих политерапию антиконвульсантами при различных видах эпилепсии.
Технический результат заключается в создании способа количественного определения группы антиконвульсантов в рамках одного хроматографического анализа с высокой воспроизводимостью и точностью, а также заключается в существенном ускорении и упрощении исполнения терапевтического лекарственного мониторинга.
Технический результат достигается тем, что создан способ количественного определения антиконвульсантов в плазме крови больных эпилепсией, включающий их выделение из плазмы крови с последующей хромато-масс-спектрометрией продуктов экстракции и регистрацию масс-спектров анализируемых средств, при этом выделение антиконвульсантов из плазмы крови и очистку экстракта осуществляют путем депротеинизации, добавляя к образцу плазмы крови внутренний стандарт N-адамантил-гексаметиленимина в ацетонитриле, образовавшуюся смесь встряхивают в течение 5 минут на вортекс-миксере, а затем центрифугируют на скорости 13000 об/мин в течение 30 мин для осаждения белков, после чего надосадочную жидкость переносят в хроматографическую виалу, которую помещают в автосамплер хроматографа для дальнейшего масс-спектрометрического детектирования, а содержание антиконвульсантов определяют методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием на жидкостном хромато-масс-спектрометре, с детектором типа квадрупольная ионная ловушка, анализируемые вещества разделяют на колонке XTerra MS С18 (4.6*150) 5μm с насосом Surveyor LC Pump и с подвижной фазой - раствором ацетата аммония с концентрацией 10 миллимоль/л (раствор А) и ацетонитрилом с добавлением 10 мМ ацетата аммония (90:10) (раствор Б), при скорости потока подвижной фазы - 0,7 мл/мин и скорости потока газа-небулайзера азота 5 л/мин, давлении на распылителе - 100 psi, в режиме электрораспылительной ионизации с мониторингом выбранных реакций (МБР), причем характеристический МВР-переход для карбамазепина по отношению массы к заряду (m/z) составляет 237.00→194.08, МВР-переход для ламотриджина - m/z 256.09→211.00, МВР-переход для леветирацетама - m/z 171.11→153.81, МВР-переход для 10-гидроксикарбазепина - m/z 237.00→194.07 и для внутреннего стандарта N-адамантил-гексаметиленимин - m/z 234,00→135,02 в режиме положительной электрораспылительной ионизации, а характеристический МВР-переход для зонисамида m/z составляет 211.06→147.01 в режиме отрицательной электрораспылительной ионизации, при этом градиентное элюирование смеси антиконвульсантов проводят по следующей схеме: исходное соотношение растворов с 0,00 по 3,00 мин - 60% раствор А:40% раствору Б, с 5,00 по 7,00 мин - 10% раствор А:90% раствору Б, с 8,00 по 11,00 мин - 60% раствор А:40% раствору Б, а количественное определение концентрации для каждого из соединений антиконвульсантов в плазме крови осуществляют по формуле: как отношение площадей пиков анализируемого антиконвульсанта и внутреннего стандарта, выраженных в условных единицах интегрирования умноженное на значение коэффициента пересчета наклона калибровочной кривой к оси абсцисс и суммированное с коэффициентом пересчета точки пересечения кривой с осью ординат.
Способ осуществляется следующим образом.
Образцы крови пациентов собирают в гепаринизированные вакуумные пробирки объемом 5 мл. Для получения плазмы крови производят центрифугирование на гематологической центрифуге при комнатной температуре на скорости 3500 оборотов в минуту. Полученная плазма может хранится до момента анализа в низкотемпературном холодильнике при температуре -70°С. Для выделения антиконвульсантов из плазмы крови и очистки экстракта используют метод депротеинизации. К образцу плазмы крови объемом 200 мкл добавляют 600 мкл внутреннего стандарта N-адамантил-гексаметиленимина, 20 мкг/мл в ацетонитриле. Образовавшуюся смесь встряхивают в течение 5 минут на вортекс-миксере Heidolph Ultra, а затем центрифугируют на скорости 13000 об/мин в течение 30 мин для осаждения белков. Надосадочную жидкость переносят в хроматографическую виалу, которую помещают в автосамплер хроматографа для дальнейшего хромато-масс-спектрометрического анализа. Раствор инжектируют в петлю хроматографа в объеме 10 мкл. Содержание антиконвульсантов определяют методом высокоэффективной жидкостной хроматографии с масс-спетрометрическим детектированием (ВЭЖХ-МС) на жидкостном хромато-масс-спектрометре, Thermo Fischer Scientific LCQ Fleet с детектором типа квадрупольная ионная ловушка или на хроматографе с аналогичными характеристиками. Изучаемые вещества разделяют на колонке XTerra MS С18 (4.6*150) 5μm (Waters, USA), или на колонке с аналогичными характеристиками. Насос Surveyor LC Pump (Waters, США), скорость потока подвижной фазы - 0,7 мл/мин, время анализа - 11 мин. Подвижная фаза - раствор ацетата аммония с концентрацией 10 миллимоль/л (раствор А) и ацетонитрил с добавлением 10 мМ ацетата аммония соответственно в соотношении (90:10) (раствор Б). Все используемые в работе растворители имели степень чистоты не ниже х.ч. Для детектирования анализируемых соединений был подобран метод мониторинга выбранных ионов с положительной ионизацией для карбамазепина, ламотриджина, леветирацетама, 10-гидроксикарбазепина и гимантана и отрицательной ионизацией для зонисамида типа электроспрей, создаваемый напряжением в 5 кВ. Скорость потока газа-небулайзера (азота) составляла: 5 л/мин, давление на распылителе - 100 psi.
Осуществляют также подбор условий градиентного элюирования, оптимального для разделения сложной смеси антиконвульсантов, таких как карбамазепин, ламотриджин, леветирацетам, зонисамид, и метаболит окскарбазепина (10-гидроксикарбазепин). В результате наиболее оптимальной схемой элюирования стала следующая: исходное соотношение растворов (с 0,00 по 3,00 мин) - 60% А:40% Б, с 5,00 по 7,00 мин -10% А.90% Б, с 8,00 по 11,00 мин - 60% А.40%Б. В этих условиях продолжительность анализа сократилась до 11 минут, которых достаточно для оптимального разделения указанной смеси антиконвульсантов (см. фиг. 1). На фиг. 1 показана масс-хроматограмма смеси антиконвульсантов по ионному току от выделенных характеристических продукт-ионов (сверху вниз: верхнее окно - хроматографические пики карбамазепина и 10-гидроксикарбазепина, хроматограмма, окно второе сверху - пик ламотриджина, третье сверху окно - пик зонисамида, четвертое окно сверху - пик леветирацетама, нижнее окно - пик внутреннего стандарта N-адамантил-гексаметиленимина (гимантана).
Затем осуществляют подбор и оптимизацию параметров масс-спектрометрического определения для каждого из указанных соединений, в связи с чем, для каждого из целевых соединений снимают масс-спектр второго порядка (MS2). По результатам проведенного масс-спектрометрического исследования подобрана база МВР-переходов, причем для каждого МВР-перехода был проведен отдельный подбор параметров энергии фрагментации. Данные полученные в ходе подбора условий хроматографирования и оптимизации условий масс-спектрометрического анализа были занесены в настройки метода. Полученные параметры суммированы в таблице 1.
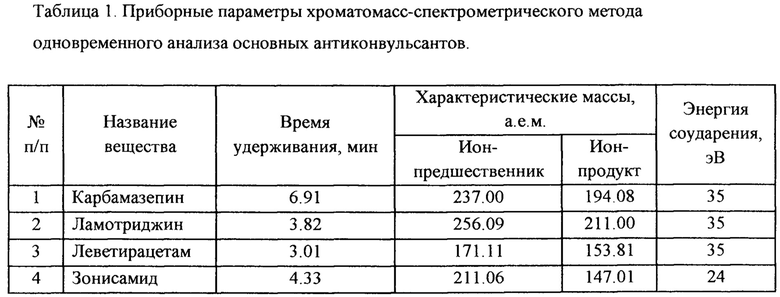
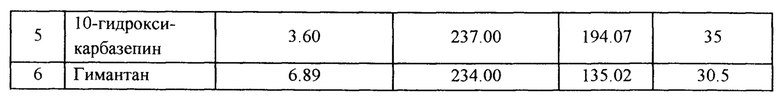
Таким образом, в режиме электрораспылительной ионизации с мониторингом выбранных реакций (МБР) характеристический МВР-переход для карбамазепина по отношению массы к заряду (m/z) составил 237.00→194.08, МВР-переход для ламотриджина - m/z 256.09→211.00, МВР-переход для леветирацетама - m/z 171.11→153.81, МВР-переход для 10-гидроксикарбазепина - m/z 237.00→194.07 и для внутреннего стандарта N-адамантил-гексаметиленимина (гимантана) - m/z 234,00→135,02 в режиме положительной электрораспылительной ионизации, а характеристический МВР-переход для зонисамида m/z составил 211.06→147.01 в режиме отрицательной электрораспылительной ионизации.
Далее производят калибровку метода количественного анализа для каждого из соединений с помощью калибровочных растворов аналитических стандартов указанных веществ. Основные калибровочные характеристики для каждого из соединений представлены в таблице 2.
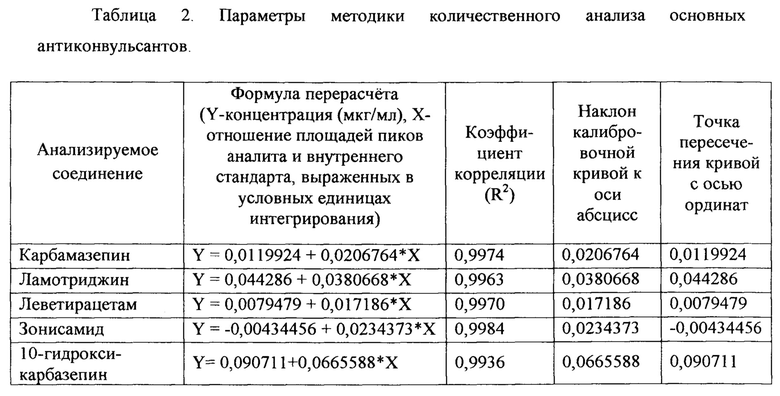
Для каждого из анализируемых соединений была рассчитана градуировочная зависимость, позволяющая производить количественный анализ содержания препаратов в области оптимального концентрационного диапазона, свойственному терапевтическому коридору соответствующего антиконвульсанта.
Таким образом, количественное определение концентрации для каждого из соединений антиконвульсантов в плазме крови осуществляют по следующей формуле: отношения площадей пиков анализируемого антиконвульсанта и внутреннего стандарта, выраженных в условных единицах интегрирования умноженное на значение коэффициента пересчета наклона калибровочной кривой к оси абсцисс и суммированное с коэффициентом пересчета точки пересечения кривой с осью ординат.
После базовых валидационных работ с использованием образцов контроля качества было установлено, что ошибка определения для всех антиконвульсантов не превышает 15%, что говорит о надлежащей аналитической аппроксимации и пригодности метода для анализа содержания антиконвульсантов в плазме крови больных эпилепсией. С помощью указанного метода было произведено измерение содержания антиконвульсантов в образцах плазмы крови 50 пациентов, страдающих эпилепсией. По итогам измерения концентраций, пациентам были даны соответствующие рекомендации по оптимизации режима дозирования соответствующих антиконвульсантов. Кроме того метод хроматографического разделения данной смеси антиконвульсантов позволяет проводить анализ пяти соединений с высокой степенью экспрессности (11 минут на 1 анализ).
Следовательно, разработан, валидирован и внедрен в клиническую практику метод одновременного хроматомасс-спектрометрического определения пяти наиболее востребованных антиконвульсантов, применяемых в современной клинической практике при лечении эпилепсии. В процессе разработки метода были сняты масс-спектры карбамазепина, ламотриджина, леветирацетама, зонисамида, 10,11-дигидро-10-гидроксикарбазепина. На основании полученных спектров были выбраны характеристические фрагмент-ионы, создана база данных МВР-переходов, для каждого перехода подобраны параметры фрагментации.
Таким образом, заявленный способ позволяет с надлежащей воспроизводимостью и точностью производить в рамках одного хроматографического анализа количественное определение антиконвульсантов с существенным ускорением и упрощением исполнения терапевтического лекарственного мониторинга.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения ликарбазепина в плазме крови | 2017 |
|
RU2660364C1 |
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| Способ количественного определения леводопы в плазме крови | 2017 |
|
RU2665164C1 |
| Способ определения топирамата в плазме крови | 2016 |
|
RU2631613C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ МЕТИЛДОПЫ В ПЛАЗМЕ КРОВИ ЧЕЛОВЕКА | 2016 |
|
RU2642593C1 |
| ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ ЭСЛИКАРБАЗЕПИНА | 2008 |
|
RU2488397C2 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИМА ПИНОСТРОБИНА В ПЛАЗМЕ КРОВИ | 2015 |
|
RU2568876C1 |
| Способ контроля содержания противотуберкулёзных препаратов основного ряда и их токсичных метаболитов в плазме крови | 2018 |
|
RU2702998C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФОСФОРОРГАНИЧЕСКИХ ПЕСТИЦИДОВ В РАСТИТЕЛЬНОМ СЫРЬЕ | 2023 |
|
RU2827397C1 |

Изобретение относится к медицине и касается способа количественного определения антиконвульсантов в плазме крови больных эпилепсией, включающего их выделение из плазмы крови с последующей хромато-масс-спектрометрией продуктов экстракции и регистрацию масс-спектров анализируемых средств, где выделение антиконвульсантов из плазмы крови и очистку экстракта осуществляют путем депротеинизации, добавляя к образцу плазмы крови внутренний стандарт N-адамантил-гексаметиленимина в ацетонитриле, образовавшуюся смесь встряхивают, а затем центрифугируют, после чего надосадочную жидкость переносят в хроматографическую виалу, которую помещают в автосамплер хроматографа для дальнейшего масс-спектрометрического детектирования, а содержание антиконвульсантов определяют методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием на жидкостном хромато-масс-спектрометре, анализируемые вещества разделяют на колонке, а количественное определение концентрации для каждого из соединений антиконвульсантов в плазме крови осуществляют по формуле как отношение площадей пиков анализируемого антиконвульсанта и внутреннего стандарта, выраженных в условных единицах интегрирования, умноженное на значение коэффициента пересчета наклона калибровочной кривой к оси абсцисс и суммированное с коэффициентом пересчета точки пересечения кривой с осью ординат. Изобретение обеспечивает высокую воспроизводимость и точность, а также существенное ускорение и упрощение исполнения терапевтического лекарственного мониторинга. 1 пр., 1 ил., 2 табл.
Способ количественного определения антиконвульсантов в плазме крови больных эпилепсией, включающий их выделение из плазмы крови с последующей хромато-масс-спектрометрией продуктов экстракции и регистрацию масс-спектров анализируемых средств, отличающийся тем, что выделение антиконвульсантов из плазмы крови и очистку экстракта осуществляют путем депротеинизации, добавляя к образцу плазмы крови внутренний стандарт N-адамантил-гексаметиленимина в ацетонитриле, образовавшуюся смесь встряхивают в течение 5 минут на вортекс-миксере, а затем центрифугируют на скорости 13000 об/мин в течение 30 мин для осаждения белков, после чего надосадочную жидкость переносят в хроматографическую виалу, которую помещают в автосамплер хроматографа для дальнейшего масс-спектрометрического детектирования, а содержание антиконвульсантов определяют методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием на жидкостном хромато-масс-спектрометре с детектором типа квадрупольная ионная ловушка, анализируемые вещества разделяют на колонке XTerra MS С18 (4.6*150) 5 μm с насосом Surveyor LC Pump и с подвижной фазой - раствором ацетата аммония с концентрацией 10 миллимоль/л (раствор А) и ацетонитрилом с добавлением 10 мМ ацетата аммония (90:10) (раствор Б) - при скорости потока подвижной фазы 0,7 мл/мин и скорости потока газа-небулайзера азота 5 л/мин, давлении на распылителе 100 psi в режиме электрораспылительной ионизации с мониторингом выбранных реакций (МБР), причем характеристический МВР-переход для карбамазепина по отношению массы к заряду (m/z) составляет 237.00→194.08, МВР-переход для ламотриджина - m/z 256.09→211.00, МВР-переход для леветирацетама - m/z 171.11→153.81, МВР-переход для 10-гидроксикарбазепина - m/z 237.00→194.07 и для внутреннего стандарта N-адамантил-гексаметиленимингимантана - m/z 234,00→135,02 в режиме положительной электрораспылительной ионизации, а характеристический МВР-переход для зонисамида m/z составляет 211.06→147.01 в режиме отрицательной электрораспылительной ионизации, при этом градиентное элюирование смеси антиконвульсантов проводят по следующей схеме: исходное соотношение растворов с 0,00 по 3,00 мин - 60% раствор А:40% раствор Б, с 5,00 по 7,00 мин - 10% раствор А:90% раствор Б, с 8,00 по 11,00 мин - 60% раствор А:40% раствор Б, а количественное определение концентрации для каждого из соединений антиконвульсантов в плазме крови осуществляют по формуле как отношение площадей пиков анализируемого антиконвульсанта и внутреннего стандарта, выраженных в условных единицах интегрирования, умноженное на значение коэффициента пересчета наклона калибровочной кривой к оси абсцисс и суммированное с коэффициентом пересчета точки пересечения кривой с осью ординат.
| RU 2159430 C1, 20.11.2000 | |||
| ОЛИГОНУКЛЕОТИДНЫЕ ПРАЙМЕРЫ, СПОСОБ И ТЕСТ-СИСТЕМА ДЛЯ ВЫЯВЛЕНИЯ ГЕНОМА ВИРУСА БОЛЕЗНИ НАЙРОБИ ОВЕЦ МЕТОДОМ ОБРАТНОЙ ТРАНСКРИПЦИИ - ПОЛИМЕРАЗНОЙ ЦЕПНОЙ РЕАКЦИИ | 2009 |
|
RU2416647C1 |
| KOLOCOURI F | |||
| et al | |||
| Dried plasma spots as an alternative sample collection technique for the quantitative LC-MS/MS determination of gabapentin | |||
| Anal Bioanal Chem | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| LEI YIN et al | |||
| et al | |||
| Simultaneous determination of ten | |||

