Настоящая заявка с частичным продолжением в соответствии с § 120 раздела 35 Сводов законов США испрашивает приоритет обычной патентной заявки США 13/365824, поданной 3 февраля 2012 года, заявки с частичным продолжением, которая испрашивает приоритет патентной заявки PCT/GB2011/052115, поданной 31 октября 2011 года, международной патентной заявки, которая испрашивает приоритет заявки GB 1018289.7, поданной 29 октября 2010 года, и испрашивает приоритет патентной заявки США 13/365828, поданной 3 февраля 2012 года, и испрашивает приоритет заявки GB 1113730.4, поданной 10 августа 2011 года, заявки GB 1113729.6, поданной 10 августа 2011 года, заявки GB 1113728.8, поданной 10 августа 2011 года, и заявки GB 1101937.9, поданной 4 февраля 2011 года, и эта заявка с частичным продолжением в соответствии с § 119 раздела 35 Сводов законов США испрашивает приоритет предварительной патентной заявки США 61/752309, поданной 14 февраля 2013 года, и предварительной патентной заявки США 61/752356, поданной 14 января 2013 года, содержание каждой из которых включено в настоящий документ посредством ссылки во всей своей полноте.
Воспаление включает активацию иммунной системы в ответ на воздействие вредных раздражителей, таких как, например, патоген, инфекция, раздражающее вещество, или на повреждение клеток. В качестве стереотипной реакции, воспаление представляет собой механизм врожденного иммунитета по сравнению с адаптивным иммунитетом, который является специфичным для каждого патогена. Воспаление может быть классифицировано либо как острое, либо хроническое. В общем случае, острое воспаление опосредуется гранулоцитами, в то время как хроническое воспаление опосредуется одноядерными клетками, такими как моноциты и лимфоциты.
Острое воспаление представляет собой первоначальную ответную защитную реакцию организма, направленную на удаления вредного раздражителя путем поддержания целостности тканей и содействия восстановлению тканей. Оно является частью природной защитной системы организма против повреждений и болезней, и при отсутствии острого воспаления никогда бы не заживали раны и не излечивались инфекции, и прогрессирующее разрушение ткани могло бы представлять угрозу для выживания организма в целом.
Процесс острого воспаления инициируется уже присутствующими во всех тканях клетками, в основном резидентными макрофагами, дендритными клетками, гистиоцитами, клетками Купфера, мастоцитами, сосудистыми эндотелиальными клетками и клетками гладкой мускулатуры сосудов. При воздействии вредного раздражителя, эти клетки активируются и высвобождают опосредующие воспаление и сенсибилизирующие молекулы, такие как, например, провоспалительные цитокины, провоспалительные простагландины, лейкотриены, гистамин, серотонин, нейтральные протеазы, брадикинин и оксид азота. Эти молекулы воспаления модулируют сложный ряд биологических явлений, в которых принимают участие клеточные и бесклеточные компоненты локальной сосудистой системы, иммунной системы и поврежденного участка ткани, приводящих к распространению и наступлению ответной воспалительной реакции. Эти явления являются ответственными за проявление острой ответной воспалительной реакции, которая обычно характеризуется 1) расширением кровеносных сосудов, что увеличивает приток крови в ткани, приводящей к эритеме (покраснению и повышению температуры), которая может распространяться за пределы этого места (мгновенной ответной реакцией); 2) проницаемостью кровеносных сосудов, что увеличивает истечение плазмы в ткань, вызывая тем самым отек (припухлость); 3) изменением возбудимости некоторых сенсорных нейронов, вызывая повышенную чувствительность и боль; 4) стимулированием высвобождения молекул, индуцирующих воспаления, таких как, например, нейропептиды, такие как вещество Р (SP) и кальцитонин-ген-связанный пептид (CGRP), простагландины и аминокислоты, такие как глутамат, из периферических нервных окончаний; и 5) увеличением миграции лейкоцитов, в основном гранулоцитов, из кровеносных сосудов в ткани. Для поддержания острой ответной воспалительной реакции требуется постоянная стимуляция, и ее следует активно прерывать, если в ней нет больше необходимости. Следовательно, острое воспаление прекращается сразу же, как только заканчивается воздействия вредного раздражителя.
Однако сильная или длительная вредная стимуляция вызывает в результате хроническую ответную воспалительную реакцию, которая приводит к прогрессирующему сдвигу в типе клеток, присутствующих в месте повреждения ткани. Хроническое воспаление может быть охарактеризовано как одновременное разрушение и заживление ткани при воспалительном процессе, конечным результатом которого является повреждение ткани, а не ее восстановление. По этой причине, хроническое воспаление представляет собой заболевание. Так как ответная воспалительная реакция может происходить в любой части организма, то хроническое воспаление вовлечено в патофизиологию широкого спектра в явном виде не связанных между собой заболеваний, которые составляют основу большой и разнообразной группы заболеваний человека. Например, хроническое воспаление вовлечено в такие разнообразные заболевания, как сердечнососудистые заболевания, рак, аллергии, ожирение, сахарный диабет, заболевания пищеварительной системы, дегенеративные заболевания, аутоиммунные нарушения и болезнь Альцгеймера.
Попытки лечения хронического воспаления привели к ограниченному успеху. В частности, это обусловлено тем фактом, что этиология хронического воспаления представляет собой сложную ответную реакцию, основанную частично на различных молекулах, индуцирующих воспаление, и на множестве опосредующих воспаление и сенсибилизирующих молекулах, которые, по-видимому, вызывают воспаление по избыточному механизму. Кроме того, помимо блокирования провоспалительных молекул, многие противовоспалительные лекарственные средства также ингибируют регуляторные петли, которые высвобождают эндогенные противовоспалительные молекулы. Так, нестероидные противовоспалительные препараты уменьшают воспаление путем блокирования ферментативной активности циклооксигеназы, ключевого фермента, который катализирует превращение арахидоновой кислоты в простагландины и лейкотриены. Таким образом нестероидные противовоспалительные препараты уменьшают воспаление путем предотвращения синтеза всех простагландинов. Однако нестероидные противовоспалительные препараты не только предотвращает синтез провоспалительных простагландинов, эти соединения также предотвращают синтез противовоспалительных простагландинов. Следовательно, применение нестероидных противовоспалительных препаратов приводит к ограниченному успеху, так как они блокируют эндогенную ответную противовоспалительную реакцию, что в некоторых случаях может пролонгировать хроническое воспаление. Поэтому, при лечении хронического воспаления было бы весьма желательно применять соединения, композиции и способы, которые преимущественно ингибируют ответные провоспалительные реакции.
В настоящем изобретении раскрываются фармацевтические композиции твердых растворов. Описанные в изобретении фармацевтические композиции приготавливают способом, который дает возможность получить систему доставки липид-адъювант, позволяющую доставить обладающее противовоспалительной активностью терапевтическое соединение таким образом, чтобы оно более эффективно ингибировало ответную провоспалительную реакцию. Итогом является улучшение результатов лечения хронического воспаления.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Аспекты настоящего изобретения раскрывают, помимо всего прочего, фармацевтическую композицию твердого раствора, включающую терапевтическое соединение, один или более твердых при комнатной температуре липидов. Раскрытая в изобретении фармацевтическая композиция твердого раствора может дополнительно включать один или более жидких при комнатной температуре липидов, один или более стабилизаторов, один или более нейтрализаторов или любую их комбинацию. Терапевтическое соединение может обладать противовоспалительной активностью.
Другие аспекты настоящего изобретения раскрывают, помимо всего прочего, способ приготовления раскрытой в изобретении фармацевтической композиции твердого раствора. Раскрытый в изобретении способ включает стадии a) контактирования раскрытого в изобретении терапевтического соединения с одним или более жидкими при комнатной температуре липидами при условиях, которые способствуют растворению терапевтического соединения в липидах; и b) контактирования раствора соединение/липид с одним или более твердыми при комнатной температуре липидами при условиях, которые способствуют образованию композиции твердого раствора. В аспектах этого способа, для того чтобы получить раствор, применяют нагревания для растворения терапевтического соединения в одном или более жидких при комнатной температуре липидах. В других аспектах этого способа, стадия (a) включает контактирование раскрытого в изобретении терапевтического соединения с одним или более жидкими при комнатной температуре липидами и/или одним или более стабилизаторами и/или одним или более нейтрализаторами при условиях, которые способствуют растворению терапевтического соединения в липидах.
Другие аспекты настоящего изобретения раскрывают способ лечения индивидуума, страдающего хроническим воспалением, где способ включает стадию введения индивидууму, если он в этом нуждается, раскрытую в изобретении фармацевтическую композицию твердого раствора, где введение приводит в результате к уменьшению интенсивности симптома, связанного с хроническим воспалением, вследствие чего происходит лечение индивидуума.
Другие аспекты настоящего изобретения раскрывают применение раскрытой в изобретении фармацевтической композиции твердого раствора в производстве лекарственного препарата для лечения хронического воспаления.
Другие аспекты настоящего изобретения раскрывают описанную в изобретении фармацевтическую композицию твердого раствора для применения при лечении хронического воспаления.
Другие аспекты настоящего изобретения раскрывают применение раскрытой в изобретении фармацевтической композиции твердого раствора для лечения хронического воспаления.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 представлены кривые дифференциальной сканирующей калориметрии (ДСК) раскрытых в изобретении композиций твердых растворов, включающих ибупрофен. Фигура 1A представляет собой кривую ДСК отдельно взятого ибупрофена, характеризующегося температурой плавления в диапазоне от 75°C до 78°C; фигура 1B представляет собой кривую ДСК отдельно взятого GELUCIE® 43/01, характеризующегося температурой плавления в диапазоне от 41°C до 45°C; фигура 1С представляет собой кривую ДСК среды, включающей GELUCIE® 43/01, MAISINE® 35-1 и ПЭГ 400, характеризующейся температурой плавления в диапазоне от 32°C до 38°C и от 41°C до 45°C; фигура 1D представляет собой кривую ДСК композиции ибупрофена LA 35-1, характеризующейся температурой плавления в диапазоне от 32°C до 44°C; фигура 1E представляет собой кривую ДСК композиции ибупрофена LA 35-2, характеризующейся температурой плавления в диапазоне от 32°C до 43°C; фигура 1F представляет собой кривую ДСК композиции ибупрофена LA 35-1, характеризующейся температурой плавления в диапазоне от 32°C до 42°C; фигура 1G представляет собой кривую ДСК композиции ибупрофена LA 35-1, характеризующейся температурой плавления в диапазоне от 32°C до 38°C; фигура 1H представляет собой кривую ДСК композиции ибупрофена LA 35-1, характеризующейся температурой плавления в диапазоне от 32°C до 42°C.
На фигуре 2 представлена кривая ДСК композиции артеметера LA 2-15-1, характеризующейся температурой плавления в диапазоне от 35°C до 40°C.
На фигуре 3 представлена кривая ДСК композиции аспирина LA 3-86-3, характеризующейся температурой плавления в диапазоне от 35°C до 40°C.
На фигуре 4 представлена кривая ДСК композиции дантролена LA 3-104-2, характеризующейся температурой плавления в диапазоне от 34°C до 39°C.
На фигуре 5 представлена кривая ДСК композиции диклофенака LA 3-103, характеризующейся температурой плавления в диапазоне от 35°C до 40°C.
На фигуре 6 представлена кривая ДСК композиции фенофибрата LA 2-19, характеризующейся температурой плавления в диапазоне от 34°C до 39°C.
На фигуре 7 представлена кривая ДСК композиции лидокаина LA 3-101-2, характеризующейся температурой плавления в диапазоне от 34°C до 40°C.
На фигуре 8 представлена кривая ДСК композиции набуметона LA 3-105-1, характеризующейся температурой плавления в диапазоне от 35°C до 40°C.
На фигуре 9 представлена кривая ДСК композиции напроксена LA 1-23-5, характеризующейся температурой плавления в диапазоне от 30°C до 39°C.
На фигуре 10 представлена кривая ДСК композиции сальбутамола LA 1, характеризующейся температурой плавления в диапазоне от 32°C до 40°C.
На фигуре 11 представлена кривая ДСК композиции салметерола LA 1-23-7, характеризующейся температурой плавления в диапазоне от 34°C до 43°C.
На фигуре 12 представлена кривая ДСК композиции симвастатина LA 3-83-3, характеризующейся температурой плавления в диапазоне от 32°C до 43°C.
На фигуре 13 представлена кривая ДСК композиции телмисартана LA 1, характеризующейся температурой плавления в диапазоне от 34°C до 43°C.
ОПИСАНИЕ
Настоящее изобретение раскрывает композиции твердых растворов, применяемых для получения лекарственных форм большого числа терапевтических соединений. Композиции твердых растворов представляют собой твердые кристаллические вещества, включающие матрицу из материала растворителя (который может быть твердым при нормальных температурах) и растворенные вещества, молекулы которых расположены случайным образом и не в упорядоченном положении. Раскрытые в изобретении фармацевтические композиции твердых растворов выполняют функцию системы доставки, которая позволяет более эффективно доставить или таргетировать раскрытое в изобретение терапевтическое соединение в конкретный тип клеток, ткань, орган или область тела способом, который более эффективно ингибирует ответную провоспалительную реакцию. Это ингибирование позволяет улучшить результаты лечения хронического воспаления.
Например, раскрытая в изобретении фармацевтическая композиция может облегчить доставку раскрытого в изобретении терапевтического соединения в макрофаги. Макрофаги существуют на пересечении двух фундаментальных путей, являясь главными клетками в иммунной системе, а также в липидном метаболизме. Что касается иммунной системы, то большинство патогенов имеют компонент поверхности, содержащий липид, который макрофаг распознает и затем поглощает. Один возможный механизм, который позволяет достигать этого селективного поиска тканей-мишеней, заключается в том, что раскрытые в изобретении фармацевтические композиции могут предназначаться для использования преимуществ активности хиломикронов. Хиломикроны представляют собой относительно крупные липопротеиновые частицы, имеющие диаметр от 75 нм до 1200 нм. Включая в себя триглицериды (85-92%), фосфолипиды (6-12%), холестерин (1-3%) и аполипопротеины (1-2%), хиломикроны переносят липиды пищи из кишечника в другие места в организме. Хиломикроны представляют собой одну из пяти основных групп липопротеинов, при этом другие группы образуют VLDL, IDL, липопротеины низкой плотности (LDL), липопротеины высокой плотности (HDL), которые позволяют жирам и холестерину передвигаться внутри раствора на водной основе кровотока.
В процессе пищеварения, жирные кислоты и холестерин подвергаются обработке в желудочно-кишечном тракте в результате воздействия панкреатических соков, включающих липазы, и эмульгированию с помощью солей желчных кислот с образованием мицелл. Эти мицеллы обеспечивают абсорбцию липида в виде свободных жирных кислот абсорбирующими клетками тонкого кишечника, называемыми энтероцитами. В результате попадания в энтероциты, триглицериды и холестерин объединяются в образующиеся хиломикроны. Образующиеся хиломикроны состоят в основном из триглицеридов (85%) и содержат некоторое количество холестерина и холестериновых эфиров. Основной компонент аполипопротеина представляет собой аполипопротеин B-48 (APOB48). Эти образующиеся хиломикроны высвобождаются в результате экзоцитоза из энтероцитов в млечные капилляры, лимфатические сосуды, берущие начало в ворсинках тонкого кишечника, и затем секретируются в кровоток в месте соединения грудного лимфатического протока с левой подключичной веной.
При циркуляции в лимфе и крови, хиломикроны обмениваются компонентами с HDL. HDL служит донором аполипопротеина C-II (APOC2) и аполипопротеина E (APOE) для образующегося хиломикрона и таким образом превращает его в зрелый хиломикрон (часто называемый просто "хиломикроном"). APOC2 является кофактором активности липопротеинлипазы (LPL). Как только запасы триглицеридов распределены, хиломикрон возвращает APOC2 в HDL (но сохраняет APOE), и в результате становится остаточным хиломикроном, имеющим в этот момент размер только 30-50 нм. APOB48 и APOE имеют важное значение для идентификации остаточного хиломикрона в печени при эндоцитозе и распаде на липопротеины (VLDL, LDL и HDL). Эти липопротеины подвергаются обработке и хранятся компетентными клетками, включающими, например, гепациты, адипоциты и макрофаги. Таким образом, не приводя в качестве доказательства какую-либо теорию, тем не менее, можно предположить, что, при пероральном введении, раскрытая в изобретении фармацевтическая композиция может превращаться в мицеллу при нахождении в желудочно-кишечном тракте, абсорбироваться энтероцитами и объединяться в возникающие хиломикроны, оставаться связанной с остаточными хиломикронами, захваченными печенью, и, в конечном счете, может загружаться в макрофаги, которые присутствуют в воспаленных тканях.
В качестве другого примера, раскрытая в изобретении фармацевтическая композиция может облегчать доставку раскрытого в изобретении терапевтического соединения в дендритные клетки. Один возможный механизм, который позволяет достигать этого селективного поиска тканей-мишеней, заключается в том, что раскрытые в изобретении фармацевтические композиции могут предназначаться для использования преимуществ эндоцитической/фагоцитической активности дендритных клеток. Дендритные клетки представляют собой иммуноциты, образующие часть иммунной системы млекопитающего. Главной функцией дендритных клеток является переработка антигенного материала и передача его на поверхность для других клеток иммунной системы. Таким образом, дендритные клетки функционируют как антиген представляющие клетки, которые действуют в качестве мессенджеров между врожденным и приобретенным иммунитетом. Дендритные клетки присутствуют в тканях, находящихся в контакте с внешней средой, таких как, например, кожа (где существует специальный тип дендритных клеток, называемый клетками Лангерганса) и внутренняя поверхность носа, легких, желудка и кишечника. Эти клетки можно также обнаружить в незрелом состоянии в крови. После активации, они мигрируют в лимфатические узлы, где они взаимодействуют с Т-клетками и В-клетками и в результате инициируют и формируют адаптивную ответную иммунную реакцию. Известно, что дендритные клетки подвергают эндоцитозу и фагоцитозу липидные частицы в качестве части их процессов мониторинга состояния окружающей среды и представления антигена. Не приводя в качестве доказательства какую-либо теорию, тем не менее, можно предположить, что при местном или ингаляционном введении, раскрытая в изобретении фармацевтическая композиция может проникать в кожу или внутреннюю поверхность носа, легких, желудка и кишечника, подвергаться эндоцитозу/фагоцитозу под воздействием дендритных клеток и, в конечном счете, загружаться в Т-клетки и/или В-клетки, которые присутствуют в воспаленных тканях.
Помимо таргетированной доставки раскрытого в изобретении терапевтического соединения, раскрытая в изобретении фармацевтическая композиция твердого раствора обладает преимуществом, связанным с различными температурами плавления разнообразных используемых липидов. Путем подбора типов и количеств добавляемых липидов, может быть приготовлена раскрытая в изобретении фармацевтическая композиция, которая является практически твердым веществом при комнатной температуре, но плавится, когда ее температура достигает температуры человеческого тела, как в случае, например, после ее проглатывания. Получающаяся расплавленная композиция легко образует мицеллы, которые абсорбируются кишечником, объединяются в хиломикроны и, в конечном счете, абсорбируются макрофагами или поглощаются дендритными клетками, как описано выше.
Аспекты настоящего изобретения раскрывают, помимо прочего, композицию твердого раствора. Раскрытую в изобретении композицию твердого раствора обычно вводят в виде фармацевтически приемлемой композиции. Используемый в изобретении термин "фармацевтически приемлемая" относится к любому молекулярному образованию или композиции, которая не вызывает побочного, аллергического или другого неблагоприятного или нежелательного ответного действия при введении индивидууму. Используемый в изобретении термин "фармацевтически приемлемая композиция" является синонимом "фармацевтической композиции" и означает терапевтически эффективную концентрацию активного ингредиента, такого как, например, любого из раскрытых в изобретении терапевтических соединений. Раскрытую в изобретении фармацевтическую композицию применяют в медицинских и ветеринарных целях. Фармацевтическая композиция может быть введена индивидууму только сама по себе, или в комбинации с другими дополнительными активными ингредиентами, средствами, лекарственными средствами или гормонами.
Для того чтобы влиять на фармакодинамику раскрытого в изобретении терапевтического соединения, композиция твердого раствора должна обладать тремя характерными особенностями. Во-первых, по меньшей мере, один липид в композиции твердого раствора должен быть получен из, по меньшей мере, одной жирной кислоты, в которой длина углеродной цепи составляет более 12 и менее 24 углеродных атомов и, поэтому, она подходит для абсорбции на протяжении путей энтероцитов. Жирные кислоты с длиной цепи меньше чем C12-C24 не образуют матрицу липид-лекарственное средство, и, поэтому, лекарственное средство поглощается организмом в результате обычного процесса абсорбции. Жирные кислоты с длиной цепи больше чем C12-C24, хотя и образуют матрицы липид-лекарственное средство, но не могут быть абсорбированы, и лекарственное средство вымывается из композиции твердого раствора и удаляется из организма через желудочно-кишечный тракт.
Во-вторых, терапевтическое соединение само по себе должно обладать липофильностью, которая позволяет ему образовывать матрицу твердого раствора с C12-C24 липидом. Как будет указано ниже, эта липофильность может быть присущей для терапевтического соединения (составы растворимого в липиде лекарственного средства), или могут быть использованы некоторые добавки, которые позволяют получать широкий спектр растворимых в липиде лекарственных средств в матрице (составы лекарственного средства в форме свободной кислоты/свободного основания, составы солевой формы лекарственного средства и комбинированные составы лекарственного средства).
В-третьих, терапевтическое соединение само по себе должно влиять на биологию некоторых типов клеток, которые подвергают контакту с композицией твердого раствора с липид-адъювантными свойствами, и которые, в конце концов, циркулирует в организме. Такие структуры включают хиломикрон, частицы LDL и частицы HDL. Подвергаемые контактированию типы клеток могут включать макрофаги, дендритные клетки и липоциты и раковые клетки. Ткани, которые имеют высокое поверхностное содержание липидов, могут также быть преимущественными мишенями. Они включают нервные ткани и мозг.
Настоящее изобретение раскрывает четыре общих типа композиций твердых растворов, а именно: составы растворимого в липиде лекарственного средства, составы лекарственного средства в форме свободной кислоты/свободного основания, составы солевой формы лекарственного средства и комбинированные составы лекарственного средства. Композиции твердых растворов, приготовленные с использованием состава растворимого в липиде лекарственного средства, требуют только липидный компонент для введения раскрытого в изобретении терапевтического соединения в композицию твердого раствора. Не приводя в качестве доказательства какую-либо теорию, тем не менее, можно предположить, что растворимые в липидах лекарственные средства обычно будут растворяться в липиде при нагревании. При охлаждении, можно полагать, что липидный компонент и лекарственное средство будут образовывать матрицы липид-лекарственное средство, сформированные так, что лекарственное средство заключено в липидную оболочку. Так как имеют место только гидрофобные взаимодействия, то отсутствует упорядоченное расположение этих матриц липид-лекарственное средство, что приводит к композиции твердого раствора (то есть отсутствует кристаллизация в классическую твердую форму).
Обычно, в составе растворимого в липиде лекарственного средства используют терапевтические соединения, имеющие величину logP приблизительно 3,0 или более. Не ограничивающие примеры включают артемизинин, такой как артеэфир, артеметер, артемизинин, артесунат и дигидроартемизинин; фибрат, такой как безафибрат, ципрофибрат, клофибрат, фенофибрат и гемфиброзил; и статин, такой как аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин и симвастатин.
В составе растворимого в липиде лекарственного средства не используется или не требуется поверхностно-активное вещество. Кроме того, в составе растворимого в липиде лекарственного средства не используется или не требуется растворитель не на основе липида.
Композиции твердых растворов, приготовленных с использованием состава лекарственного средства в форме свободной кислоты/свободного основания, требуют наличия стабилизатора помимо липидного компонента для введения раскрытого в изобретении терапевтического соединения в композицию твердого раствора. Терапевтическое соединение в форме свободной кислоты или свободного основания может растворяться в липиде при нагревании, но при охлаждении до комнатной температуры будет кристаллизоваться с образованием классической твердой композиции. Это происходит из-за термодинамических свойства этих смесей сохранять на более низком уровне энергию твердой фазы. Для приготовления композиции твердого раствора необходимо добавлять стабилизатор для стабилизации лекарственного средства и предотвращения перехода твердого раствора в классическую твердую фазу при охлаждении. Не приводя в качестве доказательства какую-либо теорию, тем не менее, можно предположить, что стабилизатор заключает в оболочку матрицы липид-лекарственное средство при их образовании. Эта оболочка препятствует взаимодействиям между матрицами и тем самым предотвращает упорядочения, необходимые для образования кристаллической матрицы твердофазной композиции. По этой причине, переход в твердую фазу не происходит и образуется композиция твердого раствора. Таким образом, стабилизаторы представляют собой соединение, которое обеспечивает термодинамический барьер для перехода в классическую твердую фазу или пролонгирует этот переход в такой степени, что он не происходит. Примеры стабилизаторов включают жидкие полиэтиленгликоли, диметиловый эфир изосорбида, моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол), моногидраты спиртов.
Обычно, в составе лекарственного средства в форме свободной кислоты/свободного основания используют терапевтические соединения, имеющие величину logP от приблизительно 2,2 до приблизительно 3,0. Не ограничивающие примеры включают нестероидное противовоспалительное лекарственное средство (NSAID) и эфир аминобензойной кислоты. NSAID включает салицилатное производное, парааминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты (оксикам), производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2). Эфир аминобензойной кислоты включает амилокаин, бензокаин, бутакаин, бутамбен, хлорпрокаин, диметокаин, лидокаин, меприлкаин, метабутетамин, метабутоксикаин, ортокаин, прилокаин, пропоксикаин, прокаин (новокаин), проксиметакаин, ризокаин и тетракаин.
Композиции твердых растворов, приготовленных с использованием состава солевой формы лекарственного средства, требуют наличия нейтрализатора помимо липидного компонента для введения раскрытого в изобретении терапевтического соединения в композицию твердого раствора. Солевая форма терапевтического соединения может растворяться в липиде при нагревании, но при охлаждении до комнатной температуры будет кристаллизоваться с образованием классической твердой композиции. Это происходит из-за термодинамических свойства этих смесей сохранять на более низком уровне энергию твердой фазы. Для приготовления композиции твердого раствора необходимо добавлять нейтрализатор для нейтрализации солевой формы лекарственного средства и предотвращения перехода твердого раствора в классическую твердую фазу при охлаждении. Не приводя в качестве доказательства какую-либо теорию, тем не менее, можно предположить, что нейтрализатор удаляет заряды, присутствующие на солевых формах лекарственных средств. Эта нейтрализация препятствует ионным взаимодействиям между матрицами и тем самым предотвращает упорядочения, необходимые для образования кристаллической матрицы твердофазной композиции. По этой причине, переход в твердую фазу не происходит и образуется композиция твердого раствора. Таким образом, нейтрализаторы представляют собой соединение, которое обеспечивает термодинамический барьер для перехода в классическую твердую фазу или пролонгирует этот переход в такой степени, что он не происходит.
Нейтрализаторы включают жирные кислоты для лекарственных средств в форме солей присоединения основания и триэтиламин для лекарственных средств в форме солей присоединения кислоты. Степень нейтрализации зависит от количества нейтрализатора, добавляемого к составу. Для полной нейтрализации, к составу добавляют один эквивалент нейтрализатора. Для частичной нейтрализации, добавляют меньше одного эквивалента нейтрализатора. Частичная нейтрализация предпочтительна при приготовлении состава с замедленным высвобождением. После введения, часть лекарственного средства моментально становится доступной для организма (мгновенная биодоступность), в то время как биодоступность другой части замедляется до тех пор, пока эта часть не будет нейтрализована с помощью нейтрализатора. Нейтрализатор может быть также добавлен в избыточном количестве, то есть в количестве более чем один эквивалент. Кроме нейтрализации солевой формы лекарственного средства, избыточные количества нейтрализатора могут также позволять корректировать температуру плавления композиции твердого раствора.
Обычно, в составе солевой формы лекарственного средства используют терапевтические соединения, имеющие величину logP приблизительно 2,2 или менее. Не ограничивающие примеры включают антагонисты рецептора рианодина, такие как азумолен и дантролен; и антагонисты рецепторов ангиотензина II, такие как азилсартан, кандесартан, эпросартан, ирбесартан, лосартан, олмесартан, телмисартан и валсартан.
Композиции твердых растворов могут также включать различные комбинации растворимых в липиде лекарственных средств, лекарственных средств в форме свободной кислоты/свободного основания и солевых форм лекарственных средств. В зависимости от используемых лекарственных средств, такие составы, помимо липидного компонента и лекарственного средства, могут также включать стабилизатор, нейтрализатор или и тот и другой.
Аспекты настоящего изобретения раскрывают, помимо прочего, терапевтическое соединение. Терапевтическое соединение представляет собой соединение, которое оказывает фармакологическое действие или другое непосредственное действие при постановке диагноза, курсе лечения, уменьшении отрицательных последствий, терапии или предотвращении заболевания, или воздействует на структуру или любую функцию организма человека или животных. Раскрытое в изобретении терапевтическое соединение может быть использовано в форме фармацевтически приемлемой соли, сольвата или сольвата соли, например гидрохлорида. Кроме того, раскрытое в изобретении терапевтическое соединение может быть получено в виде рацематов или в виде индивидуальных энантиомеров, включающих R- или S-энантиомер. Так, раскрытое в изобретение терапевтическое соединение может включать только R-энантиомер, только S-энантиомер или комбинацию и R-энантиомера и S-энантиомера терапевтического соединения. Раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием.
В одном варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни индуцирующих воспаление молекул. В аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни вещества P (SP), генетически родственного кальцитонину пептида (CGRP), глутамата или их комбинации. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни SP, CGRP, глутамата или их комбинации, высвобождаемых из сенсорного нейрона, например, по меньшей мере, на 10%, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90% или, по меньшей мере, на 95%. В еще одних аспектах варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни SP, CGRP, глутамата или их комбинации, высвобождаемых из сенсорного нейрона, на величину в диапазоне, например, от приблизительно 10% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 10% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 10% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, или от приблизительно 60% до приблизительно 80%, от приблизительно 10% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 40% до приблизительно 70% или от приблизительно 50% до приблизительно 70%.
Простагландины опосредуют местную ответную воспалительную реакцию и принимают участие во всех воспалительных функциях через воздействие на простагландиновые рецепторы и опосредование сигнального пути воспаления, включающего хемотаксис (макрофаги, нейтрофилы и эозинофилы), вазодилатацию и алгезию. Однако PG-опосредованная ответная воспалительная реакция является самоограничивающейся (разрешающейся). Главным фактором разрешения является простагландин, называемый 15dPGJ2, который представляет собой эндогенный агонист сигнального пути гамма-рецептора, активируемого пролифератором пероксисом (PPAR-γ). PPARγ сигнальный путь 1) индуцирует апоптоз макрофагов M1, вследствие чего снижаются уровни Th1 провоспалительных цитокинов и 2) промотирует дифференцировку моноцитов в макрофаги M2. Макрофаги M2 продуцируют и высвобождают Th2 провоспалительные цитокины.
В одном варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни индуцирующего воспаление простагландина. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни индуцирующего воспаление простагландина, высвобождающегося из сенсорного нейрона, например, по меньшей мере, на 10%, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90% или, по меньшей мере, на 95%. В еще одних аспектах варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни индуцирующего воспаление простагландина, высвобождающегося из сенсорного нейрона, на величину в диапазоне, например, от приблизительно 10% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 10% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 10% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, или от приблизительно 60% до приблизительно 80%, от приблизительно 10% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 40% до приблизительно 70% или от приблизительно 50% до приблизительно 70%.
В другом варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, практически аналогичным действию 15dPGJ2. В аспектах этого варианта осуществления, раскрытое в изобретение терапевтическое соединение имеет противовоспалительную активность, которая составляет, например, по меньшей мере, 5%, по меньшей мере, 15%, по меньшей мере, 25%, по меньшей мере, 50%, по меньшей мере, 55%, по меньшей мере, 60%, по меньшей мере, 65%, по меньшей мере, 70%, по меньшей мере, 75%, по меньшей мере, 80%, по меньшей мере, 85%, по меньшей мере, 90% или, по меньшей мере, 95% от активности, наблюдаемой для 15dPGJ2. В другом аспекте этого варианта осуществления, раскрытое в изобретение терапевтическое соединение имеет противовоспалительную активность, которая составляет величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 25% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 80% до приблизительно 90%, от приблизительно 25% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 60% до приблизительно 80%, от приблизительно 70% до приблизительно 80%, от приблизительно 25% до приблизительно 70%, от приблизительно 50% до приблизительно 70%, от приблизительно 25% до приблизительно 60%, от приблизительно 50% до приблизительно 60%, или от приблизительно 25% до приблизительно 50% от активности, наблюдаемой для 15dPGJ2.
Рецепторы, активируемые пролифератором пероксисом (PPAR) представляют собой группу белков ядерных рецепторов, которые выполняют функции факторов транскрипции, регулирующих экспрессию генов. Известно, что все PPAR гетеродимеризуются с ретиноидным X рецептором (RXR) и связываются со специфическими участками на ДНК генов-мишеней, называемыми элементами гормонального ответа пролифератора пероксисом (PPRE). PPAR играют важную роль в регуляции клеточной дифференцировки, развития и метаболизма (углевода, липида, белка), и в онкогенезе у высших организмов. Семейство включает трех представителей, PPAR-α, PPAR-γ и PPAR-δ (также называемый, как PPAR-β). PPAR-α экспрессируется в печени, почках, сердце, мышцах, жировой ткани, а также в других тканях. PPAR-δ экспрессируется во многих тканях, но значительно в мозге, жировой ткани и коже. PPAR-γ включает три альтернативно-сплайсированные формы, каждую с различным характером экспрессии. PPAR-γ1 экспрессируется практически во всех тканях, включая сердце, мышцы, толстую кишку, почки, поджелудочную железу и селезенку. PPAR-γ2 экспрессируется в основном в жировой ткани. PPAR-γ3 экспрессируется в макрофагах, толстом кишечнике и белой жировой ткани. Эндогенные лиганды для PPAR включают свободные жирные кислоты и эйкозаноиды. PPAR-γ активируется PGJ2 (простагландином), в то время как PPAR-α активируется лейкотриеном B4.
В одном варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно стимулировать все сигнальные пути PPAR. Такое терапевтическое соединение включает пан-агонист PPAR. В других вариантах осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое стимулирует один или два сигнальных пути PPAR. Такое терапевтическое соединение включает селективный агонист PPAR.
В другом варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно стимулировать сигнальный путь PPAR-α. В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение стимулирует сигнальный путь PPAR-α, например, по меньшей мере, на 5%, по меньшей мере, на 15%, по меньшей мере, на 25%, по меньшей мере, на 50%, по меньшей мере, на 60%, по меньшей мере, на 70%, по меньшей мере, на 80% или, по меньшей мере, на 90%. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение стимулирует сигнальный путь PPAR-α на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 25% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 80% до приблизительно 90%, от приблизительно 25% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 60% до приблизительно 80%, от приблизительно 70% до приблизительно 80%, от приблизительно 25% до приблизительно 70%, от приблизительно 50% до приблизительно 70%, от приблизительно 25% до приблизительно 60%, от приблизительно 50% до приблизительно 60% или от приблизительно 25% до приблизительно 50%.
В другом варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно стимулировать сигнальный путь PPAR-δ. В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение стимулирует сигнальный путь PPAR-δ, например, по меньшей мере, на 5%, по меньшей мере, на 15%, по меньшей мере, на 25%, по меньшей мере, на 50%, по меньшей мере, на 60%, по меньшей мере, на 70%, по меньшей мере, на 80% или, по меньшей мере, на 90%. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение стимулирует сигнальный путь PPAR-δ на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 25% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 80% до приблизительно 90%, от приблизительно 25% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 60% до приблизительно 80%, от приблизительно 70% до приблизительно 80%, от приблизительно 25% до приблизительно 70%, от приблизительно 50% до приблизительно 70%, от приблизительно 25% до приблизительно 60%, от приблизительно 50% до приблизительно 60% или от приблизительно 25% до приблизительно 50%.
В другом варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно стимулировать сигнальный путь PPARγ. Раскрытые в изобретении терапевтические соединения способны связываться со всеми изоформами PPAR-γ, или способны селективно связываться с одним из PPAR-γ1, PPAR-γ2, PPAR-γ3, или с любой их комбинацией из двух. В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение стимулирует сигнальный путь PPARγ, например, по меньшей мере, на 5%, по меньшей мере, на 15%, по меньшей мере, на 25%, по меньшей мере, на 50%, по меньшей мере, на 60%, по меньшей мере, на 70%, по меньшей мере, на 80% или, по меньшей мере, на 90%. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение стимулирует сигнальный путь PPARγ на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 25% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 80% до приблизительно 90%, от приблизительно 25% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 60% до приблизительно 80%, от приблизительно 70% до приблизительно 80%, от приблизительно 25% до приблизительно 70%, от приблизительно 50% до приблизительно 70%, от приблизительно 25% до приблизительно 60%, от приблизительно 50% до приблизительно 60% или от приблизительно 25% до приблизительно 50%.
Макрофаги активируются и поляризуются в отличающиеся фенотипы, экспрессирующие уникальные поверхностные молекулы клетки и секретирующие дискретные наборы цитокинов и хемокинов. Классический фенотип M1 поддерживает провоспалительные Th1 ответы, обусловленные цитокинами, такими как, например, интерлейкин-6 (IL-6), IL-12 и IL-23, в то время как альтернативный фенотип M2 обычно поддерживает противовоспалительные процессы, обусловленные IL-10. Клетки M2 могут быть дополнительно классифицированы на субпопуляции M2a, M2b, и M2c, на основе типа стимуляции и последующей экспрессии поверхностных молекул и цитокинов.
В еще одном варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно промотировать разрешение фенотипического изменения M1 в M2. В аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно индуцировать апоптоз макрофагов M1. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно промотировать дифференцировку макрофагов M2. В еще одном аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно индуцировать апоптоз макрофагов M1 и промотировать дифференцировку макрофагов M2.
В еще одном варианте осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно модулировать Th1 и Th2 цитокины. В аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни интерферон-гамма (IFNγ), фактора некроза опухолей альфа (TNF-α), IL-12 или их комбинации, высвобождаемых из клеток Th1. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни IFNγ, TNF-α, IL-12 или их комбинации, высвобождаемых из клеток Th1, например, по меньшей мере, на 10%, по меньшей мере, на 20%, по меньшей мере, на 30%, по меньшей мере, на 40%, по меньшей мере, на 50%, по меньшей мере, на 60%, по меньшей мере, на 70%, по меньшей мере, на 80% или, по меньшей мере, на 90%. В еще одних аспектах варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни IFNγ, TNF-α, IL-12 или их комбинации, высвобождаемых из клеток Th1, на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 10% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 10% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 50% до приблизительно 80% или от приблизительно 60% до приблизительно 80%, от приблизительно 10% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 40% до приблизительно 70% или от приблизительно 50% до приблизительно 70%.
В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно повышать уровни IL-10, высвобождаемого из клеток Th2. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно повышать уровни IL-10, высвобождаемого из клеток Th2, например, по меньшей мере, на 10%, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90% или, по меньшей мере, на 95%. В еще одних аспектах варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно повышать уровни IL-10, высвобождаемого из клеток Th2, на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 10% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 10% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, или от приблизительно 60% до приблизительно 80%, от приблизительно 10% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 40% до приблизительно 70% или от приблизительно 50% до приблизительно 70%.
В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни IFNγ, TNF-α, IL-12 или их комбинации, высвобождаемых из клеток Th1, и повышать уровни IL-10, высвобождаемого из клеток Th2. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни IFNγ, TNF-α, IL-12 или их комбинации, высвобождаемых из клеток Th1, например, по меньшей мере, на 10%, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90% или, по меньшей мере, на 95%, и способно повышать уровни IL-10, высвобождаемого из клеток Th2, например, по меньшей мере, на 10%, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90% или, по меньшей мере, на 95%. В еще одних аспектах варианта осуществления, раскрытое в изобретении терапевтическое соединение обладает противовоспалительным действием, которое способно снижать уровни IFNγ, TNF-α, IL-12 или их комбинации, высвобождаемых из клеток Th1, на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 10% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 10% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, или от приблизительно 60% до приблизительно 80%, от приблизительно 10% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 40% до приблизительно 70% или от приблизительно 50% до приблизительно 70%, и способно повышать уровни IL-10, высвобождаемого из клеток Th2, на величину в диапазоне, например, от приблизительно 10% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 60% до приблизительно 100%, от приблизительно 70% до приблизительно 100%, от приблизительно 80% до приблизительно 100%, от приблизительно 10% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 60% до приблизительно 90%, от приблизительно 70% до приблизительно 90%, от приблизительно 10% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, или от приблизительно 60% до приблизительно 80%, от приблизительно 10% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 40% до приблизительно 70% или от приблизительно 50% до приблизительно 70%.
Раскрытое в изобретении терапевтическое соединение может иметь величину log P, указывающую на то, что соединение растворимо в органическом растворителе. Используемый в изобретении термин "величина log P" относится к десятичному логарифму коэффициента распределения (P) для соединения и является критерием липофильности. Обычно, P определяется как отношение концентраций неионизированного соединения в двух фазах смеси из двух несмешивающихся растворителей при равновесии. Таким образом, log P=Log 10 (P), где P=[растворенное вещество в несмешивающемся растворителе 1]/[растворенное вещество в несмешивающемся растворителе 2]. По отношению к органической и водной фазам, величина log P соединения является постоянной для данной пары водного и органического растворителей, и ее значение может быть определено эмпирически одним из нескольких методов межфазного распределения, известных любому специалисту в этой области, включающих например, метод встряхиваемой колбы, метод ВЭЖХ и метод исследования поверхности раздела между двумя несмешивающимися растворами электролитов (ITIES).
В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P, указывающее на то, что соединение является практически растворимым в органическом растворителе. В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P, указывающее на то, что соединение, например, по меньшей мере, на 50% растворимо в органическом растворителе, по меньшей мере, на 60% растворимо в органическом растворителе, по меньшей мере, на 70% растворимо в органическом растворителе, по меньшей мере, на 80% растворимо в органическом растворителе или, по меньшей мере, на 90% растворимо в органическом растворителе. В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P, указывающее на то, что соединение может быть, например, от приблизительно 50% до приблизительно 100% растворимо в органическом растворителе, от приблизительно 60% до приблизительно 100% растворимо в органическом растворителе, от приблизительно 70% до приблизительно 100% растворимо в органическом растворителе, от приблизительно 80% до приблизительно 100% растворимо в органическом растворителе или от приблизительно 90% до приблизительно 100% растворимо в органическом растворителе.
В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P, например, более чем 1,1, более чем 1,2, более чем 1,4, более чем 1,6, более чем 1,8, более чем 2,0, более чем 2,2, более чем 2,4, более чем 2,6, более чем 2,8, более чем 3,0, более чем 3,2, более чем 3,4 или более чем 3,6. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P в диапазоне, например, от 1,8 до 4,0, от 2,0 до 4,0, от 2,1 до 4,0, от 2,2 до 4,0 или от 2,3 до 4,0, от 2,4 до 4,0, от 2,5 до 4,0, от 2,6 до 4,0 или от 2,8 до 4,0. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P в диапазоне, например, от 3,0 до 4,0 или от 3,1 до 4,0, от 3,2 до 4,0, от 3,3 до 4,0, от 3,4 до 4,0, от 3,5 до 4,0 или от 3,6 до 4,0. В еще одних аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь значение log P в диапазоне, например, от 2,0 до 2,5, от 2,0 до 2,7, от 2,0 до 3,0 или от 2,0 до 2,5.
Раскрытое в изобретении терапевтическое соединение может иметь участок полярной поверхности, который является гидрофобным. Используемый в изобретении термин "участок полярной поверхности" относится к суммарной поверхности над всеми полярными атомами в структуре соединения и является критерием гидрофобности. Обычно, эти полярные атомы включают, например, кислород, азот и присоединенные к ним водороды. В аспектах этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь участок полярной поверхности размером, например, менее чем 8,0 нм2, менее чем 7,0 нм2, менее чем 6,0 нм2, менее чем 5,0 нм2, менее чем 4,0 нм2 или менее чем 3,0 нм2. В другом аспекте этого варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь участок полярной поверхности, например, от 3,0 нм2 до 6,5 нм2, от 3,0 нм2 до 6,0 нм2, от 3,0 нм2 до 5,5 нм2, от 3,0 нм2 до 5,0 нм2, от 3,0 нм2 до 4,5 нм2, от 3,5 нм2 до 6,5 нм2, от 3,5 нм2 до 6,0 нм2, от 3,5 нм2 до 5,5 нм2, от 3,5 нм2 до 5,0 нм2, от 3,5 нм2 до 4,5 нм2, от 4,0 нм2 до 6,5 нм2, от 4,0 нм2 до 6,0 нм2, от 4,0 нм2 до 5,5 нм2 или от 4,0 нм2 до 5,0 нм2, от 4,0 нм2 до 4,5 нм2 или от 4,5 нм2 до 5,5 нм2. В еще одних аспектах варианта осуществления, раскрытое в изобретении терапевтическое соединение может иметь участок полярной поверхности, например, от 2,0 нм2 до 6,5 нм2, от 2,0 нм2 до 6,0 нм2, от 2,0 нм2 до 5,5 нм2, от 2,0 нм2 до 5,0 нм2, от 2,0 нм2 до 4,5 нм2, от 2,5 нм2 до 6,5 нм2, от 2,5 нм2 до 6,0 нм2, от 2,5 нм2 до 5,5 нм2, от 2,5 нм2 до 5,0 нм2 или от 2,5 нм2 до 4,5 нм2.
Раскрытое в изобретении терапевтическое соединение может представлять собой нестероидное противовоспалительное лекарственное средство (NSAID). NSAID образуют большую группу терапевтических соединений с болеутоляющими, противовоспалительными и жаропонижающими свойствами. NSAID снижают воспаление за счет блокирования циклооксигеназы. NSAID включают, без ограничения, ацеклофенак, ацеметацин, актирит, алкофенак, алминопрофен, амфенак, алоксипирин, аминофеназон, антрафенин, аспирин, азапропазон, бенорилат, беноксапрофен, бензидамин, бутибуфен, целекоксиб, хлортеноксацин, холин салицилат, клометацин, декскетопрофен, диклофенак, дифлунизал, эморфазон, эпиризол; этодолак, эторикоксиб, феклобузон, фелбинак, фенбуфен, фенклофенак, флурбипрофен, глафенин, гидроксилэтил салицилат, ибупрофен, индометацин, индопрофен, кетопрофен, кеторолак, лактил фенетидин, локсопрофен, люмиракоксиб, мефенамовую кислоту, мелоксикам, метамизол, метиазиновую кислоту, мофебутазон, мофезолак, набуметон, напроксен, нифеназон, нифлумовую кислоту, оксаметацин, фенацетин, пипебузон, пранопрофен, пропифеназон, проквазон, протизиновую кислоту, рофекоксиб, салициламид, салсалат, сулиндак, супрофен, тиарамид, тиноридин, толфенамовую кислоту, валдекоксиб и зомепирак.
NSAID могут быть классифицированы на основе их химической структуры или механизма действия. Неограничивающие примеры NSAIDs включают салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1) и селективный ингибитор циклооксигеназы 2 (COX 2). NSAID может представлять собой профен. Примеры подходящего салицилатного производного включают, без ограничения, ацетилсалициловую кислоту (аспирин), дифлунизал и салсалат. Примеры подходящего п-аиминофенольного производного включают, без ограничения, Парацетамол и фенацетин. Примеры подходящего производного пропионовой кислоты включают, без ограничения, алминопрофен, беноксапрофен, декскетопрофен, фенопрофен, флурбипрофен, Ибупрофен, индопрофен, кетопрофен, локсопрофен, напроксен, оксапрозин, пранопрофен и супрофен. Примеры подходящего производного уксусной кислоты включают, без ограничения, ацеклофенак, ацеметацин, актирит, алкофенак, амфенак, клометацин, диклофенак, этодолак, фелбинак, фенклофенак, индометацин, кеторолак, метиазиновую кислоту, мофезолак, набуметон, напроксен, оксаметацин, сулиндак и зомепирак. Примеры подходящего производного эноловой кислоты (оксикама) включают, без ограничения, дроксикам, изоксикам, лорноксикам, мелоксикам, пироксикам и теноксикам. примерыподходящего производного фенамовой кислоты включают, без ограничения, флуфенамовую кислоту, мефенамовую кислоту, меклофенамовую кислоту и толфенамовую кислоту. Примеры подходящих селективных ингибиторов COX-2 включают, без ограничения, целекоксиб, эторикоксиб, фирококсиб, люмиракоксиб, мелоксикам, парацетамол (ацетоминофен), парекоксиб, рофекоксиб и валдекоксиб.
Раскрытое в изобретении терапевтическое соединение может представлять собой агонист PPARα. Примеры подходящего агониста PPARα включают, без ограничения, пириниксик (WY 14643), GW6471, и фибрат.
Раскрытое в изобретении терапевтическое соединение представляет собой агонист PPARβ/δ. Примеры подходящего агониста PPARβ/δ включают, без ограничения, тетрадецилтиоуксусную кислоту (TTA). GSK0660, GSK3787, GW501516 (GW-501,516, GW1516, GSK-516 и эндуробол), GW0742 и GW610742X.
Раскрытое в изобретении терапевтическое соединение может представлять собой агонист PPARγ. Примеры подходящего агониста PPARγ включают, без ограничения, монасцин, тиазолидиндионы, такие как росиглитазон, пиоглитазон и троглитазон и T0070907. Другие подходящие агонисты PPARγ описаны в патентном документе Masson and Caumont-Bertrand, PPAR Agonist Compounds, Preparation and Uses, US 2011/0195993, содержание которого приводится в настоящем изобретении путем ссылки на него.
Раскрытое в изобретении терапевтическое соединение может представлять собой глитазар (двойной агонист α и γ PPAR). Примеры подходящего глитазара включают, без ограничения, алеглитазар, мураглитазар, сароглитазар и тесаглитазар.
Раскрытое в изобретении терапевтическое соединение может представлять собой иммуносупрессивное лекарственное средство. Примеры подходящего иммуносупрессивного лекарственного средства включают, без ограничения, азатиоприн и микофеноловую кислоту.
Раскрытое в изобретении терапевтическое соединение может представлять собой лекарственное средство, способствующее выведению мочевой кислоты. Примеры подходящего лекарственного средства, способствующего выведению мочевой кислоты, включают, без ограничения, бензбромарон.
Раскрытое в изобретении терапевтическое соединение может представлять собой агликон. Примеры подходящего лекарственного средства агликон включают, без ограничения, пицеатаннол, пиносилвин, птеростилбен и ресвератрол.
Раскрытое в изобретении терапевтическое соединение может представлять собой каннабидиол. Примеры подходящего лекарственного средства, способствующего выведению мочевой кислоты, включают, без ограничения, фитоканнабиноид, эндоканнабиноид и синтетический каннабиноид. Фитоканнабиноид включает тетрагидроканнабинол (такой как, например, дельта-9-тетрагидроканнабинол (δ9-THC, THC) и дельта-8-тетрагидроканнабинол (δ8-THC)), каннабидиол, каннабинол, каннабигерол, тетрагидроканнабиварин, каннабидиварин и каннабихромен. Эндоканнабиноид включает арахидоноилэтаноламин (анандамид или AEA), 2-арахидоноилглицерин (2-AG), 2-арахидоноилглицериновый эфир (ноладиновый эфир), N-арахидоноилдопамин (NADA), виродгамин (OAE) и лизофосфатидилинозитол (LPI). Синтетический каннабиноид включает дронабинол (маринол), набилон (цесамет), сативекс, римонабант (SR141716), JWH-018, JWH-073, CP-55940, диметилгептилпиран, HU-210, HU-331, SR144528, WIN 55,212-2, JWH-133, левонантрадол (нантродолум) и AM-2201.
Раскрытое в изобретении терапевтическое соединение может представлять собой средство, связывающее ядерный рецептор. Примеры подходящего средства, связывающего ядерный рецептор, включают, без ограничения, средство, связывающее рецептор ретиноевой кислоты (RAR), средство, связывающее ретиноидный X рецептор (RXR), средство, связывающее печеночный рецептор Х (LXR) и средство, связывающее витамин D.
Раскрытое в изобретении терапевтическое соединение может представлять собой антагонист рецепторов ангиотензина II. Примеры подходящего антагониста рецепторов ангиотензина II включают, без ограничения, азилсартан, кандесартан, эпросартан, ирбесартан, лосартан, олмесартан, телмисартан и валсартан.
Раскрытое в изобретении терапевтическое соединение может представлять собой ингибитор ацетилхолинэстеразы (ACE). Примеры подходящего ингибитора ACE включают, без ограничения, сульфгидрилсодержащее средство, дикарбоксилатсодержащее средство, фосфонатсодержащее средство, касокинин и лактокинин. Сульфгидрилсодержащее средство включает каптоприл (капотен) и зофеноприл. Дикарбоксилатсодержащее средство включает эналаприл (вазотек/ренитек), рамиприл (алтаце/прилаце/рамаце/рамивин/ триатек/тритаце), хинаприл (аккуприл), периндоприл (коверсил/ацеон), лизиноприл (листрил/лоприл/новатек/принивил/ зестрил), беназеприл (лотензин), имидаприл (танатрил), зофеноприл (зофекард) и трандолаприл (мавик/одрик/гоптен). Фосфонатсодержащее средство включает фосиноприл (фоситен/моноприл).
Раскрытое в изобретении терапевтическое соединение может представлять собой ингибитор фосфодиэстеразы. Примеры подходящего ингибитора фосфодиэстеразы включают, без ограничения, селективный ингибитор PDE 1, селективный ингибитор PDE 2, селективный ингибитор PDE 3, селективный ингибитор PDE 4, селективный ингибитор PDE 5 и селективный ингибитор PDE 10. Селективный ингибитор PDE 1 включает випроцетин. Селективный ингибитор PDE 2 включает BAY 60-7550 (2-[(3,4-диметоксифенил)метил]-7-[(1R)-1-гидроксиэтил]-4-фенилбутил]-5-метилимидазо[5,1-f][1,2,4]триазин-4(1H)-он), EHNA (эритро-9-(2-гидрокси-3-нонил)аденин), оксиндол и PDP (9-(6-фенил-2-оксогекс-3-ил)-2-(3,4-диметоксибензил)-пурин-6-он). Селективный ингибитор PDE3 включает анагрелид, цилостазол, эноксимон, инамринон и милринон. Селективный ингибитор PDE4 включает дротаверин, ибудиласт, лутеолин, месембрин, пикламиласт, рофлумиласт и ролипрам. Селективный ингибитор PDE5 включает аванафил, дипиридамол, икариин, 4-Метилпиперазин, пиразолопиримидин-7-1, силденафил, тадалафил, уденафил и варденафил. Селективный ингибитор PDE10 включает папаверин.
Раскрытое в изобретении терапевтическое соединение может представлять собой фибрат. Фибраты образуют класс амфипатических карбоновых кислот, способных модифицировать уровень липидов. Эти терапевтические соединения применяют при ряде метаболических нарушений. Одним неограничивающим применением является их использование в качестве антигиперлипидемического средства, которое может понижать уровни, например, триглицеридов и LDL, а также повышать уровни HDL. Примеры подходящего фибрата включают, без ограничения, безафибрат, ципрофибрат, клофибрат, гемфиброзил и фенофибрат.
Раскрытое в изобретении терапевтическое соединение может представлять собой статин. Статины (или ингибиторы HMG-CoA редуктазы) образуют класс терапевтических соединений, применяемых для понижения уровней LDL и/или холестерина путем ингибирования фермента HMG-CoA редуктазы, которая играет главную роль в продукции холестерина в печени. Для компенсации пониженной доступности холестерина, повышается синтез печеночных рецепторов LDL, что приводит к повышенному выведению частиц LDL из крови. Примеры подходящего статина включают, без ограничения, аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин и симвастатин.
Раскрытое в изобретении терапевтическое соединение может представлять собой токотриенол. Токотриенолы образуют другой класс ингибиторов HMG-CoA редуктазы, и они могут применяться для снижения уровней LDL и/или холестерина путем индуцирования активации печеночного рецептора LDL или снижения уровней LDL в плазме. Примеры подходящего токотриенола включают, без ограничения, γ-токотриенол и δ-токотриенол.
Раскрытое в изобретении терапевтическое соединение может представлять собой ниацин. Ниацины образуют класс терапевтических соединений, способных модифицировать уровни липидов. Например, ниацин может понижать LDL путем ингибирования печеночной диацилглицерол-ацилтрансферазы 2, уменьшать синтез триглицеридов и секрецию VLDL через рецептор HM74 и HM74A или GPR109A. Эти терапевтические соединения применяют при ряде метаболических нарушений. Одним неограничивающим применением является его использования в качестве антигиперлипидемического средства, которое может ингибировать распад жиров в жировой ткани. Так как ниацин блокирует распад жиров, он вызывает увеличение содержания свободных жирных кислот в крови и, вследствие этого, снижает секрецию липопротеинов очень низкой плотности (VLDL) и холестерина печенью. Путем снижения уровней VLDL, ниацин может также повышать уровень HDL в крови. Примеры подходящего ниацина включают, без ограничения, аципимокс, ниацин, никотинамид и витамин B3.
Раскрытое в изобретении терапевтическое соединение может представлять собой секвестрант желчных кислот. Секвестранты желчных кислот (называемые также смолами) образуют класс терапевтических соединений, применяемых для связывания конкретных компонентов желчи в желудочно-кишечном тракте. Они нарушают печеночно-кишечную рециркуляцию желчных кислот путем секвестирования их и предотвращения их реабсорбции из кишок. Секвестранты желчных кислот особенно эффективны для снижения LDL и холестерина путем секвестирования содержащих холестерин желчных кислот, высвобождающихся в кишечнике, и предотвращения их реабсорбции из кишечника. Кроме того, секвестранты желчных кислот могут также повышать уровни HDL. Примеры подходящего секвестранта желчных кислот включают, без ограничения, холестирамин, колесевелам и колестипол.
Раскрытое в изобретении терапевтическое соединение может представлять собой ингибитор абсорбции холестерина. Ингибиторы абсорбции холестерина образуют класс терапевтических соединений, которые ингибируют абсорбцию холестерина из кишечника. Пониженная абсорбция холестерина приводит к повышенной регуляции LDL-рецепторов на поверхности клеток и повышенному усвоению LDL-холестерина в этих клетках, таким образом понижая уровни LDL в плазме крови. Примеры подходящего ингибитора абсорбции холестерина включают, без ограничения, эзетимид, фитостерол, стерол и станол.
Раскрытое в изобретении терапевтическое соединение может представлять собой ингибитор абсорбции жира. Ингибиторы абсорбции жира образуют класс терапевтических соединений, которые ингибируют абсорбцию жира из кишечника. Пониженная абсорбция жира уменьшает калорийность потребляемой пищи. В одном аспекте, ингибитор абсорбции жира ингибирует липазу поджелудочной железы, фермент, который разрушает триглицериды в кишечнике. Примеры подходящего ингибитора абсорбции жира включают, без ограничения орлистат.
Раскрытое в изобретении терапевтическое соединение может представлять собой симпатомиметический амин. Симпатомиметические амины образуют класс терапевтических соединений, которые имитируют действия трансмиттерных веществ симпатической нервной системы, таких как катехоламины, эпинефрин (адреналин), норэпинефрин (норадреналин) и/или допамин. Симпатомиметический амин может действовать в качестве α-адренергического агониста, β-адренергического агониста, допаминергического агониста, ингибитора моноаминоксидазы (MAO) и ингибитора COMT. Такие терапевтические соединения применяют, помимо прочего, для лечения задержки очередного сердечного сокращения, низкого кровяного давления или даже для отсрочки преждевременных родов. Примеры подходящего симпатомиметического амина включают, без ограничения, кленбутерол, сальбутамол, эфедрин, псевдоэфедрин, метамфетамин, амфетамин, фенилэфрин, изопротеренол, добутамин, метилфенидат, лисдексамфетамин, катин, катинон, меткатинон, кокаин, бензилпиперазин (BZP), метилендиоксипировалерон (MDPV), 4-метиламинорекс, пемолин, фенметразин и пропилгекседрин. α-Адренергический агонист включает фенилеприн, пропилгекседрин и псевдоэфедрин. β-Адренергический агонист включает кленбутерол, добутамин, эфедрин, изопротеренол и сальбутамол. Допаминергический/норэпинефринергический агонист включает кокаин (ингибитор обратного захвата допамина (DA)/норэпинефрина (NE)), лисдексамфетамин (ингибитор обратного захвата 5HT/DA/NE), метилфенидат (ингибитор обратного захвата DA/NE) и метилендиоксипировалерон (ингибитор обратного захвата DA/NE). Средство, высвобождающее нейротрансмиттер, включает амфетамин (средство, высвобождающее DA/NE), бензилпиперазин (средство, высвобождающее DA/NE), катин (средство, высвобождающее DA/NE), катинон (средство, высвобождающее DA/NE), метамфетамин (средство, высвобождающее DA/NE), меткатинон (средство, высвобождающее DA/NE), 4-метиламинорекс (средство, высвобождающее DA/NE), пемолин, фенметразин (средство, высвобождающее DA/NE) и фенетиламин (средство, высвобождающее DA/NE).
Раскрытое в изобретении терапевтическое соединение может представлять собой антагонист рецептора рианодина. Примеры антагониста рецептора рианодина включают, без ограничения, азумолен и дантролен.
Раскрытое в изобретении терапевтическое соединение может представлять собой противоопухолевое лекарственное средство. Примеры подходящего противоопухолевого лекарственного средства включают, без ограничения, алкилирующее средство, антиметаболит, растительный алкалоид и терпеноид, ингибитор топоизомеразы и цитотоксический антибиотик. Алкилирующее средство включает карбоплатин, хлорамбуцил, цисплатин, циклофосфамид, ифосфамид, оксалиплатин и мехлорэтамин. Антиметаболит включает азатиоприн и меркаптопурин. Растительный алкалоид и терпеноид включают алкалоид барвинка, такой как винкристин, винбластин, винорелбин и виндезин, подофиллотоксин, такой как этопозид и тенипозид, и таксан, такой как доцетаксел и ортатаксел. Ингибитор топоизомеразы включает ингибитор топоизомеразы типа I, такой как камптотецины, такие, например, как экзатекан, иринотекан, луртотекан, топотекан, BNP 1350, CKD 602, DB 67 (AR67) и ST 1481, и ингибитор типа II, такой как эпидофиллотоксин, такой, например, как амсакрин, этопозид, этопозида фосфат и тенипозид. Цитотоксический антибиотик включает актиномицин, такой как актиномицин D, бацитрацин, колистин (полимиксин E) и полимиксин B, антрацендион, такой как митоксантрон и пиксантрон, и антрациклин, такой как блеомицин, доксорубицин (адриамицин), даунорубицин (дауномицин), эпирубицин, идарубицин, митомицин, пликамицин и валрубицин.
Раскрытое в изобретении терапевтическое соединение может представлять собой метформин, куркумин, глицирретиновую кислоту или 6-шогаол.
Раскрытое в изобретении терапевтическое соединение может представлять собой антибиотик. Примеры подходящего симпатомиметического амина включают, без ограничения, изониазид, рифампицин, пиразинамид и этамбутол.
Раскрытое в изобретении терапевтическое соединение может представлять собой противоглистное лекарственное средство. Примеры подходящего противоглистного лекарственного средства включают, без ограничения, абамектин, аминоацетонитрил, такой как монепантел, бензимидазол, диэтилкарбамазин, ивермектин, левамизол, никлозамид, октадепсипептид, такой как эмодепсид, фосфоновую кислоту (метрифонат), празиквантел, спироиндол, такой как дерквантел и сурамин, пирантела памоат. Бензимидазол включает альбендазол, фербендазол, флубендазол, мебендазол, тиабендазол и триклабендазол.
Раскрытое в изобретении терапевтическое соединение может представлять собой противомалярийное лекарственное средство. Примеры подходящего противомалярийного лекарственного средства включают, без ограничения, амодиахин, артемизинин, атоваквон, хлорохин, клиндамицин, доксициклин, галофантрин, мефлохин, примахин, прогуанил, пириметамин, хинин и относящееся к нему лекарственное средство, такое как хинимакс и хинидин, руфигаллол и сульфонамид, такой как сульфадоксин и сульфаметоксипиридазин. Артемизинин включает артеэфир, артеметер, артемизинин, артесунат и дигидроартемизинин.
Раскрытое в изобретении терапевтическое соединение может представлять собой антигиперлипидемическое средство. Существует несколько классов антигиперлипидемических средств (называемых также гиполипидемическими средствами). Они могут различаться как по их воздействию на холестериновый профиль, так и по побочным эффектам. Например, некоторые из них могут понижать липопротеин низкой плотности (LDL), в то время как другие могут в основном повышать липопротеин высокой плотности (HDL). В клиническом аспекте, выбор средства будет зависеть от холестеринового профиля индивидуума, кардиоваскулярных факторов риска индивидуума и/или функционирования печени и почек индивидуума. Примеры подходящего антигиперлипидемического средства включают, без ограничения, антагонист рецепторов ангиотензина II, ингибитор ACE, ингибитор фосфодиэстеразы, фибрат, статин, токотриенол, ниацин, секвестранты желчных кислот (смолу), ингибитор абсорбции холестерина, ингибитор липазы поджелудочной железы и симпатомиметический амин.
Раскрытое в изобретении терапевтическое соединение может представлять собой эфир терапевтического соединения. Эфир терапевтического соединения повышает значение logP относительно значения logP терапевтического соединения, которое не было подвергнуто превращению в эфир. Эфирная группа может быть присоединена к терапевтическому соединению, например, по карбоксильной или гидроксильной функциональной группе, присутствующей в терапевтическом соединении. Эфир терапевтического соединения может обладать повышенной гидрофобностью и по этой причине может растворяться в меньшем объеме раскрываемого в изобретении растворителя. В некоторых случаях, эфир терапевтического соединения может быть объединен непосредственно с описанным в изобретении вспомогательным веществом, в результате чего отпадает необходимость в использовании растворителя. Эфир терапевтического соединения дает возможность получать раскрытую в изобретении фармацевтическую композицию в тех ситуациях, когда неэтерифицированная форма этого же терапевтического соединения не смешивается с раскрытым в изобретении растворителем. Эфир терапевтического соединения может, при этом, быть доставлен так, что он более эффективно ингибирует ответную провоспалительную реакцию при условии, что соединение объединено с описанным в изобретении вспомогательным веществом. В одном варианте осуществления, терапевтическое соединение может быть подвергнуто взаимодействию с этиловым эфиром для образования этилового эфира терапевтического соединения.
Раскрытая в изобретении фармацевтическая композиция твердого раствора может включать терапевтическое соединение в количестве, достаточном для традиционного введения индивидууму. В аспектах этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, по меньшей мере, 5 мг, по меньшей мере, 10 мг, по меньшей мере, 15 мг, по меньшей мере, 20 мг, по меньшей мере, 25 мг, по меньшей мере, 30 мг, по меньшей мере, 35 мг, по меньшей мере, 40 мг, по меньшей мере, 45 мг, по меньшей мере, 50 мг, по меньшей мере, 55 мг, по меньшей мере, 60 мг, по меньшей мере, 65 мг, по меньшей мере, 70 мг, по меньшей мере, 75 мг, по меньшей мере, 80 мг, по меньшей мере, 85 мг, по меньшей мере, 90 мг, по меньшей мере, 95 мг или, по меньшей мере, 100 мг. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, по меньшей мере, 5 мг, по меньшей мере, 10 мг, по меньшей мере, 20 мг, по меньшей мере, 25 мг, по меньшей мере, 50 мг, по меньшей мере, 75 мг, по меньшей мере, 100 мг, по меньшей мере, 200 мг, по меньшей мере, 300 мг, по меньшей мере, 400 мг, по меньшей мере, 500 мг, по меньшей мере, 600 мг, по меньшей мере, 700 мг, по меньшей мере, 800 мг, по меньшей мере, 900 мг, по меньшей мере, 1000 мг, по меньшей мере, 1100 мг, по меньшей мере, 1200 мг, по меньшей мере, 1300 мг, по меньшей мере, 1400 мг или, по меньшей мере, 1500 мг. В еще одних аспектах варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, от приблизительно 5 мг до приблизительно 100 мг, от приблизительно 10 мг до приблизительно 100 мг, от приблизительно 50 мг до приблизительно 150 мг, от приблизительно 100 мг до приблизительно 250 мг, от приблизительно 150 мг до приблизительно 350 мг, от приблизительно 250 мг до приблизительно 500 мг, от приблизительно 350 мг до приблизительно 600 мг, от приблизительно 500 мг до приблизительно 750 мг, от приблизительно 600 мг до приблизительно 900 мг, от приблизительно 750 мг до приблизительно 1000 мг, от приблизительно 850 мг до приблизительно 1200 мг или от приблизительно 1000 мг до приблизительно 1500 мг. В еще одних аспектах этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, от приблизительно 10 мг до приблизительно 250 мг, от приблизительно 10 мг до приблизительно 500 мг, от приблизительно 10 мг до приблизительно 750 мг, от приблизительно 10 мг до приблизительно 1000 мг, от приблизительно 10 мг до приблизительно 1500 мг, от приблизительно 50 мг до приблизительно 250 мг, от приблизительно 50 мг до приблизительно 500 мг, от приблизительно 50 мг до приблизительно 750 мг, от приблизительно 50 мг до приблизительно 1000 мг, от приблизительно 50 мг до приблизительно 1500 мг, от приблизительно 100 мг до приблизительно 250 мг, от приблизительно 100 мг до приблизительно 500 мг, от приблизительно 100 мг до приблизительно 750 мг, от приблизительно 100 мг до приблизительно 1000 мг, от приблизительно 100 мг до приблизительно 1500 мг, от приблизительно 200 мг до приблизительно 500 мг, от приблизительно 200 мг до приблизительно 750 мг, от приблизительно 200 мг до приблизительно 1000 мг, от приблизительно 200 мг до приблизительно 1500 мг, от приблизительно 5 мг до приблизительно 1500 мг, от приблизительно 5 мг до приблизительно 1000 мг или от приблизительно 5 мг до приблизительно 250 мг.
В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, менее чем приблизительно 90% по массе, менее чем приблизительно 80% по массе, менее чем приблизительно 70% по массе, менее чем приблизительно 65% по массе, менее чем приблизительно 60% по массе, менее чем приблизительно 55% по массе, менее чем приблизительно 50% по массе, менее чем приблизительно 45% по массе, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 5% по массе или менее чем приблизительно 1% по массе. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, приблизительно от 1% до 90% по массе, приблизительно от 1% до 80% по массе, приблизительно от 1% до 75% по массе, приблизительно от 1% до 70% по массе, приблизительно от 1% до 65% по массе, приблизительно от 1% до 60% по массе, приблизительно от 1% до 55% по массе, приблизительно от 1% до 50% по массе, приблизительно от 1% до 45% по массе, приблизительно от 1% до 40% по массе, приблизительно от 1% до 35% по массе, приблизительно от 1% до 30% по массе, приблизительно от 1% до 25% по массе, приблизительно от 1% до 20% по массе, приблизительно от 1% до 15% по массе, приблизительно от 1% до 10% по массе, приблизительно от 1% до 5% по массе, приблизительно от 2% до 50% по массе, приблизительно от 2% до 40% по массе, приблизительно от 2% до 30% по массе, приблизительно от 2% до 20% по массе, приблизительно от 2% до 10% по массе, приблизительно от 4% до 50% по массе, приблизительно от 4% до 40% по массе, приблизительно от 4% до 30% по массе, приблизительно от 4% до 20% по массе, приблизительно от 4% до 10% по массе, приблизительно от 6% до 50% по массе, приблизительно от 6% до 40% по массе, приблизительно от 6% до 30% по массе, приблизительно от 6% до 20% по массе, приблизительно от 6% до 10% по массе, приблизительно от 8% до 50% по массе, приблизительно от 8% до 40% по массе, приблизительно от 8% до 30% по массе, приблизительно от 8% до 20% по массе, приблизительно от 8% до 15% по массе или приблизительно от 8% до 12% по массе.
В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в количестве, например, от приблизительно 0,1% до приблизительно 45% по массе, от приблизительно 0,1% до приблизительно 40% по массе, от приблизительно 0,1% до приблизительно 35% по массе, от приблизительно 0,1% до приблизительно 30% по массе, от приблизительно 0,1% до приблизительно 25% по массе, от приблизительно 0,1% до приблизительно 20% по массе, от приблизительно 0,1% до приблизительно 15% по массе, от приблизительно 0,1% до приблизительно 10% по массе, от приблизительно 0,1% до приблизительно 5% по массе, от приблизительно 1% до приблизительно 45% по массе, от приблизительно 1% до приблизительно 40% по массе, от приблизительно 1% до приблизительно 35% по массе, от приблизительно 1% до приблизительно 30% по массе, от приблизительно 1% до приблизительно 25% по массе, от приблизительно 1% до приблизительно 20% по массе, от приблизительно 1% до приблизительно 15% по массе, от приблизительно 1% до приблизительно 10% по массе, от приблизительно 1% до приблизительно 5% по массе, от приблизительно 5% до приблизительно 45% по массе, от приблизительно 5% до приблизительно 40% по массе, от приблизительно 5% до приблизительно 35% по массе, от приблизительно 5% до приблизительно 30% по массе, от приблизительно 5% до приблизительно 25% по массе, от приблизительно 5% до приблизительно 20% по массе, от приблизительно 5% до приблизительно 15% по массе, от приблизительно 5% до приблизительно 10% по массе, от приблизительно 10% до приблизительно 45% по массе, от приблизительно 10% до приблизительно 40% по массе, от приблизительно 10% до приблизительно 35% по массе, от приблизительно 10% до приблизительно 30% по массе, от приблизительно 10% до приблизительно 25% по массе, от приблизительно 10% до приблизительно 20% по массе, от приблизительно 10% до приблизительно 15% по массе, от приблизительно 15% до приблизительно 45% по массе, от приблизительно 15% до приблизительно 40% по массе, от приблизительно 15% до приблизительно 35% по массе, от приблизительно 15% до приблизительно 30% по массе, от приблизительно 15% до приблизительно 25% по массе, от приблизительно 15% до приблизительно 20% по массе, от приблизительно 20% до приблизительно 45% по массе, от приблизительно 20% до приблизительно 40% по массе, от приблизительно 20% до приблизительно 35% по массе, от приблизительно 20% до приблизительно 30% по массе, от приблизительно 20% до приблизительно 25% по массе, от приблизительно 25% до приблизительно 45% по массе, от приблизительно 25% до приблизительно 40% по массе, от приблизительно 25% до приблизительно 35% по массе или от приблизительно 25% до приблизительно 30% по массе.
Конечная концентрация раскрытого в изобретении терапевтического соединения в раскрытой в изобретении фармацевтической композиции может составлять любую из требуемых концентраций. В аспекте этого варианта осуществления, конечная концентрация терапевтического соединения в фармацевтической композиции может представлять собой терапевтически эффективное количество. В другом аспекте этого варианта осуществления, конечная концентрация терапевтического соединения в фармацевтической композиции может составлять, например, по меньшей мере, 0,00001 мг/мл, по меньшей мере, 0,0001 мг/мл, по меньшей мере, 0,001 мг/мл, по меньшей мере, 0,01 мг/мл, по меньшей мере, 0,1 мг/мл, по меньшей мере, 1 мг/мл, по меньшей мере, 10 мг/мл, по меньшей мере, 25 мг/мл, по меньшей мере, 50 мг/мл, по меньшей мере, 100 мг/мл, по меньшей мере, 200 мг/мл, по меньшей мере, 500 мг/мл, по меньшей мере, 700 мг/мл, по меньшей мере, 1000 мг/мл или, по меньшей мере, 1200 мг/мл. В другом аспекте этого варианта осуществления, концентрация раскрытого в изобретении терапевтического соединения в растворе может составлять, например, не более 1000 мг/мл, не более 1100 мг/мл, не более 1200 мг/мл, не более 1300 мг/мл, не более 1400 мг/мл, не более 1500 мг/мл, не более 2000 мг/мл, не более 2000 мг/мл или не более 3000 мг/мл. В другом аспекте этого варианта осуществления, конечная концентрация терапевтического соединения в фармацевтической композиции может составлять величину в диапазоне, например, от приблизительно 0,00001 мг/мл до приблизительно 3000 мг/мл, от приблизительно 0,0001 мг/мл до приблизительно 3000 мг/мл, от приблизительно 0,01 мг/мл до приблизительно 3000 мг/мл, от приблизительно 0,1 мг/мл до приблизительно 3000 мг/мл, от приблизительно 1 мг/мл до приблизительно 3000 мг/мл, от приблизительно 250 мг/мл до приблизительно 3000 мг/мл, от приблизительно 500 мг/мл до приблизительно 3000 мг/мл, от приблизительно 750 мг/мл до приблизительно 3000 мг/мл, от приблизительно 1000 мг/мл до приблизительно 3000 мг/мл, от приблизительно 100 мг/мл до приблизительно 2000 мг/мл, от приблизительно 250 мг/мл до приблизительно 2000 мг/мл, от приблизительно 500 мг/мл до приблизительно 2000 мг/мл, от приблизительно 750 мг/мл до приблизительно 2000 мг/мл, от приблизительно 1000 мг/мл до приблизительно 2000 мг/мл, от приблизительно 100 мг/мл до приблизительно 1500 мг/мл, от приблизительно 250 мг/мл до приблизительно 1500 мг/мл, от приблизительно 500 мг/мл до приблизительно 1500 мг/мл, от приблизительно 750 мг/мл до приблизительно 1500 мг/мл, от приблизительно 1000 мг/мл до приблизительно 1500 мг/мл, от приблизительно 100 мг/мл до приблизительно 1200 мг/мл, от приблизительно 250 мг/мл до приблизительно 1200 мг/мл, от приблизительно 500 мг/мл до приблизительно 1200 мг/мл, от приблизительно 750 мг/мл до приблизительно 1200 мг/мл, от приблизительно 1000 мг/мл до приблизительно 1200 мг/мл, от приблизительно 100 мг/мл до приблизительно 1000 мг/мл, от приблизительно 250 мг/мл до приблизительно 1000 мг/мл, от приблизительно 500 мг/мл до приблизительно 1000 мг/мл, от приблизительно 750 мг/мл до приблизительно 1000 мг/мл, от приблизительно 100 мг/мл до приблизительно 750 мг/мл, от приблизительно 250 мг/мл до приблизительно 750 мг/мл, от приблизительно 500 мг/мл до приблизительно 750 мг/мл, от приблизительно 100 мг/мл до приблизительно 500 мг/мл, от приблизительно 250 мг/мл до приблизительно 500 мг/мл, от приблизительно 0,00001 мг/мл до приблизительно 0,0001 мг/мл, от приблизительно 0,00001 мг/мл до приблизительно 0,001 мг/мл, от приблизительно 0,00001 мг/мл до приблизительно 0,01 мг/мл, от приблизительно 0,00001 мг/мл до приблизительно 0,1 мг/мл, от приблизительно 0,00001 мг/мл до приблизительно 1 мг/мл, от приблизительно 0,001 мг/мл до приблизительно 0,01 мг/мл, от приблизительно 0,001 мг/мл до приблизительно 0,1 мг/мл, от приблизительно 0,001 мг/мл до приблизительно 1 мг/мл, от приблизительно 0,001 мг/мл до приблизительно 10 мг/мл или от приблизительно 0,001 мг/мл до приблизительно 100 мг/мл.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, один или более липидов. Липид может быть определен в широком смысле как гидрофобная или амфифильная малая молекула. Амфифильная природа некоторых липидов позволяет им образовывать такие структуры, как везикулы, липосомы или мембраны, в водной среде. Неограничивающие примеры липидов включают жирные кислоты, глицеролипиды, фосфолипиды, сфинголипиды, стероидные липиды, пренольные липиды, сахаролипиды и поликетиды.
Липид, применяемый в раскрытых в изобретении фармацевтических композициях может представлять собой фармацевтически приемлемую жирную кислоту. Жирная кислота включает карбоновую кислоту с длинной неразветвленной углеводородной цепью, которая может быть либо насыщенной, либо ненасыщенной. Такое строение позволяет жирной кислоте иметь полярный гидрофильный конец и неполярный гидрофобный конец, который нерастворим в воде. Большинство природных жирных кислот имеют углеводородную цепь с четным числом углеродных атомов, обычно от 4 до 24 углеродов, и к ним могут быть присоединены функциональные группы, содержащие кислород, галогены, азот и серу. Синтетические или неприродные жирные кислоты могут иметь углеводородную цепь с любым числом углеродных атомов от 3 до 40 углеродов. В случае наличия двойной связи, может наблюдаться либо цис-, либо транс- геометрический изомеризм, который в значительной степени влияет на конфигурацию молекулы. Цис-двойные связи позволяют цепи жирной кислоты изгибаться, эффект, который проявляется тем больше, чем больше двойных связей присутствуют в цепи. Большинство природных жирных кислот имеют цис-конфигурацию, хотя транс-форма обязательно присутствует в некоторых природных и частично гидрированных жирах и маслах. Примеры of жирные кислоты включают, без ограничения, каприловую кислоту (8:0), пеларгоновую кислоту (9:0), каприновую кислоту (10:0), ундециловую кислоту (11:0), лауриновую кислоту (12:0), тридециловую кислоту (13:0), миристиновую кислоту (14:0), миристолеиновую кислоту (14:1), пентадециловую кислоту (15:0), пальмитиновую кислоту (16:0), пальмитолеиновую кислоту (16:1), сапиеновую кислоту (16:1), маргариновую кислоту (17:0), стеариновую кислоту (18:0), олеиновую кислоту (18:1), элаидиновую кислоту (18:1), вакценовую кислоту (18:1), линолевую кислоту (18:2), линоэлаидиновую кислоту (18:2), α-линоленовую кислоту (18:3), γ-линоленовую кислоту (18:3), стеаридоновую кислоту (18:4), нонадециловую кислоту (19:0), арахидиновую кислоту (20:0), эйкозеновую кислоту (20:1), дигомо-γ-линоленовую кислоту (20:3), мидовую кислоту (20:3), арахидоновую кислоту (20:4), эйкозапентаеновую кислоту (20:5), генэйкозановую кислоту (21:0), бегеновую кислоту (22:0), эруковую кислоту (22:1), докозагексаеновую кислоту (22:6), трикоциловую кислоту (23:0), лигноцериновую кислоту (24:0), нервоновую кислоту (24:1), пентакоциловую кислоту (25:0), церотиновую кислоту (26:0), карбоцериновую кислоту (27:0), монтановую кислоту (28:0), нонакоциловую кислоту (29:0), мелиссиновую кислоту (30:0), гентриаконтановую кислоту (31:0), лацериновую кислоту (32:0), псилластеариновую кислоту (33:0), геддовую кислоту (34:0), церопластовую кислоту (35:0) и гексатриаконтановую кислоту (36:0).
В одном варианте осуществления, липид может представлять собой фармацевтически приемлемую насыщенную или ненасыщенную жирную кислоту. В аспектах этого варианта осуществления, насыщенная или ненасыщенная жирная кислота включает, например, по меньшей мере, 8, по меньшей мере, 10, по меньшей мере, 12, по меньшей мере, 14, по меньшей мере, 16, по меньшей мере, 18, по меньшей мере, 20, по меньшей мере, 22, по меньшей мере, 24, по меньшей мере, 26, по меньшей мере, 28 или, по меньшей мере, 30 углеродных атомов. В другом аспекте этого варианта осуществления, насыщенная или ненасыщенная жирная кислота включает, например, от 4 до 24 углеродных атомов, от 6 до 24 углеродных атомов, от 8 до 24 углеродных атомов, от 10 до 24 углеродных атомов, от 12 до 24 углеродных атомов, от 14 до 24 углеродных атомов или от 16 до 24 углеродных атомов, от 4 до 22 углеродных атомов, от 6 до 22 углеродных атомов, от 8 до 22 углеродных атомов, от 10 до 22 углеродных атомов, от 12 до 22 углеродных атомов, от 14 до 22 углеродных атомов или от 16 до 22 углеродных атомов, от 4 до 20 углеродных атомов, от 6 до 20 углеродных атомов, от 8 до 20 углеродных атомов, от 10 до 20 углеродных атомов, от 12 до 20 углеродных атомов, от 14 до 20 углеродных атомов или от 16 до 20 углеродных атомов. В случае наличия ненасыщенности, жирная кислота может иметь, например, 1 или более, 2 или более, 3 или более, 4 или более, 5 или более, или 6 или более двойных связей.
Липид, применяемый в раскрытых в изобретении фармацевтических композициях, может представлять собой фармацевтически приемлемый твердый жир. Называемый также "твердым липидом при комнатной температуре" или просто "жиром", твердый жир включает любую жирную кислоту, которая находится в твердом состоянии при нормальной комнатной температуре, такой, как например, приблизительно 20°C. Жиры состоят из широкой группы соединений, которые обычно растворимы в органических растворителях и обычно нерастворимы в воде. Примеры смесей фармацевтически приемлемых твердых жиров включают, без ограничения, смесь одного или более раскрытых в изобретении глицеролипидов, смесь одного или более раскрытых в изобретении гликолевых эфиров жирных кислот, смесь одного или более раскрытых в изобретении сложных эфиров простого полиэфира жирной кислоты, смесь одного или более раскрытых в изобретении глицеридов.
Твердый жир, применяемый в раскрытых в изобретении фармацевтических композициях, может представлять собой фармацевтически приемлемый глицеролипид. Глицеролипиды состоят в основном из моно-, ди- и тризамещенных глицеринов. Одну группу глицеролипидов образуют глицериды, в которых одна, две или все три гидроксильных группы глицерина каждая этерифицирована с помощью раскрытой в изобретении жирной кислоты с образованием моноглицеридов, диглицеридов и триглицеридов, соответственно. В этих соединениях, каждая из гидроксильных групп глицерина может быть этерифицирована одной и той же жирной кислотой или различными жирными кислотами. Кроме того, глицериды могут быть подвергнуты ацетилированию с получением ацетилированных моноглицеридов, ацетилированных диглицеридов и ацетилированных триглицеридов. В аспектах этого варианта осуществления, моноглицерид может включать насыщенную или ненасыщенную жирную кислоту с длиной углеродной цепи C12-C24. В другом аспекте этого варианта осуществления, диглицерид может включать одну насыщенную или ненасыщенную жирную кислоту с длиной углеродной цепи C12-C24 или две насыщенных или ненасыщенных жирных кислоты, каждая из которой имеет длину углеродной цепи C12-C24. В еще одних аспектах варианта осуществления, триглицерид может включать одну насыщенную или ненасыщенную жирную кислоту с длиной углеродной цепи C12-C24, две насыщенных или ненасыщенных жирных кислоты, каждая из которой имеет длину углеродной цепи C12-C24 или три насыщенных или ненасыщенных жирных кислоты, каждая из которой имеет длину углеродной цепи C12-C24.
В аспектах этого варианта осуществления, смесь фармацевтически приемлемых липидов включает смесь моно-, ди- и/или триглицеридов, имеющую температуру плавления, например, приблизительно 33°C, приблизительно 34°C, приблизительно 35°C, приблизительно 36°C, приблизительно 37°C, приблизительно 38°C, приблизительно 39°C, приблизительно 40°C, приблизительно 41°C, приблизительно 42°C, приблизительно 43°C, приблизительно 44°C, приблизительно 45°C, приблизительно 46°C, приблизительно 47°C, приблизительно 48°C, приблизительно 49°C, приблизительно 50°C. В аспектах этого варианта осуществления, смесь фармацевтически приемлемых липидов включает смесь моно-, ди- и/или триглицеридов, имеющую температуру плавления, например, от приблизительно 30°C до приблизительно 44°C, от приблизительно 30°C до приблизительно 45°C, от приблизительно 30°C до приблизительно 46°C, от приблизительно 30°C до приблизительно 47°C, от приблизительно 30°C до приблизительно 48°C, от приблизительно 30°C до приблизительно 49°C, от приблизительно 30°C до приблизительно 50°C, от приблизительно 32°C до приблизительно 44°C, от приблизительно 32°C до приблизительно 45°C, от приблизительно 32°C до приблизительно 46°C, от приблизительно 32°C до приблизительно 47°C, от приблизительно 32°C до приблизительно 48°C, от приблизительно 32°C до приблизительно 49°C, от приблизительно 32°C до приблизительно 50°C, от приблизительно 34°C до приблизительно 44°C, от приблизительно 34°C до приблизительно 45°C, от приблизительно 34°C до приблизительно 46°C, от приблизительно 34°C до приблизительно 47°C, от приблизительно 34°C до приблизительно 48°C, от приблизительно 34°C до приблизительно 49°C, от приблизительно 34°C до приблизительно 50°C, от приблизительно 36°C до приблизительно 44°C, от приблизительно 36°C до приблизительно 45°C, от приблизительно 36°C до приблизительно 46°C, от приблизительно 36°C до приблизительно 47°C, от приблизительно 36°C до приблизительно 48°C, от приблизительно 36°C до приблизительно 49°C, от приблизительно 36°C до приблизительно 50°C, от приблизительно 38°C до приблизительно 44°C, от приблизительно 38°C до приблизительно 45°C, от приблизительно 38°C до приблизительно 46°C, от приблизительно 38°C до приблизительно 47°C, от приблизительно 38°C до приблизительно 48°C, от приблизительно 38°C до приблизительно 49°C, от приблизительно 38°C до приблизительно 50°C, от приблизительно 40°C до приблизительно 44°C, от приблизительно 40°C до приблизительно 45°C, от приблизительно 40°C до приблизительно 46°C, от приблизительно 40°C до приблизительно 47°C, от приблизительно 40°C до приблизительно 48°C, от приблизительно 40°C до приблизительно 49°C, от приблизительно 40°C до приблизительно 50°C, от приблизительно 42°C до приблизительно 44°C, от приблизительно 42°C до приблизительно 45°C, от приблизительно 42°C до приблизительно 46°C, от приблизительно 42°C до приблизительно 47°C, от приблизительно 42°C до приблизительно 48°C, от приблизительно 42°C до приблизительно 49°C или от приблизительно 42°C до приблизительно 50°C.
В другом аспекте этого варианта осуществления, смесь фармацевтически приемлемых липидов включает смесь эфиров ПЭГ жирных кислот, имеющую температуру плавления, например, приблизительно 33°C, приблизительно 34°C, приблизительно 35°C, приблизительно 36°C, приблизительно 37°C, приблизительно 38°C, приблизительно 39°C, приблизительно 40°C, приблизительно 41°C, приблизительно 42°C, приблизительно 43°C, приблизительно 44°C, приблизительно 45°C, приблизительно 46°C, приблизительно 47°C, приблизительно 48°C, приблизительно 49°C, приблизительно 50°C. В аспектах этого варианта осуществления, смесь фармацевтически приемлемых липидов включает смесь эфиров ПЭГ жирных кислот, имеющую температуру плавления, например, от приблизительно 30°C до приблизительно 44°C, от приблизительно 30°C до приблизительно 45°C, от приблизительно 30°C до приблизительно 46°C, от приблизительно 30°C до приблизительно 47°C, от приблизительно 30°C до приблизительно 48°C, от приблизительно 30°C до приблизительно 49°C, от приблизительно 30°C до приблизительно 50°C, от приблизительно 32°C до приблизительно 44°C, от приблизительно 32°C до приблизительно 45°C, от приблизительно 32°C до приблизительно 46°C, от приблизительно 32°C до приблизительно 47°C, от приблизительно 32°C до приблизительно 48°C, от приблизительно 32°C до приблизительно 49°C, от приблизительно 32°C до приблизительно 50°C, от приблизительно 34°C до приблизительно 44°C, от приблизительно 34°C до приблизительно 45°C, от приблизительно 34°C до приблизительно 46°C, от приблизительно 34°C до приблизительно 47°C, от приблизительно 34°C до приблизительно 48°C, от приблизительно 34°C до приблизительно 49°C, от приблизительно 34°C до приблизительно 50°C, от приблизительно 36°C до приблизительно 44°C, от приблизительно 36°C до приблизительно 45°C, от приблизительно 36°C до приблизительно 46°C, от приблизительно 36°C до приблизительно 47°C, от приблизительно 36°C до приблизительно 48°C, от приблизительно 36°C до приблизительно 49°C, от приблизительно 36°C до приблизительно 50°C, от приблизительно 38°C до приблизительно 44°C, от приблизительно 38°C до приблизительно 45°C, от приблизительно 38°C до приблизительно 46°C, от приблизительно 38°C до приблизительно 47°C, от приблизительно 38°C до приблизительно 48°C, от приблизительно 38°C до приблизительно 49°C, от приблизительно 38°C до приблизительно 50°C, от приблизительно 40°C до приблизительно 44°C, от приблизительно 40°C до приблизительно 45°C, от приблизительно 40°C до приблизительно 46°C, от приблизительно 40°C до приблизительно 47°C, от приблизительно 40°C до приблизительно 48°C, от приблизительно 40°C до приблизительно 49°C, от приблизительно 40°C до приблизительно 50°C, от приблизительно 42°C до приблизительно 44°C, от приблизительно 42°C до приблизительно 45°C, от приблизительно 42°C до приблизительно 46°C, от приблизительно 42°C до приблизительно 47°C, от приблизительно 42°C до приблизительно 48°C, от приблизительно 42°C до приблизительно 49°C или от приблизительно 42°C до приблизительно 50°C.
В другом аспекте этого варианта осуществления, смесь фармацевтически приемлемых липидов включает смесь моно-, ди- и/или триглицеридов и эфиров ПЭГ жирных кислот, имеющую температуру плавления, например, приблизительно 33°C, приблизительно 34°C, приблизительно 35°C, приблизительно 36°C, приблизительно 37°C, приблизительно 38°C, приблизительно 39°C, приблизительно 40°C, приблизительно 41°C, приблизительно 42°C, приблизительно 43°C, приблизительно 44°C, приблизительно 45°C, приблизительно 46°C, приблизительно 47°C, приблизительно 48°C, приблизительно 49°C, приблизительно 50°C. В аспектах этого варианта осуществления, смесь фармацевтически приемлемых липидов включает смесь моно-, ди- и/или триглицеридов и эфиров ПЭГ жирных кислот, имеющую температуру плавления, например, от приблизительно 30°C до приблизительно 44°C, от приблизительно 30°C до приблизительно 45°C, от приблизительно 30°C до приблизительно 46°C, от приблизительно 30°C до приблизительно 47°C, от приблизительно 30°C до приблизительно 48°C, от приблизительно 30°C до приблизительно 49°C, от приблизительно 30°C до приблизительно 50°C, от приблизительно 32°C до приблизительно 44°C, от приблизительно 32°C до приблизительно 45°C, от приблизительно 32°C до приблизительно 46°C, от приблизительно 32°C до приблизительно 47°C, от приблизительно 32°C до приблизительно 48°C, от приблизительно 32°C до приблизительно 49°C, от приблизительно 32°C до приблизительно 50°C, от приблизительно 34°C до приблизительно 44°C, от приблизительно 34°C до приблизительно 45°C, от приблизительно 34°C до приблизительно 46°C, от приблизительно 34°C до приблизительно 47°C, от приблизительно 34°C до приблизительно 48°C, от приблизительно 34°C до приблизительно 49°C, от приблизительно 34°C до приблизительно 50°C, от приблизительно 36°C до приблизительно 44°C, от приблизительно 36°C до приблизительно 45°C, от приблизительно 36°C до приблизительно 46°C, от приблизительно 36°C до приблизительно 47°C, от приблизительно 36°C до приблизительно 48°C, от приблизительно 36°C до приблизительно 49°C, от приблизительно 36°C до приблизительно 50°C, от приблизительно 38°C до приблизительно 44°C, от приблизительно 38°C до приблизительно 45°C, от приблизительно 38°C до приблизительно 46°C, от приблизительно 38°C до приблизительно 47°C, от приблизительно 38°C до приблизительно 48°C, от приблизительно 38°C до приблизительно 49°C, от приблизительно 38°C до приблизительно 50°C, от приблизительно 40°C до приблизительно 44°C, от приблизительно 40°C до приблизительно 45°C, от приблизительно 40°C до приблизительно 46°C, от приблизительно 40°C до приблизительно 47°C, от приблизительно 40°C до приблизительно 48°C, от приблизительно 40°C до приблизительно 49°C, от приблизительно 40°C до приблизительно 50°C, от приблизительно 42°C до приблизительно 44°C, от приблизительно 42°C до приблизительно 45°C, от приблизительно 42°C до приблизительно 46°C, от приблизительно 42°C до приблизительно 47°C, от приблизительно 42°C до приблизительно 48°C, от приблизительно 42°C до приблизительно 49°C или от приблизительно 42°C до приблизительно 50°C.
Раскрытая в изобретении фармацевтическая композиция твердого раствора может включать фармацевтически приемлемый твердый при комнатной температуре липид (твердый жир) в количестве, достаточном для образования раскрытой в изобретении композиции твердого раствора. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать фармацевтически приемлемый твердый при комнатной температуре липид (твердый жир) в количестве, например, по меньшей мере, 10% по массе, по меньшей мере, 20% по массе, по меньшей мере, 30% по массе, по меньшей мере, 35% по массе, по меньшей мере, 40% по массе, по меньшей мере, 45% по массе, по меньшей мере, 50% по массе, по меньшей мере, 55% по массе, по меньшей мере, 60% по массе, по меньшей мере, 65% по массе, по меньшей мере, 70% по массе, по меньшей мере, 75% по массе, по меньшей мере, 80% по массе, по меньшей мере, 85% по массе, по меньшей мере, 90% по массе, по меньшей мере, 95% по массе или, по меньшей мере, 99% по массе. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать фармацевтически приемлемый твердый при комнатной температуре липид (твердый жир) в количестве, например, от приблизительно 30% до приблизительно 99% по массе, от приблизительно 35% до приблизительно 99% по массе, от приблизительно 40% до приблизительно 99% по массе, от приблизительно 45% до приблизительно 99% по массе, от приблизительно 50% до приблизительно 99% по массе, от приблизительно 30% до приблизительно 98% по массе, от приблизительно 35% до приблизительно 98% по массе, от приблизительно 40% до приблизительно 98% по массе, от приблизительно 45% до приблизительно 98% по массе, от приблизительно 50% до приблизительно 98% по массе, от приблизительно 30% до приблизительно 95% по массе, от приблизительно 35% до приблизительно 95% по массе, от приблизительно 40% до приблизительно 95% по массе, от приблизительно 45% до приблизительно 95% по массе или от приблизительно 50% до приблизительно 95% по массе. В еще одних аспектах варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать фармацевтически приемлемый твердый при комнатной температуре липид (твердый жир) в количестве, например, от приблизительно 70% до приблизительно 97% по массе, от приблизительно 75% до приблизительно 97% по массе, от приблизительно 80% до приблизительно 97% по массе, от приблизительно 85% до приблизительно 97% по массе, от приблизительно 88% до приблизительно 97% по массе, от приблизительно 89% до приблизительно 97% по массе, от приблизительно 90% до приблизительно 97% по массе, от приблизительно 75% до приблизительно 96% по массе, от приблизительно 80% до приблизительно 96% по массе, от приблизительно 85% до приблизительно 96% по массе, от приблизительно 88% до приблизительно 96% по массе, от приблизительно 89% до приблизительно 96% по массе, от приблизительно 90% до приблизительно 96% по массе, от приблизительно 75% до приблизительно 93% по массе, от приблизительно 80% до приблизительно 93% по массе, от приблизительно 85% до приблизительно 93% по массе, от приблизительно 88% до приблизительно 93% по массе, от приблизительно 89% до приблизительно 93% по массе или от приблизительно 90% до приблизительно 93% по массе.
Производимые промышленностью смеси фармацевтически приемлемых глицеролипидов включают, без ограничения, масло какао, смеси стеарата ПЭГ-6 и пальмитостеарата этиленгликоля и стеарата ПЭГ-32 (TEFOSE® 1500; TEFOSE® 63), смеси трицетеарет-4 фосфата и пальмитостеарата этиленгликоля и пальмитостеарата диэтиленгликоля (SEDEFOS® 75), смеси глицеролмоностеарата и стеарата ПЭГ-75 (GELOT®), смеси цетилового спирта и этоксилированных жирных спиртов (seteth-2-, steareth-20) (EMULCIRE®), смеси насыщенных C10-C18 триглицеридов, имеющих температуру плавления приблизительно 33°C (GELUCIRE® 33/01), смеси насыщенных C10-C18 триглицеридов, имеющих температуру плавления приблизительно 39°C (GELUCIRE® 39/01), смеси насыщенных C10-C18 триглицеридов, имеющих температуру плавления приблизительно 43°C (GELUCIRE® 43/01), смеси глицеролмоностеарата 40-55 (тип I) и диглицеридов (GELEOL® моно и диглицериды) и смеси триглицеридов со средней длиной цепи (LABRAFAC® Lipophile WL 1349).
Твердый жир, применяемый в раскрытых в изобретении фармацевтических композициях, может представлять собой фармацевтически приемлемый гликолевый эфир жирной кислоты. Фармацевтически приемлемый гликолевый эфир жирной кислоты может представлять собой моноэфир гликоля, диэфир гликоля или триэфир гликоля. Гликолевый эфир жирной кислоты включают, без ограничения, этиленгликолевый эфир жирной кислоты, диэтиленгликолевый эфир жирной кислоты, пропиленгликолевый эфир жирной кислоты и дипропиленгликолевый эфир жирной кислоты. Неограничивающие примеры гликолевых эфиров жирных кислот включают, например, каприлат этиленгликоля, пеларгонат этиленгликоля, капрат этиленгликоля, ундецелат этиленгликоля, лаурат этиленгликоля, тридецилат этиленгликоля, миристат этиленгликоля, миристолат этиленгликоля, пентадецилат этиленгликоля, пальмитат этиленгликоля, пальмитолеат этиленгликоля, сапиенат этиленгликоля, маргарат этиленгликоля, стеарат этиленгликоля, пальмитостеарат этиленгликоля, олеат этиленгликоля, элаидат этиленгликоля, вакцинат этиленгликоля, линолеат этиленгликоля, линоэлаидат этиленгликоля, α-линоленат этиленгликоля, γ-линоленат этиленгликоля, стеаридонат этиленгликоля, каприлокапрат этиленгликоля, дикаприлокапрат этиленгликоля, дикаприлат этиленгликоля, дипеларгонат этиленгликоля, дикапрат этиленгликоля, диундецелат этиленгликоля, дилаурат этиленгликоля, дитридецилат этиленгликоля, димиристат этиленгликоля, димиристолат этиленгликоля, дипентадецилат этиленгликоля, дипальмитат этиленгликоля, дипальмитолеат этиленгликоля, дисапиенат этиленгликоля, димаргарат этиленгликоля, дистеарат этиленгликоля, дипальмитостеарат этиленгликоля, диолеат этиленгликоля, диэлаидат этиленгликоля, дивакцинат этиленгликоля, дилинолеат этиленгликоля, дилиноэлаидат этиленгликоля, ди-α-линоленат этиленгликоля, ди-γ-линоленат этиленгликоля, дистеаридонат этиленгликоля, дикаприлокапрат этиленгликоля, дидикаприлокапрат этиленгликоля, каприлат пропиленгликоля, пеларгонат пропиленгликоля, капрат пропиленгликоля, ундецилат пропиленгликоля, лаурат пропиленгликоля, тридецилат пропиленгликоля, миристат пропиленгликоля, миристолат пропиленгликоля, пентадецилат пропиленгликоля, пальмитат пропиленгликоля, пальмитолеат пропиленгликоля, сапиенат пропиленгликоля, маргарат пропиленгликоля, стеарат пропиленгликоля, пальмитостеарат пропиленгликоля, олеат пропиленгликоля, элаидат пропиленгликоля, вакцинат пропиленгликоля, линолеат пропиленгликоля, линоэлаидат пропиленгликоля, α-линоленат пропиленгликоля, γ-линоленат пропиленгликоля, стеаридонат пропиленгликоля, каприлокапрат пропиленгликоля, дикаприлокапрат пропиленгликоля, дикаприлат пропиленгликоля, дипеларгонат пропиленгликоля, дикапрат пропиленгликоля, диундецилат пропиленгликоля, дилаурат пропиленгликоля, дитридецилат пропиленгликоля, димиристат пропиленгликоля, димиристолат пропиленгликоля, дипентадецилат пропиленгликоля, дипальмитат пропиленгликоля, дипальмитолеат пропиленгликоля, дисапиенат пропиленгликоля, димаргарат пропиленгликоля, дистеарат пропиленгликоля, дипальмитостеарат пропиленгликоля, диолеат пропиленгликоля, диэлаидат пропиленгликоля, дивакцинат пропиленгликоля, дилинолеат пропиленгликоля, дилиноэлаидат пропиленгликоля, ди-α-линоленат пропиленгликоля, ди-γ-линоленат пропиленгликоля, дистеаридонат пропиленгликоля, дикаприлокапрат пропиленгликоля, дидикаприлокапрат пропиленгликоля или любую их комбинацию.
Производимые промышленностью фармацевтически приемлемые гликолевые эфиры жирных кислот включают, без ограничения, монопальмитостеарат пропиленгликоля (MONOSTEOL®), дикаприлокапрат пропиленгликоля (LABRAFAC® PG), монолауран пропиленгликоля (тип I) (LAUROGLYCOL® FCC), монолауран пропиленгликоля (тип II) (LAUROGLYCOL® 90), монокаприлат пропиленгликоля (тип I) (CAPRYOL® PGMC) и монокаприлат пропиленгликоля (тип II) (CAPRYOL® 90).
Твердый жир, применяемый в раскрытых в изобретении фармацевтических композициях, может представлять собой фармацевтически приемлемый простой полиэфир сложного эфира жирной кислоты. Фармацевтически приемлемый простой полиэфир сложного эфира жирной кислоты может представлять собой простой полиэфир сложного моноэфира жирной кислоты, простой полиэфир сложного диэфира жирной кислоты или простой полиэфир сложного триэфира жирной кислоты. Простой полиэфир сложного эфира жирной кислоты включает, без ограничения, ПЭГ сложный эфир жирной кислоты, ПЭГ глицерил жирной кислоты, ПЭГ глицерид сложного эфира жирной кислоты, ППГ сложный эфир жирной кислоты, ППГ глицерил жирной кислоты и ППГ глицерид сложного эфира жирной кислоты. Молекулярные массы ПЭГ или ППГ могут составлять, например, 5-20000. Неограничивающие примеры простого полиэфира сложного эфира жирной кислоты включают, например, ПЭГ каприлат, ПЭГ пеларгонат, ПЭГ капрат, ПЭГ ундецилат, ПЭГ лаурат, ПЭГ тридецилат, ПЭГ миристат, ПЭГ миристолат, ПЭГ пентадецилат, ПЭГ пальмитат, ПЭГ пальмитолеат, ПЭГ сапиенат, ПЭГ маргарат, ПЭГ стеарат, ПЭГ польмитостеарат, ПЭГ олеат, ПЭГ элаидат, ПЭГ вакцинат, ПЭГ линолеат, ПЭГ линоэлаидат, ПЭГ α-линоленат, ПЭГ γ-линоленат, ПЭГ стеаридонат, ПЭГ каприлокапрат, ПЭГ дикаприлокапрат, ПЭГ глицерил каприлат, ПЭГ глицерил пеларгонат, ПЭГ глицерил капрат, ПЭГ глицерил ундецилат, ПЭГ глицерил лаурат, ПЭГ глицерил тридецилат, ПЭГ глицерил миристат, ПЭГ глицерил миристолат, ПЭГ глицерил пентадецилат, ПЭГ глицерил пальмитат, ПЭГ глицерил пальмитолеат, ПЭГ глицерил сапиенат, ПЭГ глицерил маргарат, ПЭГ глицерил стеарат, ПЭГ глицерил пальмитостеарат, ПЭГ глицерил олеат, ПЭГ глицерил элаидат, ПЭГ глицерил вакцинат, ПЭГ глицерил линолеат, ПЭГ глицерил линоэлаидат, ПЭГ глицерил α-линоленат, ПЭГ глицерил γ-линоленат, ПЭГ глицерил стеаридонат, ПЭГ глицерил каприлокапрат, ПЭГ глицерил дикаприлокапрат, каприлоил ПЭГ глицерид, пеларгоноил ПЭГ глицерид, капроил ПЭГ глицерид, ундецилоил ПЭГ глицерид, лауроил ПЭГ глицерид, тридецилоил ПЭГ глицерид, миристоил ПЭГ глицерид, миристолоил ПЭГ глицерид, пентадециклоил ПЭГ глицерид, пальмитоил ПЭГ глицерид, пальмитолеоил ПЭГ глицерид, сапиеноил ПЭГ глицерид, маргароил ПЭГ глицерид, стеароил ПЭГ глицерид, пальмитостеароил ПЭГ глицерид, олеоил ПЭГ глицерид, элаидоил ПЭГ глицерид, вакциноил ПЭГ глицерид, линолеоил ПЭГ глицерид, линоэлаидоил ПЭГ глицерид, α-линоленоил ПЭГ глицерид, γ-линоленоил ПЭГ глицерид, стеаридоноил ПЭГ глицерид, каприлокапроил ПЭГ глицерид, дикаприлокапроил ПЭГ глицерид, ППГ каприлат, ППГ пеларгонат, ППГ капрат, ППГ ундецилат, ППГ лаурат, ППГ тридецилат, ППГ миристат, ППГ миристолат, ППГ пентадецилат, ППГ пальмитат, ППГ пальмитолеат, ППГ сапиенат, ППГ маргарат, ППГ стеарат, ППГ пальмитостеарат, ППГ олеат, ППГ элаидат, ППГ вакцинат, ППГ линолеат, ППГ линоэлаидат, ППГ α-линоленат, ППГ γ-линоленат, ППГ стеаридонат, ППГ каприлокапрат, ППГ дикаприлокапрат, ППГ глицерил каприлат, ППГ глицерил пеларгонат, ППГ глицерил капрат, ППГ глицерил ундецилат, ППГ глицерил лаурат, ППГ глицерил тридецилат, ППГ глицерил миристат, ППГ глицерил миристолат, ППГ глицерил пентадецилат, ППГ глицерил пальмитат, ППГ глицерил пальмитолеат, ППГ глицерил сапиенат, ППГ глицерил маргарат, ППГ глицерил стеарат, ППГ глицерил пальмитостеарат, ППГ глицерил олеат, ППГ глицерил элаидат, ППГ глицерил вакцинат, ППГ глицерил линолеат, ППГ глицерил линоэлаидат, ППГ глицерил α-линоленат, ППГ глицерил γ-линоленат, ППГ глицерил стеаридонат, ППГ глицерил каприлокапрат, ППГ глицерил дикаприлокапрат, каприлоил ППГ глицерид, пеларгоноил ППГ глицерид, капроил ППГ глицерид, ундецилоил ППГ глицерид, лауроил ППГ глицерид, тридецилоил ППГ глицерид, миристоил ППГ глицерид, миристолоил ППГ глицерид, пентадециклоил ППГ глицерид, пальмитоил ППГ глицерид, пальмитолеоил ППГ глицерид, сапиеноил ППГ глицерид, маргароил ППГ глицерид, стеароил ППГ глицерид, пальмитостеароил ППГ глицерид, олеоил ППГ глицерид, элаидоил ППГ глицерид, вакциноил ППГ глицерид, линолеоил ППГ глицерид, линоэлаидоил ППГ глицерид, α-линоленоил ППГ глицерид, γ-линоленоил ППГ глицерид, стеаридоноил ППГ глицерид, каприлокапроил ППГ глицерид, дикаприлокапроил ППГ глицерид или любую их комбинацию.
Выпускаемые промышленностью фармацевтически приемлемые простые полиэфиры сложного эфира жирной кислоты включают, без ограничения, каприлокапроил макрогол-8 глицериды (LABRASOL®), ПЭГ-8 воск (APIFIL®), лауроил макрогол-32 глицериды (GELUCIRE 44/14), стеароил макрогол-32 глицериды (GELUCIRE 50,13), линолеоил макрогол-6 глицериды (LABRAFIL® M2125CS), олеоил макрогол-6 глицериды (LABRAFIL® M1944CS) и лауроил макрогол-6 глицериды (LABRAFIL® M2130CS).
Другой липид, применяемый в раскрытых в изобретении фармацевтических композициях, может представлять собой фармацевтически приемлемый жидкий при комнатной температуре липид. Называемый также "жидким жиром", жидкий при комнатной температуре липид включает любую жирную кислоту, которая является жидкой при нормальной комнатной температуре, такой как, например, приблизительно 20°C. Жидкий при комнатной температуре липид включает обширную группу соединений, которые обычно растворимы в органических растворителях и обычно не растворимы в воде. Примеры смесей фармацевтически приемлемых жидких при комнатной температуре липидов включают, без ограничения, смесь одной или более раскрытых в изобретении жирных кислот, смесь одного или более частично гидролизованного жира и смесь одного или более частично гидрированного жира.
Фармацевтически приемлемый жидкий при комнатной температуре липид включает фармацевтически приемлемый частично гидрированный жир. Процесс гидрирования добавляет атомы водорода к ненасыщенному липиду, удаляет двойные связи и позволяет получить частично или полностью насыщенный липид. Частичное гидрирование является химическим процессом, а не ферментативным, в результате которого происходит превращение части цис-изомеров в тран-ненасыщенные липиды вместо их полного гидрирования. На первой стадии реакции, добавляется один водород, при этом другой координационно ненасыщенный углерод образует связь с катализатором. Вторая стадия представляет собой добавление водорода к оставшемуся углероду с образованием насыщенной жирной кислоты. Первая стадия является обратимой, вследствие чего водород реадсорбируется на катализаторе и повторно образуется двойная связь. Промежуточное соединение только с одним добавленным водородом не содержит двойной связи и может свободно вращаться. Благодаря этому, может повторно образовываться двойная связь либо в виде цис-, либо в виде транс-, из которых транс- является предпочтительной, независимо от исходного материала.
Раскрытая в изобретении фармацевтическая композиция может включать жидкий при комнатной температуре липид в количестве, достаточном для растворения раскрытого в изобретении терапевтического соединения. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать жидкий при комнатной температуре липид в количестве, например, менее чем приблизительно 90% по массе, менее чем приблизительно 80% по массе, менее чем приблизительно 70% по массе, менее чем приблизительно 65% по массе, менее чем приблизительно 60% по массе, менее чем приблизительно 55% по массе, менее чем приблизительно 50% по массе, менее чем приблизительно 45% по массе, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 5% по массе или менее чем приблизительно 1% по массе. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать жидкий при комнатной температуре липид в количестве в диапазоне, например, приблизительно от 1% до 90% по массе, приблизительно от 1% до 80% по массе, приблизительно от 1% до 70% по массе, приблизительно от 1% до 60% по массе, приблизительно от 1% до 50% по массе, приблизительно от 1% до 40% по массе, приблизительно от 1% до 30% по массе, приблизительно от 1% до 20% по массе, приблизительно от 1% до 10% по массе, приблизительно от 2% до 50% по массе, приблизительно от 2% до 40% по массе, приблизительно от 2% до 30% по массе, приблизительно от 2% до 20% по массе, приблизительно от 2% до 10% по массе, приблизительно от 4% до 50% по массе, приблизительно от 4% до 40% по массе, приблизительно от 4% до 30% по массе, приблизительно от 4% до 20% по массе, приблизительно от 4% до 10% по массе, приблизительно от 6% до 50% по массе, приблизительно от 6% до 40% по массе, приблизительно от 6% до 30% по массе, приблизительно от 6% до 20% по массе, приблизительно от 6% до 10% по массе, приблизительно от 8% до 50% по массе, приблизительно от 8% до 40% по массе, приблизительно от 8% до 30% по массе, приблизительно от 8% до 20% по массе, приблизительно от 8% до 15% по массе или приблизительно от 8% до 12% по массе.
Примеры фармацевтически приемлемых жидких при комнатной температуре липидов включают моноглицериды, в том числе, без ограничения, глицерина мономиристолеат, глицерина монопальмитолеат, глицерина моносапиенат, глицерина моноолеат, глицерина моноэлаидат, глицерина моновакценат, глицерина монолинолеат, глицерина монолиноэлаидат, глицерина монолиноленат, глицерина моностеаридонат, глицерина моноэйкозеноат, глицерина мономеадат, глицерина моноарахидонат, глицерина моноэйкозапентаеноат, глицерина моноэрукат, глицерина монодокозагексаеноат и глицерина мононервонат.
Производимые промышленностью фармацевтически приемлемые жидкие при комнатной температуре липиды включают, без ограничения, глицерил дибегенат (COMPRITOL® 888), глицерина дибегенат (COMPRITOL® E ATO), глицерина дипальмитостеарат (Biogapress Vegetal BM297ATO), глицерина дистеарат (тип I) (PRECIROL® ATO 5) и глицерина монолинолеат (MAISINE™ 35-1).
Аспекты настоящего изобретения раскрывают, помимо всего прочего, стабилизатор. Стабилизатор представляет собой соединение, которое взаимодействует со свободной кислотой или основанием, присутствующими на раскрытом в изобретении терапевтическом соединении, для экранирования зарядов, тем самым препятствуя ионным взаимодействиям между матрицами терапевтическое соединение/липид и предотвращая их конфигурации, необходимые для образования кристаллических матриц твердофазной композиции. Следовательно, стабилизатор предотвращает термодинамический переход композиции в классическую твердую фазу или растягивает во времени этот переход в такой степени, что он не происходит. Примеры стабилизаторов включают жидкий полимер гликоля, одноатомный спирт, диметиловый эфир изосорбида и моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол) (TRANSCUTOL®).
Раскрытая в изобретении фармацевтическая композиция может включать стабилизатор в количестве, достаточном для стабилизации свободной кислоты или основания, присутствующих на раскрытом в изобретении терапевтическом соединении. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать стабилизатор в количестве, например, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 19% по массе, менее чем приблизительно 18% по массе, менее чем приблизительно 17% по массе, менее чем приблизительно 16% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 14% по массе, менее чем приблизительно 13% по массе, менее чем приблизительно 12% по массе, менее чем приблизительно 11% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 9% по массе, менее чем приблизительно 8% по массе, менее чем приблизительно 7% по массе, менее чем приблизительно 6% по массе, менее чем приблизительно 5% по массе, менее чем приблизительно 4% по массе, менее чем приблизительно 3% по массе, менее чем приблизительно 2% по массе или менее чем приблизительно 1%. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать стабилизатор в количестве, например, от приблизительно 1% до приблизительно 5% по массе, от приблизительно 1% до приблизительно 7% по массе, от приблизительно 1% до приблизительно 10% по массе, от приблизительно 1% до приблизительно 12% по массе, от приблизительно 1% до приблизительно 15% по массе, от приблизительно 1% до приблизительно 18% по массе, от приблизительно 1% до приблизительно 20% по массе, от приблизительно 2% до приблизительно 5% по массе, от приблизительно 2% до приблизительно 7% по массе, от приблизительно 2% до приблизительно 10% по массе, от приблизительно 2% до приблизительно 12% по массе, от приблизительно 2% до приблизительно 15% по массе, от приблизительно 2% до приблизительно 18% по массе, от приблизительно 2% до приблизительно 20% по массе, от приблизительно 3% до приблизительно 5% по массе, от приблизительно 3% до приблизительно 7% по массе, от приблизительно 3% до приблизительно 10% по массе, от приблизительно 3% до приблизительно 12% по массе, от приблизительно 3% до приблизительно 15% по массе, от приблизительно 3% до приблизительно 18% по массе, от приблизительно 3% до приблизительно 20% по массе, от приблизительно 4% до приблизительно 5% по массе, от приблизительно 4% до приблизительно 7% по массе, от приблизительно 4% до приблизительно 10% по массе, от приблизительно 4% до приблизительно 12% по массе, от приблизительно 4% до приблизительно 15% по массе, от приблизительно 4% до приблизительно 18% по массе, от приблизительно 4% до приблизительно 20% по массе, от приблизительно 5% до приблизительно 7% по массе, от приблизительно 5% до приблизительно 10% по массе, от приблизительно 5% до приблизительно 12% по массе, от приблизительно 5% до приблизительно 15% по массе, от приблизительно 5% до приблизительно 18% по массе, от приблизительно 5% до приблизительно 20% по массе, от приблизительно 6% до приблизительно 7% по массе, от приблизительно 6% до приблизительно 10% по массе, от приблизительно 6% до приблизительно 12% по массе, от приблизительно 6% до приблизительно 15% по массе, от приблизительно 6% до приблизительно 18% по массе, от приблизительно 6% до приблизительно 20% по массе, от приблизительно 7% до приблизительно 10% по массе, от приблизительно 7% до приблизительно 12% по массе, от приблизительно 7% до приблизительно 15% по массе, от приблизительно 7% до приблизительно 18% по массе, от приблизительно 7% до приблизительно 20% по массе, от приблизительно 8% до приблизительно 10% по массе, от приблизительно 8% до приблизительно 12% по массе, от приблизительно 8% до приблизительно 15% по массе, от приблизительно 8% до приблизительно 18% по массе, от приблизительно 8% до приблизительно 20% по массе, от приблизительно 9% до приблизительно 10% по массе, от приблизительно 9% до приблизительно 12% по массе, от приблизительно 9% до приблизительно 15% по массе, от приблизительно 9% до приблизительно 18% по массе, от приблизительно 9% до приблизительно 20% по массе, от приблизительно 10% до приблизительно 12% по массе, от приблизительно 10% до приблизительно 15% по массе, от приблизительно 10% до приблизительно 18% по массе или от приблизительно 10% до приблизительно 20% по массе.
Раскрытый в изобретении стабилизатор не является растворителем, так как он используется в количестве, которое не приводит к существенному растворению растворяемого вещества. По этой причине, количество стабилизатора, используемое в раскрытой в изобретении композиции твердого раствора, приводит к не более чем 85% растворению раскрытого в изобретении терапевтического соединения. В аспектах этого варианта осуществления, количество стабилизатора, используемое в раскрытой в изобретении композиции твердого раствора, приводит к, например, не более чем 80%, не более чем 75%, не более чем 70%, не более чем 65%, не более чем 60%, не более чем 55%, не более чем 50%, не более чем 45%, не более чем 40%, не более чем 35%, не более чем 30%, не более чем 25%, не более чем 20%, не более чем 15%, не более чем 10% или не более чем 5% растворению раскрытого в изобретении терапевтического соединения.
В одном варианте осуществления, полимер гликоля может включать фармацевтически приемлемый полимер ПЭГ. Полимеры ПЭГ, также называемые полимерами полиэтиленоксида (PEO) или полимерами полиоксиэтилена (POE), получают полимеризацией оксида этилена и они производятся промышленностью с широким диапазоном молекулярных масс от 100 г/моль до 10000000 г/моль. Полимеры ПЭГ с низкой молекулярной массой представляют собой жидкости или низкоплавкие твердые вещества, тогда как полимеры ПЭГ с высокой молекулярной массой являются твердыми веществами. В аспекте этого варианта осуществления, полимер ПЭГ, используемый в качестве стабилизатора, является жидким полимером ПЭГ. В аспектах этого варианта осуществления, полимер ПЭГ имеет молекулярную массу, например, не более чем 100 г/моль, не более чем 200 г/моль, не более чем 300 г/моль, не более чем 400 г/моль, не более чем 500 г/моль, не более чем 600 г/моль, не более чем 700 г/моль, не более чем 800 г/моль, не более чем 900 г/моль или не более чем 1000 г/моль.
Полимер ПЭГ включают, без ограничения, ПЭГ 100, ПЭГ 200, ПЭГ 300, ПЭГ 400, ПЭГ 500, ПЭГ 600, ПЭГ 700, ПЭГ 800, ПЭГ 900, ПЭГ 1000, ПЭГ 1100, ПЭГ 1200, ПЭГ 1300, ПЭГ 1400, ПЭГ 1500, ПЭГ 1600, ПЭГ 1700, ПЭГ 1800, ПЭГ 1900, ПЭГ 2000, ПЭГ 2100, ПЭГ 2200, ПЭГ 2300, ПЭГ 2400, ПЭГ 2500, ПЭГ 2600, ПЭГ 2700, ПЭГ 2800, ПЭГ 2900, ПЭГ 3000, ПЭГ 3250, ПЭГ 3350, ПЭГ 3500, ПЭГ 3750, ПЭГ 4000, ПЭГ 4250, ПЭГ 4500, ПЭГ 4750, ПЭГ 5000, ПЭГ 5500, ПЭГ 6000, ПЭГ 6500, ПЭГ 7000, ПЭГ 7500, ПЭГ 8000, ПЭГ 8500, ПЭГ 9000, ПЭГ 9500, ПЭГ 10000, ПЭГ 11000, ПЭГ 12000, ПЭГ 13000, ПЭГ 14000, ПЭГ 15000, ПЭГ 16000, ПЭГ 17000, ПЭГ 18000, ПЭГ 19000 или ПЭГ 20000.
В другом варианте осуществления, полимер гликоля может включать фармацевтически приемлемый полимер полипропиленгликоля (ППГ). Полимеры ППГ, называемые также полимерами полипропилен оксида (ППО) или полимерами полиоксипропилена (ПОП), получают полимеризацией оксида пропилена и они производятся промышленностью с широким диапазоном молекулярных масс от 100 г/моль до 10000000 г/моль. Полимеры ППГ с низкой молекулярной массой представляют собой жидкости или низкоплавкие твердые вещества, тогда как полимеры ППГ с высокой молекулярной массой являются твердыми веществами. В аспекте этого варианта осуществления, Полимер ППГ, используемый в качестве стабилизатора, является жидким полимером ППГ. В аспектах этого варианта осуществления, полимер ППГ имеет молекулярную массу, например, не более чем 100 г/моль, не более чем 200 г/моль, не более чем 300 г/моль, не более чем 400 г/моль, не более чем 500 г/моль, не более чем 600 г/моль, не более чем 700 г/моль, не более чем 800 г/моль, не более чем 900 г/моль или не более чем 1000 г/моль.
Полимер ППГ включает, без ограничения, ППГ 100, ППГ 200, ППГ 300, ППГ 400, ППГ 500, ППГ 600, ППГ 700, ППГ 800, ППГ 900, ППГ 1000, ППГ 1100, ППГ 1200, ППГ 1300, ППГ 1400, ППГ 1500, ППГ 1600, ППГ 1700, ППГ 1800, ППГ 1900, ППГ 2000, ППГ 2100, ППГ 2200, ППГ 2300, ППГ 2400, ППГ 2500, ППГ 2600, ППГ 2700, ППГ 2800, ППГ 2900, ППГ 3000, ППГ 3250, ППГ 3350, ППГ 3500, ППГ 3750, ППГ 4000, ППГ 4250, ППГ 4500, ППГ 4750, ППГ 5000, ППГ 5500, ППГ 6000, ППГ 6500, ППГ 7000, ППГ 7500, ППГ 8000, ППГ 8500, ППГ 9000, ППГ 9500, ППГ 10000, ППГ 11000, ППГ 12000, ППГ 13000, ППГ 14000, ППГ 15000, ППГ 16000, ППГ 17000, ППГ 18000, ППГ 19000 или ППГ 20000.
В качестве стабилизатора может быть также использован одноатомный спирт. В аспектах этого варианта осуществления, одноатомный спирт может представлять, например, С2-4 спирт, С1-4 спирт, С1-5 спирт, С1-7 спирт, С1-10 спирт, С1-15 спирт или С1-20 спирт. Примеры одноатомного спирта включают, без ограничения, метанол, этанол, пропанол, бутанол, пентанол и 1-гексадеканол.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, нейтрализатор. Нейтрализатор представляет собой соединение, которое взаимодействует с раскрытом в изобретении терапевтическим соединением в его солевой форме для нейтрализации ионных зарядов, образующихся при растворении терапевтического соединения, тем самым препятствуя ионным взаимодействиям между матрицами терапевтическое соединение/липид и предотвращая их конфигурации, необходимые для образования кристаллических матриц твердофазной композиции. Следовательно, нейтрализатор предотвращает термодинамический переход композиции в классическую твердую фазу или растягивает во времени этот переход в такой степени, что он не происходит. Примеры нейтрализатора включают раскрытые в изобретении жирные кислоты для лекарственных средств в форме солей присоединения основания и ацетат натрия или триэтаноламин для лекарственных средств в форме солей присоединения кислоты.
Используемое количество нейтрализатора зависит от требуемой степени нейтрализации заряда. Для полной нейтрализации, к составу добавляют один эквивалент нейтрализатора по отношению к терапевтическому соединению. Для частичной нейтрализации, добавляют меньше одного эквивалента нейтрализатора. Частичная нейтрализация предпочтительна при приготовлении состава с замедленным высвобождением. После введения, часть терапевтического средства мгновенно становится доступной для организма (мгновенная биодоступность), в то время как биодоступность другой части задерживается до тех пор, пока терапевтическое соединение нейтрализовано в результате действия нейтрализатора. Нейтрализатор может быть также добавлен в избыточном количестве, то есть в количестве более чем один эквивалент по отношению к терапевтическому соединению. Кроме нейтрализации солевой формы лекарственного средства, избыточные количества нейтрализатора могут также позволять корректировать температуру плавления композиции твердого раствора.
В аспектах этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, по меньшей мере, 5 мг, по меньшей мере, 10 мг, по меньшей мере, 15 мг, по меньшей мере, 20 мг, по меньшей мере, 25 мг, по меньшей мере, 30 мг, по меньшей мере, 35 мг, по меньшей мере, 40 мг, по меньшей мере, 45 мг, по меньшей мере, 50 мг, по меньшей мере, 55 мг, по меньшей мере, 60 мг, по меньшей мере, 65 мг, по меньшей мере, 70 мг, по меньшей мере, 75 мг, по меньшей мере, 80 мг, по меньшей мере, 85 мг, по меньшей мере, 90 мг, по меньшей мере, 95 мг или, по меньшей мере, 100 мг. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, по меньшей мере, 5 мг, по меньшей мере, 10 мг, по меньшей мере, 20 мг, по меньшей мере, 25 мг, по меньшей мере, 50 мг, по меньшей мере, 75 мг, по меньшей мере, 100 мг, по меньшей мере, 200 мг, по меньшей мере, 300 мг, по меньшей мере, 400 мг, по меньшей мере, 500 мг, по меньшей мере, 600 мг, по меньшей мере, 700 мг, по меньшей мере, 800 мг, по меньшей мере, 900 мг, по меньшей мере, 1000 мг, по меньшей мере, 1100 мг, по меньшей мере, 1200 мг, по меньшей мере, 1300 мг, по меньшей мере, 1400 мг или, по меньшей мере, 1500 мг. В еще одних аспектах варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, от приблизительно 5 мг до приблизительно 100 мг, от приблизительно 10 мг до приблизительно 100 мг, от приблизительно 50 мг до приблизительно 150 мг, от приблизительно 100 мг до приблизительно 250 мг, от приблизительно 150 мг до приблизительно 350 мг, от приблизительно 250 мг до приблизительно 500 мг, от приблизительно 350 мг до приблизительно 600 мг, от приблизительно 500 мг до приблизительно 750 мг, от приблизительно 600 мг до приблизительно 900 мг, от приблизительно 750 мг до приблизительно 1000 мг, от приблизительно 850 мг до приблизительно 1200 мг или от приблизительно 1000 мг до приблизительно 1500 мг. В еще одних аспектах этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, от приблизительно 10 мг до приблизительно 250 мг, от приблизительно 10 мг до приблизительно 500 мг, от приблизительно 10 мг до приблизительно 750 мг, от приблизительно 10 мг до приблизительно 1000 мг, от приблизительно 10 мг до приблизительно 1500 мг, от приблизительно 50 мг до приблизительно 250 мг, от приблизительно 50 мг до приблизительно 500 мг, от приблизительно 50 мг до приблизительно 750 мг, от приблизительно 50 мг до приблизительно 1000 мг, от приблизительно 50 мг до приблизительно 1500 мг, от приблизительно 100 мг до приблизительно 250 мг, от приблизительно 100 мг до приблизительно 500 мг, от приблизительно 100 мг до приблизительно 750 мг, от приблизительно 100 мг до приблизительно 1000 мг, от приблизительно 100 мг до приблизительно 1500 мг, от приблизительно 200 мг до приблизительно 500 мг, от приблизительно 200 мг до приблизительно 750 мг, от приблизительно 200 мг до приблизительно 1000 мг, от приблизительно 200 мг до приблизительно 1500 мг, от приблизительно 5 мг до приблизительно 1500 мг, от приблизительно 5 мг до приблизительно 1000 мг или от приблизительно 5 мг до приблизительно 250 мг.
В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, менее чем приблизительно 90% по массе, менее чем приблизительно 80% по массе, менее чем приблизительно 70% по массе, менее чем приблизительно 65% по массе, менее чем приблизительно 60% по массе, менее чем приблизительно 55% по массе, менее чем приблизительно 50% по массе, менее чем приблизительно 45% по массе, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 5% по массе или менее чем приблизительно 1% по массе. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, приблизительно от 1% до 90% по массе, приблизительно от 1% до 80% по массе, приблизительно от 1% до 75% по массе, приблизительно от 1% до 70% по массе, приблизительно от 1% до 65% по массе, приблизительно от 1% до 60% по массе, приблизительно от 1% до 55% по массе, приблизительно от 1% до 50% по массе, приблизительно от 1% до 45% по массе, приблизительно от 1% до 40% по массе, приблизительно от 1% до 35% по массе, приблизительно от 1% до 30% по массе, приблизительно от 1% до 25% по массе, приблизительно от 1% до 20% по массе, приблизительно от 1% до 15% по массе, приблизительно от 1% до 10% по массе, приблизительно от 1% до 5% по массе, приблизительно от 2% до 50% по массе, приблизительно от 2% до 40% по массе, приблизительно от 2% до 30% по массе, приблизительно от 2% до 20% по массе, приблизительно от 2% до 10% по массе, приблизительно от 4% до 50% по массе, приблизительно от 4% до 40% по массе, приблизительно от 4% до 30% по массе, приблизительно от 4% до 20% по массе, приблизительно от 4% до 10% по массе, приблизительно от 6% до 50% по массе, приблизительно от 6% до 40% по массе, приблизительно от 6% до 30% по массе, приблизительно от 6% до 20% по массе, приблизительно от 6% до 10% по массе, приблизительно от 8% до 50% по массе, приблизительно от 8% до 40% по массе, приблизительно от 8% до 30% по массе, приблизительно от 8% до 20% по массе, приблизительно от 8% до 15% по массе или приблизительно от 8% до 12% по массе.
В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может включать нейтрализатор в количестве, например, от приблизительно 0,1% до приблизительно 45% по массе, от приблизительно 0,1% до приблизительно 40% по массе, от приблизительно 0,1% до приблизительно 35% по массе, от приблизительно 0,1% до приблизительно 30% по массе, от приблизительно 0,1% до приблизительно 25% по массе, от приблизительно 0,1% до приблизительно 20% по массе, от приблизительно 0,1% до приблизительно 15% по массе, от приблизительно 0,1% до приблизительно 10% по массе, от приблизительно 0,1% до приблизительно 5% по массе, от приблизительно 1% до приблизительно 45% по массе, от приблизительно 1% до приблизительно 40% по массе, от приблизительно 1% до приблизительно 35% по массе, от приблизительно 1% до приблизительно 30% по массе, от приблизительно 1% до приблизительно 25% по массе, от приблизительно 1% до приблизительно 20% по массе, от приблизительно 1% до приблизительно 15% по массе, от приблизительно 1% до приблизительно 10% по массе, от приблизительно 1% до приблизительно 5% по массе, от приблизительно 5% до приблизительно 45% по массе, от приблизительно 5% до приблизительно 40% по массе, от приблизительно 5% до приблизительно 35% по массе, от приблизительно 5% до приблизительно 30% по массе, от приблизительно 5% до приблизительно 25% по массе, от приблизительно 5% до приблизительно 20% по массе, от приблизительно 5% до приблизительно 15% по массе, от приблизительно 5% до приблизительно 10% по массе, от приблизительно 10% до приблизительно 45% по массе, от приблизительно 10% до приблизительно 40% по массе, от приблизительно 10% до приблизительно 35% по массе, от приблизительно 10% до приблизительно 30% по массе, от приблизительно 10% до приблизительно 25% по массе, от приблизительно 10% до приблизительно 20% по массе, от приблизительно 10% до приблизительно 15% по массе, от приблизительно 15% до приблизительно 45% по массе, от приблизительно 15% до приблизительно 40% по массе, от приблизительно 15% до приблизительно 35% по массе, от приблизительно 15% до приблизительно 30% по массе, от приблизительно 15% до приблизительно 25% по массе, от приблизительно 15% до приблизительно 20% по массе, от приблизительно 20% до приблизительно 45% по массе, от приблизительно 20% до приблизительно 40% по массе, от приблизительно 20% до приблизительно 35% по массе, от приблизительно 20% до приблизительно 30% по массе, от приблизительно 20% до приблизительно 25% по массе, от приблизительно 25% до приблизительно 45% по массе, от приблизительно 25% до приблизительно 40% по массе, от приблизительно 25% до приблизительно 35% по массе или от приблизительно 25% до приблизительно 30% по массе.
Раскрытая в изобретении фармацевтическая композиция может необязательно включать фармацевтически приемлемый носитель, который облегчает переработку активного ингредиента в фармацевтически приемлемые композиции. Используемый в изобретении термин "фармацевтически приемлемый носитель" является синонимом "фармакологического носителя" и означает любой носитель, который практически не обладает длительным или постоянным вредным воздействием при введении, и включает в себя такие термины, как "фармакологически приемлемые среда, стабилизатор, разбавитель, добавка, вспомогательное вещество или наполнитель". Такой носитель обычно смешивают с активным соединением или растворяют активное соединение в нем или заключают в него, и такой носитель может представлять собой твердое, полутвердое или жидкое вещество. Подразумевается, что активные ингредиенты могут быть растворены или могут быть введены в виде суспензии в требуемый носитель и разбавитель. Может быть использован любой из множества фармацевтически приемлемых носителей, включая, без ограничения, водные среды, такие как, например, вода, физиологический раствор, глицин, гиалуроновая кислота и другие подобные среды; твердые носители, такие как, например, маннит, лактоза, крахмал, стеарат магния, сахарин натрия, тальк, целлюлоза, сахароза, карбонат магния и другие подобные вещества; растворители; дисперсионные среды; покрытия; антибактериальные и противогрибковые средства; изотонические и замедляющие абсорбцию средства или другие неактивные ингредиенты. Выбор фармакологически приемлемого носителя может зависеть от способа введения. За исключением тех случаев, когда какой-либо фармацевтически приемлемый носитель не совместим с активным ингредиентом, для его использования в фармацевтически приемлемых композициях нет ограничений. Неограничивающие примеры конкретного применения таких фармацевтических носителей можно найти в следующих монографиях и справочниках: Pharmaceutical Dosage Forms and Drug Delivery Systems (Howard C. Ansel et al., eds., Lippincott Williams & Wilkins Publishers, 7th ed. 1999); REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (Alfonso R. Gennaro ed., Lippincott, Williams & Wilkins, 20th ed. 2000); Goodman & Gilman's The Pharmacological Basis of Therapeutics (Joel G. Hardman et al., eds., McGraw-Hill Professional, 10th ed. 2001); and Handbook of Pharmaceutical Excipients (Raymond C. Rowe et al., APhA Publications, 4th edition 2003). Эти протоколы представляют собой общепринятые методики, и любые их модификации могут быть осуществлены любым специалистом в этой области и исходя из идеи настоящего изобретения.
Раскрытая в изобретении фармацевтическая композиция может необязательно включать, без ограничения, другие фармацевтически приемлемые компоненты (или фармацевтические компоненты), в том числе, без ограничения, буферы, консерванты, вещества, регулирующие тоничность, соли, антиоксиданты, вещества, корректирующие осмоляльность, физиологические вещества, фармакологические вещества, объемообразующие вещества, эмульгаторы, увлажняющие средства, подсластители или ароматизаторы и другие подобные вещества. Для приготовления раскрытой в изобретении фармацевтической композиции могут быть использованы различный буферы и средства для корректировки рН, при условии, что полученный препарат будет фармацевтически приемлемым. Такие буферы включают, без ограничения, ацетатные буферы, цитратные буферы, фосфатные буферы, нейтральный забуференный физиологический раствор, забуференный фосфатом физиологический раствор и боратные буферы. Подразумевается, что, в случае необходимости, для корректировки рН композиции могут быть использованы кислоты и основания. Фармацевтически приемлемые антиоксиданты включают, без ограничения, метабисульфит натрия, тиосульфат натрия, ацетилцистеин, бутилированный гидроксианизол и бутилированный гидрокситолуол. Подходящие консерванты включают, без ограничения, бензалконий хлорид, хлорбутанол, тимеросал, ацетат фенилртути, нитрат фенилртути, стабилизированная оксихлорсодерожащая композиция и хелатообразующие соединения, такие как, например, диэтилентриаминпентауксусная кислота (DTPA) или DTPA-бисамид, кальций DTPA и CaNaDTPA-бисамид. Вещества, регулирующие тоничность, применяемые в фармацевтической композиции включают, без ограничения, соли, такие как, например, хлорид натрия, хлорид калия, маннит или глицерин и другие фармацевтически приемлемые вещества, регулирующие тоничность. Фармацевтическая композиция может быть приготовлена в виде соли, и соль может быть образована многими кислотами, включая, но этим не ограничивая, хлористоводородную, серную, уксусную, молочную, винную, яблочную, янтарную и другие кислоты. Соли, как правило, более растворимы в водных или других протонных растворителях, чем соответствующие формы свободного основания. Подразумевается, что эти и другие вещества, известные в области фармакологии, могут быть включены в фармацевтическую композицию.
В одном варианте осуществления, раскрытая в изобретении фармацевтическая композиция является твердой при комнатной температуре. В аспектах этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция может быть приготовлена так, чтобы быть твердой при температуре, например, приблизительно 35°C или ниже, приблизительно 34°C или ниже, приблизительно 33°C или ниже, приблизительно 32°C или ниже, приблизительно 31°C или ниже, приблизительно 30°C или ниже, приблизительно 29°C или ниже, приблизительно 28°C или ниже, приблизительно 27°C или ниже, приблизительно 26°C или ниже, приблизительно 25°C или ниже, приблизительно 24°C или ниже, приблизительно 23°C или ниже, приблизительно 22°C или ниже, приблизительно 21°C или ниже, приблизительно 20°C или ниже, приблизительно 19°C или ниже, приблизительно 18°C или ниже, приблизительно 17°C или ниже, приблизительно 16°C или ниже, приблизительно 15°C или ниже, приблизительно 14°C или ниже, приблизительно 13°C или ниже, приблизительно 12°C или ниже, приблизительно 11°C или ниже, приблизительно 10°C или ниже, приблизительно 9°C или ниже, приблизительно 8°C или ниже, приблизительно 7°C или ниже, приблизительно 6°C или ниже, приблизительно 5°C или ниже, приблизительно 4°C или ниже, приблизительно 3°C или ниже, приблизительно 2°C или ниже, приблизительно 1°C или ниже или приблизительно 0°C или ниже.
В другом аспекте этого варианта осуществления, раскрытая фармацевтическая композиция имеет температуру плавления, например, 5ºC или выше, 10ºC или выше, 15ºC или выше, 16ºC или выше, 17ºC или выше, 18ºC или выше, 19ºC или выше, 20ºC или выше, 21ºC или выше, 22ºC или выше, 23ºC или выше, 24ºC или выше, 25ºC или выше, 26ºC или выше, 27ºC или выше, 28ºC или выше, 29ºC или выше, 30ºC или выше, 31ºC или выше, 32ºC или выше, 33ºC или выше, 34ºC или выше, 35ºC или выше, 36ºC или выше или 37ºC или выше. В другом аспекте этого варианта осуществления, раскрытая фармацевтическая композиция имеет температуру плавления в диапазоне, например, от приблизительно 5ºC до приблизительно 24ºC, от приблизительно 10ºC до приблизительно 24ºC, от приблизительно 22ºC до приблизительно 24ºC, от приблизительно 23ºC до приблизительно 25ºC, от приблизительно 24ºC до приблизительно 26ºC, от приблизительно 25ºC до приблизительно 27ºC, от приблизительно 26ºC до приблизительно 28ºC, от приблизительно 27ºC до приблизительно 29ºC, от приблизительно 28ºC до приблизительно 30ºC, от приблизительно 29ºC до приблизительно 31ºC, от приблизительно 30ºC до приблизительно 32ºC, от приблизительно 31ºC до приблизительно 33ºC, от приблизительно 32ºC до приблизительно 34ºC, от приблизительно 33ºC до приблизительно 35ºC, от приблизительно 34ºC до приблизительно 36ºC или от приблизительно 35ºC до приблизительно 37ºC. В другом аспекте этого варианта осуществления, раскрытая фармацевтическая композиция имеет температуру плавления в диапазоне, например, от приблизительно 22ºC до приблизительно 26ºC, от приблизительно 24ºC до приблизительно 28ºC, от приблизительно 26ºC до приблизительно 30ºC, от приблизительно 28ºC до приблизительно 32ºC, от приблизительно 30ºC до приблизительно 34ºC, от приблизительно 32ºC до приблизительно 36ºC или от приблизительно 34ºC до приблизительно 38ºC.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, способ приготовления раскрытой в изобретении фармацевтической композиции твердого раствора. В одном варианте осуществления, раскрытый в изобретении способ включает стадии a) контактирования раскрытого в изобретении терапевтического соединения с одним или более жидкими при комнатной температуре липидами при условиях, которые способствуют растворению терапевтического соединения в одном или более липидах; и b) контактирования раствора соединение/липид с одним или более раскрытыми в изобретении твердыми при комнатной температуре липидами при условиях, которые способствуют образованию композиции твердого раствора.
В одном варианте осуществления, раскрытый в изобретении способ включает стадии a) контактирования раскрытого в изобретении терапевтического соединения с одним или более жидкими при комнатной температуре липидами и одним или более стабилизаторами при условиях, которые способствуют растворению терапевтического соединения в одном или более липидах; и b) контактирования раствора соединение/липид с одним или более раскрытыми в изобретении твердыми при комнатной температуре липидами при условиях, которые способствуют образованию композиции твердого раствора.
В одном варианте осуществления, раскрытый в изобретении способ включает стадии a) контактирования раскрытого в изобретении терапевтического соединения с одним или более жидкими при комнатной температуре липидами и одним или более нейтрализаторами при условиях, которые способствуют растворению терапевтического соединения в одном или более липидах; и b) контактирования раствора соединение/липид с одним или более раскрытыми в изобретении твердыми при комнатной температуре липидами при условиях, которые способствуют образованию композиции твердого раствора.
В одном варианте осуществления, раскрытый в изобретении способ включает стадии a) контактирования раскрытого в изобретении терапевтического соединения с одним или более жидкими при комнатной температуре липидами, одним или более стабилизаторами и одним или более нейтрализаторами при условиях, которые способствуют растворению терапевтического соединения в одном или более липидах; и b) контактирования раствора соединение/липид с одним или более раскрытыми в изобретении твердыми при комнатной температуре липидами при условиях, которые способствуют образованию композиции твердого раствора.
Раскрытый в изобретении способ осуществляют при условиях, которые способствуют растворению терапевтического соединения в других компонентах. В аспектах этого варианта осуществления, раскрытый в изобретении способ может быть осуществлен при температуре, достаточной для растворения терапевтического соединения в одном или более жидких при комнатной температуре липидах, и/или одном или более стабилизаторах, и/или одном или более нейтрализаторах с получением раствора. В другом аспекте этого варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, приблизительно 40°C, приблизительно 45°C, приблизительно 50°C, приблизительно 55°C, приблизительно 60°C, приблизительно 65°C, приблизительно 70°C или приблизительно 75°C. В еще одних аспектах варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, по меньшей мере, 40°C, по меньшей мере, 45°C, по меньшей мере, 50°C, по меньшей мере, 55°C, по меньшей мере, 60°C, по меньшей мере, 65°C, по меньшей мере, 70°C или, по меньшей мере, 75°C. В еще одних аспектах этого варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, не более 40°C, не более 45°C, не более 50°C, не более 55°C, не более 60°C, не более 65°C, не более 70°C или не более 75°C. В другом аспекте этого варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, от приблизительно 40°C до приблизительно 45°C, от приблизительно 40°C до приблизительно 50°C, от приблизительно 40°C до приблизительно 55°C, от приблизительно 40°C до приблизительно 60°C, от приблизительно 40°C до приблизительно 65°C, от приблизительно 40°C до приблизительно 70°C, от приблизительно 40°C до приблизительно 75°C, от приблизительно 45°C до приблизительно 50°C, от приблизительно 45°C до приблизительно 55°C, от приблизительно 45°C до приблизительно 60°C, от приблизительно 45°C до приблизительно 65°C, от приблизительно 45°C до приблизительно 70°C, от приблизительно 45°C до приблизительно 75°C, от приблизительно 50°C до приблизительно 55°C, от приблизительно 50°C до приблизительно 60°C, от приблизительно 50°C до приблизительно 65°C, от приблизительно 50°C до приблизительно 70°C, от приблизительно 50°C до приблизительно 75°C, от приблизительно 55°C до приблизительно 60°C, от приблизительно 55°C до приблизительно 65°C, от приблизительно 55°C до приблизительно 70°C, от приблизительно 55°C до приблизительно 75°C, от приблизительно 60°C до приблизительно 65°C, от приблизительно 60°C до приблизительно 70°C, от приблизительно 60°C до приблизительно 75°C, от приблизительно 65°C до приблизительно 70°C, от приблизительно 65°C до приблизительно 75°C или от приблизительно 70°C до приблизительно 75°C.
В другом аспекте этого варианта осуществления, раскрытый в изобретении способ может быть осуществлен при температуре, достаточной для введения одного или более твердых при комнатной температуре липидов в раствор, содержащий терапевтическое соединение. В другом аспекте этого варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, приблизительно 40°C, приблизительно 45°C, приблизительно 50°C, приблизительно 55°C, приблизительно 60°C, приблизительно 65°C, приблизительно 70°C или приблизительно 75°C, для введения одного или более твердых при комнатной температуре липидов. В еще одних аспектах варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, по меньшей мере, 40°C, по меньшей мере, 45°C, по меньшей мере, 50°C, по меньшей мере, 55°C, по меньшей мере, 60°C, по меньшей мере, 65°C, по меньшей мере, 70°C или, по меньшей мере, 75°C, для введения одного или более твердых при комнатной температуре липидов. В еще одних аспектах этого варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, не более 40°C, не более 45°C, не более 50°C, не более 55°C, не более 60°C, не более 65°C, не более 70°C или не более 75°C, для введения одного или более твердых при комнатной температуре липидов. В другом аспекте этого варианта осуществления, раскрытый в изобретении способ включает нагревание смеси, содержащей раскрытое в изобретении терапевтическое соединение, до температуры, например, от приблизительно 40°C до приблизительно 45°C, от приблизительно 40°C до приблизительно 50°C, от приблизительно 40°C до приблизительно 55°C, от приблизительно 40°C до приблизительно 60°C, от приблизительно 40°C до приблизительно 65°C, от приблизительно 40°C до приблизительно 70°C, от приблизительно 40°C до приблизительно 75°C, от приблизительно 45°C до приблизительно 50°C, от приблизительно 45°C до приблизительно 55°C, от приблизительно 45°C до приблизительно 60°C, от приблизительно 45°C до приблизительно 65°C, от приблизительно 45°C до приблизительно 70°C, от приблизительно 45°C до приблизительно 75°C, от приблизительно 50°C до приблизительно 55°C, от приблизительно 50°C до приблизительно 60°C, от приблизительно 50°C до приблизительно 65°C, от приблизительно 50°C до приблизительно 70°C, от приблизительно 50°C до приблизительно 75°C, от приблизительно 55°C до приблизительно 60°C, от приблизительно 55°C до приблизительно 65°C, от приблизительно 55°C до приблизительно 70°C, от приблизительно 55°C до приблизительно 75°C, от приблизительно 60°C до приблизительно 65°C, от приблизительно 60°C до приблизительно 70°C, от приблизительно 60°C до приблизительно 75°C, от приблизительно 65°C до приблизительно 70°C, от приблизительно 65°C до приблизительно 75°C или от приблизительно 70°C до приблизительно 75°C, для введения одного или более твердых при комнатной температуре липидов.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, контактирование раскрытого в изобретении терапевтического соединения с одним или более липидами. В аспектах этого варианта осуществления, один или более липидов включают один, два, три, четыре или пять раскрытых в изобретении различных липидов. В другом аспекте этого варианта осуществления, один или более липидов включают два или более, три или более, четыре или более или пять или более раскрытых в изобретении различных липидов. В еще одних аспектах варианта осуществления, один или более липидов включают от приблизительно одного до приблизительно пяти раскрытых в изобретении различных липидов, от приблизительно двух до приблизительно пяти раскрытых в изобретении различных липидов, от приблизительно трех до приблизительно пяти раскрытых в изобретении различных липидов, от приблизительно одного до приблизительно четырех раскрытых в изобретении различных липидов, от приблизительно двух до приблизительно четырех раскрытых в изобретении различных липидов или от приблизительно двух до приблизительно трех раскрытых в изобретении различных липидов.
В другом аспекте этого варианта осуществления, один или более липидов включают один или более твердых при комнатной температуре липидов и один или более жидких при комнатной температуре липидов. В аспектах этого варианта осуществления, один или более липидов включают один, два, три, четыре или пять раскрытых в изобретении различных твердых жиров и один, два, три, четыре или пять раскрытых в изобретении различных жидких при комнатной температуре липидов. В другом аспекте этого варианта осуществления, один или более липидов включают два или более, три или более, четыре или более или пять или более раскрытых в изобретении различных твердых при комнатной температуре липидов и два или более, три или более, четыре или более или пять или более раскрытых в изобретении жидких при комнатной температуре липидов. В еще одних аспектах варианта осуществления, один или более липидов включают от приблизительно одного до приблизительно пяти раскрытых в изобретении различных твердых жиров, от приблизительно двух до приблизительно пяти раскрытых в изобретении различных твердых при комнатной температуре липидов, от приблизительно трех до приблизительно пяти раскрытых в изобретении различных твердых при комнатной температуре липидов, от приблизительно одного до приблизительно четырех раскрытых в изобретении различных твердых при комнатной температуре липидов, от приблизительно двух до приблизительно четырех раскрытых в изобретении различных твердых при комнатной температуре липидов или от приблизительно двух до приблизительно трех раскрытых в изобретении различных твердых при комнатной температуре липидов и от приблизительно одного до приблизительно пяти раскрытых в изобретении различных жидких при комнатной температуре липидов, от приблизительно двух до приблизительно пяти раскрытых в изобретении различных жидких при комнатной температуре липидов, от приблизительно трех до приблизительно пяти раскрытых в изобретении различных жидких при комнатной температуре липидов, от приблизительно одного до приблизительно четырех раскрытых в изобретении различных жидких при комнатной температуре липидов, от приблизительно двух до приблизительно четырех раскрытых в изобретении различных жидких при комнатной температуре липидов или от приблизительно двух до приблизительно трех раскрытых в изобретении различных жидких при комнатной температуре липидов.
В аспектах этого варианта осуществления, в раскрытом в изобретении способе может использоваться твердый при комнатной температуре липид и жидкий при комнатной температуре липид при соотношении твердый липид:жидкий липид, например, по меньшей мере, 1:1, по меньшей мере, 2:1, по меньшей мере, 3:1, по меньшей мере, 4:1, по меньшей мере, 5:1, по меньшей мере, 6:1, по меньшей мере, 7:1, по меньшей мере, 8:1, по меньшей мере, 9:1, по меньшей мере, 10:1, по меньшей мере, 15:1 или, по меньшей мере, 20:1. В другом аспекте этого варианта осуществления, в раскрытом в изобретении способе может использоваться твердый при комнатной температуре липид и жидкий при комнатной температуре липид при соотношении твердый липид:жидкий липид, например, от приблизительно 1:1 до приблизительно 20:1, от приблизительно 5:1 до приблизительно 20:1, от приблизительно 2:1 до приблизительно 15:1, от приблизительно 5:1 до приблизительно 15:1, от приблизительно 4:1 до приблизительно 12:1 или от приблизительно 6:1 до приблизительно 10:1.
В аспектах этого варианта осуществления, в раскрытом в изобретении способе может использоваться множество твердых при комнатной температуре липидов и множество жидких при комнатной температуре липидов при соотношении суммарное количество твердых липидов:суммарное количество жидких липидов, например, по меньшей мере, 1:1, по меньшей мере, 2:1, по меньшей мере, 3:1, по меньшей мере, 4:1, по меньшей мере, 5:1, по меньшей мере, 6:1, по меньшей мере, 7:1, по меньшей мере, 8:1, по меньшей мере, 9:1, по меньшей мере, 10:1, по меньшей мере, 15:1 или, по меньшей мере, 20:1. В другом аспекте этого варианта осуществления, в раскрытом в изобретении способе может использоваться множество твердых при комнатной температуре липидов и множество жидких при комнатной температуре липидов при соотношении суммарное количество твердых липидов:суммарное количество жидких липидов, например, от приблизительно 1:1 до приблизительно 20:1, от приблизительно 5:1 до приблизительно 20:1, от приблизительно 2:1 до приблизительно 15:1, от приблизительно 5:1 до приблизительно 15:1, от приблизительно 4:1 до приблизительно 12:1 или от приблизительно 6:1 до приблизительно 10:1.
Контактирование терапевтического соединения и одного или более липидов может включать, например, перемешивание, инверсию, обработку ультразвуком или встряхивание. Смешение может быть проведено в течение, например, по меньшей мере, 1 секунды, по меньшей мере, 5 секунд, по меньшей мере, 10 секунд, по меньшей мере, 20 секунд, по меньшей мере, 30 секунд, по меньшей мере, 45 секунд, по меньшей мере, 60 секунд или более, до тех пор, пока терапевтическое соединение полностью не растворится в смеси липид/полимер гликоля.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, контактирование раствора соединение/липид с одним или более раскрытыми в изобретении полимерами гликоля.
Раскрытый в изобретении способ осуществляют при условиях, которые способствуют отвердеванию композиции твердого раствора. В аспектах этого варианта осуществления, способ может быть осуществлен при температуре, достаточной для охлаждения композиции до температуры, при которой раствор затвердевает.
В конкретных вариантах осуществления, для снижения температуры после образования раскрытой в изобретении фармацевтической композиции может быть использована стадия быстрого охлаждения. Например, стадия быстрого охлаждения может быть использована при подходах, когда применяют температуры выше, чем комнатная температура, для обеспечения полного растворения терапевтического соединения в фармацевтически приемлемом растворителе и/или для того чтобы раствор, содержащий терапевтическое соединение, мог образовать фармацевтическую композицию. В аспектах этого варианта осуществления, стадия быстрого охлаждения позволяет понизить температуру, например, на приблизительно 30°C через 20 минут, на приблизительно 25°C через 20 минут, на приблизительно 20°C через 20 минут, на приблизительно 15°C через 20 минут, на приблизительно 30°C через 15 минут, на приблизительно 25°C через 15 минут, на приблизительно 20°C через 15 минут, на приблизительно 15°C через 15 минут, на приблизительно 30°C через 10 минут, на приблизительно 25°C через 10 минут, на приблизительно 20°C через 10 минут, на приблизительно 15°C через 10 минут, на приблизительно 30°C через 5 минут, на приблизительно 25°C через 5 минут, на приблизительно 20°C через 5 минут, на приблизительно 15°C через 5 минут. В другом аспекте этого варианта осуществления, стадия быстрого охлаждения позволяет понизить температуру, например, на величину от приблизительно 20°C до приблизительно 30°C через 20 минут, на величину от приблизительно 20°C до приблизительно 30°C через 15 минут, на величину от приблизительно 20°C до приблизительно 30°C через 10 минут, на величину от приблизительно 20°C до приблизительно 30°C через 5 минут, на величину от приблизительно 15°C до приблизительно 25°C через 20 минут, на величину от приблизительно 15°C до приблизительно 25°C через 15 минут, на величину от приблизительно 15°C до приблизительно 25°C через 10 минут, на величину от приблизительно 15°C до приблизительно 25°C через 5 минут, на величину от приблизительно 10°C до приблизительно 20°C через 20 минут, на величину от приблизительно 10°C до приблизительно 20°C через 15 минут, на величину от приблизительно 10°C до приблизительно 20°C через 10 минут или на величину от приблизительно 10°C до приблизительно 20°C через 5 минут.
В еще одних аспектах этого варианта осуществления, стадия быстрого охлаждения позволяет понизить температуру со скоростью, например, приблизительно 2,0°C/минута, приблизительно 1,9°C/минута, приблизительно 1,8°C/минута, приблизительно 1,7°C/минута, приблизительно 1,6°C/минута, приблизительно 1,5°C/минута, приблизительно 1,4°C/минута, приблизительно 1,3°C/минута, приблизительно 1,2°C/минута, приблизительно 1,1°C/минута, приблизительно 1,0°C/минута, приблизительно 0,9°C/минута, приблизительно 0,8°C/минута, приблизительно 0,7°C/минута, приблизительно 0,6°C/минута, приблизительно 0,5°C/минута, приблизительно 0,4°C/минута, приблизительно 0,3°C/минута, приблизительно 0,2°C/минута или приблизительно 0,1°C/минута. В еще одних аспектах варианта осуществления, стадия быстрого охлаждения позволяет понизить температуру со скоростью, например, от приблизительно 0,1°C до приблизительно 0,4°C/минута, от приблизительно 0,2°C до приблизительно 0,6°C/минута, от приблизительно 0,4°C до приблизительно 0,8°C/минута, от приблизительно 0,6°C до приблизительно 1,0°C/минута, от приблизительно 0,8°C до приблизительно 1,2°C/минута, от приблизительно 1,0°C до приблизительно 1,4°C/минута, от приблизительно 1,2°C до приблизительно 1,6°C/минута, от приблизительно 1,4°C до приблизительно 1,8°C/минута, от приблизительно 1,6°C до приблизительно 2,0°C/минута, от приблизительно 0,1°C до приблизительно 0,5°C/минута, от приблизительно 0,5°C до приблизительно 1,0°C/минута, от приблизительно 1,0°C до приблизительно 1,5°C/минута, от приблизительно 1,5°C до приблизительно 2,0°C/минута, от приблизительно 0,5°C до приблизительно 1,5°C/минута или от приблизительно 1,0°C до приблизительно 2,0°C/минута.
Раскрытый в изобретении состав позволяет сформировать твердый раствор липидов и терапевтического соединения. Такие составы не образуют липосомальных эмульсий и/или мицелярных частиц и/или многофазных композиций другого типа. Кроме того, такие составы не требуют применения гидрофильного растворителя, такого как, например, вода или забуферированный раствор. По этой причине, раскрытую в изобретении фармацевтическую композицию готовят без применения гидрофильного растворителя. В одном варианте осуществления, раскрытая в изобретении фармацевтическая композиция не включает фармацевтически приемлемый гидрофильный растворитель.
Количество терапевтического соединения, жидкого при комнатной температуре липида, твердого при комнатной температуре липида (твердого жира), стабилизатора и нейтрализатора, используемых в раскрытом изобретением способе, может представлять собой любое требуемое количество. Факторы, принимаемые во внимание при определении количества каждого используемого компонента, включают, без ограничения, требуемое конечное количество терапевтического соединения в фармацевтической композиции, требуемая концентрация терапевтического соединения в растворе, гидрофобность терапевтического соединения, липофильность терапевтического соединения, требуемое конечное количество фармацевтической композиции и условия, используемые для получения фармацевтической композиции твердого раствора.
В одном варианте осуществления, фармацевтическая композиция включает одно и более терапевтических соединений, твердые при комнатной температуре липиды или твердые жиры и один или более жидких при комнатной температуре липидов, но не включает фармацевтически приемлемый гидрофильный растворитель.
В одном варианте осуществления, фармацевтическая композиция включает одно и более терапевтических соединений, твердые при комнатной температуре липиды или твердые жиры и один или более жидких при комнатной температуре липидов и один или более стабилизаторов, но не включает фармацевтически приемлемый гидрофильный растворитель. В аспектах этого варианта осуществления, фармацевтическая композиция твердого раствора включает терапевтическое соединение, твердый при комнатной температуре липид или твердый жир, жидкий при комнатной температуре липид и жидкий полимер гликоля и/или одноатомный спирт, и/или диметиловый эфир изосорбида, и/или моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол), но не включает фармацевтически приемлемый гидрофильный растворитель. В другом аспекте этого варианта осуществления, фармацевтическая композиция твердого раствора включает терапевтическое соединение, смесь триглицеридов, смесь моноглицеридов и жидкий полимер ПЭГ и/или С1-C5 одноатомный спирт, и/или диметиловый эфир изосорбида, и/или моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол), но не включает фармацевтически приемлемый гидрофильный растворитель. В еще одних аспектах варианта осуществления, фармацевтическая композиция включает терапевтическое соединение, GELUCIRE® 43/01 (Gattefosse), воскообразное твердое вещество (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающее смесь насыщенных C10-C18 триглицеридов, MAISINE® 35-1 (Gattefosse), глицерил монолинолеат и жидкий полимер ПЭГ и/или С1-C5 одноатомный спирт, и/или диметиловый эфир изосорбида, и/или моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол), но не включает фармацевтически приемлемый гидрофильный растворитель.
В аспектах этого варианта осуществления, композиция твердого раствора включает от приблизительно 1% до приблизительно 55% по массе терапевтического соединения, от приблизительно 40% до приблизительно 90% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 10% по массе жидкого при комнатной температуре липида и от приблизительно 1% до приблизительно 5% стабилизатора. В аспектах этого варианта осуществления, композиция твердого раствора включает от приблизительно 1% до приблизительно 55% по массе терапевтического соединения, от приблизительно 40% до приблизительно 90% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 10% по массе жидкого при комнатной температуре липида и от приблизительно 1% до приблизительно 5% жидкого полимера гликоля и/или одноатомного спирта, и/или диметилового эфира изосорбида, и/или моноэтилового эфира диэтиленгликоля (2-(2-этоксиэтокси)этанола). В другом аспекте этого варианта осуществления, композиция твердого раствора включает от приблизительно 5% до приблизительно 50% по массе терапевтического соединения, от приблизительно 40% до приблизительно 70% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 7% по массе жидкого при комнатной температуре липида и от приблизительно 1% до приблизительно 3% жидкого полимера гликоля и/или одноатомного спирта, и/или диметилового эфира изосорбида, и/или моноэтилового эфира диэтиленгликоля (2-(2-этоксиэтокси)этанола).
В одном варианте осуществления, фармацевтическая композиция включает одно или более терапевтических соединений, один или более твердых при комнатной температуре липидов или твердых жиров, один или более жидких при комнатной температуре липидов и один или более нейтрализаторов, но не включает фармацевтически приемлемый гидрофильный растворитель. В другом аспекте этого варианта осуществления, фармацевтическая композиция твердого раствора включает терапевтическое соединение, твердый при комнатной температуре липид или твердый жир, жидкий при комнатной температуре липид и жирную кислоту, но не включает фармацевтически приемлемый гидрофильный растворитель. В еще одних аспектах варианта осуществления, фармацевтическая композиция твердого раствора включает терапевтическое соединение, смесь триглицеридов, смесь моноглицеридов и С16-C18 жирную кислоту, но не включает фармацевтически приемлемый гидрофильный растворитель. В еще одних аспектах этого варианта осуществления, фармацевтическая композиция включает терапевтическое соединение, GELUCIRE® 43/01 (Gattefosse), воскообразное твердое вещество (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающее смесь насыщенных C10-C18 триглицеридов, MAISINE® 35-1 (Gattefosse), глицерил монолинолеат и стеариновую кислоту, но не включает фармацевтически приемлемый гидрофильный растворитель.
В аспектах этого варианта осуществления, композиция твердого раствора включает от приблизительно 1% до приблизительно 55% по массе терапевтического соединения, от приблизительно 30% до приблизительно 70% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 10% по массе жидкого при комнатной температуре липида и от приблизительно 1% до приблизительно 55% нейтрализатора. В другом аспекте этого варианта осуществления, композиция твердого раствора включает от приблизительно 1% до приблизительно 55% по массе терапевтического соединения, от приблизительно 30% до приблизительно 70% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 10% по массе жидкого при комнатной температуре липида и от приблизительно 1% до приблизительно 55% жирной кислоты. В еще одних аспектах варианта осуществления, композиция твердого раствора включает от приблизительно 5% до приблизительно 50% по массе терапевтического соединения, от приблизительно 30% до приблизительно 60% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 7% по массе жидкого при комнатной температуре липида и от приблизительно 5% до приблизительно 50% жирной кислоты.
В другом аспекте этого варианта осуществления, фармацевтическая композиция твердого раствора включает одно и более терапевтических соединений, один или более твердых при комнатной температуре липидов или твердых жиров, один или более жидких при комнатной температуре липидов и триэтаноламин, но не включает фармацевтически приемлемый гидрофильный растворитель. В еще одних аспектах варианта осуществления, фармацевтическая композиция твердого раствора включает терапевтическое соединение, смесь триглицеридов, смесь моноглицеридов и триэтаноламин, но не включает фармацевтически приемлемый гидрофильный растворитель. В еще одних аспектах этого варианта осуществления, фармацевтическая композиция включает терапевтическое соединение, GELUCIRE® 43/01 (Gattefosse), воскообразное твердое вещество (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающее смесь насыщенных C10-C18 триглицеридов, MAISINE® 35-1 (Gattefosse), глицерил монолинолеат и триэтаноламин, но не включает фармацевтически приемлемый гидрофильный растворитель.
В аспектах этого варианта осуществления, композиция твердого раствора включает от приблизительно 1% до приблизительно 55% по массе терапевтического соединения, от приблизительно 30% до приблизительно 70% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 10% по массе жидкого при комнатной температуре липида и от приблизительно 1% до приблизительно 55% триэтаноламина. В другом аспекте этого варианта осуществления, композиция твердого раствора включает от приблизительно 5% до приблизительно 50% по массе терапевтического соединения, от приблизительно 30% до приблизительно 60% по массе твердого при комнатной температуре липида или твердого жира, от приблизительно 1% до приблизительно 7% по массе жидкого при комнатной температуре липида и от приблизительно 5% до приблизительно 50% триэтаноламина.
В одном варианте осуществления, фармацевтическая композиция включает одно или более терапевтических соединений, твердые при комнатной температуре липиды или твердые жиры, один или более жидких при комнатной температуре липидов, один или более стабилизаторов и один или более нейтрализаторов, но не включает фармацевтически приемлемый гидрофильный растворитель.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, способ лечения индивидуума, страдающего от хронического воспаления. В одном варианте осуществления, способ включает стадию введения индивидууму, если это ему необходимо, раскрытую в изобретении фармацевтическую композицию, где введение облегчает симптом, связанный с хроническим воспалением, вследствие чего происходит лечение индивидуума.
Аспекты настоящего изобретения раскрывают, помимо прочего, раскрытую в изобретении композицию твердого раствора для применения при лечении хронического воспаления.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, применение раскрытой в изобретении композиции твердого раствора для лечения хронического воспаления.
Аспекты настоящего изобретения раскрывают, помимо всего прочего, лечение индивидуума, страдающего от хронического воспаления. Используемый в изобретении термин "лечение" относится к ослаблению или устранению у индивидуума клинического симптома хронического воспаления; или к отсрочке или предотвращению у индивидуума возникновения клинического симптома хронического воспаления. Например, термин "лечение" может означать ослабление симптома состояния, характеризующегося хроническим воспалением, например, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90%, по меньшей мере, на 95% или, по меньшей мере, на 100%. Конкретные симптомы, связанные с хроническим воспалением, хорошо известны и могут быть определены любым специалистом в данной области с учетом факторов, включающих, без ограничения, локализацию хронического воспаления, причину хронического воспаления, тяжесть хронического воспаления, и/или ткань или орган, пораженный хроническим воспалением. Специалистам в данной области известны соответствующие симптомы или признаки, связанные с конкретным типом хронического воспаления и им известно, как определить, является ли индивидуум потенциальным кандидатом для описанного в изобретении лечения.
Симптомы хронического воспаления включают, без ограничения, отек, гиперемию, эритему, кровоподтеки, болезненную чувствительность, ригидность, припухлость, лихорадочное состояние, озноб, заложенность носа, ощущение заложенности в голове, проблемы с дыханием, задержку жидкости, кровяные сгустки, потерю аппетита, учащенное сердцебиение, образование гранулем, фибринозной гнойной невязкой серозной жидкости или изъязвлений и возникновение боли. Конкретные симптомы, связанные с хроническим воспалением, хорошо известны и могут быть определены любым специалистом в данной области с учетом факторов, включающих, без ограничения, локализацию воспаления, причину воспаления, тяжесть воспаления, пораженные ткань или орган и связанное с воспалением заболевание.
Конкретные особенности хронического воспаления наблюдаются в конкретных ситуациях, которые возникают в организме, например, когда воспаление возникает на поверхности эпителия или когда в процесс вовлечены пиогенные бактерии. Например, гранулематозное воспаление представляет собой воспаление, которое является результатом образования гранулем, возникающих при ограниченном, но разноплановом числе заболеваний, включающих, без ограничения, туберкулез, проказу, саркоидоз и сифилис. Гнойное воспаление представляет собой воспаление, приводящее к образованию большого количества гноя, который состоит из нейтрофилов, погибших клеток и жидкости. Инфицирование пиогенными бактериями, такими как стафилококки, является характерной чертой этого вида воспаления. Серозное воспаление представляет собой воспаление, которое является результатом обильной эффузия невязкой серозной жидкости, обычно продуцируемой мезотелиальными клетками серозных оболочек, но она может образовываться и из плазмы крови. Волдыри на коже являются примером этого типа воспаления. Язвенное воспаление представляет собой воспаление, которое является результатом некротической потери ткани с поверхности эпителия, что приводит к обнажению нижних слоев и образованию язвы.
Симптом хронического воспаления может быть связан с широкой группой неродственных нарушений, которые лежат в основе различных заболеваний и расстройств. В хронические воспалительные расстройства часто вовлечена иммунная система, которая проявляет себя как в виде аллергических реакций, так и в виде некоторых миопатий, при этом многие расстройства иммунной системы приводят к патологическому воспалению. Неиммунные заболевания, которые могут вызывать хронические воспалительные процессы, включают рак, атеросклероз и ишемическое заболевание сердца. Неограничивающие примеры нарушений, которые проявляются в виде симптома хронического воспаления, включают, без ограничения, угри, кислотный рефлюкс/изжогу, возрастную дегенерацию желтого пятна (AMD), аллергию, аллергический ринит, болезнь Альцгеймера, боковой амиотрофический склероз, анемию, аппендицит, артериит, артрит, астму, атеросклероз, аутоиммунные заболевания, баланит, блефарит, бронхиолит, бронхит, буллезный пемфигоид, ожог, бурсит, рак, задержку очередного сердечного сокращения, кардит, глютенчувствительную целиакию, целлюлит, цервицит, холангит, холецистит, хориоамнионит, хроническое обструктивное заболевание легких (COPD), цирроз, колит, хроническую сердечную недостаточность, конъюнктивит, индуцированный циклофосфамидом цистит, кистозный фиброз, цистит, простуду, дакриоаденит, деменцию, дерматит, дерматомиозит, диабет, диабетическую нейропатию, диабетическую ретинопатию, диабетическую нефропатию, диабетическое изъязвление, заболевание системы пищеварения, экзему, эмфизему, энцефалит, эндокардит, эндометрит, энтерит, энтероколит, эпикондилит, эпидидимит, фасциит, фибромиалгию, фиброз, фиброзит, гастрит, гастроэнтерит, гингивит, гломерулонефрит, глоссит, заболевание сердца, дисфункцию сердечного клапана, гепатит, гнойный гидраденит, болезнь Гентингтона, гиперлипидемический панкреатит, гипертензию, илеит, инфекцию, воспалительное заболевание кишечника, воспалительную кардиомегалию, воспалительную невропатию, резистентность к инсулину, интерстициальный цистит, интерстициальный нефрит, ирит, ишемию, ишемическое заболевание сердца, кератит, кератоконъюнктивит, ларингит, волчаночный нефрит, мастит, мастоидит, менингит, метаболический синдром (синдром X), мигрень, множественный склероз, миелит, миокардит, миозит, нефрит, неалкогольный стеатогепатит, ожирение, омфалит, оофорит, орхит, остеохондрит, остеопению, остеомиелит, остеопороз, остит, отит, панкреатит, болезнь Паркинсона, паротит, воспалительное заболевание тазовых органов, обыкновенную пузырчатку, перикардит, перитонит, фарингит, флебит, плеврит, пневмонию, поликистозный нефрит, проктит, простатит, псориаз, пульпит, пиелонефрит, пилефлебит, почечную недостаточность, реперфузионное повреждение, ретинит, острую ревматическую лихорадку, ринит, сальпингит, саркоидоз, сиаладенит, синусит, синдром раздраженной толстой кишки, стеноз, стоматит, инсульт, послеоперационные осложнения, синовит, тендинит, тендиноз, теносиновит, тромбофлебит, тонзиллит, травму, травматическое повреждение головного мозга, отторжение трансплантата, тригонит, туберкулез, опухоль, уретрит, бурсит, увеит, вагинит, васкулит и вульвит. Смотрите также, патентный документ Eric R. First, Application of Botulinum Toxin to the Management of Neurogenic Inflammatory Disorders, U.S. Patent 6063768, содержание которого приводится в настоящем изобретении путем ссылки на него.
В одном варианте осуществления, хроническое воспаление включает воспаление ткани. Воспаление ткани представляет собой хроническое воспаление, которое ограничивается конкретной тканью или органом. В аспекте этого варианта осуществления, воспаление ткани включает, например, воспаление кожи, воспаление мышцы, воспаление сухожилия, воспаление связки, воспаление кости, воспаление хряща, воспаление легкого, воспаление сердца, воспаление печени, воспаление поджелудочной железы, воспаление почек, воспаление мочевого пузыря, воспаление желудка, воспаление кишечника, воспаление нейронов и воспаление головного мозга.
В другом варианте осуществления, хроническое воспаление включает системное воспаление. Несмотря на то, что процессы, вовлеченные в системное воспаление, идентичны процессам при воспалении ткани, тем не менее, системное воспаление не ограничивается конкретной тканью, а фактически поражает весь организм, вовлекая эндотелий и другие системы органов. Когда системное воспаление обусловлено инфекцией, применяется термин "сепсис", при этом термин "бактериемия" используют конкретно для бактериального сепсиса, а термин "виремия" конкретно для вирусного сепсиса. Вазодилатация и дисфункция органа являются серьезными проблемами, связанными с обширной инфекцией, которая может приводить к септическому шоку и смерти.
В другом варианте осуществления, хроническое воспаление включает артрит. Артрит включает группу состояний, вовлеченных в поражение суставов тела вследствие воспаления синовиальной оболочки, в том числе, без ограничения, остеоартрит, ревматоидный артрит, ювенильный идиопатический артрит, спондилоартропатии, такие как анкилозирующий спондилит, реактивный артрит (синдром Рейтера), псориатический артрит, энтеропатический артрит, связанный с воспалительным заболеванием кишечника, болезнь Уиппла и болезнь Бехчета, септический артрит, подагра (также называемая подагрическим артритом, кристаллическим синовитом, метаболическим артритом), псевдоподагра (болезнь отложения пирофосфата кальция) и болезнь Стилла. Артрит может поражать один сустав (моноартрит), от двух до четырех суставов (олигоартрит) или пять или более суставов (полиартрит) и может представлять собой или аутоиммунное заболевание, или неаутоиммунное заболевание.
В другом варианте осуществления, хроническое воспаление включает аутоиммунное заболевание. Аутоиммунные заболевания могут быть в целом подразделены на системные и органоспецифические аутоиммунные заболевания, в зависимости от основных клинико-патологических признаков каждого заболевания. Системные аутоиммунные заболевания включают, без ограничения, системную красную волчанку (SLE), синдром Шегрена, склеродермию, ревматоидный артрит и полимиозит. Очаговые аутоиммунные заболевания могут быть эндокринологическими (сахарный диабет первого типа, тиреоидит Хашимото, болезнь Аддисона и другие подобные), дерматологическими (обыкновенная пузырчатка), гематологическими (аутоиммунная гемолитическая анемия), невральными (множественный склероз) или могут вовлекать практически любую ограниченную массу ткани организма. Типы аутоиммунных заболеваний включают, без ограничения, острый рассеянный энцефаломиелит (ADEM), болезнь Аддисона, аллергию или реактивность, боковой амиотрофический склероз, антифосфолипидный синдром (APS), артрит, аутоиммунную гемолитическую анемию, аутоиммунный гепатит, аутоиммунное заболевание внутреннего уха, аутоиммунный панкреатит, буллезный пемфигоид, глютенчувствительную целиакию, болезнь Шагаса, хроническое обструктивное заболевание легких (COPD), сахарный диабет первого типа (IDDM), эндометриоз, фибромиалгию, синдром Гудпасчера, болезнь Грейвса, синдром Гийена - Барре (GBS), тиреоидит Хашимото, гнойный гидраденит, идиопатическую тромбоцитопеническую пурпуру, воспалительное заболевание кишечника, интерстициальный цистит, волчанку (в том числе дискоидную красную волчанку, лекарственную красную волчанку, волчаночный нефрит, неонатальную волчанку, подострую кожную красную волчанку и системную красную волчанку), очаговую склеродермию, множественный склероз (MS), тяжелую миастению, миопатии, нарколепсию, нейромиотонию, обыкновенную пузырчатку, злокачественную анемию, первичный биллиарный цирроз, рецидивирующий рассеянный энцефаломиелит (мультифазный диссеминированный энцефаломиелит), острую ревматическую лихорадку, шизофрению, склеродермию, синдром Шегрена, теносиновит, васкулит и витилиго. Смотрите патентный документ Pamela D. Van Schaack & Kenneth L. Tong, Treatment of Autoimmune Disorder with a Neurotoxin, U.S. Patent Publication 2006/138059, содержание которого приводится в настоящем изобретении путем ссылки на него.
В другом варианте осуществления, хроническое воспаление включает миопатию. Миопатии возникают тогда, когда иммунная система несообразно атакует компоненты мышцы, что приводит к воспалению мышцы. Миопатия включает воспалительную миопатию и аутоиммунную миопатию. Миопатии включают, без ограничения, дерматомиозит, миозит с включенными тельцами и полимиозит.
В другом варианте осуществления, хроническое воспаление включает васкулит. Васкулиты представляют собой разнородную группу расстройств, характеризующихся воспалением стенки сосуда, в том числе лимфатических сосудов и кровеносных сосудов, таких как вены (флебит), артерии (артериит) и капилляры, вследствие миграции лейкоцитов и возникшего повреждения. Воспаление может поражать кровеносный сосуд любого размера в любом месте организма. Оно может поражать и артерии и/или вены. Воспаление может быть очаговым, когда оно поражает только одно место внутри сосуда; или оно может быть обширным, когда области воспаления рассеяны по всему конкретному органу или ткани или даже поражая более чем одну систему органа в организме. Васкулиты включают, без ограничения, болезнь Бюргера (облитерирующий тромбангиит), церебральный васкулит (васкулит центральной нервной системы), артериит Черга-Страуса, криоглобулинемию, эссенциальный криоглобулинемический васкулит, гигантоклеточный (темпоральный) артериит, васкулит игрока в гольф, пурпуру Геноха-Шенлейна, аллергический васкулит, болезнь Кавасаки, микроскопический полиартериит/полиангиит, нодозный полиартериит, ревматическую полимиалгию (PMR), ревматоидный васкулит, артериит Такаясу, гранулематоз Вегенера и васкулит, вторичный по отношению к заболеванию соединительных тканей, такой как системная красная волчанка (SLE), ревматоидный артрит (RA), рецидивирующий полихондрит, болезнь Бехчета или другие поражения соединительной ткани, васкулит, вторичный по отношению к вирусной инфекции.
В другом варианте осуществления, хроническое воспаление включает заболевание кожи. Заболевания кожи включают, без ограничения, угри, в том числе угри обыкновенные, буллезный пемфигоид, дерматит, в том числе атопический дерматит и хронический фотохимически активный дерматит, экзему, такую как атопическая экзема, контактная экзема, ксеротическая экзема, себорейный дерматит, дисгидроз, монетовидная экзема, венозная экзема, герпетиформный дерматит, нейродерматит и аутоэкзематизация и статис дерматит, гнойный гидраденит, красный плоский лишай, псориаз, в том числе бляшечный псориаз, псориаз ногтей, каплевидный псориаз, псориаз волосистой части головы, обратный псориаз, пустулезный псориаз, псориатическая эритродермия и псориатический артрит, розовые угри и склеродермию, в том числе очаговую склеродермию.
В другом варианте осуществления, хроническое воспаление включает желудочно-кишечное заболевание. Желудочно-кишечное заболевание включает, без ограничения, синдром раздраженного кишечника, воспалительное заболевание кишечника, в том числе болезнь Крона, и язвенный колит, такой как язвенный проктит, левосторонний колит, панколит и скоротечный колит.
В другом варианте осуществления, хроническое воспаление включает сердечнососудистое заболевание. При отложении холестерина LDL на стенках артерий, он может вызывать ответную иммунную реакцию. Хроническое воспаление с течением времени может повредить артерии, что может привести к их перфорации. Сердечнососудистое заболевание представляет собой любое из ряда специфических заболеваний, которые поражают само сердце и/или систему кровеносных сосудов, в частности, вены и артерии, ведущие к сердцу или выходящие из сердца. Известно более 60 типов сердечнососудистых заболеваний, включающих, без ограничения, гипертензию, эндокардит, миокардит, дисфункцию сердечного клапана, хроническую сердечную недостаточность, инфаркт миокарда, диабетические состояния сердца, воспаление кровеносных сосудов, такое как артериит, флебит, васкулит; окклюзионное поражение артерии, такое как артериосклероз и стеноз, воспалительную кардиомегалию, заболевание периферических артерий, аневризму, эмболию, расслоение, псевдоаневризму, сосудистую патологию, каверному, тромбоз, тромбофлебит, варикоз вен, инсульт. Симптомы сердечнососудистого заболевания, поражающего сердце, включают, без ограничения, боль в груди или ощущение дискомфорта в груди (стенокардию), боль в одной или обеих руках, в левом плече, в шее, в челюсти или спине, одышку, головокружение, учащенное сердцебиение, тошноту, аномальные сердцебиения, ощущение усталости. Симптомы сердечнососудистого заболевания, поражающего мозг, включают, без ограничения, внезапное онемение или ослабленность лица, руки или ноги, особенно, на одной стороне тела, внезапную спутанность или затрудненность речи или проблемы с пониманием речи, внезапное нарушение зрения в одном или обоих глазах, внезапное головокружение, затрудненность походки или потерю равновесия или координации, внезапную сильную головную боль по неизвестной причине. Симптомы сердечнососудистого заболевания, поражающего ногу, таз и/или руку включают, без ограничения, хромоту, которая представляет собой боль, болезненность или спазм в мышцах и ощущение холода или онемения в ступнях или пальцах ног, особенно, ночью.
В другом варианте осуществления, хроническое воспаление включает рак. Воспаление образует вокруг опухолей микросреду, которая способствует пролиферации, выживанию и миграции раковых клеток. Например, фибринозное воспаление возникает в результате значительного увеличения проницаемости сосудов, что дает возможность фибрину проходить через кровеносные сосуды. При наличии соответствующего прокоагуляционного раздражителя, такого как раковые клетки, происходит отложение фибринозного экссудата. Он обычно обнаруживается в серозных полостях, где может происходить превращение фибринозного экссудата в рубец между серозными оболочками, что ограничивает их функции. В другом примере, рак представляет собой воспалительный рак, такой как NF-κB-опосредуемый воспалительный рак.
В другом варианте осуществления, хроническое воспаление включает лекарственно-индуцированное воспаление. Известно, что некоторые лекарственные средства или экзогенные химические соединения вызывают воспаление. Например, недостаток витамин A вызывает усиление ответной воспалительной реакции. Некоторые запрещенные лекарственные средства, такие как кокаин и экстези, могут проявлять некоторые из своих отрицательных воздействий в результате активации факторов транскрипции, непосредственно принимающих участие в воспалении (например, NF-κB).
В другом варианте осуществления, хроническое воспаление включает инфекцию. Возбудитель инфекции может выйти за границы, непосредственно зараженной им ткани через кровеносную систему или лимфатическую систему, в результате чего он может распространиться в другие части организма. Если микроорганизм не локализуется под воздействием острого воспаления, он может попасть в лимфатическую систему через расположенные рядом лимфатические сосуды. Инфицирование лимфатических сосудов называют лимфангитом, а инфицирование лимфатического узла называют лимфаденитом. Патоген может попасть в кровоток в результате лимфодренажа внутри кровеносной системы. Инфекции включают, без ограничения, бактериальный цистит, бактериальный энцефалит, пандемический грипп, вирусный энцефалит и вирусный гепатит (A, B и C).
В другом варианте осуществления, хроническое воспаление включает повреждение ткани или органа. Повреждения ткани или органа включают, без ограничения, ожог, разрыв, рану, прокол или травму.
В другом варианте осуществления, хроническое воспаление включает отторжение трансплантата. Отторжение трансплантата возникает, когда трансплантированный орган или ткань не воспринимаются организмом реципиента трансплантата в силу того, что иммунная система реципиента атакует трансплантированный орган или ткань. Адаптивная ответная иммунная реакция отторжения трансплантата опосредуется как через механизмы, опосредованные T-клетками, так и через гуморальные иммунные механизмы (через антитела). Отторжение трансплантата может быть классифицировано как сверхострое отторжение, острое отторжение или хроническое отторжение. Хроническое отторжение трансплантированного органа или ткани происходит тогда, когда отторжение является следствием недостаточно изученного хронического воспаления и ответной иммунной реакции против трансплантированной ткани. Термин "отторжение трансплантата" также включает в себя реакцию "трансплантат против хозяина" (GVHD). GVHD представляет собой часто встречающееся осложнение при трансплантации генетически отличающегося костного мозга, при которой отвечающие за иммунный ответ клетки в трансплантированном костном мозге распознают реципиента в качестве "инородного" и предпринимают иммунологическую атаку. Это может также происходить в случае переливания крови при определенных обстоятельствах. GVHD подразделяют на острую и хроническую формы. По-видимому, острая и хроническая GVHD включают в себя разные субпопуляции иммунных клеток, различные цитокиновые профили, до некоторой степени отличающиеся мишени хозяина и разную ответную реакцию на лечение.
В другом варианте осуществления, хроническое воспаление включает Th1-опосредованное воспалительное заболевание. В нормально функционирующей иммунной системе, ответная иммунная реакция должна вызывать хорошо сбалансированные провоспалительную Th1 реакцию и противовоспалительную Th2 реакцию, которые предназначаются для решения проблемы иммунного выбора. В общем случае, сразу после того как инициируется провоспалительная Th1 реакция, организм полагается на вызываемую Th2 противовоспалительную реакцию, которая противодействует этой Th1 реакции. Эта противодействующая реакция включает высвобождение цитокинов типа Th2, таких как, например, IL-4, IL-5 и IL-13, которые связаны с промотированием IgE и эозинофильными реакциями при атопическом заболевание, а также IL-10, который обладает противовоспалительной реакцией. Th1-опосредованное воспалительное заболевание включает в себя избыточную провоспалительную реакцию, продуцируемую клетками которая приводит к хроническому воспалению. Th1-опосредованное заболевание может быть вызвано вирусами, бактериями или химическими веществами (например, в результате воздействия окружающей среды). Например, вирус, вызывающий Th1-опосредованное заболевание, может вызывать хроническую или острую инфекцию, которая может вызывать расстройство деятельности органов дыхания или грипп.
В другом варианте осуществления, хроническое воспаление включает хроническое нейрогенное воспаление. Хроническое нейрогенное воспаление относится к воспалительной реакции, инициируемой и/или поддерживаемой через высвобождение молекул воспаления, таких как SP или CGRP, которые высвобождаются из периферических чувствительных нервных окончаний (то есть, эфферентное функционирование, в отличие от нормальной афферентной передачи сигналов в спинной мозг в этих нервах). Хроническое нейрогенное воспаление включает как первичное воспаление, так и вторичное нейрогенное воспаление. Используемый в изобретении термин "первичное нейрогенное воспаление" относится к воспалению ткани (симптомам воспаления), которое инициируется путем или в результате высвобождения веществ из первичных чувствительных нервных окончаний (таких как C и A-дельта волокна). Используемый в изобретении термин "вторичное нейрогенное воспаление” относится к воспалению ткани в результате воздействия ненейрональных источников (например, экстравазация из сосудистого русла или ткани, образовавшейся из интерстициальной ткани, например, из тучных клеток или иммунных клеток) медиаторов воспаления, таких как пептиды или цитокины, стимулирующих чувствительные нервные окончания и вызывающие высвобождение медиаторов воспаления из нервов. Совокупный эффект обеих форм (первичной и вторичной) хронического нейрогенного воспаления должен приводить к воспалительному состоянию, которое поддерживается путем сенсибилизации периферических чувствительных нервных волокон. Физиологические последствия возникающего хронического нейрогенного воспаления зависят от соответствующей ткани, вызывающей эти последствия, такие как, например, боль на коже (аллодиния, гипералгезия), боль в суставах и/или артрит, висцеральная боль и дисфункция, легочная дисфункция (астма, COPD) и дисфункция мочевого пузыря (боль, гиперактивность мочевого пузыря).
Композицию или соединение вводят индивидууму. Индивидуум обычно представляет собой человека. Обычно, любой индивидуум, который является потенциальным кандидатом для традиционного лечения хронического воспаления, является потенциальным кандидатом и для раскрытого в изобретении лечения хронического воспаления. Обследование перед проведением лечения обычно включает ознакомление с историей болезни и медицинский осмотр, и, кроме того, получение от пациента полного информированного согласия, в котором раскрыты все релевантные риски и положительные результаты методики лечения.
Раскрытая в изобретении фармацевтическая композиция может включать терапевтическое соединение в терапевтически эффективном количестве. Используемый в изобретении термин "эффективное количество" является синонимом терминов "терапевтически эффективное количество, "эффективная доза" или "терапевтически эффективная доза", и когда он используется в отношении лечения хронического воспаления, этот термин означает минимальную дозу раскрытого в изобретении терапевтического соединения, необходимую для достижения требуемого терапевтического эффекта, и включает дозу, достаточную для ослабления симптома, связанного с хроническим воспалением. Эффективность раскрытого в изобретении терапевтического соединения при лечении хронического воспаления определяется путем обнаружения улучшения у индивидуума, судя по одному или более клиническим симптомам и/или физиологическим показателям, связанным с этим состоянием. На улучшение состояния при хроническом воспалении может также указывать уменьшение необходимости в параллельно проводимой терапии.
Соответствующее эффективное количество раскрытого в изобретении терапевтического соединения, которое вводится индивидууму при конкретном хроническом воспалении, может быть определено любым специалистом в данной области с учетом факторов, включающих, без ограничения, тип хронического воспаления, локализацию хронического воспаления, причину хронического воспаления, тяжесть хронического воспаления, степень требуемого облегчения, продолжительность требуемого облегчения, используемое конкретное терапевтическое соединение, скорость экскреции используемого терапевтического соединения, фармакодинамику используемого терапевтического соединения, свойства других соединений, включенных в композицию, конкретный состав, конкретный способ введения, конкретные характеристики, историю болезни и факторы риска пациента, такие как, например, возраст, масса тела, общее состояние здоровья и другие подобные показатели или их комбинация. Кроме того, когда используют повторное введение терапевтического соединения, эффективное количество терапевтического соединения будет дополнительно зависеть от факторов, включающих, без ограничения, частоту введения, период полувыведения терапевтического соединения или любую их комбинацию. Для обычного специалиста в этой области в этой области является известным, что эффективное количество раскрытого в изобретении терапевтического соединения может быть определено путем экстраполяции данных in vitro исследований и данных изучения in vivo введения на животных моделях перед введением людям.
В аспектах этого варианта осуществления, терапевтически эффективное количество раскрытого в изобретении терапевтического соединения облегчает симптом, связанный с хроническим воспалением, например, по меньшей мере, на 10%, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90%, по меньшей мере, на 95% или, по меньшей мере, на 100%. В другом аспекте этого варианта осуществления, терапевтически эффективное количество раскрытого в изобретении терапевтического соединения облегчает симптом, связанный с хроническим воспалением, например, не более чем на 10%, не более чем на 15%, не более чем на 20%, не более чем на 25%, не более чем на 30%, не более чем на 35%, не более чем на 40%, не более чем на 45%, не более чем на 50%, не более чем на 55%, не более чем на 60%, не более чем на 65%, не более чем на 70%, не более чем на 75%, не более чем на 80%, не более чем на 85%, не более чем на 90%, не более чем на 95% или не более чем 100%. В еще одних аспектах варианта осуществления, терапевтически эффективное количество раскрытого в изобретении терапевтического соединения облегчает симптом, связанный с хроническим воспалением, например, на от приблизительно 10% до приблизительно 100%, на от приблизительно 10% до приблизительно 90%, на от приблизительно 10% до приблизительно 80%, на от приблизительно 10% до приблизительно 70%, на от приблизительно 10% до приблизительно 60%, на от приблизительно 10% до приблизительно 50%, на от приблизительно 10% до приблизительно 40%, на от приблизительно 20% до приблизительно 100%, на от приблизительно 20% до приблизительно 90%, на от приблизительно 20% до приблизительно 80%, на от приблизительно 20% до приблизительно 20%, на от приблизительно 20% до приблизительно 60%, на от приблизительно 20% до приблизительно 50%, на от приблизительно 20% до приблизительно 40%, на от приблизительно 30% до приблизительно 100%, на от приблизительно 30% до приблизительно 90%, на от приблизительно 30% до приблизительно 80%, на от приблизительно 30% до приблизительно 70%, на от приблизительно 30% до приблизительно 60% или на от приблизительно 30% до приблизительно 50%.
В еще одних аспектах варианта осуществления, терапевтически эффективное количество раскрытого в изобретении терапевтического соединения обычно составляет величину в диапазоне от приблизительно 0,001 мг/кг/сутки до приблизительно 100 мг/кг/сутки. В аспектах этого варианта осуществления, эффективное количество раскрытого в изобретении терапевтического соединения может составлять, например, по меньшей мере, 0,001 мг/кг/сутки, по меньшей мере, 0,01 мг/кг/сутки, по меньшей мере, 0,1 мг/кг/сутки, по меньшей мере, 1,0 мг/кг/сутки, по меньшей мере, 5,0 мг/кг/сутки, по меньшей мере, 10 мг/кг/сутки, по меньшей мере, 15 мг/кг/сутки, по меньшей мере, 20 мг/кг/сутки, по меньшей мере, 25 мг/кг/сутки, по меньшей мере, 30 мг/кг/сутки, по меньшей мере, 35 мг/кг/сутки, по меньшей мере, 40 мг/кг/сутки, по меньшей мере, 45 мг/кг/сутки или, по меньшей мере, 50 мг/кг/сутки. В другом аспекте этого варианта осуществления, эффективное количество раскрытого в изобретении терапевтического соединения может составлять величину в диапазоне, например, от приблизительно 0,001 мг/кг/сутки до приблизительно 10 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 15 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 20 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 25 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 30 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 35 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 40 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 45 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 50 мг/кг/сутки, от приблизительно 0,001 мг/кг/сутки до приблизительно 75 мг/кг/сутки или от приблизительно 0,001 мг/кг/сутки до приблизительно 100 мг/кг/сутки. В еще одних аспектах варианта осуществления, эффективное количество раскрытого в изобретении терапевтического соединения может составлять величину в диапазоне, например, от приблизительно 0,01 мг/кг/сутки до приблизительно 10 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 15 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 20 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 25 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 30 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 35 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 40 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 45 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 50 мг/кг/сутки, от приблизительно 0,01 мг/кг/сутки до приблизительно 75 мг/кг/сутки или от приблизительно 0,01 мг/кг/сутки до приблизительно 100 мг/кг/сутки. В еще одних аспектах этого варианта осуществления, эффективное количество раскрытого в изобретении терапевтического соединения может составлять величину в диапазоне, например, от приблизительно 0,1 мг/кг/сутки до приблизительно 10 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 15 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 20 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 25 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 30 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 35 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 40 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 45 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 50 мг/кг/сутки, от приблизительно 0,1 мг/кг/сутки до приблизительно 75 мг/кг/сутки или от приблизительно 0,1 мг/кг/сутки до приблизительно 100 мг/кг/сутки.
В другом аспекте этого варианта осуществления, эффективное количество раскрытого в изобретении терапевтического соединения может составлять величину в диапазоне, например, от приблизительно 1 мг/кг/сутки до приблизительно 10 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 15 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 20 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 25 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 30 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 35 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 40 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 45 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 50 мг/кг/сутки, от приблизительно 1 мг/кг/сутки до приблизительно 75 мг/кг/сутки или от приблизительно 1 мг/кг/сутки до приблизительно 100 мг/кг/сутки. В еще одних аспектах варианта осуществления, эффективное количество раскрытого в изобретении терапевтического соединения может составлять величину в диапазоне, например, от приблизительно 5 мг/кг/сутки до приблизительно 10 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 15 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 20 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 25 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 30 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 35 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 40 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 45 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 50 мг/кг/сутки, от приблизительно 5 мг/кг/сутки до приблизительно 75 мг/кг/сутки или от приблизительно 5 мг/кг/сутки до приблизительно 100 мг/кг/сутки.
Дозирование может осуществляться в виде разовой дозы или в виде кумулятивной дозы (последовательное дозирование), и оно может быть легко определено любым специалистом в этой области. Например, лечение хронического воспаления может включать разовое введение эффективной дозы раскрытой в изобретении фармацевтической композиции. В качестве варианта, лечение хронического воспаления может включать многоразовое введение эффективной дозы фармацевтической композиции, осуществляемое в течение временных интервалов, таких как, например, один раз в день, два раза в день, три раза в день, один раз в несколько дней или один раз в неделю. Временные интервалы введения могут изменяться от индивидуума к индивидууму в зависимости от таких факторов, как тяжесть симптомов у индивидуума. Например, эффективная доза раскрытой в изобретении фармацевтической композиции может быть введена индивидууму один раз в день в течение неопределенного периода времени или до тех пор, когда индивидуум больше не будет нуждаться в лечении. Для обычного специалиста в этой области является очевидным, что состояние индивидуума может постоянно контролироваться на протяжении всего курса лечения и что эффективное количество раскрытой в изобретении фармацевтической композиции, которую вводят индивидууму, может быть соответственно скорректировано.
Различные способы введения могут применяться для введения раскрытого в изобретении терапевтического соединения, в соответствии с раскрытым в изобретении способом лечения хронического воспаления. Фармацевтическая композиция может быть введена индивидууму любым из многочисленных способов, выбор которых зависит от типа хронического воспаления, подвергаемого лечению, локализации хронического воспаления, подвергаемого лечению, используемого конкретного терапевтическое соединение или композиции или другого соединения, вводимого в композицию, и истории болезни, факторов риска и симптомов индивидуума. По этой причине, может применяться местное, энтеральное или парентеральное введение для лечения раскрытого в изобретении хронического воспаления, и такие способы включают как местную, так и системную доставку раскрытого в изобретении терапевтического соединения или композиции. Композиции, включающие либо только одно раскрытое в изобретении терапевтическое соединение, либо два или более раскрытых в изобретении терапевтических соединений, предназначены для ингаляционного, местного, интраназального, сублингвального, инъекционного, инфузионного, инстилляционного, ректального и/или вагинального введения, и они могут быть приготовлены любым известным в фармацевтике методом приготовления фармацевтических композиций.
В одном варианте осуществления, после введения индивидууму, фармацевтическая композиция, включающая раскрытое в изобретении терапевтическое соединение, позволяет в результате достигать биораспределения терапевтического соединения, которое отличается от биораспределения терапевтического соединения, включенного в такую же фармацевтическую композицию, но не содержащую раскрытого в изобретении вспомогательного вещества.
В другом варианте осуществления, после введения индивидууму, терапевтическое соединение раскрытой в изобретении фармацевтической композиции доставляется в макрофаг. Макрофаги являются одними из ключевых типов клеток, которые, как считается, принимают участие в регуляции ответной воспалительной реакции. Достигаемый высокий уровень присутствующего в макрофагах терапевтического соединения, обладающего противовоспалительным действием, обеспечивает в клиническом аспекте лечение хронического воспаления. В аспекте этого варианта осуществления, после введения индивидууму, терапевтически эффективное количество терапевтического соединения раскрытой в изобретении фармацевтической композиции преимущественно доставляется в макрофаг. В другом аспекте этого варианта осуществления, после введения индивидууму, терапевтическое соединение раскрытой в изобретении фармацевтической композиции в основном доставляется в макрофаг. В еще одном аспекте этого варианта осуществления, после введения индивидууму, количество терапевтического соединения раскрытой в изобретении фармацевтической композиции, доставляемой в макрофаг, составляет, например, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 15%, по меньшей мере, 20%, по меньшей мере, 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90%, по меньшей мере, на 95% или, по меньшей мере, 100% от суммарного количества терапевтического соединения, содержащегося в веденной фармацевтической композиции. В еще одних аспектах этого варианта осуществления, после введения индивидууму, количество терапевтического соединения раскрытой в изобретении фармацевтической композиции, доставляемой в макрофаг, составляет величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 15% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 25% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 35% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 45% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 5% до приблизительно 90%, от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 45% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 5% до приблизительно 80%, от приблизительно 10% до приблизительно 80%, от приблизительно 15% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 25% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 35% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 45% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 5% до приблизительно 70%, от приблизительно 10% до приблизительно 70%, от приблизительно 15% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 25% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 35% до приблизительно 70%, от приблизительно 40% до приблизительно 70%, от приблизительно 45% до приблизительно 70%, или от приблизительно 50% до приблизительно 70% от суммарного количества терапевтического соединения, содержащегося в веденной фармацевтической композиции.
В другом варианте осуществления, после введения индивидууму, терапевтическое соединение раскрытой в изобретении фармацевтической композиции доставляется в дендритные клетки. Дендритные клетки представляют собой один из типов ключевых клеток, которые, как считается, координируют взаимодействие между врожденным и приобретенным иммунитетом. Достигаемый высокий уровень присутствующего в дендритных клетках терапевтического соединения, обладающего противовоспалительным действием, обеспечивает в клиническом аспекте лечение хронического воспаления. В аспекте этого варианта осуществления, после введения индивидууму, терапевтически эффективное количество терапевтического соединения раскрытой в изобретении фармацевтической композиции доставляется преимущественно в дендритные клетки. В другом аспекте этого варианта осуществления, после введения индивидууму, терапевтическое соединение раскрытой в изобретении фармацевтической композиции в основном доставляется в дендритные клетки. В еще одном аспекте этого варианта осуществления, после введения индивидууму, количество терапевтического соединения раскрытой в изобретении фармацевтической композиции, доставляемое в дендритные клетки, составляет, например, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 15%, по меньшей мере, 20%, по меньшей мере, 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90%, по меньшей мере, на 95% или, по меньшей мере, 100% от суммарного количества терапевтического соединения, содержащегося в веденной фармацевтической композиции. В еще одних аспектах этого варианта осуществления, после введения индивидууму, количество терапевтического соединения раскрытой в изобретении фармацевтической композиции, доставленное в дендритные клетки, составляет величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 15% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 25% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 35% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 45% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 5% до приблизительно 90%, от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 45% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 5% до приблизительно 80%, от приблизительно 10% до приблизительно 80%, от приблизительно 15% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 25% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 35% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 45% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 5% до приблизительно 70%, от приблизительно 10% до приблизительно 70%, от приблизительно 15% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 25% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 35% до приблизительно 70%, от приблизительно 40% до приблизительно 70%, от приблизительно 45% до приблизительно 70%, или от приблизительно 50% до приблизительно 70% от суммарного количества терапевтического соединения, содержащегося в веденной фармацевтической композиции.
В другом варианте осуществления, после введения индивидууму, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение желудка. В аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция существенно уменьшает раздражение желудка. В еще одном варианте осуществления, после введения индивидууму, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение желудка в отличии от такой же раскрытой в изобретении фармацевтической композиции, но не содержащей фармацевтически приемлемого вспомогательного вещества. В аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция существенно уменьшает раздражение желудка в отличии от такой же раскрытой в изобретении фармацевтической композиции, но не содержащей фармацевтически приемлемого вспомогательного вещества. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение желудка, например, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 15%, по меньшей мере, 20%, по меньшей мере, 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, по меньшей мере, на 40%, по меньшей мере, на 45%, по меньшей мере, на 50%, по меньшей мере, на 55%, по меньшей мере, на 60%, по меньшей мере, на 65%, по меньшей мере, на 70%, по меньшей мере, на 75%, по меньшей мере, на 80%, по меньшей мере, на 85%, по меньшей мере, на 90%, по меньшей мере, на 95% или, по меньшей мере, 100%. В еще одних аспектах варианта осуществления, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение желудка на величину в диапазоне, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 15% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 25% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 35% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 45% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 5% до приблизительно 90%, от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 45% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 5% до приблизительно 80%, от приблизительно 10% до приблизительно 80%, от приблизительно 15% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 25% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 35% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 45% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 5% до приблизительно 70%, от приблизительно 10% до приблизительно 70%, от приблизительно 15% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 25% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 35% до приблизительно 70%, от приблизительно 40% до приблизительно 70%, от приблизительно 45% до приблизительно 70%, или от приблизительно 50% до приблизительно 70%.
В другом варианте осуществления, после введения индивидууму, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение в кишечнике. В аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция существенно уменьшает раздражение в кишечнике. В еще одном варианте осуществления, после введения индивидууму, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение в кишечнике в отличии от такой же раскрытой в изобретении фармацевтической композиции, но не содержащей фармацевтически приемлемого вспомогательного вещества. В аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция существенно уменьшает раздражение в кишечнике в отличии от такой же раскрытой в изобретении фармацевтической композиции, но не содержащей фармацевтически приемлемого вспомогательного вещества. В другом аспекте этого варианта осуществления, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение в кишечнике, например, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 20%, по меньшей мере, 30%, по меньшей мере, 40%, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70%, по меньшей мере, 80%, по меньшей мере, 90% или, по меньшей мере, 100% в отличии от такой же раскрытой в изобретении фармацевтической композиции, но не содержащей фармацевтически приемлемого вспомогательного вещества. В еще одних аспектах варианта осуществления, раскрытая в изобретении фармацевтическая композиция уменьшает раздражение в кишечнике, например, от приблизительно 5% до приблизительно 100%, от приблизительно 10% до приблизительно 100%, от приблизительно 15% до приблизительно 100%, от приблизительно 20% до приблизительно 100%, от приблизительно 25% до приблизительно 100%, от приблизительно 30% до приблизительно 100%, от приблизительно 35% до приблизительно 100%, от приблизительно 40% до приблизительно 100%, от приблизительно 45% до приблизительно 100%, от приблизительно 50% до приблизительно 100%, от приблизительно 5% до приблизительно 90%, от приблизительно 10% до приблизительно 90%, от приблизительно 15% до приблизительно 90%, от приблизительно 20% до приблизительно 90%, от приблизительно 25% до приблизительно 90%, от приблизительно 30% до приблизительно 90%, от приблизительно 35% до приблизительно 90%, от приблизительно 40% до приблизительно 90%, от приблизительно 45% до приблизительно 90%, от приблизительно 50% до приблизительно 90%, от приблизительно 5% до приблизительно 80%, от приблизительно 10% до приблизительно 80%, от приблизительно 15% до приблизительно 80%, от приблизительно 20% до приблизительно 80%, от приблизительно 25% до приблизительно 80%, от приблизительно 30% до приблизительно 80%, от приблизительно 35% до приблизительно 80%, от приблизительно 40% до приблизительно 80%, от приблизительно 45% до приблизительно 80%, от приблизительно 50% до приблизительно 80%, от приблизительно 5% до приблизительно 70%, от приблизительно 10% до приблизительно 70%, от приблизительно 15% до приблизительно 70%, от приблизительно 20% до приблизительно 70%, от приблизительно 25% до приблизительно 70%, от приблизительно 30% до приблизительно 70%, от приблизительно 35% до приблизительно 70%, от приблизительно 40% до приблизительно 70%, от приблизительно 45% до приблизительно 70%, или от приблизительно 50% до приблизительно 70% в отличии от такой же раскрытой в изобретении фармацевтической композиции, но не содержащей фармацевтически приемлемого вспомогательного вещества.
Раскрытая в изобретении фармацевтическая композиция может быть также введена индивидууму в комбинации с другими терапевтическими соединениями с целью повышения суммарного терапевтического эффекта лечения. Использование нескольких соединений для лечения по показанию врача может увеличить положительное воздействие, уменьшая при этом побочные эффекты.
Аспекты настоящего изобретения могут быть также описаны, как изложено ниже:
1. Фармацевтическая композиция твердого раствора, включающая: a) терапевтическое соединение, которое обладает противовоспалительным действием; b) твердый при комнатной температуре липид; и c) жидкий при комнатной температуре липид.
2. Фармацевтическая композиция твердого раствора по варианту осуществления 1, где композиция дополнительно включает стабилизатор.
3. Фармацевтическая композиция твердого раствора по варианту осуществления 1 или 2, где композиция дополнительно включает нейтрализатор.
4. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-3, где фармацевтическая композиция твердого раствора не включает поверхностно-активного вещества.
5. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-4, где фармацевтическая композиция твердого раствора не включает гидрофильного растворителя.
6. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-5, где противовоспалительное действие снижает уровень молекул, индуцирующих воспаление.
7. Фармацевтическая композиция твердого раствора по варианту осуществления 6, где молекула, индуцирующая воспаление, включает вещество P (SP), кальцитонин-ген-связанный пептид (CGRP), глутамат или их комбинацию.
8. Фармацевтическая композиция твердого раствора по варианту осуществления 7, где противовоспалительное действие снижает уровень SP, CGRP, глутамата или их комбинации, по меньшей мере, на 10%.
9. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-8, где противовоспалительное действие снижает уровень простагландинов, индуцирующих воспаление.
10. Фармацевтическая композиция твердого раствора по варианту осуществления 9, где уровень воспаления, индуцированного простагландином, уменьшается, по меньшей мере, на 10%.
11. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-10, где противовоспалительное действие стимулирует сигнальный путь PPAR.
12. Фармацевтическая композиция твердого раствора по варианту осуществления 11, где сигнальный путь PPAR стимулируется, по меньшей мере, на 10%.
13. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-12, где противовоспалительное действие индуцирует апоптоз макрофагов M1, промотирует дифференцировку макрофагов M2 или осуществляет и то и другое.
14. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-13, где противовоспалительное действие снижает уровни интерферона-гамма (IFNγ), фактора некроза опухолей альфа (TNF-α), интерлейкина-12 (IL-12) или их комбинации, высвобождаемых из клеток Th1, повышает уровни IL-10, высвобождаемого из клеток Th2, или осуществляет и то и другое.
15. Фармацевтическая композиция твердого раствора по варианту осуществления 14, где уровни IFNγ, TNF-α, IL-12 или их комбинации, высвобождаемых из клеток Th1, снижаются, по меньшей мере, на 10%.
16. Фармацевтическая композиция твердого раствора по варианту осуществления 14, где уровни IL-10, высвобождаемого из клеток Th2, повышаются, по меньшей мере, на 10%.
17. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-16, где терапевтическое соединение имеет значение logP 3,0 или более.
18. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-16, где терапевтическое соединение имеет значение logP от приблизительно 2,2 до приблизительно 3,0.
19. Фармацевтическая композиция твердого раствора по вариантам осуществления 1-16, где терапевтическое соединение имеет значение logP приблизительно 2,0 или менее.
20. Фармацевтическая композиция по вариантам осуществления 1-19, где терапевтическое соединение имеет участок полярной поверхности, который является гидрофобным.
21. Фармацевтическая композиция по вариантам осуществления 1-20, где терапевтическое соединение имеет участок полярной поверхности, который составляет менее чем 8,0 нм2.
22. Фармацевтическая композиция по вариантам осуществления 1-20, где терапевтическое соединение имеет участок полярной поверхности, который составляет менее чем 6,0 нм2.
23. Фармацевтическая композиция по вариантам осуществления 1-22, где терапевтическое соединение включает нестероидное противовоспалительно лекарственное средство (NSAID).
24. Фармацевтическая композиция по варианту осуществления 23, где NSAID включает салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или их комбинацию.
25. Фармацевтическая композиция по вариантам осуществления 1-24, где терапевтическое соединение включает агонист PPARα, агонист PPARβ/δ, агонист PPARγ или глитазар.
26. Фармацевтическая композиция по вариантам осуществления 1-25, где терапевтическое соединение включает иммуносупрессивное лекарственное средство, средство, способствующее выведению мочевой кислоты, агликон или каннабидиол.
27. Фармацевтическая композиция по вариантам осуществления 1-26, где терапевтическое соединение включает антагонист рецептора рианодина.
28. Фармацевтическая композиция по варианту осуществления 27, где антагонист рецептора рианодина представляет собой азумолен или дантролен.
29. Фармацевтическая композиция по вариантам осуществления 1-28, где терапевтическое соединение включает средство, связывающее ядерный рецептор.
30. Фармацевтическая композиция по варианту осуществления 29, где средство, связывающее ядерный рецептор, включает средство, связывающее рецептор ретиноевой кислоты (RAR), средство, связывающее ретиноидный X рецептор (RXR), средство, связывающее печеночный рецептор Х (LXR), средство, связывающее витамин D или их комбинацию.
31. Фармацевтическая композиция по вариантам осуществления 1-30, где терапевтическое соединение включает антигиперлипидемическое средство.
32. Фармацевтическая композиция по варианту осуществления 31, где антигиперлипидемическое средство включает антагонист рецептора ангиотензина II, ингибитор ACE, ингибитор фосфодиэстеразы, фибрат, статин, токотриенол, ниацин, секвестранты желчных кислот (смолу), ингибитор абсорбции холестерина, ингибитор липазы поджелудочной железы, симпатомиметический амин или их комбинацию.
33. Фармацевтическая композиция по варианту осуществления 32, где антагонист рецептора ангиотензина II включает азилсартан, кандесартан, эпросартан, ирбесартан, лосартан, олмесартан, телмисартан и валсартан.
34. Фармацевтическая композиция по варианту осуществления 32, где ингибитор ACE включает сульфгидрилсодержащее средство, дикарбоксилатсодержащее средство, фосфонатсодержащее средство, касокинин и лактокинин.
35. Фармацевтическая композиция по варианту осуществления 32, где ингибитор фосфодиэстеразы включает селективный ингибитор PDE 1, селективный ингибитор PDE 2, селективный ингибитор PDE 3, селективный ингибитор PDE 4, селективный ингибитор PDE 5 или селективный ингибитор PDE 10.
36. Фармацевтическая композиция по варианту осуществления 32, где фибрат включает безафибрат, ципрофибрат, клофибрат, гемфиброзил, фенофибрат или их комбинацию.
37. Фармацевтическая композиция по варианту осуществления 32, где статин включает аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин, симвастатин или их комбинацию.
38. Фармацевтическая композиция по варианту осуществления 32, где ниацин включает аципимокс, ниацин, никотинамид, витамин B3 или их комбинацию.
39. Фармацевтическая композиция по варианту осуществления 32, где секвестрант желчных кислот включает холестирамин, колесевелам, колестирол или их комбинацию.
40. Фармацевтическая композиция по варианту осуществления 32, где ингибитор абсорбции холестерина включает эзетиниб, фитостерол, стерол, станол или их комбинацию.
41. Фармацевтическая композиция по варианту осуществления 32, где ингибитор абсорбции жира включает орлистат.
42. Фармацевтическая композиция по варианту осуществления 32, где симпатомиметический амин включает кленбутерол, сальбутамол, эфедрин, псевдоэфедрин, метамфетамин, амфетамин, фенилэфрин, изопротеренол, добутамин, метилфенидат, лисдексамфетамин, катин, катинон, меткатинон, кокаин, бензилпиперазин (BZP), метилендиоксипировалерон (MDPV), 4-метиламинорекс, пемолин, фенметразин, пропилгекседрин или их комбинацию.
43. Фармацевтическая композиция по вариантам осуществления 1-42, где терапевтическое соединение включает противоопухолевое лекарственное средство.
44. Фармацевтическая композиция по варианту осуществления 43, где противоопухолевое лекарственное средство включает алкилирующее средство, антиметаболит, растительный алкалоид и терпеноид, ингибитор топоизомеразы или цитотоксический антибиотик.
45. Фармацевтическая композиция по вариантам осуществления 1-44, где терапевтическое соединение включает метформин, куркумин, глицирретиновую кислоту или 6-шогаол.
46. Фармацевтическая композиция по вариантам осуществления 1-44, где терапевтическое соединение включает антибиотик.
47. Фармацевтическая композиция по варианту осуществления 46, где антибиотик включает изониазид, рифампицин, пиразинамид или этамбутол.
48. Фармацевтическая композиция по вариантам осуществления 1-47, где терапевтическое соединение включает противоглистное лекарственное средство.
49. Фармацевтическая композиция по варианту осуществления 48, где противоглистное лекарственное средство включает абамектин, аминоацетонитрил, такой как монепантел, бензимидазол, диэтилкарбамазин, ивермектин, левамизол, никлозамид, октадепсипептид, такой как эмодепсид, фосфоновую кислоту (метрифонат), празиквантел, спироиндол, такой как дерквантел, или сурамин, пирантела памоат.
50. Фармацевтическая композиция по вариантам осуществления 1-49, где терапевтическое соединение включает противомалярийное лекарственное средство.
51. Фармацевтическая композиция по варианту осуществления 50, где противомалярийное лекарственное средство включает амодиахин, артемизинин, атоваквон, хлорохин, клиндамицин, доксициклин, галофантрин, мефлохин, примахин, прогуанил, пириметамин, хинин и относящееся к нему лекарственное средство, такое как хинимакс и хинидин, руфигаллол и сульфонамид, такой как сульфадоксин или сульфаметоксипиридазин.
52. Фармацевтическая композиция по варианту осуществления 51, где артемизинин включает артеэфир, артеметер, артемизинин, артесунат или дигидроартемизинин.
53. Фармацевтическая композиция по вариантам осуществления 1-52, где терапевтическое соединение включает эфир терапевтического соединения.
54. Фармацевтическая композиция по вариантам осуществления 1-53, где терапевтическое соединение включает эфир терапевтического соединения по вариантам осуществления 23-53.
55. Фармацевтическая композиция по вариантам осуществления 1-54, где терапевтическое соединение присутствует в количестве менее чем приблизительно 90% по массе, менее чем приблизительно 80% по массе, менее чем приблизительно 70% по массе, менее чем приблизительно 65% по массе, менее чем приблизительно 60% по массе, менее чем приблизительно 55% по массе, менее чем приблизительно 50% по массе, менее чем приблизительно 45% по массе, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 5% по массе или менее чем приблизительно 1% по массе или приблизительно от 1% до 90% по массе, приблизительно от 1% до 80% по массе, приблизительно от 1% до 75% по массе, приблизительно от 1% до 70% по массе, приблизительно от 1% до 65% по массе, приблизительно от 1% до 60% по массе, приблизительно от 1% до 55% по массе, приблизительно от 1% до 50% по массе, приблизительно от 1% до 45% по массе, приблизительно от 1% до 40% по массе, приблизительно от 1% до 35% по массе, приблизительно от 1% до 30% по массе, приблизительно от 1% до 25% по массе, приблизительно от 1% до 20% по массе, приблизительно от 1% до 15% по массе, приблизительно от 1% до 10% по массе, приблизительно от 1% до 5% по массе, приблизительно от 2% до 50% по массе, приблизительно от 2% до 40% по массе, приблизительно от 2% до 30% по массе, приблизительно от 2% до 20% по массе, приблизительно от 2% до 10% по массе, приблизительно от 4% до 50% по массе, приблизительно от 4% до 40% по массе, приблизительно от 4% до 30% по массе, приблизительно от 4% до 20% по массе, приблизительно от 4% до 10% по массе, приблизительно от 6% до 50% по массе, приблизительно от 6% до 40% по массе, приблизительно от 6% до 30% по массе, приблизительно от 6% до 20% по массе, приблизительно от 6% до 10% по массе, приблизительно от 8% до 50% по массе, приблизительно от 8% до 40% по массе, приблизительно от 8% до 30% по массе, приблизительно от 8% до 20% по массе, приблизительно от 8% до 15% по массе или приблизительно от 8% до 12% по массе.
56. Фармацевтическая композиция по вариантам осуществления 1-55, где терапевтическое соединение присутствует в количестве от приблизительно 0,1% до приблизительно 45% по массе, от приблизительно 0,1% до приблизительно 40% по массе, от приблизительно 0,1% до приблизительно 35% по массе, от приблизительно 0,1% до приблизительно 30% по массе, от приблизительно 0,1% до приблизительно 25% по массе, от приблизительно 0,1% до приблизительно 20% по массе, от приблизительно 0,1% до приблизительно 15% по массе, от приблизительно 0,1% до приблизительно 10% по массе, от приблизительно 0,1% до приблизительно 5% по массе, от приблизительно 1% до приблизительно 45% по массе, от приблизительно 1% до приблизительно 40% по массе, от приблизительно 1% до приблизительно 35% по массе, от приблизительно 1% до приблизительно 30% по массе, от приблизительно 1% до приблизительно 25% по массе, от приблизительно 1% до приблизительно 20% по массе, от приблизительно 1% до приблизительно 15% по массе, от приблизительно 1% до приблизительно 10% по массе, от приблизительно 1% до приблизительно 5% по массе, от приблизительно 5% до приблизительно 45% по массе, от приблизительно 5% до приблизительно 40% по массе, от приблизительно 5% до приблизительно 35% по массе, от приблизительно 5% до приблизительно 30% по массе, от приблизительно 5% до приблизительно 25% по массе, от приблизительно 5% до приблизительно 20% по массе, от приблизительно 5% до приблизительно 15% по массе, от приблизительно 5% до приблизительно 10% по массе, от приблизительно 10% до приблизительно 45% по массе, от приблизительно 10% до приблизительно 40% по массе, от приблизительно 10% до приблизительно 35% по массе, от приблизительно 10% до приблизительно 30% по массе, от приблизительно 10% до приблизительно 25% по массе, от приблизительно 10% до приблизительно 20% по массе, от приблизительно 10% до приблизительно 15% по массе, от приблизительно 15% до приблизительно 45% по массе, от приблизительно 15% до приблизительно 40% по массе, от приблизительно 15% до приблизительно 35% по массе, от приблизительно 15% до приблизительно 30% по массе, от приблизительно 15% до приблизительно 25% по массе, от приблизительно 15% до приблизительно 20% по массе, от приблизительно 20% до приблизительно 45% по массе, от приблизительно 20% до приблизительно 40% по массе, от приблизительно 20% до приблизительно 35% по массе, от приблизительно 20% до приблизительно 30% по массе, от приблизительно 20% до приблизительно 25% по массе, от приблизительно 25% до приблизительно 45% по массе, от приблизительно 25% до приблизительно 40% по массе, от приблизительно 25% до приблизительно 35% по массе или от приблизительно 25% до приблизительно 30% по массе.
57. Фармацевтическая композиция по вариантам осуществления 1-56, где фармацевтически приемлемый твердый при комнатной температуре липид представляет собой фармацевтически приемлемый глицеролипид, фармацевтически приемлемый гликолевый эфир жирной кислоты, фармацевтически приемлемый простой полиэфир сложного эфира жирной кислоты, смесь фармацевтически приемлемых липидов или любую их комбинацию.
58. Фармацевтическая композиция по вариантам осуществления 1-57, где фармацевтически приемлемые глицеролипиды представляют собой масло какао, смеси стеарата ПЭГ-6 и пальмитостеарата этиленгликоля и стеарата ПЭГ-32 (TEFOSE® 1500; TEFOSE® 63), смеси трицетеарет-4 фосфата и пальмитостеарата этиленгликоля и пальмитостеарата диэтиленгликоля (SEDEFOS® 75), смеси глицеролмоностеарата и стеарата ПЭГ-75 (GELOT®), смеси цетилового спирта и этоксилированных жирных спиртов (seteth-2-, steareth-20) (EMULCIRE®), смеси насыщенных C10-C18 триглицеридов, имеющих температуру плавления приблизительно 33°C (GELUCIRE® 33/01), смеси насыщенных C10-C18 триглицеридов, имеющих температуру плавления приблизительно 39°C (GELUCIRE® 39/01), смеси насыщенных C10-C18 триглицеридов, имеющих температуру плавления приблизительно 43°C (GELUCIRE® 43/01), смеси глицеролмоностеарата 40-55 (тип I) и диглицеридов (GELEOL® моно и диглицериды) и смеси триглицеридов со средней длиной цепи (LABRAFAC® Lipophile WL 1349).
59. Фармацевтическая композиция по вариантам осуществления 1-58, где фармацевтически приемлемый гликолевый эфир жирной кислоты представляют собой этиленгликолевый эфир жирной кислоты, диэтиленгликолевый эфир жирной кислоты, пропиленгликолевый эфир жирной кислоты и дипропиленгликолевый эфир жирной кислоты, каприлат этиленгликоля, пеларгонат этиленгликоля, капрат этиленгликоля, ундецелат этиленгликоля, лаурат этиленгликоля, тридецилат этиленгликоля, миристат этиленгликоля, миристолат этиленгликоля, пентадецилат этиленгликоля, пальмитат этиленгликоля, пальмитолеат этиленгликоля, сапиенат этиленгликоля, маргарат этиленгликоля, стеарат этиленгликоля, пальмитостеарат этиленгликоля, олеат этиленгликоля, элаидат этиленгликоля, вакцинат этиленгликоля, линолеат этиленгликоля, линоэлаидат этиленгликоля, α-линоленат этиленгликоля, γ-линоленат этиленгликоля, стеаридонат этиленгликоля, каприлокапрат этиленгликоля, дикаприлокапрат этиленгликоля, дикаприлат этиленгликоля, дипеларгонат этиленгликоля, дикапрат этиленгликоля, диундецелат этиленгликоля, дилаурат этиленгликоля, дитридецилат этиленгликоля, димиристат этиленгликоля, димиристолат этиленгликоля, дипентадецилат этиленгликоля, дипальмитат этиленгликоля, дипальмитолеат этиленгликоля, дисапиенат этиленгликоля, димаргарат этиленгликоля, дистеарат этиленгликоля, дипальмитостеарат этиленгликоля, диолеат этиленгликоля, диэлаидат этиленгликоля, дивакцинат этиленгликоля, дилинолеат этиленгликоля, дилиноэлаидат этиленгликоля, ди-α-линоленат этиленгликоля, ди-γ-линоленат этиленгликоля, дистеаридонат этиленгликоля, дикаприлокапрат этиленгликоля, дидикаприлокапрат этиленгликоля, каприлат пропиленгликоля, пеларгонат пропиленгликоля, капрат пропиленгликоля, ундецилат пропиленгликоля, лаурат пропиленгликоля, тридецилат пропиленгликоля, миристат пропиленгликоля, миристолат пропиленгликоля, пентадецилат пропиленгликоля, пальмитат пропиленгликоля, пальмитолеат пропиленгликоля, сапиенат пропиленгликоля, маргарат пропиленгликоля, стеарат пропиленгликоля, пальмитостеарат пропиленгликоля, олеат пропиленгликоля, элаидат пропиленгликоля, вакцинат пропиленгликоля, линолеат пропиленгликоля, линоэлаидат пропиленгликоля, α-линоленат пропиленгликоля, γ-линоленат пропиленгликоля, стеаридонат пропиленгликоля, каприлокапрат пропиленгликоля, дикаприлокапрат пропиленгликоля, дикаприлат пропиленгликоля, дипеларгонат пропиленгликоля, дикапрат пропиленгликоля, диундецилат пропиленгликоля, дилаурат пропиленгликоля, дитридецилат пропиленгликоля, димиристат пропиленгликоля, димиристолат пропиленгликоля, дипентадецилат пропиленгликоля, дипальмитат пропиленгликоля, дипальмитолеат пропиленгликоля, дисапиенат пропиленгликоля, димаргарат пропиленгликоля, дистеарат пропиленгликоля, дипальмитостеарат пропиленгликоля, диолеат пропиленгликоля, диэлаидат пропиленгликоля, дивакцинат пропиленгликоля, дилинолеат пропиленгликоля, дилиноэлаидат пропиленгликоля, ди-α-линоленат пропиленгликоля, ди-γ-линоленат пропиленгликоля, дистеаридонат пропиленгликоля, дикаприлокапрат пропиленгликоля, дидикаприлокапрат пропиленгликоля, монопальмитостеарат пропиленгликоля (MONOSTEOL®), дикаприлокапрат пропиленгликоля (LABRAFAC® PG), монолауран пропиленгликоля (тип I) (LAUROGLYCOL® FCC), монолауран пропиленгликоля (тип II) (LAUROGLYCOL® 90), монокаприлат пропиленгликоля (тип I) (CAPRYOL® PGMC), монокаприлат пропиленгликоля (тип II) (CAPRYOL® 90) или любую их комбинацию.
60. Фармацевтическая композиция по вариантам осуществления 1-59, где фармацевтически приемлемый простой полиэфир сложного эфира жирной кислоты представляет собой ПЭГ сложный эфир жирной кислоты, ПЭГ глицерил жирной кислоты, ПЭГ глицерид сложного эфира жирной кислоты, ППГ сложный эфир жирной кислоты, ППГ глицерил жирной кислоты или ППГ глицерид сложного эфира жирной кислоты.
61. Фармацевтическая композиция по вариантам осуществления 1-60, где фармацевтически приемлемый твердый при комнатной температуре липид присутствует в количестве, достаточном для образования композиции твердого раствора.
62. Фармацевтическая композиция по вариантам осуществления 1-61, где фармацевтически приемлемый твердый при комнатной температуре липид присутствует в количестве, по меньшей мере, 10% по массе, по меньшей мере, 20% по массе, по меньшей мере, 30% по массе, по меньшей мере, 35% по массе, по меньшей мере, 40% по массе, по меньшей мере, 45% по массе, по меньшей мере, 50% по массе, по меньшей мере, 55% по массе, по меньшей мере, 60% по массе, по меньшей мере, 65% по массе, по меньшей мере, 70% по массе, по меньшей мере, 75% по массе, по меньшей мере, 80% по массе, по меньшей мере, 85% по массе, по меньшей мере, 90% по массе, по меньшей мере, 95% по массе или, по меньшей мере, 99% по массе или от приблизительно 30% до приблизительно 99% по массе, от приблизительно 35% до приблизительно 99% по массе, от приблизительно 40% до приблизительно 99% по массе, от приблизительно 45% до приблизительно 99% по массе, от приблизительно 50% до приблизительно 99% по массе, от приблизительно 30% до приблизительно 98% по массе, от приблизительно 35% до приблизительно 98% по массе, от приблизительно 40% до приблизительно 98% по массе, от приблизительно 45% до приблизительно 98% по массе, от приблизительно 50% до приблизительно 98% по массе, от приблизительно 30% до приблизительно 95% по массе, от приблизительно 35% до приблизительно 95% по массе, от приблизительно 40% до приблизительно 95% по массе, от приблизительно 45% до приблизительно 95% по массе или от приблизительно 50% до приблизительно 95% по массе или от приблизительно 70% до приблизительно 97% по массе, от приблизительно 75% до приблизительно 97% по массе, от приблизительно 80% до приблизительно 97% по массе, от приблизительно 85% до приблизительно 97% по массе, от приблизительно 88% до приблизительно 97% по массе, от приблизительно 89% до приблизительно 97% по массе, от приблизительно 90% до приблизительно 97% по массе, от приблизительно 75% до приблизительно 96% по массе, от приблизительно 80% до приблизительно 96% по массе, от приблизительно 85% до приблизительно 96% по массе, от приблизительно 88% до приблизительно 96% по массе, от приблизительно 89% до приблизительно 96% по массе, от приблизительно 90% до приблизительно 96% по массе, от приблизительно 75% до приблизительно 93% по массе, от приблизительно 80% до приблизительно 93% по массе, от приблизительно 85% до приблизительно 93% по массе, от приблизительно 88% до приблизительно 93% по массе, от приблизительно 89% до приблизительно 93% по массе или от приблизительно 90% до приблизительно 93% по массе.
63. Фармацевтическая композиция по вариантам осуществления 1-62, где фармацевтически приемлемый жидкий при комнатной температуре липид представляет собой моноглицерид.
64. Фармацевтическая композиция по варианту осуществления 63, где моноглицерид представляет собой глицерина мономиристолеат, глицерина монопальмитолеат, глицерина моносапиенат, глицерина моноолеат, глицерина моноэлаидат, глицерина моновакценат, глицерина монолинолеат, глицерина монолиноэлаидат, глицерина монолиноленат, глицерина моностеаридонат, глицерина моноэйкозеноат, глицерина мономеадат, глицерина моноарахидонат, глицерина моноэйкозапентаеноат, глицерина моноэрукат, глицерина монодокозагексаеноат, глицерина мононервонат, глицерил дибегенат (COMPRITOL® 888), глицерина дибегенат (COMPRITOL® E ATO), глицерина дипальмитостеарат (Biogapress Vegetal BM297ATO), глицерина дистеарат (тип I) (PRECIROL® ATO 5) и глицерина монолинолеат (MAISINE™ 35-1).
65. Фармацевтическая композиция по вариантам осуществления 1-64, где фармацевтически приемлемый жидкий при комнатной температуре липид присутствует в количестве, достаточном для растворения терапевтического соединения.
66. Фармацевтическая композиция по вариантам осуществления 1-65, где фармацевтически приемлемый жидкий при комнатной температуре липид присутствует в количестве менее чем приблизительно 90% по массе, менее чем приблизительно 80% по массе, менее чем приблизительно 70% по массе, менее чем приблизительно 65% по массе, менее чем приблизительно 60% по массе, менее чем приблизительно 55% по массе, менее чем приблизительно 50% по массе, менее чем приблизительно 45% по массе, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 5% по массе или менее чем приблизительно 1% по массе или приблизительно от 1% до 90% по массе, приблизительно от 1% до 80% по массе, приблизительно от 1% до 70% по массе, приблизительно от 1% до 60% по массе, приблизительно от 1% до 50% по массе, приблизительно от 1% до 40% по массе, приблизительно от 1% до 30% по массе, приблизительно от 1% до 20% по массе, приблизительно от 1% до 10% по массе, приблизительно от 2% до 50% по массе, приблизительно от 2% до 40% по массе, приблизительно от 2% до 30% по массе, приблизительно от 2% до 20% по массе, приблизительно от 2% до 10% по массе, приблизительно от 4% до 50% по массе, приблизительно от 4% до 40% по массе, приблизительно от 4% до 30% по массе, приблизительно от 4% до 20% по массе, приблизительно от 4% до 10% по массе, приблизительно от 6% до 50% по массе, приблизительно от 6% до 40% по массе, приблизительно от 6% до 30% по массе, приблизительно от 6% до 20% по массе, приблизительно от 6% до 10% по массе, приблизительно от 8% до 50% по массе, приблизительно от 8% до 40% по массе, приблизительно от 8% до 30% по массе, приблизительно от 8% до 20% по массе, приблизительно от 8% до 15% по массе или приблизительно от 8% до 12% по массе.
67. Фармацевтическая композиция по вариантам осуществления 1-66, где стабилизатор представляет собой жидкий полимер гликоля, одноатомный спирт, диметиловый эфир изосорбида и моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол) (TRANSCUTOL®).
68. Фармацевтическая композиция по варианту осуществления 67, где жидкий полимер гликоля представляет собой жидкий полимер ПЭГ и/или жидкий полимер ППГ.
69. Фармацевтическая композиция по варианту осуществления 68, где одноатомный спирт представляет собой этанол, пропанол, бутанол, пентанол или 1-гексадеканол.
70. Фармацевтическая композиция по вариантам осуществления 1-69, где фармацевтически приемлемый стабилизатор присутствует в количестве, достаточном для стабилизации свободной кислоты или основания, присутствующих в терапевтическом соединении.
71. Фармацевтическая композиция по вариантам осуществления 1-70, где фармацевтически приемлемый стабилизатор присутствует в количестве менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 19% по массе, менее чем приблизительно 18% по массе, менее чем приблизительно 17% по массе, менее чем приблизительно 16% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 14% по массе, менее чем приблизительно 13% по массе, менее чем приблизительно 12% по массе, менее чем приблизительно 11% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 9% по массе, менее чем приблизительно 8% по массе, менее чем приблизительно 7% по массе, менее чем приблизительно 6% по массе, менее чем приблизительно 5% по массе, менее чем приблизительно 4% по массе, менее чем приблизительно 3% по массе, менее чем приблизительно 2% по массе или менее чем приблизительно 1% или от приблизительно 1% до приблизительно 5% по массе, от приблизительно 1% до приблизительно 7% по массе, от приблизительно 1% до приблизительно 10% по массе, от приблизительно 1% до приблизительно 12% по массе, от приблизительно 1% до приблизительно 15% по массе, от приблизительно 1% до приблизительно 18% по массе, от приблизительно 1% до приблизительно 20% по массе, от приблизительно 2% до приблизительно 5% по массе, от приблизительно 2% до приблизительно 7% по массе, от приблизительно 2% до приблизительно 10% по массе, от приблизительно 2% до приблизительно 12% по массе, от приблизительно 2% до приблизительно 15% по массе, от приблизительно 2% до приблизительно 18% по массе, от приблизительно 2% до приблизительно 20% по массе, от приблизительно 3% до приблизительно 5% по массе, от приблизительно 3% до приблизительно 7% по массе, от приблизительно 3% до приблизительно 10% по массе, от приблизительно 3% до приблизительно 12% по массе, от приблизительно 3% до приблизительно 15% по массе, от приблизительно 3% до приблизительно 18% по массе, от приблизительно 3% до приблизительно 20% по массе, от приблизительно 4% до приблизительно 5% по массе, от приблизительно 4% до приблизительно 7% по массе, от приблизительно 4% до приблизительно 10% по массе, от приблизительно 4% до приблизительно 12% по массе, от приблизительно 4% до приблизительно 15% по массе, от приблизительно 4% до приблизительно 18% по массе, от приблизительно 4% до приблизительно 20% по массе, от приблизительно 5% до приблизительно 7% по массе, от приблизительно 5% до приблизительно 10% по массе, от приблизительно 5% до приблизительно 12% по массе, от приблизительно 5% до приблизительно 15% по массе, от приблизительно 5% до приблизительно 18% по массе, от приблизительно 5% до приблизительно 20% по массе, от приблизительно 6% до приблизительно 7% по массе, от приблизительно 6% до приблизительно 10% по массе, от приблизительно 6% до приблизительно 12% по массе, от приблизительно 6% до приблизительно 15% по массе, от приблизительно 6% до приблизительно 18% по массе, от приблизительно 6% до приблизительно 20% по массе, от приблизительно 7% до приблизительно 10% по массе, от приблизительно 7% до приблизительно 12% по массе, от приблизительно 7% до приблизительно 15% по массе, от приблизительно 7% до приблизительно 18% по массе, от приблизительно 7% до приблизительно 20% по массе, от приблизительно 8% до приблизительно 10% по массе, от приблизительно 8% до приблизительно 12% по массе, от приблизительно 8% до приблизительно 15% по массе, от приблизительно 8% до приблизительно 18% по массе, от приблизительно 8% до приблизительно 20% по массе, от приблизительно 9% до приблизительно 10% по массе, от приблизительно 9% до приблизительно 12% по массе, от приблизительно 9% до приблизительно 15% по массе, от приблизительно 9% до приблизительно 18% по массе, от приблизительно 9% до приблизительно 20% по массе, от приблизительно 10% до приблизительно 12% по массе, от приблизительно 10% до приблизительно 15% по массе, от приблизительно 10% до приблизительно 18% по массе или от приблизительно 10% до приблизительно 20% по массе.
72. Фармацевтическая композиция по вариантам осуществления 1-71, где фармацевтически приемлемый стабилизатор не используется в качестве растворителя.
73. Фармацевтическая композиция по вариантам осуществления 1-72, где фармацевтически приемлемый стабилизатор приводит к не более чем 85%, не более чем 80%, не более чем 75%, не более чем 70%, не более чем 65%, не более чем 60%, не более чем 55%, не более чем 50%, не более чем 45%, не более чем 40%, не более чем 35%, не более чем 30%, не более чем 25%, не более чем 20%, не более чем 15%, не более чем 10% или не более чем 5% растворению терапевтического соединения.
74. Фармацевтическая композиция по вариантам осуществления 1-73, где фармацевтически приемлемый нейтрализатор присутствует в количестве, достаточном для нейтрализации ионных зарядов, образующихся при растворении терапевтического соединения.
75. Фармацевтическая композиция по вариантам осуществления 1-74, где фармацевтически приемлемый нейтрализатор присутствует в количестве одного эквивалента по отношению к терапевтическому соединению.
76. Фармацевтическая композиция по вариантам осуществления 1-75, где фармацевтически приемлемый нейтрализатор присутствует в количестве менее чем один эквивалент по отношению к терапевтическому соединению.
77. Фармацевтическая композиция по вариантам осуществления 1-76, где фармацевтически приемлемый нейтрализатор присутствует в количестве более чем один эквивалент по отношению к терапевтическому соединению.
78. Фармацевтическая композиция по вариантам осуществления 1-77, где нейтрализатор присутствует в количестве менее чем приблизительно 90% по массе, менее чем приблизительно 80% по массе, менее чем приблизительно 70% по массе, менее чем приблизительно 65% по массе, менее чем приблизительно 60% по массе, менее чем приблизительно 55% по массе, менее чем приблизительно 50% по массе, менее чем приблизительно 45% по массе, менее чем приблизительно 40% по массе, менее чем приблизительно 35% по массе, менее чем приблизительно 30% по массе, менее чем приблизительно 25% по массе, менее чем приблизительно 20% по массе, менее чем приблизительно 15% по массе, менее чем приблизительно 10% по массе, менее чем приблизительно 5% по массе или менее чем приблизительно 1% по массе или приблизительно от 1% до 90% по массе, приблизительно от 1% до 80% по массе, приблизительно от 1% до 75% по массе, приблизительно от 1% до 70% по массе, приблизительно от 1% до 65% по массе, приблизительно от 1% до 60% по массе, приблизительно от 1% до 55% по массе, приблизительно от 1% до 50% по массе, приблизительно от 1% до 45% по массе, приблизительно от 1% до 40% по массе, приблизительно от 1% до 35% по массе, приблизительно от 1% до 30% по массе, приблизительно от 1% до 25% по массе, приблизительно от 1% до 20% по массе, приблизительно от 1% до 15% по массе, приблизительно от 1% до 10% по массе, приблизительно от 1% до 5% по массе, приблизительно от 2% до 50% по массе, приблизительно от 2% до 40% по массе, приблизительно от 2% до 30% по массе, приблизительно от 2% до 20% по массе, приблизительно от 2% до 10% по массе, приблизительно от 4% до 50% по массе, приблизительно от 4% до 40% по массе, приблизительно от 4% до 30% по массе, приблизительно от 4% до 20% по массе, приблизительно от 4% до 10% по массе, приблизительно от 6% до 50% по массе, приблизительно от 6% до 40% по массе, приблизительно от 6% до 30% по массе, приблизительно от 6% до 20% по массе, приблизительно от 6% до 10% по массе, приблизительно от 8% до 50% по массе, приблизительно от 8% до 40% по массе, приблизительно от 8% до 30% по массе, приблизительно от 8% до 20% по массе, приблизительно от 8% до 15% по массе или приблизительно от 8% до 12% по массе.
79. Фармацевтическая композиция по вариантам осуществления 1-78, где нейтрализатор присутствует в количестве от приблизительно 0,1% до приблизительно 45% по массе, от приблизительно 0,1% до приблизительно 40% по массе, от приблизительно 0,1% до приблизительно 35% по массе, от приблизительно 0,1% до приблизительно 30% по массе, от приблизительно 0,1% до приблизительно 25% по массе, от приблизительно 0,1% до приблизительно 20% по массе, от приблизительно 0,1% до приблизительно 15% по массе, от приблизительно 0,1% до приблизительно 10% по массе, от приблизительно 0,1% до приблизительно 5% по массе, от приблизительно 1% до приблизительно 45% по массе, от приблизительно 1% до приблизительно 40% по массе, от приблизительно 1% до приблизительно 35% по массе, от приблизительно 1% до приблизительно 30% по массе, от приблизительно 1% до приблизительно 25% по массе, от приблизительно 1% до приблизительно 20% по массе, от приблизительно 1% до приблизительно 15% по массе, от приблизительно 1% до приблизительно 10% по массе, от приблизительно 1% до приблизительно 5% по массе, от приблизительно 5% до приблизительно 45% по массе, от приблизительно 5% до приблизительно 40% по массе, от приблизительно 5% до приблизительно 35% по массе, от приблизительно 5% до приблизительно 30% по массе, от приблизительно 5% до приблизительно 25% по массе, от приблизительно 5% до приблизительно 20% по массе, от приблизительно 5% до приблизительно 15% по массе, от приблизительно 5% до приблизительно 10% по массе, от приблизительно 10% до приблизительно 45% по массе, от приблизительно 10% до приблизительно 40% по массе, от приблизительно 10% до приблизительно 35% по массе, от приблизительно 10% до приблизительно 30% по массе, от приблизительно 10% до приблизительно 25% по массе, от приблизительно 10% до приблизительно 20% по массе, от приблизительно 10% до приблизительно 15% по массе, от приблизительно 15% до приблизительно 45% по массе, от приблизительно 15% до приблизительно 40% по массе, от приблизительно 15% до приблизительно 35% по массе, от приблизительно 15% до приблизительно 30% по массе, от приблизительно 15% до приблизительно 25% по массе, от приблизительно 15% до приблизительно 20% по массе, от приблизительно 20% до приблизительно 45% по массе, от приблизительно 20% до приблизительно 40% по массе, от приблизительно 20% до приблизительно 35% по массе, от приблизительно 20% до приблизительно 30% по массе, от приблизительно 20% до приблизительно 25% по массе, от приблизительно 25% до приблизительно 45% по массе, от приблизительно 25% до приблизительно 40% по массе, от приблизительно 25% до приблизительно 35% по массе или от приблизительно 25% до приблизительно 30% по массе.
80 Способ лечения индивидуума, страдающего от хронического воспаления, где способ включает стадию: введения индивидууму, если ему это необходимо, фармацевтической композиции по вариантам осуществления 1-72, где введение приводит в результате к уменьшению интенсивности симптома, связанного с хроническим воспалением, вследствие чего происходит лечение индивидуума.
81. Применение фармацевтической композиции по вариантам осуществления 1-79 в производстве лекарственного препарата для лечения хронического воспаления.
82. Применение фармацевтической композиции по вариантам осуществления 1-79 для лечения хронического воспаления.
83. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с угрями, кислотным рефлюксом/изжогой, возрастной дегенерацией желтого пятна (AMD), аллергией, аллергическим ринитом, болезнью Альцгеймера, боковым амиотрофическим склерозом, анемией, аппендицитом, артериитом, артритом, астмой, атеросклерозом, аутоиммунными заболеваниями, баланитом, блефаритом, бронхиолитом, бронхитом, буллезным пемфигоидом, ожогом, бурситом, раком, задержкой очередного сердечного сокращения, кардитом, глютенчувствительной целиакией, целлюлитом, цервицитом, холангитом, холециститом, хориоамнионитом, хроническим обструктивным заболеванием легких (COPD), циррозом, колитом, хронической сердечной недостаточностью, конъюнктивитом, индуцированным циклофосфамидом циститом, кистозным фиброзом, циститом, простудой, дакриоаденитом, деменцией, дерматитом, дерматомиозитом, диабетом, диабетической нейропатией, диабетической ретинопатией, диабетической нефропатией, диабетическим изъязвлением, заболеванием системы пищеварения, экземой, эмфиземой, энцефалитом, эндокардитом, эндометритом, энтеритом, энтероколитом, эпикондилитом, эпидидимитом, фасциитом, фибромиалгией, фиброзом, фиброзитом, гастритом, гастроэнтеритом, гингивитом, гломерулонефритом, глосситом, заболеванием сердца, дисфункцией сердечного клапана, гепатитом, гнойным гидраденитом, болезнью Гентингтона, гиперлипидемическим панкреатитом, гипертензией, илеитом, инфекцией, воспалительным заболеванием кишечника, воспалительной кардиомегалией, воспалительной невропатией, резистентностью к инсулину, интерстициальным циститом, интерстициальным нефритом, иритом, ишемией, ишемическим заболеванием сердца, кератитом, кератоконъюнктивитом, ларингитом, волчаночным нефритом, маститом, мастоидитом, менингитом, метаболическим синдромом (синдромом X), мигренью, множественным склерозом, миелитом, миокардитом, миозитом, нефритом, неалкогольным стеатогепатитом, ожирением, омфалитом, оофоритом, орхитом, остеохондритом, остеопенией, остеомиелитом, остеопорозом, оститом, отитом, панкреатитом, болезнью Паркинсона, паротитом, воспалительным заболеванием тазовых органов, обыкновенной пузырчаткой, перикардитом, пиритонитом, фарингитом, флебитом, плевритом, пневмонией, поликистозным нефритом, проктитом, простатитом, псориазом, пульпитом, пиелонефритом, пилефлебитом, почечной недостаточностью, реперфузионным повреждением, ретинитом, острой ревматической лихорадкой, ринитом, сальпингитом, саркоидозом, сиаладенитом, синуситом, синдромом раздраженной толстой кишки, стенозом, стоматитом, инсультом, послеоперационным осложнением, синовитом, тендинитом, тендинозом, теносиновитом, тромбофлебитом, тонзиллитом, травмой, травматическим повреждением головного мозга, отторжением трансплантата, тригонитом, туберкулезом, опухолью, уретритом, бурситом, увеитом, вагинитом, васкулитом или вульвитом.
84. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление представляет собой воспаление ткани.
85. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление представляет собой системное воспаление.
86. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление представляет собой артрит.
87. Способ или применение по варианту осуществления 83 или 86, где артрит представляет собой моноартрит, олигоартрит или полиартрит.
88. Способ или применение по варианту осуществления 140 или 143, где артрит представляет собой аутоиммунное заболевание или неаутоиммунное заболевание.
89. Способ или применение по варианту осуществления 83 или 86, где артрит представляет собой остеоартрит, ревматоидный артрит, ювенильный идиопатический артрит, септический артрит, спондилоартропатию, подагру, псевдоподагру или болезнь Стилла.
90. Способ или применение по варианту осуществления 89, где спондилоартропатия представляет собой анкилозирующий спондилит, реактивный артрит (синдром Рейтера), псориатический артрит, энтеропатический артрит, связанный с воспалительным заболеванием кишечника, болезнь Уиппла или болезнь Бехчета.
91. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление представляет собой аутоиммунное заболевание.
92. Способ или применение по варианту осуществления 83 или 91, где аутоиммунное заболевание представляет собой системное аутоиммунное заболевание или органоспецифическое аутоиммунное заболевание.
93. Способ или применение по варианту осуществления 83 или 91, где аутоиммунное заболевание представляет собой острый рассеянный энцефаломиелит (ADEM), болезнь Аддисона, аллергию, антифосфолипидный синдром (APS), аутоиммунную гемолитическую анемию, аутоиммунный гепатит, аутоиммунное заболевание внутреннего уха, буллезный пемфигоид, глютенчувствительную целиакию, болезнь Шагаса, хроническое обструктивное заболевание легких (COPD), сахарный диабет первого типа (IDDM), эндометриоз, синдром Гудпасчера, болезнь Грейвса, синдром Гийена - Барре (GBS), тиреоидит Хашимото, гнойный гидраденит, идиопатическую тромбоцитопеническую пурпуру, воспалительное заболевание кишечника, интерстициальный цистит, волчанку (в том числе дискоидную красную волчанку, лекарственную красную волчанку, волчаночный нефрит, неонатальную волчанку, подострую кожную красную волчанку, системную красную волчанку), очаговую склеродермию, множественный склероз (MS), тяжелую миастению, миопатию, нарколепсию, нейромиотонию, обыкновенную пузырчатку, злокачественную анемию, первичный биллиарный цирроз, рецидивирующий рассеянный энцефаломиелит, острую ревматическую лихорадку, шизофрению, склеродермию, синдром Шегрена, теносиновит, васкулит или витилиго.
94. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление представляет собой миопатию.
95. Способ или применение по варианту осуществления 93 или 94, где миопатия представляет собой дерматомиозит, миозит с включенными тельцами или полимиозит.
96. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление представляет собой васкулит.
97. Способ или применение по варианту осуществления 83, 93 или 96, где васкулит представляет собой болезнь Бюргера, артериит, церебральный васкулит, артериит Черджа-Штрауса, криоглобулинемию, эссенциальный криоглобулинемический васкулит, гигантоклеточный артериит, васкулит игрока в гольф, пурпуру Геноха-Шенлейна, аллергический васкулит, болезнь Кавасаки, флебит, микроскопический полиартериит/полиангиит, нодозный полиартериит, ревматическую полимиалгию (PMR), ревматоидный васкулит, артериит Такаясу, тромбофлебит, гранулематоз Вегенера или васкулит, вторичный по отношению к заболеванию соединительной ткани, или васкулит, вторичный по отношению к вирусной инфекции.
98. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с заболеванием кожи.
99. Способ или применение по варианту осуществления 98, где заболевание кожи представляет собой дерматит, экзему, статис дерматит, гнойный гидраденит, псориаз, розовые угри или склеродермию.
100. Способ или применение по варианту осуществления 99, где экзема представляет собой атопическую экзему, контактную экзему, ксеротическую экзему, себорейный дерматит, дисгидроз, монетовидную экзему, венозную экзему, герпетиформный дерматит, нейродерматит или аутоэкзематизацию.
101. Способ или применение по варианту осуществления 99, где псориаз представляет собой бляшечный псориаз, псориаз ногтей, каплевидный псориаз, псориаз волосистой части головы, обратный псориаз, пустулезный псориаз или псориатическую эритродермию.
102. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с жулудочно-кишечным заболеванием.
103. Способ или применение по варианту осуществления 102, где желудочно-кишечное заболевание представляет собой синдром раздраженного кишечника или воспаление кишечника.
104. Способ или применение по варианту осуществления 103, где воспаление кишечника представляет собой болезнь Крона или язвенный колит.
105. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с сердечнососудистым заболеванием.
106. Способ или применение по варианту осуществления 105, где сердечнососудистое заболевание представляет собой гипертензию, дисфункцию сердечного клапана, хроническую сердечную недостаточность, инфаркт миокарда, диабетические состояния сердца, воспаление кровеносных сосудов, окклюзионное поражение артерии, заболевание периферических артерий, аневризму, эмболию, расслоение, псевдоаневризму, сосудистую патологию, каверному, тромбоз, тромбофлебит, варикоз вен или инсульт.
107. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с раком.
108. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с фармакологически индуцированным воспалением.
109. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с инфекцией.
110. Способ или применение по варианту осуществления 83 или 109, где инфекция представляет собой бактериальный цистит, бактериальный энцефалит, пандемический грипп, вирусный энцефалит, вирусный гепатит A, вирусный гепатит B или вирусный гепатит C.
111. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с повреждением ткани или органа.
112. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с отторжением трансплантата или реакцией "трансплантант против хозяина".
113. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с Th1-опосредованным воспалительным заболеванием.
114. Способ по варианту осуществления 80 или применение по варианту осуществления 81 или 82, где хроническое воспаление связано с хроническим нейрогенным воспалением.
115. Способ по вариантам осуществления 80 or 83-114 или применение по вариантам осуществления 81-114, где после введения индивидууму, фармацевтическая композиция, включающая терапевтическое соединение по вариантам осуществления 1-79 позволяет в результате достигать биораспределения терапевтического соединения, которое отличается от биораспределения терапевтического соединения, включенного в такую же фармацевтическую композицию, но не содержащую фармацевтически приемлемого вспомогательного вещества.
116. Способ по вариантам осуществления 80 или 83-115 или применение по вариантам осуществления 81-115, где после введения индивидууму, количество терапевтического соединения фармацевтической композиции по вариантам осуществления 1-79, доставляемое в макрофаг, составляет, по меньшей мере, 5% от суммарного количества терапевтического соединения, содержащегося в веденной фармацевтической композиции.
117. Способ по вариантам осуществления 80 или 83-116 или применение по вариантам осуществления 81-116, где после введения индивидууму, фармацевтическая композиция по вариантам осуществления 1-79 уменьшает раздражение в кишечнике, по меньшей мере, на 5% в отличии от фармацевтической композиции по вариантам осуществления 1-79, но не содержащей фармацевтически приемлемого вспомогательного вещества.
118. Способ по вариантам осуществления 80 или 83-117 или применение по вариантам осуществления 81-117, где после введения индивидууму, фармацевтическая композиция по вариантам осуществления 1-79 уменьшает раздражение желудка, по меньшей мере, на 5% в отличии от фармацевтической композиции по вариантам осуществления 1-79, но не содержащей фармацевтически приемлемого вспомогательного вещества.
ПРИМЕРЫ
Следующие неограничивающие примеры приводятся только с целью иллюстрации, для того чтобы облегчить более полное понимание раскрываемого предмета изобретения. Эти примеры не следует истолковывать в качестве ограничения любого из вариантов осуществления, описанных в настоящем изобретении, включая примеры, относящиеся к фармацевтическим композициям, способам приготовления фармацевтических композиций или способам или применениям лечения хроническое воспаление или заболевания, связанного с хроническим воспалением.
Пример 1
Дифференциальная сканирующая калориметрия
Этот пример иллюстрирует приготовление раскрытой в изобретении фармацевтической композиции твердого раствора, включающей терапевтическое соединение.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора использовали следующую общую методику. Смешивали вместе все ингредиенты, кроме твердого при комнатной температуре липида. Эту смесь нагревали до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C при перемешивании, для того чтобы растворить терапевтическое соединение и в результате получить раствор. Затем к раствору добавляли твердый при комнатной температуре липид, и смесь перемешивали до тех пор, пока не растворялся липид. Смесь затем отверждали путем охлаждения до комнатной температуры. Типичные составы, содержащие различные терапевтические соединения, приведены в таблице 1.
LA 3-51
LA 3-57
LA 35-1
LA 35-2
LA 35-3
b M35-1 обозначает MAISINE® 35-1
Смесь затем отверждали путем охлаждения до комнатной температуры. Отвержденные композиции затем оценивали по внешнему виду и с помощью дифференциальной сканирующей калориметрии (ДСК).
В процессе анализа с помощью ДСК, наблюдалась общая тенденция, заключавшаяся в том, что состав, который после отверждения был матовым на вид, образовывал классическую твердую композицию с кристаллической структурой, в то время как состав, который после отверждения был прозрачным на вид, образовывал композицию твердого раствора с аморфной структурой. Кроме того, отвержденные композиции, которые были прозрачными на вид, затем переплавляли, и при этом не происходило выпадения в осадок фазы, что являлось доказательством наличия композиции твердого раствора с аморфной структурой.
С помощью анализа внешнего вида и результатов переплавления было оценено большое число составов на их способность образовывать композицию твердого раствора. Состав, который не образовывал твердое вещество или образовывал матовое на вид твердое вещество, далее не анализировали и отбрасывали. Аналогично, далее не анализировали и отбрасывали составы, если после переплавления отвержденных прозрачных на вид композиций происходило выпадения в осадок фазы. Составы, которые образовывали прозрачное на вид твердое вещество, затем анализировали методом ДСК.
Данные для типичных составов ибупрофена, отверждающихся с прозрачным внешним видом, приведены на фигуре 1. Анализ методом ДСК показал, что индивидуальные компоненты характеризуются острыми четко выраженными пиками. Например, ибупрофен обладал четко выраженной температурой плавления в диапазоне от 75°C до 78°C (фигура 1А). Аналогично, твердый при комнатной температуре липид или твердый жир, такой как GELUCIRE® 43/01, имел четко выраженную температуру плавления в диапазоне от 41°C до 45°C (фигура 1В). Эти острые четко выраженные пики в диапазоне температур плавления являются признаком того, что композиция находится в классической твердой переходной фазе, имеющей четко выраженную кристаллическую структуру. MAISINE® 35-1 и ПЭГ 400 являются жидкостями при комнатной температуре и, поэтому, имеют температуру плавления ниже 20°C. Например, MAISINE® 35-1 имеет температуру плавления в диапазоне от приблизительно 14°C до приблизительно 16°C, а ПЭГ 400 имеет температуру плавления в диапазоне от приблизительно 4°C до приблизительно 8°C.
Неожиданно было обнаружено, что, когда среду, включающую твердый при комнатной температуре липид (твердый жир), жидкий при комнатной температуре липид и стабилизатор, исследовали методом ДСК, появляется новый пик температуры плавления. Например, помимо пика от 41°C до 45°C для GELUCIRE® 43/01, методом ДСК обнаруживали новый расширенный диапазон температуры плавления от приблизительно 32°C до приблизительно 38°C (фигура 1C). Этот температурный диапазон отличался от диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01, от 14°C до 16°C для MAISINE® 35-1 и от 4°C до 8°C для ПЭГ 400. Эти результаты указывают на то, что часть композиции превращалась в фазу твердого раствора, а не в классическую твердую фазу.
Неожиданно было обнаружено, что состав, включающий терапевтическое соединение с этой средой, характеризовался широким диапазоном плавления с температурой плавления, отличающейся от температур плавления индивидуальных компонентов (фигуры 1D-1H). Более того, не было обнаружено пиков, относящихся ни к отдельно взятому терапевтическому соединению, ни к отдельно взятому твердому жиру. Например, ибупрофен имеет диапазон температуры плавления от 75°C до 78°C, GELUCIRE® 43/01 имеет температуру плавления от 41°C до 45°C, MAISINE® 35-1 имеет температуру плавления от 14°C до 16°C и ПЭГ400 имеет температуру плавления от 4°C до 8°C. Однако состав, включающий эти компоненты, дает в результате композицию с температурой плавления в диапазоне от 35°C до 40°C, в зависимости от используемых количеств и соотношений. В случае классической смешанной фазы четко различимых твердых компонентов, должен был бы проявиться каждый индивидуальный пик, то есть, от 75°C до 78°C для ибупрофена, и от 41°C до 45°C для GELUCIRE® 43/01. Но этого не наблюдалось, эти пики полностью исчезали. Присутствие единственного нового пика температуры плавления и одновременное исчезновение пиков температур плавления индивидуальных компонентов для отвержденной композиции указывает на то, что образовывалась новая структура твердого раствора терапевтического соединения и твердого при комнатной температуре липида (твердого жира).
Пример 2
Фармацевтические композиции твердых растворов, включающих артеметер
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую артеметер.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием артеметера, применяли следующий метод. Приблизительно 1,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, и приблизительно 0,5 мл трибутирина (в качестве жидкого при комнатной температуре липида) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 40°C до приблизительно 50°C при перемешивании до тех пор, пока не были смешены все компоненты смеси. Приблизительно 40 мг артеметера добавляли к приблизительно 0,8 мл этой объединенной смеси и перемешивали до растворения. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 35°C до приблизительно 40°C (фигура 2). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 86°C до 90°C для артеметера. Трибутирин представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий артеметер.
Пример 3
Фармацевтические композиции твердых растворов, включающие аспирин
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую аспирин.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием аспирина, применяли следующий метод. Приблизительно 120 мг аспирина и приблизительно 0,5 мл диметилового эфира изосорбида (в качестве стабилизатороа) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 35°C до приблизительно 40°C (фигура 3). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 138°C до 140°C для аспирина. Диметиловый эфир изосорбида представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий аспирин.
Пример 4
Фармацевтические композиции твердых растворов, включающие дантролен
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую дантролен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием дантролена, применяли следующий метод. Добавляли приблизительно 232,7 мг стеариновой кислоты (в качестве нейтрализатора) в реакционный сосуд и нагревали до температуры в диапазоне от приблизительно 70°C до приблизительно 75°C при перемешивании до расплавления. Добавляли приблизительно 250,2 мг дантролена и 20 мл диметилового эфира изосорбида (в качестве стабилизатора) к расплавленной стеариновой кислоте и перемешивали до тех пор, пока не достигалась однородная консистенция. Приблизительно 20,02 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 34°C до приблизительно 39°C (фигура 4). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse), 70°C для стеариновой кислоты и от 279°C до 280°C для дантролена. Диметиловый эфир изосорбида представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий дантролен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием дантролена, применяли следующий метод. Добавляли приблизительно 309,8 мг стеариновой кислоты (в качестве нейтрализатора) в реакционный сосуд и нагревали до температуры в диапазоне от приблизительно 70°C до приблизительно 75°C при перемешивании до расплавления. Добавляли приблизительно 59,6 мг натриевой соли дантролена к расплавленной стеариновой кислоте и перемешивали до тех пор, пока не достигалась однородная консистенция. Добавляли приблизительно 0,75 мл диметилового эфира изосорбида (в качестве стабилизатора) и перемешивали смесь до тех пор, пока не растворялись все компоненты. Приблизительно 760,3 мг GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий дантролен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием дантролена, применяли следующий метод. Добавляли приблизительно 250,2 мг стеариновой кислоты (в качестве нейтрализатора) в реакционный сосуд и нагревали до температуры в диапазоне от приблизительно 70°C до приблизительно 75°C при перемешивании до расплавления. Добавляли приблизительно 50,3 мг натриевой соли дантролена к расплавленной стеариновой кислоте и перемешивали до тех пор, пока не достигалась однородная консистенция. Добавляли 5,0 мл диметилового эфира изосорбида (в качестве стабилизатора) и перемешивали смесь до тех пор, пока не растворялись все компоненты. Приблизительно 5,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий дантролен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием дантролена, применяли следующий метод. Добавляли приблизительно 225,0 мг стеариновой кислоты (в качестве нейтрализатора) в реакционный сосуд и нагревали до температуры в диапазоне от приблизительно 70°C до приблизительно 75°C при перемешивании до расплавления. Добавляли приблизительно 25,1 мг натриевой соли дантролена к расплавленной стеариновой кислоте и перемешивали до тех пор, пока не достигалась однородная консистенция. Добавляли 2,0 мл диметилового эфира изосорбида (в качестве стабилизатора) и перемешивали смесь до тех пор, пока не растворялись все компоненты. Приблизительно 2,07 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий дантролен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием дантролена, применяли следующий метод. Добавляли приблизительно 224,9 мг стеариновой кислоты (в качестве нейтрализатора) в реакционный сосуд и нагревали до температуры в диапазоне от приблизительно 70°C до приблизительно 75°C при перемешивании до расплавления. Добавляли приблизительно 25,1 мг натриевой соли дантролена к расплавленной стеариновой кислоте и перемешивали до тех пор, пока не достигалась однородная консистенция. Добавляли приблизительно 0,75 мл диметилового эфира изосорбида (в качестве стабилизатора) и перемешивали смесь до тех пор, пока не растворялись все компоненты. Приблизительно 304,1 мг GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий дантролен.
Пример 5
Фармацевтические композиции твердых растворов, включающие диклофенак
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую диклофенак.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием диклофенака, применяли следующий метод. Приблизительно 119 мг диклофенака, приблизительно 1,0 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 0,3 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 35°C до приблизительно 40°C (фигура 5). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 157°C до 158°C для диклофенака. MAISINE® 35-1 и диметиловый эфир изосорбида представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий диклофенак.
Пример 6
Фармацевтические композиции твердых растворов, включающие фенофибрат
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую фенофибрат.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием фенофибрата, применяли следующий метод. Приблизительно 400 мг фенофибрата и 4,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 45°C до приблизительно 55°C при перемешивании до тех пор, пока не были смешены все компоненты смеси. Приблизительно 0,76 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли к этой смеси и перемешивали до тех пор, пока не происходило смешение. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 34°C до приблизительно 39°C (фигура 6). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 80°C до 85°C для фенофибрата. Диметиловый эфир изосорбида представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий фенофибрат.
Пример 7
Фармацевтические композиции твердых растворов, включающие гемфиброзил
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую гемфиброзил.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием гемфиброзила, применяли следующий метод. Приблизительно 1 г гемфиброзила, приблизительно 0,9 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 0,4 мл ПЭГ 400 (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,9 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий гемфиброзил.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием гемфиброзила, применяли следующий метод. Приблизительно 1 г гемфиброзила и 7,5 г масла какао, воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 34°C до 38°C и включающего смесь насыщенных C16-C18 триглицеридов, добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока не были смешены все компоненты смеси. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий гемфиброзил.
Пример 8
Фармацевтические композиции твердых растворов, включающие ибупрофен
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую Ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 1 г натриевой соли ибупрофена, приблизительно 0,9 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 0,4 мл ПЭГ 400 добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,9 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 44°C (фигура 1D). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 75°C до 78°C для ибупрофена. MAISINE® 35-1 и ПЭГ 400 представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 1 г натриевой соли ибупрофена, приблизительно 0,5 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 0,4 мл ПЭГ 400 добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 2,3 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 43°C (фигура 1E). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 75°C до 78°C для ибупрофена. MAISINE® 35-1 и ПЭГ 400 представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 1 г натриевой соли ибупрофена, приблизительно 0,2 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 0,4 мл ПЭГ 400 добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 2,6 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 42°C (фигура 1F). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 75°C до 78°C для ибупрофена. MAISINE® 35-1 и ПЭГ 400 представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 5 г натриевой соли ибупрофена, приблизительно 9,5 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 1,0 мл ПЭГ 400 добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 4,0 GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 38°C (фигура 1G). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 75°C до 78°C для ибупрофена. MAISINE® 35-1 и ПЭГ 400 представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 5 г натриевой соли ибупрофена, приблизительно 6,5 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 1,5 мл ПЭГ 400 добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 6,5 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 42°C (фигура 1H). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 75°C до 78°C для ибупрофена. MAISINE® 35-1 и ПЭГ 400 представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 1 г натриевой соли ибупрофена, приблизительно 0,9 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида), приблизительно 0,4 мл ПЭГ 400 и приблизительно 0,3 мл пропиленгликоля добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,9 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием ибупрофена, применяли следующий метод. Приблизительно 5 г ибупрофена в форме свободной кислоты, приблизительно 5 г натриевой соли ибупрофена, приблизительно 8 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида), приблизительно 3 мл ПЭГ 400 и приблизительно 1 мл пропиленгликоля добавляли в реакционный сосуд, нагретый до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C, при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 19 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий ибупрофен.
Пример 9
Фармацевтические композиции твердых растворов, включающие лидокаин
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую лидокаин.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием лидокаина, применяли следующий метод. Приблизительно 200 мг лидокаина в форме основания и приблизительно 2,0 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 8,8 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 34°C до приблизительно 40°C (фигура 7). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 66°C до 69°C для лидокаина. Диметиловый эфир изосорбида представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий лидокаин.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием лидокаина, применяли следующий метод. Приблизительно 250,1 мг лидокаина в форме основания и приблизительно 1,74 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 8,72 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием лидокаина, применяли следующий метод. Приблизительно 500,4 мг лидокаина в форме основания и приблизительно 1,74 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 8,5 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием лидокаина, применяли следующий метод. Приблизительно 250,4 мг лидокаина в форме основания и приблизительно 0,87 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока не были смешены все компоненты смеси. Отдельно, приблизительно 250,1 мг прилокаин HCl основания, 0,13 мл триэтаноламина (в качестве нейтрализатора) и приблизительно 0,87 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока не были смешены все компоненты смеси. Смеси лидокаина и прилокаина объединяли и нагревали до температуры в диапазоне от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты не растворялись. Приблизительно 8,49 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Пример 10
Фармацевтические композиции твердых растворов, включающие набуметон
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую набуметон.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием набуметона, применяли следующий метод. Приблизительно 126 мг набуметона и приблизительно 0,5 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида), добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 35°C до приблизительно 40°C (фигура 8). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 80°C до 81°C для набуметона. MAISINE® 35-1 представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий набуметон.
Пример 11
Фармацевтические композиции твердых растворов, включающие напроксен
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую напроксен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием напроксена, применяли следующий метод. Приблизительно 250,1 мг напроксена и приблизительно 0,75 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида), добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 750,9 мг GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий напроксен.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием напроксена, применяли следующий метод. Приблизительно 650,5 мг напроксена и приблизительно 1,2 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида), добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,234 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 30°C до приблизительно 39°C (фигура 9). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от153°C до 154°C для напроксена. MAISINE® 35-1 представляет собой жидкость при комнатной температуре и, в силу этого, имеет температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий напроксен.
Пример 12
Фармацевтические композиции твердых растворов, включающие пентоксифиллин
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую пентоксифиллин.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием пентоксифиллина, применяли следующий метод. Приблизительно 208 мг пентоксифиллина, приблизительно 1,0 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 0,2 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 1,0 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий пентоксифиллин.
Пример 13
Фармацевтические композиции твердых растворов, включающие сальбутамол
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую сальбутамол.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием сальбутамола, применяли следующий метод. Приблизительно 61 мг сальбутамола, приблизительно 0,6 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида), 1,0 мл диметилового эфира изосорбида (в качестве стабилизатора) и приблизительно 1,0 мл абсолютного этанола (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 10 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 40°C (фигура 10). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 157°C до 158°C для сальбутамола. MAISINE® 35-1, диметиловый эфир изосорбида и абсолютный этанол представляют собой жидкости при комнатной температуре и, в силу этого, все они имеют температуры плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий сальбутамол.
Пример 14
Фармацевтические композиции твердых растворов, включающие салметерол
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую салметерол.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием салметерола, применяли следующий метод. Приблизительно 11 мг салметерола ксинафоата, приблизительно 1,0 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 1,0 мл абсолютного этанола (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 2,04 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 34°C до приблизительно 43°C (фигура 11). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 137°C до138°C для салметерола. MAISINE® 35-1 и абсолютный этанол представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий салметерол.
Пример 15
Фармацевтические композиции твердых растворов, включающие симвастатин
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую симвастатин.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием симвастатина, применяли следующий метод. Приблизительно 200 мг симвастатина и приблизительно 2,5 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 5 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 32°C до приблизительно 42°C (фигура 12). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 135°C до 138°C для симвастатина. MAISINE® 35-1 и абсолютный этанол представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий симвастатин.
Пример 16
Фармацевтические композиции твердых растворов, включающие телмисартан
Этот пример иллюстрирует, как приготовить раскрытую в изобретении фармацевтическую композицию, включающую телмисартан.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием телмисартана, применяли следующий метод. Приблизительно 60,1 мг телмисартанаа и приблизительно 2,0 мл диметилового эфира изосорбида (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 2,03 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы. Состав отверждали в твердое вещество, прозрачное на вид, которое переплавлялось без образования осадка фазы. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий телмисартан.
Для приготовления раскрытой в изобретении фармацевтической композиции твердого раствора с использованием телмисартана, применяли следующий метод. Приблизительно 160,2 мг телмисартана, приблизительно 1,0 мл MAISINE® 35-1 (Gattefosse), глицерил монолинолеата (в качестве жидкого при комнатной температуре липида) и приблизительно 1,0 мл абсолютного этанола (в качестве стабилизатора) добавляли в реакционный сосуд и нагревали в диапазоне температур от приблизительно 50°C до приблизительно 60°C при перемешивании до тех пор, пока все компоненты смеси не растворялись. Приблизительно 2,03 г GELUCIRE® 43/01 (Gattefosse), воскообразного твердого вещества (в качестве твердого при комнатной температуре липида) с температурой плавления от 41°C до 45°C, включающего смесь насыщенных C10-C18 триглицеридов, добавляли к этому раствору до тех пор, пока он не смешивался. Нагретую смесь затем охлаждали до температуры в диапазоне от приблизительно 37°C до приблизительно 40°C и разливали порциями в формы и охлаждали до комнатной температуры. В качестве варианта, смесь может быть охлаждена до комнатной температуры и затем соответственно повторно нагрета до температуры в диапазоне от приблизительно 40°C до приблизительно 45°C для разлива порциями в формы.
Состав отверждали в твердое вещество, прозрачное на вид. Кроме того, метод ДСК показал наличие нового широкого диапазона температур плавления от приблизительно 34°C до приблизительно 43°C (фигура 13). Этот температурный диапазон отличался от температурного диапазона для отдельно взятых индивидуальных компонентов: от 41°C до 45°C для GELUCIRE® 43/01 (Gattefosse) и от 261°C до 263°C для телмисартана. MAISINE® 35-1 и абсолютный этанол представляют собой жидкости при комнатной температуре и, в силу этого, имеют температуру плавления ниже 20°C. Эти результаты указывают, что образовывался раскрытый в изобретении состав твердого раствора, включающий телмисартан.
Пример 17
Эксперимент по поглощению макрофагами
Этот пример иллюстрирует, что раскрытая в изобретении фармацевтическая композиция твердого раствора преимущественно целенаправленно доставляет терапевтическое соединение в иммунную систему.
Культуры моноцитов клеточной линии U937 культивировали в среде RPMI-1640 с добавлением 10% фетальной телячьей сыворотки (FCS) до тех пор, пока клетки не образовывали монослой с 90% конфлюэнтностью. Эти клетки затем обрабатывали форболмиристацетатом (PMA) и инкубировали при 37°C в инкубаторе в 5% двуокиси углерода до тех пор, пока клетки не дифференцировались в макрофаги. Монослои макрофагов промывали свежей средой, и затем добавляли 3 мл одного из следующих испытуемых растворов: А) раскрытый в изобретении состав твердого раствора, включающий ибупрофен, Gelucire® 43/01 (Gattefosse), MAISINE® 35-1 (Gattefosse) и ПЭГ 400; Б) раскрытый в изобретении жидкий состав, включающий ибупрофен, рапсовое масло и этанол; С) ибупрофен в форме свободной кислоты; и D) среду без терапевтического соединения. После инкубации в течение 45 минут, надосадочные жидкости испытуемого раствора удаляли и сохраняли для анализа, а клетки затем промывали несколько раз в забуференном фосфатом физиологическом растворе (PBS) и лизировали, используя два цикла замораживание-размораживание. Концентрацию терапевтического соединения, присутствующего в испытуемом растворе, надосадочную жидкость испытуемого раствора и фракции клеточного лизата анализировали с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Процент терапевтического соединения, поглощенного макрофагами, рассчитывали по следующей формуле: % адсорбированного терапевтического соединения =100×(масса соединения, извлеченного из клеточного лизата)/(масса соединения, доставленного в испытуемом растворе - масса соединения, извлеченная из надосадочной жидкости испытуемого раствора). Результаты приведены в таблице 1. Эти результаты показывают, что среднее поглощение терапевтического соединения макрофагами увеличивается на 550% или более при использовании заявленных в изобретении составов фармацевтических композиций по сравнению с композициями, которые не приготавливали этим способом.
Пример 18
Экспериментальная модель эрозии желудка на животном
Для оценки того, уменьшает ли раскрытая в изобретении фармацевтическая композиция раздражение желудка, были проведены эксперименты с использованием экспериментальной модели эрозии желудка на крысах.
Крысы линии Sprague-Dawley были разделены на семь опытных групп, содержащих по пять животных в каждой. После голодания в течение ночи, животных подвергали одной из семи различных обработок. Группа А представляла собой контрольную группу, в которой каждой крысе перорально вводили только 1% метилцеллюлоза/0,5% полисорбат 80 в качестве плацебо. Группа В представляла собой контрольную группу, в которой каждой крысе перорально вводили только растворитель/вспомогательное вещество (принудительно 10% этанол и 90% рапсового масла) в качестве плацебо. Группа C представляла собой контрольную группу, в которой каждой крысе перорально вводили 150 мг/кг аспирина. Группа D представляла собой контрольную группу, в которой каждой крысе перорально вводили 100 мг/кг ибупрофена, суспендированного в 1% метилцеллюлоза/0,5% полисорбат 80. Группа E представляла собой группу эксперимента, в которой каждой крысе вводили раскрытую в изобретении фармацевтическую композицию (BC1054-100), включающую 100 мг/кг ибупрофена, 10% этанола и 90% рапсового масла. Группа F представляла собой контрольную группу, в которой каждой крысе перорально вводили 100 мг/кг ибупрофена, суспендированного в 1% метилцеллюлоза/0,5% полисорбат 80. Группа G представляла собой группу эксперимента, в которой каждой крысе вводили раскрытую в изобретении фармацевтическую композицию (BC1054-200), включающую 200 мг/кг ибупрофена, 10% этанола и 90% рапсового масла. Через 4 часа после обработки, животных умерщвляли, и проводили оценку их желудков с точки зрения степени кровоизлияния и тяжести эрозионных повреждений слизистой оболочки. Раздражение желудка оценивали в баллах следующим образом: 0 - отсутствии поражений; 1 - гиперемия; 2 - одно или два небольших повреждения; 3 - более чем два незначительных повреждения или тяжелые поражения; и 4 - очень тяжелые поражения. Показатель, составляющей 50% или более по сравнению с группой С (контрольной группой, обработанной аспирином), показатель которой принимали за 100%, считали указывающим на раздражение желудка.
Результаты приведены в таблице 4. В группе D (контрольной группе, обработанной 100 мг/кг ибупрофена) и группе F (контрольной группе, обработанной 200 мг/кг ибупрофена) возникали поражения желудка, тяжесть которых составляла 75% и 95%, соответственно, от тяжести поражения, возникающего в группе C (контрольной группе, обработанной аспирином). При этом, в группе E (группе эксперимента, обработанной BC1054-100) и группе G (группе эксперимента, обработанной BC1054-200), возникали поражения желудка, тяжесть которых составляла 20% и 40%, соответственно, от тяжести поражения, возникающего в группе C (контрольной группе, обработанной аспирином). Эти результаты показывают, что раскрытая в изобретении фармацевтическая композиция уменьшает степень поражения слизистой оболочки и раздражения желудка, которую может вызвать терапевтическое соединение.
Пример 19
Экспериментальная модель воспалительного заболевания кишечника на животном
Для оценки эффективности раскрытой в изобретении фармацевтической композиции при лечении воспалительного заболевания кишечника, были проведены эксперименты с использованием экспериментальной модели индуцированного тринитробензолсульфоновой кислотой (TNBS) колита на мышах.
Самцов мышей линии C57BI/6 (в возрасте 6-7 недель) разделяли на семь опытных групп, содержащих, по меньшей мере, по десять животных в каждой. В день 0, вызывали колит у мышей из групп B-G путем прямого введения в прямую кишку 100 мкл TNBS (4 мг) в 50% этаноле при анестезии изофлураном. Животных подвергали либо один раз, либо три раза в день в период от дня -1 до 5-го дня одной из семи различных обработок. Контрольная группа А представляла собой группу, в котором каждой мыши перорально вводили только этанол в качестве плацебо. Контрольная группа B представляла собой группу, в котором каждой мыши перорально вводили только 1% метилцеллюлозу в качестве плацебо. Контрольная группа C представляла собой группу, в котором каждой мыши перорально вводили только растворитель/вспомогательное вещество (принудительно 10% этанол и 90% рапсового масла) в качестве плацебо. Контрольная группа D представляла собой группу, в которой каждой мыши перорально вводили 3 мг/кг преднизолона. Контрольная группа E представляла собой группу, в которой каждой мыши перорально вводили 20 мг/кг ибупрофена, суспендированного в 1% метилцеллюлозе (1 мл/кг) (без вспомогательного вещества). Группа F представляла собой группу эксперимента, в которой каждой мыши вводили раскрытую в изобретении фармацевтическую композицию (BC1054-20), содержащую 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла. Группа G представляла собой группу эксперимента, в которой каждой мыши вводили раскрытую в изобретении фармацевтическую композицию (BC1054-30), содержащую 30 мг/кг ибупрофена, 10% этанола и 90% рапсового масла. Всех животных каждый день взвешивали и оценивали визуально на наличие диареи и/или кровянистого стула. На 3-й день и на 5-й день у всех животных оценивали тяжесть колита с использованием видео-эндоскопии и по полученным изображениям визуально оценивали слепым методом тяжесть колита в баллах от 0 до 4 следующим образом: 0 - нормальное состояние; 1 - потеря васкулярности; 2 - потеря васкулярности и хрупкость; 3 - хрупкость и эрозии; и 4 - изъязвления и кровотечение. После эндоскопии на 5-й день животных умерщвляли, удаляли толстую кишку и измеряли ее длину и массу. Получали образцы сыворотки, и толстую кишку фиксировали в 10% формалине. Собирали дополнительный кусочек ткани толстой кишки, взвешивали его и быстро замораживали в жидком азоте.
Результаты этих экспериментов приведены в таблице 5. Группа B (контрольная группа, подвергнутая обработке TNBS) показала статистически значимое различие в изменении средней массы по сравнению с группой А (контрольная группа, подвергнутая обработке этанолом), все остальные сравнимые группы не показали никакой разницы в изменении средней массы. Группа B (контрольная группа, подвергнутая обработке TNBS) показала статистически значимое уменьшение средней длины толстой кишки по сравнению с группой А (контрольная группа, подвергнутая обработке этанолом). Кроме того, группа D (контрольная группа, подвергнутая обработке преднизолоном), группа F (группа эксперимента, подвергнутая обработке BC1054-20) и группа G (группа эксперимента, подвергнутая обработке BC1054-30) все показали статистически значимое увеличение средней длины толстой кишки по сравнению с группой B (контрольной группой, подвергнутой обработке TNBS). Несмотря на то, что группа B (контрольная группа, подвергнутая обработке TNBS) показала статистически значимое увеличение средней массы толстой кишки по сравнению с группой А (контрольной группой, подвергнутой обработке этанолом), тем не менее, все сравнения остальных групп не показали никакого различия в средней массе толстой кишки. Что касается оценки в баллах колита на основе эндоскопии, то группа D (контрольная группа, подвергнутая обработке преднизолоном) показала статистически значимое уменьшение средних баллов по оценку колита как на 3-й день, так и на 5-ый день, по сравнению с группой B (контрольной группой, подвергнутой обработке TNBS). Аналогичным образом, как группа F (группа эксперимента, подвергнутая обработке BC1054-20), так и группа G (группа эксперимента, подвергнутая обработке BC1054-30) показали статистически значимое снижение среднего оценки колита на 5-й день по сравнению с группой B (контрольной группой, подвергнутой обработке TNBS)). Эти результаты указывают на то, что раскрытая в изобретении фармацевтическая композиция была эффективной при лечении воспалительного заболевания кишечника.
2Статистически значимое различие по сравнению с группой A (p = 0,001).
3Статистически значимое различие по сравнению с группой B (p = 0,001).
4Статистически значимое различие по сравнению с группой B (p = 0,001).
5Статистически значимое различие по сравнению с группой B (p = 0,034).
6Статистически значимое различие по сравнению с группой A (p = 0,009).
7Статистически значимое различие по сравнению с группой B (p = 0,005).
8Статистически значимое различие по сравнению с группой B (p = 0,002).
9Статистически значимое различие по сравнению с группой B (p = 0,045).
10Статистически значимое различие по сравнению с группой B (p = 0,002).
Пример 20
Экспериментальная модель системного артрита на животном
Для оценки эффективности раскрытой в изобретении фармацевтической композиции при лечении артрита, были проведены эксперименты с использованием экспериментальной модели индуцированного антителами α-коллагена (ACAIA) артрита у мышей, которая имитирует системный артрит, такой как ревматоидный артрит.
Самцы мышей линии BALB/с были разделены на восемь групп, каждая из которых содержала по 10 животных. Для того чтобы индуцировать симптомы артрита, мышам из всех восьми групп внутривенно вводили 200 мкл раствора антител, включающего 2 мг коктейля из четырех моноклональных антител α-коллагена II (ARTHRITOMAB™, MD Biosciences) на день исследования 0 (день начала исследования), и затем внутрибрюшинно вводили 200 мкл раствора, содержащего 100 мкг липополисахарида (LPS) на день 3-ий день исследования. Каждую группу подвергали ежедневной контрольной или испытуемой обработке, применяемой в дни исследования 0-11 следующим образом: группу мышей 1 (1M) подвергали пероральной обработке препаратом плацебо, содержащим 1% метилцеллюлозы, вводимым три раза в день; группу мышей 2 (2М) подвергали внутрибрюшинной обработке препаратом, использующегося в качестве положительного контроля, содержащим 10 мг/кг этанерцепта (ENBREL®, Wyeth), вводимым один раз в день; группу мышей 3 (3M) подвергали пероральной обработке в дозе 20 мг/кг испытуемой жидкой композицией, содержащей ибупрофен и рапсовое масло (BC1054 LF-RO), один раз в день; группу мышей 4 (4М) подвергали пероральной обработке в дозе 20 мг/кг испытуемой жидкой композицией, содержащей ибупрофен и рапсовое масло (BC1054 LF-RO), три раза в день; группу мышей 5 (5M) подвергали пероральной обработке в дозе 20 мг/кг испытуемой жидкой композицией, содержащей ибупрофен и глицерил монолинолеат (MAISINE® 35-1, Gattefosse) (BC1054 LF-МА), три раза в день; группу мышей 6 (6M) подвергали пероральной обработке в дозе 20 мг/кг испытуемой жидкой композицией, содержащей ибупрофен и теобромное масло (BC1054 LF-К), три раза в день; группу мышей 7 (7M) подвергали пероральной обработке контрольным препаратом 1, содержащим 20 мг/кг ибупрофена, три раза в день; и группу мышей 8 (8M) подвергали пероральной обработке в дозе 20 мг/кг испытуемым твердым составом, содержащим ибупрофен и воскообразное твердое вещество, имеющее температуру плавления в интервале от 37°С до 41°С и включающего смесь насыщенных С10-С18 триглицеридов (Gelucire® 39/01, Gattefosse) (BC1054 LF-GE), три раза в день (таблица 6). Вводимую дозу рассчитывали на основе предположения, что масса каждого животного составляла, в среднем, 20 грамм. Каждой мыши вводили фиксированный объем в размере 100 мкл, за исключением получающих положительный контроль животных (2М), которым вводили 200 мкл.
PO=перорально
N/A=Не используется
Для всех мышей постоянно проводили наблюдение развития артрита и клинические обследования, начиная с 0 дня исследования, незадолго до индуцирования артрита, и затем в дни исследования 3-7, 9, 10 и 12 (день окончания исследования). Для оценки развития артрита, проводилась как оценка артрита в баллах, так и измерения толщины лапы (плетизмография). Оценка артрита в баллах основывалась на визуальной оценке артритных реакций с использованием шкалы баллов от 0 до 4 в порядке возрастания тяжести, где 0 баллов указывал на отсутствие артритной реакции; 1 балл указывал на легкое, но определяемое, покраснение и отек лодыжки/запястья или на явное покраснение и отек, ограниченные отдельными пальцами, независимо от количества пораженных пальцев; 2 балла указывали на покраснение и отек лодыжки/запястья от умеренных до тяжелых; 3 балла указывали на покраснение и отек всей лапы, включая пальцы; и 4 балла указывали на максимальное воспаление конечности с вовлечением нескольких суставов. Толщину лапы измеряли для обеих задних лап выше подушечки лапы и ниже пяточной кости с помощью циферблатного штангенциркуля (Kroeplin, Munich, Germany). Были определены средние значения измеренной толщины лапы и, когда это требовалось, применяли дисперсионный анализ ANOVA с процедурой множественных сравнений по критерию Тьюки для определения значимости эффектов лечения.
Клинические обследования включали регистрацию изменений массы тела, состояния кожи, меха, глаз, слизистых оболочек, возникновения выделений секрета и экскреций (например, диареи) и автономной активности (например, слезотечения, слюнотечения, пилоэрекции, изменения размера зрачка, необычного способа дыхания). Также регистрировались изменения в походке, осанке и в реакции на обращение, а также наличие аномального поведения, тремора, судорог, комы и спячки. Сыворотку собирали по окончанию исследования.
Заболеваемость артритом увеличивалась во всех группах, начиная с 3 дня. В группе животных 1М пик заболеваемости приходился на 7-й день с артритными реакциями у 9 из 10 животных, которые оставались относительно неизменными до конца исследования. В группе мышей 2М, обработанных этанерцептом, пик заболеваемости приходился на 6-ой день с признаками заболевания у 9 из 10 животных, но этот показатель снижался до 1 из 10 животных на 12-ый день. Пик заболеваемости артритом в группе животных 3М и группе животных 4М, подвергавшихся обработке BC1054 LS-RO один раз или три раза в день, проявлялся на 7-ой день (9 из 10 и 7 из 10 животных, соответственно), и этот показатель уменьшался до 4 из 10 мышей в обеих группах на 12-ый день. Заболеваемость артритом достигала максимума на 6-й день в группе животных 5М, подвергавшихся обработке BC1054 LS-MA, с проявлением у 8 из 10 животных, и заболеваемость колебалась от 6 до 8 животных до конца исследования. На 6-ой день, артрит проявлялся у 9 из 10 животных группы 6М, подвергавшихся обработке BC1054 SF-TO, но этот показатель также колебался и в конечном итоге составлял 7 из 10 животных на 12-ый день. В группы животных 7М, подвергавшихся обработке ибупрофеном, пик заболеваемости артритом был зафиксирован на 6 день у 8 из 10 животных, и этот показатель оставался относительно неизменным до конца исследования. В группе мышей 8M, подвергавшихся обработке BC1054 LS-GE, пик заболеваемости приходился на 6-й день у 9 из 10 животных, проявляющих признаки артрита, но этот показатель уменьшался до 4 из 10 животных на 12-ый день.
Клинические признаки, связанные с введением липополисахаридов (LPS), развивались во всех группах после стимуляции LPS на 3-ий день. Они исчезали во всех группах на 12-ый день. В процессе этого исследования не было смертельных случаев или случаев существенного различия в массе тела между группой животных, подвергнутых обработке плацебо, и группой животных, подвергнутых обработке испытуемыми композициями.
Результаты измерений средней толщины лапы приведены в таблице 7. Средняя толщина задней лапы в группе животных 1М (подвергнутых обработке плацебо) составляла 1,72±0,01 на день 0. Толщина увеличивалась и достигала максимума на 9-й день, составляя 2,33±0,15, и в конечном итоге составляла 2,17±0,11 на 12-ый день. В группе мышей 2М, подвергнутой обработке этанерцептом, среднее значение толщины лапы составляло 1,70±0,02 на день 0. Оно увеличивалось, достигая максимума 1,96±0,05 на 6-й день и затем снижалось до 1,77±0,02 на 12-ый день. Обработка этанерцептом приводила в результате к значительному уменьшению объема лапы по сравнению с группой мышей для положительного контроля на 9-ый, 10-ый и 12-ый день. В группе 3М, которую подвергали обработке BC1054 LS-RO один раз в день, толщина задней лапы составляла 1,71±0,02 на день 0. На 7-ый день отек в этой группе достигал своего максимума, составляя 1,96±0,05, и затем оставался относительно неизменным. Наблюдались значительные уменьшения среднего значения отека лапы на 6-ой и 9-ый дни после введения BC1054 LS-RO. Средняя толщина задней лапы в группе 4М, которая получила BC1054 LS-RO три раза в день, увеличилась до 1,97±0,08 на 10-ый день (от 1,70±0,03 в день 0), начиная с 10-го дня, величина объемов лап оставалась относительно постоянной до конца исследования. Введение три раза в день BC1054 LS-RO приводило к значительному уменьшению средней толщины лапы по сравнению с обработанными плацебо мышами (группа 1M) на 6-ой, 7-ой и 9-ый день. Группа мышей 5М, подвергавшаяся обработке BC1054 LS-МA, имела максимальное значение объема лапы на 7-й день (1,97±0,05 при 1,69 ±± 0,02 на день 0), эта группа имела значительно более низкие результаты измерений на 6-й день и 9-й день по сравнению с группой животных 1M, подвергавшихся обработке плацебо. В группе животных 6М, подвергавшихся обработке BC1054 LS-ТО, средняя толщина задней лапы составляла 1,74±0,01 на день 0. Она увеличилась до максимального значения 2,05±0,10 на 7-й день и затем уменьшалась до 1,94±0,06 на 12-ый день. Не было зарегистрировано никаких существенных различий между группой животных (группой 6M), обработанных BC1054 LS-TО, и контрольной группой животных (группой 1М), получавших плацебо. В группе (группе 7М), которая получила ибупрофен, толщина задней лапы составляла 1,71±0,02 на день 0. На 7-ой день размер опухоли в этой группе был максимальным 2,15±0,10, и затем уменьшался до 2,02±0,08 на 12-ый день. Не отмечалось никаких существенных различий при сравнении этой группы с контрольной группой 1М. Группа животных 8M, подвергавшаяся обработке BC1054 LS-GE, характеризовалась незначительным увеличением толщины задней лапы от 1,72±0,02 на день 0 до 1,85±0,06 на 7-й день, и она оставалась относительно постоянной, составляя 1,77±0,03, на 12-ый день. Введение BC1054 LS-GE приводило к значительному уменьшению отека лапы у животных (группы 8М) по сравнению с контрольной группой (группой 1M), в которой вводили плацебо, на 6-ой, 7-ой, 9-ый, 10-ый и 12-ый дни.
Принимая во внимание приведенные выше результаты, можно констатировать проявление значительной противоартритной активности в группе животных 3М, которым вводили один раз в день BC1054 LS-RO, в группе животных 4M, которым вводили три раза в день BC1054 LS-RO, в группе животных 5M, которым вводили три раза в день BC1054 LS-МА, и в группе животных 8M, которым вводили три раза в день BC1054SF-GE.
Пример 21
Описание конкретных случаев лечения хронического воспаления
Женщине в возрасте 47 лет с диагнозом реактивный артрит в одном колене был проведен курс лечения с помощью раскрытой в изобретении фармацевтической композиции (BC1054), содержащей 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (1200 мг стандартная начальная доза), в течение 3 дней, и было обнаружено, что припухлость и боль стали исчезать через 1 день, и через 3 дня состояние полностью улучшилось. Соответственно, было решено отказаться от стандартного способа лечения ибупрофеном. Обследование через 3 месяца не выявили признаков реактивного артрита.
Мужчине в возрасте 50 лет был поставлен диагноз хронически воспаленной лодыжки после перелома лодыжки типа Мезоннева. Пациент принимал 30 мг кодеина вместе с 500 мг парацетамола два раза в день и 10 мг диклофенака три раза в день в течение 8 месяцев для снятия боли. Ему был проведен 5-дневный курс лечения раскрытой в изобретении фармацевтической композицией (BC1054), содержащей 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (600 мг два раза в день), и через 2 дня было отмечено значительное уменьшение боли и затем через 3 дня пациент сообщил, что боль практически исчезла. Соответственно, он перестал далее принимать кодеин, парацетамол и диклофенак, и через 2 месяца после этого, пациент все еще не чувствовал боли.
Женщина в возрасте 33 лет с диагнозом вызванной стрессом экземы страдала от острого обострения экземы средней тяжести на руках и груди. Раскрытую в изобретении фармацевтическую композицию (BC1054), содержащую 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (1200 мг стандартную начальную дозу), вводили в течение 7 дней. Через два часа прекратился зуд на месте поражения, через 1 день было отмечено заметное уменьшение отечности пораженного участка. Еще через 2-3 дня на пораженных экземой участках пропала эритема и через 7 дней участки поражения полностью исчезли. Ранее, пациентка использовала смягчающие средства для кожи и кремы с гидрокортизоном, которые только ухудшали состояние пораженных участков и могли бы привести к необходимости проведения курса лечения антибиотиками. Пациентка отмечала, что ее ответная реакция на лечение с помощью BC1054 была быстрой и полной и приводила к явному улучшению по сравнению с ранее использовавшимися медикаментозными терапиями.
Мужчина в возрасте 85 лет был поставлен диагноз реактивного остеоартрита, сопровождающегося заметной припухлостью и сильной болью в обоих коленах. Для борьбы с артритом пациенту был предписан прием в течение 1 года преднизолона и NSAIDS, но это не дало никакого эффекта. Кроме того, пациент принимал каждый день глюкозамин. Несмотря на прием значительного количества лекарственных препаратов, пациент регулярно страдал от обострения болезни, приводящего к значительному ограничению подвижности. Пациенту был проведен 10-дневный курс лечения раскрытой в изобретении фармацевтической композицией (BC1054), содержащей 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (400 мг три раза в день), и пациент чувствовал существенное улучшение на 3-ый день и полное прекращение воспалительных явлений после завершения курса лечения. Пациент сообщал, что его подвижность вернулась к норме и ремиссия сохранялась на момент обследования через 1 месяц.
Мужчина в возрасте 38 лет с реактивным остеоартритом в 1 колене на протяжении 6 месяцев (боль и опухание). В течение этого периода, пациент испробовал широкий спектр медикаментозных средств: преднизолон, хумира и сульфасалазин, наряду с NSAIDs, для снятия боли. Только сульфасалазин давал более или менее ощутимый эффект, однако пациент плохо переносил его побочные действия, поэтому он попросил отменить сульфасалазин. Через 2 недели после прекращения приема сульфасалазина, пациент испытал обострение реактивного артрита и прошел 4-дневный курс лечения раскрытой в изобретении фармацевтической композицией (BC1054), содержащей 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (600 мг два раза в день), в результате которого достигалась полная ремиссия при артрите. Через 2 недели после прекращения приема лекарственного средства, опять произошло обострение артрита, и пациенту в течение еще 4 дней был проведен курс лечения с помощью фармацевтической композиции BC1054, в результате которого опять достигалась полная ремиссия. Через 1 неделю опять произошло обострение артрита. С учетом этого, пациенту был проведен последний 10-дневный курс лечения фармацевтической композицией BC1054. В соответствии с данными последнего обследования, в результате была достигнута ремиссия для артрита в течение 11 месяцев.
Мужчине в возрасте 49 лет с диагнозом гиперхолестеринемии (LDL 4,35 ммоль/л) был проведен курс лечения с помощью раскрытой в изобретении фармацевтической композиции (BC1054), содержащей 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (600 мг два раза в день) в течение 7 дней. Через 5 дней лечения, уровни LDL у пациента нормализовались до 3,89 ммоль/л. Нормализация уровня LDL сохранялась в течение 2 месяцев после прекращения приема BC1054, что было определено при заключительном обследовании.
Мужчине в возрасте 60 лет с недавно диагностированной гиперхолестеринемией (LDL 4,31 ммоль/л) был проведен курс лечения с помощью раскрытой в изобретении фармацевтической композиции (BC1054), содержащей 20 мг/кг ибупрофена, 10% этанола и 90% рапсового масла (стандартная начальная доза 1200 мг) с целью снижения уровня LDL до диапазона нормальных значений. Через 5 дней лечения, уровни LDL пациента снизились до 3,36 ммоль/л. Пациент находился под наблюдением в течение 1 месяца и уровень LDL оставался в пределах диапазона нормальных значений, несмотря на то, что дальнейший прием фармацевтической композиции BC1054 не производился.
Пример 22
Лечение хронического воспаления
Женщина в возрасте 62 лет жалуется на скованность в суставе и опухание, и ей ставят диагноз ревматоидного артрита. Врач определяет, что скованность в суставе и опухание является следствием хронического воспаления. Женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, женщине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние женщины постоянно контролируют, и примерно через 3 дня лечения, у женщины наблюдается уменьшение скованности в суставе и опухания. Обследования, проведенные через один и три месяца, показывают, что у женщины сохранилась уменьшенная скованность в суставе и опухание в подвергнутой лечению области. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с любым моноартритом, олигоартритом или полиартритом, таким как, например, остеоартрит, ювенильный идиопатический артрит, септический артрит, спондилоартропатия (в том числе анкилозирующий спондилит, реактивный артрит (синдром Рейтера), псориатический артрит, энтеропатический артрит, связанный с воспалительным заболеванием кишечника, болезнь Уиппла или болезнь Бехчета), синовитом, подагрой, псевдоподагрой или болезнью Стилла, так же как и с бурситом, острой ревматической лихорадкой или теносиновитом. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Мужчина в возрасте 58 лет жалуется на затруднение дыхания и ему ставят диагноз хронического обструктивного заболевания легких (COPD). Врач определяет, что затруднение дыхания является следствием хронического воспаления. Мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, мужчине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние мужчины постоянно контролируют, и примерно через 3 дня лечения, у мужчины наблюдается улучшение его дыхательной способности. Обследования, проведенные через один и три месяца, показывают, что у мужчины сохранилась улучшение его дыхательной способности. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с астмой, бронхиолитом, бронхитом, эмфиземой, ларингитом, фарингитом, плевритом, пневмонией, ринитом, синуситом или любым другим типом хронического респираторного заболевания. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Мужчина в возрасте 67 лет жалуется на болезненные ощущения в мышцах и ему ставят диагноз дерматомиозита. Врач определяет, что болезненные ощущения является следствием хронического воспаления. Мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, мужчине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние мужчины постоянно контролируют, и примерно через 3 дня лечения, у мужчины наблюдается ослабление болезненных ощущений. Обследования, проведенные через один и три месяца, показывают, что у мужчины сохранилась улучшенная мышечная моторика и ослабление болевых ощущений. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с миозитом с включенными тельцами, тяжелой миастенией, полимиозитом или любым другим типом воспалительной миопатии, так же как и с фасциитом, фиброзитом, миозитом, нейромиотонией, тендинозом или тендинитом. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Женщина в возрасте 73 лет жалуется на хрипы при дыхании, и ей ставят диагноз артериита Черга-Страуса. Врач определяет, что хрипы при дыхании являются следствием хронического воспаления. Женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, женщине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние женщины постоянно контролируют, и примерно через 3 дня лечения, у женщины пропадают хрипы при дыхании. Обследования, проведенные через один и три месяца, показывают, что хрипы при дыхании у женщины все еще отсутствуют. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с любым васкулитом, таким как, например, болезнь Бюргера, церебральный васкулит, криоглобулинемия, эссенциальный криоглобулинемический васкулит, гигантоклеточный артериит, васкулит игрока в гольф, пурпура Геноха-Шенлейна, аллергический васкулит, болезнь Кавасаки, микроскопический полиартериит/полиангиит, нодозный полиартериит, ревматическая полимиалгия (PMR), ревматоидный васкулит, артериит Такаясу или гранулематоз Вегенера, так же как и с артериитом, кардитом, эндокардитом, заболеванием сердца, высоким кровяным давлением, воспалительной кардиомегалией, ишемическим заболеванием сердца, миокардитом, перикардитом, флебитом, пилефлебитом или тромбофлебитом. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Мужчина в возрасте 37 лет жалуется на покраснение кожи, и ему ставят диагноз розовых угрей. Врач определяет, что покраснение является следствием хронического воспаления. Мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, мужчине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние мужчины постоянно контролируют, и примерно через 3 дня лечения, у мужчины наблюдается уменьшение покраснения кожи. Обследования, проведенные через один и три месяца, показывают, что у мужчины сохранился улучшенный цвет кожи и уменьшение покраснения. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с угрями, цервицитом, дерматитом, экземой (в том числе атопической экземой, контактной экземой, ксеротической экземой, себорейным дерматитом, дисгидрозом, монетовидной экземой, венозной экземой, герпетиформным дерматитом, нейродерматитом или аутоэкзематизацией), эндометритом, гингивитом, глосситом, гнойным гидраденитом, кератитом, кератоконъюнктивитом, маститом, псориазом (в том числе бляшечным псориазом, псориазом ногтей, каплевидным псориазом, псориазом волосистой части головы, обратным псориазом, пустулезным псориазом или псориатической эритродермией), склеродермией, статис дерматитом, стоматитом, тонзиллитом, вагинитом, витилиго или вульвитом. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Женщина в возрасте 33 лет жалуется на боль в животе и диарею, и ей ставят диагноз болезни Крона. Врач определяет, что боль в животе и диарея являются следствием хронического воспаления. Женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, женщине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние женщины постоянно контролируют, и примерно через 3 дня лечения, у женщины наблюдается уменьшение боли в животе и у нее пропадает диарея. Обследования, проведенные через один и три месяца, показывают, что у женщины сохраняется уменьшение боли в животе и отсутствие диареи. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от нейрогенного воспаления, связанного с любым воспалительным заболеванием кишечника, такого как, например, язвенный колит (в том числе язвенный проктит, левосторонний колит, панколит и скоротечный колит), с любым синдромом раздраженного кишечника, так же как и с колитом, энтеритом, энтероколитом, гастритом, гастроэнтеритом, метаболическим синдромом (синдромом X), синдромом раздраженной толстой кишки или с любым другим желудочно-кишечным заболеванием. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Мужчина в возрасте 46 лет жалуется на лихорадочное состояние, боли в суставах и утомление, и ему ставят диагноз системной красной волчанки. Врач определяет, что эти симптомы являются следствием хронического воспаления. Мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, мужчине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние мужчины постоянно контролируют, и примерно через 3 дня лечения, у мужчины наблюдается улучшение состояния его здоровья, лихорадочное состояние прекратилось, боль в суставах уменьшилась и он не чувствует усталости. Обследования, проведенные через один и три месяца, показывают, что у мужчины сохранилась уменьшение боли в суставах и он не страдает от лихорадочного состояния или от состояния утомления. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с любым другим системным аутоиммунным заболеванием, включающим, без ограничения, антифосфолипидный синдром (APS), буллезный пемфигоид, болезнь Шагаса, дискоидную красную волчанку, лекарственную красную волчанку, синдром Гудпасчера, синдром Гийена-Барре, идиопатическую тромбоцитопеническую пурпуру, тяжелую миастению, неонатальную волчанку, пернициозную анемию, ревматическую полимиалгию, ревматоидный артрит, склеродермию, синдром Шегрена, подострую кожную красную волчанку или гранулематоз Вегенера. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Мужчина в возрасте 58 лет жалуется на депрессивное состояние, чувствительность к холоду, увеличение массы тела, забывчивость и констипацию, и ему ставят диагноз тиреоидита Хашимото. Врач определяет, что эти симптомы являются следствием хроническое воспаление. Мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, мужчине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, мужчине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние мужчины постоянно контролируют, и примерно через 3 дня лечения, у мужчины наблюдается ослабление всех симптомов, на которые он жаловался. Обследования, проведенные через один и три месяца, показывают, что мужчина больше не испытывает депрессивного состояния, чувствительности к холоду, не набирает вес, не страдает забывчивостью и констипацией. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с любым другим локализованным аутоиммунным заболеванием, включающим, без ограничения, острый рассеянный энцефаломиелит (ADEM), болезнь Аддисона, аутоиммунную гемолитическую анемию, аутоиммунный гепатит (в том числе первичный биллиарный цирроз), аутоиммунное заболевание внутреннего уха, глютенчувствительную целиакию, болезнь Крона, сахарный диабет первого типа, эндометриоз, гигантоклеточный артериит, болезнь Грейвса, интерстициальный цистит, волчаночный нефрит, множественный склероз, очаговую склеродермию, обыкновенную пузырчатку, рецидивирующий рассеянный энцефаломиелит, склерозирующий холангит, язвенный колит или витилиго. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
Женщина в возрасте 59 лет жалуется на скованность в суставе и опухание, и ей ставят диагноз реактивного артрита. Врач определяет, что скованность в суставе и опухание является следствием хронического воспаления. Женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей ибупрофен. В качестве варианта, женщине предписано пероральное введение три раза в день раскрытой в изобретении фармацевтической композиции, включающей аспирин. В качестве варианта, женщине предписано пероральное введение два раза в день раскрытой в изобретении фармацевтической композиции, включающей напроксен. Состояние женщины постоянно контролируют, и примерно через 3 дня лечения, у женщины наблюдается уменьшение скованности в суставе и опухания. Обследования, проведенные через один и три месяца, показывают, что у женщины сохраняется уменьшение скованности в суставе и опухания в подвергнутой лечению области. Это ослабление симптомов хронического воспаления указывает на успешные результаты лечения с помощью раскрытой в изобретении фармацевтической композиции. Аналогичный способ перорального введения раскрытой в изобретении фармацевтической композиции может быть использован для лечения пациента, страдающего от хронического воспаления, связанного с любым моноартритом, олигоартритом или полиартритом, таким как, например, остеоартрит, ювенильный идиопатический артрит, септический артрит, спондилоартропатия (в том числе анкилозирующий спондилит, реактивный артрит (синдром Рейтера), псориатический артрит, энтеропатический артрит, связанный с воспалительным заболеванием кишечника, болезнь Уиппла или болезнь Бехчета), синовит, подагра, псевдоподагра или болезнь Стилла, так же как и с бурситом, острой ревматической лихорадкой или теносиновитом. Аналогичным образом, любое из терапевтических соединений, такое как, например, салицилатное производное, п-аминофенольное производное, производное пропионовой кислоты, производное уксусной кислоты, производное эноловой кислоты, производное фенамовой кислоты, неселективный ингибитор циклооксигеназы (COX), селективный ингибитор циклооксигеназы 1 (COX 1), селективный ингибитор циклооксигеназы 2 (COX 2) или фибрат, может быть включено в фармацевтическую композицию и введено пациенту, как описано выше.
В заключение, следует иметь в виду, что, несмотря на то, что аспекты настоящего изобретения иллюстрируются путем ссылки на конкретные варианты осуществления, тем не менее, для специалистов в данной области техники является очевидным, что эти раскрытые варианты осуществления являются только иллюстрацией сущности раскрытого предмета изобретения. Поэтому, следует иметь в виду, что раскрытый предмет изобретения никоим образом не ограничивается описанными в настоящем изобретении конкретной методикой, протоколом и/или реагентом и так далее. В связи с этим, могут быть сделаны различные модификации или изменения или альтернативных конфигураций раскрытого предмета изобретения в соответствии с идеями изобретения без отступления от сущности настоящего изобретения. И наконец, используемая в изобретении терминология предназначена только для описания конкретных вариантов осуществления, а не для ограничения объема настоящего изобретения, который определяется исключительно только формулой изобретения. Соответственно, настоящее изобретение не ограничивается тем, что конкретно продемонстрировано и описано.
В настоящем изобретении описаны конкретные варианты осуществления, в том числе наилучший способ осуществления изобретения, известный его авторам. Несомненно, что, после ознакомления с приведенным выше описанием, для специалистов в данной области будет очевидна возможность внесения изменений в эти описанные варианты осуществления. Авторы изобретения предполагают, что квалифицированные специалисты могут вносить такие изменения, когда это целесообразно, и авторы не исключают того, что настоящее изобретение может быть осуществлено иначе, чем конкретно описано в настоящем изобретении. Соответственно, это изобретение включает все модификации и эквиваленты предмета изобретения, изложенного в пунктах прилагаемой формулы изобретения, в соответствии с действующим законодательством. Кроме того, любая комбинация описанных выше вариантов осуществления со всеми возможными их изменениями входит в объем изобретения, если в изобретении не указано иное или если это явно не противоречит контексту. Объединение в группы альтернативных вариантов осуществления, элементов или стадий настоящего изобретения не должно истолковываться в качестве ограничений. Каждый представитель группы может быть указан и заявлен по отдельности или в любой комбинации с другими раскрытыми в изобретении представителями группы. Предполагается, что один или несколько представителей группы могут быть включены или удалены из группы из соображений удобства и/или патентоспособности. Когда производится такое включение или удаление, то считают, что описание содержит группу, модифицированную так, что она удовлетворяет письменному описанию всех групп Маркуша, используемых в приложенной формуле изобретения.
Если не указано иное, то все используемые в настоящем изобретении и формуле изобретения числа, выражающие характеристику, предмет, количество, параметр, свойство, термин и так далее, следует воспринимать во всех случаях в качестве модифицированных с помощью термина "приблизительно". Используемый в изобретении термин "приблизительно" означает, что численно определяемые характеристика, предмет, количество, параметр, свойство или термин, охватывают диапазон плюс или минус десять процентов выше и ниже указанного значения для характеристики, предмета, количества, параметра, свойства или термина. Соответственно, если не указано иное, то числовые параметры, приведенные в описании и прилагаемой формуле изобретения, являются приближенными значениями, которые могут изменяться. По крайней мере, и не в качестве попытки ограничить применение теории эквивалентов к объему формулы изобретения, каждое указание в числовой форме должно, по меньшей мере, истолковываться в свете количества приведенных значащих цифр и с применением обычных методов округления. Несмотря на то, что числовые диапазоны и значения, устанавливающие широкие границы объема изобретения, являются приближенными, тем не менее, числовые диапазоны и значения, установленные в конкретных примерах, приводятся как можно точнее. Однако любой численный диапазон или значение по своей природе содержит определенные ошибки, являющиеся неизбежным результатом стандартного отклонения, обнаруживаемого при их соответствующих тестовых измерениях. Перечисление в изобретении числовых диапазонов значений предназначено лишь для того, чтобы кратко индивидуально обозначить каждое отдельное численное значение, находящееся внутри диапазона. Если не указано иное, то каждое отдельное значение числового диапазона включено в объем настоящего изобретения, как если бы оно было индивидуально указано в изобретении.
Следует считать, что формы единственного числа, используемые в контексте описания настоящего изобретения (в частности, в контексте следующей далее формулы изобретения), охватывают как единственное, так и множественное число, если не указано иное в настоящем изобретении или это явно не противоречит контексту. Все описанные в изобретении способы могут быть осуществлены в любом подходящем порядке, если не указано иное или это явно не противоречит контексту. Использование любых примеров или пояснительных слов (например, "такой как") в настоящем изобретении предназначено только для лучшего раскрытия настоящего изобретения, и они не ограничивают объем настоящего изобретения, если не заявлено иное. Ни одна формулировка в настоящем изобретении не должно истолковываться как указание на любой незаявленный элемент, необходимый для практического осуществления изобретения.
Раскрытые в изобретении конкретные варианты осуществления могут быть дополнительно ограничены в формуле изобретения путем использования формулировок "состоящий из" или "состоящий в основном из". Использование в формуле изобретения, которая была зарегистрирована или в которую были внесены поправки, связывающего термина "состоящий из" исключает какой-либо элемент, стадию или ингредиент, не указанные в формуле изобретения. Связывающий термин "состоящий в основном из" ограничивает объем формулы изобретения в отношении указанных материалов или стадий и тех параметров, которые существенно не влияют на основную и новую характеристику (характеристики). Поэтому, заявленные варианты осуществления настоящего изобретения по существу или в явном виде описаны и включены в настоящее изобретение.
Содержание патентов, патентных публикаций и других публикаций, на которые ссылаются и которые указаны в тексте, приводится в настоящем изобретении путем ссылки на них, с целью описания и раскрытия, например, композиций и методик, описанных в таких публикациях, которые могут быть использованы в связи с настоящим изобретением. Эти публикации приводятся исключительно для их раскрытия до даты регистрации настоящей заявки. В этом отношении, ничто не должно быть истолковано в качестве признания того, что авторы изобретения не имеют права относить к более ранней дате такое изобретение ввиду предшествующего изобретения или по любой другой причине. Все констатации, касающиеся даты или представлений, так же как и содержания этих документов, основываются на информации, доступной для заявителей, и они не представляют собой какого-либо подтверждения в правильности дат или содержания этих документов.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ ТВЕРДЫХ РАСТВОРОВ И ИХ ПРИМЕНЕНИЕ ПРИ ХРОНИЧЕСКОМ ВОСПАЛЕНИИ | 2014 |
|
RU2675241C2 |
| КОМПОЗИЦИИ ТВЕРДЫХ РАСТВОРОВ И ИХ ПРИМЕНЕНИЕ ПРИ СЕРДЕЧНО-СОСУДИСТОМ ЗАБОЛЕВАНИИ | 2014 |
|
RU2674982C2 |
| КОМПОЗИЦИИ ТВЕРДЫХ РАСТВОРОВ И ИХ ПРИМЕНЕНИЕ ПРИ СИЛЬНОЙ БОЛИ | 2014 |
|
RU2667977C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2680801C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2635188C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ СИЛЬНОЙ БОЛИ | 2014 |
|
RU2677346C2 |
| ПЭГИЛИРОВАННАЯ ЛИПИДНАЯ НАНОЧАСТИЦА С БИОАКТИВНЫМ ЛИПОФИЛЬНЫМ СОЕДИНЕНИЕМ | 2016 |
|
RU2737893C2 |
| КОМПОЗИЦИЯ ПРОЛЕКАРСТВА ДЛЯ БОРЬБЫ С ВИРУСОМ ГЕПАТИТА С | 2006 |
|
RU2435592C2 |
| КОМПОЗИЦИЯ В ФОРМЕ МАЗИ, СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ ВИТАМИНА D | 2007 |
|
RU2448713C2 |
| СТАБИЛЬНЫЕ ПРОТИВОВОСПАЛИТЕЛЬНЫЕ РАСТВОРЫ ДЛЯ ИНЪЕКЦИИ | 2012 |
|
RU2635521C2 |
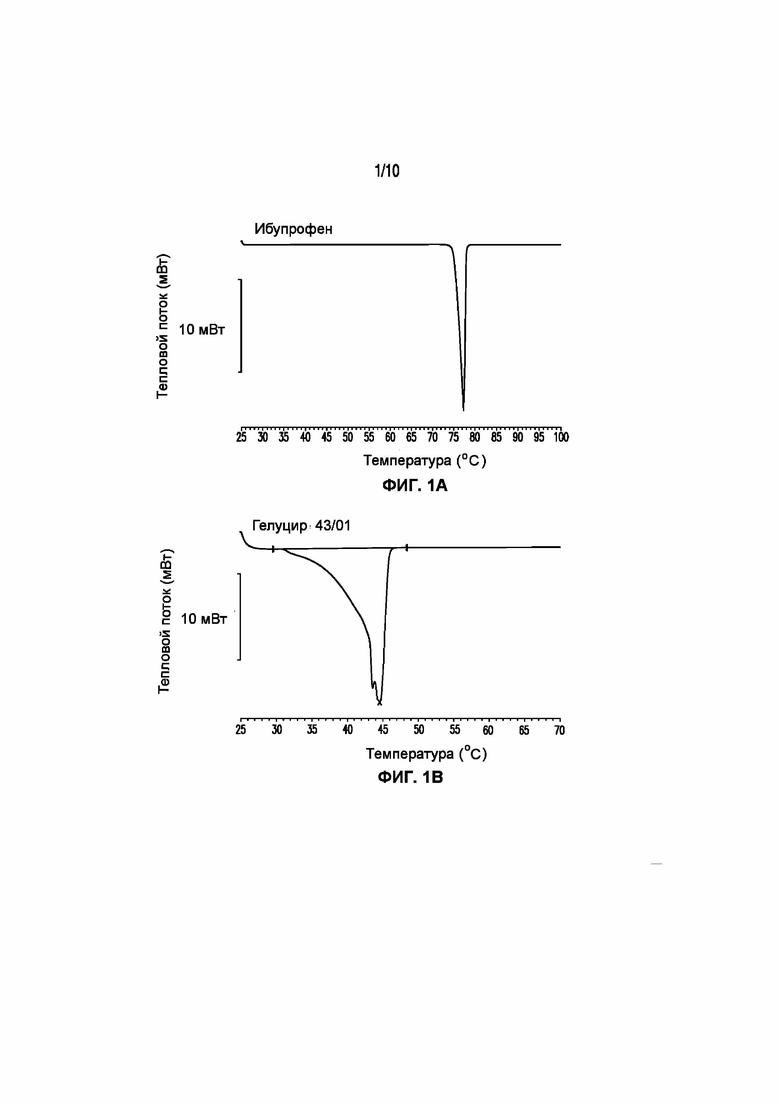
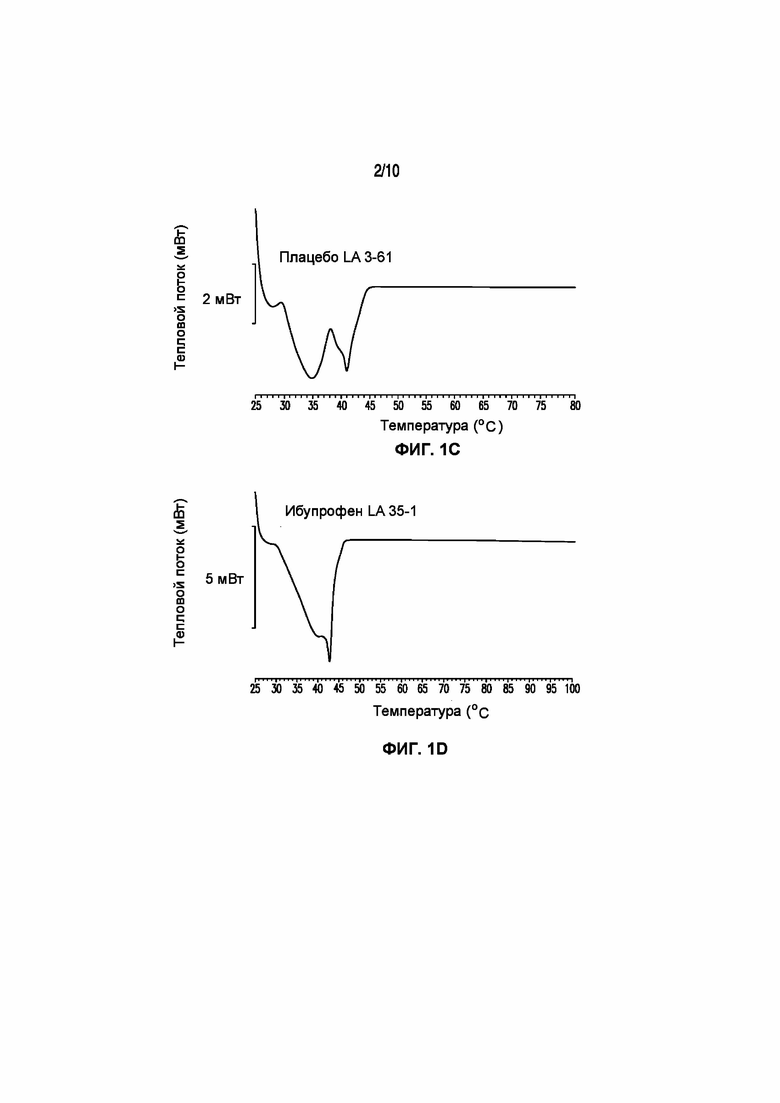
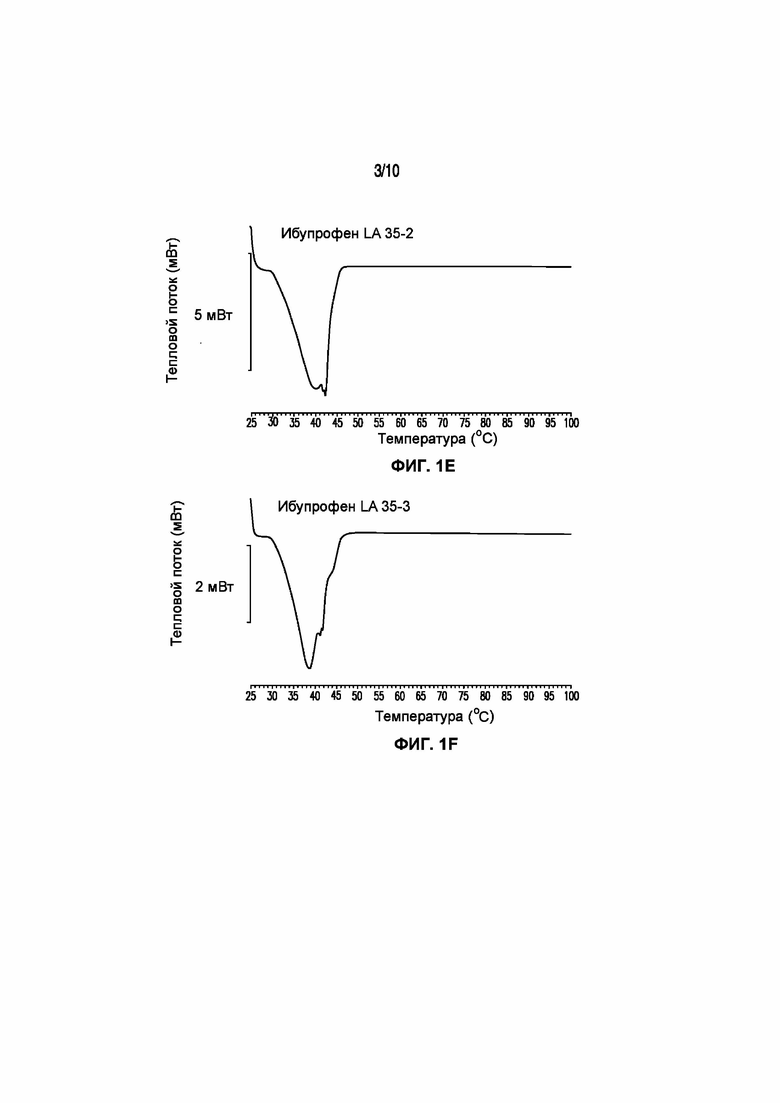
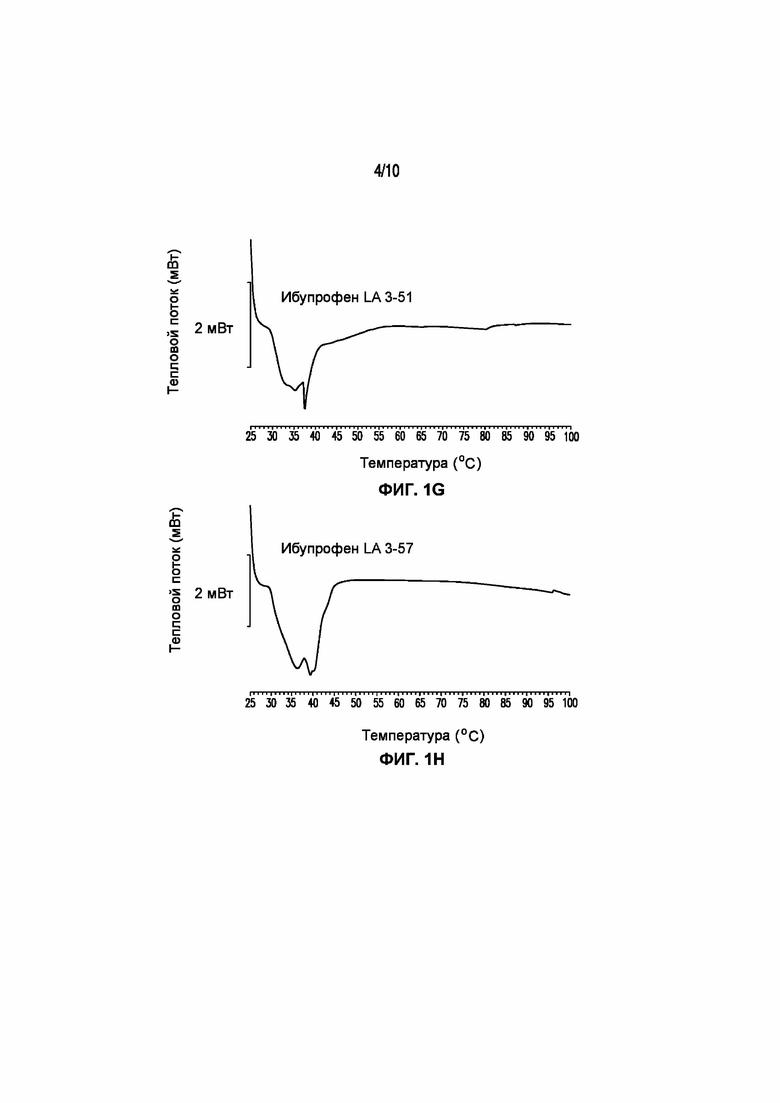
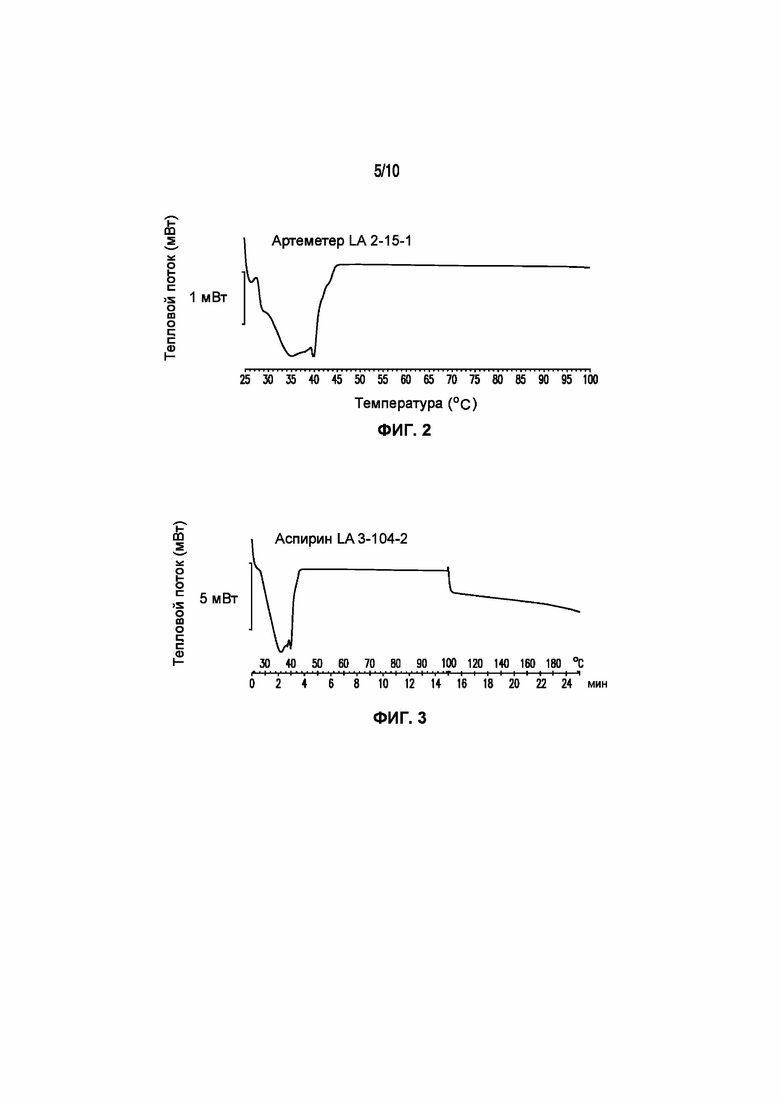
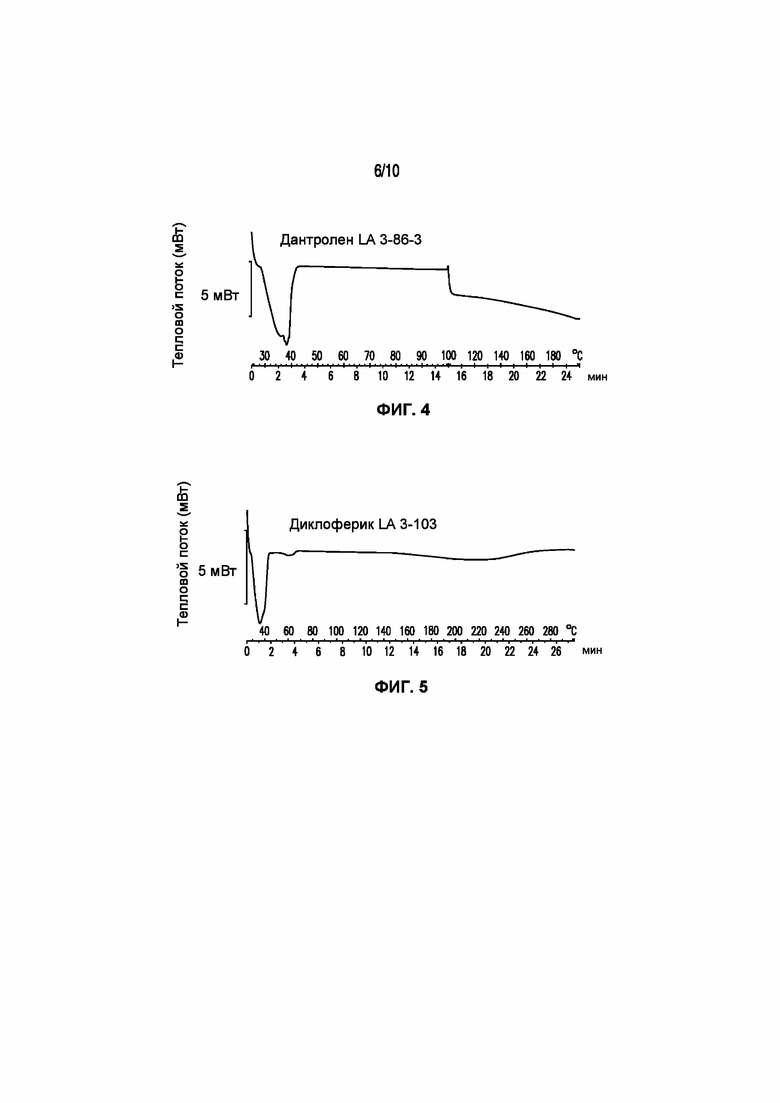
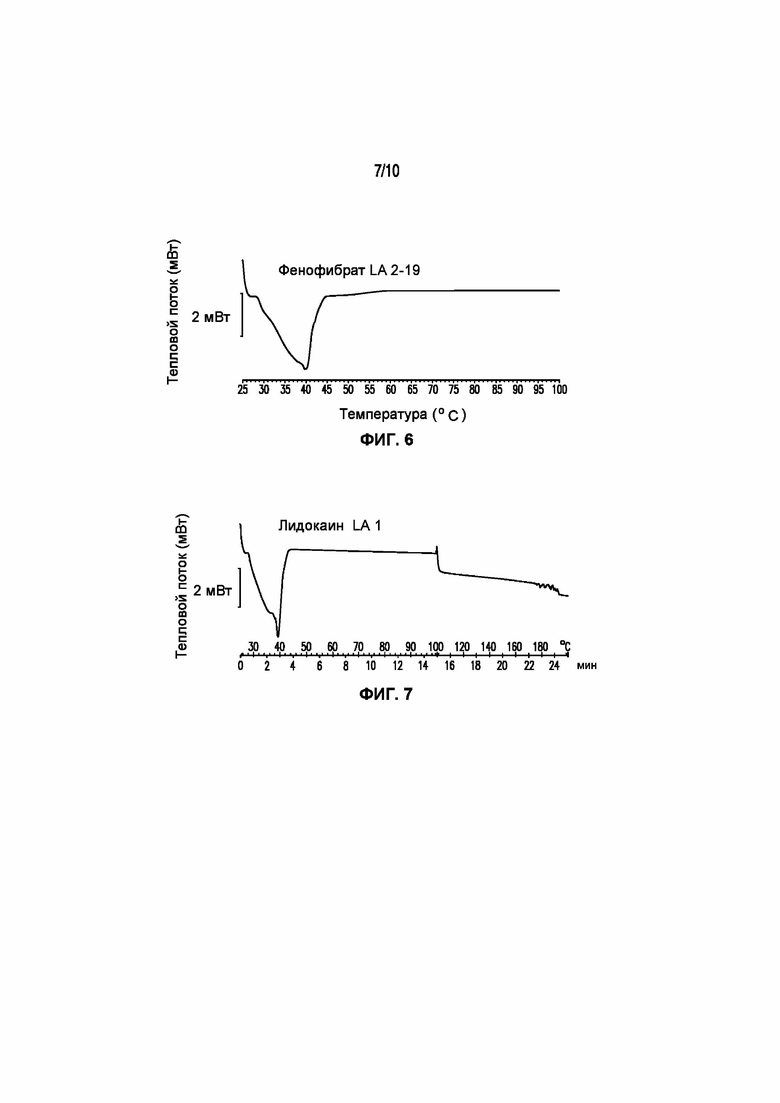
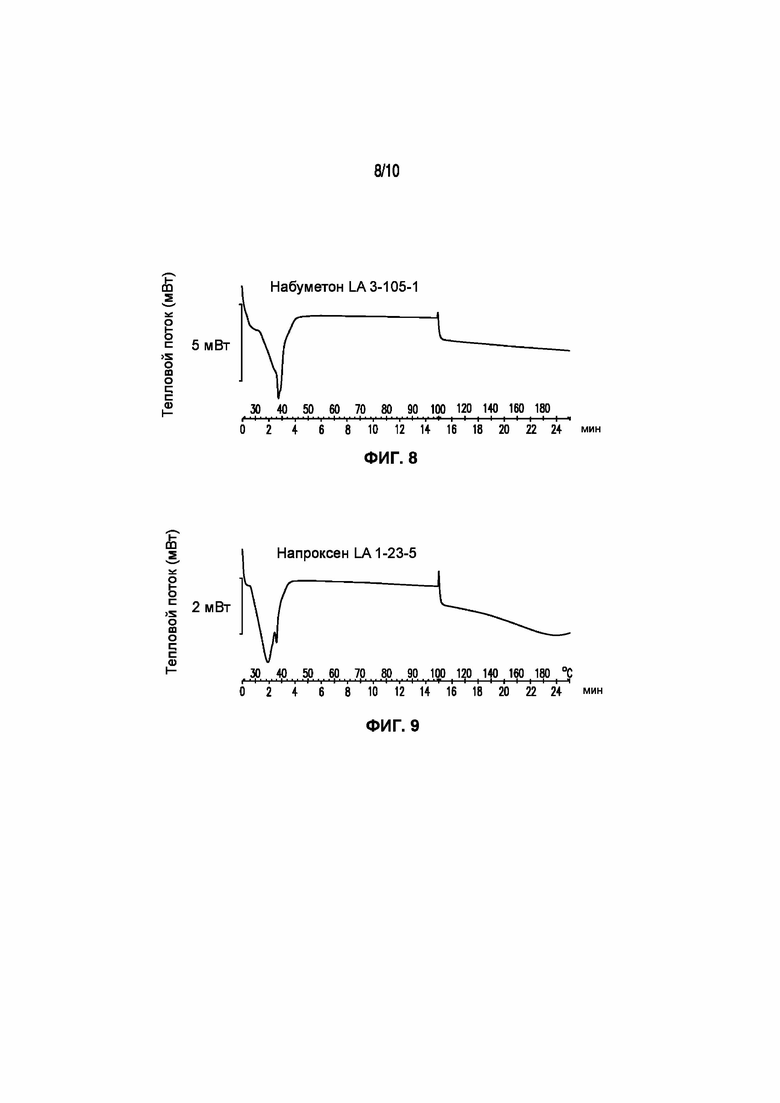
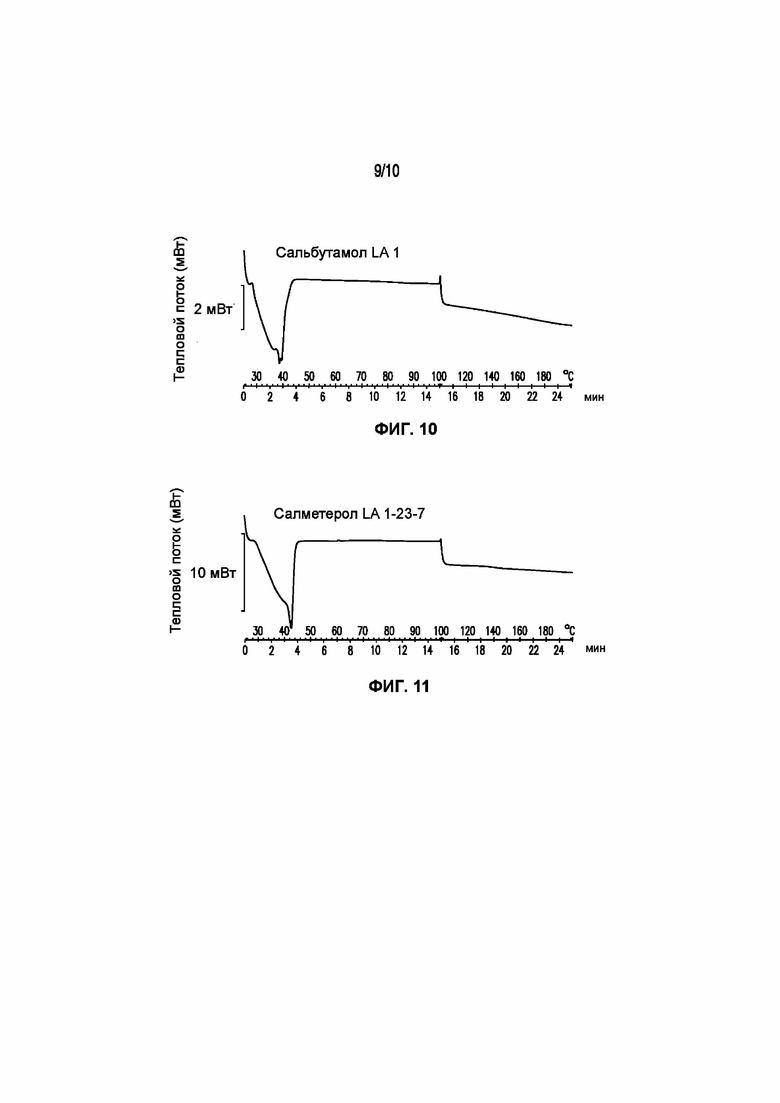
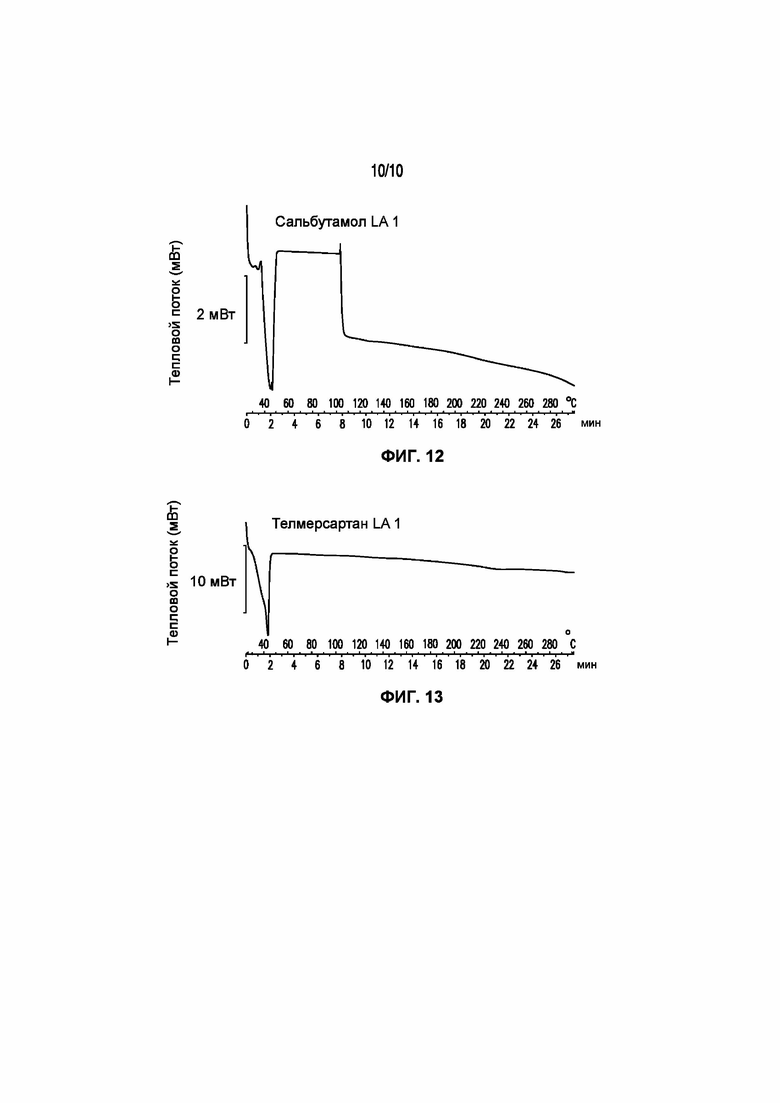
Группа изобретений относится к области медицины и раскрывает фармацевтическую композицию твердого раствора, обладающую противовоспалительной активностью, состоящую из: a) одного или более нестероидных противовоспалительных средств (NSAID) в количестве от 20 до 40% по массе (масс./масс.) от массы фармацевтической композиции; b) одного или более фармацевтически приемлемых твердых жиров в количестве от 20 до 65% по массе (масс./масс.) от массы фармацевтической композиции, где один или более твердых жиров включают один или более глицеролипидов; и c) одного или более фармацевтически приемлемых жидких жиров в количестве от 5 до 50% по массе (масс./об.) от массы фармацевтической композиции, где один или более жидких жиров включают смесь одного или более частично гидролизованного жира, включающего один или более моноглицеридов или ацетилированных моноглицеридов; d) один или более стабилизаторов, при этом общее количество компонентов (a)-(d) составляет до 100% по массе композиции твердого раствора, и где фармацевтическая композиция твердого раствора приготовлена так, что она является твердой при температуре 15°C или ниже и имеет температуру плавления 30°C или выше. Также группа изобретений относится к применению фармацевтической композиции твердого раствора (варианты) и к способу лечения индивидуума, страдающего хроническим воспалением. Техническим результатом группы изобретений является обеспечение системы доставки липид-адъювант, позволяющей доставить обладающее противовоспалительной активностью терапевтическое соединение таким образом, чтобы оно более эффективно ингибировало ответную провоспалительную реакцию. Итогом является улучшение результатов лечения хронического воспаления. 4 н. и 32 з.п. ф-лы, 6 табл., 22 пр., 13 ил.
1. Фармацевтическая композиция твердого раствора, обладающая противовоспалительной активностью, состоящая из:
a) одного или более нестероидных противовоспалительных средств (NSAID) в количестве от 20 до 40% по массе (масс./масс.) от массы фармацевтической композиции;
b) одного или более фармацевтически приемлемых твердых жиров в количестве от 20 до 65% по массе (масс./масс.) от массы фармацевтической композиции, где один или более твердых жиров включают один или более глицеролипидов; и
c) одного или более фармацевтически приемлемых жидких жиров в количестве от 5 до 50% по массе (масс./об.) от массы фармацевтической композиции, где один или более жидких жиров включают смесь одного или более частично гидролизованного жира, включающего один или более моноглицеридов или ацетилированных моноглицеридов;
d) один или более стабилизаторов,
при этом общее количество компонентов (a)-(d) составляет до 100% по массе композиции твердого раствора, и
где фармацевтическая композиция твердого раствора приготовлена так, что она является твердой при температуре 15°C или ниже и имеет температуру плавления 30°C или выше.
2. Фармацевтическая композиция твердого раствора по п.1, где количество одного или более NSAID составляет от 25 до 35% по массе (масс./масс.) от массы фармацевтической композиции.
3. Фармацевтическая композиция твердого раствора по п.1, где одно или более NSAID включают неселективный ингибитор циклооксигеназы, селективный ингибитор циклооксигеназы 1, селективный ингибитор циклооксигеназы 2 или любую их комбинацию.
4. Фармацевтическая композиция твердого раствора по п.3, где одно или более NSAID включают фармацевтически приемлемую форму салицилатного производного, фармацевтически приемлемую форму п-аминофенольного производного, фармацевтически приемлемую форму производного пропионовой кислоты, фармацевтически приемлемую форму производного уксусной кислоты, фармацевтически приемлемую форму производного эноловой кислоты, фармацевтически приемлемую форму производного фенамовой кислоты, эфира NSAID или любую их комбинацию.
5. Фармацевтическая композиция твердого раствора по п.4, где производное пропионовой кислоты включает фармацевтически приемлемую форму альминопрофена, фармацевтически приемлемую форму беноксапрофена, фармацевтически приемлемую форму декскетопрофена, фармацевтически приемлемую форму фенопрофена, фармацевтически приемлемую форму флурбипрофена, фармацевтически приемлемую форму ибупрофена, фармацевтически приемлемую форму индопрофена, фармацевтически приемлемую форму кетопрофена, фармацевтически приемлемую форму локсопрофена, фармацевтически приемлемую форму напроксена, фармацевтически приемлемую форму оксапрозина, фармацевтически приемлемую форму пранопрофена или фармацевтически приемлемую форму супрофена.
6. Фармацевтическая композиция твердого раствора по п.5, где производное пропионовой кислоты представляет собой фармацевтически приемлемую форму ибупрофена.
7. Фармацевтическая композиция твердого раствора по п.1, где количество одного или более фармацевтически приемлемых твердых жиров составляет от 35 до 45% по массе (масс./масс.) от массы фармацевтической композиции.
8. Фармацевтическая композиция твердого раствора по п.1, где один или более фармацевтически приемлемых твердых жиров имеют температуру плавления от 40 до 50°C.
9. Фармацевтическая композиция твердого раствора по п.1, где один или более глицеролипидов включают триглицерид с одной насыщенной или ненасыщенной жирной кислотой, имеющей длину углеродной цепи C12-C24, двумя насыщенными или ненасыщенными жирными кислотами, каждая из которых имеет длину углеродной цепи C12-C24, или тремя насыщенными или ненасыщенными жирными кислотами, каждая из которых имеет длину углеродной цепи C12-C24.
10. Фармацевтическая композиция твердого раствора по п.1, где один или более глицеролипидов включают смесь насыщенных C10-C18 триглицеридов, имеющих температуру плавления в диапазоне от 41 до 45°C.
11. Фармацевтическая композиция твердого раствора по п.1, где количество одного или более фармацевтически приемлемых жидких жиров составляет от 8 до 30% по массе (масс./об.) от массы фармацевтической композиции.
12. Фармацевтическая композиция твердого раствора по п.11, где количество одного или более фармацевтически приемлемых жидких жиров составляет менее чем 25% по массе (масс./об.) от массы фармацевтической композиции.
13. Фармацевтическая композиция твердого раствора по п.1, где один или более фармацевтически приемлемых моноглицеридов или ацетилированных моноглицеридов включают глицерина мономиристолеат, глицерина монопальмитолеат, глицерина моносапиенат, глицерина моноолеат, глицерина моноэлаидат, глицерина моновакценат, глицерина монолинолеат, глицерина монолиноэлаидат, глицерина монолиноленат, глицерина моностеаридонат, глицерина моноэйкозеноат, глицерина мономеадат, глицерина моноарахидонат, глицерина моноэйкозапентаеноат, глицерина моноэрукат, глицерина монодокозагексаеноат, глицерина мононервонат.
14. Фармацевтическая композиция твердого раствора по п.13, где один или более фармацевтически приемлемых моноглицеридов или ацетилированных моноглицеридов включают глицерина монолинолеат.
15. Фармацевтическая композиция твердого раствора по п.1, где один или более фармацевтически приемлемых жидких жиров дополнительно содержат диглицерид.
16. Фармацевтическая композиция твердого раствора по п.1, где один или более стабилизаторов включают жидкий полимер гликоля, одноатомный спирт, диметиловый эфир изосорбида и моноэтиловый эфир диэтиленгликоля (2-(2-этоксиэтокси)этанол).
17. Фармацевтическая композиция твердого раствора по п.16, где жидкий полимер гликоля представляет собой жидкий полимер полиэтиленгликоля (ПЭГ) и/или жидкий полимер полипропиленгликоля (ППГ).
18. Фармацевтическая композиция твердого раствора по п.17, где жидкий полимер гликоля составляет количество от 5 до 15% по массе (масс./об.) от массы фармацевтической композиции.
19. Фармацевтическая композиция твердого раствора по п.18, где жидкий полимер гликоля составляет количество от 9 до 12% по массе (масс./об.) от массы фармацевтической композиции.
20. Фармацевтическая композиция твердого раствора по п.17, где молекулярная масса полимера ПЭГ составляет не более чем 1000 г/моль.
21. Фармацевтическая композиция твердого раствора по п.20, где полимер ПЭГ включает ПЭГ 100, ПЭГ 200, ПЭГ 300, ПЭГ 400, ПЭГ 500, ПЭГ 600, ПЭГ 700, ПЭГ 800, ПЭГ 900, ПЭГ 1000 или любую их комбинацию.
22. Применение фармацевтической композиции твердого раствора по любому одному из пп.1-21 в производстве лекарственного препарата для лечения хронического воспаления.
23. Применение по п.22, где хроническое воспаление связано с угрями, кислотным рефлюксом/изжогой, возрастной дегенерацией желтого пятна (AMD), аллергией, аллергическим ринитом, болезнью Альцгеймера, боковым амиотрофическим склерозом, анемией, аппендицитом, артериитом, артритом, астмой, атеросклерозом, аутоиммунными заболеваниями, баланитом, блефаритом, бронхиолитом, бронхитом, буллезным пемфигоидом, ожогом, бурситом, раком, задержкой очередного сердечного сокращения, кардитом, глютенчувствительной целиакией, целлюлитом, цервицитом, холангитом, холециститом, хориоамнионитом, хроническим обструктивным заболеванием легких (COPD), циррозом, колитом, хронической сердечной недостаточностью, конъюнктивитом, индуцированным циклофосфамидом циститом, кистозным фиброзом, циститом, простудой, дакриоаденитом, деменцией, дерматитом, дерматомиозитом, диабетом, диабетической нейропатией, диабетической ретинопатией, диабетической нефропатией, диабетическим изъязвлением, заболеванием системы пищеварения, экземой, эмфиземой, энцефалитом, эндокардитом, эндометритом, энтеритом, энтероколитом, эпикондилитом, эпидидимитом, фасциитом, фибромиалгией, фиброзом, фиброзитом, гастритом, гастроэнтеритом, гингивитом, гломерулонефритом, глосситом, заболеванием сердца, дисфункцией сердечного клапана, гепатитом, гнойным гидраденитом, болезнью Гентингтона, гиперлипидемическим панкреатитом, гипертензией, илеитом, инфекцией, воспалительным заболеванием кишечника, воспалительной кардиомегалией, воспалительной невропатией, резистентностью к инсулину, интерстициальным циститом, интерстициальным нефритом, иритом, ишемией, ишемическим заболеванием сердца, кератитом, кератоконъюнктивитом, ларингитом, волчаночным нефритом, маститом, мастоидитом, менингитом, метаболическим синдромом (синдромом X), мигренью, множественным склерозом, миелитом, миокардитом, миозитом, нефритом, неалкогольным стеатогепатитом, ожирением, омфалитом, оофоритом, орхитом, остеохондритом, остеопенией, остеомиелитом, остеопорозом, оститом, отитом, панкреатитом, болезнью Паркинсона, паротитом, воспалительным заболеванием тазовых органов, обыкновенной пузырчаткой, перикардитом, пиритонитом, фарингитом, флебитом, плевритом, пневмонией, поликистозным нефритом, проктитом, простатитом, псориазом, пульпитом, пиелонефритом, пилефлебитом, почечной недостаточностью, реперфузионным повреждением, ретинитом, острой ревматической лихорадкой, ринитом, сальпингитом, саркоидозом, сиаладенитом, синуситом, синдромом раздраженной толстой кишки, стенозом, стоматитом, инсультом, послеоперационным осложнением, синовитом, тендинитом, тендинозом, теносиновитом, тромбофлебитом, тонзиллитом, травмой, травматическим повреждением головного мозга, отторжением трансплантата, тригонитом, туберкулезом, опухолью, уретритом, бурситом, увеитом, вагинитом, васкулитом или вульвитом.
24. Применение по п.22, где хроническое воспаление представляет собой воспаление ткани или системное воспаление.
25. Применение по п.22, где хроническое воспаление представляет собой аутоиммунное заболевание или неаутоиммунное заболевание.
26. Применение по п.22, где хроническое воспаление связано с артритом, миопатией, васкулитом, кожным заболеванием, желудочно-кишечным заболеванием, сердечно-сосудистым заболеванием, раком, воспалением, вызванным приемом лекарственного средства, инфекцией, повреждением ткани или органа, отторжением трансплантата, реакцией "трансплантант против хозяина", Th1-опосредованным воспалительным заболеванием, хроническим нейрогенным воспалением.
27. Применение фармацевтической композиции твердого раствора по любому одному из пп.1-21 для лечения хронического воспаления.
28. Применение по п.27, где хроническое воспаление связано с угрями, кислотным рефлюксом/изжогой, возрастной дегенерацией желтого пятна (AMD), аллергией, аллергическим ринитом, болезнью Альцгеймера, боковым амиотрофическим склерозом, анемией, аппендицитом, артериитом, артритом, астмой, атеросклерозом, аутоиммунными заболеваниями, баланитом, блефаритом, бронхиолитом, бронхитом, буллезным пемфигоидом, ожогом, бурситом, раком, задержкой очередного сердечного сокращения, кардитом, глютенчувствительной целиакией, целлюлитом, цервицитом, холангитом, холециститом, хориоамнионитом, хроническим обструктивным заболеванием легких (COPD), циррозом, колитом, хронической сердечной недостаточностью, конъюнктивитом, индуцированным циклофосфамидом циститом, кистозным фиброзом, циститом, простудой, дакриоаденитом, деменцией, дерматитом, дерматомиозитом, диабетом, диабетической нейропатией, диабетической ретинопатией, диабетической нефропатией, диабетическим изъязвлением, заболеванием системы пищеварения, экземой, эмфиземой, энцефалитом, эндокардитом, эндометритом, энтеритом, энтероколитом, эпикондилитом, эпидидимитом, фасциитом, фибромиалгией, фиброзом, фиброзитом, гастритом, гастроэнтеритом, гингивитом, гломерулонефритом, глосситом, заболеванием сердца, дисфункцией сердечного клапана, гепатитом, гнойным гидраденитом, болезнью Гентингтона, гиперлипидемическим панкреатитом, гипертензией, илеитом, инфекцией, воспалительным заболеванием кишечника, воспалительной кардиомегалией, воспалительной невропатией, резистентностью к инсулину, интерстициальным циститом, интерстициальным нефритом, иритом, ишемией, ишемическим заболеванием сердца, кератитом, кератоконъюнктивитом, ларингитом, волчаночным нефритом, маститом, мастоидитом, менингитом, метаболическим синдромом (синдромом X), мигренью, множественным склерозом, миелитом, миокардитом, миозитом, нефритом, неалкогольным стеатогепатитом, ожирением, омфалитом, оофоритом, орхитом, остеохондритом, остеопенией, остеомиелитом, остеопорозом, оститом, отитом, панкреатитом, болезнью Паркинсона, паротитом, воспалительным заболеванием тазовых органов, обыкновенной пузырчаткой, перикардитом, перитонитом, фарингитом, флебитом, плевритом, пневмонией, поликистозным нефритом, проктитом, простатитом, псориазом, пульпитом, пиелонефритом, пилефлебитом, почечной недостаточностью, реперфузионным повреждением, ретинитом, острой ревматической лихорадкой, ринитом, сальпингитом, саркоидозом, сиаладенитом, синуситом, синдромом раздраженной толстой кишки, стенозом, стоматитом, инсультом, послеоперационным осложнением, синовитом, тендинитом, тендинозом, теносиновитом, тромбофлебитом, тонзиллитом, травмой, травматическим повреждением головного мозга, отторжением трансплантата, тригонитом, туберкулезом, опухолью, уретритом, бурситом, увеитом, вагинитом, васкулитом или вульвитом.
29. Применение по п.27, где хроническое воспаление представляет собой воспаление ткани или системное воспаление.
30. Применение по п.27, где хроническое воспаление представляет собой аутоиммунное заболевание или неаутоиммунное заболевание.
31. Применение по п.27, где хроническое воспаление связано с артритом, миопатией, васкулитом, кожным заболеванием, желудочно-кишечным заболеванием, сердечно-сосудистым заболеванием, раком, воспалением, вызванным приемом лекарственного средства, инфекцией, повреждением ткани или органа, отторжением трансплантата, реакцией "трансплантант против хозяина", Th1-опосредованным воспалительным заболеванием, хроническим нейрогенным воспалением.
32. Способ лечения индивидуума, страдающего хроническим воспалением, где способ включает стадии введения индивидууму терапевтически эффективного количества фармацевтической композиции твердого раствора по любому из пп.1-21, где введение приводит в результате к уменьшению интенсивности симптома, связанного с хроническим воспалением, вследствие чего происходит лечение индивидуума.
33. Способ по п.32, где хроническое воспаление связано с угрями, кислотным рефлюксом/изжогой, возрастной дегенерацией желтого пятна (AMD), аллергией, аллергическим ринитом, болезнью Альцгеймера, боковым амиотрофическим склерозом, анемией, аппендицитом, артериитом, артритом, астмой, атеросклерозом, аутоиммунными заболеваниями, баланитом, блефаритом, бронхиолитом, бронхитом, буллезным пемфигоидом, ожогом, бурситом, раком, задержкой очередного сердечного сокращения, кардитом, глютенчувствительной целиакией, целлюлитом, цервицитом, холангитом, холециститом, хориоамнионитом, хроническим обструктивным заболеванием легких (COPD), циррозом, колитом, хронической сердечной недостаточностью, конъюнктивитом, индуцированным циклофосфамидом циститом, кистозным фиброзом, циститом, простудой, дакриоаденитом, деменцией, дерматитом, дерматомиозитом, диабетом, диабетической нейропатией, диабетической ретинопатией, диабетической нефропатией, диабетическим изъязвлением, заболеванием системы пищеварения, экземой, эмфиземой, энцефалитом, эндокардитом, эндометритом, энтеритом, энтероколитом, эпикондилитом, эпидидимитом, фасциитом, фибромиалгией, фиброзом, фиброзитом, гастритом, гастроэнтеритом, гингивитом, гломерулонефритом, глосситом, заболеванием сердца, дисфункцией сердечного клапана, гепатитом, гнойным гидраденитом, болезнью Гентингтона, гиперлипидемическим панкреатитом, гипертензией, илеитом, инфекцией, воспалительным заболеванием кишечника, воспалительной кардиомегалией, воспалительной невропатией, резистентностью к инсулину, интерстициальным циститом, интерстициальным нефритом, иритом, ишемией, ишемическим заболеванием сердца, кератитом, кератоконъюнктивитом, ларингитом, волчаночным нефритом, маститом, мастоидитом, менингитом, метаболическим синдромом (синдромом X), мигренью, множественным склерозом, миелитом, миокардитом, миозитом, нефритом, неалкогольным стеатогепатитом, ожирением, омфалитом, оофоритом, орхитом, остеохондритом, остеопенией, остеомиелитом, остеопорозом, оститом, отитом, панкреатитом, болезнью Паркинсона, паротитом, воспалительным заболеванием тазовых органов, обыкновенной пузырчаткой, перикардитом, перитонитом, фарингитом, флебитом, плевритом, пневмонией, поликистозным нефритом, проктитом, простатитом, псориазом, пульпитом, пиелонефритом, пилефлебитом, почечной недостаточностью, реперфузионным повреждением, ретинитом, острой ревматической лихорадкой, ринитом, сальпингитом, саркоидозом, сиаладенитом, синуситом, синдромом раздраженной толстой кишки, стенозом, стоматитом, инсультом, послеоперационным осложнением, синовитом, тендинитом, тендинозом, теносиновитом, тромбофлебитом, тонзиллитом, травмой, травматическим повреждением головного мозга, отторжением трансплантата, тригонитом, туберкулезом, опухолью, уретритом, бурситом, увеитом, вагинитом, васкулитом или вульвитом.
34. Способ по п.32, где хроническое воспаление представляет собой воспаление ткани или системное воспаление.
35. Способ по п.32, где хроническое воспаление представляет собой аутоиммунное заболевание или неаутоиммунное заболевание.
36. Способ по п.32, где хроническое воспаление связано с артритом, миопатией, васкулитом, кожным заболеванием, желудочно-кишечным заболеванием, сердечно-сосудистым заболеванием, раком, воспалением, вызванным приемом лекарственного средства, инфекцией, повреждением ткани или органа, отторжением трансплантата, реакцией "трансплантант против хозяина", Th1-опосредованным воспалительным заболеванием, хроническим нейрогенным воспалением.
| WO 2012104655 A2, 09.08.2012 | |||
| WO 1992009272 A1, 11.06.1992 | |||
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНЕЙ СУСТАВОВ | 2007 |
|
RU2353349C2 |
| WO 2012056251 A1, 03.05.2012 | |||
| WO 2012104654 A1, 09.08.2012 | |||
| WO 2000067728 A2, 16.11.2000. | |||

