Перекрестная ссылка на родственные заявки
Данная заявка испрашивает приоритет предварительной заявки США № 62/430,183, поданной 5 декабря 2016 г., предварительной заявки США № 62/532,230, поданной 13 июля 2017, описание которых включено в настоящий документ посредством ссылки в полном объеме.
Область техники
Настоящее изобретение относится к классу производных пиримидина, которые имеют иммуномодулирующие свойства, которые действуют через TLR7, и которые полезны при лечении вирусных или аллергических заболеваний и раковых заболеваний.
Уровень техники
Настоящее изобретение относится к производным пиримидина, способам их получения, фармацевтическим композициям, содержащим их, и их применению в терапии.
Иммунная система состоит из врожденного и приобретенного иммунитета, оба из которых работают совместно, чтобы защитить хозяина от микробных инфекций. Было показано, что врожденный иммунитет может распознавать законсервированные патоген–ассоциированные молекулярные исходники через toll–подобные рецепторы (TLR), экспрессируемые на клеточной поверхности иммунных клеток. Распознавание патогенных микроорганизмов затем запускает выработку цитокинов (включая альфа–интерферон (IFNα)) и активацию костимулирующих молекул на фагоцитах, что приводит к модуляции функции Т–клеток. Таким образом, врожденный иммунитет тесно связан с приобретенным иммунитетом и может влиять на развитие и регуляцию приобретенного ответа.
TLR представляют собой семейство трансмембранных рецепторов типа I, характеризующихся NH2–терминальным внеклеточным богатым лейцином повторным доменом (LRR) и COOH–терминальным внутриклеточным хвостом, содержащим консервированный участок, называемый домен гомологии Toll/IL–1 рецептора (TIR). Внеклеточный домен содержит различное количество LRR, которые, как полагают, участвуют в связывании лиганда. На сегодняшний день были описаны у людей и мышей одиннадцать TLR. Они отличаются друг от друга специфичностью лигандов, профилями экспрессии и генами–мишенями, которые они могут индуцировать.
Были разработаны лиганды, которые действуют через TLR (также известные как модификаторы иммунного ответа (IRMS)), например, производные имидазохинолина, описанные в патенте США № 4689338, которые включают продукт Имиквимод для лечения генитальных бородавок, и производные аденина, описанные в WO 98/01448 и WO 99/28321.
Соединения со свойством нацеливания на печень являются желательными. Некоторые фрагменты, которые помогают в нацеливании на печень, которые были описаны в ссылках, включают кислотные фрагменты. (Tu et al., Current Topics in Medicinal Chemistry, 2013, 13, 857–866; Oballa et al., J. Med. Chem., 2011, 54, 5082–5096; Pfefferkorn et al., J. Med. Chem., 2012, 55, 1318–1333, содержание которых включено в данный документ посредством ссылки во всей их полноте).
Краткое описание сути изобретения
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
 (1)
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3,  ; и A,
; и A,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, , или A;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, 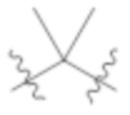 ,
, 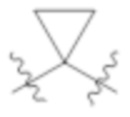 , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
, –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из  ,
,  ,
,  ,
,  ,
, 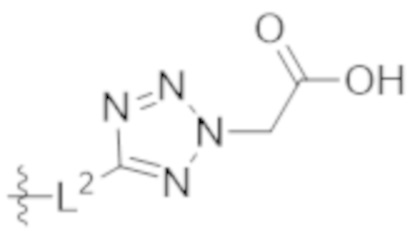 ,
,  , и
, и  ;
;
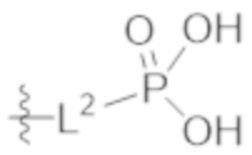
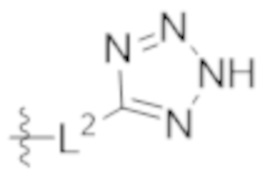
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , ,  , –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А.
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, и ,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, или ;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А.
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –COOH, и –SO2CH3, где алкил необязательно замещен –OH, –NH2, –COOH, или –SO2CH3;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А.
В некоторых вариантах реализации соединение, имеющее структуру Формулы (1), может иметь любой один или несколько из следующих признаков:
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда А не является –L2–COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда А не является –L2–COOH, кроме случаев, когда R1a содержит –COOH;
когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда Х не является –CH3;
когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда Х не является –CH3, кроме случаев, когда R1a содержит –COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является –CH2–;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является –CH2–, кроме случаев, когда R1a содержит –COOH;
когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–;
когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–, кроме случаев, когда R1a содержит –COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда R1a содержит COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; и R1a не содержит COOH; тогда A не представляет собой –L2–COOH.
В некоторых вариантах реализации соединение, имеющее структуру Формулы (1), может иметь любую одну или несколько из следующих признаков:
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда А не является –L2–COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда Х не является –CH3;
когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда X не является –CH3, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2не является –CH2–;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является –CH2–, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–;
когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда R1a содержит COOH или –SO2CH3;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; и R1a не содержит COOH или –SO2CH3; тогда A не представляет собой –L2–COOH;
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда R1a не является H или алкилом замещенным –OH.
В некоторых вариантах реализации соединение, имеющее структуру Формулы (1), может иметь любую одну или несколько из следующих признаков:
(aa) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(bb) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(cc) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; L2 представляет собой–CH2–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(dd) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(ee) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2(CF2)–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(ff) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; тогда A не представляет собой ;
(gg) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой ;
(hh) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой ;
(ii) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –O–(CH2)2–O(CH2)2–; тогда A не представляет собой ;
(jj) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –O–(CH2)2–O(CH2)2(CF2)–; тогда A не представляет собой ;
(kk) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(ll) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(mm) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; L2 представляет собой –CH2–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(nn) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(oo) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2(CF2)–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой .
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1а),
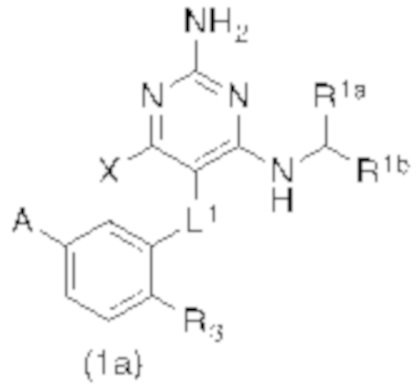
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–,  , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
, , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1а),
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–,, , и–CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1а),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из 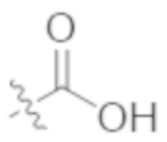 ,
, 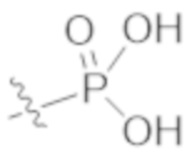 ,
, 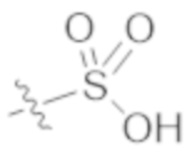 ,
, 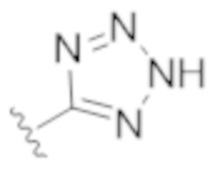 ,
, 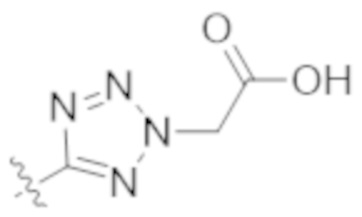 ,
,  , и
, и  ;
;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1а),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1b),
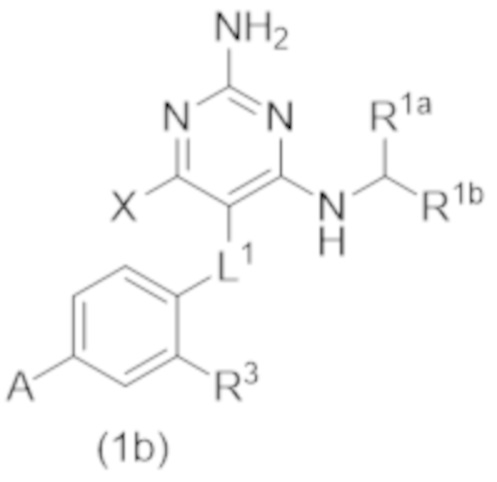
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1b),
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1b),
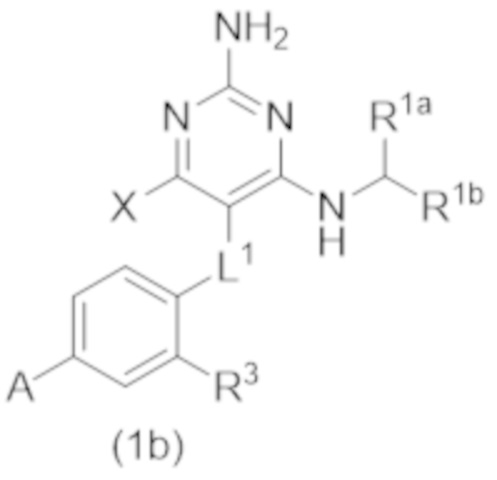
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1b),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1с),

где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1с),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1d),
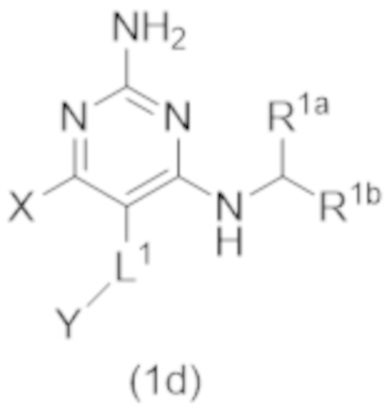
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –CH2CH2, и –S–; и
Y представляет собой H или C1–C3 алкил.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1d),
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1e),

где
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1f),
 .
.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1g),
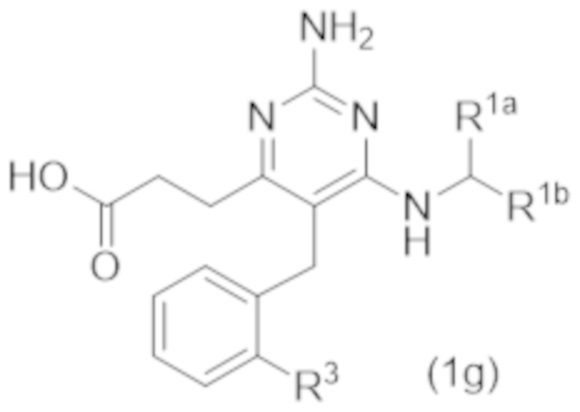
где
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1h),
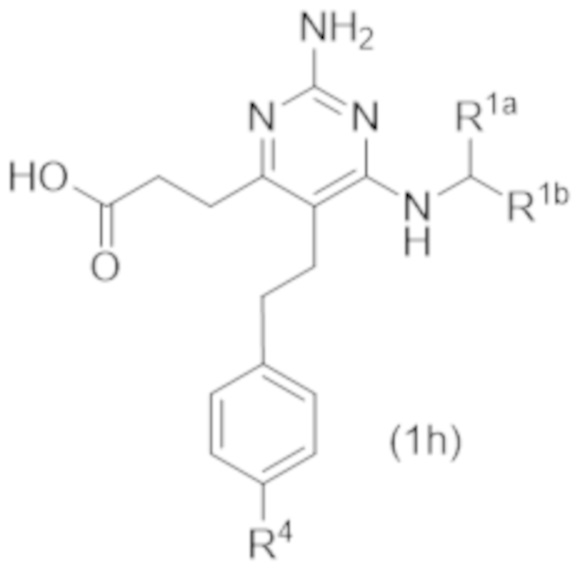
где
R4 представляет собой H или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1i),

где
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –CH2CH2–, и –S–; и
Y представляет собой H или C1–C3 алкил.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1i),
где
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1j),
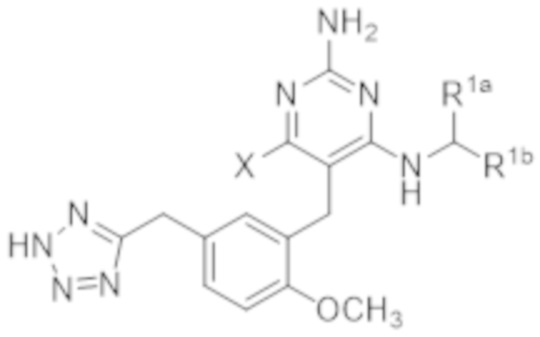 (1j).
(1j).
Настоящее изобретение относится к соединению Формулы (1), которое представляет собой соединение Формулы (1k),
 (1k),
(1k),
Настоящее описание относится к фармацевтическим композициям, содержащим соединение настоящего изобретения или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Настоящее описание относится к способам лечения состояния, связанного с модуляцией TLR7 у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. В некоторых вариантах реализации состояние представляет собой вирусную инфекцию или рак.
Настоящее описание относится к соединению настоящего изобретения или его фармацевтически приемлемой соли для применения при лечении состояния, связанного с модуляцией TLR7. В некоторых вариантах реализации состояние представляет собой вирусную инфекцию или рак.
Настоящее описание относится к использованию соединения настоящего описание или его фармацевтически приемлемой соли при изготовлении лекарственного средства для лечения состояния, связанного с модуляцией TLR7. В некоторых вариантах реализации состояние представляет собой вирусную инфекцию или рак.
Подробное описание сути изобретения
Хотя конкретные варианты реализации сути настоящего изобретения проиллюстрированы и описаны подробно, изобретение не ограничивается ими. Подробное описание приведено в качестве примера настоящего изобретения и не должно рассматриваться как составляющие какого–либо ограничения изобретения. Модификации будут очевидны для специалистов в данной области техники, и все модификации, которые не выходят за рамки сути изобретения, предназначены для включения в объем прилагаемой формулы изобретения.
Определения
Следующие определения предназначены для уточнения, а не для ограничения, определенных терминов. Если конкретный термин, используемый в данном документе, конкретно не определен, такой термин не следует считать неопределенным. Скорее, термины используются в их принятых значениях.
Используемый в данном документе термин «алкил» относится к насыщенному углеводороду с прямой или разветвленной цепью. Например, алкильная группа может иметь от 1 до 8 атомов углерода (т.e., (C1–C8)алкил) или от 1 до 6 атомов углерода (т.e., (C1–C6 алкил) или от 1 до 4 атомов углерода.
Используемый в данном документе термин «алкилен» относится к насыщенному углеводородному радикалу с прямой или разветвленной цепью, имеющему два одновалентных радикальных центра, полученных путем удаления двух атомов водорода из одинаковых или двух разных атомов углерода исходного алкана. Например, алкиленовая группа может иметь от 1 до 8 атомов углерода, от 1 до 6 атомов углерода или от 1 до 4 атомов углерода.
Используемый в данном документе термин «алкокси» относится к алкильной группе, присоединенной к исходному молекулярному фрагменту через атом кислорода.
Используемый в данном документе термин «гало» или «галоген» относится к фтору, хлору, брому и йоду.
Используемый в данном документе термин «арил» относится к единственному полностью углеродному ароматическому кольцу или множественной конденсированной полностью углеродной кольцевой системе, где по меньшей мере одно из колец является ароматическим. Например, арильная группа может иметь от 6 до 20 атомов углерода, от 6 до 14 атомов углерода или от 6 до 12 атомов углерода. Арил содержит фенильный радикал. Арил также содержит несколько конденсированных кольцевых систем (например, кольцевые системы, содержащие 2, 3 или 4 кольца), имеющих примерно от около 9 до 20 атомов углерода, в которых по меньшей мере одно кольцо является ароматическим и где другие кольца могут быть ароматическими или неароматическими (т. е. карбоцикл). Такие множественные конденсированные кольцевые системы могут быть необязательно замещены одной или несколькими (например, 1, 2 или 3) оксогруппами в любой карбоциклической части множественной конденсированной кольцевой системы. Кольца системы с несколькими конденсированными кольцами могут быть соединены друг с другом посредством конденсированных, спиро и мостиковых связей, если это допускается требованиями валентности. Следует понимать, что точка присоединения множественной конденсированной кольцевой системы, как определено выше, может находиться в любом положении кольцевой системы, включая ароматическую или карбоциклическую часть кольца.
Используемый в данном документе термин «гетероарил» относится к одному ароматическому кольцу, которое имеет по меньшей мере один атом, отличный от углерода в кольце, где атом выбирают из группы, состоящей из кислорода, азота и серы; термин также включает множество конденсированных кольцевых систем, которые имеют, по меньшей мере, одно такое ароматическое кольцо, которое для множества конденсированных кольцевых систем дополнительно описано ниже. Таким образом, термин включает отдельные ароматические кольца с около 1–6 атомами углерода и около 1–4 гетероатомами, выбранными из группы, состоящей из кислорода, азота и серы в кольцах. Атомы серы и азота также могут присутствовать в окисленной форме, при условии, что кольцо является ароматическим. Термин также включает несколько конденсированных кольцевых систем (например, кольцевые системы, содержащие 2, 3 или 4 кольца), в которых гетероарильная группа, как определено выше, может конденсироваться с одним или несколькими кольцами, выбранными из гетероарилов (с образованием, например, нафтиридинила, такого как 1,8–нафтиридинил), гетероциклов (с образованием, например, 1,2,3,4–тетрагидронафтиридинила, такого как 1,2,3,4–тетрагидро–1,8–нафтиридинил), карбоциклов (с образованием, например, 5,6,7,8–тетрагидрохинолила) и арилов (с образованием, например, индазолила), с образованием поликонденсированной кольцевой системы. Таким образом, гетероарил (одно ароматическое кольцо или система поликонденсированных колец) имеет около 1–20 атомов углерода и около 1–6 гетероатомов в гетероарильном кольце. Такие поликонденсированные кольцевые системы могут быть необязательно замещены одной или несколькими (например, 1, 2, 3 или 4) оксогруппами на карбоциклических или гетероциклических частях конденсированного кольца. Кольца системы с несколькими конденсированными кольцами могут быть соединены друг с другом посредством конденсированных, спиро и мостиковых связей, если это допускается требованиями валентности. Следует понимать, что отдельные кольца системы с множеством конденсированных колец могут быть соединены в любом порядке относительно друг друга. Также следует понимать, что точка присоединения поликонденсированной кольцевой системы (как определено выше для гетероарила) может находиться в любом положении поликонденсированной кольцевой системы, включая гетероарильную, гетероциклическую, арильную или карбоциклическую часть поликонденсированной кольцевой системы и у любого подходящего атома системы с поликонденсированными кольцами, включая атом углерода и гетероатом (например, азот).
Используемый в данном документе термин «фармацевтически приемлемый» относится к формам носителей, разбавителей, наполнителей или солей, которые совместимы с другими ингредиентами композиции и не являются вредными для реципиента фармацевтической композиции.
Используемый в данном документе термин «фармацевтическая композиция» относится к соединению настоящего изобретения, необязательно смешанного с одним или несколькими фармацевтически приемлемыми носителями, разбавителями, наполнителями или адъювантами. Фармацевтические композиции предпочтительно проявляют степень устойчивости к условиям окружающей среды, чтобы сделать их пригодными для целей производства и коммерциализации.
Используемый в данном документе термин «эффективное количество», «терапевтическое количество» или «эффективная доза» относится к количеству активного ингредиента, достаточному для достижения желаемых фармакологических или терапевтических эффектов, что приводит к эффективной профилактике или лечению расстройства. Профилактика расстройства может проявляться задержкой или предотвращением прогрессирования расстройства, а также задержкой или предотвращением появления симптомов, связанных с расстройством. Лечение расстройства может проявляться уменьшением или устранением симптомов, подавлением или устранением прогрессирования расстройства, а также любым другим вкладом в благополучие пациента.
Эффективная доза может варьироваться в зависимости от таких факторов, как состояние пациента, серьезность симптомов расстройства и способ введения фармацевтической композиции. Как правило, для введения в эффективной дозе, соединения необходимо вводить в количестве менее 30 мг/кг веса пациента. Часто, соединения могут быть введены в количестве от менее чем около 1 мг/кг веса пациента до менее чем около 100 мкг/кг веса пациента и иногда от около 10 мкг/кг до менее чем 100 мкг/кг веса пациента. Вышеуказанные эффективные дозы обычно представляют собой такое количество, которое вводят в виде разовой дозы или в виде одной или нескольких доз, вводимых в течение 24 часов. Для пациентов–людей эффективная доза соединений может потребовать введения соединения в количестве, по меньшей мере, около 1 мг/24 часа/пациент, но не более около 2400 мг/24 часа/пациент и часто не более около 500 мг/24 часа/пациент.
Соединения
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, ; и A,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, , или A;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А.
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, и ,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, или ;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А.
Как описано выше, R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, и , где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, или . В некоторых вариантах реализации, R1a представляет собой –COOH, который является выбором среди заместителей А. В некоторых вариантах реализации, R1a представляет собой , который является выбором среди заместителей А. В некоторых вариантах реализации, R1a представляет собой C1–C4 алкил, где алкил замещен –COOH, который является выбором среди заместителей A. В некоторых вариантах реализации, R1a представляет собой C1–C4 алкил, где алкил замещен , который является выбором среди заместителей А.
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –COOH, и –SO2CH3, где алкил необязательно замещен –OH, –NH2, –COOH, или –SO2CH3;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, и–[O(CH2CH2)]n–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А.
В некоторых вариантах реализации, R1b представляет собой C3 алкил, C4 алкил, или C5 алкил. В некоторых вариантах реализации, R1b представляет собой –(CH2)2CH3. В некоторых вариантах реализации, R1b представляет собой –(CH2)3CH3.
В некоторых вариантах реализации, R1a представляет собой H. В некоторых вариантах реализации, R1a представляет собой C1–C4 алкил, необязательно замещенный –ОН. В некоторых вариантах реализации, R1a представляет собой C1–C3 алкил, необязательно замещенный –ОН. В некоторых вариантах реализации, R1a представляет собой –CH2C(CH3)2OH. В некоторых вариантах реализации,  представляет собой
представляет собой 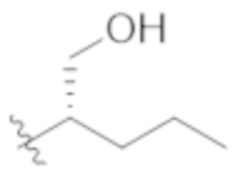 или
или  . В некоторых вариантах реализации, представляет собой , ,
. В некоторых вариантах реализации, представляет собой , ,  ,
,  ,
, 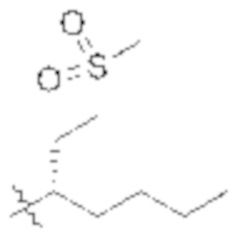 , или
, или  .
.
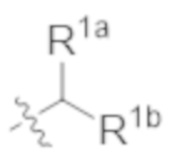
В некоторых вариантах реализации, стереоцентр углерода несущего R1A и R1B имеет (S)–конфигурацию. В некоторых вариантах реализации, стереоцентр углерода несущего R1A и R1B имеет (R)–конфигурацию.
В некоторых вариантах реализации, R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH. В некоторых вариантах реализации, R1a представляет собой C1–C3 алкил, необязательно замещенный –COOH. В некоторых вариантах реализации, представляет собой 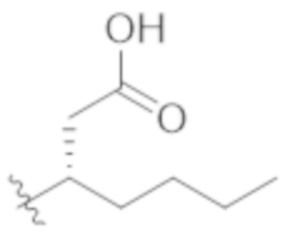 .
.
В некоторых вариантах реализации, R1a представляет собой C1–C4 алкил, где алкил необязательно замещен –OH, –NH2, –COOH, или –SO2CH3. В некоторых вариантах реализации, R1a представляет собой –NH2, –COOH, или –SO2CH3. В некоторых вариантах реализации, R1a представляет собой C1–C4 алкил, необязательно замещенный –OH, –OCH3, –SCH3, или –SO2CH3.
В некоторых вариантах реализации, X представляет собой C1–C4 алкил, где алкил замещен А. В некоторых вариантах реализации, X представляет собой C1–C4 алкил, где алкил замещен , где L2 представляет собой связь. В некоторых вариантах реализации, X представляет собой CH3.
В некоторых вариантах реализации, X представляет собой H. В некоторых вариантах реализации, X представляет собой C1–C4 алкил.
В некоторых вариантах реализации, L1 представляет собой –CH2–, –CH2CH2–, или –O–. В некоторых вариантах реализации, L1 представляет собой –CH2–. В некоторых вариантах реализации, L1 представляет собой –CH2–, –CH2CH2–, –O–, или –S–.
В некоторых вариантах реализации, L1 представляет собой связь, –CF2–, , , –S–, –SO2–, или –NH–.
В некоторых вариантах реализации, Y представляет собой C1–C3 алкил или арил. В некоторых вариантах реализации, Y представляет собой арил, где арил замещен C1–C3 алкокси. В некоторых вариантах реализации, Y представляет собой арил, где арил замещен А. В некоторых вариантах реализации, Y представляет собой арил, где арил замещен C1–C3 алкокси и A.
В некоторых вариантах реализации, Y представляет собой гетероарил, где гетероарил необязательно замещен 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси. В некоторых вариантах реализации, Y представляет собой C1–C3 алкил, где алкил необязательно замещен 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси. В некоторых вариантах реализации, представляет собой арил, где арил необязательно замещен 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкоски.
В некоторых вариантах реализации, A представляет собой . В некоторых вариантах реализации, A представляет собой . В некоторых вариантах реализации, A представляет собой . В некоторых вариантах реализации, A представляет собой . В некоторых вариантах реализации, A представляет собой . В некоторых вариантах реализации, A представляет собой .
В некоторых вариантах реализации, L2 представляет собой –(CH2)n–. В некоторых вариантах реализации, L2 представляет собой  . В некоторых вариантах реализации, L2 представляет собой . В некоторых вариантах реализации, L2 представляет собой –C(O)NH(CH2)n–.
. В некоторых вариантах реализации, L2 представляет собой . В некоторых вариантах реализации, L2 представляет собой –C(O)NH(CH2)n–.
В некоторых вариантах реализации, L2 представляет собой связь, , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, или –[O(CH2CH2)]n–OCH2CH2CF2–.
В некоторых вариантах реализации, n представляет собой один или два. В некоторых вариантах реализации, n представляет собой один. В некоторых вариантах реализации, n представляет собой два. В некоторых вариантах реализации, n представляет собой три. В некоторых вариантах реализации, n представляет собой четыре.
В некоторых вариантах реализации, m представляет собой ноль. В некоторых вариантах реализации, m представляет собой один. В некоторых вариантах реализации, m представляет собой два. В некоторых вариантах реализации, m представляет собой три. В некоторых вариантах реализации, m представляет собой четыре.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру Формулы (1а), и его фармацевтически приемлемым солям,
.
В некоторых вариантах реализации Формулы (1а),
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
В другом варианте реализации Формулы (1а)
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–,, , и–CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Как описано выше, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a выбирают из группы, состоящей из , , , , , , и или A1a выбирают из группы, состоящей из , , , , , и . В вышеупомянутом, X содержит A1a, который выбирают среди заместителей А.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , , и ; и
L2 представляет собой связь.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , и ; и
L2 представляет собой связь.
В другом варианте реализации Формулы (1а)
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
В другом варианте реализации Формулы (1а)
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1b), и его фармацевтически приемлемым солям,
.
В некоторых вариантах реализации Формулы (1b),
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
В другом варианте реализации Формулы (1b)
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Как описано выше, В некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a выбирают из группы, состоящей из , , , , , , и или A1a выбирают из группы, состоящей из , , , , , и . В вышеупомянутом, X содержит A1a, который выбирают среди заместителей А.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , , и ; и
L2 представляет собой связь.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , и ; и
L2 представляет собой связь.
В другом варианте реализации Формулы (1b)
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
В другом варианте реализации Формулы (1b)
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1с), и его фармацевтически приемлемым солям,
.
Как описано выше, В некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a выбирают из группы, состоящей из , , , , , , и или A1a выбирают из группы, состоящей из , , , , , и . В вышеупомянутом, X содержит A1a, который выбирают среди заместителей А.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , , и ; и
L2 представляет собой связь.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , и ; и
L2 представляет собой связь.
В некоторых вариантах реализации Формулы (1c),
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
В другом варианте реализации Формулы (1c)
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1d), и его фармацевтически приемлемым солям,
Как описано выше, В некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a выбирают из группы, состоящей из , , , , , , и или A1a выбирают из группы, состоящей из , , , , , и . В вышеупомянутом, X содержит A1a, который выбирают среди заместителей А.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , , и ; и
L2 представляет собой связь.
Соответственно, в некоторых вариантах реализации, X может быть –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a; где A1a представляет собой A;
где A выбирают из группы, состоящей из , , , , , и ; и
L2 представляет собой связь.
В некоторых вариантах реализации Формулы (1d),
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –CH2CH2, и –S–; и
Y представляет собой H или C1–C3 алкил.
В другом варианте реализации Формулы (1d)
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1e), и его фармацевтически приемлемым солям,
где R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1f), и его фармацевтически приемлемым солям,
.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1g), и его фармацевтически приемлемым солям,
где R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1h), и его фармацевтически приемлемым солям,
где R4 представляет собой H или C1–C3 алкокси.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1i), и его фармацевтически приемлемым солям,
где
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –CH2CH2–, и –S–; и
Y представляет собой H или C1–C3 алкил.
В другом варианте реализации Формулы (1i)
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1j), и его фармацевтически приемлемым солям,
(1j).
В некоторых вариантах реализации Формулы (1j), Х представляет собой Н или CH3.
В некоторых вариантах реализации Формулы (1j), R1a выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, или .
В некоторых вариантах реализации Формулы (1j), R1a выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен –COOH.
Настоящее изобретение относится к соединению Формулы (1), имеющему структуру формулы (1k), и его фармацевтически приемлемым солям,
(1k),
В некоторых вариантах реализации Формулы (1k), X представляет собой H или CH3.
В некоторых вариантах реализации Формулы (1k), R1b представляет собой C4 алкил.
В некоторых вариантах реализации Формулы (1k),
Y представляет собой арил или гетероарил, где арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , и ; и
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, и–[O(CH2CH2)]n–OCH2CH2CF2.
В некоторых вариантах реализации Формулы (1k),
Y представляет собой арил, где арил необязательно замещен 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , и ; и
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, и–[O(CH2CH2)]n–OCH2CH2CF2.
В некоторых вариантах реализации Формулы (1k),
Y представляет собой арил, где арил необязательно замещен 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси;
A представляет собой ; и
L2 выбирают из группы, состоящей из связи, –(CH2)n–, , , и .
В одном варианте Формулы (1), R1b представляет собой –(CH2)2CH3 и R1a представляет собой H. В одном варианте Формулы (1), R1b представляет собой –(CH2)2CH3 и R1a представляет собой C1–C4 алкил, необязательно замещенный –ОН. В одном варианте Формулы (1), R1b представляет собой –(CH2)2CH3 и R1a представляет собой –CH2C(CH3)2OH. В одном варианте Формулы (1), представляет собой или .
В одном варианте Формулы (1), Y представляет собой арил, где арил замещен C1–C3 алкокси и A. В одном варианте Формулы (1), представляет собой арил, где арил замещен –OCH3 и A.
В одном варианте Формулы (1), R1b представляет собой –(CH2)3CH3 и R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH. В одном варианте Формулы (1), R1b представляет собой –(CH2)2CH3 и R1a представляет собой –CH2COOH.
Настоящее описание относится к соединению формулы (1)
(1),
имеющему одну, две, три или более из следующих признаков:
R1b представляет собой –(CH2)2CH3;
R1a представляет собой H;
X представляет собой –CH3;
L1 представляет собой –CH2–; и
Y представляет собой арил, необязательно замещенный 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси.
Настоящее описание относится к соединению формулы (1)
(1),
имеющему одну, две, три или более из следующих признаков:
R1b представляет собой –(CH2)2CH3 или –(CH2)3CH3;
R1a представляет собой H; и
X представляет собой C1–C4 алкил, где алкил замещен А.
Настоящее описание относится к соединению формулы (1)
(1),
имеющему одну, две, три или более из следующих признаков:
R1b представляет собой –(CH2)2CH3 или –(CH2)3CH3;
R1a представляет собой C1–C4 алкил, замещенный –OH, –NH2, –COOH, или –SO2CH; и
X представляет собой C1–C4 алкил, где алкил замещен А.
Настоящее описание относится к соединению формулы (1)
(1),
имеющему один, два, три или более из следующих признаков:
R1b представляет собой –(CH2)2CH3 или –(CH2)3CH3;
R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH;
X представляет собой –CH3;
L1 представляет собой –CH2–; и
Y представляет собой арил, необязательно замещенный 1–5 заместителями, которые независимо выбраны из А, C1–C3 алкила, и C1–C3 алкокси.
Настоящее описание относится к соединению формулы (1)
(1),
имеющему одну, две, три или более из следующих признаков:
a) R1b представляет собой –(CH2)2CH3 или –(CH2)3CH3;
b) R1a представляет собой H или C1–C4 алкил, необязательно замещенный –COOH;
c) X представляет собой –CH3;
d) L1 представляет собой –CH2–; и
e) Y представляет собой арил, необязательно замещенный 1–5 заместителями, которые независимо выбраны из и C1–C3 алкокси.
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), и его фармацевтически приемлемым солям,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –COOH, и –SO2CH3, где алкил необязательно замещен –OH, –NH2, –COOH, или –SO2CH3;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A или галогеном;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А,
с одним или несколькими из следующих признаков,
(a) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH;
(b) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH;
(c) когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда X не является –CH3;
(d) когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда X не является –CH3, кроме случаев, когда R1a содержит –COOH;
(e) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является–CH2–;
(f) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является –CH2–, кроме случаев, когда R1a содержит –COOH;
(g) когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–;
(h) когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–, кроме случаев, когда R1a содержит –COOH;
(i) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда R1a содержит COOH;
(j) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH;
(k) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH;
(l) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; и R1a не содержит COOH; тогда A не представляет собой –L2–COOH.
В некоторых вариантах реализации соединение, имеющее структуру Формулы (1), может иметь любой один или несколько из следующих признаков:
(m) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH;
(n) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
(o) когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда X не является –CH3;
(p) когда L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда X не является –CH3, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
(q) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является –CH2–;
(r) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; тогда L2 не является –CH2–, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
(s) когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–;
(t) когда X представляет собой –CH3; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда L1 не является –CH2–, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
(u) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда R1a содержит COOH или –SO2CH3;
(v) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH;
(w) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3;
(x) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный А и –OCH3; и L2 представляет собой –CH2–; и R1a не содержит COOH или –SO2CH3; тогда A не представляет собой –L2–COOH;
(y) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и A представляет собой –L2–COOH; и L2 представляет собой –CH2–; тогда R1a не является H или алкилом замещенным –OH.
В некоторых вариантах реализации соединение, имеющее структуру Формулы (1), может иметь любой один или несколько из следующих признаков:
(aa) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(bb) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(cc) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; L2 представляет собой–CH2–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(dd) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(ee) когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2(CF2)–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу;
(ff) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; тогда A не представляет собой ;
(gg) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой ;
(hh) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; и L2 представляет собой –CH2–; тогда A не представляет собой ;
(ii) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –O–(CH2)2–O(CH2)2–; тогда A не представляет собой ;
(jj) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –O–(CH2)2–O(CH2)2(CF2)–; тогда A не представляет собой ;
(kk) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(ll) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(mm) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A и –OCH3; L2 представляет собой –CH2–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(nn) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой ;
(oo) когда X представляет собой –CH3; R1a представляет собой H; R1b представляет собой C4алкил; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –O–(CH2)2–O(CH2)2(CF2)–; и A и L1 находятся в пара–положении по отношению друг к другу; тогда A не представляет собой .
В некоторых вариантах реализации, настоящее описание относится к соединению формулы (1), которое не описано в WO 2009/067081 (PCT/SE2008/051334). В некоторых вариантах реализации, настоящее описание относится к соединению формулы (1), которое не описано в WO 2012/031140 (PCT/US2011/050231).
В некоторых вариантах реализации, настоящее описание относится к соединению формулы (1), которое не является 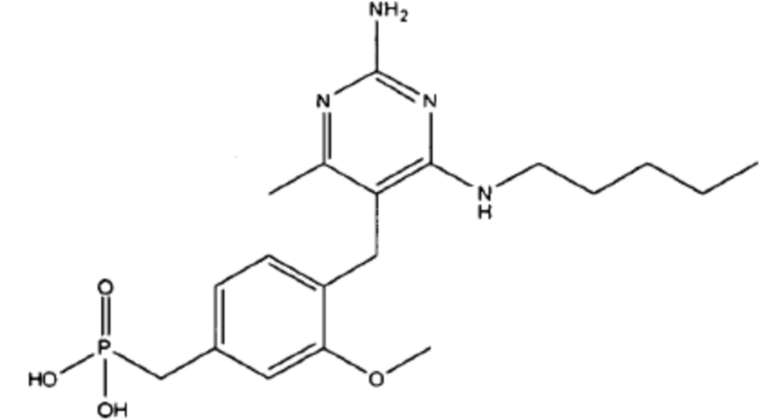 ,
, 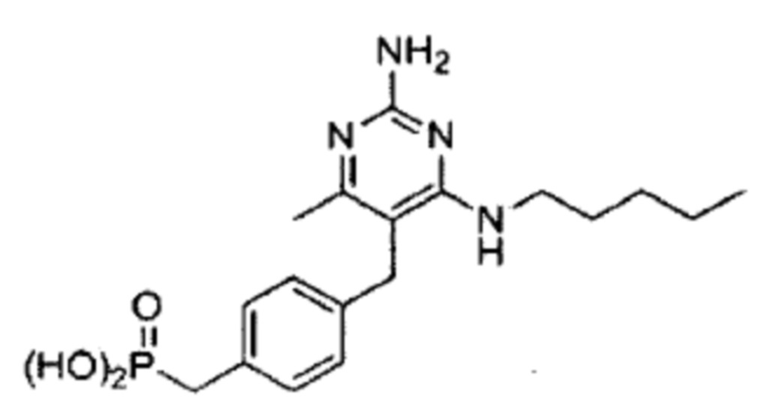 ,
,  , или
, или 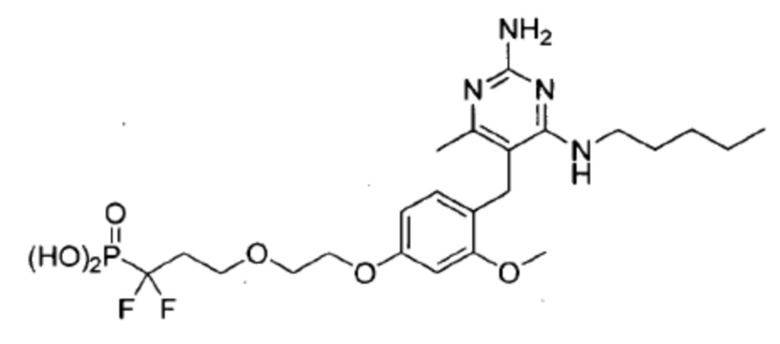 .
.
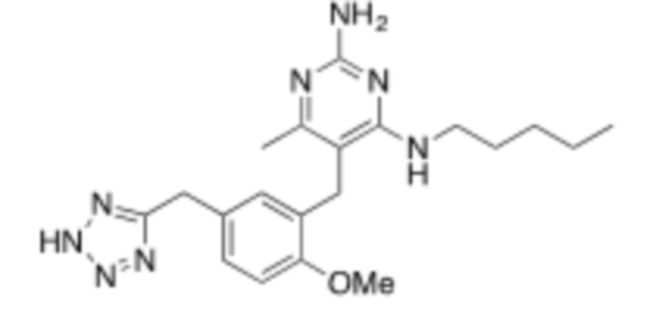
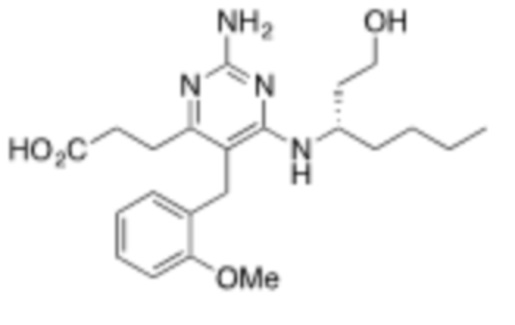
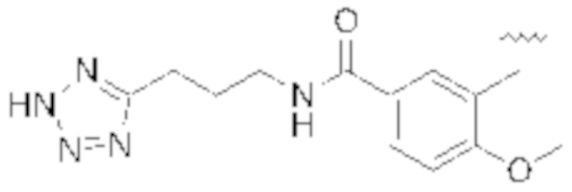
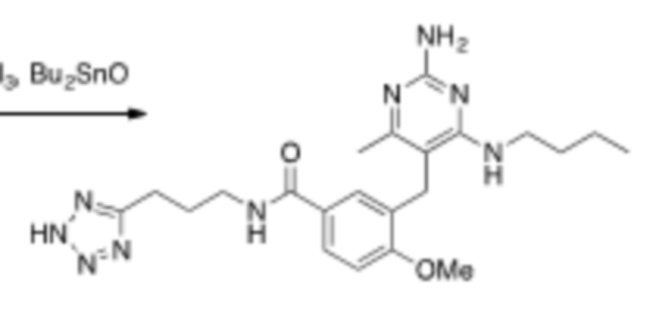
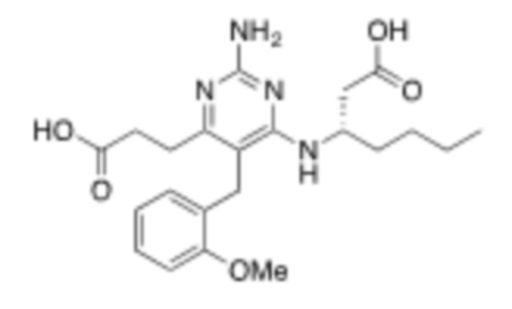
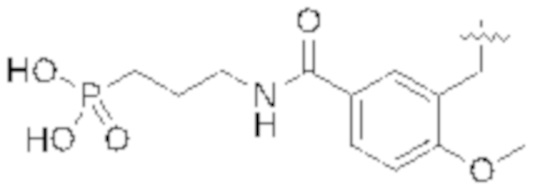
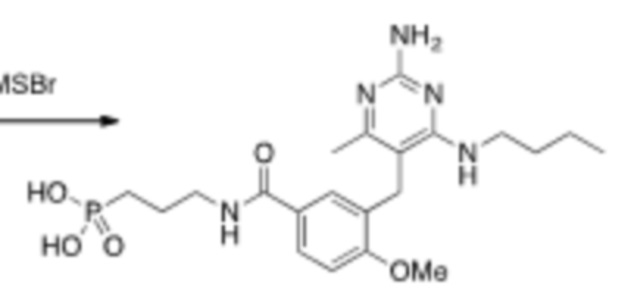
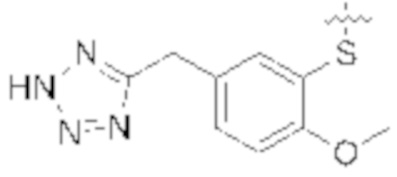
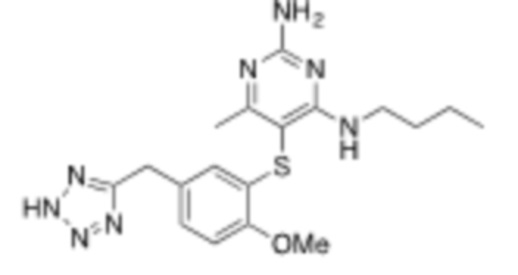
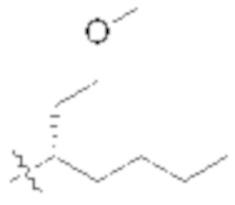
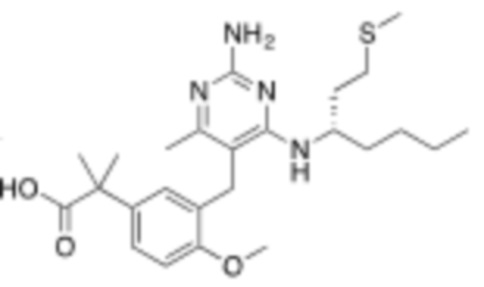
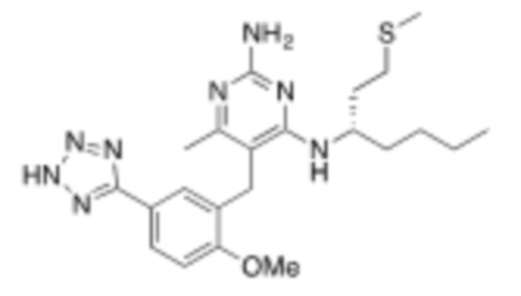
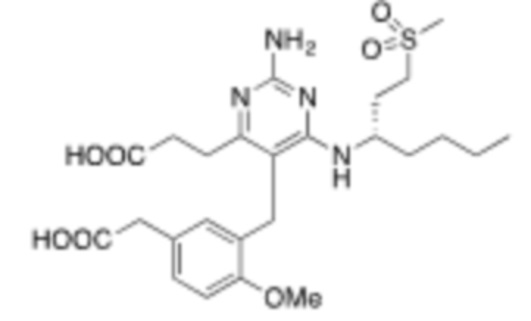
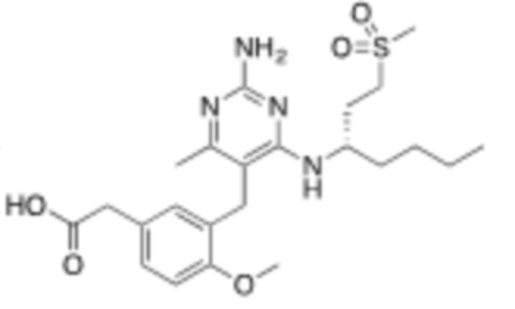
Настоящее описание относится к следующим соединениям и их фармацевтически приемлемым солям.
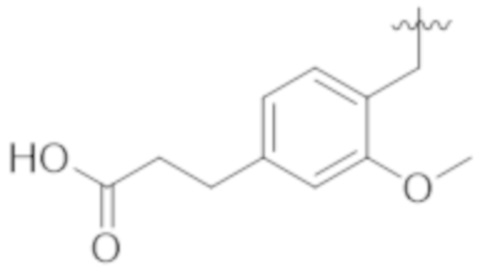

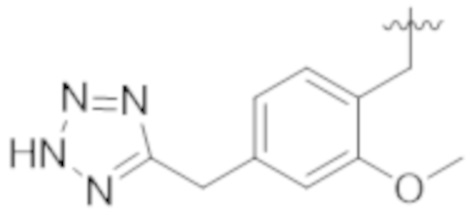
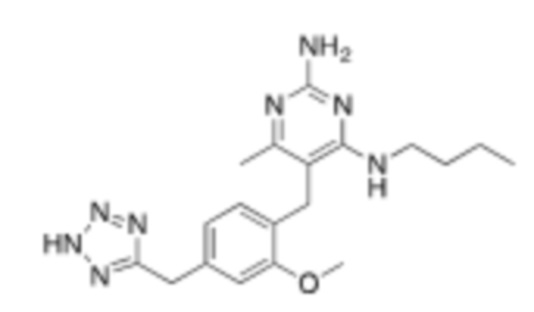
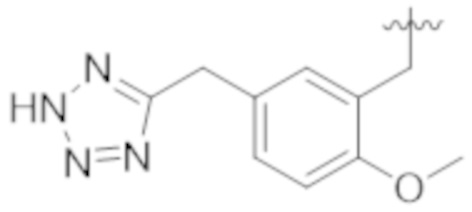
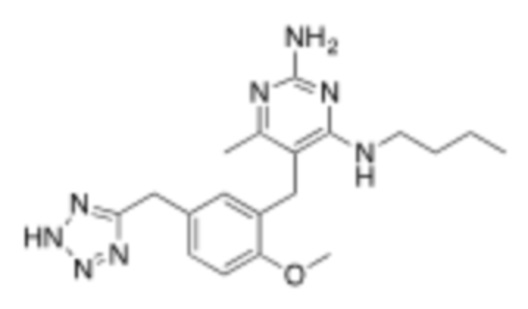

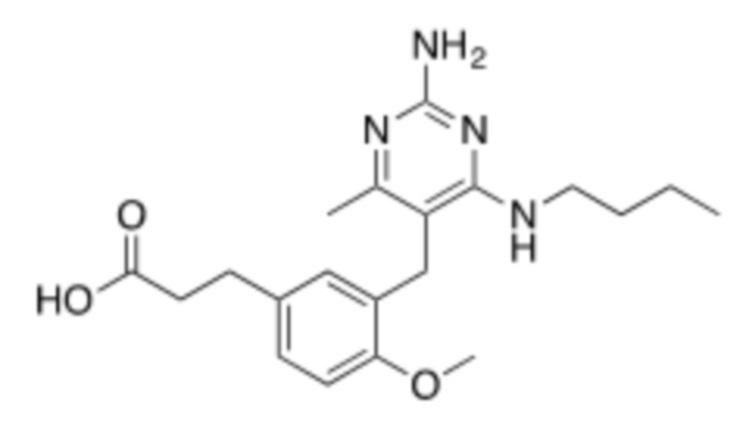
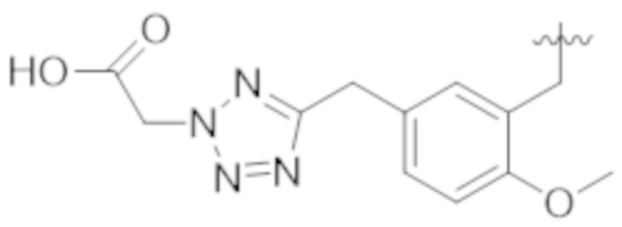

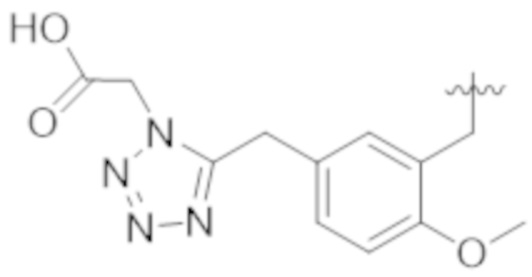
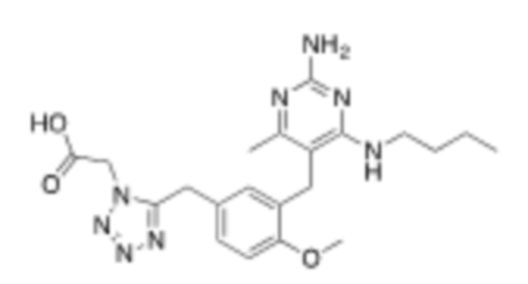



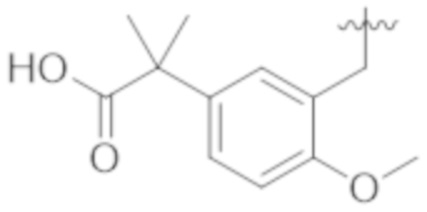
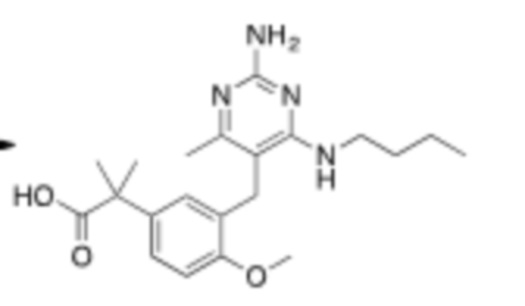
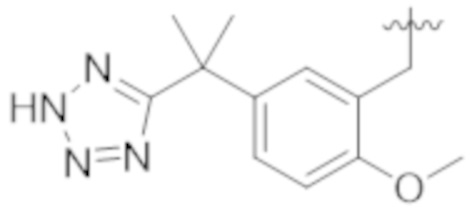
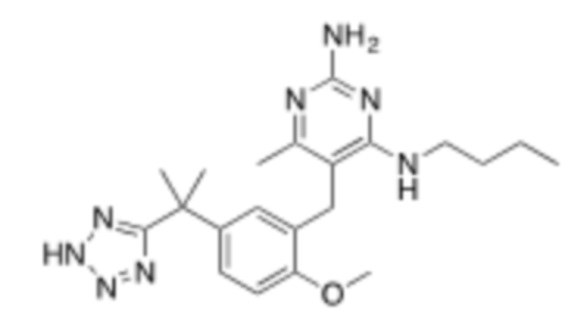
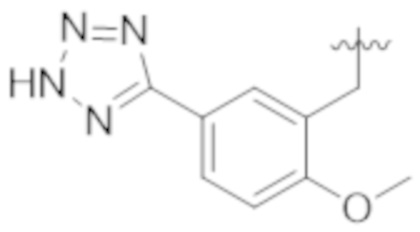
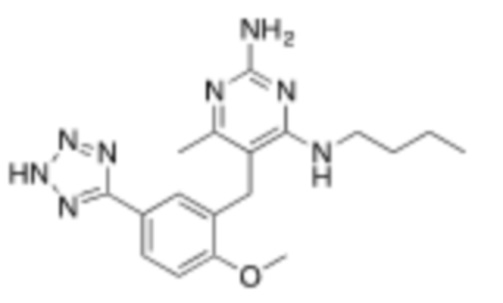
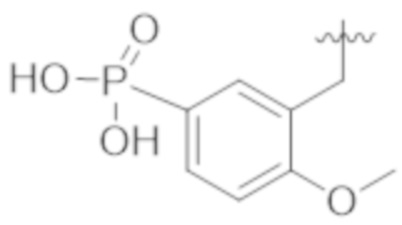
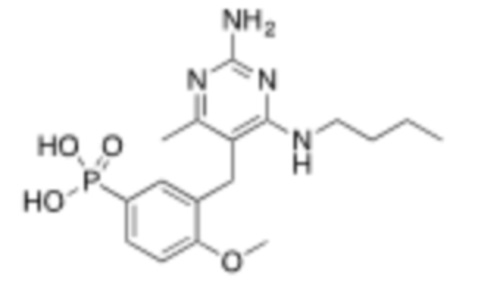
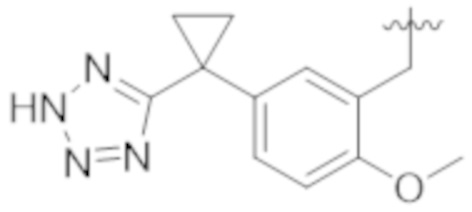
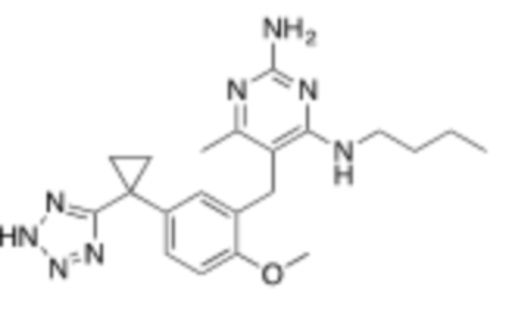






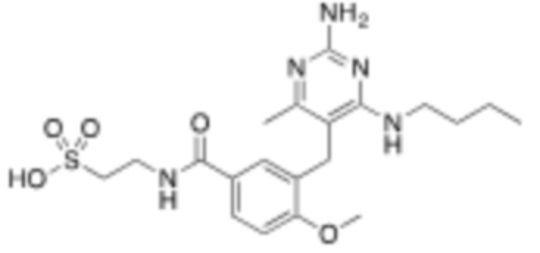




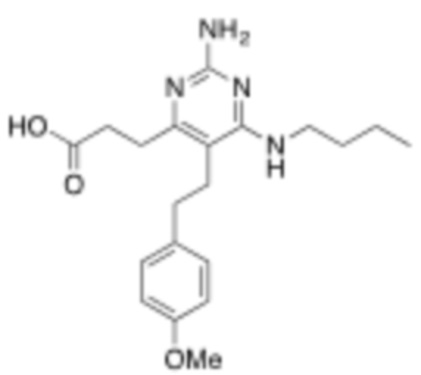
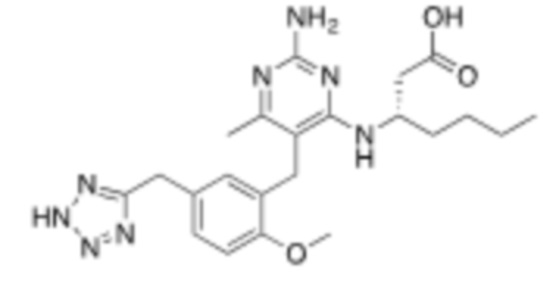


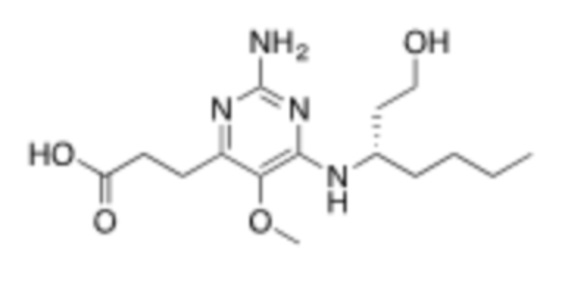
Настоящее описание относится к следующим соединениям и их фармацевтически приемлемым солям.

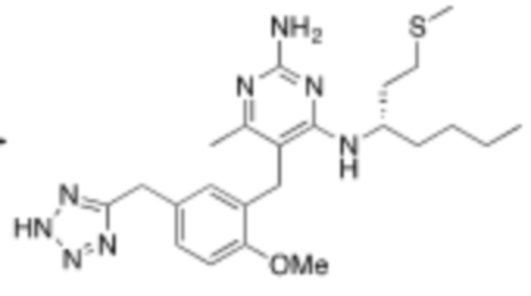


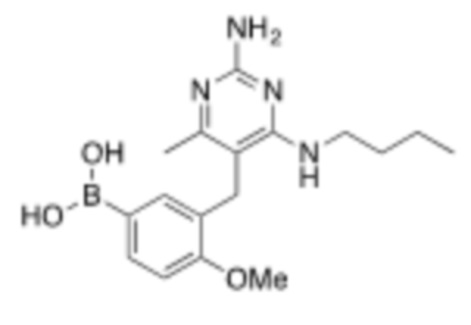
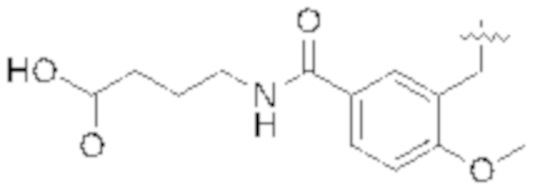
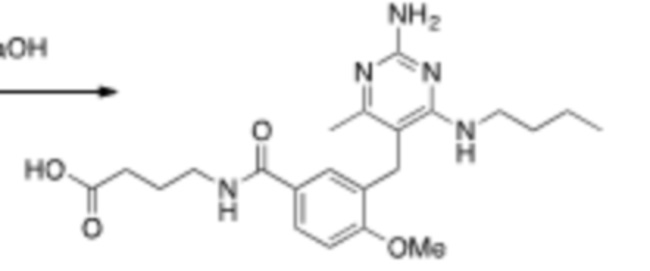


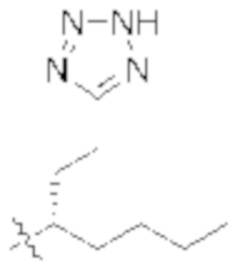

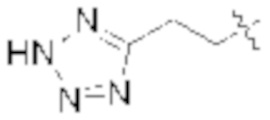





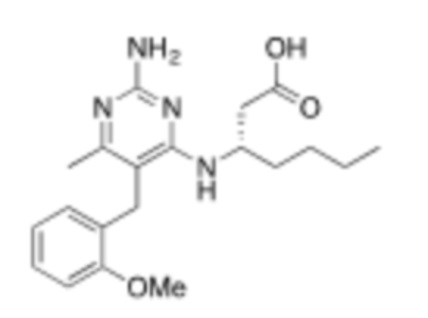


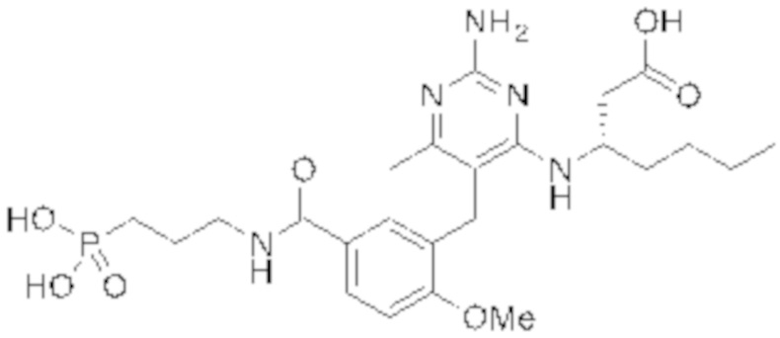
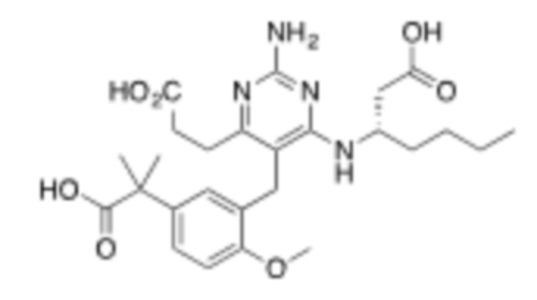
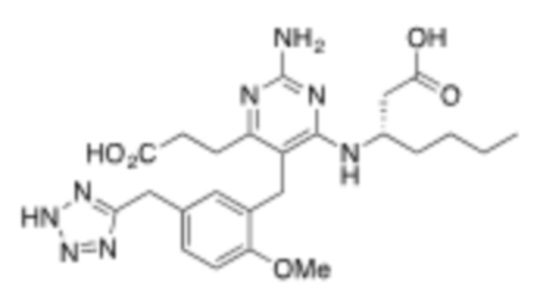

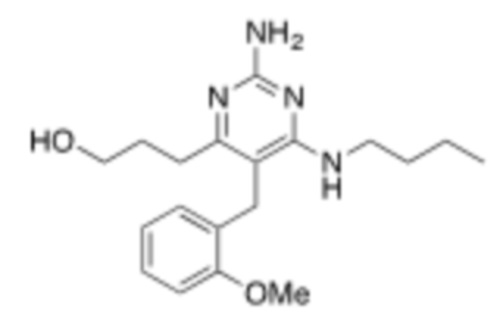
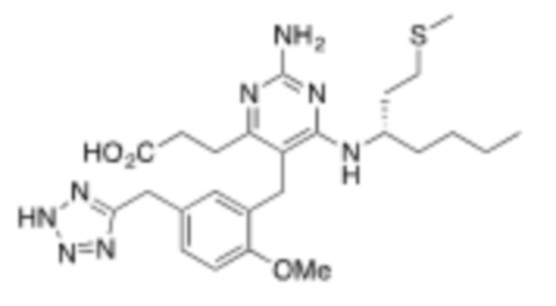


Если не указано иное, структуры, изображенные в данном документе, также подразумевают включение соединений, которые отличаются только присутствием одного или нескольких изотопобогащенных атомов. Например, соединения, имеющие настоящую структуру, за исключением замены атома водорода дейтерием или тритием, или замены атома углерода на 13C или 14C, или замены атома азота на 15N, или замены атома кислорода на 17O или 18O находятся в рамках настоящего изобретения. Такие изотопмеченные соединения полезны в качестве инструментов исследования или диагностики.
Общие способы синтеза
Как отмечено в данном документе, настоящее описание включает конкретные репрезентативные соединения, которые идентифицированы в данном документе с особенностью. Соединения настоящего изобретения могут быть получены различными способами, включая хорошо известные стандартные способы синтеза. Иллюстративные общие способы синтеза изложены ниже, и тогда конкретные соединения настоящего изобретения получают в рабочих Примерах.
Во всех примерах, описанных ниже, защитные группы для чувствительных или реакционноспособных групп используются при необходимости в соответствии с общими принципами синтетической химии. Защитные группы манипулируют в соответствии со стандартными способами органического синтеза (T. W. Green и P. G. M. Wuts, Protecting Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, New York (1999)). Эти группы удаляются на удобной стадии синтеза соединения с использованием способов, которые очевидны для специалистов в данной области. Выбор процессов, а также условия реакции и порядок их выполнения должны соответствовать получению соединений настоящего изобретения.
Типичный синтез для рассматриваемых соединений показан на Схеме 1.
Схема 1

На Схеме 1, соединение формулы (G) представляет собой вариант реализации, где L1 представляет собой –CH2– и представляет собой арил, который соответственно замещен. Также на Схеме 1, LG является уходящей группой; и R представляет собой H или алкил. Соединения формулы (А) и (В) являются коммерчески доступными исходными материалами. Альтернативно, соединения формулы (A) и (B) могут быть синтезированы различными способами синтеза с использованием коммерчески доступных исходных материалов и/или исходных материалов, полученных обычными способами синтеза.
Со ссылкой на Cхему 1 соединения формулы (C) можно получить взаимодействием соединения формулы (A) с основанием, таким как гидрид натрия, в подходящем растворителе, таком как тетрагидрофуран или N,N–диметилформамид, при температуре, например, от 0 °С до комнатной температуры (20 °С), с последующим добавлением соединения формулы (В). Реакционную смесь затем предпочтительно нагревают при температуре, например, от 50 °С до 100 °С, необязательно, в присутствии добавки, такой как йодид калия.
Соединения формулы (D) могут быть получены взаимодействием соединения формулы (C) с гуанидином или гуанидинкарбонатом в подходящем растворителе, таком как метанол или этанол, при температуре, например, в диапазоне от 50 °С до 150 °С.
Соединения формулы (Е) могут быть получены взаимодействием соединения формулы (D) с оксихлоридом фосфора при температуре, например, от 50 °С до 110 °С.
Соединения формулы (F) могут быть получен взаимодействием соединения формулы (Е) с избытком амина формулы R1aR1bNH, в подходящем растворителе, таком как NMP, бутанол или 1,2–диоксан, при температуре, например, от 50 °С до 150 °С. Альтернативно, реакцию можно проводить в микроволновой печи при температуре, например, от 50 °С до 200 °С.
Соединения формулы (G) могут быть получены взаимодействием соединения формулы (F) с восстановителем, таким как алюмогидрид лития, в подходящем растворителе, таком как тетрагидрофуран, при температуре, например, от 0 °С до 60 °С.
Типичный синтез для рассматриваемых соединений показан на Схеме 2.
Схема 2
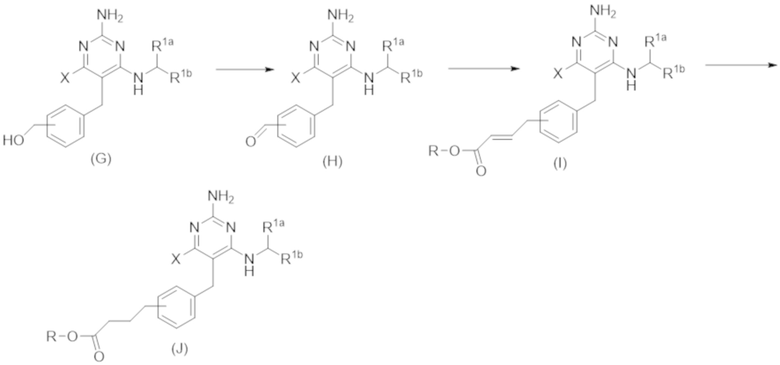
На схеме 2 соединение формулы (G) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен. Также на Схеме 2, R представляет собой H или алкил.
Продолжая ссылаться на Схему 2, соединения формулы (H) могут быть получены взаимодействием соединения формулы (G) с окислителем, таким как оксид марганца, в подходящем растворителе, таком как тетрагидрофуран или N,N–диметилформамид, при температуре, например, от 40 °С до 100 °С.
Соединения формулы (I) могут быть получены взаимодействием соединения формулы (H) посредством реакции Виттига с RO–C(O)–CH=PPh3. Реакция может проводиться в подходящем растворителе, таком как тетрагидрофуран, при температуре, например, от 50 °С до 150 °С.
Соединения формулы (J) могут быть получены восстановлением соединения формулы (I) в условиях гидрирования. Реакцию можно проводить с катализатором, таким как палладий на углероде, в атмосфере водорода в подходящем растворителе, таком как этилацетат, при температуре, например, от 20 °С до 100 °С.
Типичный синтез для рассматриваемых соединений показан на Схеме 3.
Схема 3
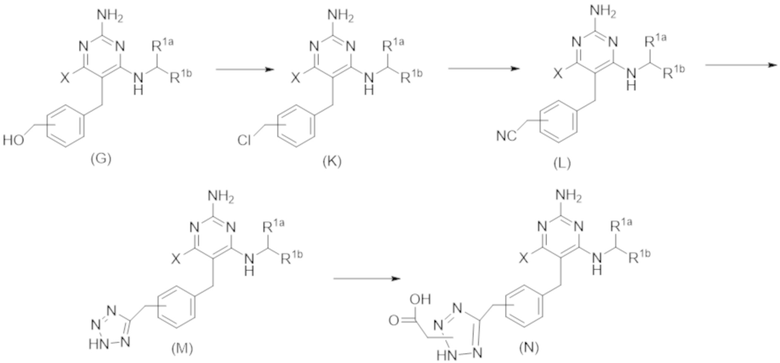
На Схеме 3 соединение формулы (G) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен.
Продолжая ссылаться на Схему 3, соединения формулы (K) могут быть получены взаимодействием соединения формулы (G) с хлорирующим реагентом, таким как тионилхлорид, в подходящем растворителе, таком как метиленхлорид, при температуре, например, от комнатной температуры до 50 °С.
Соединения формулы (L) могут быть получены взаимодействием соединения формулы (K) с цианидной солью, такой как цианид калия, в подходящем растворителе, таком как диметилсульфоксид или N,N–диметилформамид (или смесь обоих растворителей) при температуре, например, от комнатной температуры до 50 °С.
Соединения формулы (M) могут быть получены взаимодействием соединения формулы (L) с азиднынм реагентом, таким как триметилсилилазид, в реакции азид–нитрильного циклоприсоединения. Реакцию можно проводить в подходящем растворителе, таком как NMP или диоксан, при температуре, например, от 50 °С до 150 °С. Реакция может быть проведена в присутствии катализатора, такого как оксид дибутилолова.
Соединения формулы (N) могут быть получены взаимодействием соединения формулы (М) с алкилирующим агентом, таким как 2–бромацетат, в подходящем растворителе, таком как ацетон, при температуре, например, от 0 °С до 60 °С.
Схема 4
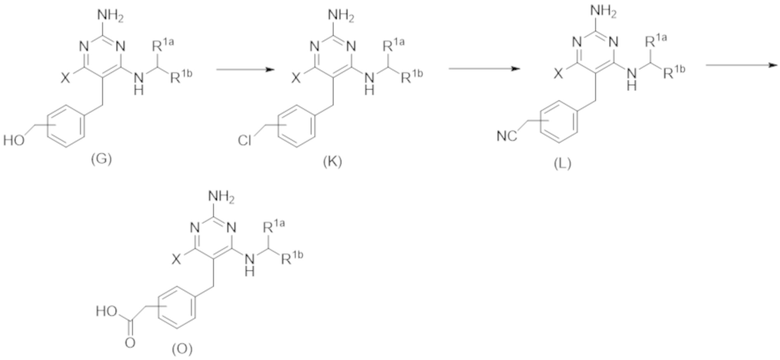
На Схеме 4 соединение формулы (G) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен. Получение соединений формулы (K) и (L) описано выше.
Продолжая ссылаться на Схему 4, соединения формулы (O) можно получить гидролизом соединения формулы (L), например, с использованием основания, такого как гидроксид калия, в подходящем растворителе, таком как этан–1,2–диол и вода (или их смеси) при температуре, например, от 50 °С до 200 °С.
Типичный синтез для рассматриваемых соединений показан на Схеме 5.
Схема 5

На Схеме 5 соединение формулы (G) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен. Получение соединения формулы (K) описано выше.
Продолжая ссылаться на Схему 5, соединения формулы (P) могут быть получены взаимодействием соединения формулы (K) с триэтилфосфитом в подходящем растворителе или без растворителя при температуре, например, от 50 °С до 150 °С.
Соединения формулы (Q) могут быть получены взаимодействием соединения формулы (P) с реагентами для удаления этильных групп, таких как бромтриметилсилан, в подходящем растворителе, таком как метиленхлорид, при температуре, например, от комнатной температуры до 60 °С.
Типичный синтез для рассматриваемых соединений показан на Схеме 6.
Схема 6

На Схеме 6, соединение формулы (F’) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен. Также на Схеме 6, LG1 и LG2 уходящей группой. Соединения формулы (А’) и (В’) являются коммерчески доступными исходными материалами. Альтернативно, соединения формулы (A’) и (B’) могут быть синтезированы различными способами синтеза с использованием коммерчески доступных исходных материалов и/или исходных материалов, полученных обычными способами синтеза.
Продолжая ссылаться на Схему 6, соединения формулы (C’) могут быть получены взаимодействием соединения формулы (A’) с основанием, таким как гидрид натрия, в подходящем растворителе, таком как тетрагидрофуран или N,N–диметилформамид, при температуре, например, от 0 °С до комнатной температуры (20 °С), с последующим добавлением соединения формулы (B). Реакционную смесь затем предпочтительно нагревают при температуре, например, от 50 °С до 100 °С, необязательно, в присутствии добавки, такой как йодид калия.
Соединения формулы (D’) могут быть получены взаимодействием соединения формулы (C’) с гуанидином или гуанидинкарбонатом в подходящем растворителе, таком как метанол или этанол, при температуре, например, в диапазоне от 50 °С до 150 °С.
Соединения формулы (E’) могут быть получены взаимодействием соединения формулы (D’) с оксихлоридом фосфора при температуре, например, от 50 °С до 110 °С.
Соединения формулы (F’) могут быть получены взаимодействием соединения формулы (E’) с избытком амина формулы R1aR1bNH, в подходящем растворителе, таком как NMP, бутанол или 1,2–диоксан, при температуре, например, от 50 °С до 150 °С. Альтернативно, реакцию можно проводить в микроволновой печи при температуре, например, от 50 °С до 200 °С.
Типичный синтез для рассматриваемых соединений показан на Схеме 7.
Схема 7

На Схеме 7, соединение формулы (F’) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен. Также на Схеме 7 LG2 является уходящей группой.
Продолжая ссылаться на Схему 7, соединения формулы (G’) могут быть получены взаимодействием соединения формулы (F’) с азидным реагентом, таким как триметилсилилазид, в реакции азид–нитрильного циклоприсоеднения. Реакцию можно проводить в подходящем растворителе, таком как NMP или диоксан, при температуре, например, от 50 °С до 150 °С. Реакция может быть проведена в присутствии катализатора, такого как оксид дибутилолова.
Типичный синтез для рассматриваемых соединений показан на Схеме 8.
Схема 8
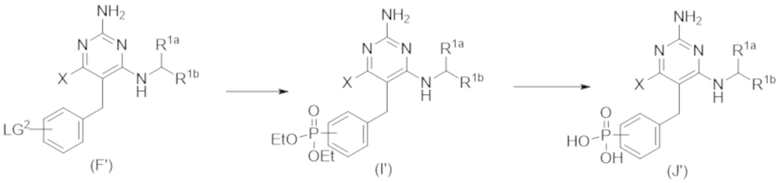
На Схеме 8, соединение формулы (F’) представляет собой вариант реализации, в котором L1 представляет собой –CH2– и Y представляет собой арил, который соответственно замещен. Также на Схеме 8 LG2 является уходящей группой.
Продолжая ссылаться на Схему 8, соединения формулы (I’) могут быть получены взаимодействием соединения формулы (F’) с триэтилфосфитом в подходящем растворителе или без растворителя при температуре, например, от 50 °С до 150 °С.
Соединения формулы (J’) могут быть получены взаимодействием соединения формулы (I’) с реагентами для удаления этильных групп, таких как бромтриметилсилан, в подходящем растворителе, таком как метиленхлорид, при температуре, например, от комнатной температуры до 60 °С.
Способ лечения
Соединения формулы (1) и их фармацевтически приемлемые соли имеют активность в качестве фармацевтических средств, в частности в качестве модуляторов активности toll–подобного рецептора (особенно TLR7), и, таким образом, могут использоваться при лечении:
1. дыхательных путей: обструктивные заболевания дыхательных путей, в том числе: астма, включая бронхиальную, аллергическую, внутреннюю, внешнюю, вызванную физическими нагрузками, лекарственную (включая аспирин и НПВП) и пылевую астму, как перемежающуюся, так и постоянную, и всех степеней тяжести и другие причины гиперчувствительности дыхательных путей; хроническое обструктивное заболевание легких (ХОБЛ); бронхит, в том числе инфекционный и эозинофильный бронхит; эмфизема; бронхоэктазы; кистозный фиброз; саркоидоз; легкое фермера и сопутствующие заболевания; гиперчувствительный пневмонит; фиброз легких, в том числе криптогенный фиброзирующий альвеолит, идиопатическая интерстициальная пневмония, фиброз представляет собой осложняющую антинеопластическую терапию и хроническую инфекцию, включая туберкулез и аспергиллез и другие грибковые инфекции; осложнения трансплантации легких; васкулитные и тромботические расстройства сосудистой сети легких и легочная гипертензия; противокашлевая активность, включая лечение хронического кашля, связанного с воспалительными и секреторными состояниями дыхательных путей, и ятрогенного кашля; острый и хронический ринит, включая медикаментозный ринит, и вазомоторный ринит; многолетний и сезонный аллергический ринит, включая нервный ринит (сенная лихорадка); носовой полипоз; острая вирусная инфекция, включая простуду, и инфекция, вызванная респираторно–синцитиальным вирусом, гриппом, коронавирусом (включая SARS) и аденовирусом;
2. кожы: псориаз, атопический дерматит, контактный дерматит или другие экзематозные дерматозы и реакции гиперчувствительности замедленного типа; фито– и фотодерматит; себорейный дерматит, герпетиформный дерматит, красный плоский лишай, лишай склероатрофический, гангренозная пиодермия, саркоидоз кожи, дискоидная красная волчанка, пузырчатки, пемфигоид, врожденный буллезный эпидермолиз, крапивница, ангионевротический отек, васкулиты, токсические эритемы, кожные эозинофилии, гнездная алопеция, облысение по мужскому типу, синдром Свита, синдром Вебера–Кристиана, мультиформная эритема; целлюлит, как инфекционный, так и неинфекционный; панникулит; кожные лимфомы, немеланомный рак кожи и другие диспластические поражения, незлокачественный рак кожи, базальноклеточный рак; старческий кератоз; вызванные лекарствами ми расстройства, включая постоянную медикаментозную сыпь;
3. глаз: блефарит; конъюнктивит, в том числе многолетний и весенний аллергический конъюнктивит; ирит; передний и задний увеит; хориоидит; аутоиммунные, дегенеративные или воспалительные расстройства, поражающие сетчатку; офтальмит, включая симпатический офтальмит; саркоидоз; инфекции, включая вирусные, грибковые и бактериальные;
4. мочеполовой системы: нефрит, включая интерстициальный и гломерулонефрит; нефротический синдром; цистит, включая острый и хронический (интерстициальный) цистит и язву Ханнера; острый и хронический уретрит, простатит, эпидидимит, оофорит и сальпингит; вульвовагинит; Болезнь Пейрони; эректильная дисфункция (как у мужчин, так и у женщин);
5. отторжение аллотрансплантата: острое и хроническое последующее, например, трансплантация почки, сердца, печени, легкого, костного мозга, кожи или роговицы или после переливания крови; или хроническое заболевание трансплантат против хозяина;
6. других аутоиммунных и аллергических расстройств, включая ревматоидный артрит, синдром раздраженного кишечника, синдром воспалительного кишечника, колит, болезнь Крона, системную красную волчанку, рассеянный склероз, тиреоидит Хашимото, болезнь Грейвса, болезнь Аддисона, сахарный диабет, идиопатический тромбоцитофитный тромбоцитопенит, атонический фасциит, синдром гипер–IgE, антифосфолипидный синдром и сазарский синдром;
7. онкологических заболеваний: лечение распространенных видов рака, включая печень, легкие, мочевой пузырь, желудочно–кишечный тракт (включая желудок, колоректальный, пищеводный и ректальный), голову и шею, простату, грудь, легкое, яичник, поджелудочную железу, кишечник и толстую кишку, желудок, кожу и мозг опухоли и злокачественные новообразования, поражающие костный мозг (включая лейкемии) и лимфопролиферативные системы, такие как лимфома Ходжкина и неходжкинская лимфома; включая профилактику и лечение метастатической болезни и рецидивов опухоли, и паранеопластических синдромов; и
8. инфекционных заболеваний: вирусные заболевания, такие как генитальные бородавки, общие бородавки, подошвенные бородавки, гепатит B, гепатит C, вирус простого герпеса, контагиозный моллюск, натуральная оспа, вирус иммунодефицита человека (ВИЧ), вирус папилломы человека (ВПЧ), цитомегаловирус (ЦМВ), вирус ветряной оспы (ВВО), риновирус, аденовирус, коронавирус, грипп, парагрипп; бактериальные заболевания, такие как туберкулез и микобактерии avium, проказа; другие инфекционные заболевания, такие как грибковые заболевания, хламидии, Кандиды, аспергиллез, криптококковый менингит, пневмоцистис Carnii, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция и лейшманиоз.
Настоящее описание обеспечивает применение соединения формулы (1) или его фармацевтически приемлемой соли в качестве вакцинного адъюванта, используемого вместе с одним или несколькими антигенами против следующих заболеваний: ВГВ, ВПЧ, менингит, TDaP, грипп, бешенство, туберкулез, туберкулез, малярия, инфекция Staphylococcus aureus и раковые заболевания (ассоциированный с опухолью антиген или нео–антиген).
Таким образом, настоящее описание относится к соединению формулы (1) или его фармацевтически приемлемой соли, как определено выше, для применения в терапии.
В дополнительном аспекте, настоящее описание относится к применению соединения формулы (1) или его фармацевтически приемлемой соли, как определено выше, при изготовлении лекарственного средства для применения в терапии.
Настоящее описание относится к способам лечения состояния, связанного с модуляцией TLR7 у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли. Настоящее описание относится к соединению настоящего изобретения или его фармацевтически приемлемой соли для применения при лечении состояния, связанного с модуляцией TLR7. Настоящее описание относится к использованию соединения настоящего описание или его фармацевтически приемлемой соли при изготовлении лекарственного средства для лечения состояния, связанного с модуляцией TLR7. В некоторых вариантах реализации состояние представляет собой вирусную инфекцию или рак.
В контексте настоящего описания термин "терапия" также включает "профилактику", если конкретно не указано противоположное. Термины "терапевтический" и "терапевтически" следует рассматривать соответствующим образом.
Можно ожидать, что профилактика является особенно важной для лечения индивидуумов, которые ранее перенесли эпизод данного нарушения или состояния или же рассматриваются как имеющие повышенный риск нарушения или состояния. Пациенты с риском развития определенного состояния обычно включают тех, у которых имеется семейный анамнез состояния, или тех, у которых генетическим тестированием или скринингом было установлено, что они являются особенно восприимчивыми к развитию состояния.
В частности, соединения настоящего изобретения (включая фармацевтически приемлемые соли) могут быть использованы при лечении астмы, ХОБЛ, аллергического ринита, аллергического конъюнктивита, атопического дерматита, рака, гепатита B, гепатита С, ВИЧ, ВПЧ, бактериальных инфекций и дерматоз.
Настоящее описание еще дополнительно обеспечивает способ лечения или снижения риска заболевания или состояния, включающего или возникающего в результате аномального клеточного роста (например, рака), причем этот способ включает введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединение формулы (1) или его фармацевтически приемлемой соли, как определено выше.
Настоящее описание также относится к способу лечения или уменьшения риска обструктивного заболевания или состояния дыхательных путей (например, астмы или ХОЗЛ), который включает введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (1) или его фармацевтически приемлемой соли, как определено выше.
Для вышеупомянутого терапевтического применения вводимая доза, разумеется, может быть различной в зависимости от используемого соединения, способа введения, желаемого лечения и указанного нарушения. Например, дневная дозировка соединения согласно настоящему изобретению при введении путем ингаляции может находиться в диапазоне от 0,05 микрограмма на килограмм массы тела (мкг/кг) до 100 микрограммов на килограмм массы тела (мкг/кг). В качестве альтернативы, если соединение вводят перорально, то дневная дозировка соединения согласно настоящему изобретению может находиться в диапазоне от 0,01 микрограмма на килограмм массы тела (мкг/кг) до 100 миллиграммов на килограмм массы тела (мг/кг).
Фармацевтические композиции
Соединения формулы (1) и их фармацевтически приемлемые соли могут быть использованы сами по себе, но, как правило, вводятся в форме фармацевтической композиции, в которой соединение/соль формулы (1) (активный ингредиент) связано с фармацевтически приемлемым адъювантом, разбавителем или носителем. Традиционные способы выбора и получения подходящих фармацевтических составов описаны, например, в "Pharmaceuticals – The Science of Dosage Form Designs", M. E. Aulton, Churchill Livingstone, 1988.
В некоторых вариантах реализации введение может быть пероральным, внутривенным, подкожным, внутримышечным, внутриопухолевым, внутрикожным, интраназальным, ингаляционным, внутрипузырным, местным, подъязычным, буккальным, интраректальным, интратекальным, внутричерепным или другими формами локальной доставки.
В зависимости от способа введения, фармацевтическая композиция будет составлять от около 0,05 до около 99 мас. % (процентов по массе), более конкретно, от около 0,05 до около 80 мас. %, еще более конкретно, от около 0,10 до около 70 мас. %, и еще более конкретно от около 0,10 до около 50 мас. % активного ингредиента, причем все массовые проценты основаны на общей композиции.
Настоящее описание также обеспечивает фармацевтическую композицию, содержащую соединение формулы (1) или его фармацевтически приемлемую соль, как определено выше, в сочетании с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Настоящее описание дополнительно обеспечивает способ получения фармацевтической композиции настоящего изобретения, который включает смешивание соединения формулы (1) или его фармацевтически приемлемой соли, как определено выше, с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтические композиции могут вводиться местно (например, в кожу или в легкие и/или дыхательные пути) в форме, например, кремов, растворов, суспензий, аэрозолей гептафтораторкана (HFA) и составов сухого порошка, например, составов в ингаляторном устройство, известном как Turbuhaler®; или системно, например пероральным введением в форме таблеток, капсул, сиропов, порошков или гранул; или путем парентерального введения в форме стерильного раствора, суспензии или эмульсии для инъекций (включая внутривенное, подкожное, внутримышечное, внутрисосудистое или инфузионное); или путем ректального введения в форме суппозиториев.
Сухие порошковые составы и аэрозоли HFA соединений настоящего изобретения (включая фармацевтически приемлемые соли) могут вводиться путем оральной или назальной ингаляции. Для ингаляции соединение желательно тонко разделить. Тонкоизмельченное соединение предпочтительно имеет массовый средний диаметр менее 10 микрометров (мкм) и может быть суспендировано в смеси пропеллентов с помощью диспергатора, такого как C8–C20 жирная кислота или ее соль (например, олеиновая кислота), желчная соль, фосфолипид, алкилсахарид, перфторированное или полиэтоксилированное поверхностно–активное вещество или другой фармацевтически приемлемый диспергатор.
Соединения настоящего изобретения могут также вводиться с помощью ингалятора сухого порошка. Ингалятор может быть ингалятором с одной или несколькими дозами и может быть ингалятором сухого порошка, приводимым в действие дыханием.
Одна возможность состоит в том, чтобы смешать тонкоизмельченное соединение настоящего изобретения с веществом–носителем, например, моно–, ди– или полисахаридом, сахарным спиртом или другим полиолом. Подходящими носителями являются сахара, например, лактоза, глюкоза, рафиноза, мелезитоза, лактит, мальтит, трегалоза, сахароза, маннит; и крахмал. Альтернативно, тонкоизмельченное соединение может быть покрыто другим веществом. Порошковая смесь также может быть помещена в твердые желатиновые капсулы, каждая из которых содержит желаемую дозу активного соединения.
Другая возможность состоит в том, чтобы переработать тонкоизмельченный порошок в сферы, которые распадаются во время процедуры ингаляции. Этот сферонизированный порошок может быть загружен в резервуар с лекарством многодозового ингалятора, например, известного как Turbuhaler® , в котором дозирующее устройство отмеряет желаемую дозу, которая затем вдыхается пациентом. С помощью этой системы активный ингредиент с веществом–носителем или без него доставляется пациенту.
Для перорального введения соединение настоящего изобретения может быть смешано с адъювантом или носителем, например, лактозой, сахарозой, сорбитом, маннитом; крахмалом, например, крахмал картофельный, кукурузный крахмал или амилопектин; производным целлюлозы; связующим, например, желатин или поливинилпирролидон; и/или смазывающим веществом, например, стеарат магния, стеарат кальция, полиэтиленгликоль, воск, парафин и тому подобное, и затем спрессованное в таблетки. Если требуются таблетки с покрытием, ядра, полученные, как описано выше, могут быть покрыты концентрированным раствором сахара, который может содержать, например, гуммиарабик, желатин, тальк и диоксид титана. Альтернативно, таблетка может быть покрыта подходящим полимером, растворенным в легколетучем органическом растворителе.
Для получения мягких желатиновых капсул соединение настоящего изобретения может быть смешано, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы соединения, используя упомянутые выше наполнители для таблеток. Также жидкие или полутвердые композиции соединения настоящего изобретения могут быть помещены в твердые желатиновые капсулы.
Жидкие препараты для перорального применения могут быть в форме сиропов или суспензий, например, растворы, содержащие соединение настоящего изобретения, остаток которого представляет собой сахар, и смесь этанола, воды, глицерина и пропиленгликоля. Необязательно такие жидкие препараты могут содержать красители, ароматизаторы, сахарин и/или карбоксиметилцеллюлозу в качестве загустителя или другие наполнители, известные специалистам в данной области.
Комбинированная терапия
Соединения настоящего изобретения (то есть, соединения формулы (1) и их фармацевтически приемлемые соли) также можно вводить в комбинации с другими соединениями, используемыми для лечения вышеуказанных состояний.
Настоящее изобретение, следовательно, дополнительно относится к комбинированной терапии, в которой соединение настоящего изобретения или фармацевтическую композицию или композицию, содержащую соединение настоящего изобретения, вводят одновременно или последовательно или в виде комбинированного препарата с другим терапевтическим агентом или агентами для лечения одного или несколько из перечисленных состояний.
Противораковое лечение, определенное выше, может применяться в качестве единственной терапии или может включать, помимо соединения настоящего изобретения, обычную хирургию или лучевую терапию или химиотерапию. Такая химиотерапия может включать одну или несколько из следующих категорий противоопухолевых агентов:
(i) другие антипролиферативные/противоопухолевые лекарства и их комбинации, используемые в медицинской онкологии, такие как алкилирующие агенты (например, цис–платин, оксалиплатин, карбоплатин, циклофосфамид, азотная горчица, мелфалан, хлорамбуцил, бусульфан, темозоламид и нитрозомочевины); антиметаболиты (например, гемцитабин и антифолаты, такие как фторпиримидины, такие как 5–фторурацил и тегафур, ралтитрексед, метотрексат, цитозина арабинозид и гидроксимочевина); противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин–С, дактиномицин и митрамицин); антимитотические агенты (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как таксол, таксотер и ингибиторы полокиназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецин);
(ii) цитостатические агенты, такие как антиоэстрогены (например, тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен и йодоксифен), антиандрогены (например, бикалутамид, флутамид, нилутамид и ацетат ципротерона), антагонисты LHRH или агонисты LHRH (например гозерелин, лейпрорелин и бусерелин), прогестагены (ацетат мегестрола), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и экземестан) и ингибиторы 5α–редуктазы, такие как финастерид;
(iii) антиинвазивные средства (например, ингибиторы семейства c–Src–киназы, такие как 4–(6–хлор–2,3–метилендиоксианилино)–7–[2–(4–метилпиперазин–1–ил)этокси]–5–тетрагидропиран–4– илоксихиназолин (AZD0530; Международная заявка на патент WO 01/94341) и N–(2–хлор–6–метилфенил)–2–{6–[4–(2–гидроксиэтил)пиперазин–1–ил]–2–метилпиримидин–4 иламино} тиазол–5–карбоновая кислота (dasatinib, BMS–354825; J. Med. Chem.,2004, 47, 6658–6661), и ингибиторы металлопротеиназы, такие как маримастат, ингибиторы функции рецептора активатора плазминогена урокиназы или антитела Гепараназы);
(iv) ингибиторы функции фактора роста: например, такие ингибиторы включают антитела фактора роста и антитела рецептору фактора роста (например, антитело анти–erbB2 трастузумаб [Herceptin™], антитело анти–EGFR панитумумаб, антитело анти–erbBl цетуксимаб [Erbitux, C225] и любые антитела фактора роста или рецепторы фактора роста, описанные Stern et al. Critical reviews in oncology/hematology, 2005, Vol. 54, ppl 1–29); такие ингибиторы также включают ингибиторы тирозинкиназы, например ингибиторы семейства факторов роста эпидермиса (например, ингибиторы тирозинкиназы семейства EGFR, такие как N–(3–хлор–4–фторфенил)–7–метокси–6–(3–морфолинопропокси)хиназолин–4–амин(гефитиниб, ZD 1839), N–(3–этинилфенил)–6,7–бис(2–метоксиэтокси)хиназолин–4–амин(эрлотиниб, OSI–774) и 6–акриламидо–N–(3–хлор–4–фторфенил)–7– (3–морфолинопропокси)хиназолин–4–амин (CI 1033), ингибиторы тирозинкиназы erbB2, такие как лапатиниб, ингибиторы семейства факторов роста гепатоцитов, ингибиторы семейства тромбоцитарных факторов роста такие как иматиниб, ингибиторы серин/треонинкиназ (например, ингибиторы передачи сигналов Ras/Raf, такие как ингибиторы фарнезилтрансферазы, например сорафениб (BAY 43–9006)), ингибиторы клеточной передачи сигналов через киназы MEK и/или AKT, ингибиторы фактора роста гепатоцитов, ингибиторы c–kit, ингибиторы abl–киназы, рецептор IGF (инсулин–подобный фактор роста) ингибиторы киназы; ингибиторы авроракиназы (например, AZD1 152, PH739358, VX–680, MLN8054, R763, MP235, MP529, VX–528 и AX39459) и ингибиторы циклинзависимой киназы, такие как ингибиторы CDK2 и/или CDK4;
(v) антиангиогенные агенты, такие как те, которые ингибируют действие фактора роста эндотелия сосудов [например, антитело фактора роста эндотелиальных клеток сосудов бевацизумаба (Avastin™) и ингибиторы тирозинкиназы рецептора VEGF, такие как 4–(4–бром–2–фторанилино)–6–метокси–7–(1–метилпиперидин–4–илметокси)хиназолин (ZD6474; Пример 2 в WO 01/32651), 4–(4–фтор–2–метилиндол–5–илокси)–6–метокси–7–(3–пирролидин–1–илпропокси)хиназолин (AZD2171; Пример 240 в WO 00/47212), ваталаниб (PTK787; WO 98/35985) и SUl 1248 (сунитиниб; WO 01/60814), соединения, такие как соединения, описанные в Международных патентных заявках WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354 и соединения, которые работают по другим механизмам (например, линомид, ингибиторы функции интегрина αvβ3 и ангиостатин)];
(vi) повреждающие сосуды агенты, такие как комбретастатин А4 и соединения, описанные в Международных патентных заявках WO 99/02166, WO 00/40529, WO 00/41669,WO 01/92224, WO 02/04434 и WO 02/08213;
(vii) антисмысловые терапии, например, те, которые направлены на цели, перечисленные выше, такие как ISIS 2503, антисмысловая антирас;
(viii) подходы генной терапии, включая например подходы для замены аберрантных генов, такие как аберрантные р53 или аберрантные BRCA1 или BRCA2, GDEPT (генно–направленная ферментная лекарственная терапия) подходы, такие как с использованием цитозин–деаминазы, тимидинкиназы или фермента бактериальной нитроредуктазы, и подходы для повышения терпимости пациента к химиотерапии или лучевой терапии, такой как генная терапия с множественной лекарственной устойчивостью;
(ix) иммунотерапевтические подходы, в том числе, например, подходы ex–vivo и in–vivo для повышения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или колониестимулирующий фактор гранулоцитов–макрофагов, подходы для снижения анергии Т–клеток, подходы с использованием трансфицированных иммунных клеток, таких как цитокин–трансфицированные дендритные клетки, подходы с использованием трансфицированных цитокинами линий опухолевых клеток и подходы с использованием антиидиотипических антител;
(x) Ингибиторы контрольных точек, включая, но не ограничиваясь, антитела для PD–1/PD–L1, CTLA–4, TIM–3, LAG–3, OX–40, GITR, VISTA, 4–1BB, CD40, TIGIT, BTLA;
(xi) Ингибиторы киназы, включая, но не ограничиваясь ими, ингибиторы низкомолекулярных или моноклональных антител BRAF, EGFR, ALK, RAS, RAF, VEGF, HER, c–MET, MEK, FGFR, BCR–ABL, PI3K;
(xii) Ингибиторы рака/иммунного обмена, включая, но не ограничиваясь, ингибиторы IDO, TDO, GLS, IDH, аргиназа, рецептор аденозина, CD73, CD39;
(xiii) Эпигенетические модуляторы, включая, но не ограничиваясь ими, ингибиторы HDAC, бромодомен, метилтрансферазу;
(xiv) Модулятор пути развития, включая, помимо прочего, Smo, Wnt, YAP;
(xv) Другие противораковые или иммунно–онкологические биопрепараты, включая, но не ограничиваясь, онколитический вирус, BCG, CART, цитокины; и
(xv) Антитела, включая, но не ограничиваясь, антитело к PD–1 и антитело к PD–L1.
Кроме того, для лечения воспалительных заболеваний, ХОЗЛ, астмы и аллергического ринита соединения настоящего изобретения можно комбинировать с такими агентами, как ингибиторы фактора альфа некроза (TNF–альфа), такими как моноклональные антитела анти–TNF (например, Remicade, CDP–870 и адалимумаб) и рецептор TNF–иммуноглобулина (такие как Энбрел); неселективные ингибиторы COX–1/COX–2 циклооксигеназы, применяемые местно или системно (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак азапропазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин), ингибиторы COX–2 (такие как мелоксикам, целекоксиб, рофекоксиб, вальдекоксиб, люмарококсиб, парекоксиб и эторикоксиб); кортикостероиды; глюкокортикостероиды (вводимые местным, пероральным, внутримышечным, внутривенным или внутрисуставным путем); бета–агонисты; антигистамины; метотрексат, лефуномид; гидроксихлорохин, д–пеницилламин, ауранофин или другие парентеральные или пероральные препараты золота.
Для лечения инфекционных заболеваний, соединения настоящего изобретения можно комбинировать с такими агентами, как нуклеозидные противовирусные препараты; ненуклеозидные противовирусные препараты, включая, но не ограничиваясь ими, ингибиторы проникновения, полимеразу, обратную транскриптазу, протеазу, интегразу; моноклональные антитела против специфических вирусов; миРНК терапии; антибиотики; и противогрибковые.
Для лечения HBV соединения настоящего изобретения могут быть объединены с такими агентами, как антивирусные нуклеозиды.
Настоящее изобретение также дополнительно относится к другим врожденным иммунным агонистам, нацеленным на следующие классы рецепторов, включая, но не ограничиваясь ими, TLR (Toll–подобный рецептор); NLR (Nod–подобный рецептор); CLR (рецептор лектина С–типа); RLR (RIG–I–подобный рецептор); и STING (стимулятор гена интерферона).
Настоящее изобретение также дополнительно относится к комбинации соединения настоящего изобретения и ингибитора биосинтеза лейкотриена, ингибитора 5–липоксигеназы (5–LO) или антагониста белка, активирующего 5–липоксигеназу (FLAP), такого как; зилеутон; АВТ–761; фенлейтон; тепоксалин; Ebbott–79175; Ebbott –85761; N–(5–замещенный)тиофен–2–алкилсульфонамид; 2,6–ди–трет–бутилфенол гидразоны; метокситетрагидропиран, такой как Zeneca ZD–2138; соединение SB–210661; пиридинилзамещенное 2–цианонафталиновое соединение, такое как L–739,010; соединение 2–цианохинолина, такое как L–746,530; или соединение индола или хинолина, такое как МК–591, МК–886 и BAY x 1005.
Настоящее изобретение дополнительно относится к комбинации соединения настоящего изобретения и антагониста рецептора лейкотриенов (LTB4, LTC4, LTD4 и LTE4), выбранных из группы, состоящей из фенотиазин–3–онов, таких как L–651,392; амидиносоединений, таких как CGS–25019c; бензоксаламины, такие как онтазоласт; бензолкарбоксимидамиды, такие как BIIL 284/260; и такие соединения, как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК–679), RG–12525, Ro–245913, иралукаст (CGP 45715A) и BAY x 7195.
Настоящее изобретение также дополнительно относится к комбинации соединения настоящего изобретения и ингибитора фосфодиэстеразы (PDE), такого как метилксантанин, включая теофиллин и аминофиллин; селективный ингибитор изофермента PDE, включающий ингибитор PDE4, ингибитор изоформы PDE4D или ингибитор PDE5.
Настоящее изобретение также относится к комбинации соединения настоящего изобретения и антагониста рецептора гистамина типа 1, такого как цетиризин, лоратадин, дезлоратадин, фексофенадин, акривастин, терфенадин, астемизол, азеластин, левокабастин, хлорфенирамин, прометазин, циклизин или мизоластин; применяется перорально, местно или парентерально.
Настоящее изобретение также дополнительно относится к комбинации соединения настоящего изобретения и гастропротекторного антагониста рецептора гистамина типа 2.
Настоящее изобретение также относится к комбинации соединения настоящего изобретения и антагониста рецептора гистамина типа 4.
Настоящее изобретение также дополнительно относится к комбинации соединения настоящего изобретения и альфа–1/альфа–2–адренорецепторного агониста вазоконстрикторного симпатомиметического агента, такого как пропилгекседрин, фенилэфрин, фенилпропаноламин, эфедрин, псевдоэфедрин, нафазолин гидрохлорид, оксиметазолина гидрохлорид,гидрохлорид тетрагидрозолина, гидрохлорид ксилометазолина, гидрохлорид трамазолина или гидрохлорид этилнорепинефрина.
Настоящее изобретение дополнительно относится к комбинации соединения настоящего изобретения и антихолинергического агента, включая антагонисты мускариновых рецепторов (M1, M2 и M3), такие как атропин, гиосцин, гликопирролат, ипратропия бромид, тиотропия бромид, окситропия бромид, пирензепин или телензепин.
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения вместе с агонистом бета–адренорецепторов (включая подтипы бета–рецепторов 1–4), такими как изопреналин, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, мезилат битолтерола и пирбутерол.
Настоящее изобретение дополнительно относится к комбинации соединения настоящего изобретения и хромона, такого как хромогликат натрия или недокромил натрия.
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения с миметиком инсулиноподобного фактора роста I типа (IGF–I).
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения и глюкокортикоида, такого как флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, пропионат флутиказона, циклесонид или мометазона фуроат.
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения с ингибитором матричных металлопротеаз (ММР), то есть стромелизинов, коллагеназ и желатиназ, а также аггреканазы; особенно коллагеназа–1 (ММР–I), коллагеназа–2 (ММР–8), коллагеназа–3 (ММР–13), стромелизин–1 (ММР–3), стромелизин–2 (ММР–IO) и стромелизин–3 (MMP–11) и MMP–9 и MMP–12.
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения вместе с модуляторами функции рецептора хемокинов, такими как антагонисты CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR1O и CCR11 (для семейства CC); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства C–X–C) и CX3CR1 для семейства C–X3–C.
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения вместе с цитокином или модулятором функции цитокинов, включая альфа–, бета– и гамма–интерферон; интерлейкины (IL), включая ILl до 15, и антагонисты или ингибиторы интерлейкина, включая агенты, которые действуют на пути передачи сигналов цитокинов.
Настоящее изобретение еще дополнительно относится к комбинации соединения настоящего изобретения с иммуноглобулином (Ig) или препаратом Ig или антагонистом или антителом, модулирующим функцию Ig, таким как анти–IgE (омализумаб).
Настоящее изобретение дополнительно относится к комбинации соединения настоящего изобретения и другого системно или местноприменяемого противовоспалительного агента, такого как талидомид или его производное, ретиноид, дитранол или кальципотриол.
Настоящее изобретение дополнительно относится к комбинации соединения настоящего изобретения с антибактериальным агентом, таким как производное пенициллина, тетрациклин, макролид, бета–лактам, фторхинолон, метронидазол, ингаляционный аминогликозид; противовирусное средство, включая ацикловир, фамцикловир, валацикловир, ганцикловир, цидофовир, амантадин, римантадин, рибавирин, занамавир и осельтамавир; ингибитор протеазы, такой как индинавир, нелфинавир, ритонавир и саквинавир; нуклеозидный ингибитор обратной транскриптазы, такой как диданозин, ламивудин, ставудин, зальцитабин или зидовудин; или ненуклеозидный ингибитор обратной транскриптазы, такой как невирапин или эфавиренц.
В дополнительном аспекте настоящее изобретение предлагает комбинацию (например, для лечения ХОЗЛ, астмы или аллергического ринита) соединения формулы (1) или его фармацевтически приемлемой соли, как определено выше, и одного или нескольких агентов, независимо выбранных из:
•агонист нестероидного глюкокортикоидного рецептора (GR–рецептор);
•селективный агонист β2–адренорецептора (такой как метапротеренол, изопротеренол,изопреналин, альбутерол, сальбутамол, формотерол, сальметерол, тербуталин,орципреналин, мезилат битолтерола, пирбутерол или индакатерол);
•ингибитор фосфодиэстеразы (такой как ингибитор PDE4);
• ингибитор протеазы (такой как ингибитор нейтрофильной эластазы или матриксной металлопротеазы ММР–12);
•глюкокортикоид;
•антихолинергический агент;
• модулятор функции хемокинового рецептора (такой как антагонист рецептора CCR1); и
• ингибитор функции киназы (такой как киназы p38 или IKK).
Настоящее изобретение также обеспечивает фармацевтический продукт, содержащий в комбинации препарат первого активного ингредиента, который представляет собой соединение формулы (1) или его фармацевтически приемлемую соль, как определено выше, и препарат второго активного ингредиента, который представляет собой
•агонист нестероидного глюкокортикоидного рецептора (GR–рецептор);
•селективный агонист β2–адренорецептора;
•ингибитор фосфодиэстеразы;
•ингибиторы протеазы;
• глюкокортикоид;
• антихолинергический агент;
• модулятор функции хемокиновых рецепторов; или
• ингибитор функции киназы;
для одновременного, последовательного или раздельного использования в терапии.
В другом аспекте настоящее изобретение предлагает набор, содержащий препарат первого активного ингредиента, который представляет собой соединение формулы (1) или его фармацевтически приемлемую соль, как определено выше, и препарат второго активного ингредиента, который представляет собой
•агонист нестероидного глюкокортикоидного рецептора (GR–рецептор);
•селективный агонист β2–адренорецептора;
• ингибитор фосфодиэстеразы; ингибиторы протеазы;
• глюкокортикоид;
• антихолинергический агент;
•модулятор функции хемокиновых рецепторов; или
• ингибитор функции киназы;
И инструкции по одновременному, последовательному или раздельному введению препаратов пациенту, нуждающемуся в этом.
Примерные варианты реализации
Вариант реализации I–1. Соединение, имеющее структуру Формулы (1), или его фармацевтически приемлемая соль,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –COOH, и –SO2CH3, где алкил необязательно замещен –OH, –NH2, –COOH, или –SO2CH3;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилeн)]–, и–[O(CH2CH2)]n–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А; и
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH.
Вариант реализации I–2. Соединение по варианту реализации I–1 или его фармацевтически приемлемая соль, где R1b представляет собой –(CH2)2CH3.
Вариант реализации I–3. Соединение по варианту реализации I–1 или его фармацевтически приемлемая соль, где R1b представляет собой –(CH2)3CH3.
Вариант реализации I–4. Соединение по любому из вариантов реализации I–1–I–3 или его фармацевтически приемлемая соль, где R1a представляет собой C1–C4 алкил, необязательно замещенный –ОН.
Вариант реализации I–5. Соединение по любому из вариантов реализации I–1–I–4 или его фармацевтически приемлемая соль, где R 1a представляет собой –CH2C(CH3)2OH.
Вариант реализации I–6. Соединение по любому из вариантов реализации I–1–I–4 или его фармацевтически приемлемая соль, где представляет собой или .
Вариант реализации I–7. Соединение по любому из вариантов реализации I–1–I–3 или его фармацевтически приемлемая соль, где R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH.
Вариант реализации I–8. Соединение по любому из вариантов реализации I–1–I–3 и I–7 или его фармацевтически приемлемая соль, где представляет собой .
Вариант реализации I–9. Соединение по любому из вариантов реализации I–1–I–3 или его фармацевтически приемлемая соль, где R1a представляет собой H.
Вариант реализации I–10. Соединение по любому из вариантов реализации I–1–I–9 или его фармацевтически приемлемая соль, где X представляет собой C1–C4 алкил, где алкил замещен А.
Вариант реализации I–11. Соединение по любому из вариантов реализации I–1–I–10 или его фармацевтически приемлемая соль, где X представляет собой C1–C4 алкил, где алкил замещен , где L2 представляет собой связь.
Вариант реализации I–12. Соединение по любому из вариантов реализации I–1–I–9 или его фармацевтически приемлемая соль, где X представляет собой CH3.
Вариант реализации I–13. Соединение по любому из вариантов реализации I–1–I–9 или его фармацевтически приемлемая соль, где X представляет собой H.
Вариант реализации I–14. Соединение по любому из вариантов реализации I–1–I–13 или его фармацевтически приемлемая соль, где L1 представляет собой –CH2–, –CH2CH2–, или –O–.
Вариант реализации I–15. Соединение по любому из вариантов реализации I–1–I–14 или его фармацевтически приемлемая соль, где L1 представляет собой –CH2–.
Вариант реализации I–16. Соединение по любому из вариантов реализации I–1–I–15 или его фармацевтически приемлемая соль, где Y представляет собой C1–C3 алкил или арил.
Вариант реализации I–17. Соединение по любому из вариантов реализации I–1–I–16 или его фармацевтически приемлемая соль, где Y представляет собой арил, где арил замещен C1–C3 алкокси.
Вариант реализации I–18. Соединение по любому из вариантов реализации I–1–I–17, или его фармацевтически приемлемая соль, где Y представляет собой арил, где арил замещен А.
Вариант реализации I–19. Соединение по любому из вариантов реализации I–1–I–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации I–20. Соединение по любому из вариантов реализации I–1–I–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации I–21. Соединение по любому из вариантов реализации I–1–I–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации I–22. Соединение по любому из вариантов реализации I–1–I–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации I–23. Соединение по любому из вариантов реализации I–1–I–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации I–24. Соединение по любому из вариантов реализации I–1–I–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации I–25. Соединение по любому из вариантов реализации I–1–I–24, или его фармацевтически приемлемая соль, где L2 представляет собой –(CH2)n–.
Вариант реализации I–26. Соединение по любому из вариантов реализации I–1–I–25, или его фармацевтически приемлемая соль, где n представляет собой один или два.
Вариант реализации I–27. Соединение по любому из вариантов реализации I–1–I–24, или его фармацевтически приемлемая соль, где L2 представляет собой .
Вариант реализации I–28. Соединение по любому из вариантов реализации I–1–I–24, или его фармацевтически приемлемая соль, где L2 представляет собой .
Вариант реализации I–29. Соединение по любому из вариантов реализации I–1–I–24, или его фармацевтически приемлемая соль, где L2 представляет собой –C(O)NH(CH2)n–.
Вариант реализации I–30. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1a),
где
X представляет собой H или CH3
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–,, , и–CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации I–31. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1a),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации I–32. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1b),
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации I–33. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1b),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации I–34. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1c),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
Вариант реализации I–35. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1d),
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
Вариант реализации I–36. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1e),
где
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации I–37. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1f),
.
Вариант реализации I–38. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1g),
где
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации I–39. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1h),
где
R4 представляет собой H или C1–C3 алкокси.
Вариант реализации I–40. Соединение варианта реализации I–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1i),
где
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
Вариант реализации I–41. Соединение или фармацевтически приемлемая соль, выбранные из группы, состоящей из:
Вариант реализации I–42. Фармацевтическая композиция, содержащая соединение по любому из вариантов реализации I–1–I–41 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Вариант реализации I–43. Способ лечения состояния, связанного с модуляцией TLR7, у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения по любому из вариантов реализации I–1–I–41 или его фармацевтически приемлемой соли.
Вариант реализации I–44. Способ по варианту реализации I–43, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации I–45. Соединение по любому из вариантов реализации I–1–I–41 или его фармацевтически приемлемая соль для применения при лечении состояния, связанного с модуляцией TLR7.
Вариант реализации I–46. Соединение варианта реализации I–45, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации I–47. Применение соединения по любому из вариантов реализации I–1–I–41 или его фармацевтически приемлемой соли при получении лекарственного средства для лечения состояния, связанного с модуляцией TLR7.
Вариант реализации I–48. Применение варианта реализации I–47, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации II–1. Соединение, имеющее структуру Формулы (1), или его фармацевтически приемлемая соль,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, и ,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, или ;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилена)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А; и
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH.
Вариант реализации II–2. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где R1b представляет собой –(CH2)2CH3.
Вариант реализации II–3. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где R1b представляет собой –(CH2)3CH3.
Вариант реализации II–4. Соединение по любому из вариантов реализации II–1–II–3, или его фармацевтически приемлемая соль, где R1a представляет собой C1–C4 алкил, необязательно замещенный –ОН, –OCH3, –SCH3, –SO2CH3
Вариант реализации II–5. Соединение по любому из вариантов реализации II–1–II–4, или его фармацевтически приемлемая соль, где R1a представляет собой –CH2C(CH3)2OH.
Вариант реализации II–6. Соединение по любому из вариантов реализации II–1–II–4, или его фармацевтически приемлемая соль, где представляет собой , , , , , или
Вариант реализации II–7. Соединение по любому из вариантов реализации II–1–II–3, или его фармацевтически приемлемая соль, где R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH.
Вариант реализации II–8. Соединение по любому из вариантов реализации II–1–II 3 и II–7, или его фармацевтически приемлемая соль, где представляет собой .
Вариант реализации II–9. Соединение по любому из вариантов реализации II–1–II–3, или его фармацевтически приемлемая соль, где R1a представляет собой H.
Вариант реализации II–10. Соединение по любому из вариантов реализации II–1–II–9, или его фармацевтически приемлемая соль, где X представляет собой C1–C4 алкил, где алкил замещен А.
Вариант реализации II–11. Соединение по любому из вариантов реализации II–1–II–10, или его фармацевтически приемлемая соль, где X представляет собой C1–C4 алкил, где алкил замещен , где L2 представляет собой связь.
Вариант реализации II–12. Соединение по любому из вариантов реализации II–1–II–9, или его фармацевтически приемлемая соль, где X представляет собой CH3.
Вариант реализации II–13. Соединение по любому из вариантов реализации II–1–II–9, или его фармацевтически приемлемая соль, где X представляет собой H.
Вариант реализации II–14. Соединение по любому из вариантов реализации II–1–II–13, или его фармацевтически приемлемая соль, где L1 представляет собой –CH2–, –CH2CH2–, –O–, или –S–.
Вариант реализации II–15. Соединение по любому из вариантов реализации II–1–II–14, или его фармацевтически приемлемая соль, где L1 представляет собой –CH2–.
Вариант реализации II–16. Соединение по любому из вариантов реализации II–1–II–15, или его фармацевтически приемлемая соль, где Y представляет собой C1–C3 алкил или арил.
Вариант реализации II–17. Соединение по любому из вариантов реализации II–1–II–16, или его фармацевтически приемлемая соль, где Y представляет собой арил, где арил замещен C1–C3 алкокси.
Вариант реализации II–18. Соединение по любому из вариантов реализации II–1–II–17, или его фармацевтически приемлемая соль, где Y представляет собой арил, где арил замещен А.
Вариант реализации II–19. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–20. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–21. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–22. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–23. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–24. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–25. Соединение по любому из вариантов реализации II–1–II–18, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации II–26. Соединение по любому из вариантов реализации II–1–II–25, или его фармацевтически приемлемая соль, где L2 представляет собой –(CH2)n–.
Вариант реализации II–27. Соединение по любому из вариантов реализации II–1–II–26, или его фармацевтически приемлемая соль, где n представляет собой один или два.
Вариант реализации II–28. Соединение по любому из вариантов реализации II–1–II–25, или его фармацевтически приемлемая соль, где L2 представляет собой .
Вариант реализации II–29. Соединение по любому из вариантов реализации II–1–II–25, или его фармацевтически приемлемая соль, где L2 представляет собой .
Вариант реализации II–30. Соединение по любому из вариантов реализации II–1–II–25, или его фармацевтически приемлемая соль, где L2 представляет собой –C(O)NH(CH2)n–.
Вариант реализации II–31. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1a),
где
X представляет собой H или CH3
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации II–32. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1a),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации II–33. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1b),
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации II–34. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1b),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации II–35. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1c),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
Вариант реализации II–36. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1d),
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2, –S–; и
Y представляет собой H или C1–C3 алкил.
Вариант реализации II–37. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1e),
где R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации II–38. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1f),
.
Вариант реализации II–39. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1g),
где R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации II–40. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1h),
где R4 представляет собой H или C1–C3 алкокси.
Вариант реализации II–41. Соединение варианта реализации II–1, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1i),
где
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –CH2CH2–, и –S–; и
Y представляет собой H или C1–C3 алкил.
Вариант реализации II–42. Соединение или фармацевтически приемлемая соль, выбранные из группы, состоящей из:
Вариант реализации II–43. Фармацевтическая композиция, содержащая соединение по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Вариант реализации II–44. Способ лечения состояния, связанного с модуляцией TLR7, у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемой соли.
Вариант реализации II–45. Способ по варианту реализации II–44, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации II–46. Способ варианта реализации II–45, где введение представляет собой пероральную, внутривенную, подкожную, внутримышечную, внутриопухолевую, внутрикожную, интраназальную, ингаляционную, внутрипузырную, местную, подъязычную, буккальную, интраректальную, интратекальную, внутричерепную или другие формы локальной доставки.
Вариант реализации II–47. Соединение по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
Вариант реализации II–48. Соединение по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемая соль для применения при лечении состояния, связанного с модуляцией TLR7.
Вариант реализации II–49. Соединение варианта реализации II–48, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации II–50. Применение соединения по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемой соли при получении лекарственного средства для лечения состояния, связанного с модуляцией TLR7.
Вариант реализации II–51. Применение варианта реализации II–50, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации II–52. Фармацевтическая композиция варианта реализации II–43, дополнительно включающая, по меньшей мере, один или несколько дополнительных терапевтических агентов.
Вариант реализации II–53. Фармацевтическая композиция варианта реализации II–52, где, по меньшей мере, один или несколько дополнительных терапевтических агентов представляет собой антивирусный нуклеозид.
Вариант реализации II–54. Фармацевтическая композиция варианта реализации II–52, где, по меньшей мере, один или несколько дополнительных терапевтических агентов представляет собой антитело к PD–1 или антитело к PD–L1.
Вариант реализации II–55. Способ лечения HBV у субъекта, нуждающегося в этом, включающий введение соединения по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемой соли в комбинации с противовирусным нуклеозидом.
Вариант реализации II–56. Способ лечения рака у субъекта, нуждающегося в этом, включающий введение соединения по любому из вариантов реализации II–1–II–42 или его фармацевтически приемлемой соли в комбинации с антителом к PD–1 или антителом к PD–L1.
Вариант реализации III–1. Соединение, имеющее структуру Формулы (1), или его фармацевтически приемлемая соль,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, ; и A,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, , или A;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех;
n представляет собой целое число от одного до четырех;
где соединение замещено, по меньшей мере, одним А; и
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3; и
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу.
Вариант реализации III–2. Соединение, имеющее структуру Формулы (1), или его фармацевтически приемлемая соль,
(1)
где
R1a выбирают из группы, состоящей из H, C1–C4 алкила, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, и ,
где алкил необязательно замещен –OH, –NH2, –NHAc, –COOH, –SO2CH3, –SCH3, –OCH3, или ;
R1b представляет собой C2–C5 алкил;
X выбирают из группы, состоящей из H и C1–C4 алкила, где алкил необязательно замещен A, –OH, или –C(CH3)2OH;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –S–, –SO2–, –NH–, и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила, арила и гетероарила, где алкил, арил и гетероарил необязательно замещены 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–, , , , –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; и –С(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–;
m представляет собой целое число от нуля до четырех; и
n представляет собой целое число от одного до четырех; и
где соединение замещено, по меньшей мере, одним А; и
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; и L2 представляет собой –CH2–; тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3; и
когда X представляет собой –CH3; L1 представляет собой –CH2–; Y представляет собой арил замещенный A; L2 представляет собой –CH2–, –O–(CH2)2–O(CH2)2–, или –O–(CH2)2–O(CH2)2(CF2)–; и A представляет собой ; тогда A и L1 не находятся в пара–положении по отношению друг к другу.
Вариант реализации III–3. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где R1b представляет собой –(CH2)2CH3.
Вариант реализации III–4. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где R1b представляет собой –(CH2)3CH3.
Вариант реализации III–5. Соединение по любому из вариантов реализации III–1–III–4, или его фармацевтически приемлемая соль, где R1a представляет собой C1–C4 алкил, необязательно замещенный –ОН, –OCH3, –SCH3, или –SO2CH3.
Вариант реализации III–6. Соединение по любому из вариантов реализации III–1–III–5, или его фармацевтически приемлемая соль, где R1a представляет собой –CH2C(CH3)2OH.
Вариант реализации III–7. Соединение по любому из вариантов реализации III–1–III–6, или его фармацевтически приемлемая соль, где представляет собой , , , , , или .
Вариант реализации III–8. Соединение по любому из вариантов реализации III–1–III–4, или его фармацевтически приемлемая соль, где R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH.
Вариант реализации III–9. Соединение по любому из вариантов реализации III–1–III–4 и III–8, или его фармацевтически приемлемая соль, где представляет собой .
Вариант реализации III–10. Соединение по любому из вариантов реализации III–1–IIII–4, или его фармацевтически приемлемая соль, где R1a представляет собой H.
Вариант реализации III–11. Соединение по любому из вариантов реализации III–1–III–10, или его фармацевтически приемлемая соль, где X представляет собой C1–C4 алкил, где алкил замещен А.
Вариант реализации III–12. Соединение по любому из вариантов реализации III–1–III–11, или его фармацевтически приемлемая соль, где X представляет собой C1–C4 алкил, где алкил замещен , где L2 представляет собой связь.
Вариант реализации III–13. Соединение по любому из вариантов реализации III–1–III–10, или его фармацевтически приемлемая соль, где X представляет собой CH3.
Вариант реализации III–14. Соединение по любому из вариантов реализации III–1–III–10, или его фармацевтически приемлемая соль, где X представляет собой H.
Вариант реализации III–15. Соединение по любому из вариантов реализации III–1–III–14, или его фармацевтически приемлемая соль, где L1 представляет собой –CH2–, –CH2CH2–, –O–, или –S–.
Вариант реализации III–16. Соединение по любому из вариантов реализации III–1–III–15, или его фармацевтически приемлемая соль, где L1 представляет собой –CH2–.
Вариант реализации III–17. Соединение по любому из вариантов реализации III–1–III–16, или его фармацевтически приемлемая соль, где Y представляет собой C1–C3 алкил или арил.
Вариант реализации III–18. Соединение по любому из вариантов реализации III–1–III–17, или его фармацевтически приемлемая соль, где Y представляет собой арил, где арил замещен C1–C3 алкокси.
Вариант реализации III–19. Соединение по любому из вариантов реализации III–1–III–18, или его фармацевтически приемлемая соль, где Y представляет собой арил, где арил замещен А.
Вариант реализации III–20. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–21. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–22. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–23. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–24. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–25. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–26. Соединение по любому из вариантов реализации III–1–III–19, или его фармацевтически приемлемая соль, где A представляет собой .
Вариант реализации III–27. Соединение по любому из вариантов реализации III–1–III–26, или его фармацевтически приемлемая соль, где L2 представляет собой –(CH2)n–.
Вариант реализации III–28. Соединение по любому из вариантов реализации III–1–III–27, или его фармацевтически приемлемая соль, где n представляет собой один или два.
Вариант реализации III–29. Соединение по любому из вариантов реализации III–1–III–26, или его фармацевтически приемлемая соль, где L2 представляет собой .
Вариант реализации III–30. Соединение по любому из вариантов реализации III–1–III–26, или его фармацевтически приемлемая соль, где L2 представляет собой .
Вариант реализации III–31. Соединение по любому из вариантов реализации III–1–III–26, или его фармацевтически приемлемая соль, где L2 представляет собой –C(O)NH(CH2)n–.
Вариант реализации III–32. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1a),
где
X представляет собой H или CH3
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации III–33. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1a),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации III–34. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1b),
где
X представляет собой H или CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–,, , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , и , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации III–35. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1b),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
A выбирают из группы, состоящей из , , , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , , –C(O)NH(CH2)n–, –[O(CH2CH2)]n–, –[O(C1–C4 алкилен)]–, и –[O(CH2CH2)]n–OCH2CH2CF2–; –C(O)NHCH2CH2–[O(CH2CH2)]m–; C(O)NHCH2CH2–[O(CH2CH2)]m–OCH2CH2CF2–; и
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации III–36. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1c),
где
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O–, –S–, –CF2–, , , и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
Вариант реализации III–37. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1d),
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a, или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, и –CH2CH2, –S–; и
Y представляет собой H или C1–C3 алкил.
Вариант реализации III–38. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1e),
где R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации III–39. Соединение варианта реализации III–1 или III–22, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1f),
.
Вариант реализации III–40. Соединение варианта реализации III–1 или III–22, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1g),
где R3 представляет собой H, C1–C3 алкил, или C1–C3 алкокси.
Вариант реализации III–41. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1h),
где R4 представляет собой H или C1–C3 алкокси.
Вариант реализации III–42. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1i),
где
L1 выбирают из группы, состоящей из связи, –CH2–, –CF2–, , , –O–, –CH2CH2–, и –S–; и
Y представляет собой H или C1–C3 алкил.
Вариант реализации III–43. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1j),
(1j).
Вариант реализации III–44. Соединение варианта реализации III–1 или III–2, или его фармацевтически приемлемая соль, где соединение Формулы (1) представляет собой соединение Формулы (1k),
(1k).
Вариант реализации III–45. Соединение или фармацевтически приемлемая соль, выбранные из группы, состоящей из:
Вариант реализации III–46. Фармацевтическая композиция, содержащая соединение по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Вариант реализации III–47. Способ лечения состояния, связанного с модуляцией TLR7, у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемой соли.
Вариант реализации III–48. Способ варианта реализации III–47, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации III–49. Способ варианта реализации III–47 или III–48, где введение представляет собой пероральную, внутривенную, подкожную, внутримышечную, внутриопухолевую, внутрикожную, интраназальную, ингаляционную, внутрипузырную, местную, подъязычную, буккальную, интраректальную, интратекальную, внутричерепную или другие формы локальной доставки.
Вариант реализации III–50. Соединение по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
Вариант реализации III–51. Соединение по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемая соль для применения при лечении состояния, связанного с модуляцией TLR7.
Вариант реализации III–52. Соединение варианта реализации III–51, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации III–53. Применение соединения по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемой соли при получении лекарственного средства для лечения состояния, связанного с модуляцией TLR7.
Вариант реализации III–54. Применение варианта реализации III–53, где состояние представляет собой вирусную инфекцию или рак.
Вариант реализации III–55. Фармацевтическая композиция варианта реализации III–46, дополнительно включающая, по меньшей мере, один или несколько дополнительных терапевтических агентов.
Вариант реализации III–56. Фармацевтическая композиция варианта реализации III–55, где, по меньшей мере, один или несколько дополнительных терапевтических агентов представляет собой антивирусный нуклеозид.
Вариант реализации III–57. Фармацевтическая композиция варианта реализации III–55, где, по меньшей мере, один или несколько дополнительных терапевтических агентов представляет собой антитело к PD–1 или антитело к PD–L1.
Вариант реализации III–58. Способ лечения HBV у субъекта, нуждающегося в этом, включающий введение соединения по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемой соли в комбинации с противовирусным нуклеозидом.
Вариант реализации III–59. Способ лечения рака у субъекта, нуждающегося в этом, включающий введение соединения по любому из вариантов реализации III–1–III–45 или его фармацевтически приемлемой соли в комбинации с антителом к PD–1 или антителом к PD–L1.
Примеры
Следующие примеры приведены для иллюстрации настоящего описания и не должны рассматриваться как его ограничение. В этих примерах все части и проценты являются массовыми, если не указано иное. Сокращения в примерах указаны ниже.
Сокращения
Пример 1: 3–(4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)пропановая кислота (Соединение 1)

Стадия 1: метил 4–(2–(этоксикарбонил)–3–оксобутил)–3–метоксибензоат
Гидрид натрия (60% в минеральном масле, 1,25 экв.) добавляли порциями в течение 10 мин к раствору этил 3–оксобутаноата (1,2 экв.) В THF (0,8 М) при 0 oC. Полученную суспензию перемешивали при 0°C в течение 10 минут и добавляли раствор метил 4–(бромметил)–3–метоксибензоата (1,0 экв.) в THF (0,5 М) по каплям в течение 10 мин. Смесь нагревали до 70°C и перемешивали в течение 3 часов. Смеси давали остыть и затем выливали в ледяную воду и перемешивали в течение 30 мин. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=от 100/1 до 5/1) с получением указанного в заголовке соединения.
Стадия 2: метил 4–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–3–метоксибензоат
Смесь метил 4–(2–(этоксикарбонил)–3–оксобутил)–3–метоксибензоата (1,0 экв.) и гуанидинкарбоната (1,0 экв.) в MeOH (0,2 М) перемешивали в течение ночи при 65°C и позволяли остыть до комнатной температуры. Осадок собирали фильтрованием и суспендировали в воде. Твердое вещество собирали фильтрованием и промывали МеОН и EA с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: метил 4–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–3–метоксибензоат
Mетил 4–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–3–метоксибензоат (1,0 экв.) в POCl3 (0,5М) перемешивали в течение ночи при 100°C в атмосфере N2. Смесь охлаждали до комнатной температуры и растворитель удаляли. К остатку добавляли воду и рН смеси доводили до 7 с помощью твердого вещества NaHCO3. Полученную смесь нагревали при 50°C в течение 1 часа и затем оставляли охлаждаться до комнатной температуры. Твердое вещество собирали, промывали водой, EA и сушили в вакууме с получением указанных в заголовке соединений.
Стадия 4: метил 4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксибензоат
В суспензию метил 4–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–3–метоксибензоата (1,0 экв.) в NMP (0,3 М) добавляли бутан–1–амин (4,5 экв.). Полученную смесь перемешивали в течение ночи при 125°C и смесь охлаждали до комнатной температуры. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=от 20:1 до 1:5) с получением указанного в заголовке соединения.
Стадия 5: (4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)метанол
К перемешиваемому раствору метила 4–((2–амино–4– (бутиламино)–6–метилпиримидин–5–ил)метил)–3метоксибензоата (1,0 экв.) в THF (0,1 М) по каплям добавляли LiAlH4 (1 М в THF, 2,0 экв.). Полученную смесь перемешивали при 0°C в течение 10 минут и при комнатной температуре в течение 1 часа. Смесь разбавляли EA и гасили 2 н. NaOH. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 6: 4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксибензальдегид
К перемешиваемому раствору (4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)метанола (1,0 экв.) в THF (0,06 М) добавляли MnO2 (5,0 экв.). Полученную смесь перемешивали в течение ночи при 50 oC. Смесь фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения.
Стадия 7: этил (Е)–3–(4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)акрилат
Смесь 4 –((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксибензальдегида (1,0 экв.) и этил 2–(трифенил–λ5–фосфанилиден)ацетата (1,2 экв.) в THF (0,1 М) перемешивали при 70°C в течении 5 ч. Смесь концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент 0–5% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 8: этил 3–(4 –((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)пропаноат
К перемешиваемому раствору этил (E)–3–(4 –((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)акрилата (1,0 экв.) в EA (0,02 М) добавляли Pd/C (20 мас. %). Полученную смесь перемешивали в течение ночи при 50°C под H2. Смесь фильтровали. Фильтрат концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент 0–5% МеОН в DCM) с получением указанного в заголовке соединения.
Стадия 9: 3–(4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)пропановая кислота
К перемешиваемому раствору этил 3–(4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)пропаноата (1,0 экв.) в МеОН (0,03 М) добавляли 1 н. NaOH ( 5,0 экв.). Полученную смесь перемешивали в течение ночи при 45 oC. Растворитель удаляли. Остаток нейтрализовали 2 н. HCl до pH 7. Суспензию фильтровали. Твердое вещество собирали и растворяли в HCl/диоксане. Раствор концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль HCl).
ЖХ–МС: [M+H] = 373,4
1H ЯМР (400 МГц, DMSO–d6) δ 12,32 (ш с, 1H), 12,11 (ш с, 1H), 7,83 (ш с, 1H), 7,41 (ш с, 2H), 6,90 (с,1H), 6,70 (с, 2H), 3,83 (с, 3H), 3,64 (с, 2H), 3,36–3,30 (м, 2H), 2,82–2,77 (м, 2H), 2,54–2,50 (м, 2H), 2,11 (с, 3H), 1,50–1,43 (м, 2H), 1,23–1,15 (м, 2H), 0,85 (т, J=7,2 Гц, 3H).
Пример 2: 5–(4–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин (Соединение 2)
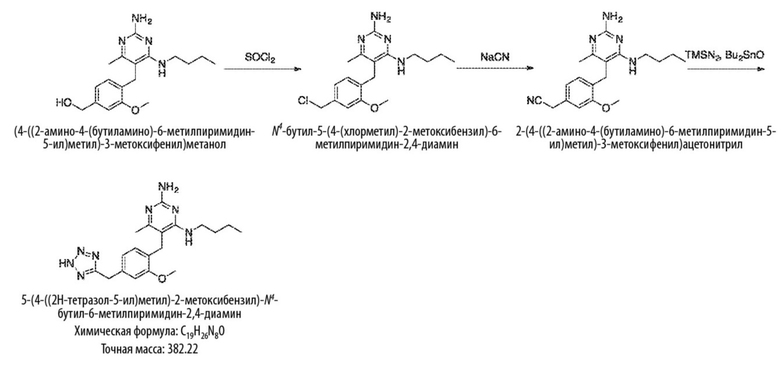
Стадия 1: N4–бутил–5–(4–(хлорметил)–2–метоксибензил)–6–метилпиримидин–2,4–диамин
К перемешиваемому раствору (4 –((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)метанола (1,0 экв., Пример 1 – Стадия 5) в DCM (0,14 M) добавляли SOCl2 (1,8 экв.) при комнатной температуре под N2. Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разделяли между DCM/насыщ. водн. NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия 2: 2–(4 –((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)ацетонитрил
Смесь N4–бутил–5–(4–(хлорметил)–2–метоксибензил)–6–метилпиримидин–2,4–диамина (1,0 экв.) в 1:1 DMSO/DMF (0,14 M) и NaCN (2,8 экв.) перемешивали в течение ночи при комнатной температуре. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент 0–5% MeOH в DCM) с получением указанного в заголовке соединения.
Стадия 3: 5–(4–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин
К перемешиваемой смеси 2–(4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)ацетонитрила (1,0 экв.), Bu2SnO (2,0 экв.) в NMP (0,06 M) добавляли TMSN3 (10,0 экв.). Полученную смесь перемешивали в течении ночи при 125°C под N2. Смесь охлаждали до комнатной температуры. Смесь фильтровали с получением указанного в заголовке соединения в виде белого порошка.
ЖХ–МС: [M+H] = 383,5
1H ЯМР (400 МГц, DMSO–d6) δ 12,19 (с, 1H), 7,85 (ш с, 1H), 7,34 (ш с, 2H), 7,01 (с, 1H), 6,74 (с, 2H), 4,24 (с, 2H), 3,84 (с, 3H), 3,65 (с, 2H), 3,37–3,34 (м, 2H), 2,09 (с, 3H), 1,48–1,41 (м, 2H), 1,23–1,16 (м, 2H), 0,83 (т, J=7,2 Гц, 3H).
Пример 3A: 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин (Соединение 3)

Стадия A: метил 3–(бромметил)–4–метоксибензоат
К смеси метил 4–метокси–3–метилбензоата (1,0 экв.) в CCl4 (0,37 M), NBS (1,1 экв.) и AIBN (0,2 экв.) перемешивали в течении ночи при 95°C в атмосфере азота. Полученную смесь концентрировали и разделяли между DCM и водой, органический слой сушили над Na2SO4, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=от 100:1 до 20:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Получение (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола
При получении (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола следовали Примеру 1 – Стадии 1–5, но с использованием метил 3–(бромметил)–4–метоксибензоата вместо метил 4–(бромметил)–3–метоксибензоата на Стадии 1.
Получение 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина
При получении 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина следовали Примеру 2 – Стадии 1–3, но с использованием (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола вместо 4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)метанола на Стадии 1. Указанное в заголовке соединение очищали с помощью препаративной ВЭЖХ с использованием ацетонитрила/воды в качестве элюента с получением твердого вещества белого цвета.
ЖХ–МС: [M+H] = 383,3
1H ЯМР (400 МГц, DMSO–d6) δ 7,03 (д, J=7,6 Гц, 1H), 6,90 (д, J=8,4 Гц, 1H), 6,73 (с, 1H), 6,60 (ш с, 1H), 6,31 (с, 2H), 3,97 (с, 2H), 3,80 (с, 3H), 3,60 (с, 2H), 3,29–3,24 (м, 2H), 2,02 (с, 3H), 1,43–1,37 (м, 2H), 1,22–1,17 (м, 2H), 0,84 (т, J=7,2 Гц, 3H).
Пример 3B: 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин (Соединение 3)
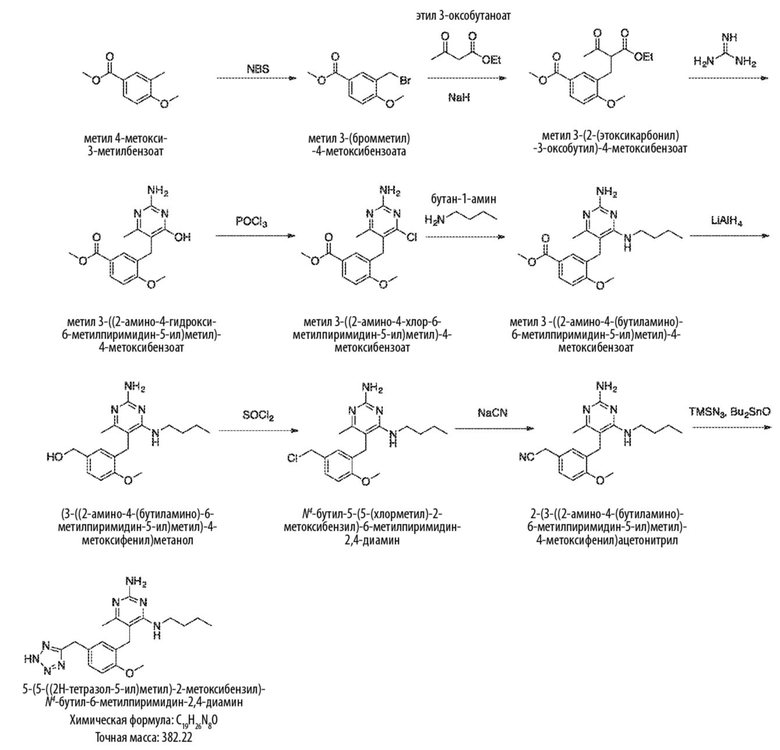
Стадия 1: метил 3–(бромметил)–4–метоксибензоат
Смесь метил 4–метокси–3–метилбензоата (1,0 экв.) в CCl4 (0,37 M), NBS (1,1 экв.) и AIBN (0,2 экв.) перемешивали в течении ночи при 95°C в атмосфере азота. Полученную смесь концентрировали и разделяли между DCM и водой, органический слой сушили над Na2SO4, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=от 100:1 до 20:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: метил 3–(2–(этоксикарбонил)–3–оксобутил)–4–метоксибензоат
К смеси этил 3–оксобутаноата (1,2 экв.) в THF (0,35 M) при 0°C добавляли порциями 60% NaH (1,25 экв.) в N2. Полученную суспензию перемешивали при 0°C в течение 10 мин, затем по каплям в течение 10 мин добавляли раствор метил 3–(бромметил)–4–метоксибензоата (1,0 экв.) в THF (2 M). Полученную смесь перемешивали в течение ночи при 70 oC. Смесь охлаждали до комнатной температуры и добавляли ледяную воду. Смесь экстрагировали этилацетатом. Органический слой разделяли и сушили над Na2SO4, концентрировали и очищали флэш хроматографией на диоксиде кремния (элюент PE/EA=50:1–5:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 3: метил 3–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–4–метоксибензоат
К раствору метил 3–(2–(этоксикарбонил)–3–оксобутил)–4–метоксибензоата (1,0 экв.) в MeOH (0,2 M) добавляли гуанидинкарбонат (1,0 экв.). Полученную смесь перемешивали при 65°C в течение 16 ч и затем охлаждали до комнатной температуры. Осадок собирали фильтрованием, твердое вещество растирали с водой и фильтровали. Осадок на фильтре промывали MeOH и EtOAc, сушили в вакууме, получая указанное в заголовке соединение в виде белого твердого вещества.
Стадия 4: метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоат
Суспензию метил 3–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в POCl3 (0,55 M) перемешивали при 100°C в течение 15 ч. Реакционной смеси давали остыть до комнатной температуры и POCl3 упаривали досуха при пониженном давлении. Остаток разбавляли водой и доводили pH до 7 с помощью насыщенного NaHCO3. Затем смесь нагревали при 50°C в течение 1 ч, охлаждали до комнатной температуры. Осадок собирали фильтрованием и осадок на фильтре промывали водой и EtOAc, сушили в вакууме с получением указанного в заголовке соединения..
Стадия 5: метил 3 –((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоат
Смесь бутан–1–амин (4,5 экв.) и метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в NMP (0,3 M) перемешивали в течение ночи при 150 oC. Реакционной смеси давали остыть, разбавляли EtOAc, промывали водой и солевым раствором. Органическую фазу высушивали и упаривали при пониженном давлении. Остаток суспендировали в диэтиловом эфире и твердое вещество собирали фильтрованием, получая указанное в заголовке соединение в виде твердого вещества.
Стадия 6: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанол
К перемешиваемому раствору метил 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в THF (0,3 M) добавляли 2,5M LiAlH4 (2,0 экв.) в THF при 0°C в атмосфере N2. Полученную смесь перемешивали при 0°C в течение 30 мин и затем при комнатной температуре в течение 2 часов. Смесь разбавляли EtOAc и гасили 2 н. NaOH. Суспензию фильтровали и фильтрат разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали и концентрировали, получая указанное в заголовке соединение в виде твердого вещества белого цвета.
Стадия 7: N4–бутил–5–(5–(хлорметил)–2–метоксибензил)–6–метилпиримидин–2,4–диамин
К перемешиваемому раствору (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола (1,0 экв.) в DCM (0,34 M) добавляли SOCl2 (1,3 экв.) при комнатной температуре в атмосфере N2. Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разделяли между DCM и насыщенным водным NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия 8: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)ацетонитрил
Смесь N4–бутил–5–(5–(хлорметил)–2–метоксибензил)–6–метилпиримидин–2,4–диамина (1,0 экв.) в 1:1 DMSO/DMF (0,34 M) и NaCN (2,8 экв.) перемешивали в течение ночи при комнатной температуре. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент 0–5% MeOH в DCM) с получением указанного в заголовке соединения.
Стадия 9: 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин
К перемешиваемой смеси 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)ацетонитрила (1,0 экв.) в NMP (0,06 M) и Bu2SnO (2,0 экв.) добавляли TMSN3 (10,0 экв.). Полученную смесь перемешивали в течении ночи при 125°C в атмосфере N2. Смесь охлаждали до комнатной температуры, и смесь концентрировали и очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения в виде твердого вещества.
ЖХ–МС: [M+H]+ = 383,3
1H ЯМР (400 МГц, DMSO–d6) δ 7,03 (д, J=7,6 Гц, 1H), 6,90 (д, J=8,4 Гц, 1H), 6,73 (с, 1H), 6,60 (ш с, 1H), 6,31 (с, 2H), 3,97 (с, 2H), 3,80 (с, 3H), 3,60 (с, 2H), 3,29–3,24 (м, 2H), 2,02 (с, 3H), 1,43–1,37 (м, 2H), 1,22–1,17 (м, 2H), 0,84 (т, J=7,2 Гц, 3H).
Пример 4: 3–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)пропановая кислота (Соединение 4)
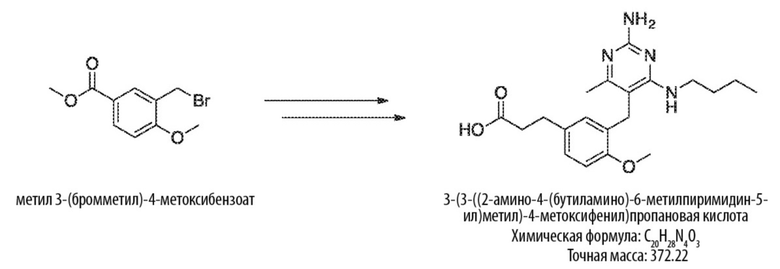
Указанное в заголовке соединение получали в соответствии с методиками, описанными, в Примере 1 – Стадии 1–9, но используя метил 3–(бромметил)–4–метоксибензоат вместо метил 4–(бромметил)–3–метоксибензоата на Стадии 1.
ЖХ–МС: [M+H] = 373,2
1H ЯМР (400 МГц, DMSO–d6) δ 12,47 (с, 1H), 7,81 (т, J=4,8 Гц, 1H), 7,46 (ш с, 2H), 7,06 (д, J=8,4 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,72 (с, 1H), 3,81 (с, 3H), 3,76 (с, 2H), 3,40–3,36 (м, 2H), 2,68 (т, J=7,2 Гц, 2H), 2,41 (т, J=7,2 Гц, 2H), 2,13 (с, 3H), 1,50–1,44 (м, 2H), 1,24–1,18 (м, 2H), 0,85 (т, J=7,2 Гц, 3H).
Пример 5: 2–(5–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)–2Н–тетразол–2–ил)уксусная кислота (Соединение 5)
Пример 6: 2–(5–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)–1Н–тетразол–1–ил)уксусная кислота (Соединение 6)
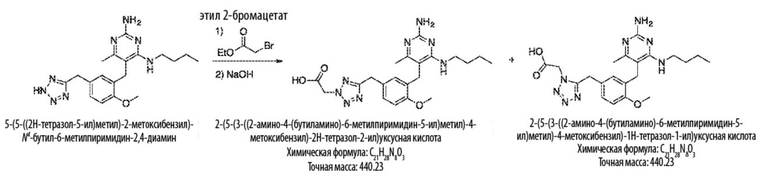
К перемешиваемому раствору 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин (1,0 экв., Пример 3) в ацетоне (0,05 M) добавляли K2CO3 (1,5 экв.) при 0°C в N2. Полученную смесь перемешивали при 0°C в течение 30 минут и по каплям добавляли этил 2–бромацетат (1,5 экв.). Смесь медленно нагревали до комнатной температуры и перемешивали в течение 5 часов. Смесь гасили нас. NH4Cl. Смесь разделяли в DCM/вода. Органическую фазу сушили над Na2SO4, концентрировали и очищали с помощью препаративной ВЭЖХ с получением двух региоизомеров в соотношении 3:2 в виде белых порошков. К перемешиваемому раствору каждого изомера (1,0 экв.) в MeOH (0,02 M) добавляли 1 н. NaOH (5,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 16 ч. Реакцию контролировали с помощью ТСХ и ЖХ–МС. Смесь нейтрализовали 1 н. HCl и очищали с получением указанных в заголовке соединений.
Изомер 1: 2–(5–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)–2H–тетразол–2–ил)уксусная кислота
ЖХ–МС: [M+H] = 441,5
1H ЯМР (400 МГц, DMSO–d6) δ 12,62 (ш с, 1H), 7,82 (ш с, 1H), 7,50 (ш с, 2H), 7,11 (д, J=7,6 Гц, 1H), 6,95 (д, J=8,8 Гц, 1H), 6,79 (с, 1H), 5,31 (с, 2H), 4,16 (с, 2H), 3,82 (с, 3H), 3,66 (с, 2H), 3,39–3,30 (м, 2H), 2,10 (с, 3H), 1,45–1,38 (м, 2H), 1,3–1,2 (м, 2H), 0,85 (т, J=7,2 Гц, 3H).
Изомер 2: 2–(5–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)–1H–тетразол–1–ил) уксусная кислота
ЖХ–МС: [M+H] = 441,5
1H ЯМР (400 МГц, DMSO–d6) δ 12,69 (с, 1H), 7,85 (ш с, 1H), 7,51 (ш с, 2H), 7,10 (д, J=8,0 Гц, 1H), 6,95 (д, J=8,4 Гц, 1H), 6,82 (с, 1H), 5,58 (с, 2H), 4,11 (с, 2H), 3,81 (с, 3H), 3,67 (с, 2H), 3,38–3,35 (м, 2H), 2,11 (с, 3H), 1,49–1,42 (м, 2H), 1,24–1,18 (м, 2H), 0,85 (т, J=7,2 Гц, 3H).
Пример 7: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)фосфоновая кислота (Соединение 7)
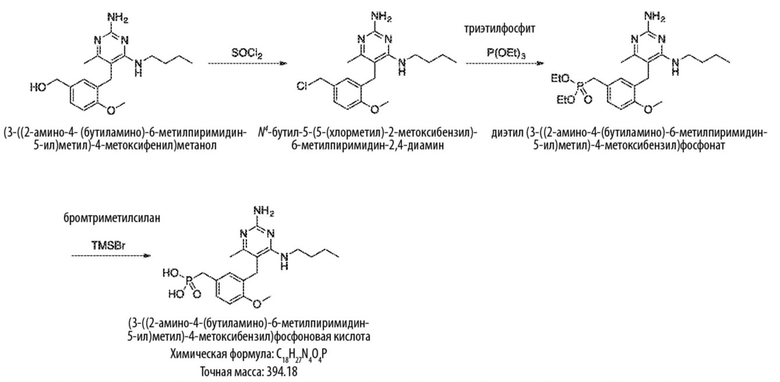
Стадия 1: N4–бутил–5–(5–(хлорметил)–2–метоксибензил)–6–метилпиримидин–2,4–диамин
Указанное в заголовке соединение получали в соответствии с методиками, описанными в Примере 2 – Стадия 1, но с использованием (3–((2–амино–4– (бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола вместо (4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)метанола.
Стадия 2: диэтил (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)фосфонат
N 4–бутил–5–(5–(хлорметил)–2–метоксибензил)–6–метилпиримидин–2,4–диамин (1,0 экв.) в неразбавленном триэтилфосфите (0,1 M) перемешивали при 140°C в течение 2 ч. Смесь концентрировали с получением указанного в заголовке соединения.
Стадия 3: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)фосфоновая кислота
К перемешиваемому раствору диэтил (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензил)фосфоната (1,0 экв.) в DCM (0,04 М) добавляли бромтриметилсилан (4,0 экв.). Полученную смесь перемешивали в течение 2 ч. при комнатной температуре и тогда разделяли между DCM/водой. Органический слой сушили над Na2SO4, концентрировали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/NH3) и лиофилизировали с получением белого порошка, который повторно растворяли в MeCN/H2O/1 н. HCl (4:2:1) и лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль HCl).
ЖХ–МС: [M+H] = 395,1
1H ЯМР (400 МГц, DMSO–d6) δ 12,64 (ш с, 2H), 7,82 (т, J=5,6 Гц, 1H), 7,48 (ш с, 2H), 7,08 (д, J=8,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,74 (с, 1H), 3,81 (с, 3H), 3,66 (с, 2H), 3,37–3,34 (м, 2H), 2,80 (д, J=20,9 Гц, 2H), 2,12 (с, 3H), 1,50–1,45 (м, 2H), 1,27–1,22 (м, 2H), 0,86 (т, J=7,2 Гц, 3H).
Пример 8: (S)–2–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)пентан–1–ол (Соединение 8)
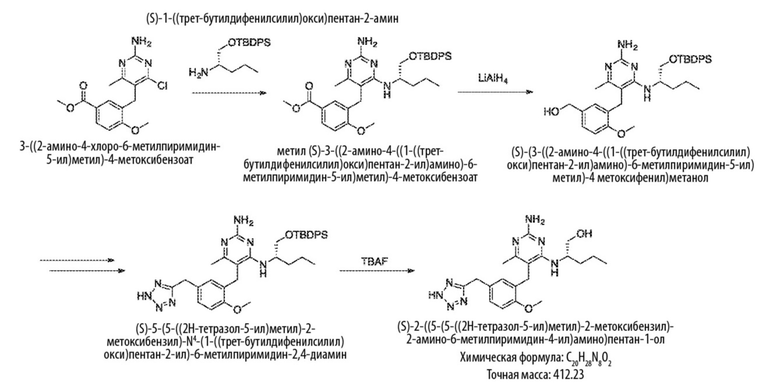
Стадия A: (S)–1–((трет–бутилдифенилсилил)окси)пентан–2–амин
К смеси (S)–2–аминопентан–1–ола (1,0 экв.) в DCM (0,2 M), TEA (5,0 экв.) и DMAP (0,3 экв.) при 0°C добавляли TBDPSCl (1,5 экв.). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь разбавляли водой и DCM, разделяли и водный слой дважды экстрагировали DCM. Объединенные органические слои сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 200:1 до 80:1) с получением указанного в заголовке соединения в виде коричневого масла.
Получение метил (S)–3–((2–амино–4–((1–((трет–бутилдифенилсилил)окси)пентан–2–ил)амино)–6–метилпиримидин–5–ил)метил)–4 –метоксибензоата
Получение метил (S)–3–((2–амино–4–((1–((трет–бутилдифенилсилил)окси)пентан–2–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоат, следуя Примеру 1 – Стадия 4, но используя метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоат вместо метил 4–((2– амино–4–хлор–6–метилпиримидин–5–ил)метил)–3–метоксибензоата и (S)–1–((трет–бутилдифенилсилил)окси)пентан–2–амин вместо бутан–1–амина.
Получение (S)–(3–((2–амино–4–((1–((трет–бутилдифенилсилил)окси)пентан–2–ил)амино)–6–метилпиримидин–5–ил)метил)–4 метоксифенил)метанола
Получение (S)–(3–((2–амино–4–((1–((трет–бутилдифенилсилил)окси)пентан–2–ил)амино)–6–метилпиримидин–5–ил)метил )–4–метоксифенил)метанола, следуя Примеру 1 – Стадия 5, но используя метил (S)–3–((2–амино–4–((1–((трет–бутилдифенилсилил)окси)пентан–2–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоат вместо метила 4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксибензоата.
Получение (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–(1–((трет–бутилдифенилсилил)окси)пентан–2–ил)–6–метилпиримидин–2,4–диамина
Получение (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–(1–((трет–бутилдифенилсилил)окси)пентан–2–ил )–6–метилпиримидин–2,4–диамина по Примеру 2 – Стадии с 1 по 3, но с использованием (S)–(3–((2–амино–4–((1–((трет–бутилдифенилсилил)окси)пентан)–2–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола вместо (4–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–3–метоксифенил)метанола.
Получение (S)–2–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)пентан–1–ола
К раствору (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–(1–((трет–бутилдифенилсилил)окси)пентан–2–ил)–6–метилпиримидин–2,4–диамина (1,0 экв.) в безводном THF (0,05 M) при 0°C добавляли TBAF (10 экв.). Полученную смесь перемешивали при комнатной температуре в течение 16 ч. ЖХМС показала завершение реакции. Затем смесь концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты)
ЖХ–МС: [M+H] = 413,2
1H ЯМР (400 МГц, DMSO) δ 8,19 (с, 1H), 7,05 (д, J=8,4 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,85 (с, 1H), 6,68–6,60 (м, 2H), 6,39–6,34 (м, 1H), 4,23–4,20 (м, 1H), 3,99 (с, 2H), 3,81 (с, 3H), 3,69–3,60 (с, 2H), 3,41–3,33 (м, 2H), 2,11 (с, 3H), 1,48–1,45 (м, 1H), 1,35–1,28 (м, 1H), 1,13–1,09 (м, 2H), 0,79 (т, J=7,2 Гц, 3H).
Пример 9: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропановая кислота (Соединение 9)
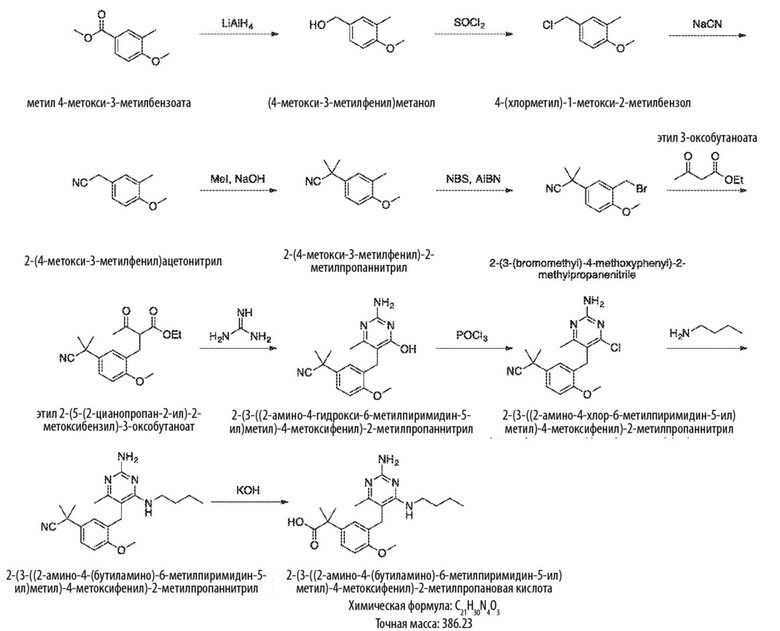
Стадия 1: (4–метокси–3–метилфенил)метанол
К раствору метил–4–метокси–3–метилбензоата (1,0 экв.) в безводном THF (0,3 М) при 0°C добавляли по каплям LiAlH4 (2,5 М, 2,5 экв.) в атмосфере N2. Полученную смесь перемешивали при комнатной температуре в течение 2 ч., затем смесь гасили 1,25 M раствором NaOH при 0°C и трижды экстрагировали EtOAc. Органический слой сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое использовали непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: 4–(хлорметил)–1–метокси–2–метилбензол
К раствору (4–метокси–3–метилфенил)метанола (1,0 экв.) в DCM (0,3 M) при 0°C добавляли SOCl2 (2,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь гасили раствором NaHCO3 и экстрагировали DCM. Органический слой сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла.
Стадия 3: 2–(4–метокси–3–метилфенил)ацетонитрил
К раствору 4–(хлорметил)–1–метокси–2–метилбензола (1,0 экв.) в 1:1 DMSO/DMF (0,5 M) добавляли NaCN (3,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 16 ч., затем добавляли воду и смесь экстрагировали EtOAc. Органический слой промывали рассолом дважды, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: PE/EA=от 100:1 до 30:1) с получением указанного в заголовке соединения в виде желтоватого масла.
Стадия 4: 2–(4–метокси–3–метилфенил)–2–метилпропаннитрил
К смеси 2–(4–метокси–3–метилфенил)ацетонитрила (1,0 экв.) в DMSO (0,4 M) при 0°C добавляли иодметан (10 экв.) и раствор 50% NaOH (6,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Затем смесь разбавляли водой и EtOAc, водный слой снова дважды экстрагировали EtOAc. Органический слой объединяли, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: PE/EA=от 100:1 до 30:1) с получением указанного в заголовке соединения в виде слегка желтоватого масла.
Стадия 5: 2–(3–(бромметил)–4–метоксифенил)–2–метилпропаннитрил
К раствору 2–(4–метокси–3–метилфенил)–2–метилпропаннитрила (1,0 экв.) в CCl4 (0,2 M) добавляли NBS (1,1 экв.) и AIBN (0,2 экв.). Полученную смесь перемешивали при 95°C в течение 16 ч. Затем смесь фильтровали и фильтрат концентрировали при пониженном давлении, с получением неочищенного сырого продукта, который очищали с помощью колоночной хроматографии на диоксиде кремния (элюент: PE/EA=от 100:1 до 30:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 6: этил 2–(5–(2–цианопропан–2–ил)–2–метоксибензил)–3–оксобутаноат
К раствору этил 3–оксобутаноата (1,2 экв.) в безводном THF (0,17 M) при 0°C добавляли порциями NaH (60% в минеральном масле, 1,3 экв.). После перемешивания в течение 10 минут раствор 2–(3–(бромметил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв.) в THF (0,3 М) добавляли по каплям в вышеуказанную смесь в течение 10 минут. Полученную смесь перемешивали при 70°C в течение 16 ч, гасили водой и смесь экстрагировали EtOAc, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на диоксиде кремния (элюент: PE/EA=от 50:1 до 10:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 7: 2–(3–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрил
Смесь этил 2–(5–(2–цианопропан–2–ил)–2–метоксибензил)–3–оксобутаноата (1,0 экв.) и гуанидинкарбоната (1,0 экв.) в MeOH (0,3 M) перемешивали при 65°C в течение 16 ч. Осажденное твердое вещество собирали фильтрованием. Твердое вещество промывали водой и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 8: 2–(3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрил
Раствор 2–(3–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв.) в POCl3 (0,3 M) перемешивали при 100°C в течение 2 ч. Реакционной смеси давали остыть до комнатной температуры и POCl3 упаривали досуха при пониженном давлении. Остаток разбавляли водой и pH доводили до 8 твердым NaHCO3. Затем смесь перемешивали при 50°C в течение 1 ч., охлаждали до комнатной температуры и выпавшее в осадок твердое вещество собирали фильтрованием. Остаток после фильтрования промывали водой, сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 9: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрил
К смеси 2–(3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв.) в NMP (0,2 M) добавляли бутан–1–амин (4,5 экв.) и перемешивали при 110°C в течение 16 ч. Затем в вышеуказанную смесь добавляли воду и EtOAc, органический слой сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 100:1 до 10:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 10: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропановая кислота
К смеси 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв.) в 1:1 этан–1,2–диол/H2O (0,075 M) добавляли KOH (10 экв.) и перемешивал при 150°C в течение 2 ч. Затем смеси давали остыть до комнатной температуры и pH доводили до 8 1M раствором HCl. Выпавшее в осадок твердое вещество фильтровали и трижды промывали водой. Остаток после фильтрования растворяли в MeCN, H2O и 4M растворе HCl в диоксане, затем раствор лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль HCl).
ЖХ–МС: [M+H] = 387,2
1H ЯМР (400 МГц, DMSO) δ 12,34 (с, 1H), 7,92 (с, 1H), 7,45 (ш с, 2H), 7,20 (д, J=8,4 Гц, 1H), 6,96 (д, J=8,4 Гц, 1H), 6,85 (с, 1H), 3,82 (с, 3H), 3,70 (с, 2H), 3,5–3,4 (м, 2H), 2,13 (с, 3H), 1,51–1,45 (м, 2H), 1,36 (с, 6H), 1,27–1,18 (м, 2H), 0,86 (т, J=7,2 Гц, 3H).
Пример 10: 5–(5–(2–(2H–тетразол–5–ил)пропан–2–ил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин (Соединение 10)

Смесь 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв., Пример 9 – Стадия 9) в диоксане (0,06 M), TMSN3 (10 экв.) и Bu2SnO (2,0 экв.) запечатывали в трубчатый реактор. Смесь перемешивали при 125°C в течение 16 ч. ЖХМС показала завершение реакции. Затем смесь концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты)
ЖХ–МС: [M+H]+ = 411,2
1H ЯМР (400 МГц, DMSO) δ 6,97 (д, J=6,4 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 6,72 (с, 1H), 6,44 (ш с, 1H), 6,16 (ш с, 2H), 3,79 (с, 3H), 3,58 (с, 2H), 3,27–3,25 (м, 2H), 1,98 (с, 3H), 1,61 (с, 6H), 1,44–1,40 (м, 2H), 1,24–1,18 (м, 2H), 0,85 (т, J=7,2 Гц, 3H).
Пример 11: N4–бутил–5–(2–метокси–5–(2H–тетразол–5–ил)бензил)–6–метилпиримидин–2,4–диамин (Соединение 11)
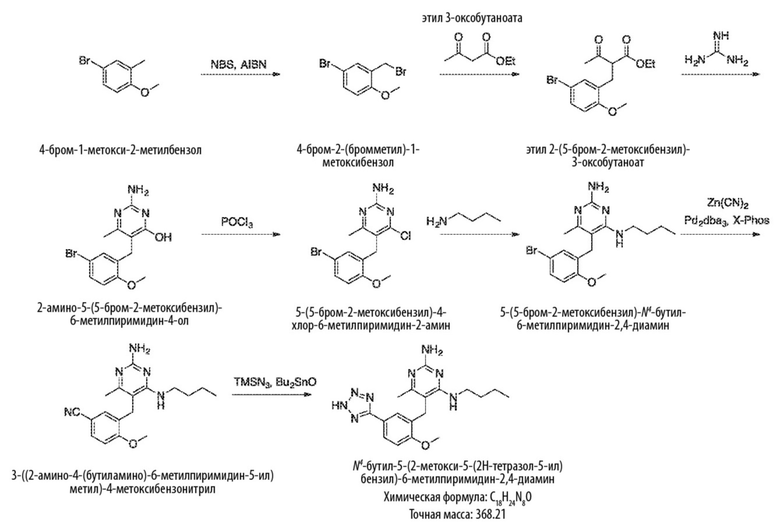
Стадия 1: 4–бром–2–(бромметил)–1–метоксибензол
Смесь 4–бром–1–метокси–2–метилбензола (1,0 экв.) в CCl4 (0,3 M), NBS (1,1 экв.), и AIBN (0,2 экв.) перемешивали при 95°C в течение 3 ч. Затем смесь фильтровали и фильтрат концентрировали при пониженном давлении, получая сырой продукт, который очищали с помощью колоночной хроматографии на силикагеле (элюент: PE) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: этил 2–(5–бром–2–метоксибензил)–3–оксобутаноат
К перемешиваемому раствору этил 3–оксобутаноата (1,2 экв.) в безводном THF (0,5 M) при 0°C добавляли порциями 60% NaH (1,3 экв.). После перемешивания в течение 10 минут раствор 4–бром–2–(бромметил)–1–метоксибензола (1,0 экв.) в THF (0,8 М) добавляли по каплям в вышеуказанную смесь в течение 10 минут. Затем полученную смесь перемешивали при 70°C в течение 16 ч, добавляли воду и экстрагировали EtOAc. Объединенную органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: PE/EA=от 100:1 до 10:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 3: 2–амино–5–(5–бром–2–метоксибензил)–6–метилпиримидин–4–ол
Смесь этил 2–(5–бром–2–метоксибензил)–3–оксобутаноата (1,0 экв.) и гуанидинкарбоната (1,0 экв.) в MeOH (0,3 M) перемешивали при 65°C в течение 16 ч. После завершения реакции смесь охлаждали до комнатной температуры. Осажденное твердое вещество собирали фильтрованием и остаток после фильтрования промывали водой, сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 4: 5–(5–бром–2–метоксибензил)–4–хлор–6–метилпиримидин–2–амин
Раствор 2–амино–5–(5–бром–2–метоксибензил)–6–метилпиримидин–4–ола (1,0 экв.) в POCl3 (0,5 M) перемешивали при 100°C в течение 16 ч. Реакционной смеси давали остыть до комнатной температуры и POCl3 упаривали досуха при пониженном давлении. Остаток разбавляли водой и pH доводили до 8 твердым NaHCO3, и затем смесь перемешивали при 50°C в течение 1 ч, охлаждали до комнатной температуры. Осажденное твердое вещество собирали фильтрованием. Остаток после фильтрования промывали водой и MeOH, сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 5: 5–(5–бром–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин
Смесь 5–(5–бром–2–метоксибензил)–4–хлор–6–метилпиримидин–2–амина (1,0 экв.) в NMP (0,4 M) и бутан–1–амина (4,5 экв.) перемешивали при 130°C в течение 16 ч. Смесь разбавляли водой и EtOAc, органический шар промывали рассолом и сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 500:1 до 50:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 6: 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензонитрил
Смесь 5–(5–бром–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина (1,0 экв.) в DMF (0,1 M), Zn(CN)2 (1,5 экв.), Pd2(dba)3 (0,08 экв.) и X–Phos (0,2 экв.) перемешивали при 110°C в течение 1 ч. в атмосфере аргона. Полученную смесь охлаждали до комнатной температуры и разбавляли водой и EtOAc. Органический слой разделяли, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 100:1 до 10:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 7: N4–бутил–5–(2–метокси–5–(2H–тетразол–5–ил)бензил)–6–метилпиримидин–2,4–диамин
Смесь 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензонитрила (1,0 экв.) в диоксане (0,07 M), TMSN3 (10 экв.) и Bu2SnO (2,0 экв.) перемешивали при 120°C в течение 4 ч. ЖХМС показала завершение реакции. Затем смесь концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты).
ЖХ–МС: [M+H] = 369,1
1H ЯМР (400 МГц, DMSO) δ 7,84 (д, J=8,4 Гц, 1H), 7,56 (ш с, 1H), 7,44 (с, 1H), 7,09 (д, J=8,4 Гц, 1H), 7,05 (ш с, 2H), 3,90 (с, 3H), 3,73 (с, 2H), 3,4–3,3 (м, 2H), 2,13 (с, 3H), 1,49–1,41 (м, 2H), 1,19–1,12 (м, 2H), 0,76 (т, J=7,6 Гц, 3H).
Пример 12: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)фосфоновая кислота (Соединение 12)

Получение N4–бутил–5–(5–йод–2–метоксибензил)–6–метилпиримидин–2,4–диамина
При получении N4–бутил–5–(5–йод–2–метоксибензил)–6–метилпиримидин–2,4–диамина следовали методикам, описанным в Примере 11, Стадии 1–5, но используя 4–йод–1–метокси–2–метилбензола вместо 4–бром–1–метокси–2–метилбензола на Стадии 1.
Стадия 1: диэтил (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)фосфонат
К раствору N4–бутил–5–(5–йод–2–метоксибензил)–6–метилпиримидин–2,4–диамина (1,0 экв.) в толуоле (0,06 М), диэтилфосфита (3,5 экв.) и триэтиламина (6,0 экв.) добавляли Pd(dppf)Cl2 (0,05 экв.). Смесь перемешивали при 110°C в течение 16 ч. Реакционной смеси давали остыть до комнатной температуры и упаривали досуха при пониженном давлении с получением неочищенного продукта, который очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 50:1 до 10:1) с получением указанного в заголовке соединения.
Стадия 2: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)фосфоновая кислота
К раствору диэтил(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)фосфоната (1,0 экв.) в DCM (0,08 M) добавляли по каплям TMSBr (20 экв.). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли водой и DCM, органический шар промывали рассолом и сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты).
ЖХ–МС: [M+H] = 381,3
1H ЯМР (400 МГц, DMSO–d6) δ 12,00 (с, 1H), 7,99 (т, J=4,8 Гц, 1H), 7,59–7,53 (м, 1H), 7,47 (ш с, 2H), 7,10–7,06 (м, 2H), 3,89 (с, 3H), 3,72 (с, 2H), 3,40–3,35 (м, 2H), 2,10 (с, 3H), 1,53–1,45 (м, 2H), 1,25–1,19 (м, 2H), 0,87 (т, J=7,2 Гц, 3H).
Пример 13: 5–(5–(1–(2H–тетразол–5–ил)циклопропил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамин (Соединение 13)
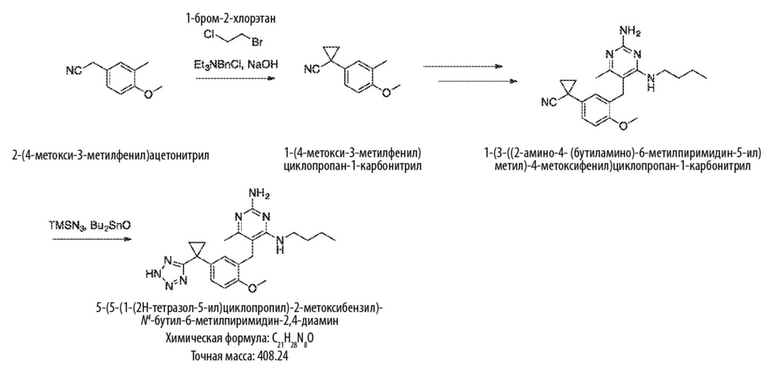
Стадия 1: 1–(4–метокси–3–метилфенил)циклопропан–1–карбонитрил:
В колбу, содержащую 2–(4–метокси–3–метилфенил)ацетонитрил (1,0 экв., Пример 9 – Стадия 3), 1–бром–2–хлорэтан (1,5 экв.), Et3NBnCl (0,02 экв.) добавляли 0,77 г/мл раствора NaOH (6,0 экв.). Полученную смесь перемешивали в течение ночи при 50 oC. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=100:1~20:1) с получением указанного в заголовке соединения.
Получение 1–(3–((2–амино–4– (бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)циклопропан–1–карбонитрила
При получении 1–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)циклопропан–1–карбонитрила следовали методикам, описанным в Примере 9 – Стадии 5–9, но используя 1–(4–метокси–3–метилфенил)циклопропан–1–карбонитрил вместо 2–(4–метокси–3–метилфенил)–2–метилпропаннитрила на Стадии 5.
Получение 5–(5–(1–(2H–тетразол–5–ил)циклопропил)–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина:
Смесь 1–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)циклопропан–1–карбонитрила (1,0 экв.) в диоксане (0,1 M), TMSN3 (10,0 экв.), Bu2SnO (2,0 экв.) перемешивали в течение ночи при 120 oC. Реакцию контролировали с помощью ЖХ–МС. Смесь концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого порошка (соль муравьиной кислоты).
ЖХ–МС: [M+H] =409,2
1H ЯМР (400 МГц, DMSO–d6) δ 8,19 (с, 1H), 7,16 (д, J=8,8 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,77 (с, 1H), 6,69 (ш с, 1H), 6,42 (ш с, 2H), 3,82 (с, 3H), 3,61 (с, 2H), 3,30–3,25 (м, 2H), 2,03 (с, 3H), 1,43–1,35 (м, 4H), 1,21–1,11 (м, 4H), 0,83 (т, J=7,2 Гц, 3H).
Пример 14: 1–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)циклопропан–1–карбоновая кислота (Соединение 14)
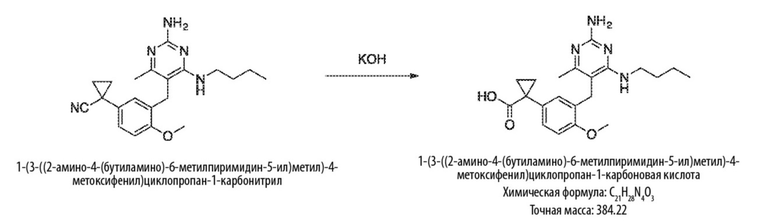
Смесь 1–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)циклопропан–1–карбонитрила (1,0 экв.) в 1:1 этиленгликоль/H2O (0,1 M) и KOH (10 экв.) перемешивали в течение 3 ч. при 150 oC. Реакцию контролировали ЖХ–MС до завершения. Смесь нейтрализовали 1 н. HCl до pH=7. Полученную суспензию фильтровали. Остаток после фильтрования промывали водой. К твердому веществу добавляли MeCN, H2O и 4N HCl/диоксан. Раствор лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль HCl).
ЖХ–МС: [M+H] =385,2
1H ЯМР (400 МГц, DMSO–d6) δ 12,21 (с, 1H), 12,16 (с, 1H), 7,90 (т, J=5,2 Гц, 1H), 7,41 (ш с, 2H), 7,16 (д, J=6,0 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,81 (с, 1H), 3,81 (с, 3H), 3,68 (с, 2H), 3,4–3,3 (м, 2H), 2,12 (с, 3H), 1,51–1,43 (м, 2H), 1,38 (с, 2H), 1,26–1,17 (м, 2H), 0,99 (с, 2H), 0,86 (т, J=7,2 Гц, 3H).
Пример 15: 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропановая кислота (Соединение 15)

Стадия 1: 1–(бромметил)–2–метоксибензол
К раствору 1–метокси–2–метилбензола (1 экв.) в CCl4 (0,9 M) добавляли AIBN (0,2 экв.) и NBS (1,1 экв.). Реакционную смесь нагревали с обратным холодильником в течение 2 ч. После завершения реакции, реакционную смесь концентрировали и разбавляли водой. Водную фазу экстрагировали DCM. Объединенные органические слои промывали водой, затем рассолом, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения в виде светло–желтого масла.
Стадия 2: диметил 2–(2–метоксибензил)малонат
К раствору диметилмалоната (1,2 экв.) в THF (0,4 M) добавляли NaH (1,25 экв.) при 0 °C порциям в азоте. Раствор перемешивали при 0 °C в течение 15 мин., и 1–(бромметил)–2–метоксибензол (1,0 экв.) добавляли. Реакционную смесь перемешивали в течение 2 ч. при комнатной температуре. После завершения реакции, реакционную смесь гасили добавлением воды. Водную фазу экстрагировали EA. Объединенные органические слои промывали водой, затем рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=40:1) с получением указанного в заголовке соединения в виде светло–желтого масла.
Стадия 3: 2–амино–5–(2–метоксибензил)пиримидин–4,6–диол
К раствору диметил 2–(2–метоксибензил)малоната (1,0 экв.) в MeOH (0,4 M) добавляли гуанидинкарбонат (1,0 экв.). Реакцию нагревали при 75°C в течение 16 ч. в азоте. После охлаждения твердые частицы отфильтровывали и собирали из раствора с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 4: 4,6–дихлор–5–(2–метоксибензил)пиримидин–2–амин
К суспензии 2–амино–5–(2–метоксибензил)пиримидин–4,6–диола (1,0 экв.) в POCl3 (0,4 M) добавляли 2 капли DMF. Реакцию нагревали при 100°C в течение 15 ч. в азоте. После охлаждения, раствор выливали в ледяную воду. Твердые частицы отфильтровывали и собирали из раствора с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 5: этил (Е)–3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)акрилат
К раствору 4,6–дихлор–5–(2–метоксибензил)пиримидин–2–амина (1,0 экв.) в 3:1 диоксан/H2O (0,14 M) добавляли этил (E)–3–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)акрилат (1,2 экв.), K2CO3 (2,0 экв.) и Pd(dppf)Cl2 (0,1 экв.). Смесь перемешивали при 100°C в течение 3 ч. в азоте. Реакционную смесь разбавляли водой, и водную фазу экстрагировали EA. Объединенные органические слои промывали водой, затем рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=40:3) с получением указанного в заголовке соединения в виде светло–желтого твердого вещества.
Стадия 6: этил (Е)–3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)акрилат
К раствору этил (E)–3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)акрилата (1,0 экв.) в NMP (0,2 M) добавляли бутан–1–амин (3,0 экв.) и DIEA (3,0 экв.) и смесь перемешивали при 150 °C в течение 2 ч. После завершения реакции реакционную смесь разбавляли водой. Водную фазу экстрагировали EA. Объединенные органические слои промывали водой, затем рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (DCM/MeOH=10:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 7: этил 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропаноат
К раствору этил (E)–3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)акрилата (1,0 экв.) в MeOH (0,04 M) добавляли Pd/C (1/3 мас. экв.). Смесь перемешивали при комнатной температуре в течение 1 ч. в водороде. После завершения реакции, твердые частицы отфильтровывали и фильтрат концентрировали с получением указанного в заголовке соединения в виде желтого масла.
Стадия 8: 3–(2–амино–6–(бутиламино)–5– (2–метоксибензил)пиримидин–4–ил)пропановая кислота
К раствору этил 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропаноата (1 экв.) в 1:1 MeOH/H2O (0,04 M) добавляли NaOH (5,0 экв.). Раствор перемешивали при комнатной температуре в течение 1 ч. После завершения реакции, рН доводили до 7 с использованием уксусной кислоты и раствор концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза: NH3.H2O/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества (соль аммония).
ЖХ–МС: [M+H] =359,4
1H ЯМР(400 МГц, MeOD) δ 7,23 (т, J=6,8 Гц, 1H), 6,99 (д, J=8,0 Гц, 1H), 6,91–6,88 (м, 2H), 3,90 (с, 3H), 3,80 (с, 2H), 3,44 (т, J=6,8 Гц, 2H), 2,85 (т, J=6,0 Гц, 2H), 2,42 (т, J=6,0 Гц 2H), 1,51–1,46 (м, 2H), 1,25–1,19 (м, 2H), 0,88 (т, J=6,8 Гц, 3H).
Пример 16: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамидо)этан–1–сульфоновая кислота (Соединение 16)
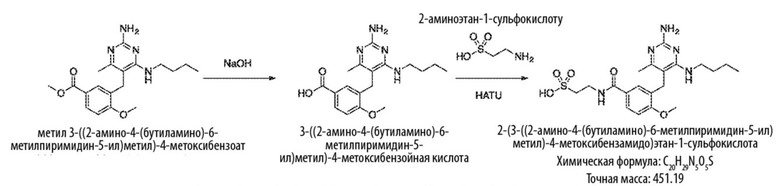
При получении метил 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоата следовали Примеру 1 – Стадии 1–4, но используя метил 3–(бромметил)–4 –метоксибензоат в качестве исходного материала на Стадии 1.
Стадия 1: 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойная кислота
К суспензии метил 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в MeOH (0,3 М) добавляли 1 н. NaOH (5,0 экв.). Полученную смесь перемешивали при 60°C в течение 2 ч. Ход реакции контролировали методом ТСХ. Смесь охлаждали до комнатной температуры. Смесь нейтрализовали 2 н. HCl и очищали с получением указанного в заголовке соединения.
Стадия 2: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамидо)этан–1–сульфоновая кислота
К раствору 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойной кислоты (1,0 экв.) в DMF (0,6 M) добавляли HATU (1,0 экв.), 2–аминоэтан–1–сульфокислоту (1,1 экв.) и DIEA (3,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь фильтровали и фильтрат очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), затем лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H]+ = 452,2
1H ЯМР (400 МГц, DMSO) δ 11,89 (с, 1H), 8,35 (т, J=3,2 Гц, 1H), 7,90 (с, 1H), 7,68 (д, J=8,4 Гц, 1H), 7,40 (ш с, 2H), 7,26 (с, 1H), 7,09 (д, J=8,4 Гц, 1H), 3,90 (с, 3H), 3,71 (с, 2H), 3,48–3,42 (м, 2H), 3,39–3,33 (м, 2H), 2,62 (т, J=6,8 Гц, 2H), 2,12 (с, 3H), 1,49–1,41 (м, 2H), 1,23–1,15 (м, 2H), 0,82 (т, J=7,2 Гц, 3H).
Пример 17: 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензолсульфоновая кислота (Соединение 17)
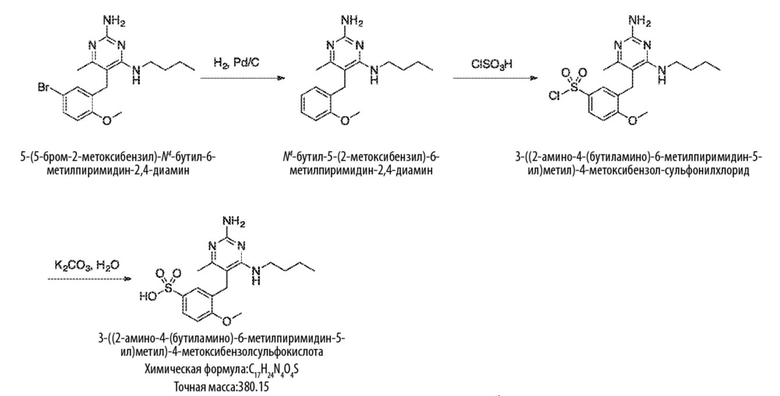
При получении 5–(5–бром–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина следовали Примеру 1 – Стадии 1–4, но с 4–бром–2–(бромметил)–1–метоксибензолом в качестве исходного вещества на Стадии 1.
Стадия 1: N4–бутил–5–(2–метоксибензил)–6–метилпиримидин–2,4–диамин
К перемешиваемому раствору 5–(5–бром–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина (1,0 экв.) в MeOH (0,1 М) добавляли Pd/C (10% мас. экв.). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. в атмосфере H2 и затем фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения.
Стадия 2: 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензол–сульфонилхлорид
N 4–бутил–5–(2–метоксибензил)–6–метилпиримидин–2,4–диамин (1,0 экв.) в ClSO3H (0,7 M) перемешивали при комнатной температуре в течение 2 ч., и затем выливали в ледяную воду. Смесь разделяли в DCM/вода. Органический слой сушили над Na2SO4, фильтровали, концентрировали с получением указанного в заголовке соединения.
Стадия 3: 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензолсульфоновая кислота
К перемешиваемому раствору 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензол–сульфонилхлорида (1,0 экв.) в 1: 1 MeCN/H2O (0,025 M) добавляли K2CO3 (2,0 экв.). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения.
ЖХ–МС: [M–H]– = 379,2
1H ЯМР (400 МГц, DMSO–d6) δ 11,92 (ш с, 1H), 7,79 (ш с, 1H), 7,46 (д, J=6,8 Гц, 1H), 7,22 (ш с, 2H), 7,04 (с, 1H), 6,95 (д, J=8,4 Гц, 1H), 3,85 (с, 3H), 3,68 (с, 2H), 3,37–3,34 (м, 2H), 2,08 (с, 3H), 1,54–1,44 (м, 2H), 1,26–1,23 (м, 2H), 0,86 (т, J=7,2 Гц, 3H).
Пример 18: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептан–1–ол (Соединение 18)

Указанное в заголовке соединение получают в соответствии с методиками, описанными в Примере 8, но с использованием (S)–3–аминогептан–1–ола (из J. Med. Chem,2016, 59, 7936−7949) вместо (S)–2–аминопентан–1–ола в Стадии A. На последней стадии неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), затем лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты).
ЖХ–МС: [M+H]+ = 441,3
1H ЯМР (400 МГц, DMSO) δ 8,22 (с, 1H), 7,03 (д, J=8,8 Гц, 1 H), 6,90 (д, J=8,4 Гц, 1 H), 6,78 (с, 1H), 6,52 (с, 2H), 6,35 (д, J=8,0 Гц, 1 H), 4,27–4,21 (м, 1H), 3,99–3,91 (м, 2H), 3,80 (с, 3H), 3,62 (с, 2H), 3,39–3,26 (м, 3H), 2,07 (с, 3H), 1,64–1,50 (м, 2H), 1,48–1,37 (м, 2H), 1,24–1,14 (м, 2H), 1,12–1,04 (м, 2H), 0,76 (т, J=7,2 Гц, 3 H).
Пример 19: 3–(2–амино–6–(бутиламино)–5– (4–метоксифенетил)пиримидин–4–ил)пропановая кислота (Соединение 19)

Стадия 1: 4,6–дихлорпиримидин–2–амин
Раствор 2–аминопиримидин–4,6–диола (1,0 экв.) в POCl3 (1 M) перемешивали при 100°C в течение 4 ч. Реакционной смеси давали остыть до комнатной температуры и смесь выливали в ледяную воду. Осажденное твердое вещество собирали фильтрованием. Твердое вещество промывали водой и сушили в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2: этил (Е)–3–(2–амино–6–хлорпиримидин–4–ил)акрилат
К раствору 4,6–дихлорпиримидин–2–амина (1,0 экв.) в 3:1 диоксан/H2O (0,3 M) добавляли этил (E)–3–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)акрилат (1,1 экв.), K2CO3 (2,0 экв.) и Pd(dppf)Cl2 (0,1 экв.). Смесь перемешивали при 100°C в течение 2 ч. в азоте. Реакционную смесь разбавляли водой, и водный слой экстрагировали EA. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 3: этил (Е)–3–(2–амино–6–(бутиламино)пиримидин–4–ил)акрилат
К раствору этил (E)–3–(2–амино–6–хлорпиримидин–4–ил)акрилата (1,0 экв.) в NMP (0,4 M) добавляли бутан–1–амин (3,0 экв.) и DIEA (3,0 экв.). Смесь перемешивали при 150°C в течение 3 ч. Реакционную смесь разбавляли водой, и водную фазу экстрагировали EA. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 4: этил 3–(2–амино–6–(бутиламино)пиримидин–4–ил)пропаноат
К раствору этил (E)–3–(2–амино–6–(бутиламино)пиримидин–4–ил)акрилата (1,0 экв.) в MeOH (0,2 M) добавляли Pd/C (20% мас. экв.). Смесь перемешивали при 25°C в течение 2 ч. в H2 и затем фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения в виде окрашеного твердого вещества.
Стадия 5: этил 3–(2–амино–6–(бутиламино)–5–йодпиримидин–4–ил)пропаноат
К раствору этил 3–(2–амино–6–(бутиламино)пиримидин–4–ил)пропаноата (1,0 экв.) в 2:1 DCM/H2O (0,05 M) добавляли NaOH (2,0 экв.), с последующим добавлением I2 (1,0 экв.). Смесь перемешивали при 25°C в течение 16 ч. Реакционную смесь разбавляли водой, и водную фазу экстрагировали EA. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4, и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=4:3) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 6: этил 3–(2–амино–6– (бутиламино)–5–((4–метоксифенил)этинил)пиримидин–4–ил)пропаноат
К раствору этил 3–(2–амино–6–(бутиламино)–5–йодпиримидин–4–ил)пропаноата (1,0 экв.) в DMF (0,1 M) добавляли 1–этинил–4–метоксибензол (1,2 экв.), TEA (1/5ый объема реакции), CuI (0,2 экв.) и Pd(PPh3)2Cl2 (0,1 экв.). Смесь перемешивали при 50°C в течение 16 ч. в азоте. Реакционную смесь разбавляли водой, и водную фазу экстрагировали EA. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1:1) с получением указанного в заголовке соединения в виде коричневого масла.
Стадия 7: этил 3–(2–амино–6–(бутиламино)–5–(4–метоксифенетил)пиримидин–4–ил)пропаноат
К раствору этил 3–(2–амино–6–(бутиламино)–5–((4–метоксифенил)этинил)пиримидин–4–ил)пропаноата (1,0 экв.) в MeOH (0,08 M) добавляли Pd/C (30% мас. экв.). Смесь перемешивали при 50°C в течение 2 ч. в H2 и затем фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения в виде желтого масла.
Стадия 8: 3–(2–амино–6–(бутиламино)–5–(4–метоксифенетил)пиримидин–4–ил)пропановая кислота
К раствору этил 3–(2–амино–6–(бутиламино)–5–(4–метоксифенетил)пиримидин–4–ил)пропаноата (1,0 экв.) в 1:1 MeOH/H2O (0,2 M) добавляли NaOH (5,0 экв.). Полученную смесь перемешивали при 25°C в течение 1 ч. pH доводили до 7 с использованием уксусной кислоты, и раствор концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза:NH4OH/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества (соль аммония).
ЖХ–МС: [M+H]+ = 373,4
1H ЯМР (400 МГц, CD3OD) δ 7,00 (д, J=8,4 Гц, 2H), 6,81 (д, J=8,8 Гц, 2H), 3,75 (с, 3H), 3,48 (т, J=7,6 Гц, 2H), 2,75–2,71 (м, 4H), 2,43 (т, J=6,0 Гц, 2H), 2,18 (т, J=6,4 Гц, 2H), 1,60–1,54 (м, 2H), 1,40–1,35 (м, 2H), 0,97 (т, J=6,8 Гц, 3H)
Пример 20A: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептановая кислота (Соединение 20)
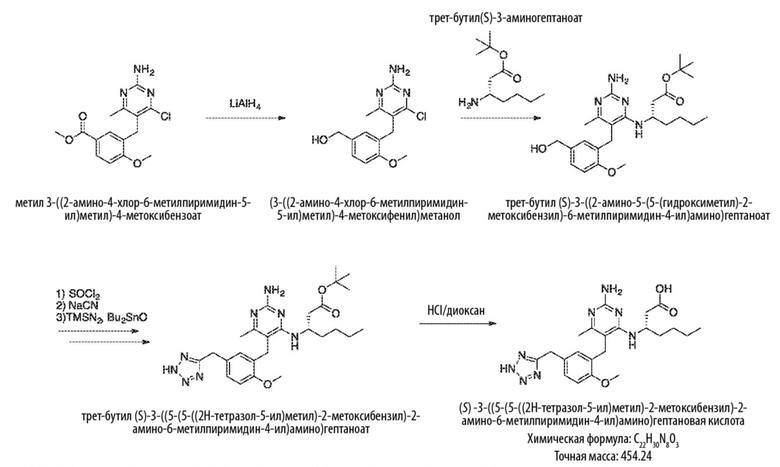
Метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоат получали в соответствии с Примером 1 – Стадии 1–3, но используя метил 3–(бромметил)–4–метоксибензоат в качестве исходного материала вместо метил 4–(бромметил)–3–метоксибензоата.
Стадия 1: (3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанол
К перемешиваемому раствору метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в THF (0,2 M) при 0°C добавляли по каплям 2,5M LiAlH4 (2,5 экв.). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Затем смесь гасили 1 М раствором NaOH и фильтровали. Фильтрат экстрагировали EtOAc. Органический слой сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент DCM/MeOH=от 100:1 до 10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2: трет–бутил (S)–3–((2–амино–5–(5–(гидроксиметил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептаноат
Смесь трет–бутил(S)–3–аминогептаноата (4,0 экв.; получен в соответствии с методикой представленной в J. Med. Chem,2016, 59, 7936−7949) и (3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола (1,0 экв.) в NMP (0,8 M) перемешивали при 120°C в течение 16 ч. Затем в реакционную смесь добавляли воду и EtOAc. Органический слой разделяли, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 100:1 до 20:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадии 3–5: трет–бутил (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептаноат
Указанное в заголовке соединение получали в соответствии с Примером 2 – Стадии 1–3, но с использованием трет–бутил (S)–3–((2–амино–5–(5–(гидроксиметил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептаноата в качестве исходного вещества. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), и лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты).
ЖХ–МС: [M+H]+ = 511,4
1H ЯМР (400 МГц, DMSO) δ 8,20 (с, 1 H), 7,04 (дд, J=8,4, 2,0 Гц, 1H), 6,93 (д, J=8,0 Гц, 1H), 6,77 (с, 1H), 6,60 (ш с, 2H), 6,39 (ш с, 1H), 4,55–4,52 (м, 1H), 4,02 (с, 2H), 3,83 (с, 3H), 3,67–3,57 (м, 2H), 2,37 (д, J=6,8 Гц, 2H), 2,07 (с, 3H), 1,46–1,38 (м, 2H), 1,32 (с, 9H), 1,26–1,13 (м, 2H), 1,09–1,01 (м, 2H), 0,77 (т, J=7,2 Гц, 3H).
Стадия 6: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептановая кислота
К трет–бутил (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептаноату (1 экв.) добавляли 3,0 M HCl/диоксан (20 экв.) при 0°C и нагревали до комнатной температуры. После перемешивания в течение 1 ч., смесь концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H]+ = 455,2
1H ЯМР (400 МГц, DMSO) δ 7,35 (ш с, 1H), 7,07 (ш с, 2H), 6,96 (д, J=7,6 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 6,76 (с, 1H), 4,56–4,54 (м, 1H), 4,07–3,97 (м, 2H), 3,79 (с, 3H), 3,68–3,66 (м, 2H), 2,46–2,38 (м, 2H), 2,05 (с, 3H), 1,53–1,50 (м, 2H), 1,27–1,16 (м, 4H), 0,82 (т, J=6,8 Гц, 3H).
Пример 20B: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептановая кислота (Соединение 20)

Стадия 1: трет–бутил (E)–гепт–2–еноат
Смесь пентаналя (1,0 экв.) и трет–бутил 2–(трифенил–λ5–фосфанилидин)ацетата (1,05 экв.) перемешивали при 50°C в течение 16 ч. Затем смесь концентрировали при пониженном давлении и добавляли петролейный эфир (PE). Твердые вещества отфильтровывали и фильтрат упаривали досуха, который очищали колоночной флэш–хроматографией (элюент: 0–1% EA в PE) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 2: трет–бутил (S)–3–(бензил((S)–1–фенилэтил)амино)гептаноат
К раствору (S)–N–бензил–1–фенилэтан–1–амина (1,3 экв.) в THF (0,9 M) при –78°C добавляли по каплям 2,5M н–BuLi (1,2 экв.) более 20 минут. Смесь перемешивали при –78°C в течение 10 минут, затем в вышеуказанную смесь добавляли по каплям трет–бутил (E)–гепт–2–еноат (1,0 экв.) в THF (0,7 M). Полученную смесь перемешивали при –78°C в течение 30 минут. Затем смесь гасили водн. NH4Cl и экстрагировали EtOAc. Органический слой сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: 0–1% EA в PE) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 3: трет–бутил (S)–3–аминогептаноат
К раствору трет–бутил (S)–3–(бензил((S)–1–фенилэтил)амино)гептаноата (1,0 экв.) в MeOH (0,5 M) добавляли 10% мас. Pd/C (1/10 мас. эквивалента). Полученную смесь перемешивали при 50 °С в течение 16 ч. при H2. Твердое вещество отфильтровывали и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде коричневого масла, которое используют непосредственно на следующей стадии без дальнейшей очистки..
Стадия 4: (3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанол
К перемешиваемому раствору метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.; из Примера 3B – Стадия 4) в THF (0,2 М) при 0°C добавляли по каплям 2,5M LiAlH4 (2,5 экв.). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Затем смесь гасили 1 М раствором NaOH и фильтровали. Фильтрат экстрагировали EtOAc. Органический слой сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент DCM/MeOH=от 100:1 до 10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 5: трет–бутил (S)–3–((2–амино–5–(5–(гидроксиметил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептаноат
Смесь трет–бутил (S)–3–аминогептаноата (4,0 экв.) и (3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола (1,0 экв.) в NMP (0,8 М) перемешивали при 120°C в течение 16 ч. Затем воду и EtOAc добавляли в реакционную смесь. Органический слой разделяли, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 100:1 до 20:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 6: трет–бутил (S)–3–((2–амино–5–(5–(хлорметил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептаноат
К перемешиваемому раствору трет–бутил (S)–3–((2–амино–5–(5–(гидроксиметил)–2–метоксибензил)–6–метил–пиримидин–4–ил)амино)гептаноата (1,0 экв.) в DCM (0,05 M) при 0°C добавляли SOCl2 (2,0 экв.). Полученную смесь перемешивали при 0°C в течение 1 ч. Затем смесь гасили раствором NaHCO3 и экстрагировали DCM. Органический слой сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла.
Стадия 7: трет–бутил (S)–3–((2–амино–5–(5–(цианометил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептаноат
Смесь трет–бутил (S)–3–((2–амино–5–(5–(хлорметил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептаноата (1,0 экв.) в 1:1 DMSO/DMF (0,12 M) и NaCN (3,0 экв.) перемешивали при комнатной температуре в течение 16 ч. Полученную смесь разделяли в EtOAc и воде. Органический слой промывали рассолом, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=100:1 до 20:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 8: трет–бутил (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептаноат
Смесь трет–бутил (S)–3–((2–амино–5–(5–(цианометил)–2–метоксибензил)–6–метил–пиримидин–4–ил)амино)гептаноата (1,0 экв.) в диоксане (1,2 M), TMSN3 (10 экв.) и Bu2SnO (2,0 экв.) перемешивали при 120°C в течение 2 ч. Затем смесь концентрировали при пониженном давлении с получением неочищенного продукта в виде коричневого масла, которое использовали непосредственно до следующей стадии без дальнейшей очистки.
Стадия 9: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептановая кислота
К трет–бутил (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептаноату (1 экв.) добавляли 3,0 M HCl/диоксан (20 экв.) при 0°C и нагревали до комнатной температуры. После перемешивания в течение 1 ч., смесь концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты).
ЖХ–МС: [M+H]+ = 455,2
1H ЯМР (400 МГц, DMSO) δ 7,35 (ш с, 1H), 7,07 (ш с, 2H), 6,96 (д, J=7,6 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 6,76 (с, 1H), 4,56–4,54 (м, 1H), 4,07–3,97 (м, 2H), 3,79 (с, 3H), 3,68–3,66 (м, 2H), 2,46–2,38 (м, 2H), 2,05 (с, 3H), 1,53–1,50 (м, 2H), 1,27–1,16 (м, 4H), 0,82 (т, J=6,8 Гц, 3H).
Пример 20C: (R)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–метилпиримидин–4–ил)амино)гептановая кислота (Соединение 20C)

Указанное в заголовке соединение получали, следуя методике, описанной в примере 20B, но с использованием (R)–N–бензил–1–фенилэтан–1–амина вместо (S)–N–бензил–1–фенилэтан–1–амина на Стадии 2. Указанное в заголовке соединение показало идентичные данные ЯМР Н и ЖХМС, как в примере 20А/20В.
Для демонстрации хиральности образцы Примера 20A/20B (например, (S)–энантиомер) и Примера 20C (например, (R)–энантиомер) вводили в колонку Superchiral S–OZ (0,46 см В. Д. x 25 см Д) и элюировали смесью гексан/EtOH/MeOH/диэтиламин (об./об./об./об. = 80/6,6/13,4/0,05) со скоростью 0,8 мл/мин. (R)–энантиомер показал время удерживания 12,2 мин, тогда как Пример 20А/20В (например, (S)–энантиомер) показал время удерживания 13,2 мин.
Пример 21: (S)–3–(2–амино–6–((1–гидроксигептан–3–ил)амино)–5–метоксипиримидин–4–ил)пропановая кислота (Соединение 21)

Указанное в заголовке соединение может быть получено в соответствии со стадиями Примера 15.
Пример 22: (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–6–метил–N4–(1–(метилтио)гептан–3–ил)пиримидин–2,4–диамин (Соединение 22)

Стадия 1: метил (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензоат
Смесь метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв., из Примера 3B – Стадия 4) в NMP (0,8 M), (S)–1–(метилтио)гептан–3–амина (1,0 экв.; полученный в соответствии с методиками, описанными в WO2014/128189, стр. 8, соединение D) и DIEA (3,0 экв.) перемешивали в течение 48 ч. при 120°C в атмосфере N2. Реакционной смеси давали остыть, разбавляли EtOAc, промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюент 0–5% MeOH в DCM) с получением указанного в заголовке соединения.
Стадия 2: (S)–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)метанол
К перемешиваемому раствору метил (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)–пиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в THF (0,18 M) добавляли по каплям 2,5M LiAlH4 (2,0 экв.) при 0 oC. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. и затем гасили EA и 1 н. NaOH. Смесь фильтровали и фильтрат разделяли между EA/вода. Органический слой сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюент 0–5% MeOH в DCM) с получением указанного в заголовке соединения.
Стадия 3: (S)–5–(5–(хлорметил)–2–метоксибензил)–6–метил–N4–(1–(метилтио)гептан–3–ил)пиримидин–2,4–диамин
К перемешиваемому раствору (S)–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)–пиримидин–5–ил)метил)–4–метоксифенил)метанола (1,0 экв.) в DCM (0,1 M) добавляли SOCl2 (5,0 экв.) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 2 ч. и затем гасили насыщ. NaHCO3. Смесь разделяли между DCM/вода. Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения.
Стадия 4: (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)ацетонитрил
К перемешиваемому раствору (S)–5–(5–(хлорметил)–2–метоксибензил)–6–метил–N4–(1–(метилтио)–гептан–3–ил)пиримидин–2,4–диамина (1,0 экв.) в 1:1 DMSO/DMF (0,1 M) добавляли NaCN (3,0 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч. и затем гасили насыщ. NaHCO3. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, концентрировали и очищали с получением указанного в заголовке соединения.
Стадия 5: (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–6–метил–N4–(1–(метилтио)–гептан–3–ил)пиримидин–2,4–диамин
К смеси (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)ацетонитрила (1,0 экв.) в диоксане (0,15 M) добавляли TMSN3 (10,0 экв.), Bu2SnO (2,0 экв.) и перемешивали в течение ночи при 120 oC. Смесь концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/ HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого порошка (соль муравьиной кислоты).
ЖХМС: [M+H]+ =471,3
1H ЯМР (400 МГц, DMSO) δ 7,03 (д, J=8,4 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,79 (с, 1H), 6,44 (с, 2H), 6,24 (д, J=8,8 Гц, 1H), 4,25–4,22 (м, 1H), 4,00 (с, 2H), 3,82 (с, 3H), 3,64 (с, 2H), 2,29–2,27 (м, 2H), 2,08 (с, 3H), 1,95 (с, 3H), 1,68–1,64 (м, 2H), 1,44–1,34 (м, 2H), 1,19–1,16 (м, 2H), 1,12–1,02 (м, 2H), 0,78 (т, J=7,2 Гц, 3H).
Пример 23: (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–6–метил–N4–(1–(метилсульфонил)гептан–3–ил)пиримидин–2,4–диамин (Соединение 23)
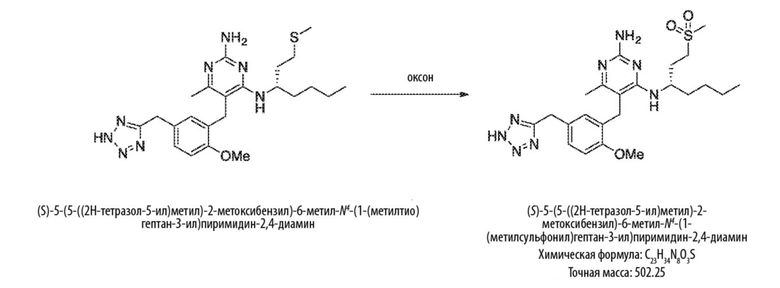
К перемешиваемому раствору (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–6–метил–N4–(1–(метилтио)гептан–3–ил)пиримидин–2,4–диамина (1,0 экв., Пример 22) в ацетоне (0,02 M) добавляли оксон (2,0 экв.) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 3 ч. перед тем фильтровали и очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого порошка (соль муравьиной кислоты).
ЖХМС: [M+H]+ =503,3
1H ЯМР (400 МГц, DMSO) δ 7,05 (д, J=8,4 Гц, 1H), 6,93 (д, J=8,4 Гц, 1H), 6,76 (с, 1H), 6,47 (с, 2H), 6,38 (д, J=6,4 Гц, 1H), 4,25–4,23 (м, 1H), 4,04 (с, 2H), 3,82 (с, 3H), 3,66 (с, 2H), 3,01–2,85 (м, 5H), 2,05 (с, 3H), 1,92–1,90 (м, 1H), 1,81–1,80 (м, 1H), 1,49–1,34 (м, 2H), 1,18–1,15 (м, 2H), 1,11–0,99 (м, 2H), 0,77 (т, J=7,2 Гц, 3H).
Пример 24: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)борная кислота (Соединение 24)

Стадия 1: N4–бутил–5–(2–метокси–5–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)бензил)–6–метилпиримидин–2,4–диамин
Смесь 5–(5–бром–2–метоксибензил)–N4–бутил–6–метилпиримидин–2,4–диамина (1,0 экв., Пример 11 – Стадия 5) в диоксане (0,03 M), 4,4,4',4',5,5,5',5'–октаметил–2,2'–би(1,3,2–диоксаборолан) (1,2 экв.), KOAc (3,0 экв.) и Pd(dppf)Cl2 (0,1 экв.) перемешивали при 90°C в течение 4 ч. Затем смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюент: 0–5% MeOH в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–борная кислота
К смеси N4–бутил–5–(2–метокси–5–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)бензил)–6–метилпиримидин–2,4–диамина (1,0 экв.) в MeOH (0,05 M) добавляли iBuB(OH)2 (2,0 экв.) и 6 н. HCl (1/10 от общего объема реакции). Смесь перемешивали при комнатной температуре в течение 16 ч. в атмосфере азота. Смесь концентрировали при пониженном давлении и очищали с помощью препаративной ВЭЖХ (подвижная фаза: FA/H2O/CH3CN) с получением указанного в заголовке соединения в виде твердого вещества (соль муравьиной кислоты).
ЖХМС: [M+H]+ = 345,3
1H ЯМР (400 МГц, DMSO–d6) δ 7,76 (с, 2H), 7,66 (д, J=7,6 Гц, 1H), 7,25 (с, 1H), 6,96 (д, J=8,0 Гц, 1H), 6,76 (ш с, 1H), 6,56 (ш с, 2H), 3,86 (с, 3H), 3,63 (с, 2H), 3,30–3,28 (м, 2H), 2,06 (с, 3H), 1,44–1,41 (м, 2H), 1,18–1,15 (м, 2H), 0,82 (т, J=7,2 Гц, 3H).
Пример 25: 4–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамидо)бутановая кислота (Соединение 25)
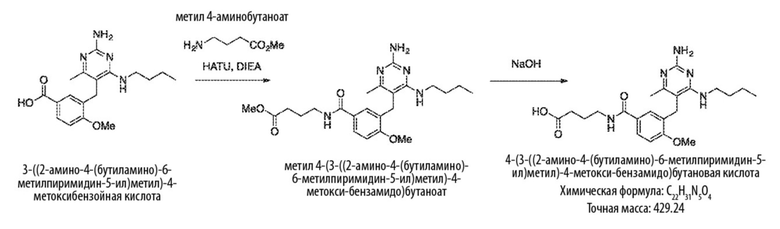
Стадия 1: метил 4–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метокси–бензамидо)бутаноат
Смесь 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойной кислоты (1,0 экв., Пример 16 – Стадия 1) в DMF (0,06 M), метил 4–аминобутаноате (1,0 экв.), HATU (1,5 экв.) и DIEA (4,0 экв.) перемешивали при 50°C в течение 5 ч. Смесь охлаждали до комнатной температуры и добавляли воду. Водный слой экстрагировали DCM. Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: DCM/MeOH=100:1 до 10:1) с получением указанного в заголовке соединения в виде коричневого твердого вещества.
Стадия 2: 4–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метокси–бензамидо)бутановая кислота
К перемешиваемому раствору метил 4–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамидо)бутаноата (1,0 экв.) в MeOH (0,025 M) добавляли 1 н. NaOH (6 экв.). Полученную смесь перемешивали при 50°C в течение 2 ч. Смесь охлаждали до комнатной температуры и доводили до рН 7 используя 1 н. HCl. Осажденные твердые вещества собирали фильтрованием и растирали с водой, сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХМС: [M+H] + = 430,3
1H ЯМР (400 МГц, DMSO) δ 8,28 (ш с, 1H), 7,70 (д, J=9,2 Гц, 1H), 7,29 (с, 1H), 7,02 (д, J=8,8 Гц, 1H), 5,99 (ш с, 1H), 5,81–5,79 (м, 2H), 3,89 (с, 3H), 3,62 (с, 2H), 2,36–3,16 (м, 4H), 2,21 (т, J=7,2 Гц, 2H), 1,98 (с, 3H), 1,72–1,67 (м, 2H), 1,42–1,38 (м, 2H), 1,20–1,14 (м, 2H), 0,81 (т, J=7,6 Гц, 3H).
Пример 26: 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–6–метил–N4–пентилпиримидин–2,4–диамин (Соединение 26)

Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 3, но с использованием пентан–1–амина вместо бутан–1–амина в Стадии 5. Полученный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/HCOOH), лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (соль муравьиной кислоты).
ЖХ–МС: [M+H]+ = 397,2
1H ЯМР (400 МГц, DMSO) δ 7,03 (д, J=8,8 Гц, 1H), 6,91 (д, J=8,0 Гц, 1H), 6,74 (с, 1H), 6,64 (ш с, 1H), 6,48 (ш с, 2H), 3,97 (с, 2H), 3,81 (с, 3H), 3,61 (с, 2H), 3,30–3,24 (м, 2H), 2,04 (с, 3H), 1,46–1,40 (м, 2H), 1,29–1,21 (м, 2H), 1,20–1,39 (м, 2H), 0,83 (т, J=6,8 Гц, 3H).
Пример 27: (S)–3–(2–амино–6–((1–гидроксигептан–3–ил)амино)–5–(2–метоксибензил)–пиримидин–4–ил)пропановая кислота (Соединение 27)

Стадия 1: диэтил 3–оксогександиоат
К раствору 4–этокси–4–оксобутановой кислоты (1,0 экв.) в безводном THF (0,5 M) при комнатной температуре добавляли карбонилдиимидазол (1,2 экв.) в N2. Полученную смесь перемешивали при комнатной температуре в течение 1 ч., затем MgCl2 (1,0 экв.) и в вышеуказанный раствор при комнатной температуре добавляли 3–этокси–3–оксопропаноат калия (1,0 экв.) и нагревали при 60oC в течение 2ч. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на диоксиде кремния (элюент: PE/EA=от 100:1 до 3:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 2: диэтил 2–(2–метоксибензил)–3–оксогександиоат
К раствору диэтил 3–оксогександиоата (1,2 экв.) в безводном THF (1,2 M) добавляли 60% NaH (1,3 экв.) порциями при 0 oC. После перемешивания в течение 10 мин, раствор 1–(бромметил)–2–метоксибензола (1,0 экв.; Пример 15 – Стадия 1) в THF (1 M) добавляли по каплям в вышеупомянутую смесь. Полученную смесь перемешивали при 60°C в течение 16 ч. и гасили водой. Смесь экстрагировали EA и объединенный органические слои сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали флэш–хроматографией на диоксиде кремния (элюент: PE/EA=от 50:1 до 10:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 3: 3–(2–амино–6–гидрокси–5–(2–метоксибензил)пиримидин–4–ил)пропановая кислота
Смесь диэтил 2–(2–метоксибензил)–3–оксогександиоат (1,0 экв.) и гуанидинкарбоната (1,0 экв.) в MeOH (0,3 M) перемешивали при 90°C в течение 16 ч. Осадок собирали фильтрованием, промывали водой и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 4: 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропановая кислота
Раствор 3–(2–амино–6–гидрокси–5–(2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв.) в POCl3 (0,26 M) перемешивали при 100°C в течение 2 ч. в азоте. Реакционную смесь охлаждали до комнатной температуры и POCl3 упаривали при пониженном давлении. Остаток разбавляли водой и pH доводили до 7 добавлением твердого NaHCO3. Смесь перемешивали при 50°C в течение 1 ч., затем охлаждали до комнатной температуры и осадок собирали фильтрованием. Осадок на фильтре промыли водой, высушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 5: (S)–3–(2–амино–6–((1–гидроксигептан–3–ил)амино)–5–(2–метоксибензил)пиримидин–4–ил)пропановая кислота
Раствор 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв.) в неразбавленном (S)–3–аминогептан–1–оле (1,6M; полученный согласно J. Med. Chem, 2016, 59, 7936−7949) перемешивали при 120°C в течение 2 ч. Смесь разбавляли водой и водную фазу экстрагировали EA. Объединенные органические слои промывали 1 н. HCl, рассолом, сушили над Na2SO4, и концентрировали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза: NH4OH/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХМС: [M+H] + = 417,3
1H ЯМР (400 МГц, CD3OD) δ 7,24 (т, J=8,0 Гц, 1H), 7,02–6,96 (м, 2H), 6,89 (т, J=7,2 Гц, 1H), 4,42–4,40 (м, 1H), 3,92 (с, 3H), 3,82 (с, 2H), 3,46–3,38 (м, 2H), 2,95–2,90 (м, 2H), 2,48 (т, J=7,2 Гц, 2H), 1,8 –1,0 (м, 8H), 0,80 (т, J=7,2 Гц, 3H).
Пример 28: N–(3–(2H–тетразол–5–ил)пропил)–3–((2–амино–4–(бутиламино)–6–метил–пиримидин–5–ил)метил)–4–метоксибензамид (Соединение 28)

Стадия 1: 4–(1,3–диоксоизоиндолин–2–ил)бутаннитрил
Смесь 4–бромбутаннитрила (1,5 экв.) и 1,3–диоксоизоиндолин–2–ида (1,0 экв.) в DMF (0,3 M) перемешивали при 100°C в течение 8 ч. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали флэш–хроматографией на диоксиде кремния (элюент: PE/EA=от 100:1 до 1:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: 4–аминобутаннитрил
К раствору 4–(1,3–диоксоизоиндолин–2–ил)бутаннитрила (1,0 экв.) в EtOH (0,26 M) добавляли NH2NH2–H2O (2,0 экв.) и перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали, затем разбавляли водой. Водную фазу экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде коричневого масла.
Стадия 3: 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–N–(3–цианопропил)–4–метоксибензамид
Смесь 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойной кислоты (1,0 экв.; Пример 16 – Стадия 1) в DMF (0,09 M), 4–аминобутанитрила (2,0 экв.), HATU (2,0 экв.) DIEA (3,0 экв.) перемешивали при 50°C в течение 5 ч. Смесь охлаждали до комнатной температуры, затем добавляли воду и водную фазу экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали флэш–хроматографией на диоксиде кремния (PE/EA= от 50:1 до 1:2) с получением указанного в заголовке соединения в виде коричневого твердого вещества.
Стадия 4: N–(3–(2H–тетразол–5–ил)пропил)–3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамид
К перемешиваемому раствору 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–N–(3–цианопропил)–4–метоксибензамида (1,0 экв.) в NMP (0,08 M) добавляли Bu2SnO (2,0 экв.) и TMSN3 (10,0 экв.). Полученную смесь перемешивали в течение ночи при 125°C в атмосфере N2. Смесь охлаждали до комнатной температуры, концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза: CH3CN/H2O/ 0,1%NH4OH) с получением указанного в заголовке соединения в виде твердого вещества.
ЖХ–МС: [M+H]+ = 454,5
1H ЯМР (400 МГц, DMSO) δ 8,45 (с, 1H), 7,74 (д, J=8,0 Гц, 1H), 7,32 (с, 1H), 7,04 (д, J=8,8 Гц, 1H), 6,57 (ш с, 1H), 6,30–6,20 (м, 2H), 3,90 (с, 3H), 3,65 (с, 2H), 3,30–3,26 (м, 4H), 2,82 (т, J=7,2 Гц, 2H), 2,03 (с, 3H), 1,90 (т, J=6,8 Гц, 2H), 1,46–1,38 (м, 2H), 1,23–1,12 (м, 2H), 0,80 (т, J=7,2 Гц, 3H).
Пример 29: (S)–3–((2–амино–6–(2–карбоксиэтил)–5–(2–метоксибензил)пиримидин–4–ил)амино)гептановая кислота (Соединение 29)
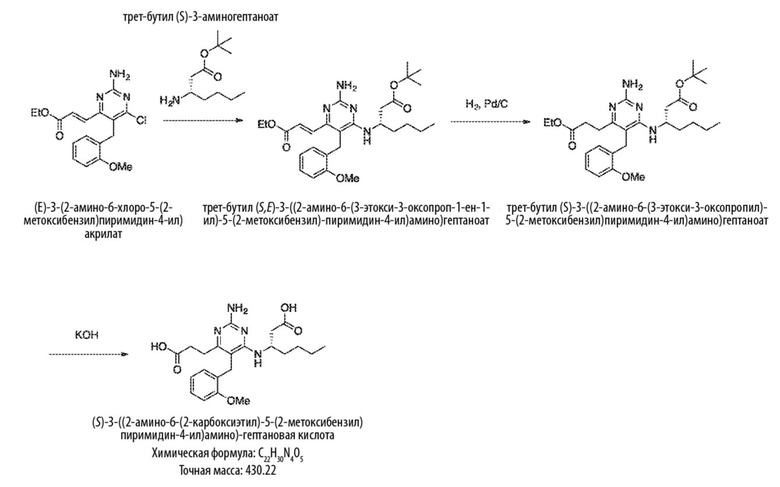
Стадия 1: трет–бутил (S,E)–3–((2–амино–6–(3–этокси–3–оксопроп–1–ен–1–ил)–5–(2–метоксибензил)–пиримидин–4–ил)амино)гептанoат
Раствор этил (E)–3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)акрилата (1,0 экв., Пример 15 – Стадия 5) в неразбавленном трет–бутил (S)–3–аминогептаноате (0,6M, Пример 20B – Стадия 3) перемешивали при 120°C в течение 2 ч. Реакционный раствор разбавляли водой и водный слой экстрагировали EA. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (DCM/MeOH=10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2: трет–бутил (S)–3–((2–амино–6–(3–этокси–3–оксопропил)–5–(2–метоксибензил)пиримидин–4–ил)амино)гептанoат
К раствору трет–бутил (S,E)–3–((2–амино–6–(3–этокси–3–оксопроп–1–ен–1–ил)–5–(2–метокси–бензил)–пиримидин–4–ил)амино)гептанoaта (1,0 экв.) в MeOH (0,04 M) добавляли Pd/C (1/3 мас. экв.). Смесь перемешивали в атмосфере H2 при комнатной температуре в течение 2 ч. Pd/C отфильтровывали и фильтарт концентрировали с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 3: (S)–3–((2–амино–6–(2–карбоксиэтил)–5–(2–метоксибензил)пиримидин–4–ил)амино)–гептановая кислота
К раствору трет–бутил (S)–3–((2–амино–6–(3–этокси–3–оксопропил)–5–(2–метоксибензил)–пиримидин–4–ил)амино)гептанoaта (1,0 экв.) в 4:1 EtOH/H2O (0,06 M) добавляли KOH (5,0 экв.). Раствор перемешивали при 100°C в течение 3 ч. pH доводили до 7 используя уксусную кислоту и раствор концентрировали и затем очищали с помощью препаративной ВЭЖХ (подвижная фаза: NH4OH/MeCN/ H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + =431,4
1H ЯМР (400 МГц, CD3OD) δ 7,23 (т, J=7,2 Гц, 1H), 7,01–6,97 (м, 2H), 6,89 (т, J=7,2 Гц, 1H), 4,61 (м, 1H), 3,91 (с, 3H), 3,82 (с, 2H), 2,94–2,92 (м, 2H), 2,47–2,41 (м, 2H), 1,6–0,9 (м, 8H), 0,80 (т, J=7,2 Гц, 3H).
Пример 30: (3–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамидо)пропил)фосфоновая кислота (Соединение 30)

Стадия 1: диэтил (3–(1,3–диоксоизоиндолин–2–ил)пропил)фосфонат
Смесь диэтил (3–бромпропил)фосфоната (1,2 экв.) и 1,3–диоксоизоиндолин–2–ида (1,0 экв.) в DMF (0,2 M) перемешивали при 100°C в течение 8 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: диэтил (3–аминопропил)фосфонат
Смесь диэтил (3–(1,3–диоксоизоиндолин–2–ил)пропил)фосфонат (1,0 экв.) в EtOH (0,25 M) и NH2NH2 H2O (2,0 экв.) перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали, фильтрат выливали в воду, и водную фазу экстрагировали DCM. Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде коричневого масла.
Стадия 3: диэтил (3–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метокси–бензамидо)пропил)фосфонат
Смесь 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойной кислоты (1,0 экв.; Пример 16 – Стадия 1) в DMF (0,1 M), диэтил (3–аминопропил)фосфонате (2,0 экв.), HATU (2,0 экв.) и DIEA (3,0 экв.) перемешивали при 50°C в течение 5 ч. в азоте. Смесь охлаждали до комнатной температуры, разбавляли водой и водную фазу экстрагировали DCM. Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде коричневого твердого вещества.
Стадия 4: (3–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метокси–бензамидо)пропил)фосфоновая кислота
Раствор диэтил (3–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)метил)–4–метоксибензамид)пропил)фосфоната (1,0 экв.) в DCM (0,08 M) и TMSBr (10,0 экв.) перемешивали в течение ночи при комнатной температуре в азоте. Смесь концентрировали, очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/0,1%NH4OH) с получением указанного в заголовке соединения в виде твердого вещества.
ЖХ–МС: [M+H] + = 466,4
1H ЯМР (400 МГц, DMSO) δ 8,59 (с, 1H), 7,76 (д, J=8,0 Гц, 1H), 7,27 (с, 1H), 7,20 (ш с, 1H), 7,03 (д, J=8,4 Гц, 1H), 3,88 (с, 3H), 3,64 (с, 2H), 3,35–3,28 (м, 2H), 3,25–3,16 (м, 2H), 2,07 (с, 3H), 1,74–1,62 (м, 2H), 1,48–1,33 (м, 4H), 1,22–1,11 (м, 2H), 0,81 (т, J=7,2 Гц, 3H).
Пример 31: 5–((5–((2H–тетразол–5–ил)метил)–2–метоксифенил)тио)–N4–бутил–6–метилпиримидин–2,4–диамин (Соединение 31)

Стадия 1: метил 3–((диметилкарбамотиоил)окси)–4–метоксибензоат
К перемешиваемому раствору метил 3–гидрокси–4–метоксибензоата (1,0 экв.) в DMF (0,55 M) добавляли NaH (1,1 экв.) при 0 oC. Смесь нагревали до комнатной температуры и перемешивали в течение 30 минут и затем обрабатывали диметилкарбамотиоидхлоридом (1,1 экв.). Смесь перемешивали в течение ночи и разделяли в эфир/вода. Полученную суспензию фильтровали. Твердое вещество собирали и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: метил 3–((диметилкарбамоил)тио)–4–метоксибензоат
Метил 3–((диметилкарбамоил)окси)–4–метоксибензоат (1,0 экв.) в PhOPh (1 M) перемешивали в течение 16 ч. при 260 oC. Смесь охлаждали до комнатной температуры и смесь очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=100:1–2:1) с получением указанного в заголовке соединения.
Стадия 3: метил 3–меркапто–4–метоксибензоат
К перемешиваемому раствору метил 3–((диметилкарбамоил)тио)–4–метоксибензоата (1,0 экв.) в THF (0,3 M) добавляли MeONa (2,0 экв.) при комнатной температуре. Полученную смесь перемешивали при 60°C в течение 2 ч. Смесь охлаждали до комнатной температуры и гасили 1 н. HCl. Смесь разделяли между EA/водой. Органическую фазу сушили над Na2SO4 и концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=100:1~5:1) с получением указанного в заголовке соединения.
Стадия 4: метил 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)тио)–4–метокси–бензоат
Смесь метил 3–меркапто–4–метоксибензоата (1,0 экв.) в диоксане (0,3 M), N4–бутил–5–йодо–6–метилпиримидин–2,4–диамина (1,5 экв.), Cs2CO3 (2,0 экв.), CuI (2,0 экв.) перемешивали в течение ночи при 100 oC. Смесь разделяли в EA/NH4OH. Органический слой промывали рассолом, сушили над Na2SO4, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=100:1~1:1) с получением указанного в заголовке соединения.
Стадия 5: (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)тио)–4–метоксифенил)–метанол
К перемешиваемому раствору метил 3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)тио)–4–метоксибензоата (1,0 экв.) в THF (0,2 M) добавляли 1M LiAlH4 (2,0 экв.) по каплям при 0 oC. Полученную смесь перемешивали при 0°C в течение 10 минут и при комнатной температуре в течение 1 часа. Смесь разбавляли EA и гасили 2 н. NaOH. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия 6: N4–бутил–5–((5–(хлорметил)–2–метоксифенил)тио)–6–метилпиримидин–2,4–диамин
К перемешиваемому раствору (3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)тио)–4–метоксифенил)метанола (1,0 экв.) в DCM (0,2 M) добавляли SOCl2 (2,0 экв.) при комнатной температуре в N2. Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разделяли между DCM и насыщенным водным NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия 7: 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)тио)–4–метоксифенил)ацетонитрил
Смесь N4–бутил–5–((5–(хлорметил)–2–метоксифенил)тио)–6–метилпиримидин–2,4–диамина (1,0 экв.) в 1:1 DMSO/DMF (0,1 M) и NaCN (3,0 экв.) перемешивали в течение ночи при комнатной температуре. Смесь разделяли между EA/водой. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент 0–5% MeOH в DCM) с получением указанного в заголовке соединения.
Стадия 8: 5–((5–((2H–тетразол–5–ил)метил)–2–метоксифенил)тио)–N4–бутил–6–метилпиримидин–2,4–диамин
Смесь 2–(3–((2–амино–4–(бутиламино)–6–метилпиримидин–5–ил)тио)–4–метоксифенил)ацетонитрила (1,0 экв.) в NMP (0,1 M), Bu2SnO (2,0 экв.), и TMSN3 (10,0 экв.) перемешивали в течение 3 ч. при 120°C в N2. Смесь охлаждали до комнатной температуры и смесь концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/NH4OH), лиофилизировали с получением указанного в заголовке соединения в виде белого порошка.
ЖХМС: [M+H]+ = 401,4
1H ЯМР (400 МГц, DMSO) δ 7,03–6,92 (м, 2H), 6,46–6,42 (м, 2H), 6,39 (с, 2H), 4,06 (с, 2H), 3,83 (с, 3H), 3,26–3,21 (м, 2H), 2,14 (с, 3H), 1,39–1,32 (м, 2H), 1,17–1,1,10 (м, 2H), 0,81 (т, J=7,2 Гц, 3H).
Пример 32: (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–(1–метоксигептан–3–ил)–6–метилпиримидин–2,4–диамин (Соединение 32)
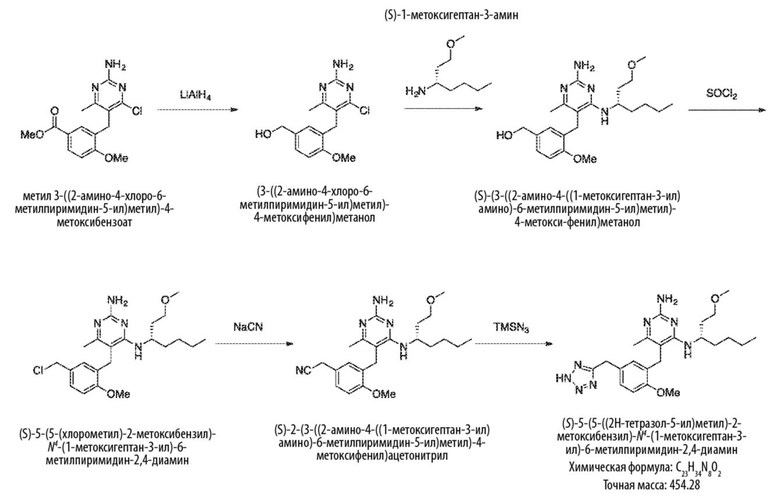
Стадия 1: (3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанол
К раствору метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв., Пример 3B – Стадия 4) в THF (1,6 M) добавляли LiAlH4 (3,0 экв.) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор разбавляли водой. Водный слой экстрагировали DCM. Органический слой промывали водой, рассолом, разделяли, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения в виде бледного твердого вещества.
Стадия 2: (S)–(3–((2–амино–4–((1–метоксигептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метокси–фенил)метанол
Смесь (3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)метанола (1,0 экв.) в NMP (0,4 M) и (S)–1–метоксигептан–3–амина (2,0 экв.) перемешивали при 120°C в течение 16 ч. в азоте. Реакционный раствор разбавляли водой. Водную фазу экстрагировали EA. Органический слой промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (DCM/MeOH=50:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 3: (S)–5–(5–(хлорметил)–2–метоксибензил)–N4–(1–метоксигептан–3–ил)–6–метилпиримидин–2,4–диамин
К перемешиваемому раствору (S)–(3–((2–амино–4–((1–метоксигептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метокси–фенил)метанола (1,0 экв.) в DCM (0,3 M) добавляли SOCl2 (2,0 экв.) по каплям. Полученную смесь перемешивали при 20°C в течение 1 ч., затем гасили H2O. Смесь разделяли в DCM/вода. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде коричневого масла.
Стадия 4: (S)–2–(3–((2–амино–4–((1–метоксигептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)ацетонитрил
Смесь (S)–5–(5–(хлорметил)–2–метоксибензил)–N4–(1–метоксигептан–3–ил)–6–метилпиримидин–2,4–диамина (1,0 экв.) в DMSO (0,4 M) и KCN (3,0 экв.) перемешивали при 80°C в течение 4 ч., затем охлаждали до комнатной температуры. Добавляли воду и раствор экстрагировали DCM. Органическую фазу сушили над Na2SO4, концентрировали, и очищали с помощью колоночной хроматографии (DCM/MeOH=40:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 5: (S)–5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–N4–(1–метоксигептан–3–ил)–6–метилпиримидин–2,4–диамин
К перемешиваемой смеси (S)–2–(3–((2–амино–4–((1–метоксигептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксифенил)ацетонитрила (1,0 экв.) в диоксане (0,2 M) и Bu2SnO (2,0 экв.) добавляли TMSN3 (10,0 экв.). Полученную смесь перемешивали при 110°C в течение 4 ч. в N2, затем охлаждали до комнатной температуры. Смесь концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза 0,1%NH3. H2O/CH3CN) с получением указанного в заголовке соединения в виде твердого вещества.
ЖХ–МС: [M+H]+ = 455,4
1H ЯМР (400 МГц, CDCl3) δ 6,86 (с, 1H), 6,84 (д, J=8,4 Гц, 1H), 6,50 (д, J =8,4 Гц, 1H), 4,28–4,23 (м, 3H), 3,90 (с, 3H), 3,58 (с, 2H), 3,19–3,16 (м, 2H), 3,14 (с, 3H), 2,50 (с, 3H), 1,83–1,81 (м, 1H), 1,59–1,06 (м, 7H), 0,83 (т, J=7,2 Гц, 3H).
Пример 33: (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропановая кислота (Соединение 33)
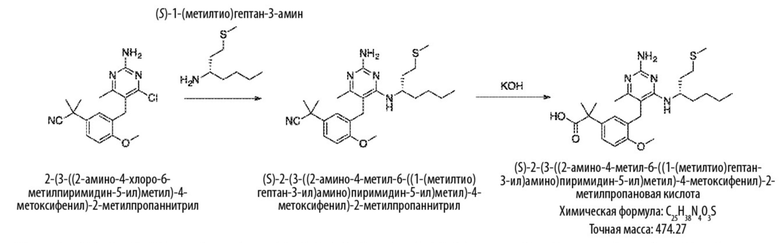
Стадия 1: (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрил
Смесь 2–(3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв., Пример 9 – Стадия 8) в NMP (0,6 M), (S)–1–(метилтио)гептан–3–амина (1,5 экв.; получен в соответствии с методиками WO2014/128189, стр. 8, соединение D) и DIEA (3,0 экв.) перемешивали при 120°C в течение 48 ч. К смеси добавляли воду и экстрагировали EtOAc. Органический слой сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш–хроматографией на диоксиде кремния (элюент: DCM/MeOH=100:1~20:1) с получением указанного в заголовке соединения в виде коричневого масла.
Стадия 2: (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропановая кислота
К перемешиваемому раствору (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропаннитрила (1,0 экв.) в 1:1 этан–1,2–диол/H2O (0,1 M) добавляли KOH (10 экв.) и нагревали в запаянной трубке при 150°C в течение 16 ч. Смесь подкисляли 1 н. раствором HCl до pH=2–3 и фильтровали. Фильтрат очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/NH3. H2O), затем лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H]+ = 475,3
1H ЯМР (400 МГц, DMSO–d6) δ 7,14 (дд, J=8,4, 2,0 Гц, 1H), 6,91 (д, J=8,8 Гц, 1H), 6,86 (с, 1H), 5,91 (ш с, 2H), 5,72 (д, J=9,2 Гц, 1H), 4,25–4,15 (м, 1H), 3,82 (с, 3H), 3,62 (с, 2H), 2,31–2,25 (м, 2H), 2,02 (с, 3H), 1,95 (с, 3H), 1,70–1,52 (м, 2H), 1,44–1,34 (м, 2H), 1,31 (с, 6H), 1,25–1,05 (м, 4H), 0,78 (т, J=6,8 Гц, 3H).
Пример 34: (S)–5–(2–метокси–5–(2H–тетразол–5–ил)бензил)–6–метил–N4–(1–(метилтио)гептан–3–ил)пиримидин–2,4–диамин (Соединение 34)
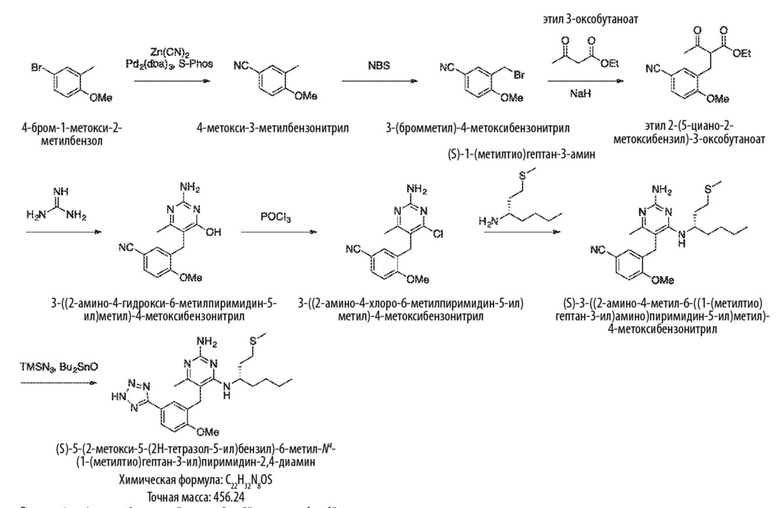
Стадия 1: 4–метокси–3–метилбензонитрил
Смесь 4–бром–1–метокси–2–метилбензола (1,0 экв.) в DMF (1 M), Zn(CN)2 (1,5 экв.), Pd2(dba)3 (0,02 экв.) и S–phos (0,05 экв.) перемешивали при 80°C в течение 16 ч. Полученную смесь концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюент PE/EA=100:1~20:1) с получением указанного в заголовке соединения.
Стадия 2: 3–(бромметил)–4–метоксибензонитрил
Смесь 4–метокси–3–метилбензолнитрила (1,0 экв.) в CCl4 (0,7 M), NBS (1,1 экв.) и AIBN (0,1 экв.) перемешивали при 95°C в течение 16 ч в азоте. Полученную смесь концентрировали и разделяли в DCM и воде. Органический слой сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюент PE/EA=100:1~20:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: этил 2–(5–циано–2–метоксибензил)–3–оксобутаноат
К раствору 3–(бромметил)–4–метоксибензонитрила (1,5 экв.) в THF (0,6 M) при 0°C добавляли порциями 60% NaH (1,5 экв.) в N2. Полученную суспензию перемешивали при 0°C в течение 10 минут, затем раствор этил 3–оксобутанoата (1,0 экв.) в THF (1 M) добавляли по каплям в течение 10 минут при 0 oC. Полученную смесь перемешивали при 70°C в течение 15 ч. Реакционную смесь охлаждали до комнатной температуры , и ледяную воду добавляли. Смесь экстрагировали этилацетатом. Органический слой сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюент PE/EA=50:1~5:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 4: 3–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–4–метоксибензонитрил
К раствору этил 2–(5–циано–2–метоксибензил)–3–оксобутанoата (1,0 экв.) в MeOH (0,7 M) добавляли гуанидинкарбонат (1,0 экв.). Полученную смесь перемешивали при 70°C в течение 16 ч. Смесь концентрировали и остаток суспендировали в EtOAc и затем фильтровали. Остаток после фильтрования промывали водой и EtOAc. Полученное твердое вещество собирали и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 5: 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензонитрил
Суспензию 3–((2–амино–4–гидрокси–6–метилпиримидин–5–ил)метил)–4–метоксибензонитрила (1,0 экв.) в POCl3 (0,3 M) перемешивали при 100°C в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и растворитель упаривали при пониженном давлении. Остаток разбавляли водой и pH доводили до 7 используя твердый NaHCO3. Осадок собирали фильтрованием и остаток после фильтрования промывали водой и EtOAc, сушили в вакууме с получением указанного в заголовке соединения.
Стадия 6: (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензонитрил
Смесь 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензонитрила (1,0 экв.) в NMP (0,3 M) и (S)–1–(метилтио)гептан–3–амина (1,5 экв., получен в соответствии с методиками WO2014/128189, стр. 8, соединение D) перемешивали при 150°C в течение 4 ч в азоте. Реакционный раствор разбавляли водой и водную фазу экстрагировали EA. Органический слой промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (DCM/MeOH=50:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 7: (S)–5–(2–метокси–5–(2H–тетразол–5–ил)бензил)–6–метил–N4–(1–(метилтио)гептан–3–ил)пиримидин–2,4–диамин
К перемешиваемой смеси (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензонитрила (1,0 экв.) в диоксане (0,4 M), Bu2SnO (2,0 экв.) добавляли TMSN3 (10,0 экв.). Полученную смесь перемешивали при 110°C в течение 4 ч. в N2. Смесь охлаждали до комнатной температуры и смесь концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза 0,1%NH3. H2O/CH3CN) с получением указанного в заголовке соединения.
ЖХ–МС: [M+H]+= 457,3
1H ЯМР (400 МГц, DMSO–d6) δ 7,82 (д, J=8,8 Гц, 1H), 7,50 (с, 1H), 7,07 (д, J =8,4 Гц, 1H), 6,55 (ш, 3H), 4,28–4,26 (м, 1H), 3,89 (с, 3H), 3,73 (с, 2H), 2,27 (т, J=7,6 Гц, 2H), 2,14 (с, 3H), 1,88 (с, 3H), 1,67–1,65 (м, 2H), 1,41–1,39 (м, 2H), 1,12–1,04 (м, 4H) , 0,69 (т, J=6,8 Гц, 3H).
Пример 35: (S)–(3–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензамидо)пропил)фосфоновая кислота (Соединение 35)
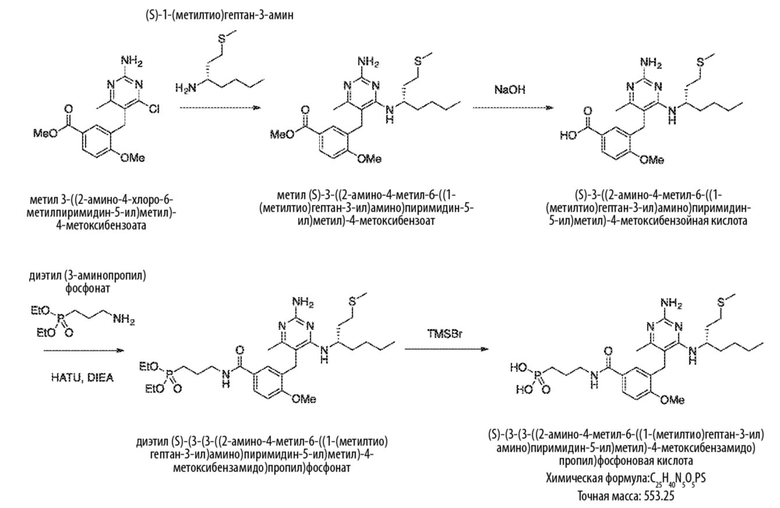
Стадия 1: метил (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензоат
Смесь метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв., Пример 3B – Стадия 4) в NMP (0,16 M) и (S)–1–(метилтио)гептан–3–амина (1,5 экв., получен в соответствии с методиками WO2014/128189, стр. 8, соединение D) перемешивали при 150°C в течение 4 ч. в азоте. Смесь разбавляли водой. Водную фазу экстрагировали EA. Органический слой промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Остаток очищали с помощью колоночной хроматографии (DCM/MeOH=50:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2: (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензойная кислота
Смесь метил (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в 1:1 EtOH/H2O (0,02 M) и NaOH (10,0 экв.) перемешивали при 70oC в течение 16 ч. Смесь нейтрализировали используя 1 н. HCl и полученную суспензию фильтровали. Остаток после фильтрования промывали водой с получением твердого вещества, которое растворяли в MeCN/H2O. Добавляли 4 н. HCl/диоксан и полученный раствор лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: диэтил (S)–(3–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензамидо)пропил)фосфонат
Смесь (S)–3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензойной кислоты (1,0 экв.) в DMF (0,1 M), диэтил (3–аминопропил)фосфоната (1,1 экв., Пример 30 – Стадия 2), HATU (1,5 экв.) и DIEA (2,0 экв.) перемешивали при 30°C в течение 16 ч. Смесь охлаждали до комнатной температуры и добавляли воду. Смесь экстрагировали DCM. Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла.
Стадия 4: (S)–(3–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензамидо)пропил)фосфоновая кислота
Смесь диэтил (S)–(3–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксибензамидо)пропил)фосфоната (1,0 экв.) в DCM (0,05 M) и TSMBr (10,0 экв.) перемешивали при 35°C в течение 5 ч. в N2. Смесь охлаждали и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/NH3. H2O) с получением указанного в заголовке соединения.
ЖХ–МС: [M+H] + = 554,3
1H ЯМР (400 МГц, CDCl3) δ 13,93 (с, 1H), 10,36 (с, 1H), 10,12 (с, 1H), 7,94 (д, J=8,0 Гц, 1H), 7,80 (с, 1H), 6,96 (д, J=8,8 Гц, 1H), 5,94 (д, J=8,4 Гц, 1H), 4,23–4,21 (м, 1H), 3,98 (с, 3H), 3,73–3,30 (м, 4H), 2,76 (с, 3H), 2,43–2,41 (м, 1H), 2,05–1,80 (м, 7H), 1,70 (с, 3H), 1,55–1,20 (м, 6H), 0,86 (т, J=4,0 Гц, 3H).
Пример 36: (S)–N4–(1–(2H–тетразол–5–ил)гептан–3–ил)–5–(2–метоксибензил)–6–метилпиримидин–2,4–диамин (Соединение 36)

Стадия 1: (S)–3–(бензил((S)–1–фенилэтил)амино)гептил метансульфонат
К перемешиваемому раствору (S)–3–(бензил((S)–1–фенилэтил)амино)гептан–1–ола (1,0 экв., полученного из J. Med. Chem. 2016, 59, 7936–7949) и DIEA (1,1 экв.) в DCM (0,5 M) добавляли MsCl (1,1 экв.) при 0 oC. Полученную смесь нагревали до комнатной температуры более 3 ч. Растворитель удаляли, и остаток разбавляли EA. Органический слой промывали 1 н. HCl, 1 н. NaOH, рассолом, сушили и концентрировали с получением указанного в заголовке соединения.
Стадия 2: (S)–4–(бензил((S)–1–фенилэтил)амино)октаннитрил
Смесь (S)–3–(бензил((S)–1–фенилэтил)амино)гептил метансульфоната (1,0 экв.) в DMSO (0,3 M) и KCN (7,5 экв.) перемешивали при 30°C в течение 16 ч. Реакционную смесь разбавляли водой. Водный слой экстрагировали EA. Органический слой разделяли, сушили и концентрировали с получением указанного в заголовке соединения в виде светло–желтого масла.
Стадия 3: (S)–N–бензил–N–((S)–1–фенилэтил)–1–(2H–тетразол–5–ил)гептан–3–амин
Смесь (S)–4–(бензил((S)–1–фенилэтил)амин)октаннитрила (1,0 экв.) в диоксане (0,2 M), TMSN3 (2,5 экв.) и Bu2SnO (2,0 экв.) перемешивали при 120°C в запаянной трубке в течение 18 ч. Смесь разбавляли EA и затем промывали водой. Органический слой сушили и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (элюент PE:EA=10:1) с получением указанного в заголовке соединения в виде светло–коричневого масла.
Стадия 4: (S)–1–(2H–тетразол–5–ил)гептан–3–амин
К раствору (S)–N–бензил–N–((S)–1–фенилэтил)–1–(2H–тетразол–5–ил)гептан–3–амина (1,0 экв.) в MeOH (0,2 M) добавляли 50% Pd/C (0,3 мас. экв.) в азоте. Реакционную смесь перемешивали при 40°C в атмосфере H2 в течение 16 ч. Pd/C отфильтровывали, и фильтрат концентрировали с получением указанного в заголовке соединения в виде светло–желтого твердого вещества.
Стадия 5: (S)–N4–(1–(2H–тетразол–5–ил)гептан–3–ил)–5–(2–метоксибензил)–6–метилпиримидин–2,4–диамин
Раствор 4–хлор–5–(2–метоксибензил)–6–метилпиримидин–2–амина (1,0 экв., Пример 41 – Стадия 3) в NMP (0,4 M) и (S)–1–(2H–тетразол–5–ил)гептан–3–амина (2,0 экв.) перемешивали при 150°C в течение 3 ч в азоте. Реакционную смесь разбавляли водой и воду экстрагировали EA. Органические слои объединяли и промывали водой, рассолом, сушили и концентрировали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза 0,1%NH3H2O/CH3CN) с получением указанного в заголовке соединения.
ЖХ–МС: [M+H]+ = 411,4
1H ЯМР (400 МГц, MeOD) δ 7,17 (т, J=7,2 Гц, 1H), 6,96 (д, J=8,8 Гц, 2H), 6,85 (т, J=7,2 Гц, 1H), 4,24–4,20 (м, 1H), 3,91 (с, 3H), 3,78–3,63 (м, 2H), 3,19–3,16 (м, 1H), 2,72 (т, J=8,0 Гц, 2H), 2,28 (с, 3H), 2,28–0,80 (м, 8H), 0,77 (т, J=7,2 Гц, 3H).
Пример 37: 6–(2–(2H–тетразол–5–ил)этил)–N4–бутил–5–(2–метоксибензил)пиримидин–2,4–диамин (Соединение 37)

Стадия 1: этил 5–циано–3–оксопентаноат
К раствору 3–цианопропановой кислоты (1,0 экв.) в THF (0,5 M) добавляли CDI (1,2 экв.). Реакционный раствор перемешивали при 25°C в течение 1 ч. MgCl2 (1,0 экв.) и калий 3–этокси–3–оксопропаноат (1,0 экв.) добавляли к вышеуказанному раствору и перемешивали при 60°C в течение 2 ч. Раствор охлаждали до комнатной температуры и гасили H2O. Водную фазу экстрагировали EA. Комбинированный органический слой промывали водой, а затем рассолом. Органический слой разделяли и сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии (PE/EA=3:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 2: диэтил 2–(2–метоксибензил)–3–оксогександиоат
К раствору этил 5–циано–3–оксопентаноата (1,2 экв.) в THF (0,3 M) добавляли порциями NaH (1,3 экв.) при 0°C в азоте. Раствор перемешивали при 0°C в течение 15 минут, затем добавляли 1–(бромметил)–2–метоксибензол (1,0 экв.). Реакционную смесь перемешивали при 40°C в течение 16 ч. Реакционную смесь гасили водой. Водный слой экстрагировали EA. Комбинированный органический слой промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5:1) с получением указанного в заголовке соединения в виде светло–желтого масла.
Стадия 3: 3–(2–амино–6–гидрокси–5–(2–метоксибензил)пиримидин–4–ил)пропаннитрил
К раствору диэтил 2–(2–метоксибензил)–3–оксогександиоата (1,0 экв.) в MeOH (0,6 M) добавляли гуанидинкарбонат (1,0 экв.). Реакцию нагревали при 70°C в течение 16 ч. в азоте. После охлаждения реакционный раствор концентрировали при пониженном давлении и добавляли 1:1 H2O/EA. Осадки фильтровали и собирали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества.
Стадия 4: 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропаннитрил
Смесь 3–(2–амино–6–гидрокси–5–(2–метоксибензил)пиримидин–4–ил)пропаннитрила (1,0 экв.) и POCl3 (0,3 M) перемешивали при 100°C в течение 2 ч. в азоте. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду. pH доводили до 7 используя раствор NaHCO3. Водный раствор экстрагировали EA. Органический слой промывали водой, рассолом, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения в виде серого твердого вещества.
Стадия 5: 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропаннитрил
К раствору 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропаннитрила (1,0 экв.) в NMP (0,16 M) добавляли бутан–1–амин (3,0 экв.) и DIEA (3,0 экв.). Смесь перемешивали при 100°C в течение 16 ч. в азоте. Реакционный раствор разбавляли водой и экстрагировали EA. Органический слой промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (DCM/MeOH=50:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 6: 6–(2–(2H–тетразол–5–ил)этил)–N4–бутил–5–(2–метоксибензил)пиримидин–2,4–диамин
К раствору 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропаннитрила (1,0 экв.) в диоксане (0,06 M) добавляли TMSN3 (2,0 экв.) и Bu2SnO (2,0 экв.). Смесь перемешивали при 120°C в течение 3 ч. Раствор концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза: NH4HCO3/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + =383,4
1H ЯМР (400 МГц, DMSO–d6) δ 7,18 (т, J=8,4 Гц, 1H), 6,98 (д, J=8,0 Гц, 1H), 6,81 (т, J=7,2 Гц, 1H), 6,73 (д, J=7,6 Гц, 1H), 6,59 (т, J=5,6 Гц, 1H), 6,27 (ш с, 2H), 3,84 (с, 3H), 3,65 (с, 2H), 3,30–3,25 (м, 2H), 3,02 (т, J=7,6 Гц, 2H), 2,76 (т, J=7,6 Гц, 2H), 1,49–1,37 (м, 2H), 1,23–1,16 (м, 2H), 0,83 (т, J=7,6 Гц, 3H).
Пример 38: (S)–3–(2–амино–5–(2–метоксибензил)–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–4–ил)пропановая кислота (Соединение 38)

Стадия 1: (S)–3–(2–амино–5–(2–метоксибензил)–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–4–ил)пропановая кислота
Смесь 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв., Пример 27 – Стадия 4) и неразбавленного (S)–1–(метилтио)гептан–3–амина (4,0 экв., получен в соответствии с методиками WO2014/128189, стр. 8, соединение D) перемешивали при 120°C в течение 24 ч. Реакционную смесь разбавляли водой и водный раствор экстрагировали EA. Органические слои объединяли и промывали 1 н. HCl, рассолом, сушили над Na2SO4, концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза:0,1% NH3,H2O/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H]+ = 447,3
1H ЯМР (400 МГц, MeOD) δ 7,23 (т, J=8,0 Гц, 1H), 7,02 (д, J=8,0 Гц, 1H), 6,97 (д, J=8,0 Гц, 1H), 6,92 (т, J=8,0 Гц, 1H), 4,41–4,34 (м, 1H), 3,92 (с, 3H), 3,83 (с, 2H), 2,93 (т, J=7,2 Гц, 2H), 2,49 (т, J=6,8 Гц, 2H), 2,25 (т, J=7,2 Гц, 2H), 1,96 (с, 3H), 1,76–0,90 (м, 8H), 0,81 (т, J=7,6 Гц, 3H).
Пример 39: 5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–6–метил–N4–(пентан–2–ил)пиримидин–2,4–диамин (Соединение 39)
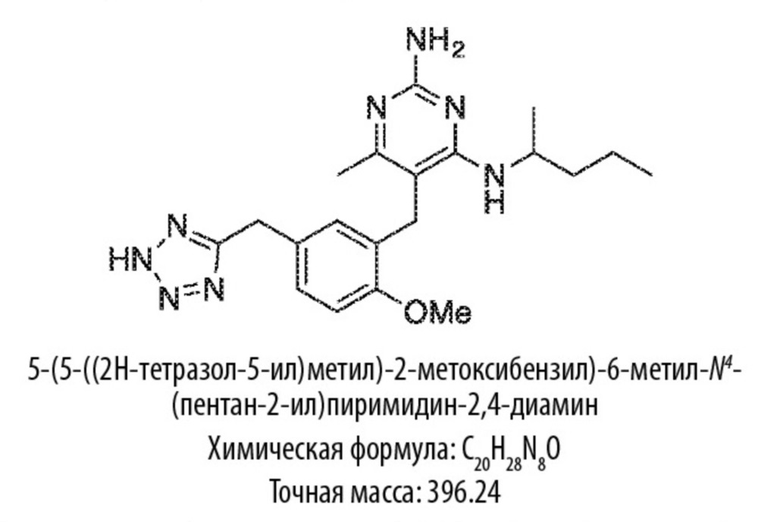
Указанное в заголовке соединение получали в соответствии с описанными методиками Примера 3, но используя пентан–2–амин вместо бутан–1–амина в Стадии 5.
ЖХ–МС: [M+H]+= 397,4
1H ЯМР (400 МГц, CDCl3) δ 7,26 (д, J=8,4 Гц, 1H), 6,92 (с, 1H), 6,85 (д, J=8,4 Гц, 1H), 6,49 (д, J =6,8 Гц, 1H), 4,26 (с, 2H), 4,20–4,18 (м, 1H), 3,92 (с, 3H), 3,58 (с, 2H), 2,59 (с, 3H), 1,48–1,46 (м, 2H), 1,40–1,38 (м, 2H), 1,08 (д, J=6,4 Гц, 3H), 0,85 (т, J=7,2 Гц, 3H).
Пример 40: 3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(бутиламино)пиримидин–4–ил)пропановая кислота (Соединение 40)

Стадия 1: (4–метокси–3–метилфенил)метанол
К перемешиваемому раствору метил 4–метокси–3–метилбензоата (1,0 экв.) в THF (0,6 M) добавляли LiAlH4 (1,2 экв.) порциями при 4°C в азоте. Полученную смесь нагревали до комнатной температуры в течение 2 часов. Смесь гасили водным раствором Na2SO4. Раствор фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения в виде бесцветного масла.
Стадия 2: 4–(хлорметил)–1–метокси–2–метилбензол
К перемешиваемому раствору (4–метокси–3–метилфенил)метанола (1,0 экв.) в DCM (1,4 M) добавляли SOCl2 (1,5 экв.) при комнатной температуре в N2. Смесь перемешивали при комнатной температуре в течение 1 ч. и затем концентрировали. Остаток разделяли в DCM и насыщенном водном растворе NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия 3: 2–(4–метокси–3–метилфенил)ацетонитрил
Смесь 4–(хлорметил)–1–метокси–2–метилбензола (1,0 экв.) в 1:1 DMSO/DMF (0,9 M) и NaCN (2,0 экв.) перемешивали в течение 16 ч. при комнатной температуре. Смесь разделяли в EA и воде. Органический слой сушили над Na2SO4, фильтровали, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент PE/EA=100:1~20:1) с получением указанного в заголовке соединения.
Стадия 4: 2–(3–(бромметил)–4–метоксифенил)ацетонитрил
К раствору 2–(4–метокси–3–метилфенил)ацетонитрила (1,0 экв.) в CCl4 (0,35 M) добавляли NBS (1,2 экв.) и AIBN (2,0 г, 13 ммоль, 0,1 экв.). Полученную смесь нагревали с обратным холодильником в течение 1 ч. Затем смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который очищали флэш–хроматографией на диоксиде кремния (элюент: PE/EA=от 40:1 до 4:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 5: диэтил 2–(5–(цианометил)–2–метоксибензил)–3–оксогександиоат
К раствору 2–(3–(бромметил)–4–метоксифенил)ацетонитрила (1,0 экв.) в безводном THF (0,73 M) при 0°C добавляли порциями 60% NaH (1,2 экв.). После перемешивания в течение 10 мин, раствор диэтил 3–оксогександиоата (1,1 экв., Пример 27 – Стадия 1) в THF (2,4 M) добавляли по каплям в вышеуказанную смесь в течение 10 мин. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 20 ч. Реакцию гасили водой. Смесь экстрагировали EtOAc, сушили над Na2SO4, концентрировали при пониженном давлении. Остаток очищали флэш–хроматографией на диоксиде кремния (элюент: PE/EA=от 50:1 до 10:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 6: 3–(2–амино–5–(5–(цианометил)–2–метоксибензил)–6–гидроксипиримидин–4–ил)пропановая кислота
Смесь диэтил 2–(5–(цианометил)–2–метоксибензил)–3–оксогександиоата (1,0 экв.) в MeOH (0,3 M) и гуанидинкарбоната (1,5 экв.) перемешивали при 70°C в течение 16 ч. Растворитель удаляли и остаток разбавляли водой и подкисляли, добавляя 1 н. HCl до рН 5. Полученный осадок фильтровали и сушили с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 7: 3–(2–амино–6–хлор–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропановая кислота
Смесь 3–(2–амино–5–(5–(цианометил)–2–метоксибензил)–6–гидроксипиримидин–4–ил)пропановой кислоты (1,0 экв.) в POCl3 (1,9 M) перемешивали при 100°C в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и POCl3 упаривали при пониженном давлении. Остаток разбавляли водой. pH доводили до 7 твердым NaHCO3. Осадок собирали фильтрованием, промывали водой, и сушили в вакууме с получением указанного в заголовке соединения в виде зеленоватого твердого вещества.
Стадия 8: 3–(2–амино–6–(бутиламино)–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропановая кислота
Смесь 3–(2–амино–6–хлор–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв.) в EtOH (0,3 M), бутан–1–амина (4 экв.), и DIEA (5 экв.) перемешивали при 85°C в течение 48 ч. Растворитель удаляли, и остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=40:1) с получением указанного в заголовке соединения в виде бледного твердого вещества.
Стадия 9: 3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(бутиламино)пиримидин–4–ил)пропановая кислота
Смесь 3–(2–амино–6–(бутиламино)–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв.) в диоксане (0,1 M), TMSN3 (5 экв.) и Bu2SnO (2,0 экв.) нагревали в запаянной трубке при 70°C в течение 3 ч. Смесь концентрировали при пониженном давлении, и неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза: 0,1% NH3,H2O в CH3CN) с получением указанного в заголовке соединения в виде твердого вещества.
ЖХ–МС: [M+H] + = 441,2
1H ЯМР (400 МГц, CDCl3) δ 7,46 (с, 1H), 7,24 (д, J=8,4 Гц,1H), 6,84 (д, J=8,4 Гц, 1H), 5,78 (ш с, 1H), 4,17 (с, 2H), 3,90 (с, 3H), 3,70 (с, 2H), 3,36–3,31 (м, 2H), 3,17 (т, J=6,0 Гц, 2H), 2,77 (т, J=6,0 Гц, 2H), 1,41–1,38 (м, 2H), 1,18–1,13 (м, 2H), 0,84 (т, J=6,8 Гц, 3H).
Пример 41: (S)–3–((2–амино–5–(2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептановая кислота (Соединение 41)
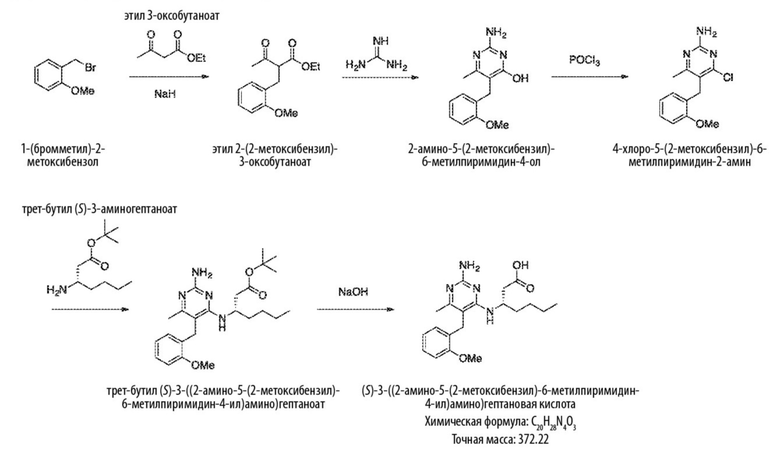
Стадия 1: этил 2–(2–метоксибензил)–3–оксобутаноат
К раствору 1–(бромметил)–2–метоксибензола (1,2 экв.) в THF (0,6 M) при 0°C добавляли порциями 60% NaH (1,25 экв.) в N2. Полученную суспензию перемешивали при 0°C в течение 10 минут, затем по каплям в течение 10 минут добавляли раствор этил 3–оксобутаноата (1,0 экв.) в THF (5 M). Полученную смесь перемешивали в течение 15 ч. при 70 oC. Смесь охлаждали до комнатной температуры и добавляли ледяную воду. Смесь экстрагировали этилацетатом. Органический слой сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюент PE/EA=50:1~5:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 2: 2–амино–5–(2–метоксибензил)–6–метилпиримидин–4–ол
К раствору этил 2–(2–метоксибензил)–3–оксобутаноата (1,0 экв.) в MeOH (0,9 M) добавляли гуанидинкарбонат (1,0 экв.). Полученную смесь перемешивали в течении ночи при 65 oC. Смесь концентрировали досуха. Остаток суспендировали в EtOAc и затем фильтровали. Остаток после фильтрования промывали водой и EtOAc, и затем сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: 4–хлор–5–(2–метоксибензил)–6–метилпиримидин–2–амин
Суспензию 2–амино–5–(2–метоксибензил)–6–метилпиримидин–4–ола (1,0 экв.) в POCl3 (1 M) перемешивали при 100°C в течение 24 ч. Смесь охлаждали до комнатной температуры и POCl3 упаривали досуха при пониженном давлении. Остаток разбавляли водой и pH доводили до 7 используя твердый NaHCO3. Осадок собирали фильтрованием, промывали водой и EtOAc и сушили в вакууме с получением указанного в заголовке соединения.
Стадия 4: трет–бутил (S)–3–((2–амино–5–(2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептанoaт
Смесь 4–хлор–5–(2–метоксибензил)–6–метилпиримидин–2–амина (1,0 экв.) в NMP (1,3 M) и трет–бутил (S)–3–аминогептанoaта (4,5 экв., Пример 20B – Стадия 3) перемешивали при 120°C в течение 3 ч. Смесь разделяли в воде и EtOAc. Органический слой промывали рассолом, сушили над Na2SO4, и концентрировали при пониженном давлении. Остаток очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=от 100:1 до 30:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 5: (S)–3–((2–амино–5–(2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептановая кислота
Смесь трет–бутил (S)–3–((2–амино–5–(2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептанoaта (1,0 экв.) в 1:1 EtOH/H2O (0,1 M) и NaOH (10,0 экв.) нагревали при 100°C в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, и растворитель удаляли. Остаток разбавляли H2O и подкисляли используя 1 н. HCl до pH 2. Полученную суспензию экстрагировали DCM. Объединенные органические слои сушили и концентрировали. Неочищенный продукт очищали с помощью препаративной ТСХ (CHCl3:MeOH=10:1) с получением масла, которое растворяли в 1:1 CH3CN/H2O и добавляли 4 н. HCl/диоксан. Раствор лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + = 373,2
1H ЯМР (400 МГц, DMSO–d6) δ 12,20 (ш, 1H), 7,20–7,17 (м, 1H), 6,99 (д, J=8,4 Гц, 1H), 6,82–6,79(м, 2H), 6,68 (ш, 2H), 4,53–4,50 (м, 1H), 3,85 (с, 3H), 3,66 (с, 2H), 2,50–2,37 (м, 2H), 2,07 (с, 3H), 1,51–1,38 (м, 2H), 1,24–0,98 (м, 4H), 0,77 (т, J=6,8 Гц, 3H).
Пример 42: (S)–3–((2–амино–5–(5–(2–карбоксипропан–2–ил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептановая кислота (Соединение 42)

Стадия 1: трет–бутил (S)–3–((2–амино–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептанoат
Смесь 2–(3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксифенил)–2–метил–пропаннитрила (1,0 экв., Пример 9 – Стадия 8) в NMP (0,9 M) и трет–бутил (S)–3–аминогептанoaта (1,3 экв., Пример 20B – Стадия 3) перемешивали при 120°C в течение 16 ч. в азоте. Реакционную смесь разбавляли водой. Водную фазу экстрагировали EA. Объединенный органический слой промывали водой, затем рассолом. Органический слой разделяли и сушили над Na2SO4, концентрировали и очищали флэш–хроматографией на диоксиде кремния (элюент DCM/MeOH=50:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2: (S)–3–((2–амино–5–(5–(2–карбоксипропан–2–ил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептановая кислота
К смеси трет–бутил (S)–3–((2–амино–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)–6–метил–пиримидин–4–ил)амино)гептанoaта (1,0 экв.) в 1:1 этилeнгликоль/H2O (0,1 M) добавляли NaOH (10,0 экв.) и перемешивали при 150°C в течение 16 ч. Смесь нейтрализовали добавлением 1 н. HCl. Полученную суспензию фильтровали. Остаток после фильтрования промывали водой и очищали с помощью препаративной ВЭЖХ (подвижная фаза: 0,1%HCOOH/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + = 459,3
1H ЯМР (400 МГц, DMSO–d6) δ 7,19 (ш, 1H), 7,18 (дд, J=8,4, 2,4 Гц, 1H), 6,96 (д, J=8,4 Гц, 1H), 6,84 (д, J=2,0 Гц, 1H), 4,58–4,56 (м, 1H), 3,83 (с, 3H), 3,79 (с, 2H), 2,48–2,42 (м, 2H) 2,13 (с, 3H), 1,52–1,50 (м, 2H), 1,38 (с, 3H), 1,35 (с, 3H), 1,25–1,18 (м, 2H), 1,24–1,07 (м, 2H), 0,91 (т, J=7,2 Гц, 3H).
Пример 43: 2–(3–((2–амино–4–(бутиламино)–6–(2–карбоксиэтил)пиримидин–5–ил)метил)–4–метокси–фенил)–2–метилпропановая кислота (Соединение 43)
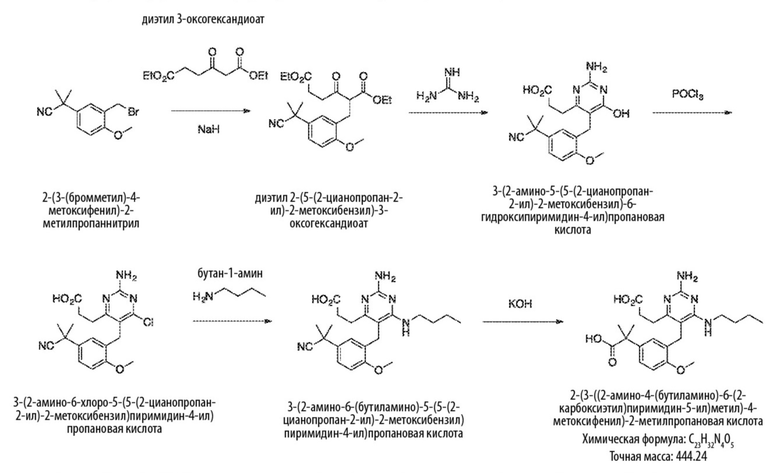
Стадия 1: диэтил 2–(5–(2–цианопропан–2–ил)–2–метоксибензил)–3–оксогександиоат
К раствору 2–(3–(бромметил)–4–метоксифенил)–2–метилпропаннитрила (1,1 экв., Пример 9 – Стадия 5) в THF (0,3 M) добавляли порциями NaH (1,2 экв.) при 0°C в азоте. Смесь перемешивали при 0°C в течение 10 минут. Раствор диэтил 3–оксогександиоата (1,0 экв., Пример 27 – Стадия 1) в THF (0,75 M) добавляли к вышеуказанному раствору и реакционную смесь перемешивали при 60°C в течение 16 ч. Реакционную смесь гасили водой. Водный раствор экстрагировали EA. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4, и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10:1) с получением указанного в заголовке соединения в виде желтого масла.
Стадия 2: 3–(2–амино–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)–6–гидроксипиримидин–4–ил)пропановая кислота
К раствору диэтил 2–(5–(2–цианопропан–2–ил)–2–метоксибензил)–3–оксогександиоата (1,0 экв.) в MeOH (0,7 M) добавляли гуанидинкарбонат (1,0 экв.). Реакцию нагревали при 65°C в течение 16 ч. в азоте. После охлаждения, реакционный раствор концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (DCM/MeOH=5:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: 3–(2–амино–6–хлор–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановая кислота
Раствор 3–(2–амино–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)–6–гидроксипиримидин–4–ил)пропановой кислоты (1,0 экв.) в POCl3 (0,3 M) перемешивали при 100°C в течение 16 ч. в азоте. После охлаждения смесь концентрировали и выливали в воду. Значение рН доводили до 8, добавляя NaHCO3. Водную фазу экстрагировали EA. Объединенные органические слои сушили над Na2SO4, концентрировали и очищали с помощью колоночной хроматографии (DCM/MeOH=10:1) с получением указанного в заголовке соединения в виде коричневого твердого вещества.
Стадия 4: 3–(2–амино–6–(бутиламино)–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановая кислота
Раствор 3–(2–амино–6–хлор–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв.) в неразбавленном бутан–1–амине (0,15 M) перемешивали при 120°C в течение 3 ч. в азоте. Реакционную смесь очищали с помощью колоночной хроматографии (DCM/MeOH=10:1) с получением указанного в заголовке соединения в виде коричневого масла.
Стадия 5: 2–(3–((2–амино–4–(бутиламино)–6–(2–карбоксиэтил)пиримидин–5–ил)метил)–4–метоксифенил)–2–метилпропановая кислота
К раствору 3–(2–амино–6–(бутиламино)–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв.) в 1:1 этилeнгликоль/H2O (0,07 M) добавляли KOH (20 экв.). Смесь перемешивали при 150°C в течение 16 ч. После охлаждения рН доводили до 6, добавляя 4 н. HCl. Смесь экстрагировали EA. Органический слой промывали рассолом, сушили над Na2SO4, концентрировали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза: NH4HCO3/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + = 445,3
1H ЯМР (400 МГц, CD3OD) δ 7,27 (дд, J =8,4, 2,4Гц, 1H), 6,95 (д, J=8,4Гц, 1H), 6,93 (с, 1H), 3,89 (с, 3H), 3,78 (с, 2H), 3,45 (т, J=7,0 Гц, 2H), 2,84 (т, J=6,0 Гц, 2H), 2,42 (т, J=6,4 Гц, 2H), 1,54–1,47 (м, 2H), 1,45 (с, 6H), 1,26–1,21 (м, 2H), 0,88 (т, J=7,2 Гц, 3H).
Пример 44: (S)–3–((2–амино–5–(2–метокси–5–((3–фосфонопропил)карбамоил)бензил)–6–метил–пиримидин–4–ил)амино)гептановая кислота (Соединение 44)

Стадия 1: метил (S)–3–((2–амино–4–((1–(трет–бутокси)–1–оксогептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоат
Смесь метил 3–((2–амино–4–хлор–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв., Пример 3B – Стадия 4) в NMP (2 M) и трет–бутил (S)–3–аминогептанoaта (2,0 экв., Пример 20B – Стадия 3) перемешивали при 120°C в течение 16 ч. в азоте. Реакционный раствор разбавляли водой и экстрагировали EA. Органический слой промывали водой, рассолом, сушили над Na2SO4, и концентрировали. Остаток очищали с помощью колоночной хроматографии (DCM/MeOH=50:1) с получением указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2: (S)–3–((2–амино–4–((1–(трет–бутокси)–1–оксогептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойная кислота
Смесь метил (S)–3–((2–амино–4–((1–(трет–бутокси)–1–оксогептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксибензоата (1,0 экв.) в 1:1 этанол/H2O (0,1 M) и NaOH (10,0 экв.) перемешивали при 30°C в течение 4 ч. Смесь нейтрализовали добавлением 1 н. HCl до pH 7. Полученную суспензию фильтровали. Остаток после фильтрования промывали водой с получением указанного в заголовке соединения.
Стадия 3: трет–бутил (S)–3–((2–амино–5–(5–((3–(диэтоксифосфорил)пропил)карбамоил)–2–метоксибензил)–6–метилпиримидин–4–ил)амино)гептанoaт
Смесь (S)–3–((2–амино–4–((1–(трет–бутокси)–1–оксогептан–3–ил)амино)–6–метилпиримидин–5–ил)метил)–4–метоксибензойной кислоты (1,0 экв.) в DMF (0,1 M), диэтил (3–аминопропил)фосфоната (2,0 экв., Пример 30 – Стадия 2), HATU (2,0 экв.) и DIEA (3,0 экв.) перемешивали при 50°C в течение 16 ч. Смесь охлаждали до комнатной температуры и добавляли воду. Смесь экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4, и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла.
Стадия 4: (S)–3–((2–амино–5–(2–метокси–5–((3–фосфонопропил)карбамоил)бензил)–6–метил–пиримидин–4–ил)амино)гептановая кислота
К раствору трет–бутил (S)–3–((2–амино–5–(5–((3–(диэтоксифосфорил)пропил)карбамоил)–2–метокси–бензил)–6–метилпиримидин–4–ил)амино)гептанoaта (1,0 экв.) в DCM (0,08 M) добавляли TSMBr (10,0 экв.) и перемешивал при 35°C в течение 16 ч. в N2. Смесь концентрировали и остаток очищали растиранием и препаративной ВЭЖХ (подвижная фаза CH3CN/H2O/NH3H2O) с получением указанного в заголовке соединения.
ЖХ–МС: [M+H] + = 538,3
1H ЯМР (400 МГц, DMSO) δ 8,69 (ш, 1H), 7,72 (д, J=8,8 Гц, 1H), 7,50 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,63(ш, 1H), 4,47–4,45 (м, 1H), 3,85 (с, 3H), 3,27–3,25 (м, 4H), 2,4 (д, J=6,0 Гц, 2H), 2,13 (с, 3H), 1,72–1,68 (м, 2H), 1,46–1,26 (м, 4H), 1,12–0,96 (м, 2H), 0,96–0,94 (м, 2H), 0,70 (т, J=7,2 Гц, 3H).
Пример 45: (S)–3–((2–амино–6–(2–карбоксиэтил)–5–(5–(2–карбоксипропан–2–ил)–2–метокси–бензил)пиримидин–4–ил)амино)гептановая кислота (Соединение 45)

Стадия 1: (S)–3–(2–амино–6–((1–(трет–бутокси)–1–оксогептан–3–ил)амино)–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановая кислота
К раствору 3–(2–амино–6–хлор–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв., Пример 43 – Стадия 3) в NMP (0,15 M) добавляли трет–бутил (S)–3–аминогептанoaт (5,0 экв., Пример 20B – Стадия 3) и K2CO3 (3,0 экв.). Смесь перемешивали при 150°C в течение 16 ч. Реакционный раствор разбавляли водой/EA и концентрировали с получением указанного в заголовке соединения в виде неочищенного масла.
Стадия 2: (S)–3–((2–амино–6–(2–карбоксиэтил)–5–(5–(2–карбоксипропан–2–ил)–2–метоксибензил)–пиримидин–4–ил)амино)гептановая кислота
К раствору (S)–3–(2–амино–6–((1–(трет–бутокси)–1–оксогептан–3–ил)амино)–5–(5–(2–цианопропан–2–ил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (неочищенный) в 1:1 этиленгликоль/H2O добавляли избыток KOH и перемешивали при 150°C в течение 16 ч. После охлаждения, реакционную смесь разбавляли водой/EA. pH доводили до 6 добавлением 4 н. HCl, затем экстрагировали 3:1 CHCl3/IPA. Объединенный органический слой промывали рассолом, сушили над Na2SO4, и концентрировали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (подвижная фаза:NH4OH/MeCN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + = 517,3
1H ЯМР (400 МГц, CD3OD) δ 7,27 (дд, J =8,0, 2,4Гц, 1H), 7,00–6,95 (м, 2H), 4.,61–4,57 (м, 1H), 3,90 (с, 3H), 3,80 (с, 2H), 2,94–2,87 (м, 2H), 2,50–2,38 (м, 4H), 1,56–1,53 (м, 1H), 1,48 (с, 3H), 1,45 (с, 3H), 1,46–1,44 (м, 1H), 1,27–1,17 (м, 2H), 1,08– 1,03 (м, 2H), 0,79 (т, J=7,4 Гц, 3H).
Пример 46: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(2–карбоксиэтил)–пиримидин–4–ил)амино)гептановая кислота (Соединение 46)

Стадия 1: трет–бутил (S)–3–(3–(2–амино–6–(((S)–1–(трет–бутокси)–1–оксогептан–3–ил)амино)–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропанамидо)гептанoат
Смесь 3–(2–амино–6–хлор–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв., Пример 40 – Стадия 7) и трет–бутил (S)–3–аминогептанoaта (3 экв., Пример 20B – Стадия 3) перемешивали при 140°C в течение 3 ч. Неочищенный продукт использовали непосредственно на следующей стадии.
Стадия 2: трет–бутил (S)–3–(3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(((S)–1–(трет–бутокси)–1–оксогептан–3–ил)амино)пиримидин–4–ил)пропанамидо)гептанoaт
Смесь трет–бутил (S)–3–(3–(2–амино–6–(((S)–1–(трет–бутокси)–1–оксогептан–3–ил)амино)–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропанамидо)гептанoaта (1,0 экв.) в диоксане (0,2 M), TMSN3 (3 экв.) и Bu2SnO (2,0 экв.) нагревали при 110°C в запаянной трубку в течение 6 ч. Смесь разбавляли EA и промывали водой. Органический слой сушили, концентрировали и остаток очищали с помощью колоночной хроматографии (DCM:MeOH=100:1~10:1) с получением указанного в заголовке соединения в виде светло–коричневого твердого вещества.
Стадия 3: (S)–3–((5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(2–карбоксиэтил)–пиримидин–4–ил)амино)гептановая кислота
К раствору трет–бутил (S)–3–(3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(((S)–1–(трет–бутокси)–1–оксогептан–3–ил)амино)пиримидин–4–ил)пропанамидо)гептанoaта (1,0 экв.) в этан–1,2–диоле (0,1 M) добавляли 10M водный KOH (44 экв.) и нагревали при 150°C в течение 5 дней. Твердое вещество отфильтровывали, и фильтрат концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза: 0,1% NH3,H2O, CH3CN) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + = 513,2
1H ЯМР (400 МГц, MeOD) δ 7,09 (дд, J=8,4, 2,0 Гц, 1H), 6,95 (с, 1H), 6,89 (д, J=8,4 Гц, 1H), 4,51–4,49 (м, 1H), 4,04 (с, 2H), 3,87 (с, 3H), 3,77 (с, 2H), 2,92–2,85 (м, 2H), 2,52–2,21 (м, 4H), 1,5–0,9 (м, 6H), 0,73 (т, J=7,6 Гц, 3H).
Пример 47: 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропан–1–ол (Соединение 47)

Стадия 1: 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропан–1–ол
К раствору 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв., Пример 27 – Стадия 4) в THF (0,16 M) добавляли LAH (3,0 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 2 ч., и затем гасили водой. Водный слой экстрагировали DCM. Объединенные органические слои промывали водой, рассолом, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения в виде бледного твердого вещества.
Стадия 2: 3–(2–амино–6–(бутиламино)–5–(2–метоксибензил)пиримидин–4–ил)пропан–1–ол
Раствор 3–(2–амино–6–хлор–5–(2–метоксибензил)пиримидин–4–ил)пропан–1–ола (1,0 экв.) в EtOH (0,2 M), бутан–1–амина (4,0 экв.) и DIEA (5 экв.) перемешивали при 85°C в течение 3 дней. Реакционный раствор концентрировали, разбавляли 1 н. HCl и экстрагировали DCM. Объединенные органические слои промывали рассолом, сушили над Na2SO4, концентрировали, и очищали колоночной флэш–хроматографией (элюент: DCM/MeOH=100:1~20:1) с получением указанного в заголовке соединения в виде бледного твердого вещества.
ЖХ–МС: [M+H] + = 345,4
1H ЯМР (400 МГц, DMSO) δ 7,80 (ш, 1H), 7,40 (ш, 2H), 7,22 (т, J=8,0 Гц, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,84 (т, J=7,6 Гц, 1H), 6,78 (д, J=8,4 Гц, 1H), 3,85 (с, 3H), 3,72 (с, 2H), 3,38–3,34 (м, 2H), 2,50–2,44 (м, 2H), 1,61–1,54 (м, 2H), 1,50–1,42 (м, 2H), 1,34–1,15 (м, 4H), 0,84(т, J=7,6 Гц, 3H).
Пример 48: (S)–3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–((1–(метилтио)–гептан–3–ил)амино)пиримидин–4–ил)пропановая кислота (Соединение 48)

Стадия 1: 3–(2–амино–5–(5–(цианометил)–2–метоксибензил)–6–(((S)–1–(метилтио)гептан–3–ил)амино)–пиримидин–4–ил)–N–((S)–1–(метилтио)гептан–3–ил)пропанамид
Раствор 3–(2–амино–6–хлор–5–(5–(цианометил)–2–метоксибензил)пиримидин–4–ил)пропановой кислоты (1,0 экв., Пример 40 – Стадия 7) в EtOH (0,4 M) и (S)–1–(метилтио)гептан–3–амине (2,3 экв.) перемешивали при 90°C в течение 3 дней. Растворитель удаляли, остаток очищали с помощью колоночной хроматографии (элюент DCM:MeOH=20:1) с получением указанного в заголовке соединения в виде светло–коричневого масла.
Стадия 2: 3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–(((S)–1–(метилтио)гептан–3–ил)амино)пиримидин–4–ил)–N–((S)–1–(метилтио)гептан–3–ил)пропанамид
Смесь 3–(2–амино–5–(5–(цианометил)–2–метоксибензил)–6–(((S)–1–(метилтио)гептан–3–ил)амино)–пиримидин–4–ил)–N–((S)–1–(метилтио)гептан–3–ил)пропанамида (1,0 экв.) в диоксане (0,04 M), TMSN3 (3,8 экв.) и Bu2SnO (2,5 экв.) нагревали при 90°C в течение 6 ч. в запаянной трубке. Реакционный раствор охлаждали и использовали непосредственно в следующей стадии без какой–либо очистки.
Стадия 3: (S)–3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–4–ил)пропановая кислота
Сырой продукт из предыдущей стадии растворяли в этан–1,2–диоле и добавляли водный КОН (избыток). Реакцию нагревали при 150°C в течение 5 дней. Твердое вещество фильтровали и фильтрат концентрировали и очищали с помощью препаративной ВЭЖХ (подвижная фаза: 0,1% TFA/CH3CN/H2O) с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H] + = 529,3
1H ЯМР (400 МГц, MeOD) δ 7,20 (д, J=7,6 Гц, 1H), 7,01 (д, J=8,8 Гц, 1H), 6,93 (с, 1H), 4,43–4,40 (м, 1H), 4,19(с, 2H), 3,91 (с, 3H), 3,83 (с, 2H), 2,92 (т, J= 6,8 Гц, 2H), 2,56 (т, J= 7,6 Гц, 2H), 2,24 (т, J=7,6 Гц, 2H), 1,93 (с, 3H), 1,78–1,75 (м, 1H), 1,67–1,63 (м, 1H), 1,51–1,49 (м, 1H), 1,41–1,39 (м, 1H), 1,26–1,19 (м, 2H), 1,10–1,04 (м, 2H), 0,80 (т, J=7,6 Гц, 3H).
Пример 49: (S)–3–(5–(5–((2H–тетразол–5–ил)метил)–2–метоксибензил)–2–амино–6–((1–метоксигептан–3–ил)амино)пиримидин–4–ил)пропановая кислота (Соединение 49)
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 48, но с использованием (S)–1–метоксигептан–3–амина вместо (S)–1–(метилтио)гептан–3–амина на Стадии 1.
ЖХ–МС: [M+H] + = 513,3
1H ЯМР (400 МГц, CDCl3) δ 7,21 (д, J=8,4 Гц, 1H), 7,17 (с, 1H), 6,82 (д, J=8,4 Гц, 1H), 6,14 (д, J=8,0 Гц, 1H), 4,26–4,05 (м, 2H), 4,03 (д, J =15,2 Гц, 1H), 3,88 (с, 3H), 3,74–3,61 (м, 2H), 3,33–3,25 (м, 2H), 3,17–3,05 (м, 2H), 3,04 (с, 3H), 2,72–2,68 (м, 1H), 2,53–2,48 (м, 1H), 1,81–1,76 (м, 1H), 1,55–1,50 (м, 1H), 1,41–1,15 (м, 4H), 1,03–0,95 (м, 2H), 0,78 (т, J=7,6 Гц, 3H).
Пример 50: (S)–3–(2–амино–5–(5–(карбоксиметил)–2–метоксибензил)–6–((1–(метилсульфонил)гептан–3–ил)амино)пиримидин–4–ил)пропановая кислота (Соединение 50)

Стадия 1: 3–(2–амино–5–(5–(цианометил)–2–метоксибензил)–6–(((S)–1–(метилтио)гептан–3–ил)амино)–пиримидин–4–ил)–N–((S)–1–(метилтио)гептан–3–ил)пропанамид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 48 – Стадия 1.
Стадия 2: (S)–3–(2–амино–5–(5–(карбоксиметил)–2–метоксибензил)–6–((1–(метилсульфонил)гептан–3–ил)амино)пиримидин–4–ил)пропановая кислота
К раствору KOH в 10% H2O в nBuOH (10 M) добавляли3–(2–амино–5–(5–(цианометил)–2–метоксибензил)–6–(((S)–1–(метилтио)гептан–3–ил)амино)–пиримидин–4–ил)–N–((S)–1–(метилтио)гептан–3–ил)пропанaмид (1,0 экв.). Полученную смесь нагревали в запаянной трубке при 150°C в течение 16 ч. Смесь охлаждали до комнатной температуры и фильтровали. К фильтрату добавляли оксон (5,0 экв.) и перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь концентрировали при пониженном давлении до 1/3 объема. Нерастворимое твердое вещество отфильтровывали и фильтрат очищали с помощью препаративной ВЭЖХ (подвижная фаза: 0,1%HCOOH/MeCN/H2O) и лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества.
ЖХ–МС: [M+H]+ = 537,4
Пример 51: (S)–2–(3–((2–амино–4–метил–6–((1–(метилсульфонил)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)уксусная кислота (Соединение 51)
Стадия 1: (S)–2–(3–((2–амино–4–метил–6–((1–(метилсульфонил)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)ацетонитрил
К раствору (S)–2–(3–((2–амино–4–метил–6–((1–(метилтио)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)ацетонитрила (1 экв., Пример 22 – Стадия 4) в 1:1:1 THF/MeOH/H2O (0,2 M) добавляли оксон (1,2 экв.) порциями при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч., и затем разбавляли DCM. Органический слой промывали водой и рассолом, сушили, и концентрировали с получением светло–желтого твердого вещества, которое использовалось непосредственно в следующей стадии.
Стадия 2: (S)–2–(3–((2–амино–4–метил–6–((1–(метилсульфонил)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)уксусная кислота
К раствору (S)–2–(3–((2–амино–4–метил–6–((1–(метилсульфонил)гептан–3–ил)амино)пиримидин–5–ил)метил)–4–метоксифенил)ацетонитрила (1,0 экв.) в 1:1 MeOH/H2O (0,1 M) добавляли KOH (7,5 экв.). Реакционную смесь перемешивали при 120°C в течение 4 ч. Растворитель удаляли и HCl добавляли для достижения pH 9. Смесь очищали с помощью препаративной ВЭЖХ (0,1%NH3.H2O/CH3CN) с получением указанного в заголовке соединения в виде светло–желтого твердого вещества.
ЖХМС: [M+H]+ = 479,3
1H ЯМР (400 МГц, CD3OD) δ 7,16 (дд, J=8,4, 2,0 Гц, 1H), 6,98 (д, J=2,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 4,30–4,22 (м, 1H), 3,89 (с, 3H), 3,77–3,68 (м, 2H), 3,32 (с, 2H), 2,91–2,70 (м, 5H), 2,32 (с, 3H), 2,08–1,95 (м, 1H), 1,81–1,69 (м, 1H), 1,56–1,05 (м, 6H), 0,83 (т, J=7,2 Гц, 3H).
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Биологический Пример 1: Анализ HEK TLR7
HEK–BlueTM TLR7 клетки были приобретены у Invivogen (Сан–Диего, Калифорния). Следующее описание было взято из информационного листа продукта.
"Клетки HEK–Blue™ hTLR7 предназначены для изучения стимуляции TLR7 человека (hTLR7) путем мониторинга активации NF–kB. Клетки HEK–Blue™ hTLR7 получали путем совместной трансфекции гена hTLR7 и оптимизированного секретируемого репортерного гена эмбриональной щелочной фосфатазы (SEAP) в клетки HEK293. Репортерный ген SEAP находится под контролем минимального промотора IFN–b, слитого с пятью сайтами связывания NF–kB и AP–1. Стимуляция лигандом TLR7 активирует NF–kB и AP–1, которые индуцируют выработку SEAP, которая обнаруживается средой для культивирования клеток HEK–Blue™ Detection".
Типичный протокол анализа включал следующие стадии:
1. Клетки культивировали в соответствии с информацией о продукте.
2. 10 мМ исходного соединения в DMSO сначала разбавляли до 3 мМ, а затем 3–кратно серийно разбавляли, используя DMSO, с получением 10–пт разбавление.
3. 3 мкл разбавленного ДМСО добавляли к 57 мкл детектирующей среды HEK–Blue™ в течение еще 20–кратного разведения.
4. 10 мкл разведенного соединения в среде для анализа добавляли в 40 мкл клеточной культуры (в среде детекции HEK–Blue ™) в 384–луночный планшет. Конечная концентрация клеток=8000 клеток на лунку.
5. Планшеты инкубировали при 37°С в 5% СО2 в течение 16 ч. SEAP определяли с использованием спектрофотометра при 620–655 нм.
Таблица активности HEK–TLR7 ниже дает результаты.
(S–энантиомер)
(R–энантиомер)
EC50 категория:
A = ˂100 нM
B=100–1000 нM
C=1000–10000 нM
Как обсуждалось в данном документе, R1A и R1B в формуле (1) могут образовывать хиральный центр. Соединения 20 и 20C отличаются стереоцентром углерода несущего R1A и R1B.
Как показано выше, (S)–энантиомер (например, Соединение 20) более чем в 10 раз более эффективен, чем (R)–энантиомер (например, Соединение 20C) в анализе HEK–TLR7. Другие примеры могут следовать той же тенденции.
Биологический Пример 2:Фармакокинетические эксперименты
Мышам BALB/с перорально вводили 10 мг/кг соединения, приготовленного в 0,5% карбоксиметилцеллюлозы+0,5% суспензии Tween–80. У мышей ретро–орбитально отбирали кровь в разные моменты времени после введения дозы. Из крови получали сыворотку путем центрифугирования с последующим осаждением белка, элюированием в градиенте обращенной фазы и обнаружением MRM с помощью масс–спектрометрии ESI+ для определения концентрации соединения. Животных умерщвляли после окончательного забора крови (5 часов), печень удаляли и быстро замораживали в жидком азоте для тканевых PK–анализов. Концентрация в печени:сыворотке (П:С) через 5 часов после введения дозы является одним из параметров для выявления соединений с потенциальным накоплением в печени.
Приведенная ниже таблица концентрации печень:сыворотка мыши (П:С) дает результаты.
П:С категория:
A = >100 представляет высокое обогащение печени
B=10–100 представляет умеренное обогащение печени
C = <10 представляет низкое обогащение печени
ЭКВИВАЛЕНТЫ
Хотя настоящее изобретение было описано в связи с конкретными вариантами реализации, изложенными выше, многие альтернативы, модификации и другие их варианты будут очевидны для специалистов в данной области техники. Предполагается, что все такие альтернативы, модификации и варианты находятся в пределах сути и объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| АНТИГИПЕРТЕНЗИВНЫЕ СОЕДИНЕНИЯ ДВОЙНОГО ДЕЙСТВИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2476427C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛО-ГЕТЕРОАРИЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2748833C2 |
| МОДУЛЯТОРЫ АНДРОГЕНОВОГО РЕЦЕПТОРА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2797622C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2605557C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| ПРОТИВОГЕЛЬМИНТНЫЕ ДЕПСИПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2707298C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2016 |
|
RU2720678C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2808165C2 |
| СТИЛЬБЕНОВОЕ ПРОИЗВОДНОЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2017 |
|
RU2776328C2 |
Настоящее изобретение относится к соединению, имеющему структуру Формулы (1), или к его фармацевтически приемлемой соли, обладающим иммуномодулирующими свойствами, которые действуют через TLR7 и которые полезны при лечении вирусных инфекций и раковых заболеваний.
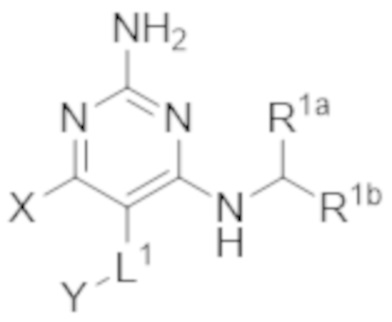 (1),
(1),
где R1a выбирают из группы, состоящей из H, C1–C4 алкила и A, где алкил необязательно замещен –COOH, –SO2CH3, –SCH3, –OCH3,  или A; R1b представляет собой C2–C5 алкил; X представляет собой C1–C4 алкил, где алкил необязательно замещен A или –OH; L1 выбирают из группы, состоящей из связи, –CH2–, -О- и –CH2CH2–; Y выбирают из группы, состоящей из C1–C3 алкила и С6 арила, где С6 арил необязательно замещен 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси; A выбирают из группы, состоящей из
или A; R1b представляет собой C2–C5 алкил; X представляет собой C1–C4 алкил, где алкил необязательно замещен A или –OH; L1 выбирают из группы, состоящей из связи, –CH2–, -О- и –CH2CH2–; Y выбирают из группы, состоящей из C1–C3 алкила и С6 арила, где С6 арил необязательно замещен 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила, и C1–C3 алкокси; A выбирают из группы, состоящей из  ,
,  ,
,  ,
,  ,
,  и
и  ; L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–,
; L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–,  ,
,  и
и 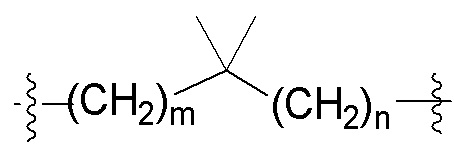 ; m представляет собой целое число от нуля до четырех; n представляет собой целое число от одного до четырех; где соединение замещено по меньшей мере одним А. При этом соблюдаются следующие условия: когда X представляет собой –CH3, L1 представляет собой –CH2–, Y представляет собой С6 арил, замещенный A, и L2 представляет собой –CH2–, тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3; и, когда X представляет собой –CH3, L1 представляет собой –CH2–, Y представляет собой С6 арил, замещенный A, L2 представляет собой –CH2– и A представляет собой , тогда A и L1 не находятся в пара–положении по отношению друг к другу. 6 н. и 49 з.п. ф-лы, 1 табл., 53 пр.
; m представляет собой целое число от нуля до четырех; n представляет собой целое число от одного до четырех; где соединение замещено по меньшей мере одним А. При этом соблюдаются следующие условия: когда X представляет собой –CH3, L1 представляет собой –CH2–, Y представляет собой С6 арил, замещенный A, и L2 представляет собой –CH2–, тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3; и, когда X представляет собой –CH3, L1 представляет собой –CH2–, Y представляет собой С6 арил, замещенный A, L2 представляет собой –CH2– и A представляет собой , тогда A и L1 не находятся в пара–положении по отношению друг к другу. 6 н. и 49 з.п. ф-лы, 1 табл., 53 пр.
1. Соединение, имеющее структуру Формулы (1), или его фармацевтически приемлемая соль,
 (1),
(1),
где R1a выбирают из группы, состоящей из H, C1–C4 алкила и A,
где алкил необязательно замещен –COOH, –SO2CH3, –SCH3, –OCH3,  или A;
или A;
R1b представляет собой C2–C5 алкил;
X представляет собой C1–C4 алкил, где алкил необязательно замещен A или –OH;
L1 выбирают из группы, состоящей из связи, –CH2–, -О- и –CH2CH2–;
Y выбирают из группы, состоящей из C1–C3 алкила и С6 арила, где С6 арил необязательно замещен 1–5 заместителями, которые независимо выбирают из A, C1–C3 алкила и C1–C3 алкокси;
A выбирают из группы, состоящей из 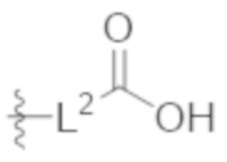 ,
, 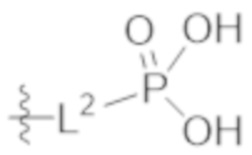 ,
,  ,
, 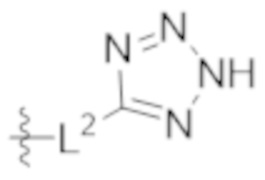 ,
, 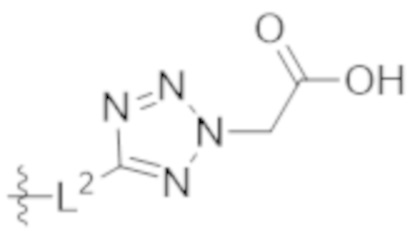 и
и  ;
;
L2 выбирают из группы, состоящей из связи, –(CH2)n–, –C(O)NH(CH2)n–,  ,
,  и
и  ;
;
m представляет собой целое число от нуля до четырех;
n представляет собой целое число от одного до четырех;
где соединение замещено по меньшей мере одним А; и,
когда X представляет собой –CH3, L1 представляет собой –CH2–, Y представляет собой С6 арил, замещенный A, и L2 представляет собой –CH2–, тогда A не представляет собой –L2–COOH, кроме случаев, когда R1a содержит –COOH или –SO2CH3; и,
когда X представляет собой –CH3, L1 представляет собой –CH2–, Y представляет собой С6 арил, замещенный A, L2 представляет собой –CH2– и A представляет собой , тогда A и L1 не находятся в пара–положении по отношению друг к другу.
2. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что R1b представляет собой –(CH2)2CH3.
3. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что R1b представляет собой –(CH2)3CH3.
4. Соединение по любому из пп. 1–3, или его фармацевтически приемлемая соль, отличающееся тем, что R1a представляет собой C1–C4 алкил, необязательно замещенный –OCH3, –SCH3 или –SO2CH3.
5. Соединение по любому из пп. 1–4, или его фармацевтически приемлемая соль, отличающееся тем, что  представляет собой
представляет собой 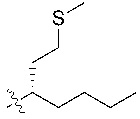 ,
,  ,
, 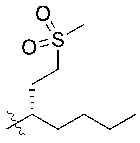 или
или 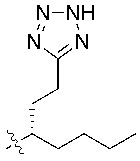 .
.
6. Соединение по любому из пп. 1–4, или его фармацевтически приемлемая соль, отличающееся тем, что R1a представляет собой C1–C4 алкил, необязательно замещенный –COOH.
7. Соединение по любому из пп. 1–6, или его фармацевтически приемлемая соль, отличающееся тем, что представляет собой  .
.
8. Соединение по любому из пп. 1–3, или его фармацевтически приемлемая соль, отличающееся тем, что R1a представляет собой H.
9. Соединение по любому из пп. 1–8, или его фармацевтически приемлемая соль, отличающееся тем, что X представляет собой C1–C4 алкил, где алкил замещен А.
10. Соединение по любому из пп. 1–9, или его фармацевтически приемлемая соль, отличающееся тем, что X представляет собой C1–C4 алкил, где алкил замещен , где L2 представляет собой связь.
11. Соединение по любому из пп. 1–8, или его фармацевтически приемлемая соль, отличающееся тем, что X представляет собой CH3.
12. Соединение по любому из пп. 1–11, или его фармацевтически приемлемая соль, отличающееся тем, что L1 представляет собой –CH2–, –CH2CH2– или –O–.
13. Соединение по любому из пп. 1–12, или его фармацевтически приемлемая соль, отличающееся тем, что L1 представляет собой –CH2–.
14. Соединение по любому из пп. 1–13, или его фармацевтически приемлемая соль, отличающееся тем, что Y представляет собой C1–C3 алкил или арил.
15. Соединение по любому из пп. 1–14, или его фармацевтически приемлемая соль, отличающееся тем, что Y представляет собой арил, где арил замещен C1–C3 алкокси.
16. Соединение по любому из пп. 1–14, или его фармацевтически приемлемая соль, отличающееся тем, что Y представляет собой арил, где арил замещен А.
17. Соединение по любому из пп. 1–16 или его фармацевтически приемлемая соль, отличающееся тем, что A представляет собой .
18. Соединение по любому из пп. 1–16, или его фармацевтически приемлемая соль, отличающееся тем, что A представляет собой .
19. Соединение по любому из пп. 1–16, или его фармацевтически приемлемая соль, отличающееся тем, что A представляет собой .
20. Соединение по любому из пп. 1–16, или его фармацевтически приемлемая соль, отличающееся тем, что A представляет собой .
21. Соединение по любому из пп. 1–16, или его фармацевтически приемлемая соль, отличающееся тем, что A представляет собой .
22. Соединение по любому из пп. 1–16, или его фармацевтически приемлемая соль, отличающееся тем, что A представляет собой .
23. Соединение по любому из пп. 1–22, или его фармацевтически приемлемая соль, отличающееся тем, что L2 представляет собой –(CH2)n–.
24. Соединение по любому из пп. 1–23, или его фармацевтически приемлемая соль, отличающееся тем, что n представляет собой один или два.
25. Соединение по любому из пп. 1–22, или его фармацевтически приемлемая соль, отличающееся тем, что L2 представляет собой  .
.
26. Соединение по любому из пп. 1–22, или его фармацевтически приемлемая соль, отличающееся тем, что L2 представляет собой .
27. Соединение по любому из пп. 1–22, или его фармацевтически приемлемая соль, отличающееся тем, что L2 представляет собой –C(O)NH(CH2)n–.
28. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1a)
 ,
,
где X представляет собой CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O– и –CH2CH2–;
A выбирают из группы, состоящей из , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–,  , и –C(O)NH(CH2)n–; и
, и –C(O)NH(CH2)n–; и
R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси.
29. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1a)
,
где X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из 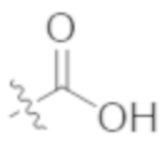 ,
, 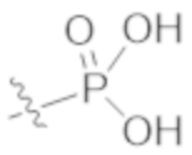 ,
, 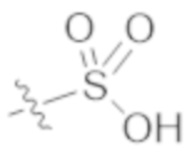 ,
,  ,
,  или
или  ;
;
L1 выбирают из группы, состоящей из связи, –CH2–, –O– и –CH2CH2–;
A выбирают из группы, состоящей из , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , и –C(O)NH(CH2)n–; и
R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси.
30. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1b)
 ,
,
где X представляет собой CH3;
L1 выбирают из группы, состоящей из связи, –CH2–, –O– и –CH2CH2–;
A выбирают из группы, состоящей из , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , и –C(O)NH(CH2)n–; и
R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси.
31. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1b)
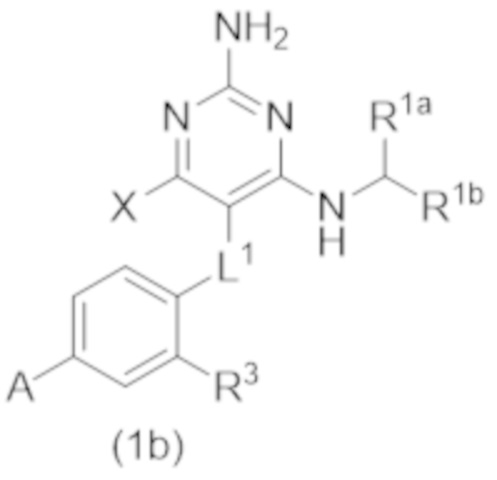 ,
,
где X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O– и –CH2CH2–;
A выбирают из группы, состоящей из , , , , и ;
L2 выбирают из группы, состоящей из связи, –CH2–, –CH2CH2–, , и –C(O)NH(CH2)n–; и
R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси.
32. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1с)
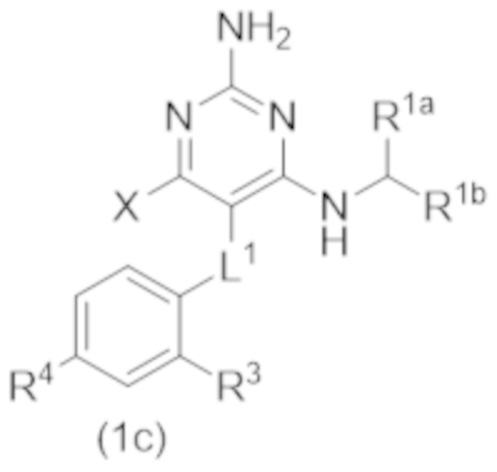 ,
,
где X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , и;
L1 выбирают из группы, состоящей из связи, –CH2– и –CH2CH2–;
R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси; и
R4 представляет собой H или C1–C3 алкокси.
33. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1d)
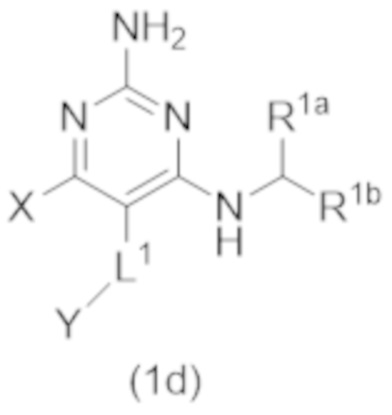 ,
,
X представляет собой –CH2–A1a, –CH2CH2–A1a, –CH2CH2CH2–A1a или –CH2C(CH3)2–A1a;
A1a выбирают из группы, состоящей из , , , , и ;
L1 выбирают из группы, состоящей из связи, –CH2–, –O– и –CH2CH2; и
Y представляет собой C1–C3 алкил.
34. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1e)
 ,
,
где R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси.
35. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1f)
 .
.
36. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1g)
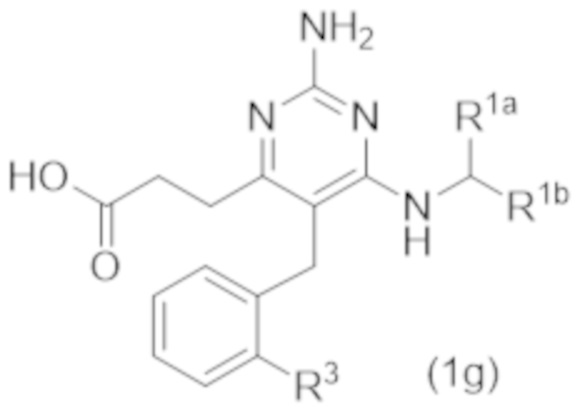 ,
,
где R3 представляет собой H, C1–C3 алкил или C1–C3 алкокси.
37. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1h)
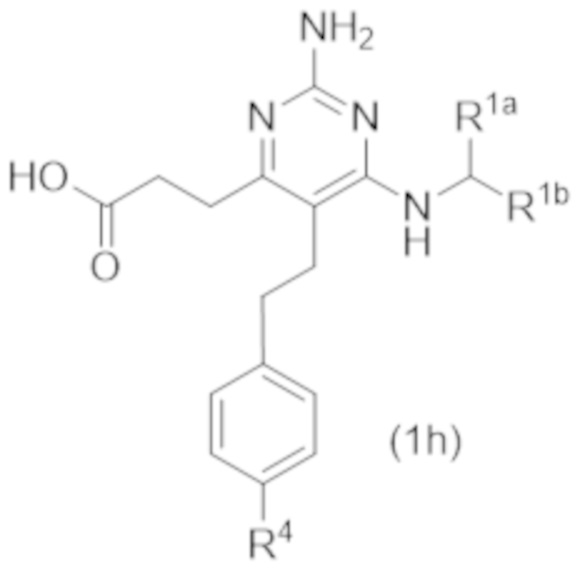 ,
,
где R4 представляет собой H или C1–C3 алкокси.
38. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1i)
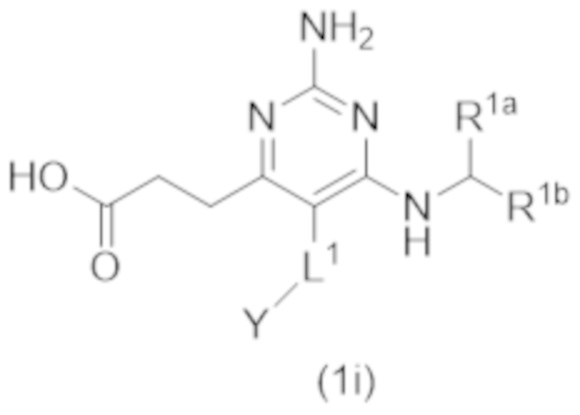 ,
,
где L1 выбирают из группы, состоящей из связи, –CH2–, –O– и –CH2CH2–; и
Y представляет собой H или C1–C3 алкил.
39. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1j)
 (1j).
(1j).
40. Соединение по п. 1, или его фармацевтически приемлемая соль, отличающееся тем, что соединение Формулы (1) представляет собой соединение Формулы (1k)
 (1k).
(1k).
41. Соединение по п. 1 или его фармацевтически приемлемая соль, выбранные из группы, состоящей из:




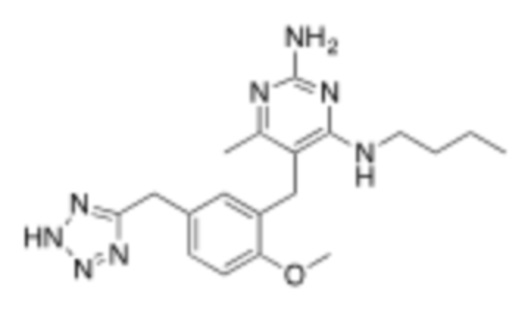

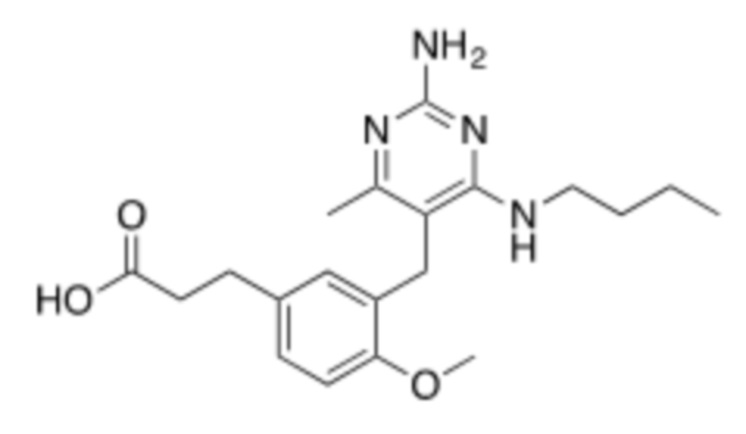
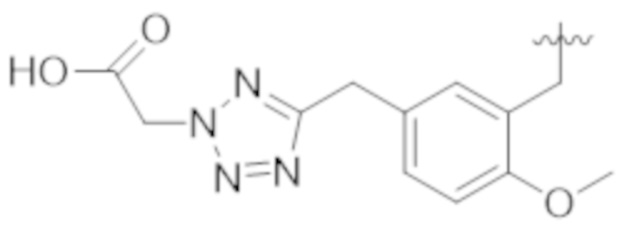

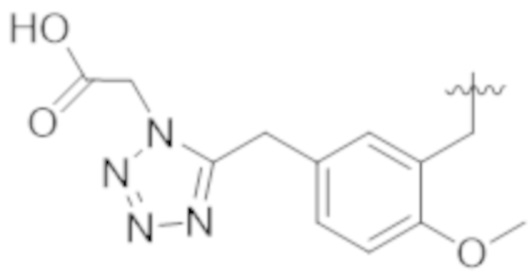
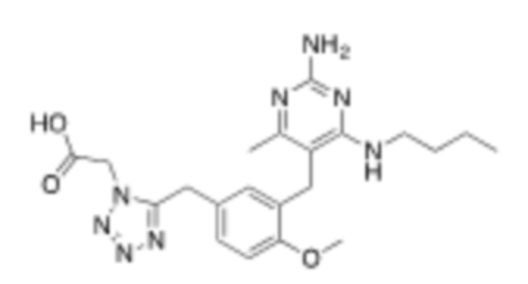


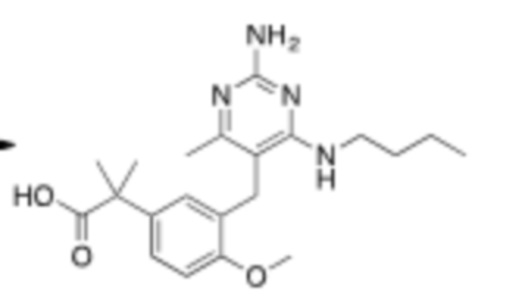

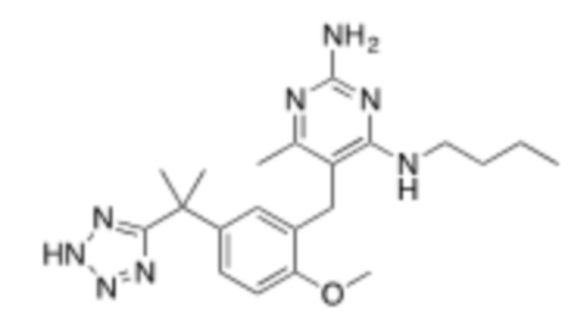
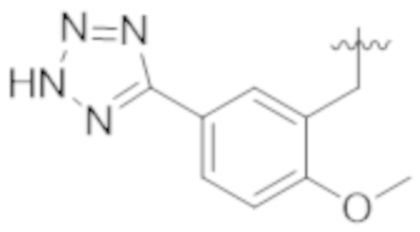
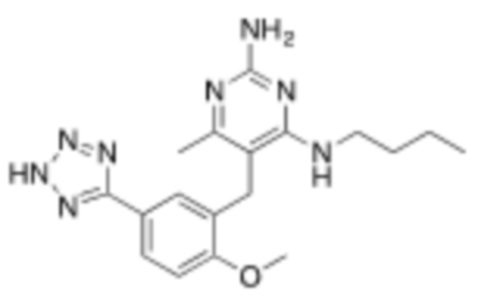


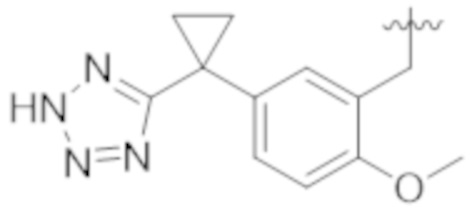

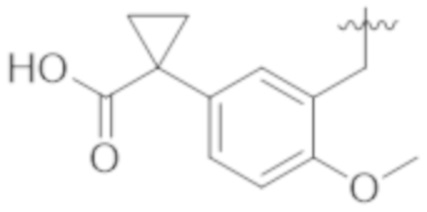
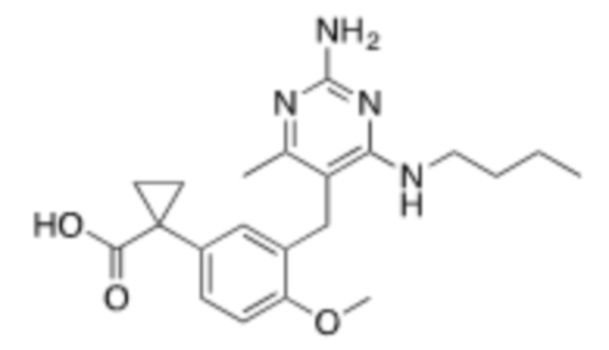

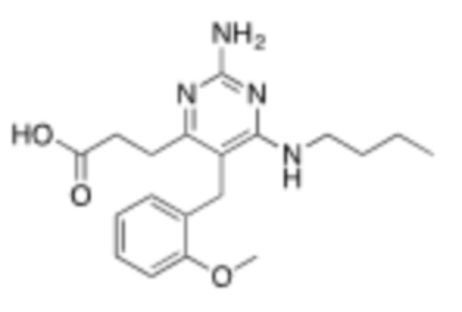

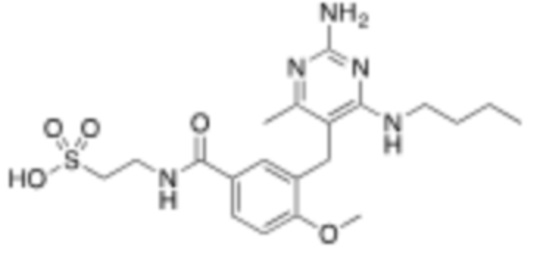
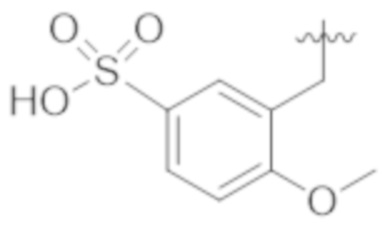
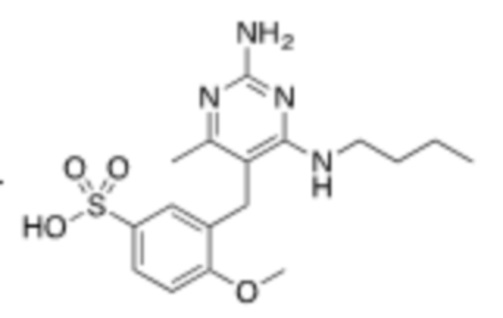
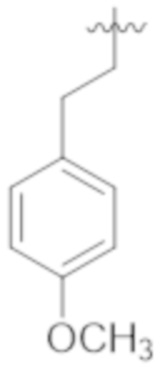

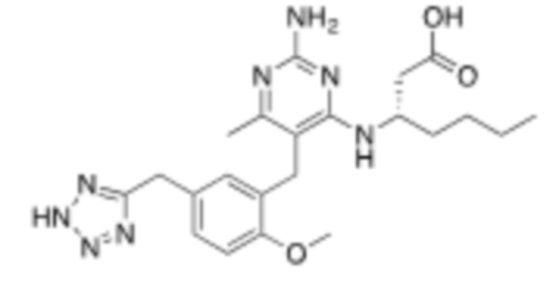

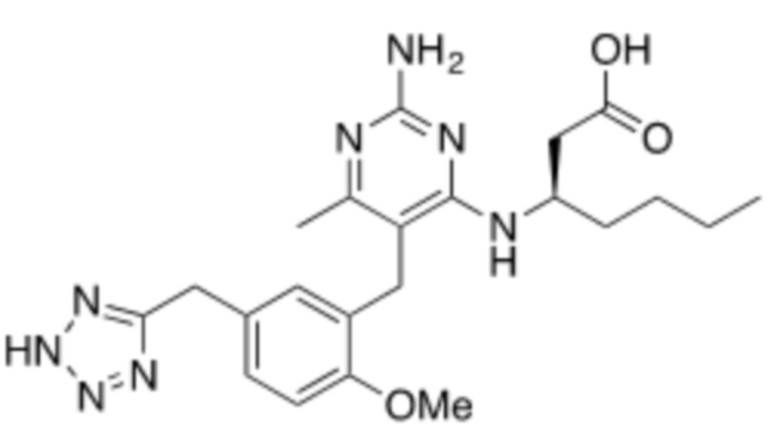





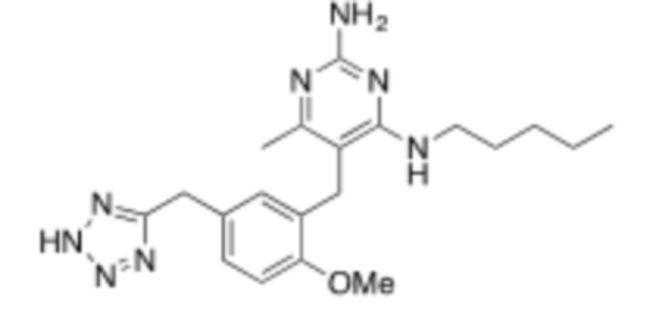
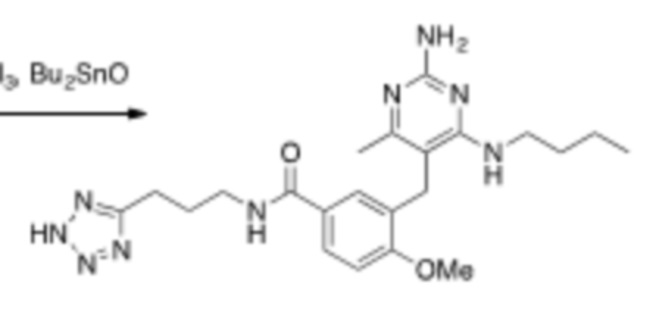
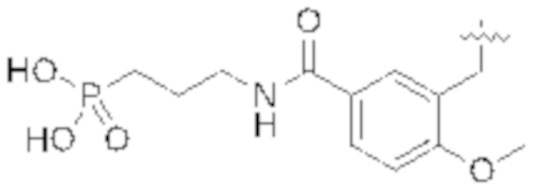
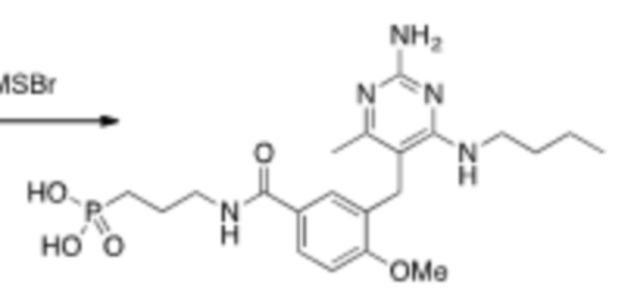


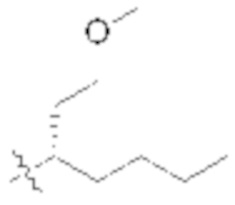
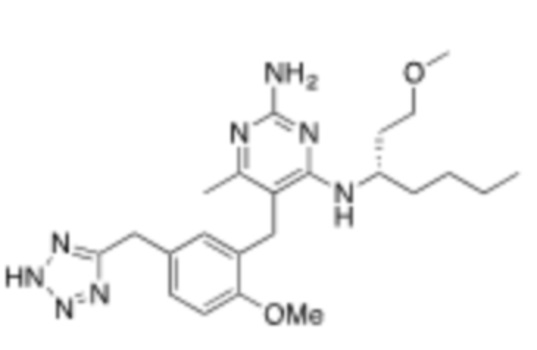
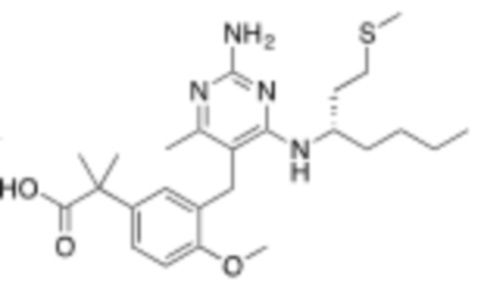




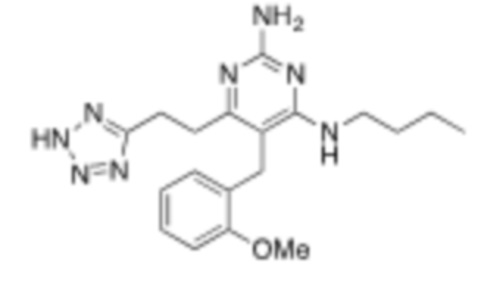


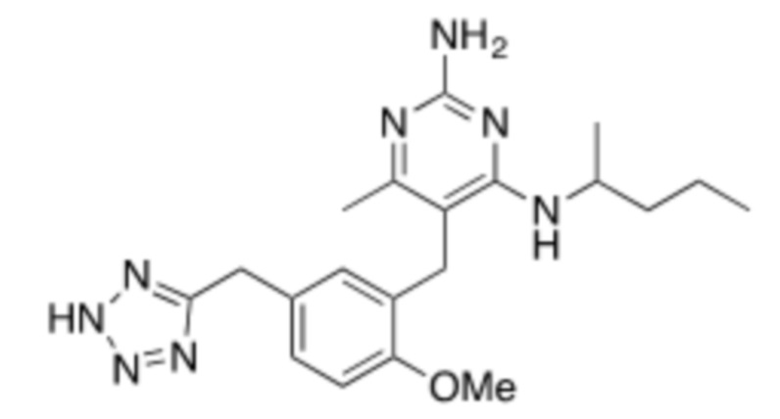
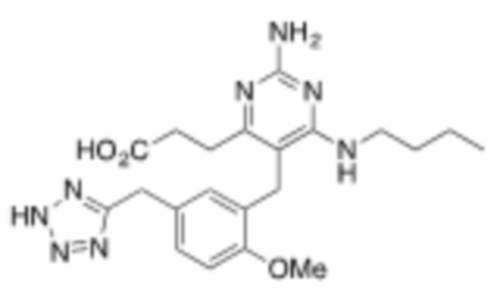



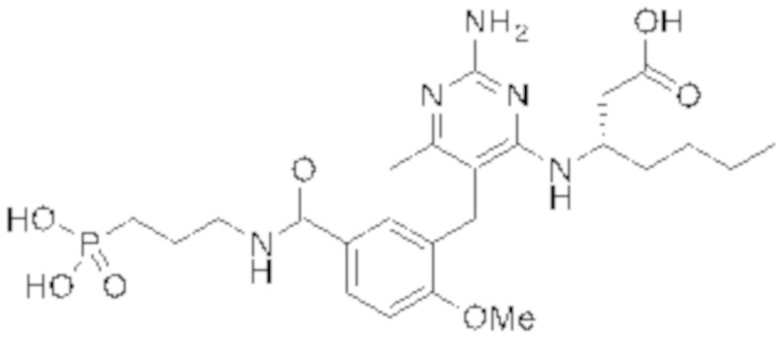

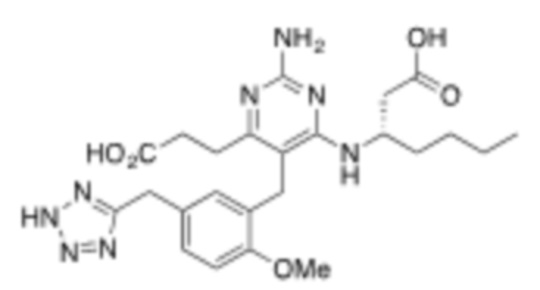


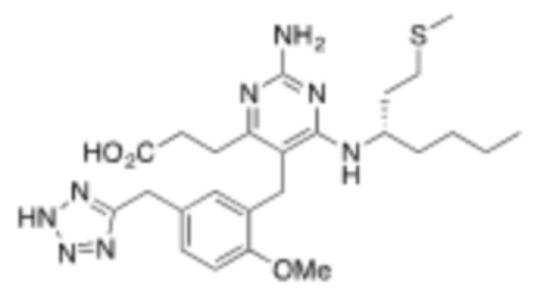
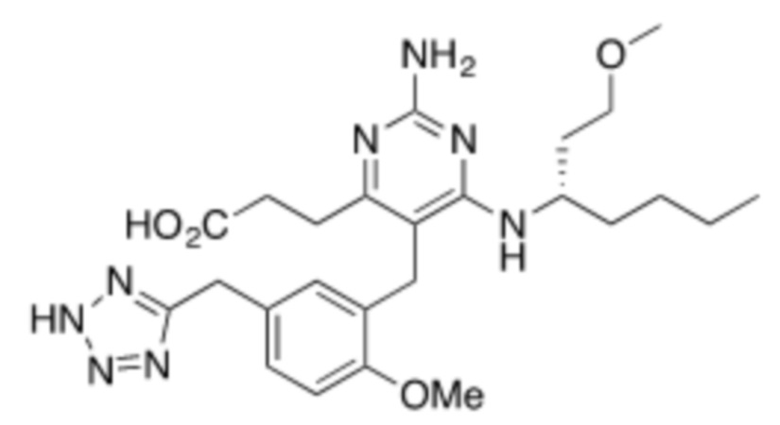


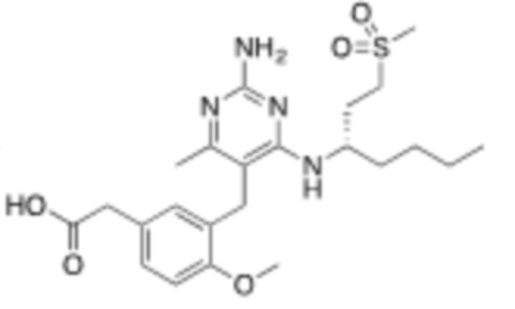
42. Фармацевтическая композиция для модуляции Toll-подобного рецептора TLR7, содержащая эффективное количество соединения по любому из пп. 1–41 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
43. Способ лечения состояния, связанного с модуляцией TLR7, у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения по любому из пп. 1–41 или его фармацевтически приемлемой соли.
44. Способ по п. 43, отличающийся тем, что состояние представляет собой вирусную инфекцию или рак.
45. Способ по п. 43 или 44, отличающийся тем, что введение представляет собой пероральную, внутривенную, подкожную, внутримышечную, внутриопухолевую, внутрикожную, интраназальную, ингаляционную, внутрипузырную, местную, подъязычную, буккальную, интраректальную, интратекальную, внутричерепную или другие формы локальной доставки.
46. Соединение по любому из пп. 1–41, или его фармацевтически приемлемая соль, для модуляции Toll-подобного рецептора TLR7.
47. Соединение по любому из пп. 1–41, или его фармацевтически приемлемая соль, для применения при лечении состояния, связанного с модуляцией TLR7.
48. Соединение по п. 47, отличающееся тем, что состояние представляет собой вирусную инфекцию или рак.
49. Применение соединения по любому из пп. 1–41 или его фармацевтически приемлемой соли при изготовлении лекарственного средства для лечения состояния, связанного с модуляцией TLR7.
50. Применение по п. 49, отличающееся тем, что состояние представляет собой вирусную инфекцию или рак.
51. Фармацевтическая композиция по п. 42, дополнительно содержащая, по меньшей мере, один или несколько дополнительных терапевтических агентов.
52. Фармацевтическая композиция по п. 51, отличающаяся тем, что, по меньшей мере, один или несколько дополнительных терапевтических агентов представляют собой антивирусный нуклеозид.
53. Фармацевтическая композиция по п. 51, отличающаяся тем, что, по меньшей мере, один или несколько дополнительных терапевтических агентов представляют собой антитело к PD–1 или антитело к PD–L1.
54. Способ лечения HBV у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения по любому из пп. 1–41 или его фармацевтически приемлемой соли в комбинации с противовирусным нуклеозидом.
55. Способ лечения рака у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения по любому из пп. 1–41 или его фармацевтически приемлемой соли в комбинации с антителом к PD–1 или антителом к PD–L1.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ АГОНИСТА TLR И КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2011 |
|
RU2603467C2 |

