[Область техники]
Настоящее изобретение относится к металлолигандному комплексу, каталитической композиции для полимеризации на основе этилена, включающей в себя указанный комплекс, и способу получения полимера на основе этилена с использованием указанного комплекса.
[Уровень техники]
Обычно при получении сополимера этилена и α-олефина или сополимера этилена и олефин-диена, используют так называемый катализатор Циглера-Натта, включающий в себя в качестве основного каталитического компонента соединение титана или ванадия и в качестве компонента-сокатализатора алкильное соединение алюминия.
Патенты США №№ 3594330 и 3676415 раскрывают улучшенный катализатор Циглера-Натта. Каталитическая система Циглера-Натта обладает высокой активностью в отношении полимеризации этилена, но из-за гетерогенного каталитического активного центра имеет место такой недостаток, как широкое молекулярно-массовое распределение обычно получаемого полимера и, в частности, неоднородное распределение состава сополимера этилена и α-олефина.
В дальнейшем были проведены различные исследования металлоценовой каталитической системы, состоящей из металлоценового соединения переходного металла 4 группы периодической таблицы, такого как цирконий и гафний, и метилалюмоксана в качестве сокатализатора, которая представляет собой гомогенный катализатор с каталитическим активным центром одного типа и может давать полиэтилен, имеющий узкое молекулярно-массовое распределение и однородное распределение состава по сравнению с обычным катализатором Циглера-Натта.
Например, в европейских патентных публикациях №№ 320762 и 372632 или выложенных японских патентных публикациях №№ (Sho) 63-092621, (Hei) 02-84405 или (Hei) 03-2347 раскрыто, что металлоценовое соединение активируется сокатализатором метилалюмоксаном в Cp2TiCl2, Cp2ZrCl2, Cp2ZrMeCl, Cp2ZrMe2, этилен(IndH4)2ZrCl2 и т.п. с полимеризацией этилена с высокой активностью, что приводит к получению полиэтилена с молекулярно-массовым распределением (Mw/Mn) в диапазоне 1,5-2,0.
Однако с помощью этой каталитической системы трудно получить высокомолекулярный полимер.
То есть, известно, что, когда эту каталитическую систему применяют к способу полимеризации в растворе, проводимому при высокой температуре, активность полимеризации быстро снижается, а преобладающей является реакция β-дегидрирования, что делает каталитическую систему неподходящей для получения высокомолекулярного полимера.
Между тем, были проведены различные исследования для улучшения растворимости катализатора в алифатическом углеводороде, так чтобы каталитическая эффективность в коммерческом процессе была максимизирована, а объем транспортировки катализатора был уменьшен, что значительно увеличивает затраты на хранение и транспортировку в таком процессе. Кроме того, поскольку концентрация катализатора ограничена, еще большей проблемой становится порча катализатора из-за естественного присутствия примесей в растворителе.
Соответственно, в настоящее время необходимы катализатор и предшественник катализатора, обладающие улучшенными характеристиками, потребность в которых сохраняется в химической промышленности.
[Документ предшествующего уровня техники]
[Патентный документ]
(Патентный документ 1) Патент США № 3594330
(Патентный документ 2) Патент США № 3676415
(Патентный документ 3) Европейская патентная публикация № 320762.
(Патентный документ 4) Европейская патентная публикация № 372632.
(Патентный документ 5) Японская выложенная патентная публикация № (Sho) 63-092621
(Патентный документ 6) Японская выложенная патентная публикация № (Hei) 02-84405
(Патентный документ 7) Японская выложенная патентная публикация № (Hei) 03-2347
[Раскрытие изобретения]
[Техническая проблема]
Целью настоящего изобретения является разработка металлолигандного комплекса с определенным заместителем и каталитической композиции для полимеризации на основе этилена, включающей в себя указанный комплекс.
Другой целью настоящего изобретения является разработка способа получения полимера на основе этилена с использованием каталитической композиции для полимеризации на основе этилена.
[Техническое решение]
В одном общем аспекте металлолигандный комплекс со значительно улучшенной растворимостью в органическом растворителе, в частности алифатическом углеводороде, представлен следующей химической формулой 1:
[Химическая формула 1]
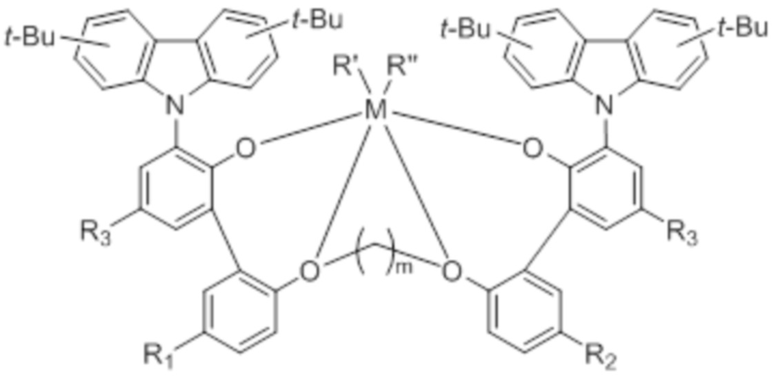
где
M представляет собой переходный металл 4 группы периодической таблицы;
R’ и R” независимо друг от друга представляют собой (C1-C20)алкил;
R1 и R2 независимо друг от друга представляют собой галоген, (C1-C20)алкил, (C1-C20)алкокси или галоген(C1-C20)алкил;
R3 представляет собой (C8-C20)алкил с прямой цепью; и
m представляет собой целое число от 2 до 4.
Предпочтительно, в химической формуле 1 в соответствии с типичным вариантом осуществления настоящего изобретения R3 может представлять собой (C8-C20)алкил с прямой цепью.
Более предпочтительно, в химической формуле 1 в соответствии с типичным вариантом осуществления настоящего изобретения R3 может представлять собой (C8-C20)алкил с прямой цепью, M может представлять собой титан, цирконий или гафний, R’ и R” могут независимо друг от друга представлять собой (C1-C5)алкил, R1 и R2 могут представлять собой идентичные друг другу галоген, (C1-C8)алкил или (C1-C8)алкокси, и m может представлять собой целое число 3.
Предпочтительно, химическая формула 1 в соответствии с типичным вариантом осуществления настоящего изобретения может быть представлена следующей химической формулой 2:
[Химическая формула 2]
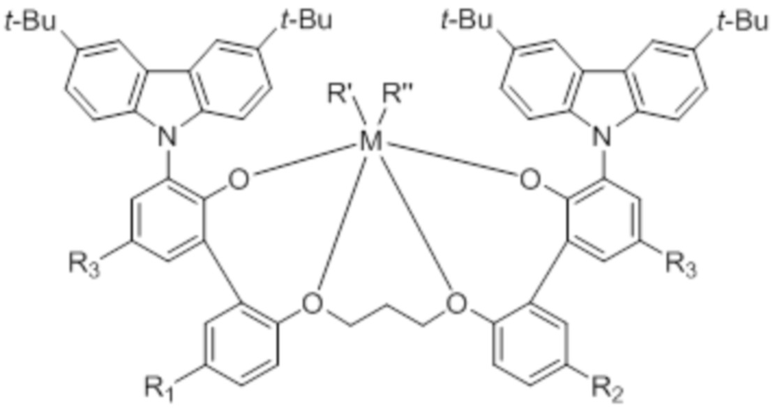
где
M представляет собой титан, цирконий или гафний;
R’ и R” независимо друг от друга представляют собой (C1-C5)алкил;
R1 и R2 независимо друг от друга представляют собой галоген, (C1-C8)алкил или (C1-C8)алкокси; и
R3 представляет собой (C8-C12)алкил с прямой цепью.
В другом общем аспекте каталитическая композиция для полимеризации на основе этилена включает в себя металлолигандный комплекс настоящего изобретения и сокатализатор.
Предпочтительно, сокатализатор в соответствии с типичным вариантом осуществления настоящего изобретения может представлять собой сокатализатор из соединения алюминия, сокатализатор из соединения бора или их смесь, и его можно использовать в количестве от 0,5 до 10000 моль на 1 моль металлолигандного комплекса.
В еще одном общем аспекте способ получения полимера на основе этилена включает в себя: полимеризацию этилена или этилена и α-олефина в присутствии каталитической композиции для полимеризации на основе этилена с получением полимера на основе этилена.
Предпочтительно, полимеризация в соответствии с типичным вариантом осуществления настоящего изобретения может проводиться при 170-250°C.
[Полезные эффекты]
Металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения обладает значительно улучшенной растворимостью в растворителе благодаря введению определенной функциональной группы с контролируемым число атомов углерода и контролируемую форму, что более эффективно улучшает способ полимеризации.
Кроме того, металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения обладает превосходной активностью катализатора благодаря введению определенной функциональной группы в определенное положение и, кроме того, обеспечивает полимеризацию даже при высокой температуре полимеризации и не снижает активность катализатора.
Кроме того, металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения обладает хорошей реакционной способностью в отношении олефинов для легкой полимеризации олефинов и может давать полимер на основе этилена с высокой молекулярной массой при высокой температуре полимеризации.
Соответственно, металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения и каталитическая композиция, включающая в себя указанный комплекс, могут быть очень полезными для получения полимера на основе этилена, имеющего превосходные физические свойства.
[Наилучший способ осуществления изобретения]
Далее описаны металлолигандный комплекс настоящего изобретения, каталитическая композиция для полимеризации на основе этилена, включающая в себя указанный комплекс, и способ получения полимера на основе этилена с использованием указанного комплекса, при этом технические термины и научные термины, используемые в настоящем документе, имеют обычное значение, понятное специалистам в области техники, к которой относится настоящее изобретение, если не определено иное, а описание известных функций и конфигураций, которое согло бы затруднять понимание настоящего изобретения, в нижеследующем описании опущено.
"Алкил", "алкокси" и другие заместители, содержащие "алкильный" фрагмент, описанные в настоящем документе, включают формы как с прямой, так и с разветвленной цепью, и, если не указано иное, содержат от 1 до 20 атомов углерода, предпочтительно от 1 до 15, более предпочтительно от 1 до 10 атомов углерода.
Кроме того, "(C8-C20)алкил, содержащий один или несколько атомов азота, атомов кислорода, атомов серы и атомов фосфора" представляет собой заместитель, содержащий один или несколько атомов азота, атомов кислорода, атомов серы и атомов фосфора в алкильной группе, содержащей от 8 до 20 атомов углерода, причем атом азота, атом кислорода, атом серы и атом фосфора могут присутствовать между атомами углерода алкильной группы или в качестве заместителя алкильной группы. В качестве примера можно упомянуть, но без ограничения ими, алкокси, аминоалкил, вторичные амины, эфирные соединения и т.п.
Галогеналкил, описанный в настоящем документе представляет собой алкильную группу, в которой один или несколько атомов водорода замещены галогеном, и может содержать один или несколько атомов галогена в алкильной группе.
Настоящее изобретение предлагает металлолигандный комплекс, представленный следующей химической формулой 1, который обладает улучшенной растворимостью, превосходной термической стабильностью и высокой каталитической активностью при высокой температуре полимеризации, благодаря введению заместителя, имеющего контролируемую форму и контролируемое число атомов углерода, в определенное положение, и, таким образом, может быть очень полезен для полимеризации на основе этилена с высокой молекулярной массой.
[Химическая формула 1]
где
M представляет собой переходный металл 4 группы периодической таблицы;
R’ и R” независимо друг от друга представляют собой (C1-C20)алкил;
R1 и R2 независимо друг от друга представляют собой галоген, (C1-C20)алкил, (C1-C20)алкокси или галоген(C1-C20)алкил;
R3 представляет собой (C8-C20)алкил с прямой цепью; и
m представляет собой целое число от 2 до 4.
Металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения представлен химической формулой 1 выше, и благодаря введению (C8-C20)алкила с прямой цепью, который задуман как контролируемый заместитель, в R3 в химической формуле 1, растворимость в органическом растворителе, в частности алифатическом углеводороде, значительно улучшена, а каталитическая активность очень высока даже при высокой температуре полимеризации.
Соответственно, металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения обладает превосходной термической стабильностью для поддержания высокой каталитической активности даже при высокой температуре, а также обладает хорошей реакционной способностью к полимеризации в отношении других олефинов и может давать высокомолекулярный полимер с высоким выходом, и, следовательно, имеет высокую коммерческую практичность по сравнению с уже известными металлоценовыми и неметаллоценовыми катализаторами, имеющими один активный центр.
Предпочтительно, в химической формуле 1 в соответствии с типичным вариантом осуществления настоящего изобретения R3 может представлять собой (C8-C20)алкил с прямой цепью, более предпочтительно (C8-C12)алкил с прямой цепью.
Предпочтительно, в химической формуле 1 в соответствии с типичным вариантом осуществления настоящего изобретения R3 может представлять собой (C8-C20)алкил с прямой цепью, M может представлять собой титан, цирконий или гафний, R’ и R” могут независимо друг от друга представлять собой (C1-C5)алкил, R1 и R2 могут представлять собой идентичные друг другу галоген, (C1-C8)алкил или (C1-C8)алкокси, и m может представлять собой целое число 3.
С точки зрения улучшения растворимости, каталитической активности и реакционной способности в отношении олефинов химическая формула 1 в соответствии с типичным вариантом осуществления настоящего изобретения может быть, предпочтительно, представлена следующей химической формулой 2:
[Химическая формула 2]
где
M представляет собой титан, цирконий или гафний;
R’ и R” независимо друг от друга представляют собой (C1-C5)алкил;
R1 и R2 независимо друг от друга представляют собой галоген, (C1-C8)алкил или (C1-C8)алкокси; и
R3 представляет собой (C8-C12)алкил с прямой цепью.
С точки зрения более высокой растворимости, каталитической активности и реакционной способности в отношении олефинов R3 в химической формуле 2 в соответствии с типичным вариантом осуществления настоящего изобретения может, предпочтительно, представлять собой (C8-C12)алкил с прямой цепью и, более предпочтительно, н-октил, н-нонил, н-децил, н-ундецил или н-додецил.
В частности, металлолигандный комплекс в соответствии с типичным вариантом осуществления настоящего изобретения может представлять собой соединения, выбранные из группы, состоящей из следующих структур, но без ограничения:
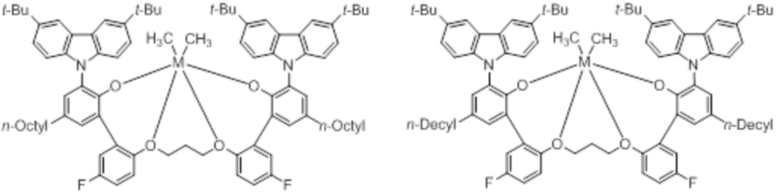
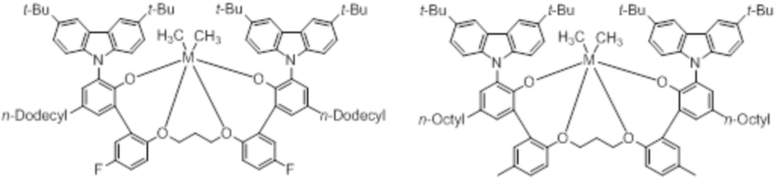
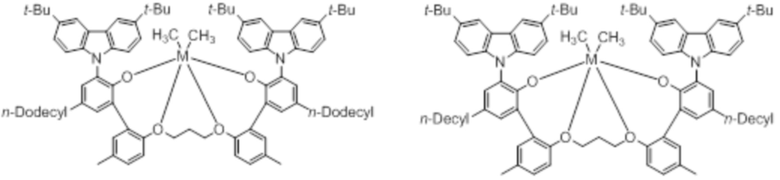
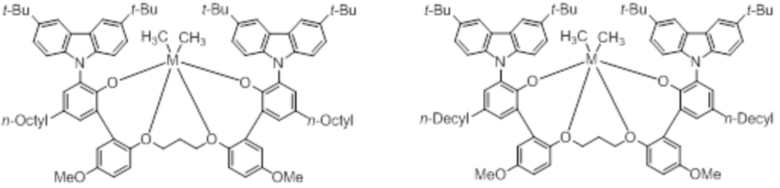
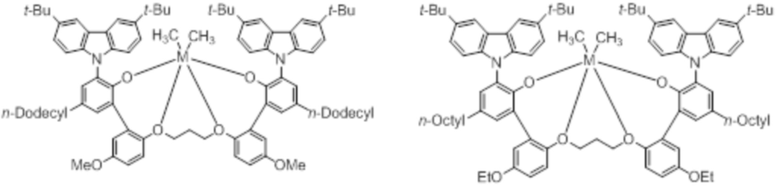
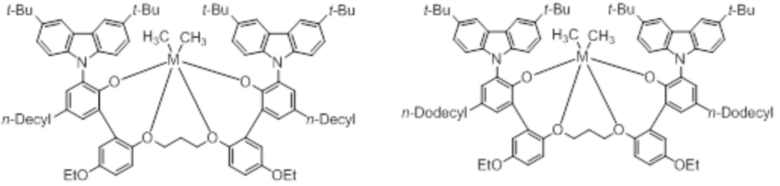
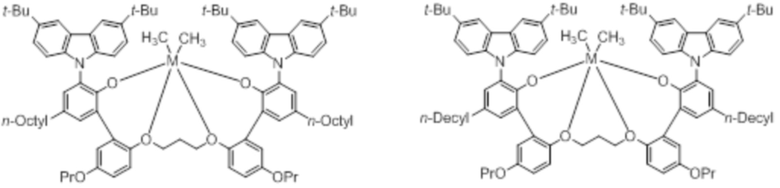
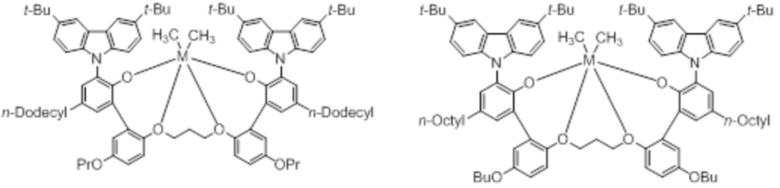
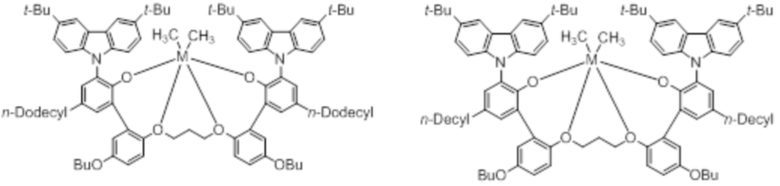
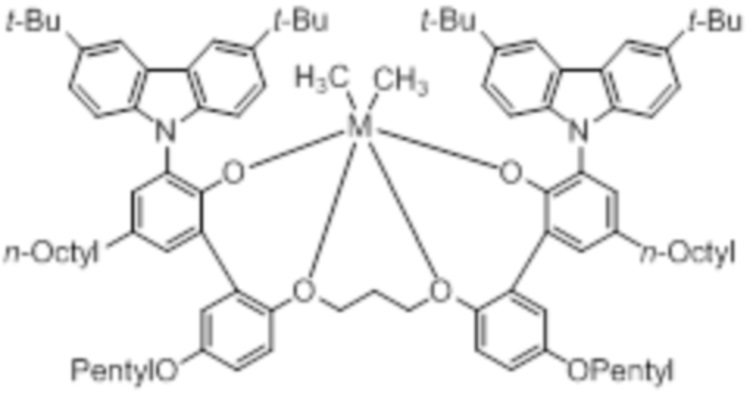
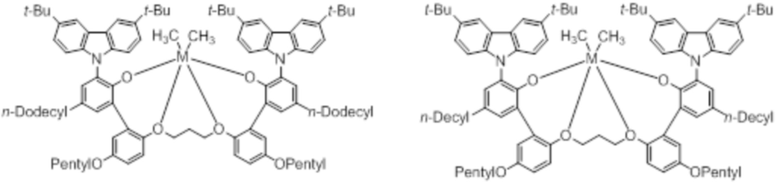
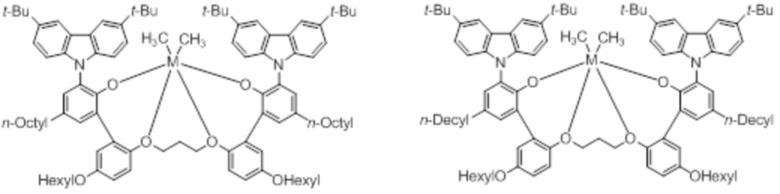
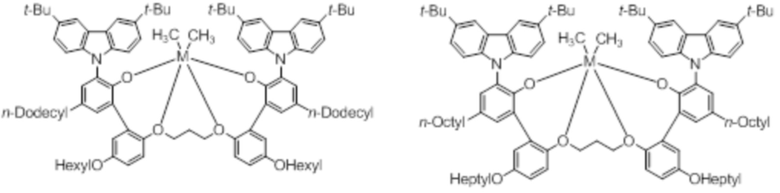
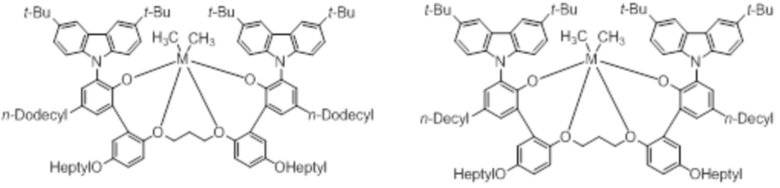
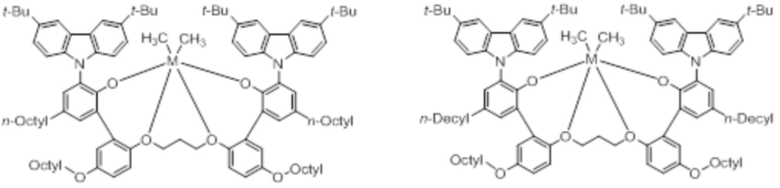
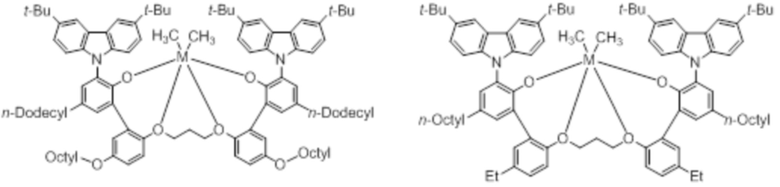
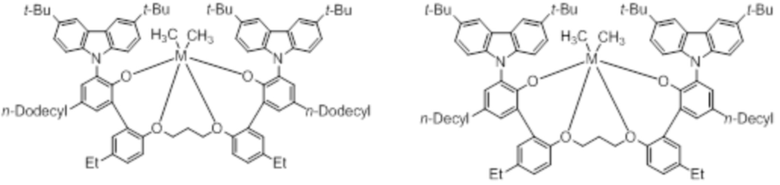
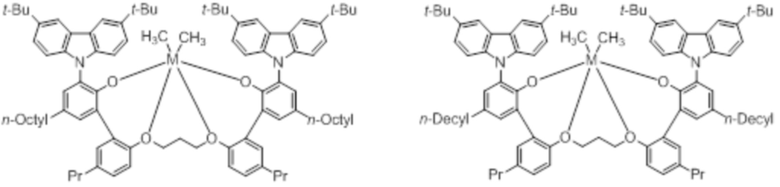
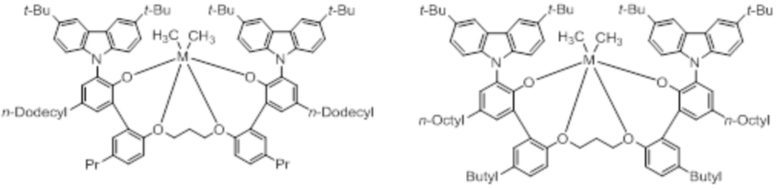
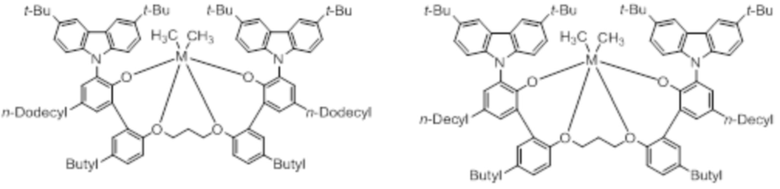
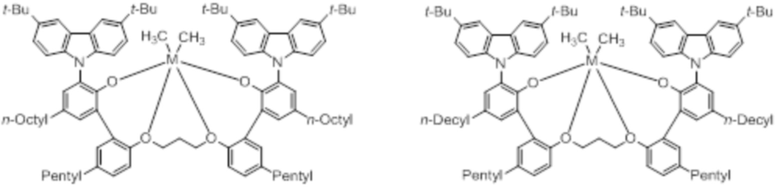
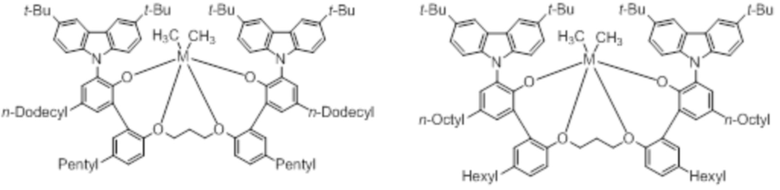
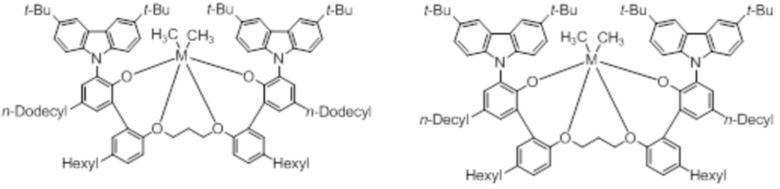
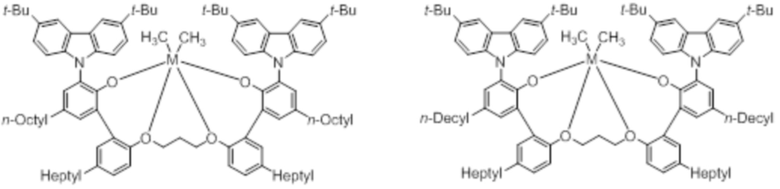
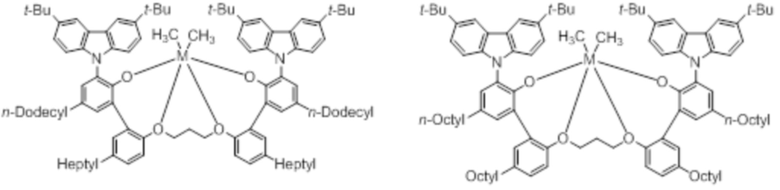
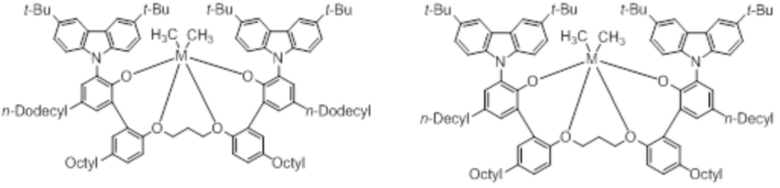
где M представляет собой титан, цирконий или гафний; и PrO-, BuO-, PentylO-, HexylO-, HeptylO-, OctylO-, Pr-, Butyl-, Pentyl-, Hexyl-, Heptyl- и Octyl- включают формы как с прямой, так и с разветвленной цепью.
Кроме того, настоящее изобретение предлагает каталитическую композицию для полимеризации на основе этилена, включающую в себя металлолигандный комплекс настоящего изобретения и сокатализатор.
Сокатализатор в соответствии с типичным вариантом осуществления настоящего изобретения может представлять собой сокатализатор из соединения бора, сокатализатор из соединения алюминия и их смесь.
Сокатализатор в соответствии с типичным вариантом осуществления настоящего изобретения может быть включен в мольном соотношении от 0,5 до 10000 на 1 моль металлолигандного комплекса.
Соединение бора, которое можно использовать в качестве сокатализатора в настоящем изобретении, может включать соединения бора, известные из патента США № 5198401, и, в частности, может быть выбрано из группы, состоящей из соединений, представленных следующими химическими формулами с 11 по 13.
[Химическая формула 11]
B(R21)3
[Химическая формула 12]
[R22]+[B(R21)4]-
[Химическая формула 13]
[(R23)pZH]+[B(R21)4]-
где B представляет собой атом бора; R21 представляет собой фенильную группу, а фенильная группа может быть дополнительно замещена 3-5 заместителями, выбранными из группы, состоящей из атома фтора, (C1-C20)алкильной группы, (C1-C20)алкильной группы, замещенной атомом фтора, (C1-C20)алкоксигруппы или (C1-C20)алкоксигруппы, замещенной атомом фтора; R22 представляет собой (C5-C7)ароматический радикал или (C1-C20)алкил(C6-C20)арильный радикал, (C6-C20)арил(C1-C20)алкильный радикал, например, трифенилметилиевый радикал; Z представляет собой атом азота или фосфора; R23 представляет собой (C1-C20)алкильный радикал или анилиниевый радикал, замещенный двумя (C1-C20)алкильными группами вместе с атомом азота; и p представляет собой целое число 2 или 3.
К предпочтительным примерам основанного на боре сокатализатора могут относиться тритилтетракис(пентафторфенил)борат, трис(пентафторфенил)боран, трис(2,3,5,6-тетрафторфенил)боран, трис(2,3,4,5-тетрафторфенил)боран, трис(3,4,5-трифторфенил)боран, трис(2,3,4-трифторфенил)боран, фенилбис(пентафторфенил)боран, тетракис(пентафторфенил)борат, тетракис(2,3,5,6-тетрафторфенил)борат, тетракис(2,3,4,5-тетрафторфенил)борат, тетракис(3,4,5-трифторфенил)борат, тетракис(2,2,4-трифторфенил)борат, фенилбис(пентафторфенил)борат или тетракис(3,5-бистрифторметилфенил)борат. Кроме того, некоторые примеры их комбинаций могут включать в себя ферроцения тетракис(пентафторфенил)борат, 1,1'-диметилферроцения тетракис(пентафторфенил)борат, серебра тетракис(пентафторфенил)борат, трифенилметилия тетракис(пентафторфенил)борат, трифенилметилия тетракис(3,5-бистрифторметилфенил)борат, триэтиламмония тетракис(пентафторфенил)борат, трипропиламмония тетракис(пентафторфенил)борат, три(н-бутил)аммония тетракис(пентафторфенил)борат, три(н-бутил)аммония тетракис(3,5-бистрифторметилфенил)борат, N, N-диметиланилиния тетракис(пентафторфенил)борат, N, N-диэтиланилиния тетракис(пентафторфенил)борат, N, N-2,4,6-пентаметиланилиния тетракис(пентафторфенил)борат, N, N-диметиланилиния тетракис(3,5-бистрифторметилфенил)борат, диизопропиламмония тетракис(пентафторфенил)борат, дициклогексиламмония тетракис(пентафторфенил)борат, трифенилфосфония тетракис(пентафторфенил)борат, три(метилфенил)фосфония тетракис(пентафторфенил)борат или три(диметилфенил)фосфония тетракис(пентафторфенил)борат, а наиболее предпочтительными среди них являются любые один или два или более, выбранные из группы, состоящей из трифенилметилия тетракис(пентафторфенил)бората, N, N-диметиланилиния тетракиспентафторфенилбората, трифенилметилия тетракиспентафторфенилбората и триспентафторборана.
Пример соединения алюминия, которое можно использовать в качестве сокатализатора в каталитической композиции в соответствии с типичным вариантом осуществления настоящего изобретения, может включать алюмоксановое соединение химической формулы 14 или 15, алюминийорганическое соединение химической формулы 16 или алюминийорганическое алкилоксидное или алюминийорганическое арилоксидное соединение химической формулы 17 или 18:
[Химическая формула 14]
(-Al(R24)-O-)m
[Химическая формула 15]
(R24)2Al-(-O(R24)-)q-(R24)2
[Химическая формула 16]
(R25)rAl(E)3-r
[Химическая формула 17]
(R26)2AlOR27
[Химическая формула 18]
R26Al(OR27)2
где R24 представляет собой (C1-C20)алкильную группу, предпочтительно метильную группу или изобутильную группу; m и q независимо друг от друга представляют собой целое число от 5 до 20; R25 и R26 независимо друг от друга представляют собой (C1-C20)алкильную группу; E представляет собой атом водорода или атом галогена; r представляет собой целое число от 1 до 3; и R27 представляет собой (C1-C20)алкильную группу или (C6-C30)арильную группу.
Конкретные примеры соединения, которое можно использовать в качестве соединения алюминия, могут включать алюмоксановые соединения, такие как метилалюмоксан, модифицированный метилалюмоксан и тетраизобутилалюмоксан; алюминийорганические соединения, например триалкилалюминий, такой как триметилалюминий, триэтилалюминий, трипропилалюминий, триизобутилалюминий и тригексилалюминий; диалкилалюминийхлорид, такой как диметилалюминийхлорид, диэтилалюминийхлорид, дипропилалюминийхлорид, диизобутилалюминийхлорид и дигексилалюминийхлорид; алкилалюминийдихлорид, такой как метилалюминийдихлорид, этилалюминийдихлорид, пропилалюминийдихлорид, изобутилалюминийдихлорид и гексилалюминийдихлорид; диалкилалюминийгидрид, такой как диметилалюминийгидрид, диэтилалюминийгидрид, дипропилалюминийгидрид, диизобутилалюминийгидрид и дигексилалюминийгидрид; алкилалкоксиалюминий, такой как метилдиметоксиалюминий, диметилметоксиалюминий, этилдиэтоксиалюминий, диэтилэтоксиалюминий, изобутилдибутоксиалюминий, диизобутилбутоксиалюминий, гексилдиметоксиалюминий, дигексилметоксиалюминий и диоктилметоксиалюминий, предпочтительно соединение, выбранное из группы, состоящей из метилалюмоксана, модифицированного метилалюмоксана, тетраизобутилалюмоксана, триалкилалюминия, триэтилалюминия и триизобутилалюминия по отдельности или в их комбинации, более предпочтительно триалкилалюминий, еще более предпочтительно триэтилалюминий и триизобутилалюминий.
Предпочтительно, в каталитической композиции в соответствии с типичным вариантом осуществления настоящего изобретения, когда в качестве сокатализатора используют соединение алюминия, предпочтительным диапазоном соотношения между металлолигандным комплексом настоящего изобретения и сокатализатором может быть соотношение переходный металл (M):атом алюминия (Al) от 1:50 до 1:5000 в пересчете на мольное соотношение.
Предпочтительно, в каталитической композиции в соответствии с типичным вариантом осуществления настоящего изобретения, когда в качестве сокатализатора используют как соединение алюминия, так и соединение бора, предпочтительным диапазоном соотношения между металлолигандным комплексом настоящего изобретения и сокатализатором может быть соотношение переходный металл (M):атом бора:атом алюминия 1:0,1-100:10-1000, более предпочтительно 1:0,5-5:25-500, в пересчете на мольное соотношение.
В качестве другого аспекта в соответствии с типичным вариантом осуществления настоящего изобретения способ получения полимера этилена с использованием каталитической композиции для полимеризации на основе этилена может быть осуществлен путем приведения металлолигандного комплекса, сокатализатора и этилена и, если необходимо, сомономера на основе винила в контакт в присутствии соответствующего органического растворителя. При этом, компонент-предкатализатор, который представляет собой металлолигандный комплекс, и компонент-сокатализатор могут быть добавлены в реактор по отдельности, или каждый компонент может быть предварительно смешан и добавлен в реактор, и условия смешивания, такие как порядок добавления, температура или концентрация, конкретно не ограничены.
Предпочтительными органическими растворителями, которые можно использовать в способе получения, могут быть C3-C20 углеводороды, и их конкретные примеры могут включать бутан, изобутан, пентан, гексан, гептан, октан, изооктан, нонан, декан, додекан, циклогексан, метилциклогексан, бензол, толуол, ксилол и т.п.
В частности, при получении сополимера этилена и α-олефина в качестве сомономера можно использовать C3-C18 α-олефины вместе с этиленом, и, предпочтительно, α-олефины можно выбирать из группы, состоящей из пропилена, 1-бутена, 1-пентена, 4-метил-1-пентена, 1-гексена, 1-октена, 1-децена, 1-додецена, 1-гексадецена и 1-октадецена. Более предпочтительно, могут быть сополимеризованы 1-бутен, 1-гексен, 1-октен или 1-децен и этилен. В этом случае предпочтительными давлением этилена и температурой реакции полимеризации могут быть давление 1-1000 атм, более предпочтительно 10-150 атм, и, для эффективной реакции полимеризации, температура реакции полимеризации 170-250°C, предпочтительно 180-200°C.
Кроме того, сополимер, получаемый в соответствии со способом настоящего изобретения, обычно содержит 50 вес.% или более, предпочтительно 60 вес.% или более, и, более предпочтительно, 60-99 вес.% этилена.
Как описано выше, линейный полиэтилен низкой плотности (LLDPE), полученный с использованием C4-C10 α-олефинов в качестве сомономера, имеет диапазон плотности 0,940 г/см3 или менее с возможностью расширения в диапазон полиэтилена очень низкой плотности (VLDPE) или полиэтилена сверхнизкой плотности (ULDPE) с плотностью 0,900 г/см3 или олефинового эластомера. Кроме того, при получении сополимера этилена в соответствии с настоящим изобретением в качестве регулятора молекулярной массы для корректировки молекулярной массы можно использовать водород, и сополимер обычно имеет средневесовую молекулярную массу (Mw) от 80000 до 500000.
Сополимер этилен-пропилен-диен в качестве конкретного примера сополимера олефин-диен, получаемого с помощью каталитической композиции в соответствии с типичным вариантом осуществления настоящего изобретения, может иметь содержание этилена от 30 до 80 вес.%, содержание пропилена от 20 до 70 вес.%, и содержание диена от 0 до 15 вес.%. Диеновый мономер, который можно использовать в настоящем изобретении, имеет две или более двойные связи, и его примеры могут включать 1,4-гексадиен, 1,5-гексадиен, 1,5-гептадиен, 1,6-гептадиен, 1,6-октадиен, 1,7-октадиен, 1,7-нонадиен, 1,8-нонадиен, 1,8-декадиен, 1,9-декадиен, 1,12-тетрадекадиен, 1,13-тетрадекадиен, 3-метил-1,4-гексадиен, 3-метил-1,5-гексадиен, 3-этил-1,4-гексадиен, 3-этил-1,5-гексадиен, 3,3-диметил-1,4-гексадиен, 3,3-диметил-1,5-гексадиен, 5-винил-2-норборнен, 2,5-норборнадиен, 7-метил-2,5-норборнадиен, 7-этил-2,5-норборнадиен, 7-пропил-2,5-норборнадиен, 7-бутил-2,5-норборнадиен, 7-фенил-2,5-норборнадиен, 7-гексил-2,5-норборнадиен, 7,7-диметил-2,5-норборнадиен, 7-метил-7-этил-2,5-норборнадиен, 7-хлор-2,5-норборнадиен, 7-бром-2,5-норборнадиен, 7-фтор-2,5-норборнадиен, 7,7-дихлор-2,5-норборнадиен, 1-метил-2,5-норборнадиен, 1-этил-2,5-норборнадиен, 1-пропил-2,5-норборнадиен, 1-бутил-2,5-норборнадиен, 1-хлор-2,5-норборнадиен, 1-бром-2,5-норборнадиен, 5-изопропил-2-норборнен, 1,4-циклогексадиен, бицикло(2,2,1)гепта-2,5-диен, 5-этилиден-2-норборнен, 5-метилен-2-норборнен, бицикло(2,2,2)окта-2,5-диен, 4-винилциклогекса-1-ен, бицикло(2,2,2)окта-2,6-диен, 1,7,7-триметилбицикло-(2,2,1)гепта-2,5-диен, дициклопентадиен, метилтетрагидроинден, 5-арилбицикло(2,2,1)гепта-2-ен, 1,5-циклооктадиен, 1,4-диарилбензол, бутадиен, изопрен, 2,3-диметил-1,3-бутадиен, 1,2-бутадиен, 4-метил-1,3-пентадиен, 1,3-пентадиен, 3-метил-1,3-пентадиен, 2,4-диметил-1,3-пентадиен, 3-этил-1,3-пентадиен и т.п., и, наиболее предпочтительно, диеновый мономер может представлять собой 5-этилиден-2-норборнен и дициклопентадиен. Диеновый мономер может быть выбран в зависимости от технологических характеристик сополимера этилен-пропилен-диен, и, если необходимо, два или более диеновых мономера могут быть смешаны.
В этом случае предпочтительными давлением и температурой в реакторе могут быть давление 1-1000 атм, более предпочтительно 5-100 атм, и, для эффективной реакции полимеризации, температура реакции полимеризации 170-250°C, предпочтительно 180-200°C.
Сополимер этилен-олефин-диен, получаемый в соответствии с типичным вариантом осуществления настоящего изобретения, может иметь содержание этилена от 30 до 80 вес.%, содержание олефина от 20 до 70 вес.% и содержание диена от 0 до 15 вес.%.
Как правило, в случае получения сополимера этилен-пропилен-диен молекулярная масса сополимера снижается с увеличением содержания пропилена, однако в случае сополимера этилен-пропилен-диен в соответствии с настоящим изобретением продукт с относительно высокой молекулярной массой может быть получен без снижения молекулярной массы даже при снижении содержания пропилена до 50%.
Поскольку каталитическая композиция, представленная в настоящем изобретении, представлена в гомогенной форме в реакторе для полимеризации, предпочтительно применять каталитическую композицию к способу полимеризации в растворе, который проводят при температуре, равной или превышающей точку плавления полимера. Однако, как раскрыто в патенте США № 4752597, каталитическую композицию можно использовать в способе суспензионной полимеризации или газофазной полимеризации в форме гетерогенной каталитической композиции, получаемой путем нанесения предкатализатора, который представляет собой металлолигандный комплекс, и сокатализатора на подложку из пористого оксида металла.
Далее настоящее изобретение подробно описано с помощью следующих примеров, однако объем настоящего изобретения не ограничен ими.
Если не указано иное, все эксперименты по синтезу лигандов и катализаторов проводили с использованием стандартной технологии Шленка или перчаточного бокса в атмосфере азота, и органический растворитель, используемый в реакции, использовали после нагрева растворителя с обратным холодильником над металлическим натрием и бензофеноном для удаления влаги и перегонки растворителя непосредственно перед употреблением. 1H-ЯМР-анализ синтезированных лиганда и катализатора проводили с использованием Bruker 400 или 500 МГц при комнатной температуре.
ИК-анализ проводили с 5 мг соединения в твердом состоянии с использованием Bruker ATR-IR.
Циклогексан в качестве растворителя для полимеризации использовали после достаточного удаления из него влаги, кислорода и других материалов, отравляющих катализатор, путем пропускания циклогексана через молекулярное сито 5 Å и трубку, заполненную активным оксидом алюминия, и барботирования циклогексана азотом высокой чистоты. Полимеризованный полимер с помощью способов, описанных ниже:
1. Индекс текучести расплава (MI)
измеряли при 190°C под нагрузкой 2,16 кг с использованием способа анализа ASTM D1238.
2. Плотность
измеряли с помощью способа анализа ASTM D792.
3. Молекулярная масса и молекулярно-массовое распределение
измеряли с помощью гель-хроматографии с колонкой с перемешиванием с тремя ступенями.
Используемый в настоящем изобретении растворитель представлял собой 1,2,4-трихлорбензол, а температура измерения составляла 120°C.
[Пример получения 1] Синтез 1,3-бис(4-фтор-2-йодфенокси)пропана (соединение A)
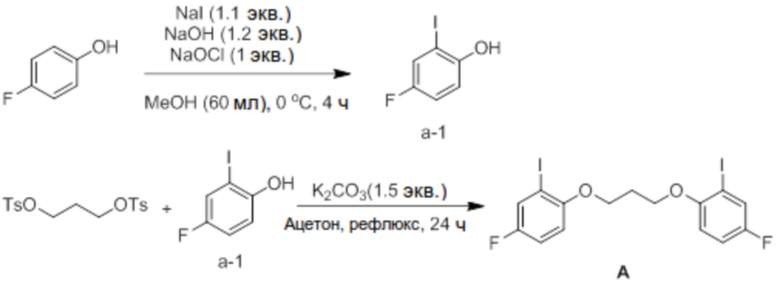
Синтез 4-фтор-2-йодфенола (соединение a-1)
4-Фторфенол (40 ммоль, 4,9 г), NaI и NaOH добавляли в соответствии с эквивалентами в круглодонную колбу, добавляли туда MeOH (60 мл), и температуру доводили до 0°C с использованием ледяной бани. Добавляли NaOCl (1 экв.) в течение 1 часа с использованием шприцевого насоса. После добавления всего NaOCl реактанты перемешивали при той же температуре в течение 3 часов. После этого добавляли туда 10% раствор тиосульфата натрия (30 мл), снова доводили pH до 5-6 с помощью 5% раствора HCl, и проводили экстракцию диэтиловым эфиром. Экстрагированный фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/30 об/об) с получением соединения a-1 в виде светло-желтого твердого вещества (5,2 г, 54%).
1H ЯМР (CDCl3): δ 7,39 (d, 1H), 7,01 (m, 1H), 6,98 (m, 1H), 5,10 (s, 1H).
Синтез 1,3-бис(4-фтор-2-йодфенокси)пропана (соединение A)
Пропан-1,3-диилбис(4-метилбензолсульфонат) (2,5 ммоль, 961 мг) и соединение a-1 (5 ммоль, 1,2 г) добавляли в круглодонную колбу, добавляли туда K2CO3 (1,5 экв.) и ацетон (20 мл), и реактанты нагревали с обратным холодильником в течение 24 часов. После завершения реакции реактанты охлаждали до комнатной температуры, ацетон удаляли перегонкой при пониженном давлении, и проводили экстракцию с помощью MC (метиленхлорид) и воды. Экстрагированный фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/10 об/об) с получением соединения A в виде желтого твердого вещества (1,03 г, 80%).
1H ЯМР (CDCl3): δ 7,49 (d, 2H), 7,02 (m, 2H), 6,80 (m, 2H), 4,25 (t, 4H), 2,34 (q, 2H).
[Пример 1] Синтез предкатализатора WC03
Синтез 2-йод-4-октилфенола (соединение 3-1)

4-н-Октилфенол (20 ммоль, 4,13 г), NaI (1,1 экв.) и NaOH (1,2 экв.) добавляли в круглодонную колбу, добавляли туда MeOH (120 мл), и температуру доводили до 0° с использованием ледяной бани. Туда медленно добавляли NaOCl (1 экв.) в течение 1 часа с использованием шприцевого насоса, и реактанты дополнительно перемешивали при той же температуре в течение 3 часов. После добавления туда 10% раствора тиосульфата натрия (30 мл) pH доводили до 5-6 с использованием 5% раствора HCl, и проводили экстракцию диэтиловым эфиром. Растворитель удаляли из экстрагированного фильтрата, и использовали колоночную хроматографию (элюент EA/n-Hex=1/30 об/об) с получением соединения 3-1 в виде бесцветной жидкости (2,9 г, 65%).
Синтез 2-(2-йод-4-октилфенокси)тетрагидро-2H-пирана (соединение 3-2)
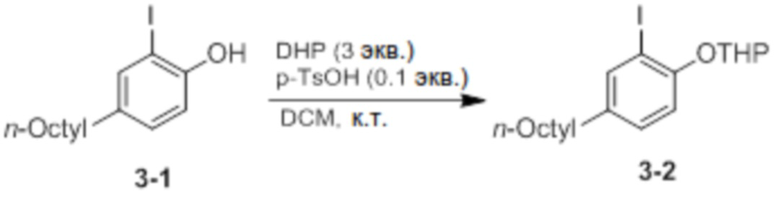
Соединение 3-1 и p-TsOH (0,1 экв.) добавляли в круглодонную колбу, добавляли DCM (50 мл), добавляли туда DHP (дигидропиран, 3 экв.), и затем реактанты перемешивали при комнатной температуре в течение 30 минут. После этого проводили экстракцию 1 М раствором K2CO3, и проводили колоночную хроматографию (элюент EA/n-Hex=1/30 об/об) с получением соединения 3-2 в виде бесцветной жидкости (9,2 г, 85%).
1H ЯМР (CDCl3): δ 7,46 (s, 1H), 7,05 (d, 1H), 6,90 (d, 1H), 5,13 (s, 1H), 2,49 (t, 3H), 1,55 (m, 2H), 1,28 (m, 10H), 0,88 (t, 3H).
Синтез 3,6-ди-трет-бутил-9-(5-октил-2-((тетрагидро-2H-пиран-2-ил)окси)фенил)-9H-карбазола (соединение 3-3)
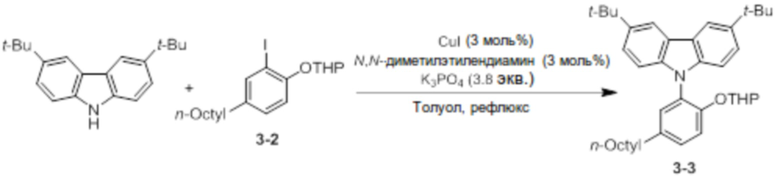
3,6-Ди-трет-бутил-9H-карбазол (5 ммоль, 1,4 г) и соединение 3-2 (1,8 экв.) добавляли в двухгорлую круглодонную колбу, добавляли туда CuI (3 моль%), N, N-диметилэтилендиамин (3 моль%) и K3PO4 (3,8 экв.), и использовали линию Шленка для создания условий с азотом. После этого добавляли туда толуол (25 мл), и реактанты нагревали с обратным холодильником при 125°C в течение 4 дней. После завершения реакции продукт охлаждали до комнатной температуры, и катализатор удаляли оттуда с помощью силикагелевого фильтра. В данном случае в качестве раствора для промывания использовали THF. Растворитель удаляли перегонкой при пониженном давлении, и продукт перекристаллизовывали с использованием ацетонитрила (5 мл), получая при этом соединение 3-3 в виде бесцветного твердого вещества (0,94 г, 51%).
1H ЯМР (CDCl3): δ 8,13 (s, 2H), 7,43 (m, 2H), 7,31 (m, 2H), 7,29 (m, 1H), 7,17 (m, 1H), 7,13 (m, 1H), 5,21 (m, 1H), 4,15 (m, 1H), 3,74 (m, 1H), 3,70 (m, 1H), 2,62 (m, 2H), 1,62 (m, 2H), 1,47 (s, 20H), 1,38 ~ 1,25 (m, 18H), 0,88 (m, 3H).
Синтез 3,6-ди-трет-бутил-9-(2-((тетрагидро-2H-пиран-2-ил)окси)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5-(2,4,4-триметилпентан-2-ил)фенил)-9H-карбазола (соединение 3-4)
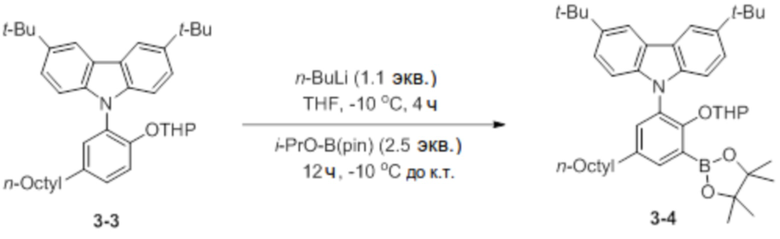
В атмосфере азота соединение 3-3 (4 ммоль, 2,3 г) добавляли в круглодонную колбу, добавляли туда THF (45 мл), очень медленно добавляли туда n-BuLi (1,1 экв.) при -10°C, и затем реактанты перемешивали в течение 4 часов. Через 4 часа очень медленно добавляли туда 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (i-PrO-B(pin), 2,5 экв.), и затем температуру поднимали до комнатной температуры, и реактанты перемешивали в течение 12 часов. После завершения реакции добавляли туда холодный насыщ. раствор NaHCO3 (водн.) (35 мл), реактанты перемешивали в течение 10 минут, и проводили экстракцию с использованием DCM. После экстракции полученное твердое вещество отфильтровывали через короткую колонку на силикагеле с этилацетатом, и затем растворитель выпаривали с получением белого твердого вещества, которое добавляли в ацетонитрил (10 мл) и перемешивали в течение 1 часа. После перемешивания соединение, полученное фильтрацией, сушили, получая при этом соединение 3-4 в виде бесцветного твердого вещества (1,1 г, 41%).
1H ЯМР (CDCl3): δ 8,13 (s, 2H), 7,94 (s, 1H), 7,55 (s, 1H), 7,45 (d, 2H), 7,35 (s, 1H), 7,17 (m, 1H), 7,13 (d, 2H), 2,58 (t, 2H), 1,61 (m, 3H), 1,44 ~ 1,37 (m, 20H), 1,35 (s, 12H), 1,27 (m, 10H), 0,87 (m, 3H).
Синтез лиганда WC03L
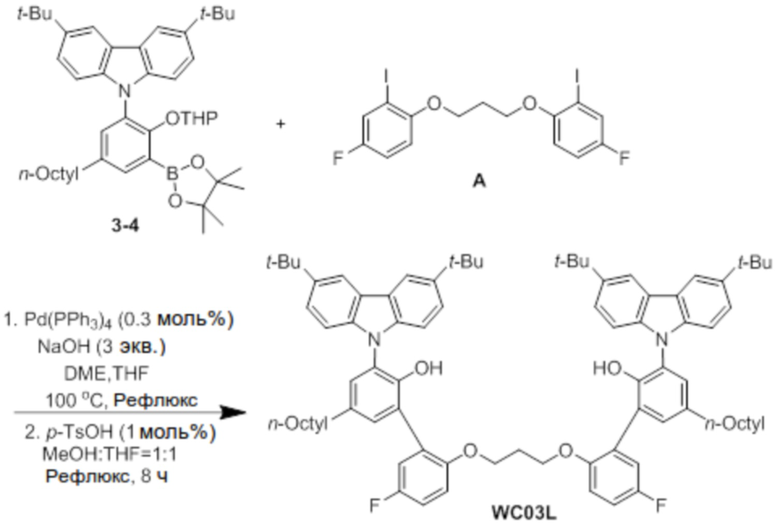
В атмосфере азота соединение 3-4 (2 ммоль), соединение A (0,5 экв.), Pd(PPh3)4 (0,3 моль%) и NaOH (3 экв.) добавляли в двухгорлую круглодонную колбу, добавляли туда DME (40 мл), THF (20 мл) и H2O (10 мл) в указанном порядке, и реактанты нагревали с обратным холодильником при 100°C в течение 48 часов. После завершения реакции продукт охлаждали до комнатной температуры, и катализатор удаляли оттуда с помощью силикагелевого фильтра. Растворитель удаляли перегонкой при пониженном давлении, и оставшийся растворитель удаляли с помощью вакуума.
Остаток, из которого удаляли растворитель, добавляли в круглодонную колбу, добавляли туда p-TsOH (1 моль%), затем добавляли туда по 100 мл MeOH и THF, и реактанты нагревали с обратным холодильником при 80°C в течение 8 часов. После завершения реакции продукт охлаждали до комнатной температуры, и проводили экстракцию с помощью воды. Экстрагированный фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/50 об/об), и затем продукт перекристаллизовывали с использованием EA (5 мл), получая при этом лиганд WC03L (0,7 г, 29%).
1H ЯМР (CDCl3): δ 8,22 (s, 4H), 7,43 (m, 4H), 7,36 (s, 2H), 7,32(s, 2H), 7,12(m, 6H), 7,03(s, 2H), 6,54 (m, 2H), 6,05 (m, 2H), 5,47 (s, 2H), 3,83 (m, 4H), 2,59 (m, 4H), 2,08 (m, 2H), 1,35 ~ 1,27 (m, 66H), 0,90 (m, 6H); 13C-{1H}-ЯМР: 156,2, 151,2, 148,2, 142,9, 139,9, 135,7, 131,1, 128,9, 128,0, 127,9, 126,7, 124,9, 123,6, 123,5, 118,4, 118,2, 116,4, 115,5, 115,3, 113,1, 113,0, 109,5, 65,2, 35,1, 34,8, 32,2, 32,0, 31,6, 29,5, 29,44, 29,41, 29,0, 22,8, 14,2.
Синтез предкатализатора WC03
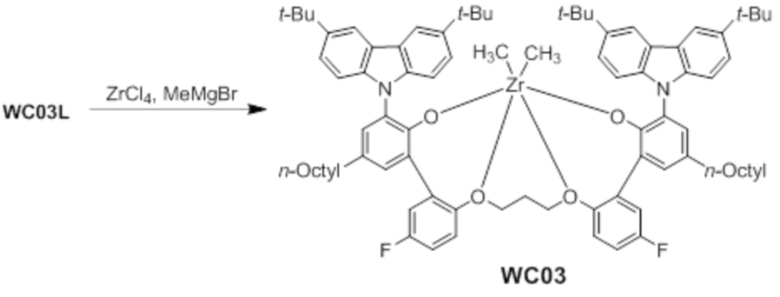
Реакцию проводили в перчаточном боксе в атмосфере азота. ZrCl4 (0,45 г, 1,93 ммоль) и толуол (80 мл) добавляли в колбу объемом 100 мл для получения суспензии. Суспензию охлаждали до -20°C в течение 30 минут в холодильнике с перчаточным боксом. К суспензии добавляли 3,0 М метилмагнийбромид (2,6 мл, 10,2 ммоль) в диэтиловом эфире при низкой температуре при перемешивании. Смесь интенсивно перемешивали в течение 30 минут. Твердое вещество растворялось, но реакционный раствор приобретал мутный желтый цвет. К смеси медленно добавляли лиганд WC03L (2,0 г, 1,6 ммоль) в виде твердого вещества. После завершения реакции колбу нагревали до комнатной температуры, реактанты перемешивали в течение 12 часов, и реакционную смесь фильтровали через шприц, соединенный с мембранным фильтром. Отфильтрованный раствор сушили под вакуумом, получая при этом предкатализатор WC03 в виде коричневого твердого вещества (2,04 г, выход: 93,2%).
1H ЯМР (CDCl3): δ 8,31 (s, 2H), 8,02 s, 2H), 7,56 (s, 2H), 7,50 (s, 2H), 7,41(d, 2H), 7,25 (d, 2H), 7,23(s, 2H), 6,95 (m, 2H), 6,25 (t, 2H), 4,61 (t, 2H), 3,84 (t, 2H), 3,32 (t, 2H), 2,0 ~ 1,3 (m, 34H), 1,78 (s, 2H), 1,72 (s, 18H), -1,62 (s, 6H).
[Пример 2] Синтез предкатализатора WC04
Синтез 2-йод-4-додецилфенола (соединение 4-1)
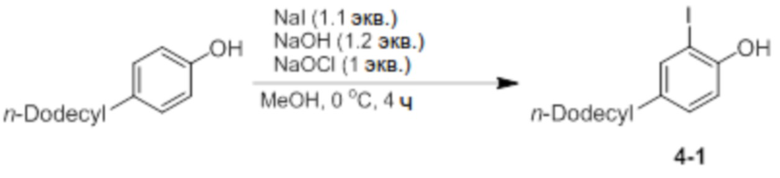
4-н-Додецилфенол (40 ммоль, 10,5 г), NaI (1,1 экв.) и NaOH (1,2 экв.) добавляли в круглодонную колбу, добавляли туда MeOH (120 мл), и температуру доводили до 0°C с использованием ледяной бани. Туда медленно добавляли NaOCl (1 экв.) в течение 1 часа с использованием шприцевого насоса, и затем реактанты дополнительно перемешивали при той же температуре в течение 3 часов. После этого добавляли туда 10% раствор тиосульфата натрия (30 мл), pH снова доводили до 5-6 с использованием 6,5% раствора HCl, и проводили экстракцию диэтиловым эфиром. Экстрагированный фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/30 об/об) с получением соединения 4-1 в виде бесцветной жидкости (8 г, 65%).
Синтез 2-(2-йод-4-додецилфенокси)тетрагидро-2H-пирана (соединение 4-2)
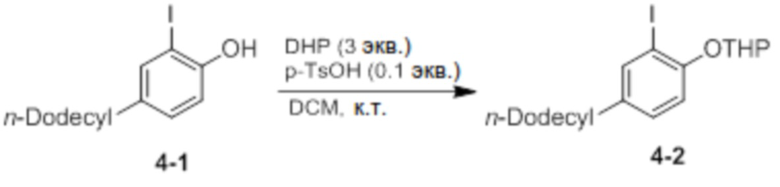
Соединение 4-1 и p-TsOH (0,1 экв.) добавляли в круглодонную колбу, и добавляли туда DCM (50 мл). После добавления DHP (3 экв.) реактанты перемешивали при комнатной температуре в течение 30 минут. После завершения реакции проводили экстракцию 1 М раствором K2CO3, и продукт отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/60 об/об) с получением соединения 4-2 в виде бесцветной жидкости (9,4 г, 95%).
Синтез 3,6-ди-трет-бутил-9-(5-додецил-2-((тетрагидро-2H-пиран-2-ил)окси)фенил)-9H-карбазола (соединение 4-3)
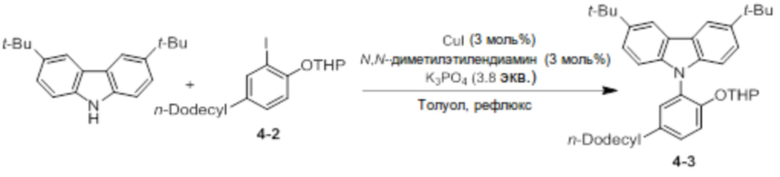
3,6-Ди-трет-бутил-9H-карбазол (5 ммоль, 1,4 г) и соединение 4-2 (1,8 экв.) добавляли в двухгорлую круглодонную колбу, добавляли туда CuI (3 моль%), N, N-диметилэтилендиамин (3 моль%) и K3PO4 (3,8 экв.), и использовали линию Шленка для создания условий с азотом. После добавления туда толуола (25 мл), реактанты нагревали с обратным холодильником при 125°C в течение 4 дней. После завершения реакции продукт охлаждали до комнатной температуры, и катализатор удаляли оттуда с помощью силикагелевого фильтра. В данном случае в качестве раствора для промывания использовали THF. После удаления растворителя перегонкой при пониженном давлении продукт перекристаллизовывали с использованием ацетонитрила (5 мл), получая при этом соединение 4-3 в виде бесцветного твердого вещества (0,94 г, 30%).
Синтез 3,6-ди-трет-бутил-9-(5-додецил-2-((тетрагидро-2H-пиран-2-ил)окси)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-9H-карбазола (соединение 4-4)
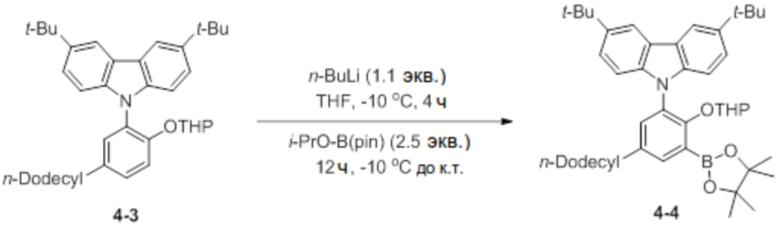
В атмосфере азота соединение 4-3 (4 ммоль, 2,3 г) добавляли в круглодонную колбу, добавляли туда THF (45 мл), очень медленно добавляли туда n-BuLi (1,1 экв.) при -10°C, и затем реактанты перемешивали в течение 4 часов. Через 4 часа очень медленно добавляли туда 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,5 экв.), и затем температуру поднимали до комнатной температуры, и реактанты перемешивали в течение 12 часов. После завершения реакции добавляли туда холодный насыщ. раствор NaHCO3 (водн.) (35 мл), реактанты перемешивали в течение 10 минут, и проводили экстракцию с использованием DCM. После этого продукт использовали в состоянии неочищенной смеси в следующей реакции.
Синтез лиганда WC04L
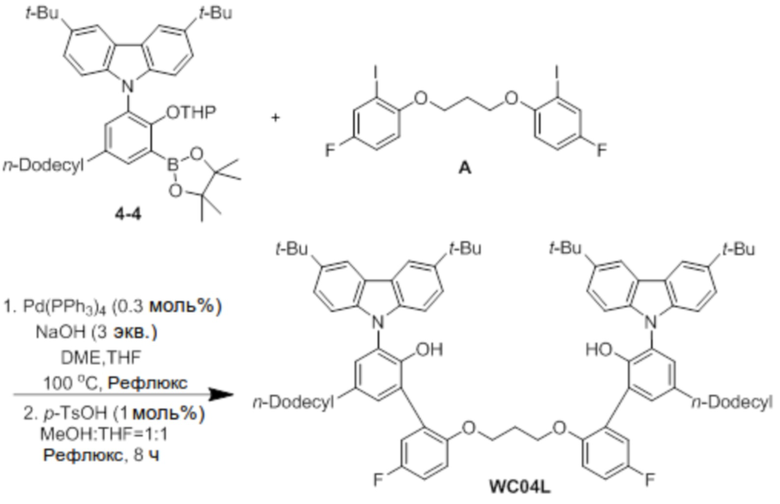
В атмосфере азота соединение 4-4 (2 ммоль), соединение A (0,5 экв.), Pd(PPh3)4 (0,3 моль%) и NaOH (3 экв.) добавляли в двухгорлую круглодонную колбу, добавляли туда DME (40 мл), THF (20 мл) и H2O (10 мл) в указанном порядке, и реактанты нагревали с обратным холодильником при 100°C в течение 48 часов. После завершения реакции продукт охлаждали до комнатной температуры, катализатор удаляли оттуда с помощью силикагелевого фильтра, растворитель удаляли перегонкой при пониженном давлении, и оставшийся растворитель удаляли с помощью вакуума.
Остаток, из которого удаляли растворитель, добавляли в круглодонную колбу, и добавляли туда p-TsOH (1 моль%). Снова добавляли туда по 100 мл MeOH и THF, и затем реактанты нагревали с обратным холодильником при 80°C в течение 8 часов. После завершения реакции продукт охлаждали до комнатной температуры, и проводили экстракцию с помощью воды. После этого продукт отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/50 об/об), и затем продукт снова перекристаллизовывали с использованием EA (5 мл), получая при этом лиганд WC04L (0,5 г).
1H ЯМР (CDCl3): δ 8,17 (s, 4H), 7,49 ~ 6,92 (m, 18H), 6,54 (m, 2H), 6,05 (m, 2H), 5,47 (s, 2H), 1,50 ~ 0,65 (m, 88H).
Синтез предкатализатора WC04

Предкатализатор WC04 получали таким же образом, как в примере 1, за исключением того, что использовали лиганд WC04L вместо лиганда WC03L.
1H ЯМР (CDCl3): δ 8,35 (s, 2H), 8,03 s, 2H), 7,54 ~ 6,95 (m, 18H), 6,54 (m, 2H), 6,05 (m, 2H), 5,47 (s, 2H), 1,50 ~ 0,65 (m, 88H), -1,63 (s, 6H).
[Сравнительный пример 1] Синтез предкатализатора WC01
Получение 2-йод-4-(2,4,4-триметилпентан-2-ил)фенола (соединение 1-1)
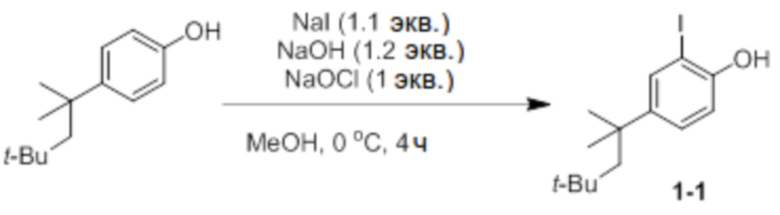
4-(2,4,4-триметилпентан-2-ил)фенол (50 ммоль, 10,32 г), NaI (1,1 экв.) и NaOH (1,2 экв.) добавляли в круглодонную колбу. Добавляли туда MeOH (120 мл), температуру доводили до 0°C с использованием ледяной бани, и медленно добавляли туда NaOCl (1 экв.) в течение 1 часа с использованием шприцевого насоса. После этого реактанты перемешивали при той же температуре в течение 3 часов, и добавляли туда 10% раствор тиосульфата натрия (30 мл). Снова доводили pH до 5-6 с использованием 5% раствора HCl, проводили экстракцию диэтиловым эфиром, и затем продукт отделяли и очищали с помощью колоночной хроматографии (элюент: EA/n-Hex=1/30 об/об) с получением соединения 1-1 в виде бесцветной жидкости (8,64 г, 52%).
Получение 2-(2-йод-4-(2,4,4-триметилпентан-2-ил)фенокси)тетрагидро-2H-пирана (соединение 1-2)
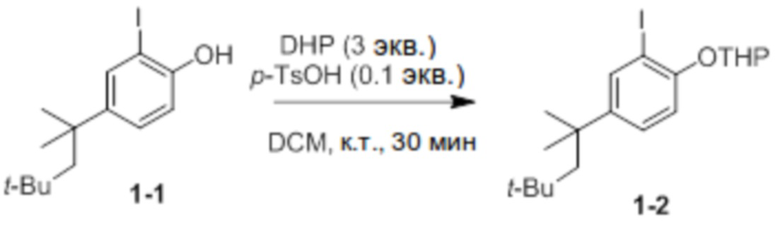
Соединение 1-1 и p-TsOH (0,1 экв.) добавляли в круглодонную колбу и растворяли в DCM (50 мл). После добавления туда DHP (3 экв.) добавляли p-TsOH (0,1 экв.), и реактанты перемешивали при комнатной температуре в течение 30 минут. После этого реакционную смесь экстрагировали 1 М раствором K2CO3, и продукт отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/30 об/об) с получением соединения 1-2 в виде бесцветной жидкости (9,2 г, 85%).
Синтез 3,6-ди-трет-бутил-9-(2-((тетрагидро-2H-пиран-2-ил)окси)-5-(2,4,4-триметилпентан-2-ил)фенил)-9-карбазола (соединение 1-3)
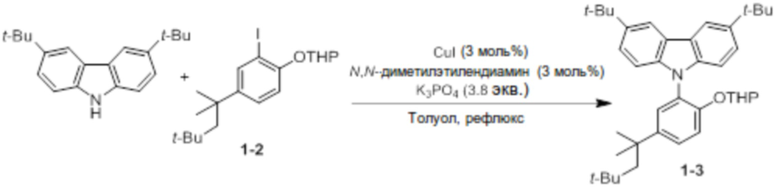
3,6-Ди-трет-бутил-9H-карбазол (9,5 ммоль, 2,7 г) и соединение 1-2 (1,8 экв.) добавляли в двухгорлую круглодонную колбу, добавляли туда CuI (3 моль%), N, N-Диметилэтилендиамин (3 моль%) и K3PO4 (3,8 экв.), и использовали линию Шленка для создания условий с азотом. После нового добавления туда толуола (50 мл) реактанты нагревали с обратным холодильником при 125°C в течение 4 дней. После завершения реакции продукт охлаждали до комнатной температуры, и катализатор удаляли оттуда с помощью силикагелевого фильтра. В данном случае в качестве раствора для промывания использовали THF. Растворитель удаляли выпариванием, продукт отфильтровывали через короткую колонку на силикагеле с этилацетатом, и перекристаллизовывали с ацетонитрилом (10 мл), получая при этом соединение 1-3 в виде бесцветного твердого вещества (3,7 г, 80%).
1H ЯМР (CDCl3): δ 8,13 (s, 2H), 7,47 (s, 1H), 7,42 (m, 3H), 7,31 (d, 1H), 7,15 (d, 1H), 7,09 (d, 1H), 5,22 (s, 1H), 3,74 (m, 1H), 3,49 (m, 1H), 1,74 (s, 2H), 1,47 (s, 18H), 1,38 (s, 6H), 0,8 (s, 9H)
Синтез 3,6-ди-трет-бутил-9-(2-((тетрагидро-2H-пиран-2-ил)окси)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5-(2,4,4-триметилпентан-2-ил)фенил)-9H-карбазола (соединение 1-4)
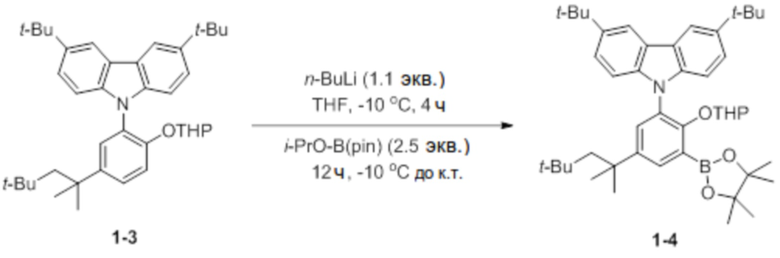
В присутствии азота соединение 1-3 (6,5 ммоль, 3,6 г) добавляли в круглодонную колбу, добавляли туда THF (45 мл), очень медленно добавляли туда n-BuLi (1,1 экв.) при -10°C, и затем реактанты перемешивали в течение 4 часов. После этого очень медленно добавляли туда 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,5 экв.), и затем температуру поднимали до комнатной температуры, и реактанты перемешивали в течение 12 часов. После завершения реакции добавляли туда холодный насыщ. раствор NaHCO3 (водн.) (35 мл), реактанты перемешивали в течение 10 минут, и проводили экстракцию с использованием DCM. Полученное твердое вещество отфильтровывали через короткую колонку на силикагеле с этилацетатом, и затем растворитель выпаривали с получением белого твердого вещества, который добавляли в ацетонитрил (10 мл) и перемешивали в течение 1 часа. После перемешивания соединение, полученное фильтрацией, сушили, получая при этом соединение 1-4 в виде бесцветного твердого вещества (4,3 г, 83%).
1H ЯМР (CDCl3): δ 8,15 (s, 1H), 8,09 (s, 2H), 8,0 (s, 1H), 7,71~ 7,55(dd, 1H), 7,45 (m, 4H), 7,14 (d, 1H), 7,10 (d, 1H), 4,84 (s, 1H), 2,74 (m, 1H), 2,60 (m, 1H), 1,65 (s, 3H), 1,60 (s, 2H), 1,55 (s, 5H), 1,45 (s, 27H), 1,38 (m, 27H), 0,81 (s, 5H), 0,77 (s, 9H)
Синтез 4-фтор-2-йод-6-метилфенола (соединение 1-5)
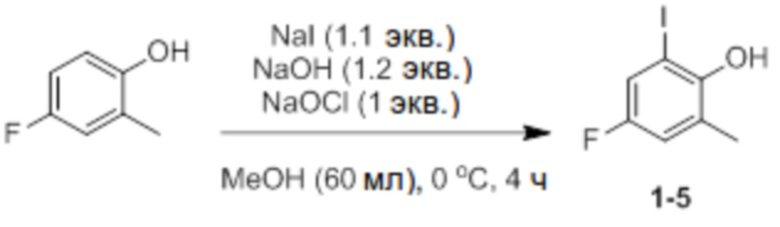
4-Фтор-2-метилфенол (20 ммоль, 2,5 г), NaI и NaOH добавляли в соответствии с эквивалентом в круглодонную колбу. Добавляли туда MeOH (60 мл), температуру доводили до 0°C с использованием ледяной бани, и медленно добавляли туда NaOCl (1 экв.) в течение 1 часа с использованием шприцевого насоса. После добавления всего NaOCl реактанты перемешивали при той же температуре в течение 3 часов. После этого добавляли туда 10% раствор тиосульфата натрия (30 мл), pH снова доводили до 5-6 5% раствором HCl, и проводили экстракцию диэтиловым эфиром. Экстрагированный фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/30 об/об) с получением соединения 1-5 в виде светло-желтого твердого вещества (4,3 г, 80%).
Синтез пропан-13-диилбис(4-метилбензолсульфоната) (соединение 1-6)
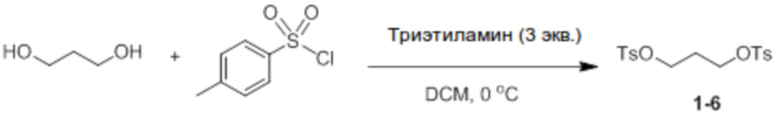
Пропан-1,3-диол (25 ммоль, 1,9 г) и 4-метилбензол-1- сульфонилхлорид (55 ммоль, 10,5 г) добавляли в круглодонную колбу, и затем добавляли туда DCM (200 мл), и температуру доводили до 0°C с использованием ледяной бани. После добавления туда триэтиламина (75 ммоль, 7,6 г) реактанты перемешивали при комнатной температуре в течение 12 часов после добавления триэтиламина. После завершения реакции проводили экстракцию с использованием DCM, и продукт отфильтровывали через короткую колонку на силикагеле с этилацетатом, а затем перекристаллизовывали с использованием ацетона, и белое твердое вещество отфильтровывали, получая при этом соединение 1-6 (7,7 г, 80%).
Синтез 1,3-бис(4-фтор-2-йод-6-метилфенокси)пропана (соединение 1-7)

Соединение 1-6 (2,5 ммоль, 961 мг) и соединение 1-5 (5 ммоль, 1,3 г) добавляли в круглодонную колбу, и затем добавляли туда K2CO3 (1,5 экв.) и ацетон (20 мл), и реактанты нагревали с обратным холодильником в течение 24 часов. После завершения реакции продукт охлаждали до комнатной температуры, ацетон удаляли перегонкой при пониженном давлении, и проводили экстракцию с помощью MC и воды. Фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент: EA/n-Hex=1/10 об/об) с получением соединения 1-7 в виде желтого твердого вещества (1,4 г, 84%).
Синтез лиганда WC01L
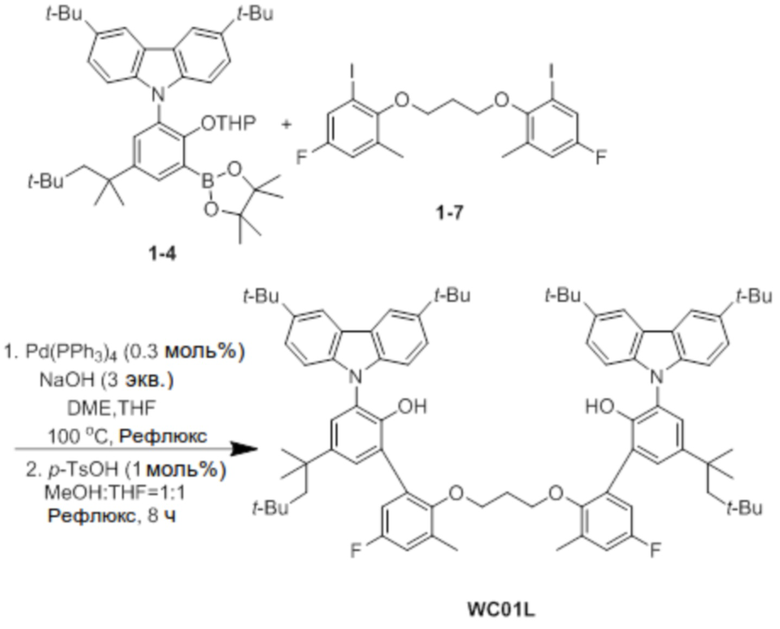
Под азотом соединение 1-4 (2,5 ммоль), соединение 1-7 (0,5 экв.), Pd(PPh3)4 (0,3 моль%) и NaOH (3 экв.) добавляли в двухгорлую круглодонную колбу, и затем добавляли туда DME (40 мл), THF (20 мл) и H2O (10 мл) в указанном порядке, и реактанты нагревали с обратным холодильником при 100°C в течение 48 часов. После завершения реакции продукт охлаждали до комнатной температуры, и катализатор удаляли оттуда с помощью силикагелевого фильтра. Фильтрат удаляли перегонкой при пониженном давлении, и оставшийся растворитель удаляли с помощью вакуума.
Остаток, из которого удаляли растворитель, добавляли в круглодонную колбу, и добавляли туда p-TsOH (1 моль%). Снова добавляли туда по 100 мл MeOH и THF, и затем реактанты нагревали с обратным холодильником при 80°C в течение 8 часов. После завершения реакции продукт охлаждали до комнатной температуры, и проводили экстракцию с помощью воды. После этого экстрагированный фильтрат отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/50 об/об), и затем перекристаллизовывали с использованием EA (5 мл), получая при этом лиганд WC01L (0,88 г, 68%).
1H ЯМР (CDCl3): δ 8,14 (s, 1H), 7,43 (s, 2H), 7,37 (m, 6H), 7,02(d, 4H), 6,95 (dd, 2H), 6,85 (dd, 2H), 6,47 (s, 2H), 3,64 (t, 4H), 2,03 (s, 6H), 1,73 (s, 6H), 1,55 (s, 18H), 1,43 (s, 36H), 1,34 (s, 12H), 0,76 (s, 18H); 13C-{1H}-ЯМР: 159,97, 158,03, 149,88, 147,55, 143,17, 142,57, 139,69, 128,69, 127,49, 126,45, 125,58, 123,53, 123,32, 117,15, 116,38, 116,22, 109,20, 70,9, 57,06, 38,24, 34,72, 32,44, 32,05, 31,86, 31,63, 30,61, 16,45.
Синтез предкатализатора WC01
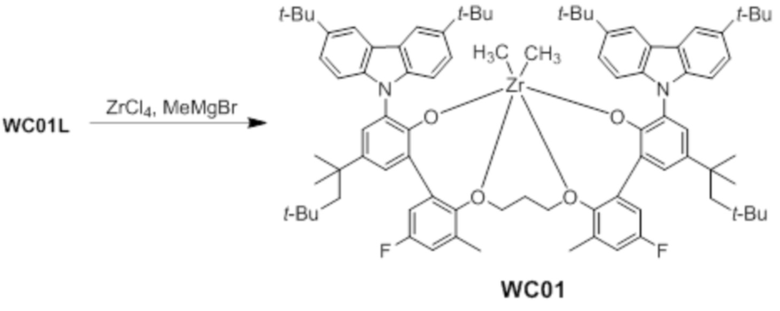
Предкатализатор WC01 получали таким же образом, как в примере 1, за исключением того, что использовали лиганд WC01L вместо лиганда WC03L.
1H ЯМР (CDCl3): δ 8,31 (s, 2H), 8,02 s, 2H), 7,56 (s, 2H), 7,50 (s, 2H), 7,41(d, 2H), 7,25 (d, 2H), 7,23(s, 2H), 6,95 (m, 2H), 6,25 (t, 2H), 4,61 (t, 2H), 3,76 (t, 2H), 3,38 (t, 2H), 2,35 (s, 12H), 1,85 (s, 4H), 1,83 (s, 36H), 1,52(s, 18H), 1,45 (s, 3H), 0,98(s, 12H), -1,65 (s, 3H)
[Сравнительный пример 2] Синтез предкатализатора WC02
Синтез лиганда WC02L
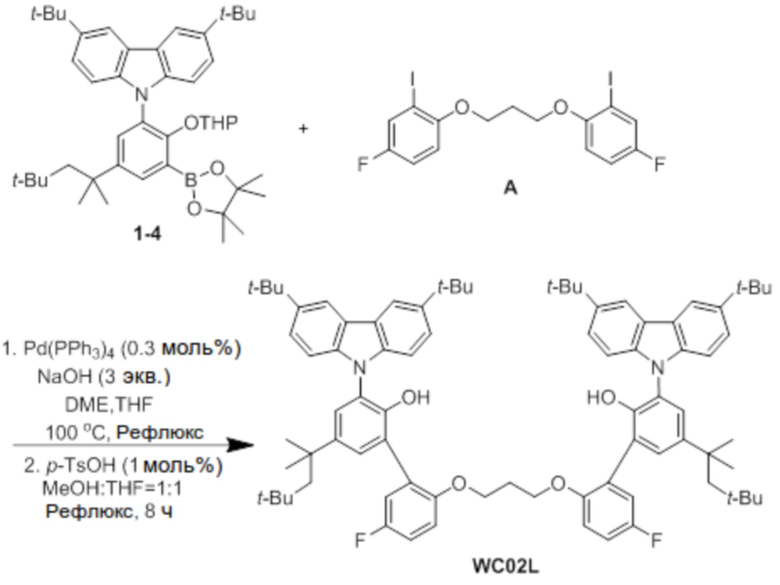
В атмосфере азота соединение 1-4 (2,5 ммоль), соединение A (0,5 экв.), Pd(PPh3)4 (0,3 моль%) и NaOH (3 экв.) добавляли в двухгорлую круглодонную колбу, добавляли туда DME (40 мл), THF (20 мл) и H2O (10 мл) в указанном порядке, и реактанты нагревали с обратным холодильником при 100°C в течение 48 часов. После завершения реакции продукт охлаждали до комнатной температуры, катализатор удаляли оттуда с помощью силикагелевого фильтра, продукт перегоняли при пониженном давлении для удаления растворителя, и оставшийся растворитель удаляли с помощью вакуума.
Остаток, из которого удаляли растворитель, добавляли в круглодонную колбу, добавляли туда p-TsOH (1 моль%), затем добавляли туда по 100 мл MeOH и THF, и реактанты нагревали с обратным холодильником при 80°C в течение 8 часов. После завершения реакции продукт охлаждали до комнатной температуры, проводили экстракцию с помощью воды, и продукт отделяли и очищали с помощью колоночной хроматографии (элюент EA/n-Hex=1/50 об/об), и затем перекристаллизовывали с использованием EA (5 мл), получая при этом лиганд WC02L (1,1 г, 70%).
1H ЯМР (CDCl3): δ 8,23 (s, 4H), 7,45 (d, 4H), 7,43 (s, 2H), 7,39(d, 2H), 7,25 (d, 4H), 6,95 (d, 2H), 6,53 (m, 2H), 5,95 (m, 2H), 5,40 (s, 2H), 3,80 (t, 4H), 1,99 (m, 2H), 1,71 (s, 4H), 1,49 (s, 36H), 1,36 (s, 12H), 0,80 (s, 18H); 13C-{1H}-ЯМР: 158,0, 156,1, 151,2, 147,8, 139,9, 129,1, 128,1, 128,0, 127,2, 126,0, 124,2, 123,6, 123,3, 118,2, 118,0, 116,3, 115,3, 115,2, 112,8, 112,8, 109,2, 64,9, 57,1, 38,2, 34,7, 32,4, 32,1, 31,8, 31,5, 29,0.
Синтез предкатализатора WC02

Предкатализатор WC02 получали таким же образом, как в примере 1, за исключением того, что использовали лиганд WC02L вместо лиганда WC03L.
1H ЯМР (CDCl3): δ 8,31 (s, 2H), 8,02 s, 2H), 7,56 (s, 2H), 7,50 (s, 2H), 7,41(d, 2H), 7,25 (d, 2H), 7,23(s, 2H), 6,95 (m, 2H), 6,25 (t, 2H), 4,61 (t, 2H), 3,76 (t, 2H), 3,38 (t, 2H), 2,35 (s, 4H), 1,85 (s, 4H), 1,83 (s, 36H), 1,52(s, 18H), 1,45 (s, 3H), 0,98(s, 12H), -1,65 (s, 3H)
[Сравнительный пример 3] Синтез предкатализатора WB02
Синтез лиганда WB02L
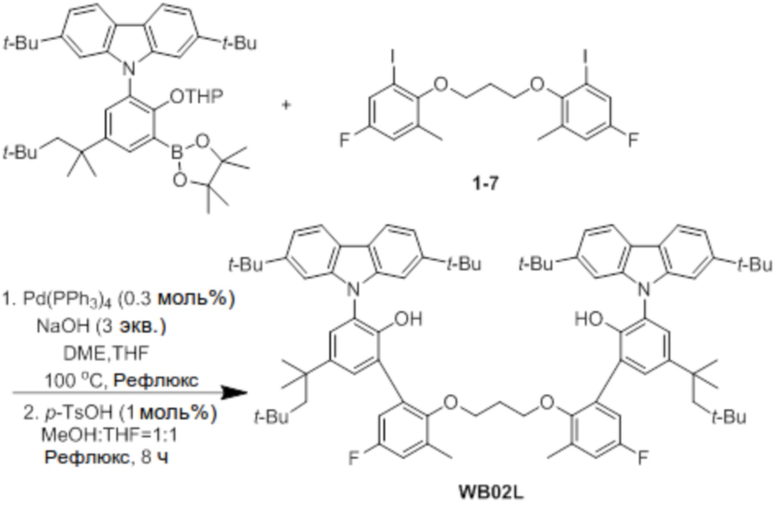
(1) Сочетание Сузуки
2,7-Ди-трет-бутил-9-(2-((тетрагидро-2H-пиран-2-ил)окси)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5-(2,4,4-триметилпентан-2-ил)фенил)-9H-карбазол (2,5 ммоль), соединение 1-7 (0,5 экв.), Pd(PPh3)4 (0,3 моль%) и NaOH (3 экв.) добавляли в двухгорлую круглодонную колбу, и создавали условия с азотом. Добавляли туда DME (40 мл), THF (20 мл) и H2O (10 мл) в указанном порядке, и нагревали реактанты с обратным холодильником при 100°C в течение 48 часов. После завершения реакции продукт охлаждали до комнатной температуры, катализатор удаляли оттуда с помощью силикагелевого фильтра, продукт перегоняли при пониженном давлении для удаления растворителя, и оставшийся растворитель удаляли с помощью вакуума.
(2) Защита THP
К остатку, полученному удалением растворителя, добавляли p-TsOH (1 моль%), добавляли туда по 100 мл MeOH и THF, и реактанты нагревали с обратным холодильником при 80°C в течение 8 часов. После завершения реакции продукт охлаждали до комнатной температуры, проводили экстракцию с помощью воды для удаления растворителя, и затем продукт отделяли и очищали с помощью колоночной хроматографии (элюент: EA/n-Hex=1/50 об/об), получая при этом лиганд WB02L в виде бесцветного твердого вещества (2,2 г, 70%).
1H ЯМР (CDCl3): δ 8,02 (d, 4H), 7,41 (d, 4H), 7,31 (s, 4H), 7,26(s, 4H), 7,06 (d, 2H), 6,85 (d, 2H), 6,21 (s, 2H), 3,61 (t, 4H), 2,05 (s, 6H), 1,92 (m, 6H), 1,42 (s, 12H), 1,27 (s, 38H), 0,79 (s, 18H); 13C-{1H}-ЯМР: 159,8, 157,8, 150,14, 150,12, 149,7, 149,1, 147,8, 142,8, 141,7, 129,1, 127,5, 126,3, 124,9, 121,0, 119,6, 119,5, 118,2, 117,7, 117,3, 117,1, 106,3, 106,2, 70,87, 57,1, 38,2, 35,2, 35,0, 32,5, 32,4, 31,9, 31,8, 31,7, 31,6, 30,7, 29,7, 16,4.
Синтез предкатализатора WB02
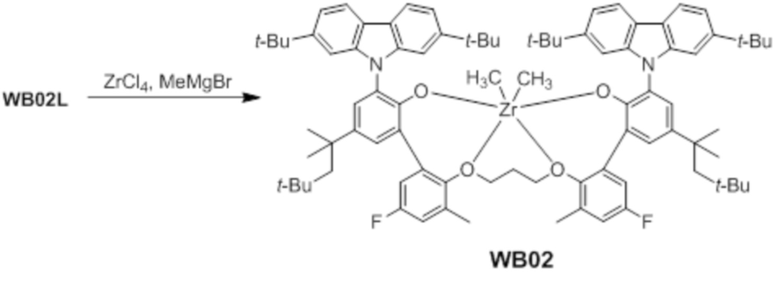
Предкатализатор WB02 получали таким же образом, как в примере 1, за исключением того, что использовали лиганд WC02L вместо лиганда WC03L.
1H ЯМР (C6D6): δ 8,25 (s, 2H), 8,03 s, 2H), 7,96 (s, 2H), 7,85 (s, 2H), 7,64 (s, 2H), 7,51(d, 2H),7,36(d, 2H), 7,31(s, 2H), 6,87 (m, 2H), 6,05 (m, 2H), 3,58 (t, 2H), 3,23 (t, 2H), 1,78 (s, 2H), 1,58 (s, 18H), 1,33 (s, 18H), 1,25 (s, 3H), 1,01(s, 3H),0,85(s, 12H).
[Сравнительный пример 4] Синтез предкатализатора WD01
Синтез лиганда WD01L
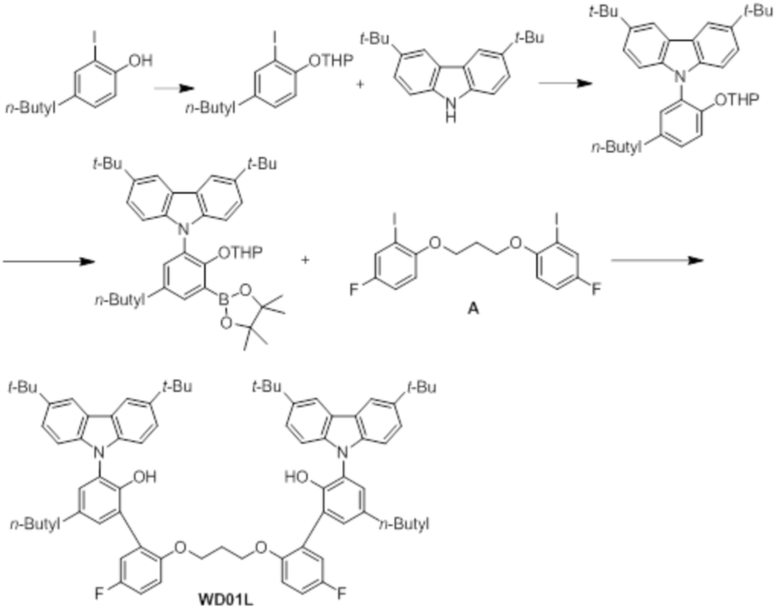
Лиганд WD01L получали таким же образом, как в примере 1, за исключением того, что использовали 2-йод-4-н-бутилфенол вместо 2-йод-4-октилфенола (соединение 3-1).
1H ЯМР (CDCl3): δ 8,20 (s, 4H), 7,41 (d, 4H), 7,26 (s, 2H), 7,06(d, 6H), 7,04 (d, 2H), 6,52 (m, 2H), 5,95 (m, 2H), 5,98 (m, 2H), 3,81 (t, 4H), 2,57 (t, 4H), 2,03 (d, 2H), 1,58 (m, 4H), 1,46 (s, 36H), 1,36 (m, 4H), 0,91 (m, 6H); 13C-{1H}-ЯМР: 158,16, 156,25, 151,24, 151,22, 148,19, 142,94, 139,93, 135,73, 131,15, 128,93, 127,99, 126,69, 124,9, 123,67, 123,52, 118,4, 118,21, 116,43, 115,52, 115,35, 113,06, 113,0, 109,51, 65,21, 34,89, 34,82, 33,79, 32,2, 28,89, 22,5, 14,01.
Синтез предкатализатора WD01
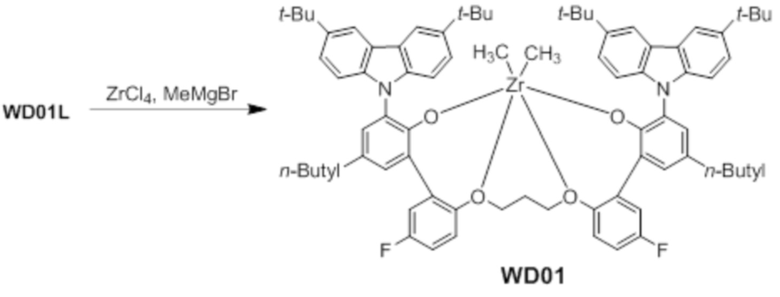
Предкатализатор WD01 получали таким же образом, как в примере 1, за исключением того, что использовали лиганд WC01L вместо лиганда WC03L.
1H ЯМР (CDCl3): δ 8,15 (s, 2H), 8,02 s, 2H), 7,56 (s, 2H), 7,50 (s, 2H), 7,23(d, 2H), 7,21 (d, 2H), 7,19(s, 2H), 6,95 (m, 2H), 6,21 (t, 2H), 4,61 (t, 2H), 3,84 (t, 2H), 3,32 (t, 2H), 2,0 ~ 1,3 (m, 42H), 1,36 (s, 4H), 0,92 (m, 6H), -1,62 (s, 6H).
[Сравнительный пример 5] Синтез предкатализатора WD02
Синтез лиганда WD02L
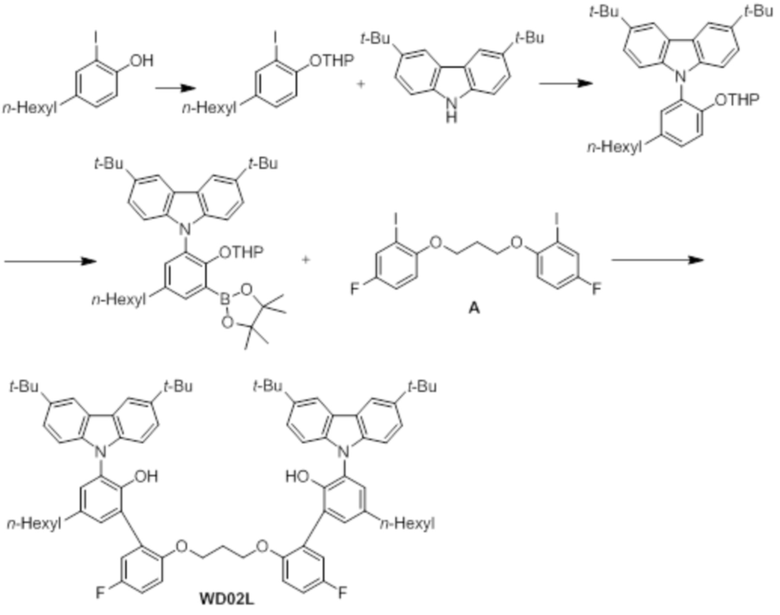
Лиганд WD02L получали таким же образом, как в примере 1, за исключением того, что использовали 2-йод-4-н-гексилфенол вместо 2-йод-4-октилфенола (соединение 3-1).
1H ЯМР (CDCl3): δ 8,20 (s, 4H), 7,43 (d, 4H), 7,28 (s, 2H), 7,08(d, 6H), 7,04 (d, 2H), 6,52 (m, 2H), 6,02 (m, 2H), 5,45 (s, 2H), 3,84 (t, 4H), 2,59 (t, 4H), 2,04 (d, 2H), 1,62 (m, 4H), 1,49 (s, 36H), 1,31 (m, 12H), 0,90 (m, 6H); 13C-{1H}-ЯМР: 158,16, 156,25, 151,24, 148,21, 142,94, 139,93, 135,78, 131,14, 128,94, 126,70, 124,89, 123,67, 123,53, 118,39, 118,21, 116,43, 115,53, 115,35, 113,07, 113,0, 109,52, 65,21, 35,14, 34,89, 32,2, 31,79, 31,59, 29,11, 29,00, 22,73, 14,21.
Синтез предкатализатора WD02
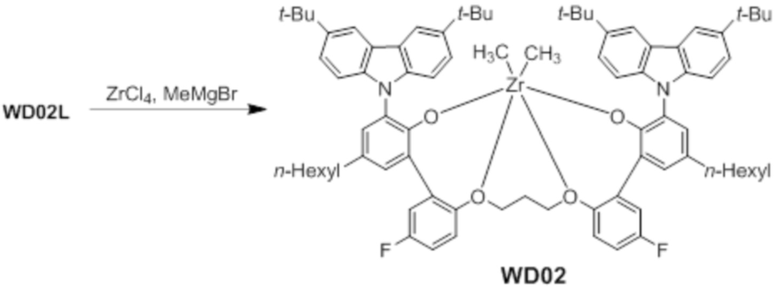
Предкатализатор WD02 получали таким же образом, как в примере 1, за исключением того, что использовали лиганд WC02L вместо лиганда WC03L.
1H ЯМР (CDCl3): δ 8,15 (s, 2H), 8,00 (s, 2H), 7,32 (s, 2H), 7,21 (s, 2H), 7,07 (d, 2H), 7,04 (d, 2H), 7,00(s, 2H), 6,95 (s, 2H), 6,51 (m, 2H), 6,21 (t, 2H), 4,61 (t, 2H), 3,84 (t, 2H), 3,32 (t, 2H), 1,61 (m, 4H), 1,5 ~ 1,3 (m, 48H), 0,92 (m, 6H), -1,62 (s, 6H).
[Пример 3] Сополимеризация этилена и 1-октена
Полимеризацию проводили в реакторе непрерывной полимеризации с регулируемой температурой, оборудованном механической мешалкой. В реактор объемом 1,0 л с непрерывным перемешиванием, предварительно нагретый до температуры 130-200°C, подавали растворитель метилциклогексан, 1-октен и этиленовый мономер под давлением 40 бар. Реакцию сополимеризации проводили путем подачи в реактор WC03 (пример 1) в качестве катализатора полимеризации и TTB (тритил тетракис-пентафторфенилборат) в качестве сокатализатора из резервуара для хранения катализатора и TiBAO (тетраизобутилалюмоксан) в качестве поглотителя. Метилциклогексан в качестве реакционного растворителя вводили в реактор в количестве 5 кг в час со временем пребывания в реакторе приблизительно 8 минут, а этилен вводили в реактор с отношением C2/MCH, установленным на 10, в количестве в диапазоне приблизительно 400-600 г в час. Полимеризацию проводили при относительно высокой температуре 180°C, 190°C и 200°C, катализатор вводили в реактор, корректируя количество вводимого катализатора на количество для поддержания разности температур между температурой подачи и температурой реактора, и давление раствора полимера, образованного в результате реакции сополимеризации, снижали до 3 бар в задней части реактора, а затем передавали его в сепаратор растворителя, тем самым удаляя большую часть растворителя в процессе отделения растворителя.
Условия реакции полимеризации и результаты полимеризации, проведенной, как описано выше, приведены ниже в таблице 1.
[Пример 4]
Способ осуществляли таким же образом, как в примере 3, за исключением того, что для полимеризации использовали предкатализатор WC04, синтезированный в примере 2. Максимальная температура 158°C была достигнута за время реакции 3 минуты, в результате чего было получено 77 г высокомолекулярного полимера, и условия реакции полимеризации и результаты полимеризации показаны ниже в таблице 1.
[Сравнительный пример 6]
Способ осуществляли таким же образом, как в примере 3, за исключением того, что в качестве предкатализатора использовали WB02, синтезированный в сравнительном примере 3. Условия и результаты реакции полимеризации показаны ниже в таблице 2.
[Сравнительный пример 7]
Способ осуществляли таким же образом, как в примере 3, за исключением того, что в качестве предкатализатора использовали WC01, синтезированный в сравнительном примере 1. Условия и результаты реакции полимеризации показаны ниже в таблице 2.
[Сравнительный пример 8]
Способ осуществляли таким же образом, как в примере 3, за исключением того, что в качестве предкатализатора использовали WC02, синтезированный в сравнительном примере 2. Условия и результаты реакции полимеризации показаны ниже в таблице 2.
[Сравнительный пример 9]
Способ осуществляли таким же образом, как в примере 3, за исключением того, что в качестве предкатализатора использовали WD01, синтезированный в сравнительном примере 4. Условия и результаты реакции полимеризации показаны ниже в таблице 2.
[Сравнительный пример 10]
Способ осуществляли таким же образом, как в примере 3, за исключением того, что в качестве предкатализатора использовали WD02, синтезированный в сравнительном примере 5. Условия и результаты реакции полимеризации показаны ниже в таблице 2.
[Таблица 1]
[Таблица 2]
(Сравнительный пример 3)
(Сравнительный пример 1)
(Сравнительный пример 2)
(Сравнительный пример 4)
(Сравнительный пример 5)
Как описано в результатах полимеризации в таблицах 1 и 2, было неожиданно обнаружено, что предкатализаторы настоящего изобретения (WC03 и WC04), в которых в R3 в химической формуле 1 введен заместитель, имеющий контролируемое число атомов углерода и определенную форму, имеют превосходные каталитическую активность и реакционную способность в отношении сомономера по сравнению с обычными катализаторами WB02, WC01, WC02, WD01 и WD02, которые являются сравнительными примерами. В частности, было обнаружено, что даже при высокотемпературной полимеризации при 180°C или более предкатализаторы примеров настоящего изобретения могут давать сополимер этилена и 1-октена с высокой молекулярной массой по сравнению с предкатализатором сравнительных примеров.
То есть в случае WC01 и WC02 молекулярная масса сополимеров, полученных сополимеризацией этилена и 1-октена при 180°C составляла 37000 г/моль и 95000 г/моль соответственно, тогда как молекулярная масса сополимеров, полученных в результате полимеризации с использованием WC03 и WC04 настоящего изобретения в качестве предкатализатора составляла 131000 г/моль и 135000 г/моль, соответственно, и, таким образом, может быть получен сополимер со значительно большей молекулярной массой по сравнению со сравнительными примерами.
Кроме того, при полимеризации при 190°C, что выше чем 180°C, катализаторы WB02 и WC02 сравнительных примеров также давали сополимеры с молекулярной массой 69000 г/моль и 67000 г/моль соответственно, тогда как сополимеры, полученные с использованием предкатализаторов настоящего изобретения (WC03 и WC04) имели высокую молекулярную массу 100000 г/моль или более.
Кроме того, при тех же условиях полимеризации при сравнении результатов полимеризации с использованием в качестве предкатализатора WD01 (сравнительный пример 9) и WD02 (сравнительный пример 10), в которых число атомов углерода в положении R3 меньше, чем 8, с результатами полимеризации с использованием в качестве предкатализатора WC03 (пример 3), который замещен н-октилом с прямой цепью с 8 атомами углерода в положении R3, WC03 настоящего изобретения имел более высокие каталитическую активность и реакционную способность в отношении сомономера при 200°C, что является высокой температурой полимеризации, тем самым демонстрируя превосходные характеристики при производстве продукта с низкой плотностью. То есть для расхода катализатора, отражающего каталитическую активность, WD01 и WD02 показали значения 2,2 и 2,3 при 200°C, в то время как WC03 настоящего изобретения показал значение 1,7, что является относительно более низким расходом катализатора, и, таким образом, отражает превосходную каталитическую активность. Кроме того, было обнаружено, что полимер, полученный при температуре полимеризации 200°C с использованием WD01 и WD02 в качестве предкатализатора, имел плотность 0,901 г/см3 и 0,900 г/см3 соответственно, тогда как полимер, полученный при тех же условиях полимеризация с использованием WD03 настоящего изобретения в качестве предкатализатора, имел более низкую плотность 0,8895 г/см3.
Как показано в приведенных выше примерах и сравнительных примерах, когда в качестве предкатализатора используют металлолигандный комплекс, который представляет собой соединение с определенной функциональной группой, введенной в определенное положение, превосходная активность неожиданно проявляется даже при высокой температуре. Кроме того, когда в качестве предкатализатора используют металлолигандный комплекс настоящего изобретения, реакционная способность в отношении олефинов является превосходной, а также полимер с высокой молекулярной массой и низкой плотностью может быть получен легко по сравнению с металл-лигандным предкатализатором сравнительного примера.
Как описано выше, хотя примеры настоящего изобретения были подробно описаны, специалист в данной области техники может внисить различные изменения в настоящее изобретение без отхода от объема настоящего изобретения, как он определен в формуле изобретения, которая следует ниже. Соответственно, любая будущая модификация примеров настоящего изобретения не может отходить от методики настоящего изобретения.
Изобретение относится к металлолигандному комплексу, выбранному из соединений, представленных следующими химическими формулами:
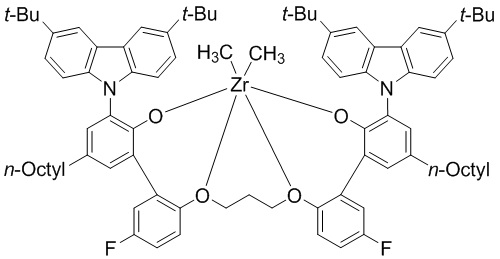
(предкатализатор WC03),
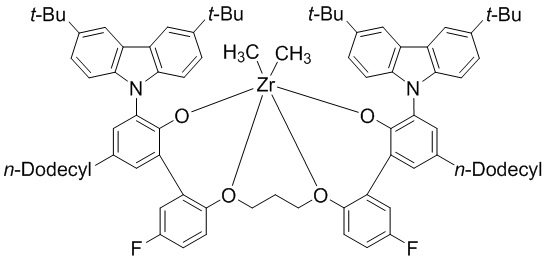
(предкатализатор WC04).
Также предложена каталитическая композиция для полимеризации на основе этилена и способ получения полимера на основе этилена с использованием указанного комплекса. Предложенные комплексы обеспечивают полимеризацию даже при высокой температуре, не снижают активность катализатора и позволяют получить сополимеры с высокой молекулярной массой. 3 н. и 3 з.п. ф-лы, 2 табл., 15 пр.
1. Металлолигандный комплекс, выбранный из соединений, представленных следующими химическими формулами:
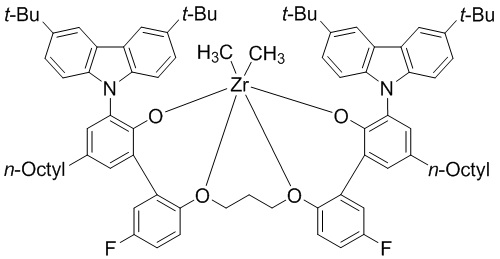
(предкатализатор WC03);
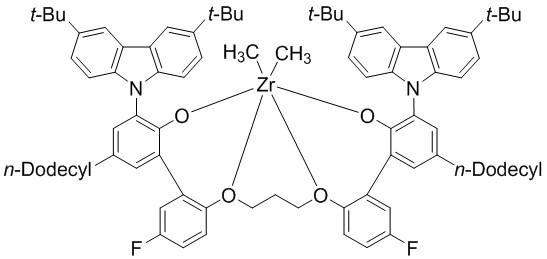
(предкатализатор WC04).
2. Каталитическая композиция для полимеризации на основе этилена, содержащая:
металлолигандный комплекс по п. 1 и
сокатализатор.
3. Каталитическая композиция по п. 2, в которой сокатализатор представляет собой сокатализатор из соединения алюминия, сокатализатор из соединения бора или их смесь.
4. Каталитическая композиция по п. 2, в которой сокатализатор используют в количестве от 0,5 до 10000 моль на 1 моль металлолигандного комплекса.
5. Способ получения полимера на основе этилена, включающий:
полимеризацию этилена или этилена и α-олефина в присутствии каталитической композиции для полимеризации на основе этилена по п. 2 с получением полимера на основе этилена.
6. Способ по п. 5, в котором полимеризацию проводят при 170-250°C.
| US 20130144018 A1, 06.06.2013 | |||
| WO 2013101375 A1, 04.07.2013 | |||
| WO 2014105415 A1, 03.07.2014 | |||
| WO 2014105411 A1, 03.07.2014 | |||
| WO 2014105412 A1, 03.07.2014 | |||
| RU 2015134543 A, 27.02.2017. |

