Настоящее изобретение относится к лиофилизированным (также называемым лиофильно высушенными) лекарственным наносуспензиям. Композиция лиофилизированной лекарственной наносуспензии по настоящему изобретению обладает приемлемой стабильностью гранулометрического состава при хранении, в том числе при длительном хранении. Стабильность гранулометрического состава при длительном хранении включает стабильность в течение периода 2 недели, в частности, периода один месяц, более конкретно, периода два месяца, и еще более конкретно, периода три месяца или периода шесть месяцев, или периода от по меньшей мере шести месяцев до двух лет. Стабильность гранулометрического состава при длительном хранении является важным свойством, поскольку она представляет собой неотъемлемую часть каждой процедуры разработки состава.
Наночастицы или наносуспензии известны из предшествующего уровня техники и были описаны, например, в EP-A-0499299.
Состав лекарственной наносуспензии, который является составом-предшественником для состава лиофилизированной лекарственной наносуспензии по настоящему изобретению, представляет собой жидкий состав, где лекарственное средство суспендировано в форме наночастиц. Жидкая или дисперсионная среда представляет собой предпочтительно водную среду, такую как вода.
Состав-предшественник лекарственной наносуспензии обычно содержит стерический стабилизатор, предпочтительно, поверхностно-активное вещество (например, полимерное поверхностно-активное вещество) или полимер. Стерический стабилизатор адсорбируется или прикрепляется на поверхности лекарственных наночастиц и обеспечивает широкий и плотный стерический барьер, препятствующий силам ван-дер-ваальсового притяжения, и, следовательно, стерический стабилизатор уменьшает агрегацию, агломерацию или даже слияние частиц. Эта методика допускает исключительно высокие дозовые нагрузки наносуспензии (например, вплоть до 400 мг лекарственного средства/мл). Стерические стабилизаторы предпочтительно представляют собой наполнители, которые являются фармацевтически приемлемыми.
Наночастицы лекарственного средства, как правило, имеют среднюю величину размера частиц меньше 1 мкм и d99-величину меньше 5 мкм. Более конкретно, d95-величина составляет 0,9 мкм (d99 составляет X, или d95 составляет X означает, что 99% или 95% частиц по весу (или по другим подходящим методикам измерения, как например, по объему или числу) меньше этого размера X).
В частности, средний размер частиц или наночастиц наносуспензий, подлежащих лиофилизации, или наносуспензии, полученной в результате восстановления лиофилизированной лекарственной наносуспензии, может быть меньше примерно 1000 нм, или меньше примерно 500 нм, или меньше примерно 400 нм, или меньше примерно 300 нм, или меньше примерно 200 нм. Предпочтительно, средний размер частиц составляет примерно 200 нм, или примерно 400 нм, или примерно 800 нм, более предпочтительно, примерно 200 нм.
Возможный подход к получению лекарственной наносуспензии заключается в превращении в порошок субстанции лекарственного средства в мельнице с перемешиваемой средой посредством мокрого измельчения с помощью шариков.
Лекарственные наносуспензии могут улучшать растворимость, скорость растворения, биологическую доступность. Лекарственные наносуспензии также могут применяться как состав с замедленным или отсроченным высвобождением (депо). Такие составы могут применяться для длительного лечения или длительной профилактики, например, при введении парентерально, например, внутримышечно или подкожно. Это означает, что состав может обеспечивать эффективные уровни в плазме крови - уровни в плазме крови, выше минимальной терапевтической концентрации - в течение определенного периода, например, в течение по меньшей мере 1 недели, или по меньшей мере двух недель, или по меньшей мере 1 месяца, или по меньшей мере двух месяцев, или по меньшей мере трех месяцев. Состав также может обеспечивать уровень в плазме крови, который ниже пороговой величины, производящей побочный эффект. Пороговая величина означает уровень в плазме крови в течение значительного периода времени, например, в течение больше 15 минут, в зависимости от конкретного лекарственного средства, выше которой пациенты могут испытывать нежелательные побочные эффекты, или наоборот, означает величину уровня в плазме крови, ниже которой общая переносимость состава, о котором идет речь, остается приемлемой. Пороговой величиной не считаются временные, высокие уровни в плазме крови в течение короткого периода времени, например, в течение меньше 15 минут, в зависимости от конкретного лекарственного средства, которые вызваны, например, неожиданным “взрывным высвобождением” активного ингредиента.
Оба вышеприведенные признаки - уровни в плазме крови выше минимальной терапевтической концентрации, но ниже пороговой величины, производящей побочной эффект - считаются основными требованиями, которым должен удовлетворять современный состав-депо, чтобы быть приемлемым для предусматриваемых пациентов. Ограничение числа введений лекарственного средства и частоты нежелательных побочных эффектов после каждого введения несомненно улучшит соблюдение пациентом терапии. Однако, кроме этих основных требований, можно отметить ряд дополнительных требований, которые будут дополнительно улучшать соблюдение пациентами схемы приема лекарственных средств, при этом два наиболее заметных представляют собой местную переносимость и легкость введения.
Что касается инъецируемых препаратов, хорошая местная переносимость означает минимальное раздражение и воспаление в месте инъекции; легкость введения относится к размеру иглы и промежутку времени, требуемому для введения дозы конкретного лекарственного состава.
Существенным недостатком жидкой лекарственной наносуспензии является ее ограниченная длительная стабильность. Оседание или эффекты Оствальдовского созревания представляют собой общепризнанные проблемы нестабильности.
Поэтому, лиофилизированные лекарственные наносуспензии могут представлять собой привлекательную альтернативу. В этом случае, срок годности состава с лекарственным средством в диапазоне наночастиц может быть увеличен. Предпочтительно, лиофилизированная лекарственная наносуспензия сама по себе обладает приемлемой длительной стабильностью, особенно с точки зрения среднего размера частиц или гранулометрического состава. После хранения и непосредственно после восстановления лиофилизированной лекарственной наносуспензии средний размер частиц, гранулометрический состав, d50, d90, d95 или d99 сохраняется или остается приемлемым.
Эти лиофилизированные лекарственные наносуспензии можно восстанавливать по мере надобности в жидкую наносуспензию, которую затем можно вводить. Введение указанной восстановленной наносуспензии включает пероральное введение или парентеральное введение, такое как, например, внутривенное, внутримышечное или подкожное введение. Введение представляет собой, предпочтительно, парентеральное введение, такое как, например, внутримышечное или подкожное введение. Следует обратить внимание, что восстановленная суспензия остается равномерно диспергированной, или что ее легко диспергировать при встряхивании, что предоставляет возможность для введения однородной суспензии.
Графические материалы
Фигура 1: Сравнение гранулометрических составов исходной лекарственной наносуспензии, стабилизированной с помощью 50 мг/мл Cremophor EL, непосредственно после лиофилизации и на стадии 3 месяцев хранения при условиях окружающей среды (штриховая линия). Граница 5 мкм представляет собой верхний предел размера частиц для парентеральной лекарственной наносуспензии.
Фигура 2: Сравнение гранулометрических составов исходной наносуспензии рилпивирина, стабилизированной с помощью 50 мг/мл полоксамера 338, непосредственно после лиофилизации и на стадии 3 месяцев хранения при условиях окружающей среды (штриховая линия).
Фигура 3: Сравнение гранулометрических составов исходной лекарственной наносуспензии, стабилизированной с помощью 50 мг/мл Cremophor EL, после агрессивных условий способа лиофилизации (сплошная линия, температуру полки при первичной сушке после замораживания сразу повысили до 40°C) и умеренных условий процесса лиофилизации (штриховая линия, температура полки 0°C). Следует обратить внимание, что при обоих режимах измеренные температуры продукта были значительно выше CFT (критической температуры состава, которая обозначается как температура стеклования максимально замороженного концентрированного раствора Tg', как правило, оцениваемой с помощью дифференциальной сканирующей калориметрии (DSC) (Pikal M J. 2002. Freeze drying, in: J. Swarbrick (Ed.), Encyclopedia of Pharmaceutical Technology, vol. 2, Marcel Dekker, New York (2002) 1299-1326).
Фигура 4: Сравнение индексов повторной диспергируемости (RDI) для исходных лекарственных наносуспензий после 3 месяцев хранения при условиях окружающей среды, стабилизированных либо с помощью Cremophor EL (Cr EL), либо с помощью полоксамера 338 (P338), в зависимости от высокого или низкого содержания остаточной влажности (R. M.).
Фигура 5: Сравнение гранулометрических составов лекарственной наносуспензии, стабилизированной с помощью 50 мг/мл Cremophor EL, и с 50 мг/мл трегалозы в качестве лиопротектора, непосредственно после лиофилизации и на стадии 3 месяцев хранения при условиях окружающей среды, в зависимости от высокого или низкого содержания остаточной влажности. Следует обратить внимание, что изначальная стабильность наночастиц не зависела от влагосодержания (сплошная в сравнении с пунктирной линией), но после хранение содержание остаточной влажности ниже 1% (штриховая линия) показало лучшую стабильность частиц по сравнению с влагосодержанием выше 1% (штрих-пунктирная линия).
Фигура 6: Сравнение гранулометрических составов высококонцентрированной лекарственной наносуспензии (200 мг/мл рилпивирина), стабилизированной с помощью 50 мг/мл полоксамера 338, и с 50 мг/мл PVP K15 в качестве лиопротектора показывает, что первоначальный гранулометрический состав после лиофилизации (сплошная линия) полностью сохранился через 3 месяца хранения при 25°C (штриховая линия), а также при 40°C (пунктирная линия).
Описание изобретения
Таким образом, настоящее изобретение относится к лиофилизированной лекарственной наносуспензии, под которой понимают лиофилизированную наносуспензию, содержащую лекарственное средство, или водную наносуспензию, содержащую лекарственное средство, лиофилизированную до твердой композиции.
Подходящие лекарственные средства или активные фармацевтические ингредиенты, которые могут применяться в лиофилизированных наносуспензиях по настоящему изобретению, включают без ограничения:
- анальгезирующие и противовоспалительные лекарственные средства (NSAID, фентанил, индометацин, ибупрофен, кетопрофен, набуметон, парацетамол, пироксикам, трамадол, ингибиторы ЦОГ-2, такие как целекоксиб и рофекоксиб);
- антиаритмические лекарственные средства (прокаинамид, хинидин, верапамил);
- антибактериальные и антипротозойные средства (амоксициллин, ампициллин, бензатин пенициллин, бензилпенициллин, цефаклор, цефадроксил, цефпрозил, цефуроксим аксетил, цефалексин, хлорамфеникол, хлорохин, ципрофлоксацин, кларитромицин, клавулановая кислота, клиндамицин, доксициклин, эритромицин, флуклоксациллин натрия, галофантрин, изониазид, канамицина сульфат, линкомицин, мефлохин, миноциклин, нафциллин натрия, налидиксовая кислота, неомицин, норфлоксацин, офлоксацин, оксациллин, феноксиметилпенициллин калия, пириметамин-сульфадоксим, стрептомицин);
- антикоагулянты (варфарин);
- антидепрессанты (амитриптилин, амоксапин, бутриптилин, кломипрамин, дезипрамин, дотиепин, доксепин, флуоксетин, ребоксетин, аминептин, селегилин, гепирон, имипрамин, карбонат лития, миансерин, милнаципран, нортриптилин, пароксетин, сертралин; 3-[2-[3,4-дигидробензофуро[3,2-c]пиридин-2(1H)-ил]этил]-2-метил-4H-пиридо[1,2-a]пиримидин-4-он);
- антидиабетические лекарственные средства (глибенкламид, метформин);
- противоэпилептические лекарственные средства (карбамазепин, клоназепам, этосуксимид, габапентин, ламотриджин, леветирацетам, фенобарбитал, фенитоин, примидон, тиагабин, топирамат, вальпромид, вигабатрин);
- противогрибковые средства (амфотерицин, клотримазол, эконазол, флуконазол, флуцитозин, гризеофульвин, итраконазол, кетоконазол, миконазола нитрат, нистатин, тербинафин, вориконазол);
- антигистаминные средства (астемизол, циннаризин, ципрогептадин, декарбоэтоксилоратадин, фексофенадин, флунаризин, левокабастин, лоратадин, норастемизол, оксатомид, прометазин, терфенадин);
- гипотензивные лекарственные средства (каптоприл, эналаприл, кетансерин, лизиноприл, миноксидил, празозин, рамиприл, резерпин, теразозин);
- антимускариновые средства (атропина сульфат, гиосцин);
- противоопухолевые средства и антиметаболиты (соединения платины, такие как цисплатин, карбоплатин; таксаны, такие как паклитаксел, доцетаксел; теканы, такие как камптотецин, иринотекан, топотекан; алкалоиды барвинка, такие как винбластин, виндецин, винкристин, винорелбин; производные нуклеозидов и антагонисты фолиевой кислоты, такие как 5-фторурацил, капецитабин, гемцитабин, меркаптопурин, тиогуанин, кладрибин, метотрексат; алкилирующие средства, такие как азотистые иприты, например, циклофосфамид, хлорамбуцил, хлорметин, ифосфамид, мелфалан, или нитрозомочевины, например, кармустин, ломустин, или другие алкилирующие средства, например, бусульфан, дакарбазин, прокарбазин, тиотепа; антибиотики, такие как даунорубицин, доксорубицин, идарубицин, эпирубицин, блеомицин, дактиномицин, митомицин; антитела к HER 2, такие как трастузумаб; производные подофиллотоксина, такие как этопозид, тенипозид; ингибиторы фарнезилтрансферазы; производные антрахинона, такие как митоксантрон; абиратерон и его сложные эфиры, такие как абиратерона ацетат);
- лекарственные средства против мигрени (алнидитан, наратриптан, суматриптан);
- лекарственные средства против болезни Паркинсона (бромокриптина мезилат, леводопа, селегилин);
- антипсихотические, снотворные и седативные средства (алпразолам, буспирон, хлордиазепоксид, хлорпромазин, клозапин, диазепам, флупентиксол, флуфеназин, флуразепам, 9-гидроксирисперидон, лоразепам, мазапертин, оланзапин, оксазепам, пимозид, пипамперон, пирацетам, промазин, рисперидон, селфотел, сероквел, сертиндол, сульпирид, темазепам, тиотиксен, триазолам, трифлуперидол, зипразидон, золпидем);
- противоинсультные средства (лубелузол, лубелузола оксид, рилузол, аптиганел, элипродил, ремацемид);
- противокашлевые препараты (декстрометорфан, леводропропизин);
- противовирусные препараты (ацикловир, ганцикловир, ловирид, тивирапин, зидовудин, ламивудин, зидовудин + ламивудин, диданозин, залцитабин, ставудин, абакавир, лопинавир, ампренавир, невирапин, эфавиренз, делавирдин, индинавир, нелфинавир, ритонавир, саквинавир, адефовир, гидроксимочевина, этравирин, дапивирин, рилпивирин, дарунавир, тенофовир или тенофовира дизопроксил фумарат, эмтрицитабин);
- блокаторы бета-адренорецептора (атенолол, карведилол, метопролол, небиволол, пропанолол);
- сердечные инотропные средства (амринон, дигитоксин, дигоксин, милринон);
- кортикостероиды (беклометазона дипропионат, бетаметазон, будезонид, дексаметазон, гидрокортизон, метилпреднизолон, преднизолон, преднизон, триамцинолон);
- дезинфицирующие средства (хлоргексидин);
- диуретики (ацетазоламид, фрусемид, гидрохлортиазид, изосорбид);
- эфирные масла (анетол, анисовое масло, тмин, кардамон, масло кассии, цинеол, коричное масло, гвоздичное масло, кориандровое масло, дементолизированное масло мяты, укропное масло, эвкалиптовое масло, эвгенол, имбирь, лимонное масло, горчичное масло, неролиевое масло, масло мускатного ореха, апельсиновое масло, масло мяты перечной, шалфей, мята колосковая, терпинеол, тимьян);
- желудочно-кишечные средства (циметидин, цизаприд, клебоприд, дифеноксилат, домперидон, фамотидин, лансопразол, лоперамид, лоперамида оксид, месалазин, метоклопрамид, мозаприд, низатидин, норцизаприд, олсалазин, омепразол, пантопразол, перпразол, прукалоприд, рабепразол, ранитидин, ридогрел, сульфасалазин);
- гемостатические средства (аминокапроновая кислота);
- средства, регулирующие уровень липидов (аторвастатин, ловастатин, правастатин, пробукол, симвастатин);
- местные анестетики (бензокаин, лигнокаин);
- опиоидные анальгетики (бупренорфин, кодеин, декстроморамид, дигидрокодеин, гидрокодон, оксикодон, морфин);
- парасимпатомиметики и лекарственные средства против деменции (AIT-082, эптастигмин, галантамин, метрифонат, миламелин, неостигмин, физостигмин, такрин, донепезил, ривастигмин, сабкомелин, талсаклидин, ксаномелин, мемантин, лазабемид);
- половые гормоны (эстрогены: конъюгированные эстрогены, этинилэстрадиол, местранол, эстрадиол, эстриол, эстрон; прогестогены; хлормадинона ацетат, ципротерона ацетат, 17-деацетил норгестимат, дезогестрел, диеногест, дидрогестерон, этинодиола диацетат, гестоден, 3-кетодезогестрел, левоноргестрел, линестренол, медроксипрогестерона ацетат, мегестрол, норэтиндрон, норэтиндрона ацетат, норэтистерон, норэтистерона ацетат, норэтинодрел, норгестимат, норгестрел, норгестриенон, прогестерон, квингестанола ацетат);
- стимулирующие средства (силденафил);
- вазодилататоры (амлодипин, буфломедил, амилнитрит, дилтиазем, дипиридамол, глицерилтринитрат, изосорбида динитрат, лидофлазин, молсидомин, никардипин, нифедипин, окспентифиллин, пентаэритритола тетранитрат);
в том числе их стереохимически изомерные формы;
их N-оксиды, их фармацевтически приемлемые соли присоединения кислоты или основания, или их сольваты.
Фармацевтически приемлемые соли присоединения кислоты включают формы солей присоединения кислоты, которые легко можно получить путем обработки формы основания активного ингредиента соответствующими органическими и неорганическими кислотами, например, галогенводородными кислотами, к примеру, хлористоводородной, бромистоводородной и т.п.; серной кислотой; азотной кислотой; фосфорной кислотой и т.п.; или органическими кислотами, например, уксусной, пропионовой, гидроксиуксусной, 2-гидроксипропионовой, 2-оксопропионовой, щавелевой, малоновой, янтарной, малеиновой, фумаровой, яблочной, винной, 2-гидрокси-1,2,3-пропантрикарбоновой, метансульфоновой, этансульфоновой, бензолсульфоновой, 4-метилбензолсульфоновой, циклогексансульфаминовой, 2-гидроксибензойной, 4-амино-2-гидроксибензойной кислотами и подобными кислотами. И, наоборот, при обработке щелочью солевую форму можно превратить в форму свободного основания.
Активные ингредиенты, содержащие кислотные протоны, можно превращать в их терапевтически активные формы солей присоединения нетоксичного металла или амина путем обработки соответствующими органическими и неорганическими основаниями. Соответствующие формы солей основания включают, например, соли аммония, соли щелочных и щелочно-земельных металлов, например, соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например, с первичными, вторичными и третичными алифатическими и ароматическими аминами, такими как метиламин, этиламин, пропиламин, изопропиламин, четыре изомера бутиламина, диметиламин, диэтиламин, диэтаноламин, дипропиламин, диизопропиламин, ди-н-бутиламин, пирролидин, пиперидин, морфолин, триметиламин, триэтиламин, трипропиламин, хинуклидин, пиридин, хинолин и изохинолин, бензатин, N-метил-D-глюкамин, 2-амино-2-(гидроксиметил)-1,3-пропандиол, гидрабаминовые соли и соли с аминокислотами, такими как, например, аргинин, лизин и т.п. И, наоборот, при обработке кислотой солевую форму можно превращать в форму свободного основания.
Термин “сольваты” включает гидраты и формы присоединения растворителя, которые способны образовывать активные ингредиенты или их фармацевтически приемлемые соли. Примерами таких форм являются, например, гидраты, алкоголяты и т.п.
Формы N-оксидов активных ингредиентов включают такие активные ингредиенты, где один или несколько третичных атомов азота окислены до так называемого N-оксида.
Термин “стехиометрически изомерные формы” обозначает все возможные стереоизомерные формы, которыми могут обладать активные ингредиенты. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию, а активные ингредиенты, содержащие одну или несколько двойных связей, могут иметь E- и Z-конфигурацию.
В одном варианте осуществления лекарственное средство или активный фармацевтический ингредиент представляет собой противогрибковое средство, такое как, например итраконазол, или противовирусное средство, в частности, средство против ВИЧ, более конкретно ненуклеозидный ингибитор обратной транскриптазы (NNRTI), такой как, например, дапивирин, этравирин или рилпивирин.
Концентрация лекарственного средства в наносуспензии, подлежащей лиофилизации, может изменяться в диапазоне от 1 до 500 мг/мл, или от 1 до 400 мг/мл, или от 50 до 200 мг/мл, или от 50 до 100 мг/мл, или от 10 до 100 мг/мл, или от 10 до 75 мг/мл, или от 10 до 50 мг/мл, или от 20 до 50 мг/мл, или составляет примерно 200 мг/мл или составляет примерно 300 мг/мл.
Высокая концентрация наночастиц улучшает механическую стабильность лиофилизата.
Лиофилизированную лекарственную наносуспензию можно восстанавливать до жидкой лекарственной наносуспензии с такой же концентрацией лекарственного средства, что и в наносуспензии, которую лиофилизировали, или с отличающейся концентрацией лекарственного средства (более или менее концентрированной).
В одном варианте осуществления настоящее изобретение относится к лиофилизированной лекарственной наносуспензии, где лекарственное средство является малорастворимым, очень малорастворимым или практически нерастворимым согласно USP 33, общие указания, 5. Компоненты фармакопейной статьи (USP 33, general notes, 5. Monograph components). В частности, малорастворимое, очень малорастворимое или практически нерастворимое лекарственное средство выбрано из малорастворимых, очень малорастворимых или практически нерастворимых лекарственных средств, перечисленных в списке выше. Предпочтительные лекарственные средства выбирают из итраконазола, этравирина, дапивирина, рилпивирина. Лекарственным средством предпочтительно является активный фармацевтический ингредиент с небольшой химической молекулой (отличающийся от крупной молекулы, такой как, например, пептид, или белок, или последовательность ДНК/РНК).
В одном варианте осуществления настоящее изобретение относится к лиофилизированной наносуспензии, содержащей лекарственное средство, в частности, малорастворимое, очень малорастворимое или практически нерастворимое лекарственное средство, и дополнительно содержащей стерический стабилизатор. В одном варианте осуществления стерический стабилизатор представляет собой твердое вещество при комнатной температуре.
В одном варианте осуществления стерический стабилизатор представляет собой кристаллическое твердое вещество при комнатной температуре, в частности, стерический стабилизатор представляет собой кристаллическое твердое вещество при комнатной температуре и характеризуется температурой плавления, равной или выше 30°C, или температурой плавления, равной или выше 50°C, или температурой плавления, равной или выше 75°C, или температурой плавления, равной или выше 90°C.
В одном варианте осуществления стерический стабилизатор представляет собой аморфное твердое вещество при комнатной температуре, в частности, стерический стабилизатор представляет собой аморфное твердое вещество при комнатной температуре и обладает температурой стеклования (Tg), равной или выше 30°C, или Tg, равной или выше 50°C, или Tg, равной и выше 75°C, или Tg, равной или выше 90°C.
Стерический стабилизатор обеспечивает приемлемую стабильность гранулометрического состава при хранении, в том числе при длительном хранении лиофилизированной лекарственной наносуспензии. Было обнаружено, что скорость замораживания не является решающим фактором для сохранения первоначального гранулометрического состава наночастиц в лиофилизированной лекарственной наносуспензии.
Определение стабильности гранулометрического состава лиофилизированной лекарственной наносуспензии можно выполнить путем расчета индекса повторной диспергируемости (RDI) или путем определения величин d50, или d90, или d99 непосредственно после лиофилизации (T0) и после периода хранения, например, через 1, или 2, или 3 месяца хранения (T1, T2, T3). RDI определяется как D0/D, где D0 представляет собой средневзвешенную по объему среднюю величину размера частиц после лиофилизации в T0, а D представляет собой соответствующую величину после периода хранения, например, через 3 месяца хранения. Следовательно, RDI 100% будет означать, что хранимая лиофилизированная лекарственная наносуспензия может полностью превращаться в частицы исходного размера при T0 после регидратации. Лиофилизированная лекарственная наносуспензия обладает приемлемой стабильностью в отношении размера частиц при хранении, если RDI через 3 месяца при 25°C составляет по меньшей мере 90%, в частности, по меньшей мере 92%, или 94%, или 96%, или 98%. В частности, лиофилизированная лекарственная наносуспензия обладает приемлемой стабильностью в отношении размера частиц при хранении, если RDI через 3 месяца при 40°C составляет по меньшей мере 90%, в частности, по меньшей мере 92%, или 94%, или 96%, или 98%.
В одном варианте осуществления стерический стабилизатор представляет собой твердое, кристаллическое или аморфное вещество при комнатной температуре и является полимером или поверхностно-активным веществом (например, полимерным поверхностно-активным веществом). В предпочтительном варианте осуществления стерический стабилизатор представляет собой полоксамер 338, особенно в лиофилизированных наносуспензиях для составов для парентерального введения. В предпочтительном варианте осуществления стерический стабилизатор представляет собой гидроксипропил метилцеллюлозу, особенно в лиофилизированных наносуспензиях для перорального введения.
Концентрация стерического стабилизатора в наносуспензии, подлежащей лиофилизации, может находиться в диапазоне от 1 до 200 мг/мл, или от 10 до 100 мг/мл, или от 10 до 75 мг/мл, или от 10 до 50 мг/мл, или от 20 до 50 мг/мл или составлять примерно 33,3 мг/мл или примерно 50 мг/мл.
В одном варианте осуществления настоящее изобретение относится к лиофилизированной лекарственной наносуспензии, содержащей стерический стабилизатор, как описано в любом из вышеприведенных вариантов осуществления, и дополнительно содержащей криопротектор или лиопротектор. Криопротектор представляет собой соединение, которое стабилизирует компоненты, подлежащие лиофилизации, во время этапа замораживания. Лиопротектор представляет собой соединение, которое стабилизирует компоненты, подлежащие лиофилизации, во время этапа дегидратации. Многие наполнители могут служить как криопротекторами, так и лиопротекторами.
В одном варианте осуществления криопротектором или лиопротектором является сахарид, в частности, моно- или дисахарид, такой как, например, сахароза, трегалоза, маннит.
В одном варианте осуществления криопротектором или лиопротектором является полимер, такой как, например, поливинилпирролидон, а именно, PVP K12, PVP K15 или PVP K17, при этом предпочтительными являются PVP K15 и PVP K17.
В одном варианте осуществления криопротектором или лиопротектором является смесь сахарида и полимера, например, смесь PVP и трегалозы.
Криопротектор или лиопротектор могут дополнительно повышать стабильность гранулометрического состава при хранении, в том числе при длительном хранении лиофилизированной лекарственной наносуспензии.
Концентрация криопротектора или лиопротектора в наносуспензии, подлежащей лиофилизации, может находиться в диапазоне от 1 до 200 мг/мл, или от 10 до 100 мг/мл, или от 10 до 75 мг/мл, или от 10 до 50 мг/мл, или от 20 до 50 мг/мл или составляет примерно 12,5 мг/мл, или примерно 25 мг/мл, или примерно 50 мг/мл, или примерно 75 мг/мл.
В предпочтительном варианте осуществления концентрация лиопротектора или криопротектора является насколько возможно низкой для предотвращения сморщивания или разрушения лиофилизата, но достаточно высокой для обеспечения стабилизации наночастиц, а концентрация стерического стабилизатора является минимальной концентрацией, которая, как правило, выше минимальной концентрации, необходимой для достижения стабильности первоначальных наночастиц непосредственно после измельчения для получения наносуспензии.
В одном варианте осуществления лиофилизированная лекарственная наносуспензия, как описано в любом из вышеприведенных вариантов осуществления, имеет содержание остаточной влажности, равное или ниже 2% вес/вес, или равное или ниже 1% вес/вес, или равное или ниже 0,5% вес/вес. Такое ограниченное содержание остаточной влажности дополнительно улучшает стабильность гранулометрического состава при хранении, в том числе при длительном хранении лиофилизированной наносуспензии.
В одном варианте осуществления настоящее изобретение относится к лиофилизированной лекарственной наносуспензии, как описано в любом из вышеприведенных вариантов осуществления, для применения в получении лекарственного препарата, в частности, жидкой лекарственной наносуспензии, более конкретно водной лекарственной наносуспензии, для перорального или парентерально введения, в частности, парентерального введения, более конкретно подкожного или внутримышечного введения.
Настоящее изобретение также относится к водной наносуспензии, получаемой путем восстановления лиофилизированной лекарственной наносуспензии, как описано в данном документе выше или ниже, с помощью жидкой или дисперсионной среды, в частности, с помощью водной дисперсионной среды, например, воды или воды для инъекций.
Настоящее изобретение также относится к способу получения жидкой наносуспензии, в частности, водной наносуспензии, отличающемуся восстановлением лиофилизированной лекарственной наносуспензии, как описано в данном документе выше или ниже, с помощью жидкой или дисперсионной среды, в частности, с помощью водной дисперсионной среды.
Лиофилизированная лекарственная наносуспензия по настоящему изобретению может также дополнительно содержать фармацевтически приемлемые ингредиенты. Последние включают любые ингредиенты для применения в инъецируемых составах, или в суспензионных составах, или в пероральных составах. Эти ингредиенты можно выбирать из одного или нескольких из суспендирующего средства, буфера, средства, регулирующего рН, консерванта, изотонирующего средства и подобных ингредиентов. В одном варианте осуществления указанные ингредиенты выбирают из одного или нескольких из суспендирующего средства, буфера, средства, регулирующего рН, и, необязательно, консерванта и изотонирующего средства. Конкретные ингредиенты могут действовать в качестве двух или более из таких средств одновременно, например, ведут себя подобно консерванту и буферу или ведут себя подобно буферу и изотонирующему средству.
Примеры фармацевтически приемлемых ингредиентов дополнительно описаны ниже.
В одном варианте осуществления настоящее изобретение относится к лиофилизированной наносуспензии, как описано в любом из вышеприведенных вариантов осуществления, где лекарственное средство представляет собой лекарственное средство против ВИЧ, в частности, ненуклеозидный ингибитор обратной транскриптазы (NNRTI), такой как, например, дапивирин, этравирин или рилпивирин, в частности, рилпивирин. Указанная наносуспензия может предназначаться для применения в получении лекарственного препарата, в частности, жидкой наносуспензии средства против ВИЧ, более конкретно водной наносуспензии лекарственного средства против ВИЧ для парентерального введения, в частности, подкожного или внутримышечного введения, в частности, для длительной профилактики или длительного лечения ВИЧ-инфекции.
Вследствие фармакокинетических свойств и необходимости поддерживать уровни в плазме крови выше минимального уровня, для применяемых в настоящее время лекарственных средств против ВИЧ требуется частое введение относительно высоких доз. Число и/или объем лекарственных форм, которые необходимо ввести, обычно рассматривают как показатель общего количества применяемых единиц дозирования (так называемый "pill burden"). Высокий "pill burden" нежелателен по многим причинам, таким как частота приема, зачастую сочетающаяся с неудобством, заключающимся в глотании большого количества лекарственных форм, а также необходимостью хранить и транспортировать большое количество или объем пилюль. Высокий "pill burden" повышает вероятность того, что пациенты не будут принимать их полную дозу, тем самым будут не способны соблюдать предписанную схему приема лекарственных средств. Помимо снижения эффективности лечения, это также приводит к появлению вирусной устойчивости. Проблемы, ассоциированные с высоким "pill burden", выражены в терапии против ВИЧ, где пациент должен принимать большое количество различных средств против ВИЧ.
Поэтому, было бы целесообразно обеспечить ВИЧ-ингибирующую терапию, которая снижает "pill burden", поскольку она предполагает введение лекарственных форм с относительно небольшим размером, и, кроме того, не требуется частый прием. Заманчиво было бы обеспечить терапию против ВИЧ, включающую введение лекарственных форм на длительные промежутки времени, такие как одна неделя или дольше, или даже один месяц или дольше.
На сегодняшний день нельзя полностью избавиться от ВИЧ, так что люди, инфицированные ВИЧ, представляют собой постоянную угрозу инфицирования других людей. После первичного инфицирования до проявления первых симптомов СПИДа проходит длительное время. Люди могут годами жить с инфекцией, не испытывая каких-либо ее воздействий, из-за чего не подозревают о риске дальнейшего переноса вируса в других людей. Поэтому профилактика передачи ВИЧ является крайне важной. На сегодняшний день профилактика направлена на то, чтобы избежать передачи половым путем, в частности, с помощью использования презервативов группой риска в отношении инфицирования ВИЧ, на тщательный контроль проб крови на наличие ВИЧ и на то, чтобы избежать контакта с кровью потенциально инфицированных субъектов.
Несмотря на эти меры, всегда существует неизбежный риск заражения отдельных лиц, контактирующих с ВИЧ-инфицированными людьми. В частности, это касается тех, кто оказывает медицинскую помощь инфицированным пациентам или пациентам, подверженным риску заразиться, а именно врачей, медсестер или стоматологов.
В связи с этим существует потребность в дополнительных средствах, обеспечивающих профилактику передачи ВИЧ. Существует особая потребность в эффективных средствах профилактики, которые были бы простыми в применении. Обеспечение таких средств профилактики представляет собой другую цель настоящего изобретения.
Следовательно, один вариант осуществления настоящего изобретения представляет собой лиофилизированную наносуспензию, как описано в любом из вышеприведенных вариантов осуществления, лекарственного средства против ВИЧ с 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилом, или его стереоизомерной формой, или его фармацевтически приемлемой солью.
Один вариант осуществления настоящего изобретения представляет собой лиофилизированную наносуспензию, как описано, во всех возможных случаях, в любом из вышеприведенных вариантов осуществления, лекарственного средства против ВИЧ с 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрилом или его стереоизомерной формой; или его фармацевтически приемлемой солью, при этом указанная лиофилизированная наносуспензия дополнительно содержит стерический стабилизатор, в частности, полоксамер 338, и дополнительно необязательно содержит криопротектор, в частности, поливинилпирролидон, более конкретно PVP K15 или PVP K17. Что касается концентраций лекарственного средства против ВИЧ, стерического стабилизатора и криопротектора в наносуспензии, подлежащей лиофилизации, для получения такой лиофилизированной наносуспензии, настоящим ссылаются на соответствующие разделы данного документа выше.
Указанную лиофилизированную наносуспензию можно восстанавливать в водную наносуспензию путем разбавления водной дисперсионной средой, в частности водой или водой для инъекции. Указанная восстановленная наносуспензия может применяться как состав-депо, в частности, как инъецируемый состав-депо, который может найти применение при лечении ВИЧ-инфекции, а также при профилактике передачи ВИЧ.
Такие восстановленные наносуспензии можно вводить периодически с интервалами времени в одну неделю или дольше, что дает уровни в плазме крови, которые могут быть достаточными для подавления размножения (репликации) ВИЧ. Этим предусматривается сниженное число введений, что, таким образом, является благоприятным с точки зрения “pill burden” и соблюдения пациентом схемы приема лекарственных средств. Поэтому, составы из наночастиц 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли по настоящему изобретению могут быть пригодны при длительном лечении ВИЧ-инфекции.
Периодическое введение восстановленной наносуспензии из 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли; с интервалами времени в одну неделю или дольше, кроме того, приводит к уровням в плазме крови, которые могут быть достаточными для обеспечения профилактики передачи ВИЧ. Также в этом случае требуется сниженное число введений, что опять-таки является выгодным с точки зрения “pill burden” и соблюдения отдельными лицами, подверженными риску инфицирования, схемы приема лекарственных средств. Таким образом, составы из наночастиц 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли по настоящему изобретению могут быть пригодны при длительной профилактике ВИЧ-инфекции.
В одном варианте осуществления настоящее изобретение касается фармацевтической композиции для введения путем внутримышечной или подкожной инъекции, содержащей терапевтически эффективное количество 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли в форме восстановленной наносуспензии 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли в фармацевтически приемлемом водном носителе; где наносуспензия восстановлена из лиофилизированной наносуспензии, содержащей:
(a) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, или его стереоизомерную форму, или его фармацевтически приемлемую соль; и
(b) стерический стабилизатор, представляющий собой твердое вещество при комнатной температуре; и
(c) необязательно криопротектор или лиопротектор.
В одном варианте осуществления стерический стабилизатор в вышеупомянутой восстановленной наносуспензии представляет собой кристаллическое твердое вещество при комнатной температуре, в частности, стерический стабилизатор представляет собой кристаллическое твердое вещество при комнатной температуре и характеризуется температурой плавления, равной или выше 30°C, или температурой плавления, равной или выше 50°C, или температурой плавления, равной или выше 75°C, или температурой плавления, равной или выше 90°C.
В одном варианте осуществления стерический стабилизатор в вышеупомянутой восстановленной наносуспензии представляет собой аморфное твердое вещество при комнатной температуре, в частности, стерический стабилизатор представляет собой аморфное твердое вещество при комнатной температуре и обладает температурой стеклования (Tg), равной или выше 30°C, или Tg, равной или выше 50°C, или Tg, равной и выше 75°C, или Tg, равной или выше 90°C.
В одном варианте осуществления стерический стабилизатор в вышеупомянутой восстановленной наносуспензии представляет собой полоксамер 338.
В одном варианте осуществления вышеупомянутая восстановленная наносуспензия дополнительно содержит криопротектор или лиопротектор, такой как, например, поливинилпирролидон, а именно PVP K12, PVP K15 или PVP K17, или смесь PVP и сахарида, такого как, например, трегалоза.
Настоящее изобретение дополнительно касается способа лечения субъекта, инфицированного ВИЧ, при этом указанный способ включает введение, в частности, путем внутримышечной или подкожной инъекции, фармацевтической композиции, как определено в данном документе выше или ниже. Или, альтернативно, настоящее изобретение касается применения фармацевтической композиции, как определено в данном документе выше или ниже, для изготовления лекарственного препарата для лечения ВИЧ-инфекции. Или, альтернативно, настоящее изобретение касается фармацевтической композиции, как определено в данном документе выше или ниже, для применения в лечении ВИЧ-инфекции.
В другом аспекте предусматривается способ длительного лечения ВИЧ-инфекции, при этом указанный способ включает введение субъекту, инфицированному ВИЧ, эффективного количества фармацевтической композиции, как определено в данном документе выше или ниже, путем внутримышечной или подкожной инъекции; где композицию вводят, или она подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет. Или, альтернативно, настоящее изобретение касается применения фармацевтической композиции, как определено в данном документе выше или ниже, для изготовления лекарственного препарата для длительного лечения ВИЧ-инфекции и для введения путем внутримышечной или подкожной инъекции, где лекарственный препарат вводят, или он подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет. Или, альтернативно, настоящее изобретение касается фармацевтической композиции, как определено в данном документе выше или ниже, для применения в длительном лечении ВИЧ-инфекции, где композиция предназначена для введения путем внутримышечной или подкожной инъекции, и где композицию вводят, или она подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет.
Настоящее изобретение дополнительно касается способа профилактики ВИЧ-инфекции у субъекта, подверженного риску инфицирования ВИЧ, при этом указанный способ включает введение указанному субъекту эффективного количества, которое эффективно для профилактики ВИЧ-инфекции, фармацевтической композиции, как определено в данном документе выше или ниже. Или, альтернативно, настоящее изобретение касается применения фармацевтической композиции, как определено в данном документе выше или ниже, для изготовления лекарственного препарата для профилактики ВИЧ-инфекции у субъекта, подверженного риску инфицирования ВИЧ. Или, альтернативно, настоящее изобретение касается фармацевтической композиции, как определено в данном документе выше или ниже, для применения в профилактике ВИЧ-инфекции у субъекта, подверженного риску инфицирования ВИЧ.
В другом аспекте настоящее изобретение относится к способу длительной профилактики ВИЧ-инфекции у субъекта, подверженного риску инфицирования ВИЧ, при этом указанный способ включает введение указанному субъекту эффективного количества, которое эффективно в профилактике ВИЧ-инфекции, фармацевтической композиции, как определено в данном документе выше или ниже, путем внутримышечной или подкожной инъекции; где композицию вводят, или она подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет. Или, альтернативно, настоящее изобретение относится к применению фармацевтической композиции, как определено в данном документе выше или ниже, для изготовления лекарственного препарата для длительной профилактики ВИЧ-инфекции у субъекта, подверженного риску инфицирования ВИЧ, и для введения путем внутримышечной или подкожной инъекции, где лекарственный препарат вводят, или он подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет. Или, альтернативно, настоящее изобретение относится к фармацевтической композиции, как определено в данном документе выше или ниже, для применения в длительной профилактике ВИЧ-инфекции у субъекта, подверженного риску инфицирования ВИЧ, где композиция предназначена для введения путем внутримышечной или подкожной инъекции, и где композицию вводят, или она подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет.
В одном варианте осуществления настоящее изобретение касается применения, или способа, или фармацевтической композиции для применения, как определено в данном документе, где фармацевтическую композицию вводят, или она подлежит введению с интервалом времени, который находится в диапазоне от одной недели до одного месяца, или в диапазоне от одного месяца до трех месяцев, или в диапазоне от трех месяцев до шести месяцев, или в диапазоне от шести месяцев до двенадцати месяцев, или в диапазоне от 12 месяцев до 24 месяцев.
В другом варианте осуществления настоящее изобретение касается применения, или способа, или фармацевтической композиции для применения, как определено в данном документе, где фармацевтическую композицию вводят или она подлежит введению раз в каждые две недели, или раз в каждый месяц, или раз в каждые три месяца.
4-[[4-[[4-(2-Цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил можно применять в форме основания или в виде подходящей фармацевтически приемлемой формы соли присоединения, такой как форма соли присоединения кислоты. Подразумевается, что фармацевтически приемлемые соли присоединения включают терапевтически активные нетоксические солевые формы. Формы солей присоединения кислоты могут быть получены путем обработки формы основания соответствующими кислотами, такими как неорганические кислоты, например, галогенводородные кислоты, к примеру, хлористоводородная, бромистоводородная и т.п.; серная кислота; азотная кислота; фосфорная кислота и т.п.; или органические кислоты, например, уксусная, пропионовая, гидроксиуксусная, 2-гидроксипропионовая, 2-оксопропионовая, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, 2-гидрокси-1,2,3-пропантрикарбоновая, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная и подобные кислоты. Предпочтительной формой для применения в настоящем изобретении является активный ингредиент 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в форме основания.
4-[[4-[[4-(2-Цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил встречается в стереоизомерных формах, более конкретно в виде E- и Z-изомерных форм. Оба изомера можно применять в настоящем изобретении. Всякий раз, когда в данном документе упоминается 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, подразумевается, что включается E- или Z-форма, а также любая смесь обоих форм. Предпочтительной формой 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила для применения в настоящем изобретении является E-изомер, т.е. (E)-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]-амино]-2-пиримидинил]амино]-бензонитрил, особенно E-изомер в форме основания, который может называться рилпивирин. Также может применяться Z-изомер 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, т.е. (Z)-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]-амино]-2-пиримидинил]-амино]-бензонитрил.
Всякий раз, когда в данном документе упоминается E-форма 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, подразумевается, что она включает чистый E-изомер или любую смесь изомеров E- и Z-форм, в которой главным образом присутствует E-форма, т.е. смесь изомеров, содержащую больше 50% или, в частности, больше 80% E-формы, или даже больше 90% E-формы. Особый интерес представляет E-форма, практически не содержащая Z-форму. “Практически не содержащая” в данном контексте относится к E-Z-смесям без или почти без Z-формы, например, к смесям изомеров, содержащим не менее 90%, в частности, 95% или даже 98%, или 99% E-формы. В то же время, всякий раз, когда в данном документе упоминается Z-форма 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, подразумевается, что она включает чистый Z-изомер или любую смесь изомеров Z- и E-форм, в которой главным образом присутствует Z-форма, т.е. смесь изомеров, содержащую больше 50% или, в частности, больше 80% Z-формы, или даже больше 90% Z-формы. Z-форма, практически не содержащая E-формы, также можно применять. “Практически не содержащая” в данном контексте относится к E-Z-смесям без или почти без E-формы, например, к смесям изомеров, содержащим не менее 90%, в частности, 95%, или даже 98%, или 99% Z-формы. В одном варианте осуществления активный ингредиент 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила применяют в E-форме, в частности, в E-форме основания 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила.
Также подразумевается, что в настоящем изобретении могут применяться соли стереоизомерных форм 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности, упомянутые выше соли Z- или E-изомерной формы 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, такие как, например, соль хлористоводородной кислоты E-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила.
Всякий раз, при использовании в данном документе ниже, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил также включает его стереоизомерную форму или его фармацевтически приемлемую соль, если не указано иное.
Было обнаружено, что физико-химические свойства 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила создают возможность для изготовления наносуспензий, которые обладают уникальными фармакокинетическими свойствами, так что наносуспензии могут применяться для длительного лечения ВИЧ-инфекции, а также для длительной профилактики ВИЧ-инфекции, при этом для этой цели требуется всего лишь небольшое число введений лекарственного средства. Это является благоприятным с точки зрения “pill-burden”, а также соблюдения пациентом предписанной схемы приема лекарственного средства.
Используемое в данном документе выражение “лечение ВИЧ-инфекции” относится к лечению субъекта, инфицированного ВИЧ. Выражение “лечение ВИЧ-инфекции” также относится к лечению заболеваний, ассоциированных с ВИЧ-инфекцией, например, СПИДа, или других состояний, ассоциированных с ВИЧ-инфекцией, в том числе тромбоцитопении, саркомы Капоши и инфекции центральной нервной системы, характеризующейся прогрессивным разрушением миелинового слоя, что приводит к деменции и симптомам, таким как прогрессивная дизартрия, атаксия и дезориентация, и дополнительных состояний, с которыми также была ассоциирована ВИЧ-инфекция, таких как периферическая невропатия, прогрессивная генерализованная лимфаденопатия (PGL) и СПИД-ассоциированный комплекс (ARC).
Выражение “профилактика ВИЧ-инфекции” относится к профилактике или предотвращению у субъекта инфицирования ВИЧ. Источники инфицирования могут быть различными, например, материал, содержащий ВИЧ, в частности, биологическая жидкость, которая содержит ВИЧ, такая как кровь или сперма, или другой субъект, который инфицирован ВИЧ. Профилактика ВИЧ-инфекции относится к профилактике передачи вируса от материала, содержащего ВИЧ, или от ВИЧ-инфицированных индивидуумов неинфицированному человеку, или относится к профилактике попадания вируса в тело неинфицированного человека. Передача ВИЧ может произойти посредством любого известного процесса переноса ВИЧ, как, например, передачи половым путем или при контакте с кровью инфицированного субъекта, например, у медицинского персонала, оказывающего помощь инфицированным субъектам. Перенос ВИЧ также может происходить при контакте с ВИЧ-инфицированной кровью, например, при манипуляциях с пробами крови или при переливании крови. Он также может происходить при контакте с инфицированными клетками, например, при выполнении лабораторных экспериментов с ВИЧ-инфицированными клетками.
Выражения “лечение ВИЧ-инфекции”, “терапия против ВИЧ”, а также подобные выражения относятся к лечению, при котором снижается вирусная нагрузка ВИЧ (представленная как число копий вирусной РНК в определенном объеме сыворотки крови). Чем эффективнее лечение, тем ниже вирусная нагрузка. Предпочтительно, что вирусная нагрузка должна снижаться до наиболее низких возможных уровней, например, ниже примерно 200 копий/мл, в частности, ниже примерно 100 копий/мл, более конкретно ниже 50 копий/мл, если это возможно, ниже предела обнаружения вируса. Снижение вирусной нагрузки на один, два или даже три порядка величины (например, снижение порядка от примерно 10 до примерно 102 или больше, например, до примерно 103) служит показателем эффективности лечения. Другим параметром для измерения эффективности лечения ВИЧ-инфекции является количество CD4-клеток, которое у здоровых взрослых находится в диапазоне от 500 до 1500 клеток на мкл. Сниженные количества CD4-клеток служат показателем ВИЧ-инфекции и если составляют ниже примерно 200 клеток на мкл, то может развиваться СПИД. Увеличение количества CD4-клеток, например, на примерно 50, 100, 200 или больше клеток на мкл, также является показателем эффективности лечения ВИЧ-инфекции. Количество CD4-клеток, в частности, должно увеличиваться до уровня выше примерно 200 клеток на мкл или выше примерно 350 клеток на мкл. Вирусную нагрузку или количество CD4-клеток, или и то, и другое, можно использовать для определения степени ВИЧ-инфекции.
Выражения “эффективное лечение ВИЧ-инфекции” и подобные выражения относятся к такому лечению, которое уменьшает вирусную нагрузку, или увеличивает количество CD4-клеток, или и то, и другое, как описано выше. Выражения “эффективная профилактика ВИЧ-инфекции” и подобные выражения относятся к такой ситуации, при которой имеет место снижение относительного числа вновь инфицированных субъектов в популяции при контакте с источником ВИЧ-инфекции, таким как материал, содержащий ВИЧ, или ВИЧ-инфицированный субъект. Эффективную профилактику можно оценивать, например, путем оценки того происходит ли в смешанной популяции из ВИЧ-инфицированных и неинфицированных индивидуумов снижение относительного числа вновь инфицированных индивидуумов, при этом сравнивая неинфицированных индивидуумов, которых подвергали лечению с помощью фармацевтической композиции по настоящему изобретению, и неинфицированных индивидуумов, которых не подвергали лечению. Это снижение можно оценивать с помощью статистического анализа числа инфицированных и неинфицированных индивидуумов в данной популяции с течением времени.
Выражения “терапевтически эффективное количество”, “эффективное количество”, “количество, эффективное в профилактике ВИЧ-инфекции” и подобные выражения относятся к количествам активного ингредиента 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, которые приводят к действенным уровням в плазме крови. Под “действенными уровнями в плазме крови” подразумевают такие уровни в плазме крови вещества, подавляющего ВИЧ, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, которые обеспечивают эффективное лечение или эффективную профилактику ВИЧ-инфекции.
Выражение “субъект” относится, в частности, к человеку.
Средний размер частиц для наночастиц наносуспензий, подлежащих лиофилизации, или наносуспензии, полученной в результате восстановления лиофилизированной наносуспензии, может быть ниже примерно 1000 нм, или ниже примерно 500 нм, или ниже примерно 400 нм, или ниже примерно 300 нм, или ниже примерно 200 нм. Предпочтительно, средний размер частиц составляет примерно 200 нм, или примерно 400 нм, или примерно 800 нм, более предпочтительно, примерно 200 нм.
Используемое в данном документе выражение средний размер частиц имеет общепринятое значение, известное специалисту в данной области, и его можно измерять с помощью известных в данной области техники методик измерения размера частиц, таких как, например, седиментационное проточное фракционирование в силовом поле, фотон-корреляционная спектроскопия, лазерная дифракция или центрифугирование на дисковой центрифуге. Средние размеры частиц, упомянутые в данном документе, могут быть связаны с объемными распределениями частиц. В таком случае под "средним размером частиц меньше примерно 50 мкм" подразумевается, что по меньшей мере 50% частиц по объему имеют размер частицы меньше примерно 50 мкм, и то же самое относится к другим упомянутым размерам частиц. Подобным образом средние размеры частиц могут быть связаны с весовыми распределениями частиц. В таком случае под "средним размером частиц меньше примерно 50 мкм" подразумевается, что по меньшей мере 50% частиц по весу имеют размер частицы меньше примерно 50 мкм, и то же самое относится к другим упомянутым размерам частиц. Обычно объемное и весовое распределение дают одинаковую или примерно одинаковую величину для среднего размера частиц.
Фармацевтические композиции по настоящему изобретению обеспечивают высвобождение активного ингредиента, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в течение продолжительного периода времени и поэтому они также могут называться композициями с замедленным или отсроченным высвобождением. После введения композиции по настоящему изобретению остаются в организме и непрерывно высвобождают 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, при этом поддерживают такие уровни этого активного ингредиента в системе пациента в течение продолжительного периода времени, что обеспечивают терапию ВИЧ-инфекции или профилактику ВИЧ-инфекции в течение указанного периода. В связи с тем, что фармацевтические композиции по настоящему изобретению остаются в организме и непрерывно высвобождают 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, их можно рассматривать как фармацевтические композиции, пригодные в качестве составов-депо.
Под используемым в данном документе выражением “продолжительный период времени” подразумевают срок (или период времени), который может находиться в диапазоне от одной недели до одного года или до двух лет, или срок в диапазоне от одной до двух недель, или от двух до трех недель, или от трех до четырех недель, или срок в диапазоне от одного до двух месяцев, или от двух до трех месяцев, или от трех до четырех месяцев, или от трех до шести месяцев, или от шести месяцев до 12 месяцев, или от 12 месяцев до 24 месяцев, или срок, который находится в пределах нескольких дней, например, 7, 10 или 12 дней, или нескольких недель, например, 2, 3 или 4 недель, или одного месяца, или нескольких месяцев, например, 2, 3, 4, 5 или шести месяцев, или даже дольше, например, 7, 8, 9 или 12 месяцев.
Фармацевтические композиции по настоящему изобретению можно использовать при длительном лечении или длительной профилактике ВИЧ-инфекции или, другими словами, их можно применять при лечении ВИЧ-инфекции или в профилактике ВИЧ-инфекции в течение продолжительного периода времени. Композиции по настоящему изобретению эффективны в терапии против ВИЧ или в профилактике ВИЧ-инфекции в течение продолжительного периода времени, например, в течение по меньшей мере примерно одной недели или дольше или в течение примерно 1 месяца или дольше. Под фразой "эффективная в течение по меньшей мере примерно одной недели или дольше" подразумевается, что уровень в плазме крови активного ингредиента, 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, должен быть выше пороговой величины. В случае применения в терапии указанная пороговая величина представляет собой самый низкий уровень в плазме крови, при котором 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил обеспечивает эффективное лечение ВИЧ-инфекции. В случае применения в профилактике ВИЧ-инфекции указанная пороговая величина представляет собой самый низкий уровень в плазме крови, при котором 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил эффективен в профилактике передачи ВИЧ-инфекции.
Под “длительным”, например, как используется в отношении “длительной профилактики ВИЧ-инфекции” или “длительного лечения ВИЧ-инфекции”, или подобной терминологии, подразумевают сроки, которые могут находиться в диапазоне от одной недели до одного года, или до двух лет, или дольше, например, до пяти или 10 лет. В частности, в случае лечения ВИЧ-инфекции такие сроки будут длинными, порядка одного - семи лет. Такие сроки также могут быть относительно короткими, в частности, в случае профилактики. Более короткими сроками являются такие, которые составляют несколько дней, например, 7, 10 или 12 дней, или несколько недель, например, 2, 3 или 4 недели, или один месяц, или несколько месяцев, например, 2, 3, 4, 5 или шесть месяцев или даже дольше, например, 7, 8, 9 или 12 месяцев. В одном варианте осуществления способы и применения в соответствии с настоящим изобретением предназначены для профилактики ВИЧ-инфекции в течение одного месяца, или нескольких месяцев, например 2, 3, 4, 5 или шести месяцев, или даже дольше, например, 7, 8, 9 или 12 месяцев. В одном варианте осуществления способы и применения в соответствии с настоящим изобретением предназначены для лечения ВИЧ-инфекции в течение одного месяца, или нескольких месяцев, например, 2, 3, 4, 5 или шести месяцев, или даже дольше, например, 7, 8, 9 или 12 месяцев.
Фармацевтические композиции по настоящему изобретению можно вводить с различными интервалами времени. При применении в профилактике ВИЧ-инфекции фармацевтические композиции по настоящему изобретению можно вводить всего один раз или ограниченное число раз, как например, два раза, три, четыре, пять или шесть раз, или больше. Это может быть рекомендовано в тех случаях, когда профилактика необходима в течение ограниченного периода времени, такого как период, в течение которого существует риск инфицирования.
Фармацевтические композиции по настоящему изобретению можно вводить с упомянутыми выше интервалами времени, такими как интервал времени, который находится в диапазоне от одной недели до одного месяца, или в диапазоне от одного месяца до трех месяцев, или в диапазоне от трех месяцев до шести месяцев, или в диапазоне от шести месяцев до двенадцати месяцев. В одном варианте осуществления фармацевтическую композицию можно вводить раз в каждые две недели, или раз в каждый месяц, или раз в каждые три месяца. В другом варианте осуществления интервал времени находится в диапазоне от одной до двух недель, или от двух до трех недель, или от трех до четырех недель, или интервал времени находится в диапазоне от одного до двух месяцев, или от двух до трех месяцев, или от трех до четырех месяцев, или от трех до шести месяцев, или от шести месяцев до 12 месяцев, или от 12 месяцев до 24 месяцев. Интервал времени может составлять по меньшей мере одну неделю, но также может составлять несколько недель, например, 2, 3, 4, 5 или 6 недель, или интервалы времени в один месяц, или несколько месяцев, например, 2, 3, 4, 5 или 6 месяцев или даже дольше, например, 7, 8, 9 или 12 месяцев. В одном варианте осуществления фармацевтические композиции по настоящему изобретению вводят с интервалом времени в один, два или три месяца. Такие более длинные периоды между каждым введением фармацевтических композиций по настоящему изобретению обеспечивают дополнительные улучшения с точки зрения “pill burden” и соблюдения терапевтических рекомендаций. Для дополнительного улучшения соблюдения терапевтических рекомендаций пациентам следует давать указания принимать медикамент в определенный день недели, если композицию вводят по еженедельному графику, или в определенный день месяца в случае ежемесячного графика.
Длина интервалов времени между каждым введением композиции по настоящему изобретению может меняться. Например, указанные интервалы времени могут выбираться в зависимости от уровней в плазме крови. Интервалы могут быть короче, если уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила признают слишком низкими, например, если они приближаются к минимальному уровню в плазме крови, установленному в данном документе ниже. Интервалы могут быть более длинными, если уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила признают слишком высокими. В одном варианте осуществления композиции по настоящему изобретению вводят с равными интервалами времени. Композиции можно вводить без каких-либо промежуточных дополнительных введений или, другими словами, композиции можно вводить в конкретные моменты времени, отделенные друг от друга периодом времени, переменной или равной длины, например, периодом времени по меньшей мере одна неделя или любым другим периодом времени, установленным в данном документе, во время которого дополнительный 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил не вводят. При интервалах времени одинаковой продолжительности преимущество заключается в том, что график введения является простым, например, введение происходит в один и тот же день недели или один и тот же день месяца. Следовательно, такой график введения предполагает ограниченное “pill burden”, тем самым благотворно влияя на соблюдение пациентом предписанной схемы приема лекарственных средств.
Концентрацию (или “C”) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в плазме крови субъекта, которого лечат с помощью него, как правило, выражают как отношение массы к единице объема, типично, нанограммов на миллилитр (нг/мл). Для удобства, эту концентрацию в данном документе можно называть “концентрацией лекарственного средства в плазме крови” или “концентрацией в плазме крови”.
Доза (или количество) вводимого 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила зависит от количества лекарственного средства в фармацевтических композициях по настоящему изобретению или от количества указанной композиции, которое вводится. Если требуются более высокие уровни в плазме крови, можно либо вводить композицию с более высокой концентрацией 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, либо вводить большее количество указанной композиции, или же применять и то, и другое. Противоположно действуют, если в плазме крови требуются более низкие уровни. Также, чтобы добиться определенных требуемых уровней в плазме крови, можно выбирать комбинацию из переменных интервалов времени и переменных дозировок.
Доза (или количество) вводимого 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила также зависит от частоты введений (т.е. интервала времени между каждым введением). Обычно доза будет более высокой при менее частых введениях. Все эти параметры можно применять для того, чтобы уровни в плазме крови достигали требуемых значений.
Режим дозирования также зависит от того, предусматривается профилактика или лечение ВИЧ-инфекции. В случае терапии дозу вводимого 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила или частоту дозирования, или и то, и другое выбирают так, чтобы концентрация 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в плазме крови поддерживалась выше минимального уровня в плазме крови. Выражение “минимальный уровень в плазме крови” (или Cmin) в данном контексте относится к уровню в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, который обеспечивает эффективное лечение ВИЧ. В частности, уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила поддерживается на уровне, выше минимального уровня в плазме крови, составляющего примерно 10 нг/мл, или выше примерно 15 нг/мл, или выше примерно 20 нг/мл, или выше примерно 40 нг/мл, или выше примерно 50 нг/мл, или выше примерно 90 нг/мл, или выше примерно 270 нг/мл, или выше примерно 540 нг/мл. В одном варианте осуществления уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила поддерживается выше уровня примерно 90 нг/мл. Или же уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила может поддерживаться в определенных диапазонах, в частности, диапазонах, начинающихся от минимального уровня в плазме крови, выбранного из упомянутых выше уровней, и заканчивающихся на более высоких уровнях в плазме крови, выбранных из упомянутых выше уровней и выбранных из 500 нг/мл и 1000 нг/мл (например, от 10 до 15, от 10 до 20, от 10 до 40 и т.д., или от 15 до 20, или от 15 до 40, или от 15 до 90 и т.д., или от 20 до 40, от 20 до 90, или от 20 до 270 и т.д., или от 40 до 90, от 40 до 270, или от 40 до 540 и т.д., причем каждый раз от примерно указанной величины в нг/мл до примерно указанной величины в нг/мл). В одном варианте осуществления указанный диапазон составляет от примерно 10 до примерно 20, от примерно 20 до примерно 90, от примерно 90 до примерно 270, от примерно 270 до примерно 540, от примерно 540 до примерно 1000, причем каждый раз от примерно указанной величины в нг/мл до примерно указанной величины в нг/мл.
Уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила следует поддерживать выше вышеупомянутых минимальных уровней в плазме крови, поскольку при более низких уровнях вирус более не может достаточно подавляться, так что он может размножаться с дополнительным риском возникновения мутаций.
В случае профилактики ВИЧ выражение “минимальный уровень в плазме крови” (или Cmin) относится к самому низкому уровню в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, который обеспечивает эффективную профилактику ВИЧ-инфекции. В случае передачи ВИЧ от материала, содержащего ВИЧ, или от субъекта, инфицированного ВИЧ, субъекту, неинфицированному ВИЧ, он представляет собой наиболее низкий уровень в плазме крови, который эффективен в ингибировании указанной передачи.
В частности, в случае профилактики ВИЧ уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила может поддерживаться на уровне, выше упомянутого ранее минимального уровня в плазме крови по отношению к терапии. Однако, при профилактике уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила может поддерживаться на более низком уровне, например, на уровне выше примерно 4 нг/мл, или примерно 5 нг/мл, или примерно 8 нг/мл. Уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, предпочтительно, должны поддерживаться выше этих минимальных уровней в плазме крови, так как при более низких уровнях лекарственное средство больше не может быть эффективным, тем самым повышается риск передачи ВИЧ-инфекции. Уровни в плазме крови TMC278 могут поддерживаться на несколько более высоких уровнях, чтобы иметь резерв безопасности. Такие более высокие уровни начинаются от примерно 50 нг/мл или больше, или от примерно 90 нг/мл или больше. Уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила может поддерживаться на уровне, который находится в диапазонах, указанных выше по отношению к терапии, но где нижние границы включают уровни в плазме крови примерно 4 нг/мл, или примерно 5 нг/мл, или примерно 8 нг/мл.
Преимущество 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила заключается в том, что его можно применять вплоть до относительно высоких уровней в плазме крови без каких-либо существенных побочных эффектов. Концентрации в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила могут достигать довольно высоких уровней, но как и в случае с любым лекарственным средством не должны превышать максимального уровня в плазме крови (или Cmax), который представляет собой уровень в плазме крови, при котором 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил вызывает существенные побочные эффекты. Используемое в данном документе выражение “существенные побочные эффекты” означает, что в соответствующей популяции пациентов наблюдаются побочные эффекты в таком масштабе, что побочные эффекты влияют на нормальное функционирование организма пациентов. Cmax для 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила можно определять по экстраполяции результатов испытаний в анализах на клетках или по итоговой оценке клинического исследования, и предпочтительно он не должен превышать величину примерно 500 нг/мл или 1000 нг/мл. В варианте осуществления количество и частоту введений 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, подлежащего введению, выбирают так, чтобы концентрации в плазме крови поддерживаются в течение длительного времени на уровне в пределах от максимального уровня в плазме крови (или Cmax, как установлено выше) до минимального уровня в плазме крови (или Cmin, как установлено выше).
В некоторых случаях может быть целесообразно поддерживать уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила на относительно низких уровнях, например, как можно ближе к минимальным уровням в плазме крови, установленным в данном документе. Это будет давать возможность снижать частоту введений и/или количество вводимого 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила при каждом введении. Это также позволит избежать нежелательных побочных эффектов, что будет способствовать приемлемости лекарственных форм для большинства целевых групп популяции, которые являются здоровыми людьми, подверженными риску инфицирования и, следовательно, менее расположены переносить побочные эффекты. В случае профилактики уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила могут поддерживаться на относительно низких уровнях. Один вариант осуществления касается применений или способов профилактики ВИЧ-инфекции, как указано выше или ниже в данном документе, где минимальный уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила является таким, как указано в данном документе, а максимальный уровень в плазме крови является примерно равным самому низкому уровню в плазме крови, что заставляет ингибитор RT действовать терапевтически, так же, как указано в данном документе.
В других вариантах осуществления уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила поддерживается на уровне, ниже нижнего максимального уровня в плазме крови, составляющего примерно 10 нг/мл, более конкретно примерно 15 нг/мл, еще конкретнее примерно 20 нг/мл, еще более конкретно примерно 40 нг/мл. В конкретном варианте осуществления уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила поддерживается ниже уровня примерно 13,5 нг/мл. В одном варианте осуществления уровень в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила поддерживается в интервале от нижнего максимального уровня в крови, установленного выше, до минимальных уровней в плазме крови, упомянутых по отношению к профилактике. Например, уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила поддерживаются ниже примерно 10 нг/мл и выше минимального уровня, составляющего примерно 4 нг/мл.
В других случаях может быть целесообразно поддерживать уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила на относительно более высоких уровнях, например, если существует высокий риск инфекции, а более частые и/или более высокие дозы не представляют собой проблему. В таких случаях минимальный уровень в плазме крови может быть равен самому низкому уровню в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, который обеспечивает эффективное лечение ВИЧ, такому как конкретные уровни, упомянутые в данном документе.
В случае профилактики дозу 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности, рилпивирина, подлежащего введению, следует рассчитывать, исходя из от примерно 0,2 мг/день до примерно 50 мг/день, или от 0,5 мг/день до примерно 50 мг/день, или от примерно 1 мг/день до примерно 10 мг/день, или от примерно 10 мг/день до примерно 20 мг/день, или от примерно 15 мг/день до примерно 25 мг/день, или от примерно 20 мг/день до примерно 25 мг/день, или от примерно 2 мг/день до примерно 5 мг/день, например, примерно 3 мг/день, или примерно 5 мг/день, или примерно 20 мг/день. Это соответствует еженедельной дозе от примерно 1,5 мг до примерно 350 мг, в частности, от примерно 3,5 мг до примерно 350 мг, в частности, от примерно 7 мг до примерно 70 мг, или от примерно 70 мг до примерно 140 мг, или от примерно 105 мг до примерно 175 мг, или от примерно 140 мг до примерно 175 мг, или от примерно 14 мг до примерно 35 мг, например, примерно 21 мг, или примерно 35 мг, или примерно 140 мг, или ежемесячной дозе от 6 мг до примерно 3000 мг, в частности, от примерно 15 мг до примерно 1500 мг, более конкретно от примерно 30 мг до примерно 300 мг, или от примерно 300 мг до примерно 600 мг, или от примерно 450 мг до примерно 750 мг, или от примерно 600 мг до примерно 750 мг, или от примерно 60 мг до примерно 150 мг, например, примерно 90 мг, или примерно 150 мг, или примерно 600 мг. Дозы для других режимом дозирования можно легко рассчитать путем умножения ежедневной дозы на число дней между каждым введением.
В случае терапии доза 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности, рилпивирина, подлежащего введению, должна быть несколько выше, и ее следует рассчитывать, исходя из от примерно 1 мг/день до примерно 150 мг/день, или от примерно 2 мг/день до примерно 100 мг/день, или от примерно 5 мг/день до примерно 50 мг/день, или от примерно 10 мг/день до примерно 25 мг/день, или от примерно 15 мг/день до примерно 25 мг/день, или от примерно 20 мг/день до примерно 25 мг/день, например, примерно 15 мг/день, или примерно 20 мг/день, или примерно 25 мг/день. Соответствующие еженедельные или ежемесячные дозы можно рассчитывать, как изложено выше. Для применений в профилактике дозы могут быть ниже, хотя можно применять такие же дозировки, что и для применений в терапии.
В одном варианте осуществления ежемесячная доза 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности, рилпивирина, составляет 600 мг. В одном варианте осуществления концентрация 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности, рилпивирина, в водной восстановленной наносуспензии составляет 200 мг/мл или 300 мг/мл.
Было обнаружено, что при однократном введении уровни в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила являются более или менее стабильными, т.е. они колеблются в пределах ограниченных границ. Как было обнаружено, уровни в плазме крови достигают более или менее стабильного состояния или приближаются к более или менее скорости высвобождения нулевого порядка в течение продолжительного периода времени. Под “стабильным состоянием” подразумевают такую ситуацию, при которой количество лекарственного средства, присутствующего в плазме крови субъекта, остается на более или менее одинаковом уровне в течение продолжительного периода времени. Для уровней в плазме крови 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, как правило, не показаны какие-либо падения ниже минимального уровня в плазме крови, при котором лекарственное средство является эффективным. Выражение “остается на более или менее одинаковом уровне” не исключает того, что могут быть небольшие отклонения концентраций в плазме крови в пределах приемлемого диапазона, например, отклонения в диапазоне примерно +/- 30%, или примерно +/- 20%, или примерно +/- 10%, или примерно +/- 10%.
В некоторых случаях после введения может наблюдаться первоначальная пиковая концентрация в плазме крови, после чего уровни в плазме крови достигают “стабильного состояния”, как упоминается выше в данном документе.
Композиции по настоящему изобретению демонстрируют хорошую местную переносимость и легкость введения. Хорошая местная переносимость относится к минимальному раздражению и воспалению в месте инъекции; легкость введения относится к размеру иглы и промежутку времени, требуемому для введения дозы конкретного лекарственного состава.
В одном варианте осуществления наночастицы в композициях по настоящему изобретению включают в основном кристаллический 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил и стерический стабилизатор, при этом их объединенное количество может составлять по меньшей мере примерно 50%, или по меньшей мере примерно 80%, или по меньшей мере примерно 90%, или по меньшей мере примерно 92%, или по меньшей мере примерно 95%, или по меньшей мере примерно 98%, или по меньшей мере примерно 99%, или по меньшей мере примерно 99,5% по весу наночастиц.
Восстановленные фармацевтические композиции согласно настоящему изобретению содержат носитель, в частности, водный носитель, который предпочтительно является фармацевтически приемлемым. Указанный водный носитель включает стерильную воду необязательно в смеси с другими фармацевтически приемлемыми ингредиентами. Последние включают любые ингредиенты для применения в инъецируемых составах, или суспензионных составах I, или в пероральных составах. Такие ингредиенты могут быть выбраны из одного или нескольких из суспендирующего средства, буфера, средства, регулирующего рН, консерванта, изотонирующего средства и подобных ингредиентов. В одном варианте осуществления указанные ингредиенты выбирают из одного или нескольких из суспендирующего средства, буфера, средства, регулирующего рН, и необязательно консерванта и изотонирующего средства. Конкретные ингредиенты могут действовать в качестве двух или более из этих средств одновременно, например, ведут себя подобно консерванту и буферу, или ведут себя подобно буферу и изотонирующему средству. Как сказано выше, другие фармацевтически приемлемые ингредиенты также могут присутствовать в лиофилизированной лекарственной наносуспензии как таковой. Или же другие фармацевтически приемлемые ингредиенты могут присутствовать как в лиофилизированной лекарственной наносуспензии как таковой, так и в носителе, в частности, в водном носителе. Предпочтительно, в лиофилизированной лекарственной наносуспензии как таковой присутствуют другие фармацевтически приемлемые ингредиенты.
Подходящие буферные средства и средства, регулирующие pH, следует применять в количестве, достаточном для переведения дисперсии в состояние от нейтрального до очень слабощелочного (до pH 8,5), предпочтительно, в диапазоне pH 7-7,5. Конкретные буферы представляют собой соли слабых кислот. Буферные средства и средства, регулирующие pH, которые можно добавлять, можно выбирать из винной кислоты, малеиновой кислоты, глицина, лактата натрия/молочной кислоты, аскорбиновой кислоты, лимонной кислоты, цитратов натрия/лимонной кислоты, ацетата натрия/уксусной кислоты, бикарбоната натрия/угольной кислоты, сукцината натрия/янтарной кислоты, бензоата натрия/бензойной кислоты, фосфатов натрия, трис(гидроксиметил)аминометана, бикарбоната натрия/карбоната натрия, гидроксида аммония, бензолсульфоновой кислоты, бензоата натрия/кислоты, диэтаноламина, глюконо-дельта-лактона, хлористоводородной кислоты, бромистого водорода, лизина, метансульфоновой кислоты, моноэтаноламина, гидроксида натрия, трометамина, глюконовой, глицериновой, глутаровой, глутаминовой, этилендиаминтетрауксусной (EDTA), триэтаноламина, в том числе их смесей.
Консерванты включают противомикробные вещества и антиоксиданты, которые можно выбирать из группы, состоящей из бензойной кислоты, бензилового спирта, бутилированного гидроксианизола (BHA), бутилированного гидрокситолуола (BHT), хлорбутола, галлата, гидроксибензоата, EDTA, фенола, хлоркрезола, метакрезола, бензетония хлорида, миристил-γ-пиколиния хлорида, фенилмеркурацетата и тимеросала. Акцепторы радикалов включают BHA, BHT, витамин E и аскорбилпальмитат, а также их смеси. Поглотители кислорода включают аскорбат натрия, сульфит натрия, L-цистеин, ацетилцистеин, метионин, тиоглицерин, ацетонбисульфит натрия, изоаскорбиновую кислоту, гидроксипропила циклодекстрин. Хелатирующие средства включают цитрат натрия, натрий EDTA и яблочную кислоту. Лимонную кислоту можно применять в качестве антиоксиданта, буфера и изотонирующего средства.
Изотонирующее средство или изотонирующая добавка могут присутствовать для обеспечения изотоничности фармацевтических композиций по настоящему изобретению, и они включают сахара, такие как глюкоза, декстроза, сахароза, фруктоза, трегалоза, лактоза; многоатомные сахарные спирты, предпочтительно трехатомные сахарные спирты или спирты с большим количеством гидроксильных групп, такие как глицерин, эритритол, арабит, ксилит, сорбит и маннит. Альтернативно, для обеспечения изотоничности растворов можно применять хлорид натрия, сульфат натрия или другие подходящие неорганические соли. Эти изотонирующие добавки можно применять по отдельности или в комбинации. Для удобства суспензии включают от 0 до 10% (вес/объем), в частности, от 0 до 6% изотонирующего средства. Интерес представляют неионные изотонирующие добавки, например, глюкоза или трегалоза, поскольку электролиты могут влиять на стабильность коллоида.
Необходимым признаком для фармацевтической композиции по настоящему изобретению является легкость введения. Вязкость фармацевтических композиций по настоящему изобретению предпочтительно должна быть достаточно низкой, чтобы обеспечить введение путем инъекции. В частности, их следует разрабатывать таким образом, чтобы их можно было легко отбирать в шприц (например, из флакона), инъецировать через тонкую иглу (например, через иглу 20 G 1½, 21 G 1½, 22 G 2 или 22 G 1¼) в течение не слишком длительного промежутка времени. В одном варианте осуществления вязкость композиций по настоящему изобретению составляет ниже примерно 75 мПа·с или ниже 60 мПа·с. Водные суспензии с такой вязкостью или ниже, как правило, отвечают вышеуказанным критериям.
В идеальном случае, восстановленные водные суспензии согласно настоящему изобретению будут содержать столько 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, насколько это допустимо, чтобы свести инъецируемый объем к минимуму, в частности, от 3 до 50% (вес/объем), или от 3 до 40% (вес/объем), или от 3 до 30% (вес/объем), или от 3 до 20% (вес/объем), или от 10 до 40% (вес/объем), или от 10 до 30% (вес/объем) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила. В одном варианте осуществления восстановленные водные суспензии по настоящему изобретению содержат примерно 10% (вес/объем) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или примерно 20% (вес/объем) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или примерно 30% (вес/объем) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила.
В одном варианте осуществления восстановленные водные наносуспензии могут содержать по весу, исходя из общего объема композиции:
(a) от 3% до 50% (вес/объем), или от 10% до 40% (вес/объем), или от 10% до 30% (вес/объем), или 10% (вес/объем), или 20% (вес/объем), или 30% (вес/объем) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, в частности, рилпивирина;
(b) от 0,5% до 10 %, или от 0,5% до 2% (вес/объем), или 3% (вес/объем), или 5% (вес/объем) стерического стабилизатора согласно настоящему изобретению, например, полоксамера, например, полоксамера 338;
(c) от 0 до 20% (вес/объем), или от 0 до 10% (вес/объем), или 5% (вес/объем) криопротектора или лиопротектора, например, PVP;
(d) от 0% до 10%, или от 0% до 5%, или от 0% до 2%, или от 0% до 1% одного или нескольких буферных средств;
(e) от 0% до 10% или от 0% до 6% (вес/объем) изотонирующего средства;
(f) от 0% до 2% (вес/объем) консерванта и
(g) воду для инъекции в достаточном количестве до 100% (вес/объем).
В суспензии необязательно можно добавлять некоторое количество кислоты или основания, чтобы довести pH до величины pH примерно 7. Подходящими кислотами или основаниями являются любые из физиологически приемлемых, например, HCl, HBr, серная кислота, гидроксиды щелочных металлов, такие как NaOH.
Для лечения ВИЧ-инфекции может быть достаточно введения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила согласно настоящему изобретению, хотя в ряде случаев может рекомендоваться совместное введение других ингибиторов ВИЧ. Последние предпочтительно включают ингибиторы ВИЧ из других классов, в частности, выбранные из NRTI- ингибиторов, ингибиторов PI и ингибиторов слияния. В одном варианте осуществления другой ингибитор ВИЧ, который вводится совместно, представляет собой ингибитор PI. Ингибиторы ВИЧ, которые можно вводить совместно, предпочтительно, представляют собой ингибиторы, применяемые в комбинациях HAART, включающих NNRTI. Например, можно вводить совместно два дополнительные NRTI или NRTI и PI. Такое совместное введение можно осуществлять путем перорального введения или парентерально, в том числе парентеральным введением при длительном лечении ВИЧ-инфекции или длительной профилактике ВИЧ-инфекции.
В некоторых случаях лечение ВИЧ-инфекции может ограничиваться только введением композиции 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в соответствии с настоящим изобретением, т.е. монотерапией без совместного введения дополнительных ингибиторов ВИЧ. Этот способ лечения можно рекомендовать, например, если вирусная нагрузка является относительно низкой, например, если вирусная нагрузка (выраженная как число копий вирусной РНК в установленном объеме сыворотки крови) составляет меньше примерно 200 копий/мл, в частности, меньше примерно 100 копий/мл, более конкретно меньше 50 копий/мл, главным образом, меньше предела обнаружения вируса. В одном варианте осуществления такой тип монотерапии применяют после первоначального лечения с помощью комбинации лекарственных средств против ВИЧ, в частности, с помощью любой из комбинаций HAART в течение определенного периода времени, пока вирусная нагрузка в плазме крови достигает вышеуказанного низкого уровня вируса.
В дополнительном аспекте настоящее изобретение относится к применению восстановленной фармацевтической композиции, содержащей эффективное против вируса количество 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в соответствии с настоящим изобретением, для изготовления лекарственного препарата для поддерживающей терапии субъекта, инфицированного ВИЧ, где композицию вводят или она подлежит введению периодически с интервалом времени, который находится в диапазоне от одной недели до одного года или от одной недели до двух лет.
Таким образом, в дополнительном аспекте настоящее изобретение предусматривает способ длительного лечения пациента, инфицированного ВИЧ, при этом указанный способ включает
(i) лечение указанного пациента с помощью комбинации ингибиторов ВИЧ с последующим
(ii) периодическим введением фармацевтической композиции, содержащей эффективное против вируса количество 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила в соответствии с настоящим изобретением, где композицию вводят с интервалом времени по меньшей мере в одну неделю, или по меньшей мере 2 недели, или раз в каждый месяц, или раз в каждые 3 месяца.
Используемое в данном документе слово "практически" не исключает "полностью", например, композиция, которая "практически не содержащая" Y, может быть полностью не содержащей Y. При необходимости слово "практически" может быть опущено из определения по настоящему изобретению. Подразумевается, что выражение “примерно” в связи с числовым значением имеет его обычное значение в контексте числового значения. При необходимости слово “примерно” может заменяться числовым значением ±10%, или ±5%, или ±2%, или ±1%.
Все документы, упоминаемые в данном документе, включены посредством ссылки во всей их полноте.
Экспериментальная часть 1
Следующие примеры предназначены для иллюстрации настоящего изобретения и не должны истолковываться как ограничивающие настоящее изобретение.
МАТЕРИАЛЫ и СПОСОБЫ
Два слаборастворимых в воде кристаллических API: итраконазол и рилпивирин. 0,3 мм шарики диоксида циркония, стабилизированного иттрием. Lutrol F108 Prill (полоксамер 338) и Cremophor EL (BASF, Людвигсхафен, Германия). PVP K15, трегалозу и сахарозу аналитической степени чистоты (Sigma Chemical Company, Мюнхен, Германия) использовали, не подвергая обработке. Воду для инъекций приобрели у Braun (Braun-Melsungen AG, Мельзунген, Германия) для получения наносуспензий. Деминерализованную воду профильтровали через мембранные фильтры Sartobran PH2O (Sartorius stedim biotech, Германия) и использовали для лазерного дифракционного анализа.
Первичный упаковочный материал
Флаконы 15R от Lutz GmbH (Вертайм, Германия) и 20 мм пробки для лиофилизации West Flurotec® от West Pharmaceutical Services, Inc. (Lionville, PA, USA) использовали, не подвергая обработке. Все флаконы помещали на полку с применением лотка без дна.
Получение наносуспензий
100 мг/мл итраконазола смешивали либо с 50 мг/мл полоксамера 338, либо с 50 мг/мл Cremophor EL и получали путем мокрого измельчения с помощью шариков в мельнице со средой с высоким усилием сдвига (Netzsch MiniCer®). Для размалывания применяли 0,3 мм шарики диоксида циркония, стабилизированного иттрием. С целью сравнения данных 100 мг/мл рилпивирина стабилизировали с помощью 50 мг/мл полоксамера 338 и размалывали с применением такой же процедуры, как упомянуто выше. Затем концентрацию рилпивирина дополнительно повышали до 200 мг/мл и вновь стабилизировали с помощью 50 мг/мл полоксамера 338. Полученные в результате исходные наносуспензии смешивали с 50 мг/мл PVP K15, трегалозой или сахарозой.
Лазерный дифракционный анализ
Malvern MasterSizer® применяли, чтобы обеспечить определение гранулометрического состава в диапазоне, в котором можно обнаружить даже агломераты. В зависимости от стерического стабилизатора, присутствующего в составе, в качестве среды применяли водный раствор 5 мг/мл полоксамера 338 или Cremophor EL, поскольку стабильность частиц в ходе измерения в чистой воде была недостаточной. Основа для расчета размера частиц представляла собой теория Ми с коэффициентом преломления продукта 1,65 и абсорбцией продукта 0,001. Полученная аппроксимирующая кривая, выполненная программным обеспечением MasterSizer 2000, свидетельствовала о правильности оптической модели. Соблюдали время интегрирования для фона и измерения 60 секунд, при этом измерения проводили в трех повторностях (n=3) на единичную пробу.
Процедура лиофилизации
По 1 мл каждого состава наливали во флаконы 15R. Затем проводили лиофильную сушку на лабораторном лиофилизаторе (VirTis Advantage Plus, SP Scientific, США). Замораживание проводили при -40°C (температура начала полки) в течение 60 минут, включая этапы уравновешивания при +5°C и -5°C в течение 15 минут каждый. Для облегчения кристаллизации полоксамера 338 в составе осуществляли этап нормализации при -20°C (температура начала полки) в течение 90 минут. Скорости линейного изменения температуры полки от заданного значения замораживания до температурного режима полки при первичной сушке составляли 1°C/мин на протяжении всего исследования. Заданное значение температуры начала полки в ходе первичной и вторичной сушки составляло 40°C. Время выдержки (период выдерживания) на этом этапе составляло либо 60 минут, либо 600 минут для обеспечения изменения во влагосодержании в образцах. На протяжении экспериментов давление в камере в ходе первичной и вторичной сушки контролировали при 100 мТорр. Необходимо отметить, что циклы лиофилизации проводили в двух повторностях (n=2) на состав.
Температуры продукта в ходе лиофилизации измеряли с помощью откалиброванных T-типа медно/константановых термопар 30 калибра от Omega (Omega Engineering, Стэмфорд, Коннектикут). Каждую термопару вводили через пробку и размещали в центре дна флакона для получения как репрезентативного контроля температуры в продукте, так и для точного выявления конечной точки фазы сублимации льда.
Дифференциальная сканирующая калориметрия, DSC
Определение тепловых переходов во всех смесях составов проводили с применением MettlerDSC822e (Mettler Toledo, Грайзензее, Швейцария). Сбор данных проводили в температурном диапазоне от 5°C до 150°C. Применяемая скорость нагрева составляла 5°C/мин.
Измерения остаточной влажности по Карлу Фишеру (KF)
Остаточную влажность лиофилизированных образцов измеряли с применением кулонометра Metrohm Karl Fischer 831 KF вместе с установкой Metrohm Thermoprep 832. Примерно 50 мг продукта взвешивали в специальном стеклянном флаконе и затем помещали в установку для нагревания после продувки флакона с образцом с помощью сухого азота. Продукт нагревали до 140°C в течение определенного периода времени, и влага собиралась в растворителе для титрования. Остаточное влагосодержание описывали в процентах (%), исходя из веса образца.
Процедура тестирования на стабильность
Сразу после лиофилизации продукты (n=4) запечатывали и хранили при 25°C и 40°C/75RH, соответственно. Через 1, 2 и 3 месяца образцы анализировали в отношении среднего размера частиц и гранулометрического состава. Измерения с помощью DSC и по Карлу Фишеру проводили после лиофилизации, а также через 3 месяца хранения.
d50, d95 и RDI-величины получали сразу после завершения лиофилизации и после 3 месяцев хранения при 25°C и 40°C, соответственно. Состав: наночастицы рилпивирина с концентрацией 200 мг/мл стабилизировали 50 мг/мл полоксамера 338 отдельно или в комбинации с 50 мг/мл поливинилпирролидона (PVP типа K15), 50 мг/мл трегалозы или 50 мг/мл сахарозы. Необходимо отметить, что рассчитанные стандартные отклонения (n=4) были небольшими, что указывает на очень однородные d50, d95 и RDI-величины. Сокращения представляют собой P338 = полоксамер 338, Tr = трегалоза, Su = сахароза, PVP = PVP K15, T0 = d50, d95 и RDI-величины, определенные сразу после лиофилизации, T3 = d50, d95 и RDI-величины после 3 месяцев хранения.
Дополнительные данные показаны на фигурах 1-6.
Экспериментальная часть 2
a) Лекарственные наносуспензии с PVP K12, или PVP K17, или трегалозой
Получение лекарственных наносуспензий 1-5a
Получение концентрированной наносуспензии:
112,5 г полоксамера 338 растворили в воде для инъекций. Добавляли 450 г рилпивирина и суспендировали. Добавляли воду для инъекций до необходимого конечного веса (300 мг рилпивирина и 75 мг полоксамера/мл). Полученную в результате суспензию измельчали в Netzsch Pharma Labstar (камера измельчения 526 мл) с 80% загрузкой шариками из 300 мкм шариков диоксида циркония, стабилизированного иттрием. Измельчение проводили при скорости перемешивающего устройства 2000 оборотов в минуту до достижения соответствующего размера частиц (измеряли на Malvern).
После измельчения:
Для лекарственной наносуспензии 1, брали 133,3 мл (или 144 г) концентрированной наносуспензии и добавляли 50 мл 200 мг/мл исходного раствора PVP K17. Добавляли воду для инъекций до 200 мл (или 212,6 г) и смесь перемешивали до однородного состояния.
Для лекарственной наносуспензии 2, брали 133,3 мл (или 144 г) концентрированной наносуспензии, и добавляли 25 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до 200 мл (или 212,6 г) и смесь перемешивали до однородного состояния.
Для лекарственной наносуспензии 3, брали 133,3 мл (или 144 г) концентрированной наносуспензии и добавляли 50 мл 200 мг/мл исходного раствора PVP K12. Добавляли воду для инъекций до 200 мл (или 212,6 г) и смесь перемешивали до однородного состояния.
Для лекарственной наносуспензии 4, брали 133,3 мл (или 144 г) концентрированной наносуспензии и добавляли 25 мл 200 мг/мл исходного раствора PVP K12. Добавляли воду для инъекций до 200 мл (или 212,6 г) и смесь перемешивали до однородного состояния.
Для лекарственной наносуспензии 5, брали 133,3 мл (или 144 г) концентрированной наносуспензии и добавляли 50 мл 200 мг/мл исходного раствора трегалозы. Добавляли воду для инъекций до 200 мл (или 212,6 г) и смесь перемешивали до однородного состояния.
Для лекарственной наносуспензии 5a, брали 133,3 мл (или 144 г) концентрированной наносуспензии и добавляли 25 мл 200 мг/мл исходного раствора PVP K17. Добавляли 25 мл 200 мг/мл исходного раствора трегалозы. Добавляли воду для инъекций до 200 мл (или 212,6 г) и смесь перемешивали до однородного состояния.
Получение лекарственных наносуспензий 6 и 7
Получение наносуспензии 6:
16,65 г полоксамера 338 растворяли в воде для инъекций. Добавляли 100 г рилпивирина и суспендировали. Добавляли воду для инъекций до необходимого конечного веса. Полученную в результате суспензию измельчали в Netzsch Pharma Labstar (камера измельчения 137 мл) с 80% загрузкой шариками из 300 мкм шариков диоксида циркония, стабилизированного иттрием. Измельчение проводили при скорости перемешивающего устройства 2000 оборотов в минуту до достижения соответствующего размера частиц (измеряли на Malvern). Добавляли 25 г порошка PVP K17 и перемешивали до растворения.
Получение наносуспензии 7:
25 г полоксамера 338 растворяли в воде для инъекций. Добавляли 150 г рилпивирина и суспендировали. Добавляли воду для инъекций до необходимого конечного веса. Полученную в результате суспензию измельчали в Netzsch Pharma Labstar (камера измельчения 137 мл) с 80% загрузкой шариками из 300 мкм шариков диоксида циркония, стабилизированного иттрием. Измельчение проводили при скорости перемешивающего устройства 2000 оборотов в минуту до достижения соответствующего размера частиц (измеряли на Malvern). К 215,92 мл указанной суспензии добавляли 10,80 г порошка PVP K17 и перемешивали до растворения.
Получение наносуспензии 7a:
25 г полоксамера 338 растворяли в воде для инъекций. Добавляли 150 г рилпивирина и суспендировали. Добавляли воду для инъекций до необходимого конечного веса. Полученную в результате суспензию измельчали в Netzsch Pharma Labstar (камера измельчения 137 мл) с 80% загрузкой шариками из 300 мкм шариков диоксида циркония, стабилизированного иттрием. Измельчение проводили при скорости перемешивающего устройства 2000 оборотов в минуту до достижения соответствующего размера частиц (измеряли на Malvern). К 206,92 мл указанной суспензии добавляли 15,52 г порошка PVP K17 и перемешивали до растворения.
Лекарственные наносуспензии 1-7a лиофилизировали следующим образом:
По 3 мл каждого из составов (для лекарственной наносуспензии 7 и 7a использовали 2 мл объем заливки) заливали в 8 мл флаконы для лиофилизации. Затем проводили лиофильную сушку на опытной установке лиофилизатора HOF. Цикл лиофилизации проводили следующим образом: загрузка при 20°C; замораживание в течение 45 мин. при 5°C, 20 мин. при -5°C, 2 ч. 40 мин. при -40°C, 2 ч. 10 мин. при -20°C, 40 мин. при -40°C; под вакуумом (0,08 мбар), после периода 2 мин. при -40°C, сушка в течение 110 ч. 21 мин. при 40°C и 40 мин. дополнительная сушка при 20°C.
d10, d50, d90 и d99 (мкм), определенные перед лиофилизацией (FD), и определенные сразу после лиофилизации (T0), и через 2 недели, 1 месяц, 3 месяца и 5 месяцев хранения при 40°C, приведены в таблице 2. Восстановление проводили в воде (стабильность до 1 месяца и через 1 месяц включительно) или в воде с 5% глюкозы (стабильность через 3 и 5 месяцев).
T0 после FD
2 недели 40°C
1 месяц 40°C
3 месяца 40°C
5 месяцев 40°C
0,071
0,074
0,073
0,073
0,073
0,163
0,179
0,176
0,175
0178
0,637
0,701
0,717
0,688
0,722
1,199
1,412
1,406
1,284
1,319
T0 после FD
2 недели 40°C
1 месяц 40°C
3 месяца 40°C
5 месяцев 40°C
0,072
0,073
0,073
0,073
0,074
0,169
0,175
0,177
0,175
0,181
0,647
0,682
0,713
0,707
0,736
1,215
1,253
1,312
1,310
1,309
1 месяц 40°C
0,073
0,172
0,695
1,283
T0 после FD
2 недели 40°C
1 месяц 40°C
3 месяца 40°C
5 месяцев 40°C
0,073
0,073
0,073
0,073
0,073
0,171
0,176
0,174
0,176
0,178
0,660
0,713
0,746
0,743
0,752
1,258
1,346
1,424
4,414
1,377
T0 после FD
2 недели 40°C
1 месяц 40°C
3 месяца 40°C
5 месяцев 40°C
0,073
0,074
0,073
0,072
0,073
0,171
0,181
0,173
0,172
0,179
0,663
0,740
0,733
0,732
0,778
1,267
1,429
1,362
1,408
1,442
T0 после FD
2 недели 40°C
0,073
0,074
0,171
0,194
0,667
1,403
1,239
2,97
T0 после FD
2 недели 40°C
1 месяц 40°C
3 месяца 40°C
5 месяцев 40°C
0,072
0,071
0,073
0,073
0,073
0,169
0,163
0,176
0,174
0,178
0,650
0,688
0,786
0,749
0,780
1,221
1,384
1,549
1,413
1,448
T0 после FD
2 недели 40°C
1 месяц 40°C
3 месяца 40°C
5 месяцев 40°C
0,072
0,073
0,070
0,073
0,073
0,163
0,172
0,161
0,174
0,179
0,657
0,692
0,700
0,709
0,764
1,549
1,331
1,765
1,408
1,440
T0 после FD
2 недели 40°C
3 месяца 40°C
5 месяцев 40°C
0,073
0,078
0,073
0,073
0,173
0,199
0,177
0,178
0,759
0,801
0,818
0,882
2,492
2,994
2,095
1,811
T0 после FD
2 недели 40°C
3 месяца 40°C
5 месяцев 40°C
0,072
0,078
0,073
0,073
0,166
0,215
0,176
0,178
0,747
0,921
0,769
0,806
2,803
4,200
1,563
1,556
b) Скрининг различных концентраций PVPK17
Получение лекарственных наносуспензий 8-11
Получение концентрированной наносуспензии:
50 г полоксамера 338 растворили в воде для инъекций. Добавляли 300 г рилпивирина и суспендировали. Добавляли воду для инъекций до необходимого конечного веса (300 мг рилпивирина и 50 мг полоксамера/мл). Полученную в результате суспензию измельчали в Netzsch Pharma Labstar (камера измельчения 526 мл) с 80% загрузкой шариками из 300 мкм шариков диоксида циркония, стабилизированного иттрием. Измельчение проводили при скорости перемешивающего устройства 2000 оборотов в минуту до достижения соответствующего размера частиц (измеряли на Malvern).
После измельчения:
Для лекарственной наносуспензии 8, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 6,1 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Для лекарственной наносуспензии 9, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 12,2 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Для лекарственной наносуспензии 10, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 18,3 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Для лекарственной наносуспензии 11, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 24,4 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Лекарственные наносуспензии 8-11 лиофилизировали следующим образом:
По 3 мл каждого состава заливали в 8 мл флаконы для лиофилизации. Затем проводили лиофильную сушку на опытной установке лиофилизатора HOF. Цикл лиофилизации проводили следующим образом: загрузка при 20°C; замораживание в течение 45 мин. при 5°C, 20 мин. при -5°C, 2 ч. 40 мин. при -50°C, 2 ч. 10 мин. при -20°C, 40 мин. при -50°C; под вакуумом (0,08 мбар), после периода 2 мин. при -50°C, сушка в течение 42 ч. 21 мин. при 40°C и 40 мин. дополнительной сушки при 20°C.
d10, d50, d90 и d99 (мкм), определенные перед лиофилизацией (FD), и определенные сразу после лиофилизации (T0), и через 1 и 3 месяца хранения при 40°C, приведены в таблице 3.
Наносуспензии восстанавливали в воде с 5% глюкозы до соответствующей концентрации 300 мг рилпивирина/мл.
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,072
0,073
0,073
0,163
0,176
0,176
0,587
0,706
0,719
1,119
1,416
1,470
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,072
0,072
0,072
0,163
0,166
0,171
0,583
0,629
0,662
1,106
1,188
1,211
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,072
0,073
0,072
0,165
0,172
0,169
0,584
0,611
0,625
1,104
1,144
1,183
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,072
0,073
0,073
0,165
0,171
0,170
0,579
0,613
0,626
1,104
1,195
1,185
c) Скрининг различных средств тоничности
Получение лекарственных наносуспензий 12-15
Получение концентрированной наносуспензии:
50 г полоксамера 338 растворяли в воде для инъекций. Добавляли 300 г рилпивирина и суспендировали. Добавляли воду для инъекций до необходимого конечного веса (300 мг рилпивирина и 50 мг полоксамера/мл). Полученную в результате суспензию измельчали в Netzsch Pharma Labstar (камера измельчения 526 мл) с 80% загрузкой шариками из 300 мкм шариков диоксида циркония, стабилизированного иттрием. Измельчение проводили при скорости перемешивающего устройства 2000 оборотов в минуту до достижения соответствующего размера частиц (измеряли на Malvern).
После измельчения:
Для лекарственной наносуспензии 12, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 12,2 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли 16,3 мл 200 мг/мл исходного раствора глюкозы (220 мг/мл глюкозы моногидрата) и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Для лекарственной наносуспензии 13, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 12,2 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли 11,2 мл 200 мг/мл исходного раствора глюкозы (220 мг/мл глюкозы моногидрата) и смесь перемешивали до однородного состояния. Добавляли 1,6 мл 300 мг/мл исходного раствора лимонной кислоты, pH 5,5, и смесь перемешивали до однородного состояния. Добавляли раствор NaOH до pH 6. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Для лекарственной наносуспензии 14, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 12,2 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли 5,2 мл 300 мг/мл исходного раствора лимонной кислоты, pH 5,5, и смесь перемешивали до однородного состояния. Добавляли раствор NaOH до pH 6. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Для лекарственной наносуспензии 15, брали 65 мл (или 70,4 г) концентрированной наносуспензии, и добавляли 12,2 мл 200 мг/мл исходного раствора PVP K17, и смесь перемешивали до однородного состояния. Добавляли 5,9 мл 100 мг/мл исходного раствора NaCl и смесь перемешивали до однородного состояния. Добавляли воду для инъекций до конечного объема (97,5 мл) или конечного веса (103,6 г).
Лекарственные наносуспензии 12-15 лиофилизировали следующим образом:
По 3 мл каждого состава заливали в 8 мл флаконы для лиофилизации. Затем проводили лиофильную сушку на опытной установке лиофилизатора HOF. Цикл лиофилизации проводили следующим образом: загрузка при 20°C; замораживание в течение 45 мин. при 5°C, 20 мин. при -5°C, 2 ч. 40 мин. при -50°C, 2 ч. 10 мин. при -20°C, 40 мин. при -50°C; под вакуумом (0,08 мбар), после периода 2 мин. при -50°C, сушка в течение 42 ч. 21 мин. при 40°C и 40 мин. дополнительной сушки при 20°C.
d10, d50, d90 и d99 (мкм), определенные перед лиофилизацией (FD), и определенные сразу после лиофилизации (T0), и через 1 и 3 месяца хранения при 40°C, приведены в таблице 4.
Наносуспензии восстанавливали в воде до соответствующей концентрации 300 мг рилпивирина/мл (гипертонические).
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,072
0,073
0,074
0,166
0,176
0,180
0,577
0,722
0,811
1,127
1,338
1,658
T0 после FD
0,072
0,167
0,595
1,128
3 месяца 40°C
0,073
0,176
0,705
1,392
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,072
0,073
0,074
0,167
0,173
0,185
0,595
0,816
1,225
1,110
2,609
3,848
T0 после FD
1 месяц 40°C
3 месяца 40°C
0,073
0,073
0,072
0,172
0,177
0,171
0,602
0,782
0,708
1,129
4,298
1,565
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИСПЕРГИРУЕМЫЕ КОМПОЗИЦИИ | 2017 |
|
RU2826218C2 |
| ПОРОШКИ ДЛЯ СОЗДАНИЯ ЖИДКОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ | 2008 |
|
RU2477133C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА 4-[[4-[[4-(2-ЦИАНОЭТЕНИЛ)-2,6-ДИМЕТИЛФЕНИЛ]АМИНО]-2-ПИРИМИДИНИЛ]АМИНО]БЕНЗОНИТРИЛА | 2008 |
|
RU2498979C2 |
| Комбинация, ее применение и способы лечения с использованием указанной комбинации | 2018 |
|
RU2755710C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(1,6-ДИГИДРО-6-ОКСО-2-ПИРИМИДИНИЛ)АМИНО-БЕНЗОНИТРИЛА | 2006 |
|
RU2458056C2 |
| ЛЕЧЕНИЕ И ПРЕДУПРЕЖДЕНИЕ ВИЧ-ИНФЕКЦИИ | 2010 |
|
RU2569058C2 |
| СПОСОБ ЛЕЧЕНИЯ ВИЧ КАБОТЕГРАВИРОМ И РИЛПИВИРИНОМ | 2020 |
|
RU2840867C1 |
| Фармацевтическая наносуспензия для терапии ВИЧ-инфекции | 2017 |
|
RU2665383C1 |
| ИМПЛАНТИРУЕМЫЕ УСТРОЙСТВА ДЛЯ ЛЕЧЕНИЯ ВИЧ | 2009 |
|
RU2546529C2 |
| Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения | 2019 |
|
RU2717101C1 |
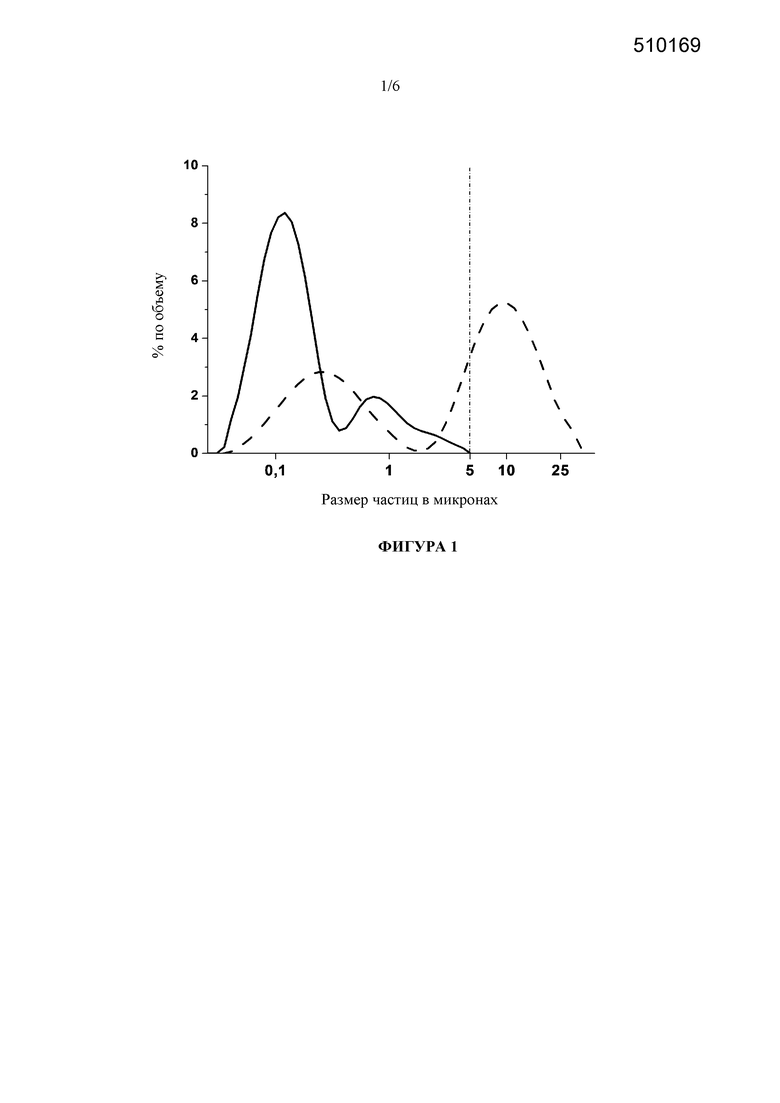
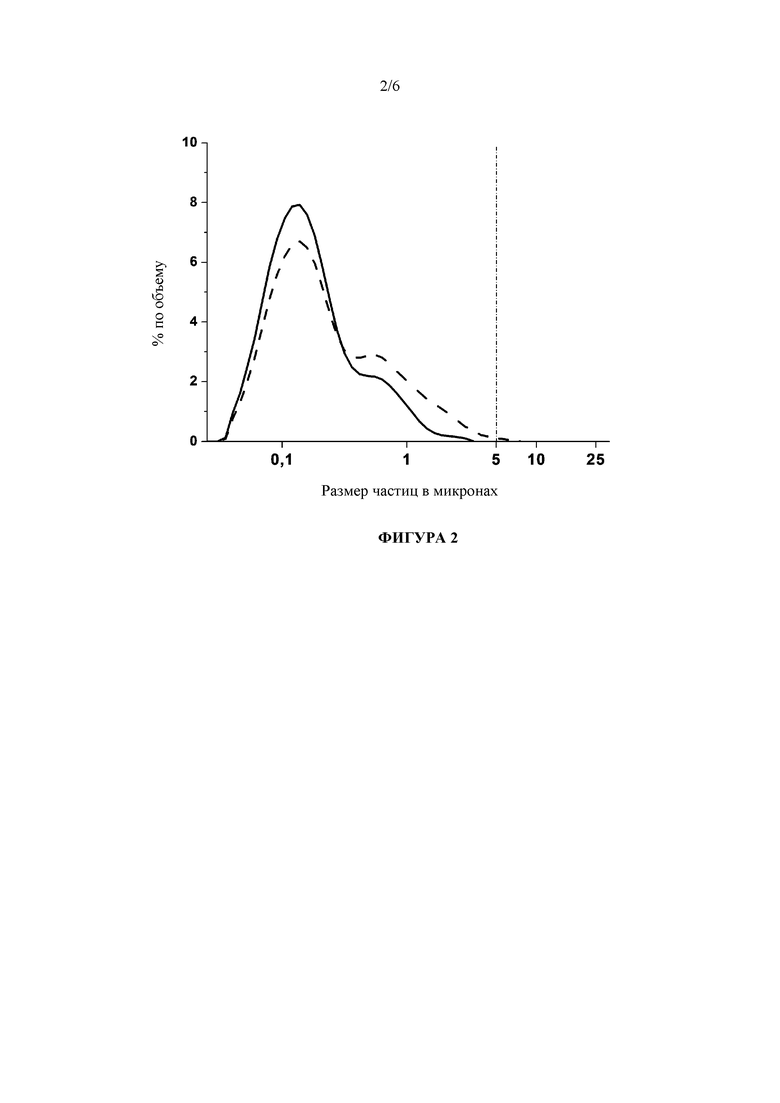
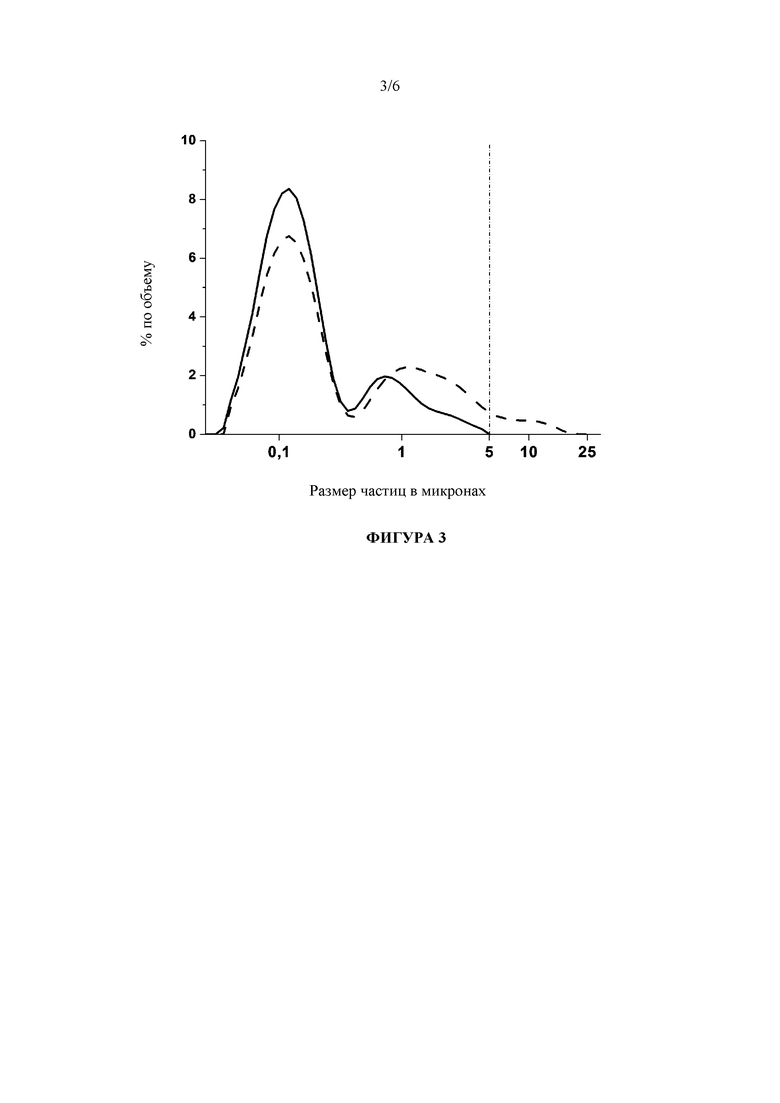
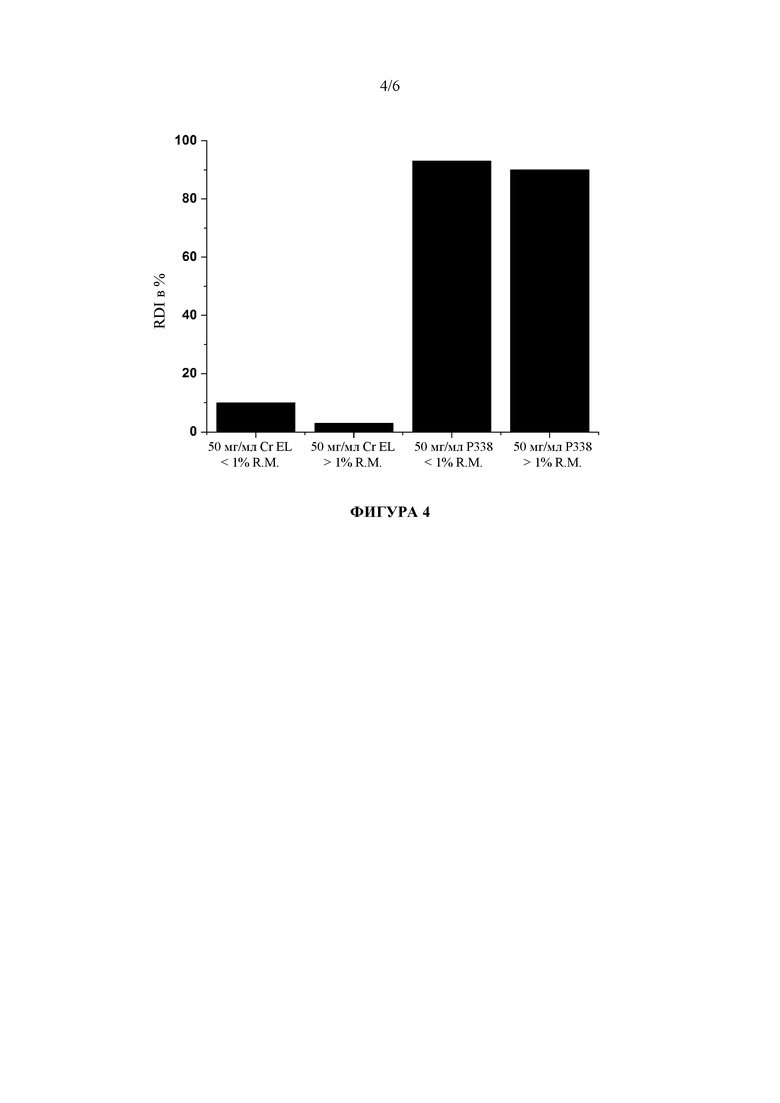
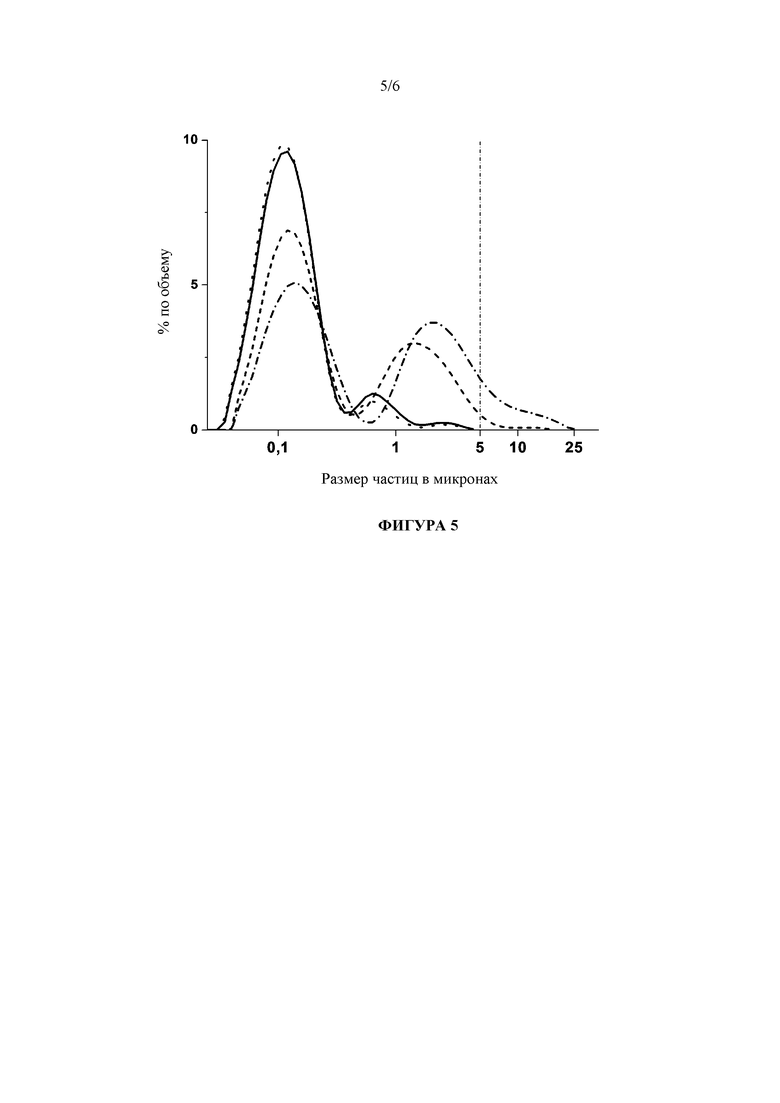
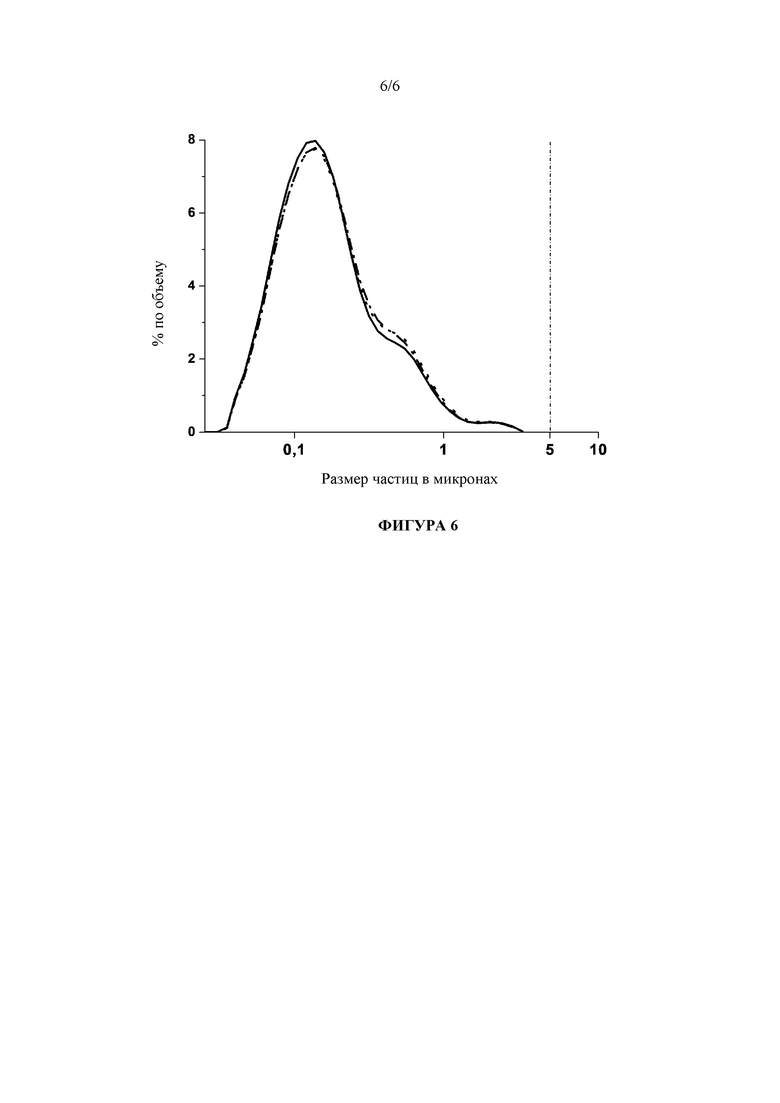
Настоящее изобретение относится к лиофилизированной (также называемой лиофильно высушенной) лекарственной наносуспензии, содержащей 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, или его стереоизомерную форму, или его фармацевтически приемлемую соль и стерический стабилизатор, представляющий собой твердое вещество при комнатной температуре и поливинилпирролидон. Композиция лиофилизированной лекарственной наносуспензии по настоящему изобретению обладает приемлемой стабильностью гранулометрического состава при хранении, в том числе при длительном хранении. 5 н. и 20 з.п. ф-лы, 6 ил., 7 табл., 2 пр.
1. Лиофилизированная лекарственная наносуспензия, содержащая 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил или его стереоизомерную форму, или его фармацевтически приемлемую соль и стерический стабилизатор, представляющий собой твердое вещество при комнатной температуре и поливинилпирролидон, где в наносуспензии, которая подвергается лиофилизации,
концентрация 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила или его стереоизомерной формы, или его фармацевтически приемлемой соли находится в диапазоне от 1 до 500 мг/мл;
концентрация стерического стабилизатора находится в диапазоне от 1 до 200 мг/мл; и
концентрация поливинилпирролидона находится в диапазоне от 1 до 200 мг/мл.
2. Лиофилизированная наносуспензия по п. 1, где стерический стабилизатор представляет собой кристаллическое твердое вещество при комнатной температуре.
3. Лиофилизированная наносуспензия по п. 1, где стерический стабилизатор представляет собой аморфное твердое вещество при комнатной температуре.
4. Лиофилизированная наносуспензия по п. 1, где стерический стабилизатор выбран из полимера или поверхностно-активного вещества.
5. Лиофилизированная наносуспензия по п. 4, где стерический стабилизатор представляет собой поверхностно-активное вещество.
6. Лиофилизированная наносуспензия по п. 5, где поверхностно-активное вещество представляет собой полоксамер.
7. Лиофилизированная наносуспензия по п. 6, где полоксамер представляет собой полоксамер 338.
8. Лиофилизированная наносуспензия по любому одному из пп. 1-7, где лекарственное средство представляет собой 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрильное основание.
9. Лиофилизированная наносуспензия по пп. 1-7, где лекарственное средство представляет собой Е-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил.
10. Лиофилизированная наносуспензия по любому одному из пп. 1-7, где в наносуспензии, которая подвергается лиофилизации,
концентрация 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила или его стереоизомерной формы, или его фармацевтически приемлемой соли находится в диапазоне от 1 до 400 мг/мл; или от 50 до 200 мг/мл, или от 50 до 100 мг/мл, или от 10 до 100 мг/мл, или от 10 до 75 мг/мл, или от 10 до 50 мг/мл, или от
20 до 50 мг/мл, или составляет около 200 мг/мл, или около 300 мг/мл.
11. Лиофилизированная наносуспензия по п. 10, где в наносуспензии, которая подвергается лиофилизации,
концентрация 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила или его стереоизомерной формы, или его фармацевтически приемлемой соли находится в диапазоне от 50 до 200 мг/мл.
12. Лиофилизированная наносуспензия по п. 10, где в наносуспензии, которая подвергается лиофилизации,
концентрация 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила или его стереоизомерной формы, или его фармацевтически приемлемой соли составляет около 200 мг/мл, или около 300 мг/мл.
13. Лиофилизированная наносуспензия по любому одному из пп. 1-7, 11, 12, где в наносуспензии, которая подвергается лиофилизации,
концентрация стерического стабилизатора находится в диапазоне от 10 до 100 мг/мл, или от 10 до 75 мг/мл, или от 10 до 50 мг/мл, или от 20 до 50 мг/мл, или составляет около 33,3 мг/мл, или около 50 мг/мл.
14. Лиофилизированная наносуспензия по п. 13, где в наносуспензии, которая подвергается лиофилизации,
концентрация стерического стабилизатора находится в диапазоне от 20 до 50 мг/мл.
15. Лиофилизированная наносуспензия по любому одному из пп. 1-7, 11, 12, 14, где в наносуспензии, которая подвергается лиофилизации,
концентрация поливинилпирролидона находится в диапазоне от 10 до 100 мг/мл, или от 10 до 75 мг/мл, или от 10 до 50 мг/мл, или от 20 до 50 мг/мл, или составляет около 12,5 мг/мл, или около 25 мг/мл, или около 50 мг/мл, или около 75 мг/мл.
16. Лиофилизированная наносуспензия по п. 15, где в наносуспензии, которая подвергается лиофилизации,
концентрация поливинилпирролидона находится в диапазоне от 20 до 50 мг/мл.
17. Водная лекарственная наносуспензия, полученная путем восстановления лиофилизированной наносуспензии по любому из пп. 1-16 водной дисперсионной средой.
18. Водная лекарственная наносуспензия, полученная путем восстановления лиофилизированной наносуспензии по п. 1 водной дисперсионной средой, где восстановленная наносуспензия содержит по весу, исходя из общего объема композиции:
(a) от 3% до 50% (вес/объем), или от 10% до 40% (вес/объем), или от 10% до 30% (вес/объем), или 10% (вес/объем), или 20% (вес/объем) или 30% (вес/объем) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила;
(b) от 0,5% до 10%, или от 0,5% до 2% (вес/объем), или 3% (вес/объем), или 5% (вес/объем) стерического стабилизатора, который является твердым веществом при комнатной температуре;
(c) от 5% до 20% (вес/объем), или от 5% до 10% (вес/объем), или 5% (вес/объем) поливинилпирролидона;
(d) от 0% до 10%, или от 0% до 5%, или от 0% до 2%, или от 0% до 1% одного или нескольких буферных средств;
(e) от 0% до 10% или от 0% до 6% (вес/объем) изотонирующего средства;
(f) от 0% до 2% (вес/объем) консервантов и
(g) воду для инъекции в достаточном количестве до 100%.
19. Водная наносуспензия по п. 18, содержащая рилпивирин.
20. Водная наносуспензия по п. 18 или 19, где стерический стабилизатор представляет собой полоксамер.
21. Водная наносуспензия по п. 20, где полоксамер представляет собой полоксамер 338.
22. Способ получения водной наносуспензии, отличающийся восстановлением лиофилизированной наносуспензии по любому из пп. 1-16 водной дисперсионной средой.
23. Фармацевтическая композиция для введения путем внутримышечной или подкожной инъекции, содержащая терапевтически эффективное количество 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли в форме восстановленной наносуспензии 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, или его стереоизомерной формы, или его фармацевтически приемлемой соли в фармацевтически
приемлемом водном носителе, где наносуспензия восстановлена из лиофилизированной наносуспензии, содержащей:
(a) 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, или его стереоизомерную форму, или его фармацевтически приемлемую соль; и
(b) стерический стабилизатор, представляющий собой твердое вещество при комнатной температуре; и
(c) поливинилпирролидон.
24. Фармацевтическая композиция по п. 23, содержащая рилпивирин.
25. Фармацевтическая композиция по п. 24, содержащая полоксамер 338.
| M | |||
| Nakarani et.el | |||
| Itraconazole nanosuspension for oral delivery: Formulation, characterization and in vitro comparison with marketed formulation / Daru: journal of Faculty of Pharmacy, Tehran University of Medical Sciences, 2010, 18(2), pages 84-90 | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Aramwit P | |||
| Et al | |||
| In vitro plasma compatibility study of a nanosuspension formulation / PDA Journal of Pharmaceutical Science and Technology, 2006 Jul-Aug; 60(4), pages 211-217 | |||
| Gerben van ′t Klooster et al | |||
| ПАРОВАЯ ИЛИ ГАЗОВАЯ ТУРБИНА | 1914 |
|
SU278A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| RU 2005122198 А, 27.02.2006. | |||

