Изобретение относится к области анализа органических соединений и может быть использовано в целях решения задач экологического контроля, связанных с хранением, перетариванием, транспортировкой и уничтожением ортохлорбензилиденмалонодинитрила (CS), хлорацетофенона (CN), дибензоксазечина (CR) и морфолида пеларгоновой кислоты (МПК).
Актуальность изобретения заключается в том, что в Указе Президента Российской Федерации от 11 марта 2019 г. №97 «Об Основах государственной политики Российской Федерации в области обеспечения химической и биологической безопасности на период до 2025 года и дальнейшую перспективу» к приоритетным направлениям государственной политики в области обеспечения химической и биологической безопасности отнесены, в том числе: и разработка методик проведения анализа опасных органических веществ, а также разработка процедур проведения химического анализа токсикантов в окружающей среде [1].
Известен способ хемолюминесцентного определения хлорацетофенона в органических экстрактах (Патент RU №2386128 С2, опубл. 10.04.2010) [2]. Сущность изобретения заключается в обработке раствора пробы раствором гидроокиси калия, щелочным раствором люминола и раствором гемина в водно-спиртовой среде при температуре 293 К и при их объемном соотношении, соответственно, 1:1:1:1, с последующим определением интенсивности хемилюминесцентного свечения пробы по сравнению с контролем и определением содержания хлорацетофенона по градуировочному графику.
Известен способ количественного колориметрического определения динитрила ортохлорбензилиденмалоновой кислоты в экстрактах. Сущность изобретения заключается в обработке анализируемого экстракта растворами 1-хлор-2,4-динитробензола /ХДНБ/ и бикарбоната натрия /БКН/ в течении 5 мин при температуре 333 К, используя 0,40%-ный спиртовой раствор ХДНБ, 1,5%-ный водный раствор БКН при объемном соотношении пробы и реагентов 1:1:2 и замеряя оптическую плотность образующего красителя на спектрофотометре (Патент RU №2096765 С1, опубл. 20.11.1997) [3]. Недостатками представленных способов являются трудоемкая пробоподготовка, а также невозможность одновременного количественного определения смеси ирритантов.
Наиболее близким к предлагаемому способу по технической сущности является известный способ, описанный в Методических указаниях МУК 4.1.059 - 08 «Методика выполнения измерения массовой концентрации ортохлорбензилиденмалонодинитрила, хлорацетофенона, дибензоксазепина, капсаициноидов, морфолида пеларгоновой кислоты в спиртовом растворе методом высокоэффективной жидкостной хроматографии» [4].
Согласно известному способу подготовка образцов к анализу заключается в извлечении целевых компонентов из образца методом жидкостной экстракции этиловым спиртом. Далее производят введение подготовленной пробы в пробоотборное устройство жидкостного хроматографа, проводят анализ экстракта в количестве 10 мкл на обращенно-фазной колонке длиной 250 мм и внутренним диаметром 4,6 мм, заполненной сорбентом типа ODS С18, скорость потока подвижной фазы, состоящей из водного раствора метанола 70/30 об.%, - 1,0 см3/мин, длина волны УФ-детектора 230 нм, время удерживания хлорацетофенона - 4,8 мин; ортохлорбензилиденмалонодинитрила - 7,5 мин; дибензоксазепина - 9,0 мин; капсаицина - 13,0 мин; дегидрокапсаицина - 19,3 мин; морфолида пеларгоновой кислоты - 15,9 мин. Определяют содержание каждого компонента с использованием ранее построенных градуировочных графиков. Диапазон измеряемых концентраций в спиртовом растворе для хлорацетофенона, ортохлорбензилиденмалонодинитрила и дибензоксазепина составляет 0,001-1,0 г/дм3; капсаициноидов и морфолида пеларгоновой кислоты - 0,01-1,0 г/дм3.
Недостатками известного способа являются: использование достаточно длинной колонки, что приводит к увеличению продолжительности анализа и увеличению расхода элюента, невозможность применения известного способа для анализа смеси ортохлорбензилиденмалонодинитрила, хлорацетофенона, дибензоксазепина, капсаициноидов, морфолида пеларгоновой кислоты в спиртовом растворе на уровне концентраций ниже 0,001 мг/дм3, а также необходимость постороения градуировочных зависимость для каждого определяемого вещества.
Признаки прототипа, совпадающие с признаками изобретения, состоят в разделении на хроматографической колонке, заполненной обращенно-фазовом носителем, содержащим привитую фазу С18, использование воды в качестве одного из компонентов подвижной фазы, детектировании с использованием диодно-матричного детектора при длине волны 230 нм.
Техническим результатом, достигаемым предлагаемым способом, является обеспечение универсальности условий одновременного определения ирритантов в спиртовых экстрактах, возможность проведения количественного анализа с использованием вещества CS в качестве внутреннего стандарта, расширение диапазона определяемых массовых концентраций при одновременном уменьшении общего времени анализа, экономия химических реактивов за счет использования хроматографической колонки меньшей длины, а также снижение токсичности способа определения за счет перехода на менее токсичный элюент (ПДК ацетонитрила в воздухе рабочей зоны составляет 10,0 мг/м3, ПДК метилового спирта - 5,0 мг/м3). Актуальность снижения токсичности метода связана с профессиональной деятельностью работников исследовательских лабораторий, условия труда которых характеризуются воздействием комплекса химических веществ, обуславливающих риск нарушения здоровья.
Поставленный технический результат достигается предлагаемым Способом определения содержания ирритантов в спиртовых экстрактах методом обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФЖХ), заключающимся в том, что осуществляют пробоподготовку пробы путем жидкостной экстракции и проводят последующий анализ экстракта пробы методом ОФЖХ, для этого осуществляют элюирование спиртового экстракта в объеме 20 мкл смесью ацетонитрила и дистиллированной воды в соотношении 80/20 об.% в изократическом режиме подачи элюента, который осуществляют путем пропускания со скоростью 1,0 см3/мин через хромато-графическую колонку Zorbax SB - C18 длиной 150 мм при температуре термостата 26°С в течение 5 мин, а количество CS, CR, ХАФ и МПК в спиртовом экстракте устанавливают по градуировочному графику вещества CS с использованием относительных массовых градуировочных коэффициентов Kg для каждого вещества.
В предлагаемых условиях анализа определены пределы количественного определения (ПКО) и времена удерживания ирритантов (Таблица 1).

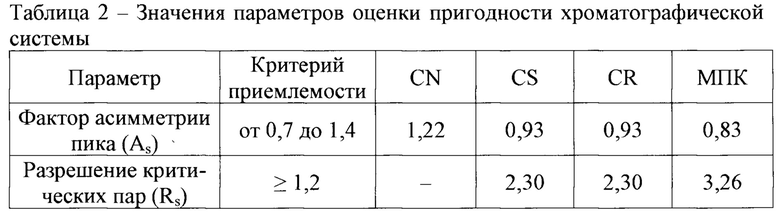
Оценку пригодности хроматографической системы проводили посредством программного обеспечения, получая значения фактора асимметрии хроматографических пиков веществ и разрешения критических пар [5].
Хроматографическую систему считают пригодной, если
- фактор асимметрии пика (As) находится в интервале от 0,7 до 1,4;
- разрешение критических пар (Rs) составляет не менее 1,2.
Результаты оценки пригодности хроматографической системы представлены в таблице 2.
Для возможности проведения количественного анализа без предварительной градуировки прибора с использованием стандартного образца каждого анализируемого ирританта в выбранных условиях необходимо получить массовые градуировочные коэффициенты относительно вещества, рассматриваемого в качестве внутреннего стандарта.
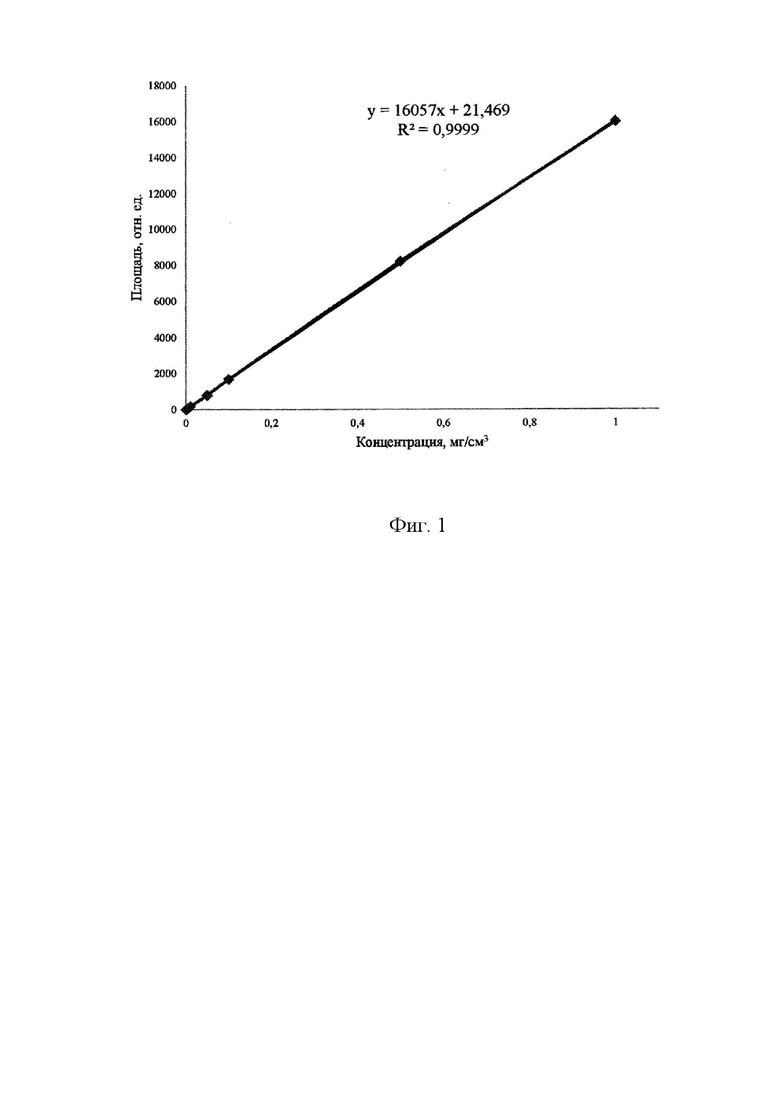
По градуировочному графику вещества CS, представленному на рисунке 1, видно, что в выбранном диапазоне концентраций зависимость площадей пиков от их концентрации носит линейный характер, при этом коэффициент апроксимации стремится к 1, что доказывает линейность графика и возможность использовать CS в качестве внутреннего стандарта.
На основнии значений площадей пиков ирритантов были рассчитаны относительные массовые градуировочные коэффициенты Ks (формула 1) каждого ирританта для каждой концентрации по веществу CS, (Таблица 3):

где SИ - площадь хроматографического пика ирританта, у. ед;
SCS - площадь хроматографического пика CS, у.ед;
CИ - концентрация ирританта, мг/см3;
CCS - концентрация CS, мг/см3.
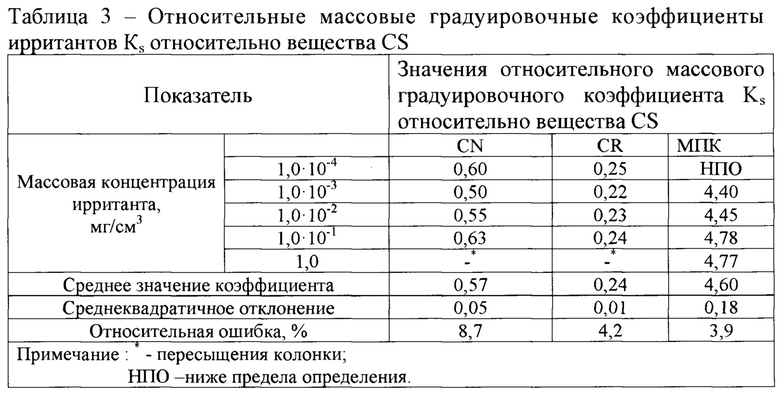
Из данных, представленных в таблице 3, видно, что рассчитанные значения Ks имеют относительную погрешность от 3,9 до 8,7% для всех апробированных концентраций ирритантов.
Для реализации изобретения были использованы следующие химические реактивы, оборудование и расходные материалы:
- стандартные образцы ирритантов, с массовой долей основного вещества не менее 92,0%;
- ацетонитрил для хроматографии фирмы «Merck», Германия;
- спирт этиловый ректиф. технический 1 сорт, ГОСТ 18300-72;
- вода дистиллированная, ГОСТ 6709-72;
- хроматограф жидкостной Agilent-1290 («Agilent Technologist, США) с фотодиодно-матричным детектором с погрешностью определения площади хроматографического пика, не превышающей 1,0%;
- колонка хроматографическая металлическая Zorbax SB - C18 («Agilent Technologist, США) длиной 150 мм и внутренним диаметром 4,6 мм, с зернением частиц 5,0 мкм;
- ультразвуковая ванна лабораторная Trans-sonic 460 «Elma»;
- весы лабораторные типа «Sartorius» 1712 МР-8, Германия с погрешностью взвешивания 0,0005 г.
Проверку работоспособности предлагаемого способа осуществляют следующим образом.
Для установления точности и относительной погрешности заявляемого способа использовали метод «введено-найдено».
1. Для приготовления градуировочных растворов навески CS, CN, CR, МПК растворяют в этиловом спирте при ультразвуковом воздействии для получения базовых растворов с концентрациями 1,0 мг/см3. Градуировочные растворы анализируемых соединений готовят методом последовательного разбавления до концентрации 1,0⋅10-4 мг/см3 (1,0⋅10-3 мг/см3 для МПК).
Модельный раствор, с массовыми концентрациями CS, CN, CR, МПК 0,25 мг/см3 готовят путем смешения при ультразвуковом воздействии соответствующих базовых растворов.
Производят введение в пробоотборное устройство жидкостного хроматографа аликвоту модельного раствора в количестве 20 мкл. Хроматографическое разделение исследуемых веществ проводят в приведенных выше режимных параметрах. Определяют площади хроматографических пиков компонентов пробы.
2. Концентрацию CS, CR, CN и МПК в модельных растворах определяют с помощью градуировочного графика CS с использованием соответствующих градуировочных коэффициентов пересчета Kg согласно данным таблицы 3.
Полученные результаты измерений концентраций ирритантов приведены в таблице 4.
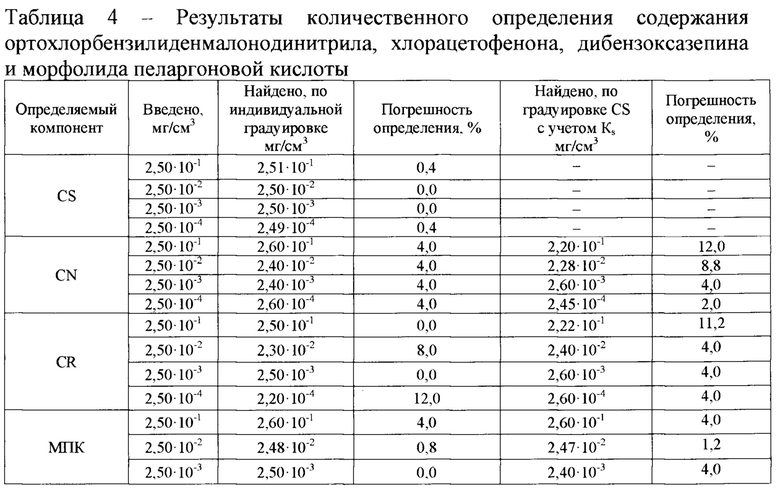
Как видно из приведенных данных, погрешность определения по градуировочной завистимости вещества CS для всех ирритантов не превышает 12,0%.
Таким образом, разработанный способ позволяет с высокой точностью и чувствительностью одновременно в одной пробе определять ортохлорбензилиденмалонодинитрил, хлорацетофенон, дибензоксазепин и морфолид пеларгоновой кислоты в спиртовых экстрактах методом высокоэффективной жидкостной хроматографии без предварительной градуировки прибора с использованием стандартного образца каждого анализируемого ирританта, при уменьшении общего времени и сокращении затрат на проведение анализа пробы.
Список литературы
1. Указ Президента РФ от 11 марта 2019 г. №97 «Об основах государственной политики Российской Федерации в области обеспечения химической ибиологической безопасности на период до 2025 года и дальнейшую перспективу» [Электронный ресурс]. - Электрон. версия печ. публ. - Доступ с сайта справ.-правовой системы. «КонсультантПлюс». URL:http://www.consultant.ru.
2. Пат. RU 2386128, МПК G01N 31/22, G01N 21/76. Способ хемолюминесцентного определения ω-хлорацетофенона [Электронный ресурс] / Дружинин А.А. [и др.]; патентообладатель ФГУ «27 Научный центр Министерства обороны Российской Федерации». - заявл. 12.10.07; опубл. 10.04.10. - Электрон, версия печ. публ. - Доступ с сайта Патентный поиск. URL: http//www.findpatent.ru/.
3. Пат. RU 2386128, МПК G01N 31/22, G01N 21/76. Способ хемолюминесцентного определения ω-хлорацетофенона [Электронный ресурс] / Дружинин А.А. [и др.]; патентообладатель ФГУ «27 Научный центр Министерства обороны Российской Федерации». - заявл. 12.10.07; опубл. 10.04.10. - Электрон, версия печ. публ. - Доступ с сайта Патентный поиск. URL: http//www.findpatent.ru/.
4. Пат. RU 2096765, МПК G01N 21/78, G01N 31/10. Способ количественного колориметрического определения динитрила ортохлорбензилиденмалоновой кислоты в экстрактах [Электронный ресурс] / Дружинин А.А. [и др.]; патентообладатель Военная академия химической защиты им. Маршала Тимошенко С.К.. - заявл. 19.09.94; опубл 20.11.97. - Электрон, версия печ. публ. - Доступ с сайта Патентный поиск. URL: http//www findpatent.ru/.
5. Методика выполнения измерения массовой концентрации ортохлор-бензилиденмалонодинитрила, хлорацетофенона, дибензоксазепина, капсаициноидов, морфолида пеларгоновой кислоты в спиртовом растворе методом высокоэффективной жидкостной хроматографии. Методические указания по методам контроля [утв. и введены в действие Главным государственным санитарным врачом по обслуживаемым организациям и обслуживаемым территориям 23 декабря 2008 г.] / М.Л. Александрова, Г.Н. Кульбицкий, Е.В. Бабаина, Л.А. Муковский. - М.: ФМБА России. - 2008. - 18 с.
6. Долгоносов A.M. Неспецифическая селективность в проблеме моделирования высокоэффективной хроматографии / A.M. Долгоносов - М.: Книжный дом «ЛИБРОКОМ», 2013. - 256 с.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКТ ПЕРВОЙ ПОМОЩИ ПРИ ПОРАЖЕНИИ СРЕДСТВАМИ РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ | 2011 |
|
RU2489135C2 |
| ЖИДКИЙ СОСТАВ РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ | 2005 |
|
RU2284311C1 |
| ЖИДКИЙ СОСТАВ РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ ДЛЯ СРЕДСТВ САМОЗАЩИТЫ | 2008 |
|
RU2381205C1 |
| ЖИДКАЯ РЕЦЕПТУРА ДЛЯ БАЛЛОНЧИКОВ АЭРОЗОЛЬНЫХ МАЛОГАБАРИТНЫХ (ВАРИАНТЫ) | 2000 |
|
RU2197456C2 |
| ГЕЛЕВЫЙ СОСТАВ СЛЕЗОТОЧИВОГО РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ ДЛЯ СРЕДСТВ САМООБОРОНЫ (ВАРИАНТЫ) | 2014 |
|
RU2562993C1 |
| ЖИДКИЙ СОСТАВ РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ (ВАРИАНТЫ) | 2009 |
|
RU2483051C2 |
| ЖИДКИЙ СОСТАВ РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ ДЛЯ СРЕДСТВ САМООБОРОНЫ | 2013 |
|
RU2528000C1 |
| Способ количественного газохроматографического анализа хлорацетофенона в воде методом внутреннего стандарта | 2019 |
|
RU2715378C1 |
| ЖИДКАЯ РЕЦЕПТУРА РАЗДРАЖАЮЩЕГО ДЕЙСТВИЯ ДЛЯ СРЕДСТВ САМОЗАЩИТЫ (ВАРИАНТЫ) | 2001 |
|
RU2213083C2 |
| Способ определения депрессорно-диспергирующих присадок в дизельном топливе | 2021 |
|
RU2756706C1 |
Изобретение относится к аналитической химии. Способ определения содержания ортохлорбензилиденмалонодинитрила (CS), хлорацетофенона (CN), дибензоксазепина (CR), морфолида пеларгоновой кислоты (МПК) в спиртовых экстрактах методом обращенно-фазовой высокоэффективной жидкостной хроматографии включает пробоподготовку путем жидкостной экстракции и последующее разделение на хроматографической колонке, заполненной обращенно-фазовым носителем, содержащим привитую фазу С18, детектирование с использованием диодно-матричного детектора при длине волны 230 нм. Одновременное количественное определение CS, ХАФ, CR, МПК проводится в ходе одного анализа по градуировочному графику вещества CS, с применением рассчитанных значений относительных массовых градуировочных коэффициентов для каждого вещества, с использованием хроматографической колонки длиной 150 мм и внутренним диаметром 4,6 мм с зернением частиц 5,0 мкм, используя в качестве элюента бинарную смесь ацетонитрила и дистиллированной воды в соотношении 80/20 об.%, при температуре термостата колонки 26°С. Техническим результатом является разработка универсального способа одновременного определения ортохлорбензилиденмалонодинитрила, хлорацетофенона, дибензоксазепина и морфолида пеларгоновой кислоты в спиртовых экстрактах методом высокоэффективной жидкостной хроматографии с расширением диапазона определяемых массовых концентраций и уменьшением общего времени анализа. 1 ил., 4 табл.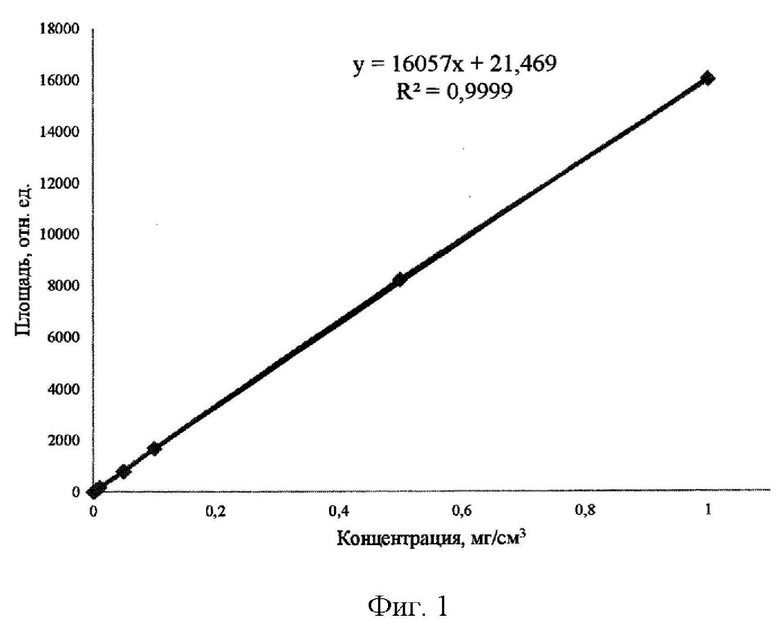
Способ определения содержания ортохлорбензилиденмалонодинитрила (CS), хлорацетофенона (CN), дибензоксазепина (CR), морфолида пеларгоновой кислоты (МПК) в спиртовых экстрактах методом обращенно-фазовой высокоэффективной жидкостной хроматографии, включающий пробоподготовку путем жидкостной экстракции и последующее разделение на хроматографической колонке, заполненной обращенно-фазовым носителем, содержащим привитую фазу С18, детектирование с использованием диодно-матричного детектора при длине волны 230 нм, отличающийся тем, что одновременное количественное определение CS, ХАФ, CR, МПК проводится в ходе одного анализа по градуировочному графику вещества CS, с применением рассчитанных значений относительных массовых градуировочных коэффициентов для каждого вещества, с использованием хроматографической колонки длиной 150 мм и внутренним диаметром 4,6 мм с зернением частиц 5,0 мкм, используя в качестве элюента бинарную смесь ацетонитрила и дистиллированной воды в соотношении 80/20 об.%, при температуре термостата колонки 26°С.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| СПОСОБ ХЕМИЛЮМИНЕСЦЕНТНОГО ОПРЕДЕЛЕНИЯ ω-ХЛОРАЦЕТОФЕНОНА | 2007 |
|
RU2386128C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО КОЛОРИМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ ДИНИТРИЛА ОРТОХЛОРБЕНЗИЛИДЕНМАЛОНОВОЙ КИСЛОТЫ В ЭКСТРАКТАХ | 1994 |
|
RU2096765C1 |
| RU | |||

