Область техники
Изобретение относится к области фармацевтической химии, и описывает способ получения медетомидина, и его производных.
Уровень техники
Медетомидин – синтетический лекарственный препарат, представляющий собой рацемическую смесь двух стереоизомеров 4-[1-(2,3-диметилфенил)этил]-1H-имидазола. Медетомидин является мощным агонистом α2-адренорецепторов, что обуславливает его дозозависимое анальгезирующее и/или седативное действие. Благодаря воздействию на связанные с G-белком октопаминовые рецепторы беспозвоночных, медетомидин ингибирует процесс прикрепления моллюсков, что используется для предотвращения биологического обрастания подводной части корпусов морских судов.
Большинство описанных методов получения медетомидина основано на использовании предшественников, уже содержащих имидазольный цикл, таких как метиловый эфир 4(5)-имидазолкарбоновой кислоты (US4544664A, 1985), N-(триметилсилил)имидазол (WO2009/53709, 2009; CN105254567, 2016; CN106749027, 2017; CN106588777, 2017; CN111253316, 2020 и CN112979552, 2021), 4(5)-ацетилимидазол (CN105884691, 2016), 4(5)-йодимидазол (CN106083724, 2016), (1-тритил-1H-имидазол-4-ил)бороновая кислота (CN108147999, 2018), 4(5)-имидазолкарбальдегид (CN107857731, 2018 и KR2021/12112, 2021), 1-диметилсульфамоил-2-(трет-бутилдиметилсилил)имидазол (CN109608400, 2019), 1H-имидазол-4-карбонитрил (WO2021/89878, 2021).
Однако получение вышеперечисленных предшественников является отдельным многостадийным процессом, часто требующим специальных условий проведения реакции, так как в большинстве случаев данные соединения коммерчески не доступны.
В частности, патент CN109608400 описывает проведение реакции с н-бутиллитием при -78°С для получения ключевого предшественника (1-диметилсульфамоил-2-(трет-бутилдиметилсилил)имидазола), что требует использования низкотемпературных криогенных установок и реакторов специальной конструкции.
(1-тритил-1H-имидазол-4-ил)бороновая кислота, используемая в методе получения медетомидина описанном в патенте CN108147999, так же является коммерчески не доступным предшественником, получение которого является отдельным многостадийным технологическим процессом.
Недостатком методов синтеза медетомидина, основанных на взаимодействии N-(триметилсилил)имидазола с 1-(1-хлорэтил)-2,3-диметилбензолом (CN105254567, 2016; CN106749027, 2017; CN111253316, 2020 и CN112979552, 2021), или 1-(2,3-диметилфенил)этанолом (WO2009/53709, 2009 и CN106588777, 2017) по реакции Фриделя-Крафтса, является использование большого избытка кислоты Льюиса, в качестве которой, в рассматриваемых патентах используется тетрахлорид титана.
Вторым синтетическим подходом к получению медетомидина является создание имидазольного цикла из предшественников, изначально его не содержащих. В этом случае как правило используются линейные, а не конвергентные схемы синтеза, и имидазольный цикл как правило создается на последних стадиях. В частности, указанный подход применительно к получению медетомидина описан в настоящем изобретении.
В настоящее время имеется только очень небольшое число документов, в которых раскрывается данный подход применительно к получению медетомидина. В частности - патенты FI77858C (1982), WO2011070069A1 (2011), WO2012/172122 (2012), WO2012/172120 (2012), WO2013/14428 (2013), WO2016/120635 (2016), CN106518812 (2017) и CN111217756 (2020).
Один из первых методов получения медетомидина включающий создание имидазольного цикла описан в патенте FI77858C (1982), и основан на реакции Бредерика (Hellmut Bredereck. Chem. Ber. 1953, 86, 88). Несмотря на умеренный выход (около 30% на последней стадии), реакция проста в исполнении, и не требует особых условий проведения. Однако, в указанном патенте не описан метод синтеза ключевого предшественника, который вводится в реакцию Бредерика, но на основании известных на момент публикации патента FI77858C подходов к синтезу α-галогенкетонов (исключая прямое галогенирование), наиболее широко использовался метод, основанный на взаимодействии диазометана с галогенангидридами карбоновых кислот, с последующим взаимодействием полученного диазокетона с соответствующим галогеноводородом one pot (Nierenstein M. The action of diazomethane on some aromatic acyl chlorides. J. Chem. Soc. Trans. 1915, 107, 1491-1494). Поскольку диазометан и диазокетоны являются чрезвычайно взрывоопасными, а также токсичными соединениями, получение медетомидина по данной технологии имеет серьезные недостатки, в частности высокую опасность взрыва, возникающую при масштабировании процесса.
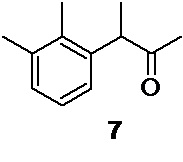
Получение авторами данного предшественника прямым галогенированием соответствующего карбонильного соединения, по нашему мнению, исключается, потому что до настоящего времени в литературе не было описано практичного подхода к синтезу 3-(2,3-диметилфенил)-2-бутанона 7. Единственной публикацией, в которой 3-(2,3-диметилфенил)-2-бутанон 7 был упомянут как компонент сложной трудноразделимой смеси изомерных кетонов, образующейся при перегруппировке 1,5,8-триметил-6-метилиден-трицикло[3.2.1.02,7]окт-3-ен-8-эндо-ола является статья Gabriele Mukherjee-Müller et al. (Helv. Chim. Acta 1977, 60, 1758-1780).
Патент WO2011070069A1 (2011) описывает способ синтеза, при котором имидазольное кольцо создается в ходе многостадийного процесса, начиная с коммерчески доступной 2,3-диметилбензойной кислоты. Рассматриваемый метод, однако, имеет существенный недостаток, заключающийся в необходимости проведения реакции гидрирования под давлением (реакция гидрирования в данном синтезе используется на 2-х стадиях), что требует для реализации использования специального оборудования (стальной реактор), а также газообразного водорода.
Патенты WO2013/14428 (2013) и WO2016/120635 (2016) описывают метод получения медетомидина, основанный на конденсации 2-бром-3-(2,3-диметилфенил)бутаналя с ацетатом формамидина в избытке 25% водного раствора аммиака и этанола. Описанный метод включает небольшое число стадий, практичен, но также требует проведения реакции под давлением.
Способ получения медетомидина, включающий создание имидазольного цикла, описанный в патенте WO2012/172122 (2012) и WO2012/172120 (2012), и основанный на взаимодействии 2-(2,3-диметилфенил)-1-пропаналя с толуолсульфонилметил изоцианидом по реакции Ван Лойзена ((а) Albert M. Van Leusen. Tetrahedron Lett. 1972, 2369-2372; (в) J. Org. Chem. 1977, 42, 7, 1153-1159) имеет существенные недостатки и ограничения. В частности, необходимость в хроматографической очистке основания медетомидина, и использовании токсичного цианида натрия. Кроме того, аналогично патенту WO2013/14428 (2013) рассмотренному выше, реакция, приводящая к формированию имидазольного цикла так же проводится под давлением.
Метод синтеза медетомидина, описываемый в патенте CN106518812 (2017), также основан на реакции Ван Лойзена и имеет недостатки аналогичные патентам WO2012/172122 (2012) и WO2012/172120 (2012), рассмотренным выше.
Таким образом, в настоящее время существует потребность в разработке новых синтетических путей получения медетомидина, которые являются более безопасными и достаточно дешевыми при внедрении в промышленное производство. Преимуществом разработанного нами синтетического пути являются широкие возможности по модификации молекулы конечного продукта (медетомидин) и получение различных производных обладающих биологической активностью.
Кроме того, предлагаемый нами новый способ получения медетомидина и его производных выгодно отличаются от способов, опубликованных в патентах WO2013/14428 (2013) и WO2016/120635 (2016) по следующим параметрам:
1. Синтез 3-(2,3-диметилфенил)-2-бутанона 7, - ключевого предшественника медетомидина, осуществляется с помощью однореакторной методологии, в которой реакция кросс-сочетания, позволяющая существенно усложнить углеродный скелет, и снятие защитной группы проводится в одну стадию, что позволяет сильно сократить общее число стадий синтеза.

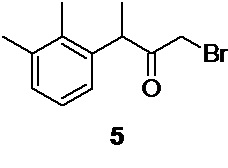
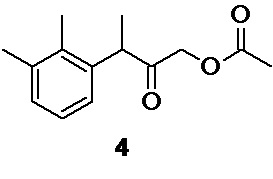
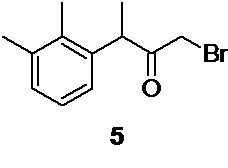
2. Промежуточные интермедиаты: 1-бром-3-(2,3-диметилфенил)бутан-2-он 5, 3-(2,3-диметилфенил)-2-оксобутил ацетат 4, и N-[3-(2,3-диметилфенил)-2-оксобутил]-N-формилформамид 22 могут быть использованы на последующих стадиях без дополнительной очистки

3. Так как описываемый способ синтеза основан на создании имидазольного цикла, нет необходимости в использовании труднодоступных предшественников, уже содержащих цикл имидазола
4. Все реакции, описываемые в предлагаемом способе получения медетомидина, проводятся при атмосферном давлении, что исключает необходимость в специальном оборудовании, таком, как реактора высокого давления
5. Реакции, описываемые в предлагаемом способе получения медетомидина, требующие охлаждения или нагрева, проводятся при умеренно низких (-15°С) или умеренно высоких температурах (≤130°С пути А и В, и ≤160°С путь С), что исключает необходимость в специальном оборудовании, таком, как низкотемпературные криогенные системы и реактора специальной конструкции.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Способ, согласно настоящему изобретению, раскрыт на следующих реакционных схемах:


Исходным соединением для получения 3-(2,3-диметилфенил)бутан-2-она 7 служит коммерчески доступный 2,3-диметилбромбензол 11,

который также может быть получен из 2,3-диметиланилина 13
по литературным методикам (синтез по модифицированной методике из J. Chem. Soc. 1940, 16-18 описан ниже).
Проведение реакции начинается с получения гидробромида 2,3-диметиланилина из 2,3-диметиланилина 13 и 40% бромоводородной кислоты. Соль без выделения и очистки вводят в реакцию с азотистой кислотой, генерируемой in situ из нитрита натрия и бромоводородной кислоты, взятой в избытке.
Так как процесс диазотирования протекает экзотермично, необходимо тщательно контролировать внутреннюю температуру в реакторе, не допуская ее повышения выше 0°С, что достигается изменением скорости добавления раствора нитрита натрия.
Превышение указанной температуры приводит к значительному падению выхода продукта, за счет протекания побочных процессов и разложения диазониевой соли 12,

которое в некоторой степени наблюдается уже при 0°С.
В раствор 2,3-диметилфенилдиазоний бромида 12 вносят свежеосажденную порошкообразную медь, и повышают температуру раствора выше 28°С, что вызывает разложение диазониевой соли с выделением газообразного азота и образованием 2,3-диметилбромбензола 11.
Проведение процесса разложения диазониевой соли 12 при достаточной скорости перемешивания позволяет избежать вспенивания реакционной смеси за счет выделения азота, и обеспечить спокойное течение процесса.
После самопроизвольного прекращения роста температуры, реакционную смесь кратковременно (20-30 минут) нагревают до 70-75°С для завершения процесса, охлаждают, и экстрагируют легким органическим растворителем, таким как гексан, гептан, или легкая фракция петролейного эфира. Использование пентана не рекомендуется по причине низкой температуры кипения, что приводит к значительным потерям растворителя во время процесса обработки и рекуперации.
Так как в процессе диазотирования образуется значительное количество 2,3-диметилфенола 25,
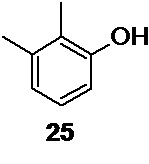
промывка экстракта водными растворами сильных оснований, таких как гидроксид натрия или гидроксид калия, является необходимой операцией. Отделение продукта 11 от смолистых примесей достигается перегонкой с водяным паром из щелочного раствора.
Использование тяжелых растворителей (метиленхлорид, хлороформ) для экстракции 2,3-диметилбромбензола 11 не рекомендуется, из-за их плотности, близкой к плотности реакционной смеси, а также, из-за склонности последних давать стойкую эмульсию в щелочной среде, что сильно осложняет процесс промывки органической фазы щелочными растворами для удаления примеси 2,3-диметилфенола 25.
Соединение структуры 14,
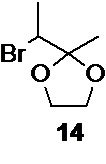
используемое на следующей стадии, может быть синтезировано из коммерчески доступного 3-бром-2-бутанона (структура 15)

по стандартной процедуре, описанной, например, в Greene's Protective Groups in Organic Synthesis 2006.
Исходный 3-бром-2-бутанон 15 так же может быть получен бромированием легко доступного метилэтилкетона 17
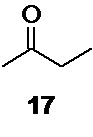
молекулярным бромом в водном растворе уксусной кислоты по литературным методикам (например, Rec. Trav. Chim. Pays-Bas 1946, 65, 691).
Так как реакция бромирования метилэтилкетона 17 приводит к образованию двух региоизомеров, 3-бром-2-бутанона 15 и 1-бром-2-бутанона 16,
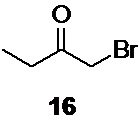
перед постановкой защитной группы смесь должна быть разделена вакуумной дистилляцией на эффективной ректификационной колонке. Колонка высотой 800 мм и диаметром 30 мм, заполненная кольцами Рашига обеспечивает получение 3-бром-2-бутанона 15, содержащего ≤5% региоизомера 16 по данным NMR. Для оценки соотношения региоизомеров 15:16 в дистилляте также может быть использована HPLC, или GC.
Взаимодействие 3-бром-2-бутанона 15 с этиленгликолем в присутствии каталитических количеств моногидрата пара-толуолсульфокислоты в толуоле, с одновременной азеотропной отгонкой образующейся в ходе реакции воды, приводит к образованию этиленкеталя 14.
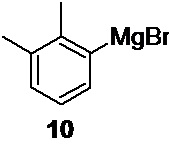
Следующая стадия включает получение 2,3-диметилфенилмагний бромида 10

из 2,3-диметилбромбензола 11, взаимодействие полученного магнийорганического реагента с 2-(1-бромэтил)-2-метил-1,3-диоксоланом 14 и снятие защитной группы.
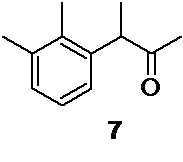
Перечисленные реакции проводятся в одном реакционном сосуде (one pot), без выделения и очистки промежуточных интермедиатов, и приводят к получению кетона 7
Получение реагента Гриньяра 10 начинается с прямой реакции 2,3-диметилбромбензола 11 и металлического магния в сухом тетрагидрофуране (THF) при температуре не выше 50°С (оптимально 48-50°С). Для завершения реакции требуется кратковременный нагрев (70-75°С, 45-60 минут)
Предварительная подготовка растворителя (THF) включает дистилляцию в атмосфере аргона, и сушку активированными молекулярными ситами 3А. Для удаления гидропероксидов из THF могут быть использованы стандартные методы, описанные в литературе, такие как сорбция активированным оксидом алюминия, обработка анионообменной смолой Dowex-1, а также перегонка с предварительным добавлением гидрохинона ((а) Захаров Л.Н. Техника безопасности в химических лабораториях 1991; (в) Donald E. Clark. Peroxides and peroxide-forming compounds. Chem. Health Saf. 2001, 8, 5, 12-22).
Оптимальная концентрация 2,3-диметилфенилмагний бромида 10 в растворе составляет 0.8-1 М. Выше этой концентрации вязкость раствора возрастает, что усложняет процесс его добавления при проведении кросс-сочетания.
Определение концентрации и выхода 2,3-диметилфенилмагний бромида 10 в приготовленном растворе может быть выполнено с помощью титрования точной навески йода, растворенной в насыщенном растворе хлорида лития в THF до исчезновения коричневой окраски раствора (Paul Knochel. Convenient Titration Method for Organometallic Zinc, Magnesium, and Lanthanide Reagents. Synthesis 2006, 5, 890-891).
Установление концентрации магнийорганического реагента в растворе через определение степени конверсии исходного 2,3-диметилбромбензола 11 с помощью GC, с последующей экстраполяцией полученных данных, не может расцениваться как корректный метод для оценки выхода магнийорганического реагента, по причине протекания побочной реакции с образованием бифенила 9

Взаимодействие 2,3-диметилфенилмагний бромида 10 с 2-(1-бромэтил)-2-метил-1,3-диоксоланом 14 в присутствии 10 моль% ацетилацетоната (III) кобальта и 10 моль% N,N,N',N'-тетраметилэтилендиамина (TMEDA) в THF при 0°С приводит к формированию новой С-С связи, и образованию циклического кеталя 8
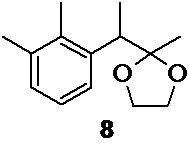
Циклический кеталь 8 легко расщепляется в кислой среде (10% соляная кислота, скорость гидролиза<5 min, мониторинг UPLC), что позволяет провести процесс снятия защитной группы одновременно с гашением реакционной смеси.
В результате оптимизации реакции кросс-сочетания было обнаружено, что снижение количества катализатора c 10 до 5 моль% приводит к падению выхода кетона 7 с 87.2 до 60%.
После экстракции неполярным растворителем (гептан) и концентрирования в вакууме кетон 7 содержит значительное количество (≥20% по данным NMR) этиленгликоля, который образуется в результате расщепления защитной группы, и перегоняется в вакууме вместе с продуктом, что приводит к уширению температурного интервала перегонки до 10°С, и как следствие, к снижению выхода, за счет необходимости отделения большего количества предгона, до начала сбора основной фракции.
Удаление примеси этиленгликоля из 3-(2,3-диметилфенил)бутан-2-она 7 перед вакуумной дистилляцией, осуществляется многократной промывкой продукта водой. Из-за плотности кетона 7, близкой к плотности воды, процесс промывки осложняется формированием стойкой эмульсии. Предварительное разбавление сырого кетона 7 гексаном, или другим легким неполярным растворителем (~1:1.5 об.), позволяет полностью избежать формирования эмульсии в процессе промывки. Полноту удаления примеси этиленгликоля контролируют с помощью NMR спектроскопии, по исчезновению сигнала метиленовых групп (3.76 м.д., s, CDCl3), либо с помощью GC.
Вторым побочным продуктом, образующимся в ходе реакции кросс-сочетания, является 2,2′,3,3′-тетраметилбифенил 9, который также должен быть отделен до процесса дистилляции, так как он летуч в вакууме, и служит источником загрязнения целевого кетона 7 в конце процесса дистилляции.
Эффективное удаление большей части 2,2′,3,3′-тетраметилбифенила (>95% по HPLC) достигается разбавлением сырого кетона 7 метанолом (1:1 об.), с последующей кристаллизацией бифенила 9 при -15…-20°С в течение 2 суток. Время кристаллизации может быть сокращено примерно до 20 часов, за счет внесения в раствор, полученного из кубового остатка кристаллического 9 в качестве затравки.
Удаление следов соединений кобальта может быть осуществлено сорбцией последних активированным (120°С, 20 ч) силикагелем с размером частиц 40-63 мкм в неполярном органическом растворителе, таком как гексан.
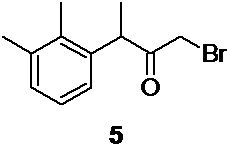
На следующей стадии полученный 3-(2,3-диметилфенил)бутан-2-он 7 бромируется молекулярным бромом по терминальной метильной группе с образованием 1-бром-3-(2,3-диметилфенил)бутан-2-она 5
Оптимальным растворителем, обеспечивающим наибольшую региоселективность является метанол. Процесс бромирования протекает в широком диапазоне температур (от -15°С, до комнатной), однако количество побочных продуктов (ди- и трибромпроизводных), образующихся при реакции, а также ее скорость, чрезвычайно сильно зависят как от температуры, так и от концентрации исходного кетона в растворе.
Бромирование при комнатной температуре (20-25°С) протекает уже в момент добавления брома, и заканчивается за несколько минут. Однако, при таком режиме бромирования сильно возрастает количество примесей полибромированных продуктов (HPLC area целевого продукта 5 ≤49%).
При проведении реакции бромирования при -12°С, скорость образования полибромированных примесей значительно замедляется (HPLC area 5 81%, HPLC area дибромкетона<5%) однако для достижения степени конверсии исходного кетона>95% (HPLC) требуется около 28 часов.
Диапазон оптимальных температур для проведения бромирования находится в пределах от -5 до -8°С.
Использование стехиометрического соотношения бром: кетон 7 приводит только к ≤85% конверсии последнего (HPLC). Для достижения полной конверсии исходного кетона требуется не менее 1.15 экв брома, оптимально 1.20-1.25 экв брома.
Контроль за протеканием побочных процессов достигается также степенью разбавления раствора. Использование менее 10 мл растворителя на 1 г кетона приводит к резкому росту побочных процессов, сопровождающихся образованием полибромированных примесей. Оптимальная концентрация кетона 7 в растворе, находится в диапазоне 0.18-0.22 моль/л.
Добавление брома к метанольному раствору кетона следует вести максимально быстро, желательно одной порцией, так как при медленном добавлении, или добавлении по каплям, образуется заметное количество (>10% HPLC area) примеси региоизомерного продукта бромирования 20

Для завершения реакции может потребоваться повышение температуры до 0°С (примерно после 17-18 часов), и выдержка при этой температуры до полного исчезновения окраски брома. В случае неполной конверсии исходного кетона 7 может потребоваться добавление дополнительных порций брома, что позволяет добиться>97% (HPLC) конверсии 7.
Образующийся в ходе реакции бромирования диметилацеталь 1-бром-3-(2,3-диметилфенил)бутан-2-она 6

легко расщепляется one pot без выделения (скорость гидролиза при рН 3<2 мин, UPLC) избытком бромоводорода, содержащегося в реакционной смеси после разбавления водой, что приводит к образованию 1-бром-3-(2,3-диметилфенил)бутан-2-она 5, который используется на следующей стадии без дополнительной очистки. Продукт 5 нестабилен при комнатной температуре, и быстро темнеет, но может храниться при -18°С в течение нескольких недель без существенного разложения.
Использование в качестве растворителей уксусной кислоты, сложных эфиров, таких как этилацетат, или галогенированных углеводородов, приводит к образованию преимущественно наиболее замещенного 3-бром-3-(2,3-диметилфенил)бутан-2-она 20.
Использование в качестве бромирующего агента бромида пербромида пиридиния (PHBP, 19) в THF, этилацетате или ледяной уксусной кислоте, также приводит к получению почти исключительно 3-бром-3-(2,3-диметилфенил)бутан-2-она 20, и небольшого количества примеси полибромированных соединений.
3-(2,3-диметилфенил)-2-оксобутил ацетат (структура 4), являющийся предшественником для формирования имидазольного цикла в молекуле медетомидина, в случае проведения синтеза по пути А (Схема 1)

получают взаимодействием 1-бром-3-(2,3-диметилфенил)бутан-2-она 5 с ацетатом калия (~3 экв) в подходящем органическом растворителе (ацетонитрил, DMF, DMSO) при комнатной температуре.
В случае использования низкокипящих растворителей (типа ацетонитрила) 4 может быть выделен из реакционной смеси простым концентрированием раствора в вакууме, после предварительной фильтрации осадка бромида калия и избытка ацетата калия.
Удаление следов ацетонитрила достигается промывкой раствора 4 в неполярном растворителе (гексан, гептан, МТBE, этилацетат и т.д.) водой, с последующим концентрированием. Полученный 3-(2,3-диметилфенил)-2-оксобутил ацетат 4 достаточно чист, и может быть использован на следующей стадии синтеза без дополнительной очистки.
Ацетонитрил, регенерированный (>90%) при обработке реакционной смеси может быть использован без дополнительной подготовки на следующей загрузке (на данной стадии синтеза), без ухудшения качества продукта 4.
Проведение реакции в метаноле при комнатной температуре приводит к низкой конверсии исходного 1-бром-3-(2,3-диметилфенил)бутан-2-она (<60% за 43.5 часа). Повышение температуры реакции в случае использования в качестве растворителя метанола, для увеличения конверсии, приводит к резкому росту побочных процессов, что дает продукт, непригодный для использования на следующей стадии без дополнительной очистки.
Преимуществом проведения реакции в DMF или DMSO является сокращение времени реакции с 18-20 часов (в случае ацетонитрила) до 5-6 часов (DMF), однако, в этом случае, для выделения продукта необходимо разбавление реакционной смеси большим объемом воды (3-5 об.), с последующей экстракцией продукта низкокипящим органическим растворителем (гексан, гептан, МТBE, этилацетат и т.д.).
Заключительная стадия процесса включает формирование имидазольного цикла, с образованием медетомидина в виде медного (I) комплекса 3,

выделение свободного основания медетомидина 2,
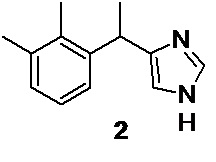
удаление следов меди, и получение фармацевтически приемлемой соли, такой как гидрохлорид медетомидина 1

Взаимодействие 3-(2,3-диметилфенил)-2-оксобутил ацетата 4 с формальдегидом, аммиаком и ацетатом меди (II) в водно-спиртовом растворе по реакции Вейденхагена (Weidenhagen R. Chem. Ber. 1935, 68, 1953) приводит к образованию медного (I) комплекса медетомидина 3.
Оптимальным спиртовым сорастворителем для проведения реакции, является н-пропанол, который обеспечивает полную растворимость исходного 3-(2,3-диметилфенил)-2-оксобутил ацетата 4 в водно-аммиачном растворе. Оптимальное количество н-пропанола составляет от 10 до 15 мл на 1 г 3-(2,3-диметилфенил)-2-оксобутил ацетата 4. В этом случае осаждение медного (I) комплекса медетомидина во время реакции происходит в виде хорошо оформленного кристаллического осадка.
Использование меньшего объема н-пропанола приводит к неполному растворению 4, и как следствие к образованию эмульсии, что в свою очередь ведет к осаждению медного комплекса медетомидина на стенках реактора и на мешалке в виде плотного налета, что затрудняет процесс фильтрации. Кроме того, за счет протекания реакции на поверхности капель, а не в гомогенной среде, происходит не полная конверсия исходного 4, и, как следствие, падение выхода продукта и загрязнение его непрореагировавшим предшественником.
Использование большего объема н-пропанола нецелесообразно по причине растворимости комплекса 3, что может привести к снижению его выхода. Реакция протекает при температуре кипения раствора (62-72°С), и заканчивается за 2 часа. Меньшее время нагрева (<1.5 часа) приводит к падению выхода медного комплекса 3 с 80.9 до 58.5% на данной стадии. Использование в качестве сорастворителей метанола, этанола или изо-пропанола также приводит к осаждению медного комплекса медетомидина на стенках реактора, по причине их недостаточной растворяющей способности по отношению к 4.
Традиционный метод выделения свободных имидазолов из медного комплекса заключается в осаждение меди в виде сульфида меди (I) под действием газообразного сероводорода. Однако, данный метод не может рассматриваться в качестве масштабируемого промышленно-приемлемого процесса, по причине высокой токсичности сероводорода.
Описываемый нами метод позволяет избежать использования сероводорода, и основан на применении нетоксичных комплексонов.
Оптимальным хелатирующим реагентом является пентанатриевая соль диэтилентриаминпентауксусной кислоты (Na5DTPA). Оптимальное мольное соотношение металл: комплексообразователь в этом случае находиться в диапазоне 1: 1.2-1.5.
Тетранатриевая соль этилендиаминтетрауксусной кислоты (Na4EDTA) обладает недостаточной хелатирующей способностью по отношению к медному (I) комплексу медетомидина, что приводит к низкой конверсии исходного комплекса и неполному высвобождению основания медетомидина даже при использовании 3-5 кратного мольного избытка хелатирующего агента и продолжительном времени проведения реакции (>4 суток).
Смещение равновесия процесса достигается также непрерывной экстракцией основания медетомидина в органическую фазу, что облегчает протекание реакции за счет постоянного удаления продукта с поверхности кристаллов медного комплекса. Для увеличения скорости реакции необходимо обеспечить тщательное перемешивание фаз.
Использование в качестве хелатирующих реагентов свободных кислот, таких как EDTA или DTPA, а также их не полностью замещенных солей, таких как динатриевая соль этилендиаминтетрауксусной кислоты (Na2EDTA), для выделения свободного основания медетомидина 2 из медного комплекса 3, приводит к образованию смолообразных продуктов сложного состава, вероятно, за счет наличия свободных карбоксильных групп, одновременно взаимодействующих с основным атомом азота имидазольного цикла и ионом меди.
Формирование фармацевтически приемлемой соли, такой как гидрохлорид медетомидина 1, происходит при взаимодействии основания медетомидина 2 с кислотой, такой как хлороводородная, в подходящем органическом растворителе. По причине высокой растворимости гидрохлорида медетомидина 1 в воде, при использовании концентрированной (35-38%) хлороводородной кислоты в ацетоне, возможно возникновение проблем с кристаллизацией продукта. В этом случае, для инициации кристаллизации необходимо добавление затравки, в виде небольшого количества кристаллов чистого гидрохлорида медетомидина. Внесение затравки на стадии формирования соли благоприятно сказывается на процессе кристаллизации, который наступает сразу, или почти сразу после добавления концентрированной соляной кислоты. Для полного завершения процесса кристаллизации 1, требуется около суток. Выход гидрохлорида медетомидина может быть повышен с 56.2 до 60-65% (в расчете на 2 стадии) за счет концентрирования маточника (приблизительно до 1/3 от исходного объема) после фильтрации основной порции кристаллов, с последующим охлаждением водно-ацетонового раствора в течение 20-25 часов в морозильной камере.
Замена формальдегида на стадии формирования медного комплекса другими альдегидами является удобным способом получения производных медетомидина, имеющих заместитель в положении 2 имидазольного цикла.
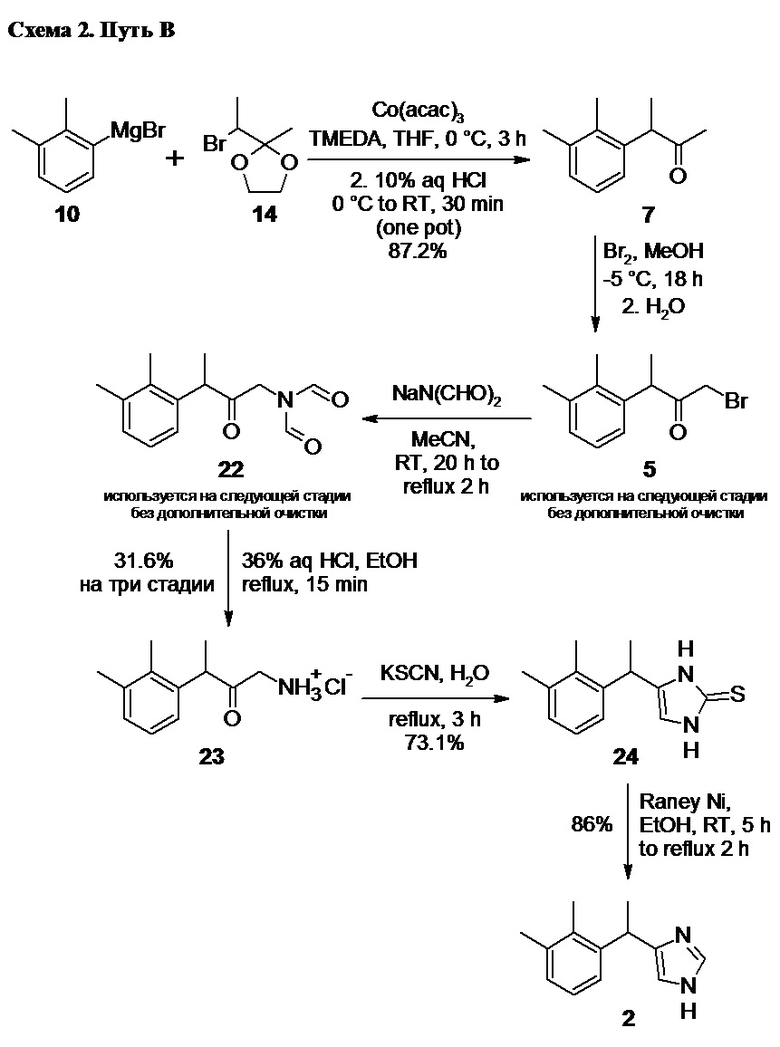
Альтернативным методом формирования имидазольного фрагмента в молекуле медетомидина, является путь В (Схема 2).
В этом случае 1-бром-3-(2,3-диметилфенил)бутан-2-он 5 вводят в реакцию с диформиламидом натрия 21

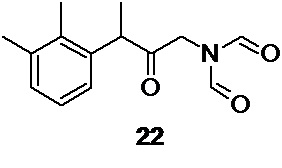
в подходящем органическом растворителе, таком как ацетонитрил, что приводит к образованию промежуточного соединения 22,
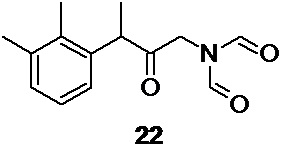
которое может быть использовано на следующей стадии без дополнительной очистки.
Исходный, коммерчески доступный диформиламид натрия 21, используемый в синтезе соединения 22, так же может быть получен по литературным методикам из метилата натрия и формамида, с высоким выходом (например, (а) Allenstein E. Chem. Ber. 1967, 100, 3551; (b) US5599986A).
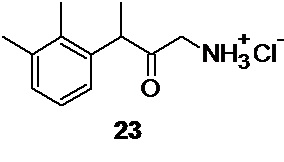
Кислотный гидролиз соединения 22 приводит к образованию гидрохлорида аминокетона 23

Заключительная стадия процесса (Путь В; Схема 2) включает формирование 2-меркаптозамещенного имидазольного цикла, и последующую десульфуризацию, приводящую к образованию медетомидина 2.
Взаимодействие 1-амино-3-(2,3-диметилфенил)бутан-2-она гидрохлорида 23 с тиоцианатом калия в водном растворе по реакции Марквальда (Marckwald W. Ber. Dtsch. Chem. Ges. 1892, 25, 2354) приводит к образованию 4-[1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тиона 24
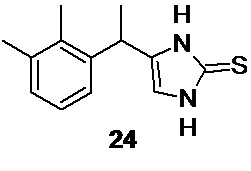
Реакция гладко протекает при нагревании гидрохлорида аминокетона 23 с избытком (4-5 экв) тиоцианата калия в водном растворе при температуре кипения. Оптимальная продолжительность нагрева составляет от 2.5 до 3.5 часов. Продукт реакции 24 практически не растворим в воде, в отличии от исходных реагентов, и выкристаллизовывается из раствора по мере протекания реакции, что существенно упрощает процесс его выделения. В случае необходимости, 4-[1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тион 24 может быть очищен кристаллизацией из водного этанола (~65% об.).
Мягкое удаление меркаптогруппы в соединении 24 с помощью никеля Ренея в спиртовой среде приводит к образованию медетомидина 2 с высоким выходом. Наилучшие результаты достигаются при использовании никеля Ренея марок W-4 и W-5 (классификацию см. Robert L. Augustine. Heterogeneous Catalysis for the Synthetic Chemist 1996).
Создание имидазольного цикла в молекуле медетомидина так же может быть выполнено непосредственно в одну стадию при взаимодействии 1-амино-3-(2,3-диметилфенил)бутан-2-она гидрохлорида 23 с формамидом (Схема 3. Путь С). Несмотря на умеренный выход (~40%) реакция очень проста в исполнении. Так как формамид используется в избытке (~10 экв), и выступает в реакции и как растворитель и как реагент, а также, имеет высокую температуру кипения (210°C), перед экстракцией основания медетомидина из реакционной смеси целесообразно провести гидролиз избытка формамида с помощью концентрированной соляной кислоты (1 час, комнатная температура).
Экспериментальная часть
Список сокращений
кислоты
кислоты
кислоты
Общие методы
Исходный 2,3-диметиланилин 13 был приобретен в Sigma-Aldrich (99%, CAS 87-59-2), и использовался без дополнительной очистки. Все эксперименты, в которых использовалась инертная атмосфера, были выполнены по стандартной технике с использованием линии Шленка. В качестве инертного газа был использован аргон 99.999% (Linde Gas) без дополнительной очистки. Все растворители и реагенты были приобретены у коммерческих поставщиков, и использовались без дополнительной очистки и подготовки, за исключением THF и метанола. THF (ACS 99.6%) перед использованием был перегнан в атмосфере аргона (с добавкой ~0.1% гидрохинона), и высушен активированными молекулярными ситами 3А (100 г/л) в течение 65 часов. Метанол используемый в реакции бромирования был высушен активированными молекулярными ситами 3А (45 г/л) в течение 168 часов. Активация молекулярных сит проводилась нагреванием в вакууме (0.01 mmHg) при 300-320°С в течение 3 часов. Для синтеза магнийорганического реагента была использована магниевая стружка марки Turnings for Grignard Reaction от Fisher Chemical, хранившаяся в сушильном шкафу при 120°С. Определение концентрации 2,3-диметилфенилмагний бромида было выполнено с помощью прямого йодометрического титрования в насыщенном растворе хлорида лития в сухом THF по методу Пауля Кнохеля. Детали стеклянных установок (за исключением реакторов большого объема), используемые на стадии приготовления магнийорганического реагента 10 и кросс-сочетания были высушены в сушильном шкафу при 120°С в течение 24 часов. Стеклянные реактора были высушены перед работой с чувствительными к влаге соединениями в вакууме пластинчато-роторного насоса (0.01 mmHg) с помощью прогрева теплоносителем в течение 2 часов при 90°С, и заполнены аргоном в три цикла откачки/заполнения. Спектры 1H и 13C NMR были записаны на спектрометре Agilent 400 MHz (400 МГц для 1H и 100 МГц для 13C) в CDCl3 и D6-DMSO. Химические сдвиги выражаются в миллионных долях относительно остаточных сигналов применяемых дейтерированных растворителей. Определение остаточного содержания тяжелых металлов проводилось на атомно-абсорбционном спектрометре Agilent Technologies 240 AA. Для подготовки проб к определению тяжелых металлов использовалась микроволновая система разложения MARS One. В качестве окислителя для минерализации проб использовалась смесь 70% азотной кислоты и 30% пероксида водорода (10:1 об., 180°С, 25 мин). ТСХ анализ проводился на алюминиевых пластинах ALUGRAM Xtra SIL G/UV254 (Macherey-Nagel). Пластины визуализировали с помощью ультрафиолетовой лампы 254 нм. Мониторинг полноты протекания реакций и оценка хроматографической чистоты интермедиатов осуществлялись с помощью HPLC системы Alliance (Waters) с матричным фотодиодным детектором (PDA).
Аналитические условия
Метод А
Прибор: Waters Alliance HPLC; Колонка XBridge C18, 3.5 μm, 4.6 мм х 150 мм; Элюент А: MeCN; Элюент В: H2O+0.15% об. 50% H3PO4; Соотношение элюентов А:В: 50:50; Изократический режим элюирования; Скорость потока 1.50 мл/мин; Температура 40°С; Объем вкола 10 мкл; Длина волны детектирования: 190-350 нм.
Метод В
Прибор: Waters Alliance HPLC; Колонка XBridge C18, 3.5 μm, 4.6 мм х 150 мм; Элюент А: MeCN; Элюент В: H2O+0.15% об. 50% H3PO4; Соотношение элюентов А:В: 30:70; Изократический режим элюирования; Скорость потока 1.00 мл/мин; Температура 40°С; Объем вкола 10 мкл; Длина волны детектирования: 190-350 нм.
Метод С
Прибор: Waters Alliance HPLC; Колонка XBridge C18, 3.5 μm, 4.6 мм х 150 мм; Элюент А: MeCN; Элюент В: H2O+0.15% об. 50% H3PO4; Градиент: 0-1.20 мин 30% А, 1.20-12.00 мин 30-90% А, 12.00-15.00 мин 90% А, 15.01-18.00 мин 90-30% А; Скорость потока 1.20 мл/мин; Температура 40°С; Объем вкола 10 мкл; Длина волны детектирования: 190-350 нм.
Экспериментальная часть
2,3-диметилбромбензол (11)
(Модифицированная процедура J. Chem. Soc. 1940, 16-18)

В 10 л стеклянный реактор с термостатируемой рубашкой, снабженный механической мешалкой, предварительно охлажденный до -10°С, загружают 3747.5 г (22.23 моль; 4.0 экв) 48% бромоводородной кислоты и 750 мл деионизированной воды. Раствор охлаждают до 4…5°С, и при перемешивании (250-300 об/мин), из капельной воронки в течение 45 минут добавляют 673.45 г (5.55 моль; 1.0 экв) 2,3-диметиланилина 13, с такой скоростью, чтобы температура реакционной смеси удерживалась в диапазоне 4…5°С. Полученную суспензию кристаллического гидробромида 2,3-диметиланилина бежевого цвета охлаждают до -1…-2°С, и из капельной воронки в течение 2 часов добавляют раствор 421.8 г (6.12 моль; 1.10 экв) нитрита натрия в 680 мл деионизированной воды, с такой скоростью что бы температура реакционной смеси удерживалась в диапазоне 0…-2°С (температура теплоносителя в рубашке реактора -15°С). После добавления раствора нитрита натрия температуру реакционной смеси повышают до 0°С, и желто-коричневый раствор 2,3-диметилфенилдиазоний бромида 12 перемешивают в течение 15 минут для завершения процесса диазотирования. Далее, увеличивают число оборотов мешалки (до 450-500 об/мин), вносят в реактор суспензию 100 г свежеосажденного медного порошка в 350 мл деионизированной воды, и повышают температуру теплоносителя в рубашке реактора до 30°С. При прогреве реакционной смеси до 28…30°С начинается экзотермический процесс разложения соли диазония с самопроизвольным ростом температуры реакционной смеси до 60°С в течение 4-5 минут. После прекращения самопроизвольного роста температуры запускают прогрев и циркуляцию теплоносителя в рубашке реактора, и реакционную смесь фиолетово-черного цвета нагревают при 70°С в течение 30 минут. Реакционную смесь охлаждают до комнатной температуры, и экстрагируют 2×1000 мл легкого петролейного эфира (40-70). Нижний водный слой сливают через донный спуск, и органическую фазу темного цвета последовательно промывают в реакторе 4 л деионизированной воды (10 минут), и 4×4 л 2.5% раствора гидроксида натрия (по 10 минут) для удаления примеси 2,3-диметилфенола. Органическую фазу отделяют, и концентрируют в вакууме (<40°С). Полученное масло оранжево-красного цвета перегоняют с паром из раствора, содержащего 50 г гидроксида натрия в 1 л деионизированной воды, собирая 3.5 л дистиллята. Прозрачный нижний слой светло-оранжевого цвета отделяют, и интенсивно перемешивают в течение 40 минут с 100 мл концентрированной серной кислоты на магнитной мешалке. Нижний сернокислотный слой, окрашенный в темно-красный цвет отделяют в делительной воронке, а верхний, светло-желтый органический слой промывают 2×600 мл насыщенного раствора гидрокарбоната натрия. Водную фазу экстрагируют 3×100 мл метиленхлорида. Объединенную органическую фазу сушат безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Полученный 2,3-диметилбромбензол подвергают фракционной перегонке в вакууме с использованием дефлегматора Вигре высотой 300 мм с вакуумированной рубашкой, собирая фракцию с t кип 76.2-76.5°С/6 mmHg.
Выход 391.8 г (38.1%). Бесцветная жидкость с характерным запахом, напоминающим запах ксилола. 1H NMR (400 MHz, CDCl3) δ 7.43 (d, 1H), 7.11 (d, 1H), 6.97 (t, 1H), 2.40 (s, 3H), 2.36 (s, 3H); 13С (100 MHz, СDCl3) δ 138.61, 136.24, 130.34, 128.97, 126.88, 125.67, 21.39, 19.51; tR 11.33 мин (HPLC Area 99.7%, метод А).
3-бром-2-бутанон (15)
(модифицированная процедура Rec. Trav. Chim. Pays-Bas 1946, 65, 691).
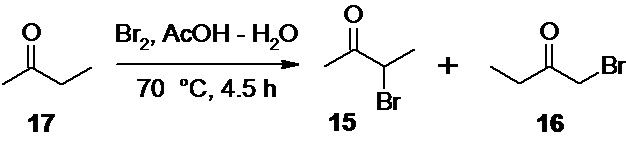
В 3 л стеклянный реактор с термостатируемой рубашкой, снабженный механической мешалкой с герметичным затвором, капельной воронкой с трубкой для выравнивания давления и обратным холодильником загружают 1200 мл деионизированной воды, 300 мл ледяной уксусной кислоты и 360.5 г (447.8 мл; 5.0 моль) метилэтилкетона 17. В капельную воронку загружают 800.0 г (257.9 мл; 5.0 моль; 1.0 экв) брома. Раствор в реакторе нагревают до 70°С, и по каплям добавляют бром в течение 4 часов, с такой скоростью, что бы возникающее вначале желтое окрашивание успевало исчезать при добавлении последующих порций брома. В процессе добавления брома образуется эмульсия, а затем реакционная смесь расслаивается на 2 фазы. После добавления всего брома реакционную смесь перемешивают при 70°С еще 30 минут, затем охлаждают до комнатной температуры, и разбавляют 1 л холодной воды (2°С). Нижний слой светло-желтого цвета отделяют через донный спуск реактора, и промывают 3х400 мл насыщенного раствора гидрокарбоната натрия (на магнитной мешалке, в неплотно закрытой конической колбе на 1000 мл), до прекращения выделения углекислого газа, а затем 400 мл деионизированной воды. Полученную смесь региоизомерных бромбутанонов 15/16 (546.1 г) сушат безводным гранулированным хлоридом кальция, фильтруют, и разделяют вакуумной дистилляцией на ректификационной колонне (заполненной кольцами Рашига) высотой 800 мм с вакуумированной рубашкой. Первую порцию дистиллята (около 10 мл) с tкип 58-60°С/38 mmHg отделяют, затем собирают основную фракцию 3-бром-2-бутанона 15 с tкип 61-63°С/38 mmHg, далее промежуточную фракцию с tкип 64-77°С/38 mmHg, и в заключении фракцию 1-бром-2-бутанона 16 с tкип 77-78°С/38 mmHg. Проведение процесса вакуумной дистилляции при слишком низкой скорости приводит к окрашиванию дистиллята в желтый цвет.
3-бром-2-бутанон (15)
Выход 279.74 г (37.0%). Жидкость светло-желтого цвета. 1H NMR (400 MHz, CDCl3) δ 4.37 (q, 1H), 2.34 (s, 3H), 1.71 (d, 3H); 13С (100 MHz, СDCl3) δ 202.11, 48.28, 26.04, 20.20. Содержание региоизомерного 1-бромбутан-2-она 16<5% (NMR). tR 5.35 мин (HPLC Area 96.9%, метод В).
1-бром-2-бутанон (16)
Выход 48.15 г (6.4%). Жидкость желтого цвета. 1H NMR (400 MHz, CDCl3) δ 3.87 (s, 2H), 2.65 (q, 2H), 1.09 (t, 3H); 13С (100 MHz, СDCl3) δ 209.92, 52.65, 29.13, 9.35; tR 4.64 мин (HPLC Area 88.9%, метод В).
2-(1-бромэтил)-2-метил-1,3-диоксолан (14)

В 2 л круглодонную колбу загружают 259.12 г (1.71 моль) свежеперегнанного 3-бром-2-бутанона 15, 117.2 г (1.88 моль; 1.10 экв) этиленгликоля, 1.58 г (8.30 ммоль; 0.0048 экв) моногидрата пара-толуолсульфокислоты и 800 мл толуола. Колбу снабжают насадкой Дина-Старка объемом 50 мл с обратным холодильником, и кипятят реакционную смесь при перемешивании на магнитной мешалке до прекращения отделения капелек воды в приемнике (31 мл; 3.5 часа). По мере протекания реакции нижний слой этиленгликоля исчезает.Прозрачный бесцветный раствор охлаждают до комнатной температуры, промывают 500 мл 2% раствора гидрокарбоната натрия, и 2х150 мл деионизированной воды. Органический слой отделяют, сушат безводным сульфатом натрия, фильтруют и концентрируют на роторном испарителе (35°С/10 mmHg), ориентируясь на объем отгона (760-780 мл). Продукт перегоняют в вакууме, собирая фракцию с tкип 43.5-44.5°С/0.016 mmHg.
Выход 271.93 г (81.2%). Бесцветная жидкость, не обладающая лакриматорным действием. 1H NMR (400 MHz, CDCl3) δ 4.05 (q, 1H), 3.99 (m, 4H), 1.67 (d, 3H), 1.45 (s, 3H); 13С (100 MHz, СDCl3) δ 109.96, 64.74, 53.22, 21.22, 20.65; tR 8.11 мин (HPLC Area 88.2%, метод В).
3-(2,3-диметилфенил)бутан-2-он (7)

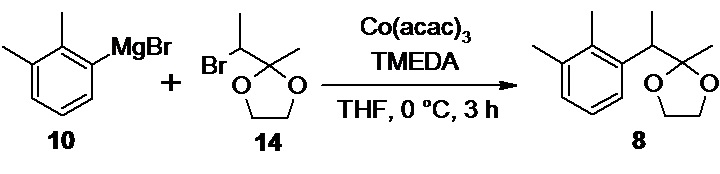

В двугорлую круглодонную колбу объемом 2 л, высушенную в сушильном шкафу при 120°С в течение ночи, снабженную капельной воронкой объемом 1 л с трубкой для выравнивания давления, и термопарой в стеклянной оболочке, загружают 33.90 г (1.39 моль; 1.05 экв) магниевой стружки. Колбу вакуумируют и заполняют сухим аргоном в 3 цикла. В колбу вносят около 0.8 г йода, и прогревают колбу горячим воздухом до начала возгонки йода. После охлаждения колбы до комнатной температуры магний покрывают 147 мл сухого THF и помещают в колбу магнитный якорь. В капельную воронку загружают раствор 245.67 г (1.32 моль; 1.0 экв) 2,3-диметилбромбензола 11 в 1000 мл сухого THF. К магниевой стружке приливают около 40-50 мл раствора из капельной воронки одной порцией. Реакция запускается сразу же после добавления первой порции раствора из капельной воронки, окраска йода исчезает, и температура быстро (<1 мин) растет до 48°С. После прекращения роста температуры реакционной смеси, сразу же начинают добавление раствора из капельной воронки при активном перемешивании (1300-1500 об/мин), с такой скоростью, что бы температура реакционной смеси поддерживалась в диапазоне 47-49°С без внешнего обогрева или охлаждения (около 5 часов). После добавления всего 2,3-диметилбромбензола реакционный раствор темного цвета нагревают с обратным холодильником в течение 45 минут при 70-75°С для завершения реакции. Раствор охлаждают в атмосфере аргона. Концентрация 2,3-диметилфенилмагний бромида 10 в полученном растворе определенная по методу Пауля Кнохеля составляет 0.99 моль/л. Выход 2,3-диметилфенилмагний бромида 92.7%. Полученный раствор 10 непосредственно используют на следующей стадии без дополнительной подготовки.
Стеклянный реактор объемом 5 л с термостатируемой рубашкой, снабженный механической мешалкой с герметичным затвором, термопарой и краном для ввода инертного газа откачивают (<0.1 mmHg) и сушат в течение 2 часов при 90°С. Реактор охлаждают до комнатной температуры, и заполняют сухим аргоном (3 цикла откачки/заполнения). В высушенный реактор в противотоке аргона последовательно загружают 41.93 г (10 моль%; 117.7 ммоль) ацетилацетоната кобальта (III), 520 мл сухого THF, 17.65 мл (13.67 г; 10 моль%; 117.7 ммоль) ТМЕDА, и 229.67 г (1.177 моль; 1.0 экв) 2-(1-бромэтил)-2-метил-1,3-диоксолана 14. Суспензию охлаждают до 0…-1°С, и при перемешивании (300-400 об/мин) в течение 180 минут с помощью перистальтического насоса добавляют в реактор раствор 2,3-диметилфенилмагний бромида 10, приготовленный на предыдущей стадии, со скоростью 6.9-7 мл/мин, тщательно поддерживая внутреннюю температуру в диапазоне от 0 до -1°С (температура теплоносителя в рубашке реактора, обеспечивающая хороший контроль реакции от -10 до -12°С). По мере добавления раствора реагента Гриньяра окраска реакционной смеси меняется с зеленой на сине-голубую, и из раствора выкристаллизовывается объемный осадок бромида магния. После добавления всего 2,3-диметилфенилмагний бромида (1.05 экв) температуру реакционной смеси повышают до 20°С, и продолжают перемешивание в течение 1 часа. К реакционной смеси при хорошем перемешивании (400 об/мин) медленно (в течение 2-3 минут) приливают 1460 мл 9% соляной кислоты, и продолжают перемешивание в течение 40 минут. В процессе добавления соляной кислоты происходит быстрое изменение цвета реакционной смеси с сине-голубого на зеленый, а затем оранжевый цвет. При этом весь кристаллический осадок бромида магния переходит в раствор. HPLC анализ аликвоты реакционной смеси, взятой по прошествии 30 минут перемешивания, показывает 100% конверсию промежуточного кеталя 8 в кетон 7. Реакционную смесь, состоящую из 2 фаз (верхней, светло-желтой органической, и нижней розово-красной водной) разбавляют 500 мл н-гептана, фазы перемешивают 15 минут, и сливают нижний водный слой через донный спуск реактора. Органическую фазу последовательно промывают 2х500 мл деионизированной воды и 500 мл насыщенного раствора гидрокарбоната натрия. Органическую фазу сушат сульфатом натрия, фильтруют и концентрируют в вакууме (45°С). Полученное оранжево-желтое масло (224.0 г) разбавляют 240 мл метанола, и вносят в раствор 100 мг 2,2′,3,3′-тетраметилбифенила 9 в качестве затравки. Раствор выдерживают в морозильной камере (-18°С) в течение суток. Суспензию кристаллов выпавшего 2,2′,3,3′-тетраметилбифенила 9 фильтруют на воронке Шотта, и промывают 2×50 мл холодного метанола. Фильтрат концентрируют в вакууме, остаток разбавляют 300 мл гексана, и последовательно промывают для удаления примеси этиленгликоля 1 л деионизированной воды (10 мин), 1 л 1% водного раствора Na2EDTA (2 часа), 1 л насыщенного водного раствора хлорида натрия (10 мин). Полноту удаления этиленгликоля контролируют с помощью 1Н NMR по исчезновению сигнала метиленовых групп (3.76 м.д., s, CDCl3). Органическую фазу отделяют, сушат безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Продукт перегоняют в вакууме с использованием дефлегматора Вигре высотой 300 мм с вакуумированной рубашкой. В качестве сборника дистиллята используют приемник Перкина. Фракцию с t кип до 68-69°С/ 0.024 mmHg отделяют, и собирают основную фракцию продукта, с t кип 69-72°С/ 0.024 mmHg.
Полученный 3-(2,3-диметилфенил)бутан-2-он 7 янтарного цвета разбавляют 250 мл гексана, и перемешивают в течение 1.5 часа с 10 г силикагеля (40-63 мкм, высушен при 120°С в течение 20 ч). Силикагель окрашенный в оранжевый цвет фильтруют на воронке Шотта, промывают 3х20 мл гексана, и фильтрат концентрируют в вакууме на роторном испарителе. Остаток выдерживают в вакууме (<0.1 mmHg, RT, 30 мин) для удаления следов гексана. Выход 161.22 г (77.7%). Еще 19.63 г продукта может быть выделено при экстракции объединенной водной фазы (4 л), полученной после гашения реакционной смеси 2х200 мл гептана, с последующей очисткой, описанной выше.
3-(2,3-диметилфенил)бутан-2-он (7)
Общий выход 180.85 г (87.2%). Жидкость светло-желтого цвета. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, 2H), 6.91 (t, 1H), 3.97 (q, 1H), 2.32 (s, 3H), 2.27 (s, 3H), 2.01 (s, 3H), 1.35 (d, 3H); 13С (100 MHz, СDCl3) δ 209.63, 139.22, 137.61, 134.50, 128.90, 126.18, 124.99, 50.58, 28.44, 21.14, 16.93, 15.33; tR 4.88 мин (HPLC Area 99.2%, метод А).
2,2′,3,3′-тетраметилбифенил (9)
Выход 10.08 г (7.2%). Белое кристаллическое вещество. 1H NMR (400 MHz, CDCl3) δ 7.14 (m, 4H), 6.96 (d, 2H), 2.34 (s, 6H), 1.96 (s, 6H); 13С (100 MHz, СDCl3) δ 142.48, 136.79, 134.67, 128.63, 127.41, 125.19, 20.68, 16.55; tR 42.3 мин (HPLC Area 98.8%, метод А).
1-бром-3-(2,3-диметилфенил)бутан-2-он (5)

Стеклянный реактор объемом 5 л с термостатируемой рубашкой, снабженный механической мешалкой с герметичным затвором, термопарой и краном для ввода инертного газа откачивают (<0.1 mmHg) и сушат в течение 2 часов при 90°С.Реактор охлаждают до комнатной температуры, и заполняют сухим аргоном (3 цикла откачки/заполнения). В высушенный реактор в противотоке аргона загружают 114.57 г (0.65 моль; 1.0 экв) 3-(2,3-диметилфенил)бутан-2-она 7 в 3420 мл абсолютированного метанола. Раствор охлаждают до -10°С и при перемешивании (250 об/мин) из капельной воронки сливают (за 30-35 с) в реактор свежеприготовленный, предварительно охлажденный до -5…-6°С раствор 124.65 г (0.78 моль; 1.20 экв) брома в 592 мл метанола (температура повышается до -9°С). Температуру реакционной смеси плавно повышают до -5…-6°С, и продолжают перемешивание при этой температуре в течение 18 часов, контролируя протекание реакции с помощью HPLC (метод А). Далее, температуру реакционной смеси (светло-оранжевого цвета) плавно повышают до 0°С, добавляют дополнительно 5.0 г (0.03 моль) брома и продолжают перемешивание в течение 2.5-3 часов для завершения реакции, отслеживая конверсию исходного кетона с помощью HPLC (метод А). После полного завершения реакции (конверсия исходного кетона>97%) прозрачный раствор сливают в 10 л реактор с мешалкой, содержащий 7200 мл предварительно охлажденной до 0.5°С деионизированной воды. Образовавшуюся эмульсию белого цвета перемешивают в течение 30 минут, и экстрагируют 4×300 мл метиленхлорида. Органический экстракт отделяют через донный спуск реактора, объединяют, и промывают 1 л насыщенного водного раствора гидрокарбоната натрия (300 об/мин, 30 минут), 1 л деионизированной воды (300 об/мин, 30 мин), и сушат безводным сульфатом натрия. Раствор фильтруют, и растворитель удаляют в вакууме (при температуре<30°С). Остаток используют непосредственно на следующем этапе без дополнительной очистки.
Выход 171.57 г (84.8%). Светло-желтое прозрачное масло, не обладающее лакриматорным действием. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, 2H), 6.85 (t, 1H), 4.39 (q, 1H), 3.79 (dd, 2H), 2.33 (s, 3H), 2.31 (s, 3H), 1.40 (d, 3H); 13С (100 MHz, СDCl3) δ 202.70, 138.10, 138.01, 134.54, 129.40, 126.43, 124.95, 46.75, 33.25, 21.04, 17.29, 15.34; tR 8.16 мин (HPLC Area 82.0%, метод А).
3-бром-3-(2,3-диметилфенил)бутан-2-он (20)

В стеклянный реактор с термостатируемой рубашкой, объемом 100 мл снабженный механической мешалкой с широкими лопастями и герметичным затвором, краном для ввода инертного газа загружают 8.0 г (45.4 ммоль; 1.0 экв) 3-(2,3-диметилфенил)бутан-2-она 7 в 80 мл этилацетата. Раствор охлаждают до 5°С, пускают слабый ток аргона (100 мл/мин) и при хорошем перемешивании (300 об/мин), через пластиковую воронку порциями примерно по 500-600 мг присыпают 15.25 г (47.6 ммоль; 1.05 экв) бромида пербромида пиридиния 19, с интервалом примерно в 1 мин. Окраска реагента мгновенно исчезает, и из раствора начинает выпадать кристаллический белый хлопьевидный осадок гидробромида пиридиния. На добавление всего реагента уходит около 30 минут.Реакционную смесь перемешивают в течение 40 минут при 5°С, а далее добавляют дополнительно 1.5 г бромида пербромида пиридиния (общее кол-во бромирующего реагента 16.75 г). Реакционную смесь перемешивают 10 минут и разбавляют 100 мл деионизированной воды. Перемешивание продолжают в течение 5 минут для растворения осадка гидробромида пиридиния, сливают нижнюю водную фазу и повторяют промывку органической фазы еще 100 мл деионизированной воды, активно перемешивая слои 5 минут.Водную фазу сливают через донный спуск реактора, и повторяют промывку 75 мл насыщенного раствора гидрокарбоната натрия. Слои активно перемешивают 40 минут при комнатной температуре, разделяют, и этилацетатный светло-желтый слой в заключении промывают 50 мл деионизированной воды. Водную фазу экстрагируют 20 мл этилацетата, объединенный органический экстракт сушат безводным сульфатом натрия, фильтруют, и концентрируют в вакууме при 40°С.Остаток выдерживают в вакууме для удаления следов этилацетата.
Выход 12.0 г (72.5%). Светло-оранжевое масло, кристаллизующееся в морозильной камере (-18°С) в иглы желтого цвета. 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 2H), 7.16 (m, 1H), 2.27 (s, 3H), 2.24 (s, 3H), 2.22 (s, 3H), 2.09 (s, 3H); 13С (100 MHz, СDCl3) δ 203.10, 138.75, 138.57, 133.86, 130.64, 126.19, 126.11, 73.48, 29.79, 25.29, 20.84, 17.16; tR 7.58 мин (HPLC Area 70%, метод А)
3-(2,3-диметилфенил)-2-оксобутил ацетат 4 (Путь А)

В круглодонную колбу объемом 2 л загружают 191.4 г (1.95 моль; 3.5 экв) ацетата калия и 1000 мл ацетонитрила. В колбу одной порцией добавляют раствор 171.57 г сырого 1-бром-3-(2,3-диметилфенил)бутан-2-она (расчетное содержание чистого 5 140.68 г; 0.55 моль), в 850 мл ацетонитрила, и перемешивают суспензию на магнитной мешалке (1500 об/мин) при комнатной температуре (21°С), контролируя ход реакции с помощью HPLC (метод А). После завершения реакции (19 часов, степень конверсии>99%) кристаллический осадок бромида калия, и избытка ацетата калия фильтруют на воронке Шотта, и промывают на фильтре 2х200 мл ацетонитрила. Прозрачный фильтрат светло-желтого цвета концентрируют в вакууме (<40°С), и остаток разбавляют 300 мл МТBE. Органическую фазу промывают 2х500 мл деионизированной воды, водную фазу дополнительно экстрагируют 2х50 мл МТBE, и объединенную органическую фазу сушат безводным сульфатом натрия. Раствор фильтруют, и растворитель удаляют в вакууме (при температуре<35°С). Остаток используют непосредственно на следующем этапе без дополнительной очистки.
Выход 158.07 г (84.8%). Желтое масло. tR 4.54 мин (HPLC Area 69.3%, метод А).
Образец соединения 4 очищали для ЯМР-анализа с помощью флеш-хроматографии (силикагель 60, гексан-этилацетат 9:1, Rf 0.16). Очищенное соединение 4 представляет собой белую воскообразную массу. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, 2H), 6.91 (t, 1H), 4.53 (dd, 2H), 4.04 (q, 1H), 2.31 (s, 3H), 2.27 (s, 3H), 2.09 (s, 3H), 1.39 (d, 3H); 13С (100 MHz, СDCl3) δ 204.59, 170.26, 137.86, 137.80, 134.34, 129.27, 126.36, 125.13, 67.08, 46.93, 21.10, 20.48, 16.73, 15.30.
Медетомидина гидрохлорид 1 (Путь А)

В 10 л стеклянный реактор с термостатируемой рубашкой, снабженный механической мешалкой, термопарой и эффективным обратным холодильником, загружают раствор 158.07 г сырого 3-(2,3-диметилфенил)-2-оксобутил ацетата (расчетное содержание чистого 4 109.54 г; 0.467 моль) в 1700 мл н-пропанола. В реактор при перемешивании (160 об/мин) одной порцией добавляют раствор 268.72 г (1.34 моль) моногидрата ацетата меди (II) в 2529 мл (2293.9 г; 33.6 моль) 25% водного раствора аммиака. Гомогенный раствор темно-синего цвета перемешивают в течение 3 минут, и одной порцией добавляют в реактор 490 мл (6.64 моль) 37% формалина. Реакционную смесь нагревают до кипения (температура обогрева 100°С) при перемешивании (160 об/мин) в течение 2 часов. Примерно через 10-15 минут после начала нагрева начинается осаждение кристаллического медного (I) комплекса медетомидина 3. По прошествии 2 часов нагрева реакционную суспензию охлаждают до 16-18°С в течение 1 часа, и продолжают перемешивание еще 30 минут при 16-18°С.Осадок фильтруют на воронке Шотта, и последовательно промывают 1 л 25% водного раствора н-пропанола, 1 л деионизированной воды и 2х250 мл ацетона. Полученный темно-желтый мелкокристаллический порошок сушат в вакууме (6 mmHg) при комнатной температуре в течение 12 часов. Выход медного (I) комплекса медетомидина 99.68 г (58.1% на 3 стадии).
В 10 л стеклянный реактор, снабженный механической мешалкой, загружают 2850 г (0.566 моль; 1.50 экв) 10% водного раствора пентанатриевой соли диэтилентриаминпентауксусной кислоты (Na5DTPA), и 99.68 г (0.377 моль) полученного медного (I) комплекса медетомидина 3, суспензированного в 2 л этилацетата. Фазы перемешивают (300-350 об/мин) при комнатной температуре (23°С) в течение 24 часов до полного растворения всех твердых частиц. Этилацетатный слой темного цвета отделяют от водной фазы, окрашенной в интенсивно-синий цвет.Водную фазу экстрагируют 2х200 мл этилацетата. Объединенную органическую фазу, содержащую основание медетомидина, переносят в 5 л стеклянный реактор с мешалкой, и последовательно промывают 2х500 мл 0.5% водного раствора Na5DTPA (по 30 мин), и 500 мл насыщенного водного раствора хлорида натрия (30 мин). Медетомидин извлекают из этилацетатной фазы последовательной экстракцией 500 мл, и 2х350 мл 10% водного раствора уксусной кислоты (перемешивание при 300-400 об/мин, по 10 минут на каждую порцию). Этилацетатный слой, обедненный по медетомидину концентрируют в вакууме до ~200 мл, и дополнительно извлекают 2х300 мл 10% водного раствора уксусной кислоты. Объединенный водный экстракт (1800 мл), содержащий ацетат медетомидина переносят в 5 л стеклянный реактор с мешалкой, и последовательно экстрагируют 3х200 мл MTBE для извлечения неосновных примесей. При этом водная фаза светлеет. Темный органический экстракт (МТВЕ) однократно промывают 100 мл деионизированной воды, водную фазу объединяют с экстрактом, содержащим ацетат медетомидина, и органическую фазу отбрасывают.Объединенную водную фазу (1900 мл) светло-желтого цвета переносят в 5 л стеклянный реактор с термостатируемой рубашкой, снабженный механической мешалкой, охлаждают до 8-10°С и нейтрализуют при перемешивании 350 мл 25% водного раствора аммиака. Выделившееся основание медетомидина 2 экстрагируют 3х250 мл этилацетата, органический слой однократно промывают 250 мл насыщенного водного раствора хлорида натрия, сушат безводным сульфатом натрия, фильтруют, и концентрируют в вакууме (осторожно, возможно вспенивание). Продукт - субстанция медообразной консистенции, янтарного цвета, вспенивающаяся в вакууме и застывающая в виде хрупкой перламутровой пены. Выход сырого основания медетомидина 65.74 г.
Основание медетомидина 2 растворяют в 250 мл сухого ацетона, непосредственно в той колбе, в которой проводилось концентрирование. Прозрачный раствор темного цвета охлаждают до -10°С, и вносят 1.0 г кристаллического гидрохлорида медетомидина в качестве затравки. Охлажденный раствор при перемешивании (500-600 об/мин) по каплям подкисляют 28.0 мл 36% соляной кислоты до рН 2-3. При этом начинается кристаллизация хорошо оформленного осадка гидрохлорида медетомидина 1. Процессу кристаллизации дают завершиться при -18…-20°С в течение 23 часов, продукт фильтруют на воронке Шотта, промывают 3х100 мл холодного (2-3°С) сухого ацетона, и сушат в вакууме при 40°С в течение 12 часов.
Выход гидрохлорида медетомидина 50.25 г (56.2% на 2 стадии). Не совсем белый мелкокристаллический порошок (HPLC Area 95.3%, метод С). Остаточное содержание меди 3.7 ppm (AAS).
Для очистки продукт кристаллизуют из ацетона, содержащего 3.85% (об.) воды (приблизительно 9.5 мл растворителя на 1 г гидрохлорида медетомидина). Выход 90% (одна стадия очистки, HPLC Area 98.5%, метод С). После двукратной кристаллизации выход составляет 78-82% (HPLC area 99.5%, метод С). Остаточное содержание меди после перекристаллизации<1 ppm (AAS).
Белый мелкокристаллический порошок. 1H NMR (400 MHz, D6-DMSO) δ 9.02 (s, 1H), 7.48 (s, 1H), 7.04 (m, 2H), 6.83 (m, 1H), 4.51 (q, 1H), 2.26 (s, 3H), 2.24 (s, 3H), 1.51 (d, 3H); 13С (100 MHz, D6-DMSO) δ 141.04, 137.36, 136.71, 134.07, 133.78, 128.30, 125.69, 124.05, 115.80, 31.88, 20.61, 20.31, 14.51; tR 3.11 мин (HPLC метод С).
Диформиламид натрия 21 (Путь В)
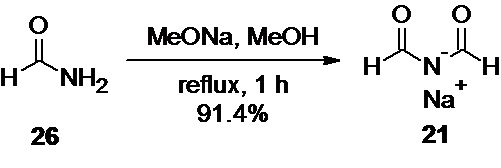
В круглодонную колбу объемом 500 мл, заполненную аргоном, загружают 28.42 г (526 ммоль) порошкообразного метилата натрия, и 100 мл сухого метанола (осторожно, разогрев!), и перемешивают до полного растворения. К полученному раствору добавляют по каплям в течение 2-3 мин 47.39 г (1.052 моль; 2.0 экв) формамида. Колбу снабжают обратным холодильником, и раствор нагревают при температуре кипения в течение 1 часа, при перемешивании на магнитной мешалке. Далее, раствор немного охлаждают, и меняют обратный холодильник на перегонный мост с нисходящим холодильником, и насадкой Клайзена, снабженной термометром, и капельной воронкой, содержащей 132 мл толуола. Метанол отгоняют, измеряя объем дистиллята мерным цилиндром, и после сбора около 80 мл дистиллята, начинают добавление толуола из капельной воронки, с такой скоростью, чтобы поддерживать постоянный объем раствора в отгонной колбе. Сбор дистиллята ведут до достижения температуры паров в насадке Клайзена 110°С (общий объем дистиллята около 150 мл). Суспензию белого цвета охлаждают до комнатной температуры, и фильтруют на воронке Шотта. Продукт промывают 80 мл ТГФ, и сушат в вакууме (0.05 mmHg), в течение 1 часа при комнатной температуре. Полученный продукт используют непосредственно на следующем этапе без дополнительной очистки.
Выход 45.68 г (91.4%). Белый мелкокристаллический порошок.
N-[3-(2,3-диметилфенил)-2-оксобутил]-N-формилформамид 22 (Путь В)
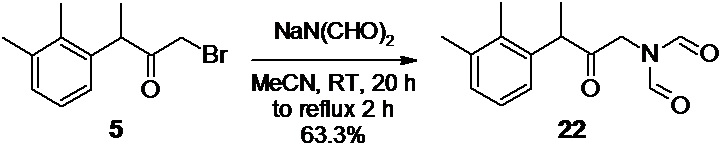
В двугорлую круглодонную колбу объемом 2 л, заполненную аргоном (3 цикла откачки/заполнения), загружают 38.46 г (404.7 ммоль; 1.4 экв) диформиламида натрия 21, и 400 мл ацетонитрила (содержание воды<0.05%). К полученной суспензии при перемешивании на магнитной мешалке одной порцией добавляют раствор 87.50 г сырого 3-(2,3-диметилфенил)-1-бромбутан-2-она 5 (хроматографическая чистота 82%; 281.2 ммоль) в 400 мл ацетонитрила. Реакционную смесь перемешивают на магнитной мешалке в атмосфере аргона при комнатной температуре в течение 20 часов, после чего снабжают колбу обратным холодильником, и нагревают при температуре кипения в течение 2 часов для достижения конверсии исходного соединения 5>99% (HPLC метод А). Горячий раствор темно-желтого цвета фильтруют на воронке Шотта, и осадок неорганических солей промывают 50 мл ацетонитрила. Прозрачный фильтрат концентрируют в вакууме (40°С), и полученное масло темно-желтого цвета промывают 350 мл деионизированной воды при перемешивании на магнитной мешалке в течение 10 минут.Продукт экстрагируют 300 мл метиленхлорида, органическую фазу отделяют, и промывают 350 мл деионизированной воды. Водную фазу повторно экстрагируют 30 мл метиленхлорида, и объединенную органическую фазу концентрируют в вакууме. Полученное желтое вязкое масло разбавляют 250 мл гексана, непосредственно в той же колбе, в которой проводилось концентрирование, и нагревают колбу на масляной бане (90°С) с обратным холодильником при перемешивании на магнитной мешалке в течение 30 минут.Смесь охлаждают, верхний гексановый слой осторожно сливают и отбрасывают, а оставшееся масло повторно промывают 100 мл гексана. Гексановый слой повторно сливают, и удаляют остаток гексана из продукта в вакууме. Остаток используют непосредственно на следующем этапе без дополнительной очистки.
Выход 51.24 г (63.3%). Желтое вязкое масло. 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 2H), 7.10 (m, 2H), 6.95 (m, 1H), 4.30 (dd, 2H), 4.03 (q, 1H), 2.33 (s, 3H), 2.26 (s, 3H), 1.42 (d, 3H); 13С (100 MHz, СDCl3) δ 209.80, 163.27, 138.01, 137.73, 134.60, 129.50, 126.53, 125.59, 48.58, 46.11, 21.10, 16.79, 15.39; tR 3.15 мин (HPLC Area 85.9%, метод А).
1-амино-3-(2,3-диметилфенил)бутан-2-он гидрохлорид 23 (Путь В)
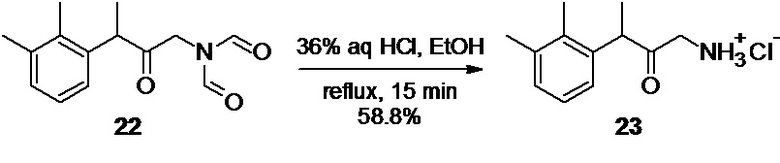
47.26 г сырого N-[3-(2,3-диметилфенил)-2-оксобутил]-N-формилформамида 22 (хроматографическая чистота 85.9%; 164.1 ммоль), полученного на предыдущей стадии растворяют в 600 мл 99% этанола, и к полученному прозрачному раствору желтого цвета одной порцией добавляют 65.0 мл 36% (764 ммоль; 4.6 экв) соляной кислоты. Колбу снабжают обратным холодильником, и раствор нагревают до кипения в атмосфере аргона, при перемешивании на магнитной мешалке. HPLC анализ аликвоты реакционной смеси, взятой по прошествии 15 минут, показывает>99% конверсии исходного соединения 22 в аминокетон 23 (HPLC метод А). Раствор фиолетово-красного цвета концентрируют в вакууме, и кристаллический остаток сушат при 40°С/2 mmHg в течение 1 часа (на роторном испарителе). Остаток суспензируют в 350 мл диэтилового эфира, суспензию кристаллов перемешивают 10-15 минут на магнитной мешалке, фильтруют на воронке Шотта, и промывают 3х100 мл ацетона. Полученный гидрохлорид аминокетона 23 (24.91 г) перекристаллизовывают из смеси 315 мл ацетона и 30 мл деионизированной воды, и сушат в вакууме (6 mmHg) при комнатной температуре в течение 12 часов.
Выход 22.0 г (58.8%). Бесцветные блестящие иголочки. 1H NMR (400 MHz, D6-DMSO) δ 8.40 (s, 3H), 7.08 (m, 2H), 6.88 (m, 1H), 4.25 (q, 1H), 3.73 (dd, 2H), 2.26 (s, 3H), 2.21 (s, 3H), 1.29 (d, 3H); 13С (100 MHz, D6-DMSO) δ 204.41, 137.95, 137.22, 134.53, 128.83, 125.92, 125.00, 46.37, 45.45, 20.66, 16.58, 15.01; tR 3.10 мин (HPLC Area 97.3%, метод В).
4-[1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тион 24 (Путь В)
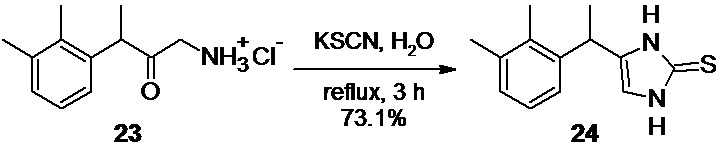
В круглодонную колбу объемом 250 мл, загружают 12.63 г (55.4 ммоль) 1-амино-3-(2,3-диметилфенил)бутан-2-она гидрохлорида 23, и 70 мл деионизированной воды. Суспензию кристаллов нагревают на масляной бане до 50-55°С, и перемешивают в течение 5 минут.К суспензии при перемешивании на магнитной мешалке одной порцией добавляют 26.95 г (277.3 ммоль; 5.0 экв) тиоцианата калия, колбу снабжают обратным холодильником, и нагревают реакционную смесь при 125-130°С (температура бани). Примерно после 10 минут нагрева все твердые вещества растворяются с образованием прозрачного раствора желтоватого цвета, из которого при дальнейшем нагреве (приблизительно через 30-40 минут) начинается выпадение продукта. После завершения реакции (общее время нагрева 3 часа 20 минут) суспензию кристаллов охлаждают до комнатной температуры, а далее до 0°С.Кристаллы фильтруют на воронке Шотта, промывают 3х100 мл деионизированной воды, и минимальным количеством холодного 96% этанола. Продукт сушат в вакууме (6 mmHg) при комнатной температуре в течение 12 часов.
Выход 9.42 г (73.1%). Светло-желтый мелкокристаллический блестящий порошок без запаха. 1H NMR (400 MHz, D6-DMSO) δ 11.83 (s, 1H), 11.70 (s, 1H), 7.02 (m, 2H), 6.92 (m, 1H), 6.50 (s, 1H), 4.15 (q, 1H), 2.24 (s, 3H), 2.20 (s, 3H), 1.38 (d, 3H); 13С (100 MHz, D6-DMSO) δ 141.71, 136.26, 133.71, 133.58, 133.44, 127.89, 125.36, 123.93, 111.40, 31.79, 20.61, 19.92, 14.39; tR 9.49 мин (HPLC Area 98.9%, метод B).
Медетомидин 2 (Путь В)

В колбу Шленка объемом 500 мл, заполненную аргоном, загружают 7.90 г (34.0 ммоль) 4-[1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тиона 24 и 50 мл 99% этанола. В противотоке аргона переносят в колбу 35 мл свежеприготовленной суспензии никеля Ренея W-4 в 99% этаноле (содержание никеля в суспензии 0.28 г/мл; 9.8 г; 167 ммоль; 4.9 экв), и перемешивают содержимое в течение 1.5 часа при комнатной температуре на магнитной мешалке. По прошествии 1.5 часа перемешивания добавляют вторую порцию (35 мл; 4.9 экв) суспензии никеля Ренея W-4 в 99% этаноле, и продолжают перемешивание при комнатной температуре в течение 3.5 часов. По прошествии указанного времени добавляют третью порцию (10 мл; 2.8 г; 47.7 ммоль; 1.4 экв) суспензии никеля Ренея W-4 в 99% этаноле, колбу снабжают обратным холодильником, и нагревают реакционную смесь при перемешивании в инертной атмосфере, в течение 2 часов при 95°С, для полного завершения реакции (конверсия исходного соединения 24 ≥99.5%). Суспензию черного цвета охлаждают до комнатной температуры, и фильтруют на воронке Шотта. Прозрачный фильтрат светло-желтого цвета дополнительно фильтруют через GFA фильтр, и концентрируют в вакууме. Полученное светло-желтое прозрачное вязкое масло основания медетомидина кристаллизуется в пористую легкую массу после удаления следов этанола в вакууме (0.05 mmHg) при комнатной температуре в течение 20 часов.
Выход 5.85 г (86%). 1H NMR (400 MHz, CDCl3) δ 10.56 (s, 1H), 7.30-6.62 (m, 5H), 4.35 (q, 1H), 2.26 (s, 3H), 2.17 (s, 3H), 1.56 (d, 3H); 13С (100 MHz, CDCl3) δ 143.38, 141.31, 136.87, 134.60, 134.16, 128.04, 125.65, 124.77, 117.26, 34.23, 21.01, 20.85, 14.76; tR 3.12 мин (HPLC Area 98.0%, метод С).
Медетомидин 2 (Путь С)

В стеклянной виале объемом 30 мл взвешивают 1.29 г (5.66 ммоль) 1-амино-3-(2,3-диметилфенил)бутан-2-она гидрохлорида 23, и 2.55 г (56.6 ммоль; 10 экв) формамида. Виалу продувают аргоном, помещают внутрь магнитный якорь, закрывают крышкой, и нагревают реакционную смесь при 160°С в течение 2 часов (анализ аликвоты реакционной смеси по прошествии указанного времени показывает>99% конверсии исходного 23). Реакционную смесь охлаждают до комнатной температуры, и разбавляют 9.63 мл 36% соляной кислоты. Гомогенный прозрачный раствор желтого цвета перемешивают на магнитной мешалке при комнатной температуре в течение 1 часа для гидролиза избытка формамида, суспензию выпавшего хлорида аммония разбавляют 10 мл деионизированной воды, и водную фазу экстрагируют 2х5 мл этилацетата для удаления не основных примесей. Водную фазу охлаждают до -3…-5°С, и нейтрализуют 12 мл 25% водного раствора аммиака. Выделившееся масло экстрагируют 2х10 мл этилацетата, объединенный органический экстракт промывают 10 мл деионизированной воды, сушат безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Полученное основание медетомидина в виде масла светло-желтого цвета вспенивается в вакууме, и после полного удаления следов этилацетата кристаллизуется в виде объемной пористой массы кремового цвета.
Продукт может быть очищен кристаллизацией из смеси циклогексан-толуол (9:1 об.), или переведен в гидрохлорид по стандартной процедуре, описанной выше.
Выход 0.43 г (38%). 1H NMR (400 MHz, CDCl3) δ 10.56 (s, 1H), 7.30-6.60 (m, 5H), 4.36 (q, 1H), 2.27 (s, 3H), 2.18 (s, 3H), 1.56 (d, 3H); 13С (100 MHz, CDCl3) δ 143.38, 141.31, 136.87, 134.60, 134.16, 128.04, 125.65, 124.77, 117.26, 34.23, 21.01, 20.85, 14.76; tR 3.12 мин (HPLC Area 95.2%, метод С).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения дексмедетомидина и его фармацевтически приемлемых солей | 2023 |
|
RU2824994C1 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2690853C2 |
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| БЕНЗОТИА(ДИ)АЗЕПИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ЖЕЛЧНЫХ КИСЛОТ | 2019 |
|
RU2785867C2 |
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| {3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ | 2015 |
|
RU2601410C1 |
| Способ синтеза триметилзамещенных фуродигидрохинолинов | 2016 |
|
RU2614248C1 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
Группа изобретений относится к области фармацевтической химии, а именно к способу получения медетомидина и его биологически активных производных. Представлено соединение 1-бром-3-(2,3-диметилфенил)бутан-2-он для получения медетомидина и его производных структурной формулы 5. В другом воплощении обеспечиваются способ синтеза соединения 5 для получения медетомидина и его производных, способ получения медетомидина в виде медного (I) комплекса формулы 3, способы получения медетомидина формулы 2, способ получения производного медетомидина 4-[1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тиона формулы 24. Группа изобретений обеспечивает новые синтетические пути получения медетомидина, которые являются более безопасными и достаточно дешевыми при внедрении в промышленное производство. Кроме того, преимуществами разработанного синтетического пути являются широкие возможности по модификации молекулы конечного продукта – медетомидина и получение различных производных, обладающих биологической активностью. 7 н. и 1 з.п. ф-лы.

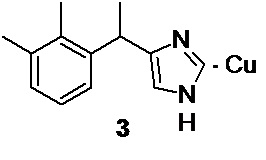


1. Соединение 1-бром-3-(2,3-диметилфенил)бутан-2-он для получения медетомидина и его производных структурной формулы 5
 .
.
2. Способ синтеза соединения 5 для получения медетомидина и его производных, включающий кобальт-катализируемое кросс-сочетание замещенного арилмагнийгалогенида 10
с защищенным по карбонильной группе 3-бром-2-бутаноном 14

снятие защитной группы one pot для получения 3-(2,3-диметилфенил)бутан-2-она 7
с последующим галогенированием 3-(2,3-диметилфенил)бутан-2-она 7 молекулярным бромом для получения исходного 1-бром-3-(2,3-диметилфенил)бутан-2-она 5
 .
.
3. Способ получения производного медетомидина в виде медного (I) комплекса формулы 3,
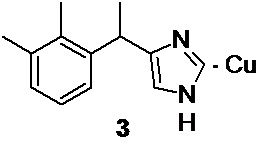
включающий взаимодействие соединения 5 по п. 1 с ацетатом калия для получения 3-(2,3-диметилфенил)-2-оксобутил ацетата 4,

и многокомпонентную реакцию соединения 4 с формальдегидом и ацетатом меди (II) в водно-спиртовом растворе аммиака.
4. Способ получения медетомидина формулы 2,

в котором из медного (I) комплекса 3 по п. 2 выделяют основание медетомидина 2 путем реакции 3 с пентанатриевой солью диэтилентриаминпентауксусной кислоты (Na5DTPA).
5. Способ по п. 4, в котором получают фармацевтически приемлемые соли медетомидина 2 реакцией свободного основания медетомидина 2 с кислотами в подходящем органическом растворителе, например гидрохлорид медетомидина 1
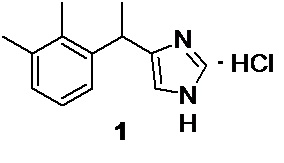
реакцией свободного основания медетомидина 2 с хлороводородной кислотой в ацетоне.
6. Способ получения производного медетомидина 4-[1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тиона формулы 24,

включающий взаимодействие соединения 5 по п. 1 c диформиламидом натрия 21

для получения N-[3-(2,3-диметилфенил)-2-оксобутил]-N-формилформамида 22,
кислотный гидролиз соединения 22 для получения 1-амино-3-(2,3-диметилфенил)бутан-2-она гидрохлорида 23,
и взаимодействие 23 с тиоцианатом калия.
7. Способ получения медетомидина формулы 2

включающий десульфуризацию производного 24 по п. 5.
8. Способ получения медетомидина формулы 2,
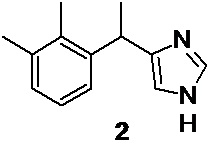
включающий взаимодействие соединения 5 по п. 1 c диформиламидом натрия 21 для получения N-[3-(2,3-диметилфенил)-2-оксобутил]-N-формилформамида 22, кислотный гидролиз 22 для получения 1-амино-3-(2,3-диметилфенил)бутан-2-она гидрохлорида 23 и его взаимодействие с формамидом.
| WO 2013014428 A1, 31.01.2013 | |||
| US 10053420 B2, 21.08.2018 | |||
| WO 2012172122 A2, 20.12.2012 | |||
| WO 2012172120 A2, 20.12.2012 | |||
| УЛУЧШЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ МЕДЕТОМИДИНА ИЛИ ЕГО НЕТОКСИЧНЫХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 2010 |
|
RU2448094C1 |

