Область техники
Изобретение относится к области фармацевтической химии, и описывает способ получения дексмедетомидина и его фармацевтически приемлемых солей
Уровень техники
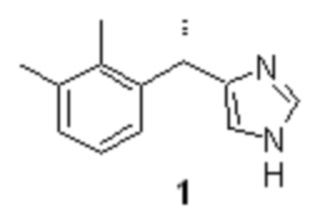
Дексмедетомидин (1), фармакологически активный S-энантиомер медетомидина, являющийся высокоселективным агонистом α2-адренорецепторов.
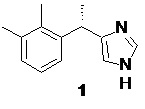
Дексмедетомидин был одобрен FDA в конце 1999 года для использования у людей (в форме гидрохлорида) в качестве лекарства для анальгезии и седации в отделениях интенсивной терапии. Дексмедетомидин обладает сильным симпатолитическим эффектом благодаря снижению высвобождения норадреналина из окончаний симпатических нервов. Седативный эффект обусловлен торможением возбуждения голубого пятна, основного норадренергического ядра, которое находится в стволе мозга (locus coeruleus). Именно через эту систему реализуется механизм естественного медленного сна. Таким образом, воздействие дексмедетомидина максимально точно соответствует естественному механизму сна человека. В отличие от опиоидов и других седативных средств, таких как пропофол, бензодиазепины и барбитураты, дексмедетомидин не вызывает дозозависимого подавления дыхания даже при 10-кратном превышение терапевтических дозировок (Venn, R.; Hell, J.; Grounds, M.; Respiratory effects of dexmedetomidine in the surgical patient requiring intensive care. Crit. Care 2000, 4, 302-08; Ebert, T.; Hall, J.; Barney, J. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology 2000, 93, 382-394). Набор этих уникальных свойств дексмедетомидина обусловили высокий интерес к данному препарату в госпитальной терапии, особенно в отделениях нейрохирургического профиля (Aryan, H.; Box, K.; Ibrahim, D. et al. Safety and efficacy of dexmedetomidine in neurosurgical patients. Brain Inj. 2006, 20, 791-798).
В настоящее время описано два основных способа производства субстанции дексмедетомидина: разделение рацемического медетомидина на энантиомеры (классическая технология) и асимметричное гидрирование (синтез нового поколения).
Классическое разделение рацемического основания медетомидина (2) на энантиомеры осуществляется с помощью L-(+)-винной кислоты (L-(+)-TA), (-)-О,О'-дибензоил-L-винной кислоты (L-DBTA) и (+)-O,O′-ди-p-толуоил-D-винной кислоты (D-DTTA).
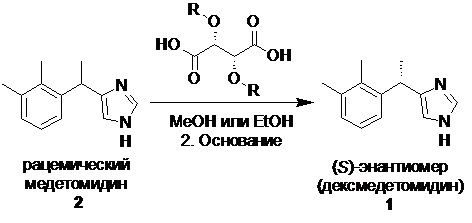
Однако, данный метод разделения как правило позволяет выделить нужный S-энантиомер с очень скромными выходами (Таблица 1)
Таблица 1. Выход дексмедетомидина (1) при расщеплении рацемата с помощью энантиомерно чистых винных кислот
Таким образом средний выход при разделении рацемата (по приведенным выше источникам информации) составляет 37-40%. Фактически, это означает что из 1 кг рацемического основания 2 при выходе 42% может быть получено только 0.21 кг S-энантиомера (в расчете на содержание S-энантиомера 0.5 кг на 1 кг рацемической смеси). Невысокая степень извлечения дексмедетомидина при классическом подходе обусловлена прежде всего, специфическими для данной молекулы свойствами, заключающимися в небольшой разнице в растворимости образуемых диастереомерных солей.
Более высокий выход продукта может быть достигнут при последовательной кристаллизации сперва с D-(-)-винной кислотой (обогащение), а затем с L-(+)-винной кислотой (4 перекристаллизации, выход 46.7%; WO2013069025A1, 2013), или, в другом варианте, сперва с D-(+)-DBTA, а затем L-(-)-DBTA (выход 55%; CN101671305A, 2010.). Однако, в обоих случаях используется дорогостоящий D-энантиомер винной кислоты или его бис-бензоилированное производное. Кроме того, из-за высокой растворимости D-винной кислоты в воде (596 г/л при 25°С; Yalkowsky, S.H.; Yan, H. Handbook of aqueous solubility data; Taylor & Francis Group, CRC Press; 2010; pp. 100.), и склонности к образованию пересыщенного раствора, процесс регенерации разделяющего агента трудноосуществим.
Также сообщалось об использовании других разделяющих агентов, таких как (S)-(+)-BNDHP и (R)-(−)-BNDHP (CN101671305A, 2010), однако в связи с высокой стоимостью последних, процесс разделения становиться коммерчески не приемлемым.
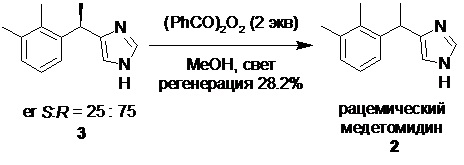
Вторым очень существенным недостатком классического метода разделения медетомидина (2) на энантиомеры с помощью оптически активных кислот, является образование отходов, обогащенных R-энантиомером (левомедетомидин, 3), который не может быть рацемизован до равновесной смеси энантиомеров безопасно масштабируемым методом.
Описанный в патенте US8877941B2 (2013) метод рацемизации отходов после выделения дексмедетомидина, содержащих до 75% R-изомера, помимо низкого процента регенерации рацемического медетомидина (28.2%), по нашему мнению, имеет существенный недостаток, заключающийся в использовании взрывоопасного пероксида бензоила в субстехиометрических количествах (2.34 кг пероксида бензоила на 1 кг обогащенной левомедетомидином смеси) согласно ниже приведенной схеме процесса рацемизации «хиральных отходов», обогащенных левомедетомидином 3:
Так как большинство описанных методов синтеза стартового рацемического медетомидина включают большое число стадий, и из-за особенностей строения молекулы (в частности стерически загруженного ароматического цикла) позволяют получать целевой продукт с выходом <30%, низкий выход S-энантиомера на стадии разделения и невозможность рециклизации отходов, с последующим повторным разделением в сумме существенно удорожает процесс производства субстанции дексмедетомидина, а так-же приводит к образованию нежелательных «хиральных отходов».
Другим методом получения дексмедетомидина, является асимметричное каталитическое гидрирование олефинового предшественника 4 (Таблица 2).
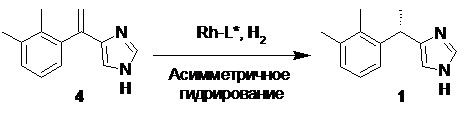
Таблица 2. Синтез дексмедетомидина с помощью асимметричного гидрирования
(R,R)-Et-DuPHOSa
MonoPhosb
Josiphos SL-J002-2c
(1R,1′R,2S,2′S)-DuanPhosd
Josiphos SL-J002-2c
a CAS 136705-64-1. b (S,S,S)-(+)-(3,5-dioxa-4-phosphacyclohepta[2,1-a:3,4-a′]dinaphthalen-4-yl)bis(1-phenylethyl)amine, CAS 380230-02-4; c CAS 277306-29-3. d CAS 528814-26-8.
Данный подход имеет важное преимущество перед классическим разделением энантиомеров медетомидина с помощью хиральных кислот, заключающееся в минимизации образования левомедетомидина, который, как мы ранее упомянули не может быть рацемизован удобным и безопасным способом. В патенте CN108147999B (2018) авторы не сообщают о полученном энантиомерном избытке непосредственно после гидрирования, а выделяют продукт с помощью кристаллизации с L-винной кислотой, что позволяет достичь высокой оптической чистоты (ee>99%). Hayashida et al. в патенте JP2018039757A (2018) сообщают о ee 89.2% в случае лиганда класса MonoPhos (0.15 ммоль субстрата), и ee 94.8% при использовании Josiphos SL-J002-2 (6.81 mmol субстрата). Однако, при масштабировании процесса до 520 г олефина 4, авторами патента WO2021089878A1 (2021) были получены более низкие значения ee (79.3%). Наиболее высокий энантиомерный избыток (>99%) сообщается в патенте CN109912508A (2019). В этом случае авторы используют 2.1 вес% лиганда и 2.05 вес% родиевого катализатора. В патенте CN112979553A (2021) сообщается об успешной попытке провести асимметричное гидрирование с использованием в качестве катализатора Pd/C, и лиганда (R,S)-DuanPhos (ee>99%), однако в этом случае авторам потребовалось использовать 70 вес% лиганда. С учетом того, что (R,S)-DuanPhos в настоящее время относиться к одним из самых дорогостоящих лигандов, использование даже нескольких весовых процентов лиганда приводит к очень высокой общей стоимости производственного процесса.
Несмотря на минимизацию образования нежелательного R-энантиомера в процессе асимметричного гидрирования, и формирование относительно небольшого количества «хиральных отходов» после финальной очистки (менее 0.3 кг на 1 кг предварительно обогащенного до 90% S-энантиомером продукта) недостатком метода является высокая стоимость лигандов и катализаторов.
Так как использование других, более дешевых лигандов в процессе синтеза не позволяет обеспечить надлежащую оптическую чистоту конечной субстанции в соответствии с требованиями USP (не более 1% левомедетомидина в финальном API), необходимо дальнейшее обогащение продукта S-энантиомером с помощью многократной кристаллизации диастереомерных солей.
Сообщалось так же о ферментативном способе разделения рацемического медетомидина. Так, авторы патента CN106749028B (2017) сообщают о способе разделения энантиомеров с помощью энантиоселективного ферментативного гидролиза предварительно сформированных амидов, в частности иммобилизованной липазой Candida antarctica (Novozym 435). Несмотря на высокий выход продукта на стадии разделения, и элегантность процесса, данный подход имеет существенные недостатки, в частности, включает образование амидов с использованием токсичных и аллергенных конденсирующих агентов, таких как N,N-дициклогексилкарбодиимид, и аналогично классическому подходу к разделению рацемического медетомидина с помощью хиральных кислот, приводит к образованию «хиральных отходов».
Для повышения степени извлечения дексмедетомидина из рацемической смеси, так же предпринимались попытки предварительной модификации молекулы медетомидина, с помощью введения вспомогательных функциональных групп. В частности, авторы патента CN106632053 (2017) сообщают о методе, основанном на предварительном сульфировании рацемического медетомидина, с последующим разделением с помощью энантиомерно чистого этилового эфира лизина, с последующим щелочным гидролизом полученной диастереомерной соли, с одновременным удалением вспомогательной сульфогруппы. Помимо увеличения количества стадий синтеза, возникающего за счет необходимости введение вспомогательных групп, в методе используется опасный, легколетучий, токсичный и неудобный в обращении жидкий триоксид серы. Кроме проблем, связанных с безопасностью процесса, несмотря на высокую степень извлечения S-энантиомера (по данным авторов от 87.9 до 94.2%), с учетом максимального содержания дексмедетомидина в равновесной рацемической смеси (0.50 кг на 1.0 кг рацемата) разделение данным способом приводит к образованию 0.53-0.56 кг отходов, обогащенных левомедетомидином, аналогично ферментативному способу разделения.
Таким образом, по-прежнему существует потребность в разработке максимально безотходного и недорогого способа синтеза дексмедетомидина, не требующего использования дорогостоящих хиральных фосфиновых лигандов и катализаторов на основе родия, опасных в обращении реагентов, и приводящего к образованию минимального количества отходов, обогащенных левомедетомидином.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Техническая проблема, решаемая изобретением, заключается в разработке максимально безотходного и недорогого способа синтеза дексмедетомидина.
Технический результат заключается в повышении эффективности за счет уве-личения степени извлечения дексмедетомидина из рацемической смеси.
Результат достигается за счет предварительного обогащения медетомидина S-энантиомером на ранних стадиях синтеза, эффективным методом регенерации «хиральных отходов», а также разделяющего амина.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Изобретение поясняется иллюстративным материалом, где
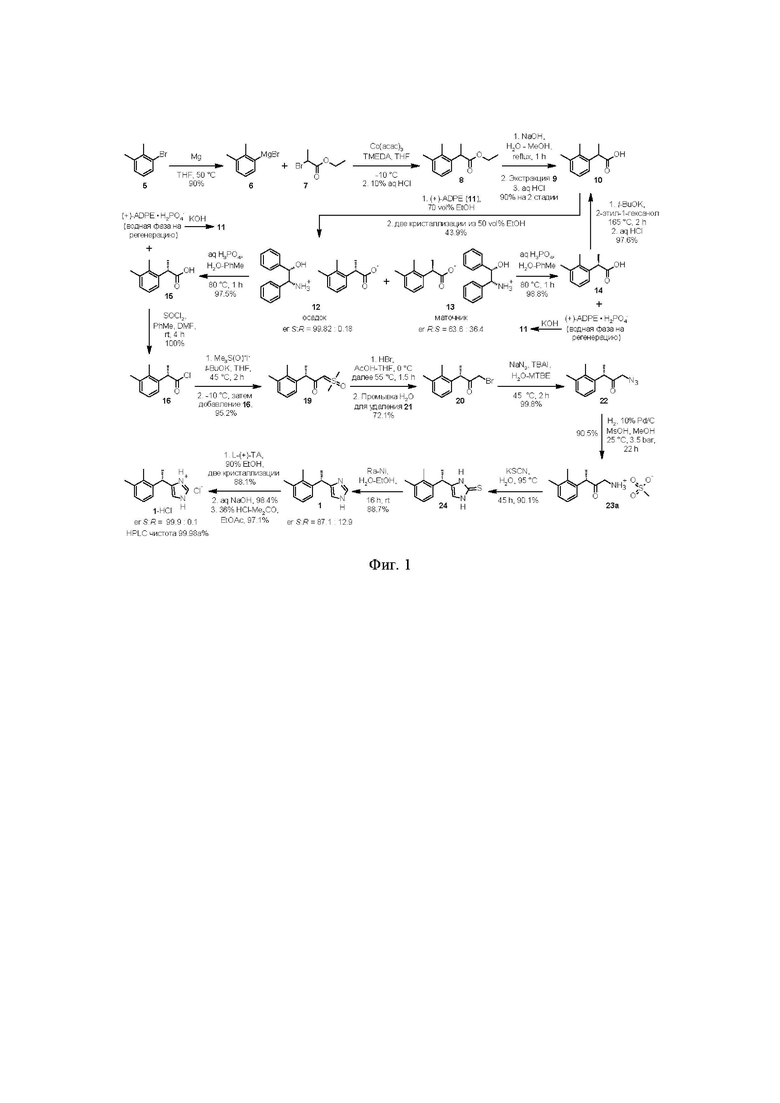
на фиг. 1 - приведена общая схема процесса синтеза субстанции дексмедетомидина (1) и дексмедетомидина гидрохлорида (1⋅HCl) реакционная схема, поясняющая заявляемый способ,
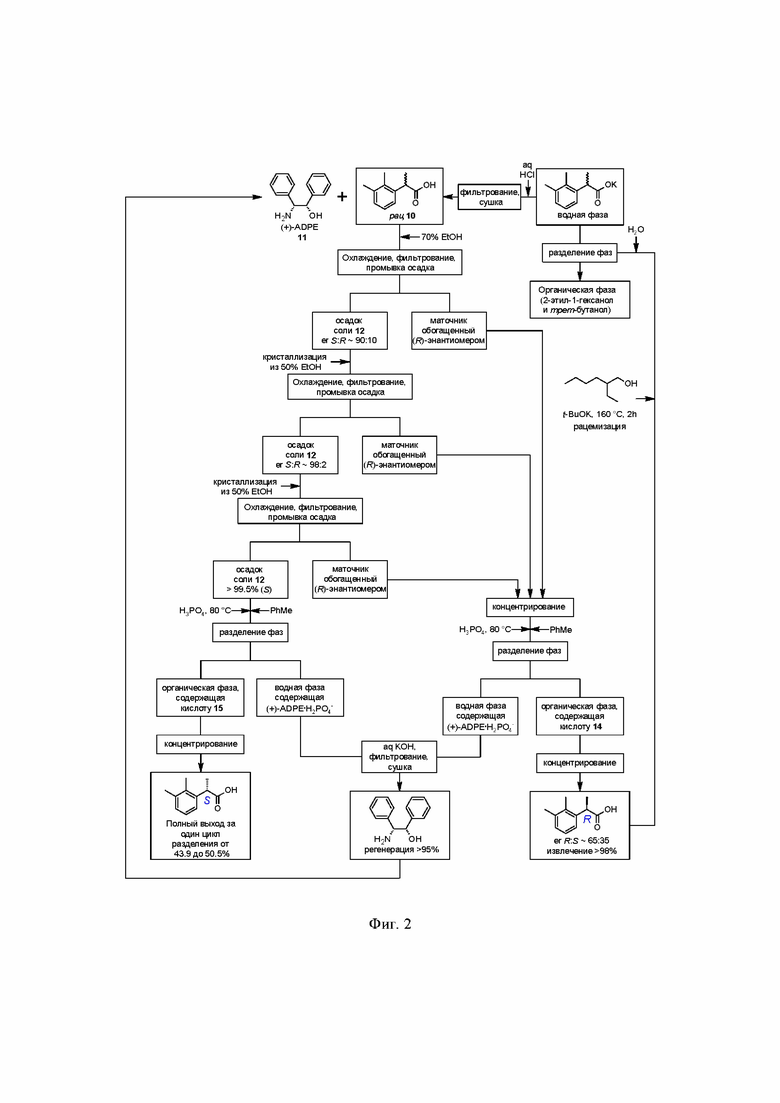
на фиг.2 приведена потоковая диаграмма процесса разделения кислоты 10 на энантиомеры, включающая регенерацию (+)-ADPE 11 и рацемизацию обогащенной R-энантиомером кислоты 14.
Список сокращений
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
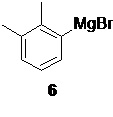
Процесс синтеза дексмедетомидина (1) начинается с получения магнийорганического реагента 6 (фиг. 1)
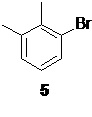
Исходным соединением для получения 6 служит коммерчески доступный 2.3-диметилбромбензол 5,
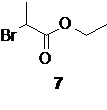
взаимодействие которого с магниевой стружкой в подходящем органическом растворителе, таком как THF, приводит к образованию реагента 6 с высоким выходом (90%). Последующее взаимодействие полученного реагента 6 с этил 2-бромпропионатом 7
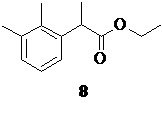
в присутствии катализатора (Co(acac)3/TMEDA; от 2 до 10 моль%) приводит к образованию этилового эфира 2-арилпропионовой кислоты 8.
Данная каталитическая система ранее была описана в литературе (Cahiez, G.; Chaboche, C.; Duplais, C.; Moyeux, A.A new efficient catalytic system for the chemoselective cobalt-catalyzed cross-coupling of aryl Grignard reagents with primary and secondary alkyl bromides. Org. Lett. 2009, 11(2), 277-280).
Оптимальный диапазон температуры при этом составляет от 0 до -15°С, предпочтительно от -5 до -10°С. Получение магнийорганического соединения 6, и последующее кросс-сочетание выполняется в атмосфере инертного газа, такого как азот или аргон, предпочтительно аргон.
Последующее гашение выполняется разбавленными водными растворами минеральных кислот, таких как H2SO4, или HCl, предпочтительно HCl.
Экстракция продукта кросс-сочетания 8 может быть выполнена не смешивающимся с водой органическим растворителем, таким как этилацетат, пропилацетат, бутилацетат, гексан, гептан, петролейный эфир, или толуол, предпочтительно гептаном. Перед экстракцией погашенной реакционной смеси, растворитель в котором проводилась реакция может быть удален в вакууме, что облегчает процесс экстракции и снижает количество отходов.
Удаление следов кобальта из сырого продукта кросс-сочетания 8 достигается кратковременной (~10 мин) промывкой 36% соляной кислотой, с последующей промывкой водными растворами комплексонов, таких как EDTA, Na2EDTA или Na4EDTA. Достигаемый уровень остаточного Co в сыром продукте 8 при этом составляет <1 ppm (ICP-OES).
Полученный сырой эфир 8, содержащий примесь побочного продукта гомосочетания 9
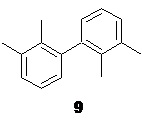
может быть использован на следующей стадии без дополнительной очистки.
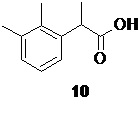
Следующая стадия включает щелочной гидролиз сырого эфира 8, содержащего примесь бифенила 9, под действием гидроксидов щелочных металлов, таких как NaOH или КОН, оптимально NaOH, в водно-спиртовой среде, оптимально водно-метанольной или водно-этанольной, с последующей отгонкой водно-спиртовой смеси. При этом происходит образование легко растворимой в воде натриевой, или соответственно калиевой соли рацемической кислоты 10,
и осаждение практически нерастворимого в воде побочного бифенила 9, который может быть легко удален фильтрованием или экстракцией подходящим органическим растворителем, таким как петролейный эфир, гептан или толуол, предпочтительно петролейный эфир.
Подкисление водной фазы разбавленными водными растворами минеральных кислот, таких как H2SO4, или HCl, предпочтительно HCl, приводит к выпадению кристаллического осадка продукта 10, который может быть отделен фильтрованием или центрифугированием, с последующей сушкой. Общий выход продукта на две стадии составляет 90%. Чистота полученной кислоты 10 составляет >99а% по данным HPLC.
Полученная кислота 10 далее подвергается классическому процессу разделения на энантиомеры посредством образования диастереомерных солей с хиральными аминами, оптимально с (1S,2R)-(+)-1,2-дифенил-2-аминоэтанолом 11
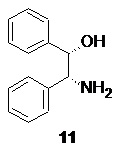
Оптимальным растворителем для кристаллизации при этом является этанол. Нагревание спиртового раствора рацемической кислоты 10 и аминоспирта 11 до полного растворения всех твердых веществ с последующим плавным охлаждением приводит к кристаллизации соли 12
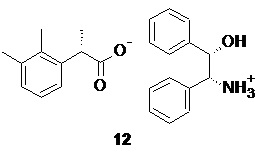
процесс кристаллизации повторяют в общей сложности 2-3 раза, контролируя энантиомерную чистоту после каждой кристаллизации с помощью хиральной HPLC, до достижения содержания индивидуального S-энантиомера кислоты 10 в соли не менее 98.5%.
Далее, полученную соль разлагают разбавленными водными растворами минеральных кислот, предпочтительно H3PO4, в двухфазной системе вода-органический растворитель, такой как этилацетат, н-пропилацетат, н-бутилацетат или толуол, предпочтительно толуол. Оптимальный диапазон температуры при этом составляет от 60 до 80°С, предпочтительно 80°С.
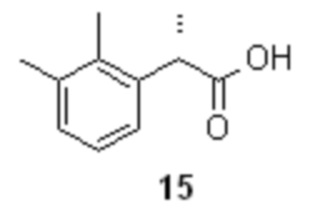
После разделения органическую фазу концентрируют в вакууме, получая S-энантиомер кислоты 15
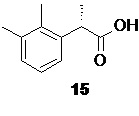
В случае использования в качестве растворителя на стадии выделения кислоты 15 толуола, концентрирование органической фазы можно проводить не досуха, так как толуол является подходящим растворителем для проведения следующей стадии. В этом случае толуольный раствор концентрируется до содержания кислоты 15 ~30 вес%, и используется на следующей стадии без дополнительной подготовки. Использование толуола в этом случае благоприятно сказывается на остаточном содержании воды, благодаря образованию азеотропной смеси.
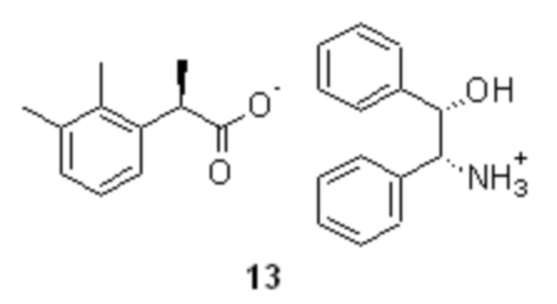
Объединенный спиртовой маточник полученный на стадии кристаллизации содержащий соль 13, обогащенную R-энантиомером кислоты 10 (er R:S ~ 65:35) концентрируют в вакууме.
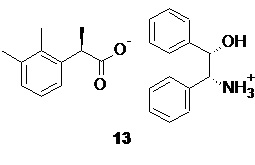
Регенерированный этанол далее вновь используют на стадии кристаллизации диастереомерных солей после корректировки крепости, а влажную соль 13 разлагают разбавленными водными растворами минеральных кислот в двухфазной системе вода-органический растворитель, аналогично процедуре, описанной выше для соли 12, непосредственно в реакторе, в котором проводилось концентрирование,
После разделения фаз объединенную водную фазу с обоих процессов, содержащую дигидрофосфат (+)-ADPE 11 направляют на регенерацию. Нейтрализация кислого раствора соли аминоспирта 11 гидроксидами щелочных металлов, такими как NaOH или KOH, предпочтительно KOH, или раствором аммиака, приводит к осаждению кристаллического основания 11 без потери энантиомерной чистоты. Достигаемая степень регенерации разделяющего хирального амина в этом случае составляет ≥95%.
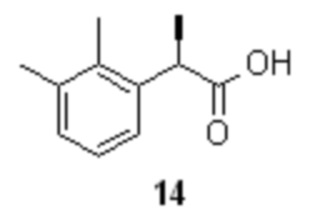
Органическую фазу, содержащую преимущественно R-энантиомер 14 (от 63 до 65%)
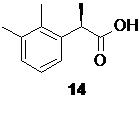
концентрируют в вакууме, и полученный твердый продукт без дополнительной очистки направляют на стадию рацемизации.
Рацемизация кислоты 14 может быть выполнена с помощью сильных оснований, таких как t-BuOK, NaOH или KOH, предпочтительно с помощью t-BuOK, в среде высококипящих спиртов, таких как н-бутанол, i-бутанол, н-амиловый спирт, i-амиловый спирт, н-гексанол, н-гептанол, н-октанол, 2-этил-1-гексанол (изооктанол), пропиленгликоль, предпочтительно в изооктаноле. Оптимальный диапазон температуры при этом составляет от 100 до 170°С, предпочтительно 165°С. Время реакции зависит от температуры проведения процесса и составляет от 2 часов (в случае изооктанола) до 18 часов (в случае н-бутанола). Последующее разбавление реакционной смеси водой приводит к переходу соответствующей натриевой или калиевой соли в водную фазу, из которой после подкисления получают рацемическую кислоту 10 высокой чистоты.
Кроме того, процесс рацемизации обогащенной R-энантиомером кислоты 14 может быть выполнен без растворителя, при нагревании расплава в течение 3 часов при 225°С. На фиг. 2 представлена потоковая диаграмма описанного выше технологического процесса, включающего разделение рацемической кислоты 10 на энантиомеры, регенерацию разделяющего амина 11, и рацемизацию нежелательного R-энантиомера кислоты 14.
Рацемизованная кислота 10 далее вновь запускается в цикл разделения, что позволяет достичь почти полное преобразование рацемической кислоты 10 в S-энантиомер в несколько циклов.
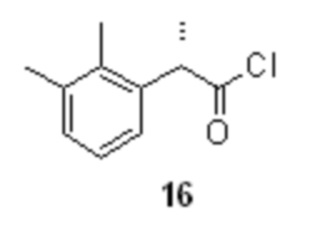
Взаимодействие кислоты 15 с небольшим избытком (1.25 экв) тионилхлорида в подходящем органическом растворителе, предпочтительно толуоле (0.5 л на 1 моль кислоты 15), при комнатной температуре в присутствии каталитического количества DMF (от 2.5 до 5 моль%) приводит к чистому преобразованию кислоты 15 в хлорангидрид 16
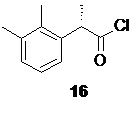
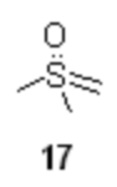
который после удаления растворителя в вакууме используется без дополнительной очистки на следующей стадии, включающей реакцию 16 с диметилсульфоксонийметилидом 17
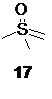
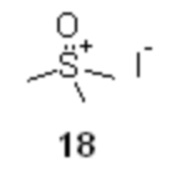
генерируемым в подходящем органическом растворителе, таком как THF из солей триметилсульфоксония, например, йодида триметилсульфоксония 18
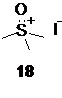
под действием сильных оснований, предпочтительно трет-бутилата калия. Оптимальный диапазон температуры на стадии генерации илида 17 составляет от 65 до 45°С, предпочтительно 45°С.
Полученный илид 17 далее вводиться в реакцию с хлорангидридом 16, для получения соединения 19
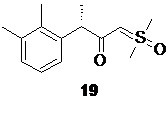
Оптимальный диапазон температуры на стадии реакции соединения 16 и илида 17 составляет от 0 до -15°С, предпочтительно -10°С. Оптимальные мольное соотношение 16 : 17 составляет 1 : 2.1. Как генерация илида 17, так и последующий синтез 19 выполняется в атмосфере инертного газа, такого как азот или аргон, предпочтительно аргон.
Образующийся в качестве побочного продукта триметилсульфоксоний хлорид непосредственно в реакционной смеси взаимодействует с избытком йодид ионов, что приводит к обмену анионами и выпадению в осадок менее растворимого триметилсульфоксоний йодида, таким образом регенерируя исходную соль 18 с выходом до 80%, которая может быть отделена фильтрованием, и снова использована в следующем цикле синтеза.
На следующей стадии полученное соединение 19 вступает в реакцию с бромоводородом (в виде раствора в АсОН) в подходящем органическом растворителе, таком как THF или 2-MeTHF, что приводит к образованию α-галогенкетона 20
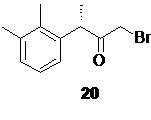
Оптимальный диапазон температуры на стадии реакции соединения 19 с HBr составляет от 0 до 2°С в момент смешивания компонентов, после чего температура должна быть повышена от 55 до 65°С, предпочтительно 55°С.
Процесс синтеза α-галогенкетона 20 сопровождается образованием побочного продукта в виде смеси диастереомерных сульфоксидов 21
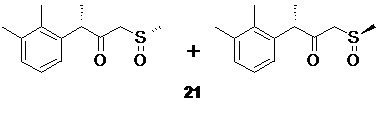
которые далее удаляются из продукта в ходе водной промывки раствора сырого α-галогенкетона 20 в углеводородных растворителях, таких как гексан, гептан или петролейный эфир, предпочтительно гептан.
Бромкетон 20 не стабилен при хранении при комнатной температуре, и полностью разлагается в течение <2 недель, однако он легко может быть стабилизирован добавкой MgO (0.1 вес%) и хранением при низкой температуре. Стабилизированный MgO продукт может храниться течение >3 месяцев при -20°С без заметных признаков разложения и рацемизации.
На следующей стадии соединение 20 вводят в реакцию с азидом щелочного металла, таким как азид натрия или азид калия, предпочтительно с азидом натрия, для получения соединения формулы 22
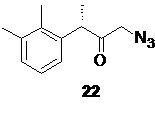
Реакция может быть проведена как в условиях межфазного катализа (NaN3, 5 моль% TBAI, вода-МТБЕ, 45°С, 2 часа), так и в индивидуальном растворителе, таком как THF (NaN3, 5 моль% TBAI, 25°С, 20 часов). Последующее удаление растворителя в вакууме дает сырой продукт, пригодный для использования на следующей стадии без дополнительной очистки. Азидокетон 22 стабилен при хранении, и при механическом воздействии.
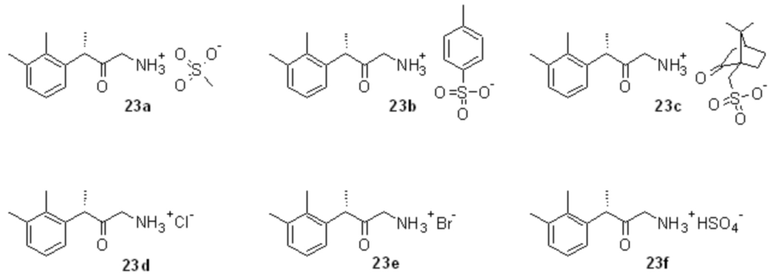
Восстановление азидокетона 22 может быть выполнено известными способами, в частности, в данном патенте раскрывается способ, основанный на каталитическом гидрировании 22 в присутствии Н2-Pd/C и органических (предпочтительно метансульфоновой, п-толуолсульфоновой, камфорсульфоновой) или минеральных (соляная, бромоводородная, серная) кислот, не ограничиваясь перечисленными кислотами, для получения соответствующих солей аминокетона 23a-23f
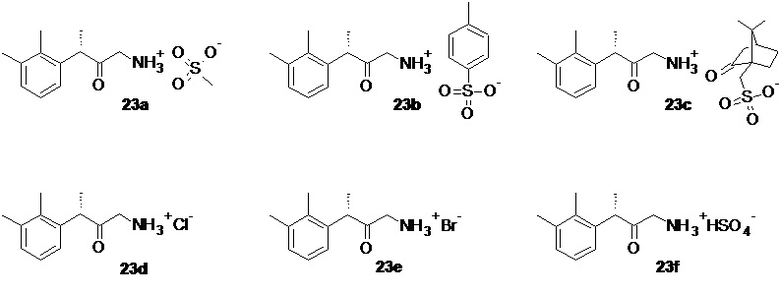
Оптимальными растворителями для проведения процесса гидрирования являются низшие спирты, такие как метанол, этанол и н-пропанол, предпочтительно метанол. Оптимальный диапазон давления составляет от 3 до 5 бар.
На следующей стадии соль аминокетона 23 вводят в реакцию с тиоцианатом щелочного металла, таким как тиоцианат натрия, тиоцианат калия, или тиоцианатом аммония, что приводит к образованию тиона 24 с высоким выходом
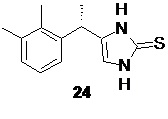
Оптимальным растворителем для проведения данной реакции является вода. Оптимальный диапазон температуры на стадии циклизации солей аминокетона составляет от 80 до 100°С, предпочтительно 95°С. Практически нерастворимый в воде тион 24 при этом осаждается в виде твердого вещества, которое может быть отделено фильтрованием или центрифугированием.
Десульфуризация соединения 24, например, с помощью никеля Ренея приводит к образованию обогащенного S-энантиомером продукта 1
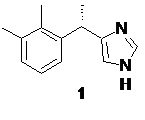
Так как в ходе процесса синтеза начиная со стадии получения илида 19, на каждой стадии наблюдается небольшая рацемизация стереоцентра, общий вклад каждой стадии приводит к получению продукта 1 содержащего как правило от 87 до 93% дексмедетомидина.
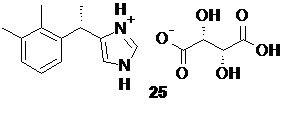
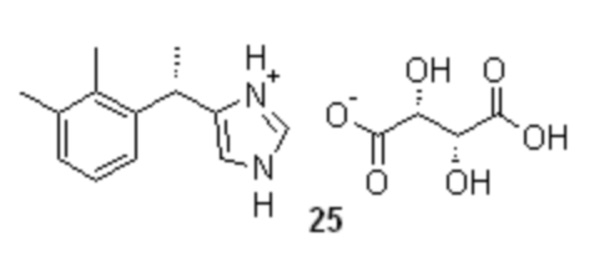
Повышение энантиомерной чистоты полученного основания дексмедетомидина 1 с 87-93% до >99% может быть выполнено с помощью кристаллизации обогащенного S-энантиомером продукта с хиральными органическими кислотами, например в виде соли с L-(+)-винной кислотой 25
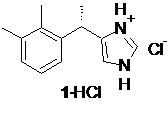
Получение финального API, включает осаждение очищенного основания дексмедетомидина 1 из соли с органической кислотой под действием щелочей, фильтрование продукта, и сушку, с последующим взаимодействием с хлороводородом в подходящем органическом растворителе, таком как ацетон или этилацетат, для получения фармацевтически приемлемой соли 1⋅HCl
Предлагаемый нами новый способ получения дексмедетомидина в значительной мере позволяет:
1. Благодаря предварительному обогащению продукта S-энантиомером (до ~90%) на ранних стадиях синтеза, снизить количество не регенерируемых «хиральных отходов» обогащенных левомедетомидином приблизительно в 4 раза (с 0.78-0.84 кг отходов с 1 кг рацемического медетомидина при классическом разделении, до 0.20-0.23 кг отходов с 1 кг продукта, предварительно обогащенного S-энантиомером в предлагаемом способе), что сравнимо по эффективности с методом каталитического асимметричного гидрирования, приводящего к образованию продукта с преобладанием нужного S-энантиомера.
2. В отличии от методов, связанных с использованием каталитического асимметричного гидрирования, предлагаемый нами новый подход полностью исключает необходимость в использовании дорогостоящих и труднодоступных хиральных фосфиновых лигандов, а также катализаторов на основе родия.
3. Использование в качестве интермедиата соединения из хорошо изученного и синтетически легкодоступного класса 2-арилпропионовых кислот, способных к легкой енолизации, и, следовательно, рацемизации хирального центра в α-положении к карбоксильной группе при действии сильных оснований, в частности дешевых гидроксидов и алкоголятов щелочных металлов, обеспечивает возможность простой регенерации «хиральных отходов» без использования взрывоопасного пероксида бензоила.
4. Эффективный, безопасный и высокодоходный (>95% за один цикл), процесс рециклизации отходов кислоты 10 обогащенных R-энантиомером, а также высокий процент регенерации разделяющего амина 11, обеспечивает почти количественное извлечение (S)-2-(2,3-диметилфенил)пропионовой кислоты 15 из рацемата за 2-3 цикла.
5. Метод не требует введения, и соответственно последующего снятия защитных или вспомогательных функциональных групп на протяжении всего процесса
6. Все промежуточные интермедиаты за исключением стадий, связанных с кристаллизацией диастереомерных солей, используются на последующих стадиях без дополнительной очистки
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Общие методы
Стартовый 2.3-диметилбромбензол (5; CAS 576-23-8) был приобретен у коммерческих поставщиков и перед использованием был дополнительно дистиллирован в вакууме. Магниевая стружка марки Turnings for Grignard Reaction от Fisher Chemical, была высушена в сушильном шкафу при 120°С в течение 20 часов, и активирована йодом (2 г на 200 г магния). Все растворители и реагенты были приобретены у коммерческих поставщиков, и использовались без дополнительной очистки и подготовки, за исключением THF. THF (ACS 99.6%) перед использованием был перегнан в атмосфере аргона (с добавкой ~0.1% гидрохинона), и высушен активированными молекулярными ситами 3А (100 г/л) в течение 3 суток. Активация молекулярных сит проводилась нагреванием в муфельной печи при 320°С в течение 8 часов с периодической продувкой сухим воздухом. Для создания инертной атмосферы использовался аргон марки 5.0 без дополнительной очистки. Определение концентрации 2,3-диметилфенилмагний бромида было выполнено с помощью прямого йодометрического титрования в насыщенном растворе хлорида лития в сухом THF по методу Пауля Кнохеля (Krasovskiy, A.; Knochel, P. Convenient titration method for organometallic zinc, magnesium, and lanthanide reagents. Synthesis 2006, (5), 0890-0891). Вся посуда, и так же детали стеклянных установок, используемые на стадии приготовления магнийорганического реагента, кросс-сочетания и синтеза илида 19 были высушены в сушильном шкафу при 120°С в течение 20 часов. Стеклянные реактора (15 и 30 л) были высушены перед работой с чувствительными к влаге соединениями с помощью циркуляции горячего теплоносителя (120°С) в течение 20 часов, с непрерывной подачей тока сухого азота (100 мл/мин) через донный клапан. Спектры 1H и 13C NMR были записаны на спектрометре Agilent 400 MHz (400 МГц для 1H и 101 МГц для 13C). Химические сдвиги выражены в миллионных долях относительно остаточных сигналов применяемых дейтерированных растворителей. Определение остаточного содержания тяжёлых металлов проводилось на оптико-эмиссионном спектрометре с индуктивно-связанной плазмой iCAP 6300 Duo (Thermo Scientific). Измерение остаточного содержания воды в интермедиатах выполнялось с помощью KF титратора V20S (Mettler Toledo). Мониторинг полноты протекания реакций и оценка хроматографической чистоты интермедиатов осуществлялись с помощью HPLC системы Alliance (Waters) с матричным фотодиодным детектором (PDA).
Аналитические условия
Метод A
Прибор: Waters Alliance HPLC;
Колонка: SunFire C18, 3.5 μm, 2.1 mm × 150 mm;
Элюент A: MeCN;
Элюент B: H2O + H3PO4 (600 μl 85% H3PO4 на 1000 мл H2O)
Режим элюирования: градиентный;
Соотношение элюентов А и В:
Температура термостата колонки: 40°C;
Длина волны детектирования: 215 nm.
Метод В
Прибор: Waters Alliance HPLC;
Колонка: CHIRALCEL OJ-H, 5 μm, 4.6 mm × 250 mm;
Элюент A: n-гексан;
Элюент B: изопропанол;
Соотношение элюентов А и В: 94:6;
Режим элюирования: изократический;
Скорость потока: 1.00 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Примечание: перед анализом диастереомерных солей, 2-3 мг соответствующей соли встряхивается в пробирке с 1 мл гексана и 500 мкл 10% HCl до полного растворения, после чего гексановая фаза непосредственно анализируется HPLC.
Метод С
Прибор: Waters Alliance HPLC;
Колонка: SunFire C18, 3.5 μm, 2.1 mm × 150 mm;
Элюент A: MeCN;
Элюент B: H2O + H3PO4 (600 μl 85% H3PO4 на 1000 мл H2O)
Режим элюирования: градиентный;
Соотношение элюентов А и В:
Температура термостата колонки: 40°C;
Длина волны детектирования: 215 nm.
Метод D
Прибор: Waters Alliance HPLC;
Колонка: CHIRALCEL OJ-H, 5 μm, 4.6 mm × 250 mm;
Элюент: 100% n-гептан;
Режим элюирования: изократический;
Скорость потока: 0.70 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Примечание: перед анализом (S)-2-(2,3-диметилфенил)пропионилхлорид (16; 5 мкл) растворяли в метаноле (100 мкл), и раствор выдерживали 15 минут при комнатной температуре. Полученный раствор метил (S)-2-(2,3-диметилфенил)пропионата разбавляли водой (300 мкл) и н-гептаном (300 мкл). Смесь встряхивали 1-2 мин на шейкере, гептановый слой отделяли, разбавляли до 1.5 мл н-гептаном, и анализировали в условиях, приведенных для метода D.
Метод E
Прибор: Waters Alliance HPLC;
Колонка: CHIRALCEL OJ-H, 5 μm, 4.6 mm × 250 mm;
Элюент A: n-гептан;
Элюент B: изопропанол;
Режим элюирования: изократический;
Соотношение элюентов А и В: 95:5;
Скорость потока: 1.00 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Примечание: перед анализом (S)-2-(2,3-диметилфенил)пропионилхлорид (16; 5 мкл) растворяли в МТБЭ (200 мкл), и полученный раствор по каплям добавляли к раствору н-пропиламина (50 мкл) в МТБЭ (200 мкл). Суспензию перемешивали 1-2 мин, гасили водой (300 мкл), и встряхивали 1-2 мин на шейкере. Органический слой (100 мкл) отделяли, и упаривали током воздуха в виале на 1.5 мл. Кристаллический амид растворяли в изопропаноле (100 мкл), разбавляли до 1.5 мл н-гептаном, и анализировали в условиях, приведенных для метода E.
Метод F
Прибор: Waters Alliance HPLC;
Колонка: SunFire C18, 3.5 μm, 2.1 mm × 150 mm;
Элюент A: MeCN;
Элюент B: H2O + H3PO4 (600 μl 85% H3PO4 на 1000 мл H2O)
Режим элюирования: градиентный;
Соотношение элюентов А и В:
Температура термостата колонки: 40°C;
Длина волны детектирования: 215 nm.
Метод G
Прибор: Waters Alliance HPLC;
Колонка: CHIRALCEL OJ-H, 5 μm, 4.6 mm × 250 mm;
Элюент: 100% n-пентан;
Режим элюирования: изократический;
Скорость потока: 0.60 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Метод H
Прибор: Waters Alliance HPLC;
Колонка: CHIRALCEL OJ-H, 5 μm, 4.6 mm × 250 mm;
Элюент A: n-гексан;
Элюент B: изопропанол;
Режим элюирования: изократический;
Соотношение элюентов А и В: 75:25;
Скорость потока: 1.00 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Примечание: перед анализом соли аминокетона 23 (10 мг) суспензировали в этилацетате (1.0 мл), добавляли N,N-диметилкарбамоил хлорид (16 мкл), и DIPEA (50 мкл). Смесь перемешивали при 25°С отслеживая конверсию стартового аминокетона в N,N-диметилкарбамоильное производное. Через 2 часа HPLC анализ показывал >99% конверсии исходного аминокетона (Метод C). Аликвоту раствора (50 мкл) разбавляли этилацетатом (300 мкл) и 5% соляной кислотой (300 мкл). Смесь встряхивали на шейкере 5 мин, органический слой (350 мкл) отделяли, и упаривали током воздуха в виале на 1.5 мл. Остаток растворяли в изопропаноле (300 мкл), разбавляли н-гексаном до 1.5 мл, и анализировали в условиях, приведенных для метода H. Аналогичным образом готовили дериват рацемического 1-амино-3-(2,3-диметилфенил)бутан-2-она, для использования в качестве стандартного образца сравнения для идентификации времени удерживания минорного энантиомера.
Метод I
Прибор: Waters Alliance HPLC;
Колонка: CHIRALCEL OJ-H, 5 μm, 4.6 mm × 250 mm;
Элюент A: n-гексан;
Элюент B: изопропанол;
Режим элюирования: изократический;
Соотношение элюентов А и В: 90:10;
Скорость потока: 1.00 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Метод J (USP 43-NF38, Dexmedetomidine hydrochloride, p. 1302)
Прибор: Waters Alliance HPLC;
Колонка: CHIRALPAK AGP, 5 μm, 4.0 mm × 150 mm;
Элюент A: MeCN;
Элюент B: Буфер: в химический стакан вместимостью 2000 мл помещают 1.0 л раствора Na2HPO4⋅2H2O (5.34 г/л) и доводят значение рН раствором KH2PO4 (4.08 г/л; около 700-800 мл) до 7.0;
Режим элюирования: изократический;
Соотношение элюентов А и В: 17.5:82.5;
Скорость потока: 1.00 мл/мин;
Температура термостата колонки: 25°C;
Длина волны детектирования: 220 nm.
Метод K (USP 43-NF38, Dexmedetomidine hydrochloride, p. 1302)
Прибор: Waters Alliance HPLC;
Колонка: XBridge BEH C18, 3.5 μm, 4.6 mm × 150 mm;
Элюент A: MeOH;
Элюент B: Буфер: растворяют 0.89 г Na2HPO4⋅2H2O в 900 мл воды и доводят значение рН раствором NaH2PO4⋅2H2O (16.0 г/л) до 7.0. Полученный раствор количественно переносят в мерную колбу вместимостью 1000 мл и доводят объём раствора водой до метки.
Режим элюирования: изократический;
Соотношение элюентов А и В: 60:40;
Скорость потока: 1.00 мл/мин;
Температура термостата колонки: 40°C;
Длина волны детектирования: 220 nm.
2,3-диметилфенилмагний бромид (6).
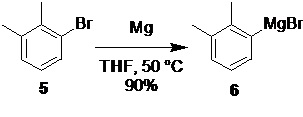
В сухой стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, обратным холодильником, термопарой, клапанами для ввода инертного газа и подачи реагентов, заполненный аргоном, загружали предварительно активированную магниевую стружку (189.6 г; 7.8 моль; 1.2 экв) и сухой ТГФ (1.0 л). В реактор с помощью перистальтического насоса добавляли 80-100 мл раствора 2.3-диметилбромбензола (5; 1.20 кг; 6.5 моль; 1.0 экв) в сухом ТГФ (4.85 л). После запуска реакции (от 3 до 5 мин) и начала роста температуры, при перемешивании (от 230 до 250 об/мин) добавляли оставшуюся часть раствора арилбромида 5 в ТГФ с такой скоростью, чтобы поддерживать внутреннюю температуру без внешнего нагрева или охлаждения в диапазоне от 48 до 52°С (расход раствора 5 около 0.85 л/час). После добавления всего раствора 5 реакционную смесь перемешивали 1 час при температуре от 50 до 55°С, охлаждали до комнатной температуры, и сливали приготовленный раствор с избытка магния в емкость для хранения. Концентрация 6 в полученном растворе (6.50 л) по результатам титрования 0.90 М. Выход 90%.
Этил 2-(2,3-диметилфенил)пропионат (8).
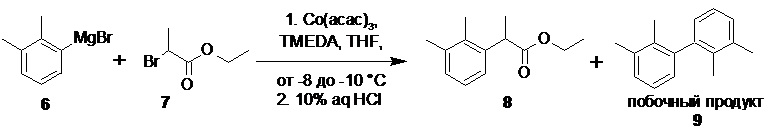
В сухой стеклянный реактор объемом 30 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, термопарой, клапанами для ввода инертного газа и подачи реагентов, в противотоке аргона загружали ацетилацетонат кобальта (III) (99.0 г; 0.278 моль; 5.0 моль%), этил 2-бромпропионат (7; 1006.0 г; 1 экв; 5.56 моль), TMEDA (32.3 г; 0.278 моль; 5.0 моль%), и сухой ТГФ (2.45 л). Реакционную смесь охлаждали до -10°С и при перемешивании (230 об/мин) добавляли раствор 6 (6.50 л; 0.90 М; 5.85 моль; 1.05 экв) с помощью перистальтического насоса со скоростью 1 л/час поддерживая внутреннюю температуру в пределах от -8 до -10°С. После добавления всего раствора 6 реакционную смесь сине-зеленого цвета, содержащую обильный осадок бромида магния перемешивали при -10°С еще 1 час. В реактор загружали 10% соляную кислоту (10.0 кг) предварительно охлажденную до 0°С, хлорид натрия (3.0 кг) и н-гептан (4.0 л). Фазы перемешивали 15-20 мин, органическую фазу светло-зеленого цвета отделяли, а водную повторно экстрагировали н-гептаном (2х2.0 л). Объединенный органический экстракт изумрудно-зеленого цвета без дополнительной обработки концентрировали в вакууме (до объема ~ 3 л), и последовательно промывали 36% соляной кислотой (1.0 л, 10 мин), водой (1.0 л, 5 мин), 5% раствором Na2EDTA (2х1.0 л, по 20 мин), водой (1.0 л, 5 мин), и остаток н-гептана удаляли на роторном испарителе. Сырой продукт использовали на следующей стадии без дополнительной очистки. Выход 1217 г (теоретический выход 1146.3 г). Прозрачная светло-желтая жидкость с легким приятным запахом. 1H NMR (400 MHz, CDCl3) δ 7.18-7.05 (m, 3H), 4.23-4.09 (m, 2H), 4.03 (q, J = 7.1 Hz, 1H), 2.33 (s, 3H), 2.29 (s, 3H), 1.49 (d, J = 7.1 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 175.1, 139.3, 137.1, 134.3, 128.7, 125.8, 124.4, 60.7, 42.0, 21.1, 18.2, 15.2, 14.3; tR 12.85 мин (чистота 82.78a%, HPLC метод A). Остаточное содержание кобальта 0.71 ppm (ICP-OES).
рац-2-(2,3-диметилфенил)пропионовая кислота (10).
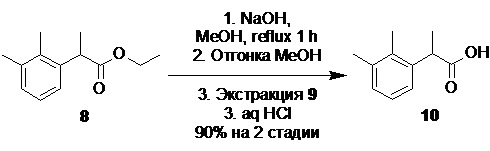
В стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором и обратным холодильником загружали воду (2.5 л) и гидроксид натрия (556.0 г; 13.9 моль; 2.5 экв). После растворения щелочи в реактор загружали раствор сырого этил 2-(2,3-диметилфенил)пропионата (8; 1.21 кг) в метаноле (2.50 л). Эмульсию нагревали до кипения (температура теплоносителя в рубашке реактора 100°С) при перемешивании в течение 1 часа, после чего температуру обогрева повышали до 110°С, и не прерывая процесс из реактора отгоняли ~2.8 л дистиллята (при атмосферном давлении). Прозрачный светло-желтый водный раствор, содержащий натриевую соль кислоты 10 охлаждали до 23°С, выпавший кристаллический осадок побочного 2,2',3,3'-тетраметилбифенила (9) экстрагировали петролейным эфиром (фракция 40-70; 2х1.0 л), разбавляли водой (10.0 л), охлаждали до 3°С и при хорошем перемешивании по каплям подкисляли 36% соляной кислотой (1.42 кг; 14.0 моль), поддерживая температуру смеси в диапазоне от 8 до 12°С. Выпавший белый кристаллический осадок кислоты 10 фильтровали, промывали на фильтре водой (4х3.0 л), и сушили в вакуумном шкафу при 55°С/5 mmHg в течение 48 часов. Выход 886.5 г (90.0% на 2 стадии). Остаточное содержание воды по результатам титрования по Карлу Фишеру (KF) <0.15%. Белый кристаллический порошок практически не растворимый в воде. tпл 91.5-93.5°С; 1H NMR (400 MHz, CDCl3) δ 7.22-7.04 (m, 3H), 4.07 (q, J = 7.1 Hz, 1H), 2.32 (s, 3H), 2.29 (s, 3H), 1.51 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 181.4, 138.4, 137.3, 134.6, 129.1, 126.0, 124.6, 41.7, 21.2, 17.8, 15.3; tR 8.49 мин (чистота 99.55a%, HPLC метод A). Остаточное содержание кобальта 0.06 ppm (ICP-OES). Концентрирование органического экстракта в вакууме дало 40.23 г побочного 2,2',3,3'-тетраметилбифенила (9). Бесцветные прозрачные крупные призмы. tпл 116-118°C; 1H NMR (400 MHz, CDCl3) δ 7.17-7.08 (m, 4H), 6.99-6.94 (m, J = 7.0 Hz, 2H), 2.34 (s, 6H), 1.96 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 142.5, 136.8, 134.7, 128.6, 127.4, 125.2, 20.7, 16.6; tR 15.54 мин (чистота 99.72a%, HPLC метод А).
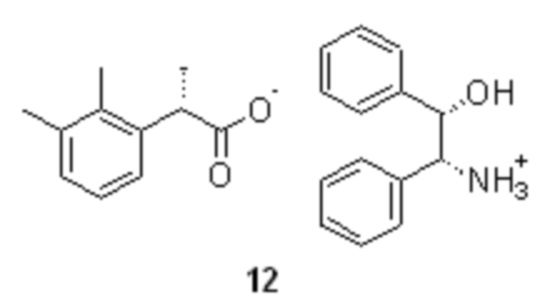
(S)-2-(2,3-диметилфенил)пропионат (1S,2R)-(+)-2-амино-1,2-дифенилэтанола (12).
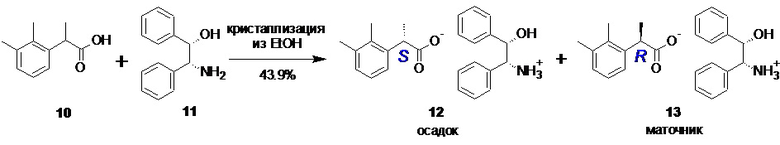
В стеклянный реактор объемом 30 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой загружали рац-2-(2,3-диметилфенил)пропионовую кислоту (10; 850.2 г; 4.77 моль), (1S,2R)-(+)-2-амино-1,2-дифенилэтанол (11; 1017.4 г; 4.77 моль) и этанол (70 об.%; 16.58 л). Реакционную смесь нагревали (от 40 до 43°С) и перемешивали (80 об/мин) до полного растворения всех твердых веществ (20-30 мин). Прозрачный раствор охлаждали до 10±0.2°С, останавливали перемешивание и в реактор вносили затравку чистой соли 12 (0.5 г; er S:R = 99.8 : 0.2). Раствор выдерживали 3 часа при 10±0.2°С, после чего плавно понижали температуру с 10 до 0°С в течение 8 часов (скорость охлаждения ~0.02°С/мин). После охлаждения до 0°С процессу кристаллизации давали завершиться в течение 12 часов при 0°С (общее время 23 часа), продукт фильтровали, промывали на фильтре холодным (0°С) этанолом (50 об.%; 3.0 л), и сушили при 50°С/0.03 mmHg в течение 20 часов. Выход 552 г (59.1%; er S:R = 90.5 : 9.5). Не совсем белый легкий порошок. Полученную соль 12 (552 г) растворяли при перемешивании (120 об/мин) в нагретом до 75°С этаноле (50 об.%; 5.52 л), прозрачный раствор охлаждали до 60±0.5°С, останавливали перемешивание и в реактор вносили затравку чистой соли 12 (0.5 г). Раствор выдерживали 1 час при 60±0.5°С, после чего плавно понижали температуру с 60 до -5°С в течение 18 часов (скорость охлаждения ~0.06°С/мин). После охлаждения до -5°С процессу кристаллизации давали завершиться в течение 5 часов при -5°С (общее время 24 часа), продукт фильтровали, промывали на фильтре холодным (-6°С) этанолом (50 об.%; 2х1.0 л), и сушили при 50°С/0.03 mmHg в течение 20 часов. Выход 445.8 г (47.7%; er S:R = 98.75 : 1.25). Тонкие иголочки не совсем белого цвета. Полученную соль 12 (445.6 г) растворяли при перемешивании (80 об/мин) в нагретом до 75°С этаноле (50 об.%; 3.12 л), прозрачный раствор охлаждали до 60±0.5°С, останавливали перемешивание и в реактор вносили затравку чистой соли 12 (0.1 г). Раствор выдерживали 2 часа при 60±0.5°С, после чего плавно понижали температуру с 60 до -10°С в течение 14.5 часов (скорость охлаждения ~0.08°С/мин). После охлаждения до -10°С процессу кристаллизации давали завершиться в течение 8 часов при -10°С (общее время 24.5 часа), продукт фильтровали, промывали на фильтре холодным (-12°С) этанолом (50 об.%; 1.0 л), и сушили при 50°С/0.03 mmHg в течение 16 часов. Выход 409.8 г (43.9%). Длинные тонкие иголочки белого цвета. tпл 97.5-101.5°С; [α]25D = +91.5° (c 1.0, MeOH); er S:R = 99.82 : 0.18 (tR 8.46 мин (R-энантиомер 14), tR 10.13 мин (S-энантиомер 15); HPLC метод B). 1H NMR (400 MHz, DMSO-d6) δ 7.25-7.09 (m, J = 26.5, 9.3 Hz, 10H), 7.09-6.98 (m, J = 14.5, 6.9 Hz, 3H), 6.20 (s, 4H), 4.82 (d, J = 3.8 Hz, 1H), 4.10 (d, J = 3.9 Hz, 1H), 3.84 (q, J = 13.5, 6.8 Hz, 1H), 2.24 (s, 3H), 2.19 (s, 3H), 1.31 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 176.9, 142.5, 140.9, 140.7, 136.1, 134.0, 128.2, 127.7, 127.5, 127.3, 126.8, 126.7, 125.3, 124.4, 75.7, 60.9, 42.4, 20.7, 18.5, 14.9. tR 1.23 мин (амин), 8.52 мин (кислота) (чистота 99.85a%, HPLC метод А).
Регенерация обогащенной R-энантиомером 2-(2,3-диметилфенил)пропионовой кислоты (14) и (+)-ADPE (11).
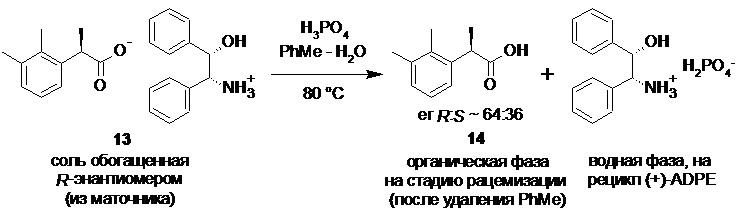
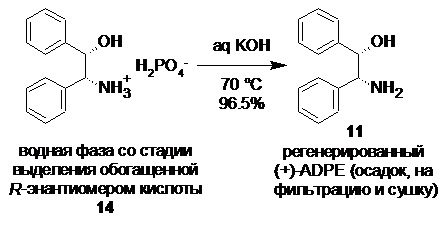
Объединенный спиртовой маточник со стадии кристаллизации (31.2 л) концентрировали в вакууме (50 mmHg) при 55-60°С до ~10 л. Полученную густую суспензию выпавшей соли разбавляли водой (10.0 л) и толуолом (3.0 л), нагревали до 80°С при перемешивании (при этом вся выпавшая соль растворяется в толуоле, и образуются две прозрачные фазы), и в течение 15 мин добавляли смесь 85% ортофосфорной кислоты (644.0 г; 5.58 моль; 1.5 экв) и воды (650 мл). Фазы перемешивали при 80°С в течение 1 часа, разделяли, и водную фазу повторно экстрагировали толуолом (3.0 л). Объединенный органический экстракт промывали 5% раствором ортофосфорной кислоты (2х1.0 л), насыщенным водным раствором хлорида натрия (2.0 л) и концентрировали в вакууме. Кристаллический остаток сушили при 45°С/5 mmHg в течение 16 часов. Продукт использовали на следующей стадии рацемизации без дополнительной очистки. Выход 2-(2,3-диметилфенил)пропионовой кислоты 655.5 г (98.8%). Светло-желтый кристаллический порошок. tпл 87.5-91.5°С; [α]25D = -33.4° (c 4.0, MeOH); er R:S = 63.6 : 36.4 (tR 8.61 мин (R-энантиомер 14), tR 10.19 мин (S-энантиомер 15); HPLC метод B); 1H и 13C NMR данные аналогичны приведённым для 10 выше; tR 8.42 мин (чистота 99.60a%, HPLC метод A).
Водную фазу содержащую дигидрофосфат (+)-ADPE охлаждали до 70°С, и перекачивали в 30 л стеклянный реактор (скорость подачи 4 л/мин), содержащий предварительно нагретый до 70°С, перемешиваемый (200-250 об/мин) раствор гидроксида калия (1.166 кг; 86 мас% KOH; 17.87 моль; 3.2 экв) в воде (4.0 л). Суспензию выпавшего кристаллического основания (+)-ADPE (11) охлаждали до 5°С, перемешивали на низких оборотах (80 об/мин) в течение 16 часов, фильтровали, промывали водой (3х2.0 л), и сушили при 50°С/0.03 mmHg в течение 48 часов. Выход 766.8 г (регенерация 96.5%). Белый кристаллический порошок, практически не растворимый в воде. tпл 141-141.6°С; [α]25D = +6.0° (c 1.0, EtOH); 1H NMR (400 MHz, DMSO-d6) δ 7.27-7.13 (m, 10H), 5.26 (d, J = 3.8 Hz, 1H), 4.63-4.57 (m, 1H), 3.95 (d, J = 5.9 Hz, 1H), 1.60 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 143.7, 143.1, 127.8, 127.4, 127.3, 127.0, 126.8, 126.3, 77.6, 61.4; tR 6.53 мин (чистота 99.92a%, HPLC метод C).
Рацемизация обогащенной R-энантиомером 2-(2,3-диметилфенил)пропионовой кислоты (14).
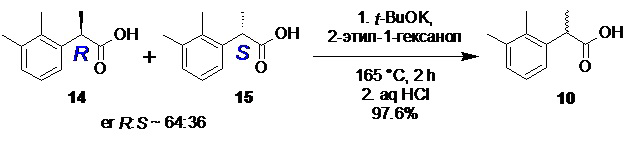
В стальной реактор объемом 30 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой и обратным холодильником, загружали обогащенную R-энантиомером 2-(2,3-диметилфенил)пропионовую кислоту (14; 655.5 г; 3.68 моль; 1.0 экв; er R:S = 63.6 : 36.4) и 2-этилгексанол (3.68 л). Смесь перемешивали при 50°С до полного растворения кислоты 14 (5-10 мин), реактор заполняли сухим аргоном, и загружали порошкообразный трет-бутилат калия (619.0 г; 5.52 моль; 1.5 экв). Температуру обогрева реактора повышали до 165-170°С, и продолжали перемешивание (60-70 об/мин) в течение 2 часов поддерживая в реакторе небольшое избыточное давление аргона (20-30 mmHg) для защиты реакционной смеси от углекислоты воздуха. По прошествии 2 часов HPLC анализ показал полную рацемизацию (Метод B). Реакционную смесь охлаждали до ~80°С, добавляли воду (7.36 л), перемешивали 20-30 минут, прозрачный нижний водный слой, содержащий калиевую соль кислоты 10 отделяли, охлаждали до 30-35°С и подкисляли 36% соляной кислотой (517 мл; 1.1 экв). Белый кристаллический осадок фильтровали, промывали водой (3х3.0 л), и сушили при 50°С/0.03 mmHg в течение 16 часов. Выход 640.1 г (97.6%). Степень восстановления кислоты 10 за 1 цикл разделения/регенерации/рацемизации (RRR) 96.5%. Белый кристаллический порошок. tпл 93-94.8°С; er R:S = 50.1 : 49.9 (tR 8.57 мин (R-энантиомер 14), tR 10.01 мин (S-энантиомер 15); HPLC метод B); 1H и 13C NMR данные аналогичны приведённым для 10 выше; tR 8.47 мин (чистота 99.80a%, HPLC метод A).
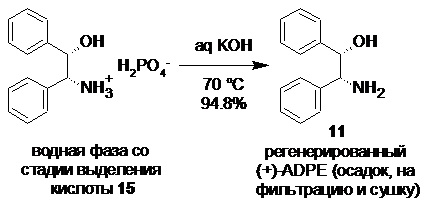
Выделение (S)-2-(2,3-диметилфенил)пропионовой кислоты (15) из соли и регенерация (+)-ADPE (11).
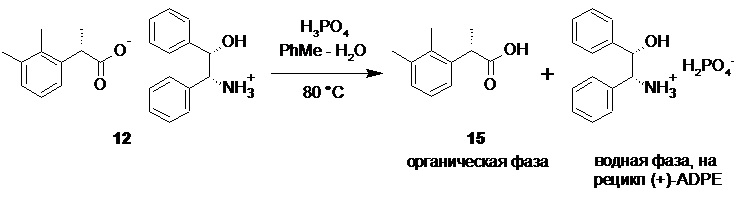
В стеклянный реактор объемом 30 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой загружали (S)-2-(2,3-диметилфенил)пропионат (1S,2R)-(+)-2-амино-1,2-дифенилэтанола (12; 1308.7 г; 3.34 моль), воду (10.0 л) и толуол (6.0 л). Смесь нагревали до 80°С, и при перемешивании (200-250 об/мин) в течение 15 мин добавляли смесь 85% ортофосфорной кислоты (578.1 г; 5.01 моль; 1.5 экв) и воды (580 мл). Фазы перемешивали при 80°С в течение 1 часа, разделяли, и водную фазу повторно экстрагировали толуолом (2х1.0 л). Объединенный органический экстракт промывали раствором ортофосфорной кислоты (50 г 85% H3PO4) в воде (1.0 л), насыщенным водным раствором хлорида натрия (3.0 л), и концентрировали в вакууме при 50°С. Полученное светло-желтое масло кристаллизовалось при стоянии в течение ночи. Продукт использовали на следующей стадии без дополнительной очистки. Выход (S)-2-(2,3-диметилфенил)пропионовой кислоты 580.8 г (97.5%). Светло-желтые кристаллы. tпл 46.7-47.2°С; [α]25D = +107.5° (c 1.0, MeOH); er S:R = 99.5 : 0.5 (tR 8.43 мин (R-энантиомер 14), tR 10.11 мин (S-энантиомер 15); HPLC метод B); 1H и 13C NMR данные аналогичны приведённым для 10 выше; tR 8.48 мин (чистота 98.0a%, HPLC метод A).
Объединенную водную фазу содержащую дигидрофосфат (+)-ADPE охлаждали до 70°С, и перекачивали в 30 л стеклянный реактор (скорость подачи 4 л/мин), содержащий предварительно нагретый до 70°С, перемешиваемый (200-250 об/мин) раствор гидроксида калия (1.137 кг; 86 мас% KOH; 17.43 моль; 3.2 экв) в воде (3.75 л). Суспензию выпавшего кристаллического основания (+)-ADPE (11) охлаждали до 2°С, перемешивали на низких оборотах (80 об/мин) в течение 16 часов, фильтровали, промывали водой (4х3.0 л), и сушили при 60°С/0.03 mmHg в течение 48 часов. Выход 676.0 г (94.8%). Белый кристаллический порошок, практически не растворимый в воде. tпл 141.7-142.5°С; [α]25D = +6.0° (c 1.0, EtOH); 1H и 13C NMR данные аналогичны приведённым для 11 выше; tR 6.67 min (чистота 99.93a%, HPLC метод С).
(S)-2-(2,3-диметилфенил)пропионилхлорид (16).

В стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, заполненный аргоном загружали (S)-2-(2,3-диметилфенил)пропионовую кислоту (15; 578.0 г; 3.24 моль; 1.0 экв), сухой толуол (1.62 л) и N,N-диметилформамид (5.92 г; 81.0 ммоль; 2.5 моль%). Смесь перемешивали до полного растворения, и одной порцией добавляли тионилхлорид (482.3 г; 4.05 моль; 1.25 экв). При этом начиналось плавное выделение газов без вспенивания, и каких-либо признаков разогревания (Осторожно! Использовать газовый скруббер для поглощения HCl и SO2 или отводить газы в вытяжное устройство!). Прозрачный раствор перемешивали при комнатной температуре до полного прекращения выделения газов. HPLC анализ аликвоты погашенной н-пропанолом по прошествии 4 часов показал >99.7% конверсии (Метод А). Раствор концентрировали в вакууме при 40°С. Сырой продукт использовали на следующей стадии без дополнительной очистки. Выход 715.8 г (100%; теоретический выход 637.8 г). Прозрачная светло-желтая жидкость. er S:R = 99.5 : 0.5 (дериватизация метанолом; tR 22.62 мин (метил (R)-2-(2,3-диметилфенил)пропионат), tR 25.47 мин (метил (S)-2-(2,3-диметилфенил)пропионат); HPLC метод D); Аналогичное соотношение энантиомеров получено при дериватизации н-пропиламином (tR 7.65 мин ((R)-2-(2,3-диметилфенил)-N-пропилпропионамид), tR 8.33 мин ((S)-2-(2,3-диметилфенил)-N-пропилпропионамид); HPLC метод E); 1H NMR (400 MHz, CDCl3) δ 7.16-7.14 (m, 2H), 7.09-7.04 (m, 1H), 4.43 (q, J = 7.0 Hz, 1H), 2.34 (s, 3H), 2.30 (s, 3H), 1.57 (d, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 176.0, 137.8, 136.4, 134.7, 129.9, 126.3, 124.9, 54.1, 21.2, 18.5, 15.4.
(3S)-1-[диметил(оксидо)-λ6-сульфанилиден]-3-(2,3-диметилфенил)бутан-2-он (19).
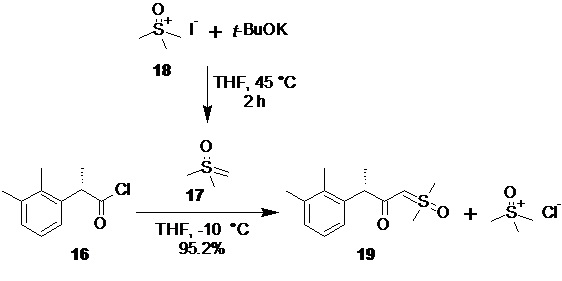

В сухой стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, заполненный аргоном загружали триметилсульфоксоний иодид (18; 1.50 кг; 6.81 моль; 2.1 экв), сухой ТГФ (6.13 л), и при перемешивании добавляли трет-бутилат калия (764.8 г; 6.81 моль; 2.1 экв). Реактор оборачивали алюминиевой фольгой (для защиты от света), реакционную смесь нагревали до 45°С и перемешивали в течение 2 часов. Суспензию охлаждали до -10°С, и добавляли при перемешивании (500 об/мин) раствор сырого (S)-2-(2,3-диметилфенил)пропионилхлорида (16; 715.1 г; расчетное содержание чистого вещества 637.1 г; 3.24 моль; 1 экв) в сухом ТГФ (2.23 л) со скоростью около 1 л/час, поддерживая внутреннюю температуру смеси в пределах от -9.8 до -10.2°С. Анализ аликвоты по прошествии 1 часа перемешивания при -10°С после добавления 16 показал >99% конверсии 16 (Метод А). Суспензию концентрировали в вакууме (при температуре не выше 35°С), разбавляли водой (7.2 л) и этилацетатом (5.0 л), перемешивали 10 минут, фазы разделяли, водную фазу фильтровали от осадка триметилсульфоксоний йодида, и повторно экстрагировали этилацетатом (3.0 и 2.0 л). Объединенный органический экстракт сушили сульфатом натрия (250 г) и концентрировали в вакууме при 35°С. Кристаллический остаток сушили в вакууме при 25°С в течение 8 часов. Выход 778.3 г (95.2%). Светло-желтый мелкокристаллический порошок. tпл 91.2-92.7°С; er S:R = 96.2 : 3.8 (tR 28.85 мин (R-энантиомер), tR 30.65 мин (S-энантиомер); HPLC метод B); 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 7.5 Hz, 1H), 7.09-7.00 (m, 2H), 4.16 (s, 1H), 3.78 (q, J = 7.1 Hz, 1H), 3.35 (s, 3H), 3.31 (s, 3H), 2.28 (s, 3H), 2.22 (s, 3H), 1.43 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 192.8, 141.5, 136.8, 134.7, 128.2, 125.7, 124.9, 68.8, 47.2, 42.3, 42.2, 21.1, 18.2, 15.4; tR 6.96 мин (чистота 96.40a%, HPLC метод F). Выход восстановленного триметилсульфоксоний йодида 558.8 г (78.3%). Светло-желтый мелкокристаллический порошок. Массовая доля 18 по результатам аргентометрического титрования 99.1%.
(S)-1-бром-3-(2,3-диметилфенил)бутан-2-он (20).
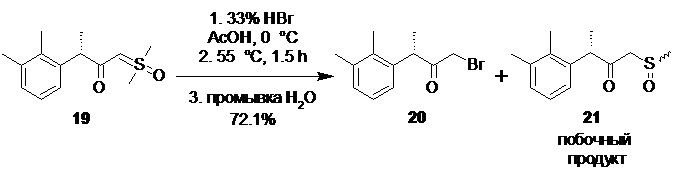
В сухой стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, заполненный аргоном загружали раствор соединения 19 (776.5 г; 3.07 моль; 1.0 экв) в сухом ТГФ (7.76 л). Раствор охлаждали до 0°С, и в течение 40 мин добавляли 33% раствор бромоводорода в ледяной уксусной кислоте (829.8 г; 3.38 моль; 1.1 экв) поддерживая внутреннюю температуру в пределах от 0.5 до 1.5°С. Полученную светло-желтую суспензию перемешивали еще 20 минут при 0°С, после чего нагревали до 55°С. По прошествии 1.5 часа нагрева при 55°С HPLC анализ аликвоты показал >99.5% конверсии стартового 19 (Метод А). Прозрачный раствор желтого цвета концентрировали в вакууме при 30°С, и остаток распределяли между водой (6.0 л) и гептаном (6.0 л). Фазы перемешивали 15 мин, водный слой отделяли, а гептановый экстракт повторно промывали водой (6х5.0 л) контролируя удаление побочного сульфоксида 21 с помощью HPLC (Метод А). Раствор концентрировали в вакууме при 35°С. Сырой продукт использовали на следующей стадии без дополнительной очистки. Выход 566.0 г (72.1%). Прозрачное темно-желтое масло, не обладающее лакриматорным действием, кристаллизующееся при стоянии в длинные иглы. er S:R = 94.9 : 5.1 (tR 19.33 мин (R-энантиомер), tR 20.72 мин (S-энантиомер); HPLC метод D); 1H NMR (400 MHz, CDCl3) δ 7.12-7.04 (m, 2H), 6.88-6.81 (m, J = 6.8, 2.2 Hz, 1H), 4.39 (q, J = 6.8 Hz, 1H), 3.79 (dd, J = 39.8, 12.7 Hz, 2H), 2.33 (s, 3H), 2.31 (s, 3H), 1.39 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 202.8, 138.1, 138.0, 134.6, 129.4, 126.4, 124.9, 46.7, 33.3, 21.2, 17.3, 15.4; tR 12.79 мин (чистота 95.34a%, HPLC метод A).
Объединенную водную фазу (~36 л), содержащую побочный сульфоксид 21 экстрагировали хлороформом (5х2.0 л), экстракт концентрировали в вакууме при 35°С, остаток растворяли в этилацетате (2.0 л), промывали насыщенным водным раствором бикарбоната натрия (2.0 л), сушили сульфатом натрия, фильтровали и этилацетат удаляли в вакууме при 35°С. Выход 186.43 г (25.4%; смесь диастереомеров). Прозрачное светло-оранжевое масло с сернистым запахом. 1H NMR (400 MHz, CDCl3) δ 7.11-7.04 (m, 2H), 6.81 (t, J = 6.5 Hz, 1H), 4.14 (q, J = 6.8 Hz, 1H), 3.80-3.40 (dd, 2H), 2.64 (s, 3H), 2.28 (d, 6H), 1.36 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 202.7, 138.2, 137.3, 134.9, 129.5, 126.4, 125.3, 60.9, 52.3, 38.7, 21.2, 16.5, 15.3 (Преобладающий диастереомер); 1H NMR (400 MHz, CDCl3) δ 7.11-7.04 (m, 2H), 6.81 (t, J = 6.5 Hz, 1H), 4.08 (q, J = 6.8 Hz, 1H), 3.80-3.40 (dd, 2H), 2.57 (s, 3H), 2.31 (d, 6H), 1.36 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 203.2, 138.3, 137.5, 134.7, 129.5, 126.4, 125.2, 64.6, 51.4, 39.7, 21.2, 16.5, 15.4 (Минорный диастереомер); tR 7.22 мин (чистота 99.73a%, HPLC метод A);
(S)-1-азидо-3-(2,3-диметилфенил)бутан-2-он (22).
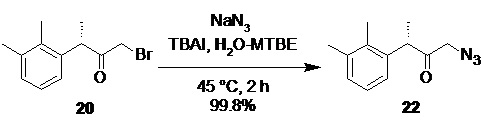
В стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, и обратным холодильником загружали раствор азида натрия (171.2 г; 2.63 моль; 1.2 экв) в воде (2.20 л), TBAI (40.5 г; 5 моль%; 109.7 ммоль) и раствор бромкетона 20 (560.0 г; 2.19 моль; 1.0 экв) в МТБЭ (2.2 л). Двухфазную систему перемешивали (300 об/мин) при 45°С. Через 2 часа HPLC анализ показал 100% конверсию исходного бромкетона 20 в азидокетон 22 (Метод А). Органическую фазу отделяли, промывали водой (2.0 л), 1% водным раствором тиосульфата натрия (1.0 л), водой (2.0 л), и концентрировали в вакууме при 30°С. Сырой продукт использовали на следующей стадии гидрирования без дополнительной очистки. Выход 475.8 г (99.8%). Прозрачное желтое масло. er S:R = 94.8 : 5.2 (tR 36.64 мин (S-энантиомер), tR 39.39 мин (R-энантиомер); HPLC метод G); 1H NMR (400 MHz, CDCl3) δ 7.12-7.05 (m, 2H), 6.92-6.84 (m, J = 5.8, 3.3 Hz, 1H), 4.06 (q, J = 6.9 Hz, 1H), 3.80 (dd, J = 3.4 Hz, 2H), 2.32 (s, 3H), 2.28 (s, 3H), 1.40 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 205.4, 138.0, 137.7, 134.3, 129.4, 126.5, 124.9, 56.1, 47.6, 21.1, 16.8, 15.3; tR 12.24 мин (чистота 94.64a%, HPLC метод A).
(S)-1-амино-3-(2,3-диметилфенил)бутан-2-он мезилат (23а).
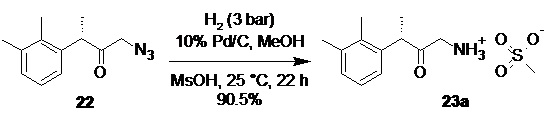
В стальной реактор для гидрирования объемом 30 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой загружали раствор азидокетона 22 (475.5 г; 2.18 моль; 1.0 экв) в метаноле (4.0 л). Реактор заполняли аргоном, и последовательно вносили раствор метансульфоновой кислоты (216.7 г; концентрация 99%; 2.23 моль; 1.02 экв) в метаноле (0.75 л), и 10% палладий на угле (47.5 г; 10 вес%). В реактор закачивали водород до давления 3.5 бар, после чего стравливали давление до 0.2 бар. Промывку реактора водородом повторяли еще 2 раза, реактор термостатировали (25°С), запускали перемешивание (100 об/мин), и продолжали процесс гидрирования под давлением 3.5 бара. Через 22 часа HPLC анализ показал >99% конверсии исходного азидокетона 22 в мезилат аминокетона 23а (Метод C). Катализатор фильтровали через GFA фильтр, промывали метанолом (0.3 л) и прозрачный светло-желтый фильтрат концентрировали досуха в вакууме при 35°С. К кристаллическому остатку добавляли МТБЭ (2.0 л) и н-гептан (0.50 л), суспензию перемешивали 20 мин, фильтровали, осадок промывали на фильтре смесью МТБЭ (2.0 л) и н-гептана (0.50 л), а затем чистым н-гептаном (2.0 л). Продукт сушили в вакууме (3 mmHg) при 25°С в течение 18 часов. Выход 569.2 г (90.5%). Бледно-желтый перламутровый кристаллический порошок. tпл 145-147°С; er S:R = 94.4 : 5.6 (Дериватизация N,N-диметилкарбамоил хлоридом; tR 4.73 мин (R-энантиомер), tR 5.63 мин (S-энантиомер); HPLC метод H); 1H NMR (400 MHz, DMSO-d6) δ 8.01 (s, 3H), 7.12-7.05 (m, 2H), 6.91-6.84 (m, J = 6.7, 2.3 Hz, 1H), 4.25 (q, J = 6.9 Hz, 1H), 3.79 (dd, J = 136.2, 18.0 Hz, 2H), 2.31 (s, 3H), 2.27 (s, 3H), 2.21 (s, 3H), 1.30 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 204.5, 137.8, 137.2, 134.6, 128.9, 125.9, 124.9, 46.2, 45.8, 39.7, 20.7, 16.7, 15.0; tR 6.89 мин (чистота 99.14a%, HPLC метод C).
4-[(S)-1-(2,3-диметилфенил)этил]-1,3-дигидро-2H-имидазол-2-тион (24).
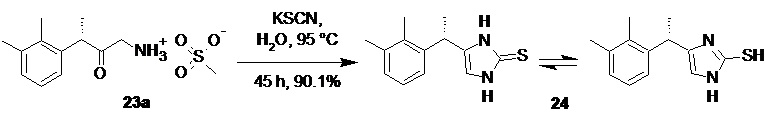
В стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, и обратным холодильником загружали мезилат аминокетона (23а; 567.3 г; 1.97 моль; 1.0 экв), тиоцианат калия (959.2 г; 9.87 моль; 5.0 экв) и воду (1.97 л). Реактор продували аргоном, и смесь нагревали при перемешивании (200-250 об/мин), поддерживая внутреннюю температуру от 93 до 95°С в течение 45 часов. Суспензию охлаждали до 20°С, разбавляли водой (5.0 л), фильтровали, осадок промывали водой (3х3.0 л), охлажденным до 0°С изопропанолом (1.0 л), и смесью изопропанола и н-гептана (1:2 по объему; 2х1.5 л). Продукт сушили в вакууме (0.03 mmHg) при 40°С в течение 18 часов. Выход 413.3 г (90.1%). Бледно-желтый, блестящий, перламутровый мелкокристаллический порошок без запаха. tпл 244-245°С (с разл.); [α]23D = +10.5° (c 1.0, MeOH); er S:R = 87.4 : 12.6 (tR 6.32 мин (S-энантиомер), tR 9.56 мин (R-энантиомер); HPLC метод I); 1H NMR (400 MHz, DMSO-d6) δ 11.83 (s, 1H), 11.70 (s, 1H), 7.03-7.00 (m, 2H), 6.95-6.90 (m, J = 8.5, 4.4 Hz, 1H), 6.50 (s, 1H), 4.15 (q, J = 7.1 Hz, 1H), 2.24 (s, 3H), 2.20 (s, 3H), 1.38 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.4, 141.7, 136.3, 133.7, 133.6, 127.9, 125.4, 123.9, 111.4, 31.8, 20.6, 19.9, 14.4; tR 7.00 мин (чистота 99.86a%, HPLC метод A).
Сырое основание дексмедетомидина (1).
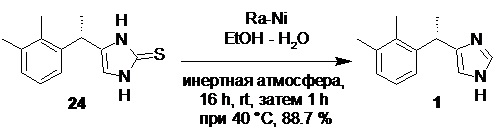
В стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, заполненный аргоном, содержащий свежеприготовленную суспензию никеля Ренея (~0.94 кг; 16 моль; 10 экв) в этаноле (4.62 л; 96 об.%) загружали тион 24 (370.0 г; 1.59 моль). Реактор продували аргоном, и суспензию перемешивали (350 об/мин) в атмосфере аргона при 27°С отслеживая конверсию с помощью HPLC. По прошествии 16 часов перемешивания анализ показывал 99.8% конверсии (содержание стартового 24 по HPLC 0.2а%; метод C). Для завершения реакции суспензию нагревали 1 час при 40°С (конверсия >99.9%, содержание стартового 24 по HPLC 0.06а%), охлаждали до 25°С, суспензии давали отстояться (15-20 мин), и сифонировали раствор продукта с осадка небольшим давлением аргона (0.2 бар). В реактор заливали свежую порцию этанола (4.0 л; 96 об.%), суспензию перемешивали 1 час, осадку давали осесть, и спиртовую фазу сливали. Промывку повторяли еще 1 раз (Осторожно! Шлам состоящий из сульфида никеля (II) и непрореагировавшего никеля Ренея все еще сохраняет пирофорность! Избегать высыхания и контакта шлама с воздухом!), объединенный спиртовой раствор продукта фильтровали через слой целита, концентрировали на роторном испарителе, и остаток сушили в вакууме (5 mmHg) в течение 12 часов. Выход 282.9 г (88.7%). Не совсем белый кристаллический порошок. tпл 130.5-132°С; [α]23D = +55.5° (c 1.0, MeOH); er S:R = 87.1 : 12.9 (tR 4.37 мин (R-энантиомер, левомедетомидин), tR 6.49 мин (S-энантиомер, дексмедетомидин); HPLC метод J); 1H NMR (400 MHz, CDCl3) δ 10.36 (s, 1H), 7.23 (s, 1H), 7.04-6.97 (m, 2H), 6.94-6.88 (m, J = 6.5, 2.5 Hz, 1H), 6.65 (s, 1H), 4.37 (q, J = 7.1 Hz, 1H), 2.25 (s, 3H), 2.18 (s, 3H), 1.55 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.3, 141.2, 136.9, 134.6, 134.2, 128.1, 125.7, 124.8, 117.3, 34.2, 21.0, 20.8, 14.8; tR 6.36 мин (чистота 99.94a%, HPLC метод K). Остаточное содержание металлов: Co 0.02 ppm, Pd 0.59 ppm, Ni 10.2 ppm (ICP-OES).
Дексмедетомидин L-(+)-тартрат (25).
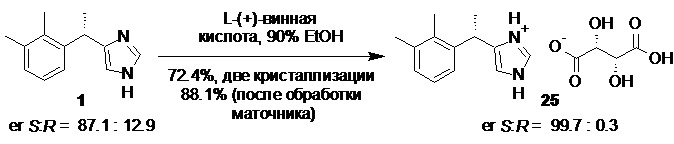
Сырое основание дексмедетомидина (1; 276.0 г; 1.37 моль; er S:R = 87.1 : 12.9) растворяли в 90% этаноле (2.0 л) непосредственно в испарительной колбе роторного испарителя при легком нагревании (<50°С), и полученный желтый раствор фильтровали через GF/A фильтр. Прозрачный фильтрат переносили в стеклянный реактор объемом 10 л с термостатируемой рубашкой, снабженный верхнеприводной мешалкой с герметичным затвором, и обратным холодильником. В реактор с раствором сырого основания 1 вносили L-(+)-винную кислоту (206.8 г; 1.37 моль; 1.0 экв), и 90% этанол (1.70 л), суспензию нагревали при перемешивании (температура теплоносителя 80°С) 30-40 мин, после чего небольшими порциями (по 10 мл) добавляли воду до достижения почти полного растворения (около 100 мл). Перемешивание останавливали, раствор охлаждали с 80 до 20°С со скоростью 0.25°С/мин, после чего процессу кристаллизации давали завершиться в течение 14 часов. Крупный кристаллический осадок фильтровали, промывали на фильтре охлажденным до 10°С этанолом (90 об.%; 1.0 л), и сушили при 50°С в течение 16 часов. Выход 343.3 г (81.6%; er S:R = 97.5 : 2.5). Полученную соль 25 (343.3 г) растворяли в горячем 90% этаноле (1.80 л; температура обогрева 85°С), перемешивание останавливали, раствор охлаждали с 85 до 20°С со скоростью 0.25°С/мин, после чего процессу кристаллизации давали завершиться в течение 18 часов. Кристаллический осадок фильтровали, промывали на фильтре охлажденным до -10°С этанолом (96 об.%; 1.0 л), и сушили при 50°С в течение 16 часов. Выход 304.55 г (72.4%; er S:R = 99.76 : 0.24).
Объединенный спиртовой маточник со всех кристаллизаций концентрировали в вакууме досуха. Остаток (178.2 г; er S:R = 62.8 : 37.2) растворяли в горячем 90% этаноле (0.675 л; температура обогрева 80°С), перемешивание останавливали, и раствор охлаждали с 80 до 20°С со скоростью 0.1°С/мин (10 часов). В диапазоне температур от 75 до 60°С прозрачный раствор засевали затравкой чистого L-(+)-тартрата дексмедетомидина (1.0 г), и после охлаждения до 20°С процессу кристаллизации давали завершиться в течение 8 часов. Кристаллический осадок фильтровали, промывали на фильтре охлажденным до -10°С этанолом (90 об.%; 0.40 л), и сушили при 50°С в течение 16 часов. Выход 92.52 г (22.0%; er S:R = 82.3 : 17.7). Полученную соль (92.52 г) растворяли в горячем 90% этаноле (0.375 л; температура обогрева 80°C), перемешивание останавливали, раствор охлаждали с 80 до 20°С со скоростью 0.1°С/мин (10 часов), после чего процессу кристаллизации давали завершиться в течение 10 часов. Кристаллический осадок фильтровали, промывали на фильтре охлажденным до -10°С этанолом (90 об.%; 0.25 л), и сушили при 50°С в течение 16 часов. Выход 75.12 г (17.9%; er S:R = 98.1 : 1.9). Полученную соль (75.12 г) суспензировали в 90% этаноле (0.35 л), и нагревали (85°С) при перемешивании в течение 2 часов. Температуру раствора стабилизировали до 80°С, перемешивание останавливали, раствор охлаждали с 80 до 20°С со скоростью 0.25°С/мин, после чего процессу кристаллизации давали завершиться в течение 12 часов. Кристаллический осадок фильтровали, промывали на фильтре охлажденным до -10°С этанолом (90 об.%; 0.20 л), и сушили при 50°С в течение 16 часов. Выход 65.88 г (15.7%; er S:R = 99.72 : 0.28). Общий выход 370.43 г (88.1%). Крупнокристаллический перламутровый блестящий порошок белого цвета. tпл 180-181°С; [α]23D = +58.0° (c 1.0, MeOH); tR 4.87 мин (левомедетомидин), tR 7.19 мин (дексмедетомидин); HPLC метод J); 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 5H), 7.91 (s, 1H), 6.99 (d, J = 4.6 Hz, 2H), 6.91-6.86 (m, 2H), 4.37 (q, J = 7.0 Hz, 1H), 4.23 (s, 2H), 2.24 (s, 3H), 2.21 (s, 3H), 1.46 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 173.7, 143.1, 139.9, 136.3, 134.6, 133.6, 127.7, 125.4, 124.3, 116.6, 72.1, 33.1, 20.8, 20.7, 14.5; tR 6.35 мин (чистота 99.84a%, HPLC метод K). Остаточное содержание металлов: Co 0.02 ppm, Pd ниже предела обнаружения (<0.02 ppm), Ni 1.27 ppm (ICP-OES).
Очищенное основание дексмедетомидина (1).
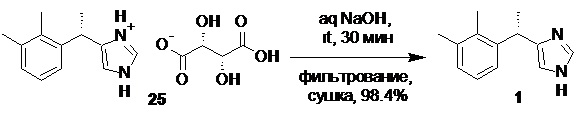
В стеклянный реактор объемом 5 л, снабженный верхнеприводной мешалкой, загружали L-(+)-тартрат дексмедетомидина (25; 368.25 г; 1.05 моль; er S:R = 99.7 : 0.3) и воду (2.0 л), и при перемешивании (120-140 об/мин) добавляли раствор гидроксида натрия (92.5 г; 2.31 моль; 2.2 экв) в воде (0.80 л) в течение 30 минут. Полученную суспензию основания дексмедетомидина перемешивали еще 10 минут, фильтровали, осадок промывали на фильтре водой (3х1.0 л), и сушили при 50°С/0.05 mmHg в течение 16 часов. Выход 207.2 г (98.4%). Белый кристаллический порошок, легко растворимый в метаноле и этаноле, слабо растворимый в ацетоне, практически не растворимый в воде. tпл 148.5-150.5°С; [α]22D = +75.5° (c 1.0, MeOH); er S:R = 99.74 : 0.26 (tR 4.80 мин (R-энантиомер, левомедетомидин), tR 7.41 мин (S-энантиомер, дексмедетомидин); HPLC метод J); 1H и 13C NMR данные аналогичны приведённым для 1 выше; tR 6.35 мин (чистота 99.95a%, HPLC метод K). Остаточное содержание металлов: Co 0.02 ppm, Pd ниже предела обнаружения, Ni 0.5 ppm (ICP-OES);
Дексмедетомидин гидрохлорид (1⋅HCl).

В стеклянный реактор объемом 3 л, снабженный верхнеприводной мешалкой, загружали основание дексмедетомидина (1; 205.0 г; 1.02 моль; er S:R = 99.74 : 0.26) и ацетон (2.0 л). Суспензию охлаждали (от 2.5 до 3°С), и при перемешивании по каплям добавляли 36% соляную кислоту (96 мл; 1.12 моль; 1.1 экв). Полученный бесцветный раствор фильтровали через GF/A фильтр, и концентрировали в вакууме на роторном испарителе. Полученное масло засевали затравкой кристаллического гидрохлорида дексмедетомидина (0.30 г), и выдерживали в вакууме (<10 mmHg) при комнатной температуре в течение 1 часа во вращающейся (20 об/мин) испарительной колбе. К закристаллизовавшемуся остатку добавляли этилацетат (2.0 л), и отгоняли ~1 л дистиллята на роторном испарителе при атмосферном давлении (температура бани 85°С) для удаления воды. Суспензию кристаллов разбавляли этилацетатом (1.0 л), охлаждали до комнатной температуры, фильтровали, остатки кристаллов смывали со стенок этилацетатом (0.25 л), и продукт быстро переносили на стеклянные противни для сушки. Продукт сушили в вакууме при 50°С в течение 88 часов. Выход 235.3 г (97.1%). Микроскопические гигроскопичные иголочки белого цвета, чрезвычайно легко растворимые в воде. tпл 155-157°С; [α]22D = +54.0° (c 1.0, H2O); er S:R = 99.90 : 0.10 (tR 4.47 мин (R-энантиомер, левомедетомидин), tR 6.86 мин (S-энантиомер, дексмедетомидин); HPLC метод J); 1H NMR (400 MHz, DMSO-d6,) δ 9.06 (s, 1H), 7.45 (s, 1H), 7.06-7.00 (m, 2H), 6.89-6.85 (m, 1H), 4.52 (q, J = 7.1 Hz, 1H), 2.24 (d, J = 4.3 Hz, 6H), 1.52 (d, J = 7.1 Hz, 3H); 13C NMR (DMSO-d6, 101 MHz) δ 141.1, 137.4, 136.7, 133.9, 133.8, 128.3, 125.7, 124.1, 115.7, 31.8, 20.7, 20.4, 14.6; tR 6.40 min (чистота 99.98a%, HPLC метод K). Остаточное содержание металлов: Co ниже предела обнаружения, Pd ниже предела обнаружения, Ni 0.5 ppm (ICP-OES).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения медетомидина и производных | 2022 |
|
RU2791397C1 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| БЕНЗОТИА(ДИ)АЗЕПИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ЖЕЛЧНЫХ КИСЛОТ | 2019 |
|
RU2785867C2 |
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОФОСБУВИРА И ФОСФОРАМИДАТЫ | 2020 |
|
RU2740058C1 |
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2769715C1 |
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| СПОСОБЫ ФОРМИРОВАНИЯ ГЛИКОЗИДНЫХ СВЯЗЕЙ, ХИМИЧЕСКАЯ КОМПОЗИЦИЯ, ГЛИКОЗИД И ГЛИКОЗИДНАЯ БИБЛИОТЕКА | 1994 |
|
RU2134693C1 |
Изобретение относится к области фармацевтической химии, и описывает способ получения дексмедетомидина и его фармацевтически приемлемых солей. Техническая проблема, решаемая изобретением, заключается в разработке максимально безотходного и недорогого способа синтеза дексмедетомидина. Технический результат заключается в повышении эффективности за счет увеличения степени извлечения дексмедетомидина из рацемической смеси. Результат достигается за счет предварительного обогащения медетомидина S-энантиомером на ранних стадиях синтеза, эффективным методом регенерации «хиральных отходов», а также разделяющего амина. 2 з.п. ф-лы, 2 ил., 2 табл., 1 пр.
1. Способ получения дексмедетомидина структурной формулы 1
или его фармацевтически приемлемой соли, включающий получение магнийорганического реагента структурной формулы 6
путем реакции 2.3-диметилбромбензола формулы 5
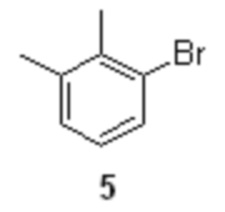
с металлическим магнием, последующее кросс-сочетание реагента 6 с этил 2-бромпропионатом 7
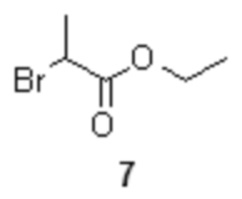
с получением соединения формулы 8
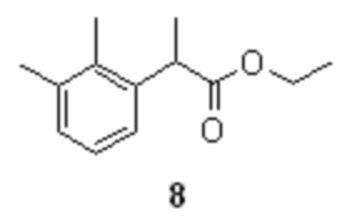
щелочной гидролиз полученного соединения 8 и последующее подкисление водной фазы для осаждения рацемической кислоты формулы 10
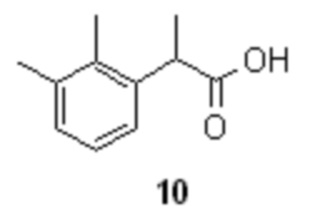
кристаллизацию рацемической кислоты 10 с (+)-ADPE формулы 11
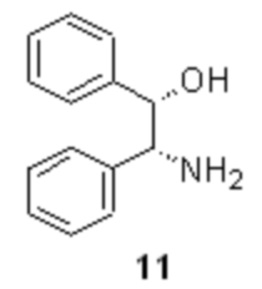
с получением диастереомерной соли формулы 12
разложение диастереомерной соли 12 в кислой среде для выделения желательного S-энантиомера кислоты формулы 15
разложение диастереомерной соли 13, содержащей нежелательный R-энантиомер, полученный на стадии кристаллизации рацемической кислоты 10
для выделения обогащенной R-энантиомером кислоты формулы 14
регенерацию кислоты 10 путем рацемизации кислоты 14, обогащенной нежелательным R-энантиомером под действием сильных оснований до равновесной смеси энантиомеров, и повторное использование рацемизованной кислоты 10 в цикле разделения кристаллизацией с (+)-ADPE формулы 11, реакцию кислоты 15 с тионилхлоридом для получения хлорангидрида формулы 16
генерацию диметилсульфоксоний метилида формулы 17 in situ
из солей триметилсульфоксония, например йодида триметилсульфоксония формулы 18
и сильного основания, с последующим добавлением хлорангидрида 16, для получения соединения формулы 19
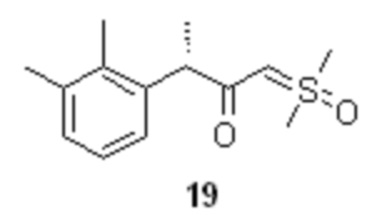
реакцию соединения 19 с бромоводородом в смеси уксусной кислоты и THF, для получения α-бромкетона формулы 20
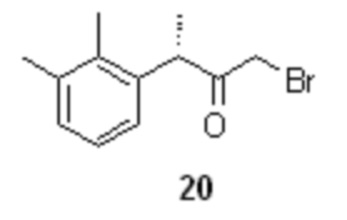
с последующей водной промывкой раствора α-бромкетона 20 в углеводородном растворителе, реакцию α-бромкетона 20 с азидом щелочного металла для получения α-азидокетона формулы 22
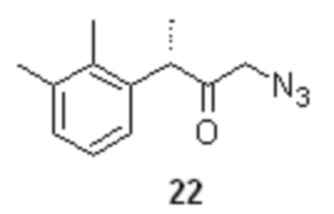
восстановление α-азидокетона 22, с помощью Н2-Pd/C в присутствии органических или минеральных кислот, для получения соответствующих солей аминокетона, циклизацию солей аминокетона с тиоцианатом калия, для получения соединения формулы 24
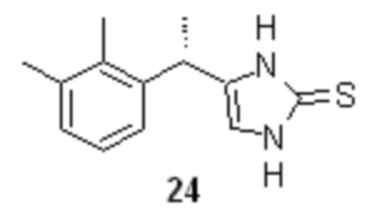
десульфуризацию соединения 24, с помощью никеля Ренея с получением продукта, содержащего от 87 до 93% дексмедетомидина.
2. Способ по п. 1, в котором в качестве органических кислот выбирают метансульфоновую или п-толуолсульфоновую или камфорсульфоновую кислоту, а в качестве минеральных кислот - соляную или бромоводородную или серную кислоту с получением соответствующих солей аминокетона формул 23a-23f
3. Способ по п. 1, включающий повышение энантиомерной чистоты до ≥99% полученного дексмедетомидина 1, путем получения соли дексмедетомидина 1 с L-(+)-винной кислотой 25 и ее кристаллизации,
осаждение очищенного основания дексмедетомидина 1 из соли 25 под действием щелочи, с последующим взаимодействием с хлороводородом в ацетоне, упаривание и азеотропную сушку с помощью этилацетата с получением фармацевтически приемлемой соли гидрохлорида дексмедетомидина формулы 1
| WO 2021089878 A1, 14.05.2021 | |||
| CN 111217756 A, 02.06.2020 | |||
| CN 106632052 A, 10.05.2017 | |||
| Способ получения медетомидина и производных | 2022 |
|
RU2791397C1 |

