Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к промежуточному соединению, применяемому для синтеза ингибитора SGLT (ингибитор натрий-зависимого переносчика глюкозы), а также к способу получения ингибитора SGLT с применением указанного промежуточного соединения.
Предшествующий уровень техники настоящего изобретения
В выложенной для всеобщего ознакомления публикации корейского патента №2017-0142904 (патентный документ 1) раскрыт способ получения производного дифенилметана, обладающего ингибирующей активностью в отношении SGLT2 (натрий-зависимый переносчик глюкозы 2 типа). В соответствии с документом, поскольку производное дифенилметана получают методом конвергентного синтеза для индивидуального синтезирования и связывания производного на основе каждой основной группы, способ синтеза может быть упрощен, а выход продукта может быть повышен по сравнению с методом линейного синтеза, раскрытым в известном уровне техники, тем самым снижая факторы риска, которые характерны для способа линейного синтеза.
Однако способ получения производного дифенилметана в соответствии с выложенной для всеобщего ознакомления публикацией корейского патента №2017-0142904 имеет недостаток, заключающийся в том, что родственные вещества, образующиеся на ранней стадии, не удаляются, поскольку важные стадии взаимодействия с3-с7, как показано на схеме 1, осуществляются при четырехстадийном процессе взаимодействия, причем качество родственных веществ не может контролироваться от стадии к стадии, поскольку одно взаимодействие оказывает влияние на последующее взаимодействие, тем самым затрудняя осуществление манипуляций с родственными веществами. Также способ имеет недостатки, заключающиеся в том, что, поскольку очистка родственных веществ, образующихся при непрерывном процессе, осуществляется на стадии с7, то, для контроля качества родственных веществ, родственные вещества необходимо очищать два и более раза, что приводит к стоимостным затратам, обусловленным процессом очистки, а также невозможности контролировать качество от стадии к стадии, осуществляя несколько стадий in situ.

Раскрытие настоящего изобретения
Техническая задача
Настоящее изобретение относится к промежуточному соединению, применяемому для синтеза ингибитора SGLT, а также к способу получения ингибитора SGLT с применением указанного промежуточного соединения.
Решение технической задачи
Способ получения производного дифенилметана, обладающего ингибирующей активностью в отношении SGLT2, как описано в выложенной для всеобщего ознакомления публикации корейского патента №2017-0142904, осуществляется в процессе, состоящем в общей сложности из шести стадий, включая стадию in situ, как показано на схеме 1. После того, как соединение c1 соединяется с с2 для синтеза соединения с3, с3 сначала десилилируется добавлением c-HCl/MeOH с получением соединения с4. При осуществлении взаимодействия, соединение с4 медленно метоксилируется с образованием соединения с5. В этом случае было трудно кристаллизовать соединение с5 вследствие его физических свойств, причем условия кристаллизации с5 определяются в присутствии толуола/гексана путем анализа нескольких условий кристаллизации. Однако родственные вещества не удаляются, а лишь затвердевают. Поскольку родственные вещества не очищаются, родственные вещества переносятся для последующего взаимодействия в форме in situ и очищаются на стадии с7. Большинство родственных веществ, включая основное родственное вещество, могут быть удалены на стадии с7. Однако, поскольку предыдущая стадия осуществляется без какой-либо очистки, процесс очистки следует проводить два или более раз, чтобы обеспечить качество соответствующих веществ. Поскольку процесс очистки осуществляют два или более раз, выход продукта на этой стадии может быть снижен, и могут возникнуть стоимостные затраты, обусловленные процессом очистки. Кроме того, поскольку с5 трудно хранить в неочищенном состоянии вследствие его пониженной химической стабильности, непрерывное осуществление синтеза непосредственно до с7, начиная с получения с5, связано с большими затратами.
Соответственно, авторы настоящего изобретения разработали новое промежуточное соединение и, таким образом, разработали способ очистки и контроля количества родственных веществ в конечном продукте (то есть производном дифенилметана). Следовательно, настоящее изобретение было осуществлено на основе этих фактов.
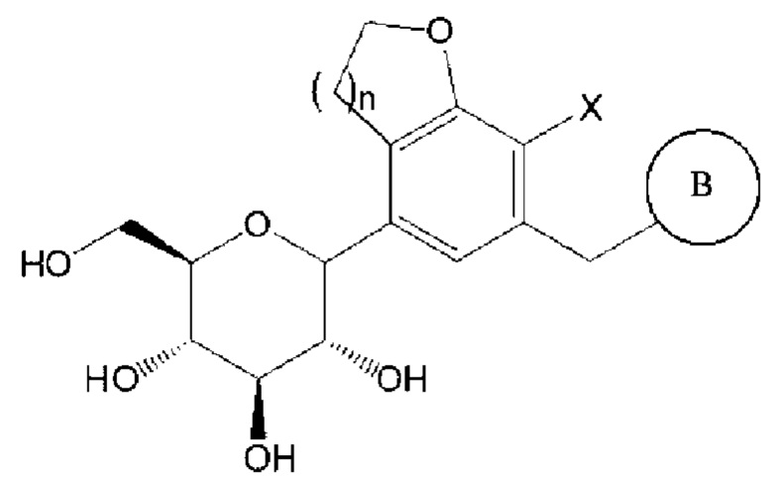
Соединение химической формулы 1, которое является конечным целевым соединением и активным ингредиентом, применяемым в качестве ингибитора SGLT, является следующим:
Химическая формула 1
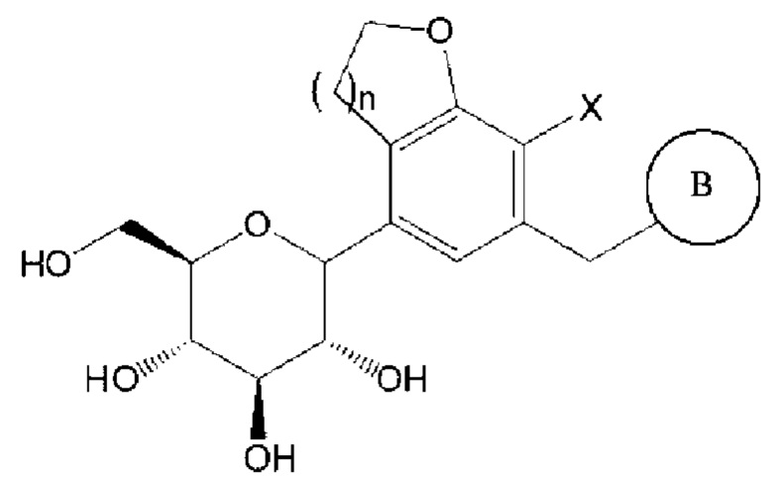
где
n равно 1 или 2,
X представляет собой галоген (например, F, Cl, Br или I),
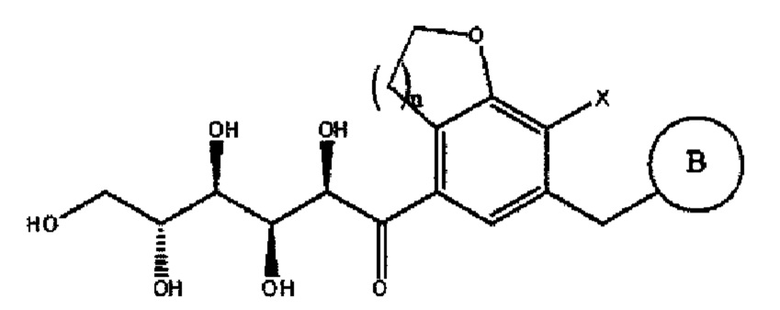
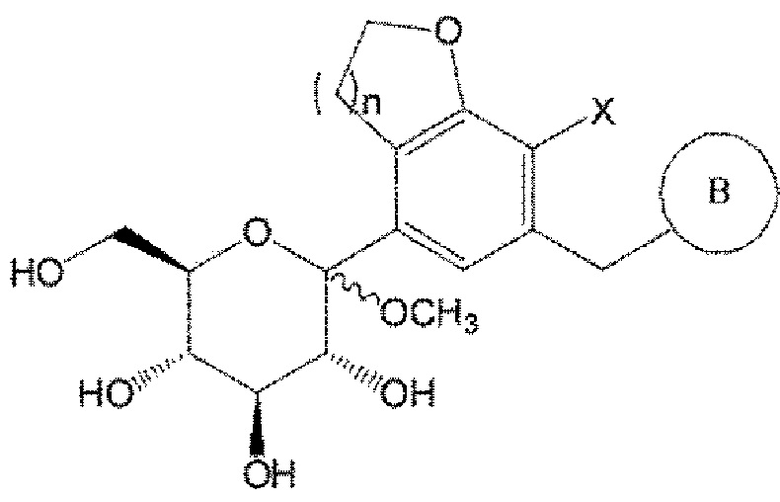
В представляет собой  или
или 
где Ra, Rb, Rc и Rd, каждый независимо, представляют собой водород, галоген, гидрокси, меркапто, циано, нитро, амино, карбокси, оксо, С1-7 алкил, С1-7 алкилтио, С2-7 алкенил, С2-7 алкинил, С1-7 алкокси, С1-7 алкокси-С1-7 алкил, С2-7 алкенил-С1-7 алкилокси, С2-7 алкинил-С1-7 алкилокси, С3-10 циклоалкил, С3-7 циклоалкилтио, С5-10 циклоалкенил, С3-10 циклоалкилокси, С3-10 циклоалкилокси-С1-7 алкокси, фенил-С1-7 алкил, С1-7 алкилтио-фенил, фенил-С1-7 алкокси, моно- или ди-С1-7 алкиламино, моно-или ди-С1-7 алкиламино-С1-7 алкил, С1-7 алканоил, С1-7 алканоиламино, С1-7 алкилкарбонил, C1-7-алкоксикарбонил, карбамоил, моно- или ди-С1-7 алкилкарбамоил, С1-7 алкилсульфониламино, фенилсульфониламино, С1-7 алкилсульфинил, С6-14 арилсульфанил, С6-14 арилсульфонил, С6-14 арил, 5-13-членный гетероарил, 5-10-членный гетероциклоалкил, 5-10-членный гетероциклоалкил-С1-7-алкил или 5-10-членный гетероциклоалкил-С1-7-алкокси;
кольцо С представляет собой С3-10 циклоалкил, С5-10 циклоалкенил, С6-14 арил, 5-13-членный гетероарил или 5-10-членный гетероциклоалкил;
алкил, алкенил, алкинил и алкокси, каждый независимо, незамещены или имеют один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, С1-7 алкила и С2-7 алкинила;
циклоалкил, циклоалкенил, арил, гетероарил и гетероциклоалкил, каждый независимо, незамещены или имеют один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, С1-4 алкила и С1-4 алкокси; и
каждый гетероарил и гетероциклоалкил, каждый независимо, содержат один или несколько гетероатомов, выбранных из группы, состоящей из N, S и О.
Согласно приведенному в качестве примера варианту осуществления настоящего изобретения кольцо В-1 может быть выбрано из группы, состоящей из следующего:

где R7 представляет собой водород или С1-7 алкил; R8a и R8b, каждый независимо, представляют собой С1-7 алкил, или взятые вместе образуют 5-10-членный гетероциклоалкил (содержащий один или несколько гетероатомов, выбранных из группы, состоящей из N, S и О).
В соответствии с другим примером варианта осуществления кольцо В-2 может быть выбрано из группы, состоящей из следующего:
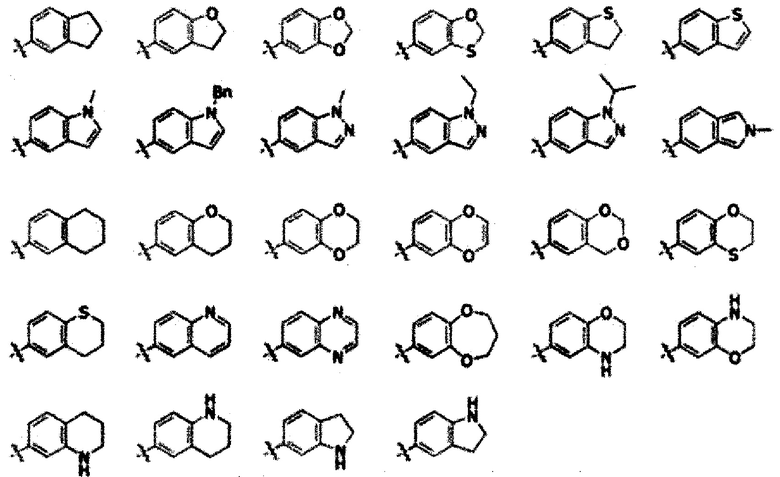
Согласно одному предпочтительному примеру соединения химической формулы 1 n может быть равно 1; X может быть галогеном; и В может представлять собой фенил, незамещенный или замещенный одним или двумя заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, С1-7 алкила, С3-10 циклоалкила и С1-7 алкокси.
Кроме того, соединение химической формулы 1 может быть соединением, в котором участок связывания гетероциклоалкильного кольца с производным дифенилметана находится в его α-форме, β-форме или рацемической форме.
Например, соединение химической формулы 1 может быть соединением следующей химической формулы 1а:
Химическая формула 1а
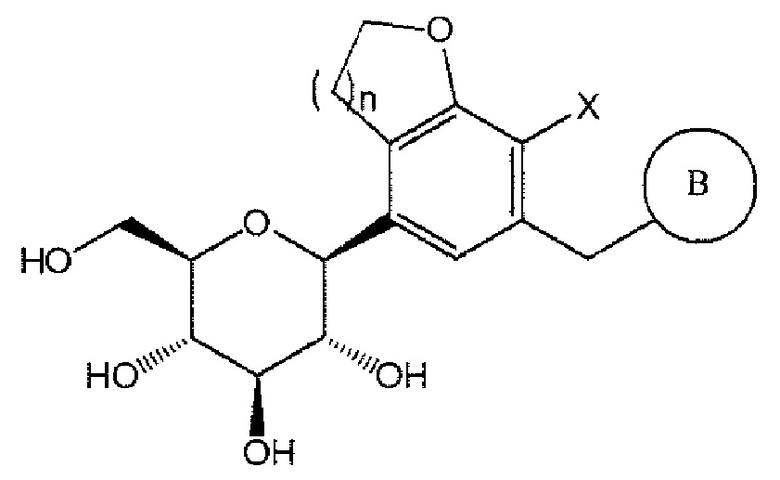
где В, n и X определены выше.
Настоящее изобретение относится к способу получения соединения химической формулы 5, которое является промежуточным соединением, применяемым для получения производного дифенилметана химической формулы 1.
Для получения соединения химической формулы 5 соединение химической формулы 4 можно синтезировать описанным ниже способом и применять, однако настоящее изобретение этим не ограничивается.
Соединение химической формулы 2 может взаимодействовать с соединением химической формулы 3 в присутствии н-бутиллития, втор-бутиллития, трет-бутиллития или хлорида изопропилмагния с получением соединения следующей химической формулы 4:
Химическая формула 2
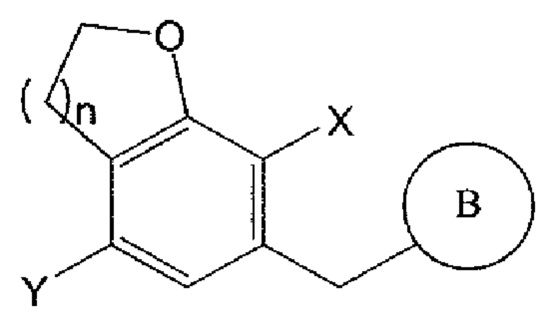
Химическая формула 3

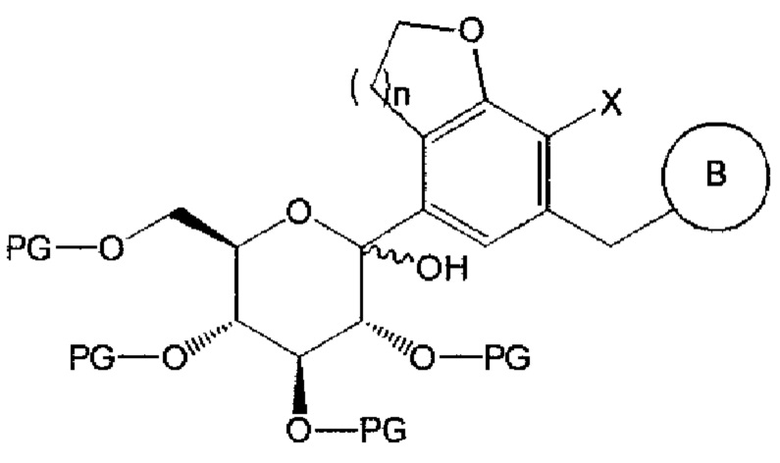
Химическая формула 4
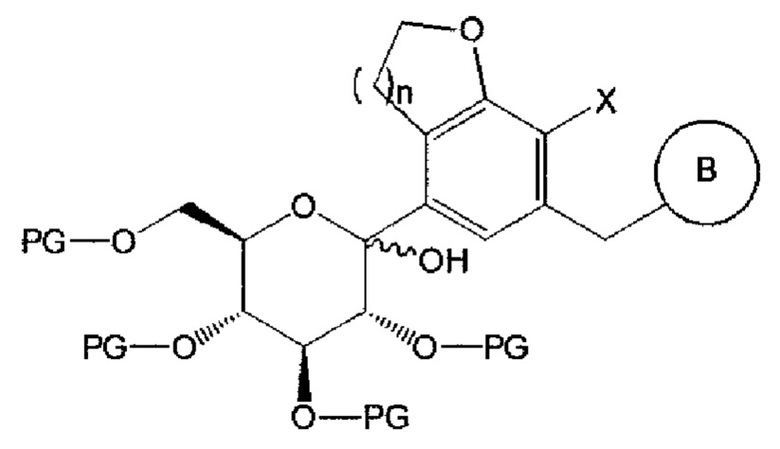
где n, В и X имеют указанные выше значения, Y представляет собой галоген; и PG представляет собой защитную группу.
Взаимодействие соединения химической формулы 2 с соединением химической формулы 3 можно осуществлять в присутствии н-бутиллития, втор-бутиллития, трет-бутиллития, хлорида изопропилмагния (изо-PrMgCl) и тому подобное.
Соединение химической формулы 2 может взаимодействовать с соединением химической формулы 3 с получением соединения следующей химической формулы 4:
Химическая формула 4
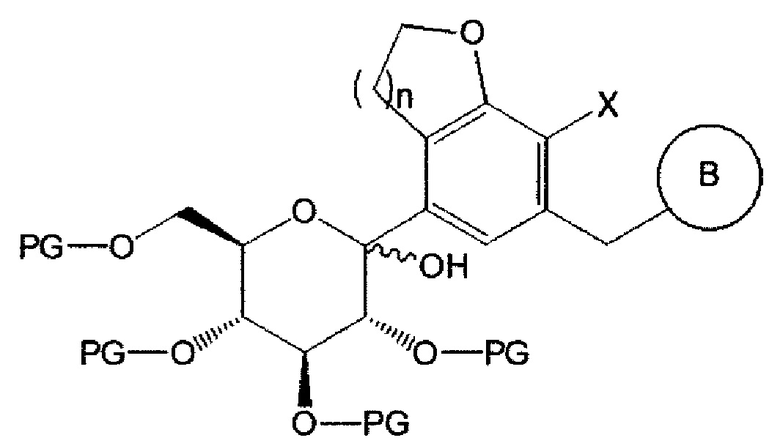
где n равно 1 или 2; X представляет собой галоген; PG представляет собой защитную группу; и В является таким, как определено выше в химической формуле 1.
На стадиях взаимодействия соединения химической формулы 2 с соединением химической формулы 3 сначала осуществляют реакцию связывания. В этом случае каждое соединение химической формулы 3 и реакционный реагент (то есть н-бутиллитий, втор-бутиллитий, трет-бутиллитий или хлорид изопропилмагния) можно применять в диапазоне от 1,5 до 2,5 эквивалентов, более предпочтительно в диапазоне от 1,7 до 2,3 эквивалента и, в частности, в количестве приблизительно 2,0 эквивалента на один эквивалент соединения химической формулы 2. В этом случае взаимодействие можно осуществлять в диапазоне температур от -80°С до от -10°С, более предпочтительно от -70°С до -60°С в течение от 1 до 12 часов или от 1 до 3 часов. Кроме того, в качестве реакционного растворителя можно применять простой растворитель на основе тетрагидрофурана или эфира, смешанный растворитель, состоящий из тетрагидрофурана и толуола (1:1) или тому подобное.
Настоящее изобретение относится к способу получения соединения химической формулы 5, который включает следующее: соединение химической формулы 4 подвергается реакциям снятия защиты и раскрытия кольца в кислотной среде в присутствии воды с получением соединения следующей химической формулы 5:
Химическая формула 5

где n, В и X определены выше.
Соединения химической формулы 4 подвергают реакциям снятия защиты и раскрытия кольца с получением соединения следующей химической формулы 5, что может быть осуществлено в кислотных условиях.
Примеры применяемой в настоящем документе кислоты включают соляную кислоту, серную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, п-толуолсульфоновую кислоту, газообразный хлористый водород и тому подобное, которые можно применять в диапазоне от 2 до 5 эквивалентов, более предпочтительно в количестве 3 эквивалентов на один эквивалент соединения химической формулы 4. В этом случае взаимодействие можно осуществлять в диапазоне температур от 0 до 40°С, более предпочтительно в диапазоне температур от 20 до 30°С в течение от 2 до 24 часов или от 3 до 6 часов.
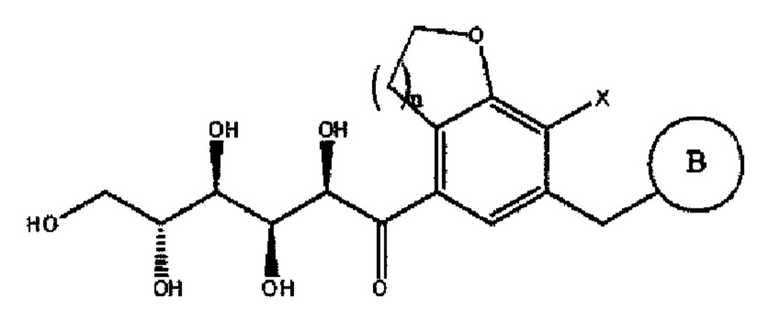
Соединение химической формулы 5 имеет форму с открытой цепью, как и соединение с4 схемы 1, применяемое в предшествующем уровне техники. В процессе получения соединения химической формулы 5 из соединения химической формулы 4, получают продукт взаимодействия, в котором соединение химической формулы 5 и соединение следующей химической формулы 5R находятся в равновесном состоянии вследствие кольчато-цепной таутомерии.
Химическая формула 5R

Поскольку физические свойства соединения химической формулы 5 отличаются от свойств соединения химической формулы 5R, только соединение химической формулы 5 может быть кристаллизовано методом кристаллизации с применением изменений физических свойств двух соединений. В следующих примерах кристаллизацию соединения химической формулы 5 осуществляли, используя разницу в растворимости между соединением химической формулы 5 и соединением химической формулы 5R в растворителе для кристаллизации.
Следовательно, в соответствии с одним из вариантов варианта осуществления настоящего изобретения, способ получения соединения химической формулы 5 может включать: кристаллизацию продукта взаимодействия, который получают, подвергая соединение химической формулы 4 реакциям снятия защиты и раскрытия кольца в кислотных условиях в присутствии воды с получением соединения химической формулы 5.
Кристаллизацию можно осуществлять путем обработки растворителем для кристаллизации, способным растворять соединение химической формулы 5, и перекристаллизацией соединения химической формулы 5.
В качестве растворителя для кристаллизации можно применять толуол, дихлорметан и так далее, однако настоящее изобретение ими не ограничивается. Растворитель для кристаллизации можно применять в количестве в 1-30 раз, предпочтительно в 10-20 раз, больше количества соединения химической формулы 5.
С другой стороны, температура кристаллизации может находиться в диапазоне от 20 до 80°С, предпочтительно от 40 до 50°С, а время кристаллизации может находиться в диапазоне от 6 до 24 часов, предпочтительно от 6 до 12 часов, однако настоящее изобретение этим не ограничивается.
В процессе кристаллизации соединения химической формулы 5 можно удалить большинство родственных веществ, включая основное родственное вещество. Следовательно, в отличие от предшествующего уровня техники, в котором невозможно удалить родственные вещества в промежуточном процессе получения соединения химической формулы 1, которое является конечным целевым материалом, технической целью настоящего изобретения является разработка промежуточного соединения, способного удалять сопутствующие вещества в процессе.
Кроме того, настоящее изобретение относится к способу получения соединения химической формулы 1 с применением соединения химической формулы 5 в качестве промежуточного соединения. В отличие от предшествующего уровня техники, в котором соединение химической формулы 1, являющееся конечным целевым материалом, получают без какой-либо очистки родственных веществ, в соответствии с настоящим изобретением можно ожидать высокий выход и качество, когда соединение химической формулы 5, из которого удалены родственные вещества, применяют в качестве промежуточного продукта для получения соединения химической формулы 1.
В частности, настоящее изобретение относится к способу получения соединения химической формулы 1, который предусматривает:
циклизацию и метоксилирование соединения химической формулы 5 в кислотной среде в присутствии реакционного растворителя с получением соединения химической формулы 6; и
получение соединения химической формулы 1 из соединения химической формулы 6:
Химическая формула 5

Химическая формула 6

Химическая формула 1
где
n равно 1 или 2,
X представляет собой галоген (например, F, Cl, Br или I),
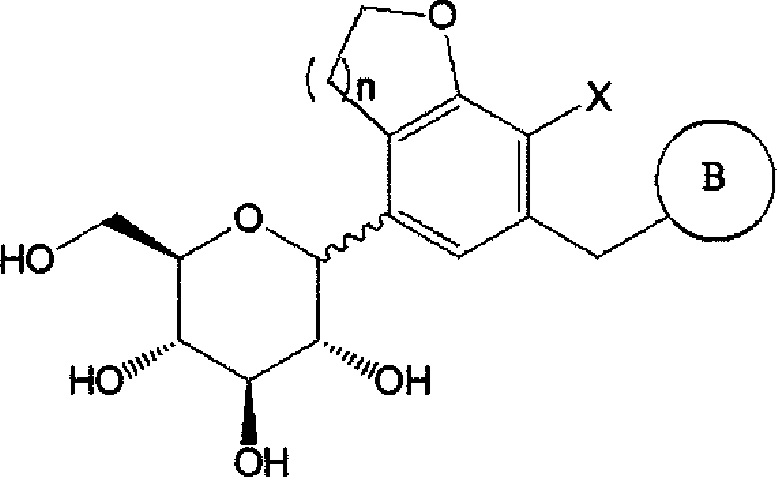
B представляет собой  (B-1) или
(B-1) или 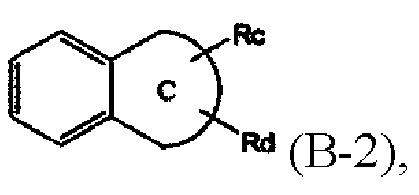
где Ra, Rb, Rc и Rd каждый независимо представляет собой водород, галоген, гидрокси, меркапто, циано, нитро, амино, карбокси, оксо, C1-7 алкил, C1-7 алкилтио, C2-7 алкенил, C2-7 алкинил, C1-7 алкокси, C1-7 алкокси-C1-7 алкил, C2-7 алкенил-C1-7 алкилокси, C2-7 алкинил-C1-7 алкилокси, C3-10 циклоалкил, C3-7 циклоалкилтио, C5-10 циклоалкенил, C3-10 циклоалкилокси, C3-10 циклоалкилокси-C1-7 алкокси, фенил-C1-7 алкил, C1-7 алкилтио-фенил, фенил-C1- 7 алкокси, моно- или ди-C1-7 алкиламино, моно- или ди-C1-7 алкиламино-C1-7 алкил, C1-7 алканоил, C1-7 алканоиламино, C1-7 алкилкарбонил, C1-7 алкоксикарбонил, карбамоил, моно- или ди-C1-7 алкилкарбамоил, C1-7 алкилсульфониламино, фенилсульфониламино, C1-7 алкилсульфинил, C6-14 арилсульфанил, C6-14 арилсульфонил, C6-14 арил, 5-13-членный гетероарил, 5-10- членный гетероциклоалкил, 5-10-членный гетероциклоалкил-C1-7-алкил или 5-10- членный гетероциклоалкил-C1-7 алкокси;
кольцо С представляет собой С3-10 циклоалкил, С5-10 циклоалкенил, С6-14 арил, 5-13-членный гетероарил или 5-10-членный гетероциклоалкил;
алкил, алкенил, алкинил и алкокси, каждый независимо, незамещены или имеют один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, С1-7 алкила и С2-7 алкинил;
циклоалкил, циклоалкенил, арил, гетероарил и гетероциклоалкил, каждый независимо, незамещены или имеют один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, С1-4 алкила и С1-4 алкокси; и
гетероарил и гетероциклоалкил, каждый независимо, содержат один или несколько гетероатомов, выбранных из группы, состоящей из N, S и О.
Соединение химической формулы 5 может быть подвергнуто циклизации и метоксилированию с получением соединения химической формулы 6. В этом случае процесс получения соединения химической формулы 1 из соединения химической формулы 6 также подробно описан в выложенной для всеобщего ознакомления публикации корейской патентной заявке №2017-0142904.
Циклизацию и метоксилирование соединения химической формулы 5 с получением соединения химической формулы 6 можно осуществлять в кислотных условиях в присутствии реакционного растворителя.
Примеры применяемой в настоящем документе кислоты включают соляную кислоту, серную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, п-толуолсульфоновую кислоту, газообразный хлористый водород и тому подобное, которые можно применять в диапазоне от 2 до 5 эквивалентов, более предпочтительно в количестве 3 эквивалентов на один эквивалент соединения химической формулы 5. В этом случае реакцию можно проводить в диапазоне температур от 0 до 40°С, более предпочтительно в диапазоне температур от 20 до 30°С в течение от 2 до 24 часов или от 3 до 6 часов. Также в качестве реакционного растворителя можно применять метанол.
Способ получения последующего соединения химической формулы 1 из соединения химической формулы 6 осуществляют, например, следующим образом, однако настоящее изобретение этим не ограничивается.
Согласно одному приведенному в качестве примера варианту осуществления настоящего изобретения получение соединения химической формулы 1 из соединения химической формулы 6 может включать:
восстановление соединения химической формулы 6 с получением соединения следующей химической формулы 7; и
введение защитной группы в соединение химической формулы 7 и перекристаллизацию, а также снятие защиты с соединения с введенной защитной группой с получением соединения химической формулы 1:
Химическая формула 7

где n, В и X определены выше.
При восстановлении соединения химической формулы 6 с получением соединения химической формулы 7 реакцию восстановления можно проводить с применением восстанавливающего агента и кислоты. Примеры восстанавливающего агента, который можно применять в настоящем документе, включают триэтилсилан, триизопропилсилан, трет-бутилдиметилсилан, боргидрид натрия и тому подобное, а примеры кислоты, которую можно применять в настоящем документе, включают диэтиловый эфир трифторида бора, триметилсилилтрифторметансульфонат, хлорид алюминия, трифторуксусную кислоту, трифторметансульфокислоту и тому подобное. Восстановитель можно применять в количестве от 2 до 5 эквивалентов, более предпочтительно, приблизительно в количестве 3 эквивалентов, а кислоту можно применять в количестве от 1,5 до 3 эквивалентов, более предпочтительно, приблизительно в количестве 2 эквивалентов. В этом случае взаимодействие можно осуществлять в диапазоне температур от -50°С до 0°С, более предпочтительно в диапазоне температур от -20°С до -10°С в течение от 2 до 12 часов или от 2 до 5 часов. Кроме того, в качестве реакционного растворителя может быть применяться простой растворитель на основе дихлорметана, 1,2-дихлорэтана, ацетонитрила или тому подобного, смешанный растворитель, состоящий из дихлорметана и ацетонитрила (1:1) или 1,2-дихлорэтана и ацетонитрила (1:1) или тому подобного.
Введение защитной группы в соединение химической формулы 7 и перекристаллизацию, а также снятие защиты с соединения с введенной защитной группой можно осуществить путем введения защитной группы в соединение химической формулы 7 и последующего нагревания соединения с введенной защитной группой в растворителе для кристаллизации, таком как спирт, этилацетат или дихлорметан, с отделением полученного осадка и снятия защиты с осадка.
Согласно еще одному приведенному в качестве примера варианту осуществления настоящего изобретения получение соединения химической формулы 1 из соединения химической формулы 6 может быть осуществлено посредством стадий, включающих:
восстановление соединения химической формулы 6 с получением соединения следующей химической формулы 7;
введение защитной группы в соединение химической формулы 7 и перекристаллизацию соединения с введенной защитной группой с выделением соединения химической формулы 8; и
снятие защиты с соединения химической формулы 8 с получением соединения химической формулы 1:
Химическая формула 8

где PG представляет собой защитную группу; и n, В и X являются такими, как определено выше.
Осуществляется введение защитной группы в соединение химической формулы 7 с отделением и снятием защиты только с соединения (β-формы) химической формулы 8. В этом случае реакцию можно осуществлять с применением ацетилирующего агента и основания. Примеры ацетилирующего агента включают ацетилхлорид, ацетилбромид, безводную уксусную кислоту и тому подобное, а примеры основания включают гидроксид натрия, карбонат натрия, триэтиламин, диизопропилэтиламин, пиридин, лутидин, 4-диметиламинопиридин и тому подобное. Ацетилирующий агент можно применять в количестве от 4 до 12 эквивалентов, более предпочтительно приблизительно в количестве 8 эквивалентов, а основание можно применять в количестве от 1 до 4 эквивалентов, более предпочтительно приблизительно в количестве 1,5 эквивалента. В этом случае взаимодействие можно проводить в диапазоне температур от 0 до 50°С, более предпочтительно в диапазоне температур от 20 до 30°С в течение от 1 до 12 часов или от 1 до 3 часов. Также в качестве реакционного растворителя можно применять ацетон, этилацетат, тетрагидрофуран, диметилформамид, диметилацетамид, дихлорметан, 1,2-дихлорэтан, хлороформ и тому подобное. Затем осуществляется реакция снятия защиты. В этом случае реагент, такой как гидроксид лития, гидроксид натрия, гидроксид калия, метоксид натрия, этоксид натрия и тому подобное, можно применять в диапазоне от 2 до 12 эквивалентов, более предпочтительно приблизительно в количестве 5 эквивалентов. В этом случае реакцию можно проводить в диапазоне температур от 0 до 50°С, более предпочтительно в диапазоне температур от 20 до 30°С в течение от 1 до 12 часов или от 1 до 3 часов. В качестве реакционного растворителя могут применяться смеси метанол/вода (1:1 - 3:1), дихлорметан/метанол (от 1:1 до 1:2), дихлорметан/этанол (1:1 - 1:2), тетрагидрофуран/метанол (1:1 - 1:2), тетрагидрофуран/этанол (1:1 - 1:2), тетрагидрофуран/метанол/вода (1:1:3 - 2:1:3), тетрагидрофуран/этанол/вода (1:1:3 - 2:1:3) и тому подобное.
Соединение химической формулы 1 по настоящему изобретению может быть получено в кристаллической форме, или аморфной форме, или в виде смеси этих форм, при этом соединение, имеющее кристаллическую форму, может быть предпочтительным в том смысле, что оно обладает физико-химическими свойствами, которые облегчают получение лекарственной формы, поскольку обладает превосходными свойствами с точки зрения стабильности и негигроскопичности.
Соответственно, способ получения соединения химической формулы 1 по настоящему изобретению может предусматривать: кристаллизацию продукта реакции с применением различных растворителей после того, как соединение химической формулы 2 вступит во взаимодействие с соединением химической формулы 3, с последующим снятием защиты и восстановлением. В этом случае возможно образование различных кристаллических форм. Соединения в различных кристаллических формах и способы их получения подробно описаны в выложенной для всеобщего ознакомления публикации корейского патента №2017-0142904.
В качестве одного примера, кристаллизация может быть осуществлена с применением растворителя. В этом случае растворитель, применяемый для кристаллизации, может быть выбран из группы, состоящей из толуола; этилацетата; дихлорметана; ацетона; ацетонитрила; 2-пропанола, тетрагидрофурана; н-гексана и их смеси (например, смеси тетрагидрофурана и дихлорметана или смесь тетрагидрофурана и н-гексана).
В качестве другого примера, растворитель, применяемый для кристаллизации, может быть выбран из смеси метанола и дистиллированной воды; смеси метанола и н-гексана; и смеси метанола, дихлорметана и н-гексана.
В качестве еще одного примера, растворитель, применяемый для кристаллизации, может быть выбран из смеси этанола, дистиллированной воды и н-гексана; и смеси тетрагидрофурана и толуола.
В качестве еще одного примера, растворитель, применяемый для кристаллизации, может представлять собой смесь этанола и н-гексана.
В качестве одного предпочтительного примера, растворитель, применяемый для кристаллизации, может быть выбран из группы, состоящей из толуола, этилацетата, дихлорметана, смеси тетрагидрофурана и дихлорметана и смеси тетрагидрофурана и н-гексана.
Один приведенный в качестве примера вариант осуществления соединения химической формулы 5, который представляет собой новое промежуточное соединение по настоящему изобретению, представляет собой соединение следующей химической формулы А.
Химическая формула А

Название соединения химической формулы А: [(2R,3S,4R,5R)-1-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-2,3,4,5,6-пентагидроксигексан-1-он].
Настоящее изобретение также относится к соединению химической формулы А и его кристаллической форме.
В следующем примере будут подробно описаны способ получения кристаллической формы [(2R,3S,4R,5R)-1-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-2,3,4,5,6-пентагидроксигексан-1-он), а также характеристики кристаллической формы.
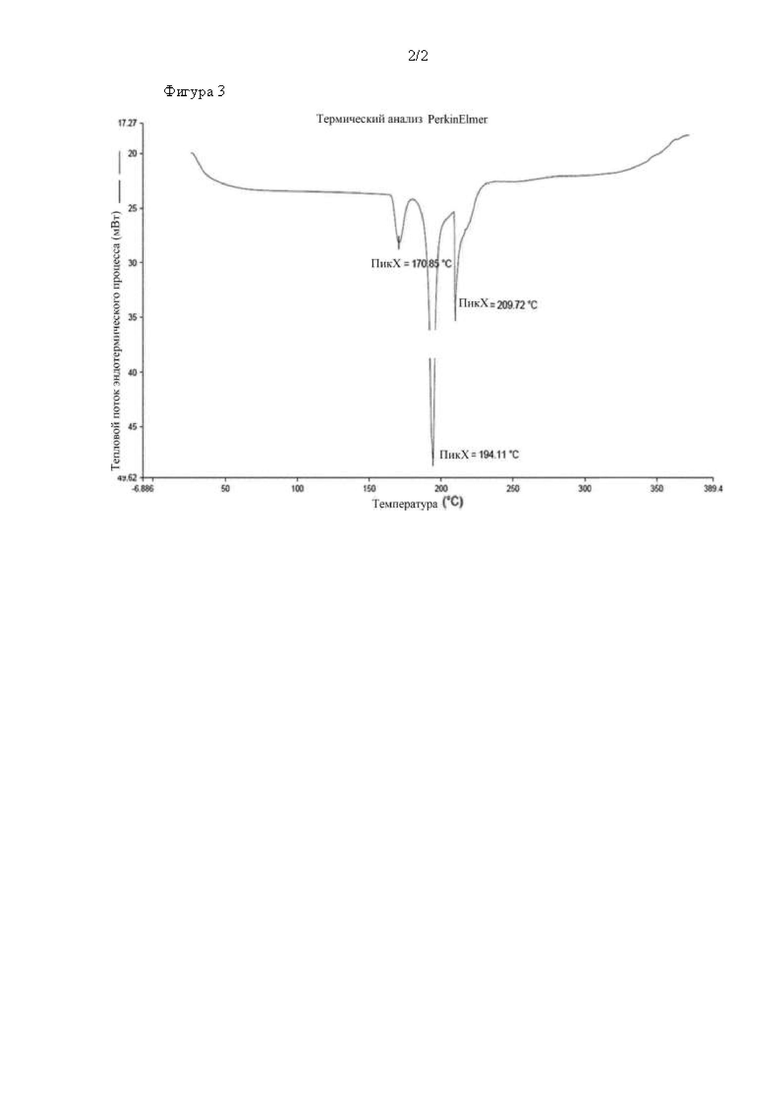
Настоящее изобретение относится к кристаллической форме соединения химической формулы А, которая характеризуется порошковой дифракционной рентгенограммой, содержащей 6 или более дифракционных пиков при значении углового положения 2[θ], выбранном из 7,8±0,2, 8,9±0,2,15,1±0,2, 16,6±0,2, 17,9±0,2, 19,4±0,2, 20,2±0,2, 21,1±0,2, 22,5±0,2, 22,9±0,2, 24,5±0,2, 26,0±0,2,28,7±0,2.
Кристаллическая форма соединения химической формулы А может быть кристаллической формой, характеризуемой порошковой рентгеновской дифрактограммой, содержащей дифракционные пики при значении углового положения 2[θ], выбранном из 7,8±0,2, 8,9±0,2, 15,1±0,2, 16,6±0,2, 17,9.±0,2 и 19,4±0,2.
Кроме того, кристаллическая форма соединения химической формулы А может быть кристаллической формой, характеризующейся кривой дифференциальной сканирующей калориметрии, определенной при скорости нагревания 1°С в минуту, которая показывает максимальный эндотермический пик при температуре от 190°С до 200°С.
Положительные эффекты изобретения
В соответствии с настоящим изобретением, путем разработки соединения химической формулы 5, соответствующего новому промежуточному соединению, проблема очистки может быть решена посредством известных процессов, требования к качеству родственных веществ могут быть удовлетворены только за одну стадию очистки, а проблема контроля качества на каждой стадии может быть решена путем осуществления нескольких стадий in situ. Способ синтеза соединения химической формулы 1 с применением соединения химической формулы 5 по настоящему изобретению обеспечивает очистку на стадии синтеза соединения химической формулы 5, тем самым решая проблемы существующих способов синтеза, в которых требования к качеству родственных веществ было трудно контролировать от стадии к стадии в непрерывном процессе, а также минимизировать количество родственных веществ в конечном продукте. Кроме того, по мере увеличения количества стадий очистки, способ может быть упрощен, поскольку очистку не обязательно осуществлять два или более раз на одной стадии, что описано в предшествующем уровне техники, тем самым повышая до максимума выход производного дифенилметана в соответствии с химической формулой 1.
Описание фигур
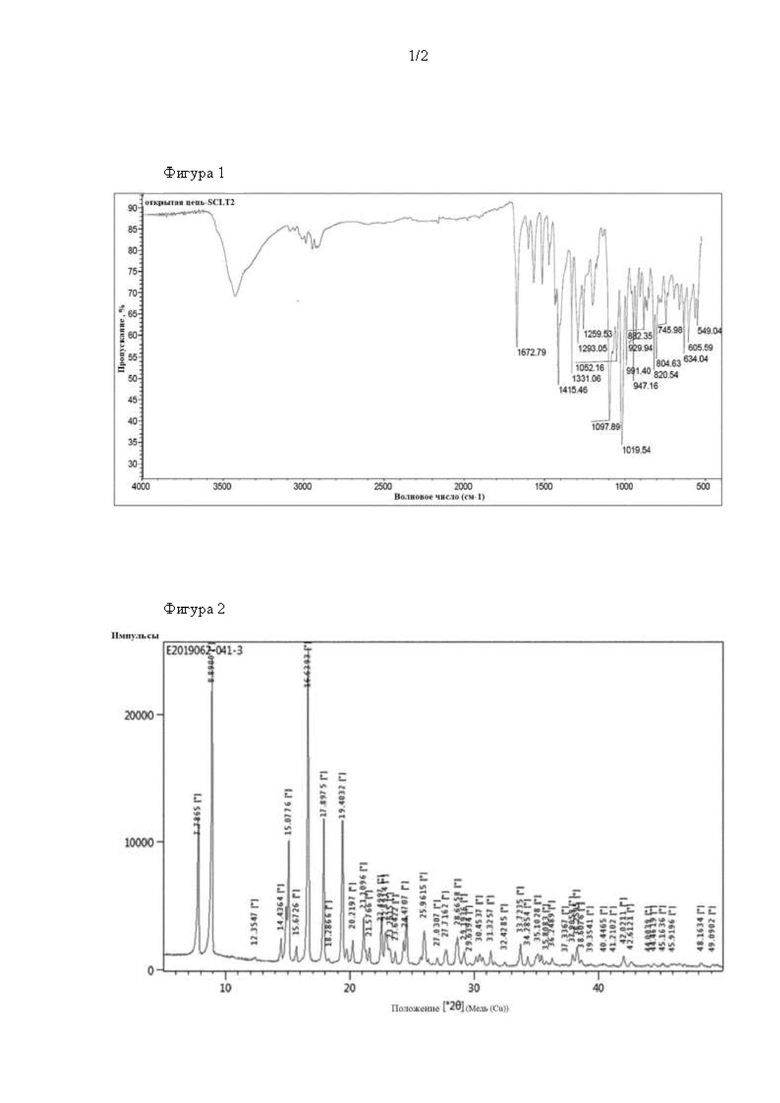
На фиг. 1 показаны результаты измерений методом инфракрасной спектроскопии, показывающие, что кристалл, полученный на стадии 1 примера 1, представляет собой соединение 4.
На фиг. 2 показаны результаты анализа соединения химической формулы А с применением рентгеновской порошковой дифрактометрии.
На фиг. 3 показана дифференциальная сканирующая калориметрия (DSC) определенного соединения химической формулы А с применением дифференциального сканирующего калориметра.
Лучший вариант осуществления изобретения
Пример 1: Получение целевого соединения
Схема 2

Стадия 1: (2R,3S,4R,5R)-1-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-2,3,4,5,6-пентагидроксигексан-1-он (соединение 4)

Соединение 1 (10,0 г, 1,0 экв.) и соединение 2 (24,4 г, 1,9 экв.) добавляли к безводному ТГФ (80 мл) при комнатной температуре в атмосфере азота, растворяли и затем охлаждали до -78°С. К раствору, в котором были растворены соединения 1 и 2, медленно по каплям добавляли 2,5 М раствор н-BuLi (23 мл, 2,1 экв.), в течение 20 минут при температуре -60°С или ниже. После завершения добавления по каплям полученный реакционный раствор перемешивали в течение 5 минут. После этого к реакционному раствору добавляли раствор, полученный добавлением с-HCl (10,2 мл, 4,2 экв.) к воде (100 мл). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 ч. После того как завершение реакции подтверждали посредством тонкослойной хроматографии (ТСХ), к реакционному раствору (рН 6-8) добавляли насыщенный раствор NaHCO3, чтобы остановить реакцию, и реакционный раствор дважды экстрагировали толуолом (30 мл). Органический слой дважды экстрагировали водой (30 мл), к полученному органическому слою дополнительно добавляли толуол (100 мл) и кристаллизовали при перемешивании при температуре от 40 до 50°С в течение 12 часов. Полученные кристаллы отфильтровывали и сушили с получением соединения 4 (11,1 г, 87,4%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, ДМСО): δ 7,02-7,06 (м, 3Н), 6,92-6,94 (м, 2Н), 6,27 (д, 1Н), 4,84 (д, 1Н), 4,66 (д, 1Н), 4,47-4,52 (м, 3Н), 4,36 (м, 1Н), 3,90-3,98 (м, 2Н), 3,62-3,65 (м, 2Н), 3,50-3,56 (м, 3Н), 3,32-3,35 (м, 1H), 3,22-3,27 (м, 1Н), 3,08-3,11 (м, 1Н), 1,82 (м, 1Н), 0,86-0,88 (м, 2Н), 0,57-0,59 (м, 2Н); ЖХ-МС: [М-Н] - 461, т.п. 195°С.
Стадия 2: (3R,4S,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триол (соединение 5)

Соединение 4 (5,0 г, 1,0 экв.) добавляли к МеОН (40 мл), растворяли и затем охлаждали до 0°С. Потом добавляли c-HCl (0,5 мл, 0,5 экв.) и далее перемешивали при комнатной температуре в течение 3 часов. После того, как завершение реакции было подтверждено посредством ТСХ, к реакционному раствору добавляли 3% раствор NaHCO3 для прекращения реакции, реакционный раствор концентрировали в вакууме с удалением МеОН и экстрагировали этилацетатом. Органический слой, полученный путем экстракции, сушили над безводным сульфатом магния, фильтровали, а затем конденсировали в вакууме с получением соединения 5 (5,2 г, 100%). Продукт непосредственно применяли на следующей стадии без какой-либо очистки.
Стадия 3: (3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол (соединение 6)
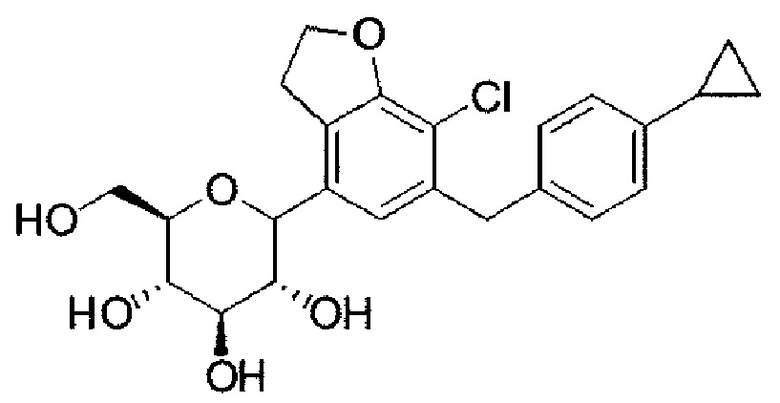
К раствору (3R,4S,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола (соединение 5) (5,2 г, 1,0 экв.) в смеси дихлорметана (50 мл) и ацетонитрила (50 мл) последовательно добавляли Et3SiH (4,0 мл, 3,0 экв.) и BF3OEt2 (5,2 мл, 3,0 экв.) при -50°С. Реакционную смесь перемешивали при температуре от -50 до -10°С в течение 2 ч и перемешивали при температуре от -10 до 0°С в течение 3 ч. После подтверждения завершения реакции посредством ТСХ к реакционному раствору добавляли насыщенный водный раствор NaHCO3 (100 мл) для прекращения реакции, и реакционный раствор экстрагировали этилацетатом. Органический слой, полученный путем экстракции, сушили над безводным сульфатом магния, фильтровали, а затем конденсировали в вакууме с получением соединения 6 (4,9 г, 100%). Продукт непосредственно применяли на следующей стадии без какой-либо очистки.
Стадия 4: (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2Н-пиран-3,4,5-триилтриацетат (соединение 7)

Соединение 6 (4,9 г, 1,0 экв.) добавляли к дихлорметану (75 мл) и растворяли в нем. Затем добавляли 4-диметиламинопиридин (DMAP) (1,6 г, 1,2 экв.) и уксусный ангидрид (8,3 мл, 8,0 экв.) при комнатной температуре в атмосфере азота и полученную смесь перемешивали в течение 2 часов. Завершение реакции подтверждали посредством ТСХ, для прекращения реакции добавляли 1н HCl (50 мл) и реакционный раствор экстрагировали дихлорметаном. Органический слой, полученный путем экстракции, сушили безводным сульфатом магния, фильтровали и к нему добавляли метанол (10 мл). Затем полученный реакционный раствор конденсировали в вакууме. Конденсированный остаток добавляли к метанолу (50 мл) и кристаллизовали при перемешивании в течение часа. Полученные кристаллы отфильтровывали и сушили с получением соединения 7 (4,5 г, 67,2%) в виде твердого вещества белого цвета.
1Н ЯМР (500 МГц, CDCl3): δ 1Н ЯМР (400 МГц, CDCl3) δ 7,04-7,02 (м, 2Н), 6,98-6,95 (м, 2Н), 6,53 (с, 2Н), 5,29-5,24 (м, 1Н), 5,18-5,12 (м, 2Н), 4,71-4,65 (м, 2Н), 4,31-4,26 (м, 1Н), 4,25-4,22 (м, 1Н), 4,15-4,11 (м, 1Н), 4,15-4,11 (м, 1Н), 4,05-3,91 (м, 2Н), 3,79-3,74 (м, 1Н), 3,40-3,35 (м, 2Н), 2,60 (с, 3Н), 2,05 (с, 3Н), 1,99 (с, 3Н), 1,88-1,81 (м, 1Н), 1,66 (с, 3Н), 0,94-0,89 (м, 2Н), 0,66-0,61 (м, 2Н); [M+Na]+ 637.
Стадия 5: (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол (соединение 8)

К соединению 7 (3,5 г, 1,0 экв.) добавляли ТГФ (17,5 мл) и метанол (17,5 мл). К раствору добавляли 4н раствор NaOH (7,1 мл, 5,0 экв.) в виде суспензии при комнатной температуре и полученную смесь перемешивали при температуре от 30 до 35°С в течение 2 ч. Завершение реакции подтверждали посредством ТСХ, реакционный раствор охлаждали до 0°С и значение рН доводили до 6,8 добавлением 1н HCl. Применяемые в реакции ТГФ и МеОН удаляли концентрированием и реакционный раствор экстрагировали этилацетатом. Органический слой, полученный путем экстракции, сушили над безводным сульфатом магния, фильтровали и затем концентрировали в вакууме. Конденсированный остаток добавляли к этилацетату (40 мл), полностью растворяли при 70°С, охлаждали до 33°С и затем перемешивали при температуре 33°С в течение одного часа. Затем по каплям добавляли IPE (65 мл) в течение 30 минут, полученную смесь охлаждали до 0°С, перемешивали при 0°С в течение часа и затем выдерживали в течение одного часа. Полученные кристаллы отфильтровывали и сушили с получением соединения 8 (2,4 г, 94,5%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ 7,02 (д, J=8,0 Гц, 2Н), 6,92 (д, J=8,0 Гц, 2Н), 6,81 (с, 1H), 4,59 (t, J=8,8 Гц, 2Н), 4,11 (д, J=9,2 Гц, 1Н), 3,96 (ABq, ΔvAB=19,0 Гц, JAB=15,2 Гц, 2Н), 3,87-3,84 (м, 1H), 3,67-3,63 (м, 1H), 3,47-3,37 (м, 3Н), 3,35-3,33 (м, 3Н), 1,85-1,79 (м, 1H), 0,91-0,86 (м, 2Н), 0,61-0,57 (м, 2Н); [M+Na]+ 469
Экспериментальный пример 1: Подтверждение кристаллизации соединения химической формулы 5
Как описано выше, соединение химической формулы 5, как и соединение с4 на схеме 1, применяемое в предшествующем уровне техники, имеет форму с открытой цепью. В процессе получения соединения химической формулы 5 из соединения химической формулы 4 получали продукт реакции, в котором соединение химической формулы 5 и соединение последующей химической формулы 5R находятся в равновесном состоянии вследствие кольчато-цепной таутомерии.
На стадии 1 примера 1, посредством измерений методом инфракрасной спектроскопии, было подтверждено, что только соединение 4 кристаллизовалось в равновесном состоянии соединения с4 (соответствует соединению химической формулы 5R) и соединения 4 (соответствует соединению химической формулы 5) на схеме 1.
Кристалл, осажденный в результате кристаллизации, исследовали посредством инфракрасного излучения (IR). В результате, как показано на фиг. 1, видно, что осажденный кристалл представлял собой соединение химической формулы 5, имеющее форму с открытой цепью, содержащую карбонильную группу в молекуле, поскольку характерный пик, соответствующий карбонильному пику, отчетливо наблюдался при 1672 см-1.
Экспериментальный пример 2: Получение и анализ кристаллической формы
После соединения, полученного способом по настоящему изобретению, в частности, неочищенный (2R,3S,4R,5R)-1-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-2,3,4,5,6-пентагидроксигексан-1-он (соединение 4) получали в соответствии со стадией 1 примера 1, и кристаллы получали путем кристаллизации с применением различных растворителей и затем анализировали.
Спектр рентгенодифракционного анализа (XRD) получали при облучении кристаллов Cu-Kα-излучением (длина волны (λ)=1,54056 Å) стандартным методом с применением рентгенодифракционного анализатора для определения порошковой рентгеновской дифракции. Осуществляли дифференциальную сканирующую калориметрию (DSC) с применением дифференциального сканирующего калориметра при скорости +1°С/мин.
(1) Получение кристалла с применением растворителя на основе толуола
Кристаллизацию с применением толуола осуществляли так, как описано в конце процедуры стадии 1 в примере 1. В частности, соединение 4 в растворенном состоянии дополнительно добавляли к толуолу (10-кратная масса соединения 4) и смесь кристаллизовали путем нагревания при температуре 40-50°С в течение 12 часов. Полученный кристалл отфильтровали, промыли толуолом (2-кратный объем фильтрата), а затем сушили в вакуумной печи (50°С, 12 часов) с получением кристалла белого цвета (выход: 87,4%).
XRD-спектр полученного кристалла показывает кристаллическую форму (кристаллическая форма А), представленную на фиг. 2, а углы дифракции (26), межплоскостные расстояния (d) и относительные интенсивности (I/Io х 100) характеристических пиков приведены в таблице 1 ниже.

Как показано на фиг. 3, можно увидеть, что на DSC-спектре наблюдаются эндотермические пики плавления соответствующих кристаллов.
(2) Получение кристалла с применением растворителя на основе дихлорметана.
Неочищенное соединение 4 дополнительно добавляли к дихлорметану (10-кратная масса соединения 4) и смесь кристаллизовали нагреванием при температуре от 40 до 50°С в течение 12 часов. Полученные кристаллы отфильтровывали, промывали дихлорметаном (2-кратный объем фильтрата), а затем сушили в вакуумной печи (50°С, 12 часов) с получением кристаллов белого цвета (выход: 88,1%).
Результаты спектрального XRD-анализа полученных кристаллов показывают, что кристалл белого цвета имел ту же кристаллическую форму (кристаллическая форма А), что и (1) экспериментального примера 2.
Экспериментальный пример 3: Анализ содержания родственных веществ
Способ получения производного дифенилметана, раскрытый в выложенной для всеобщего ознакомления публикации корейского патента №2017-0142904, представляет собой четырехстадийный непрерывный процесс, состоящий из важных реакционных стадий с3-с7, показанных на схеме 1 выше, и очистка родственных веществ осуществляется на стадии с7.
Чистоту и содержание примесей с7, полученного с 1-ой по 3-ю очистку на стадии с 7, как показано на схеме 1 в выложенной для всеобщего ознакомления публикации корейского патента №2017-0142904, сравнивали с чистотой и содержанием примесей с7 (соответствует соединению 7 на схеме 2), полученного путем однократной очистки на стадии с4 (соответствует соединению 4 на схеме 2) и другой однократной очистки на стадии с7 (соответствует соединению 7 на схеме 2) на схеме 2 настоящего изобретения. Результаты представлены в таблице 2 ниже.

Как показано в таблице 2, процесс очистки, в соответствии со способом получения по настоящему изобретению, можно проводить во время синтеза соединения химической формулы 5. Таким образом, даже без осуществления трехкратного процесса очистки на одной стадии, как описано в выложенной для всеобщего ознакомления публикации корейского патента №2017-0142904, количество родственных веществ в конечном продукте может быть сведено к минимуму.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ SGLT2 | 2009 |
|
RU2530494C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ КОМПЛЕКСОВ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SGLT2, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2641905C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗИЛБЕНЗОЛЬНЫХ ИНГИБИТОРОВ SGLT2 | 2013 |
|
RU2625795C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ СТЕРИЧЕСКИХ СОЕДИНЕНИЙ | 2007 |
|
RU2481326C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАК ИНГИБИТОРЫ КАТЕПСИНА S | 2005 |
|
RU2424239C2 |
| АЛЬТЕРНАТИВНЫЕ СПОСОБЫ СИНТЕЗА ИНГИБИТОРОВ РЕНИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2411230C2 |
| ПИРАЗОЛОПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2009 |
|
RU2538041C2 |
Изобретение относится к промежуточному соединению химической формулы 5, где n равно 1, X представляет собой галоген, В представляет собой 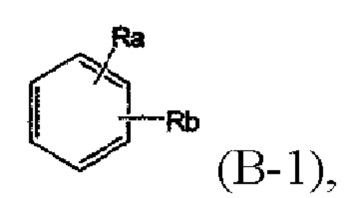 где Ra и Rb, каждый независимо, представляют собой водород или С3-6 циклоалкил. Изобретение также относится к способу получения соединения химической формулы 5 путем снятия защиты и раскрытия кольца в кислотной среде в присутствии воды с соединения химической формулы 4. Изобретение также относится к способу получения соединения химической формулы 1, который предусматривает циклизацию и метоксилирование соединения химической формулы 5 в кислотной среде в присутствии реакционного растворителя с получением соединения химической формулы 6; и получение соединения химической формулы 1 из соединения химической формулы 6. Технически результат – разработан способ получения соединения химической формулы 1, которое является ингибитором SGLT, с использованием нового промежуточного соединения химической формулы 5 с высоким выходом. 4 н. и 6 з.п. ф-лы, 3 ил., 2 табл., 1 пр.
где Ra и Rb, каждый независимо, представляют собой водород или С3-6 циклоалкил. Изобретение также относится к способу получения соединения химической формулы 5 путем снятия защиты и раскрытия кольца в кислотной среде в присутствии воды с соединения химической формулы 4. Изобретение также относится к способу получения соединения химической формулы 1, который предусматривает циклизацию и метоксилирование соединения химической формулы 5 в кислотной среде в присутствии реакционного растворителя с получением соединения химической формулы 6; и получение соединения химической формулы 1 из соединения химической формулы 6. Технически результат – разработан способ получения соединения химической формулы 1, которое является ингибитором SGLT, с использованием нового промежуточного соединения химической формулы 5 с высоким выходом. 4 н. и 6 з.п. ф-лы, 3 ил., 2 табл., 1 пр.
Химическая формула 5
Химическая формула 4

Химическая формула 6

Химическая формула 1
1. Соединение следующей химической формулы 5:
Химическая формула 5
где
n равно 1,
X представляет собой галоген,
В представляет собой 
где Ra и Rb, каждый независимо, представляют собой водород или С3-6 циклоалкил.
2. Способ получения соединения следующей химической формулы 5, который включает следующее: соединение следующей химической формулы 4 подвергается реакциям снятия защиты и раскрытия кольца в кислотной среде в присутствии воды с получением соединения химической формулы 5:
Химическая формула 4
Химическая формула 5

где
n равно 1,
Х представляет собой галоген,
PG представляет собой защитную группу,
В представляет собой 
где Ra и Rb, каждый независимо, представляют собой водород или С3-6 циклоалкил.
3. Способ по п. 2, дополнительно включающий кристаллизацию продукта реакции, который получают, подвергая соединение химической формулы 4 реакциям снятия защиты и раскрытия кольца в кислотной среде в присутствии воды с получением соединения химической формулы 5.
4. Способ по п. 3, причем кристаллизацию проводят путем обработки растворителем для кристаллизации, способным растворять соединение химической формулы 5, и перекристаллизацией соединения химической формулы 5.
5. Способ получения соединения химической формулы 1, предусматривающий:
циклизацию и метоксилирование соединения химической формулы 5 в кислотной среде в присутствии реакционного растворителя с получением соединения химической формулы 6; и
получение соединения химической формулы 1 из соединения химической формулы 6:
Химическая формула 5

Химическая формула 6

Химическая формула 1

где
n равно 1,
X представляет собой галоген,
В представляет собой 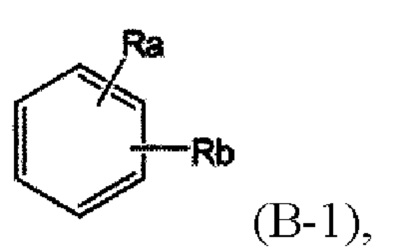
где Ra и Rb, каждый независимо, представляет собой водород или С3-6 циклоалкил.
6. Способ по п. 5, причем получение соединения химической формулы 1 из соединения химической формулы 6 предусматривает:
восстановление соединения химической формулы 6 с получением соединения следующей химической формулы 7; и
введение защитной группы в соединение химической формулы 7, а также перекристаллизацию и снятие защиты с соединения с введенной защитной группой с получением соединения химической формулы 1:
Химическая формула 6
Химическая формула 7

Химическая формула 1

где n, В и X являются такими, как определено в п. 5.
7. Способ по п. 5, причем получение соединения химической формулы 1 из соединения химической формулы 6 осуществляют посредством следующих стадий:
восстановление соединения химической формулы 6 с получением соединения следующей химической формулы 7;
введение защитной группы в соединение химической формулы 7 и перекристаллизацию соединения с введенной защитной группой с выделением соединения химической формулы 8; и
снятие защиты с соединения химической формулы 8 с получением соединения химической формулы 1, где соединение химической формулы 1 представляет собой соединение химической формулы 1а:
Химическая формула 6

Химическая формула 7
Химическая формула 8

Химическая формула 1

Химическая формула 1а

где
PG представляет собой защитную группу; и
n, В и X являются такими, как определено в п. 5.
8. Кристаллическая форма соединения химической формулы А, которая характеризуется порошковой дифракционной рентгенограммой, содержащей 6 или более дифракционных пиков при значении углового положения 2[θ], выбранном из 7,8±0,2, 8,9±0,2, 15,1±0,2, 16,6±0,2, 17,9±0,2, 19,4±0,2, 20,2±0,2, 21,1±0,2, 22,5±0,2, 22,9±0,2, 24,5±0,2, 26,0±0,2 и 28,7±0,2:
Химическая формула А

9. Кристаллическая форма по п. 8, причем порошковая дифракционная рентгенограмма имеет дифракционные пики при значении углового положения 2[θ], выбранном из 7,8±0,2, 8,9±0,2, 15,1±0,2, 16,6±0,2, 17,9±0,2 и 19,4±0,2.
10. Кристаллическая форма по п. 8, причем кристаллическая форма соединения химической формулы А характеризуется кривой дифференциальной сканирующей калориметрии, определенной при скорости нагревания 1°С в минуту, которая показывает максимальный эндотермический пик при температуре от 190°С до 200°С.
| WO 2016098016 A1, 23.06.2016 | |||
| US 9340521 B2, 17.05.2016 | |||
| CN 108530408 A, 14.09.2018 | |||
| WO 2012165914 A2, 06.12.2012 | |||
| RU 2018127488 А, 28.01.2020 | |||
| Устройство для автоматического поворота лопастей пропеллера | 1930 |
|
SU23781A1 |
| RU 2016144355 А, 19.06.2018. | |||
