Перекрестная ссылка на родственные заявки
Для настоящего изобретения испрашивается приоритет по заявке PCT/CN 2012/073697, поданной 10 апреля 2012, содержание которой включено в настоящую заявку во всех возможных аспектах.
Предшествующий уровень техники
Натрий-зависимые (''активные'') переносчики глюкозы (SGLT), включая SGLT1 (находится главным образом в щеточной каемке кишечника) и SGLT2 (локализован в почечных проксимальных канальцах) активно изучались в уровне техники. В частности, было обнаружено, что SGLT2 ответственен за основной объем обратного захвата глюкозы в почках. Подавление почечного SGLT в настоящее время рассматривается как многообещающий подход к лечению гипергликемии посредством увеличения количества глюкозы, выводимой с мочой (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999). Потенциал такого терапевтического подхода дополнительно подтверждается недавно полученными результатами, говорящими о том, что мутации гена SGLT2 наблюдаются в случаях наследственной почечной глюкозурии, несомненно доброкачественного симптома, характеризуемого выделением глюкозы с мочой в сочетании с нормальными уровнями глюкозы в крови и отсутствием общего нарушения работы почек или других заболеваний (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Поэтому соединения, подавляющие SGLT, в частности SGLT2, являются многообещающими кандидатами для применения в качестве противодиабетических лекарственных средств (см. обзор Washburn WN, Expert Opin Ther Patents 19:1485-99, 2009). Кроме того, поскольку раковые клетки демонстрируют повышенное поглощение глюкозы в сравнении с их нормальными аналогами, подавление SGLT было предложено в качестве метода лечения рака посредством недостаточного питания раковых клеток. Например, в некоторых исследованиях выдвигалась гипотеза участия SGLT2 в обратном захвате глюкозы в метастазах рака легких (Ishikawa N, et al., Jpn J Cancer Res 92:874-9, 2001). Таким образом, ингибиторы SGLT2 могут также найти применение в качестве противораковых средств.
Помимо фармацевтической активности как таковой дополнительными факторами для успешной разработки лекарственного средства являются параметры, связанные с физической природой самого действующего вещества. Некоторыми такими параметрами являются устойчивость действующего вещества в различных условиях, устойчивость действующего вещества при производстве фармацевтического препарата и устойчивость действующего вещества в финальной фармацевтической композиции. Для обеспечения требуемой устойчивости фармацевтически активное вещество, используемое в лекарственном средстве, должно обладать максимально возможной степенью чистоты, обеспечивающей устойчивость соединения при длительном хранении в разных условиях.
Соединения, полученные согласно настоящему изобретению, получались ранее по методикам, описанным в WO 2001/027128, US 2004/0230045, US 2005/0124555, US 2006/0122126, US 2007/0238866, US 2007/0275907, US 2008/0242596, US 2008/0132563, US 2008/0318874, WO 2008/034859, US 2009/0030006, US 2009/0030198, US 2009/0118201, US 2009/0156516, US 2010/0056618, US 2010/0063141 и WO 2010/147430. Целью настоящего изобретения является разработка улучшенных способов получения таких соединений.
Краткое описание изобретения
В одном варианте осуществления в настоящем изобретении описан способ получения соединения формулы I:
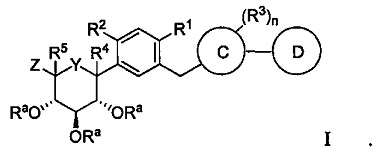
Способ получения соединения формулы I включает получение первой реакционной смеси, содержащей соединение формулы II:
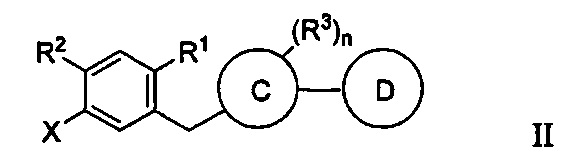
Первая реакционная смесь также включает алкил-магниевый комплекс, такой как C1-C4 алкилмагния хлорид, C1-C4 алкилмагния бромид, ди(C1-C4 алкил)магний, C3-C7 циклоалкилмагния хлорид, C3-C7 циклоалкилмагния бромид или ди(C3-C7 циклоалкил)магний, и первый органический растворитель, где соотношение алкил-магниевого комплекса к соединению формулы II равно 1,0 (моль/моль) или меньше, и где первая реакционная смесь находится при температуре ниже примерно -50°C, что приводит к получению промежуточного соединения.
Данный способ также включает получение второй реакционной смеси из промежуточного соединения, второго органического растворителя и соединения формулы III:
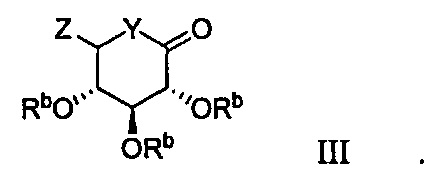
Таким образом получают соединение формулы I.
X в формуле I может представлять собой бром или иод. Y в формуле I может представлять собой CHRc, C(=O), O или S. Z в формуле I может представлять собой CH2ORa, ORa, SRa или S(O)m-Ra.
R1 в формуле I может представлять собой хлор. Каждый R2 и R3 в формуле I может независимо представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, -CH2ORa, C2-C4 алкенил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
По меньшей мере один из R2 и R3 в формуле I может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси или C3-C6 циклоалкил, и по меньшей мере один из R2 и R3 может представлять собой C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
R4 в формуле I может представлять собой Н или OR4a, где R4a может представлять собой Н или C1-C3 алкил. Альтернативно, R2 и R4 объединены с атомами, к которым они присоединены, с образованием 5-6-членного циклоалкила или гетероциклоалкила.
R5 в формуле I может представлять собой Н или -CH2ORa. Альтернативно, R4 и R5 могут быть объединены с атомами, к которым они присоединены, с образованием 5-6-членного гетероциклоалкила.
Каждый Ra в формуле I может независимо представлять собой Н, C1-C3 алкил или Rb. Rb в формуле I может представлять собой защитную группу.
Rc в формуле I может представлять собой Н, ОН или C1-C3 алкокси. Альтернативно, Rc может быть объединен с R4 или R5 с образованием связи.
Цикл C в формуле I может представлять собой арил или гетероарил. Цикл D в формуле I может отсутствовать или представлять собой гетероарил.
Индекс m в формуле I может представлять собой целое число от 1 до 2. Индекс n в формуле I может представлять собой целое число от 1 до 4.
Алкил, алкокси, циклоалкил, алкенилокси, алкинилокси, циклоалкокси, гидроксиалкокси и гетероциклоалкокси группы или их части необязательно могут быть частично или полностью фторированы, и один или больше атомов водорода в соединении формулы I необязательно могут быть заменены на атомы дейтерия.
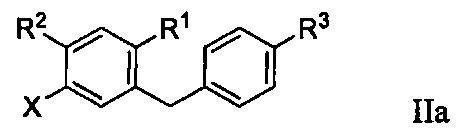
Во втором варианте осуществления в настоящем изобретении описан способ получения соединения формулы IIa:
Способ получения соединения формулы IIa включает получение первой реакционной смеси, содержащей соединение формулы IV:
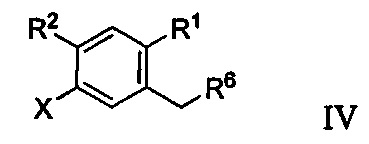
Первая реакционная смесь также включает соединение формулы V:
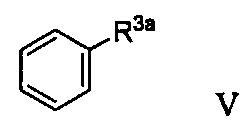
Способ получения соединения формулы IIa осуществляют в условиях, подходящих для получения соединения формулы IIa.
R1 в формуле IIa может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил или C1-C3 алкокси. R2 и R3 в формуле IIa каждый независимо могут представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
R3a в формуле IIa может представлять собой ОН. R6 в формуле IIa может представлять собой ОН или Br. X в формуле IIa может представлять собой бром или иод.
[0001] Алкил, алкокси, циклоалкил, алкенилокси, алкинилокси, циклоалкокси, гидроксиалкокси и гетероциклоалкокси группы или их части в формуле IIa необязательно могут быть частично или полностью фторированы, и один или больше атомов водорода в формуле IIa необязательно могут быть заменены на атомы дейтерия.
В третьем варианте осуществления, в настоящем изобретении описано соединение, имеющее структуру:

В другом варианте осуществления, в настоящем изобретении описана композиция, содержащая соединение формулы Ia, имеющее структуру:
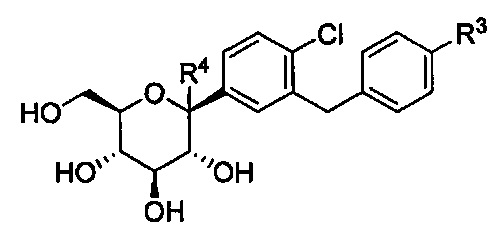
в количестве, составляющем по меньшей мере 95% композиции. Композиция может также включать побочный продукт А, имеющий структуру:
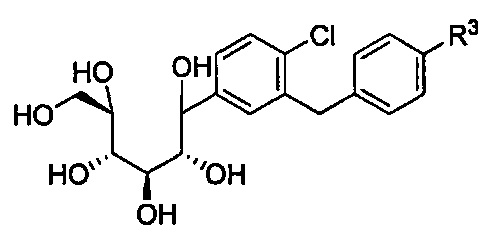
в количестве, составляющем меньше примерно 1% композиции. Композиция может также включать побочный продукт В, имеющий структуру:
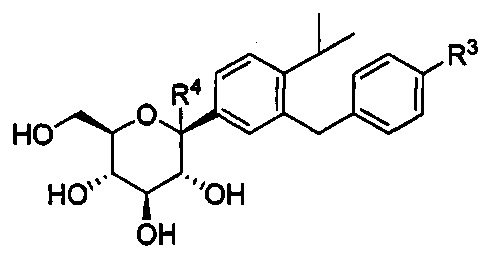
в количестве, составляющем меньше примерно 3% композиции. Данную композицию можно приготовить способами по настоящему изобретению. Соединения в данной композиции представляют собой соединения, в которых R3 может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси или C3-C6 циклоалкил, и по меньшей мере один из R2 и R3 может представлять собой C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси. Кроме того, R4 может представлять собой Н или OR4a, где R4a может представлять собой Н или C1-C3 алкил.
Краткое описание чертежей
На Фиг. 1 изображен общий метод синтеза соединений по настоящему изобретению.
Ошибка! Источник ссылки не найден. А, В, С и D показано применение арилмагний-опосредуемого сочетания для получения аналогов соединения 6, включая О-спиро и S-спиро соединения.
На Фиг. 3 изображена общая схема получения SGLT ингибиторов по настоящему изобретению, содержащих гетероарильное кольцо.
На Фиг. 4 изображен общий метод синтеза тетрагидротиопирановых соединений по настоящему изобретению.
На Фиг. 5А, В, С и D показано, как SGLT ингибиторы можно получить из тригидрокси-6-(метокси)тетрагидро-2Н-пиран-2-она или тригидрокси-6-(метилтио)тетрагидро-2Н-пиран-2-она.
На Фиг. 6А и В изображен общий метод синтеза циклогексановых, циклогексеновых и циклогексаноновых соединений по настоящему изобретению.
На Фиг. 7 показан общий синтез нескольких арилиодидных прекурсоров соединений по настоящему изобретению.
Подробное описание изобретения
Общая часть
В настоящем изобретении описаны способы получения соединений, оказывающих ингибирующее действие на натрий-зависимый переносчик глюкозы SGLT. Описанный способ включает применение реактива Гриньяра или активированного реагента Гриньяра, такого как турбо-реагент Гриньяра, реагента для сочетания бензольной системы с углеводной частью финальной молекулы. В настоящем изобретении описаны также синтетические интермедиаты, которые могут применяться для получения таких соединений.
Определения
При использовании в настоящем тексте если не указано иное, термин ''алкил'' отдельно или в комбинации означает одновалентный насыщенный алифатический углеводородный радикал, содержащий указанное число атомов углерода. Данный радикал может иметь линейную или разветвленную цепь и, где это указано, и необязательно замещен 1-3 подходящими указанными выше заместителями. Иллюстративные примеры алкильных групп включают (но не ограничены только ими) метил, этил, н-пропил, н-бутил, н-пентил, н-гексил, изопропил, изобутил, изопентил, амил, втор-бутил, трет-бутил, трет-пентил, н-гептил, н-октил, н-нонил, н-децил, н-додецил, н-тетрадецил, н-гексадецил, н-октадецил, н-эйкозил и т.п.. Предпочтительные алкильные группы включают метил, этил, н-пропил и изопропил. Предпочтительные необязательно замещенные заместители включают галоген, метокси, этокси, циано, нитро и амино-группы.
При использовании в настоящем тексте термин ''гало'' или ''галоген'' означает одновалентный галогеновый радикал или атом, выбранный из фтора, хлора, брома и иода. Предпочтительные галогеновыми группами являются фтор, хлор и бром.
При использовании в настоящем тексте если не указано иное, термин ''галогеналкил'' означает описанный выше алкильный радикал, замещенный одним или несколькими галогенами. Иллюстративные примеры галогеналкильных групп включают (но не ограничены только ими) хлорметил, дихлорметил, фторметил, дифторметил, трифторметил, 2,2,2-трихлорэтил и т.п.
При использовании в настоящем тексте если не указано иное, термин ''алкенил'' отдельно или в комбинации означает одновалентный алифатический углеводородный радикал, содержащий указанное число атомов углерода и по меньшей мере одну углерод-углеродную двойную связь. Данный радикал может иметь линейную или разветвленную цепь, может находиться в Е или Z форме, и где это указано, необязательно замещен 1-3 подходящими указанными выше заместителями. Иллюстративные примеры алкенильных групп включают (но не ограничены только ими) винил, 1-пропенил, 2-пропенил, изопропенил, 1-бутенил, 2-бутенил, изобутенил, 2-метил-1-пропенил, 1-пентенил, 2-пентенил, 4-метил-2-пентенил, 1,3-пентадиенил, 2,4-пентадиенил, 1,3-бутадиенил и т.п.. Предпочтительные алкенильные группы включают винил, 1-пропенил и 2-пропенил. Предпочтительные необязательно замещенные заместители включают галоген, метокси, этокси, циано, нитро и амино группы.
При использовании в настоящем тексте если не указано иное, термин ''алкинил'' отдельно или в комбинации означает одновалентный алифатический углеводородный радикал, содержащий указанное число атомов углерода и по меньшей мере одну углерод-углеродную тройную связь. Данный радикал может иметь линейную или разветвленную цепь и, где это указано, необязательно замещен 1-3 подходящими указанными выше заместителями. Иллюстративные примеры алкинильных групп включают (но не ограничены только ими) этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-метил-1-пентинил, 3-пентинил, 1-гексинил, 2-гексинил, 3-гексинил и т.п.. Предпочтительные алкинильные группы включают этинил, 1-пропинил и 2-пропинил. Предпочтительные необязательно замещенные заместители включают галоген, метокси, этокси, циано, нитро и амино группы.
При использовании в настоящем тексте если не указано иное, термины ''алкокси'' и ''алкилокси'' отдельно или в комбинации означают алифатический радикал вида алкил-О-, где алкил соответствует данному выше определению. Иллюстративные примеры алкокси-групп включают (но не ограничены только ими) метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентокси, изопентокси, неопентокси, трет-пентокси, гексокси, изогексокси, гептокси, октокси и т.п. Предпочтительные алкокси-группы включают метокси и этокси группы.
При использовании в настоящем тексте если не указано иное, термины ''гидроксиалкокси'' и ''гидроксиалкилокси'' отдельно или в комбинации означают алифатический радикал вида НО-алкокси-, где алкокси соответствует данному выше определению. Иллюстративные примеры гидроксиалкокси-групп включают (но не ограничены только ими) гидроксиметокси, гидроксиэтокси, гидроксиэтокси, гидроксипропокси, гидроксиизопропокси, гидроксибутокси, гидроксиизобутокси, гидрокси-трет-бутокси, гидроксипентокси, гидроксиизопентокси, гидроксигексокси, гидроксиизогексокси, гидроксигептокси, гидроксиоктокси и т.п.
При использовании в настоящем тексте если не указано иное, термин ''алкенилокси'' отдельно или в комбинации означает алифатический радикал вида алкенил-О-, где алкенил соответствует данному выше определению. Иллюстративные примеры алкенилокси-групп включают (но не ограничены только ими) винилокси, 1-пропенилокси, 2-пропенилокси, изопропенилокси, 1-бутенилокси, 2-бутенилокси, 3-бутенилокси, 1-изобутенилокси, 2-изобутенилокси, 1-пентенилокси, 2-пентенилокси, 3-пентенилокси, 4-пентенилокси и т.п.
При использовании в настоящем тексте если не указано иное, термин ''алкинилокси'' отдельно или в комбинации означает алифатический радикал вида алкинил-O-, где алкинил соответствует данному выше определению. Иллюстративные примеры алкинилокси-групп включают (но не ограничены только ими) этинилокси, 1-пропинилокси, 2-пропинилокси, 1-бутинилокси, 2-бутинилокси, 3-бутинилокси, 1-пентинилокси, 2-пентинилокси, 3-пентинилокси, 4-пентинилокси, 1-гексинилокси, 2-гексинилокси, 3-гексинилокси и т.п.
При использовании в настоящем тексте если не указано иное, термин ''галогеналкокси'' означает описанный выше алкокси радикал, замещенный одним или больше галогенами. Иллюстративные примеры галогеналкокси-групп включают (но не ограничены только ими) трифторметокси, дифторметокси и т.п.
При использовании в настоящем тексте если не указано иное, термин ''циклоалкил'' отдельно или в комбинации означает одновалентный алициклический насыщенный углеводородный радикал, содержащий три или более атомов углерода, образующих карбоциклическое кольцо, и, где это указано, необязательно замещенный 1-3 подходящими указанными выше заместителями. Иллюстративные примеры циклоалкильных групп включают (но не ограничены только ими) циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил и т.п. Предпочтительные необязательно замещенные заместители включают галоген, метил, этил, метокси, этокси, циано, нитро и амино группу.
При использовании в настоящем тексте если не указано иное, термин ''циклоалкокси'' отдельно или в комбинации означает алифатический радикал вида циклоалкил-О-, где циклоалкил соответствует данному выше определению. Иллюстративные примеры циклоалкокси-групп включают (но не ограничены только ими) циклопропокси, циклобутокси и циклопентокси группы.
При использовании в настоящем тексте если не указано иное, термин ''гетероциклоалкил'' отдельно или в комбинации означает описанную выше циклоалкильную группу, в которой один или больше атомов углерода в цикле замещены гетероатомом, выбранным из N, S и О. Иллюстративные примеры гетероциклоалкильных групп включают (но не ограничены только ими) пирролидинил, тетрагидрофуранил, пиперазинил, тетрагидропиранил и т.п.
При использовании в настоящем тексте если не указано иное, термин ''гетероциклоалкокси'' отдельно или в комбинации означает алифатический радикал вида гетероциклоалкил-О-, где гетероциклоалкил соответствует данному выше определению. Иллюстративные примеры гетероциклоалкокси-групп включают (но не ограничены только ими) тетрагидрофуранокси, пирролидинокси и тетрагидротиофенокси.
При использовании в настоящем тексте термин ''арил'' означает моноциклическую или сконденсированную бициклическую, трициклическую или большую ароматическую циклическую систему, содержащую 6-16 атомов углерода в цикле. Например, арил может представлять собой фенил, бензил или нафтил, предпочтительно фенил. ''Арилен'' означает двухвалентный радикал, являющийся производным арильной группы. Арил группы могут быть моно-, ди- или три-замещенными одним, двумя или тремя радикалами, выбранными из следующих: алкил, алкокси, арил, гидрокси-группа, галоген, циано, амино, аминоалкил, трифторметил, алкилендиокси и окси-C2-C3-алкилен; которые все могут быть необязательно замещенными, например как описано выше в настоящем тексте; или 1- или 2-нафтил; или 1- или 2-фенантренил. Алкилендиокси представляет собой двухвалентный заместитель, присоединенный к двум соседним атомам углерода в фениле, например метилендиокси или этилендиокси. Окси-C2-C3-алкилен также представляет собой двухвалентный заместитель присоединенный к двум соседним атомам углерода в фениле, например оксиэтилен или оксипропилен. Пример для окси- C2-C3-алкилен-фенил представляет собой 2,3-дигидробензофуран-5-ил.
Предпочтительным в качестве арила является нафтил, фенил или фенил, моно- или дизамещенный алкокси-группой, фенилом, галогеном, алкилом или трифторметилом, в особенности фенил или фенил, моно- или дизамещенный алкокси-группой, галогеном или трифторметилом, и в частности фенил.
При использовании в настоящем тексте термин ''гетероарил'' означает моноциклическую или конденсированную бициклическую или трициклическую ароматическую кольцевую систему, содержащую 5-16 атомов в цикле, где 1-4 атомов в цикле каждый представляют собой гетероатом N, О или S. Например, гетероарил включает пиридил, индолил, индазолил, хиноксалинил, хинолинил, изохинолинил, бензотиенил, бензофуранил, фуранил, пирролил, тиазолил, бензотиазолил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, имидазолил, тиенил или любой другой радикал, и может иметь заместители, в особенности быть моно- или дизамещенным, например алкилом, нитро-группой или галогеном. Пиридил представляет собой 2-, 3- или 4-пиридил, преимущественно 2- или 3-пиридил. Тиенил представляет собой 2- или 3-тиенил. Хинолинил представляет собой предпочтительно 2-, 3- или 4-хинолинил. Изохинолинил представляет собой предпочтительно 1-, 3- или 4-изохинолинил. Бензопиранил, бензотиопиранил представляет собой предпочтительно 3-бензопиранил или 3-бензотиопиранил, соответственно. Тиазолил представляет собой предпочтительно 2- или 4-тиазолил и, наиболее предпочтительно, 4-тиазолил. Триазолил предпочтительно представляет собой 1-, 2- или 5-(1,2,4-триазолил). Тетразолил предпочтительно представляет собой 5-тетразолил.
Предпочтительно, гетероарил представляет собой пиридин, индолил, хинолинил, пирролил, тиазолил, изоксазолил, триазолил, тетразолил, пиразолил, имидазолил, тиенил, фуранил, бензотиазолил, бензофуранил, изохинолинил, бензотиенил, оксазолил, индазолил, или любой из перечисленных радикалов, имеющий заместители, в особенности моно- или дизамещенный.
При использовании в настоящем тексте термин ''подходящий заместитель'' означает химически и фармацевтически приемлемую группу, т.е. фрагмент, который не влияет заметным образом на получение и не оказывает отрицательного действия на эффективность соединений по настоящему изобретению. Такие подходящие заместители могут быть подобраны квалифицированными специалистами в данной области техники. Подходящие заместители могут быть выбраны из группы, состоящей из следующих заместителей: галоген, C1-C6 алкил, C2-C6 алкенил, C1-C6 галогеналкил, C1-C6 алкокси, C1-C6 галогеналкокси, C2-C6 алкинил, C3-C8 циклоалкенил, (C3-C8 циклоалкил)C1-C6 алкил, (C3-C8 циклоалкил)C2-C6 алкенил, (C3-C8 циклоалкил)C1-C6 алкокси, C3-C7 гетероциклоалкил, (C3-C7 гетероциклоалкил)C1-C6 алкил, (C3-C7 гетероциклоалкил)C2-C6 алкенил, (C3-C7 гетероциклоалкил)C1-C6 алкокси, гидрокси-группа, карбокси, оксо-группа, сульфанил, C1-C6 алкилсульфанил, арил, гетероарил, арилокси, гетероарилокси, аралкил, гетероаралкил, аралкокси, гетероаралкокси, нитро, циано, амино, C1-C6 алкиламино, ди-(C1-C6 алкил)амино, карбамоил, (C1-C6 алкил)карбонил, (C1-C6 алкокси)карбонил, (C1-C6 алкил)аминокарбонил, ди-(C1-C6 алкил)аминокарбонил, арилкарбонил, арилоксикарбонил, (C1-C6 алкил)сульфонил и арилсульфонил. Группы, перечисленные выше как подходящие заместители, соответствуют данным в настоящем тексте определениям, за исключением того, что подходящий заместитель не может быть дополнительно необязательно замещенным.
При использовании в настоящем тексте термин ''получение реакционной смеси'' означает процесс введения в контакт по меньшей мере двух разных веществ с тем, чтобы они могли смешиваться и реагировать друг с другом. Следует понимать, однако, что результирующий продукт реакции может быть получен напрямую по реакции между добавленными реагентами или из интермедиата, образовавшегося из одного или более из добавленных реагентов, который может образоваться в реакционной смеси.
При использовании в настоящем тексте термин ''алкил-магниевый комплекс'' означает комплекс, содержащий металлический магний, алкильную группу, такую как C1-6 алкил или C3-7 циклоалкил, и, при необходимости, галогенид. Репрезентативные алкил-магниевые комплексы включают (но не ограничены только ими) C1-C4 алкилмагния хлорид, C1-C4 алкилмагния бромид, ди(C1-C4 алкил)магний, C3-C7 циклоалкилмагния хлорид, C3-C7 циклоалкилмагния бромид или ди(C3-C7 циклоалкил)магний.
При использовании в настоящем тексте термин ''органический растворитель'' означает растворители, такие как диэтиловый эфир, тетрагидрофуран, пентаны, гексаны, гептан, метиленхлорид, хлороформ, этилацетат, метанол, этанол, и т.п. Предпочтительные органические растворители включают тетрагидрофуран и гептан.
При использовании в настоящем тексте термин ''защитная группа'' означает соединение, которое делает функциональную группу нереакционноспособной, но также может быть удалено для восстановления функциональной группы в своем первоначальном виде. Такие защитные группы хорошо известны квалифицированному специалисту в данной области и включают соединения, описанные в книге ''Protective Groups in Organic Synthesis'', 4th edition, T.W. Greene and P.G.M. Wuts, John Wiley & Sons, New York, 2006, которая включена в настоящий текст в полном объеме посредством ссылки. Защитные группы можно подобрать таким образом, чтобы они были лабильными в определенных условиях, таких как, среди прочего, основные или кислотные условия. Кислотно-лабильными защитными группами являются те, которые устойчивы в основных и других условиях, но отщепляются в кислых условиях. Аналогично, реагент для удаления защитной группы зависит от условий удаления. Когда применяется кислотно-лабильная защитная группа, реагентом для удаления защитной группы является кислота, такая как сильная кислота.
При использовании в настоящем тексте термин ''фторированный'' означает замену по меньшей мере одного атома водорода в группе по настоящему изобретению на атом фтора. Любая группа по настоящему изобретению может быть фторированной, включая (но не ограничиваясь только ими) алкил, алкокси, циклоалкил, алкенилокси, алкинилокси, циклоалкокси, гидроксиалкокси и гетероциклоалкокси группы.
При использовании в настоящем тексте термин ''активирующий агент'' означает агент, который ускоряет реакцию компонентов в реакционной смеси. Активирующие агенты, которые могут применяться в настоящем изобретении, представляют собой агенты, ускоряющие реакцию Гриньяра.
При использовании в настоящем тексте термин ''уходящая группа'' означает группу, которая сохраняет связывающую электронную пару при гетеролитическом расщеплении связи. Например, уходящая группа легко замещается во время реакции нуклеофильного замещения. Подходящие защитные группы включают (но не ограничены только ими) хлорид, бромид, тозилат, трифлат и т.д. Квалифицированный специалист в данной области может подобрать другие уходящие группы, которые могут применяться в настоящем изобретении.
При использовании в настоящем тексте термин ''восстановитель'' означает агент, способный восстанавливать атом из более высокой степени окисления до более низкой степени окисления. Восстановители могут также использоваться как защитные группы по настоящему изобретению. Восстановители, которые могут применяться в настоящем изобретении, включают (но не ограничены только ими) триалкилсиланы, такие как триметилсилан и триэтилсилан.
При использовании в настоящем тексте термин ''практически не содержащий магния'' означает содержание менее 0,1 эквивалентов по сравнению с количеством соединения формулы Ia в реакционной смеси. Соединение формулы Ia может представлять собой кеталь.
При использовании в настоящем тексте термин ''кислота Льюиса'' означает любое вещество, являющееся акцептором неподеленной электронной пары. Определение кислоты Льюиса по номенклатуре ИЮПАК включает «любую молекулярную частицы (и соответствующие химические вещества), которые являются акцептором электронной пары». Репрезентативные кислоты Льюиса включают (но не ограничены только ими) ZnCl2.
При использовании в настоящем тексте термин ''сильная кислота'' означает любую кислоту, которая полностью ионизуется в водном растворе, и таким образом имеет рKа<-1,74. Подходящие сильные кислоты включают (но не ограничены только ими) соляную кислоту, серную кислоту и перхлорную кислоту.
При использовании в настоящем тексте термин ''реакционный сосуд'' означает любой сосуд для проведения реакции. Реакционный сосуд может представлять собой круглодонную колбу объемом от 5 мл до 5 л или реактор масштаба килограммов или сотен литров.
При использовании в настоящем тексте термин ''пролекарство'' означает соединение-предшественник, которое после введения высвобождает биологически активное соединение in vivo в результате химических или физиологических процессов (например, пролекарство при достижении физиологического уровня рН или после воздействия ферментов превращается в биологически активное соединение). Само пролекарство может обладать или не обладать желаемой биологической активностью.
При использовании в настоящем тексте термин ''активированный реагент Гриньяра'' означает комплекс из активирующего агента и реагента Гриньяра, представляющего собой алкил-магниевый комплекс. Активированные реагенты Гриньяра имеют добавки, которые придают данным реагентам усиленную кинетическую основность, способствующую обмену магний-галоген при нуклеофильном присоединении. Активирование происходит также вследствие увеличения растворимости веществ. Другие аспекты активированных реагентов заключаются в том, что они минимизируют протекание побочных реакций. Активированные реагенты Гриньяра включают (но не ограничены только ими) комплекс LiCl и изопропилмагния хлорида или втор-бутилмагния хлорида, коммерчески доступные Турбо-реагенты Гриньяра. Другие активированные реагенты Гриньяра включают комбинации хлорида лития со вторичными алкилмагния хлоридами, такими как циклические алкилмагния хлориды, т.е. циклопропилмагния хлорид, циклобутилмагния хлорид, циклопентилмагния хлорид, циклогексилмагния хлорид, циклогептилмагния хлорид и т.д. Другие вторичные алкилмагния хлориды включают (но не ограничены только ими) 2-пентилмагния хлорид, 3-пентилмагния хлорид, 2-гексилмагния хлорид, 3-гексилмагния хлорид, 2-гептилмагния хлорид, 3-гептилмагния хлорид, 4-гептилмагния хлорид и их изомеры. Другие подоходящие для применения алкилмагния хлориды включают бис(триметилсилил)метилмагния хлорид и триметилсилилметилмагния хлорид. Другие соли могут использоваться вместо хлорида лития или как добавка к нему, для дальнейшей регулировки реакционной способности.
Соединения
В некоторых вариантах осуществления способами настоящему изобретению можно получить соединение формулы I:
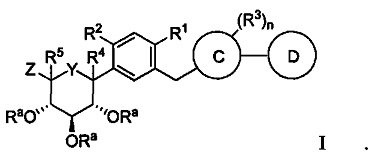
Y в формуле I может представлять собой CHRc, С(=O), О или S. Z в формуле I может представлять собой CH2ORa, ORa, SRa или S(O)m-Ra.
R1 в формуле I может представлять собой хлор. Каждый R2 и R3 в формуле I могут независимо представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, -CH2ORa, C2-C4 алкенил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
По меньшей мере один из R2 и R3 в формуле I может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси или C3-C6 циклоалкил, и по меньшей мере один из R2 и R3 может представлять собой C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
R4 в формуле I может представлять собой Н или OR4a, где R4a может представлять собой Н или C1-C3 алкил. Альтернативно, R2 и R4 объединены с атомами, к которым они присоединены, с образованием 5-6-членного циклоалкила или гетероциклоалкила.
R5 в формуле I может представлять собой Н или -CH2ORa. Альтернативно, R4 и R5 могут быть объединены с атомами, к которым они присоединены, с образованием 5-6-членного гетероциклоалкила.
Каждый Ra в формуле I может независимо представлять собой Н, C1-C3 алкил или Rb. Rb в формуле I может представлять собой защитную группу.
Rc в формуле I может представлять собой Н, ОН или C1-C3 алкокси. Альтернативно, Rc может быть объединен cR4 или R5 с образованием связи.
Цикл C в формуле I может представлять собой арил или гетероарил. Цикл D в формуле I может отсутствовать или представлять собой гетероарил.
Индекс m в формуле I может представлять собой целое число от 1 до 2. Индекс n в формуле I может представлять собой целое число от 1 до 4.
Алкил, алкокси, циклоалкил, алкенилокси, алкинилокси, циклоалкокси, гидроксиалкокси и гетероциклоалкокси группы или их части в формуле I необязательно могут быть частично или полностью фторированы. И один или больше атомов водорода в соединениях формулы I необязательно могут быть заменены на атомы дейтерия.
В некоторых вариантах осуществления соединения по настоящему изобретению представляют собой такие, в которых R1 может представлять собой галоген. В других вариантах осуществления R1 может представлять собой F, Cl, Br или I. В некоторых других вариантах осуществления R1 может представлять собой Cl.
В некоторых вариантах осуществления R2 может представлять собой Н.
В некоторых вариантах осуществления R3 может представлять собой C1-C6 алкил, C1-C3 алкокси, (C1-C3 алкокси)C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси. В других вариантах осуществления R3 может представлять собой C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси. В некоторых других вариантах осуществления R3 может представлять собой этокси, циклопропил или 2-циклопропоксиэтокси. В других вариантах осуществления R3 может представлять собой 2-циклопропоксиэтокси.
В некоторых вариантах осуществления R4 может представлять собой Н, ОН или C1-C3 алкокси. В других вариантах осуществления R4 может представлять собой ОН. В некоторых других вариантах осуществления R4 может представлять собой C1-C3 алкокси. В других вариантах осуществления R4 может представлять собой метокси, этокси или пропокси. В других вариантах осуществления R4 может представлять собой метокси. В других вариантах осуществления R4 может представлять собой Н.
Цикл С может представлять собой любой подходящий арильный или гетероарильный цикл. Арильные циклы, которые могут использоваться в качестве цикла С, включают (но не ограничены только ими) фенил, нафтил и бифенил. Гетероарильные циклы, которые могут использоваться в качестве цикла С, включают (но не ограничены только ими) пиррол, пиридин, пиран, тиофен, тиопиран, тиазол, имидазол, тиадиазол, пиразин, пиримидин, пиридазин, индол и бензотиофен. В некоторых вариантах осуществления цикл С может представлять собой фенил, тиадиазол или бензотиофен. В других вариантах осуществления цикл С может представлять собой фенил. В некоторых других вариантах осуществления цикл С может представлять собой тиадиазол.
Цикл D может отсутствовать или представлять собой любой подходящий гетероарильный цикл. Гетероарильные циклы, которые могут использоваться в качестве цикла D включают (но не ограничены только ими) пиррол, пиридин, пиран, тиофен, тиопиран, тиазол, имидазол, тиадиазол, пиразин, пиримидин, пиридазин, индол и бензотиофен. В некоторых вариантах осуществления цикл D может отсутствовать. В других вариантах осуществления цикл D может представлять собой фуран, тиофен или пиразин.
В некоторых вариантах осуществления цикл С может представлять собой фенил, и цикл D может отсутствовать. В других вариантах осуществления цикл С может представлять собой бензотиофен, и цикл D может отсутствовать. В некоторых других вариантах осуществления цикл С может представлять собой тиадиазол, и цикл D может представлять собой фуран, тиофен или пиразин.
В некоторых вариантах осуществления соединение, полученное согласно настоящему изобретению, представляет собой соединение формулы Ia:
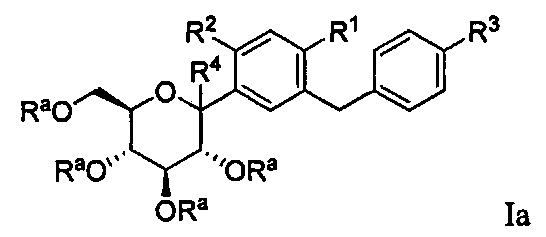
где R2 в формуле Ia может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
R4 в формуле Ia может представлять собой Н, ОН и C1-C3 алкокси.
В некоторых вариантах осуществления R1 может представлять собой F, Cl, Br или I. В других вариантах осуществления R1 может представлять собой Cl.
В некоторых вариантах осуществления R4 может представлять собой Н. В других вариантах осуществления R4 может представлять собой ОН. В некоторых других вариантах осуществления R4 может представлять собой метокси, этокси или пропокси. В других вариантах осуществления R4 может представлять собой метокси.
В некоторых вариантах осуществления каждый Ra может независимо представлять собой Н или Rb. В других вариантах осуществления каждый Ra может представлять собой Н. В некоторых других вариантах осуществления каждый Ra может представлять собой Rb. Защитные группы, которые могут применяться в соединениях по настоящему изобретению, включают любые подходящие защитные группы, такие как гидрокси или тиольная защитная группа. Такие защитные группы хорошо известны квалифицированному специалисту в данной области и включают соединения, описанные в книге ''Protective Groups in Organic Synthesis'', 4th edition, T. W. Greene and P.G.M. Wuts, John Wiley & Sons, New York, 2006, которая включена в настоящий текст в полном объеме посредством ссылки. В некоторых вариантах осуществления защитные группы в Rb представляют собой кислотно-лабильные защитные группы. Подходящие кислотно-лабильные защитные группы включают любые защитные группы, которые могут быть удалены в присутствии кислоты, и включают (но не ограничены только ими) силильные защитные группы и t-BOC защитные группы. Силильные защитные группы включают (но не ограничены только ими) триметисилан.
В некоторых вариантах осуществления соединения, полученные согласно настоящему изобретению, представляют собой такие, в которых R2 может представлять собой Н; R3 может представлять собой C1-C6 алкил, C1-C3 алкокси, (C1-C3 алкокси)C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси; и R4 может представлять собой Н, ОН или C1-C3 алкокси. В других вариантах осуществления R2 может представлять собой Н; R3 может представлять собой C1-C6 алкил, C1-C3 алкокси, (C1-C3 алкокси)C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси; и R4 может представлять собой метокси.
В других вариантах осуществления R1 может представлять собой хлор; и R2 может представлять собой Н. В некоторых других вариантах осуществления R3 может представлять собой C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси. В других вариантах осуществления R3 может представлять собой этокси, циклопропил или 2-циклопропоксиэтокси.
Соединения, полученные согласно настоящему изобретению, включают полукетали, где Y представляет собой О. и R4 представляет собой ОН. В некоторых вариантах осуществления R4 может представлять собой ОН. В других вариантах осуществления R2 может представлять собой Н; R3 может представлять собой этокси, циклопропил или 2-циклопропоксиэтокси; и R4 может представлять собой ОН. В некоторых вариантах осуществления R4 может представлять собой ОН; и каждый Ra может представлять собой Rb, как в следующей структуре:
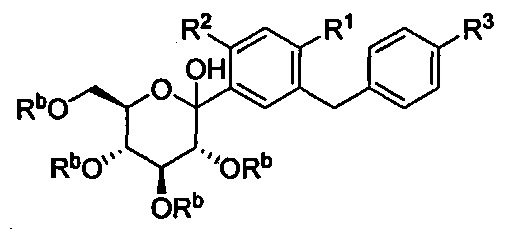
В некоторых вариантах осуществления соединение формулы I имеет структуру:
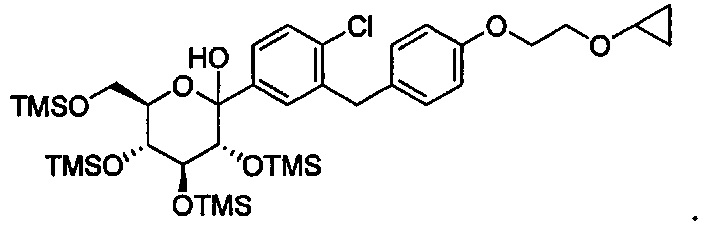
Соединения, полученные согласно настоящему изобретению включают кетали, в которых Y представляет собой О, и R4 представляет собой C1-C3 алкокси. В некоторых вариантах осуществления R4 может представлять собой C1-C3 алкокси; и каждый Ra может независимо представлять собой Н или Rb. В других вариантах осуществления каждый Rb в соединении формулы I может представлять собой кислотно-лабильную защитную группу. В некоторых вариантах осуществления кислотно-лабильная защитная группа представляет собой триметилсилан. В других вариантах осуществления каждый Ra может представлять собой Н. В некоторых других вариантах осуществления R4 может представлять собой метокси, этокси или пропокси. В других вариантах осуществления R4 может представлять собой метокси.
В некоторых вариантах осуществления соединение формулы I имеет структуру:
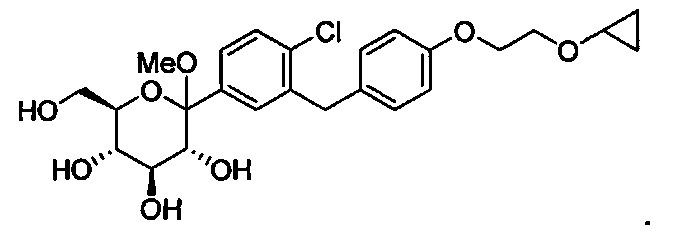
В некоторых вариантах осуществления R4 может представлять собой Н. В других вариантах осуществления соединения, полученные согласно настоящему изобретению, представляют собой такие, в которых R2 может представлять собой Н; R3 может представлять собой C1-C6 алкил, C1-C3 алкокси, (C1-C3 алкокси)C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси; R4 может представлять собой Н; и каждый Ra может представлять собой Н. В некоторых других вариантах осуществления R2 может представлять собой Н; R3 может представлять собой этокси, циклопропил или 2-циклопропоксиэтокси; R4 может представлять собой Н; и каждый Ra может представлять собой Н.
В некоторых вариантах осуществления соединение, полученное согласно настоящему изобретению, имеет структуру: 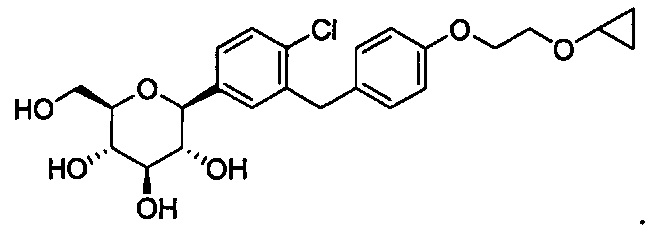
В некоторых вариантах осуществления соединение формулы I может иметь следующую структуру:
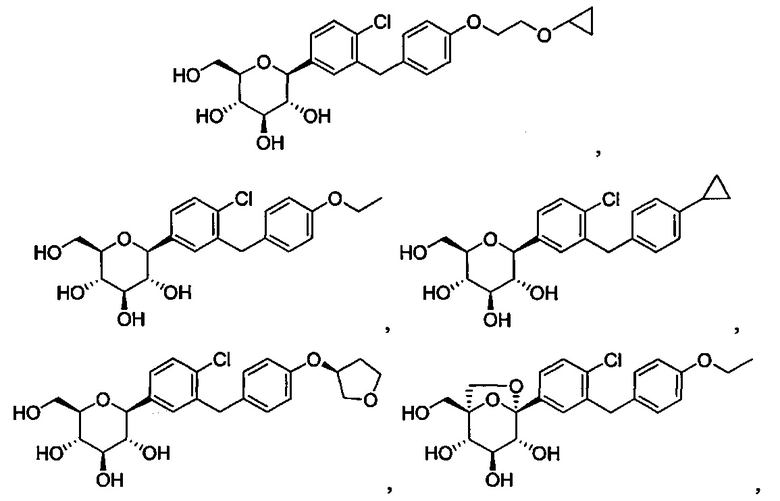
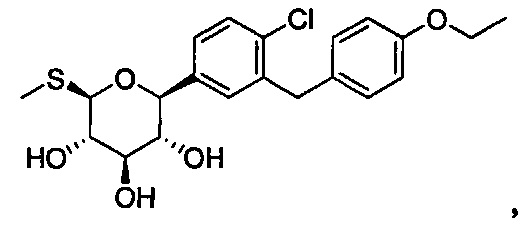 или
или 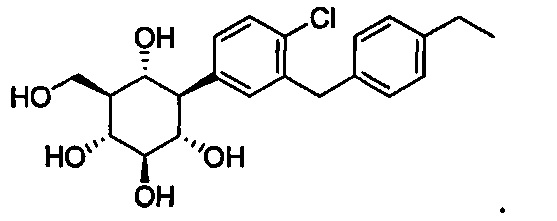
[0002] В настоящем изобретении описаны также соединения, которые могут использоваться в качестве синтетических интермедиатов в получении соединения формулы I. В некоторых вариантах осуществления в настоящем изобретении описано соединение формулы II:
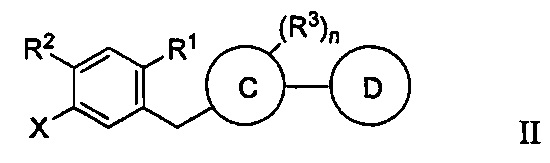
где X представляет собой бром или иод.
В некоторых вариантах осуществления соединение имеет формулу IIa:

где R1 в формуле IIa может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил или C1-C3 алкокси. R2 и R3 в формуле IIa каждый независимо могут представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси.
X в формуле IIa может представлять собой бром или иод.
Алкил, алкокси, циклоалкил, алкенилокси, алкинилокси, циклоалкокси, гидроксиалкокси и гетероциклоалкокси группы или части в формуле IIa необязательно могут быть частично или полностью фторированы, и один или больше атомов водорода в формуле IIa необязательно могут быть заменены на атомы дейтерия.
В некоторых вариантах осуществления соединения формулы IIa включают такие, в которых R1 может представлять собой галоген. В других вариантах осуществления R1 может представлять собой F, Cl, Br или I. В некоторых других вариантах осуществления R1 может представлять собой Cl.
В некоторых вариантах осуществления соединения формулы IIa включают такие, в которых R2 может представлять собой Н.
В некоторых вариантах осуществления соединения формулы IIa включают такие, в которых R3 может представлять собой C1-C6 алкил, C1-C3 алкокси, (C1-C3 алкокси)C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси. В других вариантах осуществления R3 может представлять собой C1-C3 алкокси, C3-C6 циклоалкил или (C3-C6 циклоалкокси)C1-C3 алкокси. В некоторых других вариантах осуществления R3 может представлять собой этокси, циклопропил или 2-циклопропоксиэтокси. В других вариантах осуществления R3 может представлять собой 2-циклопропоксиэтокси.
В некоторых вариантах осуществления соединение формулы IIa имеет структуру, в которой R1 может представлять собой иод, хлор; R2 может представлять собой Н; и X может представлять собой иод. В других вариантах осуществления R3 может представлять собой гидрокси. В некоторых других вариантах осуществления соединение формулы IIa имеет структуру:
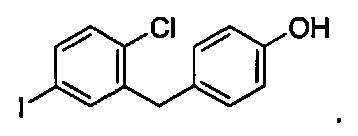
В некоторых вариантах осуществления R в формуле IIa может представлять собой C1-C3 алкокси, C3-C6 циклоалкилокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, C1-C3 гидроксиалкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C3-C4 алкенилокси или (C3-C6 циклоалкил)C3-C4 алкинилокси.
[0003] В некоторых вариантах осуществления формула IIa имеет структуру, где R1 может представлять собой галоген; R2 может представлять собой Н; и R3 может представлять собой C1-C3 алкокси или (C3-C6 циклоалкокси)C1-C3 алкокси. В других вариантах осуществления формула IIa имеет структуру, где R1 может представлять собой хлор; R2 может представлять собой Н; и R3 может представлять собой этокси или 2-циклопропоксиэтокси.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру:
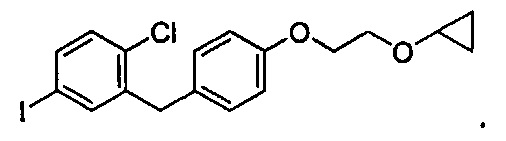
В некоторых вариантах осуществления соединение по настоящему изобретению имеет формулу III:
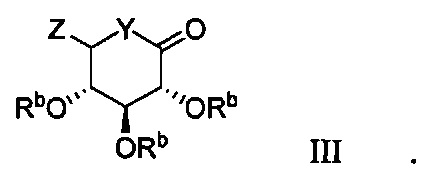
В некоторых вариантах осуществления радикал Z в формуле III может представлять собой -ОМе или -SMe.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет формулу IIIa:
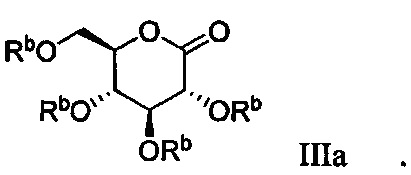
В некоторых вариантах осуществления соединение формулы III имеет структуру:
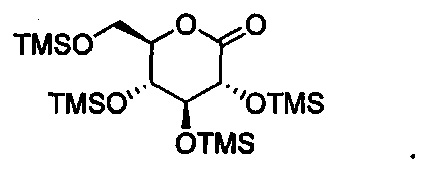
В некоторых вариантах осуществления соединение по настоящему изобретению имеет формулу IV:
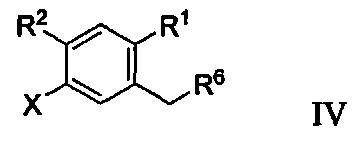
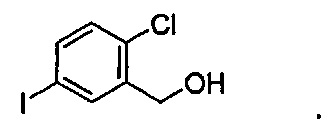
где R6 может представлять собой ОН или Br. В других вариантах осуществления R6 может представлять собой ОН. В некоторых других вариантах осуществления R6 может представлять собой Br. В других вариантах осуществления соединение формулы IV имеет структуру, в которой R1 может представлять собой хлор; R2 может представлять собой Н; и X может представлять собой иод. В других вариантах осуществления соединение формулы IV имеет структуру:
В некоторых вариантах осуществления в настоящем изобретении описано соединение формулы V:
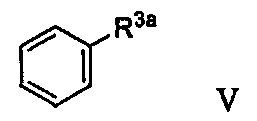
где R3a представляет собой ОН. В некоторых вариантах осуществления соединение формулы V имеет структуру:
В некоторых вариантах осуществления в настоящем изобретении описано соединение формулы VI:
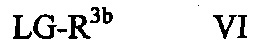
где R3b может представлять собой C1-C3 алкил, C3-C6 циклоалкил, C3-C6 гетероциклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкил, (C3-C6 гетероциклоалкокси)C1-C3 алкил, (C3-C6 циклоалкил)C3-C4 алкенил или (C3-C6 циклоалкил)C3-C4 алкинил; и LG может представлять собой уходящую группу.
Уходящая группа LG может представлять собой любую подходящую уходящую группу, такую как хлорид, бромид, иодид, гидроксил (при использовании сочетания по Мицунобу, Swamy, K.C.K., et al., Mitsunobu and Related Reactions: Advances and Applications. Chemical Reviews, 2009. 109(6): p. 2551-2651., Connolly, T.J., et al., Development of a Pilot-Plant-Scale Synthesis of an Alkylated Dihydrobenzothiazol S,S-Dioxide: Incorporation of a Late-Stage Mitsunobu Reaction Organic Process Research & Development, 2010. 14(4): p. 868-877), оксониевые ионы, нонафлаты, трифлат, фторсульфонат, тозилат, мезилат, нитраты, фосфаты, феноксиды, такие как активированные феноксиды, спирты, карбоновые кислоты, ацильные группы и т.д. В некоторых вариантах осуществления уходящая группа может быть связана с остальной частью молекулы через атом кислорода, такие как трифлат, нонафлат, фторсульфонат, тозилат, мезилат, сложные эфиры, феноксиды, такие как активированные феноксиды, карбоновые кислоты и сложные эфиры. В других вариантах осуществления уходящая группа LG может представлять собой хлорид, бромид, иодид, гидрокси-группу, тозилат или мезилат. В некоторых других вариантах осуществления уходящая группа LG может представлять собой хлорид, бромид или иодид. В других вариантах осуществления уходящая группа LG может представлять собой гидрокси. В других вариантах осуществления уходящая группа LG может представлять собой тозилат или мезилат. В других вариантах осуществления уходящая группа LG может представлять собой хлорид, бромид или тозилат. В других вариантах осуществления уходящая группа представляет собой тозилат.
В некоторых вариантах осуществления R3b формулы VI может представлять собой C1-C3 алкил или (C3-C6 циклоалкокси)C1-C3 алкил. В других вариантах осуществления R3b в формуле VI может представлять собой (C3-C6 циклоалкокси)C1-C3 алкил. В некоторых других вариантах осуществления R3b может представлять собой этил или 2-циклопропоксиэтил. В других вариантах осуществления R3b может представлять собой 2-циклопропоксиэтил.
Любая комбинация уходящей группы LG и R3b подходит для соединения формулы VI. В некоторых вариантах осуществления уходящая группа LG может представлять собой хлорид, бромид, иодид, гидрокси-группу, тозилат или мезилат, и R3b может представлять собой C1-C3 алкил или (C3-C6 циклоалкокси)C1-C3 алкил. В других вариантах осуществления уходящая группа LG может представлять собой хлорид, бромид или тозилат, и R3b может представлять собой этил или 2-циклопропоксиэтии. В некоторых вариантах осуществления соединение формулы VI имеет структуру:
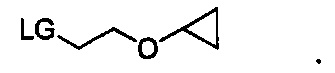
В других вариантах осуществления соединение формулы VI имеет структуру:
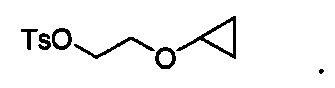
Настоящее изобретение включает все таутомеры и стереоизомеры соединений по настоящему изобретению, как в смеси, так и в чистой или практически чистой форме. Соединения по настоящему изобретению могут иметь асимметричные центры на атомах углерода, и поэтому соединения по настоящему изобретению могут существовать в диастереомерных или энантиомерных формах или в виде их смесей. Все конформационные изомеры (например, цис- и транс-изомеры) и все оптические изомеры (например, энантиомеры и диастереомеры), рацемические, диастереомерные и другие смеси таких изомеров, а также сольваты, гидраты, изоморфы, полиморфы и таутомеры также входят в объем настоящего изобретения. Соединения по настоящему изобретению можно получать с использованием диастереомеров, энантиомеров или рацемических смесей в качестве исходных веществ. Кроме того, диастереомерные и энантиомерные продукты можно разделить посредством хроматографии, дробной кристаллизации или другими методами, известными квалифицированным специалистам в данной области.
Настоящее изобретение также включает изотопно-меченые соединения по настоящему изобретению, где один или больше атомов заменены на один или больше атомов, имеющих специфические атомные массы или массовые числа. Примеры изотопов, которые можно вводить в соединения по настоящему изобретению, включают (но не ограничены только ими) изотопы атомов водорода, углерода, азота, кислорода, фтора, серы и хлора (такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 18F, 35S и 36Cl). Изотопно-меченые соединения по настоящему изобретению могут использоваться в анализе распределения в тканях описанных соединений и их пролекарств и метаболитов; предпочтительные изотопы для таких исследований включают 3H и 14C. Кроме того, в некоторых обстоятельствах замена на более тяжелые изотопы, такие как дейтерий (2H), может обеспечить повышенную метаболическую стабильность, что дает терапевтические преимущества, такие как увеличенное время полужизни in vivo или уменьшение дозировки. Изотопно-меченые соединения по настоящему изобретению в целом можно получать согласно описанным в настоящем тексте способам, заменяя изотопно-меченые реагенты на не-изотопно-меченые.
Опционально, соединения формулы I можно вводить в реакцию с комплексообразующим реагентом, таким как D или L энантиомер природной аминокислоты, в подходящем растворителе, с образованием соответствующего кристаллического комплекса, такого как аминокислотный комплекс, с соединением формулы I. Аминокислотные комплексы соединений формулы I можно получить смешиванием аминокислоты с очищенным соединением в подходящем растворителе или с неочищенной реакционной смесью, содержащей данное соединение и другие реагенты. Для получения комплекса можно использовать любую подходящую аминокислоту, включая природные и синтетические аминокислоты, а также аналоги аминокислот и миметики аминокислоты, которые работают аналогично природным аминокислотам. Природными аминокислотами являются те, которые кодируются генетически, и они включают аланин (А), глицин (G), аспарагиновую кислоту (D), глутаминовую кислоту (Е), аспарагин (N), глутамин (Q), аргинин (R), лизин (K), изолейцин (I), лейцин (L), метионин (М), валин (V), фенилаланин (F), тирозин (Y), триптофан (W), серин (S), треонин (Т), цистеин (С) и метионин (М). Также могут использоваться модифицированные формы природных аминокислот, такие как гидроксипролин, γ-карбоксиглутамат и O-фосфосерин. Также можно использовать аналоги аминокислот и неприродные аминокислоты. Например, можно использовать L-пироглутаминовую кислоту для получения сокристаллических соединений с соединениями по настоящему изобретению.
Способы получения
В настоящем изобретении описаны способы получения соединений формул I и IIa.
Соединения формулы I
Соединения формулы I можно получить разнообразными методами сочетания, включая методы с реагентом Гриньяра и активированным реагентом Гриньяра, такие как методы с турбо-реагентом Гриньяра.
В некоторых вариантах осуществления в настоящем изобретении описан способ получения соединения формулы I путем получения первой реакционной смеси, содержащей соединение формулы II, алкил-магниевый комплекс, такой как C1-C4 алкилмагния хлорид, C1-C4 алкилмагния бромид, ди(C1-C4 алкил)магний, C3-C7 циклоалкилмагния хлорид, C3-C7 циклоалкилмагния бромид или ди(C3-C7 циклоалкил)магний, и первый органический растворитель, где соотношение алкил-магниевого комплекса к соединению формулы II меньше или равно 1,0 (моль/моль), и где первая реакционная смесь находится при температуре ниже примерно -50°C, давая промежуточное соединение. Данный способ также включает получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы III. Таким образом можно получить соединение формулы I.
В некоторых вариантах осуществления в настоящем изобретении описан способ получения соединения формулы Ia, путем получения первой реакционной смеси, содержащей соединение формулы IIa, алкил-магниевый комплекс и первый органический растворитель, с получением промежуточного соединения. Данный способ также включает получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы IIIa. Таким образом можно получить соединение формулы Ia.
Алкил-магниевый комплекс может представлять собой любой подходящий алкил-магниевый комплекс, включая (но не ограничиваясь только ими) C1-C4 алкилмагния хлорид, C1-C4 алкилмагния бромид, ди(C1-C4 алкил)магний, C3-C7 циклоалкилмагния хлорид, C3-C7 циклоалкилмагния бромид или ди(C3-C7 циклоалкил)магний. В некоторых вариантах осуществления алкил-магниевый комплекс может представлять собой (изопропил)MgXl.
Первый и второй органические растворители могут представлять собой любой подходящий органический растворитель, такой как толуол, тетрагидрофуран (ТГФ), гексан, пентан, метил-третбутиловый эфир (МТБЭ), 1,4-диоксан, 2-метилтетрагидрофуран (рацемический) или их смеси. Первый и второй органический растворитель могут быть одинаковыми или разными.
Промежуточное соединение, формируемое по способу согласно настоящему изобретению, может быть выделено или использовано далее без дополнительного выделения или очистки. В некоторых вариантах осуществления промежуточное соединение может иметь следующую структуру:
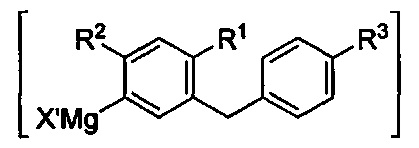
где X' представляет собой C1-C4 алкил, C3-C7 циклоалкил или галоген из алкил-магниевого комплекса. В некоторых вариантах осуществления X' может представлять собой Cl или Br.
Соединение формулы I можно получить с применением любого подходящего соотношения алкил-магниевого комплекса к соединению формулы II. Например, соединение формулы II может присутствовать в эквимольном количестве или в избытке по сравнению с алкил-магниевым комплексом. Предпочтительными соотношениями для минимизации реакций кросс-сочетания и других побочных реакций являются такие, при которых соединение формулы II находится в небольшом мольном избытке к алкил-магниевому комплексу. Подходящие соотношения алкил-магниевого комплекса к соединению формулы II включают значения меньше или равные 1,0, или от примерно 0,90 до 1,0, или от примерно 0,95 до 1,0 (моль/моль). Другие подходящие соотношения алкил-магниевого комплекса к соединению формулы II включают 0,9, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96, 0,97, 0,98, 0,99 и 1,0 (моль/моль). В некоторых вариантах осуществления соотношение алкил-магниевого комплекса к соединению формулы II может составлять от примерно 0,95 до 1,0 (моль/моль).
Способ по настоящему изобретению может также включать активирующий агент в первой реакционной смеси. Активирующий агент может представлять собой любой подходящий реагент, который улучшает работу реактивов Гриньяра, включая добавление следовых количеств иода, метилиодида, дибромэтана, или получение Mg in situ с помощью метода Рике. Также есть методы улучшения работы магния путем восстановления поверхностной пленки MgO, которая служит барьером высвобождения магния. Активирующий агент включает (но не ограничивается только ими) хлорид лития, лития бромид, лития ацетилацетонат, лития перхлорат, магния хлорид, хлорид цинка, хлорид алюминия, хлорид церия, хлорид лантана (и другие хлориды редкоземельных элементов), хлорид олова, хлорид индия, хлорид кадмия, хлорид железа, хлорид меди, хлорид марганца, диизобутиалюминий гидрид, (натрия бис(2-метоксиэтокси)алюминий, гидрид) (Organic Process Research & Development, 2001, vol 6 p 906), иод (Synthesis 1981, 585), магний по Рике (J. Am. Chem. Soc. 1972, 94, 7178; J. Chem. Soc., Chem. Commun. 1973, 879; J. Am. Chem. Soc. 1974, 96, 1775), TMSCl (Organic Process Research & Development 2008, 12, 1188-1194; Org. Process Res. DeV. 2001, 5, 479), 2,2'-оксибис(N,N-диметилэтанамин) (Organic Letters 2005, 8(2): 305-307). Могут применяться другие агенты для нарушения олигомеризации реагента Гриньяра и повышения скорости реакции, такие как фосфорамид, полиамины или полиаминовые простые эфиры или простоэфирные полиамины (N,N,N',N'-тетраметилэтилендиамин, бис[2-(N,N-диметиламино)-этиловый] эфир, N,N,N',N'',N''-пентаметилдиэтилентриамин, трис[2-(2-метоксиэтокси)этил]амин, диаминоалкиловые спирты (2-(N,N-диметил)этанол) дигидроксидисульфонамиды, саленовые катализаторы и другие (см. Synthesis, 2008. 2008(11): р. 1647, 1675).
В некоторых вариантах осуществления активирующий агент может представлять собой LiCl, ZnCl2, диизобутилалюминий гидрид, натрия бис(2-метоксиэтокси)алюминий гидрид, три-метилсилил хлорид или 2,2'-оксибис(N,N-диметилэтанамин). В других вариантах осуществления активирующий агент может представлять собой LiCl. В некоторых других вариантах осуществления активирующий агент формирует комплекс с алкил-магниевым комплексом. Например, когда алкил-магниевый комплекс представляет собой (изопропил)MgXl и активирующий агент представляет собой LiCl, комплекс активирующего агента и алкил-магниевого комплекса может представлять собой LiCl-(изопропил)MgXl. В других вариантах осуществления активирующий агент может представлять собой ZnCl2. В других вариантах осуществления активирующий агент может представлять собой LiCl или ZnCl2. В других вариантах осуществления активирующий агент может представлять собой комбинацию LiCl и ZnCl2.
Активирующий агент может присутствовать в любом подходящем количестве, и может присутствовать в том же или в другом соотношении, как соотношение алкил-магниевого комплекса к соединению формулы II. Подходящие соотношения активирующего агента к соединению формулы II включают значения меньше или равные 1,0, или от примерно 0,90 до 1,0, или от примерно 0,95 до 1,0 (моль/моль). Другие подходящие соотношения активирующего агента к соединению формулы II включают 0,9, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96, 0,97, 0,98, 0,99 и 1,0 (моль/моль). Активирующий агент может также присутствовать в любом подходящем соотношении к алкил-магниевому комплексу, таком как от примерно 0,9 до примерно 1,1 (моль/моль), включая примерно 0,9, 0,95, 1,0, 1,05 и примерно 1,1 (моль/моль). В некоторых вариантах осуществления соотношение активирующего агента к алкил-магниевому комплексу составляет около 1,0 (моль/моль).
Первая реакционная смесь может находиться при любой подходящей температуре. Подходящие температуры для первой реакционной смеси включают значения меньше около -50°C, или от примерно -75°C до примерно -50°C, или от примерно -60°C до примерно -50°C. Подходящие температуры для первой реакционной смеси также включают значения около -100°C, -90, -80, -75, -70, -65, -60, -55 и около -50°C. В некоторых вариантах осуществления первая реакционная смесь находится при температуре ниже примерно 50°C. В других вариантах осуществления первая реакционная смесь находится при температуре от примерно -60 до примерно -50°C.
В некоторых вариантах осуществления вторая реакционная смесь также может включать дополнительный алкил-магниевый комплекс. Дополнительный алкил-магниевый комплекс может присутствовать в любом подходящем соотношении к соединению формулы II, таком как от примерно 0,01 до примерно 0,1 (моль/моль), включая около 0,01 (моль/моль), 0,015, 0,02, 0,025, 0,03, 0,035, 0,04, 0,045, 0,05, 0,055, 0,06, 0,065, 0,07, 0,075, 0,08, 0,085, 0,09, 0,095 и 0,1 (моль/моль). В некоторых вариантах осуществления соотношение дополнительного алкил-магниевого комплекса во второй реакционной смеси к соединению формулы II составляет от примерно 0,01 до примерно 0,1 (моль/моль). Количество дополнительного алкил-магниевого комплекса может зависисить от разных факторов, таких как количество влаги в растворе (Соединения формулы III. В некоторых случаях, количество дополнительного алкил-магниевого комплекса определяют титрованием раствора соединения формулы III, таким как титрование по методу Карла Фишера. Предпочтительными количествами второй реакционной смеси для подавления реакций кросс-сочетания и других побочных реакций являются такие, при которых дополнительный алкил-магниевый комплекс не превышает в мольном количестве остаток соединения формулы II.
Вторая реакционная смесь может находиться при любой подходящей температуре. Подходящие температуры для второй реакционной смеси включают значения от примерно -100°C до примерно 0°C, или от примерно -75°C до примерно -25°C, или от примерно -60°C до примерно -25°C, или от примерно -60°C до примерно -50°C, или от примерно -60°C до примерно -10°C. Подходящие температуры для второй реакционной смеси также включают значения примерно -100°C, -90, -80, -75, -70, -65, -60, -55, -50, -45, -40, -35, -30, -25, -20, -15, -10, -5 и около 0°C. В некоторых вариантах осуществления вторая реакционная смесь находится при температуре от примерно -60 до примерно -25°C. В других вариантах осуществления вторая реакционная смесь находится при температуре от примерно -60 до примерно -10°C.
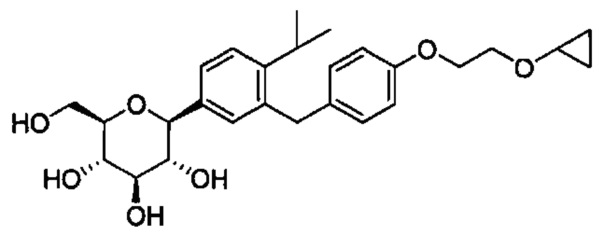
В некоторых вариантах осуществления соединение формулы I имеет структуру:
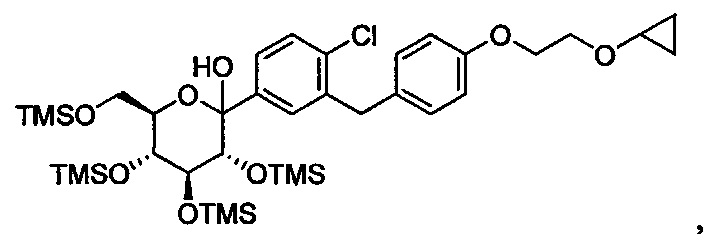
и получено способом, включающим получение первой реакционной смеси, содержащей соединение формулы II, имеющее структуру:
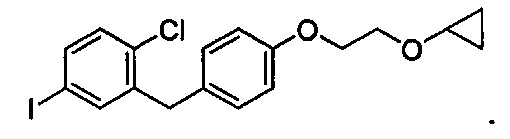
Первая реакционная смесь также включает изопропилмагния хлорид, хлорид лития, тетрагидрофуран и гептан, где соотношение изопропилмагния хлорида к соединению формулы II составляет от примерно 0,95 до 1,0 (моль/моль), и соотношение изопропилмагния хлорида к LiCl составляет примерно 1,0 (моль/моль), где первая реакционная смесь находится при температуре ниже примерно 50°C, с получением промежуточного соединения. Данный способ также включает получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы III, имеющее структуру:
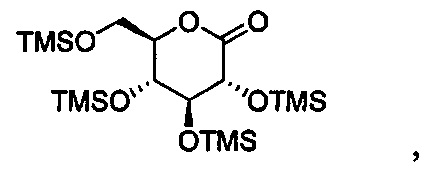
Таким образом получают соединение формулы I.
В некоторых вариантах осуществления промежуточное соединение имеет формулу:
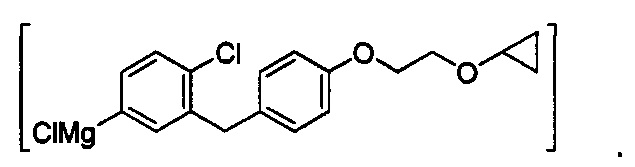
В некоторых вариантах осуществления вторая реакционная смесь также включает дополнительный изопропилмагния хлорид и хлорид лития, где соотношение дополнительного изопропилмагния хлорида к соединению формулы II составляет от примерно 0,01 до примерно 0,1 (моль/моль), и соотношение дополнительного изопропилмагния хлорид к дополнительному LiCl составляет 1,0 (моль/моль).
Первая и вторая реакционные смеси могут быть получены в отдельных реакционных сосудах или в одном и том же реакционном сосуде. В некоторых вариантах осуществления первая и вторая реакционные смеси получены в отдельных реакционных сосудах. В других вариантах осуществления первая и вторая реакционные смеси получены в одном и том же реакционном сосуде.
Способ по настоящему изобретению может включать различные другие стадии. Например, соединения, в которых R4 представляет собой ОН (полукеталь в некоторых вариантах осуществления), можно превращать в соединение, в котором R4 представляет собой C1-C3 алкокси (кеталь в некоторых вариантах осуществления).
В некоторых вариантах осуществления данный способ также включает получение третьей реакционной смеси, содержащей C1-C3 алкилгидрокси, сильную кислоту и соединение формулы I, где R4 представляет собой ОН и каждый Ra представляет собой Rb, давая соединение формулы I, где R4 представляет собой C1-C3 алкокси, и каждый Ra может независимо представлять собой Н или Rb.
Сильные кислоты, которые могут применяться в третьей реакционной смеси, включают (но не ограничены только ими) соляную кислоту, уксусную кислоту, серную кислоту и азотную кислоту. В некоторых вариантах осуществления сильная кислота представляет собой соляную кислоту.
Защитные группы Rb в формуле I в третьей реакционной смеси могут быть удалены за одну или несколько разных стадий. Удаление защитных групп можно осуществить разными способами, известными квалифицированному специалисту в данной области и описаными в книге ''Protective Groups in Organic Synthesis'', 4th edition, T.W. Greene and P.G.M. Wuts, John Wiley & Sons, New York, 2006. В некоторых вариантах осуществления каждый Rb в соединении формулы I в третьей реакционной смеси представляет собой кислотно-лабильную защитную группу, так что происходит удаление кислотно-лабильных защитных групп в третьей реакционной смеси и получение соединения формулы I, где каждый Ra представляет собой Н. Подходящие кислотно-лабильные группы и способы их удаления описаны выше.
Третья реакционная смесь может находиться при любой подходящей температуре. Подходящие температуры для третьей реакционной смеси включают значения от примерно -50°C до примерно 50°C, или от примерно -25°C до примерно 25°C, или от примерно -15°C до примерно 25°C. Подходящие температуры для третьей реакционной смеси также включают значения около -50°C, -45, -40, -35, -30, -25, -20, -15, -10, -5, 0, 5, 10, 15, 20, 25, 30, 35, 40, 45 или около 50°C. В некоторых вариантах осуществления третья реакционная смесь находится при температуре от примерно -10 до примерно 25°C. В других вариантах осуществления третья реакционная смесь находится при температуре около 0°C.
Сходным образом, соединения, в которых R4 представляет собой C1-C3 алкокси, можно превратить в соединения, где R4 представляет собой Н. В некоторых вариантах осуществления данный способ также включает получение четвертой реакционной смеси, содержащей восстановитель и соединение формулы Ia, где R4 представляет собой C1-C3 алкокси, и где реакционная смесь практически не содержит магния, тем самым давая соединение формулы Ia, в котором R4 представляет собой Н. Например, магний может присутствовать в количестве меньше примерно 0,1, 0,05, 0,01, 0,005 или 0,001 эквивалентов относительно количества соединения формулы Ia. В некоторых вариантах осуществления реакция, практически не содержащая магния, может включать менее чем около 0,1 эквивалентов магния относительно количества соединения формулы Ia.
В способе по настоящему изобретению может использоваться любой подходящий восстановитель. Например, восстановители включают (но не ограничены только ими) триалкилсиланы, такие как триметилсилан и триэтилсилан. Другие восстановители известны квалифицированному специалисту в данной области, такие как описанные в книге ''Comprehensive Organic Transformations'', 1st edition, Richard С. Larock, VCH Publishers, New York, 1989.
Защитные группы Rb в формуле I в четвертой реакционной смеси могут быть удалены в ходе одной или разных стадий. В некоторых вариантах осуществления любые защитные группы Rb удаляются восстановителем в четвертой реакционной смеси.
Четвертая реакционная смесь может находиться при любой подходящей температуре. Подходящие температуры для четвертой реакционной смеси включают значения от примерно -50°C до примерно 0°C, или от примерно -40°C до примерно -10°C, или от примерно -30°C до примерно -20°C, или от примерно -25°C до примерно -22°C. Подходящие температуры для четвертой реакционной смеси также включают значения примерно -50°C, -45, -40, -35, -30, -25, -20, -15, -10, -5 и около 0°C. В некоторых вариантах осуществления четвертая реакционная смесь находится при температуре от примерно -40 до примерно -10°C. В других вариантах осуществления четвертая реакционная смесь находится при температуре от примерно -25 до примерно -22°C.
Способы получения соединений формулы Ia дают соединения формулы Ia с высокой степенью чистоты. Соединения формулы Ia, полученные способами по настоящему изобретению, можно получить с любой подходящей чистотой, включая (но не ограничиваясь только ими) значения выше примерно 80% чистоты, примерно 85, 90, 91, 92, 93, 94, 95, 96, 97, 98 или выше примерно 99% чистоты. Процент чистоты можно определить на основе веса продукта или процента площади под кривой в хроматограмме, такой как хроматограмма, полученная методом жидкостной хроматографии (ВЭЖХ) или газовой хроматографии (ГХ). Некоторые побочные продукты могут формироваться по способу согласно настоящему изобретению, и они присутствуют в количестве меньше около 10%, 5, 4, 3, 2 или около 1% от получаемого продукта.
Побочные продукты в способе по настоящему изобретению включают (но не ограничены только ими) побочный продукт А:
Побочный продукт А может включать следующие структуры:
и 
Дополнительные побочные продукты включают побочный продукт В:
Побочный продукт В может включать следующие структуры:
и 
Радикал R3 в побочных продуктах А и В может представлять собой описанные выше. Радикал R4 в побочном продукте В может представлять собой Н или OR4a, где R4a может представлять собой Н или C1-C3 алкил. В некоторых вариантах осуществления R4 может представлять собой Н, ОН или C1-C3 алкокси. В других вариантах осуществления R4 может представлять собой Н. В некоторых других вариантах осуществления R4 может представлять собой метокси. В других вариантах осуществления R4 может представлять собой ОН.
В некоторых вариантах осуществления в настоящем изобретении описана композиция, содержащая соединение формулы Ia, имеющее структуру:
в количестве, составляющем по меньшей мере 95% композиции. Композиция может также включать побочный продукт А, имеющий структуру:
в количестве, составляющем меньше примерно 1% композиции. Композиция может также включать побочный продукт В, имеющий структуру:
в количестве, составляющем меньше примерно 3% композиции. Данную композицию можно приготовить способами по настоящему изобретению. Соединения в данной композиции представляют собой соединения, где R3 может представлять собой атом водорода, галоген, гидрокси-группу, C1-C3 алкил, C1-C3 алкокси или C3-C6 циклоалкил, и по меньшей мере один из R2 и R3 может представлять собой C1-C3 алкил, C1-C3 алкокси, C3-C6 циклоалкил, (C1-C3 алкокси)C1-C3 алкил, (C1-C3 галогеналкокси)C1-C3 алкил, (C2-C4 алкенилокси)C1-C3 алкил, (C2-C4 алкинилокси)C1-C3 алкил, (C3-C6 циклоалкокси)C1-C3 алкил, C1-C3 гидроксиалкокси, C3-C6 циклоалкокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C1-C3 алкокси, (C3-C6 циклоалкил)C2-C4 алкенилокси или (C3-C6 циклоалкил)C2-C4 алкинилокси. Кроме того, R4 может представлять собой Н или OR4a, где R4a может представлять собой Н или C1-C3 алкил.
В некоторых вариантах осуществления в настоящем изобретении описана композиция, содержащая соединение формулы Ia, имеющее структуру:
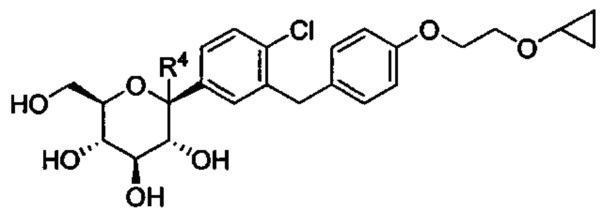
в количестве, составляющем по меньшей мере 95% композиции. Композиция может также включать побочный продукт А, имеющий структуру:
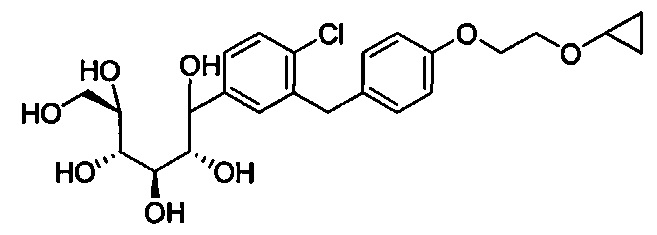
в количестве, составляющем меньше примерно 1% композиции. Композиция может также включать побочный продукт В, имеющий структуру:

в количестве, составляющем меньше примерно 3% композиции. Данную композицию можно приготовить способами по настоящему изобретению. В некоторых вариантах осуществления R4 может представлять собой Н, ОН или C1-C3 алкокси. В других вариантах осуществления R4 может представлять собой Н. В некоторых других вариантах осуществления R4 может представлять собой метокси. В других вариантах осуществления R4 может представлять собой ОН. Другие побочные продукты также могут формироваться согласно описанному способу. Например, в случае наличия, побочный продукт С может присутствовать в композиции в количестве, составляющем меньше примерно 1% композиции.
В некоторых вариантах осуществления в настоящем изобретении описана композиция, содержащая соединение формулы Ia, имеющее структуру:
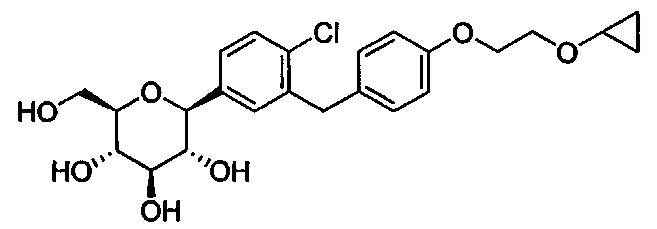
в количестве, составляющем по меньшей мере 95% композиции. Композиция может также включать побочный продукт А, имеющий структуру:
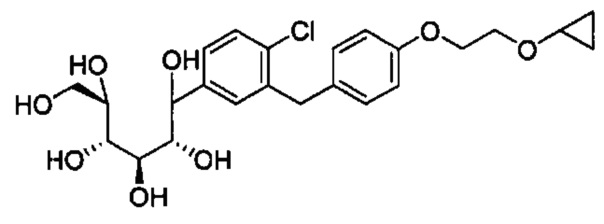
в количестве, составляющем меньше примерно 1% композиции. Композиция может также включать побочный продукт В, имеющий структуру:
в количестве, составляющем меньше примерно 3% композиции. Данную композицию можно приготовить способами по настоящему изобретению.
Композицию можно получить описанными выше способами. Например, описанный способ может включать получение первой реакционной смеси, содержащей соединение формулы II, алкил-магниевый комплекс, такой как C1-C4 алкилмагния хлорид, C1-C4 алкилмагния бромид, ди(C1-C4 алкил)магний, C3-C7 циклоалкилмагния хлорид, C3-C7 циклоалкилмагния бромид или ди(C3-C7 циклоалкил)магний, и первый органический растворитель, где соотношение алкил-магниевого комплекса к соединению формулы II меньше или равно 1,0 (моль/моль), и где первая реакционная смесь находится при температуре ниже примерно -50°C, давая промежуточное соединение. Данный способ может также включать получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы III. Таким образом можно получить соединение формулы I. Данный способ может также включать получение третьей реакционной смеси, содержащей C1-C3 алкилгидрокси, сильную кислоту и соединение формулы I, где R4 представляет собой ОН, и каждый Ra представляет собой Rb, давая соединение формулы I, где R4 представляет собой C1-C3 алкокси, и каждый Ra может независимо представлять собой Н или Rb. Данный способ может также включать получение четвертой реакционной смеси, содержащей восстановитель и соединение формулы Ia, где R4 представляет собой C1-C3 алкокси, и где реакционная смесь практически не содержащий магния, давая соединение формулы Ia, в котором R4 представляет собой Н.
Соединения формулы IIa
Соединения формулы IIa можно получить любым способом, известным квалифицированному специалисту в данной области. В некоторых вариантах осуществления соединение формулы IIa можно получить любыми из описанных ниже способов.
В некоторых вариантах осуществления в настоящем изобретении описан способ получения соединения формулы IIa, включающий получение первой реакционной смеси, содержащей описанное выше соединение формулы IV и описанное выше соединение формулы V, в условиях, подходящих для получения соединения формулы IIa.
Способ получения соединения формулы IIa может включать использование разных других соединений, известных квалифицированному специалисту в данной области, включая (но не ограничиваясь только ими) кислоту Льюиса и бромирующий агент. В некоторых вариантах осуществления первая реакционная смесь также включает кислоту Льюиса. В других вариантах осуществления кислота Льюиса может представлять собой BF3⋅Et2O, BCl3, BBr3, B(C6F5)3, SnCl4, I2, FeCl3, FeBr3, TMSOTf-AgClO4, AgOTf, Cu(OTf)2, Bi(OTf)3, In(OTf)3, Zn(NTf2)2, AuCl3, HgCl2, HgSO4, Hg(OCOCF3)2, PdCl2, Pd(OAc)2, ZnCl2, ZnBr2, ZnI2, триметилсилиловый эфир полифосфорной кислоты, AlCl3, AlBr3, AlI3, Al(OiPr)3, Al(OPh)3, TiCl4, TiCl2(OiPr)2, Ti(OiPr)4, PBr3, BeCl2, CdCl2, CeCl3, DyCl3, EuCl3, Eu(OTf)3, ErCl3, Er(OTf)3, GaCl3, GdCl3, Gd(OTf)3, HoCl3, LaCl3, La(OTf)3, LuCl3, Lu(OTf)3, Mg(ClO4)2, MgCl2, MgBr2, MgI2, NdCl3, Nd(OTf)3, PCl3, PBr3, PrCl3, Pr(OTf)3, PmCl3, Pm(OTf)3, Sc(OTf)3, SnCl4, SbCl5, SmCl3, Sm(OTf)3, Tf2O, TbCl3, Tb(OTf)3, TmCl3, Tm(OTf)3, YbCl3, Yb(OTf)3, ZrCl4 или Cp2ZrCl2. В некоторых других вариантах осуществления кислота Льюиса может представлять собой ZnCl2.
Бромирующие агенты, которые могут применяться в способах по настоящему изобретению, известны квалифицированному специалисту в данной области, и включают (но не ограничены только ими) газообразную бромистоводородную кислоту и Br2 (см. Tetrahedron Letters 52(17), 2235; и Tetrahedron 2007 63(41), 10185). В некоторых вариантах осуществления бромирующий агент представляет собой газообразную бромистоводородную кислоту.
В некоторых вариантах осуществления способ получения соединения формулы IIa включает получение первой реакционной смеси, содержащей соединение формулы IV, имеющее структуру:
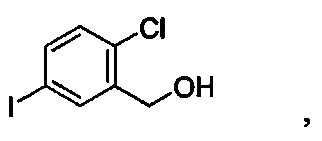
соединение формулы V, имеющее структуру:
газообразную бромистоводородную кислоту и ZnCl2, с получением соединения формулы IIa, имеющего структуру:
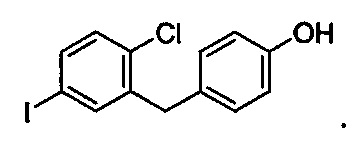
В некоторых вариантах осуществления способ получения соединения формулы IIa также включает получение второй реакционной смеси, содержащей соединение формулы IIa, где R3 представляет собой ОН, и описанное выше соединение формулы VI, формируя тем самым соединение формулы IIa, где R3 может представлять собой C1-C3 алкокси, C3-C6 циклоалкилокси, C3-C6 гетероциклоалкокси, (C1-C3 алкокси)C1-C3 алкокси, (C1-C3 галогеналкокси)C1-C3 алкокси, (C2-C4 алкенилокси)C1-C3 алкокси, (C2-C4 алкинилокси)C1-C3 алкокси, (C3-C6 циклоалкокси)C1-C3 алкокси, C1-C3 гидроксиалкокси, (C3-C6 гетероциклоалкокси)C1-C3 алкокси, (C3-C6 циклоалкил)C3-C4 алкенилокси или (C3-C6 циклоалкил)C3-C4 алкинилокси.
В некоторых вариантах осуществления способ получения соединения формулы IIa включает получение второй реакционной смеси, содержащей соединение формулы VI, имеющее структуру:
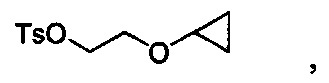
и соединение формулы IIa, имеющее структуру:
в условиях, пригодных для получения соединения формулы IIa, имеющего структуру:
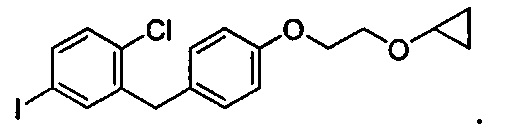
Схемы
На Фиг. 1 показано получение кристаллического 6c сочетанием глюконолактона 3 с арилиодидом 1 после магний-йодного обмена. К арилиодиду 1 добавляли комплекс изопропилмагния хлорид-хлорид лития при температуре ниже -50°C, и полученный арилмагний вводили в реакцию сочетания с персилилированным глюконолактоном 3, полученным из глюконолактона 2. Соединение 3 можно заранее обработать небольшими количествами комплекса изопропилмагния хлорид-хлорид лития для гарантии сухости вещества. После реакции сочетания, нагрева и обработки, полукеталь 4 (частично десилилированный) обрабатывали активированным углем с последующей обработкой соляной кислотой в смеси метанол/ТГФ, получая полностью десилилированный метилкеталь 5. Раствор метилкеталя 5 добавляли к силану и эфиратному комплексу фторида бора при температуре ниже -15°C, после обработки получая неочищенный продукт 6a. Полученный неочищенный продукт затем очищали сокристаллизацией с L-пролином в смеси этанол/вода/гексан/н-гептан, получая 7 в виде белого твердого вещества. В зависимости от остаточного количества примесей, более полярных чем 6a, дополнительной кристаллизацией в метаноле с разбавленным водным раствором гидроксида натрия получали чистый 6b. Если требуется более высокая чистота, перекристаллизацией из смеси метанол/вода с добавлением или без добавления затравочных кристаллов получали целевой финальный продукт 6с высокой чистоты.
На Фиг. 2А проиллюстрировано использование сочетания с арилмагнием для получения аналогов соединения 6. После получения соединения А на Фиг. 2A, в зависимости от применяемых защитных групп Rb, его можно превратить в описанный выше кеталь, применяя сильную кислоту и спирт (главным образом, для Rb=TMS), или его можно восстановить до соединения В, сохраняя защитные группы, или до конечного продукта, удаляя все защитные группы.
На Фиг. 2B проиллюстрировано получение O-спирокетальных соединений реакцией с продуктом сочетания, где R2 представляет собой CH2ORb, давая целевой продукт после обработки кислотой и удаления защитной группы (Lv, В., В. Xu, et al. Bioorganic & Medicinal Chemistry Letters 2009, 19(24), 6877-6881).
На Фиг. 2C проиллюстрировано получение C-спиро продукта из прекурсора A, где R2 представляет собой винильную группу и применяются восстановительные условия для замыкания цикла (Lv, В., Y. Feng, et al. ChemMedChem 2010, 5(6) 827-831).
На Фиг. 2D показано превращение продукта сочетания A (WO 2010023594) в C-5-спироциклический C-гликозид посредством манипуляций с защитными группами в целях селективного окисления первичного спирта и проведения в одном реакторе реакции альдольной конденсации-Канниццаро для добавления другого гидроксилметила в гликозид, с последующей внутримолекулярной циклизацией и снятием защиты, с получением спиро-соединения (Mascitti, V., R.P. Robinson, et al. Tetrahedron Letters 2010, 51(14), 1880-1883).
На Фиг. 3 изображена общая схема получения ингибиторов SGLT, содержащих в продукте гетероарильный цикл. Присоединение арилмагния к защищенному подходящим образом лактону с последующей кетализацией, восстановлением и снятием защиты, или кетализацией с сопутствующим восстановлением/снятием защиты, прямым восстановлением полукеталя и снятием защиты или прямым восстановлением полукеталя со снятием защиты дает целевой конечный продукт.
На Фиг. 4 проиллюстрирован синтез ингибиторов SGLT с использованием тиолактона. Процесс синтеза аналогичен описанному выше с применением надлежащим образом защищенного тиоглюконолактона (Kakinuma, Н., Т. Oi, et al. Journal of Medicinal Chemistry 2010, 53(8), 3247-3261). Радикал W может представлять собой CH или N, и радикал V может представлять собой NH, О или S, и таким образом формируются пиррольное, фурановое, тиофеновое, диазольное, оксазольное или тиазольное кольцо.
На Фиг. 5 показано получение SGLT ингибиторов из тригидрокси-6-(метокси)тетрагидро-2Н-пиран-2-она или тригидрокси-6-(метилтио)тетрагидро-2Н-пиран-2-она. На Фиг. 5A и 5B соответственно показано, как 2-(гидроксиметил)-6-метокситетрагидро-2Н-пиран-3,4,5-триол и 2-(гидроксиметил)-6-метилтиотетрагидро-2Н-пиран-3,4,5-триол были получены из L-глюкозы с помощью различных описанных в литературе способов (Bulletin de la Societe Chimique de France, 33, 469-471; 1905; Organic & Biomolecular Chemistry, 6(18), 3362-3365; 2008).
На Фиг. 5C показано, как оба пирантриола можно превратить в целевые лактоны путем иодирования первичного спирта, элиминирования и окислительного расщепления с получением целевых лактонов после соответствующей защиты (WO 2011058245).
На Фиг. 5D проиллюстрировано, как полученные лактоны можно ввести в реакцию сочетания с арилмагнием, получая целевые SGLT ингибиторы после кетализации, восстановления и снятия защиты.
На Фиг. 6 описано применение арилмагния для получения бифенилциклогексановых SGLT ингибиторов. На Фиг. 6A показано получение циклогексенового аналога, а на 6B показано, как можно снять защиту с полученного циклогексенового производного или далее окислить его посредством гидроборирования, и как полученный продукт можно далее окислить, получая циклогексаноны.
На Фиг. 7 показан общий синтез многих арилиодидных прекурсоров арилмагниевых соединений по настоящему изобретению. На Фиг. 7A показано, как некоторые диарилметаниодидные соединения можно получить из иодбензойной кислоты путем восстановления кислоты комбинацией боргидрида натрия и иода, с последующим цинк-опосредованным селективным сочетанием с надлежащим образом замещенным фенильным производным. На Фиг. 7B, когда R3 в исходном веществе представляет собой ОН, данное свободное фенольное соединение можно ввести в реакцию сочетания с подходящим алкилирующим агентом, получая целевой арилиодид.
На Фиг. 7C гетероциклические аналоги получают, сначала превращая кислоты в амид Вайнреба и затем вводя их в реакцию сочетания с надлежащим образом активированными гетероциклами. Полученные кетоны затем можно восстановить, получая дизамещенный метилен.
Примеры
Описанные далее примеры предложены в иллюстративных целях и не предназначены для ограничения объема настоящего изобретения каким-либо образом. Квалифицированным специалистам в данной области будут очевидны разные некритичные параметры, которые можно изменять или модифицировать, получая практически те же результаты.
Названия соединений, представленных в описанных далее примерах, были сгенерированы для показанных структур с использованием алгоритма CambridgeSoft Struct = Name, примененного в программе ChemDraw Ultra version 10.0. Если не указано иное, структуры синтезированных соединений в описанных ниже примерах были подтверждены посредством следующих методик:
(1) Если не указано иное, спектры газовой хроматографии/масс-спектрометрии с ионизацией методом электроспрея (MS ESI) получали на масс-спектрометре Agilent 5973N, оснащенном газовым хроматографом Agilent 6890 с колонкой НР-5 MS (0,25 мкм покрытие; 30 м×0,25 мм). Источник ионов имел температуру 230°C, и спектры получали сканированием в диапазоне 25-500 атомных единиц массы со скоростью 3,09 сек/скан. Газовую хроматографию (ГХ-0007) проводили на газовом хроматографе Shimadzu 2010 с колонкой DBM-5 MS (0,25 мкм покрытие; 30 м×0,25 мм). Температура инжектора 180°C, сплит 50:1; температура детектора 280°C; 40°C в течение 5 мин; градиент до 200°C в течение 12 мин; с использованием смеси водород/азот и воздуха.
(2) Если не указано иное, спектры методом высокоэффективной жидкостной хроматографии/масс-спектрометрии (LC-MS) получали на разделительном модуле Waters 2695, оснащенном детектором с фотодиодной матрицей Waters 2996, колонкой Waters XTerra (2,1×50 мм, 3,5 мкм) и детектором Waters Micromass ZQ с ионизацией методом электроспрея. Спектры сканировали в диапазоне 80-2000 атомных единиц массы с разным временем ионов согласно числу ионов в источнике. Элюентами служили A: 0,03%-ный раствор муравьиной кислоты в ацетонитриле, и B: 0,03%-ный раствор муравьиной кислоты в воде Milli-Q. Градиентное элюирование от 50 до 60% А в течение 0,5 минут при скорости потока 0,8 мл/мин, затем 4 мин градиента от 60 до 100% А, и в финале 100% А в течение 2 минут. Общее время 1 эксперимента 6,5 мин. Использовали также следующие условия (LCMS-0013): LC-MS (колонка Waters XTerra C18 3,5 мкм, 50×2,1 мм, 0,8 мл/мин, детектирование при 225 нм; градиент 10-95% растворителя А в течение 4,5 мин, удерживание 1,5 минуты при 95% А. Растворитель А: 0,03%-ный раствор муравьиной кислоты в ацетонитриле).
(3) Рутинную одномерную ЯМР спектроскопию проводили при частоте 400 МГц или 300 МГц на спектрометре Varian Mercury-Plus. Образцы растворяли в дейтерированных растворителях, полученных от Qingdao Tenglong Weibo Technology Co., Ltd., и переносили в 5 мм ID ЯМР ампулы. Спектры снимали при 293 K. Значения химсдвигов регистрировали в шкале м.д. относительно сигналов соответствующих растворителей, таких как 2,49 м.д. для ДМСО-d6, 1,93 м.д. для CD3CN, 3,30 м.д. для CD3OD, 5,32 м.д. для CD2Cl2 и 7,26 м.д. для CDCl3 в спектрах 1H.
(4) Высокоэффективную жидкостную хроматографию (ВЭЖХ-0001) проводили на модуле разделения Waters 2695, оснащенном УФ-детектором Waters 2487 с рабочей длиной волны 225 нм и колонкой Waters Sunfire C18 (5 мкм, 250 мм×4,6 мм). Элюирование в градиенте от 25 до 45% А в течение 5 минут при скорости потока 1,0 мл/мин, затем 15 минут в градиенте от 45 до 90% А, в финале 90% А в течение 10 минут. Применялись элюенты А: 99,95% ацетонитрил + 0,05% муравьиной кислоты, и В: Milli-Q вода + 0,05% муравьиной кислоты. Высокоэффективную жидкостную хроматографию (ВЭЖХ-0002) проводили на модуле разделения Waters 2695, оснащенном УФ-детектором Waters 2487 с рабочей длиной волны 225 нм и колонкой Waters Sunfire C18 (5 мкм, 250 мм×4,6 мм). Элюирование в градиенте от 50 до 100% А в течение 20 минут при скорости потока 1,0 мл/мин, затем 100% А в течение 19,5 минут. Применялись элюенты А: 99,95% ацетонитрил + 0,05% муравьиной кислоты, и В: Milli-Q вода + 0,05% муравьиной кислоты.
(5) Высокоэффективную жидкостную хроматографию (ВЭЖХ-0006) проводили на модуле разделения Waters 2695, оснащенном УФ-детектором Waters 2487 с рабочей длиной волны 280 нм и колонкой Zorbax SB-фенил (3,5 мкм, 150 мм×3 мм) при 50°C. Элюирование в градиенте от 25 до 50% А в течение 5 минут при скорости потока 0,8 мл/мин, затем 5 минут в градиенте от 50 до 90% А, затем 5 минут в градиенте до 100% А и в конце 100% А в течение 10 минут. Применялись элюенты А: 100% ацетонитрил и В: Milli-Q вода.
При использовании в настоящем тексте следующие сокращения и аббревиатуры имеют указанное значение: ACN, ацетонитрил; BF3⋅Et2O, эфират трифторида бора; Bu, бутил; вычисл., вычислено; CD3OD, метанол-d4; CDCl3, хлороформ-d; (COCl)2, оксалил хлорид; Cp2ZrCl2, бис(циклопентадиенил) циркония дихлорид; ДХМ, дихлорметан; DIBAL-H, диизобутилалюминий гидрид; ДМФА, N,N-диметилформамид; ДМСО, диметилсульфоксид; ЕА, этилацетат; экв, эквиваленты; ESI, ионизация методом электроспрея; Et, этил; ГХ, газовая хроматография; ч, часы; 1H ЯМР, протонный магнитный резонанс; ВЭЖХ, высокоэффективная жидкостная хроматография; IPC, контроль в ходе процесса; iPr, изопропил; LC-MS, жидкостная хроматография/масс-спектрометрия; Me, метил; МеОН, метанол; мин, минуты; кПа, килопаскали; MS, масс-спектрометрия; NMM, N-метилморфолин; OTf, трифторметансульфонат; РЕ, петролейный эфир; Ph, фенил; PMHS, полиметилгидросилоксан, Rf, коэффициент удерживания; насыщ., насыщенный; TBAI, тетрабутил аммония иодид; ТГФ, тетрагидрофуран; TIPS, триизопропилсилил; ТСХ, тонкослойная хроматография; TMS, триметилсилил.
Пример 1 Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола
В этом примере описано получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола с использованием избытка реактива Гриньяра и глюконолактоновой реакционной смеси, содержащей 0,04 экв. дополнительного реактива Гриньяра.
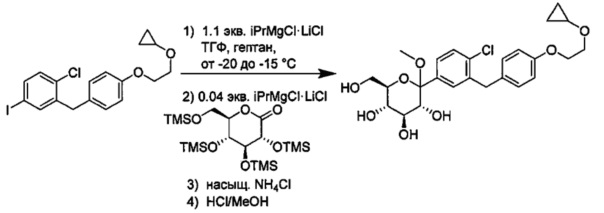
Раствор глюконолактона: В 500-литровый реактор со стеклянной внутренней поверхностью загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-он (11,4 кг) и н-гептан (12,2 кг), и смесь охлаждали до -15°C в токе азота 1 час. Прикапывали iPrMgCl⋅LiCl (0,5 кг, 1,3 М раствор в ТГФ) и полученную смесь перемешивали в течение 30 минут при -15°C.
Получение арилмагния: В 200-литровый реактор со стеклянной внутренней поверхностью оснащенный термометром, холодильником и напорным баком, загружали безводный ТГФ (15,3 кг), 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (7,5 кг). Полученную смесь перемешивали, продували азотом и охлаждали до -15°C. В полученный раствор прикапывали iPrMgCl⋅LiCl (14,55 кг, 1,3 М раствор в ТГФ) в течение 20 минут при температуре между -20 и -15°C. Полученную смесь перемешивали еще 1 час при температуре от -20 до -15°C.
Сочетание с арилмагнием: Охлажденный раствор глюконолактона прикапывали к арилмагнию в течение 100 минут при температуре между -20 и -15°C. После того как добавление было закончено, полученную смесь перемешивали в течение 5 час при температуре от -12 до -6°C.
Реакционную смесь медленно гасили насыщенным водным раствором хлорида аммония (45 кг) при -10°C, смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 7 час. Добавляли деионизованную воду (52,5 кг) и разделяли фазы. Водную фазу экстрагировали этилацетатом (3×49 кг), органические слои объединяли и промывали деионизованной водой (70 кг) и насыщенным раствором хлорида натрия (104 кг) перед осушкой над сульфатом натрия. Растворитель удаляли при пониженном давлении (~35°C, 10 кПа), добавляли метанол (15 кг), и смесь снова упаривали, получая масло.
Получение метилкеталя: Полученный остаток от упаривания растворяли в метаноле (56 кг) и тетрагидрофуране (22 кг). После охлаждения до температуры от -5°C до -10°C, прикапывали в реакционную смесь заранее охлажденный (0°C) раствор концентрированной соляной кислоты (1,74 кг), поддерживая температуру на уровне между -5 и 0°C. Полученную смесь нагревали до 12°C и перемешивали в течение 17 часов.
Реакционную смесь осторожно гасили добавлением воды (50 кг), экстрагировали петролейным эфиром (60-90°C, 15 кг) и удаляли органический слой. Водный слой осторожно нейтрализовали насыщенным водным раствором бикарбоната натрия (~28 кг). Летучие растворители удаляли при пониженном давлении (30°C, 10 кПа) в течение 1,5 час. Полученную смесь экстрагировали этилацетатом (3×64 кг). Объединенные органические слои промывали деионизованной водой (70 кг), насыщенным раствором хлорида натрия (70 кг) и деионизованной водой (70 кг), сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая неочищенный продукт.
Добавляли дихлорметан (12 кг), и смесь снова упаривали, получая неочищенный целевой продукт (7,35 кг, выход: 84,9%, >92% чистота согласно ВЭЖХ) в виде светло-желтого стекловидного твердого вещества.
LC-MS (LCMS-0013), 3,02 мин; ВЭЖХ-0001, 11,2 мин, 92% чистота. 1H ЯМР (400 МГц, CD3OD)δ=7,57 (д, J=2 Гц, 1H), 7,48 (дд, J=2, 8,4 Гц, 1Н), 7,38 (д, J=8,4 Гц, 1Н), 7,12 (д, J=8,8 Гц, 2Н), 6,84 (д, J=8,8 Гц, 2Н), 4,11 (д, J=15,2 Гц, 1Н), 4,07-4,06 (м, 2Н), 4,02 (д, J=15,2 Гц, 1Н), 3,95 (дд, J=2,0, 12 Гц, 1Н), 3,86-3,80 (м, 3Н), 3,78 (т, J=9,2 Гц, 1Н), 3,61 (ддд, J=2, 5,6, 10 Гц, 1H), 3,45 (т, J=10 Гц, 1Н), 3,43-3,39 (м, 1Н), 3,12 (д, J=9,6 Гц, 1Н), 3,09 (с, 3Н), 0,60-0,53 (м, 2Н), 0,52-0,45 м.д. (м, 2Н); MS (ESI, m/z) вычислено для C25H31ClO8: 494, найдено: 512 [М+NH4]+, 539 [М+НСОО]-.
Пример 2. Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола
В этом примере описано получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола с использованием менее чем одного эквивалента реактива Гриньяра, и глюконолактоновой реакционной смеси без добавления дополнительного реактива Гриньяра.
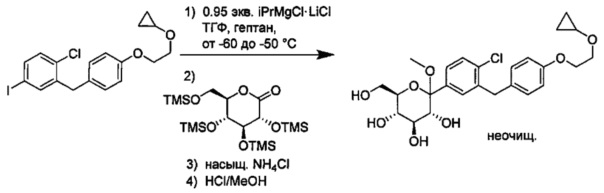
Раствор глюконолактона: В 5-литровый реактор со стеклянной внутренней поверхностью загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-он (1,52 кг) и н-гептан (1,63 кг), и перемешивали в течение 10 мин в токе азота. После продувки, смесь охлаждали до температуры от -30 до -20°C в атмосфере азота, перемешивали 30 минут и затем переносили в охлажденную капельную воронку.
Получение арилмагния: Трехгорлую колбу (10 л, стеклянный реактор), оснащенную термометром, магнитной мешалкой, холодильником и капельной воронкой, продували азотом и загружали в нее безводный ТГФ (1,67 кг) и 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (1,00 кг). После перемешивания и продувки азотом в течение 30 минут при температуре окружающей среды, смесь охлаждали до -60°C в атмосфере азота. В полученный раствор в течение 45 минут в атмосфере азота добавляли iPrMgCl⋅LiCl (1,76 кг, 0,95 экв.) через подходящую капельную воронку с такой скоростью, чтобы температура поддерживалась на уровне ниже -50°C. Полученную смесь перемешивали еще 30 минут при температуре от -60 до -50°C.
Сочетание с арилмагнием: Холодный раствор глюконолактона из охлажденной (-25°C) капельной воронки прикапывали к раствору арилмагния с такой скоростью, чтобы поддерживать температуру ниже -50°C в течение 35 минут. После того как добавление было закончено, смесь медленно нагревали до температуры от -15 до -10°C в течение одного часа и перемешивали в течение 4 часов.
Реакционную смесь медленно гасили продутым азотом (10 мин) насыщенным водным раствором хлорида аммония (5,6 кг) при температуре от -15 до 0°C через капельную воронку в течение 0,5 часа. Полученную смесь оставляли нагреваться до температуры 15°C в течение 2,5 часов и перемешивали в течение 6,5 часов. Отделяли верхний органический слой. Добавляли деионизованную воду (2,8 кг) к водному слою в реакторе с помощью капельной воронки. Водные фазы экстрагировали этилацетатом (3×3,78 кг). Органические слои объединяли и промывали деионизованной водой (4,65 кг) и насыщенным раствором хлорида натрия (16,7% вес/вес, 4,65 кг). Этилацетатный слой обрабатывали активированным углем (0,35 кг,) 1 час при 20°C, после чего фильтровали через фильтровальную бумагу. Органический слой упаривали при температуре 35°C в вакууме (~1 кПа), получая масло. Добавляли метанол (2 кг), и раствор снова упаривали при 35°C в вакууме (~1 кПа), получая масло.
Получение метилкеталя: Полученное масло растворяли в метаноле (7,47 кг) и тетрагидрофуране (2,89 кг) при механическом перемешивании (240 об/мин). Полученную смесь охлаждали до -10°C в течение 40 минут. Заранее охлажденный (0°C) раствор концентрированной соляной кислоты (0,256 кг) прикапывали в реакционную смесь, поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 20°C и перемешивали в течение 16 часов.
Реакционную смесь медленно гасили добавлением очищенной воды (2,32 кг), поддерживая температуру на уровне от 15 до 20°C. В полученную смесь загружали н-гептан (3,18 кг). После перемешивания в течение 30 минут (240 об/мин) и выдерживания в течение 15 мин, водный слой медленно гасили насыщенным водным раствором бикарбоната натрия (~3,8 кг) до слабоосновного уровня рН (рН около 8). Летучие органические вещества удаляли при пониженном давлении (~1 кПа) при температуре 30°C. Полученный остаток разбавляли очищенной водой (4,65 кг) и экстрагировали этилацетатом (3×4,2 кг). Объединенные органические слои промывали деионизованной водой (4,65 кг), насыщенным раствором хлорида натрия (4,65 кг) и деионизованной водой (4,65 кг). Органический слой упаривали в подходящем стеклянном реакторе в вакууме (~1 кПа) при температуре 30°C. Добавляли в реактор дихлорметан (1,6 кг) и снова упаривали (20-30°C, ~1 кПа) до полного удаления растворителя, получая целевой продукт (1,09 кг, выход: 94,8%, 89,7% чистота согласно ВЭЖХ - 0001) в виде светло-желтого стекловидного твердого вещества.
Пример 3. Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола
В этом примере описано получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола с использованием 0,95 экв. реактива Гриньяра с магний-иодным обменом при температуре от -60 до -50°C.
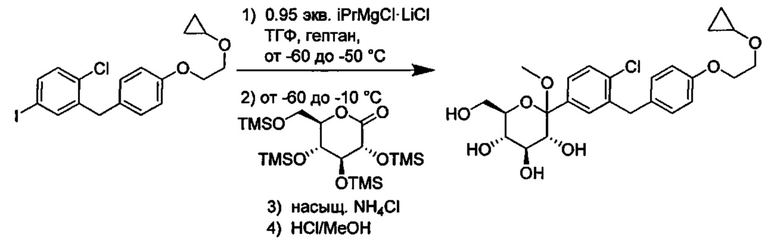
Раствор глюконолактона: В 10-литровый стеклянный реактор загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-он (4,58 кг) и н-гептан (4,89 кг), и смесь охлаждали до температуры от -30 до -20°C в токе азота 30 минут.
Получение арилмагния: В 50-литровый реактор со стеклянной внутренней поверхностью, оснащенный термометром, холодильником и напорным баком, загружали безводный ТГФ (5,2 кг), 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (3,0 кг). Полученную смесь перемешивали и продували азотом и охлаждали до -65°C. В полученный раствор прикапывали iPrMgCl⋅LiCl (5,3 кг, ~1,3 М раствор в ТГФ) с такой скоростью, чтобы поддерживать температуру ниже -50°C (~50 мин). iPrMgCl-LiCl был свежеоттитрован по методу Пакетта (Lin, H. - S. and L.A. Paquette, 1994, Synthetic Communication 24(17):2503-2506). Полученную смесь перемешивали еще 40 минут при температуре от -60 до -50°C.
Сочетание с арилмагнием: Охлажденный раствор глюконолактона прикапывали к арилмагнию в течение 1 часа при температуре ниже -50°C. После того как добавление было закончено, смесь медленно нагревали и перемешивали в течение 5 час при температуре от -15 до -10°C.
Реакционную смесь медленно гасили (~1 час) насыщенным водным раствором хлорида аммония (продували азотом 10 минут перед добавлением, 16,8 кг) при температуре от -15 до 0°C, смесь оставляли нагреваться до температуры 15°C (~2,5 часа) и перемешивали в течение 7 час. Добавляли деионизованную воду (8,4 кг) и разделяли фазы. Водную фазу экстрагировали этилацетатом (3×11,4 кг), органические слои объединяли и промывали деионизованной водой (14 кг) и насыщенным раствором хлорида натрия (14 кг).
Обработка активированным углем: Этилацетатный слой обрабатывали активированным углем (1,05 кг, СХ-700 от Zhuxi Со.) 1 час при 20°C, после чего фильтровали через фильтровальную бумагу. Осадок на фильтре промывали этилацетатом (2×1,5 кг). Растворитель удаляли при пониженном давлении (~35°C, 10 кПа), добавляли метанол (6 кг) и смесь снова упаривали, получая светло-желтое масло (6,31 кг).
Получение метилкеталя: Полученный остаток от упаривания растворяли в метаноле (22,4 кг) и тетрагидрофуране (8,7 кг). После охлаждения до -10°C, прикапывали в реакционную смесь заранее охлажденный (0°C) раствор концентрированной соляной кислоты (0,8 кг), поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 20°C и перемешивали в течение 17 часов.
Реакционную смесь осторожно гасили добавлением воды (7 кг), поддерживая температуру на уровне между 15 и 20°C. В полученную смесь загружали н-гептан (9,5 кг), перемешивали в течение 30 минут и удаляли органический слой. Водный слой осторожно нейтрализовали водной суспензией бикарбоната натрия (~1,7 кг бикарбоната натрия в 9,7 кг воды) до значения рН ~8. Летучие растворители удаляли при пониженном давлении (35°C, 10 кПа). Полученную смесь разбавляли водой (14 кг) и экстрагировали этилацетатом (3×12,6 кг). Объединенные органические слои промывали деионизованной водой (14 кг), насыщенным раствором хлорида натрия (14 кг) и деионизованной водой (14 кг), и органический слой упаривали на роторном испарителе в вакууме (10 кПа) при температуре 35°C до почти полной остановки конденсации растворителя в приемнике. Для подготовки к следующей стадии в реактор добавляли ацетонитрил (2 кг) и снова упаривали (20-30°C, 10 кПа) до почти полной остановки конденсации растворителя в приемнике, и повторяли стадии добавления и упаривания ацетонитрила, получая неочищенный продукт в виде светло-желтого стекловидного твердого вещества (2,73 кг, выход: 79,1%, 92,9% чистота согласно ВЭЖХ-0001). Полученный неочищенный продукт напрямую использовали в следующей стадии. LC-MS (LCMS-0013), 3,02 мин; ВЭЖХ-0001, 11,2 мин, 92,9% чистота.
Пример 4. Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола
В этом примере описано получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола с использованием 10 кг исходного вещества и 1,0 экв. реактива Гриньяра с магний-иодным обменом при температуре от -56 до -52°С.
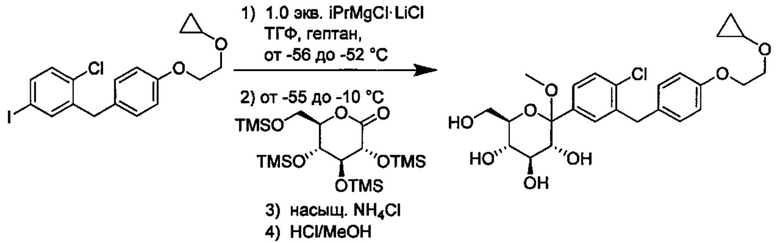
Раствор глюконолактона: Все операции, за исключением особо оговоренных случаев, осуществляли в атмосфере азота. (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-он (15,114 кг) загружали в 40-литровый криогенный реактор, добавляли смесь изомеров гептана (26 л, петролейный эфир фракция 90-100°C), и раствор охлаждали до -26°C.
Получение арилмагния: 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (10,14 кг) загружали в 200-литровый криогенный реактор. Затем добавляли ТГФ (19 л), и смесь перемешивали при 16°C до получения раствора (~15 мин). Раствор охлаждали до -58°C в течение 65 мин и прикапывали комплекс изопропилмагнийхлорида с хлоридом лития в ТГФ (3,40 кг) (в течение 50 мин), удерживая температуру на уровне между -52 и -56°C. iPrMgCl-LiCl был свежеоттитрован по методу Пакетта (Lin, H. - S. and L.A. Paquette, 1994, Synthetic Communication 24(17): 2503-2506). Аликвотную пробу анализировали методом ВЭЖХ (LCMS-0013), и полученную смесь перемешивали еще 10 мин, и новую аликвотную пробу анализировали методом ВЭЖХ (LCMS-0013) чтобы проверить, прошла ли реакция приемлемый критерий, при котором последовательные анализы должны давать отклонение ±5% друг от друга по общей площади пика. Во время анализа реакционную смесь продолжали перемешивать при той же температуре. Прикапывали комплекс изопропилмагнийхлорида с хлоридом лития (3,40 кг), удерживая температуру на уровне между -52 и -53°C в течение 20 минут. Анализировали аликвотную пробу (ВЭЖХ, LCMS-0013) чтобы выяснить, достигнут ли критерий конверсии исходного вещества ≥95 и <99%. Полученную смесь перемешивали еще 10 мин. Анализировали другую аликвотную пробу методом ВЭЖХ и обнаруживали соответствие указанному критерию. Раствор глюконолактона добавляли к раствору арилмагния через специальную линию для переноса в течение 50 минут при температуре от -50 до -55°C. 40-литровый реактор промывали гептаном (2,5 л) и добавляли гептановый смыв в 200-литровый реактор. Реакционную смесь оставляли нагреваться до температуры -10°C в течение ночи. Насыщенный водный раствор хлорида аммония (53 л) добавляли в полученную смесь в течение 40 минут, получая бежевую эмульсию/суспензию, поддерживая температуру на уровне от -10 до -5°C. Реакционную смесь оставляли нагреваться до температуры 20°C в течение ночи. Водную фазу разбавляли водой (27 л) и добавляли этилацетат (43 л). Органическую фазу отделяли, и водную фазу снова промывали этилацетатом (43 л). Органические фазы объединяли и промывали водой (45 л). Органическую фазу промывали насыщенным раствором хлорида натрия (45 л).
Обработка активированным углем: Этилацетатный раствор фильтровали через картридж с активированным углем и встроенным фильтром (картридж с активированным углем Zeta Carbon R55SP+встроенный фильтр 5,0/10 мкм). Объединенные фильтраты промывали этилацетатом (10 л). Полученный раствор упаривали при пониженном давлении. Добавляли метанол (58 л) и продолжали упаривание. Добавляли еще 54 л метанола и упаривали смесь.
Получение метилкеталя: Реактор продували азотом и устанавливали температуру внешней рубашки на уровне 20°C. Добавляли метанол (45 л), затем ТГФ (33 л). Смесь охлаждали до -6°C (заданное значение: -10°C) и добавляли концентрированную соляную кислоту (37%, 2,585 кг) в течение 19 мин, поддерживая температуру ниже -5°C. Полученную смесь оставляли перемешиваться при 20°C в течение ночи (13 час). Добавляли воду (24 л) в течение 15 минут при 15°C, в полученную желтую смесь добавляли гептан (47 л). Фазы разделяли и органическую фазу отбрасывали. Водный раствор бикарбоната натрия (7,4%, 37 л) добавляли в водную фазу до достижения рН=8. Реакционную смесь упаривали при пониженном давлении до удаления основного количества органических растворителей. Добавляли воду (47 л), затем этилацетат (30 л) и разделяли фазы. Значение рН водной фазы по-прежнему составляло 8. Водную фазу снова промывали этилацетатом (30 л). Объединенные органические экстракты промывали водой (47 л) и насыщенным раствором хлорида натрия (43 л). Органическую фазу упаривали при пониженном давлении. Добавляли ацетонитрил (2,65 л) и отгоняли растворитель, получая масло (10,928 кг), 88,4% чистота согласно ВЭЖХ.
Пример 5. Получение ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)Тетрагидро-2Н-Пиран-3,4,5-триола
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола восстановлением аномерного ОМе и/или ОН.
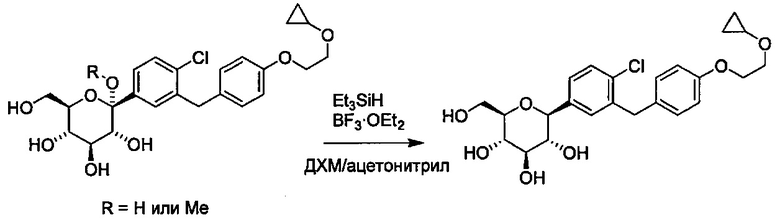
Раствор (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола: В 30-литровый стеклянный реактор, оснащенный термометром, загружали неочищенный (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (2,7 кг), ДХМ (5,4 кг) и ацетонитрил (3,2 кг), и смесь перемешивали на магнитной мешалке до полного растворения всех твердых веществ в токе азота.
Раствор триэтилсилана: BF3⋅Et2O (2,34 кг) добавляли к холодному (от -21 до -15°C) раствору триэтилсилана (2,55 кг), дихлорметана (5,4 кг) и ацетонитрила (3,2 кг) в атмосфере азота.
Раствор (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола добавляли к холодному раствору триэтилсилана с такой скоростью, чтобы температура находилась на уровне от -20 до -25°C (3 часа).
Реакционную смесь перемешивали еще 4 часа при температуре от -22 до -25°C и затем гасили добавлением водного раствора бикарбоната натрия (7,4% вес/вес, 18,3 кг) поддерживая температуру в реакционной среде ниже -10°C. Добавляли твердый бикарбонат натрия (1,35 кг), доводя значение рН до ~7,5. Растворители удаляли при пониженном давлении (температура ниже 40°C). Полученный остаток разделяли между этилацетатом (18 кг) и водой (9,2 кг). Слои разделяли, и водный слой экстрагировали этилацетатом (2×9 кг). Объединенные органические слои промывали насыщенным раствором хлорида натрия (2×9 кг) и удаляли растворители при пониженном давлении при температуре ниже 40°C до практически полного прекращения конденсации растворителя в приемнике. Добавляли безводный этанол (9 кг) и упаривали, получая неочищенное указанное в заголовке соединение (2,5 кг, 90% выход, 90,8% Чистота по ВЭЖХ, ВЭЖХ-0001) в виде твердой пены.
Пример 6. Получение ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)Тетрагидро-2Н-Пиран-3,4,5-триол
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола восстановлением аномерного ОМе и/или ОН.
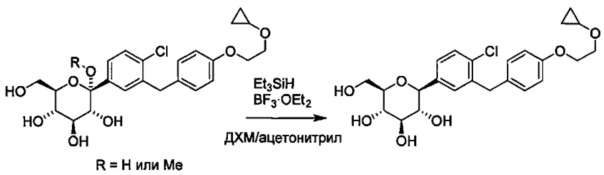
Раствор (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола: В 30-литровый стеклянный реактор, оснащенный термометром, загружали неочищенный (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триол (1,35 кг), ДХМ (2,7 кг) и ацетонитрил (1,6 кг), и смесь перемешивали на магнитной мешалке до полного растворения всех твердых веществ в токе азота.
Раствор триэтилсилана: BF3⋅Et2O (1,16 кг) добавляли к холодному (-25°C) раствору триэтилсилана (1,27 кг), дихлорметана (2,7 кг) и ацетонитрила (1,6 кг) в атмосфере азота, и температура в реакционной смеси поднималась до -14°C.
Раствор (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола добавляли к холодному раствору триэтилсилана с такой скоростью, чтобы температура находилась на уровне от -22 до -25°C (3 часа).
Реакционную смесь перемешивали еще 4 часа при температуре около -25°C и затем гасили добавлением водного раствора бикарбоната натрия (7,4% вес/вес, 9,2 кг), поддерживая температуру в реакционной среде ниже -10°C. Добавляли твердый бикарбонат натрия (0,67 кг), доводя значение рН до ~7,5. Растворители удаляли при пониженном давлении (температура ниже 40°C). Полученный остаток разделяли между этилацетатом (8,1 кг) и водой (4,6 кг). Слои разделяли, и водный слой экстрагировали этилацетатом (2×9 кг). Объединенные органические слои промывали насыщенным раствором хлорида натрия (2×4,5 кг), и растворители удаляли при пониженном давлении при температуре ниже 40°C до практически полного прекращения конденсации растворителя в приемнике. Добавляли безводный этанол (2×3,3 кг) и упаривали, получая неочищенное указанное в заголовке соединение (1,14 кг, 90% выход, 84,5% ВЭЖХ-0001) в виде не совсем белого твердого вещества.
Пример 7. Получение ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)Тетрагидро-2Н-Пиран-3,4,5-триола
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагадро-2Н-пиран-3,4,5-триола удалением аномерного ОН или ОМе.
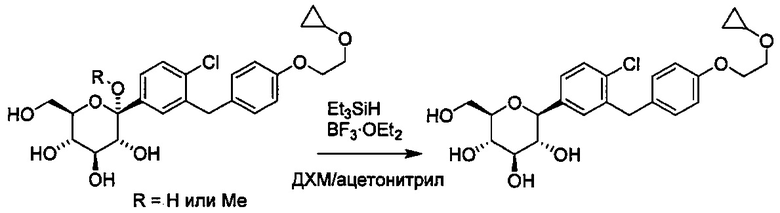
Раствор (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола: В 30-литровый стеклянный реактор, оснащенный термометром, загружали неочищенный (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагадро-2Н-пиран-3,4,5-триол (1,15 кг), ДХМ (2,3 кг) и ацетонитрил (1,4 кг), и смесь перемешивали на магнитной мешалке до полного растворения всех твердых веществ в токе азота. Раствор охлаждали до -15°C.
Раствор триэтилсилана: BF3⋅Et2O (1,2 кг) добавляли к холодному (от -20 до -15°C) раствору триэтилсилана (1,08 кг), дихлорметана (2,3 кг) и ацетонитрила (1,4 кг) в токе азота.
Холодный раствор (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола добавляли к холодному раствору триэтилсилана с такой скоростью, чтобы температура находилась на уровне от -20 до -15°C (~2-3 часа).
Реакционную смесь перемешивали еще 2-3 часа и затем гасили добавлением водного раствора бикарбоната натрия (7,4% вес/вес, 7,8 кг), и реакционную смесь перемешивали примерно 15 минут. Растворители удаляли при пониженном давлении (2 часа, температура ниже 40°C). Полученный остаток разделяли между этилацетатом (6,9 кг) и водой (3,9 кг). Слои разделяли, и водный слой экстрагировали этилацетатом (2×3,5 кг). Объединенные органические слои промывали насыщенным раствором хлорида натрия (2×3,8 кг), и растворители удаляли при пониженном давлении. Добавляли безводный этанол (2,3 кг) и упаривали, получая неочищенное указанное в заголовке соединение (1 кг, 90% выход, 90% ВЭЖХ-0001) в виде желтого твердого вещества.
Пример 8. Получение ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)Тетрагидро-2Н-Пиран-3,4,5-триола
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола восстановлением аномерного ОМе и/или ОН, с использованием примерно 22 кг исходного вещества. Все операции, за исключением особо оговоренных случаев, осуществляли в атмосфере азота.
Раствор (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола: Неочищенный (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триол (21,91 кг, [19,44 кг с корректировкой на чистоту]) растворяли в дихлорметане (32 л) и ацетонитриле (31 л) с помощью ротороного испарителя на 20 литров. Растворение достигалось с несколькими порциями смеси растворителей при температуре от 35 до 42°C. Слабо опалесцирующий раствор хранили в бочке до использования в реакции.
Раствор триэтилсилана: триэтилсилан (18,8 кг), дихлорметан (30 л) и ацетонитрил (30 л) помещали в криогенный реактор, и смесь охлаждали до -22°C в течение 1 часа. Добавляли комплекс трифторида бора с диэтиловым эфиром (17,01 кг) и промывали дихлорметаном (1 л) линии подачи и емкость для реагента.
Восстановление: Раствор исходного соединения (70 л) добавляли в охлажденную реакционную смесь при -24°C в течение 4 час 15 мин. Линии подачи и емкость для реагента промывали оставшимися 4 литрами смеси растворителей (1:1). Полученную смесь перемешивали в течение 3,5 часов при температуре -24°C, охлаждали до -29°C и перемешивали в течение ночи (12,5 часов). Смесь охлаждали до -39°C. Полученную смесь переносили в течение 35 минут через полиэтиленовую линию (15-20 м) в заранее охлажденный (0°C) раствор, состоящий из очищенной воды (120 л) и бикарбоната натрия (19,2 кг) в перемешиваемом 630-литровом реакторе. Криогенный реактор и линию промывали 12 литрами дихлорметана, который тоже добавляли в реакционную смесь. рН составлял 6-7 (цель: 7,5±0,5), поэтому добавляли бикарбонат натрия (3,0 кг), доводя рН до 7. Полученную смесь упаривали при пониженном давлении для удаления основного количества органических растворителей. Добавляли этилацетат (127 л) и еще воды (68 л), смесь экстрагировали, и ярко-желтую водную фазу снова экстрагировали этилацетатом (66 л). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (60 л). Оранжевый органический слой снова промывали насыщенным раствором хлорида натрия (60 л) и разделяли фазы. Органическую фазу упаривали при пониженном давлении, и остаток разбавляли этанолом (82 л) и упаривали при пониженном давлении. Добавляли еще этанол (82 л), упаривали до ~49 л и добавляли этанол (70 л) для подготовки к следующей стадии. Судя по анализу методом потери веса при сушке, в растворе присутствовало 19,98 кг продукта, и чистота по ВЭЖХ (ВЭЖХ-0001) составляла 89,4%.
Пример 9. Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол, бис(L-пролин) комплекса
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол, бис(L-пролин) комплекса совместной кристаллизацией ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола с L-пролином в смеси растворителей этанол/вода/н-гептан.

Неочищенный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (2,5 кг) добавляли в стеклянный реактор, содержащий этанол (95%, 16 кг) и L-пролин (1,24 кг), и смесь кипятили 1 час. Поддерживая температуру выше 60°C, добавляли н-гептан (8,5 кг) в течение 40 минут. Полученную смесь медленно охлаждали д температуры между 25 и 20°C и перемешивали при этой температуре 10 часов. Полученную смесь фильтровали и твердый осадок промывали холодным (-5°C) этанолом (95%, 2×2,5 л) и н-гептаном (2×5 л) и полученное твердое вещество сушили при пониженном давлении при температуре от 55 до 65°C в течение 20 часов, получая белое твердое вещество (3,03 кг, 81% выход, 99,4% чистота согласно ВЭЖХ-0001).
Пример 10. Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол, бис(L-пролин) комплекса
Все операции, за исключением особо оговоренных случаев, осуществляли в атмосфере азота. Неочищенный этанольный раствор из описанного выше Примера 8 (19,98 кг в 138,2 кг этанола) загружали в 630-литровый реактор. Примерно 21 л этанола отгоняли при 100°C и немного пониженном давлении. Добавляли воду (7 л), затем L-пролин (10,003 кг) и смесь кипятили (100°C) в течение 1 часа. Полученную смесь кипятили 0,5 часа, получая прозрачный раствор. Устанавливали температуру внешней рубашки 80°C. Добавляли гептан (102 л) в полученный раствор в течение 35 минут. Температура кипения смеси понижалась с 78°C до 70°C, и температуру внешней рубашки повышали до 90°C во время добавления, чтобы поддерживать кипение реакционной смеси. Часть раствора (550 мл) отбирали для выращивания затравочных кристаллов в лаборатории. В раствор добавляли затравочные кристаллы 25 мг пролинового комплекса и получали густую желтую суспензию. Кипящую смесь охлаждали до 50°C в течение 60 минут и вносили суспензию затравочных кристаллов, и полученную суспензию охлаждали до 20°C в течение ночи. Суспензию фильтровали 4 часа. Полученное твердое вещество вымывали из реактора с помощью 30 л маточного раствора. Полученное твердое вещество промывали дважды смесью этанол/вода (26 л/1 л и 26 л/1 л) и полученное твердое вещество дополнительно промывали гептаном (2×41 л). Чистота составляла 99,59% (ВЭЖХ-0001) и полученное твердое вещество сушили при пониженном давлении и 60°C в токе азота на фильтре-сушителе, получая 22,508 кг не совсем белого твердого вещества.
Пример 11. Получение кристаллов (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагадро-2Н-пиран-3,4,5-триола кристаллизацией ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол бис(L-пролин) комплекса в смеси растворителей метанол/вода.
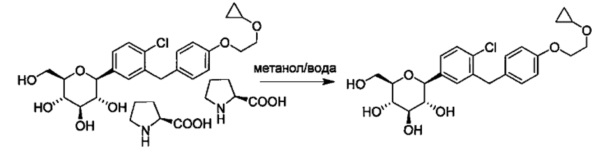
(2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетратидро-2H-пиран-3,4,5-триол (1,05 кг) помещали в полипропиленовую бочку (25 л), добавляли метанол (3,3 кг) и воду (1,05 кг), и смесь перемешивали до растворения всех твердых веществ. Полученный раствор фильтровали через мембранный фильтр (Millipore, 0,45 мкм) в чистый стеклянный реактор (20 л). Полученную смесь кипятили 30 минут и добавляли воду (4,83 кг) в течение 1,5 часа, поддерживая температуру на уровне между 50 и 65°C. Полученную смесь медленно охлаждали до ~20°C и перемешивали еще 5 часов. Отфильтровывали твердый осадок, суспендировали его на фильтре в воде и фильтровали (3×2,1 кг). Осадок на фильтре сушили при пониженном давлении 24 часа до момента, пока потери при сушке не оказывались ниже 0,5%, получая белое твердое вещество (620 г, 88,3% выход, 99,8% чистота согласно ВЭЖХ-0001).
Пример 12. Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола
В этом примере описано получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола с использованием 1,02 экв. реактива Гриньяра с магний-иодным обменом при температуре от -60 до -50°C с длительным инкубационным периодом.
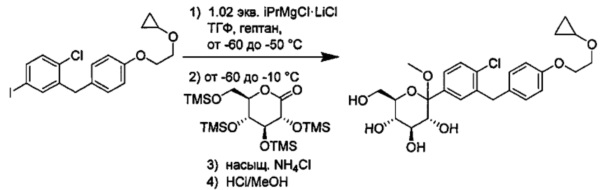
Раствор глюконолактона: В 5-литровый стеклянный реактор загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2H-пиран-2он(2,0 кг) и н-гептан (2,14 кг), и смесь охлаждали до температуры от -30 до -20°C в токе азота 30 минут.
Получение арилмагния: В 10-литровый стеклянный реактор, оснащенный термометром, холодильником и напорным баком, загружали безводный ТГФ (2,2 кг), 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (1,31 кг). Полученную смесь перемешивали и продували азотом и охлаждали до -65°C. В полученный раствор прикапывали iPrMgCl⋅LiCl (2,49 кг, ~1,3 М раствор в ТГФ) с такой скоростью, чтобы поддерживать температуру ниже -50°C (~45 мин). iPrMgCl-LiCl был свежеоттитрован по методу Пакетта (Lin, H. - S. and L.A. Paquette, 1994, Synthetic Communication 24(17): 2503-2506). Полученную смесь перемешивали еще 85 минут при температуре от -60 до -50°C.
Сочетание с арилмагнием: Охлажденный раствор глюконолактона прикапывали к арилмагнию в течение 40 минут при температуре ниже -50°C. После того как добавление было закончено, смесь медленно нагревали (1 час) и перемешивали в течение 5 часов при температуре от -15 до -10°C.
Реакционную смесь медленно гасили (~30 часов) насыщенным водным раствором хлорида аммония (продували азотом 10 минут перед добавлением, 7,3 кг) при температуре от -15 до 0°C, и смесь оставляли нагреваться до температуры 15°C (~2,5 часа) и перемешивали в течение 7 часов. Добавляли деионизованную воду (3,7 кг) и разделяли фазы. Водную фазу экстрагировали этилацетатом (3×4,95 кг), органические слои объединяли и промывали деионизованной водой (6,1 кг) и насыщенным раствором хлорида натрия (6,1 кг).
Обработка активированным углем: Этилацетатный слой обрабатывали активированным углем (0,46 кг, СХ-700 от Zhuxi Со.) 1 час при 20°C. после чего фильтровали через фильтровальную бумагу. Осадок на фильтре промывали этилацетатом (0,65 кг). Растворитель удаляли при пониженном давлении (~35°C, 16 кПа), добавляли метанол (2×2,6 кг), и смесь снова упаривали, получая светло-желтое масло.
Получение метилкеталя: Полученный остаток от упаривания растворяли в метаноле (9,8 кг) и тетрагидрофуране (3,8 кг). После охлаждения до -10°C, заранее охлажденный (0°C) раствор концентрированной соляной кислоты (0,34 кг) прикапывали в реакционную смесь, поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 20°C и перемешивали в течение 18 часов.
Реакционную смесь осторожно гасили добавлением воды (3 кг), поддерживая температуру на уровне между 15 и 20°C. В полученную смесь загружали н-гептан (4,2 кг), перемешивали в течение 30 минут и удаляли органический слой. Водный слой осторожно нейтрализовали водной суспензией бикарбоната натрия (~0,65 кг бикарбоната натрия в 3,1 кг воды) до рН ~8. Летучие растворители удаляли при пониженном давлении (38°C, 15 кПа). Полученную смесь разбавляли водой (6 кг) и экстрагировали этилацетатом (3×4,7 кг). Объединенные органические слои промывали деионизованной водой (6 кг), насыщенным раствором хлорида натрия (6 кг) и деионизованной водой (6 кг), и органический слой упаривали на роторном испарителе в вакууме (5 кПа) при температуре 35°C до почти полной остановки конденсации растворителя в приемнике. Для подготовки к следующей стадии в реактор добавляли ацетонитрил (0,9 кг) и снова упаривали (20-30°C, 5 кПа) до почти полной остановки конденсации растворителя в приемнике, и повторяли добавление ацетонитрила и упаривание, получая неочищенный продукт в виде светло-желтого стекловидного твердого вещества (1,35 кг, выход: 89,4%, 86,6% чистота согласно ВЭЖХ-0001). Полученный неочищенный продукт напрямую использовали в следующей стадии.
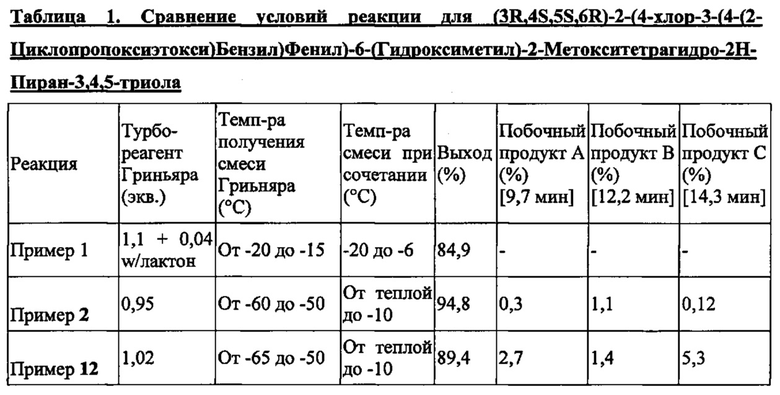
Пример 13. Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол, бис(L-пролин) комплекса
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагадро-2Н-пиран-3,4,5-триол, бис(L-пролин) комплекса совместной кристаллизацией ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола с L-пролином в смеси растворителей этанол/вода/н-гептан.
Неочищенный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (1,09 кг) добавляли в стеклянный реактор, содержащий этанол (95%, 7 кг) и L-пролин (0,54 кг), и смесь кипятили 1 час. Поддерживая температуру на уровне между 55 и 60°C, добавляли н-гептан (3,7 кг) в течение 1,5 часов. Полученную смесь перемешивали в течение 2 час при температуре 60-70°C и медленно охлаждали (в течение 12 часов, ~10°C/час) до -5°C и перемешивали при этой температуре 5 часов. Полученную смесь фильтровали, и твердый осадок промывали холодным (-5°C) этанолом (95%, 2×0,9 кг) и н-гептаном (2×1,5 кг), и полученное твердое вещество сушили при пониженном давлении при температуре от 55 до 65°C 20 часов, получая белое твердое вещество (1,34 кг, 82% выход, 98,2% чистота согласно ВЭЖХ-0001).
Пример 14. Получение кристаллов (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола кристаллизацией ((2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол бис(L-пролин) комплекса в смеси растворителей метанол/вода.
(2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (1,3 кг) добавляли в полипропиленовую бочку (25 л), добавляли метанол (3,6 кг) и воду (1,3 кг), и смесь перемешивали до растворения всех твердых соединений. Полученный раствор фильтровали через мембранный фильтр (Millipore, 0,45 мкм) в чистый стеклянный реактор (50 л). Полученную смесь кипятили 30 минут и добавляли воду (7,2 кг) в течение 1,0 часа, поддерживая температуру на уровне между 50 и 65°C. Полученную смесь медленно охлаждали до ~42°C в течение 2 часов. Добавляли суспензию затравочных кристаллов (26 г) в холодной (-5°C) смеси метанол/вода (78 мл, 2,8/6,5 (вес/вес) и продолжали медленное охлаждение до -5°C в течение 12 часов. Суспензию перемешивали еще 5 часов и фильтровали. Твердый осадок суспендировали в холодной воде и фильтровали (0-5°C, 3×2,6 кг). Осадок на фильтре сушили при пониженном давлении 24 часа до момента, пока потери при сушке не оказывались ниже 0,5%, получая белое твердое вещество (825 г, 92% выход, 99,3% чистота согласно ВЭЖХ-0001).
Пример 15. Получение 4-(2-хлор-5-иодбензил)фенола
В данном примере описано получение 4-(2-хлор-5-иодбензил)фенола с использованием газообразной бромистоводородной кислоты.
Получение (2-хлор-5-иодфенил)метан-1-ола.
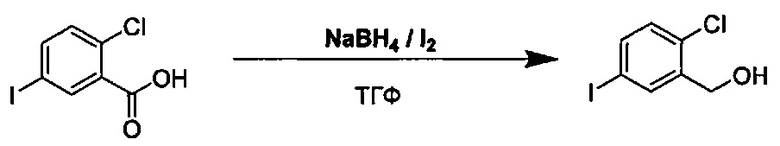
В 250-миллилитровую 4-горлую колбу, оснащенную термометром и механической мешалкой, загружали NaBH4 (4,16 г, 0,11 моль) и ТГФ (60 мл) в атмосфере аргона. После охлаждения до 0-5°C при перемешивании, медленно прикапывали раствор иода в ТГФ (12,7 г I2 в 25 мл ТГФ) в течение 30 минут и поддерживали температуру реакции ниже 10°C. После того как добавление было закончено, раствор 2-хлор-5-иодбензойной кислоты (15,0 г, 50 ммоль) в ТГФ (20 мл) прикапывали в течение 30 минут и поддерживали температуру реакции ниже 10°C. После перемешивания в течение еще 3 часов при температуре 20~25°C, реакционную смесь кипятили еще 16 часов и мониторили методом ТСХ (петролейный эфир/этилацетат = 1:1, Rf=0,2). Смесь охлаждали до 20~25°C и выливали в ледяную воду (100 мл), экстрагировали этилацетатом (2×100 мл), промывали водой (2×100 мл), насыщенным раствором хлорида натрия (100 мл), упаривали и остаток очищали методом флэш-хроматографии (элюент петролейный эфир/этилацетат = 20:1, 200 мл) получая не совсем белое твердое вещество. Выход: 10,0 г (70%) MS ESI (m/z): 269 [M+l]+.
Получение 4-(2-хлор-5-иодбензил)фенола
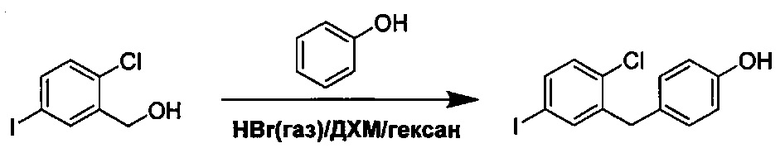
В 100-миллилитровую 4-горлую колбу, оснащенную термометром и механической мешалкой, загружали (2-хлор-5-иодфенил)метанол (268,5 мг, 1 ммоль), безводный ZnCl2 (136,3 мг, 1 ммоль), дихлорметан (5,0 мл) и н-гексан (29 мл) в атмосфере аргона. После перемешивания в течение 10 минут при 20-25°C, HBr (газ) барботировали через смесь 10 минут, и прикапывали раствор фенола (197,6 мг, 2,1 ммоль) в сухом дихлорметане (3,0 мл) в течение 30 минут. После барботирования HBr в течение еще 2 часов, смесь кипятили 3 дня. Конверсия составила около 65%. Полученную смесь гасили ледяной водой (50 мл), экстрагировали этилацетатом (2×30 мл), промывали водой (2×30 мл), насыщенным раствором хлорида натрия (30 мл), упаривали и очищали остаток методом флэш-хроматографии (элюент: петролейный эфир/этилацетат = 25:1, 200 мл), получая не совсем белое твердое вещество. Выход: 180 мг (52%). 1H ЯМР (CDCl3, 400 МГц):δ 7,44 (д, J=8,4 Гц, 2Н), 7,03~7,09 (м, 3Н), 6,77 (д, J=8,4 Гц, 2Н), 4,76 (с, 1Н), 3,95 (с, 2Н), 3,82 (с, 2Н). MS ESI (m/z): 345 [М+1]+. 13C ЯМР (CDCl3, 100 МГц):δ 154,1, 141,4, 139,5, 136,6, 134,2, 131,2, 130,9, 130,1, 115,5, 91,67, 38,07.
Пример 16. Получение 2-(4-(2-Циклопропоксиэтокси)Бензил)-1-хлор-4-иодбензола
В этом примере описано получение 2-(4-(2-циклопропоксиэтокси)бензил)-1-хлор-4-иодбензола сочетанием 4-(2-хлор-5-иодбензил)фенола с 2-циклопропоксиэтил 4-метилбензолсульфонатом.
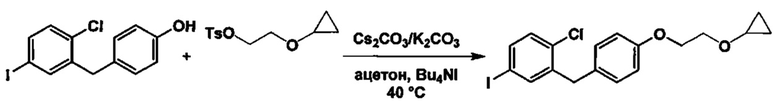
В атмосфере азота в 500-литровый реактор со стеклянной внутренней поверхностью загружали ацетон (123 кг) при перемешивании (120 об/мин), 4-(2-хлор-5-иодбензил)фенол (19,37 кг, 0,056 кмоль), 2-циклопропоксиэтил 4-метилбензолсульфонат (15,85 кг, 0,062 кмоль), порошок карбоната цезия (18,31 кг, 0,0562 кмоль), порошок карбоната калия (23,3 кг, 0,169 кмоль) и TBAI (4,15 кг, 0,011 кмоль). После перемешивания в течение 40~45 час при температуре 40°C, ТСХ (петролейный эфир/этилацетат = 4:1, Rf=0,3) показала, что исходное вещество израсходовано. Смесь охлаждали до 20~25°C.
Реакционную смесь фильтровали через диатомовую землю (28 кг) и осадок на фильтре промывали ацетоном (2×31 кг). Объединенные фильтраты переносили в 500-литровый реактор со стеклянной внутренней поверхностью и упаривали. Полученный остаток растворяли в этилацетате (175 кг), промывали водой (2×97 кг) и упаривали до объема около 100 литров, переносили в 200-литровый реактор со стеклянной внутренней поверхностью и продолжали упаривать, получая около 22,5 кг неочищенного продукта.
Неочищенный продукт растворяли в смеси метанол/н-гексан (10:1, 110 кг) при кипячении в течение 30 минут при перемешивании (100 об/мин) до получения прозрачного раствора. Смесь охлаждали до 5-10°C и добавляли затравочные кристаллы (20 г). Суспензию перемешивали еще 5 часов при температуре 5-10°C. Полученную смесь фильтровали при 0-5°C. и осадок на фильтре промывали заранее охлажденной смесью метанол/н-гексан (10:1, 5°C, 2×11 кг). Осадок на фильтре сушили при 15-20°C 15 часов, получая твердый продукт от не совсем белого до белого цвета. Выход: 18,1 кг, 75%. Температура плавления: 31°C (ДСК). 1H ЯМР (CDCl3, 400 МГц):δ 7,45~7,50 (м, 2Н), 7,09~7,12 (м, 3Н), 6,88 (д, J=8,8 Гц, 2Н), 4,11 (т, J=5,2 Гц, 2Н), 3,99 (с, 2Н), 3,88 (т, J=5,2 Гц, 2Н), 3,40~3,44 (м, 1Н), 0,63~0,67 (м, 2Н), 0,49~0,54 (м, 1Н). MS ESI (m/z):429 [M+1]+. 13C ЯМР (CDCl3, 100 МГц): δ 157,5, 141,5, 139,5, 136,6, 134,2, 131,2, 130,8, 129,9, 114,9, 91,66, 69,00, 67,13, 53,72, 38,08, 5,63.
Аналогичные методики применяли для получения следующих соединений вместо 2-(4-(2-циклопропоксиэтокси)бензил)-1-хлор-4-иодбензола:
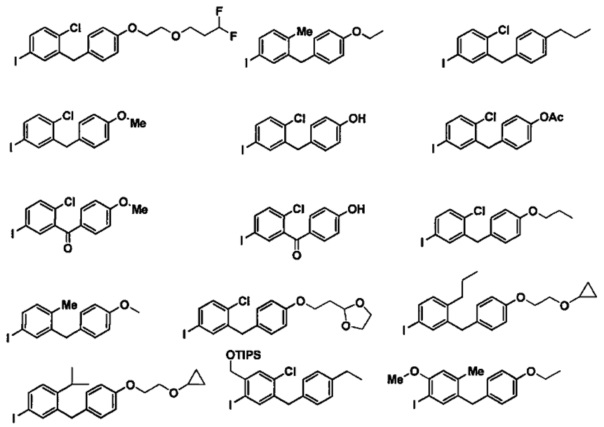
Пример 17. Получение 2-(4-Метоксибензил)-1-хлор-4-иодбензола
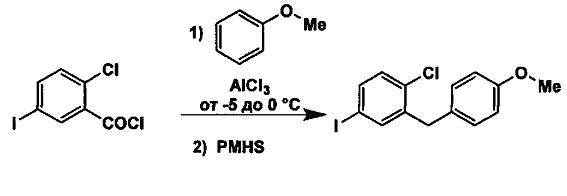
В 250-миллилитровую 4-горлую колбу, оснащенную внутренним термометром и холодильником, помещали анизол (5,7 г, 52,0 ммоль) и дихлорметан (17 мл), и смесь охлаждали до -3°C. Добавляли в полученный раствор хлорид алюминия (III) (7,4 г, 55,0 ммоль) в течение 1 часа поддерживая внутреннюю температуру ниже 5°C. После того как добавление было закончено, полученную смесь перемешивали в течение 30 минут при 0~5°C, и прикапывали раствор 2-хлор-5-иодбензоил хлорида (15,0 г, 0,05 моль) в дихлорметане (15 мл) в течение 1 часа, поддерживая внутреннюю температуру ниже 5°C. Полученную смесь перемешивали еще 1 час при 0~5°C и нагревали до 10~15°C. Прикапывали PMHS (15,0 г, 0,25 моль), поддерживая внутреннюю температуру ниже 25°C. После перемешивания в течение 10 часов при 25°C, добавляли в полученную смесь еще PMHS (9,0 г, 0,15 моль). После перемешивания в течение еще 16 часов при 30°C, смесь охлаждали до 5~10°C и медленно прикапывали ледяную воду (100 мл) в течение 1 часа при перемешивании. Замечание: При добавлении первой порции воды имел место сильный разогрев. Полученную смесь фильтровали, и осадок на фильтре суспендировали в дихлорметане (100 мл), содержащем диатомовую землю (30 г). Полученную смесь фильтровали, и осадок на фильтре промывали дихлорметаном (2×50 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл). После удаления летучих компонентов, остаток перекристаллизовывали из абсолютного этанола (58 мл), получая 12,0 г 1-хлор-4-иод-2-(4-метоксибензил)бензола в виде белого твердого вещества (выход, 67%, ВЭЖХ-0002: 98,7%). Замечание: Чистоту можно повысить, проводя вторую перекристаллизацию 1-хлор-4-иод-2-(4-метоксибензил)бензола, чистота по ВЭЖХ может составить до 99,5% при выходе 75~80%. 1H ЯМР (CDCl3, 400 МГц): δ 7,50 (д, J=8,4 Гц, 2Н), 7,10~7,13 (м, 3Н), 6,88 (д, 7=8,4 Гц, 2Н), 4,00 (с, 2Н), 3,82 (с, 3Н). MS ESI (m/z): 357 [М+1]+. 13C ЯМР (CDCl3, 100 МГц): δ 158,3, 141,5, 139,5, 136,6, 134,2, 131,2, 130,6, 129,9, 114,1, 91,71, 55,29, 38,09.
Пример 18. Крупномасштабное получение 4-(2-хлор-5-иодбензил)фенола
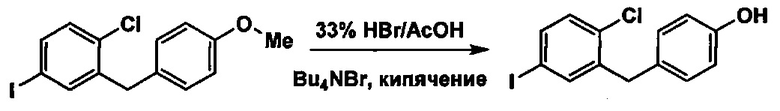
В 500-литровый реактор со стеклянной внутренней поверхностью, оснащенный уловителем кислых газов на основе гидроксида натрия, загружали 33% вес/вес раствор бромоводорода в уксусной кислоте (120 кг, 4,8 вес/вес) и 2-(4-метоксибензил)-1-хлор-4-иодбензол (25,0 кг, 69,7 моль), добавляли тетра(н-бутил)аммония бромид (1,92 кг, 6,9 моль), и смесь кипятили 10 часов. Добавляли еще раствор бромоводорода в уксусной кислоте (60 кг, 2,4 вес/вес), кипятили еще 7 часов и мониторили методом ТСХ (петролейный эфир/этилацетат = 10:1, Rf=0,8). Когда реакция заканчивалась, смесь охлаждали до 60°C и добавляли воду (60,8 кг). Чтобы гидролизовать весь 4-(2-хлор-5-иодбензил)фенил ацетат, смесь кипятили 8-10 часов и мониторили методом ТСХ (петролейный эфир/этилацетат = 10:1, Rf=0,8) или ВЭЖХ. Смесь охлаждали до 20-30°C. В другой 1000-литровый реактор со стеклянной внутренней поверхностью загружали воду (560 кг) и охлаждали до 0-5°C. Полученную в 500-литровом реакторе смесь медленно переносили в 1000-литровый реактор в течение 1 часа. После перемешивания в течение 1 часа при 10-20°C, смесь фильтровали и осадок на фильтре суспендировали в воде (175 кг) и петролейном эфире (50 кг). Полученное твердое вещество сушили при 50-55°C 8 часов, получая 21,1 кг продукта в виде не совсем белого твердого вещества. Полученный твердый продукт помещали в 500-литровый реактор со стеклянной внутренней поверхностью, содержащий этилацетат (9,6 кг) и петролейный эфир (19,1 кг). После кипячения в течение 30 минут при механическом перемешивании (100 об/мин), прикапывали петролейный эфир (81,4 кг) в 200-литровом полипропиленовом сосуде в течение 2 часов, полученную смесь перемешивали еще 1 час при температуре 45-50°C, и смесь охлаждали до 10-15°C и перемешивали еще 8 часов. Полученную смесь фильтровали, и осадок на фильтре промывали заранее охлажденной (0-5°C) смесью петролейный эфир/этилацетат (20:1, 2×22,6 кг), сушили под вакуумом при 50-55°C 8 часов, получая 18,2 кг (выход, 76%, Чистота по ВЭЖХ, ВЭЖХ-0002: 99,8%). 1H ЯМР (CDCl3, 400 МГц): δ 7,44 (д, J=8,4 Гц, 2Н), 7,03~7,09 (м, 3Н), 6,77 (д, J=8,4 Гц, 2Н), 4,76 (с, 1Н), 3,95 (с, 2H), 3,82 (с, 2Н). MS ESI (m/z): 345 [М+1]+. 13C ЯМР (CDCl3, 100 МГц): δ 154,1, 141,4, 139,5, 136,6, 134,2, 131,2, 130,9, 130,1, 115,5, 91,67, 38,07.
Пример 19. Получение (3R,4S,5R,6R)-3,4,5-трис(Триметилсилилокси)-6-((Триметилсилилокси)Метил)Тетрагидро-2Н-пиран-2-она
В данном примере описано получение защищенного глюконолактона.
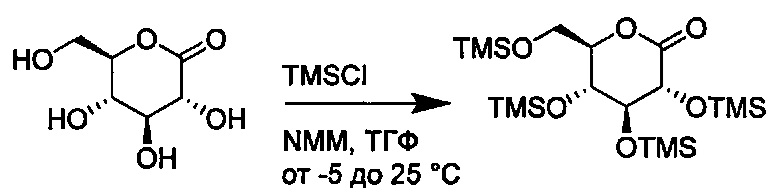
К перемешиваемому холодному (-5°C) раствору глюконолактона (10 кг, 56,2 моль) и N-метилморфолина (45,4 кг, 449,6 моль) в 93 кг ТГФ (безводный; KF<0,01%) в атмосфере азота добавляли триметилсилил хлорид (36,4 кг, 337,2 моль) через капельную воронку с такой скоростью, чтобы температура не превышала 5°C. После того как добавление было закончено, реакционную смесь медленно нагревали до 20~25°C и перемешивали смесь в течение ночи (17 час).
Смесь охлаждали до температуры 0-5°C и разбавляли 130 кг толуола, после чего осторожно добавляли 300 кг воды с такой скоростью, чтобы температура не превышала 10°C (2,7 часа). После перемешивания, фазы оставляли для разделения, и органическую фазу промывали насыщенным водным раствором дигидрофосфата натрия (132 кг), водой (45 кг) и насыщенным раствором хлорида натрия (45 кг). Органический слой упаривали в вакууме (~1 кПа), удерживая температуру ниже 35°C, получая целевой продукт (24,7 кг, 94,1 выход, 97,4% ГХ-0007, ГXMS (m/z): 466). Содержание воды ~80 м.д. по методу Карла Фишера. 1H ЯМР (CDCl3, 400 МГц): δ 4,14 (дт, J=2,4, 7,6 Гц, 1Н), 3,97 (д, J=8,0 Гц, 1Н), 3,88 (т, J=7,6, Гц, 1Н), 3,80~3,72 (м, 2Н), 3,72 (т, J=7,6, Гц, 1Н), 0,17 (с, 9Н), 0,15 (с, 9Н), 0,13 (с, 9Н), 0,10 (с, 9Н).
Альтернативная методика с циклогексаном в роли азеотропного осушающего растворителя. В перемешиваемый холодный (-5°C) раствор глюконолактона (17,8 г, 0,1 моль) и N-метилморфолина (88 мл, 0,8 моль) в 180 мл ТГФ (безводный; KF<0,01%) в атмосфере аргона добавляли триметилсилил хлорид (76 мл, 0,6 моль) через капельную воронку с такой скоростью, чтобы температура не превышала 5°C. После того как добавление было закончено, реакционную смесь медленно нагревали до 20-25°C и перемешивали смесь в течение ночи (17 час).
После разбавления циклогексаном (270 мл), смесь охлаждали до температуры 0-5°C, после чего осторожно добавляли воду (530 мл) с такой скоростью, чтобы температура не превышала 10°C. После перемешивания фазы оставляли для разделения, и органическую фазу промывали насыщенным водным раствором дигидрофосфата натрия (150 мл), водой (80 мл), насыщенным раствором хлорида натрия (80 мл) и деионизованной водой (100×2 мл). Органический слой упаривали в вакууме с помощью роторного испарителя с баней, удерживая температуру ниже 30°C, полученное светло-желтое масло дважды растворяли в 100 мл циклогексана, снова упаривали, получая 50 г указанного в заголовке соединения в виде светло-желтого масла (выход: количественный, чистота по ГХ, ГХ-0007: 92,4%).
Пример 20. Получение (3R,4S,5R,6R)-3,4,5-трис(Триметилсилилокси)-6-((Триметилсилилокси)Метил)Тетрагидро-2Н-пиран-2-она с гептаном
Все операции, за исключением особо оговоренных случаев, осуществляли в атмосфере азота. Устройство для улавливания конденсата, заправленное водой, присоединяли к газоотводному выходу из реактора и запускали. Глюконолактон (8,73 кг) загружали в 630-литровый реактор, после чего в суспензию загружали ТГФ (72 л) и N-метилморфолин (36 л) и промывали соединительные линии тетрагидрофураном (1 л). Смесь охлаждали до -5°C в течение 45 минут. Хлортриметилсилан (23,52 кг) загружали в танк для подачи и промывали соединительные линии частью тетрагидрофурана (~2 л), который добавляли в хлортриметилсилан. Полученную смесь добавляли в суспензию глюконолактона в течение 23 минут при температуре от -1 до -5°C. Танк для подачи промывали оставшимся количеством тетрагидрофурана (~2 л), которое добавляли в реакционную смесь, и полученную суспензию нагревали до 19°C в течение 1,5 часов. Реакционную смесь еще перемешивали при той же температуре 18,5 часов. Суспензию охлаждали до -7°C и добавляли гептан (петролейный эфир фракция 90-100°C, 132 л). Добавляли воду (208 л) в полученную смесь (экзотермично!), начиная при -10°C, в течение 70 минут, поддерживая температуру ниже 10°C. Полученную смесь еще перемешивали в течение 10 минут при температуре рубашки 20°C и разделяли фазы. Органическую фазу промывали водой (37 л) и насыщенным раствором хлорида натрия (31 л). Органическую фазу упаривали в реакторе при пониженном давлении при температуре рубашки 45°C. Полученное масло (22,108 кг) использовали в следующей стадии.
Пример 21. Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-Циклопропоксиэтокси)Бензил)Фенил)-6-(Гидроксиметил)-2-Метокситетрагидро-2Н-Пиран-3,4,5-триола в одном реакторе
В этом примере описано получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагадро-2Н-пиран-3,4,5-триола путем получения арилмагниевого реагента и сочетания с глюконолактоном в одном реакционном сосуде.
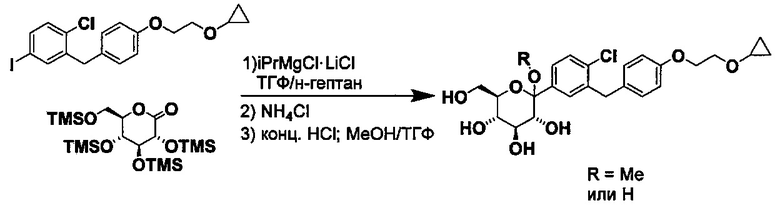
Одновременное добавление iPrMgCl⋅LiCl и (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-она.
Трехгорлую колбу (500 мл), оснащенную термометром, магнитной мешалкой, холодильником и капельной воронкой, продували азотом и загружали в нее безводный ТГФ (80 мл) и 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (43 г, 0,1 моль). После охлаждения смеси до -60°C в атмосфере азота, к описанному выше раствору почти одновременно добавляли iPrMgCl⋅LiCl (79 г, 13,05% в ТГФ, 0,1 моль) и (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-он (65,4 г, 0,14 моль) в растворе н-гептана (100 мл) с такой скоростью, чтобы температура поддерживалась на уровне ниже -50°C, в атмосфере азота. После того как добавление было закончено, смесь медленно нагревали до температуры от -15 до -10°C и перемешивали в течение 6,5 часов. Реакционную смесь медленно гасили насыщенным водным раствором хлорида аммония (240 г) при -10°C и оставляли нагреваться до 15°C. Отделяли верхний органический слой. Добавляли деионизованную воду (120 г) и водные фазы экстрагировали этилацетатом (3×162 г). Органические слои объединяли и промывали деионизованной водой (200 г) и насыщенным раствором хлорида натрия (200 г). Органический слой упаривали при температуре 35°C в вакууме, получая масло. Полученный остаток растворяли в метаноле (321,2 г) и тетрагидрофуране (125 г). После охлаждения до -10°C, концентрированную соляную кислоту (11 г) прикапывали в реакционную смесь, поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 20°C и перемешивали в течение 16 часов. Реакционную смесь медленно гасили добавлением очищенной воды (100 г). Полученную смесь медленно гасили насыщенным водным раствором бикарбоната натрия до значения рН немного выше 8. Летучие органические компоненты удаляли при пониженном давлении при температуре между 10 и 30°C. Полученный остаток разбавляли очищенной водой (200 г) и экстрагировали этилацетатом (3×180 г). Объединенные органические слои промывали деионизованной водой (200 г), насыщенным раствором хлорида натрия (200 г) и деионизованной водой (200 г). Органический слой упаривали, получая неочищенное целевое соединение (46,7 г, выход: 94%, 91% чистота согласно ВЭЖХ-0001) в виде светло-желтого стекловидного твердого вещества.
Добавление iPrMgCl⋅LiCl в смесь 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензола и (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-она.
Трехгорлую колбу (100 мл), оснащенную термометром и магнитной мешалкой, продували азотом и загружали в нее безводный ТГФ (8 мл) и 1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)-4-иодбензол (4,3 г, 0,01 моль). После охлаждения смеси до -60°C в атмосфере азота, добавляли (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-он (6,56 г, 0,014 моль) в растворе н-гептана (10 мл). В полученную смесь прикапывали iPrMgCl⋅LiCl (7,54 г, 13,05% в ТГФ, 0,95 моль) с такой скоростью, чтобы температура поддерживалась на уровне ниже -50°C в атмосфере азота. После того как добавление было закончено, смесь медленно нагревали до температуры от -15 до -10°C и перемешивали в течение 4 часов. Реакционную смесь медленно гасили насыщенным водным раствором хлорида аммония (24 г) при -10°C и оставляли нагреваться до 15°C. Отделяли верхний органический слой. Добавляли деионизованную воду (12 г) и водные фазы экстрагировали этилацетатом (3×16 г). Органические слои объединяли и промывали деионизованной водой (20 г) и насыщенным раствором хлорида натрия (20 г). Органический слой упаривали при температуре 35°C в вакууме, получая масло. Полученный остаток растворяли в метаноле (32 г) и тетрагидрофуране (13 г). После охлаждения до -10°C, прикапывали в реакционную смесь концентрированную соляную кислоту (1,1 г), поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 20°C и перемешивали в течение 16 часов. Реакционную смесь медленно гасили добавлением очищенной воды (10 г). Полученную смесь медленно гасили насыщенным водным раствором бикарбоната натрия до слабоосновного уровня рН (рН около 8). Летучие органические вещества удаляли при пониженном давлении при температуре между 10 и 30°C. Полученный остаток разбавляли очищенной водой (20 г) и экстрагировали этилацетатом (3×18 г). Объединенные органические слои промывали деионизованной водой (20 г), насыщенным раствором хлорида натрия (20 г) и деионизованной водой (20 г). Органический слой упаривали, получая неочищенное целевое соединение (4,1 г, выход: 84%, 74% чистота согласно ВЭЖХ-0001) в виде светло-желтого стекловидного твердого вещества.
Пример 22. Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола
В этом примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола по реакции Гриньяра.
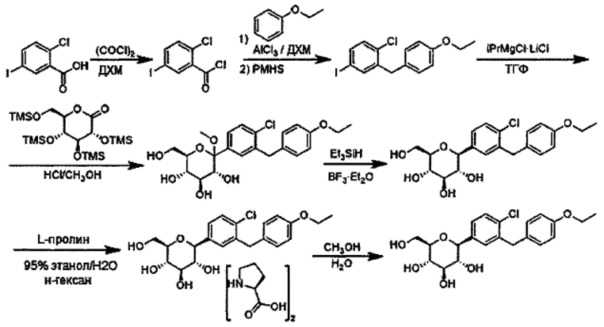
Получение 2-хлор-5-иодбензоил хлорида
В 1-литровую 4-горлую колбу, оснащенную термометром и механической мешалкой (частота 150 об/мин), загружали 2-хлор-5-иодбензойную кислоту (14,1 г, 0,05 моль), ДХМ (70,5 мл) и оксалилхлорид (5,5 мл, 0,06 моль). После перемешивания в течение 10 мин, смесь охлаждали до 10-15°C и добавляли ДМФА (0,15 мл, 1,92 ммоль) шприцем в течение 10 мин в две порции (0,1 и 0,05 мл), поддерживая температуру реакции ниже 20°C. После того как добавление было закончено, смесь нагревали до 25°C и перемешивали в течение 16 часов. Полученную смесь упаривали, и остаток сушили в вакууме при 30°C 5 часов, получая 15,0 г продукта в виде белого твердого вещества. Выход: 100%. LCMS-0013: 99% чистота. 1Н ЯМР (CDCl3, 400 МГц): 8,33 (д, J=2,4 Гц, 1Н), 7,81~7,84 (дд, J=2,4 Гц, 8,4 Гц, 1Н), 7,23 (д, J=8,4 Гц, 1Н).
Получение 1-хлор-2-(4-этоксибензил)-4-иодбензола
В 250-миллилитровую 4-горлую колбу, оснащенную внутренним термометром и холодильником, добавляли этоксибензол (6,4 г, 52,5 ммоль) и дихлорметан (19,2 мл), и смесь охлаждали до -5°C. Алюминий (III) хлорид (7,4 г, 55 ммоль) добавляли в течение 1 часа, поддерживая внутреннюю температуру ниже 0°C. После того как добавление было закончено, полученную смесь перемешивали в течение 30 минут при 0~5°C, и прикапывали раствор 2-хлор-5-иодбензоил хлорида (15,0 г, 50 ммоль) в дихлорметане (21 мл) в течение 1 часа, поддерживая внутреннюю температуру ниже 5°C. Полученную смесь перемешивали еще 1 час при 0~5°C и нагревали до 10~15°C. Прикапывали полиметилгидроксисилан (PMHS) (15,0 г, 0,25 моль), поддерживая внутреннюю температуру ниже 25°C. После перемешивания в течение 10 часов при 25°C, в полученную смесь добавляли еще PMHS (9,0 г, 0,15 моль). После перемешивания в течение еще 16 часов при 30°C, смесь охлаждали до 5~10°C и медленно прикапывали ледяную воду (50 мл) в течение 1 часа при перемешивании. Полученную смесь фильтровали, и осадок на фильтре суспендировали в дихлорметане (100 мл), содержащем диатомовую землю (20 г). Полученную смесь фильтровали, и осадок на фильтре промывали дихлорметаном (2×50 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл). После удаления летучих компонентов, остаток растворяли в абсолютном этаноле (45 мл), кипятили при механическом перемешивании (100 об/мин) и охлаждали до 0°C. После перемешивания в течение еще 16 часов при температуре 0~5°C, смесь фильтровали и осадок на фильтре промывали заранее охлажденным (0~5°C) этанолом (2×5 мл), сушили в вакууме при 40°C 12 часов, получая 14,2 г 1-хлор-2-(4-этоксибензил)-4-иодбензола в виде белого твердого вещества. Полученный твердый продукт перекристаллизовывали из этанола (42,6 мл), получая 12,5 г 1-хлор-2-(4-этоксибензил)-4-иодбензола в виде белого твердого вещества. Выход 67%, Чистота по ВЭЖХ, ВЭЖХ-0002: 99,5%. 1Н ЯМР (CDCl3, 400 МГц): δ 7,21~7,29 (м, 3Н), 7,11 (д, J=8,8 Гц, 2Н,), 6,85 (д, J=8,8 Гц, 2Н,), 3,99~4,07 (м, 4Н), 1,43 (т, J=7,2 Гц, 3Н).
Получение (3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триола
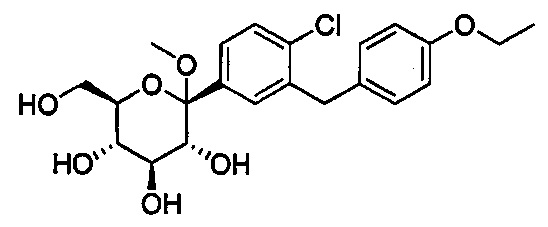
Получение арилмагния: В трехгорлую круглодонную колбу, оснащенную термометром и капельной воронкой с рубашкой, загружали раствор 1-хлор-2-(4-этоксибензил)-4-иодбензола (7,45 г, 20 ммоль) в ТГФ (15 мл), и смесь перемешивали на магнитной мешалке в атмосфере аргона. В полученный раствор прикапывали iPrMgCl⋅LiCl (17,7 мл, 1,3 М раствор в ТГФ, 23 ммоль) в течение 30 минут при температуре между -5 и 0°C. Полученную смесь перемешивали еще 1,5 часа при температуре от -5 до 0°C.
Раствор глюконолактона: В 100-миллилитровую круглодонную колбу загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидропиран-2-он (12,1 г, 26 ммоль) и н-гептан (18,5 мл), и смесь охлаждали до -5°С в атмосфере аргона. Прикапывали iPrMgCl⋅LiCl (0,8 мл, 1,3 М раствор в ТГФ, 1 ммоль), и полученную смесь перемешивали в течение 30 минут при температуре от -5 до 0°C. Охлажденный раствор глюконолактона прикапывали к арилмагнию в течение 30 минут при температуре между -5 и 0°C. После того как добавление было закончено, полученную смесь перемешивали в течение 2 часов при температуре -5°C. Заранее охлажденный (0°C) раствор концентрированной соляной кислоты (6,7 мл, 80 ммоль) в метаноле (35 мл) прикапывали в реакционную смесь, поддерживая температуру ниже 0°C. Полученную смесь оставляли нагреваться до температуры 15-20°C и перемешивали еще 16 часов. Полученную смесь медленно гасили насыщенным водным раствором бикарбоната натрия (~20 мл) до слабоосновного уровня рН, и смесь экстрагировали этилацетатом (2×80 мл). Объединенные органические слои промывали деионизованной водой (100 мл), насыщенным раствором хлорида натрия (100 мл), сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая 7,87 г продукта в виде светло-желтого стекловидного твердого вещества. Выход: ~90%. Чистота (LCMS-0013) 3,0 мин, 80% (UV); MS ESI (m/z) 439[М+1]+, вычисл. 438.
Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола
Раствор (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола (7,87 г, неочищенный, ~17,9 ммоль) в дихлорметане (59 мл) и ацетонитриле (59 мл) охлаждали до -30°C в атмосфере аргона. Добавляли в реакционный раствор триэтилсилан (11,5 мл, 71,6 ммоль), затем эфират трехфтористого бора (6,8 мл, 53,7 ммоль) так чтобы температура не превышала -10°C. По окончании добавления реакционный раствор перемешивали еще 1,5 часа и затем гасили 5%-ным раствором бикарбоната натрия до достижения значения рН 7,5. Органическую фазу отделяли, и водную фазу экстрагировали этилацетатом (2×80 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (2×80 мл) и сушили над безводным сульфатом натрия. Образец упаривали при пониженном давлении, получая 6,8 г указанного в заголовке соединения в виде опалесцирующего твердого вещества, которое использовали в следующей стадии без очистки. Выход: 93%. Чистота (LCMS-0013) 2,9 мин, 82% (UV); MS ESI (m/z) 409[М+1]+, вычисл. 408.
Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол, бис(L-пролин) сокристалла
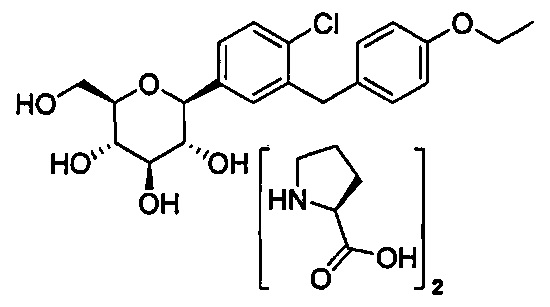
В 500-миллилитровую 4-горлую колбу загружали описанный выше неочищенный продукт (6,8 г, 82% чистота), затем L-пролин (3,8 г, 33,2 ммоль), этанол (57,4 мл) и воду (3,8 мл). Полученную смесь кипятили 30 минут при интенсивном механическом перемешивании. Прикапывали н-гексан (102 мл) к описанному выше раствору в течение 30 минут. По окончании добавления, реакционную смесь медленно охлаждали до комнатной температуры и перемешивали еще 16 часов. Полученную смесь фильтровали и осадок на фильтре промывали холодной смесью 95% этанол/вода (0°C, 2×3,4 мл) и н-гексаном (2×13,6 мл), и сушили в вакууме при 65°C, получая целевой продукт в виде белого твердого вещества (4,5 г). Этот неочищенный продукт (4,5 г) растворяли в смеси этанол/вода (95%, 22,5 мл) при 75°C при механическом перемешивании. Полученную смесь кипятили 30 минут при интенсивном механическом перемешивании. Прикапывали н-гексан (45 мл) к описанному выше раствору в течение 30 минут. По окончании добавления, реакционную смесь медленно охлаждали до комнатной температуры и перемешивали еще 16 часов. Полученную смесь фильтровали, и осадок на фильтре промывали н-гексаном (2×9 мл), и сушили в вакууме при 65°C, получая 3,8 г целевого продукта в виде белого твердого вещества. Чистота (ВЭЖХ-0001) 99,0% (УФ). 1Н ЯМР (CD3OD, 400 МГц): δ 7,34~7,25 (м, 3Н), 7,08 (д, J=8,8 Гц, 2Н), 6,78 (д, J=8,8 Гц, 2Н), 4,10 (д, J=9,2 Гц, 1Н), 4,06~3,95 (м, 6Н), 3,88~3,85 (м, 1Н), 3,72~3,68 (м, 1Н), 3,47~3,37 (м, 5Н), 3,32~3,20 (м, 3Н), 2,33~2,26 (м, 2Н), 2,16~2,08 (м, 2Н), 2,01~1,95 (м, 4Н), 1,35 (т, J=7,2 Гц, 3Н); MS ESI (m/z): 409 [M+1]+.
Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола (чистого)
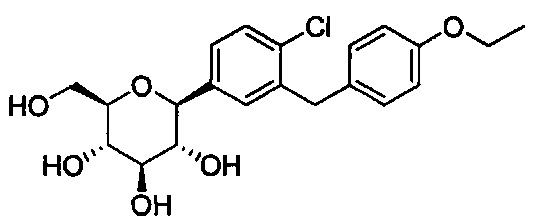
В трехгорлую круглодонную колбу, оснащенную термометром, холодильником и капельной воронкой, загружали (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол, бис(L-пролин) комплекс (3,8 г, 5,96 ммоль) и метанол (15,2 мл). После кипячения в течение 20 минут при перемешивании на магнитной мешалке (100 об/мин), прикапывали воду (76 мл) в течение 40 минут. После того как добавление было закончено, смесь охлаждали до 20~25°C и перемешивали еще 16 часов. Полученную смесь фильтровали, и осадок на фильтре промывали водой (2×7,6 мл), сушили в вакууме при температуре 45-50°C 12 часов, получая 2,3 г продукта в виде белого твердого вещества. Выход: 94%. Чистота (ВЭЖХ-0001), 99,3% (УФ); 1H ЯМР (CD3OD, 400 МГц): δ 7,34~7,25 (м, 3Н), 7,08 (д, J=8,8 Гц, 2Н), 6,78 (д, J=8,8 Гц, 2Н), 4,10 (д, J=9,2 Гц, 1Н), 4,06~3,95 (м, 4Н), 3,88~3,85 (м, 1Н), 3,69~3,65 (м, 1Н), 3,47~3,37 (м, 3Н), 3,27 (м, 1Н), 1,35 (т, J=7,2 Гц, 3Н); MS ESI (m/z): 409 [M+1]+.
Пример 23. Получение сокристаллов (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол; бис(L-Пролин)
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол; L-Пролин; L-Пролин по реакции Гриньяра.
Получение (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола
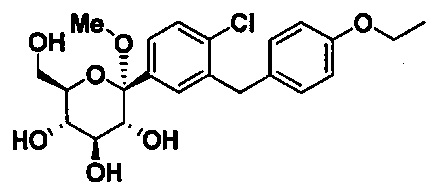
Раствор глюконолактона. В колбу объемом 100 мл загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагид-2H-пиран-2-он (6,54 г) и н-гептан (10,2 мл), и перемешивали в течение 10 минут в атмосфере аргона. Смесь охлаждали до температуры от -20°С до -30°C в атмосфере азота. Полученный раствор добавляли в подходящую охлажденную капельную воронку и готовили для добавления к арилмагнию.
Получение арилмагния. 4-горлую 100-миллилитровую колбу, оснащенную термометром, механической мешалкой, холодильником и капельной воронкой, продували азотом и загружали в нее безводный ТГФ (7 мл) и 1-хлор-2-(4-этоксибензил)-4-иодбензол (3,73 г, 10 ммоль). После перемешивания и продувки азотом в течение 30 минут при температуре окружающей среды, смесь охлаждали до -20°C в атмосфере азота. В полученный раствор добавляли iPrMgCl⋅LiCl (Aldrich, оттированная концентрация 12,9% вес/вес, 9,58 г) (в зависимости от титра реагента, 1,2 экв.) через подходящую капельную воронку с такой скоростью, чтобы температура поддерживалась на уровне между -20°C и -10°C в течение 30 минут в атмосфере азота. Полученную смесь перемешивали еще 10 минут при температуре от -20 до -10°C. Конверсию исходного вещества в арилмагний отслеживали, гася аликвоту насыщенным водным раствором хлорида аммония, и аликвоту экстрагировали этилацетатом и анализировали на приборе ВЭЖХ-0001.
Присоединение арилмагния с получением аномерного полукеталя. Холодный раствор глюконолактона из охлажденной (-15°C) капельной воронки прикапывали к раствору арилмагния с такой скоростью, чтобы поддерживать температуру между -20°C и -10°C, в течение 40 минут. После того как добавление было закончено, полученную смесь перемешивали в течение 5 часов при температуре от -20 до -10°C.
Реакционную смесь медленно гасили продутым азотом (10 мин) насыщенным водным раствором хлорида аммония (30 г) при температуре от -15°C до 0°C через капельную воронку в течение 20 минут. Полученную смесь оставляли нагреваться до температуры 10-15°C в течение 2,5 часов и перемешивали в течение 10 часов. Отделяли верхний органический слой. Добавляли деионизованную воду (10 г) в водный слой. Водные фазы экстрагировали этилацетатом (3×15 мл). Органические слои объединяли и промывали деионизованной водой (20 мл) и насыщенным раствором хлорида натрия (16,7% вес/вес, 20 г). Этилацетатный слой обрабатывали активированным углем (1,32 г, 30% вес/вес из расчета на вес ожидаемого продукта, СХ-700 от Zhuxi Со.) 1 час при 20°C, после чего фильтровали через фильтровальную бумагу. Органический слой упаривали при температуре 35°C в вакууме (0,01 МПа) до почти полной остановки конденсации растворителя в приемнике. Добавляли метанол (10 мл) и смесь снова упаривали при 35°C в вакууме (0,01 МПа) до почти полной остановки конденсации растворителя в приемнике.
Получение кеталя из полукеталя. Полученный остаток растворяли в метаноле (34 мл) и тетрагидрофуране (17 мл) при механическом перемешивании (240 об/мин). Полученную смесь охлаждали до -10°C в течение 40 минут. Заранее охлажденный (0°C) раствор концентрированной соляной кислоты (1,0 мл) прикапывали в реакционную смесь, поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 10-15°C и перемешивали в течение 18 часов.
Реакционную смесь медленно гасили добавлением очищенной воды (25 мл) поддерживая температуру ниже 20°C. В полученную смесь загружали н-гептан (15 мл). После перемешивания в течение 30 минут (240 об/мин) и отстаивания в течение 15 мин, нижний водный слой переносили в колбу. Верхний органический слой переносили в другую подходящую делительную воронку и экстрагировали смесью вода-метанол (1:1, 10 мл). Водные слои объединяли и осторожно гасили водной суспензией бикарбоната натрия (20 г) до слабоосновного уровня рН (рН примерно 7,5-8). Летучие органические вещества удаляли при пониженном давлении (0,01 МПа) при внешней температуре 30°C. Полученный остаток экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали деионизованной водой (40 мл), насыщенным раствором хлорида натрия (40 мл) и деионизованной водой (40 мл). Органический слой сушили над сульфатом натрия (15 г). Полученную суспензию фильтровали через фильтровальную бумагу, и осадок на фильтре промывали этилацетатом (10 мл). Органический слой упаривали на роторном испарителе в вакууме (0,01 МПа) при температуре 30°С до почти полной остановки конденсации растворителя в приемнике. Органический слой упаривали (20-30°C, 0,01 МПа), получая неочищенный (3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2Н-пиран-3,4,5-триол (3,56 г, выход: 81,1%, 77,1% чистота согласно ВЭЖХ-0001) в виде светло-желтого стекловидного твердого вещества. Полученный неочищенный продукт напрямую использовали в следующей стадии.
Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(-4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола

В 100-миллилитровую 3-горлую колбу, оснащенную магнитной мешалкой, в атмосфере аргона последовательно добавляли дихлорметан (7,0 мл), ацетонитрил (7,0 мл) и триэтилсилан (5,09 мл, 31,9 ммоль) при комнатной температуре. Полученную смесь охлаждали до температуры между -20 и -25°C, и добавляли BF3⋅Et2O (3,03 мл, 23,9 ммоль) в один прием. В другую 100-миллилитровую колбу загружали неочищенный (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (3,5 г, 7,97 ммоль), дихлорметан (7,0 мл) и ацетонитрил (7,0 мл), и полученную смесь встряхивали 20 минут при температуре окружающей среды до получения прозрачного раствора. В атмосфере азота раствор (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола в дихлорметане и ацетонитриле переносили в капельную воронку и медленно добавляли к раствору BF3-Et2O и триэтилсилана в течение 1 часа поддерживая внутреннюю температуру между -15 и -20°C. После того как добавление было закончено, смесь перемешивали при температуре между -15 и -20°C.
Реакцию гасили добавлением водного раствора бикарбоната натрия (7,4% вес/вес, 25 г) через капельную воронку, поддерживая температуру в реакционной среде ниже -5°C. Добавляли еще твердый бикарбонат натрия (1,7 г), доводя значение рН до ~7,5. Летучие растворители удаляли при пониженном давлении при температуре ниже 40°C. После охлаждения до температуры ниже комнатной, остаток разделяли между этилацетатом (30 мл) и водой (15 мл). Органический слой отделяли, и водный слой дважды экстрагировали этилацетатом (2×15 мл). Объединенные органические слои промывали 10%-ным насыщенным раствором хлорида натрия (2×20 мл). Объединенные экстракты упаривали при пониженном давлении при температуре ниже 40°C до почти полной остановки конденсации растворителя в приемнике. Полученный остаток сушили на масляном насосе (Р=0,1 мм рт. ст.), получая 3,30 г не совсем белого твердого вещества (100% выход, 77,2% чистота согласно ВЭЖХ-0001).
Получение сокристаллов (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол; бис(L-Пролин)
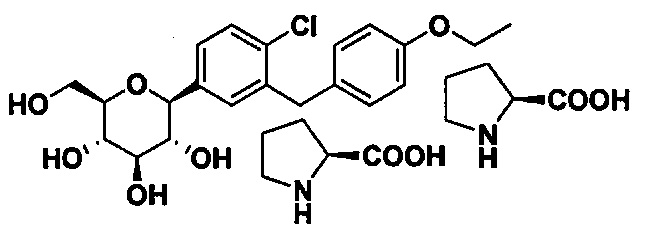
В 100-миллилитровую 3-горлую колбу загружали неочищенный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол (3,2 г, 77% чистота), L-пролин (1,8 г, 15,6 ммоль), 95% этанол (25,6 мл), и смесь кипятили 30 минут при интенсивном перемешивании на магнитной мешалке. Прикапывали в смесь гептан (16 мл) в течение 20 минут, и по окончании добавления реакционную смесь медленно охлаждали до температуры 10-15°C при скорости охлаждения 10-15°C в час. После перемешивания в течение еще 12 часов при 10-15°C, реакционную смесь фильтровали, и осадок на фильтре промывали заранее охлажденной 95%-ной смесью этанол/вода (-5 - 0°C, 2×3,2 мл) и н-гептаном (2×6,4 мл), сушили в вакууме при 50-55°C в течение 8 часов, получая не совсем белое твердое вещество. Выход: 3,0 г (60%). Чистота (ВЭЖХ-0001) 10,0 мин, 97,4% (УФ).
Пример 24. Получение сокристаллов (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол: бис(L-Пролин)
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол; L-Пролин; L-Пролин.
Получение (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола
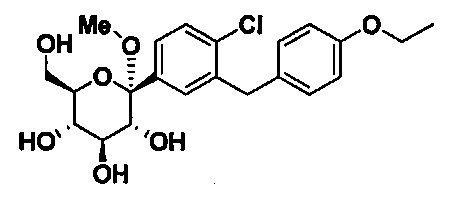
Раствор глюконолактона. В колбу объемом 100 мл загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2он (6,54 г) и н-гептан (10,2 мл), и перемешивали в течение 10 минут, барботируя аргон. Смесь охлаждали до температуры от -30°C до -20°C в атмосфере азота. Полученный раствор помещали в подходящую капельную воронку и хранили до добавления к арилмагнию.
Получение арилмагния. 4-горлую колбу объемом 100 мл, оснащенную термометром, механической мешалкой, холодильником и капельной воронкой, продували азотом и загружали в нее безводный ТГФ (7 мл) и 1-хлор-2-(4-этоксибензил)-4-иодбензол (3,73 г, 10 ммоль). После перемешивания и продувки азотом в течение 30 минут при температуре окружающей среды, смесь охлаждали до -60°C в атмосфере азота. В полученный раствор добавляли iPrMgCl⋅LiCl (Aldrich, оттитрованная концентрация 12,9% вес/вес, 7,58 г) (0,95 экв. согласно титрованию) через подходящую капельную воронку с такой скоростью, чтобы температура поддерживалась на уровне между -50°C и -60°C, в течение 30 минут в атмосфере азота. Полученную смесь перемешивали еще 10 минут при температуре от -60 до -50°C. Конверсию 1-хлор-2-(4-этоксибензил)-4-иодбензола в арилмагний отслеживали, гася аликвотные пробы насыщенным водным раствором хлорида аммония, экстрагирования аликвоты этилацетатом и анализируя методом ВЭЖХ-0001.
Присоединение арилмагния с получением аномерного полукеталя. Холодный раствор глюконолактона из охлажденной (-25°C) капельной воронки прикапывали к раствору арилмагния с такой скоростью, чтобы поддерживать температуру между -50°C и -60°C, в течение 40 минут. После того как добавление было закончено, полученную смесь перемешивали в течение 5 часов при температуре от -50 до -60°C.
Реакционную смесь медленно гасили продутым азотом (10 мин) насыщенным водным раствором хлорида аммония (30 г) при температуре от -15°C до 0°C через капельную воронку в течение 20 минут. Полученную смесь оставляли нагреваться до температуры 10-15°C в течение 2,5 часов и перемешивали в течение 10 часов. Отделяли верхний органический слой. Деионизованную воду (10 г) добавляли в водный слой.
Водные фазы экстрагировали этилацетатом (3×15 мл). Органические слои объединяли и промывали деионизованной водой (20 мл) и насыщенным раствором хлорида натрия (16,7% вес/вес, 20 г). Этилацетатный слой обрабатывали активированным углем (1,32 г, 30% вес/вес, из расчета на вес ожидаемого продукта (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола, СХ-700 от Zhuxi Со.) 1 час при 20°C, после чего фильтровали через фильтровальную бумагу. Органический слой упаривали при температуре 35°C в вакууме (0,01 МПа) до почти полной остановки конденсации растворителя в приемнике. Добавляли метанол (10 мл), и смесь снова упаривали при 35°C в вакууме (0,01 МПа) до почти полной остановки конденсации растворителя в приемнике.
Получение кеталя из полукеталя. Полученный остаток растворяли в метаноле (34 мл) и тетрагидрофуране (17 мл) при механическом перемешивании (240 об/мин). Полученную смесь охлаждали до -10°C в течение 40 минут. Заранее охлажденный (0°C) раствор концентрированной соляной кислоты (1,0 мл) прикапывали в реакционную смесь, поддерживая температуру на уровне между -10 и 0°C. Полученную смесь нагревали до 10-15°C и перемешивали в течение 18 часов.
Реакционную смесь медленно гасили добавлением очищенной воды (25 мл), поддерживая температуру ниже 20°C. В полученную смесь загружали н-гептан (15 мл). После перемешивания в течение 30 минут (240 об/мин) и отстаивании в течение 15 мин, нижний водный слой переносили в колбу. Верхний органический слой переносили в другую подходящую делительную воронку и экстрагировали смесью вода-метанол (1:1, 10 мл). Водные слои объединяли и осторожно гасили водной суспензией бикарбоната натрия (20 г) до слабоосновного уровня рН (рН примерно 7,5-8). Летучие органические вещества удаляли при пониженном давлении (0,01 МПа) при внешней температуре 30°C. Полученный остаток экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали деионизованной водой (40 мл), насыщенным раствором хлорида натрия (40 мл) и деионизованной водой (40 мл). Органический слой сушили над сульфатом натрия (15 г). Суспензию фильтровали через фильтровальную бумагу, и осадок на фильтре промывали этилацетатом (10 мл). Органический слой упаривали на роторном испарителе в вакууме (0,01 МПа) при температуре 30°C до почти полной остановки конденсации растворителя в приемнике. Органический слой упаривали (20-30°C, 0,01 МПа), получая неочищенный (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (3,65 г, выход: 83,1%, 90,4% чистота согласно ВЭЖХ-0001) в виде светло-желтого стекловидного твердого вещества. Полученный неочищенный продукт напрямую использовали в следующей стадии.

Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола
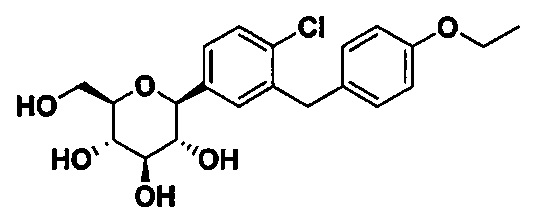
В 100-миллилитровую 3-горлую колбу, оснащенную магнитной мешалкой, в атмосфере аргона последовательно добавляли дихлорметан (7,0 мл), ацетонитрил (7,0 мл) и триэтилсилан (5,09 мл, 31,9 ммоль) при комнатной температуре. Полученную смесь охлаждали до температуры между -20 и -25°C, и добавляли BF3⋅Et2O (3,03 мл, 23,9 ммоль) в один прием. В другую 100-миллилитровую колбу загружали неочищенный (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (3,5 г, 7,97 ммоль), дихлорметан (7,0 мл) и ацетонитрил (7,0 мл), и полученную смесь встряхивали 20 минут при температуре окружающей среды до получения прозрачного раствора. В атмосфере азота раствор (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола в дихлорметане и ацетонитриле переносили в капельную воронку и медленно добавляли к раствору BF3-Et2O и триэтилсилана в течение 1 часа, поддерживая внутреннюю температуру между -15 и -20°C. После того как добавление было закончено, смесь перемешивали при температуре между -15 и -20°C.
Реакцию гасили добавлением водного раствора бикарбоната натрия (7,4% вес/вес, 25 г) через капельную воронку, поддерживая температуру в реакционной среде ниже -5°C. Добавляли еще твердый бикарбонат натрия (1,7 г), доводя значение рН до ~7,5. Летучие растворители удаляли при пониженном давлении при температуре ниже 40°C. После охлаждения до температуры ниже комнатной, остаток разделяли между этилацетатом (30 мл) и водой (15 мл). Органический слой отделяли, и водный слой дважды экстрагировали этилацетатом (2×15 мл). Объединенные органические слои промывали 10%-ным насыщенным раствором хлорида натрия (2×20 мл). Объединенные экстракты упаривали при пониженном давлении при температуре ниже 40°C до снижения скорости конденсации и почти полной остановки отгонки (остаток не пенится). Полученный остаток сушили на масляном насосе (Р=0,1 мм рт. ст.), получая 3,25 г не совсем белого твердого вещества (99,7% выход, 89,3% чистота согласно ВЭЖХ-0001).
Получение сокристаллов (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол; бис(L-Пролин)
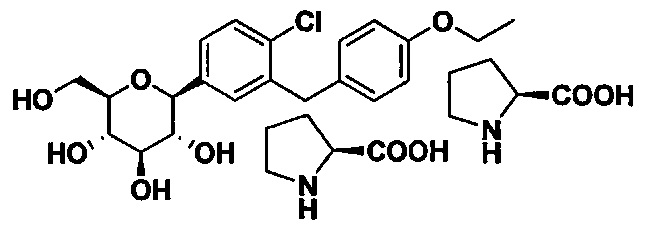
В 100-миллилитровую 3-горлую колбу загружали неочищенный (3S,6S,2R,4R,5R)-6-{4-хлор-3-[(4-этоксифенил)метил]фенил}-2-(гидроксиметил)-2Н-3,4,5,6-тетрагидропиран-3,4,5-триол (3,2 г, 89,3% чистота), L-пролин (1,8 г, 15,6 ммоль), 95%-ный этанол (25,6 мл), и смесь кипятили 30 минут при интенсивном перемешивании на магнитной мешалке. Прикапывали в смесь гептан (16 мл) в течение 20 минут, и по окончании добавления реакционную смесь медленно охлаждали до температуры 10-15°C при скорости охлаждения 10-15°C в час. После перемешивания в течение еще 12 часов при 10-15°C, реакционную смесь фильтровали, и осадок на фильтре промывали заранее охлажденной 95%-ной смесью этанол/вода (-5 - 0°C, 2×3,2 мл) и н-гептаном (2×6,4 мл), сушили в вакууме при 50-55°C в течение 8 часов, получая не совсем белое твердое вещество. Выход: 3,6 г (72%). Чистота (ВЭЖХ-0001) 10,0 мин, 98,6% (УФ).
Пример 25. Получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-циклопропилбензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола
В данном примере описано получение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-циклопропилбензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола альтернативными способами.
Способ А: реагент Гриньра Mg & DIBAL-H
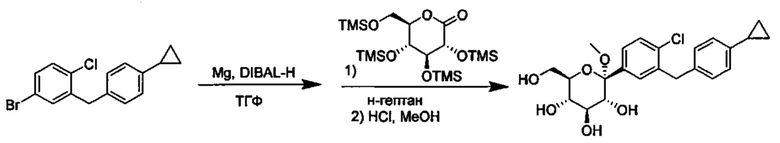
К активированному хлориду лития, полученному сушкой химически чистого безводного хлорида лития путем нагревания в вакууме (150°C, 0,5 мм рт. ст., 12 час) и всушенному на пламени горелки непосредственно перед использованием (JOC 1999, 64, 3322-3327), 93 мг, 2,2 ммоль, и магнию (57 мг, 2,4 ммоль) добавляли раствор 4-бром-1-хлор-2-(4-циклопропилбензил)бензола (644 мг, 2,0 ммоль) в безводном тетрагидрофуране (2,0 мл) в атмосфере аргона. Полученную смесь нагревали до 40°C и добавляли диизобутилалюминий гидрид (0,02 мл, 1 М). Полученную смесь перемешивали в течение 2,5 часов при температуре 40°C. Полученную черную суспензию фильтровали. Половину полученного арилмагния прикапывали к (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2Н-пиран-2-ону (606 мг, 1,3 ммоль) в н-гептане (2,0 мл) при 20°C. После того как добавление было закончено, полученную смесь перемешивали в течение 3 часов при температуре 20°C. Заранее охлажденный (0°C) раствор концентрированной соляной кислоты (0,38 мл, 4 ммоль) в метаноле (2,0 мл) прикапывали в реакционную смесь при комнатной температуре, и полученную смесь перемешивали еще 16 часов. Полученную смесь медленно гасили насыщенным водным раствором бикарбоната натрия (~4 мл) до слабоосновного уровня рН, и смесь экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали деионизованной водой (10 мл), насыщенным раствором хлорида натрия (10 мл), сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая 259 мг продукта в виде светло-желтого стекловидного твердого вещества. Выход: ~60%.
Способ В: реагент Гриньяра iPrMgCl⋅LiCl
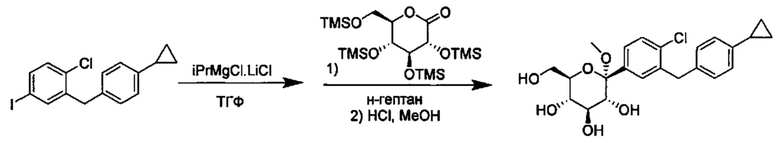
Получение арилмагния: В колбу загружали раствор 1-хлор-2-(4-циклопропилбензил)-4-иодбензола (0,736 г, 2 ммоль) и ТГФ (3 мл), и смесь перемешивали на магнитной мешалке в атмосфере аргона. В полученный раствор прикапывали iPrMgCl⋅LiCl (2 мл, 1,3 М раствор в ТГФ, 2,6 ммоль) в течение 10 минут при 0°C. Полученную смесь перемешивали еще 2 часа при температуре 0°C.
Раствор глюконолактона: В колбу загружали (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2H-пиран-2-он (1,31 г, 2,8 ммоль) и н-гептан (3,0 мл), и смесь охлаждали до 0°C. Охлажденный раствор глюконолактона прикапывали к арилмагнию в течение 30 минут при температуре между -5 и 0°C. После того как добавление было закончено, полученную смесь перемешивали в течение 3 часов при 0°C. Заранее охлажденный (0°C) раствор концентрированной соляной кислоты (0,67 мл, 8 ммоль) в метаноле (3,5 мл) прикапывали в реакционную смесь, поддерживая температуру ниже 0°C. Полученную смесь оставляли нагреваться до температуры 15-20°C и перемешивали еще 16 часов. Полученную смесь медленно гасили насыщенным водным раствором бикарбоната натрия (~4 мл) до слабоосновного уровня рН, и смесь экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали деионизованной водой (10 мл), насыщенным раствором хлорида натрия (10 мл), сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая 478 мг продукта в виде светло-желтого стекловидного твердого вещества. Выход: ~55%.
Восстановление кеталя
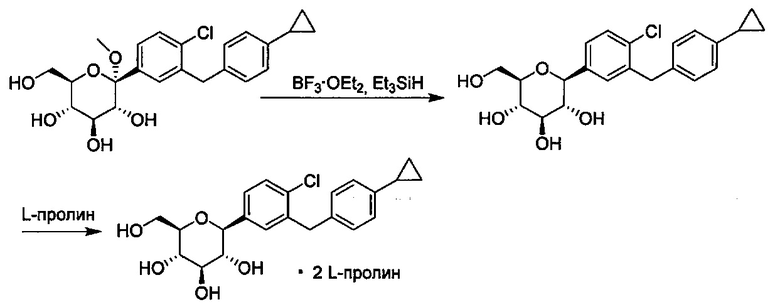
Стадию восстановления комплексом трифторида бора с диэтиловым эфиром и триэтилсиланом с получением неочищенного (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-циклопропилбензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триола и его сокристаллизацию с L-пролином, с получением комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-циклопропилбензил)фенил)-6-(гидроксиметил)тетрагидро-2Н-пиран-3,4,5-триол бис(L-пролин) проводили согласно методике, описанной в патенте WO 2010/022313.
Несмотря на то, что изложенное выше изобретение было подробно описано с использованием иллюстраций и примеров для ясности понимания, квалифицированный специалист в данной области техники поймет, что в рамках объема формулы изобретения могут иметь место определенные изменения и модификации. Кроме того, каждая приведенная в настоящем тексте ссылка на источник информации включена в настоящий текст в полном объеме посредством ссылки, как если бы каждый отдельный источник информации был отдельно включен в данный текст посредством ссылки. В случае противоречия между настоящей заявкой и приведенной в тескте описания ссылкой, приоритет имеет настоящая заявка.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ КОМПЛЕКСОВ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SGLT2, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2641905C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ SGLT2 | 2009 |
|
RU2530494C2 |
| ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ПРИМЕНЯЕМОЕ ДЛЯ СИНТЕЗА ИНГИБИТОРА SGLT, И СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА SGLT С ПРИМЕНЕНИЕМ УКАЗАННОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2021 |
|
RU2802443C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БЕНЗИЛ-БЕНЗОЛЬНОГО ИНГИБИТОРА SGLT | 2011 |
|
RU2569491C2 |
| ПРОИЗВОДНЫЕ С-АРИЛГЛЮКОЗИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2606501C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛФЕНИЛЦИКЛОГЕКСАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2505521C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛБЕНЗОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2497526C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ГИДРОКСИ-БЕНЗИЛБЕНЗОЛА | 2014 |
|
RU2671493C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
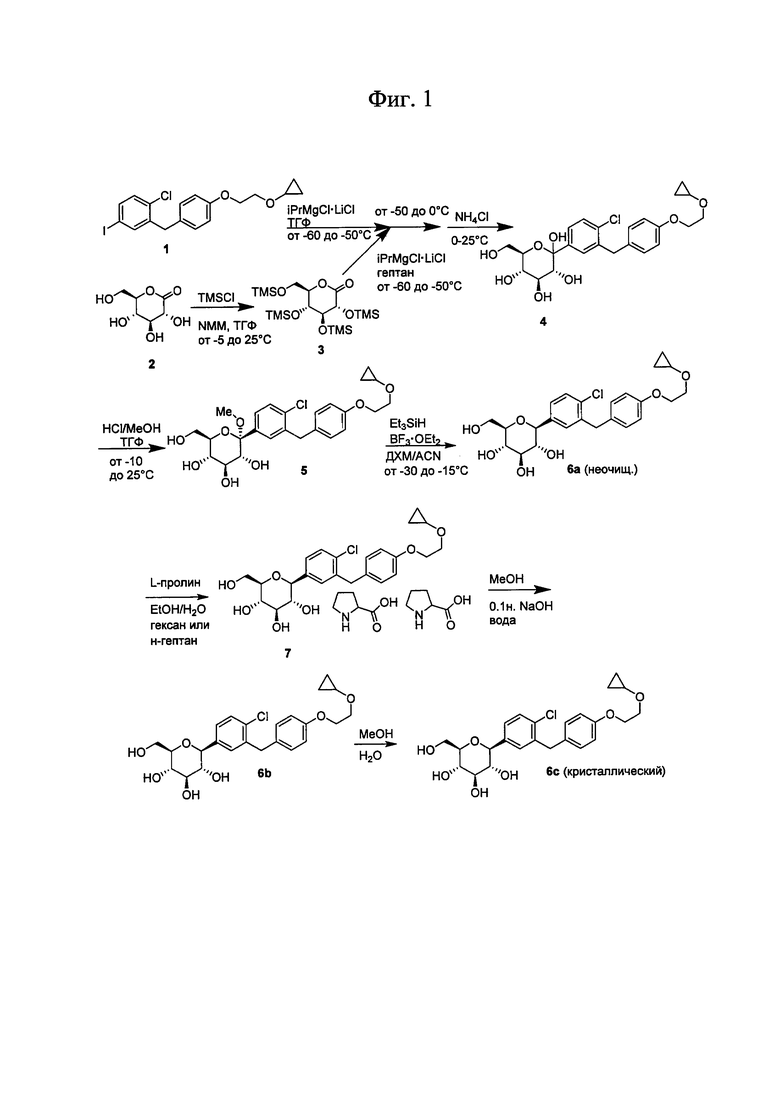
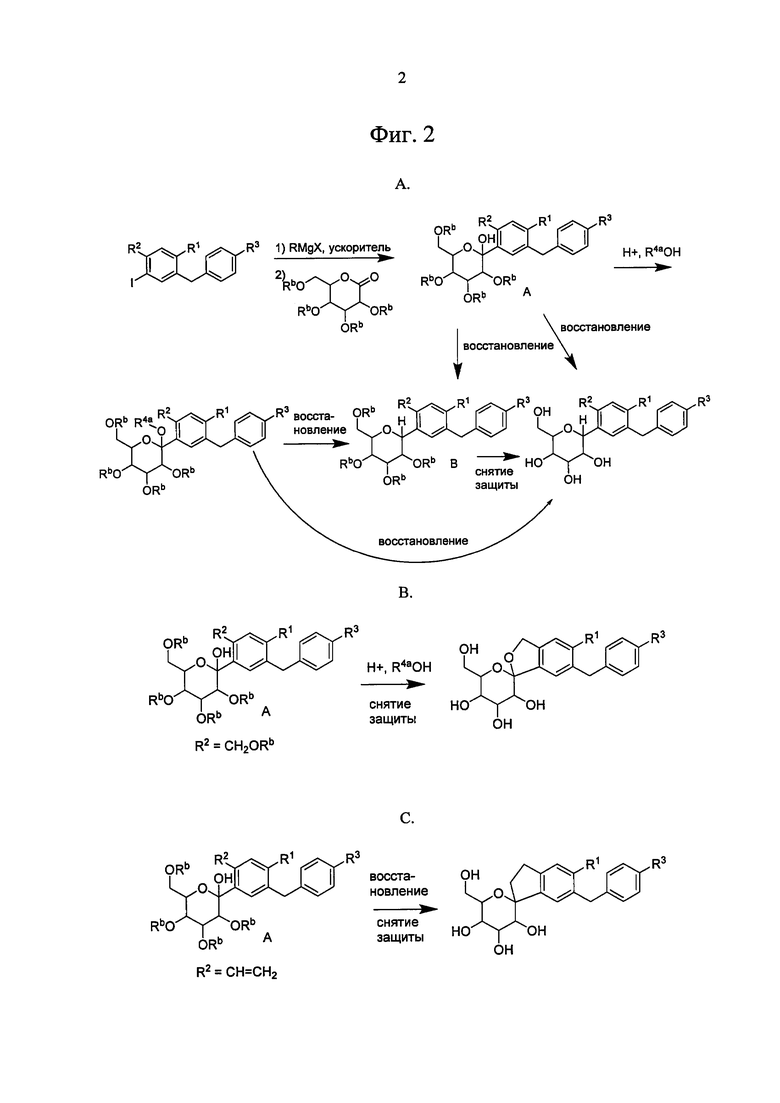
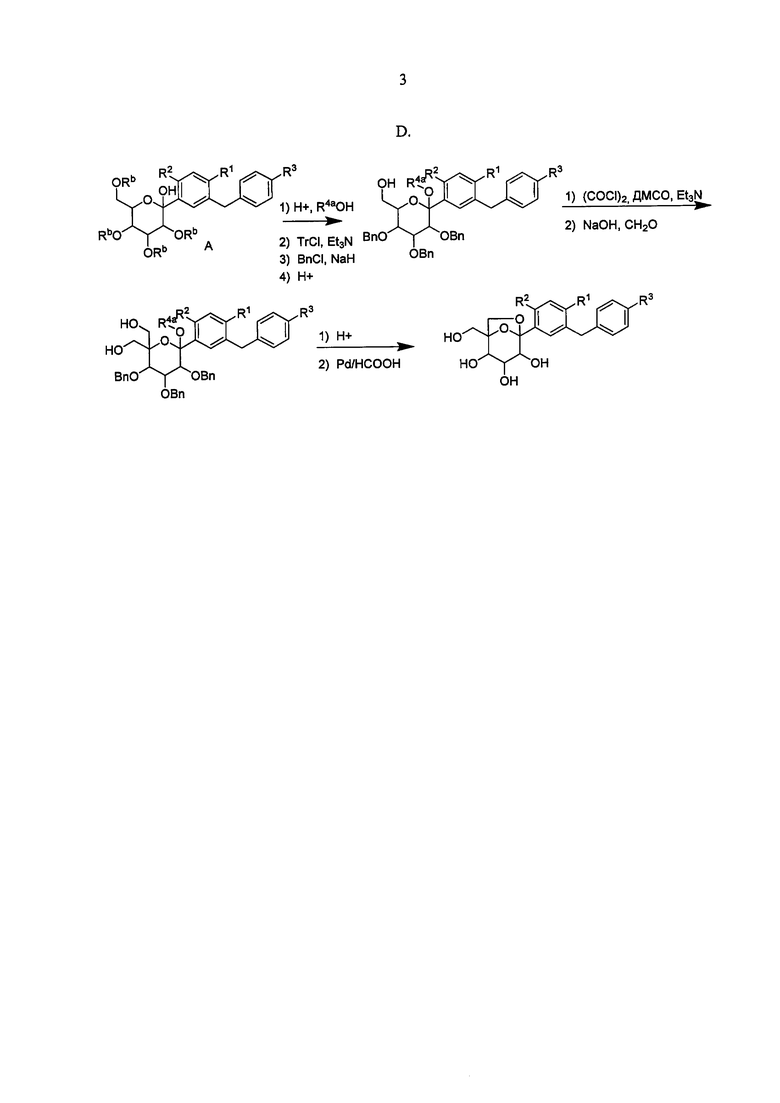
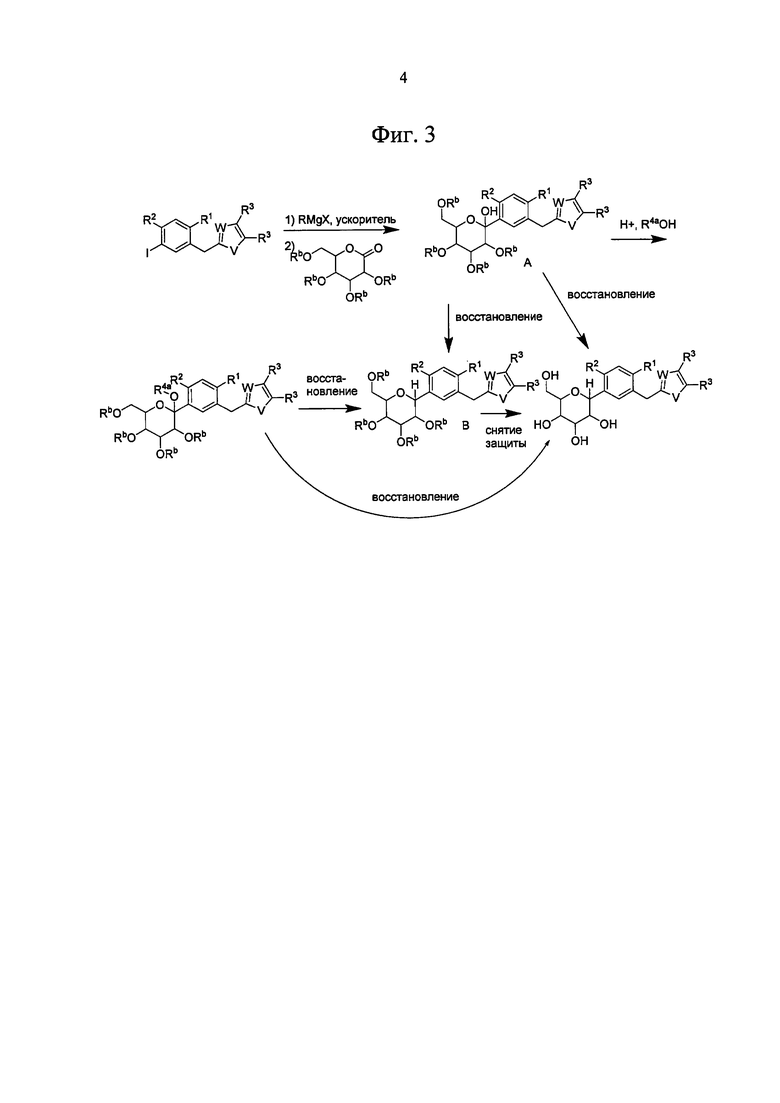
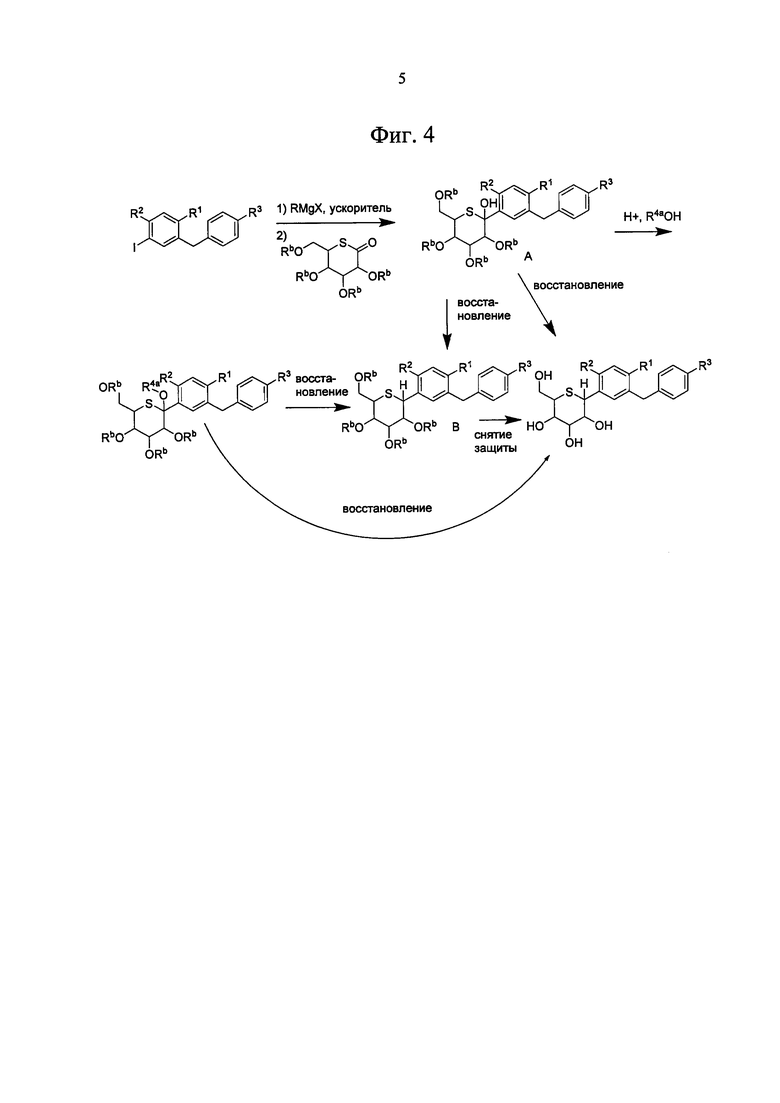
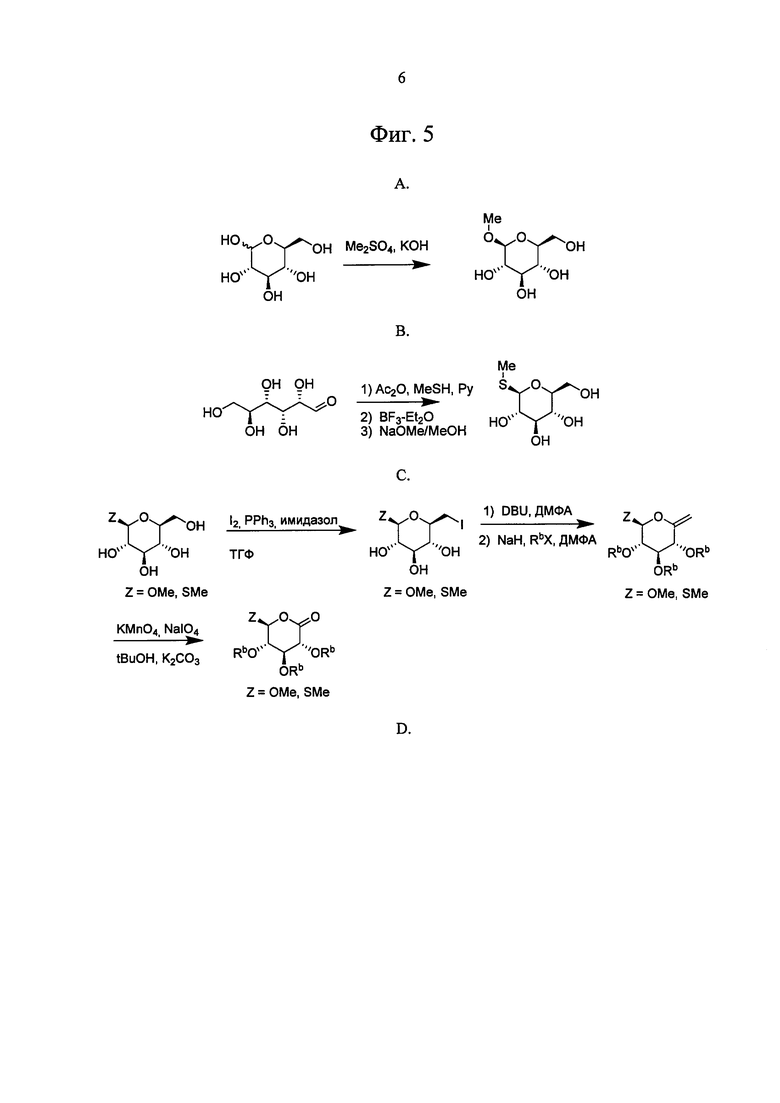
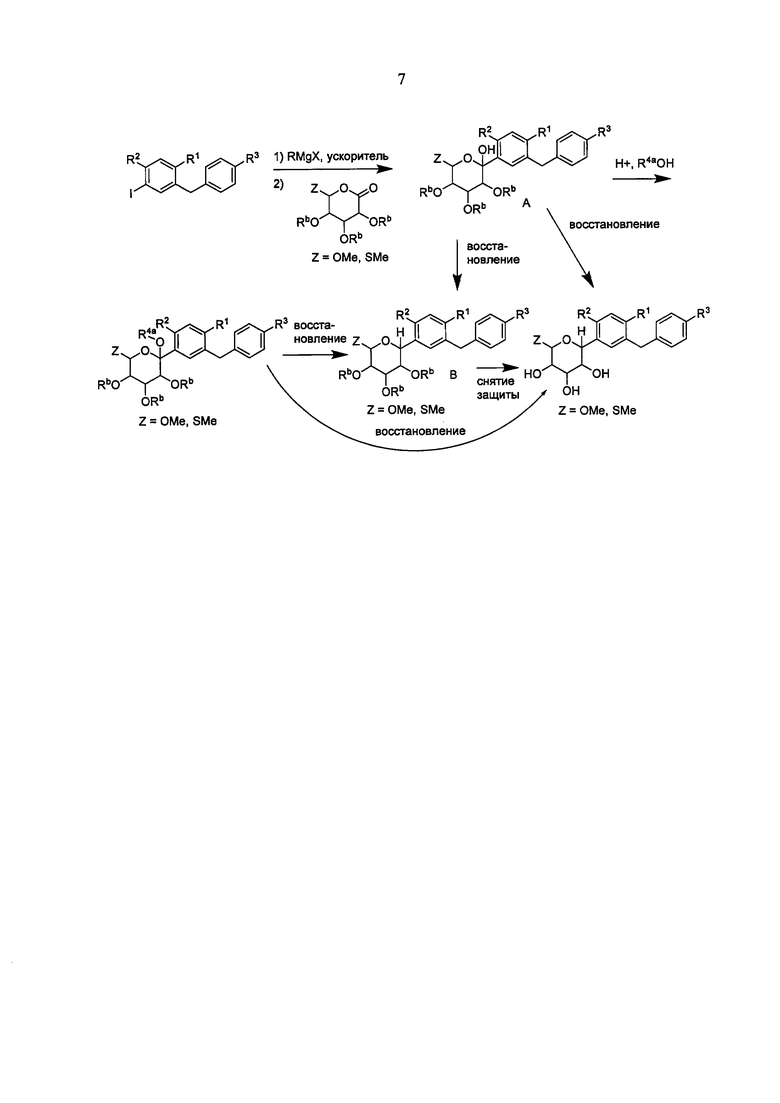
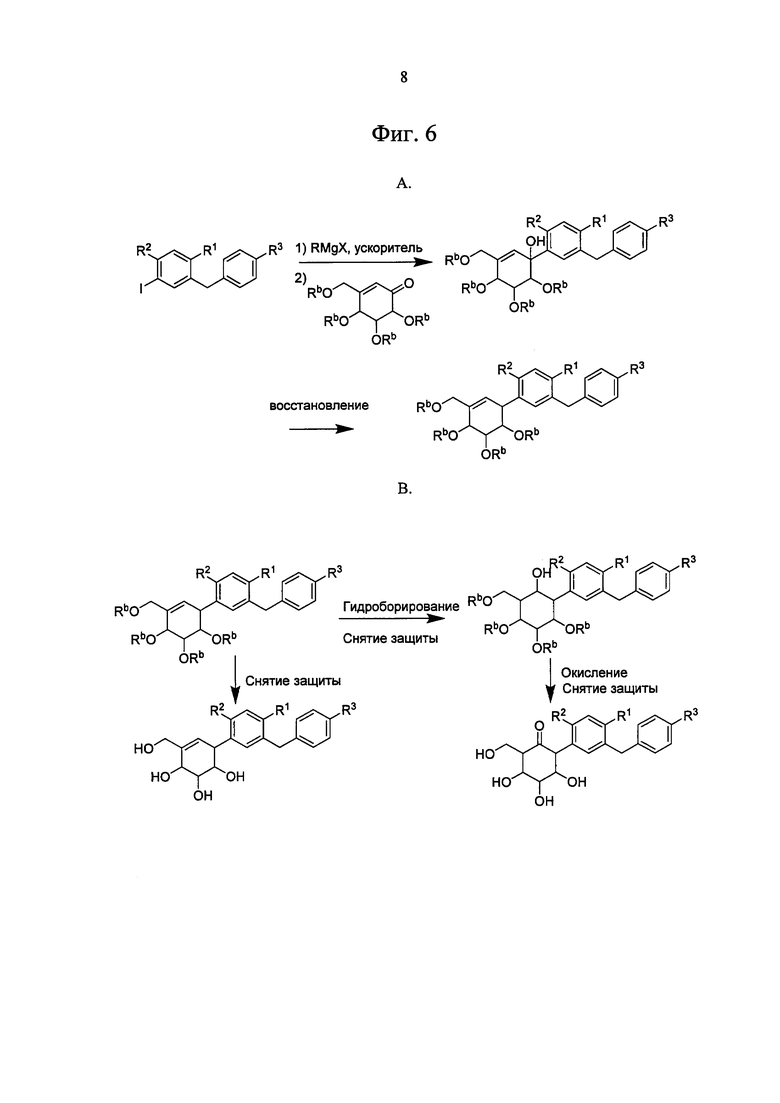

Изобретение относится к способу получения бензилбензольных ингибиторов SGLT2, оказывающих ингибирующее действие на натрий-зависимый переносчик глюкозы соединения формулы Ia, включающему (a) получение первой реакционной смеси, содержащей соединение формулы IIa: алкил-магниевый комплекс, выбранный из группы, состоящей из С1-С4 алкилмагния хлорида, С1-С4 алкилмагния бромида, ди(С1-С4 алкил)магния, С3-С7 циклоалкилмагния хлорида, С3-С7 циклоалкилмагния бромида и ди(С3-С7 циклоалкил)магния, и первый органический растворитель, где соотношение алкил-магниевого комплекса и соединения формулы IIa выбрано из 0,9, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96 и 0,97 (моль/моль), где первая реакционная смесь находится при температуре -50°С или ниже, с образованием промежуточного соединения; и (b) получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы IIIa: с образованием соединения формулы Ia, где R4 представляет собой ОН и каждый Ra представляет собой Rb; (c) получение третьей реакционной смеси, содержащей C1-С3 алкилгидрокси, сильную кислоту и соединение формулы Ia, где R4 представляет собой ОН, и каждый Ra представляет собой Rb, с образованием соединения формулы Ia, где R4 представляет собой C1-С3 алкокси, и каждый Ra независимо выбран из группы, состоящей из Н и Rb; и (d) получение четвертой реакционной смеси, содержащей восстановитель и соединение формулы Ia, где R4 представляет собой C1-С3 алкокси, и где реакционная смесь содержит менее 0,1 эквивалентов магния по отношению к количеству соединения формулы Ia, с образованием соединения формулы Ia, где R4 представляет собой Н, где X представляет собой иод, R1 представляет собой хлор, R2 представляет собой Н, R3 представляет собой (С3-С6 циклоалкокси)С1-С3 алкокси, R4 выбран из группы, состоящей из Н, ОН и C1-С3 алкокси, каждый Ra независимо выбран из группы, состоящей из Н, C1-С3 алкила и Rb, Rb представляет собой защитную группу, где алкил, алкокси и циклоалкил группы или их части могут необязательно быть полностью или частично фторированными, и один или больше атомов водорода могут быть необязательно заменены на атомы дейтерия, а также описаны синтетические интермедиаты, которые могут применяться для получения таких соединений. Технический результат заключается в увеличении выхода целевого продукта и снижении количества побочных продуктов. 2 н. и 36 з.п. ф-лы, 2 табл., 7 ил., 25 пр.
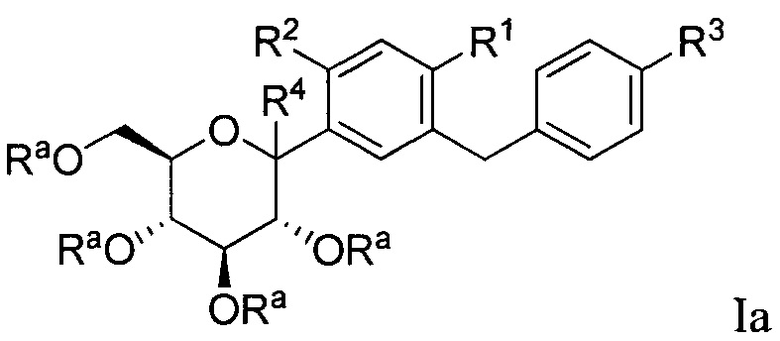
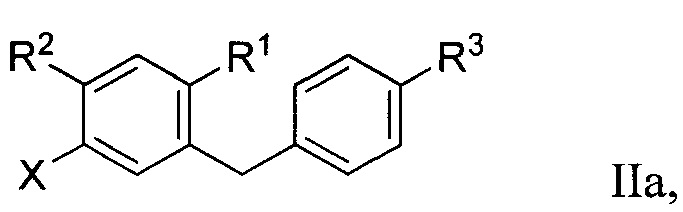

1. Способ получения соединения формулы Ia
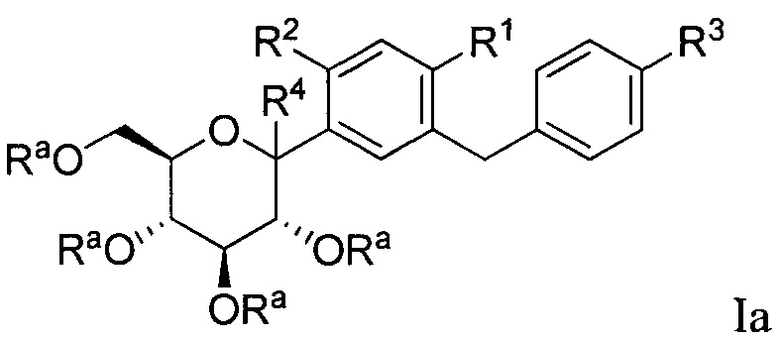 ,
,
включающий:
(a) получение первой реакционной смеси, содержащей соединение формулы IIa
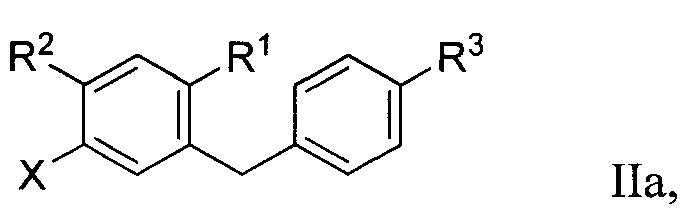
алкил-магниевый комплекс, выбранный из группы, состоящей из С1-С4 алкилмагния хлорида, С1-С4 алкилмагния бромида, ди(С1-С4 алкил)магния, С3-С7 циклоалкилмагния хлорида, С3-С7 циклоалкилмагния бромида и ди(С3-С7 циклоалкил)магния, и
первый органический растворитель,
где соотношение алкил-магниевого комплекса и соединения формулы IIa выбрано из 0,9, 0,91, 0,92, 0,93, 0,94, 0,95, 0,96 и 0,97 (моль/моль),
где первая реакционная смесь находится при температуре -50°С или ниже,
с образованием промежуточного соединения; и
(b) получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы IIIa
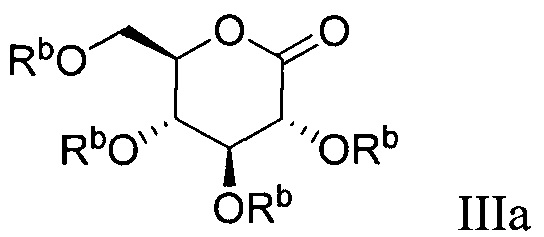
с образованием соединения формулы Ia, где R4 представляет собой ОН и каждый Ra представляет собой Rb;
(c) получение третьей реакционной смеси, содержащей C1-С3 алкилгидрокси, сильную кислоту и соединение формулы Ia, где R4 представляет собой ОН, и каждый Ra представляет собой Rb, с образованием соединения формулы Ia, где R4 представляет собой C1-С3 алкокси, и каждый Ra независимо выбран из группы, состоящей из Н и Rb; и
(d) получение четвертой реакционной смеси, содержащей восстановитель и соединение формулы Ia, где R4 представляет собой C1-С3 алкокси, и где реакционная смесь содержит менее 0,1 эквивалентов магния по отношению к количеству соединения формулы Ia, с образованием соединения формулы Ia, где R4 представляет собой Н,
где
X представляет собой иод,
R1 представляет собой хлор,
R2 представляет собой Н,
R3 представляет собой (С3-С6 циклоалкокси)С1-С3 алкокси,
R4 выбран из группы, состоящей из Н, ОН и C1-С3 алкокси,
каждый Ra независимо выбран из группы, состоящей из Н, C1-С3 алкила и Rb,
Rb представляет собой защитную группу,
где алкил, алкокси и циклоалкил группы или их части могут необязательно быть полностью или частично фторированными, и
один или больше атомов водорода могут быть необязательно заменены на атомы дейтерия.
2. Способ по п. 1, где R3 представляет собой 2-циклопропоксиэтокси.
3. Способ по любому из пп. 1, 2, где соотношение алкил-магниевого комплекса на стадии (а) и соединения формулы IIa составляет примерно 0,95 (моль/моль).
4. Способ по любому из пп. 1, 2, где первая реакционная смесь дополнительно содержит активирующий агент, выбранный из группы, состоящей из LiCl, ZnCl2, диизобутилалюминий гидрида, натрия бис(2-метоксиэтокси)алюминий гидрида, три-метилсилил хлорида и 2,2'-окси-бис(N,N-диметилэтанамина).
5. Способ по п. 4, где первая реакционная смесь дополнительно содержит LiCl.
6. Способ по любому из пп. 4, 5, где соотношение активирующего агента и алкил-магниевого комплекса составляет около 1,0 (моль/моль).
7. Способ по любому из пп. 1, 2, где первая реакционная смесь находится при температуре от примерно -60 до примерно -50°С.
8. Способ по любому из пп. 1, 2, где вторая реакционная смесь дополнительно содержит дополнительное количество алкил-магниевого комплекса.
9. Способ по п. 8, где соотношение дополнительного алкил-магниевого комплекса во второй реакционной смеси и соединения формулы IIa составляет от примерно 0,01 до примерно 0,1 (моль/моль).
10. Способ по любому из пп. 1, 2, где вторая реакционная смесь находится при температуре от примерно -60 до примерно 25°С.
11. Способ по любому из пп. 1, 2, где вторая реакционная смесь находится при температуре от примерно -60 до примерно -10°С.
12. Способ по любому из пп. 1, 2, где стадии (a)-(d) включают:
(a) получение первой реакционной смеси, содержащей соединение формулы IIa, имеющее структуру
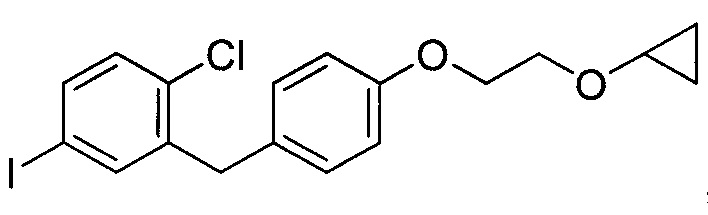 ,
,
изопропилмагния хлорид, хлорид лития, тетрагидрофуран и гептан, где соотношение изопропилмагния хлорида и соединения формулы IIa составляет примерно 0,95 (моль/моль), соотношение изопропилмагния хлорида и LiCl составляет 1,0 (моль/моль), где первая реакционная смесь находится при температуре -50°С или ниже, с образованием промежуточного соединения; и
(b) получение второй реакционной смеси, содержащей промежуточное соединение, второй органический растворитель и соединение формулы III, имеющее структуру
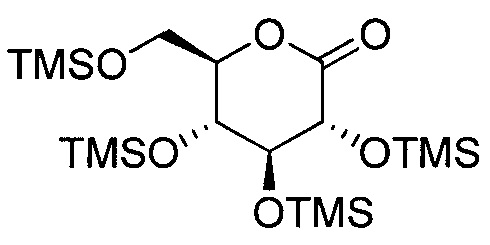 ,
,
с образованием соединения формулы Ia, в которой
R1 представляет собой хлор,
R2 представляет собой Н,
R3 представляет собой 2-циклопропоксиэтокси,
R4 представляет собой ОН, и
Ra представляет собой TMS;
(c) получение третьей реакционной смеси, содержащей C1-С3 алкилгидрокси, сильную кислоту и соединение формулы Ia, где R4 представляет собой ОН, и каждый Ra представляет собой TMS, с образованием соединения формулы Ia, где R4 представляет собой C1-С3 алкокси, и каждый Ra представляет собой Н; и
(d) получение четвертой реакционной смеси, содержащей восстановитель и соединение формулы Ia, где R4 представляет собой C1-С3 алкокси, и где реакционная смесь содержит менее 0,1 эквивалентов магния по отношению к количеству соединения формулы Ia, с образованием соединения формулы Ia, где R4 представляет собой Н.
13. Способ по п. 12, где вторая реакционная смесь дополнительно содержит дополнительное количество изопропилмагния хлорида и хлорида лития, где соотношение дополнительного изопропилмагния хлорида и соединения формулы IIa составляет от примерно 0,01 до примерно 0,1 (моль/моль), и соотношение дополнительного изопропилмагния хлорида и дополнительного LiCl составляет 1,0 (моль/моль).
14. Способ по любому из пп. 1, 2, где первую и вторую реакционные смеси получают в одном и том же реакционном сосуде.
15. Способ по любому из пп. 1, 2, где каждый Rb в соединении формулы Ia представляет собой кислотно-лабильную защитную группу, и при удалении защитных групп образуется соединение формулы Ia, в котором каждый Ra представляет собой Н.
16. Способ по любому из пп. 1, 2, где третья реакционная смесь находится при температуре от примерно -10 до примерно 25°С.
17. Способ по п. 16, где третья реакционная смесь находится при температуре около 0°С.
18. Способ по любому из пп. 1, 2, где четвертая реакционная смесь находится при температуре от примерно -40 до примерно -10°С.
19. Способ по п. 18, где четвертая реакционная смесь находится при температуре от примерно -25 до примерно -22°С.
20. Способ по любому из пп. 1, 2, где соединение формулы Ia имеет структуру
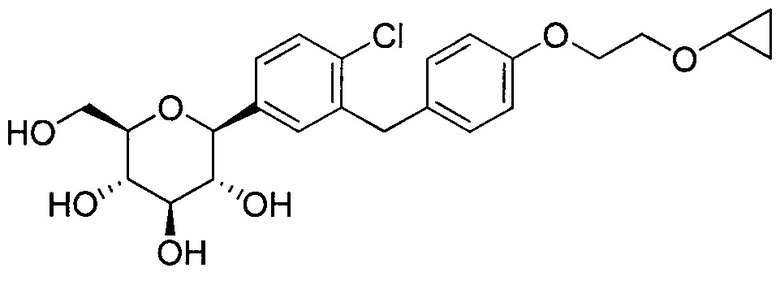
21. Способ получения соединения формулы IIa
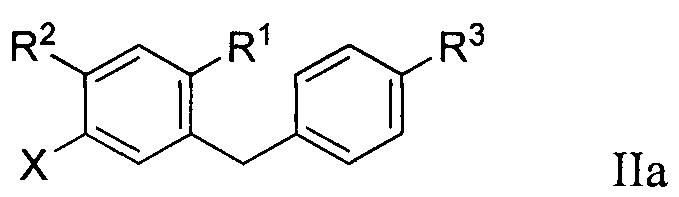 ,
,
включающий получение первой реакционной смеси, содержащей соединение формулы IV
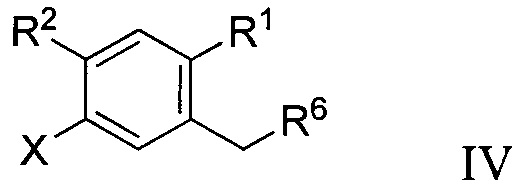
и соединение формулы V
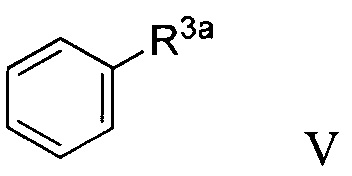
в условиях, подходящих для получения соединения формулы IIa,
где
R1 выбран из группы, состоящей из атома водорода, галогена, гидрокси-группы, C1-С3 алкила и C1-С3 алкокси,
R2 и R3 каждый независимо выбран из группы, состоящей из следующих представителей: атом водорода, галоген, гидрокси-группа, C1-С3 алкил, C1-С3 алкокси, С3-С6 циклоалкил, (C1-С3 алкокси)C1-С3 алкил, (C1-С3 галогеналкокси)C1-С3 алкил, (С2-С4 алкенилокси)C1-С3 алкил, (С2-С4 алкинилокси)C1-С3 алкил, (С3-С6 циклоалкокси)C1-С3 алкил, C1-С3 гидроксиалкокси, С3-С6 циклоалкокси, С3-С6 гетероциклоалкокси, (C1-С3 алкокси)C1-С3 алкокси, (C1-С3 галогеналкокси)C1-С3 алкокси, (С2-С4 алкенилокси)C1-С3 алкокси, (С2-С4 алкинилокси)C1-С3 алкокси, (С3-С6 циклоалкокси)C1-С3 алкокси, (С3-С6 гетероциклоалкокси)C1-С3 алкокси, (С3-С6 циклоалкил)C1-С3 алкокси, (С3-С6 циклоалкил)С2-С4 алкенилокси и (С3-С6 циклоалкил)С2-С4 алкинилокси,
R3a представляет собой ОН,
R6 выбран из группы, состоящей из ОН и Br,
X представляет собой бром или иод,
где алкил, алкокси, циклоалкил, алкенилокси, алкинилокси, циклоалкокси, гидроксиалкокси и гетероциклоалкокси группы или их части могут необязательно быть частично или полностью фторированными, и
один или больше атомов водорода могут необязательно быть заменены на атомы дейтерия.
22. Способ по п. 21, где первая реакционная смесь дополнительно содержит кислоту Льюиса.
23. Способ по п. 22, где кислота Льюиса выбрана из группы, состоящей из BF3⋅Et2O, BCl3, BBr3, B(C6F5)3, SnCl4, I2, FeCl3, FeBr3, TMSOTf-AgClO4, AgOTf, Cu(OTf)2, Bi(OTf)3, In(OTf)3, Zn(NTf2)2, AuCl3, HgCl2, HgSO4, Hg(OCOCF3)2, PdCl2, Pd(OAc)2, ZnCl2, ZnBr2, ZnI2, триметилсилилового эфира полифосфорной кислоты, AlCl3, AlBr3, AlI3, Al(OiPr)3, Al(OPh)3, TiCl4, TiCl2(OiPr)2, Ti(OiPr)4, PBr3, BeCl2, CdCl2, CeCl3, DyCl3, EuCl3, Eu(OTf)3, ErCl3, Er(OTf)3, GaCl3, GdCl3, Gd(OTf)3, HoCl3, LaCl3, La(OTf)3, LuCl3, Lu(OTf)3, Mg(ClO4)2, MgCl2, MgBr2, MgI2, NdCl3, Nd(OTf)3, PCl3, PBr3, PrCl3, Pr(OTf)3, PmCl3, Pm(OTf)3, Sc(OTf)3, SnCl4, SbCl5, SmCl3, Sm(OTf)3, Tf2O, TbCl3, Tb(OTf)3, TmCl3, Tm(OTf)3, YbCl3, Yb(OTf)3, ZrCl4 и Cp2ZrCl2.
24. Способ по любому из пп. 22, 23, где кислота Льюиса представляет собой ZnCl2.
25. Способ по любому из пп. 21-23, где
R1 представляет собой хлор;
R2 представляет собой Н; и
X представляет собой иод.
26. Способ по любому из пп. 21-23, где R3 представляет собой гидрокси-группу.
27. Способ по любому из пп. 21-23, где соединение формулы IIa имеет структуру
 .
.
28. Способ по любому из пп. 21-23, где указанный способ включает получение первой реакционной смеси, содержащей соединение формулы IV, имеющее структуру
 ,
,
соединение формулы V, имеющее структуру
 ,
,
газообразную бромисто-водородную кислоту и ZnCl2, с образованием соединения формулы IIa, имеющего структуру
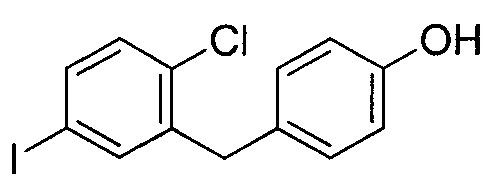 .
.
29. Способ по любому из пп. 21-23, где указанный способ дополнительно включает получение второй реакционной смеси, содержащей соединение формулы IIa, имеющее структуру
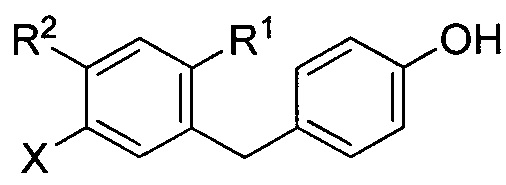 ,
,
и соединение формулы VI
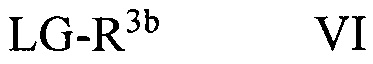 ,
,
где
R3b выбран из группы, состоящей из следующих представителей: C1-С3 алкил, С3-С6 циклоалкил, С3-С6 гетероциклоалкил, (C1-С3 алкокси)C1-С3 алкил, (C1-С3 галогеналкокси)C1-С3 алкил, (С2-С4 алкенилокси)C1-С3 алкил, (С2-С4 алкинилокси)C1-С3 алкил, (С3-С6 циклоалкокси)C1-С3 алкил, C1-С3 гидроксиалкил, (С3-С6 гетероциклоалкокси)C1-С3 алкил, (С3-С6 циклоалкил)С3-С4 алкенил и (С3-С6 циклоалкил)С3-С4 алкинил, и
LG представляет собой уходящую группу,
с образованием соединения формулы IIa
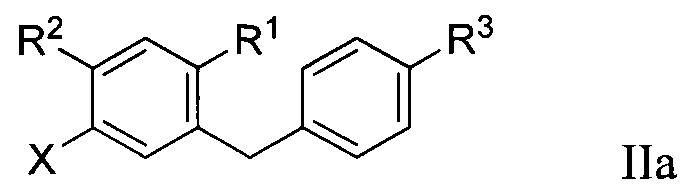 ,
,
где
R3 выбран из группы, состоящей из следующих представителей: C1-С3 алкокси, С3-С6 циклоалкилокси, С3-С6 гетероциклоалкокси, (C1-С3 алкокси)C1-С3 алкокси, (C1-С3 галогеналкокси)C1-С3 алкокси, (С2-С4 алкенилокси)C1-С3 алкокси, (С2-С4 алкинилокси)C1-С3 алкокси, (С3-С6 циклоалкокси)C1-С3 алкокси, C1-С3 гидроксиалкокси, (С3-С6 гетероциклоалкокси)C1-С3 алкокси, (С3-С6 циклоалкил)С3-С4 алкенилокси и (С3-С6 циклоалкил)С3-С4 алкинилокси.
30. Способ по п. 29, где
R1 представляет собой галоген;
R2 представляет собой Н; и
R3 выбран из группы, состоящей из C1-С3 алкокси и (С3-С6 циклоалкокси)C1-С3 алкокси.
31. Способ по п. 29, где
R1 представляет собой хлор;
R2 представляет собой Н; и
R3 выбран из группы, состоящей из этокси и 2-циклопропоксиэтокси.
32. Способ по п. 29, где
LG выбран из группы, состоящей из хлорида, бромида, иодида, гидрокси-группы, тозилата и мезилата; и
R3 выбран из группы, состоящей из C1-С3 алкила и (С3-С6 циклоалкокси)С1-С3 алкила.
33. Способ по пп. 29 и 31, 32, где
LG выбран из группы, состоящей из хлорида, бромида и тозилата; и
R3 выбран из группы, состоящей из этила и 2-циклопропоксиэтила.
34. Способ по п. 29, где соединение формулы VI имеет структуру
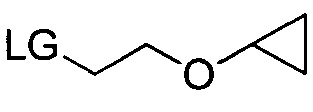 .
.
35. Способ по п. 29, включающий получение второй реакционной смеси, содержащей соединение формулы VI, имеющее структуру
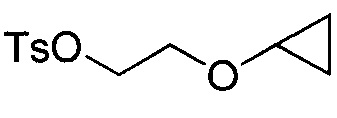 ,
,
и соединение формулы IIa, имеющее структуру
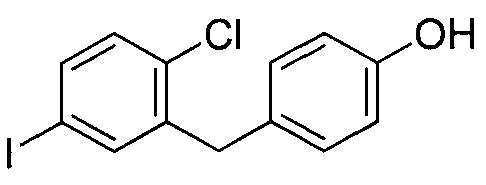 ,
,
с образованием соединения формулы IIa, имеющего структуру
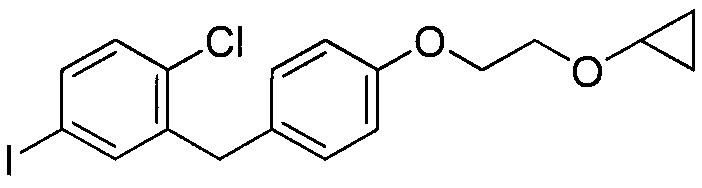 .
.
36. Способ по любому из пп. 1, 2, где соединение формулы IIa получено способом по любому из пп. 21-35.
37. Способ по п. 1, в котором осуществление стадий (a)-(d) приводит к получению смеси продуктов, содержащей по меньшей мере 95% соединения формулы Ia, имеющего структуру
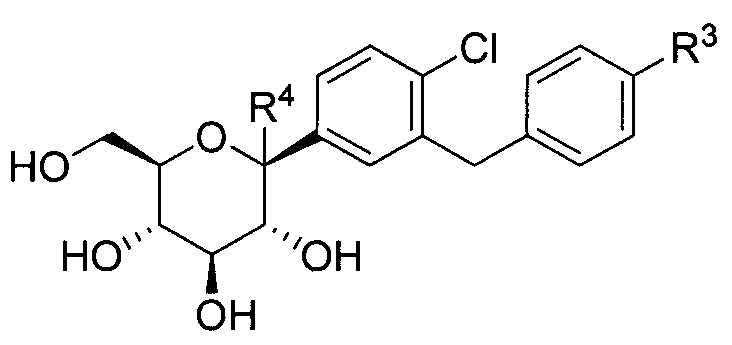 ,
,
побочный продукт А, имеющий структуру
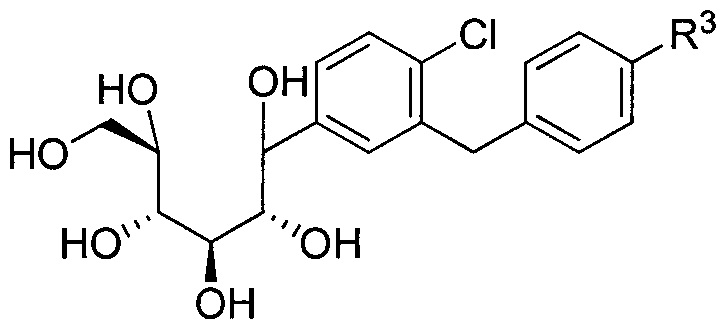 ,
,
в количестве, составляющем менее примерно 1% от смеси продуктов, и
побочный продукт В, имеющий структуру
 ,
,
в количестве, составляющем менее примерно 3% от смеси продуктов.
38. Способ по п. 12, в котором осуществление стадий (a)-(d) приводит к получению смеси продуктов, содержащей по меньшей мере 95% соединения формулы Ia, имеющего структуру
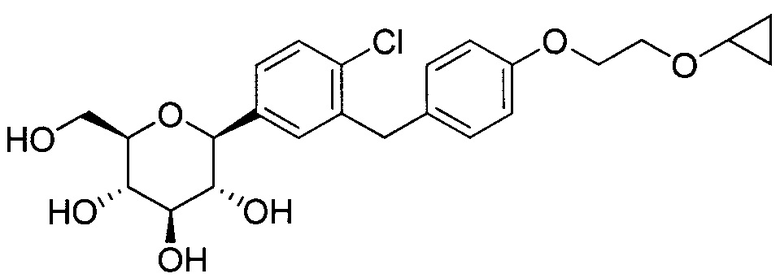 ,
,
побочный продукт А, имеющий структуру
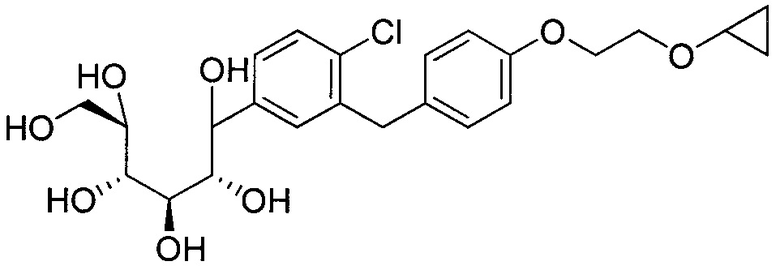 ,
,
в количестве, составляющем менее примерно 1% от смеси продуктов, и
побочный продукт В, имеющий структуру
 ,
,
в количестве, составляющем менее примерно 3% от смеси продуктов.
| WO 2009026537 A1 26.02.2009 | |||
| EP 1845095 A1 17.10.2007 | |||
| RU 2007113701 A 27.10.2008 | |||
| RU 20072109477 A 20.10.2004. |

