Область техники
Данное изобретение относится к области химии технологических процессов и, в частности, касается получения аммиака электролитическими способами и предназначенных для этого новых катализаторов.
Введение
Аммиак является одним из химических веществ, производимых в мире в наиболее высоком объеме. Промышленный синтез аммиака, называемый в настоящее время процессом Габера-Боша, является первой гетерогенной каталитической системой, ключевым элементом глобального промышленного производства азотных удобрений. На сегодняшний день аммиак также привлекает внимание в качестве возможного энергоносителя и потенциального топлива для транспорта с высокой удельной энергоемкостью, но без выбросов CO2. Централизованный и энергоемкий процесс Габера-Боша требует высокого давления (150-350 атм) и высокой температуры (350-550 °С) для непосредственной диссоциации и объединения молекул газа азота и водорода над катализатором на основе рутения или железа с образованием аммиака по следующей реакции:
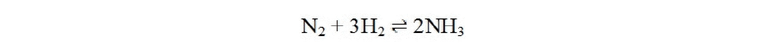
Недостатком этого промышленного подхода является высокая температура и давление, необходимые ввиду кинетических и термодинамических причин. Другой и более серьезный недостаток заключается в том, что газообразный водород получают из природного газа. Этот многостадийный процесс занимает большую часть всего химического производства и является наиболее дорогостоящим и неблагоприятным для окружающей среды. Это обстоятельство представляет собой главную причину, по которой необходима разработка самодостаточного процесса, поскольку в какой-то момент запасы природного газа будут исчерпаны. В этой связи большое значение будет иметь мелкомасштабная система для децентрализованного производства аммиака, в которой используется меньше энергии и условия окружающей среды. Кроме того, для оптимизации эффективности синтеза аммиака будут высоко цениться новые катализаторы, способные гидрировать молекулярный азот с разумной скоростью, но в более мягких условиях.
Тройная связь в молекулярном азоте N2 очень прочная и, как следствие, азот является очень неактивным и часто используется в качестве инертного газа. Она разрывается в жестких условиях процесса Габера-Боша, однако она также разрывается при условиях окружающей среды в ходе природного процесса, осуществляемого микроорганизмами с помощью фермента нитрогеназы. Активным центром нитрогеназы является кластер MoFe7S9N, катализирующий образование аммиака из сольватированных протонов, электронов и атмосферного азота посредством электрохимической реакции
N2 + 8H+ + 8e- → 2NH3 + H2
Вдохновленная природой, биологическая фиксация азота в качестве альтернативы процессу Габера-Боша для синтеза аммиака при условиях окружающей среды привлекает большое внимание. Много сил было вложено в исследования, направленные на разработку аналогичных электрохимических процессов. В последние десятилетия были исследованы различные способы синтеза аммиака при условиях окружающей среды. (Giddey S, Int J Hydrogen Energy 2013, 38, 14576-14594; Amar A, J Solid State Electrochem 2011, 15, 1845-60; Shipman MA, Catalysis Today 2017, 286, 57-68). Хотя такие исследования позволили понять процесс образования аммиака, их кинетика все еще является слишком медленной для практического применения, и в большинстве случаев газообразный водород в основном образуется быстрее, чем происходит протонирование азота. Восстановление молекулярного азота протонами и электронами для селективного образования аммиака при комнатной температуре и давлении оказалось гораздо сложнее, чем ожидалось.
Авторы изобретения ранее установили (WO 2015/189865), что некоторые катализаторы на основе нитридов металлов могут использоваться в электрохимических процессах получения аммиака. Однако остается неясным, могут ли другие или дополнительные соединения металлов быть эффективными при получении аммиака. Другие попытки искусственного синтеза аммиака с использованием электрохимических методов привели к относительно низким коэффициентам использования тока (CE). В случае многих из них регенерация активного азотфиксирующего комплекса оказалась проблематичной, и скорости получения далеки от рентабельности.
Краткое описание изобретения
Вышеуказанные признаки вместе с дополнительными деталями изобретения дополнительно описаны в приведенных ниже примерах, которые предназначены для дополнительной иллюстрации изобретения, но не предназначены для ограничения его объема каким-либо образом.
Авторы настоящего изобретения обнаружили, что определенные катализаторы на основе оксидов переходных металлов могут быть использованы в электрохимических процессах получения аммиака. Это привело к созданию настоящего изобретения, которое делает возможным получение аммиака при комнатной температуре окружающей среды и атмосферном давлении.
Настоящее изобретение относится к способу получения аммиака, включающему подачу N2 в электролитическую ячейку, содержащую по меньшей мере один источник протонов; обеспечение возможности контакта N2 с поверхностью катодного электрода в электролитической ячейке, где поверхность катодного электрода содержит каталитическую поверхность, содержащую по меньшей мере один оксид переходного металла; и пропускание тока через указанную электролитическую ячейку, в результате чего азот реагирует с протонами с образованием аммиака.
Изобретение также относится к системе для генерирования аммиака, в частности, системе, которая осуществляет способ получения аммиака, раскрытый в настоящем документе. Таким образом, изобретение относится к системе для генерирования аммиака, содержащей по меньшей мере одну электрохимическую ячейку, содержащую по меньшей мере один катодный электрод, имеющий каталитическую поверхность, где указанная каталитическая поверхность заполнена по меньшей мере одним катализатором, содержащим один или более оксидов переходных металлов.
В способе и системе в соответствии с изобретением оксид переходного металла может быть выбран из группы, состоящей из оксида титана, оксида хрома, оксида марганца, оксида ниобия, оксида тантала, оксида рутения, оксида родия, оксида платины, оксида осмия, оксида рения и оксида иридия. В некоторых предпочтительных вариантах осуществления оксид выбран из группы, состоящей из оксида рения, оксида тантала и оксида ниобия.
Каталитическая поверхность может содержать по меньшей мере одну поверхность, имеющую структуру рутила, в частности, структуру рутила, имеющую грань (110), на которой происходят каталитические реакции. В некоторых вариантах осуществления катализатор содержит поверхность, имеющую мостиковые участки между шестикратно координированными атомами переходного металла, покрытые атомами водорода.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Специалисту в данной области будет ясно, что фигуры, описанные ниже, предназначены только для целей иллюстрации. Фигуры никоим образом не предназначены для ограничения объема настоящего изобретения.
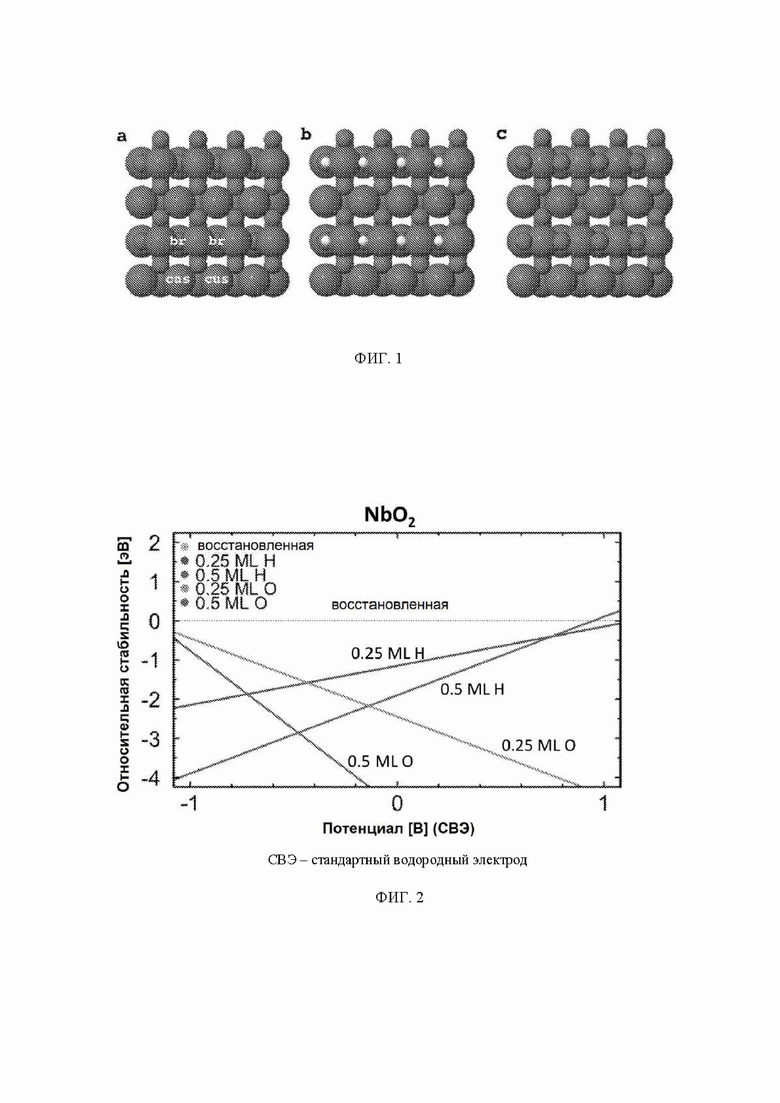
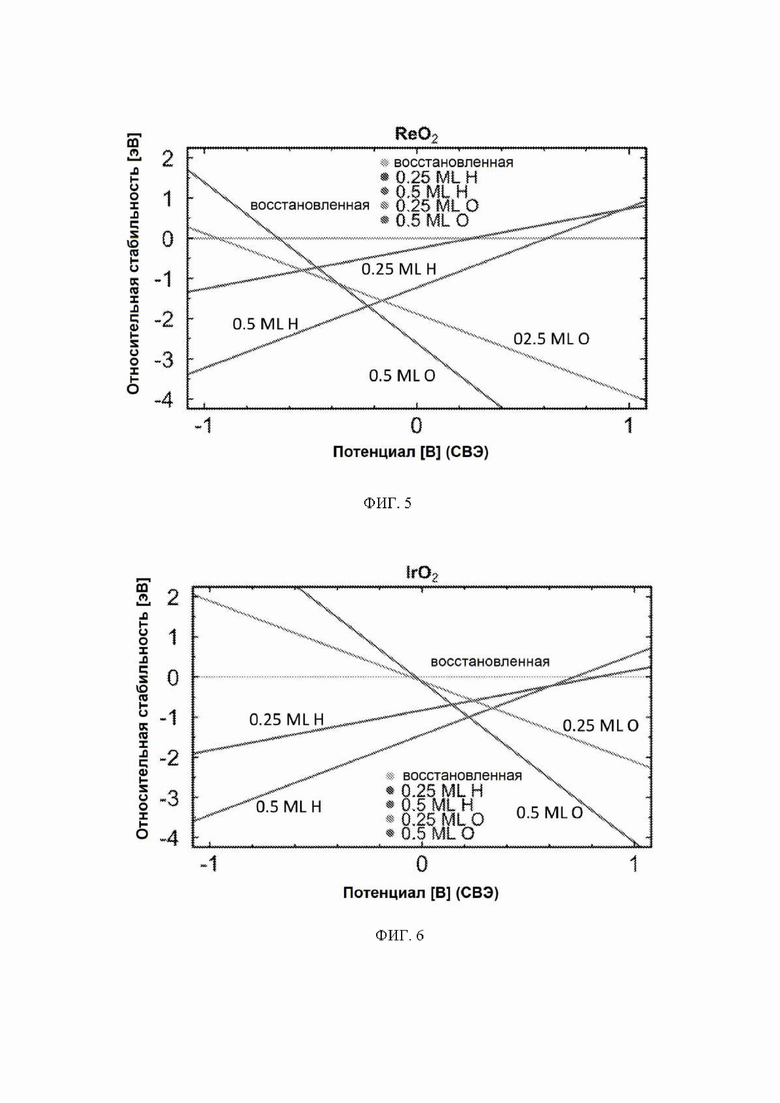
На ФИГ. 1 показаны три разные поверхности на грани (110). (а) Восстановленная поверхность находится слева, где мостиковые участки между шестикратно координированными атомами металла остаются вакантными. Мостиковые участки и участки cus отмечены на рисунке восстановленной поверхности. (b) Поверхность, модифицированная 0,5 ML (монослой) водорода, представляет собой структуру в центре, где атомы водорода занимают br-участки. (c) Поверхности, модифицированные 0,5 ML кислорода, где кислород занимает br-участки вместо водорода, изображены справа.
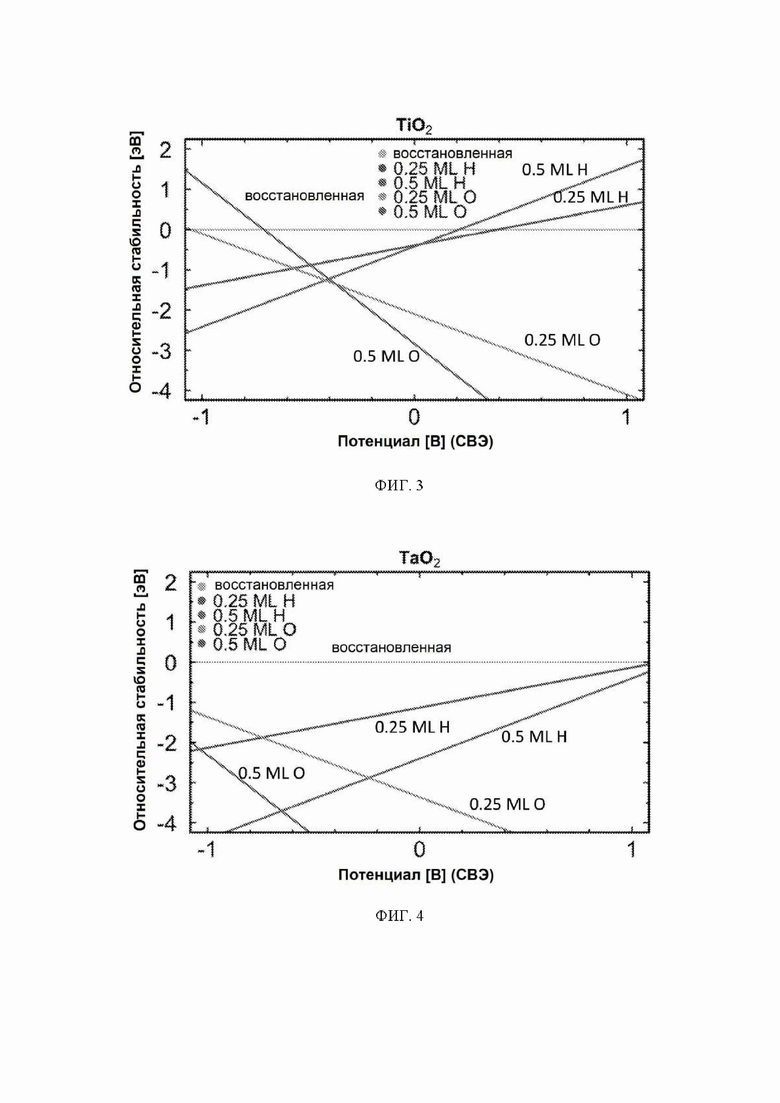
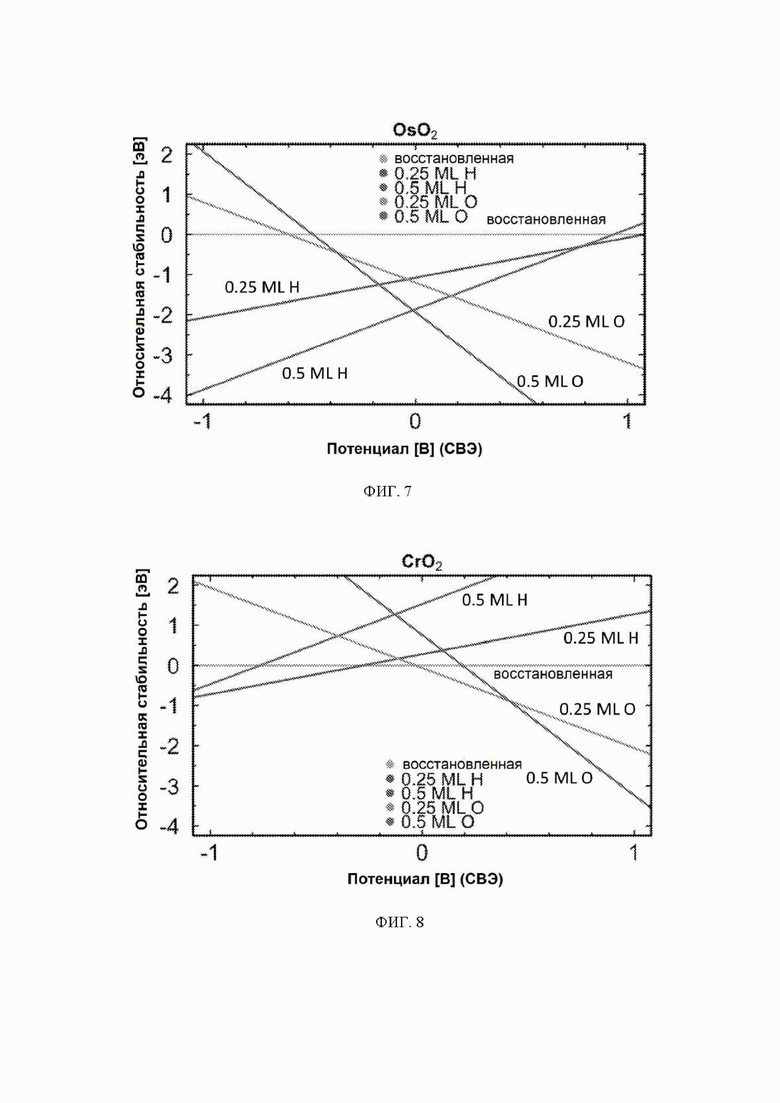
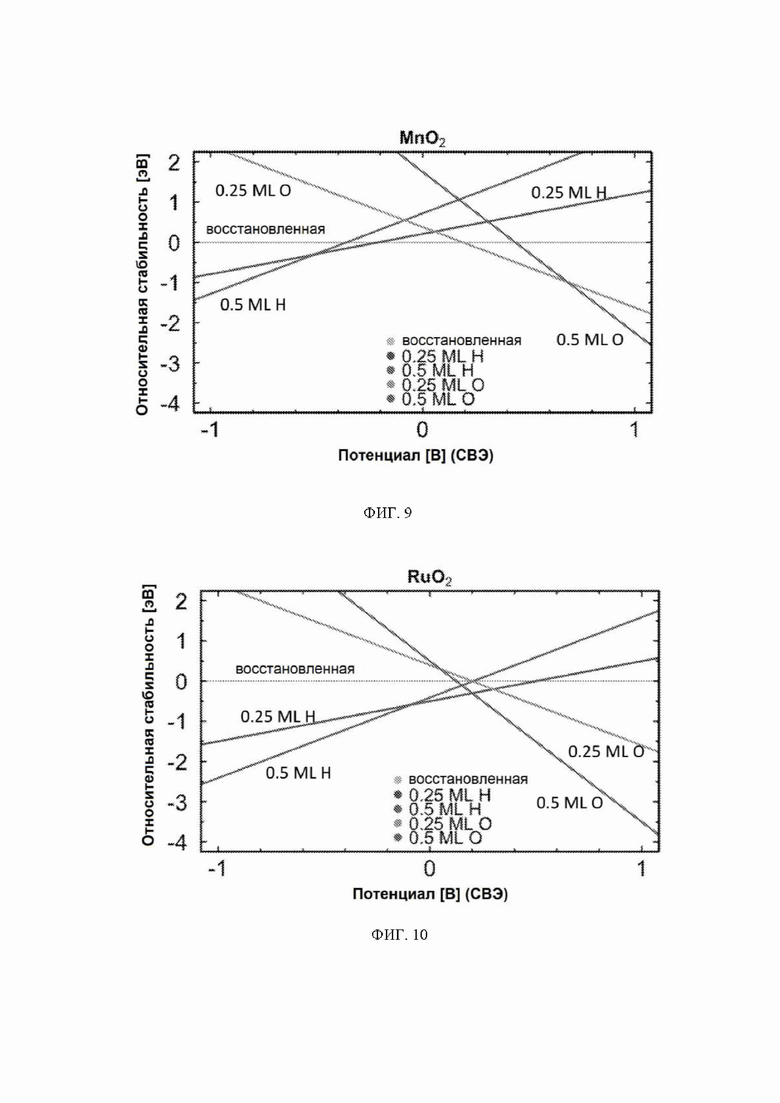
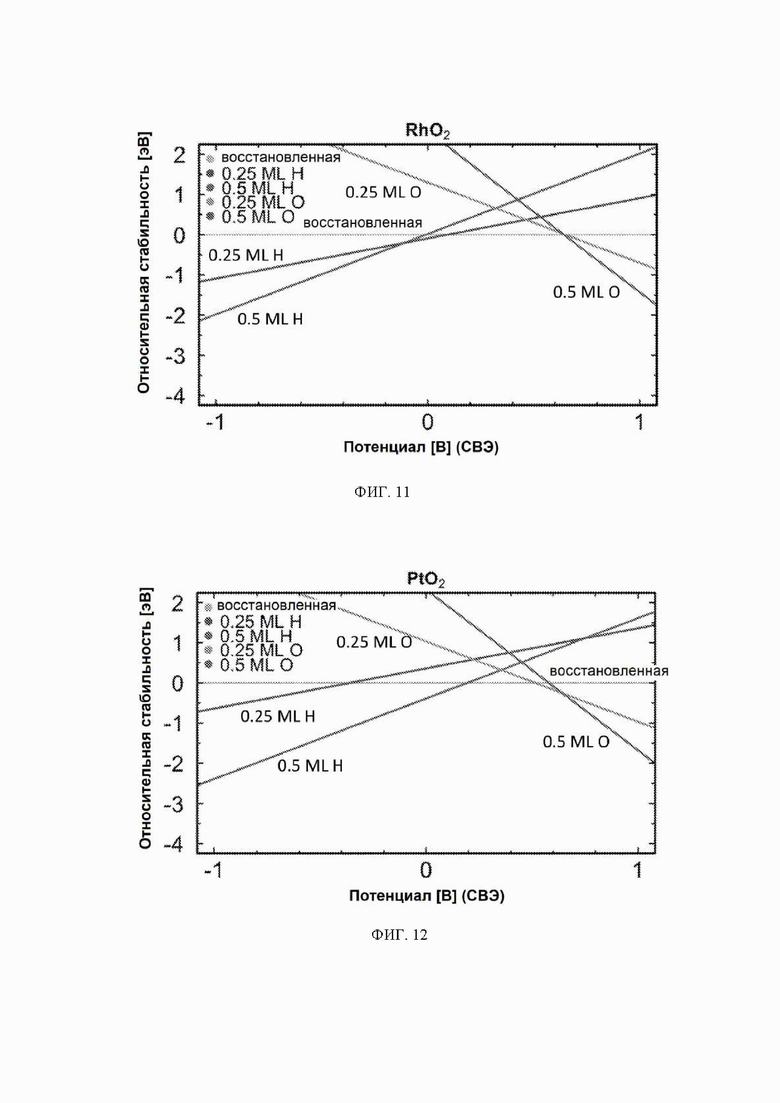
На ФИГ. 2-12 показана относительная стабильность адсорбатов, образованных восстановлением протонов и окислением воды на различных поверхностях оксидов переходных металлов (110) в структуре рутила. Верхняя (плоская) линия на каждой диаграмме показывает восстановленную поверхность, используемую в качестве эталона, где все атомы кислорода мостика были восстановлены до H2O. Показана относительная стабильность адсорбатов для граней (110) NbO2, TiO2, TaO2, ReO2, IrO2, OsO2, CrO2, MnO2, RuO2, RhO2 и PtO2, соответственно.
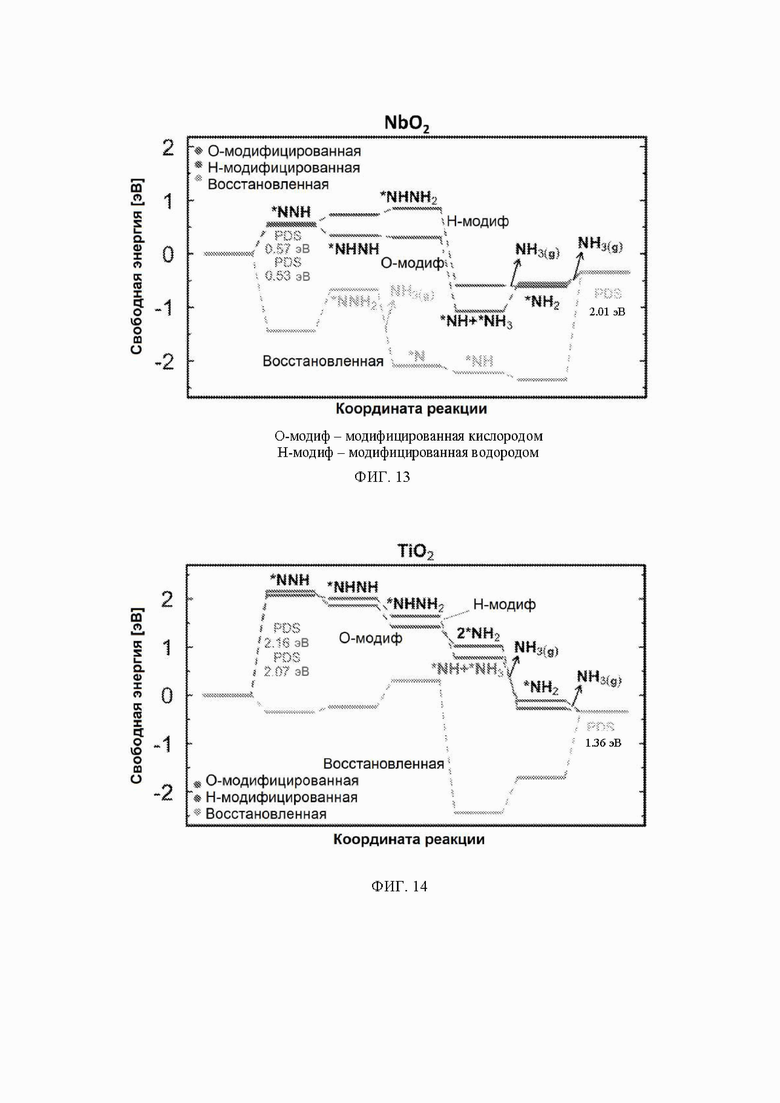
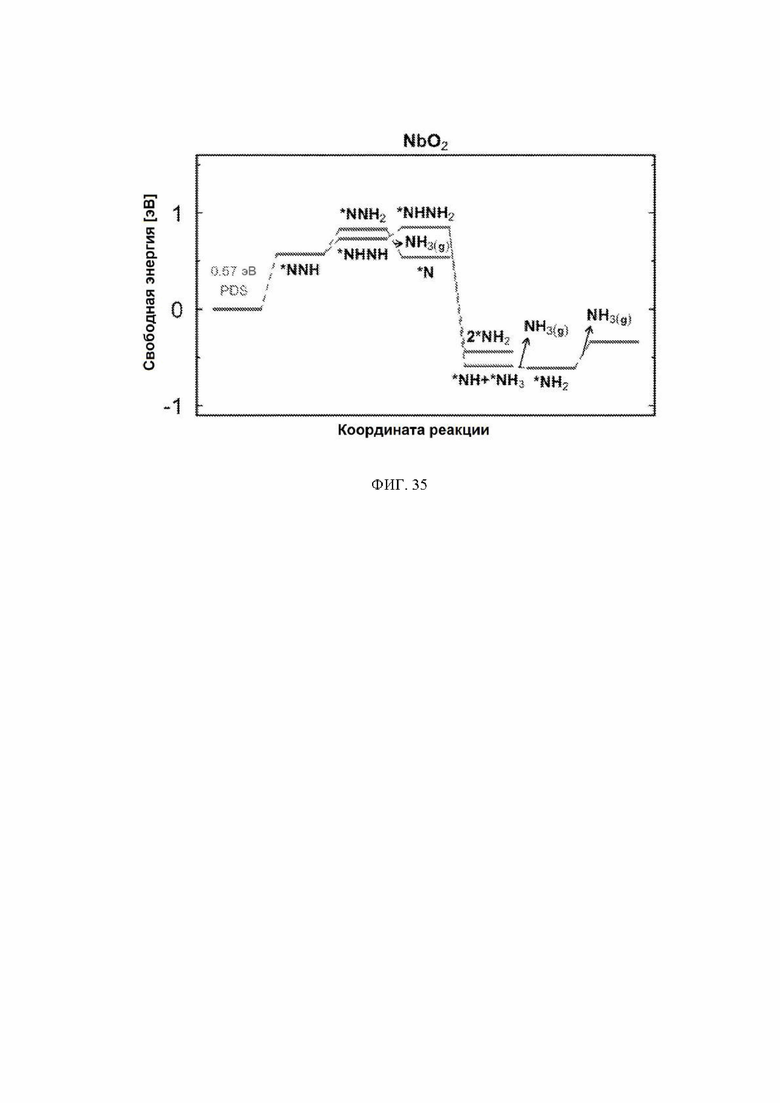
На ФИГ. 13 показана диаграмма свободной энергии для образования NH3 на грани (110) рутила NbO2. Стадия определения потенциала (PDS) для поверхности, модифицированной водородом, является первой стадией протонирования, тогда как PDS для поверхности, модифицированной кислородом, и восстановленной поверхности является последней стадией протонирования от *NH2 до NH3. Все стадии реакции относятся к чистой поверхности и N2 и H2 в газовой фазе. Десорбция газообразного аммиака обозначена стрелками. Промежуточное соединение черного цвета относится к поверхностям, где не указано другое промежуточное соединение, тогда как промежуточное соединение другого цвета относится только к этой конкретной поверхности.
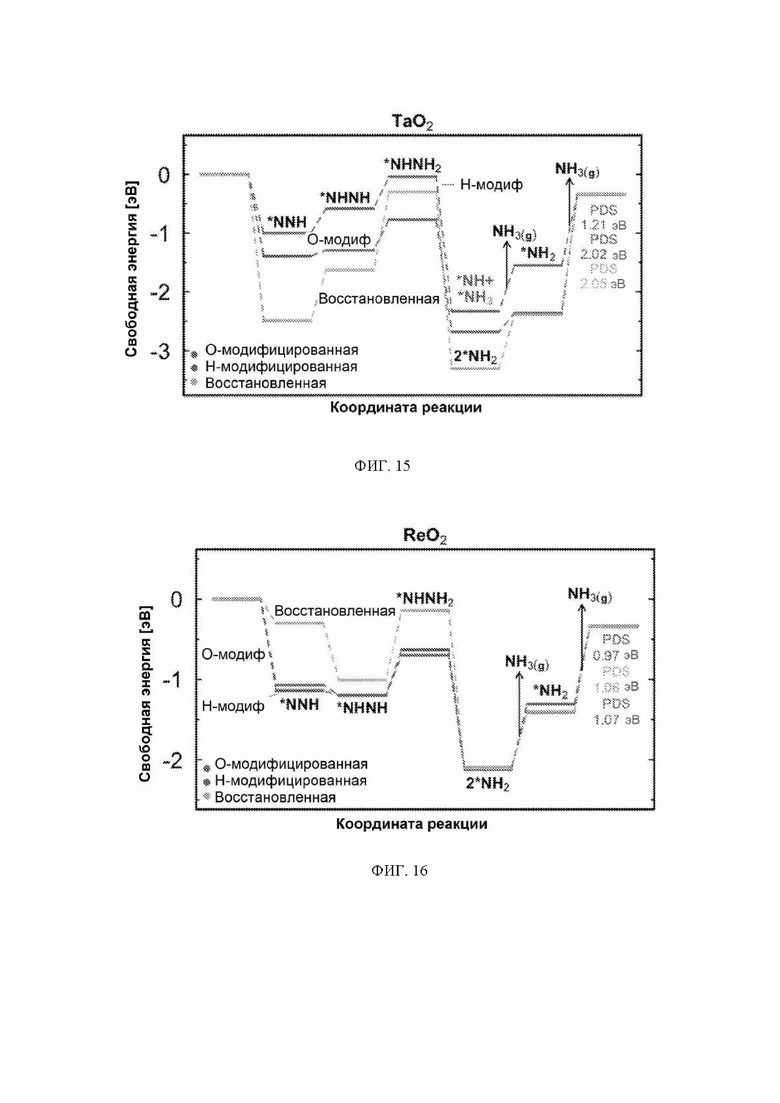
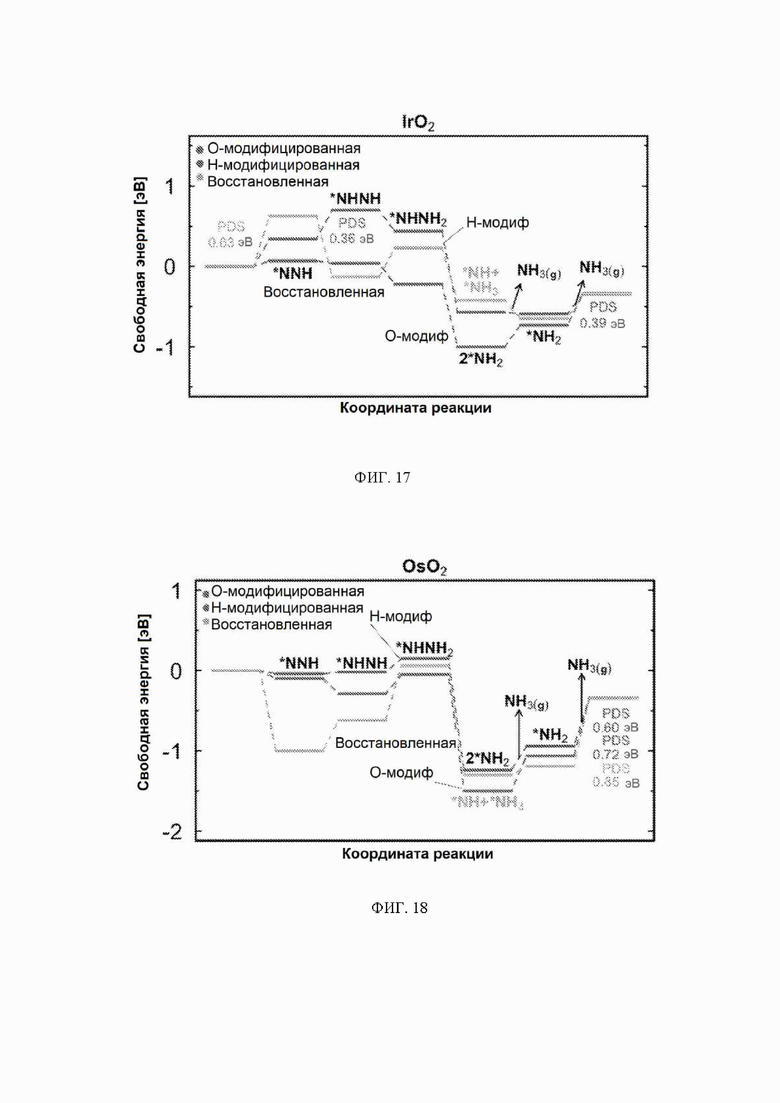
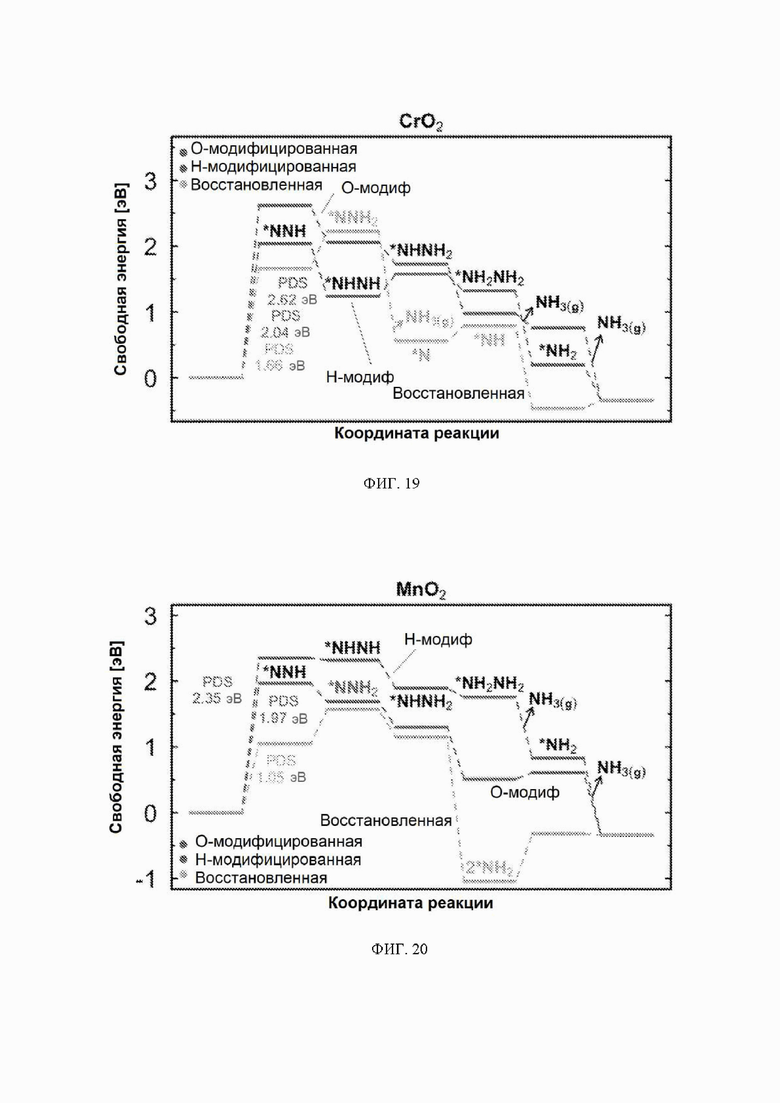
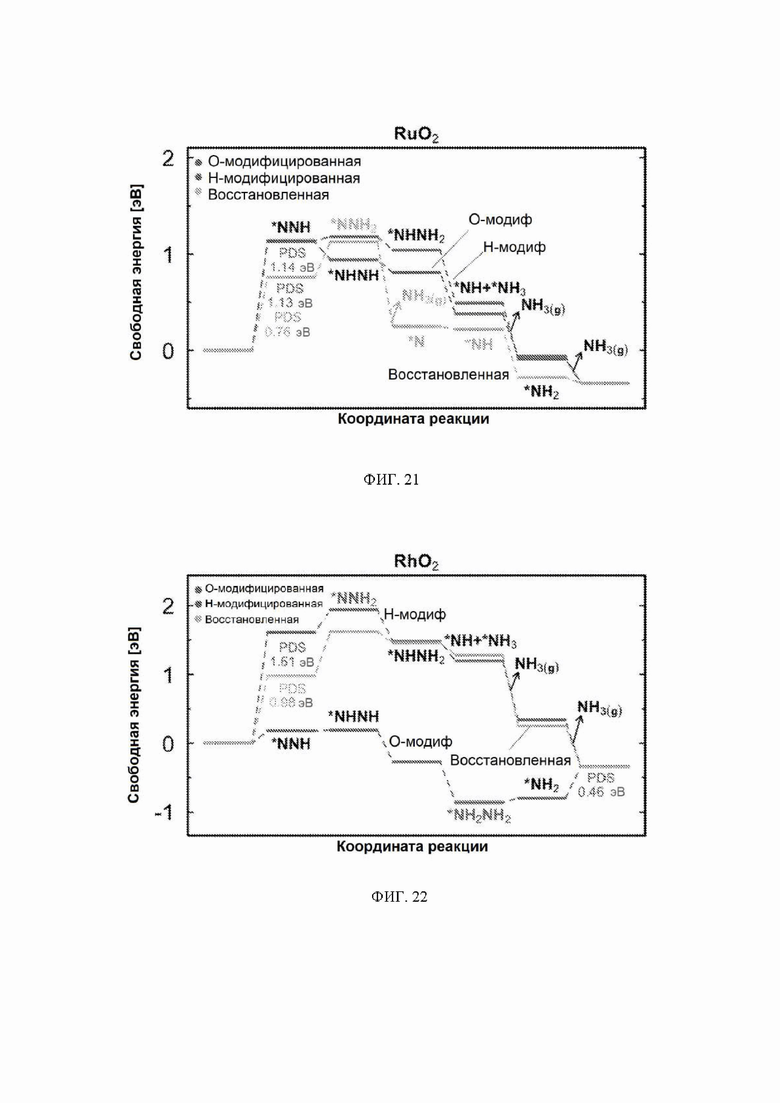
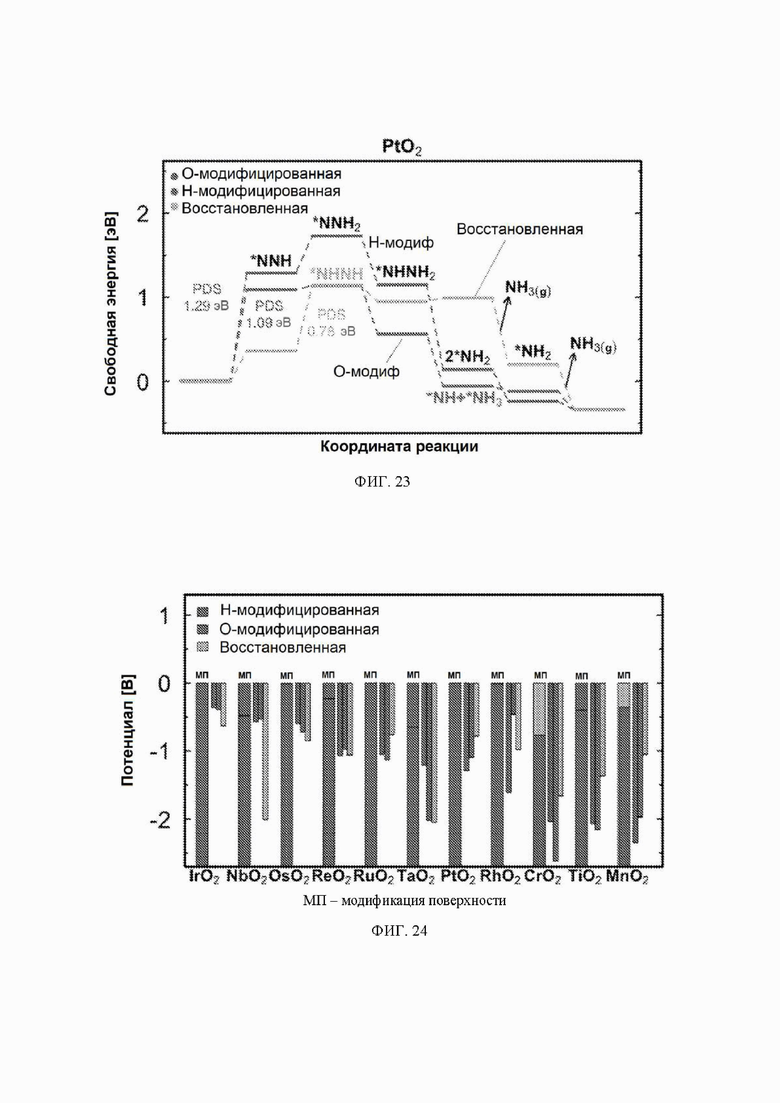
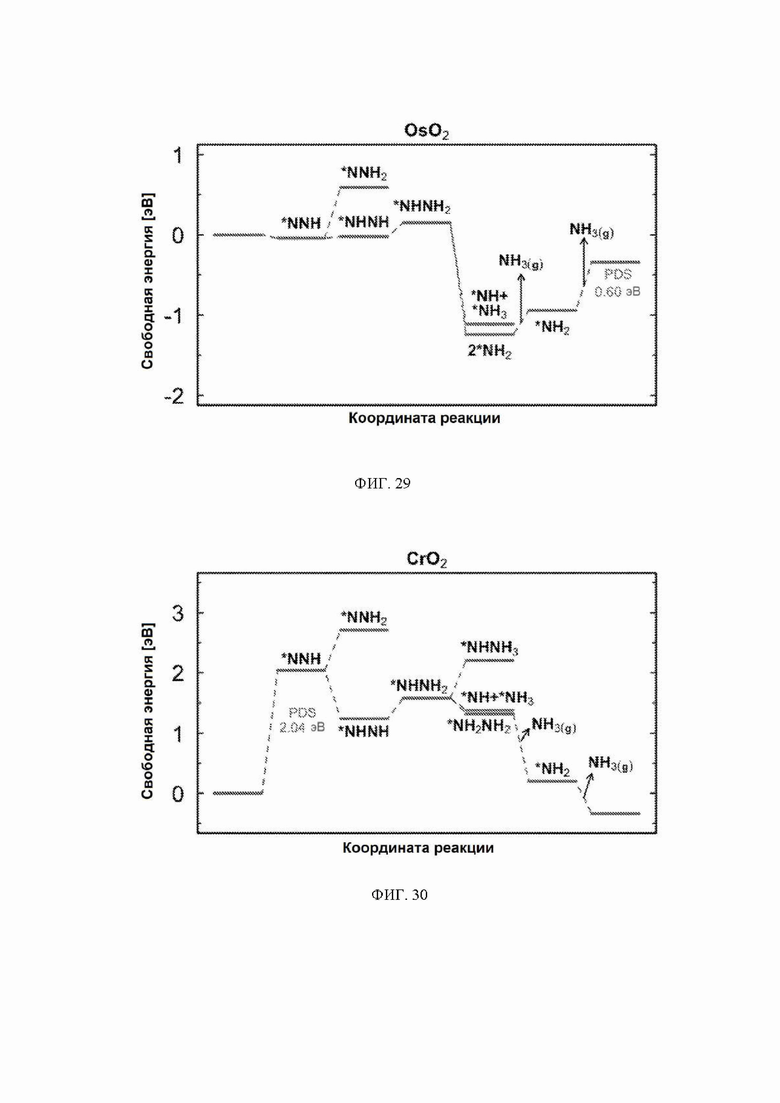
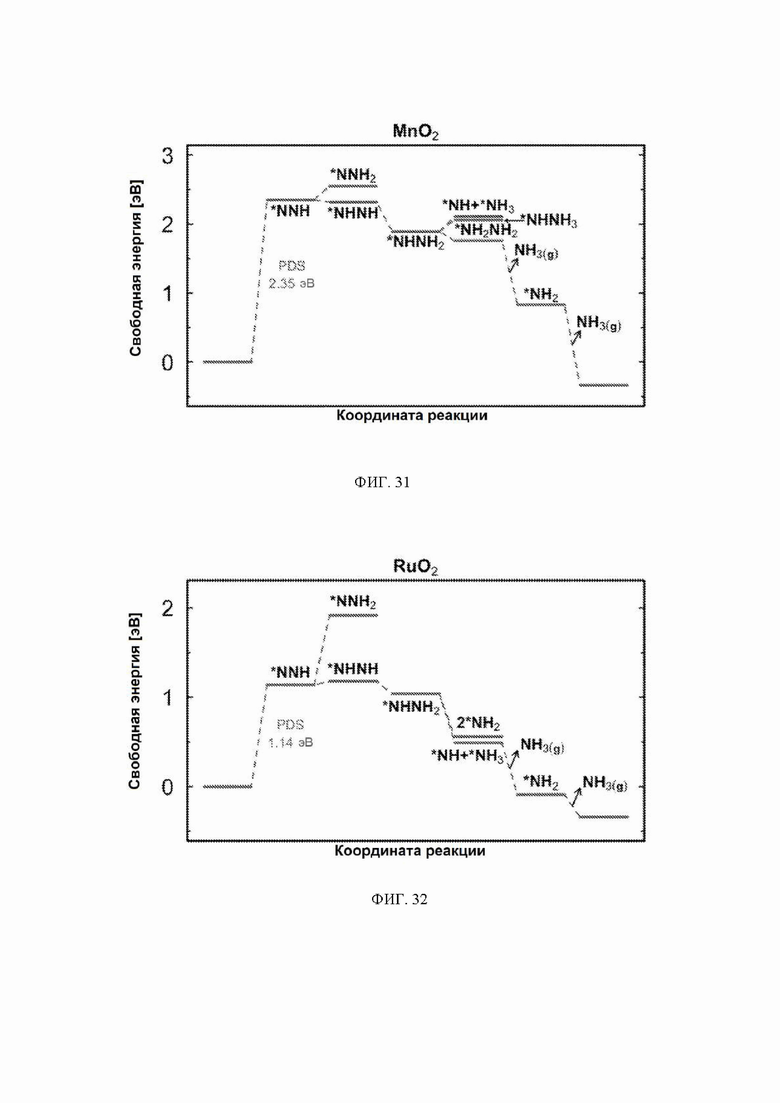
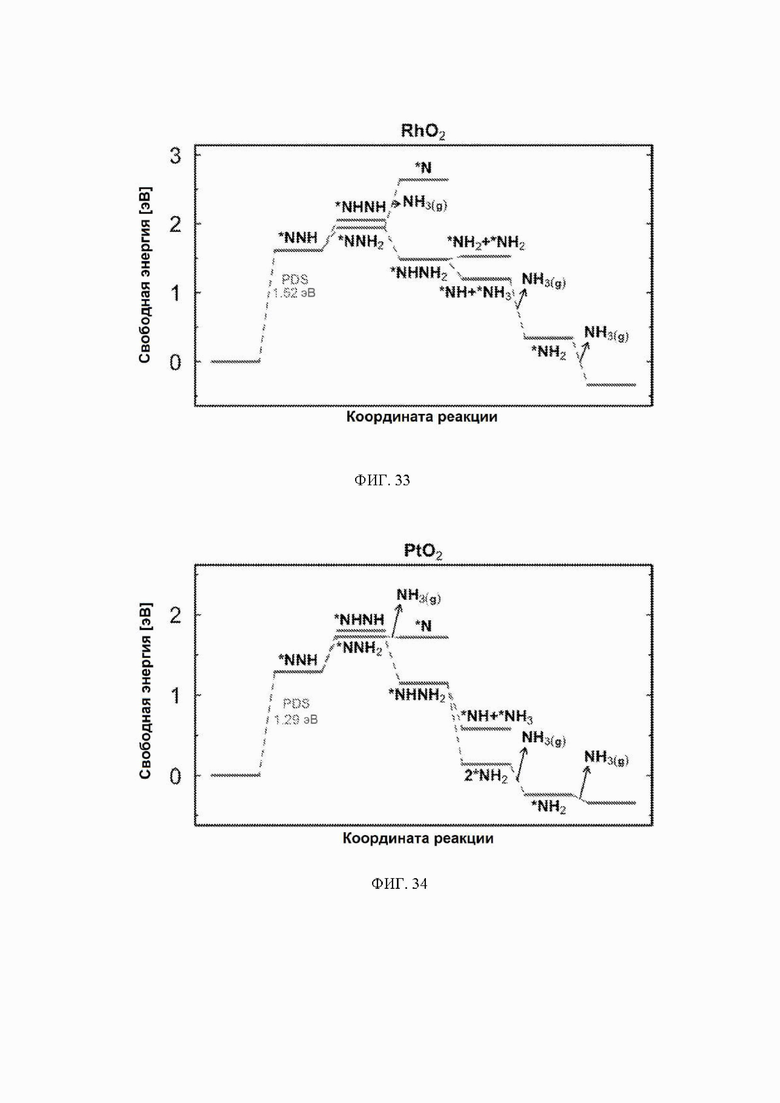
На ФИГ. 14-23 показаны диаграммы свободной энергии для электрохимического образования аммиака на гранях (110) TiO2, TaO2, ReO2, IrO2, OsO2, CrO2, MnO2, RuO2, RhO2 и PtO2, соответственно, в структуре рутила.
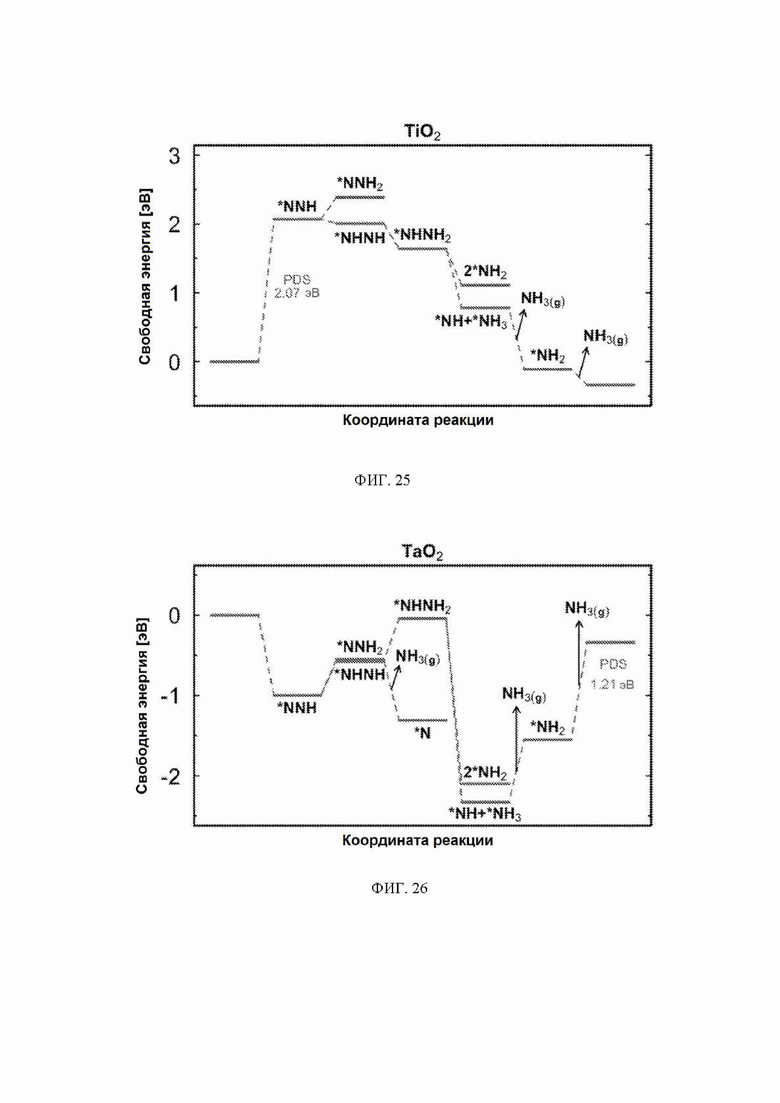
На ФИГ. 24 показаны прогнозируемые начальные потенциалы (ΔG = -U) для каждой из оксидных поверхностей, модифицированных различным образом; модифицированной водородом, модифицированной кислородом и восстановленной. Более широкие столбцы (слева от начальных потенциалов для каждого оксида) отражают предпочтительную модификацию поверхности (МП) при изменении приложенного потенциала, где потенциал показан на оси y. Оксиды расположены слева направо по возрастающей величине стадии определения потенциала (PDS) на поверхности, модифицированной водородом.
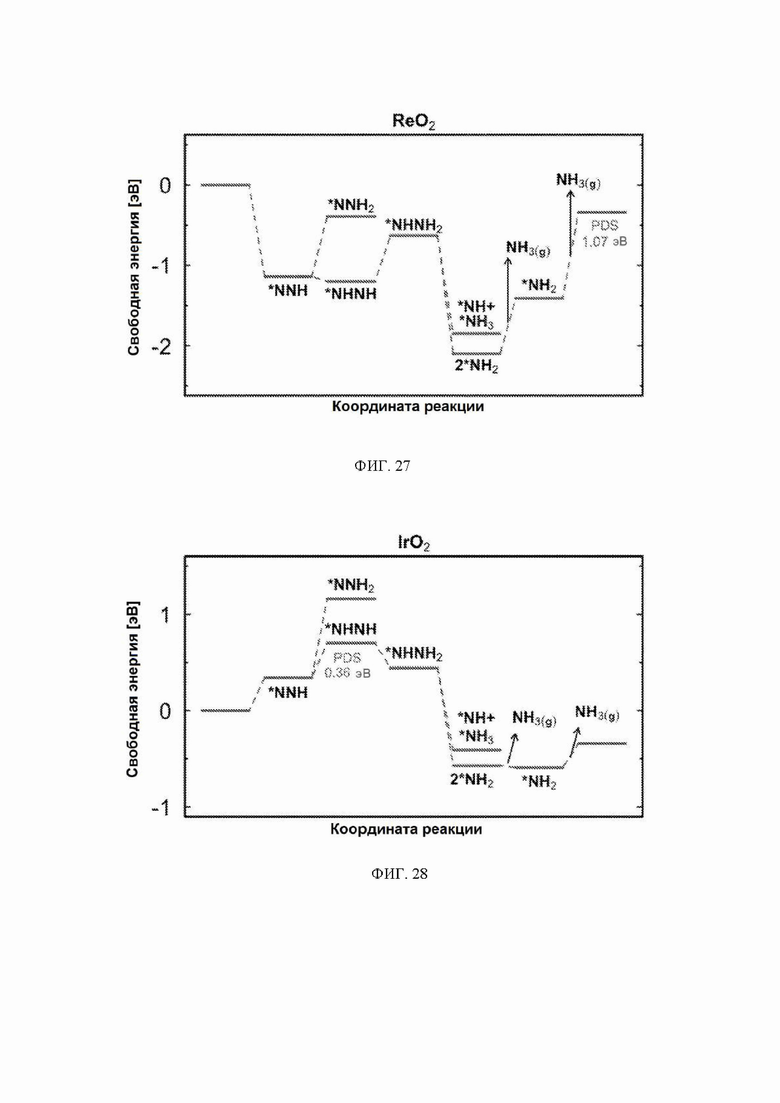
На ФИГ. 25-35 показаны диаграммы свободной энергии для образования NH3 на H-модифицированной поверхности 11 различных рутиловых оксидов. Все стадии реакции относятся к чистой поверхности и N2 и H2 в газовой фазе. Для каждой стадии реакции показаны все стабильные промежуточные соединения.
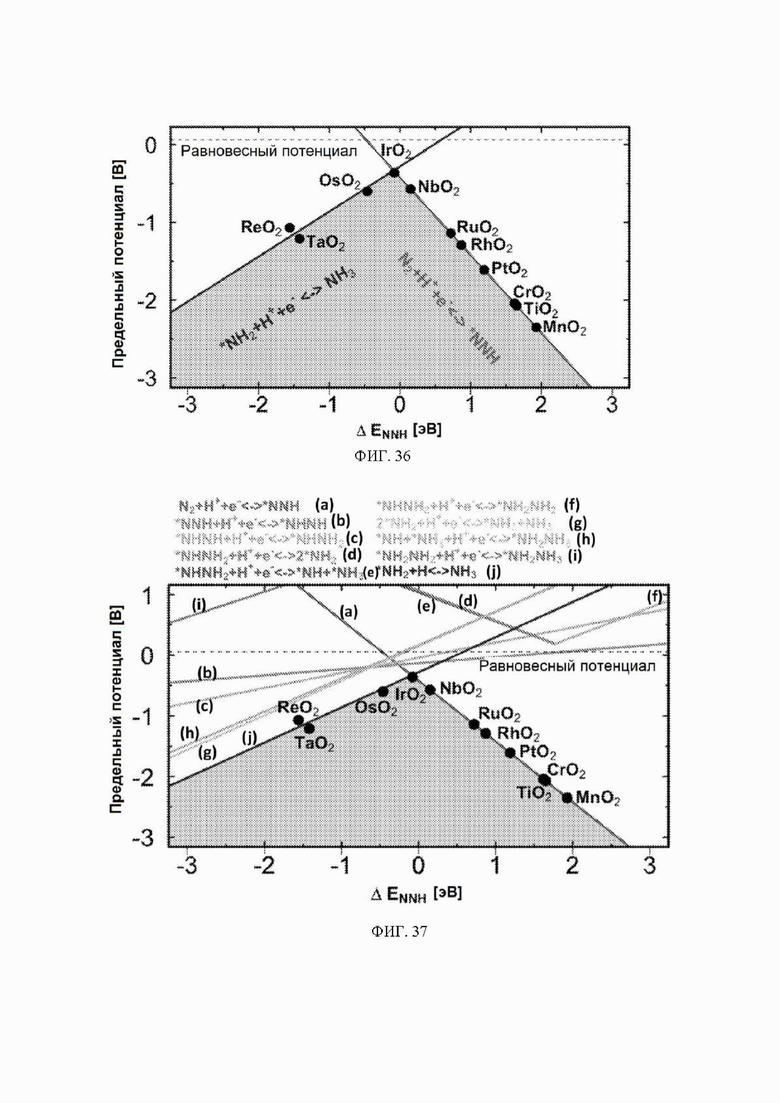
На ФИГ. 36 показана стадия определения потенциала электрохимического образования аммиака на каждом оксиде металла в зависимости от энергии связи N2H на металле. Линии рассчитаны с использованием масштабных соотношений, показанных на ФИГ. 26-28.
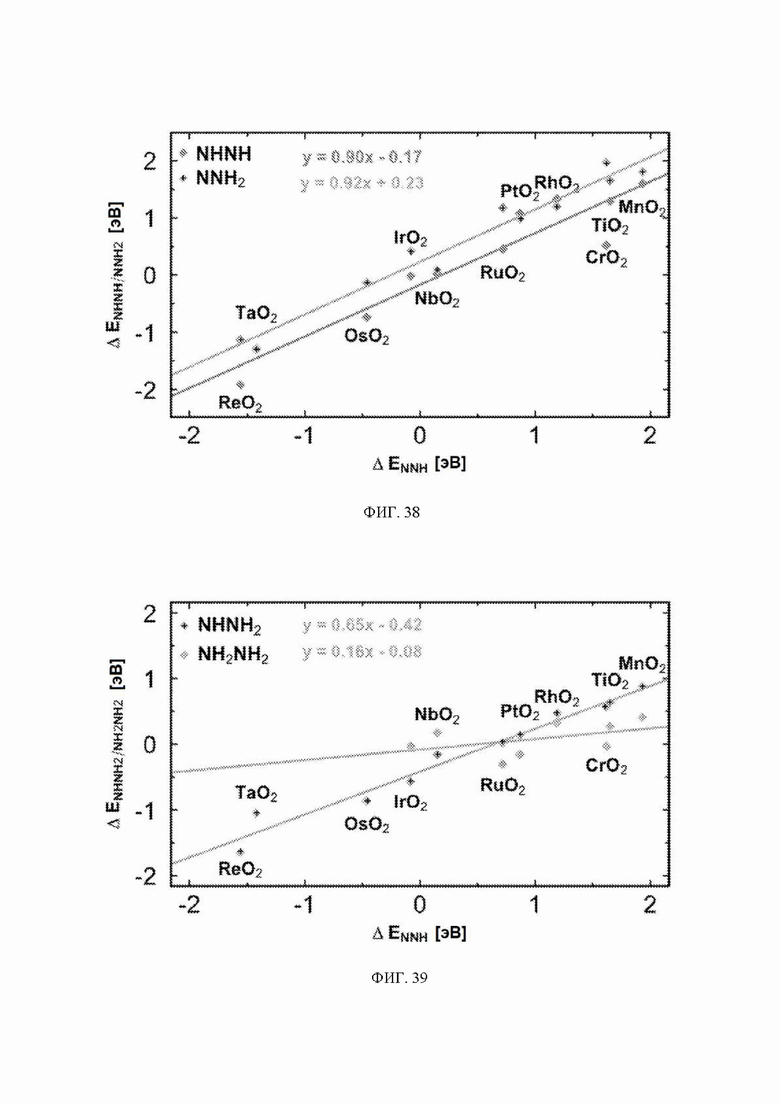
На ФИГ. 37 показана стадия определения потенциала электрохимического образования аммиака на каждом оксиде металла в зависимости от энергии связи N2H на металле. Линии рассчитаны с использованием масштабных соотношений, показанных на ФИГ. 38-43. На фигуре показана диаграмма в форме вулкана, аналогичная ФИГ. 36, но на ФИГ. 37 показаны все стадии реакции.
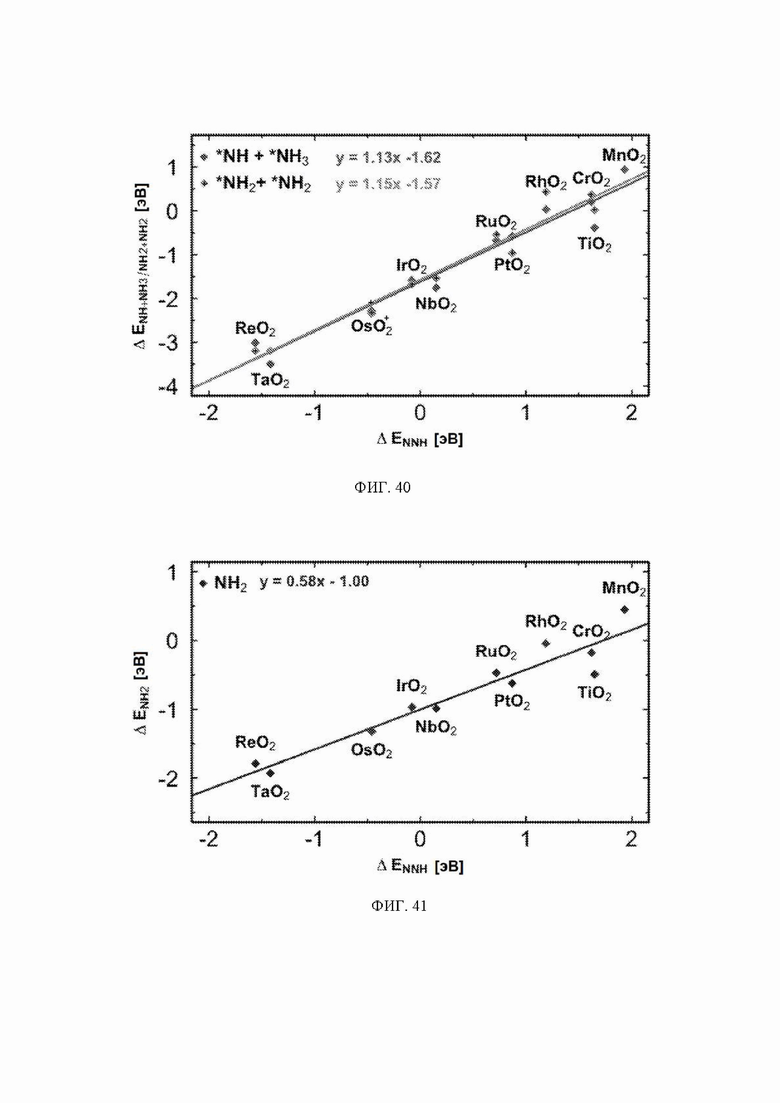
На ФИГ. 38-41 показаны энергии адсорбции NHNH, NNH2, NHNH2, NH2NH2, NH2+NH2, NH+NH3 и NH2 в зависимости от энергии хемосорбции NNH. Для каждого адсорбата приведено уравнение лучшей линии для набора данных.
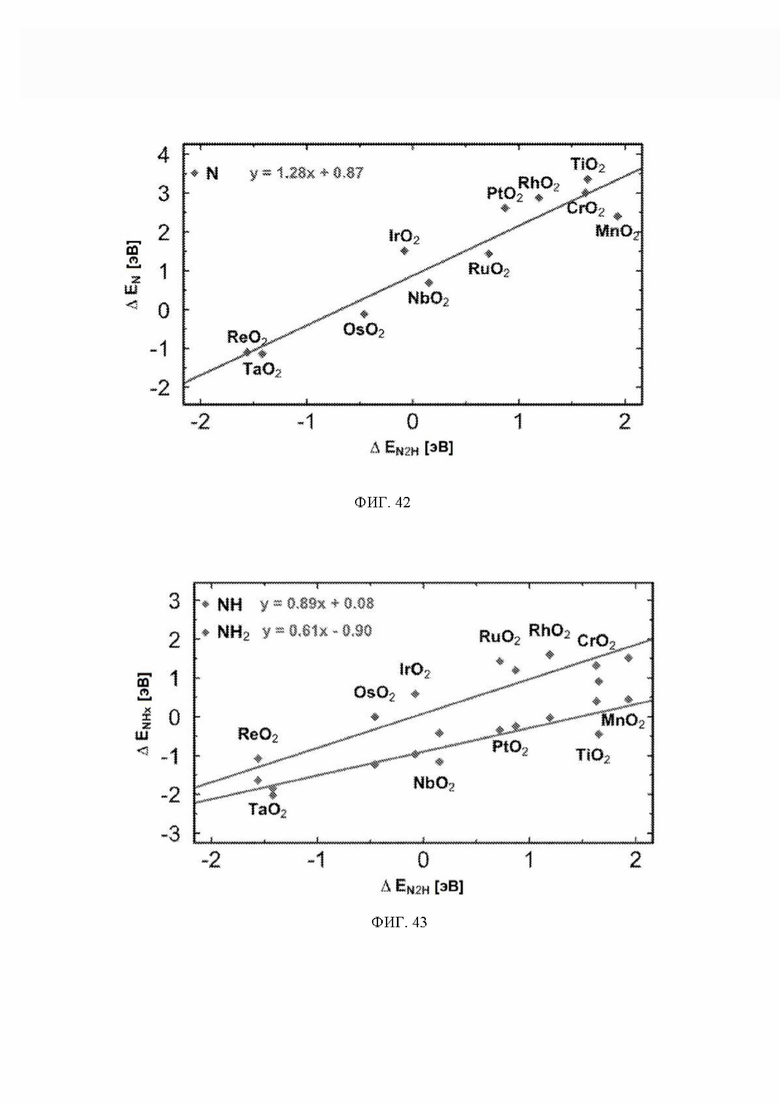
На ФИГ. 42 показаны энергии адсорбции N в зависимости от энергии хемосорбции N2H. Для каждого адсорбата приведено уравнение лучшей линии для набора данных.
На ФИГ. 43 показаны энергии адсорбции NH и NH2 в зависимости от энергии хемосорбции N2H. Для каждого адсорбата приведено уравнение лучшей линии для набора данных.
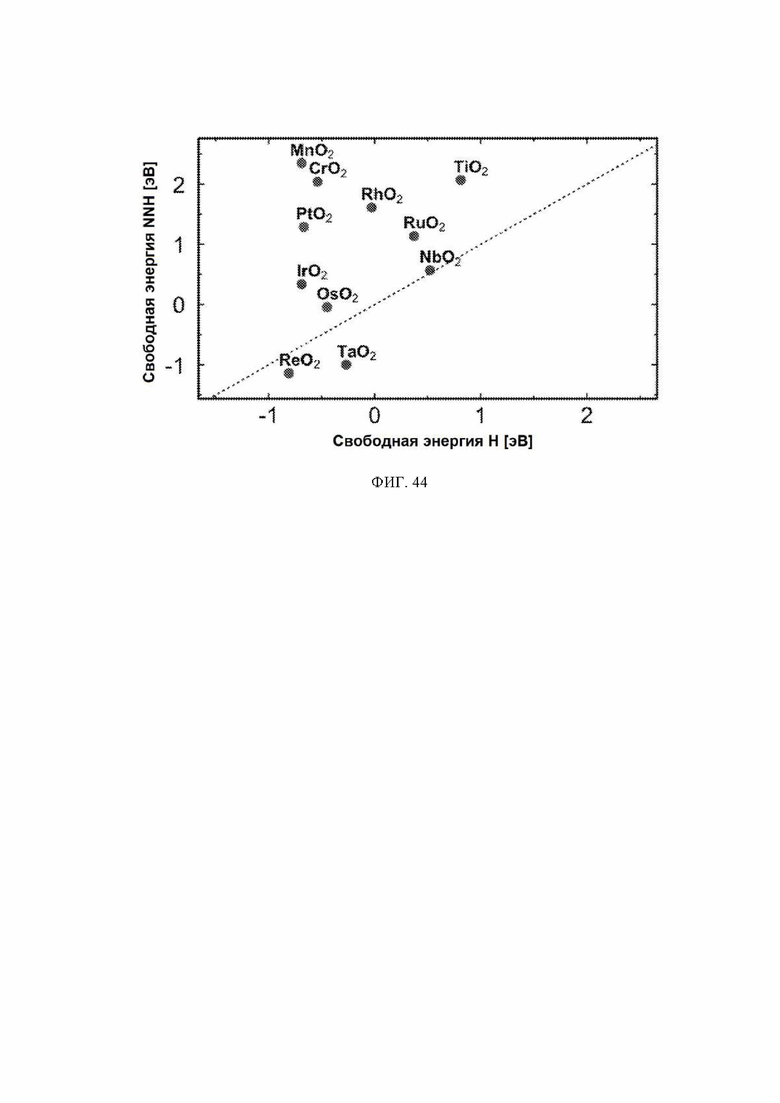
На ФИГ. 44 показана стадия определения потенциала для каждой стадии реакции электрохимического образования аммиака на оксидах металлов, изображенная в зависимости от энергии связи N2H на металле.
Описание изобретения
Далее будут описаны иллюстративные варианты осуществления изобретения со ссылкой на графические материалы. Эти примеры приведены для обеспечения лучшего понимания изобретения без ограничения его объема.
В нижеследующем описании описана последовательность стадий. Специалисту в данной области техники должно быть ясно, что, если только этого не требует контекст, порядок стадий не является существенно важным для получаемой конфигурации и ее эффекта. Кроме того, специалисту в данной области техники должно быть очевидно, что независимо от порядка стадий между все или некоторые из описанных стадий могут быть проведены с временной задержкой или без нее.
В контексте настоящего документа, в том числе в формуле изобретения, термины в единственном числе следует толковать как включающие также форму множественного числа и наоборот, если контекст не указывает на иное. Таким образом, следует отметить, что в контексте настоящего документа термины в единственном числе включают соответствующие термины во множественном числе, если контекст явным образом не требует обратного.
Во всем описании и формуле изобретения термины «содержащий», «включающий» и «имеющий», и их вариации следует толковать как означающие «включая, но не ограничиваясь перечисленным», и они не подразумевают исключение других компонентов.
Настоящее изобретение также охватывает точные термины, признаки, значения и диапазоны и т. д. в случае, если эти термины, признаки, значения и диапазоны и т. д. используются в сочетании с такими терминами, как приблизительно, около, в целом, по существу, в принципе, по меньшей мере и т. д. (то есть «приблизительно 3» также должно охватывать ровно 3, или «по существу постоянное» также должно охватывать точно постоянное).
Термин «по меньшей мере один» следует толковать как означающий «один или более», и поэтому он включает оба варианта осуществления, включающие один или несколько компонентов. Кроме того, зависимые пункты формулы изобретения, которые ссылаются на независимые пункты формулы изобретения, описывающие признаки с использованием выражения «по меньшей мере один», имеют одинаковое значение, когда элемент упоминается как «указанный» и «по меньшей мере один».
Следует иметь в виду, что в вышеописанных вариантах осуществления изобретения могут быть сделаны изменения без отклонения от объема изобретения. Признаки, раскрытые в описании, если не указано иное, могут быть заменены альтернативными признаками, служащими той же, эквивалентной или аналогичной цели. Таким образом, если не указано иное, каждый раскрытый признак представляет собой один пример общей серии эквивалентных или аналогичных признаков.
Указание на примеры, такое как «к примеру», «такой как», «например» и тому подобное, предназначено лишь для лучшей иллюстрации изобретения и не обладает ограничительным характером в отношении объема изобретения, если это не заявлено в формуле изобретения. Любые стадии, описанные в описании, могут быть проведены в любом порядке или одновременно, если контекст явно не указывает на иное.
Все признаки и/или стадии, раскрытые в описании, могут быть объединены в любой комбинации, за исключением комбинаций, в которых по меньшей мере некоторые из признаков и/или стадий являются взаимоисключающими. В частности, предпочтительные признаки изобретения применимы ко всем аспектам изобретения и могут быть использованы в любой комбинации.
Настоящее изобретение основано на неожиданном открытии, что на поверхности некоторых катализаторов на основе оксидов переходных металлов возможно образование аммиака при температуре окружающей среды и давлении с низким приложенным потенциалом. Учитывая важность аммиака, в частности, в производстве удобрений, а также энергоемкие и неблагоприятные для окружающей среды условия, которые обычно используются при его производстве, изобретение находит важное применение в различных отраслях промышленности.
Таким образом, изобретение обеспечивает способы и системы для генерирования аммиака при температуре и давлении окружающей среды. Температура окружающей среды обычно может относиться к обычным температурам как внутреннего, так и наружного воздуха. Соответственно, в некоторых вариантах осуществления способ и систему согласно изобретению применяют при температуре от приблизительно минус 10° до приблизительно 40 °C, например, от приблизительно 0° до приблизительно 40 °C, например, от приблизительно минус 10°, или от приблизительно минус 5 °C, или от приблизительно 0°, или от приблизительно 4 °C, или от приблизительно 5 °C, или от приблизительно 8 °C, или от приблизительно 10 °C до приблизительно 40 °C, или до приблизительно 30 °C, например, в диапазоне приблизительно 20-30 °C или в диапазоне приблизительно 20-25 °C. Давление окружающей среды обычно относится к атмосферному давлению. В способе и системе согласно настоящему изобретению используется электролитическая ячейка, которая может представлять собой любую из ряда традиционных коммерчески пригодных и практически реализуемых конструкций электролитической ячейки, которые могут вмещать катод специального назначения в соответствии с изобретением. Таким образом, ячейка и система могут в некоторых вариантах осуществления иметь одну или более катодных ячеек и одну или более анодных ячеек.
Электролитическая ячейка в данном контексте представляет собой электрохимическую ячейку, претерпевающая окислительно-восстановительную реакцию при подаче электрической энергии на ячейку.
Специалисту в данной области техники должно быть ясно, что химические соединения, как описано в настоящем документе, имеют свою химическую формулу независимо от их фазы или состояния. В частности, соединения, которые присутствуют в своем газообразном состоянии, находясь в чистом и выделенном виде при комнатной температуре (такие как N2, H2 и NH3), описаны в настоящем документе их химической формулой. Например, молекулярный азот в настоящем документе описывается как N2, независимо от того, присутствует ли он в виде газообразного азота, в виде отдельных молекул, в кластерах, связан с поверхностями или представлен в виде растворенных веществ, и то же самое относится к другим молекулярным частицам, описанным в настоящем документе.
Донором протонов может быть любое подходящее вещество, способное отдавать протоны в электролитической ячейке. Донор протонов может, например, представлять собой кислоту, такую как любая подходящая органическая или неорганическая кислота. Донор протонов может быть представлен в кислых, нейтральных или щелочных водных растворах. Донор протонов также, или в качестве альтернативы, может быть обеспечен при окислении H2 на аноде. То есть, водород можно рассматривать как источник протонов:
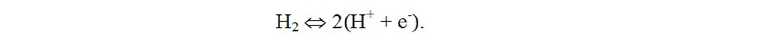
Электролитическая ячейка содержит по меньшей мере три основные части или компонента: катодный электрод, анодный электрод и электролит. Общая реакция на катоде может быть представлена как
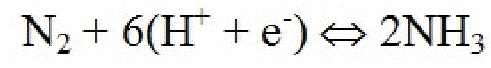 .
.
Поверхность катализатора может быть гидрирована путем добавления одного атома водорода за раз, представляющего собой протон из раствора и электрон с поверхности электрода. Механизм реакции может быть показан в уравнениях ниже, где звездочкой обозначен участок на поверхности:
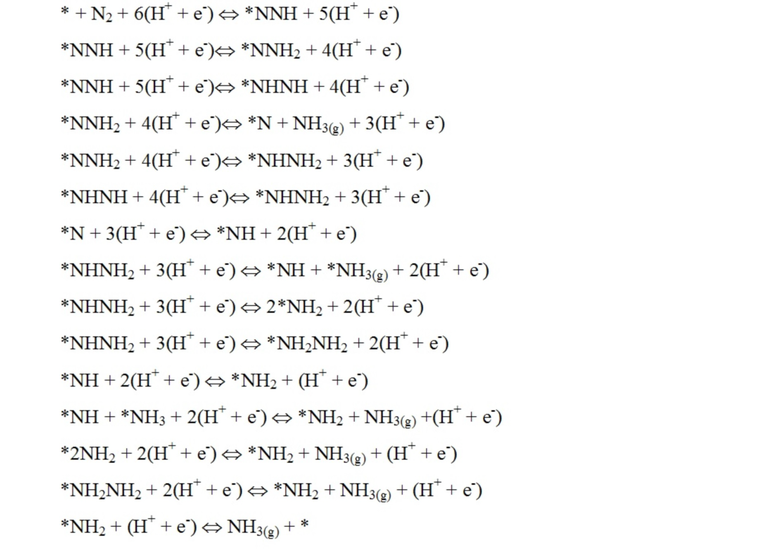
После добавления 3(H+ + e-) образуется одна молекула аммиака, и вторая образуется после добавления 6(H+ + e-).
Различные части или компоненты могут быть предоставлены в отдельных контейнерах, или они могут быть предоставлены в одном контейнере. Преимущество настоящего изобретения состоит в том, что способ и система могут подходящим образом применяться с использованием водных электролитов, таких как предпочтительно водные растворы с растворенными электролитами (солями). Таким образом, в предпочтительных вариантах осуществления способа и системы электролитическая ячейка содержит один или более водных электролитических растворов в одном или более отсеках ячейки. Растворы электролита могут содержать любые из различных обычных неорганических или органических солей, таких как, не ограничиваясь перечисленным, растворимые хлориды, нитраты, хлораты, бромиды и т. д., например, хлорид натрия, хлорид калия, хлорид кальция, хлорид аммония и другие подходящие соли. Водные растворы электролитов могут также содержать любой гидроксид щелочных или щелочноземельных металлов или их комбинацию, таких как гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид кальция, гидроксид рубидия и гидроксид цезия. Водный раствор электролита может также дополнительно или в качестве альтернативы содержать одну или несколько органических или неорганических кислот. Неорганические кислоты могут включать минеральные кислоты, включающие, не ограничиваясь перечисленным, соляную кислоту, азотную кислоту, фосфорную кислоту, серную кислоту, борную кислоту, фтористоводородную кислоту, бромистоводородную кислоту, тетрафторуксусную кислоту, уксусную кислоту и хлорную кислоту. Соответственно, водный раствор может быть нейтральным, щелочным или кислым раствором. В некоторых вариантах осуществления водный раствор представляет собой кислый раствор. Электролит также может представлять собой расплав соли, например, соль хлорида натрия. В некоторых вариантах осуществления электролитическая ячейка содержит электролитический раствор, содержащий органический протонный или апротонный растворитель или их смешивающуюся смесь, предпочтительно смешивающийся с водой органический растворитель, такой как, например, не ограничиваясь перечисленным, этанол, этиленгликоль, бутандиол, глицерин, диэтаноламин, диметоксиэтан, 1,4-диоксан и их смеси. Электролитический раствор может представлять собой раствор, содержащий воду и один или более смешивающихся с водой органических растворителей, таких как, не ограничиваясь перечисленным, один или более из указанных выше растворителей.
В общих чертах, катализатор на поверхности электрода в идеале должен иметь следующие характеристики: он должен (а) быть химически стабильным, (b) не окисляться и не расходоваться иным образом во время электролитического процесса, он должен способствовать образованию аммиака, и (d) использование катализатора должно приводить к получению минимального количества газообразного водорода. Как будет описано далее, каталитические оксиды согласно изобретению удовлетворяют этим характеристикам.
Преимущество настоящего изобретения состоит в том, что способ может подходящим образом применяться с использованием водных электролитов, таких как предпочтительно водные растворы с растворенными электролитами (солями). Таким образом, в предпочтительных вариантах осуществления способа и системы электролитическая ячейка содержит один или более водных электролитических растворов в одном или более отсеках ячейки. Водные растворы электролита могут содержать любые из различных обычных неорганических или органических солей, таких как, ограничиваясь перечисленным, растворимые хлориды, нитраты, хлораты, бромиды и т. д., например, хлорид натрия, хлорид калия, хлорид кальция, хлорид аммония и другие подходящие соли. Водные растворы электролитов могут также содержать любой оксид щелочных или щелочноземельных металлов или их комбинацию, таких как гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид кальция, гидроксид рубидия и гидроксид цезия. Водный раствор электролита может также дополнительно или в качестве альтернативы содержать одну или более органических или неорганических кислот. Неорганические кислоты могут включать минеральные кислоты, включающие, не ограничиваясь перечисленным, соляную кислоту, азотную кислоту, фосфорную кислоту, серную кислоту, борную кислоту, фтористоводородную кислоту, бромистоводородную кислоту и хлорную кислоту.
Как следует из настоящего документа, существенный признак настоящего изобретения касается состава и структуры катодного электрода. Оксиды переходных металлов характеризуются широким спектром структур поверхности, которые влияют на поверхностную энергию этих соединений и влияют на их химические свойства. На относительную кислотность и основность атомов, присутствующих на поверхности оксидов металлов, также влияет координация катиона металла и аниона кислорода, которые изменяют каталитические свойства этих соединений.
В отдельных вариантах осуществления катализатор на основе оксида переходного металла на поверхности катодного электрода выбран из одного или более из следующих: оксид титана, оксид хрома, оксид марганца, оксид ниобия, оксид тантала, оксид рутения, оксид родия, оксид платины, оксид осмия, оксид рения и оксид иридия. Предпочтительно катализатор включает один или более из оксида рения, оксида тантала и оксида ниобия.
В зависимости от состава вещества катализатора подходящая поверхностная кристаллическая структура может быть предпочтительной. Для оксидов переходных металлов существуют различные кристаллические структуры, и различные структуры могут быть получены при различных условиях роста. Выбор подходящих поверхностных кристаллических структур находится в пределах компетенции специалиста в данной области техники.
Может быть предпочтительным, чтобы катализатор содержал по меньшей мере одну поверхность, имеющую структуру рутила. Также возможны другие кристаллические структуры, известные в данной области техники (например, структура галита, структура сфалерита, структура анатаза, структура перовскита) (см., например, International Tables for Crystallography - Международные таблицы по кристаллографии; http://it.iucr.org).
Для данной кристаллической структуры может существовать несколько различных поверхностных граней (поликристаллические поверхности). Грань (110) рутила имеет наименьшую свободную поверхностную энергию и поэтому в целом она термодинамически наиболее стабильна. Соответственно, в некоторых вариантах осуществления оксиды переходных металлов могут иметь структуру рутила с гранью (110), образующей каталитическую поверхность. В качестве альтернативы, могут быть выбраны грани (100) и/или (111) структуры галита.
Таким образом, в некоторых вариантах осуществления каталитическая поверхность представляет собой поверхность рутила переходного металла. Поверхность может иметь любую подходящую грань, включая, но не ограничиваясь, грань (110). В некоторых вариантах осуществления поверхностная грань содержит или состоит из грани (110) оксида переходного металла, выбранного из группы, состоящей из оксида титана, оксида хрома, оксида марганца, оксида ниобия, оксида тантала, оксида рутения, оксида родия, оксида платины, оксида осмия, оксида рения и оксида иридия.
В некоторых предпочтительных вариантах осуществления катализатор содержит грань (110) структуры рутила одного или более оксидов, выбранных из оксида рения, оксида тантала и оксида ниобия.
Поверхность рутилового оксида металла, имеющая грань (110), содержит атомы металлов двух разных координационных сред, где ряды шестикратно координированных атомов металла чередуются с рядами пятикратных координированных атомов металла по направлению [001]. В то время как шестикратно координированные атомы металла имеют примерно такую же геометрию, что и основная масса, пятикратно координированные атомы металла имеют ненасыщенную связь, перпендикулярную поверхности. Таким образом, имеют место два различных участка поверхности: координационно ненасыщенные участки (cus-участки) поверх 5-кратно координированных атомов металла и мостиковые участки (br-участки) между двумя шестикратно координированными атомами металла. Было обнаружено, что br-участки обычно связывают адсорбаты сильнее, чем cus-участки. На стехиометрической поверхности (110) br-участки заняты кислородом, в то время как cus-участки вакантны. Поверхность (110) может называться как модифицированная кислородом (O-модифицированная). Мостиковые атомы кислорода недостаточно скоординированы и могут быть восстановлены на поверхности:
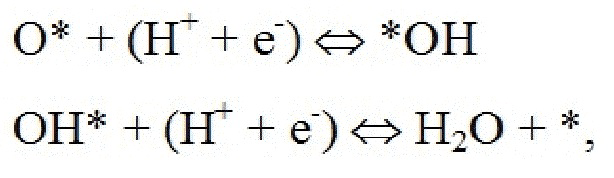
оставляя br-участки вакантными. Эти вакантные br-участки могут затем быть дополнительно протонированы:
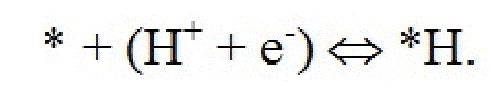
Как следствие, поверхность 110 может быть восстановлена, в результате чего мостиковые участки между шестикратно координированными атомами металла остаются вакантными. Мостиковые участки также могут быть частично или полностью заняты другими атомами, такими как атомы кислорода или водорода, как показано на ФИГ. 1 в настоящем документе.
Было обнаружено, что каталитическая активность поверхности (110) также может зависеть от занятости br-участков. Таким образом, может быть термодинамически выгодным восстановить мостиковые атомы кислорода на оксидах переходных металлов до H2O и дополнительно покрыть br-участки атомами водорода.
Соответственно, в некоторых вариантах осуществления мостиковые участки между шестикратно координированными атомами переходного металла на поверхностях оксида переходного металла покрыты атомами водорода.
Как должно быть очевидно специалисту в данной области техники, катализатор согласно изобретению может содержать один оксид переходного металла. Катализатор также может содержать или состоять из смеси двух или более таких оксидов. Такие смешанные оксиды могут содержать единую структуру, например, структуру рутила. Смешанные оксиды металлов также могут содержать смесь оксидов, имеющих различные кристаллические структуры, и/или оксидов с различными каталитическими гранями. Соответственно, такие смешанные оксиды могут дополнительно содержать одну грань или их смесь. Катализаторы на основе смешанных оксидов могут быть выращены или изготовлены отдельно и затем собраны в смешанные катализаторы, содержащие разные оксиды металлов, где оксиды в смеси имеют одинаковую или разные кристаллические структуры.
Как описано более подробно в настоящем документе, пропускание тока через электролитическую ячейку приводит к химической реакции, в которой азот реагирует с протонами с образованием аммиака. Пропускание тока достигается путем подачи напряжения на ячейку. Изобретение делает возможным электролитическое получение аммиака при низком электродном потенциале, что выгодно с точки зрения энергоэффективности и требований к оборудованию.
Не ограничиваясь какой-либо конкретной теорией, считается, что оксидные катализаторы способны сместить «узкое место» синтеза аммиака с расщепления N2 на последовательное образование азотно-водородных частиц (*NH,*NH2 или *NH3), из-за чего ожидается более простое, но при этом с более высоким выходом образование аммиака.
В некоторых пригодных вариантах осуществления изобретения аммиак может быть образован при электродном потенциале менее чем приблизительно минус 1,1 В, менее чем приблизительно минус 1,0 В, менее чем приблизительно минус 0,9 В, менее чем приблизительно минус 0,8 В, менее чем приблизительно минус 0,7 В, менее чем приблизительно минус 0,6 В, менее чем приблизительно 0,5 В или менее чем приблизительно минус 0,4 В. В некоторых вариантах осуществления аммиак может быть образован при электродном потенциале от приблизительно минус 0,2 В до приблизительно минус 1,0 В, например, от приблизительно минус 0,3 В до приблизительно минус 0,8 В, например, от приблизительно минус 0,4 В до приблизительно минус 1,0 В или от приблизительно минус 0,5 В до приблизительно минус 1,0 В. Верхний предел диапазона может составлять приблизительно минус 0,6 В, приблизительно минус 0,7 В, приблизительно минус 0,8 В, приблизительно минус 1,0 В или приблизительно минус 1,1 В. Нижний предел диапазона может составлять приблизительно минус 0,2 В, приблизительно минус 0,3 В, приблизительно минус 0,4 В, приблизительно минус 0,5 В или приблизительно минус 0,6 В.
В некоторых вариантах аммиак может быть образован с использованием катализатора на основе оксида ниобия при электродном потенциале от приблизительно минус 0,4 В до приблизительно минус 0,5 В. В некоторых вариантах аммиак может быть образован с использованием катализатора на основе оксида рения при электродном потенциале от приблизительно минус 0,8 В до приблизительно минус 0,9 В. В некоторых вариантах аммиак может быть образован с использованием катализатора на основе оксида тантала при электродном потенциале от минус 1,0 В до приблизительно минус 1,1 В.
Преимуществом настоящего изобретения является эффективность образования NH3 по сравнению с образованием H2, что было проблемой в исследованиях и испытаниях предшествующего уровня техники. В отдельных вариантах осуществления изобретения образуется менее чем приблизительно 50 % молей H2 по сравнению с количеством молей образовавшегося NH3 и предпочтительно менее чем приблизительно 40 % молей H2, менее чем приблизительно 30 % молей H2, менее чем приблизительно 20 % молей H2, менее чем приблизительно 10 % молей H2, менее чем приблизительно 5 % молей H2, менее чем приблизительно 2 % молей H2 или менее чем приблизительно 1 % молей H2.
Система согласно изобретению подходяще разработана таким образом, чтобы соответствовать одной или более из перечисленных выше особенностей способа. Преимуществом изобретения является то, что система может быть выполнена небольшой, надежной и дешевой, например, для применения на месте для производства удобрений вблизи предполагаемого места применения.
Аммиак может применяться в качестве удобрения путем введения его в почву в виде газа, хотя это требует инвестиций фермеров в резервуары для хранения под давлением и оборудование для введения. Аммиак также может применяться для образования мочевины, обычно путем взаимодействия с диоксидом углерода. Аммиак может реагировать с образованием азотной кислоты, которая, в свою очередь, легко реагирует с образованием нитрата аммония. Соответственно, системы и способы согласно настоящему изобретению могут быть легко объединены с существующими решениями для взаимодействия полученного аммиака с другими желаемыми продуктами, такими как, не ограничиваясь перечисленным, вышеупомянутые.
NOx и SOx являются общими терминами для обозначения моноазотных и моносерных аксоидов, таких как NO, NO2, SO, SO2 и SO3. Эти газы образуются при сгорании, особенно при высоких температурах. В районах с интенсивным автомобильным движением количество этих загрязнителей может быть значительным.
Соответственно, полезный аспект изобретения относится к системе для удаления NOx и/или SOx из потока газа путем взаимодействия потока газа с аммиаком, который генерируется in situ в потоке или в системе, которая может иметь жидкостное сообщение с потоком газа. Система может содержать систему для генерирования аммиака, как описано в настоящем документе, в частности, систему, содержащую электролитическую ячейку, содержащую катализатор на основе оксида переходного металла, как описано в настоящем документе. В этом контексте следует толковать in situ как означающее, что аммиак генерируется внутри системы, например, внутри потока газа или в отсеке внутри системы, который имеет жидкостное сообщение с потоком газа. Генерируемый при этом аммиак при контакте с потоком газа будет реагировать с NOx и/или SOx в потоке газа, в результате чего эти токсичные соединения превращаются в другие молекулярные соединения, такие как N2, H2O и (NH4)2SO4. В некоторых вариантах осуществления система может быть предназначена для применения в выхлопных газах автомобильных двигателей или в других двигателях, где аммиак может генерироваться in situ способом согласно настоящему изобретению и затем использован для уменьшения SOx и/или NOx в выхлопных газах двигателя. В такой системе может соответствующим образом использоваться электрический ток, получаемый преобразованием от автомобильного двигателя. Таким образом, с использованием электрического тока от автомобильного двигателя, аммиак может быть генерирован in situ, и генерируемый таким образом аммиак может реагировать с SOx и/или NOx из выхлопных газов автомобиля. Аммиак может генерироваться в автомобиле и затем подаваться в выхлопные газы автомобиля. Аммиак также может генерироваться in situ внутри выхлопной системы автомобиля. Таким образом, NOx и/или SOx удаляются из выхлопных газов автомобиля, уменьшая количество загрязняющих веществ в выхлопных газах.
Варианты осуществления изобретения включают следующие неограничивающие пункты:
1. Способ получения аммиака, включающий:
подачу N2 в электролитическую ячейку, содержащую по меньшей мере один источник протонов;
обеспечение возможности контакта N2 с поверхностью катодного электрода в электролитической ячейке, где поверхность катодного электрода содержит каталитическую поверхность, содержащую по меньшей мере один оксид переходного металла; и
пропускание тока через указанную электролитическую ячейку, в результате чего азот реагирует с протонами с образованием аммиака.
2. Способ по п. 1, где катализатор содержит один или более оксидов переходных металлов, выбранных из группы, состоящей из оксида титана, оксида хрома, оксида марганца, оксида ниобия, оксида тантала, оксида рутения, оксида родия, оксида платины, оксида осмия, оксида рения и оксида иридия.
3. Способ по п. 1 или п. 2, где катализатор содержит один или более оксидов, выбранных из группы, состоящей из оксида рения, оксида тантала и оксида ниобия.
4. Способ по любому из пп. 1-3, где каталитическая поверхность содержит по меньшей мере одну поверхность, имеющую структуру рутила.
5. Способ по любому из пп. 1-4, где каталитическая поверхность содержит по меньшей мере одну поверхность, имеющую грань (110).
6. Способ по любому из пп. 1-5, где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 1,0 В, более предпочтительно менее чем приблизительно минус 0,8 В и еще более предпочтительно менее чем приблизительно минус 0,5 В.
7. Способ по любому из пп. 1-6, где катализатор содержит оксид ниобия, и где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 0,5 В.
8. Способ по любому из пп. 1-7, где катализатор содержит оксид рения, и где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 0,9 В.
9. Способ по любому из пп. 1-8, где катализатор содержит оксид тантала, и где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 1,0 В.
10. Способ по любому из пп. 1-9, где образуется менее 50 % молей H2 по сравнению с количеством молей образовавшегося NH3, предпочтительно менее 20 % и еще более предпочтительно менее 10 %.
11. Способ по любому из пп. 1-10, где указанная электролитическая ячейка содержит один или более водных электролитических растворов.
12. Способ по любому из пп. 1-11, где источником протонов при образовании аммиака является расщепление воды на электроде или реакция окисления H2 на аноде.
13. Система для генерирования аммиака, содержащая по меньшей мере одну электрохимическую ячейку, содержащую по меньшей мере один катодный электрод, имеющий каталитическую поверхность, где каталитическая поверхность заполнена по меньшей мере одним катализатором, содержащим один или более оксидов переходных металлов.
14. Система по п. 13, где по меньшей мере один оксид переходного металла выбран из группы, состоящей из оксида титана, оксида хрома, оксида марганца, оксида ниобия, оксида тантала, оксида рутения, оксида родия, оксида платины, оксида осмия, оксида рения и оксида иридия.
15. Система по п. 13 или п. 14, где по меньшей мере один оксид переходного металла выбран из группы, состоящей из оксида рения, оксида тантала и оксида ниобия.
16. Система по п. 14 или п. 15, где каталитическая поверхность содержит по меньшей мере одну поверхность, имеющую структуру рутила.
17. Система по любому из пп. 14-16, где каталитическая поверхность содержит по меньшей мере одну поверхность, имеющую грань (110).
18. Система по любому из пп. 14-17, где указанная электролитическая ячейка дополнительно содержит один или более электролитических растворов.
19. Система по п. 18, где электролитический раствор представляет собой кислый водный раствор.
Изобретение будет далее проиллюстрировано следующими неограничивающими примерами, дополнительно описывающими конкретные преимущества и варианты осуществления настоящего изобретения.
Пример 1
Поправка на энергию нулевой точки и разность энтропии между адсорбированными частицами и молекулой газа рассчитывают для всех промежуточных соединений в реакции в гармоническом приближении. Эти значения приведены в таблице 1.
Таблица 1. Энергия нулевой точки (ZPE) и энтропийные вклады в свободную энергию газовой фазы и адсорбированных молекул при 300 К для оксидов рутила (в эВ).
восстановленная поверхность
O-модифицированная/H-модифицированная поверхности
Значения для молекул газовой фазы взяты из Weast (Handbook of Chemistry and Physics, 49th edn., p. D109, The Chemical Rubber Company, Cleveland 1968-1969) и Atkins (Physical Chemistry, Oxford University Press, 6th edn., 1998), и значения для адсорбированных молекул получены из расчетов DFT (теории функционала плотности) колебательных нормальных мод.
Пример 2
Введение
Были испытаны различные электролиты и электродные материалы для того, чтобы смягчить термодинамические требования и оптимизировать скорость образования аммиака с использованием гетерогенного катализа. (Amar 2011; Denvir 2008 (US7314544 B2); Ouzoounidou 2007; Pappenfus 2009; Amar 2011; Song 2004). Электролитические ячейки на основе твердых электролитов и полимерных электролитических мембран (ПЭМ) были предметом многих из этих исследований, поскольку они упрощают разделение подаваемого газообразного водорода и получаемого аммиака. Самая высокая скорость образования аммиака, полученная с мембраной Nafion (Нафион), составляет 1,13×10-8 моль с-1 см-2 с фарадеевской эффективностью, равной приблизительно 90 %, где в качестве подаваемого газа использовали влажный H2. (Xu 2009) Использование воздуха и воды в качестве подаваемых газов является интересной возможностью, однако, самая высокая скорость образования аммиака, достигнутая таким образом, была намного ниже, 1,14×10-9 моль с-1 см-2. (Lan 2013) Получали только 1 % CE, что требовало перенапряжения минус 1,6 В. Несмотря на то, что эти результаты являются многообещающими, они все еще далеки от скоростей получения, близких к коммерчески рентабельным (4,3-8,7×10-7 моль с-1 см-2). (Giddey, Badwal, 2013)
До сих пор экспериментаторы продолжают поиски нового пути синтеза аммиака. Однако в последнее время предметом теоретических исследований стало электрокаталитическое восстановление N2, где с помощью расчетов согласно теории функционала плотности (DFT) предложены материалы, способствующие восстановлению N2 и подавляющие выделение водорода. (Skúlason 2012, Abghoui 2015 enabling, 2016 ACS Catalysis, 2016 Catalysis Today, 2016 Catalysis Today, 2017 Phys. Chem. C, J Howalt 2013, J Howalt 2014, Hargreaves 2014, Zelinapour-Yazdi 2016) Применение вычислительных методов позволяет сканировать большие группы потенциальных катализаторов без необходимости их производства в лаборатории, что позволяет сэкономить время и деньги. В 2012 году Skúlason et al. провели детальный DFT-анализ каталитической активности как плоских, так и ступенчатых поверхностей из чистого переходного металла, в котором они определили тренды (динамику изменения) и рассчитали профиль свободной энергии для восстановления азота. Так называемые графики в форме вулканов полезны при оценке трендов каталитической реакционной способности, и Skúlason et al. обнаружили, что ранние переходные металлы были более активны в отношении восстановления N2, чем более поздние переходные металлы, а также были менее склонны к выделению водорода. (Skúlason 2012) Abghoui et al. провели аналогичное исследование на мононитридах переходных металлов и обнаружили, что нитриды ранних переходных металлов являются многообещающими кандидатами для восстановления N2, причем, предсказали, что ZrN и VN будут образовывать аммиак при потенциалах минус 0,76 В и минус 0,51 В по сравнению с СВЭ (стандартным водородным электродом), соответственно. (2015 PCCP, 2015 Proceedia computer science, 2016 ACS Catalysis). Позже они также предположили, что NbN и CrN могут вести себя одинаково. (Abghoui Skulason, 2017, MVK). Дальнейшие исследования дали многообещающие результаты, в которых, как сообщалось, грани (110) сфалеритовой структуры RuN катализировали образование аммиака при очень малом начальном потенциале, равном приблизительно минус 0,23 В по сравнению с СВЭ. (Abghoui 2017) Сравнение механизма Марса-ван Кревелена (MvK) с традиционными ассоциативными и диссоциативными механизмами показало, что MvK должен быть более благоприятным механизмом реакции на поверхности этих нитридов. (Abghoui and Skulason, 2017)
В настоящем исследовании оксиды переходных металлов в структуре рутила исследуют в качестве возможных кандидатов для катализа электрохимического образования аммиака при условиях окружающей среды. Расчеты DFT использовали для построения диаграммы стабильности для каждого оксида металла, где стабильность грани (110), покрытой различными адсорбированными частицами, рассчитывали как функцию потенциала и определяли наиболее стабильную грань для каждого потенциала. Далее авторы изучили термодинамику катодной реакции и построили диаграммы свободной энергии для электрохимического протонирования адсорбированных соединений азота. Энергетический баланс между адсорбцией протонов и соединений N2H исследовали для всех потенциальных катализаторов для того, чтобы увидеть тенденцию к восстановлению N2 по сравнению с конкурирующей реакцией выделения водорода (HER). Затем авторы оценили начальные потенциалы, необходимые для образования аммиака на поверхности этих оксидов. Влияние внешнего потенциала учитывали с помощью расчетного стандартного водородного электрода (СВЭ) (Norskov, Jonsson 2004), и для каждого оксида металла оценивали самый низкий начальный потенциал, необходимый для восстановления N2 до аммиака. Наконец, получали диаграмму в форме вулкана, на которой показана каталитическая активность различных оксидов с использованием энергии связи N2H в качестве общего показателя (Rossmeisl 2007, Skúlason 2012).
Методика
Расчеты DFT: в настоящем исследовании грань (110) одиннадцати оксидов переходных металлов, естественным образом присутствующих в структуре рутила, рассматривали с акцентом на электрохимическое образование аммиака, поскольку поверхность (110) является наиболее стабильной из граней структуры рутила с малыми индексами. Оксиды, стабильные в структуре рутила в условиях окружающей среды, представляют собой следующие: TiO2, NbO2, TaO2, ReO2, IrO2, OsO2, CrO2, MnO2, RuO2, RhO2 и PtO2. (Harold Kung) Поверхности оксидов моделировали с помощью 48 атомов в четырех слоях, каждый из которых состоял из 4 атомов металла и 8 атомов кислорода. Два нижних слоя оставались неподвижными, тогда как два верхних слоя и адсорбированные частицы находились в релаксированном состоянии. Граничные условия являлись периодическими в направлениях x и y, и поверхности разделяли вакуумом 12 Å в направлении z. Оптимизацию структуры считали приближенной, когда силы в любом направлении на всех подвижных атомах составляли меньше 0,01 эВ/Å. Константы решетки RPBE оптимизировали для каждого оксида и учитывали спиновую поляризацию. Константы решетки RPBE, оптимизированные для оксидов в данном исследовании, представляют собой следующие: TiO2: a=4,65 Å, c=2,98 Å; NbO2: a=5,11 Å, c=3,00 Å; TaO2: a=4,98 Å, c=3,02 Å; ReO2: a=4,77 Å, c=3,14 Å; IrO2: a=4,58 Å, c=3,13 Å; OsO2: a=4,58 Å, c=3,18 Å; CrO2: a=4,50 Å, c=2,96 Å; MnO2: a=4,86 Å, c=2,85 Å; RuO2: a=4,58 Å, c=3,2 Å; RhO2: a=4,55 Å, c=3,06 Å и PtO2: a=4,65 Å, c=3,19 Å.
Расчеты выполняли с помощью DFT с использованием обменного корреляционного функционала RPBE. Базис плоских волн с отсечкой энергии 350 эВ использовали для представления валентных электронов с PAW-представлением электронов внутренней оболочки, как это реализовано в коде VASP (VASP1-VASP4). Самосогласованную плотность электронов определяли итерационной диагонализацией гамильтониана Кона-Шэма, причем занятость состояний Кона-Шэма была размыта по распределению Ферми-Дирака с параметром размытия kBT=0,1 эВ. Для всех поверхностей использовали выборку k-точек 4×4×1, полученную с помощью схемы Монхорста-Пака, и для уменьшения числа k-точек в расчетах применяли максимальную симметрию.
Электрохимические реакции и моделирование: источником протонов в реакции может быть либо расщепление воды, либо реакция окисления H2 на аноде. Чтобы связать абсолютный потенциал с СВЭ, авторы в настоящем документе ссылаются на H2 только как на удобный источник протонов и электронов.

где протоны сольватированы в электролите, и электроны переносятся на катод через проволоку. Общая реакция восстановления N2 представляет собой следующую:
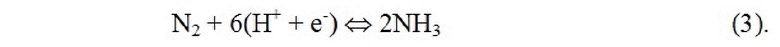
Поверхность гидрирована путем добавления одного атома водорода за раз, представляющего собой протон из раствора и электрон с поверхности электрода. Изученный ассоциативный механизм реакции показан в уравнениях ниже, где звездочкой обозначен участок на поверхности:
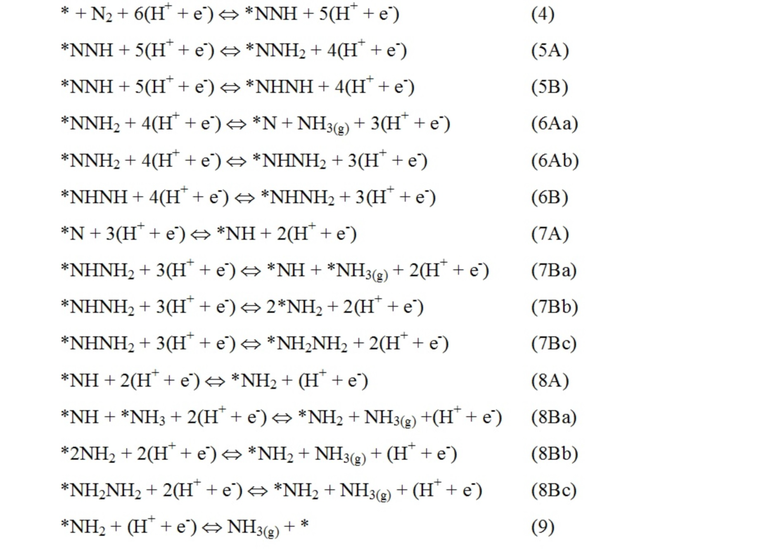 .
.
Первая молекула аммиака образуется после добавления трех-пяти протонов в зависимости от предпочтительного пути реакции. Вторая молекула аммиака затем образуется после добавления шести протонов. Свободную энергию адсорбции N2H сравнивают с энергией адсорбции протонов, чтобы определить, является ли поверхность селективной в отношении образования аммиака или выделения водорода.
В качестве первого приближения предполагают, что барьеры активации между устойчивыми минимумами являются низкими, или что они следуют соотношению Бренстеда-Эванса-Поляни и поэтому ими можно пренебрегать во время электрохимических реакций в контексте настоящего изобретения. Свободная энергия каждой элементарной стадии оценивают при pH = 0 и T = 298K согласно следующему:

где ΔE представляет собой энергию, рассчитанную с использованием DFT. ΔEZPE и ΔS представляют собой различия в энергии нулевой точки и энтропии, соответственно, между адсорбированными частицами и молекулами газовой фазы. Их рассчитывали в гармоническом приближении, и значения приведены в ESI. Эффект прикладываемого смещения U включен для всех стадий электрохимической реакции путем смещения свободной энергии для реакции с участием n электронов на минус neU, так что свободная энергия каждой элементарной стадии определяется как:
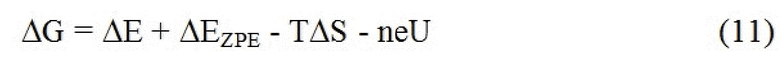 .
.
Явное включение воды в моделирование значительно увеличило бы трудоемкость вычислений и поэтому не было включено в настоящее исследование. Известно, что присутствие воды стабилизирует некоторые частицы посредством образования водородных связей. Например, ожидается, что *NH2 будет несколько более стабильным вблизи воды, тогда как на *N водный слой не окажет воздействия. В предыдущих публикациях стабилизирующее действие воды оценивали как составляющее менее 0,1 эВ на водородную связь (Montoya 2015). Таким образом, авторы оценили, что включение водородной связи изменило бы начальные потенциалы, рассчитанные в данном исследовании, менее чем на 0,1 эВ, представляющее собой поправку, не включенную в настоящее исследование.
Результаты и обсуждение
Стабильность: поверхность рутила (110) содержит атомы металла двух разных координационных сред, см. ФИГ. 1. Ряды шестикратно координированных атомов металла чередуются с рядами пятикратно координированных атомов металла по направлению [001]. В то время как шестикратно координированные атомы металла имеют примерно такую же геометрию, что и основная масса, пятикратно координированные атомы металла имеют ненасыщенную связь, перпендикулярную поверхности. (Morgan 2007) Таким образом, имеют место два различных участка поверхности: координационно ненасыщенные участки (cus-участки) поверх пятикратно координированных атомов металла и мостиковые участки (br-участки) между двумя шестикратно координированными атомами металла. Согласно наблюдениям авторов br-участки обычно связывают адсорбаты сильнее, чем cus-участки, и каталитическая активность поверхности зависит от занятости br-участков. Поэтому для каждого оксида рутила проводили систематическое исследование различных покрытий кислорода и водорода на br-участках, и на диаграммах стабильности представлена относительная стабильность грани (110) с различными покрытиями, см. ФИГ. 2 в качестве примера (но все остальные показаны в ESI). На стехиометрической поверхности (110) br-участки заняты кислородом, в то время как cus-участки являются вакантными. Авторы называют стехиометрическую поверхность (110) поверхностью, модифицированной кислородом (O-модифицированная), см. ФИГ. 1c. Мостиковые атомы кислорода O-модифицированной поверхности недостаточно скоординированы и могут быть восстановлены с поверхности в условиях эксперимента и эксплуатации. Чтобы рассчитать свободную энергию восстановления O-модифицированной поверхности, авторы следовали методике, разработанной Nørskov et al., где восстановление поверхности происходит посредством обмена воды и протонов с электролитом:
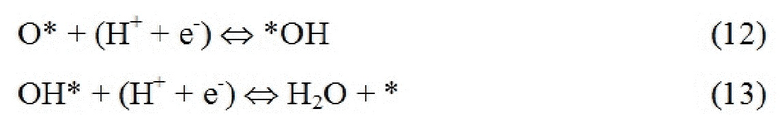 ,
,
оставляя br-участки вакантными. Авторы называют эту поверхность восстановленной, см. ФИГ. 1a. Эти вакантные br-участки могут затем быть дополнительно протонированы:
 .
.
Авторы называют эту поверхность поверхностью, модифицированной водородом (H-модифицированная), см. ФИГ. 1b. Авторы также рассматривали конфигурацию поверхности, где половина br-участков покрыта либо водородом, либо кислородом, и половина сайтов вакантна. Эти поверхности называются поверхностями 0,25 монослоя (ML).
На ФИГ. 2 показан пример диаграммы стабильности, построенной для NbO2. Ось x представляет свободную энергию восстановленной поверхности, которая используется в качестве эталона, к которой нормированы другие поверхности. Из диаграммы стабильности на ФИГ. 2 видно, что при потенциалах ниже минус 0,5 В относительно СВЭ термодинамически выгодно восстанавливать мостиковые атомы кислорода на NbO2 до H2O и далее протонировать br-участки до покрытия 0,5 ML H. Диаграммы стабильности для других металлов показаны на ФИГ. 3-12. Для всех оксидов покрытия 0,25 ML H или O не являются предпочтительными при любом соответствующем потенциале и поэтому были исключены из всех дальнейших расчетов. Следует отметить, что этот анализ основан на термодинамике и не включает энергии активации.
Каталитическая активность: каталитическую активность всех оксидов рутила в отношении электрохимического образования аммиака рассчитывали с использованием DFT. Для каждого рутила профиль свободной энергии рассчитывали для трех различных поверхностей: восстановленной, H-модифицированной и O-модифицированной поверхностей. Свободную энергию каждого промежуточного соединения находили по уравнению (10), относящемуся к N2 и H2 в газовой фазе. Как видно из диаграмм стабильности, поверхности 0,25 ML не являются предпочтительными в каком-либо значимом диапазоне потенциалов и поэтому были исключены из всех дальнейших расчетов.
На ФИГ. 13 показан профиль свободной энергии образования аммиака на NbO2, где определена соответствующая стадия определения потенциала (PDS) для каждой поверхности, то есть, стадия с наибольшим изменением свободной энергии, которое определяет начальный потенциал, необходимый для того, чтобы все стадии реакции имели снижающуюся свободную энергию. Авторы определяют эту стадию как меру активности оксида по отношению к образованию аммиака. Диаграммы свободной энергии для других оксидов рутила представлены на ФИГ. 14-23. На ФИГ. 13 видно, что изменение свободной энергии PDS, прогнозируемое для образования аммиака на O-модифицированной и на восстановленной поверхности, составляет минус 0,53 эВ и минус 2,01 эВ, соответственно.
С учетом диаграммы стабильности, представленной на ФИГ. 2 для NbO2, предпочтительная поверхность с начальным потенциалом, необходимым для образования аммиака, для обоих случаев представляет собой H-модифицированную поверхность. Это исключает возможность образования аммиака на O-модифицированной и восстановленной поверхности для NbO2. Аналогичный анализ был проведен для всех оксидов, и результаты представлены на ФИГ. 24. Для всех оксидов рутила требуемый приложенный потенциал находится в диапазоне потенциалов, где предпочтительной является H-модифицированная поверхность, и, по-видимому, исключает возможность образования аммиака на поверхности, модифицированной кислородом, и восстановленной поверхности. Изменение свободной энергии PDS для модифицированных водородом поверхностей IrO2, NbO2, OsO2, ReO2, RuO2, TaO2, PtO2, RhO2, CrO2, TiO2 и MnO2 составляет 0,36 эВ, 0,57 эВ, 0,60 эВ, 1,07 эВ, 1,14 эВ, 1,21 эВ, 1,29 эВ, 1,61 эВ, 2,04 эВ, 2,07 эВ и 2,35 эВ, соответственно. Три из этих поверхностей, IrO2, NbO2 и OsO2, имеют необходимое перенапряжение схожее или более низкое, чем перенапряжение, необходимое для восстановления азота нитрогеназой, но считается, что оно составляет приблизительно 0,63 В. Диаграммы свободной энергии, показывающие все возможные промежуточные соединения для H-модифицированной поверхности всех кандидатов, показаны на ФИГ. 25-35.
Масштабные соотношения и диаграмма в форме вулкана: с использованием масштабных соотношений между энергиями связи различных промежуточных соединений механизма восстановления N2 был построен график в форме вулкана, как показано на ФИГ. 36. На этом графике показаны только три из шести стадий электрохимической реакции механизма образования аммиака. Три стадии, которые не включены, имеют точку пересечения с осью y выше нуля и, следовательно, не влияют на выводы, сделанные на основании графика.
Масштабные соотношения, использованные для вычисления линий на ФИГ. 36-37, показаны на ФИГ. 38-43, и график, демонстрирующий все стадии реакции механизма образования аммиака, показан на ФИГ. 37.
Подобные диаграммы были ранее построены для чистых переходных металлов, где использовали энергию связи N с поверхностью, а не энергию связи N2H. (Skulason 2012, Montoya 2015)
Энергия связи *N является эндотермической для всех кандидатов в данном исследовании (за исключением ReO2 и TaO2), и поэтому диссоциация N2 будет эндотермической, а также будет иметь высокую энергию активации. Однако энергия связи *N на ReO2 и TaO2 составляет приблизительно минус 1 эВ, и поэтому диссоциация N2 на этих кандидатах может иметь низкий барьер, поскольку в этом случае энергия реакции составляет приблизительно минус 2 эВ. Однако включение пути диссоциации не изменит приведенных PDS ни для одного из кандидатов, поскольку *NH2 → NH3 представляет собой PDS на левой «ноге» вулкана, где расположены ReO2 и TaO2 Стадии реакции *N → *NH и *NH → *NH2 также включены в предложенные авторами пути, и эти стадии никогда не становятся PDS (см. ФИГ. 43).
Первое протонирование: до этого момента авторы не рассматривали конкурирующие реакции, такие как реакция выделения водорода. В качестве первой стадии в направлении включения конкурирующих реакций авторы рассчитали свободную энергию первой стадии реакции образования аммиака, которая включает адсорбцию N2H, и сравнили ее со свободной энергией адсорбции водорода на поверхности. Атому водорода позволили найти наиболее благоприятный участок связывания на поверхности. Это было сделано только для H-модифицированных поверхностей, так как поверхности, модифицированные иным образом, уже исключены из-за нестабильности в условиях эксплуатации. Результаты этого анализа приведены на ФИГ. 6. По-видимому, два оксида ReO2 и TaO2 способствуют адсорбции N2H по сравнению с протоном и, следовательно, являются перспективными для электрохимического образования аммиака с более высокими выходами. NbO2 связывает N2H и H с одинаковой силой, поэтому ожидается, что оба соединения будут находиться на этой поверхности, и, следовательно, на NbO2 можно ожидать образования как аммиака, так и газообразного водорода. IrO2 в высокой степени благоприятствует адсорбции протонов по сравнению NNH, что вызывает сожаление, поскольку IrO2 в действительности обладает самым низким прогнозируемым начальным потенциалом из одиннадцати оксидов, рассмотренных в данном исследовании. ReO2 и TaO2, однако, могут быть активными и селективными для электровосстановления N2, но с немного большим перенапряжением.
Вывод
Расчеты DFT использовали для изучения возможности активации азота для электрохимического получения аммиака в условиях окружающей среды на грани (110) рутиловой структуры NbO2, RuO2, RhO2, TaO2, ReO2, TiO2, OsO2, MnO2, CrO2, IrO2 и PtO2. Относительную стабильность граней с различными адсорбатами рассчитывали в зависимости от приложенного потенциала. Исследовали каталитическую активность каждой поверхности и обнаружили стадию, определяющую потенциал. При приложенном потенциале, необходимом для того, чтобы все электрохимические стадии имели снижающуюся свободную энергию, все оксиды оказались наиболее стабильными с адсорбированным на участках водородом. Из этих оксидов ReO2 и TaO2 способствуют адсорбции N2H.
Цитируемые источники:
1. Giddey, S.; Badwal, S. P. S.; Kulkarni, A., Review of electrochemical ammonia production technologies and materials. Int J Hydrogen Energ 2013, 38 (34), 14576-14594.
2. Amar, I. A.; Lan, R.; Petit, C. T. G.; Tao, S. W., Solid-state electrochemical synthesis of ammonia: a review. J Solid State Electr 2011, 15 (9), 1845-1860.
3. Smil, V., Detonator of the population explosion. Nature 1999, 400 (6743), 415-415.
4. Klerke, A.; Christensen, C. H.; Norskov, J. K.; Vegge, T., Ammonia for hydrogen storage: challenges and opportunities. J Mater Chem 2008, 18 (20), 2304-2310.
5. Smil, V., Global population and the nitrogen cycle. Sci Am 1997, 277 (1), 76-81.
6. Jennings, J. R., Catalytic ammonia synthesis : fundamentals and practice. Plenum Press: New York, 1991.
7. Burgess, B. K.; Lowe, D. J., Mechanism of molybdenum nitrogenase. Chem Rev 1996, 96 (7), 2983-3011.
8. Berg, J. M.; Tymoczko, J. L.; Stryer, L., Biochemistry. 5th ed.; W.H. Freeman: New York, 2002.
9. Shipman, M. A.; Symes, M. D., Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal Today 2017, 286, 57-68.
10. Becker, J. Y.; Posin, B., Nitrogen-Fixation .2. Nitrogen Reduction by Electrochemically Generated Vanadium(II) Promoted by Various Organic-Ligands in Basic Methanol. J Electroanal Chem 1988, 250 (2), 385-397.
11. Becker, J. Y.; Avraham, S., Nitrogen-Fixation .3. Electrochemical Reduction of Hydrazido (-NNH2) Mo and W-Complexes - Selective Formation of NH3 under Mild Conditions. J Electroanal Chem 1990, 280 (1), 119-127.
12. Pickett, C. J.; Talarmin, J., Electrosynthesis of Ammonia. Nature 1985, 317 (6038), 652-653.
13. Sclafani, A.; Augugliaro, V.; Schiavello, M., Dinitrogen Electrochemical Reduction to Ammonia over Iron Cathode in Aqueous-Medium. J Electrochem Soc 1983, 130 (3), 734-735.
14. Yandulov, D. V.; Schrock, R. R., Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301 (5629), 76-78.
15. Furuya, N.; Yoshiba, H., Electroreduction of Nitrogen to Ammonia on Gas-Diffusion Electrodes Loaded with Inorganic Catalyst. J Electroanal Chem 1990, 291 (1-2), 269-272.
16. Marnellos, G.; Stoukides, M., Ammonia synthesis at atmospheric pressure. Science 1998, 282 (5386), 98-100.
17. Marnellos, G.; Zisekas, S.; Stoukides, M., Synthesis of ammonia at atmospheric pressure with the use of solid state proton conductors. J Catal 2000, 193 (1), 80-87.
18. Murakami, T.; Nishikiori, T.; Nohira, T.; Ito, Y., Electrolytic synthesis of ammonia in molten salts under atmospheric pressure. J Am Chem Soc 2003, 125 (2), 334-335.
19. Murakami, T.; Nohira, T.; Goto, T.; Ogata, Y. H.; Ito, Y., Electrolytic ammonia synthesis from water and nitrogen gas in molten salt under atmospheric pressure. Electrochim Acta 2005, 50 (27), 5423-5426.
20. Murakami, T.; Nohira, T.; Araki, Y.; Goto, T.; Hagiwara, R.; Ogata, Y. H., Electrolytic synthesis of ammonia from water and nitrogen under atmospheric pressure using a boron-doped diamond electrode as a nonconsumable anode. Electrochem Solid St 2007, 10 (4), E4-E6.
21. Chatt, J.; Pearman, A. J.; Richards, R. L., Reduction of Mono-Coordinated Molecular Nitrogen to Ammonia in a Protic Environment. Nature 1975, 253 (5486), 39-40.
22. Pool, J. A.; Lobkovsky, E.; Chirik, P. J., Hydrogenation and cleavage of dinitrogen to ammonia with a zirconium complex. Nature 2004, 427 (6974), 527-530.
23. Avenier, P.; Taoufik, M.; Lesage, A.; Solans-Monfort, X.; Baudouin, A.; de Mallmann, A.; Veyre, L.; Basset, J. M.; Eisenstein, O.; Emsley, L.; Quadrelli, E. A., Dinitrogen dissociation on an isolated surface tantalum atom. Science 2007, 317 (5841), 1056-1060.
24. Amar, I. A.; Lan, R.; Petit, C. T. G.; Arrighi, V.; Tao, S. W., Electrochemical synthesis of ammonia based on a carbonate-oxide composite electrolyte. Solid State Ionics 2011, 182 (1), 133-138.
25. Murphy, O. J.; Denvir, A. J.; Teodorescu, S. G.; Uselton, K. B., Electrochemical synthesis of ammonia. Google Patents: 2008.
26. Ouzounidou, M.; Skodra, A.; Kokkofitis, C.; Stoukides, M., Catalytic and electrocatalytic synthesis of NH3 in a H+ conducting cell by using an industrial Fe catalyst. Solid State Ionics 2007, 178 (1-2), 153-159.
27. Pappenfus, T. M.; Lee, K.-m.; Thoma, L. M.; Dukart, C. R., Wind to Ammonia: Electrochemical Processes in Room Temperature Ionic Liquids. ECS Transactions 2009, 16 (49), 89-93.
28. Song, Z.; Cai, T. H.; Hanson, J. C.; Rodriguez, J. A.; Hrbek, J., Structure and reactivity of Ru nanoparticles supported on modified graphite surfaces: A study of the model catalysts for ammonia synthesis. J Am Chem Soc 2004, 126 (27), 8576-8584.
29. Xu, G. C.; Liu, R. Q.; Wang, J., Electrochemical synthesis of ammonia using a cell with a Nafion membrane and SmFe0.7Cu0.3-x Ni (x) O-3 (x=0-0.3) cathode at atmospheric pressure and lower temperature. Sci China Ser B 2009, 52 (8), 1171-1175.
30. Lan, R.; Irvine, J. T. S.; Tao, S. W., Synthesis of ammonia directly from air and water at ambient temperature and pressure. Sci Rep-Uk 2013, 3.
31. Skulason, E.; Bligaard, T.; Gudmundsdottir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jonsson, H.; Norskov, J. K., A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys Chem Chem Phys 2012, 14 (3), 1235-1245.
32. Abghoui, Y.; Garden, A. L.; Hlynsson, V. F.; Bjorgvinsdottir, S.; Olafsdottir, H.; Skulason, E., Enabling electrochemical reduction of nitrogen to ammonia at ambient conditions through rational catalyst design. Phys Chem Chem Phys 2015, 17 (7), 4909-4918.
33. Abghoui, Y.; Garden, A. L.; Howat, J. G.; Vegge, T.; Skulason, E., Electroreduction of N2 to Ammonia at Ambient Conditions on Mononitrides of Zr, Nb, Cr, and V: A DFT Guide for Experiments. Acs Catal 2016, 6 (2), 635-646.
34. Abghoui, Y.; Skulason, E., Electrochemical synthesis of ammonia via Mars-van Krevelen mechanism on the (111) facets of group III-VII transition metal mononitrides. Catal Today 2017, 286, 78-84.
35. Abghoui, Y.; Skulason, E., Onset potentials for different reaction mechanisms of nitrogen activation to ammonia on transition metal nitride electro-catalysts. Catal Today 2017, 286, 69-77.
36. Abghoui, Y.; Skulason, E., Computational Predictions of Catalytic Activity of Zincblende (110) Surfaces of Metal Nitrides for Electrochemical Ammonia Synthesis. J Phys Chem C 2017, 121 (11), 6141-6151.
37. Howalt, J. G.; Vegge, T., Electrochemical ammonia production on molybdenum nitride nanoclusters. Phys Chem Chem Phys 2013, 15 (48), 20957-20965.
38. Howalt, J. G.; Vegge, T., The role of oxygen and water on molybdenum nanoclusters for electro catalytic ammonia production. Beilstein J Nanotech 2014, 5, 111-120.
39. Abghoui, Y.; Skúlasson, E., Transition Metal Nitride Catalysts for Electrochemical Reduction of Nitrogen to Ammonia at Ambient Conditions. Procedia Computer Science 2015, 51, 1897-1906.
40. Norskov, J. K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J. R.; Bligaard, T.; Jonsson, H., Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J Phys Chem B 2004, 108 (46), 17886-17892.
41. Kung, H. H., Transition metal oxides : surface chemistry and catalysis. Elsevier ;
Distributors for the U.S. and Canada, Elsevier Science Pub. Co.: Amsterdam, The Netherlands, ; New York
New York, NY, U.S.A., 1989.
42. Hammer, B.; Hansen, L. B.; Norskov, J. K., Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys Rev B 1999, 59 (11), 7413-7421.
43. Blochl, P. E., Projector Augmented-Wave Method. Phys Rev B 1994, 50 (24), 17953-17979.
44. Kresse, G.; Hafner, J., Abinitio Molecular-Dynamics for Liquid-Metals. Phys Rev B 1993, 47 (1), 558-561.
45. Kresse, G.; Hafner, J., Ab-Initio Molecular-Dynamics Simulation of the Liquid-Metal Amorphous-Semiconductor Transition in Germanium. Phys Rev B 1994, 49 (20), 14251-14269.
46. Kresse, G.; Furthmuller, J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp Mater Sci 1996, 6 (1), 15-50.
47. Kresse, G.; Furthmuller, J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 1996, 54 (16), 11169-11186.
48. Skulason, E.; Tripkovic, V.; Bjorketun, M. E.; Gudmundsdottir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jonsson, H.; Norskov, J. K., Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J Phys Chem C 2010, 114 (42), 18182-18197.
49. Tripkovic, V.; Skulason, E.; Siahrostami, S.; Norskov, J. K.; Rossmeisl, J., The oxygen reduction reaction mechanism on Pt(111) from density functional theory calculations. Electrochim Acta 2010, 55 (27), 7975-7981.
50. Montoya, J. H.; Tsai, C.; Vojvodic, A.; Norskov, J. K., The Challenge of Electrochemical Ammonia Synthesis: A New Perspective on the Role of Nitrogen Scaling Relations. Chemsuschem 2015, 8 (13), 2180-2186.
51. Morgan, B. J.; Watson, G. W., A DFT+U description of oxygen vacancies at the TiO2 rutile (110) surface. Surf Sci 2007, 601 (21), 5034-5041.
52. Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T. R.; Moses, P. G.; Skulason, E.; Bligaard, T.; Norskov, J. K., Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys Rev Lett 2007, 99 (1).
Изобретение относится к способу получения аммиака, включающему: подачу N2 в электролитическую ячейку, содержащую по меньшей мере один источник протонов; обеспечение возможности контакта N2 с поверхностью катодного электрода в электролитической ячейке, где поверхность катодного электрода содержит каталитическую поверхность, содержащую по меньшей мере один оксид переходного металла, выбранный из группы, состоящей из оксида ниобия, оксида тантала, оксида осмия, оксида рения и оксида иридия, где по меньшей мере один оксид переходного металла содержит по меньшей мере одну поверхность, имеющую грань; и пропускание тока через указанную электролитическую ячейку, в результате чего азот реагирует с протонами с образованием аммиака. Также изобретение относится к системе. Изобретение дает возможность получения аммиака при комнатной температуре окружающей среды и атмосферном давлении. 2 н. и 20 з.п. ф-лы, 44 ил., 1 табл., 2 пр. 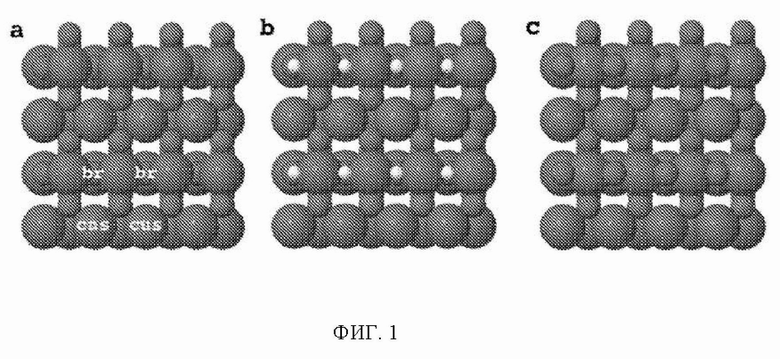
1. Способ получения аммиака, включающий:
подачу N2 в электролитическую ячейку, содержащую по меньшей мере один источник протонов;
обеспечение возможности контакта N2 с поверхностью катодного электрода в электролитической ячейке, где поверхность катодного электрода содержит каталитическую поверхность, содержащую по меньшей мере один оксид переходного металла, выбранный из группы, состоящей из оксида ниобия, оксида тантала, оксида осмия, оксида рения и оксида иридия, где по меньшей мере один оксид переходного металла содержит по меньшей мере одну поверхность, имеющую грань (110); и
пропускание тока через указанную электролитическую ячейку, в результате чего азот реагирует с протонами с образованием аммиака.
2. Способ по п. 1, где каталитическая поверхность содержит по меньшей мере одну поверхность, имеющую структуру рутила.
3. Способ по п. 1 или 2, где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 1,0 В, более предпочтительно менее чем приблизительно минус 0,8 В и еще более предпочтительно менее чем приблизительно минус 0,5 В по сравнению со стандартным водородным электродом (СВЭ).
4. Способ по п. 1 или 2, где катализатор содержит оксид ниобия, и где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 1,1 В по сравнению с СВЭ.
5. Способ по п. 1 или 2, где катализатор содержит оксид рения, и где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 0,7 В по сравнению с СВЭ.
6. Способ по п. 1 или 2, где катализатор содержит оксид тантала, и где аммиак образуется в электролитической ячейке при электродном потенциале менее чем приблизительно минус 0,7 В по сравнению с СВЭ.
7. Способ по любому из пп. 1-6, где образуется менее 50 % молей H2 по сравнению с количеством молей образовавшегося NH3, предпочтительно менее 20 % и еще более предпочтительно менее 10 %.
8. Способ по любому из пп. 1-7, где указанная электролитическая ячейка содержит один или более водных электролитических растворов.
9. Способ по любому из пп. 1-7, где указанная электролитическая ячейка содержит электролитический раствор, содержащий органический протонный или апротонный растворитель или их смешивающуюся смесь, предпочтительно смешивающийся с водой органический растворитель.
10. Способ по п. 8 или 9, где указанный азот подают в электролитическую ячейку путем барботирования газообразного азота в электролитический раствор, контактирующий с указанной поверхностью катодного электрода.
11. Способ по любому из пп. 1-10, где источником протонов при образовании аммиака является расщепление воды на аноде или реакция окисления H2 на аноде.
12. Способ по любому из пп. 1-11, который осуществляют при температуре от приблизительно минус 10 °С до приблизительно 40 °С и предпочтительно от приблизительно 0 до 40 °С.
13. Способ по любому из пп. 1-12, который осуществляют при комнатной температуре окружающей среды и атмосферном давлении.
14. Система для генерирования аммиака, содержащая по меньшей мере одну электрохимическую ячейку, содержащую по меньшей мере один катодный электрод, имеющий каталитическую поверхность, где каталитическая поверхность заполнена по меньшей мере одним катализатором, содержащим один или более оксидов переходных металлов, выбранных из группы, состоящей из оксида ниобия, оксида тантала, оксида осмия, оксида рения и оксида иридия, где оксид переходного металла содержит по меньшей мере одну поверхность, имеющую грань (110).
15. Система по п. 14, где каталитическая поверхность содержит по меньшей мере одну поверхность, имеющую структуру рутила.
16. Система по п. 14 или 15, где указанная электролитическая ячейка дополнительно содержит один или более электролитических растворов.
17. Система по п. 16, где указанная электролитическая ячейка содержит кислый, нейтральный или щелочной водный раствор.
18. Система по п. 17, где указанная электролитическая ячейка содержит электролитический раствор, содержащий органический протонный или апротонный растворитель или их смешивающуюся смесь, предпочтительно смешивающийся с водой органический растворитель.
19. Система по любому из пп. 14-18, сконструированная для получения аммиака в электролитической ячейке при электродном потенциале менее чем приблизительно минус 1,0 В, более предпочтительно менее чем приблизительно минус 0,8 В и еще более предпочтительно менее чем приблизительно минус 0,5 В по сравнению с СВЭ.
20. Система по п. 19, где катализатор содержит оксид ниобия, и где система сконструирована для получения аммиака в электролитической ячейке при электродном потенциале менее чем приблизительно минус 1,1 В по сравнению с СВЭ.
21. Система по п. 19, где катализатор содержит оксид рения, и где система сконструирована для получения аммиака в электролитической ячейке при электродном потенциале менее чем приблизительно минус 0,7 В по сравнению с СВЭ.
22. Система по п. 19, где катализатор содержит оксид тантала, и где система сконструирована для получения аммиака в электролитической ячейке при электродном потенциале менее чем приблизительно минус 0,7 В по сравнению с СВЭ.
| WO 2015009155 A1, 22.01.2015 | |||
| WO 2017132721 А1, 10.08.2017 | |||
| US 2016138176 A1, 19.05.2016 | |||
| ЭЛЕКТРОХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ АЗОТНЫХ УДОБРЕНИЙ | 2008 |
|
RU2479558C2 |
| RU 2008102379 A, 27.07.2009. | |||
