Настоящее изобретение относится к новому способу получения пиперазинил-этоксибромфенила и производных пиперазинил-
этоксифенилбороновой кислоты и к их применению в получении содержащих их соединений.
Более конкретно, настоящее изобретение относится к новому способу получения 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина и 1-{2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил- 1,3,2-диоксаборолан-2-ил)фенокси]этил}-
4- метилпиперазина и к их применению в получении содержащих их соединений.
Еще более конкретно настоящее изобретение относится к новому способу получения 1-{2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил}-4-метилпиперазина и к его применению в получении 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(5-фторфуран-2-ил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[1-(2,2,2-трифторэтил)-1H-пиразол-5-ил]метокси}фенил)пропановой кислоты, упоминается здесь как «Соединение 1», и 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановой кислоты, упоминается здесь как «Соединение 2».
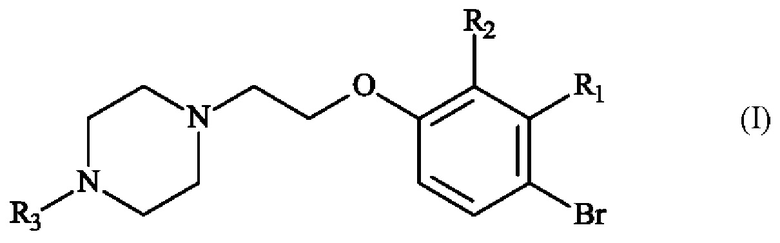
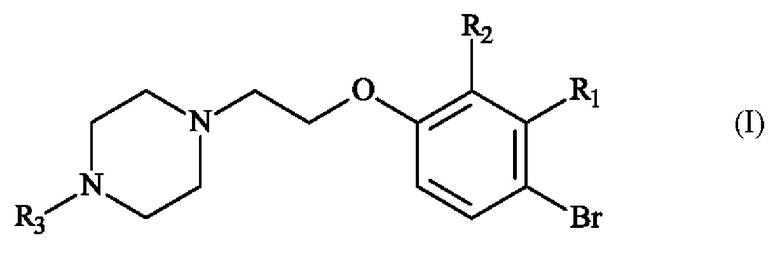
В частности, настоящее изобретение относится к способу получения пиперазинил-этоксибромфенильного соединения формулы (I):
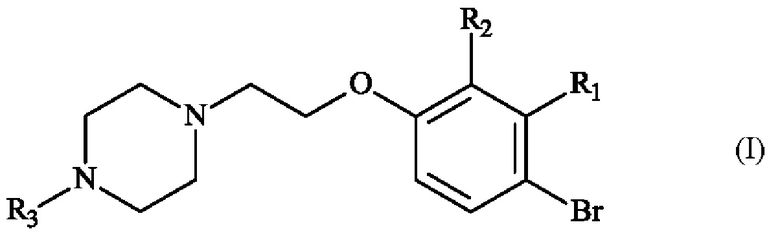
в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
♦ R3 представляет собой линейную или разветвленную (С1-С6)алкильную группу.
В частности, настоящее изобретение относится к способу получения соединения формулы (I) в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена или линейную или разветвленную (С1-С6)алкильную группу,
♦ R3 представляет собой линейную или разветвленную (С1-С6)алкильную группу.
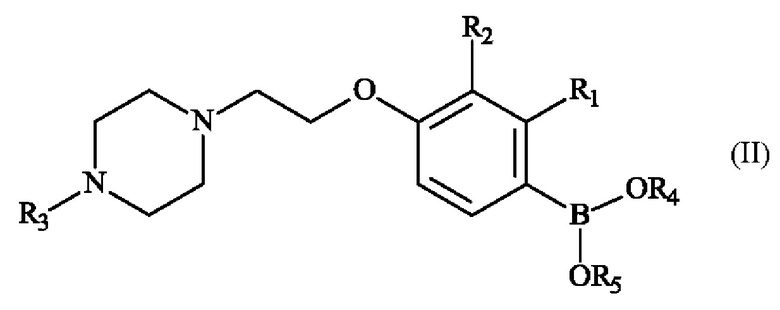
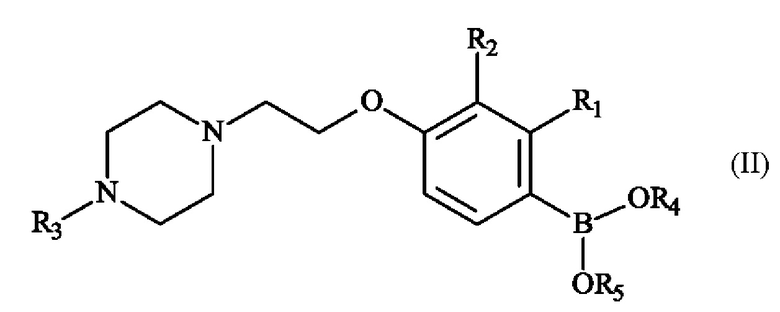
Настоящее изобретение также относится к способу получения пиперазинил-этоксифенилбороновой кислоты - соединение формулы (II):
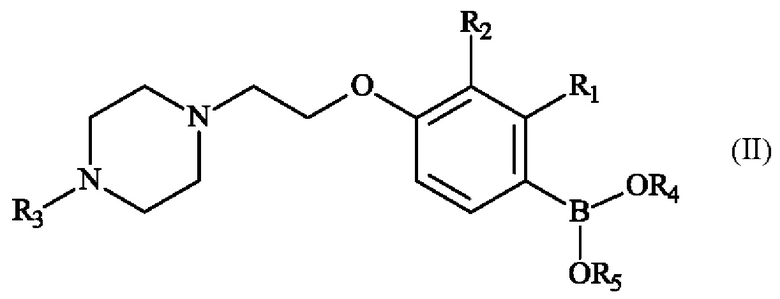
в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
♦ R3 представляет собой линейную или разветвленную (С1-С6)алкильную группу,
♦ R4 и R5 представляют собой водород, линейную или разветвленную (С1-С6)алкильную группу, или R4 и R5 образуют с несущими их атомами кислорода кольцо, которое может быть замещено одной-четырьмя линейными или разветвленными (С1-С6)алкильными группами.
В частности, настоящее изобретение относится к способу получения соединения формулы (II), в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена или линейную или разветвленную (С1-С6)алкильную группу,
♦ R3 представляет собой линейную или разветвленную (С1-С6)алкильную группу,
♦ R4 и R5 представляют собой водород, линейную или разветвленную (С1-С6)алкильную группу, или R4 и R5 образуют с несущими их атомами кислорода кольцо, которое может быть замещено одной-четырьмя линейными или разветвленными (С1-С6)алкильными группами.
Более конкретно настоящее изобретение относится к способу получения 1-{2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил}-4-метилпиперазина формулы (III):
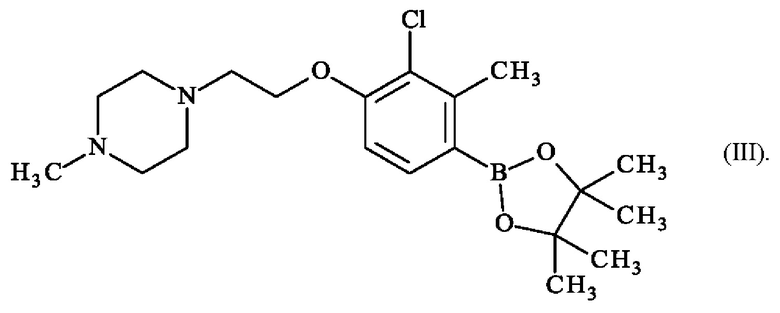
Соединения формул (I), (II) и (III), полученные в соответствии со способом, предлагаемым в изобретении, пригодны для синтеза Соединения 1 или для синтеза Соединения 2 а также их близкие по структуре аналоги.
В частности, Соединение 1 и Соединение 2 обладают проапоптотическими свойствами, а именно, они способны ингибировать белок Mcl-1, антиапоптотический член семейства Bcl-2, который сверхэкспрессируется при различных типах рака, что делает возможным использование Соединение 1 и Соединение 2 при патологиях, включающих дефект апоптоза, таких как, например, лечение рака, а также иммунных и аутоиммунных заболеваний.
Принимая во внимание фармацевтическую ценность этих соединений, важно иметь возможность получать их с помощью эффективного способа синтеза, который легко переносится в промышленный масштаб и который обеспечивает Соединение 1 или Соединение 2 с хорошим выходом и превосходной чистотой, исходя из экономичных и легко доступных исходных веществ.
Получение Соединения 1 и его фармакологические эффекты на различных моделях рака описаны в литературных источниках (Kotschy и соавт. Nature 2016, 538, 477-482 и соответствующая дополнительная информация, которая включена в качестве ссылки). Кроме того, Соединение 1, Соединение 2 и их близкие по структуре аналоги, их получение, их использование в качестве ингибиторов Mcl-1 для лечения рака и их фармацевтические составы описаны в WO 2015/097123. В частности, способ синтеза соединения формулы (III) конкретно раскрыт в Способе получения 5b заявки WO 2015/097123, в котором соединение формулы (III) получают в ходе пяти стадий, исходя из 4-бром-2-хлор-фенола. Не так давно в CN 107573360 также раскрыто альтернативное получение соединения формулы (III) из 4-бром-2-хлор-фенола за пять стадий. Помимо этого, соединение формулы (III) и его получение также конкретно раскрыты в заявках WO 2016/207226, WO 2016/207217, WO 2016/207216 и WO 2017/125224. Однако при переходе к промышленному масштабу быстро выявились трудности в реализации этого способа: в частности, риск использования легковоспламеняющихся и потенциально взрывоопасных реагентов на стадии защиты, отсутствие селективности во время реакции метилирования, а также низкий выход и многочисленные побочные продукты во время реакций борилирования и Мицунобу.
Более того, альтернативный способ синтеза соединений формулы (II) конкретно раскрыт в заявке WO 2015/097123, в которой соединения формулы (II) получают в три стадии, исходя из 2,3-дизамещенного фенола. Однако при переходе к промышленному масштабу также быстро выявились трудности в реализации этого способа: в частности, низкий выход на стадии бромирования, низкий выход и многочисленные побочные продукты во время реакции Мицунобу и низкий выход во время стадии борилирования.
Следовательно, поиск новых эффективных путей синтеза все еще продолжается, и заявитель продолжил свои исследования с целью разработки нового синтеза, который обеспечивает соединения формул (I), (II) или (III) воспроизводимым способом, с превосходными выходами и без необходимости трудоемкой очистки с чистотой, совместимой с его использованием в качестве фармацевтически приемлемого промежуточного соединения.
В частности, в настоящее время заявителем был разработан новый способ синтеза, позволяющий получать соединения формул (I) и (II) воспроизводимым способом без необходимости трудоемкой очистки, используя производные 1,2-дизамещенного-3-бромфенила в качестве исходного вещества. Это новое исходное вещество имеет то преимущество, что является простым и легко доступным в больших количествах по более низкой цене. В частности, заявителем был разработан новый способ промышленного синтеза, позволяющий получать соединения формулы (III) воспроизводимым способом без необходимости трудоемкой очистки с использованием 3-бром-2-хлортолуола в качестве исходного вещества. 3-Бром-2-хлортолуол также имеет преимущество наличия в своей структуре метальной группы, что позволяет избежать включения стадии неселективного метилирования в синтез - стадии, которая была проблематичной при переносе в промышленный масштаб.
Предлагаемый в изобретении новый способ имеет преимущество использования эффективной реакции региоселективного монобромирования, выдающейся реакции раскрытия цикла 1-алкил-1-азониабицикло[2.2.2] октанового соединения и эффективной реакции борилирования. Реакция бромирования соединения формулы (VI), в частности 2-фтор-3-метил-фенола, с использованием N-бромсукцинимида (NBS) в качестве реагента уже была описана в заявке WO 2015/162515. Тем не менее, было обнаружено, что использование реагента NBS дает нежелательные дибромированные побочные продукты и более низкий выход. Реакция раскрытия цикла 1-алкил-1-азониабицикло[2.2.2]
октанового соединения уже описана в литературных источникых (Maras и соавт. Organic and Biomolecular Chemistry 2012, 10, 1300-1310, которая включена в качестве ссылки). Однако заявитель обнаружил неожиданные экспериментальные условия, о которых говорится в публикации Maras.
Краткое описание способа согласно изобретению показано на схеме 1, см. ниже.
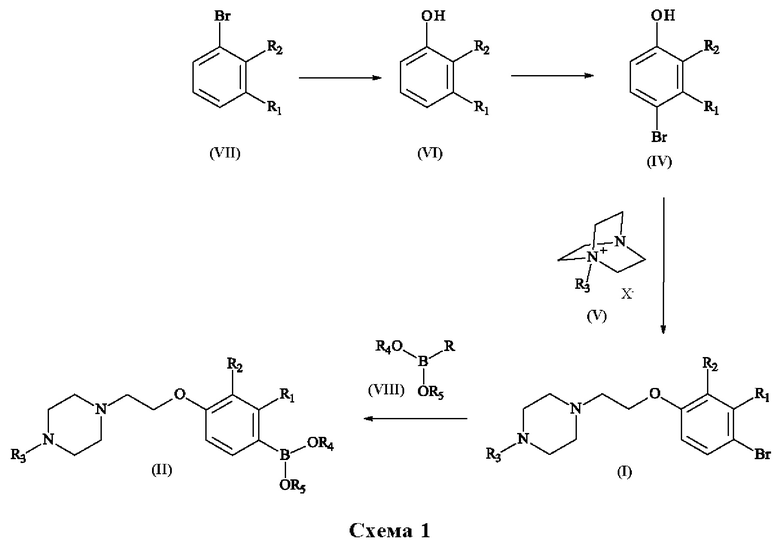
Реакции раскрытия цикла производного 1-алкил-1-азониабицикло[2.2.2] октана: (IV)+(V) -> (1)
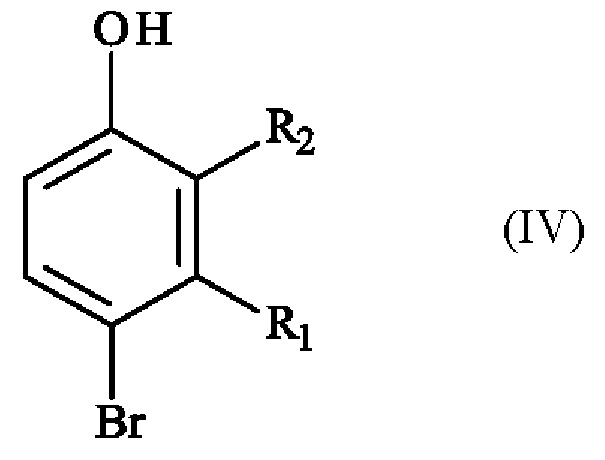
Конкретный вариант осуществления настоящего изобретения относится к способу получения соединения формулы (I):
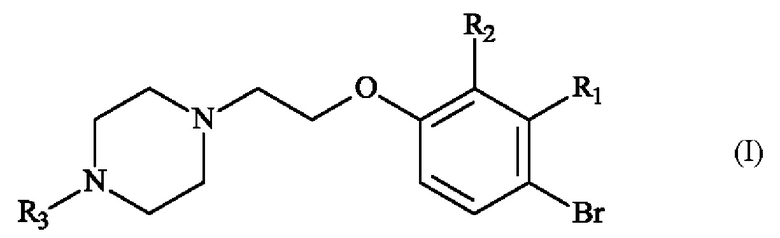
в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
♦ R3 представляет собой линейную или разветвленную (С1-С6)алкильную группу,
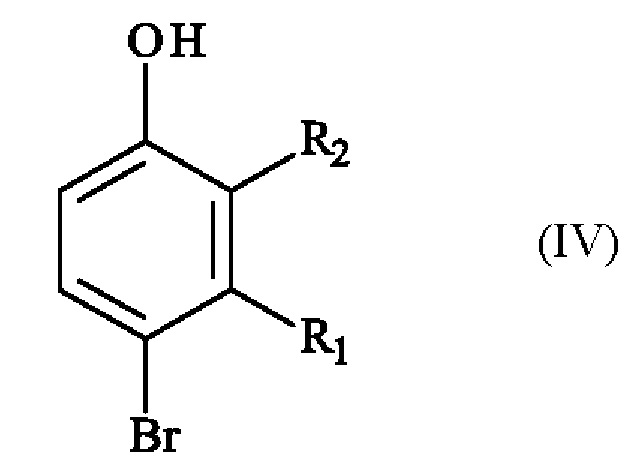
включающему в себя стадию реакции соединения формулы (IV):
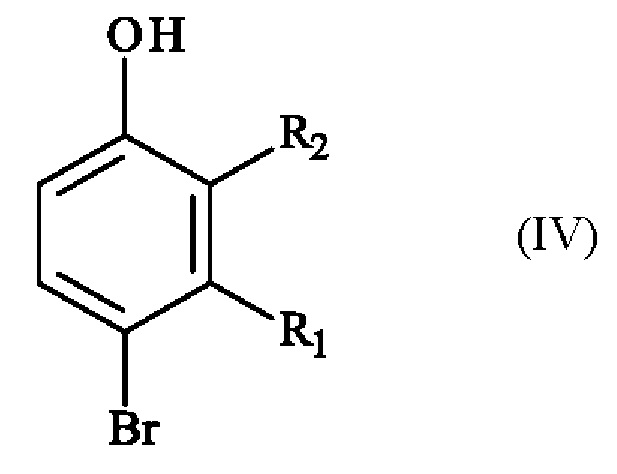
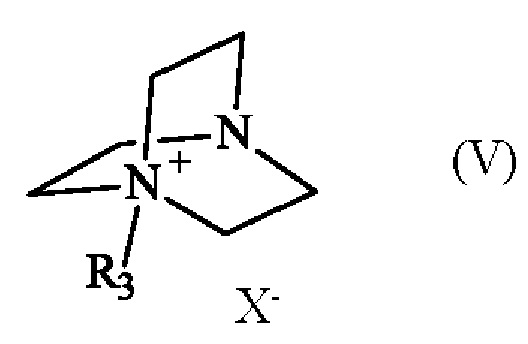
в которой R1 и R2 имеют приведенные выше определения, с соединением формулы (V):
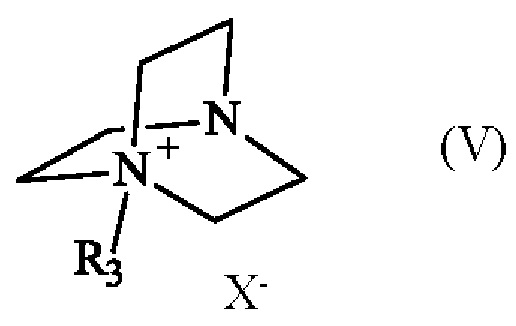
в которой R3 имеет приведенное выше определение, и X- представляет собой одновалентный анионный противоион,
в растворителе, при высокой температуре в присутствии основания.
В одном варианте осуществления растворитель, который можно использовать для проведения превращения соединения формулы (IV) с образованием соединения формулы (I), предпочтительно представляет собой полярный апротонный растворитель. Среди полярных апротонных растворителей, которые можно использовать для проведения превращения соединения формулы (IV) с образованием соединения формулы (I), можно упомянуть, без каких-либо ограничений, анизол, пиридин, N-метил-2-пирролидон, N,N-диметилформамид, N,N-диметилацетамид, диглим, диметилсульфоксид, ацетонитрил, тетрагидрофуран, 2-метилтетрагидрофуран, полиэтиленгликоль, сульфолан.
Растворитель, используемый для проведения превращения соединения формулы (IV) с образованием соединения формулы (I), также может состоять из смеси двух или более растворителей из числа вышеупомянутых растворителей.
Растворителем, предпочтительно используемым для проведения превращения соединения формулы (IV) с образованием соединения формулы (I) является анизол.
Предпочтительно реакцию превращения соединения формулы (IV) в соединение формулы (I) проводят при температуре выше 135°С, более предпочтительно от 140°С до 150°С. Одним из предпочтительных вариантов превращения соединения формулы (IV) в соединение формулы (I) является проведение реакции при температуре от 135°С до 145°С. Еще один предпочтительный вариант превращения соединения формулы (IV) в соединение формулы (I) заключается в проведении реакции при 140°С.
В числе оснований, которые можно использовать для осуществления превращения соединения формулы (IV) с образованием соединения формулы (I), можно упомянуть, без каких-либо ограничений, трет-бутоксид калия, трет-бутоксид лития, ацетат калия, этоксид лития, карбонатные соли, такие как карбонат цезия, карбонат калия, карбонат натрия, карбонат лития.
Основание, предпочтительно используемое для проведения превращения соединения формулы (IV) с образованием соединения формулы (I), представляет собой карбонатную соль, более предпочтительно карбонат цезия.
Соединение формулы (I) может быть выделено в виде свободного основания, моногидрогалогенидной соли или дигидрогалогенидной соли. Предпочтительно соединение формулы (I) может быть выделено в виде моногидрогалогенидной соли или дигидрогалогенидной соли. Более предпочтительно соединение формулы (I) выделяют в виде дигидрогалогенидной соли, даже более предпочтительно в виде дигидрохлоридной соли.
Выделение соединения формулы (I) в виде моногидрогалогенидной соли предпочтительно проводят в трет-бутилметиловом эфире, диоксане, толуоле, циклогексане, циклопентилметиловом эфире или этилацетате, более предпочтительно в трет-бутилметиловом эфире.
Выделение соединения формулы (I) в виде дигидрогалогенидной соли предпочтительно проводят в воде.
Соединение формулы (V) получают из 1,4-диазабицикло[2.2.2]октана (также известного как DABCO; CAS номер: 280-57-9). Соединение формулы (V) может быть синтезировано путем реакции 1,4-диазабицикло[2.2.2]октана с алкилирующим агентом, выбранным из алкилгалогенида, алкилтозилата, алкилсульфата или алкилмезилата. В частности, соединение формулы (V) определено следующим образом:
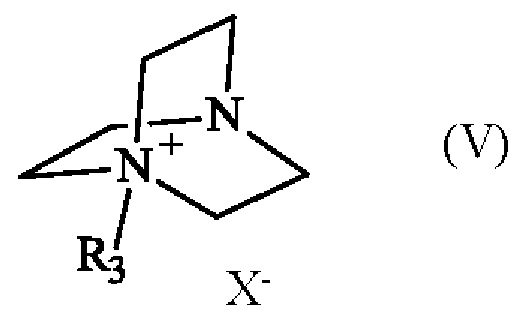
в которой R3 имеет приведенное выше определение, а X- представляет собой одновалентный анионный противоион, выбранный из галогенида, тозилата, сульфата или мезилата.
Преимущественно соединение формулы (V) определяется следующим образом:
в которой R3 представляет собой метальную группу и X- представляет собой противоион тозилата.
Предпочтительно соединение формулы (V) может быть синтезировано с метилирующим агентом, выбранным из метилгалогенида, метилтозилата (также известного как 4-метилбензол-1-сульфонат), метилсульфата или метилмезилата, более предпочтительно метилтозилата.
Соединение формулы (V) может быть синтезировано отдельно или in situ, предпочтительно in situ.
В конкретном варианте осуществления соединение формулы (I) получают с использованием 1,4-диазабицикло[2.2.2]октана.
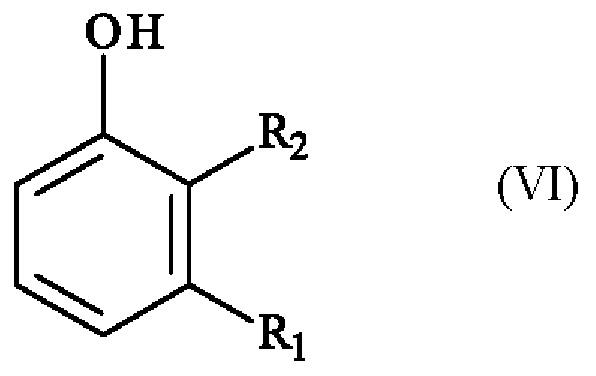
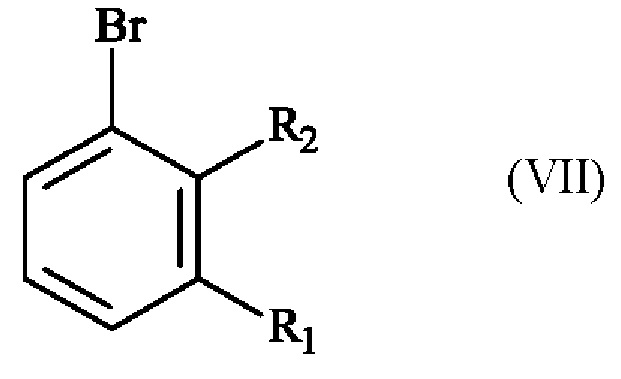
Реакция региоселективного монобромирования: (VI) -> (IV)
Конкретный вариант осуществления настоящего изобретения относится к способу, в котором соединение формулы (IV):
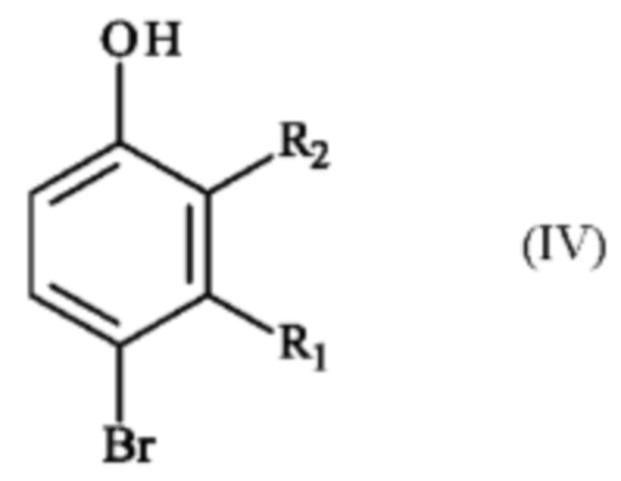
в которой R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
получают реакцией региоселективного монобромирования соединения формулы (VI):
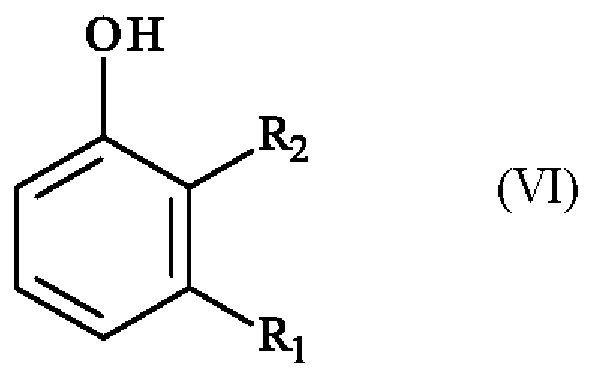
в которой R1 и R2 имеют приведенные выше определения,
в растворителе в присутствии бромирующего агента.
В предлагаемом в изобретении способе реакцию превращения соединения формулы (VI) в соединение формулы (IV) проводят в присутствии 1 эквивалента бромирующего агента.
Среди бромирующих агентов, которые могут быть использованы для осуществления превращения соединения формулы (VI) с образованием соединения формулы (IV), можно упомянуть, без каких-либо ограничений, N-бромсукцинимид, бром, бромид натрия/трихлоризоциануровая кислота, бром/ацетат натрия, бромтрихлорметан, 1,2-дибром-1,1,2,2-тетрахлорэтан, тетрабромметан, тетрабромид углерода, трибромид тетрабутиламмония, трибромид триметилфениламмония, трибромид бензилтриметиламмония, пербромид бромида пиридиния, пербромид бромида 4-диметиламинопиридиния, трибромид 1-бутил-3-метилимидазолия, тригидробромид 1,8-диазабицикло[5.4.0]-7-ундецен, N-бромфталимид, N-бромосахарин, N-бромацетамид, 2-бром-2-циано-N,N-диметилацетамид, 1,3-дибром-5,5-диметилгидантоин, дибромизоциануровая кислота, гидрат мононатрия бромизоцианурата, трибромид бора (17% в дихлорметане, прибл. 1 моль/л), трибромид бора (29% в гептане, прибл. 1 моль/л), трибромид фосфора, бромид бромдиметилсульфония, 5,5-диброммельдрумовая кислота, гексафторфосфат 2,4,4,6-тетрабром-2,5-циклогексадиенон, бис(2,4,6-триметилпиридин)-бромония.
Бромирующий агент, предпочтительно используемый для проведения превращения соединения формулы (VI) с образованием соединения формулы (IV) представляет собой N-бромсукцинимид, бром, бромид натрия/трихлоризоциануровую кислоту или бром/ацетат натрия, более предпочтительно бром, бромид натрия/трихлоризоциануровую кислоту или бром/ацетат натрия, еще более предпочтительно, бром.
Среди растворителей, которые можно использовать для превращения соединения формулы (VI) с образованием соединения формулы (IV), можно упомянуть, без каких-либо ограничений, дихлорметан, 1,2-дихлорэтан, тетрагидрофуран, ацетонитрил, ацетон, диметилформамид, вода, метанол, уксусная кислота, серная кислота, бромистоводородная кислота.
Растворитель, используемый для проведения превращения соединения формулы (VI) с образованием соединения формулы (IV), также может состоять из смеси двух или более растворителей из числа вышеупомянутых органических растворителей.
Растворителем, предпочтительно используемым для проведения превращения соединения формулы (VI) с образованием соединения формулы (IV), является уксусная кислота, дихлорметан, смесь метанола и серной кислоты или смесь уксусной кислоты и дихлорметана. В предпочтительном варианте осуществления растворитель, используемый для проведения превращения соединения формулы (VI) с образованием соединения формулы (IV), представляет собой смесь уксусной кислоты и дихлорметана, более предпочтительно смесь от 10% об./об. до 100% об./об. уксусной кислоты в дихлорметане, еще более предпочтительно, смесь от 15% об./об. до 30% об./об. уксусной кислоты в дихлорметане. Преимущественно растворитель, используемый для проведения превращения соединения формулы (VI) с образованием соединения формулы (IV), представляет собой смесь 25% об./об. уксусной кислоты и дихлорметана.
Предпочтительно реакцию превращения соединения формулы (VI) в соединение формулы (IV) проводят при температуре от -20°С до 30°С, более предпочтительно от -15°С до 5°С, еще более предпочтительно от -15°С до -5°С. В другом предпочтительном варианте осуществления реакцию превращения соединения формулы (VI) в соединение формулы (IV) проводят при температуре от -5°С до 5°С.
Предпочтительно реакцию бромирования можно проводить путем разбавления соединения формулы (VI) с примерно от 10 до примерно 20, более предпочтительно от примерно 10 до примерно 15, даже более предпочтительно примерно 10 объемов органических растворителей или смесей органических растворителе.
Реакция гидроксилирования: (VII) -> (VI)
Конкретный вариант осуществления настоящего изобретения относится к способу, в котором соединение формулы (VI):
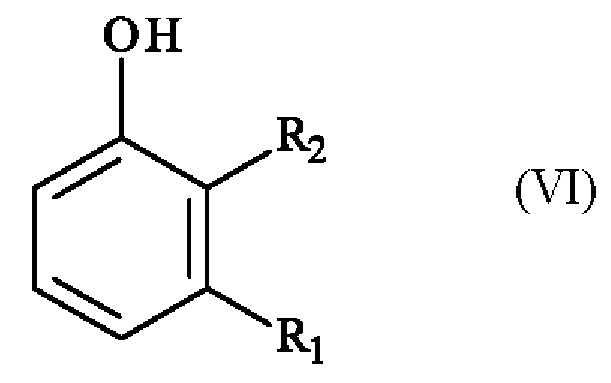
в которой R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
получают реакцией гидроксилирования соединения формулы (VII):
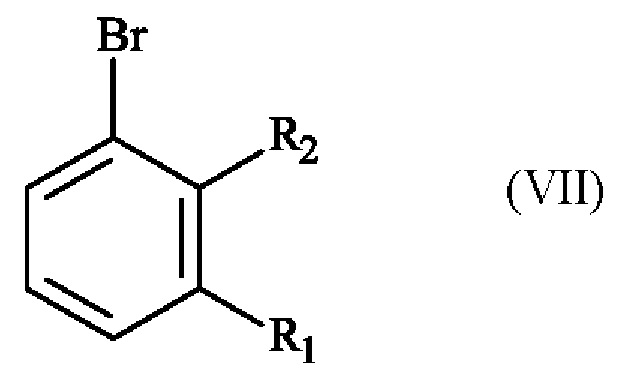
в которой R1 и R2 имеют приведенные выше определения,
в растворителе в присутствии комплекса переходного металла и основания.
В предлагаемом в изобретении способе реакция превращения соединения формулы (VII) в соединение формулы (VI) может быть проведена с помощью различных реакций гидроксилирования, катализируемых металлами (Maleczka и соавт., J. Am. Chem. Soc. 2003, 125, 7792-7793; Willis, Angew. Chem. Int. Ed. 2007, 46, 3402-3404; Alonso и соавт., Chem. Eur. J. 2010, 16, 5274-5284; Enthaler и соавт., Chem. Soc. Rev. 2011, 40, 4912-4924; Xia и соавт., J. Am. Chem. Soc. 2016, 138, 13493-13496, которые включены в качестве ссылки). Преимущественно в предлагаемом в изобретении способе реакцию превращения соединения формулы (VII) в соединение формулы (VI) можно проводить в присутствии комплекса переходного металла, который представляет собой комплекс палладия, содержащий палладиевый катализатор и лиганд.
Среди палладиевых катализаторов, которые можно использовать для превращения соединения формулы (VII) с образованием соединения формулы (VI), можно упомянуть, без каких-либо ограничений, трис(дибензилиденацетон) дипалладий Pd2(dba)3, ацетат палладия(II) Pd(OAc)2, палладий на угле Pd/C, тетракис(трифенилфосфин)палладий Pd(PPh3)4.
Палладиевый катализатор, предпочтительно используемый для проведения превращения соединения формулы (VII) с образованием соединения формулы (VI) представляет собой Pd2(dba)3.
Среди лигандов, которые могут быть использованы для осуществления превращения соединения формулы (VII) с образованием соединения формулы (VI), можно упомянуть, без каких-либо ограничений, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил XPhos, 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил t-BuXPhos.
Лиганд, предпочтительно используемый для превращения соединения формулы (VII) с образованием соединения формулы (VI) представляет собой t-BuXPhos.
В предлагаемом в изобретении способе реакцию превращения соединения формулы (VII) в соединение формулы (VI) проводят в присутствии по меньшей мере 0,01 эквивалента палладиевого катализатора, более предпочтительно по меньшей мере 0,0075 эквивалента. Реакцию превращения соединения формулы (VII) в соединение формулы (VI) проводят в присутствии по меньшей мере 0,03 эквивалента лиганда, более предпочтительно по меньшей мере 0,02 эквивалента. Преимущественно реакцию превращения соединения формулы (VII) в соединение формулы (VI) проводят в присутствии по меньшей мере 0,01 эквивалента палладиевого катализатора и по меньшей мере 0,03 эквивалента лиганда. Более предпочтительно, реакцию превращения соединения формулы (VII) в соединение формулы (VI) проводят в присутствии 0,01 эквивалента палладиевого катализатора и 0,04 эквивалента лиганда.
Среди оснований, которые можно использовать для проведения превращения соединения формулы (VII) с образованием соединения формулы (VI), можно упомянуть, без каких-либо ограничений, ацетат калия, трет-бутоксид натрия, бикарбонат натрия, карбонат калия, гидроксидные соли, такие как гидроксид калия, гидроксид натрия, гидроксид цезия, гидроксид лития.
Основание, предпочтительно используемое для проведения превращения соединения формулы (VII) с образованием соединения формулы (VI) представляет собой гидр оксидную соль, более предпочтительно гидроксид калия, гидроксид натрия, гидроксид цезия, гидроксид лития, еще более предпочтительно гидроксид калия.
Среди растворителей, которые могут быть использованы для проведения превращения соединения формулы (VII) с образованием соединения формулы (VI), можно упомянуть, без каких-либо ограничений, 1,4-диоксан, циклопентилметиловый эфир, толуол, гептан, воду, ацетонитрил, диметилсульфоксид, N,N-диметилформамид, N-метил-2-пирролидон, N,N-диметилацетамид, тетрагидрофуран, 2-метилтетрагидрофуран, трет-бутилметиловый эфир.
Растворитель, используемый для проведения превращения соединения формулы (VII) с образованием соединения формулы (VI), также может состоять из смеси двух или более растворителей из числа вышеупомянутых органических растворителей, или смеси воды и растворителя из числа упомянутых выше органических растворителей.
Растворителем, предпочтительно используемым для проведения превращения соединения формулы (VII) с образованием соединения формулы (VI) является 1,4-диоксан или смесь воды и 1,4-диоксана, более предпочтительно смесь воды и 1,4-диоксана. Преимущественно доля 1,4-диоксана в воде составляет по меньшей мере 5%, более предпочтительно по меньшей мере 15%, еще более предпочтительно по меньшей мере 25%.
Преимущественный вариант осуществления относится к последовательности реакций гидроксилирования и региоселективного монобромирования, превращающих соединение формулы (VII) в соединение формулы (IV) без выделения соединения формулы (VI). Во время такого выгодного варианта осуществления органический растворитель, используемый для проведения превращения невыделенного соединения формулы (VI) в соединение формулы (IV) состоит из смеси растворителей, предпочтительно, смеси 1,4-диоксана, уксусной кислоты и дихлорметана, в которой 1,4-диоксан представляет собой остаточный растворитель, поступающий с указанной стадии гидроксилирования (т.е. стадии превращения соединения формулы (VII) в соединение формулы(VI)).
Реакция борилирования: (I) -> (II)
Конкретный вариант осуществления настоящего изобретения относится к способу получения соединения формулы (II):
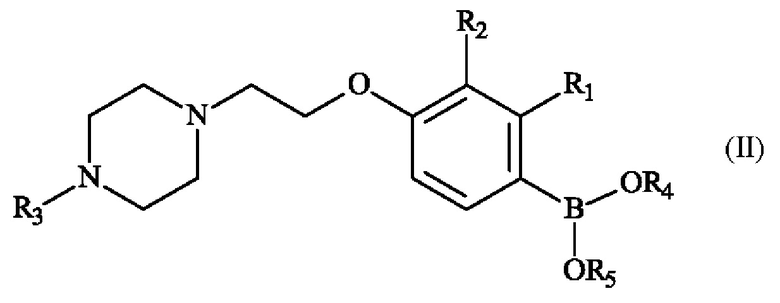
в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
♦ R3 представляет собой линейную или разветвленную (С1-С6)алкильную группу,
♦ R4 и R5 представляют собой водород, линейную или разветвленную (С1-С6)алкильную группу, или R4 и R5 образуют с несущими их атомами кислорода кольцо, которое может быть замещено одной-четырьмя линейными или разветвленными (С1-С6)алкильными группами,
включающему в себя стадию реакции соединения формулы (I),
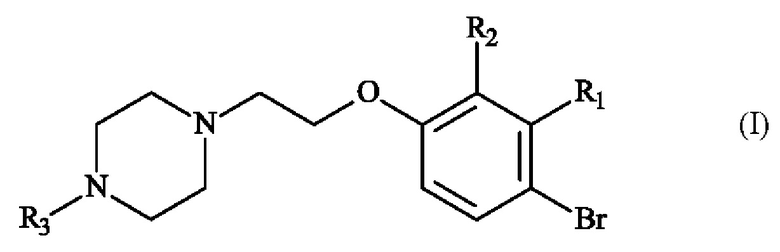
в которой R1, R2 и R3 имеют приведенные выше определения,
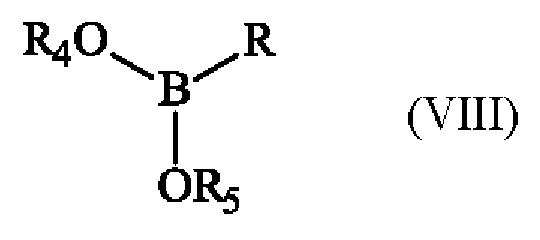
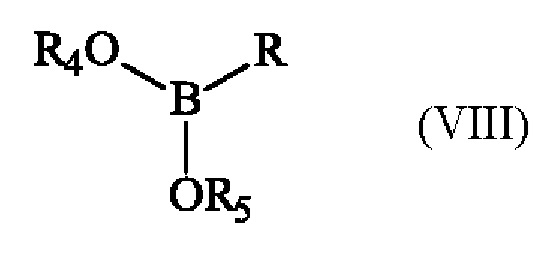
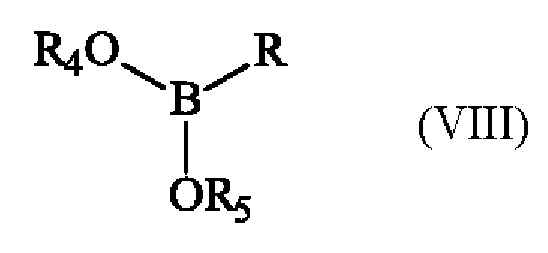
с эфиром бороновой кислоты формулы (VIII):

в которой R4 и R5 имеют приведенные выше определения и R представляет собой атом водорода, гидрокси-группу, линейную или разветвленную (С1-С6)алкокси-группу, или группу (С0-С6)алкил-В(OR4)(OR5).
В предлагаемом в изобретении способе превращение соединения формулы (I) в соединение формулы (II) состоит из действия соединения формулы (VIII), в которой R представляет собой атом водорода, гидрокси-группу, или линейную или разветвленную (С1-С6)алкокси-группу, в органическом растворителе или смеси органических растворителей в присутствии основания. Преимущественно реакцию превращения соединения формулы (I) в соединение формулы (II) проводят в тетрагидроф уране или 2-метилтетрагидрофуране, более предпочтительно 2-метилтетрагидрофуране. Предпочтительно, реакцию превращения соединения формулы (I) в соединение формулы (II) проводят в присутствии н-бутиллития.
В качестве альтернативы, в предлагаемом в изобретении способе превращение соединения формулы (I) в соединение формулы (II) заключается в действии соединения формулы (VIII), в которой R представляет собой группу (С0-С6)алкил-В(OR4)(OR5), в органическом растворителе или смеси органических растворителей в присутствии основания и комплекса палладия (борилирование Мияуры). Предпочтительно указанный комплекс палладия представляет собой дихлорид бис(трифенилфосфин)палладия (II) Pd(PPh3)2Cl2.
Соединение формулы (I) предпочтительно используют в качестве свободного основания для его превращения в соединение формулы (II). Когда соединение формулы (I) представляет собой соль дигидрогалогенида, два дополнительных эквивалента указанного основания предпочтительно добавляют в реакционную смесь для осуществления превращения соединения формулы (I) с образованием соединения формулы (II).
Чтобы осуществить превращение соединения формулы (I) с образованием соединения формулы (II), соединение формулы (I) преимущественно получают реакцией соединения формулы (IV) с соединением формулы (V).
Преимущественно настоящее изобретение относится к способу получения соединения формулы (II):
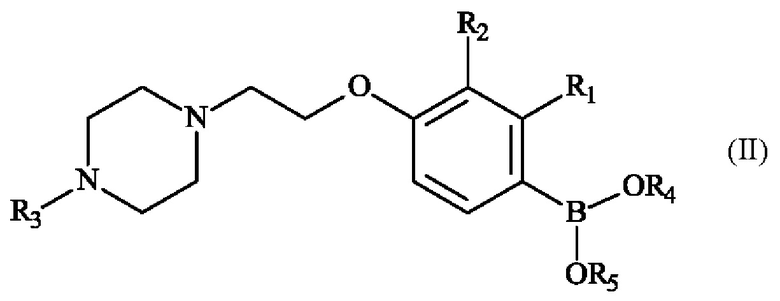
в которой:
♦ R1 и R2 независимо друг от друга представляют собой атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С1-С6)алкокси-группу, линейную или разветвленную (С1-С6)алкокси(С1-С6)алкокси-группу, гидроксильную группу или цианогруппу,
♦ R3 представляет собой линейную или разветвленную (C1-С6)алкильную группу,
♦ R4 и R5 представляют собой водород, линейную или разветвленную (С1-С6)алкильную группу, или R4 и R5 образуют с несущими их атомами кислорода кольцо, которое может быть замещено одной-четырьмя линейными или разветвленными (С1-С6)алкильными группами,
отличающемуся чет, что соединение формулы (VII):
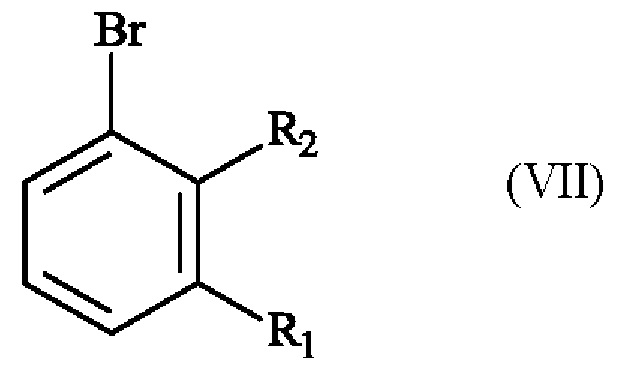
в которой R1 и R2 имеют приведенные выше определения,
подвергают реакции гидроксилирования в присутствии комплекса переходного металла и основания в растворителе,
с получением соединения формулы (VI):
в которой R1 и R2 имеют приведенные выше определения,
при этом соединение формулы (VI) подвергают реакции региоселективного монобромирования в присутствии бромирующего агента в растворителе,
с получением соединения формулы (IV):
в которой R1 и R2 имеют приведенные выше определения,
при этом соединение формулы (IV) реагирует в растворителе при высокой температуре в присутствии основания и соединения формулы (V):

в которой R3 имеет приведенное выше определение, и X- представляет собой одновалентный анионный противоион,
с получением соединения формулы (I):
в которой R1, R2 и R3 имеют приведенные выше определения,
при этом соединение формулы (I) подвергают реакции борилирования в присутствии эфира бороновой кислоты формулы (VIII):
в которой R4 и R5 имеют определения, приведенные для формулы (II) и R представляет собой атом водорода, гидрокси-группу, линейную или разветвленную (С1-С6)алкокси-группу, или группу (С0-С6)алкил-В(OR4)(OR5),
с получением соединения формулы (II).
В конкретном варианте осуществления R1 предпочтительно представляет собой атом галогена или линейную или разветвленную (С1-С6)алкильную группу, более предпочтительно атом фтора, атом хлора, этильную группу или метальную группу, еще более предпочтительно метальную группу. R2 представляет собой преимущественно атом галогена или линейную или разветвленную (С1-С6)алкильную группу, более предпочтительно атом хлора или метальную группу, еще более предпочтительно атом хлора. В частности, R3 представляет собой метальную группу. Более конкретно, R1 представляет собой линейную или разветвленную (С1-С6)алкильную группу, R2 представляет собой атом галогена и R3 представляет собой метальную группу. Еще более конкретно R1 и R3 представляют собой метальную группу и R2 представляет собой атом хлора. Предпочтительно R4 и R5 образуют с атомами кислорода, несущими их, кольцо, которое может быть диоксаборетаном, диоксабороланом, диоксаборинаном или диоксаборепаном, более предпочтительно диоксаборолановое кольцо. Преимущественно R4 и R5 образуют с несущими их атомами кислорода кольцо, которое может быть замещено одной-четырьмя линейными или разветвленными (С1-С6)алкильными группами. Более предпочтительно R4 и R5 с атомами кислорода, несущими их, образуют 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо.
Преимущественно, настоящее изобретение относится к способу получения соединения формулы (III):
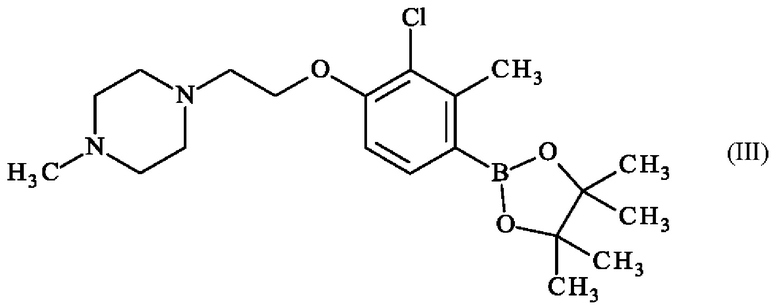
отличающемуся тем, что соединение формулы (VII):
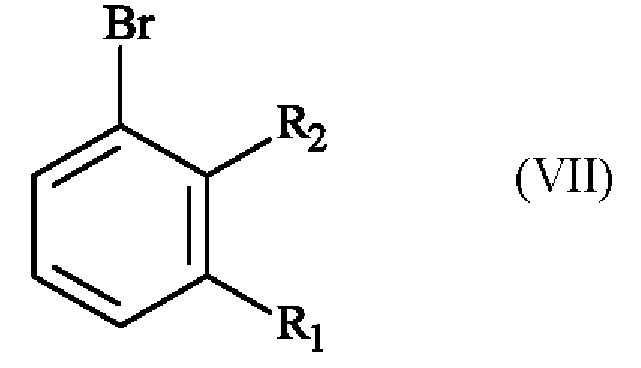
в которой R1 представляет собой метальную группу и R2 представляет собой атом хлора,
подвергают реакции гидроксилирования в присутствии комплекса переходного металла и основания в растворителе,
с получением соединения формулы (VI):
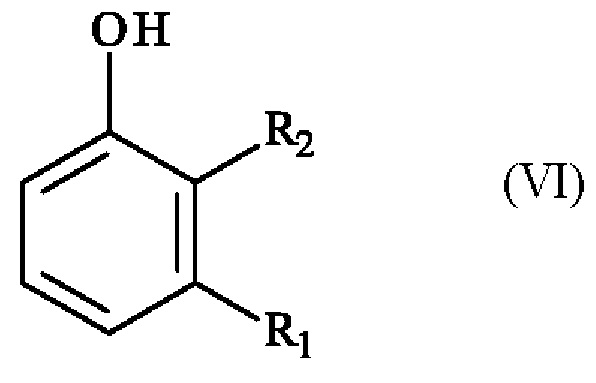
в которой R1 и R2 имеют приведенные выше определения,
при этом соединение формулы (VI) подвергают реакции региоселективного монобромирования в присутствии бромирующего агента в растворителе,
с получением соединения формулы (IV):
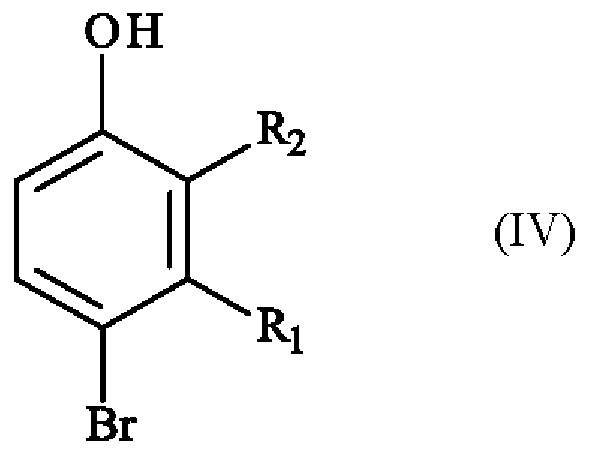
в которой R1 и R2 имеют приведенные выше определения,
при этом соединение формулы (IV) реагирует в растворителе при высокой температуре в присутствии основания и соединения формулы (V):
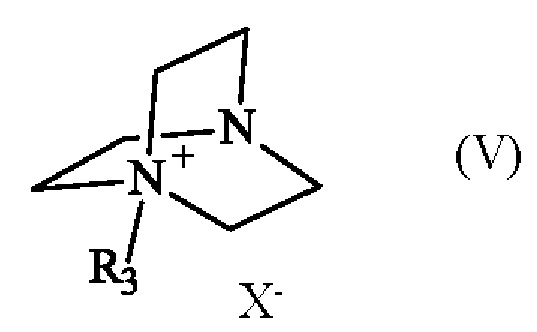
в которой R3 представляет собой метальную группу, а X- представляет собой одновалентный анионный противоион, с получением соединения формулы (I):
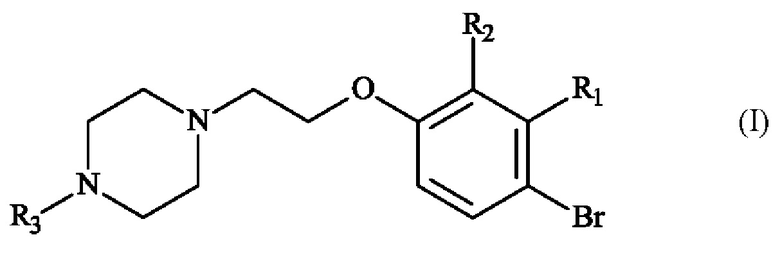
в которой R1, R2 и R3 имеют приведенные выше определения,
при этом соединение формулы (I) подвергают реакции борилирования в присутствии эфира бороновой кислоты формулы (VIII):

в которой R4 и R5 с атомами кислорода, несущими их, образуют 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо и R представляет собой атом водорода, гидрокси-группу, линейную или разветвленную (С1-С6)алкокси-группу, или группу (С0-С6)алкил-В(OR4)(OR5),
с получением соединения формулы (III).
Соединение формулы (V), (VII) и (VIII) коммерчески доступны или легко могут быть получены специалистом в данной области техники с использованием химических реакций, которые являются обычными или описаны в литературных источниках.
Данный способ особенно выгоден по следующим причинам:
- он позволяет получать соединение формулы (I) в промышленном масштабе с превосходными выходами, исходя из простого и недорогого исходного вещества без необходимости трудоемкой очистки;
- он позволяет получать соединение формулы (II), более конкретно соединение формулы (III), в промышленном масштабе с превосходными выходами, исходя из простого и недорогого исходного вещества без необходимости трудоемкой очистки;
- позволяет избежать летучих промежуточных соединений, а также использования легковоспламеняющихся и потенциально взрывоопасных реагентов;
- позволяет достичь высокого уровня чистоты с использованием стандартных методов кристаллизации.
Настоящее изобретение также относится к применению соединения формулы (VII) для синтеза соединения формулы (I) или соединения формулы (II). Альтернативно, настоящее изобретение также относится к применению соединения формулы (VII) в которой R1 представляет собой метальную группу и R2 представляет собой атом хлора для синтеза соединения 1 или Соединение 2.
Настоящее изобретение также относится к применению соединения формулы (V) для синтеза соединения формулы (I) или соединение формулы (II). Альтернативно, настоящее изобретение также относится к применению соединения формулы (V) в которой R3 представляет собой метальную группу для синтеза Соединения 1 или Соединения 2.
Соединение формулы (II) или соединение формулы (III), полученное таким образом, впоследствии подвергают ряду обычных химических реакций, таких как описанные в заявке WO 2015/097123, с получением Соединения 1 или Соединения 2, а также их близких по структуре аналогов. Преимущественно, соединение формулы (III), полученное согласно настоящему изобретению, можно использовать в реакции перекрестного сочетания, такой как реакция перекрестного сочетания типа Сузуки, для получения Соединения 1 или Соединения 2.
Преимущественно, Соединение 1 или Соединение 2 получают с использованием 1,4-диазабицикло[2.2.2]октана во время способа получения соединения формулы (II) или соединения формулы (III).
Чтобы правильно подтвердить пути реакции, промежуточные соединения синтеза были систематически выделены и охарактеризованы. Однако можно значительно оптимизировать процедуры, ограничив количество выделяемых промежуточных соединений.
Предпочтительно реагенты перемешивают во время реакции с использованием подходящих механических перемешивателей или мешалок. Реакции можно проводить от примерно 2 до примерно 24 часов или более, в зависимости от температур, объемов разбавления, катализаторов, концентраций и/или природы веществ в реакционных смесях. Используемый в настоящей заявке термин «примерно» означает +/- 5%, в частности +/- 2%, более конкретно +/- 1%.
Структуры описанных соединений подтверждено обычными спектроскопическими методами. Например, данные 1Н ЯМР представлены в виде дельта-значений, выраженных в частях на миллион (част. на млн.), с использованием остаточного пика растворителя (7.26 част. на млн. для CDCl3) в качестве внутреннего стандарта. Паттерны расщепления обозначены как: s (синглет), d (дублет), t (триплет), m (мультиплет), br или brs (широкий синглет).
Приведенные ниже примеры поясняют изобретение, но никоим образом не ограничивают его.
ПРИМЕР 1: Получение 2-хлор-3-метилфенола (реакция гидроксилирования)
Раствор 1-бром-2-хлор-3-метилбензола (5.00 г; 24,33 ммоль) в диоксане (12,5 мл) и раствор гидроксида калия (2,25 г; 40,14 ммоль) в воде (12,5 мл) дегазировали азотом в течение 15 минут. Растворы объединяли. Добавляли t-BuXPhos (827 мг; 1.95 ммоль) и Pd2(dba3) (446 мг; 0.48 ммоль) и реакционную смесь нагревали в запечатанной пробирке при 100°С в течение 35 минут. Реакционную смесь охлаждали до 20°С и промывали с помощью трет-бутил-метилового эфира. Водную фазу снова экстрагировали посредством 1 н. раствора NaOH, подкисляли до рН 4 посредством 3 н. раствора соляной кислоты и экстрагировали дихлорметаном. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (2,8 г, 80% выход).
1Н ЯМР (400 МГц, CDCl3): δ част. на млн. 6.97-7.11 (m, 1Н); 6.73-6.90 (m, 2Н); 5.88 (brs, 1Н); 2.37 (s, 3Н)
ПРИМЕР 2: Получение 4-бром-2-хлор-3-метилфенола (реакция региоселективного монобромирования)
Раствор брома (1089 г; 6.82 моль) в дихлорметане (1,94 л; 2 об.) добавляли при 0°С к раствору 2-хлор-3-метилфенола (972 г; 6,82 моль), который можно получить, как описано в Примере 1 выше, в смеси дихлорметана (5,35 л; 5,5 об.) и уксусной кислоты (2,43 л; 2.5 об.). После перемешивания в течение 15 минут при 0°С, реакционную смесь нагревали при комнатной температуре и промывали водой и посредством 5% раствора KHCO3, затем сушили над сульфатом натрия. После фильтрации продукт получали путем концентрации досуха и переводили как есть в следующую стадию (1.44 кг; 95%).
1Н ЯМР (400 МГц, CDCl3): δ част. на млн. 7.35 (d, J = 8.8 Гц, 1Н); 6.78 (d, J = 8.6 Гц, 1Н); 5.64 (brs, 1Н); 2.49 (s, 3Н)
13С ЯМР (101 МГц, CDCl3): δ част. на млн. 150.7, 135.9, 131.2, 121.1, 115.3, 114.5, 20.8
ЖХ-МС [ЭРИ-] m/z: 219.0, 219.8 [М+Н]+
ПРИМЕР 3: Получение 4-бром-2-хлор-3-метилфенола (реакция региоселективного монобромирования - другие условия)
Раствор бромирующего агента (1 экв.) в растворителе добавляли при 0°С к раствору 2-хлор-3-метилфенола (100 мг) в растворителе. После перемешивания в течение 15 минут при 0°С, реакционную смесь промывали водой и посредством 5% раствора KHCO3, затем сушили над сульфатом натрия. После фильтрации, продукт получали путем концентрации досуха и переводили как есть в следующую стадию. Состав ожидаемого продукта подтвержден с помощью 1Н ЯМР и значения дельты являются такими же, как найденные для Примера 2 выше.
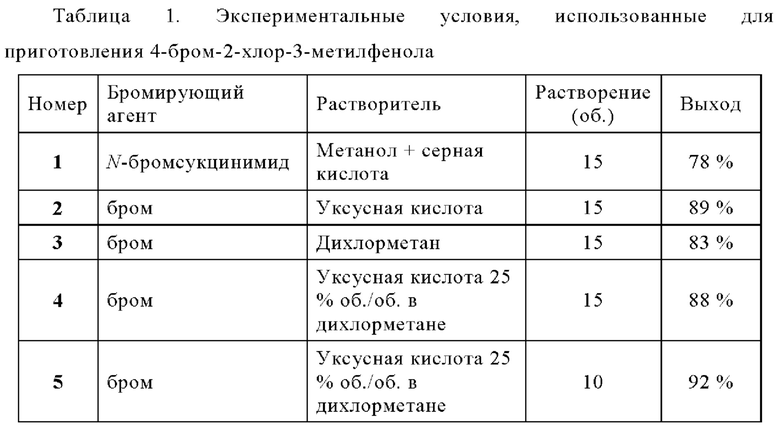
ПРИМЕР 4: Получение 4-бром-2-хлор-3-метилфенола (гидроксилирование в одном реакторе и реакция региоселективного монобромирования)
Раствор 1-бром-2-хлор-3-метилбензола (354 г; 1.72 моль) и гидроксида калия (242 г; 4.30 моль) в 1,4-диоксане (710 мл; 2.0 об.) и воде (2150 мл; 6.0 об.) дегазировали, при перемешивании, с азотом в течение 15 минут. Добавляли t-BuXphos (29.2 г; 0.069 моль) и Pd2(dba)3 (15.8 г; 0,017 моль) и суспензию нагревали кипячения с обратным холодильником (90-95°С) в течение 60 минут. Завершение реакции было подтверждено с помощью ВЭЖХ. Полученную суспензию охлаждали до 20-25°С. Добавляли тирет-бутил-метиловый эфир (800 мл) и двухфазную смесь перемешивали в течение 10-15 минут. Остаток катализатора удаляли фильтрованием через подушку из целита, и осадок ополаскивали с помощью трет-бутил-метилового эфира и 1 н. раствора гидроксида калия. Водную фазу промывали три раза посредством трет-бутил-метилового эфира, затем подкисляли до рН 1-2 с помощью 12 н. раствора соляной кислота. Раствор экстрагировали три раза дихлорметаном, затем объем раствора был скорректирован (1153 мл; 5.0 об. относительно фенола) с дихлорметаном чтобы сжать при соответствующей концентрации на следующей стадии. Концентрацию 2-хлор-3-метилфенола в растворе определяли количественным анализом GC-FID (128.1 мг/мл; 230.6 г; 1.617 моль.).
Раствор 2-хлор-3-метилфенола загружали в 5 л реактор и добавляли уксусную кислоту (584 мл; 2,5 об. относительно фенола). Раствор затем охлаждали до -10°С / -15°С под азотом и добавляли раствор брома (258.5 г; 1.617 моль) в дихлорметане (477 мл; 2.1 об. относительно фенола) в течение 70 минут между -13°С и -7°С. Добавляли еще бром (3,5 г; 0.022 моль) в дихлорметан (20 мл; 0.06 об.). Добавляли воду (1,4 л) в течение 10 минут между -11°С и 2°С. Раствор нагревали до 20-25°С и добавляли бисульфит натрия (50 г; 0,48 моль). Раствор перемешивали в течение 15-20 минут. Фазы разделяли, и затем водную фазу экстрагировали дихлорметаном. Объединенные органические фазы дважды промывали водой, два раза 10% раствором бикарбоната калия и рассолом. Раствор сушили над сульфатом магния. Осадок промывали дихлорметаном. Растворители выпаривали под вакуумом и остаточный 1,4-диоксан был подвергнут азеотропной перегонке с гептанами, чтобы получить продукт, указанный в заголовке в виде бледно-коричневого твердого вещества (353 г, выход сырого продукта: 92,6%).
1Н ЯМР (400 МГц, CDCl3): δ част. на млн. 7.35 (d, J = 8.8 Гц, 1Н); 6.78 (d, J = 8.6 Гц, 1Н); 5.64 (brs, 1Н); 2.49 (s, 3Н)
13С ЯМР (101 МГц, CDCl3): δ част. на млн. 150.7, 135.9, 131.2, 121.1, 115.3, 114.5, 20.8
ЖХ-МС [ЭРИ"] m/z: 219.0, 219.8 [М+Н]+
ПРИМЕР 5: Получение 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина (раскрытие цикла 1-алкил-1-азониабицикло[2.2.2]октана)
Раствор метил-4-метилбензол-1-сульфоната (592 г; 3.18 моль) в анизоле (320 мл) добавляли в течение 15 минут к раствору 1,4-диазабицикло[2.2.2]октана (389 г; 3.47 моль) в анизоле (6,4 л). После перемешивания в течение 1 часа при 70°С, при сильном перемешивании Cs2CO3 (1130 г; 3,466 моль) добавляли порциями в течение 5 минут. Раствор 4-бром-2-хлор-3-метилфенола (640 г; 2,89 моль), полученный как описано в Примерах 2 или 3 выше, в анизоле (0.64 л) добавляли в течение 10 минут. Реакционную смесь перемешивали в течение 6 часов при 140°С. После охлаждения до комнатной температуры добавляли тирет-бутил-метиловый эфир и этилацетат, а смесь промывали водой и рассолом, сушили с сульфатом натрия и полученный раствор продукта сохраняли для следующей стадии.
Смесь из трет-бутил-метилового эфира (1,28 л) и этанола (219 мл; 3,75 моль) добавляли в течение 30 минут к раствору ацетилхлорида (272 г; 3.46 моль) поддерживая температуру смеси ниже 25°С. После перемешивания в течение 30 минут, полученный раствор добавляли к органической фазе, полученной выше в течение 1 часа при комнатной температуре. После перемешивания полученной суспензии в течение 1 часа, продукт собирали фильтрацией и промывали с помощью трет-бутил-метилового эфира. Твердое вещество растворяли в дихлорметане и добавляли 1 н. водный раствор NaOH до щелочной среды. После разделения водный слой промывали дихлорметаном, и объединеннные органические слои сушили с сульфатом натрия и выпаривали. После добавления 2-метилтетрагидрофурана и фильтрации через подушку из целита, осадок промывали 2-метилтетрагидрофураном, и растворитель выпаривали с получением масла янтарного цвета (894 г; 89%).
1Н ЯМР (400 МГц, CDCl3): δ част. на млн. 7.33 (d, J = 8.8 Гц, 1Н); 6.63 (d, J = 8.8 Гц, 1Н); 4.09 (t, J = 5.8 Гц, 2Н); 2.83 (t, J = 5.8 Гц, 3Н); 2.63 (brs, 4Н); 2.47 (s, 4Н); 2.37-2.45 (m, 2Н); 2.25 (s, 3Н)
ПРИМЕР 6: Получение 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина в виде моногидрохлоридной соли (раскрытие цикла 1-алкил-1-азониабицикло[2.2.2]октана)
Раствор метил-4-метилбензол-1-сульфоната (435 г; 2.34 моль) в анизоле (235 мл) добавляли в течение 15 минут к раствору 1,4-диазабицикло[2.2.2]октана (286 г; 2.55 моль) в анизоле (4,7 л). Густую белую суспензию нагревали до 70°С в течение 60 минут. Добавляли карбонат цезия (831 г; 2,55 моль) одной порцией, затем раствор 4-бром-2-хлор-3-метилфенола (470 г; 2.12 моль), полученный, как описано в Примерах 2 или 3 выше, в анизоле (470 мл) добавляли за 12 минут при 70°С. Коричневую суспензию нагревали до 140°С в течение 6 часов и завершение реакции было подтверждено с помощью ВЭЖХ. Добавляли воду, тирет-бутилметиловый эфир и этилацетат, и двухфазную смесь перемешивали в течение 10 минут. Слои разделяли, а затем водную фазу экстрагировали смесью 1:1 трет-бутилметилового эфира и этилацетата. Объединенные органические фазы промывали рассолом, затем сушили над сульфатом натрия в течение примерно 30 минут. Суспензию фильтровали через фильтр Бюхнера, а затем осадок промывали трет-бутилметиловым эфиром. Раствор свободного основания хранили в стороне.
Ацетилхлорид (200 г; 2.55 моль) добавляли к охлажденной (0-5°С) смеси этанола (127 г; 2.76 моль) и трет-бутилметилового эфира (940 мл) за 35 минут между 3°С и 12°С. Раствор перемешивали в течение 30 минут, затем его добавляли к раствору свободного основания в течение 60 минут между 20°С и 25°С. Белую суспензию перемешивали в течение 60 минут при 20-25°С, затем твердое вещество собирали фильтрацией через фильтр Бюхнера и осадок промывали два раза трет-бутилметиловым эфиром. Осадок загружали обратно в колбу и растирали в тирет-бутилметиловом эфире в течение 60 минут. Суспензию фильтровали через фильтр Бюхнера и осадок два раза промывали трет-бутилметиловым эфиром. Твердое вещество сушили в вакууме при 70-75°С до тех пор, пока не наблюдали постоянную массу, с получением указанного в заголовке продукта в виде не совсем белого твердого вещества (761 г, выход: 93,2%) с чистотой 97,1% по данным GC-FID.
1Н ЯМР (400 МГц, ДМСО-d6): δ част. на млн. 11.10 (brs, 1Н); 7.54 (d, J = 8.8 Гц, 1Н); 7.01 (d, J = 9.1 Гц, 1Н); 4.27 (brs, 2Н); 3.39 (brs, 10Н); 2.72 (brs, 3Н); 2.44 (s, 3Н)
ПРИМЕР 7: Получение 1-[2-(4-Бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина в виде дигидрохлоридной соли
В круглодонную колбу объемом 22 л в режиме дистилляции загружали 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазин, соль HCl (1490 г; 3,88 моль), полученную как описано в Примере 6 выше, и воду (14,9 л). Воду частично перегоняли (2,98 л) для удаления остаточного анизола азеотропом при 50-55°С и 40-45 Торр. Раствор охлаждали до 45°С, затем добавляли 12 н. соляную кислоту (646 мл; 7.76 моль) за 5 минут. Раствор давали медленно остыть до 20-25°С в течение выходных. Затем суспензию охлаждали до 0-5°С и фильтровали через фильтр Бюхнера, и колбу ополаскивали холодной (0-5°С) водой (250 мл). Осадок промывали два раза ацетоном. Твердое вещество снова загружали в колбу и растирали в ацетоне в течение 90 минут. Суспензию фильтровали через фильтр Бюхнера, и осадок промывали два раза ацетоном. Твердое вещество сушили в вакууме при 75-80°С в течение 24 часов с получением продукта, указанного в заголовке в виде твердого вещества белого цвета (1471 г, выход: 90,2%) с чистотой 99,9% по данным GC-FID.
1Н ЯМР (400 МГц, ДМСО-d6): δ част. на млн. 10.91-13.60 (m, 2Н); 7.56 (d, J = 8.8 Гц, 1Н); 7.03 (d, J = 9.1 Гц, 1Н); 4.45 (brs, 2Н); 3.58 (brs, 10Н); 2.79 (brs, 3Н); 2.44 (s, 3Н)
13С ЯМР (101 МГц, CD3OD, D2O): δ част. на млн. 153.7, 137.9, 132.0, 124.6, 118.0, 113.7, 65.3, 56.9, 51.2, 50.8, 43.7, 20.8
ПРИМЕР 8: Получение 1-{2-[2-хлор-3-метил-4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил}-4-метилпиперазина (реакция борилирования)
1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазин (800.0 г; 2,30 моль), полученный как описано в Примере 5 (или полученный преобразованием Примеров 6 или 7 в свободное основание), и 2-метилтетрагидрофуран (5,6 л) загружали в трехгорлую круглодонную колбу на 12 л в атмосфере азота. Раствор охлаждали до между -72°С и -76°С, используя баню ацетон-сухой лед. Раствор 2,5 М н-бутиллития в гексане (1196 мл; 2,99 моль) добавляли в течение 1,5 часа, поддерживая температуру между -62°С и -74°С. Полученный желтый раствор перемешивали при температуре между -72°С и -76°С в течение 1 часа. Затем в течение 45 минут добавляли 4,4,5,5-тетраметил-2-(пропан-2-илокси)-1,3,2-диоксаборолан (556 г; 2,99 моль), поддерживая температуру реакционной смеси между -65°С и -76°С. Реакционную смесь перемешивали при температуре от -65°С до -76°С в течение 1 часа. Завершение реакции наблюдали с помощью ВЭЖХ. Реакционную смесь затем нагревали до -25°С. Затем добавляли метанол (200 мл) в течение 15 минут. Раствор выливали в раствор хлорида аммония (369 г; 6.90 моль) в воде (4 л). Фазы разделяли. Органическую фазу промывали водой, а затем упаривали непосредственно досуха, получая бесцветное масло. Добавляли гептан (2,80 л) для разбавления масла при 35-40°С и вскоре произошла кристаллизация. Суспензию перемешивали в течение 1 часа при 35-40°С, затем охлаждали до 5°С в течение 1 часа. Твердые вещества собирали фильтрацией, затем промывали гептанами. Влажный осадок сушили в высоком вакууме при 40-50°С до постоянной массы с получением продукта, указанного в заголовке в виде твердого вещества белого цвета (2.200 кг, выход 85% в целом за 3 партии).
1Н ЯМР (400 МГц, CDC13): δ част. на млн. 7.61 (d, J = 8.3 Гц, 1Н); 6.72 (d, J = 8.3 Гц, 1Н); 4.14 (t, J = 5.9 Гц, 2Н); 2.85 (t, J = 5.9 Гц, 2Н); 2.64 (brs, 3Н); 2,58 (s, 4Н); 2.38-2.50 (m, 4Н); 2.25 (s, 3Н); 1.30 (s, 12Н)
ПРИМЕР 9: Получение 1-{2-[2-хлор-3-метил-4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил}-4-метилпиперазина (реакция борилирования типа Мияуры)
Раствор 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина (20,1 г; 58 ммоль), полученный как описано в Примере 5 (или полученный из трансформации Примеров 6 или 7 в свободное основание), в 1,4-диоксане (200 мл) дегазировали с азотом в течение 20 минут. Добавляли ацетат калия (19,3 г; 197 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолан (17,8 г; 70 ммоль) и суспензию дегазировали снова в течение 20 минут. Добавляли Pd(PPh3)2Cl2 (814 мг; 1.16 ммоль) и суспензию нагревали до 100°С в течение двух часов. Завершение реакции было подтверждено с помощью ВЭЖХ. Суспензию охлаждали до 20-25°С и добавляли толуол (100 мл). Суспензию фильтровали через целит (15 г) и осадок ополаскивали толуолом (40 мл). К раствору добавляли активированный уголь (4.0 г), и перемешивали в течение 1 часа. Суспензию фильтровали через целит (15 г) и силикагель (15 г), затем осадок промывали толуолом (40 мл). Раствор концентрировали досуха, добавляли гептан (100 мл), концентрировали досуха и эту операцию повторяли еще раз. Остаток растворяли в гептане (150 мл) и обрабатывали активированным углем (4.0 г) в течение 60 минут. Суспензию фильтровали через целит (15 г) и осадок промывали два раза гептаном (2×20 мл). Раствор концентрировали досуха, к остатку добавляли гептан (40 мл) и продукт кристаллизовали при 20-25°С течение четырех часов. Суспензию охлаждали до 0-5°С в течение одного часа и продукт собирали фильтрацией. Осадок промывали холодным (0-5°С) гептаном (20 мл) и твердое вещество сушили при 35-40°С до постоянной массы с получением 10,1 г продукта в виде твердого вещества белого цвета. Маточные растворы концентрировали досуха, затем к остатку добавляли гептан (20 мл) и продукт кристаллизовали при 20-25°С в течение четырех часов. Суспензию охлаждали до 0-5°С в течение одного часа и продукт собирали фильтрацией. Осадок промывали холодным (0-5°С) гептаном (10 мл), затем твердое вещество сушили при 35-40°С до постоянной массы с получением 5,6 г продукта в виде твердого вещества белого цвета. Два вещества объединяли, получив в сумме 15,7 г (выход 69%).
1Н ЯМР (400 МГц, CDCl3): δ част. на млн. 7.64 (d, J = 8.3 Гц, 1Н); 6.76 (d, J = 8.3 Гц, 1Н); 4.18 (t, J = 5.8 Гц, 2Н); 2.88 (t, J = 5.9 Гц, 2Н); 2.25-2.83 (m, 14Н); 1.34 (s, 12Н)
ПРИМЕР 10: Получение 1-{2-[2-хлор-3-метил-4-(тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил}-4-метилпиперазина (реакция борилирования типа Мияуры)
В раствор дигидрохлоридной соли 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина (1000 г; 1 экв.; получено как описано в Примере 7) в этилацетате (10 об.), добавляли под азотом 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолан (784 г; 1.3 экв.), ацетат калия (1284 г; 5.5 экв.) и Pd(PPh3)2Cl2 (50 г; 0,03 экв.). При перемешивании, суспензию нагревали кипячения с обратным холодильником в течение 16 часов. После охлаждения до 20°С, реакционную смесь затем фильтровали и осадок промывали этилацетатом (1.5 об). Затем органический слой промывают 5% водным раствором L-ацетилцистеина, забуференным при рН 7 посредством AcOK (10 об.). После разделения слоев органический слой концентрировали до 2 объемов, а затем переходили к замене растворителя на ацетонитрил при 30°С в вакууме. Затем температуру снизили до -10°С, и произошла кристаллизация. После фильтрации, твердое вещество сушили при 40°С с получением указанного в заголовке продукта в виде твердого вещества белого цвета (48% выход).
1Н ЯМР (400 МГц, CDCl3): δ част. на млн. 7.64 (d, J = 8.3 Гц, 1Н); 6.76 (d, J = 8.3 Гц, 1Н); 4.18 (t, J = 5.8 Гц, 2Н); 2.88 (t, J = 5.9 Гц, 2Н); 2.25-2.83 (m, 14Н); 1.34 (s, 12Н)
ПРИМЕР 11: Получение 4-бром-2-хлор-3-метилфенола в больших масштабах (однореакторное гидроксилирование и реакция региоселективного монобромирования)
В реактор добавляли воду (390 л, 6.0 об.) и гидроксид калия (52,2 кг, 790.8 моль) и растворяли. Когда теплота растворения снижалась, затем загружали 1,4-диоксан (130 л, 2 об.) и 3-бром-2-хлортолуол (65 кг, 316,3 моль), раствор дегазировали, при перемешивании, с азотом в течение 30 минут. Добавляли t-BuXphos (5,38 кг, 12.65 моль) и Pd2(dba)3 (2.90 кг, 3.16 моль), и суспензию нагревали с обратным холодильником в течение 90 минут. Завершение реакции подтверждали с помощью ГХ, затем реакционную смесь охлаждали до 20~25°С. Добавляли т-бутилметиловый эфир (146 л) и двухфазную смесь перемешивали в течение 20 минут. Реакционную смесь фильтровали через подушку из целита, осадок на фильтре промывали т-бутилметиловым эфиром (39 л, 0.6 об.) и 1 н. раствором гидроксида калия (78 л, 1.2 об.), затем фазы разделяли. Водную фазу промывали три раза с помощью т-бутилметилового эфира (3×110,5 л, 3×1,7 об.) затем подкисляли до рН 1~2 с помощью 12 н. соляной кислоты при 25~30°С. Раствор экстрагировали три раза дихлорметаном (1×110,5 л, 1.7 об. и 2×42,3 л, 2 х 0.65 об.). Объединенный органический слой переносили в реактор.
Уксусную кислоту (107.3 л, 1.65 об.) добавляли к раствору 2-хлор-3-метилфенола. Раствор охлаждали до -10 ~ -5°С под азотом и раствор брома (51,1 кг, 319,5 моль) в дихлорметане (88 л, 1,35 об.) добавляли в течение 1,5 часов между -10°С и -2°С. Добавляли воду (260 л, 4.0 об.) и смесь нагревали до 20~25°С. Добавляли бисульфит натрия (9,9 кг, 94.9 моль), затем раствор перемешивали в течение 20 минут. Фазу разделяли, затем водную фазу экстрагировали дихлорметаном. Объединенные органические фазы два раза промывали водой, два раза 10% раствором бикарбоната калия и 20% раствором хлорида натрия. Раствор сушили над сульфатом магния, затем фильтровали и осадок промывали дихлорметаном. Растворители удаляли вакуумной перегонкой. Остаточный 1,4-диоксан подвергали азеотропной перегонке с гептаном с получением 70,1 кг указанного в заголовке продукта, (выход сырого продукта: 100,1%).
1H ЯМР (600 МГц, CDCl3): 2.50 (s, 3Н), 5.57 (s, 1Н), 6.78 (d, 1Н), 7.35 (d, 1Н)
ПРИМЕР 12: Получение моногидрохлорида 1-[2-(4-Бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина в больших масштабах (раскрытие цикла 1-алкил-1-азониабицикло [2.2.2] октана)
В реактор загружали анизол (701 л, 10.0 об.) и 1,4-диазабицикло[2.2.2]октан (42,6 кг, 379,6 моль) и перемешивали под азотом. Порциями добавляли метил-n-толуолсульфонат (64,8 кг, 348.0 моль). Реакционную смесь нагревали до 70°С в течение 1 часа. Добавляли карбонат цезия (123.7 кг, 379.6 моль) одной порцией, затем раствор 4-бром-2-хлор-3-метилфенола (70.1 кг, 316.33 моль; полученный как описано в Примере 11) в анизоле (50 кг) добавляли при 70°С. Раствор коричневого цвета нагревали до 140°С в течение 6 часов и завершение реакции подтверждали с помощью ГХ. После охлаждения реакционной смеси до комнатной температуры добавляли воду, т-бутилметиловый эфир и этилацетат, и двухфазный раствор перемешивали в течение 10 минут. Слои разделяли, и органические фазы промывали 20% водным раствором хлорида натрия, затем сушили над сульфатом магния. Суспензию фильтровали через фильтр Бюхнера, и затем осадок промывали т-бутилметиловым эфиром. Раствор свободного основания загружали в реактор и оставляли на потом.
В реактор загружали т-бутилметиловый эфир (140,2 л, 2.0 об.) и этанол (19.0 кг, 412,4 моль) и охлаждали до 0~5°С. Ацетилхлорид (29.8 кг, 379,6 моль) добавляли при температуре 10-15°С. Раствор перемешивали в течение 30 минут затем его добавляли к раствору свободного основания при температуре от 15°С до 25°С. Белую суспензию перемешивали в течение 60 минут при 20-25°С, затем фильтровали через фильтр Бюхнера, и осадок промывали посредством т-бутилметилового эфира. Осадок на фильтре и т-бутилметиловый эфир загружали обратно в реактор и перемешивали в течение 60 минут. Суспензию фильтровали через фильтр Бюхнера и осадок промывали т-бутилметиловым эфиром. Твердое вещество сушили в вакууме при 70-75°С в течение 16 часов с получением продукта, указанного в заголовке в виде твердого вещества белого цвета (101 кг, выход: 83,1%).
1Н ЯМР (600 МГц, ДМСО-d6): 2.42 (s, 3Н), 2.70 (s, 3Н), 2.8-3.8 (br, 10Н), 4.25 (br, 2Н), 6.95 (d, 1Н), 7.52 (d, 1Н)
ПРИМЕР 13: Получение дигидрохлорида 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина в больших масштабах
В реактор загружали воду (1010 л, 10 об.) и моногидрохлорид 1-[2-(4-бром-2-хлор-3-метилфенокси)этил]-4-метилпиперазина (101 кг, 262,9 моль; полученный как описано в Примере 12). Воду частично перегоняли для удаления остаточного анизола азеотропом при 45~50°С и 55~60 Торр. К водному раствору добавляли 12 н. соляную кислоту (43.8 л, 525.8 моль) при 45°С. Раствор медленно охлаждали до 15~20°С в течение 3 часов и дополнительно перемешивали в течение 12 часов. Суспензию фильтровали через фильтр Бюхнера, и осадок промывали холодной водой (17 л, 0.17 об.) и ацетоном (200 л, 2 об.). Твердое вещество снова загружали в реактор, затем добавляли ацетон и суспензию перемешивали в течение 60 минут и фильтровали через фильтр Бюхнера. Осадок на фильтре промывали ацетоном и сушили в вакууме при 75~80°С в течение 24 часов с получением продукта, указанного в заголовке (99,5 кг, 90.0%) в виде твердого вещества белого цвета с чистотой 99,4% по данным GC-FID.
1Н ЯМР (600 МГц, ДМСО-d6): 2.43 (s, 3Н), 2.78 (s, 3Н), 3.2-3.9 (br, 10Н), 4.47 (br, 2Н), 7.02 (d, 1Н), 7.56 (d, 1Н)
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| НОВЫЕ ГИДРОКСИКИСЛОТНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2745430C1 |
| ПРОИЗВОДНЫЕ СУЛЬФОНАМИДА И СПОСОБ БОРЬБЫ С НЕЖЕЛАТЕЛЬНОЙ РАСТИТЕЛЬНОСТЬЮ | 1990 |
|
RU2047295C1 |
| НОВЫЕ АМИНОКИСЛОТНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2747673C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| СПОСОБ И СЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ПОЛУЧЕНИЯ ЦЕФАЛОСПОРИНОВ | 2001 |
|
RU2237671C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ГИДРОКСИ-4-ТИОМЕТИЛПИРАЗОЛЬНОГО СОЕДИНЕНИЯ | 2005 |
|
RU2386619C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ИЛ-МЕТОКСИБЕНЗИЛГИДРОКСИМОЧЕВИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И 4-(ХИНОЛИН-2-ИЛ-МЕТОКСИ)ФЕНИЛ-ЦИКЛОАЛКИЛКЕТОН В КАЧЕСТВЕ ИСХОДНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИН-2-ИЛ-МЕТОКСИБЕНЗИЛГИДРОКСИМОЧЕВИНЫ | 1992 |
|
RU2048466C1 |
| АМИНОБЕНЗОФЕНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНТЕРЛЕЙКИНА IL-1БЕТА И ФАКТОРА НЕКРОЗА ОПУХОЛЕЙ TNF-АЛЬФА | 2000 |
|
RU2240995C2 |
Изобретение относится к способу получения соединения формулы (I), в которой R1 представляет собой метальную группу и R2 представляет собой атом хлора, R3 представляет собой метальную группу, включающему в себя стадию реакции соединения формулы (IV) с соединением формулы (V), в которой R3 имеет приведенное выше определение и X- представляет собой тозилатный противоион, в анизоле при температуре, превышающей 135°С, в присутствии карбонатной соли. Изобретение также относится к способам получения соединения формулы (II), в которой R1 представляет собой метальную группу и R2 представляет собой атом хлора, R3 представляет собой метальную группу, R4 и R5 образуют с несущими их атомами кислорода и атомом бора 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо, путем реакции соединения формулы (I) с эфиром бороновой кислоты формулы (VIII), в которой R4 и R5 имеют приведенные выше определения и R представляет собой атом водорода, гидрокси-группу, линейную или разветвленную (C1-С6)алкокси-группу или группу (С0-С6)алкил-В(OR4)(OR5). Также изобретение относится к применению соединений формул (VII) и (V) в синтезе соединений формулы (I) или (II). Технический результат – разработан новый способ получения соединений формул (I) и (II) с высокими выходами и чистотой, которые могут найти применение в качестве промежуточных продуктов для синтеза 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(5-фторфуран-2-ил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[1-(2,2,2-трифторэтил)-1H-пиразол-5-ил]метокси}фенил)пропановой кислоты и 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановой кислоты. 5 н. и 17 з.п. ф-лы, 1 табл., 13 пр.
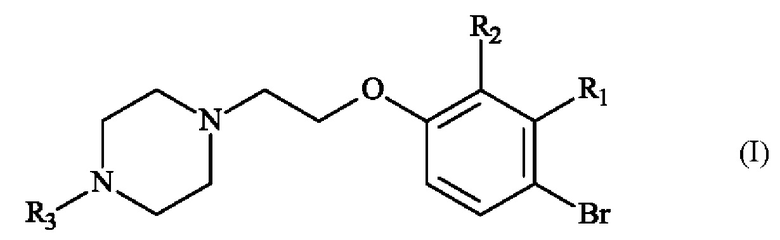
1. Способ получения соединения формулы (I)
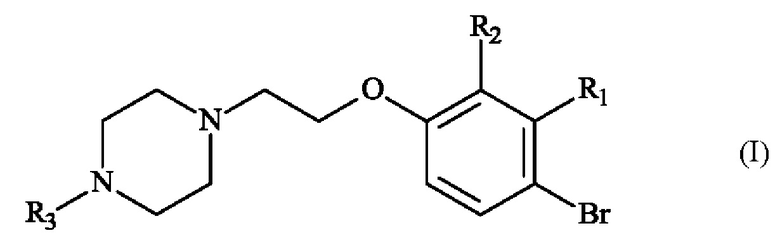 ,
,
в которой:
- R1 представляет собой метальную группу и R2 представляет собой атом хлора,
- R3 представляет собой метальную группу,
включающий в себя стадию реакции соединения формулы (IV):
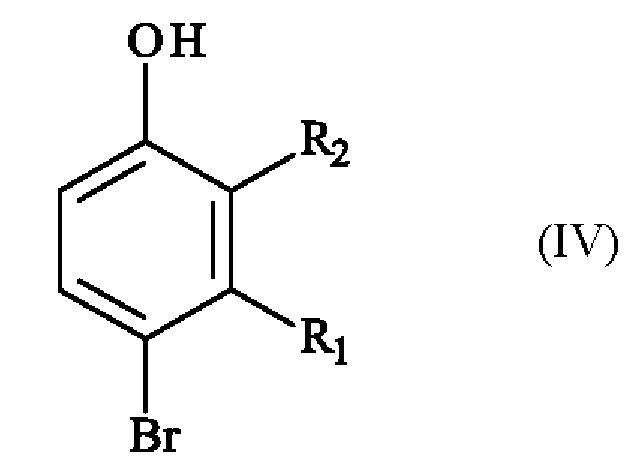 ,
,
в которой R1 и R2 имеют приведенные выше определения,
с соединением формулы (V)
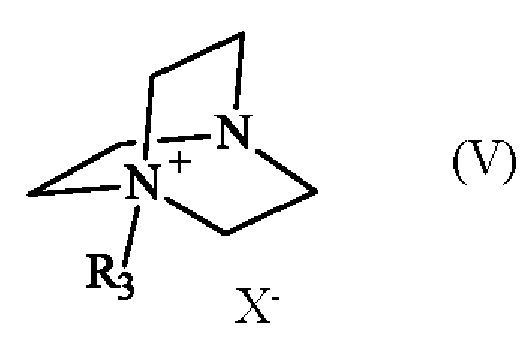 ,
,
в которой R3 имеет приведенное выше определение и X- представляет собой тозилатный противоион,
в анизоле при температуре, превышающей 135°С, в присутствии карбонатной соли.
2. Способ по п. 1, в котором соединение формулы (I) выделяют в виде моногидрогалогенидной соли или дигидрогалогенидной соли.
3. Способ по п. 1, в котором соединение формулы (V) получают из 1,4-диазабицикло[2.2.2]октана.
4. Способ по п. 1, в котором соединение формулы (I) получают с использованием 1,4-диазабицикло[2.2.2]октана.
5. Способ по любому из пп. 1-4, в котором соединение формулы (IV)
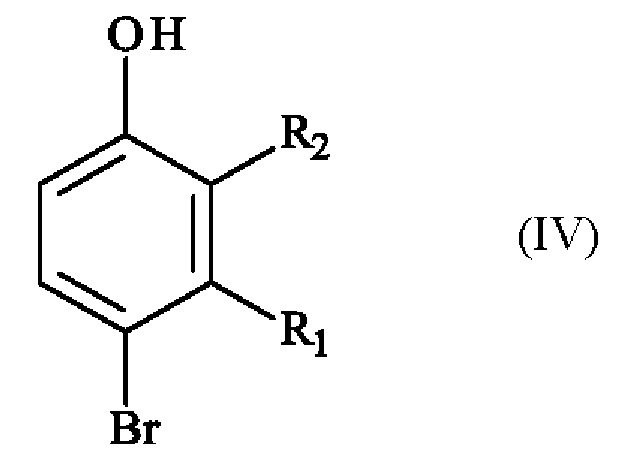 ,
,
в которой R1 представляет собой метальную группу и R2 представляет собой атом хлора,
получают реакцией региоселективного монобромирования соединения формулы (VI)
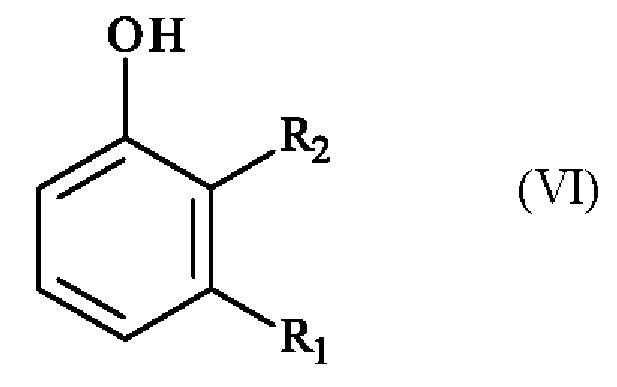 ,
,
в которой R1 и R2 имеют приведенные выше определения,
в растворителе в присутствии бромирующего агента, выбранного из брома, бромида натрия/трихлоризоциануровой кислоты или брома/ацетата натрия.
6. Способ по п. 5, в котором реакцию проводят в присутствии 1 эквивалента бромирующего агента.
7. Способ по п. 5, в котором бромирующий агент представляет собой бром.
8. Способ по п. 5, в котором растворитель выбирают из уксусной кислоты, дихлорметана, смеси метанола и серной кислоты, смеси уксусной кислоты и дихлорметана.
9. Способ по п. 5, в котором реакцию проводят путем разбавления соединения формулы (VI) с примерно от 10 до примерно 20 объемами органических растворителей или смесью органических растворителей.
10. Способ по любому из пп. 5-9, в котором соединение формулы (VI)
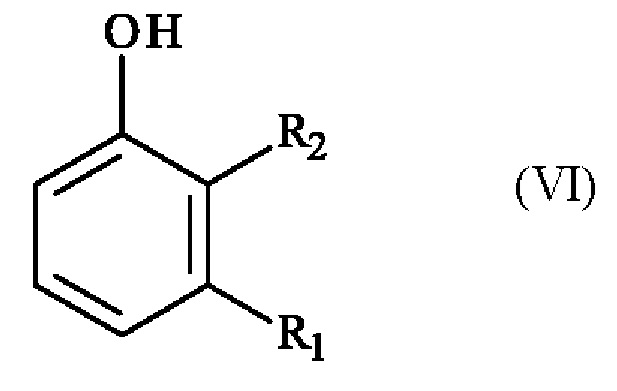 ,
,
в которой R1 представляет собой метальную группу и R2 представляет собой атом хлора,
получают реакцией гидроксилирования соединения формулы (VII)
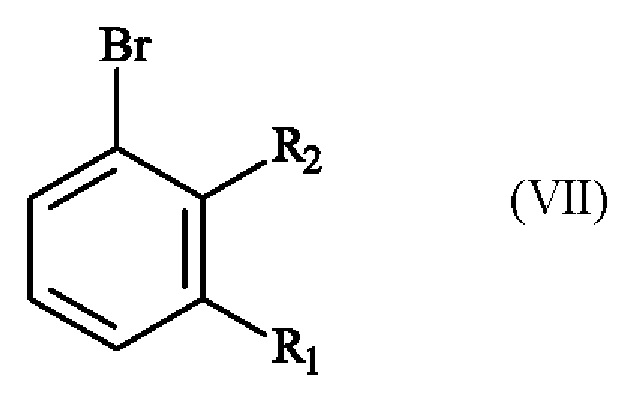 ,
,
в которой R1 и R2 имеют приведенные выше определения,
в растворителе в присутствии комплекса переходного металла и основания.
11. Способ по п. 10, в котором комплекс переходных металлов представляет собой комплекс палладия, содержащий палладиевый катализатор и лиганд.
12. Способ по п. 11, в котором реакцию проводят в присутствии по меньшей мере 0,01 эквивалента палладиевого катализатора.
13. Способ по п. 11, в котором реакцию проводят в присутствии по меньшей мере 0,03 эквивалента лиганда.
14. Способ по п. 10, в котором основание представляет собой гидроксидную соль.
15. Способ по п. 10, в котором растворитель представляет собой 1,4-диоксан или смесь воды и 1,4-диоксана.
16. Способ по любому из пп. 5-15, в котором превращение соединения формулы (VII) в соединение формулы (IV) осуществляют непосредственно без выделения соединения формулы (VI).
17. Способ получения соединения формулы (II)
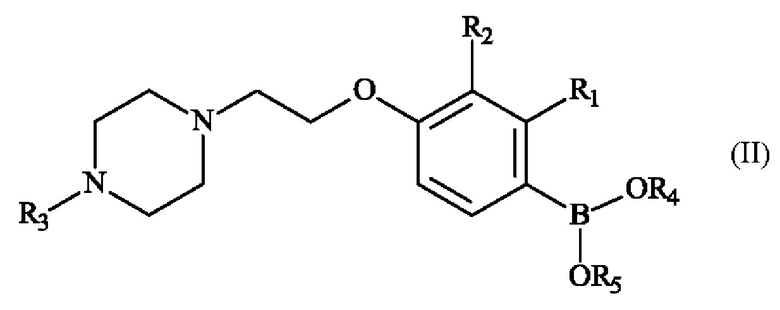
в которой:
- R1 представляет собой метальную группу и R2 представляет собой атом хлора,
- R3 представляет собой метальную группу,
- R4 и R5 образуют с несущими их атомами кислорода и атомом бора 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо,
включающий в себя стадию реакции соединения формулы (I), полученного способом по п. 1,
 ,
,
в которой R1, R2 и R3 имеют приведенные выше определения,
с эфиром бороновой кислоты формулы (VIII)
 ,
,
в которой R4 и R5 имеют приведенные выше определения и R представляет собой атом водорода, гидрокси-группу, линейную или разветвленную (C1-С6)алкокси-группу или группу (С0-С6)алкил-В(OR4)(OR5).
18. Способ по п. 17, в котором реакция состоит из действия соединения формулы (VIII), в которой R представляет собой атом водорода, гидрокси-группу или линейную или разветвленную (С1-С6)алкокси-группу, в органическом растворителе или смеси органических растворителей в присутствии основания.
19. Способ по п. 17, в котором реакция состоит из действия соединения формулы (VIII), в которой R представляет собой группу (С0-С6)алкил-B(OR4)(OR5), в органическом растворителе или смеси органических растворителей в присутствии основания и комплекса палладия.
20. Способ получения соединения формулы (II)
 ,
,
в которой:
- R1 представляет собой метальную группу и R2 представляет собой атом хлора,
- R3 представляет собой метальную группу,
- R4 и R5 образуют с несущими их атомами кислорода и атомом бора 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо,
отличающийся тем, что соединение формулы (VII)
 ,
,
в которой R1 и R2 имеют приведенные выше определения, подвергают реакции гидроксилирования в присутствии комплекса переходного металла и основания в растворителе с получением соединения формулы (VI)
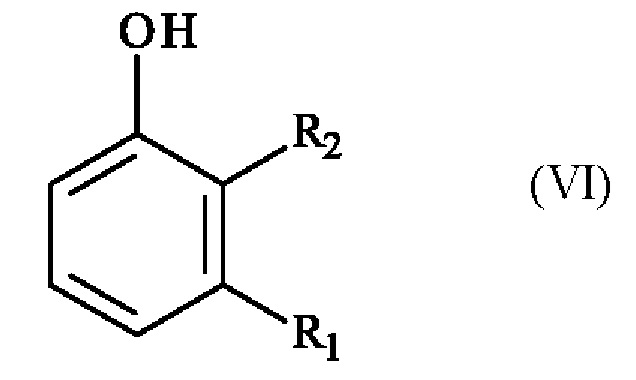 ,
,
в которой R1 и R2 имеют приведенные выше определения, при этом соединение формулы (VI) подвергают реакции региоселективного монобромирования в присутствии бромирующего агента в растворителе с получением соединения формулы (IV)
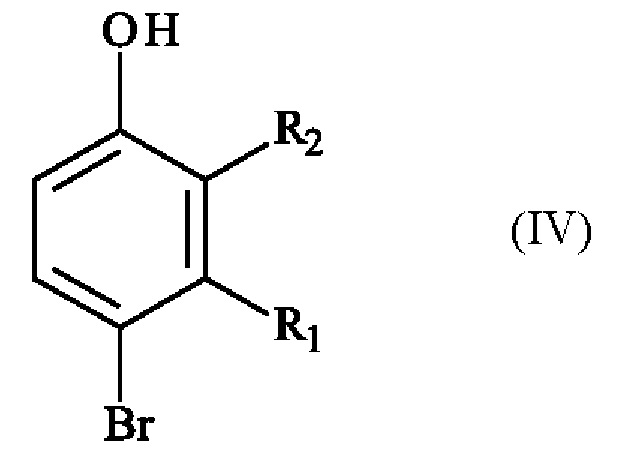 ,
,
в которой R1 и R2 имеют приведенные выше определения,
при этом соединение формулы (IV) реагирует в анизоле при температуре, превышающей 135°С, в присутствии карбонатной соли, и соединения формулы (v)
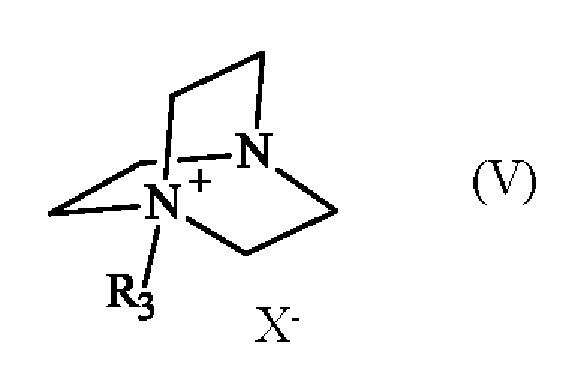 ,
,
в которой R3 имеет приведенное выше определение и X- представляет собой тозилатный противоион,
с получением соединения формулы (I)
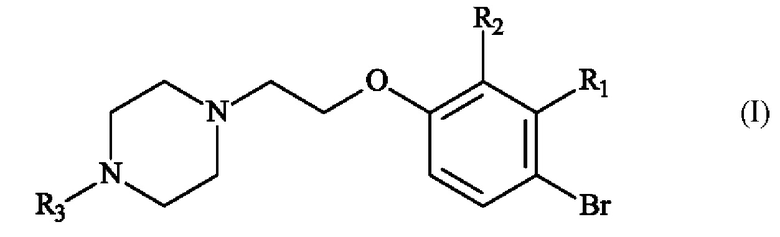 ,
,
в которой R1, R2 и R3 имеют приведенные выше определения, при этом соединение формулы (I) подвергают реакции борилирования в присутствии эфира бороновой кислоты формулы (VIII)
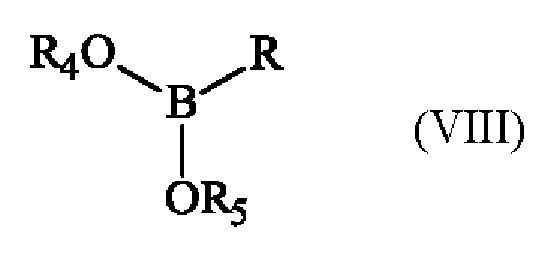 ,
,
в которой R4 и R5 имеют определения, приведенные для формулы (II), и R представляет собой атом водорода, гидрокси-группу, линейную или разветвленную (С1-С6)алкокси-группу или группу (С0-С6)алкил-В(OR4)(OR5), с получением соединения формулы (II).
21. Применение соединения формулы (VII)
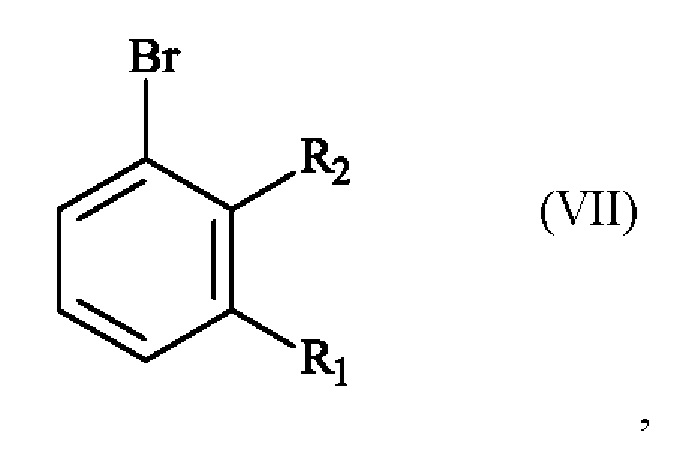
где R1 представляет собой метальную группу и R2 представляет собой атом хлор,
в синтезе соединения формулы (I)
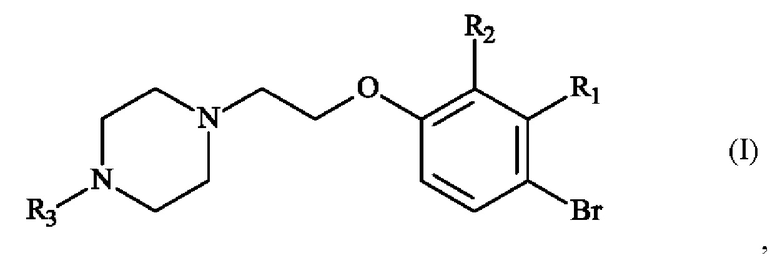
где R3 представляет собой метальную группу, или соединения формулы (II)
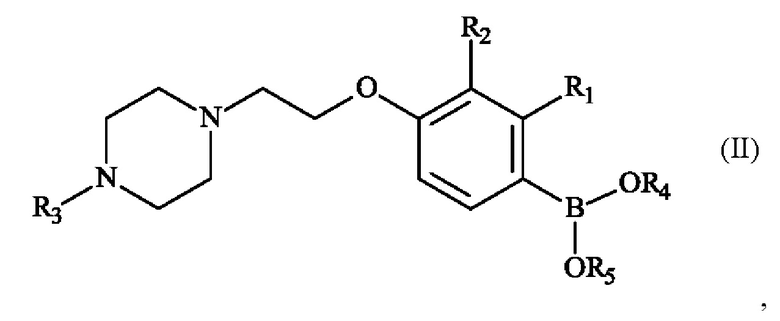
где R4 и R5 образуют с несущими их атомами кислорода и атомом бора 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо.
22. Применение соединения формулы (V)
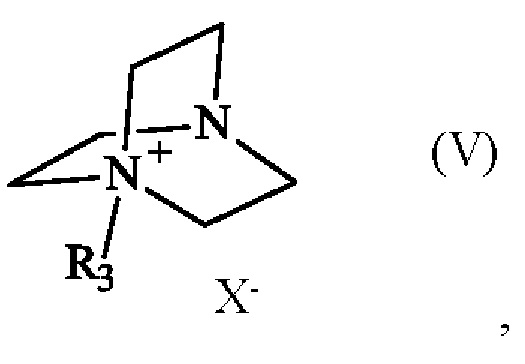
где R3 представляет собой метальную группу и X- представляет собой тозилатный противоион,
в синтезе соединения формулы (I)
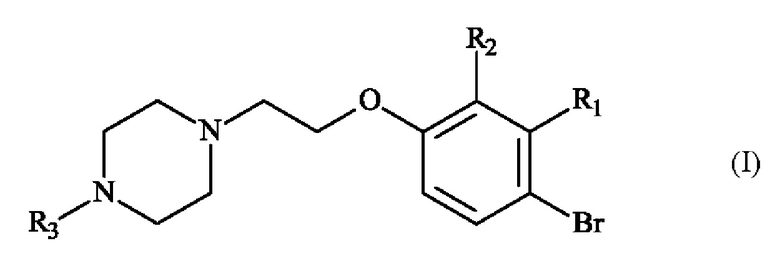
или соединения формулы (II)
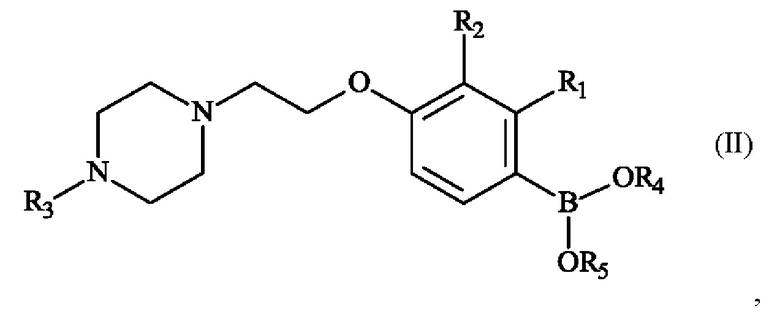
где R4 и R5 образуют с несущими их атомами кислорода и атомом бора 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ильное кольцо.
| Станок для распрямления и насадка на колодку валеного сапога | 1932 |
|
SU29601A1 |
| NENAD MARAS ET AL, Organic & Biomolecular Chemistry, vol | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| WO 2015162515 A1, 29.10.2015 | |||
| WO 2016207226 A1, 29.12.2016 | |||
| WO 2016207217 A1, 29.12.2016 | |||
| WO 2016207216 A1, 29.12.2016 | |||
| WO 2017125224 A1, 27.07.2017. | |||
