Настоящее изобретение относится к новым фосфатным соединениям, к способу их получения и к фармацевтическим композициям, содержащим их.
Соединения настоящего изобретения являются новыми и обладают чрезвычайно ценными фармакологическими и фармакокинетическими характеристиками для применения в области апоптоза и онкологии.
Апоптоз, или запрограммированная гибель клеток, является ключевым физиологическим процессом для эмбрионального развития и поддержания тканевого гомеостаза.
Гибель клеток по типу апоптоза вызывает морфологические изменения, такие как конденсация ядра, фрагментация ДНК, а также биохимический феномен, такой как активация каспаз, которые вызывают повреждение ключевых структурных компонентов клетки, вызывая, таким образом, ее разборку и смерть. Регуляция процесса апотоза является комплексной и задействует активацию или репрессию нескольких внутриклеточных путей передачи сигналов (Cory S. и др., Nature Review Cancer, 2002, 2, 647-656).
Дерегулирование апоптоза вовлечено в определенные патологии. Повышенный апоптоз связан с нейродегенеративными заболеваниями, такими как болезнь Паркинсона, болезнь Альцгеймера и ишемия. Наоборот, недостаточности осуществления апоптоза играют важную роль в развитии злокачественных новообразований и их резистентности к химиотерапии, при аутоиммунных заболеваниях, воспалительных заболеваниях и вирусных инфекциях. Следовательно, отсутствие апоптоза является одним из характерных фенотипических признаков злокачественного новообразования (Hanahan D. и др., Cell 2000, 100, 57-70).
Антиапоптозные белки семейства Bcl-2 связаны с многочисленными патологиями. Задействование белков семейства Bcl-2 описано для многочисленных видов злокачественных новообразований, таких как колоректальный рак, рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак мочевого пузыря, рак яичников, рак предстательной железы, хронический лимфоидный лейкоз, фолликулярная лимфома, миелома, и т.д. Сверхэкспрессия антиапоптозных белков семейства Bcl-2 связана с онкогенезом, с устойчивостью к химиотерапии и с клиническим прогнозом пациентов, страдающих злокачественным новообразованием. Таким образом, существует терапевтическая потребность в соединениях, которые ингибируют антиапоптотическую активность белков семейства Bcl-2.
Помимо того, что соединения настоящего изобретения являются новыми, они обладают фармакологическими и фармакокинетическими свойствами, которые позволяют их применение при патологиях, в которые вовлечен дефект апоптоза, как, например, для лечения злокачественного новообразования, аутоиммунных заболеваний и заболеваний иммунной системы.
Более конкретно, настоящее изобретение относится к фосфатному соединению формулы (I):
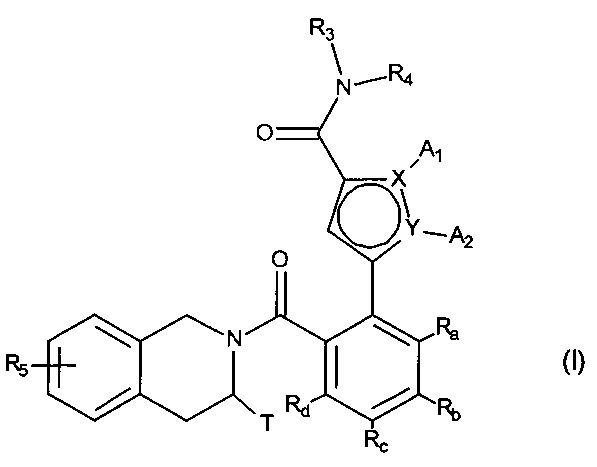
в которой:
- X и Y представляют собой атом углерода или атом азота, при этом предполагается, что они не могут одновременно представлять собой два атома углерода или два атома азота,
- A1 и A2, вместе с атомами, несущими их, образуют необязательно замещенный, ароматический или неароматический гетероцикл Het, состоящий из 5, 6 или 7 кольцевых членов, который может содержать, в дополнение к азоту, представленному посредством X или Y, от 1 до 3 гетероатомов, независимо выбранных из кислорода, серы и азота, при этом предполагается, что рассматриваемый азот может быть замещен группой, представляющей собой атом водорода, линейную или разветвленную (C1-C6) алкильную группу или группу -C(O)-O-Alk, в которой Alk означает линейную или разветвленную (C1-C6) алкильную группу,
или A1 и A2 независимо друг от друга представляют собой атом водорода, линейную или разветвленную (C1-C6)полигалогеналкильную, линейную или разветвленную (C1-C6) алкильную группу или циклоалкил,
- T представляет собой атом водорода, линейную или разветвленную (C1-C6) алкильную группу, необязательно замещенную одним-тремя атомами галогена, группу (C1-C4)алкил-NR1R2, или группу (C1-C4)алкил-OR6,
- R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (C1-C6) алкильную группу,
или R1 и R2 с атомом азота, несущим их, образуют гетероциклоалкил,
- R3 представляет собой линейную или разветвленную (C1-C6) алкильную группу, линейную или разветвленную (C2-C6) алкенильную группу, линейную или разветвленную (C2-C6) алкинильную группу, циклоалкильную группу, (C3-C10)циклоалкил-(C1-C6) алкильную группу, где алкильный фрагмент является линейным или разветвленным, гетероциклоалкильную группу, арильную группу или гетероарильную группу, при этом предполагается, что один или несколько атомов углерода предыдущих групп, или их возможных заместителей, могут быть дейтерированными,
- R4 представляет собой арильную группу, гетероарильную группу, циклоалкильную группу или линейную или разветвленную (C1-C6) алкильную группу, при этом предполагается, что один или несколько атомов углерода предыдущих групп, или их возможных заместителей, могут быть дейтерированными,
- R5 представляет собой водород или атом галогена, линейную или разветвленную (C1-C6) алкильную группу, или линейную или разветвленную (C1-C6)алкокси группу,
- R6 представляет собой атом водорода или линейную или разветвленную (C1-C6) алкильную группу,
- Ra, Rb, Rc и Rd каждый независимо от других представляет собой R7, атом галогена, линейную или разветвленную (C1-C6) алкокси группу, гидрокси группу, линейную или разветвленную (C1-C6) полигалогеналкильную группу, трифторметокси группу, группу 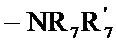
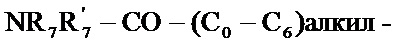
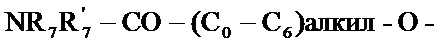
- R7 и R7' независимо друг от друга представляют собой водород, линейный или разветвленный (C1-C6)алкил, линейный или разветвленный (С2-C6)алкенил, линейный или разветвленный (С2-C6)алкинил, арил или гетероарил, или R7 и R7' вместе с атомом азота, несущим их, образуют гетероцикл, состоящий из 5-7 кольцевых членов,
соединение формулы (I) является таким, что по меньшей мере один из атомов углерода, содержащихся в нем, замещен одной из следующих фосфатных групп: -ОРО(ОМ)(ОМ'), 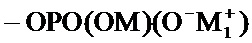
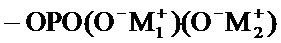
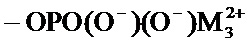
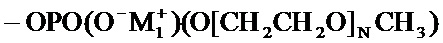
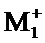
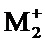
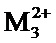
при этом предполагается, что:
- "арил" означает фенильную, нафтильную, бифенильную или инденильную группу,
- "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, имеющую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из кислорода, серы и азота (включая четвертичные атомы азота),
- "циклоалкил" означает любую моно- или бициклическую, неароматическую, карбоциклическую группу, содержащую от 3 до 10 кольцевых членов,
- "гетероциклоалкил" означает любую моно- или бициклическую, неароматическую, конденсированную или спиро группу, состоящую из 3-10 кольцевых членов и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы, SO, SO2 и азота,
причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, таким образом определенные, и группы алкил, алкенил, алкинил и алкокси могут быть замещены посредством 1-3 групп, выбранных из необязательно замещенного, линейного или разветвленного (C1-C6)алкила, (C3-C6)спиро, линейного или разветвленного, необязательно замещенного (C1-C6)алкокси, (C1-C6)алкил-S-, гидрокси, оксо (или N-оксида, в соответствующих случаях), нитро, циано, -COOR', -OCOR', NR'R", линейного или разветвленного (С1-C6)полигалогеналкила, трифторметокси, (C1-C6)алкилсульфонила, галогена, необязательно замещенного арила, гетероарила, арилокси, арилтио, циклоалкила, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена или алкильными группами, при этом предполагается, что R' и R" независимо друг от друга представляют собой атом водорода или необязательно замещенную, линейную или разветвленную (С1-C6) алкильную группу,
причем Het группа, определенная в формуле (I), может быть замещена одной-тремя группами, выбранными из линейного или разветвленного (C1-C6)алкила, гидрокси, линейного или разветвленного (С1-C6)алкокси, 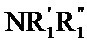
к его энантиомерам и диастереоизомерам и к его солям присоединения с фармацевтически приемлемой кислотой или основанием.
Среди фармацевтически приемлемых кислот могут быть упомянуты, без какого-либо ограничения, соляная кислота, бромистоводородная кислота, серная кислота, фосфоновая кислота, уксусная кислота, трифторуксусная кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, глутаровая кислота, фумаровая кислота, винная кислота, малеиновая кислота, лимонная кислота, аскорбиновая кислота, щавелевая кислота, метансульфоновая кислота, камфорная кислота и т.д.
Среди фармацевтически приемлемых оснований могут быть упомянуты, без какого-либо ограничения, гидроксид натрия, гидроксид калия, триэтиламин, трет-бутиламин и т.д.
Предпочтительные соединения изобретения включают соединения формулы (I), в которой R4 представляет собой фенил, замещенный в пара-положении группой формулы -ОРО(ОМ)(ОМ'),
Предпочтение отдают соединениям формулы (I), где R4 представляет собой фенильную или пиримидин-5-ильную группу, обе замещенные в пара-положении группой формулы
Предпочтительно, X представляет собой атом углерода и Y представляет собой атом азота. Еще более предпочтительно, группа:
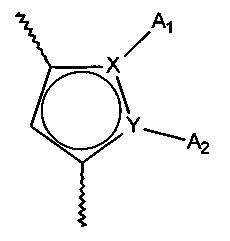
представляет собой 5,6,7,8-тетрагидроиндолизин, индолизин или диметилированный пиррол.
Т предпочтительно представляет собой метальную, (морфолин-4-ил)метильную или 3-(морфолин-4-ил)пропильную группу.
В предпочтительных соединениях изобретения, Ra и Rd каждый представляет собой атом водорода и (Rb,Rc), вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу или 1,4-диоксановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой водород или галоген.
В другом варианте осуществления изобретения, Ra и Rd каждый представляет собой атом водорода, Rb представляет собой атом галогена и Rc представляет собой метокси группу.
Альтернативно, Ra, Rb и Rd каждый предпочтительно представляет собой атом водорода и Rc представляет собой группу
Кроме того, R3 предпочтительно представляет собой группу, выбранную из фенила, 1H-индола, 1H-пирроло[2,3-6]пиридина, пиридина, 1H-пиразола, 1H-пиррола и 2,3-дигидро-1H-пирроло[2,3-b]пиридина, причем эти группы необязательно имеют один или несколько заместителей, выбранных из линейного или разветвленного (С1-C6)алкила (более предпочтительно метила), циано и тридейтерометила.
Среди предпочтительных соединений изобретения могут быть упомянуты:
-4-[{[3-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат,
-4-({[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}[1-(тридейтерометил)-1H-пиразол-4-ил]амино)фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1,2-диметил-1H-пиррол-3-ил)амино]фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1-метил-1H-пиррол-3-ил)амино]фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат,
-4-[(5-циано-1,2-диметил-1H-пиррол-3-ил){[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}амино]фенил динатрийфосфат,
-4-[{[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат,
их энантиомеры и диастереоизомеры и их соли присоединения с фармацевтически приемлемой кислотой или основанием.
Фармакокинетическое исследование фосфатных соединений формулы (I) показало, что они in vivo были превращены в соединения формулы (I'), отличающиеся тем, что фосфатная функция была метаболизирована в гидрокси функцию. Соответственно, соединения формулы (I) ведут себя в качестве пролекарств соединений формулы (I'), имеющих следующую формулу:
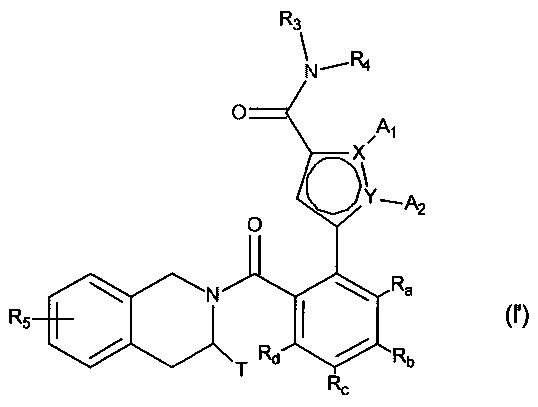
в которой:
- X и Y представляют собой атом углерода или атом азота, при этом предполагается, что они не могут одновременно представлять собой два атома углерода или два атома азота,
- A1 и A2, вместе с атомами, несущими их, образуют необязательно замещенный, ароматический или неароматический гетероцикл Het, состоящий из 5, 6 или 7 кольцевых членов, который может содержать, в дополнение к азоту, представленному посредством X или Y, от 1 до 3 гетероатомов, независимо выбранных из кислорода, серы и азота, при этом предполагается, что рассматриваемый азот может быть замещен группой, представляющей собой атом водорода, линейную или разветвленную (C1-C6) алкильную группу или группу -C(O)-O-Alk, в которой Alk означает линейную или разветвленную (C1-C6) алкильную группу,
или A1 и A2 независимо друг от друга представляют собой атом водорода, линейную или разветвленную (C1-C6)полигалогеналкильную, линейную или разветвленную (C1-C6) алкильную группу или циклоалкил,
- T представляет собой атом водорода, линейную или разветвленную (C1-C6) алкильную группу, необязательно замещенную одним-тремя атомами галогена, группу (C1-C4)алкил-NR1R2, или группу (C1-C4)алкил-OR6,
- R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (C1-C6) алкильную группу,
или R1 и R2 с атомом азота, несущим их, образуют гетероциклоалкил,
- R3 представляет собой линейную или разветвленную (C1-C6) алкильную группу, линейную или разветвленную (C2-C6) алкенильную группу, линейную или разветвленную (С2-C6) алкинильную группу, циклоалкильную группу, (C3-C10)циклоалкил-(C1-C6) алкильную группу, где алкильный фрагмент является линейным или разветвленным, гетероциклоалкильную группу, арильную группу или гетероарильную группу, при этом предполагается, что один или несколько атомов углерода предыдущих групп, или их возможных заместителей, могут быть дейтерированными,
- R4 представляет собой арильную группу, гетероарильную группу, циклоалкильную группу или линейную или разветвленную (C1-C6) алкильную группу, при этом предполагается, что один или несколько атомов углерода предыдущих групп, или их возможных заместителей, могут быть дейтерированными,
- R5 представляет собой водород или атом галогена, линейную или разветвленную (C1-C6) алкильную группу, или линейную или разветвленную (C1-C6) алкокси группу,
- R6 представляет собой атом водорода или линейную или разветвленную (C1-C6) алкильную группу,
- Ra, Rb, Rc и Rd каждый независимо от других представляет собой R7, атом галогена, линейную или разветвленную (C1-C6) алкокси группу, гидрокси группу, линейную или разветвленную (C1-C6) полигалогеналкильную группу, трифторметокси группу, группу
- R7 и 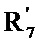
при этом предполагается, что:
- "арил" означает фенильную, нафтильную, бифенильную или инденильную группу,
- "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, имеющую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из кислорода, серы и азота (включая четвертичные атомы азота),
- "циклоалкил" означает любую моно- или бициклическую, неароматическую, карбоциклическую группу, содержащую от 3 до 10 кольцевых членов,
- "гетероциклоалкил" означает любую моно- или бициклическую, неароматическую, конденсированную или спиро группу, состоящую из 3-10 кольцевых членов и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы, SO, SO2 и азота,
причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, таким образом определенные, и группы алкил, алкенил, алкинил и алкокси могут быть замещены посредством 1-3 групп, выбранных из необязательно замещенного, линейного или разветвленного (C1-C6)алкила, (C3-C6)спиро, линейного или разветвленного, необязательно замещенного (C1-C6)алкокси, (C1-C6)алкил-S-, гидрокси, оксо (или N-оксида, в соответствующих случаях), нитро, циано, -COOR', -OCOR', NR'R", линейного или разветвленного (C1-C6)полигалогеналкила, трифторметокси, (C1-C6)алкилсульфонила, галогена, необязательно замещенного арила, гетероарила, арилокси, арилтио, циклоалкила, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена или алкильными группами, при этом предполагается, что R' и R" независимо друг от друга представляют собой атом водорода или необязательно замещенную, линейную или разветвленную (C1-C6) алкильную группу,
причем Het группа, определенная в формуле (I'), может быть замещена одной - тремя группами, выбранными из линейного или разветвленного (C1-C6)алкила, гидрокси, линейного или разветвленного (C1-C6)алкокси, 
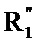
их энантиомеров и диастереоизомеров и их солей присоединения с фармацевтически приемлемой кислотой или основанием.
Соединения формулы (I') обладают проапоптотическими свойствами и в результате этого имеют важное терапевтическое значение для лечения злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы. В настоящем изобретении показано, что, путем введения фосфатных соединений формулы (I), in vivo воздействие соединений формулы (I') оптимизировалось. Фактически растворимость соединений формулы (I) является значительно большей, чем растворимость соединений формулы (I'). Следовательно, применение соединений формулы (I) для изготовления фармацевтических композиций с точки зрения галеновых препаратов особенно предпочтительно.
Изобретение относится также к способу получения соединений формулы (I), который отличается тем, что в качестве исходного вещества используют соединение формулы (II):
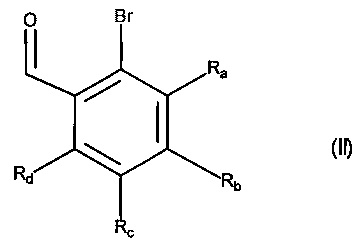
в которой Ra, Rb, Rc и Rd принимают значения, определенные для формулы (I'),
причем соединение формулы (II) подвергают реакции Хека, в водной или органической среде, в присутствии палладиевого катализатора, основания, фосфина и соединения формулы (III):
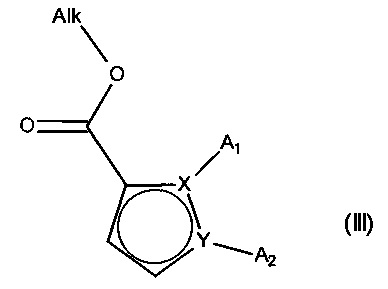
в которой группы A1, A2, X и Y принимают значения, определенные для формулы (I'), и Alk представляет собой линейный или разветвленный (C1-C6)алкил,
с получением соединения формулы (IV):

в которой A1, A2, X, Y, Ra, Rb, Rc и Rd принимают значения, определенные для формулы (I'), и Alk принимает значения, определенные выше,
альдегидную функцию соединения формулы (IV) окисляют до карбоновой кислоты с образованием соединения формулы (V):
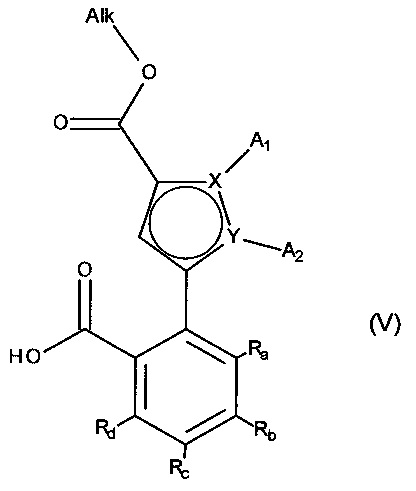
в которой A1, A2, X, Y, Ra, Rb, Rc и Rd принимают значения, определенные для формулы (I'), и Alk принимает значения, определенные выше,
соединение формулы (V) затем подвергают пептидному сочетанию с соединением формулы (VI):

в которой T и R5 принимают значения, определенные для формулы (I'),
с получением соединения формулы (VII):
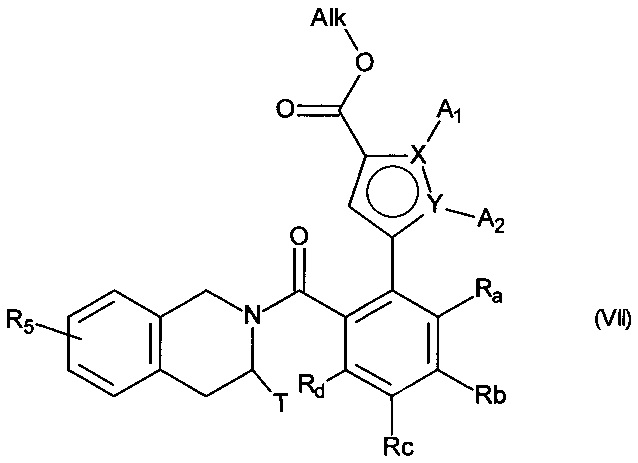
в которой A1, A2, X, Y, Ra, Rb, Rc, Rd, T и R5 принимают значения, определенные для формулы (I'), и Alk принимает значения, определенные выше,
сложноэфирную функцию соединения формулы (VII) гидролизуют с получением соответствующей карбоновой кислоты или карбоксилата, которую(-ый) можно превратить в производное по кислотной группе, такое как соответствующий ацилхлорид или ангидрид, перед сочетанием с амином NHR3R4, где R3 и R4 имеют те же значения, что и для формулы (I'), прежде чем подвергнуть действию пирофосфатного, фосфонатного или фосфорильного соединения в основных условиях, причем соединение, полученное таким образом, может быть необязательно гидролизовано или гидрогенолизировано с получением соединения формулы (I),
соединение формулы (I) может быть очищено в соответствии с обычными методами разделения, превращено, при необходимости, в его соли присоединения с фармацевтически приемлемой кислотой или основанием, и необязательно разделено на его изомеры в соответствии с обычными методами разделения,
при этом предполагается, что, в любое время, признанное подходящим в ходе описанного выше способа, определенные группы (гидрокси, амино…) реагентов или промежуточных соединений синтеза могут быть защищены и затем лишены защиты в соответствии с требованиями синтеза.
Соединения формул (II), (III), (VI) и амин NHR3R4 либо доступны для приобретения, либо могут быть получены специалистом в данной области техники с использованием обычных химических реакций, описанных в литературе.
Более конкретно, фосфатные соединения формулы (I) в соответствии с изобретением будут полезны для лечения хемо- или радиорезистентных злокачественных новообразований, а также при злокачественных заболеваниях крови и мелкоклеточном раке легкого.
Среди намеченных для лечения злокачественных заболеваний могут быть упомянуты, без какого-либо ограничения, рак мочевого пузыря, головного мозга, молочной железы и матки, хронические лимфоидные лейкозы, колоректальный рак, рак пищевода и печени, лимфобластные лейкозы, неходжкинские лимфомы, меланомы, злокачественные заболевания крови, миелы, рак яичников, немелкоклеточный рак легкого, рак предстательной железы и мелкоклеточный рак легкого. Среди неходжкинских лимфом, более предпочтительно, могут быть упомянуты фолликулярные лимфомы, лимфомы из клеток мантийной зоны, диффузные B-крупноклеточные лимфомы, мелкоклеточные лимфоцитарные лимфомы и B-клеточные лимфомы из клеток маргинальной зоны.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение формулы (I) в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями.
Среди фармацевтических композиций в соответствии с изобретением могут быть упомянуты особенно те, которые подходят для перорального, парентерального, назального, чрес- или транскожного, ректального, перлингвального, офтальмологического или респираторного введения, главным образом таблетки или драже, сублингвальные таблетки, саше, пакеты, капсулы, глоссеты, лепешки, суппозитории, кремы, мази, кожные гели, и питьевые или инъекционные ампулы.
Дозировка варьируется в зависимости от пола, возраста и веса пациента, пути введения, природы терапевтического показания, или каких-либо связанных лечений, и находится в диапазоне от 0.01 мг до 1 г в 24 часа за одно или несколько введений.
Кроме того, настоящее изобретение также относится к ассоциации соединения формулы (I) с противоопухолевым средством, выбранным из генотоксичных средств, митотических ядов, антиметаболитов, ингибиторов протеосом, ингибиторов киназ и антител, а также к фармацевтическим композициям, содержащим такой тип ассоциации, и к их применению для изготовления лекарственных средств для применения для лечения злокачественного новообразования.
Соединения настоящего изобретения также могут использоваться в ассоциации с радиотерапией для лечения злокачественного новообразования.
Следующие методики получения и примеры иллюстрируют изобретение, не ограничивая его каким-либо образом.
Методика получения 1: 6-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Стадия A: 1-Формил-2-пиперидинкарбоновая кислота
К раствору 40 г рацемической смеси 2-пиперидинкарбоновой кислоты (0.310 ммоль) в 300 мл муравьиной кислоты, помещенному на баню с температурой 0°C, по каплям добавляют 200 мл (2.15 ммоль) уксусного ангидрида. Загрузочную порцию затем перемешивают при температуре окружающей среды в течение ночи. Затем, реакционную смесь охлаждают до 0°C, гидролизуют путем добавления 250 мл воды, и перемешивают в течение получаса при 0°C перед выполнением концентрирования досуха. Полученное таким образом масло переносят в 200 мл метанола и затем концентрируют досуха. Указанный в заголовке продукт получают в виде масла с выходом 98%. Его используют непосредственно, без очистки иным способом, на следующей стадии.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 13.0 (m, 1H ОН); 8.0-8.05 (2s, 1Н альдегид); 4.9-4.5 (2d, 1Н α к N и СООН); 4.1-2.6 (m, 2Н α к N); 2.2-1.2 (m, 6Н пиперидин)
ИК: ν: -ОН: 2000-3000 см-1 кислота; ν:>С=0 1703 см-1 широкая полоса
Стадия B: Метил 5,6,7,8-тетрагидро-1-индолизинкарбоксилат
К раствору 10 г карбоновой кислоты, полученной на стадии A, (63.6 ммоль) в 65 мл дихлорэтана последовательно добавляют 13.4 г тозилхлорида (70.4 ммоль), 11.5 мл метил 2-хлоракрилата (113.5 ммоль) и затем, по каплям, 17.8 мл N,N,N-триэтиламина (127.2 ммоль). Реакционную смесь затем нагревают с обратным холодильником в течение 1 часа 30 минут. Ее затем доводят до температуры окружающей среды, и затем добавляют 5 мл метил 2-хлоракрилата (48.9 ммоль) и, по каплям, 9 мл N,N,N-триэтиламина (64 ммоль). Загрузочную порцию нагревают с обратным холодильником в течение ночи.
Реакционную смесь затем разбавляют метиленхлоридом, последовательно промывают 1М раствором HCl, насыщенным водным раствором NaHCO3 и затем соляным раствором до тех пор, пока не достигают нейтрального значения pH. Затем органическую фазу сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на силикагеле (градиент гептан/AcOEt). Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (400 МГц; CDCl3; 300 K): 6.55-6.40 (d, 2Н тетрагидроиндолизин); 3.91 (t, 3Н сложный метиловый эфир); 3.78 (s, 3Н тетрагидроиндолизин); 3.08 (t, 2Н тетрагидроиндолизин); 1.95-1.85 (m, 4Н тетрагидроиндолизин)
ИК: ν:>C=O 1692 см-1 сложный эфир
Стадия C: Метил 3-(6-формил-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидро-1-индолизинкарбоксилат
К раствору 6.4 г сложного эфира, полученного на стадии B, (35.7 ммоль) в 12 мл N,N-диметилацетамида последовательно добавляют 12.3 г 6-бром-1,3-бензодиоксол-5-карбальдегида (53.6 ммоль) и 7 г ацетата калия (71.4 ммоль), и затем загрузочную порцию перемешивают под аргоном в течение 20 минут. К ней затем добавляют 1.3 г палладиевого катализатора - дихлорбис(трифенилфосфин)палладия(II) (PdCl2(PPh3)2) (1.8 ммоль). Реакционную смесь затем нагревают при 130°C в течение одного часа перед добавлением к ней 139 мкл H2O. Нагревание продолжают, поддерживая такую же температуру в течение ночи. Температуре смеси позволяют вернуться к температуре окружающей среды и ее затем разбавляют с помощью AcOEt. Добавляют животный уголь (25 г на г продукта), и загрузочную порцию перемешивают при температуре окружающей среды в течение 1 часа и затем фильтруют. Затем органическую фазу промывают водой, сушат над сульфатом магния и концентрируют досуха. Полученный таким образом сырой продукт очищают с помощью хроматографии на силикагеле (градиент гептан/AcOEt). Указанный в заголовке продукт получают в виде масла.
1H ЯМР: δ:(400 МГц; дмсо-d6; 353 K): 9.65 (s, 1Н, Н альдегид); 7.3-7.15 (2s, 2Н, ароматические Н); 6.45 (s, 1H тетрагидроиндолизин); 6.20 (s, 2Н метилендиокси); 3.70 (s, 3Н сложный метиловый эфир); 3.5-4.0 (m, 2Н тетрагидроиндолизин); 3.05 (m, 2Н тетрагидроиндолизин); 1.85 (m, 4Н тетрагидроиндолизин)
ИК: ν:>C=O 1695 см-1 сложный эфир; ν:>C=O 1674 см-1
Стадия D: 6-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Получают раствор, содержащий 3.37 г соединения, полученного на стадии C, (10.3 ммоль) в 9.3 мл ацетона и 8.8 мл (80.24 ммоль) 2-метил-2-бутена, и помещают его на баню с температурой 0°C. К нему по каплям добавляют 9.3 мл водного раствора, содержащего смесь 3.3 г хлорита натрия (NaClO2) (36.05 ммоль) и 3.6 г моногидрата дигидрофосфата натрия (NaH2PO4) (25.75 ммоль). Загрузочную порцию затем перемешивают при температуре окружающей среды в течение 7 часов. Реакционную смесь затем концентрируют для удаления ацетона. Полученное твердое вещество затем отфильтровывают, промывают водой и затем сушат при 40°C в вакууме в течение ночи. Указанный в заголовке продукт получают в виде твердого вещества, которое впоследствии используют без очистки иным способом.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 12.10 (m, 1Н, Н карбоновая кислота); 7.40-6.88 (2s, 2Н, ароматические Н); 6.20 (s, 1Н, Н тетрагидроиндолизин); 6.18 (s, 2Н, Н метилендиокси); 3.70 (s, 3Н сложный метиловый эфир); 3.55 (t, 2Н тетрагидроиндолизин); 3.00 (t, 2Н тетрагидроиндолизин); 1.80 (m, 4Н, Н тетрагидроиндолизин)
ИК: ν: -ОН: 3000-2000 см-1 кислота; ν:>C=O 1686-1676 см-1 сложный эфир + кислота; ν:>C=C<1608 см-1
Методика получения 2: 2-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует протоколу, описанному в методике получения 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на стадии C, на 2-бромбензальдегид.
Методика получения 3: 6-[1-(Метоксикарбонил)-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Стадия A: Бромид 1-(карбоксиметил)-1,2-дигидропиридиния
К раствору 16.2 мл пиридина (200 ммоль) в 120 мл этилацетата порциями добавляют 27.8 г (200 ммоль) бромуксусной кислоты. Загрузочную порцию затем перемешивают при температуре окружающей среды в течение ночи. Полученный таким образом осадок отфильтровывают и затем промывают холодным этилацетатом. После сушки, указанный в заголовке продукт получают в виде порошка, который непосредственно используют на следующей стадии.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 9.15 (d, 2Н, ароматические Н, пиридин); 8.7 (t, 1Н, ароматический Н); 8.25 (t, 2Н, ароматический Н); 5.65 (s, 2Н, Н СН2СООН)
ИК: ν: C=O: 1732 см-1; -ОН кислота: 2800 см-1
Стадия B: Метил 1-индолизинкарбоксилат
К суспензии 6.55 г соли пиридиния, полученной на стадии A, (30 ммоль) в 240 мл толуола последовательно добавляют 16.7 мл метилакрилата (150 ммоль), 4.2 мл триэтиламина (30 ммоль) и затем, порциями, 20.9 г MnO2 (240 ммоль). Загрузочную порцию затем нагревают при 90°C в течение 3 часов. После охлаждения, реакционную смесь фильтруют через слой целита и концентрируют досуха. Указанный в заголовке продукт затем выделяют путем очистки на силикагеле (градиент гептан / AcOEt: 0-10%) в виде масла, которое кристаллизуется в холодном состоянии.
1Н ЯМР: δ (300 МГц; дмсо-d6; 300 K): 8.5 (d, 1Н, Н индолизин); 8.05 (d, 1Н, Н индолизин); 7.6 (s, 1Н, Н индолизин); 7.15 (m, 2Н, Н индолизин); 6.85 (m, 1Н, Н индолизин); 4.25 (q, 2Н, -С(O)СН2СН3); 1.35 (t, 3Н, -С(O)СН2СН3)
ИК: ν: C=O сложный эфир: 1675 см-1; ароматический C=C фрагменты: 1634 см-1
Стадия C: 6-[1-(Метоксикарбонил)-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Методика соответствует протоколу, описанному на стадиях С и D методики получения 1.
Методика получения 4: 4-Хлор-2-[4-(этоксикарбонил)-1,5-диметил-1H-пиррол-2-ил]бензойная кислота
Стадия A: Этил 1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору 10 г этил 2-метил-1H-пиррол-3-карбоксилата (65.3 ммоль) и 8.95 мл (130.6 ммоль) метилйодида в 70 мл диметилформамида, помещенному на баню с температурой 0°C, тремя порциями добавляют 2.61 г (65.3 ммоль) 60%-ного гидрида натрия. Загрузочную порцию затем перемешивают при 0°C в течение 1 часа. Затем, реакционную смесь гидролизуют путем добавления 420 мл ледяной воды. Реакционную смесь затем разбавляют этилацетатом, последовательно промывают 0.1М раствором HCl, насыщенным водным раствором LiCl и затем соляным раствором. Затем органическую фазу сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на силикагеле (градиент петролейный эфир/AcOEt).
1H ЯМР: δ (400 МГц; дмсо-d6; 300 K): 6.65 (d, 1Н пиррол); 6.3 (1d, 1Н пиррол); 4.1 (1 q, 2Н, ОСН2СН3); 3.5 (s, 3Н N-пиррол); 2.4 (s, 3Н пиррол); 1.5 (1t, 3Н ОСН2СН3)
ИК: ν:>C=O: 1688 см-1; ν: C-O-C: 1172 см-1
Стадия B: Этил 5-(5-хлор-2-формилфенил)-1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору 10.5 г соединения, полученного на стадии A, (62.8 ммоль) в 65 мл N,N-диметилацетамида последовательно добавляют 15.2 г 2-бром-4-хлорбензальдегида (69 ммоль), 12.3 г ацетата калия (125.6 ммоль), и затем загрузочную порцию перемешивают под аргоном в течение 20 минут. К ней затем добавляют 2.2 г палладиевого катализатора PdCl2(PPh3)2 (3.14 ммоль). Реакционную смесь затем нагревают при 130°C в течение ночи. Температуре смеси позволяют вернуться к температуре окружающей среды, и ее затем разбавляют дихлорметаном. Добавляют животный уголь (30 г), и загрузочную порцию перемешивают при температуре окружающей среды в течение 1 часа и затем фильтруют. Затем органическую фазу промывают водой, сушат над сульфатом магния и концентрируют досуха. Полученный таким образом сырой продукт очищают с помощью хроматографии на силикагеле (градиент петролейный эфир/AcOEt). Указанный в заголовке продукт получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 9.8 (s, 1H формил); 7.91-7.69-7.61 (d, 3Н, ароматические Н); 6.5 (s, 1Н пиррол); 4.2 (q, 2Н, ОСН2СН3); 3.4 (s, 3Н, СН3-N-пиррол); 2.55 (s, 3Н пиррол); 1.28 (t, 3Н, ОСН2СН3)
Стадия C: 4-Хлор-2-[4-(этоксикарбонил)-1,5-диметил-1Н-пиррол-2-ил/бензойная кислота
Получают раствор, содержащий 12.85 г соединения, полученного на стадии B, (42 ммоль) и 35.7 мл (336 ммоль) 2-метил-2-бутена в смеси, содержащей 20 мл ацетона и 20 мл тетрагидрофурана. К нему по каплям добавляют 200 мл водного раствора, содержащего смесь 13.3 г хлорита натрия (NaClO2) (147 ммоль) и 14.5 г моногидрата дигидрофосфат натрия (NaH2PO4H2O) (105 ммоль). Загрузочную порцию затем энергично перемешивают при температуре окружающей среды в течение 7 часов. Реакционную смесь затем концентрируют для удаления ацетона. Добавляют этилацетат, и органическую фазу промывают водой и затем концентрируют досуха. Остаток затем переносят в минимальное количество этилового эфира. Полученное твердое вещество затем отфильтровывают, промывают эфиром и затем сушат в вакууме при 40°C в течение ночи. Указанный в заголовке продукт получают в виде твердого вещества, которое впоследствии используют без очистки иным способом.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 13 (m, 1Н СООН); 7.85-7.6-7.41(d,dd, wd, 3Н, ароматические Н); 6.3 (s, 1Н, Н пиррол); 4.15 (q, 2Н, ОСН2СН3); 3.25 (s, 3Н, СН3-N-пиррол); 2.5 (s, 3Н, СН3-пиррол); 1.25 (t, 3Н, ОСН2СН3)
ИК: ν: -ОН: 3100-2500 см-1 кислота; ν:>C=O: 1681 см-1 сложный эфир + кислота
Методика получения 5: 6-[4-(Этоксикарбонил)-1,5-диметил-1H-пиррол-2-ил]-1,3-бензодиоксол-5-карбоновая кислота
Методика соответствует методике получения 4, с заменой 2-бром-4-хлорбензальдегида, используемого на стадии B, на 6-бром-1,3-бензодиоксол-5-карбальдегид.
Методика получения 6: 4-Фтор-3-метокси-2-[4-(этоксикарбонил)-1,5-диметил-1H-пиррол-2-ил] бензойная кислота
Методика соответствует методике получения 4, с заменой 2-бром-4-хлорбензальдегида, используемого на стадии B, на 2-бром-4-фтор-3-метоксибензальдегид.
Методика получения 7: 4-Фтор-2-[4-(этоксикарбонил)-1,5-диметил-1H-пиррол-2-ил]бензойная кислота
Методика соответствует методике получения 4, с заменой 2-бром-4-хлорбензальдегида, используемого на стадии B, на 2-бром-4-фторбензальдегид.
Методика получения 8: 7-[4-(Метоксикарбонил)-1,5-диметил-1H-пиррол-2-ил]-2,3-дигидро-1,4-бензодиоксин-6-карбоновая кислота
Методика соответствует методике получения 4, с заменой этил 2-метил-1H-пиррол-3-карбоксилата на стадии A на метил 2-метил-1H-пиррол-3-карбоксилат, и 2-бром-4-хлорбензальдегида, используемого на стадии B, на 7-бром-2,3-дигидро-1,4-бензодиоксин-6-карбальдегид.
Методика получения 9: 5-Бензилокси-2-(1-метоксикарбонил-5,6,7,8-тетрагидроиндолизин-3-ил)бензойная кислота
Стадия A: Метил 3-(4-бензилокси-2-формилфенил)-5,6,7,8-тетрагидро индол из ин-1-карбоксилат
5-Бензилокси-2-бромбензальдегид (12.3 г, 42.2 ммоль) вводят в колбу в присутствии ацетата калия (8.3 г; 84.2 ммоль) и 120 мл диметилацетамида. После дегазирования под аргоном, добавляют дихлорбис(трифенилфосфин)палладий (II) (1.04 г, 1.5 ммоль), и смесь дегазируют в атмосфере аргона перед осуществлением нагревания при 100°C в течение 16 часов. После возвращения к температуре окружающей среды, реакционную смесь выливают в 200 мл этилацетата, фильтруют через целит, и промывают водой и затем соляным раствором. Объединенные водные фазы экстрагируют этилацетатом. Органические фазы сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток очищают с помощью хроматографии на силикагеле с целью получения указанного в заголовке продукта.
Стадия В: 5-Бензилокси-2-(1-метоксикарбонил-5,6,7,8-тетрагидроиндолизин-3-ил)бензойная кислота
К раствору соединения, полученного на стадии B, (4.63 г, 11.89 ммоль) в 300 мл ацетона добавляют 2-метил-2-бутен (6.31 мл, 59 ммоль). Затем по каплям вливают раствор моногидрата дигидрофосфата натрия (6.56 г, 47.6 ммоль) и хлорита натрия (2.69 г, 23.8 ммоль) в 40 мл воды, при поддержании температуры ниже 20°C. После перемешивания в течение 30 минут при температуре окружающей среды, смесь подкисляют с помощью 2М раствора HCl, и затем фазы разделяют. Водную фазу экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия, фильтруют и упаривают досуха с обеспечением ожидаемого соединения.
Методика получения 1': (3S)-3-(4-Морфолинилметил)-1,2,3,4-тетрагидроизохинолин
Стадия A: Бензил (3S)-3-(4-морфолинилкарбонил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилат
К раствору 5 г (3S)-2-[(бензилокси)карбонил]-1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты (16 ммоль) в 160 мл дихлорметана добавляют 1.5 мл морфолина (17.6 ммоль), затем 9 мл N,N,N-триэтиламина (64 ммоль), 3.3 г 1-этил-3-(3'-диметиламинопропил)-карбодиимида (EDC) (19.2 ммоль) и 2.6 г гидроксибензотриазола (HOBt) (19.2 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение ночи; затем ее выливают в водный раствор хлорида аммония и экстрагируют этилацетатом. Затем органическую фазу сушат над сульфатом магния, и затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол). Продукт получают в виде пены.
1Н ЯМР: δ (400 МГц; дмсо-d6; 353 K): 7.30 (m, 5Н бензил); 7.15 (m, 4Н, ароматические Н); 5.2-5.0 (m, 3Н, 2Н бензил, 1Н дигидроизохинолин); 4.75-4.5 (2d, 2Н дигидроизохинолин); 3.55-3.3 (m, 8Н морфолин); 3.15-2.9 (2dd, 2Н дигидроизохинолин)
ИК: ν:>C=O: 1694; 1650 см-1
Стадия B: Бензил (3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилат
К раствору 5.3 г продукта, полученного на стадии A, (13.9 ммоль) в 278 мл тетрагидрофурана добавляют 14 мл комплекса боран-диметилсульфид (BH3Me2S) (27.8 ммоль) при температуре окружающей среды. Загрузочную порцию нагревают в течение 4 часов при 80°C. Температуре позволяют вернуться к температуре окружающей среды, и затем добавляют 7 мл (14 ммоль) BH3Me2S. Реакционную смесь вновь нагревают при 80°C в течение 2 часов. Затем упаривают тетрагидрофуран, и затем медленно добавляют метанол и затем 5.6 мл 5М соляной кислоты (27.8 ммоль). Смесь перемешивают при температуре окружающей среды в течение ночи, и затем при 80°C в течение 1 часа. Затем к реакционной смеси, помещенной на баню с температурой 0°C, добавляют насыщенный водный раствор NaHCO3 до тех пор, пока не достигают значения pH 8, и затем осуществляют экстрагирование этилацетатом. Затем органическую фазу сушат над сульфатом магния, и затем фильтруют и упаривают досуха. Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (400 МГц; дмсо-d6; 353 K): 7.43-7.30 (неразрешенный пик, 5Н бензил); 7.19 (m, 4Н, ароматические Н); 5.16 (m, 2Н, 2Н бензил); 4.79-4.29 (d, 2Н дигидроизохинолин); 4.58 (m, 1Н дигидроизохинолин); 3.50 (m, 4Н морфолин); 3.02-2.80 (dd, 2Н дигидроизохинолин); 2.42-2.28 (неразрешенный пик, 5Н, 4Н морфолин, 1Н морфолин); 2.15 (dd, 1Н морфолин)
ИК: ν:>СН: 2810 см-1; ν:>C=O: 1694 см-1; ν:>C-O-C<: 1114 см-1; ν:>CH-Ar: 751; 697 см-1
Стадия C: (3S)-3-(4-Морфолинилметил)-1,2,3,4-тетрагидроизохинолин
К раствору 4.9 г соединения стадии B (13.4 ммоль) в 67 мл этанола добавляют 0.980 г дигидроксида палладия (20 мас. %) при температуре окружающей среды. Реакционную смесь помещают под давление водорода 1.2 бар при температуре окружающей среды на 4 часа. Ее затем пропускают через фильтр Whatman и палладий затем промывают несколько раз этанолом. Фильтрат упаривают досуха. Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: 8 (400 МГц; дмсо-d6; 300 K): 7.12-7.0 (неразрешенный пик, 4Н, ароматические Н); 3.92 (s, 2Н тетрагидроизохинолин); 3.60 (t, 4Н морфолин); 2.98 (m, 1Н тетрагидроизохинолин); 2.68 (dd, 1Н тетрагидроизохинолин); 2.5-2.3 (неразрешенный пик, 8Н, 1Н тетрагидроизохинолин, 6Н морфолин, 1Н NH)
ИК: ν:>NH: 3322 см-1; ν:>С-O-С<: 1115 см-1; ν:>СН-Ar: 742 см-1
Методика получения 2': Гидрохлорид (3R)-3-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: {(3S)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил 4-метилбензолсульфонат
К раствору 30.2 г [(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]метанола (185 ммоль) в 750 мл дихлорметана последовательно добавляют 91.71 г тозилхлорида (481 ммоль) и затем, по каплям, 122.3 мл N,N,N-триэтиламина (740 ммоль). Реакционную смесь затем перемешивают при температуре окружающей среды в течение 20 часов. Ее затем разбавляют дихлорметаном, последовательно промывают 1М раствором HCl, насыщенным водным раствором NaHCO3 и затем соляным раствором до нейтральной среды. Затем органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. Полученное твердое вещество затем растворяют в минимальном объеме дихлорметана, и затем, до тех пор, пока не образуется осадок, добавляют циклогексан. Этот осадок затем отфильтровывают и промывают циклогексаном. После сушки, указанный в заголовке продукт получают в виде кристаллов.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 7.75 (d, 2Н, ароматические Н, орто О-тозил); 7.6 (d, 2Н, ароматические Н, орто N-тозил); 7.5 (d, 2Н, ароматические Н, мета О-тозил); 7.3 (d, 2Н, ароматические Н, мета N-тозил); 7.15-6.9 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.4-4.15 (dd, 2Н, алифатические Н, тетрагидроизохинолин); 4.25 (m, 1Н, алифатический Н, тетрагидроизохинолин); 4.0-3.8 (2dd, 2Н, алифатические Н, СН2-O-тозил); 2.7 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 2.45 (s, 3Н, O-SO2-Ph- СН3); 2.35 (s, 3Н, N-SO2-Ph- СН3)
ИК: ν: -SO2: 1339-1165 см-1
Стадия B: (3R)-3-Метил-2-[(4-метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин
К суспензии 8.15 г (214.8 ммоль) LiAlH4 в 800 мл метил-трет-бутилового эфира (МТВЕ) добавляют 101.2 г дитозильного соединения, полученного на стадии A, (214.8 ммоль), растворенного в 200 мл МТВЕ. Загрузочную порцию затем нагревают при 50°C в течение 2 часов. Ей позволяют охладиться и помещают на баню с температурой 0°C, и затем, по каплям, добавляют 12 мл 5М раствора NaOH. Загрузочную порцию перемешивают при температуре окружающей среды в течение 45 минут. Полученное таким образом твердое вещество затем отфильтровывают и промывают с помощью МТВЕ и затем дихлорметаном. Фильтрат затем концентрируют досуха. Указанный в заголовке продукт затем получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 7.70 (d, 2Н, ароматические Н, орто N-тозил); 7.38 (d, 2Н, ароматические Н, мета N-тозил); 7.2-7.0 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.4 (m, 2Н, алифатические Н, тетрагидроизохинолин); 4.3 (m, 1Н, алифатический Н, тетрагидроизохинолин); 2.85-2.51 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 2.35 (s, 3Н, N-SO2-Ph- СН3); 0.90 (d, 3Н, тетрагидроизохинолин-СН3)
ИК: ν: -SO2: 1332-1154 см-1
Стадия C: (3R)-3-Метил-1,2,3,4-тетрагидроизохинолин
К раствору 31.15 г (103.15 ммоль) монотозильного соединения, полученного на стадии B, в 500 мл безводного метанола порциями добавляют 3.92 г (161 ммоль) магниевых стружек. Загрузочную порцию перемешивают под воздействием ультразвука в течение 96 часов. Реакционную смесь затем фильтруют, и твердое вещество промывают несколько раз метанолом. Фильтрат затем концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент дихлорметан /EtOH/NH4OH), указанный в заголовке продукт получают в виде масла.
1H ЯМР: δ (400 МГц; дмсо-d6; 300 K): 7.05 (m, 4Н, ароматические Н, тетрагидроизохинолин); 3.90 (m, 2Н, алифатические Н, тетрагидроизохинолин); 2.85 (m, 1Н, алифатический Н, тетрагидроизохинолин); 2.68-2.4 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 1.12 (d, 3Н, тетрагидроизохинолин-СН3); 2.9-2.3 (m, широкий, 1Н, HN (тетрагидроизохинолин))
ИК: ν: -NH: 3248 см-1
Стадия D: Гидрохлорид (3R)-3-метил-1,2,3,4-тетрагидроизохинолина
К раствору 14.3 г (97.20 ммоль) соединения, полученного на стадии C, в 20 мл безводного этанола по каплям добавляют 100 мл 1М раствора HCl в эфире. Загрузочную порцию перемешивают при температуре окружающей среды в течение 1 часа и затем фильтруют. Полученные таким образом кристаллы промывают этиловым эфиром. После сушки, указанный в заголовке продукт получают в виде кристаллов.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 9.57 (m, широкий, 2Н, 
ИК: ν: 
Методика получения 3': (3R)-3-[3-(Морфолин-4-ил)пропил]-1,2,3,4-тетрагидроизохинолин
Стадия A: {(3S)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил 4-метилбензолсульфонат
Методика является такой же, что и на стадии A методики получения 2'.
Стадия B: трет-Бутил 2-({(3R)-2-[(4-метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил)-3-(морфолин-4-ил)-3-оксопропаноат
К суспензии 1 г NaH (60%) (25.08 ммоль) в 30 мл МТВЕ по каплям добавляют раствор 5 г трет-бутип 3-морфолино-3-оксопропаноата (21.81 ммоль) в 20 мл безводного МТВЕ. Эту суспензию перемешивают при температуре окружающей среды в течение 1 часа, и затем добавляют соединение, полученное на стадии A, в виде порошка. Загрузочную порцию перемешивают при 60°C в течение 30 часов. Добавляют 100 мл насыщенного водного раствора хлорида аммония. Получающийся раствор экстрагируют дихлорметаном. Затем органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент дихлорметан/МеОН), ожидаемый продукт получают в виде масла.
1Н ЯМР (500 МГц, дмсо-d6) δ м.д.: 7.63/7.59 (2d, 2Н), 7.3/7.26 (2d, 2Н), 7.13 (m, 2Н), 7.09/6.97 (2t, 2Н), 4.64/4.55/4.36/4.28 (2АВ, 2Н), 4.25/4.11 (2 m, 1Н), 3.81 (m, 1Н), 3.73-3.48 (m, 4Н), 3.57-3.32 (m, 4Н), 2.51 (m, 2Н), 2.32/2.31 (2s, 3Н), 1.88/1.79 (2m, 2H), 1.39/1.38 (2s, 9Н)
ИК (ATR) см-1: ν:>С=O: 1731 (сложный эфир); ν:>С=O: 1644 (амид); ν: -SO2: 1334-1156; ν:>С-O-С<: 1115; γ:>CH-Ar: 815-746-709
Стадия C: 2-({(3R)-2-[(4-Метилфенил)сулъфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил)-3-(морфолин-4-ил)-3-оксопропановая кислота
К раствору 9.5 г (17.97 ммоль) соединения, полученного на стадии B, в 40 мл диоксана по каплям добавляют 20 мл 4М раствора HCl в диоксане. Загрузочную порцию перемешивают при температуре окружающей среды в течение 48 часов, и затем раствор концентрируют досуха. После сушки, ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц, дмсо-d6) δ м.д.: 12.75 (m, 1Н), 7.6 (2*d, 2Н), 7.3 (2*d, 2Н), 7.1/6.95 (2*m, 4Н), 4.7-4.2 (d, 2Н), 4.25/4.12 (2*m, 1Н), 3.9-3.3 (m, 9Н), 2.55 (d, 2Н), 2.3 (2*s, 3Н), 1.8 (t, 2Н)
ИК (ATR) см-1: ν: -ОН: 3500-2000; ν:>С=O: 1727 (кислота); ν:>С=O: 1634 (амид); ν: -SO2: 1330-1155
Стадия D: 3-{(3R)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}-1-(морфолин-4-ил)пропан-1-он
К раствору 7.80 г (16.51 ммоль) соединения, полученного на стадии C, в 100 мл ДМСО добавляют 1.16 г (19.83 ммоль) твердого хлорида натрия и затем, по каплям, 5 мл воды. Загрузочную порцию перемешивают при 130°C в течение 1 часа, и затем раствор концентрируют до ¾. Реакционную смесь затем разбавляют дихлорметаном, и последовательно промывают насыщенным водным раствором хлорида лития и затем соляным раствором. Затем органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент циклогексан/этилацетат), ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц, дмсо-d6) δ м.д.: 7.65 (d, 2Н), 7.3 (d, 2Н), 7.15/7 (2m, 4Н), 4.6 (d, 1Н), 4.25 (d, 1Н), 4.2 (m, 1Н), 3.5 (m, 4Н), 3.4 (2m, 4Н), 2.6 (2dd, 2Н), 2.35 (s, 3Н), 2.3 (m, 2Н), 1.5 (quad., 2Н)
ИК (ATR)см-1:ν:>С=O: 1639; ν: -SO2: 1331-1156; γ:>CH-Ar: 815-675
Стадия E: (3R)-2-[(4-Метилфенил)сульфонил]-3-[3-(морфолин-4-ил)пропил]-1,2,3,4-тетрагидроизохинолин
К раствору 6.0 г (14.0 ммоль) соединения, полученного на стадии D, в 60 мл МТВЕ и 14 мл дихлорметана в течение 5 минут порциями добавляют 1.06 г (28 ммоль) LAH. Загрузочную порцию перемешивают при температуре окружающей среды в течение 15 часов. К ней по каплям добавляют 1.5 мл воды, и перемешивание осуществляют в течение 15 минут. Затем по каплям добавляют 1.5 мл 5М раствора гидроксида натрия, и перемешивание осуществляют в течение 15 минут. Реакционную смесь затем разбавляют с помощью МТВЕ и дихлорметана. Суспензию затем фильтруют и осадок промывают с помощью МТВЕ и дихлорметана. Затем органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент дихлорметан/EtOH/NH4OH), ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц, дмсо-d6) δ м.д.: 7.68 (d, 2Н), 7.32 (d, 2Н), 7.1 (неразрешенный пик, 4Н), 4.65/4.23 (АВ, 2Н), 4.2 (m, 1Н), 3.55 (t, 4Н), 2.7/2.6 (АВх, 2Н), 2.35 (s, 3Н), 2.25 (t, 4Н), 2.2 (t, 2Н), 1.4/1.3 (2m, 4Н).
ИК (ATR) см-1: ν: -SO2: 1333-1158
Стадия F: (3R)-3-[3-(Морфолин-4-ил)пропил]-1,2,3,4-тетрагидроизохинолин
К раствору 1.50 г (3.62 ммоль) соединения, полученного на стадии E, в 20 мл безводного метанола порциями добавляют 2.0 г (82.3 ммоль) магниевых стружек. Загрузочную порцию перемешивают под воздействием ультразвука в течение 96 часов. Реакционную смесь затем фильтруют, твердое вещество промывают несколько раз метанолом, и фильтрат концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент дихлорметан/EtOH/NH4OH), ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц, дмсо-d6) δ м.д.: 7.3 (d, 2Н), 7.1 (t, 2Н), 7.1 (d+t, 3Н), 7 (d, 2Н), 3.9 (s, 2Н), 3.55 (t, 4Н), 2.75 (m, 1Н), 2.72/2.45 (dd, 2Н), 2.35 (t, 4Н), 2.25 (t, 2Н), 1.6 (m, 2Н), 1.45 (m, 2Н)
ИК (ATR) см-1: ν:>NH2+/NH+: 3500-2300; ν:>C-O-C<: 1115
Масс-спектрометрия высокого разрешения (ESI+-/FIA/HR):
Эмпирическая формула: C16 Н24 N2 О
[М+Н]+ рассчитано: 261.1961
[М+Н]+ найдено: 261.1959
Методика получения 4': (3R)-3-(4-Морфолинилметил)-1,2,3,4-тетрагидроизохинолин
Методика соответствует методике получения 1', с заменой (3S)-2-[(бензилокси)карбонил]-1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты, используемой на стадии A, на (3R)-2-[(бензилокси)карбонил]-1,2,3,4-тетрагидро-3-изохинолинкарбоновую кислоту.
Методика получения 1”: 4-{[трет-Бутил(диметил)силил]окси}-N-фениланилин
К раствору 12 г 4-анилинофенола (64.7 ммоль) в 200 мл ацетонитрила при температуре окружающей среды добавляют 6.7 г имидазола (97.05 ммоль) и 11.7 г трет-бутил(хлор)диметилсилана (77.64 ммоль). Загрузочную порцию перемешивают при 70°C в течение 4 часов. Реакционную смесь затем выливают в воду и экстрагируют эфиром. Затем органическую фазу сушат над сульфатом магния, затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент петролейный эфир/дихлорметан). Указанный в заголовке продукт получают в виде порошка.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 7.84 (s, 1Н NH); 7.17 (t, 2Н анилин); 6.98 (d, 2Н фенокси); 6.94 (d, 2Н анилин); 6.76 (d, 2Н фенокси); 6.72 (t, 1Н анилин); 0.95 (s, 9Н трет-бутил); 0.15 (s, 6Н диметил)
ИК: ν:>NH: 3403 см-1; ν:>Ar: 1597 см-1
Методика получения 2”: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-индол-5-амин
Методика соответствует методике получения 5”, с заменой 4-бром-1-метила-пиразола, используемого на стадии B, на 5-бром-1-метил-1H-индол.
Методика получения 3”: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-пирроло[2,3-b]пиридин-5-амин
Методика соответствует методике получения 5”, с заменой 4-бром-1-метил-1H-пиразола, используемого на стадии B, на 5-бром-1-метил-1H-пирроло[2,3-b] пиридин (полученный в соответствии с протоколом из литературы: Heterocycles, 60(4), 865, 2003).
ИК: ν: -NH-: 3278 см-1; ν: ароматические -С=С- фрагменты: 1605 см-1
Методика получения 4”: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)пиридин-4-амин
Методика соответствует методике получения 5”, с заменой 4-бром-1-метил-1H-пиразола, используемого на стадии B, на 4-бромпиридин.
ИК: ν -NH-: 3200 и 2500 см-1; ν -Si-O-: 902 см-1; ν -Si-C-: 820 см-1
Методика получения 5": N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-пиразол-4-амин
Стадия A: 4-{[трет-Бутил(диметил)силил]окси}анилин
Указанное в заголовке соединение получают исходя из 4-аминофенола в ТГФ в присутствии имидазола и трет-бутил(хлор)диметилсилана в соответствии с протоколом, описанным в литературе (S. Knaggs и др., Organic & Biomolecular Chemistry, 3(21), 4002-4010; 2005).
1Н ЯМР: δ (400 МГц; дмсо-d6; 300 K): 6.45-6.55 (dd, 4Н, ароматические Н); 4.60 (m, 2Н, NH2-Ph); 0.90 (s, 9Н, Si (СН2)2СН(СН3)2); 0.10 (s, 6Н, Si (СН2)2СН(СН3)2)
ИК: ν:
Стадия B: N-[4-трет-Бутил(диметил)силил/оксифенил]-1-метилпиразол-4-амин
К раствору 30.8 г (0.137 моль) соединения стадии A в 525 мл безводного толуола последовательно добавляют 29.8 г трет-бутилата натрия (0.310 моль), 4.55 г Pd2(dba)3 (также называемого трис(дибензилиденацетон)дипалладием(0)) (4.96 ммоль), 4.81 г 2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенила (9.91 ммоль) и 12.8 мл 4-бром-1-метил-1H-пиразола (0.124 моль). Загрузочную порцию дегазируют под аргоном в течение 30 минут и затем нагревают с обратным холодильником в течение 3 часов. Ей позволяют охладиться. Реакционную смесь концентрируют досуха и затем переносят в дихлорметан, фильтруют через целит и затем вновь концентрируют досуха. Остаток затем очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/AcOEt) с обеспечением ожидаемого продукта в виде твердого вещества.
1Н ЯМР: δ (400 МГц; дмсо-d6; 300K): 7.55 (s, 1Н пиразол); 7.23 (s, 1H пиразол); 7.18 (широкий s, 1Н, NH2-Ph); 6.64 (m, 4Н, ароматические Н); 3.77 (s, 3Н, CH3-пиразол); 0.90 (s, 9Н, Si (СН2)2СН(СН3)2); 0.12 (s, 6Н, Si (СН2)2СН(СН3)2)
ИК: ν -NH+: 3275 см-1; ν Ar и C=N: 1577 и 1502 см-1; ν -Si-C-: 1236 см-1;
ν -Si-O-: 898 см-1; ν -Si-C-: 828, 774 см-1
Методика получения 6”: N-{4-[(трет-Бутилдиметилсилил)окси]фенил}-1-тридейтерометил-1H-пиразол-4-амин
Стадия A: 4-Бром-1-тридейтерометил-1Н-пиразол
4-Бром-1H-пиразол (9.05 г, 61.6 ммоль) порциями добавляют к суспензии гидрида натрия (60%-ного в масле) (2.83 г, 70.8 ммоль) в тетрагидрофуране (90 мл), охлажденной на ледяной бане. После удаления ледяной бани, раствор перемешивают при температуре окружающей среды в течение 0.5 часов. Его вновь охлаждают на ледяной бане и добавляют йодметан-d3 (5.0 мл, 80.3 ммоль). Раствор перемешивают при температуре окружающей среды в течение 19 часов. Суспензию затем концентрируют. Остаток после упаривания растирают с трет-бутилметиловым эфиром (90 мл) и фильтруют. Фильтрат концентрируют в вакууме с получением ожидаемого соединения в виде масла.
1Н ЯМР (400 МГц, CDCl3) δ м.д.: 7.37 (s, 1Н), 7.43 (s, 1Н)
Стадия B: N-{4-[(трет-Бутилдиметилсилил)окси]фенил}-1-тридейтерометил-1Н-пиразол-4-амин
4-Бром-1-тридейтерометил-1H-пиразол (9.6 г, 58.5 ммоль), 4-[(трет-бутилдиметилсилил)окси]анилин (14.4 г, 64.6 ммоль) и толуол (150 мл) добавляют в 500-мл трехгорлую колбу. Раствор дегазируют азотом в течение 15 минут, и затем последовательно добавляют трет-бутилат натрия (11.4 г, 0.12 моль), 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0.77 г, 1.81 ммоль) и трис(дибензилиденацетон)дипалладий(0) (1.64 г, 1.79 ммоль). Суспензию нагревают при 85°C в течение 1.5 часов. Реакционную смесь затем охлаждают до температуры окружающей среды и добавляют воду (270 мл). Смесь перемешивают в течение 30 минут. Затем добавляют целит (30 г), и суспензию фильтруют через набивку целита. Фазы фильтрата разделяют и водную фазу экстрагируют этилацетатом (3×200 мл). Объединенные органические фазы сушат над сульфатом натрия и фильтруют. Продукт очищают с помощью хроматографии на силикагеле (градиент этилацетат/гептан). Полученный продукт перекристаллизовывают их гептана (80 мл) с получением ожидаемого соединения.
1Н ЯМР (400 МГц, CDCl3) δ м.д.: 0.16 (s, 6Н), 0.97 (s, 9Н), 4.92 (s, 1Н), 6.61-6.73 (m, 4Н), 7.25 (s, 1Н), 7.36 (s, 1Н)
13С ЯМР (100 МГц, CDCl3) δ м.д.: -4.37, 18.28, 25.86, 38.67 (sept., 1JC-D=21.0 Гц), 115.12, 120.73, 123.76, 126.52, 134.74, 141.07, 148.43
МС (ESI): [М+Н]+ 307.08
Методика получения 7": 4-({4-[(трет-Бутилдиметилсилил)окси]фенил}амино)-1,5-диметил-1H-пиррол-2-карбонитрил
Стадия A: 4-Бром-1,5-диметил-1Н-пиррол-2-карбонитрил
Раствор брома (6.58 мл, 0.13 моль) в уксусной кислоте (60 мл) добавляют по каплям с помощью капельной воронки к раствору 1,5-диметил-1H-пиррол-2-карбонитрила (15.0 г, 0.12 моль) в уксусной кислоте (300 мл). Загрузочную порцию перемешивают при температуре окружающей среды в течение 24 часов. Реакционную смесь затем выливают в лабораторный стакан, содержащий 300 мл воды. Образованное твердое вещество отфильтровывают и промывают водой. Его затем растворяют в дихлорметане (300 мл) и органическую фазу промывают соляным раствором, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением ожидаемого продукта в виде твердого вещества.
1Н ЯМР (CDCl3) δ м.д.: 2.25 (s, 3Н), 3.67 (s, 3Н), 6.74 (s, 1Н)
Стадия B: 4-({4-[(трет-Бутилдиметилсилил)окси]фенил}амино)-1,5-диметил-1Н-пиррол-2-карбонитрил
Раствор соединения вышеприведенной стадии (1.5 г, 7.53 ммоль), 4-[{трет-бутилдиметилсилил)окси]анилин (2.02 г, 9.04 ммоль), трет-бутилат натрия (1.45 г, 15.06 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0.13 г, 0.30 ммоль) в толуоле (20 мл) продувают азотом. Добавляют трис(дибензилиденацетон)дипалладий(0) (0.28 г, 0.30 ммоль), и затем реакционную смесь нагревают при 90°C до тех пор, пока реакция (контролируемая с помощью ТСХ) не завершится. Нагревание прекращают и температуре смеси позволяют вернуться к температуре окружающей среды. Добавляют воду (75 мл), и смесь экстрагируют этилацетатом (3×75 мл). Объединенные органические фазы промывают соляным раствором и затем концентрируют. Сырой продукт очищают с помощью флэш-хроматографии на силикагеле (градиент этилацетат/гептан). Полученный таким образом продукт растворяют в гептане в горячем состоянии и позволяют выпасть в осадок при перемешивании, при температуре окружающей среды и затем при 0°C. Твердое вещество отфильтровывают и операцию повторяют на фильтрате с получением ожидаемого соединения в виде твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ м.д.: 0.15 (s, 6Н), 0.97 (s, 9Н), 2.13 (s, 3Н), 3.66 (s, 3Н), 4.68 (br. s, 1Н), 6.49 (d, J=8.5 Гц, 2Н), 6.64 (s, 1Н), 6.66 (d, J=8.7 Гц, 2Н)
13С ЯМР (100 МГц, CDCl3) δ м.д.: 4.34, 9.72, 18.30, 25.88, 32.94, 101.27, 114.37, 114.70, 116.41, 120.73, 124.52, 131.23, 141.54, 148.27
МС (ESI+): [М+Н]+ найдено: 342.3
Методика получения 8”: 4-[(4-{[трет-Бутил(диметил)силил]окси}фенил)амино]-1-метил-1H-пиррол-2-карбонитрил
Стадия A: 1-Метил-1Н-пиррол-2-карбонитрил
N,N-Диметилформамид (3 мл) и 1,4-диазабицикло[2.2.2]октан (0.49 г, 4.3 ммоль) добавляют к раствору пиррол-2-карбонитрила (4 г, 43.4 ммоль) в диметилкарбонате (56 мл). Раствор перемешивают при 90°C в течение 15 часов, и затем нагревают при 110°C в течение 8 часов. Смесь охлаждают до температуры окружающей среды, и затем добавляют этилацетат (80 мл). Фазы разделяют и органическую фазу промывают водой (2×80 мл) и 1М водным раствором соляной кислоты (1×80 мл). Объединенные водные фазы вновь экстрагируют этилацетатом (1×80 мл). Объединенные органические фазы промывают соляным раствором (1×80 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением ожидаемого продукта в виде жидкости.
1Н ЯМР (400 МГц, CDCl3) δ м.д.: 3.78 (m, 2Н), 6.12-6.18 (m, 1Н), 6.74-6.82 (m, 1Н)
Стадия B: 4-Бром-1-метил-1Н-пиррол-2-карбоиитрил
N-Бромсукцинимид (6.2 г, 34.9 ммоль) добавляют к раствору 1-метил-1Н-пиррол-2-карбонитрила (3.7 г, 34.9 ммоль) в N,N-диметилформамиде (150 мл). Раствор перемешивают в течение 15 часов при температуре окружающей среды. Добавляют иное количество N-бромсукцинимида (2.0 г, 11 ммоль), и смесь перемешивают в течение 3 часов. Продукт очищают с помощью хроматографии на силикагеле (градиент этилацетат/гептан) с получением ожидаемого продукта в виде твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 3.77 (s, 3Н), 6.75 (d, J=1.7 Гц, 1Н), 6.80 (d, J=1.7 Гц, 1Н)
Стадия C: 4-[(трет-Бутилдиметилсилил)окси]фенил}амино)-1-метил-1Н-пиррол-2-карбонитрил
Через раствор 4-бром-1-метил-1H-пиррол-2-карбонитрила (2.82 г, 15.2 ммоль) и 4-[(трет-бутилдиметилсилил)окси]анилина (4.08 г, 18.3 ммоль) в толуоле (55 мл) в течение 5 минут барботируют азот. Затем к реакционной смеси добавляют трет-бутилат натрия (2.92 г, 30.4 ммоль), трис(дибензилиденацетон)дипалладий(0) (556 мг, 0.6 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (255 мг, 0.6 ммоль). Смесь перемешивают в течение 1 часа при 80°C под азотом. Суспензию затем охлаждают до температуры окружающей среды и фильтруют через целит. Осадок на целите затем промывают этилацетатом. Фильтрат промывают водой и затем соляным раствором. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Продукт очищают дважды с помощью хроматографии на силикагеле (градиент AcOEt/гептан), и затем с помощью растирания в гептане с получением ожидаемого продукта в виде твердого вещества.
lH ЯМР (400 МГц, CDCl3) δ м.д.: 0.16 (s, 6Н), 0.97 (s, 9Н), 3.73 (s, 3Н), 6.57 (d, J=1.9 Гц, 1Н), 6.64-6.66 (m, 1Н), 6.70 (s, 4Н); ЯМР
13С ЯМР (100 МГц, CDCl3) δ м.д.: -4.48, 18.17, 25.72, 35.46, 103.01, 113.56, 113.69, 115.92, 119.55, 120.67, 129.04, 139.94, 148.85
МС (ESI+): [М+Н]+ 328.25
Амины NHR3R4, где R3 и R4 каждый независимо от другого представляет собой арильную или гетероарильную группу, получают в соответствии со способами, описанными в литературе (Surry D.S. и др., Chemical Science, 2011, 2, 27-50, Charles M.D. и др., Organic Letters, 2005, 7, 3965-3968). Реакция защиты гидроксильной функции 4-анилинофенола, описанная в методике получения 1”, может использоваться для различных вторичных аминов NHR3R4 (как определено выше), содержащих одну или несколько гидроксильных функций, когда они являются коммерчески доступными. Альтернативно, вторичные амины, содержащие по меньшей мере один гидрокси заместитель могут быть синтезированы непосредственно в защищенной форме, то есть исходя из реагентов, чья гидроксильной функция была защищена заранее. Среди защитных групп, трет-бутил(диметил)силилокси и бензилокси являются особенно предпочтительными.
Среди аминов NHR3R4, содержащих гидроксильный заместитель, которые используют для синтеза соединений согласно изобретению, могут быть упомянуты: 4-(4-толуидино)фенол, 4-(4-хлоранилино)фенол, 4-(3-фтор-4-метиланилино)фенол, 4-[4-(трифторметокси)анилино]фенол, 4-[4-гидроксианилино]фенол, {4-[(1-метил-1H-индол-6-ил)амино]фенил}метанол, 4-(2,3-дигидро-1H-индол-6-иламино)фенол, 4-[(1-метил-2,3-дигидро-1H-индол-6-ил)амино]фенол, 4-[(1-метил-1H-индол-6-ил)амино]фенол, 4-[(1-метил-1H-индол-6-ил)амино]циклогексанол, 4-[(1-метил-1,2,3,4-тетрагидро-6-хинолинил)амино]фенол, 4-[(4-метил-3,4-дигидро-2H-1,4-бензоксазин-7-ил)амино]фенол, 4-[4-(диэтиламино)анилино]фенол, 4-(2,3-дигидро-1H-инден-5-иламино)фенол, 4-[(1-метил-1H-индазол-5-ил)амино]фенол, 4-[(1'-метил-1',2'-дигидроспиро[циклопропан-1,3'-индол]-5'-ил)амино]фенол, 4-[(1,3,3-триметил-2,3-дигидро-1H-индол-5-ил)амино]фенол, 4-[4-метокси-3-(трифторметил)анилино]фенол, 4-[4-(метилсульфанил)-3-(трифторметил)анилино]фенол, 2-фтор-4-[(1-метил-1Н-индол-5-ил)амино]фенол, 4-[(1-этил-1H-индол-5-ил)амино]фенол, 4-[(1-этил-2,3-дигидро-1H-индол-5-ил)амино]фенол, 4-[(1-изопропил-2,3-дигидро-1H-индол-5-ил)амино]фенол, 4-(бутиламино)фенол, 3-[(1-метил-1H-индол-5-ил)амино]-1-пропанол, 4-[(1-метил-1H-индол-5-ил)амино]-1-бутанол, 4-[(3-фтор-4-метилфенил)амино]фенол, 4-[(3-хлор-4-метилфенил)амино]фенол, 4-[(4-фторфенил)амино]фенол, 4-[(1-метил-1H-пирроло[2,3-6]пиридин-5-ил)амино]фенол, 4-[(4-фторфенил)амино]фенол, 4-[(2-фторфенил)амино]фенол, 4-[(3-фторфенил)амино]фенол, 4-[(2,4-дифторфенил)амино]фенол, 4-[(3,4-дифторфенил)амино]фенол, 3-[(4-гидроксифенил)амино]бензонитрил, 4-[(3-метоксифенил)амино]фенол, 4-[(3,5-дифторфенил)амино]фенол, 4-[(3-метилфенил)амино]фенол, 4-[(4-гидроксифенил)амино]бензонитрил, 4-[(3-хлорфенил)амино] фенол, 4-(пиримидин-2-иламино)фенол, 4-[(циклобутилметил)амино]фенол, 2-[(4-гидроксифенил)амино]бензонитрил, 4-{[(1-метил-1H-пиразол-4-ил)метил]амино}фенол, 4-[(циклопропилметил)амино]фенол, 4-{[(1-метил-1H- пиразол-3-ил)метил]амино}фенол, 4-(бут-2-ин-1-иламино)фенол, 4-(пиразин-2-иламино)фенол, 4-(пиридин-2-иламино)фенол, 4-(пиридазин-3-иламино)фенол, 4-(пиримидин-5-иламино)фенол, 4-(пиридин-3-иламино)фенол, 4-[(3,5-дифтор-4-метоксифенил)амино]фенол, 4-(пиридин-4-иламино)фенол, 4-[(3-фтор-4-метоксифенил)амино]фенол, 2-(фениламино)пиримидин-5-ол, 5-[(4-гидроксифенил)амино]-2-метоксибензонитрил и 4-{[3-(трифторметил)фенил]амино} фенол.
Гидроксильную(-ые) функцию(-и) вторичных аминов, перечисленных выше, защищают заранее с помощью подходящих защитных групп перед любым сочетанием с производным соединения формулы (VII) по кислотной группе, как определено в предшествующем общем способе.
Пример 1. 4-[{[3-(6-{[(3S)-3-(Морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат
Стадия A: Метил 3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-5,6,7,8-тетрагидро-1-индолизинкарбоксилат
К раствору 2 г соединения методики получения 1 (5.83 ммоль) в 20 мл дихлорметана при температуре окружающей среды добавляют 5.5 мл N,N,N-триэтиламина (6.96 ммоль), 2.12 г соединения методики получения 1' (6.96 ммоль), и затем 0.94 г гидроксибензотриазола (НОВТ) и 1.34 г 1-этил-3-(3'-диметиламинопропил)-карбодиимида (EDC) (6.96 ммоль). Реакционную смесь затем перемешивают при температуре окружающей среды в течение ночи, и затем ее выливают в насыщенный водный раствор хлорида аммония и экстрагируют этилацетатом. Затем органическую фазу сушат над сульфатом магния и затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент гептан/AcOEt). Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (500 МГц; дмсо-d6; 300 K): 7.2-6.9 (m, 4Н, ароматические Н); 7.04-7.03-7.00 (m, 1Н, ароматический Н); 6.85 (m, 1Н, ароматический Н); 6.35-6.26-6.06 (m, 1H, H тетрагидроиндолизин); 6.15-6.12 (m, 2H, H метилендиокси); 5.06-4.84 (m, 1Н, Н дигидроизохинолин); 4.86-4.17 (m, 2Н, Н дигидроизохинолин); 3.65-3.6-3.55 (m, 3Н, Н сложный метиловый эфир); 3.43-4.26 (m, 2Н, Н тетрагидроиндолизин); 3.58-3.5 (m, 4Н, Н морфолин); 2.37-3.05 (m, 4Н, 2Н дигидроизохинолин, 2Н тетрагидроиндолизин); 1.68-2.56 (m, 4Н, Н морфолин); 1.4-2.0 (m, 4Н, Н тетрагидроиндолизин)
ИК: ν:>С=O 1695 см-1 сложный эфир; ν:>С=O 1625 см-1 амид; ν:>С-O-С<1214-1176-1115 см-1; >СН-Ar 772-744 см-1
Стадия B: 3-[6-[(3S)-3-(Морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]-1,3-бензодиоксол-5-ил]-5,6,7,8-тетрагидро-1-индолизинкарбоксилат лития
К раствору 4.6 г соединения стадии A (8.26 ммоль) в 24 мл диоксана добавляют раствор гидроксида лития (675 мг, 16.1 ммоль). Загрузочную порцию помещают в микроволновую печь при мощности 140 Вт, 100°C на период 2 часа 30 минут. Реакционную смесь затем фильтруют и упаривают. Полученное таким образом твердое вещество сушат при 40°C в сушильном шкафу в присутствии P2O5.
1H ЯМР: δ (400 МГц; дмсо-d6; 353 K): 6.7-7.15 (неразрешенный пик, 6Н, ароматические Н); 6.21 (s, 1Н, ароматический Н); 6.03 (s, 2Н, Н метилендиокси); 4.0-5.0 (неразрешенный пик, 3Н дигидроизохинолин); 3.4-3.6 (неразрешенный пик, 3Н тетрагидроиндолизин, 3Н морфолин); 2.5-3.1 (неразрешенный пик, 4Н, 2Н тетрагидроиндолизин, 2Н морфолин); 1.5-2.4 (неразрешенный пик, 10Н морфолин)
ИК: ν:>С=O широкий 1567 см-1 ацетат; ν: 1236 см-1
Стадия C: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-зохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамид
К раствору 2.6 г соединения стадии B (4.73 ммоль) в 47 мл дихлорметана по каплям добавляют 1.2 мл оксалилхлорида (14.2 ммоль) при 0°C. Реакционную смесь перемешивают при температуре окружающей среды в течение 11 часов, и затем несколько раз упаривают совместно с дихлорметаном. Полученный таким образом продукт суспендируют в 37 мл дихлорметана, и затем добавляют к раствору 2.1 г соединения, полученного в методике получения 1” (7.1 ммоль) в 10 мл дихлорметана в присутствии 0.6 мл пиридина (7.1 ммоль). Загрузочную порцию перемешивают при температуре окружающей среды в течение ночи.
Реакционную смесь концентрируют и очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол). Указанный в заголовке продукт получают в виде пены.
1Н ЯМР: δ (500 МГц; дмсо-d6; 300 K): 6.9-7.3 (9Н, ароматические Н); 6.88 (2Н, ароматические Н); 6.72-6.87 (2Н, ароматические Н); 6.64 (2Н, ароматические Н); 6.13 (2Н метилендиокси); 5.05-4.74 (1Н дигидроизохинолин); 4.25-4.13 (2Н дигидроизохинолин); 3.44-3.7 (4Н морфолин); 3.62-3.52 (2Н тетрагидроиндолизин); 3.0-2.6 (4Н, 2Н тетрагидроиндолизин, 2Н дигидроизохинолин); 2.54-1.94 (6Н морфолин); 1.91-1.53 (4Н тетрагидроиндолизин); 0.92 (9Н трет-бутил); 0.17 (6Н диметил)
ИК: ν:>C=O: 1632 см-1; ν:>С-O-С<: 1237 см-1; ν: -Si-O-C-: 1035 см-1; -Si-C-: 910 см-1; >CH-Ar: 806 см-1
Стадия D: Гидрохлорид N-(4-гидроксифенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамида
К раствору 1.9 г соединения, полученного на стадии C (2.3 ммоль), в 4 мл метанола добавляют 0.646 г (11.5 ммоль) гидроксида калия, растворенного в 8 мл метанола. Загрузочную порцию перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь затем разбавляют дихлорметаном и последовательно промывают 1М раствором HCl, насыщенным водным раствором NaHCO3 и затем соляным раствором до тех пор, пока не будет достигнуто нейтральное значение pH. Затем органическую фазу сушат над сульфатом магния, фильтруют и упаривают. Полученный таким образом сырой продукт очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол). Твердое вещество затем растворяют в дихлорметане, и добавляют 2 мл 1М эфирной HCl. Загрузочную порцию перемешивают в течение 1 часа и затем упаривают досуха. Полученный таким образом гидрохлорид растворяют в смеси вода/ацетонитрил до тех пор, пока растворение не завершится, и затем лиофилизируют.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=69.11:68.95; %Н=5.8:5.46; %N=7.5:7.51; %Cl-=4.74:4.48
Оптическое вращение: (α) D20=+50.8° (c=9 мг/мл, МеОН)
Стадия E: 4-[{[3-(6-{[(3S)-3-(Морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил дибензилфосфат
К суспензии 82 мг гидрида натрия (2.06 ммоль) в 10 мл безводного ТГФ порциями и при 0°C добавляют 700 мг соединения стадии D. После перемешивания в течение 30 минут при 0°C и в течение 30 минут при температуре окружающей среды, при 0°C добавляют тетрабензилпирофосфат, и реакционную смесь перемешивают в течение ночи при температуре окружающей среды. После упаривания растворителя, сырой продукт реакции разбавляют дихлорметаном (30 мл), промывают насыщенным водным раствором NaHCO3 и затем соляным раствором. Затем органическую фазу сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН). Указанный в заголовке продукт затем получают в виде твердого вещества.
1Н ЯМР: δ (500 МГц; ДМСО-d6; 300 K): 7.34 (m, 10Н фенил); 7.30-6.71 (m, 15Н арил); 6.06 (s, 1Н метилендиокси); 5.30-4.97 (m, 1Н пиррол); 5.11 (m, 4Н бензил): 5.03-3.64 (m, 1Н, третичный С THIQ); 4.91-4.09 (m, 2Н, вторичный С THIQ); 3.99-3.48 (m, 2Н, вторичный С THIQ); 3.54-3.44 (m, 4Н морфолин); 2.89-2.65 (m, 3Н, вторичный С THIQ); 2.51-1.87 (m, 4Н, вторичный С THID); 2.36-1.85 (m, 2Н, вторичный С THIQ); 1.91-1.45 (m, 4Н, вторичный С THID)
Стадия F: 4-[{[3-(6-{[(3S)-3-(Морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат
50 мг Pd(OH)2 добавляют к раствору продукта, полученного на стадии Е, (505 мг; 0.52 ммоль) в метаноле (10 мл), и затем реакционную смесь помещают в атмосферу водорода (1 бар) на 5 часов. После отфильтровывания катализатора и концентрирования досуха, сырой продукт реакции растворяют в метаноле (5 мл) и обрабатывают 0.95 мл 1М раствора гидроксида натрия. Растворители затем упаривают, и сырой продукт реакции очищают с помощью хроматографии на фазе OASIS® (градиент ацетонитрил/H2O) с получением белого твердого вещества.

ИК: ν: -С=O: 1628 см-1; ν: С-О-С: 1234 см-1; ν: Р=O: 115 см-1; ν: Р-О: 985 см-1; ν: CH-Ar: 876 см-1
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: С43 Н41 N4 Na2 О9 Р
[М+Н]+ рассчитано: 835.2479
[М+Н]+ найдено: 835.2467
Пример 2. 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат
Стадия A: N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой продукта методики получения 1' на стадии A на соединение методики получения 2', при этом предполагается, что полученный таким образом продукт не подвергают стадии превращения в солевую форму в присутствии HCl в эфире.
Элементный микроанализ: (%, теоретическое значение:найдено)
%074.86:74.88; %Н=5.64:5.31; %N=6.72:6.78
Стадия B: 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях Е и F примера 1.
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C39H36N3O8P
[М+Н]+ рассчитано: 706.2313
[М+Н]+ найдено: 706.2324
Пример 3. 4-[(1-Метил-1H-индол-5-ил){[3-(2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 3-{5-хлор-2-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинил)карбонил]фенил}-1H-(4-гидроксифенил)-1H-(1-метил-1Н-индол-5-ил)-5,6,7,8-тетрагидро-1-индолизин карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 2, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 2”.

Оптическое вращение: (α) D20=+55.9° (c=7 мг/мл, МеОН)
Стадия B: 4-[(1-Метил-1Н-индол-5-ил){[3-(2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях Е и F примера 1.
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C45H44N5Na2O7P
[M-2Na+3H]+ рассчитано: 800.3208
[M-2Na+3H]+ найдено: 800.3211
Пример 4. 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)амино] фенил динатрийфосфат
Стадия A: Гидрохлорид N(4-гидроксифенил)-3-(6-{[(3R-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединений методик получения 1 и 1', используемых на стадии A, на соединения методик получения 3 и 2', и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 3”.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=69.14:70.09; %Н=4.81:4.55; %N=9.83:10.09; %С1-=4.98:3.26
Стадия B: 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)амино]фенил диэтилфосфат
К суспензии соединения, полученного на стадии A, (1.5 ммоль) в 10 мл безводного CH2Cl2 добавляют триэтиламин (0.42 мл; 3 ммоль), и затем, по каплям, диэтилцианофосфонат (0.24 мл; 1.65 ммоль) при температуре окружающей среды. После перемешивания в течение ночи при температуре окружающей среды, реакционную смесь разбавляют с помощью CH2Cl2, промывают насыщенным водным раствором NaHCO3 и затем соляным раствором. Затем органическую фазу сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН). Указанный в заголовке продукт затем получают в виде твердого вещества.
Стадия C: 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)амино]фенил динатрийфосфат
0.4 мл триметилсилилбромида (3 ммоль) добавляют по каплям при температуре окружающей среды к раствору полученного на стадии B продукта (0.78 ммоль) в CH2Cl2 (12 мл). Реакционную смесь перемешивают в течение 5 часов, и затем медленно добавляют раствор Na2CO3 (580 мг) в воде (4 мл) при 0°C. После перемешивания в течение 30 минут, реакционную смесь концентрируют досуха, разбавляют безводным метанолом (25 мл) и подвергают микрофильтрованию. Фильтрат сушат и очищают с помощью хроматографии на фазе OASIS® (градиент ацетонитрил/H2O).
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C45H44N5Na2O7P
[M-2Na+3H]+ рассчитано: 800.3211
[M-2Na+3H]+ найдено: 800.3201
Пример 5. 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)индолизин-1-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединений методик получения 1 и 1', используемых на стадии A, на соединения методик получения 3 и 2', и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 4”.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=69.24:69.12; %Н=4.74:4.23; %N=8.5:8.45; %Cl-=5.38:5.2
Стадия B: 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(пиридин-4-ил)амино]фенил диэтилфосфат
К суспензии 950 мг соединения, полученного на стадии A, (1.5 ммоль) в 10 мл безводного CH2Cl2 добавляют триэтиламин (0.42 мл; 3 ммоль), и затем, по каплям, диэтилцианофосфонат (0.24 мл; 1,65 ммоль) при температуре окружающей среды. После перемешивания в течение ночи при температуре окружающей среды, реакционную смесь разбавляют с помощью CH2Cl2, промывают насыщенным водным раствором NaHCO3 и затем соляным раствором. Затем органическую фазу сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН). Указанный в заголовке продукт затем получают в виде твердого вещества.
1Н ЯМР: δ (500 МГц; ДМСО-d6; 300K): 8.5-8. 0 (m, 5Н); 7.2-7.1 (m, 1H); 6.85-6.65 (m, 1Н); 7.3-6.8 (m, 10Н); 6.25-6.10 (bs, 1Н); 6.2 (bs, 2Н); 5.1-3.7 (6d, 2Н); 4.7-3.8 (m, 1Н); 4.15 (m, 4Н); 3.0-1.7 (m, 2Н); 1.25 (m, 6Н); 0.85-0.24 (m, 3Н)
Стадия С: 4-[{[3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат
0.4 мл триметилсилилбромида (3 ммоль) добавляют по каплям при температуре окружающей среды к раствору полученного на стадии B продукта (591 мг; 0.78 ммоль) в CH2Cl2 (12 мл). Реакционную смесь перемешивают в течение 5 часов, и затем медленно добавляют раствор Na2CO3 (580 мг) в воде (4 мл) при 0°C. После перемешивания в течение 30 минут, реакционную смесь концентрируют досуха, разбавляют безводным метанолом (25 мл) и подвергают микрофильтрованию. Фильтрат сушат и очищают с помощью хроматографии на фазе OASIS® (градиент ацетонитрил/Н2О).
1Н ЯМР: δ (500 МГц; D2O; 300K): 8.23-7.98 (m, 2Н пиридил); 7.01-6.97 (m, 2Н пиридил); 7.88-7.80 (m, 1Н индолизин); 7.18-6.57 (m, 13Н, ароматические Н, THIQ+арил+индолизин+фенол); 6.17-6.15 (m, 1Н индолизин): 5.96 (m, 2Н метилендиокси); 4.61-3.76 (m, 1Н, тернарный С THIQ); 4.16 (m, 2Н, вторичный С THIQ); 2.86-2.31 (m, 2Н, вторичный С THIQ); 0.94-0.76 (m, 3Н, первичный С THIQ)
ИК: ν: -С=O: 1620 см-1; ν: С-О-С: 1218 см-1; ν: Р=O: 1107 см-1 ν: Р-О: 981 см-1 ν: CH-Ar: 881-741 см-1
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C38H29N4Na2O8P
[M-2Na+3H]+ рассчитано: 703.1952
[M-2Na+3H]+ найдено: 703.1951
Пример 6. 4-[{[3-(6-{[(3S)-3-(Морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил дибензилфосфат
Методика соответствует протоколу, описанному на стадиях A-E примера 1.
1Н ЯМР: δ (500 МГц; ДМСО-d6; 300K): 7.34 (m, 10Н фенил); 7.30-6.71 (m, 15Н арил); 6.06 (s, 1Н метилендиокси); 5.30-4.97 (m, 1Н пиррол); 5.11 (m, 4Н бензил): 5.03-3.64 (m, 1Н, третичный С THIQ); 4.91-4.09 (m, 2Н, вторичный С THIQ); 3.99-3.48 (m, 2Н, вторичный С THIQ); 3.54-3.44 (m, 4Н морфолин); 2.89-2.65 (m, 3Н, вторичный С THIQ); 2.51-1.87 (m, 4Н, вторичный С THID); 2.36-1.85 (m, 2Н, вторичный С THIQ); 1.91-1.45 (m, 4Н, вторичный С THID)
Пример 7. Диэтил 4-[{[3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}(пиридин-4-ил)амино]фенил фосфат
Методика соответствует протоколу, описанному на стадиях A и B примера 5.
1Н ЯМР: δ (500 МГц; ДМСО-d6; 300K): 8.5-8. 0 (m, 5Н); 7.2-7.1 (m, 1Н); 6.85-6.65 (m, 1Н); 7.3-6.8 (m, 10Н); 6.25-6.10 (bs, 1Н); 6.2 (bs, 2Н); 5.1-3.7 (6d, 2Н); 4.7-3.8 (m, 1Н); 4.15 (m, 4Н); 3.0-1.7 (m, 2Н); 1.25 (m, 6Н); 0.85-0.24 (ш, 3Н)
Пример 8. Гидрохлорид 4-[{[3-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил дигидрофосфата
100 мг Pd(OH)2 добавляют к раствору полученного на стадии E примера 1 продукта (500 мг; 0.51 ммоль) в метаноле (10 мл), и затем реакционную смесь помещают в атмосферу водорода (1 бар) на 5 часов. После отфильтровывания катализатора и концентрирования досуха, сырой продукт реакции незамедлительно очищают с помощью хроматографии на фазе С18 (градиент ацетонитрил/Н2О+0.2% HCl) с получением твердого вещества.
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C43H43N4O9P
[М+Н]+ рассчитано: 791.2846
[М+Н]+ найдено: 791.2852
Пример 9. 4-[{[5-(5-Хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(4-гидроксифенил)-1,2-диметил-N-(пиридин-4-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединений методик получения 1 и 1', используемых на стадии A, на соединения методик получения 4 и 2', и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 4”.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=66.99:66.88; %Н=5.14:5.28; %N=8.93:8.87; %Cl-=5.65:4.98
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C35H32ClN4O3
[М+Н]+ рассчитано: 591.2157
[М+Н]+ найдено: 591.2178
Стадия B: 4-[{[5-(5-Хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях B и C примера 4.
Пример 10. 4-[{[1,2-Диметил-5-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1H-пиррол-3-ил]карбонил}(1-метил-1H-пирроло[2,3-6]пиридин-5-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид N-(4-гидроксифенил)-1,2-диметил-N-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 5, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 3”.
Элементный микроанализ: (%, найдено (теоретическое значение))
%C=66.41(66.62);%H=5.08(5.59);%N=10.85(10.84);%Cl-=4.68(4.57)
Стадия B: 4-[{[1,2-Диметил-5-(6-{[(3D)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1Н-пиррол-3-ил]карбонил}(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях B и C примера 4.
Пример 11. 4-[{[1,2-Диметил-5-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид N-(4-гидроксифенил)-1,2-диметил-N-(1-метил-1Н-пиразол-4-ил)-5-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 5, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 5”.
Элементный микроанализ: (%, найдено (теоретическое значение))
%C=64.25(64.59);%H=5.4(5.7);%N=11.41(11.59);%Cl-=4.93(4.89)
Стадия B: 4-[{[1,2-Диметил-5-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1Н-пиррол-3-ил]карбонил}(1-метил-1Н-пиразол-4-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях Е и F примера 1.
ИК (см-1): ν: С=O: 1628; ν: (фосфат; простой эфир): 1238, 1143,1113, 985; γ:>CH Ar: 740
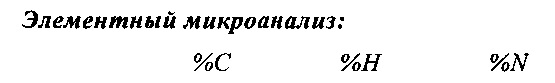
Масс-спектрометрия высокого разрешения (ESI+-/FIA/HR);
Эмпирическая формула: C39H39ClN6Na2O9P
[M-Na+H]+ рассчитано: 791.2565
[M-Na+H]+ найдено: 791.2564
Пример 12. 4-[{[1,2-Диметил-5-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1H-пиррол-3-ил]карбонил}(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид N-(4-гидроксифенил)-1,2-диметил-N-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединений методик получения 1 и 1', используемых на стадии A, на соединения методик получения 5 и 3', и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 3”.
Элементный микроанализ: (%, найдено(теоретическое значение))
%C=67.63(68.06);%H=5.27(5.95);%N=10.08(10.13);%Cl-=4.53(4.27)
Масс-спектрометрия высокого разрешения (ESI+): Эмпирическая формула: C35H32ClN4O3
[М+Н]+ рассчитано: 793.3708
[М+Н]+ найдено: 793.3704
Стадия B: 4-[{[1,2-Диметил-5-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1Н-пиррол-3- ил]карбонил}(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях Е и F примера 1.
Если не указано иное, соединения следующих примеров синтезируют в соответствии со способом примера 1, используя: (i) подходящую кислоту, полученную в соответствии с одной из методик получения 1-9, и (ii) подходящее тетрагидроизохинолиновое соединение, полученное в соответствии с одной из методик получения 1'-4' и, на стадии C: (iii) подходящий NHR3R4 амин (неполный перечень предлагается в методиках получения 1” - 8”).
Пример 13. 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(4-гидроксифенил)-1,2-диметил-N-(пиридин-4-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 4, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 4”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии 1М HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C39H38ClN5O4
[М+Н]+ рассчитано: 676.2685
[М+Н]+ найдено: 676.2684
Стадия B: 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях B и C примера 16.
31Р ЯМР (500 МГц, D2O) δ м.д.: -0.05
ИК (см-1): ν: С=O: 1631; ν: (фосфат; простой эфир): 1243, 1136, 1112, 982; γ:>СН Ar: 883, 745

Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C39H37ClN5Na2O7P
[M-Na+2H]+ рассчитано: 778.2168
[M-Na+2H]+ найдено: 778.2169
Пример 14. 4-[{[5-(5-Фтор-4-метокси-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат
Пример 15. 4-[{[5-(5-Фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(4-гидроксифенил)-1,2-диметил-N-(1-метил-1Н-пиразол-4-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 7, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 5”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Элементный микроанализ: (%, найдено (теоретическое значение))
%C=65.69(65.28); %H=5.38(5.77); %N=11.18(12.02); %Cl-=5.61(5.07)
Стадия B: 4-[{[5-(5-Фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях B и C примера 16.
31Р ЯМР (400/500 МГц, CD3OD) δ м.д.: -0.5
ИК (см-1): ν: С=O: 1628; ν: (фосфат; простой эфир): 1238, 1114, 983

Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C38H38FN6Na2O7P
[M-2Na+3H]+ рассчитано: 743.2752
[M-2Na+3H]+ найдено: 743.2760
Пример 16. 4-[{[1,2-Диметил-5-(7-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-2,3-дигидро-1,4-бензодиоксин-6-ил)-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид N-(4-гидроксифенил)-1,2-диметил-N-(1-метил-1Н-пиразол-4-ил)-5-(7-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-2,3-дигидро-1,4-бензодиоксин-6-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 8, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 5”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии 1М HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=64.99:64.67; %Н=5.86:5.67; %N=11.37:11.27; %Cl-=4.8:4.71
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C40H43N6O6
[М+Н]+ рассчитано: 703.3236
[М+Н]+ найдено: 703.3239
Стадия B: 4-[{[1,2-Диметил-5-(7-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-2,3-дигидро-1,4-бензодиоксин-6-ил)-1Н-пиррол-3-ил]карбонил}(1-метил-1Н-пиразол-4-ил)амино]фенил N,N,N',N'-тетраметилфосфородиамидат
К раствору 125 мг соединения стадии A (0.18 ммоль) в дихлорметане (6 мл) добавляют 55 мкл диазабицикло[5.4.0]ундец-7-ена (DBU; 0,36 ммоль), и затем 33 мкл бисдиметиламинофосфорилхлорида (0.19 ммоль) и 2 мг диметиламино-4-пиридина (0.02 ммоль). Реакционную смесь перемешивают в течение 15 часов, разбавляют дихлорметаном и затем насыщенным водным раствором карбоната натрия. Водную фазу экстрагируют дихлорметаном; органические фазы затем объединяют, промывают водой и соляным раствором и сушат над сульфатом магния. После упаривания растворителей, сырой продукт реакции непосредственно используют на следующей стадии.
Стадия C: 4-[{[1,2-Диметил-5-(7-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-2,3-дигидро-1,4-бензодиоксин-6-ил)-1Н-пиррол-3-ил]карбонил}(1-метил-1Н-пиразол-4-ил)амино]фенил динатрийфосфат
4 мл трифторуксусной кислоты добавляют по каплям к раствору 125 мг соединения стадии B (0.15 ммоль) в 1:1 смеси ацетонитрила и воды (5 мл). После перемешивания в течение 20 часов при температуре окружающей среды, реакционную смесь упаривают досуха, поддерживая температуру водяной бани ниже 40°C, и затем остаток обрабатывают раствором карбоната натрия (95 мг; 0.9 ммоль) в воде (4 мл). После перемешивания в течение 2 часов при температуре окружающей среды, реакционную смесь упаривают досуха, и затем добавляют 6 мл безводного этанола. Твердое вещество отфильтровывают, и фильтрат концентрируют досуха, и затем очищают на фазе OASIS® (градиент ацетонитрил/вода).
31Р ЯМР (500 МГц, D2O) δ м.д.: 0.9
ИК (см-1): ν: С=O: 1623; ν: (фосфат; простой эфир): 1235, 1162, 1115, 1065, 985; γ:>СН Ar:745
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C40H41N6Na2O9P
[M-2Na+3H]+ рассчитано: 783.2902
[M-2Na+3H]+ найдено: 783.2907
Пример 17. 5-[{[5-(5-Фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]пиримидин-2-ил динатрийфосфат
Пример 18. 5-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]пиримидин-2-ил динатрийфосфат
Пример 19. 4-({[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}[1-(тридейтерометил)-1H-пиразол-4-ил]амино)фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(4-гидроксифенил)-1,2-диметил-N-[1-(тридеитерометил)-1Н-пиразол-4-ил]-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 4, и, с другой стороны, соединения методики получения 1”, используемого на стадии C, на соединение методики получения 6”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии 1М HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=63.51:63.41; %Н=5.63:5.42; %N=11.69:11.61; %Cl-=4.93:4.85
Масс-спектрометрия высокого разрешения (ESI+-/FIA/HR, ESI-/FIA):
Эмпирическая формула: C38H36ClD3N6O4
[М+Н]+ рассчитано: 682.2982
[М+Н]+ найдено: 682.2986
Стадия B: 4-({[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}[1-(тридейтерометил)-1Н-пиразол-4-ил]амино)фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
31Р ЯМР (500 МГц, D2O) δ м.д.: 4.8
ИК (см-1): ν: С=O: 1626; ν: (фосфат; простой эфир): 1243, 1141, 1115, 982; γ:>СН Ar:880, 831
Масс-спектрометрия высокого разрешения (ESI/FIA/HR и MC/MQ):
Эмпирическая формула: C38H35ClD3N6Na2O7P
[М+Н]+ рассчитано: 806.2285
[М+Н]+ найдено: 806.2280
Пример 20. 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1,2-диметил-1H-пиррол-3-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(5-циано-1,2-диметил-1Н-пиррол-3-ил)-N-(4-гидроксифенил)-1,2-диметил-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии A, на соединение методики получения 4, и, с другой стороны, соединения методики получения 1”, используемого на стадии С, на соединение методики получения 7”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии 1М HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
1Н ЯМР (500 МГц, дмсо-d6) δ м.д.: 11.2 (bs, 1H), 9.39 (bs, 1H), 7.83 (d, 1Н), 7.54 (d, 1Н), 7.33 (s, 1Н), 7.14 (m, 2Н), 7 (m, 2Н), 6.8 (d, 2Н), 6.62 (d, 2Н), 6.57 (bs, 1Н), 5.26 (s, 1Н), 5.26 (m, 1Н), 4.64/4.03 (АВ, 2Н), 4.01/3.92 (2m, 4Н), 3.75/3.43/3.15/3.02 (4m, 4Н), 3.59 (s, 3Н), 3.3/3.15 (2m, 2Н), 2.97 (s, 3Н), 2.69/2.52 (dd+d, 2Н), 2.06 (s, 3Н), 1.91 (s, 3Н)
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=65.34:65.50; %Н=5.62:5.15; %N=11.15:10.84%Cl-=4.70:4.44
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C41H41ClN6O4
[М+Н]+ рассчитано: 717.2952
[М+Н]+ найдено: 717.2951
Стадия B: 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}(5-циано-1,2-диметил-1Н-пиррол-3-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
31Р ЯМР (500 МГц, дмсо-d6) δ м.д.: 3.7
ИК (см-1): ν: -CN: 2210 см-1; ν: С=O: 1623; ν: (фосфат; простой эфир): 1227, 1133, 1110, 982; γ:>СН Ar:884-741

Масс-спектрометрия высокого разрешения (ESI+-/FIA/HR):
Эмпирическая формула: C41H40ClN6Na2O7P
[M-2Na+H]+ рассчитано: 797.2614
[M-2Na+H]+ найдено: 797.2618
Пример 21. 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1-метил-1H-пиррол-3-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N(5-циано-1-метил-1Н-пиррол-3-ил)-N-(4-гидроксифенил)-1,2-диметил-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии А, на соединение методики получения 4, и, с другой стороны, соединения методики получения 1”, используемого на стадии С, на соединение методики получения 8”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии 1М HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=64.95:65.09; %Н=5.45:5.20; %N=11.36:11.26; %Cl-=4.79:4.62
Масс-спектрометрия высокого разрешения (ESI/+):
Эмпирическая формула: C40H39ClN6O4
[М+Н]+ рассчитано: 703.2794
[М+Н]+ найдено: 703.2789
Стадия В: 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}(5-циано-1-метил-1Н-пиррол-3-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
31Р ЯМР (500 МГц, дмсо-d6) δ м.д.: 4.5
ИК (см-1): ν: -CN: 2215 см-1; ν: С=O 1626; ν: (фосфат; простой эфир): 1227, 1141, 1112, 982; γ>СН Ar: 826-742
Масс-спектрометрия высокого разрешения (ESI+-/FIA/HR):
Эмпирическая формула: C40H39ClN6Na2O7P
[M-2Na+3H]+ рассчитано: 783.2457
[M-2Na+3H]+ найдено: 783.2462
Пример 22. 4-[{[3-(6-{[(3R)-3-(Морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид N(4-гидроксифенил)-3-{6-[((3R)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой соединения методики получения 1', используемого на стадии А, на соединение методики получения 4'. Твердое вещество затем растворяют в дихлорметане, и добавляют 2 мл 1М HCl в эфире. Загрузочную порцию перемешивают в течение 1 часа и затем упаривают досуха. Полученный таким образом гидрохлорид растворяют в смеси вода/ацетонитрил до тех пор, пока растворение не завершится, и затем лиофилизируют.

Оптическое вращение: (α) D20=- 45.1° (с=9 мг/мл, МеОН)
Стадия В: 4-[{[3-(6-{[(3R)-3-(Морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
31Р ЯМР (400/500 МГц, дмсо-d6) δ м.д.: 2.6

Масс-спектрометрия высокого разрешения (ESI+-/FIA/HR):
Эмпирическая формула: C43H41N4Na2O9P
[M-2Na+H]+ рассчитано: 791.2840
[M-2Na+H]+ найдено: 791.2845
Пример 23. 4-[(1-Метил-2,3-дигидро-1H-пирроло[2,3-b]пиридин-5-ил){[3-(2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-4-[2-оксо-2-(пиперидин-1-ил)этокси]фенил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}амино]фенил динатрийфосфат
Стадия A: Метил 3-[4-бензилокси-2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]фенил]-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К раствору 14.19 г (35.0 ммоль) соединения, полученного в методике получения 9, в 200 мл диметилформамида последовательно добавляют 4-[[(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]метил]морфолин (методика получения 1'; 8.13 г; 35.0 ммоль), гидроксибензотриазол (6.15 г; 45,5 ммоль), гидрохлорид 1-этил-3-(3'-диметиламинопропил)-карбодиимида (6.70 г; 45.5 ммоль) и триэтиламин (21.95 мл; 0.16 моль). Загрузочную порцию затем перемешивают в течение ночи при температуре окружающей среды. Реакционную смесь затем выливают в 400 мл этилацетата и затем последовательно промывают насыщенным водным раствором NaHCO3, водой и соляным раствором. Объединенные водные фазы экстрагируют этилацетатом. Получающиеся органические фазы сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный продукт очищают с помощью хроматографии на силикагеле с обеспечением указанного в заголовке соединения.
1H ЯМР (500 МГц, дмсо-d6, 300 К) δ м.д.: 7.5-7.3 (m, 5Н), 7.38 (d, 1Н), 7.2-6.9 (m, 4Н), 7.15 (dd, 1Н), 6.9 (d, 1Н), 6.35/6.25/6.08 (3*s, 1Н), 5.21/5.12 (3*s, 2Н), 5.09/4.85/3.7 (3*m, 1Н), 4.9-3.8 (8*d, 2Н), 4.2-3.4 (m, 2Н), 3.65/3.6/3.55 (3*s, 3Н), 3.6-3.4 (m, 4Н), 3-2.4 (m, 2Н), 2.9-1.8 (6*dd, 2Н), 2.5-1.95 (4*m, 4Н), 2.35-1.7 (6*m, 2Н), 2-1.45 (6*m, 4Н)
Стадия B: 3-[4-Бензилокси-2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]фенил]-5,6,7,8-тетрагидроиндолизин-1-карбоновая кислота
40.1 мл 1М водного раствора LiOH добавляют к раствору 12.7 г (20 ммоль) соединения, полученного на стадии выше, в 40 мл диоксана. Загрузочную порцию нагревают при 100°C в течение ночи. Реакционную смесь выливают в воду и затем экстрагируют этиловым эфиром. Эфирную фазу экстрагируют еще раз водой. Объединенные водные фазы подкисляют до pH 4 путем добавления порошкообразной лимонной кислоты, и затем экстрагируют дихлорметаном. Дихлорметановую фазу промывают соляным раствором, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде безе.
1Н ЯМР (500 МГц, дмсо-d6, 300K) δ м.д.: 11.35 (bs, 1Н), 7.5-7.3 (m, 5Н), 7.38 (m, 1Н), 7.2-6.9 (m, 4Н), 7.15 (m, 1Н), 6.9 (m, 1Н), 6.31/6.25/6.1 (3*s, 1Н), 5.22/5.2/5.15 (3*s, 2Н), 5.1/4.82/3.7 (3*m, 1Н), 4.85-3.8 (8*d, 2Н), 4.2-3.4 (m, 2Н), 3.6-3.45 (m, 4Н), 3-2.3 (m, 2Н), 2.9-1.8 (m, 2Н), 2.5-1.9 (6*m, 4Н), 2.35-1.8 (6*m, 2Н), 1.9-1.3 (m, 4Н)
Стадия С: 3-[4-Бензилокси-2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]фенил]-N[4-[трет-бутил(диметил)силил]оксифенил]-N-(1-метилпирроло[2,3-b]пиридин-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Кислоту, полученную на стадии B, (9 г, 11.8 ммоль) растворяют в 90 мл 1,2-дихлорэтана. К раствору добавляют 1.9 мл 1-хлор-N,N,2-триметилпропениламина (14 ммоль). После перемешивания в течение 3 часов при температуре окружающей среды добавляют 90 мл толуола и 4.62 г N-[4-[трет-бутил(диметил)силил]оксифенил]-1-метил-1H-пирроло[2,3-b]пиридин-5-амина (методика получения 3”, 13 ммоль). Реакционную смесь нагревают при 110°C в течение 20 часов. После возвращения к температуре окружающей среды, реакционную смесь промывают соляным раствором, сушат над Na2SO4, фильтруют и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле (градиент дихлорметан/этанол) с получением ожидаемого продукта.
1Н ЯМР (500 МГц, дмсо-d6, 300K) δ м.д.: 7.95/7.8/7.75 (3*d, 1Н), 7.68/7.65/7.4 (3*d, 1Н), 7.4/7.3 (2*d, 1Н), 7.25-6.8 (m, 9Н), 7.05/6.9 (2*m, 1Н), 7-6.6 (3*bd, 2Н), 6.9 (m, 1Н), 6.75-6.45 (3*bd, 2Н), 6.7 (m, 1Н), 6.3 (2*d, 1Н), 5.15-4.95 (m, 2Н), 5.15/5.1/4.8 (3*s, 1Н), 4.95/4.6/3.5 (3*m, 1Н), 4.9-3.7 (8*d, 2Н), 3.8-3.3 (3*m, 2Н), 3.75/3.7/3.5 (3*s, 3Н), 3.45/3.3 (2*m, 4Н), 3-2.5 (3*m, 2Н), 3-2.3 (m, 2Н), 2.4-1.75 (5*m, 4Н), 2.25-1.7 (6*m, 2Н), 1.75-1.3 (m, 4Н), 0.7 (bs, 9Н), 0.1 (m, 6Н)
Стадия D: N[4-[трет-Бутил(диметил)силил]оксифенил]-3-[4-гидрокси-2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]фенил]-N-(1-метилпирроло[2,3-b]пиридин-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К раствору 8.88 г (8.4 ммоль) соединения, полученного на стадии С, в 100 мл этанола, при барботировании через него аргона добавляют 0.9 г Pd/C 10%. Реакционную смесь помещают под давление водорода 1.2 бар при температуре окружающей среды на 15 часов. Катализатор отфильтровывают, и растворитель упаривают при пониженном давлении с обеспечением указанного в заголовке соединения.
1Н ЯМР (500 МГц, дмсо-d6, 300K) 5 м.д.: 8.06/7.92/7.87 (3*d, 1Н), 7.75/7.5/7.39 (3*d, 1Н), 7.5 (m, 1Н), 7.28-6.9 (m, 5Н), 6.87/6.7 (2*m, 2Н), 6.76 (m, 1Н), 6.75/6.67/6.62 (3*m, 2Н), 6.67/6.46 (m, 1Н), 6.4/6.36 (2*m, 1Н), 5.19/5.13/4.9 (3*bs, 1Н), 5.06/4.7/3.6 (3*m, 1Н), 4.97/4.2/4.15/4.07 (4*m, 2Н), 4.87/4.81 (bs, 1H), 3.86/3.56/3.39 (3*m, 2Н), 3.78/3.57 (2*m, 3Н), 3.59/3.44 (2*m, 4Н), 2.96-2.61 (2*m, 2Н), 2.88/2.6 (2*m, 2Н), 2.59-1.81 (m, 6Н), 1.87-1.42 (m, 4Н), 0.89 (s, 9Н), 0.12 (m, 6Н)
Стадия E: N-[4-[трет-Бутил(диметил)силил]оксифенил]-N-(1-метилпирроло[2,3-b]пиридин-5-ил)-3-[2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]-4-[2-оксо-2-(1-пиперидил)этокси]фенил]-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Соединение стадии D (3.0 г, 2.9 ммоль) растворяют в 100 мл толуола. К раствору добавляют 1.53 г (5.8 ммоль) трифенилфосфина и 0.62 г (4.3 ммоль) 2-гидрокси-1-(1-пиперидил)этанона. Смесь нагревают до 50°C, и затем добавляют 1.01 г (4.3 ммоль) ди-трет-бутилазодикарбоксилата. Реакционную смесь перемешивают при 50°C в течение 1 часа, и затем температуре позволяют вернуться к температуре окружающей среды перед добавлением 1 мл трифторуксусной кислоты. После перемешивания в течение ночи при температуре окружающей среды, смесь последовательно промывают водой, насыщенным раствором NaHCO3 и соляным раствором. Объединенные водные фазы экстрагируют этилацетатом. Получающиеся органические фазы сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный сырой продукт очищают с помощью хроматографии на силикагеле (дихлорметан/этанол 98/2) с получением ожидаемого соединения.
1Н ЯМР (500 МГц, дмсо-d6, 300K) δ м.д.: 8.06/7.92/7.87 (3*d, 1Н), 7.75/7.51/7.4 (3*d, 1Н), 7.49 (2*d, 1Н), 7.29-6.89 (m, 5Н), 6.93 (m, 1Н), 6.88/6.7 (m, 2Н), 6.75/6.67 (m, 1Н), 6.75/6.68/6.59 (3*m, 2Н), 6.4/6.36 (2*m, 1Н), 5.2/5.16/4.92 (3*m, 1Н), 5.06/4.69/3.58 (3*m, 1Н), 4.97/4.25/4.16/4.03 (4*d, 2Н), 4.89/4.81 (2*m, 2Н), 3.79/3.59 (2*m, 3Н), 3.59/3.43/3.4 (3*m, 6Н), 3.58/3.43 (2*m, 4Н), 3.03-2.61 (m, 2Н), 2.97-2.65 (m, 2Н), 2.57-1.74 (m, 6Н), 1.89-1.3 (m, 10Н), 0.89 (2bs, 9Н), 0.11 (m, 6Н)
Стадия F: N(4-Гидроксифенил)-N-(1-метилпирроло[2,3-b]пиридин-5-ил)-3-[2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]-4-[2-оксо-2-(1-пиперидил)этокси]фенил]-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
1М раствор фторида тетрабутиламмония (3.14 мл, 3 ммоль) в тетрагидрофуране добавляют к раствору соединения, полученного на стадии Е, (2.92 г, 2.9 ммоль) в 30 мл тетрагидрофурана при температуре окружающей среды. После перемешивания в течение 5 минут, реакционную смесь выливают в 50/50 смесь этилацетата и насыщенного водного раствора NaHCO3. Отделенную органическую фазу промывают водой, и затем соляным раствором. Объединенные водные фазы экстрагируют этилацетатом. Получающиеся органические фазы сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный сырой продукт очищают с помощью хроматографии на силикагеле (градиент дихлорметан/этанол/аммиак) с получением указанного в заголовке соединения.
1H ЯМР (500 МГц, дмсо-d6, 300K) δ м.д.: 9.4 (m, ОН), 8.1-7.8 (3*d, 1Н), 7.7-7.3 (2*m, 1Н), 7.5/7.4 (2*m, 1Н), 7.3-6.9 (m, 4Н), 7.2 (m, 1Н), 6.9 (m, 1Н), 6.8-6.5 (m, 2Н), 6.7-6.5 (m, 2Н), 6.7 (m, 1Н), 6.4 (m, 1Н), 5.3-5 (m, 1Н), 5.1/4.7/3.6 (3*m, 1Н), 5-3.6 (m, 2Н), 5-3.6 (m, 2Н), 4.8 (m, 2Н), 3.8-3.6 (m, 3Н), 3.6/3.4 (m, 2Н), 3.4 (m, 6Н), 3.1-2.5 (m, 2Н), 2.9-1.9 (m, 2Н), 2.5-1.7 (m, 4Н), 1.8-1.4 (m, 6Н), 1.6-1.3 (m, 4Н)
Стадия G: N-(4-Гидроксифенил)-N-(1-метил-2,3-дигидропирроло[2,3-b]пиридин-5-ил)-3-[2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]-4-[2-оксо-2-(1-пиперидил)этокси]фенил]-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
0.71 г (11 ммоль) цианоборогидрида натрия добавляют к раствору соединения, полученного на стадии F, (2.0 г, 2.2 ммоль) в 20 мл уксусной кислоты. После перемешивания в течение 14 часов при температуре окружающей среды вновь добавляют 0.36 г (5.5 ммоль) цианоборогидрида натрия, и затем реакционную смесь нагревают при 50°C в течение 3 часов перед вторым добавление 0.1 экв. цианоборогидрида натрия для завершения реакции в течение 30 минут при 50°C. Уксусную кислоту упаривают при пониженном давлении, и остаток затем переносят в дихлорметан, и промывают насыщенным водным раствором NaHCO3, водой и соляным раствором. Объединенные водные фазы экстрагируют дихлорметаном. Получающиеся органические фазы сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный сырой продукт очищают с помощью хроматографии на силикагеле (градиент дихлорметан/этанол/аммиак) с получением указанного в заголовке соединения в виде безе.
1H ЯМР (500 МГц, дмсо-d6, 300K) δ м.д.: 9.3 (bs, 1Н), 7.5/7.4/7.3 (3*m, 1Н), 7.2/6.7 (2*m, 1Н), 7.2 (m, 1Н), 7.1-6.8 (m, 4Н), 6.9/6.7 (m, 1Н), 6.9 (m, 1Н), 6.8-6.5 (m, 2Н), 6.7-6.5 (m, 2Н), 5.3-5.1 (2*d, 1Н), 5.1/4.7/3.6 (3*m, 1Н), 4.9/4.2-3.5 (2*m, 1Н), 4.9/4.2-3.5 (2*, 1Н), 4.9-4.8 (m, 2Н), 3.6/3.4 (2*m, 4Н), 3.4/3.3 (m, 2Н), 3.4 (m, 6Н), 3.1-2.5 (m, 4Н), 3-2.4 (m, 2Н), 2.8/2.6 (m, 3Н), 2.6-1.7 (m, 6Н), 1.9-1.3 (m, 10Н)
Стадия H: Гидрохлорид N-(4-гидроксифенил)-N-(1-метил-2,3-дигидропирроло[2,3-b]пиридин-5-ил)-3-[2-[(3S)-3-(морфолинометил)-3,4-дигидро-1Н-изохинолин-2-карбонил]-4-[2-оксо-2-(1-пиперидил)этокси]фенил]-5,6,7,8-тетрагидро индол из ин-1-карбоксамида
Основание, полученное на стадии G, (0.60 г, 0.69 ммоль) растворяют в ацетонитриле, и затем превращают в солевую форму, используя 0.7 мл (0.7 ммоль) 1 н. раствор HCl. Раствор фильтруют, замораживают и затем лиофилизируют с обеспечением указанного в заголовке соединения в виде порошка.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=68.02:68.06; %Н=6.49:6.21; %N=10.89:10.87; %Cl=4.14:3.94
Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C51H57N7O6
[М+Н]+ рассчитано: 864.4445
[М+Н]+ найдено: 864.4443
Стадия I: 4-[(1-Метил-2,3-дигидро-1Н-пирроло[2,3-b]пиридин-5-ил){[3-(2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-4-[2-оксо-2-(пиперидин-1-ил)этокси]фенил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
ИК (см-1): ν: С=O: 1625; ν: (фосфат; простой эфир): 1229, 1138, 1115, 982; γ:>СН Ar: 880-748-745

Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C51H56N7Na2O9P
[M-2Na+3H]+ рассчитано: 944.4106
[M-2Na+3H]+ найдено: 944.4116
Пример 24. 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат
Стадия A: Гидрохлорид 5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(4-гидроксифенил)-1,2-диметил-N-(1-метил-1Н-пиразол-4-ил)-1Н-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии А, на соединение методики получения 4, и, с другой стороны, соединения методики получения 1”, используемого на стадии С, на соединение методики получения 5”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Элементный микроанализ: (%, теоретическое значение:найдено)
%С=63.77:62.83; %Н=5.63:5.83; %N=11.74:11.29; %Cl-=4.95:5.42
Стадия В: 4-[{[5-(5-Хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-ил]карбонил}(1-метил-1Н-пиразол-4-ил)амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
ИК (см-1): ν: С=O: 1625; ν: (фосфат; простой эфир): 1241, 1146, 1112, 983

Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C38H38ClN6Na2O7P
[M-2Na+3H]+ рассчитано: 759.2457
[M-2Na+3H]+ найдено: 759.2465
Пример 25. 4-[(5-Циано-1,2-диметил-1H-пиррол-3-ил){[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}амино]фенил динатрийфосфат
Стадия А: Гидрохлорид N-(5-циано-1,2-диметил-1H-пиррол-3-ил)-5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-1,2-диметил-1H-пиррол-3-карбоксамида
Методика соответствует протоколу, аналогичному описанному на стадиях A-D примера 1, с заменой, с одной стороны, соединения методики получения 1, используемого на стадии А, на соединение методики получения 7, и, с другой стороны, соединения методики получения 1”, используемого на стадии С, на соединение методики получения 7”. Полученный продукт в заключение подвергают стадии превращения в солевую форму в присутствии HCl в эфире. После фильтрования и лиофилизации в смеси ацетонитрил/вода, получают ожидаемое соединение.
Масс-спектрометрия высокого разрешения (ESI/FIA/HR и МС/МС):
Эмпирическая формула: C41H41FN6O4
[М+Н]+ рассчитано: 701.3246
[М+Н]+ найдено: 701.3282
Стадия В: 4-[(5-Циано-1,2-диметил-1H-пиррол-3-ил){[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}амино]фенил динатрийфосфат
Методика соответствует протоколу, аналогичному описанному на стадиях В и С примера 16.
31Р ЯМР (400/500 МГц, CD3OD) δ м.д.: -0.5
ИК (см-1): ν: -CN: 2211 см-1; ν: С=O: 1629; ν: (фосфат; простой эфир): 1236, 1114, 984

Масс-спектрометрия высокого разрешения (ESI+):
Эмпирическая формула: C41H40FN6Na2O7P
[M-2Na+3H]+ рассчитано: 781.2909
[M-2Na+3H]+ найдено: 781.2898
ФАРМАКОЛОГИЧЕСКИЕ И ФАРМАКОКИНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ
В целях ясности, и где-либо далее, соединения формулы (1') будут называться "лекарственным средством примера х", из которого они были получены. Например, N-(4-гидроксифенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1H)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизинкарбоксамид будет называться "лекарственное средство примера 1".
ПРИМЕР А1: Индукция активности каспазы in vivo соединениями формулы (I')
Способность соединений формулы (I') активировать каспазу 3 оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11.
1×107 клеток RS4;11 трансплантируют под кожу иммуносупрессивным мышам (штамм SCID). Через 25-30 дней после трансплантации, животные перорально получают лечение различными соединениями. Через шестнадцать часов после введения, опухолевые массы извлекают и лизируют, и в опухолевых лизатах измеряют активность каспазы 3.
Ферментативное измерение осуществляют путем анализа появления флуоригенного продукта расщепления (DEVD-азная активность, Promega). Оно выражается в виде фактора активации, который равняется соотношению между двумя активностями каспазы: активности у мышей, получающих лечение, деленной на активность у контрольных мышей.
Исследовали N-(4-гидроксифенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1H)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамид (также называемый лекарственным средством примера 1). В дозе 100 мг/кг п/о, in vivo фактор активации каспазы составляет 29.3.
Полученные результаты показывают, что соединения формулы (1') способны индуцировать апоптоз in vivo в опухолевых клетках RS4;11.
ПРИМЕР А2: Количественное определение расщепленной формы каспазы 3 in vivo, обусловленной соединениями формулы (I').
Способность соединений формулы (1') активировать каспазу 3 оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11.
1×107 клеток RS4;11 трансплантируют под кожу иммуносупрессивным мышам (штамм SCID). Через 25-30 дней после трансплантации, животные перорально получают лечение различными соединениями. После лечения, опухолевые массы извлекают и лизируют, и количественно определяют расщепленную (активированную) форму каспазы 3 в опухолевых лизатах.
Количественное определение осуществляют с использованием "Meso Scale Discovery (MSD) ELISA platform" исследования, которое предназначено для проведения специфического анализа на расщепленную форму каспазы 3. Оно выражается в виде фактора активации, который равняется соотношению между количеством расщепленной каспазы 3 у мышей, получающих лечение, и количеством расщепленной каспазы 3 у контрольных мышей.
Результаты показывают, что соединения формулы (I') способны индуцировать апоптоз in vivo в опухолевых клетках RS4;11.
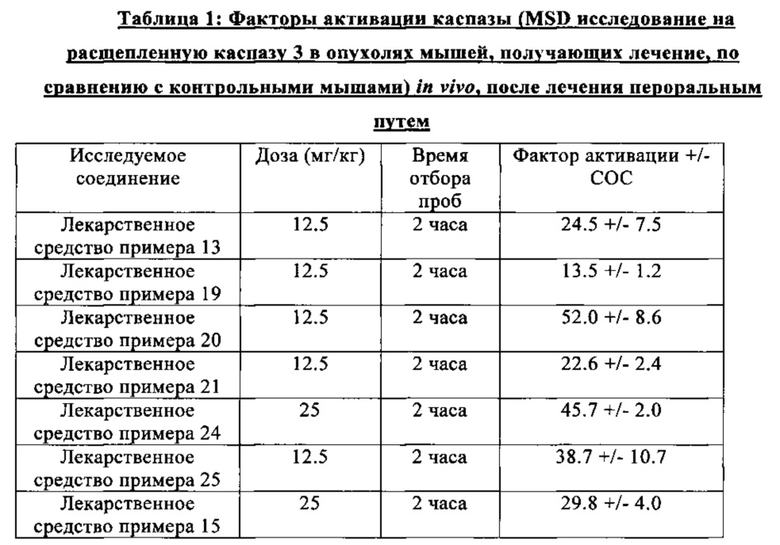
ПРИМЕР A3: Количественное определение расщепленной формы каспазы 3 in vivo, обусловленной соединениями формулы (I).
Способность соединений формулы (I) активировать каспазу 3 оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11 в соответствии с протоколом, приведенным в примере А2.
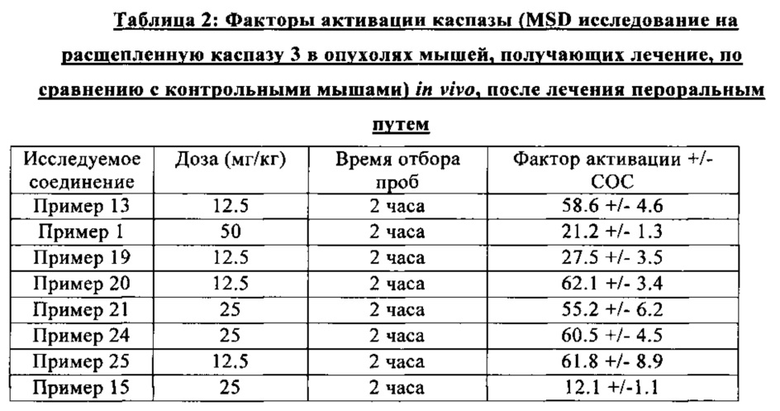
ПРИМЕР В: Растворимость соединений формулы (I)
Растворимость соединений формулы (I) в воде измеряли и сравнивали с растворимостью соединений формулы (I').
Более конкретно, 4-[{[3-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат (также называемый соединением примера 1) исследовали и сравнивали с N-(4-гидроксифенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамидом (также называемым лекарственным средством примера 1).
Растворимость соединения примера 1 в воде превышает или равна 10 мг/мл (12.6 мМ), тогда как сопряженного лекарственного средства только 40 мкг/мл (56.2 мкМ). Растворимости соединений также измеряли в среде, забуференной до физиологического значения pH (см. также таблицу 3).


Результаты показывают, что соединения формулы (I) гораздо лучше растворимы, чем соединения формулы (I'). Только соединения формулы (I) демонстрируют растворимости выше или равные 100 мкМ.
ПРИМЕР С: In vivo превращение соединений формулы (I)
Фармакокинетический профиль фосфатных соединений формулы (I) оценивают в липидном составе и в водном растворе на самках SCID мышей. Его сравнивают с фармакокинетическим профилем соединений формулы (I') в липидном составе. Более конкретно, 4-[{[3-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат (также называемый соединением примера 1) исследовали и сравнивали с N-(4-гидроксифенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1Н)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамидом (также называемым лекарственным средством примера 1).
Липидный состав соединения примера 1
Соединение примера 1 приготовляют в смеси безводный этанол/полиэтиленгликоль 300/вода (10/40/50, об./об./об.), предназначенной для введения п/о путем. Исследование осуществляют на 2 группах SCID мышей, которым соединение примера 1 вводят при следующих условиях:
- группа 1: 3 мг/кг п/о (желудочный зонд, 10 мл/кг),
- группа 2: 25 мг/кг п/о (желудочный зонд, 10 мл/кг).
Пробы крови отбирают в следующие моменты времени (3 пробы на животное и 3 животных на каждый момент времени): через 0.25 ч, 0.5 ч, 1 ч, 2 ч, 6 ч и 24 ч после перорального введения.
Водный состав соединения примера 1
Соединение примера 1 также вводят SCID мышам пероральным путем в водной среде при следующих условиях:
- группа 1: 30 мг/кг п/о, растворенных в 1 мМ растворе карбоната натрия (желудочный зонд, 10 мл/кг),
- группа 2: 100 мг/кг п/о в воде (желудочный зонд, 10 мл/кг).
Пробы крови отбирают в следующие моменты времени (3 животных на каждый момент времени и 1 образец на животное): через 0.25 ч, 0.5 ч, 1 ч, 2 ч, 4 ч, 6 ч и 24 ч после перорального введения.
В случае всех составов соединения примера 1, собранную таким образом кровь центрифугируют, и плазму переносят в пробирки, содержащие 1М соляную кислоту. Концентрации в плазме фосфатного соединения (пролекарства) и его гидроксилированного гомолога (лекарственного средства) определяют одновременно, используя метод жидкостной хроматографии, сопряженной с масс-спектрометрическим детектированием (ТТХ-ЖХ-МС/МС). Граница детектирования для обоих соединений составляет 0.5 нг/мл.
Липидный состав лекарственного средства из соединения примера 1 Лекарственное средство примера 1 приготовляют в смеси безводный этанол/полиэтиленгликоль 300/вода (10/40/50, об./об./об.), предназначенной для введения п/о путем. Исследование осуществляют на нескольких группах SCID мышей, которым вводят лекарственное средство примера 1 при следующих условиях:
- группа 1: 3 мг/кг п/о (желудочный зонд, 10 мл/кг),
- группа 2: 30 мг/кг п/о (желудочный зонд, 10 мл/кг),
- группа 3: 25 мг/кг п/о (желудочный зонд, 10 мл/кг),
- группа 4: 100 мг/кг п/о (желудочный зонд, 10 мл/кг).
Пробы крови отбирают в следующие моменты времени (3 животных на каждый момент времени и 3 пробы на животное для групп 1-2 и 1 проба на животное для групп 3 и 4):
- п/о: перед введением и затем через 0.25 ч, 0.5 ч, 0.75 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 16 ч и 24 ч после перорального введения в дозе 3 и 30 мг/кг,
- п/о: через 0.5 ч, 2 ч, 6 ч, 16 ч и 24 ч после перорального введения в дозе 25 мг/кг и через 0.5 ч, 1 ч, 2 ч, 6 ч, 16 ч, 30 ч и 48 ч после перорального введения в дозе 100 мг/кг.
Плазму из проб крови, собранных после введения липидных составов лекарственного средства из соединения примера 1, анализируют с помощью жидкостной хроматографии, сопряженной с масс-спектрометрическим детектированием. Граница количественного определения лекарственного средства из соединения примера 1 является меньшей или равна 0.5 нг/мл.
Некомпартментный фармакокинетический анализ осуществляют на средних значениях концентрации в плазме исследуемых соединений. Результаты приведены в таблицах 4 и 5 ниже.
Результаты показывают, что, при любой дозе (от 3 до 100 мг/кг) и любом носителе (липидный или водный состав), основная часть пролекарства формулы (I) быстро превращается in vivo в соответствующее лекарственное средство формулы (I') (см. таблицу 4). Содержание в плазме крови пролекарства (Cmax, AUC) является низким по сравнению с содержанием соответствующего лекарственного средства. Результаты также показывают, что концентрация в плазме лекарственного средства, найденная подобным образом (после введения пролекарства), является эквивалентной или даже более высокой, чем концентрация, измеренная после прямого введения лекарственного средства пероральным путем (см. таблицу 5).
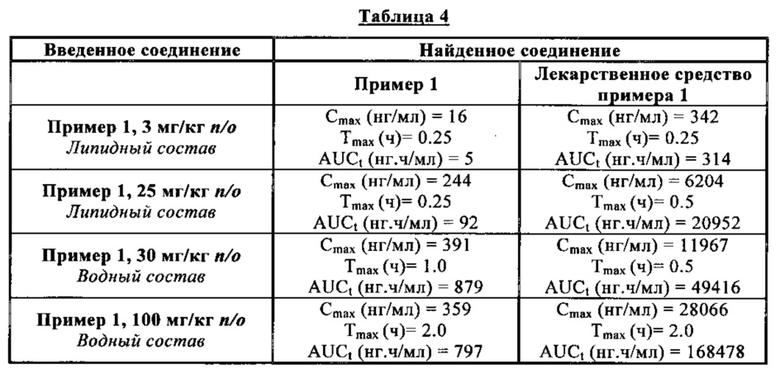

Более конкретно, п/о введение пролекарства в водном носителе позволяет достичь концентраций лекарственного средства в плазме, которые эквивалентны или даже выше концентраций, полученных после прямого п/о введения лекарственного средства в липидном носителе. Следовательно, пролекарство дает преимущество в легкости получения состава, в сравнении с соответствующим лекарственным средством, особенно в водной среде, что очень полезно в расчете на клинические исследования. Действительно, как показывает пример D, из лекарственного средства примера 1 трудно приготовить состав в водной среде.
Водный состав соединений примеров 20 и 25
Соединения примеров 20 и 25 вводят пероральным путем в водной среде SCID мышам при следующих условиях:
- группа 1: 3 мг/кг п/о в растворе в 1М карбонате натрий (желудочный зонд, 10 мл/кг),
- группа 2: 25 мг/кг п/о в растворе в 1М карбонате натрия (желудочный зонд, 10 мл/кг).
Пробы крови отбирают в следующие моменты времени (3 животных на каждый момент времени): через 0.25 ч, 0.5 ч, 1 ч, 2 ч, 6 ч и 24 ч после перорального введения.
Собранную таким образом кровь центрифугируют, и плазму переносят в пробирки, содержащие 1М соляную кислоту. Концентрации в плазме фосфатного соединения (пролекарства) и его гидроксилированного гомолога (лекарственного средства) определяют одновременно, используя метод жидкостной хроматографии, сопряженной с масс-спектрометрическим детектированием (ТТХ-ЖХ-МС/МС). Граница детектирования для обоих соединений составляет 0.5 нг/мл.
Липидный состав лекарственного средства из соединений примеров 20 и 25 Лекарственные средства из примерев 20 и 25 приготовляют в смеси полиэтиленгликоль 300/этанол/Phosal 50PG (30/10/60, об./об./об.), предназначенной для введения п/о путем SCID мышам при следующих условиях:
- группа 1: 3 мг/кг п/о (желудочный зонд, 10 мл/кг),
- группа 2: 25 мг/кг п/о (желудочный зонд, 10 мл/кг).
Пробы крови отбирают в следующие моменты времени (3 животных на каждый момент времени): через 0.25 ч, 0.5 ч, 1 ч, 2 ч, 6 ч и 24 ч после перорального введения.
Собранную таким образом кровь центрифугируют, и плазму переносят в пробирки, содержащие 1М соляную кислоту. Концентрации в плазме лекарственного средства определяют, используя метод жидкостной хроматографии, сопряженной с масс-спектрометрическим детектированием (ТТХ-ЖХ-МС/МС). Граница количественного определения составляет 0.5 нг/мл.
Осуществляют некомпартментный фармакокинетический анализ. Средние результаты приведены в таблицах 6, 7, 8 и 9 ниже.
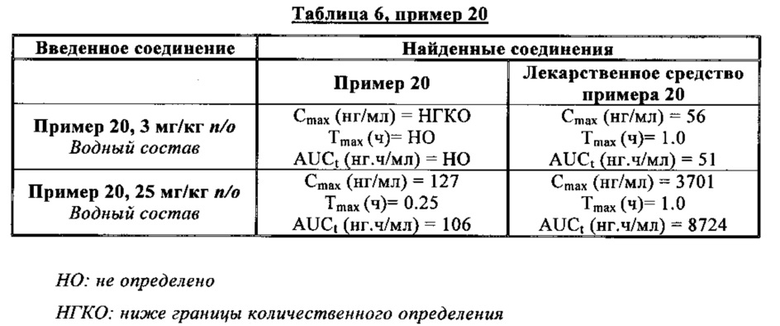


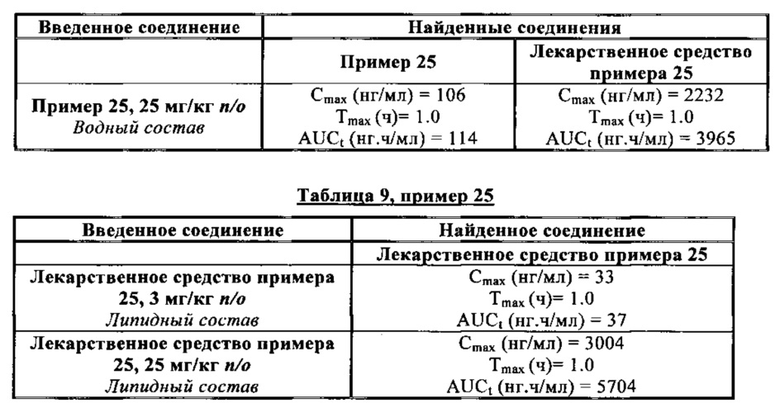
Результаты показывают, что, при любой дозе (3 или 25 мг/кг), основная часть пролекарств формулы (I) быстро превращается in vivo в соответствующие лекарственные средства формулы (I') (см. таблицы 6, 7, 8 и 9). Содержание в плазме крови пролекарств (Cmax, AUC) является низким по сравнению с содержанием в плазме соответствующих лекарственных средств. Результаты также показывают, что концентрации в плазме лекарственных средств, найденные подобным образом (после введения пролекарств), эквивалентны концентрациям, найденным после прямого введения лекарственных средств пероральным путем (см. таблицы 7 и 9).
ПРИМЕР D: In vivo фармакокинетический профиль соединений формулы (I')
Фармакокинетический профиль N-(4-гидроксифенил)-3-{6-[((3S)-3-(4-морфолинилметил)-3,4-дигидро-2(1H)-изохинолинил)карбонил]-1,3-бензодиоксол-5-ил}-N-фенил-5,6,7,8-тетрагидро-1-индолизин карбоксамида (также называемого лекарственным средством примера 1) также оценивают в липидном и водном составе на крысе Вистар.
Лекарственное средство примера 1 приготовляют в водной суспензии в гидроксиэтилцеллюлозе 1% (мас./об.) в воде и сравнивают с липидным составом, состоящим из смеси безводный этанол/полиэтиленгликоль 400/Phosal 50PG (10/30/60, об./об./об.). Два состава вводят пероральным путем самцам крыс Вистара (3 крысы на состав) в дозе 100 мг/кг п/о (желудочный зонд, 10 мл/кг).
Пробы крови отбирают в следующие моменты времени у каждого животного (3 животных/момент времени): через 0.25 ч, 0.5 ч, 0.75 ч, 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после перорального введения.
Концентрации в плазме исследуемого соединения определяют после экстрагирования с последующим использованием жидкостной хроматографии, сопряженной с масс-спектрометрическим детектированием. Граница количественного определения составляет 0.1 нг/мл. Результаты представлены в таблице ниже:
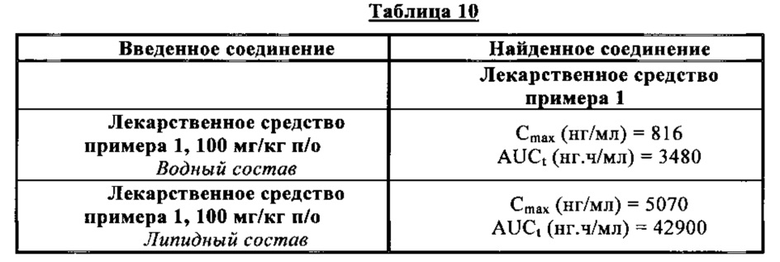
Результаты показывают, что липидный состав позволяет достичь значительно лучшего содержания лекарственного средства примера 1 в плазме крови, чем водный состав.
ПРИМЕР Е: In vitro исследование на клетках Сасо-2 человека.
Клеточный транспорт от А к В (от апикального к базолатеральному) фосфатных соединений формулы (I) и соединений формулы (I') (соответствующих лекарственных средств) изучают на клетках Сасо-2 человека. Каждое соединение депонируют апикально при концентрациях 1 или 3 мкМ (в двух повторностях), и затем инкубируют в течение 120 минут.
В ходе эксперимента отбирают несколько образцов;
- апикальный: непосредственно после депонирования (t=0) и на 120 минуте
- базолатеральный: в конце эксперимента (120 минут)
Концентрации фосфатного соединения (пролекарства) и/или его гидроксилированного гомолога (лекарственного средства) определяют с помощью жидкостной хроматографии, сопряженной с масс-спектрометрическим детектированием (ЖХ-МС/МС). Граница количественного определения для обоих соединений составляет 2 нг/мл.
Кажущуюся проницаемость (Рарр) и прогнозированную абсорбированную фракцию (Fabs) У людей рассчитывают для пролекарства, для лекарственного средства после инкубирования пролекарства и для лекарственного средства после инкубирования лекарственного средства (Hubatsch и др., Nat Protoc. 2007; 2(9), 2111-2119).
Также рассчитывают экспериментальный выход, который соответствует соотношению (в процентах) общего количества соединения, найденного в конце эксперимента, к инкубированному.
Результаты были сведены в таблицу 11. Они показывают, что пролекарства соединений формулы (I) заметно разлагаются в ходе эксперимента (экспериментальные выходы <1.5%), таким образом, везде вызывая образование в значительных количествах сопряженных лекарственных средств.
В конечном итоге, прогнозированная абсорбированная фракция у людей для лекарственных средств, образованных после инкубирования пролекарств, аналогична таковой, полученной после инкубирования лекарственных средства.


ПРИМЕР F: Антиопухолевая активность in vivo.
Антиопухолевую активность соединений настоящего изобретения оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11.
1×107 клеток RS4;11 трансплантируют под кожу иммуносупрессивным мышам (штамм SCID). Через 25-30 дней после трансплантации, когда масса опухоли достигнет приблизительно 150 мм3, мышей лечат пероральным путем различными соединениями в двух различных режимах (ежедневное введение в течение пяти дней в неделю в течение двух недель, или два введения в неделю в течение двух недель). Массу опухоли измеряют дважды в неделю с начала лечения.
Соединения настоящего изобретения обладают антиопухолевой активностью при пероральном пути введения на модели лейкоза RS4;11 (острый лимфобластный лейкоз). Полученные результаты показывают, что соединения настоящего изобретения способны индуцировать значительную регрессию опухолей.
ПРИМЕР G: Фармацевтическая композиция: Таблетки
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693629C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
| НОВЫЕ МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2018 |
|
RU2808689C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ БЕНЗОПИПЕРАЗИН, В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНОВ ВЕТ | 2014 |
|
RU2720237C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| НОВЫЕ (ГЕТЕРО)АРИЛ-ЗАМЕЩЕННЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2742271C2 |
Изобретение относится к соединению формулы (I), его энантиомерам и фармацевтически приемлемым солям и композициям на их основе, которые могут применяться в онкологии:
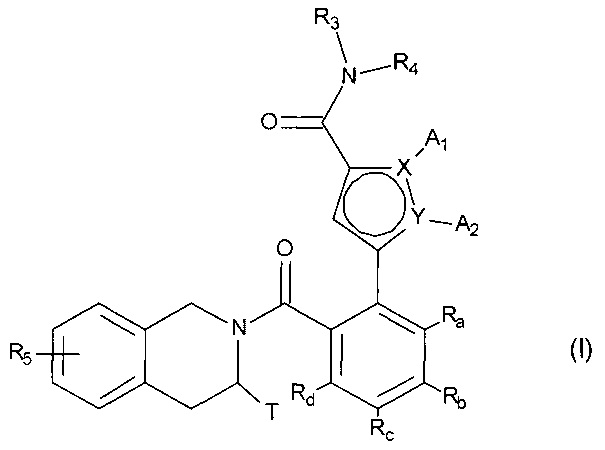
где X и Y представляют собой С или N, но не могут быть одинаковыми, A1 и А2 вместе с атомами, несущими их, образуют гетероцикл Het, выбранный из 5,6,7,8-тетрагидроиндолизина или индолизина, или A1 и А2 представляют собой Н, (С1-С6)полигалогеналкил или (С1-С6)алкил, Т представляет собой Н, (С1-С6)алкил, необязательно замещенный одним-тремя атомами галогена, (C1-C4)алкил-NR1R2, или (С1-С4)алкил-OR6, R1 и R2 представляют собой Н или (С1-С6)алкил, или R1 и R2 с атомом азота, несущим их, образуют гетероциклоалкил, R3 представляет собой циклоалкил, гетероциклоалкил, арил или гетероарил, R4 представляет собой фенил, замещенный в пара-положении группой -ОРО(ОМ)(ОМ'), -ОРО(ОМ)(O-М1+), -OPO(O-M1+)(O-M2+), -ОРО(O-)(O-)М32+, -ОРО(ОМ)(O[CH2CH2O]nCH3), или -ОРО(O-М1+)(O[CH2CH2O]nCH3), где М и М' представляют собой Н, (С1-С6)алкил, (С2-С6)алкенил, (С2-С6)алкинил, циклоалкил или гетероциклоалкил из 5 или 6 членов, тогда как М1+, М2+ и М32+ представляют собой фармацевтически приемлемые катионы, n равен от 1 до 5, при этом атомы углерода предыдущих групп или их возможных заместителей могут быть дейтерированными, R5 представляет собой Н или галоген, (С1-С6)алкил или (C1-С6)алкокси, R6 представляет собой Н или (С1-С6)алкил, Ra, Rb, Rc и Rd представляет собой R7, галоген, (С1-С6)алкокси, гидрокси, NR7R7'-CO-(C0-C6)алкил-O-, или заместители пары (Rb,Rc) вместе с атомами углерода, несущими их, образуют кольцо из 5-7 членов с 1 или 2 гетероатомами, выбранными из О и S, при этом один или несколько атомов углерода кольца могут быть дейтерированными или замещенными 1-3 группами, выбранными из галогена и (С1-С6)алкила, R7 и R7' представляют собой H, (С1-С6)алкил, или R7 и R7' вместе с атомом азота, несущим их, образуют гетероцикл, состоящий из 5-7 членов, "арил" означает фенил, нафтил, бифенил или инденил, "гетероарил" означает моно- или бициклическую группу, состоящую из 5-10 членов c по меньшей мере одним ароматическим фрагментом и 1-4 гетероатомами, выбранными из O, S и N, "циклоалкил" означает моно- или бициклическую, неароматическую, карбоциклическую группу из 3-10 членов, "гетероциклоалкил" означает моно- или бициклическую, неароматическую, конденсированную или спирогруппу из 3-10 кольцевых членов с 1-3 гетероатомами, выбранными из О, S, SO, SO2 и N, причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, алкил, алкенил, алкинил и алкокси могут быть замещены посредством 1-3 групп, выбранных из (С1-С6)алкила, (С3-С6)спиро, (С1-С6)алкокси, (С1-С6)алкил-S-, гидрокси, оксо или N-оксида, нитро, циано, -COOR', -OCOR', NR'Rʺ, (С1-С6)полигалогеналкила, трифторметокси, (С1-С6)алкилсульфонила, галогена, арила, гетероарила, арилокси, арилтио, циклоалкила, гетероциклоалкила, необязательно замещенного одним или несколькими галогенами или алкилами, R' и Rʺ представляют собой Н или (C1-С6)алкил, Het группа может быть замещена 1-3 группами, выбранными из (C1-С6)алкила, гидрокси, (С1-С6)алкокси, NR1'R1ʺ и галогена, при этом R1' и R1ʺ принимают значения, определенные для групп R' и Rʺ. Предложены новые эффективные противоопухолевые агенты и эффективный способ их получения. 7 н. и 26 з.п. ф-лы, 25 пр., 11 табл.
1. Фосфатное соединение формулы (I):
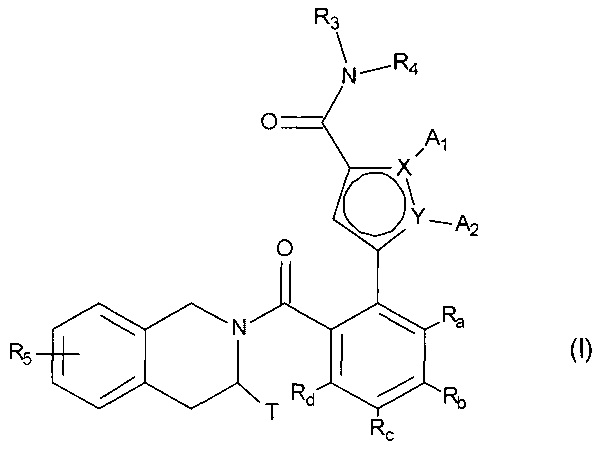
в которой:
♦ X и Y представляют собой атом углерода или атом азота, при этом предполагается, что они не могут одновременно представлять собой два атома углерода или два атома азота,
♦ A1 и А2 вместе с атомами, несущими их, образуют необязательно замещенный, ароматический или неароматический гетероцикл Het, состоящий из 6 кольцевых членов, так что в результате группа:
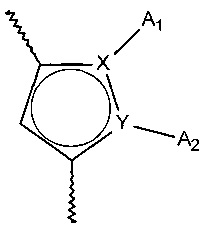
представляет собой 5,6,7,8-тетрагидроиндолизин или индолизин,
или A1 и А2 независимо друг от друга представляют собой атом водорода, линейную или разветвленную (С1-С6)полигалогеналкильную или линейную или разветвленную (С1-С6)алкильную группу,
♦ Т представляет собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, необязательно замещенную одним-тремя атомами галогена, группу (C1-C4)алкил-NR1R2 или группу (С1-С4)алкил-OR6,
♦ R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
или R1 и R2 с атомом азота, несущим их, образуют гетероциклоалкил,
♦ R3 представляет собой циклоалкильную группу, гетероциклоалкильную группу, арильную группу или гетероарильную группу, при этом предполагается, что один или несколько атомов углерода предыдущих групп или их возможных заместителей могут быть дейтерированными,
♦ R4 представляет собой фенильную группу, замещенную в пара-положении одной из следующих фосфатных групп: -ОРО(ОМ)(ОМ'), -ОРО(ОМ)(O-М1+), -OPO(O-M1+)(O-M2+), -ОРО(O-)(O-)М32+, -ОРО(ОМ)(O[CH2CH2O]nCH3), или -ОРО(O-М1+)(O[CH2CH2O]nCH3), где М и М' независимо друг от друга представляют собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С2-С6)алкенильную группу, линейную или разветвленную (С2-С6)алкинильную группу, циклоалкил или гетероциклоалкил, оба состоящие из 5 или 6 кольцевых членов, тогда как М1+и М2+независимо друг от друга представляют собой фармацевтически приемлемый однозарядный катион, и М32+ представляет собой фармацевтически приемлемый двухзарядный катион, и n означает целое число от 1 до 5, при этом предполагается, что один или несколько атомов углерода предыдущих групп или их возможных заместителей могут быть дейтерированными,
♦ R5 представляет собой водород или атом галогена, линейную или разветвленную (С1-С6)алкильную группу или линейную или разветвленную (C1-С6)алкокси группу,
♦ R6 представляет собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
♦ Ra, Rb, Rc и Rd каждый независимо от других представляет собой R7, атом галогена, линейную или разветвленную (С1-С6)алкокси группу, гидрокси группу, NR7R7'-CO-(C0-C6)алкил-O-, или заместители пары (Rb,Rc) вместе с атомами углерода, несущими их, образуют кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от 1 до 2 гетероатомов, выбранных из кислорода и серы, при этом также предполагается, что один или несколько атомов углерода кольца, определенного выше, могут быть дейтерированными или замещенными 1-3 группами, выбранными из галогена и линейного или разветвленного (С1-С6)алкила,
♦ R7 и R7' независимо друг от друга представляют собой водород, линейный или разветвленный (С1-С6)алкил, или R7 и R7' вместе с атомом азота, несущим их, образуют гетероцикл, состоящий из 5-7 кольцевых членов,
при этом предполагается, что:
- "арил" означает фенильную, нафтильную, бифенильную или инденильную группу,
- "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, имеющую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из кислорода, серы и азота (включая четвертичные атомы азота),
- "циклоалкил" означает любую моно- или бициклическую, неароматическую, карбоциклическую группу, содержащую от 3 до 10 кольцевых членов,
- "гетероциклоалкил" означает любую моно- или бициклическую, неароматическую, конденсированную или спирогруппу, состоящую из 3-10 кольцевых членов и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы, SO, SO2 и азота,
причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, таким образом определенные, и группы алкил, алкенил, алкинил и алкокси могут быть замещены посредством 1-3 групп, выбранных из линейного или разветвленного (С1-С6)алкила, (С3-С6)спиро, линейного или разветвленного, (С1-С6)алкокси, (С1-С6)алкил-S-, гидрокси, оксо (или N-оксида в соответствующих случаях), нитро, циано, -COOR', -OCOR', NR'Rʺ, линейного или разветвленного (С1-С6)полигалогеналкила, трифторметокси, (С1-С6)алкилсульфонила, галогена, арила, гетероарила, арилокси, арилтио, циклоалкила, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена или алкильными группами, при этом предполагается, что R' и Rʺ независимо друг от друга представляют собой атом водорода или линейную или разветвленную (C1-С6)алкильную группу,
причем Het группа, определенная в формуле (I), может быть замещена одной-тремя группами, выбранными из линейного или разветвленного (C1-С6)алкила, гидрокси, линейного или разветвленного (С1-С6)алкокси, NR1'R1ʺ и галогена, при этом предполагается, что R1' и R1ʺ принимают значения, определенные для групп R' и Rʺ, упомянутых выше,
его энантиомеры и его соли присоединения с фармацевтически приемлемой кислотой или основанием.
2. Соединение формулы (I) по п. 1, в которой R4 представляет собой фенил, замещенный в пара-положении группой формулы -ОРО(ОМ)(ОМ'), -OPO(OM)(O-M1+), -OPO(O-M1+)(O-M2+), -ОРО(O-)(O-)М32+, -ОРО(ОМ)(O[CH2CH2O]nCH3), или -OPO(O-M1+)(O[CH2CH2O]nCH3), где М и М' независимо друг от друга представляют собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С2-С6)алкенильную группу, линейную или разветвленную (С2-С6)алкинильную группу, циклоалкил или гетероциклоалкил, оба состоящие из 5 или 6 кольцевых членов, тогда как M1+ и М2+ независимо друг от друга представляют собой фармацевтически приемлемый однозарядный катион, и М32+ представляет собой фармацевтически приемлемый двухзарядный катион, и n означает целое число от 1 до 5, при этом предполагается, что фенильная группа необязательно может быть замещена одним или несколькими атомами галогенов.
3. Соединение формулы (I) по п. 1, в которой R4 представляет собой фенил, замещенный в пара-положении группой формулы -OPO(O-Na+)(O-Na+).
4. Соединение формулы (I) по любому из пп. 1-3, в которой X представляет собой атом углерода и Y представляет собой атом азота.
5. Соединение формулы (I) по любому из пп. 1-3, в которой группа:
представляет собой 5,6,7,8-тетрагидроиндолизин, индолизин или диметилированный пиррол.
6. Соединение формулы (I) по любому из пп. 1-3, в которой Т представляет собой метильную, (морфолин-4-ил)метильную или 3-(морфолин-4-ил)пропильную группу.
7. Соединение формулы (I) по любому из пп. 1-3, в которой Ra и Rd каждый представляет собой атом водорода и (Rb,Rc) вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу или 1,4-диоксановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой водород или галоген.
8. Соединение формулы (I) по любому из пп. 1-3, в которой Ra и Rd каждый представляет собой атом водорода, Rb представляет собой атом галогена и Rc представляет собой метокси группу.
9. Соединение формулы (I) по любому из пп. 1-3, в которой Ra, Rb и Rd каждый предпочтительно представляет собой атом водорода и Rc представляет собой группу NR7R7'-CO-(C0-C6)алкил-O-.
10. Соединение формулы (I) по любому из пп. 1-3, в которой R3 предпочтительно представляет собой группу, выбранную из фенила, 1H-индола, 1H-пирроло[2,3-b]пиридина, пиридина, 1H-пиразола, 1H-пиррола и 2,3-дигидро-1H-пирроло[2,3-b]пиридина, причем эти группы необязательно имеют один или несколько заместителей, выбранных из линейного или разветвленного (C1-С6)алкила, циано и тридейтерометила.
11. Соединение формулы (I) по п. 1, выбранное из следующего списка:
-4-[{[3-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат,
-4-({[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}[1-(тридейтерометил)-1H-пиразол-4-ил]амино)фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1,2-диметил-1H-пиррол-3-ил)амино]фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1-метил-1H-пиррол-3-ил)амино]фенил динатрийфосфат,
-4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1Н-пиразол-4-ил)амино]фенил динатрийфосфат,
-4-[(5-циано-1,2-диметил-1H-пиррол-3-ил){[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}амино]фенил динатрийфосфат,
-4-[{[5-(5-фтор-2-{[(35)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1Н-пиразол-4-ил)амино]фенил динатрийфосфат,
их энантиомеры и их соли присоединения с фармацевтически приемлемой кислотой или основанием.
12. Соединение формулы (I) по п. 1, которое представляет собой 4-[{[3-(6-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-ил]карбонил}(фенил)амино]фенил динатрийфосфат.
13. Соединение формулы (I) по п. 1, которое представляет собой 4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(пиридин-4-ил)амино]фенил динатрийфосфат.
14. Соединение формулы (I) по п. 1, которое представляет собой 4-({[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}[1-(тридейтерометил)-1H-пиразол-4-ил]амино)фенил динатрийфосфат.
15. Соединение формулы (I) по п. 1, которое представляет собой 4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1,2-диметил-1H-пиррол-3-ил)амино]фенил динатрийфосфат.
16. Соединение формулы (I) по п. 1, которое представляет собой 4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(5-циано-1-метил-1H-пиррол-3-ил)амино]фенил динатрийфосфат.
17. Соединение формулы (I) по п. 1, которое представляет собой 4-[{[5-(5-хлор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат.
18. Соединение формулы (I) по п. 1, которое представляет собой 4-[(5-циано-1,2-диметил-1H-пиррол-3-ил){[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}амино]фенил динатрийфосфат.
19. Соединение формулы (I) по п. 1, которое представляет собой 4-[{[5-(5-фтор-2-{[(3S)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-1,2-диметил-1H-пиррол-3-ил]карбонил}(1-метил-1H-пиразол-4-ил)амино]фенил динатрийфосфат.
20. Способ получения соединений формулы (I) по п. 1, который отличается тем, что в качестве исходного вещества используют соединение формулы (II):
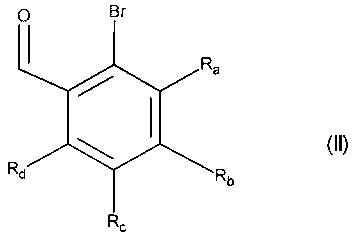
в которой Ra, Rb, Rc и Rd принимают значения, определенные в п. 1,
причем соединение формулы (II) подвергают реакции Хека, в водной или органической среде, в присутствии палладиевого катализатора, основания, фосфина и соединения формулы (III):
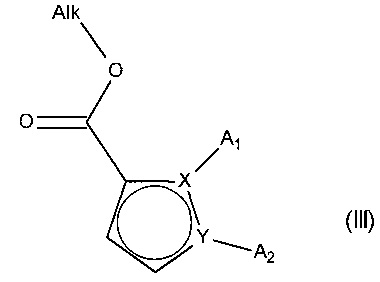
в которой А1, А2, X и Y принимают значения, определенные в п. 1, и Alk представляет собой линейную или разветвленную (С1-С6)алкильную группу,
с получением соединения формулы (IV):
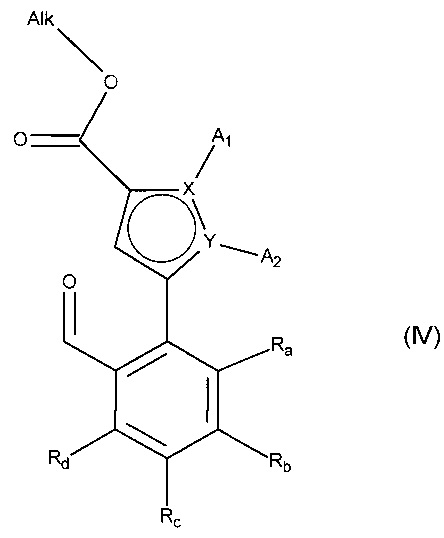
в которой A1, А2, X, Y, Ra, Rb, Rc и Rd принимают значения, определенные в п. 1, и Alk принимает значения, определенные выше,
альдегидную функцию соединения формулы (IV) окисляют до карбоновой кислоты с образованием соединения формулы (V):
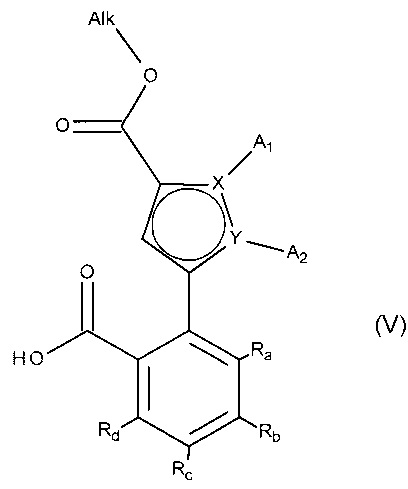
в которой A1, А2, X, Y, Ra, Rb, Rc и Rd принимают значения, определенные в п. 1, и Alk принимает значения, определенные выше,
соединение формулы (V) затем подвергают пептидному сочетанию с соединением формулы (VI):
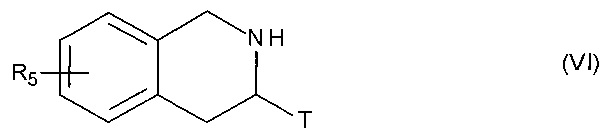
в которой Т и R5 принимают значения, определенные в п. 1,
с получением соединения формулы (VII):
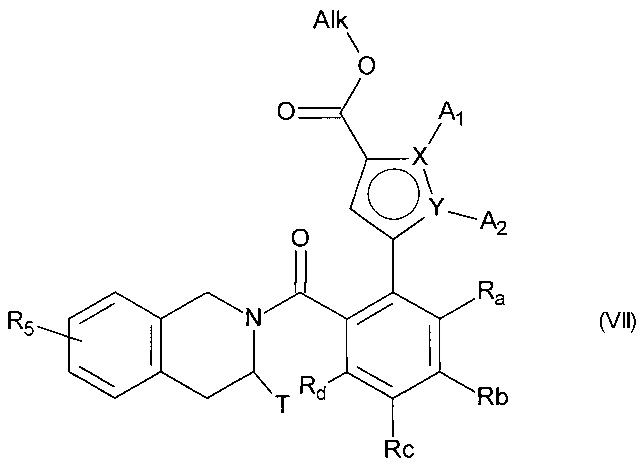
в которой A1, А2, X, Y, Ra, Rb, Rc, Rd, T и R5 принимают значения, определенные в п. 1, и Alk принимает значения, определенные выше,
сложноэфирную функцию соединения формулы (VII) гидролизуют с получением соответствующей карбоновой кислоты или карбоксилата, которую(-ый) можно превратить в производное по кислотной группе, такое как соответствующий ацилхлорид или ангидрид, перед сочетанием с амином NHR3R4, где R3 и R4 имеют те же значения, что и в п. 1, прежде чем подвергнуть действию пирофосфатного, фосфонатного или фосфорильного соединения, выбранного из тетрабензилпирофосфата, диэтилцианофосфоната и бисдиметиламинофосфорилхлорида, в основных условиях, причем соединение, полученное таким образом, может быть необязательно гидролизовано или гидрогенолизировано с получением соединения формулы (I),
соединение формулы (I) превращено, при необходимости, в его соли присоединения с фармацевтически приемлемой кислотой или основанием.
21. Способ по п. 20 получения соединений формулы (I), где соединение формулы (I) очищено в соответствии с обычными методами разделения или разделено на его изомеры.
22. Способ по п. 20 получения соединений формулы (I), где гидроксильные и аминогруппы реагентов или промежуточных соединений синтеза защищены и затем лишены защиты в соответствии с требованиями синтеза.
23. Фармацевтическая композиция для применения для лечения злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы, содержащая соединение формулы (I) по любому из пп. 1-19 или его соль присоединения с фармацевтически приемлемой кислотой или основанием в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями.
24. Фармацевтическая композиция по п. 23 для применения для лечения рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого.
25. Применение фармацевтической композиции по п. 23 для изготовления лекарственного средства, предназначенного для лечения злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы.
26. Применение по п. 25 фармацевтической композиции для изготовления лекарственного средства, предназначенного для лечения рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого.
27. Соединение формулы (I) по любому из пп. 1-3 или 11-19 или его соль присоединения с фармацевтически приемлемой кислотой или основанием для применения для лечения рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого.
28. Применение соединения формулы (I) по любому из пп. 1-19 или его соли присоединения с фармацевтически приемлемой кислотой или основанием для изготовления лекарственного средства, предназначенного для лечения рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого.
29. Фармацевтическая комбинация соединения формулы (I) по любому из пп. 1-19 с противоопухолевым средством, выбранным из генотоксичных средств, митотических ядов, антиметаболитов, ингибиторов протеосом, ингибиторов киназ и антител.
30. Фармацевтическая композиция, содержащая комбинацию по п. 29 в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями.
31. Фармацевтическая комбинация по п. 29 для применения для лечения злокачественных новообразований.
32. Применение фармацевтической комбинации по п. 29 для изготовления лекарственного средства для применения для лечения злокачественных новообразований.
33. Соединение формулы (I) по любому из пп. 1-3 или 11-19 для применения в комбинации с радиотерапией для лечения злокачественных новообразований.
| Porter J | |||
| et al, Bioorganic Chemistry Letters, 2009, vol | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Porter J | |||
| et al, Bioorganic & Medicinal Chemistry Letters, 2009, vol | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Heidi L | |||
| Perez et al, Bioorganic & Medicinal Chemistry Letters, 2012, vol | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Ana L | |||
| Simplicio et al, Molecules, 2008, 13, 519-547 | |||
| RU 96105060 A, 27.09.1998 | |||
| WO 2004004727 A1, 15.01.2004 | |||
| Bungaard H., Textbook of drug design and development, 1991, 113-191. | |||

