Настоящее изобретение относится к способу модификации материала полимерного носителя, предназначенного для его использования в качестве стационарной фазы в способе аналитического или препаративного разделения, причем указанный способ включает в себя стадии: обеспечения материала полимерного носителя, по меньшей мере частично полученного из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя; генерирование гидроксильных групп на/в материале полимерного носителя способом, который включает в себя окислительную обработку материала полимерного носителя и последующую восстановительную или гидролитическую обработку продукта реакции; введения во взаимодействие продукта предшествующей стадии с полифункциональным соединением. Кроме того, изобретение относится к материалу полимерного носителя для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, в частности, в хроматографическом способе, полученному способом согласно настоящему изобретения.
Ионообменники обычно образованы из твердых частиц, несущих на своей поверхности заряд, который позволяет им удерживать ионы. В случае анионообменников они часто представляют собой катионные соединения аммония, хотя известны также ионы фосфония и арсония. Обменная группа является монокатионной. В чисто электростатических взаимодействиях время удерживания определяется законом Кулона. Согласно этому закону, время удерживания удерживаемого аниона должно определяться только его зарядом.
Однако в ионной хроматографии с использованием водного раствора можно идентифицировать другие факторы, влияющие на характер удерживания, такие как гидратирование аниона и гидратирование обменной группы. Кроме того, играет роль и поляризуемость участвующих ионов, и слабые вторичные взаимодействия между анализируемыми веществами и субстратом обменника. Поскольку гидрофильность базового полимера, содержащего обменную группу, также влияет на ее гидратацию, характер удерживания ионообменника можно изменить модификацией материала носителя при той же обменной группе. Непосредственные заместители катионной группы также оказывают влияние на характер удерживания.
Настоящий уровень техники уже включает способы регулирования гидрофильности частицы для ионообменной хроматографии. Другие желательные параметры, такие как балансная емкость, большое число теоретических тарелок или химическая инертность ионообменного материала также обсуждаются выборочно.
В US20050181224 сшивающие слои, содержащие обменные группы, нанесены на частицу сульфированного гидрофильного носителя посредством циклической реации между диэпоксидами и аминами. При каждом цикле емкость ионобменника возрастает. Гидрофильность в принципе гарантирована. Однако вследствие чередования эпоксидных/аминных функциональных групп, дополнительной гидрофильности нельзя достигнуть независимо от емкости.
В EP 3248678 на пористую частицу дивинилбензола наносят покрытие из модифицированных полисахаридов (продукт реакции агарозы и глицидилфенилового эфира), а затем модифицированный полисахарид сшивают с полифункциональным сшивающим агентом (например, диглицидиловым эфиром этиленгликоля), получая макромолекулу, содержащую на поверхности гидроксильные группы. После этого в субстрат добавляют гидрохлорид диэтиламиноэтилхлорида для получения действительной ионообменной группы. В данной заявке также проводят поиск гидрофильной ионообменной частицы, обладающей повышенной прочностью (прочностью на разрыв). Вследствие сил чистой адсорбции, действующих между частицей и первоначально гелеобразным покрытием, носитель также оказывается менее прочным.
В EP1217012 исходят из гидрофобного полимера сложного эфира винилового спирта. Сначала проводят гидролиз содержащихся сложных эфиров с высвобождением спиртовых групп. Полимер становится гидрофильным. ОН-группы вводят во взаимодействие с диэпоксидом, а затем с амином для получения покрытия на субстрате. Гидрофильность можно дополнительно повысить, подвергнув основной полимер более интенсивному гидролизу на первой стадии. Однако это привело бы к потере механической прочности частицы. Может появиться нежелательное набухание.
В публикации Çaglayan et al. (J Sep Sci 2006, 29. 940) предпринята попытка оптимизировать некоторые из упомянутых параметров, такие как число теоретических тарелок, размер пор и расширение поверхности, за счет использования субстрата из частиц поли(винилацетат-со-дивинилбензола) различного состава. Согласно данной публикации, увеличенное количество винилацетата и, следовательно, увеличенное количество ОН групп в гидролизованной частице приводит к резкому повышению давления флегмы, когда колонку, заполненную данным субстратом, подвергают увеличенной скорости потока. Это нежелательное обратное давление объясняется недостаточной механической прочностью частиц, которые деформируются в процессе разделения.
В US5503933 раскрыты гидрофильные покрытия, ковалентно связанные с гидрофобными поверхностями, и способы их получения. Соединение, содержащее гидрофобный домен, включающий ненасыщенную группу, и гидрофильный домен, предоставлено для получения поверхностей с покрытием. Кроме того, предоставлена гидрофобная поверхность, содержащая ненасыщенные группы. Молекулы соединения адсорбируются гидрофобной поверхностью, а затем ненасыщенные группы в гидрофобных доменах молекул соединения ковалентно связываются с ненасыщенными группами на гидрофобной поверхности посредством свободно-радикальной реакции. В одном из вариантов осуществления гидрофильные покрытия могут быть ковалентно связаны с полистиролом, сшитым дивинилбензолом. Однако образующиеся частицы подходят только для разделения макромолекул. Для использования в других хроматографических методах, в частности, в ионной хроматографии, частицы имеют неудовлетворительное число теоретических тарелок вследствие своего большого диаметра. В свою очередь, в случае уменьшенного размера частиц, механическая удельная нагрузка макропористого материала будет недостаточной для давлений, создаваемых в таких хроматографических процессах.
Цель настоящего изобретения заключается в преодолении упомянутых выше недостатков предшествующего уровня техники. В настоящее время отсутствует способ, при помощи которого на полимерный субстрат можно нанести покрытие таким образом, чтобы гидрофильность поверхности полимерного субстрата можно было регулировать независимо от содержания кислорода в сердцевине полимерного субстрата и в результате образовывались механически стабильные и прочные частицы. В то же время ионообменный субстрат на его основе должен быть в значительной степени химически инертным, и должна быть возможность конфигурировать гидрофильность и емкость или селективность и емкость независимо друг от друга.
Данную задачу решают способом, который имеет признаки пункта 1 формулы изобретения. Он относится к способу модификации материала полимерного носителя, предназначенного для использования в качестве стационарной фазы в способе аналитического или препаративного разделения. Изобретение также относится к материалу полимерного носителя, полученному согласно способу изобретения, предназначенному для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, в частности, хроматографическом способе. Изобретение относится к хроматографической колонке, заполненной материалом полимерного носителя согласно изобретению, к способу разделения анализируемых веществ с использованием материала полимерного носителя согласно изобретению, и к применению материала полимерного носителя согласно изобретению для аналитического и препаративного разделения анализируемых веществ.
Данный способ включает в себя стадии: обеспечения материала полимерного носителя, по меньшей мере частично полученного из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, предпочтительно по меньшей мере частично полученного из мономеров дивинилбензола (стадия a); генерирования гидроксильных групп на/в материале полимерного носителя способом, который включает в себя стадии: окислительной обработки материала полимерного носителя (стадия b.1); последующей восстановительной или гидролитической обработки продукта реакции стадии b.1 (стадия b.2); необязательно: реакции продукта стадии b.2. с полифункциональным соединением, в частности, соединением, содержащим по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, предпочтительно, галоидную группу, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, предпочтительно, эпоксигруппу (стадия c).
Безусловно, функциональная группа, реакционноспособная с аминами, является или может быть одновременно реакционноспособной с другими элементоорганическими соединениями 5-ой основной группы, а именно с арсинами или фосфинами.
Как будет дополнительно объяснено далее, в частности, в отношении способа изобретения, стадии согласно изобретению влияют на содержание кислорода на поверхности материала полимерного носителя. Таким образом, гидрофильность поверхности материала полимерного носителя не зависит от содержания кислорода в сердцевине субстрата полимерного носителя.
Материал полимерного носителя, обеспеченный на стадии a, обычно обеспечивают в виде частицы, предпочтительно в виде сферической частицы, особенно предпочтительно в виде сферической частицы со средним размером частиц (медианой) от 1 до 50 мкм, еще более предпочтительно со средним размером частиц от 2 до 25 мкм, особенно предпочтительно со средним размером частиц от 3 до 9 мкм. Однако возможны другие материалы полимерного носителя, в частности, материалы полимерного носителя в виде мембран или монолитов.
Содержание кислорода на поверхности материала полимерного носителя возрастает за счет первоначального окисления и восстановления или гидролиза. Модификация также способна привести к появлению атомов кислорода на/в сердцевине материала полимерного носителя, в которой отсутствует обнаруживаемое содержание кислорода. Увеличенное содержание кислорода влияет на природу и проявление вторичных взаимодействий, в частности, на гидрофобность образующегося материала полимерного носителя. Кроме того, при помощи ряда стадий, следующих за первоначальным окислением и восстановлением/гидролизом, емкость можно регулировать независимо от содержания кислорода на поверхности.
Под поверхностью полимерного носителя или поверхностью материала полимерного носителя в настоящем описании, в частности, подразумевают доступную для контакта с раствором внешнюю поверхность структуры материала полимерного носителя, а также слой от 1 до 30 нм, непосредственно примыкающий к данной внешней поверхности, при этом доступная для контакта с раствором внешняя поверхность может частично располагаться на микроструктурах, например, пористых структур. В частности, имеется в виду доступная для контакта с раствором внешняя поверхность частиц материала полимерного носителя, имеющих пористую или непористую структуру.
Первоначальный вклад в химическую и механическую стабильность вносит ковалентная связь, образущаяся между материалом полимерного носителя и покрытием. Это отличается от ситуации с ионообменниками на основе латекса, где латексные частицы и, следовательно, обменные группы на субстрате удерживаются за счет чисто электростатических взаимодействий. Кроме того, наличие ковалентных связей обеспечивает высокую химическая инертность. Второй вклад в механическую стабильность вносит тот факт, что сердцевина материала полимерного носителя образована, по меньшей мере частично, ароматическими углеводородными соединениями, содежащими по меньшей мере два винильных или аллильных заместителя, предпочтительно, по меньшей мере частично, мономерами дивинилбензола. Стадии b.1 и b.2 не оказывают влияния на стабильность данной сердцевины материала полимерного носителя. Сердцевина материала полимерного носителя предпочтительно является монодисперсной.
Стадия c, т.е. реакция продукта стадии b.2. с полифункциональным соединением, в частности, с соединением, содержащим по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксильными группами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксильными группами, необязательна. При исключении данной стадии частицы подходят для использования, например, в эксклюзионной хроматографии.
В частности, получение ионообменного материала для использования в анионообменной хроматографии или катионообменной хроматографии на основе материала полимерного носителя, модифицированного согласно настоящему изобретению, описано ниже. Однако использованием данных частиц никоим образом не ограничено этим. Частицы можно также использовать в других методах аналитического и препаративного разделения, таких как другие методы адсорбционной хроматографии, HILIC (жидкостная хроматография гидрофильного взаимодействия), хроматография с обращенной фазой, твердофазная экстракция и т.д.
В предпочтительном варианте осуществления способ включает в себя описанные выше стадии a, b и c, и дополнительные стадии, следующие за стадией c, а именно, осуществление ряда циклов нанесения покрытия (стадия d). Один цикл нанесения покрытия, стадия d, включает в себя: введение или генерирование гидроксигрупп за счет введения во взаимодействие второй функциональной группы, реакционноспособной с аминами и/или гидроксигруппами, предпочтительно эпоксигруппой, введенной на стадии с, с полифункциональным соединением, содержащим гидроксигруппы, в частности, с полиолом, или за счет гидролиза, или их комбинации (стадия d.1); и введение во взаимодействие продукта стадии d.1. с полифункциональным соединением, в частности, соединением, содержащим по меньшей мере первую фукнциональную группу, реакционноспособную с гидроксигруппами, предпочительно галоидную группу, и по меньшей мере вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, предпочтительно эпоксигруппу (стадия d.2). Число циклов нанесения покрытия составляет от 0 до 20.
Если число циклов нанесения покрытия составляет по меньшей мере 2, введение или генерирование гидроксигрупп при первом повторе и далее, очевидно, больше не относится или, по крайней мере, больше не относится исключительно к функциональным группам, введенным на стадии c, а в основном относится к соответствующим функциональным группам, введенным на стадии d.2. Это особенно верно, если условия выбраны таким образом, что вторые функциональные группы со стадии c реагируют практически полностью, когда первый цикл нанесения покрытия проводят на стадии d.1.
За счет увеличения числа циклов нанесения покрытия содержание кислорода на поверхности материала полимерного носителя можно дополнительно повысить, и можно увеличить гидрофильность ионообменного материала на основе данного материала полимерного носителя. Путем выбора соответствующей степени гидрофильности можно усилить взаимодействия ионообменного материала с сильно гидратированными ионами (такими как фторид) и ослабить взаимодействия с ионами, которые гидратированы слишком слабо (такими как бромат, нитрат, хлорат). Таким образом, например, можно оказать влияние на порядок удерживания бромата и хлорида, чтобы на хроматограмме бромат количественно опережал хлорид. Кроме того, можно гарантировать, что фторид отделен от пика ввода, в частности, также при использовании карбонатного элюента.
Кроме того, способом настоящего изобретения можно получить ионообменный материал с числом тарелок > 50000 TТ/м теоретических тарелок на метр колонки для семи стандартных анионов (фторида, хлорида, нитрита, бромида, нитрата, фосфата и сульфата) с высокой симметрией сигнала (асимметрия < 1,5). Все упомянутые выше ионы проявляются на хроматограмме, отделенные друг от друга базовой линией, с совокупным небольшим общим временем хроматографии.
Преимущества высокой прочности частиц становятся особенно очевидными, когда колонку, которая заполнена материалом полимерного носителя, полученным описанным выше способом, подвергают испытанию на расход под давлением. В таком нагрузочном тесте изменение давления в колонке определяют как функцию от непрерывно увеличивающегося расхода потока. В колонке в соответствии с изобретением давление линейно зависит от расхода потока. Это противоречит результатам, полученным с использованием обычных колонок, заполненных гидрофильным субстратом pDVB, имеющим высокое содержание винилацетата. В случае обычных колонок давление возрастает более чем линейно в зависимости от расхода потока. Например, может возникнуть гиперболический наклон функции. Это следует, например, из цитированной вначале публикации Çaglayan et al. (J Sep Sci 2006, 29, 940).
Характеристики колонки согласно изобретению после проведения испытания под давлением также лучше по сравнению с обычными колонками. С одной стороны, в данной колонке наблюдается лишь небольшое увеличение перепада давления после выполнения нагрузочного теста по сравнению с обычными колонками. Кроме того, число теоретических тарелок уменьшается в ходе нагрузочных испытаний колонки согласно изобретению в гораздо меньшей степени, чем в случае обычных колонок (Çaglayan et al., J Sep Sci 2006, 29, 940). Поддержание соотношения давления и количества тарелок даже при высоких расходах потока обеспечивает эффективность и высокую производительность процессов разделения.
Далее способ может дополнительно включать в себя стадию e - введение ионообменных групп в продукт реакции стадии c или d.2. На стадии е ионообменный материал, который реализует преимущества, связанные с материалом полимерного носителя, получают из материала полимерного носителя. Гидрофильность и емкость или селективность и емкость являются независимо регулируемыми в данном ионообменном материале согласно изобретению. Ионообменный материал согласно изобретению также характеризуется большим числом теоретических тарелок.
Под ионообменными группами подразумевают заряженные группы на поверхности материала полимерного носителя, в частности заряженные аминные, арсиновые или фосфиновые группы.
Под материалом полимерного носителя, который по меньшей мере частично получен из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, в контексте настоящего изобретения подразумевают, что данный материал полимерного носителя является получаемым реакцией полимеризации с участием по меньшей мере ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя. Предпочтительно, данный материал полимерного носителя получен реакциями полимеризации с участием дивинилбензола. Однако возможны также реакции с участием тривинилбензола и дивинилнафталина, а также соединений, известных специалистам в данной области техники в качестве эквивалентов.
Способ может отличаться тем, что материал полимерного носителя на стадии а, который по меньшей мере частично получен из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, в дополнение к этому частично получен из мономеров, которые выбраны из группы, состоящей из этилвинилбензола, винилацетата, стирола и их комбинаций. В связи с этим доля ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, предпочтительно составляет по меньшей мере 50 масс.%. Например, материал полимерного носителя на стадии а может содержать по меньшей мере 50 масс.% звеньев дивинилбензола. Такой материал полимерного носителя обладает выгодными свойствами в отношении структуры пор и, в частности, характеризуется большим числом доступных двойных связей для последующей модификации поверхности.
Способ может отличаться тем, что окислительная обработка на стадии b.1 представляет собой обработку пероксикислотой, предпочтительно выбранной из группы, состоящей из мета-хлорпербензойной кислоты (m-CPBA), пероксимуравьиной кислоты, пероксиуксусной кислоты, трифторпероксиуксусной кислоты, обработку KMnO4, обработку кислородной плазмой, или их комбинации. При обработке полимера пероксикислотой происходит окисление любых имеющихся двойных связей, и они становятся доступными для последующего восстановления или гидролиза. Безусловно, данного эффекта в принципе можно достигнуть и при помощи других методов окисления, которые известны специалисту в данной области техники, например, озонолизом.
Особенно предпочтительно использовать пероксикислоту на стадии b.1. Преимущество пероксикислот заключается в том, что они позволяют добиться более высокого содержания кислорода, чем, например, в результате обработки плазмой. Содержание кислорода 2,0% можно получить при помощи кислородной плазмы, тогда как содержание кислорода 3,2%, определенное методом элементного анализа, можно достигнуть при использовании, например, mCPBA. При использовании пероксикислоты ее можно добавить к суспензии полимера или получить in situ из кислоты и перекиси водорода. Предпочтительно, m-CPBA прибавляют к суспендированному материалу полимерного носителя, например, материалу полимерного носителя PS/DVB, поскольку его обработка в твердом состоянии не представляет сложности.
В предпочтительном варианте осуществления восстановительную обработку продукта реакции, полученного на окислительной стадии b.1, осуществляют на стадии b.2 при использовании реагента для восстановления полярных связей, предпочтительно, гидрида металла. Он может представлять собой, например, NaBH4, BH3, LAH, NaH, CaH. Преимущество использования гидридов заключается в том, что растворенный реагент способен проникать в поры частицы. Например, в случае палладия на активированном угле это невозможно. По сравнению с гидролизом под действием хлористоводородной кислоты, в результате которого эпоксиды можно превратить в гидроксилен (см. пример 3), при восстановлении гидридами металлов можно также превратить карбонильную и карбоксильную группы в гидроксилен. В результате окисления образующиеся продукты окисления превращаются в спирты. Предпочтительно используют раствор алюмогидрида лития в диэтиловом эфире. В одном из вариантов осуществления представлена 1-20% масс./об. суспензия полимера в сухом диэтиловом эфире, к которой прибавляют алюмогидрид лития в количестве 5-100% масс./масс. из расчета на массу полимера в сухом состоянии. Особенно предпочтительно использование 5-15% масс./об. суспензии полимера в сухом диэтиловом эфире, к которой прибавляют алюмогидрид лития в количестве 5-20% масс./масс. из расчета на массу полимера в сухом состоянии. Можно выбрать температуру 25-70°C, особенно предпочтительна температура кипения диэтилового эфира, и продолжительность реакции от 1 мин до 72 ч, особенно предпочтительно от 3 ч до 48 ч.
Теперь OH группы, генерированные на поверхности полимерного носителя описанным выше способом, доступны в достаточном количестве для модификации на стадии с. В качестве альтернативы условиям восстановления можно также выбрать гидролитические условия.
Продукт реакции, полученный на стадии b.2, вводят во взаимодействие с полифункциональным соединением, в частности, с соединением, содержащим по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, предпочтительно галоидную группу, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, предпочтительно эпоксигруппу.
Первая функциональная группа, реакционноспособная с гидроксигруппами, может представлять собой структуру, способную подвергаться нуклеофильной атаке OH группами или аминогруппами, такую как галогенуглерод, эпоксид, тозилат, метилсульфид или их смесь. Вторая функциональная группа, реакционноспособная с аминами и/или гидроксигруппами, может представлять собой, например, эпоксигруппу. Предпочтительно, полифункциональное соединении на стадии с представляет собой эпихлоргидрин (ECH). Например, субстрат можно суспендировать в эпихлоргидрине (предпочтительно, 5-30% масс./об. твердого вещества в ECH, более предпочтительно 10-20% масс./об. твердого вещества в ECH). После этого его можно ввести во взаимодействие с основанием, таким как водный раствор гидроксида щелочного и щелочноземельного металла. Оказалось, что для этой цели подходят водные растворы NaOH и KOH, особенно предпочтительно 10-50% масс./масс.NaOH в соотношении ECH:NaOH(водн.)=1:(0,1-10). Особенно предпочтительно, реакцию проводят с использованием четвертичной аммониевой соли в качестве катализатора межфазного переноса. Альтернативным образом, субстрат, суспендированный в ECH, можно ввести во взаимодействие с гидроксидом четвертичного аммония. Найдено, что для этой цели подходит гидроксид тетраметиламмония. Предпочтительно, к суспензии полимера в ECH, полученной, как описано выше, прибавляют такое же количество диметилсульфоксида (ДМСО), как и ЕСН, и предпочтительно, на грамм использованного полимера прибавляют от 1 до 10 ммоль гидроксида тетраметиламмония в виде концентрированного водного раствора, более предпочтительно, от 2 до 5 ммоль гидроксида тетраметиламмония (водн.).
Однако полифункциональное соединение, используемое на стадии с, содержащее по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, может также представлять собой спейсерную молекулу.
В контексте настоящей заявки спейсерная молекула (спейсер для краткости) означает молекулу, содержащую упомянутые выше по меньшей мере две функциональных группы, при этом данная молекула обеспечивает расстояние по меньшей мере в 3 атома, предпочтительно от 3 до 20 атомов между поверхностью модифицированного материала полимерного носителя и предназначенными для введения ионообменными группами. Спейсерная молекула в конечном ионообменном материале связывается, с одной стороны, с модифицированным материалом полимерного носителя, а с другой стороны – с обменной группой. Функциональные группы спейсерной молекулы могут представлять собой структуры, способные подвергаться нуклеофильной атаке OH группами или аминогруппами, такие как галогенуглероды, эпоксиды, тозилаты, метилсульфиды или их смесь. Спейсерные атомы могут представлять собой углеродные цепи, но также могут включать гетероатомы, например, эфирные группы или тиоэфиры. Спейсерная молекула обеспечивает расстояние между субстратом и ионообменной группой. Функция спейсера состоит в предотвращении взаимодействия между ионами и субстратом. Это препятствует уширению нежелательного пика на хроматограмме. Спейсерные углеродные цепи, содержащие эфирные группы, предпочтительны благодаря их более высокой гидрофильности. Особенно предпочтительно, когда спейсер содержит глицидиловые группы, способные взаимодействовать с аминами, фосфинами, арсинами и/или гидроксильными группами. Особенно предпочтительной спейсерной молекулой является диглицидиловый эфир 1,4-бутандиола.
При введении на стадии d.1 гидроксигрупп посредством реакции второй функциональной группы, введенной на стадии c (или, как вариант, на стадии d.2), с полифункциональными соединениями, содержащими гидроксигруппы, предпочтительно использовать диол. Особенно предпочтителен бутандиол. Данный диол можно использовать в качестве растворителя и реагента, а реакция может протекать в условиях основного катализа при повышенной температуре. Особенно предпочтительно использование 0,1-1 моль/л KOH при 60-160°C в течение 1-48 ч. Наиболее предпочтительно, температура составляет 100-130°C, а продолжительность реакции составляет 3-36 ч. При добавлении такого соединения эпоксиды, ранее связанные с субстратом, превращаются в цепи, содержащие OH группы.
В одном из вариантов осуществления полифункциональное соединение, используемое на стадии d.2, которое содержит по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, представляет собой эпигалогенгидрин, предпочтительно эпихлоргидрин. В связи с этим частицу полимерного носителя можно сначала суспендировать в эпихлоргидрине (предпочтительно: 5-30% масс./об. твердого вещества в ECH, более предпочтительно, 10-30% масс./об. твердого вещества в ECH). После этого, полученную суспензию можно ввести во взаимодействие с основанием, таким как водные растворы гидроксида щелочного или щелочноземельного металла. Оказалось, что для этой цели подходят водные растворы NaOH и KOH, при этом особенно предпочтителен 10-50% масс./масс. NaOH при соотношении ECH:NaOH(водн.)=1:(0,1-10). Особенно предпочтительно, реакцию проводят с использованием четвертичной аммониевой соли в качестве катализатора межфазного переноса.
Однако в качестве альтернативы спейсерную молекулу можно использовать на стадии d.2. Особенно предпочтительно, когда спейсерную молекулу используют по меньшей мере при последнем осуществления цикла нанесения покрытия на стадии d.2.
Предпочтительной особенностью настоящего изобретения является то, что стадии d.1 и d.2, совместно называемые также циклом нанесения покрытия, можно повторять. Число циклов нанесения покрытия может составлять от 0 до 20, предпочтительно от 0 до 10. Однако предпочтительно, чтобы число циклов нанесения покрытия составляло от 0 до 5, более предпочтительно от 1 до 3.
Гидрофильность материала полимерного носителя возрастает с каждым циклом. Выбрав соответствующее количество циклов, можно оптимально отрегулировать гидрофильность для подлежащей разделению смеси. В частности, за счет выбора соответствующей степени гидрофильности можно усилить гидрофильные взаимодействия между субстратом и сильно гидратированными ионами (такими как фторид), а взаимодействия со слабо гидратированными ионами (такими как бромат, нитрат, хлорат) можно ослабить. Это может повлиять на порядок удерживания. Образование каждого слоя также ослабляет вторичные взаимодействия между поляризуемыми ионами, такими как хлорат или бромат, с одной стороны, и субстратом, с другой стороны. Значительно уменьшается образование сигналами «хвостов», наблюдаемое вследствие подобных вторичных взаимодействий, так что даже поляризуемые ионы элюируются симметрично. Общая емкость обменного материала уменьшается.
В предпочтительном варианте осуществления в качестве полифункционального соединения на стадии d.2 при последнем осуществлении цикла d нанесения покрытия используют спейсерную молекулу, предпочтительно диэпоксид, предпочтительно диглицидиловый эфир бутандиола. Преимущества спейсерной молекулы описаны выше применительно к стадии c. Взаимодействие со спейсерной молекулой предпочтительно осуществляют при добавлении к реакционной смеси полярного растворителя, особенно предпочтительно, ДМСО в объемном соотношении 1:(0,1-5), более предпочтительно в соотношении 1:(0,5-1,5) относительно диглицидилового эфира. Также предпочтительно использовать четвертичную аммониевую соль в качестве катализатора межфазного переноса, особенно предпочтительно, бромид тетрабутиламмония, в интервале концентраций предпочтительно 1-100 ммоль/л, особенно предпочтительно 10-50 ммоль/л из расчета на общий объем реакционной смеси. В качестве основания можно использовать гидроксиды щелочных и щелочноземельных металлов, предпочтительно, водные растворы NaOH и KOH, особенно предпочтительно NaOH в интервале концентраций 0,1-5 моль/л, наиболее предпочтительно в интервале концентраций 0,1-1 моль/л. Объемное соотношение относительно диглицидилового эфира составляет 1:(0,1-5), предпочтительно 1:(0,5-1,5). Предпочтительная температура реакции составляет 0-50°C, особенно предпочтительно 20-30°C, при предпочтительной продолжительности реакции 2-40 ч, особенно предпочтительно 15-25 ч.
В предпочтительном варианте осуществления ионообменную группу вводят в результате взаимодействия соединения, полученного на стадии c или d.2, с элементоорганическим соединением элемента 5-ой основной группы, предпочтительно амином, особенно предпочтительно третичным амином. Однако ионообменная группа может включать фосфин или арсин вместо амина. Особенно подходящим оказалось соединение, содержащее амин, включающий от 1 до 3 органических радикалов, содержащих от 1 до 10 атомов в радикале, включая также циклические соединения. Циклические соединения могут быть замещенными. Примеры подходящих соединений включают в себя азотсодержащие гетероциклы, например, пиридины, содержащие или не содержащие углеводородных или гидроксильных заместителей, монозамещенные алкилпирролидины, монозамещенные алкилпиперидины или дизамещенные алкилпиперазины. Углеводородные радикалы в аминосоединениях также могут включать гетероатомы, например, атомы кислорода или серы, или другие заместители.
Однако можно было бы также ввести соединения, подходящие для катионообменной хроматографии или методов HILIC. Подходящие катионные функциональные группы включают сульфокислоты, карбоновые кислоты или их комбинацию. Предпочтительно, предназначенные для введения соединения могут также содержать множество функциональных групп, например, в случае введения аминокислот.
Например, для образования катионообменного центра полимер, эпоксидированный на предшествующей стадии, можно суспендировать в смеси воды и полярного растворителя, предпочтительно ДМСО, и добавить предпочтительный амин. Предпочтительная продолжительность реакции составляет от 0,5 до 48 ч при предпочтительной температуре 20-70°C.
Предпочтительно, чтобы за стадией е - введением ионообменных групп - следовала дополнительная стадия f, включающая нагревание материала полимерного носителя, снабженного ионообменными группами, в щелочном растворе. Это позволит отрегулировать селективность и емкость ионообменного материала, полученного на предшествующей стадии. Данная обработка в дальнейшем будет называться элиминированием, и она заключается, в частности, в нагревании частиц, содержащих обменные группы, в водном растворе щелочи, особенно предпочтительно, в нагревании в водном растворе гидроксида или карбоната щелочного или щелочноземельного металла, например, в растворе гидроксида натрия. Предпочтительная концентрация NaOH составляет от 0,1 до 5 моль/л основания, а особенно предпочтительна концентрация от 0,2 до 2 моль/л основания. Температура реакции может составлять 20-100°C, особенно предпочтительно 90-100°C, при времени обработки 0,1-150 ч, особенно предпочтительно 2-6 ч.
В результате стадии элиминирования изменяется относительная интенсивность взаимодействия субстрата с отдельными ионами. В частности, можно уменьшить вторичные взаимодействия с поляризуемыми анализируемыми веществами. Образование «хвостов» у пиков, которое наблюдается вследствие подобных вторичных взаимодействий, уменьшается, так что поляризуемые ионы также элюируются симметрично. В то же время после элиминирования снижается также способность колонки к электростатическому взаимодействию, и уменьшается общая емкость ионообменного материала.
Следующий аспект изобретения относится к материалу полимерного носителя, предназначенному для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, в частности, хроматографическом способе, который является получаемым способом, включающим в себя описанные выше стадии, по меньшей мере стадии а, b и c.
Следующий аспект изобретения относится к материалу полимерного носителя, предназначенному для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, в частности, хроматографическом способе, который является получаемым способом, включающим описанные выше стадии, по меньшей мере стадии a и b, предпочтительно по меньшей мере стадии a и b и c, при этом материал полимерного носителя, обеспеченный на стадии a, является гидрофобным и микропористым или мезопористым. Под гидрофобным здесь имеют в виду, что данный материал полимерного носителя является неполярным, т.е. не содержит мономерных звеньев с дипольным моментом > 0,2 D. Под микропористым или мезопористым здесь подразумевают, что средний диаметр пор материала полимерного носителя составляет не более 50 нм.
Следующий аспект изобретения относистя к модифицированному материалу полимерного носителя, предназначенному для использования в качестве стационарной фазы в ионообменной хроматографии, когда способ включает в себя по меньшей мере стадии a, b, c и e.
Модифицированный материал полимерного носителя, получаемый таким образом, отличается тем, что
- в поперечном сечении материала полимерного носителя методом рентгеновской фотоэлектронной спектроскопии (РФЭС) являются определяемыми различия, составляющие по меньшей мере 20%, предпочтительно различия, составляющие по меньшей мере 50%, и особенно предпочтительно различия, составляющие 100%, в каждом случае начиная со значения максимального содержания O;
- кислородсодержащие группы ковалентно связаны с ядром материала полимерного носителя;
- материал полимерного носителя механически стабилен, так что перепад давления вдоль колонки, заполненной указанным материалом полимерного носителя, возрастает только линейно как функция от возрастающего расхода потока;
- материал полимерного носителя не имеет заряда при нейтральном значении рН;
- содержание азота в материале полимерного носителя составляет меньше 2%.
В частности, содержание кислорода на поверхности модифицированного материала полимерного носителя выше, чем в ядре. Модифицированный материал полимерного носителя, полученный данным способом, также в значительной степени химически инертен. Полученный таким образом материал полимерного носителя является регулируемым в том, что он может иметь в большей или меньшей степени кислородсодержащую поверхность. Благодаря своей структуре и свойствам поверхности, данный материал особенно подходит для использования в качестве стационарной фазы в способах аналитического или препаративного разделения. В частности, данный субстрат подходит для дальнейшей переработки в ионообменный материал в виде частиц, полученный описанным выше способом, который также является частью изобретения. Однако материал полимерного носителя можно также в дальнейшем переработать для использования в других способах абсорбционной хроматографии, способах HILIC, хроматографии с обращенной фазой, твердофазной экстракции и т.д. Вследствие своей микропористости или мезопористости, модифицированный материал полимерного носителя подходит для получения колонки с большим числом теоретических тарелок с соответственно хорошими характеристиками разделения, поскольку микропористые и мезопористые частицы механически стабильны даже при небольших диаметрах и таким образом диффузные пути могут быть короче.
Следующий аспект изобретения относится к материалу полимерного носителя, модифицированному согласно изобретению, предназначенному для использования в качестве стационарной фазы в ионообменной хроматографии, получаемому способом, который включает в себя описанные выше стадии, в котором ионообменные группы дополнительно вводят в материал полимерного носителя согласно стадии е.
Ионообменный материал, получаемый данным способом, может быть охарактеризован тем, что
- в поперечном сечении материала полимерного носителя методом рентгеновской фотоэлектронной спектроскопии РФЭС являются определяемыми различия, составляющие по меньшей мере 20%, предпочтительно различия, составляющие по меньшей мере 50%, и особенно предпочтительно различия, составляющие 100%, в каждом случае начиная со значения максимального содержания O,
- кислородсодержащие группы ковалентно связаны с сердцевиной материала полимерного носителя;
- ионообменный материал механически стабилен, так что перепад давления в колонке, заполненной указанным материалом полимерного носителя, возрастает только линейно как функция возрастающего расхода потока;
- ионообменный материал, за исключением введенных ионообменных групп, не основан на модификациях посредством реакций между эпоксидом и амином, которые могут быть обнаружены элиминированием по Гофману;
- необязательно число тарелок ионообменного материала > 50000 ТТ/м;
- необязательно селективность и емкость являются дополнительно регулируемыми с помощью стадии f элиминирования.
В частности, содержание кислорода на поверхности ионообменного материала выше, чем в его сердцевине. Например, содержание кислорода на поверхности модифицированного материала полимерного носителя, полученного способом изобретения до стадии с включительно, на 50% больше, предпочтительно, на 60% больше, исходя из значения максимального содержания О, чем во внутренних областях частиц, что определяется методом РФЭС.
Преимущество ионообменного материала согласно изобретению заключается в том, что гидрофильность и емкость или селективность и емкость можно настраивать на отдельных стадиях, т.е. независимо друг от друга. Для ионообменного материала, полученного способом изобретения, проявляются только слабые вторичные взаимодействия с поляризуемыми ионами и возрастает время удерживания сильно гидратированных ионов. Колонка, заполненная гидрофилизированным ионообменным субстратом, демонстрирует желаемую селективность. Данный материал не набухает и демонстрирует благоприятные свойства в нагрузочных тестах. Особенно эффективная колонка может быть заполнена данным ионообменным материалом.
Материал полимерного носителя, описанный выше, подходит для использования в качестве стационарной фазы в способе ионной хроматографии, в частности, способе хроматографии для разделения стандартных ионов, таких как фторид, хлорид, нитрит, бромид, нитрат, фосфат и сульфат ионы. Высокая ионообменная способность колонки особенно необходима для разделения небольших, одно- или двухзарядных ионов. Напротив, конвективный или перфузионный массоперенос раствора анализируемого вещества, достигаемый в случае макропористых структур, нежелателен. Макропористые структуры также часто связаны с меньшей механической допустимой нагрузкой.
Изобретение также относится к модифицированному материалу полимерного носителя, описанному выше, в котором материал полимерного носителя, обеспеченный на стадии а, по существу полностью образован из мономерных звеньев, выбранных из группы:
- мономерных звеньев, полученных из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, предпочтительно полученных из дивинилбензола;
- мономерных звеньев, полученных из этилвинилбензола;
- мономерных звеньев, полученных из стирола;
- их комбинации.
Под выражением «по существу полностью образованный» в описании подразумевается, что общая доля перечисленных мономерных звеньев в материале полимерного носителя, обеспеченном на стадии а, составляет по меньшей мере 95 масс. %, предпочтительно по меньшей мере 98 масс. %, особенно предпочтительно по меньшей мере 99 масс. %. Другими словами, мономерные звенья могут быть получены по существу полностью из соединений, не содержащих атомов кислорода, так что образуется сердцевина гидрофобной частицы.
Следующий аспект изобретения относится к описанному выше модифицированному материалу полимерного носителя, в котором материал полимерного носителя, обеспеченный на стадии а, имеет средний радиус пор от 1 до 50 нм, предпочтительно от 2 до 25 нм, более предпочтительно от 2 до 10 нм, измеренный путем сорбции азота с использованием модели BJH. Измерение проводили, как описано ниже в Примере 5.
Следующий аспект изобретения относится к описанному выше модифицированному материалу полимерного носителя, в котором материал полимерного носителя, обеспеченный на стадии а, имеет удельную площадь поверхности от 80 до 1000 м2/г, предпочтительно от 100 до 800 м2/г, еще более предпочтительно от 200 до 600 м2/г, измеренную путем сорбции азота с использованием модели ВЕТ. Измерение проводили, как описано ниже в Примере 5. Большая удельная площадь поверхности повышает емкость и разрешающую способность колонки, особенно при разделении ионов малого размера, например, стандартных ионов.
Следующий аспект изобретения относится к описанному выше модифицированному материалу полимерного носителя, в котором материал полимерного носителя, обеспеченный на стадии а, имеет устойчивость к давлению до 220 бар, пpедпочтительно до 250 бар. Под устойчивостью к давлению здесь имеется в виду, что увеличение давления как функция расхода потока ведет себя только линейно. Измерение проводили, как описано ниже в Примере 6. Высокая устойчивость к давлению является результатом малого диаметра частиц и микро/мезопористой структуры частиц.
Предпочтительно, модифицированный материал полимерного носителя представлен в виде частиц, предпочтительно, в виде сферических частиц, более предпочтительно, в виде сферических частиц со средним размером частицы (медианой) от 1 до 50 мкм. Особенно предпочтительно, данные частицы представлены в диапазоне размеров от 2 до 25 мкм и особенно предпочтительно в диапазоне размеров от 3 до 9 мкм. Размер частиц в данном случае представляет собой среднюю величину между самой длинной и самой короткой прямой линиями, проходящими через центр частицы, что можно измерить методом сканирующей электронной микроскопии (SEM) с автоматической оценкой изображения.
Размер частиц можно регулировать за счет подходящей скорости перемешивания, выбора растворителя, концентрации полимера в растворителе и т.д. Эти способы известны специалисту в данной области техники. Полимер-носитель такой формы и размера имеет соотношение объем/площадь поверхности, которое, как было обнаружено, особенно выгодно для обменной емкости. Он демонстрирует высокий коэффициент диффузии в поры и легко подвергается уплотнению.
Один из аспектов изобретения относится к описанному выше модифицированному материалу полимерного носителя, в котором данный модифицированный материал полимерного носителя стабилен в интервале рН от 0 до 14. Под pH стабильностью в описании подразумевают, что время удерживания сульфата в колонке, заполненной модифицированным материалом полимерного носителя после промывания 1M раствором NaOH и/или промывания 1M раствором HCl, отличается не более чем на 8%, предпочтительно не более чем на 5%, более предпочтительно не более чем на 3% от времени удерживания сульфата в колонке, заполненной модифицированным материалом полимерного носителя, который перед этим не подвергался воздействию среды со значениям рН 0 и/или 14. Метод измерения стабильности pH основан на приведенном ниже варианте осуществления Примера 8.
Следующий аспект изобретения относится к колонке для ионнообменной хроматографии, заполненной модифицированным материалом полимерного носителя, предпочтительно, дисперсным модифицированным материалом полимерного носителя, который является получаемым способом настоящего изобретения.
Кроме того, изобретение относится к способу хроматографического разделения анализируемых веществ, отличающемуся тем, что раствор, содержащий анализируемые вещества, приводят в контакт с модифицированным материалом полимерного носителя согласно изобретению, в частности, пропускают через колонку для ионообменной хроматографии согласно изобретению.
Таким образом, изобретение относится к применению материала полимерного носителя, получаемого способом изобретения, для аналитического или препаративного разделения анализируемых веществ, в частности, к применению в анионообменной хроматографии, катионообменной хроматографии и/или HILIC (жидкостной хроматографии гидрофильного взаимодействия).
Для дополнительной иллюстрации изобретения описаны следующие примеры вариантов осуществления. Примеры вариантов осуществления не ограничивают содержания раскрытия и формулу изобретения.
В изобретении представлены следующие фигуры:
Фиг 1: Схематическое изображение иллюстративной последовательности модификации материала полимерного носителя;
Фиг 2: Схематическое изображение стадий b.1 и b.2 модификации;
Фиг 3: Схематическое изображение стадий b.1 и b.2 модификации с последующими иллюстративными стадиями c и d.1 модификации;
Фиг 4: Схематическое изображение альтернативной стадии c или d.2 модификации;
Фиг 5: Схематическое изображение результата стадии d.1 модификации с последующей реакцией с BDGE (диглицидиловый эфир бутандиола);
Фиг 6: Схематическое представление примера стадии е модификации, следующей за стадией c или d. 2, введение ионообменной группы;
Фиг 7: Хроматограмма, получаемая с использованием хроматографической колонки согласно варианту осуществления 1 или 4;
Фиг 8: Хроматограммы, получаемые с использованием хроматографических колонок согласно примеру 1 или 4 варианта осуществления при увеличивающемся числе повторений стадии d модификации;
Фиг 9: Зависимость давления от расхода потока, определенная на хроматографической колонке согласно изобретению;
Фиг 10: Хроматограммы, получаемые с использованием хроматографических колонок согласно примеру 1 или 4 варианта осуществления при увеличении продолжительности стадии f;
Фиг 11: Зависимость давления от расхода потока, определенная на хроматографической колонке, заполненной материалом полимерного носителя согласно изобретению со стадии b.2 способа;
Фиг 12: Результат анализа размера частиц методом SEM (число по диаметру [мкм]).
Пример 1:
Все используемые вещества имели классификацию «химически чистые» или «чда» (за исключением пероксида водорода и муравьиной кислоты), растворители отделяли от низкокипящих компонентов перегонкой на роторном испарителе.
Окисление перменганатом калия
В колбу для сульфатирования объемом 350 мл помещали 10,0 г PS/DVB (55% DVB в этилвинилбензоле (EVB)) и суспендировали в 100 мл ацетонитрила. После этого в течение 20 минут прибавляли 5,0 г KMnO4, растворенного в 100 мл воды. Доводили рН реакционной смеси до кислых значений добавлением небольшего количества кислоты. Полученную суспензию перемешивали в течение 120 ч при 25°C. Частицы обрабатывали полуконцентрированной хлористоводородной кислотой, после чего промывали водой сверхвысокой чистоты. Продукт сушили в вакуумном сушильном шкафу до постоянной массы. Конечная масса составляла 9,7 г.
Восстановление алюмогидридом лития
В реактор объемом 500 мл помещали 8,6 г высушенных окисленных частиц и перемешивали с 150 мл тетрагидрафурана (ТГФ). В атмосфере аргона и с внешним охлаждением до 5°C медленно прибавляли 1,5 г алюмогидрида лития и перемешивали еще с 50 мл ТГФ. Реактор нагревали до комнатной температуры и перемешивали смесь в течение 17 ч. Реакцию останавливали путем медленного добавления воды. После этого проводили обработку смесью вода/ацетон, подкисляли разбавленной серной кислотой и промывали водой до нейтральной реакции. После конечного промывания ацетоном полученное твердое вещество сушили в вакуумном сушильном шкафу. Получали 8,35 г частиц.
Взаимодействие с эпихлоргидрином (ECH)
В трехгорлую колбу объемом 250 мл помещали 7,6 г восстановленных, высушенных частиц. Прибавляли 35 мл эпихлоргидрина и смесь трижды вакуумировали и продували аргоном. Раствор нагревали до 45°C. После этого прибавляли 7 мл раствор катализатора межфазного переноса (3 г гидроксида тетрабутиламмония в 10 мл воды), затем добавляли 140 мл раствор гидроксида натрия. Реакционную смесь перемешивали в течение 3,5 часов, а затем реакцию останавливали, добавляя смесь вода/этанол. Обработку осуществляли смесью вода/этанол или вода/ацетон. Продукт использовали непосредственно на следующей стадии без высушивания.
Взаимодействие с бутандиолом
После этого описанный выше полимер суспендировали в 70 мл бутандиола в смеси с 1,98 г KOH при комнатной температуре, а затем перемешивали при 130°C в течение 18 ч. После этого реакционную смесь оставляли для взаимодействия на несколько минут. По окончании реакции реакционную смесь разбавляли водой и фильтровали. Полимер несколько раз промывали водой и ацетоном. Остаток на фильтре сушили в вакуумном сушильном шкафу в течение ночи. Получали 7 г полимера.
Присоединение спейсера
В 16,5 мл ДМСО и 16,5 мл диглицидилового эфира бутандиола суспендировали 6,6 г описанного выше полимера и реакционную колбу трижды вакуумировали и заполняли аргоном. После этого добавляли 1,4 мл 1M раствора бромида тетрабутиламмония и 16,5 мл 0,6M NaOH (водн.) и перемешивали при помощи механической мешалки в течение 22 ч. Реакцию останавливали, добавляя смесь воды и этанола в соотношении 1:1. Повторное промывали проводили смесью вода/этанол. В заключение продукт фильтровали досуха. Полимер использовали непосредственно на следующей стадии.
Введение анионообменных групп
После этого описанный выше полимер суспендировали в 45 мл ДМСО, не перенося его, а затем прибавляли 45 мл воды. Суспензию нагревали до 70°C и добавляли 45 мл N-метилпирролидина. По прошествии 2 ч протекания реакции, реакцию останавливали, добавляя уксусную кислоту. Полимер фильтровали и несколько раз промывали водой. Влажный полимер использовали непосредственно на следующей стадии.
Элиминирование
Описанный выше полимер суспендировали в 50 мл воды и добавляли 7,5 мл 40%-ного NaOH (водн.). После этого суспензию перемешивали в течение 4 ч при 100°C. Реакцию останавливали фильтрованием. Остаток на фильтре несколько раз промывали водой сверхвысокой чистоты, а затем помещали в полиэфирэфиркетоновую (PEEK) колонку 4 x 100 мм в соответствии с известными методиками набивки под высоким давлением.
Пример 2:
Окисление кислородной плазмой низкого давления
40 г PS/DVB (55% DVB в EVB) окисляли в плазменно-порошковой установке под действием кислородной плазмы. Частицы можно подвергнуть дальнейшей переработке непосредственно после обработки.
Восстановление алюмогидридом лития (LAH)
В реакторе объемом 1000 мл суспендировали 30 г окисленного высушенного полимера в 250 мл сухого диэтилового эфира при выравнивании давления. После выдерживания до 25°C медленно прибавляли 6 г алюмогидрида лития в атмосфере аргона, нагревали до 30°C в течение 6 ч при перемешивании и перемешивали еще 20 ч при комнатной температуре. Реакцию останавливали, охлаждая реакционную смесь до 0°C и медленно прибавляя 15 мл этилацетата. Обработку проводили с использованием воды, разбавленной серной кислоты, воды, 5 масс.% раствора NaOH, воды сверхвысокой чистоты и разбавленной хлористоводородной кислоты.
Полимер промывали водой сверхвысокой чистоты до нейтральной реакции и фильтровали до сухого состояния с использованием ацетона. Продукт сушили в вакуумном сушильном шкафу. Получали 30 г полимера.
Взаимодействие с эпихлоргидрином
В трехгорлую колбу объемом 250 мл помещали 5,0 г восстановленных высушенных частиц и прибавляли 25 мл эпихлоргидрина. Суспензию трижды вакуумировали и продували аргоном. После этого прибавляли 1,75 мл 1 M раствор катализатора межфазного переноса (бромида тетрабутиламмония в воде) и 25 мл 30%-ного раствора гидроксида натрия, нагревали до 45°C и перемешивали в течение 3,5 часов. Реакцию завершали, добавляя смесь вода/этанол. Очистку продукта осуществляли, промывая его неесколько раз смесью вода/этанол или вода/ацетон. Продукт использовали непосредственно на следующей стадии без осушки.
Взаимодействие с бутандиолом
После этого описанный выше полимер суспендировали в 50 мл бутандиола с 1,3 г KOH при комнатной температуре и нагревали до 130°C в течение 18 ч. Затем реакционную смесь оставляли для отстаивания. По окончании времени реакции к реакционной смеси добавляли 200 мл воды и фильтровали.
Остаток на фильтре промывали водой и ацетоном. Продукт сушили в вакуумном сушильном шкафу и получали 4,2 г сухого продукта.
Присоединение спейсера
В 10 мл ДМСО и 10 мл диглицидилового эфира бутандиола суспендировали 3,8 г описанного выше полимера и трижды подвергали циклическому изменению давления, повторно заполняя реакционный сосуд аргоном. Затем прибавляли 0,8 мл 1M раствора бромида тетрабутиламмония и 10 мл 0,6 M NaOH (водн.) и перемешивали в течение 22 ч. После этого реакционную смесь трижды подвергали изменениями давления. Затем к смеси прибавляли 200 мл смеси воды и этанола в соотношении 1:1 и фильтровали. Данную процедуру промывания повторяли несколько раз, продукт фильтровали до сухого состояни и использовали непосредственно на следующей стадии.
Введение ионообменных групп
Описанный выше полимер суспендировали в 30 мл ДМСО, прибавляли 30 мл волы и нагревали суспензию до 70°C. После достижения температуры реакции прибавляли 30 мл N-метилпирролидина и перемешивали в течение 2 ч при 70°C. По истечении времени реакции добавляли 60 мл концентрированной уксусной кислоты и фильтровали продукт. Остаток на фильтре промывали водой до нейтральной реакции и использовали в реакции элиминирования.
Элиминирование
Описанный выше полимер суспендировали в 100 мл воды, смешивали с 20 мл 30%-ного NaOH (водн.) и перемешивали в течение 28 ч при 100°C. По истечении времени реакции реакцию останавливали добавлением хлористоводородной кислоты, суспензию фильтровали, остаток на фильтре несколько раз промывали водой до нейтральной реакции, а затем набивали им PEEK колонку 4 x 100 мм в соответствии с известными методиками набивки под высоким давлением.
Пример 3:
Окисление мета-хлорпербензойной кислотой
В стеклянную бутылку объемом 250 мл с навинчивающейся крышкой помещали 20 г PS/DVB (55% DVB в EVB) и суспендировали его в 93 г дихлорометана. После этого прибавляли 5,5 г мета-хлорпербензойной кислоты в твердой форме и перемешивали реакционную смесь при комнатной температуре в течение 18 ч на встряхивателе. Продукт несколько раз промывали этанолом и водой и сушили в вакуумном сушильном шкафу до постоянной массы. Конечная масса составляла 19,0 г.
Гидролиз хлористоводородной кислотой
В стеклянную бутылку объемом 250 мл с навинчивающейся крышкой помещали 19 г очищенного высушенного полимера и суспендировали его в 52 г ацетона и 13 г 37%-ной хлористоводородной кислоты. Реакционную смесь перемешивали при 40°C на встряхивателе с циркуляцией воздуха в течение 21 ч. После этого смесь удаляли из бутылки. Продукт промывали водой до нейтральной реакции, затем несколько раз промывали водой и ацетоном и сушили в вакуумном сушильном шкафу до постоянной массы. Конечная масса составляла 18,5 г.
Взаимодействие с эпихлоргидрином
В трехгорлой колбе объемом 250 мл, содержащей 60 мл эпихлоргидрина, суспендировали 11,8 г гидролизованного высушенного полимера. Реакционный сосуд подвергали трем циклам вакуумирования/заполнения аргоном. Реакционную смесь нагревали до 45°C при перемешивании, а затем добавляли 3 мл 1 M (водн.) раствора бромида тетрабутиламмония. После этого прибавляли 60 мл 30%-ного (водн.) раствора гидроксида натрия и энергично перемешивали. Через 22 ч реакционную смесь разбавляли 200 мл воды и 200 мл этанола, а затем полимер выделяли фильтрованием. Полимер промывали ацетоном, водой и снова ацетоном.
Взаимодействие с бутандиолом
В трехгорлой колбе объемом 100 мл суспендировали 5 г описанного выше продукта с 1,4 г гидроксида калия и 50 мл 1,4-бутандиола и перемешивали в течение 19 ч при 130°C. После этого смесь охлаждали и перемешивали с 45 мл воды. Продукт выделяли фильтрование и промывали водой до нейтральной реакции, затем сушили до постоянной массы в вакуумном сушильном шкафу. Конечная масса составляла 4,0 г.
Взаимодействие с диглицидиловым эфиром 1,4-бутандиола
В трехгорлой колбе объемом 100 мл, содержащей 8 мл диметилсульфоксида и 8 мл диглицидилового эфира 1,4-бутандиола, суспендировали 3,1 г описанного выше продукта. Реакционный сосуд подвергали трем циклам вакуумирования/заполнения аргоном. К реакционной смеси при перемешивании прибавляли 0,8 мл 1 M (водн.) раствора бромида тетрабутиламмония и 8 мл 0,6 M раствора гидроксида натрия. Через 22 ч к реакционной смеси прибавляли 25 мл воды и 25 мл этанола, а затем ее фильтровали. Продукт один раз промывали водой и этанолом.
Введение ионообменных групп
В трехгорлой колбе объемом 100 мл описанный выше продукт суспендировали в 15 мл диметилсульфоксида, 15 мл воды и 15 мл N-метилпирролидина. Реакционную смесь перемешивали при 70°C в течение одного часа, затем охлаждали и прибавляли 30 мл уксусной кислоты. Полимер выделяли фильтрованием и промывали водой.
Элиминирование
В круглодонной колбе объемом 100 мл суспендировали описанный выше продукт в 50 мл воды и 7,5 мл 40% -ного (водн.) раствора гидроксида натрия и нагревали смесь до 100°C. Через 4 ч реакционную смесь охлаждали и фильтровали. Продукт дважды промывали водой и заполняли им PEEK колонку 4 х 100 мм в соответствии с известными методиками набивки под высоким давлением.
Пример 4:
Окисление муравьиной кислотой
В трехгорлой колбе объемом 500 мл с выравниванием давления суспендировали 25,0 г PS/DVB (55% DVB в EVB) в 188 мл муравьиной кислоты. Через капельную воронку медленно прибавляли 54 мл 35%-ного пероксида водорода и охлаждали реакционный раствор снаружи. После рассеивания теплоты реакции смесь перемешивали 65 ч при комнатной температуре. По завершении второй реакции реакционную смесь промывали водой сверхвысокой чистоты, а затем сушили в вакуумном сушильном шкафу до постоянной массы. Конечная масса составляла 27,78 г.
Восстановление алюмогидридом лития
В трехгорлой колбе объемом 500 мл, находящейся под давлением, суспендировали 27,64 г окисленного высушенного полимера в 270 мл сухого диэтилового эфира, охлаждали до 0°C на бане со льдом и осторожно прибавляли 8,8 г алюмогидрида лития при перемешивании. По завершении прибавления баню со льдом убирали и кипятили при перемешивании в течение 10 ч и перемешивали еще 24 ч при комнатной температуре. Реакцию останавливали путем внешнего охлаждения и добавления диэтилового эфира, этилацетата и воды свехвысокой очистки.
После реакции оставшегося гидрида реакционную смесь помещали на лед и прибавляли разбавленную охлажденную серную кислоту при перемешивании. Реакционную смесь промывали следующими растворами: водой, 5%-ным раствором NaOH, водой, разбавленной уксусной кислотой, водой и ацетоном. Остаток на фильтре фильтровали до сухого состояния и сушили в вакуумном сушильном шкафу. Выход составлял 26,20 г.
Взаимодействие с эпихлоргидрином
В ЕСН и ДМСО объемом по 20 мл каждый суспендировали 4,00 г полимера, подвергали воздействию ультразвука в течение 15 мин в ультразвуковой бане, а затем подвергали двум изменениям давления, повторно заполняя реакционный сосуд аргоном. После добавления 4,30 мл 25%-ного раствора гидроксида тетраметиламмония в воде перемешивание продолжали в течение 2 ч при комнатной температуре. Реакционную смесь фильтровали и промывали смесью воды и 2-пропанола в соотношении 1:1 и ацетоном. Остаток на фильтре фильтровали до сухого состояния.
Взаимодействие с бутандиолом
После этого описанный выше полимер суспендировали в 40 мл бутандиола вместе с 1,12 г KOH при комнатной температуре, а затем перемешивали при 120°C в течение 20 ч. Реакционную смесь несколько раз промывали водой и ацетоном. По истечении времени реакции реакционную смесь несколько раз промывали водой и ацетоном. Продукт фильтровали до сухого состояния. Массы сырого продукта составляла 13,36 г.
Цикл нанесения покрытия: взаимодействие с эпихлоргидрином (ЕСН)
Массу еще влажного полимера, полученного на описанной выше стадии, доводили до 13,50 г при помощи воды и прибавляли 1 мл 1M раствора бромида тетрабутиламмония и 20 мл ECH. Затем добавляли 10,5 мл 50%-ного раствора NaOH (водн.) и перемешивали в течение 5,5 ч. Послеэтого реакционную смесь промывали смесью воды и 2-пропанола в соотношении 1:1 и ацетоном. Остаток на фильтре фильтровали до сухого состояния.
Цикл нанесения покрытия: взаимодействие с бутандиолом
Описанный выше полимер суспендировали в 40 мл бутандиола вместе с 1,12 г KOH при комнатной температуре, а затем перемешивали при 120°C в течение 18 ч. Реакционную смесь несколько раз промывали водой и ацетоном. По истечении времени реакции реакционную смесь несколько раз промывали водой и ацетоном. Продукт фильтровали до сухого состояния. Полимер сушили последний раз перед следующей реакцией. Выход составлял 5,66 г.
Присоединение спейсера
В 6 мл ДМСО и 6 мл диглицидилового эфира бутандиола суспендировали 2,30 г описанного выше полимера и трижды осуществляли цикл изменения давления, повторно заполняя реакционный сосуд аргоном. Прибавляли при перемешивании 0,5 мл 1M раствора бромила тетрабутиламмония и 6 мл 0,6 M NaOH (водн.) и перемешивали в течение 22 ч. После этого реакционную смесь подвергали трехкратному изменению давления. Затем реакционную смесь промывали смесью воды и 2-пропанола в соотношении 1:1 и фильтровали до сухого состояния.
Введение анионообменных групп
Описанный выше полимер суспендировали в 5 мл ДМСО и смешивали с 5 мл воды и 5 мл N-метилпирролидина. После этого реакционную смесь перемешивали в течение 1 ч при 70°C. Реакцию останавливали, добавляя воду и разбавленную уксусную кислоту. Затем остаток на фильтре промывали разбавленной хлористоводородной кислотой, водой и ацетоном. Полученный полимер сушили при 60°C в сушильном шкафу. Выход составлял 2,54 г.
Элиминирование
Описанный выше полимер суспендировали в 50 мл воды, добавляли 5 мл 30%-ного NaOH (водн.) и перемешивали в течение 2 ч при 100°C. Реакцию останавливали фильтрованием с последующим промыванием водой, разбавленной HCl, водой и ацетоном. Остаток на фильтре фильтровали до сухого состояния, а полимера затем сушили при 60°C в сушильном шкафу. Значительной потери массы не наблюдалось.
На фигуре 1 схематически показаны различные иллюстративные последовательности последовательности модификации сердцевины материала полимерного носителя (pDVB). После первоначального окисления (b.1) и последующего восстановления (в качестве альтернативы - последующего гидролиза, b.2) получают материал полимерного носителя, содержащий на поверхности гидроксигруппы (pDVB-OH). После этого материал полимерного носителя, содержащий OH группы, (pDVB-OH), может быть введен во взаимодействие с эпихлоргидрином (ECH). В результате образуется соединение согласно стадии c. Альтернативным образом, материал полимерного носителя, содержащий OH группы, (pDVB-OH), может быть введен во взаимодействие с диглицидиловым эфиром бутандиола (BDGE) на стадии c.
Согласно изобретению, материал полимерного носителя может быть введен во взаимодействие после стадии b.2 в одном или более циклов нанесения покрытия с ECH, диолом, а затем снова с ECH или BDGE (стадии c, d.1, d.2). Кроме того, материал полимерного носителя может быть введен во взаимодействие непосредственно с BDGE после стадии b.2, например. В результате образуется модифицированный материал полимерного носителя, содержащий на поверхности подходящие реакционноспособные функциональные группы. В представленном примере они являются эпоксигруппами.
В принципе, допустима любая комбинация модификаций с использованием ECH/BDGE на стадиях c и d.2, соответственно, а также любая комбинация превращений/гидролиза диолов на стадии d.1. Однако, как описано выше, предпочтительно введение спейсерной молекулы во время последнего цикла нанесения покрытия на стадии d.2.
Модифицированный материал полимерного носителя, представленный на фигуре 1, подходит для последующего введения ионообменных групп. Содержание кислорода на поверхности модифицированного материала полимерного носителя возрастает в порядке вариантов, перечисленных в предыдущем разделе (варианты сверху вниз на фигуре). Если ионообменные группы вводят в каждый из продуктов, получают ионообенный материал, гидрофильность которого возрастает в порядке перечисленных вариантов. Повышенная гидрофильность проявляется, например, в снижении селективности σ при переходе от NO3 к Cl.
Фигуры с 2 по 6 предназначены для иллюстрации стадий способа согласно изобретению и показывают последовательность реакций в очень упрощенной форме. Они не претендуют на полноту. Основное внимание уделяется соответствующей модификации поверхности материала полимерного носителя. Часть материала полимерного носителя, не модифицированная на соответствующей стадии, упрощенно показана в виде сферической частицы.
На фигуре 2 схематически представлены стадии b.1 и b.2 модификации. В результате окисления и восстановления (в качестве альтернативы - гидролиза) получают материал полимерного носителя, содержащий ОН группы на поверхности. Специалисту в данной области техники известно, что на стадии окисления могут образовываться не только кетоны. В зависимости от обработки и, в частности, во время обработки KMnO4, помимо кетонов, могут также образовываться диолы, дикетоны, либо, в качестве продукта расщепления, дикарбоновые кислоты. Такие процессы и промежуточные соединения включены в заявленный способ и не подразумевают исключения из примеров, показанных на фигурах.
На фигуре 3 схематически показаны стадии b.1 и b.2 модификации, которые сопровождаются иллюстративными стадиями c и d.1 модификации. В представленном варианте соединение, использованное на стадии с, содержащее по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, представляет собой эпихлоргидрин. В представленном варианте осуществления полифункциональное соединение, содержащее гидроксигруппы, использованное на стадии d.1, представляет собой бутандиол. Последовательность нанесения покрытия, включающую в себя чередующиеся реакции с эпихлоргидрином и бутандиолом, можно повторять в виде d.1 и d.2. Полученные частицы снова показаны в абстрактном виде после двойной стрелки.
На фигуре 4 схематично показана альтернативная стадия c или d.2 модификации. Соединение, использованное на стадии с или d.2, содержащее по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с ионообменными группами и/или гидроксигруппами, представляет собой BDGE.
Превращение с участием BDGE может также завершать один или более циклов нанесения покрытия, как показано на фигуре 3. На фигуре 5 схематически и в качестве примера показан результат стадии d.1 модификации. После использования ECH на стадии c осуществляли стадии нанесения покрытия с использованием ECH на стадии d.2. После проведения взаимодействия с бутандиолом осуществляли последнюю стадию d.2 с использованием BDGE в качестве спейсера. В результате получали модифицированный материал полимерного носителя, содержание кислорода на поверхности которого, при прочих равных условиях, выше, чем в материалах носителя, образующихся в результате схем Фиг. 3 / Фиг. 4.
На фигуре 6 схематически представлен пример стадии e модификации, следующей за стадией c или d.2, введение ионообменной группы. В представленном примере ионообменная группа образуется посредством кватернизации 1-метилпирролидина. В результате модификации согласно изобретению получают материал полимерного носителя, содержащий на поверхности боковые цепи, такие как цепи, показанные в примере.
На фигуре 7 представлена хроматограмма, полученная с использованием хроматографической колонки согласно примеру 1 или 4. Вдоль оси х отмечено время хроматографирования в минутах. Вдоль оси y отмечена проводимость в мкСм/см. Для этого колонку 150 x 4 мм заполняли сухим субстратом. В качестве элюента использовали 6,0 ммоль/л Na2CO3 и 1,0 ммоль/л NaHCO3. Анализируемые вещества стандартного раствора присутствуют на уровне базовой линии, отделенные друг от друга, с порядком элюирования слева направо: фторид, бромат, хлорид, нитрит, бромид, хлорат, нитрат, азид, фосфат, сульфат. Бромат количественно присутствует до хлорида (пики при 5,2, 5,7 мин времени хроматографирования) и на хроматограмме появляются высокосимметричные сигналы. Общее время хроматографирования на колонке составляет 15 мин. При использовании хроматографических колонок, приготовленных в условиях примера 1, получают сопоставимую хроматограмму.
На фигуре 8 представлены три хроматограммы, полученные с использованием хроматографической колонки согласно примеру 4. В примере 1 получены очень похожие хроматограммы. Вдоль оси х отмечено время хроматографирования в минутах. Вдоль оси y отмечена проводимость в мкСм/см. Кривые представляют хроматограммы идентичного стандартного раствора, со следующим порядком элюирования слева направо: фторид, хлорид, нитрит, бромид, нитрат, фосфат и сульфат. Было увеличено (сверху вниз) число повторений стадий d.1/d.2 модификации. В случае построенной кривой A последовательность стадий d.1/d.2 осуществляли один раз. В случае кривой из точек B последовательность стадий d.1/d.2 осуществляли дважды. В случае пунктирной кривой C последовательность стадий d.1/d.2 осуществляли трижды. Таким образом, в частности, в случае изображенной кривой A использовали полимерный субстрат, который подвергали обработке окислением/восстановлением, один раз обрабатывали ЕСН, а затем один раз - 1,4-бутандиолом, а затем вводили во взаимодействие с BDGE. В случае кривой B использовали полимерный субстрат, который подвергали обработке окислением/восстановлением, обрабатывали ЕСН, а затем 1,4-бутандиолом, потом снова обрабатывали ЕСН, а затем 1,4-бутандиолом, а после этого вводили во взаимодействие с BDGE. Кривая C представляет полимерный субстрат, подвергнутый обработке окислением/восстановлением. После этого имела место следующая последовательность стадий: взаимодействие с ECH, 1,4-бутандиолом, ECH, 1,4-бутандиолом, ECH, 1,4-бутандиолом, BDGE. Во всех трех случаях модифицированный субстрат впоследствии вводили во взаимодействие с метилпирролидином. Из хроматограмм очевидно, что сигналы анионов, на которые влияют хвосты (нитрит, бромид, нитрат), приобретают симметрию с увеличением числа циклов нанесения покрытия. Селективность σ от NO3 к Cl снижается при увеличении числа циклов нанесения покрытия. Общая емкость уменьшается.
На фигуре 9 представлена зависимость давления от расхода потока, измеренная на хроматографической колонке согласно изобретению, подготовленной согласно примеру 4, определенная при комнатной температуре. Вдоль оси y отмечено давление системы в МПа. Вдоль оси х отмечен расход потока в мл/мин. Было измерено семнадцать интервалов по 20 минут каждый при постепенном увеличении расхода потока от 1 мл/мин до 2,6 мл/мин. Давление линейно зависит от расхода потока. Это отличается от результатов, полученных с использованием стандартных колонок, заполненных гидрофильным субстратом pDVB. В случае стандартных колонок давление возрастает более чем линейно в зависимости от расхода потока. Например, может проявиться гиперболический наклон функции.
На фигуре 10 представлены хроматограммы, полученные с использованием хроматографических колонок согласно изобретению, в частности, хроматографических колонок, заполненных модифицированным материалом полимерного носителя согласно примеру 1 или 4. Вдоль оси х отмечено время хроматографирования в минутах. Вдоль оси y отмечена проводимость в мкСм/см. Продолжительность стадии f была переменной. Высушенным субстратом заполняли колонку 100 x 4 мм. В каждом случае кривые представляют хроматограмму идентичного стандартного раствора со следующим порядком элюирования слева направо: фторид, хлорид, нитрит, бромид, нитрат, фосфат и сульфат. В направлении сверху вниз кривые представляют хроматограммы, полученные после элиминирования колоночного субстрата на стадии f в течение 0 мин (A), 60 мин (B), 120 мин (C), 180 мин (D), 240 мин (E) и 300 мин (F). При исключении стадии f (0 мин), пик нитрата и пик фосфата перекрываются. Общая емкость снижается при увеличении времени элиминирования. Анализируемые вещества присутствуют на уровне базовой линии, отделенные друг от друга, и на хроматограмме появляются высокосимметричные сигналы. Общее время хроматографирования на колонке невелико и составляет приблизительно от 14 до 20 минут.
Пример 5: Определение среднего радиуса пор и удельной площади поверхности материала полимерного носителя, обеспеченного на стадии a.
В примере 1 осуществляют модификацию согласно изобретению с использованием PS/DVB (55% DVB в EVB). За счет этого образующийся исходный полимерный носитель является и гидрофобным, и микропористым, или мезопористым. Образующийся исходный полимерный носитель получают следующим образом:
Получение затравочной частицы полистирола при дисперсионной полимеризации стирола в этаноле при стабилизации поливинилпирролидоном и инициировании азобис(изобутиронитрилом). Получают частицу полистирола диаметром 1,5 мкм и MN=15 кг/моль, MW=55 кг/моль. Осуществляют набухание полученной частицы полистирола в эмульсии 55% дивинилбензола (DVB)/45% этилвинилбензола (EVB) и толуола в смеси вода/изоамиловый спирт, стабилизированной поливиниловым спиртом, и последующую полимеризацию, инициированную азобис(изобутиронитрилом). Получают пористую частицу поли(DVB-со-EVB) с высокой степенью сшивки с радиусом 5 мкм и пористостью 1 см3/г.
Средний радиус пор исходного материала полимерного носителя определяли методом сорбции азота с использованием модели BJH (Баррет, Джойнер, Халенда). Удельную площадь поверхности определяли методом сорбции азота с использованием модели BET (Брунауэр, Эммет, Теллер). Для обоих анализов использовали образец полимерного носителя PS/DVB массой 0,0945 г. Плотность материала образца составляла 1,05 г/см3. Измерения проводили на приборе Autosorb iQ S/N:14713051301 в 9 мм ячейке. Температура бани составляла 77,35 K. Конечная температура дегазации составляла 60°C. Оценку результатов измерения осуществляли с использованием программного обеспечения Quantachrome ASiQwin, версия 3.01. Измерения проводили дважды, один раз с временем выдержки 80 мин, другой раз с временем выдержки 40 мин. Скорость дегазации составляла 1,0°C/мин и 20,0°C/мин, соответственно. Средний радиус пор, полученный методом BJH, исходя из объема пор, составлял 5,060 нм. Рассчитанная удельная площадь поверхности в соответствии с Multi-Point BET Plot составляла 815,0 м2/г.
Пример 6: Определение устойчивости к сжатию материала полимерного носителя, полученного на стадии b.2
В примере 4 модификацию согласно изобретению осуществляют с использованием PS/DVB (55% DVB в EVB). Обеспеченный исходный материал полимерного носителя получают, как описано в Примере 5. Как окисление пероксидом водорода, так и восстановление алюмогидридом лития проводят на исходном материале полимерного носителя, как описано в Примере 4, в соответствии с результатами стадии b.2. Полученную частицу подвергали испытанию на сопротивление сжатию. Для проведения испытания на сопротивление сжатию колонку 250 x 4 мм заполняли полученными частицами и пропускали через колонку воду при возрастающем расходепотока. На фигуре 11 показана зависимость давления от расхода потока, определенная при комнатной температуре. Вдоль оси y отмечено давление системы в барах. Вдоль оси х отмечен расход потока в мл/мин. Было измерено восемь интервалов по 30 секунд каждый при постепенном увеличении расхода потока от 0,2 мл/мин до 1,6 мл/мин. Из фигуры видно, что давление линейно зависит от расхода потока до давления давления 400 бар или 40 МПа.
Пример 7: Определение среднего размера частиц
Округлость и средний диаметр часиц определяли для образца исходного материала полимерного носителя, обеспеченного на стадии а. Образец напыляли на предметное стекло сканирующего электронного микроскопа в виде одного слоя частиц. Для этого образец наносили на предметное стекло сканирующего электронного микроскопа в виде одного слоя частиц и покрывали золотом при помощи устройства для ионного напыления LOT AutomaticSputterCoater MSC1, соединенного с выкуумным насосом Vacubrand RZ 6. При помощи сканирующего электронного микроскопа (Phenom ProX) получали серию из 27 изображений, и идентифицировали и измеряли отдельные частицы при помощи программы Olympus Imaging Solutions Scandium. Проводили анализ сферического диаметра и правильности округлой формы идентифицированных частиц. Все изображения анализировали методом пакетной обработки с использованием одних и тех же пороговых значений и настроек измерения. Всего было измерено 6039 частиц и их округлость всегда была ≥0,8. Результаты измерений представлены на фигуре 12. На ней по оси y отложено число частиц, а по оси х – диаметр в мкм. Наименьшие измеренные радиусы составили 0,8 мкм, наибольшие – до 10 мкм. Средний диаметр (медиана) составляла 4,59 мкм при относительном сверднеквадратичном отклонении 6,23%. Коэффициент полидисперсности PDI (Mw/Mn) равнялся 1,044.
Пример 8: Определение pH стабильности
pH стабильность определяют для образца дисперсного материала полимерного носителя согласно примеру применения 5, модифицированного согласно примеру 4, т.е. соответствующего полимерному материалу, который можно получить согласно стадиям от a до f. С этой целью данным образцом заполняли хроматографическую колонку (250 x 4 мм) и определяли время удерживания сульфата по результатам десяти измерения при элюировании 6 ммоль/л Na2CO3 и 1 ммоль/л NaHCO3. Затем через колонку в течение 14 ч пропускали поток элюента, содержащий 6 ммоль/л Na2CO3 и 1 моль/л NaOH (pH 14), при расходе 0,8 мл/мин. После этого вновь проводили 10 измерений времени удерживания сульфата при элюировании 6 ммоль/л Na2CO3 и 1 ммоль/л NaHCO3. Затем колонку промывали в течение 14 ч элюентом, состоящим из 6 ммоль/л Na2CO3 и 1 ммоль/л HNO3 (pH 0), при расходе 0,8 мл/мин. После этого вновь проводили 10 измерений времени удерживания сульфата при элюировании 6 ммоль/л Na2CO3 и 1 ммоль/л NaHCO3. При каждой загрузке элюента осуществляли промывание водой в течение 1 ч, чтобы избежать выпадения осадка. Возможное изменение изучали, исходя из времени удерживания и числа тарелок для сульфата; максимальное отклонение обоих параметров составляло 3% от параметров, определенных вначале после основной и кислотной обработки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СВЯЗЫВАЮЩИХ МЕТАЛЛ ЧАСТИЦ НА ОСНОВЕ ФОСФИНА | 2012 |
|
RU2598946C1 |
| КОНЪЮГАТЫ ГИДРОКСИАЛКИЛКРАХМАЛА И G-CSF | 2004 |
|
RU2370281C2 |
| КЛЕИ С ЛАТЕНТНОЙ РЕАКЦИОННОЙ СПОСОБНОСТЬЮ ДЛЯ ИДЕНТИФИКАЦИОННЫХ ДОКУМЕНТОВ | 2008 |
|
RU2496651C2 |
| ЧАСТИЦЫ В ФОРМЕ СЕРДЦЕВИНЫ В ОБОЛОЧКЕ И СПОСОБ ИХ ПРИГОТОВЛЕНИЯ | 2001 |
|
RU2286845C2 |
| НОСИТЕЛЬ ДЛЯ АФФИННОЙ ХРОМАТОГРАФИИ И СПОСОБ ОЧИСТКИ БИОЛОГИЧЕСКОГО ВЕЩЕСТВА | 2016 |
|
RU2694637C1 |
| ГЛИЦЕРИН-СВЯЗАННЫЕ ПЭГИЛИРОВАННЫЕ САХАРА И ГЛИКОПЕПТИДЫ | 2007 |
|
RU2460543C2 |
| ПОЛИМЕРНЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ПРИВИТЫЕ ПОЛИМЕРНЫЕ СЕТКИ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2017 |
|
RU2748735C2 |
| ПЕРФТОРИРОВАННЫЕ СУЛЬФОНИЛГАЛОГЕНИДЫ И АНАЛОГИЧНЫЕ ИМ СОЕДИНЕНИЯ КАК МОДИФИКАТОРЫ ПОЛИМЕРНЫХ ПОДЛОЖЕК | 2002 |
|
RU2291146C2 |
| ГИБРИДНЫЕ СИСТЕМЫ-НОСИТЕЛИ | 2008 |
|
RU2491311C2 |
| ГЛИКОПЭГИЛИРОВАННЫЙ ГРАНУЛОЦИТАРНЫЙ КОЛОНИЕСТИМУЛИРУЮЩИЙ ФАКТОР | 2004 |
|
RU2400490C2 |
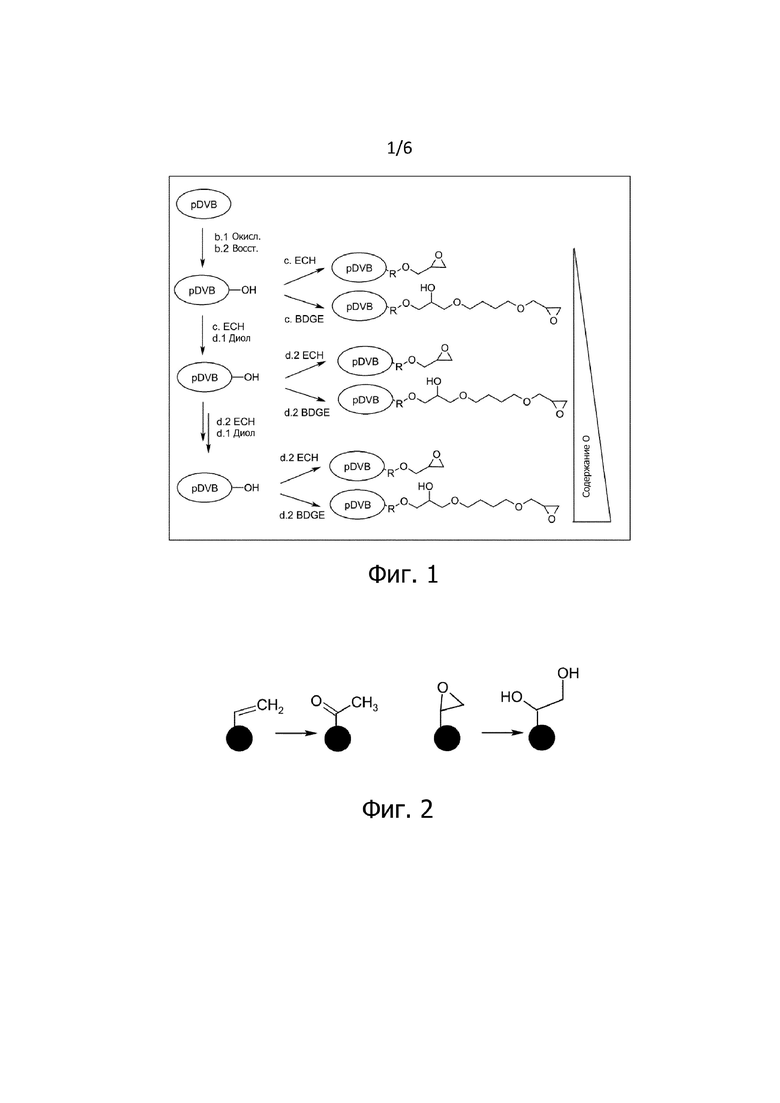
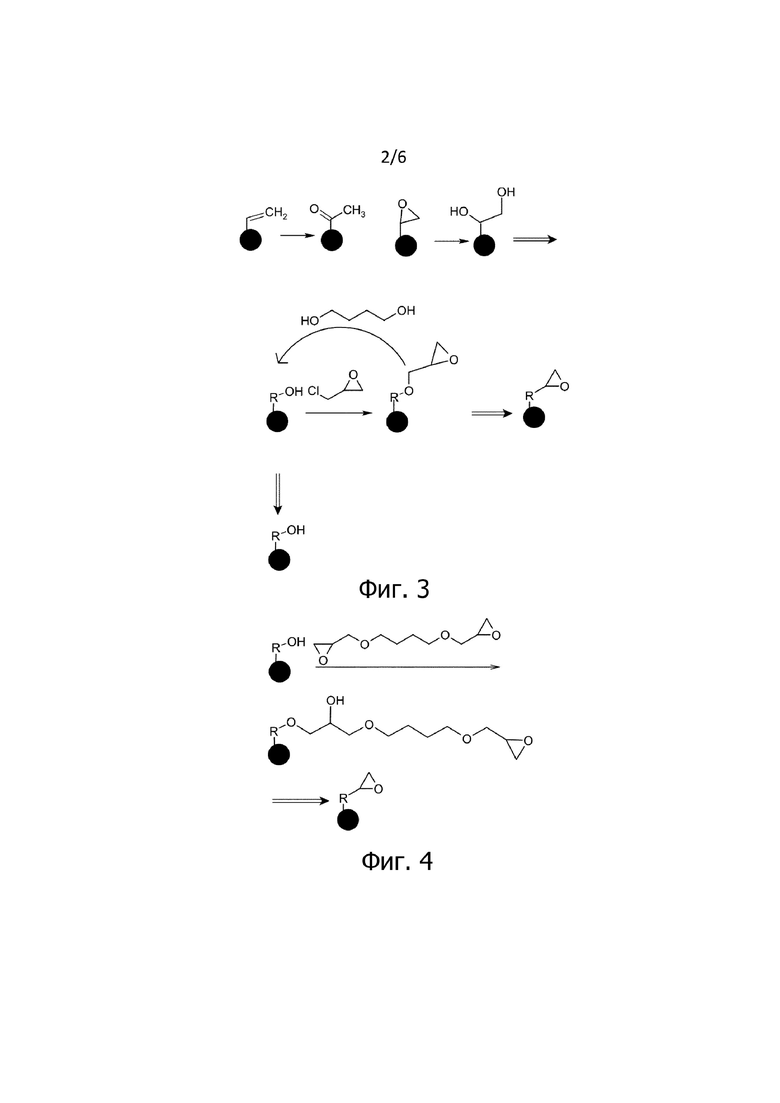
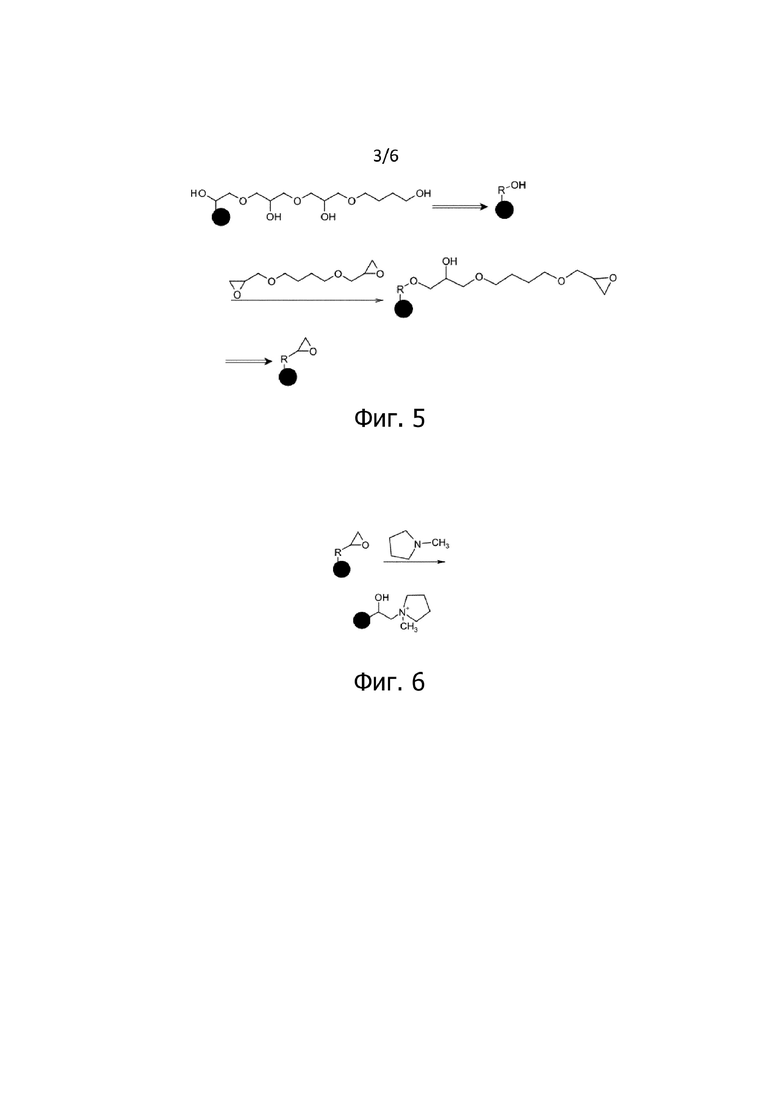
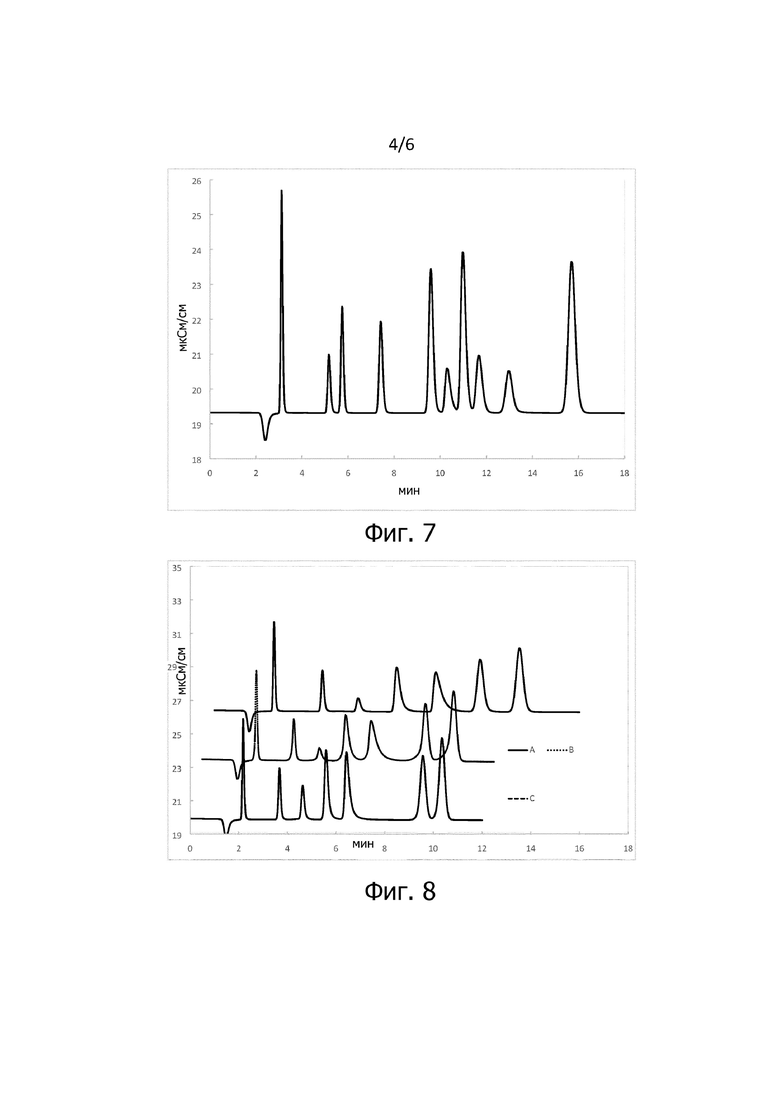
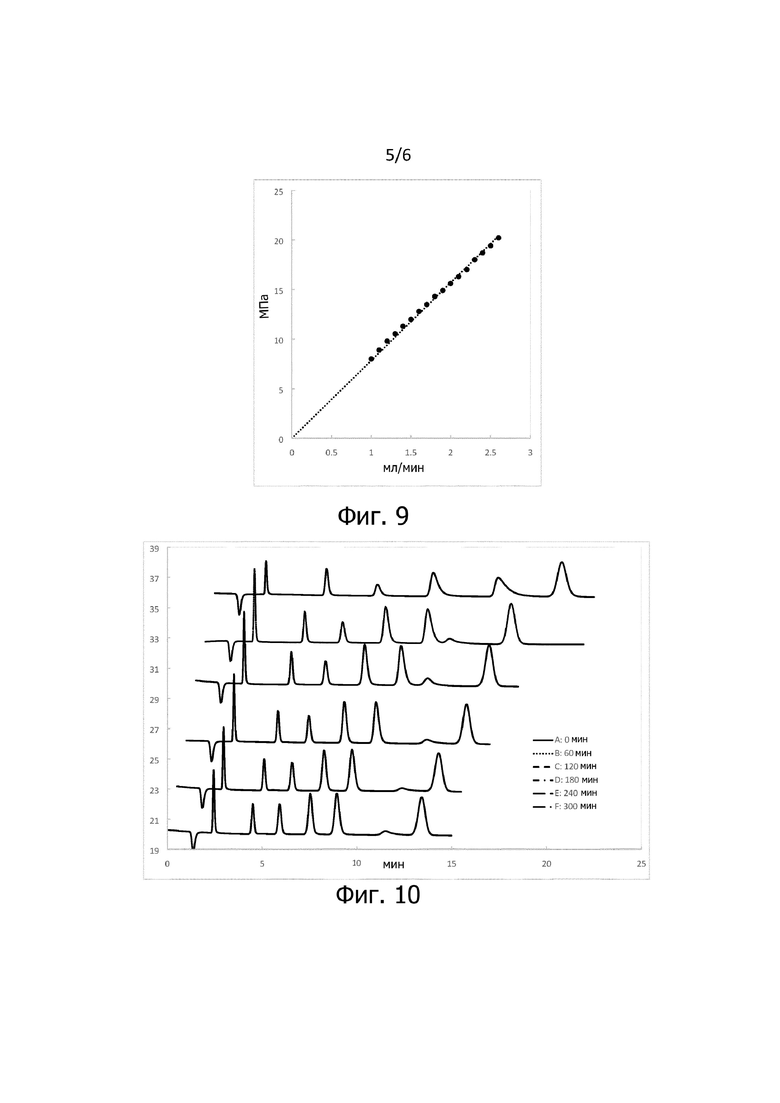
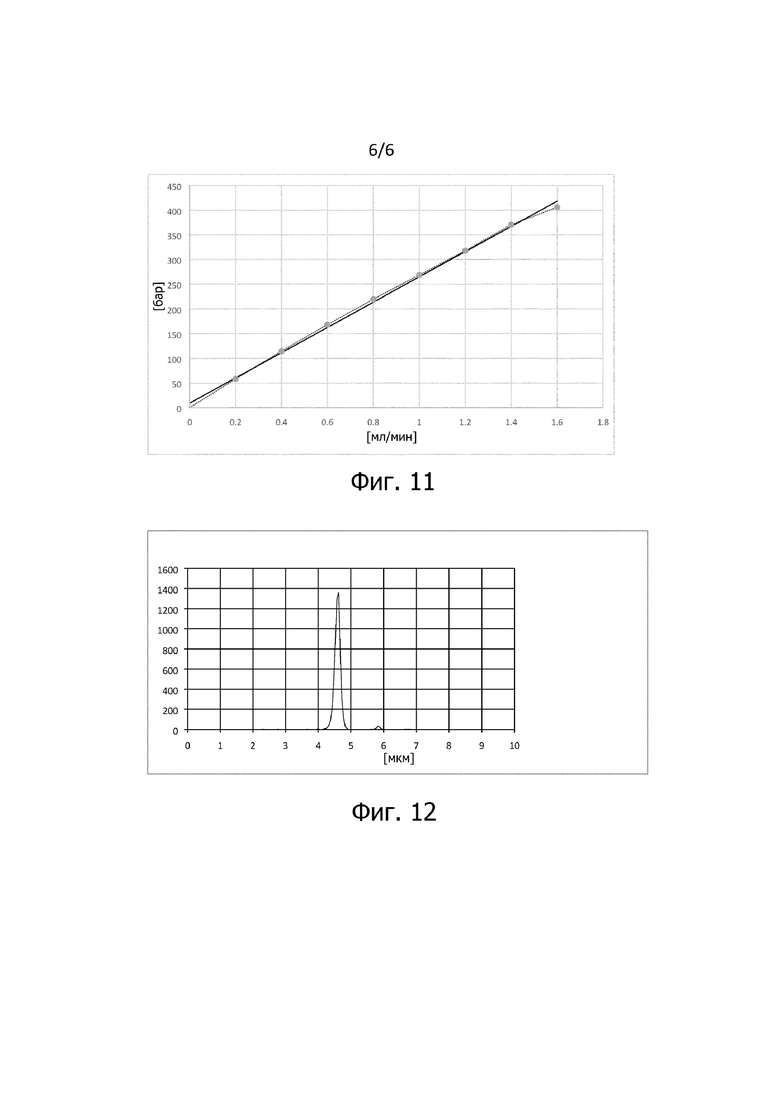
Настоящее изобретение относится к способу модификации материала полимерного носителя, предназначенного для использования в качестве стационарной фазы в способе аналитического или препаративного разделения. Модификацию полимерного носителя проводят в несколько стадий: вначале выбирают полимерный носитель, по меньшей мере частично полученный из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя. Затем на поверхности полимерного материала генерируют гидроксигруппы. Впоследствии проводят окислительную обработку материала, после чего проводят восстановительную или гидролитическую обработку. Предложенный в изобретении подход позволяет наносить на полимерный субстрат покрытие таким образом, чтобы гидрофильность поверхности полимерного субстрата можно было регулировать независимо от содержания кислорода в сердцевине полимерного субстрата и в результате получать механически стабильные и прочные частицы. Ионообменный субстрат на его основе в значительной степени химически инертный, и при этом имеется возможность конфигурировать гидрофильность и емкость или селективность и емкость независимо друг от друга. 6 н. и 22 з.п. ф-лы, 12 ил., 8 пр.
1. Способ модификации материала полимерного носителя, предназначенного для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, включающий стадии
a) обеспечения материала полимерного носителя, по меньшей мере частично полученного из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя;
b) генерирования гидроксигрупп на/в материале полимерного носителя способом, включающим стадии
b.1) окислительной обработки материала полимерного носителя, а после этого
b.2) восстановительной или гидролитической обработки продукта реакции со стадии b.1).
2. Способ по п. 1, в котором материал полимерного носителя на стадии a) по меньшей мере частично получен из мономеров дивинилбензола.
3. Способ по п. 1 или 2, дополнительно включающий стадию c) введения во взаимодействие продукта реакции со стадии b.2) с полифункциональным соединением.
4. Способ по п. 3, в котором полифункциональное соединение содержит
- по меньшей мере первую функциональную группу, реакционноспособную с гидроксигруппами,
и
- по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами.
5. Способ по п. 3 или 4, включающий стадии a), b) и c), в котором за стадией с) следует стадия d):
d) осуществление ряда циклов нанесения покрытия, включающих стадии
d.1) введения или генерирования гидроксигрупп при помощи реакции второй функциональной группы, предпочительно эпоксигруппы, введенной на стадии с), путем
- взаимодействия с полифункциональным соединением, содержащим гидроксигруппы, в частности реакции с полиолом, или
- гидролиза, или
- их комбинации;
d.2) взаимодействия продукта стадии d.1) с полифункциональным соединением, в частности соединением, содержащим
- по меньшей мере первую функциональную группу, реакционноспособную с гидроксигруппами, предпочтительно, галоидную группу,
и
- по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, предпочтительно, эпоксигруппу;
при этом число циклов нанесения покрытия составляет от 0 до 20.
6. Способ по п. 3, включающий стадии a), b) и c), или по п. 5, дополнительно включающий стадию:
e) введения ионообменных группы в продукт реакции со стадии с) или d.2).
7. Способ по любому из пп. 1-6, отличающийся тем, что материал полимерного носителя на стадии а), образованный по меньшей мере частично из ароматических углеводородных соединений, содержащих по меньшей мере два аллильных или винильных заместителя, предпочтительно образованный по меньшей мере частично из мономеров дивинилбензола, наряду с этим частично образован из мономеров, которые выбраны из группы, состоящей из
- этилвинилбензола,
- винилацетата,
- стирола и
- их комбинации;
причем процентная доля ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, в частности, процентная доля дивинилбензола, предпочтительно составляет по меньшей мере 50 масс. %.
8. Способ по любому из пп. 1-7, отличающийся тем, что окислительная обработка на стадии b.1) представляет собой обработку пероксикислотой, предпочтительно выбранной из группы, состоящей из мета-хлорпербензойной кислоты, пероксимуравьиной кислоты, пероксиуксусной кислоты, трифторпероксиуксусной кислоты, обработку KMnO4, обработку кислородной плазмой, или их комбинацию.
9. Способ по любому из пп. 1-8, отличающийся тем, что соединение, используемое на стадии с), содержащее по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, представляет собой эпигалогенгидрин, в частности, эпихлоргидрин.
10. Способ по любому из пп. 1-8, в котором полифункциональное соединение, используемое на стадии с), содержащее по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, представляет собой спейсерную молекулу, предпочтительно диэпоксид, более предпочтительно диглицидиловый эфир бутандиола.
11. Способ по любому из пп. 5-10, отличающийся тем, что полифункциональное соединение, используемое на стадии d.2), содержащее по меньшей мере одну первую функциональную группу, реакционноспособную с гидроксигруппами, и по меньшей мере одну вторую функциональную группу, реакционноспособную с аминами и/или гидроксигруппами, представляет собой эпигалогенгидрин или спейсерную молекулу.
12. Способ по любому из пп. 5-11, отличающийся тем, что число циклов нанесения покрытия составляет от 0 до 10, предпочтительно от 0 до 5, более предпочтительно от 1 до 3.
13. Способ по любому из пп. 6-12, отличающийся тем, что ионообменную группу вводят путем введения во взаимодействие соединения стадии c) или d.2) с элементоорганическим соединением 5-й основной группы, предпочтительно, амином, особенно предпочтительно, третичным амином.
14. Способ по любому из пп. 6-13, отличающийся тем, что за стадией е) следует стадия f), включающая нагревание материала полимерного носителя, содержащего ионобменные группы, в щелочном растворе.
15. Модифицированный материал полимерного носителя, предназначенный для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, который является получаемым способом по любому из пп. 1-14, при этом материал полимерного носителя, обеспеченный на стадии a), имеет средний радиус пор от 2 до 25 нм, измеренный путем сорбции азота с использованием модели BJH.
16. Модифицированный материал полимерного носителя, предназначенный для использования в качестве стационарной фазы в способе аналитического или препаративного разделения, который является получаемым способом по любому из пп. 1-14,
в котором материал полимерного носителя, обеспеченный на стадии а), является гидрофобным и микропористым.
17. Модифицированный материал полимерного носителя по п. 15 или 16, в котором материал полимерного носителя, обеспеченный на стадии а), образован по существу полностью из мономерных звеньев, выбранных из группы, состоящей из:
- мономерных звеньев, полученных из ароматических углеводородных соединений, содержащих по меньшей мере два винильных или аллильных заместителя, предпочтительно полученных из дивинилбензола,
- мономерных звеньев, полученных из этилвинилбензола;
- мономерных звеньев, полученных из стирола;
- их комбинации.
18. Модифицированный материал полимерного носителя по п. 15, в котором материал полимерного носителя, обеспеченный на стадии а), имеет средний радиус пор от 2 до 10 нм, измеренный путем сорбции азота с использованием модели BJH.
19. Модифицированный материал полимерного носителя по п. 15, в котором материал полимерного носителя, обеспеченный на стадии а), имеет удельную площадь поверхности от 80 до 1000 м2/г, предпочтительно от 100 до 800 м2/г, более предпочтительно от 200 до 600 м2/г, измеренную путем сорбции азота с использованием модели BET.
20. Модифицированный материал полимерного носителя по п. 15, в котором материал полимерного носителя, обеспеченный на стадии b.2), является устойчивым к давлению при давлении до 220 бар, предпочтительно до 250 бар, более предпочтительно до 300 бар.
21. Модифицированный материал полимерного носителя по п. 15, причем указанный материал имеет форму частиц, предпочтительно сферических частиц, более предпочтительно сферических частиц со средним размером частиц от 1 до 50 мкм, еще более предпочтительно со средним размером частиц от 2 до 25 мкм, более предпочтительно со средним размером частиц от 3 до 9 мкм.
22. Модифицированный материал полимерного носителя по п. 15, причем указанный материал стабилен в интервале рН от 0 до 14, в частности время удерживания сульфата в колонке, заполненной указанным модифицированным материалом полимерного носителя после промывания 1M раствором NaOH и/или промывания 1M раствором HNO3, отклоняется не более чем на 8%, предпочтительно не более чем на 5%, особенно предпочтительно не более чем на 3% от времени удерживания сульфата на колонке, заполненной модифицированным материалом полимерного носителя, который предварительно не подвергали действию среды с значениями рН от 0 до 14.
23. Колонка для хроматографии, заполненная модифицированным материалом полимерного носителя по любому из пп. 15-22.
24. Колонка для хроматографии по п. 23, которая представляет собой колонку для ионообменной хроматографии.
25. Способ хроматографического разделения анализируемых веществ, отличающийся тем, что раствор, содержащий данные анализируемые вещества, приводят в контакт с модифицированным материалом полимерного носителя по любому из пп. 15-22.
26. Способ хроматографического разделения анализируемых веществ по п. 25, в котором раствор, содержащий данные анализируемые вещества, пропускают через колонку для хроматографии по п. 23.
27. Применение материала полимерного носителя по любому из пп. 15-22, или получаемого способом по любому из пп. 1-14, для аналитического или препаративного разделения анализируемых веществ.
28. Применение по п. 27, в анионообменной хроматографии, катионообменной хроматографии и/или жидкостной хроматографии гидрофильного взаимодействия (HILIC).
| Fontanals N., Marcé R | |||
| M., Borrull F | |||
| New materials in sorptive extraction techniques for polar compounds //Journal of Chromatography A | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| - Т | |||
| Ленточный станок для льна | 1924 |
|
SU1152A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| - С | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| СПОСОБ ХИМИЧЕСКОГО НИКЕЛИРОВАНИЯ СТАЛЬНЫХ ИЗДЕЛИЙ | 1983 |
|
SU1217012A1 |
| US 20050181224 A1, 11.06.2007 | |||
| EP 3248678 A1, 21.08.2020 | |||
| Кузнецова О | |||
| И | |||
| и др | |||
| Новые анионообменники на основе сополимера стирола и | |||

