Изобретение относится к биотехнологии, а именно к способу обработки данных (пайплану) полногеномного секвенирования и предназначен для автоматического выполнения анализа герминальных образцов со средним покрытием 30х от набора сырых ридов до результатов, готовых для анализа клиническим интерпретатором.
Секвенирование - метод определения нуклеотидной последовательности ДНК и РНК, используемый для определения генетических повреждений (мутаций) в ДНК, являющихся причиной наследственных болезней, наследственных предрасположенностей или особенностей организма, за последние 10 лет стало распространенной процедурой в молекулярной биологии для изучения генома (полной последовательности ДНК из всех хромосом клетки) и транскриптома (совокупности активных РНК, синтезируемых клеткой).
Из уровня техники существуют различные технологии секвенирования и пайплайны обработки данных. В первом случае описываются непосредственно способы секвенирования, например, путем синтеза с обратимой терминацией (lllumina), пиросеквенирование (Roche), секвенирование путем лигирования (SOLiD), полупроводниковое секвенирование (lon torrent), во втором - пайпланы, предназначенные для автоматического выполнения анализа герминальных образцов. Можно сказать, что инструменты описывают примерно один и тот же процесс, но с разных сторон. Настоящая заявка относится к пайплайнам обработки данных полногеномного секвенирования.
Первые попытки считывать последовательность ДНК были сделаны с помощью применения методов секвенирования по Сэнгеру. Недостатком известного способа является низкая пропускная способность и дороговизна при исследовании большого объема данных. Именно эти минусы стали основой для разработки и внедрения технологии - NGS.
Необходимость разработки NGS была обусловлена стремлением к автоматизации анализа, увеличению объема получаемой информации и снижению стоимости исследования. Принцип технологии NGS основан на массовом одновременном секвенировании тысяч фрагментов ДНК на базе подготовленных однонитевых библиотек. Методика включает три этапа: подготовка библиотек; сиквенс; анализ полученных данных.
Преимущество NGS - в снижении стоимости исследования; автоматизации анализа; большом объеме получаемой информации. Методы NGS имеют большую производительность, позволяют выполнять одновременное считывание миллиардов коротких фрагментов нуклеиновых кислот. Кроме того, NGS дает возможность проводить секвенирование сразу нескольких десятков геномов за один запуск анализатора.
Поскольку геном человека состоит примерно из 3,1 миллиарда пар оснований, а каждый фрагмент последовательности, или прочитанный, обычно имеет размер от 100 до 500 или 1000 нуклеотидов, время и превышение, затрачиваемые на построение таких полноразмерных геномных последовательностей и определение их вариантов, довольно велики, часто требуют использования нескольких различных компьютерных ресурсов с использованием нескольких различных алгоритмов в течение длительных периодов времени.
Из уровня техники известен источник AU2016226288 (G06F19/00; G06N3/00; G16B30/10; G16B50/30 дата публикации 14.09.2017 г. ) в котором раскрыты система, способ и устройство для выполнения конвейера анализа последовательности данных о генетической последовательности.
Согласно изобретению, система включают интегральную схему, образованную набором жестко соединенных цифровых логических схем, которые соединены между собой физическими электрическими межсоединениями. Зашитые цифровые логические схемы организованы как набор механизмов обработки, причем каждый механизм обработки сформирован из подмножества зашитых цифровых логических схем для выполнения одного или нескольких шагов в конвейере анализа последовательности при считывании геномных данных. В различных случаях каждое подмножество проводных цифровых логических схем может быть сформировано в проводной конфигурации для выполнения одного или нескольких шагов операции вариантного вызова.
AU2016226288 описывает биоинформатический конвейер способный обрабатывать данные секвенирования человека от сырых прочтений до vcf файлов, содержащих вариации генома, относительно референса. Пайлпайн в AU2016226288 выполнена на специфическом аппаратном обеспечении FPGA, что делает такой подход не универсальным с точки зрения развития и изменений анализа, в сравнении с модульными пайплайнами, и их частной имплементацией, описанной в текущей заявке. За счет использования модульной структуры и аппаратного обеспечения общего назначения, пайплайн описанный в текущей заявке имеет расширенный функционал, в сравнении с AU2016226288, позволяя так же обнаруживать геномные вариации, гаплогруппы, hla типы и проводить другие геномные анализы.
Известен ускоритель биоинформатического анализа MegaBOLT. Он ускоряет такие алгоритмы, как SOAPnuke, Minimap2, BWA, GATK HaplotypeCaller + MuTect2, благодаря многопотоковой и высокопараллелизованной архитектуре вычислений, что способствует значительному ускорению анализа массивных данных.
MegaBOLT поддерживает анализ секвенирования всего генома (WGS), секвенирования всего экзома (WES) и панельного секвенирования данных зародышевой линии или соматических данных. Это до 300 раз быстрее, чем классический алгоритм. Интегрированный с многозадачной системой планирования собственной разработки, MegaBOLT поддерживает одновременные многозадачные вычисления на одном единственном сервере. Эффективность вычислений дополнительно повышается с 16% до 28%. (источник https://en.mgi-tech.com/products/software_info/2/)
Недостатками известного решения являются низкая скорость анализа, сложное планирование, сложность в использовании и высокая стоимость вычислений. Данное решение принято в качестве ближайшего аналога.
Информация, получаемая при использовании технологий высокопроизводительного секвенирования, представляет собой миллионы коротких (порядка 75-300 букв) прочтений. Анализ таких данных в большинстве случаев заключается в картировании полученных прочтений на референсный геном и детекции отличий от него.
Обработка данных высокопроизводительного секвенирования в рамках масштабного проекта несет в себе три основные трудности. Во-первых, она может быть выполнена множеством разных инструментов, как с открытым исходным кодом, так и проприетарных. Выбор конкретных решений требует наличия экспертизы в области биологии и технологий секвенирования, а также понимания задач, решаемых с помощью того или иного анализа. Во-вторых, большой объем анализируемых данных требует существенных вычислительных и временных затрат на обработку, а также создания автоматизированной IT системы осуществляющий контроль над анализом. В-третьих, геномные данные являются чувствительными и их анализ может требовать их обработки локально, не прибегая к облачным вычислениям.
Существующие решения для анализа, опирающиеся на академические разработки, в большинстве своем не предполагают работы с большими объемами данных и не могут обеспечить необходимых скоростей обработки. Коммерческие решения, с закрытым исходным кодом напротив не обеспечивают достаточную гибкость добавлении новых типов анализа данных, а также опираются на специализированное аппаратное обеспечение, что увеличивает их цену и усложнят ограничивает пользователя в выборе вычислительных мощностей.
Задача, решаемая изобретением, заключается в том, чтобы предложенные изменения в пайплайн обеспечивали анализ данных одного образца WGS человека с покрытием 30х примерно за 5 часов. Одновременно задача направлена на то, чтобы время расчета было дополнительно снижено за счет параллельного анализа образцов. При решении указанных задач достигается технический результат, выражающийся в повышении функциональной возможности анализа данных полного генома человека при уменьшении времени получения готовых для интерпретации результатов генетического анализа.
Благодаря изобретению по сравнению с ближайшим аналогом производительность системы анализа данных полного генома человека выше в 7,5 раз по количеству обработки в день и в 6, 6 раз при сравнении мощности обработки в месяц:
Megabolt
Предложенное решение имеет больше функциональных возможностей анализа данных полного генома человека по сравнению с ближайшим аналогом, в частности:
Megabolt
Основой для описываемого пайплайна служат рекомендации для анализа и соответствующий программный код, поддерживаемый broad institute (https://github.com/broadinstitute/warp/tree/develop/pipelines/broad/dna_seq/germline/single_sample/wgs). Пайплайн предоставляемый на приведенном репозитории осуществляет обработку одного образца WGS человека с покрытием 30х примерно за 24 часа. Тогда как предложенное решение позволяет параллельно анализировать 5 образцов со средней скоростью на образец 2,5 часа.
На вход пайплайн принимает произвольное количество пар файлов прочтений для одного образца, а также набор необходимых референсных данных для каждого анализа.
Все необходимые для работы пайплайна файлы предоставляются в виде tar.gz архива, однако так же могут быть автоматически скачаны и нужным образом отформатированы запуском скриптов hg19_dwl.sh или hg38_dwl.sh, которые создают референсные файлы для hg19 и hg38 сборок, соответственно.
Отдельные файлы, полученные специально для работы пайплайна, и создание которых в рамках простых скриптов затруднительно поставляются вместе с исходным кодом пайплайна.
Все, получающиеся в результате выполнения пайплайна выходные файлы имеют одинаковый префикс, указанный при запуске в файле WholeGenomeGermlineSingleSample.inputs.plumbing.json в поле “sample name”. Выходные файлы сохраняются в папках, указанных в полях “WholeGenomeGermlineSingleSample.copy_path”. Если папки, указанные в этих полях, не существуют, то они будут автоматически созданы по мере выполнения пайплайна. Отдельно, при необходимости, можно отключить копирование выходных файлов из временной рабочей директории Cromwell-execution, проставляя значение поля “WholeGenomeGermlineSingleSample.copy_output”.
Предлагаемое решение поясняется следующими материалами.
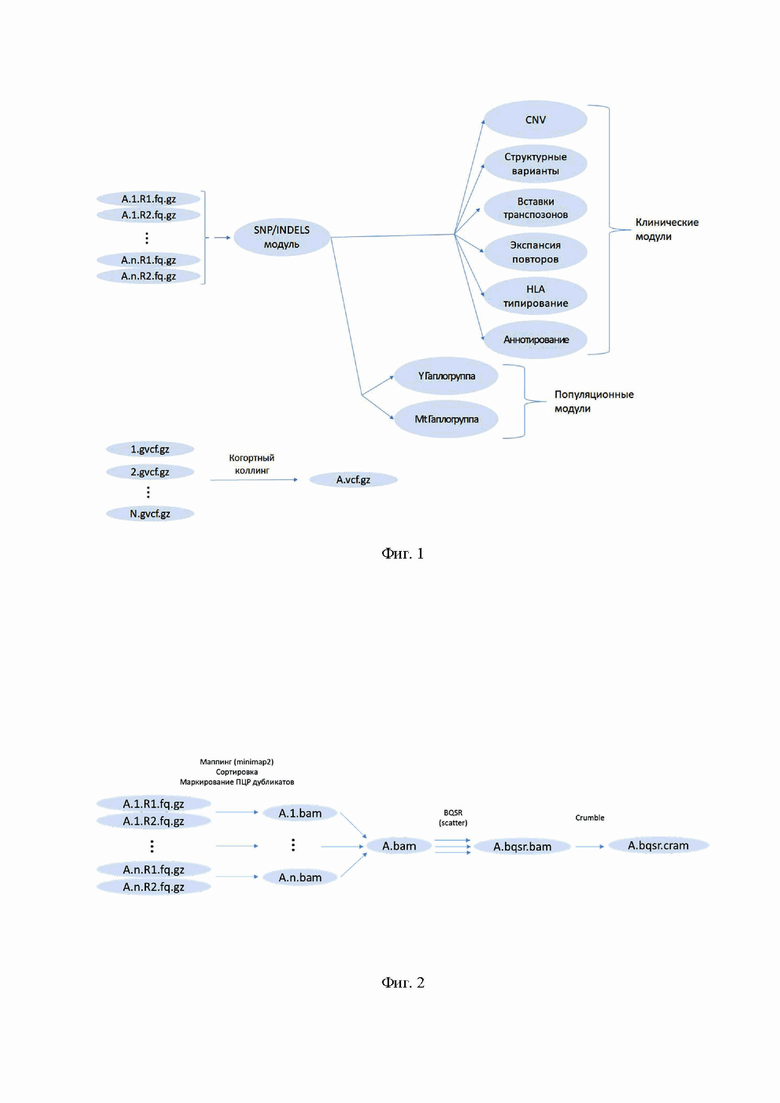
На фиг. 1 - показана общая блок-схема ускорительной системы обработки данных полногеномного секвенирования.
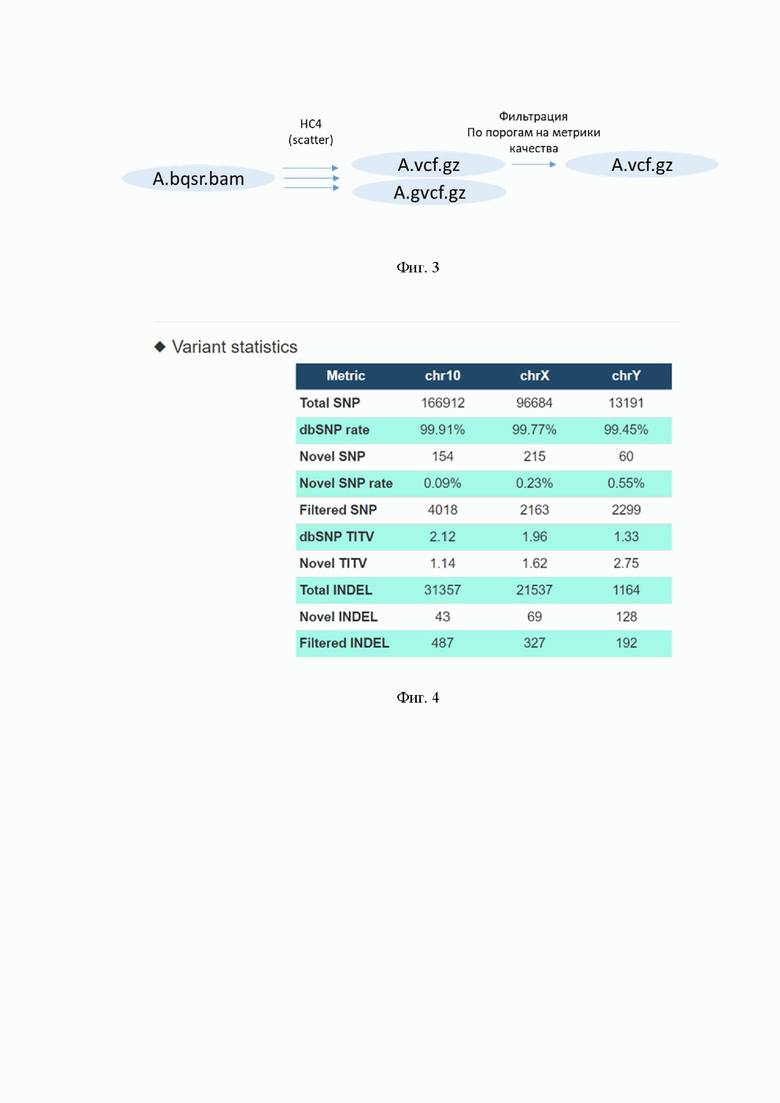
На фиг. 2 - Картирование пар fq.gz файлов на геном.
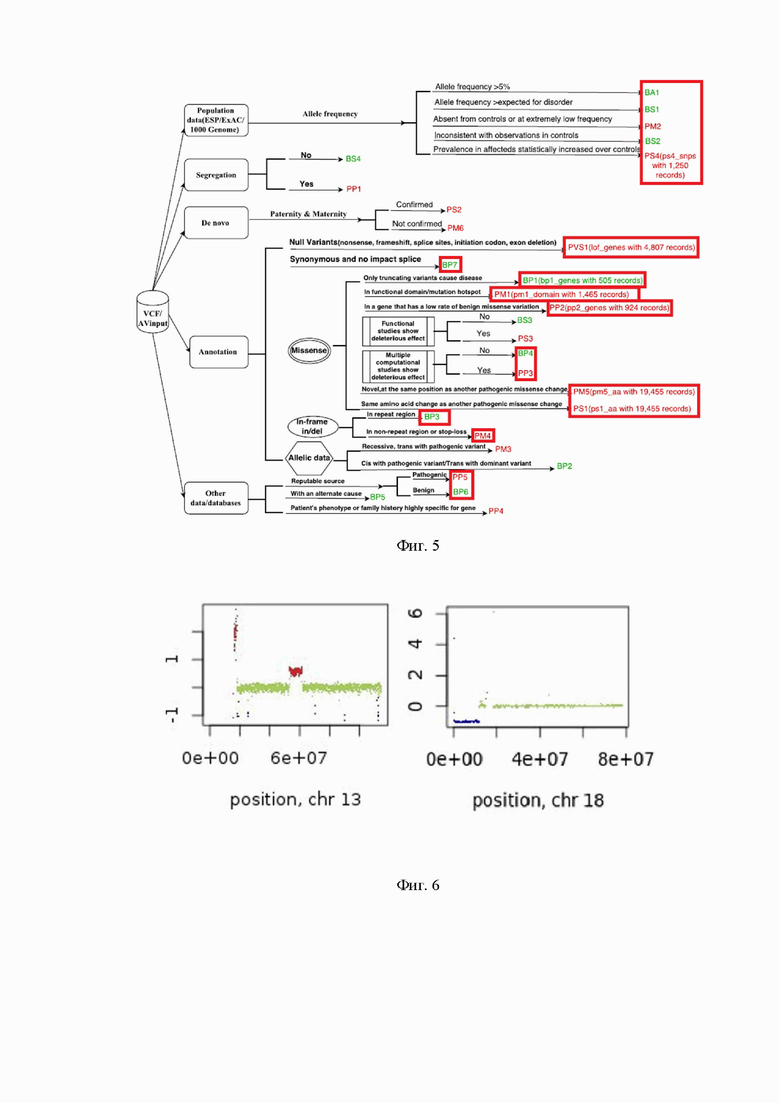
На фиг. 3 - Получение файлов vcf, gvcf.
На фиг. 4 - показан пример из отчета.
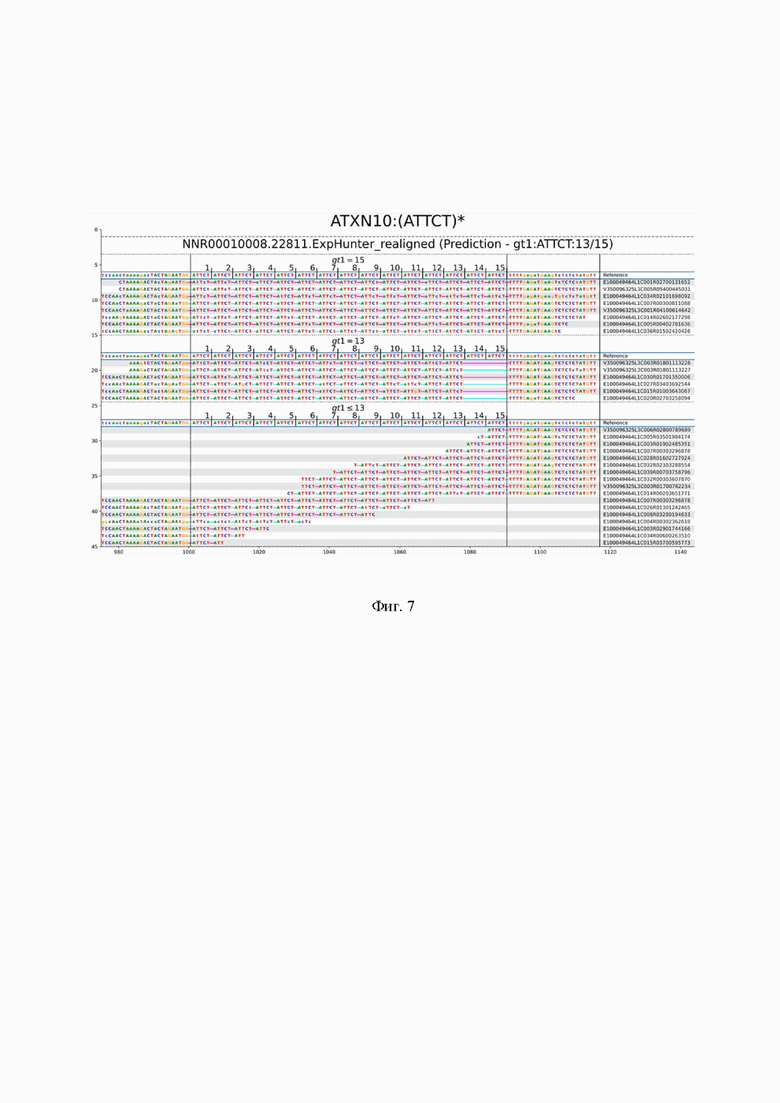
На фиг. 5 - показана реализация изобретения, где красными рамками выделены критерии, присвоение которых автоматизировано благодаря предлагаемому решению.
На фиг. 6 - показаны примеры презентации дупликации (chr13) и делеции (chr18) в результатах модуля поиска CNV.
На фиг. 7 - Пример картирования прочтений на участок гена ATXN10, содержащий повторы мотива ATTCT.
Предлагаемая система обработки данных полногеномного секвенирования состоит из модулей, причем модуль, выполняющий картирование сырых ридов и генотипирование SNP/INDELS является базовым, на который опираются все остальные типы анализов.
Запуск модулей “Аннотирование”, “Y и “MT” гаплогруппы, а также когортный коллинг в текущей реализации системы осуществляется отдельно от всех остальных модулей, представляющих собой единый пайплайн.
Далее более подробно описаны модули, образующую систему обработки данных полногеномного секвенирования.
Модуль SNP/INDELS
Модуль SNP/INDELS является базовым в пайплайне, так как осуществляет картирование прочтений на референсный геном и генотипирование vcf и INDELS. Модуль состоит из трех блоков.
Первый блок обеспечивает картирование исходных прочтений подаваемых парами в формате.fastq.gz на референсный геном человека (hg19 или hg38 по выбору) с получением файла в формате bam (фиг. 2). Для фильтрации, картирования, сортинга и маркирования дубликатов используется связка программ fastp, minimap2 и samtools. Ускорение на данном шаге обеспечивается отказом от записи промежуточных данных на диск. Этот этап хорошо параллелизуется по ядрам сервера и его скорость напрямую зависит от их количества. Если на вход подавалось несколько пар fq.gz файлов отдельные отдельные bam файлы сливаются в единый файл. Скорость слияния файлов лимитируется скоростью диска, на котором идет анализ. Полученный единый файл формата bam подвергается пересчету метрик качества коллинга нуклеотидов секвенатором (BQSR), что увеличивает точность детекции SNP/INDELS в дальнейшем анализе. Параллелизм на данном этапе обеспечивается разбитием bam файла на несколько отдельных файлов меньшего объема и их независимым анализом. Лимитирующим ресурсом на данном шаге является количество оперативной памяти сервера. Для длительного хранения финальный bam файл переводится в cram формат, с потерей информации о качестве отдельных букв рида, но с сохранением информации о картировании. Такой подход позволяет максимально экономить место, занимаемое файлом, сохраняя основную информацию, необходимую при работе с таким файлов с целью проверки результатов анализа.
Второй логический блок осуществляет коллинг SNP/INDELS и формирование vcf и, опционально gvcf, файлов, содержащих информацию по полиморфизмам анализируемого образца (фиг. 3). Коллинг осуществляется программой HaplotypeCaller из пакета GATK4. Параллелизация на данном этапе осуществляется за счет независимого анализа участков генома, и лимитируется объемом оперативной памяти сервере. Полиморфизмы в vcf файле фильтруются по предустановленным порогам метрик качества.
Третий логический блок собирает статистику по прохождению каждого шага Модуля SNP/INDELS и формирует отдельный отчет в html формате для контроля качества анализируемых образцов. Собираемая статистика характеризует:
- Входные данные (количество ридов, нуклеотидный состав, загрязнение адаптерами, качество чтений, размер вставки)
- Качество картирования ридов на референс и глубину прочтения (для одной аутосомы и половых хромосом)
- Количество обнаруженных SNP и INDELS процент полиморфизмов не входящих в dbSNP, количество трансверсий и транзиций
Модуль когортного коллинга
Модуль когортного коллинга запускается отдельно от основного пайплайна. Модуль принимает на вход набор файлов формата.gvcf, полученных после выполнения модуля поиска SNP/INDELS на нескольких образцах (например на трио, или отдельной когорте пациентов) и возвращает единый файл формата.vcf содержащий результирующий набор SNP/INDELS для всех образцов.
Когортный коллинг осуществляет генотипирование набора.gvcf файлов с помощью программы HaplotypeCaller, пакета GATK4. Использование данного модуля облегчает анализ SNP/INDELS в связанных образцах, стандартизируюя репрезентацию аллелей и увеличивая точность коллинга (для когорт от нескольких десятков пациентов).
Модуль аннотирования SNP/INDELS
Модуль аннотирование SNP/INDELS в текущей реализации пайплайна запускается отдельно, для каждого полученного файла формата.vcf. Для каждого варианта, записанного в.vcf файле, модуль ставит в соответствие информацию из различных баз данных, содержащих клинически значимую информацию о вариантах, а также агрегирует ее в ACMG ранг. Работа модуля основана на программе с открытым исходным кодом OpenCravat (https://github.com/KarchinLab/open-cravat ).
Модуль аннотирования может работать с файлами формата.vcf, полученными, как из модуля SNP/INDELS, так и из модуля когортного коллинга. Результатом работы модуля является файл формата. sqlite, предназначенный для загрузки в интерфейс программы OpenCravat. В текущей реализации модуля, аннотация осуществляется по следующим базам данных:
Частотные:
- GnomadV3
- Внутренние частоты выборки Эвоген
Клинические:
- ClinVar
- OMIM
- HPO
Структурные:
- InterPro
- Gene structures
- Информация по спалйс сайтам dbscsnv11 (из dbNSFP)
- Информация по повторяющимся последовательностям в геноме (rmsk)
In-silico аннотирование вариантов:
- metaSVM (из dbNSFP)
- Gerp ++ (из dbNSFP)
Помимо этого, реализована функция присвоения критерия патогенности по ACMG (фиг. 5), на базе программы IntereVar, интегрированной в OpenCravat и с обновленными базами данных.
Критерии ACMG
Модифицировано из InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. 2017 Полный список ACMG критериев. Критерии, присвоение которых автоматизировано в текущем пайплайне обведены в красные рамки.
Модуль поиска CNV
Модуль для поиска изменений копийности может быть включен опционально проставлением соответствующего параметра при запуске пайплайна. Модуль оценивает изменения копийности участков хромосомы, ориентировочного размера от 100 т.п.о. до целой хромосомы, а также выявлять участки с потерей гетерозиготности (LOH). Модуль работает на основе программы с открытым исходным кодом Freec (https://github.com/BoevaLab/FREEC ).
Работа Freec основана на сравнении покрытия участка генома в небольшом регионе к среднему покрытию по всему геному для оценки обычных CNV или собирая статистику по биалельной частоте вариантов, для обнаружения LOH путем сбора статистики по биалельной частоте вариантов. Детекция CNV и LOH опирается на полученные в ходе работы модуля SNP/INDELS bam и vcf файлы. Данный подход показывает хорошие результаты для детекции больших перестроек (больше 1МБ участков генома), для оценки достоверности перестроек меньше 1МБ рекомендуется учитывать частоты встречаемости данной перестройки в проанализированных ранее образцах, близость теломерных и центромерных участков, а также результаты модуля по поиску структурных вариантов (см. ниже). Результатом работы модуля по поиску CNV является таблица с обнаруженными вариациями с указанием их статистической достоверности, а также визуальное представление биаллельных частот и среднего покрытия по каждой хромосоме (фиг. 4). Примеры презентации дупликации (chr13) и делеции (chr18) в результатах модуля поиска CNV показаны на фиг. 6.
Модуль поиска структурных вариантов
Модуль по поиску структурных вариантов может быть включен опционально, проставлением соответствующего параметра при запуске пайплайна (см. Раздел “Опции запуска пайплайна”). Модуль анализирует так называемые сплит прочтения и дискордант прочтения, для выявления инсерций, делеций, транслокаций и сложных перестроек участков генома. Следует отметить, что данный подход чувствителен к среднему покрытию в анализируемом образце. Модуль основан на работе программы с открытым исходным кодом smoove (https://github.com/brentp/smoove ).
Работа модуля опираться на полученные в ходе работы модуля SNP/INDELS bam файлы. Результатом работы является таблица наиболее вероятных структурных перестроек разбитая отдельно на гомо- и гетерозиготы с указанием генов, затронутых предполагаемой перестройкой. Ассоциация перестрок с генами осуществляется с помощью инструмента VEP (https://github.com/Ensembl/ensembl-vep ). Помимо итоговой таблицы создаются отдельные bam файлы содержащие сплит и дискордант прочтения для визуальной оценки, а так же vcf файл содержащий все варианты обнаруженные с помощью smoove без дополнительной фильтрации на достоверность.
Модуль поиска экспансии повторов
Модуль для поиска экспансии повторов может быть включен опционально, проставлением соответствующего параметра при запуске пайплайна. Модуль анализирует прочтения картированные на ряд заранее обозначенных участков генома, содержащих повторы коротких последовательностей ДНК и определяет количество таких повторений в гаплотипах образца. Модуль работает на базе программы с открытым исходным кодом ExpansionHunter (https://github.com/Illumina/ExpansionHunter ).
Работа модуля опираться на полученные в ходе работы модуля SNP/INDELS bam файлы. Результатом работы модуля поиска экспансии повтором является.pdf документ, отображающий участки генома, где было обнаружено отличие количества повторов от референсного значения, а также графическое представление прочтений, перекартированных на данный регион (фиг. 7). Помимо этого сохраняется bam файл, содержащий прочтения, перекартированные программой ExpansionHunter на регионы с повторами, а так же.vcf файл, содержащий результаты по всем анализируемым повторам, включая, те, копийность которых не отличается от референсных значений.
На фиг. 7 показан пример картирования прочтений на участок гена ATXN10, содержащий повторы мотива ATTCT.
Модуль поиска вставок мобильных элементов
Модуль для поиска вставок мобильных элементов может быть включен опционально, проставлением соответствующего параметра при запуске пайплайна. Модуль ищет детектирует вставки мобильных элементов семейств ALU, SVA, LINE1 в геном и работает на базе программы с открытым исходным кодом TEMP2 (https://github.com/weng-lab/TEMP2 ).
Работа модуля опираться на полученные в ходе работы модуля SNP/INDELS bam файлы. Результатом работы модуля являются два файла формата.tsv. Первый файл содержит полный список обнаруженных инсерций, и их ассоциацию с генами полученную с помощью инструмента VEP (https://github.com/Ensembl/ensembl-vep ). Второй файл содержит те инсерции, которые, базируясь на поле IMPACT программы VEP, могут влиять на функции ассоциированных с ними генов.
Модуль HLA типирования
Модуль для типирования HLA может быть включен опционально, проставлением соответствующего параметра при запуске пайплайна. Модуль направлен на определение гаплотипов HLA и работает на базе программы с открытым исходным кодом Kourami (https://github.com/Kingsford-Group/kourami ).
Работа модуля опираться на полученные в ходе работы модуля SNP/INDELS bam файлы. Прочтения картированные на HLA регионы, а также не картированные и частично картированные прочтения агрегируются и перекартируются на базу последовательностей гаплотипов HLA локуса. Результатом работы модуля служит файл формата.tsv содержащий предсказанные типы HLA аллелей, согласно установленной номенклатуре.
Модуль определения Mt и Y гаплогрупп
Модуль аннотирования гаплогрупп в текущей реализации пайплайна запускаются отдельно, для каждого полученного файла формата.vcf. Модуль аннотирования Mt гаплогруппы использует программу с открытым исходным кодом (https://github.com/seppinho/haplogrep-cmd). Модуль аннотирования Y гаплогруппы использует модификацию программы с открытым исходным кодом y-leaf (https://github.com/genid/Yleaf ).
Для аннотирования митохондриальной (МТ) гаплогруппы используется филогенетическое древо всемирной вариации митохондриальной ДНК человека Phylotree 17-й сборки. На вход программа берет vcf-файл, затем производит классификацию и на выходе получается наиболее вероятная МТ-гаплогруппа для данного индивида. Для идентификации гаплогруппы y-leaf использует филогенетически информативные SNP из базы данных ISOGG (International Society of Genetic Genealogy). На вход y-leaf берет vcf-файл, относящийся к индивиду мужского пола, после чего производит классификацию, выдавая на выход искомую Y-гаплогруппу.
Способ обработки данных полногеномного секвенирования включает следующие этапы.
Первый этап анализа данных представляет собой выравнивание сырых прочтений на референсный геном. В отличии от признанного в академической среде решения на базе best practice от института broad (GATK) все инструменты, используемые на данном этапе выстроены в единый конвейер, передающий данные между программами без записи промежуточных файлов и часть заменена более быстрыми аналогами.
Прочтения триммируются инструментом fastp и потоком данных передаются в картировщик minimap2 откуда так же потоком данных передаются в инструмент samatools осуществляющий маркирование дубликатов и индексирование получившегося bam файла. Количество потоков для исполнения данного шага регулируется пользователем и зависит от конфигурации сервера и количества образцов, анализируемых параллельно. Данные на этом этапе читаются и записываются на nvme диск, позволяющий использовать до 130 потоков работы с данными на этом этапе.
Полученный bam файл направляется в следующий этап обработки, представляющий собой рекалибрацию метрик качества, проставляемых секвенатором (BQSR и ApplyBQSR в стандартном пайплайне обработки геномных данных GATK). В пайплайне описанном в данной заявке, использование инструментов BQSR и ApplyBSQR оптимизированно путем разбивки входных данных на равные геномные интервалы, в отличии от использования разбивки данных по отдельным хромосомам. Такой подход обеспечивает равномерную загрузку вычислительных ресурсов сервера, а также одновременное завершение обработки каждого геномного интервала и, в следствии этого, экономию вычислительного времени.
Полученный bam файл используется в качестве входных данных для других модулей, а также переводится в формат cram, для длительного хранения. Шаг трансформации bam в cram реализован посредством инструмента crumble с генерацией cram файла, без информации о метриках качества коллинга нуклеотидов, но весящего примерно в 10 раз меньше исходного bam файла, что обеспечивает существенную экономию места в файловом хранилище. Преимуществом такого подхода является то, что такой cram файл подходит для визуализации картирования и для его ручной оценки, если же необходимо получить полноценный bam файл с метриками качества для стороннего анализа, то обычно он необходим для определенного участка генома и восстанавливается из комбинации cram файла и fastq файлов. Из cram файла экстрагируются ID прочтений интересующего нас региона, а по ним из fastq файла экстрагируются оригинальные прочтения и, при необходимости повторно картируются на геном.
Блок генерации vcf и gvcf файлов использует стандартные инструменты HaplotypeCaller пакета GATK. Оптимизация скорости работы пайплайна на этом этапе достигается разбивкой генома на равномерные интервалы для анализа, аналогично этапу рекалибрации метрик качества.
Контроль качества выполнения исходных данных оценивается по результатам тримминга, картирования прочтений и коллинга SNP/INDELS. Сбор метрик качества является достаточно время затратным процессом. В описываемом пайплайне ускорение этого шага достигается сбором метрик не по полному геному, а по его репрезентативным участкам, что уменьшает необходимые вычисления. Возможные загрязнения сторонним генетическим материалом оцениваются по митохондриальной ДНК, что драматически быстрее обычной оценки уровня загрязнения по всей геномной ДНК.
Дополнительные модули анализа на основе получаемых bam файлов так же были собраны исходя из необходимости экономить общее вычислительное время.
Блоки CNV анализа и анализа инсерций транпозонов настроены аналогично блокам генерации Vcf файлов и блокам рекалибрации скоров исходный bam файл разделяется на равные блоки, содержащие равное количество информации, для паралеллизации обработки данных.
Блоки HLA типирования и определения гаплогрупп дополнительно ускорены тем, что на вход в них подаются bam файлы, содержащие только риды, из регионов, необходимых для анализа
Данные характеристики процессов актуальны при запуске анализа на сервере с конфигурацией 128 CPU, 256Gb RAM, nmve диск, объемом от 2Т.
Благодаря предложенному изобретению пайплайн способен выполнять следующие типы анализов:
- Идентификация SNP/INDELS в образце
- Аннотирование (в частности ACMG)
- Определение Y и МТ гаплогрупп
- Когортный коллинг вариантов
- Изменения копийности участков хромосомы, ориентировочного размера от 1Мб до целой хромосомы
- Анализ на структурные варианты размером ориентировочного размера от 100 до 100 тыс. пн
- Вставки мобильных элементов (ALU, SVA, LINE1)
- Экспансию повторяющихся участков генома
- HLA типирование образца
Разработанная конфигурация учитывает необходимость оптимизации времени анализа образцов. Пайплайн позволяет оптимизировать выполнение необходимых анализов и выдачу заключений специалистами в режиме поточной обработки образцов клиентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| Система обработки данных полногеномного секвенирования | 2023 |
|
RU2804535C1 |
| СПОСОБ ОБНАРУЖЕНИЯ ВАРИАЦИЙ ЧИСЛА КОПИЙ (CNV) ПО ДАННЫМ СЕКВЕНИРОВАНИЯ ПОЛНОГО ЭКЗОМА ЧЕЛОВЕКА И ГЕНОМА С НИЗКИМ ПОКРЫТИЕМ | 2023 |
|
RU2822040C1 |
| РАСЧЕТ БРЕМЕНИ ОПУХОЛЕВЫХ МУТАЦИЙ С ИСПОЛЬЗОВАНИЕМ ДАННЫХ РНК СЕКВЕНИРОВАНИЯ ОПУХОЛЕЙ И КОНТРОЛИРУЕМОГО МАШИННОГО ОБУЧЕНИЯ | 2020 |
|
RU2759205C1 |
| СИСТЕМА И СПОСОБ ИНТЕРПРЕТАЦИИ ДАННЫХ И ПРЕДОСТАВЛЕНИЯ РЕКОМЕНДАЦИЙ ПОЛЬЗОВАТЕЛЮ НА ОСНОВЕ ЕГО ГЕНЕТИЧЕСКИХ ДАННЫХ И ДАННЫХ О СОСТАВЕ МИКРОБИОТЫ КИШЕЧНИКА | 2017 |
|
RU2699284C2 |
| БИОИНФОРМАЦИОННЫЕ СИСТЕМЫ, УСТРОЙСТВА И СПОСОБЫ ДЛЯ ВЫПОЛНЕНИЯ ВТОРИЧНОЙ И/ИЛИ ТРЕТИЧНОЙ ОБРАБОТКИ | 2017 |
|
RU2799750C2 |
| Способ анализа митохондриальной ДНК для неинвазивного пренатального тестирования | 2021 |
|
RU2772912C1 |
| БИОИНФОРМАЦИОННЫЕ СИСТЕМЫ,УСТРОЙСТВА И СПОСОБЫ ВЫПОЛНЕНИЯ ВТОРИЧНОЙ И/ИЛИ ТРЕТИЧНОЙ ОБРАБОТКИ | 2017 |
|
RU2750706C2 |
| СПОСОБ ОЦЕНКИ СТАТУСА 14-ГО ЭКЗОНА ГЕНА MET ПО ДАННЫМ РНК СЕКВЕНИРОВАНИЯ | 2023 |
|
RU2817869C1 |
| СПОСОБ НЕИНВАЗИВНОЙ ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ АНЕУПЛОИДИЙ ПЛОДА | 2014 |
|
RU2583830C2 |
| Использование аллельных вариантов генов pks15 и pe_pgrs54 в качестве маркера повышенной вирулентности Mycobacterium tuberculosis в отношении людей с пониженным иммунитетом | 2019 |
|
RU2737622C1 |
Изобретение относится к биотехнологии. Описан способ обработки данных (пайплан) полногеномного секвенирования. Выравнивают сырые прочтения на референсный геном. Прочтения триммируют инструментом fastp и потоком данных передают в картировщик minimap2, откуда также потоком данные передают в инструмент samatools, осуществляющий маркирование дубликатов и индексирование получившегося bam файла. Полученный bam файл рекалибруют по метрикам качества, проставляемых секвенатором. Ускорение контроля качества выполнения исходных данных оценивают по результатам тримминга, картирования прочтений и коллинга SNP/INDELS. Изобретение обеспечивает повышение функциональной возможности анализа данных полного генома человека при уменьшении времени получения готовых для интерпретации результатов генетического анализа и позволяет выполнять анализ герминальных образцов со средним покрытием 30х от набора сырых ридов до результатов, готовых для анализа клиническим интерпретатором. 7 ил.
Способ обработки данных полногеномного секвенирования, характеризующийся тем, что включает этап анализа данных, представляющий собой выравнивание сырых прочтений на референсный геном, прочтения триммируются инструментом fastp и потоком данных передаются в картировщик minimap2, откуда так же потоком данных передаются в инструмент samatools, осуществляющий маркирование дубликатов и индексирование получившегося bam файла, полученный bam файл направляется в следующий этап обработки, представляющий собой рекалибрацию метрик качества, проставляемых секвенатором, при этом ускорение контроля качества выполнения исходных данных оценивают по результатам тримминга, картирования прочтений и коллинга SNP/INDELS, что достигается сбором метрик по репрезентативным участкам генома.
| RU 2021109321 A, 07.10.2022 | |||
| WO 2021045947 A1, 11.03.2021 | |||
| WO 2017009770 A1, 19.01.2017 | |||
| Sarah A | |||
| Gagliano, Sebanti Sengupta et al., Relative impact of indels versus SNPs on complex disease, Genet Epidemiol | |||
| Станок для придания концам круглых радиаторных трубок шестигранного сечения | 1924 |
|
SU2019A1 |
