РОДСТВЕННАЯ ЗАЯВКА
Настоящая заявка заявляет преимущество даты подачи согласно 35 USC. § 119 (e) предварительной заявки США № 62/758819, поданной 12 ноября 2018 г. Все содержание упомянутой выше заявки включено в данный документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым способам получения производных цитотоксического индолинобензодиазепина.
УРОВЕНЬ ТЕХНИКИ
Было показано, что конъюгаты клеточно-связывающего агента с димерами индолинобензодиазепина, которые имеют одну иминную функциональную группу и одну аминную функциональную группу, демонстрируют гораздо более высокий терапевтический индекс (отношение максимальной переносимой дозы к минимальной эффективной дозе) in vivo по сравнению с ранее описанными производными бензодиазепина, содержащими две иминные функциональные группы. См., например, WO 2012/128868. Раскрытый ранее способ получения димеров индолинобензодиазепина с одной иминной функциональной группой и одной аминной функциональной группой включает частичное восстановление димеров индолинобензодиазепина, имеющих две иминные функциональные группы. Стадия частичного восстановления обычно приводит к образованию полностью восстановленного побочного продукта и не вступившего в реакцию исходного материала, что требует громоздких стадий очистки и приводит к низкому выходу.
Таким образом, существует потребность в улучшенных способах получения димеров индолинобензодиазепина, которые являются более эффективными и пригодными для крупномасштабного производства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает новые способы получения димерных соединений индолинобензодиазепина и их синтетических предшественников.
В одном варианте осуществления настоящего изобретения предложен способ получения соединения формулы (IIIа):

или его соли, включающий стадии:
(a) введения в реакцию соединения формулы (Iа):
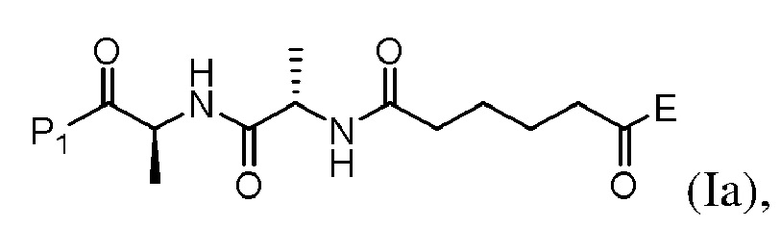
или его соли с соединением формулы (а):
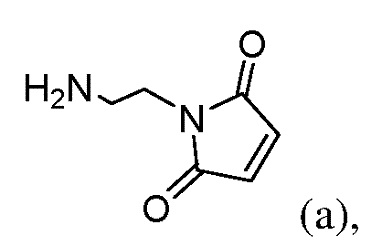
или его соли с образованием соединения формулы (IIа):
 и
и
(b) введения в реакцию соединения формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa) или его соли, где E представляет собой -OH, галогенид, или -C(=O)E1 представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
В другом варианте осуществления настоящее изобретение предусматривает способ получения соединения формулы (Va):

включающий стадии:
(a) введения в реакцию соединения формулы (Iа):

или его соли с соединением формулы (а):
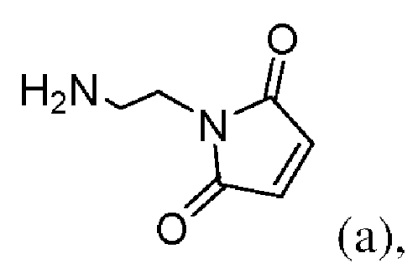
или его соли с образованием соединения формулы (IIа):

(b) введения в реакцию соединения формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa):
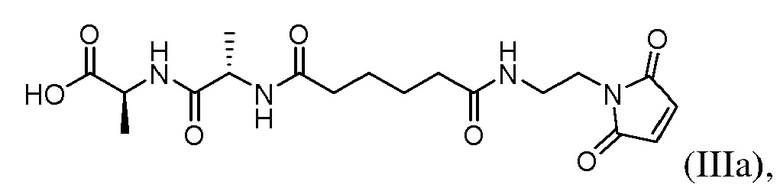
или его соли; и
(с) введения в реакцию соединения формулы (IIIа) с соединением формулы (IV):
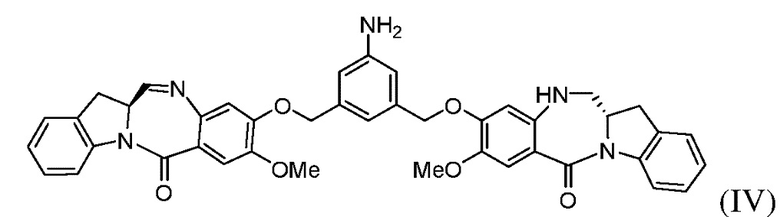
с образованием соединения формулы (Va), где E представляет собой -OH, галогенид, или -C(=O)E1 представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Теперь будет сделана подробная ссылка на определенные варианты осуществления изобретения, примеры которых проиллюстрированы в прилагаемых структурах и формулах. Хотя изобретение будет описано в связи с пронумерованными вариантами осуществления, следует понимать, что они не предназначены для ограничения изобретения этими вариантами осуществления. Напротив, изобретение предназначено для охвата всех альтернатив, модификаций и эквивалентов, которые могут быть включены в объем настоящего изобретения, как определено формулой изобретения. Специалист в данной области распознает множество способов и материалов, подобных или эквивалентных описанным в данном документе, которые могут быть использованы в практике настоящего изобретения.
Следует понимать, что любой из вариантов осуществления, описанных в данном документе, может быть объединен с одним или несколькими другими вариантами осуществления изобретения, если явно не оговорено или это не является неправильным. Комбинация вариантов осуществления не ограничивается теми конкретными комбинациями, заявленными в нескольких зависимых пунктах формулы изобретения.
ОПРЕДЕЛЕНИЯ
«Алкил» в контексте настоящего описания относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу. В предпочтительных вариантах осуществления алкил с прямой или разветвленной цепью имеет тридцать или менее атомов углерода (например, C1-C30 для алкильной группы с прямой цепью и C3-C30 для разветвленного алкила) и более предпочтительно - двадцать или менее атомов углерода. Еще более предпочтительно, алкил с прямой или разветвленной цепью имеет десять или менее атомов углерода (т.е. C1-C10 для алкильной группы с прямой цепью и C3-C10 для алкила с разветвленной цепью). В других вариантах осуществления алкил с прямой или разветвленной цепью имеет шесть или менее атомов углерода (т.е. C1-C6 для алкильной группы с прямой цепью или C3-C6 для алкила с разветвленной цепью). Примеры алкила включают, но не ограничиваются этим, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-метил-1-пропил, -CH2CH(CH3)2), 2-бутил, 2-метил-2-пропил, 1-пентил, 2-пентил 3-пентил, 2-метил-2-бутил, 3-метил-2-бутил, 3-метил-1-бутил, 2-метил-1-бутил, 1-гексил), 2-гексил, 3-гексил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 3-метил-3-пентил, 2-метил-3-пентил, 2,3-диметил-2-бутил, 3,3-диметил-2-бутил, 1-гептил, 1-октил и тому подобное. Кроме того, термин «алкил», используемый в описании, примерах и формуле изобретения, предназначен для включения как «незамещенных алкилов», так и «замещенных алкилов», последний из которых относится к алкильным группам, имеющим заместители, замещающие водород на одном или более атомах углерода углеводородного каркаса. Используемый в настоящем документе термин (Cx-Cxx)алкил или Cx-xx алкил означает линейный или разветвленный алкил, имеющий x-xx атомы углерода.
Используемый в настоящем документе термин «активированный сложный эфир» относится к сложноэфирной группе, которая легко замещается гидроксильной группой или аминогруппой. Примеры активированных сложных эфиров включают, но не ограничиваются ими, сложный эфир N-гидроксисукцинимида, сложный эфир нитрофенила (например, 2- или 4-нитрофенила), сложный эфир динитрофенила (например, 2,4-динитрофенила), сложный эфир сульфо-тетрафторфенила (например, 4-сульфо-2,3,5,6-тетрафторфенила), сложный эфир пентафторфенила, сложный эфир нитропиридила (например, 4-нитропиридила), трифторацетат и ацетат.
Термин «галогенид» относится к F, Cl, Br или I. В одном варианте осуществления галогенид представляет собой Cl. В одном варианте осуществления галогенид представляет собой Br. В одном варианте осуществления галогенид представляет собой I. В одном варианте осуществления галогенид представляет собой F.
Термин «соединение» предназначен для включения соединений, для которых структура или формула или любое их производное были раскрыты в настоящем изобретении, или их структура, формула или любое их производное, которые включены в качестве ссылки. Термин также включает стереоизомеры, геометрические изомеры или таутомеры. Конкретное перечисление «стереоизомеров», «геометрических изомеров», «таутомеров», «солей» в определенных аспектах изобретения, описанных в данной заявке, не должно интерпретироваться как предполагаемое исключение этих форм в других аспектах изобретения, где термин «соединение» используется без перечисления этих других форм.
Термин «предшественник» данной группы относится к любой группе, которая может привести к образованию этой группы посредством любого из снятия защиты, химической модификации или реакции сочетания.
Термин «хиральный» относится к молекулам, которые обладают свойством неналожения зеркального изображения партнера, тогда как термин «ахиральный» относится к молекулам, которые являются налагающимися на зеркальное изображение их партнера.
Термин «стереоизомер» относится к соединениям, которые имеют идентичное химическое строение и связность, но разные ориентации их атомов в пространстве, которые не могут быть взаимно преобразованы вращением вокруг одинарных связей.
Термин «диастереомер» относится к стереоизомеру с двумя или более центрами хиральности, молекулы которых не являются зеркальным отображением друг друга. Диастереомеры имеют разные физические свойства, например, точки плавления, точки кипения, спектральные свойства и реакционную способность. Смеси диастереомеров можно разделить с помощью аналитических процедур с высоким разрешением, таких как кристаллизация, электрофорез и хроматография.
Термин «энантиомеры» относится к двум стереоизомерам соединения, которые являются несовпадающими при наложении зеркальными отображениями друг друга.
Стереохимические определения и условные обозначения, используемые в данном документе, обычно следуют из S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. и Wilen, S., “Stereochemistry of Organic Compounds,” John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать асимметричные или хиральные центры и, следовательно, существовать в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, помимо прочего, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. Многие органические соединения существуют в оптически активных формах, то есть они обладают способностью вращать плоскость плоско-поляризованного света. При описании оптически активного соединения приставки D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(-ов). Приставки d и 1 или (+) и (-) используются для обозначения знака вращения плоско-поляризованного света соединением, при этом (-) или 1 означает, что соединение является левовращающим. Соединение с приставкой (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры идентичны, за исключением того, что они являются зеркальным отображением друг друга. Конкретный стереоизомер также может называться энантиомером, и смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называется рацемической смесью или рацематом, которая может возникать в тех случаях, когда в химической реакции или процессе.не было стерео-селекции или стереоспецифичности Термины «рацемическая смесь» и «рацемат» относятся к эквимолярной смеси двух энантиомерных видов, лишенной оптической активности.
Термин «таутомер» или «таутомерная форма» относится к структурным изомерам различной энергии, которые являются взаимопревращаемыми посредством низкого энергетического барьера. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимные превращения посредством миграции протона, такой как изомеризация кето-енола и имин-енамина. Валентные таутомеры включают взаимопревращения за счет реорганизации некоторых связывающих электронов.
Термин «защитная группа» или «защитный фрагмент» относится к заместителю, который обычно используется для блокирования или защиты конкретной функциональности при взаимодействии других функциональных групп в соединении, его производном или его конъюгате.
«Защитная группа карбоновой кислоты» представляет собой заместитель, присоединенный к карбонильной группе, который блокирует или защищает функциональную группу карбоновой кислоты в соединении. Такие группы хорошо известны в данной области (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 5, J. Wiley & Sons, NJ). Подходящие защитные группы карбоновой кислоты включают, но не ограничиваются ими, алкиловый эфир (например, метиловый эфир или трет-бутиловый эфир), бензиловый эфир, тиоэфир (например, трет-бутилтиоэфир), силиловый эфир (например, триметилсилиловый эфир), 9-флуоренилметиловый эфир, (2-триметилсилил)этоксиметиловый эфир, 2-(триметилсилил)этиловый эфир, дифенилметиловый эфир или оксазолин. В определенных вариантах осуществления защитная группа карбоновой кислоты представляет собой метиловый эфир, трет-бутиловый эфир, бензиловый эфир или триметилсилиловый эфир. В определенных вариантах осуществления защитная группа карбоновой кислоты представляет собой трет-бутиловый эфир.
Используемый в настоящем документе термин «агент для снятия защиты карбоновой кислоты» относится к реагенту, который способен расщеплять защитную группу карбоновой кислоты с образованием свободной карбоновой кислоты. Такие реагенты хорошо известны в данной области техники (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 5, J. Wiley & Sons, NJ) и зависят от используемой защитной группы карбоновой кислоты. Например, когда защитной группой карбоновой кислоты является трет-бутиловый эфир, он может быть расщеплен кислотой. В определенном варианте осуществления агент для снятия защиты карбоновой кислоты представляет собой трифторуксусную кислоту.
Термин «спирт-активирующий агент» относится к реагенту, который увеличивает реакционную способность гидроксильной группы, тем самым делая гидроксильную группу группой, лучше уходящей. Примеры таких спиртовых активирующих агентов включают п-толуолсульфонилхлорид, тионилхлорид, трифторметансульфоновый ангидрид, мезилхлорид, мезилангидрид, трифенилфосфин, ацилхлорид, 4-диметиламинопиридин и другие. В определенных вариантах осуществления спирт-активирующий агент представляет собой тионилхлорид. В определенном варианте осуществления спирт-активирующий агент представляет собой трифенилфосфин.
Используемая здесь фраза « соль» относится к органическим или неорганическим солям соединения изобретения. Типичные соли включают, но не ограничиваются ими, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат «мезилат», этансульфонат, бензолсульфонат, п-толуолсульфонат, соли памоата (например, 1,1’-метилен-бис-(2-гидрокси-3-нафтаноат)), соли щелочных металлов (например, натрия и калия), соли щелочноземельных металлов (например, магния) и соли аммония. Соль может включать включение другой молекулы, такой как ацетат-ион, сукцинат-ион или другой противоион. Противоион может быть любым органическим или неорганическим фрагментом, который стабилизирует заряд исходного соединения. Кроме того, соль может иметь более одного заряженного атома в своей структуре. В случаях, когда несколько заряженных атомов являются частью соли, может присутствовать несколько противоионов. Следовательно, соль может иметь один или более заряженных атомов и/или один или более противоионов.
Если соединение изобретения представляет собой основание, желаемая соль может быть получена любым подходящим способом, доступным в данной области, например обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидрокси кислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или тому подобное.
Если соединение изобретения представляет собой кислоту, желаемую соль можно получить любым подходящим способом, например обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксидом щелочного металла или гидроксидом щелочноземельного металла или тому подобное. Иллюстративные примеры подходящих солей включают, но не ограничиваются ими, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиак, первичные, вторичные и третичные амины, и циклические амины, такие как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
В определенных вариантах осуществления соль представляет собой фармацевтически приемлемую соль. Фраза «фармацевтически приемлемый» указывает на то, что вещество или композиция должны быть химически и/или токсикологически совместимыми с другими ингредиентами, составляющими препарат, и/или млекопитающим, которое ими лечат.
Как используется в данном документе, объемное количество (V) означает отношение объема растворителя (в мл) к весовому количеству (в г) соединения. Например, объем 40 означает, что на 1 г соединения используется 40 мл растворителя.
СПОСОБЫ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Настоящее изобретение предлагает новые синтетические способы получения димерных соединений индолинобензодиазепина и предшественников.
В первом варианте осуществления настоящего изобретения предложен способ получения соединения формулы (III):

или его соли, включающий стадии:
(a) введения в реакцию соединения формулы (I):

или его соли с соединением формулы (а):

или его соли (например, соль HCl или соль ТФУ) с образованием соединения формулы (II):
 и
и
(b) введения в реакцию соединения формулы (II) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (III), где E представляет собой -OH, галогенид, или -C(=O)E представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
В 1-м варианте осуществления настоящее изобретение предусматривает способ получения
соединения формулы (IIIa):

или его соли, включающий стадии:
(a) введения в реакцию соединения формулы (Iа):

или его соли с соединением формулы (а):
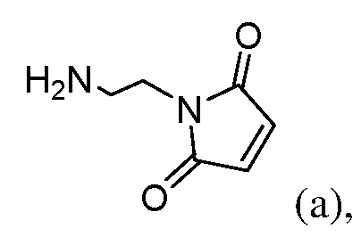
или его соли (например, соль HCl или соль ТФУ) с образованием соединения формулы (IIa):
 и
и
(b) введения в реакцию соединения формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa), где E представляет собой -OH, галогенид, или C(=O)E1 представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
В одном варианте осуществления для способа, описанного в первом варианте осуществления или 1ом конкретном варианте осуществления, E представляет собой -ОН, и соединение формулы (I) или (Ia) получают путем гидролиза соединения формулы (I”) или (Ia”):

В конкретном варианте осуществления гидролиз проводят в присутствии основания. В другом конкретном варианте осуществления основание выбрано из LiOH, KOH, NaOH. В еще другом конкретном варианте осуществления основание представляет собой LiOH.
Во втором варианте осуществления способ по первому варианту осуществления дополнительно включает введение в реакцию соединения формулы (III) с соединением формулы (IV):
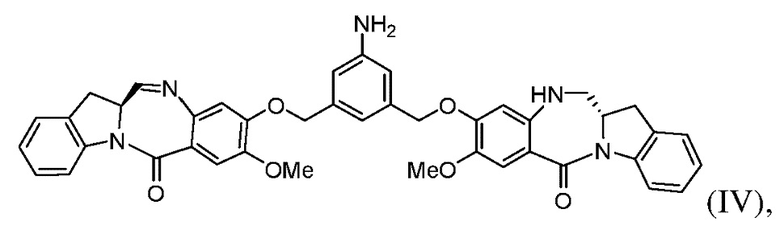
с образованием соединения формулы (V):
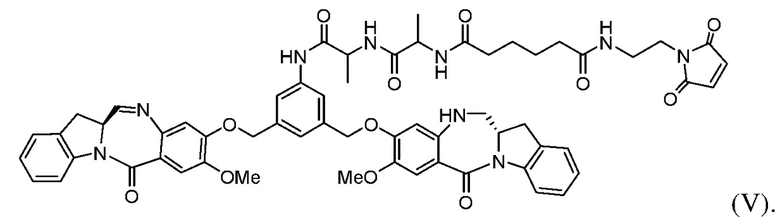
Во 2ом конкретном варианте осуществления способ по 1-му конкретному варианту осуществления дополнительно включает введение в реакцию соединения формулы (IIIa) с соединением формулы (IV):

с образованием соединения формулы (Va):

В третьем варианте осуществления настоящего изобретения предложен способ получения соединения формулы (V):

включающий стадии:
(a) введения в реакцию соединения формулы (I):

или его соли с соединением формулы (а):
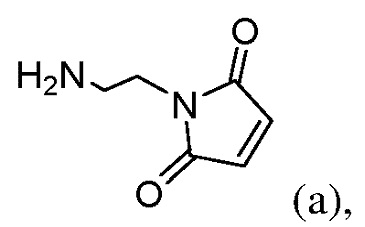
или его соли (например, соль HCl или соль ТФУ) с образованием соединения формулы (II):
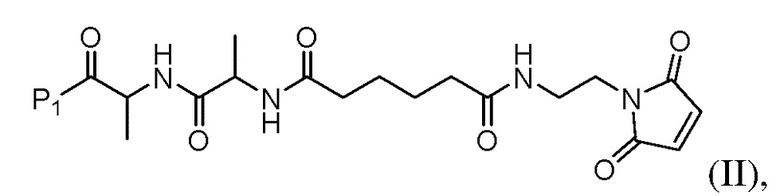
(b) введения в реакцию соединения формулы (II) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (III):
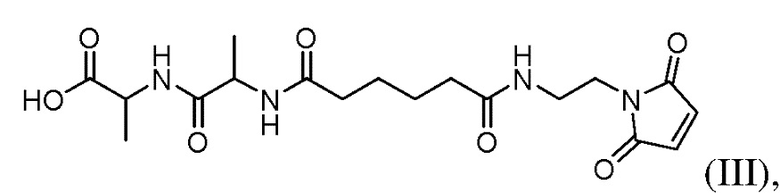
или его соли; и
(с) введения в реакцию соединения формулы (III) с соединением формулы (IV):
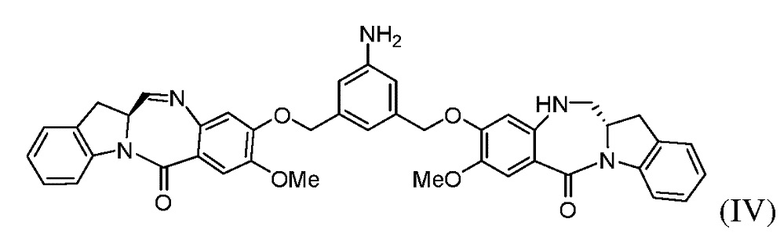
с образованием соединения формулы (V), где E представляет собой -OH, галогенид, или -C(=O)E1 представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
В 3ем конкретном варианте осуществления настоящего изобретения предложен способ получения соединения формулы (Va):
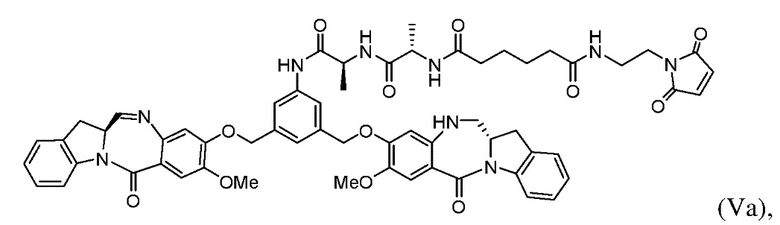
включающий стадии:
(а) введения в реакцию соединения формулы (Iа):
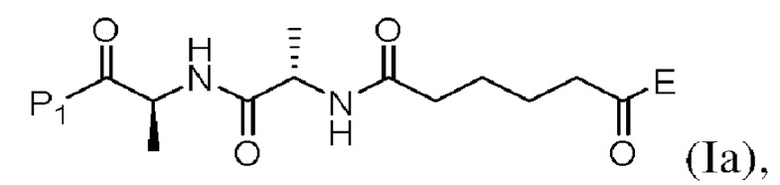
или его соли с соединением формулы (а):

или его соли (например, соль HCl или соль ТФУ) с образованием соединения формулы (IIa):
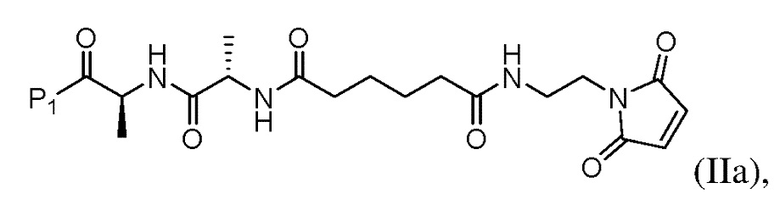
(b) введения в реакцию соединения формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa):

или его соли; и
(b) введения в реакцию соединения формулы (IIIа) с соединением формулы (IV):
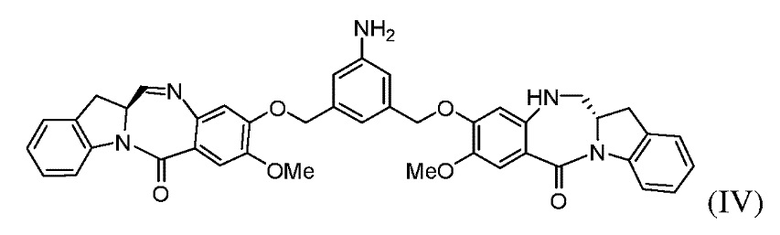
с образованием соединения формулы (Va), где E представляет собой -OH, галогенид, или -C(=O)E1 представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
В четвертом варианте осуществления для способа, описанного в первом, втором или третьем варианте осуществления или 1ом, 2ом или 3ем конкретном варианте осуществления, P1 может быть любой подходящей защитной группой карбоновой кислоты, известной в данной области техники. В одном варианте осуществления защитные группы карбоновой кислоты включают, но не ограничиваются ими, алкиловый эфир (например, метиловый эфир или трет-бутиловый эфир), бензиловый эфир, тиоэфир (например, трет-бутилтиоэфир), силиловый эфир (например,триметилсилиловый эфир), 9-флуоренилметиловый эфир, (2-триметилсилил)этоксиметиловый эфир, 2-(триметилсилил)этиловый эфир, дифенилметиловый эфир или оксазолин. В конкретном варианте осуществления защитная группа карбоновой кислоты представляет собой метиловый эфир, трет-бутиловый эфир, бензиловый эфир или триметилсилиловый эфир, т.е. P1 представляет собой -OMe, -OtBu, -OBn, -O-силил (например, -OSi(Me)3). В другом конкретном варианте осуществления защитная группа карбоновой кислоты представляет собой трет-бутиловый эфир, т.е. P1 представляет собой -OtBu.
В пятом варианте осуществления для способа, описанного в первом, втором, третьем или четвертом варианте осуществления или в 1ом, 2ом или 3ем конкретном варианте осуществления, E представляет собой -ОН, и реакция между соединением формулы (I) или (Ia) или его солью и соединением формулы (а) или его солью (например, солью HCl или солью ТФУ) осуществляют в присутствии активирующего агента; а остальные переменные описаны в первом, втором, третьем или четвертом вариантах осуществления или в 1ом, 2ом или 3ем конкретном варианте осуществления.
В конкретном варианте осуществления активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид, карбодиимид (например, N,N’-дициклогексилкарбодиимид (DCC) или 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC)), 1,1’-карбонилдиимидазол (CDI), уроний, активированный сложный эфир, фосфоний, 2-алкил-1-алкилкарбонил-1,2-дигидрохинолин, 2-алкокси-1-алкоксикарбонил-1,2-дигидрохинолин или алкилхлорформиат.
В другом конкретном варианте осуществления активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид. В более конкретном варианте осуществления активирующий агент представляет собой 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид (T3P).
В еще одном конкретном варианте осуществления активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид, 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксидгексафторфосфат (HATU), 1-гидрокси-7-азабензотриазол или 1H-[1,2,3]триазоло[4,5-b]пиридин-1-ол (HOAt), 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид, карбодиимид, уроний, активированный сложный эфир, фосфоний , 2-алкил-1-алкилкарбонил-1,2-дигидрохинолин, 2-алкокси-1-алкоксикарбонил-1,2-дигидрохинолин или алкилхлорформиат или их комбинацию.
В другом конкретном варианте осуществления активирующий агент представляет собой 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло [4,5-b]пиридиния 3-оксидгексафторфосфат (HATU). В более конкретном варианте осуществления активирующими агентами являются HATU и HOAt.
Любое подходящее количество активирующего агента можно использовать в реакции между соединением формулы (I) или (Ia) или его солью и соединением формулы (а) или его солью (например, солью HCl или солью ТФУ). В одном варианте осуществления в реакции используется от 1,0 до 5,0 молярных эквивалентов активирующего агента (например, HATU) по отношению к количеству соединения формулы (I) или (Ia). В конкретном варианте осуществления используется 1,0-2,0, 1,2-1,7 или 1,3-1,6 эквивалента HATU. В конкретном варианте осуществления используется 1,2, 1,3, 1,4, 1,5, 1,6 или 1,7 эквивалента HATU. В более конкретном варианте осуществления используется 1,5 эквивалента HATU.
В одном варианте осуществления реакцию между соединением формулы (I) или (Ia) или его солью и соединением формулы (a) или его солью (например, солью HCl или солью ТФУ) проводят в присутствии основания. В одном варианте осуществления основание представляет собой ненуклеофильное основание. Примеры ненуклеофильных оснований включают, помимо прочего, триэтиламин, имидазол, диизопропилэтиламин, пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или тетраметилпиперидин. В конкретном варианте осуществления основание представляет собой триэтиламин или диизопропилэтиламин.. В другом конкретном варианте осуществления основание представляет собой диизопропилэтиламин.
В другом варианте осуществления реакцию между соединением формулы (I) или (Ia) или его солью и соединением формулы (a) или его солью (например, солью HCl или солью ТФУ) проводят в присутствии активирующего агента, описанного выше, и основания, описанного выше. В конкретном варианте осуществления реакцию проводят в присутствии 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксида в качестве активирующего агента и триэтиламина или диизопропилэтиламина в качестве основания. В другом конкретном варианте осуществления реакцию проводят в присутствии 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринана-2,4,6-триоксида (T3P) и диизопропилэтиламина. В другом конкретном варианте осуществления реакцию проводят в присутствии HATU и HOAt в качестве активирующих агентов и диизопропилэтиламина в качестве основания.
Реакцию между соединением формулы (I) или (Ia) или его солью и соединением формулы (а) или его солью (например, солью HCl или солью ТФУ) можно проводить в любом подходящем органическом растворителе(-ях). В одном варианте осуществления реакцию проводят в дихлорметане.
В другом варианте осуществления реакцию между соединением формулы (I) или (Ia) или его солью и соединением формулы (a) или его солью (например, солью HCl или солью ТФУ) проводят в инертной атмосфере. В конкретном варианте осуществления инертная атмосфера достигается дегазированием реакционных растворов и продувкой реакционного сосуда азотом или аргоном.
Реакцию между соединением формулы (I) или (Ia) или его солью и соединением формулы (а) или его солью (например, солью HCl или солью ТФУ) можно проводить при подходящей температуре. В некоторых вариантах осуществления реакцию проводят при температуре от 0°C до 50°C, от 5°C до 50°C, от 10°C до 50°C, от 10°C до 40°C, от 10°C до 30° C или от 15°C до 25°C. В более конкретных вариантах осуществления реакцию проводят при 20±3°C.
В шестом варианте осуществления для способа, описанного в первом, втором, третьем, четвертом или пятом вариантах осуществления или в 1ом, 2ом или 3ем конкретном варианте осуществления, на стадии (b) можно использовать любую подходящую защитную группу карбоновой кислоты. Подходящие средства для снятия защиты, которые можно использовать, зависят от идентичности защитной группы карбоновой кислоты. Например, когда P1 представляет собой OtBu, защитная группа может быть удалена с помощью обработки кислотой, основанием или подходящим восстановителем. В некоторых вариантах осуществления можно использовать кислоту для удаления защитной группы трет-бутилового эфира. Примеры кислот включают, но не ограничиваются ими, муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту, соляную кислоту и фосфорную кислоту. В конкретном варианте осуществления в качестве агента для снятия защиты карбоновой кислоты используется трифторуксусная кислота.
В одном варианте осуществления реакцию снятия защиты можно проводить в любом подходящем(-их) органическом(-их) растворителе(-ях). Примеры органических растворителей включают, но не ограничиваются ими, DMF, CH2Cl2, дихлорэтан, ТГФ, диметилацетамид, метанол, этанол и т. д. В конкретном варианте осуществления реакцию снятия защиты проводят в дихлорметане.
В седьмом варианте осуществления настоящего изобретения предложен способ получения соединения формулы (IIIa):

включающий стадии:
(a) введения в реакцию соединения формулы (Ia1):

с соединением формулы (а):

или его соли (например, соли HCl или соли ТФУ) в присутствии активирующего агента с образованием соединения формулы (IIa1):
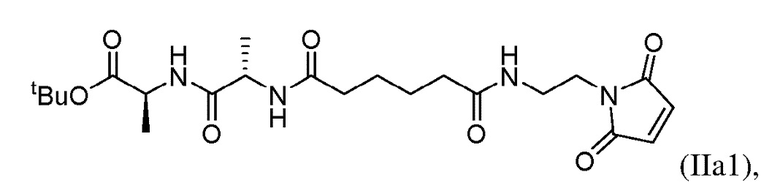
(b) введения в реакцию соединения формулы (IIa1) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa):
В одном варианте осуществления для способа, описанного в седьмом варианте осуществления, соединение формулы Ia1 получают путем гидролиза соединения формулы (Ia1”):

В конкретном варианте осуществления гидролиз проводят в присутствии основания. В другом конкретном варианте осуществления основание выбрано из LiOH, KOH, NaOH. В еще другом конкретном варианте осуществления основание представляет собой LiOH.
В восьмом варианте осуществления настоящего изобретения предложен способ получения соединения формулы (Va):
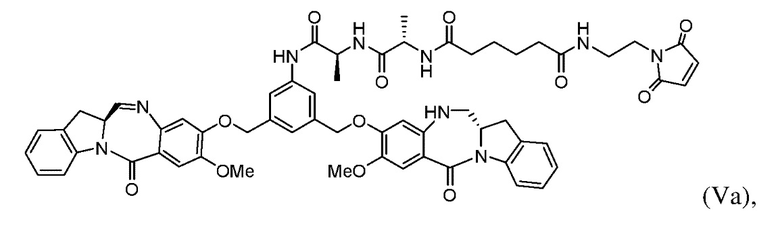
включающий стадии:
(а) введения в реакцию соединения формулы (Iа1):
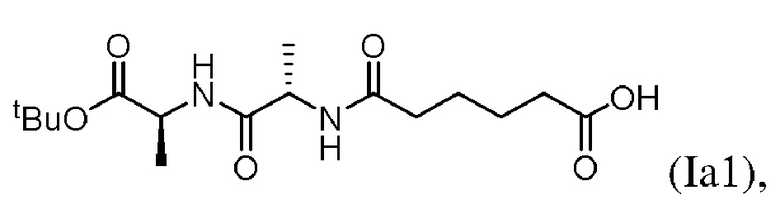
с соединением формулы (а):
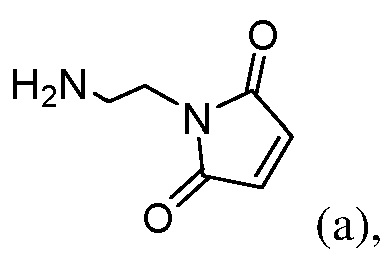
или его соли (например, соль HCl или соль ТФУ) в присутствии активирующего агента с образованием соединения формулы (IIa1):

(b) введения в реакцию соединения формулы (IIa1) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa):
 и
и
(с) введения в реакцию соединения формулы (IIIа) с соединением формулы (IV):

с образованием соединения формулы (Vа):
В девятом варианте осуществления для способа по седьмому или восьмому варианту осуществления реакцию соединения формулы (Ia1) и соединения формулы (a) или его соли (например, соли HCl или соли ТФУ) на стадии (a) проводят в присутствии 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксида (T3P) в качестве активирующего агента на стадии (а). В другом варианте осуществления реакцию соединения формулы (Ia1) и соединения формулы (a) или его соли (например, соли HCl или соли ТФУ) на стадии (a) можно проводить в присутствии основания. В одном варианте осуществления основание представляет собой триметиламин или диизопропилэтиламин. В конкретном варианте осуществления реакцию соединения формулы (Ia1) и соединения формулы (a) или его соли (например, соли HCl или соли ТФУ) на стадии (a) можно проводить в присутствии 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксида (T3P) в качестве активирующего агента и диизопропилэтиламина в качестве основания. В одном варианте осуществления реакцию можно проводить в дихлорметане.
В десятом варианте осуществления для способа, описанного в седьмом, восьмом или девятом варианте осуществления, агент для снятия защиты карбоновой кислоты на стадии (b) представляет собой трифторуксусную кислоту (ТФУ). В одном варианте осуществления реакцию снятия защиты проводят в дихлорметане.
В одиннадцатом варианте осуществления для способа, описанного во втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом или десятом варианте осуществления или во 2ом или 3ем конкретном варианте осуществления, реакцию между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью проводят в присутствии активирующего агента; а остальные переменные описаны во втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом или десятом вариантах осуществления или во 2ом или 3ем конкретном варианте осуществления.
В конкретном варианте осуществления активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан 2,4,6-триоксид, 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксидгексафторфосфат (HATU), 1-гидрокси-7-азабензотриазол или 1H-[1,2,3]триазоло[4,5-b]пиридин-1-ол (HOAt), карбодиимид (например, N,N’-дициклогексилкарбодиимид (DCC) или 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC)), 1,1’-карбонилдиимидазол (CDI), уроний, активированный сложный эфир, фосфоний, 2-алкил-1-алкилкарбонил-1,2-дигидрохинолин, 2-алкокси-1-алкоксикарбонил-1,2-дигидрохинолин или алкилхлорформиат, или их комбинацию.
В другом конкретном варианте осуществления активирующий агент представляет собой 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло [4,5-b]пиридиния 3-оксидгексафторфосфат (HATU). В более конкретном варианте осуществления активирующими агентами являются HATU и HOAt.
Любое подходящее количество активирующего агента можно использовать в реакции между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью. В одном варианте осуществления в реакции используется от 1,0 до 5,0 молярных эквивалентов HATU по отношению к количеству соединения формулы (IV). В конкретном варианте осуществления используется 1,0-2,0, 1,2-1,7 или 1,3-1,6 эквивалента HATU. В конкретном варианте осуществления используется 1,2, 1,3, 1,4, 1,5, 1,6 или 1,7 эквивалента HATU. В более конкретном варианте осуществления используется 1,5 эквивалента HATU.
В другом варианте осуществления от 0,1 до 1,0 молярного эквивалента HOAt относительно количества соединения формулы (IV) используется в реакции между соединением формулы (III) или (IIIa) или его солью и соединением формула (IV) или его солью. В конкретном варианте осуществления используется 0,2-0,8, 0,3-0,7 или 0,4-0,6 эквивалента HOAt. В другом варианте осуществления используется 0,3, 0,4, 0,5, 0,6 или 0,7 эквивалента HOAt. В более конкретном варианте осуществления используется 0,5 эквивалента HOAt.
В одном варианте осуществления реакцию между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью проводят в присутствии основания. В одном варианте осуществления основание представляет собой ненуклеофильное основание. Примеры ненуклеофильных оснований включают, помимо прочего, триэтиламин, имидазол, диизопропилэтиламин, пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или тетраметилпиперидин. В конкретном варианте осуществления основание представляет собой триэтиламин или диизопропилэтиламин.. В другом конкретном варианте осуществления основание представляет собой диизопропилэтиламин.
В другом варианте осуществления реакцию между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью проводят в присутствии активирующего агента, описанного выше, и основания, описанного выше. В конкретном варианте осуществления реакцию проводят в присутствии HATU и HOAt в качестве активирующих агентов и диизопропилэтиламина в качестве основания.
В другом варианте осуществления реакцию между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью проводят в инертной атмосфере. В конкретном варианте осуществления инертная атмосфера достигается дегазированием реакционных растворов и продувкой реакционного сосуда азотом или аргоном.
Реакцию между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью (например, солью HCl или солью ТФУ) можно проводить в любом подходящем органическом растворителе(-ях). В одном варианте осуществления реакцию проводят в дихлорметане.
Реакцию между соединением формулы (III) или (IIIa) или его солью и соединением формулы (IV) или его солью (например, солью HCl, солью ТФУ) можно проводить при подходящей температуре. В некоторых вариантах осуществления реакцию проводят при температуре от 0°C до 50°C, от 5°C до 50°C, от 10°C до 50°C, от 10°C до 40°C, от 10°C до 30°C или от 15°C до 25°C. В более конкретных вариантах осуществления реакцию проводят при 20 °C.
В одиннадцатом варианте осуществления для способа, описанного во втором, третьем, четвертом, пятом, шестом, восьмом, девятом или десятом варианте осуществления или во 2ом или 3ем конкретном варианте осуществления, соединение формулы (IV) может быть получено способом, включающим стадию введения в реакцию соединения формулы (V):

с восстанавливающим агентом с образованием соединения формулы (IV).
В двенадцатом варианте осуществления для способа, описанного во втором, третьем, четвертом, пятом, шестом, восьмом, девятом или десятом варианте осуществления или во 2ом или 3ем конкретном варианте осуществления, соединение формулы (IV) может быть получено способом, включающим стадии:
1) введения в реакцию соединения формулы (VI):
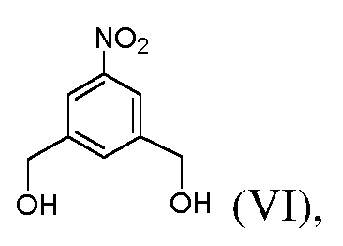
с соляной кислотой в толуоле с образованием соединения формулы (VII):

2) введения в реакцию соединения формулы (VII) с мономером соединения формулы (a1),
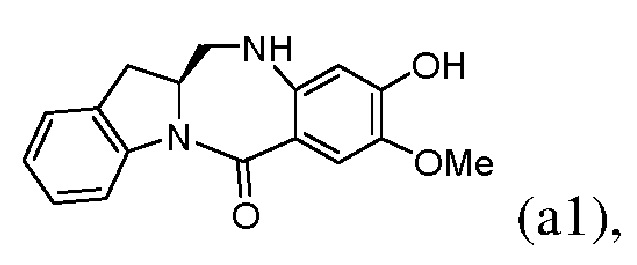
с образованием соединения формулы (VIII):
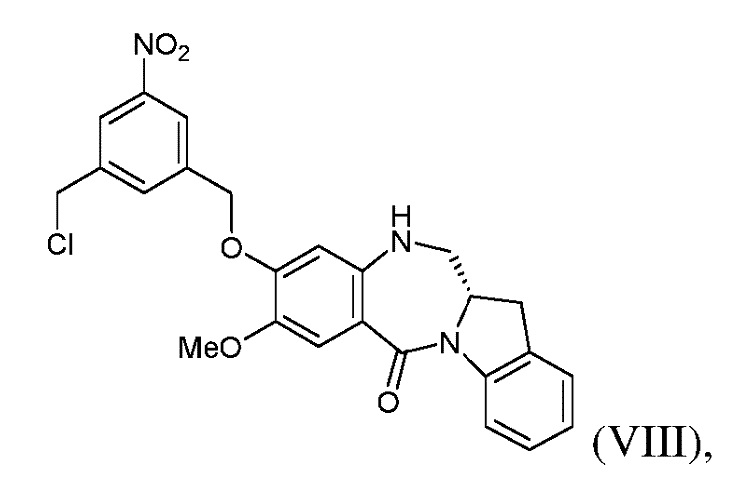
или его соли;
3) введения в реакцию соединения формулы (VIII) или его соли с мономером соединения формулы (b1):

с образованием соединения формулы (V):

или его соли; и
4) введения в реакцию соединения формулы (V) или его соли с восстанавливающим агентов с образованием соединения формулы (IV):
В одном варианте осуществления для способа, описанного в одиннадцатом или двенадцатом варианте осуществления, любой подходящий восстанавливающий агент, который может превращать нитро(-NO2)-группу в амино(-NH2)-группу, можно использовать для превращения соединения формулы (V) в соединение формулы (IV). В одном варианте осуществления восстанавливающий реагент выбран из группы, состоящей из газообразного водорода, гидросульфита натрия, сульфида натрия, хлорида двухвалентного олова, хлорида титана(II), цинка, железа и йодида самария. В другом варианте осуществления восстанавливающий реагент представляет собой Fe/NH2Cl, Fe/NH4Cl, Zn/NH2Cl, FCSO4/NH4OH или губчатый никель. В конкретном варианте осуществления восстанавливающий агент представляет собой Fe/NH4C1.
В одном варианте осуществления для стадии 1) способа, описанного в двенадцатом варианте осуществления, соединение формулы (VI) вводят в реакцию с концентрированной соляной кислотой с образованием соединения формулы (VII).
В другом варианте осуществления для стадии 2) способа, описанного в двенадцатом варианте осуществления, соединение формулы (VII) вводят в реакцию с мономерным соединением формулы (a1) в присутствии спиртового активирующего агента и азодикарбоксилата. В конкретном варианте осуществления спирт-активирующий агент представляет собой трибутилфосфин или трифенилфосфин. В другом конкретном варианте осуществления азодикарбоксилат представляет собой диэтилазодикарбоксилат (DEAD), диизопропилазодикарбоксилат (DIAD), 1,1’-(азодикарбонил)дипиперидин (ADDP) и дитретбутил-азодикарбоксилат (DTAD). В другом конкретном варианте осуществления спирт-активирующий агент представляет собой трибутилфосфин или трифенилфосфин, и азодикарбоксилат представляет собой диэтилазодикарбоксилат (DEAD), диизопропилазодикарбоксилат (DIAD), 1,1’-(азодикарбонил)дипиперидин (ADDP) и дитретбутил-азодикарбоксилат (DTAD). В еще более конкретном варианте осуществления спиртовой активирующий агент представляет собой трибутилфосфин, а азодикарбоксилат представляет собой DIAD. В другом более конкретном варианте осуществления спиртовой активирующий агент представляет собой трифенилфосфин, а азодикарбоксилат представляет собой DIAD. В одном варианте осуществления трифенилфосфин добавляют после смешивания соединения формулы (VII), мономерного соединения формулы (a1) и азодикарбоксилата. В еще более конкретном варианте осуществления трифенилфосфин добавляют после смешивания соединения формулы (VII), мономерного соединения формулы (a1) и DIAD.
В одном варианте осуществления для стадии 3) способа, описанного в двенадцатом варианте осуществления, реакцию между соединением формулы (VIII) и мономером соединения формулы (b1) проводят в присутствии основания. В одном варианте осуществления основание представляет собой карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. В конкретном варианте осуществления основание представляет собой карбонат калия. В другом варианте осуществления реакция между соединением формулы (VIII) и мономером соединения формулы (b1) дополнительно включает йодид калия. В конкретном варианте осуществления реакцию между соединением формулы (VIII) и мономером соединения формулы (b1) проводят в присутствии карбоната калия и йодида калия.
Все ссылки, цитируемые в данном документе и в следующих примерах, непосредственно включены в качестве ссылки во всей их полноте.
ПРИМЕРЫ
Следующие растворители, реагенты, защитные группы, фрагменты и другие обозначения могут упоминаться в сокращенном виде в скобках:
экв. = молярный эквивалент
об. = объем
DCM или CH2CI2 = дихлорметан
DIEA или DIPEA = N,N-диизопропилэтиламин
г = граммы
ЖХМС = жидкостная хроматография - масс-спектрометрия
мин = минуты
мг = милиграммы
мл = милилитры
ммоль = милимоли
МС = масс-спектрометрия
tBME или MTBE = метил-трет-бутиловый эфир
ЯМР = спектроскопия ядерного магнитного резонанса
T3P = 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид
ТФУ = трифторуксусная кислота
АЦН = ацетонитрил
HATU = 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксидгексафторфосфат
HAOt = 1H-[1,2,3]триазоло[4,5-b]пиридин-1-ол или 1-гидрокси-7-азабензотриазол
Пример 1. Синтез (S)-трет-бутил-2-((S)-2-(6-((2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил)амино)-6-оксогексанамидо)пропанамидо)пропаноата
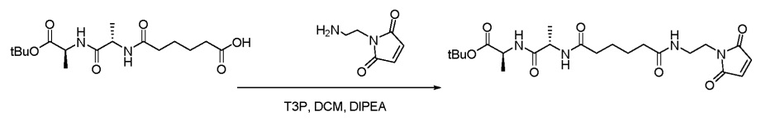
В чистую сухую круглодонную колбу объемом 100 мл с мешалкой и термопарой в атмосфере азота добавляли -(((S)-1-(((S)-1-(трет-бутокси)-1-оксопропан-2-ил)амино)-1-оксопропан-2-ил)амино)-6-оксогексановую кислоту (0,51 г, 1,48 ммоль, 1,0 эквив.), HCl 1-(2-аминоэтил)-1H-пиррол- 2,5-диона (0,27 г, 1,53 ммоль, 1,1 экв.) и дихлорметан (10,0 мл, 20 об.). Полученную смесь перемешивали и охлаждали до 5±3°C. К смеси добавляли DIPEA (0,760 мл, 4,4 ммоль, 3,0 экв.) с последующим добавлением T3P (1,3 мл, 2,22 ммоль, 1,5 экв.). Реакционную смесь медленно нагревали до 20+5°C и перемешивали в течение 2 часов. Реакционную смесь гасили добавлением воды к реакционной смеси в течение 30+5 мин. Органическую фазу отделяли, промывали полунасыщенным солевым раствором (2 × 5,0 мл, 2 × 10 об.) и водой (2 × 5,0 мл, 2 × 10 об.) и концентрировали. Добавляли толуол к концентрированной органической фазе ((2 × 5,0 мл, 2 × 10 об.). Полученный раствор концентрировали под вакуумом, чтобы удалить оставшуюся воду. Полученное твердое вещество суспендировали в ДХМ (5,0 мл, 10 об.) и tBME (5,0 мл, 10 об.), перемешивали при температуре 5+2°C и концентрировали досуха с получением продукта в виде белого твердого вещества (0,557 г, выход 80,6%). Рассчитанный m/z 466,24, найдено 467,04
Пример 2. Синтез (S)-2-((S)-2-(6-((-2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил)амино)-6-оксогексанамидо)пропанамидо)пропановой кислоты
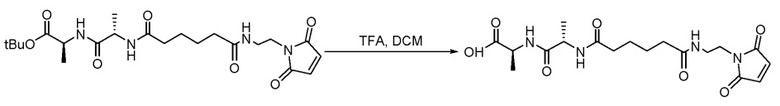
В чистую сухую круглодонную колбу объемом 100 мл с мешалкой и термопарой в атмосфере азота добавляли (S)-трет-бутил-2-((S)-2-(6-((2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил)амино)-6-оксогексанамидо)пропанамидо)пропаноат (0,41 г, 0,88 ммоль, 1,0 экв.), затем дихлорметан (4,0 мл, 10 об.). Полученную смесь перемешивали с образованием суспензии с последующим охлаждением до 5±3°C. Добавляли ТФУ (4,0 мл, 10 об.) и реакционную смесь перемешивали и обеспечивали медленное нагревание до 20±5°C. Реакционную смесь перемешивали при 20±5°C в течение 2 часов и гасили водой (несколько капель добавляли стеклянной пипеткой). Реакционную смесь концентрировали досуха и добавляли толуол. Полученный толуольный раствор концентрировали для удаления оставшейся воды. Полученное твердое вещество суспендировали в дихлорметане (5,0 мл, 10 об.) и tBME (5,0 мл, 10 об.), перемешивали при температуре 5±2°C и концентрировали досуха с получением продукта (S)-трет-бутил-2-((S)-2-(6-((2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил)амино)-6-оксогексанамидо)пропанамидо)пропаноата (0,557 г, выход 80,6%) в виде белого твердого вещества. Рассчитанный m/z 410,18, найдено 410,99
Пример 3. Синтез (S)-9-((3-(хлорметил)-5-нитробензил)окси)-8-метокси-11,12,12a,13-тетрагидро-6H-бензо[5,6][1,4]диазепино[1,2-а]индол-6-она

В сухую круглодонную колбу объемом 100 мл, обеспеченную мешалкой и термопарой в атмосфере азота, добавляли (S)-9-гидрокси-8-метокси-11,12,12a,13-тетрагидро-6H-бензо[5,6][1,4]диазепино[1,2-а]индол-6-он (1,0 г, 3,375 ммоль, 1,0 экв.), затем тетрагидрофуран (10,0 мл, 10 об.). Смесь перемешивали при 20°C+5°C с получением слегка мутного раствора. Затем к раствору добавляли 3-(хлорметил)-5-нитрофенил)метанол (0,849 г, 4,22 ммоль) и реакционную смесь перемешивали при 20°C±5°C с получением слегка мутного раствора. Реакционную смесь охлаждали до 5±3°C и через капельную воронку по каплям добавляли диизопропилазодикарбоксилат (0,930 мл, 4,725 ммоль) в течение 10±2 минуты. Трифенилфосфин (1,24 г, 7,425 ммоль) растворяли в ТГФ (3 мл, 2,4 об.), и полученный раствор добавляли к реакционной смеси по каплям для поддержания температуры ≤10°C. Реакционную смесь перемешивали при 5±3°C в течение 30 минут, затем охлаждали до 5±3°C и добавляли воду (5,0 мл, 5 об.). Полученную смесь перемешивали в течение 30 минут и добавляли ДХМ (40 мл, 40 об.). Полученную смесь переносили в делительную воронку на 250 мл и промывали водой (2 × 10 мл, 2 × 10 об.). Органическую фазу отделяли и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле, с элюированием градиентом 0-20% ДХМ/этилацетата в течение 40 минут. Фракции, содержащие чистые продукты, объединяли и концентрировали досуха с получением необходимого продукта. Растворяли концентрированные фракции в этилацетате и капали в перемешиваемый трет-бутилметиловый эфир (36,0 мл, 18 об.) с образованием бело-оранжевого твердого вещества. Смесь охлаждали до 5±5°C и перемешивали в течение 2 часов. Твердое вещество фильтровали с получением продукта (1,08 г, выход 66,6%).
Пример 4. Синтез соединения Va

Стадия 1a
Загружали ДХМ(~1,5 об.), затем соединение 7a (8,6 г, 1 экв.) и промывали с помощью ДХМ (14,5 об.). Добавляли малеимидное соединение 8 и использовали ДХМ (4 об.) для промывания. Раствор охлаждали до 5°C. Медленно добавляли DIPEA, а затем T3P, и реакционную смесь перемешивали при 20°C. Через 1 час 30 минут скорость преобразования составила 99,4%. Реакционную смесь охлаждали до 10°C и гасили медленным добавлением воды (20 об.). После отделения фазы водную фазу снова экстрагировали с помощью ДХМ (3 × 20 об.). Органические фазы объединяли и промывали 15% раствором NaCl (2 × 10 об.). Неочищенный продукт 7b хранили при 5°C перед использованием для синтеза соединения 7.
Стадия 1b
Неочищенный продукт 7b со стадии 1a концентрировали до 10 об. (по сравнению с теоретическим выходом 11,65 г). Температуру смеси снижали до 5°C. Медленно добавляли ТФУ при 5°C, обеспечивали нагревание реакционной смеси до 20°C и перемешивали. Через 1 час скорость превращения составила 99,6%. Реакционную смесь гасили добавлением воды (1V) и концентрировали досуха. Проводили совместное выпаривание с ДХМ (3 × 30 об.). Остаток растворяли в ДХМ (13 об.) и медленно добавляли к MTBE (13 об.). Температуру смеси снижали до 5°C, и суспензию перемешивали при 5°C в течение 30 минут перед фильтрованием. Твердое вещество промывали с помощью МТВЕ (2 × 2,5 об.) перед сушкой в глубоком вакууме при 35°C.
Стадия 1
Соединение 2 (1 экв.) суспендировали в ТГФ (10 об.) и добавляли соединение 1 (1,25 экв.). Реакционную смесь охлаждали до 5°C и добавляли DIAD (1,4 экв.), чтобы не превышать 10°C в массе. PPh3 (1,4 экв.) растворяли в ТГФ (2 об.) и медленно добавляли к реакционной смеси, чтобы не превышать 10°C в массе. Реакционную смесь перемешивали при 5 °C в течение 30 минут. Реакционную смесь гасили, добавляя воду (5 об.), и смесь перемешивали в течение 30 минут при 5°C. Обеспечивали нагревание реакционной смеси и перемешивание прекращали. После разделения фаз водную фазу экстрагируют с помощью ДХМ (20 об.). Органические фазы объединяли и промывали водой (2 × 10 об.) с последующим концентрированием и азеотропной перегонкой с помощью ДХМ (2 × 20 об.). Смесь концентрировали до 10 об. и раствор неочищенного продукта 3 очищали посредством хроматографии с обращенной фазой с использованием колонки YMC C18 Triart, с элюированием смесью АЦН/вода 55/45 об./об. Основной пик собирали и собранную фракцию экстрагировали с помощью ДХМ. Экстрагированные фракции объединяли и концентрировали до 10 об.
Стадия 2.
Соединение 3 (12,1 г, 1 экв.) в растворе ДХМ/АЦН со стадии 1 разбавляли в DMF (17 об.) при комнатной температуре. Затем раствор концентрировали приблизительно до 17 об. и переносили в реактор (кольцо с DMF (3 об.)). Добавляли соединение 4 (7,8 г, 1,05 экв.), затем KI (2,09 г, 0,5 экв.) и K2CO3 (7,0 г, 2 экв.). Реакционную смесь перемешивали при 35°C в течение 4 часов. Еще 0,1 экв. соединения 4 и реакционную смесь перемешивали при 35°C в течение 45 минут. Реакционную смесь охлаждали до 20°C и добавляли ДХМ (40 об.), а затем воду (20 об.). Фазы разделяли, и водную фазу снова экстрагировали с помощью ДХМ (20 об.). Органические фазы объединяли и промывали 15% раствором NaCl (2 × 20 об.), а затем водой (2 × 20 об.). Органическую фазу концентрировали до 10 об. и проводили азеотропную перегонку с помощью ДХМ (2 × 20 об.). В конце органический раствор концентрировали приблизительно до 10 об. (расчетный выход 77,7%).
Стадия 3.
Соединение 5 (18,6 г, 1 экв.) растворяли в ТГФ/МеОН/воде (12,5 об./1,7 об./0,85 об.) при комнатной температуре и переносили в реактор. В реактор добавляли NH4Cl (14,2 г, 10,5 экв.), а затем Fe (7,9 г, 5,6 экв.). Реакционную смесь перемешивали при 60°C в течение 1 часа. Реакционную смесь охлаждали до 20°C и разбавляли ДХМ, фильтровали через целит и промывали с помощью ДХМ. После концентрирования досуха остаток растворяли в ДХМ (20 об.). Органическую фазу промывали насыщенным раствором NaCl (20 об.), а затем водой (2 × 10 об.). Органическую фазу концентрировали и выпаривали совместно с ДХМ (2 × 20 об.). Неочищенный продукт очищали нормально-фазовой хроматографией на силикагеле, используя колонку Dalso SP-100-10-P, с элюированием градиентом ДХМ/MEOH из 2,4% MeOH с наклоном 0,34%. Объединенные фракции концентрировали почти досуха (расчетный выход 47,6%).
Стадия 4
Соединение 7 (1,2 экв.) суспендировали в ДХМ (24 об.) и добавляли EEDQ (2,5 экв.). Смесь перемешивали в течение 30 минут при 20°C. Добавляли MeOH (5 об.) и смесь перемешивали в течение 5 минут при 20°C. Соединение 6 (1 экв.) растворяли в ДХМ (12 об.) и раствор добавляли к реакционной смеси соединения 7 и EEDQ. Реакционную смесь перемешивали при 20°C до скорости превращения 95% или выше. Реакционную смесь промывали 1% раствором NaCl (14 об.). После разделения фаз органическую фазу концентрировали и выпаривали совместно с ДХМ (3 × 10 об.). Неочищенное соединение Va растворяли в ДХМ/MeOH 97,5/2,5 об./об. и очищали посредством хроматографии на силикагеле (Daiso SP-100-10-P), с элюированием градиентом ДХМ/MeOH от 5% до 10% MeOH. Объединенные фракции концентрировали и выпаривали совместно с ДХМ до 10 об. Раствор концентрировали, остаток растворяли в ДМСО и очищали посредством хроматографии с обращенной фазой (YMC C18 Triart), с элюированием смесью АЦН/вода 45/55 об./об. Фракции, содержащие продукт, экстрагировали с помощью ДХМ (0,4 об.) и промывали 0,5% раствором NaHCO3 (0,4 об.), а затем водой (2 × 0,4 об). Объединенные фракции концентрировали и выпаривали совместно с ДХМ почти досуха. Полученный остаток суспендировали в ДХМ и переносили в реакцию осаждения (75 об., включая промывку с помощью ДХМ). Медленно добавляли гептан (75 об.) и взвесь дополнительно перемешивали при комнатной температуре в течение 30 минут. После фильтрации твердое вещество промывали смесью ДХМ/гептан 1/1 об./об., а затем гептаном. Твердое вещество сушили при 35°C в глубоком вакууме.
Пример 5. Синтез соединения (Va)
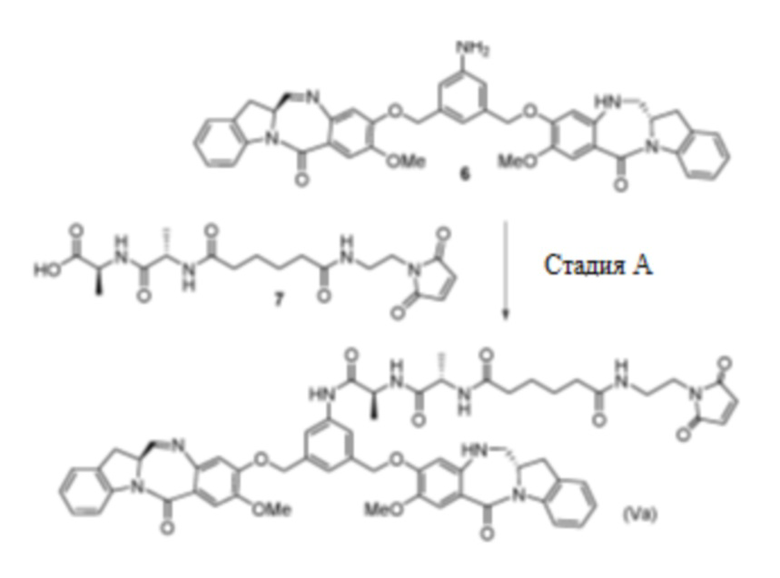
В чистую сухую круглодонную колбу (RBF) объемом 100 мл в атмосфере азота загружали соединение 7 (0,640 г, 1,528 ммоль), затем HATU (0,587 г, 1,529 ммоль), HOAt (0,587 г, 1,529 ммоль) и ДХМ (12 мл, 15 об.). Смесь перемешивали в течение 5±2 минуты при 20°C±5°C. Соединение 6 (0,82 г, 1,020 ммоль) растворяли в ДХМ (8,2 мл, 10 об.), и раствор добавляли к реакционной смеси соединения 7, HATU и HOAt. Затем добавляли МеОН (0,5 мл, 0,6 об.). Реакционный сосуд дегазировали и продували азотом. К реакционному раствору медленно добавляли DIPEA (0,268 мл, 1,529 ммоль) в ДХМ (4,1 мл, 5 об.). Смесь перемешивали в течение 30±5 минут при 20°C±5°C. К реакционной смеси добавляли нейтрализованный ДХМ (8,2 мл, 10 об., предварительно промытый раствором бикарбоната натрия), а затем 5% раствор карбоната натрия (12,3 мл, 15 об.). Полученный раствор перемешивали при 20°C±5°C в течение 30±5 минут, а затем переносили в делительную воронку и добавляли нейтрализованный ДХМ (12 мл, 15 об.). Раствор перемешивали путем дегазирования, а затем обеспечивали его разделение в течение 10±5 минут. Органическую фазу концентрировали и очищали посредством хроматографии с обращенной фазой (YMC C18 Triart), с элюированием градиентом 5-60% ацетонитрил/деионизированная вода в течение 20 минут с последующей выдержкой при 60% в течение 10 минут с повышением градиента до 95% за 10 минут и удерживанием при 95% в течение 5 минут. Фракции, содержащие продукт, объединяли и экстрагировали с помощью ДХМ (0,7 x общий объем фракций). Органический слой собирали и растворитель удаляли перегонкой c получением сухого грязно-белого мелкодисперсного порошка продукта соединения (Va) (0,6062 г, 0,529 ммоль, выход 51,9%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2018 |
|
RU2762891C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2813137C2 |
| МАКРОЦИКЛИЧЕСКИЕ АМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2013 |
|
RU2625796C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2016 |
|
RU2746322C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ, АКТИВИРУЮЩЕЙ КАНАЛЫ | 2007 |
|
RU2419625C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2016 |
|
RU2727151C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 10 И АУРИСТАТИНОВ | 2014 |
|
RU2662951C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2017 |
|
RU2773346C2 |
Изобретение относится к способу получения соединения формулы (IIIA) или его соли. Способ включает стадии (a) и (b). На стадии (a) вводят в реакцию соединение формулы (Iа) или его соль с соединением формулы (а) или его солью (например, солью HCl) с образованием соединения формулы (IIa). На стадии (b) вводят в реакцию соединение формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa), где E представляет собой -OH, галогенид или C(=O)E представляет собой активированный сложный эфир; P1 представляет собой защитную группу карбоновой кислоты. Предлагаемый способ позволяет улучшить получение димеров индолинобензодиазепина и является более эффективным и пригодным для крупномасштабного производства. Изобретение относится также к способу получения соединения формулы (Va) с использованием указанного способа. 2 н. и 30 з.п. ф-лы, 5 пр.





1. Способ получения соединения формулы (IIIA)
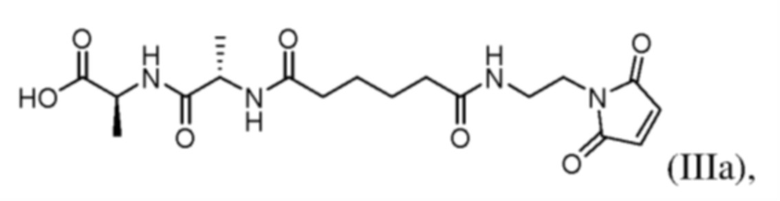
или его соли, включающий стадии:
(a) введения в реакцию соединения формулы (Iа)

или его соли с соединением формулы (а)

или его соли (например, соли HCl) с образованием соединения формулы (IIa)
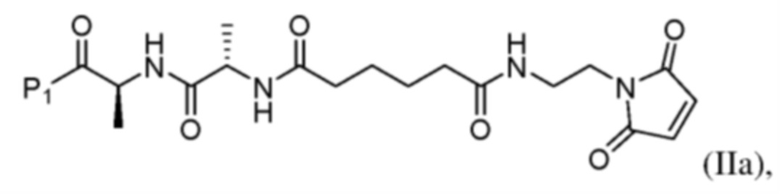 и
и
(b) введения в реакцию соединения формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa), где E представляет собой -OH, галогенид или C(=O)E представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
2. Способ по п. 1, где способ дополнительно включает введение в реакцию соединения формулы (IIIa) с соединением формулы (IV)
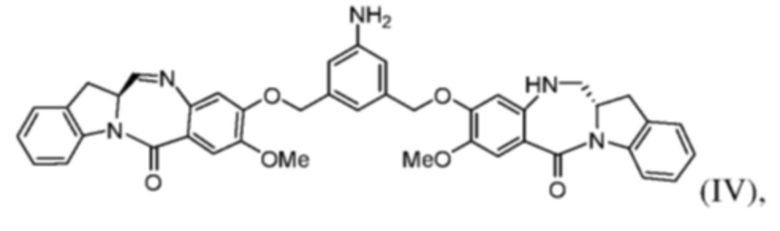
с образованием соединения формулы (Va)

3. Способ по п. 1 или 2, где P1 представляет собой -OtBu, -OMe, -OBn или -O-силил.
4. Способ по п. 3, где P1 представляет собой -OtBu.
5. Способ по любому из пп. 1-4, где E представляет собой -OH и реакцию между соединением формулы (Ia) и соединением формулы (a) проводят в присутствии активирующего агента.
6. Способ по п. 5, где активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид, карбодиимид, уроний, активированный сложный эфир, фосфоний, 2-алкил-1-алкилкарбонил-1,2-дигидрохинолин, 2-алкокси-1-алкоксикарбонил-1,2-дигидрохинолин или алкилхлорформиат.
7. Способ по п. 6, где активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид.
8. Способ по п. 7, где активирующий агент представляет собой 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид.
9. Способ по любому из пп. 1-8, где агент для снятия защиты карбоновой кислоты на стадии (b) представляет собой кислоту.
10. Способ по п. 9, где кислота представляет собой трифторуксусную кислоту (ТФУ).
11. Способ по п. 5, при этом способ включает стадии:
(a) введения в реакцию соединения формулы (Ia1)

или его соли с соединением формулы (а)

или его соли (например, соли HCl или соли ТФУ) в присутствии активирующего агента с образованием соединения формулы (IIa1)
 и
и
(b) введения в реакцию соединения формулы (IIa1) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa).
12. Способ по п. 11, где активирующий агент на стадии (а) представляет собой 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид.
13. Способ по п. 11 или 12, где агент для снятия защиты карбоновой кислоты на стадии (b) представляет собой трифторуксусную кислоту (ТФУ).
14. Способ получения соединения формулы (Va)

включающий стадии:
(а) введения в реакцию соединения формулы (Iа)

или его соли с соединением формулы (а)

или его соли (например, соли HCl или ТФУ) с образованием соединения формулы (IIa)
(b) введения в реакцию соединения формулы (IIa) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa)

или его соли; и
(c) введения в реакцию соединения формулы (IIIа) с соединением формулы (IV)

с образованием соединения формулы (Va), где E представляет собой -OH, галогенид или -C(=O)E представляет собой активированный сложный эфир; и P1 представляет собой защитную группу карбоновой кислоты.
15. Способ по п. 14, где P1 представляет собой -OtBu, -OMe, -OBn или -O-силил.
16. Способ по п. 15, где P1 представляет собой -OtBu.
17. Способ по любому из пп. 14-16, где E представляет собой -OH и реакцию между соединением формулы (Ia) и соединением формулы (a) проводят в присутствии активирующего агента.
18. Способ по п. 17, где активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид, карбодиимид, уроний, активированный сложный эфир, фосфоний, 2-алкил-1-алкилкарбонил-1,2-дигидрохинолин, 2-алкокси-1-алкоксикарбонил-1,2-дигидрохинолин или алкилхлорформиат.
19. Способ по п. 18, где активирующий агент представляет собой 2,4,6-триалкил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид.
20. Способ по п. 19, где активирующий агент представляет собой 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид.
21. Способ по любому из пп. 14-20, где агент для снятия защиты карбоновой кислоты на стадии (b) представляет собой кислоту.
22. Способ по п. 21, где кислота представляет собой трифторуксусную кислоту (ТФУ).
23. Способ по п. 17, при этом способ включает стадии:
(a) введения в реакцию соединения формулы (Ia1)

или его соли с соединением формулы (а)

или его соли (например, соли HCl или соли ТФУ) в присутствии активирующего агента с образованием соединения формулы (IIa1)

(b) введения в реакцию соединения формулы (IIa1) с агентом для снятия защиты карбоновой кислоты с образованием соединения формулы (IIIa)

или его соли; и
(с) введения в реакцию соединения формулы (IIIа) с соединением формулы (IV)
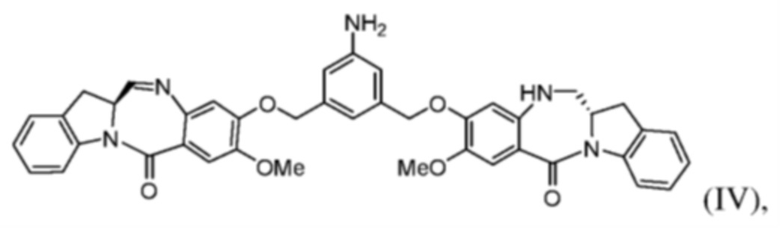
с образованием соединения формулы (Vа).
24. Способ по п. 23, где активирующий агент на стадии (а) представляет собой 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид.
25. Способ по п. 23 или 24, где агент для снятия защиты карбоновой кислоты на стадии (b) представляет собой трифторуксусную кислоту (ТФУ).
26. Способ по любому из пп. 1-25, где E, если присутствует, представляет собой -OH, а реакцию на стадии (а) проводят в присутствии активирующего агента, который представляет собой HATU и HOAt.
27. Способ по п. 26, где реакцию на стадии (а) проводят в присутствии основания.
28. Способ по п. 27, где основание представляет собой триэтиламин или диизопропилэтиламин.
29. Способ по любому из пп. 2-28, где реакцию между соединением формулы (IIIa) и соединением формулы (IV) проводят в присутствии активирующего агента.
30. Способ по п. 29, где активирующий агент представляет собой HATU и HOAt.
31. Способ по п. 29 или 30, где реакцию между соединением формулы (IIIa) и соединением формулы (IV) проводят в присутствии основания.
32. Способ по п. 31, где основание представляет собой триэтиламин или диизопропилэтиламин.
| WO 2018140435 A1, 02.08.2018 | |||
| WO 2017015496 А1, 26.01.2017 | |||
| WO 2017015495 А1, 26.01.2017 | |||
| СПОСОБЫ ПОЛУЧЕНИЯ КОНЪЮГАТОВ | 2012 |
|
RU2621035C2 |

