ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает приоритет по дате подачи заявки в соответствии с 35 U.S.C §119 (e) предварительной заявки США № 62/327973, поданной 26 апреля 2016 года, и предварительной заявки США № 62/195023, поданной 21 июля, 2015 года. Содержимое каждой из вышеперечисленных заявок включено в данный документ посредством ссылки в полном объеме.
ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к новым способам получения цитотоксических производных индолинобензодиазепина.
УРОВЕНЬ ТЕХНИКИ
Показано, что конъюгаты димеров индолинобензодиазепина с агентами, которые связываются с клетками, содержащие одну иминную функциональную группу и одну аминную функциональную группу, демонстрируют in vivo гораздо более высокий терапевтический индекс (соотношение максимально переносимой дозы к минимальной эффективной дозе) по сравнению с ранее описанными производными бензодиазепина, содержащими две иминные функциональные группы. См., например, WO 2012/128868. Ранее описанный способ получения димеров индолинобензодиазепина с одной иминной функциональной группой и одной аминной функциональной группой включает частичное восстановление димеров индолинобензодиазепина, содержащих две иминные функциональные группы. Стадия частичного восстановления обычно приводит к образованию полностью восстановленного побочного продукта и непрореагировавшего исходного материала, что требует громоздкой стадии очистки и приводит к низкому выходу.
Таким образом, существует потребность в улучшенных способах получения димеров индолинобензодиазепина, которые были бы более эффективными и пригодными для крупномасштабного производственного процесса.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В данном изобретении предложены различные способы получения димерных индолинобензодиазепиновых соединений и их синтетических предшественников. По сравнению с ранее описанным способом способы по данному изобретению могут давать желаемые димерные соединения с более высоким выходом без необходимости в громоздких стадиях очистки. Эти способы лучше подходят для крупномасштабного производственного процесса.
Данное изобретение относится к способам вариантов осуществления изобретения с первого по сорок четвертый, которые подробно описаны ниже.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1-19 показаны типичные схемы для способов по данному изобретению.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Ниже будет сделана ссылка на некоторые варианты осуществления изобретения, примеры которых проиллюстрированы в сопроводительных структурах и формулах. Хотя изобретение будет описано в сочетании с перечисленными вариантами осуществления изобретения, следует понимать, что изобретение не ограничивается этими вариантами осуществления изобретения. Напротив, изобретение включает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем данного изобретения, определенный формулой изобретения. Для специалиста в данной области техники будут очевидны многие способы и материалы, сходные или эквивалентные описанным в данном документе, которые могут быть применены в практике данного изобретения.
Следует понимать, что любой из вариантов осуществления изобретения, описанных в данном документе, может быть объединен с одним или большим количеством других вариантов осуществления изобретения, если явно не отклонено или неправильно. Комбинация вариантов осуществления изобретения не ограничивается конкретными комбинациями, заявленными в многочисленных зависимых пунктах формулы изобретения.
ОПРЕДЕЛЕНИЯ
Как используется в данном документе, термин «связывающийся с клеткой агент» или «СКА (CBA)» относится к соединению, которое может связываться с клеткой (например, с лигандом на клеточной поверхности) или связываться с лигандом, связанным с клеткой или находящимся вблизи нее, предпочтительно специфичным образом. В некоторых вариантах осуществления изобретения связывание с клеткой или лигандом на клетке или вблизи нее является специфичным. СКА (CBA) может включать пептиды и не пептиды.
«Линейный или разветвленный алкил», как используется в данном документе, относится к насыщенному одновалентному углеводородному радикалу с линейной или разветвленной цепью, содержащему от одного до двадцати атомов углерода. Примеры алкила включают, но не ограничиваясь этим, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-метил-1-пропил, -CH2CH(CH3)2), 2-бутил, 2-метил-2-пропил, 1-пентил, 2-пентил-3-пентил, 2-метил-2-бутил, 3-метил-2-бутил, 3-метил-1-бутил, 2-метил-1-бутил, 1-гексил), 2-гексил, 3-гексил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 3-метил-3-пентил, 2-метил- 3-пентил, 2,3-диметил-2-бутил, 3,3-диметил-2-бутил, 1-гептил, 1-октил и тому подобное. Предпочтительно алкил содержит от одного до десяти атомов углерода. Более предпочтительно алкил содержит от одного до четырех атомов углерода.
«Линейный или разветвленный алкенил» относится к одновалентному углеводородному радикалу с линейной или разветвленной цепью, содержащему от двух до двадцати атомов углерода и по меньшей мере одну кратную связь, т.е., двойную углерод-углеродную связь, причем алкенильный радикал включает радикалы с «цис» и «транс» ориентацией, или, в качестве альтернативы, с «E» и «Z» ориентацией. Примеры включают, но не ограничиваясь этим, этилен или винил (-CH=CH2), аллил (-CH2CH=CH2) и т.п. Предпочтительно алкенил содержит от двух до десяти атомов углерода. Более предпочтительно алкил содержит от двух до четырех атомов углерода.
«Линейный или разветвленный алкинил» относится к линейному или разветвленному одновалентному углеводородному радикалу, содержащему от двух до двадцати атомов углерода и по меньшей мере одну кратную связь, т.е., тройную углерод-углеродную связь. Примеры включают, но не ограничиваясь этим, этинил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-пентинил, гексинил и т.п. Предпочтительно алкинил содержит от двух до десяти атомов углерода. Более предпочтительно алкинил содержит от двух до четырех атомов углерода.
Термины «карбоцикл», «карбоциклил» и «карбоциклическое кольцо» относятся к одновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, содержащему от 3 до 12 атомов углерода в виде моноциклического кольца или от 7 до 12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, содержащие от 7 до 12 атомов, могут быть расположены, например, в виде системы бицикло [4,5], [5,5], [5,6] или [6,6], а бициклические карбоциклы, содержащие 9 или 10 кольцевых атомов, могут быть расположены в виде системы бицикло [5,6] или [6,6] или в виде мостиковых систем, таких как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноциклических карбоциклов включают, но не ограничиваясь этим, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и т.п.
Термины «циклический алкил» и «циклоалкил» могут использоваться взаимозаменяемо. Они относятся к одновалентному насыщенному карбоциклическому кольцевому радикалу. Предпочтительно, циклический алкил представляет собой 3-7-членный кольцевой моноциклический радикал. Более предпочтительно, циклический алкил представляет собой циклогексил.
Термин «циклический алкенил» относится к карбоциклическому кольцевому радикалу, содержащему по меньшей мере одну двойную связь в структуре кольца.
Термин «циклический алкинил» относится к карбоциклическому кольцевому радикалу, содержащему по меньшей мере одну тройную связь в структуре кольца.
«Арил» означает одновалентный ароматический углеводородный радикал, содержащий 6-18 атомов углерода, который получен путем удаления одного атома водорода при одном атоме углерода исходной ароматической системы колец. Некоторые арильные группы представлены в типичных структурах как «Ar». Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Типичные арильные группы включают, но не ограничиваясь этим, радикалы, полученные из бензола (фенил), замещенных бензолов, нафталина, антрацена, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и т.п. Предпочтительно арил является фенильной группой.
Термины «гетероцикл», «гетероциклил» и «гетероциклическое кольцо» используются в данном документе взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (т.е., содержащему одну или несколько двойных и/или тройных связей в пределах кольца) карбоциклическому радикалу, содержащему от 3 до 18 кольцевых атомов, в которых по меньшей мере один кольцевой атом представляет собой гетероатом, выбранный из азота, кислорода, фосфора и серы, причем остальные кольцевые атомы представляют собой С, и, при этом, один или большее количество кольцевых атомов необязательно и независимо замещены одним или большим количеством заместителей, описанных ниже. Гетероцикл может представлять собой моноцикл, содержащий от 3 до 7 атомов в кольце (от 2 до 6 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, O, P и S), или бицикл, содержащий от 7 до 10 атомов в кольце (от 4 до 9 атомов углерода и от 1 до 6 гетероатомов, выбранных из N, O, P и S), например: система бицикло [4,5], [5,5], [5,6] или [6,6]. Гетероциклы описаны в Paquette, Leo A.; “Principles of Modern Heterocyclic Chemistry” (W. A. Benjamin, New York, 1968), особенно Главы 1, 3, 4, 6, 7 и 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, с 1950 по данное время), в частности, Тома 13, 14, 16, 19 и 28; и J. Am. Chem. Soc. (1960) 82: 5566. Кроме того, «гетероциклил» включает радикалы, в которых гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но не ограничиваясь этим, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксапанил, тиапанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил и азабицикло[2.2.2]гексанил. Спирофрагменты также включены в это определение. Примерами гетероциклической группы, в которой атомы кольца замещены оксо (=О) фрагментами, являются пиримидинонил и 1,1-диоксотиоморфолинил.
Термин «гетероарил» относится к одновалентному ароматическому радикалу 5- или 6-членных колец и включает конденсированные системы колец (по меньшей мере одно из которых является ароматическим), содержащие 5-18 атомов, которые содержат один или большее количество гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил.
Гетероциклические или гетероарильные группы могут быть присоединены через атом углерода (связаны с углеродом) или азота (связаны с азотом), если это возможно. В качестве примера, но не ограничиваясь этим, связанные с углеродом гетероциклы или гетероарилы присоединены в положении 2, 3, 4, 5 или 6 пиридина, положении 3, 4, 5 или 6 пиридазина, положении 2, 4, 5, или 6 пиримидина, положении 2, 3, 5 или 6 пиразина, положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, положении 2, 4 или 5 оксазола, имидазола или тиазола, положении 3, 4 или 5 изоксазола, пиразола или изотиазола, положении 2 или 3 азиридина, положении 2, 3 или 4 азетидина, положении 2, 3, 4, 5, 6, 7 или 8 хинолина или положении 1, 3, 4, 5, 6, 7 или 8 изохинолина. В качестве примера, но не ограничиваясь этим, связанные с азотом гетероциклы или гетероарилы присоединены в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1Н-индазола, положении 2 изоиндола или изоиндолина, положении 4 морфолина и положении 9 карбазола или O-карболина.
Гетероатомы, присутствующие в гетероариле или гетероциклиле, включают окисленные формы, такие как NO, SO и SO2.
Термин «гало» или «галоген» относится к F, Cl, Br или I.
Алкил, алкенил, алкинил, циклический алкил, циклический алкенил, циклический алкинил, карбоциклил, арил, гетероциклил и гетероарил, описанные выше, могут быть необязательно замещены еще одним (например, 2, 3, 4, 5, 6 или более) заместителями.
Если заместитель описан как «замещенный», то вместо водородного заместителя при атоме углерода, кислорода, серы или азота заместителя присутствует неводородный заместитель. Так, например, замещенный алкильный заместитель представляет собой алкильный заместитель, в котором по меньшей мере один неводородный заместитель присутствует вместо водородного заместителя в алкильном заместителе. Для иллюстрации, монофторалкил представляет собой алкил, замещенный фтор-заместителем, а дифторалкил представляет собой алкил, замещенный двумя фтор-заместителями. Следует признать, что если на заместителе присутствует несколько заместителей, то каждый неводородный заместитель может быть таким же или другим (если обратное прямо не указано в тексте).
Если заместитель описан как «необязательно замещенный», то заместитель может быть (1) не замещенным или (2) замещенным. Если атом углерода заместителя описан как необязательно замещенный одним или большим количеством из перечня заместителей, то один или большее количество атомов водорода при атоме углерода (в той степени, в которой они присутствуют) могут по отдельности и/или совместно быть заменены независимо выбранным необязательным заместителем. Если атом азота заместителя описан как необязательно замещенный одним или большим количеством из перечня заместителей, то один или большее количество из атомов водорода при атоме азота (в той степени, в которой они присутствуют) могут быть заменены независимо выбранным необязательным заместителем. Один типичный заместитель может быть изображен как -NR’R’', где R’ и R’’ вместе с атомом азота, к которому они присоединены, могут образовывать гетероциклическое кольцо. Гетероциклическое кольцо, образованное R’ и R’’ вместе с атомом азота, к которому они присоединены, может быть частично или полностью насыщенным. В одном варианте осуществления изобретения гетероциклическое кольцо состоит из 3-7 атомов. В другом варианте осуществления изобретения гетероциклическое кольцо выбрано из группы, состоящей из пирролила, имидазолила, пиразолила, триазолила, тетразолила, изоксазолила, пиридила и тиазолила.
Если группа заместителей совместно описана как необязательно замещенная одним или большим количеством перечисленных заместителей, то группа может включать: (1) недоступные для замещения заместители, (2) доступные для замещения заместители, которые не замещены необязательными заместителями и/или (3) доступные для замещения заместители, которые замещены одним или несколькими необязательными заместителями.
Если заместитель описан как необязательно содержащий до определенного количества неводородных заместителей, то такой заместитель может быть (1) незамещенным; или (2), содержать до указанного конкретного количества неводородных заместителей или вплоть до максимального количества доступных для замещения положений на заместителе, в зависимости от того, что меньше.
Так, например, если заместитель описан как гетероарил, необязательно содержащий до 3 неводородных заместителей, то любой гетероарил с менее чем 3 доступными для замещения положениями может необязательно содержать до такого количества неводородных заместителей, сколько доступных для замещения положений в гетероариле. Такие заместители в неограничивающих примерах могут быть выбраны из линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, арила, гетероарила, гетероциклила, галогена, гуанидиния [-NH(C=NH)NH2], -OR100, NR101R102, -NO2, -NR101COR102, -SR100, сульфоксида, представленного -SOR101, сульфона, представленного -SO2R101, сульфоната -SO3M, сульфата -OSO3M, сульфонамида, представленного -SO2NR101R102, циано, азидо, -COR101, -OCOR101, -OCONR101R102 и фрагмента полиэтиленгликоля (-CH2CH2O)nR101, где М представляет собой Н или катион (такой как Na+ или K+); каждый R101, R102 и R103 независимо выбран из H, линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, фрагмента полиэтиленгликоля (-CH2CH2O)n-R104, где n представляет собой целое число от 1 до 24, арила, содержащего от 6 до 10 атомов углерода, гетероциклического кольца, содержащего от 3 до 10 атомов углерода, и гетероарила, содержащего от 5 до 10 атомов углерода; и R104 представляет собой H или линейный или разветвленный алкил, содержащий от 1 до 4 атомов углерода, где алкил, алкенил, алкинил, арил, гетероарил и гетероциклил в группах, представленных R100, R101, R102, R103 и R104, необязательно замещены одним или большим количеством (например, 2, 3, 4, 5, 6 или более) заместителей, независимо выбранных из галогена, -ОН, -CN, -NO2 и незамещенного линейного или разветвленного алкила, содержащего от 1 до 4 атомов углерода. Предпочтительно заместители для необязательно замещенного алкила, алкенила, алкинила, циклического алкила, циклического алкенила, циклического алкинила, карбоциклила, арила, гетероциклила и гетероарила, описанных выше, включают галоген, -CN, -NR102R103, -CF3, -OR101, арил, гетероарил, гетероциклил, -SR101, -SOR101, -SO2R101 и -SO3M.
Термины «соединение» или «цитотоксическое соединение», «цитотоксический димер» и «димерное цитотоксическое соединение» используются взаимозаменяемо. Они включают соединения, для которых структура или формула или любое их производное раскрыты в данном изобретении, или структура или формула или любое их производное включено путем ссылки. Кроме того, термин включает стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты, соли (например, фармацевтически приемлемые соли) и пролекарства и соли пролекарств соединения всех формул, раскрытых в данном изобретении. Термин дополнительно включает любые сольваты, гидраты и полиморфы любого из вышеперечисленного. Конкретное указание «стереоизомеров», «геометрических изомеров», «таутомеров», «сольватов», «метаболитов», «соли», «пролекарства», «соли пролекарства», «конъюгатов», «соли конъюгатов», «сольвата», «гидрата» или «полиморфа» в определенных аспектах изобретения, описанных в данной заявке, не должны интерпретироваться как предполагаемое опущение этих форм в других аспектах изобретения, в которых термин «соединение» используется без указания таких других форм.
Термин «способный к связыванию с агентом, который связывается с клетками» или «способный ковалентно связывать цитотоксическое соединение со связывающимся с клеткой агентом», как используется в данном документе, относится к описанным в данном документе соединениям или их производным, содержащим по меньшей мере одну связывающую группу или ее предшественник, подходящий для связывания указанных соединений или их производных с агентом, связывающимся с клеткой.
Термин «предшественник» указанной группы относится к любой группе, которая может давать эту группу посредством любого удаления защитной группы, химической модификации или введения в реакцию сочетания.
Термин «хиральный» относится к молекулам, изображение которых обладает свойством не совмещаться с зеркальным партнером, тогда как термин «ахиральный» относится к молекулам, изображение которых совмещается с их зеркальным партнером.
Термин «стереоизомер» относится к соединениям, обладающим идентичным химическим строением и конфигурацией связей, но разной ориентацией атомов в пространстве, которая не может быть взаимно преобразована вращением вокруг одиночных связей.
«Диастереомер» относится к стереоизомеру с двумя или большим количеством центров хиральности, молекулы которых не являются зеркальным отображением друг друга. Диастереомеры обладают разными физическими свойствами, например, температура плавления, температура кипения, спектральные свойства и реакционная способность. Смеси диастереомеров можно разделять в методиках анализа с высоким разрешением, таких как кристаллизация, электрофорез и хроматография.
«Энантиомеры» относятся к двум стереоизомерам соединения, которые не являются совмещаемыми зеркальными изображениями друг друга.
Стереохимические определения и условные обозначения, используемые в данном документе, обычно соответствуют S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. and Wilen, S., “Stereochemistry of Organic Compounds,” John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать асимметричные или хиральные центры и, следовательно, существовать в разных стереоизомерных формах, Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропизомеры, а также их смеси, такие как рацемические смеси, являются частью данного изобретения. Многие органические соединения существуют в оптически активных формах, т.е., обладают способностью вращать плоскость плоско поляризованного света. При описании оптически активного соединения приставки D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов). Приставки d и l или (+) и (-) используются для обозначения знака поворота плоско поляризованного света соединением, причем (-) или 1 означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. В случае конкретной химической структуры эти стереоизомеры идентичны, за исключением того, что они являются зеркальными изображениями друг друга. Конкретный стереоизомер можно также назвать энантиомером, а смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров 50:50 упоминается как рацемическая смесь или рацемат, который может возникать, если не применялась стереоселекция или стереоспецифичность в ходе химической реакции или способа. Термины «рацемическая смесь» и «рацемат» относятся к эквимолярной смеси двух энантиомерных форм, лишенной оптической активности.
Термин «таутомер» или «таутомерная форма» относится к структурным изомерам с различной энергией, которые взаимопревращаются через барьер с низкой энергией. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения путем миграции протона, такие как кето-енольная и имино-енаминовая изомеризация. Таутомеры валентности включают взаимопревращения путем реорганизации некоторых электронов, принимающих участие в образовании связей.
Термин «реагент, способный реагировать с имином» относится к реагенту, который способен реагировать с иминной группой. Примеры реагента, способного реагировать с имином, включают, но не ограничиваясь этим, сульфиты (H2SO3, H2SO2 или соль HSO3-, SO32- или HSO2-, образованная с катионом), метабисульфит (H2S2O5 или соль S2O52--, образованная с катионом), моно-, ди-, три- и тетратиофосфаты (PO3SH3, PO2S2H3, POS3H3, PS4H3 или соль PO3S3-, PO2S23-, POS33- или PS43-, образованная с катионом), тиофосфатные эфиры ((RiO)2PS(ORi), RiSH, RiSOH, RiSO2H, RiSO3H), различные амины (гидроксиламин (например, NH2OH), гидразин (например, NH2NH2), NH2O-Ri, Ri’NH-Ri, NH2-Ri), NH2-CO-NH2, NH2-C(=S)-NH2, тиосульфат (H2S2O3 или соль S2O32-, образованная с катионом), дитионит (H2S2O4 или соль S2O42-, образованная с катионом), фосфордитиоат (P(=S)(ORk)(SH)(OH) или его соль, образованная с катионом), гидроксамовую кислоту (RkC(=O)NHOH или соль, образованная с катионом), гидразид (RkCONHNH2), формальдегид сульфоксилата (HOCH2SO2H или соль HOCH2SO2-, образованная с катионом, такая как HOCH2SO2-Na+), гликированный нуклеотид (такой как гуанозиндифосфат, ГДФ (GDP)-манноза), флударабин или их смесь, где каждый Ri и Ri’ независимо представляет собой линейный или разветвленный алкил, содержащий от 1 до 10 атомов углерода и замещенный по меньшей мере одним заместителем, выбранным из -N(Rj)2, -CO2H, -SO3H, и -PO3H; Ri и Ri’ дополнительно могут быть необязательно замещены заместителем для алкила, описанным в данном документе; Rj представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода; и Rk представляет собой линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 10 атомов углерода, арил, гетероциклил или гетероарил (предпочтительно Rk представляет собой линейный или разветвленный алкил, содержащий от 1 до 4 атомов углерода, более предпочтительно Rk представляет собой метил, этил или пропил). Предпочтительно катион представляет собой одновалентный катион, такой как Na+ или K+. Предпочтительно реагент, способный реагировать с имином, выбирают из сульфитов, гидроксиламина, мочевины и гидразина. Более предпочтительно реагент, способный реагировать с имином, представляет собой NaHSO3 или KHSO3.
Как используется в данном документе, термин «восстанавливающий имин реагент» относится к реагенту, который способен восстанавливать иминную функциональную группу до аминной функциональной группы. В некоторых вариантах осуществления изобретения восстанавливающий имин реагент представляет собой гидридный восстановительный реагент. Примеры таких восстановительных реагентов для имина включают, но не ограничиваясь этим, боргидриды (например, боргидрид натрия, триацетоксиборгидрид натрия, цианоборгидрид натрия, боргидрид лития (LiBH4), боргидрид калия (KBH4)), газообразный водород и лития алюминия гидрид, формиат аммония, боран, 9-борабицикло[3.3.1]нонан (9-ББН (9-BBN)), диизобутилалюминия гидрид (ДИБАЛ (DIBAL)) и бис(2-метоксиэтокси)алюмогидрид натрия (Red-Al). В некоторых вариантах осуществления изобретения восстановительный реагент для имина представляет собой триацетоксиборгидрид натрия.
Термин «защитная группа» или «защитный фрагмент» относится к заместителю, который обычно используется для блокирования или защиты определенной функциональной группы при взаимодействии других функциональных групп в соединении, его производном или его конъюгате. Например, «защитная группа для аминогруппы» или «защитный фрагмент для аминогруппы» представляет собой заместитель, присоединенный к аминогруппе, который блокирует или защищает функциональную аминогруппу в соединении. Такие группы хорошо известны в данной области техники (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 7, J. Wiley & Sons, NJ), их примерами являются карбаматы, такие как метил- и этилкарбамат, 9-флуоренилметоксикарбонил, ФМОК (FMOC), замещенные этилкарбаматы, расщепленные 1,6-β-элиминированием карбаматы (также называемые «саморасщепляющимися»), производные мочевины, амиды, пептиды, алкильные и арильные производные. Подходящие защитные группы для аминогруппы включают, но не ограничиваясь этим, ацетил, трифторацетил, трет-бутоксикарбонил (БОК (BOC)), бензилоксикарбонил (БЗК (CBZ)) и 9-флуоренилметиленоксикарбонил (ФМОК (Fmoc)), 2-триметилсилилэтил, (2-фенил-2-триметилсилил)этил, триизопропилсилокси, 2-(триметилсилил)этоксиметил, аллилоксикарбонил, 9-флуоренилметоксикарбонил, 2-(триметилсилил)этоксикарбонил или 2,2,2,2-трихлорэтоксикарбонил. Общее описание защитных групп и их применение см. в P. G.M. Wuts & T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 2007.
«Защитная группа для спиртовой группы» или «защитный фрагмент для спиртовой группы» представляет собой заместитель, присоединенный к спиртовой группе, который блокирует или защищает функциональную группу спирта в соединении. Такие группы хорошо известны в данной области техники (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 2, J. Wiley & Sons, NJ). Подходящая защитная группа для спиртовой группы включает, но не ограничиваясь этим, пивалоил, метоксиметил, 2-метоксиэтоксиметил, п-метоксибензил, 3,4-диметиоксибензил, 2,6-диметиоксибензил, дифенилметил, бензилоксиметил, 2,2,2-трихлорэтоксикарбонил, тетрагидрофуранил, тетрагидропиранил, бензил, бензоил, пара-фенилбензоил, 2,4,6-триметилбензоил, пара-бромбензоил, пара-нитробензоил, пиколиноил, никотиноил, 5-дибензосуберил, тритил/трифенилметил, или трис(4-трет-бутилфенил)метил и различные силильные защитные группы (например, диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, 2-триметиэтилсилил (ТМЭС (TEOC)) или [2-(триметилсилил)этокси]метил). В некоторых вариантах осуществления изобретения защитная группа для спиртовой группы является пространственно затрудненной. В некоторых вариантах осуществления изобретения защитная группа для спиртовой группы предпочтительно представляет собой метоксиметил, тетрагидропиранил, 2-метоксиэтоксиметил, п-метоксибензил, бензилоксиметил или 2,2,2-трихлорэтоксикарбонил. Более предпочтительно, защитная группа для спиртовой группы представляет собой 2,2,2-трихлорэтоксикарбонил. В некоторых вариантах осуществления изобретения защитная группа для спиртовой группы представляет собой силильную защитную группу, предпочтительно триэтилсилил, триизопропилсилил или трет-бутилдиметилсилил. Более предпочтительно, защитная группа для спиртовой группы представляет собой трет-бутилдиметилсилил.
«Защитный реагент для спиртовой группы», как используется в данном документе, относится к реагенту, который вводит защитную группу для спиртовой группы в спиртовую группу.
«Кислотолабильная защитная группа для спиртовой группы» представляет собой защитную группу для спиртовой группы, которая является лабильной в кислой среде и высвобождает защитную группу из спиртовой группы с образованием свободного спирта. Примеры кислотолабильной защитной группы для спиртовой группы включают, но не ограничиваясь этим, ацетат, аллил, метоксиметил, тетрагидрофуранил, тетрагидропиранил, 5-дибензосуберил, 1-этоксиэтил, 1-метил-1-метоксилэтил, 2-(фенилселенил) этил, тритил/трифенилметил, трис(4-трет-бутилфенил)метил и различную силильную защитную группу (например, диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триэтилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил или 2-триметиэтилсилил (ТМЭС (TEOC)), [2-(триметилсилил)этокси]метил). В некоторых вариантах осуществления изобретения защитная группа для спиртовой группы представляет собой силильную защитную группу, предпочтительно триэтилсилил, триизопропилсилил или трет-бутилдиметилсилил. Более предпочтительно, защитная группа для спиртовой группы представляет собой трет-бутилдиметилсилил.
Как используется в данном документе, термин «реагент для удаления защитной группы спиртовой группы» относится к реагенту, который способен отщеплять защитную группу спиртовой группы с образованием свободного спирта. Такие реагенты хорошо известны в данной области техники (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 2, J. Wiley & Sons, NJ). Примеры таких реагентов для удаления защитной группы спиртовой группы включают, но не ограничиваясь этим, фторид тетра-н-бутиламмония, трис(диметиламино)сульфония дифтортриметилсиликат, фтороводород или его сольват, пиридина гидрофторид, тетрафторид кремния, гексафторкремниевую кислоту, фторид цезия, хлористоводородную кислоту, уксусную кислоту, трифторуксусную кислоту, пиридиния п-толуолсульфонат, п-толуолсульфоновую кислоту (п-TsOH), муравьиную кислоту, йодную кислоту. В некоторых вариантах осуществления изобретения реагент для удаления защитной группы спиртовой группы представляет собой хлористоводородную кислоту или тетра-н-бутиламмония фторид (ТБАФ (TBAF)). В некоторых вариантах осуществления изобретения средство для удаления защитной группы спиртовой группы представляет собой гидрофторид пиридина (HF-пиридин).
Как используется в данном документе, термин «группа для удаления защитной группы аминогруппы» относится к реагенту, который способен отщеплять защитную группу аминогруппы с образованием свободного амина. Такие реагенты хорошо известны в данной области техники (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 7, J. Wiley & Sons, NJ). Примеры таких реагентов для удаления защитной группы из аминогруппы включают, но не ограничиваясь этим, тетра-н-бутиламмония фторид, уксусную кислоту, гидрофторид пиридина, фторид цезия, пиперидин, морфолин или трифторуксусную кислоту.
Как используется в данном документе, термин «активирующий спирт агент» относится к реагенту, который повышает реакционную способность гидроксильной группы, тем самым делая гидроксильную группу лучшей уходящей группой. Примеры таких активирующих спирт агентов включают п-толуолсульфонилхлорид, тионилхлорид, трифлатный ангидрид, мезилхлорид, мезиловый ангидрид, трифенилфосфин, хлорангидрид кислоты, 4-диметиламинопиридин и др. В некоторых вариантах осуществления изобретения активирующий спирт агент представляет собой тионилхлорид. В определенном варианте осуществления изобретения активирующий спирт агент представляет собой трифенилфосфин.
Как используется в данном документе, выражение «фармацевтически приемлемая соль» относится к фармацевтически приемлемым органическим или неорганическим солям соединения по изобретению. Примеры солей включают, но не ограничиваясь этим, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат «мезилат», этансульфонат, бензолсульфонат, п-толуолсульфонат, памоатные соли (т.е., 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)), соли щелочных металлов (например, натрия и калия), соли щелочноземельных металлов (например, магния) и соли аммония. Фармацевтически приемлемая соль может включать введение другой молекулы, такой как ацетатный ион, сукцинатный ион или другой противоион. Противоионом может быть любой органический или неорганический фрагмент, который стабилизирует заряд на исходном соединении. Кроме того, фармацевтически приемлемая соль может содержать в своей структуре более одного заряженного атома. В случаях, когда несколько заряженных атомов являются частью фармацевтически приемлемой соли, может присутствовать несколько противоионов. Следовательно, фармацевтически приемлемая соль может содержать один или большее количество заряженных атомов и/или один или большее количество противоионов.
Если соединение по изобретению является основанием, то желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, доступным в данной области техники, например, обработкой свободного основания неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфокислота, такая как п-толуолсульфокислота или этансульфоновая кислота, и т.п.
Если соединение по изобретению представляет собой кислоту, то желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла и т.п. Иллюстративные примеры подходящих солей включают, но не ограничиваясь этим, органические соли, полученные с аминокислотами, такими как глицин и аргинин, аммиаком, первичным, вторичным и третичным аминами и циклическими аминами, такими как пиперидин, морфолин и пиперазин, и неорганические соли, полученные с натрием, кальцием, калием, магнием, марганцем, железом, медью, цинком, алюминием и литием.
Выражение «фармацевтически приемлемый» указывает на то, что вещество или композиция с химической и/или токсикологической точки зрения должны быть совместимы с другими ингредиентами, составляющими препарат, и/или с организмом млекопитающего, подлежащего лечению ими.
Термин «уходящая группа» относится к группе заряженного или незаряженного фрагмента, которая уходит во время нуклеофильного замещения или вытеснения. Такие уходящие группы хорошо известны в данной области техники и включают, но не ограничиваясь этим, галогены, сложные эфиры, алкокси, гидроксил, тозилаты, трифлаты, мезилаты, нитрилы, азид, карбамат, дисульфиды, тиоэфиры, тиоэфиры и соединения диазония.
Термин «бифункциональный поперечно-сшивающий агент», «бифункциональный линкер» или «поперечно-сшивающие агенты» относится к модифицирующим агентам, которые содержат две реакционноспособные группы; одна из которых способна реагировать с агентом, связывающимся с клетками, тогда как другая реагирует с цитотоксическим соединением, чтобы соединить два фрагмента вместе. Такие бифункциональные поперечно-сшивающие агенты хорошо известны в данной области техники (см., например, Isalm and Dent in Bioconjugation chapter 5, p218-363, Groves Dictionaries Inc. New York, 1999). Например, бифункциональные поперечно-сшивающие агенты, которые обеспечивают соединение посредством тиоэфирной связи, включают N-сукцинимидил-4-(N-малеинимидометил)циклогексан-1-карбоксилат (СМЦК (SMCC)) для введения малеинимидных групп или N-сукцинимидил-4-(йодацетил)-аминобензоат (СЙАБ (SIAB)) для введения йодацетильных групп. Другие бифункциональные поперечно-сшивающие агенты, которые вводят малеинимидные группы или галогенацетильные группы в агент, связывающийся с клеткой, хорошо известны в данной области техники (см. Патентные заявки США 2008/0050310, 20050169933, доступные в Pierce Biotechnology Inc. PO Box 117, Rockland, IL 61105, USA) и включают, но не ограничиваясь этим, бис-малеинимидополиэтиленгликоль (БМПЭГ (BMPEO)), БМ(ПЭГ)2 (BM(PEO)2), БМ(ПЭГ)3 (BM(PEO)3), N-(β-малеинимидопропилокси)сукцинимидный эфир (БМПС (BMPS)), γ-малеинимидомасляной кислоты N-сукцинимидиловый эфир (ГММС (GMBS)), N-гидроксисукцинимидный эфир ε-малеинимидокапроновой кислоты (ЭМКС (EMCS)), 5-малеинимидовалериановой кислоты N-гидроксисукцинимид (NHS), 1,6-гексан-бис-винилсульфон (ГБВС (HBVS)), N-сукцинимидил-4-(N-малеинимидометил)-циклогексан-1-карбокси-(6-амидокапроат), который представляет собой «длинноцепочечный» аналог СМЦК (SMCC) (ДЦ-СМЦК (LC-SMCC)), м-малеинимидобензоил-N-гидроксисукцинимидный эфир (МБС (MBS)), 4-(4-N-малеинимидофенил)-масляной кислоты гидразид или соль HCl (МФМГ (MPBH)), N-сукцинимидил-3-(бромоацетамидо)пропионат (СБАП (SBAP)), N-сукцинимидилйодацетат (СЙА (SIA)), N-сукцинимидиловый эфир κ-малеинимидоундекановой кислоты (КМУК (KMUA)), N-сукцинимидил-4-(п-малеинимидофенил)-бутират (СМФБ (SMPB)), сукцинимидил-6-(β-малеинимидопропионамидо)гексаноат (СМПГ (SMPH)), сукцинимидил-(4-винилсульфонил)бензоат (СВСБ (SVSB)), дитиобис-малеинимидоэтан (ДТМЭ (DTME)), 1,4-бис-малеинимидобутан (БМБ (BMB)), 1,4-бисмалеинимидил-2,3-дигидроксибутан (БМДБ (BMDB)), бис-малеинимидогексан (БМГ (BMH)), бис-малеинимидоэтан (БМОЭ (BMOE)), сульфосукцинимидил-4-(N-малеинимидо-метил)циклогексан-1-карбоксилат (сульфо-СМЦК (сульфо-SMCC)), сульфосукцинимидил(4-йод-ацетил)аминобензоат (сульфо-СЙАБ (сульфо-SIAB)), м-малеинимидобензоил-N-гидроксисульфосукцинимидный эфир (сульфо-МБС (сульфо-MBS)), N-(γ-малеинимидобутилокси)сульфосукцинимидный эфир (сульфо-ГМБС (сульфо-GMBS)), N-(ε-малеинимидокапроилокси)сульфосукцинимидный эфир (сульфо-ЭМКС (сульфо-EMCS)), N-(κ-малеинимидоундеканоилокси)сульфосукцинимидный эфир (сульфо-КМУС (сульфо-KMUS)) и сульфосукцинимидил-4-(п-малеинимидофенил)бутират (сульфо-СМФБ (сульфо-SMPB)).
Гетеробифункциональные поперечно-сшивающие агенты представляют собой бифункциональные поперечно-сшивающие агенты, содержащие две различные реакционноспособные группы. Гетеробифункциональные поперечно-сшивающие агенты, содержащие как способную реагировать с амином N- гидроксисукцинимидную группу (NHS-группу), так и способную реагировать с карбонилом гидразиновую группу, также могут быть применены для связывания описанных в данном документе цитотоксических соединений с агентом, связывающимся с клеткой (например, антителом). Примеры таких коммерчески доступных гетеробифункциональных поперечно-сшивающих агентов включают сукцинимидил-6-гидразинникотинамида ацетонгидразон (САНГ (SANH)), сукцинимидил-4-гидразидтерефталата гидрохлорид (СГТГ (SHTH)) и гидрохлорид никотината сукцинимидилгидразиния (СГНГ (SHNH)). Кроме того, конъюгаты, несущие кислотолабильную связь, могут быть получены с применением гидразинсодержащего бензодиазепинового производного по данному изобретению. Примеры бифункциональных поперечно-сшивающих агентов, которые можно применять, включают сукцинимидил-п-формилбензоат (СФБ (SFB)) и сукцинимидил-п-формилфеноксиацетат (СФФА (SFPA)).
Бифункциональные поперечно-сшивающие агенты, которые обеспечивают связывание агента, связывающегося с клеткой, с цитотоксическими соединениями посредством дисульфидных связей, известны в данной области техники и включают N-сукцинимидил-3-(2-пиридилдитио)пропионат (СПДП (SPDP)), N-сукцинимидил-4-(2-пиридилдитио)пентаноат (СПП (SPP)), N-сукцинимидил-4-(2-пиридилдитио)бутаноат (СПДБ (SPDB)), N-сукцинимидил-4-(2-пиридилдитио)2-сульфобутаноат (сульфо-СПДБ (сульфо-SPDB)) для введения дитиопиридильных групп. Другие бифункциональные поперечно-сшивающие агенты, которые можно применять для введения дисульфидных групп, известны в данной области техники и описаны в патентах США 6913748, 6716821 и патентных публикациях US 20090274713 и 20100129314, все из которых включены в данный документ путем ссылки. В качестве альтернативы, дополнительно можно применять такие поперечно-сшивающие агенты, как 2-иминотиолан, гомоцистеина тиолактон или S-ацетилянтарный ангидрид, которые вводят тиольные группы.
«Линкер», «линкерный фрагмент» или «связывающая группа», как определено в данном документе, относится к фрагменту, который соединяет вместе две группы, такие как агент, связывающийся с клеткой, и цитотоксическое соединение. Как правило, линкер является по существу инертным в условиях, в которых соединяются две группы, которые он соединяет. В некоторых вариантах осуществления изобретения связывающая группа является частью описанного в данном документе цитотоксического соединения. Связывающая группа может содержать реакционноспособную группу, которая может вступать в реакцию с агентом, связывающимся с клеткой, или его предшественником. Связывающий фрагмент может содержать химическую связь, которая позволяет высвобождать цитотоксический фрагмент на определенном участке. Подходящие химические связи хорошо известны в данной области техники и включают дисульфидные связи, тиоэфирные связи, кислотолабильные связи, фотолабильные связи, пептидазолабильные связи и эстеразолабильные связи (см., например, патенты США 5208020, 5475092, 6441163, 6716821, 6913748, 7276497, 7276499, 7368565, 7388026 и 7414073). Предпочтительными являются дисульфидные связи, тиоэфирные и пептидазолабильные связи. Кроме того, можно применять нерасщепляемый связывающий фрагмент.
«Реакционноспособная группа» или «реакционноспособный фрагмент», как определено в данном документе, относится к фрагменту, который легко образует ковалентную связь с агентом, связывающимся с клеткой, например, амидную связь с аминогруппой лизина на антителе, или с бифункциональным поперечно-сшивающим агентом, например, посредством тиоэфирной или дисульфидной связи. Реакционноспособная группа является инертной по отношению к реакциям, описанным в способах по данному изобретению. Кроме того, включены функциональные группы, которые могут быть превращены в реакционноспособную группу. Например, реакционноспособная группа может представлять собой N-гидроксисукцинимидный эфир или группу метилового сложного эфира, которая может быть превращена в N-гидроксисукцинимидный эфир. В другом примере реакционноспособная группа может быть тиольной (-SH) группой, которая способна с легкостью образовывать ковалентную связь с бифункциональным поперечно-сшивающим агентом посредством дисульфидной связи или тиоэфирной связи. Дополнительно она может быть алкилдисульфидом или пиридилдисульфидом (R-S-S-, где R представляет собой алкил или пиридил), которые могут быть превращены в тиольную группу.
В одном варианте осуществления изобретения связывающая группа с реакционной группой, присоединенной на одном конце, такой как реакционноспособный сложный эфир, выбрана из следующего:
-O(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-O(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-O(CR20R21)m(алкинил)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-O(CR20R21)m(пиперазино)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-O(CR20R21)m(пирроло)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-O(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-S(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-S(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-S(CR20R21)m(алкинил)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-S(CR20R21)m(пиперазино)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-S(CR20R21)m(пирроло)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-S(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-NR33(C=O)p”(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-NR33(C=O)p”(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’
(CR24R25)q(CO)tX’’,
-NR33(C=O)p”(CR20R21)m(алкинил)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q-(CO)tX’’,
-NR33(C=O)p”(CR20R21)m(пиперазино)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q
(CO)tX’’,
-NR33(C=O)p”(CR20R21)m(пирроло)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q-(CO)tX’’,
-NR33(C=O)p”(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q
(CO)tX’’,
-(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-(CR20R21)m(алкинил)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-(CR20R21)m(пиперазино)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-(CR20R21)m(CR29=N-NR30)n”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q(CO)tX’’,
-(CR20R21)m(CR29=N-NR30)n”(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’
(CR24R25)q(CO)tX’’,
-(CR20R21)m(CR29=N-NR30)n”(алкинил)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’
(CR24R25)q-(CO)tX’’,
-(CR20R21)m(CR29=N-NR30)n”A”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’(CR24R25)q
(CO)tX’’,
где:
m, n, p, q, m', n', t' представляют собой целые числа от 1 до 10 или необязательно представляют собой 0;
t, m", n" и p" представляют собой 0 или 1;
X" выбран из OR36, SR37, NR38R39, где R36, R37, R38, R39 представляют собой H или линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 20 атомов углерода и, или полиэтиленгликолевый фрагмент -(OCH2CH2)n, R37 необязательно представляет собой защитную группу для тиола, если t = 1, то COX'' образует реакционноспособный сложный эфир, выбранный из N-гидроксисукцинимидных эфиров, N-гидроксифталимидных эфиров, N-гидроксисульфосукцинимидных эфиров, пара-нитрофениловых эфиров, динитрофениловых эфиров, пентафторфениловых эфиров и их производных, где указанные производные облегчают образование амидной связи;
Y'' отсутствует или выбран из O, S, S-S или NR32, где R32 имеет такое же определение, как указано выше для R; или
если Y" не является S-S, и t = 0, то Х" выбирают из группы малеинимидо, галогенацетильной группы или SR37, где R37 имеет такое же определение, как указано выше;
А" представляет собой аминокислотный остаток или полипептид, содержащий от 2 до 20 аминокислотных остатков;
R20, R21, R22, R23, R24, R25, R26 и R27 являются одинаковыми или разными и представляют собой -H или линейный или разветвленный алкил, содержащий от 1 до 5 атомов углерода;
R29 и R30 являются одинаковыми или разными и представляют собой -Н или алкил, содержащий от 1 до 5 атомов углерода;
R33 представляет собой -Н или линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 12 атомов углерода, полиэтиленгликолевый фрагмент R-(OCH2CH2)n-, или R33 представляет собой -COR34, -CSR34, -SOR34 или -SO2R34, где R34 представляет собой H или линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 20 атомов углерода, или полиэтиленгликолевый фрагмент -(OCH2CH2)n; и
один из R40 и R41 необязательно представляет собой отрицательно или положительно заряженную функциональную группу, а другой представляет собой Н или алкил, алкенил, алкинил, содержащий от 1 до 4 атомов углерода.
Любая из вышеуказанных связывающих групп может присутствовать в любом из соединений, соединений лекарственное средство-линкер или конъюгатов по изобретению, включая замену связывающих групп любой из формул, описанных в данном документе.
Термин «аминокислота» относится к природным аминокислотам или неприродной аминокислоте. В одном варианте осуществления изобретения аминокислота представлена формулой NH2-C(Raa’Raa)-C(=O)OH, где каждый Raa и Raa’ независимо представляет собой H, необязательно замещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 10 атомов углерода, арил, гетероарил или гетероциклил, или Raa и N-концевой атом азота вместе могут образовывать гетероциклическое кольцо (например, как в пролине). Термин «аминокислотный остаток» относится к соответствующему остатку, когда один атом водорода удаляется из аминогруппы и/или карбоксильного конца аминокислоты, такому как -NH-C(Raa’Raa)-C(=O)O-.
Термин «катион» относится к иону с положительным зарядом. Катион может быть одновалентным (например, Na+, K+ и т.д.), двухвалентным (например, Ca2+, Mg2+ и т.д.) или поливалентным (например, Al3+ и т.д.). Предпочтительно катион является одновалентным.
Как используется в данном документе, термин «галогенирующий реагент» относится к реагенту, который превращает спиртовую группу в галогенидную группу. «Бромирующий реагент» представляет собой реагент, который превращает спиртовую группу в бромидную группу. «Йодирующий реагент» представляет собой реагент, который превращает спиртовую группу в йодидную группу. «Хлорирующий реагент» представляет собой реагент, который превращает спиртовую группу в хлоридную группу. Типичные бромирующие реагенты включают, но не ограничиваясь этим, бром, бромистоводородную кислоту, тетрабромид углерода, трибромид фосфора и бромид калия. Типичный йодирующий реагент включает, но не ограничиваясь этим, йодистоводородную кислоту, йод, тетрайодид углерода, трийодид фосфора, йодид натрия или йодид калия. Типичный хлорирующий реагент включает, но не ограничиваясь этим, четыреххлористый углерод, метансульфонилхлорид, сульфурилхлорид, тионилхлорид, циануровый хлорид, N-хлорсукцинимид, оксихлорид фосфора (V), пентахлорид фосфора и трихлорид фосфора. В конкретном варианте осуществления изобретения хлорирующим реагентом является метансульфонилхлорид.
Как используется в данном документе, термин «сульфонирующий реагент» относится к реагенту, который превращает спиртовую группу в сложноэфирную сульфонатную группу. Предпочтительно, сульфонирующий реагент представляет собой сульфоновый ангидрид, такой как ангидрид метансульфокислоты, или сульфонийхлорид, такой как метансульфонилхлорид (MsCl).
Как используется в данном документе, термин «активированный сложный эфир» относится к сложноэфирной группе, которая легко вытесняется гидроксильной или аминной группой. Типичные активированные сложные эфиры включают, но не ограничиваясь этим, нитрофениловый эфир (например, 2 или 4-нитрофенил), динитрофениловый эфир (например, 2,4-динитрофенил), сульфотетрафторфениловый эфир (например, 4-сульфо-2,3,5,6-тетрафторфенил), пентафторфениловый эфир, нитропиридиловый эфир (например, 4-нитропиридил), трифторацетат и ацетат.
Как используется в данном документе, термин «эстерифицирующий реагент» относится к реагенту, который превращает спиртовую группу в сложноэфирную группу. Типичный эстерифицирующий реагент включает, но не ограничиваясь этим, нитробензойную кислоту (например, 2 или 4-нитробензойную кислоту), динитробензойную кислоту (например, 2,4-динитробензойную кислоту), сульфотетрафторбензойную кислоту (например, 4-сульфо-2,3,5,6-тетрафторбензойную кислоту), пентафторбензойную кислоту, нитропиридинкарбоновую кислоту (например, 4-нитро-2-пиридинкарбоновую кислоту), трифторуксусную кислоту и уксусную кислоту или хлорангидрид кислоты, ангидрид кислоты или другие активированные производные карбоновых кислот.
СПОСОБЫ ПО ДАННОМУ ИЗОБРЕТЕНИЮ
В данном изобретении предложены новые способы получения димерных индолинобензoдиазепиновых соединений, которые содержат одну иминную функциональную группу и одну аминную функциональную группу. По сравнению со способами, известными в данной области техники, данные способы могут давать желаемые димерные соединения с более высоким выходом и без применения очистки ВЭЖХ.
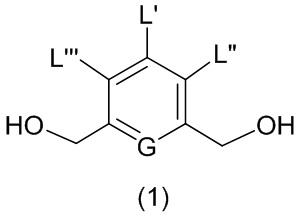
В первом варианте осуществления данного изобретения предложен способ получения соединения формулы (2),
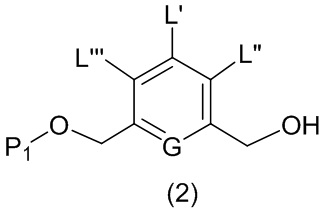
или его соли, включающий в себя введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1) путем введения соединения формулы (I) в реакцию с защитным реагентом для спиртовой группы,
где:
L’, L’’ и L’’’ являются одинаковыми или разными и независимо представляют собой -H, необязательно замещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 10 атомов углерода, полиэтиленгликолевый фрагмент -(OCH2CH2)n’-Rc, галоген, гуанидиний [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3M, -OSO3M, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’, -OCONR’R’’ или линкерную группу со связанной с ней реакционноспособной группой, которая способна ковалентно связывать цитотоксическое соединение со связывающимся с клеткой агентом, СКА (CBA), при условии, что только один из L’, L’’ и L’’’ представляет собой линкерную группу со связанной с ней реакционноспособной группой;
M представляет собой -H или катион;
R в каждом случае независимо выбран из группы, состоящей из: -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n-Rc, необязательно замещенного арила, содержащего от 6 до 18 атомов углерода, необязательно замещенного 5-18-членного гетероарильного кольца, содержащего один или большее количество гетероатомов, независимо выбранных из азота, кислорода и серы, или необязательно замещенного 3-18-членного гетероциклического кольца, содержащего от 1 до 6 гетероатомов, независимо выбранных из O, S, N и P;
каждый R’ и R’’ независимо выбран из -H, -OH, -OR, -NHR, -N(R)2, -COR, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n’-Rc и необязательно замещенного 3-18-членного гетероциклического кольца, содержащего от 1 до 6 гетероатомов, независимо выбранных из O, S, N и P;
Rc представляет собой -H или замещенный или незамещенный линейный или разветвленный алкил, содержащий от 1 до 4 атомов углерода, или линкерную группу со связанной с ней реакционноспособной группой;
n’ представляет собой целое число от 1 до 24;
G выбран из -CH- или -N-; и
P1 представляет собой защитную группу для спиртовой группы.
В одном варианте осуществления изобретения соединение формулы (1) представлено формулой, выбранной из следующего:
 (1b);
(1b); 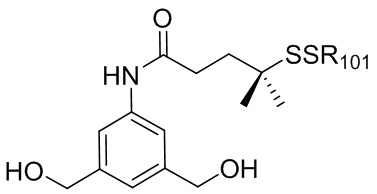 (1c); и
(1c); и
 (1A),
(1A),
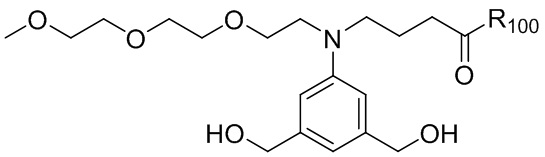
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения защитная группа для спиртовой группы является стерически затрудненной.
В другом конкретном варианте осуществления изобретения защитная группа для спиртовой группы представляет собой пивалоил, метоксиметил, 2-метоксиэтоксиметил, п-метоксибензил, 3,4-диметиоксибензил, 2,6-диметиоксибензил, дифенилметил, бензилоксиметил, 2,2,2-трихлорэтоксикарбонил, тетрагидрoфуранил, тетрагидрoпиранил, бензил, бензоил, пара-фенилбензоил, 2,4,6-триметилбензоил, пара-бромбензоил, пара-нитробензоил, пиколиноил, никотиноил, 5-дибензoсуберил, тритил/трифенилметил или трис(4-трет-бутилфенил)метил. Предпочтительно защитная группа для спиртовой группы представляет собой метоксиметил, тетрагидрoпиранил, 2-метоксиэтоксиметил, п-метоксибензил, бензилоксиметил или 2,2,2-трихлорэтоксикарбонил. Даже более предпочтительно защитная группа для спиртовой группы представляет собой 2,2,2-трихлорэтоксикарбонил.
В другом конкретном варианте осуществления изобретения защитная группа для спиртовой группы представляет собой силильную защитную группу. Например, силильная защитная группа представляет собой диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, 2-триметиэтилсилил (ТМЭС, (TEOC)), или [2-(триметилсилил)этокси]метил. Предпочтительно силильная защитная группа представляет собой триэтилсилил, триизопропилсилил или трет-бутилдиметилсилил. Более предпочтительно, силильная защитная группа представляет собой трет-бутилдиметилсилил.
Силильная защитная группа может быть введена путем введения соединения формулы (1) в реакцию с R3-Cl, R3-Br, R3-I или R3-OSO2CF3 (коллективно называются защитным реагентом для спиртовой группы) в присутствии основания, где R3 представляет собой диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил или [2-(триметилсилил)этокси]метил. В вариантах осуществления изобретения молярное соотношение защитного реагента для спиртовой группы и соединения формулы (1) находится в диапазоне 0,8-1,2, от 1 до 5, от 1 до 2, от 1 до 1,5, от 1 до 1,4, от 1 до 1,3, от 1 до 1,2 или от 1 до 1,1. В определенном варианте осуществления изобретения применяют менее чем 2 молярных эквивалента защитного реагента для спиртовой группы относительно соединения формулы (I). Предпочтительно применяют 1,5, 1,4, 1,3, 1,2, 1,1 или 1,0 молярных эквивалентов защитного реагента для спиртовой группы относительно соединения формулы (1).
В одном варианте осуществления изобретения основание может быть ненуклеофильным основанием. Примеры ненуклеофильного основания включают в себя, но не ограничиваясь этим, имидазол, триэтиламин, диизопропилэтиламин, пиридин, 2,6-лутидин, 1,8-диазабициклоундец-7-ен или тетраметилпиперидин. Предпочтительно ненуклеофильное основание представляет собой имидазол. Можно применять молярный избыток основания. В некоторых вариантах осуществления изобретения применяют более чем 2 молярных эквивалента основания (например¸ ненуклеофильного основания) относительно соединения формулы (1).
В другом варианте осуществления изобретения реакцию между соединением формулы (1) и R3-Cl, R3-Br, R3-I или R3-OSO2CF3 осуществляют в присутствии катализатора, который облегчает введение силильной защитной группы. В реакции можно применять любые подходящие катализаторы, известные в данной области техники (см., например, P. Wuts and T. Greene, 2007, Protective Groups in Organic Synthesis, Chapter 2, J. Wiley & Sons, NJ). Типичные катализаторы включают в себя, но не ограничиваясь этим, 4-диметиламинопиридин (ДМАП, (DMAP)), 1,1,3,3-тетраметилгуанидин и 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, (DBU)).
Для способов первого варианта осуществления изобретения можно применять любые подходящие органические растворители. Типичные растворители включают в себя, но не ограничиваясь этим, ДМФА, CH2Cl2, дихлорэтан, ТГФ, диметилацетамид, и т.п. В некоторых вариантах осуществления изобретения в качестве растворителя применяют ДМФА.
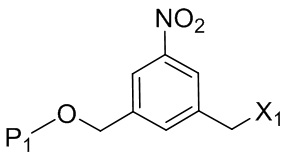
Во втором варианте осуществления данного изобретения предложен способ получения соединения формулы (3),
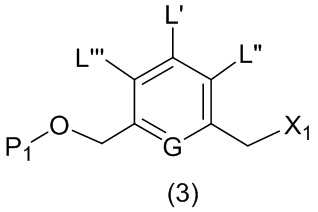
или его соли, включающий в себя введение соединения формулы (2) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом,
где L’, L”, L”’, G и P1 являются такими, как определено в первом варианте осуществления изобретения, а X1 представляет собой уходящую группу, выбранную из группы, состоящей из: -Br, -I, -Cl, сульфонатного эфира и активированного эфира.
В конкретном варианте осуществления изобретения соединение формулы (2) представлено формулой, выбранной из следующего:
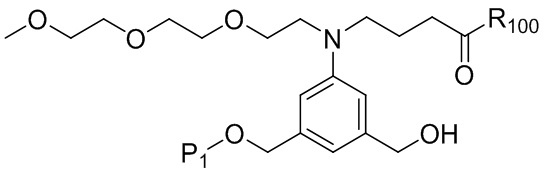 (2b);
(2b); 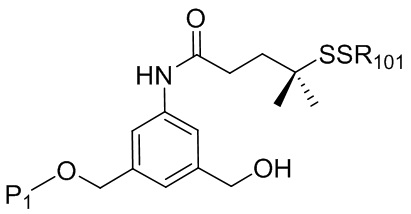 (2c); и
(2c); и
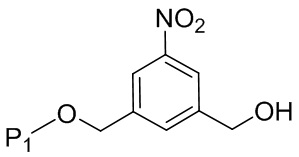 (2A),
(2A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В другом конкретном варианте осуществления изобретения для описанных выше способов получения соединения формулы (3d) или (3A), X1 представляет собой –Br, -I или сульфонатный эфир.
В другом конкретном варианте осуществления изобретения X1 представляет собой мезилат, тозилат, брозилат или трифлат. Предпочтительно X1 представляет собой мезилат.
В другом конкретном варианте осуществления изобретения способ согласно второму варианту осуществления изобретения включает введение соединения формулы (2) в реакцию с галогенирующим реагентом. Типичные галогенирующие реагенты включают в себя, но не ограничиваясь этим, бром, бромистоводородную кислоту, тетрабромид углерода, трибромид фосфора, бромид калия, йодистоводородную кислоту, йод, тетрайодид углерода, трийодид фосфора, йодид натрия или йодид калия.
В еще другом конкретном варианте осуществления изобретения способ согласно второму варианту осуществления изобретения включает введение соединения формулы (2) в реакцию с сульфонирующим реагентом. Предпочтительно сульфонирующий реагент представляет собой сульфоновый ангидрид, такой как метансульфоновый ангидрид или сульфоновый галогенангидрид, такой как метансульфонилхлорид (MsCl).
В определенном варианте осуществления изобретения реакцию между соединением формулы (2) и сульфонирующим реагентом можно осуществлять в присутствии основания. В одном варианте осуществления изобретения основание является ненуклеофильным основанием. Типичные ненуклеофильные основания включают в себя, но не ограничиваясь этим, триэтиламин, имидазол, триэтиламин, диизопропилэтиламин, пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, (DBU)) или тетраметилпиперидин. Предпочтительно основание представляет собой триэтиламин или диизопропилэтиламин.
В способе согласно второму варианту осуществления изобретения можно применять любые подходящие органические растворители. В одном варианте осуществления изобретения растворитель представляет собой дихлорметан.
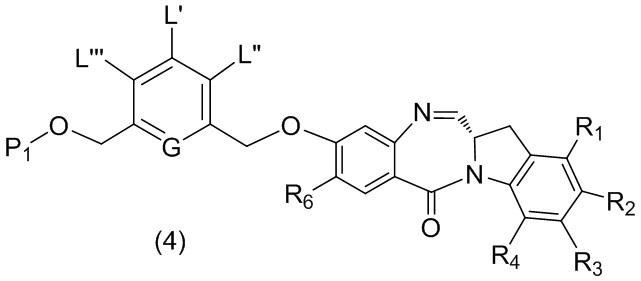
В третьем варианте осуществления данного изобретения предложен способ получения соединения формулы (4),
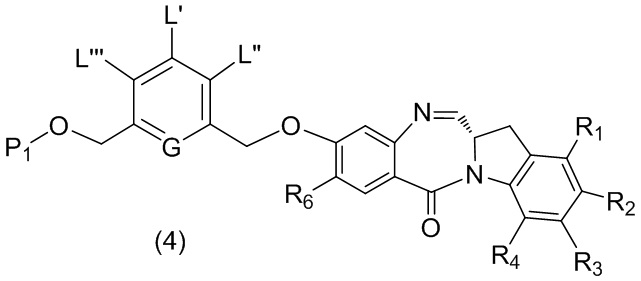
или его соли, причем указанный способ включает введение соединения формулы (3) в реакцию
с мономерным соединением формулы (a),
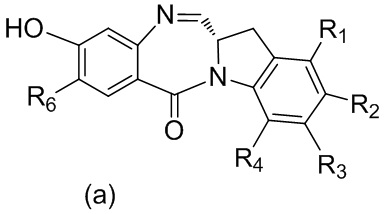 ,
,
где:
каждый R1, R2, R3 и R4 независимо выбран из группы, состоящей из: -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n’-Rc, галогена, гуанидиния [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NCO, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3-H, -OSO3H, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’ и -OCONR’R’’; и
R6 представляет собой -H, -R, -OR, -SR, -NR’R’’, -NO2 или галоген; а остальные переменные являются такими, как описано во втором варианте осуществления изобретения.
В качестве альтернативы, в третьем варианте осуществления изобретения предложен способ получения соединения формулы (8),
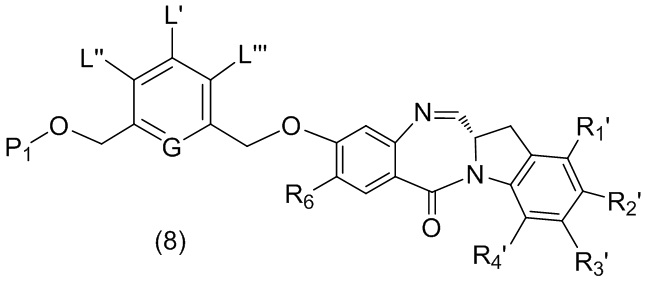
или его соли, причем указанный способ включает введение соединения формулы (3’)
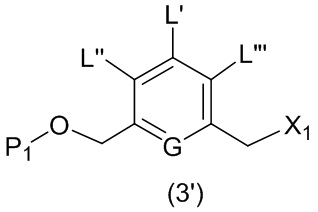
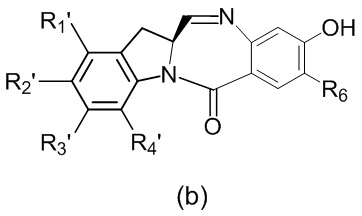
в реакцию с мономерным соединением формулы (b),
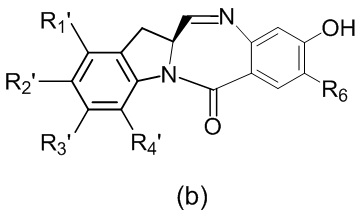
где каждый R1’, R2’, R3’ и R4’ независимо выбран из группы, состоящей из: -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n’-Rc, галогена, гуанидиния [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NCO, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3-H, -OSO3H, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’ и -OCONR’R’’; R6 представляет собой -H, -R, -OR, -SR, -NR’R’’, -NO2 или галоген; а остальные переменные являются такими, как описано во втором варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (3) представлено формулой, выбранной из следующего:
 (3b);
(3b);  (3c); и
(3c); и
 (3A),
(3A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения для способов согласно третьему варианту осуществления изобретения, X1 представляет собой -Br, -I или сульфонатный эфир.
В конкретном варианте осуществления изобретения соединение формулы (3) вводят в реакцию с мономерным соединением формулы (b) в присутствии основания. Можно применять любое подходящее основание. Типичные основания включают в себя, но не ограничиваясь этим, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. В одном варианте осуществления изобретения основание представляет собой карбонат калия.
В четвертом варианте осуществления данного изобретения предложен способ получения соединения формулы (5),
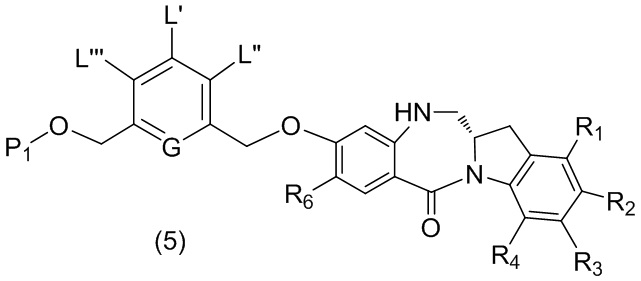
или его соли, причем указанный способ включает введение соединения формулы (4)
в реакцию с восстанавливающим имин реагентом, где переменные являются такими, как описано выше в третьем варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (4) представлено формулой, выбранной из следующего:
 (4b);
(4b);  (4c); и
(4c); и
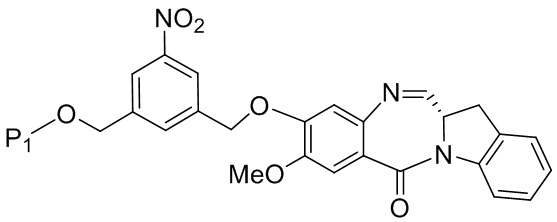 (4A),
(4A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения восстанавливающий имин реагент представляет собой гидридный восстановительный реагент.
В другом конкретном варианте осуществления изобретения восстанавливающий имин реагент представляет собой боргидрид натрия, триацетоксиборгидрид натрия, цианоборгидрид натрия, гидрид лития алюминия, газообразный водород, формиат аммония, боран, 9-борабицикло[3.3.1]нонан (9-ББН (9-BBN)), диизобутилалюминия гидрид (ДИБАЛ (DIBAL)), боргидрид лития (LiBH4), боргидрид калия (KBH4) или натрия бис(2-метоксиэтокси)алюминия гидрид (Red-Al). Предпочтительно восстанавливающий имин реагент представляет собой триацетоксиборгидрид натрия (NaBH(OAc)3).
В способе согласно четвертому варианту осуществления изобретения можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой дихлорэтан.
В пятом варианте осуществления данного изобретения предложен способ получения соединения формулы (6),
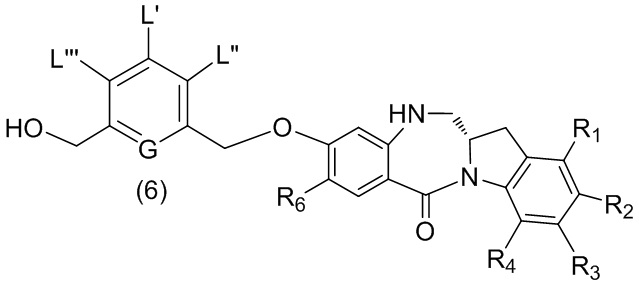
или его соли, причем указанный способ включает введение соединения формулы (5)
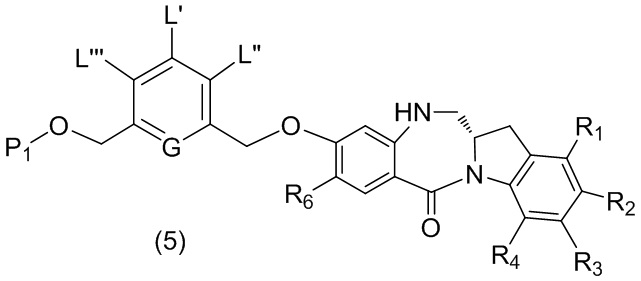
в реакцию с реагентом, удаляющим защитную группу спиртовой группы, где переменные являются такими, как описано выше в четвертом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (5) представлено формулой, выбранной из следующего:
 (5b);
(5b);  (5c); и
(5c); и
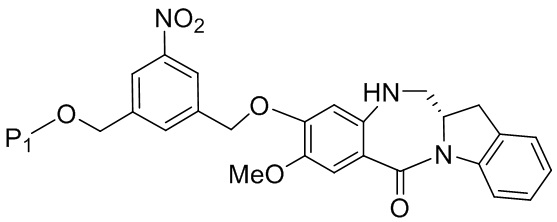 (5A),
(5A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения реагент, удаляющий защитную группу спиртовой группы, представляет собой тетра-н-бутиламмония фторид, трис(диметиламино)сульфония дифтортриметилсиликат, фтороводород или его сольват, пиридина гидрофторид, тетрафторид кремния, гексафторкремниевую кислоту, фторид цезия, хлористоводородную кислоту, уксусную кислоту, трифторуксусную кислоту, пиридиния п-толуолсульфонат, п-толуолсульфоновую кислоту (п-TsOH), муравьиную кислоту или йодную кислоту. Предпочтительно реагент, удаляющий защитную группу спиртовой группы, представляет собой хлористоводородную кислоту или тетра-н-бутиламмония фторид.
В реакции удаления защитной группы, описанной выше, можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой ТГФ.
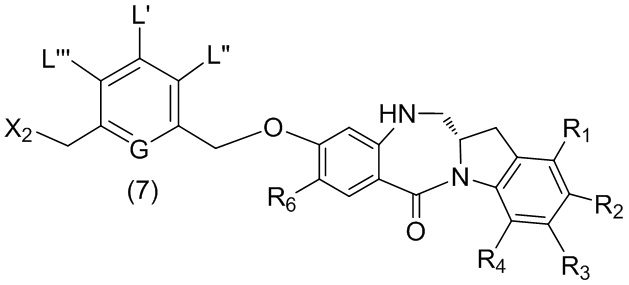
В шестом варианте осуществления данного изобретения предложен способ получения соединения формулы (7),
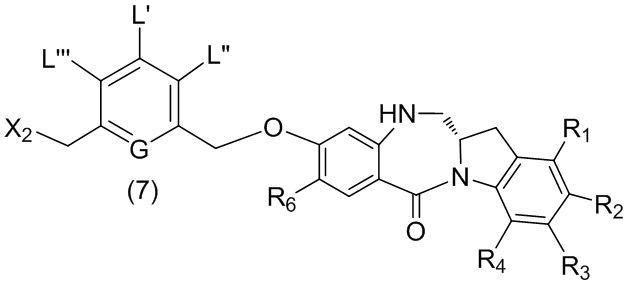
или его соли, причем указанный способ включает введение галогенирующего реагента, сульфонирующего реагента или эстерифицирующего реагента в реакцию с первичным спиртом соединением формулы (6),
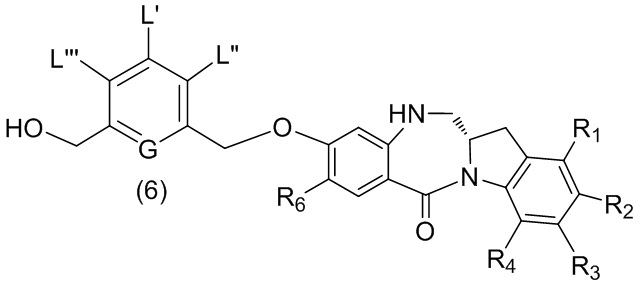
где X2 представляет собой –Br, -I, -Cl, сульфонатный эфир или активированный эфир; а остальные переменные являются такими, как описано выше в пятом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (6) представлено формулой, выбранной из следующего:
 (6b);
(6b);  (6c); и
(6c); и
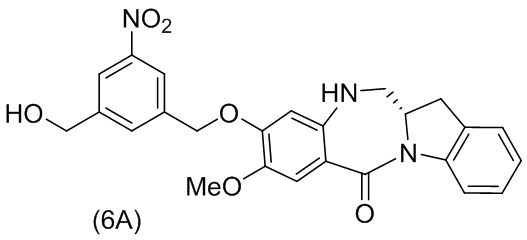 ,
,
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения для способов согласно шестом варианту осуществления изобретения X2 представляет собой –Br, -I или сульфонатный эфир.
В конкретном варианте осуществления изобретения X2 представляет собой мезилат, тозилат, брозилат или трифлат. Предпочтительно X2 представляет собой мезилат.
В другом конкретном варианте осуществления изобретения способ согласно шестому варианту осуществления изобретения включает введение соединения формулы (6) в реакцию с галогенирующим реагентом. Типичный галогенирующий реагент включают в себя, но не ограничиваясь этим, бром, бромистоводородную кислоту, тетрабромид углерода, трибромид фосфора, бромид калия, йодистоводородную кислоту, йод, тетрайодид углерода, трийодид фосфора, йодид натрия или йодид калия. В еще другом конкретном варианте осуществления изобретения способ согласно шестому варианту осуществления изобретения включает введение соединения формулы (6) в реакцию с сульфонирующим реагентом. Предпочтительно сульфонирующий реагент представляет собой сульфоновый ангидрид, такой как метансульфоновый ангидрид, или сульфоновый хлорангидрид, такой как метансульфонилхлорид (MsCl).
В одном варианте осуществления изобретения реакцию между соединением формулы (6) и сульфонирующим реагентом осуществляют в присутствии основания. Предпочтительно основание представляет собой ненуклеофильное основание. Типичное ненуклеофильное основание включают в себя, но не ограничиваясь этим, триэтиламин, имидазол, триэтиламин, диизопропилэтиламин, пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, (DBU)) или тетраметилпиперидин. Предпочтительно основание представляет собой триэтиламин или диизопропилэтиламин.
В описанной выше реакции можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой дихлорметан.
В седьмом варианте осуществления данного изобретения предложен способ получения соединения формулы (7”)
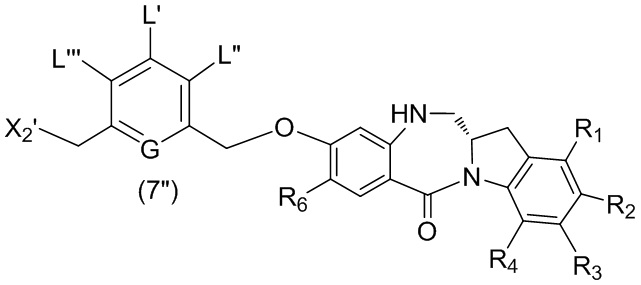 ,
,
или его соли, причем указанный способ включает введение соединения формулы (5”)

в реакцию с реагентом, удаляющим защитную группу спиртовой группы, и галогенирующим реагентом, где P1’ представляет собой кислотолабильную защитную группу для спиртовой группы; X2’ представляет собой –Br или –I; а остальные переменные являются такими, как описано выше в шестом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (5”) представлено формулой, выбранной из следующего:
 (5b”);
(5b”);  (5c”); и
(5c”); и
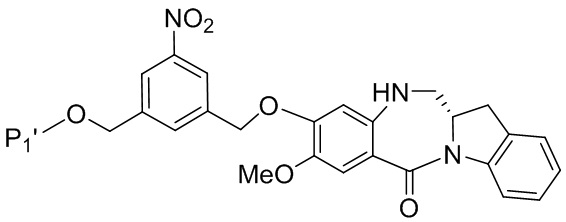 (5A”),
(5A”),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
Способ согласно седьмому варианту осуществления изобретения объединяет стадию удаления защитной группы спиртовой группы, описанную в пятом варианте осуществления изобретения, и реакцию галогенирования полученного спирта, описанную в шестом варианте осуществления изобретения, в одну стадию.
В конкретном варианте осуществления изобретения для способа согласно седьмому варианту осуществления изобретения соединение формулы (7”) представлено следующей формулой:
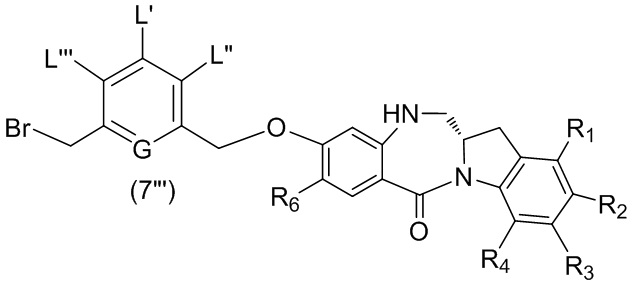 ,
,
и способ включает введение соединения формулы (5”) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, и бромирующим реагентом.
В одном варианте осуществления изобретения кислотолабильная защитная группа для спиртовой группы представляет собой ацетат, аллил, метоксиметил, тетрагидрoфуранил, тетрагидрoпиранил, 5-дибензoсуберил, 1-этоксиэтил, 1-метил-1метоксилэтил, 2-(фенилселенил)этил, тритил/трифенилметил или трис(4-трет-бутилфенил)метил.
В другом варианте осуществления изобретения кислотолабильная защитная группа для спиртовой группы представляет собой силильную защитную группу. Типичные силильные защитные группы включают в себя, но не ограничиваясь этим, диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триэтилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, 2-триметиэтилсилил (ТМЭС, (TEOC)) или [2-(триметилсилил)этокси]метил. Предпочтительно силильная защитная группа представляет собой триэтилсилил, триизопропилсилил или трет-бутилдиметилсилил. Более предпочтительно, силильная защитная группа представляет собой трет-бутилдиметилсилил.
В одном варианте осуществления изобретения реагент, удаляющий защитную группу спиртовой группы, представляет собой тетра-н-бутиламмония фторид, трис(диметиламино)сульфония дифтортриметилсиликат, фтороводород или его сольват, пиридина гидрофторид, тетрафторид кремния, гексафторкремниевую кислоту, фторид цезия, хлористоводородную кислоту, уксусную кислоту, пиридиния п-толуолсульфонат, муравьиную кислоту, йодную кислоту, трифторуксусную кислоту или п-толуолсульфоновую кислоту (п-TsOH). Предпочтительно реагент, удаляющий защитную группу спиртовой группы, представляет собой уксусную кислоту.
В еще другом варианте осуществления изобретения бромирующий реагент представляет собой HBr.
В одном конкретном варианте осуществления изобретения способ согласно седьмому варианту осуществления изобретения включает введение соединения формулы (5”) в реакцию со смесью уксусной кислоты и HBr с получением соединения формулы (7”).
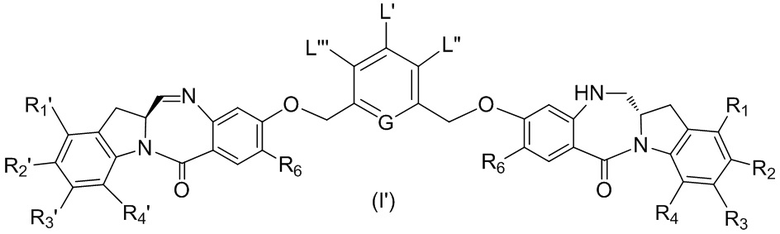
В восьмом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
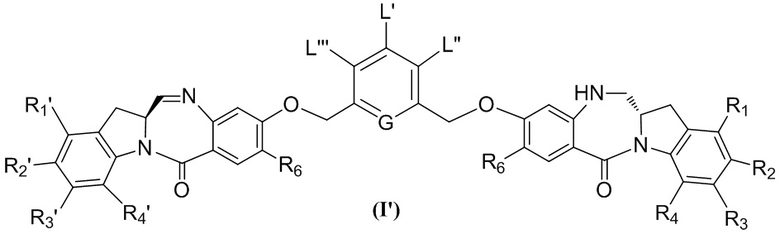
или его фармацевтически приемлемой соли, причем указанный способ включает введение соединения формулы (7) в реакцию
с мономерным соединением формулы (b),
где каждый R1’, R2’, R3’ и R4’ независимо выбран из группы, состоящей из: -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n-Rc, галогена, гуанидиния [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NCO, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3-H, -OSO3H, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’ и -OCONR’R’’; а остальные переменные являются такими, как описано выше в седьмом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (7) представлено формулой, выбранной из следующего:
 (7b);
(7b);  (7c); и
(7c); и
 (7A),
(7A),
где X2 представляет собой уходящую группу, выбранную из группы, состоящей из: -Br, -I, -Cl, сульфонатного эфира и активированного эфира; R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В одном варианте осуществления изобретения для способов согласно восьмому варианту осуществления изобретения, X2 представляет собой –Br, -I или сульфонатный эфир.
В одном варианте осуществления изобретения соединение формулы (7) вводят в реакцию с мономерным соединением формулы (b) в присутствии основания. Примеры основания включают в себя, но не ограничиваясь этим, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. В одном варианте осуществления изобретения основание представляет собой карбонат калия.
В описанных выше способах можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой ДМФА.
В девятом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1),
с получением соединения формулы (2),
;
(2) введение соединения формулы (2) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом с получением соединения формулы (3),
;
(3) введение соединения формулы (3) в реакцию с мономерным соединением формулы (a),
с получением соединения формулы (4),
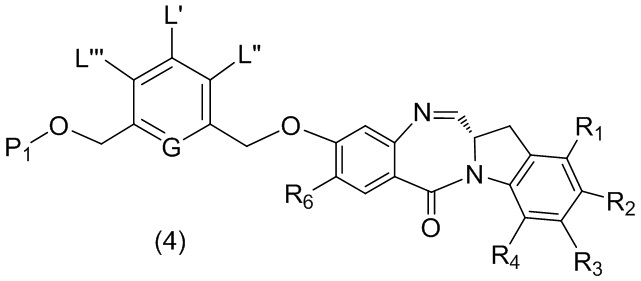 ;
;
(4) введение соединения формулы (4) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5),
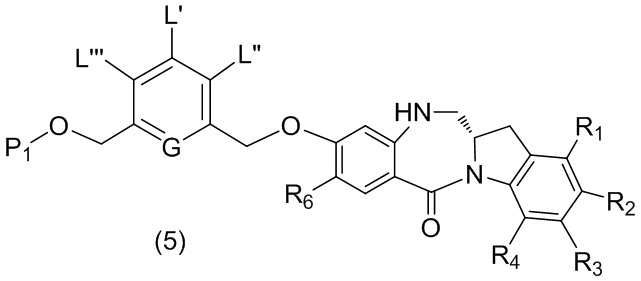 ;
;
(5) введение соединения формулы (5) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (6),
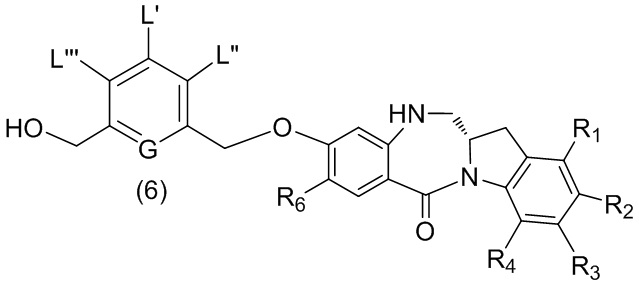 ;
;
(6) введение второго галогенирующего реагента, второго сульфонирующего реагента или второго эстерифицирующего реагента в реакцию с соединением формулы (6) с получением соединения формулы (7),
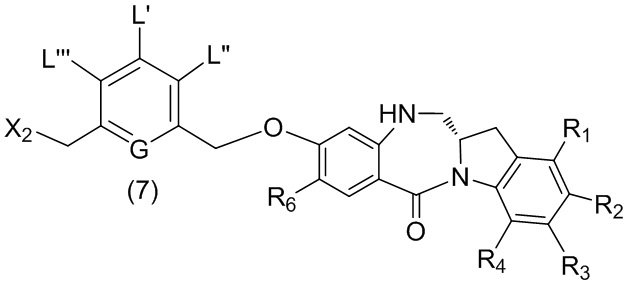 ; и
; и
(7) введение соединения формулы (7) в реакцию с мономерным соединением формулы (b),
с получением соединения формулы (I’), где переменные являются такими, как описано выше в первом, втором, третьем, четвертом, пятом, шестом и восьмом вариантах осуществления изобретения.
В одном конкретном варианте осуществления изобретения способ согласно девятому варианту осуществления изобретения включает получение соединения формулы (Ib’)
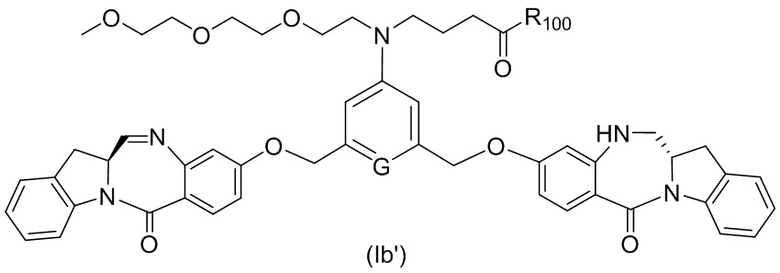 ,
,
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1b) с получением соединения формулы (2b);
(2) введение соединения формулы (2b) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом с получением соединения формулы (3b);
(3) введение соединения формулы (3b) в реакцию с мономерным соединением формулы (a) с получением соединения формулы (4b),
(4) введение соединения формулы (4b) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5b),
(5) введение соединения формулы (5b) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (6b),
(6) введение соединения формулы (6b) в реакцию со вторым галогенирующим реагентом, вторым сульфонирующим реагентом или вторым эстерифицирующим реагентом с получением соединения формулы (7b),
(7) введение соединения формулы (7b) в реакцию с мономерным соединением формулы (b) с получением соединения формулы (Ib’).
В одном конкретном варианте осуществления изобретения способ согласно девятому варианту осуществления изобретения включает получение соединения формулы (Ic’)
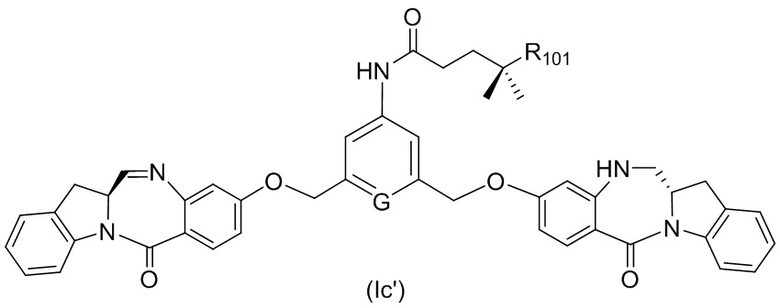 ,
,
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1c) с получением соединения формулы (2c);
(2) введение соединения формулы (2c) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом с получением соединения формулы (3c);
(3) введение соединения формулы (3c) в реакцию с мономерным соединением формулы (a) с получением соединения формулы (4c),
(4) введение соединения формулы (4c) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5c),
(5) введение соединения формулы (5c) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (6c),
(6) введение соединения формулы (6c) в реакцию со вторым галогенирующим реагентом, вторым сульфонирующим реагентом или вторым эстерифицирующим реагентом с получением соединения формулы (7c),
(7) введение соединения формулы (7c) в реакцию с мономерным соединением формулы (b) с получением соединения формулы (Ic’).
В одном конкретном варианте осуществления изобретения способ согласно девятому варианту осуществления изобретения включает получение соединения формулы (IA)
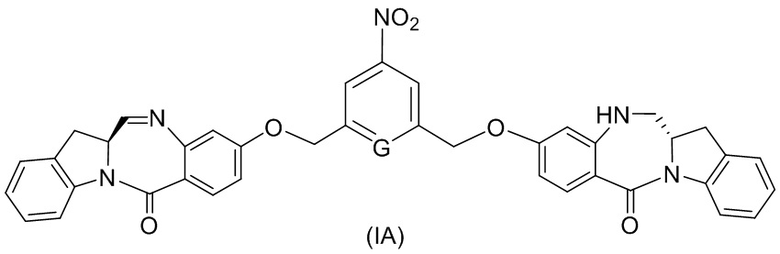 ,
,
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1A) с получением соединения формулы (2A);
(2) введение соединения формулы (2A) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом с получением соединения формулы (3A);
(3) введение соединения формулы (3A) в реакцию с мономерным соединением формулы (a) с получением соединения формулы (4A),
(4) введение соединения формулы (4A) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5A),
(5) введение соединения формулы (5A) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (6A),
(6) введение соединения формулы (6A) в реакцию со вторым галогенирующим реагентом, вторым сульфонирующим реагентом или вторым эстерифицирующим реагентом с получением соединения формулы (7A),
(7) введение соединения формулы (7A) в реакцию с мономерным соединением формулы (b) с получением соединения формулы (IA).
В одном варианте осуществления изобретения для способов согласно девятому варианту осуществления изобретения каждый X1 и X2 независимо представляет собой –Br, -Cl или сульфонатный эфир.
Условия реакции и реагенты для каждой стадии способа согласно девятому варианту осуществления изобретения являются такими, как описано в первом, втором, третьем, четвертом, пятом, шестом и/или восьмом варианте осуществления изобретения или любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В десятом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
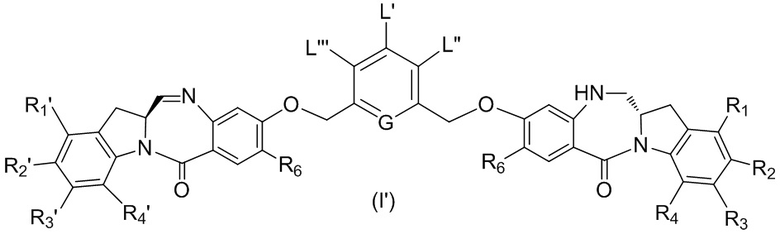
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1),
с получением соединения формулы (2”),
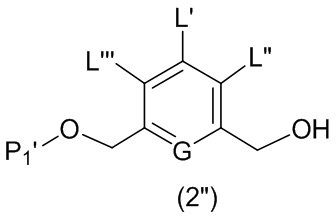 ;
;
(2) введение соединения формулы (2’’) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом с получением соединения формулы (3”),
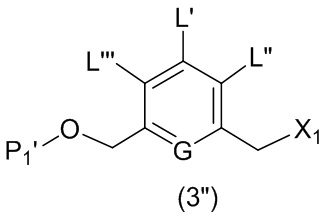 ;
;
(3) введение соединения формулы (3”) в реакцию с мономерным соединением формулы (a),
с получением соединения формулы (4”),
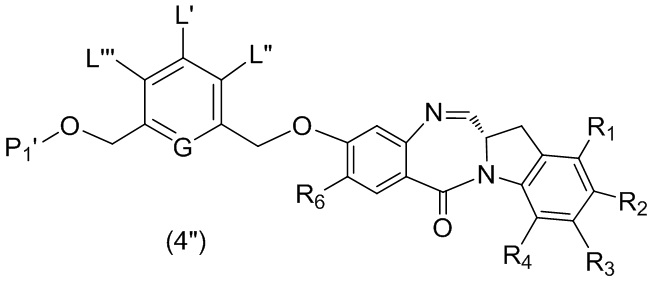 ;
;
(4) введение соединения формулы (4”) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5”),
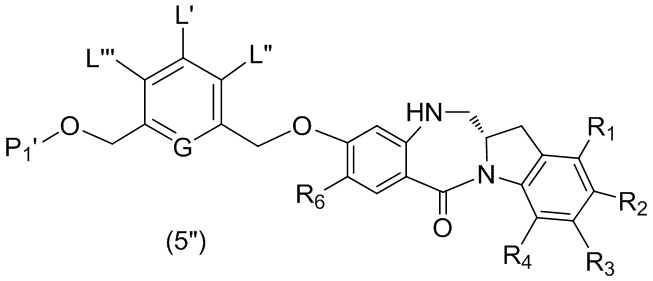 ;
;
(5) введение соединения формулы (5”) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, и галогенирующим реагентом с получением соединения формулы (7”),
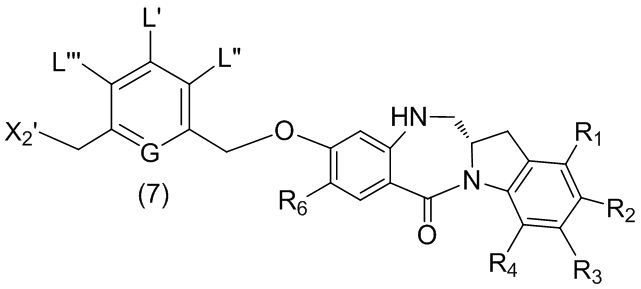 ;
;
(6) введение соединения формулы (7”) в реакцию с мономерным соединением формулы (b),
с получением соединения формулы (I’), где X2’ представляет собой –Br или –I; а остальные переменные являются такими, как описано выше в девятом варианте осуществления изобретения.
В одном варианте осуществления изобретения способ согласно десятому варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1b) с получением соединения формулы (2b”)
 (2b”);
(2b”);
(2) введение соединения формулы (2b”) в реакцию с галогенирующим реагентом или сульфонирующим реагентом с получением соединения формулы (3b”);
 (3b”)
(3b”)
(3) введение соединения формулы (3b”) в реакцию с мономерным соединением формулы (a1):
с получением соединения формулы (4b”);
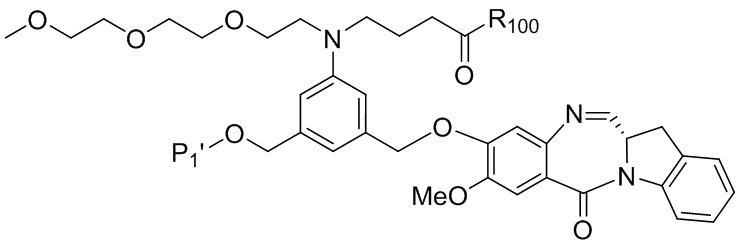 (4b”)
(4b”)
(4) введение соединения формулы (4b”) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5b”);
(5) введение соединения формулы (5b”) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, и галогенирующим реагентом с получением соединения формулы (7b”);
(6) введение соединения формулы (7b”) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (Ib’).
В другом варианте осуществления изобретения способ согласно десятому варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1c) с получением соединения формулы (2c”);
(2c”);
(2) введение соединения формулы (2c”) в реакцию с галогенирующим реагентом или сульфонирующим реагентом с получением соединения формулы (3c”);
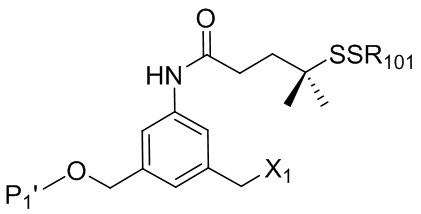 (3c”)
(3c”)
(3) введение соединения формулы (3c”) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (4c”);
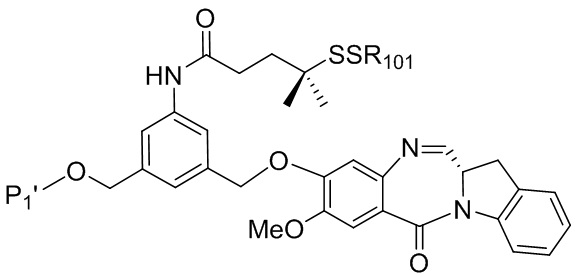 (4c”)
(4c”)
(4) введение соединения формулы (4c”) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5c”);
(5) введение соединения формулы (5c”) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, и галогенирующим реагентом с получением соединения формулы (7c”);
(6) введение соединения формулы (7c”) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно десятому варианту осуществления изобретения включает получение соединения формулы (IA),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1A) с получением соединения формулы (2A”);
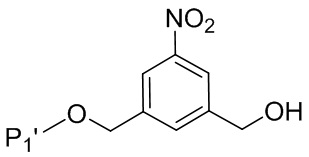 (2A”),
(2A”),
(2) введение соединения формулы (2A”) в реакцию с галогенирующим реагентом или сульфонирующим реагентом с получением соединения формулы (3A”);
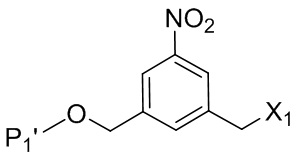 (3A”)
(3A”)
(3) введение соединения формулы (3A”) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (4A”);
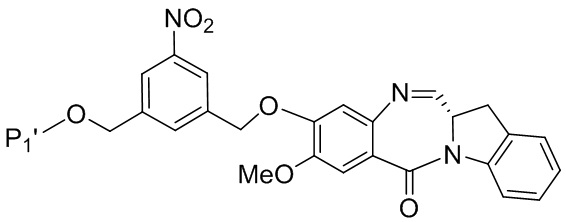 (4A”)
(4A”)
(4) введение соединения формулы (4A”) в реакцию с восстанавливающим имин агентом с получением соединения формулы (5A”);
(5) введение соединения формулы (5A”) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, и галогенирующим реагентом с получением соединения формулы (7A”);
(6) введение соединения формулы (7A”) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (IA’).
Условия и реагенты для способа согласно десятому варианту осуществления изобретения являются такими, как описано выше в первом, втором, третьем, четвертом, седьмом и/или восьмом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В одиннадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (9),
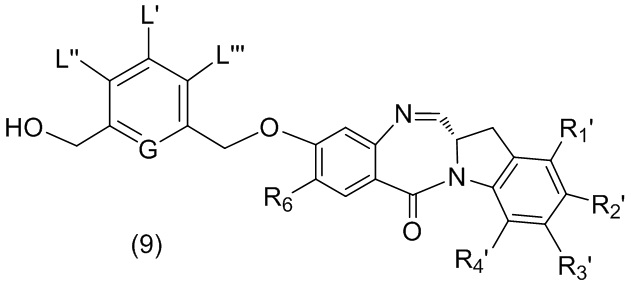
или его соли, причем указанный способ включает введение соединения формулы (8),

в реакцию с реагентом, удаляющим защитную группу спиртовой группы, где переменные являются такими, как описано выше в третьем варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (8) представлено формулой, выбранной из следующего:
(4b); (4c); и
(4A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения реагент, удаляющий защитную группу спиртовой группы, представляет собой тетра-н-бутиламмония фторид, трис(диметиламино)сульфония дифтортриметилсиликат, фтороводород или его сольват, пиридина гидрофторид, тетрафторид кремния, гексафторкремниевую кислоту, фторид цезия, хлористоводородную кислоту, уксусную кислоту, пиридиния п-толуолсульфонат, муравьиную кислоту, йодную кислоту, трифторуксусную кислоту или п-толуолсульфоновую кислоту (п-TsOH). Более конкретно, реагент, удаляющий защитную группу спиртовой группы, представляет собой хлористоводородную кислоту или тетра-н-бутиламмония фторид.
В двенадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (10),
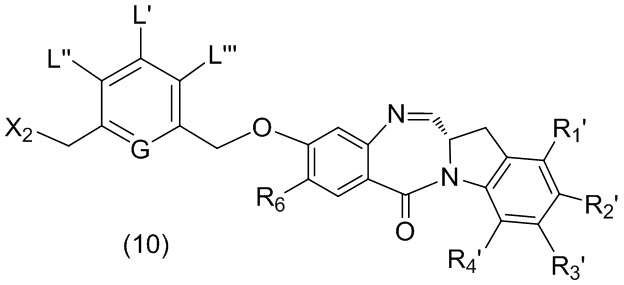
или его соли, причем указанный способ включает введение соединения формулы (9) в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом,
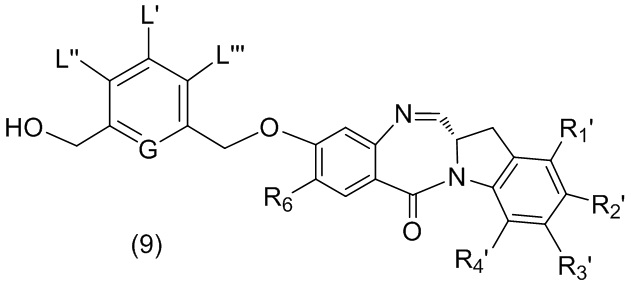
где X2 представляет собой -Br, -I, -Cl, сульфонатный эфир или активированный эфир; а остальные переменные являются такими, как описано выше в одиннадцатом варианте осуществления изобретения.
В одном варианте осуществления изобретения соединение формулы (9) представлено формулой, выбранной из следующего:
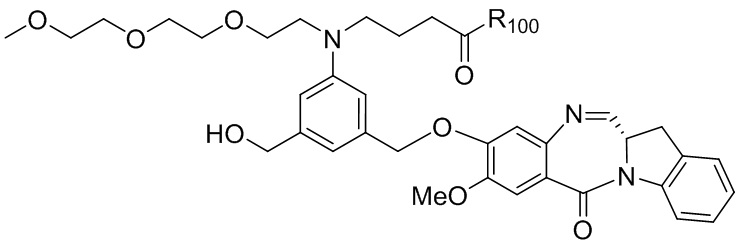 (9b);
(9b);  (9c); и
(9c); и
 (9A),
(9A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения для способов согласно двенадцатом варианту осуществления изобретения, X2 представляет собой –Br, -I или сульфонатный эфир.
В конкретном варианте осуществления изобретения X2 представляет собой мезилат, тозилат, брозилат или трифлат. Предпочтительно X2 представляет собой мезилат.
В другом конкретном варианте осуществления изобретения способ, описанный в шестом варианте осуществления изобретения, включает введение соединения формулы (9) в реакцию с галогенирующим реагентом. Типичный галогенирующий реагент включают в себя, но не ограничиваясь этим, бром, бромистоводородную кислоту, тетрабромид углерода, трибромид фосфора, бромид калия, йодистоводородную кислоту, йод, тетрайодид углерода, трийодид фосфора, йодид натрия или йодид калия.
В еще другом конкретном варианте осуществления изобретения способ согласно шестому варианту осуществления изобретения включает введение соединения формулы (9) в реакцию с сульфонирующим реагентом. Предпочтительно сульфонирующий реагент представляет собой сульфоновый ангидрид, такой как метансульфоновый ангидрид или сульфоновый хлорангидрид, такой как метансульфонилхлорид (MsCl).
В одном варианте осуществления изобретения реакцию между соединением формулы (9) и сульфонирующим реагентом осуществляют в присутствии основания. Предпочтительно основание представляет собой ненуклеофильное основание. Типичное ненуклеофильное основание включает в себя, но не ограничиваясь этим, триэтиламин, имидазол, триэтиламин, диизопропилэтиламин, пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, (DBU)) или тетраметилпиперидин. Предпочтительно основание представляет собой триэтиламин или диизопропилэтиламин.
В тринадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
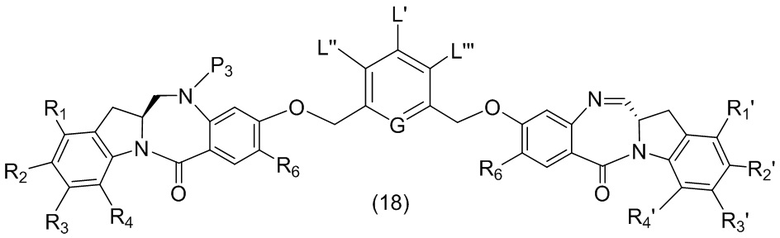
или его соли, причем указанный способ включает введение соединения формулы (10)
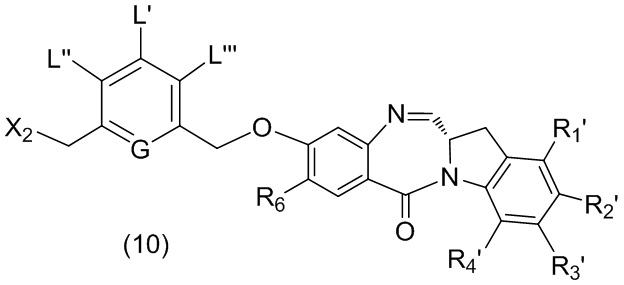
в реакцию с мономерным соединением формулы (d),
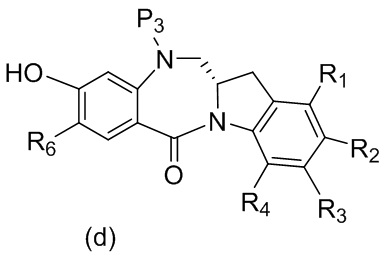
где X2 представляет собой уходящую группу, выбранную из группы, состоящей из: -Br, -I, -Cl, сульфонатного эфира и активированного эфира; P3 представляет собой H или P2; P2 представляет собой защитную группу для аминогруппы; каждый R1’, R2’, R3’ и R4’ независимо выбран из группы, состоящей из: -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n-Rc, галогена, гуанидиния [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NCO, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3-H, -OSO3H, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’ и -OCONR’R’’; а остальные переменные являются такими, как описано выше в двенадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения X2 представляет собой -Br, -I или сульфонатный эфир.
В конкретном варианте осуществления изобретения соединение формулы (10) представлено формулой, выбранной из следующего:
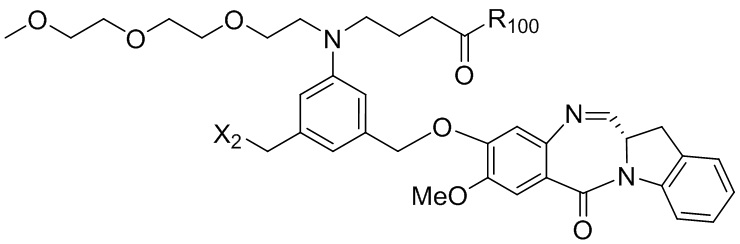 (10b);
(10b); 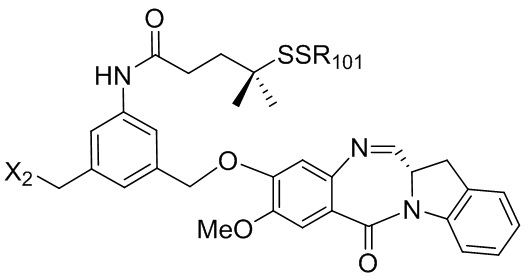 (10c); и
(10c); и
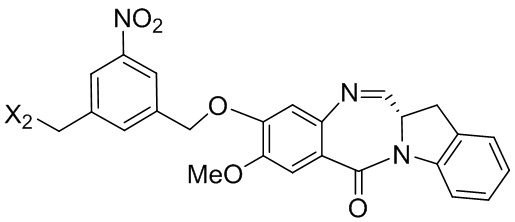 (10A),
(10A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения P3 представляет собой H, а соединение (10) вводят в реакцию с мономерным соединением (d) с получением соединения (I’):
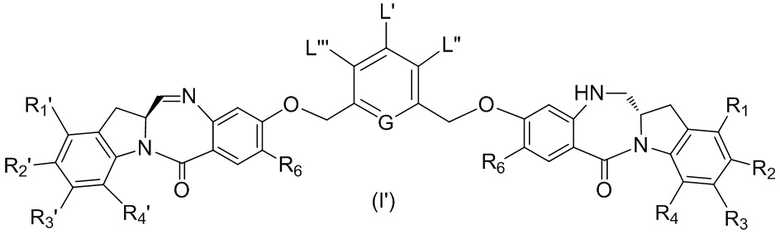 .
.
В другом конкретном варианте осуществления изобретения P3 представляет собой P2; мономерное соединение представлено формулой (c):
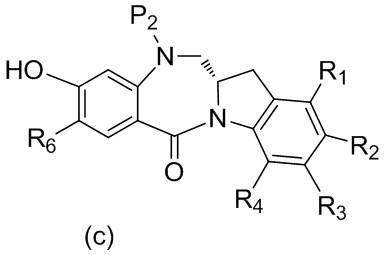 ;
;
а соединение формулы (18) представлено формулой (11),
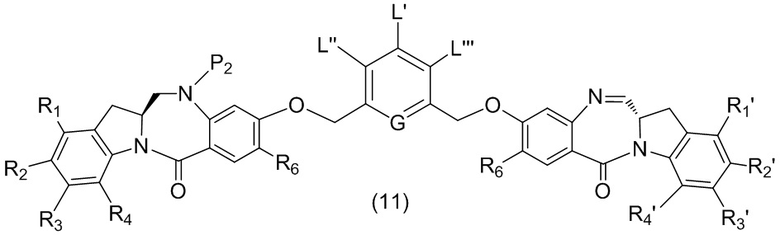 .
.
В описанном выше способе можно применять любую подходящую защитную группу для аминогруппы. В одном варианте осуществления изобретения защитная группа для аминогруппы представляет собой 2-триметилсилилэтил, (2-фенил-2-триметилсилил)этил, триизопропилсилокси, 2-(триметилсилил)этоксиметил, аллилоксикарбонил, 9-флуоренилметоксикарбонил, 2-(триметилсилил)этоксикарбонил или 2,2,2,2-трихлорэтоксикарбонил.
В конкретном варианте осуществления изобретения соединение формулы (10) вводят в реакцию с мономерным соединением формулы (d) или (c) в присутствии основания. Примеры основания включают в себя, но не ограничиваясь этим карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия.
В описанной выше реакции можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой ДМФА.
В четырнадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
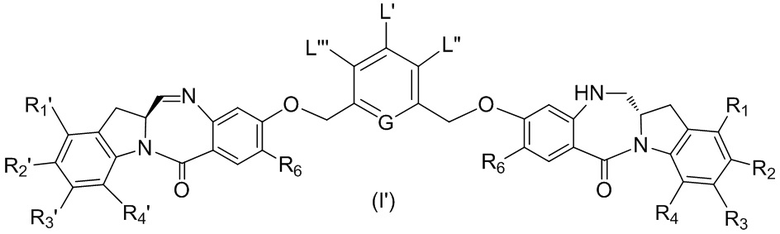
или его фармацевтически приемлемой соли, причем указанный способ включает введение соединения формулы (11),
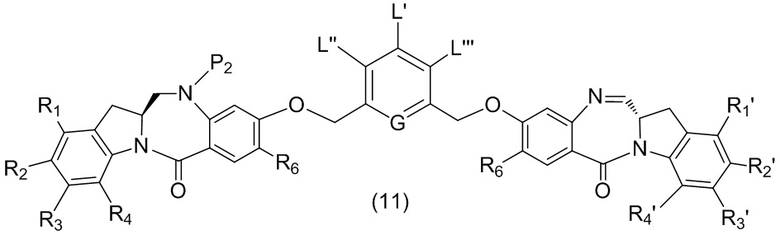
в реакцию с реагентом, удаляющим защитную группу аминогруппы, где переменные являются такими, как описано выше в тринадцатом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (11) представлено формулой, выбранной из следующего:
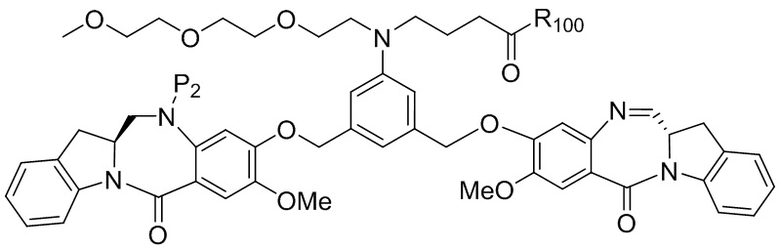 (11b);
(11b); 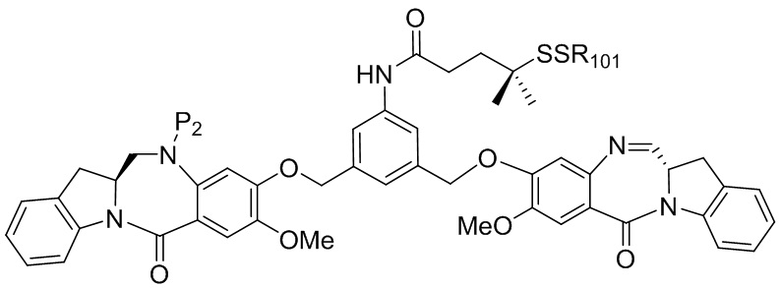 (11c); и
(11c); и
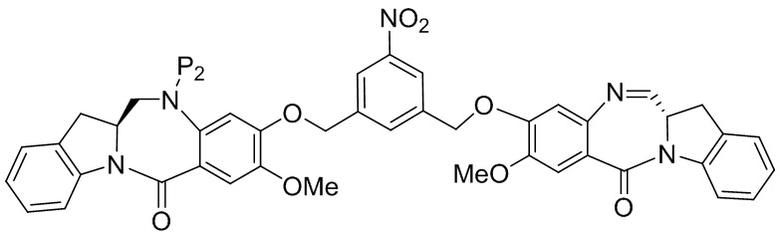 (11A),
(11A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В описанном выше способе можно применять любой подходящий реагент для удаления защитной группы аминогруппы. В одном варианте осуществления изобретения реагент, удаляющий защитную группу аминогруппы, представляет собой тетра-н-бутиламмония фторид, уксусную кислоту, пиридина гидрофторид, фторид цезия, пиперидин, морфолин или трифторуксусную кислоту.
В пятнадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1),
с получением соединения формулы (2),
;
(2) введение галогенирующего реагента, сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (2) с получением соединения формулы (3),
;
(3) введение соединения формулы (3) в реакцию с мономерным соединением формулы (b),
с получением соединения формулы (8),
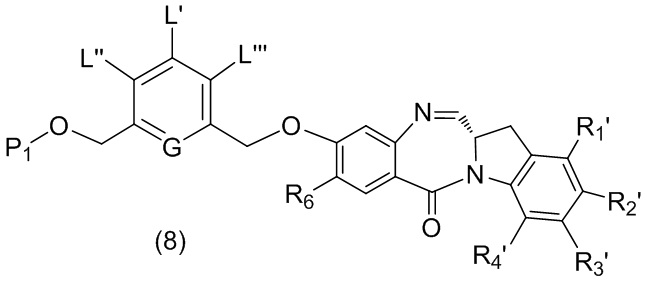 ;
;
(4) введение соединения формулы (8) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (9),
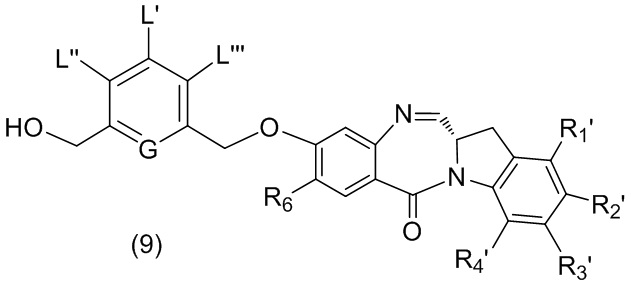
(5) введение второго галогенирующего реагента, второго сульфонирующего реагента или второго эстерифицирующего реагента в реакцию с соединением формулы (9) с получением соединения формулы (10),
;
(6) введение соединения формулы (10) в реакцию с мономерным соединением формулы (d)
с получением соединения формулы (18),
,
(7) если P3 представляет собой защитную группу для аминогруппы; то введение соединения формулы (18) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’),
где переменные являются такими, как описано выше в четырнадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения каждый X1 и X2 независимо представляет собой –Br, -I или сульфонатный эфир.
В одном варианте осуществления изобретения способ согласно пятнадцатому варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1b), с получением соединения формулы (2b);
(2) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (2b) с получением соединения формулы (3b);
(3) введение соединения формулы (3b) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (4b);
(4) введение соединения формулы (4b) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (9b);
(5) введение второго галогенирующего реагента или второго сульфонирующего реагента в реакцию с соединением формулы (9b) с получением соединения формулы (10b);
(6) введение соединения формулы (10b) в реакцию с мономерным соединением формулы (d1):
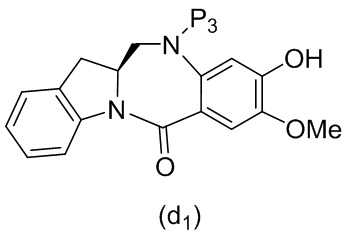
с получением соединения формулы (18b);
(7) если P3 представляет собой защитную группу для аминогруппы; то введение соединения формулы (18b):
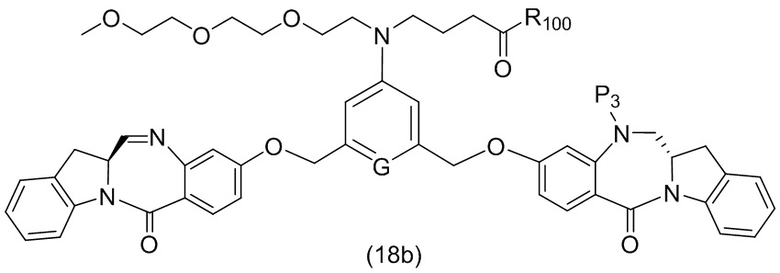
в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’),
В другом варианте осуществления изобретения способ согласно пятнадцатому варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1c), с получением соединения формулы (2c);
(2) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (2c) с получением соединения формулы (3c);
(3) введение соединения формулы (3b) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (4c);
(4) введение соединения формулы (4c) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (9c);
(5) введение второго галогенирующего реагента или второго сульфонирующего реагента в реакцию с соединением формулы (9c) с получением соединения формулы (10c);
(6) введение соединения формулы (10c) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (18c);
(7) если P3 представляет собой защитную группу для аминогруппы; то введение соединения формулы (18c):
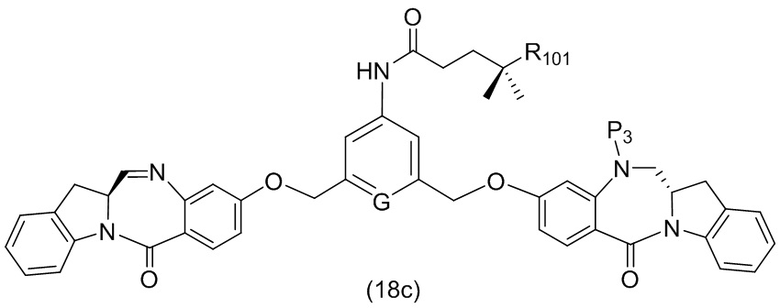
в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно пятнадцатому варианту осуществления изобретения включает получение соединения формулы (IA),
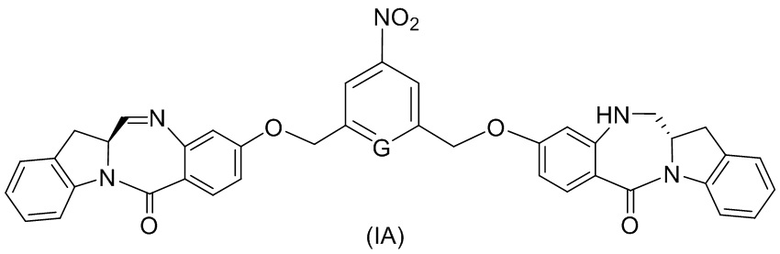
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение защитной группы для спиртовой группы в одну из первичных спиртовых групп соединения формулы (1A) с получением соединения формулы (2A);
(2) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (2A) с получением соединения формулы (3A);
(3) введение соединения формулы (3A) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (4A);
(4) введение соединения формулы (4A) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (9A);
(5) введение второго галогенирующего реагента или второго сульфонирующего реагента в реакцию с соединением формулы (9A) с получением соединения формулы (10A);
(6) введение соединения формулы (10A) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (18A):
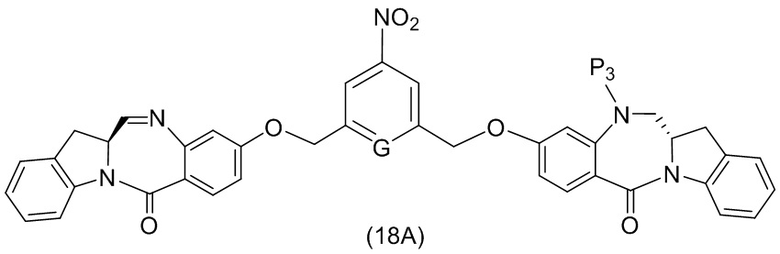 ;
;
(7) если P3 представляет собой защитную группу для аминогруппы; то введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA’).
В одном варианте осуществления изобретения P3 представляет собой H, а соединение (10) вводят в реакцию с мономерным соединением (d) с получением соединения (I’).
В другом варианте осуществления изобретения P3 представляет собой P2; мономерное соединение представлено формулой (c):
;
а соединение формулы (18) представлено формулой (11),
где P2 представляет собой защитную группу для аминогруппы.
В шестнадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (12),
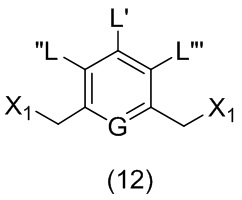 ,
,
или его соли, причем указанный способ включает введение соединения формулы (1),
в реакцию с галогенирующим реагентом, сульфонирующим реагентом или эстерифицирующим реагентом, где X1 представляет собой -Br, -I, -Cl, сульфонатный эфир или активированный эфир; и остальные переменные являются такими, как описано выше в первом варианте осуществления изобретения. В одном варианте осуществления изобретения X1 представляет собой -Br, -I или сульфонатный эфир.
В одном варианте осуществления изобретения соединение формулы (1) представлено формулой, выбранной из следующего:
(1b); (1c); и
(1A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения X1 представляет собой -Br или –I. В другом конкретном варианте осуществления изобретения X1 представляет собой -Cl.
В другом конкретном варианте осуществления изобретения галогенирующий реагент вводят в реакцию с первичными спиртами соединениями формулы (1) в присутствии активирующего спирт агента. В одном варианте осуществления изобретения активирующий спирт агент представляет собой тионилхлорид. В другом конкретном варианте осуществления изобретения галогенирующий реагент представляет собой бромид лития, бромид натрия, бромид калия, йодид калия или йодид натрия. В другом конкретном варианте осуществления изобретения галогенирующий реагент представляет собой тетрахлорид углерода/трифенилфосфин, метансульфонил(мезил)хлорид/хлорид лития или метансульфонил(мезил)хлорид/пиридин.
В еще другом конкретном варианте осуществления изобретения способ включает введение соединения формулы (1) в реакцию с LiBr в присутствии тионилхлорида.
В описанном выше способе можно применять любые подходящие растворители. Типичные растворители включают в себя, но не ограничиваясь этим, ДМФА, CH2Cl2, ТГФ, дихлорэтан, и т.п.
В семнадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (10’),
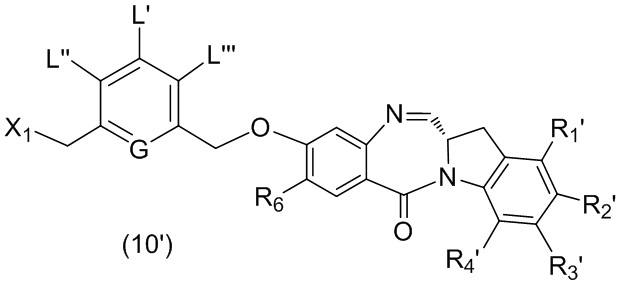
или его соли, причем указанный способ включает введение соединения формулы (12),
в реакцию с мономерным соединением формулы (b),
где L’, L”, L”’, X1, G являются такими, как описано выше в семнадцатом варианте осуществления изобретения; а R1’, R2’, R3’, R4’ и R6 являются такими, как описано выше в одиннадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения X1 представляет собой -Br, -I или сульфонатный эфир.
В конкретном варианте осуществления изобретения соединение формулы (12) представлено формулой, выбранной из следующего:
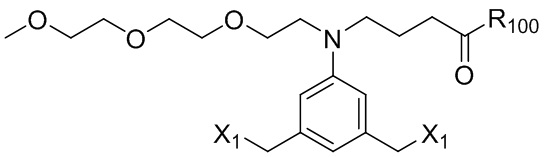 (12b);
(12b); 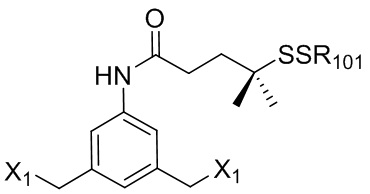 (12c); и
(12c); и
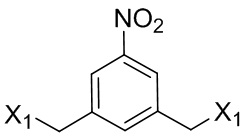 (12A),
(12A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
Кроме того, в семнадцатом варианте осуществления изобретения предложен способ получения соединения формулы (7-1)
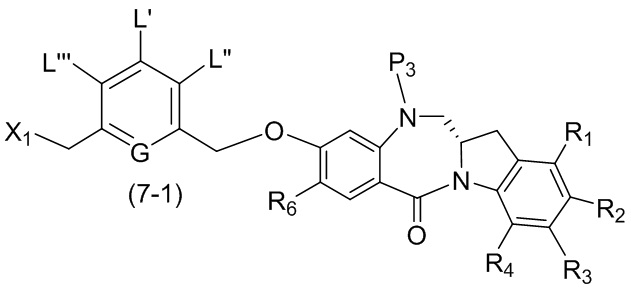
или его соли, причем указанный способ включает введение соединения формулы (12) в реакцию с мономерным соединением формулы (d), где X1 представляет собой -Br, -I, -Cl, сульфонатный эфир или активированный эфир; P3 представляет собой H или защитную группу для аминогруппы; и R100 представляет собой (C1-C3)алкокси.
В конкретном варианте осуществления изобретения соединение формулы (12) представлено формулой (12b), (12c) или (12A).
В конкретном варианте осуществления изобретения для формулы (7-1’), P3 представляет собой H. В другом конкретном варианте осуществления изобретения P3 представляет собой защитную группу для аминогруппы, как описано в данном документе.
В конкретном варианте осуществления изобретения для способов согласно семнадцатому варианту осуществления изобретения, соединение формулы (12) вводят в реакцию с мономерным соединением формулы (b) в присутствии основания. Примеры подходящего основания включают в себя, но не ограничиваясь этим, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. В одном варианте осуществления изобретения основание представляет собой карбонат калия.
В другом конкретном варианте осуществления изобретения соединение формулы (12d) или (12A) вводят в реакцию с мономерным соединением формулы (d) в присутствии основания. Примеры подходящего основания включают в себя, но не ограничиваясь этим, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. В одном варианте осуществления изобретения основание представляет собой карбонат калия.
Для способов согласно семнадцатому варианту осуществления изобретения, можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой ДМФА.
В другом конкретном варианте осуществления изобретения в реакции применяют избыток молярных эквивалентов соединения формулы (12) относительно мономера соединения формулы (b).
В восемнадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (7’),
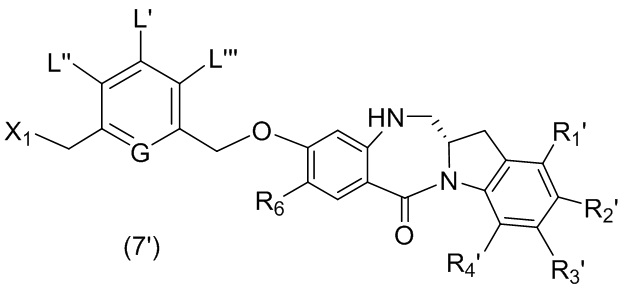 ,
,
или его соли, причем указанный способ включает введение в реакцию соединения формулы (10’),
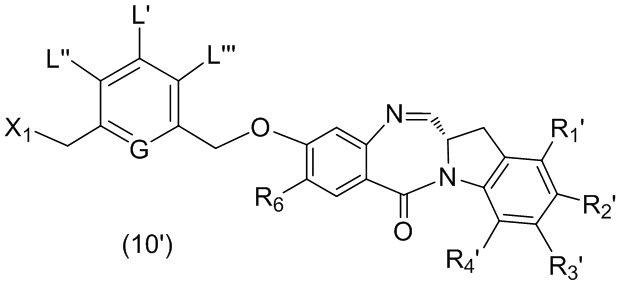
или его соли, с восстанавливающим имин агентом, где X1 представляет собой -Br, -I, -Cl, сульфонатный эфир или активированный эфир, а остальные переменные являются такими же, как описано выше в одиннадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения X1 представляет собой -Br, -I или сульфонатный эфир.
В конкретном варианте осуществления изобретения соединение формулы (10’) представлено одной из следующих формул:
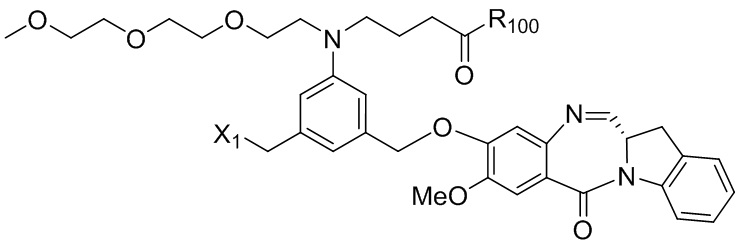 (10b’);
(10b’); 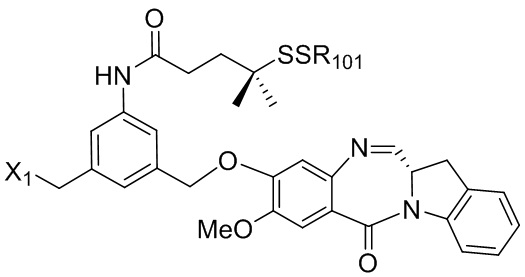 (10c’); и
(10c’); и
 (10A’),
(10A’),
В конкретном варианте осуществления изобретения для способов согласно восемнадцатому варианту осуществления изобретения, X1 представляет собой сульфонатный эфир. Предпочтительно X1 представляет собой мезилат.
В одном варианте осуществления изобретения для способов согласно восемнадцатому варианту осуществления изобретения, восстанавливающий имин реагент представляет собой гидридный восстановительный реагент. В качестве альтернативы, восстанавливающий имин реагент представляет собой боргидрид натрия, триацетоксиборгидрид натрия, цианоборгидрид натрия, гидрид лития алюминия, газообразный водород, формиат аммония, боран, 9-борабицикло[3.3.1]нонан (9-ББН (9-BBN)), диизобутилалюминия гидрид (ДИБАЛ (DIBAL)), боргидрид лития (LiBH4), боргидрид калия (KBH4) или натрия бис(2-метоксиэтокси)алюминия гидрид (Red-Al). В предпочтительном варианте осуществления изобретения восстанавливающий имин реагент представляет собой триацетоксиборгидрид натрия (NaBH(OAc)3).
В способах согласно восемнадцатому варианту осуществления изобретения можно применять любые подходящие растворители. В одном варианте осуществления изобретения растворитель представляет собой дихлорэтан.
В девятнадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента, сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (1),
с получением соединения формулы (12),
;
(2) введение соединения формулы (12) в реакцию с мономерным соединением формулы (b),
с получением соединения формулы (10’),
;
(3) введение соединения формулы (10’) в реакцию с мономерным соединением формулы (d),
с получением соединения формулы (18),
; и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’);
где X1 представляет собой –Br, -I, -Cl, сульфонатный эфир или активированный эфир; P3 представляет собой H или защитную группу для аминогруппы, а остальные переменные являются такими же, как описано в одиннадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения X1 представляет собой –Br, -I или сульфонатный эфир.
В одном варианте осуществления изобретения способ согласно девятнадцатому варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1b) с получением соединения формулы (12b);
(2) введение соединения формулы (12b) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (10b’);
(3) введение соединения формулы (10b’) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (18b); и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом варианте осуществления изобретения способ согласно девятнадцатому варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1c) с получением соединения формулы (12c);
(2) введение соединения формулы (12c) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (10c’);
(3) введение соединения формулы (10c’) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (18c); и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно девятнадцатому варианту осуществления изобретения включает получение соединения формулы (IA),
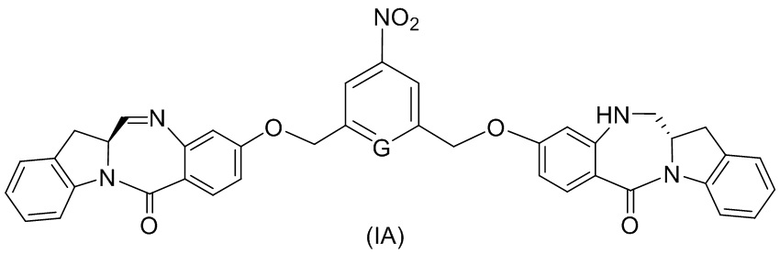
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1A) с получением соединения формулы (12A);
(2) введение соединения формулы (12A) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (10A’);
(3) введение соединения формулы (10A’) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (18A); и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA’).
В одном варианте осуществления изобретения для способов согласно девятнадцатому варианту осуществления изобретения P3 представляет собой H, а соединение (10’) вводят в реакцию с мономерным соединением (d) с получением соединения (I’).
В другом варианте осуществления изобретения для способов согласно девятнадцатому варианту осуществления изобретения, P3 представляет собой P2; мономерное соединение представлено формулой (c):
;
а соединение формулы (18) представлено формулой (11),
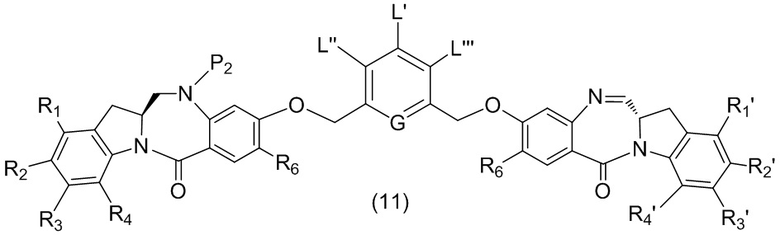
где P2 представляет собой защитную группу для аминогруппы.
Условия и реагенты для способа согласно девятнадцатому варианту осуществления изобретения являются такими, как описано выше в шестнадцатом, семнадцатом, тринадцатом и/или четырнадцатом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В двадцатом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
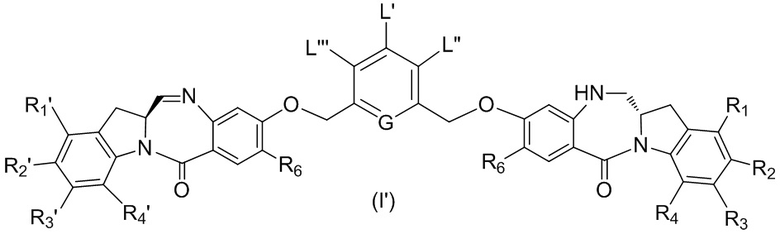
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента, сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (1),
с получением соединения формулы (12),
;
(2) введение соединения формулы (12) в реакцию с мономерным соединением формулы (b),
с получением соединения формулы (10’),
;
(3) введение соединения (10’) в реакцию с восстанавливающим имин реагентом с получением соединения (7’),
,
(4) введение соединения формулы (7’) в реакцию с мономерным соединением формулы (a),
с получением соединения формулы (I’) или его фармацевтически приемлемой соли, где X1 представляет собой –Br, -I, -Cl, сульфонатный эфир или активированный эфир, а остальные переменные являются такими же, как описано выше в одиннадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения X1 представляет собой –Br, -I или сульфонатный эфир.
В одном варианте осуществления изобретения способ согласно двадцатому варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1b) с получением соединения формулы (12b);
(2) введение соединения формулы (12b) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (10b’);
(3) введение соединения (10b’) в реакцию с восстанавливающим имин реагентом с получением соединения (7b’);
(4) введение соединения формулы (7b’) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (Ib’).
В другом варианте осуществления изобретения способ согласно двадцатому варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1c) с получением соединения формулы (12c);
(2) введение соединения формулы (12c) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (10c’);
(3) введение соединения (10c’) в реакцию с восстанавливающим имин реагентом с получением соединения (7c’);
(4) введение соединения формулы (7c’) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно двадцатому варианту осуществления изобретения включает получение соединения формулы (IA’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1A) с получением соединения формулы (12A);
(2) введение соединения формулы (12A) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (10A’);
(3) введение соединения (10A’) в реакцию с восстанавливающим имин реагентом с получением соединения (7A’);
(4) введение соединения формулы (7A’) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (IA’).
В конкретном варианте осуществления изобретения X1 представляет собой мезилат.
Условия и реагенты для способа согласно двадцатому варианту осуществления изобретения являются такими, как описано выше в шестнадцатом, семнадцатом, восемнадцатом и/или восьмом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В двадцать первом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента, сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (1),
с получением соединения формулы (12),
;
(2) введение соединения формулы (12) в реакцию с мономерным соединением формулы (d),
с получением соединения формулы (7-1),
;
(3) введение соединения формулы (7-1) в реакцию с мономерным соединением формулы (b),
с получением соединения формулы (18),
; и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’);, где X1 представляет собой –Br, -I, -Cl, сульфонатный эфир или активированный эфир; P3 представляет собой H или защитную группу для аминогруппы, а остальные переменные являются такими же, как описано выше в одиннадцатом варианте осуществления изобретения. В одном варианте осуществления изобретения X1 представляет собой –Br, -I или сульфонатный эфир.
В одном варианте осуществления изобретения способ согласно двадцать первому варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1b) с получением соединения формулы (12b);
(2) введение соединения формулы (12b) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (7b-1);
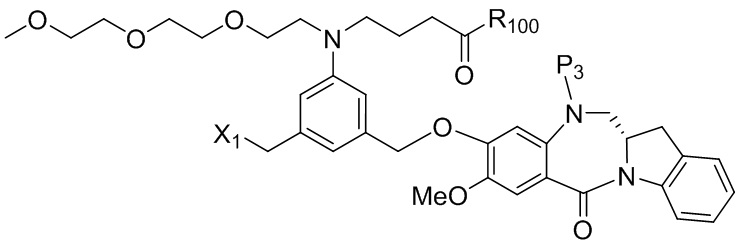 (7b-1)
(7b-1)
(3) введение соединения формулы (7b-1) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (18b); и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом варианте осуществления изобретения способ согласно двадцать первому варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1c) с получением соединения формулы (12c);
(2) введение соединения формулы (12c) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (7c-1);
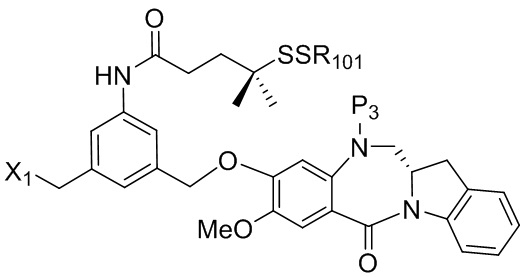 (7c-1)
(7c-1)
(3) введение соединения формулы (7c-1) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (18c); и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно двадцать первому варианту осуществления изобретения включает получение соединения формулы (IA’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента или сульфонирующего реагента в реакцию с соединением формулы (1A) с получением соединения формулы (12A);
(2) введение соединения формулы (12A) в реакцию с мономерным соединением формулы (d1) с получением соединения формулы (7A-1);
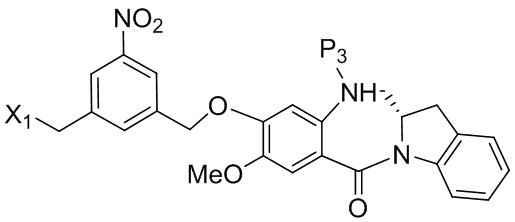 (7A-1)
(7A-1)
(3) введение соединения формулы (7A-1) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (18A); и
(4) если P3 представляет собой защитную группу для аминогруппы, то введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA’).
В одном варианте осуществления изобретения для способов согласно двадцать первому варианту осуществления изобретения P3 представляет собой H
В одном варианте осуществления изобретения P3 представляет собой H, а соединение (7-1) вводят в реакцию с мономерным соединением (b) с получением соединения (I’).
В другом варианте осуществления изобретения P3 представляет собой P2; мономерное соединение представлено формулой (c):
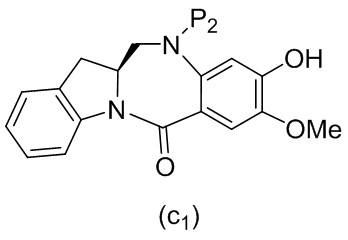 ;
;
а соединение формулы (18) представлено формулой (11),
где P2 представляет собой защитную группу для аминогруппы.
В конкретном варианте осуществления изобретения X1 представляет собой мезилат.
Условия и реагенты для способов согласно двадцать первому варианту осуществления изобретения являются такими, как описано выше в шестнадцатом, семнадцатом, восемнадцатом, восьмом и/или четырнадцатом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В двадцать втором варианте осуществления данного изобретения предложен способ получения соединения формулы (13),
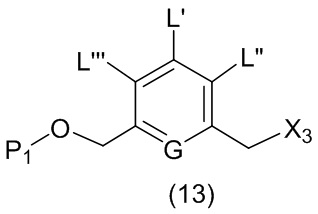
или его соли, причем указанный способ включает введение хлорирующего реагента в реакцию с соединением формулы (2),
где X3 представляет собой Cl, а остальные переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения соединение формулы (2) выбрано из группы, состоящей из:
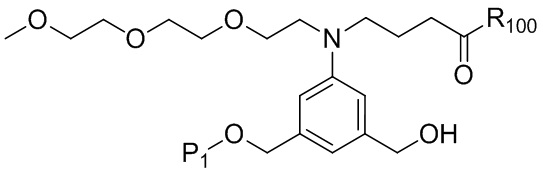 (2b);
(2b); 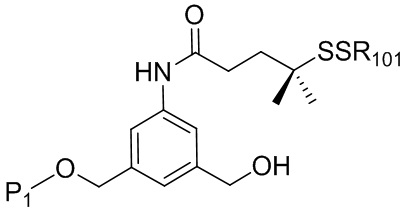 (2c); и
(2c); и  (2A),
(2A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В другом конкретном варианте осуществления изобретения защитная группа для спиртовой группы представляет собой пиволоил, метоксиметил, 2-метоксиэтоксиметил, п-метоксибензил, 3,4-диметиоксибензил, 2,6-диметиоксибензил, дифенилметил, бензилоксиметил, 2,2,2-трихлорэтоксикарбонил, тетрагидрoфуранил, тетрагидрoпиранил, бензил, бензоил, пара-фенилбензоил, 2,4,6-триметилбензоил, пара-бромбензоил, пара-нитробензоил, пиколиноил, никотиноил, 5-дибензoсуберил, тритил/трифенилметил или трис(4-трет-бутилфенил)метил. Предпочтительно защитная группа для спиртовой группы представляет собой метоксиметил, тетрагидрoпиранил, 2-метоксиэтоксиметил, п-метоксибензил, бензилоксиметил или 2,2,2-трихлорэтоксикарбонил. Даже более предпочтительно защитная группа для спиртовой группы представляет собой 2,2,2-трихлорэтоксикарбонил.
В другом конкретном варианте осуществления изобретения защитная группа для спиртовой группы представляет собой силильную защитную группу. Например, силильная защитная группа представляет собой диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, 2-триметиэтилсилил (ТМЭС, (TEOC)) или [2-(триметилсилил)этокси]метил. Предпочтительно силильная защитная группа представляет собой триэтилсилил, триизопропилсилил или трет-бутилдиметилсилил. Более предпочтительно, силильная защитная группа представляет собой трет-бутилдиметилсилил.
В одном варианте осуществления изобретения применяют основание. Основание может быть ненуклеофильным основанием. Примеры ненуклеофильных оснований включают в себя, но не ограничиваясь этим, триэтиламин, имидазол, диизопропилэтиламин, ДИПЭА (DIPEA), пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, (DBU)) или тетраметилпиперидин. Предпочтительно ненуклеофильное основание представляет собой пиридин.
Для способов согласно двадцатому варианту осуществления изобретения можно применять любые подходящие органические растворители. Типичные растворители включают в себя, но не ограничиваясь этим, ДМФА, CH2Cl2, дихлорэтан, ТГФ, диметилацетамид, и т.п. В некоторых вариантах осуществления изобретения в качестве растворителя применяют ДМФА.
В двадцать третьем варианте осуществления данного изобретения предложен способ получения соединения формулы (14),
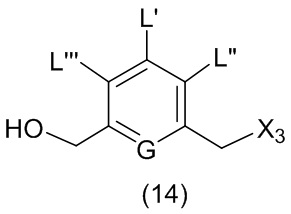
или его соли, причем указанный способ включает введение в реакцию соединения формулы (13)
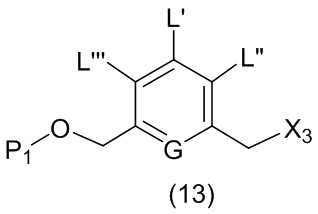
с реагентом, удаляющим защитную группу спиртовой группы, причем переменные являются такими же, как описано в двадцатом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (13) выбрано из группы, состоящей из:
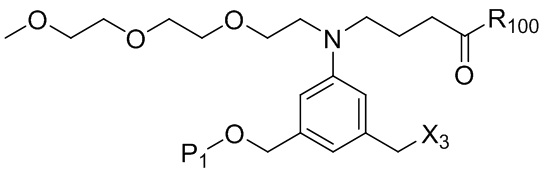 (13b);
(13b); 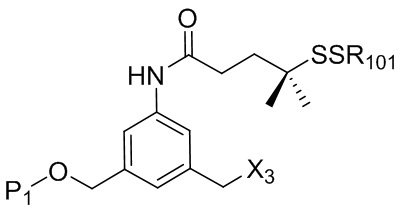 (13c); и
(13c); и  (13A),
(13A),
где R100 и R101 являются такими же, как определено выше.
В другом конкретном варианте осуществления изобретения реагент, удаляющий защитную группу спиртовой группы, представляет собой тетра-н-бутиламмония фторид, трис(диметиламино)сульфония дифтортриметилсиликат, фтороводород или его сольват, пиридина гидрофторид, тетрафторид кремния, гексафторкремниевую кислоту, фторид цезия, хлористоводородную кислоту, уксусную кислоту, трифторуксусную кислоту, пиридиния п-толуолсульфонат, п-толуолсульфоновую кислоту (п-TsOH), муравьиную кислоту или йодную кислоту. Предпочтительно реагент, удаляющий защитную группу спиртовой группы, представляет собой пиридина гидрофторид.
В двадцать четвертом варианте осуществления данного изобретения предложен способ получения соединения формулы (15):
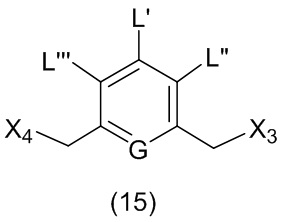
или его соли, причем указанный способ включает введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14),
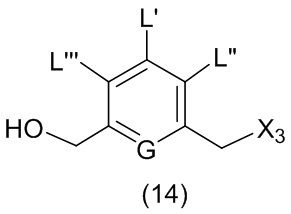 ,
,
где X4 представляет собой сульфонатный эфир или активированный эфир, а остальные переменные являются такими же, как описано в двадцатом варианте осуществления изобретения.
В конкретном варианте осуществления изобретения соединение формулы (14) выбрано из группы, состоящей из:
 (14b);
(14b);  (14c); и
(14c); и 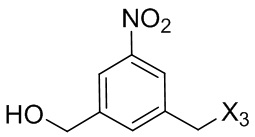 (14A),
(14A),
где R100 и R101являются такими же, как определено выше.
В конкретном варианте осуществления изобретения для способов согласно двадцать четвертому варианту осуществления изобретения, X4 представляет собой сульфонатный эфир.
В другом конкретном варианте осуществления изобретения метансуфонильный ангидрид, метансуфонилхлорид, п-толуолсульфонилхлорид, 4-бромбензолсульфонилхлорид или трифторметансульфонильный ангидрид.
В другом конкретном варианте осуществления изобретения сульфонатный эфир представляет собой мезилат, тозилат, брозилат или трифлат. Предпочтительно сульфонатный эфир представляет собой мезилат.
В другом варианте осуществления изобретения применяют основание. Основание может быть ненуклеофильным основанием. Примеры ненуклеофильных оснований включают в себя, но не ограничиваясь этим, триэтиламин, имидазол, диизопропилэтиламин, пиридин, 2,6-лутидин, диметилформамид, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, (DBU)) или тетраметилпиперидин. Предпочтительно ненуклеофильное основание представляет собой диизопропилэтиламин.
В двадцать пятом варианте осуществления данного изобретения предложен способ получения соединения формулы (20):
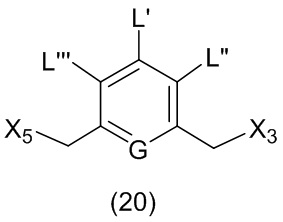
или его соли, причем указанный способ включает введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14),
где переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения соединение формулы (14) выбрано из группы, состоящей из:
(14b), (14c) и 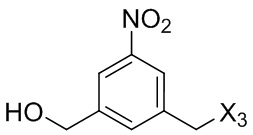 (14A)
(14A)
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения бромирующий или йодирующий реагент представляет собой бром, бромистоводородную кислоту, тетрабромид углерода, трибромид фосфора, бромид калия, йодистоводородную кислоту, йод, тетрайодид углерода, трийодид фосфора, йодид натрия или йодид калия.
В двадцать шестом варианте осуществления данного изобретения предложен способ получения соединения формулы (16):
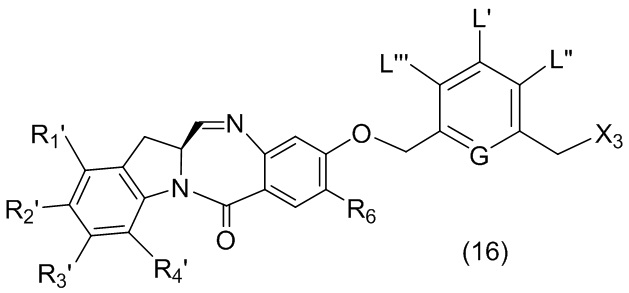 ,
,
или его соли, причем указанный способ включает введение соединения формулы (15)
в реакцию с мономерным соединением формулы (b),
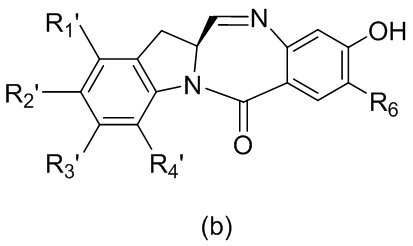 ,
,
где переменные X3, X4, L’, L’’, L’’ и G являются такими, как описано в двадцать пятом варианте осуществления изобретения, а переменные R1’, R2’, R3’, R4’ и R6 являются такими же, как описано выше. В одном варианте осуществления изобретения X4 представляет собой сульфонатный эфир
В конкретном варианте осуществления изобретения соединение формулы (15) выбрано из группы, состоящей из:
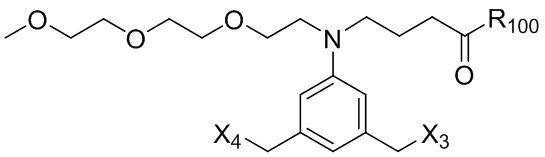 (15b);
(15b);  (15c); и
(15c); и  (15A),
(15A),
а мономерное соединение формулы (b) представлено следующей формулой:
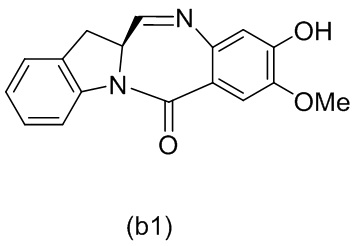 ;
;
где R100 и R101являются такими же, как определено выше.
В варианте осуществления изобретения применяют основание. В конкретном варианте осуществления изобретения основание представляет собой карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. Предпочтительно основание представляет собой карбонат калия.
Для способов согласно двадцатому варианту осуществления изобретения можно применять любые подходящие органические растворители. Типичные растворители включают в себя, но не ограничиваясь этим, ДМФА, CH2Cl2, дихлорэтан, ТГФ, диметилацетамид, и т.п. В некоторых вариантах осуществления изобретения в качестве растворителя применяют диметилацетамид.
В двадцать седьмом варианте осуществления данного изобретения предложен способ получения соединения формулы (16):
,
или его соли, причем указанный способ включает введение в реакцию соединения формулы (20)
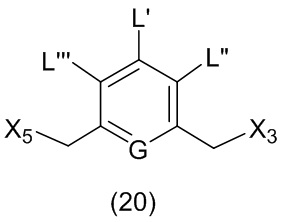
с мономерным соединением формулы (b),
,
где переменные являются такими же, как описано выше.
В одном варианте осуществления изобретения соединение формулы (20) выбрано из группы, состоящей из:
 (20b);
(20b);  (20c); и
(20c); и 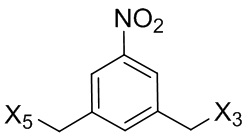 (20A),
(20A),
а мономерное соединение формулы (b) представлено следующей формулой:
где X3 представляет собой –Cl; X5 представляет собой –Br или –I; R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения, соединение формулы (20) вводят в реакцию с мономерным соединением формулы (b) в присутствии основания. Подходящие основания включают карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. В предпочтительном варианте осуществления изобретения основание представляет собой карбонат калия.
В другом варианте осуществления изобретения соединение формулы (20) вводят в реакцию с мономерным соединением формулы (b) в присутствии полярного апротонного растворителя. В предпочтительном варианте осуществления изобретения полярный апротонный растворитель представляет собой диметилацетамид.
В двадцать восьмом варианте осуществления данного изобретения предложен способ получения соединения формулы (16):
,
или его соли, причем указанный способ включает введение в реакцию соединения формулы (14)
с мономерным соединением формулы (b),
,
где переменные являются такими же, как описано выше.
В одном варианте осуществления изобретения соединение формулы (14) выбрано из группы, состоящей из:
(14b); (14c); и 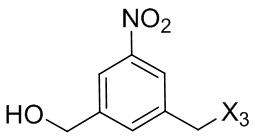 (14A),
(14A),
а мономерное соединение формулы (b) представлено следующей формулой:
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В одном варианте осуществления изобретения для способов согласно двадцать восьмому варианту осуществления изобретения соединение формулы (14) вводят в реакцию с мономером формулы (b) в присутствии активирующего спирт агента. В одном варианте осуществления изобретения активирующий спирт агент представляет собой триалкилфосфин, триарилфосфин или тригетероарилфосфин. В конкретном варианте осуществления изобретения активирующий спирт агент представляет собой триметилфосфин, трибутилфосфин, три(o-толил)фосфин, три(м-толил)фосфин, три(п-толил)фосфин, три(2-пиридил)фосфин, три(3-пиридил)фосфин, три(4-пиридил)фосфин или [4-(3.3.4.4.5.5.6.6.7.7.8.8.9.9.10.10.10-гептадекафтордецил)фенил] дифенилфосфин. В другом варианте осуществления изобретения активирующий спирт агент может представлять собой фосфин-подобный реагент, такой как (трибутилфосфоранилиден)ацетонитрил, (цианометилен)трибутилфосфоран, ЦМТФ (CMBP) или (цианометилен)триметилфосфоран, ЦММФ (CMMP). В более конкретном варианте осуществления изобретения, активирующий спирт агент представляет собой трифенилфосфин. В одном варианте осуществления изобретения активирующий спирт агент может быть связан с полимером или присоединен к полимерной подложке, например, как связанный с полимером или присоединенный к полимерной подложке триалкилфосфин, триарилфосфин (например, трифенилфосфин) или тригетероарилфосфин.
В другом варианте осуществления изобретения для способов согласно двадцать восьмому варианту осуществления изобретения, соединение формулы (14) вводят в реакцию с мономером формулы (b) в присутствии азодикарбоксилата. В одном варианте осуществления изобретения азодикарбоксилат выбран из группы, состоящей из: диэтилазодикарбоксилата (ДЭАД (DEAD)), диизопропилазодикарбоксилата (ДИАД (DIAD)), 1,1’-(азодикарбонил)дипиперидина (АДДП (ADDP)), дитрет-бутил-азодикарбоксилата, ДТАД (DTAD), 1,6-диметил-1,5,7-гексагидрo-1,4,6,7-тетразоцин-2,5-диона, ДГТД (DHTD), ди-(4-хлорбензил)азодикарбоксилата, ДХАД (DCAD), азодикарбоксилдиморфолида, N,N,N',N'- тетраметилазодикарбоксамида, ТМАД (TMAD), N,N,N',N'- тетраизопропилазодикарбоксамида, ТИПА (TIPA), 4,4′-азопиридина, бис(2,2,2-трихлорэтил)азодикарбоксилата, о-(трет-бутилдиметилсилил)-N-тозилгидроксиламина, ди-(4-хлорбензил)азодикарбоксилата, циклического 1,6-диметил-1,5,7-гексагидрo-1,4,6,7-тетразоцин-2,5-диона, ДГТД (DHTD), диметилацетилендикарбоксилата, ДМАД (DMAD), ди-2-метоксиэтилазодикарбоксилата, ди-(4-хлорбензил)азодикарбоксилата и бис(4,4,5,5,6,6,7,7,8,8,9,9,9-тридекафторнонил)азодикарбоксилата. Более конкретно, азодикарбоксилат представляет собой ДИАД (DIAD). В одном варианте осуществления изобретения азодикарбоксилат связан с полимером или присоединен к полимерной подложке, например, как присоединенный к полимерной подложке алкилазодикарбоксилат (например, связанный с полимером ДЭАД (DEAD), ДИАД (DIAD), ДТАД (DTAD) или АДДП (ADDP)).
В еще другом конкретном варианте осуществления изобретения для способов согласно двадцать восьмому варианту осуществления изобретения, соединение формулы (14) вводят в реакцию с мономером формулы (b) в присутствии трифенилфосфина и азодикарбоксилата. В одном варианте осуществления изобретения азодикарбоксилат выбран из группы, состоящей из: диэтилазодикарбоксилата (ДЭАД (DEAD)), диизопропилазодикарбоксилата (ДИАД (DIAD)), 1,1’-(азодикарбонил)дипиперидина (АДДП (ADDP)) и ди-трет-бутилазодикарбоксилата (ДТАД (DTAD)). Более конкретно, азодикарбоксилат представляет собой ДИАД (DIAD).
В двадцать девятом варианте осуществления данного изобретения предложен способ получения соединения формулы (18):
,
его фармацевтически приемлемой соли, причем указанный способ включает введение соединения формулы (16):
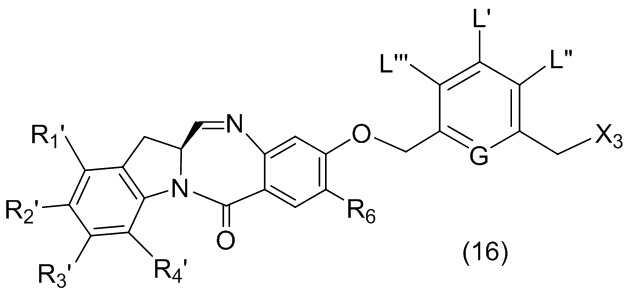
в реакцию с восстановленным мономером формулы (d):
,
где переменные R1, R2, R3, R4, R6 и P2 являются такими, как описано в двадцать третьем варианте осуществления изобретения; и где
каждый R1, R2, R3 и R4 независимо выбран из группы, состоящей из: -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n-Rc, галогена, гуанидиния [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NCO, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3-H, -OSO3H, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’ и -OCONR’R’’;
R6 представляет собой -H, -R, -OR, -SR, -NR’R’’, -NO2 или галоген; и
P3 представляет собой H или защитную группу для аминогруппы.
В конкретном варианте осуществления изобретения соединение формулы (16) выбрано из группы, состоящей из:
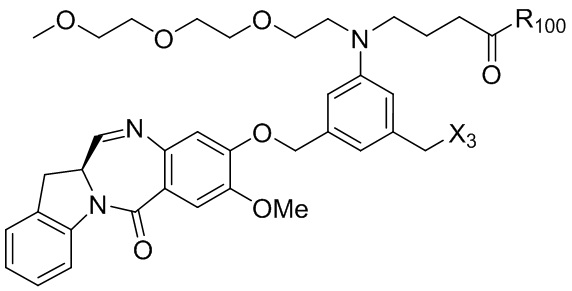 (16b);
(16b);
 (16c); и
(16c); и 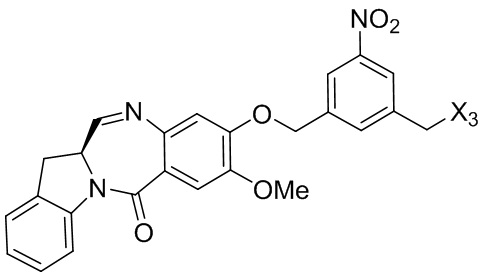 (16A),
(16A),
а восстановленный мономер формулы (d) представлен следующей формулой:
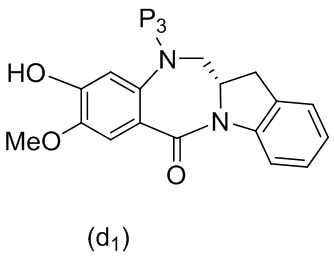
где X3 представляет собой –Cl; P3 представляет собой H или защитную группу для аминогруппы; R100 и R101 являются такими же, как определено выше.
В одном варианте осуществления изобретения для способов согласно двадцать девятому варианту осуществления изобретения, реакцию между соединением формулы (16d) или (16A) и восстановленным мономером формулы (d1) проводят в присутствии основания. В конкретном варианте осуществления изобретения основание представляет собой карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. Предпочтительно основание представляет собой карбонат калия.
Для способов согласно двадцатому варианту осуществления изобретения можно применять любые подходящие органические растворители. Типичные растворители включают в себя, но не ограничиваясь этим, диметилформамид (ДМФА), CH2Cl2, дихлорэтан, ТГФ, диметилацетамид, и т.п. В некоторых вариантах осуществления изобретения в качестве растворителя применяют диметилформамид или диметилацетамид.
В конкретном варианте двадцать четвертого варианта осуществления изобретения соединение формулы (16) вводят в реакцию с восстановленным мономером формулы (d), где P3 представляет собой H, с получением соединения формулы (I’):
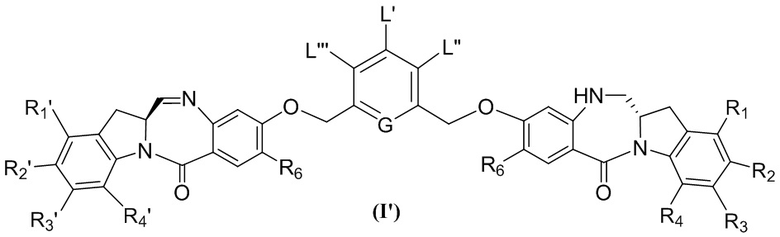 .
.
В другом конкретном варианте двадцать четвертого варианта осуществления изобретения P3 представляет собой защитную группу для аминогруппы. В описанном выше способе можно применять любую подходящую защитную группу для аминогруппы. В одном варианте осуществления изобретения защитная группа для аминогруппы представляет собой 2-триметилсилилэтил, (2-фенил-2-триметилсилил)этил, триизопропилсилокси, 2-(триметилсилил)этоксиметил, аллилоксикарбонил, 9-флуоренилметоксикарбонил, 2-(триметилсилил)этоксикарбонил или 2,2,2,2-трихлорэтоксикарбонил.
Если P3 представляет собой защитную группу для аминогруппы, то соединение формулы (18) дополнительно вводят в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’):
.
Примеры подходящих реагентов для удаления защитной группы аминогруппы включают в себя, но не ограничиваясь этим, реагент, удаляющий защитную группу аминогруппы, который выбран из группы, состоящей из: тетра-н-бутиламмония фторида, уксусной кислоты, пиридина гидрофторида, фторида цезия, пиперидина, морфолина или трифторуксусной кислоты.
В тридцатом варианте осуществления данного изобретения предложен способ получения соединение формулы (17):
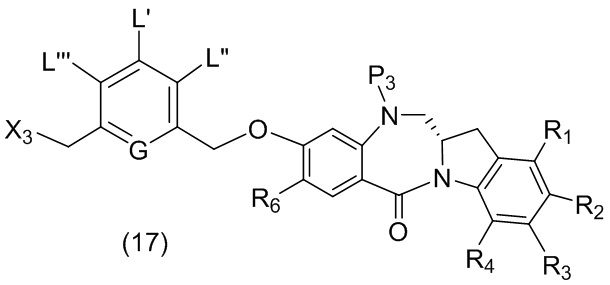
или его соли, причем указанный способ включает введение в реакцию соединения формулы (15)
с мономерным соединением формулы (d),
,
где X3 представляет собой –Cl; X4 представляет собой сульфонатный эфир или активированный эфир; P3 представляет собой H или защитную группу для аминогруппы; L’, L’’, L’’ и G являются такими, как описано в двадцать втором варианте осуществления изобретения, а переменные R1, R2, R3, R4, R6 и P3 являются такими же, как описано в двадцать четвертом варианте осуществления изобретения. В одном варианте осуществления изобретения X4 представляет собой активированный эфир.
В конкретном варианте осуществления изобретения соединение формулы (15) выбрано из группы, состоящей из:
(15b); (15c); и  (15A),
(15A),
где X3 представляет собой –Cl; X4 представляет собой сульфонатный эфир или активированный эфир; R100 и R101 являются такими же, как определено выше. В одном варианте осуществления изобретения X4 представляет собой активированный эфир.
В варианте осуществления изобретения для способов согласно тридцатому варианту осуществления изобретения применяют основание. В конкретном варианте осуществления изобретения основание представляет собой карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. Предпочтительно основание представляет собой карбонат калия.
Можно применять любые подходящие органические растворители для способов согласно тридцатому варианту осуществления изобретения. Типичные растворители включают в себя, но не ограничиваясь этим, ДМФА, CH2Cl2, дихлорэтан, ТГФ, диметилацетамид, и т.п. В некоторых вариантах осуществления изобретения в качестве растворителя применяют диметилацетамид.
В конкретном варианте тридцатого варианта осуществления изобретения, P3 представляет собой H, а соединение формулы (15) вводят в реакцию с мономерным соединением формулы (d) с получением соединения формулы (17’).
В другом конкретном варианте тридцатого варианта осуществления изобретения, P3 представляет собой защитную группу для аминогруппы, а способ дополнительно включает стадию введения соединения формулы (17) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (17’):
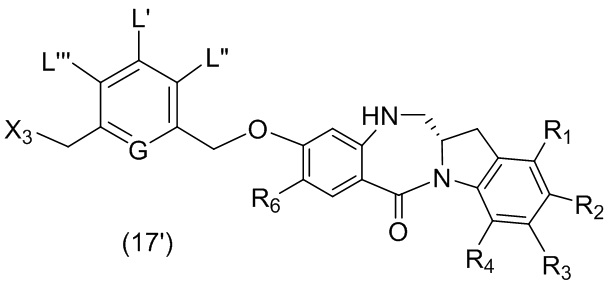 .
.
Примеры подходящего реагента для удаления защитной группы аминогруппы включают в себя, но не ограничиваясь этим, реагент, удаляющий защитную группу аминогруппы, выбранный из группы, состоящей из: тетра-н-бутиламмония фторида, уксусной кислоты, пиридина гидрофторида, фторида цезия, пиперидина, морфолина или трифторуксусной кислоты.
В конкретном варианте осуществления изобретения соединение формулы (17) выбрано из группы, состоящей из:
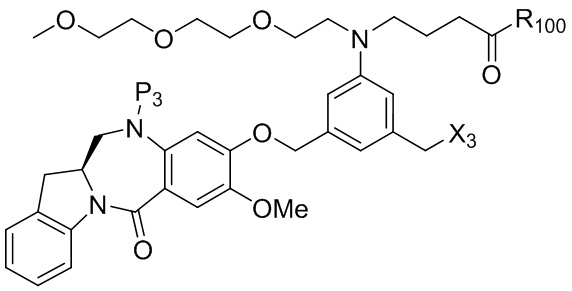 (17b);
(17b);
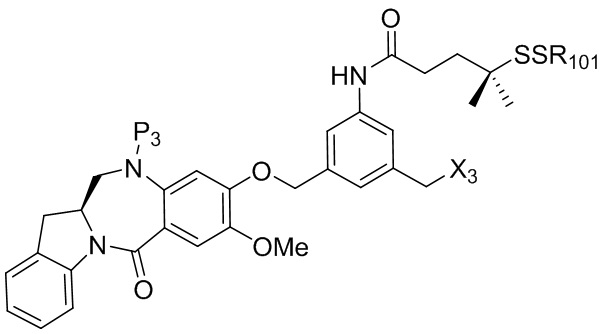 (17c); и
(17c); и 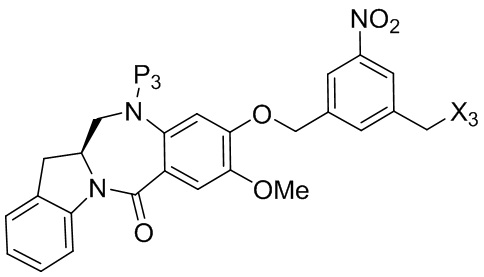 (17A),
(17A),
где R100 и R101 являются такими же, как определено выше.
В тридцать первом варианте осуществления данного изобретения предложен способ получения соединения формулы (17):
или его соли, причем указанный способ включает введение соединения формулы (14)
в реакцию с мономерным соединением формулы (d),
,
где X3 представляет собой –Cl; P3 представляет собой H или защитную группу для аминогруппы и остальные переменные являются такими, как описано выше.
В конкретном варианте осуществления изобретения соединение формулы (14) выбрано из группы, состоящей из:
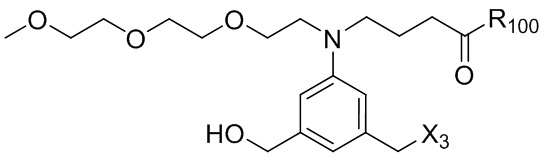 (14b);
(14b);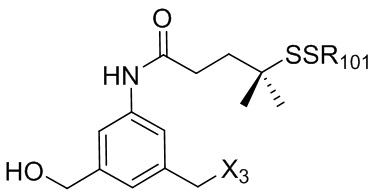 (14c);
(14c); 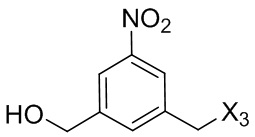 (14A),
(14A),
а мономерное соединение формулы (d) представлено формулой (d1), где X3 представляет собой –Cl; R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В конкретном варианте осуществления изобретения соединение формулы (14) вводят в реакцию с мономером формулы (b) в присутствии активирующего спирт агента. В одном варианте осуществления изобретения активирующий спирт агент представляет собой триалкилфосфин, триарилфосфин или тригетероарилфосфин. В конкретном варианте осуществления изобретения активирующий спирт агент представляет собой триметилфосфин, трибутилфосфин, три(o-толил)фосфин, три(м-толил)фосфин, три(п-толил)фосфин, три(2-пиридил)фосфин, три(3-пиридил)фосфин, три(4-пиридил)фосфин или [4-(3.3.4.4.5.5.6.6.7.7.8.8.9.9.10.10.10-гептадекафтордецил)фенил]дифенилфосфин. В другом варианте осуществления изобретения активирующий спирт агент может представлять собой фосфин-подобный реагент, такой как (трибутилфосфоранилиден)ацетонитрил, (цианометилен)трибутилфосфоран, ЦМБФ (CMBP) или (цианометилен)триметилфосфоран, ЦММФ (CMMP). В более конкретном варианте осуществления изобретения, активирующий спирт агент представляет собой трифенилфосфин. В одном варианте осуществления изобретения активирующий спирт агент может быть связан с полимером или присоединен к полимерной подложке, например, как связанный с полимером или присоединенный к полимерной подложке триалкилфосфин, триарилфосфин (например, трифенилфосфин) или тригетероарилфосфин.
В другом конкретном варианте осуществления изобретения соединение формулы (14) вводят в реакцию с мономером формулы (b) в присутствии азодикарбоксилата. В одном варианте осуществления изобретения азодикарбоксилат выбран из группы, состоящей из: диэтилазодикарбоксилата (ДЭАД (DEAD)), диизопропилазодикарбоксилата (ДИАД (DIAD)), 1,1’-(азодикарбонил)дипиперидина (АДДП (ADDP)), ди-трет-бутилазодикарбоксилата, ДТАД (DTAD), 1,6-диметил-1,5,7-гексагидрo-1,4,6,7-тетразоцин-2,5-диона, ДГТД (DHTD), ди-(4-хлорбензил)азодикарбоксилата, ДХАД (DCAD), азодикарбоксилдиморфолида, N,N,N',N'- тетраметилазодикарбоксамида, ТМАД (TMAD), N,N,N',N'- тетраизопропилазодикарбоксамида, ТИПА (TIPA), 4,4′-азопиридина, бис(2,2,2-трихлорэтил)азодикарбоксилата, о-(трет-бутилдиметилсилил)-N-тозилгидроксиламина, ди-(4-хлорбензил)азодикарбоксилата, циклического 1,6-диметил-1,5,7-гексагидрo-1,4,6,7-тетразоцин-2,5-диона, ДГТД (DHTD), диметилацетилендикарбоксилата, ДМАД (DMAD), ди-2-метоксиэтилазодикарбоксилата, ди-(4-хлорбензил)азодикарбоксилата и бис(4,4,5,5,6,6,7,7,8,8,9,9,9-тридекафторнонил)азодикарбоксилата. Более конкретно, азодикарбоксилат представляет собой ДИАД (DIAD). В одном варианте осуществления изобретения азодикарбоксилат связан с полимером или присоединен к полимерной подложке, например, как присоединенный к полимерной подложке алкилазодикарбоксилат (например, связанный с полимером ДЭАД (DEAD), ДИАД (DIAD), ДТАД (DTAD) или АДДП (ADDP)).
В еще другом конкретном варианте осуществления изобретения для способов согласно двадцать восьмому варианту осуществления изобретения, соединение формулы (14) вводят в реакцию с мономером формулы (d) в присутствии трифенилфосфина и азодикарбоксилата. В одном варианте осуществления изобретения азодикарбоксилат выбран из группы, состоящей из: диэтилазодикарбоксилата (ДЭАД (DEAD)), диизопропилазодикарбоксилата (ДИАД (DIAD)), 1,1’-(азодикарбонил)дипиперидина (АДДП (ADDP)) и ди-трет-бутилазодикарбоксилата (ДТАД (DTAD)). Более конкретно, азодикарбоксилат представляет собой ДИАД (DIAD).
В еще другом варианте осуществления изобретения соединение формулы (15) вводят в реакцию с мономерным соединением формулы (d), где P3 представляет собой H, с получением соединения формулы (17’):
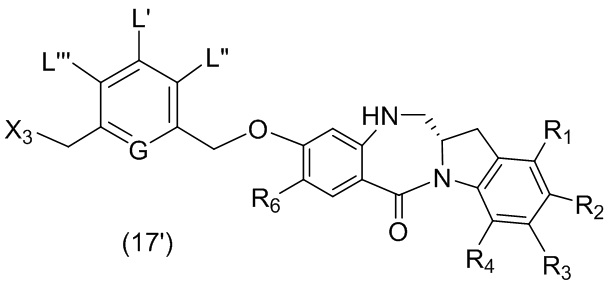 .
.
В конкретном варианте осуществления изобретения P3 представляет собой защитную группу для аминогруппы, а способ дополнительно включает стадию введения соединения формулы (17) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (17’):
.
В конкретном варианте осуществления изобретения соединение формулы (17) выбрано из группы, состоящей из:
(17b);
(17c); и 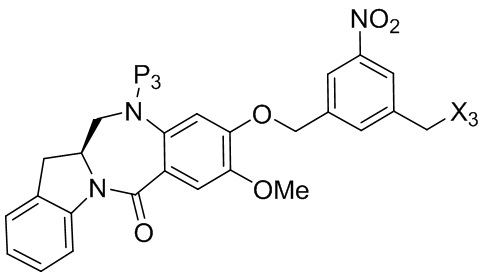 (17A),
(17A),
где R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
Примеры подходящего реагента для удаления защитной группы аминогруппы включают в себя, но не ограничиваясь этим, тетра-н-бутиламмония фторид, уксусную кислоту, пиридина гидрофторид, фторид цезия, пиперидин, морфолин или трифторуксусную кислоту.
В тридцать втором варианте осуществления данного изобретения предложен способ получения соединения формулы (17):
или его соли, причем указанный способ включает введение соединения формулы (20)
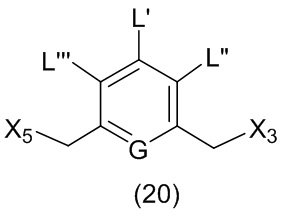
в реакцию с мономерным соединением формулы (d),
,
где X3 представляет собой –Cl; X5 представляет собой –Br или –I, P3 представляет собой H или защитную группу для аминогруппы; а остальные переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения соединение формулы (20) выбрано из группы, состоящей из:
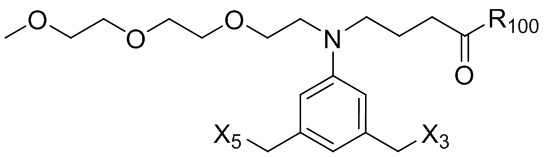 (20b);
(20b); 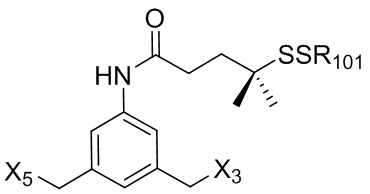 (20c); и
(20c); и  (20A),
(20A),
где X3 представляет собой –Cl; X5 представляет собой –Br или –I; R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В одном варианте осуществления изобретения соединение формулы (20) вводят в реакцию с мономерным соединением формулы (d) в присутствии основания. Можно применять любое подходящее основание. Подходящие основания включают в себя, но не ограничиваясь этим, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. Предпочтительно основание представляет собой карбонат калия.
В другом варианте осуществления изобретения соединение формулы (20) вводят в реакцию с мономерным соединением формулы (d) в присутствии полярного апротонного растворителя. Предпочтительно полярный апротонный растворитель представляет собой диметилацетамид.
В конкретном варианте осуществления изобретения соединение формулы (20) вводят в реакцию с мономерным соединением формулы (d), где P3 представляет собой H, с получением соединения формулы (17’):
.
В другом конкретном варианте осуществления изобретения P3 представляет собой защитную группу для аминогруппы, а способ дополнительно включает стадию введения в реакцию соединения формулы (17) с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (17’):
.
В одном варианте осуществления изобретения соединение формулы (17) выбрано из группы, состоящей из:
(17b);
(17c); и 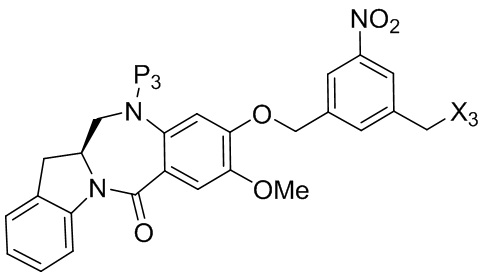 (17A),
(17A),
где X3 представляет собой –Cl; P3 представляет собой H или защитную группу для аминогруппы; R100 представляет собой (C1-C3)алкокси; и R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
Подходящие реагенты для удаления защитной группы аминогруппы включают в себя, но не ограничиваясь этим, тетра-н-бутиламмония фторид, уксусную кислоту, пиридина гидрофторид, фторид цезия, пиперидин, морфолин или трифторуксусную кислоту.
В тридцать третьем варианте осуществления данного изобретения предложен способ получения соединения формулы (17’):
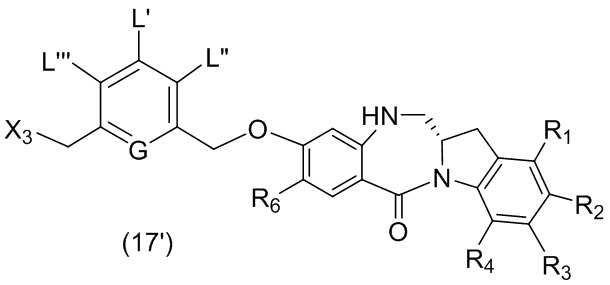 ,
,
или его соли, причем указанный способ включает введение в реакцию соединения формулы (19)
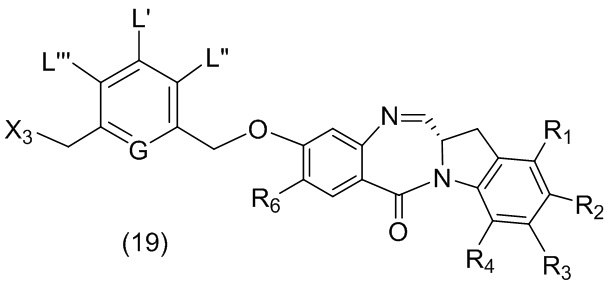 ,
,
где X3 представляет собой –Cl; остальные переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения соединение формулы (19) выбрано из группы, состоящей из:
 (16b);
(16b);
 (16c); и
(16c); и 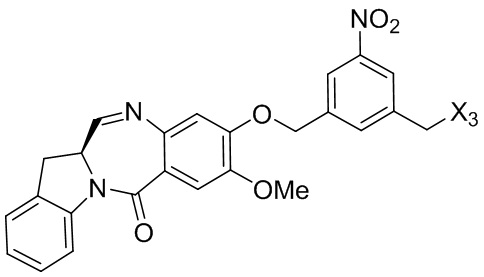 (16A)
(16A)
где X3 представляет собой –Cl; R100 и R101 являются такими же, как определено выше.
В другом конкретном варианте осуществления изобретения восстанавливающий имин агент представляет собой гидридный восстановительный агент. Примеры подходящих гидридных восстановительных агентов включают в себя, но не ограничиваясь этим, боргидрид oдия, триацетоксиборгидрид натрия, цианоборгидрид натрия, гидрид лития алюминия, газообразный водород, формиат аммония, боран, 9-борабицикло[3.3.1]нонан (9-ББН (9-BBN)), диизобутилалюминия гидрид (ДИБАЛ (DIBAL)), боргидрид лития (LiBH4), боргидрид калия (KBH4) или натрия бис(2-метоксиэтокси)алюминия гидрид (Red-Al). В одном конкретном варианте осуществления изобретения, гидридный восстановительный агент представляет собой натрия триацетоксиборгидрид (NaBH(OAc)3).
В тридцать четвертом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает введение в реакцию соединения формулы (17):
с мономером формулы (b):
где X3 представляет собой –Cl; P3 представляет собой H или защитную группу для аминогруппы; а остальные переменные являются такими же, как описано выше.
В одном варианте осуществления изобретения соединение формулы (17) вводят в реакцию с мономерным соединением формулы (b) в присутствии основания. Подходящие основания включают в себя, но не ограничиваясь этим, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия. Предпочтительно основание представляет собой карбонат калия.
В другом варианте осуществления изобретения соединение формулы (17) вводят в реакцию с мономерным соединением формулы (b) в присутствии полярного апротонного растворителя. Предпочтительный полярный апротонный растворитель включает диметилформамид или диметилацетамид.
В еще другом варианте осуществления изобретения, соединение формулы (17) вводят в реакцию с восстановленным мономером формулы (b), где P3 представляет собой H, с получением соединения формулы (I’):
.
В конкретном варианте осуществления изобретения P3 представляет собой защитную группу для аминогруппы. Подходящие защитные группы для аминогруппы включают в себя, но не ограничиваясь этим, 2-триметилсилилэтил,(2-фенил-2-триметилсилил)этил, триизопропилсилокси, 2-(триметилсилил)этоксиметил, аллилоксикарбонил, 9-флуоренилметоксикарбонил, 2-(триметилсилил)этоксикарбонил или 2,2,2,2-трихлорэтоксикарбонил.
В еще другом варианте осуществления изобретения, если P3 представляет собой защитную группу для аминогруппы, то соединение формулы (18) дополнительно вводят в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’):
.
Примеры подходящего реагента для удаления защитной группы аминогруппы включают в себя, но не ограничиваясь этим, реагент, удаляющий защитную группу аминогруппы, выбранный из группы, состоящей из: тетра-н-бутиламмония фторида, уксусной кислоты, пиридина гидрофторида, фторида цезия, пиперидина, морфолина или трифторуксусной кислоты.
В тридцать пятом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14),
с получением соединения формулы (15):
,
или его соли;
(2) введение соединения формулы (15) в реакцию с мономерным соединением формулы (b),
,
с получением соединения формулы (16):
,
или его соли;
(3) введение соединения формулы (16) в реакцию с восстановленным мономером формулы (d):
с получением соединения формулы (18), или его фармацевтически приемлемой соли, где X3 представляет собой –Cl; X4 представляет собой сульфонатный эфир или активированный эфир; P1 представляет собой защитную группу для спиртовой группы; P3 представляет собой H или защитную группу для аминогруппы; и остальные переменные являются такими же, как описано выше. В одном варианте осуществления изобретения X4 представляет собой сульфонатный эфир.
В конкретном варианте осуществления изобретения способ согласно тридцать пятому варианту осуществления изобретения включает способ получения соединения формулы (Ib’), причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (15b);
(2) введение соединения формулы (15b) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (16b);
(3) введение соединения формулы (16b) в реакцию с восстановленным мономером формулы (d) с получением соединения формулы (18b) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16b) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом конкретном варианте осуществления изобретения способ согласно тридцать пятому варианту осуществления изобретения включает способ получения соединения формулы (Ic’), причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14c) с получением соединения формулы (15c);
(2) введение соединения формулы (15c) в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (16c);
(3) введение соединения формулы (16c) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18c) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16c) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В еще другом конкретном варианте осуществления изобретения способ согласно тридцать пятому варианту осуществления изобретения включает способ получения соединения формулы (IA), причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (15A);
(2) введение в реакцию соединения формулы (15A) с мономерным соединением формулы (b1) с получением соединения формулы (16A);
(3) введение в реакцию соединения формулы (16A) с восстановленным мономером формулы (d1) с получением соединения формулы (18A) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16A) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (IA); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA).
Условия и реагенты для способов согласно тридцать пятому варианту осуществления изобретения являются такими, как описано выше в двадцать четвертом, двадцать шестом и/или двадцать девятом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В тридцать шестом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение в реакцию соединения формулы (14):
с мономерным соединением формулы (b),
,
с получением соединения формулы (16):
,
или его соли; и
(2) введение в реакцию соединения формулы (16) с восстановленным мономером формулы (d):
с получением соединения формулы (18) или его фармацевтически приемлемой соли, где X3 представляет собой –Cl; P1 представляет собой защитную группу для спиртовой группы; P3 представляет собой H или защитную группу для аминогруппы; а остальные переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения способ согласно тридцать шестому варианту осуществления изобретения включает способ получения соединения формулы (Ib’), причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14b) в реакцию с мономерным соединением формулы (b1), в присутствии активирующего спирт агента с получением соединения формулы (16b);
(2) введение соединения формулы (16b) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18b) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16b) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение в реакцию соединения формулы (18b) с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом конкретном варианте осуществления изобретения способ согласно тридцать шестому варианту осуществления изобретения включает способ получения соединения формулы (Ic’), причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14c) в реакцию с мономерным соединением формулы (b1) в присутствии активирующего спирт агента с получением соединения формулы (16c);
(2) введение соединения формулы (16c) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18c) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16c) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ic’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом конкретном варианте осуществления изобретения способ согласно тридцать шестому варианту включает способ получения соединения формулы (IA), причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14A) в реакцию с мономерным соединением формулы (b1) в присутствии активирующего спирт агента с получением соединения формулы (16A);
(2) введение соединения формулы (16A) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18A) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16A) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (IA); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA).
Условия и реагенты для способа согласно тридцать шестому варианту осуществления изобретения являются такими, как описано выше в двадцать восьмом и/или двадцать девятом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В тридцать седьмом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента в реакцию с соединением формулы (14),
с получением соединения формулы (20):
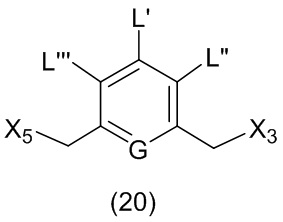 ,
,
или его соли;
(2) введение соединения формулы (20) или его соли в реакцию с мономерным соединением формулы (b),
,
с получением соединения формулы (16):
,
или его соли; и
(3) введение соединения формулы (16) в реакцию с восстановленным мономером формулы (d):
с получением соединения формулы (18) или его фармацевтически приемлемой соли, где X3 представляет собой –Cl; X5 представляет собой –Br или -I; P3 представляет собой H или защитную группу для аминогруппы; и остальные переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения способ согласно тридцать седьмому варианту включает способ получения соединения формулы (Ib’), причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (20b);
(2) введение соединения формулы (20b) или его соли в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (16b)
(3) введение соединения формулы (16b) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18b) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16b) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом конкретном варианте осуществления изобретения способ согласно тридцать седьмому варианту включает способ получения соединения формулы (Ic’), причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента в реакцию с соединением формулы (14c) с получением соединения формулы (20c);
(2) введение соединения формулы (20c) или его соли в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (16c)
(3) введение соединения формулы (16c) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18c) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (16c) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом конкретном варианте осуществления изобретения способ согласно тридцать седьмому варианту осуществления изобретения включает способ получения соединения формулы (IA), причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента в реакцию с соединением формулы (14A) с получением соединения формулы (20A);
(2) введение соединения формулы (20A) или его соли в реакцию с мономерным соединением формулы (b1) с получением соединения формулы (16A)
(3) введение соединения формулы (16A) в реакцию с восстановленным мономером формулы (d1) с получением соединения формулы (18A) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то вводят соединение (16A) в реакцию с восстановленным мономером (d1) с получением соединения формулы (IA); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA).
Условия и реагенты для способа согласно тридцать седьмому варианту осуществления изобретения являются такими, как описано выше в двадцать пятом, двадцать седьмом и/или двадцать девятом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В конкретном варианте тридцать пятого, тридцать шестого или тридцать седьмого варианта осуществления изобретения, соединение формулы (16) вводят в реакцию с восстановленным мономером формулы (d), где P3 представляет собой H, с получением соединения формулы (I’):
.
В другом конкретном варианте тридцать пятого, тридцать шестого или тридцать седьмого варианта осуществления изобретения, P3 представляет собой защитную группу для аминогруппы, а соединение формулы (18) дополнительно вводят в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’):
.
В тридцать восьмом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14),
с получением соединения формулы (15):
,
или его соли;
(2) введение соединения формулы (15) в реакцию с восстановленным мономерным соединением формулы (d),
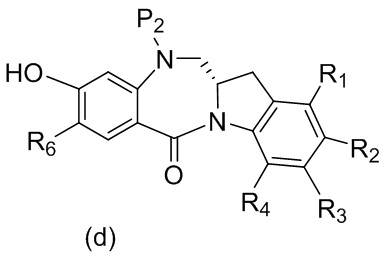 ,
,
с получением соединения формулы (17):
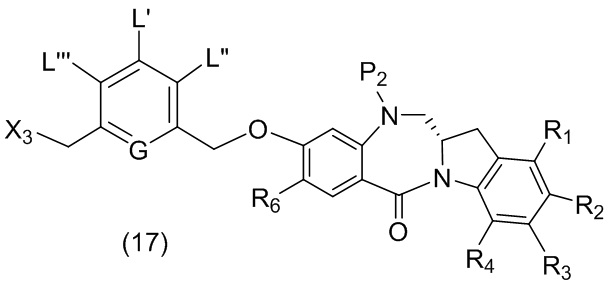 ,
,
или его соли; и
(3) введение соединения формулы (17) в реакцию с мономером формулы (b):
с получением соединения формулы (18) или его фармацевтически приемлемой соли, где X3 представляет собой –Cl; X4 представляет собой сульфонатный эфир или активированный эфир; P1 представляет собой защитную группу для спиртовой группы; P3 представляет собой H или защитную группу для аминогруппы; остальные переменные являются такими же, как описано выше. В одном варианте осуществления изобретения X4 представляет собой сульфонатный эфир.
В конкретном варианте осуществления изобретения способ согласно тридцать восьмому варианту осуществления изобретения включает способ получения соединения формулы (Ib’), причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (15b);
(2) введение соединения формулы (15b) или его соли в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17b)
(3) введение соединения формулы (17b) в реакцию с мономером формулы (b1) с получением соединения формулы (18b) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17b) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом конкретном варианте осуществления изобретения способ согласно тридцать восьмому варианту осуществления изобретения включает способ получения соединения формулы (Ic’), причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (15c);
(2) введение соединения формулы (15c) или его соли в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17c)
(3) введение соединения формулы (17c) в реакцию с мономером формулы (b1) с получением соединения формулы (18c) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17c) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ic’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом конкретном варианте осуществления изобретения способ согласно тридцать восьмому варианту осуществления изобретения включает способ получения соединения формулы (IA), причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14A) с получением соединения формулы (15A);
(2) введение соединения формулы (15A) или его соли в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17A)
(3) введение соединения формулы (17A) в реакцию с мономером формулы (b1) с получением соединения формулы (18A) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17A) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (IA); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA).
Условия и реагенты для способа согласно тридцать восьмому варианту осуществления изобретения являются такими, как описано выше в двадцать пятом, тридцатом и/или тридцать четвертом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В тридцать девятом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение в реакцию соединения формулы (14):
с восстановленным мономерным соединением формулы (d),
,
с получением соединения формулы (17):
,
или его соли; и
(2) введение в реакцию соединения формулы (17) с мономером формулы (b):
с получением соединения формулы (18), или его фармацевтически приемлемой соли, где X3 представляет собой –Cl; P1 представляет собой защитную группу для спиртовой группы; P3 представляет собой H или защитную группу для аминогруппы;, а остальные переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения способ согласно тридцать девятому варианту осуществления изобретения включает способ получения соединения формулы (Ib’), причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14b) в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17b)
(2) введение соединения формулы (17b) в реакцию с мономером формулы (b1) с получением соединения формулы (18b) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17b) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом конкретном варианте осуществления изобретения способ согласно тридцать девятому варианту осуществления изобретения включает способ получения соединения формулы (Ic’), причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14c) в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17c)
(2) введение соединения формулы (17c) в реакцию с мономером формулы (b1) с получением соединения формулы (18c) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17c) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ic’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом конкретном варианте осуществления изобретения способ согласно тридцать девятому варианту включает способ получения соединения формулы (IA), причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14A) в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17A)
(2) введение соединения формулы (17A) в реакцию с мономером формулы (b1) с получением соединения формулы (18A) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17A) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (IA’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA).
Условия и реагенты для способа согласно тридцать девятому варианту осуществления изобретения являются такими, как описано выше в тридцать первом и/или тридцать четвертом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В сороковом варианте осуществления данного изобретения предложен способ получения соединения формулы (18),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение галогенирующего реагента в реакцию с соединением формулы (14):
,
с получением соединения формулы (20):
,
или его соли;
(2) введение соединения формулы (20) в реакцию с восстановленным мономерным соединением формулы (d),
,
с получением соединения формулы (17):
,
или его соли; и
(3) введение соединения формулы (17) в реакцию с мономером формулы (b):
с получением соединения формулы (18), или его фармацевтически приемлемые соли, где X3 представляет собой –Cl; X5 представляет собой –Br или –I; P1 представляет собой защитную группу для спиртовой группы; P3 представляет собой H или защитную группу для аминогруппы; и переменные являются такими же, как описано выше.
В конкретном варианте осуществления изобретения способ согласно сороковому варианту осуществления изобретения включает способ получения соединения формулы (Ib’), причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (20b);
(2) введение соединения формулы (20b) в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17b)
(3) введение соединения формулы (17b) в реакцию с мономером формулы (b1) с получением соединения формулы (18b) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то соединение (17b) вводят в реакцию с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18b) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
В другом конкретном варианте осуществления изобретения способ согласно сороковому варианту осуществления изобретения включает способ получения соединения формулы (Ic’), причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14c) с получением соединения формулы (20c);
(2) введение соединения формулы (20c) в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17c)
(3) введение соединения формулы (17c) в реакцию с мономером формулы (b1) с получением соединения формулы (18c) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то вводят в реакцию соединение (17c) с восстановленным мономером (d1) с получением соединения формулы (Ic’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18c) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ic’).
В еще другом конкретном варианте осуществления изобретения способ согласно сороковому варианту осуществления изобретения включает способ получения соединения формулы (IA), причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14A) с получением соединения формулы (20A);
(2) введение соединения формулы (20A) в реакцию с восстановленным мономерным соединением формулы (d1) с получением соединения формулы (17A)
(3) введение соединения формулы (17A) в реакцию с мономером формулы (b1) с получением соединения формулы (18A) или его фармацевтически приемлемой соли, причем если P3 представляет собой H, то вводят в реакцию соединение (17A) с восстановленным мономером (d1) с получением соединения формулы (Ib’); а если P3 представляет собой защитную группу для аминогруппы, то способ дополнительно включает введение соединения формулы (18A) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (Ib’).
Условия и реагенты для способа согласно сороковому варианту осуществления изобретения являются такими, как описано выше в двадцать пятом, тридцать втором и/или тридцать четвертом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В конкретном варианте тридцать восьмого, тридцать девятого или сорокового варианта осуществления изобретения, соединение формулы (17) вводят в реакцию с восстановленным мономером формулы (d), где P3 представляет собой H, с получением соединения формулы (I’):
.
В другом конкретном варианте тридцать шестого, тридцать седьмого или тридцать восьмого варианта осуществления изобретения, P3 представляет собой защитную группу для аминогруппы. Если P3 представляет собой защитную группу для аминогруппы, то соединение формулы (18) дополнительно вводят в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (I’):
.
В сорок первом варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
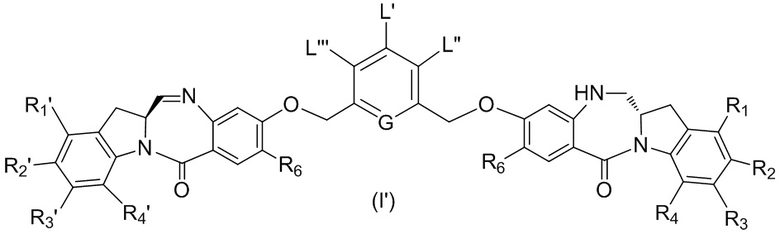
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента или эстерифицирующего реагента в реакцию с соединением формулы (14),
с получением соединения формулы (15):
,
или его соли;
(2) введение соединения формулы (15) в реакцию с мономерным соединением формулы (a),
,
с получением соединения формулы (19):
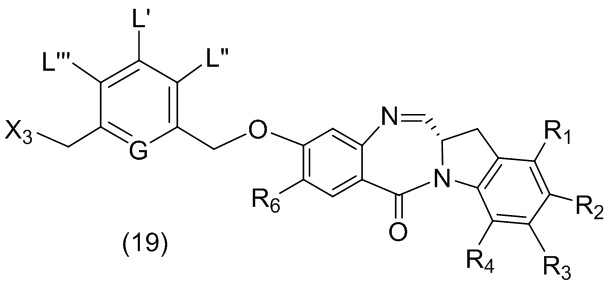 ,
,
или его соли;
(3) введение соединения формулы (19) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17’):
,
или его соли; и
(6) введение соединения формулы (17’) в реакцию с мономером формулы (B):
,
с получением соединения формулы (I’) или его фармацевтически приемлемой соли, где X3 представляет собой –Cl; X4 представляет собой сульфонатный эфир или активированный эфир; P1 представляет собой защитную группу для спиртовой группы; P2 представляет собой защитную группу для аминогруппы;, а остальные переменные являются такими же, как описано выше. В одном варианте осуществления изобретения X4 представляет собой сульфонатный эфир.
В одном варианте осуществления изобретения способ согласно сорок первому варианту осуществления изобретения включает получение соединения формулы (Ib’),или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента в реакцию с соединением формулы (14b) с получением соединения формулы (15b) или его соли;
(2) введение соединения формулы (15b) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (16b) или его соли;
(3) введение соединения формулы (16b) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17b’) или его соли; и
(6) введение соединения формулы (17b’) в реакцию с мономером формулы (a1) с получением соединения формулы (Ib’) или его фармацевтически приемлемой соли.
В другом варианте осуществления изобретения способ согласно сорок первому варианту осуществления изобретения включает получение соединения формулы (Ic’) или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента в реакцию с соединением формулы (14c) с получением соединения формулы (15c) или его соли;
(2) введение соединения формулы (15c) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (16c) или его соли;
(3) введение соединения формулы (16c) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17c’) или его соли; и
(6) введение соединения формулы (17c’) в реакцию с мономером формулы (a1) с получением соединения формулы (Ic’) или его фармацевтически приемлемой соли.
В еще другом варианте осуществления изобретения способ согласно сорок первому варианту осуществления изобретения включает получение соединения формулы (IA’) или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение сульфонирующего реагента в реакцию с соединением формулы (14A) с получением соединения формулы (15A) или его соли;
(2) введение соединения формулы (15A) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (16A) или его соли;
(3) введение соединения формулы (16A) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17A’) или его соли; и
(6) введение соединения формулы (17A’) в реакцию с мономером формулы (a1) с получением соединения формулы (IA’) или его фармацевтически приемлемой соли.
В сорок втором варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение в реакцию соединения формулы (14):
,
с мономерным соединением формулы (b),
,
с получением соединения формулы (19):
,
или его соли;
(2) введение соединения формулы (19) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17’):
,
или его соли; и
(3) введение соединения формулы (17’) в реакцию с мономером формулы (a):
,
с получением соединения формулы (I’);, где X3 представляет собой –Cl; P1 представляет собой защитную группу для спиртовой группы; переменные являются такими же, как описано выше.
В одном варианте осуществления изобретения способ согласно сорок второму варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14b) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (19b) или его соли;
(2) введение соединения формулы (19b) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17b’) или его соли; и
(3) введение соединения формулы (17b’) в реакцию с мономером формулы (a1) с получением соединения формулы (Ib’).
В другом варианте осуществления изобретения способ согласно сорок второму варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14c) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (19c) или его соли;
(2) введение соединения формулы (19c) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17c’) или его соли; и
(3) введение соединения формулы (17c’) в реакцию с мономером формулы (a1) с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно сорок второму варианту осуществления изобретения включает получение соединения формулы (IA),
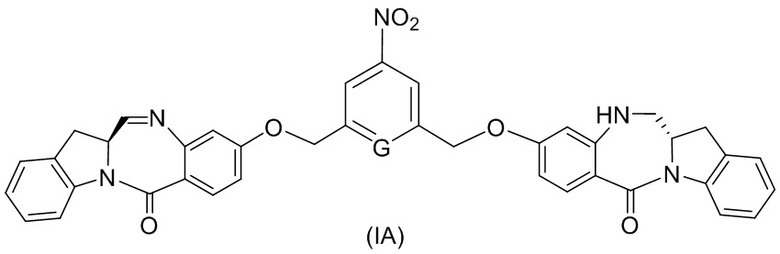
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14A) в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (19A) или его соли;
(2) введение соединения формулы (19A) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17A’) или его соли; и
(3) введение соединения формулы (17A’) в реакцию с мономером формулы (a1) с получением соединения формулы (IA’).
Условия и реагенты для способа согласно сорок второму варианту осуществления изобретения являются такими, как описано выше в двадцать восьмом, тридцать третьем и/или тридцать четвертом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В сорок третьем варианте осуществления данного изобретения предложен способ получения соединения формулы (I’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14):
,
или его солью, с получением соединения формулы (20):
,
или его соли;
(2) введение соединения формулы (20) или его соли в реакцию с мономерным соединением формулы (a):
;
с получением соединения формулы (19):
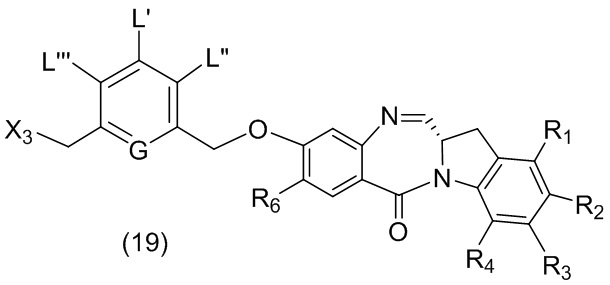 ;
;
(3) введение соединения формулы (19) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17’):
;
или его соли и
(4) введение соединения (17’) в реакцию с мономером формулы (b):
с получением соединения формулы (I’), где X3 представляет собой –Cl; X5 представляет собой –Br или –I;, а остальные переменные являются такими же, как описано выше.
В одном варианте способ согласно сорок третьему варианту осуществления изобретения включает получение соединения формулы (Ib’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14b) или его солью с получением соединения формулы (20b) или его соли;
(2) введение соединения формулы (20b) или его соли в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (19b);
(3) введение соединения формулы (19b) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17b’) или его соли и
(4) введение соединения (17b’) в реакцию с мономером формулы (a1) с получением соединения формулы (Ib’).
В другом варианте способ согласно сорок третьему варианту осуществления изобретения включает получение соединения формулы (Ic’),
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14c) или его солью с получением соединения формулы (20c) или его соли;
(2) введение соединения формулы (20c) или его соли в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (19c);
(3) введение соединения формулы (19c) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17c’) или его соли и
(4) введение соединения (17c’) в реакцию с мономером формулы (a1) с получением соединения формулы (Ic’).
В еще другом варианте осуществления изобретения способ согласно сорок третьему варианту осуществления изобретения включает получение соединения формулы (IA),
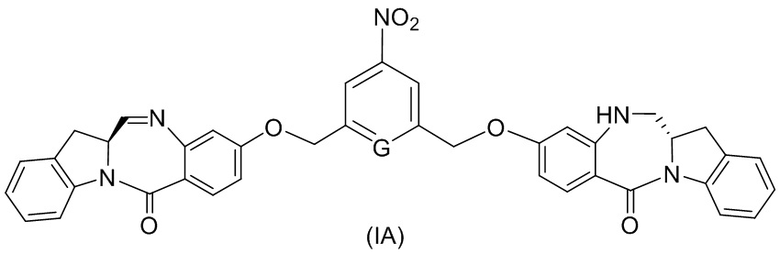
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение бромирующего или йодирующего реагента в реакцию с соединением формулы (14A) или его солью, с получением соединения формулы (20A) или его соли;
(2) введение соединения формулы (20A) или его соли в реакцию с мономерным соединением формулы (a1) с получением соединения формулы (19A);
(3) введение соединения формулы (19A) в реакцию с восстанавливающим имин агентом с получением соединения формулы (17A’) или его соли и
(4) введение соединения (17A’) в реакцию с мономером формулы (a1) с получением соединения формулы (IA’).
Условия и реагенты для способа согласно сорок третьему варианту осуществления изобретения являются такими, как описано выше в двадцать пятом, двадцать седьмом, тридцать третьем и/или тридцать четвертом варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В одном варианте любого из вариантов осуществления изобретения с тридцать пятого по сорок третий соединение формулы (14) или его соль получают по способу, который включает следующие стадии:
(1) введение хлорирующего реагента в реакцию с соединением формулы (2):
с получением соединения формулы (13):
,
или его соли; и
(2) введение соединения формулы (13) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (14) или его соли, где X3 представляет собой –Cl; и P1 представляет собой защитную группу для спиртовой группы.
В конкретном варианте осуществления изобретения соединение формулы (14b) или его соль получают по способу, который включает следующие стадии: (1) введение хлорирующего реагента в реакцию с соединением формулы (2b) с получением соединения формулы (13b) или его соли; и (2) введение соединения формулы (13b) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (14b) или его соли.
В другом конкретном варианте осуществления изобретения соединение формулы (14c) или его соль получают по способу, который включает следующие стадии: (1) введение хлорирующего реагента в реакцию с соединением формулы (2c) с получением соединения формулы (13c) или его соли; и (2) введение соединения формулы (13c) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (14c) или его соли.
В еще другом конкретном варианте осуществления изобретения соединение формулы (14A) или его соль получают по способу, который включает следующие стадии: (1) введение хлорирующего реагента в реакцию с соединением формулы (2A) с получением соединения формулы (13A) или его соли; и (2) введение соединения формулы (13A) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (14A) или его соли.
Условия и реагенты для способа получения соединения формулы (14), (14b), (14c) или (14A) выше являются такими, как описано выше в двадцать втором и/или двадцать третьем варианте(ах) осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В другом варианте осуществления изобретения соединение формулы (2) получают введением соединения формулы (1) в реакцию с защитным реагентом для спиртовой группы
.
В конкретном варианте осуществления изобретения соединение формулы (2b) получают введением соединения формулы (1b) в реакцию с защитным реагентом для спиртовой группы.
В другом конкретном варианте осуществления изобретения соединение формулы (2c) получают введением соединения формулы (1c) в реакцию с защитным реагентом для спиртовой группы.
В еще другом конкретном варианте осуществления изобретения соединение формулы (2A) получают введением соединения формулы (1A) в реакцию с защитным реагентом для спиртовой группы.
Условия и реагенты для способа получения соединения формулы (2), (2b), (2c) или (2A) выше являются такими, как описано выше в первом варианте осуществления изобретения и любых конкретных вариантах осуществления изобретения, описанных в данном документе.
В сорок четвертом варианте осуществления данного изобретения предложен способ получения соединения формулы (IB)
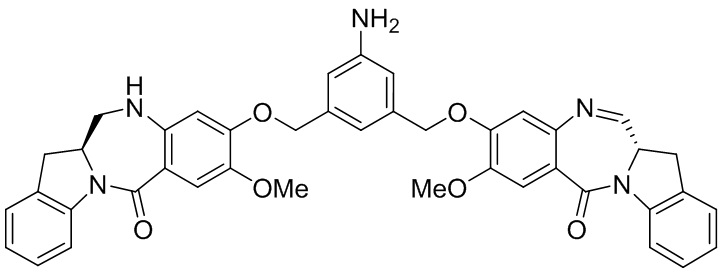 (IB)
(IB)
или его фармацевтически приемлемой соли, включающий в себя стадию введения соединения формулы (IA) в реакцию с восстановительным агентом. Можно применять любой подходящий восстановительный агент. В одном варианте осуществления изобретения восстановительный агент выбран из группы, состоящей из: газообразного водорода, гидросульфита натрия, сульфида натрия, хлорида олова, хлорида титана (II), цинка, железа и йодида самария. В конкретном варианте осуществления изобретения восстановительный агент представляет собой Fe/NH4Cl или Zn/NH4Cl.
В одном варианте осуществления изобретения соединение формулы (Ic’) может быть получено введением соединения формулы (IB) в реакцию с соединением формулы (Lc’):
 (Lc’),
(Lc’),
где E представляет собой –OH, галогенид, или -C(=O)E представляет собой активированный эфир; и R100 представляет собой (C1-C3)алкокси
В конкретном варианте осуществления изобретения E представляет собой –OH и реакцию соединения формулы (IB) и соединения формулы (Lc’) осуществляют в присутствии активирующего агента.
В одном варианте осуществления изобретения активирующий агент представляет собой карбoдиимид, уроний, активный сложный эфир, фосфоний, 2-алкил-1-алкилкарбонил-1,2-дигидрoхинолин, 2-алкокси-1-алкоксикарбонил-1,2-дигидрoхинолин или алкилхлорформиат. В конкретном варианте осуществления изобретения активирующий агент представляет собой карбoдиимид. В более конкретном варианте осуществления изобретения, активирующий агент представляет собой дициклогексилкарбoдиимид, ДЦК (DCC), 1-этил-3-(3-диметиламинопропил)карбoдиимид, ЭДК (EDC) или диизопропилкарбoдиимид, ДИК (DIC). В другом конкретном варианте осуществления изобретения активирующий агент представляет собой N-этоксикарбонил-2-этокси-1,2-дигидрoхинолин.
В одном варианте осуществления изобретения для описанных выше способов R100 представляет собой метокси, и R101 представляет собой метил.
Кроме того, способ по данному изобретению может быть любой комбинацией описанных выше способов (например, способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором или сорок третьем варианте осуществления изобретения). Например, комбинация способов первого и второго вариантов осуществления изобретения, комбинация способов первого, второго и третьего вариантов осуществления изобретения, комбинация способов четвертого и пятого вариантов осуществления изобретения, комбинация способов четвертого, пятого и шестого вариантов осуществления изобретения, комбинация способов шестого и восьмого вариантов осуществления изобретения, комбинация способов тринадцатого и четырнадцатого вариантов осуществления изобретения, комбинация способов тринадцатого, четырнадцатого и пятнадцатого вариантов осуществления изобретения и комбинация способов семнадцатого и восемнадцатого вариантов осуществления изобретения также включены в данное изобретение. Кроме того, различные определения, описанные в любом из конкретных вариантов осуществления изобретения ниже, применимы к любой комбинации описанных выше способов.
Реакции, описанные в данном документе в способах по данному изобретению, могут быть проведены в любом(ых) подходящем(их) растворителе(ях). В одном варианте осуществления изобретения растворитель представляет собой органический растворитель. Типичные органические растворители включают в себя, но не ограничиваясь этим, дихлорметан, дихлорэтан, ДМФА, ДМА, ацетон, ацетонитрил, ТГФ, ДМСО, этилацетат и т.п. или их комбинацию.
Реакции, описанные в данном документе в способах по данному изобретению, могут быть проведены при любой подходящей температуре. В одном варианте осуществления изобретения реакция может быть проведена при комнатной температуре. В другом варианте осуществления изобретения реакция может быть проведена при низкой температуре, такой как 0 °C. В еще другом варианте осуществления изобретения, реакция может быть проведена при повышенной температуре, такой как около 40 °C, около 50 °C и т.п.
В 1 конкретном варианте осуществления изобретения, для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом варианте осуществления изобретения, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором или сорок третьем варианте осуществления изобретения, один из L’, L’’ и L’’’ представлен следующей формулой:
-Z1-P-Z2-Rx-J (A)
а другие два являются одинаковыми или разными и независимо выбраны из -H, необязательно замещенного линейного, разветвленного или циклического алкила, алкенила или алкинила, содержащего от 1 до 10 атомов углерода, полиэтиленгликолевого фрагмента -(CH2CH2O)n’-Rc, галогена, гуанидиния [-NH(C=NH)NH2], -OR, -NR’R’’, -NO2, -NR’COR’’, -SR, -SOR’, -SO2R’, -SO3H, -OSO3H, -SO2NR’R’’, циано, азидо, -COR’, -OCOR’ и -OCONR’R’’;
где:
один из Z1 и Z2 представляет собой -C(=O)-, а другой представляет собой -NR5-;
P представляет собой остаток аминокислоты или пептид, содержащий от 2 до 20 остатков аминокислот;
J представляет собой фрагмент, содержащий реакционноспособную группу, способную ковалентно связывать цитотоксическое соединение со связывающимся с клеткой агентом;
Rx представляет собой необязательно замещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 10 атомов углерода; и
R5 представляет собой -H или необязательно замещенный линейный или разветвленный алкил, содержащий от 1 до 10 атомов углерода.
В более конкретном варианте осуществления изобретения один из L’, L” и L’” представлен формулой (A), а каждый из других независимо представляет собой -H, линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, галоген, -OH, (C1-C6)алкокси или -NO2.
В другом более конкретном варианте осуществления изобретения один из L’, L” и L’” представлен формулой (A), а другие представляют собой –H.
В еще другом более конкретном варианте осуществления изобретения, L’ представлен формулой (A); а L” и L’” оба представляют собой –H.
В другом более конкретном варианте осуществления изобретения для формулы (A), Rx представляет собой линейный, разветвленный или циклический алкил, содержащий от 1 до 6 атомов углерода, необязательно замещенный галогеном, -OH, (C1-C3)алкилом, (C1-C3)алкокси, галоген(C1-C3)алкилом или заряженным заместителем или ионизируемой группой Q.
В более конкретном варианте осуществления изобретения J представляет собой NHRc1, -COORc1 или -COE, где -COE представляет собой реакционноспособный эфир, а Rc1 представляет собой -H или линейный или разветвленный алкил, содержащий от 1 до 4 атомов углерода, необязательно замещенный галогеном, -OH или (C1-C3)алкокси. Даже более конкретно, J представляет собой -COORc1, а Rc1 представляет собой (C1-C3)алкил.
Во 2 конкретном варианте осуществления изобретения, для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором или сорок третьем варианте осуществления изобретения или первом конкретном варианте осуществления изобретения L’ представлен следующей формулой:
-NR5-P-C(=O)-(CRaRb)m-J (B1);
-NR5-P-C(=O)-Cy-(CRaRb)m’-J (B2);
-C(=O)-P-NR5-(CRaRb)m-J (C1) или
-C(=O)-P-NR5-Cy-(CRaRb)m’-J (C2)
где:
J представляет собой -COORc1;
Rc1 представляет собой -H или линейный или разветвленный алкил, содержащий от 1 до 4 атомов углерода, необязательно замещенный галогеном, -OH или (C1-C3)алкокси;
каждый Ra и Rb в каждом случае независимо представляет собой -H, (C1-C3)алкил или заряженный заместитель или ионизируемую группу Q;
m представляет собой целое число от 1 до 6;
m’ представляет собой 0 или целое число от 1 до 6; и
Cy представляет собой циклический алкил, содержащий 5 или 6 атомов углерода в кольце, необязательно замещенный галогеном, -OH, (C1-C3)алкилом, (C1-C3)алкокси или галоген(C1-C3)алкилом.
В более конкретном варианте осуществления изобретения Ra и Rb оба представляют собой H; Cy представляет собой циклогексан; и R5 представляет собой H или Me.
В другом более конкретном варианте осуществления изобретения m’ представляет собой 0 или 1.
В 3 конкретном варианте осуществления изобретения для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором или сорок третьем варианте осуществления изобретения или 1 конкретном варианте осуществления изобретения L’ представлен следующей формулой:
-NR5-P-C(=O)-(CRaRb)m-S-Zs (B3); или
-C(=O)-P-NR5-(CRaRb)m-S-Zs (C3),
где:
каждый Ra и Rb в каждом случае независимо представляет собой -H, (C1-C3)алкил или заряженный заместитель или ионизируемую группу Q;
m представляет собой целое число от 1 до 6;
Zs представляет собой -H, -SRd, -C(=O)Rd1 или выбран как любая одна из следующих формул:
 (a1);
(a1); (a2);
(a2); (a3);
(a3); (a4);
(a4);  (a5);
(a5); (a6);
(a6); 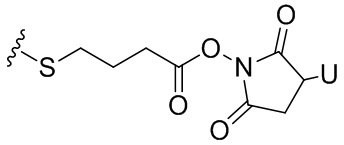 (a7);
(a7);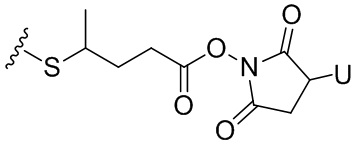 (a8);
(a8); 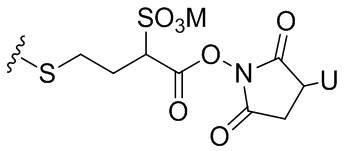 (a9);
(a9);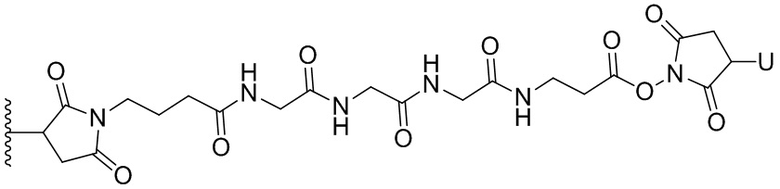 (a10);
(a10); 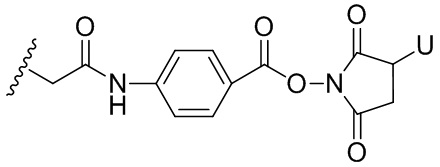 (a11);
(a11); 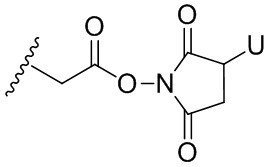 (a12);
(a12);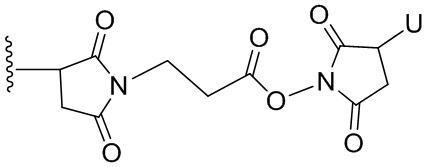 (a13);
(a13); 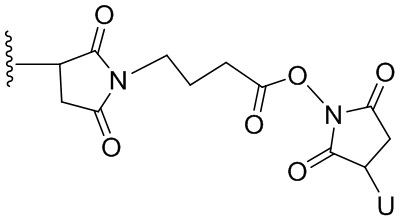 (a14); или
(a14); или
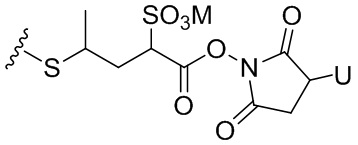 (a15),
(a15),
где:
q представляет собой целое число от 1 до 5;
n’ представляет собой целое число от 2 до 6;
U представляет собой -H или SO3M;
M представляет собой H+, Na+ или K+;
Rd представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, или выбран из фенила, нитрофенила (например, 2- или 4-нитрофенил), динитрофенила (например, 2,4-динитрофенил), карбоксинитрофенила (например, 3-карбокси-4-нитрофенил), пиридила или нитропиридила (например, 4-нитропиридил); и
Rd1 представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода
В более конкретном варианте осуществления изобретения Zs представляет собой -SRd, а Rd представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В более конкретном варианте осуществления изобретения заряженный заместитель или ионизируемая группа Q представляет собой i) -SO3H, -Z’-SO3H, -OPO3H2, -Z’-OPO3H2, -PO3H2, -Z’-PO3H2, -CO2H, -Z’-CO2H, -NR11R12 или -Z’-NR11R12 или фармацевтически приемлемую соль перечисленного; или ii) -N+R14R15R16XA- или -Z’-N+R14R15R16XA-; Z’ представляет собой необязательно замещенный алкилен, необязательно замещенный циклоалкилен или необязательно замещенный фенилен; каждый R11, R12, с R14 по R16 независимо представляет собой H или необязательно замещенный алкил; и XA- представляет собой фармацевтически приемлемый анион. Даже более конкретно, Q представляет собой SO3H или его фармацевтически приемлемую соль.
В другом более конкретном варианте осуществления изобретения Ra и Rb оба представляют собой –H, и R5 представляет собой H или Me.
В еще другом более конкретном варианте осуществления изобретения, -(CRaRb)m- представляет собой -(CH2)m”-C(Me2)-, и m” представляет собой целое число от 1 до 5.
В 4 конкретном варианте осуществления изобретения для формулы (B1) (B2), (B3), (C1), (C2) или (C3) P представляет собой пептид, содержащий от 2 до 10 остатков аминокислот; и остальные переменные являются такими, как описано выше во 2 или 3 конкретном варианте осуществления изобретения или любых более конкретных вариантах осуществления изобретения, описанных в данном документе.
В более конкретном варианте осуществления изобретения P представляет собой пептид, содержащий от 2 до 5 остатков аминокислот.
В другом более конкретном варианте осуществления изобретения P представляет собой Gly-Gly-Gly, Ala-Val, Val-Ala, Val-Cit, Val-Lys, Phe-Lys, Lys-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ile-Cit, Trp, Cit, Phe-Ala, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg, Phe-Phe-Lys, D-Phe-Phe-Lys, Gly-Phe-Lys, Leu-Ala-Leu, Ile-Ala-Leu, Val-Ala-Val, Ala-Leu-Ala-Leu, β-Ala-Leu-Ala-Leu и Gly-Phe-Leu-Gly, Val-Arg, Arg-Val, Arg-Arg, Val-D-Cit, Val-D-Lys, Val-D-Arg, D-Val-Cit, D-Val-Lys, D-Val-Arg, D-Val-D-Cit, D-Val-D-Lys, D-Val-D-Arg, D-Arg-D-Arg, Ala-Ala, Ala-D-Ala, D-Ala-Ala, D-Ala-D-Ala, Ala-Met или Met-Ala. Даже более конкретно, P представляет собой Gly-Gly-Gly, Ala-Val, Ala-Ala, Ala-D-Ala, D-Ala-Ala или D-Ala-D-Ala.
В 5 конкретном варианте осуществления изобретения для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором и сорок третьем вариантах осуществления изобретения L’ представлен следующей формулой:
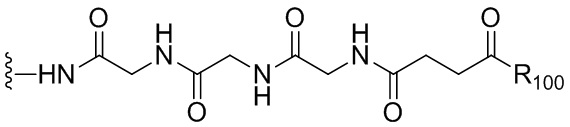 ;
;
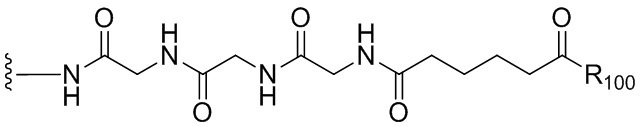 ;
;
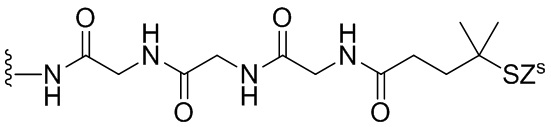 ;
;
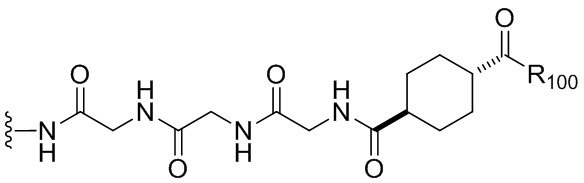 ;
;
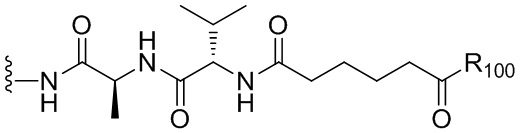 ;
;
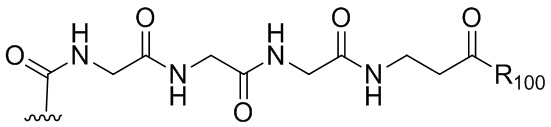 ;
;
 ;
;
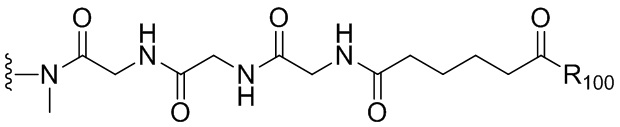 ;
;
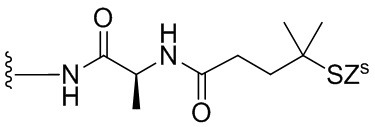 ;
;
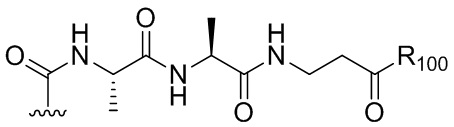
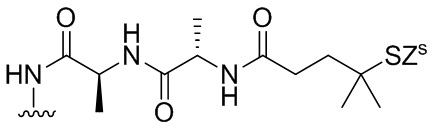 ;
;
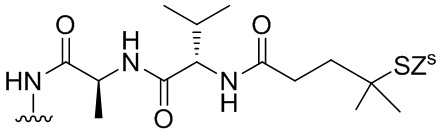 ; или
; или
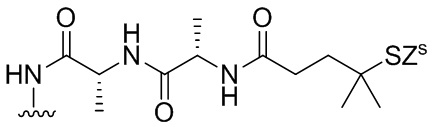 ,
,
где:
R100 представляет собой (C1-C3)алкокси;
Zs представляет собой –SR101; и
R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В 6 конкретном варианте осуществления изобретения, для способов в восьмом, девятом, десятом, пятнадцатом, шестнадцатом или девятнадцатом варианте осуществления изобретения соединения формулы (I’) представлены следующей формулой:
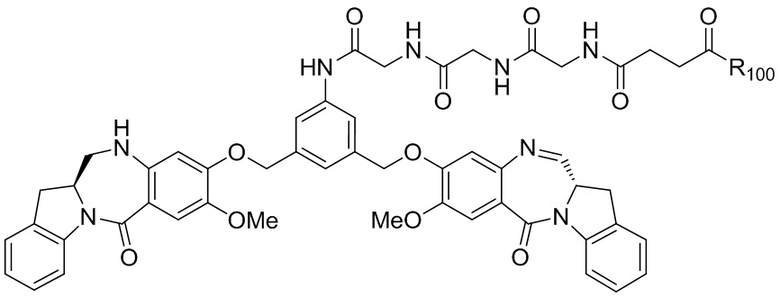 ;
;
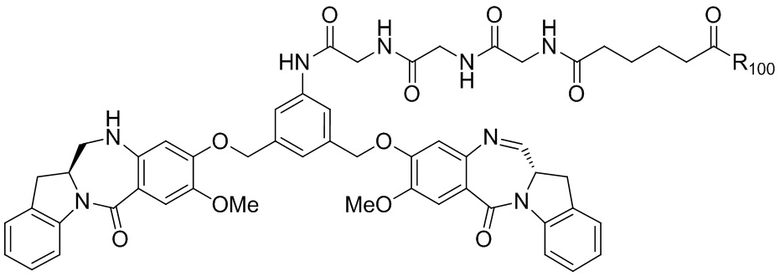 ;
;
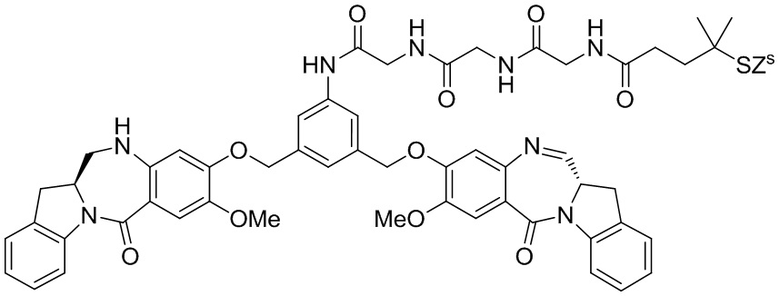 ;
;
 ;
;
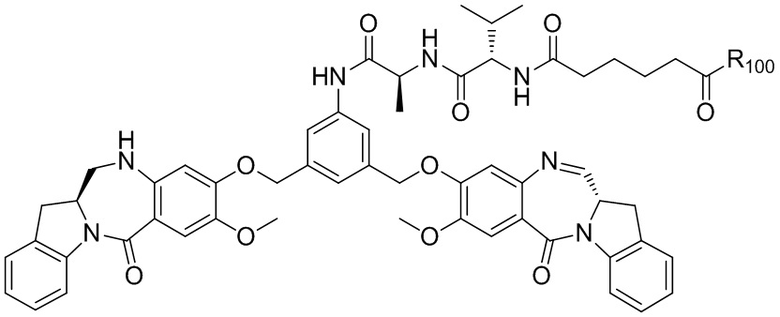 ;
;
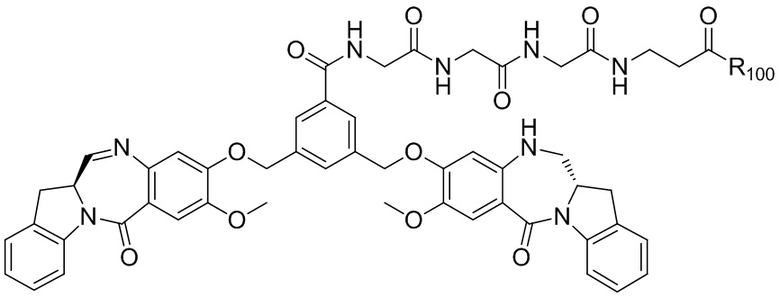 ;
;
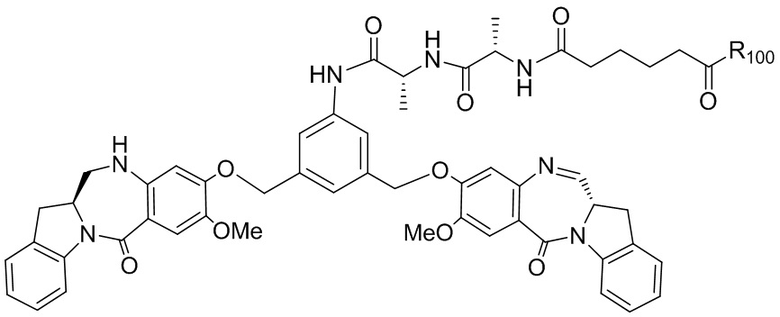 ;
;
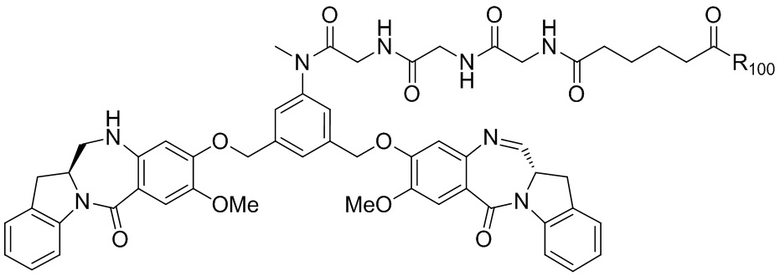 ;
;
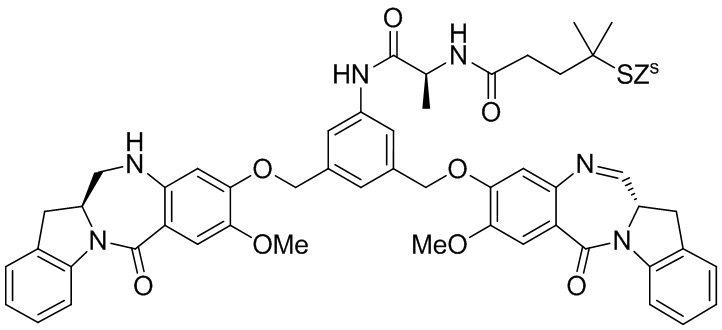 ;
;
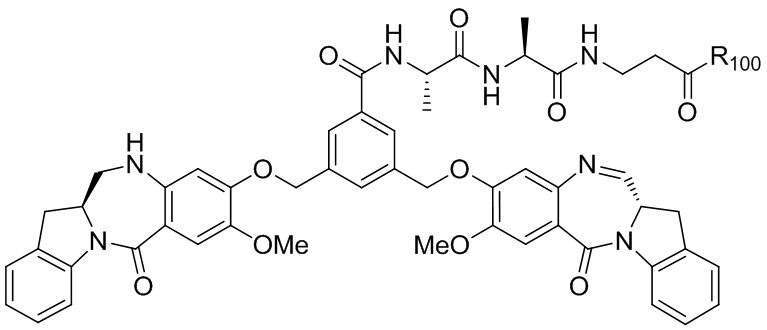 ;
;
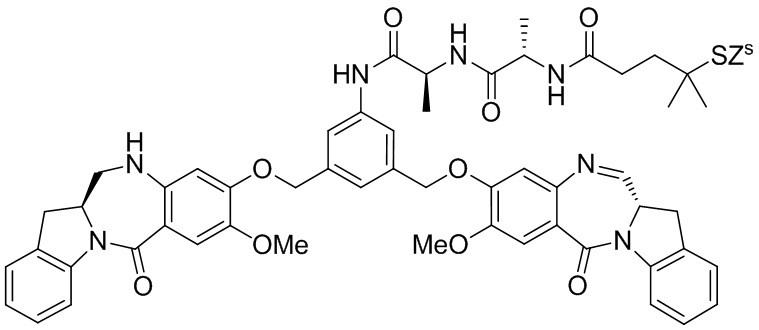 ;
;
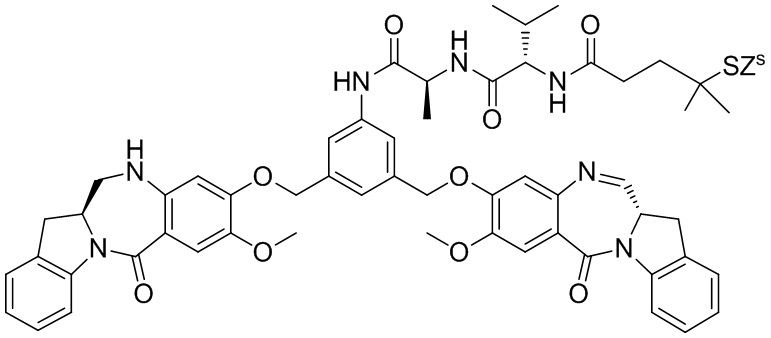 ; или
; или
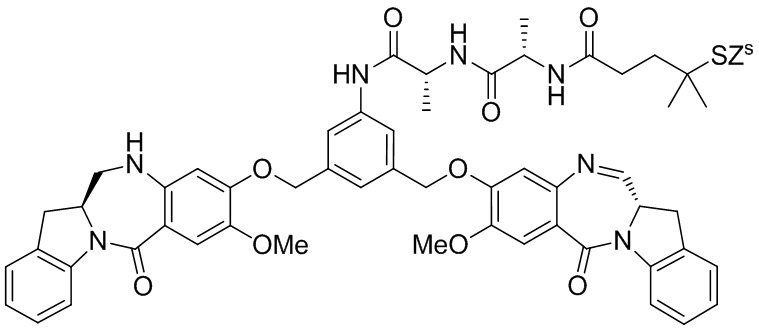 ;
;
или ее фармацевтически приемлемой солью, где:
R100 представляет собой (C1-C3)алкокси;
Zs представляет собой –SR101 ; и
R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В 7 конкретном варианте осуществления изобретения для способов, описанных в 5 или 6 конкретном варианте осуществления изобретения, R100 представляет собой –OMe, и R101 представляет собой Me или пиридил.
В 8 конкретном варианте осуществления изобретения, для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом варианте осуществления изобретения, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором и сорок третьем варианте осуществления изобретения один из L’, L’’ и L’’’ представлен следующей формулой:
-N(Re)-C(=O)-Rx-S-Zs (B);
где:
Rx представляет собой линейный или разветвленный алкилен, содержащий от 1 до 6 атомов углерода, необязательно замещенный заряженным заместителем или ионизируемой группой Q;
Q представляет собой i) -SO3H, -Z’-SO3H, -OPO3H2, -Z’-OPO3H2, -PO3H2, -Z’-PO3H2, -CO2H, -Z’-CO2H, -NR11R12 или -Z’-NR11R12 или фармацевтически приемлемую соль перечисленного; или ii) -N+R14R15R16XA- или -Z’-N+R14R15R16XA-; Z’ представляет собой необязательно замещенный алкилен, необязательно замещенный циклоалкилен или необязательно замещенный фенилен; каждый R11, R12, R14, R15 и R16 независимо представляет собой H или необязательно замещенный алкил; и XA- представляет собой фармацевтически приемлемый анион;
Re представляет собой -H или линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода;
Zs представляет собой -H, -SRd, -C(=O)Rd1 или выбран как любая одна из следующих формул:
(a1); (a2); (a3);(a4); (a5);(a6); (a7); (a8); (a9); (a10); (a11); (a12);(a13); (a14); или
(a15);
где:
q представляет собой целое число от 1 до 5;
Rd представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, или выбран из фенила, нитрофенила, динитрофенила, карбоксинитрофенила, пиридила и нитропиридила;
Rd1 представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода;
n” представляет собой целое число от 2 до 6;
U представляет собой -H или -SO3M; и
M представляет собой -H или катион.
В более конкретном варианте осуществления изобретения один из L’, L” и L’” представлен формулой (B), а каждый из остальных независимо представляет собой -H, линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, галоген, -OH, (C1-C6)алкокси или -NO2.
В другом более конкретном варианте осуществления изобретения один из L’, L” и L’” представлен формулой (B), а другие представляют собой –H.
В другом более конкретном варианте осуществления изобретения L’ представлен формулой (B); а L” и L’” оба представляют собой -H.
В другом более конкретном варианте осуществления изобретения для формулы (B) Zs представляет собой -SRd, а Rd представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В 9 конкретном варианте осуществления изобретения для формулы (B) Rx представляет собой -(CH2)p-(CRfRg)-, где каждый Rf и Rg независимо выбран из -H или линейного или разветвленного алкила, содержащего от 1 до 4 атомов углерода; и p представляет собой 0, 1, 2 или 3; а остальные переменные являются такими, как описано в 8 конкретном варианте осуществления изобретения или любых более конкретных вариантах осуществления изобретения, описанных в данном документе.
В более конкретном варианте осуществления изобретения Rf и Rg являются одинаковыми или разными и выбраны из -H и –Me.
В другом более конкретном варианте осуществления изобретения Rf и Rg оба представляют собой -Me; и p представляет собой 2.
В 10 конкретном варианте осуществления изобретения для формулы (B) Rx представляет собой линейный или разветвленный алкилен, содержащий от 1 до 4 атомов углерода, замещенный заряженным заместителем или ионизируемой группой Q; а остальные переменные являются такими, как описано в 8 конкретном варианте осуществления изобретения или любых более конкретных вариантах осуществления изобретения, описанных в данном документе.
В более конкретном варианте осуществления изобретения заряженный заместитель или ионизируемая группа Q представляет собой: i) -SO3H, -Z’-SO3H, -OPO3H2, -Z’-OPO3H2, -PO3H2, -Z’-PO3H2, -CO2H, -Z’-CO2H, -NR11R12 или -Z’-NR11R12 или фармацевтически приемлемую соль перечисленного; или ii) -N+R14R15R16XA- или -Z’-N+R14R15R16XA-; Z’ представляет собой необязательно замещенный алкилен, необязательно замещенный циклоалкилен или необязательно замещенный фенилен; каждый R11, R12, с R14 по R16 независимо представляет собой H или необязательно замещенный алкил; а XA- представляет собой фармацевтически приемлемый анион. Более конкретно, Q представляет собой -SO3H или его фармацевтически приемлемую соль.
В 11 конкретном варианте осуществления изобретения Re представляет собой -H или –Me; а остальные переменные являются такими, как описано в 9 или 10 конкретном варианте осуществления изобретения или любых более конкретных вариантах осуществления изобретения, описанных в данном документе
В 12 конкретном варианте осуществления изобретения для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором или сорок третьем варианте осуществления изобретения L’ представлен следующей формулой:
 ;
;
 ;
;
 ;
;
 ;
;
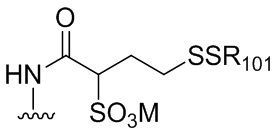 ;
;
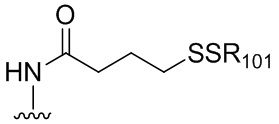 ; или
; или
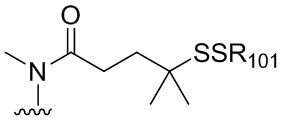 ;
;
где R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил); и M представляет собой H+, Na+ или K+.
В 13 конкретном варианте осуществления изобретения для способов в восьмом, десятом, четырнадцатом, пятнадцатом, девятнадцатом, двадцатом, двадцать первом, сорок первом, сорок втором и сорок третьем вариантах осуществления изобретения соединение формулы (I’) представлено как любое одно из следующего:
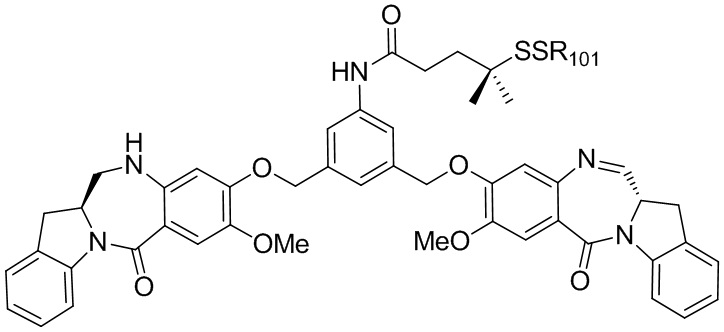 ;
;
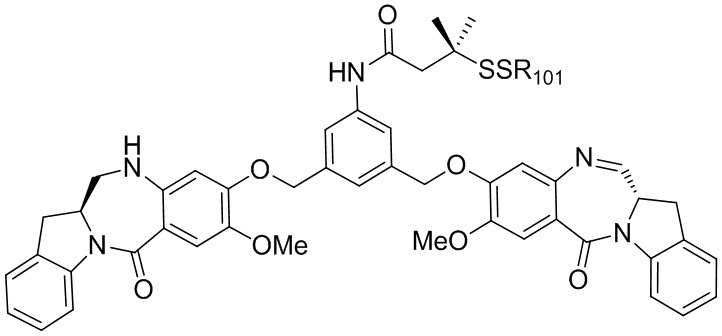 ;
;
 ;
;
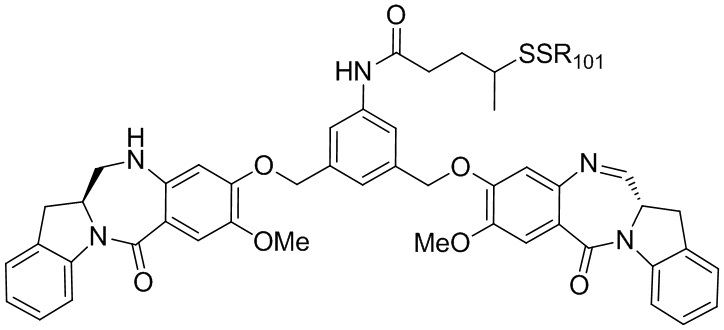 ;
;
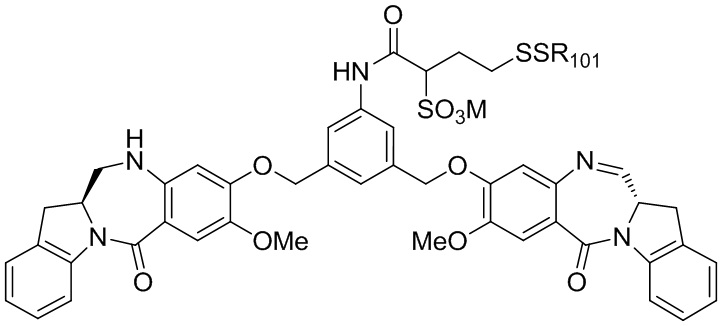 ;
;
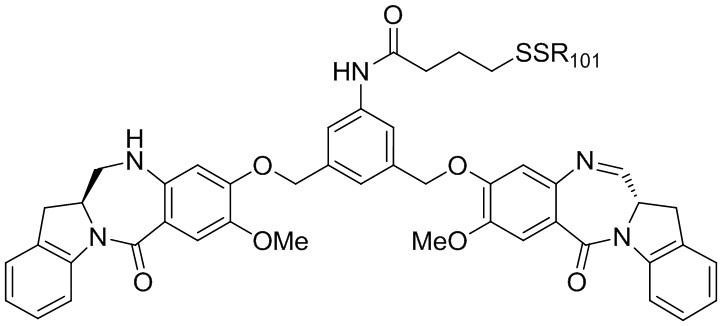 ; или
; или
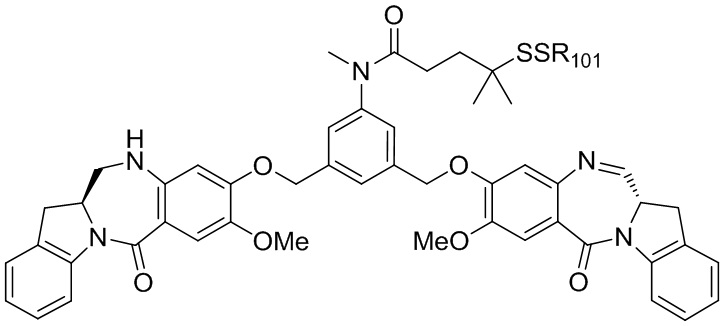 ;
;
или фармацевтически приемлемая соль перечисленного, где R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил); и M представляет собой H+, Na+ или K+.
В 14 конкретном варианте осуществления изобретения для способов, описанных в 12 или 13 конкретном варианте осуществления изобретения, R101 представляет собой метил.
В 15 конкретном варианте осуществления изобретения, для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором или сорок третьем варианте осуществления изобретения, один из L’, L’’ и L’’’ представлен следующей формулой:
-W’-Rx-S-Zs (C);
где:
W’ представляет собой -N(Re)-;
Re представляет собой -H, линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 10 атомов углерода, или -(CH2-CH2-O)n-Rk, где Rk представляет собой -H, линейный, разветвленный циклический алкил, содержащий от 1 до 6 атомов углерода, необязательно несущий вторичную аминогруппу (например, -NHR101) или третичную аминогруппу (-NR101R102) или 5- или 6-членный азотсодержащий гетероцикл, такой как пиперидин или морфолин, где каждый R101 и R102 независимо представляет собой линейный, разветвленный или циклический алкил, алкенил или алкинил, содержащий от 1 до 10 атомов углерода;
Rx представляет собой линейный, разветвленный или циклический алкил, содержащий от 1 до 10 атомов углерода;
Zs представляет собой -H, -SRd, -C(=O)Rd1 или выбран как любая одна из следующих формул:
(a1); (a2); (a3);(a4); (a5);(a6); (a7); (a8); (a9); (a10); (a11); (a12);(a13); (a14); или
(a15);
где:
Rd представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, или выбран из фенила, нитрофенила (например, 2- или 4-нитрофенил), динитрофенила (например, 2- или 4-нитрофенил), карбоксинитрофенила (например, 3-карбокси-4-нитрофенил), пиридила или нитропиридила (например, 4-нитропиридил);
Rd1 представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода;
q представляет собой целое число от 1 до 5;
n представляет собой целое число от 2 до 6;
n’ представляет собой целое число от 1 до 24;
U представляет собой -H или -SO3M; и
M представляет собой -H или катион, такой как Na+ или K+.
В более конкретном варианте осуществления изобретения один из L’, L” и L’” представлен формулой (C), а каждый из остальных независимо представляет собой -H, линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, галоген, -OH, (C1-C6)алкокси или -NO2.
В другом более конкретном варианте осуществления изобретения один из L’, L” и L’” представлен формулой (C), а другие представляют собой –H.
В еще другом более конкретном варианте осуществления изобретения, L’ представлен формулой (C); а L” и L’” оба представляют собой -H.
В другом более конкретном варианте осуществления изобретения Zs представляет собой -SRd, а Rd представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил).
В 16 конкретном варианте осуществления изобретения, Rx представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода; а остальные переменные являются такими, как описано в 14 конкретном варианте осуществления изобретения или любых более конкретных вариантах осуществления изобретения, описанных в данном документе. Более конкретно, Rx представляет собой -(CH2)p-(CRfRg) -, где каждый Rf и Rg независимо выбран из -H или линейного или разветвленного алкила, содержащего от 1 до 4 атомов углерода; и p представляет собой 0, 1, 2 или 3. Даже более конкретно, Rf и Rg являются одинаковыми или разными и выбраны из -H и -Me; и p представляет собой 1.
В 17 конкретном варианте осуществления изобретения Rk представляет собой -H или –Me, и n равно 3; а остальные переменные являются такими, как описано выше в 15 или 16 конкретном варианте осуществления изобретения.
В 18 конкретном варианте осуществления изобретения для способов в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом, двадцать восьмом, двадцать девятом, тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом, тридцать шестом, тридцать седьмом, тридцать восьмом, тридцать девятом, сороковом, сорок первом, сорок втором и сорок третьем вариантах осуществления изобретения L’ представлен следующей формулой:
где R100 представляет собой (C1-C3)алкокси; а остальные переменные являются такими, как описано выше в 1-17 конкретных вариантах осуществления изобретения. В другом варианте осуществления изобретения L’ представляет собой NO2. В еще другом варианте осуществления изобретения, L’ представлен следующей формулой:
 .
.
В определенном варианте осуществления изобретения димерное индолинобензoдиазепиновое соединение формулы (I’) может быть получено в соответствии со Схемами 1-12, представленными ниже.
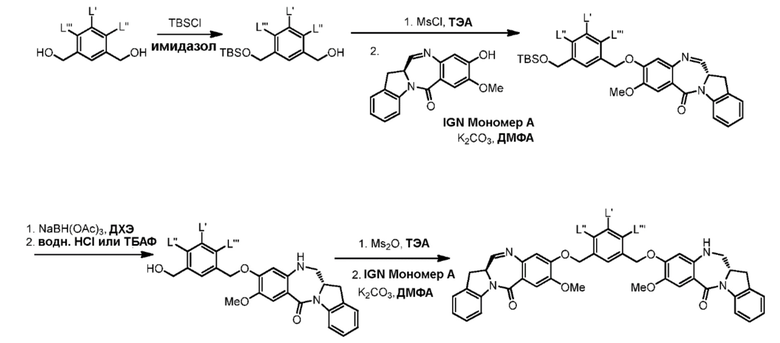
Схема 1
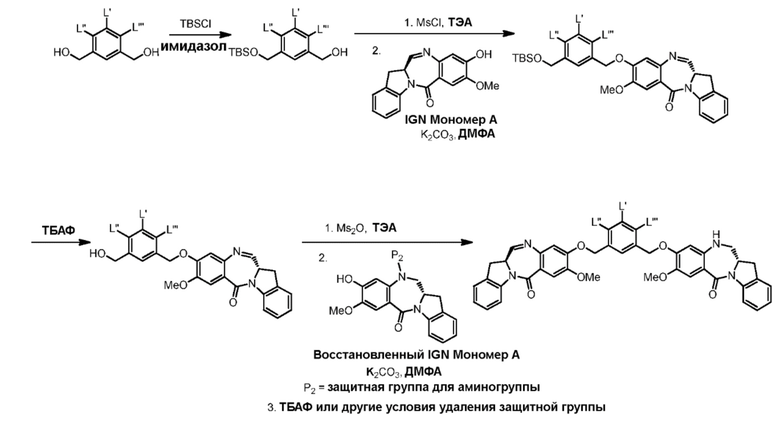
Схема 2
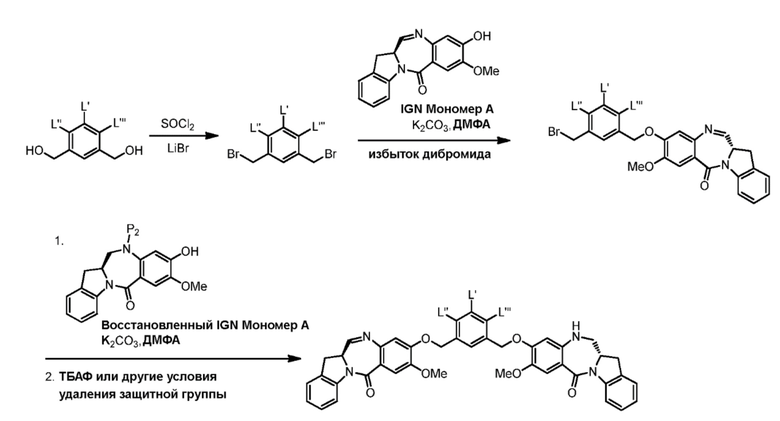
Схема 3
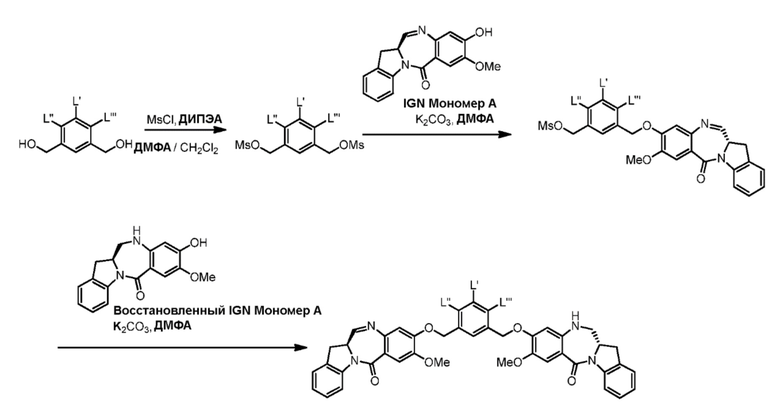
Схема 4
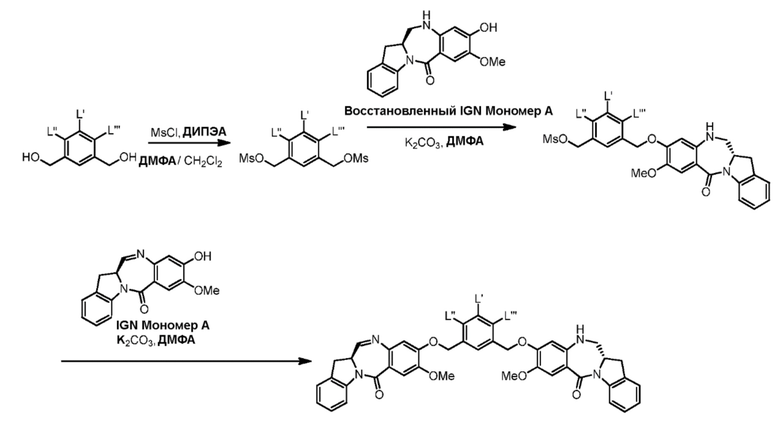
Схема 5
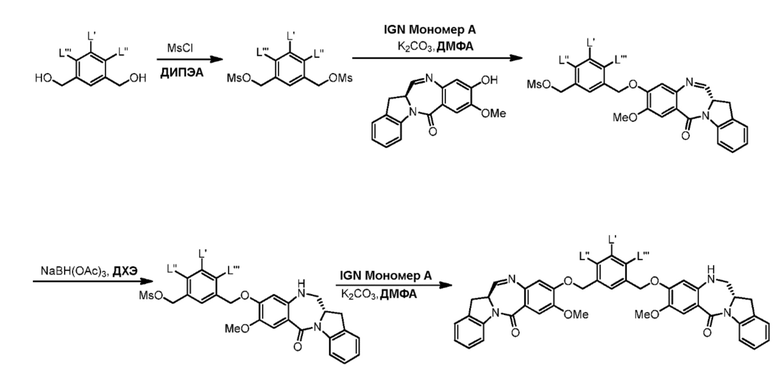
Схема 6
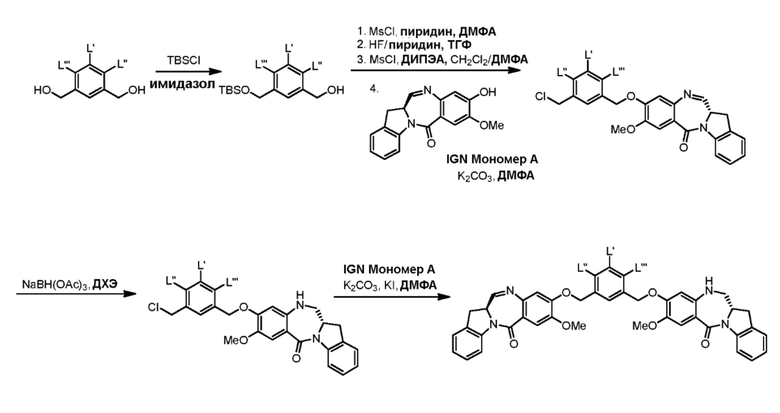
Схема 7
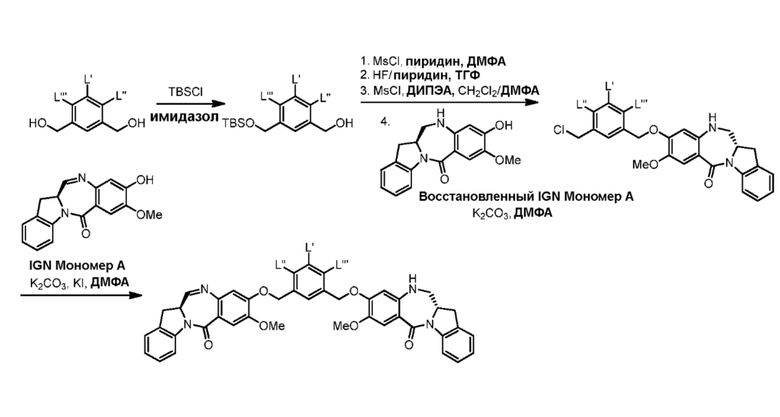
Схема 8
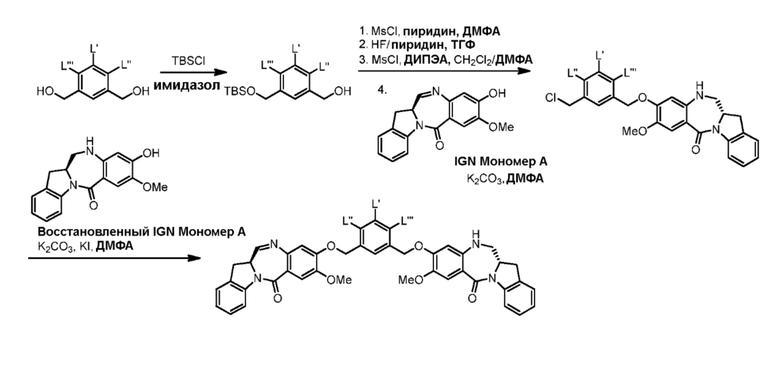
Схема 9
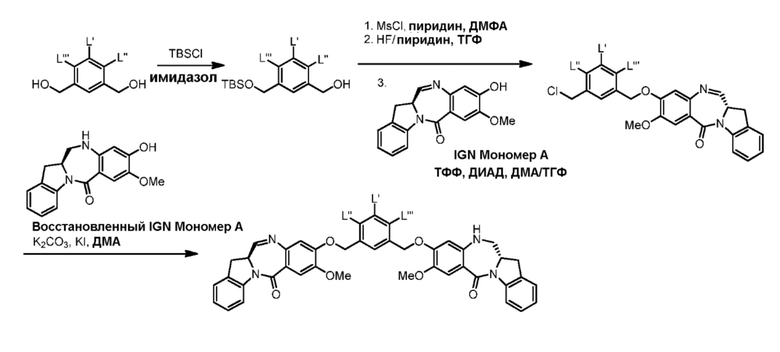
Схема 10
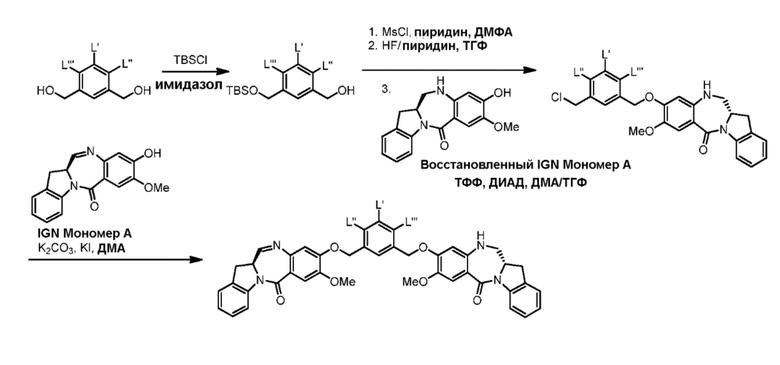
Схема 11
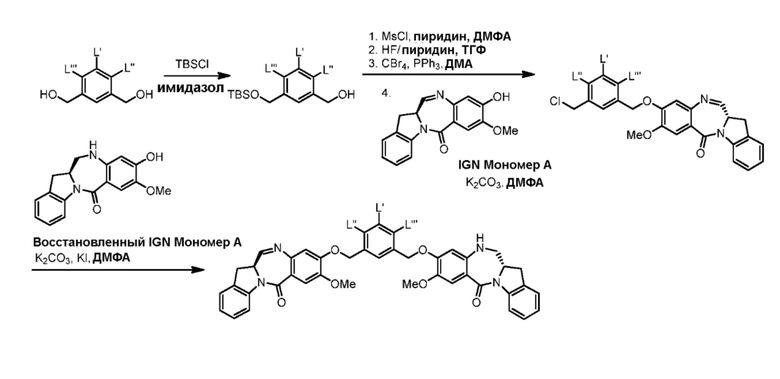
Схема 12
СОЕДИНЕНИЯ ПО ИЗОБРЕТЕНИЮ
В данном изобретении также предлагаются новые соединения, описанные в данном документе. В некоторых вариантах осуществления изобретения соединения по данному изобретению представляют собой соединения формул (1), (2), (2”), (3), (3”), (4), (4”), (5), (5”), (6), (7), (7’), (7-1), (7”), (7”’), (8), (9), (10), (10’), (11), (12), (13), (14), (15), (16), (17), (17’), (18), (19), (20), (a), (b), (d), (I’), (IA) и (IB), где переменные являются такими, как описано в вариантах осуществления изобретения с первого по сорок третий или в конкретных вариантах осуществления изобретения с 1 по 18 или любом более конкретном варианте осуществления изобретения, описанном в данном документе.
В 19 конкретном варианте осуществления изобретения для соединений по данному изобретению, R100, если он присутствует, представляет собой (C1-C3)алкокси; R101 представляет собой (C1-C3)алкил, пиридил или нитропиридил (например, 4-нитропиридил); P1 представляет собой защитную группу для спиртовой группы; X1 и X2 в каждом случае независимо представляют собой -Br, -I и сульфонатный эфир; P2 представляет собой защитную группу для аминогруппы; P3 представляет собой H или защитную группу для аминогруппы; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок третий и конкретном варианте осуществления изобретения с 1 по 18.
В 20 конкретном варианте осуществления изобретения для соединений по данному изобретению R100, если он присутствует, представляет собой –OMe; и R101 представляет собой Me или пиридил; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 19.
В 21 конкретном варианте осуществления изобретения для соединений по данному изобретению P1, если он присутствует, представляет собой силильную защитную группу; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 20. Более конкретно, силильная защитная группа представляет собой диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, триметилсилил, триизопропилсилил, трибензилсилил, трифенилсилил, 2-норборнилдиметилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил или [2-(триметилсилил)этокси]метил. Даже более конкретно, силильная защитная группа представляет собой триэтилсилил, триизопропилсилил или трет-бутилдиметилсилил. В другом даже более конкретном варианте осуществления изобретения силильная защитная группа представляет собой трет-бутилдиметилсилил.
В 22 конкретном варианте осуществления изобретения для соединений по данному изобретению, X1 и X2, если они присутствуют, в каждом случае независимо представляют собой сульфонатный эфир; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 21. Более конкретно, сульфонатный эфир представляет собой мезилат, тозилат, брозилат или трифлат. Даже более конкретно, сульфонатный эфир представляет собой мезилат.
В 23 конкретном варианте осуществления изобретения для соединений по данному изобретению X3, если он присутствует, представляет собой хлор; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 22.
В 24 конкретном варианте осуществления изобретения для соединений по данному изобретению X4, если он присутствует, представляет собой сульфонатный эфир; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 23. Более конкретно, сульфонатный эфир представляет собой мезилат, тозилат, брозилат или трифлат. Даже более конкретно, сульфонатный эфир представляет собой мезилат.
В 25 конкретном варианте осуществления изобретения для соединений по данному изобретению P2 представляет собой защитную группу для аминогруппы, выбранную из 2-триметилсилилэтила,(2-фенил-2-триметилсилил)этила, триизопропилсилокси, 2-(триметилсилил)этоксиметила, аллилоксикарбонила, 9-флуоренилметоксикарбонила, 2-(триметилсилил)этоксикарбонила и 2,2,2,2-трихлорэтоксикарбонила; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 24.
В 26 конкретном варианте осуществления изобретения для соединений по данному изобретению P3, если он присутствует, представляет собой H или защитную группу для аминогруппы, выбранную из 2-триметилсилилэтила, (2-фенил-2-триметилсилил)этила, триизопропилсилокси, 2-(триметилсилил)этоксиметила и аллилоксикарбонила, 9-флуоренилметоксикарбонила, 2-(триметилсилил)этоксикарбонила и 2,2,2,2-трихлорэтоксикарбонила; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 25.
В 27 конкретном варианте осуществления изобретения для соединений по данному изобретению X5, если он присутствует, представляет собой –Br; а остальные переменные являются такими, как описано в любом варианте осуществления изобретения с первого по сорок четвертый и конкретном варианте осуществления изобретения с 1 по 26.
В одном варианте осуществления изобретения для способов и соединений по данному изобретению, описанных в данном документе, L’ не представлен:
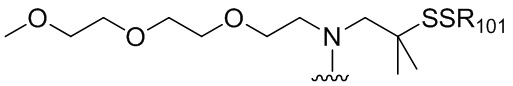 ; или
; или 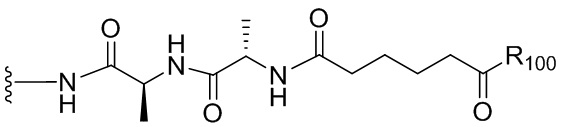 .
.
В другом варианте осуществления изобретения для способов и соединений по данному изобретению, описанных в данном документе, L’ не представлен:
; ; или NO2
Все ссылки, процитированные в данном документе и в следующих примерах явно включены путем ссылки в полном объеме.
ПРИМЕРЫ
Теперь изобретение будет проиллюстрировано путем ссылки на неограничивающие примеры. Если обратное прямо не указано в тексте, все проценты, соотношения, части и т.п. приведены по массе. Все реагенты приобретены у Aldrich Chemical Co., штат Нью-Джерси, или получены из других коммерческих источников. Спектры ядерного магнитного резонанса (1H ЯМР) зарегистрированы на приборе Bruker 400 МГц. Масс-спектры зарегистрированы на приборе Bruker Daltonics Esquire 3000, ЖХМС получены с помощью прибора Agilent 1260 Infinity LC с МС Agilent 6120 с одной квадрупольной линзой с применением ионизации электрораспылением, и СВЭЖХ получены с помощью системы Waters, Acquity с МС Zspray™ с одной квадрупольной линзой (колонка: Acquity BEH C18, 2,1 x 50 мм, 1,7 мкм, способ: 2,5 минуты, скорость потока 0,8 мл/мин, растворитель A: вода, растворитель B: MeCN, от 5 до 95 % MeCN на протяжении 2,0 минут и 95 % MeCN на протяжении 0,5 минут).
Следующие растворители, реагенты, защитные группы, фрагменты и другие обозначения могут быть приведены в виде аббревиатуры в скобках:
Me = метил; Et = этил; Pr = пропил; и-Pr = изопропил; Bu = бутил; трет-Bu = трет-бутил; Ph = фенил и Ac = ацетил
AcOH или HOAc = уксусная кислота
ACN или CH3CN = ацетонитрил
Ala = аланин
Ar = аргон
водн. = водный
Bn = бензил
Boc или BOC = трет-бутоксикарбонил
CBr4 = тетрабромид углерода
Cbz или Z = бензилоксикарбонил
ДХМ (DCM) или CH2Cl2 = дихлорметан
ДХЭ (DCE) = 1,2-дихлорэтан
ДМАП (DMAP) = 4-диметиламинопиридин
вода ДМ (DI) = деминерализованная вода
ДИБАЛ (DIBAL) = диизобутилалюминия гидрид
ДИЭА (DIEA) или ДИПЭА (DIPEA) = N,N-диизопропилэтиламин
ДМА (DMA) = N,N-диметилацетамид
ДМФА (DMF) = N,N-диметилформамид
ДМСО (DMSO) = диметилсульфоксид
ДТТ (DTT) = дитиотрейтол
ЭДК (EDC) = 1-этил-3-(3-диметиламинопропил)карбoдиимид
ЭЭДХ (EEDQ) = N-этоксикарбонил-2-этокси-1,2-дигидрoхинолин
ИЭР (ESI) или ЭР (ES) = ионизация электрораспылением
EtOAc = этилацетат
Gly = глицин
г = грамм(ы)
час = час(ы)
ГАТУ (HATU) = N,N,N’N’-тетраметил-O-(7-азабензoтриазол-1-ил)урония гексафосфат
ВЭЖХ (HPLC) = высокоэффективная жидкостная хроматография
ГОБТ (HOBT) или HOBt = 1-гидроксибензoтриазол
ЛАГ (LAH) = лития алюминия гидрид
ЖХ (LC) = жидкостная хроматография
ЖХМС (LCMS) = жидкостной хроматографии масс-спектрометрия
мин = минута(ы)
мг = миллиграмм(ы)
мл = миллилитр(ы)
ммоль = миллимоль
мкг = микрограмм(ы)
мкл = микролитр(ы)
мкмоль = микромоль
Me = метил
MeOH = метанол
MeI = метилйодид
МС (MS) = масс-спектрометрия
MsCl = метансульфонилхлорид (мезилхлорид)
Ms2O = метансульфоновый ангидрид
МТБЭ (MTBE) = метил-трет-бутиловый эфир
NaBH(OAc)3 = триацетоксиборгидрид натрия
NHS = N-гидроксисукцинамид
ЯМР (NMR) = спектроскопия ядерного магнитного резонанса
PPh3 = трифенилфосфин
ПТСХ (PTLC) = препаративная тонкослойная хроматография
рац (rac) = рацемическая смесь
Rf = коэффициент удерживания
ОФВЭЖХ (RPHPLC) или ОФ-ВЭЖХ (RP-HPLC) = обращенно-фазная высокоэффективная жидкостная хроматография
КТ (RT) или кт (rt) = комнатная температура (температура окружающей среды, около 25 ºC)
насыщ. (sat или sat’d) = насыщенный
НТАБ (STAB) = триацетоксиборгидрид натрия (NaBH(OAc)3)
TBSCl или TBDMSCl = трет-бутилдиметилсилилхлорид
ТБС (TBS) = трет-бутилдиметилсилил
ТКЭФ·HCl (TCEP·HCl) = трис(2-карбоксиэтил)фосфина гидрoхлоридная соль
ТЭА (TEA) = триэтиламин (Et3N)
ТФУ (TFA) = трифторуксусная кислота
ТГФ (THF) = тетрагидрoфуран
ТСХ (TLC) = тонкослойная хроматография
Пример 1.
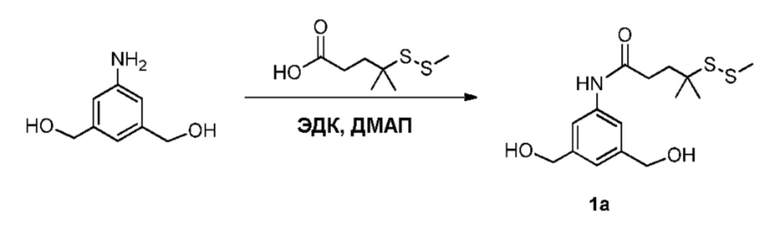
При перемешивании к раствору (5-амино-1,3-фенилен)диметанола (1,01 г, 6,59 ммоль) в безводном диметилформамиде (16,48 мл) и безводном тетрагидрoфуране (16,48 мл) добавляют 4-метил-4-(метилдисульфанил)пентановую кислоту (1,281 г, 6,59 ммоль), ЭДК·HCl (2,53 г, 13,19 ммоль) и ДМАП (0,081 г, 0,659 ммоль). Полученную смесь перемешивают в течение 18 час при комнатной температуре. Реакционную смесь гасят насыщенным раствором хлорида аммония и экстрагируют этилацетатом (3 x 50 мл). Органические экстракты промывают водой, раствором хлорида натрия и сушат над безводным сульфатом натрия. Раствор фильтруют и концентрируют под вакуумом, а полученный остаток очищают хроматографией на силикагеле (гексаны/EtOAc) с получением соединения 1a в виде твердого вещества белого цвета (0,70 г, выход 35 %). 1H ЯМР (400 МГц, ДМСО-d6): д 9,90 (с, 1H), 7,43 (с, 2H), 6,93 (с, 1H), 5,16 (т, 2H, J = 5,7 Гц), 4,44 (д, 4H, J = 5,7 Гц), 2,43 (с, 3H), 2,41-2,38 (м, 2H), 1,92-1,88 (м, 2H), 1,29 (с, 6H). МС (соотношения массы и заряда, м/з (m/z)), найдено 330,0 (M + H)+.
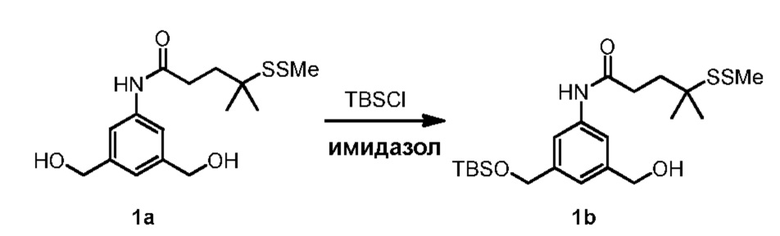
Диол 1a (1,0 г, 3,04 ммоль) растворяют в ДМФА (10,12 мл). TBSCl (503 мг, 3,34 ммоль) и имидазол (238 мг, 3,49 ммоль) добавляют к раствору, и реакционную смесь перемешивают при кт в течение ночи. Дополнительное количество TBSCl (600 мг) и имидазола (220 мг) добавляют к реакционной смеси и перемешивают при кт еще в течение 5 час. Реакционную смесь разбавляют ДХМ и промывают насыщенным раствором хлорида аммония, раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (EtOAc/гексаны, градиент) с получением 1b в виде бесцветного масла (710 мг, выход 53 %). ЖХМС (способ 8 мин) = 6,967 мин. Наблюдаемая масса (ИЭР+): 445,95 (M+H)+ и 467,90 (M+Na)+.
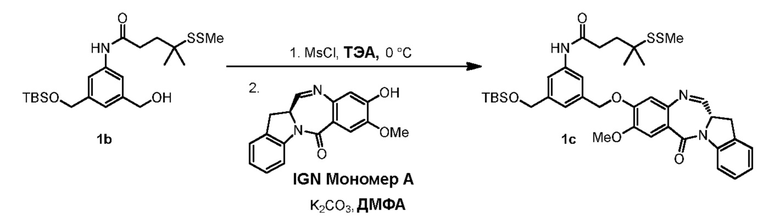
Соединение 1b (200 мг, 0,451 ммоль) растворяют в ДХМ (4,51 мл) и охлаждают до 0 °C. Et3N (82 мкл, 0,586 ммоль) добавляют к реакционной смеси в атмосфере аргона, с последующим добавлением по каплям механсульфонилхлорида (42,1 мкл, 0,541 ммоль). Реакционную смесь перемешивают при 0 °C, выдерживая при этой температуре в течение 2 час. Раствор разбавляют EtOAc и промывают холодной водой (2x). Органическую фракцию сушат над сульфатом натрия, фильтруют и концентрируют с получением мезилата. Неочищенный мезилат используют на следующей стадии без очистки. ЖХМС (способ 8 мин) = 7,444 мин. Наблюдаемая масса (ИЭР+): 521,8 (M+H)+ и 543,8 (M+Na)+. Неочищенный мезилат (210 мг, 0,402 ммоль) растворяют в ДМФА (2,68 мл). IGN мономер A (130 мг, 0,443 ммоль) и карбонат калия (111 мг, 0,805 ммоль) добавляют к смеси и перемешивают при кт в атмосфере аргона в течение ночи. Продукт осаждают добавлением воды (15 мл). Суспензию перемешивают в течение 5 мин и фильтруют. Остаток на фильтре промывают водой (3x) и сушат под вакуумом/N2 с получением соединения 1c в виде твердого вещества (270 мг, выход 93 %). ЖХМС (способ 8 мин) = 7,624 мин. Наблюдаемая масса (ИЭР+): 719,8 (M+H)+.
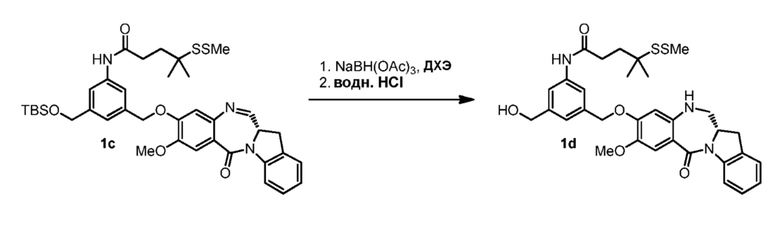
Соединение 1c (686 мг, 0,953 ммоль) растворяют в ДХЭ (6,35 мл). Натрия триацетоксиборгидрид (400 мг, 1,91 ммоль) добавляют к реакционной смеси и перемешивают при кт в течение 3 час. Реакционную смесь разбавляют ДХМ и промывают насыщенным раствором хлорида аммония, раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют с получением восстановленного иминного продукта. ЖХМС (способ 8 мин) = 4,363 мин. Наблюдаемая масса (ИЭР+): 720,75 (M+H)+.
Неочищенный восстановленный имин (680 мг, 0,942 ммоль) растворяют в ТГФ (5,23 мл). Добавляют HCl (водн. 5 М) (3,77 мл, 9,42 ммоль) и перемешивают при кт в течение 4 час. Реакционную смесь разбавляют ДХМ и промывают насыщенным раствором натрия бикарбоната, раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (градиент, MeOH/ДХМ) с получением соединения 1d (420 мг, выход 73 %, 2 стадии). ЖХМС (способ 8 мин) = 5,905 мин. Наблюдаемая масса (ИЭР+): 607,8 (M+H)+.
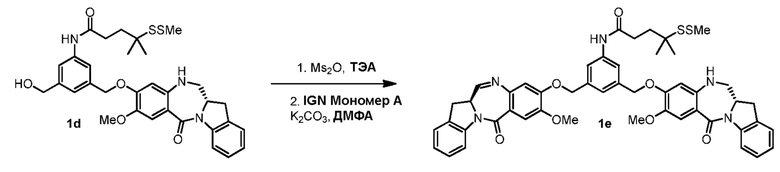
Соединение 1d (420 мг, 0,691 ммоль) растворяют в ДХМ (4,61 мл). Раствор охлаждают до минус 5 °C (на бане со льдом-раствором хлорида натрия) и в атмосфере аргона добавляют ТЭА (125 мкл, 0,898 ммоль), с последующим добавлением метансульфонового ангидрида (144 мг, 0,829 ммоль). Реакционную смесь перемешивают при минус 5 °C в атмосфере аргона в течение 1,5 час. Реакционную смесь гасят водой при минус 5 °C и нагревают до комнатной температуры. Смесь экстрагируют EtOAc (2x), и органическую фракцию промывают водой (2x). Органические фракции сушат над сульфатом натрия, фильтруют и концентрируют с получением мезилатированного продукта в виде коричневой пены. ЖХМС (способ 8 мин) = 6,380 мин. Наблюдаемая масса (ИЭР+): 685,7 (M+H)+.
Мезилат (470 мг, 0,582 ммоль) растворяют в ДМФА (3,88 мл). Добавляют IGN мономер A (189 мг, 0,641 ммоль), с последующим добавлением карбоната калия (121 мг, 0,874 ммоль) при комнатной температуре, и реакционную смесь перемешивают в течение ночи. К осажденному продукту добавляют воду (~5 мл). Суспензию перемешивают в течение 5 мин, затем фильтруют и сушат под вакуумом/N2. Неочищенный продукт очищают хроматографией на силикагеле (градиент, MeOH/ДХМ) с получением соединения 1e в виде твердого вещества желтого цвета (543 мг, выход 53 %). ЖХМС (способ 8 мин) = 6,804 мин. Наблюдаемая масса (ИЭР+): 883,7 (M+H)+.
Пример 2.
(S)-2-(((бензилокси)карбонил)амино)пропановую кислоту (5 г, 22,40 ммоль) и (S)-трет-бутил 2-аминопропанoата гидрoхлорид (4,48 г, 24,64 ммоль) растворяют в безводном ДМФА (44,8 мл). Добавляют ЭДК·HCl (4,72 г, 24,64 ммоль), HOBt (3,43 г, 22,40 ммоль) и ДИПЭА (9,75 мл, 56,0 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре в атмосфере аргона. Реакционную смесь разбавляют дихлорметаном и затем промывают насыщенным раствором хлорида аммония, насыщенным раствором бикарбоната натрия, водой и раствором хлорида натрия. Органическую фракцию сушат над сульфатом натрия и концентрируют. Неочищенное масло очищают хроматографией на силикагеле (гексаны/этилацетат) с получением соединения 2a (6,7 г, выход 85 %). 1H ЯМР (400 МГц, CDCl3): δ 7,38-7,31 (м, 5H), 6,53-6,42 (м, 1H), 5,42-5,33 (м, 1H), 5,14 (с, 2H), 4,48-4,41 (м, 1H), 4,32-4,20 (м, 1H), 1,49 (с, 9H), 1,42 (д, 3H, J = 6,8 Гц), 1,38 (д, 3H, J = 7,2 Гц).

Соединение 2a (6,7 г, 19,12 ммоль) растворяют в метаноле (60,7 мл) и воде (3,03 мл). Раствор продувают аргоном в течение пяти минут. Медленно добавляют палладий на угле (влажный, 10 %) (1,017 г, 0,956 ммоль). Реакционную смесь перемешивают в течение ночи в атмосфере водорода. Раствор фильтруют сквозь броунмиллерит, промывают метанолом и концентрируют. Осуществляют азеотропную перегонку с метанолом и ацетонитрилом, и полученное масло помещают непосредственно под глубокий вакуум с получением соединения 2b (4,02 г, выход 97 %) которое используют непосредственно на следующей стадии. 1H ЯМР (400 МГц, CDCl3): δ 7,78-7,63 (м, 1H), 4,49-4,42 (м, 1H), 3,55-3,50 (м, 1H), 1,73 (с, 2H), 1,48 (с, 9H), 1,39 (д, 3H, J = 7,2 Гц), 1,36 (д, 3H, J = 6,8 Гц).

Соединение 2b (4,02 г, 18,59 ммоль) и монометиладипат (3,03 мл, 20,45 ммоль) растворяют в безводном ДМФА (62,0 мл). Добавляют ЭДК·HCl (3,92 г, 20,45 ммоль), HOBt (2,85 г, 18,59 ммоль) и ДИПЭА (6,49 мл, 37,2 ммоль). Смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют дихлорметаном/метанолом (150 мл, 5:1) и промывают насыщенным раствором хлорида аммония, насыщенным раствором бикарбоната натрия и раствором хлорида натрия. Сушат над сульфатом натрия, фильтруют и концентрируют. Осуществляют азеотропную перегонку соединения с ацетонитрилом (5x), затем помещают под глубокий вакуум с помощью насоса при 35 °C с получением соединения 2c (6,66 г, выход 100 %). Неочищенный материал переносят на следующую стадию без очистки. 1H ЯМР (400 МГц, CDCl3): δ 6,75 (д, 1H, J = 6,8 Гц), 6,44 (д, 1H, J = 6,8 Гц), 4,52-4,44 (м, 1H), 4,43-4,36 (м, 1H), 3,65 (с, 3H), 2,35-2,29 (м, 2H), 2,25-2,18 (м, 2H), 1,71-1,60 (м, 4H), 1,45 (с, 9H), 1,36 (т, 6H, J = 6,0 Гц).
Соединение 2c (5,91 г, 16,5 ммоль) перемешивают в ТФУ (28,6 мл, 372 ммоль) и деминерализованной воде (1,5 мл) при комнатной температуре в течение трех часов. Реакционную смесь концентрируют с ацетонитрилом и помещают под глубокий вакуум с получением неочищенного соединения 2d в виде клейкого твердого вещества (5,88 г, выход 100 %). 1H ЯМР (400 МГц, CDCl3): δ 7,21 (д, 1H, J = 6,8 Гц), 6,81 (д, 1H, J = 7,6 Гц), 4,69-4,60 (м, 1H), 4,59-4,51 (м, 1H), 3,69 (с, 3H), 2,40-2,33 (м, 2H), 2,31-2,24 (м, 2H), 1,72-1,63 (м, 4H), 1,51-1,45 (м, 3H), 1,42-1,37 (м, 3H).
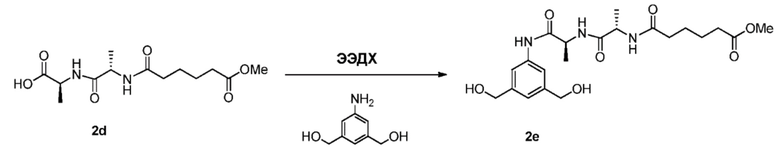
Соединение 2d (5,6 г, 18,52 ммоль) растворяют в безводном дихлорметане (118 мл) и безводном метаноле (58,8 мл). Добавляют (5-амино-1,3-фенилен)диметанол (2,70 г, 17,64 ммоль) и ЭЭДХ (8,72 г, 35,3 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают и добавляют этилацетат. Полученную суспензию фильтруют, промывают этилацетатом и сушат под вакуумом/N2 с получением соединения 2e (2,79 г, выход 36 %). 1H ЯМР (400 МГц, ДМСО-d6): δ 9,82 (с, 1H), 8,05, (д, 1H, J = 9,2 Гц), 8,01 (д, 1H, J = 7,2 Гц), 7,46 (с, 2H), 6,95 (3, 1H), 5,21-5,12 (м, 2H), 4,47-4,42 (м, 4H), 4,40-4,33 (м, 1H), 4,33-4,24 (м, 1H), 3,58 (с, 3H), 2,33-2,26 (м, 2H), 2,16-2,09 (м, 2H), 1,54-1,46 (м, 4H), 1,30 (д, 3H, J = 7,2 Гц), 1,22 (д, 3H, J = 4,4 Гц).
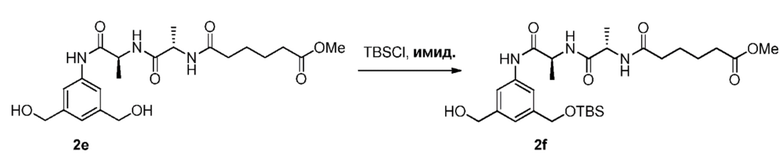
Диол 2e (1,0 г, 2,286 ммоль) растворяют в безводном ДМФА (7,6 мл). Добавляют TBSCl (0,482 г, 3,20 ммоль) и имидазол (0,467 г, 6,86 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2 час. Реакционную смесь гасят насыщенным раствором хлорида аммония и разбавляют водой и EtOAc. Водную фракцию однократно экстрагируют EtOAc, и объединенные органические фракции промывают водой и раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют. Неочищенный остаток очищают флеш-хроматографией на силикагеле (ДХМ/MeOH) с получением соединения 2f (360 мг, выход 28 %). ЖХМС (способ 8 мин, 40-98 %) = 2,35 мин. Наблюдаемая масса (ИЭР+): 574,4 (M+Na)+.

Соединение 2f (360 мг, 0,652 ммоль) растворяют в безводном дихлорметане (6,52 мл) и охлаждают на бане с ацетоном/льдом. Добавляют триэтиламин (227 мкл, 1,631 ммоль) и метансульфоновый ангидрид (146 мг, 0,816 ммоль). Реакционную смесь перемешивают при минус 10 °C на бане с ацетоном/льдом в течение 1 час. Реакционную смесь разбавляют холодным EtOAc и гасят ледяной водой. Органическую фракцию промывают ледяной водой и затем сушат над сульфатом натрия и сульфатом магния, фильтруют и концентрируют с получением неочищенного соединения 2g в виде хлопьевидного твердого вещества (390 мг, выход 95 %). ЖХМС (способ 8 мин, 40-98 %) = 2,81 мин; 5,86 мин (способ 8 мин, 5-98 %). Наблюдаемая масса (ИЭР-): 628,0 (M-H)-.
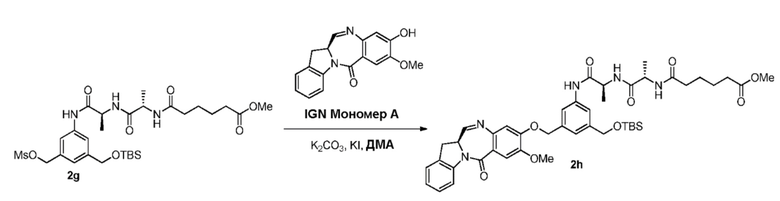
Мезилат 2g (390 мг, 0,619 ммоль) и IGN мономер A (264 мг, 0,897 ммоль) растворяют в безводном ДМА (7,47 мл). Добавляют карбонат калия (207 мг, 1,495 ммоль) и йодид калия (51,4 мг, 0,310 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь осаждают водой, фильтруют и остаток на фильтре промывают водой. Твердое вещество повторно растворяют в ДХМ, промывают водой, сушат над сульфатом магния и концентрируют с получением неочищенного соединения 2h (568 мг, выход 111 %). Продукт переносят без дополнительной очистки. ЖХМС (способ 8 мин, 5-98 %) = 6,23 мин. Наблюдаемая масса (ИЭР+): 827,8 (M+H)+.
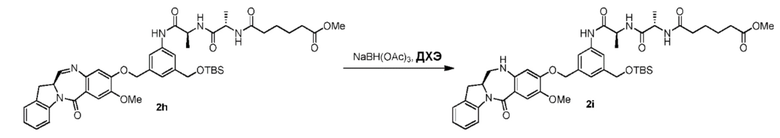
Соединение 2h (0,513 г, 0,619 ммоль) растворяют в ДХЭ (7,74 мл). Добавляют NaBH(OAc)3 (0,276 г, 1,239 ммоль), и смесь перемешивают при комнатной температуре в течение 1,5 час. Реакционную смесь разбавляют ДХМ, гасят насыщенным раствором хлорида аммония и промывают раствором хлорида натрия. Органическую фракцию сушат над сульфатом магния, фильтруют и концентрируют с получением соединения 2i. ЖХМС (15 мин способ) = 9,93 мин.
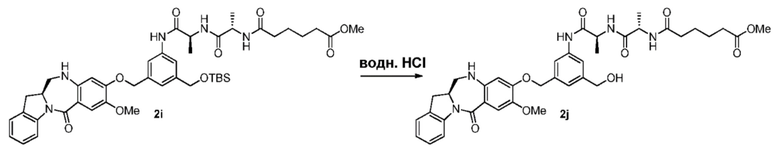
Соединение 2i (514 мг, 0,619 ммоль) растворяют в ТГФ (3,44 мл). При комнатной температуре добавляют 5 M водный HCl (1,24 мл, 6,19 ммоль), и реакционную смесь перемешивают в течение 1 час. Реакционную смесь разбавляют ДХМ/MeOH (20:1), органическую фракцию промывают насыщенным раствором бикарбоната натрия, раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Неочищенный остаток очищают хроматографией на силикагеле (ДХМ/MeOH) с получением соединения 2j (210 мг, выход 47 %). ЖХМС (способ 8 мин, 5-98 %) = 4,56 мин. Наблюдаемая масса (ИЭР+): 715,8 (M+H)+.
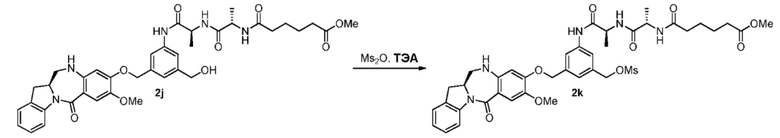
Соединение 2j (210 мг, 0,293 ммоль) растворяют в ДХМ (3,95 мл) и ДМФА (500 мкл) и охлаждают до минус 10 °C (баня со льдом/ацетоном). Добавляют ТЭА (57,2 мкл, 0,411 ммоль) и метансульфоновый ангидрид (46,6 мг, 0,260 ммоль), и реакционную смесь перемешивают в течение 3 час в атмосфере аргона. Реакционную смесь гасят холодной водой при минус 5 °C и разбавляют EtOAc. Водную фракцию экстрагируют холодным EtOAc (2×), и объединенные органические фракции промывают холодной водой (2×). Органическую фракцию сушат над безводным сульфатом натрия/магния, фильтруют и концентрируют. Неочищенный продукт 2k с помощью насоса помещают под глубокий вакуум и переносят на следующую стадию без очистки. ЖХМС (способ 8 мин, 5-98 %) = 5,06 мин. Наблюдаемая масса (ИЭР-): 791,8 (M-H)-.
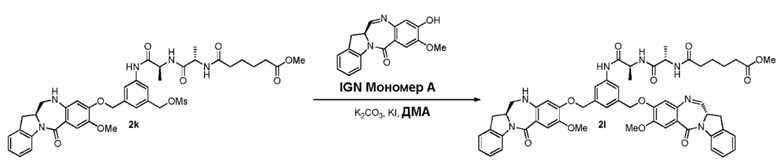
[267] Соединение 2k (233 мг, 0,293 ммоль) растворяют в ДМА (1,95 мл). IGN мономер A (103 мг, 0,352 ммоль) и карбонат калия (60,7 мг, 0,440 ммоль) добавляют при комнатной температуре, и реакционную смесь перемешивают в течение ночи. К реакционной смеси добавляют ДМ воду, и полученное твердое вещество отфильтровывают и промывают водой. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1), промывают водой, сушат над сульфатом магния, фильтруют и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (44 мг, выход 15 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 3.
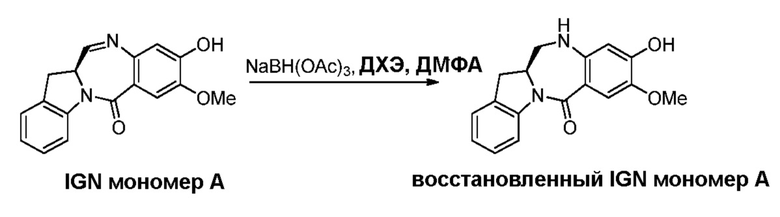
К раствору IGN мономера A (1,0 г, 3,4 ммоль) в ДХЭ (10 мл) и ДМФА (4 мл) добавляют триацетоксиборгидрид натрия (1,1 г, 5,1 ммоль, 1,5 экв.), и реакционную смесь перемешивают до тех пор, пока исходный материал полностью израсходуется. После того, как исходный материал полностью израсходовался, через 2 час при комнатной температуре, реакционную смесь гасят насыщ. раствором хлорида аммония (10 мл), и затем фракции разделяют. Водную фракцию однократно экстрагируют дихлорметаном (10 мл), объединенные органические фракции промывают водой (2 x 10 мл) и раствором хлорида натрия (10 мл). Органическую фракцию сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом с получением белого/коричневого порошка. Порошок промывают EtOAc (2 x 10 мл) и сушат под вакуумом с получением восстановленного IGN мономера A в виде твердого вещества белого цвета (0,87 г, 2,9 ммоль, выход 87 %) которое используют на следующей стадии без дополнительной очистки. Сверхвысокоэффективная ЖХМС, СВЭЖХМС (UPLCMS) (способ 2,5 мин) = 1,34 мин. Наблюдаемая масса (ИЭР+): 297,4 (M+H)+. 1H ЯМР (400 МГц, ДМСО-d6): δ 9,44 (с, 1H), 8,20 (д, J = 8,1 Гц, 1H), 7,30 – 7,23 (м, 2H), 7,22 – 7,12 (м, 1H), 7,01 (тд, J = 7,4, 1,1 Гц, 1H), 6,21 (с, 1H), 6,17 (д, J = 6,6 Гц, 1H), 4,37 (тдд, J = 10,1, 4,4, 1,9 Гц, 1H), 3,70 (с, 3H), 3,58 – 3,39 (м, 2H), 3,31 – 3,15 (м, 2H), 2,88 (дд, J = 16,9, 4,4 Гц, 1H).
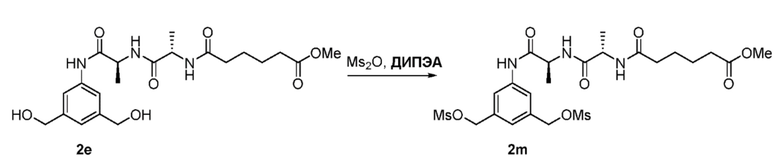
Раствор 2e (5,53 г, 12,6 ммоль) в ДХМ (81 мл) и ДМФА (64,9 мл) охлаждают до 0 oC, и затем добавляют ДИПЭА (6,13 мл, 37,9 ммоль, 3,0 экв.), с последующим добавлением по каплям раствора метансульфонового ангидрида (5,06 г, 29,1 ммоль, 2,3 экв.) в ДХМ (15 мл) / ДМФА (1 мл). Реакционную смесь перемешивают в течение 1 час, после чего гасят холодной водой. После промывания водой и раствором хлорида натрия раствор сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом с получением оранжевого масла, которое растирают с диэтиловым эфиром с получением бис-мезилата 2m (6,4 г, 10,8 ммоль, выход 85 %). ЖХМС (способ 8 мин) = 4,019 мин. Наблюдаемая масса (ИЭР+): 594,8 (M+H)+. Неочищенный материал переносят на следующую стадию без дополнительной очистки.
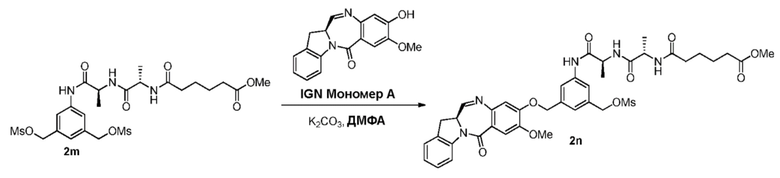
К раствору 2m (0,52 г, 0,88 ммоль) и IGN мономера A (0,18 г, 0,61 ммоль, 0,7 экв.) в ДМФА (7 мл) добавляют карбонат калия (0,24 г, 1,75 ммоль, 2,0 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакционную смесь гасят водой (30 мл) и экстрагируют ДХМ (3 x 15 мл). Органические фракции объединяют и промывают водой (3 x 60 мл), раствором хлорида натрия (60 мл), сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом с получением неочищенного желтого масла. Материал очищают хроматографией на силикагеле (ДХМ/(MeCN/MeOH (4/1) от 100/0 до 65/35) с получением желаемого продукта 2n (0,09 г, 0,12 ммоль, выход 13 %). СВЭЖХМС (способ 2,5 мин) = 1,46 мин. Наблюдаемая масса (ИЭР+): 792,6 (M+H)+.
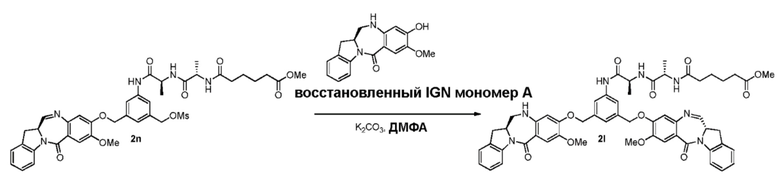
К раствору 2n (0,05 г, 0,06 ммоль) в ДМФА (0,48 мл, 6,2 ммоль) добавляют карбонат калия (0,02 г, 0,12 ммоль, 2,0 экв.), с последующим добавлением восстановленного IGN мономера A (0,02 г, 0,07 ммоль, 1,1 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакцию гасят водой, полученное твердое вещество отфильтровывают и промывают водой. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1), промывают водой, сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,03 г, 0,04 ммоль, выход 55 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+. 1H ЯМР (400 МГц, ДМСО-d6, приведено как смесь аддуктов воды): δ 10,10 (д, J = 3,7 Гц, 1H), 8,27 (д, J = 8,0 Гц, 1H), 8,21 – 8,10 (м, 1H), 8,05 (д, J = 7,4 Гц, 1H), 7,78 (дт, J = 8,5, 1,8 Гц, 2H), 7,43 – 7,13 (м, 7H), 7,16 – 6,98 (м, 2H), 6,49 (с, 1H), 6,36 (д, J = 13,1 Гц, 0,4H), 6,16 (д, J = 6,2 Гц, 0,4H), 5,80 (с, 0,4H), 5,67 (с, 0,4H), 5,57 (д, J = 5,6 Гц, 0,4H), 5,35 – 5,09 (м, 2H), 5,03 (т, J = 5,9 Гц, 2H), 4,81 – 4,72 (м, 0,4H), 4,60 (дт, J = 9,7, 5,0 Гц, 0,2H), 4,51 – 4,36 (м, 2H), 4,39 – 4,23 (м, 1H), 4,17 (тд, J = 9,7, 2,9 Гц, 0,4H), 3,93 (с, 0,4H), 3,83 – 3,74 (м, 5H), 3,62 (с, 2H), 3,75 – 3,44 (м, 2H), 3,32 (д, J = 11,6 Гц, 1H), 3,19 – 3,07 (м, 1H), 2,95 (дд, J = 17,1, 4,3 Гц, 1H), 2,38 – 2,29 (м, 1H), 2,18 (м, 1H), 1,56 (м, J = 3,9 Гц, 4H), 1,41 – 1,31 (м, 3H), 1,30 – 1,14 (м, 3H).
Пример 4.
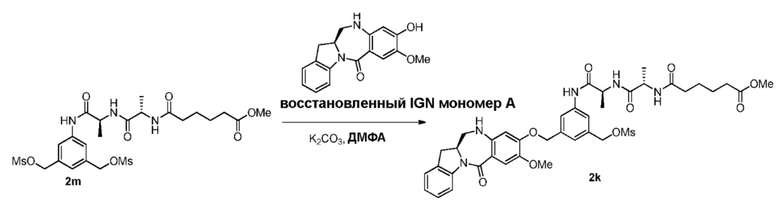
К раствору 2m (0,88 г, 1,47 ммоль) в ДМФА (11 мл) добавляют восстановленный IGN мономер A (0,26 г, 0,88 ммоль, 0,6 экв.), с последующим добавлением карбоната калия (0,41 мг, 2,95 ммоль, 2,0 экв.). После перемешивания в течение 12 час реакционную смесь разбавляют водой (50 мл) и EtOAc (30 мл). Водную фракцию экстрагируют EtOAc (3 x 10 мл). Объединенные органические фракции промывают раствором хлорида натрия (20 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют, и неочищенную смесь очищают хроматографией на силикагеле (ДХМ/MeOH) с получением желаемого продукта 2k (0,11 г, 0,14 ммоль, выход 10 %). ЖХМС (способ 8 мин) = 5,013 мин. Наблюдаемая масса (ИЭР+): 794,3 (M+H)+.
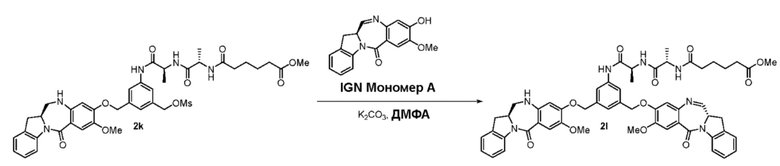
К раствору 2k (0,11 г, 0,14 ммоль) в ДМФА (2 мл) добавляют карбонат калия (0,04 г, 0,29 ммоль, 2,0 экв.) Добавляют IGN мономер A (0,04 г, 0,14 ммоль, 1,0 экв.), и реакционную смесь перемешивают при кт в течение 12 час. Реакцию гасят водой (10 мл), полученное твердое вещество отфильтровывают и промывают водой. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1), промывают водой (10 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,08 г, 0,09 ммоль, выход 59 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 5.
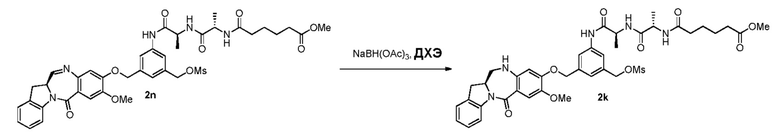
К раствору 2n (0,1 г, 0,13 ммоль) в ДХЭ (2 мл) добавляют триацетоксиборгидрид натрия (0,03г, 0,13 ммоль, 1,0 экв.), и реакционную смесь перемешивают при кт в течение 2 час. Реакцию гасят насыщенным раствором хлорида аммония (2 мл), и фракции разделяют. Водную фракцию экстрагируют ДХМ (5 мл), объединенные органические фракции промывают водой, раствором хлорида натрия, сушат над сульфатом магния и фильтруют. Неочищенное твердое вещество желтого цвета очищают с применением хроматографии на силикагеле (EtOAc/MeOH (95/5)) с получением желаемого восстановленного продукта 2k (0,035 г, 0,044 ммоль, выход 35 %). ЖХМС (способ 8 мин) = 5,021 мин. Наблюдаемая масса (ИЭР+): 794,3 (M+H)+.
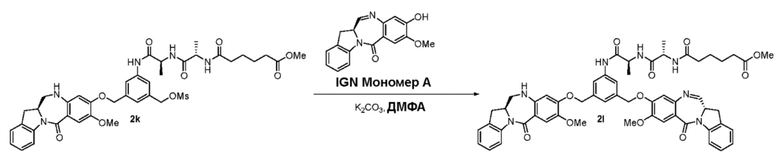
К раствору 2k (0,035 г, 0,044 ммоль) в ДМФА (1,0 мл) добавляют карбонат калия (0,013 г, 0,09 ммоль, 2,0 экв.). Добавляют IGN мономер A (0,013 г, 0,04 ммоль, 1,0 экв.), и реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакцию гасят водой (10 мл), полученное твердое вещество отфильтровывают и промывают водой. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1, 20 мл), промывают водой (20 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,017 г, 0,01 ммоль, выход 38 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин). Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 6.

К раствору 2f (8,8 г, 16,0 ммоль) в ДМФА (100 мл) добавляют пиридин (4,51 мл, 55,8 ммоль, 3,5 экв.). Реакционную смесь охлаждают до 0 oC, затем по каплям добавляют метансульфонилхлорид (2,5 мл, 31,9 ммоль, 2,0 экв.), и реакционную смесь перемешивают в течение 2 час. Смесь гасят насыщ. раствором бикарбоната натрия (30 мл), добавляют EtOAc и фракции разделяют. Водную фракцию экстрагируют EtOAc (3 x 50 мл), объединенные органические фракции промывают водой, раствором хлорида натрия, сушат над сульфатом магния и фильтруют. Растворитель удаляют, и неочищенное твердое вещество белого цвета 2o используют на следующей стадии без очистки (6,2 г, 10,9 ммоль, 68 %). СВЭЖХМС (способ 2,5 мин) = 1,96 мин. Наблюдаемая масса (ИЭР+): 570,7 (M+H)+.

К раствору 2o (1,7 г, 2,98 ммоль) в ТГФ (36,6 мл) добавляют ДИПЭА (2,1 мл, 11,9 ммоль, 4,0 экв.), с последующим добавлением HF-пиридина (0,84 мл, 6,0 ммоль, 2,0 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 3 час. Реакцию гасят насыщ. раствором бикарбоната натрия (20 мл), и затем фракции разделяют. Водную фракцию экстрагируют EtOAc (3 x 10 мл). Объединенные органические фракции промывают раствором хлорида натрия (30 мл), сушат над сульфатом магния, фильтруют и растворитель удаляют под вакуумом с получением неочищенного масла белого цвета, которое очищают хроматографией на силикагеле (ДХМ/MeOH) с получением желаемого продукта 2p в виде твердого вещества белого цвета (0,75 г, 1,6 ммоль, выход 55 %). СВЭЖХМС (способ 2,5 мин) = 1,23 мин. Наблюдаемая масса (ИЭР+): 456,4 (M+H)+.
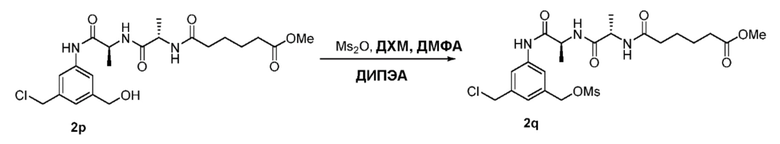
К раствору 2p (0,65 г, 1,43 ммоль) в ДХМ (10 мл) и ДМФА (2 мл) добавляют ДИПЭА (0,51 мл, 2,85 ммоль, 2,0 экв.), и реакционную смесь охлаждают до 0 oC. Медленно добавляют раствор метансульфонового ангидрида (0,3 г, 1,71 ммоль) в ДХМ (2 мл). Реакция завершается через 30 мин, реакционную смесь гасят водой (20 мл), фракции экстрагируют, водную фракцию промывают ДХМ (2 x 10 мл). Органические фракции объединяют, промывают водой (20 мл), раствором хлорида натрия (10 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом с получением желаемого продукта 2q (0,76 г, 1,42 ммоль, выход 100 %), который в неочищенном виде переносят на следующую стадию без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 1,37 мин. Наблюдаемая масса (ИЭР+): 534,4 (M+H)+.
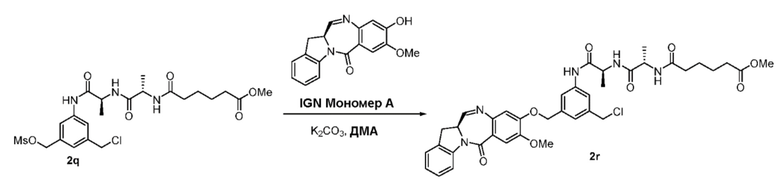
К раствору 2q (0,76 г, 1,42 ммоль) в ДМА (13 мл) добавляют карбонат калия (0,59 г, 4,27 ммоль), с последующим добавлением раствора IGN мономера A (0,5 г, 1,71 ммоль) в ДМА (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакцию гасят водой (30 мл), и смесь перемешивают в течение 10 мин. Твердое вещество отфильтровывают, затем растворяют в ДХМ/MeOH (9/1, 20 мл) и промывают раствором хлорида натрия (10 мл). Органическую фракцию отделяют и сушат над сульфатом магния, фильтруют и концентрируют под вакуумом с получением неочищенного твердого вещества желтого цвета 2r (0,76 г, 1,04 ммоль, выход 73 %), которое в неочищенном виде переносят на следующую стадию без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 1,55 мин. Наблюдаемая масса (ИЭР+): 732,9 (M+H)+.
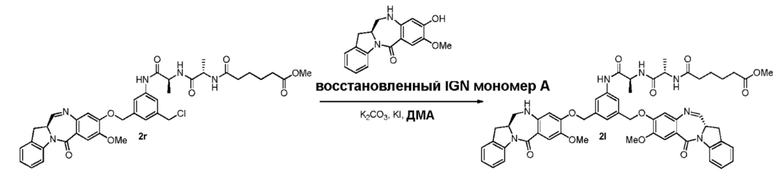
К раствору 2r (0,26 г, 0,36 ммоль) в ДМА (10 мл) добавляют йодид калия (0,06 г, 0,355 ммоль, 1,0 экв.), восстановленный IGN мономер A (0,1 г, 0,37 ммоль, 1,05 экв.) и карбонат калия (0,15 г, 1,06 ммоль, 3,0 экв.). Реакционную смесь нагревают до 40 oC и перемешивают в течение 4 час. Реакцию гасят водой (20 мл), и смесь перемешивают в течение 10 мин. Полученное твердое вещество отфильтровывают. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1, 20 мл), промывают водой (20 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,097 г, 0,097 ммоль, выход 28 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 7.

К раствору 2r (0,76 г, 1,04 ммоль) в ДХЭ (10 мл) добавляют ДМФА (3,0 мл), с последующим добавлением триацетоксиборгидрида натрия (0,33 г, 1,56 ммоль) при 0 oC. Реакционную смесь перемешивают при комнатной температуре в течение 4 час. Реакцию гасят насыщ. раствором хлорида аммония (20 мл), и фракции разделяют. Водную фракцию экстрагируют ДХМ (3 x 10 мл), объединенные органические фракции промывают водой (10 мл), раствором хлорида натрия (10 мл), сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом с получением желаемого неочищенного материала 2s в виде масла (0,65 г, 0,88 ммоль, выход 85 %), которое используют на следующей стадии без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 1,80 мин. Наблюдаемая масса (ИЭР+): 735,3 (M+H)+.

К раствору 2s (0,65 г, 0,88 ммоль) в ДМА (15 мл) добавляют карбонат калия (0,25 г, 1,78 ммоль, 2,0 экв.), с последующим добавлением йодида калия (0,073 г, 0,44 ммоль, 0,5 экв.,) и раствор IGN мономера A (0,29 г, 0,974 ммоль, 1,1 экв.) в ДМА (2 мл) добавляют к реакционной смеси при комнатной температуре. Реакционную смесь нагревают до 40 oC, выдерживая при этой температуре в течение 5 час. Реакцию гасят водой (30 мл), и затем твердое вещество отфильтровывают. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1, 30 мл), промывают водой (20 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток (0,78 г) очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,43 г, 0,43 ммоль, выход 49 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 8.
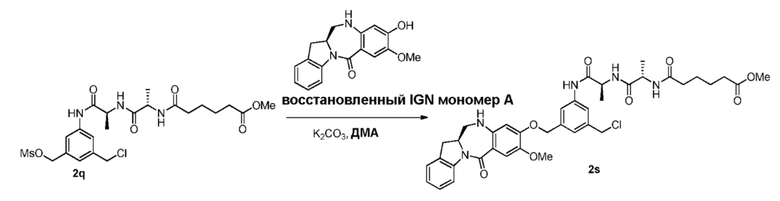
К раствору 2q (0,14 г, 0,27 ммоль) в ДМА (3 мл) добавляют карбонат калия (0,11 г, 0,81 ммоль), с последующим добавлением раствора восстановленного IGN мономера A (0,084 г, 0,28 ммоль) в ДМА (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакцию гасят водой (20 мл), и смесь перемешивают в течение 10 мин. Твердое вещество отфильтровывают, затем растворяют в ДХМ/MeOH (9/1, 20 мл) и промывают раствором хлорида натрия (10 мл). Органическую фракцию отделяют и сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом. Неочищенный материал очищают хроматографией на силикагеле с применением ДХМ (MeOH/EtOAc, 1/4) с получением желаемого продукта 2s (0,08 г, 0,11 ммоль, выход 40 %). СВЭЖХМС (способ 2,5 мин) = 1,63 мин. Наблюдаемая масса (ИЭР+): 735,2 (M+H)+
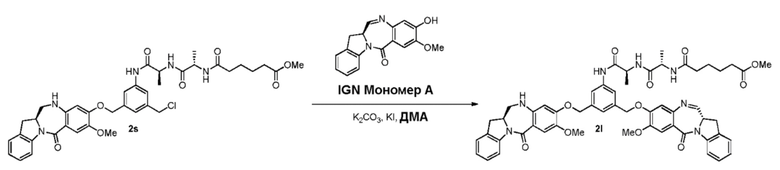
К раствору 2s (0,06 г, 0,09 ммоль) в ДМА (2 мл) добавляют карбонат калия (0,025 г, 0,18 ммоль), с последующим добавлением йодида калия (0,007 г, 0,044 ммоль). Раствор IGN мономера A (0,03 г, 0,097 ммоль) в ДМА (1 мл) добавляют к реакционной смеси при комнатной температуре. Реакционную смесь нагревают при 40 ºC в течение 5 час. Реакционную смесь охлаждают и гасят водой (20 мл), твердое вещество отфильтровывают. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1, 20 мл), промывают водой (10 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток (0,07 г) очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,035 г, 0,035 ммоль, выход 51 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 9.

К раствору 2h (0,85 г, 1,027 ммоль) в ТГФ (9 мл) добавляют ДИПЭА (0,54 мл, 3,1 ммоль, 3,0 экв.), с последующим добавлением HF-пиридина (0,3 мл, 2,053 ммоль, 2,0 экв.) при комнатной температуре. Реакционную смесь перемешивают в течение 3 часов при комнатной температуре. Реакцию гасят насыщ. раствором бикарбоната натрия (10 мл), фракции разделяют, и водную фракцию экстрагируют ДХМ (3 x 10 мл). Объединенные органические фракции промывают раствором хлорида натрия (10 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом с получением неочищенного продукта в виде твердого вещества, которое промывают EtOAc с получением желаемого продукта 2t (0,64 г, 0,89 ммоль, выход 87 %). СВЭЖХМС (способ 2,5 мин) = 1,36 мин. Наблюдаемая масса (ИЭР+): 714,6 (M+H)+.

К раствору 2t (0,23 г, 0,322 ммоль) в дихлорметане (3 мл) добавляют ДИПЭА (0,11 мл, 0,644 ммоль, 2,0 экв.), с последующим добавлением метансульфонового ангидрида (0,084 г, 0,48 ммоль, 1,5 экв.) в виде раствора в ДХМ (1 мл) при 0 oC. Реакционную смесь перемешивают в течение 1 час. Реакционную смесь гасят водой (3 мл) и разбавляют ДХМ (3 мл). Фракции разделяют, органическую фракцию промывают раствором хлорида натрия (3 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом и неочищенный материал 2n (0,25 г, 0,31 ммоль, выход 98 %) используют на следующей стадии без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 1,45 мин. Наблюдаемая масса (ИЭР+): 792,5 (M+H)+.
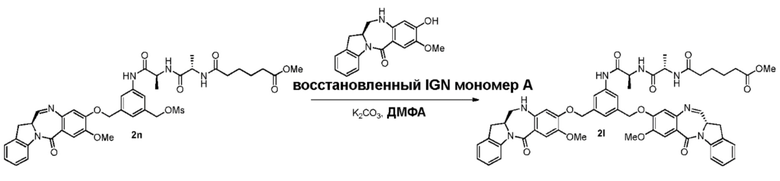
К раствору 2n (0,02 г, 0,027 ммоль) в ДМФА (0,2 мл) добавляют карбонат калия (0,007г, 0,053 ммоль, 2,0 экв.), с последующим добавлением восстановленного IGN мономера A (0,009г, 0,029 ммоль, 1,1 экв.), и реакционную смесь перемешивают при комнатной температуре в течение 18 час. К реакционной смеси добавляют воду (3 мл), полученное твердое вещество отфильтровывают. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1, 5 мл), промывают водой (5 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,005 г, 0,005 ммоль, выход 19 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 10.

К раствору 2t (0,02 г, 0,031 ммоль) в ТГФ (2 мл) добавляют ДИПЭА (0,016 мл, 0,092 ммоль, 3,0 экв.), с последующим добавлением раствора дибромтрифенилфосфорана (0,03 г, 0,062 ммоль, 2,0 экв.) в ТГФ (0,5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 12 час. Реакцию останавливают выпариванием растворителя, и затем неочищенный материал очищают хроматографией на силикагеле с получением 2u (0,006 г, 0,007 ммоль, выход 25 %). СВЭЖХМС (способ 2,5 мин) = 1,56 мин. Наблюдаемая масса (ИЭР+): 778,2 (M+H)+.
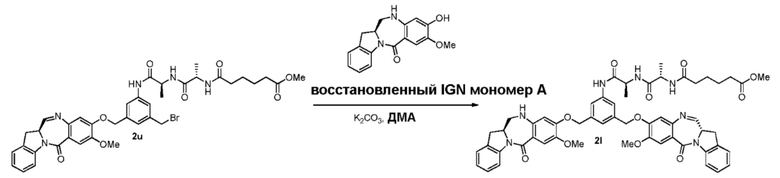
К раствору 2u (0,006 г, 7,73 мкмоль) в ДМА (1 мл) добавляют восстановленный IGN мономер A (0,003 г, 9,27 мкмоль), с последующим добавлением карбоната калия (0,002 г, 0,015 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 18 час. Воду (3 мл) добавляют к реакционной смеси, полученное твердое вещество отфильтровывают и промывают водой. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1, 5 мл), промывают водой (5 мл), сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (ACN/H2O) с получением 2l (0,001 г, 0,001 ммоль, выход 13 %). ЖХМС (способ 8 мин, 5-98 %) = 5,4 мин. Наблюдаемая масса (ИЭР+): 991,7 (M+H)+.
Пример 11.
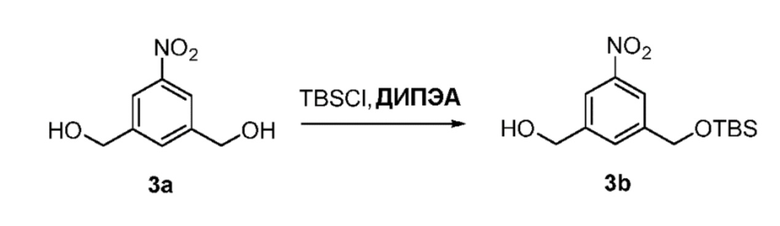
К раствору (5-нитро-1,3-фенилен)диметанола 3a (4,0 г, 21,84 ммоль) в ДХМ (40 мл) и ДМФА (5 мл) добавляют ДИПЭА (3,86 мл, 21,84 ммоль, 1,0 экв.), с последующим добавлением TBSCl (3,29 г, 21,84 ммоль, 1,0 экв.) в виде раствора в ДМФА (5 мл). Реакционную смесь перемешивают при 0 oC в течение 1 час. Реакцию гасят насыщ. раствором хлорида аммония (20 мл), и фракции разделяют. Водную фракцию экстрагируют ДХМ (2 x 20 мл), объединенные органические фракции промывают водой (2 x 50 мл), раствором хлорида натрия, сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом с получением неочищенного масла желтого цвета. Неочищенный продукт очищают хроматографией на силикагеле (ДХМ/MeOH) с получением желаемого продукта 3b (3,69 г, 12,41 ммоль, выход 57 %). СВЭЖХМС (способ 2,5 мин) = 1,96 мин. Наблюдаемая масса (ИЭР+): 298,5 (M+H)+.

К раствору 3b (2,0 г, 6,72 ммоль) в ДМФА (50 мл) добавляют пиридин (1,6 мл, 20,17 ммоль, 3,0 экв.), с последующим добавлением метансульфонилхлорида (1,1 мл, 13,45 ммоль, 2,0 экв.) при 0 oC. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 3 час. Реакцию гасят насыщ. раствором бикарбоната натрия (20 мл), и фракции разделяют. Водную фракцию экстрагируют EtOAc (3 x 30 мл). Объединенные органические фракции промывают водой (2 x 100 мл), раствором хлорида натрия (100 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом, и неочищенный материал 3c (2,0г, 6,7 ммоль, выход 94 %) переносят на следующую стадию без очистки. СВЭЖХМС (способ 2,5 мин) = 2,22 мин. Наблюдаемая масса (ИЭР+): 316,7 (M+H)+.

К раствору 3c (2,0 г, 6,33 ммоль) в ТГФ (38,9 мл) добавляют ДИПЭА (5,5 мл, 31,6 ммоль, 5,0 экв.), с последующим добавлением HF-пиридина (2,7 мл, 19,0 ммоль, 3,0 экв.), и реакционную смесь перемешивают при комнатной температуре в течение 2 час. Затем реакцию гасят насыщ. раствором бикарбоната натрия (100 мл). Фракции разделяют, и затем водную фракцию экстрагируют EtOAc (3 x 20 мл). Далее объединенные органические фракции промывают водой (30 мл), раствором хлорида натрия (30 мл), сушат над сульфатом магния и фильтруют. Избыток растворителя удаляют под вакуумом с получением желаемого продукта 3d (1,1г, 5,46 ммоль, выход 86 %). СВЭЖХМС (способ 2,5 мин) = 1,31 мин. Наблюдаемая масса (ИЭР+): 202,4 (M+H)+.
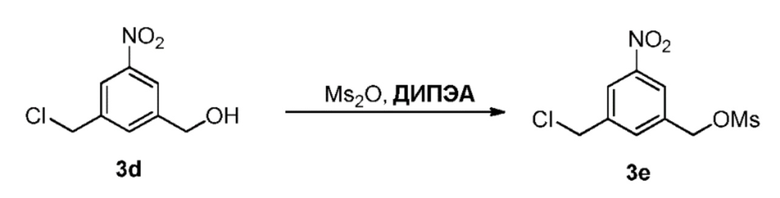
К раствору 3d (1,0 г, 4,96 ммоль) в ДХМ (10 мл) добавляют ДИПЭА (2,6 мл, 14,9 ммоль, 3,0 экв.) при 0 oC, и затем раствор метансульфонового ангидрида (1,1 г, 6,45 ммоль, 1,3 экв.) в ДХМ добавляют к реакционной смеси. Реакционную смесь перемешивают в течение 1 час. Реакцию гасят водой (10 мл), фракции разделяют и водную фракцию экстрагируют ДХМ (2 x 20 мл). Объединенные органические фракции промывают насыщ. раствором бикарбоната натрия (10 мл), раствором хлорида натрия (20 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом, и неочищенный материал 3e (1,3 г, 4,65 ммоль, выход 94 %) используют на следующей стадии без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 1,51 мин. Наблюдаемая масса (ИЭР+): 280,6 (M+H)+.
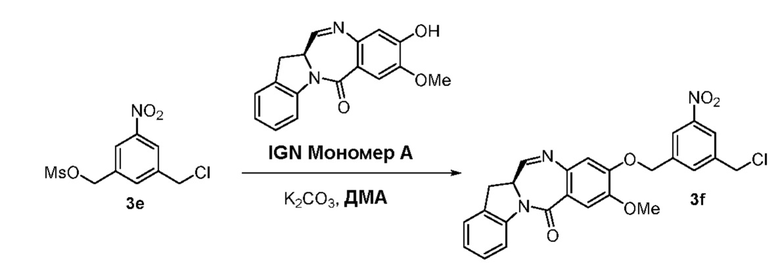
К раствору 3e (0,4 г, 1,43 ммоль) и карбоната калия (0,6 г, 4,29 ммоль, 3,0 экв.) в ДМА (13,4 мл) добавляют раствор IGN мономера A (0,46 г, 1,57 ммоль, 1,1 экв.) в ДМА (2 мл) при комнатной температуре, и реакционную смесь перемешивают в течение 5 час. Реакцию гасят водой (30 мл), фракции разделяют, и водную фракцию экстрагируют EtOAc (3 x 30 мл). Объединенные органические фракции промывают водой (30 мл), раствором хлорида натрия (30 мл), сушат над сульфатом магния, и растворитель удаляют под вакуумом. Неочищенное масло очищают хроматографией на силикагеле с применением ДХМ/MeOH, с получением соединения 3f (0,37г, 0,77 ммоль, выход 54 %). СВЭЖХМС (способ 2,5 мин) = 1,69 мин. Наблюдаемая масса (ИЭР+): 478,3 (M+H)+.
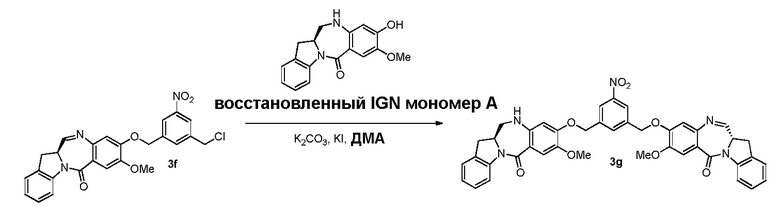
К раствору 3f (0,11 г, 0,23 ммоль) в ДМА (3,0 мл) добавляют карбонат калия (0,095 г, 0,69 ммоль, 3,0 экв.), с последующим добавлением йодида калия (0,02 г, 0,11 ммоль, 0,5 экв.). Добавляют раствор восстановленного IGN мономера A (0,07 г, 0,25 ммоль, 1,1 экв.) в ДМА (1 мл). Далее реакционную смесь осторожно нагревают при 35 oC в течение 5 час. Реакцию гасят водой, и твердое вещество отфильтровывают. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1), промывают водой, сушат с помощью сульфата магния, отфильтровывают и концентрируют. Неочищенный остаток (0,13 г) очищают ОФВЭЖХ (ACN/H2O) с получением 3g (0,063 г, 0,085 ммоль, выход 36 %). СВЭЖХМС (способ 2,5 мин) = 1,79 мин. Наблюдаемая масса (ИЭР+): 738,3 (M+H)+. 1H ЯМР (400 МГц, ДМСО-d6, приведены в виде смеси аддуктов воды) 1H ЯМР (400 МГц, ДМСО-d6): δ 8,43 – 8,36 (м, 2H), 8,27 (д, J = 8,1 Гц, 1H), 8,13 – 8,02 (м, 2H), 7,44 – 7,14 (м, 6H), 7,14 – 6,99 (м, 2H), 6,79 (с, 0,5H), 6,56 (с, 0,5H), 6,50 (д, J = 2,2 Гц, 1H), 6,39 (д, J = 6,9 Гц, 1H), 6,17 (д, J = 6,8 Гц, 0,5H), 5,69 (с, 0,5H), 5,59 (д, J = 5,7 Гц, 0,5H), 5,47 – 5,27 (м, 4H), 5,03 (т, J = 6,1 Гц, 0,5H), 4,77 (дд, J = 9,1, 6,8 Гц, 0,5H), 4,61 (дт, J = 9,7, 5,1 Гц, 0,15H), 4,50 – 4,39 (м, 0,5H), 4,27 (дд, J = 10,9, 4,2 Гц, 0,5H), 4,16 (тд, J = 9,6, 2,9 Гц, 0,5H), 3,95 (с, 0,5H), 3,89 – 3,76 (м, 6H), 3,76 – 3,44 (м, 4H), 3,20 – 3,08 (м, 1H), 2,96 (дд, J = 17,0, 4,4 Гц, 1H).
Пример 12.

К раствору 3e (0,45 г, 1,61 ммоль) в ДМА (15,1 мл) добавляют карбонат калия (0,67 г, 4,83 ммоль, 3,0 экв.), с последующим добавлением раствора восстановленного IGN мономера A (0,5 г, 1,69 ммоль, 1,1 экв.) в ДМА (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 час. Реакцию гасят водой (30 мл), и смесь перемешивают в течение 10 мин. Твердое вещество отфильтровывают, затем растворяют в ДХМ/MeOH (9/1, 30 мл) и промывают раствором хлорида натрия (20 мл). Органическую фракцию отделяют и сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом. Неочищенный материал очищают хроматографией на силикагеле с применением гексана/EtOAc, с получением соединения 3h (0,28 г, 0,58 ммоль, выход 36 %) в виде бесцветного масла. СВЭЖХМС (способ 2,5 мин) = 1,82 мин. Наблюдаемая масса (ИЭР+): 480,3 (M+H)+.
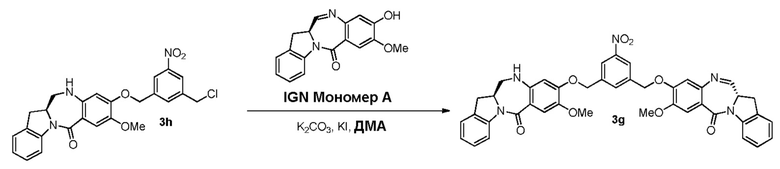
К раствору 3h (0,27 г, 0,56 ммоль) в ДМА (10 мл) добавляют карбонат калия (0,16 г, 1,12 ммоль, 2,0 экв.), с последующим добавлением йодида калия (0,05 г, 0,28 ммоль, 0,05 экв.). Раствор IGN мономера A (0,18 г, 0,62 ммоль, 1,1 экв.) в ДМА (2 мл) добавляют к реакционной смеси при комнатной температуре. Затем реакционную смесь перемешивают при 40 oC в течение 3 час. Реакцию гасят водой (20 мл), твердое вещество отфильтровывают и промывают водой. Неочищенное твердое вещество желтого цвета растворяют в ДХМ/MeOH (9/1, 30 мл), затем промывают водой (10 мл), сушат над сульфатом магния и отфильтровывают. Растворитель удаляют под вакуумом с получением неочищенного твердого вещества желтого цвета. Неочищенный продукт очищают хроматографией на силикагеле с применением ДХМ/MeOH (от 0 % до 5 % MeOH/ДХМ) с получением продукта 3g в виде желтого порошка (0,35 г, 0,48 ммоль, выход 86 %). СВЭЖХМС (способ 2,5 мин) = 1,79 мин (способ 2,5 мин). Наблюдаемая масса (ИЭР+): 738,4 (M+H)+.
Пример 13.
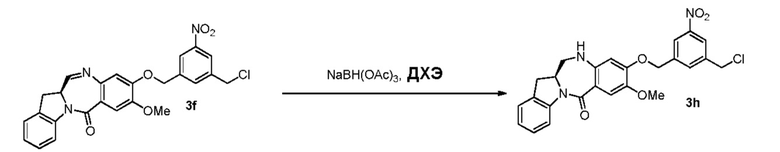
К раствору 3f (0,15 г, 0,31 ммоль) в ДХЭ (2 мл) добавляют триацетоксиборгидрид натрия (0,067 г, 0,31 ммоль, 1,0 экв.), и реакционную смесь перемешивают при комнатной температуре в течение 1 час. Реакцию гасят насыщ. раствором хлорида аммония (1 мл), и затем фракции разделяют. Водную фракцию экстрагируют ДХМ (3 x 10 мл), объединенные органические фракции промывают раствором хлорида натрия (20 мл), сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом. Неочищенное масло коричневого цвета очищают хроматографией на силикагеле с получением желаемого продукта 3h (0,08 г, 0,16 ммоль, выход 52 %). СВЭЖХМС (способ 2,5 мин) = 1,80 мин. Наблюдаемая масса (ИЭР+): 480,5 (M+H)+.
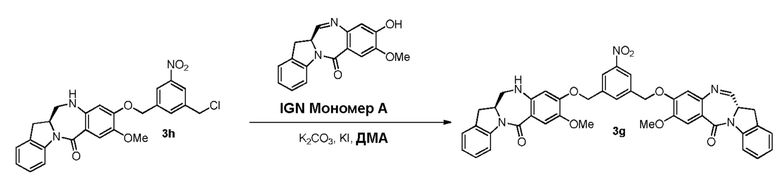
К раствору 3h (0,07г, 0,16 ммоль) в ДМА (2 мл) добавляют карбонат калия (0,07 г, 0,47 ммоль, 3,0 экв.), с последующим добавлением йодида калия (0,013 г, 0,08 ммоль, 0,05 экв.), и затем добавляют раствор IGN мономера A (0,05 г, 0,17 ммоль, 1,1 экв.) в ДМА (0,5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 12 час. К смеси добавляют воду (20 мл), и смесь перемешивают в течение 10 мин; в этой точке твердое вещество отфильтровывают. Твердое вещество солюбилизируют в ДХМ (10 мл), и затем промывают раствором хлорида натрия (10 мл). Органическую фракцию сушат над сульфатом магния и фильтруют. Растворитель удаляют с получением желтого масла (0,09 г, 0,12 ммоль, выход 80 %). СВЭЖХМС (способ 2,5 мин) = 1,79 мин (способ 2,5 мин). Наблюдаемая масса (ИЭР+): 738,5 (M+H)+.
Пример 14.
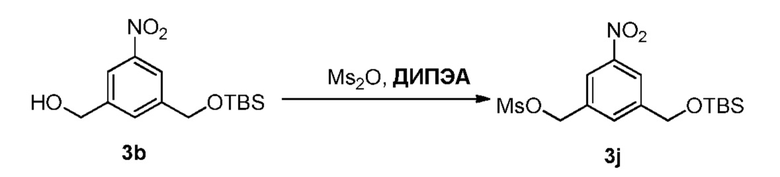
К раствору 3b (1,00 г, 3,4 ммоль) в ДХМ (33 мл) добавляют ДИПЭА (1,781 мл, 10,09 ммоль, 3,0 экв.), с последующим добавлением раствора метансульфонового ангидрида (0,703 г, 4,03 ммоль, 1,2 экв.) при 0 oC. Реакционную смесь перемешивают в течение 1 час. Растворитель выпаривают с получением неочищенного продукта 3j (1,2 г, 3,2 ммоль, выход 95 %) которое используют на следующей стадии без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 2,04 мин. Наблюдаемая масса (ИЭР+): 376,5 (M+H)+.
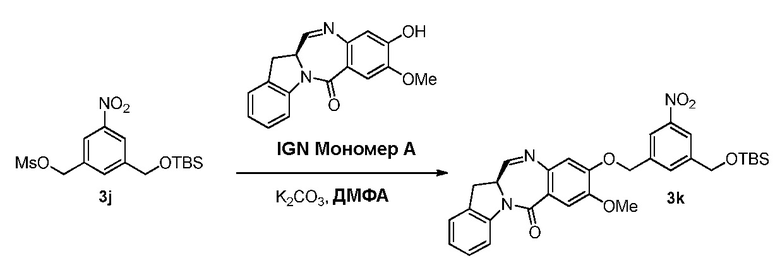
К раствору 3j (1,24 г, 3,30 ммоль) в ДМФА (26 мл) добавляют карбонат калия (0,91 г, 6,60 ммоль, 2,0 экв.), с последующим добавлением IGN мономера A (0,97 г, 3,30 ммоль, 1,0 экв.) при комнатной температуре в течение 12 час. Реакцию гасят водой (60 мл), твердое вещество отфильтровывают, и затем растворяют в ДХМ/MeOH (20/1, 20 мл). Органическую фракцию промывают раствором хлорида натрия, сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом, и неочищенный материал очищают хроматографией на силикагеле с получением желаемого продукта 3k (1,3 г, 2,27 ммоль, выход 69 %). СВЭЖХМС (способ 2,5 мин) = 2,12 мин (способ 2,5 мин). Наблюдаемая масса (ИЭР+): 574,4 (M+H)+.
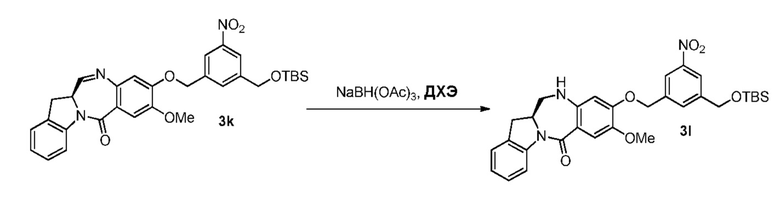
3k (0,63 г, 1,1 ммоль) растворяют в безводном ДХЭ (11 мл). Добавляют триацетоксиборгидрид натрия (0,70 г, 3,3 ммоль, 3,0 экв.), и реакционную смесь перемешивают в течение 1 час при комнатной температуре. Смесь гасят насыщ. раствором хлорида аммония (10 мл). Фракции разделяют, и водную фракцию экстрагируют ДХМ (2 x 20 мл). Объединенные органические фракции промывают раствором хлорида натрия (20 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют с получением 3l (0,58 г, 1,0 ммоль, выход 92 %). СВЭЖХМС (способ 8,0 мин) = 7,797 мин (способ 8,0 мин). Наблюдаемая масса (ИЭР+): 576,3 (M+H)+.
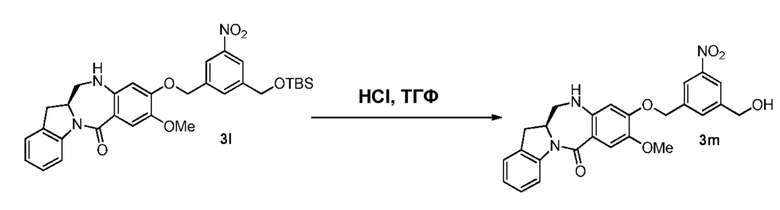
Раствор 3l (0,58 г, 1,0 ммоль) растворяют в безводном ТГФ (5 мл), и добавляют 5 М водный раствор хлористоводородной кислоты (2,01 мл, 10,07 ммоль). Смесь перемешивают при комнатной температуре в течение 2 час. Реакцию гасят насыщ. раствором бикарбоната натрия (5 мл), фракции разделяют, и водную фракцию экстрагируют ДХМ (2 x 10 мл). Объединенные органические фракции промывают раствором хлорида натрия (20 мл), сушат над сульфатом магния и концентрируют с получением твердого вещества ярко-оранжевого цвета. Полученное твердое вещество очищают хроматографией на силикагеле (ДХМ/MeOH) с получением соединения 3m (0,33 г, 0,71 ммоль, выход 71 %). СВЭЖХМС (способ 8,0 мин) = 5,166 мин. Наблюдаемая масса (ИЭР+): 462,1 (M+H)+.
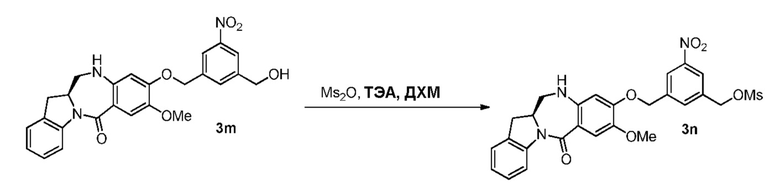
3m (0,1 г, 0,22 ммоль) растворяют в безводном ДХМ (1,5 мл) и безводном ДМФА (0,7 мл). Реакционную смесь охлаждают до 0 oC и добавляют триэтиламин (0,12 мл, 0,88 ммоль) и метансульфоновый ангидрид (0,08 г, 0,44 ммоль). Реакционную смесь перемешивают при 0 oC в течение 1 час. Реакционную смесь разбавляют этилацетатом (20 мл), промывают водой (2 x 20 мл), сушат над сульфатом магния, фильтруют и концентрируют. Соединение вначале очищают хроматографией на силикагеле (ДХМ/EtOAc), с последующей дополнительной очисткой ОФВЭЖХ (MeCN/вода), с получением желаемого продукта 3n (0,041 г, 0,076 ммоль, выход 34 %). Наблюдаемая масса (ИЭР+): 540,3 (M+H)+.
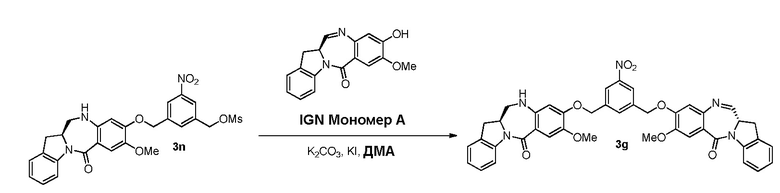
Соединение 3n (0,041 г, 0,076 ммоль) и IGN мономер A (0,027 г, 0,091 ммоль) растворяют в безводном ДМА (0,5 мл). Добавляют карбонат калия (0,012 г, 0,091 ммоль) и йодид калия (0,006 г, 0,038 ммоль), и смесь перемешивают в течение 12 час. К реакционной смеси добавляют воду (5 мл). Твердое вещество отфильтровывают, и затем повторно растворяют в ДХМ (20 мл) и промывают водой (10 мл). После сушки над сульфатом магния, фильтрации и концентрирования твердое вещество очищают ОФВЭЖХ (ACN/H2O) с получением 3g (0,012 г, 0,016 ммоль, выход 21 %). СВЭЖХМС (способ 2,5 мин) = 1,79 мин. Наблюдаемая масса (ИЭР+): 738,5 (M+H)+.
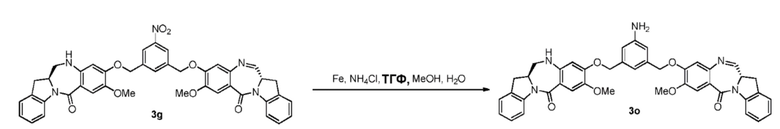
Соединение 3g (0,017 г, 0,023 ммоль) растворяют в безводном ТГФ (1 мл), безводном MeOH (0,5 мл) и воде (0,1 мл). Добавляют хлорид аммония (0,012 г, 0,23 ммоль, 10,0 экв.) и железо (0,006 г, 0,115 ммоль, 5,0 экв.). Смесь перемешивают при 60 oC в течение 2 час. Реакционную смесь охлаждают до комнатной темп., фильтруют сквозь броунмиллерит и промывают 20 % MeOH/ДХМ (10 мл). Фильтрат концентрируют, и неочищенный продукт очищают хроматографией на силикагеле (ДХМ/MeOH) с получением соединения 3o в виде твердого вещества белого цвета (0,012 г, 0,018 ммоль, выход 76 %). СВЭЖХМС (способ 2,5 мин) = 1,84 мин. Наблюдаемая масса (ИЭР+): 708,5 (M+H)+. 1H ЯМР (400 МГц, ДМСО-d6, приведено как смесь аддуктов воды, T = 330K): δ 8,26 (д, J = 7,9 Гц, 1H), 8,17 (д, J = 7,8 Гц, 1H), 8,03 (д, J = 4,5 Гц, 1H), 7,49 (с, 1H), 7,42 – 7,33 (м, 2H), 7,36 – 7,08 (м, 4H), 7,09 – 6,95 (м, 2H), 6,76 – 6,64 (м, 3H), 6,47 (с, 1H), 6,15 (д, J = 6,5 Гц, 1H), 5,11 (м, 2H), 4,98 (м, 2H), 4,58 (дт, J = 9,9, 4,7 Гц, 1H), 4,47 – 4,36 (м, 1H), 3,87 (м, 1H), 3,76 (с, 3H). 3,71 – 3,46 (м, 4H), 3,39 – 3,28 (м, 1H), 2,93 (дд, J = 16,8, 4,7 Гц, 1H).
Пример 15.
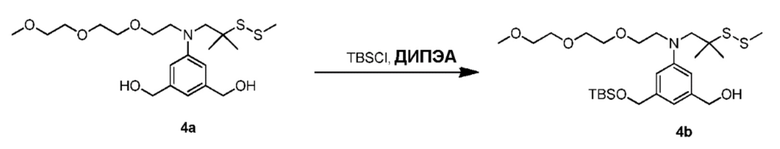
К раствору 4a (5,6 г, 12,9 ммоль, 1,0 экв.) в ДХМ (83 мл) добавляют ДИПЭА (6,77 мл, 38,7 ммоль, 3,0 экв.), с последующим добавлением раствора TBS-Cl (2,336 г, 15,50 ммоль, 1,2 экв.) в ДХМ (10 мл) при 0 oC. Реакционную смесь перемешивают при комнатной температуре в течение 3 час. Реакцию гасят насыщ. раствором хлорида аммония (30 мл), и фракции разделяют. Водный раствор экстрагируют ДХМ (2 x 30 мл), объединенные органические фракции промывают водой (2 x 50 мл), раствором хлорида натрия (50 мл), сушат над сульфатом магния, фильтруют и растворитель удаляют под вакуумом с получением неочищенного масла желтого цвета. Неочищенный продукт очищают хроматографией на силикагеле (гексан/EtOAc) с получением желаемого продукта 4b (3,0 г, 5,48 ммоль, выход 43 %). СВЭЖХМС (способ 2,5 мин) = 2,29 мин. Наблюдаемая масса (ИЭР+): 549,0 (M+H)+.
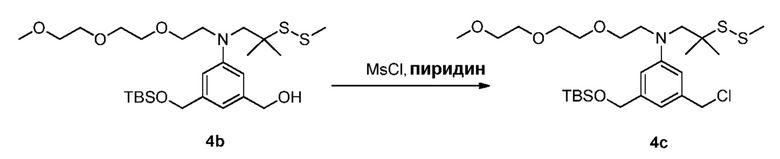
К раствору 4b (3,00 г, 5,48 ммоль, 1,0 экв.) в ДМФА (30 мл) добавляют пиридин (1,33 мл, 16,4 ммоль, 3,0 экв.), с последующим добавлением раствора метансульфонилхлорида (0,64 мл, 8,21 ммоль, 1,5 экв.) при 0 oC. Реакционную смесь перемешивают в течение 1 час, гасят насыщ. раствором бикарбоната натрия (30 мл) и разбавляют EtOAc (40 мл). Фракции разделяют, и водную фракцию экстрагируют EtOAc (2 x 30 мл). Объединенные органические фракции промывают водой (2 x40 мл), раствором хлорида натрия (40 мл), сушат над сульфатом магния, фильтруют, и растворитель удаляют под вакуумом с получением неочищенного продукта 4c (2,5 г, 4,41 ммоль, выход 81 %). СВЭЖХМС (способ 10,0 мин) = 8,23 мин. Наблюдаемая масса (ИЭР+): 567,6 (M+H)+.

К раствору 4c (2,5 г, 4,41 ммоль, 1,0 экв.) в ТГФ (43 мл) добавляют ДИПЭА (2,46 мл, 14,1 ммоль, 4,0 экв.), с последующим добавлением HF-пиридина (1,48 мл, 10,6 ммоль, 3,0 экв.), и реакционную смесь перемешивают при комнатной температуре в течение 2 час. Реакцию гасят насыщ. раствором бикарбоната натрия (100 мл), и фракции разделяют. Водную фракцию экстрагируют EtOAc (3 x 20 мл), объединенные органические фракции промывают водой (30 мл), раствором хлорида натрия (30 мл) сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом с получением желаемого продукта 4d (0,9 г, 2,0 ммоль, выход 56 %). СВЭЖХМС (способ 10,0 мин) = 5,20 мин. Наблюдаемая масса (ИЭР+): 435,4 (M+H)+.
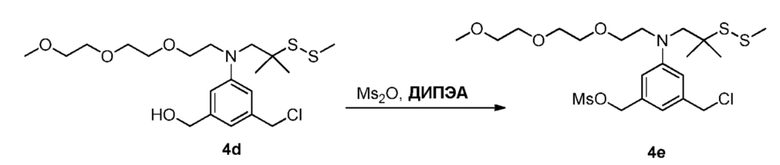
К раствору 4d (0,9 г, 2,0 ммоль, 1,0 экв.) в ДХМ (10 мл) добавляют ДИПЭА (0,69 мл, 3,98 ммоль, 2,0 экв.) при 0 oC, с последующим добавлением раствора метансульфонового ангидрида (0,52 г, 2,99 ммоль, 1,5 экв.) в ДХМ (2 мл). Реакционную смесь перемешивают в течение 1 час. Реакцию гасят водой (10 мл), фракции разделяют, и водную фракцию экстрагируют ДХМ (2 x 20 мл). Объединенные органические фракции промывают насыщ. раствором бикарбоната натрия (10 мл), раствором хлорида натрия (20 мл), сушат над сульфатом магния и фильтруют. Растворитель удаляют под вакуумом, и неочищенный материал 4e (1,0 г, 1,88 ммоль, выход 95 %) используют на следующей стадии без дополнительной очистки. СВЭЖХМС (способ 10 мин) = 5,7 мин. Наблюдаемая масса (ИЭР+): 531,4 (M+H)+.
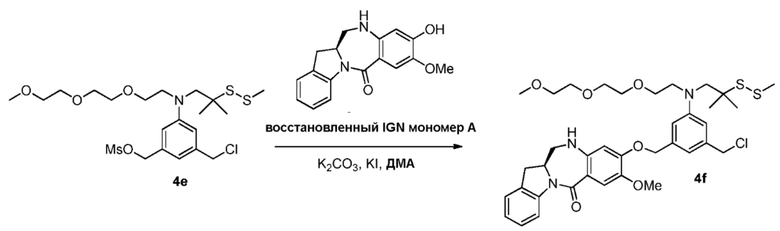
К раствору 4e (0,21 г, 0,39 ммоль, 1,0 экв.) в ДМА (2,0 мл) добавляют карбонат калия (0,16 г, 1,19 ммоль, 3,0 экв.), с последующим добавлением раствора восстановленного IGN мономера A (0,12 г, 0,41 ммоль, 1,05 экв.) в ДМА (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 час. Реакцию гасят водой (30 мл), и смесь перемешивают в течение 10 мин. Твердое вещество отфильтровывают, растворяют в ДХМ/MeOH (9/1, 30 мл) и промывают раствором хлорида натрия (20 мл). Органическую фракцию отделяют и сушат над сульфатом магния, фильтруют и растворитель удаляют под вакуумом. Неочищенный материал очищают хроматографией на силикагеле (гексан/EtOAc) с получением желаемого продукта 4f (0,11 г, 0,15 ммоль, выход 38 %) в виде бесцветного масла. СВЭЖХМС (способ 10 мин) = 6,55 мин. Наблюдаемая масса (ИЭР+): 730,9 (M+H)+.
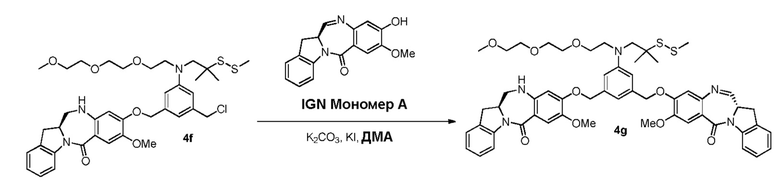
Раствор 4f (0,11 г, 0,15 ммоль, 1,0 экв.) и IGN мономера A (0,053 г, 0,18 ммоль) растворяют в безводном ДМА (1,0 мл). Добавляют карбонат калия (0,041 г, 0,30 ммоль) и йодид калия (0,025 г, 0,15 ммоль). Смесь перемешивают в течение 4 час при 40 oC. Воду (5 мл) добавляют к реакционной смеси, твердое вещество отфильтровывают, затем повторно растворяют в ДХМ (20 мл) и промывают водой (10 мл). Органическую фракцию сушат над сульфатом магния, фильтруют и концентрируют. Неочищенное твердое вещество очищают хроматографией на силикагеле (гексан/EtOAc) с получением 4g (0,099 г, 0,10 ммоль, выход 66 %). СВЭЖХМС (способ 10 мин) = 6,38 мин. Наблюдаемая масса (ИЭР+): 988,7 (M+H)+. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,22 (д, J = 7,9 Гц, 1H), 8,12 (д, J = 8,1 Гц, 1H), 8,03 (д, J = 4,5 Гц, 1H), 7,38 – 7,25 (м, 2H), 7,24 (т, J = 7,9 Гц, 2H), 7,24 – 7,06 (м, 2H), 7,11 – 6,94 (м, 1H), 6,98 (с, 1H), 6,91 (д, J = 15,2 Гц, 2H), 6,79 (с, 1H), 6,45 (с, 1H), 6,32 (д, J = 6,8 Гц, 1H), 5,18 (к, J = 12,3 Гц, 2H), 5,01 (м, 2H), 4,54 (дт, J = 9,7, 5,2 Гц, 1H), 4,37 (дт, J = 10,6, 5,4 Гц, 1H), 3,87 (с, 3H), 3,71 (с, 3H), 3,60 (м, 6H), 3,57 – 3,50 (м, 2H), 3,47 (кд, J = 4,3, 1,0 Гц, 4H), 3,47 (с, 3H), 3,42 – 3,33 (м, 2H), 3,32 – 3,16 (м, 2H), 3,21 (с, 3H), 2,97 – 2,85 (м, 1H), 2,44 (с, 3H), 1,30 (с, 6H).
Пример 16.
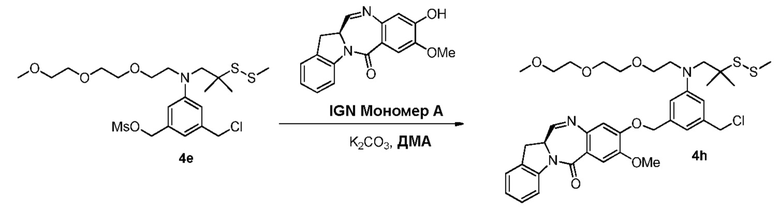
К раствору 4e (0,52 г, 0,98 ммоль, 1,0 экв.) и карбоната калия (0,41 г, 2,94 ммоль, 3,0 экв.) в ДМА (10 мл) добавляют раствор IGN мономера A (0,30 г, 1,03 ммоль, 1,05 экв.) в ДМА (2 мл) при комнатной температуре, и реакционную смесь перемешивают в течение 5 час. Реакцию гасят водой (30 мл), фракции разделяют, и водную фракцию экстрагируют EtOAc (3 x 30 мл). Объединенные органические фракции промывают водой (30 мл), раствором хлорида натрия (30 мл), сушат над сульфатом магния, и избыток растворителя удаляют под вакуумом. Неочищенное масло очищают хроматографией на силикагеле (гексан/EtOAc) с получением желаемого продукта 4h (0,35 г, 0,48 ммоль, выход 49 %). СВЭЖХМС (способ 10 мин) = 6,19 мин. Наблюдаемая масса (ИЭР+): 728,7 (M+H)+.
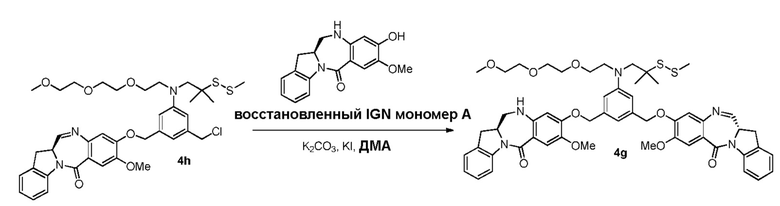
К раствору 4h (0,18 г, 0,25 ммоль, 1,0 экв.) в ДМА (5,0 мл) добавляют карбонат калия (0,10 г, 0,74 ммоль, 3,0 экв.), с последующим добавлением йодида калия (0,04 г, 0,2 ммоль, 1,0 экв.). Добавляют раствор восстановленного IGN мономера A (0,08 г, 0,27 ммоль, 1,1 экв.) в ДМА (1 мл), и затем реакционную смесь нагревают при 40 oC в течение 5 час. Реакцию гасят водой, и затем твердое вещество отфильтровывают. Твердое вещество повторно растворяют в ДХМ/MeOH (20:1), промывают водой, сушат над сульфатом магния, отфильтровывают и концентрируют. Неочищенный остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением 4g (0,05 г, 0,05 ммоль, выход 21 %). СВЭЖХМС (способ 10 мин) = 6,39 мин. Наблюдаемая масса (ИЭР+): 989,0 (M+H)+.
Пример 17.
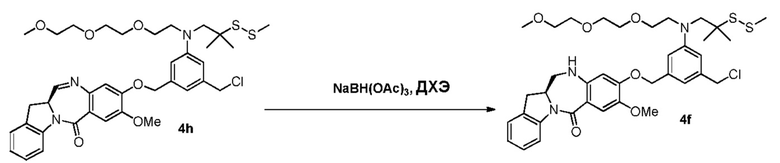
Соединение 4h (0,17 г, 0,24 ммоль, 1,0 экв.) растворяют в безводном ДХЭ (3 мл) и триацетоксиборгидрид натрия (0,10 г, 0,48 ммоль, 3,0 экв.) добавляют при комнатной температуре. Реакционную смесь перемешивают в течение 1 час. Смесь гасят насыщ. раствором хлорида аммония (10 мл). Фракции разделяют, и водную фракцию экстрагируют ДХМ (2 x 20 мл). Объединенные органические фракции промывают раствором хлорида натрия (20 мл), сушат над безводным сульфатом магния, фильтруют и концентрируют с получением 4f (0,13 г, 0,18 ммоль, выход 77 %), которое используют на следующей стадии без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 2,13 мин. Наблюдаемая масса (ИЭР+): 731,2 (M+H)+.
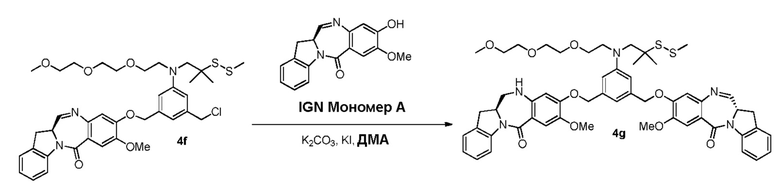
Соединение 4f (0,19 г, 0,26 ммоль, 1,0 экв.) и IGN мономер A (0,084 г, 0,28 ммоль, 1,1 экв.) растворяют в безводном ДМА (4,0 мл). Добавляют карбонат калия (0,11 г, 0,78 ммоль, 3,0 экв.) и йодид калия (0,043 г, 0,26 ммоль, 1,0 экв.). Смесь перемешивают в течение 4 час при 40 oC. Воду (5 мл) добавляют к реакционной смеси. Твердое вещество отфильтровывают, повторно растворяют в ДХМ (20 мл) и промывают водой (10 мл). После сушки над сульфатом магния, фильтрации и концентрирования, твердое вещество очищают хроматографией на силикагеле (гексан/EtOAc) с получением 4g (0,065 г, 0,06 ммоль, выход 25 %). СВЭЖХМС (способ 10 мин) = 6,38 мин. Наблюдаемая масса (ИЭР+): 988,7 (M+H)+.
Пример 18.
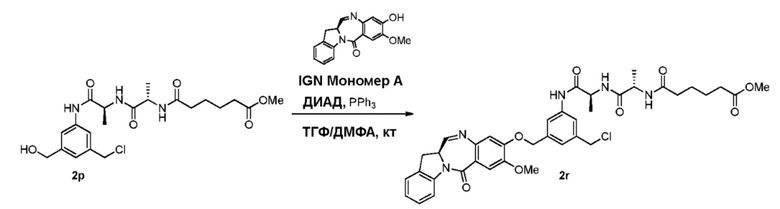
Соединение 2p (0,03 г, 0,066 ммоль, 1,0 экв.) и IGN мономер A (0,021 г, 0,072 ммоль, 1,1 экв.) растворяют в ТГФ (0,65 мл) и ДМФА (0,3 мл). Добавляют трифенилфосфин (0,021 г, 0,079 ммоль, 1,2 экв.), с последующим медленным добавлением ДИАД (0,015 мл, 0,079 ммоль, 1,2 экв.). Реакционную смесь перемешивают в течение 2 час в атмосфере аргона при кт. Реакционную смесь концентрируют, и добавляют воду (~2 мл) для растирания продукта. Осадок отфильтровывают и остаток твердого вещества промывают водой. Неочищенный остаток очищают ОФВЭЖХ (колонка C18, MeCN/вода, градиент, от 40 % до 60 %) с получением соединения 2r в виде хлопьевидного твердого вещества белого цвета (0,015 г, 0,02 ммоль, выход 31 %). СВЭЖХМС (способ 2,5 мин) = 1,62 мин. Наблюдаемая масса (ИЭР+) = 732,9 (M+H)+.
Пример 19.
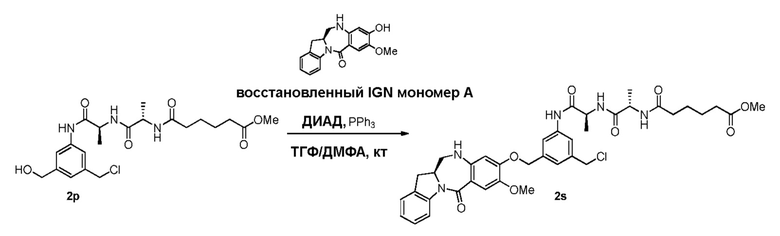
Соединение 2p (0,03г, 0,066 ммоль, 1,0 экв.) и восстановленный IGN мономер A (0,02г, 0,072 ммоль, 1,1 экв.) растворяют в ТГФ (0,66 мл) и ДМФА (0,1 мл). Добавляют трифенилфосфин (0,021 г, 0,079 ммоль, 1,2 экв.), с последующим медленным добавлением ДИАД (0,015 мл, 0,079 ммоль, 1,2 экв.). Реакционную смесь перемешивают в течение 2 час в атмосфере аргона при кт. Реакционную смесь разбавляют ДХМ и промывают водой (2x). Органическую фракцию сушат над сульфатом магния, фильтруют и концентрируют. Неочищенный остаток очищают ОФВЭЖХ (колонка C18, MeCN/вода, градиент, от 40 % до 65 %) с получением 2s в виде хлопьевидного твердого вещества белого цвета (0,017 г, 0,02 ммоль, выход 35 %). СВЭЖХМС (способ 2,5 мин) = 1,71 мин. Наблюдаемая масса (ИЭР+) = 735,4 (M+H)+.
Пример 20.
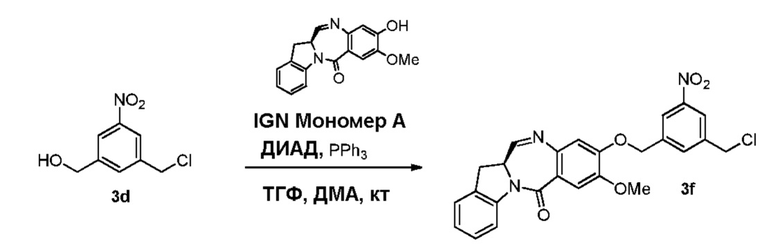
Соединение 3d (0,03 г, 0,149 ммоль, 1,0 экв.) и IGN мономер A (0,046 г, 0,156 ммоль, 1,05 экв.) растворяют в ТГФ (1,5 мл) и ДМФА (0,3 мл). Добавляют трифенилфосфин (0,047 г, 0,179 ммоль, 1,2 экв.), с последующим медленным добавлением ДИАД (0,032 мл, 0,164 ммоль, 1,1 экв.). Реакционную смесь перемешивают в течение 12 час в атмосфере аргона при кт. Реакционную смесь концентрируют, и добавляют воду (~2 мл) для растирания продукта. Осадок отфильтровывают, и остаток твердого вещества промывают водой. Неочищенный остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением соединения 3f в виде бело-желтого твердого вещества (0,013 г, 0,027 ммоль, выход 18 %). СВЭЖХМС (способ 2,5 мин) = 1,80 мин. Наблюдаемая масса (ИЭР+) = 478,4 (M+H)+.
Пример 21.
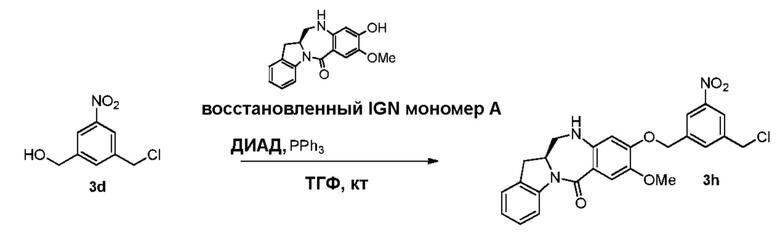
Соединение 3d (0,03 г, 0,149 ммоль, 1,0 экв.) и восстановленный IGN мономер A (0,046 г, 0,156 ммоль, 1,05 экв.) растворяют в ТГФ (1,5 мл). Добавляют трифенилфосфин (0,047 г, 0,179 ммоль, 1,2 экв.), с последующим медленным добавлением ДИАД (0,032 мл, 0,164 ммоль, 1,1 экв.). Реакционную смесь перемешивают в течение 2 час в атмосфере аргона при кт. Реакционную смесь концентрируют и упаривают с толуолом (2 x). Неочищенный остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением соединения 3h в виде оранжево-желтого твердого вещества (0,055 г, 0,115 ммоль, выход 77 %). СВЭЖХМС (способ 2,5 мин) = 1,90 мин. Наблюдаемая масса (ИЭР+) = 480,5 (M+H)+.
Пример 22.
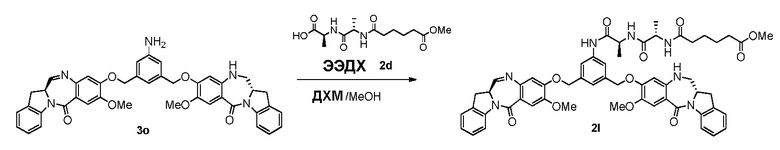
К раствору 2d (0,024 г, 0,078 ммоль, 1,1 экв.) в ДХМ (1 мл) добавляют ЭЭДХ (0,019 г, 0,078 ммоль, 1,1 экв.). Реакционную смесь перемешивают в течение 5 мин и добавляют MeOH (0,1 мл), с последующим добавлением раствора 3o (0,05 г, 0,071 ммоль) в ДХМ (1 мл). Реакционную смесь перемешивают при кт в течение 2 час или до тех пор, пока весь исходный материал не израсходуется. Реакционную смесь концентрируют с получением осадка белого цвета, к которому добавляют МТБЭ (5 мл), и полученную смесь перемешивают в течение 30 мин при комнатной температуре. Твердое вещество отфильтровывают с получением соединения 2l, которое затем очищают ОФВЭЖХ (колонка C18, MeCN/вода) с получением 2l (0,023 г, 0,023 ммоль, выход 33 %). СВЭЖХМС (способ 2,5 мин) = 1,75 мин. Наблюдаемая масса (ИЭР+) = 993,2 (M+H)+.
Пример 23.
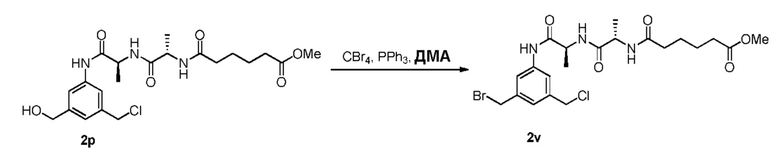
К раствору 2p (0,05 г, 0,110 ммоль, 1,0 экв.) в ДМА (1 мл), добавляют тетрабромид углерода (0,044 г, 0,132 ммоль, 1,2 экв.), с последующим добавлением трифенилфосфина (0,043 г, 0,164 ммоль, 1,5 экв.), и реакционную смесь перемешивают при комнатной температуре в течение 2 час. Растворитель удаляют с получением твердого вещества белого цвета, которое растирают с МТБЭ, и твердое вещество отфильтровывают с получением соединения 2v. (0,03 г, 0,058 ммоль, выход 57 %, чистота 52 %), которое переносят на следующую стадию без дополнительной очистки. СВЭЖХМС (способ 2,5 мин) = 1,59 мин. Наблюдаемая масса (ИЭР+) = 518,2 (M+H)+.
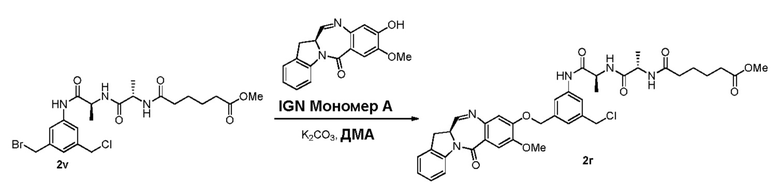
К раствору 2v (0,03 г, 0,043 ммоль, 1,0 экв.) в ДМА (0,5 мл) добавляют карбонат калия (0,012 г, 0,087 ммоль, 2,0 экв.), с последующим добавлением IGN мономера A (0,013 г, 0,046 ммоль, 1,05 экв.). Реакционную смесь перемешивают в течение 4 час при комнатной температуре. Реакционную смесь разбавляют водой (5 мл), и твердое вещество отфильтровывают. Твердое вещество растворяют в ДХМ/MeOH (9/1, 2 мл). Органическую фракцию промывают водой (10 мл), раствором хлорида натрия (10 мл) и сушат над сульфатом магния. После фильтрации и удаления растворителя неочищенный продукт очищают ОФВЭЖХ (колонка C18, MeCN/вода) с получением 2r (0,011 г, 0,015 ммоль, выход 35 %). СВЭЖХМС (способ 2,5 мин) = 1,62 мин. Наблюдаемая масса (ИЭР+) = 733,3 (M+H)+
.
Пример 24.
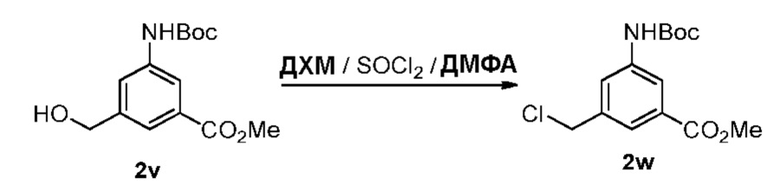
К суспензии соединения 2v (14,7 г, 0,052 моль, 1,0 экв., получено, как описано в литературе, см.: Beilstein J. Org. Chem. 2014, 10, 535-543) в ДХМ и (100 мл) ДМФА (1 мл), добавляют SOCl2 (12,6 г, 0,104 моль, 2,0 экв.) в виде одной порции. Полученный раствор перемешивают при 35 oC в течение ночи с образованием густой суспензии рыжеватого цвета. Суспензию фильтруют, и твердое вещество сушат с получением 7,5 г в виде твердого вещества почти белого цвета. ЯМР показывает отщепление защитной группы Boc. К фильтрату темного цвета добавляют твердый карбонат натрия (10,6 г, 0,1 моль), с последующей буферизацией до pH ~6-7 добавлением дополнительного количества бикарбоната натрия. К полученному раствору добавляют Boc2O (12,7 г,0,058 моль, 1,1 экв.) и перемешивают в течение 0,5 час. Отфильтрованное твердое вещество (7,5 г) добавляют к реакционной смеси, с последующим добавлением Boc2O (6,5 г, 0,030 моль, 1,7 экв.) (pH ~6), и перемешивание продолжают при кт в течение ночи. Затем добавляют насыщ. раствор бикарбоната натрия (10 мл) до достижения pH 6~7. Добавляют дополнительное количество Boc2O (9,3 г, 42,6 ммоль) и ДМАП (0,2 г, 1,63 ммоль), и перемешивание продолжают в течение ночи. Реакционную смесь темного цвета фильтруют для удаления некоторого количества осадка. Фракцию ДХМ промывают 1 н HCl для удаления продукта без Boc, который подщелачивают и экстрагируют ДХМ с получением 3,0 г бесцветного хрустящего твердого вещества (продукт без Boc). Фракцию ДХМ промывают раствором хлорида натрия и концентрируют с получением жидкой суспензии темного цвета. Неочищенный продукт очищают хроматографией на силикагеле (EtOAc/гексаны) с получением 2w в виде твердого вещества бледно-коричневого цвета (9,5 г, 0,031 ммоль, выход 62 %). 1H ЯМР (400 МГц, CDCl3): δ 7,84 (м, 2H), 7,75 (м, 1H), 6,60 (с, 1H, NH), 4,58 (с, 2H), 3,91 (с, 3H), 1,53 (с, 9H).
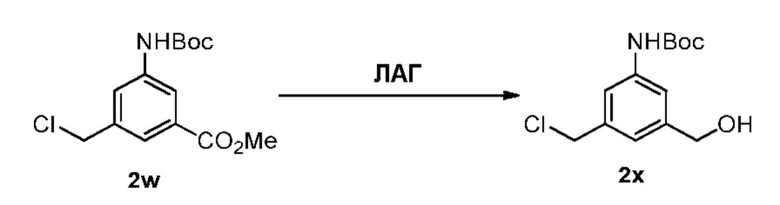
Раствор LAH/ТГФ (0,6M, 60 мл, 1,15 экв.) перемешивают при кт в течение 30 мин, и затем охлаждают до минус 65 oC с помощью бани с ацетоном-сухим льдом. Соединение 2w (9,3 г, 0,031 моль, 1,0 экв.) медленно добавляют порциями (Ti ~ минус 60oC) с получением желто-коричневой суспензии, которую перемешивают в течение 4 час. Реакцию гасят водой (1,3 мл), 15 % раствором NaOH (1,3 мл) и водой (4 мл) и перемешивают в течение 20 мин (Ti ~5oC). Реакционную смесь фильтруют и промывают этилацетатом (~90 мл). Фильтрат промывают раствором хлорида натрия и концентрируют с получением 2x (8,0 г, 0,029 моль, выход 93 %) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3): δ 7,45 (с, 1H), 7,40 (с, 1H), 7,10 (с, 1H), 6,60 (с, 1H, NH), 4,75 (с, 2H), 4,50 (с, 2H), 1,53 (с, 9H).
Соединение 2x (8,0 г, 0,029 моль, 1,0 экв.) растворяют в ДХМ (20 мл) и охлаждают на бане со льдом-водой. Добавляют 4 н HCl/диоксан (15 мл, 1,5 экв.), полученную смесь нагревают при 50 oC, выдерживая при этой температуре в течение 1 час, и затем охлаждают до комнатной температуры. Суспензию концентрируют, и растворитель заменяют на гептан. Суспензию фильтруют, промывают гексаном и сушат в печи (60 oC) с получением 2y (5,4 г, 0,026 моль, выход 88 %) в виде светло-коричневого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,45 (с, 1H), 7,25 (с, 2H), 4,76 (с, 2H), 4,52 (с, 2H).

К раствору 2d (0,969 г, 3,20 ммоль, 1,1 экв.) в ДХМ (25 мл) при комнатной температуре добавляют ЭЭДХ (0,79 г, 3,2 ммоль, 1,1 экв.). Через 8 мин по каплям добавляют раствор 2y (0,5 г, 2,91 ммоль, 1,0 экв.), ДИПЭА (0,51 мл, 2,91 ммоль, 1,0 экв.) в MeOH (5 мл) на протяжении 1 минуты. Реакционную смесь перемешивают в течение 2 час. Реакцию гасят водой (30 мл), фракции разделяют, и водную фракцию экстрагируют ДХМ (2 x 20 мл). Объединенные органические фракции промывают насыщ. раствором бикарбоната натрия (20 мл), раствором хлорида натрия (20 мл), сушат над сульфатом магния, фильтруют и концентрируют, оставляя минимальное количество растворителя. Полученное твердое вещество белого цвета разбавляют МТБЭ и отфильтровывают с получением желаемого продукта 2p в виде твердого вещества белого цвета (0,64 г, 1,40 ммоль, выход 48 %). СВЭЖХМС (способ 2,5 мин) = 1,30 мин. Наблюдаемая масса (ИЭР+) = 456,3 (M+H)+.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2016 |
|
RU2727151C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2807546C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2813137C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНИЛКАРБАМАТНЫХ ПРОИЗВОДНЫХ ЖЕЛЧНЫХ КИСЛОТ | 2018 |
|
RU2791682C2 |
| ИНГИБИТОРЫ МАТРИКСНОЙ МЕТАЛЛОПРОТЕИНАЗЫ (ММР) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2840074C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЦИТОТОКСИЧЕСКИХ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 2018 |
|
RU2762891C2 |
| ИНГИБИТОРЫ ЛИЗИНСПЕЦИФИЧНОГО ГИНГИПАИНА | 2015 |
|
RU2728785C2 |
| СИНТЕЗ ИНСУЛИНОТРОПНЫХ ПЕПТИДОВ | 2007 |
|
RU2448978C2 |
| НОВЫЕ ИНГИБИТОРЫ BRAF КАК «РАЗРУШИТЕЛИ ПАРАДОКСА» | 2020 |
|
RU2825870C1 |
| МАКРОЦИКЛИЧЕСКИЕ ФЕНИЛКАРБАМАТЫ, ИНГИБИРУЮЩИЕ HCV | 2008 |
|
RU2490261C2 |
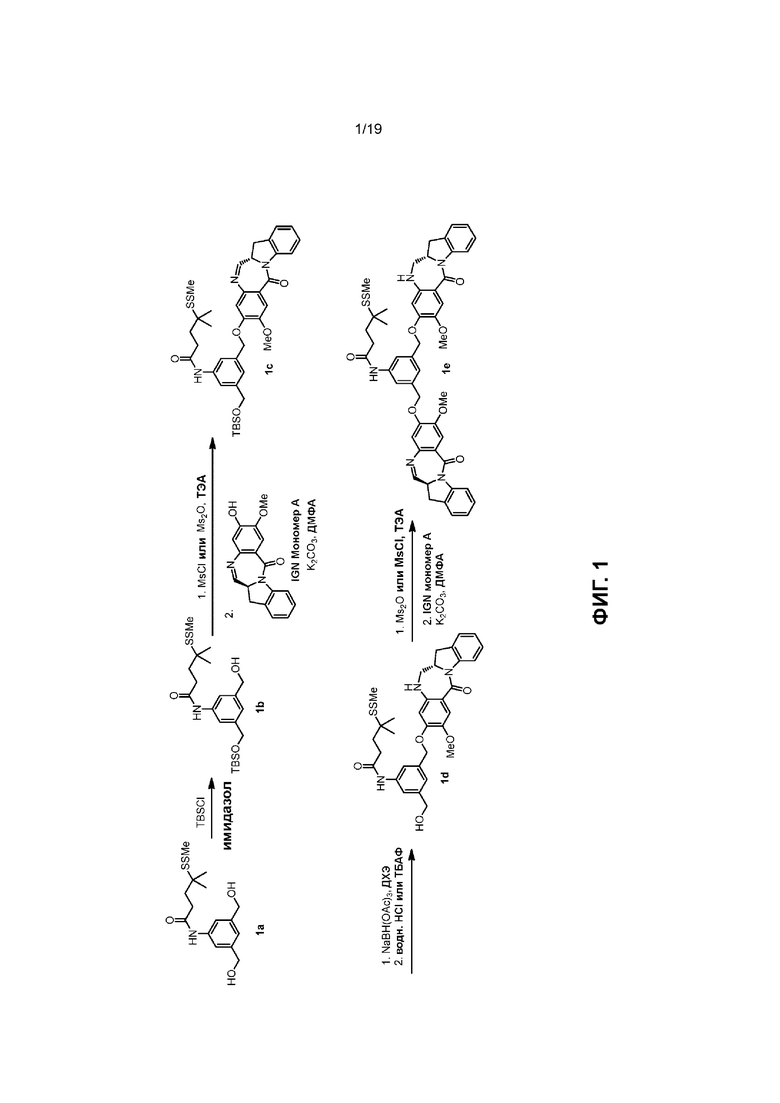

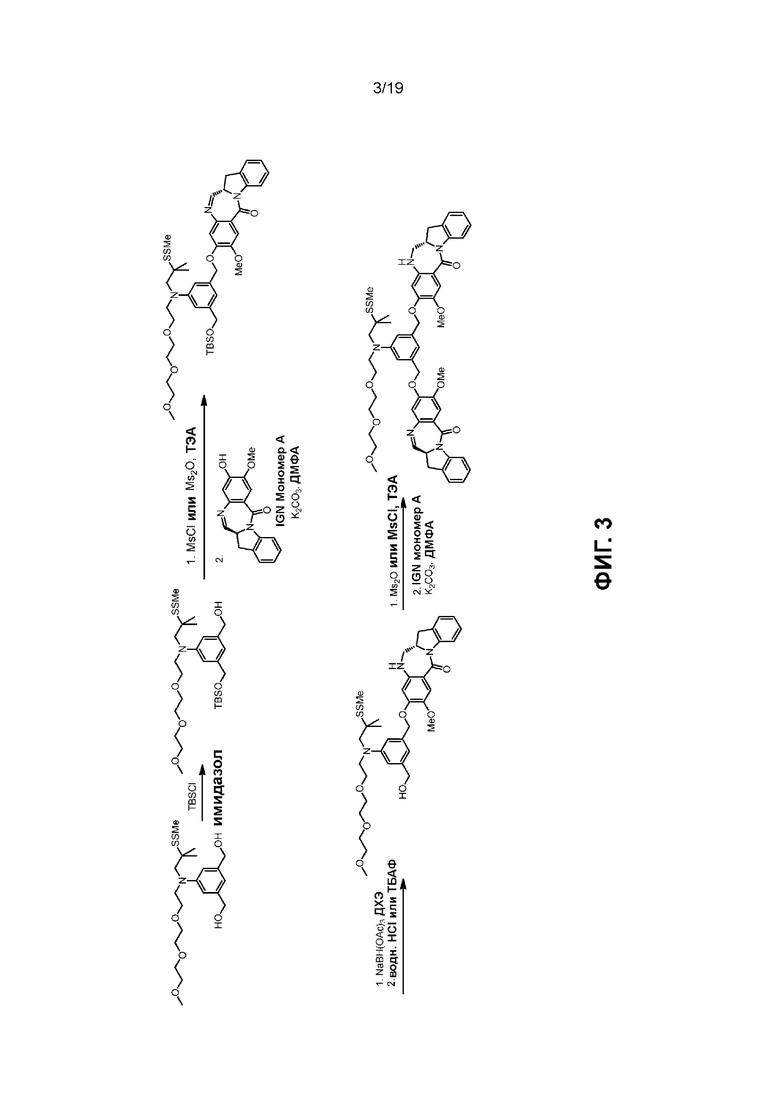
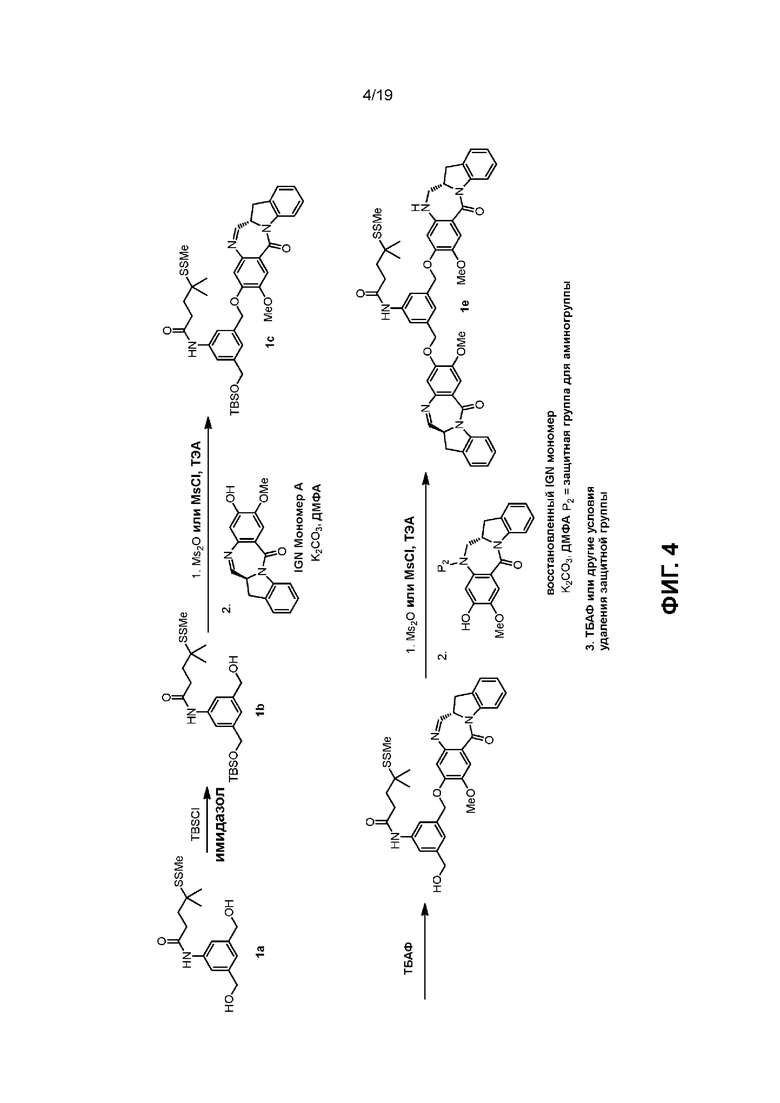
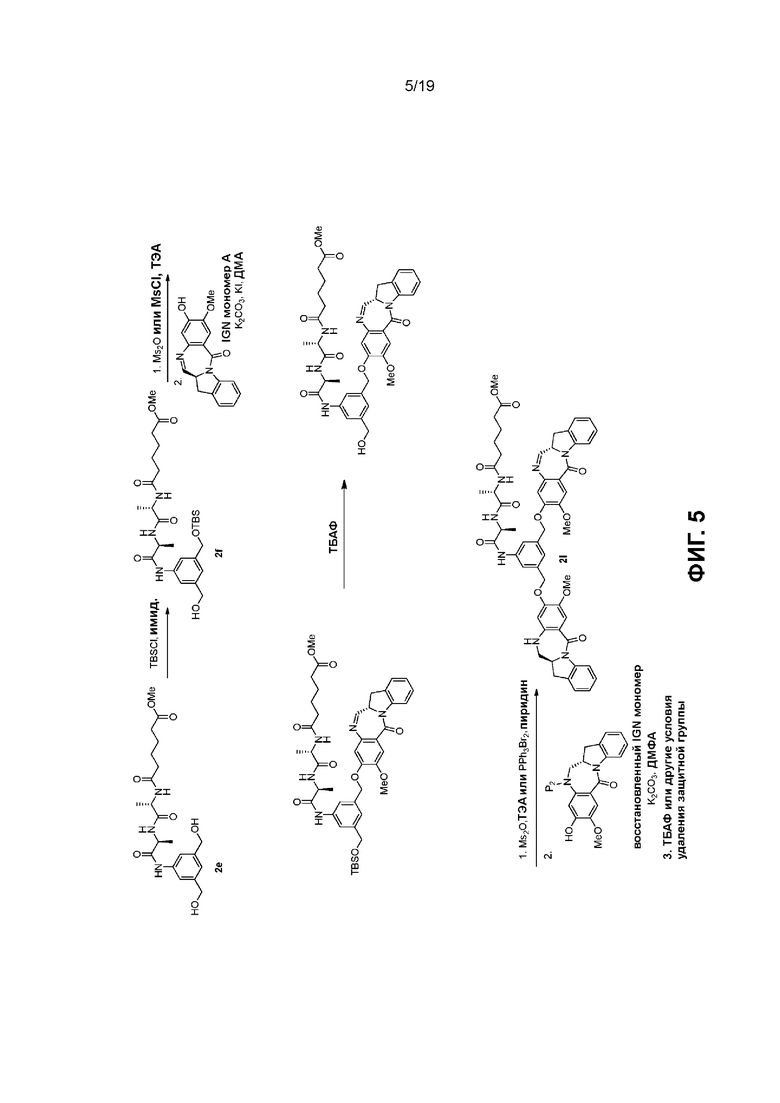
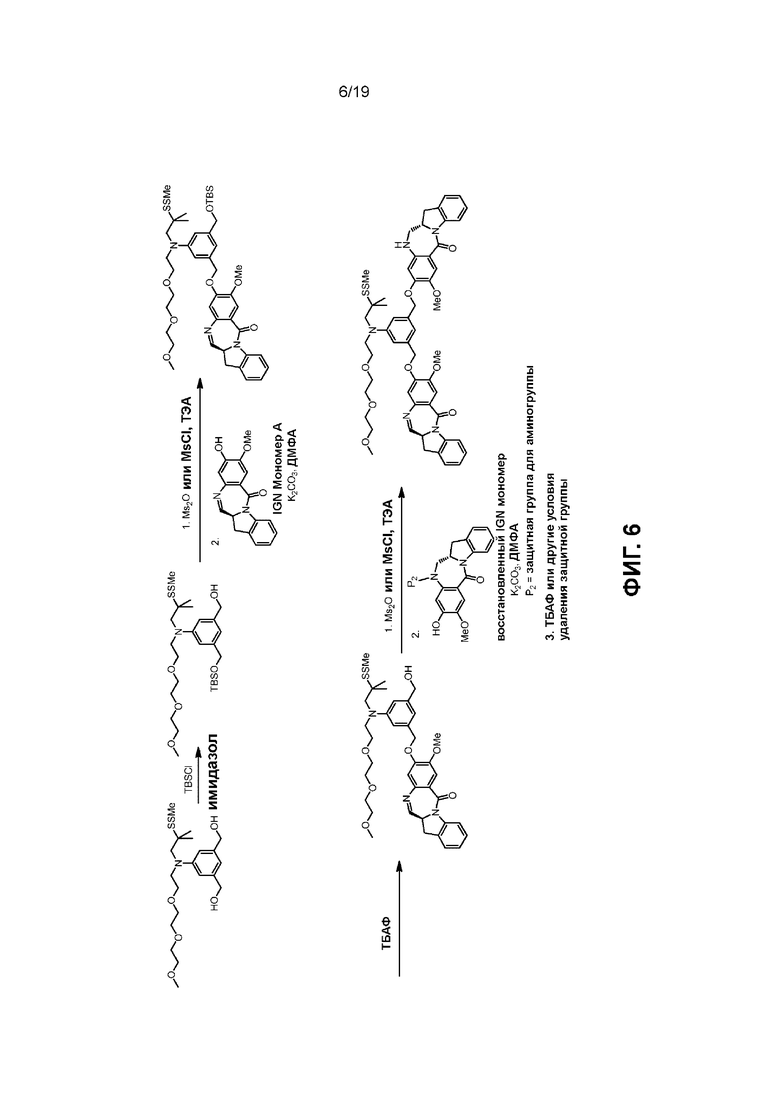
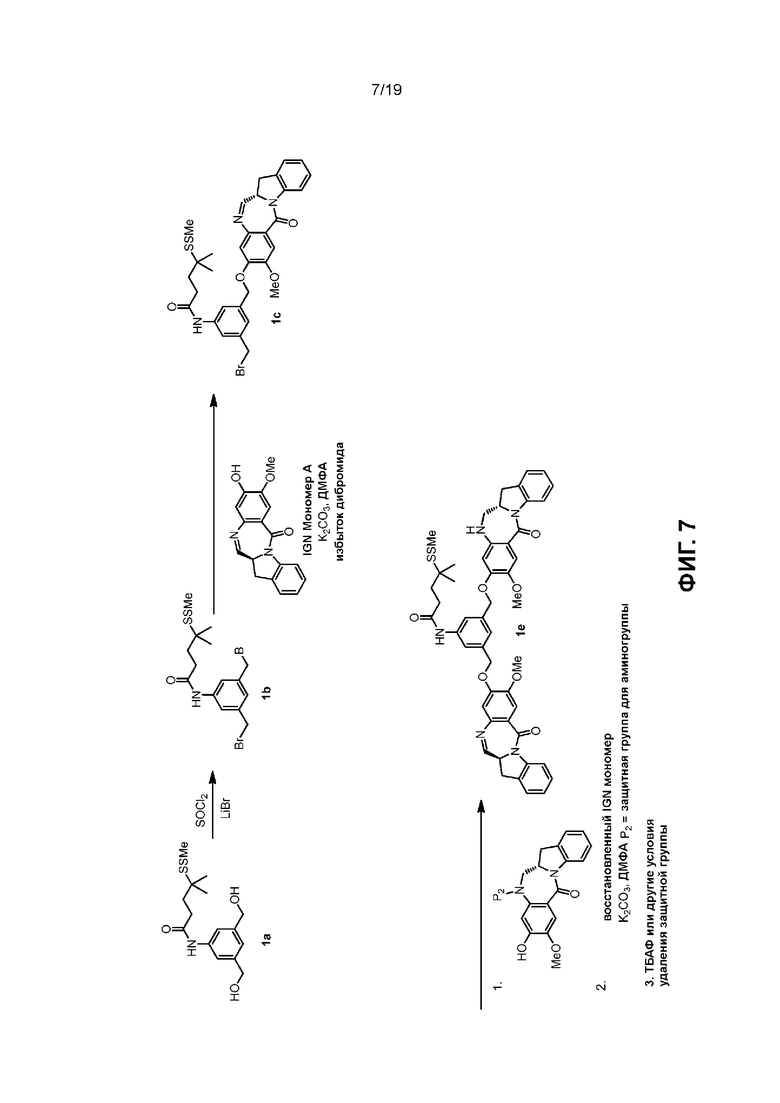
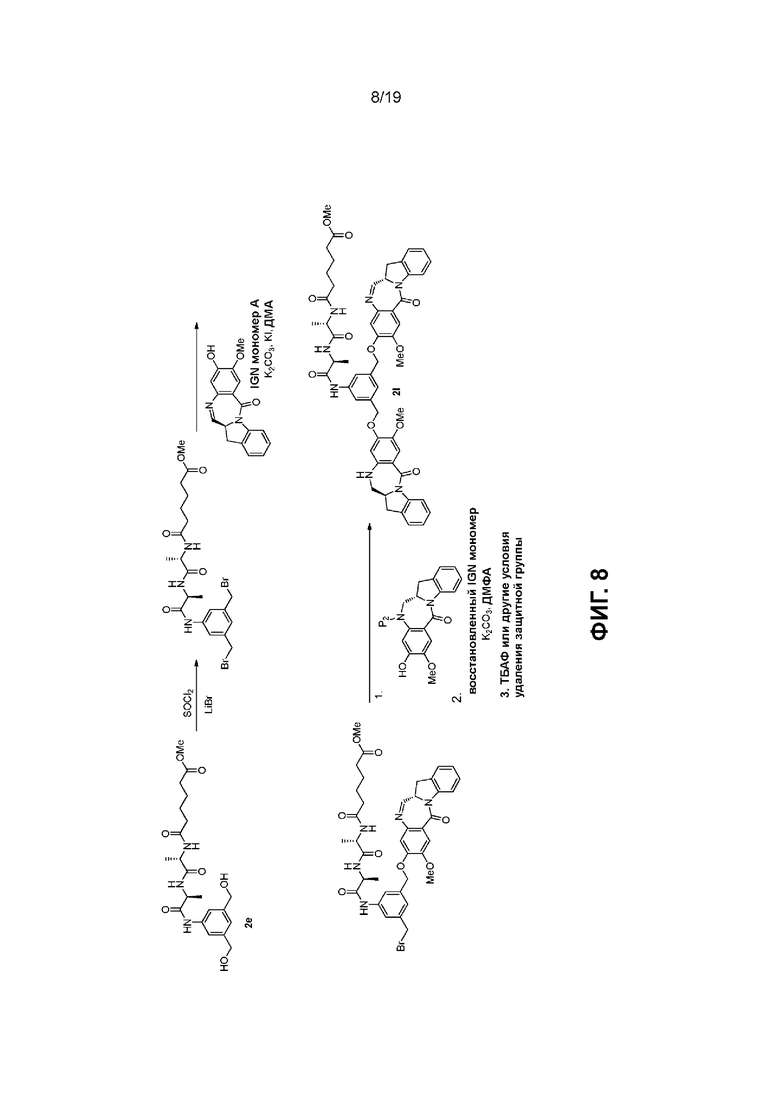
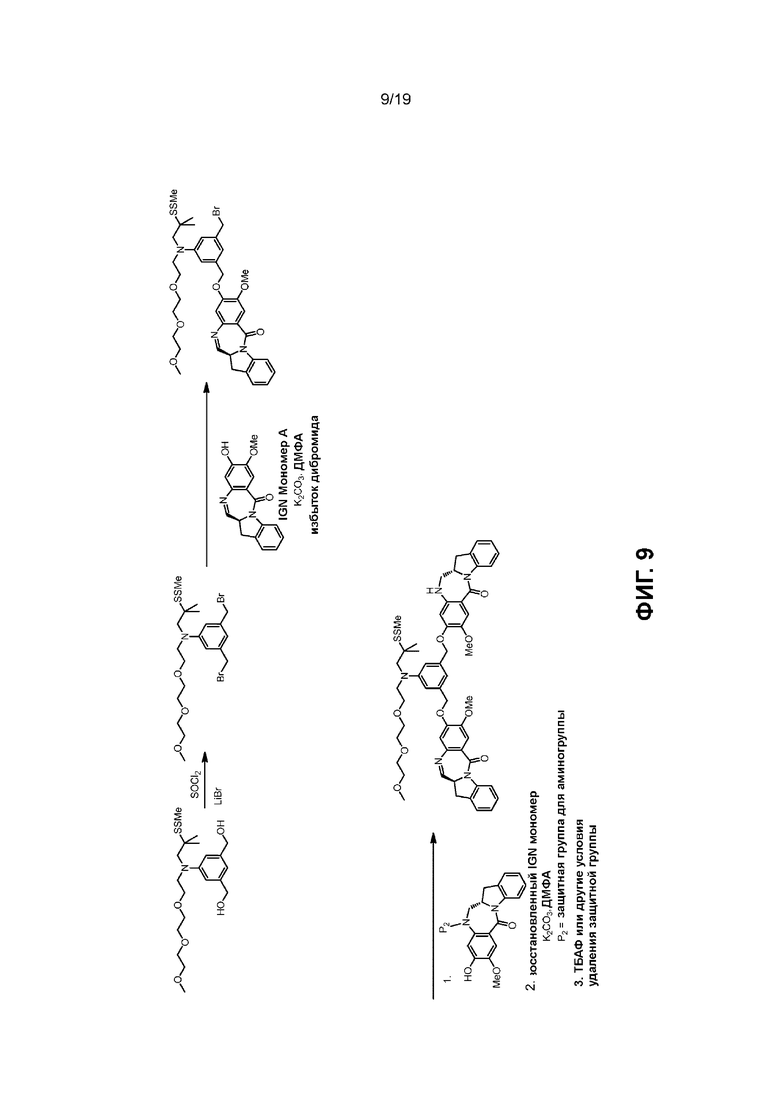
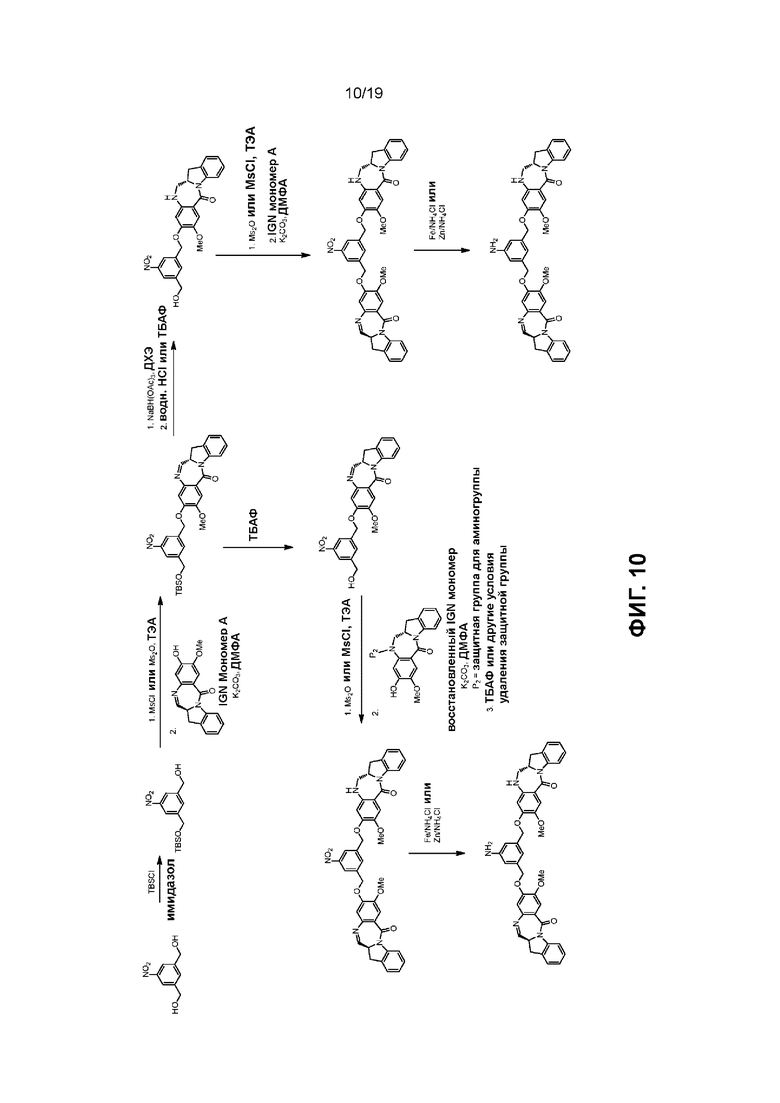

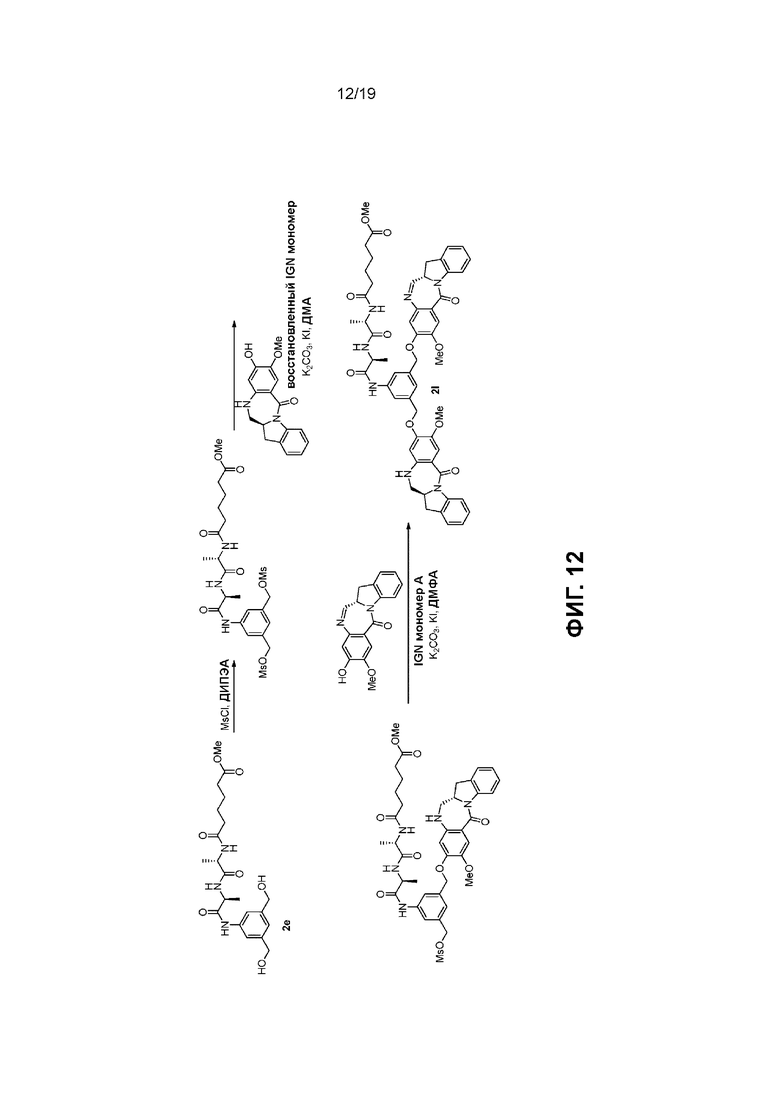
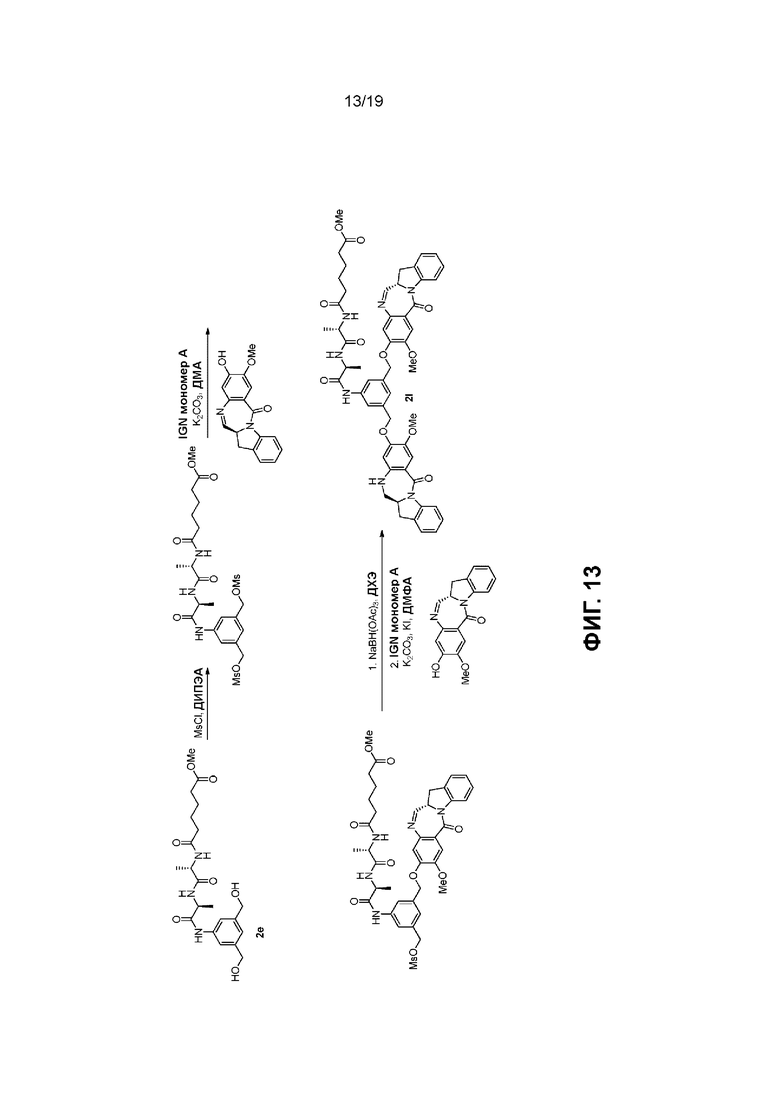
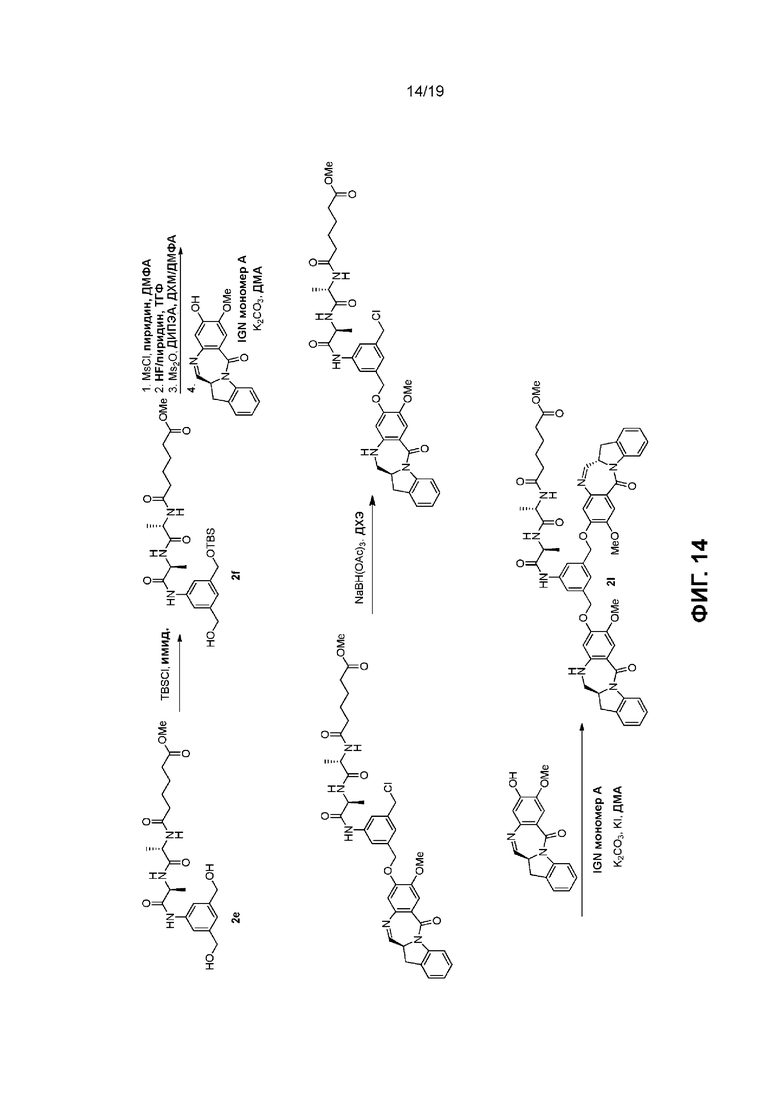
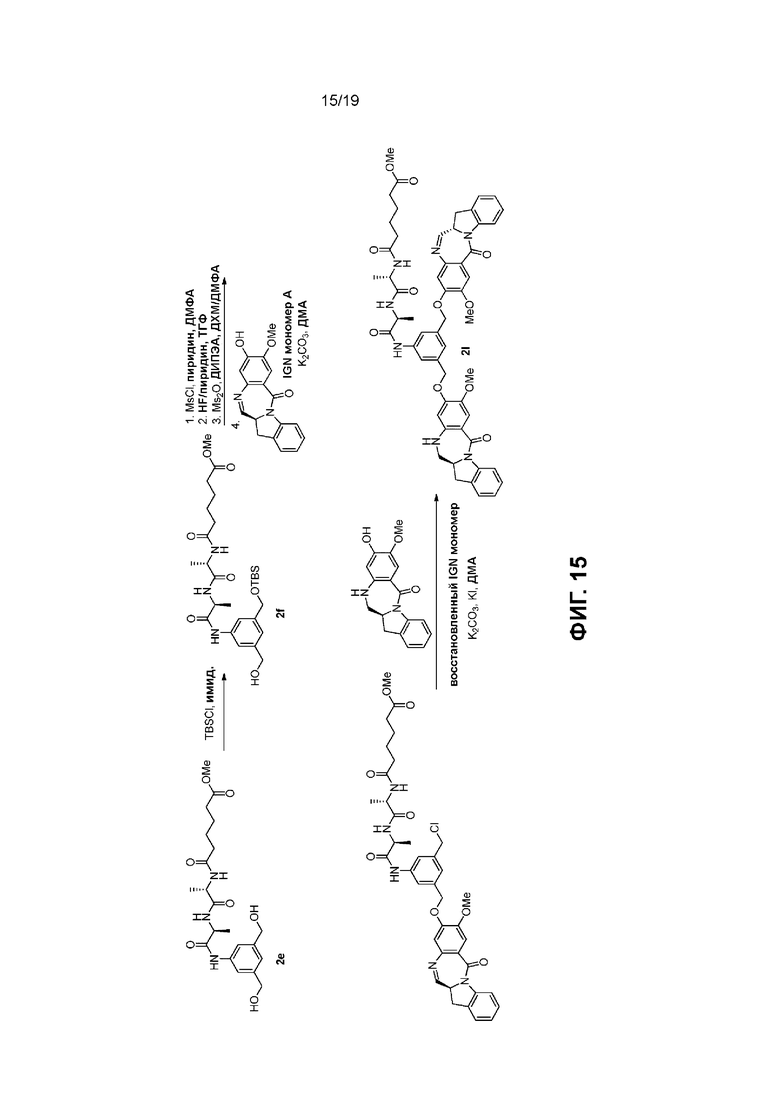
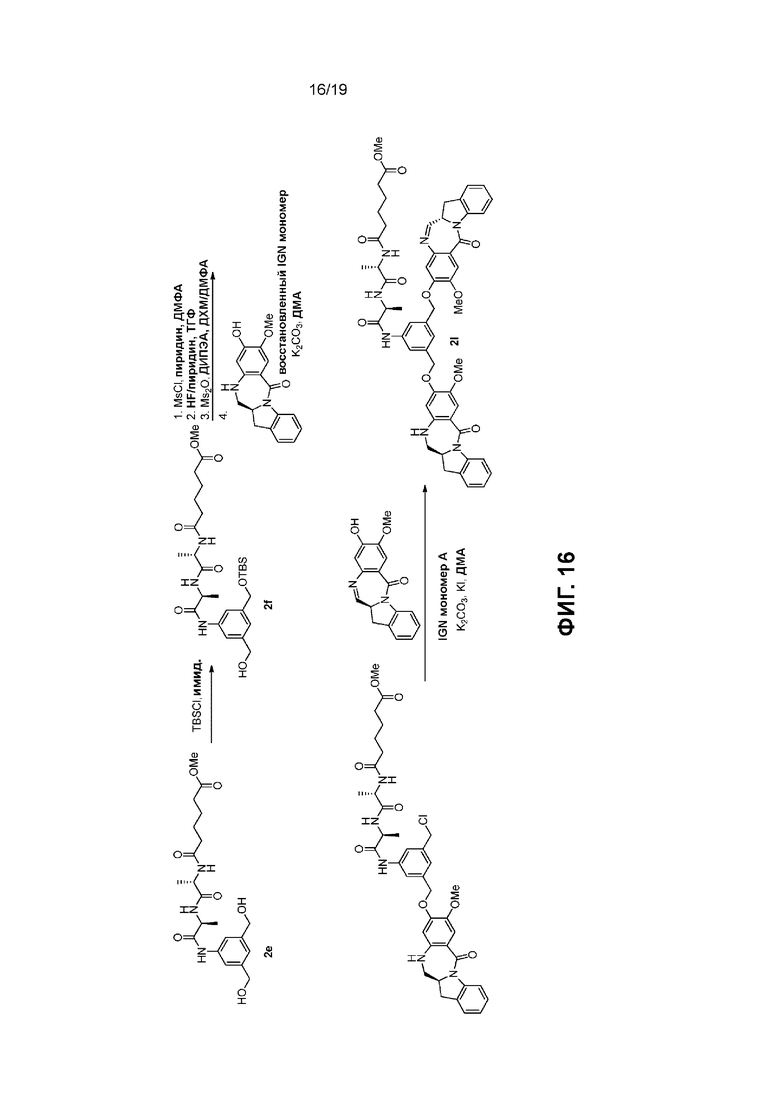
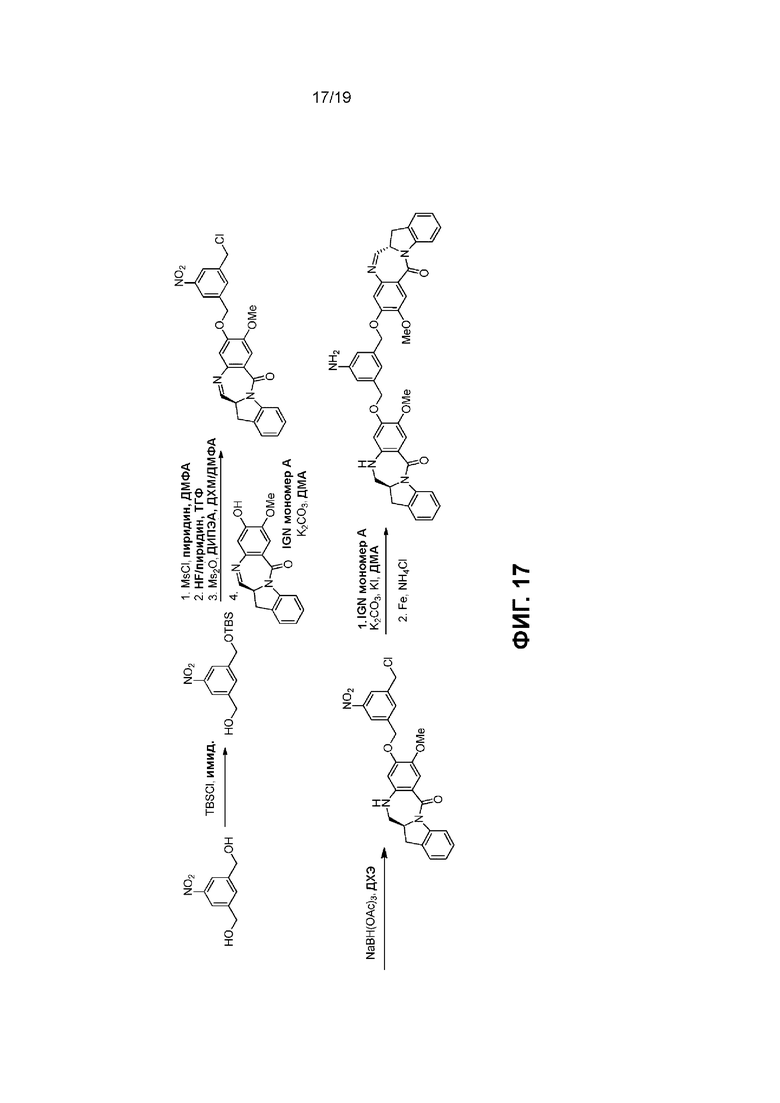
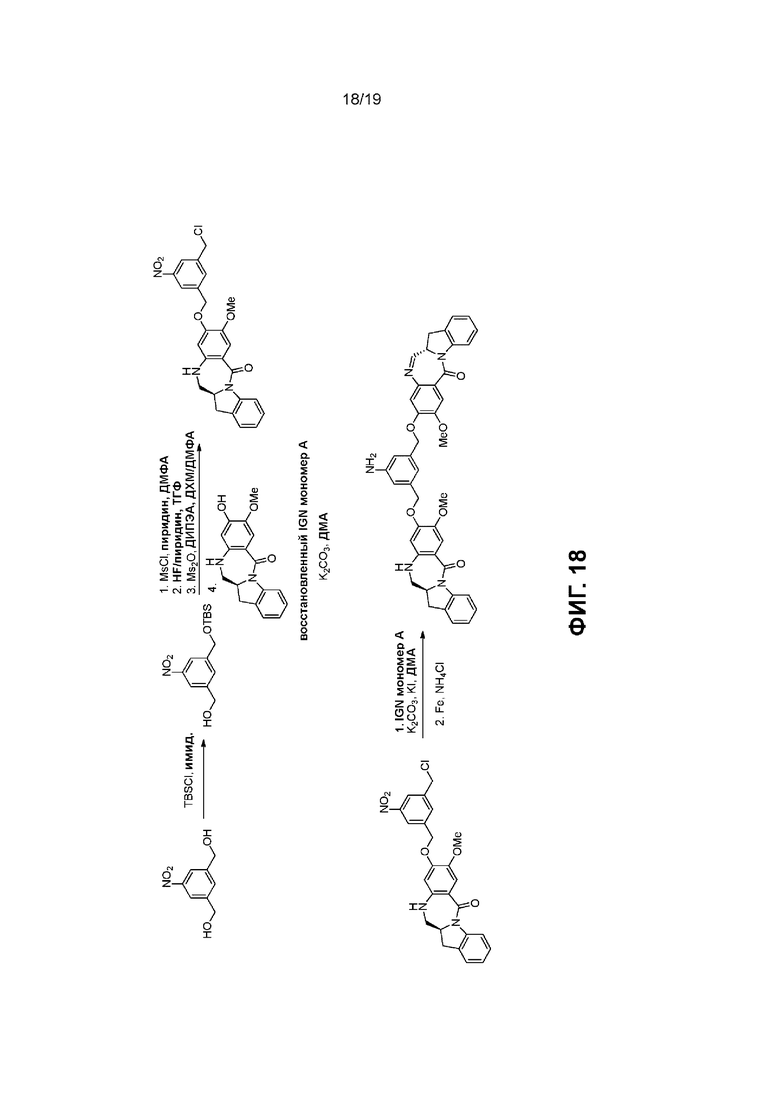
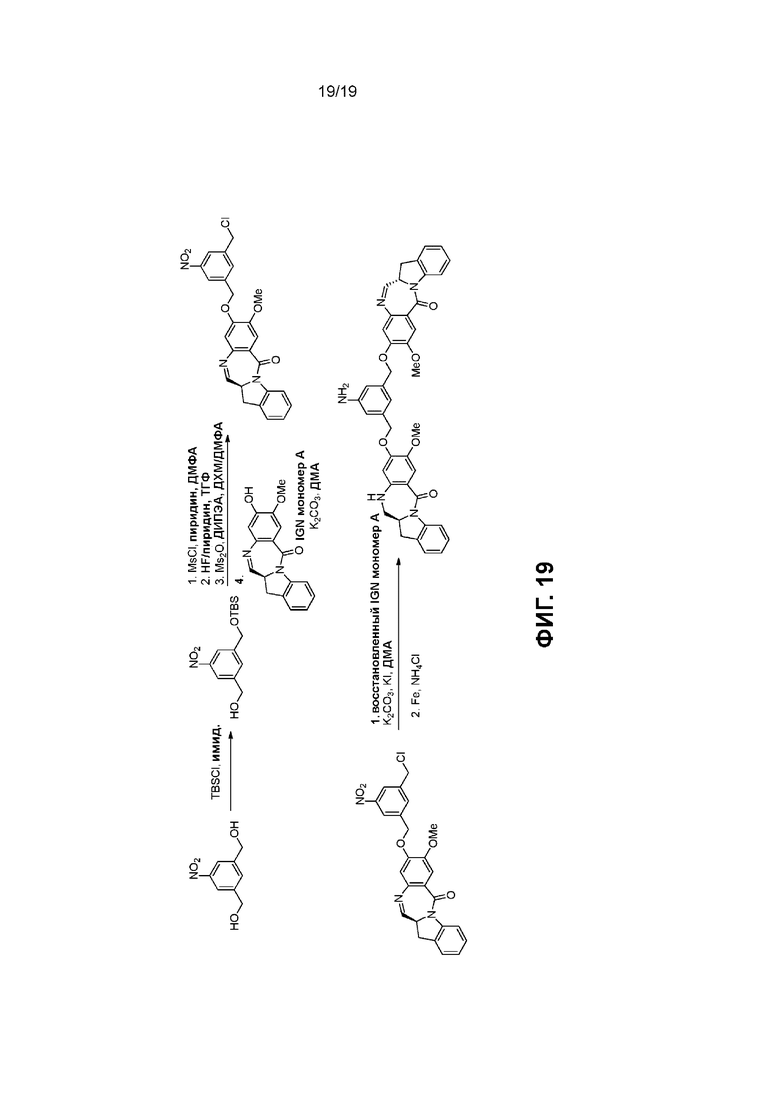
Изобретение относится к новым способам получения димерных индолинобензодиазепиновых соединений, например, формулы 18А и IA и их синтетических предшественников. Технический результат: разработаны новые улучшенные способы получения димеров индолинобензодиазепина, которые являются более эффективными и пригодными для крупномасштабного производственного процесса. 7 н. и 22 з.п. ф-лы, 24 пр., 19 ил.
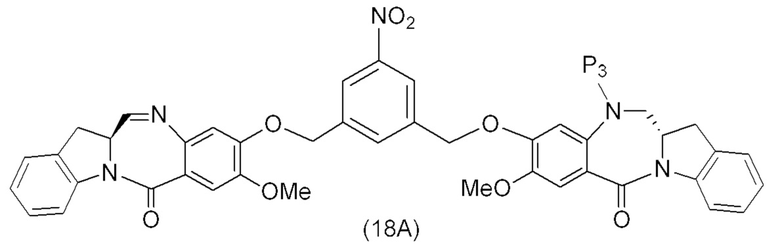
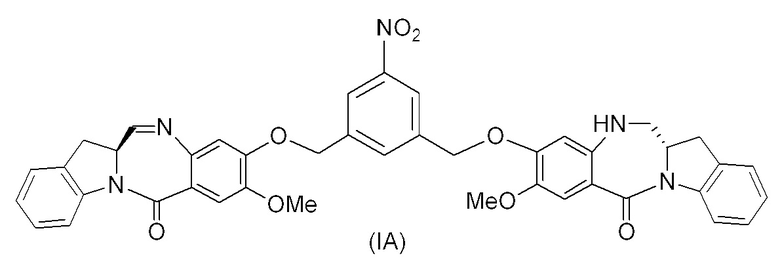
1. Способ получения соединения формулы (17А)
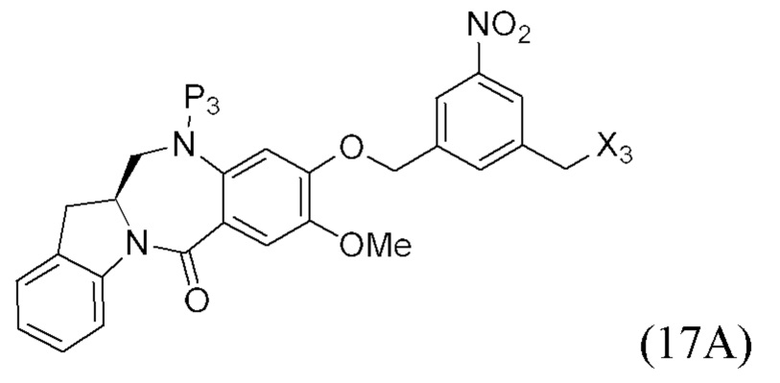
или его соли, причем указанный способ включает введение соединения формулы (14А)
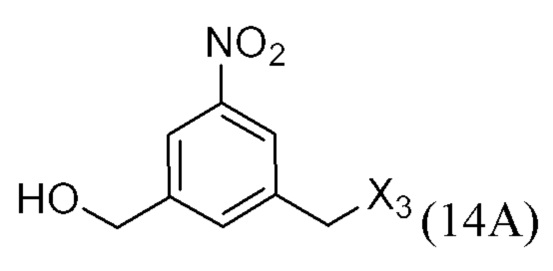
в реакцию с мономерным соединением формулы (d1)
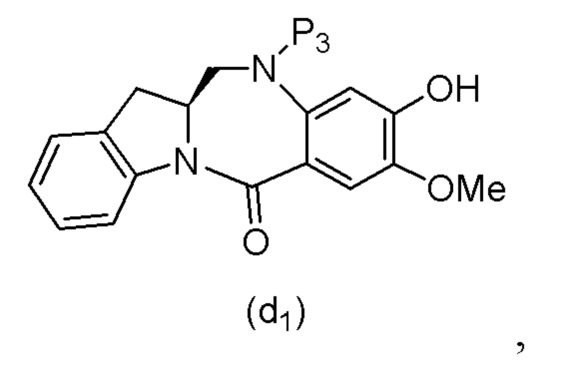
где Х3 представляет собой -Cl и
Р3 представляет собой Н или защитную группу для аминогруппы.
2. Способ по п. 1, отличающийся тем, что соединение формулы (14А) вводят в реакцию с мономером формулы (d1) в присутствии активирующего спирт агента.
3. Способ по п. 2, отличающийся тем, что активирующий спирт агент представляет собой трифенилфосфин.
4. Способ по любому из пп. 1-3, отличающийся тем, что соединение формулы (14А) вводят в реакцию с мономером формулы (d1) в присутствии азодикарбоксилата.
5. Способ по п. 4, отличающийся тем, что азодикарбоксилат выбран из группы, состоящей из диэтилазодикарбоксилата (ДЭАД (DEAD)), диизопропилазодикарбоксилата (ДИАД (DIAD)), 1,1'-(азодикарбонил)дипиперидина (АДДП (ADDP)) и дитрет-бутилазодикарбоксилата (ДТАД (DTAD)).
6. Способ по любому из пп. 1-5, отличающийся тем, что соединение формулы (14А) вводят в реакцию с мономерным соединением формулы (d1), при том что Р3 представляет собой Н, с получением соединения формулы (17А')
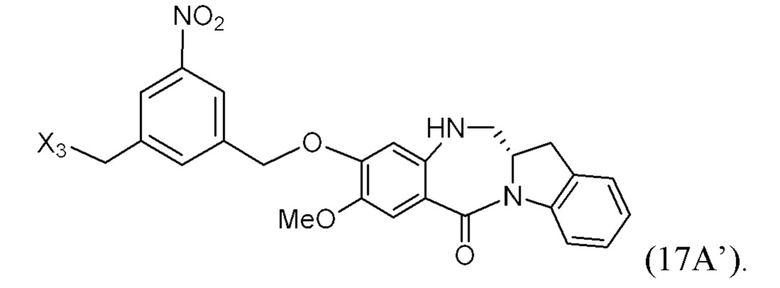
7. Способ по любому из пп. 1-5, отличающийся тем, что Р3 представляет собой защитную группу для аминогруппы.
8. Способ получения соединения формулы (17А')
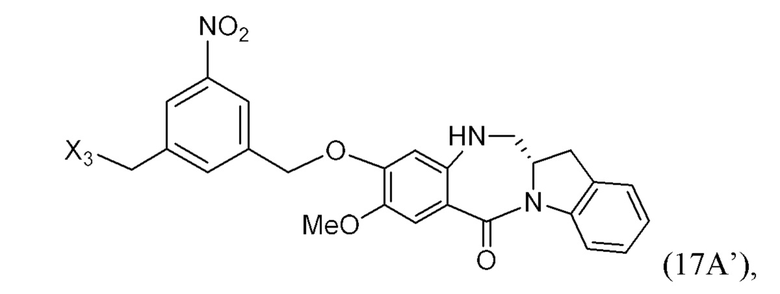
включающий стадии:
а) введения соединения формулы (14А)
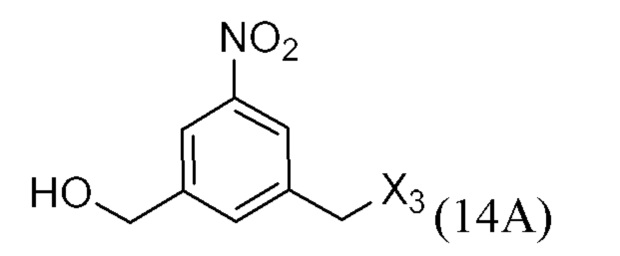
в реакцию с мономерным соединением формулы (d1)
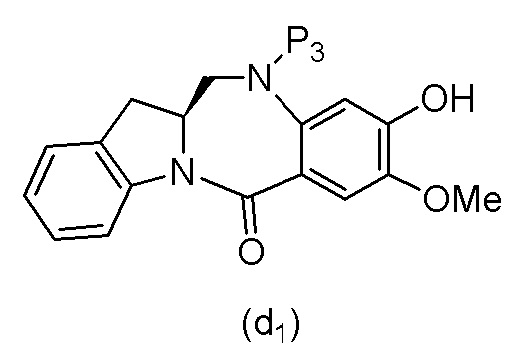
с получением соединения формулы (17А)
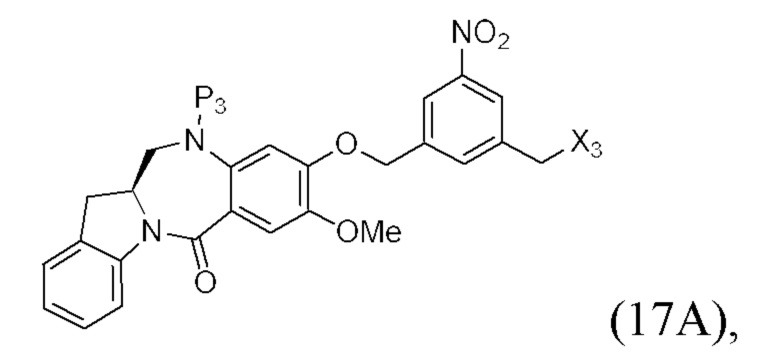
b) введения соединения формулы (17А) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (17А'),
где Х3 представляет собой -Cl и
Р3 представляет собой защитную группу для аминогруппы.
9. Способ по п. 8, отличающийся тем, что реагент, удаляющий защитную группу аминогруппы, выбран из группы, состоящей из тетра-н-бутиламмония фторида, уксусной кислоты, пиридина гидрофторида, фторида цезия, пиперидина, морфолина или трифторуксусной кислоты.
10. Способ получения соединения формулы (18А)
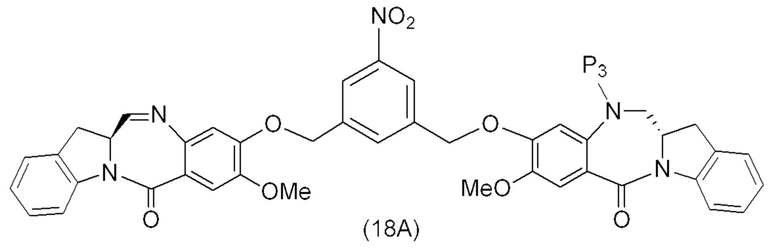
или его фармацевтически приемлемой соли, причем указанный способ включает введение соединения формулы (17А)

в реакцию с мономером формулы (b1)
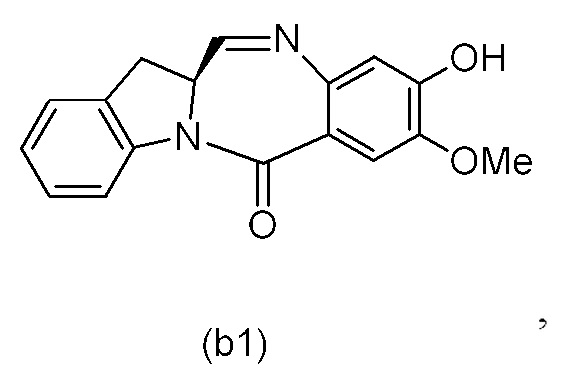
где Х3 представляет собой -Cl;
Р3 представляет собой Н или защитную группу для аминогруппы.
11. Способ по п. 10, отличающийся тем, что соединение формулы (17А) вводят в реакцию с мономерным соединением формулы (b1) в присутствии основания.
12. Способ по п. 11, отличающийся тем, что основание представляет собой карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия или гидрид калия.
13. Способ по п. 12, отличающийся тем, что основание представляет собой карбонат калия.
14. Способ по любому из пп. 10-13, отличающийся тем, что соединение формулы (17А) вводят в реакцию с мономерным соединением формулы (b1) в присутствии полярного апротонного растворителя.
15. Способ по п. 14, отличающийся тем, что полярный апротонный растворитель представляет собой диметилформамид или диметилацетамид.
16. Способ по любому из пп. 10-15, отличающийся тем, что соединение формулы (17А) вводят в реакцию с мономером формулы (b1), при том что Р3 представляет собой Н, с получением соединения формулы (IA)
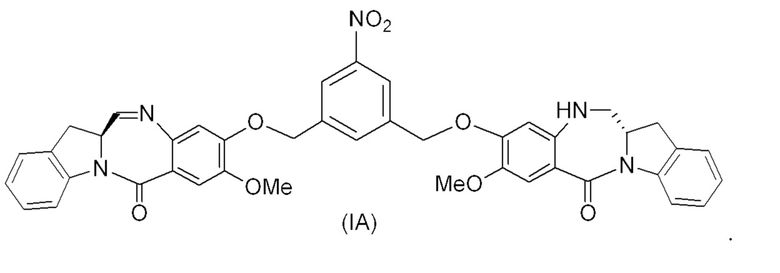
17. Способ по любому из пп. 10-15, отличающийся тем, что Р3 представляет собой защитную группу для аминогруппы.
18. Способ по п. 17, отличающийся тем, что защитная группа для аминогруппы выбрана из группы, состоящей из 2-триметилсилилэтила, (2-фенил-2-триметилсилил)этила, триизопропилсилокси, 2-(триметилсилил)этоксиметила, аллилоксикарбонила, 9-флуоренилметоксикарбонила, 2-(триметилсилил)этоксикарбонила и 2,2,2,2-трихлорэтоксикарбонила.
19. Способ получения соединения формулы (IA)
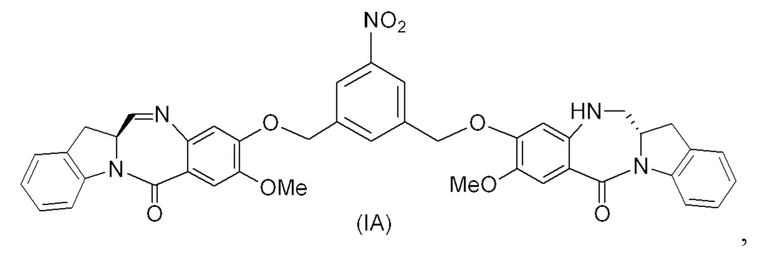
включающий стадии:
a) введения соединения формулы (17А)
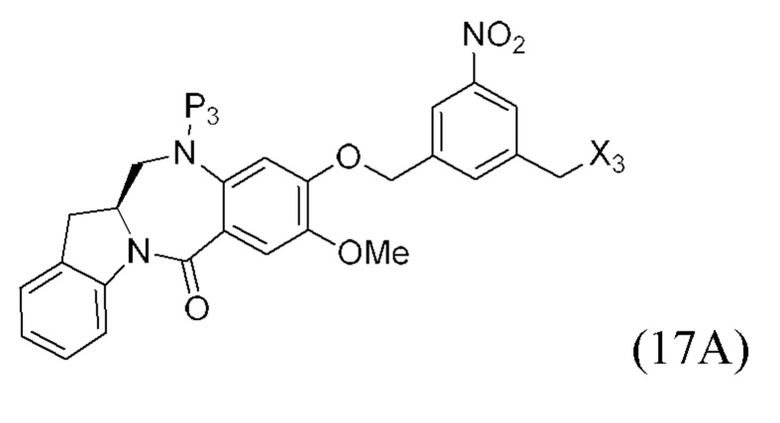
в реакцию с мономером формулы (b1)

с получением соединения формулы (18А)
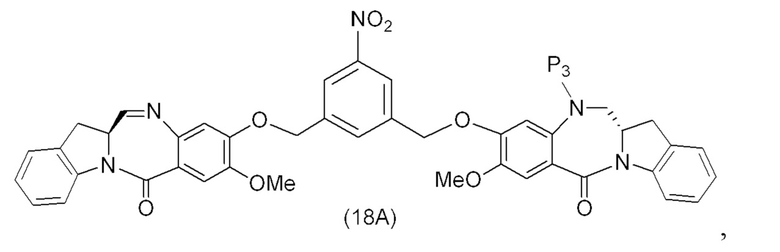
b) введения соединения формулы (18А) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA),
где Х3 представляет собой -Cl и
Р3 представляет собой защитную группу для аминогруппы.
20. Способ по п. 19, отличающийся тем, что реагент, удаляющий защитную группу аминогруппы, выбран из группы, состоящей из тетра-н-бутиламмония фторида, уксусной кислоты, пиридина гидрофторида, фторида цезия, пиперидина, морфолина или трифторуксусной кислоты.
21. Способ получения соединения формулы (18А)
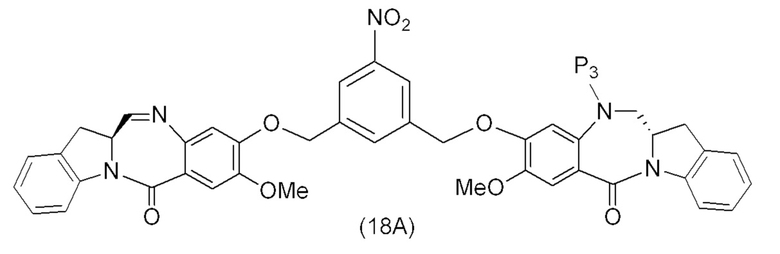
или его фармацевтически приемлемой соли, причем указанный способ включает следующие стадии:
(1) введение соединения формулы (14А)
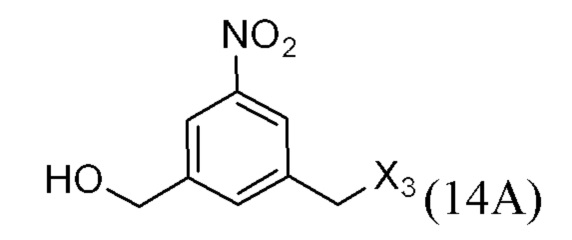
в реакцию с восстановленным мономерным соединением формулы (d1)
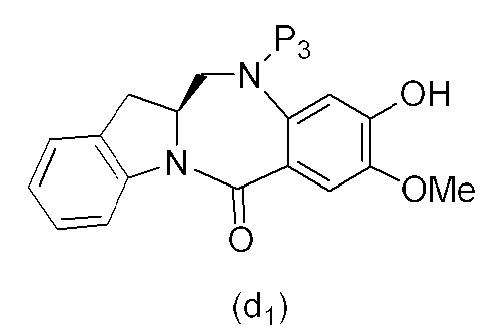
с получением соединения формулы (17А)
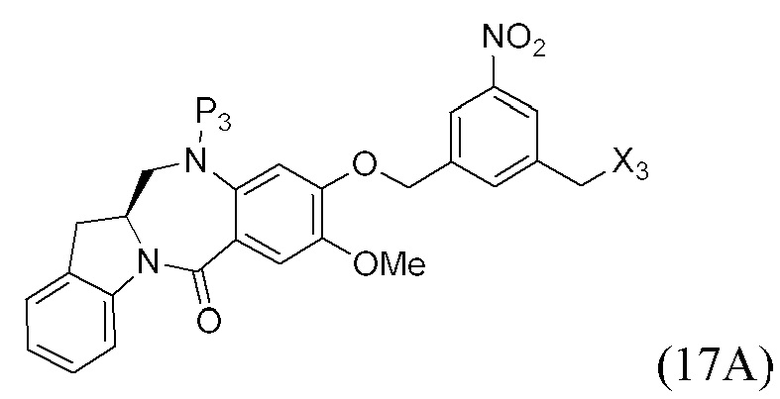
или его соли; и
(2) введение соединения формулы (17А) в реакцию с мономером формулы (b1)
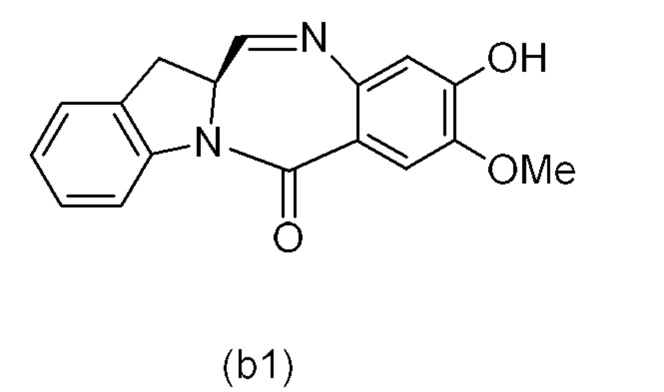
с получением соединения формулы (18А) или его фармацевтически приемлемой соли,
где Х3 представляет собой -Cl и
Р3 представляет собой Н или защитную группу для аминогруппы.
22. Способ по п. 21, отличающийся тем, что соединение формулы (17А) вводят в реакцию с мономером формулы (b1), при том что Р3 представляет собой Н, с получением соединения формулы (IA)
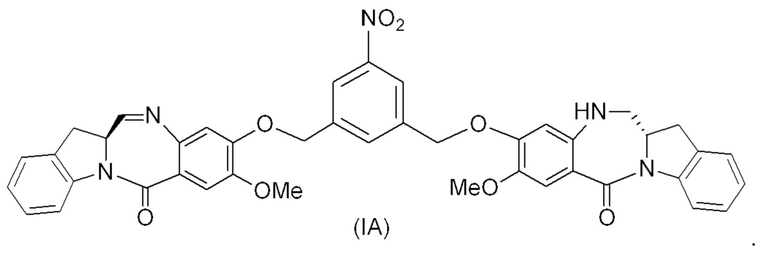
23. Способ по п. 21, отличающийся тем, что Р3 представляет собой защитную группу для аминогруппы.
24. Способ получения соединения формулы (IA)
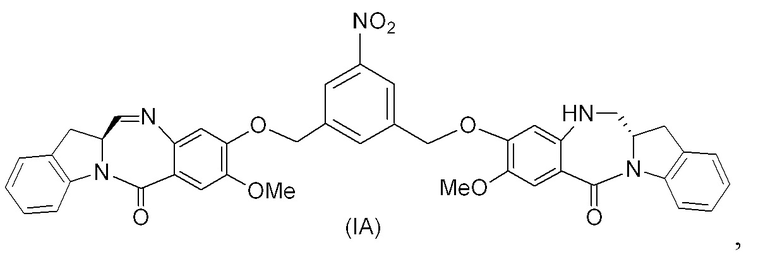
включающий следующие стадии:
(1) введение соединения формулы (14А)
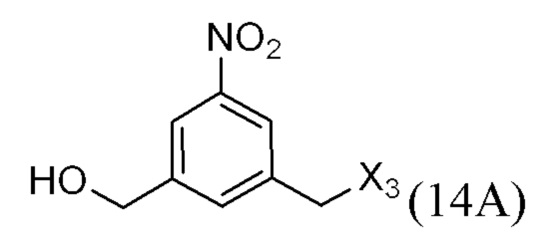
в реакцию с восстановленным мономерным соединением формулы (d1)
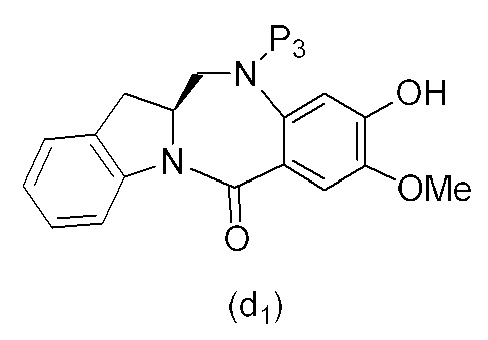
с получением соединения формулы (17А)
или его соли; и
(2) введение соединения формулы (17А) в реакцию с мономером формулы (b1)
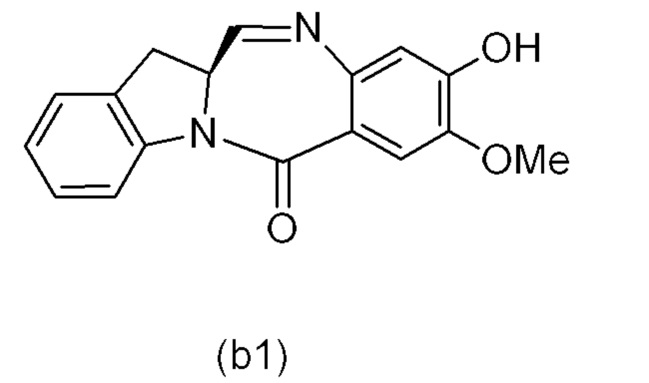
с получением соединения формулы (18А)
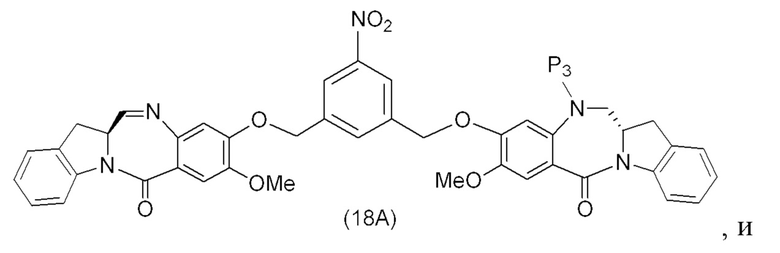
(3) введение соединения формулы (18А) в реакцию с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA),
где Х3 представляет собой -Cl и
Р3 представляет собой защитную группу для аминогруппы.
25. Способ по любому из пп. 1-9 и 21-24, отличающийся тем, что соединение формулы (14А) или его соль получают способом, включающим в себя следующие стадии:
(1) введение хлорирующего реагента в реакцию с соединением формулы (2А)
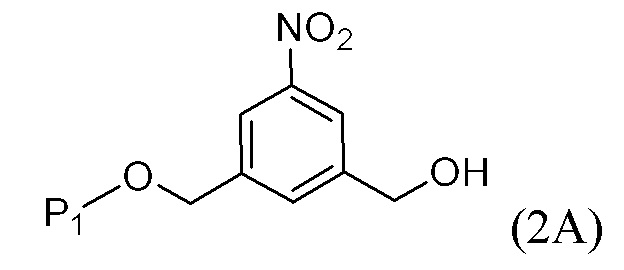
с получением соединения формулы (13А)
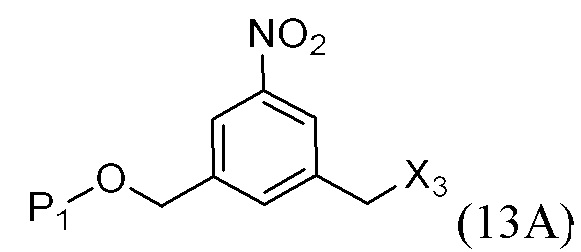
или его соли; и
(2) введение соединения формулы (13А) в реакцию с реагентом, удаляющим защитную группу спиртовой группы, с получением соединения формулы (14А) или его соли.
26. Способ по п. 25, отличающийся тем, что соединение формулы (2А) получают введением соединения формулы (1А) в реакцию с защитным реагентом для спиртовой группы
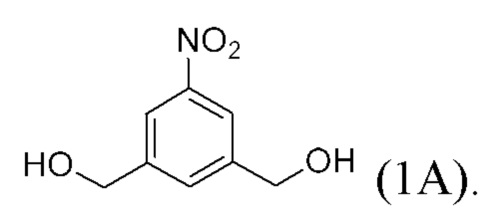
27. Способ получения соединения формулы (IB)
или его фармацевтически приемлемой соли, включающий следующие стадии:
(1) введение соединения формулы (14А)
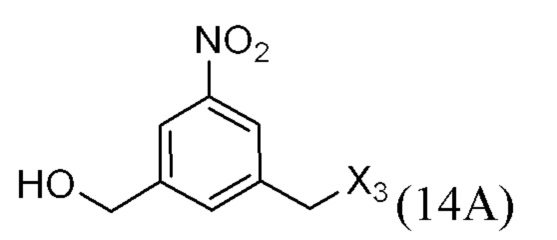
в реакцию с восстановленным мономерным соединением формулы (d1)
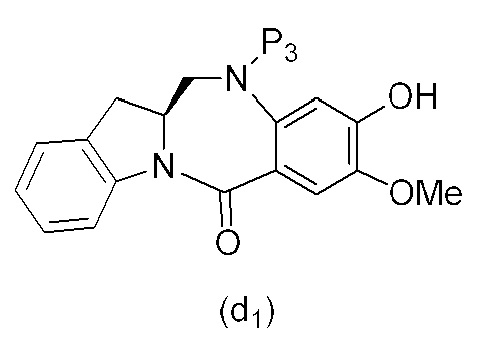
с получением соединения формулы (17А)
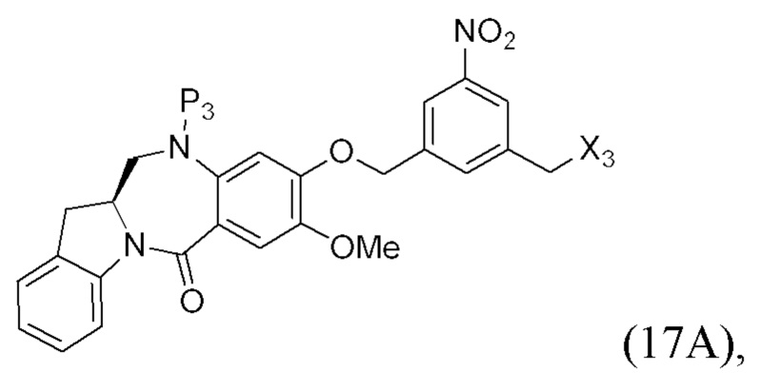
(2) введение соединения формулы (17А) в реакцию с мономером формулы (b1)
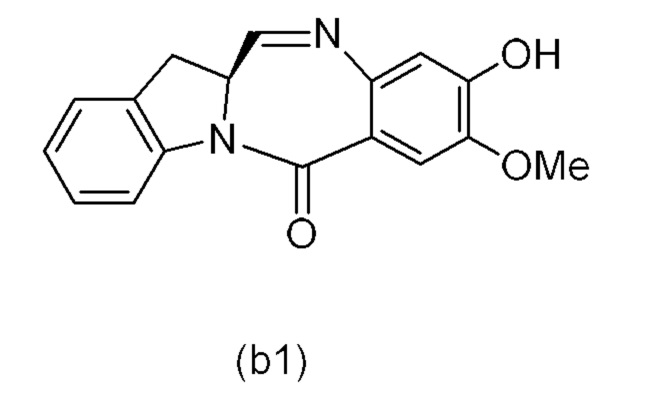
с получением соединения формулы (18А)
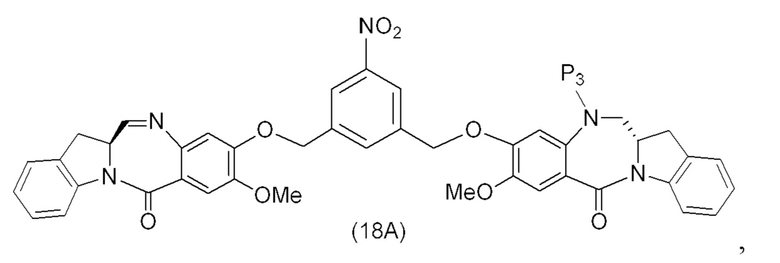
(3) если Р3 представляет собой защитную группу для аминогруппы, то введение в реакцию соединения формулы (18А) с реагентом, удаляющим защитную группу аминогруппы, с получением соединения формулы (IA)
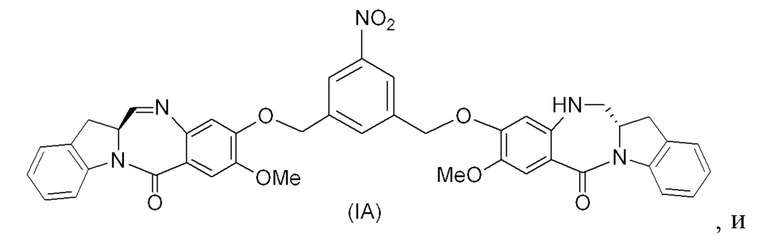
(4) введение соединения формулы (IA) в реакцию с восстановительным агентом с получением соединения формулы (IB)
или его фармацевтически приемлемой соли,
где Х3 представляет собой -Cl и
Р3 представляет собой Н или защитную группу для аминогруппы.
28. Способ по п. 27, отличающийся тем, что восстановительный агент выбран из группы, состоящей из газообразного водорода, гидросульфита натрия, сульфида натрия, хлорида олова, хлорида титана (II), цинка, железа и йодида самария.
29. Способ по п. 28, отличающийся тем, что восстановительный агент представляет собой Fe/NH4Cl или Zn/NH4Cl.
| В WO 2011064141 A1, 06.03.2011 | |||
| Kim, Yong-Chul; et al., Journal of Medicinal Chemistry, 2000, т | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ГАЗОГЕНЕРАТОР ДЛЯ ДРОВ, ТОРФА И КИЗЯКА | 1923 |
|
SU746A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2545080C2 |
| US 2007117855 A1, 24.05.2007 | |||
| WO 20057118560 A1, 15.12.2005 | |||
| WO 2002028825 А2, 11.04.2002 | |||
| US 2002042539 A1, 11.04.2002. | |||

