РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Данная заявка заявляет приоритет и преимущество по предварительной заявке США № 62/523204, поданной 21 июня 2017 года. Содержание заявки включено в данный документ посредством ссылки во всей ее полноте.
ОБЛАСТЬ ТЕХНИКИ
[0002] Данное изобретение в целом относится к кишечнорастворимым (GR) лекарственным формам для перорального применения с контролируемым высвобождением (CR), которые снижают риск пролонгации интервала QT у пациентов, которые получают лечение принимая соединение, обозначенное как 1H-изоиндол-1-он, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро, гидрохлорид, гидрат (1:1:2) и применение данных лекарственных форм для лечения шизофрении и других заболеваний.
УРОВЕНЬ ТЕХНИКИ
[0003] Интервал QT представляет собой измерение продолжительности деполяризации и реполяризации желудочков сердца. Пролонгация интервала QT, называемая пролонгацией QT, может привести к увеличению риска желудочковых аритмий, включая полиморфную желудочковую тахикардию типа «пируэт» (TdP). Поскольку было показано, что ряд лекарственных средств вызывает пролонгацию QT, разработка новых лекарственных средств обычно включает оценку их потенциала к пролонгации QT.
[0004] Minerva Neurosciences, Inc. (Уолтем, Массачусетс) разрабатывает новое исследуемое лекарственное средство гидрохлорид ролуперидона с кодовым наименованием MIN-101 для лечения негативных симптомов у пациентов с шизофренией. Активный ингредиент в MIN-101 (ранее известный как CYR-101 и MT-210) имеет химическое название 1H-изоиндол-1-он, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2). Формула I ниже демонстрирует структуру свободного основания (Соединение (I)):
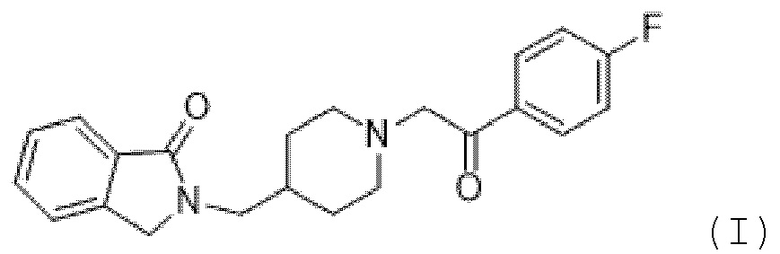
[0005] Как раскрыто в патенте США № 9458130, содержание которого полностью включено в данное описание, наблюдалась пролонгация QT у пациентов, получавших MIN-101, и, по-видимому, это было связано с уровнями Соединения (I) в плазме и, более конкретно, с метаболитом, идентифицированным как BFB-520. В патенте '130 раскрывается тот факт, что пролонгация QT, вызванная введением MIN-101, может быть уменьшена путем введения этого агента в препараты с модифицированным высвобождением (MR), который обеспечивает максимальную концентрацию (Cmax) в плазме Соединения (I) и BFB-520 ниже 80 нг/мл и 12 нг/мл соответственно. Однако существует потребность в препарате, который дополнительно снижает вероятность пролонгации QT после перорального введения MIN-101 натощак или после еды, при сохранении терапевтически эффективного уровня Соединения (I) в течение интервала дозирования.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0006] Данное изобретение основано, частично, на открытии того факта, что минимизация высвобождения Соединения (I) в течение первых четырех часов после перорального введения лекарственной формы, содержащей Соединение (I), является ключевым фактором для поддержания низких уровней BFB-520 в плазме.
[0007] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 2 до приблизительно 200 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение.
[0008] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 2 до приблизительно 200 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль плазмы для Соединения (I), который включает Tmax от приблизительно 4 ч до приблизительно 22 ч.
[0009] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение.
[0010] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль в плазме для Соединения (I), который включает Tmax от приблизительно 1 ч до приблизительно 22 ч.
[0011] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль в плазме для Соединения (I), который включает Tmax от приблизительно 1,5 ч до приблизительно 22 ч.
[0012] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль в плазме для Соединения (I), который включает Tmax от приблизительно 2 ч до приблизительно 22 ч.
[0013] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль в плазме для Соединения (I), который включает Tmax от приблизительно 2,5 ч до приблизительно 22 ч.
[0014] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль в плазме для Соединения (I), который включает Tmax от приблизительно 3 ч до приблизительно 22 ч.
[0015] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль в плазме для Соединения (I), который включает Tmax от приблизительно 3,5 ч до приблизительно 22 ч.
[0016] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль плазмы для Соединения (I), который включает Tmax от приблизительно 4 ч до приблизительно 22 ч.
[0017] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), причем количество Соединения (I) составляет от 4 мг до 8 мг, от 8 мг до 16 мг, от 16 мг до 32 мг, от 32 мг до 40 мг, от 40 мг до 64 мг, от 64 мг до 80 мг или от 80 мг до 100 мг.
[0018] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), причем количество Соединения (I) составляет 4 мг, 5 мг, 6 мг, 7 мг, 8 мг, 9 мг, 10 мг, 11 мг, 12 мг, 13 мг, 14 мг, 15 мг, 16 мг, 17 мг, 18 мг, 19 мг, 20 мг, 21 мг, 22 мг, 23 мг, 24 мг, 25 мг, 26 мг 27 мг, 28 мг, 29 мг, 30 мг, 31 мг, 32 мг, 33 мг, 34 мг, 35 мг, 36 мг, 37 мг, 38 мг, 39 мг, 40 мг, 41 мг, 42 мг, 43 мг, 44 мг, 45 мг, 46 мг, 47 мг, 48 мг, 49 мг, 50 мг, 51 мг, 52 мг, 53 мг, 54 мг, 55 мг, 56 мг, 57 мг, 58 мг, 59 мг, 60 мг, 61 мг, 62 мг, 63 мг, 64 мг, 65 мг, 66 мг, 67 мг, 68 мг, 69 мг, 70 мг, 71 мг, 72 мг, 73 мг, 74 мг, 75 мг, 76 мг , 77 мг, 78 мг, 79 мг, 80 мг, 81 мг, 82 мг, 83 мг, 84 мг, 85 мг, 86 мг, 87 мг, 88 мг, 89 мг, 90 мг, 91 мг, 92 мг, 93 мг, 94 мг, 95 мг, 96 мг, 97 мг, 98 мг, 99 мг или 100 мг.
[0019] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), причем количество Соединения (I) составляет 4 мг, 8 мг, 16 мг, 24 мг, 32 мг, 40 мг, 64 мг, 80 мг, 96 мг или 100 мг.
[0020] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. приблизительно 32 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение.
[0021] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. приблизительно 32 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль плазмы для Соединения (I), который включает Tmax от приблизительно 4 ч до приблизительно 22 ч.
[0022] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. приблизительно 64 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение.
[0023] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
i. приблизительно 64 мг Соединения (I) или эквивалентного количества его фармацевтически приемлемой соли, и/или сольвата; и
ii. по меньшей мере один агент, контролирующий высвобождение;
причем лекарственная форма дает, при пероральном введении субъекту, фармакокинетический профиль плазмы для Соединения (I), который включает Tmax от приблизительно 4 ч до приблизительно 22 ч.
[0024] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 68 ч*нг/мл.
[0025] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 16 нг/мл.
[0026] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 17 нг/мл.
[0027] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 18 нг/мл.
[0028] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 19 нг/мл.
[0029] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 20 нг/мл.
[0030] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 21 нг/мл.
[0031] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 22 нг/мл.
[0032] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а Cmax Соединения (I) менее чем приблизительно 23 нг/мл.
[0033] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а AUC0-24ч Соединения (I) находится между от приблизительно 50 ч*нг/мл до приблизительно 400 ч*нг/мл.
[0034] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а AUC0-24ч Соединения (I) находится между от приблизительно 75 ч*нг/мл до приблизительно 350 ч*нг/мл.
[0035] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а AUC0-24ч Соединения (I) находится между от приблизительно 75 ч*нг/мл до приблизительно 300 ч*нг/мл.
[0036] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, а AUC0-24ч Соединения (I) находится между от приблизительно 100 ч*нг/мл до приблизительно 300 ч*нг/мл.
[0037] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 32 мг, и фармакокинетический профиль в плазме для метаболита BFB-520 Соединения (I) включает Cmax менее чем 3,0 нг/мл, менее чем 2,5 нг/мл, менее чем 2,0 нг/мл, менее чем 1,5 нг/мл или менее чем 1,0 нг/мл.
[0038] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 50 ч*нг/мл.
[0039] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 60 ч*нг/мл.
[0040] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 70 ч*нг/мл.
[0041] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 80 ч*нг/мл.
[0042] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 90 ч*нг/мл.
[0043] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 100 ч*нг/мл.
[0044] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 110 ч*нг/мл.
[0045] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 120 ч*нг/мл.
[0046] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-4ч Соединения (I) менее чем приблизительно 130 ч*нг/мл.
[0047] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а Cmax Соединения (I) менее чем приблизительно 36 нг/мл или менее чем приблизительно 25 нг/мл.
[0048] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, а AUC0-24ч Соединения (I) находится между от приблизительно 200 ч*нг/мл до приблизительно 600 ч*нг/мл.
[0049] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая Соединение (I), раскрытое в данном документе, причем количество Соединения (I) составляет приблизительно 64 мг, и фармакокинетический профиль в плазме для метаболита BFB-520 Соединения (I) включает Cmax, которая ниже 4,0 нг/мл, ниже 3,5 нг/мл, ниже 3,0 нг/мл или ниже 2,5 нг/мл.
[0050] В одном варианте реализации, раскрытые в данном документе кишечнорастворимые лекарственные формы с контролируемым высвобождением находятся в форме таблетки, которая содержит ядро таблетки и кишечнорастворимую оболочку.
[0051] В одном варианте реализации ядро таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, и агент, контролирующий высвобождение.
[0052] В одном варианте реализации ядро таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, агент, контролирующий высвобождение, наполнитель, вещество, способствующее скольжению, и смазывающее вещество.
[0053] В одном варианте реализации ядро таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, агент, контролирующий высвобождение, наполнитель, вещество, способствующее скольжению, смазывающее вещество и оболочку.
[0054] В одном варианте реализации агент, контролирующий высвобождение, в ядре таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит одну или более гипромеллоз.
[0055] В одном варианте реализации агент, контролирующий высвобождение, в ядре таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит одну или более гипромеллоз, выбранных из группы, состоящей из Metolose® 90SH K15M 100 SR, Metolose® 90SH 100 SR, Methocel™ K100M CR, Methocel™ K15M CR, Methocel™ K4M CR и Methocel™ K100LV CR или эквивалент.
[0056] В одном варианте реализации агент, контролирующий высвобождение, в ядре таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит смесь (i) гипромеллозы с низкой вязкостью с вязкостью от приблизительно 15 милипаскаль-секунд (мПа·с) до приблизительно 100 мПа·с и (ii) гипромеллозы с высокой вязкостью с вязкостью приблизительно 100000 мПа·с, причем каждая из гипромеллоз с низкой и высокой вязкостью имеет чистоту «для контролируемого или замедленного высвобождения» и дополнительно характеризуется содержанием метоксигруппы от 19,0% до 24,0% и содержанием гидроксипропоксигруппы от 4,0% до 12,0%.
[0057] В одном варианте реализации вещество, способствующее скольжению, в ядре таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, представляет собой коллоидный безводный диоксид кремния.
[0058] В одном варианте реализации смазывающее вещество в ядре таблетки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, представляет собой стеарат магния.
[0059] В одном варианте реализации кишечнорастворимая оболочка кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит по меньшей мере один полимерный агент, контролирующий высвобождение, со свойством растворения при рН более чем рН 5,5, 6,0 или 6,5, и агент, предотвращающий слипание.
[0060] В одном варианте реализации кишечнорастворимая оболочка кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, дополнительно содержит пластификатор.
[0061] В одном варианте реализации полимерный агент, контролирующий высвобождение, кишечнорастворимой оболочки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, содержит Eudragit L30D55.
[0062] В одном варианте реализации агент, предотвращающий слипание, кишечнорастворимой оболочки кишечнорастворимых лекарственных форм с контролируемым высвобождением, раскрытых в данном документе, представляет собой Plasacryl HTP20.
[0063] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
от приблизительно 7 до приблизительно 17% масс./масс. Соединения (I) или его фармацевтически приемлемой соли, и/или сольвата;
от приблизительно 4 до приблизительно 14% масс./масс. гипромеллозы (Metolose® 90SH K15M 100 SR);
от приблизительно 17 до приблизительно 27% масс./масс. гипромеллозы (Methocel™ K100M CR);
от приблизительно 25 до приблизительно 35% масс./масс. микрокристаллической целлюлозы;
от приблизительно 13 до приблизительно 23% масс./масс. моногидрата лактозы;
от приблизительно 0,1 до приблизительно 4% масс./масс. коллоидного безводного диоксида кремния;
от приблизительно 0,1 до приблизительно 4% масс./масс. стеарата магния;
от приблизительно 1 до приблизительно 10% масс./масс. Eudragit L30D55; и
от приблизительно 0,5 до приблизительно 5% масс./масс. Plasacryl HTP20.
[0064] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
приблизительно 12% масс./масс. Соединения (I) или его фармацевтически приемлемой соли, и/или сольвата;
приблизительно 9% масс./масс. гипромеллозы (Metolose® 90SH K15M 100 SR);
приблизительно 23% масс./масс. гипромеллозы (Methocel™ K100M CR);
приблизительно 30% масс./масс. микрокристаллической целлюлозы;
приблизительно 19% масс./масс. моногидрата лактозы;
приблизительно 0,5% масс./масс. коллоидного безводного диоксида кремния;
приблизительно 1% масс./масс. стеарата магния;
приблизительно 5% масс./масс. Eudragit L30D55; и
приблизительно 1% масс./масс. Plasacryl HTP20.
[0065] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
от приблизительно 7 до приблизительно 17% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
от приблизительно 4 до приблизительно 14% масс./масс. гипромеллозы (Methocel™ K15M CR);
от приблизительно 17 до приблизительно 27% масс./масс. гипромеллозы (Methocel™ K100M CR);
от приблизительно 25 до приблизительно 35% масс./масс. микрокристаллической целлюлозы;
от приблизительно 13 до приблизительно 23% масс./масс. моногидрата лактозы;
от приблизительно 0,1 до приблизительно 4% масс./масс. коллоидного безводного диоксида кремния;
от приблизительно 0,1 до приблизительно 4% масс./масс. стеарата магния;
от приблизительно 1 до приблизительно 10% масс./масс. Eudragit L30D55;
от приблизительно 0,5 до приблизительно 5% масс./масс. Plasacryl HTP20; и
от приблизительно 0,5 до приблизительно 5% масс./масс. Surelease E-7-19040.
[0066] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
приблизительно 12% масс./масс. Соединения (I) или его фармацевтически приемлемой соли, и/или сольвата;
приблизительно 9% масс./масс. гипромеллозы (Methocel™ K15M CR);
приблизительно 23% масс./масс. гипромеллозы (Methocel™ K100M CR);
приблизительно 30% масс./масс. микрокристаллической целлюлозы;
приблизительно 19% масс./масс. моногидрата лактозы;
приблизительно 0,5% масс./масс. коллоидного безводного диоксида кремния;
приблизительно 1% масс./масс. стеарата магния;
приблизительно 5% масс./масс. Eudragit L30D55;
приблизительно 1% масс./масс. Plasacryl HTP20; и
приблизительно 1% масс./масс. Surelease E-7-19040.
[0067] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
от приблизительно 7 до приблизительно 17% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
от приблизительно 4 до приблизительно 14% масс./масс. гипромеллозы (Methocel™ K100LV CR);
от приблизительно 17 до приблизительно 27% масс./масс. гипромеллозы (Methocel™ K100M CR);
от приблизительно 25 до приблизительно 35% масс./масс. микрокристаллической целлюлозы;
от приблизительно 13 до приблизительно 23% масс./масс. моногидрата лактозы;
от приблизительно 0,1 до приблизительно 4% масс./масс. коллоидного безводного диоксида кремния;
от приблизительно 0,1 до приблизительно 4% масс./масс. стеарата магния;
от приблизительно 1 до приблизительно 10% масс./масс. Eudragit L30D55; и
от приблизительно 0,5 до приблизительно 5% масс./масс. Plasacryl HTP20.
[0068] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
приблизительно 12% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
приблизительно 9% масс./масс. гипромеллозы (Methocel™ K100LV CR);
приблизительно 23% масс./масс. гипромеллозы (Methocel™ K100M CR);
приблизительно 30% масс./масс. микрокристаллической целлюлозы;
приблизительно 19% масс./масс. моногидрата лактозы;
приблизительно 0,5% масс./масс. коллоидного безводного диоксида кремния;
приблизительно 0.5% масс./масс. стеарата магния;
приблизительно 5% масс./масс. Eudragit L30D55; и
приблизительно 1% масс./масс. Plasacryl HTP20.
[0069] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
приблизительно 12% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
приблизительно 9% масс./масс. гипромеллозы (Methocel™ K100LV CR);
приблизительно 23% масс./масс. гипромеллозы (Methocel™ K100M CR);
приблизительно 30% масс./масс. микрокристаллической целлюлозы;
приблизительно 19% масс./масс. моногидрата лактозы;
приблизительно 0,5% масс./масс. коллоидного безводного диоксида кремния;
приблизительно 1% масс./масс. стеарата магния;
приблизительно 5% масс./масс. Eudragit L30D55; и
приблизительно 1% масс./масс. Plasacryl HTP20.
[0070] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
от приблизительно 19 до приблизительно 29% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
от приблизительно 4 до приблизительно 14% масс./масс. гипромеллозы (Methocel™ K100LV CR);
от приблизительно 17 до приблизительно 27% масс./масс. гипромеллозы (Methocel™ K100M CR);
от приблизительно 19 до приблизительно 29% масс./масс. микрокристаллической целлюлозы;
от приблизительно 8 до приблизительно 18% масс./масс. моногидрата лактозы;
от приблизительно 0,1 до приблизительно 4% масс./масс. коллоидного безводного диоксида кремния;
от приблизительно 0,1 до приблизительно 4% масс./масс. стеарата магния;
от приблизительно 1 до приблизительно 10% масс./масс. Eudragit L30D55; и
от приблизительно 0,5 до приблизительно 5% масс./масс. Plasacryl HTP20.
[0071] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
приблизительно 24% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
приблизительно 9% масс./масс. гипромеллозы (Methocel™ K100LV CR);
приблизительно 23% масс./масс. гипромеллозы (Methocel™ K100M CR);
приблизительно 24% масс./масс. микрокристаллической целлюлозы;
приблизительно 13% масс./масс. моногидрата лактозы;
приблизительно 0,5% масс./масс. коллоидного безводного диоксида кремния;
приблизительно 0,5% масс./масс. стеарата магния;
приблизительно 5% масс./масс. Eudragit L30D55; и
приблизительно 1% масс./масс. Plasacryl HTP20.
[0072] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая:
приблизительно 24% масс./масс. Соединения (I) или его фармацевтически приемлемой соли и/или сольвата;
приблизительно 9% масс./масс. гипромеллозы (Methocel™ K100LV CR);
приблизительно 23% масс./масс. гипромеллозы (Methocel™ K100M CR);
приблизительно 24% масс./масс. микрокристаллической целлюлозы;
приблизительно 13% масс./масс. моногидрата лактозы;
приблизительно 0,5% масс./масс. коллоидного безводного диоксида кремния;
приблизительно 1% масс./масс. стеарата магния;
приблизительно 5% масс./масс. Eudragit L30D55; и
приблизительно 1% масс./масс. Plasacryl HTP20.
[0073] В одном аспекте в данном документе представлен способ снижения риска пролонгации QT при лечении субъекта Соединением (I) или его фармацевтически приемлемой солью, и/или сольватом, причем способ включает пероральное введение субъекту кишечнорастворимой формы с контролируемым высвобождением, описанной в данном документе.
[0074] В одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает пероральное введение субъекту кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе, причем субъект имеет диагноз расстройства, например, шизофрения.
[0075] В одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает один раз в день пероральное введение субъекту кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе, причем субъект имеет диагноз расстройства, например, шизофрения.
[0076] В одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает пероральное введение субъекту кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе, причем субъект имеет диагноз, например, шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM.
[0077] В одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает один раз в день пероральное введение субъекту кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе, причем субъект имеет диагноз, например, шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM.
[0078] В одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает пероральное введение субъекту кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе (например, включая низкую дозу Соединения (I), например приблизительно 4 мг, 5 мг, 6 мг, 7 мг, 8 мг или приблизительно 16 мг), причем субъект имеет диагноз, например, шизофрения, при этом субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM.
[0079] В одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает пероральное введение субъекту один раз в день кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе (например, включая низкую дозу Соединения (I), например приблизительно 4 мг, 5 мг, 6 мг, 7 мг, 8 мг или приблизительно 16 мг), причем субъект имеет диагноз, например, шизофрения, при этом субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM.
[0080] В одном аспекте для любого из способов, раскрытых в данном документе, субъект находится в состоянии сытости перед пероральным введением кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе.
[0081] В одном аспекте для любого из способов, раскрытых в данном документе, субъект находится в голодном состоянии перед пероральным введением кишечнорастворимой лекарственной формы с контролируемым высвобождением, описанной в данном документе.
[0082] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для использования для снижения риска пролонгации QT.
[0083] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении.
[0084] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM.
[0085] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг, 8 мг или приблизительно 16 мг.
[0086] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для использования для снижения риска пролонгации QT, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[0087] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[0088] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM, при этом кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[0089] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг, 8 мг или приблизительно 16 мг, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[0090] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении, причем субъект находится в состоянии сытости перед пероральным введением лекарственной формы.
[0091] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении, причем субъект находится в голодном состоянии перед пероральным введением лекарственной формы.
[0092] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM, причем субъект находится в состоянии сытости перед пероральным введением лекарственной формы.
[0093] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM, причем субъект находится в голодном состоянии перед пероральным введением лекарственной формы.
[0094] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг, 8 мг или приблизительно 16 мг, причем субъект находится в сытом состоянии до перорального введения лекарственной формы
[0095] В одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма с контролируемым высвобождением, раскрытая в данном документе, для применения при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг, 8 мг или приблизительно 16 мг, причем субъект находится в голодном состоянии до перорального введения лекарственной формы
[0096] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытая в данном документе, в производстве лекарственного средства для снижения риска пролонгации QT.
[0097] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении.
[0098] В одном аспекте в данном изобретении предусматривается использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM.
[0099] В одном аспекте в данном изобретении предусматривается использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытая в данном документе, в производстве лекарственного средства при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг или приблизительно 8 мг.
[00100] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытая в данном документе, в производстве лекарственного средства для снижения риска пролонгации QT, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[00101] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[00102] В одном аспекте в данном изобретении предусматривается использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрении, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[00103] В одном аспекте в данном изобретении предусматривается использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытая в данном документе, в производстве лекарственного средства при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг или приблизительно 8 мг, причем кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
[00104] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект находится в голодном состоянии перед пероральным введением лекарственной формы.
[00105] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM, причем субъект находится в голодном состоянии перед пероральным введением лекарственной формы.
[00106] В одном аспекте в данном изобретении предусматривается использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытая в данном документе, в производстве лекарственного средства при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг или приблизительно 8 мг, причем субъект находится в голодном состоянии до перорального введения лекарственной формы.
[00107] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект находится в состоянии сытости перед пероральным введением лекарственной формы.
[00108] В одном аспекте в данном изобретении предусматривает использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытой в данном документе, в производстве лекарственного средства для лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 EM, при этом субъект находится в состоянии сытости перед пероральным введением лекарственной формы.
[00109] В одном аспекте в данном изобретении предусматривается использование кишечнорастворимой лекарственной формы с контролируемым высвобождением, раскрытая в данном документе, в производстве лекарственного средства при лечении расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, например, у субъекта, имеющего диагноз шизофрения, причем субъект, имеющий диагноз шизофрения, имеет генотип CYP2D6 IM или PM, а лекарственная форма имеет низкую дозу Соединения (I), например, приблизительно 4 мг, 5 мг, 6 мг, 7 мг или приблизительно 8 мг, причем субъект находится в состоянии сытости до перорального введения лекарственной формы.
[00110] Таким образом, в одном аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма для перорального введения с контролируемым высвобождением, которая содержит (i) от приблизительно 4 мг до приблизительно 100 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и (ii) по меньшей мере один агент, контролирующий высвобождение, причем лекарственная форма при пероральном введении субъекту создает фармакокинетический (PK) профиль плазмы для Соединения (I), который содержит Tmax от приблизительно 4 до приблизительно 11 ч. В одном варианте реализации Tmax Соединения (I) в профиле ФК в плазме составляет от приблизительно 5 до приблизительно 10 ч; от приблизительно 6 до приблизительно 9 ч, от приблизительно 7 до приблизительно 9 ч или от приблизительно 6 до приблизительно 8 ч.
[00111] В некоторых вариантах реализации количество Соединения (I) в пероральной лекарственной форме составляет от 4 мг до 8 мг, от 8 до 16 мг, от 16 мг до 32 мг, от 32 мг до 40 мг, от 40 мг до 64 мг, от 64 мг до 80 мг, от 80 до 100 мг или приблизительно 4 мг, 8 мг, 16 мг, 24 мг, 32 мг, 40 мг, 64 мг, 80 мг, 96 мг или 100 мг.
[00112] В одном варианте реализации лекарственная форма содержит приблизительно 32 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 68 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 16 нг/мл, 17 нг/мл, 18 нг/мл, 19 нг/мл, 20 нг/мл, 21 нг/мл, 22 нг/мл или 23 нг/мл; и (c) AUC0-24ч от приблизительно 75 ч*нг/мл до приблизительно 350 ч*нг/мл или от приблизительно 100 ч*нг/мл до 300 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 3,0 нг/мл, менее 2,5 нг/мл, менее 2,0 нг/мл, менее 1,5 нг/мл или менее 1,0 нг/мл.
[00113] В одном варианте реализации лекарственная форма содержит приблизительно 4 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 8 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 2,5 нг/мл; и (c) AUC0-24ч от приблизительно 12 ч*нг/мл до 35 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 2,0 нг/мл, менее 1,5 нг/мл, менее 1,0 нг/мл или менее 0,5 нг/мл.
[00114] В одном варианте реализации лекарственная форма содержит приблизительно 8 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 16 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 5 нг/мл; и (c) AUC0-24ч от приблизительно 25 ч*нг/мл до 75 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 2,5 нг/мл, менее 2,0 нг/мл, менее 1,5 нг/мл или менее 1,0 нг/мл.
[00115] В одном варианте реализации лекарственная форма содержит приблизительно 16 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 32 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 10 нг/мл или менее чем приблизительно 6 нг/мл; и (c) AUC0-24ч от приблизительно 50 ч*нг/мл до 150 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 2,5 нг/мл, менее 2,0 нг/мл, менее 1,5 нг/мл или менее 1,0 нг/мл.
[00116] В одном варианте реализации лекарственная форма содержит приблизительно 40 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 80 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 24 нг/мл или менее чем приблизительно 20 нг/мл; и (c) AUC0-24ч от приблизительно 125 ч*нг/мл до 375 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 3,5 нг/мл, менее 3,0 нг/мл, менее 2,5 нг/мл или менее 2,0 нг/мл.
[00117] В одном варианте реализации лекарственная форма содержит приблизительно 64 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 50, 60, 70, 80, 90, 100, 110, 120 или 130 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 36 нг/мл или менее чем приблизительно 25 нг/мл; и (c) AUC0-24ч от приблизительно 200 ч*нг/мл до 600 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 4,0 нг/мл, менее 3,5 нг/мл, менее 3,0 нг/мл или менее 2,5 нг/мл.
[00118] В одном варианте реализации лекарственная форма содержит приблизительно 80 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 160 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 48 нг/мл или менее чем приблизительно 40 нг/мл; и (c) AUC0-24ч от приблизительно 250 ч*нг/мл до 750 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 4,5 нг/мл, менее 4,0 нг/мл, менее 3,5 нг/мл или менее 3,0 нг/мл.
[00119] В одном варианте реализации лекарственная форма содержит приблизительно 100 мг Соединения (I) или эквивалентное количество его фармацевтически приемлемой соли и/или сольвата, и профиль ФК в плазме дополнительно содержит: (a) AUC0-4ч менее чем приблизительно 220 ч*нг/мл; (b) Cmax Соединения (I) менее чем приблизительно 72 нг/мл; и (c) AUC0-24ч от приблизительно 325 ч*нг/мл до 975 ч*нг/мл. В одном варианте реализации профиль ФК в плазме для метаболита BFB-520 Соединения (I) содержит Cmax, что составляет менее 5,0 нг/мл, менее 4,5 нг/мл, менее 4,0 нг/мл или менее 3,5 нг/мл.
[00120] В некоторых вариантах реализации любой из указанных выше лекарственных форм параметры ФК в плазме представляют собой значения, определенные после двухразовых ежедневных введений одной единицы лекарственной формы. В одном варианте реализации параметр ФК определяется после 3-го или 4-го введения.
[00121] В одном варианте реализации лекарственная форма содержит приблизительно 32 мг Соединения (I) и при введении субъекту создает фармакокинетический (PK) профиль в плазме для Соединения (I), который аналогичен целевому профилю, показанному на фиг.1.
[00122] В некоторых вариантах реализации любой из указанных выше лекарственных форм профиль ФК в плазме для одного или обоих Соединений (I) и метаболита BFB-520 получают после 1-го, 2-го, 3-го или 4-го однократного ежедневного введения одной единицы лекарственной формы.
[00123] В некоторых вариантах реализации профиль ФК в плазме для одного или обоих Соединений (I) и метаболита BFB-520 продуцируется при введении субъекту натощак. В других вариантах реализации профиль ФК в плазме для одного или обоих Соединений (I) и метаболита BFB-520 продуцируется при введении субъекту в состоянии сытости.
[00124] В одном варианте реализации лекарственная форма с контролируемым высвобождением по данному изобретению содержит от приблизительно 4 до приблизительно 100 мг Соединения (I) и дает целевой профиль растворения in vitro с использованием 24-часового двухстадийного способа растворения in vitro, который включает 2-часовую кислотную стадию и 22-часовую буферную стадию. Целевой профиль растворения in vitro включает (а) не обнаруживаемое высвобождение Соединения (I) в течение первых 2,0 ч способа растворения и (b) высвобождение по меньшей мере 80% от общего количества Соединения (I) в лекарственной форме в течение 16-19 ч. В одном варианте реализации целевой профиль растворения in vitro включает высвобождение по меньшей мере 85%, 90% или 95% количества Соединения (I) в лекарственной форме за 24 ч способа растворения.
[00125] В варианте реализации целевой профиль растворения in vitro дополнительно включает высвобождение Соединения (I) со скоростью высвобождения, которая дает каждый из следующих совокупных процентов от исходного общего количества:
менее чем 0,6% через 2,5 ч;
от 0,2 до 7,9% через 3,0 ч;
от 2,5 до 19,2% через 4 ч;
от 12,7 до 34,0% через 6 ч;
от 22,8 до 44,3% через 8 ч;
от 35,5 до 75,7% через 13 ч;
от 43,3 до 89,0% через 16 ч; и
от 59,3 до 96,9% через 19 ч.
[00126] В варианте реализации целевой профиль растворения in vitro дополнительно включает высвобождение Соединения (I) со скоростью высвобождения, которая дает каждый из следующих совокупных процентов от исходного общего количества:
менее чем приблизительно 0,5% через 2,5 ч;
от приблизительно 2,8 до приблизительно 3,1% чере 3,0 ч;
от приблизительно 9,0 до приблизительно 11,0% через 4 ч;
от приблизительно 14,5 до приблизительно 18,0% через 5 ч;
от приблизительно 19,5 до приблизительно 24,5% через 6 ч;
от приблизительно 30,5 до приблизительно 38,0% через 8 ч;
от приблизительно 41,5 до приблизительно 51,0% через 10 ч;
от приблизительно 54,5 до приблизительно 67,0% через 13 ч;
от приблизительно 58,5 до приблизительно 71,5% через 14 ч;
от приблизительно 61,5 до приблизительно 75,5% через 15 ч;
от приблизительно 70,0 до приблизительно 86,0% через 18 ч; и
от приблизительно 77,5 до приблизительно 95,0% через 21 ч.
[00127] В одном варианте реализации лекарственная форма CR содержит 32 мг Соединения (I) и генерирует профили накопленной скорости растворения и растворения in vitro, которые по существу аналогичны целевому профилю, показанному на фиг.1, или целевому профилю, показанному в таблицах 6 и 7 в Примерах ниже.
[00128] В варианте реализации целевой профиль растворения in vitro дополнительно включает высвобождение Соединения (I) со скоростью высвобождения, которая дает каждый из следующих совокупных процентов от исходного общего количества:
менее чем приблизительно 0,5% через 2 ч;
от приблизительно 19 до приблизительно 29% через 4 ч;
от приблизительно 54 до приблизительно 64% через 8 ч; и
от приблизительно 83 до приблизительно 93% через 16 ч.
[00129] В варианте реализации целевой профиль растворения in vitro дополнительно включает высвобождение Соединения (I) со скоростью высвобождения, которая дает каждый из следующих совокупных процентов от исходного общего количества:
менее чем приблизительно 0,5% через 2 ч;
приблизительно 24,1% через 4 ч;
приблизительно 59,2% через 8 ч; и
приблизительно 88,6% через 16 ч.
[00130] В каждом из приведенных выше вариантов реализации способ растворения предпочтительно проводят в соответствии со способом растворения, описанным в приведенных ниже примерах.
[00131] В одном варианте реализации пероральная лекарственная форма CR представляет собой таблетку, которая содержит ядро таблетки и кишечнорастворимую оболочку. Ядро таблетки содержит желаемое количество Соединения (I), агента с контролируемым высвобождением, наполнителя, агента, предотвращающего слипание, и смазывающего вещества, и кишечнорастворимая оболочка содержит по меньшей мере один полимерный агент, контролирующий высвобождение, со свойством растворения более чем рН 5,5 и агент, предотвращающий слипание. В одном варианте реализации кишечнорастворимая оболочка растворяется при pH более чем 6,0 или 6,5.
[00132] В одном варианте реализации пероральная лекарственная форма CR представляет собой таблетку, которая содержит ядро таблетки и кишечнорастворимую оболочку. Ядро таблетки содержит желаемое количество Соединения (I) или его фармацевтически приемлемой соли и/или сольвата (например, MIN-101), агента с контролируемым высвобождением, наполнителя, агента, предотвращающего слипание, и смазывающего вещества, и кишечнорастворимая оболочка содержит по меньшей мере один полимерный агент, контролирующий высвобождение, со свойством растворения более чем рН 5,5 и агент, предотвращающий слипание. В одном варианте реализации кишечнорастворимая оболочка растворяется при pH более чем 6,0 или 6,5.
[00133] В некоторых вариантах реализации агент, контролирующий высвобождение, в ядре таблетки содержит смесь (i) гипромеллозы с низкой вязкостью приблизительно 15 милипаскаль-секунд (мПа·с) до приблизительно 100 мПа·с и (ii) гипромеллозы с высокой вязкостью приблизительно 100.000 мПа·с, причем каждая из гипромеллоз с низкой и высокой вязкостью имеет класс с контролируемым высвобождением или с замедленным высвобождением и дополнительно характеризуется содержанием метокси группы от 19,0% до 24,0% и содержание гидроксипропоксигруппы от 4,0% до 12,0%. В одном варианте реализации гипромеллоза с высокой вязкостью характеризуется содержанием метоксигруппы от 22,0% до 24,0% и содержанием гидроксипропоксигруппы от 9,5% до 11,5%. В одном варианте реализации гипромеллоза с низкой вязкостью составляет приблизительно 10% от массы ядра таблетки, а гипромеллоза с высокой вязкостью составляет приблизительно 24% от массы ядра таблетки.
[00134] В одном варианте реализации ядро таблетки с сердцевиной содержит 38,4 мг 1Н-изоиндол-1-она, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2) и агент, контролирующий высвобождение, в ядре таблетки состоит в основном из (i) 9,45% масс./масс. гипромеллозы, имеющей химические и физические характеристики гипромеллозы, выпускаемой на рынок как METOLOSE® 90 SH 100 SR от Shin-Etsu Chemical Co., Ltd. или METHOCEL™ K100LV CR; и (ii) 22,67% масс./масс. гипромеллозы, имеющей химические и физические характеристики гипромеллозы, выпускаемой как METHOCEL™ K100M CR компанией The Dow Chemical Company.
[00135] В одном варианте реализации, лекарственная форма дополнительно содержит покрытие с контролируемым высвобождением, расположенное между ядром таблетки и кишечнорастворимой оболочкой. Оболочка с контролируемым высвобождением содержит по меньшей мере один реагент, контролирующий высвобождение. В одном варианте реализации оболочка с контролируемым высвобождением содержит полупроницаемую мембрану, которая содержит этилцеллюлозу в качестве агента с контролируемым высвобождением.
[00136] В одном варианте реализации лекарственной формы, которая содержит оболочку с контролируемым высвобождением, ядро таблетки содержит 38,4 мг 1Н-изоиндол-1-она, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2) и агент, контролирующий высвобождение, в ядре таблетки состоит, по существу, из 9,36% масс./масс. гипромеллозы, имеющей химические и физические характеристики продукта гипромеллозы, продаваемой как METHOCEL™ K15M CR компанией The Dow Chemical Company, или METHOCEL™ K100LV CR; и 22,46% гипромеллозы, имеющей химические и физические характеристики продукта гипромеллозы, поставляемого на рынок как METHOCEL™ K100M CR компанией Dow Chemical, и агент, контролирующий высвобождение, в оболочке с контролируемым высвобождением состоит по существу из 0,94% масс./масс. этилцеллюлозы, имеющей химические и физические характеристики этилцеллюлозы, поставляемой на рынок как Surelease® E-7-19040 от Colorcon.
[00137] В некоторых вариантах реализации любой из указанных выше лекарственных форм кишечнорастворимая оболочка состоит по существу из смеси (i) 4,68% масс./масс. сополимера метакриловой кислоты и этилакрилата, имеющего те же физические и химические свойства, что и сополимер, продаваемый на рынке как EUDRAGIT® L 30 D-55 от Evonik Industries AG и (ii) 0,80% масс./масс. вещества, предотвращающего слипание, имеющего химические и физические характеристики продукта, предотвращающего слипание, продаваемого как PlasACRYLTM от Evonik Industries AG.
[00138] В варианте реализации изобретения, кишечнорастворимая таблетка CR по данному изобретению имеет круглую форму, овальную форму, форму капсулы или продолговатую форму. В одном варианте реализации изобретения, таблетка является круглой, с диаметром 10 мм и радиусом кривизны (R) 10.
[00139] В других аспектах в данном изобретении предлагается содержание серии и способ производства кишечнорастворимой CR лекарственной формы для перорального введения, описанной в данном документе.
[00140] В еще одном аспекте в данном документе представлен способ снижения риска пролонгации QT при лечении субъекта Соединением (I), причем способ включает введение субъекту кишечнорастворимую CR лекарственную форму для перорального введения с контролируемым высвобождением, описанной в данном документе.
[00141] В еще одном аспекте в данном документе представлен способ лечения расстройства (например, негативных симптомов шизофрении) у субъекта, нуждающегося в этом, причем способ включает один раз в день введение пациенту кишечнорастворимой CR лекарственной формы для перорального введения, описанной в данном документе. В одном варианте реализации субъект, например, пациент, имеет диагноз шизофрения. В одном варианте реализации пациент имеет диагноз шизофрения, генотип быстрого метаболизатора (БМ) CYP2D6, и пероральная лекарственная форма содержит от 32 до 64 мг Соединения (I). В другом варианте реализации пациент имеет диагноз шизофрения, генотип медленного метаболизатора (ММ) CYP2D6, и пероральная лекарственная форма с контролируемым высвобождением содержит от 4 до 16 мг Соединения (I). В другом варианте реализации пациент имеет диагноз шизофрения, генотип промежуточного метаболизатора (ПМ) CYP2D6, и пероральная лекарственная форма с контролируемым высвобождением содержит от 8 мг до 32 мг Соединения (I).
[00142] В другом аспекте в данном изобретении предлагается кишечнорастворимая лекарственная форма для перорального введения с контролируемым высвобождением описанная в данном документе для применения при лечении негативных симптомов у пациента. В одном варианте реализации изобретения, пациент имеет диагноз шизофрения. В одном варианте реализации изобретения, лекарственная форма предназначена для применения для улучшения одного или обоих негативных симптомов и когнитивных нарушений у пациентов с диагнозом шизофрения.
[00143] В другом аспекте в данном изобретении предлагается использование кишечнорастворимой лекарственной формы для перорального введения с контролируемым высвобождением, описанной в данном документе, для получения лекарственного средства для лечения негативных симптомов у пациента. В одном варианте реализации изобретения, пациент имеет диагноз шизофрения.
[00144] В другом аспекте в данном изобретении предлагается набор для применения при лечении негативных симптомов у пациента, при этом набор содержит кишечнорастворимую лекарственную форму для перорального введения с контролируемым высвобождением, описанную в данном документе, и инструкции по применению лекарственной формы. В одном варианте реализации изобретения, инструкции включают инструкции по тестированию пациента для определения CYP2D6 генотипа пациента. В одном варианте реализации изобретения, инструкции включают инструкции по введению лекарственной формы пациенту после еды или натощак.
[00145] Во всех вышеупомянутых аспектах и вариантах реализации изобретения Соединение (I) может быть представлено в кишечнорастворимой CR лекарственной форме для перорального введения в виде 1H-изоиндол-1-она, 2-[[1-[2-(4-фторфенил))-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2).
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[00146] Файл патента или заявки содержит как минимум одну фигуру, выполненную в цвете. Копии данного патента или публикации патентной заявки с цветными графическими материалами будут предоставлены Ведомством по запросу и уплате необходимой пошлины.
[00147] На графических материалах продемонстрированы различные графики, которые включают временной профиль концентрации в плазме различных Соединений, включая, например, 1H-изоиндол-1-он, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, т.е. Соединение (I). В данных графических материалах использование «MIN-101» или «MIN101» предназначено для обозначения свободного основания, то есть Соединения (I).
[00148] Вышеизложенное краткое изложение, а также последующее подробное описание изобретения будут лучше поняты при прочтении вместе с прилагаемыми графическими материалами.
[00149] На Фиг.1 проиллюстрирован типовой целевой профиль ФК в плазме для Соединения (I), полученный пероральным введением кишечнорастворимой лекарственной формы с контролируемым высвобождением, содержащей 32 мг Соединения (I) («прогнозируемый новый состав»), по сравнению с наблюдаемым ФК профиль в плазме для Соединения (I), полученного с помощью предыдущей таблетки MIN-101 по 32 мг с модифицированным высвобождением (описанной в примере 1) («MR 32 мг»). Временной профиль концентрации в плазме через 24 ч.
[00150] На Фиг.2 продемонстрировано сравнение целевых профилей растворения in vitro для Соединения (I) (мишень, красная кривая) с наблюдаемыми профилями растворения для двух типичных кишечнорастворимых таблеток CR 32 мг по данному изобретению, с левым графиком, показывающим кумулятивные профили растворения и правый график, показывающий профили скорости растворения. Временной профиль концентрации в плазме через 24 ч.
[00151] На Фиг.3 продемонстрирован другой типовой целевой ФК профиль в плазме для Соединения (I), полученного пероральным введением кишечнорастворимой лекарственной формы с контролируемым высвобождением, содержащей 32 мг Соединения (I), субъекту в после еды или натощак («Оптимальный состав») по сравнению с наблюдаемыми профилями ФК в плазме для Соединения (I), полученного с помощью предыдущей таблетки 32 мг MIN-101 с модифицированным высвобождением (описанной в примере 1), у пациентов в после еды или натощак. Временной профиль концентрации в плазме через 24 ч.
[00152] На Фиг.4 продемонстрирован график временного профиля концентраций в плазме Соединения (I) для субъектов, которым вводили таблетку по 32 мг с модифицированным высвобождением. Временной профиль концентрации в плазме через 36 ч.
[00153] На Фиг.5 продемонстрирован график временного профиля концентрации в плазме Соединения (I) для субъектов, которым вводили GR-01 таблетку. Временной профиль концентрации в плазме через 36 ч.
[00154] На Фиг.6 продемонстрирован график временного профиля концентрации в плазме Соединения (I) для субъектов, которым вводили GR-02 таблетку. Временной профиль концентрации в плазме через 36 ч.
[00155] На Фиг.7 продемонстрирован график временного профиля концентрации в плазме Соединения (I) для субъектов, которым вводили таблетку по 32 мг с модифицированным высвобождением, таблетку GR-01 или GR-02. Временной профиль концентрации в плазме через 48 ч.
[00156] На Фиг.8 продемонстрирован график скоростей увеличения или уменьшения концентраций в плазме - время Соединения (I) для субъектов, которым вводили таблетку по 32 мг с модифицированным высвобождением, таблетку GR-01 или GR-02. Временной профиль концентрации в плазме через 8 ч.
[00157] На Фиг.9 продемонстрирован график временного профиля концентрации в плазме BFB-520 для субъектов, которым вводили таблетку по 32 мг с модифицированным высвобождением. Временной профиль концентрации в плазме через 36 ч.
[00158] На Фиг.10 продемонстрирован график временного профиля концентрации в плазме BFB-520 для субъектов, которым вводили GR-01 таблетку. Временной профиль концентрации в плазме через 36 ч.
[00159] На Фиг.11 продемонстрирован график концентрация в плазме - время BFB-520 для субъектов, которым вводили GR-02 таблетку. Временной профиль концентрации в плазме через 36 ч.
[00160] На Фиг.12 продемонстрирован график концентрация в плазме - время основного BFB-520 для субъектов, которым вводили таблетку по 32 мг с модифицированным высвобождением, таблетку GR-01 или GR-02. Временной профиль концентрации в плазме через 48 ч.
[00161] На Фиг.13 продемонстрирован график скоростей увеличения или уменьшения временного профиля концентраций BFB-520 в плазме для субъектов, которым вводили таблетку по 32 мг с модифицированным высвобождением, таблетку GR-01 или GR-02. Временной профиль концентрации в плазме через 8 ч.
[00162] На Фиг. 14 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций Соединения (I) в плазме для субъектов, которым вводили 4 суточные дозы таблетки MR 32 мг или таблетки GR-01 (32 мг) на основании фактических данных, наблюдаемых после введения один раз в день. Временной профиль концентрации в плазме через 96 ч.
[00163] На Фиг. 15 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций BFB-520 в плазме для субъектов, которым вводили 4 суточные дозы таблетки MR 32 мг или таблетки GR-01 (32 мг) на основании фактических данных, наблюдаемых после введения один раз в день. Временной профиль концентрации в плазме через 96 ч.
[00164] На Фиг. 16 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций Соединения (I) в плазме для субъектов, которым вводили 4 суточные дозы 64 мг (2×32 мг) таблетки MR 32 мг или таблетки GR-01. Временной профиль концентрации в плазме через 96 ч.
[00165] На Фиг. 17 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций BFB-520 в плазме для субъектов, которым вводили 4 суточные дозы 64 мг (2×32 мг) таблетки MR 32 мг или таблетки GR-01. Временной профиль концентрации в плазме через 96 ч.
[00166] На Фиг. 18 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций Соединения (I) в плазме для субъектов, которым вводили 4 суточные дозы таблетки MR 32 мг или таблетки GR-02 (32 мг) на основании фактических данных, наблюдаемых после введения один раз в день. Временной профиль концентрации в плазме через 96 ч.
[00167] На Фиг. 19 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций BFB-520 в плазме для субъектов, которым вводили 4 суточные дозы таблетки MR 32 мг или таблетки GR-02 (32 мг) на основании фактических данных, наблюдаемых после введения один раз в день. Временной профиль концентрации в плазме через 96 ч.
[00168] На Фиг. 20 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций Соединения (I) в плазме для субъектов, которым вводили 4 суточные дозы таблетки MR 32 мг или таблетки GR-02 на основании фактических данных, наблюдаемых после введения один раз в день. Временной профиль концентрации в плазме через 96 ч.
[00169] На Фиг. 21 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций BFB-520 в плазме для субъектов, которым вводили 4 суточные дозы 64 мг (2×32 мг) таблетки MR 32 мг или таблетки GR-02. Временной профиль концентрации в плазме через 96 ч.
[00170] На Фиг.22 продемонстрирован график концентрация в плазме - время Соединения (I) для субъектов, которым вводили GR-01 таблетки в сытом состоянии. Временной профиль концентрации в плазме через 36 ч.
[00171] На Фиг.23 продемонстрирован график концентрация в плазме - время Соединения (I) для субъектов, которым вводили GR-01 таблетки натощак. Временной профиль концентрации в плазме через 36 ч.
[00172] На Фиг.24 продемонстрирован график, сравнивающий профиль концентрации в плазме - время Соединения (I) для субъектов, которым вводили GR-01 таблетки натощак. Временной профиль концентрации в плазме через 48 ч.
[00173] На Фиг.25 продемонстрирован график концентрация в плазме - время BFB-520 для субъектов, которым вводили GR-01 таблетки после еды. Временной профиль концентрации в плазме через 48 ч.
[00174] На Фиг.26 продемонстрирован график концентрация в плазме - время BFB-520 для субъектов, которым вводили GR-01 таблетки натощак. Временной профиль концентрации в плазме через 48 ч.
[00175] На Фиг.27 продемонстрирован график, сравнивающий профиль концентрации в плазме - время BFB-520 для субъектов, которым вводили GR-01 таблетки в натощак. Временной профиль концентрации в плазме через 48 ч.
[00176] На Фиг. 28 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций Соединения (I) в плазме для субъектов, которым вводили 4 суточные дозы таблетки GR-01 (32 мг) на основании фактических данных, наблюдаемых после введения один раз в день после еды и натощак. Временной профиль концентрации в плазме через 96 ч.
[00177] На Фиг. 29 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций BFB-520 в плазме для субъектов, которым вводили 4 суточные дозы таблетки GR-01 (32 мг) на основании фактических данных, наблюдаемых после введения один раз в день после еды и натощак. Временной профиль концентрации в плазме через 96 ч.
[00178] На Фиг. 30 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций Соединения (I) в плазме для субъектов, которым вводили 4 суточные дозы 64 мг (2×32 мг) таблетки GR-01 после еды и натощак. Временной профиль концентрации в плазме через 96 ч.
[00179] На Фиг. 31 продемонстрирована пара графиков прогнозируемого временного профиля установившихся концентраций BFB-520 в плазме для субъектов, которым вводили 4 суточные дозы 64 мг (2×32 мг) таблетки GR-01 (64 мг) после еды и натозак. Временной профиль концентрации в плазме через 96 ч.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[00180] В данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, составы серии и способы изготовления лекарственных форм, и применение лекарственных форм для терапевтического лечения пациентов, страдающих различными расстройствами и состояниями.
[00181] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем при пероральном введении субъекту Cmax Соединения (I) и его метаболита, BFB-520, снижается, в то время как AUC(0-tau) сохраняется по сравнению с ранее раскрытыми составами и/или лекарственными формами, например, такими, которые раскрыты в патенте США № 9458130.
[00182] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем при пероральном введении субъекту аналогичное воздействие Соединения (I) на основе AUC поддерживается по сравнению с предыдущим исследованием с ранее раскрытыми составами и/или лекарственными формами, например, такими, которые описаны в патенте США № 9458130, которое достигло своей первичной конечной точки улучшения негативных симптомов у пациентов с шизофренией с использованием обеих протестированных доз, 64 мг и 32 мг.
[00183] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем при пероральном введении субъекту t1/2 Соединения (I) является пролонгированным по сравнению с ранее раскрытыми составами и/или лекарственными формами, например, такими, которые раскрыты в патенте США № 9458130.
[00184] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем при пероральном введении субъекту Cmax BFB-520 в плазме субъекта снижается для обеспечения безопасности лекарственного средства.
[00185] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем при пероральном введении субъекту Cmax BFB-520 снижается на приблизительно 30% или более (например, 30%, 35% или 40%) по сравнению с ранее раскрытыми составами и/или лекарственными формами, например, такими, которые раскрыты в патенте США № 9458130.
[00186] В одном варианте реализации снижение Cmax BFB-520 у субъекта приводит к снижению вероятности временного увеличения QTc, наблюдаемого в предыдущем исследовании при более высокой дозе, но не при более низкой дозе. В одном примере введение новых кишечнорастворимых CR лекарственных форм для перорального применения, описанных в данном документе, содержащих Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, не приводит к наблюдаемым пролонгациям QTc.
[00187] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем введение лекарственной формы не приводит к наблюдаемому влиянию приема пищи, т.е. введение лекарственной формы может происходить с пищей или без нее без изменения ее фармакокинетических свойств.
[00188] В одном варианте реализации в данном изобретении предлагаются новые кишечнорастворимые CR лекарственные формы для перорального применения, содержащим Соединение (I) или его фармацевтически приемлемую соль, и/или сольват, причем лекарственные формы, содержащие Соединение (I), сохраняют ранее установленные общие профили безопасности и переносимости.
[00189] Новые кишечнорастворимые CR лекарственные формы для перорального применения, раскрытые в данном документе, позволяют доставлять Соединение (I) в нижнюю часть желудочно-кишечного тракта, что неожиданно снижало самую высокую концентрацию BFB-520. Этот неожиданный фармакокинетический эффект привел к отсутствию наблюдаемых пролонгаций QTc у субъектов, которым вводили данные новые кишечнорастворимые лекарственные формы для перорального применения с контролируемым высвобождением.
Определения и сокращения
[00190] Термины, используемые в данном документе, имеют свое обычное значение, и значение таких терминов является независимым при каждом их появлении. Несмотря на вышесказанное, и, если не указано иное, в описании и формуле изобретения применяются следующие определения.
[00191] Химические названия, общие названия и химические структуры могут использоваться взаимозаменяемо для описания одной и той же структуры. Если химическое Соединение упоминается с использованием как химической структуры, так и химического названия, и существует неоднозначность между структурой и названием, структура считается преобладающей.
[00192] Все ссылки на Соединение (I) в данном документе включают все фармацевтически приемлемые соли (такие как MIN-101) и/или все сольваты (например, включая гидраты) и их альтернативные физические формы, если не указано иное. Все приведенные в данном документе дозы основаны на молекулярной массе свободного основания Соединения (I), которая составляет 366,43 г/моль, а не на молекулярной массе его фармацевтически приемлемой соли или сольвата (например, гидрата) или любых вспомогательных веществ в композиции, если не указано иное.
[00193] Все количества компонента пероральной лекарственной формы, описанной в данном документе, которые указаны в расчете на % масс./масс., относятся к общей массе пероральной лекарственной формы, если не указано иное.
[00194] Термин «приблизительно» как часть количественного выражения, такого как «приблизительно Х», включает любое значение, которое на 10% выше или ниже Х, а также включает любое числовое значение, которое находится в диапазоне от Х-10% до Х+10%. Так, например, масса приблизительно 40 г включает массу от 36 до 44 г.
[00195] «Введение» относится к введению агента, такого как Соединение или лекарственная форма, описанная в данном документе, субъекту. Соответствующие термины «введение» и «введение» (и грамматические эквиваленты) относятся как к прямому введению, которое может быть введением субъекту медицинским работником или посредством самостоятельного управления субъектом, так и/или косвенным введением, которое может быть актом назначения лекарственного средства, такого как лекарственная форма, описанная в данном документе. Например, доктор, который инструктирует пациента самостоятельно вводить лекарственное средство и/или предоставляет пациенту рецепт на введение лекарственного средства пациенту.
[00196] «BFB-520» является метаболитом Соединения (I) и имеет структуру, проиллюстрированную формулой II ниже:

[00197] «BFB-999» является метаболитом Соединения (I) и имеет структуру соли малеиновой кислоты BFB-999, показанной в формуле III ниже:
[00198] «Подобный ФК профиль», используемый в данном документе, в отношении временного профиля концентрации в плазме, получаемого при пероральным введении субъекту лекарственной формы по данному изобретению, представляет собой временной профиль концентрации в плазме, который по существу аналогичен целевому профилю, показанному на фиг. 1, так что первая лекарственная форма, содержащая Соединение (I), которая дает целевой временной профиль концентрации в плазме на Фиг.1, и вторая лекарственная форма, содержащая Соединение (I), которая дает аналогичный временной профиль концентрации в плазме, приведет к характеристике ФК, такой как AUC, рассматриваемой как биоэквивалентная регулирующим органом. В одном варианте реализации регулирующим агентством является Управление по контролю за продуктами и лекарственными средствами США.
[00199] «BNSS» - краткая шкала оценки негативных симптомов.
[00200] «Включение» или «включает», применительно к конкретной лекарственной форме, композиции, способу или процессу, описанным или заявленным в данном документе, означает, что лекарственная форма, композиция или способ включает все перечисленные элементы в конкретном описании или формуле изобретения, но не исключает другие элементы. «По существу состоит из» и «состоящий по существу из» означает, что описанная или заявленная композиция, лекарственная форма, способ или процесс не исключают другие материалы или этапы, которые не оказывают существенного влияния на указанные физические, фармакологические, фармакокинетические свойства или терапевтические эффекты композиции, лекарственной формы, способа или процесса. «Состоит из» и «состоящий из» означает исключение более чем микроэлементов других ингредиентов и существенных этапов способа или процесса.
[00201] Термин «контролируемое высвобождение» или «КВ», как используется в данном документе в отношении пероральной лекарственной формы по данному изобретению, означает, что Соединение (I) высвобождается из лекарственной формы в соответствии с заранее определенным профилем, который может включать, когда и где высвобождение происходит после перорального введения и/или указанная скорость высвобождения в течение определенного периода времени.
[00202] Термин «агент, контролирующий высвобождение», используемый в настоящем документе в отношении пероральной лекарственной формы, раскрытой в описании, относится к одному или более веществам, или материалам, которые модулируют высвобождение Соединения (I) из лекарственной формы. Контролируемые разделительные средства могут быть материалами, которые являются органическими или неорганическими, встречающимися в природе или синтетическими, такими как полимерные материалы, триглицериды, производные триглицеридов, жирные кислоты и соли жирных кислот, тальк, борная кислота и коллоидный диоксид кремния.
[00203] «Аллель CYP2D6» относится к одной из более чем 100 названных версий гена CYP2D6, которые присутствуют в общей популяции и обычно классифицируются в одну из трех категорий: активные (функциональные); сниженная активность (частично активная или пониженная функция) и неактивная (нефункциональная).
[00204] Активные аллели CYP2D6 включают: *1, *2, *2A, *33, *35, *39, *48 и *53.
[00205] Снижение активности CYP2D6 аллелей включает: *9, *10, *17, *29, *41, *49, *50, *54, *55, *59, *69 и *72.
[00206] Неактивные аллели CYP2D6 включают: *3, *4, *5 (делеция), *6, *7, *8, *11, *12, *13, *14A, *14B, *15, *18, *19, *20, *21, *38, *40, *42, *44, *56, *56A, *56B и *68.
[00207] «Генотип CYP2D6 быстрый метаболизатор (EM)» применительно к субъекту означает, что субъект имеет CYP2D6, в результате чего метаболическая активность CYP2D6 считается нормальной. Генотипы CYP2D6 EM включают комбинации: (а) двух активных аллелей CYP2D6, (б) одного активного и одного аллеля CYP2D6 пониженной активностью и (в) одного активного и одного неактивного аллеля CYP2D6.
[00208] «Генотип CYP2D6 промежуточный метаболизатор (IM)» применительно к субъекту означает, что субъект имеет генотип CYP2D6, что приводит к снижению метаболической активности CYP2D6. Генотипы CYP2D6 IM включают комбинации: (a) одного активного и одного аллеля CYP2D6 с пониженной активностью и (в) двух аллелей с пониженной активностью CYP2D6.
[00209] «Генотип CYP2D6 PM» применительно к субъекту означает, что субъект имеет положительный результат теста на генотип с медленным метаболизатором CYP2D6 и, следовательно, вероятно, не обладает активностью CYP2D6. Генотип CYP2D6 PM представляет собой 2 неактивных аллеля.
[00210] «Генотип CYP2D6 UM» применительно к субъекту означает, что субъект имеет положительный результат теста на генотип сверхбыстродействующего метаболизатора CYP2D6 и, таким образом, вероятно, имеет более высокую, чем в среднем, активность CYP2D6. Генотип CYP2D6 UM представляет собой 3 или более активных аллелей.
[00211] Термин «кишечнорастворимая оболочка», используемый в данном документе в отношении лекарственной формы по настоящему изобретению, относится к pH-зависимому материалу, который окружает ядро, содержащее Соединение (I), и который остается практически интактным в кислой среде желудка, но который растворяется при рН среды кишечника.
[00212] В одном варианте реализации в лекарственных формах по настоящему изобретению наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, моногидрата лактозы, сахарозы, глюкозы и сорбита.
[00213] Используемый в данном документе термин «вещество, способствующее скольжению» относится к веществу, используемому для ускорения потока порошка путем уменьшения межчастичной когезии. В одном варианте реализации в лекарственных формах по настоящему изобретению вещество, способствующее скольжению, выбрано из группы, состоящей из безводного коллоидного диоксида кремния, крахмала и талька.
[00214] Используемый в данном документе термин «смазочный материал» относится к веществу, которое предотвращает слипание и/или комкование ингредиентов в машинах, используемых при приготовлении лекарственных форм по настоящему изобретению. В одном варианте реализации в лекарственных формах по данному изобретению смазочный материал выбран из группы, состоящей из стеарата магния, стерической кислоты и растительного стеарина.
[00215] Термин «состояние сытости» или «состояние голода», используемый для описания субъекта, означает, что субъект не принимал пищу в течение по меньшей мере 4 ч до интересующего момента времени, такого как время введения лекарственной формы, описанной в данном документе. В одном варианте реализации субъект в состоянии голодания не принимал пищу в течение по меньшей мере любого из 6, 8, 10 или 12 ч до введения лекарственной формы, описанной в настоящем документе.
[00216] Термин «состояние сытости», используемое для описания субъекта в настоящем документе, означает, что субъект принимал пищу менее чем за 4 ч до интересующего момента времени, такого как время введения лекарственной формы, описанной в данном документе. В одном варианте реализации субъект в состоянии сытости не принимал пищу в течение по меньшей мере любого из 3, 2, 1 или 0,5 ч до введения лекарственной формы, описанной в настоящем документе.
[00217] «Кишечнорастворимый» или «КР» применительно к пероральной дозированной форме с контролируемым высвобождением, описанной в данном документе, означает, что высвобождение Соединения (I) в желудок субъекта не должно превышать 5%, 2,5%, 1% или 0,5% от общего количества Соединения (I) в лекарственной форме.
[00218] «MIN-101» является кодовым названием 1Н-изоиндол-1-она, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2), с альтернативным названием 2-{1-[2-(4-фторфенил)-2-оксоэтил]пиперидин-4-илметил}-2,3-дигидроизоиндол-1-он гидрохлорид дигидрат.
[00219] Термин «лекарственная форма для перорального применения», в контексте данного документа, относится к фармацевтическому лекарственному продукту, который содержит определенное количество (дозу) Соединения (I) в качестве активного ингредиента или его фармацевтически приемлемой соли и/или сольвата, и неактивные компоненты (наполнители), сформулированы в конкретной конфигурации, которая подходит для перорального введения, такой как таблетка или капсула.
[00220] «Фармацевтически приемлемая соль», используемая в данном документе в отношении Соединения (I), означает солевую форму Соединения (I), а также гидраты солевой формы с одной или более присутствующими молекулами воды. Такие солевые и гидратированные формы сохраняют биологическую активность Соединения (I) и не являются биологически или иным образом нежелательными, то есть проявляют минимальные, если таковые имеются, токсикологические эффекты. В одном варианте реализации фармацевтически приемлемая соль Соединения (I) имеет одну молекулу HCl и две молекулы воды, то есть 1H-изоиндол-1-он, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2).
[00221] «PANSS» - шкала оценки позитивных и негативных симптомов.
[00222] «Фармакокинетический параметр» означает измерение или характеристику, которая описывает фармакокинетические свойства соединения, представляющего интерес. Параметры PK, используемые в данном документе, определены ниже.
[00223] «AUC» обозначает площадь под фармакокинетической кривой, описывающей зависимость «концентрация в плазме/время», которая является мерой воздействия интересующего соединения, и является интегралом от кривой концентрация-время после однократного приема или в стационарном состоянии. AUC выражается в единицах нг·ч/мл (нг х ч/мл).
[00224] «AUC(0-4ч)» означает AUC от 0 до 4 ч после введения однократной дозы.
[00225] «AUC(0-24ч)» означает AUC от 0 до 24 ч после введения однократной дозы.
[00226] «AUClast» означает AUC от времени 0 до последней измеряемой концентрации (Clast).
[00227] «AUC(0-tau)» означает AUC от 0 ч до конца интервала между введениями препарата.
[00228] «Cmax» означает наблюдаемую максимальную (пиковую) концентрацию в плазме указанного соединения, такого как Соединение (I), после введения дозы композиции, содержащей соединение. В одном варианте реализации Cmax измеряют после 2 или более доз композиции. В одном варианте реализации Cmax измеряется, когда указанное соединение достигает стационарного состояния.
[00229] «Cmin» означает минимальную концентрацию в плазме указанного соединения, такого как Соединение (I), после введения дозы композиции, содержащей соединение. В одном варианте реализации Cmax измеряют после 2 или более доз композиции. В одном варианте реализации Cmax измеряется, когда указанное соединение достигает стационарного состояния.
[00230] «Css» означает концентрацию в равновесном состоянии.
[00231] «Cave» означает среднюю концентрацию, которая представляет собой отношение AUC к времени.
[00232] «Cp» означает концентрацию в плазме указанного соединения, такого как Соединение (I), в любое время T после введения дозы композиции, содержащей соединение.
[00233] «Cp(last)» означает последний измеренный Cp со ссылкой на время сбора последней из серии образцов крови для анализа на указанное соединение.
[00234] «Cp(T)» означает Cp в указанное время; таким образом, Cp(4ч) и Cp(12ч) представляют собой Cp через 4 ч и 24 ч соответственно.
[00235] «Ч» означает час.
[00236] «ФK» представляет собой фармакокинетический(ие), фармакокинетика.
[00237] «Равновесное состояние» означает скорость абсорбции указанного представляющего интерес соединения, такого как Соединение (I), равна скорости элиминации соединения.
[00238] «Tau» означает интервал дозирования (H). Например, для однократной суточной дозы tau составляет 24 ч.
[00239] Tmax означает время до максимальной (или пиковой) концентрации указанного терапевтического соединения в плазме или сыворотке после введения однократной дозы композиции, содержащей соединение, и до введения второй дозы.
[00240] Vmax означает максимальную скорость поглощения (мг/ч).
[00241] Термин «субъект» и «пациент» могут использоваться в данном документе взаимозаменяемо и относиться к человеку любого возраста.
[00242] «Терапевтически эффективное количество», как используется в данном документе в отношении терапевтического применения лекарственной формы, содержащей Соединение (I) или его фармацевтическую соль и/или сольват, означает количество свободного основания (Соединения (I)), которое является достаточным для лечения, улучшения или предотвращения конкретного заболевания, симптома, расстройства или состояния заболевания, или проявления обнаруживаемого терапевтического или ингибирующего эффекта. Эффект может быть обнаружен любым способом анализа, известным в данной области техники. Эффективное количество для конкретного субъекта может зависеть от массы тела, размера и здоровья субъекта; характер и степень состояния; и следует ли вводить дополнительные терапевтические средства субъекту. Терапевтически эффективные количества для данной ситуации могут быть определены обычным экспериментом, который находится в пределах квалификации и суждения врача.
[00243] Термины «лечить», «лечение» и аналогичные термины, используемые в данном документе в отношении одного или более указанных симптомов заболевания, должны включать лечение и уход за пациентом с целью улучшения одного или более указанных симптомов, и включают введение кишечнорастворимой пероральной лекарственной формы с контролируемым высвобождением, описанной в данном документе, с частотой дозирования и в течение периода лечения, которые являются достаточными для предотвращения появления одного или более симптомов, уменьшения частоты, интенсивности или тяжести одного или более симптомов, задержки или избежания развития дополнительных симптомов или любой комбинации данных целей лечения. В одном варианте реализации эффект лечения лекарственной формой по данному изобретению оценивают путем сравнения тяжести симптомов субъекта на исходном уровне (например, до лечения) и после по меньшей мере одного периода лечения. В одном варианте реализации период лечения составляет по меньшей мере одну неделю, по меньшей мере две недели, по меньшей мере четыре недели, по меньшей мере шесть недель, по меньшей мере восемь недель, по меньшей мере 10 недель или по меньшей мере двенадцать недель или более. В одном варианте реализации симптомы, подлежащие лечению, представляют собой, по меньшей мере, один отрицательный симптом у пациента с шизофренией или без шизофрении, лекарственная форма содержит 32 мг Соединения (I), частота приема составляет один раз в день, и период лечения составляет, по меньшей мере, восемь недель.
Краткий обзор пероральных кишечнорастворимых лекарственных форм с контролируемым (CR) высвобождением
[00244] В одном варианте реализации данное изобретение относится к кишечнорастворимой пероральной CR лекарственной форме, содержащей от приблизительно 4 мг до приблизительно 100 мг Соединения (I) или эквивалентного количества фармацевтически приемлемой соли и/или сольвата Соединения (I), причем кишечнорастворимая пероральная CR лекарственная форма выбрана из группы, состоящей из 32 мг CR GR-01 таблетки, 32 мг CR GR-02 таблетки, 32 мг CR GR-01/B таблетки, 64 мг CR GR-01/B таблетки, 32 мг CR GR-01/C таблетки и 64 мг CR GR-01/C таблетки.
[00245] В одном варианте реализации 32 мг CR GR-01 таблетка имеет следующий состав:
масс.)
таблетку
(Метолоза ® 90SH 100 SR)
1Применяется поправочный коэффициент 1,2 на соль
NA: Не применимо
[00246] В одном варианте реализации 32 мг CR GR-02 таблетка имеет следующий состав:
1Применяется поправочный коэффициент 1,2 на соль
NA: Не применимо
[00247] В одном варианте реализации 32 мг CR GR-01/B таблетка имеет следующий состав:
(Methocel™ K100LV CR)
(Methocel™ K100M CR)
1Применяется поправочный коэффициент 1,2 на соль
[00248] В одном варианте реализации 32 мг CR GR-01/C таблетка имеет следующий состав:
(Methocel ™ K100LV CR)
(Methocel™ K100M CR)
[00250] В одном варианте реализации 64 мг CR GR-01/B таблетка имеет следующий состав:
(Methocel ™ K100LV CR)
(Methocel™ K100M CR)
1Применяется поправочный коэффициент 1,2 на соль
[00251] В одном варианте реализации 64 мг CR GR-01/C таблетка имеет следующий состав:
(Methocel™ K100LV CR)
(Methocel™ K100M CR)
Разработка и изготовление кишечнорастворимых лекарственных форм для перорального применения с контролируемым высвобождением
[00252] Целью данного изобретения является предоставление кишечнорастворимой лекарственной формы для перорального применения с контролируемым высвобождением, содержащей от приблизительно 4 до приблизительно 100 мг Соединения (I) или эквивалентное количество фармацевтически приемлемой соли и/или сольвата Соединения (I). Лекарственная форма составлена таким образом, чтобы при пероральном введении субъекту проявлялся конкретный желаемый профиль высвобождения для Соединения (I), который снижает максимальные концентрации BFB-520 в плазме, обеспечивая при этом терапевтически эффективное количество Соединения (I) в течение одного или более интервалов дозирования. Данный желаемый профиль высвобождения достигается двумя способами: (а) задержкой высвобождения Соединения (I) до тех пор, пока опорожнение желудка не вытолкнет лекарственную форму в тонкую кишку, и затем (b) не обеспечит замедленное высвобождение, по меньшей мере, приблизительно 90%, 95% или 100% количества Соединения (I) в лекарственной форме со скоростью, которая обеспечивает ФK профиль в плазме, который включает Tmax для Соединения (I) от приблизительно 4 ч до приблизительно 22 ч.
[00253] Указанный профиль высвобождения in vivo для Соединения (I) предназначен для снижения уровней BFB-520 в плазме у субъекта ниже порога, который коррелирует с более высоким риском пролонгации QT. В одном варианте реализации пороговое значение представляет собой Cmax для BFB-520, которая ниже 5,0 нг/мл, ниже 4,5 нг/мл, ниже 4,0 нг/мл, ниже 3,5 нг/мл, ниже 3,0 нг/мл, ниже 2,5 нг/мл, ниже 2,0 нг/мл, ниже 1,5 нг/мл, ниже 1,0 нг/мл или ниже 0,5 нг/мл.
[00254] В некоторых вариантах реализации ФК профиль в плазме для Соединения (I) дополнительно характеризуется с точки зрения одного или более дополнительных ФК параметров, таких как Cmax, AUC(0-tau), Cmin и других ФК параметров, определенных выше. Специалист в данной области поймет, что значения некоторых из этих дополнительных ФК параметров будут зависеть, по меньшей мере частично, от количества Соединения (I) в лекарственной форме.
[00255] Значения Tmax и других ФК параметров в плазме, полученных после введения лекарственной формы, описанной в данном документе, могут демонстрировать некоторые межиндивидуальные вариации в популяции субъектов. Таким образом, в некоторых вариантах реализации определенные параметры ФK в плазме выражаются в виде средних значений, определенных для популяции по меньшей мере из 2, 4, 8, 16 или более субъектов. В одном варианте реализации популяция состоит из здоровых добровольцев. В одном варианте реализации каждый субъект в популяции имеет положительный результат теста на генотип EM. В одном варианте реализации каждый субъект в популяции имеет положительный результат теста на генотип EM или генотип IM. В одном варианте реализации каждый субъект в популяции имеет положительный результат теста на генотип IM или генотип PM. В одном варианте реализации каждый субъект в популяции имеет положительный результат теста на генотип PM.
[00256] Соединение (I) может быть синтезировано с использованием стандартных синтетических способов и процедур для получения органических молекул и превращений и манипуляций с функциональными группами, включая использование защитных групп, которые могут быть найдены в соответствующей научной литературе или стандартных справочных учебниках данной области техники. Хотя и не ограниченные каким-либо одним или более источниками, признанные справочные учебники по органическому синтезу включают: Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th ed.; John Wiley & Sons: New York, 2001; и Greene, T.W.; Wuts, P.G. M. Protective Groups in Organic Synthesis, 3rd; John Wiley & Sons: New York, 1999. Способ получения Соединения (I) описан в патенте США № 7166617, содержание которого включено в данный документ в полном объеме.
[00257] В одном варианте реализации лекарственная форма Соединения (I), используемая в лекарственной форме, представляет собой дигидрат гидрохлорида Соединения (I), который имеет химическое название 1H-изоиндол-1-он, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро, гидрохлорид, гидрат (1: 1: 2), который имеет молекулярную формулу C22H23FN2O2, HCl, 2H2O и молекулярный вес 438,92. Способы получения данного Соединения (I) описаны в патентах США №№ 7166617 и 9458130. Количество данного лекарственного вещества, которое эквивалентно указанному количеству свободного основания, может быть рассчитано путем умножения указанного количества Соединения (I) на 1,2; таким образом, 38,4 мг этого лекарственного вещества эквивалентно 32,0 мг Соединения (I).
[00258] В одном варианте реализации свойства отложенного и замедленного высвобождения пероральной лекарственной формы CR могут быть обеспечены путем заключения композиции с замедленным высвобождением, содержащей желаемое количество Соединения (I) или его фармацевтически приемлемой соли и/или сольвата, в пределы кишечнорастворимой оболочки.
[00259] Различные физические и химические подходы для создания композиций с замедленным высвобождением хорошо известны в данной области техники. Любая композиция с замедленным высвобождением, которая способна высвобождать Соединение (I) для обеспечения ФК профиля в плазме in vivo, описанного в данном документе, может быть использована для приготовления лекарственной формы по данному изобретению. В одном варианте реализации композиция с замедленным высвобождением содержит по меньшей мере один полимерный материал, который модулирует высвобождение Соединения (I). Подходящие полимерные материалы включают, но не ограничиваются ими, сшитый поливинилпирролидон, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, сшитую натриевую соль карбоксиметилцеллюлозы, карбоксиметилкрахмал, крахмал и его производные, полимеры и сополимеры акриловой и метакриловой кислоты, сложные полиэфиры, полиангидриды, полиметилвиниловый эфир/ангидрид сополоимеры, калий метакрилат-дивинилбензольные сополимеры, поливиниловые спирты, глюкан, склероглюкан, маннан, бетациклодекстрины и производные циклодекстрина, содержащие линейные и/или разветвленные полимерные цепи. В одном варианте реализации полимерный материал представляет собой гидроксипропилметоцеллюлозу.
[00260] В варианте реализации смесь гипромеллозы низкой вязкости и высокой вязкости используется в качестве агента, контролируемого высвобождение в композиции с замедленным высвобождением. Вязкостные свойства подходящих гипромеллоз могут быть определены в 2% -ном по массе растворе в воде при 20 °C, как описано в USP Hypromellose Monograph, от 1 декабря 2016 г., которая доступна по адресу http://www.usp.org/usp-nf/official-text/stage-6/hypromellose-2015-11-20.
[00261] Кишечнорастворимая оболочка, которая обычно содержит чувствительный к рН полимер, начинает растворяться в водном растворе при рН более 5,5 и в одном варианте начинает растворяться в водном растворе при рН более 6,0. В одном варианте реализации рН-чувствительный полимер начинает растворяться в водном растворе при рН выше 6,5. В одном варианте рН-чувствительный полимер начинает растворяться в водном растворе при рН 6,7. В одном варианте реализации количества Соединения (I), высвобождаемого в желудке из кишечнорастворимой лекарственной формы CR, вводимой субъектам после еды или натощак, являются примерно одинаковыми (например, разница составляет менее 5%, менее 2% или менее 1%).
[00262] Состав и толщина кишечнорастворимой оболочки обычно выбираются таким образом, чтобы по существу поддерживать ее целостность в желудке, в то же время позволяя по существу всей кишечнорастворимой оболочке растворяться после того, как лекарственная форма покидает желудок. В одном варианте реализации практически вся кишечнорастворимая оболочка растворяется в течение 15 мин, 30 мин, 1 ч или 2 ч после того, как лекарственная форма покидает желудок.
[00263] Создание и изготовление кишечнорастворимых покрытий хорошо известны в области составов. Поликислоты, имеющие соответствующий диапазон рКа, могут быть использованы для приготовления кишечнорастворимых оболочек. Неограничивающими примерами подходящих материалов для кишечнорастворимой оболочки являются полимеризованный желатин, шеллак, сополимер метакриловой кислоты типа C NF, фталат бутират целлюлозы, гидрофталат целлюлозы, фталат пропаноат целлюлозы, поливинилацетатфталат (PVAP), ацетат фталат целлюлозы (CAP), ацетат целлюлозы тримеллитат (CAT), фталат гидроксипропилметилцеллюлозы, ацетат гидроксипропилметилцеллюлозы, сукцинат диоксипропилметилцеллюлозы, карбоксиметилэтилцеллюлоза (CMEC), ацетат сукцинат гидроксипропилметилцеллюлозы (HPMCAS) и полимеры и сополимеры акриловой кислоты, как правило, образованные из метилакрилата, этилакрилата, метилметакрилата, и/или этилметакрилата с сополимерами эфиров акриловой и метакриловой кислот. Например, кишечнорастворимая оболочка может содержать сополимер на основе метакриловой кислоты и этилакрилата, с коммерческим названием EUDRAGIT® L 30 D-55 от Evonik Industries AG. В одном варианте реализации оболочка содержит смесь (i) EUDRAGIT® L 30 D-55 в количестве от 4,5% до 5,0% масс. или приблизительно 4,7% масс. и (ii) PlasACRYLTM HTP20 в количестве 0,80% масс.
[00264] В другом аспекте данное изобретение предлагает состав серии и способ изготовления кишечнорастворимой пероральной лекарственной формы CR, описанной в данном документе. В одном варианте реализации состав серии для изготовления 32 мг лекарственной формы содержит компоненты, перечисленные в Таблице 4 в Примере 4 ниже. В другом варианте реализации состав серии для изготовления 32 мг лекарственной формы содержит компоненты, перечисленные в Таблице 5 в Примере 4 ниже или в Таблице в Примере 4 ниже. Примеры способов, которые подходят для изготовления указанных 32 мг лекарственных форм, описаны в блок-схемах 1 и 2 Примера 4.
Аналитические способы
[00265] А. Тестирование растворения in vitro
[00266] Для того, чтобы оценить потенциал предлагаемой кишечнорастворимой пероральной дозированной формы CR, содержащей Соединение (I), для получения желаемого профиля высвобождения in vivo, и ФK в плазме Соединения (I), может быть выполнено тестирование растворения in vitro, как описано в приведенных ниже примерах.
[00267] В одном варианте реализации дозированная форма содержит 32 мг и дает кумулятивные профили растворения и скорости растворения, которые по существу сходны с целевым профилем, профилем CR-GR-01 или профилем CR-GR-02, показанными на Фиг,1 и в Таблицах 6 и 7 ниже. В одном варианте реализации совокупное количество растворения и скорость растворения в каждый момент времени в по существу одинаковых профилях находятся в пределах +/- 10% от значений для соответствующего момента времени в целевом профиле растворения, профили растворения CR-GR-01 или CR-GR-02 представлены в Таблицах 6 и 7.
[00268] B. Обнаружение Соединения (I) и BFB-520 в плазме человека
[00269] Для того, чтобы оценить, дает ли кишечнорастворимая пероральная дозированная форма CR, содержащая Соединение (I), желаемый ФК профиль для одного или обоих Соединения (I) и BFB-520, концентрации в плазме интересующего соединения (соединений) могут быть определены в различные моменты времени после введения лекарственной формы одному субъекту, но обычно определяются в группе из двух или более субъектов. В одном варианте реализации ФК профиль определяется в группе по меньшей мере из 8, 12, 16 или 20 субъектов. В одном варианте реализации группа включает здоровых мужчин и женщин. В одном варианте реализации число субъектов в группе выбирают так, чтобы получить статистически значимую оценку того, является ли ФК профиль, полученный тестируемой пероральной лекарственной формой, биоэквивалентным ФК профилем относительно ФК профиля, показанного на Фиг,1.
[00270] Результаты открытого рандомизированного исследования с 3 последовательностями лечения, с 3 периодами исследования для оценки профиля ФK Соединения (I) и его метаболита BFB-520 после однократного перорального введения 3 составов MIN-101 (2 прототипа кишечнорастворимых (GR) CR составов (GR-01 и GR-02) и 1 состав сравнения MR (MR32)) суммированы на Схеме 1 (MIN-101) и Схеме 2 (BFB-520). Полную информацию об этих экспериментах можно найти в разделе «Примеры».
[00271] Схема 1. Краткое описание результатов ФК исследования MIN-101 для составов MR32, GR-01 и GR-02.
(от n=12 субъектов (перекрестн.)
[00272] Схема 2. Краткое описание результатов ФК исследования BFB-520 для составов MR32, GR-01 и GR-02.
(от n=12 субъектов (перекрестн.)
[00273] ФК профили MIN-101 и его метаболита BFB-520 были предсказаны для 3 составов MIN-101 (2 прототипа кишечнорастворимых (GR) CR составов (GR-01 и GR-02) и 1 состав сравнения MR (MR32)) в 2 дозировках. (32 мг и 64 мг) из расчета 4 суточных дозировки. Результаты исследования суммированы на Схеме 3 (32 мг) и Схеме 4 (64 мг). Полную информацию можно найти в разделе «Примеры».
[00274] Схема 3. Краткое описание прогнозируемых концентраций в плазме MIN-101 и BFB-520 для составов MR32, GR-01 и GR-02 (32 мг).
[00275] Схема 4. Краткое описание прогнозируемых концентраций в плазме MIN-101 и BFB-520 для составов MR32, GR-01 и GR-02 (64 мг).
[00276] ФК профили состава GR-01 у здоровых CYP2D6 EM мужчин и женщин после еды и натощак суммированы на Схеме 5 (MIN-101) и Схеме 6 (BFB-520). Субъекты, которые завершили часть 1 исследования (оценка ФК профиля MIN-101 и его метаболита BFB-520 в составах GR-01, GR-02 и MR32) вернулись и получили дополнительную однократную пероральную дозу GR-01 после еды и натощак, что позволило оценить влияние пищи путем сравнения ФК свойств с теми, которые получены в части 1 (Примеры 9-12). Был введен период отмывки 14 ± 2 дня после части 1. Полную информацию об этих экспериментах можно найти в разделе «Примеры».
[00277] Схема 5. Краткое описание результатов ФК исследования MIN-101 для состава GR-01 после еды и натощак.
[00278] Схема 6. Краткое описание результатов ФК исследования BFB-520 для состава GR-01 после еды и натощак.
[00279] ФК профили MIN-101 и его метаболита BFB-520 были предсказаны после еды и натощак для кишечнорастворимого состава GR-01 в 2 дозироваках (32 мг и 64 мг), основываясь на 4 суточных дозировках. Данные исследования суммированы на Схеме 7 (32 мг) и Схеме 8 (64 мг) соответственно. Полную информацию можно найти в разделе «Примеры».
[00280] Схема 7. Краткое описание прогнозируемых концентраций в плазме MIN-101 и BFB-520 для GR-01 (32 мг) после еды и натощак.
[00281] Схема 8. Краткое описание прогнозируемых концентраций в плазме MIN-101 и BFB-520 для GR-01 (64 мг) после еды и натощак.
[00282] Считается, что BFB-520 частично метаболизируется CYP2D6. В клинических исследованиях MIN-101, субъекты со слабым метаболизмом посредством CYP2D6 показали высокие уровни BFB-520 в плазме. Таким образом, в одном варианте реализации Cmax для BFB-520 оценивают после перорального введения тестируемой лекарственной формы, содержащей Соединение (I), только субъектам, которые имели генотип IM CYP2D6 или генотип EM CYP2D6, что было определено с использованием коммерчески доступного теста генотипа. В одном варианте реализации все субъекты имели генотип EM CYP2D6.
[00283] Уровни Соединения (I) и метаболита BFB-520, вырабатываемого в плазме после перорального введения пероральной лекарственной формы по изобретению, могут быть определены способом, описанным ниже. Ожидается, что могут быть также использованы варианты и усовершенствования данного способа.
[00284] Образцы крови от субъектов собирают в пробирки с гепарином натрия в различные представляющие интерес моменты времени. Подходящий график отбора проб включает следующее:
День 1 (D1): перед введением; 1 ч; 2 ч; 3 ч; 4 ч; 6 ч; 8ч; 10 ч; 12 ч и 16 ч;
День 2 - День 6 (D2-D6): перед введением
День 7 (D7): перед введением; 1 ч; 2 ч; 3 ч; 4 ч; 6 ч; 8ч; 10 ч; 12 ч; 16 ч; 24 ч (D8) и 48 ч (D9).
[00285] После центрифугирования крови желаемое количество аликвот (обычно 2) каждого образца плазмы готовят в подходящих контейнерах для хранения (например, в полипропиленовых пробирках, плотно закрытых для предотвращения утечки и высыхания, которые могут возникнуть во время хранения). Контейнеры с образцами плазмы хранятся при температуре -80°C до одного месяца перед анализом.
[00286] Утвержденный GLP способ обнаружения и количественного определения Соединения (I) и его метаболитов BFB-520 и BFB-999 использует жидкостный хроматографический (ЖХ) анализ в сочетании с масс-спектрометрическим обнаружением (МС/МС) после стадии экстракции жидкость/жидкость Соединения ( I) и метаболитов BFB-520 и BFB-999 из образцов плазмы.
[00287] Аналитический способ использует два внутренних стандарта (дейтерированный аналог MIN-101 (обозначенный в данном документе как [2H6]-MIN-101 или MIN-101-d6 или CYR-101-d6) и BFB-784 для BFB-520, и для BFB-999), которые были подвергнуты той же аналитической процедуре, что и MIN-101, BFB-520 и BFB-999 в образцах плазмы. MIN-101-d6 и BFB-784 имеют структуры, показанные в Формулах IV и V ниже:
[00288] Хроматограммы обрабатываются по умолчанию в автоматическом режиме. Хроматографические пики MIN-101, BFB-520, BFB-999 и внутренних стандартов (IS) идентифицируют по времени их удерживания. Зарегистрированный отклик выражается в виде отношения площадей от MIN-101 к MIN-101-d6 и BFB-520 или BFB-999 к BFB-784.
[00289] Нижний предел количественного определения (LLOQ) этого аналитического способа в плазме составляет 0,25 нг/мл для MIN-101 и его метаболитов BFB-520 и BFB-999.
[00290] Детали аналитического способа описаны в Примере 7 ниже. Ожидается, что могут быть также использованы варианты и усовершенствования данного аналитического способа.
Способы лечения
[00291] Кишечнорастворимые пероральные дозированные формы CR по данному изобретению могут быть полезны для лечения заболеваний или состояний, которые чувствительны к лечению Соединением (I). В качестве не ограничивающих примеров, полагают, что Соединение (I) потенциально может быть использовано для лечения пациентов с шизофренией и без шизофрении с одним или более из следующих симптомов или состояний: негативные симптомы, депрессивные симптомы, нарушения сна и когнитивные нарушения.
[00292] В фазе 2b исследования, MIN-101 в дозах 32 мг и 64 мг продемонстрировало быстрое, статистически значимое и клинически значимое снижение негативных симптомов у пациентов с шизофренией. Пероральные лекарственные формы, используемые в этом исследовании фазы 2b, представляли собой таблетку 32 мг MR, описанную в Примере 1 ниже, и по существу идентичную таблетку MR 64 мг. Ни одна из этих таблеток MR не имела покрытия GR, и каждая из них давала профили растворения in vitro и ФK в плазме, которые отличаются от профилей, полученных с помощью кишечнорастворимых пероральных дозированных форм CR по данному изобретению.
[00293] Отрицательные симптомы, как правило, относятся к снижению нормального функционирования и включают в себя пять основных субдоменов: притупленный аффект (аффективное выравнивание, притупленное выражение), алогия (плохая речь), амотивация (потеря воли), ангедония (снижение способности испытывать или предвкушать удовольствие) и асоциальность (социальный уход). В то время как негативные симптомы являются хорошо документированным и интенсивно изучаемым аспектом шизофрении, этот класс симптомов был выявлен у пациентов с другими психическими и неврологическими расстройствами, включая, например, болезнь Альцгеймера и другие деменции, в частности лобно-височную деменцию (FTD), аутическое спектральное расстройство (ASD), биполярное расстройство (BPD), большое депрессивное расстройство (MDD), болезнь Паркинсона, эпилепсия височной доли, инсульт и черепно-мозговая травма (TBI) (см., например, Boone et al, J. of Internat. Neuropsycol. Soc., 2003, Vol 9, pages 698-709; Bastiaansen, J. et al., J. Autism Dev. Disord. 2011, Vol 41:1256-1266; Getz, K. et al., Am. J. Psychiatry 2002, Vol 159:644-651; Winograd-Gurvich, C. et al., Brain Res. Bulletin, 2006, Vol. 70:312-321; Galynker et al., Neuropsychiatry Neuropsychol Behav Neurol 2000, Vol 13:171-176; Galynker I, et al., J. Nerv. Ment. Dis 1997, Vol 185:616-621; Chaudhury, S., et al., Indian J. of Neurotrauma 2005, Vol 2:13-21; Ameen, S et al., German J. of Psychiatry 2007). Действительно, еще в 2001 году было высказано предположение, что негативные симптомы являются общими для психических заболеваний в целом (Herbener and Harrow, Schizophrenia Bulletin 2001, Vol. 27:527-537). Кроме того, результаты нескольких популяционных исследований пришли к выводу, что у 20-22% населения в целом есть один или более негативных симптомов, и что большинство субъектов с негативными симптомами не имеют клинически диагностированного психического расстройства (Werbeloff, N. et al., PLoS ONE 2015, Vol 10:e0119852; Barrantes-Vidal, N., et al., Schizophr. Res. 2010, Vol 122:219-225).
[00294] Таким образом, целью настоящего изобретения является лечение по меньшей мере одного негативного симптома у субъекта способом введения субъекту кишечнорастворимой лекарственной формы CR, описанной в данном документе, один раз в день (QD). В одном варианте реализации субъекту поставлен диагноз шизофрения. В другом варианте реализации у субъекта нет клинического диагноза шизофрении, то есть у пациента нет шизофрении.
[00295] В целях раскрытия, охватываемого данным документом, термин «отрицательные симптомы» следует понимать как включающий первичные отрицательные симптомы, обычно связанные с шизофренией, отрицательные симптомы, измеренные в балльной шкале отрицательных симптомов PANSS, отрицательный оценочный коэффициент на основе модели пятиугольной структуры, и негативные симптомы измерянные в BNSS.
[00296] В одном варианте реализации отрицательный симптом представляет собой один из пяти основных поддоменов отрицательных симптомов: притупленный аффект, алогия, амотивация, ангедония и асоциальность. Основные характеристики каждого субдомена описаны ниже.
[00297] Притупленный аффект (аффективное выравнивание, притупленное выражение) характеризуется сниженной интенсивностью и диапазоном эмоционального выражения, что проявляется через вокальные и невербальные способы общения, включая интонацию (просодию), выражение лица, жесты рук и движения тела.
[00298] Алогия (бедность речи) характеризуется уменьшением количества речи, уменьшением спонтанной речи и потерей разговорной речи.
[00299] Амотивация (потеря воли) характеризуется недостатками в инициировании и поддержании целенаправленного поведения, такого как работа, учеба, спорт, личная гигиена и повседневные задачи, особенно когда требуются и усилия (когнитивные или физические), и значительная организация, а также дефицит в желании предпринять такие действия. Этот поддомен связан с апатией и недостатком энергии.
[00300] Ангедония (пониженная способность испытывать или предвосхищать удовольствие) характеризуется тем, что нетерпение в ожидании награды, отдыха или другого приятного опыта («ожидание») более заметно и нарушено (упреждающая ангедония), по сранению с оценкой («симпатия») опыта самой по себе (конечная ангедония).
[00301] Асоциальность (социальная изоляция) характеризуется сниженным интересом, мотивацией и оценкой социальных взаимодействий с другими, такими как семья и друзья, потеря интереса к интимным (сексуальным) отношениям, независимая от каких-либо соматических проблем, и для ребенка может включать потеря интереса к игре с другими детьми.
[00302] В некоторых вариантах реализации лекарственную форму вводят субъекту один раз в день в течение первого периода лечения, достаточного для достижения улучшения по меньшей мере одного негативного симптома. В одном варианте реализации первый период лечения составляет, по меньшей мере 2 недели, по меньшей мере 4 недели, по меньшей мере 6 недель, по меньшей мере 8 недель, по меньшей мере 10 недель или по меньшей мере 12 недель. В одном варианте реализации положительные симптомы у субъекта, которого лечат лекарственной формой, являются стабильными в течение периода лечения, то есть остаются по существу на том же уровне, что и в начале исследования. В одном из вариантов реализации уровень улучшения негативных симптомов представляет собой снижение по меньшей мере на 3 балла по пятифакторной шкале PANSS (модель пятиугольной структуры) негативных симптом-факторов после 12 недель лечения. В одном варианте реализации показатель негативных симптомов у субъекта продолжает улучшаться от 12 недель до, по меньшей мере, приблизительно 24, 36 или 48 недель лечения. Модель пятиугольной структуры PANSS описана в WHITE L, HARVEY PD, OPLER L, LINDENMAYER J. EMPIRICAL ASSESSMENT OF THE FACTORIAL STRUCTURE OF CLINICAL SYMPTOMS IN SCHIZOPHRENIA. PSYCHOPATHOLOGY. 1997;30(5):263-74.
[00303] В некоторых вариантах реализации, если у субъекта наблюдается улучшение по меньшей мере одного негативного симптома в течение первого периода лечения, тогда введение терапевтически эффективной дозы Соединения (I) продолжают в течение второго периода лечения, по меньшей мере 12 недель, по меньшей мере 24 недель, по меньшей мере 48 недель или до тех пор, пока у субъекта не будет установлено функциональное улучшение после улучшения негативных симптомов. В одном варианте реализации положительные симптомы у субъекта, которого лечат лекарственной формой, являются стабильными в течение, по меньшей мере, части второго периода лечения, то есть остаются по существу на том же уровне, что и в начале исследования.
[00304] В некоторых вариантах реализации субъект имеет диагноз шизофрения. В одном варианте реализации субъект, выбранный для лечения пероральной лекарственной формой по данному изобретению, имеет отрицательный суб-балл PANSS базовой линии, больший или равный 20. В одном варианте реализации выбранный субъект также имеет базовые оценки менее 4 по следующим элементам PANSS: возбуждение, гиперактивность, враждебность, подозрительность, отказ от сотрудничества и плохой импульсный контроль. В одном варианте реализации выбранный субъект с шизофренией проявлял стабильные положительные симптомы шизофрении в течение по меньшей мере предыдущего одного, двух или трех месяцев и проявлял отрицательные симптомы в течение по меньшей мере предыдущего одного, двух или трех месяцев.
[00305] В некоторых вариантах реализации субъект с шизофренией, которого лечат кишечнорастворимой пероральной лекарственной формой CR по данному изобретению, имеет выраженные негативные симптомы. В варианте реализации субъект с шизофренией определяется как имеющий выраженные отрицательные симптомы, когда у субъекта имеется оценка ≥4 (умеренная) по меньшей мере по трем элементам подшкалы в элементах подкласса негативных симптомов PANSS, но не по элементам подкласса позитивных подуровней PANSS. В варианте реализации у субъекта имеются как явные положительные, так и явные отрицательные симптомы, когда у субъекта имеются оценки ≥4 по пунктам как для положительных, так и для отрицательных симптомов.
[00306] До 75% пациентов с шизофренией страдают от когнитивных нарушений, и исследование фазы 2b MIN-101, рассмотренное выше, показало улучшение когнитивной функции. Таким образом, в одном аспекте введение кишечнорастворимой пероральной лекарственной формы CR по данному изобретению пациенту с диагнозом шизофрения предназначено для улучшения когнитивной функции у пациента.
[00307] В некоторых вариантах реализации субъект ранее не лечился антипсихотическим препаратом. В других вариантах реализации субъект прекратил предшествующее лечение антипсихотическим лекарственным средством вследствие переживания одного или более из следующих факторов: удовлетворительное уменьшение положительных симптомов, неадекватный ответ на отрицательные симптомы или невыносимые побочные эффекты.
[00308] Вторичным результатом исследования фазы 2b MIN-101, обсуждавшегося выше, было поведение пациентов по шкале депрессии Калгари при шизофрении (CDSS) ADDINGTON D, ADDINGTON J, MATICKA-TYNDALE E. ASSESSING DEPRESSION IN SCHIZOPHRENIA: THE CALGARY DEPRESSION SCALE. BRITISH JOURNAL OF PSYCHIATRY SUPPLEMENT 1993; (22):39-44. CDSS имеет небольшое совпадение с положительными и отрицательными симптомами и стала рекомендованной шкалой для оценки тяжести депрессии у пациентов с шизофренией. В исследовании фазы 2b тяжесть симптомов, измеренная с помощью CDSS, была снижена после лечения 32 или 64 мг MIN-101 по сравнению с плацебо. Анализ корреляции между базовыми эффектами лечения на негативные симптомы и симптомы депрессии в этой группе пациентов показал, что между эффектами MIN-101 на эти две категории симптомов была лишь небольшая корреляция. Таким образом, поскольку влияние MIN-101 на негативные симптомы и симптомы депрессии у пациентов с шизофренией было в значительной степени независимым друг от друга, MIN-101 обладает потенциалом ослабления одного или более симптомов депрессии у пациентов, которые не являются шизофрениками.
[00309] Таким образом, другой целью настоящего изобретения является лечение по меньшей мере одного симптома депрессии у субъекта, нуждающегося в этом, с помощью способа введения кишечнорастворимой лекарственной формы CR, описанной в данном документе. В одном варианте реализации субъект имеет диагноз шизофрения. В одном варианте реализации улучшение симптомов депрессии у пациента с шизофренией измеряют с использованием CDSS.
[00310] Другой целью настоящего изобретения является снижение риска пролонгации QT у субъекта, которого лечат Соединением (I), путем введения субъекту Соединения (I), составленного в кишечнорастворимой дозированной форме CR, описанной в данном документе. В одном варианте реализации субъект был идентифицирован как имеющий один или более факторов риска пролонгированного QT, вызванного лекарственным средством. В одном варианте реализации субъект прекратил предыдущее лечение соединением, отличным от Соединения (I), из-за пролонгации интервала QT. В одном варианте реализации субъект прекратил предыдущее лечение другой лекарственной формой, содержащей Соединение (I), из-за пролонгации QT. В одном варианте реализации субъект имеет диагноз, выбранный из группы, состоящей из: выраженных отрицательных симптомов шизофрении, выраженных положительных и выраженных отрицательных симптомов шизофрении, большого депрессивного расстройства (MDD), расстройства сна и когнитивных нарушений.
[00311] В некоторых вариантах реализации любого из вышеуказанных способов лечения кишечнорастворимую пероральную лекарственную форму вводят утром или вечером. В одном варианте реализации лекарственную форму вводят по меньшей мере за два часа до еды.
[00312] В некоторых вариантах реализации любого из вышеуказанных способов лечения субъекту исполнилось 12 или более лет. В некоторых вариантах реализации субъекту по меньшей мере 14, 16, 18 или 20 лет. В некоторых вариантах реализации субъекту менее чем 50, 45, 40, 35 или 30 лет. В одном варианте реализации субъекту по меньшей мере 16 лет и меньше, чем 40, 35 или 30 лет.
[00313] В некоторых вариантах реализации любого из вышеуказанных способов лечения лекарственную форму можно вводить субъекту в сочетании с другим терапевтическим агентом. В одном варианте реализации другой терапевтический агент не ингибирует активность CYP2D6. В одном варианте реализации у субъекта диагностирована шизофрения, и другим терапевтическим агентом является антипсихотическое лекарственное средство.
[00314] В некоторых вариантах реализации любого из вышеуказанных способов лечения субъект может иметь генотип IM или генотип EM. В одном из вариантов реализации субъект имеет генотип EM.
[00315] В некоторых вариантах реализации любого из вышеуказанных способов лечения пероральная лекарственная форма может содержать 32 мг Соединения (I). В одном варианте реализации пероральная лекарственная форма состоит по существу из компонентов, перечисленных в Таблице 2 или Таблице 3 ниже.
ПРИМЕРЫ
Пример 1: Описание таблетки 32 мг MR, используемой в фазе 2b испытания MIN-101.
[00316] Таблетки 32 мг MR поставляются в виде круглых (диаметр 10 мм и R=10) таблеток с белым покрытием без видимых дефектов. Каждая таблетка содержит 32 мг Соединения (I). Полное описание компонентов и количественного состава таблетки 32 мг MR приведено в Таблице 1 ниже.
Таблица 1: Состав таблетки 32 мг MR
таблетку
1Применяется поправочный коэффициент 1,2 на соль
NA: Не применимо
Пример 2: Описание типичной 32 мг кишечнорастворимой таблетки CR.
[00317] Таблетки CR GR-01 поставляются в виде круглых (диаметр 10 мм и R=10) таблеток без видимых дефектов. Каждая таблетка содержит 32 мг Соединения (I). Полное описание компонентов и количественного состава таблетки CR GR-01 приведено в Таблице 2.
Таблица 2: Состав таблетки CR GR-01
таблетку
1Применяется поправочный коэффициент 1,2 на соль
NA: Не применимо
Пример 3: Описание другой типичной 32 мг кишечнорастворимой таблетки CR.
[00318] Таблетки CR GR-02 поставляются в виде круглых (диаметр 10 мм и R=10) таблеток без видимых дефектов. Каждая таблетка содержит 32 мг Соединения (I). Полное описание компонентов и количественного состава таблетки CR GR-02 приведено в Таблице 3 ниже.
Таблица 3: Состав таблетки CR GR-02
таблетку
1Применяется поправочный коэффициент 1,2 на соль
NA: Не применимо
Пример 4: Состав на серию для таблеток CR GR-01 и CR GR-02.
[00319] Типичный размер серии таблеток CR GR-01 и CR GR-02 составляет 5400 таблеток. Составы на серию описаны в Таблицах 4 и 5 ниже.
Таблица 4: Состав на серию для таблеток CR GR-01
NA: Не применимо
Таблица 5: Состав на серию для таблеток CR GR-02
Таблица 5A: Состав на серию для таблеток GR-01/B (типичный размер состава на серию составляет 150 000 таблеток)
Пример 5: Разработка оптимизированного способа растворения in vitro
[00320] Основываясь на ФК профилях Соединения (I), полученных с помощью таблеток MIN-101 32 мг MR, используемых в клинических исследованиях, был предложен подход корреляции in vitro/in vivo (IVIVC). Подход IVIVC определяется FDA как прогнозирующая математическая модель, описывающая взаимосвязь между свойством in vitro лекарственной формы и реакцией in vivo. В этом контексте модель относится к взаимосвязи между растворением in vitro таблетки 32 мг MR и ее реакцией in vivo, такой как концентрация Соединения (I) в плазме. Основные цели модели IVIVC заключались в обосновании использования прогнозирующего способа растворения in vitro и выборе целевых оптимизированных составов. Если достоверность модели IVIVC подтверждается клиническими результатами, способ растворения in vitro можно использовать в качестве суррогатного способа для клинических исследований.
[00321] Сначала после анализа всех ФК данных для Соединения (I) из клинических исследований был определен профиль растворения in vitro таблетки 32 мг MR, описанной в Примере 1. Указанный целевой профиль растворения in vitro затем использовали для разработки оптимизированного способа растворения in vitro. Данный способ описан в следующем примере.
[00322] Вторично, и когда способ растворения in vitro рассматривался как достаточно закрытый для ожиданий, был определен целевой профиль растворения in vitro кишечнорастворимой лекарственной формы для перорального применения CR и он был использован для разработки кишечнорастворимой лекарственной формы, описанной в Примерах 2 и 3. Профили растворения для этих двух лекарственных форм GR (GR-01 и GR-02) и таблетки 32 мг MR из Примера 1, которые были получены с использованием оптимизированного способа растворения, показаны в Таблицах 6 и 7 ниже.
Таблица 6: Кумулятивные профили растворения in vitro
Таблица 7: In vitro профили скорости растворения
1 не считается
[00323] Таблетки CR GR-01 и CR GR-02 и таблетка 32 мг MR, используемые в качестве сравнения, были испытаны в клиническом исследовании (MIN-101-C06) для оценки ФК профиля в плазме каждой лекарственной формы.
Пример 6: Аналитический способ для анализа MIN-101, BFB-520 и BFB-999 в плазме человека
Подготовка растворителей и реагентов
Все растворители и реагенты перечисленные ниже имеют степень чистоты «чда» или выше (относится ко всему документу).
Объемы указаны в качестве примеров, другие объемы могут быть приготовлены при условии соблюдения пропорций .
Растворитель для разведения: 50/50 (об./об.) ацетонитрил/вода раствор
Смешайте 500 мл ацетонитрила с 500 мл воды.
Хранение: 1 месяц при комнатной температуре
Буфер: буферный раствор рН 9
Перенесите содержимое ампулы с буферным концентратом рН 9 (Merck, P/N 109889) в 500 мл мерную колбу.
Заполните колбу до метки 500 мл воды.
Хранение: 1 месяц при приблизительно +5°С
Буфер: буферный раствор 1М ацетата аммония
Растворите 7,7 г ацетата аммония в 100 мл воды.
Хранение: 3 месяца при приблизительно +5°С
Подвижная фаза: 10мМ ацетат аммония буферный раствор
Добавте 10 мл буферного раствора 1М ацетата аммония к 990 мл воды.
Или растворите 0,77 г ацетата аммония в 1л воды.
Дегазируйте при необходимости (посредством ультразвука или перемешивания при пониженном давлении)
Хранение: 5 дней при комнатной температуре
Растворитель для восстановления: 80/20 (об./об.) 10мМ ацетат аммония буферный раствор/ацетонитрил
Смейшате 400 мл 10 мМ ацетата аммония буферного раствора с 100 мл ацетонитрила.
Или добавте 4 мл 1М ацетата аммония буферного раствора к 396 мл воды и 100 мл ацетонитрила.
Хранение: 5 дней при комнатной температуре
Растворитель для промывки иглы: 80/20 (об./об.) ацетонитрил/вода раствор
Смешайте 800 мл ацетонитрила с 200 мл воды.
Дегазируйте при необходимости (посредством ультразвука или перемешивания при пониженном давлении)
Хранение: 1 месяц при комнатной температуре
Растворитель для промывки иглы: 65/35 (об./об.) ацетонитрил/вода раствор
Смешайте 650 мл ацетонитрила с 350 мл воды.
Дегазируйте при необходимости (посредством ультразвука или перемешивания при пониженном давлении)
Хранение: 1 месяц при комнатной температуре
Растворитель для промывки колонки: 90/10 (об./об.) ацетонитрил/вода раствор
Смешайте 900 мл ацетонитрила с 100 мл воды.
Дегазируйте при необходимости (посредством ультразвука или перемешивания при пониженном давлении)
Хранение: 1 месяц при комнатной температуре
ПРОБОПОДГОТОВКА И ПРОЦЕДУРА ЭКСТРАКЦИИ
Контрольные образцы и образцы плазмы оттаивали при комнатной температуре и центрифугировали при 1920 g в течение 5 минут при +4°С.
Подготовка образцов
Холостой образец
В 10 мл полипропиленовую пробирку:
1. Переносят 250 мкл воды.
Холостой и нулевой образцы
В 10 мл полипропиленовую пробирку:
1. Переносят 250 мкл контрольного образца плазмы.
Калибровочные образцы
В 1,5 мл коническую полипропиленовую пробирку:
1. Переносят 900 мкл контрольного образца плазмы,
2. Добавляют 100 мкл соответствующего WS,
3. Перемешивают на вортексе 30 секунд,
4. Переносят 250 мкл препарата в 10 мл полипропиленовую пробирку.
Контрольные образцы
В 1,5 мл коническую полипропиленовую пробирку:
1. Переносят 900 мкл контрольного образца плазмы,
2. Добавляют 100 мкл соответствующего контрольного образца WS,
3. Перемешивают на вортексе в течение 30 секунд,
4. Переносят 250 мкл препарата в 10 мл полипропиленовую пробирку.
Исследуемые образцы
В 10 мл полипропиленовую пробирку:
1. Переносят 250 мкл образеца плазмы.
20-кратно разведенные образцы [2]
В 1,5 мл коническую полипропиленовую пробирку:
1. Переносят 380 мкл контрольного образца плазмы,
2. Добавляют 20 мкл образца плазмы для разбавления,
З. Перемешивают на вортексе в течение 30 секунд,
4. Переносят 250 мкл препарата в 10 мл полипропиленовую пробирку.
Процедура экстракции
1. Добавляют 25 мкл растворителя для разбавления (холостой образец реагента, холостой образей) или 25 мкл IS-WS (другие образцы),
2. Добавляют 1 мл буферного раствора рН 9,
3. Перемешивают на вортексе в течение 10 секунд,
4. Добавляют 4 мл диэтилового эфира,
5. Перемешивают на шейкере при низкой скорости в течение 20 мин,
6. Центрифугируют при 1920 g в течение 10 мин при +4°С,
7. Переносят в пробирки при приблизительно -80°С в течение 15 мин,
8. Переносят органическую фазу (верхний слой) в 5 мл стеклянную пробирку,
9. Упаривают досуха в потоке азота при +30°С,
10. Восстанавливают в 200мкл растворителя для восстановления,
11. Перемешивают на вортексе в течение 30 секунд,
12. Центрифугируют при 1920 g в течение 5 мин при +4°С,
13. Переносят конечный экстракт в полипропиленовую виалу,
14. Закрывают виалу крышкой с тефлон/силикон/тефлоновой септой,
15. Центрифугируют при 2500 g в течение 7 мин при +4°С
16. Помещают виалы в автодозатор до анализа
Или [4]
13. Переносят конечный экстракт в 2 мл полипропиленовый 96-луночный планшет,
14. Закрывают планшет силиконовой препроткнутой крышкой,
15. Центрифугируют при 2500 g в течение 7 мин при +4°С
16. Помещают планшет в автодозатор до анализа.
Оборудование для ЖХ №1
Описание
Система обслуживания
Растворитель для промывки иглы 80/20 (об./об.) ацетонитрил/вода раствор
Программа для промывки иглы
Аналитические условия
Объем введения 5 мкл
Продолжительность последовательности 6,0 мин
Времена удержания (Rt)
Аппаратура для ЖХ № 3 [5]
Описание
Промывка после ввода: 6 секунда
Оборудование для ЖХ №2 [3]
Описание
Система обслуживания
Растворитель для промывки иглы
Сильный растворитель 80/20 (об./об.) ацетонитрил/вода раствор
2000 мкл
Слабый растворитель 65/35 (об./об.) ацетонитрил/вода раствор
4000 мкл
Аналитические условия
Объем введения 1 мкл
Способ введения частичная петля
Продолжительность последовательности 6,0 мин
Времена удержания (Rt)
Обработка данных и критерии приемлемости
Обработка данных [2]
(CYR-101)
CYR-101-d6 площадь пика
BFB-784 площадь пика
BFB-784 площадь пика
Пример 7: Краткое содержание протокола MIN-101C06
Minerva Neurosciences, Inc.
MIN-101
Первичная:
Часть 1: ФК оценка
Оценить фармакокинетические (ФK) профили MIN-101 и его основных метаболитов (BFB-520 и BFB-999) после введения 2 кишечнорастворимых составов и 1 состава сравнения с модифицированным высвобождением (MR) MIN-101 у здоровых мужчин и женщин экстенсивно метаболизирующих (ЕМ) посредством цитохрома Р450 (CYP) 2D6.
Выбрать 1 кишечнорастворимый состав МR для применения в после еды.
Часть 2: Эффект еды
Оценить влияние пищи (представленной в виде содержащей высокий уровень жира, высококалорийной еды) на биодоступность MIN-101 и его основных метаболитов, когда выбранный кигечнорастворимый состав MR вводят в виде разовой дозы 32 мг здоровым CYP2D6 EM мужчинам и женщинам.
Вторичная:
Часть 1: ФК оценка
Предоставить дополнительную информацию о безопасности и переносимости однократных доз MIN-101 у здоровых CYP2D6 EM мужчин и женщин.
Оценить взаимосвязь между уровнями в плазме MIN-101 и его основных метаболитов по параметрам ЭКГ, включая QT/QTcF
Часть 2: Эффект еды
Оценить безопасность и переносимость выбранного состава МR у здоровых CYP2D6 EM мужчин и женщин, после еды, по сравнению с приемом натощак.
Это одноцентровое двухэтапное 1 фазовое исследование.
Часть 1: ФК оценка
Часть 1 представляет собой открытое рандомизированное исследование с 3 последовательностями лечения, с 3 периодами исследования для оценки профиля ФK MIN-101 и его метаболитов (BFB-520 и BFB-999) после однократного перорального введения 3 составов MIN-101 (2 прототипа кишечнорастворимых (GR) CR составов и 1 состав сравнения MR). Каждый субъект получит одну дозу каждого из составов в течение 3 периодов. Между этими тремя периодами будет период отмывки 14 ± 2 дня.
В общей сложности 16 здоровых CYP2D6 EM субъектов мужского или женского пола (с идеальным равным гендерным разделением, но не менее 6 для каждого пола) будут дозированы для обеспечения данных по 12 оцениваемым субъектам. Чтобы быть оцениваемыми, субъекты должны были получить все 3 состава и иметь достаточные данные для основной цели части 1 исследования. Субъекты должны предоставить письменное информированное согласие на участие в исследовании до проведения оценки или сбора лабораторных образцов. Субъекты будут оцениваться на предмет соответствия критериям исследования в течение периода отбора. После получения письменного информированного согласия полная история болезни будет задокументирована. Будет проведен полный осмотр, включая измерение показателей жизненно важных функций, ЭКГ (трижды), массы тела и роста. Гематология, клиническая химия и анализ мочи будут выполнены для всех субъектов. Все субъекты должны быть готовы использовать приемлемый способ контроля рождаемости с двойным барьером со своими партнерами от скрининга и до 90 дней после последней дозы.
Часть 2: Эффект еды
Субъекты, которые завершили часть 1 исследования вернутся и получат дополнительную однократную пероральную дозу выбранного прототипа GR после еды, что позволит оценить влияние пищи путем сравнения ФК свойств с теми, которые получены в части 1. Часть 2 начнется после анализа ФK и данных по безопасности, чтобы решить, какой состав GR будет использоваться. После завершения части 1 будет период промывки в течение 14 ± 2 дней.
Оценки в конце исследования или в начале отмены будут проводиться через 5-9 дней после последней полученной дозы.
В общей сложности 16 здоровых CYP2D6 EM субъектов мужского или женского пола (с идеальным равным гендерным разделением, но не менее 6 для каждого пола) будут дозированы для обеспечения данных по 12 оцениваемым субъектам.
Субъекты, отозванные по поводу нежелательных явлений (НЯ), не связанных с ИЛП, будут заменены по мере необходимости для обеспечения 12 оцениваемых субъектов для части 1 и для части 2 в конце клинического исследования. Субъекты, отозванные в связи с НЯ, связанными с ИЛП, замене не подлежат.
Критерии включения
Субъекты должны соответствовать всем следующим критериям включения во время отбора, чтобы быть зачисленным в исследование:
1. Подтверждено, что генотип субъекта экстенсивно метаболизирующего посредством CYP 2D6 определен как генотип субъекта, у которого есть по крайней мере один функциональный аллель (* 1 или * 2), но нет нефункционального аллеля, что означает любую комбинацию * 1 и * 2, и аллель пониженной функции (* 10, * 17 или * 41) допускается с помощью документированных испытаний.
2. Субъект дал добровольное письменное информированное согласие перед выполнением любой процедуры, связанной с исследованием.
3. Должен быть от 18 до 45 лет включительно.
4. Субъект должен быть здоровым мужчиной или женщиной, как указано ниже:
Клинические химические, гематологические и анализы мочи должны быть в пределах нормальных допустимых пределов (если они выходят за пределы диапазона, за исключением калия, магния и кальция, они должны считаться клинически значимыми, чтобы быть исключающими) и проводиться в течение 21 дня после получения первой дозы исследуемого препарата.
Индекс массы тела от 18 до 30 кг/м2 включительно
Нормальные показатели жизнедеятельности после 5 мин отдыха в положении лежа:
95 мм рт. ст. <Систолическое артериальное давление <140 мм рт. ст.
50 мм рт. ст. <Диастолическое артериальное давление <90 мм рт. ст.
50 ударов в мин < частота сердечных сокращений <90 ударов в мин
Нормальная ЭКГ с 12 отведениями определяется как: P ≤120 мс, 120 мс <PR <210 мс, QRS
<120 мс, QTc (Fridericia) ≤ 430 мсек для мужчин и ≤ 440 мсек для женщин (может быть принята неполная блокада правой ножки пучка)
5. Согласие воздерживаться от всех лекарств (за исключением разрешенного контроля рождаемости, определенного в критериях включения 6), включая лекарства, отпускаемые без рецепта и по рецепту (включая витамины и натуральные или растительные лекарственные средства, например зверобой), в течение 21 дня перед первой дозой с ИЛП в период 1 до выписки из исследования (окончание медицинского периода после исследования после периода 4)
6. Субъект соглашается использовать следующие способы контроля рождаемости:
Субъекты женского пола, обладающие детородным потенциалом, должны быть готовы использовать два способа контрацепции на протяжении и до 30 дней после завершения исследования. Один из которых должен быть высокоэффективным способом, определяемым как способ, который приводит к низкой частоте отказов (то есть менее 1% в год) при последовательном и правильном использовании.
Следующие высокоэффективные способы контрацепции, приемлемые для данного исследования:
Хирургическая стерилизация (то есть двусторонняя перевязка маточных труб/сальпингэктомия, гистерэктомия для субъектов или партнеров женского пола; вазэктомия для субъектов или партнеров мужского пола)
Размещение внутриматочной спирали или внутриматочной системы
Гормональная контрацепция (имплантируемая, пластырь, инъецируемая)
ПОЖАЛУЙСТА, ОБРАТИТЕ ВНИМАНИЕ: Оральные гормональные контрацептивы не разрешены в этом исследовании.
Истинное половое воздержание, когда это соответствует предпочтительному и обычному образу жизни субъекта, периодическое воздержание (например, календарная овуляция, симптоматические, постовуляционные способы), объявление воздержания в течение всего периода испытания и абстиненция НЕ являются приемлемыми способами контрацепции)
Барьерные способы для субъектов женского пола включают в себя использование презерватива партнером или использование субъектом окклюзионного колпачка [диафрагмы или колпачка шейки/свода] со спермицидной пеной, гелем, пленкой, кремом или суппозиторием
Пациенты женского пола, которые находятся в постменопаузе (определяется как спонтанная аменорея в течение по крайней мере 1 года или спонтанная аменорея в течение по крайней мере 6 месяцев, подтвержденная результатом анализа уровня фолликулостимулирующего гормона [FSH] ≥40 МЕ/мл), имеют право на участие в этом исследовании.
Субъекты мужского пола
Субъекты мужского пола, которые были стерилизованы или имеют партнеров, не способных к деторождению (включая гомосексуальных мужчин), должны использовать один барьерный способ контрацепции. Это необходимо для предотвращения непреднамеренного воздействия на партнера исследуемого препарата через семенную жидкость (для мужчин это должен быть презерватив или использование их партнером окклюзионного колпачка [диафрагмы или колпачка шейки/свода]).
Субъекты мужского пола, у которых есть партнеры с детородным потенциалом, должны быть готовы использовать один барьерный способ контрацепции со своими партнерами на протяжении всего исследования (использование презерватива или их партнером окклюзионного колпачка [диафрагмы или колпачка шейки/свода]). Их партнеры также должны использовать высокоэффективный способ контроля над рождаемостью, определяемый как способ, который приводит к низкой частоте отказов (т.е. менее 1% в год) при последовательном и правильном использовании, например, стерилизацию, имплантацию, инъекции, комбинированные оральные контрацептивы, и внутриматочные устройства на срок до 90 дней после завершения исследования. Субъекты должны согласиться сообщить исследователю, если их партнер забеременеет в это время.
7. Должны быть готовы и способны общаться и участвовать во всем исследовании.
8. Готовы съедать всю еду, поставляемую на протяжении всего исследования
Субъекты, отвечающие любому из следующих критериев исключения, не должны участвовать в исследовании:
1. История клинически значимых заболеваний желудочно-кишечного тракта (особенно пептические язвы, желудочно-кишечные кровотечения, язвенный колит, болезнь Крона или синдром раздраженного кишечника); почечная, печеночная, неврологическая, гематологическая, эндокринная, онкологическая, легочная, иммунологическая или психиатрическая болезнь (особенно те, у которых в анамнезе были клинически значимые депрессии, суицидальные мысли или попытки самоубийства), или сердечно-сосудистые заболевания или любые другие состояния, которые, по мнению основного исследователя, поставят под угрозу безопасность субъекта или повлияют на достоверность результатов исследования
2. Острая диарея или запор за 7 дней до предполагаемого первого дня исследования. Если проверка проводится за> 7 дней до первого дня исследования, этот критерий будет определен в первый день исследования. Диарея определяется как проход жидких фекалий и/или частота стула
> 3 раз в день. Запор будет определяться как неспособность двигать кишечник чаще, чем через день.
3. Субъект сдавал кровь в течение 90 дней или плазму в течение 30 дней после введения дозы
4. Регулярное употребление алкоголя у мужчин> 21 ед. в неделю и у женщин> 14 ед. в неделю (1 ед. = ½ пинты пива, 25 мл 40% спирта или 125 мл вина)
5. Субъект имеет пограничный или длинный интервал Fridericia QTc, который определяется показаниями скрининга > 430 мсек для мужчин иь> 440 для женщин или личную или семейную историю синдрома длинного QT
6. Субъект участвовал в клиническом испытании в течение 90 дней до начала исследования
7. Женщины, которые беременны или кормят грудью
8. Субъект использовал любые рецептурные лекарства или лекарства, отпускаемые без рецепта (OTC), включая витаминные добавки, в течение 21 дня до дня -1
9. Субъекта лечили любыми известными препаратами, изменяющими ферменты P450 2D6 или 3A4 (например, бета-блокаторы, антидепрессанты, антипсихотические средства, некоторые антибиотики, такие как эритромицин, кетоконазол, рифампицин, триметоприм или кларитромицин, бензодиазепин, такой как альпразолам или мидазолам, антигистаминные лекарственные средства, такеи как хлорфенирамин, блокаторы кальциевых каналов, такие как амлодипин или дилтиазем, или ингибиторы PDE5) в течение 30 дней до исследования
10. Субъект курил или использовал никотиновые продукты в течение 2 месяцев до или во время исследования
11. Субъект обратился за советом или был направлен к терапевту или консультанту из-за злоупотребления алкоголем, немедицинскими препаратами, лекарственными препаратами или другими веществами, например, растворителями
12. Субъект имеет положительный анализ крови на ВИЧ, поверхностный антиген гепатита В (HBsAg) и антитела к гепатиту С
13. Любое текущее или предыдущее употребление наркотиков, таких как опиаты, кокаин, экстази или амфетамины для внутривенного введения, и/или положительный анализ мочи на алкоголь или наркотики. Субъекты, которые допускали случайное употребление каннабиса в прошлом, не будут исключены, если они проходят отрицательный тест на злоупотребление наркотиками и воздерживаются от употребления каннабиса в течение не менее 3 месяцев.
14. Субъект имеет текущее неконтролируемое внутреннее заболевание (то есть активную инфекцию) или имел клинически значимое заболевание в течение последних 30 дней до дня -1.
15. Субъект перенес серьезную операцию в области желудочно-кишечной хирургии в течение 28 дней после начала исследования или за 12 месяцев до исследования.
16. Невыполнение требования исследователя к пригодности к участию по любой другой причине.
Часть 1:
После голодания в течение 10 ч в течение ночи, утром в дни дозирования, исследуемое лекарственное средство будет вводиться с 240 мл негазированной воды. Субъекты получат первую еду во время обеда. Субъекты будут получать одно пероральное введение каждой из следующих схем в случайном порядке, с интервалом 14 ± 2 дня для отмывки:
Схема А: 32 мг MIN-101 текущего состава с модифицированным высвобождением (состав сравнения), идентифицированного как состав MR-32, вводят натощак
Схема В: 32 мг MIN-101 кишечнорастворимого состава CR, идентифицированного как GR-01: вводят натощак
Схема C: 32 мг MIN-101 кишечнорастворимого состава CR, идентифицированного как GR-02: вводят натощак
Часть 2:
Во второй части субъекты получат 1 пероральную дозу выбранного кишечнорастворимого состава CR (GR-01 или GR-02) после еды.
После голодания в течение 10 ч в течение ночи, утром 1-го дня, субъектам будет предоставлен с высоким содержанием жира, высококалорийный завтрак перед введением исследуемого препарата. Субъекты будут потреблять еду в течение 25 мин или меньше. После завершения приема пищи и через 30 мин после начала приема пищи вводится исследуемое лекарственное средство.
Все исследуемые препараты будут вводиться перорально с 240 мл негазированной воды. Вода будет добавляться по желанию, за исключением 1 ч до и 1 ч после приема препарата.
Не применимо. 32 мг MIN-101 действующего состава с модифицированным высвобождением для использования в качестве сравнения.
До 21 дня до начала первой дозы 1
Институционализация:
С утра дня -1 до дня 4 в течение 4 отдельных периодов
Период отмывки:
14 ± 2 дня после предыдущего периода дозирования
Последний прием исследования:
7 (± 2 дня) дней после последней дозы
Общая продолжительность исследования (включая период обследования 21 день):
До 78 дней.
Фармакокинетика:
Плазма будет храниться при температуре -80°С до анализа. Образцы плазмы будут проанализированы на наличие MIN-101 и его метаболитов BFB-520 и BFB-999 с использованием проверенного способа ЖХ-МС/МС.
Образцы крови для MIN-101 будут собраны в моменты времени 0 (до введения дозы), 0,25, 0,5, 1, 1,5, 2, 2,5, 3, 3,5, 4, 5,
6, 7, 8, 10, 12, 14, 16, 20, 24, 28, 32, 36, 48, 60 и 72 ч после введения дозы в 1-й день всех периодов. Следующие ключевые параметры ФК в плазме будут рассчитаны с использованием не компартментальных способов: Cmax, Tmax, Tlag, частичная AUC (например, AUC12, AUC24), AUClast, AUC∞, и t1/2. Дополнительные ФК параметры могут быть включены, если это будет сочтено целесообразным.
ЭКГ будет уточнена, так как будет проведена оценка ФK/ФД
Влияние на сердечно-сосудистые переменные будет оцениваться с помощью описательной статистики и частотных таблиц. Эти таблицы будут включать наблюдаемые значения и изменения от Базовых значений (ЭКГ до введения дозы будет использоваться в качестве Базового уровня), чтобы позволить обнаруживать клинически значимые изменения у людей.
Переменными ЭКГ, которые будут проанализированы, являются частота сердечных сокращений, интервал PR, интервал QRS, интервал QT и интервал QTc, скорректированные на частоту сердечных сокращений с использованием QTcF. Значения QTcF будут сведены в таблицу для их абсолютных значений, а также сведены в таблицу относительно базовых измерений, чтобы обнаружить отдельные изменения QTcF.
Описательная статистика интервалов QTcF и изменений по сравнению с базовой линией будет суммироваться в каждый запланированный момент времени. Процент субъектов с интервалом QTc> 450 мсек, > 480 мсек или ≥ 500 мсек будет суммирован, так же как процент субъектов с интервалом QTcF увеличившимся по сравнению с базовой линией от 30 до 59 мсек или ≥ 60 мсек.
Будет сообщаться о важных отклонениях в форме волны ЭКГ, которые являются изменениями по сравнению с показаниями базовой линии (например, изменения в морфологии зубца T или возникновение волн U).
Безопасность и Переносимость:
Частота нежелательных явлений и клинически значимых отклонений от нормы лабораторных показателей, показателейжизнедеятельности и ЭКГ будут регистрироваться на основании данных, полученных от исследователя, и отчетов субъектов.
Размер выборки: Размер выборки для этого исследования основан на количественном и качественном рассмотрении. В предыдущем опыте однократного приема с субъектами EM коэффициент дисперсии (CV) между субъектами для AUC в плазме и Cmax для эталонной композиции MIN-101 оценивался примерно в 30% и 50% соответственно. Следовательно, выбранный размер выборки от 12 до 16 должен быть достаточным для достижения целей данного исследования и выявления редких нежелательных явлений, если такое событие, вероятно, связано с лечением с помощью MIN-101. Будут предприняты все усилия, чтобы равное количество мужчин и женщин были зарегистрированы для исследования.
Фармакокинетика: Фармакокинетические параметры будут суммироваться по среднему значению, стандартному отклонению, стандартной ошибке среднего значения, коэффициенту вариации, минимуму, медиане и максимуму, в зависимости от ситуации, для каждого состава и между выбранным составом MR для каждого состояния пищи. 90-процентные доверительные интервалы для отношения средней логарифмированной плазменной частичной AUC, AUClast и AUC∞ и Cmax будут построены с использованием оценочных средних наименьших квадратов и дисперсии внутри субъекта из модели смешанных эффектов.
При необходимости будет проведен дополнительный анализ, включая взаимосвязь между уровнями MIN-101 в плазме и его основными метаболитами и изменениями в интервалах QTcF.
Безопасность: Безопасность и переносимость MIN-117 будут основаны на обзоре индивидуальных значений и сводной статистике. Частота возникновения нежелательных явлений, возникающих при лечении, будет сведена в таблицу по количеству и процентам. Аномалии в клинической лабораторных показателях, показателях жизнедеятельности и ЭКГ будут основаны на заранее определенных нормальных диапазонах и будут сведены в таблицу по группам доз, показывающим количество и процент субъектов.
[00324] В примерах 8-11 подробно описано открытое рандомизированное исследование с 3 последовательностями лечения, с 3 периодами исследования для оценки профиля ФK MIN-101 и его метаболита BFB-520 после однократного перорального введения 3 составов MIN-101 (2 прототипа кишечнорастворимых (GR) CR составов (GR-01 и GR-02) и 1 состав сравнения MR (MR32)). Каждый субъект получал одну дозу каждого из составов в течение 3 периодов. Между этими тремя периодами был период отмывки 14 ± 2 дня.
[00325] В примерах показаны различные таблицы, которые включают временной профиль концентраций в плазме различных соединений, включая, например, 1H-изоиндол-1-он,2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, т.е., Соединение (I). В этих таблицах использование «MIN-101» или «MIN101» предназначено для обозначения свободного основания, то есть Соединения (I).
Пример 8: Фармакокинетический анализ in vivo MIN-101 в таблетках CR GR-01, таблетках CR GR-02 и капсулах 32 мг MR (MR32) (tau=72 ч)
[00326] MR32-12 субъектов (перекрестн.)
[00327] Геом. среднее Cmax: 28,34 нг/мл
[00328] Медиана Tmax: 2,00 ч
AUC(0-tau): 291,55 нг · ч/мл
Таблица 8 MR32 Средние концентрации и параметры MIN-101 в плазме
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
6,56
19,65
23,96
24,16
24,68
22,06
20,34
19,45
21,54
15,19
11,89
10,36
12,46
12,66
10,33
7,74
4,12
3,11
2,39
1,45
0,96
0,20
0,03
0,04
0,00
7,42
7,24
7,68
7,89
6,47
5,41
5,38
5,61
10,08
6,89
5,89
4,69
5,94
5,64
4,86
3,14
2,26
1,62
2,24
1,42
1,15
0,35
0,12
0,13
0,00
6,46
5,44
6,33
5,97
5,19
4,71
4,21
4,92
7,17
4,91
3,82
3,39
4,95
4,41
3,96
2,64
1,85
1,48
1,38
0,96
0,65
0,27
0,06
0,07
0,00
113,08
36,77
32,04
32,64
26,24
24,54
26,46
28,8З
46,80
45,33
49,52
45,27
47,65
44,58
47,05
40,56
54,83
52,21
93,57
97,56
119,73
176,58
346,41
346,41
Tmax, ч
AUC0-t ч*нг/мл
2,42
291,55
1,33
66,12
0,99
53,61
54,98
22,68
[00329] Смотрите Фиг. 4.
[00330] GR-01-12 субъектов (перекрестн.)
[00331] Геом. среднее Cmax: 18,82 нг/мл
[00332] Медиана Tmax: 4,50 ч
AUC(0-tau): 284,52 нг · ч/мл
Относительная биодоступность по сравнению с MR32: F% Cmax: 69,9%, F% AUC(0-tau): 101,3%
Таблица 9. GR-01 Индивидуальные концентрации и параметры MIN-101 в плазме

Таблица 10. GR-01 Средние концентрации и параметры MIN-101 в плазме
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,35
1,25
5,75
11,71
15,54
16,04
15,99
14,62
11,35
10,33
9,39
10,69
11,26
11,76
10,58
5,49
5,57
3,96
3,10
2,42
0,77
0,30
0,15
0,00
0,00
1,22
2,77
5,57
7,46
7,84
7,01
6,08
4,60
4,17
3,58
3,93
5,39
4,65
4,67
4,87
2,33
3,30
2,80
2,06
3,33
0,92
0,51
0,24
0,00
0,00
0,65
1,95
4,67
5,46
6,08
5,89
5,27
3,26
3,35
2,64
3,25
4,42
3,48
3,33
3,96
1,85
2,58
2,04
1,50
2,05
0,77
0,40
0,20
0,00
0,00
346,41
220,78
96,91
63,69
50,45
43,68
36,00
31,47
36,70
34,63
41,81
50,45
41,34
39,73
46,01
42,48
59,31
70,67
66,68
137,50
119,51
171,48
159,83
Tmax, ч
AUC0-t ч*нг/мл
F AUC
F Cmax
6,09
284,52
101,30
69,90
3,43
60,83
29,16
23,92
3,00
41,49
20,90
17,04
57,19
21,38
28,79
34,22
[00333] Смотрите Фиг. 5.
[00334] GR-02-12 субъектов (перекрестн.)
[00335] Геом. среднее Cmax: 15,43 нг/мл
[00336] Медиана Tmax: 14,00 ч
AUC(0-tau): 253,01 нг ⋅ ч/мл
Относительная биодоступность по сравнению с MR32: F% Cmax: 54,33%, F% AUC(0-tau): 86,9%
Таблица 11. GR-02 Индивидуальные концентрации и параметры MIN-101 в плазме
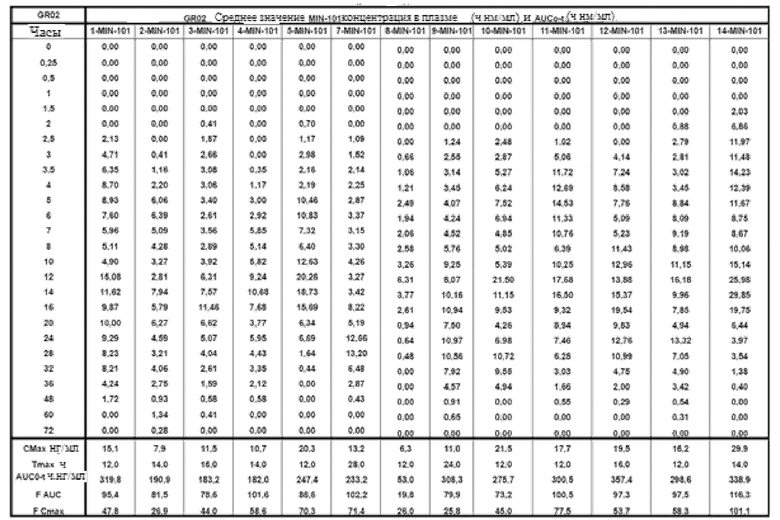
Таблица 12. GR-02 Средние концентрации и параметры MIN-101 в плазме
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,00
0,16
0,68
1,98
3,22
4,69
5,20
7,05
6,16
5,86
5,95
7,86
12,81
12,06
10,63
6,23
7,72
6,49
4,36
2,35
0,50
0,21
0,02
0,00
0,00
0,00
0,59
1,95
3,29
3,03
4,39
4,14
3,94
3,20
2,63
2,84
4,27
7,69
7,28
5,24
2,37
4,01
4,15
2,90
1,62
0,34
0,41
0,08
0,00
0,00
0,00
0,31
1,04
1,89
1,90
3,38
3,38
3,24
2,68
2,09
2,19
3,79
6,62
5,35
3,98
1,75
3,23
3,47
2,24
1,26
0,29
0,30
0,04
0,00
0,00
0,00
375,28
286,76
166,30
94,21
93,69
79,73
55,85
51,99
44,82
47,70
54,29
60,04
60,38
49,25
37,95
52,01
63,88
66,52
69,09
66,40
197,87
375,28
Tmax, ч
AUC0-t ч*нг/мл
F AUC
F Cmax
15,23
253,01
86,94
54,33
5,20
85,28
24,39
23,09
3,67
65,81
16,36
17,99
34,12
33,71
28,05
42,50
[00337] Смотрите Фиг. 6.
Таблица 13. Сравнение плазменных концентраций MR32, GR-01 и GR-02 MIN-101.
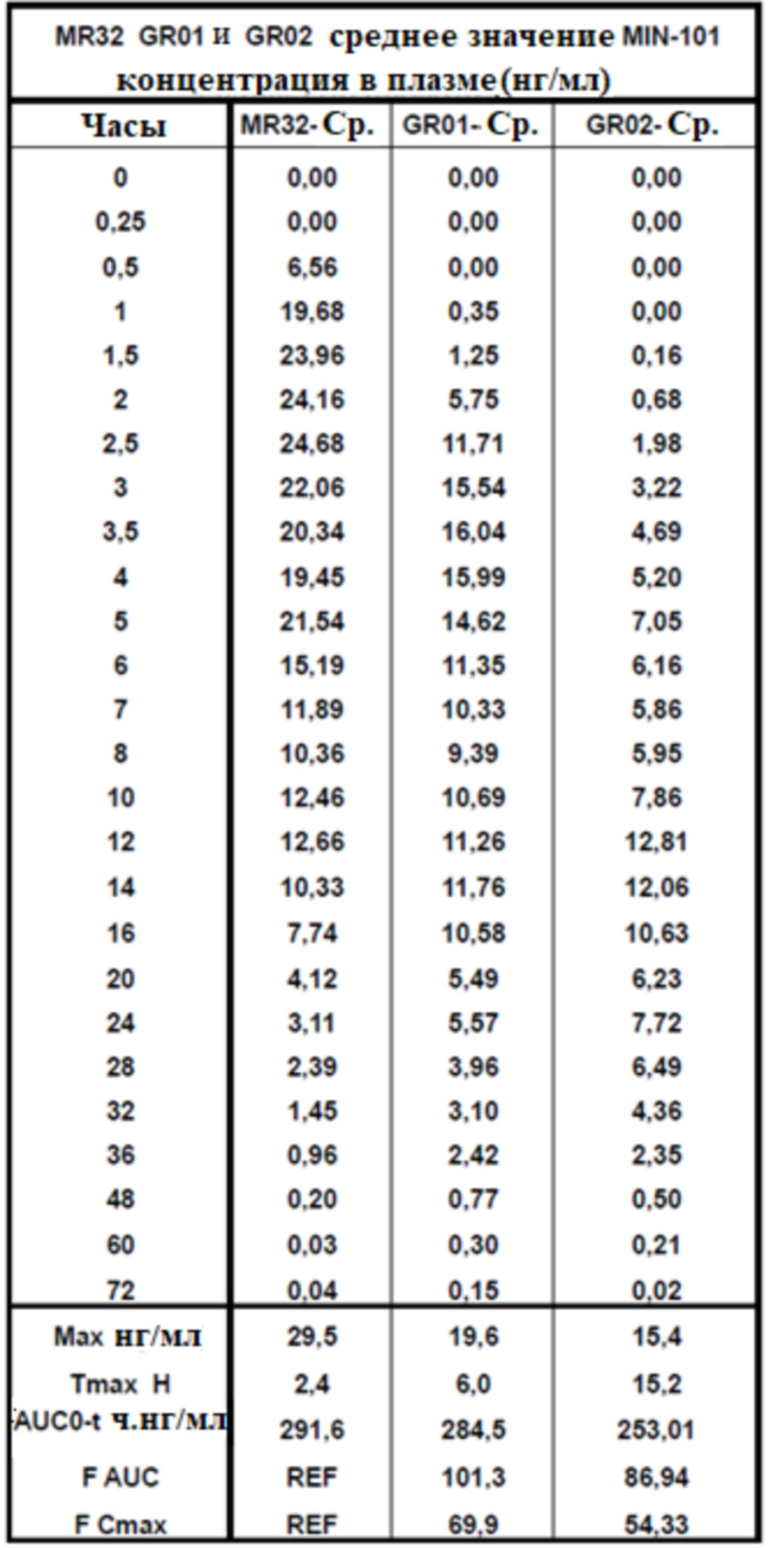
[00338] Смотрите Фиг. 7.
Таблица 14. Сравнение концентраций в плазме MR32, GR-01 и GR-02 MIN-101 - скорость увеличения и уменьшения.
Скорость увеличения и уменьшения концентраций в плазме (нг/мл)/ч
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
0,00
26,25
26,24
8,56
0,39
1,03
0,00
0,00
0,00
2,09
0,00
0,00
0,00
1,05
0,10
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,70
1,80
9,00
11,92
7,66
1,00
0,00
0,00
0,00
0,00
0,00
0,65
0,29
0,25
0,00
0,00
0,02
0,00
0,00
0,00
0,00
0,00
0,00
0,31
1,05
2,60
2,48
2,94
1,02
1,85
0,00
0,00
0,09
0,96
2,48
0,00
0,00
0,00
0,37
0,00
0,00
0,00
Время Vmax
Тест/MR32
0,50
Стандт.
2,50
0,45
3,50
0,11
[00339] В течение каждого временного интервала (dt) концентрация в плазме (Cp) MIN-101 увеличивается или уменьшается. От t=0 до Tmax скорость увеличения Vmax=d (Cp)/dt. После Vmax показатели снижаются. Смотрите Фиг. 8.
Пример 9: Фармакокинетический анализ in vivo BFB-520 в таблетках CR GR-01, таблетках CR GR-02 и капсулах 32 мг MR (MR32) (tau=72 ч)
[00340] MR32-12 субъектов (перекрестн.)
[00341] Геом. среднее Cmax: 1,77 нг/мл
[00342] Медиана Tmax: 6,00 ч
AUC(0-tau): 30,26 нг · ч/мл
Таблица 15. MR32 Индивидуальные концентрации и параметры BFB-520 в плазме

Таблица 16. MR32 Средние концентрации и параметры BFB-520 в плазме
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,42
0,81
1,04
1,21
1,46
1,49
1,55
1,59
1,55
1,44
1,51
1,48
1,21
1,40
1,01
0,85
0,50
0,35
0,15
0,05
0,00
0,00
0,00
0,33
0,51
0,45
0,68
0,58
0,48
0,67
0,69
0,68
0,75
0,66
0,63
0,77
0,44
0,34
0,22
0,26
0,26
0,18
0,28
0,36
0,36
0,46
0,43
0,40
0,54
0,55
0,57
0,57
0,53
0,46
0,60
0,36
0,27
0,18
0,19
0,21
0,09
41,40
48,77
37,60
46,84
38,99
31,20
42,36
44,45
47,23
49,53
44,91
52,49
54,91
43,42
40,36
44,39
75,95
168,74
346,41
Tmax, ч
AUC0-t ч*нг/мл
6,92
30,26
3,65
11,35
3,06
9,43
52,84
37,51
[00343] Смотрите Фиг. 9.
[00344] GR-01-12 субъектов (перекрестн.)
[00345] Геом. среднее Cmax: 1,77 нг/мл
[00346] Медиана Tmax: 6,00 ч
AUC(0-tau): 27,48 нг · ч/мл
Относительная биодоступность по сравнению с MR32: F% Cmax: 80,48%, F% AUC(0-tau): 96,1%
Таблица 17. GR-01 Индивидуальные концентрации и параметры BFB-520 в плазме

Таблица 18. GR-01 Средние концентрации и параметры BFB-520 в плазме
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,00
0,00
0,04
0,22
0,49
0,65
0,77
0,89
0,87
0,94
1,07
1,10
1,04
1,11
1,08
0,96
0,63
0,63
0,37
0,25
0,03
0,00
0,00
0,25
0,2З
0,35
0,36
0,36
0,29
0,35
0,39
0,45
0,55
0,42
0,45
0,43
0,36
0,36
0,23
0,33
0,09
0,22
0,17
0,27
0,31
0,29
0,22
0,28
0,29
0,36
0,43
0,31
0,34
0,38
0,28
0,26
0,18
0,25
0,05
112,01
45,99
53,37
47,35
40,77
33,50
37,58
36,81
40,79
53,43
37,58
41,52
45,29
57,32
57,90
60,48
133,52
346,41
Tmax, ч
AUC0-t ч*нг/мл
F AUC
F Cmax
12,50
27,48
96,10
80,48
8,04
9,46
35,77
24,60
5,08
8,07
18,95
20,65
64,32
34,41
37,22
30,56
[00347] Смотрите Фиг. 10.
[00348] GR-02-12 субъектов (перекрестн.)
[00349] Геом. среднее Cmax: 1,13 нг/мл
[00350] Медиана Tmax: 16,00 ч
AUC(0-tau): 27,53 нг ⋅ ч/мл
Относительная биодоступность по сравнению с MR32: F% Cmax: 69,48%, F% AUC(0-tau): 88,46%
Таблица 19. GR-02 Индивидуальные концентрации и параметры BFB-520 в плазме
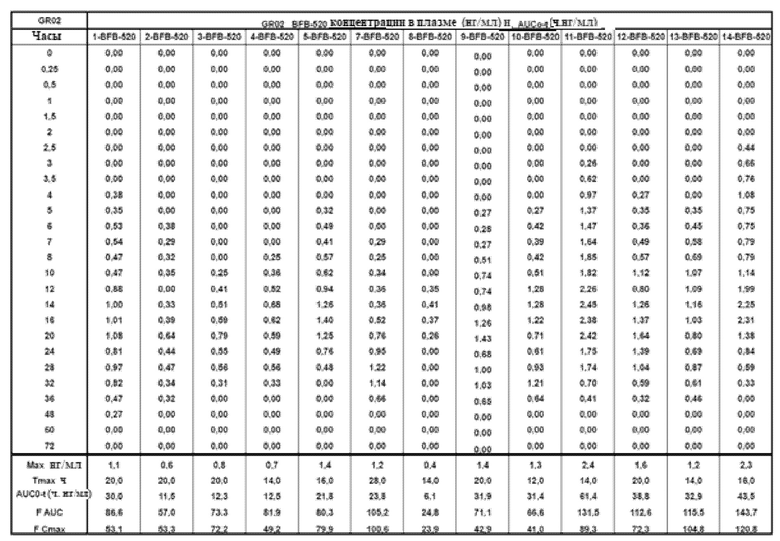
Таблица 20. GR-02 Средние концентрации и параметры BFB-520 в плазме
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,00
0,00
0,00
0,03
0,07
0,11
0,21
0,31
0,39
0,44
0,52
0,68
0,90
1,07
1,11
1,06
0,77
0,80
0,57
0,30
0,02
0,00
0,00
0,20
0,27
0,40
0,40
0,42
0,45
0,49
0,51
0,68
0,70
0,69
0,59
0,45
0,45
0,41
0,28
0,00
0,13
0,19
0,29
0,27
0,28
0,30
0,31
0,41
0,51
0,53
0,54
0,47
0,31
0,34
0,33
0,24
280,02
254,72
191,24
130,19
106,04
102,76
94,19
75,52
75,73
65,30
61,68
55,93
59,06
55,61
71,69
92,16
Tmax, ч
AUC0-t ч*нг/мл
F AUC
F Cmax
17,54
27,53
88,46
69,48
4,46
15,99
33,92
29,58
3,56
12,65
27,56
24,00
25,42
58,08
38,35
42,56
[00351] Смотрите Фиг. 11.
Таблица 21. Сравнение плазменных концентраций MR32, GR-01 и GR-02 BFB-520.

[00352] Смотрите Фиг. 12.
Таблица 22. Сравнение концентраций в плазме MR32, GR-01 и GR-02 BFB-520 - скорость увеличения и уменьшения.
Скорость увеличения и уменьшения концентраций в плазме (нг/мл)/ч
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
0,00
0,00
0,84
0,77
0,46
0,34
0,49
0,07
0,11
0,04
0,00
0,00
0,07
0,00
0,00
0,10
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,09
0,35
0,54
0,31
0,24
0,13
0,00
0,07
0,13
0,01
0,00
0,04
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,07
0,07
0,07
0,20
0,10
0,08
0,04
0,08
0,08
0,11
0,09
0,02
0,00
0,00
0,01
0,00
0,00
Время Vmax
Тест/MR32
1,00
Стандт.
3,00
0,64
4,00
0,24
[00353] В течение каждого временного интервала (dt) концентрация в плазме (Cp) BFB-520 увеличивается или уменьшается. От t=0 до Tmax скорость увеличения Vmax=d (Cp)/dt. После Vmax показатели снижаются. Смотрите Фиг. 13.
Пример 10: Прогнозируемые концентрации в плазме MIN-101 и BFB-520 в стационарном состоянии в таблетках MR32 и GR-01 (32 и 64 мг)
Таблица 23. Прогнозируемые концентрации в плазме MIN-101 в стационарном состоянии в таблетках MR32 и GR-01, 32 мг
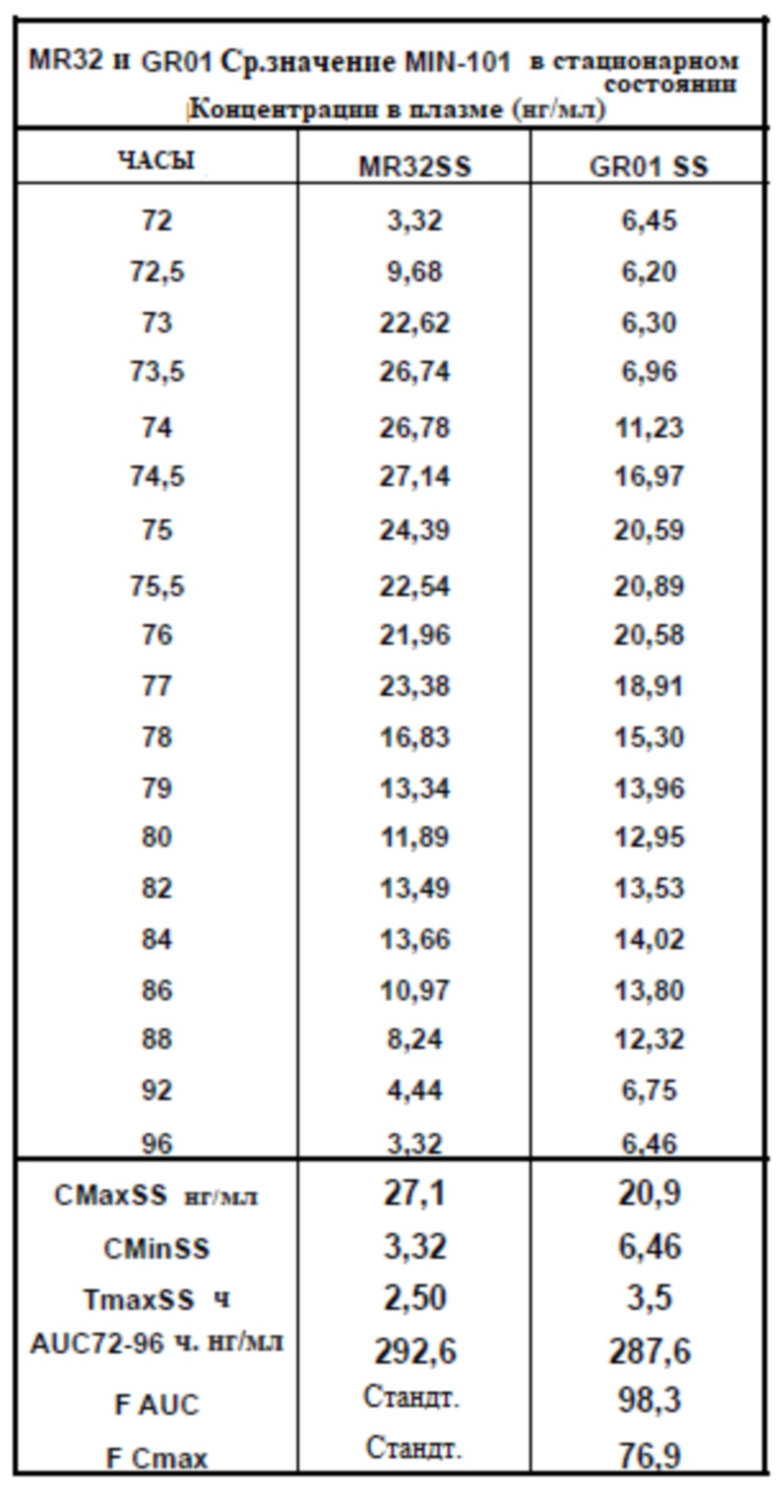
[00354] Смотрите Фиг. 14.
Таблица 24. Прогнозируемые концентрации в плазме BFB-520 в стационарном состоянии в таблетках MR32 и GR-01, 32 мг
72,5
73
73,5
74
74,5
75
75,5
76
77
78
79
80
82
84
86
88
92
96
0,44
0,81
1,15
1,35
1,48
1,70
1,71
1,89
1,74
1,66
1,54
1,66
1,53
1,26
1,41
1,03
0,85
0,50
0,63
0,60
0,57
0,58
0,72
0,97
1,10
1,42
1,27
1,21
1,24
1,46
1,31
1,29
1,25
1,19
1,03
0,66
CminSS
TmaxSS ч
AUC72-96 ч*нг/мл
F AUC
F Cmax
0,50
4,00
29,1
Стандт.
Стандт.
0,66
8,00
26,3
90,6
77,2
[00355] Смотрите Фиг. 15.
Таблица 25. Прогнозируемые концентрации в плазме MIN-101 в стационарном состоянии в таблетках MR32 и GR-01, 64 мг
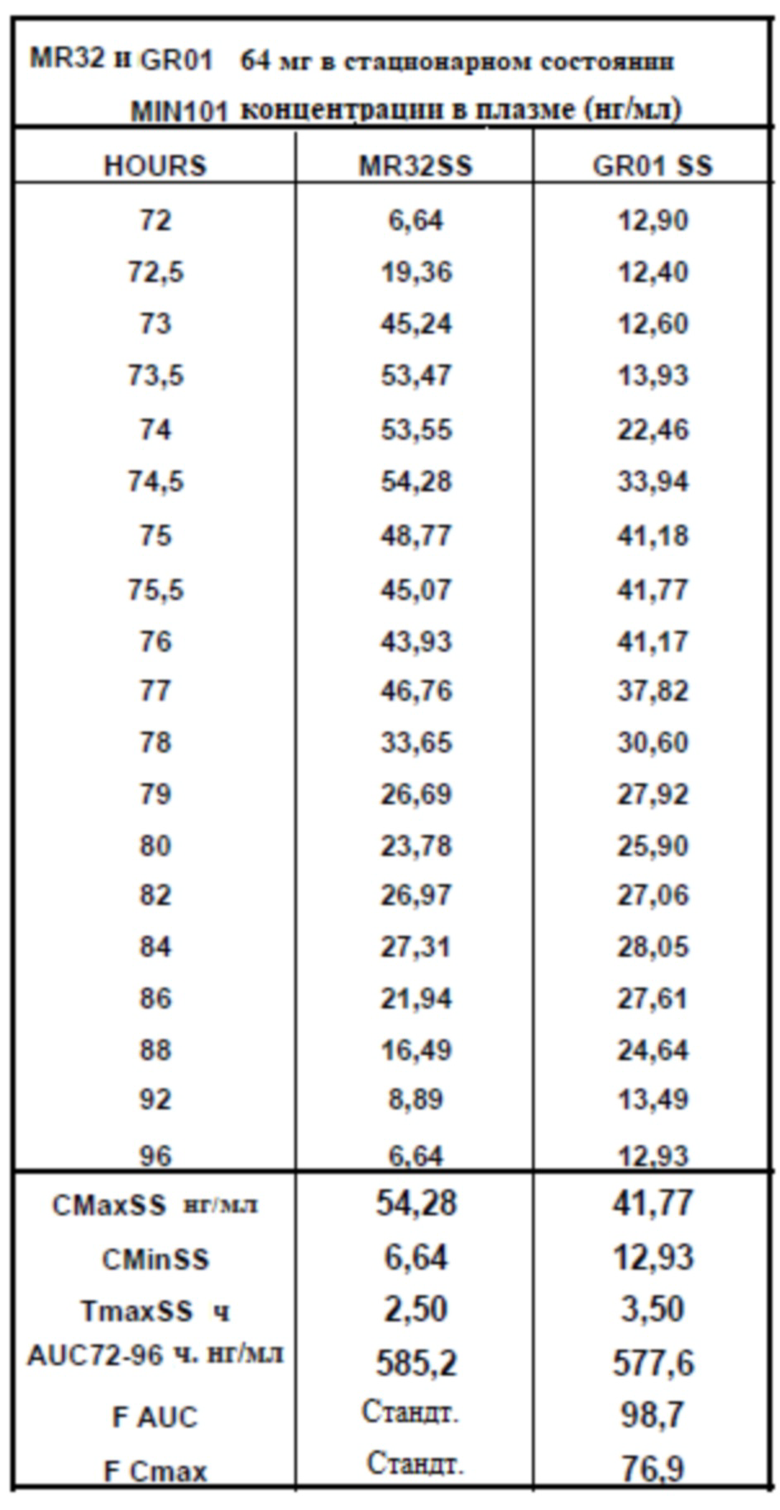
[00356] Смотрите Фиг. 16.
Таблица 26. Прогнозируемые концентрации в плазме BFB-520 в стационарном состоянии в таблетках MR32 и GR-01, 64 мг
BFB-520 концентрации в плазме (нг/мл)
72,5
73
73,5
74
74,5
75
75,5
76
77
78
79
80
82
84
86
88
92
96
0,88
1,63
2,31
2,69
2,96
3,40
3,41
3,79
3,48
3,33
3,07
3,32
3,05
2,51
2,83
2,05
1,70
0,99
1,27
1,20
1,13
1,16
1,45
1,93
2,19
2,84
2,54
2,41
2,48
2,92
2,62
2,57
2,49
2,37
2,05
1,32
CminSS
TmaxSS ч
AUC72-96 ч*нг/мл
F AUC
F Cmax
0,99
4,00
58,1
Стандт.
Стандт.
1,32
8,00
52,8
90,8
77,2
[00357] Смотрите Фиг. 17.
Пример 11: Прогнозируемые концентрации в плазме MIN-101 и BFB-520 в стационарном состоянии в таблетках MR32 и GR-02 (32 и 64 мг)
Таблица 27. Прогнозируемые концентрации в плазме MIN-101 в стационарном состоянии в таблетках MR32 и GR-02, 32 мг
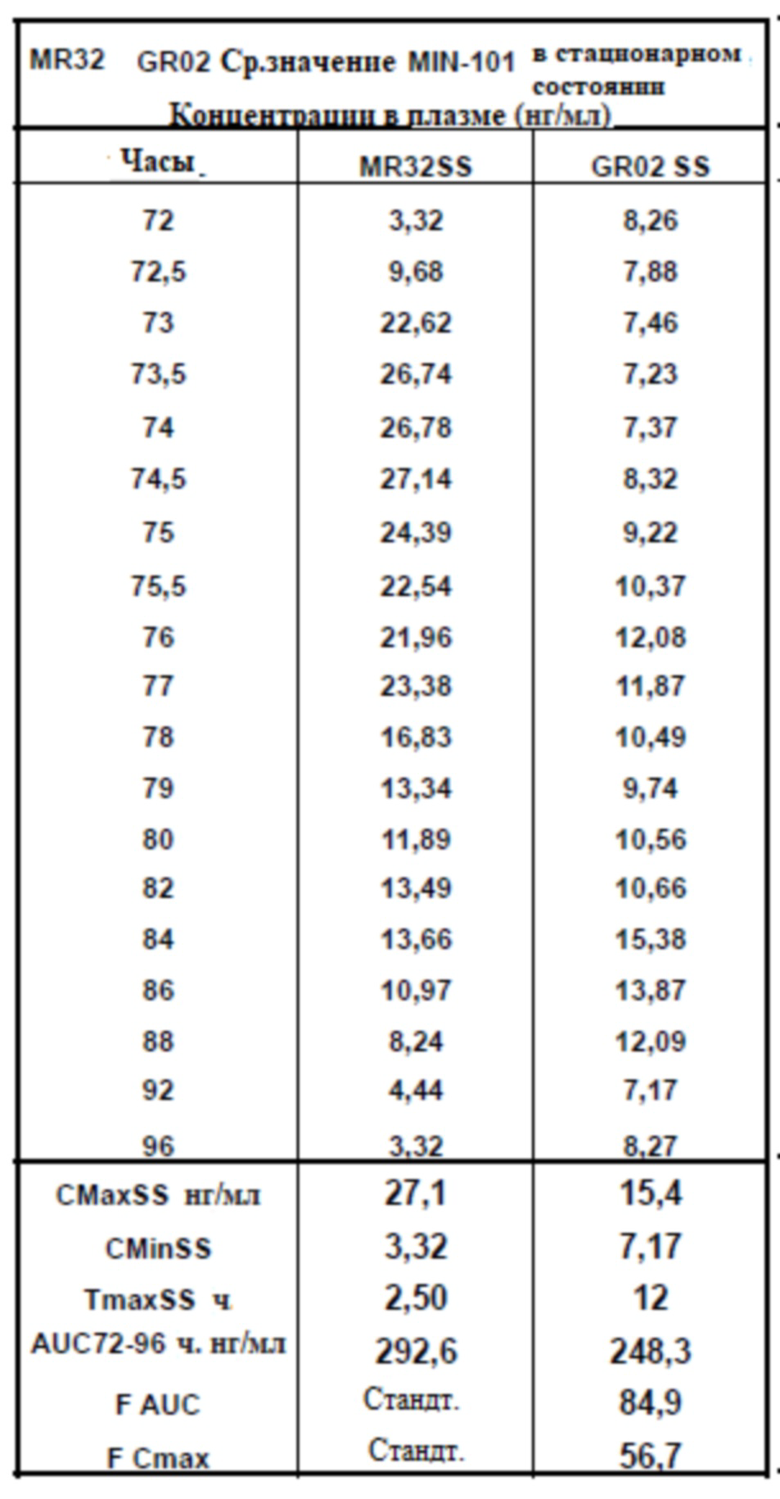
[00358] Смотрите Фиг. 18.
Таблица 28. Прогнозируемые концентрации в плазме BFB-520 в стационарном состоянии в таблетках MR32 и GR-02, 32 мг
Концентрации в плазме (нг/мл)
72,5
73
73,5
74
74,5
75
75,5
76
77
78
79
80
82
84
86
88
92
96
0,44
0,81
1,15
1,35
1,48
1,70
1,71
1,89
1,74
1,66
1,54
1,66
1,53
1,26
1,41
1,03
0,85
0,50
0,76
0,72
0,67
0,63
0,63
0,63
0,63
1,04
0,75
0,78
0,78
1,10
0,92
1,20
1,22
1,23
1,13
0,79
CminSS
TmaxSS ч
AUC72-96 ч*нг/мл
F AUC
F Cmax
0,50
4,00
29,1
Стандт.
Стандт.
0,79
16,00
23,6
81,4
64,8
[00359] Смотрите Фиг. 19.
Таблица 29. Прогнозируемые концентрации в плазме MIN-101 в стационарном состоянии в таблетках MR32 и GR-02, 64 мг
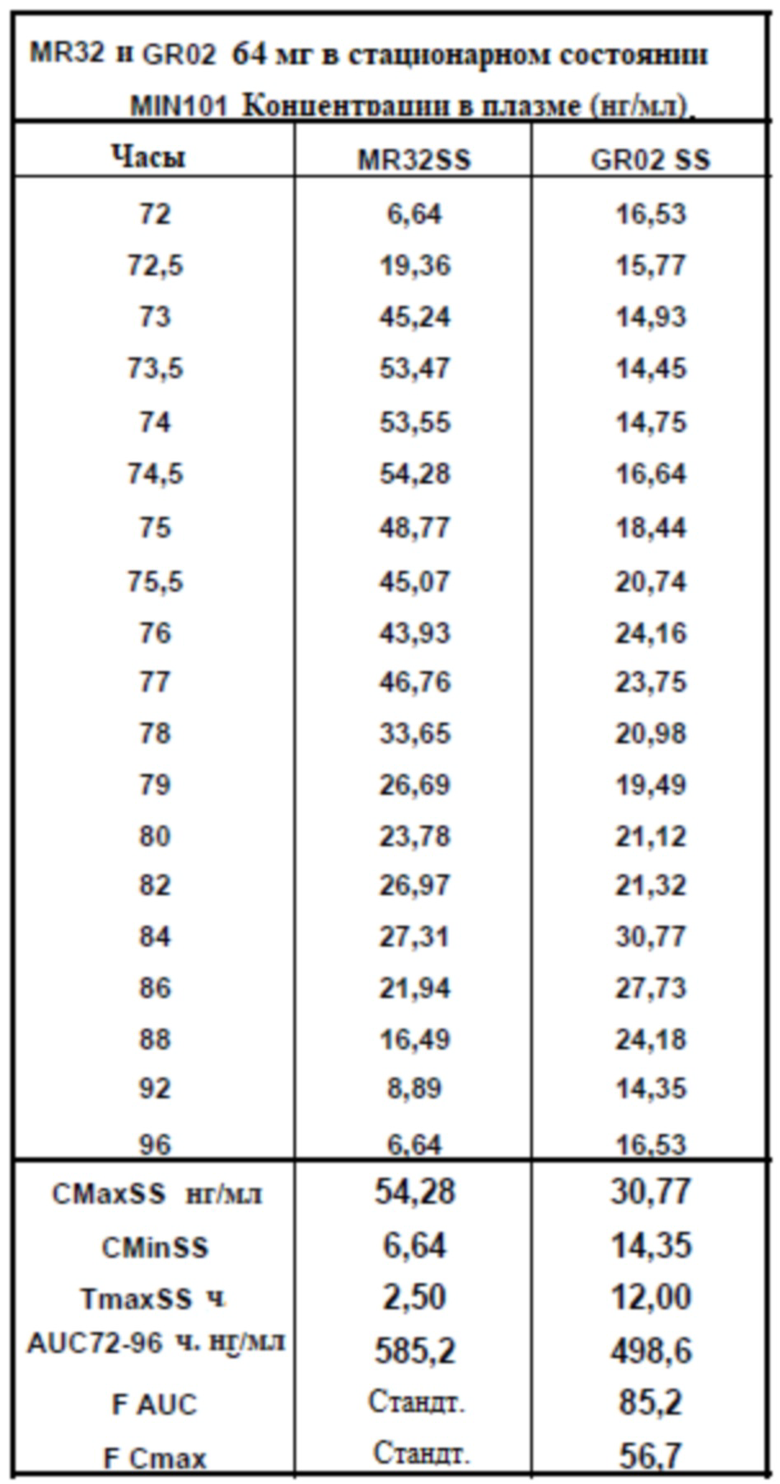
[00360] Смотрите Фиг. 20.
Таблица 30. Прогнозируемые концентрации в плазме BFB-520 в стационарном состоянии в таблетках MR32 и GR-02, 64 мг
BFB-520 концентрации в плазме (нг/мл)
72,5
73
73,5
74
74,5
75
75,5
76
77
78
79
80
82
84
86
88
92
96
0,88
1,63
2,31
2,69
2,96
3,40
3,41
3,79
3,48
3,33
3,07
3,32
3,05
2,51
2,83
2,05
1,70
0,99
1,52
1,43
1,35
1,27
1,26
1,26
1,27
2,07
1,50
1,57
1,57
2,20
1,83
2,40
2,44
2,;5
2,25
1,58
CminSS
TmaxSS ч
AUC72-96 ч*нг/мл
F AUC
F Cmax
0,99
4,00
58,1
Стандт.
Стандт.
1,58
16,00
47,4
81,6
64,8
[00361] Смотрите Фиг. 21.
[00362] Примеры 12-15 детально оценивают ФК профиль состава GR-01 у здоровых CYP2D6 EM мужчин и женщин после еды и натощак (или их прогнозы). Субъекты, которые завершили часть 1 исследования (оценка ФК профиля MIN-101 и его метаболита BFB-520 в составах GR-01, GR-02 и MR32) вернулись и получили дополнительную однократную пероральную дозу GR-01 после еды и натощак, что позволило оценить влияние пищи путем сравнения ФК свойств с теми, которые получены в части 1 (Примеры 9-12). Был введен период отмывки 14 ± 2 дня после части 1.
Пример 12: In Vivo Фармакокинетический анализ MIN-101 в таблетках CR GR-01 у субъектов после еды и натощак. (tau=72 h)
[00363] Натощак - 12 субъектов (перекрестн.)
[00364] Геом. среднее Cmax: 19,70 нг/мл
[00365] Tmax: 12,00 ч
AUC(0-tau): 269,19 нг · ч/мл
Таблица 31. CR GR-01 Индивидуальные концентрации и параметры MIN-101 в плазме (после еды)

Таблица 32. CR GR-01 Средние концентрации и параметры MIN-101 в плазме (после еды)
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,00
0,00
0,00
0,57
1,44
1,55
1,79
4,77
7,74
7,77
6,36
7,58
13,70
9,38
8,88
7,04
10,71
8,26
5,22
2,83
0,71
0,16
0,03
3,53
3,69
4,42
7,77
9,01
7,71
6,59
6,94
9,54
5,54
5,51
3,93
9,09
6,76
4,30
2,17
0,52
0,22
0,09
2,40
2,59
2,94
6,27
8,00
6,91
5,65
5,20
6,71
4,66
3,85
3,13
6,31
5,27
3,50
1,79
0,39
0,19
0,05
245,26
238,14
246,80
162,90
116,41
99,30
103,62
91,54
69,63
59,09
62,03
55,89
84,89
81,78
82,42
76,52
73,23
133,11
346,41
Tmax, ч
AUC0-t ч*нг/мл
F AUC
F Cmax
12,50
269,19
95,14
108,97
7,65
107,53
36,15
31,40
6,00
86,11
28,68
23,32
61,16
39,95
38,00
28,81
[00366] Смотрите Фиг. 22.
[00367] Натощак - 12 субъектов (перекрестн.)
[00368] Геом. среднее Cmax: 18,82 нг/мл
[00369] Медиана Tmax: 4,50 ч
AUC(0-tau): 284,52 нг · ч/мл
Таблица 33. CR GR-01 Индивидуальные концентрации и параметры MIN-101 в плазме (натощак)
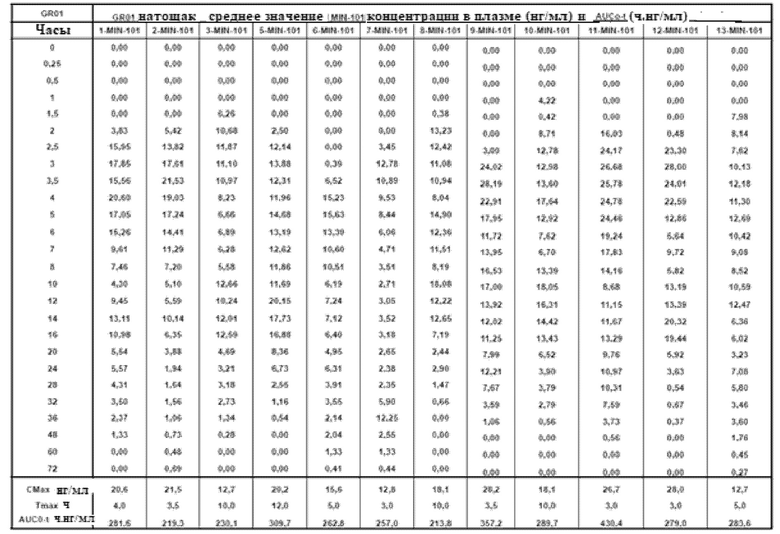
Таблица 34. CR GR-01 Средние концентрации и параметры MIN-101 в плазме (натощак)
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,35
1,25
5,75
11,71
15,54
16,04
15,99
14,62
11,35
10,33
9,39
10,69
11,26
11,76
10,58
5,49
5,57
3,96
3,10
2,42
0,77
0,30
0,15
0,00
0,00
1,22
2,77
5,57
7,46
7,84
7,01
6,08
4,60
4,17
3,58
3,93
5,39
4,66
4,67
4,87
2,33
3,30
2,80
2,06
3,33
0,92
0,51
0,24
0,00
0,00
0,65
1,95
4,67
5,46
6,08
5,89
5,27
3,26
3,35
2,64
3,25
4,42
3,48
3,33
3,96
1,85
2,58
2,04
1,50
2,05
0,77
0,40
0,20
0,00
0,00
346,41
220,78
96,91
63,69
50,45
43,68
38,00
31,47
36,70
34,63
41,81
50,45
41,34
39,73
46,01
42,48
59,31
70,67
66,68
137,50
119,51
171,48
159,83
Tmax, ч
AUC0-t ч*нг/мл
6,00
284,52
3,43
60,83
3,00
41,49
57,19
21,38
[00370] Смотрите Фиг. 23.
Таблица 35. CR GR-01 Средние концентрации и параметры MIN-101 в плазме (после еды по сравнению с натощак)
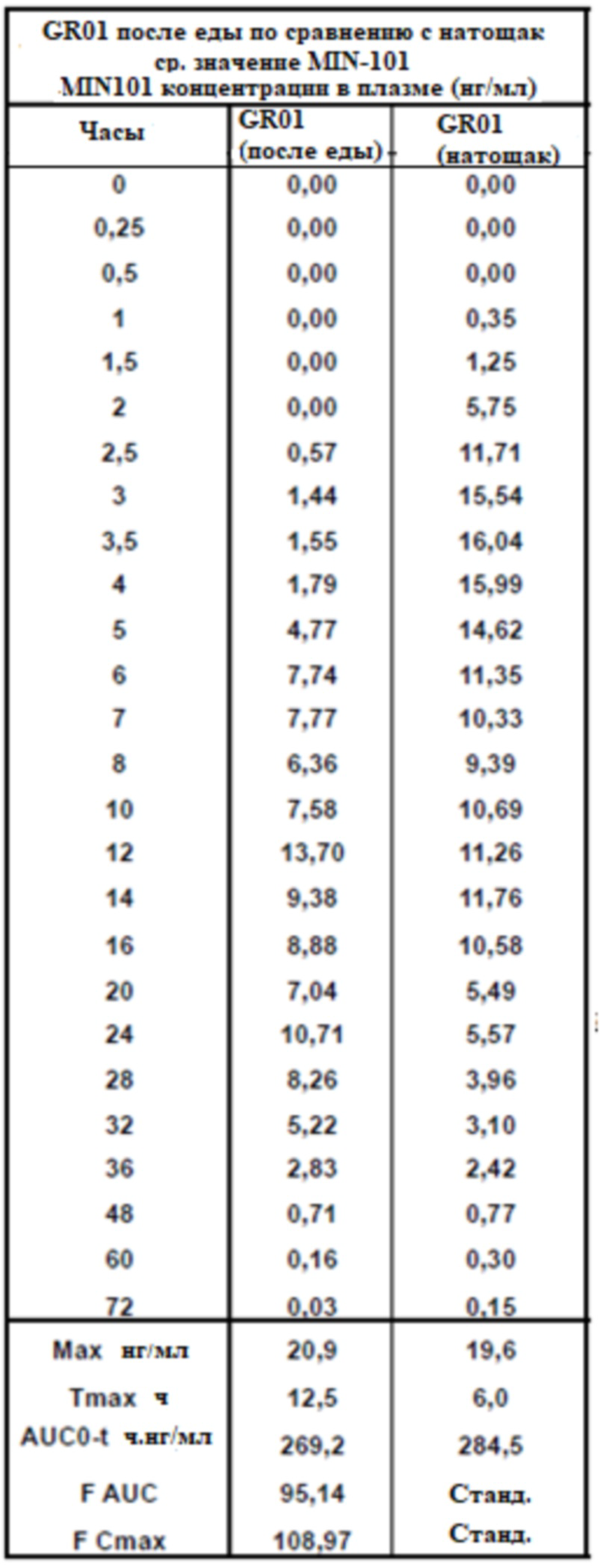
[00371] Относительная биодоступность MIN-101 в составе GR-01 после еды по сравнению с натощак:
F% Cmax: 108,97%
F% AUC(0-tau): 95,14%
[00372] Смотрите Фиг. 24.
Пример 13: In Vivo Фармакокинетический анализ BFB-520 в таблетках CR GR-01 у субъектов после еды и натощак. (tau=72 h)
[00373] Натощак - 12 субъектов (перекрестн.)
[00374] Геом. среднее Cmax: 1,54 нг/мл
[00375] Медиана Tmax: 18,00 ч
AUC(0-tau): 30,12 нг · ч/мл
Таблица 36. CR GR-01 Индивидуальные концентрации и параметры BFB-520 в плазме (после еды)

Таблица 37. CR GR-01 Средние концентрации и параметры BFB-520 в плазме (после еды)
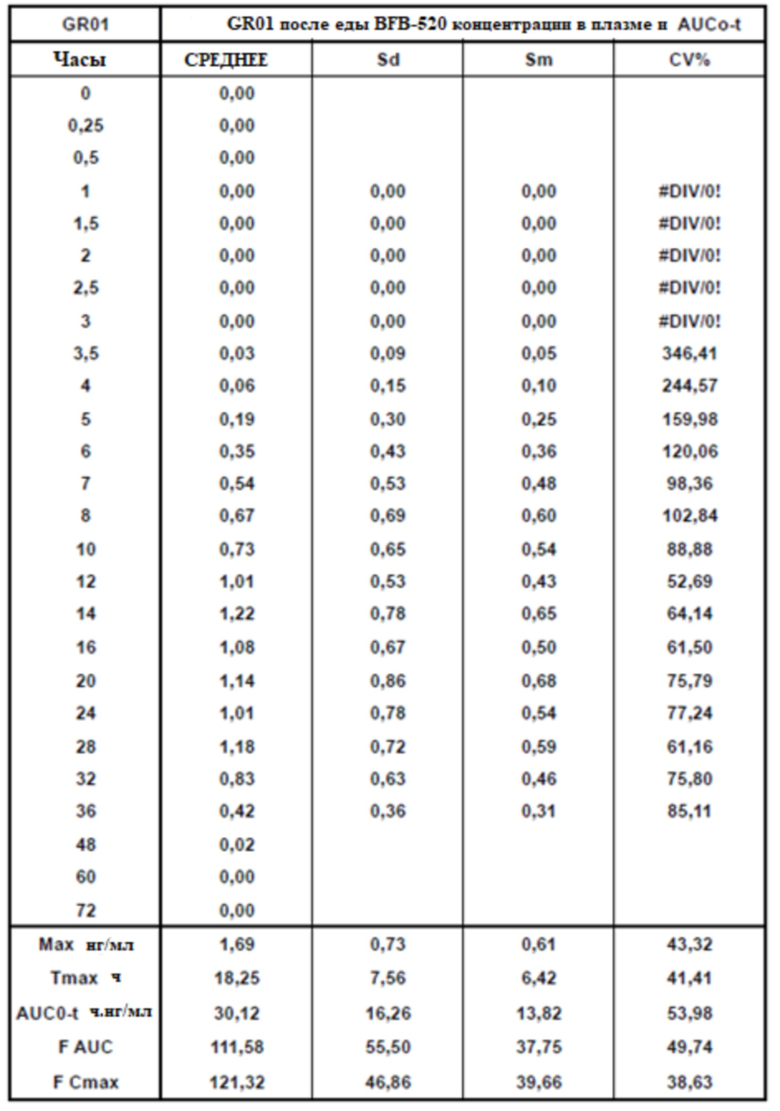
[00376] Смотрите Фиг. 25.
[00377] Натощак - 12 субъектов (перекрестн.)
[00378] Геом. среднее Cmax: 1,32 нг/мл
[00379] Медиана Tmax: 12,5 ч
AUC(0-tau): 27,48 нг · ч/мл
Таблица 38. CR GR-01 Индивидуальные концентрации и параметры BFB-520 в плазме (натощак)

Таблица 39 CR GR-01 Средние концентрации и параметры BFB-520 в плазме (натощак)

[00380] Смотрите Фиг. 26.
Таблица 40. CR GR-01 Средние концентрации и параметры BFB-520 в плазме (после еды по сравнению с натощак)
0,25
0,5
1
1,5
2
2,5
3
3,5
4
5
6
7
8
10
12
14
16
20
24
28
32
36
48
60
72
0,00
0,00
0,00
0,00
0,04
0,22
0,49
0,65
0,77
0,89
0,87
0,94
1,07
1,10
1,04
1,11
1,08
0,96
0,63
0,63
0,37
0,25
0,03
0,00
0,00
0,25
0,23
0,35
0,36
0,36
0,29
0,35
0,39
0,45
0,55
0,42
0,45
0,43
0,36
0,36
0,23
0,33
0,09
0,22
0,17
0,27
0,31
0,29
0,22
0,28
0,29
0,36
0,43
0,31
0,34
0,38
0,28
0,26
0,18
0,25
0,05
112,01
45,99
53,37
47,35
40,77
33,50
37,58
36,81
40,79
53,43
37,58
41,52
45,29
57,32
57,90
60,48
133,52
346,41
Tmax, ч
AUC0-t ч*нг/мл
F AUC
F Cmax
12,50
27,48
96,10
80,48
8,04
9,46
35,77
24,60
5,08
8,07
18,95
20,65
64,32
34,41
37,22
30,56
[00381] Относительная биодоступность BFB-520 в составе GR-01 после еды по сравнению с натощак:
F% Cmax: 121,32%
F% AUC(0-tau): 111,58%
[00382] Смотрите Фиг. 27.
Пример 14: Прогнозируемое влияние пищи на плазменные концентрации MIN-101 и BFB-520 в стационарном состоянии в таблетках GR-01 (32 мг)
[00383] Определение наклонов выведения по временным кривым средних концентраций в плазме перед численными расчетами.
[00384] MIN-101
После еды: Ke=0,119/ч
Натощак: Ke=0,082/ч
[00385] BFB-520:
После еды: от 14 до 28 ч во время фазы триггера Ke=0,014/ч; постпоглощение Ke=0,233/ч
Натощак: от 14 до 28 ч во время фазы триггера Ке=0,005545/ч; постпоглощение Ke=0,1586/ч
Таблица 41. Прогнозируемые концентрации в плазме MIN-101 в стационарном состоянии в таблетках GR-01, 32 мг (после еды и натощак)

[00386] Смотрите Фиг. 28.
Таблица 42. Прогнозируемые концентрации в плазме BFB-520 в стационарном состоянии в таблетках GR-01, 32 мг (после еды и натощак)
BFB-520 концентрации в плазме (нг/мл)
72,5
73
73,5
74
74,5
75
75,5
76
77
78
79
80
82
84
86
88
92
96
1,02
1,01
1,00
0,99
0,99
0,98
1,00
1,25
1,13
1,29
1,46
1,51
1,26
1,43
1,43
1,21
1,19
1,03
0,64
0,62
0,60
0,63
0,79
1,04
1,18
1,41
1,38
1,33
1,38
1,45
1,37
1,29
1,26
1,19
1,02
0,66
CminSS
TmaxSS ч
AUC72-96 ч*нг/мл
F AUC
F Cmax
1,03
8,00
29,5
109,4
103,7
0,66
8,00
27,0
Стандт.
Стандт.
[00387] Смотрите Фиг. 29.
Пример 15: Прогнозируемое влияние пищи на плазменные концентрации MIN-101 и BFB-520 в стационарном состоянии в таблетках GR-01 (64 мг)
[00388] Определение наклонов выведения по временным кривым средних концентраций в плазме перед численными расчетами.
[00389] MIN-101
После еды: Ke=0,119/ч
Натощак: Ke=0,082/ч
[00390] BFB-520:
После еды: от 14 до 28 ч во время фазы триггера Ke=0,014/ч; постпоглощение Ke=0,233/ч
Натощак: от 14 до 28 ч во время фазы триггера Ке=0,005545/ч; постпоглощение Ke=0,1586/ч
Таблица 43. Прогнозируемые концентрации в плазме MIN-101 в стационарном состоянии в таблетках GR-01, 64 мг (после еды и натощак)
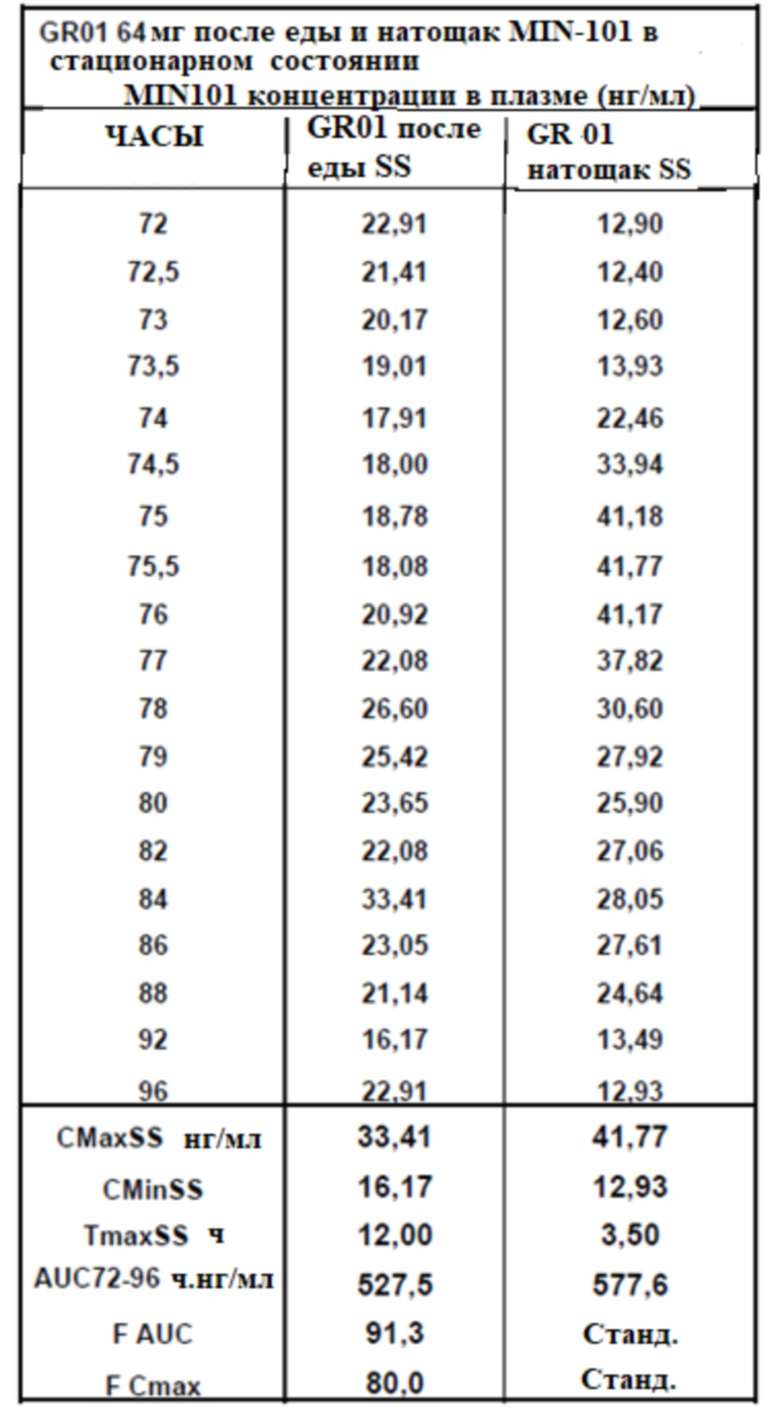
[00391] Смотрите Фиг. 30.
Таблица 44. Прогнозируемые концентрации в плазме BFB-520 в стационарном состоянии в таблетках GR-01, 64 мг (после еды и натощак)
BFB-520 концентрации в плазме (нг/мл)
72,5
73
73,5
74
74,5
75
75,5
76
77
78
79
80
82
84
86
88
92
96
2,04
2,02
2,00
1,99
1,97
1,95
1,99
2,50
2,27
2,57
2,92
3,01
2,51
2,87
2,86
2,42
2,38
2,06
1,28
1,24
1,21
1,26
1,58
2,09
2,37
2,82
2,77
2,66
2,76
2,91
2,75
2,57
2,52
2,37
2,03
1,32
CminSS
TmaxSS ч
AUC72-96 ч*нг/мл
F AUC
F Cmax
2,06
8,00
59,0
109,1
103,7
1,32
8,00
54,1
Стандт.
Стандт.
[00392] Смотрите Фиг. 31.
Пример 16: Описание 32 мг кишечнорастворимой таблетки CR (GR01/B-32 мг)
[00393] Таблетки CR GR-01/B поставляются в виде круглых (диаметр 10 мм и R=10) таблеток без видимых дефектов. Каждая таблетка содержит 32 мг Соединения (I). Полное описание компонентов и количественного состава таблеток CR GR-01/B приведено в Таблице 45.
Таблица 45: Состав таблеток CR GR-01/B 32 мг
(METHOCEL™ K100LV CR)
(Methocel™ K100M CR)
1Применяется поправочный коэффициент 1,2 на соль
Пример 17: Описание 64 мг кишечнорастворимой таблетки CR (GR-01/B-64 мг)
[00394] Таблетки CR GR-01/B поставляются в виде круглых (диаметр 10 мм и R=10) таблеток без видимых дефектов. Каждая таблетка содержит 64 мг Соединения (I). Полное описание компонентов и количественного состава таблеток CR GR-01/B приведено в Таблице 46.
Таблица 46: Состав таблетки CR GR-01/B 64 мг
(METHOCEL™ K100LV CR)
(Methocel™ K100M CR)
1Применяется поправочный коэффициент 1,2 на соль
Пример 18: Сравнение таблеток GR-01 32 мг, таблеток GR-01/B 32 мг и таблеток GR-01/B 64 мг
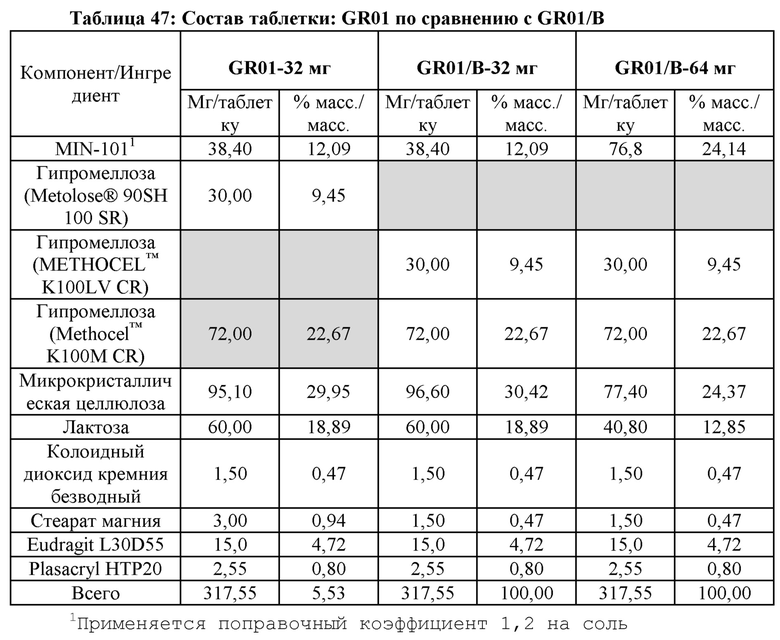
Пример 19: Данные экспериментов по стабильности: Сравнение таблеток GR-01 и GR-01/B 32 мг
[00395] «Примесь A», «2-изомер» и «PMIC» относятся к примесям, полученным во время производства MIN-101.
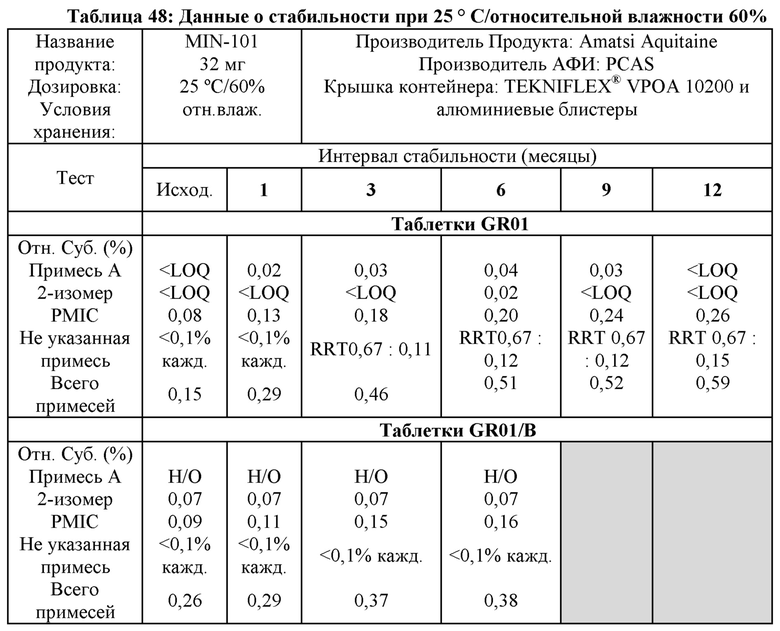
Таблица 49: Данные о стабильности при 40°C/относительной влажности 75%
Дозировка:
Условия хранения:
32 мг
40 ºC/75% отн.влаж.
Производитель АФИ: PCAS
Крышка контейнера: TEKNIFLEX® VPOA 10200 и алюминиевые блистеры
Пример 20: In vitro растворимость для таблеток GR-01/B
Таблица 50: Технические характеристики для таблеток GR-01/B
(%)
2 ч
4 ч
8 ч
16 ч
0,0%
24,1%
59,2%
88,6%
Пример 21: Описание 32 мг кишечнорастворимой таблетки CR (GR-01/C-32 мг)
[00396] Таблетки CR GR-01/C поставляются в виде круглых (диаметр 10 мм и R=10) таблеток без видимых дефектов. Каждая таблетка содержит 32 мг Соединения (I). Полное описание компонентов и количественного состава таблеток CR GR-01/C приведено в Таблице 51.
Таблица 51: Состав таблеток CR GR-01/C 32 мг
(Methocel™ K100LV CR)
(Methocel™ K100M CR)
Пример 22: Описание 64 мг кишечнорастворимой таблетки CR (GR-01/C-64 мг)
[00397] Таблетки CR GR-01/C поставляются в виде круглых (диаметр 10 мм и R=10) таблеток без видимых дефектов. Каждая таблетка содержит 64 мг Соединения (I). Полное описание компонентов и количественного состава таблеток CR GR-01/C приведено в Таблице 53.
Таблица 53: Состав таблеток CR GR-01/C 64 мг
(Methocel™ K100LV CR)
(Methocel™ K100M CR)
ЭКВИВАЛЕНТЫ И ВКЛЮЧЕНИЯ ССЫЛОК
[00398] Дозированные формы, композиции и способы по данному изобретению были описаны в данном документе со ссылкой на некоторые предпочтительные варианты реализации. Тем не менее, поскольку конкретные изменения по ним станут очевидными для специалистов в данной области техники, основываясь на изложенном в данном документе описании, описание не следует рассматривать как ограниченное представленным в данном документе.
[00399] Если не указано иное, все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понимается специалистом в области техники, к которой относится это описание. В описании и формуле изобретения формы единственного числа также включают множественное число, если контекст явно не предписывает иное.
[00400] Следует понимать, что, по меньшей мере, некоторые из описаний раскрытия были упрощены, чтобы сосредоточиться на элементах, которые имеют отношение к ясному пониманию описания, в то же время исключая, для ясности, другие элементы, которые обычные специалисты в данной области техники оценят, и могут также включать часть описания. Однако, поскольку такие элементы хорошо известны в данной области техники и поскольку они не обязательно облегчают лучшее понимание описания, описание таких элементов в данном документе не приводится.
[00401] Кроме того, в той степени, в которой способ не основывается на конкретном порядке этапов, изложенных в данном документе, конкретный порядок этапов, изложенных в формуле изобретения, не должен рассматриваться как ограничение этой формулы.
[00402] Все патенты, патентные заявки, ссылки и публикации, процитированные в данном документе, полностью и в полном объеме включены в качестве ссылки, как если бы они были изложены полностью. Такие документы не являются предшествующим уровнем техники по отношению к настоящему описанию.

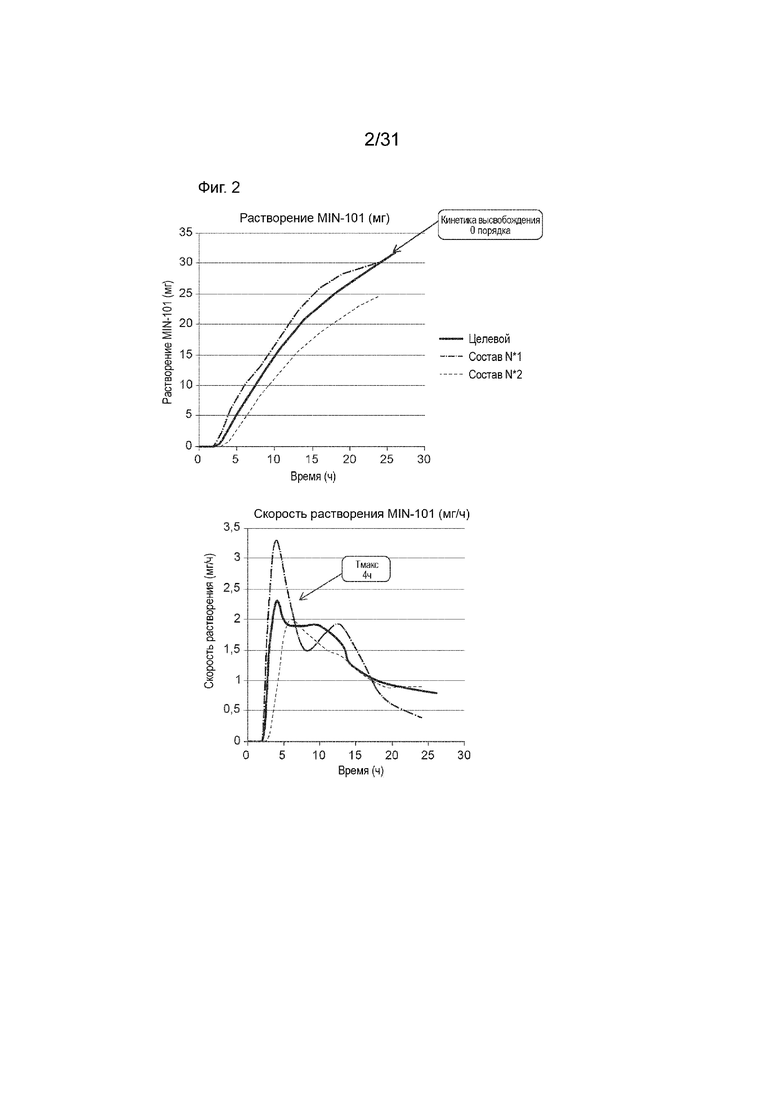
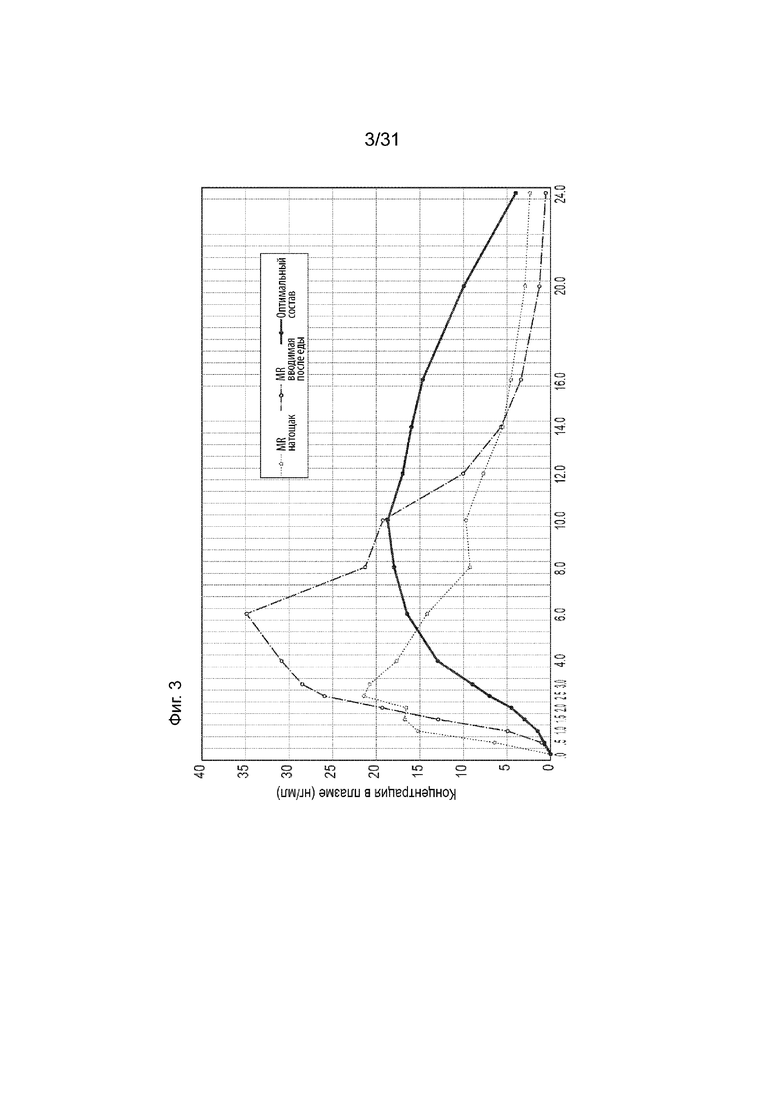
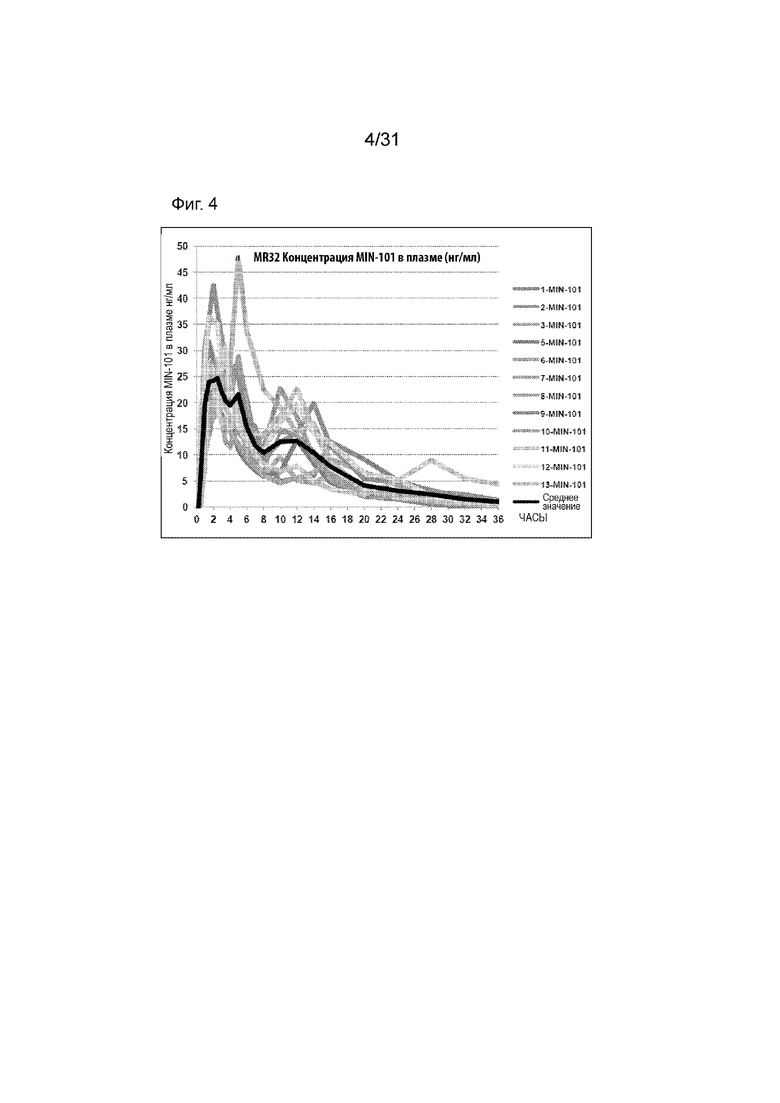
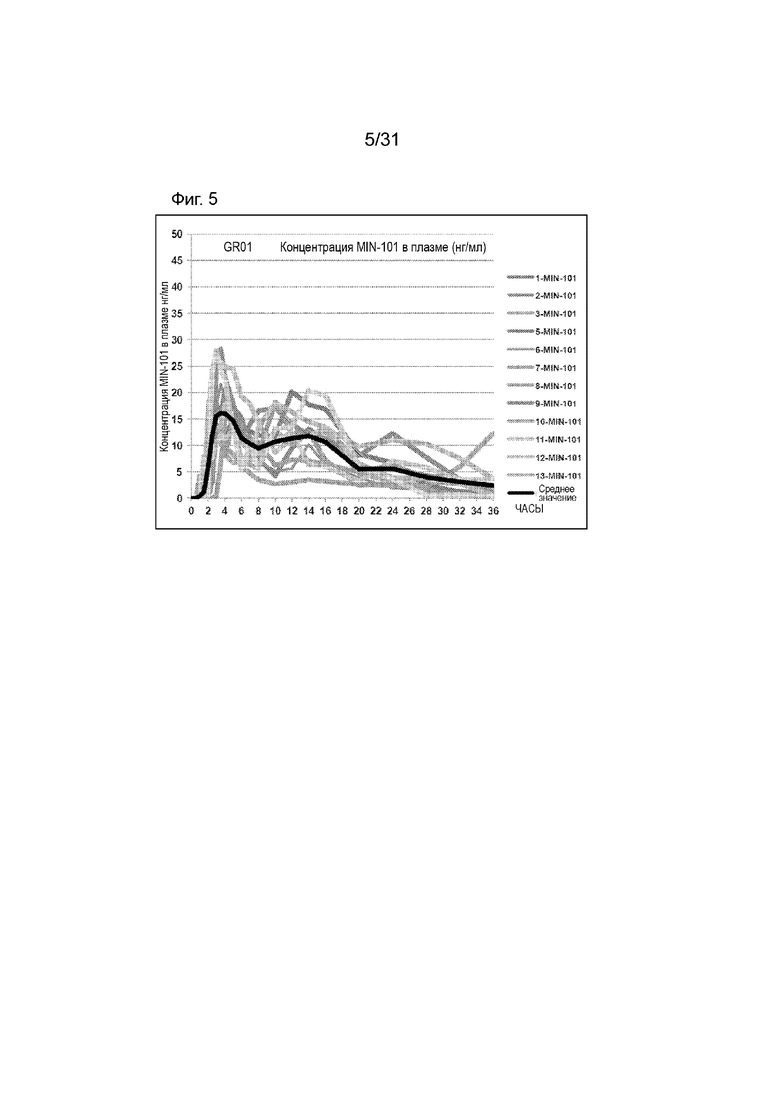

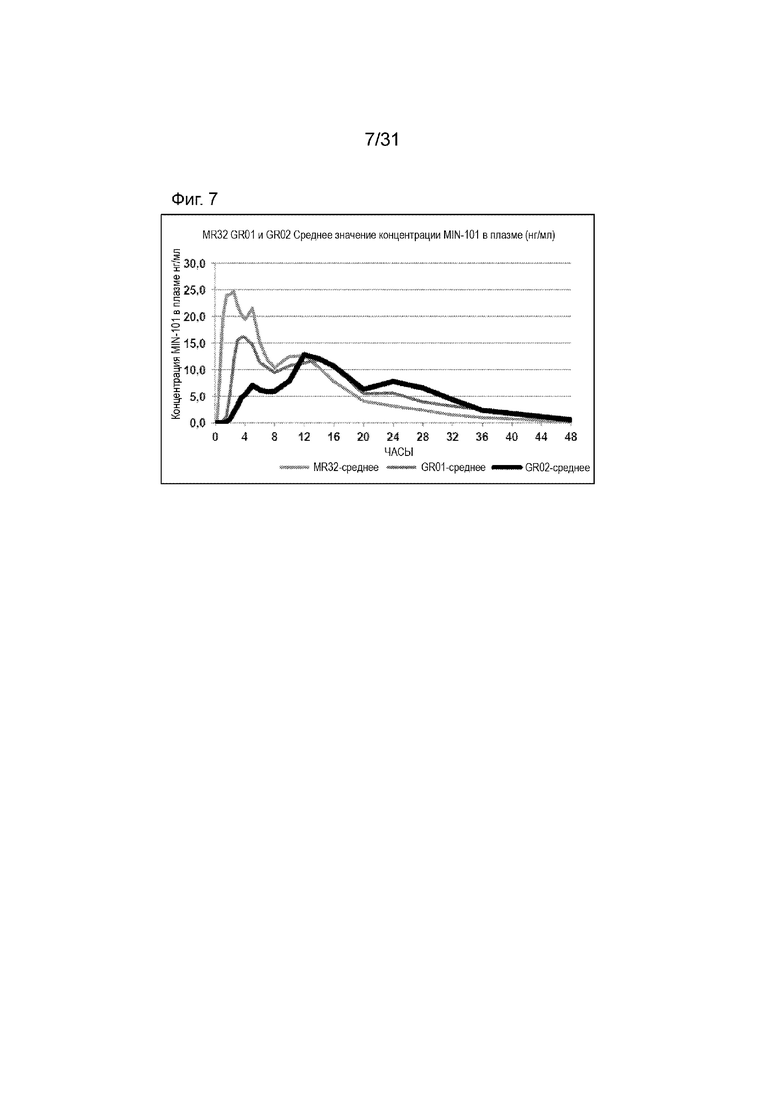
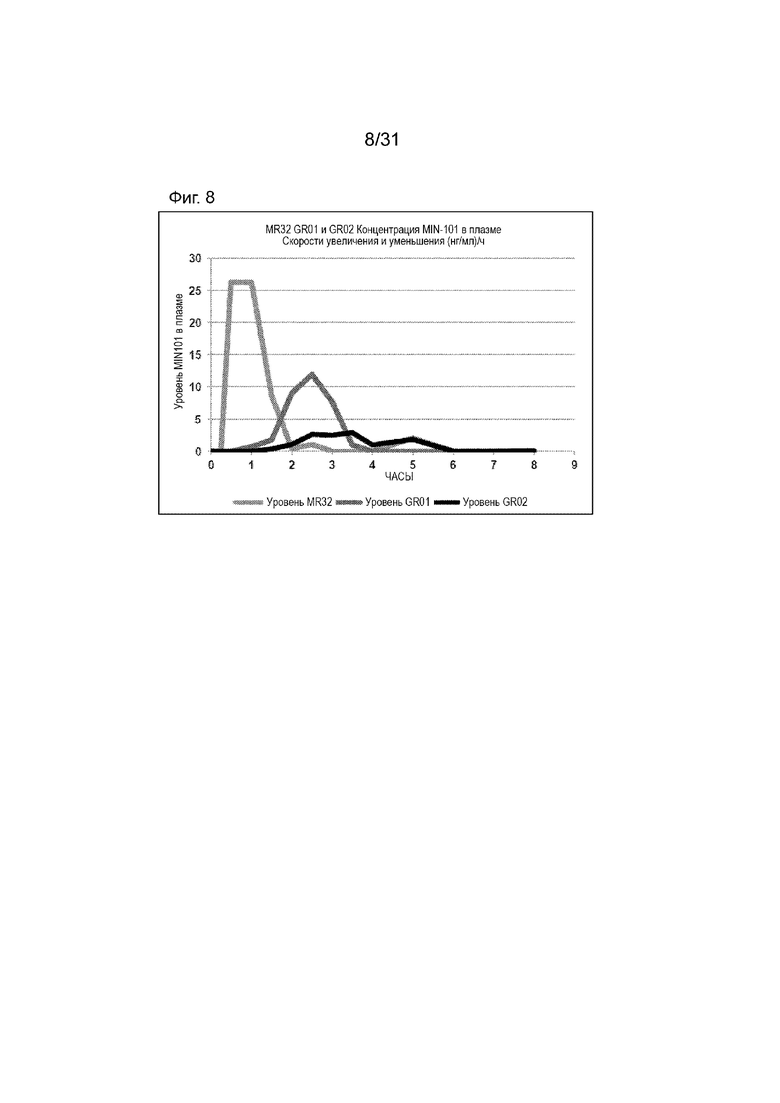
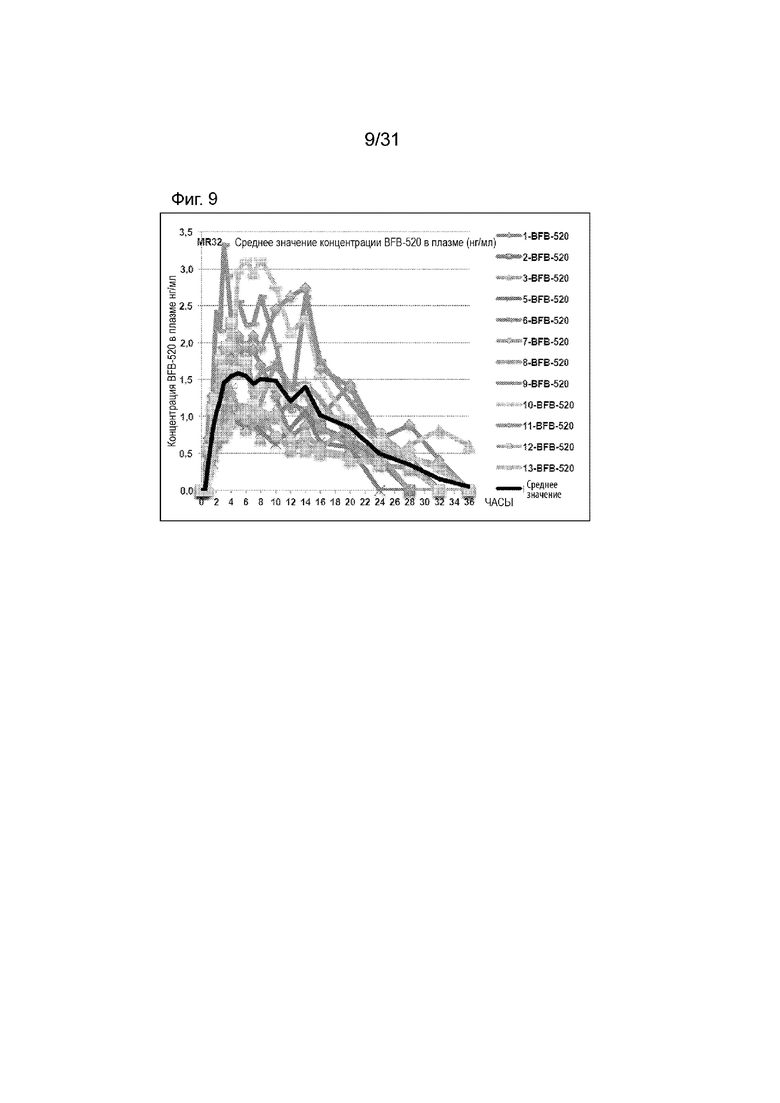

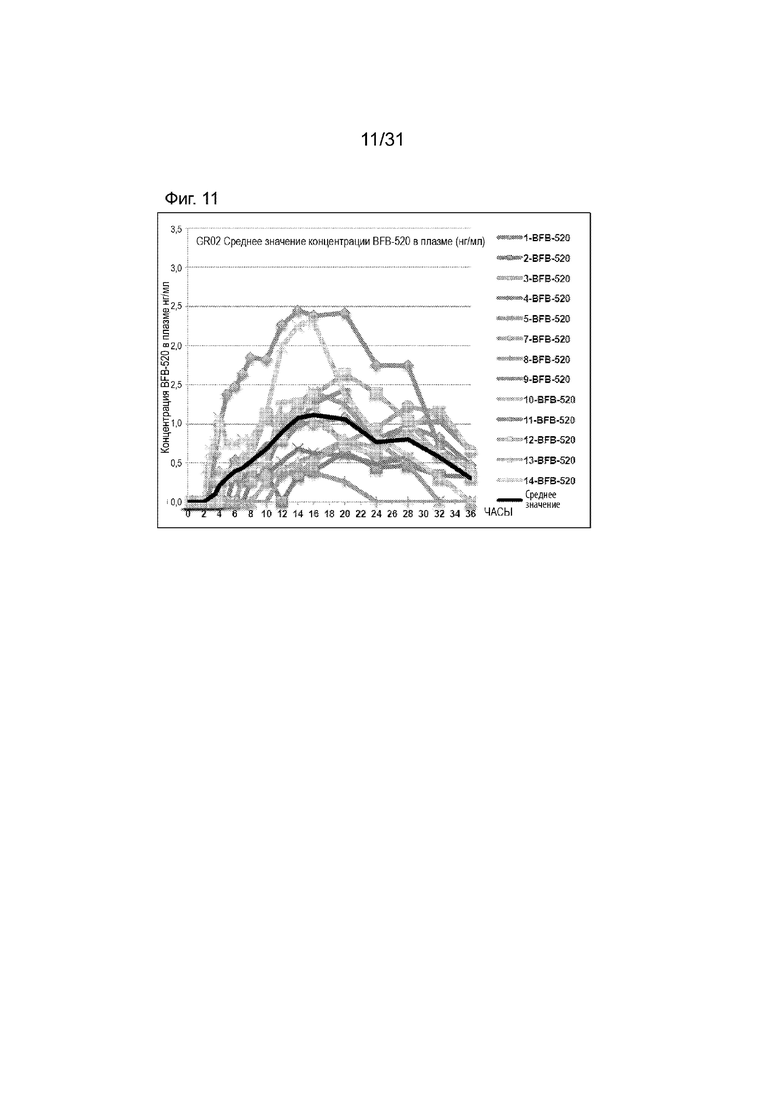

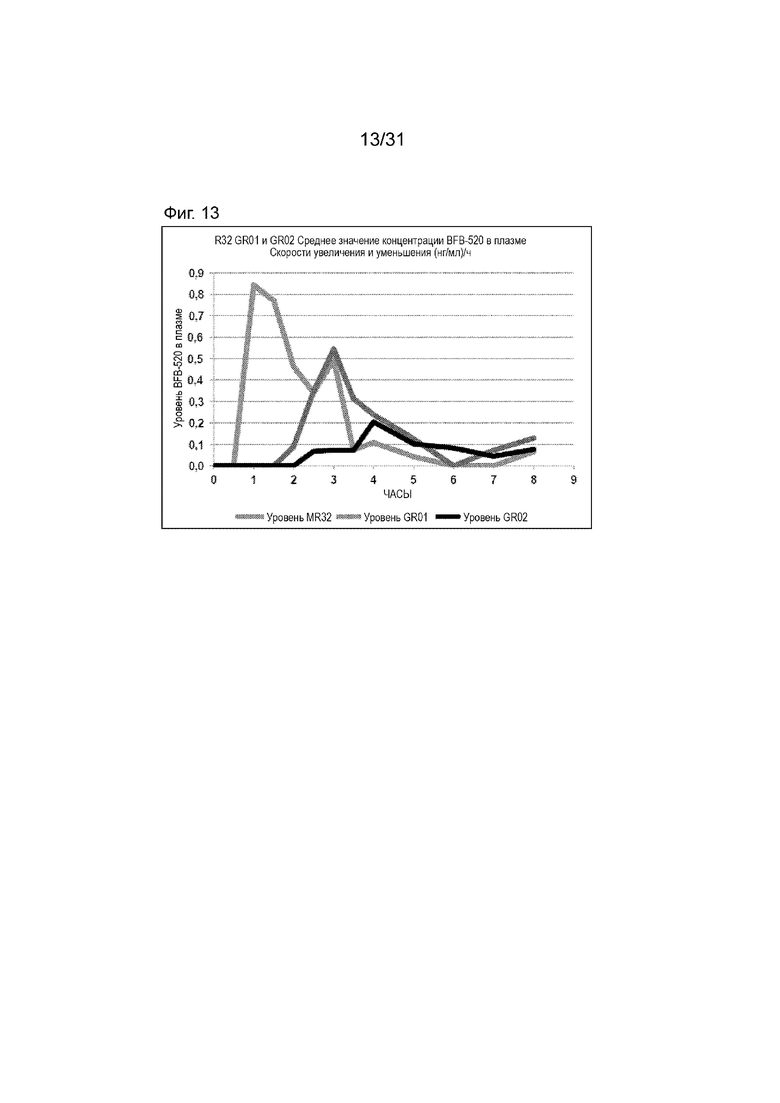
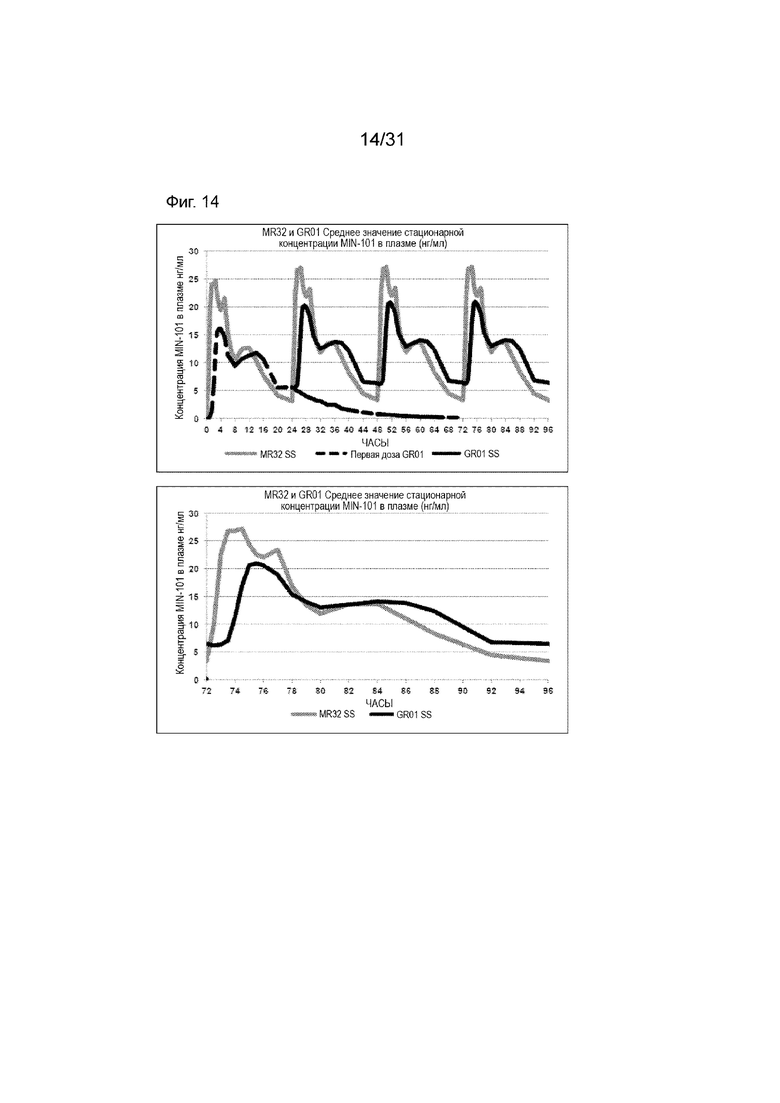
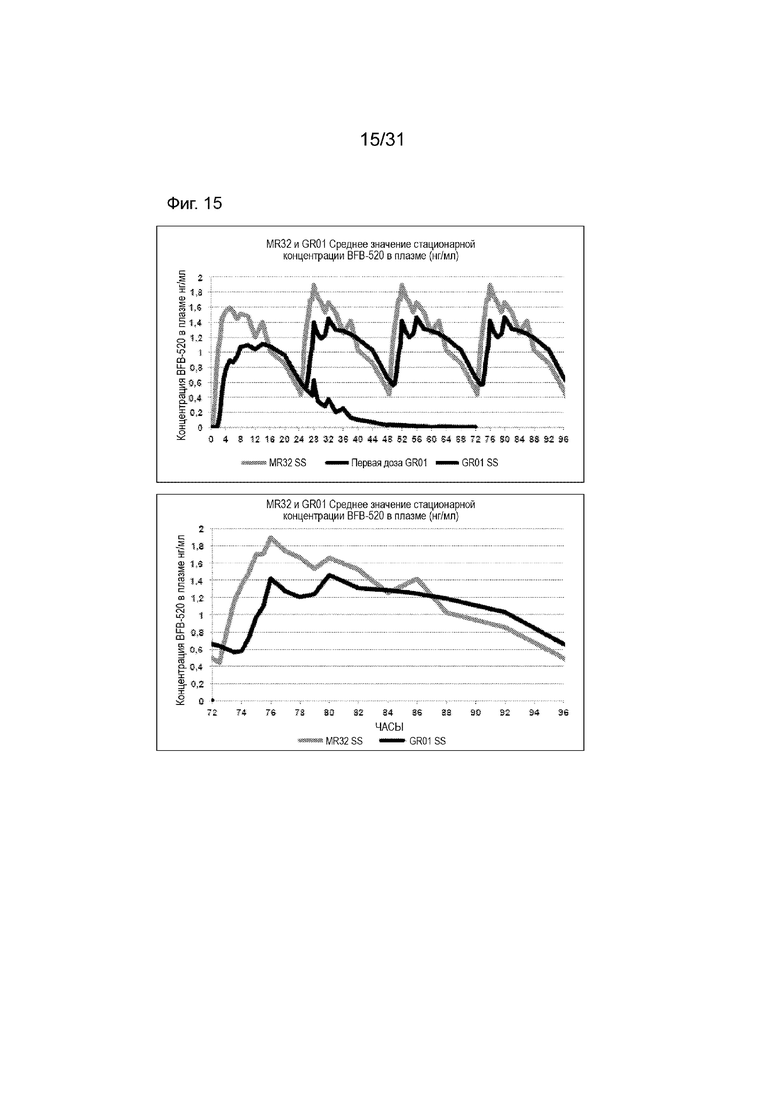
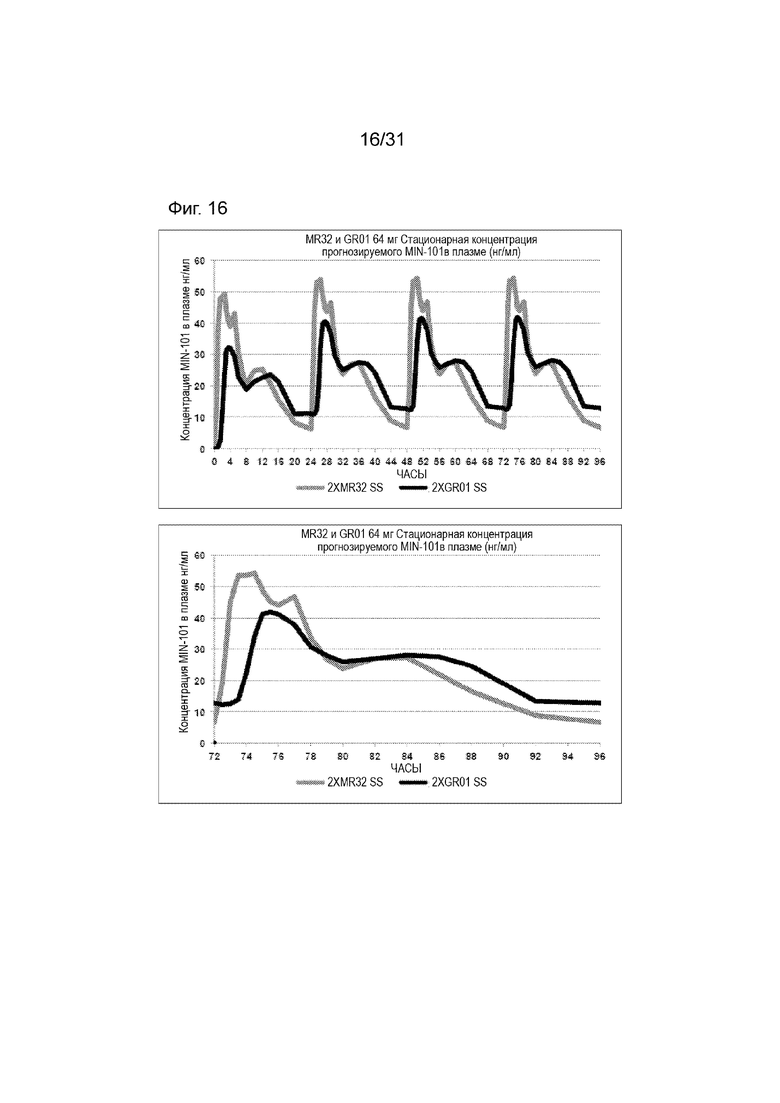

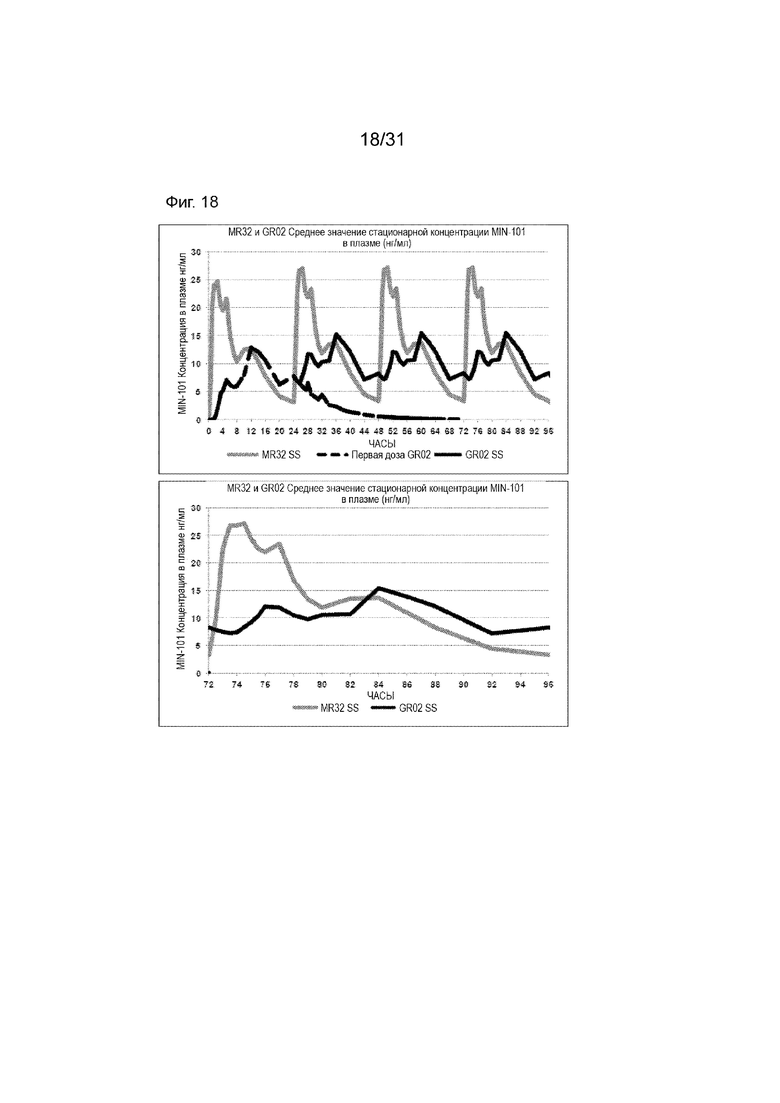
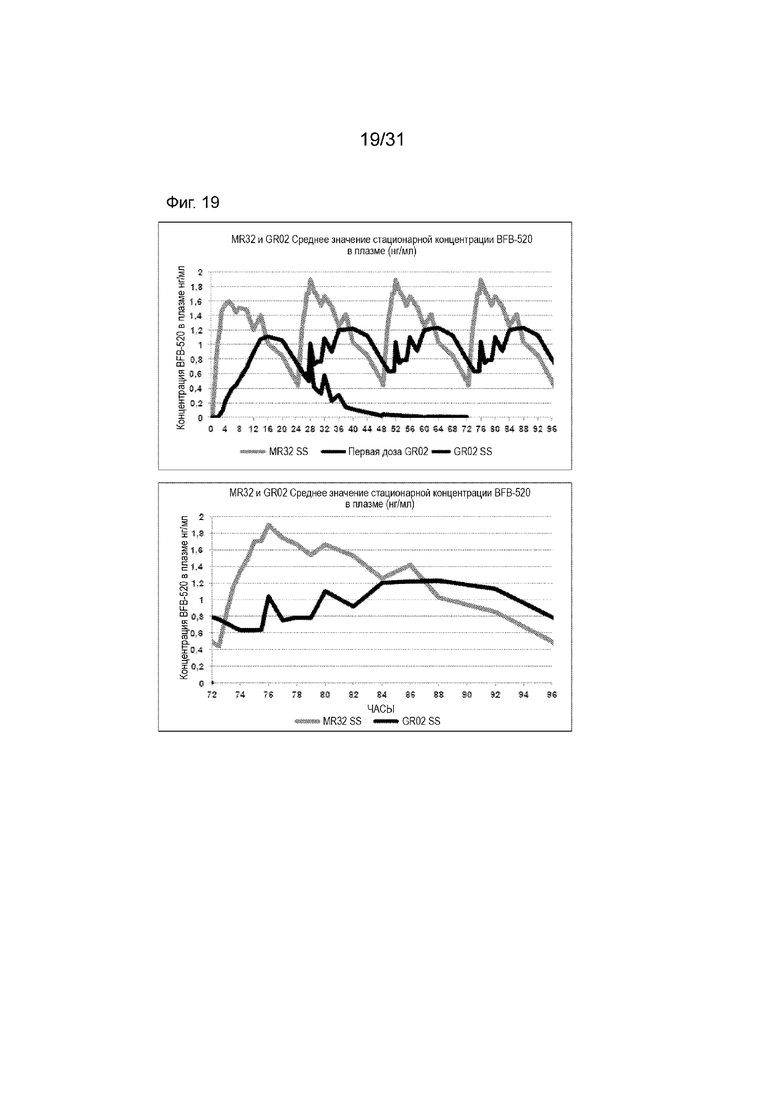
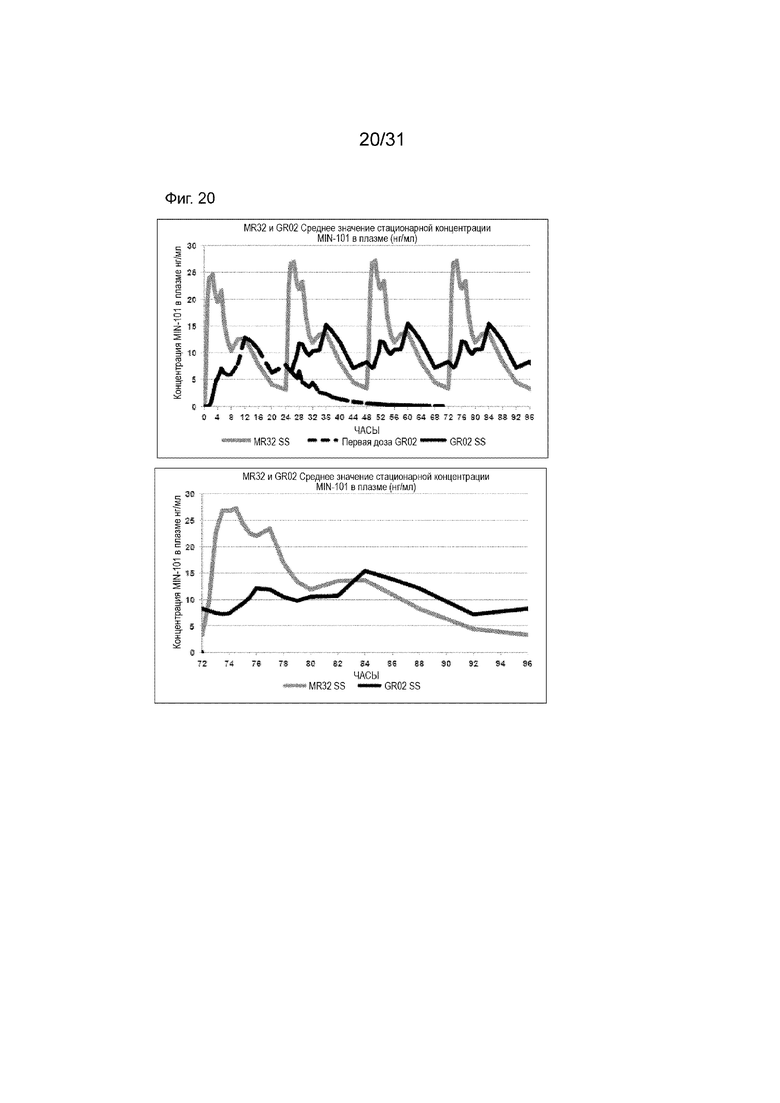







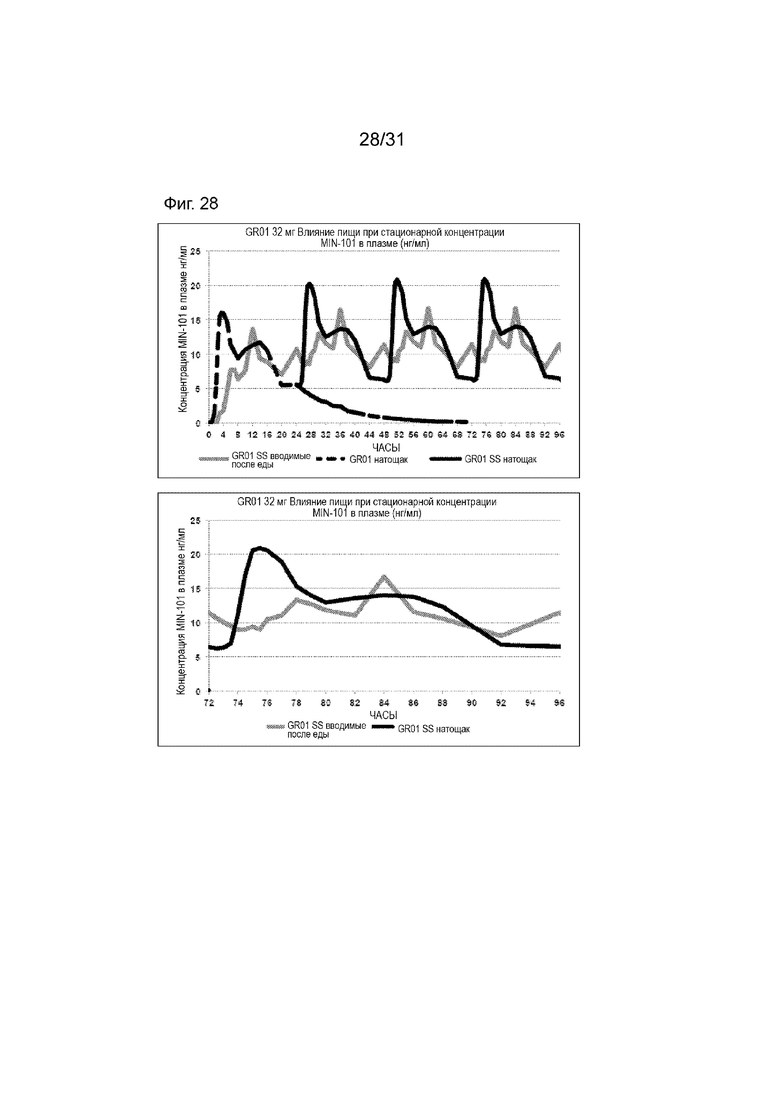
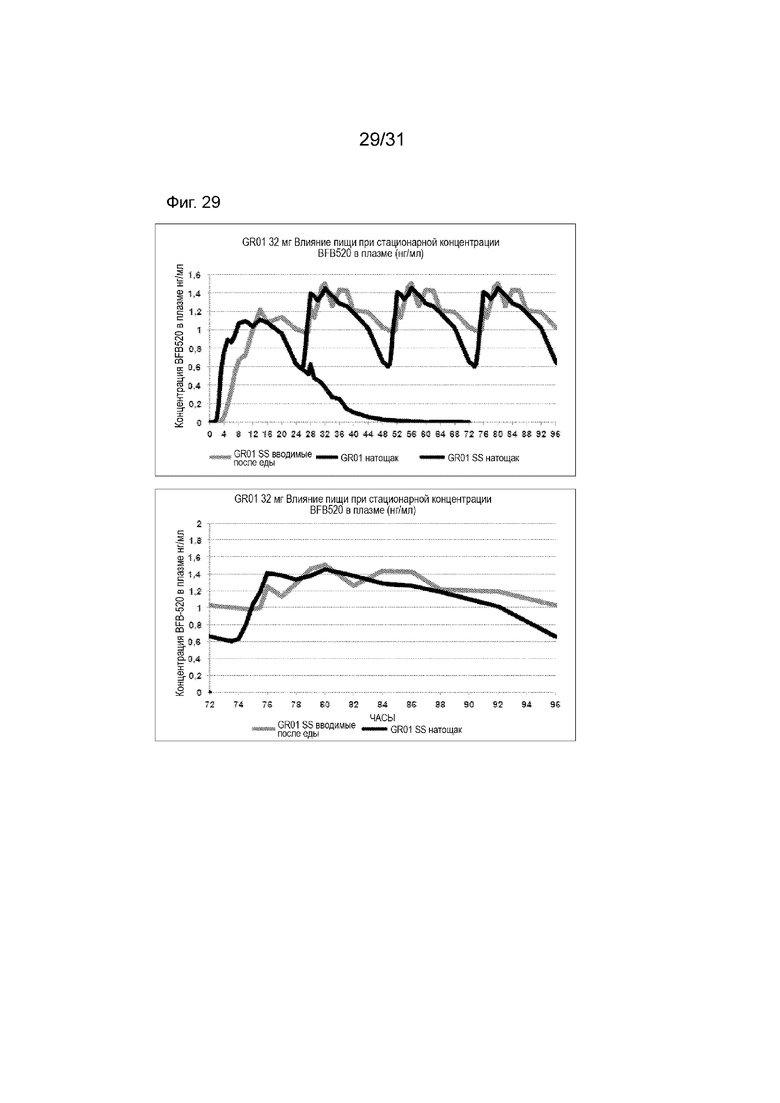
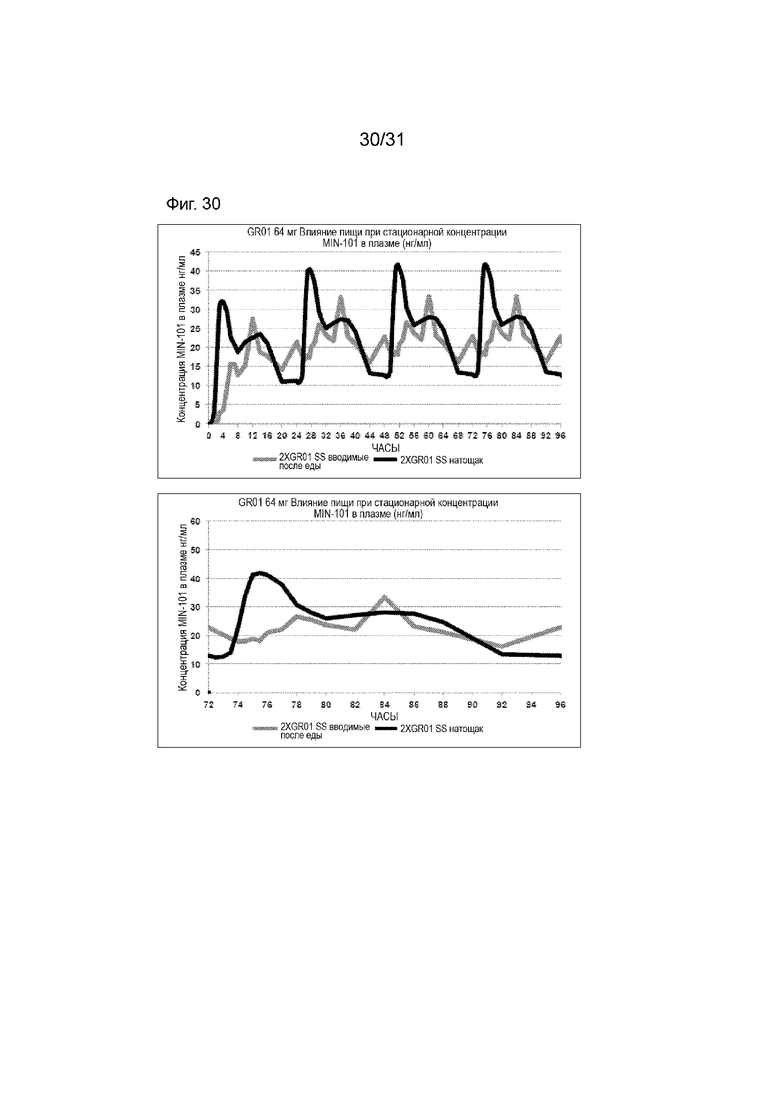

Данное изобретение относится к кишечнорастворимым лекарственным формам с контролируемым высвобождением, содержащим Соединение (I):
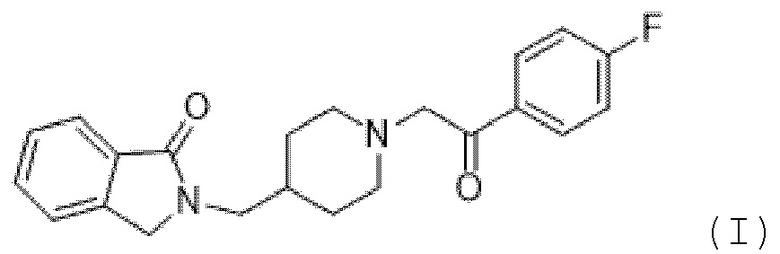
или его фармацевтически приемлемую соль, и/или сольват, фармакокинетическим свойствам этих лекарственных форм и их получению. Новые лекарственные формы, раскрытые в данном изобретении, полезны для снижения риска пролонгации QT у субъекта и для лечения расстройства у субъекта, нуждающегося в этом, например у субъекта с диагнозом шизофрения, например при лечении негативных симптомов у субъекта с диагнозом шизофрения, имеющего генотип CYP2D6 EM. 2 н. и 20 з.п. ф-лы, 31 ил., 53 табл., 22 пр.
1. Кишечнорастворимая лекарственная форма с контролируемым высвобождением, содержащая ядро таблетки и кишечнорастворимую оболочку, причем ядро таблетки содержит:
от приблизительно 4 мг до приблизительно 100 мг 1H-изоиндол-1-она, 2-[[1-[2-(4-фторфенил)-2-оксоэтил]-4-пиперидинил]метил]-2,3-дигидро-, гидрохлорид, гидрат (1:1:2) (гидрохлорид ролуперидона);
наполнитель;
вещество, способствующее скольжению;
смазывающее вещество; и
агент контролируемого высвобождения, который представляет собой смесь (i) гипромеллозы низкой вязкости с вязкостью от приблизительно 15 миллипаскаль-секунд (мПа⋅с) до приблизительно 100 мПа⋅с при 2% концентрации в воде при 20°С; и (ii) гипромеллозы высокой вязкости с вязкостью приблизительно 100000 мПа⋅с при 2% концентрации в воде при 20°С; где каждая из гипромеллоз низкой и высокой вязкости имеет чистоту для контролируемого высвобождением или замедленного высвобождения и дополнительно характеризуется содержанием метоксигруппы от 19,0% до 24,0% и содержанием гидроксипропоксигруппы от 4,0% до 12,0%.
2. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 1, где наполнителем является микрокристаллическая целлюлоза, моногидрат лактозы, сахароза, глюкоза, сорбит или их комбинация.
3. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 2, где наполнитель представляет собой комбинацию микрокристаллической целлюлозы и моногидрата лактозы.
4. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 1, где вещество, способствующее скольжению представляет собой коллоидный безводный диоксид кремния, крахмал или тальк.
5. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 4, где вещество, способствующее скольжению, агент представляет собой безводный коллоидный диоксид кремния.
6. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 1, где смазывающим веществом является стеарат магния, стериновая кислота или растительный стеарин.
7. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 6, где смазывающим веществом является стеарат магния.
8. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по любому из пп. 1-7, где кишечнорастворимая оболочка содержит дисперсию сополимера метакриловой кислоты и этилакрилата.
9. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 8, где кишечнорастворимая оболочка дополнительно содержит кишечнорастворимую оболочку, дополнительно содержащую агент, предотвращающий слипание.
10. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по любому из пп. 1-9, где ядро таблетки содержит приблизительно 38,4 мг гидрохлорида ролуперидона.
11. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по любому из пп. 1-9, где ядро таблетки содержит приблизительно 76,8 мг гидрохлорида ролуперидона.
12. Способ лечения негативных симптомов шизофрении у субъекта, включающий введение кишечнорастворимой лекарственной формы с контролируемым высвобождением по любому из пп. 1-11 субъекту.
13. Способ по п. 12, где способ приводит к снижению риска удлинения интервала QT у субъекта.
14. Способ по п. 12 или 13, где кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят один раз в день.
15. Способ по любому из пп. 12-14, где субъект имеет генотип CYP2D6 EM.
16. Способ по любому из пп. 12-15, где субъект сыт перед пероральным введением лекарственной формы.
17. Способ по любому из пп. 12-15, где субъект голоден перед пероральным введением лекарственной формы.
18. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по любому из пп. 1-11 для применения при лечении негативных симптомов шизофрении у субъекта.
19. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 18, где кишечнорастворимую лекарственную форму с контролируемым высвобождением вводят субъекту один раз в день.
20. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по п. 18 или 19, где субъект имеет генотип CYP2D6 EM.
21. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по любому из пп. 18-20, где субъект сыт перед пероральным введением лекарственной формы.
22. Кишечнорастворимая лекарственная форма с контролируемым высвобождением по любому из пп. 18-20, где субъект голоден перед пероральным введением лекарственной формы.
| WO 2016089766 A1, 2016.06.09 | |||
| СПОСОБЫ ПРИМЕНЕНИЯ ПРОИЗВОДНЫХ ЦИКЛИЧЕСКОГО АМИДА ДЛЯ ЛЕЧЕНИЯ ШИЗОФРЕНИИ | 2011 |
|
RU2576611C2 |
| SINGH DEEP HUSSAN ET AL, "A review on recent advances of enteric coating", IOSR JOURNAL OF PHARMACY, INTERNATIONAL ORGANIZATION OF SCIENTIFIC RESEARCH, IN, Vol | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| William R | |||
| O | |||
| Et al., Investigation of excipient type and level on drug release | |||

