Информация по связанным заявкам
Данная заявка притязает на приоритет предварительной заявки США №61/355164, поданной 16 июня 2010 года, содержание которой включено здесь в полном объеме в виде ссылки.
Область изобретения
Настоящее изобретение относится к новым дозированным формам, содержащим по меньшей мере один ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы. Кроме того, настоящее изобретение также относится к способам лечения некоторых заболеваний с использованием новых дозированных форм, раскрытых здесь.
Предпосылки создания изобретения
2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновая кислота (также известная как "фебуксостат" и "ТМХ-67") является мощным, непуриновым селективным ингибитором ксантиноксидоредуктазы. Фебуксостат в дозе 40 и 80 мг один раз в день (QD) одобрен в США для постоянного контроля гиперурикемии у пациентов с подагрой. Подагра представляет собой заболевание, связанное с отложением кристаллов уратов в синовиальной жидкости и других тканях, когда имеется перенасыщенность уратов в крови. Фебуксостат является мощным селективным ингибитором фермента ксантиноксидоредуктазы (или ингибитором ксантиноксидоредуктазы), который необходим для синтеза мочевой кислоты.
Фермент ксантиноксидоредуктаза может находиться в двух различных формах (см. Enroth C, et al., "Crystal structures of bovine milk xanthine dehydrogenase A and xanthine oxidase: structure-based mechanism of conversion," Proc. Natl. Acad. Sci. USA, 97(20): 10723-8 (Sept. 26, 2000)). В одной из форм фермента ксантиноксидоредуктаза синтезируется как ксантиндегидрогеназа. Эта форма фермента обладает очень низкой реакционной способностью в отношении кислорода. Тем не менее, при стрессе или в условиях болезни, такой как травматическая ишемическая реперфузия и застойная сердечная недостаточность, ксантиндегидрогеназа может подвергаться образованию внутримолекулярных дисульфидных связей или протеолитическому расщеплению, что конвертирует фермент во вторую форму, ксантиноксидазу. Ксантиноксидаза обладает высокой реакционной способностью в отношении кислорода. Таким образом, синтез мочевой кислоты из ксантина и гипоксантина за счет фермента ксантиноксидоредуктазы в виде ксантиноксидазы, связан с генерацией свободных радикалов кислорода, таких как супероксид-анион и перекись водорода. Эти свободные радикалы способны вызывать различные токсичные активности в организме, такие как инактивация белков, разрушение ДНК, перекисное окисления липидов (которое вызывает разрушение клеточных мембран) и увеличение уровня провоспалительных цитокинов.
Множество болезненных состояний связано с повышенной активностью ксантиноксидоредуктазы, в частности, с повышенной активностью ксантиноксидазы. Такие заболевания включают, но не ограничиваются перечисленными, гиперурикемию, гипертензию, метаболический синдром, диабет, ишемию миокарда, атеросклероз, инсульт, застойную сердечную недостаточность, воспалительное заболевание кишечника, прогрессирующее почечное заболевание, простатит, апноэ во сне и аутоиммунные заболевания. Гиперурикемия также связана с рядом болезненных состояний, таких как повреждение почек и гипертензия.
В лечении гиперурикемии используется аллопуринол. Было показано, что аллопуринол предотвращает повреждение почек и гипертензию, связанные с гиперурикемией путем ингибирования ксантиноксидоредуктазы, снижая тем самым уровень мочевой кислоты. В противоположность этому, было установлено, что степень защиты от повреждения почек и гипертензии у субъектов, страдающих гиперурикемией, была ниже у пациентов, получавших бензиодарон - средство, способствующее выведению мочевой кислоты. Бензиодарон не ингибирует ксантиноксидоредуктазную активность, но вместо этого снижает уровни мочевой кислоты в плазме, увеличивая выведение мочевой кислоты почками (см. Mazzali M, et al., "Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism," Hypertension, 38: 1101-1106 (2001), и Mazzali M, et al., "Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism," Am. J. Physiol Renal Physiol., 282:F991-F997 (2002)). Таким образом, в уровне техники существует необходимость в новых дозированных формах, которые не только снижают уровень мочевой кислоты у субъектов, страдающих гиперурикемией, но и способны поддерживать высокий уровень (а именно, по меньшей мере 80%) ингибирования активности ксантиноксидоредуктазы у субъекта для того, чтобы защитить пациентов, получающих эти дозированные формы на протяжении всего режима (например, интервала дозирования, который составляет, как правило, двадцать четыре часа) лечения против увеличения концентрации свободных радикалов кислорода.
Как указано выше, другим средством лечения гиперурикемии является соединение фебуксостат. Обширные фармакокинетические и фармакодинамические данные показали, что поддержание концентрации фебуксостата в плазме крови на протяжении длительного периода времени обеспечивает схожую эффективность лечения высокими дозами препарата. В целом, эти исследования показали, что поддержание концентрации 100 нг/мл фебуксостата в плазме требуется для обеспечения 95% или более ингибирования ксантиноксидазы. В настоящее время существующие коммерчески доступные препараты фебуксостата являются только препаратами немедленного высвобождения. В настоящее время нет никаких коммерчески доступных препаратов фебуксостата с продленным или отсроченным высвобождением. Таким образом, препарат фебуксостата, который поддерживает концентрацию лекарства выше критической концентрации 100 нг/мл в течение длительного периода времени, что, как ожидается, приведет к увеличению эффективности препарата, и был бы желательным вариантом лечения для контроля гиперурикемии, подагры и многих других болезненных состояний.
Сущность изобретения
В одном варианте осуществления настоящее раскрытие относится к дозированным формам с модифицированным высвобождением. Дозированные формы с модифицированным высвобождением могут содержать по меньшей мере один ингибитор ксантиноксидоредуктазы или по меньшей мере один ингибитор ксантиноксидазы.
В другом варианте дозированные формы с модифицированным высвобождением, раскрытые здесь, включают ингибитор ксантиноксидоредуктазы или его фармацевтически приемлемую соль, где указанная дозированая форма, после перорального введения субъекту, нуждающемуся в лечении, проявляет по меньшей мере одно из следующего:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение периода от приблизительно 5 часов до приблизительно 24 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл.
Альтернативно, дозированная форма с модифицированным высвобождением, после перорального введения субъекту, нуждающемуся в лечении, показывает следующее:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 24 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,0 мкг/мл до приблизительно 1,0 мкг/мл.
Еще также, в качестве альтернативы, дозированная форма с модифицированным высвобождением, после перорального введения субъекту, нуждающемуся в лечении, может проявлять каждое из следующего:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 24 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл.
В одном аспекте дозированные формы с модифицированным высвобождением, раскрытые здесь, могут содержать от приблизительно 5 до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли. В другом аспекте дозированные формы с модифицированным высвобождением, раскрытые здесь, могут содержать от приблизительно 40 до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли.
При пероральном введении пациенту, нуждающемуся в лечении, дозированные формы с модифицированным высвобождением, раскрытые здесь, могут продуцировать у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве приблизительно 2,5 мкг/мл, приблизительно 2,4 мкг/мл, приблизительно 2,3 мкг/мл, приблизительно 2,2 мкг/мл, приблизительно 2,1 мкг/мл, 2,0 мкг/мл, приблизительно 1,9 мкг/мл, приблизительно 1,8 мкг/мл, приблизительно 1,7 мкг/мл, приблизительно 1,6 мкг/мл, приблизительно 1,5 мкг/мл, приблизительно 1,4 мкг/мл, приблизительно 1,3 мкг/мл, приблизительно 1,2 мкг/мл, приблизительно 1,1 мкг/мл, приблизительно 1,0 мкг/мл, приблизительно 0,9 мкг/мл, приблизительно 0,8 мкг/мл, приблизительно 0,7 мкг/мл, приблизительно 0,6 мкг/мл или приблизительно 0,5 мкг мл. В частности, дозированные формы с модифицированным высвобождением, раскрытые здесь, при пероральном введении субъекту, нуждающемуся в лечении, могут продуцировать у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в диапазоне от приблизительно 2,5 мкг/мл до приблизительно 1,0 мкг/мл. Еще более конкретно, дозированные формы с модифицированным высвобождением, раскрытые здесь, при пероральном введении субъекту, нуждающемуся в лечении, могут продуцировать у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в диапазоне от приблизительно 2,0 мкг/мл до приблизительно 1,5 мкг/мл.
В еще одном воплощении дозированные формы с модифицированным высвобождением, раскрытые здесь, включают ингибитор ксантиноксидоредуктазы или его фармацевтически приемлемую соль, где указанная дозированная форма, после перорального введения субъекту, нуждающемуся в лечении, проявляет по меньшей мере одно из следующего:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 16 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,050 мкг/мл.
Альтернативно, дозированная форма с модифицированным высвобождением, после перорального введения субъекту, нуждающемуся в лечении, проявляет следующее:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 16 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,0 мкг/мл до приблизительно 0,075 мкг/мл.
Еще также в качестве альтернативы, дозированная форма с модифицированным высвобождением, после перорального введения субъекту, нуждающемуся в лечении, может проявлять следующее:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 16 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,050 мкг/мл.
Дозированные формы с модифицированным высвобождением, раскрытые здесь, могут содержать от приблизительно 40 до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли.
При пероральном введении пациенту, нуждающемуся в лечении, дозированные формы с модифицированным высвобождением, раскрытые здесь, могут продуцировать у субъекта Cmax ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве приблизительно 2,5 мкг/мл, приблизительно 2,4 мкг/мл, приблизительно 2,3 мкг/мл, приблизительно 2,2 мкг/мл, приблизительно 2,1 мкг/мл, 2,0 мкг/мл, приблизительно 1,9 мкг/мл, приблизительно 1,8 мкг/мл, приблизительно 1,7 мкг/мл, приблизительно 1,6 мкг/мл, приблизительно 1,5 мкг/мл, приблизительно 1,4 мкг/мл, приблизительно 1,3 мкг/мл, приблизительно 1,2 мкг/мл, приблизительно 1,1 мкг/мл, приблизительно 1,0 мкг/мл, приблизительно 0,9 мкг/мл, приблизительно 0,8 мкг/мл, приблизительно 0,7 мкг/мл, приблизительно 0,6 мкг/мл, приблизительно 0,5 мкг/мл, приблизительно 0,4 мкг/мл, приблизительно 0,3 мкг/мл, приблизительно 0,2 мкг/мл, приблизительно 0,1 мкг/мл, приблизительно 0,099 мкг/мл, приблизительно 0,098 мкг/мл, приблизительно 0,097 мкг/мл, приблизительно 0,096 мкг/мл, приблизительно 0,095 мкг/мл, приблизительно 0,094 мкг/мл, приблизительно 0,093 мкг/мл, приблизительно 0,092 мкг/мл, приблизительно 0,091 мкг/мл, приблизительно 0,090 мкг/мл, приблизительно 0,089 мкг/мл, приблизительно 0,088 мкг/мл, приблизительно 0,087 мкг/мл, приблизительно 0,086 мкг/мл, приблизительно 0,085 мкг/мл, приблизительно 0,084 мкг/мл, приблизительно 0,083 мкг/мл, приблизительно 0,082 мкг/мл, приблизительно 0,081 мкг/мл, приблизительно 0,080 мкг/мл, приблизительно 0,079 мкг/мл, приблизительно 0,078 мкг/мл, приблизительно 0,077 мкг/мл, приблизительно 0,076 мкг/мл, приблизительно 0,075 мкг/мл, приблизительно 0,074 мкг/мл, приблизительно 0,073 мкг/мл, приблизительно 0,072 мкг/мл, приблизительно 0,071 мкг/мл, приблизительно 0,070 мкг/мл, приблизительно 0,069 мкг/мл, приблизительно 0,068 мкг/мл, приблизительно 0,067 мкг/мл, приблизительно 0,066 мкг/мл, приблизительно 0,065 мкг/мл, приблизительно 0,064 мкг/мл, приблизительно 0,063 мкг/мл, приблизительно 0,062 мкг/мл, приблизительно 0,061 мкг/мл, приблизительно 0,060 мкг/мл, приблизительно 0,059 мкг/мл, приблизительно 0,058 мкг/мл, приблизительно 0,057 мкг/мл, приблизительно 0,056 мкг/мл, приблизительно 0,055 мкг/мл, приблизительно 0,054 мкг/мл, приблизительно 0,053 мкг/мл, приблизительно 0,052 мкг/мл, приблизительно 0,051 мкг/мл или приблизительно 0,050 мкг/мл. В частности, дозированные формы с модифицированным высвобождением, раскрытые здесь, при пероральном введении субъекту, нуждающемуся в лечении, могут продуцировать у субъекта Cmax ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в диапазоне от приблизительно 2,5 мкг/мл до приблизительно 0,050 мкг/мл. Еще более конкретно, дозированные формы с модифицированным высвобождением, раскрытые здесь, при пероральном введении субъекту, нуждающемуся в лечении, могут продуцировать у субъекта Cmax ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в диапазоне от приблизительно 2,0 мкг/мл до приблизительно 0,075 мкг/мл.
В еще одном варианте дозированные формы с модифицированным высвобождением, раскрытые здесь, включают ингибитор ксантиноксидоредуктазы или его фармацевтически приемлемую соль, где указанная дозированная форма, после перорального введения субъекту, нуждающемуся в лечении, проявляет по меньшей мере одно из следующего:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 14 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,090 мкг/мл.
Альтернативно, дозированная форма с модифицированным высвобождением, после перорального введения субъекту, нуждающемуся в лечении, проявляет следующее:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 14 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,0 мкг/мл до приблизительно 0,095 мкг/мл.
Еще также в качестве альтернативы, дозированная форма с модифицированным высвобождением, после перорального введения субъекту, нуждающемуся в лечении, может проявлять каждое из следующего:
(а) поддерживает у субъекта концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме на уровне более чем приблизительно 0,1 мкг/мл в течение периода от приблизительно 5 часов до приблизительно 14 часов, и
(b) продуцирует у субъекта максимальную концентрацию (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,090 мкг/мл.
Дозированные формы с модифицированным высвобождением, раскрытые здесь, могут содержать от приблизительно 40 до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли.
При пероральном введении пациенту, нуждающемуся в лечении, дозированные формы с модифицированным высвобождением, раскрытые здесь, могут продуцировать у субъекта Cmax ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в количестве приблизительно 2,5 мкг/мл, приблизительно 2,4 мкг/мл, приблизительно 2,3 мкг/мл, приблизительно 2,2 мкг/мл, приблизительно 2,1 мкг/мл, 2,0 мкг/мл, приблизительно 1,9 мкг/мл, приблизительно 1,8 мкг/мл, приблизительно 1,7 мкг/мл, приблизительно 1,6 мкг/мл, приблизительно 1,5 мкг/мл, приблизительно 1,4 мкг/мл, приблизительно 1,3 мкг/мл, приблизительно 1,2 мкг/мл, приблизительно 1,1 мкг/мл, приблизительно 1,0 мкг/мл, приблизительно 0,9 мкг/мл, приблизительно 0,8 мкг/мл, приблизительно 0,7 мкг/мл, приблизительно 0,6 мкг/мл, приблизительно 0,5 мкг/мл, приблизительно 0,4 мкг/мл, приблизительно 0,3 мкг/мл, приблизительно 0,2 мкг/мл, приблизительно 0,1 мкг/мл, приблизительно 0,099 мкг/мл, приблизительно 0,098 мкг/мл, приблизительно 0,097 мкг/мл, приблизительно 0,096 мкг/мл, приблизительно 0,095 мкг/мл, приблизительно 0,094 мкг/мл, приблизительно 0,093 мкг/мл, приблизительно 0,092 мкг/мл, или приблизительно 0,091 мкг/мл. В частности, дозированные формы с модифицированным высвобождением, раскрытые здесь, при пероральном введении субъекту, нуждающемуся в лечении, могут продуцировать у субъекта Cmax ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в диапазоне от приблизительно 2,5 мкг/мл до приблизительно 0,090 мкг/мл. Еще более конкретно, дозированные формы с модифицированным высвобождением, раскрытые здесь, при пероральном введении субъекту, нуждающемуся в лечении, могут продуцировать у субъекта Cmax ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в плазме в диапазоне от приблизительно 2,0 мкг/мл до приблизительно 0,095 мкг/мл.
Примером ингибитора ксантиноксидоредуктазы, который может быть использован в дозированных формах с модифицированным высвобождением, раскрытых здесь, представляет собой ингибиторы ксантиноксидоредуктазы, которые включают следующую формулу:

где R1 и R2, независимо друг от друга, представляют собой водород, гидроксильную группу, группу COOH, незамещенную или замещенную C1-C10 алкильную группу, незамещенную или замещенную C1-C10 алкоксигруппу, незамещенный или замещенный гидроксиалкокси, фенилсульфинильную группу или циано (-CN) группу;
где R3 и R4, независимо друг от друга, представляют собой водород или A, B, C или D, как показано ниже:
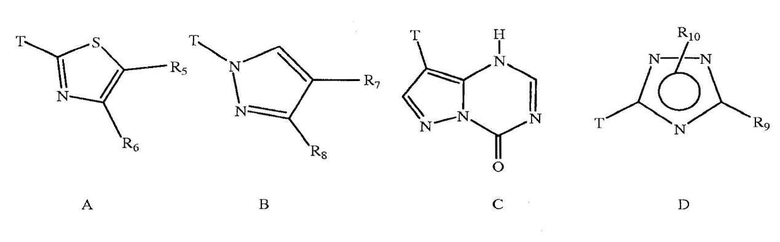
где T соединяет A, B, C или D с ароматическим кольцом, показанным выше, по R1, R2, R3 или R4;
где R5 и R6, независимо друг от друга, представляют собой водород, гидроксильную группу, группу COOH, незамещенную или замещенную C1-C10 алкильную группу, незамещенную или замещенную C1-C10 алкоксигруппу, незамещенный или замещенный гидроксиалкокси, COO-глюкуронид или COO-сульфат;
где R7 и R8, независимо друг от друга, представляют собой водород, гидроксильную группу, группу COOH, незамещенную или замещенную C1-C10 алкильную группу, незамещенную или замещенную C1-C10 алкоксигруппу, незамещенный или замещенный гидроксиалкокси, COO-глюкуронид или COO-сульфат;
где R9 представляет собой незамещенную пиридильную группу или замещенную пиридильную группу; и
где R10 представляет собой водород или низшую алкильную группу, низшую алкильную группу, замещенную пивалоилоксигруппой, и в каждом случае R10 связан с одним из атомов азота в 1,2,4-триазольном кольце, как показано выше.
Примерами соединений, имеющих приведенную выше формулу, являются: (а) 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновая кислота или ее фармацевтически приемлемая соль; (b) 2-[3-циано-4-(3-гидрокси-2-метилпропокси)фенил]-4-метил-5-тиазолкарбоновая кислота или ее фармацевтически приемлемая соль; (c) 2-[3-циано-4-(2-гидрокси-2-метилпропокси)фенил]-4-метил-5-тиазолкарбоновая кислота или ее фармацевтически приемлемая соль; (d) 2-(3-циано-4-гидроксифенил)-4-метил-5-тиазолкарбоновая кислота или ее фармацевтически приемлемая соль; (е) 2-[4-(2-карбоксипропокси)-3-цианофенил]-4-метил-5-тиазолкарбоновая кислота или ее фармацевтически приемлемая соль; (f) 1-3-циано-4-(2,2-диметилпропокси)фенил]-1H-пиразол-4-карбоновая кислота или ее фармацевтически приемлемая соль; (g) пиразоло-[1,5-а]-1,3,5-триазин-4-(1H)-он, 8-[3-метокси-4-(фенилсульфинил)фенил]-натриевая соль (±); и (h) 3-(2-метил-4-пиридил)-5-циано-4-изобутоксифенил)-1,2,4-триазол или его фармацевтически приемлемая соль.
Другой пример по меньшей мере одного ингибитора ксантиноксидоредуктазы, который может быть использован в дозированных формах с модифицированным высвобождением, раскрытых здесь, представляет собой ингибиторы ксантиноксидоредуктазы, которые имеют следующую формулу:
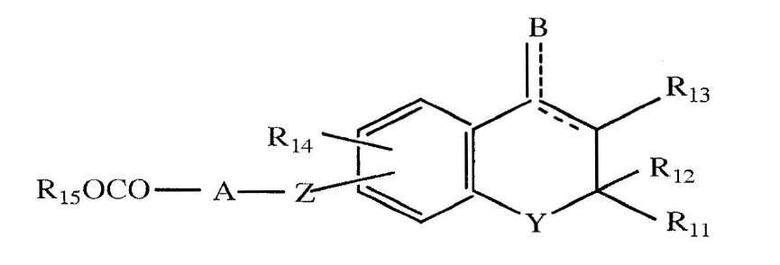
где R11 и R12, независимо друг от друга, представляют собой водород, замещенную или незамещенную низшую алкильную группу, замещенный или незамещенный фенил, или R11 и R12 могут вместе образовывать четырех-восьми-членное углеродное кольцо вместе с атомом углерода, к которому они присоединены;
где R13 представляет собой водород или замещенную или незамещенную низшую алкильную группу;
где R14 представляет собой один или два радикала, выбранных из группы, состоящей из водорода, галогена, нитрогруппы, замещенной или незамещенной низшей алкильной группы, замещенного или незамещенного фенила, -OR16 и -SO2NR17R17', где R16 представляет собой водород, замещенный или незамещенный низший алкил, фенил-замещенный низший алкил, карбоксиметил или его сложный эфир, гидроксиэтил или его простой эфир, или аллил; R17 и R17', независимо друг от друга, представляют собой водород или замещенный или незамещенный низший алкил;
где R15 представляет собой водород или фармацевтически активную эфирообразующую группу;
где А представляет собой неразветвленный или разветвленный углеводородный радикал, имеющий от одного до пяти атомов углерода;
где B представляет собой галоген, кислород или этилендитио;
где Y представляет собой кислород, серу, азот или замещенный азот;
где Z представляет собой кислород, азот или замещенный азот; и
пунктирная линия относится либо к простой связи, двойной связи, или к двум одинарным связям.
В другом варианте осуществления настоящее изобретение относится к способу лечения пациента, страдающего от подагры, гиперурикемии, простатита, воспалительного заболевания кишечника, удлинения интервала QT, инфаркта миокарда, гипертрофии сердца, гипертензии, почечнокаменной болезни, почечной недостаточности, хронического заболевания почек, метаболического синдрома, сахарного диабета, диабетической нефропатии или застойной сердечной недостаточности, и нуждающегося в их лечении. Способ включает стадию введения субъекту, страдающему от подагры, гиперурикемии, простатита, воспалительного заболевания кишечника, удлинение интервала QT, инфаркта миокарда, гипертрофии сердца, гипертензии, почечнокаменной болезни, почечной недостаточности, хронического заболевания почек, метаболического синдрома, диабета, диабетической нефропатии или застойной сердечной недостаточности и нуждающемуся в их лечении, терапевтически эффективного количества описанной выше дозированной формы с модифицированным высвобождением, содержащей по меньшей мере один ингибитор ксантиноксидоредуктазы или по меньшей мере один ингибитор ксантиноксидазы.
В еще одном варианте настоящее изобретение относится к фармацевтической композиции, которая содержит ингибитор ксантиноксидоредуктазы или его фармацевтически приемлемую соль или по меньшей мере один ингибитор ксантиноксидазы или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый полимер, где фармацевтическая композиция включает по меньшей мере одно из следующего: немедленное высвобождение компонента, отсроченное высвобождение компонента и/или контролируемое высвобождение компонента. Примерами ингибиторов ксантиноксидоредуктазы, которые могут быть включены в фармацевтическую композицию, включают все указанные выше. Примером ингибитора ксантиноксидазы является оксипуринол или аллопуринол. Кроме того, компонент немедленного высвобождения, компонент отсроченного высвобождения и/или компонент контролируемого высвобождения может содержать одну или несколько гранул с различными профилями высвобождения. Гранулы с немедленным высвобождением высвобождают ингибитор ксантиноксидоредуктазы сразу после приема, гранулы с отсроченным высвобождением высвобождают ингибитор ксантиноксидоредуктазы под воздействием внутренней среды со специфическим уровнем рН, и гранулы с контролируемым высвобождением высвобождают ингибитор ксантиноксидоредуктазы в течение длительного периода времени, по сравнению с гранулами с немедленным высвобождением. Различные гранулы включают инертное ядро, покрытое соединением ингибитора ксантиноксидоредуктазы и одним или более слоев фармацевтически приемлемого полимера.
В дополнительном варианте осуществления изобретения настоящее раскрытие охватывает единую фармацевтическую композицию, которая включает как гранулы с немедленным высвобождением, так и гранулы с отсроченным высвобождением, растворимые при значениях рН больших или равных 6,8. Фармацевтическая композиция этого варианта осуществления включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве от приблизительно 20% до приблизительно 40% (масс./масс.) от общей массы композиции, гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,8, в количестве от приблизительно 60% до приблизительно 80% (масс./масс.) от общей массы композиции. Например, в одном аспекте фармацевтическая композиция включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве приблизительно 20% (масс./масс.) от общей массы композиции, гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,8, в количестве приблизительно 80% (масс./масс.) от общей массы композиции. В еще одном аспекте фармацевтическая композиция включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве приблизительно 25% (масс./масс.) от общей массы композиции, гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,8, в количестве приблизительно 75% (масс./масс.) от общей массы композиции. В еще одном аспекте фармацевтическая композиция включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве приблизительно 30% (масс./масс.) от общей массы композиции, гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,8, в количестве приблизительно 70% (масс./масс.) от общей массы композиции. В еще одном аспекте фармацевтическая композиция включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве приблизительно 40% (масс./масс.) от общей массы композиции, гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,8, в количестве приблизительно 60% (масс./масс.) от общей массы композиции.
В еще одном варианте осуществления настоящего раскрытия фармацевтическая дозированная форма охватывает отдельную фармацевтическую композицию, в которую включены гранулы с немедленным высвобождением, гранулы с отсроченным высвобождением, растворимые при значениях рН больших или равных 6,0, и гранулы с отсроченным высвобождением, растворимые при достижении значений рН больших или равных 6,8. Фармацевтическая композиция этого варианта осуществления включает гранулы ингибитора ксантиноксидоредуктазы с немедленным высвобождением в количестве от приблизительно 25% до приблизительно 35% (масс./масс.) от общей массы композиции, гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,0, в количестве от приблизительно 25% до приблизительно 35% (масс./масс.) от общей массы композиции, и гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным высвобождением, высвобождаемые при достижении значения рН 6,8, в количестве от приблизительно 35% до приблизительно 45% (масс./масс.) от общей массы композиции.
В другом варианте осуществления настоящего раскрытия фармацевтическая композиция охватывает единую фармацевтическую композицию, в которую включены гранулы с немедленным высвобождением, гранулы с отсроченным/контролируемым высвобождением, растворимые при значении рН по меньшей мере равном 6,8, и контролируемым высвобождением через период времени приблизительно четыре-шесть часов. Фармацевтическая композиция этого варианта осуществления включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве в диапазоне от приблизительно 20% до приблизительно 40% (масс./масс.) от общей массы композиции, и гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с отсроченным/контролируемым высвобождением, растворимые при значении рН, большим или равным 6,8, и предоставляющие пролонгированное высвобождение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы через период от приблизительно 4 часов до приблизительно 6 часов, в количестве в диапазоне от приблизительно 60% до приблизительно 80% (масс./масс.) от общей массы композиции.
В еще одном варианте осуществления настоящего раскрытия фармацевтическая композиция охватывает отдельную фармацевтическую композицию, в которую включены гранулы с немедленным высвобождением, гранулы с контролируемым высвобождением, способные высвобождать действующее вещество в течение от приблизительно десяти до приблизительно двенадцати часов. Фармацевтическая композиция этого варианта осуществления в общем включает гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с немедленным высвобождением в количестве в диапазоне от приблизительно 10% до приблизительно 30% (масс./масс.) от общей массы композиции, и гранулы ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы с контролируемым высвобождением, предоставляющие пролонгированное высвобождение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы через период времени от приблизительно 10 часов до приблизительно 12 часов, в количестве в диапазоне от приблизительно 70% до приблизительно 90% (масс./масс.) от общей массы композиции.
Создание вышеописанных вариантов воплощений получено как результат длительного процесса разработки лекарственных средств. Первоначальное мультидозовое, плацебо-контролируемое, двойное слепое исследование, проведенное на двенадцати здоровых испытуемых, было предназначено для оценки безопасности и максимально переносимой дозы фебуксостата при пероральном введении. Исследование также было организовано для получения и оценки фармакокинетических и фармакодинамических профилей множественных дневных пероральных доз в диапазоне доз и режимов, в том числе прием один раз в день и прием два раза в день. В результате этого исследования была получена ценная фармакокинетическая и фармакодинамическая информация, относящаяся к биодоступности фебуксостата in vivo. Результаты этого исследования были опубликованы в статье: Reza Khosravan et al., Pharmacokinetics, Pharmacodynamics and Safety of Febuxostat, a Non-Purine Selective Inhibitor of Xanthine Oxidase, in a Dose Escalation Study in Healthy Subjects, Clinical Pharmacokinetics, 2006: 45 (8): 821-841. В частности, фармакокинетические параметры исследования, обсуждаемые на стр. 829 статьи, приведены в настоящем документе в таблице 1 примера 1.
На фазе 1 мультидозового, рандомизированного, плацебо-контролируемого, двойного слепого, одноцентрового, мультилокализированного исследования, включающего фебуксостат с повышением дозы, у здоровых субъектов изучали фармакокинетику и фармакодинамику фебуксостата. В этом исследовании пероральная доза фебуксостата с немедленным высвобождением (ингибитор ксантиноксидоредуктазы) варьировала от 10 мг один раз в день до 240 мг один раз в день (далее обозначено как "QD"), и 30 мг два раза в день (далее обозначено как "BID"). В этом исследовании было установлено, что доза 30 мг фебуксостата, вводимая дважды в день (в общей дозе 60 мг в день), была так же эффективна в снижении уровня мочевой кислоты, как доза фебуксостата 120 мг, вводимая один раз в день. Учитывая эти результаты, было установлено, что для поддержания уровней вышеуказанных лекарств минимальная концентрация имеет решающее значение для улучшенного снижения уровня мочевой кислоты. В ходе дальнейших исследований и сбора фармакокинетических данных было установлено, что поддержание концентраций фебуксостата in vivo на уровне или выше 100 нг/мл (0,1 мкг/мл) приводило к ингибированию мочевой кислоты на уровне 80% или более. В связи с этим неожиданным открытием авторы разработали препарат фебуксостата пролонгированного высвобождения, эффективного в течение максимального времени, обеспечивающий концентрацию фебуксостата выше минимальной критической концентрации, равной 100 нг/мл (0,1 мкг/мл).
Фармакокинетические данные, полученные в указанных выше клинических испытаниях, впоследствии были использованы для оценки профилей в плазме для различных препаратов фебуксостата, включая матричные таблетки пролонгированного высвобождения, препараты фебуксостата с двухстадийным высвобождением и препараты фебуксостата с трехстадийным высвобождением. Данные оценок по высвобождению препаратов были основаны на матричных формах препаратов, включающих один или несколько полимеров, и оценочные данные для препаратов с двухстадийным и трехстадийным высвобождением были основаны на формах препаратов, включающих два или более типов гранул с различными профилями высвобождения. Эта информация и методология обсуждается в примере 2. Кроме того, как часть процесса исследования препаратов фебуксостата пролонгированного высвобождения, различные области всасывания были исследованы для определения оптимальных физиологических областей всасывания препарата фебуксостата пролонгированного высвобождения. В начале доклинических исследований всасывание фебуксостата в различных областях желудочно-кишечного тракта было изучено на крысах. Тестирование на крысиной модели показало, что всасывание фебуксостата в области толстой кишки было очень плохим. С целью разработки лекарственной формы, которая обеспечит желаемый профиль концентрации в плазме во времени, изучение областей всасывания было проведено на людях. Эти данные и методология сбора информации содержатся в примере 3. Данные, касающиеся области всасывания, неожиданно показали, что всасывание фебуксостата в толстой кишке составило приблизительно только 40% по сравнению с профилем всасывания для контрольного препарата с немедленным высвобождением в проксимальном и дистальном отделах кишечника, что явилось существенно большим, чем это можно было бы ожидать из данных, полученных на крысах.
Авторы, в свете этих неожиданных экспериментальных данных, начали разработку препарата фебуксостата с пролонгированным высвобождением, который минимизирует действие фебуксостата в толстой кишке, и максимизирует действие фебуксостата в других областях, в том числе в желудке и в проксимальном и дистальном отделах кишечника. Были разработаны новые препараты фебуксостата путем создания препаратов с компонентами немедленного высвобождения, компонентами с отсроченным высвобождением в зависимости от уровней рН, и компонентами с непрерывным высвобождением, основанными на профиле высвобождения в течение длительного периода времени. Конкретные препараты описаны в примерах 4-9. Затем новые препараты фебуксостата были протестированы в модели на собаках, как описано в примере 10. Результаты тестирования в модели на собаках были ожидаемыми, принимая во внимание хорошо известные ограничения фармакокинетические испытаний в модели на собаках. Несмотря на ограничения в отношении длины желудочно-кишечного тракта собаки, препараты с отсроченным высвобождением (т.е. зависимым от рН) продемонстрировали улучшенные фармакокинетические параметры по сравнению с референсным препаратом фебуксостата с немедленным высвобождением. Эти препараты затем были протестированы на людях в однодозовом исследовании, как описано в примере 11.
Конкретные параметры и объем притязаний в отношении препаратов фебуксостата пролонгированного высвобождения более детально раскрыты в подробном описании.
Краткое описание фигур
На фигуре 1 показан временной профиль для средних концентраций фебуксостата в плазме для нескольких препаратов, содержащих 80 мг фебуксостата, изготовленных для высвобождения фебуксостата в различных частях желудочно-кишечного тракта. В частности, на фигуре 1 показаны средние концентрации фебуксостата в плазме во времени для дозированной формы, предназначенной для немедленного высвобождения фебуксостата в желудке, в проксимальном отделе тонкого кишечника, в дистальном отделе тонкого кишечника и в толстой кишке.
На фигуре 2 показано моделирование временного профиля концентрации фебуксостата для дозированной формы, содержащей 80 мг препарата фебуксостата с 3-стадийным высвобождением, где 30% дозы фебуксостата высвобождается немедленно (в момент времени = 0 часов, т.е. стадия высвобождения 1), 30% дозы фебуксостата высвобождается через 5 часов (т.е. стадия высвобождения 2), и 40% дозы фебуксостата высвобождается через 10 часов (т.е. стадия высвобождения 3). Модельные данные были рассчитаны с использованием параметров, полученных из данных по участкам поглощения, указанных и обсужденных в примере 3.
На фигуре 3 показано моделирование временного профиля концентрации фебуксостата для дозированной формы, содержащей 80 мг препарата фебуксостата с 2-стадийным высвобождением, где 20% дозы фебуксостата высвобождается немедленно (в момент времени = 0 часов, т.е. стадия высвобождения 1), 75% дозы фебуксостата высвобождается через 5 часов, и 5% препарата фебуксостата высвобождается в толстой кишке через 10 часов (высвобождение на 5 часов и 10 часов, вместе составляют стадию высвобождения 2). Модельные данные были рассчитаны с использованием параметров, полученных из данных по участкам поглощения, указанных и обсужденных в примере 3.
На фигуре 4 показано моделирование временного профиля концентрации фебуксостата для дозированной формы, содержащей 80 мг препарата фебуксостата пролонгированного высвобождения (ER), где 90% дозы фебуксостата поглощается в течение 6 часов после приема препарата, а остальные 10% дозы фебуксостата поглощаются в толстой кишке. Модельные данные были рассчитаны с использованием параметров, полученных из данных по участкам поглощения, указанных и обсужденных в примере 3.
На фигуре 5 показана таблица с описанием состава восьми матричных таблетированных препаратов фебуксостата с модифицированным высвобождением.
На фигуре 6 показаны профили растворения во времени восьми различных матричных таблетированных препаратов фебуксостата с модифицированным высвобождением. В частности, профили растворения были получены путем растворения 50 мг матричных таблетированных препаратов фебуксостата с модифицированным высвобождением в растворе с рН 6,8 и в присутствии 0,5 М фосфатного буфера.
На фигуре 7 показан временной профиль концентрации фебуксостата в плазме для нескольких дозированных форм с модифицированным высвобождением, как описано в примере 10, при испытании в модели на собаках.
На фигурах 8А и 8B показан временной профиль средней концентрации фебуксостата в плазме (в линейной и полулогарифмической форме) после перорального введения одной 80 мг дозы 4 препаратов фебуксостата пролонгированного высвобождения и препарата немедленного высвобождения (IR), как описано в примере 11. На фигурах 8А и 8B препаратами являются следующие:
Препарат А (референсный): таблетка IR фебуксостата (ULORIC) 80 мг.
Препарат B (тестируемый): капсула прототипа фебуксостата (80 мг) с двухстадийным высвобождением (TMX-67, XR препарат B).
Препарат C (тестируемый): капсула прототипа фебуксостата (80 мг) с трехстадийным высвобождением (TMX-67, XR препарат С).
Препарат D (тестируемый): капсула с комбинацией фебуксостата (80 мг) многостадийного и непрерывного высвобождения (TMX-67, препарат D).
Препарат E (тестируемый): капсула прототипа фебуксостата (80 мг) с непрерывным высвобождением (TMX-67, XR препарат E).
На фигуре 9 показано, как профиль растворения препаратов, описанных в примере 12, может варьировать в зависимости от соотношения ацетата целлюлозы и полиэтиленгликоля (PEG).
На фигуре 10 показано, что препарат, описанный в примере 12, может быть покрыт слоем лекарства (фебуксостат) с немедленным высвобождением, для того чтобы преодолеть задержку во времени.
На фигуре 11 показано, что препараты из множества частиц, описанные в примере 12, могут быть получены для того, чтобы иметь желательные характеристики высвобождения за счет варьирования количества этилцеллюлозного покрытия, содержащегося на указанных препаратах.
Подробное описание изобретения
I. Определения
Заголовки подразделов, используемые в этом разделе, и все раскрытия, приведенные здесь, не предназначены для ограничения.
Как здесь используется, формы в единственном числе включают множественное число, если контекст явно не указывает иное. При указании здесь числовых диапазонов, каждое промежуточное численное значение, указанное внутри них, охватывается с той же степенью точности. Например, для диапазона 6-9, числа 7 и 8 охватываются в дополнение к значениям 6 и 9, а для диапазона 6,0-7,0, числа 6,0, 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9 и 7,0 охватываются в явном виде.
Как здесь используется, термин "приблизительно" используется как синоним термина "примерно". В качестве иллюстрации, использование термина "приблизительно" означает, что значения немного выходят за пределы указанных значений, а именно, плюс или минус 10%. Таким образом, объемом формулы изобретения охватываются дозы с указанием условия "примерно" или "приблизительно".
Как здесь используется, термин "AUC" относится к площади под кривой зависимости концентрации активного агента в плазме от времени, и которая рассчитывается по формуле трапеций. Термин "AUCt" означает площадь под кривой зависимости концентрации в плазме от времени за период от 0 до 120 часов после введения, выраженную в единицах нг·час/мл, определенную с помощью правила трапеций. Термин "AUC∞" означает площадь под кривой зависимости концентрации в плазме от времени, от момента времени 0 до бесконечности. AUC∞ рассчитывается как AUCt+LMT/(-β), где "LMT" представляет собой последнюю измеряемую концентрацию в плазме, и β представляет собой константу скорости элиминации на конечной фазе. Если не указано иное, указываемое значение AUC является центральным значением AUC. "Центральное значение" AUC является средним значением AUC ± стандартное отклонение.
Термины "введение", "вводить" или "прием" относятся к любым способам предоставления субъекту или пациенту лекарственных средств (таких как ингибитор ксантиноксидоредуктазы или его фармацевтически приемлемая соль). Пути введения могут быть реализованы с помощью любых средств, известных специалистам в данной области. Такие средства охватывают, без ограничения перечисленными, пероральный, буккальный, внутривенный, подкожный, внутримышечный, трансдермальный, ингаляционный путь введения и тому подобное.
Термин "активный агент", используемый здесь, относится к (1) ингибитору ксантиноксидоредуктазы или к его фармацевтически приемлемой соли или к (2) ингибитору ксантиноксидазы или к его фармацевтически приемлемой соли. Термины "активный агент" и "лекарственное вещество" используются здесь взаимозаменяемо. Состояние активного агента в твердой форме, используемого при изготовлении дозированных форм с модифицированным высвобождением по настоящему изобретению, не является критическим. Например, активное вещество, используемое при изготовлении дозированных форм с модифицированным высвобождением по настоящему изобретению, может быть аморфным или кристаллическим. Итоговая дозированная форма содержит по меньшей мере детектируемое количество активного агента в кристаллической форме. Кристаллическую природу активного агента можно обнаружить с помощью анализа дифракции рентгеновских лучей на порошке, дифференциальной сканирующей калориметрии или любых других методов, известных в данной области.
Термин "Cmax" относится к максимальной наблюдаемой в плазме концентрации ингибитора ксантиноксидоредуктазы или его соли, получаемой при приеме дозированных форм настоящего изобретения. Если не указано иное, приводимое значение Cmax является центральным значением Cmax. "Центральное значение" Cmax является средним значением Cmax ± стандартное отклонение.
Как здесь используется, термин "отсроченное высвобождение" относится к типу модифицированного высвобождения, где дозированная лекарственная форма обладает временной задержкой между пероральным приемом дозированной лекарственной формы и высвобождением лекарственного вещества из указанной дозированной формы. Многостадийные системы высвобождения (также известные как системы с "импульсным высвобождением лекарственного вещества") и использование энтеросолюбильных покрытий, которые хорошо известны специалистам в данной области, приведены как примеры механизмов отсроченного высвобождения. Как правило, дозированные формы с отсроченным высвобождением высвобождают небольшое количество или не высвобождают никакого активного соединения в течение заданного периода времени или до тех пор, пока заданное условие, такое как воздействие определенного уровня рН, не будет выполнено, и тогда высвобождение активного соединения происходит немедленно.
Как здесь используется, термин "отсроченное контролируемое высвобождение" относится к типу модифицированного высвобождения, где дозированная лекарственная форма обладает пролонгированным высвобождением лекарственного вещества в течение установленного периода времени, с неинициированным высвобождением, с некоторым временем задержки, после приема дозированной формы. Как правило, дозированная форма с "отсроченным контролируемым высвобождением" высвобождает небольшое количество или не высвобождает никакого активного соединения в течение заданного периода времени или до тех пор, пока заданное условие, такое как воздействие определенного уровня рН, не будет выполнено, и тогда высвобождение активного соединения происходит в течение длительного дополнительного периода времени.
Термин "дозированная форма" относится к любым твердым объектам, полутвердой или жидкой композиции, предназначенным для размещения в них конкретного, заранее определенного количества (т.е. дозы) определенного активного агента. Подходящие дозированные формы могут быть фармацевтическими системами для доставки лекарственных средств, в том числе для перорального приема, буккального введения, ректального введения, для местной доставки или для чресслизистой доставки, или в виде подкожных имплантатов или других имплантируемых систем доставки лекарств и тому подобное. Предпочтительно, дозированные формы настоящего изобретения считаются твердыми, однако они могут содержать жидкий или полутвердый компонент. Более предпочтительно, чтобы дозированная форма представляла собой пероральную систему доставки активного вещества в желудочно-кишечный тракт пациента. Дозированная форма по настоящему изобретению демонстрирует модифицированное высвобождение активного вещества.
Под "эффективным количеством" или "терапевтически эффективным количеством" активного агента подразумевается нетоксичное, но достаточное количество активного агента, для обеспечения желаемого эффекта. Количество активного агента, которое является "эффективным", будет варьироваться от субъекта к субъекту, в зависимости от возраста и общего состояния индивидуума, конкретного активного агента или агентов, и тому подобное. Таким образом, не всегда возможно определить точное "эффективное количество". Тем не менее, соответствующее "эффективное количество" в каждом конкретном случае может быть определено специалистом в данной области с использованием обычных экспериментов.
Как здесь используется, термин "пролонгированное высвобождение" относится к лекарственному препарату, который обеспечивает постепенное высвобождение препарата в течение длительного периода времени. Термин "контролируемое" высвобождение относится к типу препарата с длительным высвобождением, в котором постепенное высвобождение лекарства контролируется или управляется в течение определенного длительного периода времени.
Термин "немедленное высвобождение" используется в его обычном смысле для обозначения дозированной формы, которая обеспечивает высвобождение активного агента сразу же после введения лекарства.
Как здесь используется, термин "модифицированный" относится к лекарственному веществу, содержащего препарат, в котором высвобождение лекарственного вещества происходит не сразу (см., например, Guidance for Industry SUPAC-MR: Modified Release Solid Oral Dosage Forms, Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution, Testing and In Vivo Bioequivalence Documentation, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research ("CDER"), September 1997 CMC 8, page 34, включено ссылкой). Для модифицированного препарата, дозированной формы с модифицированным высвобождением или модифицированной дозированной формы, введение указанного препарата или дозированной формы не приводит к немедленному высвобождению лекарственного вещества или активного агента в области поглощения. Этот термин используется как синоним термина "не немедленное высвобождение", как определено в Remington: The Science and Practice of Pharmacy, Nineteenth Ed. (Easton, Pa.: Mack Publishing Company, 1995). Как здесь используется, термин "модифицированное высвобождение" включает в себя продолжительное или контролируемое высвобождение, отсроченное высвобождение и отсроченное-контролируемое высвобождение.
Термин "фармацевтически приемлемый", например, при указании "фармацевтически приемлемый эксципиент" или "фармацевтически приемлемая добавка", означает материал, который не является биологически нежелательным или нежелательным иным образом, т.е. материал может быть включен в фармацевтическую композицию, которую вводят пациенту, не вызывая нежелательных биологических эффектов.
Термин "субъект" относится к животному, предпочтительно млекопитающему, включая человека, или не относящемуся к человеку. Термины "пациент" и "субъект" могут быть использованы взаимозаменяемо. Термин "лечение" относится к уменьшению тяжести и/или частоты симптомов, устранению симптомов и/или основной причины, профилактике появления симптомов и/или их причины, и облегчению или восстановлению повреждений. Так, например, "лечение" пациента включает профилактику конкретного нарушения или неблагоприятных физиологических событий у восприимчивого человека, так и клиническое лечение отдельных симптомов ингибированием или путем регрессии расстройства или заболевания.
Как здесь используется, термин "ксантиноксидоредуктаза" относится по меньшей мере к одной из форм фермента ксантиноксидоредуктазы, а именно - ксантиноксидазы и/или ксантиндегидрогеназы.
Как используется здесь, выражение "ингибитор ксантиноксидоредуктазы" относится к любому соединению, которое (1) является ингибитором ксантиноксидоредуктазы, такой как, но не ограничиваясь, ксантиноксидазы, и (2) химически не содержит пуриновое кольцо в своей структуру (то есть является «не пуриновым"). Выражение " ингибитор ксантиноксидоредуктазы", как определено здесь, также охватывает метаболиты, полиморфы, сольваты и пролекарства таких соединений, в том числе метаболиты, полиморфы, сольваты и пролекарства соединений, описанных в формуле I и в формуле II ниже. Примеры ингибиторов ксантиноксидоредуктазы включают, но не ограничиваются перечисленными, 2-[4-(2-карбоксипропокси)-3-цианофенил]-4-метил-5-тиазолкарбоновую кислоту и соединения, имеющие следующие формулу I или формулу II:
Соединения формулы I:
где R1 и R2, независимо друг от друга, представляют собой водород, гидроксильную группу, группу COOH, незамещенную или замещенную C1-C10 алкильную группу, незамещенную или замещенную C1-C10 алкоксигруппу, незамещенный или замещенный гидроксиалкокси, фенилсульфинильную группу или циано (-CN) группу;
где R3 и R4, независимо друг от друга, представляют собой водород или A, B, C или D, как показано ниже:
где T соединяет или прикрепляет A, B, C или D к ароматическому кольцу, показанному выше, по R1, R2, R3 или R4;
где R5 и R6, независимо друг от друга, представляют собой водород, гидроксильную группу, группу COOH, незамещенную или замещенную C1-C10 алкильную группу, незамещенную или замещенную C1-C10 алкоксигруппу, незамещенный или замещенный гидроксиалкокси, COO-глюкуронид или COO-сульфат;
где R7 и R8, независимо друг от друга, представляют собой водород, гидроксильную группу, группу COOH, незамещенную или замещенную C1-C10 алкильную группу, незамещенную или замещенную C1-C10 алкоксигруппу, незамещенный или замещенный гидроксиалкокси, COO-глюкуронид или COO-сульфат;
где R9 представляет собой незамещенную пиридильную группу или замещенную пиридильную группу; и
где R10 представляет собой водород или низшую алкильную группу, низшую алкильную группу, замещенную пивалоилоксигруппой, и в каждом случае R10 связан с одним из атомов азота в 1,2,4-триазольном кольце, как показано выше в формуле I.
Соединения формулы II:
где R11 и R12, независимо друг от друга, представляют собой водород, замещенную или незамещенную низшую алкильную группу, замещенный или незамещенный фенил (замещенный фенил в этой формуле II обозначает фенил, замещенный галогеном или низшим алкилом и тому подобное. Примеры включают, но не ограничиваются перечисленным, п-толил и п-хлорфенил), или R11 и R12 могут вместе образовывать от четырех- до восьми-членное углеродное кольцо вместе с атомом углерода, к которому они присоединены;
где R13 представляет собой водород или замещенную или незамещенную низшую алкильную группу;
где R14 представляет собой один или два радикала, выбранных из группы, состоящей из водорода, галогена, нитрогруппы, замещенной или незамещенной низшей алкильной группы, замещенного или незамещенного фенила (замещенный фенил в этой формуле II обозначает фенил, замещенный галогеном или низшим алкилом, и тому подобное. Примеры включают, но не ограничиваются перечисленным, п-толил и п-хлорфенил), -OR16 и -SO2NR17R17', где R16 представляет собой водород, замещенный или незамещенный низший алкил, фенил-замещенный низший алкил, карбоксиметил или его сложный эфир, гидроксиэтил или его простой эфир, или аллил; R17 и R17', независимо друг от друга, представляют собой водород или замещенный или незамещенный низший алкил;
где R15 представляет собой водород или фармацевтически активную эфирообразующую группу;
где А представляет собой неразветвленный или разветвленный углеводородный радикал, имеющий от одного до пяти атомов углерода;
где B представляет собой галоген, кислород или этилендитио;
где Y представляет собой кислород, серу, азот или замещенный азот;
где Z представляет собой кислород, азот или замещенный азот; и
пунктирная линия относится либо к простой связи, двойной связи, либо к двум одинарным связям (например, когда В представляет собой этилендитио, пунктирная линия, показанная в структуре кольца, может быть двумя одинарными связями).
Как здесь используется, термин "низший(е) алкил(ы)" относится к C1-C7 алкильной группе, включающей, в том числе, но не ограничиваясь перечисленным, метил, этил, н-пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, гексил, гептил и тому подобное.
Как здесь используется, термин "низший алкокси" относится к группам, образованным связыванием низшей алкильной группы с атомом кислорода, в том числе, но не ограничиваясь перечисленным, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, пентокси, гексокси, гептокси и тому подобным.
Как здесь используется, термин "низшая алкилтиогруппа" относится к группам, образованным связыванием низшего алкила с атомом серы.
Как здесь используется, термин "галоген" относится к фтору, хлору, брому и йоду.
Как здесь используется, термин "замещенный пиридил" относится к пиридильной группе, которая может быть замещена галогеном, цианогруппой, низшим алкилом, низшим алкокси или низшей алкилтиогруппой.
Как здесь используется, термин " четырех-восьми членные углеродные кольца" относится к циклобутилу, циклопентилу, циклогексилу, циклогептилу, циклооктилу и тому подобному.
Как используется здесь, выражение «фармацевтически активная эфирообразующая группа" относится к группе, которая связывается с карбоксильной группой через эфирную связь. Такие эфирообразующие группы могут быть выбраны из карбокси-защитных групп, обычно использующихся для получения фармацевтически активных веществ, особенно пролекарств. Для целей настоящего изобретения, указанная группа должна быть выбрана из тех, которые способны связываться с соединениями, имеющими структуру формулы II, где R15 представляет собой водород, связанный через эфирную связь. Полученные эфиры являются эффективными для повышения стабильности, растворимости и поглощения в желудочно-кишечном тракте соответствующих неэтерифицированных форм указанных соединений, имеющих структуру формулы II, а также продлевают их эффективные уровни в крови. Кроме того, эфирная связь может легко расщепляться при рН жидкости организма или под ферментативным воздействия в условиях in vivo, для того чтобы обеспечить биологически активную форму соединения формулы II. Предпочтительные фармацевтически активные эфирообразующие группы включают, но не ограничиваются перечисленными, 1-(кислородзамещенные)-C2-С15 алкильные группы, например, неразветвленные, разветвленные, кольцевые или частично кольцевые алканоилоксиалкильные группы, такие как ацетоксиметил, ацетоксиэтил, пропионилоксиметил, пивалоилоксиметил, пивалоилоксиэтил, циклогексанацетоксиэтил, циклогексанкарбонилоксициклогексилметил и тому подобное, C3-C15 алкоксикарбонилоксиалкильнные группы, таких как этоксикарбонилоксиэтил, изопропоксикарбонилоксиметил, изопропоксикарбонилоксипропил, трет-бутоксикарбонилоксиэтил, изопентилоксикарбонилоксипропил, циклогексилоксикарбонилоксиэтил, циклогексилметоксикарбонилоксиэтил, борнилоксикарбонилоксиизопропил и тому подобное, C2-С8 алкоксиалкилы, такие как метоксиметил, метоксиэтил и тому подобное, C4-C8 2-оксициклоалкилы, такие как тетрагидропиранил, тетрагидрофуранил и тому подобное, замещенные С8-С12 аралкилы, например, фенацил, фталидил и тому подобное, C6-С12 арил, например, фенилксилил, инданил и тому подобное, C2-С12 алкенил, например, аллил, (2-оксо-1,3-диоксолил)метил и тому подобное, и [4,5-дигидро-4-оксо-1Н-пиразол[3,4-d]пиримидин-1-ил]метил и тому подобное.
В R16 в формуле II термин "эфир", используемый во фразе "эфир карбоксиметила", относится к низшему алкильному эфиру, такому как метиловый или этиловый эфир, а термин "эфир", используемый во фразе "эфир гидроксиэтила", означает эфир, который образуется путем замещения атома водорода гидроксильной группы в гидроксиэтильной группе алифатической или ароматической алкильной группой, такой как бензил.
Карбокси-защитные группы могут быть замещены различными способами. Примеры заместителей включают атом галогена, алкильные группы, алкоксигруппы, алкилтиогруппы и карбоксильные группы.
Как здесь используется, термин "прямой или разветвленный углеводородный радикал" в определении значения А в формуле II, выше, относится к метилену, этилену, пропилену, метилметилену или изопропилену.
Как здесь используется, заместителем "замещенного азота" в определении значений Y и Z в формуле II, выше, являются водород, низший алкил или ацил.
Как здесь используется, термин "фенил-замещенный низший алкил" относится к низшей алкильной группе, замещенной фенилом, такой как бензил, фенетил или фенилпропил.
Как здесь используется, термин "пролекарство" относится к производным соединения, представленным в описанных выше формуле I и формуле II, которые имеют химически или метаболически расщепляемые группы и которые за счет сольволиза или в физиологических условиях становятся соединениями, которые являются фармакологически активными в условиях in vivo. Эфиры карбоновых кислот являются примером пролекарств, которые можно использовать в дозированных формах настоящего изобретения. Метиловые эфиры пролекарств могут быть получены реакцией соединения, имеющего описанную выше формулу, в среде, такой как метанол, с кислотным или основным катализатором этерификации (например, NaOH, H2SO4). Этиловые эфиры пролекарств готовят аналогичным образом с использованием этанола вместо метанола.
Примерами соединений, имеющих вышеуказанную формулу I, являются: 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновая кислота (также известное как "фебуксостат"), 2-[3-циано-4-(3-гидрокси-2-метилпропокси)фенил]-4-метил-5-тиазолкарбоновая кислота, 2-[3-циано-4-(2-гидрокси-2-метилпропокси)фенил]-4-метил-5-тиазолкарбоновая кислота, 2-(3-циано-4-гидроксифенил)-4-метил-5-тиазолкарбоновая кислота, 2-[4-(2-карбоксипропокси)-3-цианофенил]-4-метил-5-тиазолкарбоновая кислота, 1-(3-циано-4-(2,2-диметилпропокси)фенил)-1Н-пиразол-4-карбоновая кислота, 1-3-циано-4-(2,2-диметилпропокси)фенил]-1H-пиразол-4-карбоновая кислота, пиразоло[1,5-а]-1,3,5-триазин-4-(1H)-он, 8-[3-метокси-4-(фенилсульфинил)фенил]-натриевая соль (±) или 3-(2-метил-4-пиридил)-5-циано-4-изобутоксифенил)-1,2,4-триазол.
Предпочтительными соединениями, имеющими вышеуказанную формулу I, являются: 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновая кислота, 2-[3-циано-4-(3-гидрокси-2-метилпропокси)фенил]-4-метил-5-тиазолкарбоновая кислота, 2-[3-циано-4-(2-гидрокси-2-метилпропокси)фенил]-4-метил-5-тиазолкарбоновая кислота, 2-(3-циано-4-гидроксифенил)-4-метил-5-тиазолкарбоновая кислота, 2-[4-(2-карбоксипропокси)-3-цианофенил]-4-метил-5-тиазолкарбоновая кислота. Было также обнаружено, что эти предпочтительные соединения в терапевтически эффективном количестве не влияют на активность у пациента любого из следующих ферментов, включенных в пуриновый и пиримидиновый метаболизм: гуаниндезаминазы, гипоксантин-гуанин фосфорибозилтрансферазы, фосфорилазы пуриновых нуклеотидов, оротата фосфорибозилтрансферазы или оротидин-5-монофосфата декарбоксилазы (например, это означает, что они не являются "селективными" ни для одного из этих ферментов, которые участвуют в пуриновом и пиримидиновом метаболизме). Анализы для определения активности для каждого из описанных выше ферментов описаны в Yasuhiro Takano, et al., Life Sciences, 76: 1835-1847 (2005). Эти предпочтительные соединения также были упомянуты в литературе как непуриновые селективные ингибиторы ксантиноксидазы (NP/SIXO).
Примеры соединений, имеющих приведенную выше формулу II, описаны в патенте США № 5268386 и ЕР 0415566 A1, которые включены в настоящий документ в полном объеме.
За исключением пиразоло[1,5-а]-1,3,5-триазин-4-(1H)-он, 8-[3-метокси-4-(фенилсульфинил)фенил] натриевой соли (±), способы получения соединений, ингибирующих ксантиноксидоредуктазу, формул I и II для использования в способах настоящего изобретения, известны в данной области и описаны, например, в патентах США № 5268386, 5614520, 6225474, 7074816 и ЕР 0415566 A1, и в публикации Ishibuchi, S. et al., Bioorg. Med. Chem. Lett., 11:879-882 (2001), каждый из которых включен здесь в виде ссылки. Другие соединения, ингибирующие ксантиноксидоредуктазу, могут быть найдены с помощью ксантиноксидоредуктазы и ксантина в анализах для определения таких соединений-кандидатов, ингибирующих конверсию ксантина в мочевую кислоту. Такие анализы хорошо известны в данной области.
Пиразоло[1,5-а]-1,3,5-триазин-4-(1H)-он, 8-[3-метокси-4-(фенилсульфинил)фенил]-натриевая соль (±) является доступной в Otsuka Pharmaceutical Co. Ltd. (Токио, Япония) и описана в следующих публикациях: Uematsu T., et al., "Pharmacokinetic and Pharmacodynamic Properties of a Novel Xanthine Oxidase Inhibitor, BOF-4272, in Healthy Volunteers, J. Pharmacology and Experimental Therapeutics, 270:453-459 (August 1994); Sato, S., A Novel Xanthine Degydrogenase Inhibitor (BOF-4272). In Purine and Pyrimidine Metabolism in Man, Vol. VII, Part A, ed. By P.A. Harkness, pp.135-138, Plenum Press, New York. Пиразоло[1,5-а]-1,3,5-триазин-4-(1H)-он, 8-[3-метокси-4-(фенилсульфинил)фенил]-натриевая соль (±) может быть получена с помощью обычных методов, известных в данной области.
II. Дозированные формы
Настоящее изобретение относится к твердым дозированным формам с модифицированным высвобождением, содержащим по меньшей мере одно активное вещество. В частности, по меньшей мере одно активное вещество, содержащееся в твердых дозированных формах с модифицированным высвобождением по настоящему изобретению, представляет собой по меньшей мере один ингибитор ксантиноксидоредуктазы или по меньшей мере один ингибитор ксантиноксидазы.
Дозированные формы с модифицированным высвобождением по настоящему изобретению могут обеспечивать достижение любой одной из нескольких целей. Во-первых, дозированные формы с модифицированным высвобождением по настоящему изобретению, при введении субъекту, нуждающемуся в лечении, обеспечивают высокий уровень ингибирования ксантиноксидоредуктазы или ингибирования ксантиноксидазы при максимальной наблюдаемой концентрации в плазме (в частности, Cmax), которая существенно ниже, чем обеспечивается дозированной формой с немедленным высвобождением, содержащей по меньшей мере один ингибитор ксантиноксидоредуктазы (например, дозированная форма с немедленным высвобождением, содержащая 40 мг, 80 мг, 120 мг или 240 мг фебуксостата, которую вводят субъекту один раз в день), аналогичный или ниже, чем достигается наиболее высокими дозами ингибиторов ксантиноксидоредуктазы в настоящее время (а именно, в настоящее время дозы (например, 80 мг (в США) или 120 мг (Европа)) 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты (которая также известна как фебуксостат)) или по меньшей мере одним ингибитором ксантиноксидазы (например, дозированная форма с немедленным высвобождением, содержащая 300 мг аллопуринола, которую вводят субъекту один раз в день). Во-вторых, поскольку дозированные формы по настоящему изобретению обеспечивают ингибирование ксантиноксидоредуктазы или ингибирование ксантиноксидазы в течение длительных периодов времени (дозирования), эти твердые дозированные формы могут быть использованы для лечения различных состояний или заболеваний, таких как, но не ограничиваясь перечисленными, подагра, гиперурикемия, простатит, воспалительное заболевания кишечника, удлинение интервала QT, инфаркт миокарда, гипертрофия сердца, гипертензия, почечнокаменная болезнь, почечная недостаточность, хроническое заболевание почек, метаболический синдром, диабет, диабетическая нефропатия, застойная сердечная недостаточность и другие нарушения. В-третьих, дозированные формы с модифицированным высвобождением по настоящему изобретению защищают пациентов, получающих эти дозированные формы на протяжении всей схемы лечения, от увеличения концентрации свободных радикалов кислорода.
Для того чтобы получить эти преимущества, дозированные формы с модифицированным высвобождением по настоящему изобретению должны достигать определенного фармакокинетического профиля по сравнению с дозированными формами с немедленным высвобождением ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы.
В одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, содержащие по меньшей мере один ингибитор ксантиноксидоредуктазы, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют по меньшей мере два из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 24 часов, или (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл. В другом воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют по меньшей мере два из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 24 часов, или (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,0 мкг/мл до приблизительно 1,0 мкг/мл. В еще одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют каждое из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в течение от приблизительно 5 часов до приблизительно 24 часов, и (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл.
Как уже упоминалось ранее в настоящем документе, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут поддерживать в плазме крови пациента концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 24 часов. Более конкретно, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут поддерживать в плазме крови пациента концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в течение приблизительно 5,0 часов, приблизительно 6,0 часов, приблизительно 7,0 часов, приблизительно 8,0 часов, приблизительно 9,0 часов, приблизительно 10,0 часов, приблизительно 11,0 часов, приблизительно 12,0 часов, приблизительно 13,0 часов, приблизительно 14,0 часов, приблизительно 15,0 часов, приблизительно 16,0 часов, приблизительно 17,0 часов, приблизительно 18,0 часов, приблизительно 19,0 часов, приблизительно 20,0 часов, приблизительно 21,0 часа, приблизительно 22,0 часов, приблизительно 23,0 часов или приблизительно 24,0 часов.
Как уже упоминалось ранее в настоящем документе, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл (также как и любую комбинацию диапазонов между этими значениями, такую как, например, от приблизительно 2,5 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 1,0 мкг/мл и т.д.). Более конкретно, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве приблизительно 2,5 мкг/мл, приблизительно 2,4 мкг/мл, приблизительно 2,3 мкг/мл, приблизительно 2,2 мкг/мл, приблизительно 2,1 мкг/мл, 2,0 мкг/мл, приблизительно 1,9 мкг/мл, приблизительно 1,8 мкг/мл, приблизительно 1,7 мкг/мл, приблизительно 1,6 мкг/мл, приблизительно 1,5 мкг/мл, приблизительно 1,4 мкг/мл, приблизительно 1,3 мкг/мл, приблизительно 1,2 мкг/мл, приблизительно 1,1 мкг/мл, приблизительно 1,0 мкг/мл, приблизительно 0,9 мкг/мл, приблизительно 0,8 мкг/мл, приблизительно 0,7 мкг/мл, приблизительно 0,6 мкг/мл или приблизительно 0,5 мкг/мл.
Дозированные формы настоящего изобретения могут содержать от приблизительно 5 мг до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы. Более конкретно, дозированная форма может содержать приблизительно 5 мг, приблизительно 6,25 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, примерно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг или приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы.
В другом воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, содержащие по меньшей мере один ингибитор ксантиноксидоредуктазы, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют по меньшей мере два из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 16 часов, или (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,05 мкг/мл. В еще одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют по меньшей мере два из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 16 часов, или (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,0 мкг/мл до приблизительно 0,075 мкг/мл. В еще одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют каждое из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 16 часов, и (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,05 мкг/мл.
Как уже упоминалось ранее, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут поддерживать в плазме крови пациента концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 16 часов. Более конкретно, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут поддерживать в плазме крови пациента концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в течение приблизительно 5,0 часов, приблизительно 6,0 часов, приблизительно 7,0 часов, приблизительно 8,0 часов, приблизительно 9,0 часов, приблизительно 10,0 часов, приблизительно 11,0 часов, приблизительно 12,0 часов, приблизительно 13,0 часов, приблизительно 14,0 часов, приблизительно 15,0 часов или приблизительно 16,0 часов.
Кроме того, как уже здесь упоминалось ранее, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,05 мкг/мл (также как и любую комбинацию диапазонов между этими значениями, такую как, например, от приблизительно 2,5 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,06 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,07 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,08 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,09 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,9 мкг/мл или от приблизительно 1,5 мкг/мл до приблизительно 1,0 мкг/мл). Более конкретно, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве 2,5 мкг/мл, приблизительно 2,4 мкг/мл, приблизительно 2,3 мкг/мл, приблизительно 2,2 мкг/мл, приблизительно 2,1 мкг/мл, приблизительно 2,0 мкг/м, приблизительно 1,9 мкг/мл, приблизительно 1,8 мкг/мл, приблизительно 1,7 мкг/мл, приблизительно 1,6 мкг/мл, приблизительно 1,5 мкг/мл, приблизительно 1,4 мкг/мл, приблизительно 1,3 мкг/мл, приблизительно 1,2 мкг/мл, приблизительно 1,1 мкг/мл, приблизительно 1,0 мкг/мл, приблизительно 0,9 мкг/мл, приблизительно 0,8 мкг/мл, приблизительно 0,7 мкг/мл, приблизительно 0,6 мкг/мл, приблизительно 0,5 мкг/мл, приблизительно 0,4 мкг/мл, приблизительно 0,3 мкг/мл, приблизительно 0,2 мкг/мл, приблизительно 0,1 мкг/мл, приблизительно 0,099 мкг/мл, приблизительно 0,098 мкг/мл, приблизительно 0,097 мкг/мл, приблизительно 0,096 мкг/мл, приблизительно 0,095 мкг/мл, приблизительно 0,094 мкг/мл, приблизительно 0,093 мкг/мл, приблизительно 0,092 мкг/мл, приблизительно 0,091 мкг/мл, приблизительно 0,090 мкг/мл, приблизительно 0,089 мкг/мл, приблизительно 0,088 мкг/мл, приблизительно 0,087 мкг/мл, приблизительно 0,086 мкг/мл, приблизительно 0,085 мкг/мл, приблизительно 0,084 мкг/мл, приблизительно 0,083 мкг/мл, приблизительно 0,082 мкг/мл, приблизительно 0,081 мкг/мл, приблизительно 0,080 мкг/мл, приблизительно 0,079 мкг/мл, приблизительно 0,078 мкг/мл, приблизительно 0,077 мкг/мл, приблизительно 0,076 мкг/мл, приблизительно 0,075 мкг/мл, приблизительно 0,074 мкг/мл, приблизительно 0,073 мкг/мл, приблизительно 0,072 мкг/мл, приблизительно 0,071 мкг/мл, приблизительно 0,070 мкг/мл, приблизительно 0,069 мкг/мл, приблизительно 0,068 мкг/мл, приблизительно 0,067 мкг/мл, приблизительно 0,066 мкг/мл, приблизительно 0,065 мкг/мл, приблизительно 0,064 мкг/мл, приблизительно 0,063 мкг/мл, приблизительно 0,062 мкг/мл, приблизительно 0,061 мкг/мл, приблизительно 0,060 мкг/мл, приблизительно 0,059 мкг/мл, приблизительно 0,058 мкг/мл, приблизительно 0,057 мкг/мл, приблизительно 0,056 мкг/мл, приблизительно 0,055 мкг/мл, приблизительно 0,054 мкг/мл, приблизительно 0,053 мкг/мл, приблизительно 0,052 мкг/мл, приблизительно 0,051 мкг/мл или приблизительно 0,050 мкг/мл.
Дозированные формы настоящего изобретения могут содержать от приблизительно 5 мг до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы. Более конкретно, дозированная форма может содержать приблизительно 5 мг, приблизительно 6,25 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, примерно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг или приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы.
В еще одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, содержащие по меньшей мере один ингибитор ксантиноксидоредуктазы, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют по меньшей мере два из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 14 часов, или (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,90 мкг/мл. В еще одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют по меньшей мере два из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 14 часов, или (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,0 мкг/мл до приблизительно 0,95 мкг/мл. В еще одном воплощении дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, демонстрируют каждое из следующего: (а) поддержание у пациента в плазме крови концентрации ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 14 часов, и (b) продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,90 мкг/мл.
Как уже упоминалось ранее в настоящем документе, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут поддерживать в плазме крови пациента концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в период от приблизительно 5 часов до приблизительно 14 часов. Более конкретно, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут поддерживать в плазме крови пациента концентрацию ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли больше, чем приблизительно 0,1 мкг/мл в течение приблизительно 5,0 часов, приблизительно 6,0 часов, приблизительно 7,0 часов, приблизительно 8,0 часов, приблизительно 9,0 часов, приблизительно 10,0 часов, приблизительно 11,0 часов, приблизительно 12,0 часов, приблизительно 13,0 часов или приблизительно 14,0 часов.
Кроме того, как уже упоминалось ранее в настоящем документе, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве от приблизительно 2,5 мкг/мл до приблизительно 0,090 мкг/мл (также как и любую комбинацию диапазонов между этими значениями, такую как, например, от приблизительно 2,5 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,5 мкг/мл от приблизительно 2,5 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,5 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,4 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,3 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,2 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,1 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 2,0 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,9 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,8 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,05 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,7 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 0,9 мкг/мл, от приблизительно 1,6 мкг/мл до приблизительно 1,0 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,1 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,2 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,3 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,40 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,5 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,6 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,7 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,8 мкг/мл, от приблизительно 1,5 мкг/мл до приблизительно 0,9 мкг/мл или от приблизительно 1,5 мкг/мл до приблизительно 1,0 мкг/мл). Более конкретно, дозированные формы с модифицированным высвобождением по настоящему изобретению, после перорального введения субъекту, нуждающемуся в лечении, могут продуцировать у пациента максимальную концентрацию в плазме (Cmax) ингибитора ксантиноксидоредуктазы или его фармацевтически приемлемой соли в количестве 2,5 мкг/мл, приблизительно 2,4 мкг/мл, приблизительно 2,3 мкг/мл, приблизительно 2,2 мкг/мл, приблизительно 2,1 мкг/мл, приблизительно 2,0 мкг/мл, приблизительно 1,9 мкг/мл, приблизительно 1,8 мкг/мл, приблизительно 1,7 мкг/мл, приблизительно 1,6 мкг/мл, приблизительно 1,5 мкг/мл, приблизительно 1,4 мкг/мл, приблизительно 1,3 мкг/мл, приблизительно 1,2 мкг/мл, приблизительно 1,1 мкг/мл, приблизительно 1,0 мкг/мл, приблизительно 0,9 мкг/мл, приблизительно 0,8 мкг/мл, приблизительно 0,7 мкг/мл, приблизительно 0,6 мкг/мл, приблизительно 0,5 мкг/мл, приблизительно 0,4 мкг/мл, приблизительно 0,3 мкг/мл, приблизительно 0,2 мкг/мл, приблизительно 0,1 мкг/мл, приблизительно 0,099 мкг/мл, приблизительно 0,098 мкг/мл, приблизительно 0,097 мкг/мл, приблизительно 0,096 мкг/мл, приблизительно 0,095 мкг/мл, приблизительно 0,094 мкг/мл, приблизительно 0,093 мкг/мл, приблизительно 0,092 мкг/мл, приблизительно 0,091 мкг/мл или приблизительно 0,090 мкг/мл.
Дозированные формы настоящего изобретения могут содержать от приблизительно 5 мг до приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы. Более конкретно, дозированная форма может содержать приблизительно 5 мг, приблизительно 6,25 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, приблизительно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг или приблизительно 240 мг по меньшей мере одного ингибитора ксантиноксидоредуктазы.
Способы определения Cmax ингибиторов ксантиноксидоредуктазы и концентрации ингибиторов ксантиноксидоредуктазы в плазме хорошо известны в данной области. Для того чтобы определить процент ингибирования ксантиноксидоредуктазы, представленной в дозированной форме, может быть использовано следующее уравнение:
Процент ингибирования ("% ингибирования") активности ксантиноксидоредуктазы:
% Ингибирования ксантиноксидоредуктазы = 100 
где C = концентрация ингибитора ксантиноксидоредуктазы в плазме крови ("XORI") у пациента,
fu = свободная фракция XORI в плазме и
Ki = константа ингибирования ксантиноксидоредуктазы XORI.
Концентрацию XORI в плазме крови можно определить с помощью методов, известных в данной области, таких как высокоэффективная жидкостная хроматография с флуоресцентной детекцией или тандемная высокоэффективная жидкостная хроматография с подтверждающей масс-спектрометрией (см. Mayer, M. et al., American Journal of Therapeutics, 12:22-34 (2005)). Значение fu может быть определено с помощью анализа связывания in vitro 14C XORI при номинальной концентрации 1 мкг/мл, используя метод равновесного диализа, который хорошо известен в данной области. Например, значение fu для XORI, такого как фебуксостат, было определено как 0,9±0,2 у нормальных пациентов, и как 1,2±0,2 у пациентов с тяжелой почечной недостаточностью (см. Mayer, M. et al., American Journal of Therapeutics, 12:22-34 (2005)). В другом исследовании, с большим числом субъектов, доля свободной фракции фебуксостата в плазме была определена как 0,7±0,1 в мужской, женской, подростковой и пожилой группе испытуемых (см. Khosravan R., et al, Clinic. Pharmacology & Therapeutics, P50 (2005)).
Значение Ki для XORI может быть определено с помощью обычных методов, известных в данной области. Например, Ki для XORI, такого как фебуксостат, была определена с использованием анализа ксантиноксидазы, такого, как описано в Osada Y., et al., European J. Pharmacology, 241: 183-188 (1993). Более конкретно, Ki для фебуксостата была определена как 0,7 нМ и 0,6 нМ, соответственно (см. Osada Y., et al., European J. Pharmacology, 241: 183-188 (1993) и Takano, Y., et al., Life Sciences, 76: 1835-1847 (2005)).
В еще одном варианте воплощения дозированные формы с модифицированным высвобождением по настоящему изобретению содержат по меньшей мере один ингибитор ксантиноксидазы. Эти дозированные формы с модифицированным высвобождением, содержащие по меньшей мере один ингибитор ксантиноксидазы, после их перорального приема субъектом, как ожидается, будут поддерживать критическую концентрацию в плазме в течение более длительного периода по сравнению с препаратами немедленного высвобождения, содержащими аллопуринол, ингибируя, тем самым, целевой фермент в течение длительного периода времени. Таким образом, эти дозированные формы с модифицированным высвобождением будут более выгодным по сравнению с таблетками немедленного высвобождения, так как эти дозированные формы с модифицированным высвобождением позволят сократить различия среди пациентов, связанные с вариацией времен полужизни оксипуринола и аллопуринола, улучшая тем самым терапевтический результат.
Дозированные формы настоящего изобретения могут содержать, в дополнение к ингибитору ксантиноксидоредуктазы или ингибитору ксантиноксидазы, другие лекарственные вещества. Эти другие лекарственные вещества могут быть выбраны из любых различных классов агентов, включая, но не ограничиваясь перечисленным, нестероидные противовоспалительные средства, анальгетики, анестетики, антиангинальные средства, антиартрические средства, антиаритмические агенты, противоастматические средства, антибактериальные средства, средства против высокого давления крови (BPH), противоопухолевые агенты, антихолинергические средства, антикоагулянты, противосудорожные препараты, антидепрессанты, противодиабетические средства, антидиарейные, антиэпилептические средства, противогрибковые средства, средства против подагры, противоглистные средства, антигистаминные средства, антигипертензивные средства, противовоспалительные средства, противомалярийные средства, средства против мигрени, антимускариновые агенты, средства против тошноты, противоопухолевые средства, средства для похудения, средства против остеопороза, антипаркинсонические средства, антипротозойные средства, средства против зуда, антипсихотические средства, жаропонижающие средства, спазмолитики, антитиреоидные средства, противотуберкулезные средства, противоязвенные средства, противовоспалительные средства, средства против недержания мочи, противовирусные средства, транквилизаторы, средства для подавления аппетита, средства для лечения расстройства, связанного с дефицитом внимания (ADD), и синдрома дефицита внимания и гиперактивности (ADDH), блокаторы кальциевых каналов, сердечные инотропные средства, бета-блокаторы, стимуляторы центральной нервной системы, когнитивные усилители, кортикостероиды, ингибиторы COX-2, противоотечные средства, мочегонные средства, желудочно-кишечные средства, генетический материал, препаратов, используемых для лечения подагры (например, колхицин; урикозурические средства, такие как пробенецид, сульфинпиразон, бензиодарон; ингибиторы ксантиноксидазы, такие как оксипуринол, аллопуринол, и т.д.), антагонисты рецепторов гистамина, гормонолитики, снотворные средства, сахаропонижающие средства, иммунодепрессанты, кератолитики, ингибиторы лейкотриенов, средства, регулирующие липиды, макролиды, ингибиторы митоза, миорелаксанты, наркотические антагонисты, нейролептики, никотин, пищевые масла, производные ксантина (такие как, но не ограничиваясь перечисленными, кофеин и производные кофеина), парасимпатолитические средства, седативные средства, половые гормоны, симпатомиметики, транквилизаторы, сосудорасширяющие средства, витамины и их комбинации. Любое из вышеуказанных лекарственных средств может вводиться в сочетании с ингибиторами ксантиноксидоредуктазы или ингибиторами ксантиноксидазы, используемыми в дозированных формах настоящего изобретения.
Преимущества настоящего изобретения не ограничиваются единственным типом дозированной формы, имеющим конкретный механизм высвобождения лекарственного вещества. Этот улучшенный фармакокинетический профиль может быть получен с любой из пероральных дозированных форм с отсроченным высвобождением, известных в данной области, такими как, но не ограничиваясь перечисленными, дозированная форма с многостадийным (импульсным) высвобождением, дозированная форма с отсроченным высвобождением или дозированная форма с пролонгированным высвобождением.
Многие различные типы пероральных дозированных форм с модифицированным высвобождением на основе полимеров известны в данной области и предполагаются для использования в рамках настоящего изобретения. Примеры трех различных типов пероральных дозированных форм с модифицированным высвобождением на основе полимеров, таких как матричные системы, осмотический насос или на основе технологий контролируемых мембран (также называемые резервуарные системы), более подробно описаны ниже. Подробное обсуждение этих дозированных форм может также быть найдено в: (i) Handbook of pharmaceutical controlled release technology, ed. D. L. Wise, Marcel Dekker, Inc. New York, N.Y. (2000), и (ii) Treatise on controlled drug delivery, fundamentals, optimization, and applications, ed. A. Kydonieus, Marcel Dekker, Inc. New York, N.Y. (1992), содержание которых включено в виде ссылки.
Однако, хотя эти три пероральные дозированные формы с модифицированным высвобождением на основе полимеров описаны более подробно, другие дозированные формы с модифицированным высвобождением, известные специалистам в данной области, охватываются объемом притязаний настоящего изобретения.
Матричные системы
Матричные системы хорошо известны в данной области. В матричной системе лекарственное вещество гомогенно диспергировано в полимере в сочетании с обычными эксципиентами. Для получения таблеток эта заранее приготовленная смесь, как правило, прессуется под давлением. Лекарственное вещество высвобождается из такой таблетки за счет диффузии и эрозии. Матричные системы подробно описаны у Wise и Kydonieus, см. выше.
Матричные дозированные формы по настоящему изобретению могут содержать ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы и фармацевтически приемлемый полимер. В одном аспекте ингибитор ксантиноксидоредуктазы представляет собой 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновую кислоту. В другом аспекте ингибитор ксантиноксидазы представляет собой аллопуринол.
Фармацевтически приемлемый полимер является водорастворимым гидрофильным полимером или нерастворимым в воде гидрофобным полимером (включая воски). Примеры подходящих водорастворимых полимеров включают поливинилпирролидон, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, метилцеллюлозу, сополимеры винилацетата, полисахариды (такие как альгинат, ксантановая смола и т.п.), полиэтиленоксид, сополимеры метакриловой кислоты, сополимеры малеинового ангидрида/метилвинилового эфира, их производные и их смеси. Примеры подходящих нерастворимых в воде полимеров включают акрилаты, производные целлюлозы, такие как ацетат этилцеллюлозы или целлюлозы, полиэтилен, метакрилаты, сополимеры акриловой кислоты и поливиниловых спиртов с высоким молекулярным весом. Примеры подходящих восков включают жирные кислоты и глицериды.
В одном аспекте изобретения полимер выбран из гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы и метилцеллюлозы. В другом аспекте полимер представляет собой гидроксипропилметилцеллюлозу. В еще одном аспекте полимер представляет собой гидроксипропилметилцеллюлозу высокой вязкости, вязкость которой составляет от приблизительно 4000 сПз до приблизительно 100000 сПз. Наиболее предпочтительным полимером высокой вязкости является гидроксипропилметилцеллюлоза с вязкостью приблизительно 15000 сПз, коммерчески доступная под торговым названием Methocel® от компании Dow Chemical.
Количество полимера в дозированной форме обычно варьирует от примерно 10% до примерно 70% по массе композиции.
Дозированные формы настоящего изобретения, как правило, включают фармацевтически приемлемые эксципиенты. Как хорошо известно специалистам в данной области техники, фармацевтические эксципиенты обычно включены в твердые дозированные формы. Это делается для того, чтобы облегчить производственный процесс, а также для удобства работы с дозированной формой. Обычные эксципиенты включают разбавители или объемообразующие средства, лубриканты, связующие и т.п. Такие эксципиенты могут использоваться в дозированных формах настоящего изобретения.
Разбавители или наполнители могут быть добавлены для того, чтобы увеличить массу индивидуальных доз до размеров, пригодных для прессования таблеток. Подходящие разбавители включают сахарную пудру, фосфат кальция, сульфат кальция, микрокристаллическую целлюлозу, лактозу, маннит, каолин, хлорид натрия, сухой крахмал, сорбит и т.п.
Лубриканты могут быть включены в дозируемую форму для различных целей. Лубриканты уменьшают трение между гранулятом и стенками пресс-формы при прессовании и выталкивании. Это предотвращает прилипание гранулята к таблеточной пресс-форме, облегчает его выброс из таблеточной пресс-формы и т.п. Примеры подходящих лубрикантов, которые можно использовать, включают, но не ограничиваются перечисленными, тальк, стеариновую кислоту, растительное масло, стеарат кальция, стеарат цинка, стеарат магния и т.п.
Глиданты также могут быть включены в дозированную форму. Глидант улучшает реологические характеристики гранулята. Примеры подходящих глидантов включают, но не ограничиваются перечисленными, тальк, диоксид кремния и кукурузный крахмал.
Связующие вещества могут быть включены в дозированную форму. Связующие вещества обычно используются при изготовлении дозированной формы, если производство включает стадию грануляции. Примеры подходящих связующих веществ включают, но не ограничиваются перечисленными, повидон, поливинилпирролидон, ксантановую камедь, целлюлозные смолы, такие как карбоксиметилцеллюлоза, метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксицеллюлоза, желатин, крахмал и предварительно желатинизированный крахмал.
Другие эксципиенты, которые могут быть включены в дозированную форму, включают, но не ограничиваются перечисленным, консерванты, антиоксиданты или любой другой эксципиент, обычно используемый в фармацевтической промышленности, и т.д. Количество эксципиентов, используемых в дозированной форме, будет соответствовать обычно используемым в матричной системе. Общее количество эксципиентов, разбавителей, наполнителей и т.п. может варьироваться в пределах от примерно 10% до примерно 70% от массы дозированной формы.
Матричные дозированные формы получают, как правило, с использованием стандартных методов, хорошо известных в данной области. Как правило, их получают путем сухого смешивания полимера, наполнителя, ингибитора ксантиноксидоредуктазы, такого как 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновая кислота, или ингибитора ксантиноксидазы, такого как аллопуринол и оксипуринол, и других эксципиентов, с последующим гранулированием смеси с использованием спирта, до получения гранулята надлежащего качества. Грануляция осуществляется способами, известными в данной области. Влажные гранулы сушат в сушилке с псевдоожиженным слоем, просеивают и измельчают до соответствующего размера. Смазывающие вещества смешивают с высушенным гранулятом для получения конечной дозированной формы.
Альтернативно, матричные дозированные формы можно изготавливать с помощью прямого прессования порошковой, кристаллической или гранулированной композиции, содержащей активный(е) агент(ы) отдельно или в сочетании с одним или несколькими носителями, добавками, или тому подобными. Методы прямого прессования хорошо известны в данной области.
Дозированные формы настоящего изобретения могут быть введены перорально в виде таблеток, пилюль или гранулята, который может быть свободно помещен в капсулы. Таблетки могут быть получены способами, известными в данной области, и они содержат терапевтически эффективное количество ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, а также такие наполнители, которые необходимы для формирования таблетки соответствующим способом. Таблетки и пилюли могут быть дополнительно изготовлены с энтеросолюбильным покрытием и другими покрытиями, контролирующими высвобождение, с целью защиты от кислот, облегчения проглатывания и т.п. Покрытие может быть окрашено фармацевтически приемлемым красителем. Количество красителя и других эксципиентов в жидкости для покрытия может варьироваться, и оно не будет влиять на удобство работы с таблетками с модифицированным высвобождением. Жидкость для покрытия обычно содержит пленкообразующие полимеры, такие как, но не ограничиваясь перечисленными, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, сложные или простые эфиры целлюлозы (такие как ацетат целлюлозы или этилцеллюлозы), акриловый полимер или смесь полимеров. Раствор для нанесения покрытия, как правило, представляет собой водный раствор или раствор в органическом растворителе, дополнительно содержащий пропиленгликоль, сорбитан моноолеат, сорбиновую кислоту, наполнители, такие как диоксид титана, и фармацевтически приемлемый краситель.
Осмотические насосы
В системе осмотического насоса таблеточное ядро заключено в полупроницаемую мембрану, имеющую по меньшей мере одно отверстие. Полупроницаемая мембрана проницаема для воды, но непроницаема для лекарственного вещества. Когда система подвергается воздействию жидкостей организма, вода будет проникать через полупроницаемую мембрану в таблеточное ядро, содержащее осмотические эксципиенты и активное лекарственное вещество. Осмотическое давление увеличивается в дозированной форме, и лекарственное вещество высвобождается через отверстие для того, чтобы уравнять давление.
В более сложных насосах таблеточное ядро содержит два внутренних отдела. Первый отдел содержит лекарственное вещество. Второй отдел содержит полимер, который набухает при контакте с жидкостью. После приема препарата этот полимер набухает, воздействуя на отдел, содержащий лекарственное средство, с заданной скоростью и силой, обеспечивая вытеснение препарата из дозированной формы с такой скоростью. Такие дозированные формы часто используются, когда желателен нулевой порядок профиля высвобождения.
Осмотические насосы хорошо известны в данной области и описаны в литературе. Патенты США № 4088864, 4200098 и 5573776, которые включены здесь в виде ссылки, описывают осмотические насосы и способы их изготовления.
В качестве общей рекомендации, осмотические насосы настоящего изобретения могут быть получены путем прессования таблетки осмотически активного лекарственного вещества (или осмотически неактивного лекарственного вещества в сочетании с осмотически активными средствами или осмоагентами), с последующим покрытием таблетки полупроницаемой мембраной, которая проницаема для внешней жидкости на водной основе, но непроницаема для прохождения лекарственного средства и/или осмоагента. Могут быть созданы одно или несколько отверстий для доставки через полупроницаемую оболочку мембраны. Альтернативно, отверстие(я) в оболочке могут быть образованы in situ путем включения в оболочку материалов, формирующих выщелачиваемые поры. В процессе функционирования внешняя жидкость на водной основе впитывается через полупроницаемую мембрану оболочки и контактирует с лекарственным веществом и/или солью, формируя раствор или суспензию лекарственного вещества. Затем раствор или суспензия лекарственного вещества выдавливается через отверстие, по мере того как свежая жидкость проникает через полупроницаемую мембрану.
В одном воплощении таблетка содержит два различных отдела. Первый отдел содержит лекарственное вещество, как описано выше. Второй отдел содержит расширяемое вытесняющее средство, состоящее из слоя набухающего гидрофильного полимера, который функционирует с целью уменьшения объема, занимаемого лекарственным веществом, тем самым высвобождая лекарственное вещество из устройства с контролируемой скоростью в течение длительного периода времени.
Типичные материалы для полупроницаемой мембраны включают полупроницаемые полимеры, известные в данной области как мембраны для осмоса и обратного осмоса, такие как ацилированная целлюлоза, диацилированная целлюлоза, триацилированная целлюлоза, ацетат целлюлозы, диацетат целлюлозы, триацетат целлюлозы, ацетат агара, триацетат амилозы, ацетат бета-глюкана, ацетат диметилацетальдегида, ацетат этилкарбамат целлюлозы, полиамиды, полиуретаны, сульфированные полистиролы, ацетат фталат целлюлозы, ацетат метилкарбамат целлюлозы, ацетат сукцинат целлюлозы, ацетат диметилглицин целлюлозы, ацетат этилкарбамат целлюлозы, ацетат хлорацетат целлюлозы, дипальмитат целлюлозы, диоктаноат целлюлозы, дикаприлат целлюлозы, дипентанлат целлюлозы, ацетат валерат целлюлозы, ацетат сукцинат целлюлозы, пропионат сукцинат целлюлозы, метилцеллюлоза, ацетат п-толуолсульфонат целлюлозы, ацетат бутират целлюлозы, поперечно сшитые полимеры селективной полупроницаемости, полученные соосаждением полианиона и поликатиона, как описано в патентах США № 3173876, 3276586, 3541005, 3541006 и 3546142, полупроницаемые полимеры, как раскрыто в патенте США № 3133132 (Loeb, Sourirajan), частично поперечно сшитые производные полистирола, поперечно сшитый поли(стирол-сульфонат натрия), поли(винилбензилтриметилхлорид аммония), ацетат целлюлозы, имеющий степень замещения вплоть до 1 и с содержанием ацетила до 50%, диацетат целлюлозы, имеющий степень замещения от 1 до 2 и с содержанием ацетила от 21 до 35%, триацетат целлюлозы, имеющий степень замещения от 2 до 3 и с содержанием ацетила от 35 до 44,8%, как описано в патенте США № 4160020.
Осмотически активное средство, представленное в насосе, которое может быть использовано, когда препарат сам по себе не является осмотически активным, представляет собой осмотически эффективное соединение, растворимое в жидкости, которая поступает в устройство, и имеет градиент осмотического давления между полупроницаемой оболочкой и внешней жидкостью. Осмотически эффективные осмоагенты, полезные для данной цели, включают, но не ограничиваются перечисленными, сульфат магния, сульфат кальция, хлорид магния, хлорид натрия, хлорид лития, сульфат калия, карбонат натрия, сульфит натрия, сульфат лития, хлорид калия, сульфат натрия, d-маннит, мочевину, сорбит, инозит, раффинозу, сахарозу, глюкозу, гидрофильные полимеры, такие как целлюлозные полимеры, их смеси, и тому подобное. Осмоагент обычно присутствует в избытке, и он может быть в любой физической форме, такой как частицы, порошок, гранулы и тому подобное. Осмотическое давление для осмоагента (в атмосферах), приемлемого для целей изобретения, будет иметь значение выше нуля и обычно достигает 500 атм или выше.
Расширяемое вытесняющее средство представляет собой, как правило, набухающий гидрофильный полимер, который взаимодействует с водой и водными биологическими жидкостями, и который разбухает или расширяется до равновесного состояния. Полимеры обладают способностью к набуханию в воде и сохраняют значительную часть впитавшейся воды в структуре полимера. Полимеры набухают или расширяются до очень высокой степени, проявляя, как правило, увеличение объема от 2- до 50-кратного. Полимеры могут быть поперечно и не поперечно сшитыми. Набухающие гидрофильные полимеры могут быть частично поперечно сшитыми, при этом такие поперечные сшивки образуются за счет ковалентных ионных связей или водородных связей. Полимеры могут быть растительного, животного или синтетического происхождения. Гидрофильные полимеры, пригодные для использования в настоящем изобретении, включают, но не ограничиваются перечисленными, поли(гидроксиалкилметакрилат) с молекулярной массой от 30000 до 5000000; каппа каррагинан, поливинилпирролидон с молекулярной массой от 10000 до 360000; анионные и катионные гидрогели; полиэлектролитные комплексы; поли(виниловый спирт), имеющий низкое содержание остатков уксусной кислоты, поперечно сшитый с глиоксалем, формальдегидом или глутаральдегидом, и имеющий степень полимеризации от 200 до 30000; смесь метилцеллюлозы; поперечно сшитый агар и карбоксиметилцеллюлоза; нерастворимый в воде и набухающий в воде сополимер, полученный путем образования тонкой дисперсии сополимера малеинового ангидрида со стиролом, этиленом, пропиленом, бутиленом и изобутиленом, сшитый с от 0,001 до 0,5 молей на моль малеинового ангидрида насыщенным агентом для поперечной сшивки; набухающие в воде полимеры N-виниллактамов, и тому подобное.
Выражение "отверстие", как используется здесь, включает средства и методы, подходящие для высвобождения лекарственного вещества из системы. Выражение включает одно или несколько выходов или отверстий в полупроницаемой мембране, которые были проделаны механическими способами. Альтернативно, оно может быть получено путем включения разрушаемого элемента, такого как желатиновая пробка, в полупроницаемую мембрану. В случаях, когда полупроницаемая мембрана является достаточно проницаемой для прохождения лекарственного вещества, наличие пор в мембране может быть достаточно, чтобы высвободить агент/лекарственное вещество в терапевтически эффективных количествах. В таких случаях выражение "выходное отверстие" относится к порам в мембране оболочки, даже если нет отверстия или какие-либо другие отверстия были проделаны в ней. Подробное описание осмотических выходных отверстий, максимальные и минимальные размеры выходных отверстий раскрыты в патентах США № 3845770 и 3916899, содержание которых включено в виде ссылки.
Осмотические насосы настоящего изобретения могут быть изготовлены стандартными способами. Например, в одном варианте лекарственное вещество и другие ингредиенты, которые могут быть размещены в одном отделе у выходного отверстия, прессуются в виде твердого тела, обладающего размерами, которые соответствуют внутренним размерам отдела, который будет занимать активный агент, или активный агент и другие ингредиенты и растворитель смешиваются в твердые или полутвердые формы с помощью традиционных методов, таких как шаровой помол, каландрование, перетирание или помол с помощью валков, а затем прессуются в виде предварительно заданной формы. Затем подобным способом приводят в контакт слой гидрофильного полимера со слоем активного агента, и оба слоя окружают полупроницаемой оболочкой. Наслоения препарата активного агента и гидрофильного полимера могут быть выполнены обычными способами двухслойного прессования. Оболочки могут быть выполнены литьем, распылением или погружением выпрессованной формы в материал, образующий оболочку. Другой способ, который можно использовать для нанесения оболочки, представляет собой способ пневматического нанесения суспензии. Этот способ состоит из суспендирования и обволакивания выпрессованного активного агента гидрофильным полимером в потоке воздуха до тех пор, пока не сформируется композиция, образующая оболочку состава из активного агента и гидрофильного полимера. Способ пневматического нанесения суспензии описан в патенте США № 2799241; J. Am. Pharm. Assoc., Vol. 48, pp. 451-459, (1979). Другие стандартные способы и приемы описаны в Modern Plastics Encyclopedia, Vol. 46, pp. 62-70 (1969); и в Remington's Pharmaceutical Sciences, Fourteenth Edition, pp. 1626-1678 (1970), Mack Publishing Company, Easton, Pa.
Резервуарные полимерные системы
Резервуарные системы хорошо известны в данной области. Эта технология также обычно касается микрокапсуляции, технологии получения микрогранул или таблеток, покрытых оболочкой. Мелкие частицы препарата инкапсулируются с фармацевтически приемлемым(и) полимером(ами). Этот полимер, и его относительное количество, обеспечивает предопределенную диффузию лекарственного вещества из резервуара в желудочно-кишечный тракт. Таким образом, лекарственное вещество постепенно высвобождается из гранул в желудочно-кишечный тракт и обеспечивает желаемое контролируемое высвобождение (1) ингибитора ксантиноксидоредуктазы, такого как 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновая кислота, или (2) ингибитора ксантиноксидазы, такого как аллопуринол и оксипуринол.
Эти дозированные формы хорошо известны в данной области. Патенты США № 5286497 и 5737320, каждый из которых включен здесь в виде ссылки, описывают такие препараты и способы их получения. Специалист в данной области, принимая во внимание раскрытие настоящего изобретения и а также вышеуказанные патенты '320 и '497, может изготовить дозированную форму на гранулированной или пеллетизированной основе, формирующую фармакокинетический профиль, описанный выше.
В качестве общей рекомендации необходимо отметить, что гранула формируется с инертным сферическим ядром и ингибитором ксантиноксидоредуктазы или ингибитором ксантиноксидазы, и необязательно, в сочетании с обычными эксципиентами. Ядро указанных гранулы может необязательно включать любые материалы, обычно использующиеся в фармацевтике, и которые должны быть выбраны на основе совместимости с активным лекарственным веществом и с учетом физико-химических свойств гранул. Дополнительные компоненты могут включать, но не ограничиваясь перечисленными, связующие вещества, дезинтегранты, наполнители, поверхностно-активные вещества, солюбилизаторы, стабилизаторы и тому подобное. Кроме того, это ядро затем покрывают одним или несколькими фармацевтически приемлемыми полимерами, способными обеспечить различные характеристики высвобождения. Центральное ядро может быть получено рядом способов, известных в данной области. Как правило, ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы связан с инертным ядром обычным связующим. Инертное ядро обычно содержит крахмал, сахар или микрокристаллическую целлюлозу. Специалисту в данной области техники будет понятно, что различные сахара могут быть включены в ядро гранулы, и что при выборе соответствующего сахара следует учитывать проблемы совместимости. До того как ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы связывают с инертным ядром, их, как правило, смешивают с обычными эксципиентами для ускорения обработки и улучшения свойств конечной дозированной формы. Эти эксципиенты идентичны тем, которые описаны выше для матричных систем. Количество этих эксципиентов может варьироваться в широких пределах, но обычно они используются в обычных количествах. Затем инертное ядро подвергается обработке для соединения смеси порошкообразного ингибиторора ксантиноксидоредуктазы или ингибиторора оксидоредуктазы с твердым носителем с использованием связующего. Это может быть выполнено с помощью известных в данной области средств для получения фармацевтических гранул. Подходящие средства включают использование обычных котлов для покрытия, ванн с кипящим слоем, экструзионную сферонизацию или ротогрануляцию. Получение таких центральных ядер более подробно описано в Pharmaceutical Pelletization Technology, ed. I. Ghebre-Sellassie, Marcel Dekker, Inc. New York, N.Y. (1989), включенную здесь в виде ссылки.
Вторым основным компонентом гранул является полимерное покрытие. Как отмечалось выше, полимерное покрытие является ответственным за придание гранулам параметров пролонгированного высвобождения. Полимерное покрытие может быть нанесено на центральное ядро с использованием приемов и способов, известных в данной области. Примеры подходящих устройств для покрытия включают, но не ограничиваются перечисленными, ванны для покрытия в кипящем слое, котлы для нанесения покрытий и т.п. Применяемые способы более подробно описаны в: 1) Aqueous polymeric coatings for pharmaceutical dosage forms, ed. J. W. McGinity, Marcel Dekker, Inc. New York, N.Y. (1997); и 2) Pharmaceutical Dosage Forms: Tablets Vol. 3. ed. H. A. Lieberman, L. Lachman and J. B. Schwartz, Marcel Dekker, Inc. New York, N.Y. pp. 77-287, (1990), содержание которых включено в данное описание ссылкой.
Полимер может быть включен в гранулы как слой, прикрепленный к фармацевтически активным агентам, дистально по отношению к ядру, а также может быть предоставлен в виде несколько слоев, причем каждый слой включает различные полимеры, обеспечивающие различные параметры высвобождения в каждом слое. Один из таких полимерных слоев включает слой полимера, модифицирующего высвобождение. Фармацевтически активный агент может быть высвобожден из полимерного слоя, модифицирующего высвобождение, так, чтобы активные частицы высвобождались по мере того, как полимер становится растворимым в окружающей среде. Подходящие примеры полимеров немедленного высвобождения, которые могут быть использованы для полимерного слоя с немедленным высвобождением, включают, но не ограничиваются перечисленным, этилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, ацетат целлюлозы, пропионат целлюлозы (с низкой, средней или высокой молекулярной массой), ацетат пропионат целлюлозы, ацетат бутират целлюлозы, ацетат фталат целлюлозы, триацетат целлюлозы, поли(метилметакрилат), поли(этилметакрилат), поли(бутилметакрилат), поли(изобутилметакрилат), поли(гексилметакрилата), поли(изодецилметакрилат), поли(лаурилметакрилат), поли(фенилметакрилат), поли(метилакрилат), поли(изопропилакрилат), поли(изобутилакрилат), поли(октадецилакрилат), поли(этилен), поли(этилен) низкой плотности, поли(этилен) высокой плотности, поли(пропилен), поли(этиленоксид), поли(этилентерефталат), поли(винилизобутиловый эфир), поли(винилацетат), поли(винилхлорид), полиуретан, водные дисперсии этилцеллюлозы (Aquacoat®, Surelease®), поли(бутилметакрилат, (2-диметиламиноэтил)метакрилат, метилметакрилат), поли(метакриловая кислота, метилметакрилат), поли(метакриловая кислота, этилакрилат), поли(метилакрилат, метилметакрилат, метакриловая кислота), поли(этилакрилат, метилметакрилат, триметиламмонийэтил хлорид метакрилата), поли(этилакрилат, метилметакрилат), поли(метакриловая кислота, этилакрилат), сополимер метакриловой кислоты типа А, сополимер метакриловой кислоты типа В, сополимер метакриловой кислоты типа C, дисперсия сополимера метакриловой кислоты, водная дисперсия акрилового полимера, (соединения EUDRAGIT®), Opadry® и тому подобное, и их смеси. В одном аспекте полимер немедленного высвобождения включает гидроксипропилметилцеллюлозу.
Полимерный слой, который инкапсулирует ядро, становится растворимым и начинает высвобождение активного лекарственного вещества сразу после его приема пациентом. При определенных обстоятельствах может быть выгодно покрывать ядро полимером, укрывающим материал ядра и предоставляющим более легкий способ покрытия ядра.
Гранулы настоящего изобретения могут также содержать слой энтеросолюбильного покрытия, которое наносится как полностью или частично закрывающее покрытие на ядра традиционными способами нанесения покрытий, такими как покрытие в котле для нанесения покрытий или покрытие в ванне с кипящим слоем, с использованием растворов полимеров в воде или в подходящих органических растворителях, или с использованием водных дисперсий полимеров. Все коммерчески доступные рН-чувствительные полимеры включены в объем данного изобретения. В случае наличия слоев энтеросолюбильного покрытия, фармацевтически активное вещество не высвобождается в кислой среде желудка при значении рН, меньшем приблизительно 4,5, но не ограничивается этим значением. Фармацевтически активное вещество высвобождается, как правило, когда рН-чувствительный слой растворяется при больших значениях рН. Подходящие примеры энтеросолюбильных полимеров для отсроченного высвобождения включают, но не ограничиваются перечисленными, ацетат фталат целлюлозы, ацетат тримеллитат целлюлозы, фталат гидроксипропилметилцеллюлозы, поливинилацетатфталат, карбоксиметилэтилцеллюлозу, со-полимеризированную метакриловую кислоту и метиловые эфиры метакриловой кислоты, такие как, например, материалы, известные под торговой маркой EUDRAGIT® L12.5, L100, EUDRAGIT® S12.5, S100, или подобные соединения, используемые для получения энтеросолюбильных покрытий. Сополимеры метакриловой кислоты и метиловых эфиров метакриловой кислоты обычно включают три подкласса соединений: сополимер метакриловой кислоты типа А, сополимер метакриловой кислоты типа B и сополимер метакриловой кислоты типа C. Различные типы сополимеров представляют соединения с различными соотношениями метакриловой кислоты и метилового эфира метакриловой кислоты. Соответственно, сополимер метакриловой кислоты типа А имеет отношение метакриловой кислоты к метиловому эфиру метакриловой кислоты равное приблизительно 1:1, тип B имеет соотношение, равное приблизительно 1:2, и тип C имеет соотношение, схожее с типом А, но может включать в себя дополнительные компоненты, такие как поверхностно-активные вещества. Могут быть также применены водные коллоидные дисперсии полимеров или восстановленные дисперсии, включающие, в том числе, например, полимеры, продаваемые под торговой маркой EUDRAGIT® L30D-55, EUDRAGIT® L100-55, EUDRAGIT® S100, препарат EUDRAGIT® 4110D (Rohm Pharma); EUDAGRIT® FS30D; AQUATERIC®, AQUACOAT® CPD30 (FMC); KOLLICOAT MAE® 30D и 30DP (BASF) и EASTACRYL® 30D (Eastman Chemical). В одном аспекте энтеросолюбильный полимер с отсроченным высвобождением содержит сополимер метакриловой кислоты типа А. В еще одном аспекте энтеросолюбильный полимер с отсроченным высвобождением представляет собой смесь сополимера метакриловой кислоты типа А и сополимера метакриловой кислоты типа B.
Специалисту в данной области техники будет понятно, что дополнительные компоненты могут быть добавлены в полимер с отсроченным высвобождением без отклонения от объема раскрытия. Например, к энтеросолюбильному полимеру с отсроченным высвобождением может быть добавлен пластификатор для улучшения физических характеристик полимерного слоя с отсроченным высвобождением. Неограничивающие примеры пластификаторов включают триэтилцитрат, ацетилтриэтилцитрат, трибутилцитрат, ацетилтрибутилцитрат, тригексилцитрат, ацетилтригексилцитрат, триоктилцитрат, ацетилтриоктилцитрат, бутирилтригексилцитрат, ацетилбутирилтригексилцитрат, триметилцитрат, ацетилированные моноглицериды, и сложные эфиры алкилсульфоновых кислот и фенила. В еще одном аспекте пластификатор включает в себя триэтилцитрат.
Кроме того, энтеросолюбильные полимеры, используемые в данном изобретении, могут быть модифицированы путем смешивания с другими известными продуктами для покрытий, которые не являются рН-чувствительными. Примеры таких продуктов для покрытий включают нейтральные сложные эфиры метакриловой кислоты с небольшой частью триметиламмонийэтил метакрилат хлорида, которые в настоящее время продаются под торговыми марками EUDRAGIT® и Eudragit® RL; нейтральную дисперсию сложного эфира без каких-либо функциональных групп, которая продается под торговыми названиями EUDRAGIT® NE30D и EUDRAGIT® NE30, а также другие продукты для покрытий, которые не являются рН-чувствительными.
Кроме того, в рамках этого изобретения может быть добавлен дополнительный модифицирующий слой поверх слоя энтеросолюбильного покрытия. Этот модифицирующий слой может включать в себя барьерный водонепроницаемый слой (полупроницаемую полимер), которым может быть последовательно покрыто энтеросолюбильное покрытие для снижения скорости проникновения воды через слой энтеросолюбильного покрытия, и тем самым увеличить время задержки высвобождения препарата. Обычные покрытия для контролируемого высвобождения, известные специалистам в данной области, могут быть использованы для этой цели, наносимые путем обычных способов покрытия, такими как покрытие в котле для нанесения покрытий или покрытие в ванне с кипящим слоем, с использованием растворов полимеров в воде или в подходящих органических растворителях, или с использованием водных дисперсий полимеров. Например, следующий неограничивающий перечень полимеров для контролируемого высвобождения может быть использован в настоящем изобретении: ацетат целлюлозы, бутират ацетат целлюлозы, пропионат ацетат целлюлозы, этилцеллюлоза, гидроксипропилметилцеллюлоза, ацетат целлюлозы, пропионат целлюлозы (с низкой, средней или высокой молекулярной массой), ацетат пропионат целлюлозы, ацетат бутират целлюлозы, ацетат фталат целлюлозы, триацетат целлюлозы, поли(метилметакрилат), поли(этилметакрилат), поли(бутилметакрилат), поли(изобутилметакрилат), поли(гексилметакрилат), поли(изодецилметакрилат), поли(лаурилметакрилат), поли(фенилметакрилат), поли(метилакрилат), поли(изопропилакрилат), поли(изобутилакрилат), поли(октадецилакрилат), поли(этилен), поли(этилен) низкой плотности, поли(этилен) высокой плотности, поли(пропилен), поли(этиленоксид), поли(полиэтилентерефталат), поли(винилизобутиловый эфир), поли(винилацетат), поли(винилхлорид), полиуретан, водные дисперсии этилцеллюлозы, такие как AQUACOAT® и SURELEASE®, поли(бутилметакрилат, (2-диметиламиноэтил)метакрилат, метилметакрилат), поли(метакриловая кислота, метилметакрилат), поли(метакриловая кислота, этилакрилат), поли(метилакрилат, метилметакрилат, метакриловая кислота), поли(этилакрилат, метилметакрилат, триметиламмонийэтил хлорид метакрилата), поли(этилакрилат, метилметакрилат), поли(метакриловая кислота, этилакрилат), сополимер метакриловой кислоты типа А, сополимер метакриловой кислоты типа B, сополимер метакриловой кислоты типа C, дисперсия сополимера метакриловой кислоты, водная дисперсия акрилового полимера, (соединения типа EUDRAGIT®), OPADRY®, жирные кислоты и их эфиры, воски, зеин, водная дисперсия полимера, такая как EUDRAGIT® RS и RL 30D, EUDRAGIT® NE 30D, латекс ацетата целлюлозы. Может быть также включена комбинация вышеуказанных полимеров и гидрофильных полимеров, таких как гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза (KLUCEL®, Hercules Corp.), гидроксипропилметилцеллюлоза (METHOCEL®, Dow Chemical Corp.) и поливинилпирролидон. В одном аспекте полимер контролируемого высвобождения включает этилцеллюлозу, гидроксипропилметилцеллюлозу и их комбинации. В еще одном аспекте полимер контролируемого высвобождения включает комбинацию этилцеллюлозы и гидроксипропилметилцеллюлозы в отношении этилцеллюлозы к гидроксипропилметилцеллюлозе в диапазоне от приблизительно 0,1 до приблизительно 10, от приблизительно 0,2 до приблизительно 5, от приблизительно 0,5 до приблизительно 3, и от приблизительно 1 до приблизительно 2. В еще одном аспекте полимер контролируемого высвобождения включает комбинацию водной дисперсии этилцеллюлозы и гидроксипропилметилцеллюлозу в отношении водной дисперсии этилцеллюлозы к гидроксипропилметилцеллюлозе в диапазоне от приблизительно 0,1 до приблизительно 10, от приблизительно 0,1 до приблизительно 5, от приблизительно 0,5 до приблизительно 4, и от приблизительно 1,5 до приблизительно 3.
Фармацевтическая композиция настоящего изобретения, в одном аспекте, включающая один или несколько типов гранул, может быть включена во множественные дозированные формы, такие как капсулы, пилюли и таблетки. Капсулы, включающие твердые желатиновые капсулы, могут быть получены способами, известными в уровне техники. В целом, капсулы могут включать гранулы, обсужденные здесь, и могут необязательно включать дополнительные эксципиенты, как описано выше. Фармацевтическая композиция может также быть включена в таблетку. В общем, этот процесс охватывает включение гранул в матрицу таблетки, как описано ранее. Квалифицированный специалист знает, что, с целью придания желаемых физических характеристик, к препарату могут быть добавлены дополнительные компоненты, не отступая от объема изобретения.
Типы гранул
Имеется много типов препаратов гранул, которые входят в объем притязаний настоящего изобретения, включающих различные полимеры, в соответствии с полимерами, описанными ранее. Квалифицированный специалист может модифицировать препарат на основе гранул, чтобы придать ему определенные химические характеристики. Настоящее изобретение, в одном аспекте, описывает четыре первичных типа гранул. А именно, четыре типа гранул могут быть описаны как гранулы немедленного высвобождения, гранулы отсроченного высвобождения, гранулы контролируемого высвобождения и гранулы отсроченного/контролируемого высвобождения. Гранулы немедленного высвобождения содержат слой ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, нанесенный на инертное ядро, такое как сахарные сферы или сферы микрокристаллической целлюлозы, с использованием подходящего полимерного связующего. Полимерное связующее выполняет функцию по созданию грунтовочного слоя сцепления вокруг инертного материала ядра, улучшая показатели рыхлости инертного ядра. Полимерное связующее может включить любой из описанных ранее полимеров немедленного высвобождения. В одном аспекте полимерное связующее включает гидроксипропилметилцеллюлозу. Для целей различных композиций гранул, описанных здесь, полимерный компонент связующего будет включать такой же материал, как и у полимера с немедленным высвобождением, и процентное содержание полимера с немедленным высвобождением в композиции будет включать как полимер с немедленным высвобождением, используемым в слое немедленного высвобождения, окружающим инертное ядро, так и полимер с немедленным высвобождением, который обеспечивает грунтовочный слой сцепления для инертного ядра.
Гранула немедленного высвобождения, как правило, включает от приблизительно 5% до приблизительно 55% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 20% до приблизительно 80% (масс./масс.) инертного ядра и от приблизительно 1% до приблизительно 40% (масс./масс.) полимера немедленного высвобождения. В одном аспекте гранула немедленного высвобождения, как правило, включает от приблизительно 25% до приблизительно 35% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 40% до приблизительно 60% (масс./масс.) инертного ядра и от приблизительно 10% до приблизительно 20% (масс./масс.) полимера немедленного высвобождения. В другом аспекте ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы составляет от приблизительно 29% до приблизительно 34% (масс./масс.) от общей массы композиции, инертное ядро составляет от приблизительно 50% до приблизительно 55% (масс./масс.) от общей массы композиции, и полимер немедленного высвобождения составляет от приблизительно 14% до приблизительно 18% (масс./масс.) от общей массы композиции.
Дополнительный тип гранул, рассмотренный в настоящем изобретении, представляет собой гранулу отсроченного высвобождения. Гранулы отсроченного высвобождения представляют собой покрытые гранулы, полученные покрытием гранул немедленного высвобождения энтеросолюбильным полимером с отсроченным высвобождением в водной дисперсии или в органическом растворителе. Эти полимеры имеют растворимость, зависящую от значения pH, в зависимости от функциональных групп полимера. Для гранулы отсроченного высвобождения, покрытой подходящим количеством энтеросолюбильного полимера с отсроченным высвобождением, высвобождение лекарственного вещества не будет происходить в среде, в которой среднее значение pH не будет выше значения pH, при котором растворяет полимер. В целом, энтеросолюбильные полимеры с отсроченным высвобождением по настоящему изобретению становятся растворимыми, когда гранула подвергается воздействию среды с величиной pH, соответствующему менее кислому, чем в желудке. Конкретно, полимер отсроченного высвобождения может стать растворимым при значениях pH, больших или равных 4,5; 4,6; 4,7; 4,8; 4,9; 5,0; 5,1; 5,2; 5,3; 5,4; 5,5; 5,6; 5,7; 5,8; 5,9; 6,0; 6,1; 6,2; 6,3; 6,4; 6,5; 6,6; 6,7; 6,8; 6,9; 7,0; 7,1; 7,2; 7,3; 7,4; 7,5; 7,6; 7,7; 7,8; 7,9; 8,0; 8,1; 8,2; 8,3; 8,4; 8,5; 8,6; 8,7; 8,8; 8,9; 9,0; 9,1; 9,2; 9,3; 9,4; 9,5; 9,6; 9,7; 9,8; 9,9 и 10,0. В одном аспекте полимер отсроченного высвобождения становится растворимым при значениях pH, больших или равных 5,5, 6,0 и 6,8.
Композиция компонента немедленного высвобождения гранул отсроченного высвобождения является такой же, как описано ранее, и в целом включает ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы, инертное ядро и полимер немедленного высвобождения, как описано ранее. Гранула отсроченного высвобождения дополнительно включает чувствительный к величине pH энтеросолюбильный полимер, как описано ранее. Неограничивающий пример гранулы отсроченного высвобождения представляет собой таковой, в котором гранула имеет растворимость при уровнях величины pH, большей или равной 6,0. Эта гранула с отсроченным высвобождением до pH 6,0, как правило, включает ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы в количестве от приблизительно 5% до приблизительно 50% (масс./масс.) от полной массы гранулы отсроченного высвобождения, инертное ядро, в количестве от приблизительно 20% до приблизительно 70% (масс./масс.) от полной массы гранулы отсроченного высвобождения, полимер немедленного высвобождения, в количестве от приблизительно 1% до приблизительно 35% (масс./масс.) от полной массы гранулы отсроченного высвобождения, и энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 1% до приблизительно 35% (масс./масс.) от полной массы гранулы отсроченного высвобождения. В одном аспекте гранула отсроченного высвобождения включает от приблизительно 20% до приблизительно 30% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 40% до приблизительно 50% (масс./масс.) инертного ядра, от приблизительно 10% до приблизительно 16% (масс./масс.) полимера немедленного высвобождения, и от приблизительно 13% до приблизительно 20% (масс./масс.) полимера отсроченного высвобождения. В последующем повторении этого примера гранул отсроченного высвобождения до pH 6,0, гранула дополнительно включает от приблизительно 1% до приблизительно 3% (масс./масс.) пластификатора.
Другой неограничивающий пример гранулы отсроченного высвобождения представляет собой таковой, в котором гранула имеет растворимость при уровнях величины pH, большей или равной 6,8. Эта гранула с отсроченным высвобождением до pH 6,8, как правило, включает ингибитор ксантиноксидоредуктазы или ингибитор ксантиноксидазы в количестве от приблизительно 5% до приблизительно 50% (масс./масс.) от полной массы гранулы отсроченного высвобождения, инертное ядро, включающее от приблизительно 20% до приблизительно 70% (масс./масс.) от полной массы гранулы отсроченного высвобождения, полимер немедленного высвобождения в количестве от приблизительно 1% до приблизительно 35% (масс./масс.) от полной массы гранулы отсроченного высвобождения, и энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 1% до приблизительно 35% (масс./масс.) от полной массы гранулы отсроченного высвобождения. В одном аспекте гранула отсроченного высвобождения до pH 6,8 включают от приблизительно 20% до приблизительно 30% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 40% до приблизительно 50% (масс./масс.) инертного ядра, от приблизительно 10% до приблизительно 16% (масс./масс.) полимера немедленного высвобождения, и от приблизительно 13% до приблизительно 20% (масс./масс.) полимера отсроченного высвобождения. В одном аспекте гранула отсроченного высвобождения до pH 6,8 включают от приблизительно 23% до приблизительно 27% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 40,5% до приблизительно 43% (масс./масс.) инертного ядра, от приблизительно 12% до приблизительно 14% (масс./масс.) полимера немедленного высвобождения, и от приблизительно 17% до приблизительно 19% (масс./масс.) одного или нескольких энтеросолюбильных полимеров отсроченного высвобождения. В последующем повторении этого примера гранул отсроченного высвобождения до pH 6,8, гранула дополнительно включают от приблизительно 1% до приблизительно 3% (масс./масс.) пластификатора.
Дополнительный тип гранул, охватываемый настоящим изобретением, представляет собой гранулу контролируемого высвобождения. Гранулы контролируемого высвобождения представляют собой покрытые гранулы, полученные путем покрытия гранул немедленного высвобождения полимером контролируемого высвобождения, согласно способам, известными в настоящее время в уровне техники. В целом, гранулы контролируемого высвобождения включают один или несколько полимеров, которые снижают скорость высвобождения лекарственного вещества из гранулы таким образом, чтобы лекарственное вещество высвобождалось в течение длительного периода времени. Гранулы контролируемого высвобождения отличаются от гранул отсроченного высвобождения тем, что после воздействия среды растворения высвобождение из гранул контролируемого высвобождения является непрерывным в течение длительного времени, в то время как высвобождение из гранул отсроченного высвобождения происходит очень быстро, как только на гранулы воздействует величина pH, выше которой энтеросолюбильный полимер отсроченного высвобождения является растворимым. В целом, композиция полимерного слоя контролируемого высвобождения может быть модифицирована таким образом, что высвобождение становится возможным в течение времени в пределах от приблизительно 1 часа до приблизительно 24 часов. В частности, препарат контролируемого высвобождения может высвобождать активное лекарственное вещество в течение 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 и 24 часа.
В одном воплощении настоящего изобретения гранулы контролируемого высвобождения включают композицию, способную к высвобождению активного соединения в течение периода времени в пределах от приблизительно четырех часов до приблизительно шести часов. Гранулы контролируемого высвобождения этого воплощения в целом включают от приблизительно 5% до приблизительно 40% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 20% до приблизительно 50% (масс./масс.) инертного ядра, от приблизительно 5% до приблизительно 25% (масс./масс.) полимера немедленного высвобождения, от приблизительно 10% до приблизительно 50% (масс./масс.) полимера контролируемого высвобождения. В одном аспекте гранулы контролируемого высвобождения включают от приблизительно 20% до приблизительно 24% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 30% до приблизительно 40% (масс./масс.) инертного ядра, от приблизительно 9% до приблизительно 13% (масс./масс.) полимера немедленного высвобождения, от приблизительно 25% до приблизительно 35% (масс./масс.) полимера контролируемого высвобождения. В одном аспекте гранулы контролируемого высвобождения включают от приблизительно 25% до приблизительно 35% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 40% до приблизительно 60% (масс./масс.) инертного ядра, от приблизительно 12% до приблизительно 18% (масс./масс.) полимера немедленного высвобождения, от приблизительно 3% до приблизительно 9% (масс./масс.) полимера контролируемого высвобождения.
В дополнительном воплощении гранулы контролируемого высвобождения включают композицию, способную к высвобождению активного соединения в течение времени в пределах от приблизительно десяти часов до приблизительно двенадцати часов. Гранулы контролируемого высвобождения этого воплощения в целом включают от приблизительно 10% до приблизительно 50% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 30% до приблизительно 70% (масс./масс.) инертного ядра, от приблизительно 5% до приблизительно 25% (масс./масс.) полимера немедленного высвобождения и от приблизительно 1% до приблизительно 15% (масс./масс.) полимера контролируемого высвобождения. В одном аспекте гранулы контролируемого высвобождения включают от приблизительно 25% до приблизительно 35% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 40% до приблизительно 60% (масс./масс.) инертного ядра, от приблизительно 12% до приблизительно 18% (масс./масс.) полимера немедленного высвобождения и от приблизительно 3% до приблизительно 9% (масс./масс.) полимера контролируемого высвобождение. В одном аспекте гранулы контролируемого высвобождения включают от приблизительно 28% до приблизительно 31% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 47% до приблизительно 51% (масс./масс.) инертного ядра, от приблизительно 14% до приблизительно 16% (масс./масс.) полимера немедленного высвобождения и от приблизительно 5% до приблизительно 7% (масс./масс.) полимера контролируемого высвобождения.
Дополнительный тип гранул, охватываемый настоящим изобретением, представляет собой гранулу отсроченного/контролируемого высвобождения. Гранула отсроченного/контролируемого высвобождения объединяет признаки гранул отсроченного высвобождения, описанных выше, и гранул контролируемого высвобождения, описанных выше, с целью задержки высвобождения лекарственного вещества, пока на гранулы не воздействует pH, большая, чем величина pH, при которой полимер растворяется, и затем пролонгированного высвобождения лекарственного вещества в течение длительного периода времени. Иллюстративно, гранула контролируемого высвобождения, введенная отдельно, будет, как правило, высвобождать активный компонент в течение длительного периода времени, с высвобождение, начинающимся немедленно после приема. Гранулы отсроченного высвобождения, введенные отдельно, не будут высвобождать активный компонент, пока окружающая среда не будет иметь минимальный уровень pH. Например, уровень pH в кишечнике выше чем в желудке, поэтому гранула отсроченного высвобождения может быть создана для того, чтобы высвободить активный компонент, как только будет достигнут уровень pH, имеющийся в кишечнике, без высвобождения активного компонента в областях, где уровень pH ниже, такой как в желудке. Однако, как только уровень pH достигается, высвобождение лекарственного вещества, как правило, происходит быстро.
В настоящем воплощении слой полимера отсроченного высвобождения инкапсулирует слой полимера контролируемого высвобождения, так что пролонгированное высвобождение активного компонента, обеспечиваемое полимером контролируемого высвобождения, не будет начинаться, пока гранула отсроченного/контролируемого высвобождения не подвергнется воздействию минимального уровня pH. Соответственно, специалист в данной области техники может ожидать, что характеристики гранулы отсроченного/контролируемого высвобождения могут быть изменены так, что полимер отсроченного высвобождения не станет растворимым, пока гранула не подвергнется воздействию уровня pH, находящегося в общем случае в пределах от приблизительно 5 до приблизительно 10, как описано ранее. Кроме того, гранулы отсроченного/контролируемого высвобождения могут быть разработаны так, чтобы высвобождать активный компонент в течение периода времени в пределах от приблизительно одного часа до приблизительно двадцати четырех часов, как описано ранее.
В одном воплощении настоящего изобретения гранула отсроченного/контролируемого высвобождения включает энтеросолюбильный полимер отсроченного высвобождения с растворимостью на уровнях величины pH, большей или равной 6,8, и полимер контролируемого высвобождения, обеспечивающий доставку активного соединения с пролонгированием более чем на четыре-шесть часов. Гранулы отсроченного/контролируемого высвобождения этого воплощения в целом включают от приблизительно 5% до приблизительно 35% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 20% до приблизительно 50% (масс./масс.) инертного ядра, от приблизительно 5% до приблизительно 20% (масс./масс.) полимера немедленного высвобождения, от приблизительно 5% до приблизительно 20% (масс./масс.) полимера контролируемого высвобождение и от приблизительно 5% до приблизительно 35% (масс./масс.) энтеросолюбильного полимера отсроченного высвобождения. В одном аспекте гранулы отсроченного/контролируемого высвобождения включают от приблизительно 15% до приблизительно 25% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 30% до приблизительно 40% (масс./масс.) инертного ядра, от приблизительно 8% до приблизительно 14% (масс./масс.) полимера немедленного высвобождения, от приблизительно 8% до приблизительно 15% (масс./масс.) полимера контролируемого высвобождения и от приблизительно 13% до приблизительно 22% (масс./масс.) энтеросолюбильного полимера отсроченного высвобождения. В другом аспекте гранула отсроченного/контролируемого высвобождения включает от приблизительно 20% до приблизительно 23% (масс./масс.) ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, от приблизительно 34% до приблизительно 37% (масс./масс.) инертного ядра, от приблизительно 10% до приблизительно 12% (масс./масс.) полимера немедленного высвобождения, от приблизительно 11% до приблизительно 13% (масс./масс.) полимера контролируемого высвобождение и от приблизительно 17% до приблизительно 20% (масс./масс.) энтеросолюбильного полимера отсроченного высвобождения.
Специалист в данной области техники может понять, что различные гранулы модифицированного высвобождения настоящего изобретения могут быть изготовлены любыми способами, известными в уровне техники. Неограниченные примеры способов изготовления гранул включают обработку в кипящим слое, грануляцию в центрифуге, экструзионную сферонизацию, грануляцию при высоком усилии сдвига, экструзию с плавлением и наслаивание в растворе или суспензии. В процессе с кипящим слоем полимер немедленного высвобождения растворен в воде, и микронизированное лекарственное вещество суспендировано в растворе полимера немедленного высвобождения. Эта суспензия затем распыляется на инертные сферические гранулы основы, такие как сахарные сферы или сферы микрокристаллической целлюлозы. Альтернативно, немикронизированное лекарственное вещество может быть суспендировано в растворе полимера немедленного высвобождения, и суспензия может быть пропущена через мельницу. В способе грануляции в центрифуге инертные гранулы помещают на вращающийся диск гранулятора у основания гранулятора. Микронизированное лекарственное вещество вводят в гранулятор, и одновременно распыляют раствор полимера немедленного высвобождения. Экструзия со сферонизацией представляет собой другой промышленный способ для гранул немедленного высвобождения, в котором лекарственное вещество смешивают с сухими эксципиентами, и путем добавления раствора связующего получают влажную массу, которую экструдируют с образованием похожих на спагетти прядей. Затем экструдат рубят и превращают в плотные сферические гранулы, используя сферонизатор. Другой способ получения гранул включает гранулирование при высоком усилии сдвига. Гранулирование при высоком усилии сдвига включает сухое смешивание активного компонента и других компонентов. После этого смесь увлажняют добавлением раствора связующего в смесителе/грануляторе с высоким усилием сдвига. Гранулы формируют после увлажнения за счет комбинированного действия смешивания и помола. Затем получившиеся гранулы или пеллеты высушивают и просеивают. Дополнительный способ включает экструзию с плавлением или гранулирование с плавлением. Этот способ в целом включает плавление обычно твердого гидрофобного материала связующего, например воска или подобного вещества, и включение в него порошкообразного лекарственного вещества. Для получения дозированной формы контролируемого или пролонгированного высвобождения дополнительные гидрофобные материалы для высвобождения, например этилцеллюлоза или нерастворимый в воде акриловый полимер, могут быть включены в расплавленный воскообразный гидрофобный материал связующего. Затем осуществляют наслаивание в растворе или суспензии, включающее способ, посредством которого раствор или суспензия активного компонента, содержащий или не содержащий связующее, напыляется на исходные затравки с определенным размером частиц в аппарате с кипящим слоем или на другом приемлемом оборудовании. Таким образом, поверхность исходных затравок покрывают лекарственным веществом. Пеллеты, нагруженные лекарственным веществом, высушивают для дальнейших применений.
Фармацевтические дозированные формы
Квалифицированный специалист в данной области техники может понять, что различные типы гранул, описанные здесь, с различными профилями высвобождения активного вещества, могут быть объединены в единые или множественные фармацевтические дозированные формы для того, чтобы обеспечить многостадийную доставку лекарственного вещества, такого как ингибитор ксантиноксидоредуктазы или ингибиторов ксантиноксидазы, описанные здесь. Различные комбинации гранул немедленного высвобождения, гранул отсроченного высвобождения, гранул контролируемого высвобождения и гранул отсроченного/контролируемого высвобождения могут использоваться, чтобы создать различные профили высвобождения. Должно быть понято, что любая потенциальная комбинация гранул немедленного высвобождения, отсроченного высвобождения, контролируемого высвобождения и отсроченного/контролируемого высвобождения для распределения ингибитора ксантиноксидоредуктазы или ингибиторов ксантиноксидазы, описанных здесь, находятся в рамках настоящего изобретения.
В одном воплощении настоящее изобретение охватывает единую фармацевтическую композицию, которая включает как гранулы немедленного высвобождения, так и гранулы отсроченного высвобождения, с растворимостью при уровнях величины, pH большей или равной 6,8. Фармацевтическая композиция этого воплощения включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве в пределах от приблизительно 20% до приблизительно 40% (масс./масс.) от общей массы композиции и гранулы отсроченного высвобождения при величине pH 6,8 ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве в пределах от приблизительно 60% до приблизительно 80% (масс./масс.) от общей массы композиции.
Гранулы немедленного высвобождения включают: a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от массы гранулы немедленного высвобождения; и b) слой немедленного высвобождения, который инкапсулирует инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и связующего, такого как гидроксипропилметилцеллюлоза или гидроксипропилцеллюлоза, в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от массы гранулы немедленного высвобождения, где отношение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе или гидроксипропилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 3. Нерастворимый дезинтегрант, такой как низкозамещенная гидроксипропилцеллюлоза (L-HPC), может быть добавлен для ускорения высвобождения активного вещества.
Гранулы отсроченного высвобождения до pH 6,8 включают a) инертное ядро в количестве в пределах от приблизительно 40,5% до приблизительно 43% (масс./масс.) от массы гранулы отсроченного высвобождения; b) слой немедленного высвобождения, инкапсулирующий инертное ядро (как описано ранее) в количестве в пределах от приблизительно 35% до приблизительно 40% (масс./масс.) от массы гранулы отсроченного высвобождения, при отношении ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3; c) слой энтеросолюбильного полимера отсроченного высвобождения, инкапсулирующий слой немедленного высвобождения, включающий энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 17% до приблизительно 20% (масс./масс.) от массы гранулы отсроченного высвобождения, где указанный энтеросолюбильный полимер отсроченного высвобождения включает смесь сополимера метакриловой кислоты типа A и сополимера метакриловой кислоты типа B в соотношении в пределах от приблизительно 0,1 до приблизительно 0,5; и d) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения. В одном аспекте фармацевтической композиции ингибитор ксантиноксидоредуктазы включает фебуксостат, и пластификатор включает триэтилцитрат. В другом аспекте ингибитор ксантиноксидазы в фармацевтической композиции включает аллопуринол, и пластификатор включает триэтилцитрат.
В еще одном воплощении настоящего изобретения фармацевтическая дозированная форма включает единую фармацевтическую композицию, включающую гранулы немедленного высвобождения, гранулы отсроченного высвобождения, с растворимостью на уровнях величины pH, большей или равной 6,0, и гранулы отсроченного высвобождения, с растворимостью на уровнях величины pH, большей или равной 6,8. Фармацевтическая композиция этого воплощения включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве в пределах от приблизительно 25% до приблизительно 35% (масс./масс.) от полной массы композиции, гранулы отсроченного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы до pH 6,0 в количестве в пределах от приблизительно 25% до приблизительно 35% (масс./масс.) от полной массы композиции, и гранулы отсроченного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы до pH 6,8 в количестве в пределах от приблизительно 35% до приблизительно 45% (масс./масс.) от полной массы композиции.
Гранулы немедленного высвобождения включают: a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от массы гранулы немедленного высвобождения; и b) слой немедленного высвобождения, инкапсулирующий инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от массы гранулы немедленного высвобождения, при отношении ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3.
Гранулы отсроченного высвобождения до pH 6,0 включают: a) инертное ядро в количестве в пределах от приблизительно 40,5% до приблизительно 43% (масс./масс.) от массы гранулы отсроченного высвобождения до pH 6,0; b) слой немедленного высвобождения, инкапсулирующий инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 35% до приблизительно 40% (масс./масс.) от массы гранулы отсроченного высвобождения до pH 6,0, при отношении ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3; c) слой энтеросолюбильного полимера отсроченного высвобождения до pH 6,0, инкапсулирующий слой немедленного высвобождения, включающий энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 17% до приблизительно 19% (масс./масс.) от массы гранулы отсроченного высвобождения, где указанный энтеросолюбильный полимер отсроченного высвобождения включает сополимер метакриловой кислоты типа A; и d) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения.
Два типа гранул, а именно, гранулы немедленного высвобождения и отсроченного высвобождения до pH 6,0, могут быть для удобства объединены вместе в единую гранулу, применяя сверху покрытие немедленного высвобождения на покрытии отсроченного высвобождения до pH 6,0. Получившаяся композиция содержит a) инертное ядро в количестве в пределах от приблизительно 25% до приблизительно 35% (масс./масс.) от массы комбинированной гранулы немедленного/отсроченного высвобождения до pH 6,0; b). слой немедленного высвобождения, инкапсулирующий инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 25% до приблизительно 31% (масс./масс.) от массы комбинированной гранулы; c) слой энтеросолюбильного полимера отсроченного высвобождения до pH 6,0, инкапсулирующий слой немедленного высвобождения, включающий энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 11% до приблизительно 15% (масс./масс.) от массы гранулы отсроченного высвобождения, где указанный энтеросолюбильный полимер отсроченного высвобождения включает сополимер метакриловой кислоты типа A; d) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы немедленного/отсроченного высвобождения; и e) слой покрытия немедленного высвобождения, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 23% до приблизительно 29% (масс./масс.) от массы комбинированной гранулы.
Гранулы отсроченного высвобождения до pH 6,8 включают: a) инертное ядро в количестве в пределах от приблизительно 40,5% до приблизительно 43% (масс./масс.) от массы гранулы отсроченного высвобождения; b) слой немедленного высвобождения, инкапсулирующий инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 35% до приблизительно 40% (масс./масс.) от массы гранулы отсроченного высвобождения, при отношении ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3; c) слой энтеросолюбильного полимера отсроченного высвобождения до pH 6,8, инкапсулирующий слой немедленного высвобождения, включающий энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 17% до приблизительно 20% (масс./масс.) от массы гранулы отсроченного высвобождения до pH 6,8, где указанный энтеросолюбильный полимер отсроченного высвобождения включает смесь сополимера метакриловой кислоты типа A и сополимера метакриловой кислоты типа B в соотношении в пределах от приблизительно 0,1 до приблизительно 0,5; и d) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где пластификатор включает триэтилцитрат. В другом аспекте фармацевтической композиции ингибитор ксантиноксидоредуктазы включает фебуксостат, и пластификатор включает триэтилцитрат. В еще одном аспекте этой фармацевтической композиции ингибитор ксантиноксидазы включает аллопуринол, и пластификатор включает триэтилцитрат.
В дополнительном воплощении настоящего изобретения фармацевтическая композиция включает единую фармацевтическую композицию, которая включает гранулы немедленного высвобождения и гранулы отсроченного/контролируемого высвобождения, с полимером отсроченного высвобождения, имеющим растворимость при величине pH по меньшей мере 6,8, и контролируемое высвобождение от четырех до шести часов. Фармацевтическая композиция этого воплощения включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве в пределах от приблизительно 20% до приблизительно 40% (масс./масс.) от общей массы композиции, и гранулы отсроченного/контролируемого высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, имеющие растворимость на уровнях величины pH, большей или равной 6,8, и обеспечивающие пролонгированное высвобождение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в период, начинающийся через от приблизительно 4 часа до приблизительно 6 часов, в количестве в пределах от приблизительно 60% до приблизительно 80% (масс./масс.) от общей массы композиции. Например, в одном аспекте фармацевтическая композиция включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве приблизительно 20% (масс./масс.) от общей массы композиции, гранулы отсроченного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, высвобождающие до pH 6,8, в количестве приблизительно 80% (масс./масс.) от общей массы композиции. В еще одном аспекте фармацевтическая композиция включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве приблизительно 25% (масс./масс.) от общей массы композиции, гранулы отсроченного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, высвобождающие до pH 6,8, в количестве приблизительно 75% (масс./масс.) от общей массы композиции. В еще одном аспекте фармацевтическая композиция включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве приблизительно 30% (масс./масс.) от общей массы композиции, гранулы отсроченного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, высвобождающие до pH 6,8, в количестве приблизительно 70% (масс./масс.) от общей массы композиции. В еще одном аспекте фармацевтическая композиция включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве приблизительно 40% (масс./масс.) от общей массы композиции, гранулы отсроченного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, высвобождающие до pH 6,8, в количестве приблизительно 60% (масс./масс.) от общей массы композиции.
Гранулы немедленного высвобождения включают: a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от массы гранулы немедленного высвобождения; и b) слой немедленного высвобождения, инкапсулирующий инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от массы гранулы немедленного высвобождения, при отношении ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3.
Гранулы отсроченного/контролируемого высвобождения включают: a) инертное ядро в количестве в пределах от приблизительно 34% до приблизительно 37% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения; b) слой немедленного высвобождения, который инкапсулирует инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 31% до приблизительно 34% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где отношение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 2,5; c) слой контролируемого высвобождения, инкапсулирующий слой немедленного высвобождения, включающий полимер контролируемого высвобождения в количестве в пределах от приблизительно 10% до приблизительно 14% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где указанный полимер контролируемого высвобождения включает смесь водной дисперсии этилцеллюлозы и гидроксипропилметилцеллюлозы, при отношении дисперсии этилцеллюлозы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3; d) слой отсроченного высвобождения до pH 6,8, инкапсулирующий слой контролируемого высвобождения, включающий полимер отсроченного высвобождения до pH 6,8 в количестве в пределах от приблизительно 17,5% до приблизительно 20% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где полимер отсроченного высвобождения включает смесь сополимера метакриловой кислоты типа A и сополимера метакриловой кислоты типа B в соотношении в пределах от приблизительно 0,1 до приблизительно 0,5; и e) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где указанный пластификатор включает триэтилцитрат. В одном аспекте фармацевтической композиции ингибитор ксантиноксидоредуктазы включает фебуксостат и пластификатор включает триэтилцитрат. В еще одном аспекте этой фармацевтической композиции ингибитор ксантиноксидазы включает аллопуринол и пластификатор включает триэтилцитрат.
В другом воплощении настоящего изобретения фармацевтическая композиция включает единую фармацевтическую композицию, включающую гранулы немедленного высвобождения и гранулы контролируемого высвобождения, которые способны к высвобождению активного вещества в течение от приблизительно десяти до приблизительно двенадцати часов. Фармацевтическая композиция этого воплощения в целом включает гранулы немедленного высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в количестве в пределах от приблизительно 10% до приблизительно 30% (масс./масс.) от общей массы композиции, и гранулы контролируемого высвобождения ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы, обеспечивающие пролонгированное высвобождение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы в период, начинающийся через от приблизительно 10 часов до приблизительно 12 часов, в количестве в пределах от приблизительно 70% до приблизительно 90% (масс./масс.) от общей массы композиции.
Гранулы немедленного высвобождения включают: a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от массы гранулы немедленного высвобождения; и b) слой немедленного высвобождения, инкапсулирующий инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от массы гранулы немедленного высвобождения, при отношении ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3.
Гранулы 10-12 часового контролируемого высвобождения включают: a) инертное ядро в количестве в пределах от приблизительно 47% до приблизительно 51% (масс./масс.) от массы гранулы контролируемого высвобождения; b) слой немедленного высвобождения, который инкапсулирует инертное ядро, включающий смесь ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 42% до приблизительно 48% (масс./масс.) от массы гранулы контролируемого высвобождения, где отношение ингибитора ксантиноксидоредуктазы или ингибитора ксантиноксидазы к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 2,5; и c) слой контролируемого высвобождения, инкапсулирующий слой немедленного высвобождения, включающий полимер контролируемого высвобождения, где указанный полимер контролируемого высвобождения включает смесь этилцеллюлозы и гидроксипропилметилцеллюлозы, в количестве в пределах от приблизительно 4% до приблизительно 8% (масс./масс.) от массы гранулы контролируемого высвобождения, при отношении этилцеллюлозы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1 до приблизительно 2. В еще одном аспекте фармацевтической композиции ингибитор ксантиноксидоредуктазы включает фебуксостат. В еще одном аспекте этой фармацевтической композиции ингибитор ксантиноксидазы включает аллопуринол.
Квалифицированный специалист в данной области техники может понять, что различные воплощения и дозированные формы, описанные здесь, могут включить любую дозированную форму, известную в данной области. В одном аспекте дозированные формы включают пилюли, таблетки и капсулы. Кроме того, фармацевтические композиции могут иметь общую массу композиции в пределах от приблизительно 5 мг до приблизительно 240 мг. В одном аспекте общая масса фармацевтической композиции (общая масса) включает от приблизительно 60 мг до приблизительно 100 мг. В другом аспекте общая масса фармацевтической композиции составляет приблизительно 80 мг.
Способы лечения
Дозированные формы настоящего изобретения могут использоваться при лечении множества болезненных состояний. Когда дозированные формы настоящего изобретения содержат фебуксостат, то такие дозированные формы могут использоваться при лечении состояний, таких как, но не ограничиваясь перечисленными, подагра, гиперурикемия, простатит, воспалительное заболевание кишечника, удлинение интервала QT, инфаркт миокарда, гипертрофия сердца, гипертензия, почечнокаменная болезнь, почечная недостаточность, хроническое заболевание почек, метаболический синдром (также называемый "синдром X", и который включает по меньшей мере одно из следующего: абдоминальное ожирение, атерогенная дислипидемия, устойчивость к инсулину, непереносимость глюкозы, протромбическое состояние или провоспалительное состояние), диабет, диабетическая нефропатия, застойная сердечная недостаточность и т.д. Субъекту, страдающему от одного из вышеперечисленных болезненных состояний и нуждающемуся в их лечении, можно вводить эффективное количество (или терапевтически эффективное количество) дозированной формы настоящего изобретения для лечения указанных болезненных состояний.
Когда дозированные формы настоящего изобретения содержат аллопуринол или оксипуринол, то такие дозированные формы могут использоваться при лечении таких состояний, но не ограничиваясь перечисленными, как подагра, гиперурикемия, простатит, воспалительное заболевание кишечника, удлинение интервала QT, инфаркт миокарда, гипертрофия сердца, гипертензия, почечнокаменная болезнь, почечная недостаточность, хроническое заболевание почек, метаболический синдром (также называемый "синдром X", и который включает по меньшей мере одно из следующего: абдоминальное ожирение, атерогенная дислипидемия, устойчивость к инсулину, непереносимость глюкозы, протромбическое состояние или провоспалительное состояние), диабет, диабетическая нефропатия, застойная сердечная недостаточность и т.д. Субъекту, страдающему от одного из вышеперечисленных болезненных состояний и нуждающемуся в их лечении, можно вводить эффективное количество (или терапевтически эффективное количество) дозированной формы настоящего изобретения для лечения указанных болезненных состояний.
Далее представлены примеры, предназначенные для иллюстративных целей раскрытия настоящего изобретения, но не для ограничения.
ПРИМЕР 1: Способы получения высокого уровня ингибирования ксантиноксидазы - Сравнительный пример
У здоровых людей после перорального приема фебуксостата его абсорбция происходит в большинстве случаев приблизительно через 1 час (то есть, tmax составляет приблизительно один час). Кроме того, клиренс фебуксостата из плазмы при пероральном приеме составляет приблизительно 7,3-15,1 л/час, с временем эффективной полужизни приблизительно шесть (6) часов. Фактически, лекарственное вещество сильно связано с альбумином в крови (~99,3%) и, вероятно, имеет кажущийся объем распределения от низкого до среднего, приблизительно равный 0,7 л/кг.
Несмотря на относительно короткое время эффективной полужизни фебуксостата, клинические исследования показали, что у препарата немедленного высвобождения, содержащего всего 10 мг фебуксостата, дозирование в режиме один раз в день снижает концентрацию мочевой кислоты с минимальными колебаниями концентрации мочевой кислоты здоровых субъектов. Это происходит из-за природы фармакокинетической (PK) - фармакодинамической (PD) взаимосвязи между концентрациями фебуксостата (PK маркер) в плазме и концентрациями мочевой кислоты в сыворотке (маркер PD). Дозированная форма фебуксостата немедленного высвобождения, содержащая всего 10 мг, при дозировке в режиме один раз в день, как ожидают, эффективно уменьшит и поддержит концентрацию мочевой кислоты в сыворотке в терапевтических целях (то есть <6 мг/дл) у пациента с подагрой с низкой концентрацией мочевой кислоты в сыворотке (то есть 7 мг/дл); однако эти дозированные формы не в состоянии поддержать высокую степень ингибирования, а именно, по меньшей мере 80%-ного ингибирования фермента ксантиноксидазы в течение интервала дозирования (а именно - 24 часа) даже после многократного дозирования.
Таблица 1, ниже, показывает результаты, полученные при изучении фебуксостата на Фазе 1 мультидозового, рандомизированного, плацебо-контролируемого, двойного слепого, одноцентрового, мультилокализированного исследования с повышением дозы. Это исследование позволило оценить фармакокинетику и фармакодинамику фебуксостата у здоровых субъектов. В этом исследовании пероральные дозы дозированной формы фебуксостата немедленного высвобождения варьировались от 10 мг один раз в день до 240 мг один раз в день (в дальнейшем "QD"), а также составляли 30 мг два раза в день (в дальнейшем "BID"). Плазма, сыворотка и образцы мочи собирались для определения концентраций фебуксостата и метаболитов, мочевой кислоты, ксантина и гипоксантина. Образцы подвергали анализу методом высокоэффективной жидкостной хроматографии.
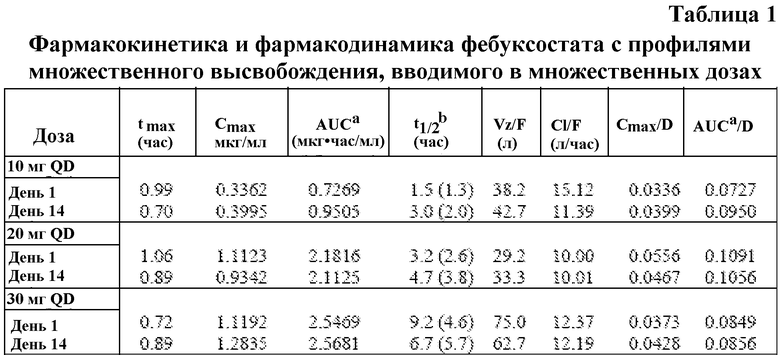

Кроме того, процент ингибирования ксантиноксидазы на 12, 16 и 24 часа (после дозирования) при многократном дозировании дозированных форм немедленного высвобождения, содержащим 70 мг и 120 мг фебуксостата, здоровым субъектам, был вычислен, используя Уравнение 1, представленное ниже. Результаты этого вычисления были включены в Уравнение 2, обсуждены ниже, и результаты Уравнения 2 перечислены в таблице 2 ниже:
Процент ингибирования ("% ингибирования") ксантиноксидазы (XOD):
% Ингибирования XOD = 100
где C = концентрация фебуксостата в плазме,
fu = свободная фракция фебуксостата в плазме, и
Ki = константа ингибирования ксантиноксидазы фебуксостатом.
Концентрация фебуксостата в плазме может быть определена, используя валидированную высокоэффективную жидкостную хроматографию с флюоресцентной детекцией (см. Biopharmaceutics Coordinating Committee in the Center for Drug Evaluation and Research (CDER). Guidance for industry: bioanalytical method validation. May 2001 и Mayer, M. et al., American Journal of Therapeutics, 12:22-34 (2005), каждый из которых включен в виде ссылки). Нижний предел количественного определения фебуксостата в пробе плазмы объемом 0,5 мл составлял 0,01 мкг/мл.
Значение fu может быть определено с помощью анализа связывания in vitro 14C фебуксостата при номинальной концентрации 1 мкг/мл, используя метод равновесного диализа, который хорошо известен в данной области. Например, доля fu для фебуксостата была определена как 0,9±0,2 у нормальных пациентов и как 1,2±0,2 у пациентов с тяжелой почечной недостаточностью (см. Mayer, M. et al., American Journal of Therapeutics, 12:22-34 (2005), включено ссылкой). В другом исследовании, с большим числом субъектов, доля свободной фракции фебуксостата в плазме была определена как 0,7±0,1 в мужской, женской, подростковой и пожилой группе испытуемых субъектов (см. Khosravan R., et al, Clinic. Pharmacology & Therapeutics, P50 (2005), включено ссылкой).
Значение Ki для фебуксостата было определено с использованием анализа ксантиноксидазы, такого как описано в Osada Y., et al., European J. Pharmacology, 241: 183-188 (1993), включено сюда в виде ссылки. Более конкретно, Ki для фебуксостата была определена как 0,7 нМ и 0,6 нМ, соответственно, (см. Osada Y., et al., European J. Pharmacology, 241: 183-188 (1993) и Takano, Y., et al., Life Sciences, 76: 1835-1847 (2005)). Значение Ki для фебуксостата, известное из уровня техники, составляет 0,6 нМ (см. Takano, Y., et al., Life Sciences, 76: 1835-1847 (2005), включено ссылкой).
Кроме того, концентрации фебуксостата, которые обеспечивают 50%, 60%, 70%, 80%, 90%, 95% и 99%-ное ингибирование активности ксантиноксидазы, были вычислены, используя приведенное ниже Уравнение 2 (основанное на вычислениях, сделанных выше с использованием Уравнения 1). Результат этого вычисления для всех режимов дозирования представлен ниже в таблице 2.
Уравнение 2

где С = концентрация фебуксостата в плазме,
fu = свободная фракция фебуксостата в плазме, и
Ki = константа ингибирования ксантиноксидазы фебуксостатом.
Как показано в таблице 2, даже когда субъекты получили высокие дозы фебуксостата (120 мг), не было достигнуто ингибирование фермента ксантиноксидазы по меньшей мере на уровне 80% через шестнадцать (16) часов. Дополнительно, другие исследования, включающие дозированные формы 120 мг фебуксостата немедленного высвобождения, продемонстрировали, что такие дозированные формы обеспечивают Cmax приблизительно 3,9 и приблизительно 4,2 мкг/мл, и показали по меньшей мере 80%-ное ингибирование ксантиноксидазы в течение приблизительно четырнадцати (14) часов. Дальнейшие исследования, включающие дозированные формы 240 мг фебуксостата немедленного высвобождения, продемонстрировали, что такие дозированные формы обеспечивают Cmax 10,2 мкг/мл, и показали по меньшей мере 80%-ное ингибирование ксантиноксидазы в течение приблизительно 22 часов. Поэтому, принимая во внимание эти результаты, авторы пришли к созданию дозированных форм с модифицированным высвобождением по настоящему изобретению. Эти дозированные формы показывают высокие уровни (по меньшей мере 80%) ингибирования ксантиноксидазы в течение периода более шестнадцати (16) часов, не подвергая пациента риску из-за несоблюдения режима лечения. Дозированные формы настоящего изобретения поддерживают высокие уровни ингибирования фермента ксантиноксидазы при сходных или более низких полных уровнях воздействия в плазме, по сравнению с высокими дозами фебуксостата (а именно, 120 мг и 240 мг QD).
ПРИМЕР 2: Оценка плазменных профилей для препаратов фебуксостата пролонгированного высвобождения
В свете фармакокинетических данных, касающихся препарата фебуксостата немедленного высвобождения, включенных в пример 1, авторы продолжили оценку плазменных профилей для различных препаратов пролонгированного высвобождения. В частности, авторы оценили плазменные профили для трех типов препаратов: препараты пролонгированного высвобождения, препараты двухстадийного высвобождения и препараты трехстадийного высвобождения. Информация по плазменным профилям для препаратов пролонгированного высвобождения основана на данных для препаратов с полимерным компонентом, который обеспечивает высвобождение фебуксостата с постоянной скоростью в течение длительного времени, и препарат может быть основан на технологии, такой как матричный препарат. Оценочные данные для препарата двухстадийного высвобождения получены для препарата фебуксостата, содержащего гранулы немедленного высвобождения и гранулы, которые высвобождают фебуксостат приблизительно через 5 часов. Оценочные данные для препарата трехстадийного высвобождения получены для препарата фебуксостата, содержащего гранулы немедленного высвобождения, гранулы, которые высвобождают фебуксостат приблизительно через 5 часов, и гранулы, которые высвобождают фебуксостат приблизительно через 10 часов. Установленные плазменные профили для различных препаратов могут быть найдены в таблице 3. Следует отметить, что информация, касающаяся плазменных профилей препаратов немедленного высвобождения, представленная в таблице 3, представляет собой фактические данные, полученные в клиническом тестировании.
(час)
(мкг/
мл)
(мкг/
мл)
(мкг·час/мл)
ПРИМЕР 3: Биодоступность фебуксостата, высвобождаемого в различных участках желудочно-кишечного тракта
Авторы проверили относительное бионакопление для 80 мг фебуксостата, высвобождаемого в проксимальной тонкой кишке, дистальной тонкой кишке и толстой кишке у 12 здоровых мужчин, по сравнению с бионакоплением для дозированной формы немедленного высвобождения.
Субъектам были произвольно присвоены эквивалентные номера последовательностей режимов от первого до четвертого, как представлено в таблице 4. В отношении субъектов применялся режим по перекрестной схеме, согласно случайному списку. Дополнительные периоды (до 2 на субъект) были добавлены, где имели место проблемы высвобождения доз, и требовалось повторение режима. Имелся перерыв по меньшей мере 7 дней между дозой в один период и следующей дозой в последующий период.
Режим B: капсулы InteliSite, содержащие 80 мг фебуксостата, где лекарственное вещество высвобождается в проксимальной тонкой кишке
Режим C: капсулы InteliSite, содержащие 80 мг фебуксостата, где лекарственное вещество высвобождается в дистальной тонкой кишке
Режим D: капсулы InteliSite, содержащие 80 мг фебуксостата, где лекарственное вещество высвобождается в толстой кишке
Образцы крови были получены до дозирования (0 часов) в день 1 для каждого периода, и до того, как лекарственный препарат, проходящий клиническое испытание, был высвобожден из капсулы InteliSite (преактивация 0 часов) для Режимов B, C и D, и через 0,25, 0,5, 1, 1,5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 30 и 36 часов после дозирования таблетки или высвобождения фебуксостата в выбранной области желудочно-кишечного тракта. Концентрации фебуксостата в плазме были определены в образцах, обработанных EDTA, используя валидированный жидкостной хроматографический анализ с масс-спектрометрической детекцией, используя положительную ионную распылительную ионизацию в приборе PPD (Richmond, VA). В способе применялось осаждение белка ацетонитрилом в аликвотах плазмы объемом 100 мкл, с нижним пределом обнаружения 10,0 нг/мл.
Фармакокинетические параметры для фебуксостата были оценены, используя стандартные модельно-независимые методы. Вычисления были выполнены с использованием WinNonlin Pro Version 5.2 (Mountain View, CA, USA). Были вычислены описательные статистические показатели для фармакокинетических оценок параметров. Дисперсионный анализ (ANOVA) был выполнен для фебуксостата в отношении Tmax и натуральных логарифмов Cmax, AUC(0-tlqc), и AUC(0-inf), с учетом факторов для последовательностей, субъектов в пределах последовательностей, периодов и режимов. Эффект высвобождения в проксимальой тонкой кишке, дистальной тонкой кишке и толстой кишке на бионакопление фебуксостата был оценен посредством точечной оценки и 90%-ных доверительных интервалов для отношения центральных значений для Cmax и AUC фебуксостата для Режима B по отношению к Режиму A, для Режима C по отношению к Режиму A и для Режима D по отношению к Режиму A, соответственно. Субъектам, у которых было отмечено неприемлемое высвобождение из капсул, разрешили повторить до двух дополнительных периодов, после того как их запланированная последовательность завершилась. Для целей ANOVA, пропущенные или неполные данные по периодам с неприемлемым высвобождением из капсул, были заменены данными, полученными в рамках соответствующих периодов повторов, используя первоначально запланированный режим периода.
Профили "средняя концентрация-время" для фебуксостата в плазме (линейные и логарифмически-нормальные форматы) для каждого режима и участка поглощения, изображены на фиг. 1. Обобщенные средние фармакокинетические оценки параметров для фебуксостата после единственной дозы 80 мг фебуксостата для всех четырех периодов показаны в таблице 5. Все концентрации фебуксостата в плазме, полученные от каждого субъекта, использовались для оценок фармакокинетических параметров; однако у субъекта 017-JDF (рандомизированный номер 112) имело место существенно низкое воздействие на плазму (Cmax и AUC) из-за очевидного отсроченного высвобождения (приблизительно 3 часа) лекарственного вещества в дистальной части тонкой кишки (Режим C), по сравнению со всеми другими субъектами. При высвобождении фебуксостата в дистальной части кишечника у субъекта 017-JDF, значение Cmax составило 5-13%, значение AUC составило 12-28% от значений, наблюдавшихся у всех других субъектов. В этой связи данные, полученные от этого субъекта в отношении высвобождения в дистальной части кишечника, не были включены в статистический анализ.
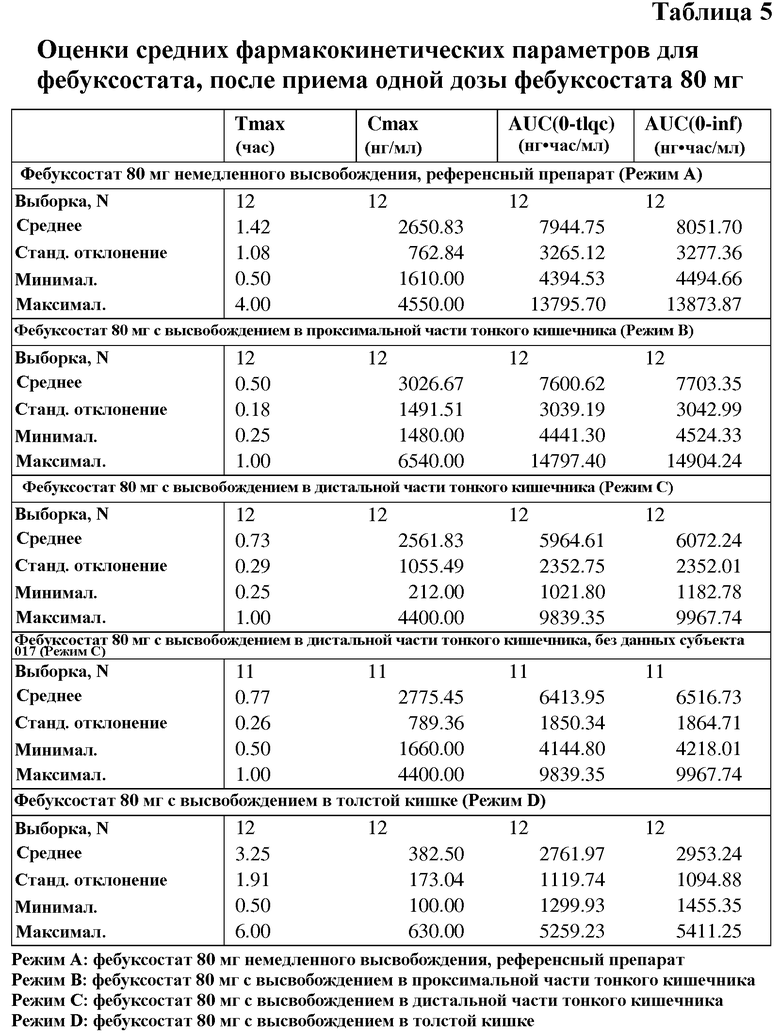
После введения 80 мг фебуксостата, высвобождаемого в проксимальной части тонкой кишки (Режим B) или в дистальной части тонкой кишки (Режим C), среднее значение Tmax составило на 65% и на 49% меньше (46% с исключенным субъектом 017-JDF) по сравнению с таблеткой немедленного высвобождения (Режим A). В толстой кишке (Режим D) среднее значение Tmax было более чем в два раза выше, чем у таблетки немедленного высвобождения (Режим A). Когда фебуксостат высвобождался в проксимальной части (Режим B) или в дистальной части (Режим C) тонкой кишки (несмотря на субъекта 017-JDF), оба средних значения Cmax были приблизительно равны среднему значению Cmax, полученного в случае таблетки немедленного высвобождения (Режим A); в тоже время, среднее значение Cmax фебуксостата для толстой кишки (Режим D) составило 14% от среднего значения Cmax для таблетки немедленного высвобождения (Режим A). Средние значения AUC фебуксостата в целом были схожи после высвобождения фебуксостата в проксимальной части тонкой кишки (Режим B), и в случае таблетки немедленного высвобождения (Режим A); однако для случая дистального высвобождения (Режим C) средние значения AUC были снижены до 75% (до 81% при исключении субъекта 017-JDF), по сравнению со средним значением AUC для таблетки немедленного высвобождения (Режим A). После высвобождения фебуксостата в толстой кишке (Режим D) средние значения AUC составили приблизительно 35% от среднего значения AUC для таблетки немедленного высвобождения (Режим A).
Статистические оценки по биодоступности принимаемого фебуксостата (Режимы B, C и D) относительно фебуксостата немедленного высвобождения (Режим A), были оценены посредством точечных оценок и 90%-ных доверительных интервалов для отношений центральных значений Cmax, AUC(0-tlqc) и AUC(0-inf), и отражены в таблице 6 (исключая данные по высвобождению в дистальном участке тонкой кишки у субъекта 017-JDF).

Основанные на данных от 12 субъектов (11 для дистального высвобождения в кишечнике - Режим C), 90%-ные доверительные интервалы для отношений средних значений, когда 80 мг фебуксостата вводились для высвобождения в проксимальной части тонкой кишки, в дистальной части тонкой кишки или в толстой кишке (Режимы B, C или D, соответственно), относительно дозирования таблетки немедленного высвобождения (Режим A), были за пределами биоэквивалентного лимита, составляющего от 0,80 до 1,25, для значений Cmax, AUC(0-tlqc) или AUC(0-inf) фебуксостата. Центральные значения Cmax фебуксостата для проксимального (Режим B) и дистального (Режим C) высвобождения были приблизительно на 7% и на 6%, соответственно, больше, чем центральное значение Cmax после приема таблетки немедленного высвобождения (Режим A). Центральные значения AUC для проксимального высвобождения (Режим B) или дистального высвобождения (Режим C) были снижены примерно на 3% и 16%, соответственно, по сравнению с показателями после приема таблетки немедленного высвобождения (Режим A). Центральные значения Cmax, AUC(0-tlqc) и AUC(0-inf) для случая высвобождения в толстой кишке (Режим D), составляли соответственно 14%, 35% и 37% от значений, полученных после приема таблетки немедленного высвобождения (Режим A).
Соответственно, было установлено, что после приема единичной пероральной дозы 80 мг фебуксостата, высвобождаемого в проксимальном участке тонкой кишки, в дистальном участке тонкой кишки или в толстой кишке (Режимы B, C или D), 12 здоровыми мужчинами (исключая субъекта 017-JDF для случая высвобождения в участке тонкой кишки), системное воздействие фебуксостата не было биоэквивалентным приему таблетки 80 мг фебуксостата немедленного высвобождения (Режим A).
Кроме того, авторы изобретения использовали данные оценок, включенные в таблицы 3, 4 и 5, для представления фиг. 2-4. Авторы вычислили оценочные фармакокинетические значения абсорбции фебуксостата в различных частях желудочно-кишечного тракта (то есть, в желудке, в проксимальном участке тонкой кишки, в дистальном участке тонкой кишки и в толстой кишке) и использовали эти параметры для создания логарифмически-линейных графиков зависимости концентрации фебуксостата в плазме от времени для различных дозированных форм, включая препарат 80 мг фебуксостата с трехстадийным высвобождением, препарат 80 мг фебуксостата с двухстадийным высвобождением и включая препарат 80 мг фебуксостата пролонгированного высвобождения (ER).
ПРИМЕР 4: Препараты матричных таблеток с модифицированным высвобождением
Дозированные формы с модифицированным высвобождением, которые непрерывно высвобождают ингибитор ксантиноксидоредуктазы, а именно, фебуксостат, были изготовлены как матричные таблетки, содержащие компоненты, показанные на фиг. 5 (ингредиенты указаны в процентах по массе). Более конкретно, таблетки были изготовлены влажным гранулированием, используя гранулятор Black & Decker Handy Chopper. Порядок, в котором были добавлены компоненты, не был важен. Для получения конечной смеси использовали V-блендер (Blend Master Lab, LC 9292659). Таблетки прессовались в моделирующем прессе, используя оснастку для круглой формы (A-2308) или для овальной формы (A-2253). Для каждой из дозированных форм, показанных на фиг. 5, были определены характеристики высвобождения лекарственного вещества, и полученные профили растворения показаны на фиг. 6. Более конкретно, в отношении характеристик высвобождения лекарственного вещества и профилей растворения, дозированные пероральные дозированные формы фебуксостата, описанные здесь, и содержащие ингредиенты, показанные на фиг. 5, были оценены по растворению в 900 мл 0,5 М фосфатного буфера, pH 6,8, уравновешенного при 37°C ± 0,5°C, используя способ с лопастной мешалкой (аппарат 2 по USP) при 50 об/мин. Пробы аликвот были взяты в различные временные интервалы и проанализированы с использованием высокоэффективной жидкостной хроматографии. Композиции, показанные на фиг. 5, представляют препараты фебуксостата пролонгированного высвобождения.
ПРИМЕР 5: Дозированные формы с трехстадийным высвобождением
Дозированные формы с трехстадийным высвобождением ингибитора ксантиноксидоредуктазы, такого как фебуксостат, могут быть изготовлены, как содержащие ингредиенты (показанные в процентах по массе), перечисленные в таблице 7 ниже. Эти дозированные формы включают три (3) набора гранул. Гранула A предназначена для высвобождения активного агента в желудке. Гранула B предназначена для высвобождения активного агента в тощей кишке. Гранула C предназначена для высвобождения активного агента в дистальной части подвздошной кишки и восходящей части ободочной кишки.
Более конкретно, слой, нагруженный активным агентом, может быть нанесен распылением водной суспензии активного агента на множество нейтральных ядер, для того чтобы получить множество гранул, несущих лекарственное вещество. Эти гранулы лекарственного вещества идентифицированы как "Гранула А". Первую часть гранул A удаляют, и затем покрывают дисперсией сополимера метакриловой кислоты (такой как Eudragit® L30D-55 или Eudragit® L100-55), которая содержит активное вещество. Дисперсия сополимера метакриловой кислоты может быть нанесена на гранулы A путем распыления водной суспензии непосредственно на гранулы A. Эти гранулы А, покрытые сополимером метакриловой кислоты, далее обозначаются как "Гранулы B". Вторую часть гранул A удаляют, и покрывают водной дисперсией, которая содержит смесь сополимера метакриловой кислоты типа А (такого как Eudragit® L100 и Eudragit® L12.5) и типа B (такого как Eudragit® S100 и Eudragit® S12.5), и активного агента. Эта водная дисперсия, содержащая смесь сополимера метакриловой кислоты типа A и типа B, может быть нанесена на гранулы А распылением этой смеси непосредственно на гранулы A. Эти покрытые гранулы A далее обозначаются как "Гранулы C". Затем остающиеся гранулы A смешивают вместе с гранулами B и C, и заполняют ими твердые желатиновые капсулы размера 0.
Фиг. 2 показывает предполагаемый плазменный профиль для дозированной формы, имеющей три типа гранул (трехстадийное высвобождение), как показано в таблице 7 выше (включающий все три типа гранул). В частности, фиг. 2 показывает предполагаемый профиль концентрации фебуксостата в плазме во времени, после множественных дозировок дозированной формы фебуксостата контролируемого высвобождения (гранулы с трехстадийным высвобождением, IR (немедленное высвобождение) = 24 мг, CR (контролируемое высвобождение) = 24 мг, высвобождаемые через 5,0 часов, CR = 32 мг, высвобождаемые через 10 часов), используя человеческие фармакокинетические данные для человека из Примеров 1-3 и других исследований и двухкомпартментную модель абсорбции первого порядка, а также сравнение с предполагаемым плазменным профилем 80 мг препарата с немедленным высвобождением.
ПРИМЕР 6: Композиция с 30% гранул фебуксостата немедленного высвобождения и 70% гранул фебуксостата отсроченного высвобождения до рН 6,8
Была создана следующая композиция, как система доставки с двухстадийным высвобождением лекарственного вещества, в которой одна капсула включает два типа гранул. Гранулы первой стадии высвобождения состоят из 24 мг фебуксостата немедленного высвобождения, где фебуксостат немедленно высвобождается после приема пациентом. Гранулы второй стадии высвобождения состоят из 56 мг фебуксостата отсроченного высвобождения до рН 6,8, где фебуксостат высвобождается, когда гранулы подвергаются воздействию pH при величине по меньшей мере 6,8. В таблицах 8 и 9 ниже перечислены компоненты гранул немедленного высвобождения и отсроченного до рН 6,8 высвобождения.
отсроченного высвобождения до рН 6,8
ПРИМЕР 7: Композиция с 30% гранул фебуксостата немедленного высвобождения, 30% гранул фебуксостата отсроченного высвобождения до рН 6,0 и 40% гранул фебуксостата отсроченного высвобождения
Была создана следующая композиция, как система доставки с трехстадийным высвобождением лекарственного вещества, в которой одна капсула включает три типа гранул. Гранулы первой стадии высвобождения состоят из 24 мг фебуксостата немедленного высвобождения, где фебуксостат немедленно высвобождается после приема пациентом. Гранулы второй стадии высвобождения состоят из 24 мг фебуксостата отсроченного высвобождения до рН 6,0, где фебуксостат высвобождается, когда гранулы подвергаются воздействию pH при величине по меньшей мере 6,0. Гранулы третьей стадии высвобождения состоят из 42 мг фебуксостата отсроченного до рН 6,8 высвобождения, где фебуксостат высвобождается, когда гранулы подвергаются воздействию pH при величине по меньшей мере 6,8. Состав гранул фебуксостата немедленного высвобождения и гранул отсроченного высвобождения до рН 6,8 представлен в таблицах 8 и 9, соответственно. Состав гранул отсроченного высвобождения до рН 6,0 представлен в таблице 10, ниже.
отсроченного высвобождения до рН 6,0
ПРИМЕР 8: Композиция с 30% гранул фебуксостата немедленного высвобождения и 70% гранул фебуксостата отсроченного/контролируемого высвобождения
Была создана следующая композиция, как система одностадийного и отсроченного/контролируемого высвобождения лекарственного вещества, в которой одна капсула включает два типа гранул фебуксостата. Гранулы первой стадии высвобождения состоят из 24 мг фебуксостата немедленного высвобождения, где фебуксостат немедленно высвобождается после приема пациентом. Оставшаяся часть гранул капсулы включает 56 мг гранул отсроченного/контролируемого высвобождения, которые за счет наиболее удаленного слоя отсроченного высвобождения становятся растворимыми на уровнях pH 6,8 или выше, и после того как наиболее удаленный слой растворяется, слой контролируемого высвобождения высвобождает фебуксостат в течение длительного периода, который составляет от четырех до шести часов. Состав гранул фебуксостата немедленного высвобождения указан в таблице 8, выше. Состав гранул отсроченного/контролируемого высвобождения указан в таблице 11, ниже.
отсроченного/контролируемого высвобождения
ПРИМЕР 9: Композиция с 20% гранул фебуксостата немедленного высвобождения и 80% гранул контролируемого высвобождения в течение 10-12 часов.
Была создана следующая композиция, как система одностадийного и контролируемого высвобождения лекарственного вещества, в которой одна капсула включает два типа гранул фебуксостата. Гранулы первой стадии высвобождения состоят из 16 мг фебуксостата немедленного высвобождения, где фебуксостат немедленно высвобождается после приема пациентом. Оставшаяся часть гранул капсулы включает 64 мг гранул контролируемого высвобождения, за счет которых фебуксостат высвобождается в течение длительного периода, составляющего от десяти до двенадцати часов, начиная сразу после приема пациентом. Состав гранул фебуксостата немедленного высвобождения указан в таблице 8, выше. Состав гранул контролируемого высвобождения в течение 10-12 часов указан в таблице 12, ниже.
контролируемого высвобождения в течение 10-12 часов
ПРИМЕР 10: Фармакокинетические данные по контролируемому высвобождения фебуксостата, полученные в модели на собаках
Было выполнено исследование, в котором восемь различных препаратов фебуксостата вводили 6 собакам перекрестным способом, и после дозирования у собак была измерена концентрация в плазме (нг/мл) в периоды времени 0,25, 0,5, 1, 2, 3, 4, 5, 6, 8, 12, 18 и 24 часа. был выполнен тест для того, чтобы определить, как различные препараты абсорбируются в модели на собаках, и для того, чтобы определить плазменные профили концентрации во времени. В частности, в исследовании использовались шесть чистокровных кобелей породы бигль, при этом каждый из восьми испытуемых препаратов вводили одной и той же группе из шести собак. Собаки были размещены индивидуально и не смешивались в течение по меньшей мере 24 часов после введения дозы, для того чтобы обеспечить мониторинг любого тестируемого эффекта, связанного с исследуемым препаратом. Испытуемые препараты вводили в виде пероральной дозированной формы, включающей капсулу или таблетку. После дозирования собаки не получали корма в течение ночи, приблизительно 4 часа. Индивидуальные дозы были вычислены на основании массы тела, измеренной в каждый день дозирования. Приблизительно за 1 час до приема тестируемого препарата каждой собаке внутримышечно вводили пентагастрин в дозе 6 мкг/кг (0,048 мл/кг). Средняя концентрация фебуксостата в плазме была измерена в образцах крови (приблизительно 2 мл), которые были отобраны из яремной вены через шприц и иглу в пробирки, содержащие K2EDTA в качестве предозы антикоагулянта, во времена, указанные выше. Затем образцы плазмы отправлялись в лабораторию для анализа. Модель на собаках была выбрана исходя из предшествующего опыта дозирования гранул отсроченного высвобождения для других лекарственных веществ собакам, и с учетом соотношениях данных, полученных на собаках, с данными у человека. В предыдущих исследованиях задержка Tmax для гранул отсроченного высвобождения, с наблюдаемым Tmax, равным приблизительно 2 часа у собак, соответствовала задержке Tmax до 8 часов у человека.
Все восемь препаратов фебуксостата включали суммарную дозу 80 мг фебуксостата для различных композиций гранул немедленного высвобождения, отсроченного высвобождения, контролируемого высвобождения и отсроченного/контролируемого высвобождения. Восемь препаратов фебуксостата включали:
(1) 80 мг таблетки немедленного высвобождения (референсный препарат) (названный "Фаза 1");
(2) 80 мг гранул отсроченного высвобождения до рН 5,5, имеющие растворимость при уровне pH по меньшей мере 5,5 (названный "Фаза 2");
(3) 24 мг гранул немедленного высвобождения и 56 мг гранул отсроченного высвобождения до рН 6,0 (названный "Фаза 3");
(4) 24 мг гранул немедленного высвобождения и 56 мг гранул отсроченного высвобождения до рН 6,8 (названный "Фаза 4");
(5) 24 мг гранул немедленного высвобождения, 24 мг гранул отсроченного высвобождения до рН 6,0 и 32 мг гранул отсроченного высвобождения до рН 6,8 (названный "Фаза 5");
(6) 24 мг гранул немедленного высвобождения и 56 мг гранул контролируемого высвобождения в течение 4-6 часов (названный "Фаза 6");
(7) 24 мг гранул немедленного высвобождения и 56 мг гранул отсроченного/контролируемого высвобождения, где слой отсроченного высвобождения растворим на уровнях pH, равных по меньшей мере 6,0, и слой контролируемого высвобождения высвобождает фебуксостат в течение 4-6 часов (названный "Фаза 7"); и
(8) 24 мг гранул немедленного высвобождения и 56 мг гранул контролируемого высвобождения в течение 10-12 часов (названный "Фаза 8").
Плазменные концентрации для различных дозированных форм показаны на фиг. 7. В частности, фиг. 7 отображает график для каждого препарата, раскрывая среднее значение концентрации фебуксостата в плазме для каждого препарата в течение шестичасового периода после приема дозированных форм. Хотя образцы плазмы были собраны вплоть до 24 часов, для большинства животных через 6-8 часов были зафиксированы очень низкие плазменные концентрации. Это совместимо с литературными данными, которые раскрывают, что длина желудочно-кишечного тракта собак короче, чем у людей. Поэтому по сравнению с людьми транзит твердых дозированных форм через желудочно-кишечный тракт собаки более быстрый. В результате препараты отсроченного высвобождения, разработанные, чтобы быстро высвобождать фебуксостат при определенных критических значениях pH, абсорбировались намного лучше, чем препараты контролируемого высвобождения, которые высвобождают лекарственное вещество в течение времени.
Кроме того, было выполнено сравнение всех восьми препаратов путем оценки средней концентрации фебуксостата в плазме для каждой фазы исследования в четыре часа, пять часов и шесть часов после дозирования. Как и ожидались, результаты в целом показали, что препараты, включающие гранулы контролируемого высвобождения, не достигали концентраций фебуксостата в плазме значительно более высоких, чем обеспечивал препарат, включающий только гранулы немедленного высвобождения. Следует повторить, что этот результат являлся ожидаемым, поскольку желудочно-кишечный тракт собак намного короче, чем у человека. Это физиологическое последствие, связанное с фармакокинетическим тестированием в моделях на собаках, обсуждено более подробно в следующих статьях: Stephen C. Sutton, Companion animal physiology and dosage form performance, Advanced Drug Delivery Reviews, 2004, vol. 56, pp. 1383-1398; и Jennifer B. Dressman, Comparison of Canine and Human Gastrointestinal Physiology, Pharmaceutical Research, 1986, vol. 3, no. 3, pp. 123-131. Соответственно, препараты контролируемого высвобождения не высвобождают полностью активный компонент в период до 4-6 или 10-12 часов после приема пищи, поскольку к тому времени, когда активное вещество начинает высвобождение, препарат уже прошел большую часть, если не весь желудочно-кишечный тракт собаки, и высокие плазменные концентрации не могут быть достигнуты. Обобщение этих результатов включено в таблицу 13 ниже.
У препарата Фазы 1, включающего 80 мг гранул немедленного высвобождения, на четвертый час после дозирования имела место средняя плазменная концентрация 289,8 нг/мл. Это значение было значительно ниже, чем средние плазменные концентрации для дозированных форм, включающих гранулы отсроченного высвобождения, таких как препараты Фазы 2, Фазы 3, Фазы 4 и Фазы 5, которые показали средние концентрации 632,6 нг/мл, 785,2 нг/мл, 448,3 нг/мл и 827,9 нг/мл, соответственно. Уровни величин pH в желудочно-кишечном тракте собаки и у человека являются схожими, и поэтому гранулы отсроченного высвобождения не зависели от длины желудочно-кишечного тракта собаки, как это наблюдалось в случае гранул контролируемого высвобождения. Дозированные формы, включающие комбинацию гранул немедленного высвобождения и гранул контролируемого высвобождения, в целом показали средние плазменные концентрации, подобные Фазе 1, как и ожидалось. Препараты Фазы 6, Фазы 7 и Фазы 8 показали средние плазменные концентрации 319,0 нг/мл, 329,8 нг/мл и 86,2 нг/мл, соответственно. Вероятно, в этих случаях было высвобождено только небольшое количество фебуксостата в гранулах контролируемого высвобождения, поскольку плазменные концентрации зависели, по меньшей мере частично, от гранул немедленного высвобождения, представленных в препаратах Фазы 6, Фазы 7 и Фазы 8.
Рассматривая средние концентрации на 5 часов после дозировании, видно, что у препарата Фазы 1 была средняя плазменная концентрация 73,2 нг/мл. В сравнении с этим, препараты отсроченного высвобождения Фазы 2, Фазы 3, Фазы 4 и Фазы 5 показали более высокие средние плазменные концентрации, равные 207,8 нг/мл, 283,7 нг/мл, 151,3 нг/мл и 249,1 нг/мл, соответственно. Препараты контролируемого высвобождения Фазы 6, Фазы 7 и Фазы 8 показали средние плазменные концентрации 124,2 нг/мл, 95,5 нг/мл и 50,2 нг/мл, соответственно.
Кроме того, средние концентрации в плазме через 6 часов после дозировании показали схожие сравнительные данные по отношению к данным через 4 часа и 5 часов после дозирования. У препарата Фазы 1 была средняя концентрации в плазме 53,8 нг/мл. Аналогично данным на 4 и 5 часов, препараты отсроченного высвобождения Фазы 2, Фазы 3, Фазы 4 и Фазы 5 показали более высокие средние концентрации в плазме, равные 79,1 нг/мл, 105,3 нг/мл, 69,3 нг/мл и 104,9 нг/мл, соответственно. Препараты контролируемого высвобождение Фазы 6, Фазы 7 и Фазы 8 показали средние плазменные концентрации 64,2 нг/мл, 44,3 нг/мл и 25,6 нг/мл, соответственно.
Таким образом, даже при том, что набор плазменных концентраций в модели на собаках безусловно ограничен более короткой длиной желудочно-кишечного тракта собаки по сравнению с желудочно-кишечным трактом человека, сравнительные результаты поддерживают улучшенные концентрации фебуксостата в плазме для препаратов модифицированного высвобождения, по сравнению с препаратами немедленного высвобождения. Препараты отсроченного высвобождения Фазы 2, Фазы 3, Фазы 4 и Фазы 5 показали более высокие средние плазменные концентрации через 4, 5 и 6 часов после дозирования для всех препаратов, по сравнению с референсным препаратом немедленного высвобождения. Препараты контролируемого высвобождения Фазы 6 и Фазы 7 (оба включающие гранулы с контролируемым высвобождением через 4-6 часов) показали улучшенные средние плазменные концентрации через 4, 5 и 6 часов после дозирования (за исключением средней концентрации для Фазы 7 на 6 часов), хотя не в той степени, как у препарата отсроченного высвобождения. Вероятно, эти результаты являются следствием того, что компонент контролируемого высвобождения для Фазы 6 и 7 был предназначен для высвобождения активного вещества более чем через 4-6 часов. Это предполагает, что препарат, возможно, высвободил только часть фебуксостата до того, как препарат прошел всю длину желудочно-кишечного тракта. Препарат контролируемого высвобождения Фазы 8 (включающий гранулы с контролируемым высвобождением через 10-12 часов) показал самую низкую среднюю плазменную концентрацию, ниже, чем у референсного препарата немедленного высвобождения. Как обсуждалось ранее, этот результат не является неожиданным, поскольку это вероятно имело место из-за того, что гранулы с контролируемым высвобождением через 10-12 часов (содержащие 70% препарата) высвободили только малую часть фебуксостата до полного прохождения через желудочно-кишечный тракт собаки.
ПРИМЕР 11: Результаты Фазы 1 исследования единичной дозы четырех препаратов фебуксостата пролонгированного высвобождения и одного препарата фебуксостата немедленного высвобождения на людях
Этот пример описывает результаты Фазы 1 одноцентрового, открытого, рандомизированного, пятистороннего перекрестного исследования. Для участия в этом эксперименте были отобраны тридцать пять взрослых мужчин и женщин в возрасте от 18 до 55 лет, включительно, с хорошим состоянием здоровья. Субъекты в равном количестве были рандомизированно распределены в группы 1-5 по последовательностям приема препаратов, как показано в таблице 14.
Препарат B (тестируемый): капсула с 80 мг фебуксостата двухстадийного высвобождения, прототип, (TMX-67, XR, Препарат B).
Препарат C (тестируемый): капсула с 80 мг фебуксостата трехстадийного высвобождения, прототип (TMX-67, XR, Препарат С).
Препарат D (тестируемый): капсула с комбинацией фебуксостата многостадийного и длительного высвобождения (TMX-67, Препарат D).
Препарат E (тестируемый): капсула с 80 мг фебуксостата длительного высвобождения, прототип (TMX-67, XR, Препарат E).
Все субъекты в исследовании получали по 5 препаратов фебуксостата (IR, 2-стадийного высвобождения, 3-стадийного высвобождения, в виде комбинации многостадийного и длительного высвобождения, а также непрерывного высвобождения) в перекрестном порядке, согласно схеме рандомизации. Схематический план исследования показан в таблице 15.
Сбор фармакокинетических проб
Образцы крови (4 мл) для определения концентрации фебуксостата в плазме были собраны в предписанных временных точках в течение 48 часов после приема 80 мг фебуксостата в периоды 1-5. Концентрации фебуксостата в плазме были определены количественно, используя валидированную тандемную жидкостную хроматографию - масс-спектрометрию (LC/MS/MS).
Фармакокинетические результаты
Обобщение средних фармакокинетических параметров, полученных для фебуксостата после приема пяти различных препаратов, содержащих 80 мг фебуксостата, представлено в таблице 16. Профили средних плазменных концентраций фебуксостата во времени (линейные и логарифмически-линейные форматы), после перорального приема таблетки с единичной дозой 80 мг фебуксостата немедленного высвобождения и четырех препаратов в виде капсул с 80 мг фебуксостата пролонгированного высвобождения, представлены на фиг. 8A и 8B. У всех субъектов фебуксостат немедленно обнаруживался в плазме, без какой-либо задержки абсорбции после перорального приема дозы. Концентрации фебуксостата в плазме падали ниже целевой концентрации, равной 100 нг/мл, приблизительно через 16 часов после приема дозы препарата пролонгированного высвобождения (Препараты B-E).

ПРИМЕР 12: Осмотические таблетки фебуксостата
Препарат для осмотической таблетки был приготовлен с использованием технологии ядра, способного к набуханию. Таблетка состоит из слоя лекарственного вещества и слоя полимера, способного к набуханию. Эта двухслойная таблетка покрыта полупроницаемой мембраной, включающей ацетат целлюлозы и полиэтиленгликоль. Отверстие на верхней поверхности таблетки было проделано при помощи лазера. Полупроницаемая мембрана позволяет воде абсорбироваться в таблетку, однако не позволяет диффундировать какому-либо другому материалу через мембрану. Слой способного к набуханию полимера набухает по мере поглощения воды и выдавливает лекарственное вещество через отверстия, проделанные лазером. Состав слоя способного к набуханию полимера и толщина полупроницаемой мембраны влияют на высвобождение лекарственного вещества. Состав таблеток из различных препаратов показан в таблице 17, ниже. Препарат 1 разработан для того, чтобы обеспечить более длительную продолжительность высвобождения, так как он содержит более низкое количество осмогенного NaCl. Более высокое количество осмогена в Препарате 2, как ожидается, будет обеспечивать более быстрое набухание полимера.
Препарат 3 был покрыт полупроницаемым покрытием, состоящим из ацетата целлюлозы (СА) и PEG 3350. Отношение CA:PEG может варьировать. Например, отношение CA:PEG может находиться в диапазоне от 5:5 до 9:1. Отношение CA:PEG, равное 6:4, приводит к более быстрому высвобождению из таблетки, по сравнению с отношением 7:3, как можно видеть на фиг. 9. Количество покрытия может меняться за счет использования обычных способов, известных в уровне техники, для того чтобы получить желаемый профиль высвобождения.
Осмотические таблетки могут быть дополнительно покрыты сверху слоем немедленного высвобождения лекарственного вещества (фебуксостата), для того чтобы преодолеть временную задержку в высвобождении, как показано на фиг. 10. Представленная на фиг. 10 таблетка, содержащая 60 мг фебуксостата (Препарат 2, выше), дополнительно покрыта сверху 20 мг фебуксостата, который, как ожидается, будет немедленно высвобождаться и, таким образом, станет доступным для абсорбции.
Осмотические мультичастицы были получены наслаиванием фебуксостата на сферы микрокристаллической целлюлозы, используя обычные способы, известные в уровне техники. Гранулы со слоями лекарственного вещества были покрыты слоем дезинтегранта (такого как кроскармеллоза натрия, кросповидон и т.д.) и затем были дополнительно покрыты сверху водной дисперсией этилцеллюлозы. Как указано на фиг. 11, можно получить мультичастицы с желательными характеристиками высвобождения, изменяя количество этилцеллюлозного покрытия. Разрываемые под действием осмоса мультичастицы могут быть объединены с гранулами без покрытия для того, чтобы обеспечить системы препарата с двухстадийным высвобождением, подобным другим двухстадийным системам, описанными здесь.
Специалист в данной области техники должен признать, что настоящее описание хорошо адаптировано для получения объектов, имеющих указанные здесь показатели и преимущества. Для специалиста будет очевидным, что изменение заместителей и модификации может быть сделано в рамках представленного раскрытия, не отступая от объема притязаний и сути раскрытия. Все патенты и публикации, упомянутые в описании, являются индикативными для специалиста в данной области техники, на которого ориентировано представленное раскрытие. Все патенты и публикации, включенные здесь в виде ссылки, представлены в той же степени, как и каждая отдельная публикация.
Изобретение, иллюстративно описанное здесь, может быть осуществлено без любого элемента или элементов, ограничения или ограничений, которые не раскрыты здесь в явном виде. Таким образом, например, в каждом случае любой из терминов "включающий", "состоящий по существу из" и "состоящий из" может быть заменен любым другим из двух терминов. Термины и выражения, которые использовались, являются описательными терминами, а не терминами ограничения, и нет никаких предпосылок для использовании таких терминов и выражений с целью исключения каких-либо показанных и описанных эквивалентов признаков или их частей, признавая при этом, что в рамках заявленного раскрытия возможны различные модификации. Таким образом, должно быть понято, что хотя настоящее изобретение было в явном виде раскрыто предпочтительными воплощениями и с необязательными признаками, то специалисту в данной области техники должно быть понятно, что модификации и изменения изобретения, изложенного здесь, могут быть выполнены в рамках этого раскрытия и должны рассматриваться в рамках раскрытия и объема притязаний, определяемого прилагаемой формулой изобретения.
Кроме того, когда признаки или аспекты раскрытия описаны через группы Маркуша, то специалисты в данной области техники должны признать, что раскрытие также описывает признаки любого индивидуального члена или любой подгруппы членов этой группы Маркуша. Например, если X описан как выбранный из группы, состоящей из брома, хлора и иода, то считается, что описана формула для X, который является бромом, и также описана формула, где X является бромом и хлором.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ОТ HELICOBACTER PYLORI | 2014 |
|
RU2671400C2 |
| ЛЕЧЕНИЕ ЭССЕНЦИАЛЬНОГО ТРЕМОРА С ПОМОЩЬЮ (R)-2-(4-ИЗОПРОПИЛФЕНИЛ)-N-(1-(5-(2,2,2-ТРИФТОРЭТОКСИ)ПИРИДИН-2-ИЛ)ЭТИЛ)АЦЕТАМИДА | 2019 |
|
RU2780318C1 |
| СОСТАВЫ L-ОРНИТИН ФЕНИЛАЦЕТАТА | 2016 |
|
RU2755904C2 |
| ОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ТОФАЦИТИНИБА С НЕПРЕРЫВНЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2674345C2 |
| СОСТАВ С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ СНИЖЕНИЯ ЧАСТОТЫ МОЧЕИСПУСКАНИЯ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2013 |
|
RU2599017C2 |
| ОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ТОФАЦИТИНИБА С НЕПРЕРЫВНЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2790166C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ИНГИБИТОРА ГИСТОНДИАЦЕТИЛАЗЫ В КОМБИНАЦИИ С БЕНДАМУТИНОМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609833C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ С ОТСРОЧЕННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩИЕ ВАЛЬПРОЕВУЮ КИСЛОТУ, И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2760304C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НАРУШЕНИЙ СО СТОРОНЫ ОРГАНА ЗРЕНИЯ | 2018 |
|
RU2761436C1 |
| Применение композиции для негормональной контрацепции и упаковка | 2019 |
|
RU2799756C2 |

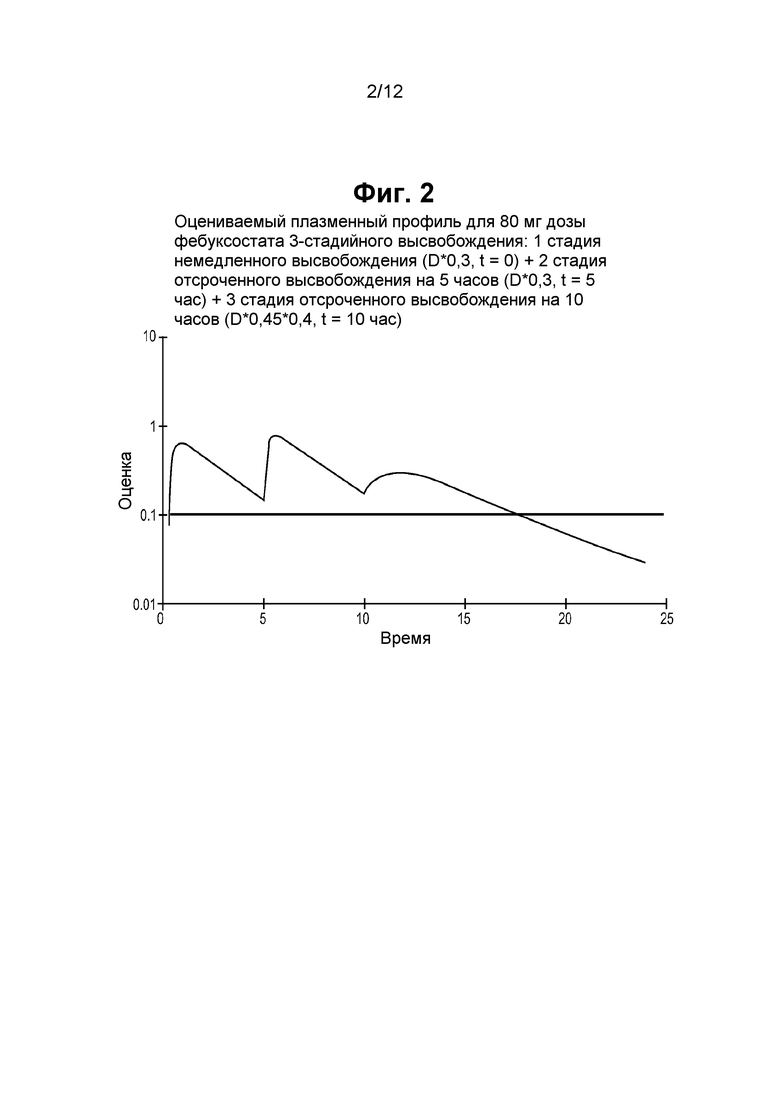
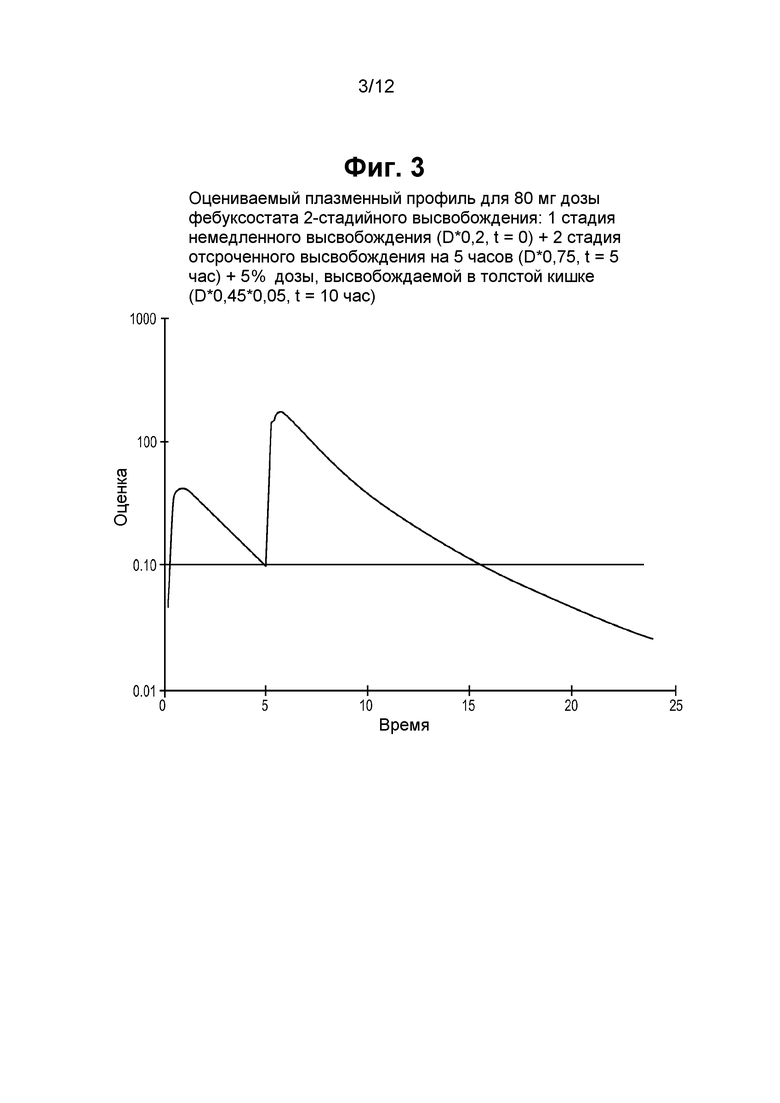

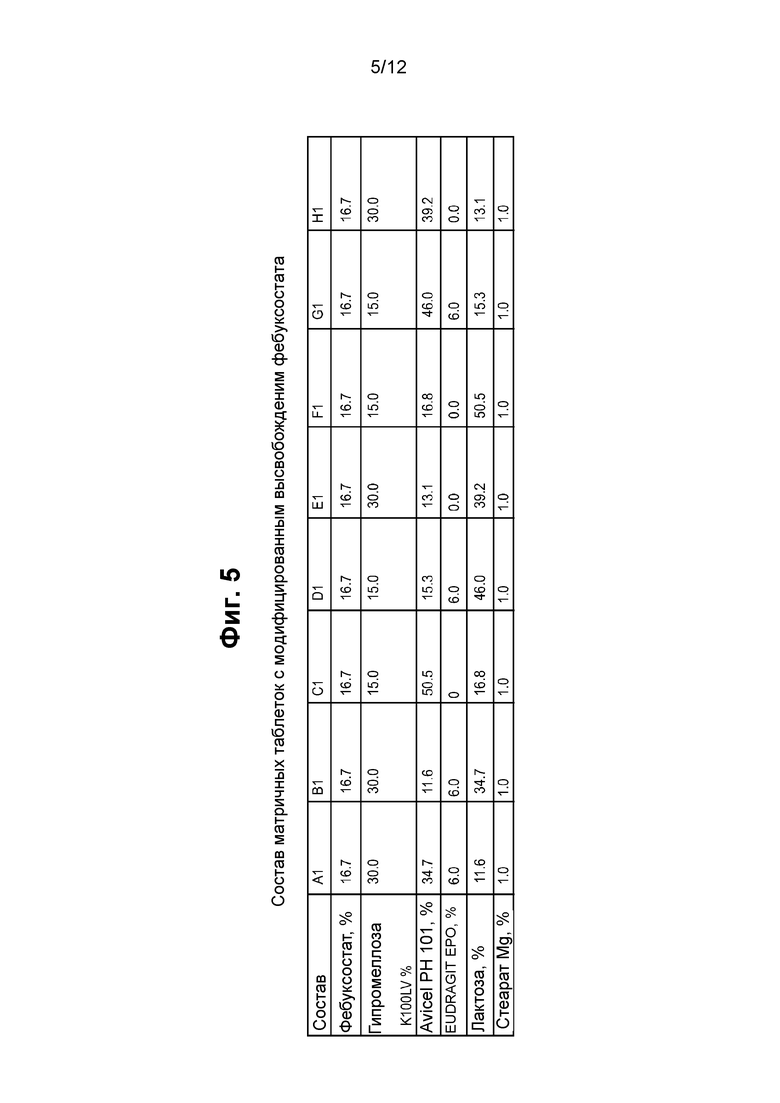
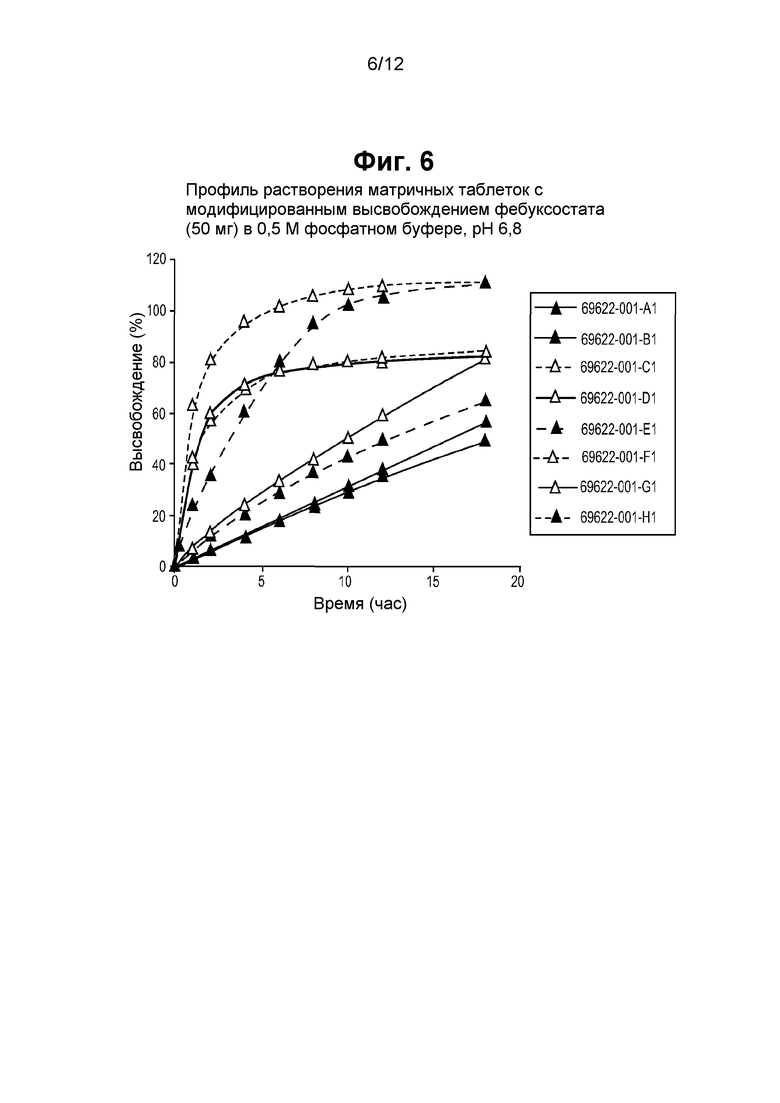

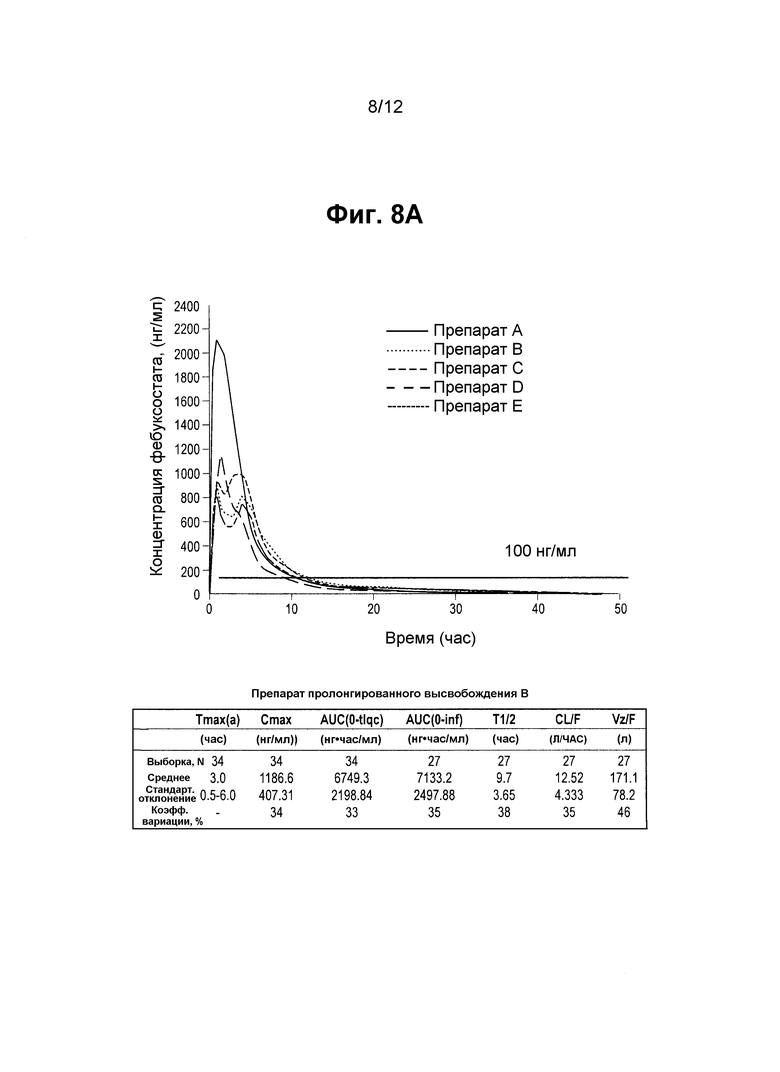
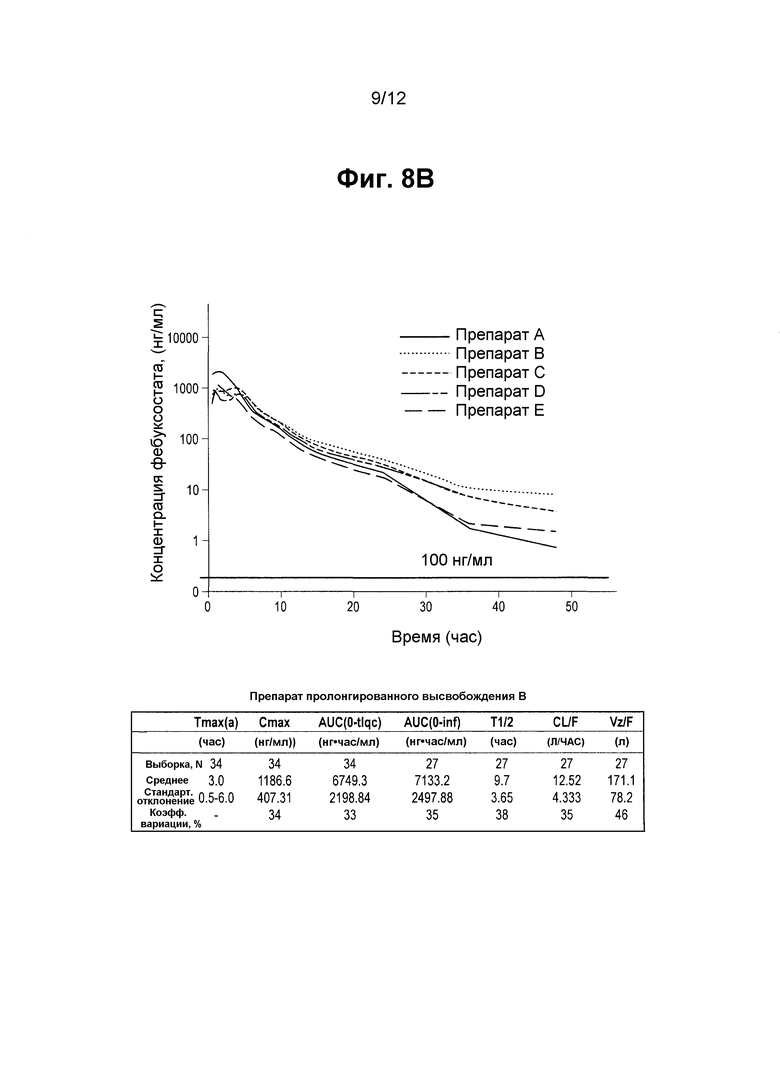
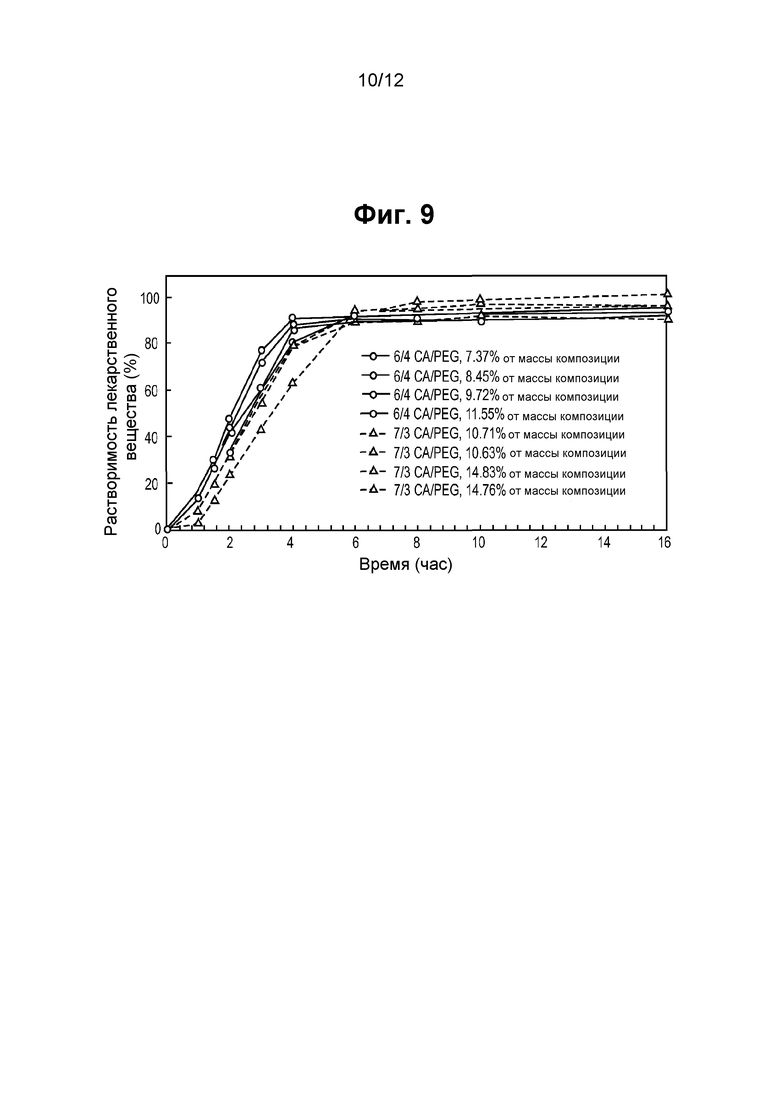
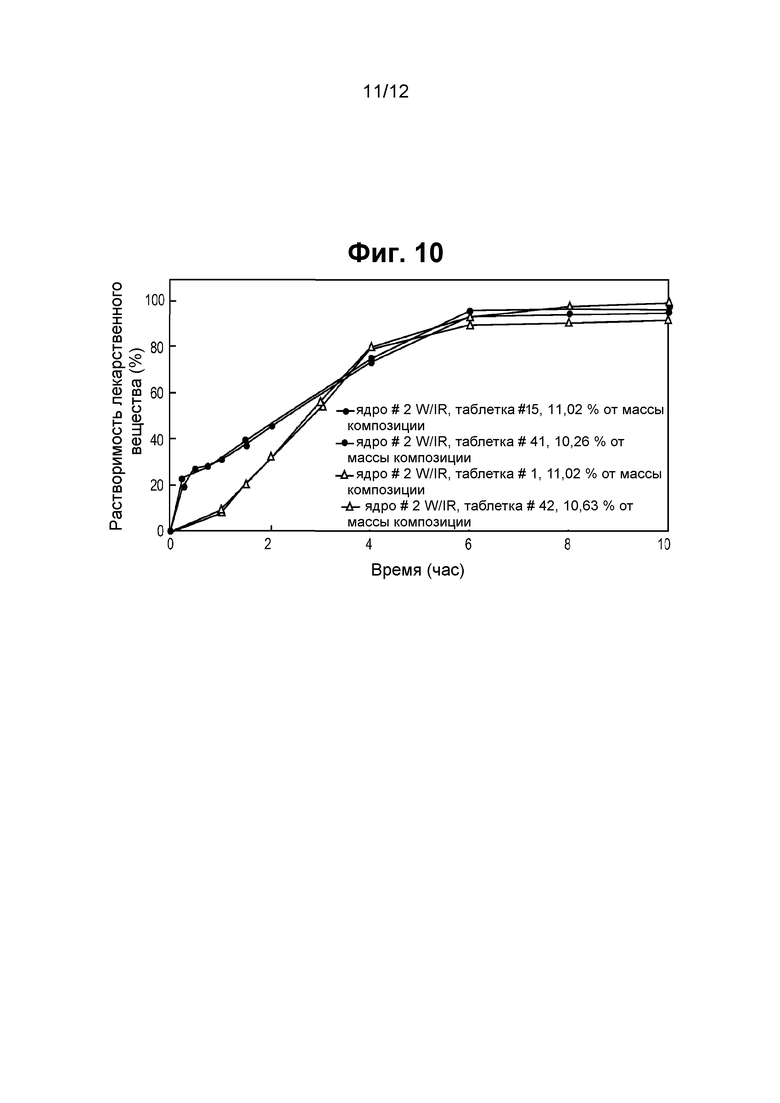

Группа изобретений относится к фармацевтике. Описано 3 варианта фармацевтических композиций с модифицированным высвобождением, включающие гранулы фебуксостата немедленного высвобождения, а также гранулы фебуксостата отсроченного, или отсроченного/контролируемого, или контролируемого высвобождения в различных соотношениях. Описаны также 2 варианта дозированной дозы, включающие указанные фармацевтические композиции, и способ лечения пациента от подагры или гиперурекемии. Группа изобретений обеспечивает высокие уровни ингибирования ксантиноксидазы при концентрации активного вещества более 0,1 мкг/мл в период от 5 до 24 часов. 6 н. и 18 з.п. ф-лы, 12 ил., 17 табл., 12 пр.
1. Фармацевтическая композиция с модифицированным высвобождением, включающая гранулы фебуксостата немедленного высвобождения в количестве в пределах от приблизительно 20% до приблизительно 40% (масс./масс.) от общей массы композиции и гранулы фебуксостата отсроченного высвобождения, растворимые при значениях рН, больших или равных 6,8, в количестве в пределах от приблизительно 60% до приблизительно 80% (масс./масс.) от общей массы композиции,
где указанные гранулы немедленного высвобождения включают
(a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от общей массы гранулы немедленного высвобождения, и
(b) слой немедленного высвобождения, который инкапсулирует инертное ядро, содержащий смесь фебуксостата и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от общей массы гранулы немедленного высвобождения, где отношение фебуксостата к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 3; и
где указанные гранулы отсроченного высвобождения включают
(a) инертное ядро в количестве в пределах от приблизительно 40,5% до приблизительно 43% (масс./масс.) от общей массы гранулы отсроченного высвобождения,
(b) слой немедленного высвобождения, который инкапсулирует инертное ядро, содержащий смесь фебуксостата и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 35% до приблизительно 40% от общей массы гранулы отсроченного высвобождения, где отношение фебуксостата к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 3,
(c) слой энтеросолюбильного полимера отсроченного высвобождения, инкапсулирующий слой немедленного высвобождения, включающий энтеросолюбильный полимер отсроченного высвобождения в количестве в пределах от приблизительно 1% до приблизительно 20% (масс./масс.) от массы гранулы отсроченного высвобождения, где указанный энтеросолюбильный полимер отсроченного высвобождения включает смесь сополимера метакриловой кислоты типа А и сополимера метакриловой кислоты типа В в соотношении в пределах от приблизительно 0,1 до приблизительно 0,5, и
(d) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где указанный пластификатор включает триэтилцитрат.
2. Фармацевтическая композиция с модифицированным высвобождением по п. 1, где общее количество фебуксостата, содержащегося в композиции, составляет 80 мг.
3. Фармацевтическая композиция с модифицированным высвобождением по п. 1, где указанные гранулы включены в пероральную дозированную форму, включающую пилюли, таблетки и капсулы.
4. Фармацевтическая композиция с модифицированным высвобождением, включающая гранулы фебуксостата немедленного высвобождения в количестве в пределах от приблизительно 20% до приблизительно 40% (масс./масс.) от общей массы композиции, и гранулы фебуксостата отсроченного/контролируемого высвобождения, растворимые при значениях рН, больших или равных 6,8, и обеспечивающие пролонгированное высвобождение фебуксостата в период через от приблизительно 4 часов до приблизительно 6 часов, в количестве в пределах от приблизительно 60% до приблизительно 80% (масс./масс.) от общей массы композиции,
где указанные гранулы немедленного высвобождения включают
(a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от общей массы гранулы немедленного высвобождения, и
(b) слой немедленного высвобождения, который инкапсулирует инертное ядро, содержащий смесь фебуксостата и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от общей массы гранулы немедленного высвобождения, где отношение фебуксостата к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 3; и
где гранулы отсроченного/контролируемого высвобождения включают
(a) инертное ядро в количестве в пределах от приблизительно 34% до приблизительно 37% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения,
(b) слой немедленного высвобождения, который инкапсулирует инертное ядро, включающий смесь фебуксостата и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 31% до приблизительно 34% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где отношение фебуксостата к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 2,5,
(c) слой контролируемого высвобождения, инкапсулирующий слой немедленного высвобождения, включающий полимер контролируемого высвобождения в количестве в пределах от приблизительно 10% до приблизительно 14% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где указанный полимер контролируемого высвобождения включает смесь водной дисперсии этилцеллюлозы и гидроксипропилметилцеллюлозы, при отношении водной дисперсии этилцеллюлозы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1,5 до приблизительно 3,
(d) слой отсроченного высвобождения рН 6,8, инкапсулирующий слой контролируемого высвобождения, включающий полимер отсроченного высвобождения рН 6,8 в количестве в пределах от приблизительно 1% до приблизительно 20% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где полимер отсроченного высвобождения рН 6,8 включает смесь сополимера метакриловой кислоты типа А и сополимера метакриловой кислоты типа В в соотношении в пределах от приблизительно 0,1 до приблизительно 0,5, и
(e) пластификатор в количестве в пределах от приблизительно 1% до приблизительно 3% (масс./масс.) от массы гранулы отсроченного/контролируемого высвобождения, где указанный пластификатор включает триэтилцитрат.
5. Фармацевтическая композиция с модифицированным высвобождением по п. 4, где общее количество фебуксостата, содержащегося в композиции, составляет 80 мг.
6. Фармацевтическая композиция с модифицированным высвобождением по п. 4, где указанные гранулы включены в пероральную дозированную форму, включающую пилюли, таблетки и капсулы.
7. Фармацевтическая композиция с модифицированным высвобождением, включающая гранулы фебуксостата немедленного высвобождения в количестве в пределах от приблизительно 10% до приблизительно 30% (масс./масс.) от общей массы композиции, и гранулы фебуксостата контролируемого высвобождения, обеспечивающие пролонгированное высвобождение фебуксостата в период через от приблизительно 10 часов до приблизительно 12 часов, в количестве в пределах от приблизительно 70% до приблизительно 90% (масс./масс.) от общей массы композиции,
где указанные гранулы немедленного высвобождения включают
(a) инертное ядро в количестве в пределах от приблизительно 50% до приблизительно 55% (масс./масс.) от общей массы гранулы немедленного высвобождения, и
(b) слой немедленного высвобождения, который инкапсулирует инертное ядро, содержащий смесь фебуксостата и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 45% до приблизительно 50% (масс./масс.) от общей массы гранулы немедленного высвобождения, где отношение фебуксостата к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 3;
где гранулы контролируемого высвобождения включают
(a) инертное ядро в количестве в пределах от приблизительно 47% до приблизительно 51% (масс./масс.) от массы гранулы контролируемого высвобождения,
(b) слой немедленного высвобождения, который инкапсулирует инертное ядро, включающий смесь фебуксостата и гидроксипропилметилцеллюлозы в количестве в пределах от приблизительно 42% до приблизительно 48% (масс./масс.) от массы гранулы контролируемого высвобождения, где отношение фебуксостата к гидроксипропилметилцеллюлозе находится в пределах от приблизительно 1,5 до приблизительно 2,5, и
(c) слой контролируемого высвобождения, инкапсулирующий слой немедленного высвобождения, включающий полимер контролируемого высвобождения, где указанный полимер контролируемого высвобождения включает смесь этилцеллюлозы и гидроксипропилметилцеллюлозы, в количестве в пределах от приблизительно 4% до приблизительно 8% (масс./масс.) от массы гранулы контролируемого высвобождения, при отношении этилцеллюлозы к гидроксипропилметилцеллюлозе в пределах от приблизительно 1 до приблизительно 2.
8. Фармацевтическая композиция с модифицированным высвобождением по п. 7, где общее количество фебуксостата, содержащегося в композиции, составляет 80 мг.
9. Фармацевтическая композиция с модифицированным высвобождением по п. 7, где указанные гранулы включены в пероральную дозированную форму, включающую пилюли, таблетки и капсулы.
10. Способ лечения пациента, страдающего от подагры или гиперурикемии и нуждающегося в их лечении, где способ включает стадию введения субъекту, страдающему от подагры или гиперурикемии и нуждающемуся в их лечении, терапевтически эффективного количества фармацевтической композиции с модифицированным высвобождением по пп. 1, 4 или 7.
11. Дозированная форма с модифицированным высвобождением, включающая фармацевтическую композицию с модифицированным высвобождением по любому из пунктов 1, 4 и 7, где указанная дозированная форма, после однократного введения пациенту-человеку, нуждающемуся в лечении, продуцирует у субъекта среднюю максимальную концентрацию (Cmax) 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты или ее фармацевтически приемлемой соли в плазме между приблизительно 0,8 мкг/мл и приблизительно 1,7 мкг/мл.
12. Дозированная форма по п. 11, где дозированная форма обеспечивает 80 мг 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты или ее фармацевтически приемлемой соли.
13. Дозированная форма по п. 11, где дозированная форма показывает среднее значение Cmax 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты между приблизительно 0,8 мкг/мл и приблизительно 1,6 мкг/мл.
14. Дозированная форма по п. 11, где дозированная форма показывает среднее значение Cmax 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты между приблизительно 0,8 мкг/мл и приблизительно 1,5 мкг/мл.
15. Дозированная форма по п. 11, где дозированная форма показывает среднее значение Cmax 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты между приблизительно 1,13 мкг/мл и приблизительно 1,29 мкг/мл.
16. Дозированная форма по п. 11, где дозированная форма показывает среднее значение Cmax 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 1,19 мкг/мл.
17. Дозированная форма по п. 11, где дозированная форма после однократного введения пациенту-человеку, нуждающемуся в лечении, обеспечивает среднее время появления пика концентрации (Tmax) для 2-[3-циано-4-(2-метилпропокси) фенил]-4-метилтиазол-5-карбоновой кислоты у субъекта между приблизительно 1,5 часами и приблизительно 3,0 часами.
18. Дозированная форма по п. 11, где дозированная форма, после однократного введения пациенту-человеку, нуждающемуся в лечении, обеспечивает среднее время появления пика концентрации (Tmax) для 2-[3-циано-4- (2-метилпропокси) фенил]-4-метилтиазол-5-карбоновой кислоты у субъекта равное приблизительно 3,0 часа.
19. Дозированная форма по п. 11, где дозированная форма, после однократного введения пациенту-человеку, нуждающемуся в лечении, продуцирует у субъекта среднюю площадь под кривой (0-inf) (AUC0-inf), составляющую между приблизительно 5334,2 нг·час/мл и приблизительно 7726,7 нг·час/мл.
20. Дозированная форма по п. 11, где дозированная форма после однократного введения пациенту-человеку, нуждающемуся в лечении, продуцирует у субъекта среднюю площадь под кривой (0-inf) (AUC0-inf), составляющую между приблизительно 5334,2 нг·час/мл и приблизительно 7133,2 нг·час/мл.
21. Дозированная форма с модифицированным высвобождением, включающая фармацевтическую композицию с модифицированным высвобождением по любому из пп. 2, 5 и 8, где указанная дозированная форма, после однократного введения пациенту-человеку, нуждающемуся в лечении, продуцирует у субъекта среднюю максимальную концентрацию (Cmax) для 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты или ее фармацевтически приемлемой соли в плазме между приблизительно 0,113 мкг/мл и приблизительно 1,29 мкг/мл, и проявляет по меньшей мере одно из следующего:
(a) средняя площадь под кривой (0-inf) (AUC0-inf) составляет между приблизительно 5334, 2 нг·час/мл и приблизительно 7726,7 нг·час/мл, или
(b) среднее время появления пика концентрации (Tmax) 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты у субъекта между приблизительно 1,5 часа и приблизительно 3,0 часа.
22. Дозированная форма по п. 21, где дозированная форма показывает среднее значение Cmax для 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 1,19 мкг/мл, и среднее время появления пика концентрации (Tmax) 2-[3-циано-4-(2-метилпропокси) фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 3,0 часа.
23. Дозированная форма по п. 21, где дозированная форма показывает среднее значение AUC0-inf 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 7133,2 нг·час/мл.
24. Дозированная форма по п. 21, где дозированная форма показывает среднее значение Cmax для 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 1,19 мкг/мл, и среднее время появления пика концентрации (Tmax) для 2-[3-циано-4-(2-метилпропокси) фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 3,0 часа, и среднее значение AUC0-inf для 2-[3-циано-4-(2-метилпропокси)фенил]-4-метилтиазол-5-карбоновой кислоты, равное приблизительно 7133,2 нг·час/мл.
| Khosravan R., Pharmacokinetics, Pharmacodynamics and Safety of Febuxostat, a Non-Purine Selective Inhibitor of Xanthine Oxidase, in a Dose Escalation Study in Healthy Subjects, Clin | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| CN 101658505 A, 03.03.2010 | |||
| CN 101716132 A, 02.06.2010 | |||
| СПОСОБ ЛЕЧЕНИЯ ПОДАГРЫ С ПОМОЩЬЮ ПЛАЗМОСОРБЦИИ | 2001 |
|
RU2219957C2 |

