Область техники, к которой относится изобретение
Настоящее изобретение относится к C-гликозидным производным, содержащим конденсированное фенильное кольцо, или их фармацевтически приемлемым солям, способу получения таковых, фармацевтической композиции, содержащей таковые, их применению, а также способу двойного ингибирования натрий-глюкозного котранспортера 1-го типа (SGLT1) и натрий-глюкозного котранспортера 2-го типа (SGLT2) с применением таковых.
Предпосылки создания изобретения
Диабет представляет собой заболевание, при котором развиваются осложнения, такие как аномалии периферических нервов и вегетативных нервов, симптомы заболевания глаз, стоп и почек, сосудистые заболевания или тому подобное, вследствие повышения уровня сахара в крови, обусловленного снижением секреции и функций инсулина.
Известно, что диабет обычно делят на два типа: I-го типа и II-го типа. I-й тип зачастую возникает у детей, главным образом вследствие врожденных факторов, и таким пациентам необходимо обеспечивать инъекции инсулина на протяжении всей жизни из-за недостаточности секреции инсулина поджелудочной железой, а также поддерживать уровень сахара в крови на соответствующем уровне посредством диетотерапии и периодических проверок. II-й тип главным образом возникает у взрослых в состоянии, когда секреция инсулина снижается или резистентность к инсулину повышается до уровня, достаточного для ограничения восприимчивости клеток к инсулину, вследствие образа жизни, предусматривающего, например, особенности питания, отсутствие физических упражнений, ожирение и т. д., а также факторов окружающей среды, при этом данный тип заболевания составляет от 90 до 95% случаев из 285 миллионов пациентов с диабетом по всему миру. Пациенты с диабетом II-го типа могут контролировать уровень сахара в крови посредством снижения веса, здорового питания и физических упражнений, но их симптомы ухудшаются вследствие особенностей данного прогрессирующего заболевания. Таким образом, у пациентов нет выбора, кроме как выполнять инъекцию инсулина, а основными симптомами являются полиурия, жажда, заторможенность, булимия, потеря веса и т. д., обусловленные высоким уровнем сахара в крови.
В качестве лекарственного средства, предназначенного для лечения диабета, обычно применяют инсулин и пероральное гипогликемическое средство. При диабете I-го типа применяют инъекцию инсулина, при этом при диабете II-го типа применяют пероральное гипогликемическое средство отдельно или в комбинации с инсулином. В качестве перорального гипогликемического средства в настоящее время применяют лекарственные средства на основе сульфонилмочевины и меглитинида для стимуляции секреции инсулина, лекарственные средства на основе бигуанида (метформина) и тиазолидиндиона (PPAR-γ) для улучшения восприимчивости к инсулину, лекарственное средство на основе ингибитора α-глюкозидазы для ингибирования усвоения углеводов, ингибитор DPP-4, который представляет собой препарат на основе инкретина, ингибитор SGLT2 для предотвращения реабсорбции глюкозы и т. д. Несмотря на назначение такого перорального гипогликемического средства, для многих пациентов снижение уровня гликозилированного гемоглобина до целевого уровня или меньше является затруднительным. В то же время в исследовании пациентов с диабетом в отношении регулирования факторов сосудистого риска только 37% данных участников были способны достичь уровня менее 7,0% гликозилированного гемоглобина (Saydah, S.H. et. al., J. Am. Med. Assoc. 2004, 291, 335-342). Кроме того, существующие пероральные гипогликемические средства проявляют побочные эффекты, такие как желудочно-кишечные проблемы, гипогликемия, увеличение веса, молочнокислый ацидоз, отек, кардиотоксичность и гепатотоксичность, наряду с ограниченной продолжительностью лечебных эффектов. Таким образом, по-прежнему остается медицинская потребность в области пероральных гипогликемических средств, где крайне необходимо разработать быстродействующее терапевтическое средство с новым механизмом действия, которое характеризуется превосходной эффективностью и продолжительностью лечебных эффектов, безопасностью и хорошей переносимостью лекарственного средства, которое, в частности, не вызывает гипогликемию. Следовательно, большое внимание было уделено разработке ингибиторов SGLT2 в виде перорального препарата с новым механизмом действия, который не относится к инсулину, но обладает соответствующей эффективностью и в то же время является способным к снижению веса.
Натрий-глюкозный котранспортер (SGLT), который представляет собой транспортер, выполняющий функцию всасывания глюкозы в нашем теле, делится на 6 подтипов и экспрессируется в нескольких частях нашего тела, при этом SGLT1 преимущественно экспрессируется в кишечнике и почках, тогда как SGLT2 преимущественно экспрессируется в почках. Кроме того, SGLT1 характеризуется высоким сродством к глюкозе, но характеризуется низкой транспортной способностью, тогда как SGLT2 характеризуется низким сродством к глюкозе, но при этом характеризуется высокой транспортной способностью. У здоровых людей реабсорбции подвергается 99% глюкозы, отфильтрованной клубочками почек, тогда как с мочой выводится лишь ее 1% или менее, при этом такая глюкоза реабсорбируется в соотношении 90% и 10% с помощью соответственно SGLT2 и SGLT1. Тем не менее, пациенты с диабетом II-го типа характеризуются высокой степенью экспрессии SGLT1 и SGLT2, таким образом, характеризуются повышением абсорбции глюкозы с помощью SGLT1 в кишечнике и реабсорбции глюкозы с помощью SGLT1/2 в почках, что обуславливает предпосылки повышения уровня сахара в крови. Таким образом, выполнили разработку гипогликемических средств с новым механизмом действия, при этом уровень сахара в крови нормализуется за счет ингибирования SGLT1/2 таким образом, чтобы восстановить секрецию инсулина поджелудочной железы и улучшить резистентность к инсулину в мышцах и печени.
Флоридзин экстрагируют из коры яблони, и он представляет собой вещество, прежде всего оцениваемое в качестве ингибитора SGLT, причем оно характеризуется противодиабетической эффективностью, но обладает низкой пероральной абсорбционной способностью и метаболизируется в кишечнике с обуславливанием желудочно-кишечных проблем или диареи, таким образом, он еще не был разработан в качестве лекарственного средства. Кроме того, в Tanabe Seiyaku разрабатывали препарат T-1095 в 1990-х годах в качестве перорально абсорбируемого лекарственного средства, действующего на SGLT2, но его разработку остановили в фазе II клинических испытаний, и разработку серглифлозина или ремоглифлозина, которые представляли собой O-глюкозид с аналогичной с ним структурой, остановили в фазе II клинических испытаний. Начали разработку C-глюкозидного лекарственного средства во избежание метаболизма посредством β-глюкозидазы, которая была слабым местом O-глюкозидного лекарственного средства. Поскольку компания Bristol-Myers Squibb начала клинический тест дапаглифлозина в 2004 году, многие фармацевтические компании начали разрабатывать лекарственное средство из этой серии. Затем было получено первое разрешение на продажу такого дапаглифлозина в Европе в 2012 году, после чего было получено первое разрешение на продажу канаглифлозина (Johnson & Johnson, Mitsubishi Tanabe) в Соединенных Штатах в 2013 году, а затем это было сделано для дапаглифлозина и эмпаглифлозина (Boehringer-Ingelheim) в США, при этом для ипраглифлозина (Astellas), лузеоглифлозина (Taisho) и тофоглифлозина (Chugai) это было сделано соответственно в Японии. В то же время известно, что SGLT1 играет важную роль в абсорбции глюкозы и галактозы в тонком кишечнике, а также в реабсорбции глюкозы в почках (Levin, R. J., Am. J. Clin. Nutr. 1994, 59(3), 690S-698S). Соответственно, считается, что абсорбцию глюкозы можно ингибировать в тонком кишечнике, а реабсорбцию глюкозы можно ингибировать в почках посредством ингибирования SGLT1 с проявлением, таким образом, эффективности в отношении контроля сахара в крови. Таким образом, двойной ингибитор SGLT1/2 может стать новым механизмом лечения диабета, причем сотаглифлозин, двойной ингибитор SGLT1/2, в настоящее время находится в фазе III клинических испытаний для диабета I-го типа и в подготовке к фазе III клинических испытаний для диабета II-го типа, при этом LIK-066, двойной ингибитор SGLT1/2 от Novartis, также в настоящее время находится в фазе II клинических испытаний.
Описание изобретения
Техническая задача
Целью настоящего изобретения является получение нового соединения или его фармацевтически приемлемых солей, проявляющих двойную ингибирующую активность в отношении SGLT1/2.
Другой целью настоящего изобретения является обеспечение способа получения таковых.
Другой целью настоящего изобретения является получение фармацевтической композиции для предупреждения или лечения заболевания, связанного с SGLT1/2, содержащей соединение по настоящему изобретению или его фармацевтически приемлемые соли в качестве эффективного компонента.
Еще одна цель настоящего изобретения предусматривает их применение для получения лекарственного средства, предназначенного для предупреждения или лечения заболевания, связанного с SGLT1/2.
Еще одной целью настоящего изобретения является обеспечение способа для предупреждения или лечения заболевания, связанного с SGLT1/2, включающего введение терапевтически эффективной дозы фармацевтической композиции по настоящему изобретению.
Решение технической задачи
Для достижения вышеуказанных целей авторы настоящего изобретения приложили усилия и определили, что C-гликозидные производные, содержащие новое синтезированное конденсированное фенильное кольцо, проявляют двойную ингибирующую активность в отношении SGLT1/2, таким образом осуществляя настоящее изобретение.
C-гликозидное производное соединение, содержащее конденсированное фенильное кольцо
В настоящем изобретении предусмотрены соединение, представленное следующей формулой 1, или его фармацевтически приемлемые соли:
[Формула 1]
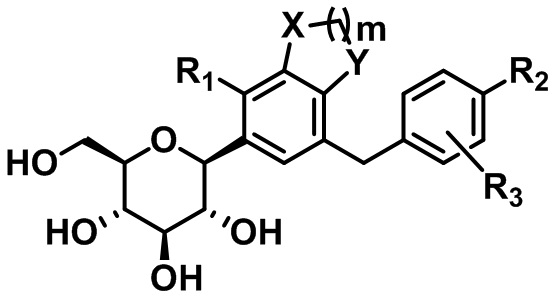 ,
,
где
каждый из X и Y независимо представляет собой -CH2-, -CH(CH3)-, -C(CH3)2-, -C(=O)-, -O-, -S- или -NH-;
m представляет собой целое число от 1 до 3;
каждый из R1 - R3 независимо представляет собой водород, галоген, C1-C4алкил, C2-C4алкенил, C2-C4алкинил, C3-C7циклоалкил, -C(=O)R4, циано, гидрокси, C1-C4алкокси, -OCF3, -SR5, -S(=O)R6, -S(=O)2R7, нитро, -NR8R9, арил, гетероарил или гетероциклил (где каждый из по меньшей мере одного атома водорода в C1-C4алкиле, C2-C4алкениле, C2-C4алкиниле и C3-C7циклоалкиле может быть независимо не замещен или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, гидрокси, циано, нитро и амино, и каждый из по меньшей мере одного атома водорода в ариле, гетероариле и гетероциклиле может быть независимо не замещен или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, C1-C4алкила, гидрокси, C1-C4алкокси, циано, нитро и амино);
R4 представляет собой гидрокси, C1-C4алкокси, амино, моно- или ди-(C1-C4алкил)амино;
R5 представляет собой водород или C1-C4алкил;
каждый из R6 и R7 независимо представляет собой C1-C4алкил или арил (где арил может быть не замещен или замещен C1-C4алкилом);
каждый из R8 и R9 независимо представляет собой водород, C1-C4алкил, -C(=O)R10 или -S(=O)2R11;
R10 представляет собой C1-C4алкил; и
R11 представляет собой C1-C4алкил или арил; (где арил может быть не замещен или замещен C1-C4алкилом);
где, если m равняется 1, R1 представляет собой галоген, C1-C4алкил, C2-C4алкенил, C2-C4алкинил, C3-C7циклоалкил, -C(=O)R4, циано, гидрокси, C1-C4алкокси, -OCF3, -SR5, -S(=O)R6, -S(=O)2R7, нитро, -NR8R9, арил, гетероарил или гетероциклил.
В соответствии с одним вариантом осуществления настоящего изобретения:
каждый из X и Y независимо представляет собой -CH2- или -O-;
m равняется 1 или 2;
каждый из R1 - R3 независимо представляет собой водород, галоген, C1-C4алкил, C2-C4алкенил, C3-C7циклоалкил, гидрокси, C1-C4алкокси, -OCF3, -SR5 или арил (где каждый из по меньшей мере одного атома водорода в C1-C4алкиле, C2-C4алкениле и C3-C7циклоалкиле может быть независимо не замещен или замещен галогеном или гидрокси, и каждый атом водорода в ариле может быть независимо не замещен или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, C1-C4алкила, гидрокси и C1-C4алкокси); и
R5 представляет собой C1-C4алкил.
В соответствии с другим вариантом осуществления настоящего изобретения:
каждый из X и Y независимо представляет собой -CH2- или -O-;
m равняется 1 или 2;
R1 представляет собой водород, галоген, C1-C4алкил, C3-C7циклоалкил или C1-C4алкокси (где каждый из по меньшей мере одного атома водорода в C1-C4алкиле может быть независимо не замещен или замещен галогеном);
каждый из R2 и R3 независимо представляет собой водород, галоген, C1-C4алкил, C2-C4алкенил, C3-C7циклоалкил, C1-C4алкокси, -OCF3, -SR5 или арил (где каждый из по меньшей мере одного атома водорода в C1-C4алкиле, C2-C4алкениле и C3-C7циклоалкиле может быть независимо не замещен или замещен галогеном, и каждый из по меньшей мере одного атома водорода в ариле может быть независимо не замещен или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, C1-C4алкила и C1-C4алкокси); и
R5 представляет собой C1-C4алкил;
В настоящем изобретении в качестве примера галогена представлен фтор, хлор, бром или йод.
В настоящем изобретении алкил может относиться к одновалентному углеводороду с линейными или разветвленными цепями, при этом пример алкила может предусматривать метил, этил, н-пропил, изопропил и бутил.
В настоящем изобретении циклоалкил может предусматривать циклопропил, циклобутил, циклопентил, 3-метилциклопентил, 2,3-диметилциклопентил, циклогексил, 3-метилциклогексил, 4-метилциклогексил, 2,3-диметилциклогексил, 3,4,5-триметилциклогексил, 4-трет-бутилциклогексил и циклогептил.
В настоящем изобретении алкокси может относиться к линейной цепи, разветвленной цепи или кольцеобразной цепи и может предусматривать метокси, этокси, н-пропокси, изопропокси, изопропилокси, н-бутокси, изобутокси, трет-бутокси и втор-бутокси.
В настоящем изобретении алкенил может относиться к одновалентному углеводороду с линейными или разветвленными цепями, при этом пример алкенильной группы может предусматривать винил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил и 3-бутенил.
В настоящем изобретении арил может относиться к моноциклическому или полициклическому арилу, при этом моноциклический арил может предусматривать фенил, бифенил и терфенил, и полициклический арил может предусматривать нафтил, антрацен, флуорен, пиренил и т. д.
В настоящем изобретении гетероарил может относиться к гетероарилу, содержащему в ариле по меньшей мере один гетероатом, отличный от углерода.
В настоящем изобретении гетероциклил может относиться к гетероциклилу, содержащему в циклоалкиле по меньшей мере один гетероатом, отличный от углерода.
В настоящем изобретении гетероатом может предусматривать O, S и N, а не углерод.
В соответствии с одним вариантом осуществления настоящего изобретения X представляет собой -CH2-.
В соответствии с другим вариантом осуществления X представляет собой -O-.
В соответствии с одним вариантом осуществления настоящего изобретения Y представляет собой -CH2-.
В соответствии с другим вариантом осуществления Y представляет собой -O-.
В соответствии с одним вариантом осуществления настоящего изобретения X и Y представляют собой -CH2-.
В соответствии с другим вариантом осуществления X и Y представляют собой -O-.
В соответствии с одним вариантом осуществления настоящего изобретения один из X и Y представляет собой -CH2-, а другой представляет собой -O-.
В соответствии с одним вариантом осуществления настоящего изобретения R1 представляет собой водород.
В соответствии с другим вариантом осуществления настоящего изобретения R1 представляет собой галоген. В частности, R1 может представлять собой хлор.
В соответствии с другим вариантом осуществления настоящего изобретения R1 представляет собой C1-C4алкокси. В частности, указанный R1 представляет собой метоксигруппу.
В соответствии с другим вариантом осуществления настоящего изобретения R1 представляет собой C1-C4алкил. C1-C4алкил может представлять собой метил, этил, пропил, бутил, изобутил или изопропил.
В соответствии с другим вариантом осуществления настоящего изобретения R1 представляет собой C3-C7циклоалкил. C3-C7циклоалкил может представлять собой циклопентил.
В соответствии с одним вариантом осуществления настоящего изобретения R4 представляет собой водород.
В соответствии с одним вариантом осуществления настоящего изобретения R2 представляет собой C1-C4алкил. C1-C4алкил может представлять собой метил, этил, пропил, бутил, изобутил или изопропил. Каждый из по меньшей мере одного атома водорода в C1-C4алкиле может быть независимо не замещен или замещен галогеном, в частности фтором.
В соответствии с другим вариантом осуществления настоящего изобретения R2 представляет собой C1-C4алкенил. C2-C4алкенил может представлять собой, в частности, винил.
В соответствии с другим вариантом осуществления настоящего изобретения R2 представляет собой C1-C4алкокси. C1-C4алкокси может представлять собой метокси, этокси или изопропокси.
В соответствии с другим вариантом осуществления настоящего изобретения R2 представляет собой -OCF3.
В соответствии с другим вариантом осуществления настоящего изобретения R2 может представлять собой галоген. В частности, R2 может представлять собой фтор или хлор.
В соответствии с другим вариантом осуществления настоящего изобретения R2 представляет собой -SR5, и R5 представляет собой C1-C4алкил.
В соответствии с другим вариантом осуществления настоящего изобретения R2 представляет собой арил. В частности, арил может представлять собой фенил.
В соответствии с одним вариантом осуществления настоящего изобретения R3 представляет собой водород.
В соответствии с другим вариантом осуществления настоящего изобретения R3 представляет собой C1-C4алкокси. В частности, C1-C4алкокси может представлять собой метокси, этокси или изопропокси.
В соответствии с другим вариантом осуществления настоящего изобретения R3 представляет собой C1-C4алкил. В частности, C1-C4алкил может представлять собой метил, этил, пропил, бутил, изобутил или изопропил.
В соответствии с одним вариантом осуществления настоящего изобретения R3 может быть замещен в 3-ем положении бензольного кольца формулы i).
В соответствии с аспектом другого варианта осуществления настоящего изобретения, R3 может быть замещен во 2-ом положении бензольного кольца формулы i).
В соответствии с аспектом предпочтительного варианта осуществления настоящего изобретения соединение, представленное вышеприведенной формулой 1, может быть выбрано из группы, состоящей из следующих соединений:
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-изопропоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-пропилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-изопропилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-винилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-трифторметил)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-трифторметокси)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(3,4-диметоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(2,4-диметоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-метилтио)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-фторбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-фтор-3-метилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-хлорбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(8-(4-этоксибензил)-2,3-дигидробензо[b][1,4]диоксин-6-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(8-(4-этилбензил)-2,3-дигидробензо[b][1,4]диоксин-6-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)бензо[d][1,3]диоксол-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-(метилтио)бензил)бензо[d][1,3]диоксол-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)бензо[d][1,3]диоксол-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-(4-этоксибензил)-5,6,7,8-тетрагидронафтален-2-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-(4-этилбензил)-5,6,7,8-тетрагидронафтален-2-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-(4-метоксибензил)-1-метил-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-метилбензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-трифторметил)бензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-трифторметокси)бензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-(метилтио)бензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-(4-хлорбензил)-1-метил-5,6,7,8-тетрагидронафтален-2-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-метил-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-(метилтио)бензил)-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-метил-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-винилбензил)-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-хлор-7-(4-этоксибензил)-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-(4-метоксибензил)-7-метил-2,3-дигидробензофуран-6-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-метил-4-(4-винилбензил)-2,3-дигидробензофуран-6-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(8-метокси-5-(4-метоксибензил)хроман-7-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(8-метокси-5-(4-метилбензил)хроман-7-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(5-(4-этоксибензил)-8-метилхроман-7-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-этилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-фторбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-хлорбензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-трифторметокси)бензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-трифторметил)бензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-изопропоксибензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-изопропилбензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(бифенил-3-илметил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метилбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-фторбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-этоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-этилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изопропил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изопропил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-циклопентил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изобутил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-изобутил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
Соединение формулы 1 согласно настоящему изобретению может быть представлено в форме фармацевтически приемлемой соли. Соль присоединения кислоты, образованная с помощью фармацевтически приемлемой свободной кислоты, является пригодной в качестве соли. В настоящем изобретении термин «фармацевтически приемлемая соль» означает любую и все соли присоединения органических или неорганических кислот указанного соединения, при этом побочный эффект, обусловленный такими солями, не снижает предпочтительную эффективность соединения, представленного формулой 1, при их концентрации, характеризующейся относительно нетоксичным и безопасным эффективным действием на пациентов.
Соли присоединения кислот получают посредством общепринятых способов, например, таким образом, при котором соединение растворяют в избыточном количестве водного раствора кислоты, а затем полученные соли осаждают с помощью смешиваемого с водой органического растворителя, например, метанола, этанола, ацетона или ацетонитрила. Одинаковые молярные количества соединения и кислоты в воде или спирте (например, простом монометиловом эфире гликоля) можно нагревать, а затем указанную смесь можно выпаривать и высушивать или осажденные соли можно фильтровать с отсасыванием.
При этом органическую кислоту и неорганическую кислоту можно применять в виде свободной кислоты, причем хлористоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, винную кислоту или т. п. можно применять в качестве неорганической кислоты, тогда как в качестве органической кислоты можно применять следующие кислоты: метансульфоновую кислоту, п-толуолсульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, малеиновую кислоту, янтарную кислоту, щавелевую кислоту, бензойную кислоту, винную кислоту, фумаровую кислоту, миндальную кислоту, пропионовую кислоту, лимонную кислоту, молочную кислоту, гликолевую кислоту, глюконовую кислоту, галактуроновую кислоту, глутаминовую кислоту, глутаровую кислоту, глюкуроновую кислоту, аспарагиновую кислоту, аскорбиновую кислоту, угольную кислоту, ванилиновую кислоту, йодистоводородную кислоту или т. п. без ограничения ними.
Кроме того, фармацевтически приемлемые соли металлов могут быть получены с помощью основания. Соль щелочного металла или соль щелочноземельного металла получают, например, таким образом, при котором соединение растворяют в избыточном количестве раствора гидроксида щелочного металла или раствора гидроксида щелочноземельного металла, а затем нерастворенную соль соединения фильтруют, после чего оставшуюся жидкую часть раствора выпаривают и высушивают. При этом в качестве соли металла фармацевтически приемлемыми для получения являются, в частности, без ограничения соли натрия, калия или кальция. Кроме того, соль серебра, соответствующая этому, может быть получена таким образом, при котором соли щелочных металлов или щелочноземельных металлов вводят в реакцию с соответствующей солью серебра (например, нитратом серебра).
Фармацевтически приемлемые соли согласно настоящему изобретению предусматривают соль кислотной или основной группы, которая может присутствовать в соединении вышеприведенной формулы 1, если не указано иное. Например, фармацевтически приемлемые соли могут предусматривать соли натрия, кальция, калия и т. п., образованные с гидроксигруппой, тогда как в качестве других фармацевтически приемлемых солей, образованных с аминогруппой, представлены гидробромидные, сульфатные, гидросульфатные, фосфатные, гидрофосфатные, дигидрофосфатные, ацетатные, сукцинатные, цитратные, тартратные, лактатные, манделатные, метансульфонатные (мезилатные), п-толуолсульфонатные (тозилатные) соли и т. п., при этом они могут быть получены посредством способа получения солей, известного из уровня техники.
Способ получения C-гликозидного производного соединения, содержащего конденсированное фенильное кольцо
Настоящее изобретение относится к способу получения C-гликозидного производного соединения, содержащего конденсированное фенильное кольцо, представленного формулой 1, или его фармацевтически приемлемых солей.
Соединение формулы I согласно настоящему изобретению может быть получено с помощью следующих стадий:
(S1) осуществление реакции соединения следующей формулы II с соединением следующей формулы III с получением соединения следующей формулы IV и
(S2) проведение восстановления с удалением защитной группы или удаления защитной группы с восстановлением для соединения вышеприведенной формулы IV с получением соединения следующей формулы I:[Формула II]
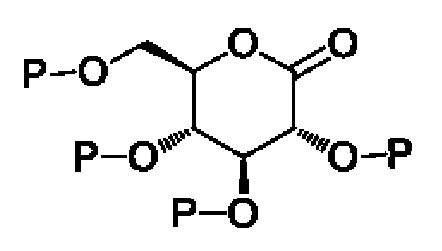 ,
,
[Формула III]
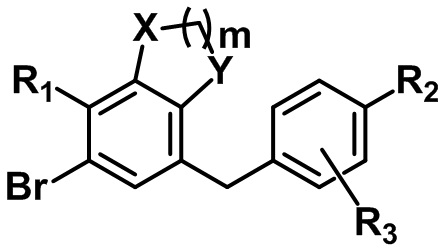 ,
,
[Формула IV]
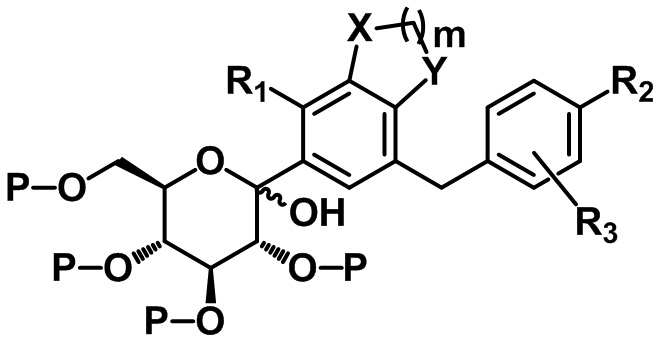 ,
,
[Формула I]
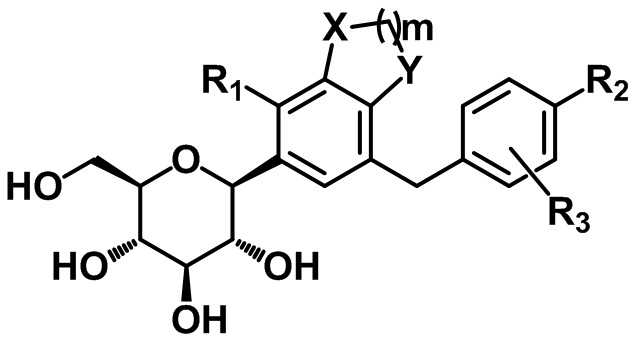 ,
,
где
X, Y, m, R1, R2 и R3 определены в данном документе, и P представляет собой триметилсилил или бензил.
В одном варианте осуществления настоящего изобретения, если P представляет собой триметилсилил, соединение следующей формулы V может быть получено посредством удаления защитной группы из соединения формулы IV, и соединение формулы I может быть получено посредством восстановления соединения формулы V:
[Формула V]
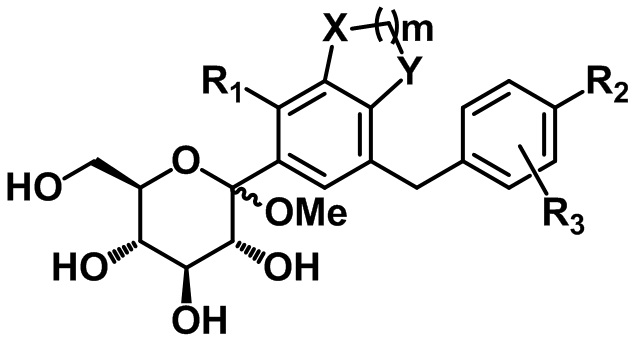 ,
,
где
X, Y, m, R1, R2 и R3 определены в данном документе.
В другом варианте осуществления настоящего изобретения, если P представляет собой бензил, соединение следующей формулы VI может быть получено посредством восстановления соединения формулы IV, и соединение формулы I может быть получено посредством удаления защитной группы из соединения формулы VI:
[Формула VI]
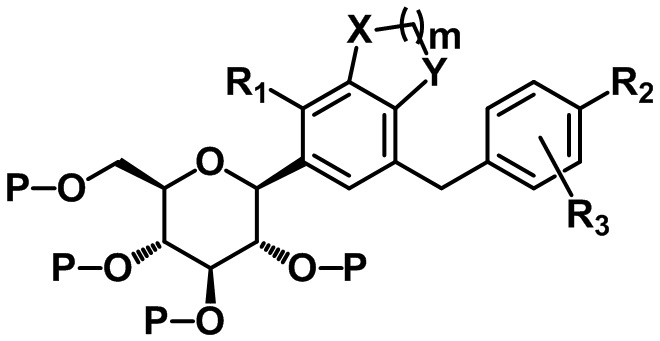 ,
,
где
X, Y, m, R1, R2, R3 и P определены в данном документе.
Подробный способ получения соединения, представленного формулой 1 согласно настоящему изобретению, или его фармацевтически приемлемых солей является таким, как показано в формуле реакции 1 и формуле реакции 2, при этом способ получения, который модифицирован для соответствия уровню специалистов в данной области техники, также включен в данный документ.
[Формула реакции 1]
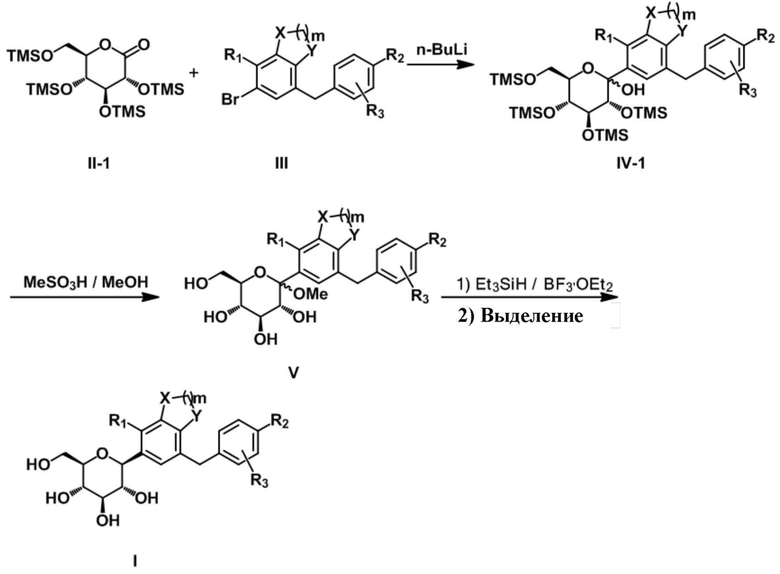
Проводили реакцию обмена литий-галоген для бромированного соединения III, после чего полученный продукт вводили в реакцию с персилилированным глюконолактоновым соединением II-1 с получением лактольной смеси IV-1. Полученную смесь обрабатывали метансульфоновой кислотой из метанола в пределах той же реакционной системы с превращением, таким образом, в десилилированное O-метиллактольное соединение V. Восстановление аномерной метоксигруппы лактольного соединения V проводили с помощью триэтилсилана и диэтилэфирата трифторида бора с получением соответствующей смеси α,β-изомеров. Необходимый β-изомер I отделяли путем селективной кристаллизации перацетилированной смеси конечного соединения или путем препаративной HPLC.
[Формула реакции 2]
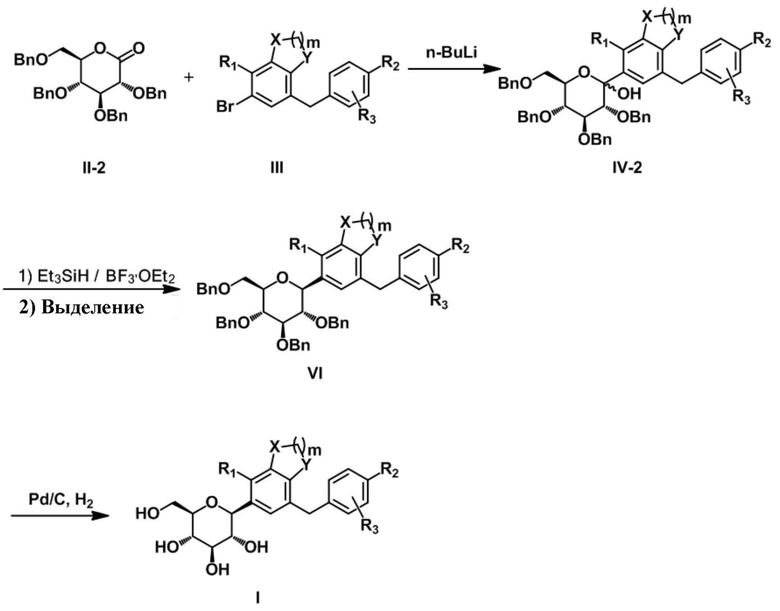
Затем лактольное соединение IV-2 получали с помощью пербензилированного глюконолактонового соединения II-2, после чего восстановление аномерной гидроксигруппы лактольного соединения IV-2 проводили с помощью триэтилсилана и диэтилэфирата трифторида бора с получением соответствующей смеси α,β-изомеров. Необходимый β-изомер соединения VI выделяли посредством селективной кристаллизации. Удаляли защитную бензильную группу из соединения VI с помощью Pd/C в атмосфере водорода с получением целевого соединения I.
Композиция, содержащая C-гликозидное производное соединение, содержащее конденсированное фенильное кольцо, ее применение и способ лечения с применением таковой
В настоящем изобретении представлена фармацевтическая композиция для предупреждения или лечения заболевания, связанного с активностью SGLT, содержащая соединение, представленное следующей формулой 1, или его фармацевтически приемлемые соли в качестве эффективного компонента.
[Формула 1]
Вышеприведенная формула 1 определена выше.
Соединение формулы 1 по настоящему изобретению или его фармацевтически приемлемые соли могут проявлять ингибирующую активность в отношении SGLT1, SGLT2 или в отношении их обоих. Таким образом, соединение формулы 1 по настоящему изобретению или его фармацевтически приемлемые соли можно с пользой применять для лечения или предупреждения диабета.
Предназначенная для введения фармацевтическая композиция по настоящему изобретению может дополнительно содержать по меньшей мере один из фармацевтически приемлемых носителей, в дополнение к соединению, представленному вышеприведенной формулой 1, или его фармацевтически приемлемым солям. Применяемые фармацевтически приемлемые носители могут представлять собой солевой раствор, стерилизованную воду, раствор Рингера, забуференный физиологический раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь по меньшей мере одного из них, при этом другие традиционные добавки, такие как антиоксидант, буферный раствор, бактериостат и т. д., могут быть также добавлены к ним, если это необходимо. Кроме того, разбавитель, диспергирующее средство, поверхностно-активное вещество, связующее средство и смазывающее вещество могут быть дополнительно добавлены в фармацевтическую композицию согласно настоящему изобретению, таким образом, она может быть составлена в виде лекарственной формы для инъекций, такой как водный раствор, суспензия, эмульсия и т. д., в виде пилюли, капсулы, гранулы, таблетки или т. п. Таким образом, композиция согласно настоящему изобретению может представлять собой пластырь, жидкость, пилюлю, капсулу, гранулу, таблетку, суппозиторий и т. д. Такие препараты могут быть получены посредством традиционного способа, применяемого для составления в уровне техники, или способа, раскрытого в Remington's Pharmaceutical Science (последнее издание), Mack Publishing Company, Истон, Пенсильвания, при этом композиция может быть составлена в виде различных препаратов в соответствии с каждым заболеванием или компонентом.
Композицию согласно настоящему изобретению можно вводить перорально или парентерально (например, применять внутривенно, подкожно, внутрибрюшинно или местно) в соответствии с целевым способом, при этом объем ее дозы варьируется в зависимости от веса, возраста, пола, состояния здоровья, режима питания пациента, времени введения, способа введения, скорости экскреции, тяжести заболевания и т. п. Суточная доза соединения, представленного формулой 1 согласно настоящему изобретению, может составлять от приблизительно 1 до 1000 мг/кг, предпочтительно от 5 до 100 мг/кг, при этом ее можно вводить один раз в сутки или разделить на несколько введений в сутки.
Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать по меньшей мере один эффективный компонент, который проявляет такой же или аналогичный медицинский эффект, в дополнение к соединению, представленному вышеприведенной формулой 1, или его фармацевтически приемлемым солям.
В настоящем изобретении представлен способ предупреждения или лечения заболевания, связанного с активностью SGLT, включающий введение терапевтически эффективного количества соединения, представленного вышеприведенной формулой 1, или его фармацевтически приемлемых солей.
Применяемый в данном документе термин «терапевтически эффективное количество» относится к количеству соединения, представленного вышеприведенной формулой 1, которое является эффективным в предупреждении или лечении заболевания, связанного с активностью SGLT.
Кроме того, решение в соответствии с настоящим изобретением может обеспечивать ингибирование SGLT1, SGLT2 или как SGLT1, так и SGLT2 посредством введения соединения, представленного вышеприведенной формулой 1, или его фармацевтически приемлемых солей млекопитающим, в том числе людям.
Заявляемый способ предупреждения или лечения заболевания, связанного с активностью SGLT, также включает лечение самого заболевания до проявления его симптомов, а также ингибирование или устранение его симптомов посредством введения соединения, представленного вышеприведенной формулой 1. При контроле течения заболеваний профилактическая или терапевтическая доза определенного активного компонента может варьироваться в зависимости от природы и тяжести заболевания или состояния и пути, которым вводят активный компонент. Доза и частота ее введения могут варьироваться в зависимости от возраста, веса и ответной реакции отдельного пациента. Подходящая доза и способ применения могут быть легко выбраны специалистами в данной области техники, естественно, с учетом таких факторов. Кроме того, заявляемый способ предупреждения или лечения заболевания, связанного с активностью SGLT, может дополнительно включать введение терапевтически эффективного количества дополнительного активного препарата, который является пригодным для лечения заболеваний, вместе с соединением, представленным вышеприведенной формулой 1, при этом такой дополнительный активный препарат может проявлять синергический эффект или вспомогательный эффект вместе с соединением вышеприведенной формулы 1.
В настоящем изобретении также представлено применение соединения, представленного вышеприведенной формулой 1, или его фармацевтически приемлемых солей с целью получения лекарственного средства для лечения заболевания, связанного с активностью SGLT. Соединение, представленное вышеприведенной формулой 1, при получении лекарственного средства может быть объединено с приемлемым вспомогательным веществом, разбавителем, носителем и т. д., и может быть подготовлено в виде комплексного препарата вместе с другими активными препаратами с получением, таким образом, синергического действия активных компонентов.
Объекты, упомянутые в применении, композиции, способе лечения согласно настоящему изобретению, применяются одинаково, если они не противоречат друг другу.
Предпочтительные эффекты
Новые С-гликозидные производные согласно настоящему изобретению могут обеспечивать двойное ингибирование SGLT1 и SGLT2, таким образом, могут быть применяться с пользой в лечении или предупреждении диабета.
Предпочтительный вариант осуществления настоящего изобретения
Далее в данном документе конфигурации и эффекты согласно настоящему изобретению будут более подробно описаны с помощью примеров. Тем не менее, следующие примеры представлены только с целью иллюстрации настоящего изобретения, и, таким образом, объем настоящего изобретения не ограничивается ими.
Пример 1. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез этил-7-метил-2,3-дигидро-1H-инден-4-карбоксилата (1-1)
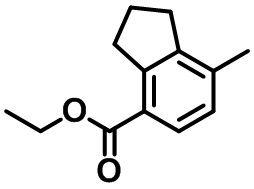 (1-1)
(1-1)
Смесь этилсорбата (25,0 мл, 170 ммоль, реагент TCI) в ксилоле (100 мл) и 1-пирролидино-1-циклопентена (24,8 мл, 170 ммоль, реагент TCI) перемешивали с обратным холодильником в течение ночи. После завершения реакции летучий растворитель выпаривали при пониженном давлении. К полученной смеси добавляли EtOAc. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение применяли на следующей стадии без дополнительной очистки. S8 (5,45 г, 170 ммоль) добавляли к неочищенному соединению. Реакционную смесь перемешивали при 250ºC в течение 2 часов. После завершения реакции полученную смесь перегоняли при пониженном давлении с получением указанного в заголовке соединения (1-1) (20,0 г, 97,9 ммоль, 58%).
1H ЯМР (400 МГц, CDCl3); δ 7,76 (d, J = 7,6 Гц, 1H), 7,03 (d, J = 8,0 Гц, 1H), 4,34 (q, J = 7,2 Гц, 2H), 3,30 (t, J = 7,6 Гц, 2H), 2,84 (t, J = 7,6 Гц, 2H), 2,30 (s, 3H), 2,12-2,05 (m, 2H), 1,38 (t, J = 7,2 Гц, 3H)
Стадия 2. Синтез этил-6-бром-7-метил-2,3-дигидро-1H-инден-4-карбоксилата (1-2)
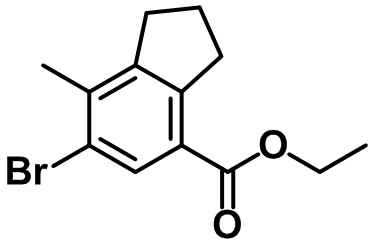 (1-2)
(1-2)
Br2 (6,0 мл, 117 ммоль) и AgNO3 (16,6 г, 97,9 ммоль) в воде (20 мл) добавляли по каплям к смеси соединения (1-1) (20,0 г, 97,9 ммоль) в AcOH (100 мл) и концентрированной HNO3 (4,4 мл) при комнатной температуре. Полученную смесь перемешивали в течение ночи при комнатной температуре. Реакцию завершали с помощью насыщенного раствора Na2S2O3 с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (1-2) (22,1 г, 78,0 ммоль, 80%).
1H ЯМР (400 МГц, CDCl3); δ 8,02 (s, 1H), 4,34 (q, J = 7,2 Гц, 2H), 3,25 (t, J = 7,6 Гц, 2H), 2,90 (t, J = 7,6 Гц, 2H), 2,37 (s, 3H), 2,11-2,07 (m, 2H), 1,39 (t, J = 7,2 Гц, 3H).
Стадия 3. Синтез 6-бром-7-метил-2,3-дигидро-1H-инден-4-карбоновой кислоты (1-3)
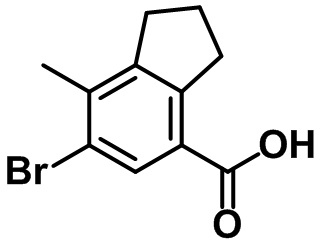 (1-3)
(1-3)
LiOH.H2O (9,82 г, 234 ммоль) добавляли в раствор соединения (1-2) (22,1 г, 78,0 ммоль) в смеси THF/MeOH/вода (120 мл/40 мл/40 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (1-3) (15,4 г, 60,4 ммоль, 77%).
1H ЯМР (400 МГц, CDCl3); δ 8,08 (s, 1H), 3,28 (t, J = 7,6 Гц, 2H), 2,91 (t, J = 7,6 Гц, 2H), 2,38 (s, 3H), 2,13-2,09 (m, 2H).
Стадия 4. Синтез (6-бром-7-метил-2,3-дигидро-1H-инден-4-ил)(4-метоксифенил)метанона (1-4)
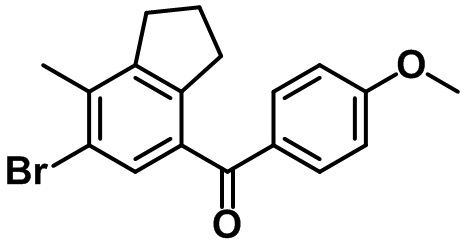 (1-4)
(1-4)
DMF (0,12 мл) и (COCl)2 (2,46 мл, 29,0 ммоль) добавляли по каплям в раствор 6-бром-7-метил-2,3-дигидро-1H-инден-4-карбоновой кислоты (1-3) (4,94 г, 19,3 ммоль) в DCM (45 мл) при 0ºC в атмосфере азота. Полученный раствор перемешивали в течение ночи при комнатной температуре, после чего полученную смесь концентрировали под вакуумом с получением неочищенного оксихлорида. 4-Метоксибензол (2,52 мл, 23,2 ммоль) и AlCl3 (3,09 г, 23,2 ммоль) по частям добавляли в раствор неочищенного оксихлорида в DCM (45 мл) при 0ºC. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов при комнатной температуре. Полученную смесь выливали в ледяную воду с проведением экстракции с помощью EtOAc. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (1-4) (4,84 г, 14,02 ммоль, 73%).
1H ЯМР (400 МГц, CDCl3); δ 7,78 (d, J = 8,8 Гц, 2H), 7,49 (s, 1H), 6,95 (d, J = 8,8 Гц, 2H), 3,89 (s, 3H), 2,97-2,91 (m, 4H), 2,39 (s, 3H), 2,11-2,03 (m, 2H)
Стадия 5. Синтез 5-бром-7-(4-метоксибензил)-4-метил-2,3-дигидро-1H-индена (1-5)
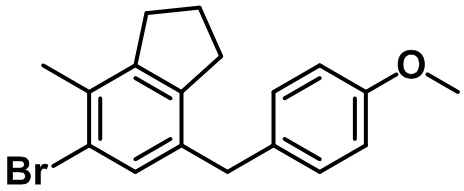 (1-5)
(1-5)
Триэтилсилан (4,61 мл, 28,0 ммоль) и BF3.OEt2 (3,55 мл, 28,0 ммоль) добавляли по каплям в раствор соединения (1-4) (4,84 г, 14,0 ммоль) в смеси DCM/ацетонитрил (20 мл/20 мл) при 0ºC в атмосфере азота. Полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи при комнатной температуре. Насыщенный водный раствор NaHCO3 медленно добавляли к полученной смеси с проведением экстракции с помощью EtOAc. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле, с получением указанного в заголовке соединения (1-5) (4,21 г, 12,7 ммоль, 91%).
1H ЯМР (400 МГц, CDCl3); δ 7,12 (s, 1H), 7,05 (d, J = 8,4 Гц, 2H), 6,81 (d, J = 8,4 Гц, 2H), 3,80 (s, 2H), 3,78 (s, 3H), 2,87 (t, J = 7,4 Гц, 2H), 2,75 (t, J = 7,6 Гц, 2H), 2,29 (s, 3H), 2,08-2,00 (m, 2H)
Стадия 6. Синтез (2R,3R,4R,5S,6S)-3,4,5-трис(бензилокси)-2-(бензилоксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пирана (1-6)
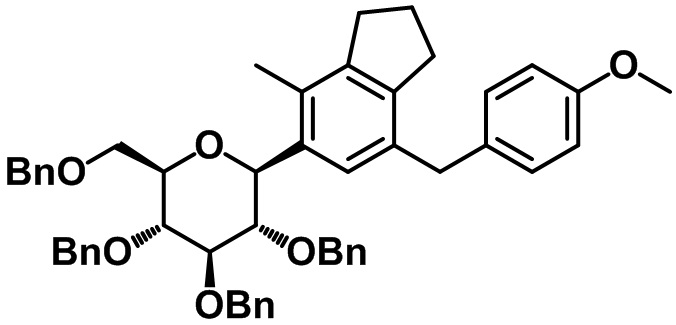 (1-6)
(1-6)
В раствор соединения (1-5) (6,82 г, 20,6 ммоль) в смеси толуол/THF (70 мл/70 мл) при -78ºC в атмосфере азота добавляли n-BuLi (12,4 мл, 30,9 ммоль, 2,5 M в н-гексане). Через 30 минут к полученной смеси при -78ºC добавляли пербензилированный глюконолактон (14,4 г, 26,8 ммоль) в толуоле (70 мл). Полученную смесь перемешивали при той же температуре в течение 2 часов. Реакцию завершали с помощью воды с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом, таким образом, получали неочищенное промежуточное соединение и его применяли без дополнительной очистки. В раствор промежуточного соединения в смеси DCM/ацетонитрил (100 мл/100 мл) при -78ºC в атмосфере азота добавляли триэтилсилан (10,1 мл, 61,8 ммоль) и BF3.OEt2 (7,83 мл, 61,8 ммоль). Полученную смесь нагревали до -60ºC в течение 1 часа. Насыщенный раствор NaHCO3 медленно добавляли к полученной смеси с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (1-6) (8,78 г, 11,32 ммоль, 55%).
1H ЯМР (400 МГц, CDCl3); δ 7,32-7,11 (m, 19H), 7,02 (d, J = 8,8 Гц, 2H), 6,87 (d, J = 6,4 Гц, 2H), 6,71 (d, J = 8,0 Гц, 2H), 4,96-4,87 (m, 3H), 4,68-4,63 (m, 2H), 4,54-4,49 (m, 2H), 4,35 (d, J = 10,4 Гц, 1H), 3,86-3,75 (m, 7H), 3,72 (s, 3H), 3,67-3,57 (m, 2H), 2,85-2,77 (m, 4H), 2,24 (s, 3H), 2,08-2,01 (m, 2H)
Стадия 7. Синтез целевого соединения
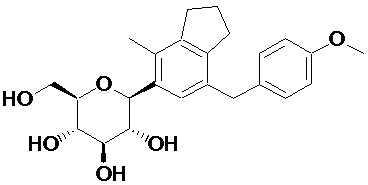
Суспензию соединения (1-6) (152 мг, 0,20 ммоль) в THF (3 мл) и MeOH (3 мл), а также Pd/C (20 вес. %, 30 мг) перемешивали при комнатной температуре в атмосфере водорода в течение 16 часов. Реакционную смесь фильтровали с помощью подушки из целита и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением целевого соединения (79 мг, 0,19 ммоль, 95%).
1H ЯМР (400 МГц, CD3OD); δ 7,11 (s, 1H), 7,04 (d, J = 8,4 Гц, 2H), 6,78 (d, J = 8,4 Гц, 2H), 4,45 (d, J = 9,2 Гц, 1H), 3,88-3,84 (m, 3H), 3,74 (s, 3H), 3,68-3,64 (m, 1H), 3,56 (t, J = 8,8 Гц, 1H), 3,52-3,47 (m, 1H), 3,40-3,38 (m, 2H), 2,84 (t, J = 7,6 Гц, 2H), 2,73 (t, J = 7,6 Гц, 2H), 2,28 (s, 3H), 2,01-1,98 (m, 2H)
Примеры 2 и 3
Целевые соединения из примеров 2 и 3 получали посредством способа, показанного в примере 1.
Пример 2. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
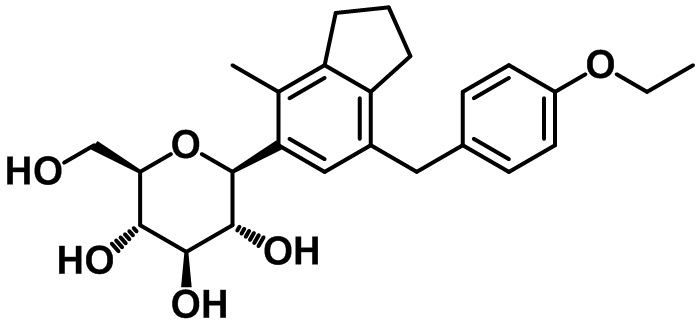
1H ЯМР (400 МГц, CD3OD); δ 7,10 (s, 1H), 7,03 (d, J = 8,4 Гц, 2H), 6,76 (d, J = 8,8 Гц, 2H), 4,45 (d, J = 8,8 Гц, 1H), 3,97 (q, J = 6,8 Гц, 2H), 3,88-3,84 (m, 3H), 3,67-3,48 (m, 4H), 3,40-3,38 (m, 2H), 2,84 (t, J = 7,6 Гц, 2H), 2,73 (t, J = 7,6 Гц, 2H), 2,28 (s, 3H), 2,02-1,97 (m, 2H), 1,35 (t, J = 6,8 Гц, 3H)
Пример 3. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-изопропоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
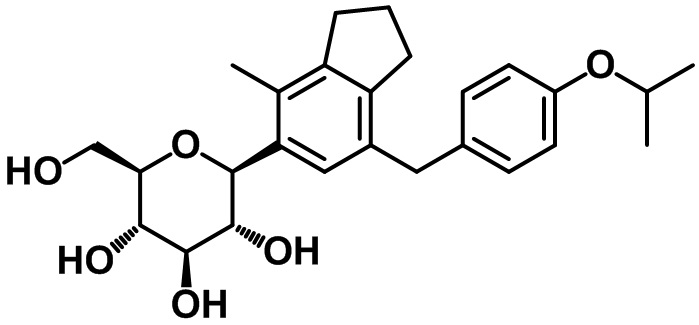
1H ЯМР (400 МГц, CD3OD); δ 7,11 (s, 1H), 7,02 (d, J = 8,4 Гц, 2H), 6,76 (d, J = 8,8 Гц, 2H), 4,54-4,48 (m, 1H), 4,45 (d, J = 9,2 Гц, 1H), 3,84-3,88 (m, 3H), 3,66 (dd, J = 11,6, 5,2 Гц, 1H), 3,59-3,48 (m, 2H), 3,41-3,38 (m, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,74 (t, J = 7,4 Гц, 2H), 2,28 (s, 3H), 2,04-1,96 (m, 2H), 1,27 (d, J = 6,0 Гц, 6H)
Пример 4. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез (6-бром-7-метил-2,3-дигидро-1H-инден-4-ил)метанола (4-1)
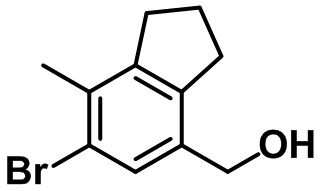 (4-1)
(4-1)
Комплекс BH3.SMe2 (13,7 мл, 137,2 ммоль, 10,0 M в метилсульфиде) медленно добавляли в раствор 6-бром-7-метил-2,3-дигидро-1H-инден-4-карбоновой кислоты (1-3) (3,50 г, 13,7 ммоль) в THF (50 мл) при 0ºC в атмосфере азота, после чего реакционную смесь перемешивали в течение ночи при комнатной температуре. Полученную реакционную смесь охлаждали при 0ºC, после чего насыщенный водный раствор NaHCO3 медленно добавляли к полученной смеси с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (4-1) (2,33 г, 9,66 ммоль, 70%).
1H ЯМР (400 МГц, CDCl3); δ 7,39 (s, 1H), 4,60 (d, J = 6,0 Гц, 2H), 2,91-2,86 (m, 4H), 2,32 (s, 3H), 2,14-2,07 (m, 2H), 1,48 (t, J = 5,8 Гц, 1H)
Стадия 2. Синтез 5-бром-7-(бромметил)-4-метил-2,3-дигидро-1H-индена (4-2)
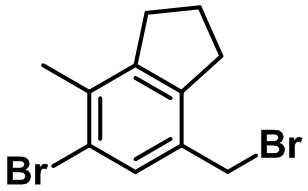 (4-2)
(4-2)
В раствор соединения (4-1) (2,33 г, 9,66 ммоль) в толуоле (45 мл) при 0ºC в атмосфере азота по каплям добавляли PBr3 (1,38 мл, 14,5 ммоль). Полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение 2 часов при комнатной температуре. Насыщенный водный раствор NaHCO3 медленно добавляли к полученной смеси с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (4-2) (2,14 г, 7,04 ммоль, 73%).
1H ЯМР (400 МГц, CDCl3); δ 7,35 (s, 1H), 4,40 (s, 2H), 2,95-2,88 (m, 4H), 2,31 (s, 3H), 2,18-2,10 (m, 2H)
Стадия 3. Синтез 5-бром-4-метил-7-(4-метилбензил)-2,3-дигидро-1H-индена (4-3)
 (4-3)
(4-3)
Соединение (4-2) (150 мг, 0,49 ммоль), 4-метилфенилбороновую кислоту (81 мг, 0,59 ммоль) и K2CO3 (136 мг, 0,99 ммоль) растворяли в смеси ацетон/вода (3 мл/1 мл), после чего к полученной смеси добавляли Pd2(dba)3 (90 мг, 0,10 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Полученную реакционную смесь фильтровали с помощью целита и распределяли между EtOAc и водой. Водный слой экстрагировали с помощью EtOAc, после чего объединенный органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (4-3) (130 мг, 0,41 ммоль, 84%).
1H ЯМР (400 МГц, CDCl3); δ 7,14 (s, 1H), 7,08 (d, J = 7,6 Гц, 2H), 7,02 (d, J = 8,0 Гц, 2H), 3,82 (s, 2H), 2,87 (t, J = 7,4 Гц, 2H), 2,75 (d, J = 7,6 Гц, 2H), 2,31 (s, 3H), 2,29 (s, 3H), 2,08-2,02 (m, 2H)
Стадия 4. Синтез целевого соединения
В раствор соединения (4-3) (130 мг, 0,41 ммоль) в смеси толуол/THF (3 мл/1,5 мл) при -78ºC в атмосфере азота добавляли n-BuLi (0,25 мл, 0,62 ммоль, 2,5 M в н-гексане). Через 30 минут TMS-защищенный глюконолактон (231 мг, 0,49 ммоль) в толуоле (3 мл) добавляли к полученной смеси при -78ºC. Полученную смесь перемешивали при той же температуре в течение 2 часов. В реакционную смесь при той же температуре добавляли метансульфоновую кислоту (0,2 мл) и MeOH (1,6 мл). Реакционную смесь перемешивали при -78ºC в течение 2 часов. Реакцию завершали с помощью насыщенного раствора NaHCO3 с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом с получением неочищенного промежуточного соединения, которое применяли без дополнительной очистки. В раствор промежуточного соединения в смеси DCM/ацетонитрил (2 мл/2 мл) при -78ºC в атмосфере азота добавляли триэтилсилан (0,14 мл, 0,82 ммоль) и BF3.OEt2 (0,11 мл, 0,82 ммоль). Полученную смесь нагревали до -50ºC в течение 1 часа. Насыщенный раствор NaHCO3 медленно добавляли к полученной смеси с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали с помощью препаративной HPLC с получением целевого соединения (5,6 мг, 0,014 ммоль, 3,4%).
1H ЯМР (400 МГц, CD3OD); δ 7,11 (s, 1H), 7,04-6,99 (m, 4H), 4,45 (d, J = 9,2 Гц, 1H), 3,88-3,86 (m, 3H), 3,66 (dd, J = 11,6, 5,6 Гц, 1H), 3,59-3,48 (m, 2H), 3,40-3,35 (m, 2H), 2,84 (t, J = 7,6 Гц, 2H), 2,72 (t, J = 7,6 Гц, 2H), 2,29 (s, 3H), 2,27 (s, 3H), 2,04-1,96 (m, 2H)
Примеры 5-16
Целевые соединения из примеров 5-16 получали посредством способа, показанного в примере 4.
Пример 5. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
1H ЯМР (400 МГц, CD3OD); δ 7,12 (s, 1H), 7,06-7,02 (m, 4H), 4,45 (d, J = 9,2 Гц, 1H), 3,88-3,86 (m, 3H), 3,68-3,48 (m, 3H), 3,40-3,39 (m, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,73 (t, J = 7,6 Гц, 2H), 2,58 (q, J = 7,6 Гц, 2H), 2,29 (s, 3H), 2,01-1,98 (m, 2H), 1,19 (t, J = 8,0 Гц, 3H)
Пример 6. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-пропилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
1H ЯМР (400 МГц, CD3OD); δ 7,11 (s, 1H), 7,03 (s, 4H), 4,45 (d, J = 9,2 Гц, 1H), 3,88-3,86 (m, 3H), 3,66 (dd, J = 11,6, 5,6 Гц, 1H), 3,59-3,48 (m, 2H), 3,41-3,39 (m, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,74 (d, J = 7,6 Гц, 2H), 2,52 (t, J = 7,6 Гц, 2H), 2,29 (s, 3H), 2,04-1,96 (m, 2H), 1,65-1,55 (m, 2H), 0,91 (t, J = 7,2 Гц, 3H)
Пример 7. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-изопропилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
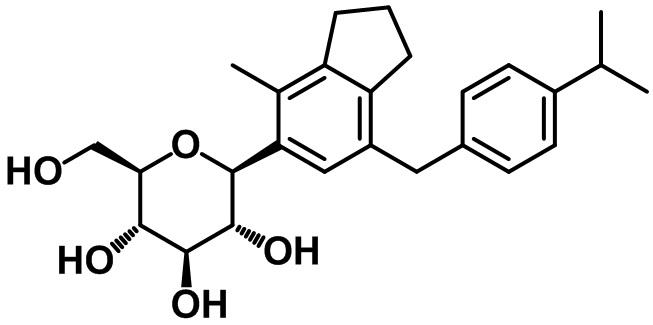
1H ЯМР (400 МГц, CD3OD); δ 7,12-7,03 (m, 5H), 4,45 (d, J = 9,2 Гц, 1H), 3,88-3,86 (m, 3H), 3,66 (dd, J = 12,0, 5,6 Гц, 1H), 3,57 (t, J = 9,2 Гц, 1H), 3,52-3,48 (m, 1H), 3,41-3,39 (m, 2H), 2,87-2,81 (m, 3H), 2,74 (t, J = 7,2 Гц, 2H), 2,29 (s, 3H), 2,04-1,98 (m, 2H), 1,21 (d, J = 7,2 Гц, 6H)
Пример 8. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-винилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
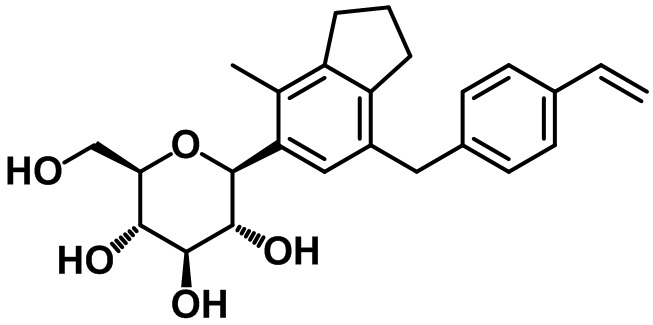
1H ЯМР (400 МГц, CD3OD); δ 7,33 (d, J = 8,4 Гц, 2H), 7,10 (d, J = 8,0 Гц, 2H), 7,03 (s, 1H), 6,66 (dd, J = 17,6, 11,2 Гц, 1H), 5,73 (d, J = 18,0 Гц, 1H), 5,17 (d, J = 10,0 Гц, 1H), 4,95-4,92 (m, 2H), 4,69 (d, J = 4,0 Гц, 1H), 4,38-4,37 (m, 1H), 4,22 (d, J = 8,4 Гц, 1H), 3,83 (s, 2H), 3,70-3,65 (m, 1H), 3,30-3,16 (m, 1H), 2,76 (t, J = 7,2 Гц, 2H), 2,71-2,66 (m, 2H), 2,17 (s, 3H), 1,94-1,90 (m, 2H)
Пример 9. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-трифторметил)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
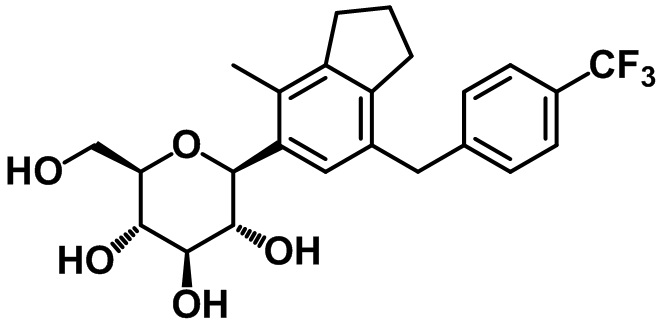
1H ЯМР (400 МГц, CD3OD); δ 7,45 (d, J = 8,0 Гц, 2H), 7,26 (d, J = 8,4 Гц, 2H), 7,08 (s, 1H), 4,39 (d, J = 8,8 Гц, 1H), 3,94 (s, 2H), 3,80 (d, J = 11,2 Гц, 1H), 3,59 (dd, J = 12,0, 5,2 Гц, 1H), 3,51-3,41 (m, 2H), 3,34-3,32 (m, 2H), 2,78 (t, J = 7,6 Гц, 2H), 2,65 (t, J = 7,6 Гц, 2H), 2,22 (s, 3H), 1,95-1,92 (m, 2H)
Пример 10. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-трифторметокси)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
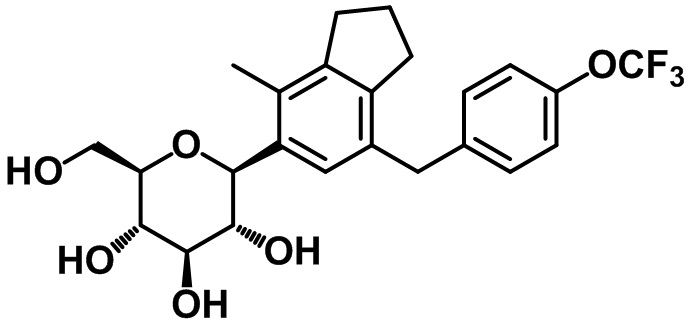
1H ЯМР (400 МГц, CD3OD); δ 7,22 (d, J = 8,4 Гц, 2H), 7,14 (s, 1H), 7,12 (d, J = 8,4 Гц, 2H), 4,46 (d, J = 9,2 Гц, 1H), 3,94 (s, 2H), 3,87 (d, J = 12,4 Гц, 1H), 3,68-3,62 (m, 1H), 3,55-3,46 (m, 2H), 3,40-3,39 (m, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,72 (d, J = 7,6 Гц, 2H), 2,29 (s, 3H), 2,02-1,99 (m, 2H)
Пример 11. Получение (2S,3R,4R,5S,6R)-2-(7-(3,4-диметоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
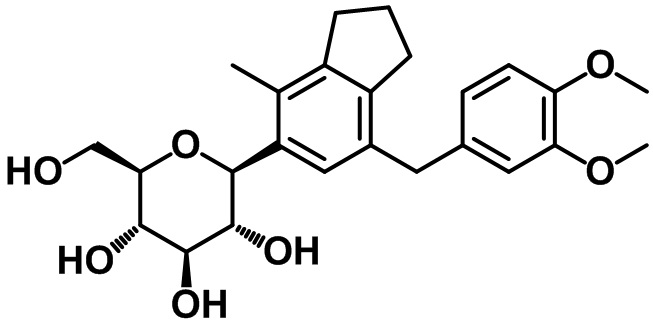
1H ЯМР (400 МГц, CD3OD); δ 7,12 (s, 1H), 6,81 (d, J = 8,0 Гц, 1H), 6,76 (s, 1H), 6,68 (d, J = 8,4 Гц, 1H), 4,45 (d, J = 8,8 Гц, 1H), 3,88-3,85 (m, 3H), 3,78 (s, 3H), 3,75 (s, 3H), 3,66 (dd, J = 12,0, 5,2 Гц, 1H), 3,59-3,48 (m, 2H), 3,41-3,39 (m, 2H), 2,85 (t, J = 7,4 Гц, 2H), 2,76 (t, J = 7,4 Гц, 2H), 2,29 (s, 3H), 2,05-1,97 (m, 2H)
Пример 12. Получение (2S,3R,4R,5S,6R)-2-(7-(2,4-диметоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
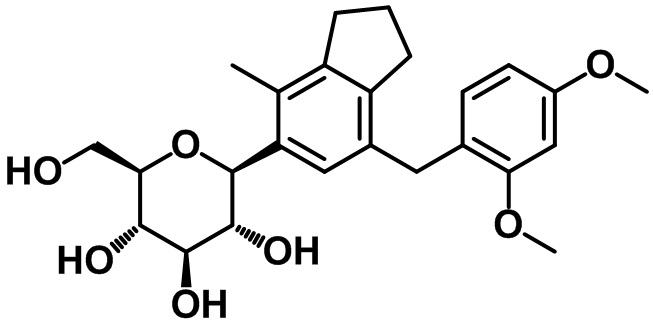
1H ЯМР (400 МГц, CD3OD); δ 7,17 (s, 1H), 6,91 (d, J = 8,0 Гц, 1H), 6,60 (d, J = 2,4 Гц, 1H), 6,47 (dd, J = 8,4, 2,4 Гц, 1H), 4,54 (d, J = 8,8 Гц, 1H), 3,97 (d, J = 12,0 Гц, 1H), 3,91 (s, 3H), 3,89 (s, 2H), 3,86 (s, 3H), 3,75 (dd, J = 12,0, 5,6 Гц, 1H), 3,67-3,57 (m, 2H), 3,50-3,45 (m, 2H), 2,96 (t, J = 7,6 Гц, 2H), 2,87 (t, J = 7,6 Гц, 2H), 2,39 (s, 3H), 2,16-2,10 (m, 2H)
Пример 13. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-метилтио)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
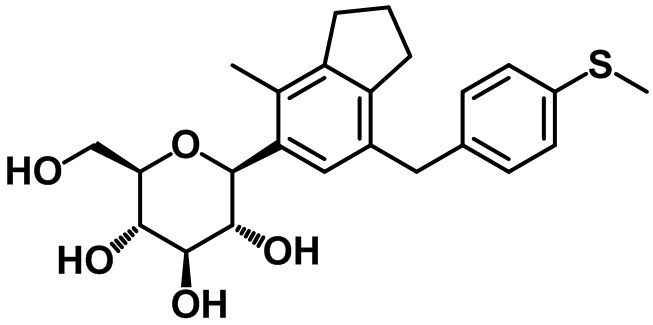
1H ЯМР (400 МГц, CD3OD); δ 7,14 (d, J = 8,0 Гц, 1H), 7,11 (s, 1H), 7,07 (d, J = 8,0 Гц, 2H), 4,46 (d, J = 9,2 Гц, 1H), 3,87-3,86 (m, 3H), 3,67 (dd, J = 11,6, 5,2 Гц, 1H), 3,59-3,48 (m, 2H), 3,41-3,39 (m, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,73 (t, J = 7,6 Гц, 2H), 2,42 (s, 3H), 2,29 (s, 3H), 2,04-1,96 (m, 2H)
Пример 14. Получение (2S,3R,4R,5S,6R)-2-(7-(4-фторбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
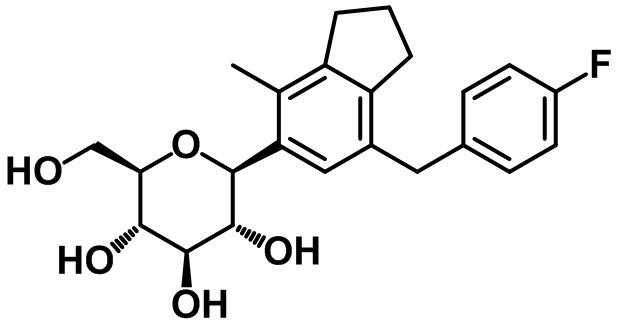
1H ЯМР (400 МГц, CD3OD); δ 7,24-7,21 (m, 3H), 7,05-7,00 (m, 2H), 4,54 (d, J = 9,2 Гц, 1H), 3,99 (s, 2H), 3,96 (d, J = 12,0 Гц, 1H), 3,79-3,73 (m, 1H), 3,65-3,59 (m, 2H), 3,49-3,48 (m, 2H), 2,94 (t, J = 7,6 Гц, 2H), 2,81 (t, J = 7,6 Гц, 2H), 2,38 (s, 3H), 2,11-2,07 (m, 2H)
Пример 15. Получение (2S,3R,4R,5S,6R)-2-(7-(4-фтор-3-метилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
1H ЯМР (400 МГц, CD3OD); δ 7,12 (s, 1H), 6,99 (d, J = 7,2 Гц, 1H), 6,96-6,93 (m, 1H), 6,88-6,84 (m, 1H), 4,47 (d, J = 9,2 Гц, 1H), 3,90-3,86 (m, 3H), 3,68-3,64 (m, 1H), 3,61-3,49 (m, 2H), 3,42-3,40 (m, 2H), 2,86 (t, J = 7,2 Гц, 2H), 2,73 (t, J = 7,2 Гц, 2H), 2,30 (s, 3H), 2,19 (s, 3H), 2,03-1,99 (m, 2H)
Пример 16. Получение (2S,3R,4R,5S,6R)-2-(7-(4-хлорбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
1H ЯМР (400 МГц, CD3OD); δ 7,21 (d, J = 8,4 Гц, 2H), 7,12 (d, J = 8,4 Гц, 2H), 7,11 (s, 1H), 4,46 (d, J = 8,8 Гц, 1H), 3,90-3,86 (m, 3H), 3,67 (dd, J = 11,6, 5,2 Гц, 1H), 3,58-3,48 (m, 2H), 3,39-3,43 (m, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,72 (t, J = 7,6 Гц, 2H), 2,29 (s, 3H), 2,04-1,97 (m, 2H)
Пример 17. Получение (2S,3R,4R,5S,6R)-2-(8-(4-этоксибензил)-2,3-дигидробензо[b][1,4]диоксин-6-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 5-бром-2,3-дигидробензойной кислоты (17-1)
 (17-1)
(17-1)
К 2,3-дигидробензойной кислоте (10,0 г, 64,9 ммоль, реагент Aldrich) в AcOH (120 мл) по каплям добавляли Br2 (3,32 мл, 64,9 ммоль), после чего полученную смесь перемешивали при комнатной температуре в течение 12 часов. Реакцию завершали с помощью насыщенного водного раствора Na2S2O3, после чего полученную смесь высушивали при пониженном давлении с удалением летучего вещества. Полученный остаток распределяли между EtOAc и водой. Водный слой экстрагировали с помощью EtOAc, после чего объединенный органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Указанное в заголовке соединение (17-1) (14,1 г, 60,3 ммоль, 93%) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 7,47 (s, 1H), 7,37 (s, 1H)
Стадия 2. Синтез метил-5-бром-2,3-дигидробензоата (17-2)
 (17-2)
(17-2)
В раствор соединения (17-1) (14,1 г, 60,3 ммоль) в MeOH (200 мл) при 0ºC в атмосфере азота по каплям добавляли SOCl2 (13,1 мл, 180,9 ммоль). Полученную смесь перемешивали с обратным холодильником в течение ночи. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (17-2) (12,5 г, 50,6 ммоль, 84%).
1H ЯМР (400 МГц, CDCl3); δ 10,85 (s, 1H), 7,51 (s, 1H), 7,23 (s, 1H), 5,69 (s, 1H), 3,96 (s, 3H)
Стадия 3. Синтез метил-7-бром-2,3-дигидробензо[b][1,4]диоксин-5-карбоксилата (17-3)
 (17-3)
(17-3)
1,2-Дибромэтан (8,2 мл, 94,9 ммоль) добавляли по каплям к смеси соединения (17-2) (15,6 г, 63,3 ммоль) в DMF (200 мл), а также K2CO3 (26,2 г, 95,0 ммоль). Реакционную смесь нагревали при 100ºC в течение ночи, после чего реакцию в ней завершали с помощью воды. Водный слой экстрагировали с помощью EtOAc, после чего объединенный органический слой промывали солевым раствором, таким образом, полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (17-3) (11,0 г, 40,4 ммоль, 64%).
1H ЯМР (400 МГц, CDCl3); δ 7,52 (d, J = 2,4 Гц, 1H), 7,16 (d, J = 2,8 Гц, 1H), 4,37-4,28 (m, 4H), 3,88 (s, 3H).
Стадия 4. Синтез 7-бром-2,3-дигидробензо[b][1,4]диоксин-5-карбоновой кислоты (17-4)
 (17-4)
(17-4)
К соединению (17-3) (5,0 г, 15,4 ммоль) в смеси THF/MeOH (20 мл/40 мл) при комнатной температуре добавляли 1 н. водный раствор NaOH (30,7 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (17-4) (3,4 г, 13,0 ммоль, 85%).
1H ЯМР (400 МГц, CDCl3); δ 8,00 (s, 1H), 7,83 (s, 1H), 4,52-4,50 (m, 4H)
Стадия 5. Синтез 7-бром-5-(4-этоксибензил)-2,3-дигидробензо[b][1,4]диоксина (17-5)
 (17-5)
(17-5)
Указанное в заголовке соединение (17-5) получали с помощью соединения (17-4) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 7,09 (d, J = 8,8 Гц, 2H), 6,88 (d, J = 2,4 Гц, 1H), 6,81 (d, J = 8,8 Гц, 2H), 6,73 (d, J = 2,4 Гц, 1H), 4,26-4,22 (m, 4H), 4,01 (q, J = 7,2 Гц, 2H), 3,81 (s, 2H), 1,40 (t, J = 6,8 Гц, 3H)
Стадия 6. Синтез целевого соединения
Целевое соединение получали с помощью соединения (17-5) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,10 (d, J = 8,4 Гц, 2H), 6,78-6,74 (m, 4H), 4,21 (dd, J = 10,0, 4,8 Гц, 4H), 4,00-3,95 (m, 3H), 3,87-3,78 (m, 3H), 3,66 (dd, J = 12,0, 5,6 Гц, 1H), 3,44-3,29 (m, 4H), 1,35 (t, J = 6,8 Гц, 3H)
Пример 18. Получение (2S,3R,4R,5S,6R)-2-(8-(4-этилбензил)-2,3-дигидробензо[b][1,4]диоксин-6-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 7-бром-5-(4-этилбензил)-2,3-дигидробензо[b][1,4]диоксина (18-1)
 (18-1)
(18-1)
Указанное в заголовке соединение (18-1) получали с помощью соединения (17-4), полученного на стадии 4 примера 17, посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,13-7,10 (m, 4H), 6,88 (d, J = 2,0 Гц, 1H), 6,75 (d, J = 2,0 Гц, 1H), 4,28-4,22 (m, 4H), 3,85 (s, 2H), 2,62 (q, J = 7,6 Гц, 2H), 1,22 (t, J = 7,6 Гц, 3H)
Стадия 2. Синтез целевого соединения
Целевое соединение получали с помощью соединения (18-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,10 (d, J = 7,6 Гц, 2H), 7,05 (d, J = 8,0 Гц, 2H), 6,78 (d, J = 2,0 Гц, 1H), 6,75 (d, J = 2,0 Гц, 1H), 4,24-4,20 (m, 4H), 3,96 (d, J = 9,2 Гц, 1H), 3,91-3,81 (m, 4H), 3,66 (dd, J = 12,0, 5,6 Гц, 1H), 3,42-3,32 (m, 3H), 2,58 (q, J = 7,6 Гц, 2H), 1,19 (t, J = 7,6 Гц, 3H)
Пример 19. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)бензо[d][1,3]диоксол-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез метил-6-бромбензо[d][1,3]диоксол-4-карбоксилата (19-1)
 (19-1)
(19-1)
Дибромметан (3,2 мл, 45,3 ммоль) добавляли по каплям к смеси соединения (17-2) (7,5 г, 30,2 ммоль), полученного на стадии 2 примера 17 в DMF (100 мл), а также K2CO3 (12,5 г, 95,0 ммоль). Реакционную смесь нагревали при 100ºC в течение ночи, после чего реакцию в ней завершали с помощью воды. Водный слой экстрагировали с помощью EtOAc, после чего объединенный органический слой промывали солевым раствором, таким образом, полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (19-1) (7,6 г, 29,3 ммоль, 97%).
1H ЯМР (400 МГц, CDCl3); δ 7,55 (d, J = 2,0 Гц, 1H), 7,08 (d, J = 1,6 Гц, 1H), 6,12 (s, 2H), 3,92 (s, 3H)
Стадия 2. Синтез 6-бромбензо[d][1,3]диоксол-4-карбоновой кислоты (19-2)
 (19-2)
(19-2)
К соединению (19-1) (7,6 г, 29,3 ммоль) в смеси THF/MeOH (40 мл/80 мл) при комнатной температуре добавляли 1 н. водный раствор NaOH (58,7 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (19-2) (7,2 г, 29,3 ммоль, 99%).
1H ЯМР (400 МГц, CDCl3); δ 7,59 (d, J = 2,0 Гц, 1H), 7,13 (d, J = 2,0 Гц, 1H), 6,16 (s, 2H).
Стадия 3. Синтез 6-бром-4-(4-метоксибензил)бензо[d][1,3]диоксола (19-3)
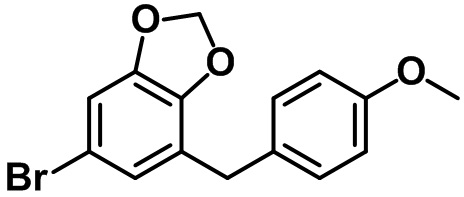 (19-3)
(19-3)
Указанное в заголовке соединение (19-3) получали с помощью соединения (19-2) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 7,13 (d, J = 8,8 Гц, 2H), 6,85-6,81 (m, 3H), 6,75 (s, 1H), 5,96 (s, 2H), 3,81 (s, 2H), 3,79 (s, 3H)
Стадия 4. Синтез целевого соединения

Целевое соединение получали с помощью соединения (19-3) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,14 (d, J = 8,4 Гц, 2H), 6,81-6,79 (m, 3H), 6,73 (s, 1H), 5,92 (s, 2H), 4,00 (d, J = 9,2 Гц, 1H), 3,87-3,82 (m, 4H), 3,75 (s, 3H), 3,69-3,64 (m, 1H), 3,42-3,32 (m, 3H)
Пример 20. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-(метилтио)бензил)бензо[d][1,3]диоксол-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 6-бром-4-(4-метилтио)бензил)бензо[d][1,3]диоксола (20-1)
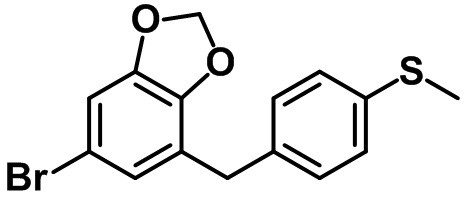 (20-1)
(20-1)
Указанное в заголовке соединение (20-1) получали с помощью соединения (19-2), полученного на стадии 2 примера 18, посредством способа, показанного на стадиях 1-3 примера 4.
1H NMR (400 МГц, CDCl3); δ 7,20 (d, J = 8,0 Гц, 2H), 7,14 (d, J = 8,4 Гц, 2H), 6,83 (s, 1H), 6,76 (s, 1H), 5,97 (s, 2H), 3,83 (s, 2H), 2,47 (s, 3H)
Стадия 2. Синтез целевого соединения
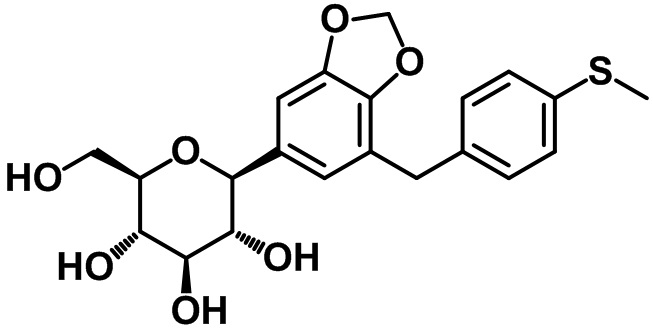
Целевое соединение получали с помощью соединения (20-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,16 (s, 4H), 6,80 (s, 1H), 6,75 (s, 1H), 5,93 (s, 2H), 4,00 (d, J = 9,2 Гц, 1H), 3,86-3,84 (m, 3H), 3,69-3,64 (m, 1H), 3,43-3,28 (m, 4H), 2,43 (s, 3H)
Пример 21. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)бензо[d][1,3]диоксол-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
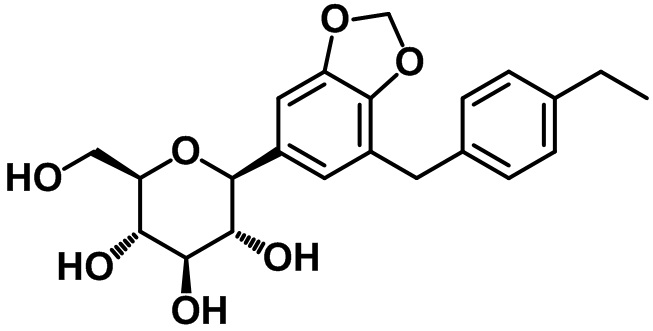
Целевое соединение получали посредством способа, показанного в примере 20.
1H ЯМР (400 МГц, CD3OD); δ 7,13 (d, J = 8,0 Гц, 2H), 7,07 (d, J = 7,6 Гц, 2H), 6,79 (s, 1H), 6,75 (s, 1H), 5,92 (s, 2H), 4,00 (d, J = 9,2 Гц, 1H), 3,90-3,81 (m, 3H), 3,67 (dd, J = 12,0, 5,6 Гц, 1H), 3,45-3,29 (m, 4H), 2,58 (q, J = 7,6 Гц, 2H), 1,19 (t, J = 7,6 Гц, 3H)
Пример 22. Получение (2S,3R,4R,5S,6R)-2-(4-(4-этоксибензил)-5,6,7,8-тетрагидронафтален-2-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез метил-5,6,7,8-тетрагидронафталин-1-карбоксилата (22-1)
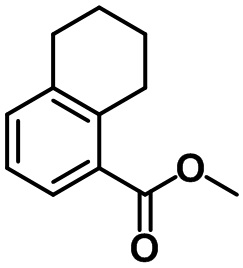 (22-1)
(22-1)
SOCl2 (4,1 мл, 56,7 ммоль) добавляли по каплям в раствор 5,6,7,8-тетрагидронафталин-1-карбоновой кислоты (2,0 г, 11,3 ммоль, реагент TCI) в MeOH (30 мл) при 0ºC в атмосфере азота. Полученную смесь перемешивали с обратным холодильником в течение ночи. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (22-1) (1,98 г, 10,4 ммоль, 92%).
1H ЯМР (400 МГц, CDCl3); δ 7,64 (d, J = 7,6 Гц, 1H), 7,21 (d, J = 7,2 Гц, 1H), 7,13 (t, J = 7,6 Гц, 1H), 3,87 (s, 3H), 3,06-3,03 (m, 2H), 2,83-2,79 (m, 2H), 1,80-1,77 (m, 4H)
Стадия 2. Синтез метил-3-бром-5,6,7,8-тетрагидронафталин-1-карбоксилата (22-2)
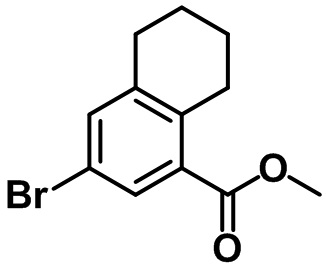 (22-2)
(22-2)
Концентрированную HNO3 (0,4 мл, 8,91 ммоль) и Br2 (3,32 мл, 64,9 ммоль) добавляли по каплям в перемешанный раствор с соединением (22-1) (1,13 г, 5,94 ммоль) в AcOH (10 мл), а также AgNO3 (1,51 г, 8,91 ммоль) в воде (5 мл), после чего полученную смесь перемешивали при комнатной температуре в течение 12 часов. Реакцию завершали с помощью насыщенного водного раствора Na2S2O3, после чего полученную смесь высушивали при пониженном давлении с удалением летучего вещества. Полученный остаток распределяли между EtOAc и водой. Водный слой экстрагировали с помощью EtOAc, после чего объединенный органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (22-2) (1,41 г, 5,24 ммоль, 88%).
1H ЯМР (400 МГц, CDCl3); δ 7,78 (d, J = 1,6 Гц, 1H), 7,36 (s, 1H), 3,87 (s, 3H), 2,99-2,96 (m, 2H), 2,81-2,77 (m, 2H), 1,81-1,74 (m, 4H)
Стадия 3. Синтез 3-бром-5,6,7,8-тетрагидронафталин-1-карбоновой кислоты (22-3)
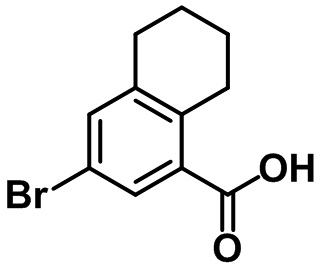 (22-3)
(22-3)
К соединению (22-2) (1,41 г, 5,24 ммоль) в смеси THF/MeOH/вода (15 мл/5 мл/5 мл) при комнатной температуре добавляли LiOH.H2O (0,67 г, 15,7 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (22-3) (1,31 г, 5,14 ммоль, 98%).
1H ЯМР (400 МГц, CD3OD); δ 7,72 (s, 1H), 7,39 (s, 1H), 3,06-3,01 (m, 2H), 2,86-2,80 (m, 2H), 1,83-1,74 (m, 4H)
Стадия 4. Синтез 7-бром-5-(4-этоксибензил)-1,2,3,4-тетрагидронафталина (22-4)
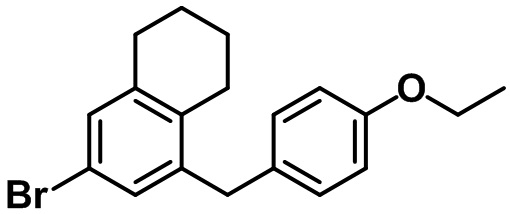 (22-4)
(22-4)
Указанное в заголовке соединение (22-4) получали с помощью соединения (22-3) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 7,12 (s, 1H), 7,03-6,98 (m, 3H), 6,81 (d, J = 8,8 Гц, 2H), 4,01 (q, J = 7,2 Гц, 2H), 3,82 (t, J = 5,6 Гц, 2H), 2,74 (t, J = 5,6 Гц, 2H), 2,53 (t, J = 6,0 Гц, 2H), 1,73-1,70 (m, 4H), 1,40 (t, J = 7,2 Гц, 3H)
Стадия 5. Синтез целевого соединения
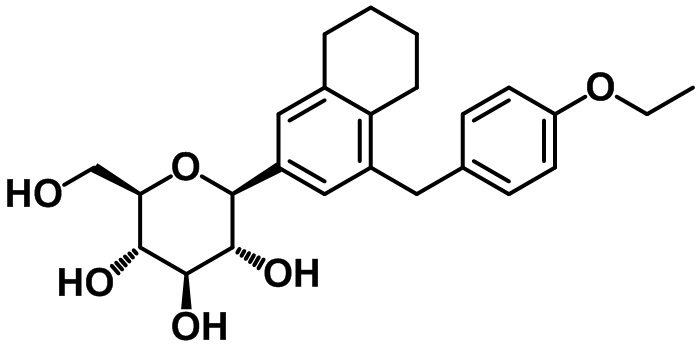
Целевое соединение получали с помощью соединения (22-4) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,02-6,99 (m, 4H), 6,77 (d, J = 8,4 Гц, 2H), 4,04 (d, J = 9,6 Гц, 1H), 3,97 (q, J = 6,8 Гц, 2H), 3,89-3,87 (m, 3H), 3,69 (dd, J = 12,0, 5,2 Гц, 1H), 3,46-3,36 (m, 4H), 2,78-2,76 (m, 2H), 2,56-2,54 (m, 2H), 1,73-1,34 (m, 4H), 1,35 (t, J = 6,8 Гц, 3H)
Пример 23. Получение (2S,3R,4R,5S,6R)-2-(4-(4-этилбензил)-5,6,7,8-тетрагидронафтален-2-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 7-бром-5-(4-этилбензил)-1,2,3,4-тетрагидронафталина (23-1)
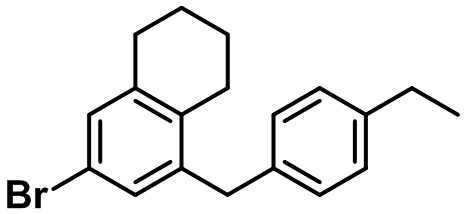 (23-1)
(23-1)
Указанное в заголовке соединение (23-1) получали с помощью соединения (22-3), полученного на стадии 3 примера 22, посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,12-7,10 (m, 3H), 7,05 (s, 1H), 7,01 (d, J = 8,4 Гц, 2H), 3,85 (s, 2H), 2,73 (t, J = 6,4 Гц, 2H), 2,62 (q, J = 7,6 Гц, 2H), 2,53 (t, J = 6,0 Гц, 2H), 1,78-1,70 (m, 4H), 1,22 (t, J = 7,6 Гц, 3H)
Стадия 2. Синтез целевого соединения
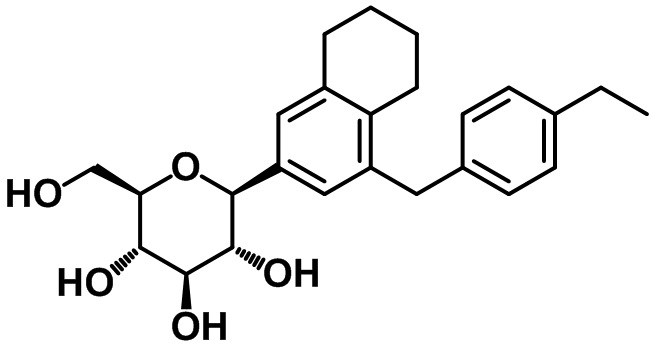
Целевое соединение получали с помощью соединения (23-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,06-6,99 (m, 6H), 4,03 (d, J = 9,2 Гц, 1H), 3,89-3,85 (m, 3H), 3,69-3,67 (m, 1H), 3,45-3,34 (m, 4H), 2,76 (s, 2H), 2,60-2,55 (m, 4H), 1,71 (s, 4H), 1,18 (t, J = 7,6 Гц, 3H)
Пример 24. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-(4-метоксибензил)-1-метил-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез этил-4-метил-5,6,7,8-тетрагидронафталин-1-карбоксилата (24-1)
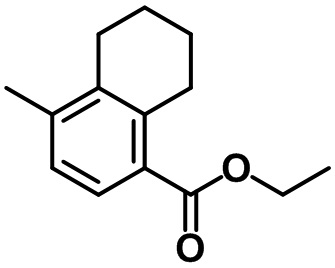 (24-1)
(24-1)
Смесь этилсорбата (49,5 мл, 0,33 моль, реагент TCI) в ксилоле (330 мл), а также 1-пирролидино-1-циклогексен (50,24 г, 0,33 моль, реагент TCI) перемешивали с обратным холодильником в течение ночи. После завершения реакции летучий растворитель выпаривали при пониженном давлении. К полученной смеси добавляли EtOAc. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение применяли на следующей стадии без дополнительной очистки. S8 (10,7 г, 0,33 моль) добавляли к неочищенному соединению. Реакционную смесь перемешивали при 250ºC в течение 2 часов. После завершения реакции полученную смесь перегоняли при пониженном давлении с получением указанного в заголовке соединения (24-1) (24,7 г, 0,11 моль, 34%).
1H ЯМР (400 МГц, CDCl3); δ 7,58 (d, J = 7,6 Гц, 1H), 7,02 (d, J = 8,0 Гц, 1H), 4,32 (q, J = 7,2 Гц, 2H), 3,06 (t, J = 6,4 Гц, 2H), 2,64 (t, J = 6,4 Гц, 2H), 2,24 (s, 3H), 1,85-1,73 (m, 4H), 1,37 (t, J = 7,2 Гц, 3H)
Стадия 2. Синтез этил-3-бром-4-метил-5,6,7,8-тетрагидронафталин-1-карбоксилата (24-2)
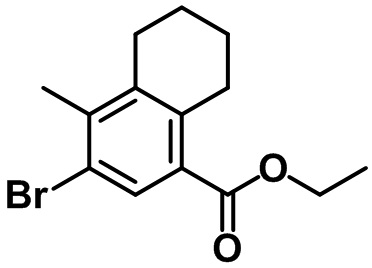 (24-2)
(24-2)
Br2 (3,5 мл, 68,6 ммоль) и AgNO3 (11,64 г, 68,6 ммоль) в воде (60 мл) добавляли по каплям к смеси соединения (24-1) (11,5 г, 68,6 ммоль) в AcOH (450 мл), а также концентрированной HNO3 (5,2 мл) при комнатной температуре. Полученную смесь перемешивали в течение ночи при комнатной температуре. Реакцию завершали с помощью насыщенного раствора Na2S2O3 с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение (24-2) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 7,87 (s, 1H), 4,32 (q, J = 7,2 Гц, 2H), 3,00 (t, J = 6,4 Гц, 2H), 2,70 (t, J = 6,4 Гц, 2H), 2,36 (s, 3H), 1,82-1,71 (m, 4H), 1,38 (t, J = 7,2 Гц, 3H).
Стадия 3. Синтез 3-бром-4-метил-5,6,7,8-тетрагидронафталин-1-карбоновой кислоты (24-3)
 (24-3)
(24-3)
LiOH.H2O (3,6 г, 86,2 ммоль) добавляли в раствор соединения (24-2) (12,8 г, 43,1 ммоль) в смеси THF/MeOH/вода (150 мл/50 мл/50 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (24-3) (9,3 г, 34,4 ммоль, 80%).
1H ЯМР (400 МГц, CDCl3); δ 8,07 (s, 1H), 3,07 (t, J = 6,4 Гц, 2H), 2,71 (t, J = 6,4 Гц, 2H), 2,39 (s, 3H), 1,83-1,72 (m, 4H).
Стадия 4. Синтез 6-бром-8-(4-метоксибензил)-5-метил-1,2,3,4-тетрагидронафталина (24-4)
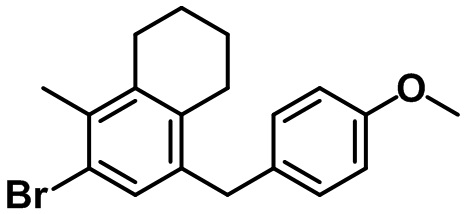 (24-4)
(24-4)
Указанное в заголовке соединение (24-4) получали с помощью соединения (24-3) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 7,16 (s, 1H), 7,02 (d, J = 8,8 Гц, 2H), 6,82 (d, J = 8,0 Гц, 2H), 3,82 (s, 2H), 3,79 (s, 3H), 2,67 (t, J = 6,4 Гц, 2H), 2,54 (t, J = 6,4 Гц, 2H), 2,31 (s, 3H), 1,77-1,68 (m, 4H).
Стадия 5. Синтез целевого соединения
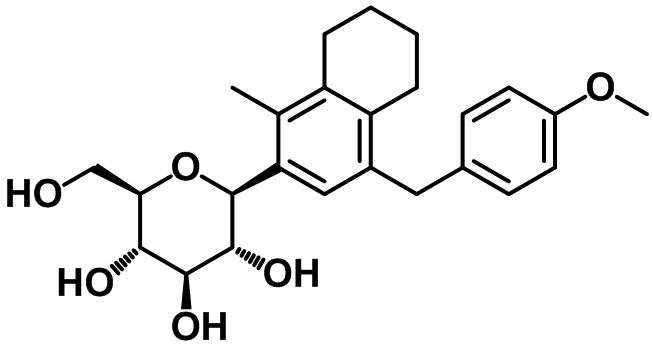
Целевое соединение получали с помощью соединения (24-4) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,13 (s, 1H), 7,00 (d, J = 9,2 Гц, 2H), 6,77 (d, J = 8,4 Гц, 1H), 4,51 (d, J = 9,6 Гц, 1H), 3,89-3,85 (m, 3H), 3,73 (s, 3H), 3,67 (dd, J = 11,6, 5,6 Гц, 1H), 3,60 (t, J = 8,8 Гц, 1H), 3,51 (t, J = 8,8 Гц, 1H), 3,41-3,39 (m, 2H), 2,65 (t, J = 6,4 Гц, 2H), 2,54 (t, J = 6,0 Гц, 2H), 2,24 (s, 3H), 1,75-1,64 (m, 4H)
Пример 25. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-метилбензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 6-бром-5-метил-8-(4-метилбензил)-1,2,3,4-тетрагидронафталина (25-1)
 (25-1)
(25-1)
Указанное в заголовке соединение (25-1) получали с помощью соединения (24-3), полученного на стадии 3 примера 24, посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,17 (s, 1H), 7,08 (d, J = 7,6 Гц, 2H), 6,99 (d, J = 8,0 Гц, 2H), 3,84 (s, 2H), 2,67 (t, J = 6,4 Гц, 2H), 2,54 (t, J = 6,4 Гц, 2H), 2,31 (s, 6H), 1,76-1,68 (m, 4H)
Стадия 2. Синтез целевого соединения
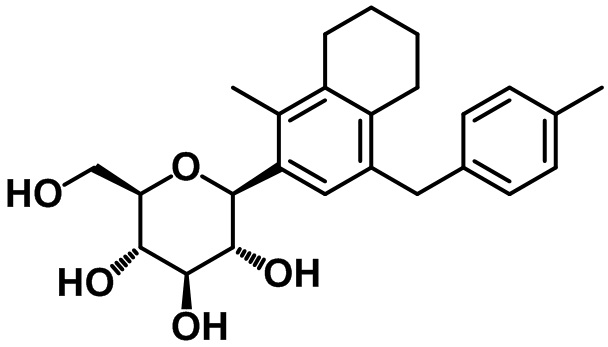
Целевое соединение получали с помощью соединения (25-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,13 (s, 1H), 7,02 (d, J = 7,6 Гц, 2H), 6,97 (d, J = 8,4 Гц, 2H), 4,51 (d, J = 9,6 Гц, 1H), 3,86-3,89 (m, 3H), 3,67 (dd, J = 11,6, 6,0 Гц, 1H), 3,60 (t, J = 9,2 Гц, 1H), 3,51 (t, J = 8,8 Гц, 1H), 3,42-3,40 (m, 2H), 2,65 (t, J = 6,4 Гц, 2H), 2,54 (t, J = 6,4 Гц, 2H), 2,26 (s, 3H), 2,24 (s, 3H), 1,75-1,64 (m, 4H)
Примеры 26-29
Целевые соединения из примеров 26-29 получали посредством способа, показанного в примере 25.
Пример 26. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-трифторметил)бензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола
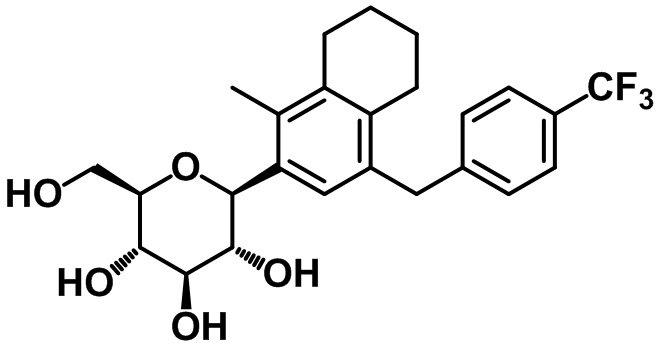
1H ЯМР (400 МГц, CD3OD); δ 7,51 (d, J = 7,6 Гц, 2H), 7,29 (d, J = 8,0 Гц, 2H), 7,18 (s, 1H), 4,52 (d, J = 9,2 Гц, 1H), 4,03 (s, 2H), 3,87 (dd, J = 11,6, 5,6 Гц, 1H), 3,69-3,65 (m, 1H), 3,59 (t, J = 8,8 Гц, 1H), 3,52 (t, J = 8,8 Гц, 1H), 3,42-3,40 (m, 2H), 2,66 (t, J = 6,0 Гц, 2H), 2,51 (t, J = 5,6 Гц, 2H), 2,25 (s, 3H), 1,76-1,65 (m, 4H)
Пример 27. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-трифторметокси)бензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола
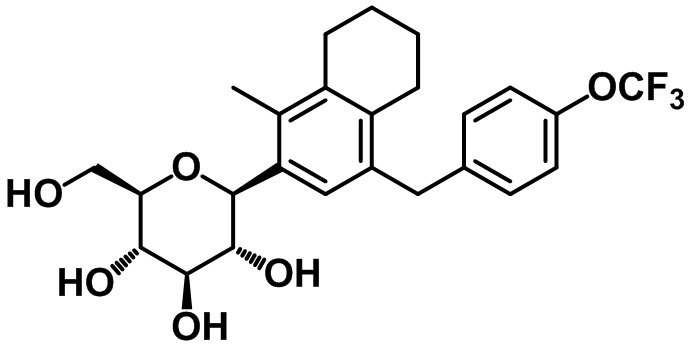
1H ЯМР (400 МГц, CD3OD); δ 7,19 (d, J = 8,4 Гц, 2H), 7,16 (s, 1H), 7,12 (d, J = 8,4 Гц, 2H), 4,52 (d, J = 9,6 Гц, 1H), 3,97 (s, 2H), 3,87 (dd, J = 11,6, 2,0 Гц, 1H), 3,67 (dd, J = 11,6, 5,6 Гц, 1H), 3,58 (t, J = 9,2 Гц, 1H), 3,51 (t, J = 7,6 Гц, 1H), 3,42-3,40 (m, 2H), 2,66 (t, J = 6,0 Гц, 2H), 2,53 (t, J = 6,0 Гц, 2H), 2,25 (s, 3H), 1,76-1,66 (m, 4H)
Пример 28. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(1-метил-4-(4-(метилтио)бензил)-5,6,7,8-тетрагидронафтален-2-ил)тетрагидро-2H-пиран-3,4,5-триола
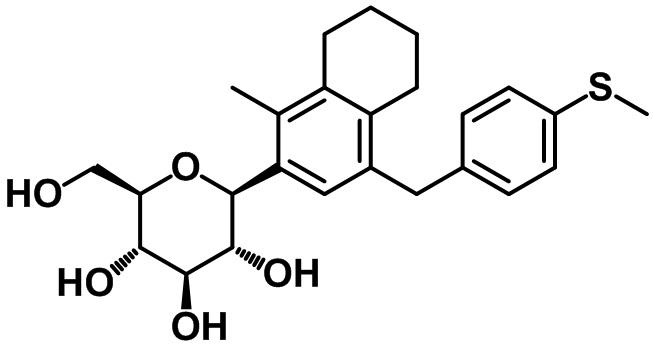
1H ЯМР (400 МГц, CD3OD); δ 7,14 (d, J = 8,0 Гц, 2H), 7,14 (s, 1H), 7,04 (d, J = 8,4 Гц, 2H), 4,51 (d, J = 9,6 Гц, 1H), 3,86-3,90 (m, 3H), 3,67 (dd, J = 11,6, 5,6 Гц, 1H), 3,59 (t, J = 9,2 Гц, 1H), 3,51 (t, J = 8,8 Гц, 1H), 3,39-3,41 (m, 2H), 2,66 (t, J = 6,0 Гц, 2H), 2,54 (t, J = 5,6 Гц, 2H), 2,42 (s, 3H), 2,25 (s, 3H), 1,75-1,65 (m, 4H)
Пример 29. Получение (2S,3R,4R,5S,6R)-2-(4-(4-хлорбензил)-1-метил-5,6,7,8-тетрагидронафтален-2-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
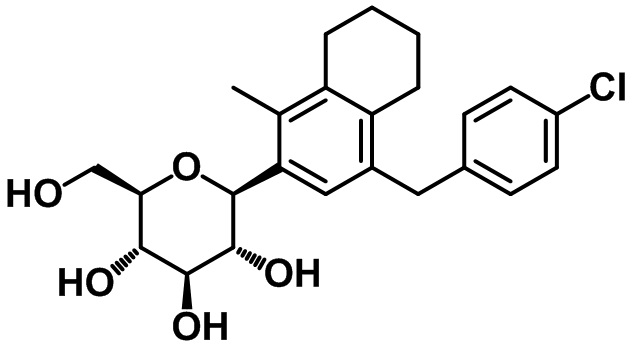
1H ЯМР (400 МГц, CD3OD); δ 7,20 (d, J = 8,4 Гц, 2H), 7,14 (s, 1H), 7,08 (d, J = 8,8 Гц, 2H), 4,51 (d, J = 9,6 Гц, 1H), 3,92 (s, 2H), 3,87 (dd, J = 11,6, 2,0 Гц, 1H), 3,67 (dd, J = 11,6, 5,6 Гц, 1H), 3,59 (t, J = 9,2 Гц, 1H), 3,52 (t, J = 8,8 Гц, 1H), 3,42-3,40 (m, 2H), 2,66 (t, J = 6,0 Гц, 2H), 2,52 (t, J = 6,0 Гц, 2H), 2,25 (s, 3H), 1,76-1,65 (m, 4H)
Пример 30. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез метил-2-гидрокси-4-метилбензоата (30-1)
 (30-1)
(30-1)
SOCl2 (10,9 мл, 150 ммоль) добавляли по каплям в раствор 4-метилсалициловой кислоты (5,0 г, 32,9 ммоль, реагент TCI) в MeOH (80 мл) при 0ºC в атмосфере азота. Полученную смесь перемешивали с обратным холодильником в течение ночи. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (30-1) (5,18 г, 31,2 ммоль, 95%).
1H ЯМР (400 МГц, CDCl3); δ 10,70 (s, 1H), 7,71 (d, J = 8,0 Гц, 1H), 6,79 (s, 1H), 6,69 (d, J = 8,4 Гц, 1H), 3,93 (s, 3H), 2,34 (s, 3H)
Стадия 2. Синтез метил-2-(аллилокси)-4-метилбензоата (30-2)
 (30-2)
(30-2)
Аллилбромид (3,2 мл, 37,4 ммоль) добавляли по каплям к смеси соединения (30-1) (5,18 г, 31,2 ммоль) в DMF (40 мл), а также K2CO3 (5,17 г, 37,4 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего реакцию в ней завершали с помощью воды. Водный слой экстрагировали с помощью EtOAc, после чего объединенный органический слой промывали солевым раствором, таким образом, полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (30-2) (6,35 г, 30,8 ммоль, 99%).
1H ЯМР (400 МГц, CDCl3); δ 7,73 (d, J = 8,0 Гц, 1H), 6,79 (d, J = 8,4 Гц, 1H), 6,76 (s, 1H), 6,15-6,02 (m, 1H), 5,53 (dd, J = 17,2, 1,6 Гц, 1H), 5,30 (dd, J = 10,4, 1,6 Гц, 1H), 4,61 (dd, J = 3,2, 1,6 Гц, 2H), 3,88 (s, 3H), 2,36 (s, 3H)
Стадия 3. Синтез метил-3-аллил-2-гидрокси-4-метилбензоата (30-3)
 (30-3)
(30-3)
Соединение (30-2) (6,65 г, 32,2 ммоль) перемешивали в микроволновом реакторе при 250ºC в течение 1 часа. Неочищенное соединение (30-3) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 11,07 (s, 1H), 7,63 (d, J = 8,4 Гц, 1H), 6,71 (d, J = 8,4 Гц, 1H), 5,99-5,88 (m, 1H), 4,99 (dd, J = 10,0, 1,6 Гц, 1H), 4,93 (dd, J = 17,2, 2,0 Гц, 1H), 3,93 (s, 3H), 3,46 (dt, J = 5,6, 1,6 Гц, 2H), 2,32 (s, 3H)
Стадия 4. Синтез метил-2-гидрокси-4-метил-3-(2-оксоэтил)бензоата (30-4)
 (30-4)
(30-4)
N-оксид N-метилморфолина (3,51 г, 30,0 ммоль) и OsO4 (1,3 мл, 0,200 ммоль, 4 вес. % в H2O) добавляли в раствор соединения (30-3) (2,06 г, 10,0 ммоль) в смеси THF/вода (24 мл/8 мл) в атмосфере азота. После перемешивания полученной реакционной смеси при комнатной температуре в течение 8 часов реакцию с реагентом завершали с помощью насыщенного водного раствора Na2S2O3 с проведением экстракции полученной смеси с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом с получением неочищенного промежуточного соединения, которое применяли без дополнительной очистки. NaIO4 (10,7 г, 50,0 ммоль) добавляли к раствору неочищенного промежуточного соединения в смеси THF/вода (48 мл/16 мл) при комнатной температуре в атмосфере азота. После перемешивания полученной реакционной смеси при комнатной температуре в течение 5 часов реакцию с реагентом завершали с помощью насыщенного водного раствора Na2S2O3 с проведением экстракции полученной смеси с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом, после чего полученный концентрат очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (30-4) (2,00 г, 9,61 ммоль, 96%).
1H ЯМР (400 МГц, CDCl3); δ 11,16 (s, 1H), 9,70 (t, J = 1,6 Гц, 1H), 7,71 (d, J = 8,0 Гц, 1H), 6,77 (d, J = 8,0 Гц, 1H), 3,95 (s, 3H), 3,81 (s, 2H), 2,29 (s, 3H)
Стадия 5. Синтез метил-2-гидрокси-3-(2-гидроксиэтил)-4-метилбензоата (30-5)
 (30-5)
(30-5)
NaBH4 (436 мг, 11,5 ммоль) добавляли в раствор соединения (30-4) (2,00 г, 9,61 ммоль) в EtOH (30 мл) при 0ºC в атмосфере азота. После перемешивания полученной реакционной смеси при 0ºC в течение 1 часа реакцию с реагентом завершали с помощью насыщенного водного раствора NH4Cl с проведением экстракции полученной смеси с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (30-5) (1,82 г, 9,04 ммоль, 94%).
1H ЯМР (400 МГц, CDCl3); δ 11,19 (s, 1H), 7,63 (d, J = 8,4 Гц, 1H), 6,73 (d, J = 8,4 Гц, 1H), 3,94 (s, 3H), 3,84 (dd, J = 12,0, 6,4 Гц, 2H), 3,01 (t, J = 6,4 Гц, 2H), 2,37 (s, 3H)
Стадия 6. Синтез метил-4-метил-2,3-дигидробензофуран-7-карбоксилата (30-6)
 (30-6)
(30-6)
DIAD (1,7 мл, 8,66 ммоль) медленно добавляли по каплям к смеси соединения (30-5) (910 мг, 4,33 ммоль) в THF (30 мл), а также PPh3 (2,27 г, 8,66 ммоль) при 0ºC в атмосфере азота. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (30-6) (813 мг, 4,23 ммоль, 98%).
1H ЯМР (400 МГц, CDCl3); δ 7,65 (d, J = 8,0 Гц, 1H), 6,70 (d, J = 8,0 Гц, 1H), 4,74 (t, J = 8,8 Гц, 2H), 3,89 (s, 3H), 3,13 (t, J = 8,8 Гц, 2H), 2,28 (s, 3H)
Стадия 7. Синтез метил-5-бром-4-метил-2,3-дигидробензофуран-7-карбоксилата (30-7)
 (30-7)
(30-7)
Br2 (0,66 мл, 12,8 ммоль) добавляли по каплям в раствор соединения (30-6) (1,23 г, 6,40 ммоль) в AcOH (20 мл) при комнатной температуре. Полученную смесь перемешивали в течение ночи при комнатной температуре, после чего реакцию с реагентом завершали с помощью насыщенного раствора Na2S2O3 с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (30-7) (1,58 г, 5,83 ммоль, 91%).
1H ЯМР (400 МГц, CDCl3); δ 7,92 (s, 1H), 4,76 (t, J = 8,8 Гц, 2H), 3,89 (s, 3H), 3,19 (t, J = 8,8 Гц, 2H), 2,33 (s, 3H)
Стадия 8. Синтез 5-бром-4-метил-2,3-дигидробензофуран-7-карбоновой кислоты (30-8)
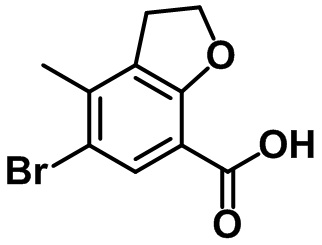 (30-8)
(30-8)
LiOH.H2O (489 мг, 11,7 ммоль) добавляли в раствор соединения (30-7) (1,58 г, 5,83 ммоль) в смеси THF/MeOH/вода (12 мл/4 мл/4 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (30-8) (1,02 г, 3,97 ммоль, 68%).
1H ЯМР (400 МГц, CD3OD); δ 7,81 (s, 1H), 4,70 (t, J = 8,8 Гц, 2H), 3,23 (t, J = 8,8 Гц, 2H), 2,34 (s, 3H)
Стадия 9. Синтез 5-бром-7-(4-метоксибензил)-4-метил-2,3-дигидробензофурана (30-9)
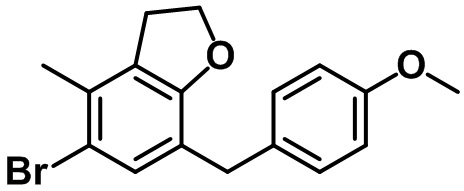 (30-9)
(30-9)
Указанное в заголовке соединение (30-9) получали с помощью соединения (30-8) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 7,13 (d, J = 8,4 Гц, 2H), 7,04 (s, 1H), 6,82 (d, J = 8,4 Гц, 2H), 4,59 (t, J = 8,4 Гц, 2H), 3,78 (s, 5H), 3,16 (t, J = 8,8 Гц, 2H), 2,25 (s, 3H)
Стадия 10. Синтез целевого соединения
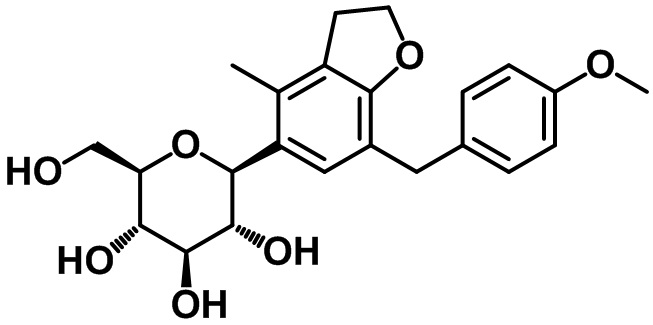
Целевое соединение получали с помощью соединения (30-9) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,11 (d, J = 8,4 Гц, 2H), 7,02 (s, 1H), 6,76 (d, J = 8,8 Гц, 2H), 4,54 (t, J = 8,8 Гц, 2H), 4,36 (d, J = 9,2 Гц, 1H), 3,86-3,83 (m, 1H), 3,77 (s, 2H), 3,73 (s, 3H), 3,66-3,62 (m, 1H), 3,51-3,44 (m, 2H), 3,37-3,35 (m, 2H), 3,14 (t, J = 8,8 Гц, 2H), 2,27 (s, 3H)
Пример 31. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-метил-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
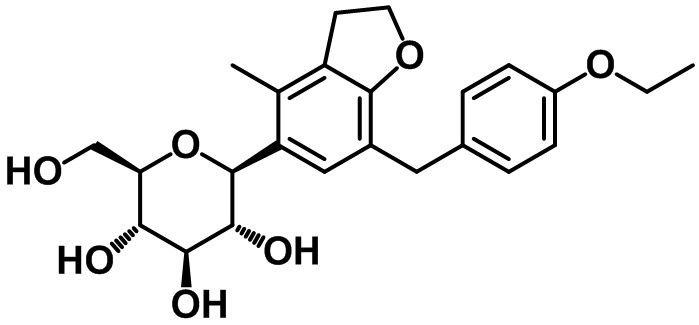
Целевое соединение получали посредством способа, показанного в примере 30.
1H ЯМР (400 МГц, CD3OD); δ 7,10 (d, J = 8,4 Гц, 2H), 7,03 (s, 1H), 6,75 (d, J = 8,8 Гц, 2H), 4,53 (t, J = 8,8 Гц, 2H), 4,36 (d, J = 9,2 Гц, 1H), 3,96 (q, J = 7,2 Гц, 2H), 3,85 (d, J = 11,6 Гц, 1H), 3,76 (s, 2H), 3,66-3,62 (m, 1H), 3,54-3,45 (m, 2H), 3,37-3,35 (m, 2H), 3,13 (t, J = 8,6 Гц, 2H), 2,26 (s, 3H), 1,34 (t, J = 6,8 Гц, 3H)
Пример 32. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-(метилтио)бензил)-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 5-бром-4-метил-7-(4-(метилтио)бензил)-2,3-дигидробензофурана (32-1)
 (32-1)
(32-1)
Указанное в заголовке соединение (32-1) получали с помощью соединения (30-8), полученного на стадии 8 примера 30, посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,18 (d, J = 8,8 Гц, 2H), 7,14 (d, J = 8,4 Гц, 2H), 7,04 (s, 1H), 4,59 (t, J = 8,8 Гц, 2H), 3,79 (s, 2H), 3,17 (t, J = 8,8 Гц, 2H), 2,46 (s, 3H), 2,25 (s, 3H)
Стадия 2. Синтез целевого соединения
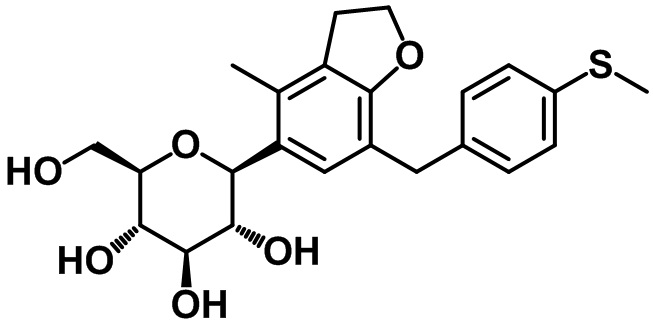
Целевое соединение получали с помощью соединения (32-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,16-7,11 (m, 4H), 7,04 (s, 1H), 4,54 (t, J = 8,4 Гц, 2H), 4,36 (d, J = 8,8 Гц, 1H), 3,85 (d, J = 12,0 Гц, 1H), 3,80 (s, 2H), 3,64 (dd, J = 12,0, 5,2 Гц, 1H), 3,54-3,45 (m, 2H), 3,38-3,36 (m, 2H), 3,14 (t, J = 8,4 Гц, 2H), 2,42 (s, 3H), 2,27 (s, 3H)
Примеры 33 и 34
Целевые соединения из примеров 33 и 34 получали посредством способа, показанного в примере 32.
Пример 33. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-метил-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
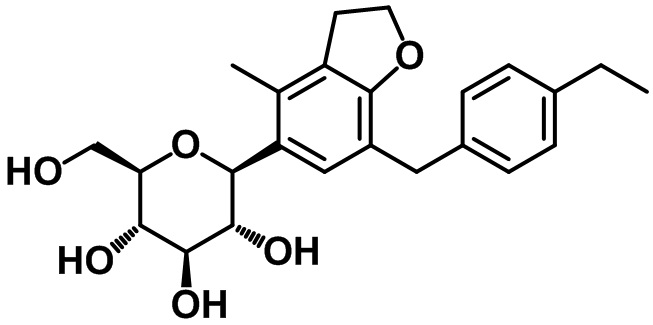
1H ЯМР (400 МГц, CD3OD); δ 7,11 (d, J = 7,2 Гц, 2H), 7,05 (s, 3H), 4,54 (t, J = 8,4 Гц, 2H), 4,36 (d, J = 9,2 Гц, 1H), 3,86-3,80 (m, 3H), 3,68-3,61 (m, 1H), 3,46-3,54 (m, 2H), 3,38 (s, 2H), 3,14 (t, J = 8,4 Гц, 2H), 2,58 (q, J = 7,6 Гц, 2H), 2,27 (s, 3H), 1,18 (t, J = 7,2 Гц, 3H)
Пример 34. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-винилбензил)-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола
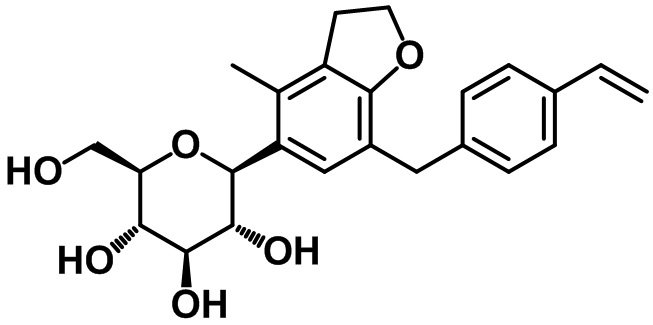
1H ЯМР (400 МГц, CD3OD); δ 7,27 (d, J = 7,6 Гц, 2H), 7,16 (d, J = 8,0 Гц, 2H), 7,04 (s, 1H), 6,66 (dd, J = 17,6, 11,2 Гц, 1H), 5,68 (dd, J = 17,6, 1,2 Гц, 1H), 5,13 (dd, J = 10,8, 0,8 Гц, 1H), 4,54 (t, J = 8,8 Гц, 2H), 4,36 (d, J = 9,2 Гц, 1H), 3,86-3,83 (m, 3H), 3,66-3,53 (m, 1H), 3,51-3,44 (m, 2H), 3,39-3,34 (m, 2H), 3,14 (t, J = 8,8 Гц, 2H), 2,27 (s, 3H)
Пример 35. Получение (2S,3R,4R,5S,6R)-2-(4-хлор-7-(4-этоксибензил)-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез метил-4-хлор-2-гидроксибензоата (35-1)
 (35-1)
(35-1)
SOCl2 (12,6 мл, 174 ммоль) добавляли по каплям в раствор 4-хлорсалициловой кислоты (10,0 г, 58,0 ммоль, реагент TCI) в MeOH (200 мл) при 0ºC в атмосфере азота. Полученную смесь перемешивали с обратным холодильником в течение 4 часов. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Насыщенный водный раствор NaHCO3 медленно добавляли к полученному остатку, после чего водный слой экстрагировали с помощью EtOAc. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (35-1) (7,6 г, 40,7 ммоль, 70%).
1H ЯМР (400 МГц, CDCl3); δ 7,76 (d, J = 8,8 Гц, 1H), 7,01 (d, J = 2,0 Гц, 1H), 6,87 (dd, J = 8,4, 2,0 Гц, 1H), 3,95 (s, 3H)
Стадия 2. Синтез метил-2-(аллилокси)-4-хлорбензоата (35-2)
 (35-2)
(35-2)
Аллилбромид (5,3 мл, 61,0 ммоль) добавляли по каплям к смеси соединения (35-1) (7,59 г, 40,7 ммоль) в DMF (114 мл), а также K2CO3 (8,43 г, 61,0 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего реакцию в ней завершали с помощью воды. Водный слой экстрагировали с помощью EtOAc, после чего органический слой промывали солевым раствором, таким образом, полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (35-2) (8,50 г, 37,5 ммоль, 92%).
1H ЯМР (400 МГц, CDCl3); δ 7,78 (d, J = 8,0 Гц, 1H), 7,26-6,95 (m, 2H), 6,07-6,02 (m, 1H), 5,53 (dd, J = 17,2, 1,6 Гц, 1H), 5,34 (dd, J = 10,4, 1,2 Гц, 1H), 4,63-4,61 (m, 2H), 3,89 (s, 3H)
Стадия 3. Синтез метил-3-аллил-4-хлор-2-гидроксибензоата (35-3)
 (35-3)
(35-3)
Соединение (35-2) (2,10 г, 9,27 ммоль) перемешивали в микроволновом реакторе при 250ºC в течение 1 часа. Неочищенное соединение (35-3) (2,01 г, 8,87 ммоль, 96%) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 11,26 (s, 1H), 7,66 (d, J = 8,8 Гц, 1H), 6,92 (d, J = 8,8 Гц, 1H), 5,98-5,91 (m, 1H), 5,07-5,02 (m, 2H), 3,95 (s, 3H), 3,59 (d, J = 6,0 Гц, 2H)
Стадия 4. Синтез метил-4-хлор-2-гидрокси-3-(2-оксоэтил)бензоата (35-4)
 (35-4)
(35-4)
N-оксид N-метилморфолина (1,55 г, 13,2 ммоль) и OsO4 (22,4 мл, 0,09 ммоль) добавляли в раствор соединения (35-3) (2,00 г, 8,82 ммоль) в смеси ацетон/вода (30 мл/3 мл) в атмосфере азота. После перемешивания полученной реакционной смеси при комнатной температуре в течение 8 часов реакцию с реагентом завершали с помощью насыщенного водного раствора Na2S2O3 с проведением экстракции полученной смеси с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом с получением неочищенного промежуточного соединения, которое применяли без дополнительной очистки. NaIO4 (5,61 г, 26,3 ммоль) добавляли к раствору неочищенного промежуточного соединения в смеси THF/вода (50 мл/30 мл) при комнатной температуре в атмосфере азота. После перемешивания полученной реакционной смеси при комнатной температуре в течение 5 часов реакцию с реагентом завершали с помощью насыщенного водного раствора Na2S2O3 с проведением экстракции полученной смеси с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом с получением неочищенного соединения (35-4) (1,90 г, 8,31 ммоль, 95%), которое применяли без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 11,32 (s, 1H), 9,73 (s, 1H), 7,75 (d, J = 8,4 Гц, 1H), 6,98 (d, J = 8,4 Гц, 1H), 3,97 (s, 5H)
Стадия 5. Синтез метил-4-хлор-2-гидрокси-3-(2-гидроксиэтил)бензоата (35-5)
 (35-5)
(35-5)
NaBH4 (628 мг, 16,6 ммоль) добавляли в раствор соединения (35-4) (1,90 г, 8,31 ммоль) в MeOH (30 мл) при 0ºC в атмосфере азота. После перемешивания полученной реакционной смеси при 0ºC в течение 1 часа реакцию с реагентом завершали с помощью насыщенного водного раствора NH4Cl с проведением экстракции полученной смеси с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (35-5) (1,45 г, 6,29 ммоль, 76%).
1H ЯМР (400 МГц, CDCl3); δ 11,37 (s, 1H), 7,67 (d, J = 8,8 Гц, 1H), 6,94 (d, J = 8,8 Гц, 1H), 3,96 (s, 3H), 3,87 (dd, J = 12,8, 6,0 Гц, 2H), 3,16 (t, J = 6,4 Гц, 2H)
Стадия 6. Синтез метил-4-хлор-2,3-дигидробензофуран-7-карбоксилата (35-6)
 (35-6)
(35-6)
DIAD (2,47 мл, 12,6 ммоль) медленно добавляли по каплям к смеси соединения (35-5) (1,45 г, 6,29 ммоль) в THF (30 мл), а также PPh3 (3,30 г, 12,6 ммоль) при 0ºC в атмосфере азота. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (35-6) (1,31 г, 6,16 ммоль, 98%).
1H ЯМР (400 МГц, CDCl3); δ 7,69 (d, J = 8,4 Гц, 1H), 6,88 (d, J = 8,4 Гц, 1H), 4,79 (t, J = 8,8 Гц, 2H), 3,90 (s, 3H), 3,27 (t, J = 8,8 Гц, 2H)
Стадия 7. Синтез метил-5-бром-4-хлор-2,3-дигидробензофуран-7-карбоксилата (35-7)
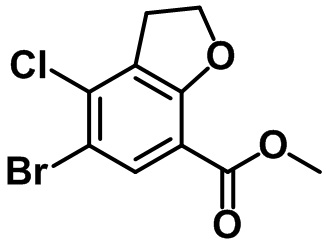 (35-7)
(35-7)
Br2 (0,4 мл, 8,01 ммоль) добавляли по каплям в раствор соединения (35-6) (1,31 г, 6,16 ммоль) в AcOH (20 мл) при комнатной температуре. Полученную смесь перемешивали в течение ночи при комнатной температуре, после чего реакцию с реагентом завершали с помощью насыщенного раствора Na2S2O3 с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение (35-7) (1,70 г, 5,83 ммоль, 95%) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 8,00 (s, 1H), 4,81 (t, J = 8,8 Гц, 2H), 3,91 (s, 3H), 3,31 (t, J = 8,8 Гц, 2H)
Стадия 8. Синтез 5-бром-4-хлор-2,3-дигидробензофуран-7-карбоновой кислоты (35-8)
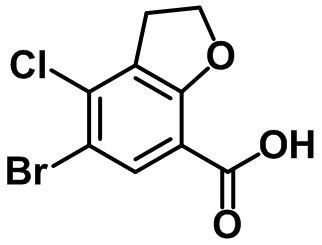 (35-8)
(35-8)
LiOH.H2O (490 мг, 11,2 ммоль) добавляли в раствор соединения (35-7) (1,70 г, 5,83 ммоль) в смеси THF/MeOH/вода (15 мл/5 мл/5 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения реакции летучее вещество удаляли при пониженном давлении. Водный раствор 1 н. HCl добавляли к остатку с проведением подкисления, в ходе которого полученную смесь перемешивали с осаждением неочищенного продукта. Неочищенный продукт фильтровали, промывали с помощью воды и высушивали в высоком вакууме с получением указанного в заголовке соединения (35-8) (1,54 г, 5,54 ммоль, 95%).
1H ЯМР (400 МГц, CD3OD); δ 7,93 (s, 1H), 4,76 (t, J = 8,8 Гц, 2H), 3,35-3,30 (m, 2H)
Стадия 9. Синтез целевого соединения
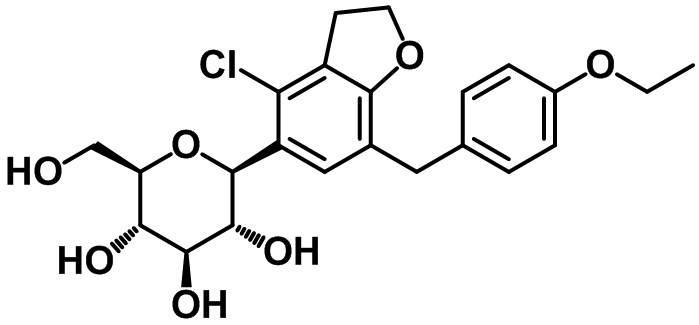
Целевое соединение получали с помощью соединения (35-8) посредством способа, показанного на стадиях 4-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,14-7,10 (m, 3H), 6,77 (d, J = 8,4 Гц, 2H), 4,63-4,59 (m, 3H), 3,97 (q, J = 6,8 Гц, 2H), 3,86-3,78 (m, 3H), 3,68-3,64 (m, 1H), 3,49-3,47 (m, 2H), 3,39-3,37 (m, 2H), 3,25 (t, J = 8,8 Гц, 2H), 1,35 (t, J = 6,8 Гц, 3H)
Пример 36. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-(4-метоксибензил)-7-метил-2,3-дигидробензофуран-6-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 6-бром-7-метил-2,3-дигидробензофуран-4-карбоновой кислоты (36-1)
 (36-1)
(36-1)
Указанное в заголовке соединение (36-1) получали с помощью 3-гидрокси-4-метилбензойной кислоты (реагент TCI) посредством способа, показанного на стадиях 1-8 примера 30.
1H ЯМР (400 МГц, CDCl3); δ 7,15 (s, 1H), 4,52 (t, J = 8,8 Гц, 2H), 3,36 (t, J = 8,8 Гц, 2H), 2,20 (s, 3H)
Стадия 2. Синтез 6-бром-4-(4-метоксибензил)-7-метил-2,3-дигидробензофурана (36-2)
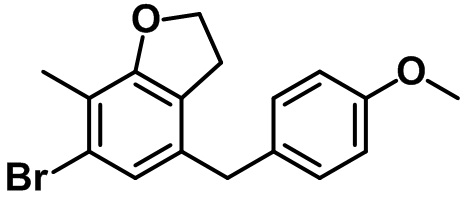 (36-2)
(36-2)
Указанное в заголовке соединение в соответствии с названием изобретения (36-2) получали с помощью соединения (36-1) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 7,18 (s, 1H), 7,05 (d, J = 8,8 Гц, 2H), 6,80 (d, J = 8,8 Гц, 2H), 4,52 (t, J = 8,8 Гц, 2H), 4,00 (s, 2H), 3,77 (s, 3H), 3,04 (t, J = 8,8 Гц, 2H), 2,16 (s, 3H)
Стадия 3. Синтез целевого соединения
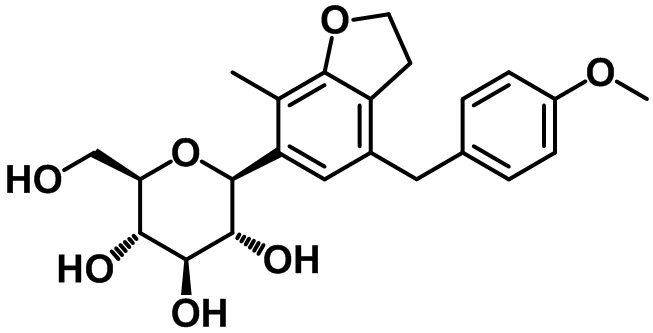
Целевое соединение получали с помощью соединения (36-2) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,11 (s, 1H), 7,04 (d, J = 9,2 Гц, 2H), 6,78 (d, J = 8,8 Гц, 2H), 4,47 (t, J = 9,2 Гц, 2H), 4,35 (d, J = 9,6 Гц, 1H), 4,13 (d, J = 12,0 Гц, 1H), 3,89 (d, J = 16,0 Гц, 1H), 3,73 (dd, J = 12,0, 2,4 Гц, 1H), 3,73 (s, 3H), 3,62-3,56 (m, 2H), 3,44-3,35 (m, 2H), 3,19-3,15 (m, 1H), 3,04-2,92 (m, 2H), 2,16 (s, 3H)
Пример 37. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-метил-4-(4-винилбензил)-2,3-дигидробензофуран-6-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 6-бром-7-метил-4-(4-винилбензил)-2,3-дигидробензофурана (37-1)
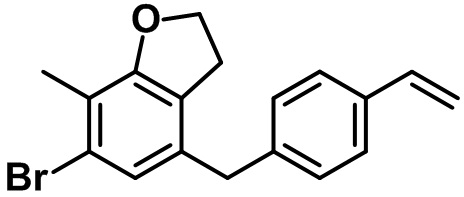 (37-1)
(37-1)
Указанное в заголовке соединение (37-1) получали с помощью соединения (36-1), полученного на стадии 1 примера 36, посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,31 (d, J = 7,2 Гц, 2H), 7,18 (s, 1H), 7,09 (d, J = 7,2 Гц, 2H), 6,68 (dd, J = 17,6, 10,8 Гц, 1H), 5,69 (d, J = 17,6 Гц, 1H), 5,41 (d, J = 10,8 Гц, 1H), 4,53 (t, J = 8,8 Гц, 2H), 4,06 (s, 2H), 3,09 (t, J = 8,8 Гц, 2H), 2,17 (s, 3H)
Стадия 2. Синтез целевого соединения
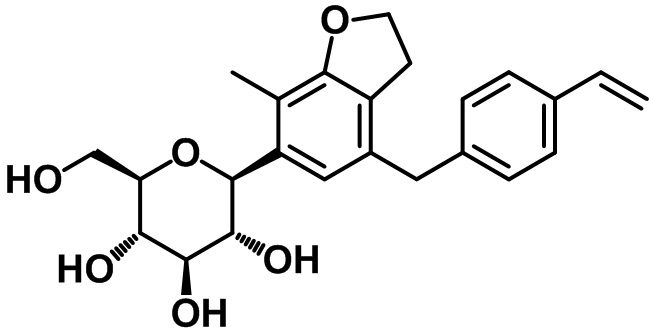
Целевое соединение получали с помощью соединения (37-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,29 (d, J = 8,4 Гц, 2H), 7,11 (s, 1H), 7,09 (d, J = 7,6 Гц, 2H), 6,67 (dd, J = 17,6, 10,8 Гц, 1H), 5,69 (d, J = 17,6 Гц, 1H), 5,14 (d, J = 10,8 Гц, 1H), 4,48 (t, J = 8,8 Гц, 2H), 4,33 (d, J = 9,6 Гц, 1H), 4,19 (d, J = 16,0 Гц, 1H), 3,95 (d, J = 16,4 Гц, 1H), 3,71 (dd, J = 12,0, 2,4 Гц, 1H), 3,61-3,52 (m, 2H), 3,43-3,35 (m, 2H), 3,17-3,14 (m, 1H), 3,07-2,92 (m, 2H), 2,16 (s, 3H)
Пример 38. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(8-метокси-5-(4-метоксибензил)хроман-7-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез метил-3-(аллилокси)-4-метоксибензоата (38-1)
 (38-1)
(38-1)
Аллилбромид (2,8 мл, 32,9 ммоль) добавляли по каплям к смеси метилизованилата (5,00 г, 27,4 ммоль, реагент TCI) в DMF (30 мл), а также K2CO3 (4,55 г, 32,9 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего реакцию в ней завершали с помощью воды. Водный слой экстрагировали с помощью EtOAc, после чего органический слой промывали солевым раствором, таким образом, полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (38-1) (5,80 г, 26,1 ммоль, 95%).
1H ЯМР (400 МГц, CDCl3); δ 7,68 (dd, J = 8,0, 2,0 Гц, 1H), 7,56 (s, 1H), 6,90 (d, J = 8,8 Гц, 1H), 6,17-6,04 (m, 1H), 5,44 (dd, J = 17,6, 1,2 Гц, 1H), 5,31 (dd, J = 10,4, 1,2 Гц, 1H), 4,66 (d, J = 5,6 Гц, 2H), 3,93 (s, 3H), 3,89 (s, 3H)
Стадия 2. Синтез метил-2-аллил-3-гидрокси-4-метоксибензоата (38-2)
 (38-2)
(38-2)
Соединение (38-1) (1,00 г, 4,50 ммоль) перемешивали в микроволновом реакторе при 250ºC в течение 1 часа. Неочищенное соединение (38-2) (0,99 г, 4,45 ммоль, 99%) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 7,52 (d, J = 8,4 Гц, 1H), 6,76 (d, J = 8,8 Гц, 1H), 6,08-5,98 (m, 1H), 5,77 (s, 1H), 5,04-4,97 (m, 2H), 3,94 (s, 3H), 3,85 (s, 3H), 3,82 (d, J = 6,0 Гц, 2H)
Стадия 3. Синтез метил-3-гидрокси-2-(3-гидроксипропил)-4-метоксибензоата (38-3)
 (38-3)
(38-3)
Комплекс BH3.SMe2 (1,0 мл, 10,0 ммоль, 10,0 M в метилсульфиде) медленно добавляли в раствор соединения (38-2) (1,91 г, 8,59 ммоль) в THF (40 мл) при -10ºC в атмосфере азота, после чего реакционную смесь перемешивали при комнатной температуре в течение 1 часа. К ней медленно добавляли раствор H2O2 (1,2 мл) в насыщенном растворе NaHCO3 (20 мл). Полученную реакционную смесь охлаждали при 0ºC и перемешивали в течение 30 минут. К полученной смеси добавляли EtOAc. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение (38-3) (2,06 г, 8,57 ммоль, 99%) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CD3OD); δ 7,44 (d, J = 8,4 Гц, 1H), 6,85 (d, J = 8,8 Гц, 1H), 3,91 (s, 3H), 3,84 (s, 3H), 3,57 (t, J = 6,8 Гц, 2H), 3,03 (t, J = 7,6 Гц, 2H), 1,85-1,77 (m, 2H)
Стадия 4. Синтез метил-8-метоксихроман-5-карбоксилата (38-4)
 (38-4)
(38-4)
DIAD (3,40 мл, 17,15 ммоль) медленно добавляли к смеси соединения (38-3) (2,06 г, 8,57 ммоль) в THF (20 мл), а также PPh3 (4,5 г, 17,2 ммоль) при 0ºC в атмосфере азота. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции летучий растворитель выпаривали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (38-4) (1,87 г, 8,41 ммоль, 98%).
1H ЯМР (400 МГц, CDCl3); δ 7,58 (d, J = 8,4 Гц, 1H), 6,74 (d, J = 8,4 Гц, 1H), 4,27 (t, J = 5,2 Гц, 2H), 3,92 (s, 3H), 3,85 (s, 3H), 3,14 (t, J = 6,4 Гц, 2H), 2,05-1,99 (m, 2H)
Стадия 5. Синтез 8-метоксихроман-5-карбоновой кислоты (38-5)
 (38-5)
(38-5)
Смесь соединения (38-4) (1,87 г, 8,41 ммоль) в THF (5 мл), а также 1 н. водного раствора NaOH (13 мл) перемешивали с обратным холодильником в течение 2 часов. Полученную реакционную смесь охлаждали при комнатной температуре, после чего полученную смесь подкисляли с помощью 1 н. раствора HCl с проведением экстракции с помощью EtOAc. Объединенный органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (38-5) (1,72 г, 8,26 ммоль, 96%).
1H ЯМР (400 МГц, CDCl3); δ 7,59 (d, J = 8,8 Гц, 1H), 6,84 (d, J = 8,8 Гц, 1H), 4,19 (t, J = 5,2 Гц, 2H), 3,86 (s, 3H), 3,12 (t, J = 6,4 Гц, 2H), 2,01-1,95 (m, 2H)
Стадия 6. Синтез 7-бром-8-метоксихроман-5-карбоновой кислоты (38-6)
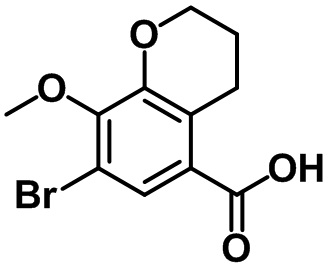 (38-6)
(38-6)
Br2 (0,28 мл, 10,7 ммоль) добавляли по каплям в раствор соединения (38-5) (1,72 г, 8,26 ммоль) в AcOH (20 мл) при комнатной температуре. Полученную смесь перемешивали в течение ночи при комнатной температуре, после чего реакцию с реагентом завершали с помощью насыщенного раствора Na2S2O3 с проведением экстракции с помощью EtOAc. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное соединение (38-6) (1,86 г, 6,18 ммоль, 75%) применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CD3OD); δ 6,99 (s, 1H), 4,19 (t, J = 5,2 Гц, 2H), 3,82 (s, 3H), 2,77 (t, J = 6,4 Гц, 2H), 2,02-1,96 (m, 2H)
Стадия 7. Синтез 7-бром-8-метокси-5-(4-метоксибензил)хромана (38-7)
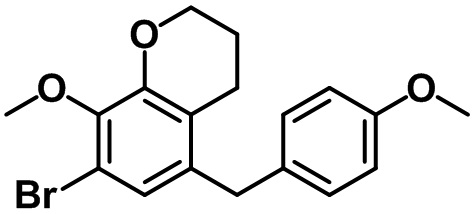 (38-7)
(38-7)
Указанное в заголовке соединение (38-7) получали с помощью соединения (38-6) посредством способа, показанного на стадиях 4-5 примера 1.
1H ЯМР (400 МГц, CDCl3); δ 6,99-6,97 (m, 3H), 6,80 (d, J = 8,8 Гц, 2H), 4,17 (t, J = 4,8 Гц, 2H), 4,06 (s, 2H), 3,87 (s, 3H), 3,77 (s, 3H), 2,60 (t, J = 6,4 Гц, 2H), 1,96-1,91 (m, 2H)
Стадия 8. Синтез целевого соединения
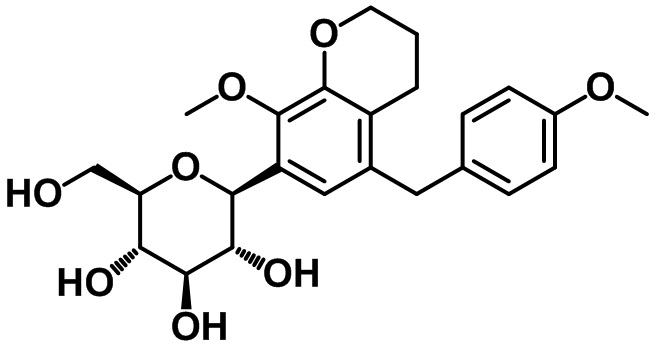
Целевое соединение получали с помощью соединения (38-7) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,01-6,99 (m, 3H), 6,78 (d, J = 8,4 Гц, 2H), 4,38 (d, J = 9,6 Гц, 1H), 4,16-4,06 (m, 3H), 3,91-3,87 (m, 1H), 3,85 (s, 3H), 3,78-3,74 (m, 1H), 3,73 (s, 3H), 3,65-3,57 (m, 2H), 3,43-3,40 (m, 2H), 3,21-3,17 (m, 1H), 2,68-2,62 (m, 1H), 2,53-2,47 (m, 1H), 1,92-1,88 (m, 2H)
Пример 39. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(8-метокси-5-(4-метилбензил)хроман-7-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 7-бром-8-метокси-5-(4-метилбензил)хромана (39-1)
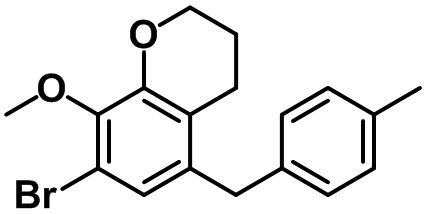 (39-1)
(39-1)
Указанное в заголовке соединение (39-1) получали с помощью соединения (38-6), полученного на стадии 6 примера 38, посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,07 (d, J = 7,6 Гц, 2H), 6,99 (s, 1H), 6,95 (d, J = 8,0 Гц, 2H), 4,17 (t, J = 5,2 Гц, 2H), 4,09 (s, 2H), 3,87 (s, 3H), 2,59 (t, J = 6,4 Гц, 2H), 2,30 (s, 3H), 1,96-1,92 (m, 2H)
Стадия 2. Синтез целевого соединения
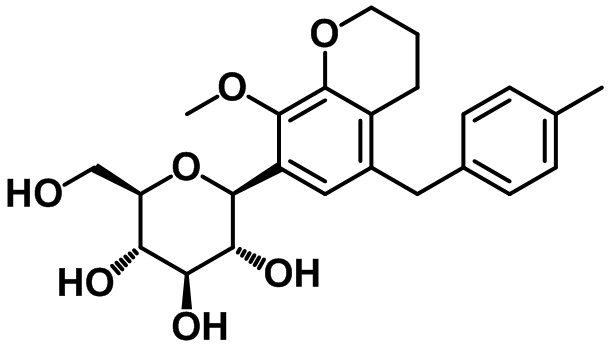
Целевое соединение получали с помощью соединения (39-1) посредством способа, показанного на стадии 4 примера 4.
1H ЯМР (400 МГц, CD3OD); δ 7,03 (d, J = 8,0 Гц, 2H), 6,99 (s, 1H), 6,96 (d, J = 8,0 Гц, 2H), 4,38 (d, J = 9,6 Гц, 1H), 4,17 (d, J = 16,8 Гц, 1H), 4,12-4,03 (m, 2H), 3,88 (d, J = 10,0 Гц, 1H), 3,85 (s, 3H), 3,75 (dd, J = 12,0, 2,4 Гц, 1H), 3,65-3,57 (m, 2H), 3,44-3,39 (m, 2H), 3,20-3,16 (m, 1H), 2,67-2,61 (m, 1H), 2,52-2,44 (m, 1H), 2,26 (s, 3H), 1,91-1,87 (m, 2H)
Пример 40. Получение (2S,3R,4R,5S,6R)-2-(5-(4-этоксибензил)-8-метилхроман-7-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез 7-бром-5-(4-этоксибензил)-8-метилхромана (40-1)
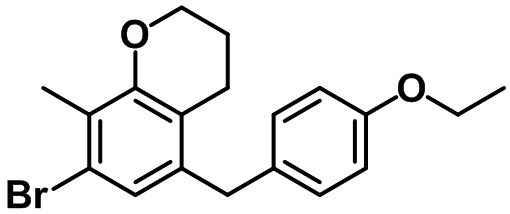 (40-1)
(40-1)
Указанное в заголовке соединение (40-1) получали с помощью 3-гидрокси-4-метилбензойной кислоты (реагент TCI) посредством способа, показанного на стадиях 1-7 примера 38.
1H ЯМР (400 МГц, CDCl3); δ 7,24 (s, 1H), 6,97 (d, J = 8,8 Гц, 2H), 6,78 (d, J = 8,8 Гц, 2H), 4,10 (t, J = 8,8 Гц, 2H), 4,08 (s, 2H), 3,96 (q, J = 6,8 Гц, 2H), 2,60 (t, J = 8,8 Гц, 2H), 2,15 (s, 3H), 1,94-1,87 (m, 2H), 1,39 (t, J = 6,8 Гц, 3H)
Стадия 2. Синтез целевого соединения
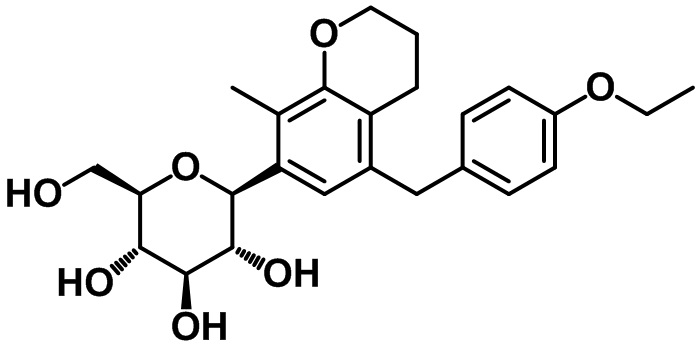
Целевое соединение получали с помощью соединения (40-1) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР (400 МГц, CD3OD); δ 7,14 (s, 1H), 6,98 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,8 Гц, 2H), 4,33 (d, J = 9,2 Гц, 1H), 4,15-4,06 (m, 3H), 3,99-3,90 (m, 3H), 3,74 (dd, J = 12,0, 2,0 Гц, 1H), 3,62-3,57 (m, 2H), 3,40-3,35 (m, 2H), 3,17-3,15 (m, 1H), 2,93 (s, 3H), 2,68-2,47 (m, 2H), 1,90-1,87 (m, 2H), 1,34 (t, J = 6,8 Гц, 3H)
Пример 41. Получение (2S,3R,4R,5S,6R)-2-(4-этил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез (E)-пент-2-еналя (41-1)
 (41-1)
(41-1)
DCM (104 мл) охлаждали при -78ºC, после чего к полученному продукту по каплям добавляли (COCl)2 (24 мл, 278,64 ммоль) и DMSO (2,06 мл, 464,44 ммоль), таким образом, полученную смесь перемешивали в течение 30 минут. Транс-2-пентен-1-ол (16,00 г, 185,76 ммоль) разбавляли в DCM (40 мл), после чего полученный раствор медленно добавляли в реакционную колбу в течение 15 минут, таким образом, полученную смесь перемешивали при той же температуре в течение 30 минут, а затем дополнительно перемешивали в течение 1 часа с повышением температуры до 0ºC. Воду выливали к полученной смеси с завершением реакции и проведением экстракции с помощью диэтилового эфира. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали с получением указанного в заголовке соединения (41-1). Полученное соединение применяли непосредственно в следующей реакции без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); δ 9,52 (d, J = 7,6 Гц, 1H), 6,86 (dt, J = 15,6, 6,2 Гц, 1H), 6,16-6,09 (m, 1H), 2,41-2,34 (m, 2H), 1,13 (t, J = 7,2 Гц, 3H)
Стадия 2. Синтез 5(2E,4E)-этилгепта-2,4-диеноата (41-2)
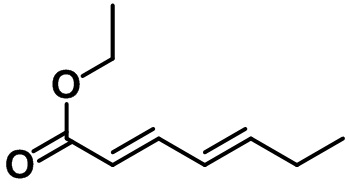 (41-2)
(41-2)
К THF (200 мл) добавляли гидрид натрия (13,00 г, 325,08 ммоль), после чего полученный раствор охлаждали при -78ºC. К полученному продукту в течение 5 минут медленно добавляли триэтилфосфоноацетат (65 мл, 325,08 ммоль), после чего полученную смесь перемешивали при той же температуре в течение 30 минут. Соединение (41-1) в THF (60 мл) медленно добавляли по каплям к полученной смеси, после чего полученную смесь перемешивали в течение 30 минут, а затем дополнительно перемешивали в течение 1 часа с повышением температуры до -40ºC. Полученный продукт разбавляли диэтиловым эфиром, после чего в полученный раствор медленно добавляли насыщенный раствор хлорида аммония, таким образом, полученную смесь перемешивали при комнатной температуре в течение 10 минут. Органический слой промывали дважды солевым раствором, после чего слой полученного продукта высушивали над безводным MgSO4, фильтровали и концентрировали. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (41-2) (21,86 г, 141,75 ммоль, 76%).
1H ЯМР (400 МГц, CDCl3); δ 7,29-7,23 (m, 1H), 6,18-6,10 (m, 2H), 5,79 (d, J = 12,8 Гц, 1H), 4,19(q, J = 6,8 Гц, 2H), 2,24-2,18 (m, 2H), 1,29 (t, J = 7,2 Гц, 3H), 1,05 (t, J = 7,2 Гц, 3H)
Стадия 3. Синтез этил-7-этил-2,3-дигидро-1H-инден-4-карбоксилата (41-3)
 (41-3)
(41-3)
Соединение (41-2) (21,80 г, 141,36 ммоль) и 1-пирролидино-1-циклопентен (22,67 мл, 155,50 мл) растворяли в ксилоле (64 мл), после чего полученный раствор перемешивали с обратным холодильником в течение 24 часов. После охлаждения при комнатной температуре в полученный раствор по каплям добавляли 1 н. HCl с проведением экстракции с помощью EtOAc, после чего полученный экстракт высушивали над безводным MgSO4, фильтровали и концентрировали. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (41-3) (12,20 г, 55,89 ммоль, 59%).
1H ЯМР (400 МГц, CDCl3); δ 7,80 (d, J = 8,0 Гц, 1H), 7,06 (d, J = 8,0 Гц, 1H), 4,34 (q, J = 7,2 Гц, 2H), 3,30 (t, J = 7,6 Гц, 2H), 2,88 (t, J = 7,6 Гц, 2H), 2,08 (q, J = 7,6 Гц, 2H), 2,12-2,04 (m, 2H), 1,38 (t, J = 7,2 Гц, 3H), 0,88 (t, J = 7,2 Гц, 3H)
Стадия 4. Синтез 7-этил-2,3-дигидро-1H-инден-4-карбоновой кислоты (41-4)
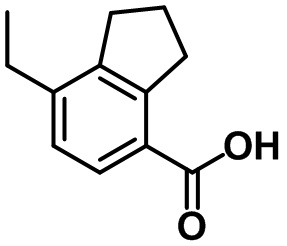 (41-4)
(41-4)
Соединение (41-3) (11,50 г, 52,68 ммоль) растворяли в метаноле (230 мл), после чего 2 н. водный раствор гидроксида натрия (115 мл) добавляли по каплям в полученный раствор, таким образом, полученную смесь перемешивали с обратным холодильником в течение 5 часов. Метанол концентрировали при пониженном давлении, после чего полученный концентрат охлаждали при 0ºC, таким образом, в него медленно добавляли по каплям 1 н. HCl, пока значение pH смешанного раствора не достигало 6. Полученное твердое вещество фильтровали и высушивали в атмосфере азота с получением указанного в заголовке соединения (41-4) (7,60 г, 39,95 ммоль, 76%).
1H ЯМР (400 МГц, CD3OD); δ 7,69 (d, J = 8,0 Гц, 1H), 7,01 (d, J = 8,0 Гц, 1H), 3,21 (t, J = 7,6 Гц, 2H), 2,85 (t, J = 7,6 Гц, 2H), 2,61 (q, J = 7,6 Гц, 2H), 2,07-2,02 (m, 2H), 1,17 (t, J = 7,6 Гц, 3H)
Стадия 5. Синтез 6-бром-7-этил-2,3-дигидро-1H-инден-4-карбоновой кислоты (41-5)
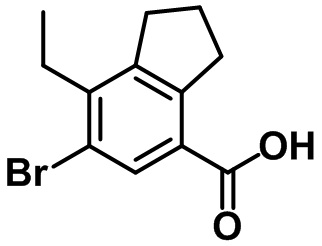 (41-5)
(41-5)
Соединение (41-4) (7,60 г, 39,95 ммоль) растворяли в уксусной кислоте (140 мл), после чего в полученный смешанный раствор по порядку по каплям добавляли азотную кислоту (4,56 мл, 59,92 ммоль) и бром (3,07 мл, 59,92 ммоль). Нитрат серебра (10,18 г, 59,92 ммоль) растворяли в воде (50 мл), после чего полученный раствор медленно добавляли по каплям в реакционную смесь, после чего полученную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь охлаждали при 0ºC, после чего насыщенный раствор тиосульфата натрия медленно добавляли по каплям к полученной смеси с завершением реакции. Полученную смесь экстрагировали дважды с помощью EtOAc, после чего органический слой высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Концентрированный раствор высушивали под вакуумом с получением указанного в заголовке соединения (41-5), которое применяли на следующей стадии без дополнительной очистки.
Стадия 6. Синтез 5-бром-4-этил-7-(4-метилбензил)-2,3-дигидро-1H-индена (41-6)
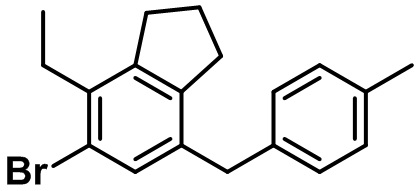 (41-6)
(41-6)
Указанное в заголовке соединение (41-6) получали с помощью соединения (41-5) посредством способа, показанного на стадиях 1-3 примера 4.
1H ЯМР (400 МГц, CDCl3); δ 7,13 (s, 1H), 7,10-7,02 (m, 4H), 3,82 (s, 2H), 2,90 (t, J = 7,6 Гц, 2H), 2,75 (t, J = 7,6 Гц, 2H), 2,71 (q, J = 7,6 Гц, 2H), 2,31 (s, 3H), 2,08-2,01 (m, 2H), 1,25 (t, J = 7,6 Гц, 3H)
Стадия 7. Синтез целевого соединения
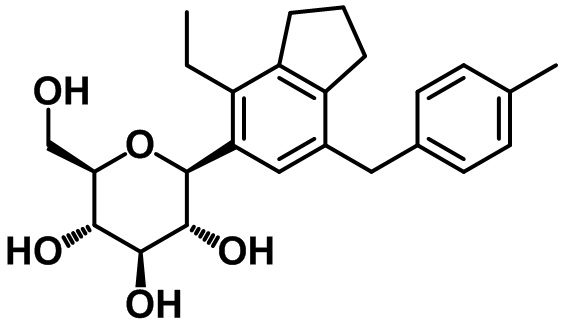
Целевое соединение получали с помощью соединения (41-6) посредством способа, показанного на стадиях 6-7 примера 1.
1H ЯМР(400 МГц, CD3OD); δ 7,08 (s, 1H), 6,98 (s, 4H), 4,39 (d, J = 9,2 Гц, 1H), 3,82-3,79 (m, 3H), 3,63-3,59 (m, 1H), 3,55 (t, J = 9,2 Гц, 1H), 3,47-3,43 (m, 1H), 3,35 (d, J = 6,0 Гц, 2H), 2,84 (t, J = 7,6 Гц, 2H), 2,78-2,71 (m, 1H), 2,67 (t, J = 7,6 Гц, 2H), 2,63-2,58 (m, 1H), 2,22 (s, 3H), 1,99-1,91 (m, 2H), 1,10 (t, J = 7,6 Гц, 3H)
Примеры 42-60
Целевые соединения примеров 42-60 получали посредством способа, показанного в примере 41.
Пример 42. Получение (2S,3R,4R,5S,6R)-2-(4-этил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
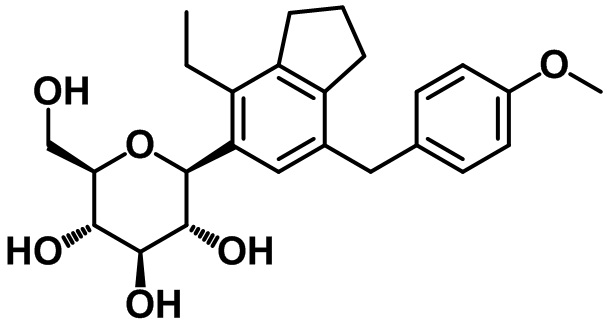
1H ЯМР(400 МГц, CD3OD); δ 7,07 (s, 1H), 7,02 (d, J = 8,8 Гц, 2H), 6,75 (d, J = 8,8 Гц, 2H), 4,41 (d, J = 9,6 Гц, 1H), 3,83-3,80 (m, 3H), 3,70 (s, 3H), 3,61 (dd, J = 11,2, 3,7 Гц, 1H), 3,55 (t, J = 9,2 Гц, 1H), 3,47-3,43 (m, 1H), 3,35 (d, J = 5,2 Гц, 2H), 2,84 (t, J = 7,6 Гц, 2H), 2,78-2,73 (m, 1H), 2,68 (t, J = 7,6 Гц, 2H), 2,64-2,58 (m, 1H), 1,99-1,92 (m, 2H), 1,11 (t, J = 7,2 Гц, 3H)
Пример 43. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
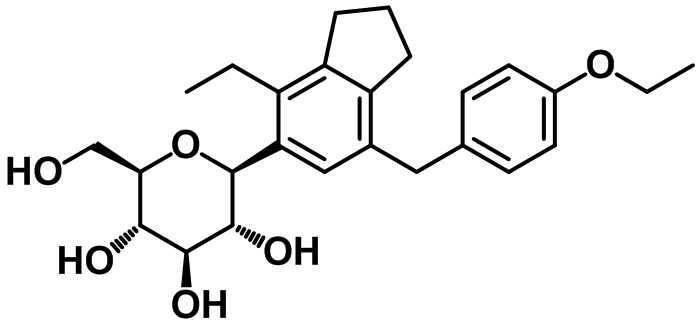
1H ЯМР (400 МГц, CD3OD); δ 7,12 (s, 1H), 7,06 (d, J = 8,4 Гц, 2H), 6,78 (d, J = 8,4 Гц, 2H), 4,45 (d, J = 9,6 Гц, 1H), 4,00 (dd, J = 7,2, 6,8 Гц, 2H), 3,67-3,54 (m, 2H), 3,51-3,48 (m, 1H), 3,43-41 (m, 2H), 2,88 (t, J = 7,6 Гц, 2H), 2,83-2,61 (m, 5H), 2,04-1,96 (m, 3H), 1,35 (t, J = 6,8 Гц, 4H), 1,15 (t, J = 7,6 Гц, 3H)
Пример 44. Получение (2S,3R,4R,5S,6R)-2-(4-этил-7-(4-этилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
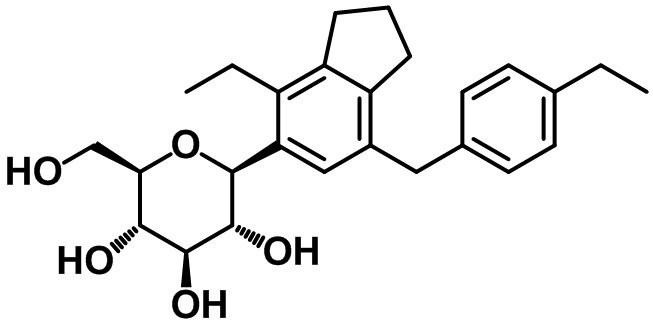
1H ЯМР (400 МГц, CD3OD); δ 7,13 (s, 1H), 7,05 (s, 4H), 4,43 (d, J = 9,6 Гц, 1H), 3,84 (d, J = 4,8 Гц, 3H), 3,76-3,57 (m, 3H), 3,52-3,48 (m, 3H), 2,89 (t, J = 7,2 Гц, 2H), 2,83-2,78 (m, 1H), 2,73 (t, J = 6,8 Гц, 2H), 2,68-2,61 (m, 1H), 2,59 (dd, J = 8,0, 7,6 Гц, 3H), 2,04-1,96 (m, 3H), 1,21-1,07 (m, 5H)
Пример 45. Получение (2S,3R,4R,5S,6R)-2-(4-этил-7-(4-фторбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола

1H ЯМР (400 МГц, CD3OD); δ 7,17-7,13 (m, 3H), 6,97-6,92 (m, 2H), 4,45 (d, J = 9,2 Гц, 1H), 3,90-3,84 (m, 3H), 3,68-3,64 (m, 1H), 3,64-3,57 (m, 1H), 3,52-3,48 (m, 1H), 3,43-3,40 (m, 2H), 2,89 (t, J = 7,4 Гц, 2H), 2,83-2,76 (m, 1H), 2,73-2,63 (m, 3H), 2,04-1,97 (m, 2H), 1,15 (t, J = 7,6 Гц, 3H)
Пример 46. Получение (2S,3R,4R,5S,6R)-2-(7-(4-хлорбензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
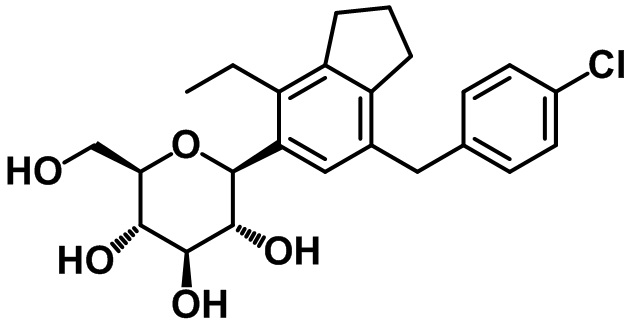
1H ЯМР (400 МГц, CD3OD); δ 7,22 (d, J = 8,0 Гц, 2H), 7,14 (d, J = 8,0 Гц, 2H), 7,13 (s, 1H), 4,45 (d, J = 9,2 Гц, 1H), 3,91 (s, 2H), 3,86 (d, J = 11,6 Гц, 1H), 3,63 (dd, J = 11,6, 4,0 Гц, 1H), 3,56 (t, J = 9,2 Гц, 1H), 3,52-3,48 (m, 1H), 3,40 (d, J = 5,2 Гц, 2H), 2,89 (t, J = 7,6 Гц, 2H), 2,84-2,78 (m, 1H), 2,71 (t, J = 7,6 Гц, 2H), 2,68-2,65 (m, 1H), 2,05-1,97 (m, 2H), 1,15 (t, J = 7,6 Гц, 3H)
Пример 47. Получение (2S,3R,4R,5S,6R)-2-(4-этил-7-(4-трифторметокси)бензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
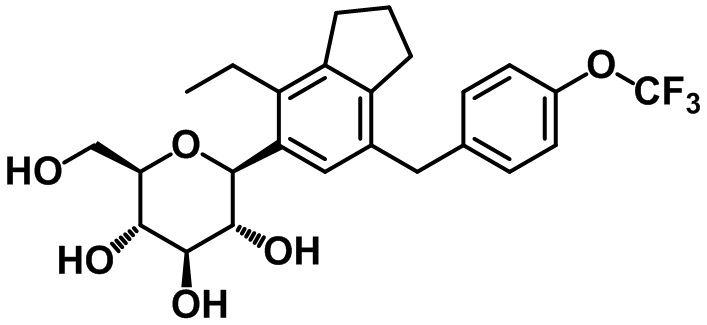
1H ЯМР (400 МГц, CD3OD); δ 7,24 (d, J = 8,4 Гц, 2H), 7,16 (s, 1H), 7,13 (d, J = 8,8 Гц, 2H), 4,45 (d, J = 9,2 Гц, 1H), 3,95 (s, 2H) 3,86 (d, J = 12,0 Гц, 1H), 3,66 (dd, J = 12,0, 4,0 Гц, 1H), 3,59 (t, J = 9,2 Гц, 1H), 3,56-3,48 (m, 1H), 3,41 (d, J = 5,6 Гц, 2H), 2,89 (t, J = 7,2 Гц, 2H), 2,84-2,79 (m, 1H), 2,72 (t, J = 7,6 Гц, 2H), 2,69-2,63 (m, 1H), 2,05-1,98 (m, 2H), 1,16 (t, J = 7,6 Гц, 3H)
Пример 48. Получение (2S,3R,4R,5S,6R)-2-(4-этил-7-(4-трифторметил)бензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
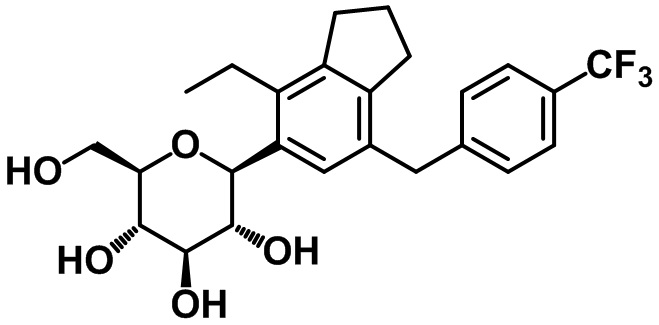
1H ЯМР (400 МГц, CD3OD); δ 7,53 (d, J = 7,6 Гц, 2H), 7,35 (d, J = 7,6 Гц, 2H), 7,17 (s, 1H), 4,46 (d, J = 9,2 Гц, 1H), 4,01 (s, 2H), 3,86 (d, J = 11,6 Гц, 1H), 3,69-3,65 (m, 1H), 3,62-3,57 (m, 1H), 3,53-3,48 (m, 1H), 3,41-3,40 (m, 2H), 2,92-2,88 (m, 2H), 2,84-2,77 (m, 1H), 2,77-2,64 (m, 3H), 2,05-1,98 (m, 2H), 1,16 (t, J = 7,6 Гц, 3H)
Пример 49. Получение (2S,3R,4R,5S,6R)-2-(7-(4-изопропоксибензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола

1H ЯМР (400 МГц, CD3OD); δ 7,12 (s, 1H), 7,05 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,8 Гц, 2H), 4,55-4,48 (m, 1H) 4,44 (d, J = 9,6 Гц, 1H), 3,87 (s, 1H), 3,68-3,57 (m, 2H), 3,52-3,48 (m, 1H), 3,43-3,39 (m, 2H), 3,31-3,18 (m, 2H), 2,88 (t, J = 7,2 Гц, 2H), 2,83-2,63 (m, 4H), 2,04-1,97 (m, 2H), 1,27 (d, J = 6,0 Гц, 6H), 1,15 (t, J = 7,2 Гц, 3H)
Пример 50. Получение (2S,3R,4R,5S,6R)-2-(7-(4-изопропилбензил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
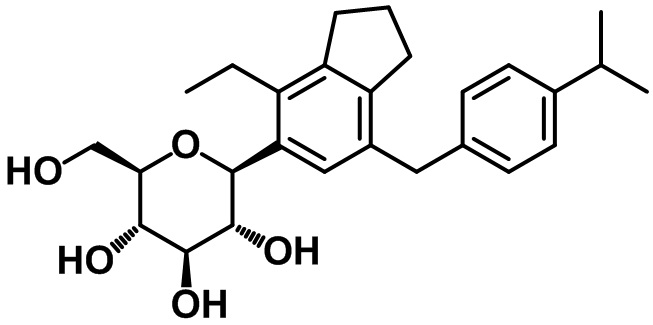
1H ЯМР (400 МГц, CD3OD); δ 7,14 (s, 1H), 7,07 (dd, J = 8,4, 4,8 Гц, 4H), 4,44 (d, J = 9,2 Гц, 1H), 3,88-3,81 (m, 2H), 3,68-3,58 (m, 2H), 3,52-3,48 (m, 1H), 3,41-3,39 (m, 2H), 3,28-3,03 (m, 2H), 2,89 (t, J = 7,2 Гц, 2H), 2,85-2,78 (m, 1H), 2,74 (t, J = 7,6 Гц, 2H), 2,68-2,63 (m, 1H), 2,04-1,98 (m, 2H), 1,21 (d, J = 6,8 Гц, 6H), 1,15 (t, J = 7,2 Гц, 3H)
Пример 51. Получение (2S,3R,4R,5S,6R)-2-(7-(бифенил-3-илметил)-4-этил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
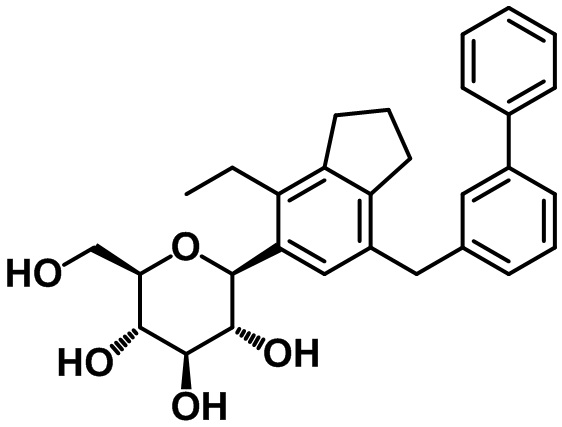
1H ЯМР (400 МГц, CD3OD); δ 7,52 (d, J = 7,6 Гц, 2H), 7,37 (t, J = 7,6 Гц, 4H), 7,27 (d, J = 7,2 Гц, 2H), 7,16 (s, 1H), 7,10 (d, J = 6,8 Гц, 1H), 4,42 (d, J = 9,2 Гц, 1H), 3,96 (s, 1H), 3,84-3,81 (dd, J = 12,4, 11,2, 1H), 3,62-3,55 (m, 2H), 3,47 (t, J = 8,4 Гц, 1H), 3,67-3,58 (m, 2H), 2,86 (t, J = 6,8 Гц, 2H), 2,80-2,73 (m, 3H), 2,65-2,60 (m, 2H), 2,00-1,95 (m, 2H), 1,12 (t, J = 7,2 Гц, 3H)
Пример 52. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
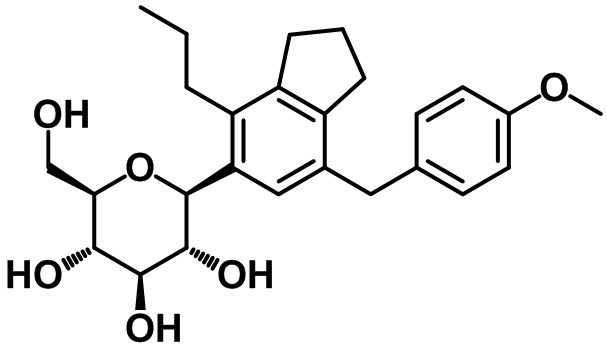
1H ЯМР (400 МГц, CD3OD); δ 7,07 (s, 1H), 7,01 (d, J = 8,8 Гц, 2H), 6,74 (d, J = 8,8 Гц, 2H), 4,38 (d, J = 9,6 Гц, 1H), 3,82-3,80 (m, 3H), 3,70 (s, 3H), 3,61 (dd, J = 12,0, 5,6 Гц, 1H), 3,55 (t, J = 8,8 Гц, 1H), 3,45 (t, J = 8,8 Гц, 1H), 3,34 (d, J = 6,8 Гц, 2H), 2,83 (t, J = 6,8 Гц, 2H), 2,75-2,66 (m, 3H), 2,57-2,50 (m, 1H), 1,99-1,91 (m, 2H), 1,55-1,48 (m, 2H), 0,96 (t, J = 7,6 Гц, 3H)
Пример 53. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метилбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
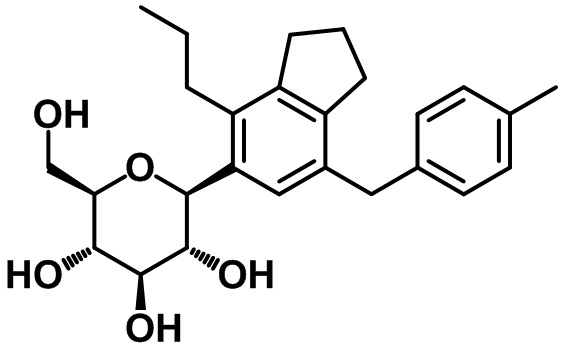
1H ЯМР (400 МГц, CD3OD); δ 7,08 (s, 1H), 6,99 (s, 4H), 4,38 (d, J = 8,8 Гц, 1H), 3,83-3,80 (m, 3H), 3,64-3,59 (m, 1H), 3,55 (t, J = 8,8 Гц, 1H), 3,45 (t, J = 8,8 Гц, 1H), 3,35 (d, J = 5,6 Гц, 2H), 2,83 (t, J = 7,2 Гц, 2H), 2,75-2,66 (m, 3H), 2,56-2,50 (m, 1H), 2,23 (s, 3H), 1,99-1,91 (m, 2H), 1,55-1,50 (m, 2H), 0,97 (t, J = 7,6 Гц, 3H)
Пример 54. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
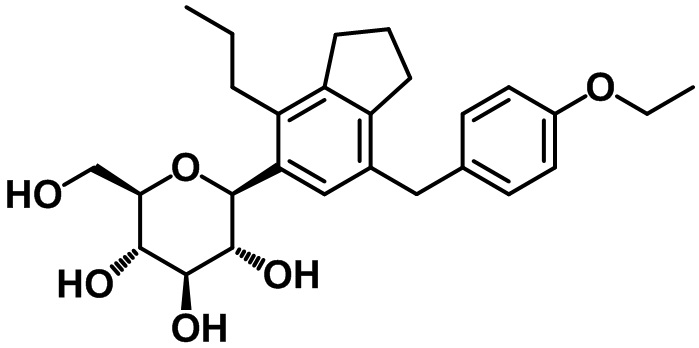
1H ЯМР (400 МГц, CD3OD); δ 7,08 (s, 1H), 7,01 (d, J = 8,8 Гц, 2H), 6,74 (d, J = 8,8 Гц, 2H), 4,40 (d, J = 9,2 Гц, 1H), 3,96 (dd, J = 7,2, 6,8 Гц, 2H), 3,83-3,82 (m, 1H), 3,62 (dd, J = 6,4, 5,2 Гц, 1H), 3,55 (t, J = 8,4 Гц, 1H), 3,45 (t, J = 8,4 Гц, 1H) 3,38-3,36 (m, 2H), 3,27 (s, 2H), 2,83 (t, J = 7,2 Гц, 2H), 2,74-2,67 (m, 3H), 2,58-2,52 (m, 1H), 1,99-1,93 (m, 2H), 1,56-1,50 (m, 2H), 1,31 (t, J = 7,2 Гц, 3H), 0,97 (t, J = 7,2 Гц, 3H)
Пример 55. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
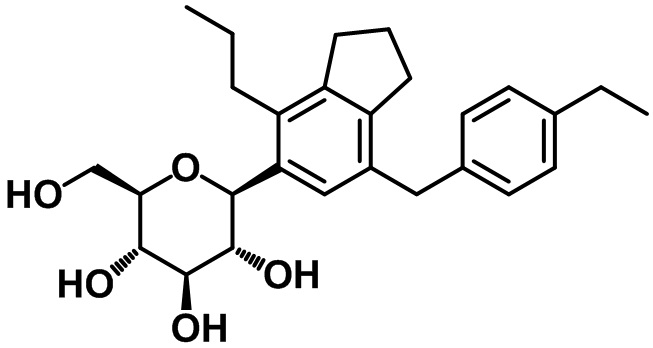
1H ЯМР (400 МГц, CD3OD); δ 7,13 (s, 1H), 7,05 (s, 4H), 4,44 (d, J = 8,4 Гц, 1H), 3,88 (s, 2H), 3,67-3,63 (m, 1H), 3,59 (t, J = 4,8 Гц, 1H), 3,49 (t, J = 7,6 Гц, 1H), 2,88 (t, J = 7,6 Гц, 2H), 2,79-2,71 (m, 3H), 2,61-2,55 (m, 4H), 2,03-1,95 (m, 3H), 1,59-1,54 (m, 3H), 1,19 (t, J = 7,6 Гц, 3H), 1,01 (t, J = 7,2 Гц, 3H)
Пример 56. Получение (2S,3R,4R,5S,6R)-2-(7-(4-фторбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
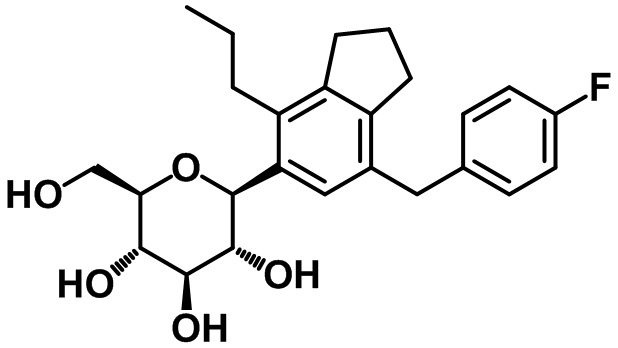
1H ЯМР (400 МГц, CD3OD); δ 7,17-7,13 (m, 3H), 6,97-6,92 (m, 2H), 4,43 (d, J = 9,6 Гц, 1H), 3,90-3,84 (m, 3H), 3,68-3,63 (m, 1H), 3,60-3,56 (m, 1H), 3,51-3,47 (m, 1H), 3,43-3,39 (m, 2H), 2,88 (t, J = 7,4 Гц, 2H), 2,80-2,70 (m, 3H), 2,62-2,54 (m, 1H), 2,03-1,96 (m, 2H), 1,59-1,52 (m, 2H), 1,01 (t, J = 7,2 Гц, 3H)
Пример 57. Получение (2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
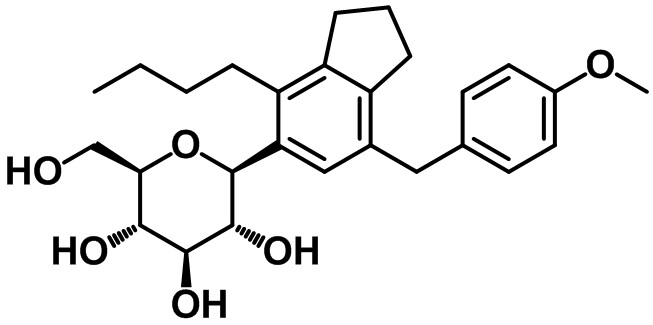
1H ЯМР (400 МГц, CDCl3); δ 7,03-7,01 (m, 3H), 6,76 (d, J = 8,4 Гц, 2H), 4,45 (d, J = 8,4 Гц, 1H), 4,19 (br s, 1H), 4,05 (br s, 1H), 3,81 (s, 2H), 3,75-3,66 (m, 6H), 3,46-3,40 (m, 1H), 2,85 (t, J = 7,2 Гц, 2H), 2,72 (t, J = 7,2 Гц, 2H), 2,70-2,63 (m, 1H), 2,60-2,51 (m, 1H), 2,03-1,97 (m, 2H), 1,46-1,35 (m, 4H), 0,92 (t, J = 6,8 Гц, 3H)
Пример 58. Получение (2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
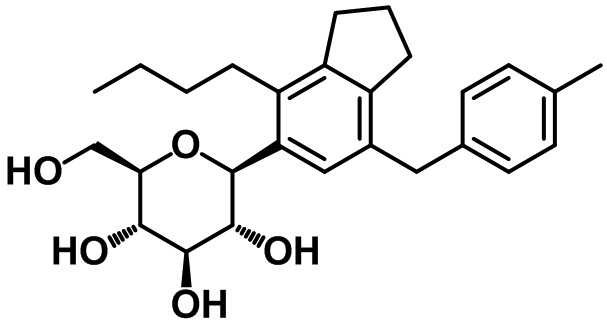
1H ЯМР (400 МГц, CDCl3); δ 7,05-6,98 (m, 5H), 4,45 (d, J = 8,4 Гц, 1H), 4,30 (br s, 1H), 4,15 (br s, 1H), 3,84 (s, 2H), 3,80-3,69 (m, 3H), 3,43 (m, 1H), 2,85 (t, J = 7,2 Гц, 2H), 2,72 (t, J = 7,2 Гц, 2H), 2,68-2,62 (m, 1H), 2,60-2,51 (m, 1H), 2,26 (s, 3H), 2,03-1,95 (m, 2H), 1,50-1,38 (m, 4H), 0,92 (t, J = 6,8 Гц, 3H)
Пример 59. Получение (2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-этоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
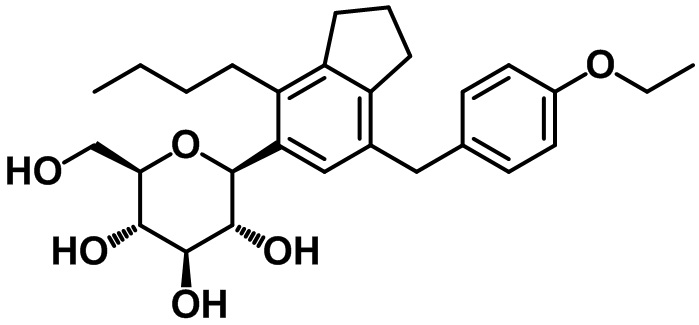
1H ЯМР (400 МГц, CD3OD); δ 7,07 (s, 1H), 7,01 (d, J = 8,4 Гц, 2H), 6,72 (d, J = 8,4 Гц, 2H), 4,38 (d, J = 9,2 Гц, 1H), 3,94 (q, J = 7,2 Гц, 2H), 3,83-3,79 (m, 3H), 3,63-3,59 (m, 1H), 3,58-3,53 (m, 1H), 3,46-3,42 (m, 1H), 3,35-3,34 (m, 2H), 2,83 (t, J = 7,6 Гц, 2H), 2,78-2,51 (m, 1H), 2,67 (t, J = 7,6 Гц, 2H), 2,59-2,52 (m, 1H), 1,99-1,93 (m, 2H), 1,52-1,44 (m, 2H), 1,44-1,37 (m, 2H), 1,31 (t, J = 6,8 Гц, 3H), 0,93 (t, J = 6,8 Гц, 3H)
Пример 60. Получение (2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-этилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
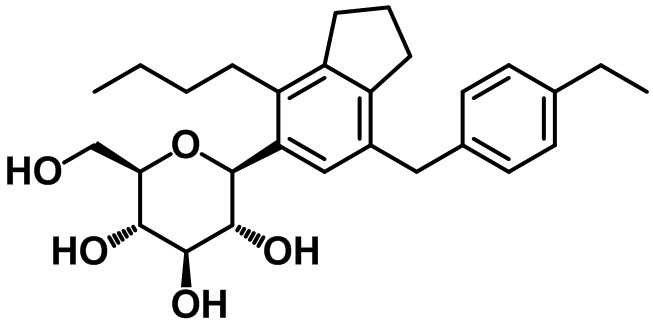
1H ЯМР (400 МГц, CD3OD); δ 7,08 (s, 1H), 7,01 (s, 4H), 4,38 (d, J = 9,2 Гц, 1H), 3,83-3,79 (m, 3H), 3,63-3,59 (m, 1H), 3,58-3,53 (m, 1H), 3,46-3,42 (m, 1H), 3,35-3,34 (m, 2H), 2,83 (t, J = 7,6 Гц, 2H), 2,78-2,67 (m, 3H), 2,59-2,50 (m, 3H), 1,98-1,91 (m, 2H), 1,54-1,44 (m, 2H), 1,44-1,34 (m, 2H), 1,14 (t, J = 7,2 Гц, 3H), 0,93 (t, J = 7,2 Гц, 3H)
Пример 61. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изопропил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез (E)-этил-4-метилпент-2-еноата (61-1)
 (61-1)
(61-1)
(Карбэтоксиметилен)трифенилфосфоран (24,10 г, 69,34 ммоль) растворяли в DCM (101 мл), после чего полученный раствор охлаждали при 0ºC, таким образом, изомасляный альдегид (5,0 г, 69,34 ммоль, Aldrich) медленно добавляли по каплям к полученному продукту, а затем полученную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель реакционной смеси концентрировали при пониженном давлении, после чего в полученный концентрат по каплям добавляли простой эфир, таким образом, полученное твердое вещество фильтровали и удаляли. Полученный фильтрат собирали и концентрировали при пониженном давлении, после чего полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (61-1) (8,48 г, 59,63 ммоль, 86%).
1H ЯМР (400 МГц, CDCl3); δ 6,95 (dd, J = 15,6, 6,8 Гц, 1H), 5,77 (dd, J = 15,6, 1,2 Гц, 1H), 4,19 (q, J = 7,2 Гц, 2H), 2,50-2,42 (m, 1H), 1,29 (t, J = 7,2 Гц, 3H), 1,06 (d, J = 6,8 Гц, 6H)
Стадия 2. Синтез (E)-4-метилпент-2-ен-1-ола (61-2)
 (61-2)
(61-2)
Алюмогидрид лития (6,79 г, 178,9 ммоль) и хлорид алюминия (7,95 г, 59,63 ммоль) разбавляли в диэтиловом эфире (500 мл), после чего полученный раствор охлаждали при -78ºC. Соединение в соответствии с названием изобретения (61-1) (8,48 г, 59,63 ммоль) в диэтиловом эфире (50 мл) медленно добавляли по каплям в реакционную смесь, после чего полученную смесь перемешивали при той же температуре в течение 2 часов. К полученной смеси медленно по каплям добавляли воду, после чего реакцию завершали, таким образом, полученное твердое вещество фильтровали и удаляли. Органический слой промывали солевым раствором, после чего полученный продукт высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Концентрированный раствор высушивали под вакуумом с получением указанного в заголовке соединения (61-2), которое применяли на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3); 5,65 (d, J = 6,4 Гц, 1H), 5,60 (t, J = 6,0 Гц, 1H), 4,09 (d, J = 5,2 Гц, 2H), 2,35-2,27 (m, 1H), 1,00 (d, J = 6,8 Гц, 6H)
Стадия 3. Синтез целевого соединения
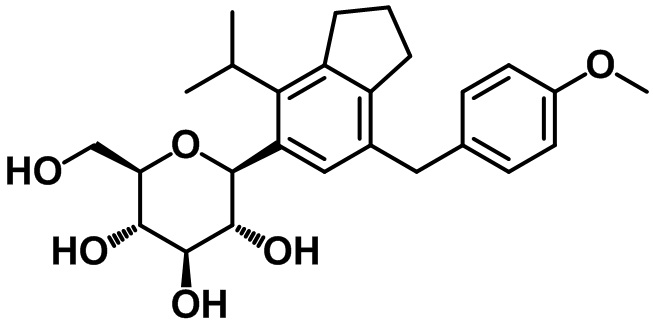
Целевое соединение получали с помощью соединения (61-2) посредством способа, показанного на стадиях 1-7 примера 41.
1H ЯМР (400 МГц, CD3OD); δ 7,13 (s, 1H), 7,05 (d, J = 8,4 Гц, 2H), 6,78 (d, J = 8,4 Гц, 2H), 4,55 (br s, 1H), 3,87-3,85 (m, 3H), 3,74 (s, 3H), 3,68-3,64 (m, 1H), 3,62-3,56 (m, 1H), 3,51-3,47 (m, 2H), 3,40-3,38 (m, 2H), 3,02 (t, J = 7,2 Гц, 2H), 2,67-2,61 (m, 2H), 1,99-1,92 (m, 2H), 1,32-1,30 (m, 6H)
Примеры 62 и 63
Целевые соединения из примеров 62 и 63 получали посредством способа, показанного в примере 61.
Пример 62. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изопропил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
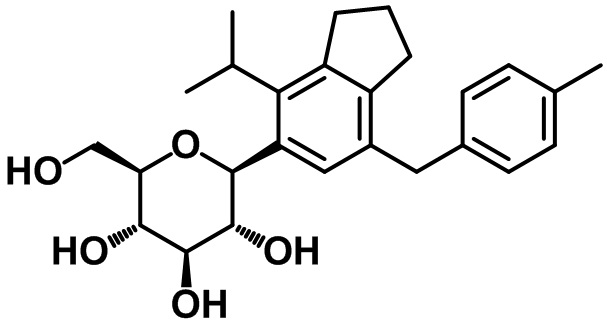
1H ЯМР (400 МГц, CD3OD); δ 7,14 (s, 1H), 7,02 (s, 4H), 4,55 (br s, 1H), 3,86-3,84 (m, 3H), 3,67-3,64 (m, 1H), 3,62-3,57 (m, 1H), 3,52-3,48 (m, 2H), 3,40-3,38 (m, 2H), 3,00 (t, J = 7,2 Гц, 2H), 2,67-2,63 (m, 2H), 2,27 (s, 3H), 1,98-1,91 (m, 2H), 1,32-1,30 (m, 6H)
Пример 63. Получение (2S,3R,4R,5S,6R)-2-(4-циклопентил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
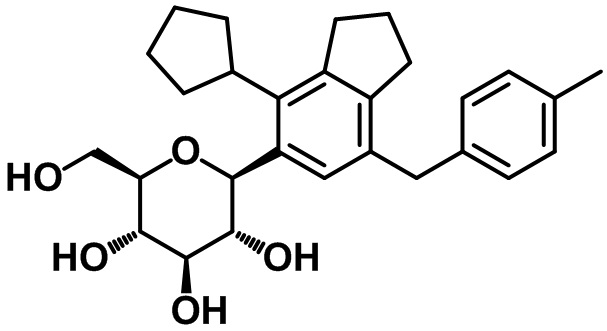
1H ЯМР (400 МГц, CD3OD); δ 7,10 (br s, 1H), 7,98 (s, 4H), 4,50 (br s, 1H), 3,82-3,79 (m, 3H), 3,63-3,59 (m, 1H), 3,58-3,48 (m, 1H), 3,46-3,42 (m, 1H), 3,35-3,33 (m, 2H), 2,89 (t, J = 7,2 Гц, 2H), 2,61 (t, J = 7,2 Гц, 2H), 2,23 (s, 3H), 1,97-1,78 (m, 9H), 1,74-1,64 (m, 2H)
Пример 64. Получение (2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изобутил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1. Синтез (2E,4E)-этил-7-метилокта-2,4-диеноата (64-1)
 (64-1)
(64-1)
В раствор изопентилтрифенилфосфония бромида (15,0 г, 36,29 ммоль) в THF (50 мл) при -78ºC в атмосфере азота добавляли n-BuLi (14,5 мл, 36,29 ммоль, 2,5 M в н-гексане), после чего полученную смесь перемешивали при той же температуре в течение 1 часа. К полученной смеси медленно по каплям добавляли этил-4-оксобут-2-еноат (1,55 г, 12,09 ммоль), после чего полученную смесь перемешивали в течение 30 минут с повышением температуры до комнатной температуры. Реакционную смесь охлаждали при 0ºC, после чего насыщенный раствор хлорида аммония добавляли по каплям к полученному продукту с завершением реакции и проведением экстракции с помощью диэтилового эфира. Органический слой высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали посредством колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (64-1) (1,89 г, 10,37 ммоль, 86%).
1H ЯМР (400 МГц, CDCl3); 5,60 (dd, J = 15,2, 11,2 Гц, 1H), 6,17 (t, J = 11,2 Гц, 1H), 5,91-5,84 (m, 2H), 4,21 (q, J = 7,2 Гц, 2H), 2,20 (t, J = 7,2 Гц, 2H), 1,72-1,65 (m, 1H), 1,29 (t, J = 7,2 Гц, 3H), 0,93 (d, J = 6,8 Гц, 6H)
Стадия 2. Синтез целевого соединения
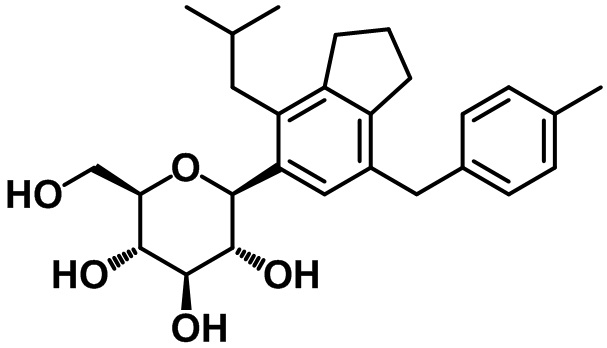
Целевое соединение получали с помощью соединения (64-1) посредством способа, показанного на стадиях 3-7 примера 41.
1H ЯМР (400 МГц, CDCl3); δ 7,07-7,01 (m, 5H), 4,49 (d, J = 8,4 Гц, 1H), 3,88-3,85 (m, 3H), 3,78-3,74 (m, 1H), 3,72-3,65 (m, 3H), 3,49-3,44 (m, 1H), 2,88 (t, J = 7,2 Гц, 2H), 2,80-2,75 (m, 2H), 2,66-2,61 (m, 1H), 2,49-2,44 (m, 1H), 2,30 (s, 3H), 2,03-1,98 (m, 2H), 1,86-1,79 (m, 1H), 0,94 (d, J = 6,4 Гц, 6H)
Пример 65. Получение (2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-изобутил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
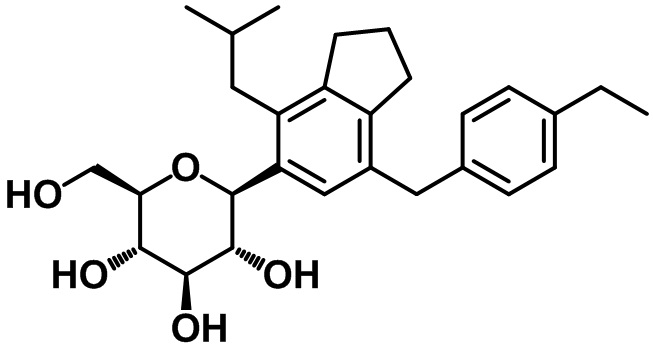
Целевое соединение получали с помощью соединения (64-1) посредством способа, показанного на стадии 2 примера 64.
1H ЯМР (400 МГц, CDCl3); δ 7,11-7,05 (m, 5H), 4,50 (d, J = 8,4 Гц, 1H), 3,89-3,87 (m, 3H), 3,81-3,74 (m, 1H), 3,73-3,66 (m, 3H), 3,51-3,46 (m, 1H), 2,88 (t, J = 7,2 Гц, 2H), 2,82-2,76 (m, 2H), 2,66-2,58 (m, 3H), 2,49-2,44 (m, 1H), 2,03-1,97 (m, 2H), 1,86-1,79 (m, 1H), 1,21 (t, J = 7,2 Гц, 3H), 0,94 (d, J = 6,4 Гц, 6H)
Экспериментальный пример 1. Клонирование генов SGLT1, SGLT2 человека и разработка клеточных линий для экспрессии SGLT1, SGLT2 человека
Гены SGLT1 человека (hSGLT1), SGLT2 человека (hSGLT2) амплифицировали из библиотеки кДНК человека Marathon-ready (Clontech) посредством метода ПЦР, после чего полученные амплифицированные последовательности объединяли с вектором pcDNA 3.1(+), который представлял собой вектор экспрессии млекопитающих, с получением рекомбинантных векторов экспрессии pcDNA3.1(+)/hSGLT1, pcDNA3.1(+)/hSGLT2. Полученные рекомбинантные векторы экспрессии трансформировали в клетки яичника китайского хомячка, после чего стабильно трансформированных клонов отбирали посредством метода отбора колоний с помощью резистентности к G418, селективному маркеру, содержащемуся в векторе. Из выбранных клонов выбирали клонов, экспрессирующих hSGLT1 и hSGLT2, на основании активности в анализе транспорта 14C-α-метил-D-глюкопиранозида (14C-AMG).
Экспериментальный пример 2. Ингибирующий эффект на активность SGLT1, SGLT2 человека
Для анализа натрий-зависимого транспорта глюкозы клетки, экспрессирующие hSGLT1 и hSGLT2, высевали при концентрации 1 x 105 клеток на лунку в 96-луночный культуральный планшет, после чего полученные клетки культивировали в среде RPMI 1640, содержащей 10% фетальной бычьей сыворотки (FBS). Через 1 сутки после культивирования полученные клетки культивировали в буферном растворе для предварительной обработки (10 мМ HEPES, 5 мМ трис, 140 мМ хлорида холина, 2 мМ KCl, 1 мМ CaCl2 и 1 мМ MgCl2, pH 7,4) при условиях 37ºC/5% CO2 в течение 10 минут. Затем полученные клетки культивировали в буферном растворе, предназначенном для измерения поглощения (10 мМ HEPES, 5 мМ трис, 140 мМ NaCl, 2 мМ KCl, 1 мМ CaCl2, 1 мМ MgCl2 и 1 мМ AMG, pH 7,4), содержащем 14C-AMG (8 мкМ) и соединение согласно настоящему изобретению или среду-носитель на основе диметилсульфоксида (DMSO) при условиях 37ºC/5% CO2 в течение 2 часов. После культивирования клетки дважды промывали промывочным буферным раствором (буферный раствор для предварительной обработки, содержащий 10 мМ AMG при комнатной температуре), после чего измеряли его излучение посредством применения жидкостного сцинтилляционного счетчика. IC50 для каждого соединения измеряли в соответствии с нелинейным регрессионным анализом посредством применения SigmaPlot (Document Analytical Biochemistry 429: 70-75, Molecular and Cellular Biochemistry 280: 91–98, 2005). Результаты in-vitro анализа SGLT1/2 показаны в следующей таблице 1.[Таблица 1]
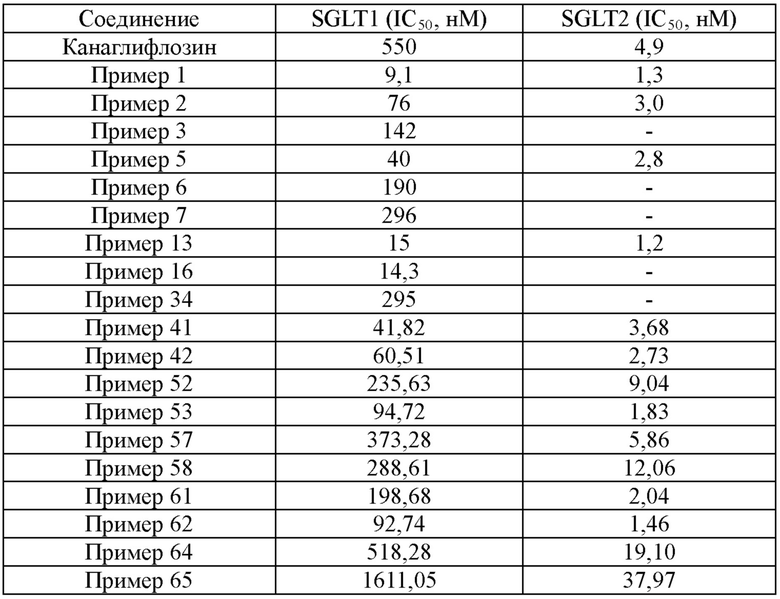
Экспериментальный пример 3. Эксперимент по измерению экскреции глюкозы с мочой (тест UGE)
Что касается фармацевтической эффективности соединения, полученного в примере, 1 мг/кг такого соединения перорально вводили обычной мыши, после чего проводили тест UGE. В результате было установлено, что соединение согласно настоящему изобретению увеличивало уровень глюкозы в моче (мг/24 ч.) и снижало уровень глюкозы в крови (мг/дл).
Соответственно ожидается, что соединение согласно настоящему изобретению будет применяться с пользой в лечении или предупреждении диабета.
Экспериментальный пример 4. Эксперимент по измерению противодиабетической активности
Что касается фармацевтической эффективности соединения, полученного в примере, 2 мг/кг такого соединения перорально вводили каждой db/db-мыши и DIO-мыши в течение 4 недель, после чего измеряли изменение уровня сахара в крови. В результате было установлено, что уровень сахара в крови значительно снижался.
Кроме того, что касается фармацевтической эффективности соединения, полученного в примере выше, такое соединение вводили OB/OB-мыши в течение 2 недель, после чего измеряли изменение уровня сахара в крови. В результате было установлено, что уровень сахара в крови значительно снижался.
Соответственно ожидается, что соединение согласно настоящему изобретению будет применяться с пользой в лечении или предупреждении диабета.
Экспериментальный пример 5. Эксперимент в виде перорального теста толерантности к глюкозе
Чтобы установить фармацевтическую эффективность соединения, полученного в примере, измеряли уровень глюкозы после приема пищи для обычной мыши. В результате было установлено, что уровень глюкозы в крови, AUC 0-4 ч.: мг·ч./дл, через 4 часа после введения соединения (1 мг/кг) в значительной степени снижался.
Такой результат также выявляли в эксперименте с db/db-мышью, таким образом, было установлено, что уровень глюкозы в крови, AUC 0-4 ч.: мг·ч./дл, через 4 часа после введения соединения (2 мг/кг) в значительной степени снижался.
Кроме того, в эксперименте с db/db-мышью также было установлено, что уровень глюкозы в крови, AUC 0-4 ч.: мг·ч./дл, через 4 часа после введения соединения (10 мг/кг) также в значительной степени снижался.
Соответственно ожидается, что соединение согласно настоящему изобретению будет применяться с пользой в лечении или предупреждении диабета.
Несмотря на то, что конкретные части настоящего изобретения были подробно описаны выше, специалистам в данной области техники очевидно, что такие подробные описания изложены лишь для иллюстрации иллюстративных вариантов осуществления, но не предназначены для ограничения объема настоящего изобретения. Таким образом, следует понимать, что фактический объем настоящего изобретения определен прилагаемыми пунктами формулы изобретения и их эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| ТВЕРДЫЕ ФОРМЫ (2S,3R,4R,5S,6R)-2-(4-ХЛОР-3-(4-ЭТОКСИБЕНЗИЛ)ФЕНИЛ)-6-(МЕТИЛТИО)ТЕТРАГИДРО-2Н-ПИРАН-3,4,5-ТРИОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2505543C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОРЫ КОТРАНСПОРТЕРОВ НАТРИЯ-ГЛЮКОЗЫ 1 И 2, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2669921C2 |
| СОЕДИНЕНИЯ С-МАННОЗИДА, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МОЧЕВЫВОДЯЩИХ ПУТЕЙ | 2019 |
|
RU2790228C2 |
| ПРОИЗВОДНЫЕ С-АРИЛГЛЮКОЗИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2606501C2 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
Изобретение относится к применимым в медицине соединениям, соответствующим общей формуле I, или их фармацевтически приемлемым солям:
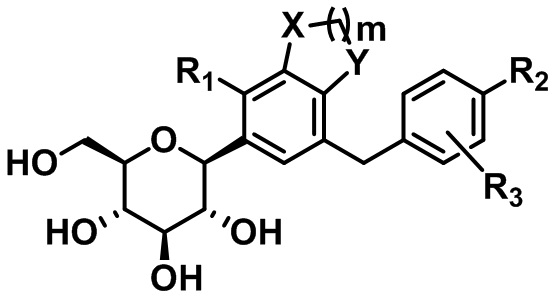 [Формула I],
[Формула I],
где X представляет собой -CH2; Y представляет собой -CH2 или -O-; m равняется 1; R1 представляет собой C1-C4алкил; R2 представляет собой галоген, C1-C3алкил, C2-C3алкенил, C1-C3алкокси, -OCF3 или -SR5, где каждый из по меньшей мере одного атома водорода в указанном C1-C3алкиле может быть независимо не замещен или замещен галогеном, и водород указанного С2-С3алкенила не замещен; R3 представляет собой водород; и R5 представляет собой C1алкил. Предложены новые соединения, эффективный способ их получения, фармацевтические композиции на их основе, эффективные для лечения или предупреждения диабета. 5 н. и 3 з.п. ф-лы, 1 табл., 70 пр.
1. Соединение, представленное следующей формулой I, или его фармацевтически приемлемая соль:
[Формула I]
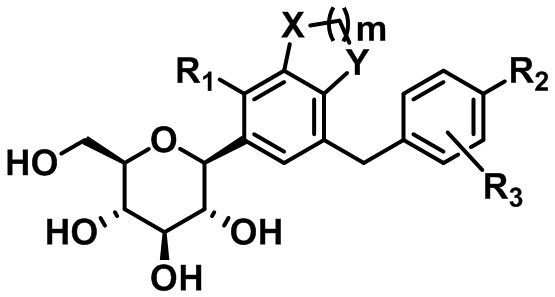 ,
,
где
X представляет собой -CH2;
Y представляет собой -CH2 или -O-;
m равняется 1;
R1 представляет собой C1-C4алкил;
R2 представляет собой галоген, C1-C3алкил, C2-C3алкенил, C1-C3алкокси, -OCF3 или -SR5, где каждый из по меньшей мере одного атома водорода в указанном C1-C3алкиле может быть независимо не замещен или замещен галогеном, и водород указанного С2-С3алкенила не замещен;
R3 представляет собой водород; и
R5 представляет собой C1алкил,
где соединение выбрано из группы, состоящей из следующих соединений:
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-изопропоксибензил)-4-метил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-пропилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-винилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-трифторметил)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-трифторметокси)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-метилтио)бензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-фторбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-хлорбензил)-4-метил-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-метил-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этоксибензил)-4-метил-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-(метилтио)бензил)-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(7-(4-этилбензил)-4-метил-2,3-дигидробензофуран-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-метил-7-(4-винилбензил)-2,3-дигидробензофуран-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-этил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метоксибензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(7-(4-метилбензил)-4-пропил-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола;
(2S,3R,4R,5S,6R)-2-(4-бутил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола;
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изопропил-7-(4-метоксибензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола; и
(2R,3S,4R,5R,6S)-2-(гидроксиметил)-6-(4-изопропил-7-(4-метилбензил)-2,3-дигидро-1H-инден-5-ил)тетрагидро-2H-пиран-3,4,5-триола.
2. Фармацевтическая композиция, предназначенная для лечения или предупреждения диабета, содержащая в качестве активного компонента терапевтически эффективное количество соединения формулы I по п. 1 или его фармацевтически приемлемой соли.
3. Фармацевтическая композиция по п. 2, где композиция обеспечивает ингибирование SGLT1, SGLT2 или как SGLT1, так и SGLT2.
4. Способ получения соединения по п. 1, представленного формулой I, или его фармацевтически приемлемой соли, где способ включает следующие стадии:
(S1) осуществление реакции соединения следующей формулы II с соединением следующей формулы III с получением соединения следующей формулы IV и
(S2) проведение восстановления с удалением защитной группы или удаления защитной группы с восстановлением для соединения вышеприведенной формулы IV с получением соединения следующей формулы I:
[Формула II]
 ,
,
[Формула III]
 ,
,
[Формула IV]
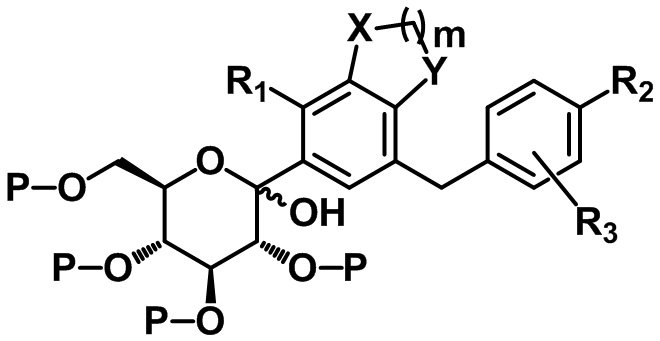 ,
,
[Формула I]
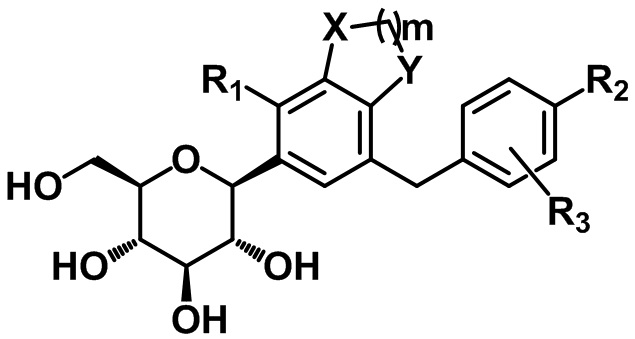 ,
,
где
X, Y, m, R1, R2 и R3 определены в п. 1, и P представляет собой триметилсилил или бензил.
5. Способ по п. 4, где, если P представляет собой триметилсилил, соединение следующей формулы V получают из соединения формулы IV в соответствии со схемой:
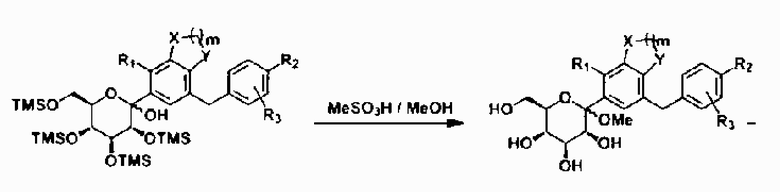
и соединение формулы I получают посредством восстановления соединения формулы V:
[Формула V]
 ,
,
где
X, Y, m, R1, R2 и R3 определены в п. 1.
6. Способ по п. 4, где, если P представляет собой бензил, соединение следующей формулы VI получают посредством восстановления соединения формулы IV и соединение формулы I получают посредством удаления защитной группы из соединения формулы VI:
[Формула VI]
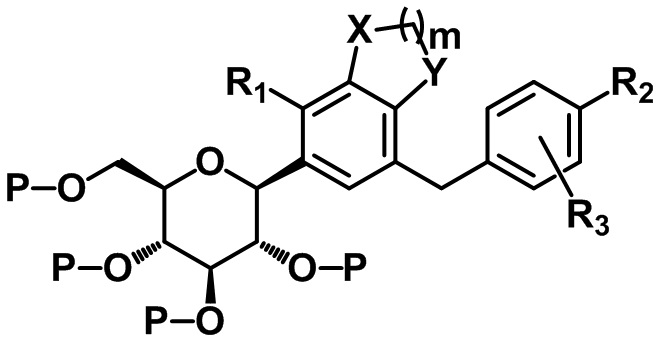 ,
,
где
X, Y, m, R1, R2 и R3 определены в п. 1, и P представляет собой бензил.
7. Способ лечения диабета, предусматривающий введение в качестве активного компонента терапевтически эффективного количества соединения формулы I по п. 1 или его фармацевтически приемлемой соли.
8. Применение соединения формулы I по п. 1 или его фармацевтически приемлемой соли для изготовления лекарственного препарата, предназначенного для лечения или предупреждения диабета.
| WO 2015174695 A1, 19.11.2015 | |||
| С-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 | 2000 |
|
RU2262507C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛБЕНЗОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2497526C2 |
| RU 2013107748 A, 20.09.2014 | |||
| WO 2012041898 A1, 05.04.2012 | |||
| US 20030114390 A1, 19.06.2003 | |||
| EA 201100266 A1, 31.10.2011 | |||
| WO 2008116195 A3, 25.09.2008 | |||
| WO 2012165356 A1, 06.12.2012 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| WO 2012033390 A2, 15.03.2012. | |||

