Изобретение относится к новым биологически активным соединениям, производным бисамидов дикарбоновых кислот или их фармацевтически приемлемым солям, обладающим способностью к комплексообразованию или хелатированию ионов металлов, а также их применению в качестве средства, обладающего антиоксидантным действием, средства для профилактики и/или лечения сердечнососудистых, вирусных, онкологических, нейродегенеративных, воспалительных заболеваний, диабета, геронтологических заболеваний, заболеваний, вызываемых токсинами микроорганизмов, а также алкоголизма, алкогольного цирроза печени, анемии, поздней порфирии, отравлений солями переходных металлов.
Уровень техники
Ионы металлов играют важную роль как в нормальном функционировании клеток и всего организма, так и в развитии патологий.
Известны лекарственные препараты, которые осуществляют свое действие благодаря способности хелатировать ионы металлов. В связи с этим в настоящее время ведется поиск нетоксичных соединений, способных высокоэффективно и селективно хелатировать ионы металлов и пригодных для биомедицинского применения.
Соединения с хелатирующей способностью обнаружены среди различных классов соединений моно- и дитиолов, дисульфидов, азосоединений, нитрозоароматических соединений, производных полиаминокарбоновых кислот, тиосемикарбазона, пиридоксаль изоникотиноилгидразона, хинолина, адамантана, пирогаллола, фенантролина, тиопирофосфатов и других. Кроме того, они отмечены и среди прочих природных соединений, таких как, например, карнозин, фитин, пектин. Наибольший интерес представляют соединения, имеющие несколько функциональных групп, способные выступать в роли доноров электронов при комплексообразовании. В связи с этим такие соединения могут представлять собой лиганды, специфически взаимодействующие с ионами металла или группы металлов.
Широко известными в настоящее время комплексообразователями являются производные полиаминокарбоновых кислот (например, ЭДТА), D-пеницилламин, полициклические криптанды, которые успешно применяются при отравлении тяжелыми металлами. В качестве хелатора железа при некоторых железоизбыточных состояниях и гематохроматозе используется деферроксамин. Помимо этого, возможно применение хелаторов при патологиях, связанных с Са-избыточными состояниями, например, при артрозах, атеросклерозе, почечно-каменной болезни. Известно также, что хелатотерапия препятствует отложению холестерина и восстанавливает его уровень в крови, понижает кровяное давление, позволяет избежать ангиопластики, подавляет нежелательные побочные эффекты некоторых сердечных препаратов, удаляет кальций из холестериновых бляшек, растворяет тромбы и восстанавливает эластичность кровеносных сосудов, нормализирует аритмию, препятствует старению, восстанавливает силу сердечной мышцы и улучшает функции сердца, увеличивает внутриклеточное содержание калия, регулирует минеральный обмен, полезна при лечении болезни Альцгеймера, препятствует возникновению рака, улучшает память и проявляет множество других положительных эффектов. Однако сильные хелаторы, применяемые в настоящее время в хелатотерапии, как правило, обладают токсическим действием, которое проявляется, в основном, в повреждении слизистой оболочки тонкой кишки и нарушении функции почек. В некоторых случаях при быстром введении больших количеств известных хелаторов возможно нарушение возбудимости мышц и свертываемости крови. Кроме того, сильные хелаторы могут взаимодействовать с полезными биоэлементами (Na, K, Ca, Mg, Ca), а также могут изменять активность жизненно важных металлоферментов [Зеленин К.Н. "Комплексоны в медицине", Соросовский Образовательный Журнал, 2001, т.7, № 1, стр.45-50].
В связи с этим проводится активный поиск новых высокоэффективных хелаторов с хелатирующей способностью, достаточной для осуществления биологического эффекта in vivo, лишенных побочных эффектов.
Особенно актуальна идея применения хелатотерапии в случае вирусных заболеваний, таких как ВИЧ, папиллома человека, герпес, гепатит С и другие.
Перспективной мишенью для этого представляется цинк-связывающие участки в структуре вирусных белков - так называемые «цинковые пальцы».
В настоящее время выявлено несколько соединений, воздействующих на «цинковые пальцы» важных белков этих вирусов.
В статье Andreas J.K., Boorganic & Medicinal chemistry, 2003, v.11, p.4599-4613, описано производное адамантана, имеющее тривиальное название бананин, которое является хелатором ионов цинка. Предполагается, что оно химически подходит для удаления цинка из белка NCp7 ВИЧ. Имеющее указанный выше механизм действия азапроизводное - азадикарбонамид находится в I/II стадии клинических испытаний против прогрессирующего СПИДа.
В статье Rice W.G., Schaeffer C.A., Harten B., Nature, 1993, v.4, p.473-475, раскрыт 3-нитрозобензамид, который удаляет цинк из NCp7 белка ВИЧ, ингибируя репликацию ВИЧ и его патогенность in vitro и in vivo.
В качестве мишени лекарственных средств был выбран также участок типа «цинковый палец» белка Е6 вируса папилломы человека. Этот вирус является возможным посредником в этиологии цервикальной карциномы.
В статье Beerheide W., Bernard H.-U., Tan Y.-J., Ganesan A.J., National Cancer Institute, 1999, v.91, № 14, 1211-1220, описаны испытания in vitro азасоединений, дисульфидных и нитрозоароматических производных. Показано, что соединения типа 4,4'-дитиодиморфолина провоцируют высвобождение ионов цинка. В результате наблюдали изменение структуры вирусного белка и нарушение его функций, связанных с биологией и патологией вируса папилломы человека. Однако клинические испытания данных соединений в этом отношении еще не закончены, и об их эффективности можно судить только на основании исследований in vitro.
Указанные выше соединения рассматриваются как перспективные для развития лекарственных средств против цервикального рака, остроконечных кондилом и латентных папилломавирусных инфекций половых органов. Вирус гепатита С принадлежит к числу наиболее широко распространенных человеческих патогенов. Современная терапия гепатита С основана практически исключительно на использовании интерферона, а также его комбинации с нуклеозидным аналогом - рибавирином [Козлов М.В., Поляков К.М., Иванов А.В., Биохимия, 2006, т.71, № 9, стр. 1253-12594]. Следует отметить невысокую эффективность такой терапии.
Что касается развития терапевтических агентов против вируса гепатита C, то одной из мишеней является NS3-сериновая протеиназа, в поддержании стабильности структуры которой важную роль играет цинковый участок [Andrea Urbani, Renzo Bazzo, Maria Chiara Nardi, Daniel Oscar Cicero, Raffaele De Francesco, J. Biol. Chem, 1998, v.273, № 30, р. 18760-18769]. Ингибирование или изменение ее активности путем применения соединений, способных к извлечению цинка, в некоторых литературных источниках оценено в качестве многообещающей стратегии управления болезнью, вызванной вирусом гепатита С.
В статье Timothy L. Tellinghuisen, Matthew S. Paulson, Charles M. Rice, J. Virology, 2006, v. 80, № 15, p. 7450-7458, описано, что металлохелаторы (ЭДТА и 1,10-фенантролин) были эффективными ингибиторами протеазы, оцененными относительно NS2/3 авторасщепления. Имеются также сведения о том, что 1,10-фенантролин действовал в этом случае именно через хелатирование цинка.
В статье Sperandio D., Gangloff A.R., Litvak J. Goldsmith R., Bioorg. Med. Chem. Lett., 2002, v.12, № 21, 3129-3133, описан скрининг группы бисбензимидазолов с целью поиска ингибиторов сериновой протеазы NS3/NS4A вируса гепатита C, который также привел к идентификации соответствующего мощного Zn2+-зависимого ингибитора.
При разработке новых подходов к терапии гепатита С другой привлекательной мишенью можно назвать РНК-зависимую РНК-полимеразу вируса гепатита С (вирусный белок NS5B), имеющий в своей структуре цинк-связывающий участок [Timothy L. Tellinghuisen, Matthew S. Paulson, Charles M. Rice, J. Virology, 2006, v. 80, № 15, p. 7450-7458].
В норме клетка печени не содержит белков, обладающих аналогичной активностью [Козлов М.В., Поляков К.М., Иванов А.В., Биохимия, 2006, т.71, № 9, стр. 1253-12594].
Известные в настоящее время ингибиторы РНК-зависимой РНК полимеразы вируса гепатита С можно условно разделить на два основных класса: производные нуклеозидов и ненуклеозидные ингибиторы различной природы [Maria Bretner, Acta biochemica polonica, 2005, v.52, № 1, p. 57-70]. Кроме того, обнаружено ингибирование активности данного фермента производными пирогаллола. Примечательно, что механизм ингибирования производными пирогаллола, как полагают, заключается в хелатировании катионов магния, принимающих участие в каталитическом акте на стадии переноса фосфорильного остатка [Козлов М.В., Поляков К.М., Иванов А.В., Биохимия, 2006, т.71, № 9, стр. 1253-12594].
Заболевания, вызываемые герпесвирусами, широко распространены. Так, известно несколько человеческих герпесвирусов - вирус простого гса 1 и 2 (HSV-1 и HSV-2), цитомегаловирус (CMV), вирус ветряной оспы, вирус Эпштейна-Барра. Деструктивные действия, которые при этом оказываются на центральную нервную систему, вызывают такие заболевания, как энцефалит и менингит. Можно отметить интерес к исследованиям влияния хелатирующих цинк соединений, например, диэтилентриаминпентауксусной кислоты, на ингибирование репликации человеческого цитомегаловируса in vitro [Kanekiyo M., Itoh N., Mano M., Antiviral Res., 2000, v. 47, p. 207-214].
Однако следует отметить и тот факт, что герпесвирусы, так же как и вышеуказанные вирусы, имеют белки, содержащие мотив типа «цинковый палец». Химические изменения в «цинковом пальце» могут приводить к высвобождению цинка и изменениям в структуре функционирования вирусных белков [Yan Chen, Christine M. Livingston, Stacy D. Carrington-Lawrence, J. of Virology, 2007, v.81, № 16, p. 8742-8751].
«Цинковые пальцы» могут служить мишенью для препаратов нового поколения антивирусного действия. Уже выявлено несколько таких соединений. Однако в настоящее время об их эффективности можно судить, только основываясь на исследованиях, проведенных in vitro.
Вышеприведенная информация позволяет утверждать о важной роли ионов металлов как в нормальном функционировании клеток и всего организма, так и в развитии патологий. Способность некоторых соединений к хелатированию ионов металлов может служить основой для создания препаратов, способных к лечению разнообразных заболеваний, в частности, вирусных.
В статье Megan Whitnall, Jonathan Howard, Prem Ponka, “A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics”, PNAS, 2006, v. 103, № 40, p. 14901-14906, раскрыты эффективные хелаторы железа, которые демонстрируют высокую антипролиферативную и противоопухолевую активность, сравнимую с активностью известных цитоcтатиков, и перспективны для клинических исследований.
В статье Kik K., Szmigiero L., “Dexrazoxane (ICRF-187)- a cardioprotectant and modulator of some anticancer drugs”, Postepy Hig Med Dosw Online, 2006, v. 60, p. 584-590, указано, что некоторые хелаторы железа могут быть использованы в качестве «помощников» при противораковой терапии, так как обладают кардиопротекторным действием.
Хелатирование ионов меди ингибирует ангиогенез и уменьшает рост опухоли [Yu Yu, Jacky Wong, David B. Loveioy, “Chelatorsat the cancer coalface: Desferrioxamine to Triapine and Beyond”, Clin. Cancer Res., 2006, v. 12, p. 6876-6883].
Хелатор цинка - клиохинол, связывая ионы Zn2+, вызывает апоптоз раковых клеток человека [Haijun Yu, Yunfeng Zhou, Stuart E. Lind, “Clioquinol targets zinc to lysosomes in human cancer cells”, Biochem. J., 2009, v. 417, p. 133-139].
В качестве ингибиторов альдегиддегидрогеназы хелаторы используются для лечения алкоголизма [Shian S.G., Kao Y.R., Wu F.Y., Wu C.W., “Inhibition of invasion and angiogenesis by zinc-chelating agent disulfiram”, Mol. Pharmacol., 2003, v. 64(5), p. 1076-84], а также при алкогольном циррозе печени, железоизбыточной анемии, поздней порфирии кожи [Schroterova L., Kaiserova H., Baliharova V., “The effect of new lipophilic chelators on the activities of cytosolic reductases and P450 cytochromes involved in the metabolism of antracyclin as antibiotics: studies in vitro”, Physiol Res., 2004, v. 53(6), p. 683-691].
Активность некоторых антиоксидантов обусловлена хелатированием ионов переходных металлов (Fe, Cu), что сопровождается снижением металлозависимого перекисного окисления липидов [Babizhayev M.A., Seguin Marie-C., Gueynej J., Evstigneev R.P., Ageyeva E.A., Zheltuchina G.A., “L-Carnosine(-alanyl-L-histidine) and carcinine f-alanylhistamine) act as natural antioxidants with hydroxyl-radical-scavenging and lipid-peroxidase activities”, Biochem. J., 1994, v. 304, p. 509-516].
Применение антиоксидантов может способствовать рассасыванию катаракты, устраняет заболевания сетчатки и понижает потребность в инсулине у диабетиков, устраняет пигментацию кожи, а также способствует устранению последствий инсульта. Хелатирование полезно также при лечении воспалительных заболеваний, таких как остеоартриты, ревматоидные артриты. [Зеленин К.Н., "Комплексоны в медицине", Соросовский Образовательный журнал, 2001, т. 7, №1, стр. 45-50].
Хелаторы могут быть использованы в медицине в качестве комплексонов для транспортировки и легкого выведения из организма мышьяка, ртути, сурьмы, кобальта, цинка, хрома, никеля [Жолнин А.В., "Комплексные соединения", Челябинск: ЧГМА, 2000, стр. 28].
Известно ингибирование ботулинического токсина посредством хелатирования ионов цинка [Anne C., Blommaert A., “Thio-derivede disulfides as potent inhibitors of botulinum neurotoxin B: implications of zinc interaction”, Bioorg. Med. Chem., 2003, v. 11(21), р. 4655-60], кроме того, хелатирование защищает при газовой гангрене [Зеленин К.Н., "Комплексоны в медицине", Соросовский Образовательный журнал, 2001, т. 7, №1, стр. 45-50].
Хелатотерапия полезна при лечении нейродегенеративных заболеваний, в частности, болезни Альцгеймера, способствуя улучшению памяти [Bossy-Wetzel E., Schwarzenbacher R., Lipton S.A., “Molecular pathways to neurodegeneration”, Nat. Med, 2004, v. 10, p. 2-9]; болезни Паркинсона [Kevin J. Barnham, Colin L. Masters, Ashley I. Bush, “Neurodegenerative diseases and oxidative stress, Nature Reviews Drug Discovery, 2004, v. 3, p. 205-214]; болезни Вильсона [Yu Yu, Jacky Wong, David B. Lovejoy, “Chelators at the Cancer Coalface: Desferrioxamine to Triapine and Beyond”, Clin. Cancer Res.? 2006, v. 12, p. 6876-6883]; болезни Гентингтона [Whitnall M., Richardson D.R., “Iron: a new target for pharmacological interention in neurodegenerative diseases”, Semin Pediatr Neurol, 2006, v. 13, p. 186-197]; бокового амиотрофического склероза [Kevin J. Bernham, Colin L. Masters, Ashley I. Bush, “Neurodegenerative diseases and oxidative stress”, Nature Reviews Drug Discovery, 2004, v. 4] и прионных заболеваний [Daniel L. Cox, Jianping Pan, Rajiv R.P. Singh, “A Mechanism for Copper Inhibition of Infection Prion Conversion”, Biophysical Journal, 2006, v. 91, L11-L13]. Хелаторы препятствуют возникновению рака [Megan Whitnall, with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics”, PNAS, 2006, v. 103, № 40, p. 14901-14906].
Значительное место в ряду известных хелаторов занимают производные гетероциклических соединений, например, имидазола, содержащие в своем составе имидо- и амидогруппы.
В статье M.A. Podyminogin, V.V. Vlassov, “Synthesis RNA-cleaving molecules mimicking ribonuclease A active center. Design and cleavage of tRNA transcrints”, Nucleic Acids Research, 1993, v. 21, №25, стр. 5950-5956, описано бисгистаминовое производное глутаровой кислоты, которое может служить моделью активного центра нуклеаз и проявляет слабую активность при расщеплении молекул РНК.
В статье Elfriede Schuhmann et al., “Bis[platinum(II)] and Bis[Palladium (II)] complexes of α,ώ-Dicarboxylic Acid Bis(1,2,4-triaminobutane-N4)-Amides”, Inorg. Chem., 1995, v. 34, p. 2316-2322, описаны бисгистаминовые производные глутаровой и адипиновой кислот, которые являются промежуточными соединениями для синтеза комплексов с платиной и палладием:

n=3-6,8
M=Pt, Pd
Способ синтеза N1,N1-глутарилбис(гистамина), включающий взаимодействие гистамина дигидрохлорида и дихлорангидрида глутаровой кислоты в диметилформамиде в присутствии 4-кратного избытка триэтиламина описан в статье Elfriede Schuhmann et al., “Bis[platinum(II)] and Bis[Palladium (II)] complexes of α,ώ-Dicarboxylic Acid Bis(1,2,4-triaminobutane-N4)-Amides”, Inorg. Chem., 1995, v. 34, p. 2316-2322.
Авторами настоящего изобретения впервые было обнаружено, что бисгистаминовое производное глутаровой кислоты, а именно - N1,N1-глутарил бис(гистамин), способно к образованию комплексов с ионами металлов.
Таким образом, целью настоящего изобретения является получение биосовместимых гетероциклических хелаторов ионов металлов и их применение в качестве лекарственного средства для лечения и/или профилактики различных заболеваний, используя способность заявляемых соединений хелатировать ионы металлов.
Задачей изобретения также является разработка простых, использующих доступные реагенты, способов получения таких соединений.
Краткое описание изобретения
Настоящее изобретение относится к производным бисамидов дикарбоновых кислот общей формулы I:

в которой
R1 представляет собой 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 2 гетероатомов, выбранных из N и/или S, необязательно конденсированную с 6-членной ненасыщенной циклической группой;
R2 представляет собой группу -С(О)-R3-C(O)-, где R3 представляет собой группу -(CH2)n-, необязательно замещенную одним или двумя С1-С6 алкилами, или фенил,
n представляет собой целое число от 0 до 4;
или их фармацевтически приемлемым солям.
Настоящее изобретение также относится к производным бисамидов дикарбоновых кислот общей формулы I, обладающим способностью хелатировать ионы металлов (Zn, Сu, Fe, Mg, Са и др.); а также их применению в качестве средства для профилактики и/или лечения сердечно-сосудистых, вирусных, онкологических, нейродегенеративных, воспалительных заболеваний, диабета, геронтологических заболеваний, а также заболеваний, вызываемых токсинами микроорганизмов, а также алкоголизма, алкогольного цирроза печени, анемии, поздней порфирии, отравлений солями переходных металлов.
Настоящее изобретение также относится к способам получения соединений общей формулы I, включающим:
взаимодействие дикарбоновой кислоты и соответствующего амина при нагревании; или
взаимодействие дикарбоновой кислоты с N-гидроксисукцинимидом в присутствии N,N'-дициклогексилкарбодиимида с получением соответствующего бис-N-оксисукцинимидного эфира, который конденсируют с амином; или
взаимодействие диэфира дикарбоновой кислоты с гидразингидратом, обработку полученного дигидразида нитритом натрия с получением соответствующего азида, который конденсируют с амином; или
нагревание раствора имида, образованного из дикарбоновой кислоты, и соответствующего амина в органическом растворителе; или
взаимодействие соответствующего амина и дикарбоновой кислоты в молярных соотношениях 2:1 в присутствии конденструющего агента.
Предлагаемые способы получения гетероциклических биспроизводных дикарбоновых кислот общей формулы I просты в осуществлении, протекают в достаточно мягких условиях, без образования побочных продуктов, технологичны, позволяют получать целевые продукты с хорошим выходом (до 82%) и высокой степенью чистоты.
Детальное описание изобретения
Предпочтительными соединениями настоящего изобретения являются соединения общей формулы I:
,
где R1 представляет собой группу, выбранную из:
 ,
,
R2 представляет собой группу, выбранную из: -С(О)-(CH2)0-C(O)-, -С(О)-(CH2)1-C(O)-, -С(О)-(CH2)2-C(O)-, -С(О)-(CH2)3-C(O)-, -С(О)-(CH2)4-C(O)-, -С(О)-CH2-СН(СН3)-СН2-C(O)-, -С(О)-CH2-С(СН3)2-СН2-C(O)-, или группу
Наиболее предпочтительными соединениями настоящего изобретения являются соединения, представленные в таблице 1.

В качестве фармацевтически приемлемых солей соединений по настоящему изобретению могут быть использованы аддитивные соли органических кислот (например, формиат, ацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и др.), аддитивные соли неорганических кислот (например, гидрохлорид, гидробромид, сульфат, фосфат и др.), соли с аминокислотами (например, соль аспарагиновой кислоты, соль глутаминовой кислоты и т.д.), предпочтительно, хлоргидраты и ацетаты.
Наиболее предпочтительными известными соединениями, которые могут быть использованы в фармацевтической композиции и способе лечения по настоящему изобретению, являются производные глутаримидов, представленные в таблице 2.

Соединения настоящего изобретения могут быть получены способом, включающим конденсацию дикарбоновой кислоты общей формулы II:
R4O-C(O)-R3-C(O)-OR4,
где R3 представляет собой группу -(CH2)n-, необязательно замещенную одним или двумя С1-С6 алкилами, или фенил,
n представляет собой целое число от 0 до 4,
R4 представляет собой водород, С1-С6 алкил,
и амина общей формулы III:
NH2-(CH2)2-R1,
где R1 представляет собой 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 2 гетероатомов, выбранных из N и/или S, необязательно конденсированную с 6-членной ненасыщенной циклической группой;
при нагревании, необязательно в присутствии растворителя.
Предпочтительным является использование диметилового эфира, и нагревание до температуры 150-170ºС, еще более предпочтительно проводить конденсацию при кипении.
В качестве растворителей могут быть использованы диглим или спирты, наиболее предпочтительно, изоамиловый спирт.
Еще одним способом получения производных бисамидов дикарбоновых кислот общей формулы I является способ, включающий взаимодействие дикарбоновой кислоты общей формулы II:
R4O-C(O)-R3-C(O)-OR4,
где R3 представляет собой группу -(CH2)n-, необязательно замещенную одним или двумя С1-С6 алкилами, или фенил,
n представляет собой целое число от 0 до 4,
R4 представляет собой водород,
с N-гидроксисукцинимидом в присутствии N,N'-дициклогексилкарбодиимида в N,N-диметилформамиде с получением соответствующего бис-N-оксисукцинимидного эфира, общей формулы IV:
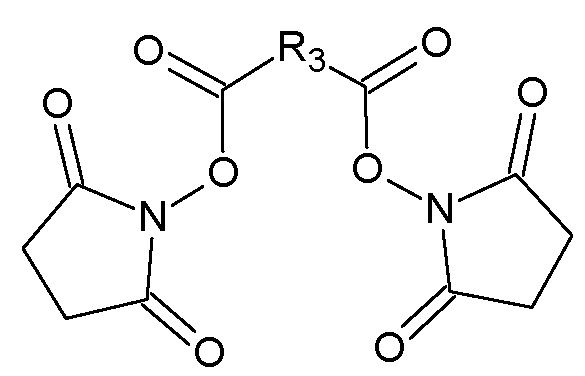 ,
,
который конденсируют с амином общей формулы III:
NH2-(CH2)2-R1,
где R1 представляет собой 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 2 гетероатомов, выбранных из N и/или S, необязательно конденсированную с 6-членной ненасыщенной циклической группой.
Предпочтительным является охлаждение до температуры 0-5ºС.
Еще одним способом получения производных бисамидов дикарбоновых кислот общей формулы I является способ, включающий взаимодействие эфира дикарбоновой кислоты общей формулы II:
R4O-C(O)-R3-C(O)-OR4,
где R3 представляет собой группу -(CH2)n-, необязательно замещенную одним или двумя С1-С6 алкилами, или фенил,
n представляет собой целое число от 0 до 4,
R4 представляет собой С1-С6 алкил,
с гидразингидратом в органическом растворителе с получением бисгидразида общей формулы V:
Н2N-NH-C(O)-R3-C(O)-NH-NH2,
обработку бисгидразида нитритом натрия в кислой среде при температуре около 0ºС с получением бисазида общей формулы VI:
N--N+=N-C(O)-R3-C(O)-N=N+-N-,
конденсацию бисазида с амином общей формулы III:
NH2-(CH2)2-R1,
где R1 представляет собой 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 2 гетероатомов, выбранных из N и/или S, необязательно конденсированную с 6-членной ненасыщенной циклической группой, в органическом растворителе.
Предпочтительно в качестве органического растворителя используют спирты, наиболее предпочтительно, изопропанол. Способ прост, но применим в случае, если количество метиленовых звеньев в исходной дикарбоновой кислоте больше или равно трем, так как получаемые в процессе синтеза бисазиды для кислот с меньшим числом метиленовых групп нестабильны.
Производные бисамидов дикарбоновых кислот общей формулы I также могут быть получены способом, включающим конденсацию имида общей формулы VII:
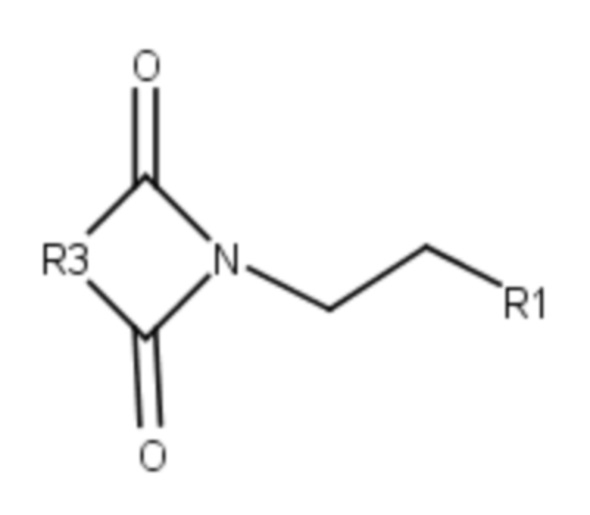 ,
,
где R3 представляет собой группу -(CH2)n-, необязательно замещенную одним или двумя С1-С6 алкилами,
n представляет собой целое число от 0 до 4,
с эквимолярным количеством амина общей формулы III:
NH2-(CH2)2-R1,
где R1 представляет собой 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 2 гетероатомов, выбранных из N и/или S, необязательно конденсированную с 6-членной ненасыщенной циклической группой,
в органическом растворителе при нагревании.
Предпочтительно в качестве органического растворителя используют спирты, наиболее предпочтительно, изопропанол и конденсацию проводят при кипячении.
Еще одним способом настоящего изобретения является способ получения производных бисамидов дикарбоновых кислот общей формулы I, включающий взаимодействие дикарбоновой кислоты общей формулы II:
R4O-C(O)-R3-C(O)-OR4,
где R3 представляет собой группу -(CH2)n-, необязательно замещенную одним или двумя С1-С6 алкилами, или фенил,
n представляет собой целое число от 0 до 4,
R4 представляет собой водород,
и амина общей формулы III:
NH2-(CH2)2-R1,
где R1 представляет собой 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 2 гетероатомов, выбранных из N и/или S, необязательно конденсированную с 6-членной ненасыщенной циклической группой;
при молярном соотношении 1:2-2,5 в растворе тетрагидрофурана в присутствии конденсирующего агента, предпочтительно, карбонилдиимидазола.
Настоящее изобретение также относится к лекарственному средству, фармацевтической композиции, содержащим производные бисамидов дикарбоновых кислот общей формулы I, и способу, включающему введение производных бисамидов дикарбоновых кислот общей формулы I, для профилактики и/или лечения у человека и животных вирусных заболеваний, в том числе заболеваний, вызванных вирусом гепатита С, вирусом папилломы человека, ВИЧ или онкогенными РНК вирусами, такими как вирус лейкемии; сердечнососудистых заболеваний, в том числе заболеваний, вызванных кардиотоксичностью цитостатиков, отложением холестерина, повышенным кровяным давлением; заболеваний, связанных с металлозависимыми реакциями свободнорадикального окисления, в том числе геронтологических заболеваний, таких как катаракта, заболевания сетчатки, пигментация кожи; последствия инсульта; атеросклероз; воспалительных заболеваний, таких как остеоартрит, ревматоидный артрит;
диабета и его сосудистых осложнений; нейродегенеративных заболеваний, в том числе болезни Альцгеймера, болезни Паркинсона, Вильсона, Гентингтона, бокового амиотрофического склероза, прионных заболеваний;
онкологических заболеваний; заболеваний, вызываемых токсинами микроорганизмов, в частности, ботулизма или газовой гангрены;
алкоголизма и алкогольного цирроза печени; железоизбыточной анемии, поздней порфирии; отравлений солями переходных металлов.
Соединения настоящего изобретения вводятся в эффективном количестве, которое обеспечивает желаемый терапевтический результат.
Соединения общей формулы (I) могут быть введены перорально, местно, парентерально, интраназально, ингаляционно и ректально в виде стандартных лекарственных форм, содержащих нетоксичные фармацевтически приемлемые носители. Используемый в настоящем описании термин «парентеральное введение» означает подкожные, внутривенные, внутримышечные или внутригрудные инъекции или вливания.
Соединения настоящего изобретения могут быть введены пациенту в дозах, составляющих от 0,1 до 100 мг/кг веса тела в день, предпочтительно, в дозах от 0,25 до 25 мг/кг один или более раз в день.
При этом следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от многих факторов, включая активность данного используемого соединения, возраст, вес тела, пол, общее состояние здоровья и режим питания пациента, время и способ введения лекарственного средства, скорость его выведения из организма, конкретно используемую комбинацию лекарственных средств, а также тяжесть заболевания у данного индивида, подвергаемого лечению.
Фармацевтические композиции по настоящему изобретению содержат соединение общей формулы (I) в количестве, эффективном для достижения желаемого результата, и могут быть введены в виде стандартных лекарственных форм (например, в твердой, полутвердой или жидкой форме), содержащих соединения настоящего изобретения в качестве активного ингредиента в смеси с носителем или наполнителем, пригодным для внутримышечного, внутривенного, перорального, сублингвального, ингаляционного, интраназального и интраректального введения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, драже, эмульсий, суспензий, мазей, гелей и любых других лекарственных форм.
В качестве наполнителей могут быть использованы различные вещества, такие как сахариды, например, глюкоза, лактоза или сахароза, маннит или сорбит, производные целлюлозы и/или фосфаты кальция, например, трикальций фосфат или кислый фосфат кальция, в качестве связующего компонента могут быть использованы, такие компоненты, как крахмальная паста, например, кукурузный, пшеничный, рисовый, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон. При необходимости могут быть использованы разрыхляющие агенты, такие как вышеупомянутые крахмалы и карбоксиметилкрахмал, поперечносшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Могут быть использованы необязательные добавки, такие как агенты, регулирующие текучесть, и смазывающие агенты, такие как диоксид кремния, тальк, стеариновая кислота и ее соли, такие как стеарат магния или стеарат кальция, и/или пропиленгликоль.
Ядро драже обычно покрывают слоем, который устойчив к действию желудочного сока. Для этой цели могут быть использованы концентрированные растворы сахаридов, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, и подходящие органические растворители или их смеси.
В качестве добавок могут быть также использованы стабилизаторы, загустители, красители и отдушки.
В качестве мазевой основы могут быть использованы углеводородные мазевые основы, такие как вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции - такие как твердый парафин и воск; абсорбтивные мазевые основы, такие как гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens); мазевые основы, смываемые водой, такие как гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы, такие как полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие.
В качестве основы для гелей могут быть использованы метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, оксипропилцеллюлоза, полиэтиленгликоль или полиэтиленоксид, карбопол.
В качестве основы для суппозитория могут быть использованы основы, не растворимые в воде, такие как масло какао; основы, растворимые в воде или смешиваемые с водой, такие как желатино-глицериновые или полиэтиленоксидные; комбинированные основы - мыльно-глицериновые.
При приготовлении стандартной лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьироваться в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства.
Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций, содержание активного агента в них составляет до 5% по массе. В качестве разбавителей могут быть использованы 0,9% раствор хлорида натрия, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы, специфические добавки для растворения. При введении в организм соединений настоящего изобретения в виде таблеток и суппозиториев, их количество составляет до 200 мг на стандартную лекарственную форму.
Лекарственные формы настоящего изобретения получают по стандартным методикам, таким как, например, процессы смешивания, гранулирования, формирование драже, растворение и лиофилизация.
Детальное описание соединений настоящего изобретения, их получения и исследования активности представлено в нижеследующих примерах, предназначенных для иллюстрации предпочтительных вариантов изобретения, и не ограничивающими его объем.
Примеры синтеза производных глутаримидов общей формулы I
Средства и методы
Индивидуальность полученных соединений проверяют методом ТСХ на пластинках “Kieselgel 60 F254” (фирмы “Merck”, Германия) в системе растворителей: пиридин-уксусная кислота-вода (20:6:11) - система А, А:этилацетат 3:1 (1), хлороформ-метанол 9:1 (2).
Электрофорез на бумаге (бумага для электрофореза Кондопожского ЦБК, 120×320 мм) проводят в буфере с рН 5,1 состава пиридин-уксусная кислота-вода 12:10:1000 в камере (150×320×150 мм) с градиентом 15 В/см в течение 1,5 часа.
Хроматограммы и электрофореграммы проявляют хлор-тетраметилбензидиновым реактивом и реактивом Паули.
Температуру плавления определяют на приборе ПТП (завод лаб. приборов, Россия, г. Клин).
Спектры ИК-Фурье снимают в таблетках КВr на приборе “Magna 750” (“Nicolet” (США)).
Спектры 1Н-ЯМР регистрируют на приборе АМХ-400 (Германия), Bruker DPX-400 (Германия).
Масс-спектры высокого разрешения получают на времяпролетном масс-спектрометре методом матриксной лазерно-десорбционной ионизации с использованием в качестве матрицы 2,5-дигидроксибензойной кислоты, на приборе Ultraflex (“Bruker”, Германия).
ЖХ/МС-система анализа многокомпонентных смесей Shimadzu Analytical HPLC SCL10Avp, масс-спектрометр PE SCIEX API 165 (150), (Канада).
Аналитическую обращенно-фазовую ВЭЖХ проводили на приборе: хроматограф HPLC Shimadzu в условиях: колонка Luna C18 (2) 100 A, 250×4,6 мм (сер.599779-23), градиент элюирования в системе фосфатный буферный раствор рН 3,0:метанол (условия А). На приборе хроматограф Beсkman (USA) «System Gold» с УФ-детектором (λ=220 нм), колонка «Gemini» С-18, 150×2,0 мм Phenomenex, подвижная фаза градиент в 0,05 М фосфатном буфере (рН 3,0) 90% MeCN в воде, скорость элюции 0,25 мл/мин, инжекция по 10 мкл (условия Б).
Пример 1
Бис-1,5-(Nβ-гистаминил)глутаровая кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]пентандиамид (соединение 11)
К 5 г (0,031 моль) диметилового эфира глутаровой кислоты прибавляют 8 г (0,072 моль) гистамина и нагревают при 170ºС в течение 3,5-4 ч до прекращения выделения паров метилового спирта. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Затем реакционную массу суспендируют в изопропиловом спирте и оставляют на 24 ч при +4ºС. Продукт отделяют, промывают изопропиловым спиртом, сушат. Выход 6,8 г (69%). Rf 0,42 (1). Е+49 мм. Т.пл. 166-168ºС. ЖХ/МС, индивидуальный пик, время удерживания 0,3 мин [М+Н]+=319. ВЭЖХ в условиях А, индивидуальный пик, время удерживания 5,58 мин. Спектр 1H-ЯМР (400,13 МГц, ДМСО-d6, δ, м.д., J/Гц): 1,70 (пент., 2H, CH2CH2CH2, J=7,3 Гц), 2,03 (т, 4H, CH2CH2CH2, J=7,3 Гц), 2,61 (т, 4H, CCH2CH2N, J=7,5 Гц), 3,26 (кв., 4H, CCH2CH2N, J=7,5 Гц), 6,76 (шир.с, 2H, CCH ), 7,50 (с, 2H, NCHN ), 7,94 (шир.т, 2H, NH ). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1651 (ν C=O амид I), 1580 (δ NH амид II), 1425 (-СН2-СО-). Найдено, %: С 56,40, Н 6,86, N 26,31. С15Н22N6O2. Вычислено, %: C 56,59, H 6,96, N 26,40.
Пример 2
Бис-1,4-(Nβ-гистаминил)янтарная кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]бутандиамид) (соединение 3)
К 5,5 г (0,046 моль) янтарной кислоты прибавляют 12,2 г (0,11 моль) гистамина и нагревают при 170ºС в течение 3,5-4 ч до прекращения выделения паров воды. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Затем реакционную массу суспендируют в 50 мл воды и оставляют на 24 ч при +4ºС. Продукт отделяют, промывают водой, сушат. Выход 10,7 г (76%). Rf 0,51 (1). Е+51 мм. Т.пл. 230-231ºС. [M+Н]+ 304,95. Спектр 1H-ЯМР (D2O): δ, м.д.: 2,48 (с, 4Н, CH2-Suc), 2,80-2,84 (т, 4Н, β-СН2-HA), 3,44-3,48 (т, 4Н, α-СН2-НА), 6,97 (с, 2Н, 5-СН-Im), 7,78 (с, 2Н, 2-СН-Im). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1634 (ν C=O амид I), 1583 (δ NH амид II), 1429 (-СН2-СО-). Найдено, %: С 55,40, Н 6,66, N 27,31. С14Н20N6O2. Вычислено, %: C 55,25, H 6,62, N 27,61.
Пример 3
Бис-1,3-(Nβ-гистаминил)малоновая кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]пропандиамид) (соединение 2)
К 6,4 г (0,039 моль) диэтилового эфира малоновой кислоты прибавляют 9,0 г (0,081 моль) гистамина и нагревают при 170ºС в течение 2,5-3 ч до прекращения выделения паров этилового спирта. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Затем реакционную массу суспендируют в 20 мл воды и оставляют на 72 ч при +4ºС. Продукт отделяют, промывают изопропиловым спиртом, сушат. Выход 5,0 г (44%). Rf 0,41 (1). Е+57 мм. Т.пл. 187-188ºС. Спектр 1H-ЯМР (D2O): δ, м.д.: 2,74-2,78 (т, 4Н, β-СН2-НА), 3,15 (с, 2Н, СН2-Mal), 3,41-3,44 (т, 4Н, α-СН2-НА), 6,78 (с, 2Н, 5-СН-Im), 7,66 (с, 2Н, 2-СН-Im). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1643 (ν C=O амид I), 1574 (δ NH амид II), 1426 (-СН2-СО-). Найдено, %: С 53,24, Н 6,51, N 28,56. С13Н18N6O2. Вычислено, %: C 53,78, H 6,25, N 28,95.
Пример 4
Бис-1,2-(Nβ-гистаминил)щавелевая кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]этандиамид) (соединение 1)
К раствору 7,3 г (0,05 моль) диэтилового эфира щавелевой кислоты в 35 мл изоамилового спирта прибавляют 12,2 г (0,11 моль) гистамина и кипятят в течение 4 ч. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Реакционную смесь оставляют на 16 ч при +4ºС. Осадок отделяют, промывают изопропиловым спиртом, сушат. Выход 10 г (72%). Rf 0,55 (1). Е+55 мм. Т.пл. 235-236ºС. Спектр 1H-ЯМР (ДМСО): δ, м.д.: 2,66-2,72 (т, 4Н, β-СН2-НА), 3,33-3,41 (м, 4Н, α-СН2-НА), 6,81 (с, 2Н, 5-СН-Im), 7,54 (с, 2Н, 2-СН-Im), 8,78-8,84 (т, 2Н, NH-CO). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1651 (ν C=O амид I), 1566 (δ NH амид II), 1425 (-СН2-СО-). Найдено, %: С 52,24, Н 5,51, N 29,97. С12Н16N6O2. Вычислено, %: C 52,17, H 5,84, N 30,42.
Пример 5
Бис-1,3-(Nβ-гистаминил)изофталевая кислота (соединение 6)
В 30 мл диметилфорамида растворяют 5 г (30 ммоль) изофталевой кислоты и 7,93 г (69 ммоль) N-гидроксисукцинимида. К полученному раствору, охлажденному до 5ºС, прибавляют охлажденный раствор 14,24 г (69 ммоль) N,N’-дициклогексилкарбодиимида в 10 мл диметилфорамида, реакционную смесь оставляют на ночь при 5ºС. Мочевину отделяют, промывают 15 мл диметилформамида и сушат. Исходя из массы полученной мочевины, выход реакции считают равным 80%. Раствор N-гидроксисукцинимидного диэфира изофталевой кислоты охлаждают до 5ºС, после чего к нему порциями приливают расплав 6,84 г (61,6 ммоль) гистамина. Реакционную смесь оставляют на 2 ч. Растворитель удаляют в вакууме. Остаток, представляющий собой желтое масло, растворяют в 120 мл воды и пропускают через колонку с Amberlite IRA-96 (27×115). Фракции, содержащие продукт, объединяют. Выпавшие кристаллы целевого продукта отделяют. Полученный технический продукт перекристаллизовывают из смеси 35 мл изопропанола с 10 мл воды. Кристаллы продукта фильтруют, промывают водой (3×30 мл) и изопропанолом (2×13 мл), сушат. Получают целевой продукт в виде белых кристаллов. Выход 1,55 г (18%, считая на гистамин, или 14% - на изофталевую кислоту). Rf 0,42 (1). Т.пл. 218-219ºС. Е+40 мм. Macc-спектр: [М+1]+ 353. Спектр 1H-ЯМР: (300 МГц, ДМCO-d6): δ, м.д.: 2,74-2.79 (т, 4Н, β-СН2-Hа), 3,46-3,52 (т, 4Н, α-СН2-Ha), 6,81 (с, 2Н, 5-СН-Im), 7,52-7,56 (м, 3Н, ArН+2-СН-Im), 7,92-7,95 (д, 2Н, ArН), 8,28 (с, 1Н, ArН), 8,65 (шир.с, 2Н, -С(О)-NН-), 11,80 (шир.с, 2Н, -NН-). ВЭЖХ в условиях Б, индивидуальный пик, время выхода 12,5 мин.
Пример 6
Бис-1,5-(Nβ-гистаминил)глутаровая кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]пентандиамид) (соединение 11)
К раствору 13,2 г (0,10 моль) глутаровой кислоты в 50 мл метанола при охлаждении и перемешивании порциями добавляют 8 мл PCl3. Растворитель из реакционной смеси удаляют в вакууме. Полученный остаток перегоняют в вакууме. Получают 14,7 г (92%) диметилового эфира глутаровой кислоты с т.кип. 110-112ºС.
К 20 мл изопропилового спирта приливают 7,5 мл гидразингидрата, нагревают до кипения и по каплям добавляют 8 г (0,05 моль) диметилового эфира глутаровой кислоты, реакционную смесь оставляют при +20ºС на 16 ч. Выпавший в осадок продукт отделяют, промывают изопропиловым спиртом. Сушат. Получают 7,3 г (91%) дигидразида глутаровой кислоты с т.пл. 210-212ºС.
К раствору 1,61 г (0,010 моль) дигидразида глутаровой кислоты в смеси 20 г льда с 2,5 мл соляной кислоты и 10 мл предварительно охлажденного хлороформа при перемешивании порциями прибавляют 1,45 г (0,021 моль) нитрита натрия. Слой хлороформа отделяют, промывают 10 мл охлажденной до +4ºС воды и приливают к охлажденному раствору 2,6 г (0,023 моль) гистамина в 5 мл изопропанола. Реакционную смесь оставляют при +20ºС на 2 ч, затем растворитель удаляют в вакууме. Полученный остаток растворяют в 20 мл воды и пропускают через колонку с 50 мл ионообменной смолы КУ-2-20 в Н+-форме. Смолу промывают 0,5%-ным раствором аммиака, собирая фракции продукта, не содержащего гистамин. Элюаты объединяют, растворитель удаляют в вакууме, полученный остаток перекристаллизовывают из 2 мл изопропанола, сушат. Получают 1,8 г (55%). Rf 0,42 (1). Е+49 мм. Т.пл. 166-168ºС. Спектр 1H-ЯМР (D2O): δ, м.д.: 1,73-1,78 (м, 2Н, β-CH2-Glt), 2,10-2,15 (т, 4Н, α-СН2-Glt), 2,78-2,82 (т, 4Н, β-СН2-НА), 3,43-3,48 (т, 4Н, α-СН2-НА), 6,94 (с, 2Н, 5-СН-Im), 7,69 (с, 2Н, 2-СН-Im). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1651 (ν C=O амид I), 1580 (δ NH амид II), 1425 (-СН2-СО-). Найдено, %: С 56,40, Н 6,86, N 26,31. С15Н22N6O2. Вычислено, %: C 56,59, H 6,96, N 26,40.
Пример 7
Бис-1,5-(Nβ-гистаминил)адипиновая кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]гександиамид) (соединение 12)
К раствору 7,40 г (0,05 моль) адипиновой кислоты в 25 мл метанола при охлаждении по каплям добавляют 4 мл треххлористого фосфора. Растворитель из реакционной смеси удаляют в вакууме, полученный остаток перегоняют в вакууме. Получают 7,5 г (86%) диметилового эфира адипиновой кислоты, т.кип. 115-117ºС.
К смеси 20 мл изопропропилового спирта и 5 мл гидразингидрата, нагретой до кипения, по каплям прибавляют 7,5 г (0,04 моль) диметилового эфира адипиновой кислоты, реакционную смесь оставляют при +20ºС на 16 ч. Выпавший в осадок продукт отделяют, промывают изопропиловым спиртом, сушат. Получают 6,2 г (83%) дигидразида адипиновой кислоты с т.пл. 182-182,5ºС.
К раствору 2,2 г (0,013 моль) дигидразида адипиновой кислоты в смеси 30 г льда с 3 мл соляной кислоты и 15 мл охлажденного хлороформа при перемешивании постепенно порциями присыпают 1,8 г (0,026 моль) нитрита натрия. Слой хлороформа отделяют, промывают 10 мл охлажденной до +4ºС воды и приливают к охлажденному раствору 3 г (0,027 моль) гистамина в 8 мл изопропанола. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Реакционную смесь оставляют на 24 ч при +4ºС, осадок отделяют, промывают изопропиловым спиртом, перекристаллизовывают из воды, сушат. Получают 2,8 г (65%). Rf 0,48 (1). Е+45 мм. Т.пл. 184-186ºС. [M+Н]+ 332,92. Спектр 1H-ЯМР (D2O): δ, м.д.: 1,37-1,41 (м, 4Н, β-CH2-Аdip), 2,11-2,15 (м, 4Н, α-СН2-Adip), 2,74-2,78 (т, 4Н, β-СН2-НА), 3,40-3,44 (т, 4Н, α-СН2-НА), 6,88 (с, 2Н, 5-СН-Im), 7,63 (с, 2Н, 2-СН-Im). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1646 (ν C=O амид I), 1581 (δ NH амид II), 1425 (-СН2-СО-). Найдено, %: С 57,45, Н 7,15, N 24,97. С16Н24N6O2. Вычислено, %: C 57,81, H 7,28, N 25,28.
В соответствии с вышеуказанной методикой получены следующие соединения:

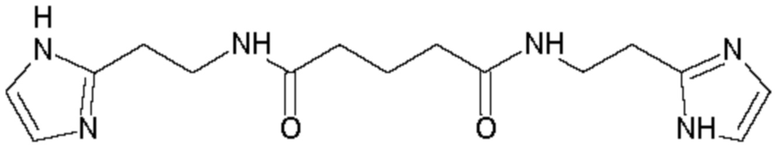
Пример 8
Бис-1,4-(Nβ-гистаминил)янтарная кислота; (N,N'-бис-[2-(1Н-имидазол-4-ил)этил]бутандиамид) (соединение 3)
К раствору 0,10 г (0,9 ммоль) гистамина в 3 мл изопропилового спирта прибавляют 0,15 г (0,78 ммоль) сукцинимидогистамина и нагревают до кипения в течение 2,5-3 ч. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Затем реакционную массу охлаждают до комнатной температуры и оставляют на 26 ч при +4ºС. Продукт отделяют, промывают изопропиловым спиртом, сушат. Выход 0,20 г (81%). Rf 0,44 (1). Е+51 мм. Т.пл. 231ºС. [M]+ 304,12. Спектр 1H-ЯМР (D2O): δ, м.д.: 2,48 (с, 4Н, CH2-Suc), 2,80-2,84 (т, 4Н, β-СН2-HA), 3,44-3,48 (т, 4Н, α-СН2-НА), 6,97 (с, 2Н, 5-СН-Im), 7,78 (с, 2Н, 2-СН-Im). Спектр ИК-Фурье (в таблице КВr, ν, см-1): 1634 (ν C=O амид I), 1583 (δ NH амид II), 1429 (-СН2-СО-). Найдено, %: С 6,57, N 27,18. С14Н20N6O2. Вычислено, %: C 55,25, H 6,62, N 27,61.
Пример 9
Бис-1,5-(N-этиламино-2-бензимидазолил)глутаровая кислота; (N,N'-бис-[2-(1Н-бензимидазол-2-ил)этил]пентандиамид) (соединение 7)
К раствору 8 г (0,06 моль) глутаровой кислоты в 500 мл безводного тетрагидрофурана прибавляют 23,4 г (0,145 моль) карбонилдиимидазола, перемешивают 2 ч. Затем к реакционной смеси добавляют 21,6 г (0,133 моль) бензимидазолил этиламин. Полученный раствор перемешивают 3ч и оставляют при комнатной температуре. Осадок продукта отделяют, промывают водой, сушат. Получают 21 г (82,9%). Rf 0,21 (2). Т.пл. 170-170,5ºС. [M+1]+ 419. Спектр 1H-ЯМР (ДМСО): δ, м.д.: 1,61-1,76 (м, 2H, СН2), 1,95-2,07 (т, 2H, CH2), 2,9-3,0 (т, 4H, 2CH2), 3,45-3,55 (кв., 4H, 2CH2), 7,06-7,14 (м, 4H, Ar), 7,39-7,54 (м, 4H, Ar), 7,98-8,08 (т, 2H, Ar), 11,6-12,7 (с, 2H, NH). ВЭЖХ в условиях Б, индивидуальный пик, время выхода 26,6 мин.
В соответствии с вышеуказанной методикой получены следующие соединения:

Исследование хелатирующей способности описываемых соединений
Предлагаемые соединения общей формулы I, как показали дальнейшие исследования, обладают способностью к комплексообразованию или хелатированию ионов металлов. Они имеют несколько функциональных групп, способных выступать в роли доноров электронов при комплексообразовании, например, карбоксильная группа, имидо-, амидогруппы, и представляют собой лиганды, специфически взаимодействующие с ионами металлов, например, с ионами цинка, меди, железа, кальция, магния.
Важной характеристикой способности лиганда к комплексообразованию являются константы диссоциации (pkn) исследуемых соединений в водном растворе. Величины pkn определяют, используя метод потенциометрического титрования. Расчет констант диссоциации проводят согласно методу Шварценбаха [Гринберг А.А., "Введение в химию комплексных соединений", изд.4-е, испр., Л., Химия, 1971].
Методы рН-потенциометрического титрования
Титрование проводилось на установке с использованием иономера лабораторного типа И 120.1. Созданная гальваническая ячейка состояла из двух электродов: индикаторного стеклянного и хлорсеребряного электрода сравнения. Значения рН определялись с точностью ±0,2.
Исходные 0,05 М растворы солей ионов металлов Mz+ готовили из нитратов цинка (Zn(NO3)2·6H2O), кальция (Ca(NO3)2·4H2O), сульфата меди (CuSO4·5H2O), хлорида магния (MgCl2·6H2O) и соли Мора ((NH4)2Fe(SO4)2·6H2O). Стандартизацию исходных растворов солей Mz+ производили методами комплексонометрического титрования с трилоном Б (Mz+=Zn2+, Cu2+, Mg2+, Ca2+), спектрофотометрически при 490 нм с помощью о-фенантролина (Mz+=Fe2+).
Типичная методика определения ступенчатых констант кислотной диссоциации (k1, . . ., kn) рН-метрическим титровнием по методу Шварценбаха
Навеску исследуемого соединения (3⋅10-4 моль) растворяли в 15,0 мл 0,5 М водного раствора KNO3. Полученный 0,02 М раствор при интенсивном перемешивании титровали 0,05 М водным раствором NaOH или HCl. Фиксировали в каждой точке титрования количество (V, мл) израсходованного титранта и рН раствора. Шаг титрования составлял 0,5 мл, вблизи точки эквивалентности - 0,2 мл. Титрование проводили до значений рН раствора 11,0-11,5. По кривой титрования рН=f(а) определяли соответствующее значению а=0,5 и/или а=1,5 значение рН раствора, а - количество прибавленных г-эквивалентов титранта в пересчете на 1 моль титруемого вещества.
Результаты определения констант кислотной диссоциации [pkn] по методу Шварценбаха исследуемых соединений представлены в таблице 3.
Значения pk исследуемых соединений в водном растворе, рассчитанные по методу Шварценбаха (С 0,02 М, ионная сила раствора 0,5 (KNO3), 22ºС)
(2) - титрование водным 0,05 М раствором NaOH.
С учетом таких параметров, как количество исследуемого вещества, необходимого для потенциометрического титрования, методика его проведения и расчета констант, наиболее подходящим, информативным и простым является метод Бьеррума [Галактионов, М.А. Чистяков, В.Г. Севастьянов и др., "Ступенчатые константы устойчивости комплексов элементов III Б группы и лантаноидов с ацетилацетоном в водном растворе и произведения растворимости их триацетилацетонатов", Ж. Физ. Хим., 2004, т. 78, №9, с. 1596-1604].
Метод основывается на представлении о ступенчатости процесса комплексообразования. Главным допущением метода Бьеррума является предположение об образовании только моноядерных комплексов. Схематически процесс комплексообразования можно описать следующим образом:
M+L↔ML K1=[ML]/[M][L]
MLn-1+L↔MLn Kn=[MLn]/[MLn-1][L]
Согласно методу Бьеррума исследуемое соединение титруется дважды раствором NaOH: в отсутствие и в присутствии иона металла. Титрование проводят при высокой постоянной концентрации индифферентного электролита, обеспечивающей постоянную ионную силу раствора.
Для этих целей применяют хорошо растворимые соли, например, нитрат калия. Экспериментально находят концентрационные ступенчатые константы устойчивости (Kn). Кроме того, возможно получение информации о составе образующихся комплексов, а именно - максимально возможном числе присоединяемых лигандов (n).
Типичная методика определения ступенчатых констант устойчивости комплексов рН-метрическим титрованием по методу Бьеррума
Навеску исследуемого соединения (3⋅10-4 моль) растворяли в 14,0 мл 0,5 М водного раствора KNO3. При интенсивном перемешивании добавляли 1,0 мл (5⋅10-4 моль) 0,05 М водного раствора соли Mz+. Исследовали системы с соотношением Mz+ к L 1:6. Фиксировали значения рН раствора при совместном присутствии лиганда и ионов металла в растворе. Титрование смеси осуществляли 0,05 М водным раствором NaOH в широком интервале рН (до значений рН 11,0-11,5). Фиксировали в каждой точке титрования количество израсходованного титранта (VNaOH, мл) и рН раствора. В момент образования осадка, помутнения раствора или изменения его окраски фиксировали значение рН, характер изменения и объем раствора NaOH, пошедшего на титрование. Шаг титрования равен 0,25 мл. По кривой Бьеррума  =f(pA) при каждом полу-целочисленном значении функции образования =n-0,5 определяли lgKn.
=f(pA) при каждом полу-целочисленном значении функции образования =n-0,5 определяли lgKn.
В настоящее время метод Бьеррума часто применяется для изучения комплексообразования. Этот метод предусматривает определение двух параметров - концентрации свободного лиганда в равновесных условиях - [L-] и функции образования , представляющей собой среднее число лигандов, входящих в комплекс. Так, в любой момент титрования, используя экспериментальные данные, можно рассчитать [L-] (а именно, -lg[L-]=рА) и по уравнению (1) - . Когда лиганд имеет несколько диссоциирующих групп, то в уравнении (1) KL соответствует значению константы диссоциации, имеющей наибольшее значение рk.
Из построенной кривой Бьеррума (кривой образования) =f(pA) по уравнению (2) при каждом полу-целочисленном значении функции образования =n-0,5 находят Kn. Таким образом, ступенчатая константа устойчивости будет равна обратной концентрации свободного лиганда в точке  =n-0,5. Соответственно, чем больше значение Kn, тем более устойчивым является комплексное соединение (хелат).
=n-0,5. Соответственно, чем больше значение Kn, тем более устойчивым является комплексное соединение (хелат).

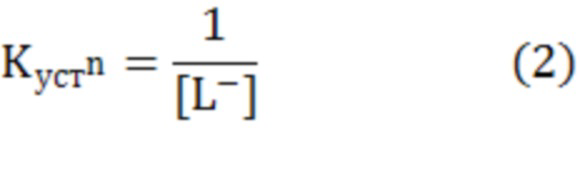
где  и
и  - общие концентрации присутствующих в растворе лиганда и металла, моль/л;
- общие концентрации присутствующих в растворе лиганда и металла, моль/л;
[L-] - концентрация свободных ионов лиганда, моль/л;
KL - константа кислотной диссоциации лиганда;
 - объем раствора NaOH (
- объем раствора NaOH ( , моль/л) в любой момент титрования, добавленный к титруемой смеси Mz+ и L, л;
, моль/л) в любой момент титрования, добавленный к титруемой смеси Mz+ и L, л;
nL,  - количество вещества лиганда и соли Mz+ в титруемом растворе, моль.
- количество вещества лиганда и соли Mz+ в титруемом растворе, моль.
Найденные по методу Бьеррума значения lgK1 для комплексов исследуемых соединений с ионами двухвалентных металлов, представлены в таблицах 4, 5. Выбор соотношения Mz+ к L, равного 1:6, обусловлен наличием данных о максимально возможном координационном числе для Zn(II) и Fe(II), равном шести, и для Cu(II) - равном четырем [Claudia A. Blindauer, M. Tahir Razi, Simon Parsons, Peter J. Sadler, Polyhedron, 2006, v. 25, р. 513-520].
Первая константа устойчивости комплексов состава 1:1 исследуемых соединений с ионами двухвалентных металлов - lgK1, найденные по методу Бьеррума (водный раствор, CL=0,02 M, соотношение Mz+ к L 1:6, ионная сила 0,5 (KNO3), 22ºС)
<0,5
Ступенчатые константы устойчивости комплексов состава 1:1 исследуемых соединений с ионами цинка, найденные по методу Бьеррума (водный раствор, CL=0,02 M, соотношение Mz+ к L 1:6, ионная сила раствора 0,5 (KNO3), 22ºС)
Таким образом, проведенные исследования показали, что испытанные соединения - производные бисамидов дикарбоновых кислот, являются эффективными комплексообразователями, для большинства из которых найдены высокие значения lgK1≥5 по отношению к ионам переходных металлов. При этом наблюдается несколько повышенная комплексообразующая способность предлагаемых соединений общей формулы I по отношению к ионам железа.
Изобретение относится к фармацевтической композиции для профилактики и/или лечения воспалительных заболеваний, включающей эффективное количество соединения формулы 11 или его фармацевтически приемлемой соли. Изобретение относится также к лекарственному средству для профилактики и/или лечения воспалительных заболеваний, включающему соединение формулы 11. Указанное соединение обладает способностью к комплексообразованию или хелатированию ионов металлов и может найти применение в качестве средства для профилактики и/или лечения воспалительных заболеваний. 2 н.п. ф-лы, 5 табл., 9 пр.
 11
11
1. Фармацевтическая композиция для профилактики и/или лечения воспалительных заболеваний, включающая эффективное количество соединения формулы 11
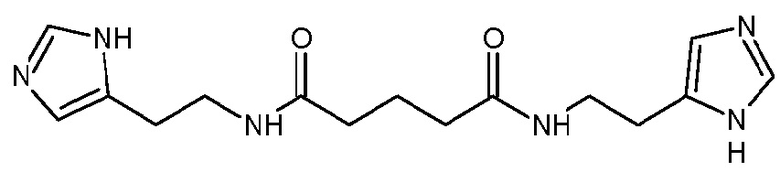
или его фармацевтически приемлемой соли.
2. Лекарственное средство для профилактики и/или лечения воспалительных заболеваний, включающее соединение формулы 11 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
| G | |||
| FERRY ET AL., A zinc chelator inhibiting gelatinases exerts potent in vitro anti-invasive effects, EUROPEAN JOURNAL OF PHARMACOLOGY, 1998, vol | |||
| Деревобетонный каток | 1916 |
|
SU351A1 |
| Синхронизирующее устройство для аппарата, служащего для передачи изображений на расстояние | 1920 |
|
SU225A1 |
| E | |||
| SCHUHMANN ET AL., Bis[platinum(II)] and Bis[palladium(II)] Complexes of α,ω-Dicarboxylic Acid Bis(1,2,4-triaminobutane-N4) Amides, INORGANIC CHEMISTRY, 1995, vol | |||
| Нивелир для отсчетов без перемещения наблюдателя при нивелировании из средины | 1921 |
|
SU34A1 |

