Изобретение относится к новым биологически активным соединениям, производным глутаримидов или их фармацевтически приемлемым солям, их применению в качестве средств для лечения заболеваний верхних дыхательных путей, а также к способам получения предлагаемых соединений.
Уровень техники
Хронические заболевания верхних дыхательных путей принадлежат к наиболее распространенным заболеваниям у детей и взрослых по всему миру. К хроническим заболеваниям верхних дыхательных путей относят, в частности, риносинусит.
Риносинусит (РС) представляет собой воспаление слизистой оболочки полости носа и околоносовых пазух (ОНП), проблема которого в настоящее время является одной из самых актуальных в оториноларингологии (Fokkens W.J., Lund V.J., Mullol J. et al., “European Position Paper on Rhinosinusitis and Nasal Polyps”, Rhinology, 2007; 45; 20: 1-139). Причиной риносинусита практически всегда является застой секрета, блок естественных соустий ОНП и нарушение их аэрации, когда страдает механизм мукоцилиарного клиренса, который является важным первичным врожденным механизмом, защищающим дыхательные пути от повреждающего действия вдыхаемых загрязняющих веществ, аллергенов и возбудителей.
Острый риносинусит представляет собой частое осложнение при острой респираторной вирусной инфекции (ОРВИ).
В настоящее время лечение риносинуситов начинают с применения кортикостероидов, т.к. они обладают выраженным противовоспалительным эффектом. Кортикостероиды применяют в качестве монотерапии или в сочетании с антибиотиками. В случае более тяжелых форм ринусинуситов применяются антибиотики. Основными кортикостероидами являются флутиказон, будесонид и мометазон. Кортикостероиды при лечении ринусинуситов назначают продолжительными курсами, что может вызвать побочные эффекты и толерантность. Побочные эффекты, как правило, являются проявлением собственно глюкокортикоидного действия этих лекарственных средств, но в степени, превышающей физиологическую норму.
В качестве антибиотиков, как правило, назначаются антибиотики группы пенициллина (амоксициклин, пенициллин V) или непенициллиновые антибиотики (макролиды, тетрациклины) (Fokkens W.J., Lund V.J., Mullol J. et al., “European Position Paper on Rhinosinusitis and Nasal Polyps”, Rhinology, 2007; 45; 20: 1-139).
Таким образом, необходимы новые препараты, которые позволили бы интенсифицировать лечение риносинусита и уменьшить воспалительную реакцию со снижением гнойного воспаления и глубоких повреждений в виде язвенно-некротических дефектов, а также препятствовать переходу заболевания в хроническую форму. Таким образом, целью настоящего изобретения является развитие и внедрение новых лекарственных препаратов для лечения риносинусита.
В публикации международной заявки WO 2007/007054 раскрыты производные сукцинимидов и глутаримидов общей формулы (I), обладающие действием, ингибирующим метилирование ДНК в клетках, в частности опухолевых клетках. Раскрытые в данной публикации соединения получают реакцией сочетания аминопроизводного соединения, содержащего углеводородную цепь, с соответствующим ангидридом или кислотой, или эфиром, с последующим, если необходимо, закрытием кольца, необязательно в присутствии основания.
Наиболее распространенным способом синтеза имидов дикарбоновых кислот является метод термической циклизации, заключающийся в нагревании дикарбоновой кислоты или ее производного, такого как ангидрид, диэфир и другие, с первичным амином или его амидом. Выход циклических имидов обычно составляет 80%, однако, поскольку процесс ведут при высоких температурах, он может быть использован только для синтеза термически стабильных имидов [Вейганд - Хильгетаг, “Методы эксперимента в органической химии”, Под ред. проф. Н.Н. Суворова, М., Химия, 1968, стр. 446].
В статье Yong Sup Lee et al., “Studies on the site-selective N-acyliminium ion cyclazation: synthesis of (±)-glochidine and (±)-glochidicine”, Heterocycles, Vol 37, No 1, 1994, раскрыто получение сукцинимида гистамина сплавлением дигидрохлорида гистамина и янтарного ангидрида при нагревании исходных реагентов до 200-230°С в течение 40 минут.
В международной публикации патентной заявки WO2007/000246 описан способ синтеза глутаримидов алкилированием пиперидин-2,6-диона и пирролидин-2,5-диона соответствующими галогенпроизводными в ДМФА, с выделением целевых замещенных имидов методом препаративной хроматографии, что неприменимо для синтеза макроколичеств.
В статье Shimotori et al., “Asymmetric synthesis of δ-lactones with lipase catalyst”, Flavour and Fragrance Journal, 2007, V. 22, №6, p. 531-539, описан способ получения циклических имидов циклизацией моноамидов соответствующих дикарбоновых кислот с использованием водоотнимающего средства в качестве реагента, активирующего карбоксильную группу, такого как уксусный ангидрид.
В статье Ito et al., “Chemoselective Hydrogenation of Imides Catalyzed by CpRu(PN) Complexes and Its Application to the Asymmetric Synthesis of Paroxetine”, Journal of the American Chemical Society, 2007, V. 129, №2, p. 290-291, описан способ получения циклических имидов циклизацией моноамидов соответствующих дикарбоновых кислот с использованием водоотнимающего средства в качестве реагента, активирующего карбоксильную группу, такого как ацетилхлорид.
В статье Polniaszek et al., “Stereoselective nucleophilic additions to the carbon-nitrogen double bond. 3. Chiral acyliminium ions”, Journal of Organic Chemistry, 1990, V. 55, №1, p. 215-223, описан способ получения циклических имидов циклизацией моноамидов соответствующих дикарбоновых кислот с использованием водоотнимающего средства в качестве реагента, активирующего карбоксильную группу, такого как карбонилдиимидазол.
В статье Ainhoa Ardeo et al., “A practical approach to the fused β-carboline system. Asymmetric synthesis of indolo[2,3-α]indolizidinones via a diastereoselective intramolecular α-amidoalkylation reaction”, Tetrahedron Letters, 2003, 44, 8445-8448, описан способ синтеза имидов из первичного амина и соответствующего ангидрида, в котором в качестве водоотнимающего средства используют избыток глутарового или янтарного ангидридов. В частности, в данной статье приведена схема синтеза глутаримидотриптамина и сукцинимидотриптамина исходя из триптамина и ангидридов соответствующих кислот при кипячении в уксусной кислоте. Глутаримидотриптамин получают по этому методу с выходом 67%, сукцинимидотриптамин - 81%.
Таким образом, целью настоящего изобретения является создание новых нетоксичных производных глутаримидов, эффективных для лечения риносинусита.
Краткое описание изобретения
Настоящее изобретение относится к производным глутаримидов общей формулы (I):
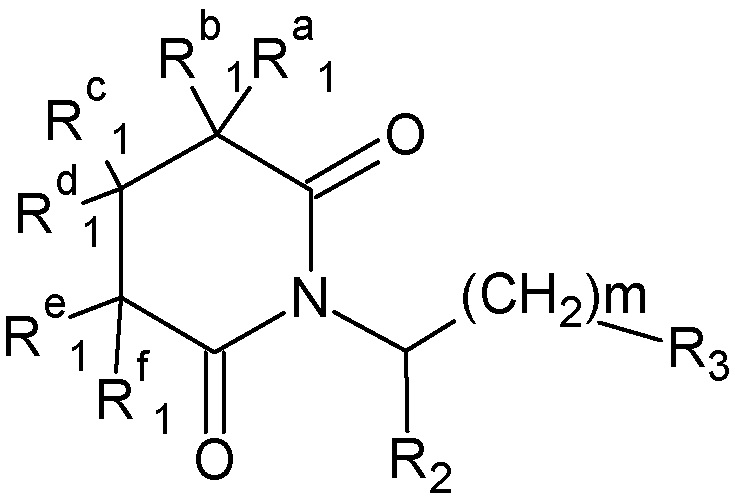
где
m представляет собой целое число от 0 до 2;
Ra1, Rb1, Rc1, Rd1, Re1, Rf1, каждый независимо представляет собой водород, С1-С6 алкил; -NH2, -NНC1-C6 алкил, гидроксигруппу, С1-С6 алкокси;
R2 представляет собой водород, С1-С6 алкил, группу -С(О)ОН, -С(О)ОС1-С6 алкил;
R3 представляет собой:
1) 5-членную насыщенную или ненасыщенную гетероциклическую группу, содержащую от 1 до 4 гетероатомов, выбранных из N, O и S, необязательно замещенную от 1 до 3 заместителей, выбранных из галогена, С1-С6 алкила, С1-С6 алкокси, группы -С(О)ОН, группы -С(О)ОС1-С6 алкил, группы -NHС(О)С1-С6 алкил, фенила или пиридинила;
2) 6-членную насыщенную или ненасыщенную гетероциклическую группу, содержащую 1 или 2 гетероатома, выбранных из N и O, необязательно замещенную группой, выбранной из галогена, С1-С6 алкила;
3) 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 3 гетероатомов, выбранных из N и S, необязательно замещенную 1 или 2 заместителями, выбранными из С1-С6 алкила, конденсированную с 6-членной ненасыщенной циклической или гетероциклической группой, содержащей атом азота, необязательно замещенной одним или двумя заместителями, выбранными из гидроксигруппы, галогена или С1-С6 алкила;
4) 6-членную ненасыщенную циклическую или гетероциклическую группу, содержащую 1 или 2 атома N, конденсированную с 5- или 6-членной ненасыщенной гетероциклической группой, содержащей от 1 до 3 гетероатомов, выбранных из N и S;
5) группу формулы
или его фармацевтически приемлемые соли,
при условии, что соединение не является соединением, в котором:
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой -C(O)OCH3, R3 не является:
 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
 ;
;
когда m=1, Ra1 представляет собой аминогруппу, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, или Re1 представляет собой аминогруппу, Ra1, Rb1, Rc1, Rd1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
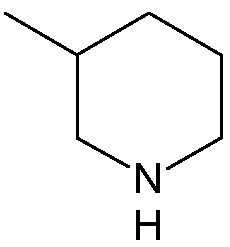 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
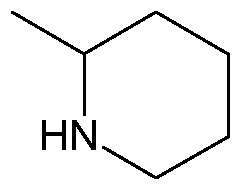 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
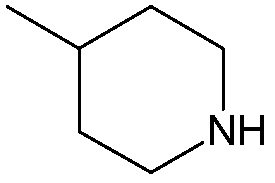 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
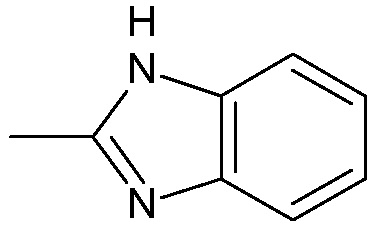 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой -С(О)ОН, R3 не является:
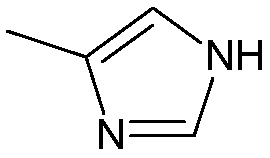 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой -С(О)ОН, R3 не является:
 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
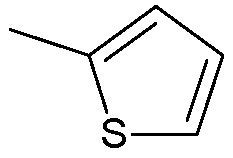 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
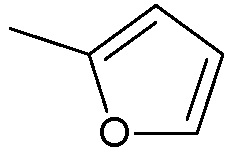 ;
;
когда m=2, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
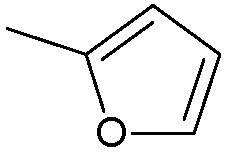 ;
;
когда m=2, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
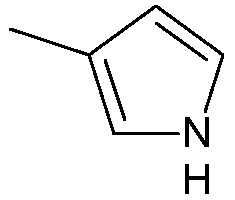 ;
;
когда m=1, Ra1, Rb1, Rc1, Rd1, Re1, Rf1 представляют собой водород, R2 представляет собой водород, R3 не является:
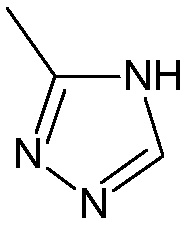 .
.
Настоящее изобретение также относится к лекарственному средству для лечения заболеваний верхних дыхательных путей, предпочтительно, риносинусита, представляющему собой производные глутаримидов общей формулы (I) или их фармацевтически приемлемые соли.
Еще одним объектом настоящего изобретения является фармацевтическая композиция для лечения заболеваний верхних дыхательных путей, предпочтительно, риносинусита, включающая эффективное количество производных глутаримидов общей формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способу лечения заболеваний верхних дыхательных путей, предпочтительно, риносинусита, включающему введение пациенту эффективного количества производных глутаримидов общей формулы (I) или его фармацевтически приемлемой соли.
Настоящее изобретение далее относится к способу получения производных глутаримидов общей формулы (I) или их фармацевтически приемлемых солей нагреванием моноамида дикарбоновой кислоты общей формулы (II).
Формула II:
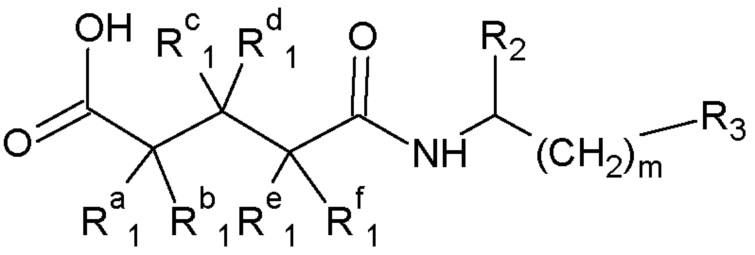
где
m представляет собой целое число от 0 до 2;
Ra1, Rb1, Rc1, Rd1, Re1, Rf1, каждый независимо представляет собой водород, С1-С6 алкил; -NH2, -NНC1-C6 алкил, гидроксигруппу, С1-С6 алкокси;
R2 представляет собой водород С1-С6 алкил, группу -С(О)ОН, -С(О)ОС1-С6 алкил;
R3 представляет собой:
1) 5-членную насыщенную или ненасыщенную гетероциклическую группу, содержащую от 1 до 4 гетероатомов, выбранных из N, O и S, необязательно замещенную от 1 до 3 заместителей, выбранных из галогена, С1-С6 алкила, С1-С6 алкокси, группы -С(О)ОН, группы -С(О)ОС1-С6 алкил, группы -NHС(О)С1-С6 алкил, фенила или пиридинила;
2) 6-членную насыщенную или ненасыщенную гетероциклическую группу, содержащую 1 или 2 гетероатома, выбранных из N и O, необязательно замещенную группой, выбранной из галогена, С1-С6 алкила;
3) 5-членную ненасыщенную гетероциклическую группу, содержащую от 1 до 3 гетероатомов, выбранных из N и S, необязательно замещенную 1 или 2 заместителями, выбранными из С1-С6 алкила, конденсированную с 6-членной ненасыщенной циклической или гетероциклической группой, содержащей атом азота, необязательно замещенной одним или двумя заместителями, выбранными из гидроксигруппы, галогена или С1-С6 алкила;
4) 6-членную ненасыщенную циклическую или гетероциклическую группу, содержащую 1 или 2 атома N, конденсированную с 5- или 6-членной ненасыщенной гетероциклической группой, содержащей от 1 до 3 гетероатомов, выбранных из N или S;
5) группу формулы
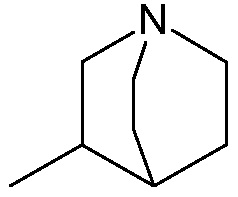
с водоотнимающим агентом в органическом растворителе.
Детальное описание изобретения
Предпочтительными соединениями настоящего изобретения являются соединения общей формулы (I), где
m представляет собой целое число от 0 до 2;
Ra1, Rb1 представляют собой водород, метил, аминогруппу, гидроксигруппу;
Rc1, Rd1 представляют собой водород, метил, аминогруппу, гидроксигруппу;
Re1, Rf1 представляют собой водород, метил;
R2 представляет собой водород, метил, карбоксильную группу, метоксикарбонил, этоксикарбонил;
R3 представляет собой:
группу, выбранную из:
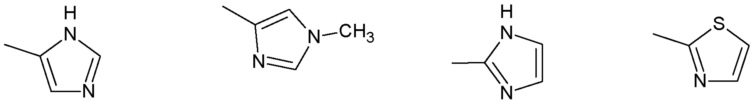

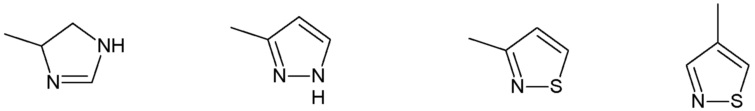



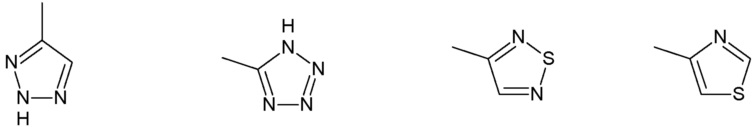

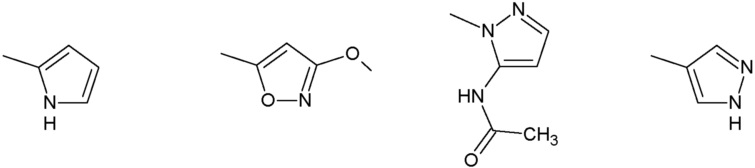

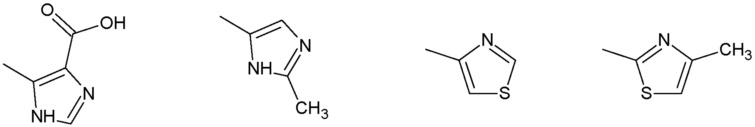

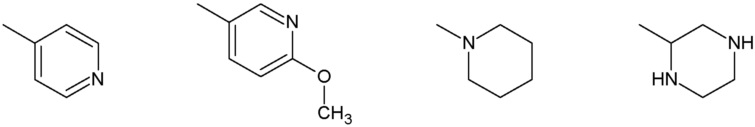
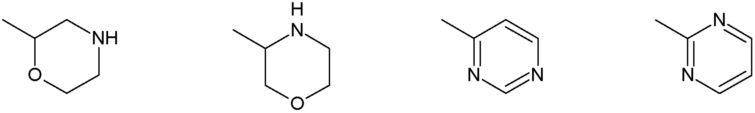

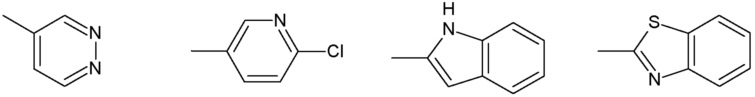
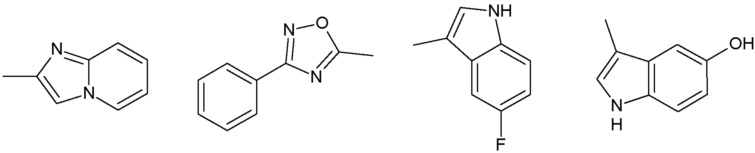
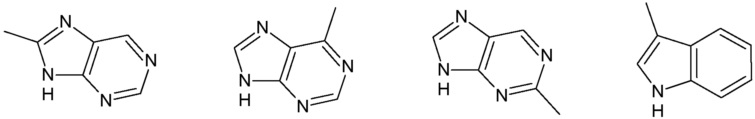
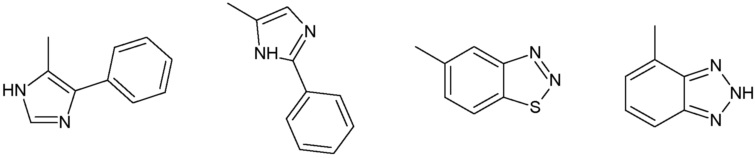
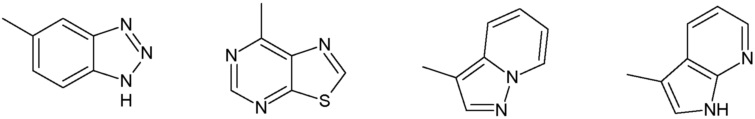
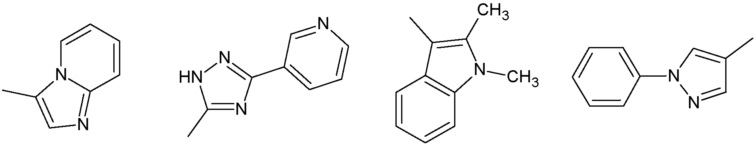
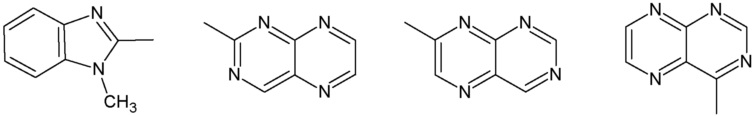
Наиболее предпочтительными соединениями настоящего изобретения являются соединения, представленные в таблице 1.
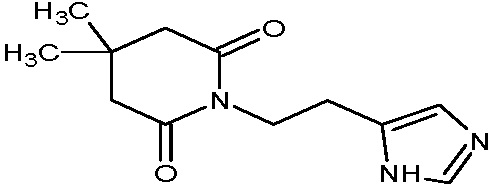
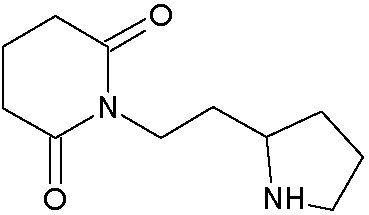
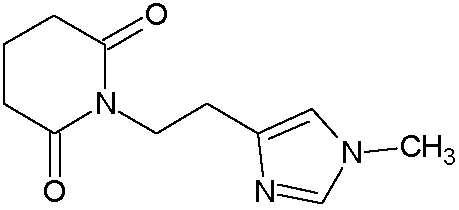
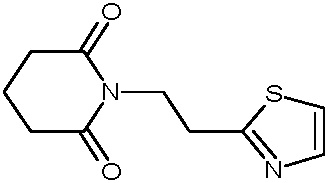
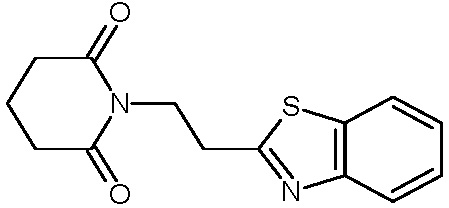
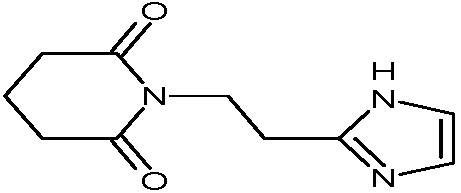
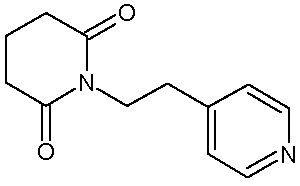
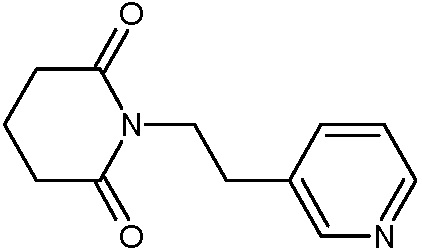
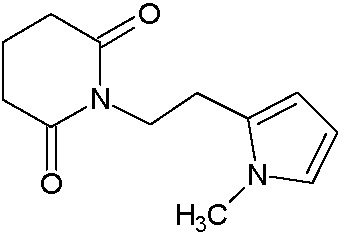
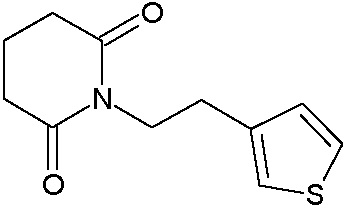
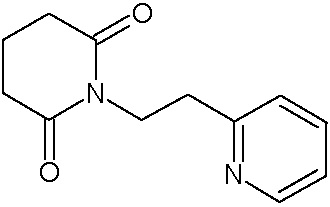
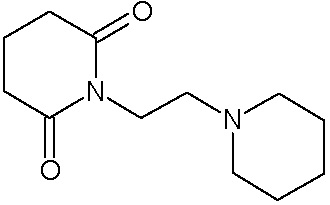
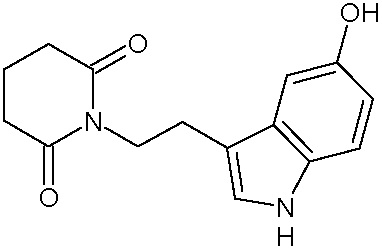

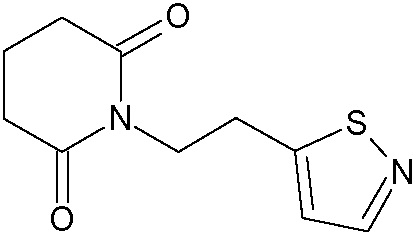
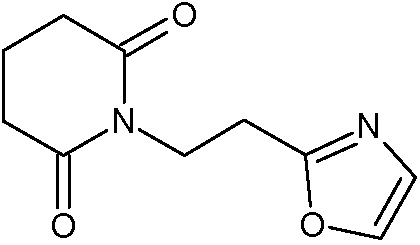
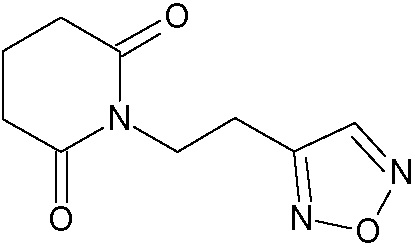
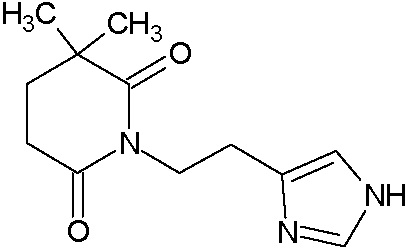
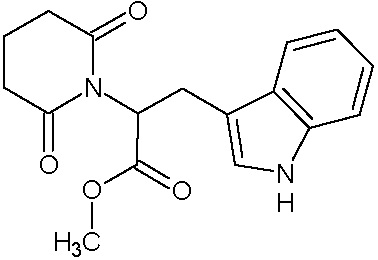
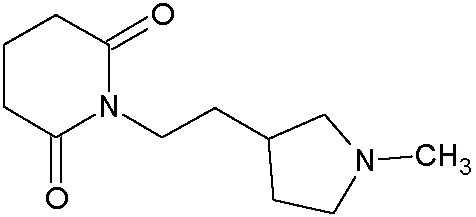
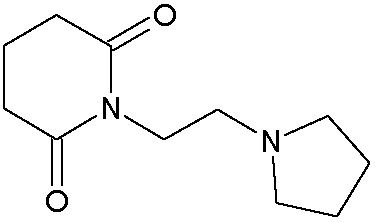

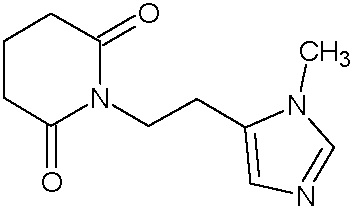
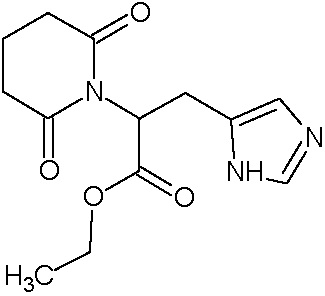
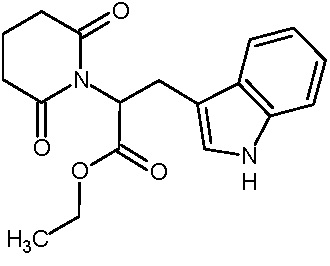
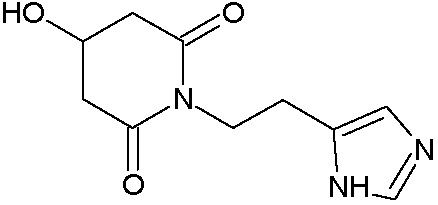
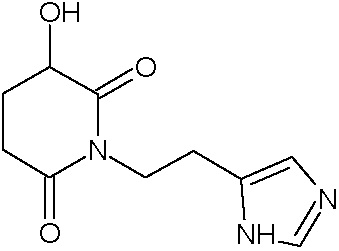
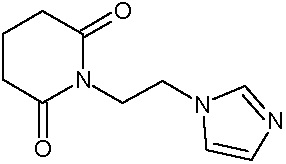
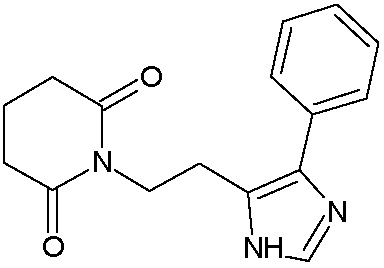
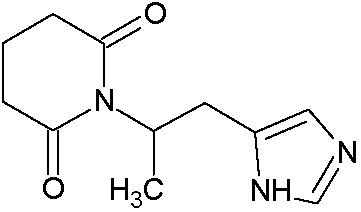
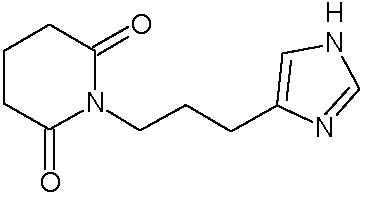
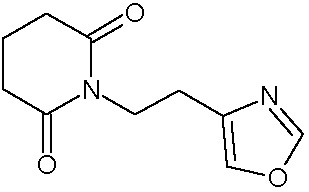

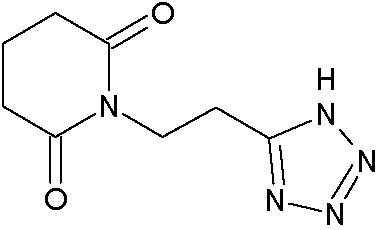
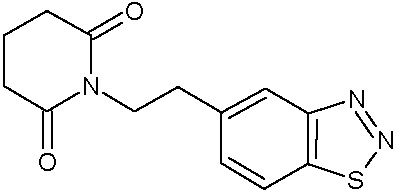
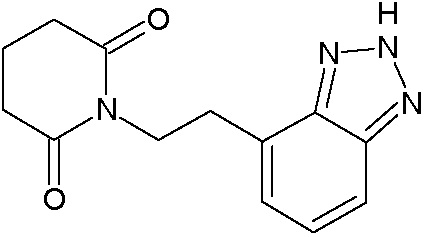
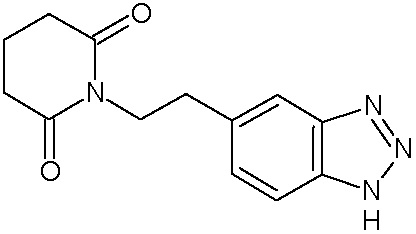
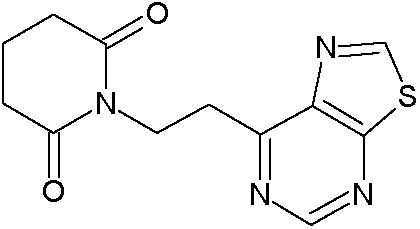

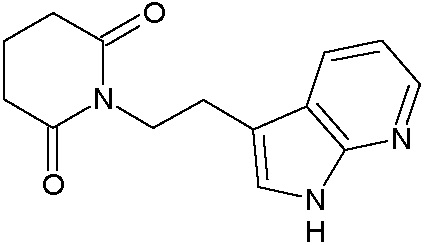
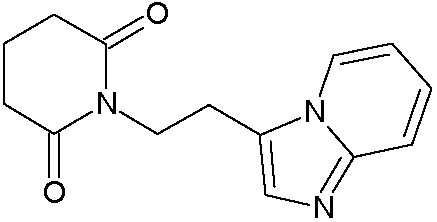
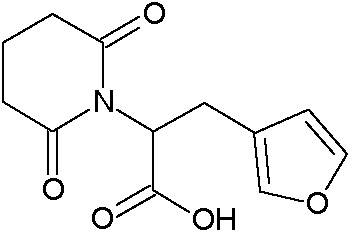
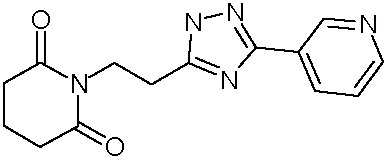
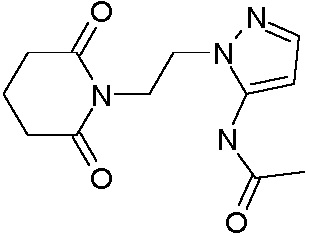
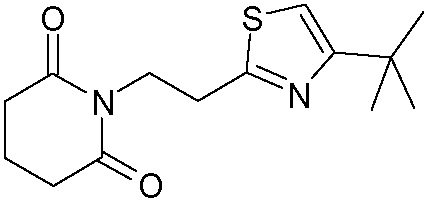
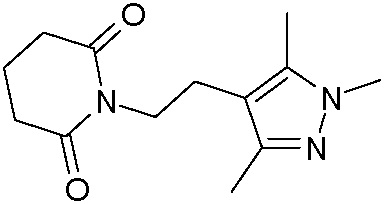
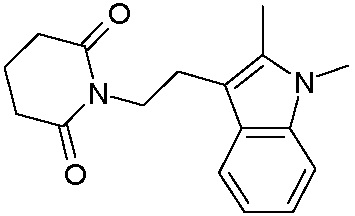
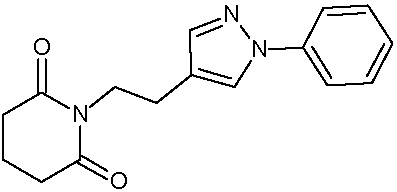
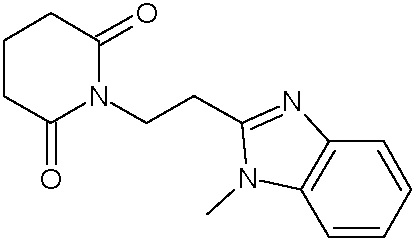
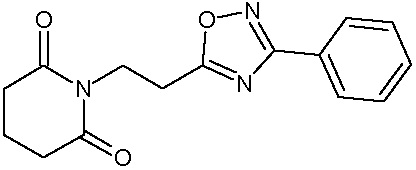
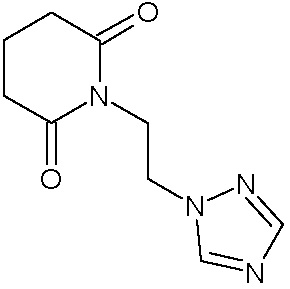
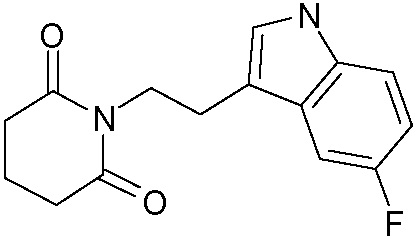
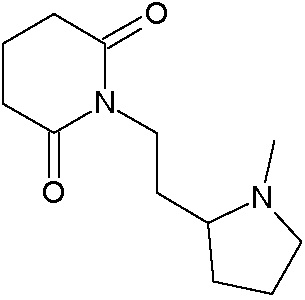

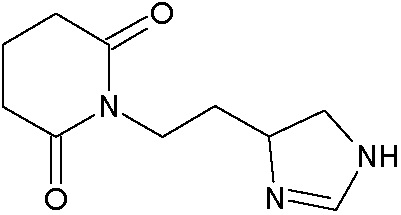
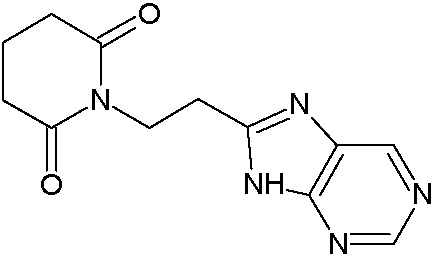
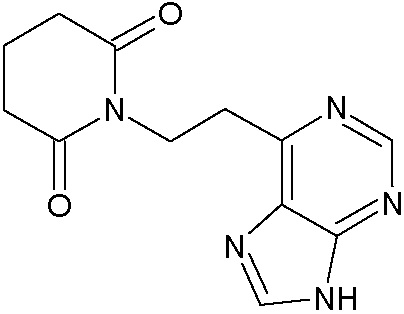
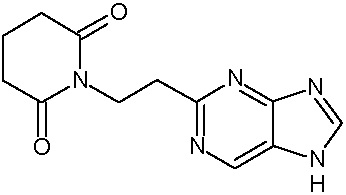
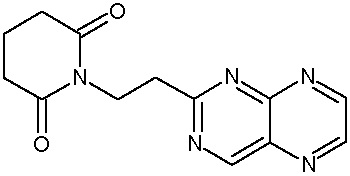
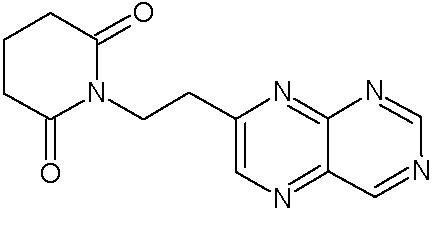
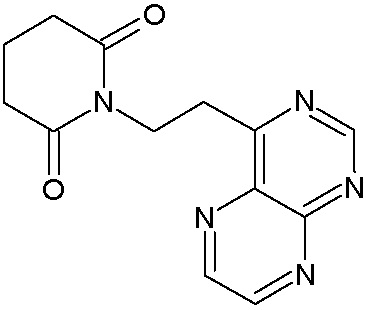
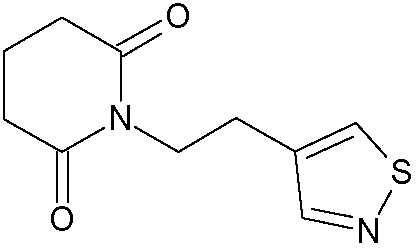
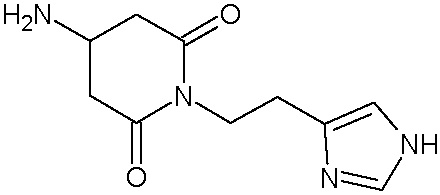
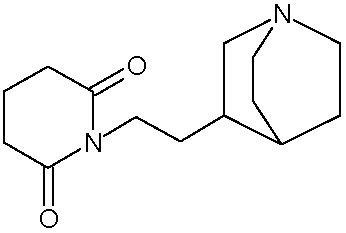
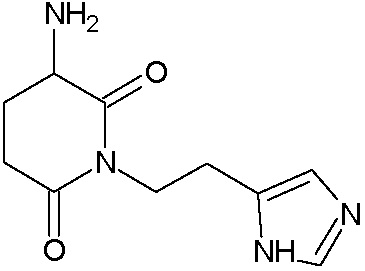
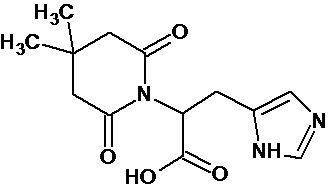
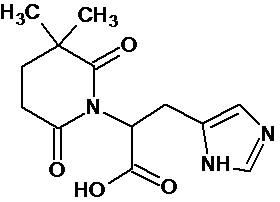
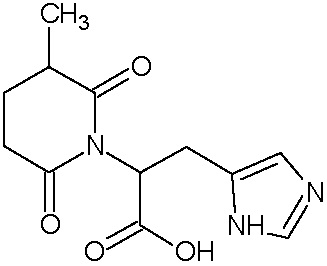
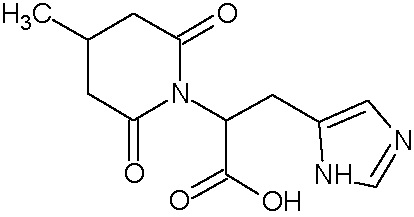
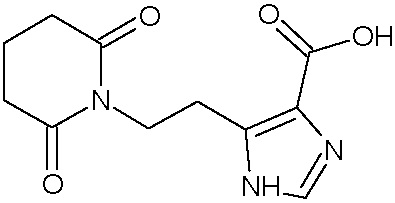
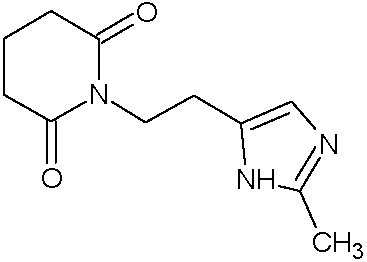
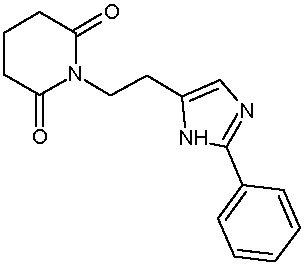
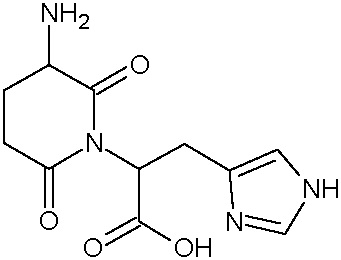
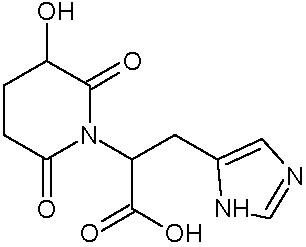
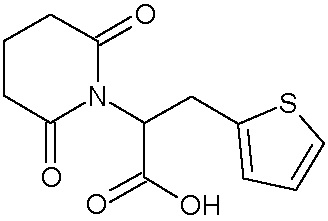
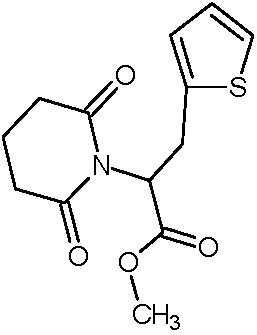
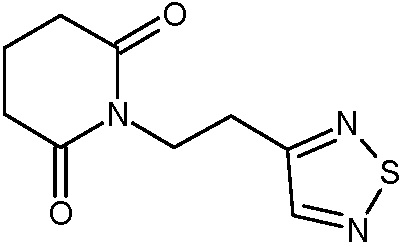
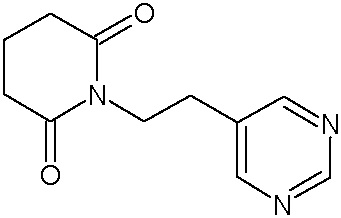
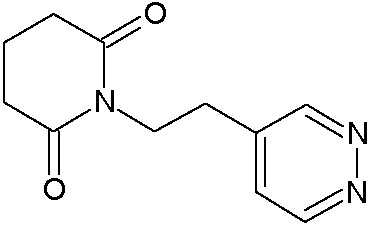
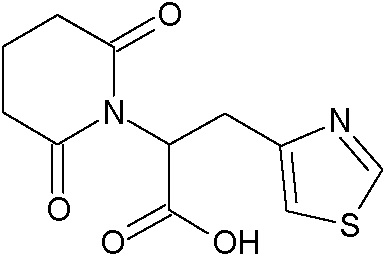
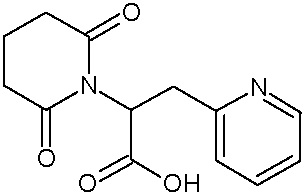
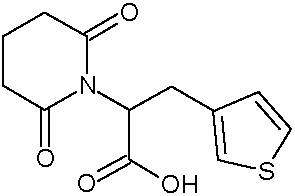
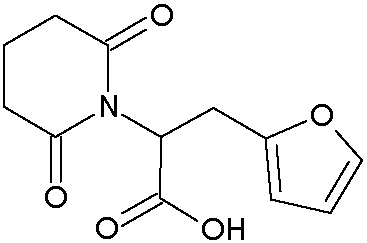

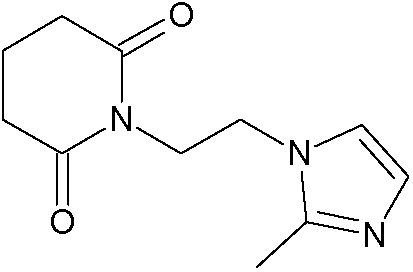
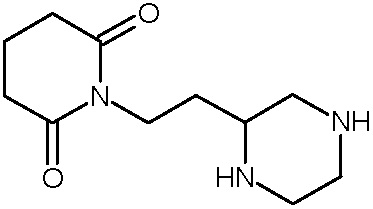
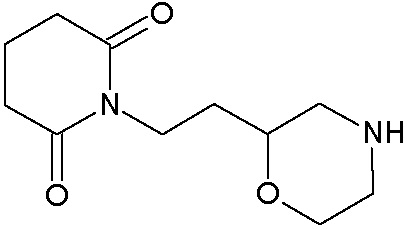
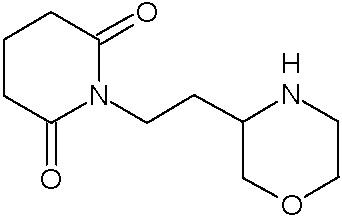
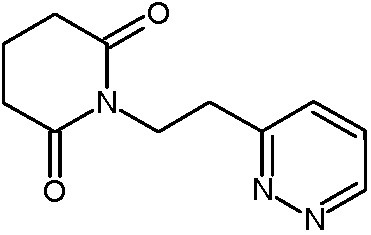

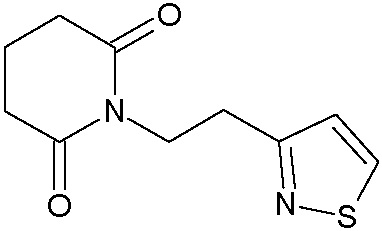
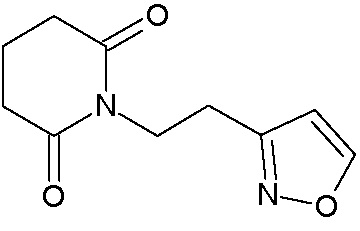
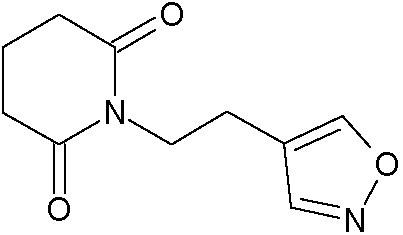
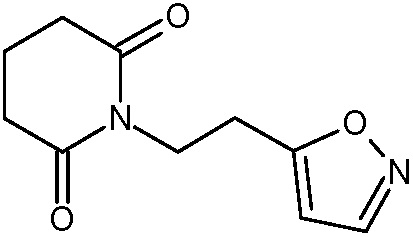
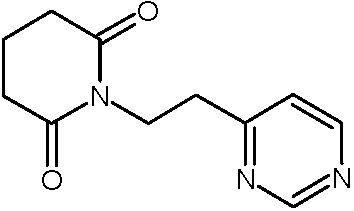
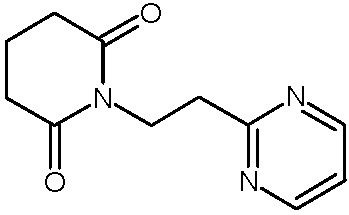
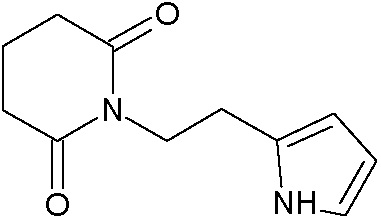
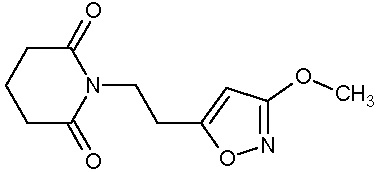
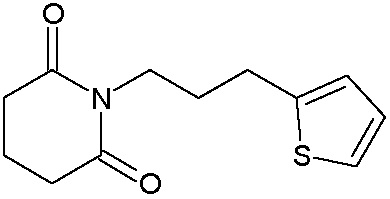
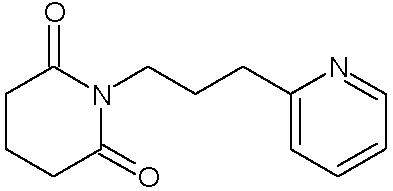
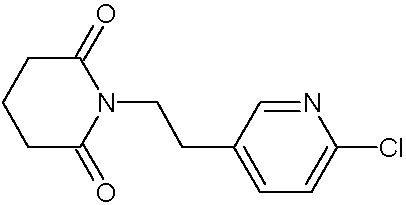
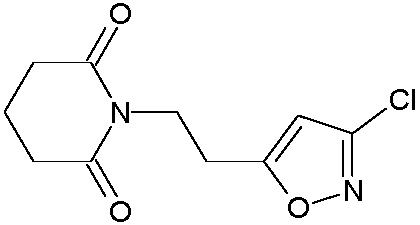

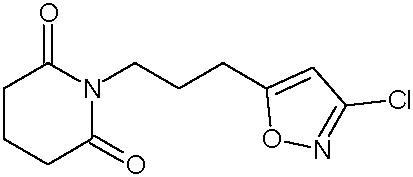

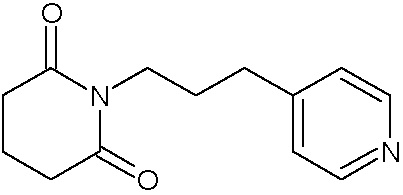

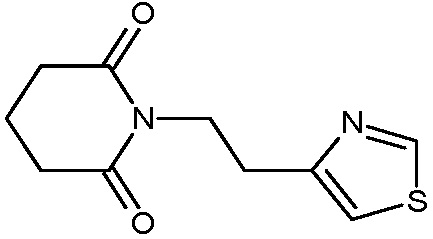
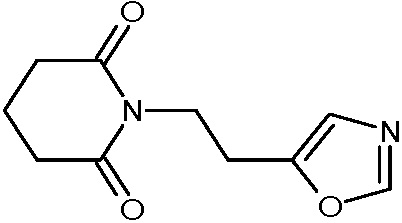

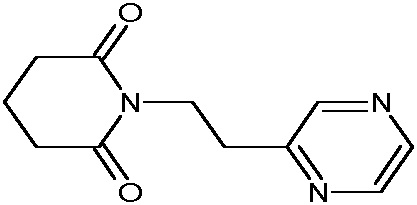
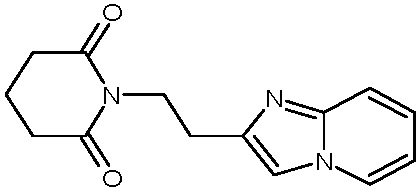
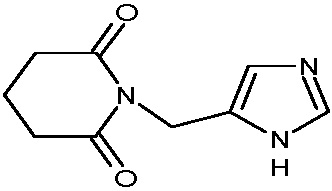
В качестве фармацевтически приемлемых солей соединений по настоящему изобретению могут быть использованы аддитивные соли органических кислот (например, формиат, ацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и др.), аддитивные соли неорганических кислот (например, гидрохлорид, гидробромид, сульфат, фосфат и др.), соли с аминокислотами (например, соль аспарагиновой кислоты, соль глутаминовой кислоты и т.д.), предпочтительно, хлоргидраты и ацетаты.
Наиболее предпочтительными известными соединениями, которые могут быть использованы в фармацевтической композиции и способе лечения по настоящему изобретению, являются производные глутаримидов, представленные в таблице 2.
по заявке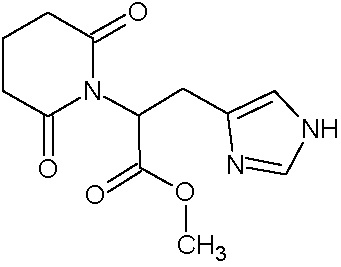
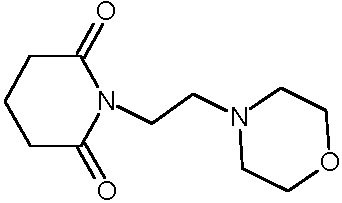
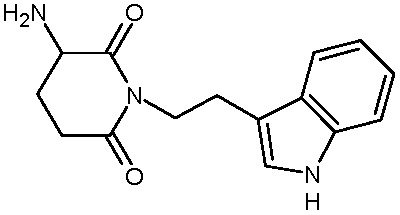

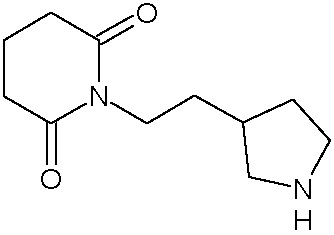
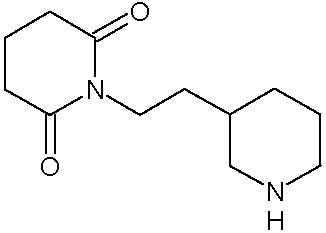
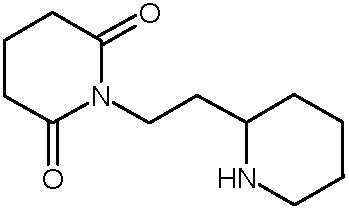
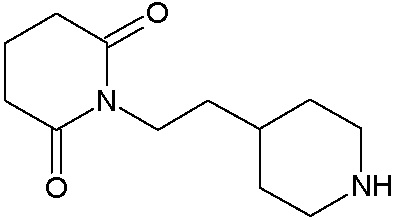
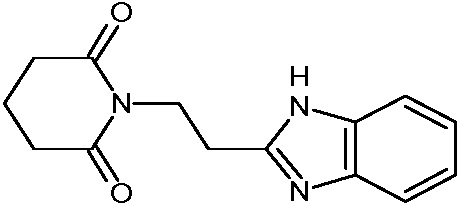

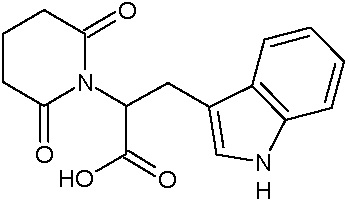
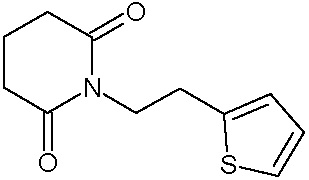


Соединения настоящего изобретения могут быть получены способом, включающим нагревание исходных моноамидов дикарбоновых кислот общей формулы (II) с водоотнимающим агентом в среде органического растворителя или в среде самого водоотнимающего агента необязательно с добавлением ацетата натрия.
Соединения общей формулы (II), а также способы их получения раскрыты в публикации международной заявки WO 1999/001103.
Предпочтительно, нагревание проводят при температуре от 90°С до 120°С, наиболее предпочтительно, при 100°С, еще более предпочтительно, при кипячении.
В качестве водоотнимающих агентов в данном способе могут быть использованы ангидриды дикарбоновых кислот, хлорангидриды органических кислот и карбонилдиимидазол.
Предпочтительными водоотнимающими агентами, которые могут быть использованы в способе, являются глутаровый ангидрид, пропионовый ангидрид, уксусный ангидрид, хлорангидрид уксусной кислоты и карбонилдиимидазол. В наиболее предпочтительном варианте используют пропионовый ангидрид в толуоле, глутаровый ангидрид, предпочтительно, в диметилформамиде, или уксусный ангидрид в диоксане, или хлорангидрид уксусной кислоты в уксусной кислоте.
Наиболее предпочтительным вариантом способа является способ с использованием в качестве водоотнимающего агента и растворителя ангидрида уксусной кислоты и нагреванием при температуре 90-100°С.
При наличии в структуре соединения дополнительных функциональных групп (например, ОН, NH2, COOH) их предварительно защищают, используя для этого обычные принятые в органическом синтезе защитные группы, такие как бензилоксикарбонильная, бензильная, ацетильная группа. По окончании синтеза эти группы, в случае необходимости, могут быть удалены, например, гидрированием.
Предлагаемые способы получения замещенных по азоту глутаримидов общей формулы (I) просты в осуществлении, протекают в достаточно мягких условиях, технологичны, позволяют получать целевые продукты с хорошим выходом и высокой степенью чистоты.
Производные глутаримидов общей формулы (I) обладают терапевтическим действием в отношении заболеваний верхних дыхательных путей.
В частности, соединения настоящего изобретения могут быть использованы для лечения риносинусита.
Соединения настоящего изобретения вводятся в эффективном количестве, которое обеспечивает желаемый терапевтический результат.
Соединения общей формулы (I) могут быть введены перорально, местно, парентерально, интраназально и ингаляционно в виде стандартных лекарственных форм, содержащих нетоксичные фармацевтически приемлемые носители. Используемый в настоящем описании термин «парентеральное введение» означает подкожные, внутривенные, внутримышечные или внутригрудные инъекции или вливания.
Соединения настоящего изобретения могут быть введены пациенту в дозах, составляющих от 0,1 до 100 мг/кг веса тела в день, предпочтительно, в дозах от 0,25 до 25 мг/кг один или более раз в день.
При этом следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от многих факторов, включая активность данного используемого соединения, возраст, вес тела, пол, общее состояние здоровья и режим питания пациента, время и способ введения лекарственного средства, скорость его выведения из организма, конкретно используемую комбинацию лекарственных средств, а также тяжесть заболевания у данного индивида, подвергаемого лечению.
Фармацевтические композиции по настоящему изобретению содержат соединение общей формулы (I) в количестве, эффективном для достижения желаемого результата, и могут быть введены в виде стандартных лекарственных форм (например, в твердой, полутвердой или жидкой формах), содержащих соединения настоящего изобретения в качестве активного ингредиента в смеси с носителем или наполнителем, пригодным для внутримышечного, внутривенного, перорального, сублингвального, ингаляционного интраназального введения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, драже, эмульсий, суспензий, мазей, гелей и любых других лекарственных форм.
В качестве наполнителей могут быть использованы различные вещества, такие как сахариды, например, глюкоза, лактоза или сахароза, маннит или сорбит, производные целлюлозы и/или фосфаты кальция, например, трикальций фосфат или кислый фосфат кальция, в качестве связующего компонента могут быть использованы, такие как крахмальная паста, например, кукурузный, пшеничный, рисовый, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон. При необходимости могут быть использованы разрыхляющие агенты, такие как вышеупомянутые крахмалы и карбоксиметилкрахмал, поперечносшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Могут быть использованы необязательные добавки, такие как агенты, регулирующие текучесть, и смазывающие агенты, такие как диоксид кремния, тальк, стеариновая кислота и ее соли, такие как стеарат магния или стеарат кальция, и/или пропиленгликоль.
Ядро драже обычно покрывают слоем, который устойчив к действию желудочного сока. Для этой цели могут быть использованы концентрированные растворы сахаридов, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, и подходящие органические растворители или их смеси.
В качестве добавок могут быть также использованы стабилизаторы, загустители, красители и отдушки.
В качестве мазевой основы могут быть использованы углеводородные мазевые основы, такие как вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции, такие как твердый парафин и воск; абсорбтивные мазевые основы, такие как гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens), мазевые основы, смываемые водой, такие как гидрофильная мазь (Unguentum hydrophylum), водорастворимые мазевые основы, такие как полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие.
В качестве основы для гелей могут быть использованы метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, оксипропилцеллюлоза, полиэтиленгликоль или полиэтиленоксид, карбопол.
При приготовлении стандартной лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьироваться в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства.
Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций, содержание активного агента в них составляет до 5% по массе. В качестве разбавителей могут быть использованы 0,9% раствор хлорида натрия, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы, специфические добавки для растворения. При введении в организм соединений настоящего изобретения в виде таблеток и суппозиториев, их количество составляет до 200 мг на стандартную лекарственную форму.
Лекарственные формы настоящего изобретения получают по стандартным методикам, таким как, например, процессы смешивания, гранулирования, формирование драже, растворение и лиофилизация.
Следует отметить, что соединения настоящего изобретения проявляют биологическую активность в дозах на два-три порядка ниже по сравнению с известными препаратами, использованными для сравнения, при практически одинаковой эффективности, и для них не выявлено отрицательных побочных действий и не обнаружено противопоказаний к применению. При этом при исследовании токсичности соединений настоящего изобретения в дозе 3000 мг/кг, перорально, не зарегистрировали гибели экспериментальных животных.
Детальное описание соединений настоящего изобретения, их получения и исследования фармакологической активности представлено в нижеследующих примерах, предназначенных для иллюстрации предпочтительных вариантов изобретения и не ограничивающих его объем.
Примеры синтеза производных глутаримидов общей формулы (I)
Средства и методы
Индивидуальность полученных соединений проверяют методом ТСХ на пластинках “Kieselgel 60 F254” (фирмы “Merck”, Германия) в системе растворителей: хлороформ-метанол 8:2 (1), хлороформ-метанол 9:1 (2).
Хроматограммы и электрофореграммы проявляют хлор-тетраметилбензидиновым реактивом и реактивом Паули.
Температуру плавления определяют на приборе ПТП (завод лаб. приборов, Россия, г. Клин).
Спектры ИК-Фурье снимают в таблетках КВr на приборе “Magna 750” (“Nicolet”, США).
ЖХ/МС-система анализа многокомпонентных смесей Shimadzu Analytical HPLC SCL10Avp, масс-спектрометр PE SCIEX API 165 (150), Канада.
Аналитическую обращенно-фазовую ВЭЖХ проводили на приборе: хроматограф HPLC Shimadzu, в условиях: колонка Symmetry C18 250×4,6 мм, градиент элюирования в системе вода с 0,1% HCOOH: ацетонитрил с 0,1% HCOOH (условия А), колонка Merk: LiChroCART 250×4 мм, 5 мкм, LiChrospher 100RP-8E 5 мкм.C8. серийный № 1.50837.0001, градиент элюирования в системе ацетатно-аммиачный буферный раствор рН 7,5:ацетонитрил (условия Б), градиент элюирования в системе буфер с 1-гексилсульфонатом натрия 0,0025 М рН=3:ацетонитрил (условия В), колонка Luna C18 (2) 100 A 250×4,6 мм (сер.599779-23), градиент элюирования в системе фосфатный буферный раствор рН 3,0:метанол (условия Г).
Спектры 1Н-ЯМР регистрируют на приборе Bruker DPX-400 (Германия).
Масс-спектры высокого разрешения получают на времяпролетном масс-спектрометре методом матриксной лазерно-десорбционной ионизации с использованием в качестве матрицы 2,5-дигидроксибензойной кислоты, на приборе Ultraflex (“Bruker”, Германия).
Пример 1
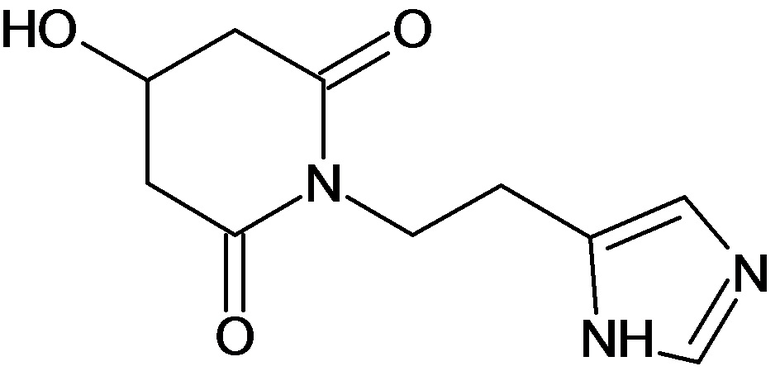
Получение 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона (соединение 1)
В плоскодонную колбу загружают 1 г (4,4 ммоль) 2-(имидазол-4-ил)этанамид пентандиовой-1,5 кислоты, растворенной в 5 мл уксусной кислоты. При перемешивании добавляют по каплям 1,5 экв. ацетилхлорида. Реакционную массу выдерживают 12 часов при перемешивании и температуре 90°С. Контроль реакции с помощью 1Н-ЯМР спектроскопии. Охлаждают реакционную массу, растворитель удаляют в вакууме. Образовавшийся осадок растворяют в минимальном количестве воды, и при охлаждении порционно добавляют карбонат натрия до pH=8-9. Выпавший осадок отфильтровывают и промывают небольшим количеством воды, сушат. Маточный раствор после фильтрации трижды экстрагируют хлористым метиленом. Объединенные органические маточники сушат сульфатом натрия, растворитель удаляют в вакууме. Образовавшийся осадок сушат, объединяют с первой порцией (после фильтрации) и получают 0,52 г с выходом 56% 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона, в виде светлого порошка. ЖХ/МС, индивидуальный пик, время удерживания 1,57 мин, [М+Н]+=208о Спектр 1Н-ЯМР (CD3ОD): δ м.д.: 1,87-1,93 (м, 2Н, 4'-CH2), 2,61-2,65 (т, 4Н, 3',5'-СН2), 2,76-2,80 (т, 2Н, 1-СН2), 3,96-4,00 (т, 2Н, 2-СН2), 6,8 (с, 1Н, 5''-СН-Im), 7,55 (с, 1Н, 2''-СН-Im).
Пример 2
Получение 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона (соединение 1)
В плоскодонную колбу загружают 1 г (4,4 ммоль) 2-(имидазол-4-ил)этанамида пентандиовой-1,5 кислоты и 10 мл пропионового ангидрида. Прибавляют 3 экв. ацетата натрия и оставляют при перемешивании и температуре 120°С в течение 12 часов. Контроль реакции с помощью 1Н-ЯМР спектроскопии. Реакционную массу разбавляют трехкратным избытком воды при охлаждении и перемешивании, порционно прибавляют карбонат натрия до pH=8-9. Реакционную смесь трижды экстрагируют этилацетатом. Объединенный органические маточники сушат сульфатом натрия, растворитель удаляют в вакууме. Получают 0,37 г 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона в виде светло-желтых кристаллов, с выходом 40%. [M]+ 207,9. Спектр 1Н-ЯМР (CD3ОD): δ м.д.: 1,85-1,91 (м, 2Н, 4'-CH2), 2,60-2,63 (т, 4Н, 3',5'-СН2), 2,73-2,77 (т, 2Н, 1-СН2), 3,95-4,00 (т, 2Н, 2-СН2), 6,8 (с, 1Н, 5''-СН-Im), 7,52 (с, 1Н, 2''-СН-Im).
Пример 3
Получение 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона (соединение 1)
В 1-литровую коническую колбу, снабженную обратным холодильником, помещают 100 г (0,44 моль) 2-(имидазол-4-ил)этанамид пентандиовой-1,5 кислоты, 80 мл (0,85 моль) уксусного ангидрида и 200 мл толуола. Полученную суспензию нагревают до растворения твердого вещества и кипятят раствор в течение 6-8 часов. Растворитель удаляют в вакууме, к полученному маслу приливают 300 мл метанола и повторно растворитель удаляют в вакууме. Остаток растворяют в 300 мл хлористого метилена и прибавляют 65 мл триэтиламина. Полученный раствор концентрируют в вакууме и оставляют на 18 ч при +4°С. Осадок отфильтровывают на воронке Бюхнера диаметром 10 см, промывают тремя порциями изопропанола, сушат при +70°С. Степень чистоты продукта контролируют с помощью ТСХ (Rf продукта 0,54; (1)). При необходимости дополнительной очистки и осветления продукта его перекристаллизовывают, с одновременной обработкой горячего раствора продукта угольной пылью/углем. Получают 73,6 г 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона, выход 80%, Т.пл. 150-151°С. [М+Н]+=208. Спектр 1Н-ЯМР (CD3ОD): δ м.д.: 1,87-1,93 (м, 2Н, 4'-CH2), 2,61-2,65 (т, 4Н, 3',5'-СН2), 2,76-2,80 (т, 2Н, 1-СН2), 3,96-4,00 (т, 2Н, 2-СН2), 6,8 (с, 1Н, 5''-СН-Im), 7,55 (с, 1Н, 2''-СН-Im).
В соответствии с вышеуказанной методикой получены следующие соединения:
Пример 4
Получение 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона (соединение 1)
К растворенному при нагревании в 25 мл N,N'-диметилформамида 4,5 г (0,020 моль) 2-(имидазол-4-ил)этанамиду пентандиовой-1,5 кислоты добавляют 3,5 г (0,031 моль) глутарового ангидрида и нагревают до 100°С в течение 4-6 часов. Полноту протекания реакции проверяют методом ТСХ или электрофореза. Растворитель удаляют в вакууме, маслообразный остаток растворяют в 50 мл воды, раствор пропускают через колонку с 70 мл Амберлита IRA-96. Элюат, содержащий целевое вещество, собирают, растворитель удаляют в вакууме. Полученный твердый остаток перекристаллизовывают из хлороформа. Выход 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона составляет 3,1 г (75,6%). Rf 0,43 (2). Т.пл. 150-151°С. [M]+ 207,9. Спектр 1Н-ЯМР (CD3ОD): δ м.д.: 1,87-1,93 (м, 2Н, 4'-CH2), 2,61-2,65 (т, 4Н, 3',5'-СН2), 2,76-2,80 (т, 2Н, 1-СН2), 3,96-4,00 (т, 2Н, 2-СН2), 6,8 (с, 1Н, 5''-СН-Im), 7,55 (с, 1Н, 2''-СН-Im).
ВЭЖХ в условиях Г: индивидуальный пик, время удерживания 15,5 мин.
Спектр ИК-Фурье (в табл. КВr, ν, см-1): 3136, 3070, 2833 (-NH- вал.), 1720, 1670 (С=О, цикл. имид), 1339, 1257 (-СН2-). Найдено, %: С 57,60; Н 6,12; N 21,17. С10Н13N3O2. Вычислено, %: C 57,96, H 6,32, N 20,28.
Пример 5
Получение 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона (соединение 1)
В 1-литровую круглодонную колбу, снабженную обратным холодильником, помещают 100 г (0,44 моль) 2-(имидазол-4-ил)этанамида пентандиовой-1,5 кислоты, 102 мл (0,80 моль) пропионового ангидрида и 200 мл толуола. Полученную суспензию нагревают до растворения твердого вещества и кипятят раствор в течение 8-9 часов. Растворитель удаляют в вакууме, к полученному маслу приливают 300 мл метанола и повторно растворитель удаляют в вакууме. Остаток растворяют в 300 мл хлористого метилена и прибавляют 65 мл триэтиламина. Полученный раствор концентрируют в вакууме, отгоняя примерно 70% хлористого метилена, и оставляют на 18 ч при температуре 0-+4°С. Осадок отфильтровывают, промывают тремя порциями изопропанола, охлажденного до 0−-5°С. Технический продукт перекристаллизовывают, с одновременной обработкой горячего раствора продукта угольной пылью/углем. Степень чистоты продукта контролируют с помощью ТСХ (Rf продукта 0,54 (1)). Проводят горячее фильтрование раствора продукта на системе фильтрации «MILLIPORE» (0,45 мкм), сушат в вакуумном сушильном шкафу при +70°С. Получают 60,0 г 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона, выход 65%. Т.пл. 150-151°С. Спектр 1H-ЯМР (400,13 МГц, ДМСО-d6, δ, м.д., J/Гц): 1,81 (м, 2H, CH2CH2CH2), 2,58 (м, 6H, CH2C, CH2CH2CH2), 3,83 (т, 2H, CH2N, J=7,8 Гц), 6,77 (шир.с, 1H, CCH ), 7.48 (шир.с, 1H, NCHN_), 11,8 (шир.с, 1H, NH_).
Пример 6
Получение 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона (соединение 1)
5,0 г (0,022 моль) Nβ-глутарилгистамина нагревают в 12 мл уксусного ангидрида до 100°С в течение 4-6 часов. Полноту протекания реакции контролируют методом ТСХ или электрофореза. Растворитель из реакционной смеси удаляют в вакууме, полученный твердый остаток перекристаллизовывают из изопропилового спирта. Выход 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона 3,7 г (80%). Rf 0,43 (2). Т.пл. 149-150°С. Найдено, %: С 57,73; Н 6,15; N 20,17. С10Н13N3O2. Вычислено, %: C 57,96, H 6,32, N 20,28.
Пример 7
1-[2-(1Н-бензотиазол-2-ил)этил]пиперидин-2,6-дион (соединение 7)
Смесь 22 г (0,075 моль) 5{[2-(1,3-бензотиазол-2-ил)этил]амино}-5-оксопентановой кислоты и 23 г (0,225 моль) уксусного ангидрида кипятят в 150 мл диоксана и течение 3 часов. Диоксан удаляют в вакууме, добавляют 200 мл воды. Нейтрализуют 30% гидроксидом натрия до нейтральной среды. Выпавшее масло затирают в кристаллы. Осадок очищают колоночной хроматографией (SiO2 60-100 мкм, элюент: этилацетат-гексан (1:1)). Получают 16,5 г 1-[2-(1Н-бензотиазол-2-ил)этил]пиперидин-2,6-диона с выходом 79,9%. ЖХ/МС, время удерживания 2,26 мин, [М+Н]+=275. T.пл.=79,5-80,5°С. ВЭЖХ в условиях В, индивидуальный пик, время удерживания 9,34 мин. Спектр 1H-ЯМР (400,13 МГц, ДМСО-d6, δ, м.д., J/Гц): 1,85 (пент. 2H, CH2CH2CH2, J=6,8 Гц), 2,59 (т, 4H, CH2CH2CH2, J=6,8 Гц), 3,24 (т, 2H, CH2C, J=7,3 Гц), 4,08 (т, 2H, CH2N, J=7,3 Гц), 7,43, 7,49 (т, 1H, Ar, J=7,6 Гц), 7,96, 8,04 (д, 1H, Ar, J=7,6 Гц).
В соответствии с вышеуказанной методикой получены следующие соединения:
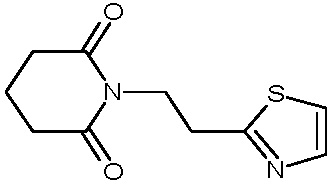
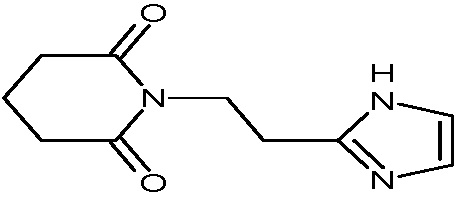
Пример 8
1-[2-(1Н-пиридил-3-ил)этил]пиперидин-2,6-дион (соединение 10)
В 200 мл уксусного ангидрида растворяют 2-(пиридил-3-ил)этанамид пентандиовой-1,5 (29,00 г, 0,12 моль) и безводный ацетат натрия (5,9 г, 0,07 моль). Реакционную смесь нагревают до слабого кипения и кипятят при перемешивании и с обратным холодильником 18 ч. После прохождения реакции растворитель удаляют в вакууме, а остаток растворяют в 500 мл дихлорметана, промывают 2 раза по 100 мл 3% раствором соды и сушат над сульфатом натрия. Растворитель удаляют в вакууме, полученное масло растворяют в диоксане. Добавляют 3 М раствор НСl в диоксане, выпавший осадок отфильтровывают и перекристаллизовывают из 12,5 мл изопропанола. Получают 25 г продукта в виде гидрохлорида с выходом 80%. ЖХ/МС, индивидуальный пик, время удерживания 0,5 мин, [М+Н]+=218. ВЭЖХ в условиях Г, индивидуальный пик, время удерживания 16,72 мин. Спектр 1H-ЯМР (400,13 МГц, ДМСО-d6, δ, м.д., J/Гц): 1,78 (пент. 2H, CH2CH2CH2, J=6,4 Гц), 2,56 (т, 4H, CH2CH2CH2, J=6,4 Гц ), 2,73 (т, 2H, CH2C, J=7,3 Гц), 3,86 (т, 2H, CH2N, J=7,3 Гц), 7,30 (дд, 1H, 5-Pyr, J=7,8, 4,5 Гц), 7,60 (д, 1H, 4-Pyr, J=7,8 Гц), 8,37 (д, 1H, 2-Pyr, J=1,5 Гц), 8,41 (дд, 1H, 6-Pyr, J=4,5, 1,5 Гц).
В соответствии с вышеуказанной методикой получены следующие соединения:

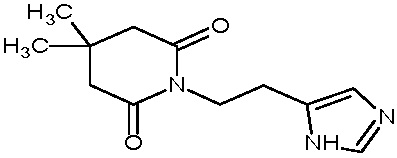
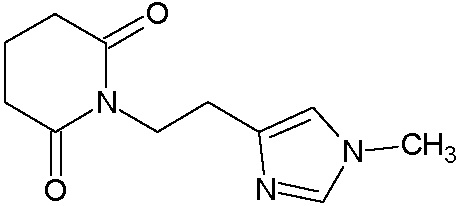
Пример 9
1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-дион (соединение 1)
В плоскодонную колбу (250 мл) загружают 60 мл N,N'-диметилформамида и 20 г 2-(имидазол-4-ил)этанамид пентандиовой-1,5 кислоты. При интенсивном перемешивании прибавляют 17,3 г (1,2 экв.) карбонилдиимидазола. Реакционную массу нагревают при перемешивании до 90°С в течение 2 часов, контроль реакции методом 1H-ЯМР (пробу, 0,5 мл, разбавляют серным эфиром, выпавший осадок растворяют в ДМСО-d6). При отсутствии исходной 2-(имидазол-4-ил)этанамид пентандиовой-1,5 кислоты в реакционной массе, ее охлаждают и выливают в трехкратный объем метил-трет-бутилового эфира (180 мл). Оставляют при перемешивании на 1 час, выпавший осадок отфильтровывают, промывают 60 мл метил-трет-бутилового эфира, сушат. Выход технического 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона составляет 12,4 г (67%).
В плоскодонную колбу на 100 мл загружают 12 г технического 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона и 36 мл изопропанола. Смесь нагревают до полного растворения осадка, затем прибавляют 1,2 г активированного угля и выдерживают при кипячении в течение часа. Раствор еще горячим быстро отфильтровывают через предварительно нагретый керамический фильтр. Осадок на фильтре промывают 6 мл горячего изопропанола. Горячий маточный раствор охлаждают до комнатной температуры и оставляют на ночь при перемешивании для кристаллизации. Выпавшие кристаллы отфильтровывают, промывают 6 мл холодного изопропанола, сушат. Выход 1-(2-(1Н-имидазол-4-ил)этил)пиперидин-2,6-диона после перекристаллизации составляет 10,1 г (84%). Продукт анализировали методом ЖХ/МС, индивидуальный пик, время удерживания 1,57 мин, [М+Н]+=208. Спектр 1Н-ЯМР (CD3ОD): δ м.д.: 1,87-1,93 (м, 2Н, 4'-CH2), 2,61-2,65 (т, 4Н, 3',5'-СН2), 2,76-2,80 (т, 2Н, 1-СН2), 3,96-4,00 (т, 2Н, 2-СН2), 6,8 (с, 1Н, 5''-СН-Im), 7,55 (с, 1Н, 2''-СН-Im).
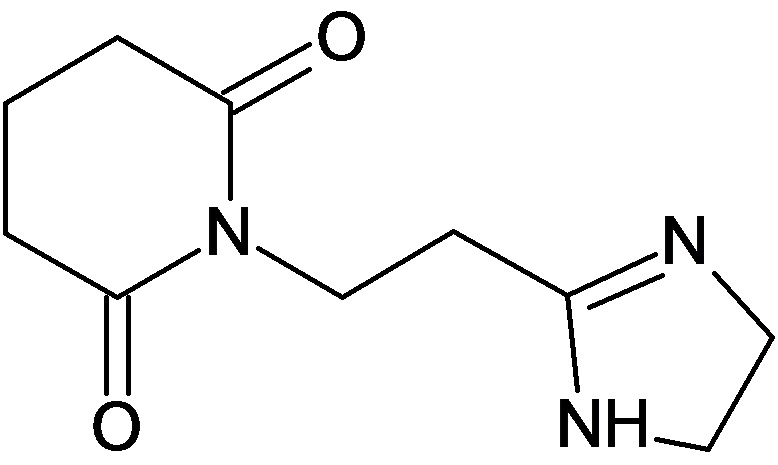
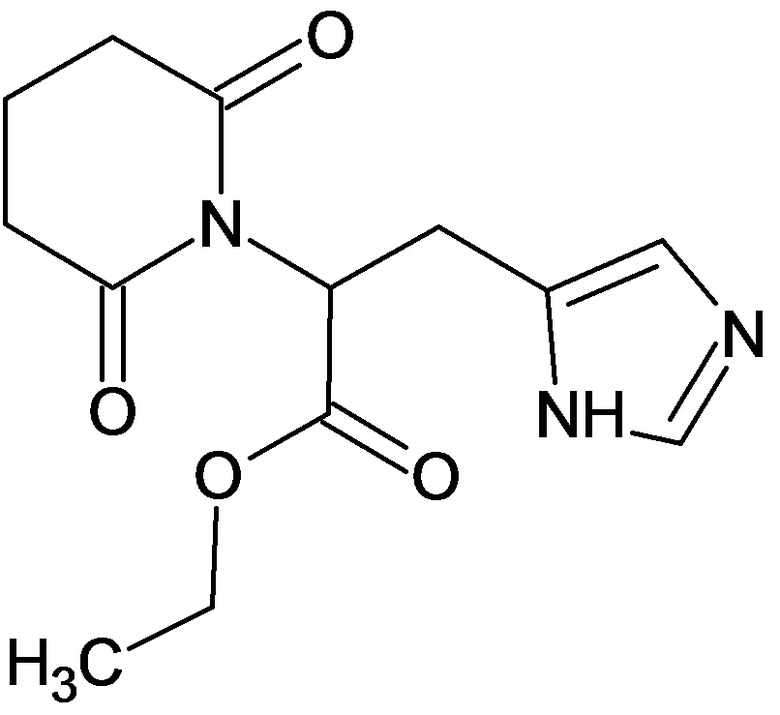
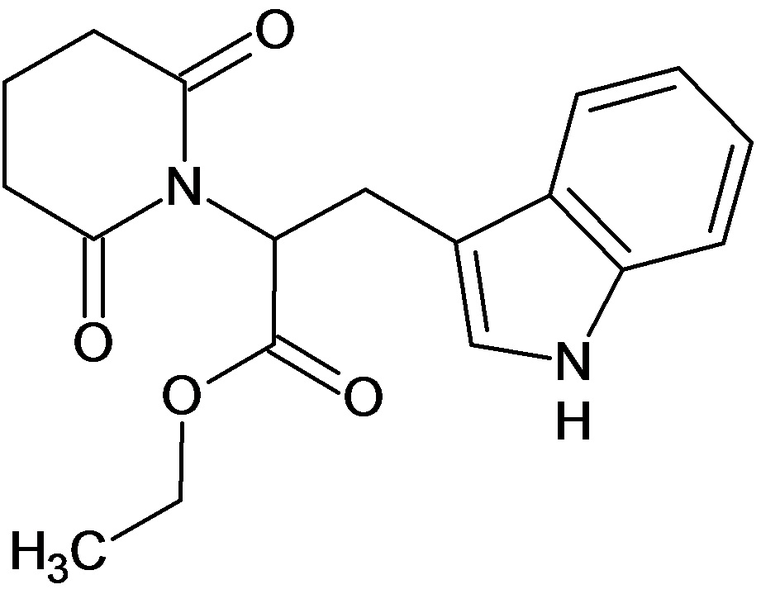
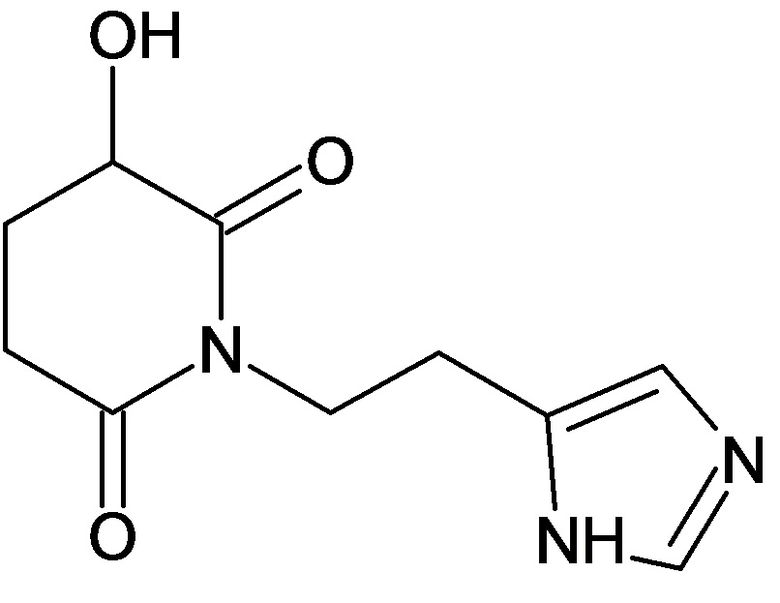
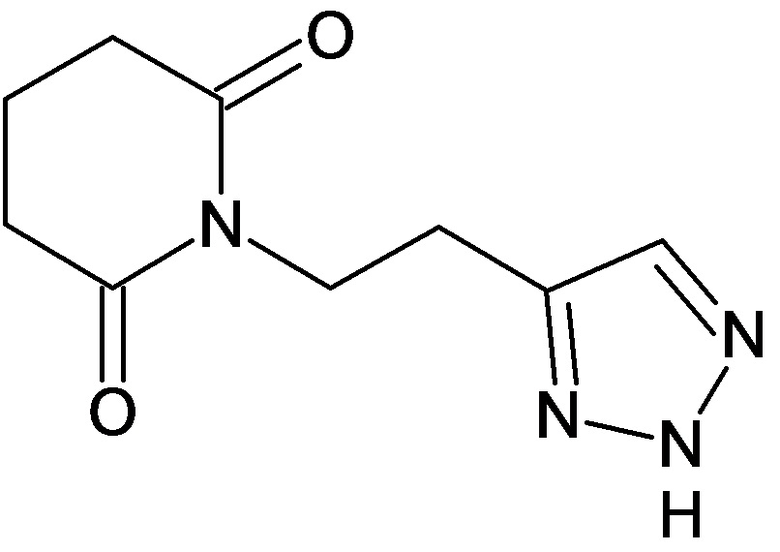
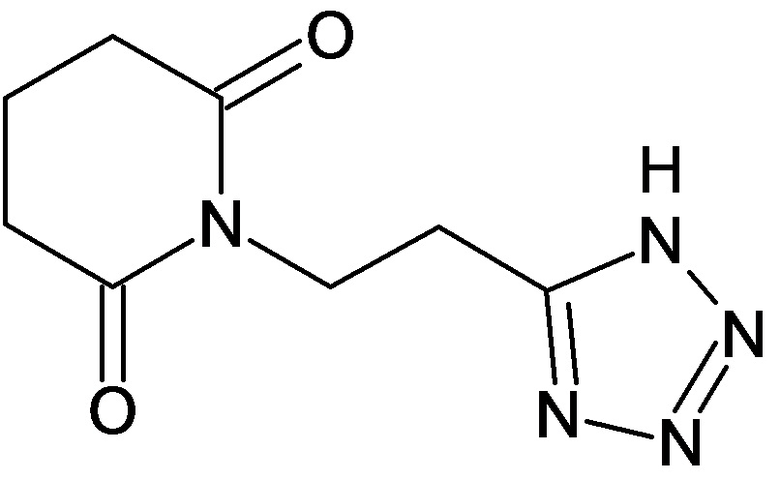
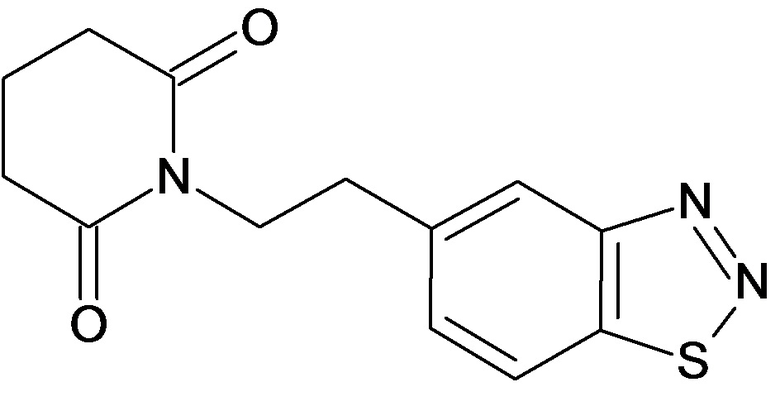
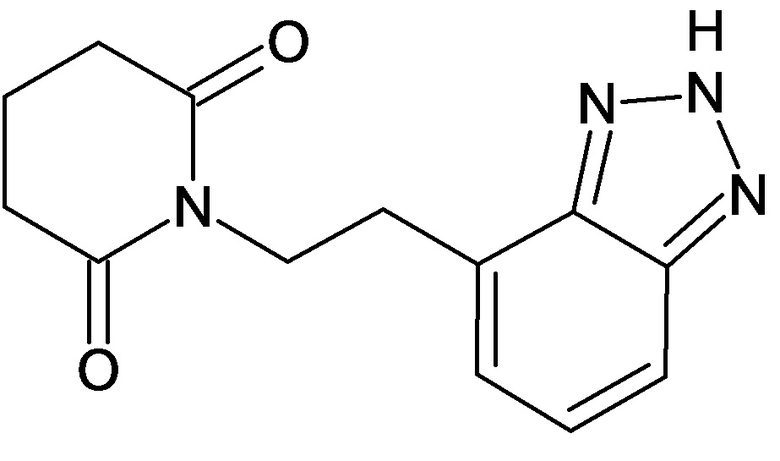
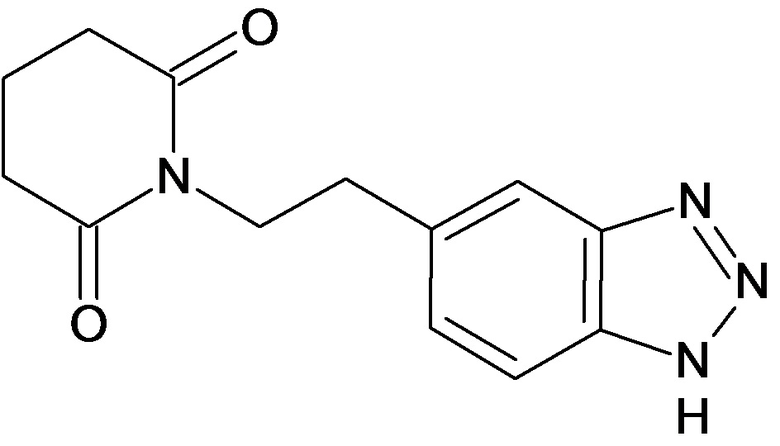
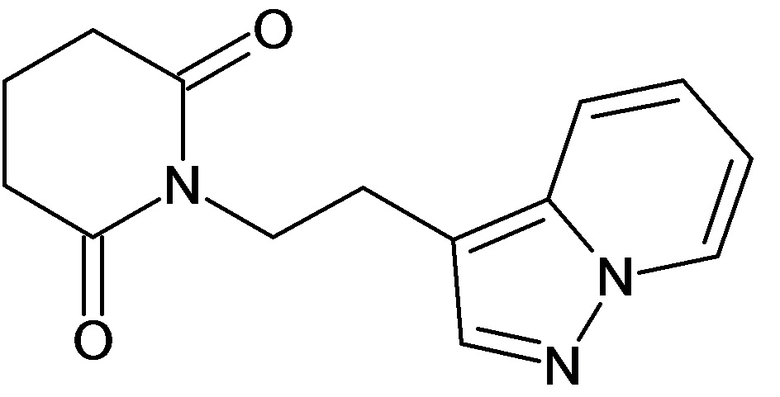
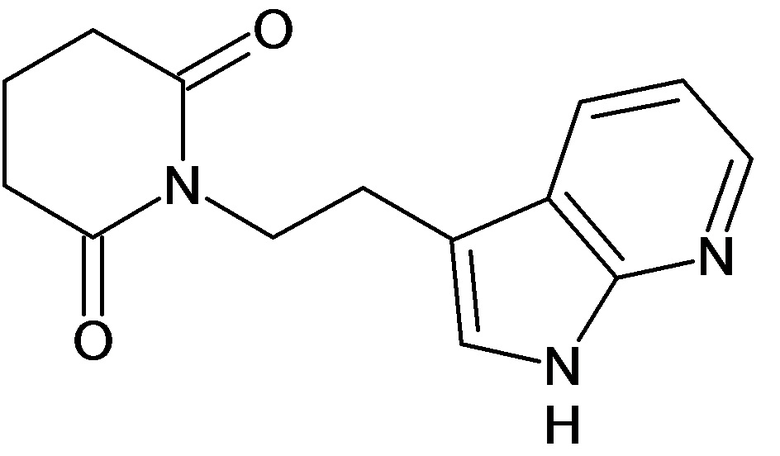
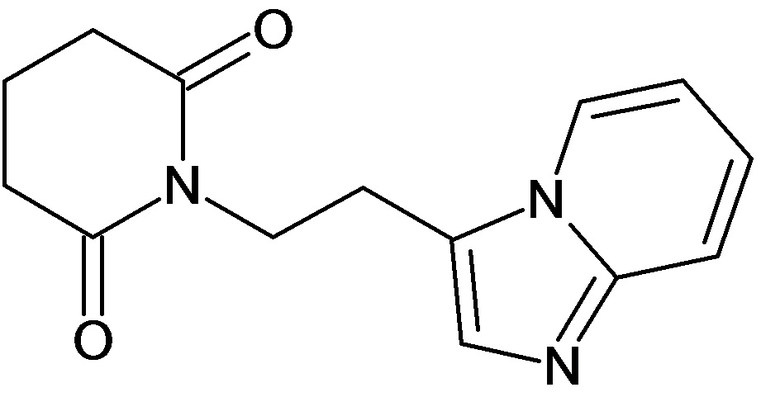
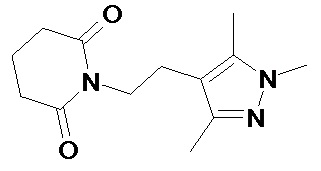
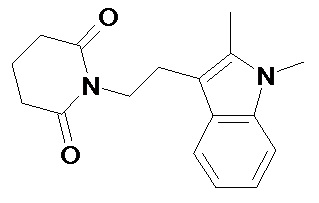
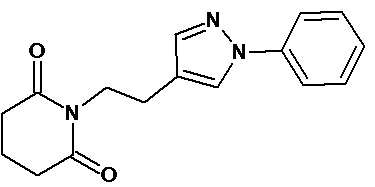
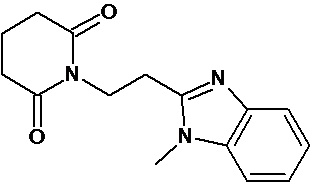
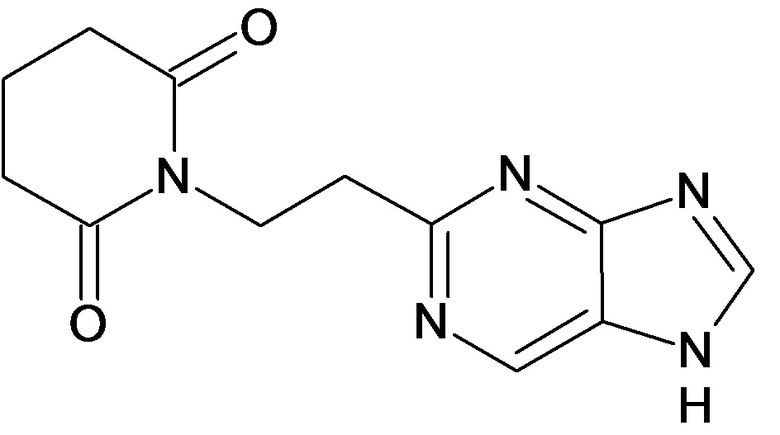
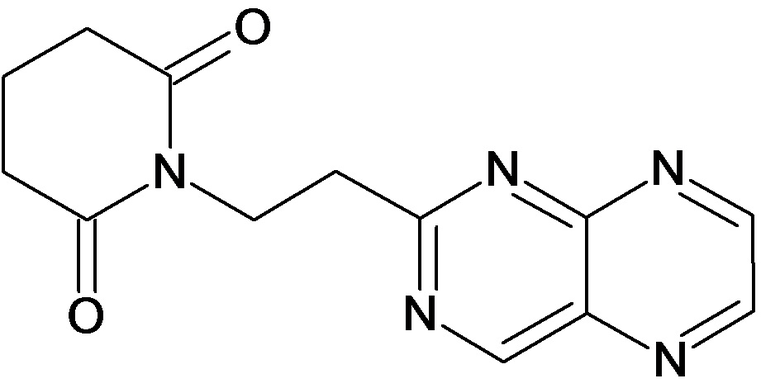
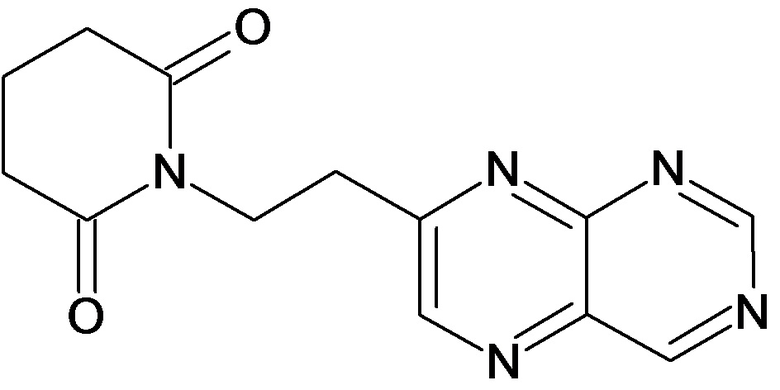
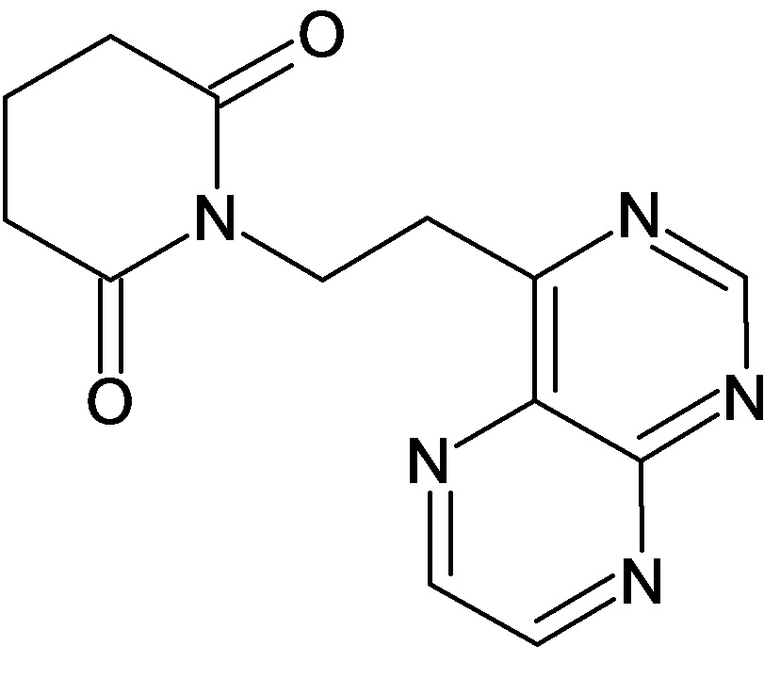
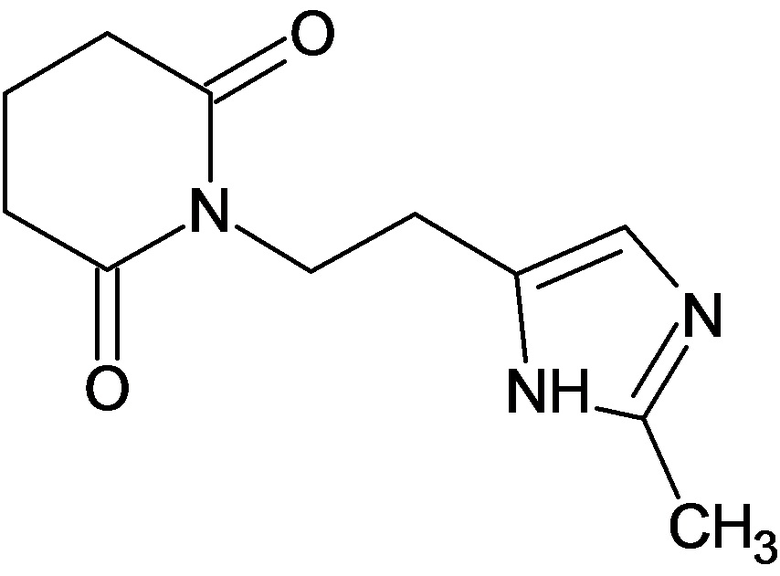
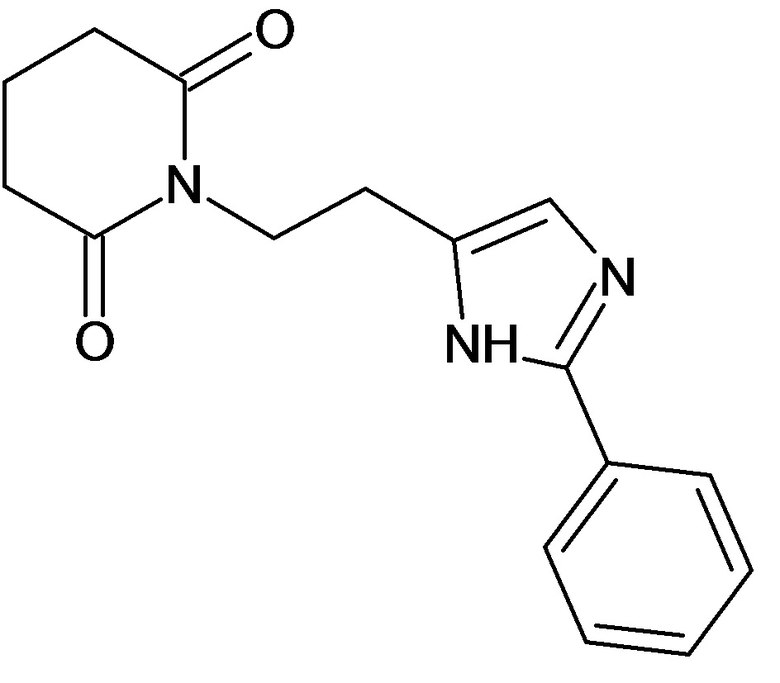
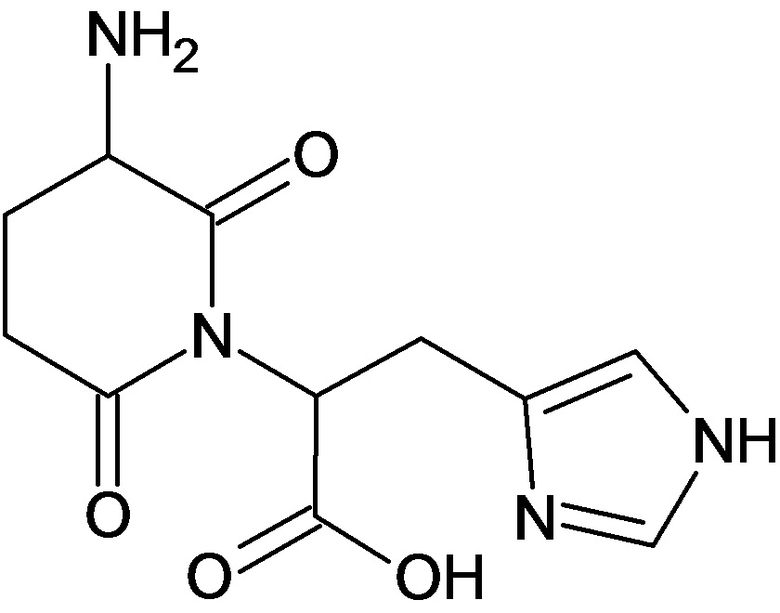
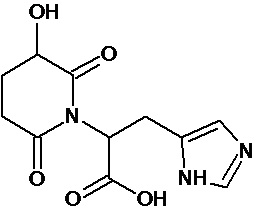
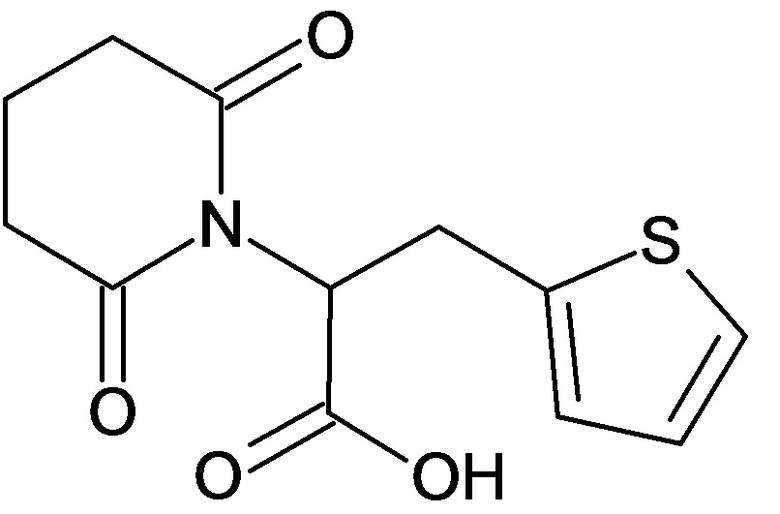
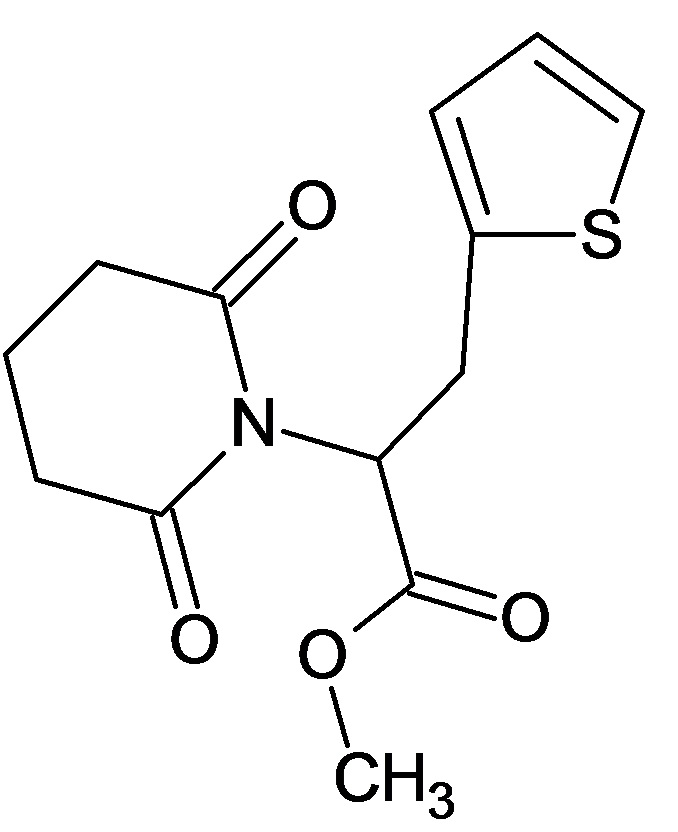
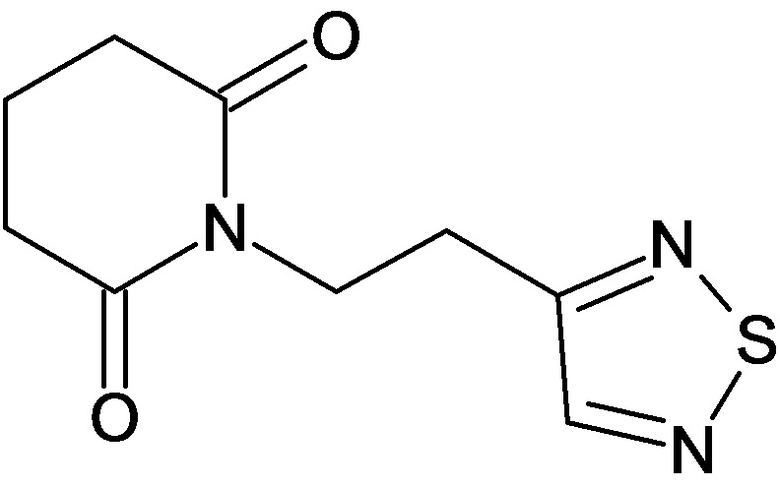
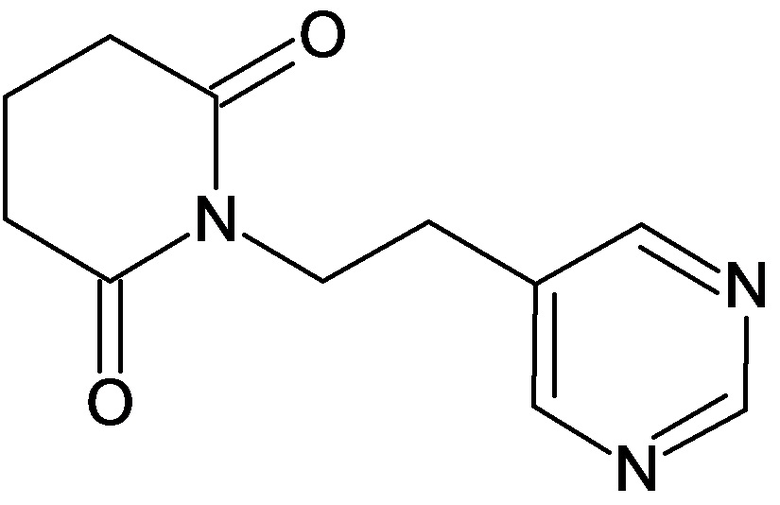
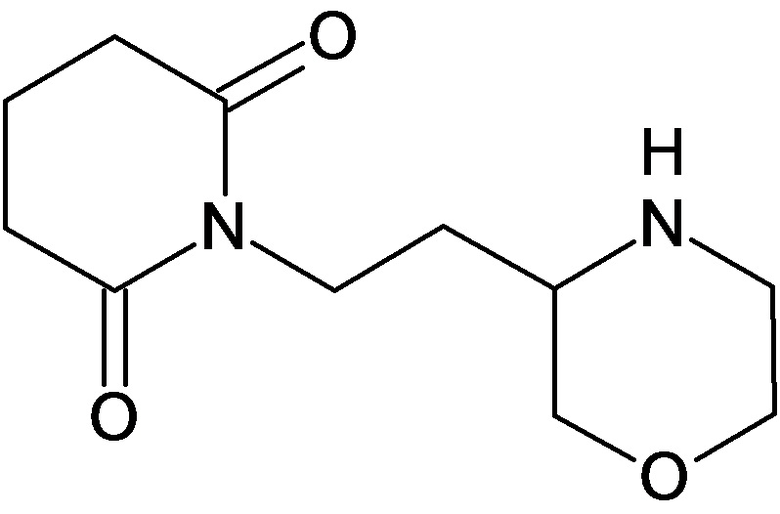
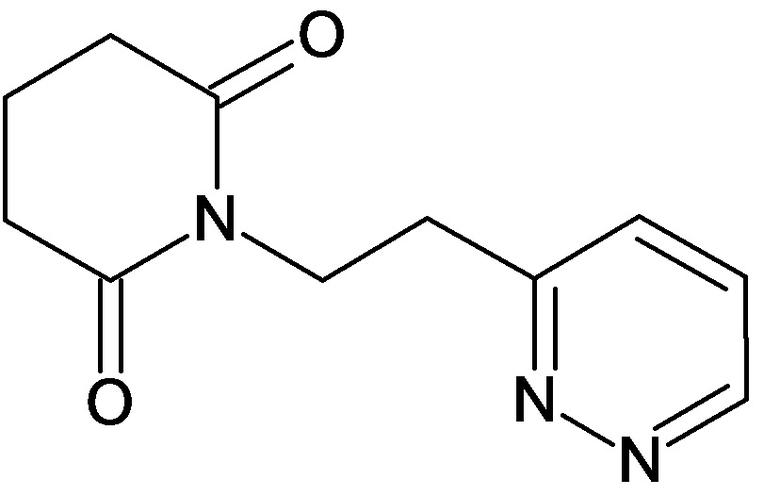
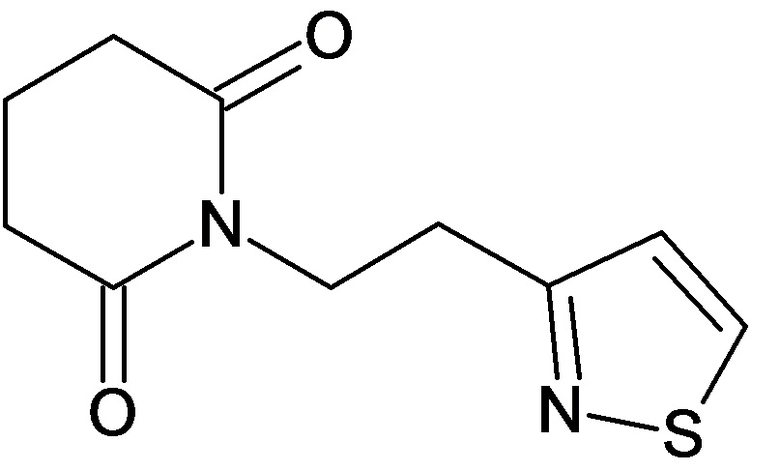
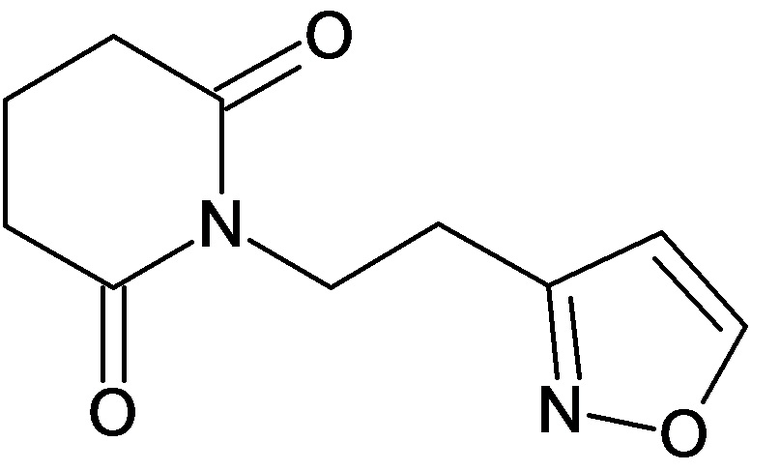
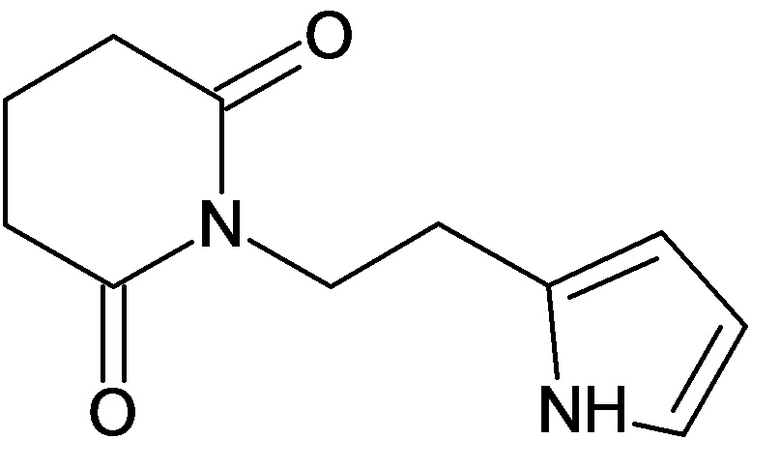
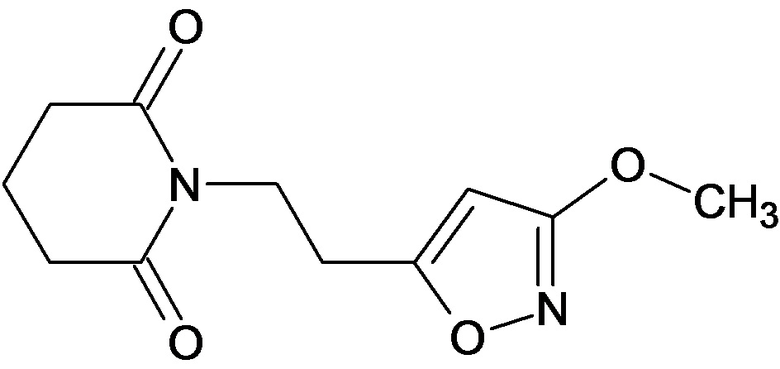
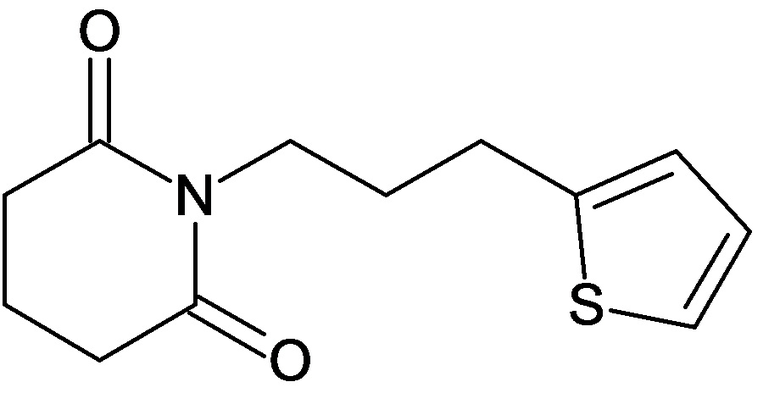
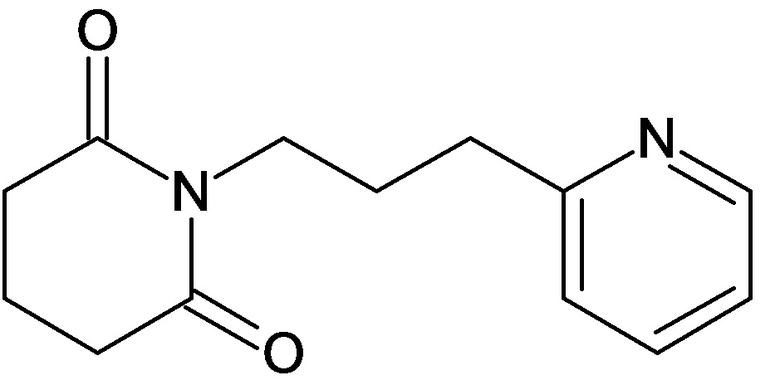
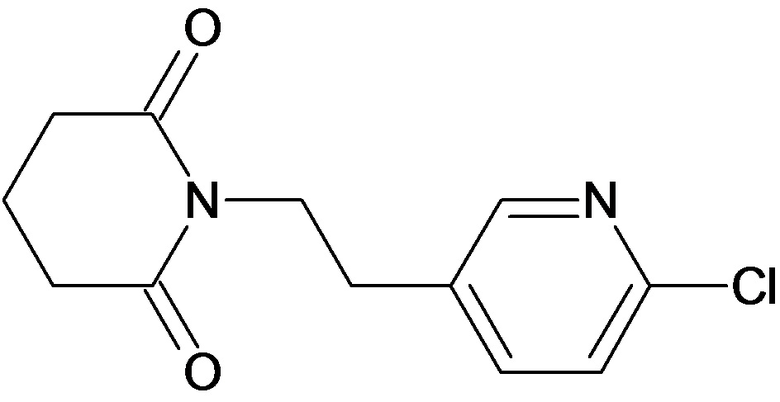
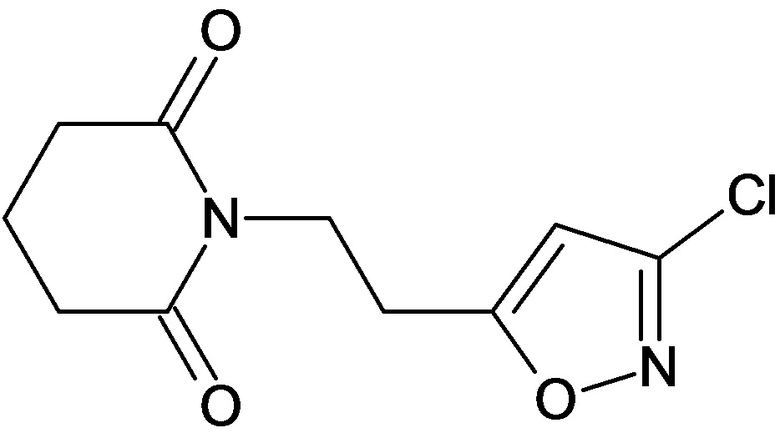
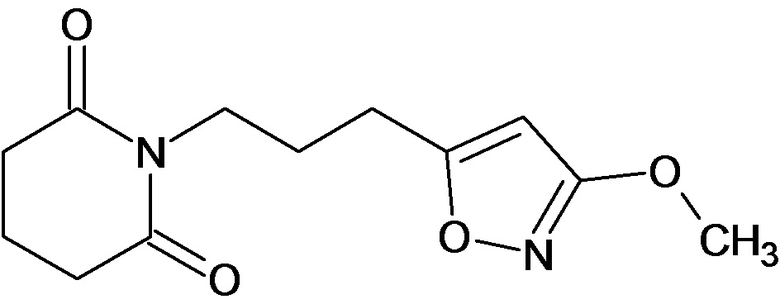
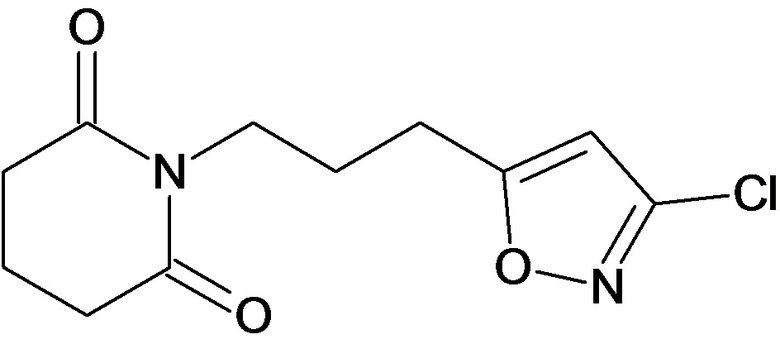
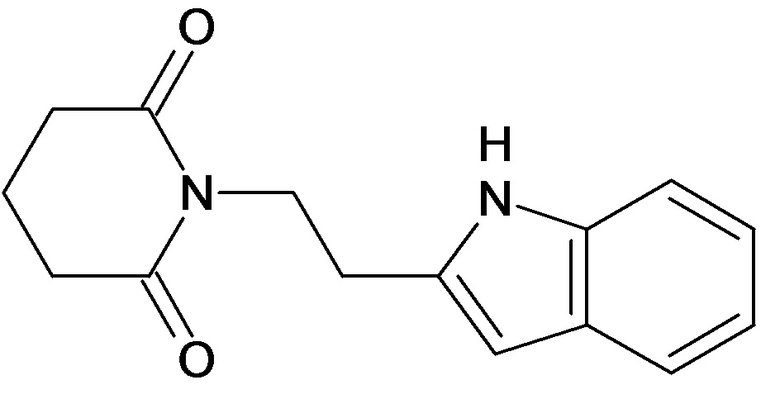
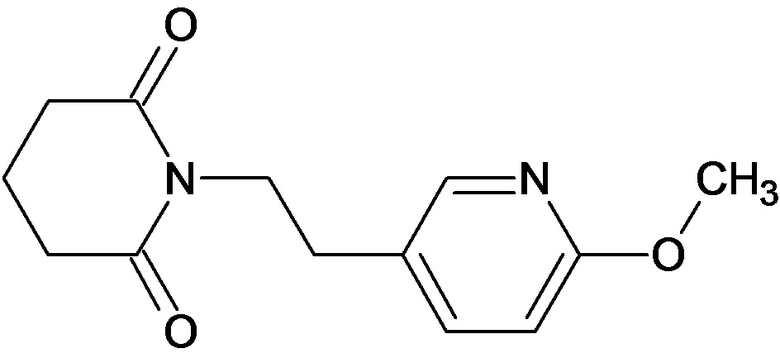
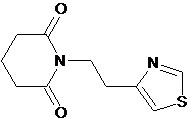
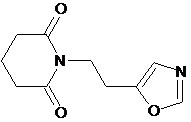
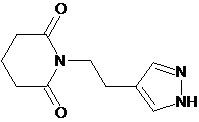
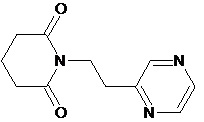
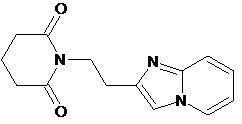
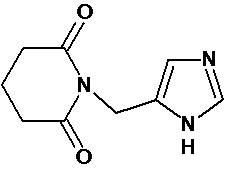
По аналогичным вышеприведенным методикам были синтезированы соединения 9, 12-115, представленные в следующей таблице:


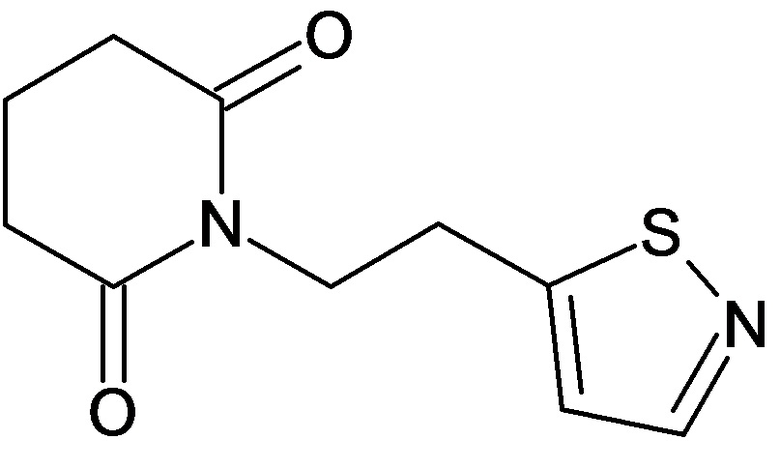
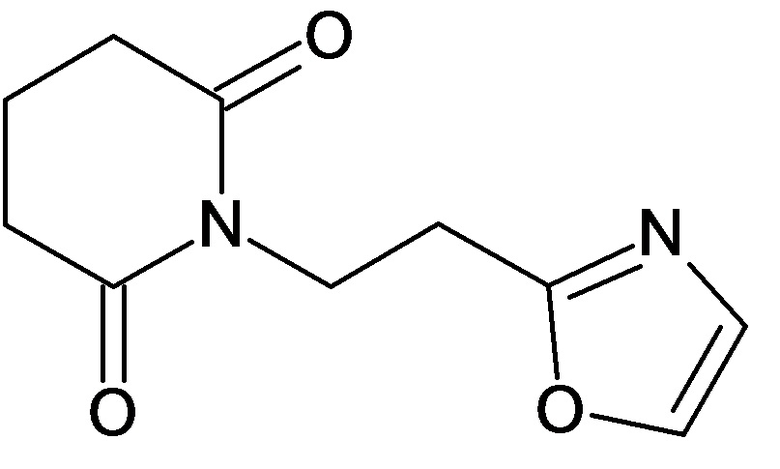
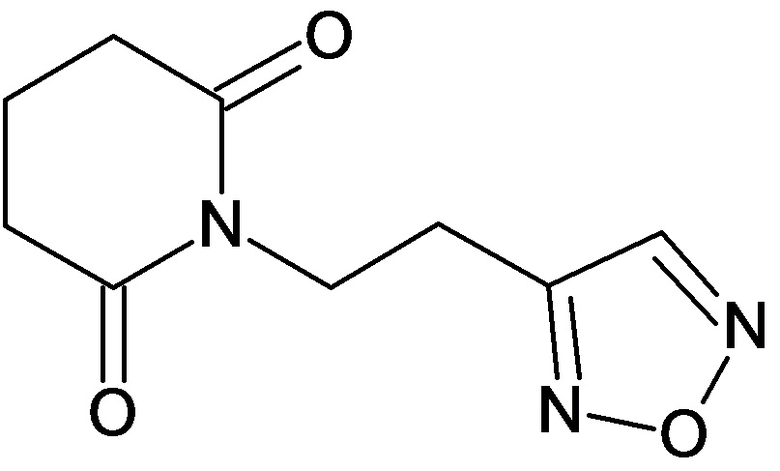
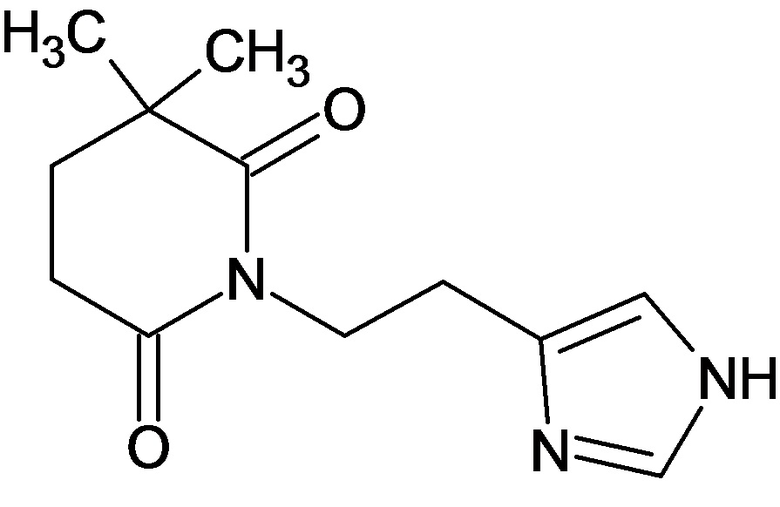
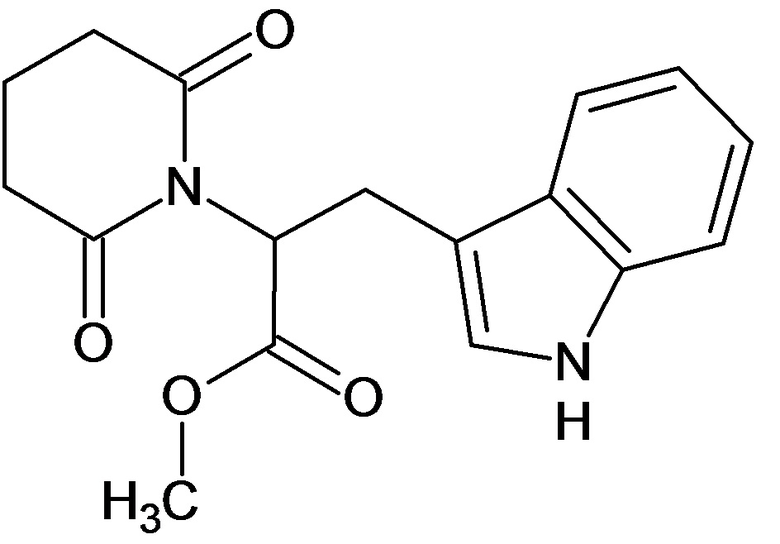
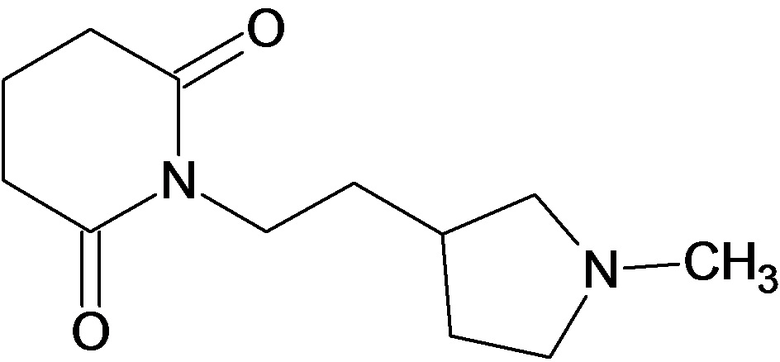
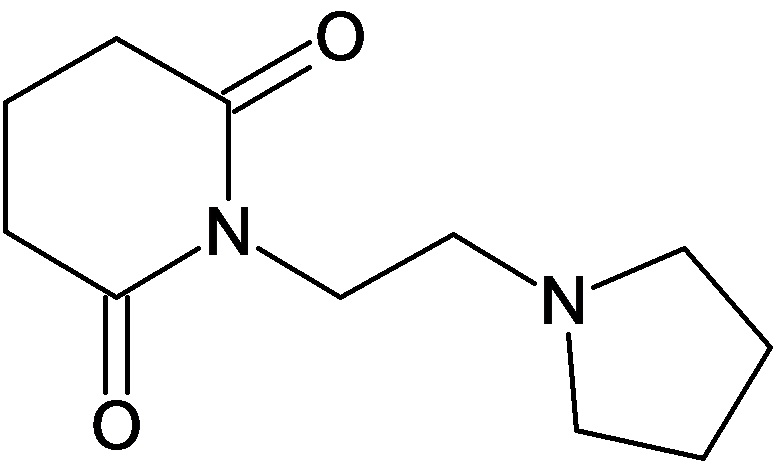
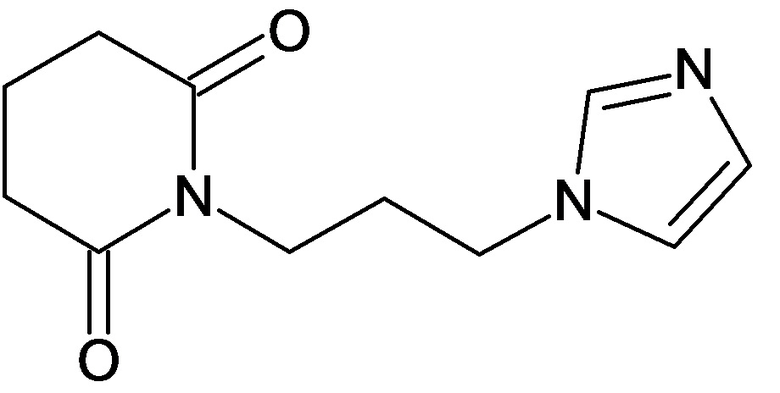

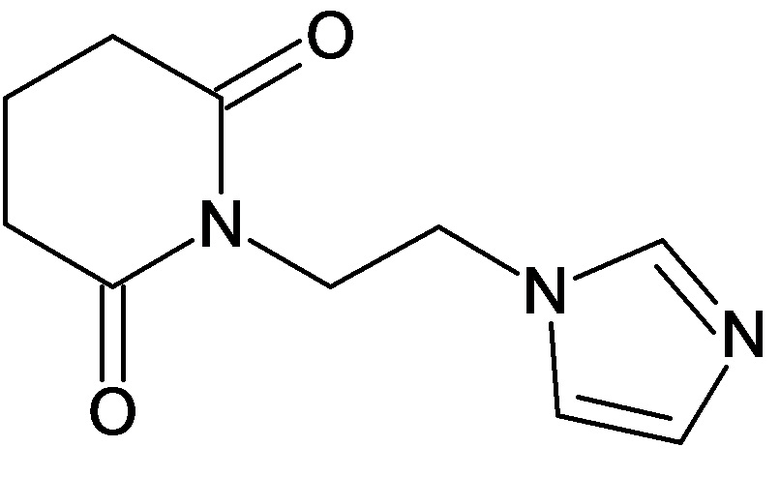
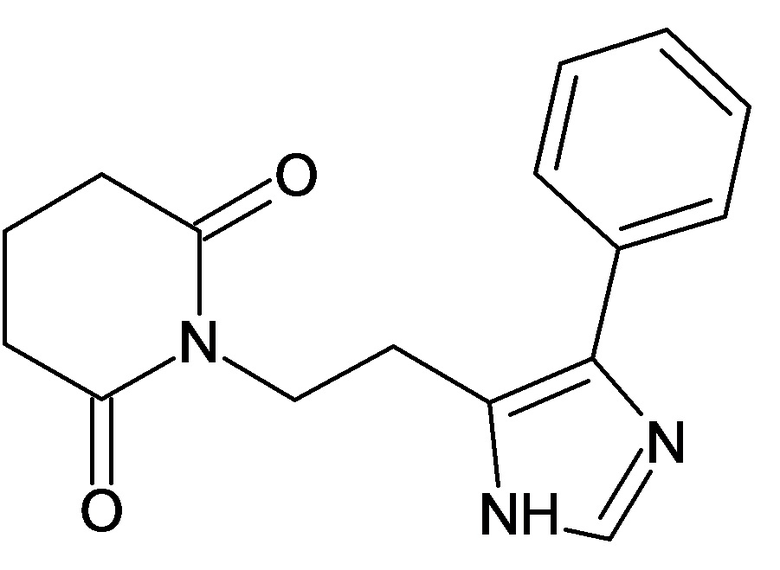
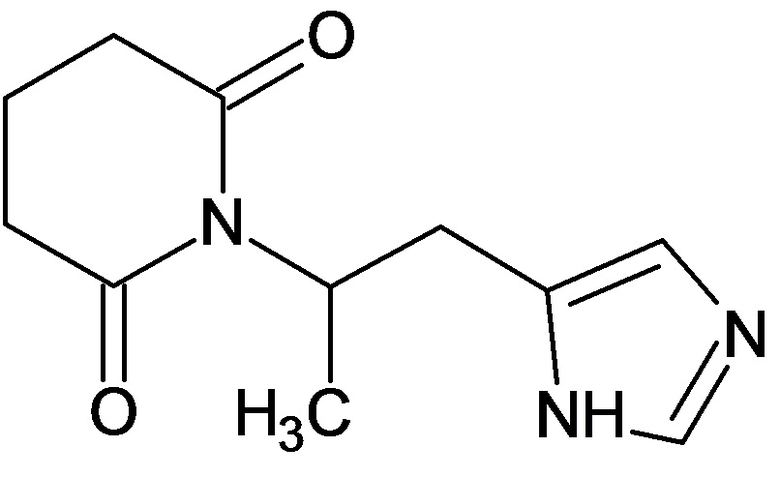

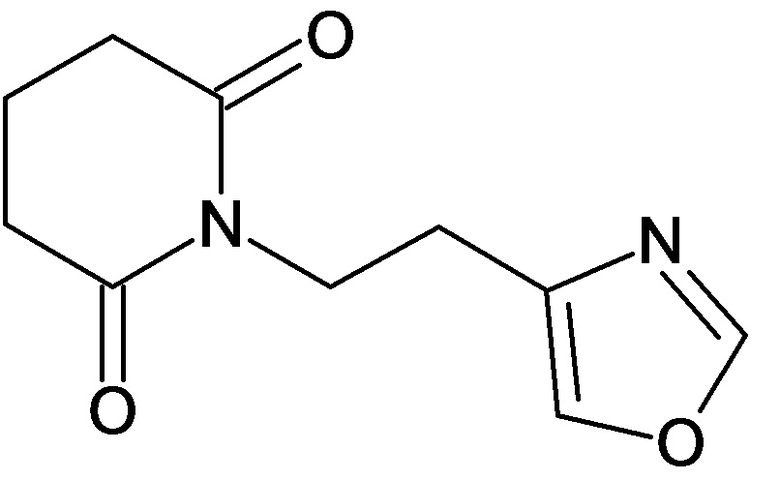

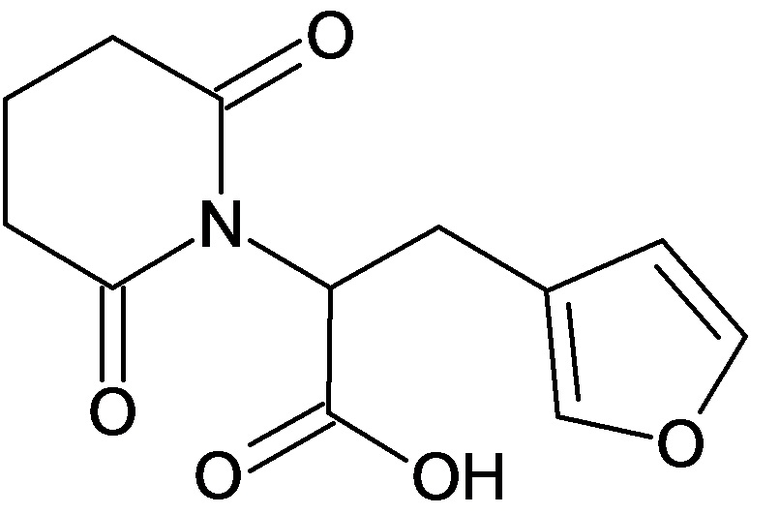
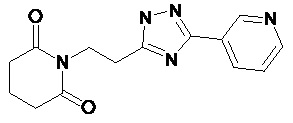
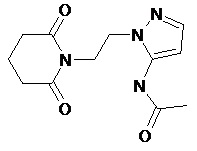
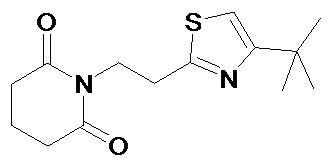
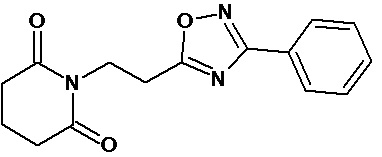
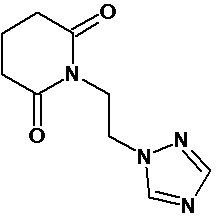
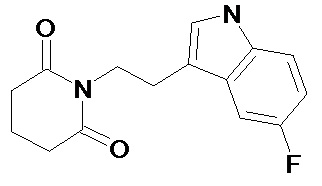
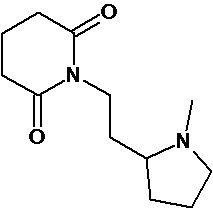
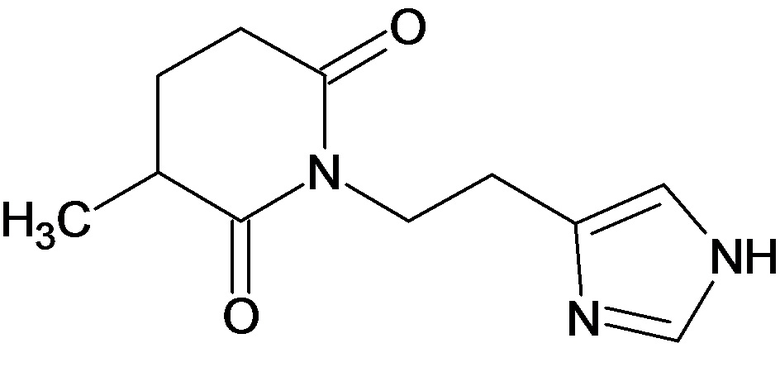
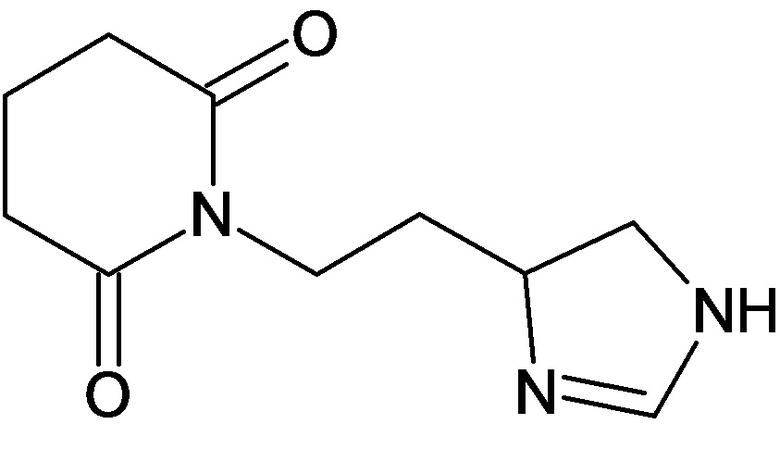
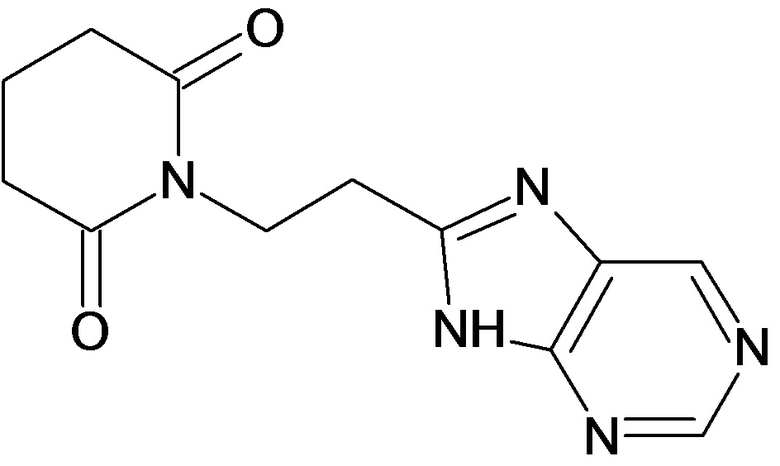
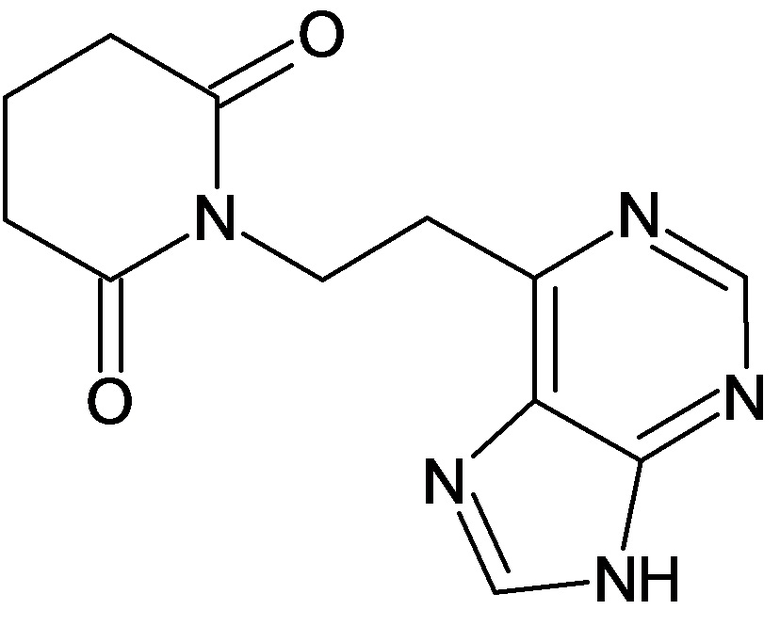
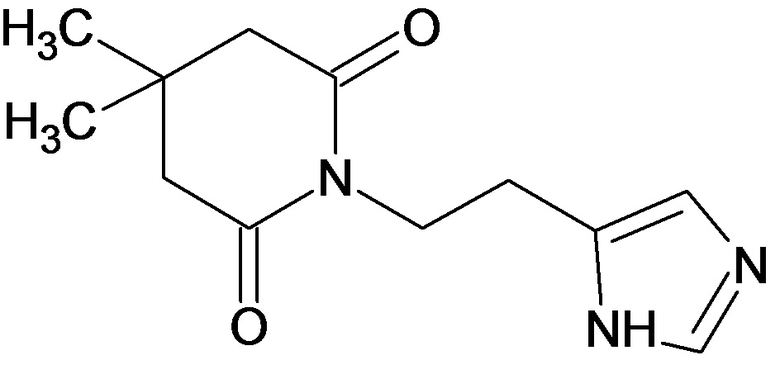
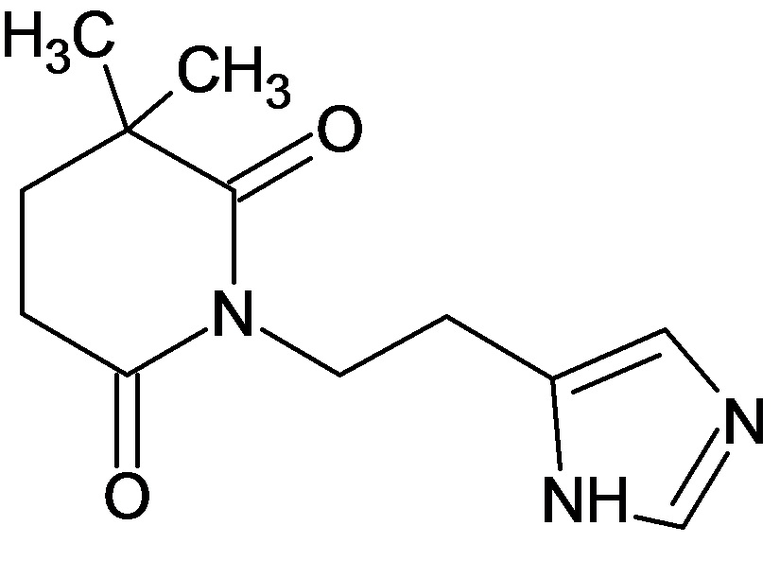
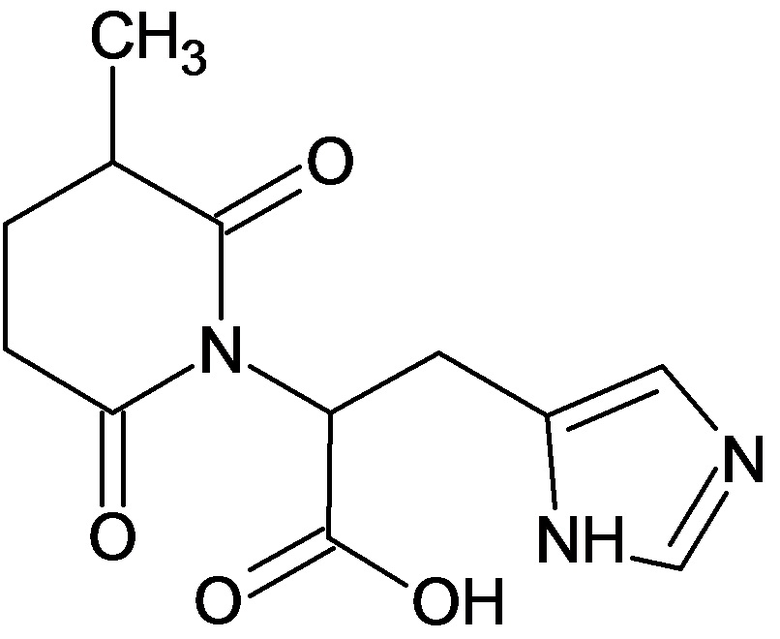

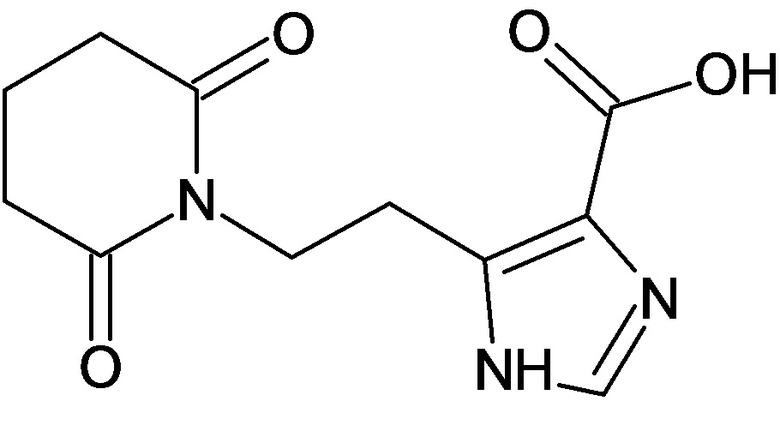
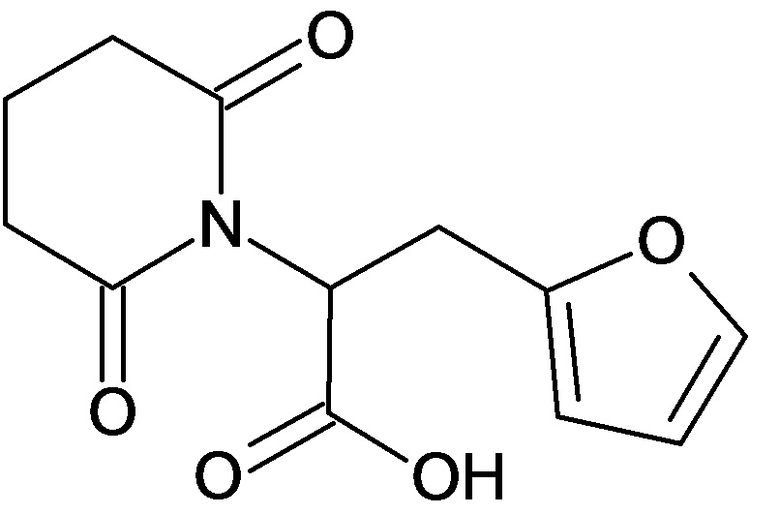
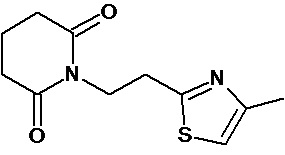
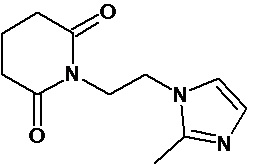

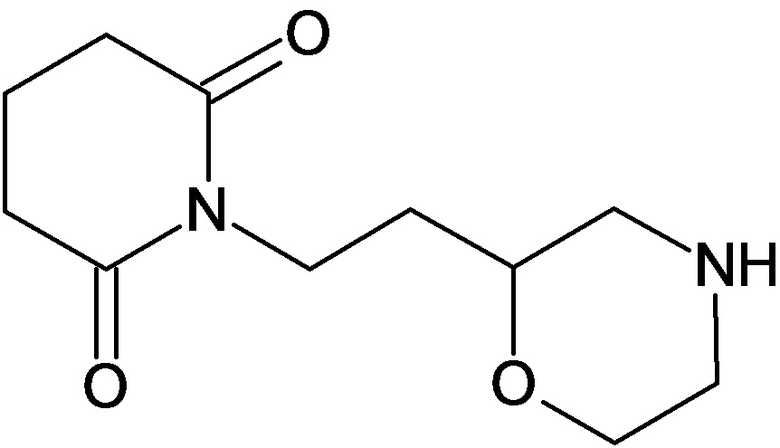

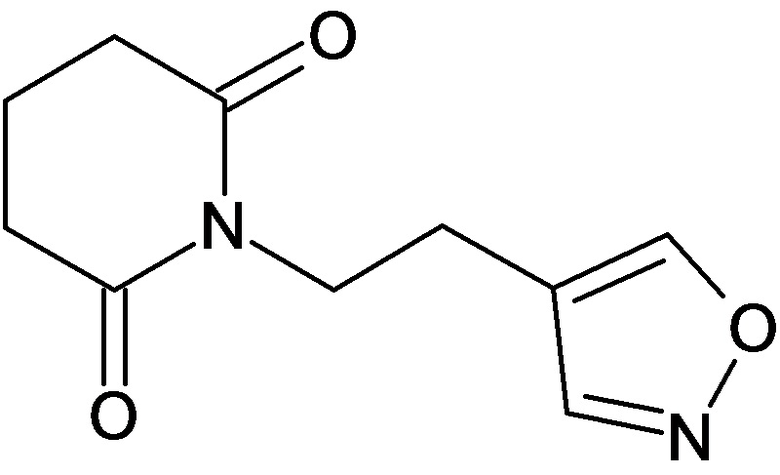
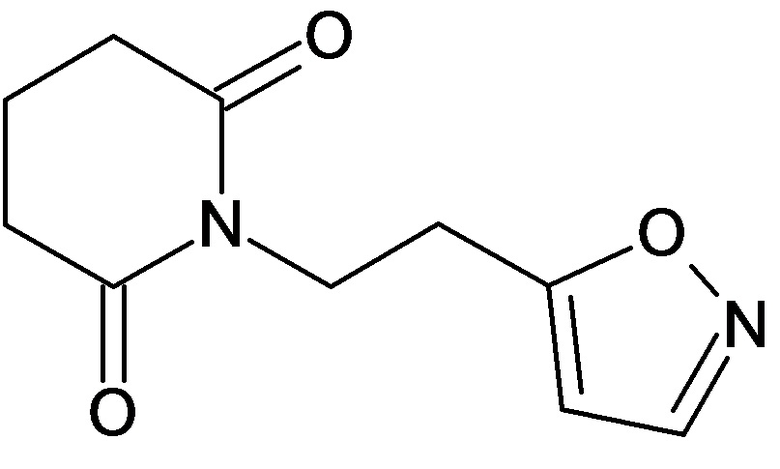
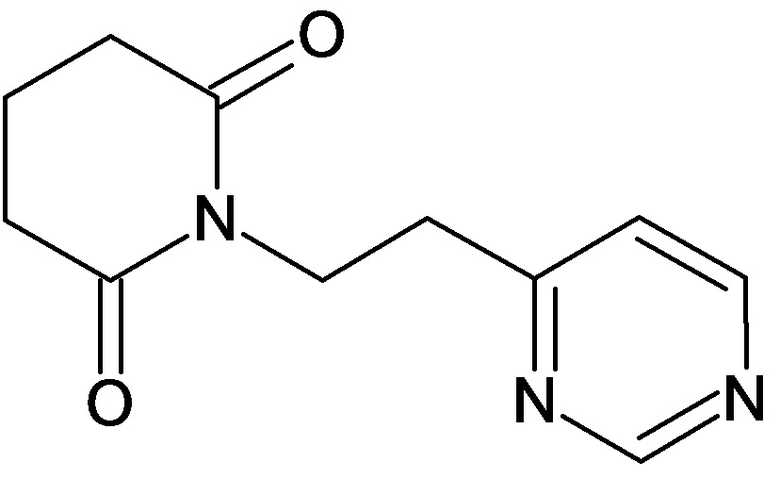
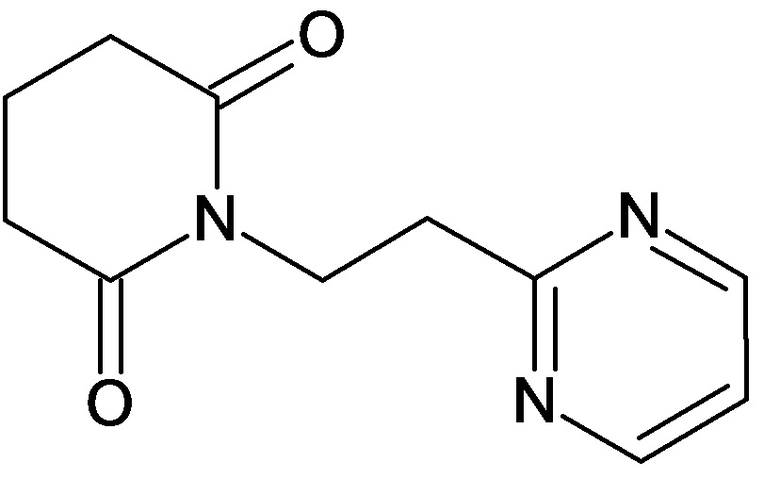

Тесты на биологическую активность
Средства и методы
Морфологическое исследование гистологических препаратов проводили при помощи светооптического микроскопа Leica DM LS (Leica Microsystems, Германия). Микроморфометрическое исследование выполняли с помощью окуляр-микрометра микроскопа Leica DM LS.
Математическую обработку полученных морфологических данных проводили методами вариационной статистики при помощи программы Statistica 6.0. Для анализа данных применяли описательную статистику: данные проверены на нормальность распределения с помощью критерия Шапиро-Уилка. Поскольку часть данных принадлежала к нормальному распределению, межгрупповые различия анализировали параметрическими методами с помощью критерия Стьюдента. Различия считали достоверными при p<0,05.
Пример 10
Оценка эффективности соединений на модели острого риносинусита у крыс
Морфологическое исследование гистологических препаратов проводилось при помощи светооптического микроскопа LeicaDMLS (LeicaMicrosystems, Германия). Микроморфометрическое исследование выполнялось с помощью окуляр-микрометра микроскопа LeicaDMLS.
Индукцию острого риносинусита проводили путем интраназального введения крысам 20 мкл 7,5% формалина (водный раствор, содержащий 40% формальдегида, 8% метилового спирта и 52% воды) в каждый носовой ход.
Введение формалина в носовые ходы крыс приводит к распространению воспаления на прилежащие ткани, в результате чего развивается клиническая картина, сходная с симптомами острого риносинусита у человека.
После акклиматизационного периода были сформированы следующие группы животных:
- интактные животные, получавшие физиологический раствор внутрижелудочно в количестве 0,2 мл, индукция острого риносинусита не проводилась;
- контрольная группа, животные, получавшие физиологический раствор внутрижелудочно в количестве 0,2 мл, в течение 7 дней после индукции острого синусита;
- животные, получавшие дексаметазон внутримышечно в дозе 0,33 мг/кг, в течение 7 дней после индукции острого синусита;
- животные, получавшие исследуемые вещества в дозе 27 мг/кг в течение 7 дней после индукции острого синусита.
Клинический осмотр каждого животного проводился ежедневно не менее 2-х раз в день.
В эксперименте на крысах линии Wistar путем индукции острого риносинусита введением в носовые ходы 7,5% раствора формалина были получены выраженные патологические изменения в контрольной группе животных, характеризующие развитие острого воспалительного процесса в слизистой оболочке носа. Вызванная патология характеризовалась полнокровием, гиперплазией и очаговым некрозом слизистой оболочки носовых ходов, увеличением количества бокаловидных клеток, выраженной инфильтрацией мнононуклеарами и лейкоцитами, гиперпродукцией слизи железами подслизистой оболочки.
С целью выявления специфической активности соединений было произведено морфологическое исследование слизистой и подслизистой оболочки обоих носовых ходов (дыхательная и обонятельная область) экспериментальных животных.
После окончания клинической фазы эксперимента материал от животных (нос, носогубный треугольник) вырезали и фиксировали в течение 24 часов в 10% растворе формалина, далее в течение 3-х суток декальцинировали в 12% смеси де Кастро, после чего материал проходил стандартную обработку в спиртах нарастающей концентрации (70-95%), ксилоле и парафине для изготовления гистологических препаратов с толщиной серийных парафиновых срезов 3-5 мкм. Для микроскопического исследования срезы окрашивались гематоксилином и эозином. С целью выявления кислых мукополисахаридов, продукция которых увеличивается при воспалении, применялось гистохимическое окрашивание препаратов альциановым синим с рН 2,5. Сопоставление и гистологическая оценка изменений проводилась в сравнении с группой интактных крыс.
После забоя во всех группах крыс была изучена макроскопическая картина воспаления в носовых ходах. При гистологическом, гистохимическом и морфометрическом исследовании у крыс в носовых ходах оценивались показатели: слизистая оболочка - полнокровие, гиперплазия и некроз эпителия, количество бокаловидных клеток на протяжении 1 мм слизистой носовой перегородки, характер воспаления.
Отражением эффективности работы мукоцилиарной системы в данном исследовании служили количество бокаловидных клеток и, как следствие, макроскопические изменения слизистой носовых ходов.
Количество бокаловидных клеток на 1 мм слизистой оболочки носовой перегородки у крыс, M±m
(приведены данные нескольких экспериментов)
Макроскопическая характеристика изменений слизистой носовых ходов у крыс различных групп
(приведены данные нескольких экспериментов)
Из представленных таблиц видно, что соединения общей формулы I эффективно сохраняют работу мукоцилиарной системы и показывают свою терапевтичекую эффективность на модели риносинусита. Фармакологическое действие исследованных соединений выражалось более выраженной регенерацией эпителия, снижением количества бокаловидных клеток и гиперсекреции слизи.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ГЛУТАРИМИДОВ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2015 |
|
RU2610275C2 |
| ПРОИЗВОДНОЕ ГЛУТАРИМИДОВ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2610276C1 |
| АМИДНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ, ВЫЗЫВАЕМЫХ РНК-СОДЕРЖАЩИМИ ВИРУСАМИ | 2017 |
|
RU2665638C1 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2721421C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2725881C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2719464C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2815401C2 |
| ПРОИЗВОДНЫЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ИХ ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2709529C1 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| НОВОЕ ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ПРИМЕНЕНИЕ ЕГО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРЕДОТВРАЩЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2474575C2 |
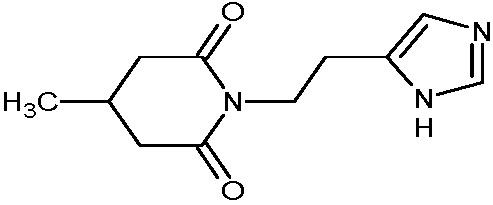
Изобретение относится к новому гетероциклическому соединению – 1-(2-(1Н-имидазол-4-ил)этил)-пиперидин-2,6-диону, лекарственному средству и фармацевтической композиции на его основе, его применению для получения лекарственного средства и способу лечения заболеваний верхних дыхательных путей. Технический результат: получено новое гетероциклическое соединение, обладающее полезным биологическим свойством. 5 н. и 4 з.п. ф-лы, 5 табл., 10 пр.
1. Соединение формулы:
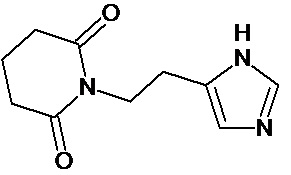
или его фармацевтически приемлемая соль.
2. Лекарственное средство для лечения заболеваний верхних дыхательных путей, представляющее собой соединение по п.1 или его фармацевтически приемлемую соль.
3. Лекарственное средство по п.2, где заболевание верхних дыхательных путей представляет собой риносинусит.
4. Фармацевтическая композиция для лечения заболеваний верхних дыхательных путей, включающая эффективное количество соединения по п.1 или его фармацевтически приемлемой соли, и фармацевтически приемлемые наполнители.
5. Фармацевтическая композиция по п.4, где заболевание верхних дыхательных путей представляет собой риносинусит.
6. Способ лечения заболеваний верхних дыхательных путей, включающий введение пациенту эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
7. Способ по п. 6, где заболевание представляет собой риносинусит.
8. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболеваний верхних дыхательных путей.
9. Применение по п.8, где заболевание представляет собой риносинусит.
| WO 2001053261 A1, 26.07.2001 | |||
| Способ получения N-(I-пиперазинил)бутилглутаримидов | 1991 |
|
SU1836365A3 |

