Область техники
Настоящее изобретение относится к соединениям формулы I и их фармацевтически приемлемым солям, к фармацевтическим композициям, содержащим такие соединения и соли, и к их применению. Соединения, соли и композиции по настоящему изобретению являются ингибиторами HPK1 и как таковые могут быть использованы для усиления активации иммунной системы при лечении или ослаблении аномальных клеточных пролиферативных нарушений, таких как рак, и для улучшения эффективности вакцинотерапии.
Уровень техники
Гемопоэтическая киназа-предшественник 1 (HPK1), также известная как митоген-активированная киназа киназы киназы протеинкиназы 1 (MAP4K1), является членом Ste20-подобного семейства серин/треонинкиназ млекопитающих, который действует через сигнальные пути,7NK и ERK. HPK1 в основном экспрессируется в гемопоэтических органах и клетках (например, Т-клетках, В-клетках и дендритных клетках), что позволяет предположить потенциальное вовлечение HPK1 в регуляцию передачи сигналов в гемопоэтических линиях, включая лимфоциты. (Shui, et al., "Hematoppietic progenitor kinase 1 negatively regulates T cell receptor signaling and T eel-mediated immune responses", Nature Immunology 8, 84-91 (2006)).
Например, стимуляция Т-клеточного рецептора (TCR) индуцирует фосфорилирование HPK1 тирозина 379 и его перемещение на плазматическую мембрану. Ферментативная активация HPK1 сопровождается фосфорилированием регуляторных сайтов в петле активации HPK1 киназы. Полная активация HPK1 зависит от аутофосфорилирования треонина 165 и фосфорилирования протеинкиназой D (PKD) серина 171 (Arnold et al., "Activation of Hematopoietic Progenitor Kinase 1 Involves Relocation, Autophosphorylation, and Transphosphorylation by Protein Kinase D1.", Mol Cell Biol 25 (6), 2364-83 (2005)). HPK1-опосредованное фосфорилирование адаптерного белка SLP76 в конечном итоге приводит к дестабилизации сигнального комплекса TCR, который препятствует и ослабляет последующие события передачи сигналов киназы митоген-активированного белка (MAP), необходимые для активации и пролиферации Т-клеток. (Hernandez, et al., "The kinase activity of hematopoietic progenitor kinase 1 is esential for the regulation of T cell function", Cell Reports 25, (1), 80-94, (2018)). Также было показано, что HPK1 киназа отрицательно регулирует передачу сигналов Т-клетками посредством рецептора PGE2 РКА-зависимым образом. Кроме того, сообщалось, что HPK1 киназа играет роль в: i) индуцированной активацией гибели клеток (AICD) и активации,7NK; ii) регуляции активации интегрина антигена-1, ассоциированного с функцией лейкоцитов (LFA-1), на Т-клетках путем прямой конкуренции с адаптерным белком, способствующим адгезии и дегрануляции (ADAP), за связывание SLP76 SH2-домена; и iii) регуляции активации посредством передачи сигналов ядерного фактора кВ (NF-кВ) путем взаимодействия с IKK-α и -β. Исследования также показали, что HPK1 отрицательно регулирует сигнальный путь киназы MAP и транскрипцию АР-1 в Т-клетках. (рассмотрено в Hernandez, et al. 2018).
Исследования, проведенные на сегодняшний день в отношении HPK1 киназ, показывают, что ингибирование HPK1 играет роль в усилении ответа дендритных и Т-клеток и, таким образом, в усилении противоопухолевого иммунитета, клиренсе вируса и ответе на вакцинотерапию.
Сущность
Настоящее изобретение предлагает, частично, соединения формулы I и их фармацевтически приемлемые соли. Такие соединения могут ингибировать активность HPK1 киназы, воздействуя тем самым на биологические функции. Также предложены фармацевтические композиции и лекарственные средства, содержащие соединения или соли по изобретению, отдельно или в комбинации с дополнительными противораковыми терапевтическими агентами или паллиативными агентами.
Настоящее изобретение также предлагает, частично, способы получения соединений, фармацевтически приемлемых солей и композиций по изобретению и способы применения вышеизложенного.
В одном варианте осуществления, изобретение предлагает соединение Формулы I:

или его фармацевтически приемлемую соль, где:
R1 представляет собой (C1-C6)алкил, галоген (C1-C6)алкил, (C1-C6)алкокси, галоген (C1-C6)алкокси,-N(R5)(R6), или (С3-С6)циклоалкил, где указанные (C1-C6)алкил, галоген (C1-C6)алкил и (С3-С6)циклоалкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано, (C1-C6)алкил или (C1-С6)алкокси, где:
R5 и R6 каждый независимо представляет собой водород или (C1-С6)алкил, замещенный 0, 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C1-C6)алкокси, циано и гидрокси, или
R5 и R6 вместе с азотом, к которому они присоединены, образуют (4-8-членный) гетероциклоалкил, который замещен 0, 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C1-C6)алкила, галоген (C1-C6)алкила, (C1-C6)алкокси и галоген (C1-C6)алкокси;
R2 представляет собой N(R7)(R8), где:
каждый из R7 и R8 независимо представляет собой водород или (C1-С6)алкил, который замещен 0 или 1 заместителем, представляющим собой галоген, (C1-C6)алкокси, циано или гидрокси; или
R7 представляет собой водород или (C1-C6)алкил, замещенный 0 или 1 заместителем, представляющим собой галоген, (C1-C6)алкокси, циано или гидрокси; и R8 вместе с азотом, к которому он присоединен, и вместе с R3a и углеродом, к которому он присоединен, образуют (4-6-членный) гетероциклоалкил, который независимо замещен 0, 1 или 2 заместителями, выбранными из галогена, гидрокси, (C1-C3)алкила, галоген (C1-C3)алкила, (С1-С3)алкокси и галоген (C1-C6)алкокси; или
R7 и R8 вместе с азотом, к которому они присоединены, образуют (4-6-членный) гетероциклоалкил, который замещен 0, 1 или 2 заместителями, независимо выбранными из галогена, гидрокси, (C1-C6)алкила, галоген (C1-C6)алкила, (C1-C6)алкокси и галоген (C1-C6)алкокси;
R3a представляет собой водород или (C1-C3)алкил, замещенный О или 1 заместителем, представляющим собой гидрокси или (С1-С3)алкокси;
R3b представляет собой водород или (C1-C3)алкил, при условии, что R3a и R3b оба не представляют собой Н, когда R4 представляет собой (R4-i);
R4 представляет собой (R4-i) или (R4-ii):


где:
R4N представляет собой (С1-С6)алкил, галоген (С1-С6)алкил или (С3-С6)циклоалкил, где указанный (C1-C6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси;
R4C представляет собой водород, (C1-C6)алкил, галоген (С1-С6)алкил, (C1-C6)алкокси, галоген (C1-C6)алкокси или (С3-С6)циклоалкил, где указанные (C1-C6)алкил и галоген (C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (C1-C6)алкокси;
R4D представляет собой водород, (C1-C6)алкил, галоген (С1-С6)алкил, (C1-C6)алкокси, галоген (C1-C6)алкокси или (С3-С6)циклоалкил, где указанные (C1-C6)алкил и галоген (C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (C1-C6)алкокси;
R4E представляет собой водород, галоген, циано, гидрокси или (C1-C6)алкил; и
R4F представляет собой водород, галоген, циано, гидрокси, (C1-C6)алкил, галоген (C1-C6)алкил, (С1-С6)алкокси, галоген (C1-С6)алкокси или (С3-С6)циклоалкил, где указанные (С1-С6)алкил и галоген (С1-С6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (С1-С6)алкокси.
Изобретение также относится к терапевтическим способам и применениям, включающим введение соединения по изобретению или его фармацевтически приемлемой соли.
В другом варианте осуществления, изобретение предлагает способ лечения аномального роста клеток, в частности рака, у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли. Соединения по изобретению можно вводить в виде отдельных агентов или можно вводить в комбинации с другими противораковыми терапевтическими агентами, в частности, стандартами лечения, подходящими для конкретного вида рака.
В другом варианте осуществления, изобретение предлагает способ лечения аномального роста клеток, в частности рака, у субъекта, нуждающегося в этом, включающий введение субъекту некоторого количества соединения по изобретению или его фармацевтически приемлемой соли, в комбинации с некоторым количеством дополнительного противоракового терапевтического агента, где количества вместе эффективны при лечении указанного аномального клеточного роста.
В другом варианте осуществления, изобретение относится к соединению по изобретению или его фармацевтически приемлемой соли для применения в качестве лекарственного средства, в частности лекарственного средства для лечения рака.
В другом варианте осуществления, изобретение относится к соединению по изобретению или его фармацевтически приемлемой соли для применения при лечении аномального роста клеток, в частности, рака, у субъекта.
В другом варианте осуществления, изобретение предлагает применение соединения по изобретению или его фармацевтически приемлемой соли для лечения аномального роста клеток, в частности, рака, у субъекта.
В другом варианте осуществления, изобретение относится к фармацевтической композиции для лечения аномального роста клеток у субъекта, нуждающегося в этом, которая содержит соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент.
В еще одном варианте осуществления, изобретение предлагает применение соединения формулы I, как описано в настоящем документе, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения аномального роста клеток или патогенов у субъекта.
В частых вариантах осуществления вышеуказанных соединений, способов и применений, аномальный рост клеток представляет собой рак.
В некоторых вариантах осуществления, предлагаемые способы и применения приводят к одному или нескольким из следующих эффектов: (1) ингибирование пролиферации раковых клеток; (2) ингибирование инвазивности раковых клеток; (3) индуцирование апоптоза раковых клеток; (4) ингибирование метастазирования раковых клеток; (5) ингибирование ангиогенеза; (6) усиление ответов Т-клеток; или (7) усиление ответов дендритных и В-клеток; (8) усиление противоопухолевой активности; (9) усиление вакцинотерапии; и (10) усиление опосредованного иммунной системой удаления патогенов, таких как вирусы, бактерии или паразиты (например, кишечные черви).
В другом варианте осуществления, изобретение предлагает способ лечения HPKl-зависимых нарушений и усиления иммунного ответа у субъекта, включающему введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, которое является эффективным для лечения указанного нарушения или усиления указанного иммунного ответа.
В некоторых вариантах осуществления, способы и применения, описанные в настоящем документе, дополнительно включают введение субъекту количества дополнительного противоракового терапевтического агента, вакцины, антибактериального агента, противовирусного агента или паллиативного агента, которые вместе эффективны для лечения указанного аномального клеточного роста или патогена. Каждый из описанных ниже вариантов осуществления соединений по настоящему изобретению можно комбинировать с одним или несколькими другими вариантами осуществления соединений по настоящему изобретению, описанными в настоящем документе, что не противоречит варианту(ам) осуществления, с которым(и) он комбинируется.
Следует понимать, что как предшествующее общее описание, так и последующее подробное описание являются только иллюстративными и пояснительными и не ограничивают заявленное изобретение.
Кроме того, каждый из вариантов осуществления, описанных ниже, включает фармацевтически приемлемые соли соединений по изобретению. Соответственно, фраза «или его фармацевтически приемлемая соль» подразумевается в описании всех описанных в настоящем документе соединений.
Подробное описание
Определения и примеры
Настоящее изобретение может быть легче понято со ссылкой на следующее подробное описание предпочтительных вариантов осуществления изобретения и включенных в него примеров. Следует понимать, что используемая в настоящем документе терминология предназначена только для целей описания конкретных вариантов осуществления и не предназначена для ограничения. Кроме того, следует понимать, что, если конкретно не указано в настоящем документе, терминология, используемая в настоящем документе, должна иметь свое традиционное значение, известное в данной области техники.
Используемые в настоящем документе формы единственного числа «а», «ап» и «the» включают ссылки во множественном числе, если не указано иное. Например, заместитель «а» включает один или несколько заместителей.
Термин «примерно» относится к относительному термину, обозначающему приближение плюс или минус 10% от номинального значения, которое, в одном варианте осуществления, означает плюс или минус 5%, в другом варианте осуществления, плюс или минус 2%. Для области этого описания, этот уровень приближения является подходящим, если значение конкретно не указано как требующее более узкого диапазона.
В различных местах настоящего описания, заместители соединений по изобретению описаны в группах или в диапазонах. В частности, предполагается, что изобретение включает каждую отдельную субкомбинацию членов таких групп и диапазонов. Например, термин «C1-6 алкил» специально предназначен для включения С1 алкила (метила), С2 алкила (этила), С3 алкила, С4 алкила, С5 алкила и С6 алкила.
Используемый в настоящем документе термин «антагонист HPK1» или «ингибитор HPK1» представляет собой молекулу, которая снижает, ингибирует или иным образом ослабляет одну или несколько биологических активностей HPK1 (например, активность серин/треонинкиназы, рекрутирование в комплекс TCR при активации TCR, взаимодействие с партнером по связыванию белка, таким как SLP76). Антагонизм с использованием антагониста HPK1 не обязательно указывает на полное устранение активности HPK1. Вместо этого, активность может снизиться на статистически значимую величину. Например, соединение по настоящему изобретению может снижать активность HPK1, по меньшей мере, от примерно 2,5% до примерно 100%, от примерно 10% до примерно 90%, от примерно 20% до примерно 70%, от примерно 30% до примерно 60%, от примерно 40% до примерно 50% по сравнению с соответствующим контролем. В некоторых вариантах осуществления, антагонист HPK1 снижает, ингибирует или иным образом снижает серин/треонинкиназную активность HPK1. В некоторых из этих вариантов осуществления, антагонист HPK1 уменьшает, ингибирует или иным образом уменьшает HPK1-опосредованное фосфорилирование SLP76 и/или Gads. Описанные в настоящем документе соединения связываются непосредственно с HPK1 и ингибируют его киназную активность.
Изобретение, описанное в настоящем документе, может быть реализовано в отсутствие какого(их)-либо элемента(ов), не описанного в настоящем документе конкретно. Так, например, в каждом случае настоящего документа любой из терминов «содержащий», «состоящий по существу из» и «состоящий из» может быть заменен любым из двух других терминов.
Термин «(Сх-Су)алкил», используемый в настоящем документе, относится к насыщенной алкильной группе с разветвленной или прямой цепью, содержащей от х до у атомов углерода. Например, «(C1-С6)алкил» представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, и включает, но не ограничен ими, метил, этил, н-пропил, изопропил, н-бутил, вгор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил и н-гексил. Термин «(С1-С3)алкил» содержит от 1 до 3 атомов углерода и включен в «(C1-С6)алкил».
Термин «галоген(Сх-Су)алкил», используемый в настоящем документе, относится к (Сх-Су)алкильной группе, как определено выше, где алкильная группа замещена одним или несколькими атомами галогена. Типовое число заместителей галогена составляет от 1 до 3 заместителей. Типовые примеры галоген (Сх-Су)алкилов включают, но не ограничены ими, фторметил, фторэтил, дифторметил, дифторэтил, трифторметил и трифторэтил.
Термин «(Сх-Су)алкокси», используемый в настоящем документе, относится к (Сх-Су)алкильной группе, как определено выше, присоединенной к основной молекулярной группе через атом кислорода. Типовые примеры (С1-С6)алкокси включают, но не ограничены ими, метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси и гексилокси.
Термин «галоген(Сх-Су)алкокси», используемый в настоящем документе, относится к (Сх-Су)алкоксигруппе, как определено выше, где алкоксигруппа замещена одним или несколькими атомами галогена. Типовое число заместителей галогена составляет от 1 до 3 заместителей. Типовые примеры галоген(Сх-Су)алкокси включают, но не ограничены ими, фторметокси, фторэтокси, дифторметокси, дифторэтокси и трифторметокси, трифторэтокси.
Используемый в настоящем документе термин «циклоалкил» относится к циклической одновалентной углеводородной группе формулы-CnH(2n-1), содержащей, по меньшей мере, три атома углерода. «(С3-Су)циклоалкил» относится к циклоалкилу, содержащему от 3 до у атомов углерода. «(С3-С6)циклоалкил» может представлять собой моноциклическое кольцо, примеры которого включают циклопропил, циклобутил, циклопентил и циклогексил.
Используемый в настоящем документе термин «гетероциклоалкил» относится к циклоалкилу, как определено выше, в котором, по меньшей мере, один из атомов углерода в кольце замещен гетероатомом, выбранным из азота, кислорода и серы. Используемый в настоящем документе термин «n-членный», где n представляет собой целое число, обычно описывает количество образующих кольцо атомов в группе, где количество образующих кольцо атомов равно n. Термин «(4-6-членный)гетероциклоалкил» означает, что гетероциклоалкильный заместитель содержит всего от 4 до 6 кольцевых атомов, по меньшей мере, один из которых является гетероатомом. Термин «(4-8-членный)гетероциклоалкил» означает, что гетероциклоалкильный заместитель содержит всего от 4 до 8 кольцевых атомов, по меньшей мере, один из которых является гетероатомом. «(6-членный)гетероциклоалкил» означает, что гетероциклоалкильный заместитель содержит всего 6 кольцевых атомов, по меньшей мере, один из которых является гетероатомом. «(5-членный)гетероциклоалкил» означает, что гетероциклоалкильный заместитель содержит всего 5 кольцевых атомов, по меньшей мере, один из которых является гетероатомом. Гетероциклоалкильный заместитель может быть присоединен через атом азота, имеющий соответствующую валентность, или через любой атом углерода в кольце. Гетероциклоалкильная группа может быть необязательно замещена одним или несколькими заместителями, такими как (С1-С6)алкил, на атоме азота, имеющего соответствующую валентность, или на любо доступном атоме углерода.
Примеры гетероциклоалкильных колец включают, но не ограничены ими, азетидинил, тетрагидрофуранил, тетрагидропиразолил, тетрагидрооксазинил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тетрагидропиранил, тетрагидрооксазолил, морфолинил и оксетанил.
«Гало» или «галоген», как используется в настоящем документе, относится к атому хлора, фтора, брома или йода.
«Гидрокси» или «гидроксил», как используется в настоящем документе, означает группу-ОН.
«Циано», как используется в настоящем документе, означает группу-CN, которая также может быть изображена: 
«Пациент» или «субъект» относится к теплокровным животным, таким как, например, свиньи, коровы, куры, лошади, морские свинки, мыши, крысы, песчанки, кошки, кролики, собаки, обезьяны, шимпанзе и люди.
«Фармацевтически приемлемый» означает, что вещество или композиция должны быть химически и/или токсикологически совместимы с другими ингредиентами, входящими в состав, и/или млекопитающим, подвергаемым лечению.
Термин «терапевтически эффективное количество», используемый в настоящем документе, относится к такому количеству вводимого соединения, которое в некоторой степени облегчит один или несколько симптомов нарушения, подвергаемого лечению. Что касается лечения опосредованного HPK1 киназой нарушения (например, рака), терапевтически эффективное количество относится к такому количеству, которое в некоторой степени облегчает (или, например, устраняет) один или несколько симптомов, связанных с нарушением, опосредованным HPK1 киназой. Например, терапевтически эффективное количество относится к такому количеству, которое имеет эффект (1) уменьшения размера опухоли, (2) ингибирования (то есть, до некоторой степени замедления, предпочтительно остановки) метастазирования опухоли, (3) ингибирования в некоторой степени (то есть, замедление, в некоторой степени, предпочтительно, прекращение) роста опухоли или инвазивности опухоли и/или (4) облегчение, в некоторой степени (или, предпочтительно, устранение) одного или нескольких признаков или симптомов, связанных с раком.
Термин «лечение», используемый в настоящем документе, если не указано иное, означает обращение вспять, облегчение, ингибирование развития или предотвращение нарушения или состояния, к которому применяется такой термин, или одного или нескольких симптомов такого нарушения или состояния. Термин «лечение», используемый в настоящем документе, если не указано иное, относится к акту лечения, поскольку «лечение» определено в настоящем документе. Термин «лечение» также включает адъювантное и неоадъювантное лечение субъекта.
«Изомер» означает «стереоизомер» и «геометрический изомер», как определено ниже.
«Стереоизомер» относится к соединениям, которые обладают одним или несколькими хиральными центрами, каждый из которых может существовать в R-или S-конфигурации. Стереоизомеры включают все диастереомерные, энантиомерные и эпимерные формы, а также рацематы и их смеси.
«Геометрический изомер» относится к соединениям, которые могут существовать в цис-, транс-, анти-, entgegen (Е) и zusammen (Z) формах, а также к их смесям.
В настоящем описании, термины «заместитель», «радикал» и «группа» используются взаимозаменяемо.
Если заместители описываются как «независимо выбранные» из группы, каждый пример заместителя выбирают независимо от любого другого. Таким образом, каждый заместитель может быть идентичен или отличаться от другого(их) заместителя(ей).
Соединения
Соединения формулы I, как описано в настоящем документе, содержат ядро азалактама (2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-она), в котором пирроло кольцо присоединено через его атом азота к пиридину, который замещен R4.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R4 представляет собой (R4-i);
R4N представляет собой (С1-С6)алкил или (С3-С6)циклоалкил; и
R4C представляет собой водород или (С1-С3)алкил; где R1, R2, R3a и R3b имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R4 представляет собой (R4-i), где R1 представляет собой N(R5)(R6) или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил представляет собой циклопропил и замещен 0 или 1 заместителем, который представляет собой (C1-C6)алкил, где указанный (С1-С6)алкил представляет собой метил;
R5 и R6 каждый независимо представляет собой водород или (С1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой (C1-C6)алкил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или (C1-C6)алкил, который представляет собой метил;
R3a представляет собой (C1-C3)алкил, замещенный 0 или 1 заместителем, который представляет собой (C1-C3)алкокси; и
R3b представляет собой водород или (C1-C3)алкил.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R4 представляет собой (R4-ii);
R4D представляет собой водород, (C1-C6)алкил или галоген(C1-С6)алкил, где указанный (C1-C6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси;
R4E представляет собой водород; и
R4F представляет собой водород, (C1-C6)алкил или галоген(C1-С6)алкил, где указанный (C1-C6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси; и где R1, R2, R3a и R3b имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R4 представляет собой (R4-ii), и где R1 представляет собой -N(R5)(R6) или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил представляет собой циклопропил и замещен 0 или 1 заместителем, который представляет собой (C1-C6)алкил, где указанный (С1-С6)алкил представляет собой метил;
R5 и R6 каждый независимо представляет собой водород или (С1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С6)алкил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или (C1-C6)алкил, который представляет собой метил;
R3a представляет собой водород или (С1-С3)алкил, замещенный О или 1 заместителем, который представляет собой (С1-С3)алкокси; и
R3b представляет собой водород или (С1-С3)алкил.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R1 представляет собой 2-метилпирролидин-1-ил или 2(R)-метилпирролидин-1-ил; и где R2, R3a, R3b и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R2 представляет собой N(R7)(R8), где каждый из R7 и R8 представляет собой водород, и где R1, R3a, R3b и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R3a представляет собой (C1-C3)алкил, замещенный 0 или 1 заместителем, который представляет собой (C1-C3)алкокси.; и где R1, R2, R3b и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R3b представляет собой (C1-C3)алкил; и R1, R2, R3a и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R2 представляет собой N(R7)(R8), где каждый из R7 и R8 представляет собой водород; и где R1, R3a, R3b и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R2 представляет собой N(R7)(R8), где R7 представляет собой водород; и R8 представляет собой (С1-С3)алкил; и где R1, R3a, R3b и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение предлагает соединение формулы IA (соединение формулы I, где R4 представляет собой (R4-i):
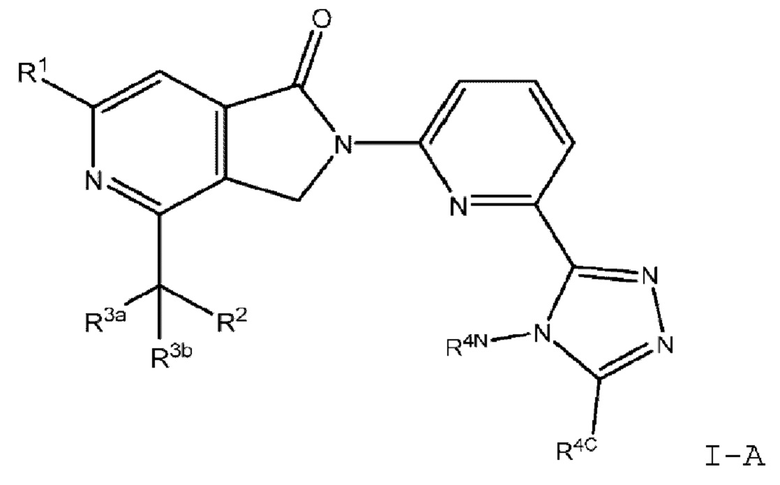
или его фармацевтически приемлемую соль, где:
R1 представляет собой -N(R5)(R6) или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил представляет собой циклопропил и замещен 0 или 1 заместителем, то есть (C1-C6)алкил, где указанный (С1-С6)алкил представляет собой метил;
R5 и R6 каждый независимо представляет собой водород или (С1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С6)алкил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или (С1-С6)алкил, который представляет собой метил;
R3a представляет собой (С1-С3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси;
R3b представляет собой водород или (С1-С3)алкил;
R4N представляет собой (C1-C6)алкил или (С3-С6)циклоалкил; и
R4C представляет собой водород или (С1-С3)алкил.
В другом варианте осуществления, изобретение относится к соединению формулы I-A или его фармацевтически приемлемой соли, где R3a представляет собой (С1-С3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси; и
R3b представляет собой (С1-С3)алкил.
В некоторых вариантах осуществления, когда R4 представляет собой (R4-i), соединение Формулы IA имеет абсолютную стереохимию, как показано в Формуле I-A-i или I-A-ii:
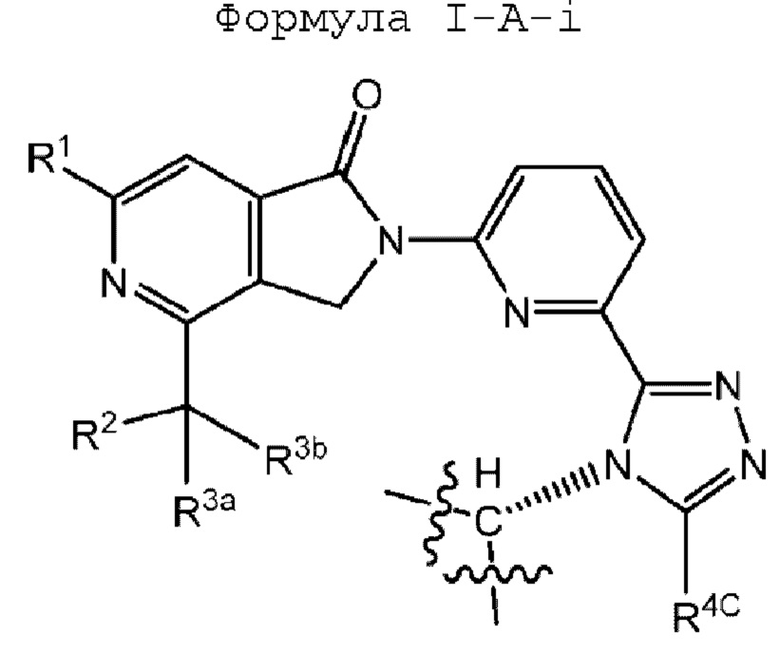

или его фармацевтически приемлемую соль, где R1, R2, R3a, R3b и R4C определены в любом варианте осуществления формулы I-A.
В другом варианте осуществления, изобретение относится к соединению формулы I-B (соединение формулы I, где R4 представляет собой (R4-ii):

или его фармацевтически приемлемая соль, где:
R1 представляет собой (C1-C6)алкил, галоген (C1-C6)алкил, (C1-С6)алкокси, галоген (С1-С6)алкокси,-N(R5)(R6), или (С3-С6)циклоалкил, где указанные (С1-С6)алкил, галоген (C1-C6)алкил и (С3-С6)циклоалкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано, (C1-C6)алкил или (С1-С6)алкокси, где:
R5 и R6 каждый независимо представляет собой водород или (С1-С6)алкил, замещенный 0, 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C1-C6)алкокси, циано и гидрокси, или
R5 и R6 вместе с азотом, к которому они присоединены, образуют (4-8-членный) гетероциклоалкил, который замещен 0, 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C1-C6)алкила, галоген (C1-C6)алкила, (C1-C6)алкокси и галоген (C1-C6)алкокси;
R2 представляет собой N(R7)(R8), где:
каждый из R7 и R8 независимо представляет собой водород или (С1-С6)алкил, который замещен 0 или 1 заместителем, представляющим собой галоген, (С1-С6)алкокси, циано или гидрокси; или
R7 представляет собой водород или (С1-С6)алкил, замещенный О или 1 заместителем, представляющим собой галоген, (С1-С6)алкокси, циано или гидрокси; и R8 вместе с азотом, к которому он присоединен, и вместе с R3a и углеродом, к которому он присоединен, образуют (4-6-членный) гетероциклоалкил, который независимо замещен 0, 1 или 2 заместителями, выбранными из галогена, гидрокси, (С1-С3)алкила, галоген (С1-С3)алкила, (С1-С3)алкокси или галоген(C1-C6)алкокси; или
R7 и R8 вместе с азотом, к которому они присоединены, образуют (4-6-членный) гетероциклоалкил, который замещен 0, 1 или 2 заместителями, независимо выбранными из галогена, гидрокси, (C1-С6)алкила, галоген (C1-C6)алкила, (C1-C6)алкокси и галоген(C1-C6)алкокси;
R3a представляет собой водород или (C1-C3)алкил, замещенный О или 1 заместителем, представляющим собой гидрокси или (C1-С3)алкокси;
R3b представляет собой водород или (C1-C3)алкил;
R4D представляет собой водород, (C1-C6)алкил, галоген(C1-С6)алкил, (C1-С6)алкокси, галоген(C1-C6)алкокси или (С3-С6)циклоалкил, где указанные (C1-C6)алкил и галоген (C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (C1-C6)алкокси;
R4E представляет собой водород, галоген, циано, гидрокси или (С1-С6)алкил; и
R4F представляет собой водород, галоген, циано, гидрокси, (С1-С6)алкил, галоген (C1-C6)алкил, (C1-C6)алкокси, галоген (C1-С6)алкокси или (С3-С6)циклоалкил, где указанные (C1-C6)алкил и галоген(С1-С6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (С1-С6)алкокси; и где R2 имеет значение, определенное в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где:
R1 представляет собой -N(R5)(R6) или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил замещен 0 или 1 заместителем, который представляет собой (С1-С3)алкил, где:
R5 и R6 каждый независимо представляет собой водород или (С1-С6)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (4-8-членный) гетероциклоалкил, замещенный 0, 1 или 2 заместителями, который представляет собой (C1-C6)алкил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или (C1-C6)алкил;
R3a представляет собой водород или (С1-С3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси;
R3b представляет собой водород или (С1-С3)алкил;
R4D представляет собой водород, (С1-С3)алкил или галоген (C1-С3)алкил, где указанный (С1-С3)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси, циано или (C1-С3)алкокси;
R4E представляет собой водород; и
R4F представляет собой водород, (С1-С3)алкил или галоген(C1-С3)алкил, где указанный (С1-С3)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где R3b представляет собой водород или (С1-С3)алкил.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где R4D представляет собой водород, (С1-С3)алкил или галоген(C1-С3)алкил, где указанный (С1-С3)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси; и где R1, R3a, R3b, R4E и R4F имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где R3a представляет собой (С1-С3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси; и
R3b представляет собой (С1-С3)алкил; и где R1, R2, R4D, R4E и R4F имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение предлагает соединение формулы I-B или его фармацевтически приемлемой соли, где:
R1 представляет собой -N(R5)(R6) или (C3-C6)циклоалкил, где (С3-С6)циклоалкил замещен 0 или 1 (С1-С3)алкилом;
R5 и R6 каждый независимо выбран из группы, состоящей из (С1-С6)алкила, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, который замещен с 0 или 1 (C1-C6)алкилом;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 представляет собой водород;
R3a представляет собой водород или (С1-С3)алкил, где указанный (C1-С3)алкил замещен 0 или 1 (С1-С3)алкоксигруппой;
R3b представляет собой водород или (С1-С3)алкил;
R4D представляет собой водород или (С1-С3)алкил, где (С1-С3)алкил замещен от 0 до 1 заместителя, выбранного из ОН и F;
R4E представляет собой водород; и
R4F представляет собой водород или (C1-C3)алкил.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где:
R1 представляет собой -N(R5)(R6) или циклопропил, замещенный 0 или 1 заместителем, представляющим собой метил;
R5 и R6 каждый независимо представляет собой водород или (C1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой метил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или метил;
R3a представляет собой (C1-C3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси; и
R3b представляет собой водород или (С1-С3)алкил; и где R4D, R4E и R4F имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R4 представляет собой (R4-ii);
R4D представляет собой водород, (C1-C6)алкил или галоген (С1-С6)алкил, где указанный (С1-С6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси;
R4E представляет собой водород; и
R4F представляет собой водород, (C1-C6)алкил или галоген(С1-С6)алкил, где указанный (C1-C6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси; и где R1, R2, R3a и R3b имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где R4 представляет собой (R4-ii);
R4D представляет собой водород, метил, фторметил, гидроксиметил или этил;
R4E представляет собой водород; и
R4F представляет собой водород; и где R1, R2, R3a и R3b имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, имеющим специфическую стереохимию в отношении ориентации R2, R3a и R3b, когда R3a отличается от R3b. Например, когда R3b представляет собой водород (R3b отсутствует), соединения Формулы I будут выглядеть следующим образом:


где R1, R3a и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I(R) или формулы I(S) или его фармацевтически приемлемой соли, где:
R1 представляет собой -N(R5)(R6) или циклопропил, замещенный одним заместителем, который представляет собой метил;
R5 и R6 каждый независимо представляет собой водород или (С1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой метил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или метил;
R3a представляет собой (C1-C3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси; R3b представляет собой водород; R4 представляет собой (R4-ii);
R4D представляет собой водород, метил, фторметил, гидроксиметил или этил;
R4E представляет собой водород; и
R4F представляет собой водород или метил.
В еще одном варианте осуществления, изобретение относится к соединениям формулы I (R) или их фармацевтически приемлемым солям, где R3b представляет собой водород (R3b отсутствует):

и где R1, R3a и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В еще одном варианте осуществления, изобретение относится к соединениям формулы I (S) или их фармацевтически приемлемым солям, где R3b представляет собой водород (R3b отсутствует):

где R1, R3a и R4 имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где R4 представляет собой 5-метил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил, (5R)-5-метил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил, (5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил, 5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил, (5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил или (5R)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил, где указанный метил замещен 0 или 1 заместитель, который представляет собой F или ОН, и где R1, R2, R3a и R3b имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
В другом варианте осуществления, изобретение относится к соединению формулы I-B или его фармацевтически приемлемой соли, где R4 представляет собой (5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил или (5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил, где указанный метил замещен 0 или 1 заместителем, который представляет собой F или ОН, и где R1, R2, R3a и R3b имеют значения, определенные в любом из описанных в настоящем документе вариантов осуществления.
Каждый из вариантов осуществления, описанных в настоящем документе в отношении формулы I, также применим к соединениям формул I-A-i, I-A-ii, I-B-i и I-B-ii.
В другом варианте осуществления, изобретение предлагает соединение, где соединение представляет собой: 4-[1-аминопропил]-2-{6-[5-метил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он;
4-[1-аминоэтил]-2-{6-5-этил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он;
4-[1-аминоэтил]-2-{6-[5-этил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он;
4-[1-аминоэтил]-2-{6-[5-этил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он;
4-[1-аминопропил]-2-{3-[5-метил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он; или
4-[1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он; или его фармацевтически приемлемую соль.
Например, соединение по изобретению представляет собой соединение, которое представляет собой 4-[(1R)-1-аминопропил]-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[{1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пиррол о[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[{2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[{1S)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[{2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1R)-1-аминопропил]-2-{3-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[{1R)-1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он; или
4-[{1S)-1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он; или его фармацевтически приемлемую соль.
В другом варианте осуществления, изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, где соединения представляют собой соединения формулы 1(R) или соединения формулы I(S,), имеющие специфическую стереохимию в отношении ориентации R2, R3a и R3b, когда R3a отличается от R3b.
В другом варианте осуществления, изобретение относится к соединению, которое представляет собой 4-[(1R)-1-аминопропил]-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он; или его фармацевтически приемлемую соль.
В другом варианте осуществления, изобретение относится к соединению, которое представляет собой 4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он; или его фармацевтически приемлемую соль.
В другом варианте осуществления, изобретение относится к соединениям, которые представляют собой 4-[(1S)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он; или его фармацевтически приемлемую соль.
В другом аспекте изобретение предлагает соединение, выбранное из группы, состоящей из соединений, приведенных в настоящем документе в качестве примеров, или их фармацевтически приемлемой соли.
Соединения по изобретению являются селективными в отношении HPK1 киназы.
«Фармацевтическая композиция» относится к смеси одного или нескольких соединений, описанных в настоящем документе, или их фармацевтически приемлемой соли, сольвата, гидрата или пролекарства в качестве активного ингредиента и, по меньшей мере, одного фармацевтически приемлемого носителя или эксципиента. В некоторых вариантах осуществления,
фармацевтическая композиция содержит два или несколько фармацевтически приемлемых носителя и/или эксципиента.
В некоторых вариантах осуществления, фармацевтическая композиция дополнительно содержит, по меньшей мере, один дополнительный противораковый терапевтический агент или паллиативный агент. В некоторых таких вариантах осуществления, по меньшей мере, один дополнительный агент представляет собой противораковый терапевтический агент, как описано ниже. В некоторых таких вариантах осуществления, комбинация предлагает аддитивное, большее, чем аддитивное, или синергетическое противораковое действие.
В одном варианте осуществления, изобретение предлагает способ лечения аномального роста клеток у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
В другом варианте осуществления, изобретение предлагает способ лечения аномального роста клеток у нуждающегося в этом субъекта, включающий введение субъекту некоторого количества соединения по изобретению или его фармацевтически приемлемой соли в комбинации с количеством дополнительного терапевтического агента (например, противоракового терапевтического агента), где количество в совокупности является эффективным при лечении указанного аномального клеточного роста.
В частых вариантах осуществления способов, предложенных в настоящем документе, аномальный рост клеток представляет собой рак. Соединения по изобретению можно вводить в виде отдельных агентов или можно вводить в комбинации с другими противораковыми терапевтическими агентами, в частности, стандартом лечения, подходящими для конкретного вида рака.
В некоторых вариантах осуществления, предложенные способы приводят к одному или нескольким из следующих эффектов: (1) ингибирование пролиферации раковых клеток; (2) ингибирование инвазивности раковых клеток; (3) индуцирование апоптоза раковых клеток; (4) ингибирование метастазирования раковых клеток; (5) ингибирование ангиогенеза; (6) усиление ответов Т-клеток; (7) усиление ответов дендритных и В-клеток; (8) усиление противоопухолевой активности; (9) усиление вакцинотерапии; и (10) усиление опосредованного иммунной системой удаления патогенов, таких как вирусы, бактерии, черви.
В другом аспекте, изобретение предлагает способ лечения нарушения, опосредованного активностью HPK1 киназы, у субъекта, такого как некоторые виды рака, включающий введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, которое эффективно для лечения указанного нарушения.
Если не указано иное, все ссылки в настоящем документе на соединения по изобретению включают ссылки на их соли, сольваты, гидраты и комплексы, а также на их сольваты, гидраты и комплексы солей, включая их полиморфы, стереоизомеры и меченные изотопами варианты.
Соединения по изобретению могут существовать в форме фармацевтически приемлемых солей, таких как, например, кислотно-аддитивные соли и основно-аддитивные соли соединений формулы I, представленных в настоящем документе. Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства исходного соединения. Фраза «фармацевтически приемлемая(ые) соль(и)», используемая в настоящем документе, если не указано иное, включает соли кислотных или основных групп, которые могут присутствовать в соединениях формулы I, описанных в настоящем документе.
Например, соединения по изобретению, которые являются основными по своей природе, способны образовывать широкий спектр солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, на практике часто желательно сначала выделить соединение по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли, и затем просто превратить последнюю обратно в соединение в виде свободного основания путем обработки щелочным реагентом, и затем превратить последнее свободное основание в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений по настоящему изобретению могут быть получены обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. После выпаривания растворителя, получают желаемую твердую соль. Желаемую кислую соль также можно осадить из раствора свободного основания в органическом растворителе путем добавления к раствору подходящей минеральной или органической кислоты.
Кислоты, которые можно использовать для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений, представляют собой такие, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат [т.е. 1,1'-метилен-бис(2-гидрокси-3-нафтоат)].
Примеры солей включают, но не ограничены ими, ацетат, акрилат, бензолсульфонат, бензоат (например, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат и метоксибензоат), бикарбонат, бисульфат, бисульфит, битартрат, борат, бромид, бутин-1,4-диоат, эдетат кальция, камзилат, карбонат, хлорид, капроат, каприлат, клавуланат, цитрат, деканоат, дигидрохлорид, дигидрофосфат, эдетат, эдисилат, эстолат, эзилат, этилсукцинат, формиат, фумарат, глюкептат, глюконат, глутамат, гликолят, гликоллиларсанилат, гептаноат, гексин-1,6-диоат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, γ-гидроксибутират, йодид, изобутират, изотионат, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, мезилат, метафосфат, метансульфонат, метилсульфат, моногидрофосфат, мукат, напсилат, нафталин-1-сульфонат, нафталин-2-сульфонат, нитрат, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фенилацетаты, фенилбутират, фенилпропионат, фталат, фосфат/дифосфат, полигалактуронат, пропансульфонат, пропионат, пропиолат, пирофосфат, пиросульфат, салицилат, стеарат, субацетат, суберат, сукцинат, сульфат, сульфонат, сульфит, таннат, тартрат, теоклат, тозилат, триэтиодод и валерат.
Иллюстративные примеры подходящих солей включают органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиак, первичные, вторичные и третичные амины и циклические амины, такие как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Соединения по изобретению, которые включают основную группу, такую как аминогруппа, могут образовывать фармацевтически приемлемые соли с различными аминокислотами в дополнение к кислотам, упомянутым выше.
Те соединения по изобретению, которые являются кислыми по своей природе, способны образовывать основные соли с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных или щелочноземельных металлов и, в частности, соли натрия и калия. Все эти соли получают обычными методами. Химические основания, которые используются в качестве реагентов для получения фармацевтически приемлемых солей оснований по настоящему изобретению, представляют собой основания, которые образуют нетоксичные основные соли с кислыми соединениями в настоящем документе. Эти соли могут быть получены любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла или подобными. Эти соли также могут быть получены обработкой соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и последующим выпариванием полученного раствора досуха, предпочтительно, при пониженном давлении. Альтернативно, их также можно приготовить путем смешивания низших алканольных растворов кислых соединений и желаемого алкоксида щелочного металла вместе с последующим выпариванием полученного раствора досуха тем же способом, что и ранее. В любом случае, предпочтительно использовать стехиометрические количества реагентов для обеспечения полноты реакции и максимального выхода желаемого конечного продукта.
Химические основания, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых основных солей соединений по изобретению, которые являются кислыми по своей природе, представляют собой те, которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают, но не ограничены ими, аддитивные соли, полученные из таких фармакологически приемлемых катионов, таких как катионы щелочных металлов (например, калия и натрия) и катионов щелочноземельных металлов (например, кальция и магния), аммония или водорастворимого амина, такие как N-метилглюкамин (меглюмин), низший алканоламмоний и другие основные соли фармацевтически приемлемых органических аминов.
Могут также образовываться полусоли кислот и оснований, например, темисульфатные и гемикальциевые соли.
Обзор подходящих солей см. в Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002). Способы получения фармацевтически приемлемых солей соединений по изобретению известны специалистам в данной области техники.
Соли по настоящему изобретению могут быть получены способами, известными специалистам в данной области техники. Фармацевтически приемлемая соль соединений по изобретению может быть легко получена путем смешивания вместе растворов соединения и желаемой кислоты или основания, в зависимости от ситуации. Соль может осаждаться из раствора и собираться фильтрованием, или может быть выделена выпариванием растворителя. Степень ионизации в соли может варьироваться от полностью ионизированной до почти не ионизированной.
Специалистам в данной области техники должно быть понятно, что соединения по изобретению в форме свободного основания, обладающие основной функциональностью, могут быть превращены в кислотно-аддитивные соли путем обработки стехиометрическим избытком соответствующей кислоты. Кислотно-аддитивные соли соединений по изобретению могут быть повторно превращены в соответствующее свободное основание обработкой стехиометрическим избытком подходящего основания, такого как карбонат калия или гидроксид натрия, обычно в присутствии водного растворителя и при температуре между примерно 0°С и 100°С. Форма свободного основания может быть выделена обычными способами, такими как экстракция органическим растворителем. Кроме того, кислотно-аддитивные соли соединений по настоящему изобретению могут быть заменены за счет использования преимущества различной растворимости солей, летучести или кислотности кислот, или путем обработки ионообменной смолой с соответствующим содержанием. Например, на обмен может влиять реакция соли соединений по изобретению с небольшим стехиометрическим избытком кислоты с более низким рК, чем кислотный компонент исходной соли. Это превращение обычно проводят при температуре примерно от 0°С до точки кипения растворителя, используемого в качестве среды для процедуры. Подобные обмены возможны с основно-аддитивными солями, как правило, через посредство формы свободного основания.
Соединения по изобретению могут существовать как в не сольватированной, так и в сольватированной формах. Когда растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, не зависящую от влажности. Однако, когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях, не стехиометрия будет нормой. Термин «сольват» используется в настоящем документе для описания молекулярного комплекса, содержащего соединение по изобретению и одну или несколько молекул фармацевтически приемлемого растворителя, например, этанола. Термин «гидрат» используется, когда растворителем является вода. Фармацевтически приемлемые сольваты по изобретению включают гидраты и сольваты, в которых растворитель кристаллизации может быть изотопно замещен, например D2O, d6-ацетон, d6-ДМСО.
Также в объем изобретения включены такие комплексы, как клатраты, комплексы включения лекарственное средство-хозяин, в которых, в отличие от вышеупомянутых сольватов, лекарственное средство и хозяин присутствуют в стехиометрических или не стехиометрических количествах. Также включены комплексы лекарственного средства, содержащие два или несколько органических и/или неорганических компонентов, которые могут находиться в стехиометрических или не стехиометрических количествах. Образовавшиеся комплексы могут быть ионизированными, частично ионизированными или не
ионизированными. Обзор таких комплексов см. в,7 Pharm Sci, 64 (8), 1269-1288, Haleblian (August 1975), описание которого полностью включено в настоящий документ посредством ссылки.
Изобретение также относится к пролекарствам соединений формулы I, представленных в настоящем документе. Таким образом, некоторые производные соединений по изобретению, которые могут иметь небольшую или не иметь фармакологическую активность, при введении пациенту, могут быть превращены в соединения по изобретению, например, путем гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в Pro-Drugs as Novel Delivery Systems, Vol.14, ACS Symposium Series (T Higuchi and W Stella) и «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (ed. E В Roche, American Pharmaceutical Association), описание которых полностью включено в настоящее описание посредством ссылки.
Пролекарства в соответствии с изобретением могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединениях по изобретению, некоторыми группами, известными специалистам в данной области техники как «про-группы», как описано, например, в "Design of Prodrugs" by H Bundgaard (Elsevier, 1985), описание которой полностью включено в настоящее описание посредством ссылки.
Некоторые неограничивающие примеры пролекарств по изобретению включают:
(i) если соединение содержит функциональную группу карбоновой кислоты (-СООН), ее сложный эфир, например, замещение водорода на (С1-C6)алкил;
(ii) если соединение содержит спиртовую функциональную группу (-ОН), ее простой эфир, например, замещение водорода на (С1-С6)алканоилоксиметил или группу фосфатного эфира; и
(iii) если соединение содержит первичную или вторичную аминогруппу (-NH2 или-NHR, где R≠Н), его амид, например, замещение одного или обоих атомов водорода подходящей метаболически лабильной группой, такой как амид, карбамат, мочевина, фосфонат, сульфонат и т.д.
Дополнительные примеры замещающих групп в соответствии с приведенными выше примерами и примеры других типов пролекарств можно найти в вышеупомянутых ссылках.
Наконец, некоторые соединения по изобретению могут сами действовать как пролекарства других соединений по изобретению.
Также в объем изобретения включены метаболиты соединений формулы I, как описано в настоящем документе, т.е. соединения, образующиеся in vivo при введении лекарственного средства.
Представленные в настоящем документе соединения формулы I могут иметь асимметричные атомы углерода. Связи углерод-углерод соединений по изобретению могут быть изображены в настоящем документе сплошной линией ( ), сплошным клином (
), сплошным клином ( ) или пунктирным клином (
) или пунктирным клином ( ). Использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что включены все возможные стереоизомеры (например, определенные энантиомеры, рацемические смеси и т.д.) на этом атоме углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода означает, что предполагается включить только показанный стереоизомер. Возможно, что соединения по изобретению могут содержать более одного асимметрического атома углерода. В этих соединениях, использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что все возможные стереоизомеры должны быть включены, и стереоцентр присоединен. Например, если не указано иное, предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для изображения связей с одним или несколькими асимметричными атомами углерода в соединении по изобретению, и использование сплошного или пунктирного клина для изображения связей с другими асимметрическими атомами углерода в том же соединении означает, что присутствует смесь диастереомеров.
). Использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что включены все возможные стереоизомеры (например, определенные энантиомеры, рацемические смеси и т.д.) на этом атоме углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода означает, что предполагается включить только показанный стереоизомер. Возможно, что соединения по изобретению могут содержать более одного асимметрического атома углерода. В этих соединениях, использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что все возможные стереоизомеры должны быть включены, и стереоцентр присоединен. Например, если не указано иное, предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для изображения связей с одним или несколькими асимметричными атомами углерода в соединении по изобретению, и использование сплошного или пунктирного клина для изображения связей с другими асимметрическими атомами углерода в том же соединении означает, что присутствует смесь диастереомеров.
Соединения по изобретению, которые имеют хиральные центры, могут существовать в виде стереоизомеров, таких как рацематы, энантиомеры или диастереомеры.
Стереоизомеры соединений Формулы I в настоящем документе могут включать цис-и транс-изомеры, оптические изомеры, такие как (R) и (S) энантиомеры, диастереомеры, геометрические изомеры, вращательные изомеры, атропоизомеры, конформационные изомеры и таутомеры соединений по изобретению, включая соединения, проявляющие более одного типа изомерии; и их смеси (такие как рацематы и диастереомерные пары).
Также включены кислотно-аддитивные соли или основно-аддитивные соли, в которых противоион является оптически активным, например, d-лактат или 1-лизин, или рацемическим, например, d-тартрат или d-аргинин.
При кристаллизации любого рацемата, возможны кристаллы двух различных типов. Первый тип представляет собой упомянутое выше рацемическое соединение (истинный рацемат), в котором образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, где две формы кристаллов образуются в эквимолярных количествах, каждая из которых содержит один энантиомер.
Соединения по изобретению могут проявлять феномен таутомерии и структурной изомерии. Например, соединения могут существовать в нескольких таутомерных формах, включая енольную и иминовую форму, а также кето- и енаминовую форму, и геометрические изомеры и их смеси. Все такие таутомерные формы включены в объем соединений по изобретению. Таутомеры существуют в виде смесей таутомерного набора в растворе. В твердой форме, обычно преобладает один таутомер. Хотя может быть описан один таутомер, настоящее изобретение включает все таутомеры представленных соединений формулы I.
Кроме того, некоторые из соединений по изобретению могут образовывать атропоизомеры (например, замещенные биарилы). Атропоизомеры представляют собой конформационные стереоизомеры, которые возникают, когда вращение вокруг простой связи в молекуле предотвращается или значительно замедляется в результате пространственных взаимодействий с другими частями молекулы, и заместители на обоих концах одинарной связи несимметричны. Взаимное превращение атропоизомеров происходит достаточно медленно, чтобы их можно было разделить и выделить в заданных условиях. Энергетический барьер для термической рацемизации может определяться пространственными затруднениями для свободного вращения одной или нескольких связей, образующих хиральную ось.
Обычные методы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ВЭЖХ) или сверхкритической жидкостной хроматографии (СЖХ).
Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например, спиртом, или, в случае, когда соединение содержит кислую или основную группу, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученную диастереомерную смесь можно разделить хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера превратить в соответствующий(ие) чистый(ые) энантиомер(ы) способами, хорошо известными специалисту в данной области техники.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно, от 2 до 20% и от 0 до 5% алкиламина, обычно 0,1% диэтиламина. Концентрация элюата дает обогащенную смесь.
Стереоизомерные конгломераты могут быть разделены обычными способами, известными специалистам в данной области техники; см., например, «Stereochemistry of Organic Compounds» E L Eliel (Wiley, New York, 1994), описание которой полностью включено в настоящее описание посредством ссылки.
Энантиомерная чистота соединений, описанных в настоящем документе, может быть описана с точки зрения энантиомерного избытка (эи), который указывает на степень, в которой образец содержит один энантиомер в большем количестве, чем другой. Рацемическая смесь имеет эи 0%, в то время как отдельный полностью чистый энантиомер имеет эи 100%. Точно так же, диастереомерная чистота может быть описана в терминах диастереомерного избытка (ди).
Настоящее изобретение также включает соединения, меченные изотопами, которые идентичны соединениям, указанным в одной из представленных Формул I, но с тем фактом, что один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречающихся в природе.
Меченые изотопами соединения по изобретению, как правило, могут быть получены обычными методами, известными специалистам в данной области техники, или способами, аналогичными описанным в настоящем документе, с использованием соответствующего меченного изотопами реагента вместо не меченого реагента, используемого в других случаях.
Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как, но не ограничиваясь ими, 2Н, 3Н, 13С, 14С, 15N, 18O, 17O, 31Р, 32Р, 35S, 18F и 36Cl. Некоторые меченые изотопами соединения по изобретению, например те, в которые включены радиоактивные изотопы, такие как 3Н и 14С, можно использовать в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития, т.е. 3Н, и углерода-14, т.е. 14С, особенно предпочтительны из-за простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличение периода полужизни in vivo или снижение требований к дозировке, и, следовательно, в некоторых обстоятельствах может быть предпочтительным. Меченые изотопами соединения по настоящему изобретению, как правило, могут быть получены путем проведения процедур, описанных на схемах и/или в приведенных ниже примерах и получениях, путем замены реагента, не меченого изотопом, реагентом, меченым изотопом.
Соединения по изобретению, предназначенные для фармацевтического применения, можно вводить в виде кристаллических или аморфных продуктов или их смесей. Их можно получить, например, в виде твердых слоев, порошков или пленок такими способами, как осаждение, кристаллизация, сушка вымораживанием, сушка распылением или сушка выпариванием. Для этой цели можно использовать микроволновую или радиочастотную сушку. Соединения по изобретению могут существовать в континууме твердых состояний в диапазоне от полностью аморфного до полностью кристаллического. Термин «аморфный» относится к состоянию, в котором в материале отсутствует дальний порядок на молекулярном уровне и, в зависимости от температуры, он может проявлять физические свойства твердого тела или жидкости. Обычно такие материалы не дают отчетливых рентгенограмм и, хотя и проявляют свойства твердого тела, более формально описываются как жидкости. При нагревании происходит переход от твердых к жидким свойствам, который характеризуется изменением состояния, как правило, второго порядка («стеклование»). Термин «кристаллический» относится к твердой фазе, в которой материал имеет правильную упорядоченную внутреннюю структуру на молекулярном уровне и дает отчетливую рентгенограмму с определенными пиками. Такие материалы при достаточном нагревании также будут проявлять свойства жидкости, но переход от твердого состояния к жидкому характеризуется фазовым переходом, обычно первого порядка («температура плавления»).
Соединения формулы I могут также существовать в мезоморфном состоянии (мезофаза или жидкие кристаллы) при воздействии на них подходящих условий. Мезоморфное состояние является промежуточным между истинным кристаллическим состоянием и истинным жидким состоянием (расплавом или раствором). Мезоморфизм, возникающий в результате изменения температуры, описывается как «термотропный», и тот, который возникает в результате добавления второго компонента, такого как вода или другой растворитель, как «лиотропный». Соединения, способные образовывать лиотропные мезофазы, описываются как «амфифильные» и состоят из молекул, обладающих ионной (такой как -COO-Na+,-СОО-K+ или -SO3-Na+) или не ионной (например,-N-N+(CH3)3) полярной головной группой. Для получения дополнительной информации см. Crystals and the Polarizing Microscope by N. H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970).
Соединения формулы I могут демонстрировать полиморфизм и/или один или несколько видов изомерии (например, оптическую, геометрическую или таутомерную изомерию). Соединения формулы I также могут быть помечены изотопами. Такая вариация подразумевается для соединений формулы I, определенных по их структурным особенностям и, следовательно, входящих в объем изобретения.
Терапевтические способы и использование
Изобретение дополнительно относится к терапевтическим способам и применениям, включающим введение соединений по изобретению, или их фармацевтически приемлемым солям, отдельно или в комбинации с другими терапевтическими агентами или паллиативными агентами.
В одном варианте осуществления, изобретение предлагает способ лечения аномального роста клеток у субъекта, включающий введение субъекту терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли. В частых вариантах осуществления, аномальный рост клеток представляет собой рак.
В другом варианте осуществления, изобретение предлагает способ лечения рака у субъекта, включающий введение субъекту некоторого количества соединения по изобретению или его фармацевтически приемлемой соли в комбинации с некоторым количеством дополнительного противоракового терапевтического агента, где эти количества вместе эффективны при лечении указанного рака.
Соединения по изобретению включают соединения формулы I, как описано в настоящем документе, или их фармацевтически приемлемые соли.
В еще одном варианте осуществления, изобретение относится к способу ингибирования пролиферации раковых клеток у субъекта, включающему введение субъекту эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
В другом варианте осуществления, изобретение относится к способу ингибирования инвазивности раковых клеток у субъекта, включающему введение субъекту эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
В другом варианте осуществления, изобретение относится к способу, вызывающему гибель раковых клеток у субъекта, включающему введение субъекту эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
В еще одном варианте осуществления, изобретение предлагает способ усиления вакцинотерапии у млекопитающего, включающему введение млекопитающему терапевтически эффективного количества вакцины и дополнительно включающему введение млекопитающему терапевтически эффективного количества соединения по любому из пунктов 1-16, или его фармацевтически приемлемой соли.
В еще одном варианте осуществления, изобретение предлагает способ улучшения способности иммунной системы уничтожать вирусную инфекцию, бактериальную инфекцию или патоген (включая паразитических червей) у субъекта, включающий введение субъекту эффективного количества соединения по изобретению или его фармацевтически приемлемой соли. Изобретение предлагает способ усиления удаления патогенов, опосредованного иммунной системой, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пунктов 1-16 или его фармацевтически приемлемой соли. Способ включает введение соединения по изобретению в виде монотерапии или в комбинации с другими агентами для лечения инфекции или патогена.
Описанные в настоящем документе соединения находят применение для ингибирования активности HPK1 киназы. HPK1, также называемая митоген-активируемой киназой киназы киназы протеинкиназы 1 или MAP4K1, является членом подсемейства киназ зародышевого центра Ste20-родственных серии/тренонинкиназ. HPK1 киназа функционирует как МАР4К путем фосфорилирования и активации белков MAP3K, включая MEKK1, MLK3 и TAK1, что приводит к активации MAPK,7nk.
Полинуклеотиды и полипептиды HPK1 известны в данной области техники (Ни et al. (1996) Genes Dev. 10: 2251-2264, которая полностью включена в настоящий документ посредством ссылки). Полипептиды HPK1 содержат множество консервативных структурных мотивов. Полипептиды HPK1 содержат амино-концевой домен Ste20-подобной киназы, который охватывает аминокислотные остатки 17-293, который включает АТФ-связывающий сайт из аминокислотных остатков 23-46. За киназным доменом следуют четыре богатых пролином (PR) мотива, которые служат сайтами связывания для SH3-содержащих белков, таких как CrkL, Grb2, HIP-55, Gads, Nek и Crk. Четыре мотива PR охватывают аминокислотные остатки 308-407, 394-402, 432-443 и 468-477, соответственно. HPK1 становится фосфорилированным и активированным в ответ на стимуляцию TCR или BCR. TCR-и BCR-индуцированное фосфорилирование тирозина в положении 381, расположенном между PR1 и PR2, опосредует связывание с SLP-76 в Т-клетках или BLNK в В-клетках через домен SLP-76 или BLNK SH2, и требуется для активации киназы. Домен гомологии цитрона, обнаруженный на С-конце HPK1, приблизительно охватывающий остатки 495-800, может действовать как регуляторный домен и может участвовать в макромолекулярных взаимодействиях.
Описанные в настоящем документе соединения связываются непосредственно с HPK1 и ингибируют его киназную активность. В некоторых вариантах осуществления описанные в настоящем документе соединения уменьшают, ингибируют или иным образом уменьшают HPK1-опосредованное фосфорилирование SLP76 и/или Gads. Описанные в настоящем документе соединения могут быть или не быть специфическим ингибитором HPK1. Специфический ингибитор HPK1 снижает биологическую активность HPK1 на величину, которая статистически больше, чем ингибирующее действие ингибитора на любой другой белок (например, другие серии/треонинкиназы). В некоторых вариантах осуществления, описанные в настоящем документе соединения специфически ингибируют серин/треонинкиназную активность HPK1.
Описанные в настоящем документе соединения могут быть использованы в способе ингибирования HPK1. Такие способы включают контакт HPK1 с эффективным количеством соединения, описанного в настоящем документе. Термин «контакт» означает приведение соединения в достаточно близкое взаимодействие с выделенным ферментом HPK1 или клеткой, экспрессирующей HPK1 (например, Т-клеткой, В-клеткой, дендритной клеткой), так что соединение способно связываться с HPK1 и ингибировать его активность. Соединение может контактировать с HPK1 in vitro или in vivo путем введения соединения субъекту.
Любой способ, известный в данной области техники для измерения киназной активности HPK1, может быть использован для определения того, была ли ингибирована HPK1, включая анализ киназы in vitro, иммуноблоты с антителами, специфичными к фосфорилированным мишеням HPK1, таким как SLP76 и Gads, или измерение последующего биологического эффекта активности HPK1 киназы, такого как привлечение белков 14-3-3 к фосфорилированным SLP7 и Gads, высвобождение комплекса SLP76-Gads-14-3-3 из LAT-содержащих микрокластеров или активация Т-или В-клеток.
Описанные в настоящем документе соединения можно использовать для лечения HPK1l-зависимого нарушения. Используемый в настоящем документе термин «зависимое от HPK1 нарушение» представляет собой патологическое состояние, при котором активность HPK1 необходима для возникновения или поддержания патологического состояния.
Описанные в настоящем документе соединения также находят применение для усиления иммунного ответа у субъекта, нуждающегося в этом. Такие способы включают введение эффективного количества описанного в настоящее время соединения (т.е. любого из соединений формулы I или его фармацевтически приемлемой соли, пролекарства, метаболита или производного). Термин «усиление иммунного ответа» относится к улучшению любого иммуногенного ответа на антиген. Неограничивающие примеры улучшения иммуногенного ответа на антиген включают усиленное созревание или миграцию дендритных клеток, усиленную активацию Т-клеток (например, CD4 Т-клеток, CD8 Т-клеток), усиление Т-клеток (например, CD4 Т-клеток, CD8 Т-клеток). пролиферация Т-клеток, усиленная пролиферация В-клеток, повышенная выживаемость Т-клеток и/или В-клеток, улучшенная презентация антигена антигенпрезентирующими клетками (например, дендритными клетками), улучшенный клиренс антигена, увеличение продуцирования цитокинов Т-клетками (например, интерлейкина-2), повышенная устойчивость к иммунодепрессии, индуцированной простагландином Е2 или аденозином, и повышенная примирующая и/или цитолитическая активность CD8 Т-клеток. В некоторых вариантах осуществления, CD8 Т-клетки у субъекта обладают повышенной активностью примирования, активации, пролиферации и/или цитолитической активности по сравнению с предшествующим введением соединения формулы I или его фармацевтически приемлемой соли, пролекарства, метаболита или производного. В некоторых вариантах осуществления, примирование CD8 Т-клеток характеризуется повышенной экспрессией CD44 и/или повышенной цитолитической активностью в CD8 Т-клетках. В некоторых вариантах осуществления, активация CD8 Т-клеток характеризуется повышенной частотой IFNy+ CD8 Т-клеток. В некоторых вариантах осуществления, CD8 Т-клетка представляет собой антигенспецифическую Т-клетку.
В некоторых вариантах осуществления, антигенпрезентирующие клетки у субъекта имеют усиленное созревание и активацию по сравнению с тем, что было до введения соединения формулы I или его фармацевтически приемлемой соли, пролекарства, метаболита или производного. В некоторых вариантах осуществления, антигенпрезентирующие клетки представляют собой дендритные клетки. В некоторых вариантах осуществления, созревание антигенпрезентирующих клеток характеризуется повышенной частотой CD83+ дендритных клеток. В некоторых вариантах осуществления, активация антигенпрезентирующих клеток характеризуется повышенной экспрессией CD80 и CD86 на дендритных клетках.
Вовлечение TCR приводит к активации HPK1, который функционирует как отрицательный регулятор пути ответа АР-1, индуцированного TCR. Считается, что HPK1 отрицательно регулирует активацию Т-клеток, уменьшая постоянство сигнальных микрокластеров путем фосфорилирования SLP76 по Ser376 (Di Bartolo et al. (2007),7EM 204:681-691) и Gads на Thr254, что приводит к рекрутингу 14-3-3, которые связываются с фосфорилированными SLP76 и Gads, высвобождая комплекс SLP76-Gads-14-3-3 из LAT-содержащих микрокластеров, что приводит к дисфункции Т-клеток, включая анергию и истощение (Lasserre et al. (2011).), 7 Cell Biol 195(5): 839-853). Термин «дисфункция» в контексте иммунной дисфункции относится к состоянию сниженного иммунного ответа на антигенную стимуляцию. Этот термин включает общие элементы как истощения, так и/или анергии, при которых может происходить распознавание антигена, но последующий иммунный ответ неэффективен для контроля инфекции или роста опухоли.
Термин «дисфункциональный», используемый в настоящем документе, также включает рефракторность или невосприимчивость к распознаванию антигена, в частности, нарушенную способность транслировать распознавание антигена в последующие эффекторные функции Т-клеток, такие как пролиферация, продуцирование цитокинов (например, IL-2, гамма-IFN) и/или уничтожение клеток-мишеней.
Термин «анергия» относится к состоянию невосприимчивости к антигенной стимуляции, возникающему из-за неполных или недостаточных сигналов, доставляемых через Т-клеточный рецептор (например, увеличение внутриклеточного Са в отсутствие ras-активации). Анергия Т-клеток также может возникать при стимуляции антигеном в отсутствие костимуляции, в результате чего клетка становится невосприимчивой к последующей активации антигеном даже в контексте костимуляции. Состояние невосприимчивости часто может быть преодолено присутствием интерлейкина-2. Анергические Т-клетки не подвергаются клональной экспансии и/или не приобретают эффекторные функции.
Термин «истощение» относится к истощению Т-клеток как состоянию дисфункции Т-клеток, которое возникает из-за устойчивой передачи сигналов TCR, которая возникает во время многих хронических инфекций и рака. Оно отличается от анергии тем, что возникает не из-за неполной или недостаточной передачи сигналов, а из-за устойчивой передачи сигналов. Оно определяется плохой эффекторной функцией, устойчивой экспрессией ингибирующих рецепторов и состоянием транскрипции, отличным от состояния функциональных эффекторных клеток или Т-клеток памяти. Истощение препятствует оптимальному контролю инфекции и опухолей. Истощение может быть результатом как внешних отрицательных регуляторных путей (например, иммунорегуляторных цитокинов), так и внутренних отрицательных регуляторных (ко-стимулирующих) путей клетки (PD-1, В7-Н3, В7-Н4 и т.д.).
«Усиление функции Т-клеток» означает индукцию, вызывание или стимулирование Т-клетки, чтобы она имела устойчивую или усиленную биологическую функцию, или обновление или реактивацию истощенных или неактивных Т-клеток. Примеры усиления функции Т-клеток включают: повышенную секрецию цитокинов (например, гамма-интерферона, IL-2, IL-12 и TNF-альфа), повышенную пролиферацию, повышенную чувствительность к антигенам (например, клиренс вирусов, патогенов или опухолей) по сравнению с такими уровнями до вмешательства, и повышенное продуцирование эффекторных гранул CD8 Т-клетками, такими как гранзим В.
Соответственно, описанные в настоящем документе соединения формулы I или их фармацевтически приемлемые соли, пролекарства, метаболиты или производные применимы для лечения нарушений, связанных с дисфункцией Т-клеток. «Т-клеточное дисфункциональное нарушение» представляет собой нарушение или состояние Т-клеток, характеризующееся сниженной реакцией на антигенную стимуляцию. В конкретном варианте осуществления, Т-клеточное дисфункциональное нарушение представляет собой нарушение, которое специфически связано с повышенной киназной активностью HPK1. В другом варианте осуществления, дисфункциональное Т-клеточное нарушение представляет собой нарушение, при котором Т-клетки являются энергическими или имеют сниженную способность секретировать цитокины, пролиферировать или осуществлять цитолитическую активность. В конкретном аспекте, сниженная реакционная способность приводит к неэффективному контролю над патогеном или опухолью, экспрессирующей иммуноген. Примеры Т-клеточных дисфункциональных нарушений, характеризующихся дисфункцией Т-клеток, включают неразрешенную острую инфекцию, хроническую инфекцию и опухолевый иммунитет.
Таким образом, описанные в настоящем документе соединения можно использовать для лечения состояний, при которых желательна повышенная иммуногенность, например, для повышения иммуногенности опухоли для лечения рака. «Иммуногенность» относится к способности конкретного вещества вызывать иммунный ответ. Опухоли являются иммуногенными, и повышение иммуногенности опухоли способствует удалению опухолевых клеток за счет иммунного ответа.
«Опухолевой иммунитет» относится к процессу, при котором опухоли избегают иммунного распознавания и очистки. Таким образом, как терапевтическая концепция, опухолевый иммунитет «лечится», когда такое избегание ослабляется, и опухоли распознаются и атакуются иммунной системой. Примеры распознавания опухоли включают связывание с опухолью, уменьшение опухоли и удаление опухоли.
В одном аспекте, в настоящем документе предложен способ лечения рака у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли, пролекарства, метаболита или производного. В некоторых вариантах осуществления, у субъекта имеется меланома. Меланома может быть на ранней или поздней стадии. В некоторых вариантах осуществления, у субъекта имеется колоректальный рак. Колоректальный рак может быть на ранней или поздней стадии. В некоторых вариантах осуществления, у субъекта имеется немелкоклеточный рак легкого. Немелкоклеточный рак легкого может быть на ранней или поздней стадии. В некоторых вариантах осуществления, у субъекта имеется рак поджелудочной железы. Рак поджелудочной железы может быть на ранней или поздней стадии. В некоторых вариантах осуществления, у субъекта имеется гематологическое злокачественное новообразование.
Гематологическое злокачественное новообразование может быть на ранней или поздней стадии. В некоторых вариантах осуществления, у субъекта имеется рак яичников. Рак яичников может быть на ранней или поздней стадии. В некоторых вариантах осуществления, у субъекта имеется рак молочной железы. Рак молочной железы может быть на ранней стадии или на поздней стадии. В некоторых вариантах осуществления, у субъекта имеется почечно-клеточная карцинома. Почечно-клеточная карцинома может быть на ранней стадии или на поздней стадии. В некоторых вариантах осуществления, рак характеризуется повышенным уровнем Т-клеточной инфильтрации.
В некоторых вариантах осуществления, лечение приводит к устойчивому ответу у субъекта после прекращения лечения. «Устойчивый ответ» относится к устойчивому эффекту снижения роста опухоли после прекращения лечения. Например, размер опухоли может оставаться таким же или меньшим по сравнению с размером в начале фазы введения.
Описанные в настоящем документе способы лечения могут привести к частичному или полному ответу. Используемый в настоящем документе термин «полный ответ» или «CR» относится к исчезновению всех поражений-мишеней; «частичный ответ» или «PR» относится к, по меньшей мере, 30-процентному уменьшению суммы самых длинных диаметров (SLD) поражений-мишеней, принимая за основу исходную SLD; и «стабильное заболевание» или «SD» не относится ни к достаточному уменьшению поражений-мишеней, чтобы квалифицировать их как PR, ни к достаточному увеличению, чтобы квалифицировать как PD, принимая в качестве эталона наименьшую SLD с момента начала лечения. Используемый в настоящем документе термин «общая частота ответа» (ORR) относится к сумме частоты полного ответа (CR) и частоты частичного ответа (PR).
Описанные в настоящем документе способы лечения могут привести к увеличению выживаемости без прогрессирования и общей выживаемости субъекта, которому вводили антагонист HPK1. Используемый в настоящем документе термин «выживаемость без прогрессирования» (PFS) относится к продолжительности времени во время и после лечения, в течение которого заболевание, подвергаемое лечению (например, рак), не ухудшается. Выживаемость без прогрессирования может включать количество времени, в течение которого у пациентов наблюдался полный или частичный ответ, а также количество времени, в течение которого у пациентов наблюдалась стабилизация заболевания.
Используемый в настоящем документе термин «общая выживаемость» относится к доле субъектов в группе, которые, вероятно, будут живы после определенного периода времени.
В некоторых вариантах осуществления способов, представленных в настоящем документе, аномальный рост клеток представляет собой рак, где рак выбран из группы, состоящей из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легких (включая NSCLC, SCLC, плоскоклеточную карциному или аденокарциному), рак пищевода, рак головы и шеи, колоректальный рак, рак почки (включая RCC), рак печени (включая НСС), рак поджелудочной железы, рак желудка (например, желудочный) и рак щитовидной железы. В дополнительных вариантах осуществления способов, представленных в настоящем документе, рак выбран из группы, состоящей из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легких, рака пищевода, рака печени, рака поджелудочной железы и рака желудка.
В некоторых вариантах осуществления, рак выбран из группы, состоящей из рака молочной железы и рака яичников.
В некоторых вариантах осуществления, рак представляет собой рак яичников.
В других вариантах осуществления, рак представляет собой рак молочной железы, включая, например, ЕИ-положительный/НИ-положительный рак молочной железы, НЕРч2-отрицательный рак молочной железы; ER-положительный/HR-положительный рак молочной железы, НЕИ2-положительный рак молочной железы; трижды отрицательный рак молочной железы (TNBC); или воспалительный рак молочной железы. В некоторых вариантах осуществления, рак молочной железы представляет собой эндокринно-резистентный рак молочной железы, рак молочной железы, резистентный к трастузумабу, или рак молочной железы, демонстрирующий первичную или приобретенную резистентность к ингибированию CDK4/CDK6. В некоторых вариантах осуществления, рак молочной железы представляет собой распространенный или метастатический рак молочной железы.
В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии первой линии. В других вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии. В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии после лечения эндокринным терапевтическим агентом и/или ингибитором CDK4/CDK6. В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии после лечения эндокринным терапевтическим агентом. В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии после лечения ингибитором CDK4/CDK6. В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии после лечения одной или несколькими схемами химиотерапии, например, включающими таксаны или препараты платины. В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии после лечения агентами, таргетирующими HER2, например, трастузумабом.
Термины «аномальный рост клеток» и «гиперпролиферативное нарушение» используются в этой заявке взаимозаменяемо.
«Аномальный рост клеток», используемый в настоящем документе, если не указано иное, относится к росту клеток, который не зависит от нормальных механизмов регуляции (например, потере контактного ингибирования). Аномальный рост клеток может быть доброкачественным (не раковым) или злокачественным (раковым).
Аномальный рост клеток включает аномальный рост опухолей, резистентных к эндокринной терапии, антагонистам HER2 или ингибированию CDK4/6.
Термин «дополнительный противораковый терапевтический агент», используемый в настоящем документе, означает любой один или несколько терапевтических агентов, кроме соединения по изобретению, которые используются или могут быть использованы для лечения рака, такие как агенты, происходящие из следующих классов: митотические ингибиторы, алкилирующие агенты, антиметаболиты, противоопухолевые антибиотики, ингибиторы топоизомеразы I и II, растительные алкалоиды, гормональные агенты и антагонисты, ингибиторы факторов роста, радиация, ингибиторы протеинтирозинкиназ и/или серии/треонинкиназ, ингибиторы клеточного цикла, модификаторы биологического ответа, ингибиторы ферментов, антисмысловые олигонуклеотиды или производные олигонуклеотидов, цитотоксические и иммуноонкологические агенты (иммуноонкологические агенты включают моноклональные антитела, биспецифические антитела, цитокины, CAR-t-клетки).
Используемый в настоящем документе термин «рак» относится к любому злокачественному и/или инвазивному росту или опухоли, вызванной аномальным ростом клеток. Рак включает солидные опухоли, названные по типу образующих их клеток, рак крови, костного мозга или лимфатической системы. Примеры солидных опухолей включают саркомы и карциномы. Рак крови включает, но не ограничен ими, лейкоз, лимфому и миелому. Рак также включает первичный рак, который возникает в определенном месте в организме, метастатический рак, который распространился из места, в котором он начался, на другие части тела, рецидив исходного первичного рака после ремиссии и второй первичный рак, который представляет собой новый первичный рак у человека, у которого в анамнезе был рак другого типа.
В некоторых вариантах осуществления способов, представленных в настоящем документе, рак выбран из группы, состоящей из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легких, рака пищевода, рака печени, рака поджелудочной железы и рака желудка.
Дозированные формы и схемы
Введение соединений по изобретению можно осуществлять любым способом, который предлагает доставку соединений к месту действия. Эти способы включают пероральный путь введения, интрадуоденальный путь введения, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую или инфузионную), местное и ректальное введение.
Схемы дозирования могут быть скорректированы для обеспечения оптимального желаемого ответа. Например, можно вводить один болюс, можно вводить несколько разделенных доз с течением времени, или доза может быть пропорционально уменьшена или увеличена в соответствии с требованиями терапевтической ситуации. Особенно выгодно составлять парентеральные композиции в виде стандартной дозированной формы для простоты введения и однородности дозировки. Стандартная дозированная форма, используемая в настоящем документе, относится к физически дискретным единицам, подходящим в качестве единичных доз для млекопитающих, подлежащих лечению; каждая единица содержит заранее определенное количество активного соединения, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с требуемым фармацевтическим носителем. Спецификация стандартных дозированных форм по изобретению диктуется и напрямую зависит от (а) уникальных характеристик химиотерапевтического агента и конкретного терапевтического или профилактического эффекта, который должен быть достигнут, и (b) ограничений, присущих искусству составления такого активного соединения для лечения чувствительности у индивидуумов.
Таким образом, специалисту в данной области техники будет понятно, основываясь на представленном в настоящем документе описании, что доза и схема дозирования корректируются в соответствии со способами, хорошо известными в области терапии. То есть, максимально переносимая доза может быть легко установлена, а также может быть определено эффективное количество, обеспечивающее поддающийся обнаружению терапевтический эффект у пациента, а также временные требования для введения каждого агента для обеспечения поддающегося обнаружению терапевтического эффекта у пациента. Соответственно, несмотря на то, что в настоящем документе приведены примеры определенных доз и схем введения, эти примеры никоим образом не ограничивают дозы и схемы введения, которые могут быть предоставлены пациенту при осуществлении настоящего изобретения.
Следует отметить, что значения дозировки могут варьироваться в зависимости от типа и тяжести состояния, подлежащего облегчению, и могут включать однократные или многократные дозы. Кроме того, следует понимать, что для любого конкретного субъекта, конкретные схемы дозирования должны корректироваться с течением времени в соответствии с индивидуальными потребностями и профессиональным мнением лица, вводящего или контролирующего введение композиций, и что диапазоны дозировок, указанные в настоящем документе, являются только примерными и не предназначены для ограничения объема или применения заявленной композиции. Например, дозы можно корректировать на основе фармакокинетических или фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты и/или лабораторные показатели. Таким образом, настоящее изобретение охватывает повышение дозы внутри пациента, как определено специалистом в данной области техники. Определение подходящих дозировок и схем введения химиотерапевтического агента хорошо известно в соответствующей области техники, и специалисту в данной области техники должно быть понятно, что они охватываются, как только будут предложены идеи, описанные в настоящем документе.
Количество вводимого соединения по изобретению будет зависеть от субъекта, которого лечат, тяжести нарушения или состояния, скорости введения, расположения соединения и усмотрения лечащего врача. Однако эффективная доза находится в диапазоне от примерно 0,001 до примерно 100 мг на кг массы тела в день, предпочтительно, от примерно 1 до примерно 35 мг/кг/день, в виде однократной или разделенной дозы. Для человека массой 70 кг это будет составлять от около 0,05 до около 7 г/день, предпочтительно, от около 0,1 до около 2,5 г/день. В некоторых случаях, уровни доз ниже нижнего предела вышеуказанного диапазона могут быть более чем достаточными, в то время как в других случаях могут использоваться еще большие дозы, не вызывающие каких-либо вредных побочных эффектов, при условии, что такие большие дозы сначала разделяются на несколько малых доз для введения в течение дня.
Составы и пути введения
Используемый в настоящем документе термин «фармацевтически приемлемый носитель» относится к носителю или разбавителю, который не вызывает значительного раздражения организма и не отменяет биологическую активность и свойства вводимого соединения.
Фармацевтически приемлемый носитель может включать любой обычный фармацевтический носитель или эксципиент. Выбор носителя и/или эксципиента будет в значительной степени зависеть от таких факторов, как конкретный способ введения, влияние носителя или эксципиента на растворимость и стабильность, и природа дозированной формы.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители (такие как гидраты и сольваты). Фармацевтические композиции могут, при желании, содержать дополнительные ингредиенты, такие как ароматизаторы, связующие вещества, эксципиенты и подобные. Таким образом, для перорального введения, таблетки, содержащие различные эксципиенты, такие как лимонная кислота, могут использоваться вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связующими агентами, такими как сахароза, желатин и аравийская камедь. Примеры эксципиентов, без ограничения, включают карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли. Кроме того, для таблетирования часто используются смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции аналогичного типа также можно использовать в мягких и твердых заполненных желатиновых капсулах. Таким образом, неограничивающие примеры материалов включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии или эликсиры, содержащееся в них активное соединение можно комбинировать с различными подсластителями или ароматизаторами, красителями или пигментами и, при желании, с эмульгаторами или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Фармацевтическая композиция может быть, например, в форме, пригодной для перорального введения, в виде таблетки, капсулы, пилюли, порошка, составов с замедленным высвобождением, суспензии в растворе, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема, или для ректального введения в виде суппозиториев.
Типовые формы для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. При желании, такие дозированные формы могут быть соответствующим образом забуферены.
Фармацевтическая композиция может быть в виде стандартных дозированных форм, пригодных для однократного введения точных дозировок.
Фармацевтические композиции, подходящие для доставки соединений по изобретению, и способы их получения будут очевидны специалистам в данной области техники. Такие композиции и способы их получения можно найти, например, в «Remington's Pharmaceutical Sciences», 19th Edition (Mack Publishing Company, 1995), описание которого полностью включено в настоящее описание посредством ссылки.
Соединения по изобретению можно вводить перорально. Пероральное введение может включать проглатывание, так что соединение попадает в желудочно-кишечный тракт, или может применяться трансбуккальное или подъязычное введение, при котором соединение попадает в кровоток непосредственно изо рта.
Составы, подходящие для перорального введения, включают твердые составы, такие как таблетки, капсулы, содержащие частицы, жидкости или порошки, пастилки (включая наполненные жидкостью), жевательные таблетки, мульти- и наночастицы, гели, твердый раствор, липосомы, пленки (включая мукоадгезив), капсулы, спреи и жидкие составы.
Жидкие составы включают суспензии, растворы, сиропы и эликсиры. Такие составы могут быть использованы в качестве наполнителей в мягких или твердых капсулах, и обычно включают носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или несколько эмульгаторов и/или суспендирующих агентов. Жидкие составы также могут быть приготовлены восстановлением твердого вещества, например, из саше.
Соединения по изобретению также можно использовать в быстрорастворимых и быстро распадающихся дозированных формах, таких как формы, описанные в Expert Opinion in Therapeutic Patents, 11 (6), 981-986 Liang and Chen (2001), описание которого полностью включено в настоящий документ посредством ссылки.
Для таблетированных дозированных форм, в зависимости от дозы, лекарственное средство может составлять от 1% масс. до 80% масс. дозированной формы, более типично, от 5% масс. до 60% масс. дозированной формы. В дополнение к лекарственному средству, таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают гликолят крахмала натрия, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшим алкилом, крахмал, прежелатинизированный крахмал и альгинат натрия. Как правило, разрыхлитель составляет от 1% масс. до 25% масс. предпочтительно, от 5% масс. до 20% масс. дозированной формы.
Связующие агенты обычно используются для придания когезивных свойств составу таблеток. Подходящие связующие агенты включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки также могут содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводный и подобные), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двухосновного фосфата кальция.
Таблетки также могут необязательно включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как диоксид кремния и тальк. Поверхностно-активные агенты, если они присутствуют, обычно находятся в количестве от 0,2% масс. до 5% масс. таблетки, и глиданты обычно составляют от 0,2% масс. до 1% масс. таблетки.
Таблетки также обычно содержат смазывающие агенты, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Смазывающие агенты обычно присутствуют в количествах от 0,25% масс. до 10% масс. предпочтительно, от 0,5% масс. до 3% масс. таблетки.
Другие традиционные ингредиенты включают антиоксиданты, красители, ароматизаторы, консерванты и агенты, маскирующие вкус.
Примеры таблеток содержат до примерно 80% масс. лекарственного средства, от примерно 10% масс. до примерно 90% масс. связующего агента, от примерно 0% масс. до примерно 85% масс. разбавителя, от примерно 2% масс. до примерно 10% масс. разрыхлителя и от примерно 0,25% масс. до примерно 10% масс, смазывающего агента.
Смеси для таблеток могут быть спрессованы напрямую или с помощью валика с образованием таблеток. Смеси для таблеток или части смесей, альтернативно, могут быть получены влажной, сухой или грануляцией расплава, замороженными из расплава или экструдированными перед таблетированием. Окончательный состав может включать один или несколько слоев, и может быть с покрытием или без покрытия; или инкапсулирован.
Состав таблеток подробно обсуждается в «Pharmaceutical Dosage Forms: Tablets, Vol.1», H. Lieberman and L. Lachman, Marcel Dekker, NY, NY, 1980 (1SBN 0-8247-6918-Х), описание которого полностью включено в настоящее описание посредством ссылки.
Твердые составы для перорального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
Подходящие составы с модифицированным высвобождением описаны в патенте США №6,106,864. Подробности других подходящих технологий высвобождения, таких как высокоэнергетические дисперсии и осмотические частицы и частицы с покрытием, можно найти в Verma et al., Pharmaceutical Technology On-line, 25(2), 1-14 (2001). Использование жевательной резинки для достижения контролируемого высвобождения описано в WO 00/35298. Описание этих ссылок полностью включено в настоящий документ посредством ссылки.
Парентеральное введение
Соединения по изобретению можно также вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие средства для парентерального введения включают внутривенное, внутриартериальное, внутрибрюшинное, подоболочечное, внутрижелудочковое, внутриуретральное, интрастернальное, внутричерепное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольчатые (включая микроиглы) инъекторы, безыгольные инъекторы и методы инфузии.
Составы для парентерального введения обычно представляют собой водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и буферные агенты (предпочтительно, с рН от 3 до 9), но для некоторых применений они могут быть более подходящим образом составлены в виде стерильного не водного раствора или в виде высушенной формы для использования в сочетании с подходящим носителем, таким как стерильная апирогенная вода.
Приготовление парентеральных составов в стерильных условиях, например, путем лиофилизации, может быть легко осуществлено с использованием стандартных фармацевтических методик, хорошо известных специалистам в данной области техники.
Растворимость соединений по изобретению, используемых при получении парентеральных растворов, может быть повышена применением подходящих методов составления, таких как введение агентов, улучшающих растворимость.
Составы для парентерального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение. Таким образом, соединения по изобретению могут быть составлены в виде твердой, полутвердой или тиксотропной жидкости для введения в виде имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких составов включают стенты с лекарственным покрытием и микросферы PGLA.
Соединения по изобретению можно также вводить местно на кожу или слизистую оболочку, то есть дермально или трансдермально. Типовые составы для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, облатки, имплантаты, губки, волокна, бинты и микроэмульсии. Можно также использовать липосомы. Типовые носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения; см., например, 7 Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999). Другие способы местного введения включают доставку посредством электропорации, ионтофореза, фонофореза, сонофореза и инъекции с помощью микроиглы или без иглы (например, Powderject™, Bioject™ и т.д.). Описание этих ссылок полностью включено в настоящий документ посредством ссылки.
Составы для местного введения могут быть составлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
Соединения по изобретению можно также вводить интраназально или путем ингаляции, как правило, в виде сухого порошка (либо отдельно, либо в виде смеси, например, в сухой смеси с лактозой, либо в виде частиц смешанного компонента, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора для сухого порошка или в виде аэрозольного спрея из баллона под давлением, насоса, спрея, распылителя (предпочтительно распылителя, использующего электрогидродинамику для получения мелкодисперсного тумана) или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения, порошок может включать биоадгезив, например, хитозан или циклодекстрин.
Контейнер под давлением, насос, спрей, распылитель или небулайзер содержат раствор или суспензию соединения(ий) по изобретению, содержащую, например, этанол, водный раствор этанола или подходящий альтернативный агент для диспергирования, солюбилизации или продления высвобождения активного вещества, пропеллент(ы) в качестве растворителя и необязательное поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота.
Перед применением в виде сухого порошка или суспензии, лекарственный продукт микронизируют до размера, подходящего для доставки путем ингаляции (обычно менее 5 микрон). Это может быть достигнуто с помощью любого подходящего способа измельчения, такого как спирально-струйное измельчение, струйное измельчение в псевдоожиженном слое, обработка сверхкритической жидкостью с образованием наночастиц, гомогенизация под высоким давлением или сушка распылением.
Капсулы (изготовленные, например, из желатина или НРМС), блистеры и картриджи для использования в ингаляторе или инсуффляторе могут быть составлены таким образом, чтобы содержать порошковую смесь соединения по изобретению, подходящую порошковую основу, такую как лактоза или крахмал, и модификатор рабочих характеристик, такой как лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата, предпочтительно в последней форме. Другие подходящие эксципиенты включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Подходящий состав раствора для использования в распылителе, использующем электрогидродинамику для получения мелкодисперсного тумана, может содержать от 1 мкг до 20 мг соединения по изобретению на одно нажатие, и рабочий объем может варьироваться от 1 мкл до 100 мкл. Типичный состав включает соединение по изобретению, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые можно использовать вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
Подходящие ароматизаторы, такие как ментол и левоментол, или подсластители, такие как сахарин или сахарин натрия, могут быть добавлены к тем композициям по изобретению, которые предназначены для ингаляционного/интраназального введения.
Составы для ингаляционного/интраназального введения могут быть составлены так, чтобы обеспечить немедленное и/или модифицированное высвобождение с использованием, например, поли(DL-молочной-когликолевой кислоты (PGLA). Композиции с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
В случае ингаляторов с сухим порошком и аэрозолей, единица дозировки определяется с помощью клапана, который подает отмеренное количество. Единицы по изобретению обычно устроены так, чтобы вводить отмеренную дозу или «пшик», содержащую желаемое количество соединения по изобретению. Общая суточная доза может быть введена в виде разовой дозы или, чаще, в виде разделенных доз в течение дня.
Соединения по изобретению можно вводить ректально или вагинально, например, в форме суппозитория, пессария или клизмы. Масло какао является традиционной основой для суппозиториев, но при необходимости могут использоваться различные альтернативы.
Составы для ректального/вагинального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
Соединения по изобретению можно также вводить непосредственно в глаз или ухо, как правило, в форме капель микронизированной суспензии или раствора в изотоническом стерильном солевом растворе с отрегулированным рН. Другие составы, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (например, рассасывающиеся гелевые губки, коллаген) и не биоразлагаемые (например, силиконовые) имплантаты, пластины, линзы и системы частиц или везикул, такие как ниосомы или липосомы. Полимер, такой как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, гелановая камедь, может быть включен вместе с консервантом, таким как хлорид бензалкония. Такие составы также могут быть доставлены с помощью ионтофореза.
Составы для глазного/ушного введения могут быть составлены для немедленного и/или модифицированного высвобождения. Препараты с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, таргетное или запрограммированное высвобождение.
Другие технологии
Соединения по изобретению можно комбинировать с растворимыми макромолекулярными соединениями, такими как циклодекстрин и его подходящие производные, или полимеры, содержащие полиэтиленгликоль, для улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильности для применения в любой из вышеупомянутых схем введения.
Комплексы лекарственное средство-циклодекстрин обычно применимы для большинства дозированных форм и путей введения. Могут быть использованы как комплексы включения, так и не включения. Альтернативно непосредственному комплексообразованию с лекарственным средством, циклодекстрин можно использовать в качестве вспомогательной добавки, т.е. в качестве носителя, разбавителя или солюбилизатора. Наиболее часто для этих целей используются альфа-, бета-и гамма-циклодекстрины, примеры которых можно найти в публикациях РСТ №№ WO 91/11172, WO 94/02518 и WO 98/55148, описание которых полностью включено в настоящий документ посредством ссылки.
Дозировка
Количество вводимого активного соединения будет зависеть от субъекта, которого лечат, тяжести нарушения или состояния, скорости введения, расположения соединения и усмотрения лечащего врача. Однако, эффективная доза обычно находится в диапазоне от примерно 0,001 до примерно 100 мг на кг массы тела в день, предпочтительно, от примерно 1 до примерно 35 мг/кг/день, в виде однократной или разделенной дозы. Для человека массой 70 кг это будет составлять от около 0,05 до около 7000 мг/день, предпочтительно, от около 0,1 до около 2500 мг/день. В некоторых случаях, уровни доз ниже нижнего предела вышеуказанного диапазона могут быть более чем достаточными, в то время как в других случаях могут использоваться еще большие дозы, не вызывающие каких-либо вредных побочных эффектов, при этом такие большие дозы обычно делятся на несколько меньших доз для введения в течение всего дня.
Наборы
Поскольку может быть желательным введение комбинации активных соединений, например, с целью лечения конкретного заболевания или состояния, в объем настоящего изобретения входят две или несколько фармацевтических композиций, по меньшей мере, одна из которых содержит соединение в соответствии с изобретением, которые могут быть удобно объединены в виде набора, подходящего для совместного введения композиций. Таким образом, набор по изобретению включает две или несколько отдельных фармацевтических композиций, по меньшей мере, одна из которых содержит соединение по изобретению, и средства для раздельного хранения указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является известная блистерная упаковка, используемая для упаковки таблеток, капсул и подобных.
Набор по изобретению особенно подходит для введения различных дозированных форм, например пероральных и парентеральных, для введения отдельных композиций с различными интервалами дозирования или для титрования отдельных композиций относительно друг друга. Для облегчения комплаентности, набор обычно включает инструкции по применению и может быть снабжен запоминающим устройством.
Комбинированная терапия
Используемый в настоящем документе термин «комбинированная терапия» относится к введению соединения по изобретению вместе с, по меньшей мере, одним дополнительным фармацевтическим или лекарственным агентом (например, противораковым агентом, вакциной, антибактериальным агентом, противовирусным агентом или противопаразитарным агентом), либо последовательно, либо одновременно.
Как отмечалось выше, соединения по изобретению можно использовать в комбинации с одним или несколькими дополнительными агентами, такими как противораковые агенты. Эффективность соединений по изобретению при некоторых опухолях может быть повышена путем комбинирования с другими одобренными или экспериментальными противораковыми терапиями, например, лучевой терапией, хирургией, химиотерапевтическими агентами, таргетной терапией, агентами, которые ингибируют другие сигнальные пути, которые не регулируются в опухолях, и другими агентами, усиливающими иммунитет, такими как антагонисты PD-1 и подобные.
Когда используется комбинированная терапия, один или несколько дополнительных агентов можно вводить последовательно или одновременно с соединением по изобретению. В одном варианте осуществления, дополнительный агент вводят млекопитающему (например, человеку) перед введением соединения по изобретению. В другом варианте осуществления, дополнительный агент вводят млекопитающему (например, человеку) после введения соединения по изобретению. В другом варианте осуществления, дополнительный агент вводят млекопитающему (например, человеку) одновременно с введением соединения по изобретению.
Изобретение также относится к фармацевтической композиции для лечения аномального роста клеток у млекопитающего, включая человека, которая содержит количество соединения по изобретению, как определено выше (включая гидраты, сольваты, полиморфы, изомеры, пролекарства и/или метаболиты указанного соединения или его фармацевтически приемлемые соли) в комбинации с одним или несколькими (предпочтительно от одного до трех) противораковыми терапевтическими агентами.
В конкретных вариантах осуществления, соединение по изобретению можно вводить в комбинации с одним или несколькими: таргетными агентами, такими как ингибиторы киназы PI3, mTOR, PARP, Kras, IDO, TDO, ALK, ROS, MEK, VEGF, FLT3, AXL, R0R2, EGFR, FGFR, Src/АЫ, RTK/Ras, Мус, Raf, PDGF, АКТ, c-Kit, erbB, CDK2, CDK4, CDK4/CDK6, CDK5, CDK7, CDK9, SMO, CXCR4, HER2, GLS1, EZH2 или Hsp90, или иммуномодулирующими агентами, такими как антагонисты PD-1, антагонисты PD-L1, антагонисты CTLA-4, агонисты ОХ40, агонисты 4-1 ВВ или агонисты CD80.
В других вариантах осуществления, соединение по настоящему изобретению можно вводить в комбинации со стандартом лечения, таким как тамоксифен, доцетаксел, паклитаксел, цисплатин, капецитабин, гемцитабин, винорелбин, экземестан, летрозол, фулвестрант, анастрозол или трастузумаб.
Способы синтеза
Соединения формулы I могут быть получены способами, описанными ниже, вместе со способами синтеза, известными в области органической химии, или модификациями и превращениями, которые известны специалистам в данной области техники. Исходные материалы, используемые в настоящем документе, являются коммерчески доступными или могут быть получены обычными способами, известными в данной области техники[такими как способы, описанные в стандартных справочниках, таких как Compendium of Organic Synthetic Methods, Vol.I-XIII (published by Wi1ey-Interscience)]. Предпочтительные способы включают, но не ограничены ими, способы, описанные ниже.
Во время любой из следующих последовательностей синтеза может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы на любой из рассматриваемых молекул. Этого можно достичь с помощью обычных защитных групп, таких как группы, описанные в Т. W. Greene, Protective Groups in Organic Chemistry,,7ohn Wiley & Sons, 1981; T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Chemistry,,7ohn Wiley & Sons, 1991; и Т. W. Greene and P. G. M. Wuts, Protective Groups in Organic Chemistry,,7ohn Wiley & Sons, 1999, которые включены в настоящий документ посредством ссылки.
Соединения формулы I или их фармацевтически приемлемые соли могут быть получены в соответствии со схемами реакций, обсуждаемыми в настоящем документе ниже. Если не указано иное, заместители на схемах имеют указанные выше значения.
Выделение и очистку продуктов осуществляют стандартными методами, известными специалисту в области химии.
Специалисту в данной области техники будет понятно, что различные символы, верхние и нижние индексы, используемые в схемах, способах и примерах, используются для удобства представления и/или для отражения порядка, в котором они представлены на схемах, и не предназначены для обязательного соответствия символам, верхним или нижним индексам в прилагаемой формуле изобретения. Кроме того, специалисту в данной области техники будет понятно, что во многих случаях эти соединения представляют собой смеси и энантиомеры, которые можно разделить на различных стадиях схем синтеза с использованием обычных методов, таких как, но не ограничиваясь ими, кристаллизация, хроматография с нормальной фазой, хроматография обращенной фазой и хиральная хроматография, для получения отдельных энантиомеров. Схемы представляют способы, используемые при синтезе соединений по настоящему изобретению. Они никоим образом не ограничивают объем изобретения.
Соединения по изобретению получают в соответствии с приведенными в настоящем документе типовыми процедурами и их модификациями, известными специалистам в данной области техники.
В примерах используются следующие сокращения: «Ас» означает ацетил, «АЦН» означает ацетонитрил, «ВОС», «Boc» или «boc» означает N-трет-бутоксикарбонил, «Bu» означает бутил, «tBu» означает трет-бутил, «ДХМ» (CH2Cl2) означает метиленхлорид, «ди» означает диастереомерный избыток, «ДМФ» означает N,N-диметилформамид, «ДМСО» означает диметилсульфоксид, «эи» означает энантиомерный избыток, «Et» означает этил, «EtOAc» означает этилацетат, «EtOH» означает этанол, «i-Pr» или «iPr» означает изопропил, «Ме» означает метил, «МеОН» означает метанол, «МС» означает масс-спектрометрию, «МТБЭ» означает метил-трет-бутиловый эфир, «Ph» означает фенил, «ТГФ» означает тетрагидрофуран, «СЖХ» означает сверхкритическую жидкостную хроматографию, «ТСХ» означает тонкослойную хроматографию, «Rf» означает коэффициент удержания, «~» означает приблизительно, «КТ» означает комнатную температуру, которая включает температуру окружающей среды (обычно от 20°С до 25°С), «ч» означает часы, «мин» означает минуты, «экв» означает эквиваленты, «насыщ.» значит насыщенный.
Соединения формулы I могут быть получены способами, описанными в общих способах, представленных ниже, или их обычными модификациями. Если не указано иное, заместители в способах являются такими, как определено в настоящем документе. Хотя R4 находится в соединениях формулы I, из промежуточных соединений, используемых в соответствующем пути синтеза, будет ясно, является ли R4 R4-i или R4-ii. Настоящее изобретение также охватывает любой один или несколько из этих способов получения соединений формулы I в дополнение к любым новым промежуточным соединениям, используемым в нем. Специалисту в данной области техники понятно, что следующие реакции можно охлаждать, нагревать термически, нагревать под действием микроволнового излучения или проводить в условиях проточной химии. Кроме того, будет понятно, что может быть необходимо или желательно проводить превращения в порядке, отличном от порядка, описанного в способах, или модифицировать одно или несколько превращений для получения желаемого соединения по изобретению.
Способ А
Способ А представляет собой получение соединений формулы I, где R3b представляет собой водород. В способе А, перекрестное сочетание или нуклеофильное ароматическое замещение между необходимым партнером сочетания или амином, при условии, что R1 имеет значения, указанные в формуле I (или его защищенном варианте), дает А-2. Для дихлорпиридина формулы А-1 V представляет собой N(CH3)2, ОН, пиперидин, морфолин, ОМе, OEt или OiPr. Затем форматирование А-2 дает альдегид А-3. Последующая конденсация с сульфинамидом Эллмана дает соединение формулы А-4. Восстановление и опосредованная основанием циклизация соединения формулы А-4 дает соединение формулы А-5. Альтернативно, соединение формулы I, где R1 представляет собой циклопропилметил, соединение формулы А-5. может быть получено с использованием способа синтеза, описанного для получения промежуточного соединения 15с, описанного в настоящем документе. На следующей стадии, опосредованное иридием и никелем декарбоксилативное окислительно-восстановительное сочетание под действием света между хлорпиридином формулы А-5 и карбоновой кислотой формулы А-6 дает соединение формулы А-7. На этой стадии, заместитель R3a в А-6 должен быть представлен той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. Кроме того, заместитель R7 и R8 может представлять собой Н, Boc, альтернативную защитную группу или алкил; или, альтернативно, заместители R3a и R8 могут быть частью циклической системы, как это желательно в конечном продукте формулы I. Сочетание соединения формулы А-7 с бромпиридинтриазолом А-8 в присутствии палладиевого или медного катализа с последующим отщеплением любой защитной группы(групп) в стандартных условиях дает пирроло[3,4-c]пиридин-1-он формулы I. На этой стадии, заместитель R4N в А-8 должен быть представлен той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте.

Гидразинолиз бромпиридинового эфира А-9 (J. Med. Chem., 60(2), 722-748; 2017) с последующей реакцией соответствующего гидразида А-10 с диметилацеталем диметилформамида дает формамидин А-11. Конденсация формамидина А-11 с амином дает триазол А-8.
Способ В
Способ В представляет собой получение соединений формулы I, где R3a и R3b представляют собой водород. На первой стадии способа В, из соединения формулы А-3 (V=N(CH3)2, ОН, пиперидин, морфолин, ОМе, OEt или OiPr) перекрестное сочетание Негиши с необходимым аминоалкилцинкатом дает соединение формулы В-1. На этой стадии, заместители R7 и R8 представляют собой либо Н, либо алкил, представленный в формуле I, или Boc, или альтернативную защитную группу. Последующая конденсация с сульфонамидом Эллмана, восстановление и опосредованная основанием циклизация дают соединение формулы В-2. Сочетание с бромпиридинтриазолом В-3 в присутствии палладиевого или медного катализа и последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-c]пиридин-1-он формулы I. На этой стадии, заместитель R4D формулы В-3 должен быть представлен той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте.

Пактам формулы В-4 превращают в О-алкилимидат формулы В-5. На этой стадии, заместитель R4D формулы В-4 должен быть представлен той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. Циклизация O-алкилимидата формулы В-5 с ацилгидразином формулы А-10 дает соединение формулы В-3.
Способ С

Способ С представляет собой получение соединений формулы I, где R3b представляет собой метил. На первой стадии способа С, опосредованное палладием перекрестное сочетание соединения формулы А-5 и сложного эфира бороновой кислоты формулы С-1 с последующим опосредованным железом гидроазидированием олефина или алкена, восстановлением и необязательной защитой полученного амина, дает соединение формулы С-2. На этой стадии, заместитель R3a формулы С-1 и заместитель R1 формулы А-5 должны быть такими, как представлено в желаемом продукте формулы I или его защищенном варианте. Перекрестное сочетание соединения формулы С-2 с бромпиридинтриазолом А-8 при катализе палладием или медью с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-с]пиридин-1-он формулы I. На этой стадии, заместитель R4N в А-8 должен быть представлен той же группой, которая требуется в R4 конечного продукта формулы I или ее защищенного варианта.
Способ D
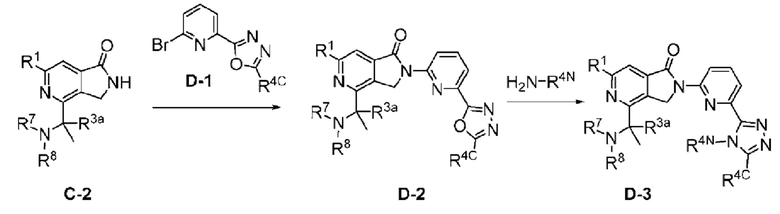
Способ D представляет получение соединений формулы I, где R3b представляет собой метил. На первой стадии способа D, сочетание соединения формулы С-2 с бромпиридином D-1 в присутствии медного или палладиевого катализа дает соединение формулы D-2. На этой стадии заместители R1, R7, R8 и R3a С-2 и заместитель R4C D-1 должны быть представлены одной и той же группой, которая требуется в конечном продукте D-3 или его защищенном варианте. На этой стадии, R7 и/или R8 также необязательно могут быть представлены защитными группами. Реакция соединения формулы D-2 с необходимым амином дает соединение формулы D-3. На этой стадии, заместитель R4N должен быть представлен той же группой, которая требуется в конечном продукте, D-3, или его защищенном варианте. Отщепление любой(ых) защитной(ых) группы(групп) в соединении формулы D-3 в конечном итоге дает пирроло[3,4-с]пиридин-1-он формулы IA.

Чтобы получить соединение формулы D-1, соединение формулы А-10 подвергают реакции с соответствующим ацилхлоридом с получением соединения формулы D-4. На этой стадии, заместитель R4C ацилхлорида представляет собой такой, как представлен в конечном продукте формулы I или его защищенном варианте. Циклизация соединения формулы D-4 дает соединение формулы D-1.
Способ Е

Способ Е представляет собой получение соединений формулы I, где R3b представляет собой водород. На первой стадии способа Е, либо
i. Опосредованное палладием перекрестное сочетание между соединением формулы А-5 и трибутил(1-этоксивинил)оловом с последующим гидролизом винилового эфира дает кетон формулы Е-1, где заместитель R3a должен быть представлен той же группой, которая требуется в конечном продукте формулы I, либо
ii. Опосредованное палладием перекрестное сочетание между соединением формулы А-5 и цианидом цинка с последующей реакцией с алкильным реагентом Гриньяра дает кетон формулы Е-1, где заместитель R3a должен быть представлен той же группой, которая требуется в конечном продукте формулы I.
На этой стадии, заместитель R1 в А-5 должен быть представлен той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте.
Из соединения формулы Е-1, либо
i. Восстановление кетона с последующей активацией в виде мезилата или хлорида, заменой азидом и восстановлением и необязательной защитой дает соединение формулы А-7 (R7 и R8 оба представляют собой Н или R7 представляет собой Н, и R8 представляет собой Boc или альтернативную защитную группу), либо
ii. Восстановление кетона с последующей активацией в виде мезилата или хлорида, замещением алкиламином и необязательной защитой дает соединение формулы А-7 (R7 и/или R8 представляют собой Н, алкил, Boc или альтернативную защитную группу), либо
iii. Конденсация с сульфинамидом с последующим восстановлением дает соединение формулы А-7 (R7 представляет собой Н и R8 представляет собой SOtBu).
Сочетание соединения формулы А-7 с бромпиридинтриазолом А-8 в присутствии палладиевого или медного катализа с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-c]пиридин-1-он из формулы I. На этой стадии, заместитель R4N в А-8 должен быть представлен той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте, в пределах R4.
Способ F
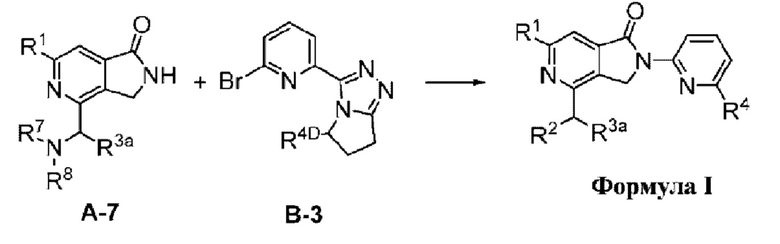
Способ F представляет собой получение соединений формулы I, где R3b представляет собой водород. В способе F, сочетание соединения формулы А-7 с бромпиридинтриазолом В-3 в присутствии палладиевого или медного катализа с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-с]пиридин-1-он из формулы I. На этой стадии, заместитель R4C в В-3 и заместители R1, R7, R8 и R3a формулы А-7 должны быть представлены той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. На этой стадии, заместители R7 и/или R8 формулы А-7 также могут быть представлены Boc или другой защитной группой.
Способ G

Способ G представляет собой получение соединений формулы I, где R3b представляет собой водород. На первой стадии способа G, бромпиридин формулы D-1 подвергают реакции с необходимым амином с получением соединения формулы G-1. На этой стадии, заместители R4C и R4N должны быть представлены той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. Сочетание соединения формулы G-1 с соединением А-7 в присутствии палладиевого или медного катализа с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-с]пиридин-1-он формулы I. На этой стадии, заместители R1, R7, R8 и R3a формулы А-7 должны быть представлены той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. На этой стадии, заместители R7 и/или R8 формулы А-7 также могут быть представлены Boc или другой защитной группой.
Способ Н

Способ Н представляет собой получение соединений формулы I, где R3b представляет собой метил. На первой стадии способа Н, сочетание соединения формулы С-2 с бромпиридинтриазолом В-3 при катализе палладием или медью с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-c]пиридин-1-он формулы I. На этой стадии, заместители R1, R7, R8 и R3a в С-2 и заместитель R4D в В-3 должны быть представлены той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. R7 и/или R8 формулы С-2 также необязательно могут быть представлены защитной группой на этой стадии.
Способ I

Способ I представляет собой получение соединений формулы I, где R3b представляет собой метил. На первой стадии способа I, сочетание соединения формулы С-2 с бромпиридинтриазолом G-1 в присутствии палладиевого или медного катализа с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-с]пиридин-1-он формулы I, где R3b представляет собой метил. На этой стадии, заместители Rl, R7, R8 и R3a С-2 и заместители R4C и R4N G-1 должны быть представлены той же группой, которая требуется в конечном продукте, формула I или его защищенном варианте. R7 и/или R8 формулы С-2 также необязательно могут быть представлены защитной группой на этой стадии.
Способ,7

Способ,7 представляет получение соединений формулы I, где R3b представляет собой водород. На первой стадии способа,7, спирт формулы,7-1 защищают с получением соединения формулы,7-2 (например, X=OTBDMS). Соединение формулы,7-2 превращают в O-алкилимидат формулы,7-3. Циклизация О-алкилимидата формулы,7-3 с ацилгидразином формулы А-10 дает соединение формулы,7-4. Соединение формулы,7-4 подвергают сочетанию с соединением формулы А-7 в атмосфере палладия или меди с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях с получением пирроло[3,4-с]пиридина-1-он из формулы I. На этой стадии, заместители R1, R7, R8 и R3a в А-7 должны быть представлены той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. На этой стадии, заместители R7 и/или R8 формулы А-7 также могут быть необязательно представлены защитной группой.
Альтернативно, для получения соединений формулы I, где R4 представляет собой R4-ii и R4D представляет собой алкил, замещенный атомом F (Х=фтор), из соединения формулы,7-4 (X представляет собой защищенную гидроксильную группу), отщепление защитной группы предлагает соединение формулы,7-4, где X представляет собой гидрокси. Затем спирт можно превратить в алкилфторид с получением соединения формулы,7-4, где X представляет собой фтор.
Способ K

Способ K представляет собой получение соединений формулы I, где R3a и R3b представляют собой водород. В способе K, соединение формулы,7-4 (X представляет собой фтор) подвергается сочетанию с соединением формулы В-2 при катализе палладием или медью с последующим отщеплением защитной(ых) группы(групп) в стандартных условиях с получением пирроло[3,4-с]пиридин-1-она формулы I. На этой стадии, заместители R1, R7 и R8 в В-2 и заместитель X в формуле,7-4 должны быть представлены той же группой, которая требуется в конечном продукте формулы I или его защищенном варианте. R7 и/или R8 формулы В-2 также необязательно могут быть представлены защитной группой на этой стадии.
Способ L

На первой стадии способа L, соединение формулы L-1 подвергают олефинированию по Виттигу с получением соединения формулы L-2. Восстановление и последующее омыление соединения формулы L-2 дает соединение формулы L-3. На этих стадиях, заместитель R4D в L-1 и заместители R4D и R4F в L-2 и L-3, а также последующие промежуточные продукты представлены в конечном продукте формулы I (как в R4) или его защищенном варианте. Альтернативно, некоторые соединения формулы L-3 являются коммерчески доступными. На следующей стадии, соединение формулы L-3 подвергают реакции с соединением формулы А-10 с получением соединения формулы L-4. Циклизация соединения формулы L-4 дает оксадиазол формулы L-5. Последующая циклизация, которой предшествует термическое или кислотное снятие защиты с соединения формулы L-5, дает соединение формулы L-6. Соединение формулы L-6 подвергают сочетанию с соединением формулы А-7, В-2 или С-2 в присутствии палладия или меди с последующим отщеплением любой(ых) защитной(ых) группы(групп) в стандартных условиях с получением пирроло[3,4-с]пиридин-1-она формулы I. Заместители R1, R2, R3a и R3b формулы I зависят от их определений в предыдущих способах для А-7, В-2 или С-2 или их защищенных вариантов.
Способ М

Способ М представляет собой получение соединений формулы I, где R3a и R3b представляют собой водород. В способе М, сочетание соединения формулы В-2 с бромпиридинтриазолом А-8 при катализе палладием или медью дает защищенный пирроло[3,4-с]пиридин-1-он формулы I. На этой стадии, заместители R1, R7 и R8 формулы В-2 и заместитель R4N формулы А-8 должны быть представлены той же группой, которая требуется в конечном продукте формулы I, или его защищенном варианте, или в виде защитной группы (например, R7=Boc). Отщепление любой(ых) защитной(ых) группы(групп) в стандартных условиях дает пирроло[3,4-с]пиридин-1-он формулы I.
Получение синтетических промежуточных соединений
Для промежуточных соединений и примеров, приготовленных в настоящем документе, где стереохимия известна, стереохимия является такой, как показано, и название обозначает конкретную стереохимию, такую как (R) или (S). Приведенная в настоящем документе стереохимия известна, поскольку соединение было синтезировано из известных хиральных исходных материалов или рацемических смесей, и стереохимия некоторых примеров или промежуточных соединений была подтверждена с использованием рентгеновской кристаллографии. В случае последнего, стереохимия других примеров или промежуточных соединений затем будет выведена на основе известной хиральности вышеупомянутых примеров или промежуточных соединений и пути синтеза. Если стереохимия неизвестна, но энантиомеры разделены, «or1» или «or2» относится к хиральному атому углерода. В названии, углерод с разрешенным, но не подтвержденным стереохимическим центром обозначен символом «ξ». Связь, изображенная в этом углероде, является представлением стереохимии; это означает, что углерод будет иметь эту конфигурацию связи (сплошной клин) или противоположную конфигурацию (заштрихованный клин). См., например, пример 6а и пример 6b. Рацемические соединения обозначены аннотацией «@1» и связь изображена как определенная стереохимия, означающая, что углерод будет иметь эту конфигурацию связи (сплошной клин) и противоположную конфигурацию (заштрихованный клин). См., например, промежуточное соединение 15.
Промежуточное соединение 1: 2-бром-6-(4-этил-4H-1,2,4-триазол-3-ил)пиридин

Стадия 1: 6-бромпиридин-2-карбогидразид (1а)

К раствору метил-6-бром-2-пиридинкарбоксилата (16,0 г, 74,0 ммоль) в МеОН (120 мл) добавляют моногидрат гидразина (5,23 г, 88,8 ммоль, 85%) и смесь перемешивают в течение 16 часов при комнатной температуре. Полученный раствор концентрируют примерно до половины объема и затем растирают, добавляя 40 мл метил-трет-бутилового эфира и перемешивая в течение 10 мин. Полученное белое твердое вещество фильтруют и сушат в вакууме с получением указанного в заголовке соединения (1а) (15 г, 94%). 1Н ЯМР (400 МГц, CDCl3) δ 8,82 (шс, 1Н), 8,13 (дд, J=0,9, 7,5 Гц, 1Н), 7,76-7,71 (м, 1Н), 7,64 (дд, J=0,9, 8,0 Гц, 1Н), 4,08 (шс, 2Н). m/z (ИЭР) для (C6H6BrN3O) 217,5 (М+Н)+.
Стадия 2: N'-[(6-бромпиридин-2-ил) карбонил]-N,N-диметилгидразоноформамид (1b)

Раствор 6-бромпиридин-2-карбогидразида (1a) (15,0 г, 69,4 ммоль) в диметилацетале диметилформамида (80 мл) перемешивают при 80°С в течение 16 часов. Полученную смесь концентрируют при пониженном давлении с получением остатка. К этому остатку добавляют метил-трет-бутиловый эфир (60 мл) и перемешивают в течение 40 мин. Полученное желтое твердое вещество фильтруют и сушат с получением указанного в заголовке соединения (1b) (16 г, 85%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,74 (с, 1Н), 8,06 (с, 1Н), 8,01-7,97 (м, 1Н), 7,91 (т, J=7,7 Гц, 1Н), 7,80 (дд, J=1,0, 7,8 Гц, 1Н), 2,84 (с, 6Н). m/z (ИЭР) для (C9H11BrN4O) 272,7 (М+Н)+.
Стадия 3: Промежуточное соединение 1
В колбу загружают N'-[(6-бромпиридин-2-ил) карбонил]-N,N-диметилгидразоноформамид (1b) (2,0 г, 7,4 ммоль), этиламин (0,5 мл, 333 мг, 7,4 ммоль), уксусную кислоту (3 мл) и MeCN (15 мл, 0,5 М). Раствор нагревают в течение 16 ч при 95°С. Добавляют EtOAc (10 мл) и H2O (10 мл). Добавляют твердый K2CO3 до тех пор, пока рН водного слоя не станет ~рН 8. Слои разделяют, и водный слой экстрагируют EtOAc (3×30 мл). Объединенные органические слои промывают насыщенным раствором соли (30 мл), сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток суспендируют с EtOAc (0,3 мл) и петролейным эфиром (3 мл) в течение 5 мин. Твердые вещества собирают фильтрацией с получением промежуточного соединения 1 (1,5 г, 80%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,75 (с, 1Н), 8,19 (дд, J=7,7, 0,9 Гц, 1Н), 7,99-7,90 (м, 1Н), 7,79 (дд, J=8,0, 0,9 Гц, 1Н), 4,47 (кв, J=7,2 Гц, 2Н), 1,38 (т, J=7,2 Гц, 3Н); m/z (ХИАД+) для (C9H9BrN4), 252,7 (М+Н)+.
Промежуточное соединение 2: (5S)-3-(6-бромпиридин-2-ил)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол

Стадия 1: трет-бутил {(2S)-5-[2-(6-бромпиридин-2-карбонил)гидразинил]-5-оксопентан-2-ил}карбамат (2а)
Раствор (4S)-4-[(трет-бутоксикарбонил)амино]пентановой кислоты (600 мг, 2,76 ммоль) в ТГФ (13,8 мл, 0,2 М) охлаждают до 0°С. Раствор ангидрида пропилфосфоновой кислоты (50% раствор в EtOAc, 3,62 мл, 6,08 ммоль) добавляют к раствору при 0°С, после чего баню удаляют, и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Затем добавляют N,N-диизопропилэтиламин (2,89 мл, 16,6 ммоль) и 6-бромпиколиногидразид (656 мг, 3,04 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 22 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию гасят водой (15 мл) и переносят в делительную воронку с EtOAc (20 мл). Слои разделяют, и органическую фазу последовательно промывают 20% лимонной кислотой (20 мл), насыщенным раствором NaHCO3 (20 мл) и насыщенным раствором соли (20 мл). Затем органический экстракт сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (2а) (1,07 г, выход 93%) в виде не совсем белого твердого вещества, которое используют без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 9,76 (шс, 1Н), 9,36 (шс, 1Н), 8,15 (дд, J=0,9, 7,5 Гц, 1Н), 7,77-7,71 (м, 1Н), 7,69-7,64 (м, 1Н), 4,45 (шд, J=1,0 Гц, 1Н), 3,91 (шс, 1Н), 2,47-2,35 (м, 2Н), 2,00-1,89 (м, 1Н), 1,75-1,66 (м, 1Н), 1,48 (с, 9Н), 1,22 (д, J=6,6 Гц, 3Н). ЖХМС m/z (ХИАД) для (C11H15BrN4O2), 315,0 (М+Н-Вос)+.
Стадия 2: трет-бутил {(2S)-4-[5-(6-бромпиридин-2-ил)-1,3,4-оксадиазол-2-ил]бутан-2-ил}карбамат (2b)

К раствору трет-бутил {[2S)-5-[2-(6-бромпиридин-2-карбонил)гидразинил]-5-оксопентан-2-ил}карбамата (2а) (290 мг, 0,698 ммоль) в ДХМ (2,8 мл, 0,25 М) добавляют триэтиламин (0,292 мл, 2,09 ммоль) и п-толуолсульфонилхлорид (160 мг, 0,838 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Добавляют этилендиамин (0,047 мл, 0,698 ммоль) для удаления избытка п-толуолсульфонилхлорида; при добавлении сразу образовывается осадок. После перемешивания при комнатной температуре в течение 30 мин, реакционную смесь промывают 20% лимонной кислотой (5 мл) и разделяют слои. Водный слой экстрагируют ДХМ (5 мл), затем объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (2b) (274 мг, 98% выход) в виде светло-желтого твердого вещества, которое используют без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 8,21 (дд, J=0,9, 7,6 Гц, 1Н), 7,76-7,69 (м, 1Н), 7,68-7,65 (м, 1Н), 4,39 (шс, 1Н)), 3,85 (шс, 1Н), 3,08-3,01 (м, 2Н), 2,17-2,03 (м, 1Н), 2,03-1,90 (м, 1Н), 1,44 (с, 9Н), 1,22 (д, J=6,6 Гц, 3Н). ЖХМС m/z (ХИАД) для (C11H13BrN4O), 297,0 (М+Н-Вос)+.
Стадия 3: Промежуточное соединение 2
В сосуд для микроволновой печи загружают трет-бутил {(2S)-4-[5-(6-бромпиридин-2-ил)-1,3,4-оксадиазол-2-ил]бутан-2-ил}карбамат (2b) (150 мг, 0,378 ммоль) и трифторэтанол (1,89 мл, 0,2 М) и герметично закрывают перед нагреванием в микроволновой печи до 180°С в течение 30 мин. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь концентрируют, и остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 1:10 MeOH/EtOAc) с получением промежуточного соединения 2 (74,3 мг, выход 71%) в виде желтовато-коричневого липкого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,38 (д, J=7,7 Гц, 1Н), 7,74 (т, J=7,8 Гц, 1Н), 7,58 (д, J=7,9 Гц, 1Н), 5,22-5,08 (м, 1Н), 3,21-3,16 (м, 1Н), 3,16-3,02 (м, 2Н), 2,55-2,44 (м, 1Н), 1,60 (д, J=6,6 Гц, 3Н). ЖХМС m/z (ХИАД) для (C11H11BrN4), 279,1 (М+Н)+. Определяют как единственный энантиомер СЖХ (10-60% метанола (0,5% NH3) в диоксиде углерода при 400-450 бар, время градиента=2 мин, скорость потока=4 мл/мин, Chiralpack IC-U 50 мм * 3 * мм колонка 1,6 мкм). Стереохимию промежуточного соединения 2 определяют на основании использования (4S)-4-[(трет-бутоксикарбонил)амино]пентановой кислоты на стадии 1.
Абсолютную конфигурацию промежуточного соединения 2 однозначно устанавливают с помощью рентгеновской кристаллографии малых молекул. Рентгеноструктурные исследования монокристаллов проводят на дифрактометре Bruker APEX II Ultra CCD, оснащенном Mo Кα излучением (λ=0,71073). Кристалл используют, как получен (выращенный диффузией паров из дихлорметана). Бесцветный кристалл размером 0,200x0,075x0,060 мм устанавливают на Cryoloop с маслом Paratone. Данные собирают в потоке газообразного азота при 100(2) К) с использованием φ и ω сканирования. Расстояние от кристалла до детектора составляет 45 мм при времени экспозиции 1 с (в зависимости от диапазона 2θ) при ширине сканирования 0,80°. Сбор данных завершают на 100,0% до 25,242° дюйма θ. Данные интегрируют с использованием программного обеспечения Bruker SAINT и масштабируют с использованием программного обеспечения SADABS. Решение с помощью прямых способов (SHELXT) позволяет получить полную фазовую модель, соответствующую предложенной структуре. Все неводородные атомы уточнены анизотропно полноматричным способом наименьших квадратов (SHELXL-2014). Все атомы водорода, связанные с углеродом, размещены с использованием модели «наездника». Их положения ограничены относительно их родительского атома с помощью соответствующей команды HFIX в SHELXL-2014. Положения атомов водорода СН, NH и ОН уточняют с использованием соответствующих команд HFIX. Кристаллографические данные суммированы в таблице 1.


Промежуточное соединение 3: (5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол
Стадия 1: трет-бутил{(2S)-1-[метокси(метил)амино]-1-оксобутан-2-ил}карбамат (3а)
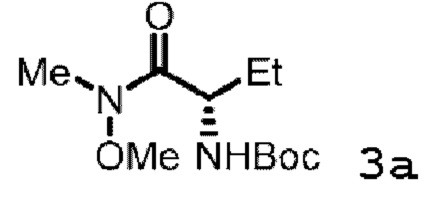
Раствор (2S)-2-[(трет-бутоксикарбонил)амино]бутановой кислоты (2,0 г, 9,84 ммоль) в ТГФ (49,2 мл, 0,2 М) охлаждают до 0°С. Раствор ангидрида пропилфосфоновой кислоты (50% раствор в EtOAc, 12,9 мл, 21,6 ммоль) добавляют к раствору при 0°С перед удалением бани, реакционной смеси дают нагреться до КТ и перемешивают в течение 30 минут. Затем добавляют N,N-диизопропилэтиламин (10,3 мл, 59,0 ммоль) и гидрохлорид метокси(метил)амина (1,06 г, 10,8 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию гасят водой (40 мл) и переносят в делительную воронку с EtOAc (40 мл). Слои разделяют, и органическую фазу последовательно промывают 20% лимонной кислотой (40 мл), насыщенным раствором NaHCO3 (40 мл) и насыщенным раствором соли (40 мл). Затем органический экстракт сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (3а) (1,29 г, выход 53%) в виде желтого масла, которое используют без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 5,28-5,12 (м, 1Н), 4,74-4,52 (м, 1Н), 3,79 (с, 3H), 3,23 (с, 3H), 1,85-1,73 (м, 1Н), 1,64-1,54 (м, 1Н), 1,46 (с, 9Н), 0,96 (т, J=7,5 Гц, 3H). ЖХМС m/z (ХИАД) для (C11H22N2O4), 247,1 (М+Н)+.
Стадия 2: трет-бутил (S)-(1-оксобутан-2-ил)карбамат (3b)

Раствор трет-бутил {(2S)-1-[метокси(метил)амино]-1-оксобутан-2-ил} карбамата (3а) (1,29 г, 5,25 ммоль) в ТГФ (26,2 мл, 0,2 М) охлаждают до 0°С. К раствору по каплям добавляют алюмогидрид лития (2,3 М раствор в 2-МеТГФ, 2,51 мл, 5,77 ммоль) при 0°С. Реакционную смесь перемешивают в течение 40 мин при 0°С. Анализ ЖХМС показал потребление исходного материала. Реакцию гасят EtOAc (10 мл) и 1 М HCl (10 мл) и переносят в делительную воронку. Слои разделяют, и водную фазу экстрагируют EtOAc (3×10 мл). Затем объединенные органические экстракты сушат над MgSO4, фильтруют и концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 100% EtOAc) с получением указанного в заголовке соединения (3b) (0,575 г, выход 59%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,60 (с, 1Н), 5,09 (шс, 1Н), 4,22 (шс, 1Н), 2,04-1,90 (м, 1Н), 1,73-1,64 (м, 2Н), 1,48 (с, 9Н), 0,99 (т, J=7,5 Гц, 3H).
Стадия 3: этил (2Е,AS)-4-[(трет-бутоксикарбонил)амино]гекс-2-еноат (3с)

К раствору трет-бутил (S)-(1-оксобутан-2-ил)карбамата (3b) (575 мг, 3,07 ммоль) в ДХМ (6,14 мл, 0,5 М) добавляют (этоксикарбонилметилен)трифенилфосфоран (1,60 г, 4,61 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 17 часов. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь концентрируют досуха, затем добавляют изопропанол (8,5 мл, 0,36 М) и хлорид цинка (1,26 г, 9,21 ммоль) для осаждения оксида трифенилфосфина. После перемешивания при комнатной температуре в течение 30 мин, реакционную смесь фильтруют и концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 100% EtOAc) с получением указанного в заголовке соединения (3с) (653 мг, выход 83%, соотношение изомеров E/Z >20:1) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 6,86 (дд, J=5,4, 15,6 Гц, 1Н), 5,94 (дд, J=1,7, 15,7 Гц, 1Н), 4,50 (шс, 1Н), 4,31-4,16 (м, 3H), 1,65-1,63 (м, 1Н), 1,60-1,51 (м, 1Н), 1,47 (с, 9Н), 1,32-1,30 (м, 3H), 0,99-0,95 (м, 3H). ЖХМС m/z (ХИАД) для (C8H15NO2), 158,2 (М+Н-Вос)+.
Стадия 4: этил (4S)-4-[(трет-бутоксикарбонил)амино]гексаноат (3d)

К раствору этилового эфира (2Е,4S)-4-[(трет-бутоксикарбонил)амино]гекс-2-еноата (3с) (653 мг, 2,54 ммоль) в метаноле (12,7 мл, 0,2 М) добавляют палладий на угле (10% масс./масс. 270 мг, 0,254 ммоль). Реакционный сосуд вакуумируют и снова заполняют Н2 в динамическом вакууме в течение 10 секунд. Затем реакционную смесь перемешивают при комнатной температуре под давлением 1 атм Н2 в течение 18 часов. Смесь перемешивают при комнатной температуре в течение 17 часов. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь фильтруют через Celite® и концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 100% EtOAc) с получением указанного в заголовке соединения (3d) (589 мг, выход 90%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 4,37-4,23 (м, 1Н), 4,20-4,11 (м, 2Н), 3,61-3,46 (м, 1Н), 2,42-2,33 (м, 2Н), 1,93-1,82 (м, 1Н), 1,71-1,62 (м, 1Н), 1,57-1,51 (м, 1Н), 1,46 (с, 9Н), 1,44-1,38 (м, 1Н), 1,30-1,26 (м, 3H), 0,94 (т, J=7,4 Гц, 3H).
Стадия 5: (4S)-4-[(трет-бутоксикарбонил)амино]гексановая кислота (3е)

К раствору этил (4S)-4-[(трет-бутоксикарбонил)амино]гексаноата (3d) (589 мг, 2,27 ммоль) в ТГФ (11,4 мл, 0,2 М) и МеОН (5,7 мл, 0,4 М) добавляют раствор LiOH (544 мг, 22,7 ммоль) в воде (2,84 мл, 0,8 М). Реакционную смесь перемешивают при КТ в течение 24 ч. Реакционную смесь концентрируют перед добавлением воды и 1М HCl до достижения рН 5. Водный слой экстрагируют EtOAc (3×20 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (3е) (260 мг, выход 50%) в виде светло-желтого масла, которое используют без дополнительной очистки.
Стадия 6: трет-бутил {(3S)-6-[2-(6-бромпиридин-2-карбонил)гидразинил]-6-оксогексан-3-ил}карбамат (3f)

Раствор (4S)-4-[(трет-бутоксикарбонил)амино]гексановой кислоты (3е) (260 мг, 1,12 ммоль) в ТГФ (5,6 мл, 0,2 М) охлаждают до 0°С. Раствор ангидрида пропилфосфоновой кислоты (50% раствор в EtOAc, 1,47 мл, 2,47 ммоль) добавляют к раствору при 0°С перед удалением бани, и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Затем добавляют N,N-диизопропилэтиламин (1,17 мл, 6,74 ммоль) и 6-бромпиколиногидразид (267 мг, 1,24 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Реакцию гасят водой (15 мл) и переносят в делительную воронку с EtOAc (20 мл). Слои разделяют, и органическую фазу последовательно промывают 20% лимонной кислотой (20 мл), насыщенным раствором NaHCO3 (20 мл) и насыщенным раствором соли (20 мл). Затем органический экстракт сушат над MgSO4, фильтруют и концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 100% EtOAc) с получением указанного в заголовке соединения (3f) (241 мг, выход 50%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 9,76 (шс, 1Н), 9,29 (шс, 1Н), 8,15 (д, J=6,8 Гц, 1Н), 7,76-7,71 (м, 1Н), 7,68-7,64 (м, 1Н), 4,45-4,31 (м, 1Н), 3,69 (шс, 1Н), 2,45-2,37 (м, 2Н), 2,04-1,94 (м, 1Н), 1,69-1,62 (м, 1Н), 1,49 (с, 9Н), 1,45 (шд, J=7,3 Гц, 2H), 0,99 (т, J=1,5 Гц, 3H). ЖХМС m/z (ХИАД) для (C12H17BrN4O2), 329,0 (М+Н-Вос)+.
Стадия 7: трет-бутил {(3S)-1-[5-(6-бромпиридин-2-ил)-1,3,4-оксадиазол-2-ил]пентан-3-ил}карбамат (3g)

К раствору трет-бутил {(33)-6-[2-(6-бромпиридин-2-карбонил)гидразинил]-6-оксогексан-3-ил}карбамата (3f) (241 мг, 0,561 ммоль) в ДХМ (2,2 мл, 0,25 М) добавляют триэтиламин (0,235 мл, 1,68 ммоль) и п-толуолсульфонилхлорид (128 мг, 0,673 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Добавляют этилендиамин (0,038 мл, 0,561 ммоль) для удаления избытка п-толуолсульфонилхлорида; при добавлении сразу же образовывается осадок. После перемешивания при комнатной температуре в течение 30 мин, реакционную смесь промывают 20% лимонной кислотой (5 мл) и разделяют слои. Водный слой экстрагируют ДХМ (5 мл), затем объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над MgSO4, фильтруют и концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 100% EtOAc) с получением указанного в заголовке соединения (3g) (178 мг, выход 77%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,22 (дд, J=0,7, 7,6 Гц, 1Н), 7,77-7,72 (м, 1Н), 7,68-7,65 (м, 1Н), 4,35 (шд, J=8,2 Гц, 1Н), 3,65 (шс, 1Н), 3,07 (дт, J=6,1, 10,0 Гц, 2Н), 2,21-2,11 (м, 1Н), 1,93-1,83 (м, 1Н), 1,65-1,61 (м, 1Н), 1,53-1,48 (м, 1Н), 1,46 (с, 9Н), 0,98 (т, J=7,4 Гц, 3H). ЖХМС m/z (ХИАД) для (C12H15BrN4O), 311,0 (М+Н-Вос)+.
Стадия 8: Промежуточное соединение 3
В сосуд для микроволновой печи загружают трет-бутил {(3S)-1-[5-(6-бромпиридин-2-ил)-1,3,4-оксадиазол-2-ил]пентан-3-ил}карбамат (3g) (178 мг, 0,431 ммоль) и трифторэтанол (2,16 мл, 0,2 М) и герметично закрывают перед нагреванием в микроволновой печи до 180°С в течение 60 мин. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь концентрируют, и остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 1:10 МеОН/EtOAc) с получением промежуточного соединения 3 (116 мг, выход 91%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,26 (дд, J=0,1, 1,1 Гц, 1Н), 7,67 (т, J=1,9 Гц, 1Н), 7,49 (дд, J=0,1, 7,8 Гц, 1Н), 4,90-4,83 (м, 1Н), 3,07-2,89 (м, 3H), 2,58-2,49 (м, 1Н), 2,04-1,97 (м, 1Н), 1,83-1,70 (м, 1Н), 0,96 (т, J=7,5 Гц, 3H). ЖХМС m/z (ХИАД) для (C12H13BrN4), 293,0 (М+Н)+. Определен 97,4% эи по СЖХ (10-60% метанола (0,5% NH3) в диоксиде углерода при 400-450 бар, время градиента=2 мин, скорость потока=4 мл/мин, Kromasil (R,R)Whelk-O колонка 50 мм * 3 мм * 1,8 мкм). Стереохимию определяют на основе использования (2S)-2-[(трет-бутоксикарбонил)амино]бутановой кислоты на первой стадии. Абсолютная конфигурация промежуточного соединения 3 однозначно установлена с помощью рентгеновской кристаллографии малых молекул.
В таблице 2 приведены результаты кристаллической рентгеновской дифракции, проводимой на дифрактометре Bruker APEX II Ultra CCD, оснащенном Mo Kα излучением (λ=0,71073). Кристаллы используют в том виде, в каком они были получены (выращены путем диффузии паров из смеси этилацетат:метанол (10:1). Бесцветные кристаллы размером 0,180×0,065×0,055 мм помещают на Cryoloop с маслом Paratone. Данные собирают в потоке азота при 100(2) К) с помощью φ и ω сканирования. Расстояние от кристалла до детектора составляет 45 мм при времени экспозиции 2 с (в зависимости от диапазона 2θ) при ширине сканирования 0,80°. Сбор данных на 100,0% завершен до 25,242° в θ. Данные интегрируют с использованием программного обеспечения Bruker SAINT и масштабируют с использованием программного обеспечения SADABS. Решение с помощью прямых способов (SHELXT) позволяет получить полную модель фазирования, соответствующую предложенной структуре. Все не водородные атомы уточнены анизотропно полноматричным способом наименьших квадратов (SHELXL-2014). Все атомы водорода, связанные с углеродом, размещают с использованием модели «наездника». Их положения ограничены относительно их родительского атома с помощью соответствующей команды HFIX в SHELXL-2014. Положения атомов водорода СН, NH и ОН уточняют с использованием соответствующих команд HFIX.


Промежуточное соединение 4: (S,S)-2-метил-N-[(1R)-1-{6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил}этил]пропан-2-сульфинамид
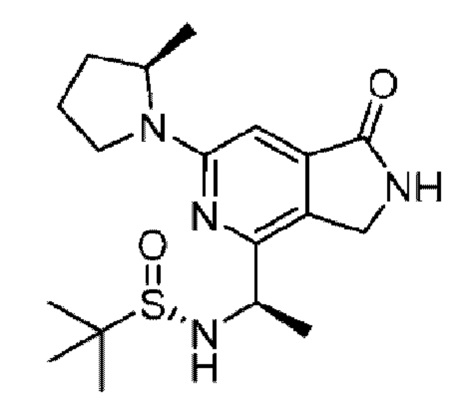
Стадия 1: {2-хлор-6-[(2R)-2-метилпирролидин-1-ил]пиридин-4-ил}(пиперидин-1-ил)метанон (4а)

Раствор (2,6-дихлорпиридин-4-ил) (пиперидин-1-ил)метанона (600 мг, 2,32 ммоль) и {2R)-2-метилпирролидина (591 мг, 6,95 ммоль) в ДМФ (1,5 мл) перемешивают при 100°С в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь охлаждают до комнатной температуры, добавляют H2O (40 мл) и реакционную смесь экстрагируют ДХМ (3×40 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (24 г SiO2, 0-20% EtOAc/гептан) с получением указанного в заголовке соединения (4а) (664 мг, выход 93%). 1Н ЯМР (400 МГц, CDCl3) δ 6,44 (д, J=1,0 Гц, 1Н), 6,21 (д, J=1,0 Гц, 1Н), 4,12 (к, J=1,1 Гц, 1Н), 3,72-3,62 (м, 2Н), 3,54 (ддд, J=10,5, 7,6, 2,9 Гц, 1Н), 3,40-3,28 (м, 2Н), 2,10-2,04 (м, 2Н), 1,75-1,62 (м, 4Н), 1,26 (т, J=7,2 Гц, 1Н), 1,21 (д, J=6,3 Гц, 2Н); m/z (ХИАД+) для (C16H22ClN3O), 308,2 (М+Н)+.
Стадия 2: 2-хлор-6-[{2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-3-карбальдегид (4b)
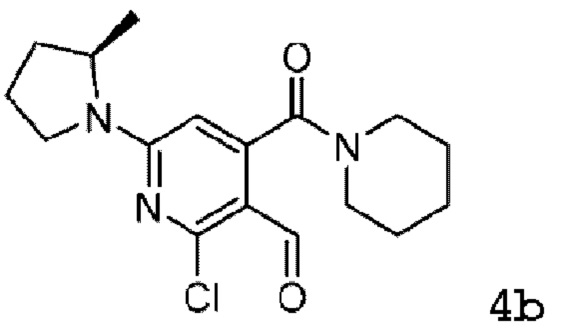
К раствору ДМФ (473 мг, 6,47 ммоль) в ДХМ (3,0 мл) добавляют POCl3 (992 мг, 6,47 ммоль). Смесь перемешивают в течение 10 мин, и затем добавляют раствор {2-хлор-6-[(2R)-2-метилпирролидин-1-ил]пиридин-4-ил}(пиперидин-1-ил)метанона (4а) (664 мг, 2,16 ммоль) в ДХМ (3,0 мл). Смесь перемешивают при температуре кипения с обратным холодильником в течение 15 часов. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь концентрируют досуха и медленно выливают в насыщенный раствор NaHCO3 (30 мл). Смесь экстрагируют ДХМ (3×30 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (24 г SiO2, 0-40% EtOAc/гептан) с получением указанного в заголовке соединения (4b) (568 мг, выход 78%). 1Н ЯМР (400 МГц, CDCl3) δ 10,07 (с, 1Н), 5,99 (с, 1Н), 4,13-4,65 (м, 1Н), 3,68-3,83 (м, 1Н), 3,55-3,68 (м, 2Н), 3,35-3,55 (м, 1Н), 2,98-3,20 (м, 2Н), 1,88-2,17 (м, 3H), 1,71-1,83 (м, 2Н), 1,55-1,67 (м, 3H), 1,46-1,55 (м, 1Н), 1,31-1,42 (м, 1Н), 1,17-1,26 (м, 3H); m/z (ХИАД+) для (C17H22ClN3O2), 336,1 (М+Н)+.
Стадия 3: N-[(Е)-{2-хлор-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-3-ил}метилиден]-2-метилпропан-2-сульфинамид (4с)

Смесь 2-хлор-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-3-карбальдегида (4b) (432 мг, 1,29 ммоль), (R)-(+)-2-метил-2-пропансульфинамид (187 мг, 1,54 ммоль) и Ti (OEt)4 (880 мг, 3,86 ммоль) в ТГФ (10,0 мл) перемешивают при 45°С в течение 16 часов. Анализ ЖХМС показал ~25% оставшегося исходного материала. Дополнительные партии (R)-(+)-2-метил-2-пропансульфинамида (62,4 мг, 0,515 ммоль) и Ti(OEt)4 (293 мг, 1,29 ммоль) и смесь перемешивают при 50°С в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию охлаждают до КТ. Смесь разбавляют ДХМ (50 мл) и промывают насыщенным раствором NaHCO3 (35 мл) и насыщенным раствором соли (35 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют, с получением указанного в заголовке соединения (4с) (495 мг, выход 88%) в виде белой смолы, которую используют без дополнительной очистки, m/z (ХИАД+) для (C21H31ClN4O2S), 440,2 (М+Н)+.
Стадия 4: 4-хлор-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он (4d)

Раствор N-[(Е)-{2-хлор-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-3-ил}метилиден]-2-метилпропан-2-сульфинамида (4 с) (495 мг, 1,13 ммоль) в ТГФ (15,0 мл) охлаждают до 0°С, и затем добавляют раствор LiBH4 (2,0 М в ТГФ, 620 мл, 1,24 ммоль). Смесь перемешивают при 0°С в течение 2 ч, затем добавляют раствор NaOMe (25% в МеОН, 2,5 мл, 10,1 ммоль). Реакционной смеси дают нагреться до комнатной температуры и затем перемешивают в течение 16 часов. Реакционную смесь разбавляют в ДХМ (60 мл) и промывают насыщенным водным раствором NH4Cl (60 мл) и насыщенным раствором соли (60 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (SiO2, 50-100% EtOAc/гептан) с получением указанного в заголовке соединения (4d) (199 мг, выход 70%) в виде бесцветной пены. 1Н ЯМР (400 МГц, CDCl3) δ 6,68 (с, 1Н), 6,45 (с, 1Н), 4,35 (с, 2Н), 4,21-4,14 (м, 1Н), 3,58 (ддд, J=10,5, 7,6, 2,8 Гц, 1Н), 3,39 (кв, J=8,9 Гц, 1Н), 2,13-1,97 (м, 2Н), 1,75 (дт, J=5,2, 2,6 Гц, 1Н), 1,23 (д, J=6,3 Гц, 3H). Предполагается, что один атом водорода скрыт пиком воды; m/z (ХИАД+) для (C12H14ClN3O), 252,3 (М+Н)+.
Стадия 5: 6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-карбонитрил (4е)

Смесь 4-хлор-6-[{2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-она (4d) (438 мг, 1,74 ммоль), Zn(CN)2 (306 мг, 2,6 ммоль), ДМФ (15 мл) и Pd(PPh3)4 (100 мг, 0,09 ммоль) нагревают до 140°С в микроволновой печи в течение 30 мин. Реакционную смесь разбавляют ДХМ и фильтруют. Осадок на фильтре промывают ДХМ. Фильтрат концентрируют в вакууме и неочищенное соединение очищают флэш-хроматографией (SiO2, 0-100% EtOAc:ДХМ/гептан, 1:1) с получением указанного в заголовке соединения (4е) в виде желтого твердого вещества (375 мг, выход 89%). 1Н ЯМР (400 МГц, CDCl3) δ 7,00-6,94 (м, 1Н), 6,46-6,32 (м, 1Н), 4,55 (с, 2H), 4,29-4,19 (м, 1Н), 3,61 (ддд, J=2,6, 7,6, 10,3 Гц, 1Н), 3,46-3,34 (м, 1Н), 2,22-2,02 (м, 3H), 1,86-1,72 (м, 1Н), 1,25 (д, J=6,4 Гц, 3H); m/z (ХИАД+) для (C13H14N4O), 243,1 (М+Н)+.
Стадия 6: 4-ацетил-6-[[2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он (4f)

К раствору 6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-карбонитрила (4е) (355 мг, 1,47 ммоль) в ТГФ (15 мл) на бане лед/вода добавляют MeMgBr (4,88 мл, 14,7 ммоль, 3,0 М в ТГФ). Полученную смесь перемешивают при этой температуре в течение 10 минут, затем нагревают до комнатной температуры и перемешивают в течение 2 часов. Смесь гасят 2 N HCl (6 мл) при 0°С и перемешивают при комнатной температуре в течение 15 мин. Смесь нейтрализуют насыщенным раствором NaHCO3 и экстрагируют ДХМ (3×40 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенное соединение очищают флэш-хроматографией (SiO2, 20-50% EtOAc/гептан) с получением указанного в заголовке соединения (4f) в виде желтого твердого вещества (190 мг, выход 50%). 1Н ЯМР (400 МГц, CDCl3) 6,98 (с, 1Н), 6,59 (шс, 1Н), 4,70 (с, 2Н), 4,28 (шт, J=5,7 Гц, 1Н), 3,64 (ддд, J=2,6, 7,5, 10,1 Гц, 1Н), 3,48-3,38 (м, 1Н), 2,70 (с, 3H), 2,19-2,10 (м, 2Н), 2,08-2,01 (м, 1Н), 1,79 (тд, J=2,6, 5,0 Гц, 1Н), 1,30 (д, J=6,2 Гц, 3H); m/z (ХИАД+) для (C14H17N3O2), 260,2 (М+Н)+.
Стадия 7: Промежуточное соединение 4
Во флакон объемом 40 мл добавляют 4-ацетил-6-[{2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он (4f) (200 мг, 0,77 ммоль), (S)-трет-бутилсульфинамид (187 мг, 1,54 ммоль), ТГФ (1,54 мл) и Ti(OEt)4 (1,54 ммоль, 0,323 мл). Флакон закрывают крышкой и нагревают до 80°С в течение 48 часов. Затем реакционную смесь охлаждают до -78°С (сухой лед/ацетоновая баня) и добавляют по каплям L-селектрид (1,54 ммоль, 1,54 мл, 1,0 М в ТГФ). Полученную смесь перемешивают при -78°С в течение 3 часов. Смесь нагревают до комнатной температуры и гасят насыщенным раствором NH4Cl (2 мл) по каплям, затем добавляют ДХМ (20 мл) и насыщенный раствор соли (20 мл). Смесь фильтруют через Celite® и промывают ДХМ (40 мл). Органический слой собирают, и водный слой экстрагируют ДХМ (20 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют.Неочищенный продукт очищают флэш-хроматографией (SiO2, растворитель 0-10% МеОН в EtOAc) с получением промежуточного соединения 4 в виде желтой пены (127 мг, выход 45%). m/z (ХИАД+) для (C18H28N4O2S), 365,3 (М+Н)+. Стереохимию определяют на основе использования {2R)-2-метилпирролидина на стадии 1 и (S)-трет-бутилсульфинамида на стадии 7.
Промежуточное соединение 5: (S,R)-2-метил-N-[(1R)-1-{6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил}пропил]пропан-2-сульфинамид
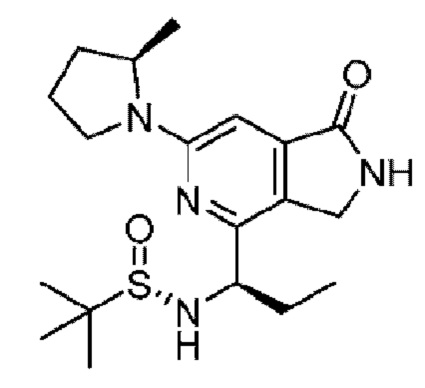
Стадия 1: 6-[(2R)-2-метилпирролидин-1-ил]-4-пропаноил-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он (5а)

Раствор 6-[{2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-карбонитрила (4е) (808 мг, 3,33 ммоль) в ТГФ (8,0 мл) охлаждают до 0°С и затем обрабатывают раствором этилмагнийбромида (3,0М в Et2O, 11,1 мл, 33,3 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа. Анализ ЖХМС показал потребление исходного материала. Реакцию охлаждают до 0°С и гасят добавлением 2 N HCl. Смесь перемешивают при комнатной температуре в течение 10 минут, нейтрализуют добавлением насыщенного раствора NaHCO3 и затем экстрагируют ДХМ (2×30 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (24 г SiO2, 0-100% EtOAc/гептан) с получением указанного в заголовке соединения (5а) (646 мг, выход 71%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,01 (с, 1Н), 6,41 (шс, 1Н), 4,73 (с, 2Н), 4,39-4,22 (м, 1Н), 3,66 (ддд, J=2,6, 7,4, 10,1 Гц, 1Н), 3,52-3,38 (м, 1Н), 3,30-3,14 (м, 2Н), 2,26-2,10 (м, 2Н), 2,06-2,01 (м, 1Н), 1,81 (тд, J=2,5, 5,1 Гц, 1Н), 1,31 (д, J=6,2 Гц, 3H), 1,24 (т, J=7,3 Гц, 3H); m/z (ХИАД+) для (C15H19N3O2), 274,2 (М+Н)+.
Стадия 2: (S,S)-2-метил-N-[(1E)-1-{6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил}пропилиден]пропан-2-сульфинамид (5b)

К раствору 6-[(2R)-2-метилпирролидин-1-ил]-4-пропаноил-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-она (5а) (640 мг, 2,34 ммоль) в ТГФ (2,0 мл) добавляют (S)-(-)-2-метил-2-пропансульфинамид (568 мг, 4,68 ммоль) и Ti (OEt)4 (2,14 г, 9,37 ммоль). Смесь перемешивают при 90°С в течение 23 часов. Анализ ЖХМС показал потребление исходного материала. Смесь охлаждают до комнатной температуры и затем добавляют солевой раствор (40 мл) и ДХМ (30 мл). Смесь перемешивают в течение 10 мин и затем фильтруют через Celite®. Слои разделяют. Водный слой экстрагируют ДХМ (30 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением указанного в заголовке соединения (5b) (779 мг, выход 88%) в виде белой пены, которую непосредственно используют на следующей стадии. m/z (ХИАД+) для (C19H28N4O2S), 377,2 (М+Н)+.
Стадия 3: Промежуточное соединение 5
Раствор (S,S)-2-метил-N-[(1E)-1-{6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил}пропилиден]пропан-2-сульфинамид (5b) (654 мг, 1,40 ммоль) в ТГФ (12,0 мл) охлаждают до -78°С, и затем обрабатывают по каплям раствором L-селектрида (1,0 М в ТГФ, 2,5 мл, 2,5 ммоль). Смесь перемешивают при -78°С в течение 1,5 часов. Добавляют дополнительное количество L-селектрида (1,0 М, 0,417 мл, 0,417 ммоль) и смесь перемешивают при -78°С еще 1,5 часа. Смесь гасят МеОН, разбавляют насыщенным раствором соли (50 мл) и экстрагируют ДХМ (2×50 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (12 г SiO2, 0-10% МеОН/ДХМ) с получением промежуточного соединения 5 (300 мг, выход 57%) в виде желтой смолы. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,66 (с, 1Н), 6,48 (с, 1Н), 5,14 (д, J=6,6 Гц, 1Н), 4,35 (с, 2Н), 4,27-4,14 (м, 2Н), 3,59-3,44 (м, 1Н), 3,29-3,18 (м, 1Н), 2,14-2,01 (м, 2Н), 1,98-1,81 (м, 3H), 1,73-1,60 (м, 1Н), 1,20 (д, J=6,2 Гц, 3H), 1,07 (с, 9Н), 0,86-0,73 (м, 3H); m/z (ХИАД+) для (C19H30N4O2S), 379,2 (М+Н)+.
Стереохимию определяют на основе использования (2R)-2-метилпирролидина на стадии 1 синтеза промежуточного соединения 4 и (S)-(-)-2-метил-2-пропансульфинамида на стадии 2. Абсолютная конфигурация промежуточного соединения 5 однозначно установлена с помощью рентгеноструктурного анализа малых молекул.
В таблице 3 представлены данные рентгеновской дифракции, подтверждающие стереохимию промежуточного соединения 5. Рентгеноструктурные исследования монокристаллов проводят на Bruker Microstar APEX II CCD дифрактометре, оснащенном Cu Кα излучением (λ=1,54178 Å). Абсолютную стереохимию определяют окончательно (Flack=0,003 (12)). Кристаллы промежуточного соединения 5 выращивают из смеси эфир/пентан при 4°С. Кусочек бесцветного кристалла размером 0,12×0,04×0,02 мм устанавливают на Cryoloop с маслом Paratone. Данные собирают в потоке газообразного азота при 100(2) К с использованием ϕ и ω сканирования. Расстояние от кристалла до детектора составляло 45 мм, и время экспозиции составляет 4, 6, 8, 10, 14 или 20 секунд в зависимости от диапазона 29 на рамку при ширине сканирования 1,25°. Сбор данных завершают на 97,0% до 67500° в θ. Данные интегрируют с использованием программного обеспечения Bruker SAINT Software и масштабируют с использованием программного обеспечения SADABS. Решение с помощью прямых способов (SHELXT) позволяет получить полную фазовую модель, соответствующую предложенной структуре. Все не водородные атомы уточняют анизотропно полноматричным способом наименьших квадратов (SHELXL-2014). Все атомы водорода, связанные с углеродом, размещены с использованием модели «наездника». Их положения ограничены относительно их родительского атома с помощью соответствующей команды HFIX в SHELXL-2014.



Промежуточное соединение 6: 4-(2-аминопропан-2-ил)-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он

К суспензии 6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-карбонитрила (4е) (213 мг, 0,88 ммоль) в ТГФ (9 мл) при комнатной температуре добавляют комплекс бис(хлорида лития) и хлорида лантана (III) (1,76 ммоль, 2,93 мл, 0,6 М). Полученную смесь перемешивают при комнатной температуре в течение 30 мин, и затем охлаждают до -78°С (баня сухой лед/ацетон) и по каплям добавляют метиллитий (3,52 ммоль, 2,20 мл, 1,6 М). Полученную смесь перемешивают при -78°С в течение 1 ч, затем нагревают до комнатной температуры. Смесь гасят насыщенным раствором NH4Cl (20 мл), затем добавляют насыщенный раствор соли (20 мл) и ДХМ (20 мл). Смесь перемешивают при комнатной температуре в течение 5 мин и фильтруют через Celite®. Органический слой собирают, и водный слой экстрагируют ДХМ (20 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (SiO2, растворитель 0-10% МеОН в ДХМ) с получением промежуточного соединения 6 в виде твердого вещества бледно-коричневого цвета (157 мг, выход 65%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,60 (с, 1Н), 6,42 (с, 1Н), 4,56 (с, 2Н), 4,17 (шт, J=5,7 Гц, 1Н), 3,50 (ддд, J=2,4, 7,2, 9,8 Гц, 1Н), 3,30-3,22 (м, 1Н), 2,07-2,02 (м, 1Н), 2,00-1,92 (м, 2Н), 1,68 (тд, J=2,4, 4,9 Гц, 1Н), 1,41 (с, 3H), 1,40 (с, 3H), 1,20 (д, J=6,2 Гц, 3H); m/z (ХИАД+) для (C15H22N4O), 275,2 (М+Н)+.
Промежуточное соединение 7: (5R)-3-(6-бромпиридин-2-ил)-5-(фторметил)-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол
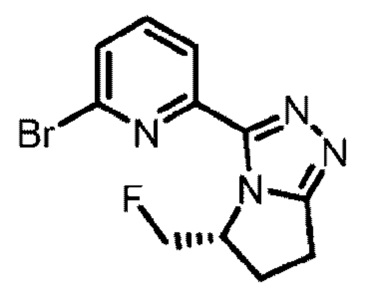
Стадия 1: (5R)-5-(фторметил)пирролидин-2-он (7а)

Раствор (5R)-5-(бромметил)пирролидин-2-она (600 мг, 3,37 ммоль) в CH3CN (6,74 мл, 0,5 М) накрывают алюминиевой фольгой для исключения света. К этому раствору одной порцией добавляют суспензию AgF (1,07 г, 8,43 ммоль) в CH3CN (12,5 мл, 0,27 М). Реакционную смесь перемешивают при комнатной температуре в темноте в течение 36 часов. Реакционную смесь фильтруют через Celite® и концентрируют досуха. Неочищенный остаток очищают флэш-хроматографией (SiO2, от 100% EtOAc до 1:1 EtOAc/ацетон) с получением указанного в заголовке соединения (7а) (173 мг, выход 44%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 7,17 (шс, 1Н), 4,49-4,17 (м, 2Н), 4,01-3,84 (м, 1Н), 2,44-2,28 (м, 2Н), 2,27-2,16 (м, 1Н), 1,88-1,75 (м, 1Н). 19F ЯМР (376 МГц, CDCl3) δ-224,58 (с, 1F).
Стадия 2: трет-бутил (2R)-2-(фторметил)-5-оксопирролидин-1-карбоксилат (7b)

К раствору (5R)-5-(фторметил)пирролидин-2-она (7а) (170 мг, 1,45 ммоль) в CH3CN (6,74 мл, 0,5 М) добавляют Boc2O (348 мг, 1,60 ммоль) и 4-(диметиламино)пиридин (17,7 мг, 0,145 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Анализ ТСХ (2:1 EtOAc:ацетон) показал потребление исходного материала. Реакционную смесь концентрируют и очищают флэш-хроматографией (SiO2, от 100% гептана до 100% EtOAc) с получением указанного в заголовке соединения (7b) (277 мг, выход 88%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 4,78-4,42 (м, 2Н), 4,40-4,25 (м, 1Н), 2,76-2,62 (м, 1Н), 2,50-2,39 (м, 1Н), 2,29-2,14 (м, 1Н), 2,13-2,03 (м, 1Н), 1,56 (с, 9Н). 19F ЯМР (376 МГц, CDCl3) δ-233,52 (с, 1F).
Стадия 3: трет-бутил[(2R)-1-фтор-5-гидразинил-5-оксопентан-2-ил]карбамат (7с)

К раствору трет-бутил (2R)-2-(фторметил)-5-оксопирролидин-1-карбоксилата (7b) (1,78 г, 8,19 ммоль) в ТГФ (41,0 мл, 0,2 М) добавляют моногидрат гидразина (1,3 мл, 41,0 ммоль) через шприц. Реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Анализ ТСХ (1:1 гептан:EtOAc) показал потребление исходного материала. Реакционную смесь концентрируют и затем добавляют EtOAc (50 мл) и насыщенный водный раствор NH4Cl (50 мл). Слои разделяют, и водный слой экстрагируют EtOAc (3×50 мл). Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (7 с) (1,6 г, выход 78%) в виде белого твердого вещества, которое используют без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 5,15-5,00 (м, 1Н), 4,52-4,44 (м, 1Н), 4,39-4,33 (м, 1Н), 3,93-3,75 (м, 1Н), 2,40 (шс, 2Н), 2,00-1,81 (м, 2Н), 1,46 (с, 9Н). 19F ЯМР (376 МГц, CDCl3) δ-232,02 (с, 1F).
Стадия 4: трет-бутил {(2R)-5-[2-(6-бромпиридин-2-карбонил)гидразинил]-1-фтор-5-оксопентан-2-ил}карбамат (7d)

Раствор 6-бромпиридин-2-карбоновой кислоты (1,4 г, 6,93 ммоль) в ТГФ (34,7 мл, 0,2 М) охлаждают до 0°С. Раствор ангидрида пропилфосфоновой кислоты (50% раствор в EtOAc, 9,1 мл, 15,2 ммоль) добавляют к раствору при 0°С, после чего баню удаляют, и реакционную смесь перемешивают в течение 30 минут при комнатной температуре. Затем добавляют N,N-диизопропилэтиламин (7,24 мл, 41,6 ммоль) и трет-бутил[(2R)-1-фтор-5-гидразинил-5-оксопентан-2-ил]карбамат (7 с) (1,90 г, 7,62 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 17 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию гасят водой (30 мл) и переносят в делительную воронку с EtOAc (50 мл). Слои разделяют и органическую фазу последовательно промывают 20% лимонной кислотой (30 мл), насыщенным раствором NaHCO3 (30 мл) и насыщенным раствором соли (30 мл). Затем органический экстракт сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (7d) (2,37 г, выход 78%) в виде желтого твердого вещества, которое используют без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 9,74 (шс, 1Н), 9,15 (шс, 1Н), 8,14 (д, J=7,5 Гц, 1Н), 7,77-7,71 (м, 1Н), 7,70-7,65 (м, 1Н), 4,95 (шд, J=9,2 Гц, 1Н), 4,62-4,36 (м, 2Н), 4,12-3,98 (м, 1Н), 2,49-2,37 (м, 2Н), 2,03-1,95 (м, 2Н), 1,49 (с, 9Н). 19F ЯМР (376 МГц, CDCl3) δ-232,01 (с, 1F). ЖХМС m/z (ХИАД) для (CnH14BrFN4O2), 333,1 (М+Н-Вос)+.
Стадия 5: трет-бутил {(2R)-4-[5-(6-бромпиридин-2-ил)-1,3,4-оксадиазол-2-ил]-1-фторбутан-2-ил}карбамат (7е)
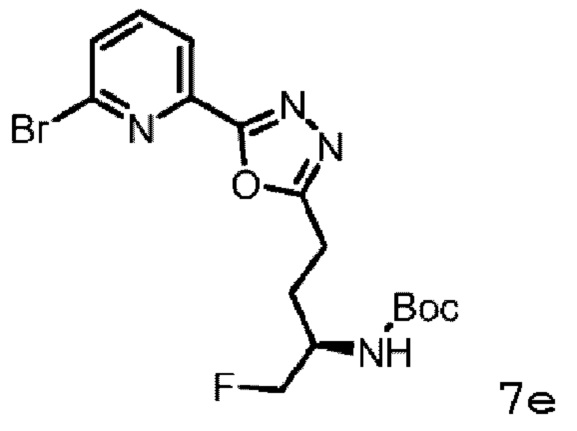
К раствору трет-бутил {(2R)-5-[2-(6-бромпиридин-2-карбонил)гидразинил]-1-фтор-5-оксопентан-2-ил}карбамата (7d) (2,3 г, 5,3 ммоль) в ДХМ (21,2 мл, 0,25 М), добавляют триэтиламин (2,22 мл, 15,9 ммоль) и л-толуолсульфонилхлорид (1,21 г, 6,37 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. Анализ ЖХМС показал потребление исходного материала. Добавляют этилендиамин (0,355 мл, 5,31 ммоль) для удаления избытка п-толуолсульфонилхлорида, во время добавления сразу же образовывался осадок. После перемешивания при комнатной температуре в течение 30 мин, реакционную смесь промывают 20% лимонной кислотой (20 мл) и разделяют слои. Водный слой экстрагируют ДХМ (40 мл), затем объединенные органические слои промывают насыщенным раствором соли (25 мл), сушат над MgSO4, фильтруют и концентрируют досуха с получением указанного в заголовке соединения (7е) (2,07 г, выход 94%).) в виде желтого твердого вещества, которое используют без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 8,21 (д, J=1,6 Гц, 1Н), 4,90-4,69 (м, 1Н), 4,55 (шд, J=2,8 Гц, 1Н), 4,43 (шс, 1Н), 4,08-3,84 (м, 1Н), 3,15-3,05 (м, 2Н), 2,29-2,06 (м, 2Н), 1,45 (с, 9Н). 19F ЯМР (376 МГц, CDCl3) δ-231,82 (с, 1F). ЖХМС m/z (ХИАД) для (C11H12BrFN4O), 315,0 (М+Н-Вос)+.
Стадия 6: Промежуточное соединение 7
В микроволновую пробирку загружают трет-бутил {(2R)-4-[5-(6-бромпиридин-2-ил)-1,3,4-оксадиазол-2-ил]-1-фторбутан-2-ил}карбамат (7е) (1,00 г, 2,41 ммоль) и трифторэтанол (10,4 мл, 0,17 М) и герметично закрывают перед нагреванием в микроволновой печи до 180°С в течение 30 мин. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь концентрируют и остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 1:10 MeOH/EtOAc) с получением промежуточного соединения 7 (564 мг, выход 78%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=7,3 Гц, 1Н), 7,79-7,73 (м, 1Н), 7,58 (д, J=1,9 Гц, 1Н), 5,26-5,01 (м, 2Н), 4,89-4,71 (м, 1Н), 3,25-3,08 (м, 3H), 2,96-2,85 (м, 1Н). 19F ЯМР (376 МГц, CDCl3) δ-235,95 (с, 1F). ЖХМС m/z (ХИАД) для (C11H10BrFN4), 297,0 (М+Н)+. Стереохимию определяют на основе использования (5R)-5-(бромметил)пирролидин-2-она на стадии 1.
Промежуточное соединение 8: трет-бутил метил({6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил}метил)карбамат

Стадия 1: трет-бутил ({3-формил-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-2-ил}метил)метилкарбамат (8а)

Раствор 2-хлор-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-3-карбальдегида (4b) (600 мг, 1,79 ммоль) и PdCl2(dppf) (261 мг, 0,357 ммоль) в 1,4-диоксане (25,0 мл) барботируют N2 в течение 5 мин и затем нагревают до 80°С. Добавляют раствор {[(трет-бутоксикарбонил)(метил)амино]метил}(хлоридо)цинка* (0,158 М в ТГФ, 39,6 мл) при 80°С и смесь перемешивают еще 35 мин при той же температуре. Анализ ЖХМС показал потребление исходного материала. Смесь охлаждают до 30°С и фильтруют через Celite®. Осадок на фильтре промывают ДХМ (5×10 мл), и фильтрат концентрируют досуха. Остаток объединяют с неочищенным продуктом, полученным в ходе параллельной реакции, идентичным образом со 100 мг 2-хлор-6-[{2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-3-карбальдегида. Смесь очищают флэш-хроматографией (SiO2, 1:1 EtOAc/петролейный эфир) с получением указанного в заголовке соединения (8а) (900 мг, выход 97%) в виде желтой смолы. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,84 (с, 1Н), 6,40-6,06 (м, 1Н), 4,91-4,45 (м, 2Н), 4,52-3,92 (м, 1Н), 3,69-3,47 (м, 3H), 3,21-3,00 (м, 2Н), 2,98-2,84 (м, 3H), 2,22-1,82 (м, 3H), 1,83-1,66 (м, 1Н), 1,59 (с, 4Н), 1,41 (с, 7Н), 1,19 (д, J=18,2 Гц, 8Н). m/z (ИЭР+) для (C24H36N4O4), 445,4 (М+Н)+.
* как получено в: Angew. Chem. Inf. Ed. 2014, 53, 2678.
Стадия 2: трет-бутил метил{[3-{{Е)-[(2-метилпропан-2-сульфинил)имино]метил}-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-2-ил]метил}карбамат (8b)

Смесь трет-бутил ({3-формил-6-[(2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-2-ил}метил)метилкарбамата (8а) (1,40 г, 3,15 ммоль), Ti (OEt)4 (1,44 г, 6,30 ммоль) и (S)-{-)-2-метил-2-пропансульфинамид (573 мг, 4,72 ммоль) в ТГФ (50,0 мл) перемешивают при 50°С в течение 18 часов. Добавляют дополнительные партии Ti(OEt)4 (359 мг, 1,57 ммоль) и (S)-(-)-2-метил-2-пропансульфинамида (115 мг, 0,945 ммоль) и смесь перемешивают при 50°С в течение дополнительных 20 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию гасят насыщенным раствором Na2CO3 (150 мл) и добавляют ДХМ (100 мл). Смесь фильтруют через Celite® и слои разделяют. Водный слой экстрагируют ДХМ (100 мл). Объединенные органические слои промывают насыщенным раствором соли (150 мл), сушат над Na2SO4, фильтруют и концентрируют с получением указанного в заголовке соединения (8b) (1,7 г, выход >99%) в виде желтого твердого вещества, m/z (ИЭР+) для (C28H45N5O4S), 548,5 (М+Н)+.
Стадия 5: Промежуточное соединение 8
К раствору трет-бутил метил{[3-{[Е)-[(2-метилпропан-2-сульфинил)имино]метил}-6-[{2R)-2-метилпирролидин-1-ил]-4-(пиперидин-1-карбонил)пиридин-2-ил]метил}карбамата (8b) (1,72 г, 3,41 ммоль) в ТГФ (20,0 мл) при 0°С добавляют LiBH4 (68,6 мг, 3,15 ммоль). Реакционную смесь перемешивают при 0°С в течение 1 часа. Анализ ТСХ показал потребление исходного материала. Смесь нагревают до комнатной температуры и добавляют раствор NaOMe (30% в МеОН, 6,24 г, 34,6 ммоль). Смесь перемешивают в течение 16 часов. Анализ ЖХМС показал образование желаемой массы продукта. Реакцию концентрируют досуха. Остаток растворяли в EtOAc (40 мл) и промывают H2O (40 мл). Водный слой экстрагируют EtOAc (30 мл). Объединенные органические слои промывают насыщенным раствором соли (60 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (SiO2, EtOAc) с получением промежуточного соединения 8 (750 мг, выход 66%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,04-6,79 (м, 1Н), 6,68 (с, 1Н), 4,56-4,43 (м, 2Н), 4,43-4,33 (м, 2Н), 4,27-4,16 (м, 1Н), 3,58 (ддд, J=2,5, 7,3, 10,0 Гц, 1Н), 3,45-3,30 (м, 1Н), 3,01-2,91 (м, 3H), 2,17-1,96 (м, 3H), 1,80-1,72 (м, 1Н), 1,53-1,37 (м, 9Н), 1,26-1,23 (м, 3H); m/z (ИЭР+) для (C19H28N4O3), 361,2 (М+Н)+.
Стереохимию определяют на основе использования (2R)-2-метилпирролидина на стадии 1 синтеза промежуточного соединения 4.
Промежуточное соединение 9: трет-бутилметил ({6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил}метил)карбамат

Стадия 1: 2-хлор-N,N-диметил-6-[метил (пропан-2-ил)амино]пиридин-4-карбоксамид (9а)

Смесь 2, 6-дихлор-N,N-диметилпиридин-4-карбоксамида (30,0 г, 137 ммоль) и N-метилпропан-2-амина (50,1 г, 685 ммоль) в MeCN (120 мл) распределяют по трем закрытым реакционным сосудам, и каждый перемешивают при 100°С в течение 60 часов. Анализ ЖХМС показал потребление исходного материала. Реакционные смеси объединяют и концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, 1:1 EtOAc/петролейный эфир) с получением указанного в заголовке соединения (9а) (30,5 г, выход 87%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 6,45 (д, J=0,9 Гц, 1Н), 6,31 (д, J=1,0 Гц, 1Н), 4,82 (п, J=6,8 Гц, 1Н), 3,08 (с, 3H), 2,97 (с, 3H), 2,83 (с, 3H), 1,16 (д, J=6,1 Гц, 6Н); m/z (ИЭР+) для (C12H18ClN3O), 255,9 (М+Н)+.
Стадия 2: 2-хлор-3-формил-N,N-диметил-6-[метил (пропан-2-ил)амино]пиридин-4-карбоксамид (9b)
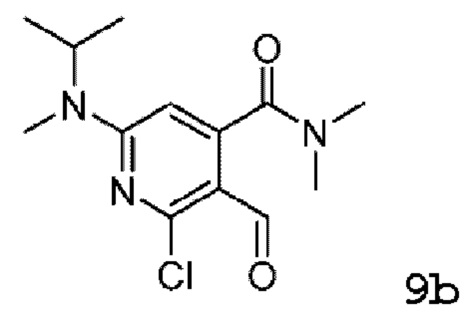
К раствору ДМФ (21,9 г, 299 ммоль) в ДХЭ (120 мл) по каплям добавляют POCl3 (45,9 г, 299 ммоль) при 5-15°С. Смесь перемешивают при комнатной температуре в течение 15 мин и добавляют 2-хлор-N,N-диметил-6-[метил(пропан-2-ил)амино]пиридин-4-карбоксамид (9а) (25,5 г, 99,7 ммоль). Реакционную смесь перемешивают при 65°С в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь охлаждают до комнатной температуры и добавляют по каплям к насыщенному водному раствору Na2CO3 (900 мл). Смесь экстрагируют ДХМ (2×300 мл). Объединенные органические слои промывают насыщенным раствором соли (5×500 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (SiO2, 1:1 EtOAc/петролейный эфир) с получением указанного в заголовке соединения (9b) (23,7 г, выход 84%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 10,19 (с, 1Н), 6,25 (шс, 1Н), 3,12 (с, 3H), 3,02-2,85 (м, 3H), 2,77 (с, 3H), 1,22 (шд, J=6,5 Гц, 6Н); m/z (ИЭР+) для (C13H18ClN3O2), 283,9 (М+Н)+.
Стадия 3: 2-хлор-N,N-диметил-3-{(Е)-[(2-метилпропан-2-сульфинил)имино]метил}-6-[метил(пропан-2-ил)амино]пиридин-4-карбоксамид (9с)

Смесь 2-хлор-3-формил-N,N-диметил-6-[метил (пропан-2-ил)амино]пиридин-4-карбоксамида (9b) (23,7 г, 83,5 ммоль), (R)-(+)-2-метил-2-пропансульфинамид (12,1 г, 100 ммоль) и Ti(OEt)4 (38,1 г, 167 ммоль) в ТГФ (250 мл) перемешивают при 50°С в течение 20 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию концентрируют досуха. Остаток перемешивают с насыщенным раствором NaHCO3 (300 мл) в течение 30 мин. Смесь фильтруют. Осадок на фильтре промывают H2O (3×80 мл) и петролейным эфиром (3×50 мл) и сушат в вакууме с получением указанного в заголовке соединения (9с) (32,3 г, выход 99%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,07 (с, 1Н), 6,74 (с, 1Н), 5,36-5,10 (м, 1Н), 3,30 (с, 3H), 3,26 (с, 3H), 3,10 (с, 3H), 1,54 (д, J=6,7 Гц, 6Н), 1,49 (с, 9Н); m/z (ИЭР+) для (C17H27ClN4O2S), 387,2 (М+Н)+.
Стадия 4: 4-хлор-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он (9d)

Раствор 2-хлор-N,N-диметил-3-{[Е)-[(2-метилпропан-2-сульфинил)имино]метил}-6-[метил(пропан-2-ил)амино]пиридин-4-карбоксамида (9 с) (32,3 г, 83,5 ммоль) в ТГФ (200 мл) охлаждают до 0°С и добавляют LiBH4 (1,82 г, 83,5 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа. Анализ ЖХМС показал потребление исходного материала. Добавляют NaOMe (165 г, 919 ммоль) и смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь фильтруют и осадок на фильтре промывают EtOAc (3×200 мл). Объединенный фильтрат концентрируют досуха. Остаток растворяют в ДХМ (300 мл) и промывают H2O (500 мл). Водный слой экстрагируют ДХМ (2×300 мл). Объединенные органические слои промывают насыщенным раствором соли (500 мл), сушат над Na2SO4, фильтруют и концентрируют. Твердое вещество суспендируют в смеси ДХМ (50 мл) и петролейного эфира (120 мл) в течение 30 мин. Твердые вещества собирают фильтрацией. Осадок на фильтре сушат в вакууме, с получением указанного в заголовке соединения (9d) (11,3 г, выход 56%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,19 (с, 1Н), 6,79 (с, 1Н), 4,82 (п, J=6,7 Гц, 1Н), 4,35 (д, J=1,2 Гц, 2Н), 2,88 (с, 3H), 1,18 (д, J=6,7 Гц, 6Н); m/z (ИЭР+) для (C11H14ClN3O), 239,9 (М+Н)+.
Стадия 5: метил-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-карбоксилат (9е)

Смесь 4-хлор-6-[метил (пропан-2-ил) амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-она (9d) (11,3 г, 47,1 ммоль), PdCl2(dppf) (2,16 г, 2,95 ммоль) и ТЭА (14,3 г, 141 ммоль) в МеОН (200 мл) перемешивают при 80°С в течение 40 ч в атмосфере СО при 50 фунт/кв. дюйм. Анализ ТСХ (1:1 EtOAc/петролейный эфир) показал потребление исходного материала. Реакцию концентрируют досуха. Остаток растворяют в H2O (200 мл) и экстрагируют ДХМ (2×150 мл). Объединенные органические слои промывают насыщенным раствором соли (200 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток суспендируют в ДХМ (50 мл) в течение 30 мин. Твердые вещества собирают фильтрацией. Осадок на фильтре промывают петролейным эфиром (3×5 мл) и сушат в вакууме. Фильтрат очищают флэш-хроматографией (SiO2, 40-70% EtOAc/ДХМ). Фракции, содержащие продукт, концентрируют досуха и объединяют с указанным выше осадком на фильтре с получением указанного в заголовке соединения (9е) (12,3 г, выход 99%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,12 (с, 1Н), 6,79 (с, 1Н), 4,89 (п, J=6,6 Гц, 1Н), 4,68 (д, J=1,1 Гц, 2Н), 3,97 (с, 3H), 2,96 (с, 3H), 1,21 (д, J=6,7 Гц, 6Н); m/z (ИЭР+) для (C13H17N3O3), 263,9 (М+Н)+.
Стадия 6: 4-(гидроксиметил)-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он (9f)
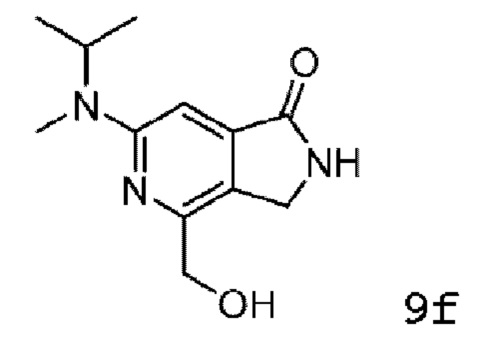
К смеси метил 6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-карбоксилата (9е) (1,0 г, 3,80 ммоль) в ТГФ (60 мл) добавляют по каплям раствор LiAlH4 (2,5 М в ТГФ, 1,67 мл, 4,18 ммоль) при 0°С. Смесь перемешивают при 0°С в течение 1 часа, и затем при 20°С в течение 16 часов. Анализ ТСХ (1:1 EtOAc/петролейный эфир) показал потребление исходного материала. Смесь гасят добавлением 20% водного раствора NaOH (0,5 мл). К смеси добавляют Na2SO4 (4 г). Смесь перемешивают в течение 30 мин и затем фильтруют.Фильтрат концентрируют досуха с получением указанного в заголовке соединения (9f) (890 мг, выход 99%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,04 (с, 1Н), 6,82 (с, 1Н), 4,85 (п, J=6,6 Гц, 1Н), 4,67 (д, J=4,7 Гц, 2Н), 4,33 (с, 2Н), 4,09 (т, J=4,6 Гц, 1Н), 2,92 (с, 3H), 1,20 (д, J=6,7 Гц, 6Н); m/z (ИЭР+) для (C12H17N3O2), 236,0 (М+Н)+.
Стадия 7: {6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил}метилметансульфонат (9g)

К смеси 4-(гидроксиметил)-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-она (9f) (890 мг, 3,78 ммоль) и ТЭА (957 мг, 9,46 ммоль) в ТГФ (20,0 мл) добавляют по каплям MsCl (953 мг, 8,23 ммоль) при 0°С в атмосфере N2. Смесь перемешивают при 0°С. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь разбавляют насыщенным водным раствором Na2CO3 (30 мл) и экстрагируют EtOAc (2×20 мл). Объединенные органические слои промывают насыщенным раствором соли (30 мл), сушат над Na2SO4, фильтруют и концентрируют с получением указанного в заголовке соединения (9g) (1,2 г, выход 99%) в виде желтого твердого вещества, которое используют без дальнейшей очистки, m/z (ИЭР+) для (C13H19N3O4S), 314,0 (М+Н)+.
Стадия 8: 4-[(метиламино)метил]-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-он (9h)

К смеси {6-[метил (пропан-2-ил) амино]-1-оксо-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил}метилметансульфоната (9g) (1,18 г, 3,78 ммоль) в ТГФ (20,0 мл) добавляют раствор метиламина (2,0 М в ТГФ, 37,8 мл, 75,6 ммоль). Смесь перемешивают в течение 1 часа. Анализ ЖХМС показал потребление исходного материала. Смесь концентрируют досуха с получением указанного в заголовке соединения (9h) (940 мг, выход 99%) в виде коричневого твердого вещества, которое используют без дополнительной очистки. m/z (ИЭР+) для (C13H20N4O), 249,0 (М+Н)+.
Стадия 9: Промежуточное соединение 9
К раствору 4-[(метиламино)метил]-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-она (9h) (940 мг, 11,4 ммоль) и ТЭА (1,15 г, 11,4 ммоль) в ДХМ (20,0 мл) добавляют Вос20 (1,65 мг, 7,57 ммоль). Смесь перемешивают в течение 30 мин. Анализ ЖХМС показал потребление исходного материала. Реакцию концентрируют досуха. Остаток очищают флэш-хроматографией (SiO2, EtOAc) с получением промежуточного соединения 9 (600 мг, выход 46%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 6,81 (с, 1Н), 6,57-6,42 (м, 1Н), 4,91 (п, J=6,6 Гц, 1Н), 4,48 (с, 2Н), 4,35 (д, J=12,4 Гц, 2Н), 2,92 (с, 3H), 2,88 (с, 3H), 1,48 (с, 5Н), 1,41 (с, 4Н), 1,17 (д, J=6,7 Гц, 6Н); m/z (ИЭР+) для (C18H28N4O3), 349,2 (М+Н)+.
Промежуточное соединение 10: (S,S)-2-метил-N-[(1R)-1-{6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил}пропил]пропан-2-сульфинамид

Стадия 1: 6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-карбонитрил
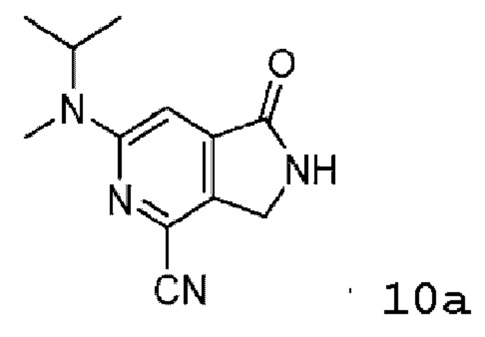
Смесь 4-хлор-6-[метил (пропан-2-ил) амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-она (9d) (706 мг, 2,95 ммоль), Zn(CN)2 (519 мг, 4,42 ммоль), ДМФ (10 мл) и Pd(PPh3)4 (170 мг, 0,147 ммоль) нагревают до 140°С в микроволновой печи в течение 30 мин. Реакционную смесь разбавляют ДХМ и фильтруют. Осадок на фильтре промывают ДХМ. Фильтрат концентрируют в вакууме с получением указанного в заголовке соединения (10а) в виде желтого твердого вещества (544 мг, выход 80%), которое используют без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО-d6) 9,01 (с, 1Н), 7,13 (с, 1Н), 4,80 (тд, J=6,7, 13,4 Гц, 1Н), 4,47 (с, 2Н), 2,89 (с, 3H), 1,15 (д, J=6,7 Гц, 6Н); m/z (ХИАД+) для (C12H14N40), 231,2 (М+Н)+.
Стадия 2: 6-[метил(пропан-2-ил)амино]-4-пропаноил-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он (10b)
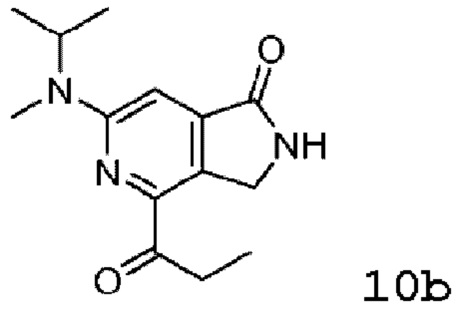
Суспензия 6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-карбонитрила (10а) (511 мг, 2,22 ммоль) в ТГФ (20,0 мл) охлаждают до 0°С и затем обрабатывают раствором этилмагнийбромида (3,0 М в Et2O, 7,40 мл, 22,2 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь охлаждают до 0°С и гасят добавлением насыщенного NH4Cl (60 мл), и затем экстрагируют ДХМ (2×80 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (24 г SiO2, 0-100% EtOAc/гептан) с получением указанного в заголовке соединения (10b) (125 мг, выход 22%) в виде желтой пены, m/z (ХИАД+) для (C14H19N3O2), 262,2 (М+Н)+.
Стадия 3: Промежуточное соединение 10
К раствору 6-[метил(пропан-2-ил)амино]-4-пропаноил-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-она (10b) (124 мг, 0,475 ммоль) в ТГФ (6,0 мл) добавляют (S)-(-)-2-метил-2-пропансульфинамид (92 мг, 0,759 ммоль) и Ti(OEt)4 (433 мг, 1,90 ммоль). Смесь перемешивают при температуре кипения с обратным холодильником в течение 42 часов. Анализ ЖХМС показал потребление исходного материала. Смесь охлаждают до -78°С и затем обрабатывают по каплям раствором L-селектрида (1,0 М в ТГФ, 1,9 мл, 1,90 ммоль). Смесь перемешивают при -78°С в течение 4 часов. Смесь гасят МеОН, разбавляют насыщенным раствором соли (30 мл) и экстрагируют ДХМ (20×2 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (12 г SiO2, 0-10% МеОН/ДХМ) с получением промежуточного соединения 10 (72 мг, выход 41%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,67 (с, 1Н), 6,64 (с, 1Н), 5,25 (д, J=6,7 Гц, 1Н), 4,94 (кв., J=6,7 Гц, 1Н), 4,34 (с, 2Н), 4,21 (кв, J=6,7 Гц, 1Н), 2,84 (с, 3H), 1,98-1,80 (м, 2Н), 1,13 (дд, J=3,6, 6,7 Гц, 6Н), 1,06 (с, 9Н), 0,83 (т, J=1,3 Гц, 3H). m/z (ХИАД+) для (C18H30N4O2S), 367,2 (М+Н)+. Стереохимию определяют на основе использования (S)-(-)-2-метил-2-пропансульфинамида на стадии 2.
Промежуточное соединение 11: трет-бутил{[6-(диметиламино)-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил]метил}метилкарбамат.

Стадия 1: 2-хлор-6-(диметиламино)-N,N-диметилпиридин-4-карбоксамид (11а)
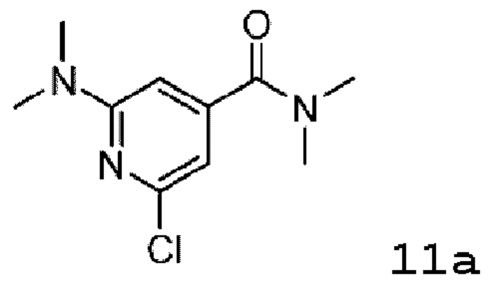
В 3,0 л круглодонную колбу, загруженную метил-2,6-дихлорпиридин-4-карбоксилатом (58,0 г, 281 ммоль) в атмосфере азота, добавляют N,N-диметиламин (38,1 г, 845 ммоль) при 0-10°С. Добавляют ТГФ (200 мл). Раствор i-PrMgCl (2,0 М в ТГФ, 352 мл, 704 ммоль) добавляют в течение 3 ч, поддерживая температуру реакции на уровне 0-10°С. Реакционную смесь перемешивают еще 10 минут при 0°С, а затем при 25°С в течение 18 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию охлаждают на бане со льдом и гасят добавлением холодного насыщенного водного раствора NH4Cl (500 мл), поддерживая температуру реакции <20°С. Добавляют EtOAc (500 мл) и слои разделяют. Водный слой экстрагируют EtOAc (500 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Полученное масло поглощали гептаном (200 мл) и концентрируют на роторном испарителе до образования твердого вещества. Суспензию перемешивают в течение 0,5 ч и собирают твердые вещества фильтрацией. Осадок на фильтре промывают гексаном (3×50 мл). Осадок на фильтре суспендируют в смеси EtOAc/петролейный эфир 1:20 (100 мл) и собирают твердые вещества фильтрацией. Осадок на фильтре промывают смесью 1:20 EtOAc/петролейный эфир (3×30 мл) и затем сушат в вакууме с получением указанного в заголовке соединения (11а) (51 г, выход 80%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 6,49 (д, J=0,7 Гц, 1Н), 6,35 (д, J=0,9 Гц, 1Н), 3,13-3,09 (м, 9Н), 2,98 (с, 3H); m/z (ИЭР+) для (C10H14ClN3O), 227,9 (М+Н)+.
Стадия 2: 2-хлор-6-(диметиламино)-3-формил-N,N-диметилпиридин-4-карбоксамид (11b)

Параллельно проводят две серии реакций. В круглодонную колбу, содержащую ДМФ (250 мл), при перемешивании добавляют POCl3 (85,9 г, 560 ммоль) при 15-25°С. Смесь перемешивают при 15-25°С в течение 15 мин и затем добавляют 2-хлор-6-(диметиламино)-N,N-диметилпиридин-4-карбоксамид (11а) (25,5 г, 112 ммоль). Смесь перемешивают при 50°С в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Две реакции объединяют и затем гасят, медленно выливая в холодный водный насыщенный раствор Na2CO3, поддерживая рН ~9. Смесь экстрагируют EtOAc (4×1,0 л). Объединенные органические слои промывают насыщенным раствором соли (5×600 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт объединяют с двумя дополнительными реакциями, проведенными аналогичным образом с 7,5 г и 5,0 г 2-хлор-6-(диметиламино)-N,N-диметилпиридин-4-карбоксамида. Материал помещают в EtOAc (200 мл) и суспендируют в течение 20 мин. Суспензию фильтруют. Осадок на фильтре промывают EtOAc (2×50 мл). Осадок на фильтре суспендируют в петролейном эфире/EtOAc (1:1) (80 мл) в течение 20 мин. Суспензию фильтруют, и осадок на фильтре промывают петролейным эфиром/EtOAc (1:1) (60 мл). Осадок на фильтре сушат под вакуумом. Объединенный фильтрат концентрируют досуха. Остаток суспендируют петролейным эфиром/EtOAc (100 мл) в соотношении 1:1 в течение 30 мин. Суспензию фильтруют и осадок на фильтре промывают петролейным эфиром/EtOAc (1:1) (2×50 мл) и сушат в вакууме. Объединенные высушенные твердые вещества суспендируют в петролейном растворе (200 мл) в течение 10 мин и собирают путем фильтрации. Осадок на фильтре промывают петролейным эфиром (100 мл) и затем концентрируют в вакууме. Объединенный фильтрат концентрируют под вакуумом до ~50 мл и затем оставляли стоять на 2 дня. Полученные твердые вещества собирают фильтрацией и осадок на фильтре промывают петролейным эфиром/EtOAc (3:2) (2×50 мл). Твердые вещества объединяют с указанным в заголовке соединением (11b) (52 г, выход 73% для объединенного набора реакций) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 10,21 (д, J=0,6 Гц, 1Н), 6,28 (д, J=0,6 Гц, 1Н), 3,19 (с, 6Н), 3,13 (с, 3H), 2,77 (с, 3H); m/z (ИЭР+) для (C11H14ClN3O2), 255,9 (М+Н)+.
Стадия 3: трет-бутил {[6-(диметиламино)-4-(диметилкарбамоил)-3-формилпиридин-2-ил]метил}метилкарбамат (11с)
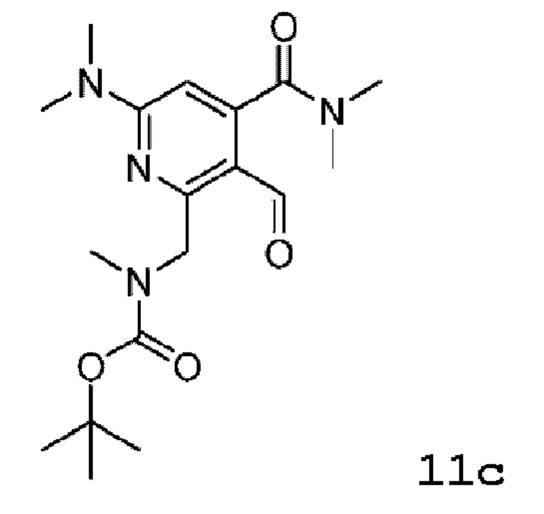
Смесь трет-бутил диметилкарбамата (3,41 г, 23,5 ммоль) и N,N,N,N-тетраметилендиамина (3,27 г, 28,2 ммоль) в 135 мл ТГФ охлаждают до -55°С в атмосфере N2. Медленно добавляют раствор s-BuLi (1,4 М в циклогексане, 20,1 мл, 28,2 ммоль), поддерживая температуру раствора ниже -52°С (внутренняя). Смесь перемешивают еще 30 мин при -55°С, и затем обрабатывают раствором ZnCl2 (1,9 М в 2-метилтетрагидрофуране, 14,8 мл, 28,2 ммоль), поддерживая температуру реакции ниже -52°С. Раствор перемешивают в течение дополнительных 40 мин при -55°С, и затем нагревают до комнатной температуры с получением раствора {[{трет-бутоксикарбонил)(метил)амино]метил}(хлоридо)цинка (С=0,195 М). Часть предварительно приготовленного раствора цинката (90,2 мл, 17,6 ммоль) переносят в высушенную в печи круглодонную колбу на 250 мл в атмосфере N2 и концентрируют досуха с получением белой пены. Колбу обратно заполняют N2. В отдельную колбу загружают 2-хлор-6-(диметиламино)-3-формил-N,N-диметилпиридин-4-карбоксамид (11b) (3,0 г, 10 ммоль), PdCl2(dppf) (0,858 г, 1,17 ммоль), 1,4-диоксан (50 мл) и H2O (0,159 г, 8,8 ммоль). Суспензию переносят в колбу, содержащую цинкат, через канюлю, и затем смесь перемешивают при 80°С в течение 80 мин. ЖХМС показала образование массы продукта с некоторым оставшимся исходным материалом. Добавляют дополнительную аликвоту раствора {[{трет-бутоксикарбонил)(метил)амино]метил}(хлоридо)цинка (2,0 мл) и смесь перемешивают при 80°С в течение 20 мин. Дополнительной конверсии не наблюдается. Реакцию охлаждают до 0°С и гасят добавлением насыщенного водного раствора NH4Cl (10 мл) и H2O (20 мл). Смесь перемешивают при 0°С в течение 20 мин и затем фильтруют через слой Celite®. Фильтрат экстрагируют EtOAc (4х). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (80 г SiO2, 0-100% EtOAc/гептан). Полученную белую пену растирают с МТБЭ и концентрируют в вакууме с получением указанного в заголовке соединения (11с) (3,8 г, выход 95%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,84 (д, J=6,8 Гц, 1Н), 6,45 (с, 1Н), 4,71 (с, 2Н), 3,15 (с, 6Н), 2,99 (с, 3H), 2,90 (с, 3H), 2,75 (с, 3H), 1,33 (д, J=69,8 Гц, 9Н), m/z (ИЭР+) для (C18H28N4O4), 365,3 (М+Н)+.
Стадия 4: трет-бутил {[6-(диметиламино)-4-(диметилкарбамоил)-3-{(Е)-[(2-метилпропан-2-сульфинил)имино]метил}пиридин-2-ил]метил}метилкарбамат (11d)
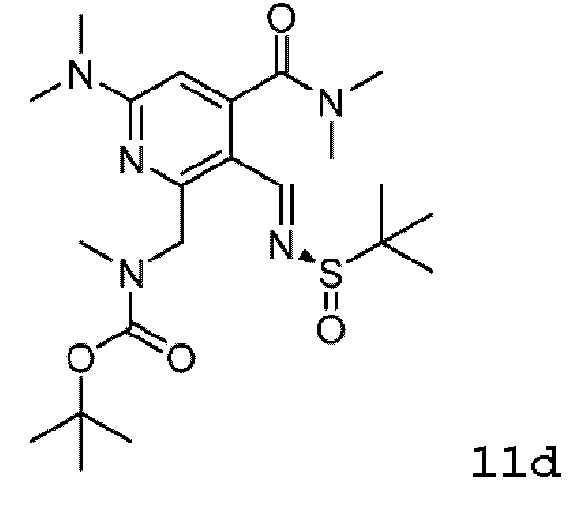
К раствору трет-бутил {[6-(диметиламино)-4-(диметилкарбамоил)-3-формилпиридин-2-ил]метил}метилкарбамата (11 с) (3,0 г, 8,0 ммоль) и добавляют (R)-(+)-2-метил-2-пропансульфинамид (1,2 г, 9,88 ммоль) в ТГФ (40 мл) Ti(OEt)4 (5,63 г, 24,7 ммоль). Смесь перемешивают при 50°С в течение ночи. Реакционную смесь охлаждают до комнатной температуры, разбавляют ДХМ (50 мл) и гасят добавлением насыщенного раствора NaHCO3 (20 мл). Раствор энергично перемешивают в течение 20 мин и затем фильтруют через слой Celite®. Celite® промывают ДХМ (3 раза). Объединенный фильтрат сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (80 г SiO2, 0-100% EtOAc/гептан) с получением указанного в заголовке соединения (lid) (3,89 г, выход 97%) в виде бесцветной пены. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,39 (с, 1Н), 6,46 (с, 1Н), 4,77-4,55 (м, 2Н), 3,12 (с, 6Н), 2,95 (с, 3H), 2,93 (с, 3H), 2,74 (с, 3H), 1,41 (с, 4Н), 1,20 (с, 5Н), 1,12 (с, 9Н); m/z (ИЭР+) для (C22H37N5O4S), 468,4 (М+Н)+.
Стадия 5: Промежуточное соединение 11
В круглодонную колбу загружают трет-бутил {[6-(диметиламино)-4-(диметилкарбамоил)-3-{(Е)-[(2-метилпропан-2-сульфинил)имино]метил}пиридин-2-ил]метил}метилкарбамат (11d) (3,89, 8,32 ммоль) в атмосфере N2 добавляют ТГФ (42 мл). Смесь охлаждают до 0°С и затем обрабатывают раствором NBH4 (2,0 М в ТГФ, 4,37 мл, 8,73 ммоль). Смесь перемешивают при 0°С в течение 1 ч, а затем при той же температуре добавляют раствор NaOMe (25% в МеОН, 17,1 мл, 74,9 ммоль). Реакционной смеси дают медленно нагреться до комнатной температуры и перемешивают в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Смесь разбавляют ДХМ и промывают насыщенным водным раствором NH4Cl и насыщенным раствором соли. Органический слой сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (80 г SiO2, 0-100% EtOAc/гептаны) с получением промежуточного соединения 11 (1,7 г, выход 64%) в виде бесцветной пены. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,68 (с, 1Н), 6,68 (с, 1Н), 4,42 (с, 2Н), 4,23 (с, 2Н), 3,06 (с, 6Н), 2,86 (с, 3H), 1,36 (м, 9Н); ЖХМС m/z (ИЭР+) для (C16H24N4O3), 321,2 (М+Н)+.
Промежуточное соединение 12: 2-бром-6-(4-пропил-4H-1,2,4-триазол-3-ил)пиридин.

Смесь N'-[(6-бромпиридин-2-ил) карбонил]-N,N-диметилгидразоноформамида (1b) (29,0 г, 106,8 ммоль) и пропан-1-амина (31,6 г, 534 ммоль) в MeCN (440 мл) и уксусную кислоту (110 мл) перемешивают при 95°С в течение 16 часов. Анализ ЖХМС показал потребление исходного материала. Реакцию концентрируют досуха. Остаток помещают в H2O (50 мл) и подщелачивают до рН ~9 с помощью 1 N NaOH (~500 мл). Смесь экстрагируют EtOAc (3×150 мл). Объединенные органические слои промывают насыщенным раствором соли (150 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток суспендируют с EtOAc (50 мл) в течение 10 мин, и твердое вещество собирают фильтрацией. Осадок на фильтре промывают петролейным эфиром (2×50 мл) и сушат в вакууме с получением указанного в заголовке соединения (промежуточное соединение 12) (21,0 г, выход 74%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (с, 1Н), 8,20 (дд, J=0,7, 7,8 Гц, 1Н), 7,98-7,91 (м, 1Н), 7,78 (дд, J=0,7, 8,0 Гц, 1Н), 4,45-4,36 (м, 2Н), 1,77 (секст, J=7,4 Гц, 2Н), 0,87 (т, J=1,4 Гц, 3H); m/z (ИЭР+) для (C10H11BrN4), 266,7 (М+Н)+.
Промежуточное соединение 13: (S,S)-2-метил-N-[(1R)-1-{6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил}этил]пропан-2-сульфинамид
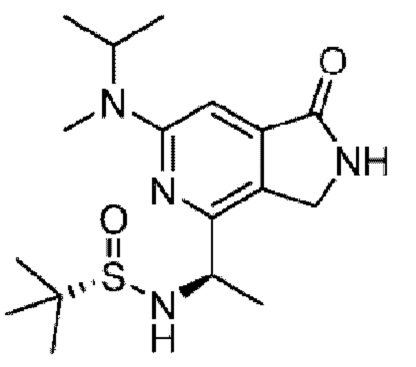
Стадия 1: 4-ацетил-6-(изопропил(метил)амино)-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он

Бромид метилмагния (9,11 г, 76,4 ммоль, 25,9 мл, 3,0 М) добавляют к раствору 6-(изопропил(метил)амино)-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-карбонитрила (промежуточное соединение 10а) (1,76 г, 7,64 ммоль) в ТГФ (70 мл, с=0,11 М) при 0°С. Смесь перемешивают при этой температуре в течение 10 минут, затем давали нагреться до комнатной температуры и перемешивают в течение 3 часов. Затем смесь гасят по каплям 2 М HCl (7 мл) при комнатной температуре и перемешивают в течение 30 мин. Добавляют насыщенный раствор NaHCO3 (70 мл) для нейтрализации раствора и экстрагируют ДХМ (3×100 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют в вакууме. Полученный остаток очищают флэш-хроматографией (40 г SiO2, 10-40% EtOAc/гептан) с получением указанного в заголовке соединения (13а) в виде желтого твердого вещества (553 мг, выход 29%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,82 (с, 1Н) 7,06 (с, 1Н) 4,88 (шд, J=6,12 Гц, 1Н) 4,49 (с, 2Н) 2,94 (с, 3H) 2,61 (с, 3H) 1,19 (д, J=6,60 Гц, 6Н); m/z (ХИАД+) для (C13H17N3O2), 248,2 (М+Н)+.
Стадия 2: Промежуточное соединение 13
Смесь 4-ацетил-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-она (13а) (8,10 г, 32,75 ммоль), (S)-трет-бутилсульфинамида (7,94 г, 65,5 ммоль) в Ti(OEt)4 (29,9 г, 131 ммоль) в ТГФ (100 мл) в атмосфере N2 нагревают при 90°С в течение 72 ч и реакцию контролируют с помощью ЖХМС. Затем реакционную смесь охлаждают до 0°С и по каплям добавляют L-селектрид (1 М в ТГФ, 131 ммоль, 131 мл) при 0°С. Смесь перемешивают при 0°С в течение 3 часов. Смесь гасят насыщенным NH4Cl (200 мл) и насыщенным раствором соли (200 мл) при 0-5°С. Суспензию фильтруют через слой Celite® и осадок на фильтре промывают ДХМ (500 мл). Слои фильтрата разделяют и водный слой экстрагируют ДХМ (2×100 мл). Объединенные органические слои промывают насыщенным раствором соли (250 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (SiO2, 0-10% МеОН/ДХМ) с получением промежуточного соединения 13 в виде желтого твердого вещества (10 г, 87%). 1Н ЯМР (400 МГц, CDCl3) δ 6,83 (шс, 1Н), 6,75 (с, 1Н), 4,89-4,76 (м, 1Н), 4,48-4,39 (м, 1Н), 4,37-4,26 (м, 3H), 2,83 (с, 3H), 1,55 (д, J=6,6 Гц, 3H), 1,19-1,07 (м, 15Н). m/z (ХИАД+) для (C17H28N4O2S), 353,2 (М+Н)+. Стереохимию определяют на основе использования (S)-трет-бутил сульфинамида на стадии 1.
Промежуточное соединение 14: (S,R)-2-метил-N-[(1S)-1-{6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил}этил]пропан-2-сульфинамид

Смесь 4-ацетил-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-она (4f) (4000 мг, 15,43 ммоль), (R)-2-метилпропан-2-сульфинамид (2240 мг, 18,5 ммоль) в Ti(OEt)4 (20 мл) перемешивают при 100°С в течение 20 часов в атмосфере N2. Анализ ЖХМС показал потребление исходного материала. Желтый раствор охлаждают до 0°С и разбавляют ТГФ (50 мл). По каплям добавляют L-селектрид (38,6 ммоль, 38,6 мл, 1М в ТГФ) при 0°С. Затем смесь перемешивают при 10°С в течение 20 часов. ЖХМС показала оставшееся исходное вещество. Добавляют по каплям дополнительное количество L-селектрида (38,6 ммоль, 38,6 мл 1М в ТГФ) при 0°С и смесь перемешивают при 10°С в течение дополнительных 2 часов. Анализ ТСХ показал потребление исходного материала. Смесь гасят насыщенным водным раствором NH4Cl (200 мл) при 0-5°С. Суспензию фильтруют через слой Celite® и осадок промывают EtOAc (2×50 мл). Слои фильтрата разделяют и водный слой экстрагируют EtOAc (2×20 мл). Объединенные органические слои промывают насыщенным раствором соли (30 мл), сушат над Na2SO4, фильтруют и концентрируют с получением остатка. Остаток сначала очищают флэш-хроматографией (SiO2, EtOAc/MeOH=10:1), и затем дополнительно очищают препаративной ВЭЖХ (ACSSH-CA; колонка способа: YMC Triart С18 250*50 мм*7 мкм, вода (0,05% гидроксида аммония об/об)-АЦН с получением промежуточного соединения 14 (2,5 г, 44%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6,70 (с, 1Н), 6,47 (шс, 1Н), 4,57-4,34 (м, 4Н), 4,20 (шт, J=5,9 Гц, 1Н), 3,64-3,53 (м, 1Н), 3,47-3,35 (м, 1Н), 2,20-1,99 (м, 3H), 1,77 (шдд, J=2,5, 4,7 Гц, 1Н), 1,64 (д, J=6,5 Гц, 3H), 1,28 (д, J=6,2 Гц, 3H), 1,22 (с, 9Н). m/z (ИЭР) для (C18H28N4O2S), 365,1 (М+Н)+ Стереохимию определяют на основе использования (R)-трет-бутилсульфинамида.
Промежуточное соединение 15: трет-бутил {2-метокси-1-[6-(1-метилциклопропил)-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил]этил}карбамат

Стадия 1: этил 3-циано-2-гидрокси-6-(1-метилциклопропил)пиридин-4-карбоксилат (15а)

Смесь 2-цианоацетамида (10,0 г, 119 ммоль) и ТЭА (12,0 г, 119 ммоль) в EtOH (50 мл) нагревают до 65°С (внутренняя температура) до растворения твердых веществ, и затем добавляют этил 3-(1-метилциклопропил)-3-оксопропаноат (24,6 г, 124 ммоль). Смесь перемешивают при 65°С в течение 2 часов. Анализ ТСХ (1:10 EtOAc/петролейный эфир) показал потребление исходного материала. Реакцию охлаждают до 10°С. Образовавшийся осадок собирают фильтрацией. Осадок на фильтре промывают МТБЭ (3×10 мл) и сушат в вакууме. Фильтрат концентрируют досуха. Добавляют EtOH (10 мл) и затем добавляют МТБЭ (30 мл). Полученные твердые вещества собирают фильтрацией. Осадок на фильтре промывают EtOH (5 мл) и МТБЭ (2×10 мл) и сушат в вакууме. Твердые вещества объединяют с получением указанного в заголовке соединения (15а) (25,0 г, выход 85%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,74 (шс, 1Н), 6,63 (шс, 1Н), 4,36 (кв, J=7,1 Гц, 2Н), 1,45-1,27 (м, 6Н)), 1,16-1,06 (м, 2Н), 0,92-0,75 (м, 2Н).
Стадия 2: этил 2-хлор-3-циано-6-(1-метилциклопропил)пиридин-4-карбоксилат (15b)

К раствору этил 3-циано-2-гидрокси-6-(1-метилциклопропил)пиридин-4-карбоксилата (15а) (24,0 г, 97,5 ммоль) в MeCN (487 мл) добавляют POCl3 (74,7 г, 487 ммоль) по каплям при 30°С. Смесь перемешивают при 65°С в течение 60 часов. Анализ ТСХ (EtOAc) показал потребление исходного материала. Раствор концентрируют для удаления остаточного POCl3. Остаток выливают на лед и подщелачивают насыщенным раствором NaHCO3 до рН ~8. Смесь экстрагируют EtOAc (2×100 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией (1:10 EtOAc/петролейный эфир) с получением указанного в заголовке соединения (15b) (21,9 г, выход 85%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,79 (с, 1Н), 4,50 (кв, J=7,2 Гц, 2Н), 1,55 (с, 3H), 1,50-1,40 (м, 5Н), 1,03 (кв, J=3,9 Гц, 2Н); m/z (ИЭР+) для (C13H13ClN2O2), 264,9 (М+Н)+.
Стадия 3: 4-хлор-6-(1-метилциклопропил)-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он (15 с)
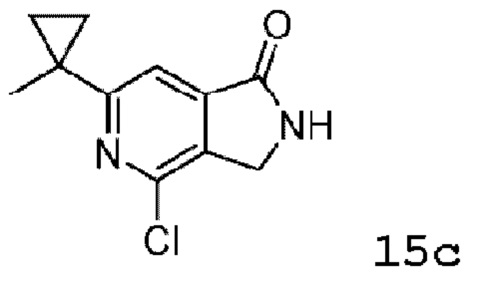
К раствору этил 2-хлор-3-циано-6-(1-метилциклопропил)пиридин-4-карбоксилата (15b) (2,5 г, 9,44 ммоль) в EtOH (500 мл) добавляют Ni Ренея (2,0 г, 34,1 ммоль). Черную смесь перемешивают при 30°С в атмосфере Н2 при 30 фунт/кв. дюйм в течение 48 часов. Анализ ТСХ (1:10 EtOAc/петролейный эфир) показал потребление исходного материала. Смесь фильтруют через слой Celite®. Осадок на фильтре промывают МеОН (250 мл). Объединенный фильтрат концентрируют досуха. Остаток суспендируют в EtOAc (5 мл) в течение 20 мин и суспензию фильтруют. Осадок на фильтре промывают EtOAc (2 мл) и сушат в вакууме с получением указанного в заголовке соединения (15 с) (1,1 г, выход 52%) в виде серого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,66 (шс, 1Н), 7,23-7,05 (м, 1Н), 4,56-4,34 (м, 2Н), 1,55 (шс, 3H), 1,39-1,11 (м, 2Н), 0,99-0,66 (м, 2Н); m/z (ИЭР+) для (C11H11ClN2O), 222,8 (М+Н)+.
Стадия 4: Промежуточное соединение 15
В 40 мл флакон загружают 4-хлор-6-(1-метилциклопропил)-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он (15 с) (200 мг, 0,898 ммоль), Cs2CO3 (644 мг, 1,98 ммоль), N-(трет-бутоксикарбонил)-О-метилсерин (394 мг, 1,80 ммоль), NiCl2⋅глим (39,5 мг, 0,18 ммоль), пиридин-2-ил-N-цианоамидин (26,3 мг, 0,180 ммоль), гексафторфосфат бис[2-(2,4-дифторфенил)-5-метилпиридин]-4,40-ди-трет-бутил-2,20-бипиридина иридия(III) (18,2 мг, 0,018 ммоль) и безводный ДМФ (27 мл). Смесь барботируют N2 в течение 2 мин и облучают светом 365 нМ при 15-25°С в течение 18 ч (скорость вентилятора 5200 об/мин, скорость перемешивания 1200 об/мин, 100% LED). Смесь концентрируют и очищают флэш-хроматографией (SiO2, 10-20%[0% МеОН в EtOAc] в петролейном эфире) с получением желтого твердого вещества. Очистка преп.ВЭЖХ (колонка: YMC Triart С18 150*25 мкм*5 мкм, вода (0,05% гидроксида аммония об./об.)-CAN) дает промежуточное соединение 15 в виде белого твердого вещества (315 мг, 32%). 1Н ЯМР (400 МГц, CDCl3) δ 7,65 (с, 1Н), 6,77 (с, 1Н), 5,66 (д, J=8,0 Гц, 1Н), 4,94 (д, J=6,3 Гц, 1Н), 4,63-4,48 (м, 2Н), 3,77 (дд, J=8,9, 5,0 Гц, 1Н), 3,58-3,44 (м, 1Н), 3,26 (с, 3H), 1,62 (с, 3H), 1,45 (с, 9Н), 1,30 (кв, J=3,6 Гц, 2Н), 0,88 (кв, J=3,4 Гц, 2Н); m/z (ИЭР+) для (C19H27N304), 362,3 (М+Н)+.
Примеры:
Полученные в настоящем документе примеры имеют стереохимию, назначенную на основе использования промежуточных соединений с подтвержденной стереохимией, например, примеры 1 и 2, или на основе использования промежуточных соединений, полученных из стереоспецифических исходных материалов, например, примеры 3 и 4.
Пример 1: 4-[(1R)-1-аминопропил]-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[{2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он

Стадия 1: {S,S)-2-метил-N-[(1R)-1-(2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)пропил]пропан-2-сульфинамид
[
Смесь (S,S)-2-метил-N-[(1R)-1-{6-[{2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-4-ил}пропил]пропан-2-сульфинамид (промежуточное соединение 5) (63,0 мг, 0,17 ммоль), (5S)-3-(6-бромпиридин-2-ил)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол (промежуточное соединение 2) (147 мг, 0,528 ммоль), K3PO4 (336 мг, 1,59 ммоль), Pd2(dba)3 (30,4 мг, 0,0528 ммоль) и XantPhos (61,1 мг, 0,106 ммоль) в 1,4-диоксане (4,5 мл, с=0,1 М) нагревают до 100°С. (температура блока) во флаконе 40 мл (закрытом) на 18 часов. Смесь фильтруют через Celite®, промывают ДХМ. Фильтрат концентрируют в вакууме, и неочищенное соединение очищают флэш-хроматографией (SiO2, 0-10% МеОН/ДХМ) с получением указанного в заголовке соединения в виде пены бледно-желтого цвета (194 мг, выход 64%). m/z (ХИАД+) для (C29H40N8O2S), 577,3 (М+Н)+.
Стадия 2: Пример 1
Раствор 4 N HCl в 1,4-диоксане (0,01 ммоль, 0,252 мл, 4 М) добавляют к суспензии (S,S)-2-метил-N-[(1R)-1-(2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-4-ил)пропил]пропан-2-сульфинамида (194 мг, 0,336 ммоль) в МеОН (12 мл). Полученную смесь перемешивают при КТ в течение 2 ч. Летучие вещества удаляют при пониженном давлении. Неочищенный продукт очищают с помощью хиральной СЖХ (колонка Phenomenex Lux Cellulose-1 4,6×100 мм, 3 мкм, 30% МеОН+10 мМ NH3 в CO2 при 120 бар, 4 мл/мин) с получением примера 1 (146 мг, выход 92%), >99% ди) в виде бледно-желтого твердого вещества. 1Н ЯМР (600 МГц, ДМСО-d6) δ 8,48 (д, J=8,3 Гц, 1Н), 8,01 (т, J=8,0 Гц, 1Н), 7,91 (д, J=7,7 Гц, 1Н), 6,67 (с, 1Н), 5,22-5,03 (м, 3H), 4,33-4,21 (м, 2Н), 3,60-3,50 (м, 1Н), 3,41-3,32 (м, 1Н), 3,06-2,98 (м, 1Н), 2,97-2,89 (м, 1Н), 2,88-2,76 (м, 1Н), 2,37-2,27 (м, 1Н), 2,09-1,86 (м, 5Н), 1,69-1,62 (м, 1Н), 1,44 (д, J=6,4 Гц, 3H), 1,15 (д, J=6,2 Гц, 3H), 0,86 (т, d=7,4 Гц, 3H); m/z (ХИАД+) для (C26H32N8O), 473,2 (М+Н)+; [α]D22=+37,0° (с=0,1 М, МеОН).
В таблице 4 представлены данные дифракции рентгеновских лучей монокристалла, подтверждающие стереохимию Примера 1. Рентгеноструктурные исследования монокристаллов проводят на дифрактометре Bruker Microstar APEX II CCD, оснащенном Cu Кα излучением (λ=1,54178 Å). Кристаллы примера 1 выращивают из ацетонитрила/пентана. Кусок бесцветного кристалла размером 0,25×0,2×0,15 мм устанавливают на Cryoloop с маслом Paratone. Данные собирают в потоке газообразного азота при 100(2) К с использованием ϕ и ω сканирования. Расстояние от кристалла до детектора составляет 40 мм, и время экспозиции составляет 2 или 10 секунд в зависимости от диапазона 2θ на рамку при ширине сканирования 1,00°. Сбор данных завершен на 99,4% до 67,679° по 9. Данные интегрируют с использованием программного обеспечения Bruker SAINT Software и масштабируют с использованием программного обеспечения SADABS. Решение с помощью прямых способов (SHELXT) позволяет получить полную фазовую модель, соответствующую предложенной структуре. Все не водородные атомы уточнены анизотропно полноматричным способом наименьших квадратов (SHELXL-2014). Все атомы водорода, связанные с углеродом, размещены с использованием модели «наездника». Их положения ограничены относительно их родительского атома с помощью соответствующей команды HFIX в SHELXL-2014.


Пример 2: 4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он
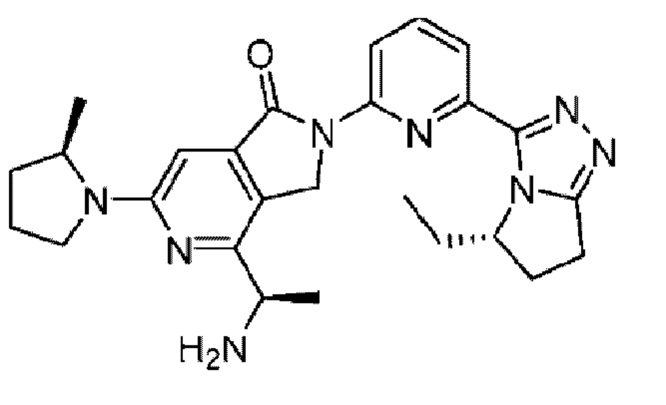
Стадия 1: (S,S)-N-[{1R)-1-(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)этил]-2-метилпропан-2-сульфинамид

К смеси (S,S)-2-метил-N-[(1R)-1-{6-[{2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-4-ил}этил]пропан-2-сульфинамида (промежуточное соединение 4) (50 мг, 0,14 ммоль), {5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазола (промежуточное соединение 3) (42,2 мг, 0,144 ммоль) и K3PO4 (87,4 мг, 0,412 ммоль) в 1,4-диоксане (3 мл) добавляют Pd2(dba)3 (12,6 мг, 0,014 ммоль) и XantPhos (15,9 мг, 0,027 ммоль) в атмосфере N2. После добавления, смесь барботируют N2 в течение 2 мин. Полученную смесь герметично закрывают и перемешивают при 85°С в течение 18 часов. Реакционную смесь концентрируют в вакууме и остаток очищают флэш-хроматографией (SiO2, 10% EtOAc/MeOH) с получением указанного в заголовке соединения (60 мг, выход 76%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,70 (д, J=8,2 Гц, 1Н), 8,18 (д, J=1,4 Гц, 1Н), 7,93 (т, J=8,0 Гц, 1Н), 6,76 (с, 1Н), 5,11-5,02 (м, 2Н), 4,99-4,93 (м, 1Н), 4,61-4,49 (м, 2Н), 4,34-4,27 (м, 1Н), 3,67-3,57 (м, 1Н), 3,46-3,35 (м, 1Н), 3,11-3,01 (м, 3H), 2,65 (тд, J=3,7, 7,9 Гц, 1Н), 2,19-2,08 (м, 3H), 2,07 (с, 1Н), 1,86-1,79 (м, 1Н), 1,79 (шд, J=2,3 Гц, 1Н), 1,69 (д, J=6,4 Гц, 3H), 1,31-1,28 (м, 3H), 1,22 (с, 9Н), 1,02 (т, J=7,5 Гц, 3H); m/z (ИЭР+) для (C30H40N8O2S), δ 77,5 (М+Н)+;[α]D22=+85,3° (с=0,1 М, МеОН).
Стадия 2: Пример 2
К раствору (S,S)-N-[(1R)-1-(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[{2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)этил]-2-метилпропан-2-сульфинамида (60 мг, 0,10 ммоль) в EtOAc (5 мл) добавляют по каплям 4 М HCl в EtOAc (3 мл) при 0°С. Смесь перемешивают при 20°С в течение 1 часа. Реакционную смесь концентрируют в вакууме. К остатку добавляют EtOAc (10 мл) и воду (10 мл). Водный слой подщелачивают насыщенным раствором NaHCO3 и экстрагируют ДХМ (10 мл × 3). Объединенные органические слои промывают насыщенным раствором соли (20 мл), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток лиофилизируют в течение 16 часов с получением соединения примера 2 (47 мг, 95%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,59 (д, J=8,3 Гц, 1Н), 8,09-8,02 (м, 1Н), 8,02-7,97 (м, 1Н), 6,58 (с, 1Н), 5,33 (д, J=17,1 Гц, 1Н), 5,10 (д, J=17,1 Гц, 1Н), 4,99 (шт, J=6,1 Гц, 1Н), 4,29-4,19 (м, 1Н), 4,12 (кв, J=6,9 Гц, 1Н), 3,55 (шт, J=8,0 Гц, 1Н), 3,04-2,85 (м, 3H), 2,58-2,55 (м, 1Н), 2,11-1,96 (м, 4Н), 1,79-1,66 (м, 2Н), 1,36 (д, J=6,5 Гц, 3H), 1,24-1,20 (м, 3H), 0,93 (т, J=7,4 Гц, 3H); m/z (ИЭР+) для (C26H32N8O), 473,4 (М+Н)+.
Пример 3: 4-[(1S)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он

Стадия 1:-N-[(1S)-1-(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)этил]-2-метилпропан-2-сульфинамид
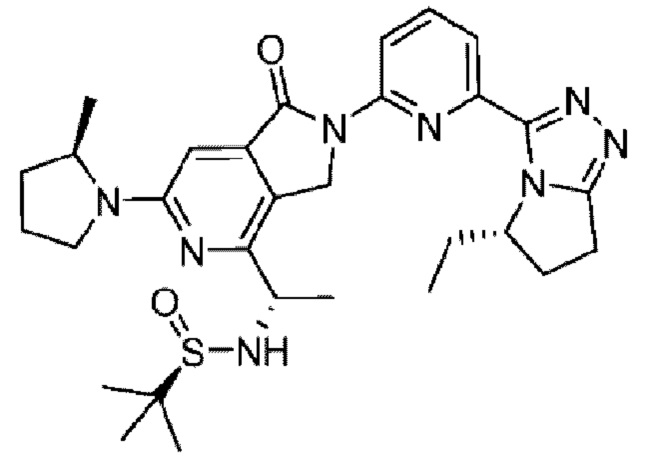
К суспензии (S,R)-2-метил-N-[(1S)-1-{6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил}этил]пропан-2-сульфинамида (промежуточное соединение 14) (2000 мг, 5,487 ммоль), (5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазола (промежуточное соединение 3) (1610 мг, 5,49 ммоль) и K3PO4 (3490 мг, 16,5 ммоль) в 1,4-диоксане (30 мл), Pd2(dba)3 (502 мг, 0,549 ммоль) и XantPhos (635 мг, 1,10 ммоль) добавляют в атмосфере азота. После добавления, смесь барботируют аргоном в течение 2 мин. Полученную смесь герметично закрывают и перемешивают при 85°С в течение 18 часов. Анализ ТСХ показал потребление исходного материала. Смесь разбавляют H2O (100 мл) при перемешивании, и полученную суспензию фильтруют. Водный фильтрат отделяют. Твердое вещество промывают ДХМ (100 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют с получением желтого остатка (3 г), который очищают флэш-хроматографией (SiO2, 0-10% МеОН в EtOAc) с получением желтого твердого вещества. Твердое вещество растворяют в ДХМ (50 мл) и добавляют диоксид кремния-SH (4 г). Желтую смесь кипятят с обратным холодильником в течение 20 мин. Смесь фильтруют, и осадок на фильтре промывают смесью ДХМ/МеОН (10:1). Фильтрат концентрируют. Обработку диоксидом кремния-SH повторяют еще три раза с получением указанного в заголовке соединения (2,3 г, 72,7%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,71 (дд, J=0,7, 8,4 Гц, 1Н), 8,18 (дд, J=0,1, 7,6 Гц, 1Н), 7,97-7,87 (м, 1Н), 6,76 (с, 1Н), 5,19-5,11 (м, 1Н), 5,03-4,94 (м, 2Н), 4,65-4,57 (м, 1Н), 4,57-4,51 (м, 1Н), 4,24 (шт, J=6,1 Гц), 1Н), 3,66-3,56 (м, 1Н), 3,48-3,39 (м, 1Н), 3,13-2,98 (м, 3H), 2,71-2,59 (м, 1Н), 2,21-2,03 (м, 4Н), 1,87-1,75 (м, 2Н), 1,68 (д, J=6,5 Гц, 4Н), 1,30 (д, J=6,2 Гц, 3H), 1,24 (с, 9Н), 1,02 (т, J=1,5 Гц, 3H). m/z (ИЭР) для (C30H40N8O2S), 577,5 (М+Н)+.
Стадия 2: Пример 3
К раствору (S,R)-N-[(1S)-1-(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)этил]-2-метилпропан-2-сульфинамида (2300 мг, 3,988 ммоль) в EtOAc (10 мл) добавляют 4М HCl в EtOAc (20 мл) при 0°С. После добавления, смесь перемешивают при 15°С в течение 1 часа. Реакцию контролируют с помощью ЖХМС. Полученную желтую суспензию концентрируют с получением остатка, который растворяют в H2O (30 мл) и экстрагируют EtOAc (25 мл). Водный слой подщелачивают насыщенным раствором NaHCO3 до рН ~8 и экстрагируют ДХМ (3×35 мл). Объединенные органические слои промывают насыщенным раствором соли (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением твердого вещества, к которому добавляют ДХМ (50 мл). Раствор фильтруют через фильтр из стеклянного микроволокна GF/F (~0,7 мкм) четыре раза. Фильтрат концентрируют с получением желтого твердого вещества, которое очищают с помощью преп.ВЭЖХ (колонка: Agela DuraShell С18 150*40 мм* 5 мкм, вода (0,05% гидроксида аммония об./об.)-CAN, время градиента 12 мин, скорость потока 25 мл/мин). Это дает соединение примера 3 (1,5 г, 79,6%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,55 (д, J=8,0 Гц, 1Н), 8,08-7,90 (м, 2Н), 6,51 (с, 1Н), 5,26 (д, J=16,8 Гц, 1Н), 5,08-4,92 (м, 2Н), 4,23 (шт, J=5,8 Гц, 1Н), 4,09 (кв, J=6,5 Гц, 1Н), 3,53 (шт, J=7,5 Гц, 1Н), 3,30-3,23 (м, 1Н), 3,07-2,83 (м, 3H), 2,62-2,53 (м, 1Н), 2,16-1,85 (м, 6Н), 1,77-1,64 (м, 2Н), 1,33 (д, J=6,5 Гц, 3H), 1,21 (д, J=6,0 Гц, 3H), 0,93 (т, J=1,3 Гц, 3H). m/z (ИЭР) для (C26H32N8O), 473,4 (М+Н)+.
Пример 4: 4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-он

Стадия 1: (S,S)-N-[(1R)-1-(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)этил]-2-метилпропан-2-сульфинамид

Раствор (S,S)-2-метил-N-[(1R)-1-{6-[метил(пропан-2-ил)амино]-1-OKCO-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил}этил]пропан-2-сульфинамида (промежуточное соединение 13) (627 мг, 1,78 ммоль), (5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазола (промежуточное соединение 3) (521 мг, 1,78 ммоль), K3PO4 (1,13 г, 5,34 ммоль), Pd2(dba)3 (102 мг, 0,178 ммоль) и XantPhos (206 мг, 0,356 ммоль) в 1,4-диоксане (17,8 мл, с=0,1 М) нагревают при 100°С в 100 мл колбе с конденсатором под N2 в течение 18 часов. Смесь охлаждают до КТ, фильтруют и промывают ДХМ (20 мл). Фильтрат концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией (24 г, SiO2, 0-10% МеОН/ДХМ) с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (808 мг, 80%). m/z (ХИАД+) для (C29H40N8O2S), 565,3 (М+Н)+.
Стадия 2: Пример 4
4 N раствор HCl в 1,4-диоксане (1,07 мл, 4,29 ммоль) добавляют к раствору {S,S)-N-[(1R)-1-(2-{6-[{5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)этил]-2-метилпропан-2-сульфинамида (808 мг, 1,43 ммоль) в МеОН (14,3 мл, с=0,1 М). Смесь перемешивают при комнатной температуре в течение 2 часов. Летучие вещества удаляют при пониженном давлении. Неочищенный продукт очищают с помощью хиральной СЖХ (колонка Phenomenex Lux Cellulose-1 21×250 мм, 30% МеОН+10 мМ NH3 в CO2, выдерживают при 120 бар, 100 мл/мин), с получением примера 4 (345 мг, 52% выход, >99% ди, >99% чистоты) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,56 (дд, J=1,0, 8,1 Гц, 1Н), 8,07-8,01 (м, 1Н), 8,00-7,93 (м, 1Н), 6,78 (с, 1Н), 5,34-5,28 (м, 1Н), 5,17-5,10 (м, 1Н), 5,02-4,95 (м, 1Н), 4,89 (тд, J=6,7, 13,4 Гц, 1Н), 4,18 (кв, J=6,7 Гц, 1Н), 3,01-2,94 (м, 2Н), 2,93 (с, 3H), 2,62-2,54 (м, 2Н), 2,09-1,97 (м, 1Н), 1,84-1,72 (м, 1Н), 1,41 (д, J=6,6 Гц, 3H), 1,20 (д, J=6,6 Гц, 6Н), 0,92 (т, J=7,5 Гц, 3H); m/z (ХИАД+) для (C25H32N8O), 461,3 (М+Н)+. [α]D22=+120,5° (с=0,1 М, МеОН).
Пример 5: 4-[(1R)-1-аминопропил]-2-{3-[(5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-он
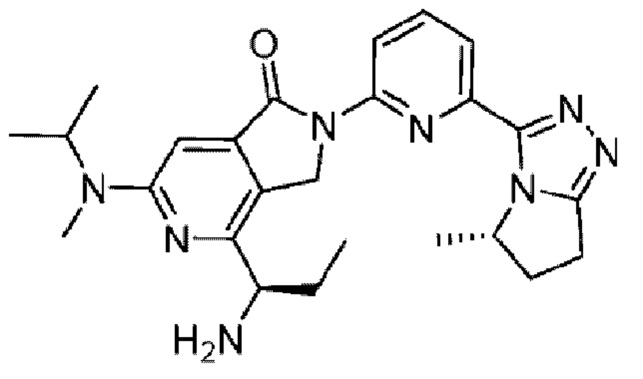
Стадия 1: (S,S)-2-метил-N-[(1R)-1-(2-{3-[(5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил)пропил]пропан-2-сульфинамид

Смесь {S,S)-2-метил-N-(1-{6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-С]пиридин-4-ил}пропил)пропан-2-сульфинамида (промежуточное соединение 10) (63 мг, 0,17 ммоль), (5S)-3-(3-бромфенил)-5-метил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазола (промежуточное соединение 2) (48 мг, 0,172 ммоль), K3PO4 (109 мг, 0,516 ммоль), Pd2(dba)3 (9,88 мг, 0,0172 ммоль) и XantPhos (19,9 мг, 0,0344 ммоль) в 1,4-диоксане (4,5 мл, с=0,1 М) нагревают при 100°С в 40 мл флаконе (закрытом крышкой) в течение 18 часов. Летучие вещества удаляют при пониженном давлении. Остаток очищают флэш-хроматографией (12 г, SiO2, 0-10% МеОН/ДХМ) с получением указанного в заголовке соединения в виде бледно-желтой пены (74,0 мг, 16%). m/z (ХИ7АД+) для (C29H40N8O2S), 565,3 (М+Н)+.
Стадия 2: Пример 5
4 N раствор HCl в 1,4-диоксане (0,164 мл, 0,655 ммоль) добавляют к раствору (S,S)-2-метил-А7-[(1R)-1-(2-{3-[(5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)пропил]пропан-2-сульфинамида (74 мг, 0,13 ммоль) в МеОН (5,0 мл, с=0,026 М). Смесь перемешивают при комнатной температуре в течение 2 часов. Летучие вещества удаляют при пониженном давлении. Неочищенный продукт очищают с помощью хиральной СЖХ (Phenomenex Lux Cellulose-1 колонка 4,6×100 мм, 3 мкм, 5-60% МеОН+10 мМ NH3 в CO2 с линейным изменением в течение 3,0 минут при 120 бар, 4 мл/мин) с получением примера 5 (11,6 мг, выход 19%, ди >99%, чистота >95%) в виде бледно-желтого твердого вещества. 1H ЯМР (600 МГц, ДМСО-d6) δ 8,54 (д, J=8,3 Гц, 1Н), 8,35 (шс, 2Н), 8,07 (т, J=8,0 Гц, 1Н), 8,00-7,90 (м, 1Н), 6,91 (д, J=0,9 Гц, 1Н), 5,27-5,07 (м, 3H), 5,01 (шс, 1Н), 4,37 (шс, 1Н), 3,14-2,97 (м, 2Н), 2,94 (д, J=1,5 Гц, 3H), 2,92-2,85 (м, 1Н), 2,42-2,35 (м, 1Н), 2,06-1,91 (м, 2Н), 1,50 (д, J=6,1 Гц), 3H), 1,19 (д, J=6,6 Гц, 3H), 1,15 (д, J=6,6 Гц, 3H), 0,92 (т, J=7,3 Гц, 3H); m/z (ХИАД+) для (C25H32N8O), 461,3 (М+Н)+. [α]D22=+75,9° (с=0,2 М, МеОН).
Пример 6: 4-[(1ξ)-1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он


Стереохимия соединений 6а и 6b изображена произвольно при том понимании, что два изомера были разделены (см. описание в предыдущем разделе о промежуточных соединениях).
Стадия 1: трет-бутил{2-метокси-1-[6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-1-оксо-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил]этил}карбамат

Раствор трет-бутил{2-метокси-1-[6-(1-метилциклопропил)-1-оксо-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-4-ил]этил}карбамата (промежуточное соединение 15) (136 мг, 0,376 ммоль), (5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазола (промежуточное соединение 2) (105 мг, 0,376 ммоль), K3PO4 (240 мг, 1,13 ммоль) и 1,4-диоксана (12 мл) барботируют азотом в течение 2 мин. К этой смеси добавляют Pd2(dba)3 (34,5 мг, 0,038 ммоль) и XantPhos (43,5 мг, 0,075 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 18 часов. Реакционную смесь охлаждают и объединяют с идентичным реакционным прогоном в масштабе 43 мг (промежуточное соединение 15). Объединенную смесь концентрируют и очищают с помощью флэш-хроматографии (SiO2, 10:1 EtOAc/MeOH) с последующей преп. ТСХ (SiO2, 10:1 EtOAc/MeOH) с получением указанного в заголовке соединения в виде желтого твердого вещества (170 мг, 61%). 1H ЯМР (400 МГц, CDCl3) δ 8,67 (дд, J=12,2, 8,3 Гц, 1Н), 8,20-8,10 (м, 1H), 8,01-7,84 (м, 1H), 5,66 (д, J=8,0 Гц, 1Н), 5,35-5,11 (м, 3H), 5,03 (д, J=11,7 Гц, 1H), 3,60-3,46 (м, 2Н), 3,27 (д, J=8,5 Гц, 3H), 3,16-2,94 (м, 4Н), 2,50 (д, J=8,7 Гц, 2Н), 2,39 (с, 2Н), 1,71 (с, 3H), 1,43 (с, 9Н), 1,33 (кв, J=2,3 Гц, 2Н), 0,92 (т, J=2,6 Гц, 2Н). m/z (ИЭР) для (C30H37N7O4), 460,2 (М+Н)+.
Стадия 2: Пример 6
К раствору трет-бутил-{2-метокси-1-[6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-1-оксо-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил]этил}карбамата (170 мг, 0,304 ммоль) в ДХМ (5 мл) добавляют по каплям 4М раствор HCl в EtOAc (3 мл) при 0°С. Реакционную смесь перемешивают при 20°С в течение 1 ч и затем концентрируют. Полученный остаток разбавляют H2O (15 мл) и подщелачивали до рН ~8 насыщенным раствором NaHCO3. Смесь экстрагируют ДХМ (3×8 мл), и объединенный органический слой промывают насыщенным раствором соли (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением желтого твердого вещества. Полученное твердое вещество очищают с помощью преп. СЖХ (колонка: Daicel Chiralpak 250 мм * 30 мм * 10 мкм, 0,1% NH3⋅H2O EtOH) с получением:
Изомера, элюирующегося первым, как в примере 6а (30 мг, 21%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,56 (д, J=l, 9 Гц, 1Н), 8,08 (т, J=7,8 Гц, 1Н), 7,99 (д, J=1,8 Гц, 1Н), 7,57 (с, 1Н), 5,48 (д, J=18,1 Гц, 1Н), 5,30 (д, J=18,1 Гц, 1Н), 5,12 (шт, d=6,5 Гц, 1Н), 4,30 (т, d=6,3 Гц, 1Н), 3,65-3,52 (м, 2Н), 3,30-3,26 (м, 3H), 3,13-2,86 (м, 3H), 2,40 (шдд, J=8,4, 11,9 Гц, 3H), 1,59-1,52 (м, 6Н), 1,31-1,24 (м, 2Н), 0,89 (д, J=2,9 Гц, 2Н). m/z (ИЭР) для (C25H29N7O), 560,3 (М+Н)+. [α]D22=+66,1° (с=0,1 М, МеОН).
Изомера, элюирующегося вторым, как в примере 6b (33 мг, 23%). 1Н ЯМР (400 МГц, ДМСО-d6) α 8,58 (д, J=7,9 Гц, 1Н), 8,07 (т, J=7,9 Гц, 1Н), 7,99 (д, J=7,3 Гц, 1Н), 7,55 (с, 1Н), 5,44 (д, J=18,1 Гц, 1Н), 5,31 (д, J=18,1 Гц, 1Н), 5,16 (шт, J=6,6 Гц, 1Н), 4,29 (т, J=6,4 Гц, 1Н), 3,55 (д, J=6,4 Гц, 2Н), 3,25 (с, 3H), 3,13-2,86 (м, 3H), 2,40 (шдд, J=8,2, 11,6 Гц, 1Н), 2,12 (шс, 2Н), 1,59-1,48 (м, 6Н), 1,30-1,23 (м, 2Н), 0,88 (д, J=3,l Гц, 2Н). m/z (ИЭР) для (C25H29N7O), 560,2 (М+Н)+. [α]D22=+41,5°.
Дополнительные соединения по настоящему изобретению получают с помощью модификаций способов, приведенных в настоящем документе в качестве примеров, и они представлены в таблицах 5, 6 и 7. Если не указано иное, все соединения, имеющие хиральные атомы углерода, получают и/или выделяют в виде одного энантиомера, как показано в структуре. На хиральность дополнительно указывает название соединения, которое обозначает конкретную стереохимию как (R) или (S) для каждого хирального углерода. Когда все хиральные атомы углерода обозначены как известная стереохимия, стереохимия основана на использовании известных хиральных исходных материалов и/или подтверждении разделенных энантиомеров с помощью рентгеновской кристаллографии. Некоторые соединения получают из рацемических промежуточных соединений и разделяют на отдельные энантиомеры подходящим способом хиральной препаративной СЖХ. Если стереохимия неизвестна, но энантиомеры разделены, «or1» или «or2» относится к хиральному атому углерода. В названии, углерод с разрешенным, но не подтвержденным стереохимическим центром обозначен символом Связь, нарисованная на этом углероде, является представлением стереохимии; это означает, что углерод будет иметь эту конфигурацию связи (сплошной клин) или противоположную конфигурацию (заштрихованный клин). См., например, примеры 101 и 102. При получении, оптическое вращение (αD22 или αD20) представлено после названия IUPAC, представленного просто как α.







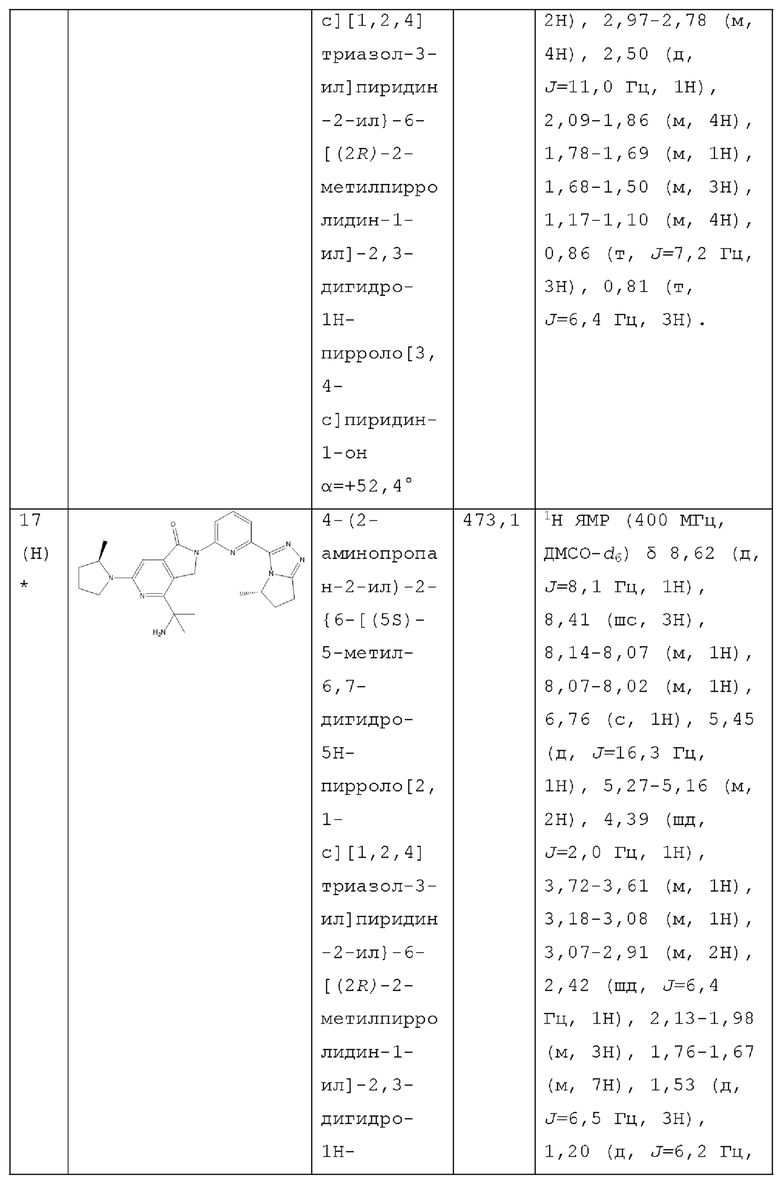


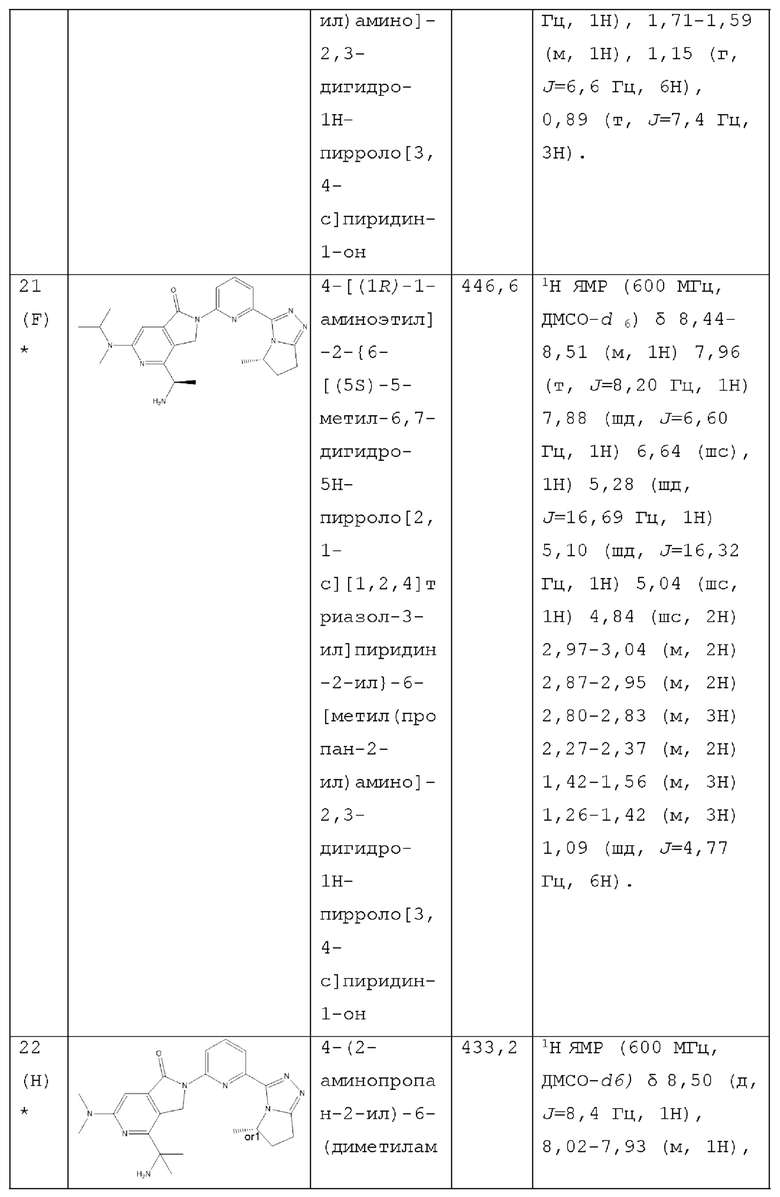





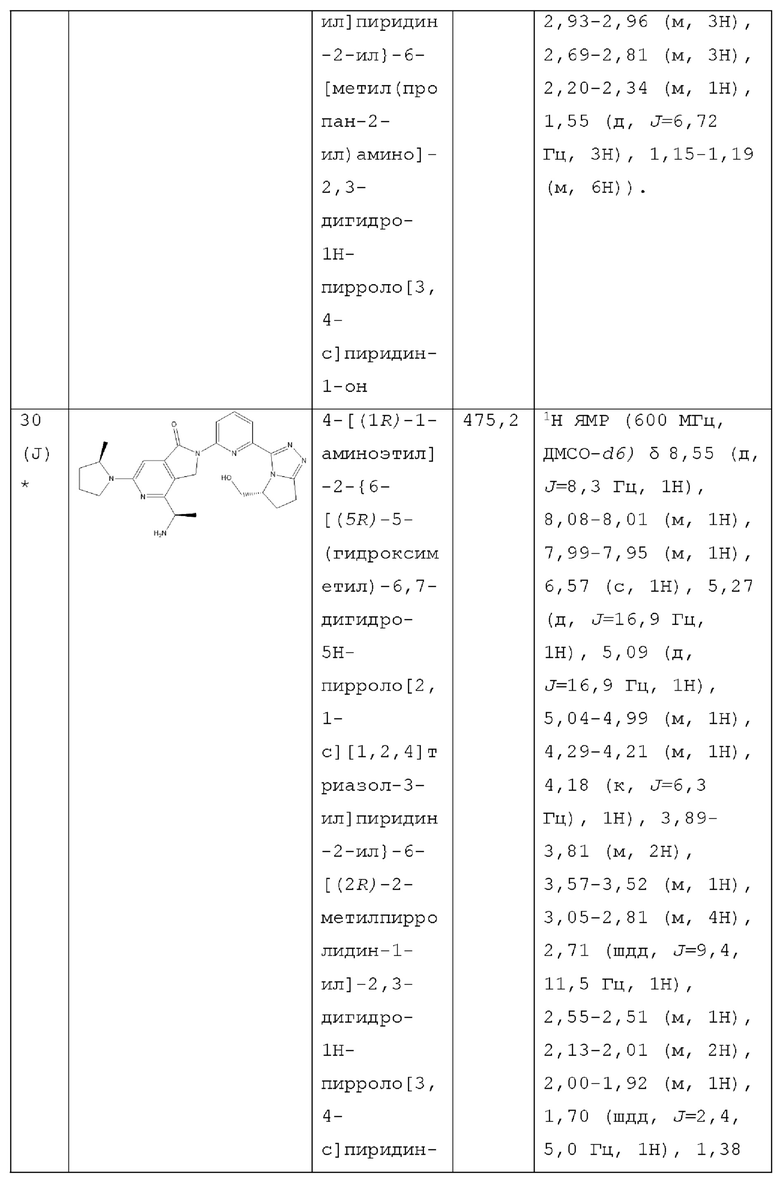





Пример 100: 4-(2-аминопропан-2-ил)-2-[6-(4-этил-4Н-1,2,4-триазол-3-ил)пиридин-2-ил]-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он

Смесь 4-(2-аминопропан-2-ил)-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридина-1-она (промежуточное соединение 6) (63,0 мг, 0,23 ммоль), 2-бром-6-(4-этил-4H-1,2,4-триазол-3-ил)пиридина (промежуточное соединение 1) (58,1 мг, 0,230 ммоль), K2CO3 (69,8 мг, 0,505 ммоль), Cul (10,9 мг, 0,058 ммоль) и N,N-диметилэтилендиамина (0,115 ммоль, 0,0125 мл) в CH3CN (3,0 мл) нагревают при 120°С с микроволновым облучением в течение 30 мин. Летучее вещество удаляют, и остаток очищают флэш-хроматографией (SiO2, 0-10% МеОН/ДХМ). Продукт дополнительно очищают с помощью ВЭЖХ (колонка Phenemonex Gemini NX С18, 150×21,2 мм, 5 мкм. подвижная среда А: вода+10 мМ ацетат аммония, подвижная среда В: ацетонитрил) с получением примера 100 в виде бледно-желтого твердого вещества (49,3 мг, 49% выход). 1H ЯМР (600 МГц, ДМСО-d6) δ 8,74 (с, 1H), 8,62 (д, J=8,4 Гц, 1Н), 8,11-8,04 (м, 1Н), 8,03-7,90 (м, 1H), 6,55 (с, 1H), 5,40 (с, 2Н), 4,65 (кв, J=7,2 Гц, 2Н), 4,24-4,19 (м, 1H), 3,29-3,33 (м, 2Н, предполагается; частично перекрыт пиком воды), 2,16-2,03 (м, 2Н), 1,98-1,89 (м, 1H), 1,71-1,69 (м, 1H), 1,50 (т, J=7,l Гц, 3H), 1,47 (д, J=5,9 Гц, 6Н), 1,22 (д, J=6,2 Гц, 3H); m/z (ХИАД+) для (C24H30N8O), 447,3 (М+Н)+.

























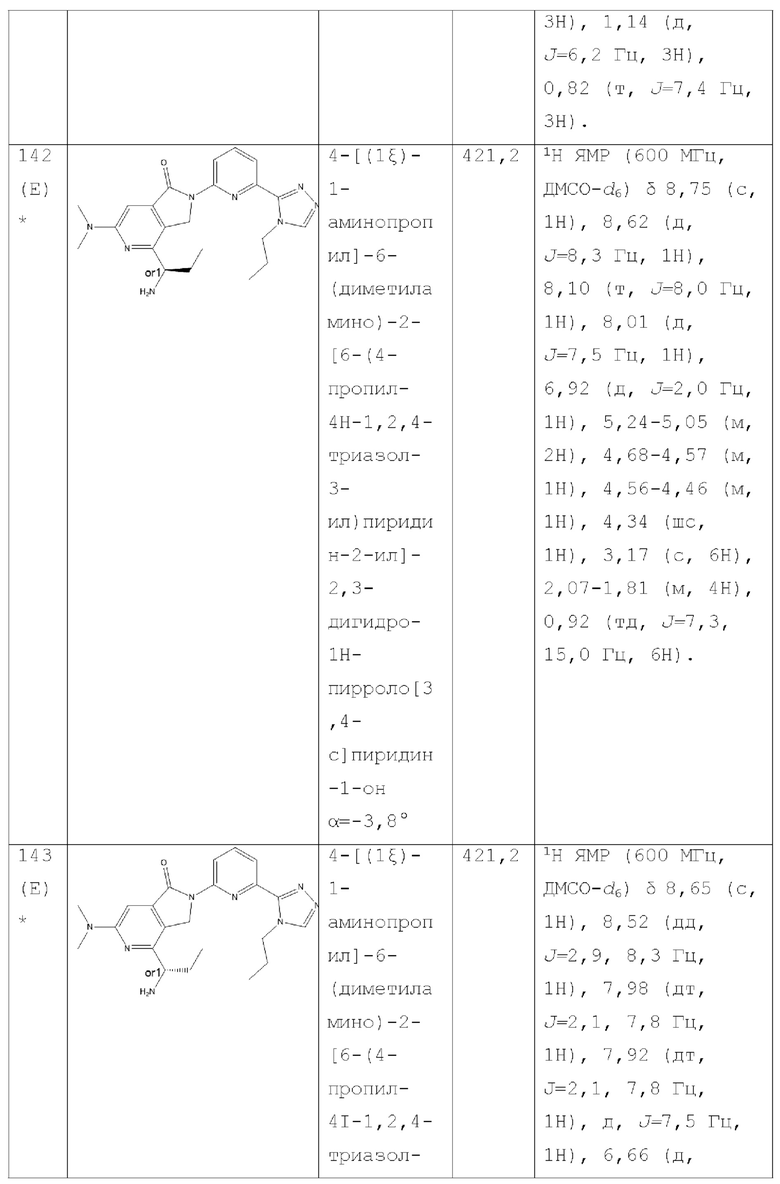



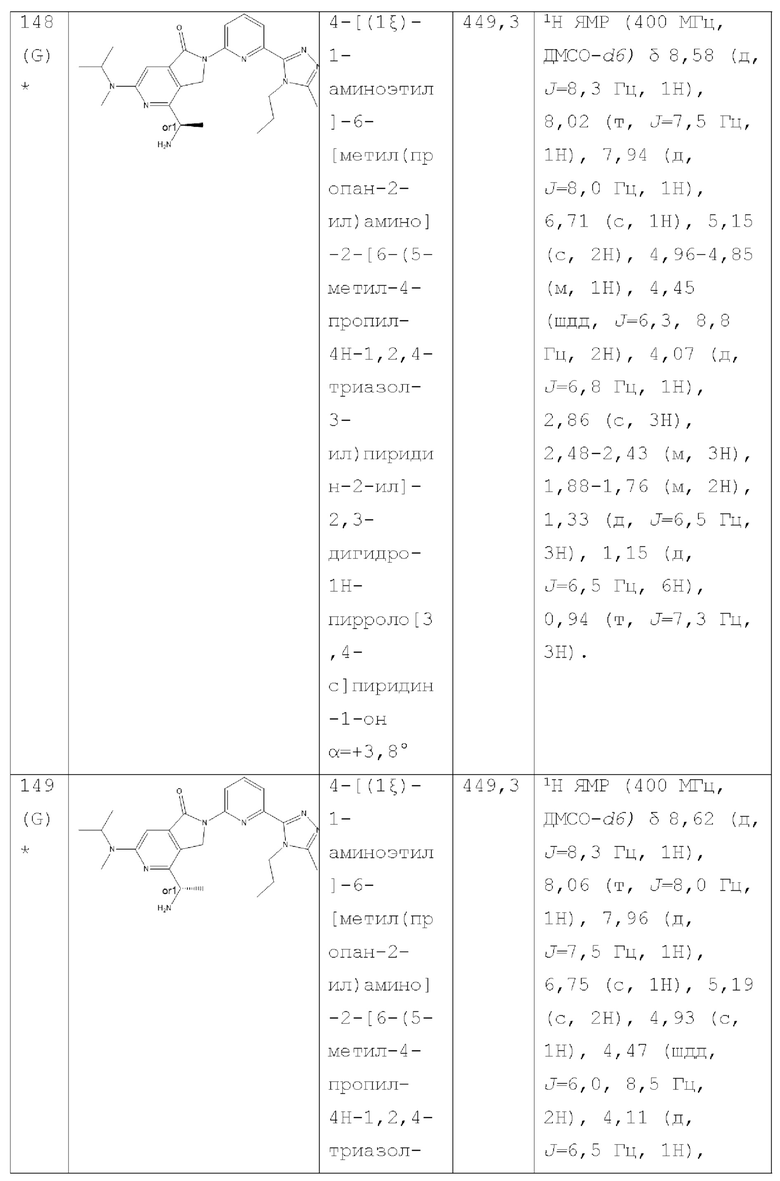

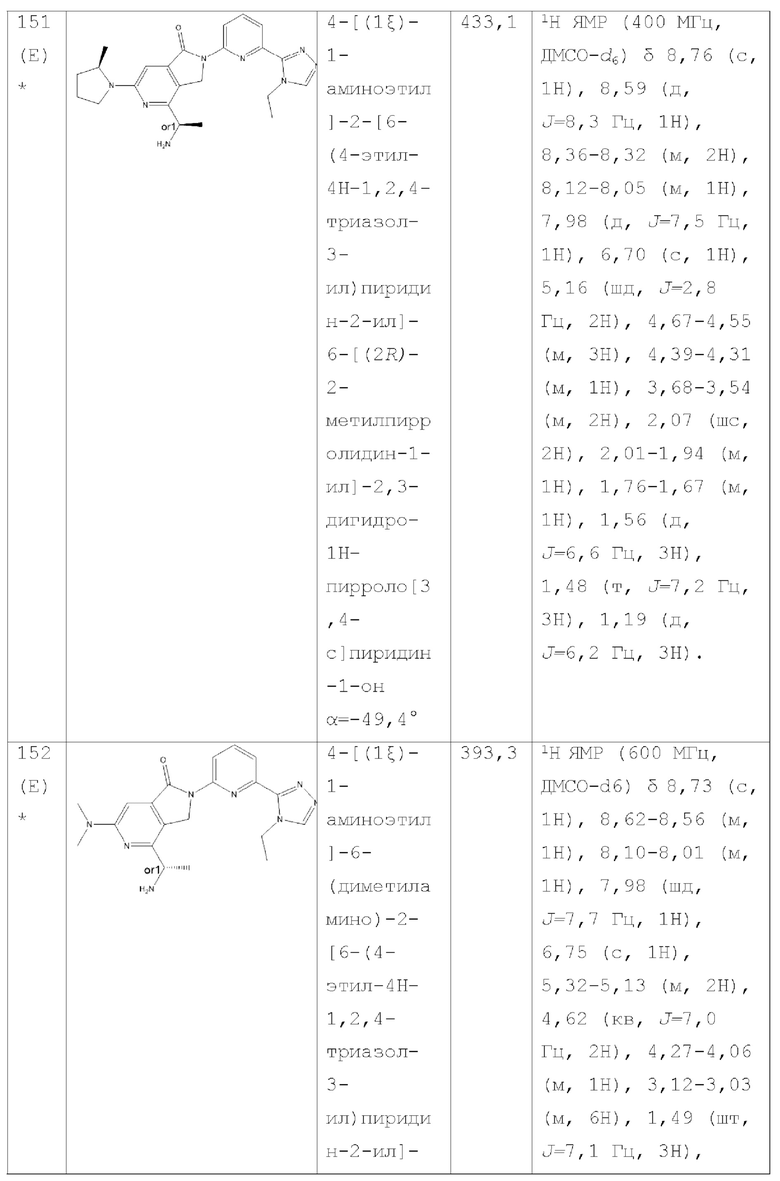


Пример 200: 2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-4-[(метиламино)метил]-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он

Стадия 1: трет-бутил[(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)метил]метилкарбамат

Смесь трет-бутил метил({6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил}метил)карбамата (промежуточное соединение 8) (752 мг, 2,09 ммоль), (5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазола (промежуточное соединение 3) (612 мг, 2,09 ммоль), K3PO4 (1,33 г, 6,26 ммоль), Pd2(dba)3 (120 мг, 0,209 ммоль), и XantPhos (241 мг, 0,417 ммоль) в 1,4-диоксане (20,9 мл, с=0,1 М) нагревают при 100°С в 100 мл колбе с конденсатором в атмосфере азота в течение 18 часов. Смесь охлаждают до КТ, фильтруют и промывают ДХМ (20 мл). Фильтрат концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией (24 г, SiO2, 0-10% МеОН/ДХМ) с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,13 г, 85%). m/z (ХИАД+) для (C31H40N8O3), 573,30 (М+Н)+.
Стадия 2: Пример 200
4 N раствор HCl в 1,4-диоксане (4,42 мл, 17,7 ммоль) добавляют к раствору трет-бутил[(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил)метил]метилкарбамата (1,01 г, 1,7 ммоль) в ДХМ (22,1 мл, с=0,08 М). Смесь перемешивают при комнатной температуре в течение 2 часов. Летучие вещества удаляют при пониженном давлении. Неочищенный продукт очищают СЖХ (колонка ZymorSPHER HADP с метанолом и 10 мМ NH3) с получением соединения примера 200 в виде бледно-желтого твердого вещества (640 мг, 77%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,97 (шс, 1Н), 8,60 (дд, J=0,9, 8,3 Гц, 1Н), 8,17-8,06 (м, 1Н), 8,04-7,95 (м, 1Н), 6,76 (с, 1Н), 5,28 (д, J=16,6 Гц, 1Н), 5,09-4,91 (м, 2Н), 4,45-4,19 (м, 3H), 3,73-3,61 (м, 1Н), 3,49-3,37 (м, 1Н), 3,09-2,87 (м, 3H), 2,76 (с, 3H), 2,61-2,54 (м, 1Н), 2,15-1,96 (м, 4Н), 1,80-1,64 (м, 2Н), 1,21 (д, J=6,2 Гц, 3H), 0,93 (т, J=7,4 Гц, 3H). m/z (ХИАД+) для (C26H32N8O), 473,3 (М+Н)+. [α]D22=+76,0° (с=0,1М, МеОН).
Пример 201: 2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-4-[(метиламино)метил]-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-он
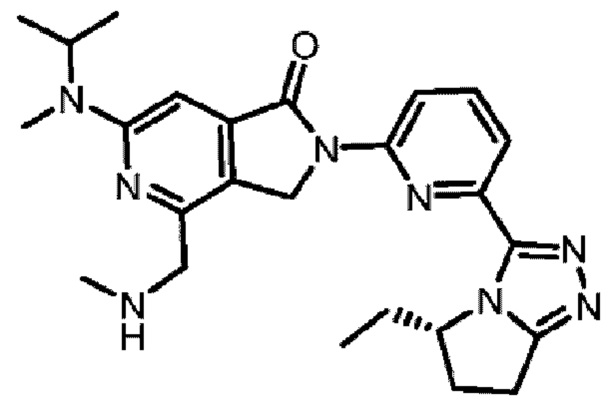
Стадия 1: трет-бутил[(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-4-ил)метил]метилкарбамат
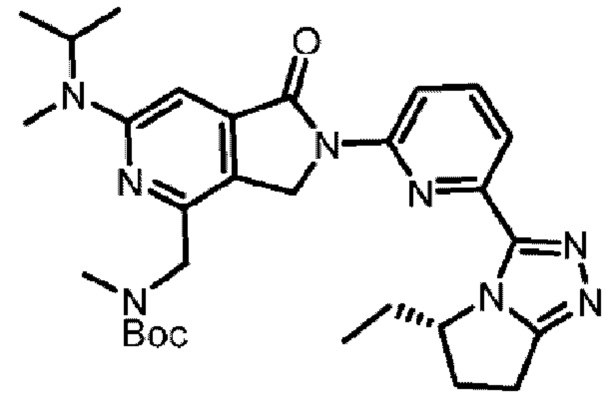
В колбу с завинчивающейся крышкой, снабженную магнитной мешалкой, добавляют трет-бутилметил({6-[метил(пропан-3-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил}метил)карбамат (промежуточное соединение 9) (294 мг, 0,84 ммоль), (5 S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол (промежуточное соединение 3) (260 мг, 0,88 ммоль), K3PO4 (537 мг, 2,53 ммоль), Pd2(dba)3 (78,0 мг, 0,084 ммоль), XantPhos (97,6 мг, 0,17 ммоль) и 1,4-диоксан (16,9 мл, 0,05 М). Смесь барботируют N2 в течение 5 минут перед закрытием колбы и нагреванием до 85°С в течение 22 часов. Анализ ЖХМС показал потребление исходного материала. Реакционную смесь концентрируют и остаток очищают флэш-хроматографией (SiO2, от 100% гептана до 1:10 MeOH/EtOAc) с получением указанного в заголовке соединения (344 мг, выход 73%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ=8,72-8,65 (м, 1Н), 8,13 (д, J=7,5 Гц, 1Н), 7,91 (т, J=7,9 Гц, 1Н), 6,89 (с, 1Н), 5,13-4,85 (м, 4Н), 4,25-4,08 (м, 1Н), 3,10-2,96 (м, 6Н), 2,96-2,93 (м, 3H), 2,66-2,58 (м, 1Н), 2,07-1,94 (м, 1Н), 1,87-1,74 (м, 1Н), 1,53 (с, 1Н), 1,50-1,35 (м, 9Н), 1,23 (дд, J=2,8, 6,6 Гц, 6Н), 1,00-0,87 (м, 3H); ЖХМС m/z (ХИАД) для (C30H40N8O3), 561,3 (М+Н)+.
Стадия 2: Пример 201
В колбу, снабженную магнитной мешалкой и содержащую трет-бутил[(2-{6-[(5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-4-ил)метил]метилкарбамат (344 мг, 0,62 ммоль) и CH2Cl2 (12,3 мл, 0,05 М) добавляют HCl (4M в 1,4-диоксане, 1,53 мл, 6,2 ммоль). После перемешивания при комнатной температуре в течение 20 ч анализ ЖХМС показал потребление исходного материала. В реакционную колбу добавляют толуол (5 мл), и реакционную смесь концентрируют. Неочищенный остаток пропускали через колонку с сильным ионообменом (SCX) для удаления остаточной HCl и концентрируют, получая соединение примера 201 (278 мг, выход 98%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ=8,59 (д, J=7,8 Гц, 1Н), 8,11-8,03 (м, 1Н), 8,00-7,96 (м, 1Н), 6,77 (с, 1Н), 5,76 (с, 1Н), 5,25 (с, 1Н), 5,08 (с, 3H), 3,84 (с, 2Н), 3,01-2,88 (м, 6Н), 2,61-2,54 (м, 1Н), 2,39 (с, 3H), 2,05-1,93 (м, 1Н), 1,80-1,67 (м, 1Н), 1,16 (д, J=6,7 Гц, 6Н), 0,92 (т, J=7,4 Гц, 3H); ЖХМС m/z (ХИАД) для (C25H32N8O), 461,3 (М+Н)+.
Пример 202: 2-{6-[(5R)-5-(фторметил)-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-4-[(метиламино)метил]-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-он

Стадия 1: трет-бутил[(2-{6-[(5R)-5-(фторметил)-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1H-пирроло[3,4-с]пиридин-4-ил)метил]метилкарбамат

К смеси трет-бутил метил({6-[(2R)-2-метилпирролидин-1-ил]-1-okco-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил}метил)карбамата (промежуточное соединение 8) (90 мг, 0,25 ммоль), (5S)-3-(6-бромпиридин-2-ил)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазола (промежуточное соединение 7) (74,2 мг, 0,250 ммоль) и K3PO4 (159 мг, 0,749 ммоль) в 1,4-диоксане (8 мл) добавляют Pd2(dba)3 (22,9 мг, 0,025 ммоль) и XantPhos (28,9 мг, 0,0499 ммоль) в атмосфере N2. После добавления, смесь барботируют N2 в течение 2 мин. Полученную смесь герметично закрывают и перемешивают при 85°С в течение 18 часов. К реакционной смеси добавляют насыщенный раствор соли (30 мл). Смесь экстрагируют EtOAc (20 мл × 2). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением коричневого твердого вещества (250 мг). Материал очищают флэш-хроматографией (SiO2, ДХМ/EtOAc=1:1) с получением указанного в заголовке соединения (120 мг, 83%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,69 (д, J=8,3 Гц, 1H), 8,16 (д, J=7,6 Гц, 1Н), 7,96-7,86 (м, 1Н), 6,72 (с, 1Н), 5,40 (д, J=30,1 Гц, 1Н), 5,01 (дд, J=48,3, 16,6 Гц, 4Н), 4,71 (д, J=14,5 Гц, 1Н), 4,26-4,05 (м, 1Н), 3,59 (т, J=8,7 Гц, 1H), 3,39 (кв, J=9,0, 8,4 Гц, 1Н), 3,22-3,05 (м, 3H), 3,02 (с, 3H), 2,92-2,82 (м, 1Н), 2,19-1,96 (м, 4Н), 1,77-1,71 (м, 1Н), 1,43 (с, 9Н), 1,26 (д, J=6,3 Гц, 3H). ЖХМС m/z (ИЭР) для (C30H37FN8O3), 577,2 (М+Н)+.
Стадия 2: Пример 202
К суспензии трет-бутил[(2-{6-[(5R)-5-(фторметил)-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[{2R)-2-метилпирролидин-1-ил]-1-оксо-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-4-ил)метил]метилкарбамат (120 мг, 0,208 ммоль) в ДХМ (8 мл) добавляют по каплям HCl в EtOAc (4 мл, 4 М) при 0°С. Смесь перемешивают при комнатной температуре в течение 2 ч и контролируют с помощью ЖХМС. Затем реакционную смесь охлаждают до 0°С и добавляют HCl в EtOAc (5,0 мл, 4 М). Смесь перемешивают при комнатной температуре в течение 30 мин. Смесь концентрируют в вакууме с получением желтого твердого вещества (100 мг). К остатку добавляют воду (10 мл) и промывают EtOAc (10 мл). Водный слой лиофилизируют в течение 16 часов с получением примера 202 (94 мг, 88%, соль HCl) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,21-9,00 (м, 2Н), 8,61 (д, J=8,3 Гц, 1Н), 8,14-8,08 (м, 1Н), 8,06-8,02 (м, 1Н)), 6,75 (с, 1Н), 5,43-5,29 (м, 1Н), 5,23 (д, J=16,6 Гц, 1Н), 5,09-4,91 (м, 3H), 4,40-4,29 (м, 3H), 3,70-3,59 (м, 1Н), 3,13-2,92 (м, 3H), 2,79-2,71 (м, 4Н), 2,15-1,96 (м, 3H), 1,77-1,70 (м, 1Н), 1,21 (д, J=6,3 Гц, 3H). ЖХМС m/z (ИЭР) для (C25H29FN8O), 477,4 (М+Н)+.






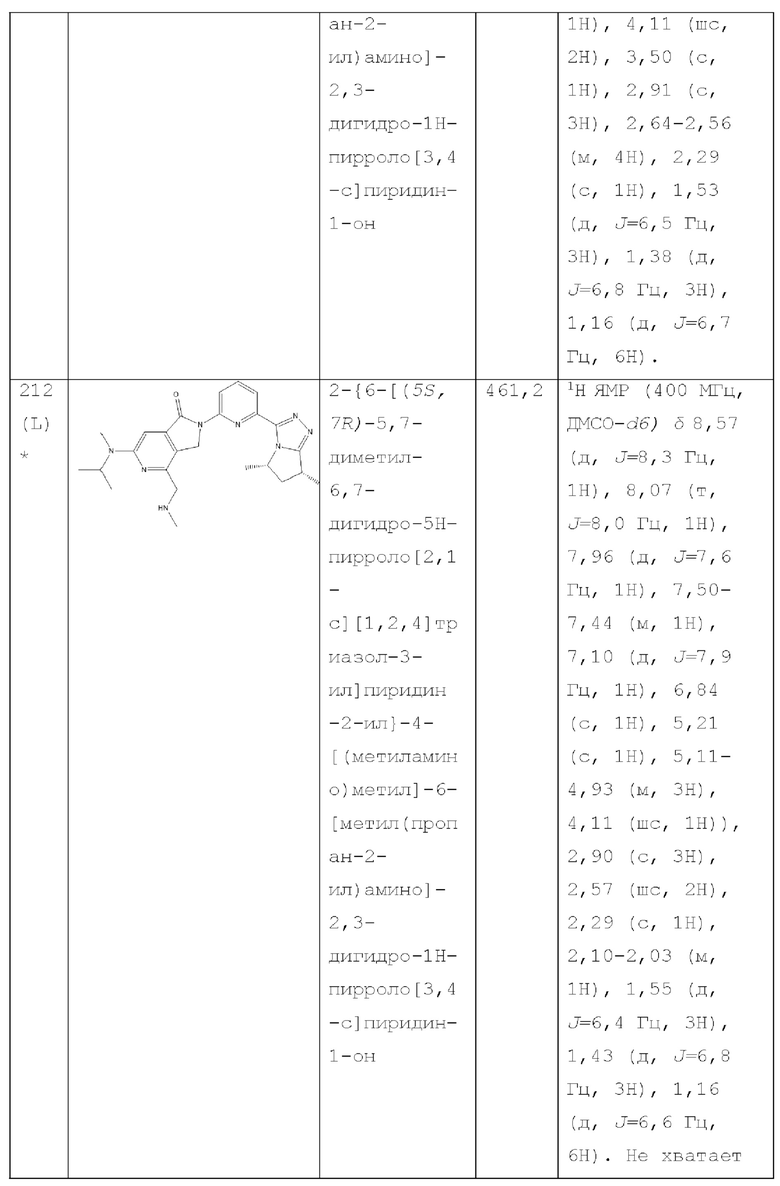
Пример 300: 6-(диметиламино)-4-[(метиламине-) метил]-2-[6-(4-пропил-4H-1,2,4-триазол-3-ил)пиридин-2-ил]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он

Стадия 1: трет-бутил({6-(диметиламино)-1-оксо-2-[6-(4-пропил-4H-1,2,4-триазол-3-ил)пиридин-2-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил}метил)метилкарбамат
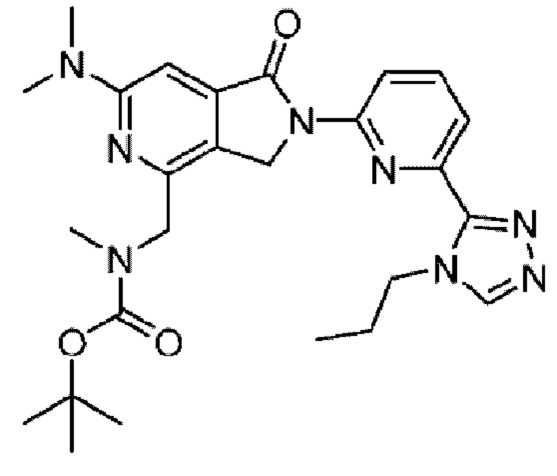
Смесь трет-бутил-{[6-(диметиламино)-1-оксо-2,3-дигидро-1H-пирроло[3,4-c]пиридин-4-ил]метил}метилкарбамата (промежуточное соединение 11) (1,33 г, 4,15 ммоль), 2-бром-6-(4-пропил-4Н-1,2,4-триазол-3-ил)пиридина (промежуточное соединение 12) (1,16 г, 4,36 ммоль), Pd2(dba)3 (380 мг, 0,415 ммоль), XantPhos (480 г, 0,830 ммоль) и K3PO4 (2,64 г, 12,5 ммоль) в 1,4-диоксане (46 мл) дегазируют азотом в течение 5 минут, и затем перемешивают при 85°С в течение 16 часов. Реакцию анализируют с помощью ЖХМС, которая показала потребление исходного материала. Смесь охлаждают до комнатной температуры, фильтруют через слой Celite® и концентрируют в вакууме. Остаток суспендируют с EtOAc (15 мл) в течение 10 мин и собирают твердые вещества фильтрацией. Осадок на фильтре промывают EtOAc (4х) и затем сушат в вакууме. Остаток очищают флэш-хроматографией (40 г SiO2, 0-100% EtOAc/гептан, затем 10% MeOH/EtOAc) с получением твердого вещества светло-желтого цвета. Материал растворяют в смеси 1:9 EtOH/ДХМ и обрабатывают сверхчистым Si-ThioSiO2 (1,59 г). Смесь перемешивают в течение 2 ч и фильтруют. Осадок на фильтре промывают смесью 1:9 EtOH/ДХМ и объединенный фильтрат концентрируют в вакууме. Остаток растворяют в смеси 1:9 EtOH/ДХМ и обрабатывают сверхчистым Si-ThioSiO2 (1,32 г). Смесь перемешивают в течение 3 ч и затем фильтруют. Осадок на фильтре промывают смесью 1:9 EtOH/ДХМ и объединенный фильтрат концентрируют. Остаток растворяют в 1:9 EtOH и обрабатывают сверхчистым Si-ThioSiO2 (1,22 г). Смесь перемешивают в течение 16 ч и затем фильтруют. Осадок на фильтре промывают смесью 1:9 EtOH/ДХМ. Объединенный фильтрат концентрируют досуха, с получением указанного в заголовке соединения (2,08 г, выход 95%) в виде твердого вещества светло-желтого цвета. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,74 (с, 1Н), 8,61 (д, J=8,4 Гц, 1Н), 8,09 (т, J=8,0 Гц, 1Н), 7,97 (д, J=7,6 Гц, 1Н), 6,82 (с, 1Н), 5,05 (д, J=11,7 Гц, 2Н), 4,56 (с, 2Н), 4,52-4,46 (м, 2Н), 3,09 (с, 6Н), 2,92 (с, 3H), 1,88-1,76 (м, 2Н), 1,40-1,19 (м, 9Н), 0,87 (т, J=7,4 Гц, 3H); ЖХМС m/z (ИЭР+) для (C26H34N8O3), 507,4 (М+Н)+.
Стадия 2: Пример 300
К суспензии трет-бутил ({6-(диметиламино)-1-оксо-2-[6-(4-пропил-4Н-1,2,4-триазол-3-ил)пиридин-2-ил]-2,3-дигидро-1Н-пирроло[3.4-с]пиридин-4-ил}метил)метилкарбамата (1,96 г, 3,87 ммоль) в МеОН (20 мл) добавляют раствор HCl (4,0 М в 1,4-диоксане, 19,3 мл, 77,4 ммоль) медленно при 0°С. Смесь перемешивают в течение 3 ч при 0°С и затем медленно нагревают до комнатной температуры. Смесь перемешивают при комнатной температуре в течение 16 часов. Реакцию концентрируют досуха. Твердые вещества растворяют в смеси 1:9 МеОН/ДХМ (80 мл), охлаждают до 0°С и затем перемешивают с насыщенным раствором Na2CO3 (25 мл) в течение 20 мин. Смесь разделяют. Водный слой экстрагируют смесью 1:19 МеОН/ДХМ (3×50 мл). Объединенные органические слои промывают H2O (2×30 мл), сушат над Na2SO4, фильтруют и концентрируют. Твердые вещества суспендируют в EtOAc при 40°С в течение 40 мин. Твердые вещества собирают фильтрацией. Осадок на фильтре промывают EtOAc, а затем сушат в течение 16 часов в вакуумной печи при 30°С с получением примера 300 (1,42 г, выход 90%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,73 (с, 1Н), 8,62 (дд, J=8,4, 1,0 Гц, 1Н), 8,11-8,04 (м, 1Н), 8,00 (дд, J=1,7, 1,0 Гц, 1Н), 6,78 (с, 1Н), 5,14 (с, 2Н), 4,57 (дд, d=7,9, 6,5 Гц, 2Н), 3,80 (с, 2Н), 3,09 (с, 6Н), 2,35 (с, 3H), 1,88 (г, J=1,4 Гц, 2Н), 0,93 (т, J=1,4 Гц, 3H); ЖХМС m/z (ИЭР+) для (C21H26N8O), 407,3 (М+Н)+.
Биологические анализы и данные
Биохимический ферментный анализ HPK1
Ингибирование фермента HPK1 измеряют с использованием основанного на флуоресценции способа флуоресценции, усиленной хелатированием (CHEF) (1), и с использованием запатентованного флуоресцентного пептидного субстрата, в котором остаток цистеина алкилируется производным на основе сульфонамидоксина с получением аминокислоты, называемой C-Sox (CSx). Реакции проводят в 50 мкл объемах в 96-луночных планшетах, и они содержат 0,5 нМ полноразмерного автофосфорилированного рекомбинантного HPK1 человека (способ получения см. ниже), 3 мкМ субстрата фосфоакцепторного пептида (субстрат пептида Ас-[CSx]HSLPRFNR-амид, также известный как AQT0178, AssayQuant Technologies Inc., Hopkinton, MA), тестируемое соединение (11 доз, 3-кратные серийные разведения, конечная концентрация 2% ДМСО) или только ДМСО, 0,002% Tween-20, 1 мМ DTT и 2,5 мМ MgCl2 в 50 мМ буфере MOPS (3-(N-морфолино)пропансульфокислота), рН 7,8, и инициируют добавлением 45 мкМ АТФ после 20-минутной предварительной инкубации. Начальные скорости реакции определяют, следя за флуоресценцией пептида (λех=360 нм, λem=500 нм) при 30°С в течение 15 минут на планшетном ридере Tecan М1000 (Tecan Group Ltd., Mannedorf, Zurich, Switzerland). Значения константы ингибирования (Ki) рассчитывают путем подгонки начальных скоростей реакции к уравнению Моррисона (2) для конкурентного ингибирования с сильным связыванием с использованием способа нелинейной регрессии и экспериментально измеренной кажущейся Km АТФ (19 мкМ). Кинетические и кристаллографические исследования показывают, что ингибиторы являются АТФ-конкурентными.
Получение аутофосфорилированного полноразмерного рекомбинантного HPK1
Последовательность ДНК, кодирующую полноразмерный HPK1 человека (Genbank NM_001042600.2), синтезируют в GenScript, и она содержит ограниченное количество замен молчащих оснований для устранения определенных сайтов узнавания рестрикционными эндонуклеазами, и слита с последовательностью ДНК, кодирующей расщепляемую вирусом травления табака (TEV) N-концевую полигистидиновая метку для очистки. Рекомбинантный бакуловирус получают с использованием способа Bac-to-Bac (InVitrogen) и используют для заражения 9 л клеток насекомых ST21. Все стадии очистки проводят при 4°С. Клетки, содержащие рекомбинантный HPK1, лизируют в 50 мМ HEPES-NaOH, рН 7,5, 250 мМ NaCl, 1 мМ трис(2-карбоксиэтил)фосфина (ТСЕР), 10 мкМ лейпептина (Sigma), 10 мкМ Е-64 (Sigma) и одну таблетку ингибитора протеазы «без ЭДТА» (Roche) на 75 мл буфера, центрифугируют при 15000хд в течение 1 часа, и надосадочную жидкость пропускали через 5 мл смолы ProBond IMAC (InVitrogen). После промывки, HPK1 элюируют 12,5 мл 50 мМ HEPES-NaOH, рН 7,5, 400 мМ NaCl, 250 мМ имидазол-HCl, рН 7,5, 1 мМ ТСЕР, 2 мкМ лейпептина и 2 мкМ Е-64. Элюированный HPK1 диализируют в течение ночи против 0,8 л 50 мМ HEPES-NaOH, рН 7,5, 400 мМ NaCl, 20 мМ имидазол-HCl, рН 7,5, 1 мМ ТСЕР, 2 мМ лейпептина, 2 мМ Е-64, 2 мМ АТФ-NaOH рН 7,3, 5 мМ MgCl2 и 1 мг TEV (эта стадия одновременно позволяет снизить концентрацию имидазола при подготовке к последующей стадии обратной IMAC-хроматографии, удалить полигистидиновую метку очистки с помощью протеазы TEV и автофосфорилировать HPK1 в присутствии МдАТФ). HPK1 с удаленной меткой и аутофосфорилированный доводят до 40 мМ имидазол-HCl, рН 7,5, и пропускают через 10 мл колонку со смолой ProBond, уравновешенную аналогичным образом. Не меченый HPK1, прошедший через колонку, концентрируют примерно до 7 мл и дополнительно очищают гель-фильтрацией через эксклюзионную колонку Superdex 26/60 (GE Healthcare), уравновешенную и обработанную в 41,7 мМ буфере HEPES-NaOH, рН 7,5, содержащем 417 мМ NaCl и 0,417 мМ ТСЕР. Пиковые фракции, содержащие HPK1, объединяют и концентрируют примерно до 2,5 мл с использованием центрифужного концентратора (Millipore). Концентрированный HPK1 доводят до 20% (об./об.) глицерина путем добавления 50% (об./об.) глицерина, в результате чего получают окончательный состав 25 мМ HEPES-NaOH, рН 7,5, 250 мМ NaCl, 0,25 мМ ТСЕР и 20% (об./об.) глицерина. Конечная концентрация белка составляет приблизительно 1 мг/мл, по данным анализа Бредфорда (реактив Пирса) и с использованием бычьего сывороточного альбумина (стандарт Пирса 2 мг/мл) в качестве стандарта белка. Анализ масс-спектрограммы показывает, что аутофосфорилированный HPK1 содержит в среднем 15 фосфатов на мономер. Аликвоты фермента мгновенно замораживают в жидком азоте и хранят при -80°С. Конечный продукт содержит глицин-сериновое удлинение на N-конце полноразмерного белка в результате стратегии клонирования.
Окончательная последовательность ДНК полноразмерного HPK1 с N-концевой меткой (SEQ ID NO. 1) включает последовательность после солидуса после удаления метки очистки (подчеркнута), которая кодирует полноразмерный HPK1 с дополнительной последовательностью 5'-GGATCC BamR I сайта клонирования (SEQ ID NO. 3):


Конечная белковая последовательность меченого на N-конце полноразмерного HPK1 (SEQ ID NO. 2) включает последовательность после солидуса после удаления метки очистки (подчеркнута) с дополнительной последовательностью GlySer на N-конце, кодируемой из сайта клонирования BamH I (SEQ ID NO. 4):

Клеточные анализы
Фосфо-SLP-76 (Ser376) гомогенный времяразрешенный анализ флуоресценции (HTRF)
Клетки,7urkat высевают по 90000 клеток/лунку в 90 мкл питательной среды RPMI1640, содержащей 10% FBS, и инкубируют при 37°С с 5% CO2 в течение ночи. На следующий день, соединения серийно разводят, начиная с максимальной дозы 10 мМ, для получения 11-точечной кривой 3-кратного разведения в ДМСО. Соединения промежуточно разводят 1:100 в среде для выращивания перед разведением 1:10 на клетках до конечной концентрации от 10 мкМ до 0,1 нМ в 0,1% ДМСО. После 30-минутной предварительной обработки соединениями, клетки стимулируют с использованием 200 мкг/мл анти-CD3 в комплексе с F(ab)2 (клон UCTH1) в течение 15 минут при 37°С с 5% CO2. Стимуляцию прекращают ледяным PBS, и клетки собирают центрифугированием перед лизисом в буфере для лизиса Cisbio (Cisbio, Bedford, MA). Лизаты переносят на белые планшеты небольшого объема, содержащие антитела против фосфо-SLP-76-криптата плюс анти-фосфо-SLP-76-d2 HTRF, и инкубируют в течение ночи при комнатной температуре в защищенном от света месте в соответствии с протоколом производителя (Cisbio, Bedford, MA). HTRF измеряют на приборе Perkin Elmer Envision, и значения IC50 рассчитывают путем подгонки кривой зависимости концентрации от реакции с использованием четырехпараметрического нелинейного регрессионного анализа.
Данные о биологической активности соединений представлены в таблице 8. Данные для HPK1 CHEF представлены в таблице 8 в столбце с заголовком HPK1 Ki (мкМ). Данные для клеточных анализов гомогенной флуоресценции с временным разрешением (HTRF) Phospho-SLP-76 (Ser376) представлены в таблице 8 в столбце с заголовком pSLP76 IC50 (мкМ). Для каждого из этих двух анализов в соответствующем соседнем столбце указано, сколько раз соединение тестировалось в анализе.
Анализ биохимической селективности киназы
Анализы киназы PRKD2 (PKD2) и RPS6KA2 (RSK3) проводят с Кт-уровнями АТФ в компании Thermo Fisher Scientific, Inc. (Madison, WI) с использованием их Invitrogen™SelectScreen™ для резонансного переноса энергии флуоресценции (FRET) Z'-LYTE™, основанной на различной чувствительности фосфорилированных и не фосфорилированных пептидов к протеолитическому расщеплению. Рекомбинантные полноразмерные меченые GST PRKD2 (PKD2) и RPS6KA2 (RSK3) человека производит компания Thermo Fisher Scientific, Inc. (Madison, WI). Киназные анализы проводят в 10-мкл реакционных смесях. Реакции PRKD2 (PKD2) содержат 0,64-5,84 нг фермента, 25 мкМ АТФ (~Km), 2 мкМ Ser/Thr 17 (пептидный субстрат Z'-LYTE™), 10 мМ MgCl2, 0,01% BRIJ-35, 1 мМ EGTA в 50 мМ НЕ PES, рН 7,5. Реакции RPS6KA2 (RSK3) проводят аналогично, и они содержат 0,5-9 нг фермента или 10 мкМ АТФ (~Km) и 2 мкМ пептида Ser/Thr 06. После 1-часовой инкубации киназной реакции, к реакциям PRKD2 (PKD2) и RPS6KA2 (RSK3), соответственно, добавляют 5 мкл раствора Z'-LYTE™ Development Reagent А в разведении 1:256 и 1:4096. Измеряют степень киназных реакций, приводящих к изменению сигнала FRET пептидного субстрата, и ингибирование каждой киназы измеряют по отношению к контролю ДМСО и представляют как среднее значение повторных измерений. Каждое определение 50% ингибирующей концентрации (IC50) основано на 10-дозовых повторных измерениях, соответствующих стандартному четырехпараметрическому уравнению IC50 с использованием программного обеспечения XLfit от IDBS (Guildford, United Kingdom). Кривая доза-ответ соответствует кривой модели номер 205 (сигмоидальная модель доза-ответ). Отношение IC50 PRKD2 к HPK1 Ki и отношение IC50 RSK3 к HPK1 Ki представлено в соответствующих столбцах, соседних со столбцами для PRKD2 IC50 и RSK3 IC50.
Альфа-разветвленные аналоги аминов, где, по меньшей мере, один из R3a и R3b отличен от водорода, значительно улучшают селективность в отношении HPK1 по сравнению с другими киназами (включая PRKD2 и RSK3) по сравнению с не-альфа-разветвленными соединениями амина, например, примером 300. Циклические, конденсированные триазолы, например, соединения формулы I-B, где R4 представляет собой (R4-ii), обеспечивают улучшение биохимической и клеточной эффективности по сравнению с не циклическими соединениями, например, примером 300.



Пустое поле в Таблице 8 означает, что данные не были получены.
Все публикации и патентные заявки, цитируемые в описании, полностью включены в настоящее описание посредством ссылки. Специалистам в данной области техники будет очевидно, что в них могут быть внесены определенные изменения и модификации без отклонения от сущности или объема прилагаемой формулы изобретения.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Pfizer Inc.
Barber, Joyann
Cho-Schultz, Sujin
Del Bel, Matthew L.
Gallego, Rebecca Anne
He, Mingying
Jalaie, Mehran
Kania, Robert Steven
McTigue, Michele Ann
Nair, Sajiv Krishnan
Schmitt, Anne-Marie Dechert
Tuttle, Jamison Bryce
Zhou, Dahui
Zhou, Ru
<120> Соединения азалактама в качестве ингибиторов HPK1
<130> PC72605A
<150> US 63/018,689
<151> 2020-05-01
<160> 4
<170> PatentIn version 3.5
<210> 1
<211> 2472
<212> ДНК
<213> Homo sapiens
<400> 1
ggatccatgg acgtcgtgga ccctgacatt ttcaatagag acccccggga ccactatgac 60
ctgctacagc ggctgggtgg cggcacgtat ggggaagtct ttaaggctcg cgacaaggtg 120
tcaggggacc tggtggcact gaagatggtg aagatggagc ctgatgatga tgtctccacc 180
cttcagaagg aaatcctcat attgaaaact tgccggcacg ccaacatcgt ggcctaccac 240
gggagttatc tctggttgca gaaactctgg atctgcatgg agttctgtgg ggctggttct 300
ctccaggaca tctaccaagt gacaggctcc ctgtcagagc tccagattag ctatgtctgc 360
cgggaagtgc tccagggact ggcctatttg cactcacaga agaagataca cagggacatc 420
aagggagcta acatcctcat caatgatgct ggggaggtca gattggctga ctttggcatc 480
tcggcccaga ttggggctac actggccaga cgcctctctt tcattgggac accctactgg 540
atggctccgg aagtggcagc tgtggccctg aagggaggat acaatgagct gtgtgacatc 600
tggtccctgg gcatcacggc catcgaactg gccgagctac agccaccgct ctttgatgtg 660
caccctctca gagttctctt cctcatgacc aagagtggct accagcctcc ccgactgaag 720
gaaaaaggca aatggtcggc tgccttccac aacttcatca aagtcactct gactaagagt 780
cccaagaaac gacccagcgc caccaagatg ctcagtcatc aactggtatc ccagcctggg 840
ctgaatcgag gcctgatcct ggatcttctt gacaaactga agaatcccgg gaaaggaccc 900
tccattgggg acattgagga tgaggagccc gagctacccc ctgctatccc tcggcggatc 960
agatccaccc accgctccag ctctctgggc atcccagatg cagactgctg tcggcggcac 1020
atggagttca ggaagctccg aggaatggag accagacccc cagccaacac cgctcgccta 1080
cagcctcccc gagacctcag gagcagcagc cccaggaagc aactgtcaga gtcgtctgac 1140
gatgactatg acgacgtgga catccccacc cctgcagagg acacacctcc tccacttccc 1200
cccaagccca agttccgttc tccatcagac gagggtcctg ggagcatggg ggatgatggg 1260
cagctgagcc cgggggtgct ggtccggtgt gccagtgggc ccccaccaaa cagcccccgt 1320
cctgggcctc ccccatccac cagcagcccc cacctcaccg cccattcaga accctcactc 1380
tggaacccac cctcccggga gcttgacaag cccccacttc tgccccccaa gaaggaaaag 1440
atgaagagaa agggatgtgc ccttctcgta aagttgttca atggctgccc cctccgcatc 1500
cacagcacgg ccgcctggac acatccctcc accaaggacc agcacctgct cctgggggca 1560
gaggaaggca tcttcatcct gaaccggaat gaccaggagg ccacgctgga aatgctcttt 1620
cctagccgga ctacgtgggt gtactccatc aacaacgttc tcatgtctct ctcaggaaag 1680
accccccacc tgtattctca tagcatcctt ggcctgctgg aacggaaaga gaccagagca 1740
ggaaacccca tcgctcacat tagcccccac cgcctactgg caaggaagaa catggtttcc 1800
accaagatcc aggacaccaa aggctgccgg gcgtgctgtg tggcggaggg tgcgagctct 1860
gggggcccgt tcctgtgcgg tgcattggag acgtccgttg tcctgcttca gtggtaccag 1920
cccatgaaca aattcctgct tgtccggcag gtgctgttcc cactgccgac gcctctgtcc 1980
gtgttcgcgc tgctgaccgg gccaggctct gagctgcccg ctgtgtgcat cggcgtgagc 2040
cccgggcggc cggggaagtc ggtgctcttc cacacggtgc gctttggcgc gctctcttgc 2100
tggctgggcg agatgagcac cgagcacagg ggacccgtgc aggtgaccca ggtagaggaa 2160
gatatggtga tggtgttgat ggatggctct gtgaagctgg tgaccccgga ggggtcccca 2220
gtccggggac ttcgcacacc tgagatcccc atgaccgaag cggtggaggc cgtggctatg 2280
gttggaggtc agcttcaggc cttctggaag catggagtgc aggtgtgggc tctaggctcg 2340
gatcagctgc tacaggagct gagagaccct accctcactt tccgtctgct tggctccccc 2400
aggcctgtag tggtggagac acgcccagtg gatgatccta ctgctcccag caacctctac 2460
atccaggaat ga 2472
<210> 2
<211> 823
<212> БЕЛОК
<213> Homo sapiens
<400> 2
Gly Ser Met Asp Val Val Asp Pro Asp Ile Phe Asn Arg Asp Pro Arg
1 5 10 15
Asp His Tyr Asp Leu Leu Gln Arg Leu Gly Gly Gly Thr Tyr Gly Glu
20 25 30
Val Phe Lys Ala Arg Asp Lys Val Ser Gly Asp Leu Val Ala Leu Lys
35 40 45
Met Val Lys Met Glu Pro Asp Asp Asp Val Ser Thr Leu Gln Lys Glu
50 55 60
Ile Leu Ile Leu Lys Thr Cys Arg His Ala Asn Ile Val Ala Tyr His
65 70 75 80
Gly Ser Tyr Leu Trp Leu Gln Lys Leu Trp Ile Cys Met Glu Phe Cys
85 90 95
Gly Ala Gly Ser Leu Gln Asp Ile Tyr Gln Val Thr Gly Ser Leu Ser
100 105 110
Glu Leu Gln Ile Ser Tyr Val Cys Arg Glu Val Leu Gln Gly Leu Ala
115 120 125
Tyr Leu His Ser Gln Lys Lys Ile His Arg Asp Ile Lys Gly Ala Asn
130 135 140
Ile Leu Ile Asn Asp Ala Gly Glu Val Arg Leu Ala Asp Phe Gly Ile
145 150 155 160
Ser Ala Gln Ile Gly Ala Thr Leu Ala Arg Arg Leu Ser Phe Ile Gly
165 170 175
Thr Pro Tyr Trp Met Ala Pro Glu Val Ala Ala Val Ala Leu Lys Gly
180 185 190
Gly Tyr Asn Glu Leu Cys Asp Ile Trp Ser Leu Gly Ile Thr Ala Ile
195 200 205
Glu Leu Ala Glu Leu Gln Pro Pro Leu Phe Asp Val His Pro Leu Arg
210 215 220
Val Leu Phe Leu Met Thr Lys Ser Gly Tyr Gln Pro Pro Arg Leu Lys
225 230 235 240
Glu Lys Gly Lys Trp Ser Ala Ala Phe His Asn Phe Ile Lys Val Thr
245 250 255
Leu Thr Lys Ser Pro Lys Lys Arg Pro Ser Ala Thr Lys Met Leu Ser
260 265 270
His Gln Leu Val Ser Gln Pro Gly Leu Asn Arg Gly Leu Ile Leu Asp
275 280 285
Leu Leu Asp Lys Leu Lys Asn Pro Gly Lys Gly Pro Ser Ile Gly Asp
290 295 300
Ile Glu Asp Glu Glu Pro Glu Leu Pro Pro Ala Ile Pro Arg Arg Ile
305 310 315 320
Arg Ser Thr His Arg Ser Ser Ser Leu Gly Ile Pro Asp Ala Asp Cys
325 330 335
Cys Arg Arg His Met Glu Phe Arg Lys Leu Arg Gly Met Glu Thr Arg
340 345 350
Pro Pro Ala Asn Thr Ala Arg Leu Gln Pro Pro Arg Asp Leu Arg Ser
355 360 365
Ser Ser Pro Arg Lys Gln Leu Ser Glu Ser Ser Asp Asp Asp Tyr Asp
370 375 380
Asp Val Asp Ile Pro Thr Pro Ala Glu Asp Thr Pro Pro Pro Leu Pro
385 390 395 400
Pro Lys Pro Lys Phe Arg Ser Pro Ser Asp Glu Gly Pro Gly Ser Met
405 410 415
Gly Asp Asp Gly Gln Leu Ser Pro Gly Val Leu Val Arg Cys Ala Ser
420 425 430
Gly Pro Pro Pro Asn Ser Pro Arg Pro Gly Pro Pro Pro Ser Thr Ser
435 440 445
Ser Pro His Leu Thr Ala His Ser Glu Pro Ser Leu Trp Asn Pro Pro
450 455 460
Ser Arg Glu Leu Asp Lys Pro Pro Leu Leu Pro Pro Lys Lys Glu Lys
465 470 475 480
Met Lys Arg Lys Gly Cys Ala Leu Leu Val Lys Leu Phe Asn Gly Cys
485 490 495
Pro Leu Arg Ile His Ser Thr Ala Ala Trp Thr His Pro Ser Thr Lys
500 505 510
Asp Gln His Leu Leu Leu Gly Ala Glu Glu Gly Ile Phe Ile Leu Asn
515 520 525
Arg Asn Asp Gln Glu Ala Thr Leu Glu Met Leu Phe Pro Ser Arg Thr
530 535 540
Thr Trp Val Tyr Ser Ile Asn Asn Val Leu Met Ser Leu Ser Gly Lys
545 550 555 560
Thr Pro His Leu Tyr Ser His Ser Ile Leu Gly Leu Leu Glu Arg Lys
565 570 575
Glu Thr Arg Ala Gly Asn Pro Ile Ala His Ile Ser Pro His Arg Leu
580 585 590
Leu Ala Arg Lys Asn Met Val Ser Thr Lys Ile Gln Asp Thr Lys Gly
595 600 605
Cys Arg Ala Cys Cys Val Ala Glu Gly Ala Ser Ser Gly Gly Pro Phe
610 615 620
Leu Cys Gly Ala Leu Glu Thr Ser Val Val Leu Leu Gln Trp Tyr Gln
625 630 635 640
Pro Met Asn Lys Phe Leu Leu Val Arg Gln Val Leu Phe Pro Leu Pro
645 650 655
Thr Pro Leu Ser Val Phe Ala Leu Leu Thr Gly Pro Gly Ser Glu Leu
660 665 670
Pro Ala Val Cys Ile Gly Val Ser Pro Gly Arg Pro Gly Lys Ser Val
675 680 685
Leu Phe His Thr Val Arg Phe Gly Ala Leu Ser Cys Trp Leu Gly Glu
690 695 700
Met Ser Thr Glu His Arg Gly Pro Val Gln Val Thr Gln Val Glu Glu
705 710 715 720
Asp Met Val Met Val Leu Met Asp Gly Ser Val Lys Leu Val Thr Pro
725 730 735
Glu Gly Ser Pro Val Arg Gly Leu Arg Thr Pro Glu Ile Pro Met Thr
740 745 750
Glu Ala Val Glu Ala Val Ala Met Val Gly Gly Gln Leu Gln Ala Phe
755 760 765
Trp Lys His Gly Val Gln Val Trp Ala Leu Gly Ser Asp Gln Leu Leu
770 775 780
Gln Glu Leu Arg Asp Pro Thr Leu Thr Phe Arg Leu Leu Gly Ser Pro
785 790 795 800
Arg Pro Val Val Val Glu Thr Arg Pro Val Asp Asp Pro Thr Ala Pro
805 810 815
Ser Asn Leu Tyr Ile Gln Glu
820
<210> 3
<211> 66
<212> ДНК
<213> Homo sapiens
<400> 3
atggcgtcgc atcaccatca ccatcacgat tacgatggtg ctacgaccga aaacctgtat 60
tttcag 66
<210> 4
<211> 22
<212> БЕЛОК
<213> Homo sapiens
<400> 4
Met Ala Ser His His His His His His Asp Tyr Asp Gly Ala Thr Thr
1 5 10 15
Glu Asn Leu Tyr Phe Gln
20
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2019 |
|
RU2801140C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| СУЛЬФОНАМИДСОДЕРЖАЩИЕ СВЯЗЫВАЮЩИЕ СИСТЕМЫ ДЛЯ ЛЕКАРСТВЕННЫХ КОНЪЮГАТОВ | 2014 |
|
RU2729194C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2687093C1 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| НОВЫЕ АКТИВАТОРЫ РАСТВОРИМОЙ ГУАНИЛАТЦИКЛАЗЫ И ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗЫ С ДВОЙНЫМ МЕХАНИЗМОМ ДЕЙСТВИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2758373C2 |
Группа изобретений относится к органической химии и включает соединение формулы I, его фармацевтически приемлемые соли, стереоизомеры, индивидуальные соединения, указанные в формуле изобретения, фармацевтическую композицию, их содержащую, и способы лечения. В формуле I R1 представляет собой (C1-C6)алкил, -N(R5)(R6), или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил замещен 0 или 1 заместителем, который представляет собой (C1-C6)алкил или (C1-C6)алкокси; R5 и R6 каждый независимо представляет собой водород или (C1-C6)алкил, или R5 и R6 вместе с азотом, к которому они присоединены, образуют (4-8-членный) гетероциклоалкил, который замещен 0, 1 или 2 заместителями, независимо выбранными из группы, состоящей из (C1-C6)алкила, галоген(C1-C6)алкила и (C1-C6)алкокси; R2 представляет собой N(R7)(R8), каждый из R7 и R8 независимо представляет собой водород или (C1-C6)алкил, который замещен 0 или 1 заместителем, представляющим собой галоген, (C1-C6)алкокси, циано или гидрокси; R3a представляет собой водород или (C1-C3)алкил, замещенный 0 или 1 заместителем, представляющим собой гидрокси или (C1-C3)алкокси; R3b представляет собой водород или (C1-C3)алкил, при условии, что R3a и R3b оба не представляют собой Н, когда R4 представляет собой (R4-i); R4 представляет собой (R4-i) или (R4-ii); R4N представляет собой (C1-C6)алкил, галоген(C1-C6)алкил или (С3-С6)циклоалкил; R4C представляет собой водород, (C1-C6)алкил, галоген(C1-C6)алкил или (C1-C6)алкокси; R4D представляет собой водород, (C1-C6)алкил, галоген(C1-C6)алкил, галоген(C1-C6)алкокси или (С3-С6)циклоалкил, где указанные (C1-C6)алкил и галоген(C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси или (C1-C6)алкокси; R4E представляет собой водород или (C1-C6)алкил; R4F представляет собой водород, галоген, (C1-C6)алкил или галоген(C1-C6)алкил, где указанные (C1-C6)алкил и галоген(C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (C1-C6)алкокси. Технический результат – производные 2-(пиридин-2-ил)-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-она формулы I, обладающие ингибирующей активностью в отношении киназы HPK1. 8 н. и 13 з.п. ф-лы, 8 табл., 300 пр.

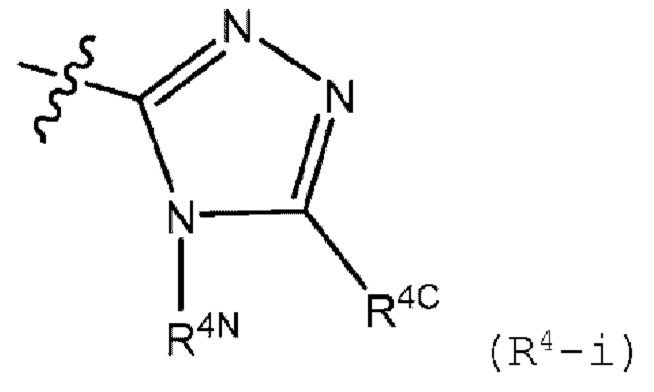
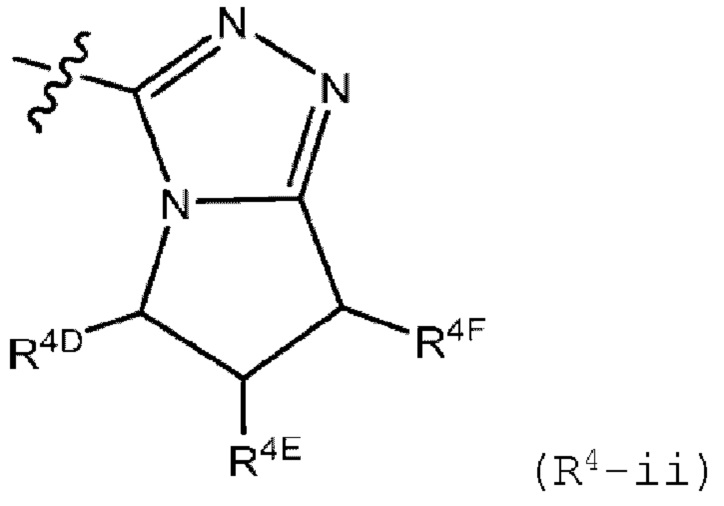
1. Соединение формулы I:

или его фармацевтически приемлемую соль или стереоизомер, где:
R1 представляет собой (C1-C6)алкил, -N(R5)(R6), или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил замещен 0 или 1 заместителем, который представляет собой (C1-C6)алкил или (C1-C6)алкокси, где:
R5 и R6 каждый независимо представляет собой водород или (C1-C6)алкил, или
R5 и R6 вместе с азотом, к которому они присоединены, образуют (4-8-членный) гетероциклоалкил, который замещен 0, 1 или 2 заместителями, независимо выбранными из группы, состоящей из (C1-C6)алкила, галоген(C1-C6)алкила и (C1-C6)алкокси;
R2 представляет собой N(R7)(R8), где:
каждый из R7 и R8 независимо представляет собой водород или (C1-C6)алкил, который замещен 0 или 1 заместителем, представляющим собой галоген, (C1-C6)алкокси, циано или гидрокси;
R3a представляет собой водород или (C1-C3)алкил, замещенный 0 или 1 заместителем, представляющим собой гидрокси или (C1-C3)алкокси;
R3b представляет собой водород или (C1-C3)алкил, при условии, что R3a и R3b оба не представляют собой Н, когда R4 представляет собой (R4-i);
R4 представляет собой (R4-i) или (R4-ii):
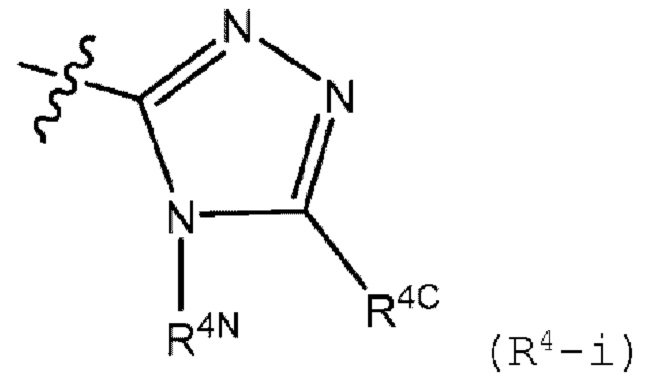

где:
R4N представляет собой (C1-C6)алкил, галоген(C1-C6)алкил или (С3-С6)циклоалкил;
R4C представляет собой водород, (C1-C6)алкил, галоген(C1-C6)алкил или (C1-C6)алкокси;
R4D представляет собой водород, (C1-C6)алкил, галоген(C1-C6)алкил, галоген(C1-C6)алкокси или (С3-С6)циклоалкил, где указанные (C1-C6)алкил и галоген(C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси или (C1-C6)алкокси;
R4E представляет собой водород или (C1-C6)алкил; и
R4F представляет собой водород, галоген, (C1-C6)алкил или галоген(C1-C6)алкил, где указанные (C1-C6)алкил и галоген(C1-C6)алкил замещены 0 или 1 заместителем, который представляет собой гидрокси, циано или (C1-C6)алкокси.
2. Соединение по п. 1 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R4 представляет собой (R4-i);
R4N представляет собой (C1-C6)алкил или (С3-С6)циклоалкил; и
R4C представляет собой водород или (C1-C3)алкил.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 представляет собой -N(R5)(R6) или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил представляет собой циклопропил и замещен 0 или 1 заместителем, который представляет собой (C1-C6)алкил, где указанный (C1-C6)алкил представляет собой метил;
R5 и R6 каждый независимо представляет собой водород или (С1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой (C1-C6)алкил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или (С1-С6)алкил, который представляет собой метил;
R3a представляет собой (С1-С3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси; и
R3b представляет собой водород или (С1-С3)алкил.
4. Соединение по п. 1 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R4 представляет собой (R4-ii);
R4D представляет собой водород, (C1-C6)алкил или галоген(С1-С6)алкил, где указанный (С1-С6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси;
R4E представляет собой водород; и
R4F представляет собой водород, (С1-С6)алкил или галоген(C1-C6)алкил, где указанный (C1-C6)алкил замещен 0 или 1 заместителем, который представляет собой гидрокси.
5. Соединение по п. 1 или 4 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 представляет собой -N(R5)(R6) или (С3-С6)циклоалкил, где указанный (С3-С6)циклоалкил представляет собой циклопропил и замещен 0 или 1 заместителем, который представляет собой (C1-C6)алкил, где указанный (C1-C6)алкил представляет собой метил;
R5 и R6 каждый независимо представляет собой водород или (C1-С3)алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют (5-членный) гетероциклоалкил, замещенный 0 или 1 заместителем, который представляет собой (C1-C6)алкил;
R2 представляет собой N(R7)(R8), где каждый из R7 и R8 независимо представляет собой водород или (C1-C6)алкил, который представляет собой метил;
R3a представляет собой водород или (С1-С3)алкил, замещенный 0 или 1 заместителем, который представляет собой (С1-С3)алкокси; и
R3b представляет собой водород или (С1-С3)алкил.
6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 представляет собой 2-метилпирролидин-1-ил или 2(R)-метилпирролидин-1-ил.
7. Соединение по любому из пп. 1-6 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что каждый из R7 и R8 представляет собой водород.
8. Соединение по любому из пп. 1-6 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R7 представляет собой водород; и R8 представляет собой (С1-С3)алкил.
9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R3b представляет собой водород, и где ориентация R2, R3a и R3b дает соединение формулы I(R) или формулы I(S):


10. Соединение по любому из пп. 1 и 4-9 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R4 представляет собой 5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил, (5R)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил, (5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил, 5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил, (5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил или (5R)-5-этил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил, где указанный метил замещен 0 или 1 заместителем, который представляет собой F или ОН.
11. Соединение по п. 10 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R4 представляет собой (5S)-5-метил-6,7-дигидро-5Н-пирроло[2,1-c][1,2,4]триазол-3-ил или (5S)-5-этил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-3-ил, где указанный метил замещен 0 или 1 заместителем, который представляет собой F или ОН.
12. Соединение, представляющее собой 4-[1-аминопропил]-2-{6-[5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[1-аминоэтил]-2-{6-[5-этил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[1-аминоэтил]-2-{6-[5-этил-6,7-дигидро-5H-пирроло[2,1-с][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[1-аминоэтил]-2-{6-[5-этил-6,7-дигидро-5H-пирроло[2,1-е][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он;
4-[1-аминопропил]-2-{3-[5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-с]пиридин-1-он; или
4-[1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он.
13. Соединение по п. 12, отличающееся тем, что соединение представляет собой 4-[(1R)-1-аминопропил]-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1S)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1R)-1-аминопропил]-2-{3-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]фенил}-6-[метил(пропан-2-ил)амино]-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он;
4-[(1R)-1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он; или
4-[(1S)-1-амино-2-метоксиэтил]-6-(1-метилциклопропил)-2-{6-[(5S)-5-метил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-2,3-дигидро-1H-пирроло[3,4-c]пиридин-1-он.
14. Соединение, имеющее структуру:

15. Соединение, представляющее собой 4-[(1R)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1Н-пирроло[3,4-с]пиридин-1-он.
16. Соединение, представляющее собой 4-[(1S)-1-аминоэтил]-2-{6-[(5S)-5-этил-6,7-дигидро-5H-пирроло[2,1-c][1,2,4]триазол-3-ил]пиридин-2-ил}-6-[(2R)-2-метилпирролидин-1-ил]-2,3-дигидро-1Н-пирроло[3,4-c]пиридин-1-он.
17. Фармацевтическая композиция для ингибирования HPK1, содержащая терапевтически эффективное количество соединения по любому из пп. 1-16 или его фармацевтически приемлемую соль или стереоизомер и фармацевтически приемлемый носитель или эксципиент.
18. Способ лечения аномального роста клеток, связанного с ингибированием активности HPK1 у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пп. 1-16 или его фармацевтически приемлемой соли или стереоизомера.
19. Способ по п. 18, отличающийся тем, что аномальный рост клеток представляет собой рак.
20. Способ лечения аномального роста клеток, связанного с ингибированием активности HPK1 у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пп. 1-16 или его фармацевтически приемлемой соли или стереоизомера, и дополнительно включающий введение количества дополнительного противоракового терапевтического агента, количества которых вместе эффективны при лечении указанного рака.
21. Способ по п. 20, где рак выбран из группы, состоящей из рака молочной железы, рака яичников, рака мочевого пузыря, рака матки, рака предстательной железы, рака легких, рака пищевода, рака головы и шеи, колоректального рака, рака почки, рака печени, рака поджелудочной железы, рака желудка и рака щитовидной железы.
| WO 2019015559 A1, 24.01.2019 | |||
| WO 2018167147 A1, 20.09.2018 | |||
| EA 201890059 A1, 31.07.2018. |
