ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет международной патентной заявки № PCT/CN2019/094634, поданной 4 июля 2019 г. Все содержание вышеупомянутой заявки включено в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Гемопоэтическая киназа-предшественник 1 (HPK1), также известная как киназа-киназа-киназа митоген-активируемой протеинкиназы-1 (MAP4K1), является протеинкиназой, которая действует выше классического 3-уровневого пути MAPK, который включает MAP3K (MAP киназа киназа), которая активирует MAP2K (MAP-киназа киназа), которая, в свою очередь, активирует двойной Thr и Tyr член семейства MAPK JNK (c-Jun N-терминальную киназу). Первоначально клонированные в гемопоэтических клетках-предшественниках, HPK1/MAP4K1 преимущественно экспрессируются в лимфоидных органах/тканях, включая костный мозг, печень плода, лимфатические узлы, плаценту, селезенку и тимус (Hu et al., Gene & Dev. 10(18): 2251-2264, 1996; Kiefer et al., The EMBO J. 15(24): 7013-7025, 1996). На клеточном уровне, HPK1 экспрессируется во всех типах клеток гемопоэтического компартмента, включая гемопоэтические клетки-предшественники, Т-клетки, В-клетки, макрофаги, дендритные клетки, нейтрофилы и тучные клетки (Hu, выше; Kiefer, выше).
HPK1/MAP4K1 является одной из шести MAP4K, включающих HPK1 (MAP4K1), GCK (MAP4K2), GLK (MAP4K3), HGK/NIK (MAP4K4), KHS/GCKR (MAP4K5) и MINK (MAP4K6). Вместе эти MAP4K являются членами примерно 26 Ste20-подобных серин/треонинкиназ млекопитающих, идентифицированных к настоящему времени, которые являются гомологами белка дрожжей sterile20 (Ste20p), предполагаемого MAP4K, который активирует MAP3K в сигнальном пути дрожжевого феромона. Эти Ste20-подобные киназы млекопитающих делятся на два подсемейства на основе доменных структур: p21-активируемые киназы (PAK) и киназы зародышевого центра (GCK). Среди подсемейства GCK некоторые из них могут активировать киназный каскад MAP3K, что приводит к активации JNK.
MAP4K очень структурно похожи с N-концевым доменом киназы (KD), за которым следуют 2-4 мотива, богатых пролином, и C-концевым доменом гомологии цитрона (CNH).
Сайт связывания ATP киназного домена HPK1 включает Lys-46. Мутация этого остатка в Met (HPK1-M46) отменяет каталитическую активацию HPK1 (Hu, выше).
Существует несколько консервативных сайтов фосфорилирования Ser/Thr в киназном домене HPK1 и консервативный сайт фосфорилирования Tyr между его первыми двумя богатыми пролином мотивами. Фосфорилирование Tyr379 (у мыши или Tyr381 у человека) посредством LCK/ZAP70, по-видимому, необходимо для активации HPK1, поскольку дефицит LCK или ZAP70 отменяет фосфорилирование Tyr-379 и киназную активность HPK1 в Т-клетках Jurkat при анти-CD3-стимуляции. (Ling et al., JBC 276(22):18908-18914, 2001; Liou et al., Immunity 12(4):399-408, 2000; Sauer et al., JBC 276(48):45207, 45216, 2001). С другой стороны, аутофосфорилирование Thr-355 регулирует убиквитинирование и деградацию HPK1. Thr-355 является PP4-таргетным сайтом дефосфорилирования; это дефосфорилирование предотвращает CUL7/Fbxw8-опосредованное убиквитинирование и протеасомную деградацию активированной HPK1 (Wang et al., Cancer Res. 69(3):1063-1070, 2009), таким образом, HPK1 также стабилизируется и активируется протеинфосфатазой 4 (PP4) (Zhou et al., JBC 279(47):49551-49561, 2004).
Сайт фосфорилирования Tyr также примыкает к сайту расщепления каспазой (DDVD). Было показано, что полноразмерный HPK1 может расщепляться каспазой-3 в этом сайте в апоптотических клетках, что приводит к повышенной каталитической активности N-концевого фрагмента HPK1 (Chen et al., Oncogene 18:7370-7377, 1999).
Четыре Pro-богатых мотива HPK1 опосредуют взаимодействие HPK1 со многими доменсодержащими белками SH3 (Boomer & Tan, JCB 95(1): 34-44, 2005).
CNH домен HPK1 может быть вовлечен в HPK1-опосредованную адгезию лимфоцитов, поскольку домен гомологии цитрона в другой Ste20-подобной киназе TNIK связывается с Rap2 и регулирует актиновый цитоскелет (Taira et al., JBC 279(47):49488-49496, 2004).
MAP4K играют важную роль в иммунной системе, особенно в лимфоцитах, через регуляцию клеточной передачи сигналов, активацию иммунных клеток, трансформацию клеток и миграцию клеток. У мышей с нокаутом (KO) HPK1 наблюдается повышенная активация Т-клеток, повышенное продуцирование цитокинов и повышенное продуцирование антител после иммунизации KLH. Мыши HPK1 KO также более восприимчивы к индукции EAE. Т-клетки и В-клетки HPK1 KO демонстрируют усиленную клеточную активацию и передачу сигналов антигенного рецептора. Дендритные клетки HPK1 KO демонстрируют более высокие уровни костимулирующих молекул и провоспалительных цитокинов (Alzabin et al., J. Immunol. 182(10):6187-6194, 2009; Shui et al., Nat. Immunol. 8(1):84-91, 2007).
Сверхэкспрессия в клеточных линиях (например, клетках HEK293 и COS-1, гемопоэтических Т-клетках Jurkat и клетках лейкоза HL-60) продемонстрировала, что HPK1 может активировать MAPK JNK (но не p38 или ERK MAP киназы) через несколько MAP3K (включая TAK1, MEKK1 и MLK3), которые активируют MAP2Ks MKK4 и MKK7, которые, в свою очередь, активируют JNK.
Интересно, что регуляторные функции MAP4K в иммунных клетках, по-видимому, в значительной степени опосредованы независимыми от JNK механизмами. Было продемонстрировано, что активация киназы HPK1 необходима для активации IKK-NF-κB, и полагают, что HPK1 делает это посредством регуляции CARMA1. CARMA1 является адаптерным белком в так называемом комплексе CBM (CARMA1/BCL10/MALT1), который способствует активации IKKβ в Т-клетках Jurkat при стимуляции анти-CD3. Активированная IKK расщепляет IκB и высвобождает ассоциированный ядерный транскрипционный фактор NF-κB. В частности, HPK1 индуцибельно ассоциирован с CARMA1 и прямо фосфорилирует CARMA1 на Ser-551, что необходимо для активации NF-kB (Brenner et al., PNAS USA, 196(34):14508-14513, 2009).
В Т-клетках, при стимуляции TCR, лимфоцитарная протеинтирозинкиназа (Lck) фосфорилирует основанные на иммунорецепторные тирозиновые активирующие мотивы (ITAM) на цитозольной стороне комплекса TCR/CD3. Затем Zap-70 рекрутируется в комплекс TCR/CD3, где он фосфорилируется и активируется. Активированный ZAP-70 фосфорилирует адапторный белок, называемый SLP-76, который транслоцируется на мембрану плазмы и способствует образованию мультибелкового комплекса сигналосомы путем связывания с рядом белков, включая HPK1. Эти белки совместно передают сигналы TCR различным эффекторным молекулам, что приводит к активации, выживанию и пролиферации Т-лимфоцитов.
Во время этого процесса, HPK1 напрямую связывается с SH2 доменом SLP-76 и, в первую очередь, служит негативным регулятором передачи сигналов TCR. Например, передача сигналов TCR усиливается в первичных Т-клетках HPK1 KO, в том, что они демонстрируют гиперпролиферацию и продуцирование IL-2 при лигировании TCR in vitro (Shui, выше). Считается, что HPK1 может подавлять передачу сигналов TCR посредством механизма отрицательной обратной связи, через фосфорилирование адаптерного белка SLP-76 на Ser-376. При фосфорилировании Ser-376 посредством HPK1, SLP-76 связывается с 14-3-3 через фосфорилированный остаток Ser-376, что приводит к убиквитинированию остатка Lys-30 (K30) SLP-76, который впоследствии таргетируется для протеасомной деградации. HPK1 также подавляет передачу сигналов TCR с помощью аналогичного механизма в других адаптерных белках, включая GADS (например, путем фосфорилирования Thr-254 GADS и способствует взаимодействию 14-3-3).
Таким образом, оказывается, что HPK1 играет двойную и противоположную роли в активации JNK и передаче сигналов TCR. Хотя было продемонстрировано, что HPK1 напрямую активирует путь JNK в различных системах сверхэкспрессии через путь MAP3K-MAP2K-MAPK, HPK1-опосредованное ингибирование активации SLP-76 также может привести к ингибированию активности JNK в передаче сигналов TCR. Это согласуется с наблюдением, что первичные Т-клетки с нокаутом HPK1 проявляют неизмененную активность JNK (Shui, выше). Точно так же, HPK1, по-видимому, регулирует активацию IKK двумя различными и контрастирующими механизмами - с одной стороны, HPK1 активирует IKK путем прямого фосфорилирования CARMA1; с другой стороны, HPK1 также негативно регулирует активацию IKK, ингибируя активацию SLP-76. Эта, казалось бы, контрастирующая двойная роль, которую играет HPK1, лучше всего понимается тем, что HPK1 облегчает активацию JNK и IKK в начальной фазе передачи сигналов TCR, но играет критическую роль в подавлении передачи сигналов TCR в поздней фазе.
HPK1 также играет аналогичную негативную регуляторную роль в BCR-индуцированной активации и пролиферации клеток в B-клетках. В-клетки используют SLP-76-подобный адаптерный белок, называемый BLNK, для трансдукции передачи сигналов BCR, включая активацию JNK и IKK. В В-клетках, Tyr киназы Syk и Lyn способствуют фосфорилированию Tyr и активации HPK1, и полученный pY379 HPK1 опосредует связывание HPK1-BLNK. Отрицательная обратная связь HPK1 BLNK осуществляется через Thr-152 BLNK. Связывание pT152 посредством 14-3-3 приводит к убиквитинированию BLNK по нескольким остаткам Lys и последующей протеасомной деградации BLNK (таким образом ослабляя передачу сигналов BCR).
Интересно, что HPK1, по-видимому, является положительным регулятором супрессивных функций регуляторных Т-клеток (Treg) (Sawasdikosol et al., J Immunol. 188(supp. 1):163, 2012). Treg мышей Foxp3+ с дефицитом HPK1 были дефектны в подавлении TCR-индуцированной пролиферации эффекторных Т-клеток и парадоксальным образом приобретали способность продуцировать IL-2 после вовлечения TCR (Sawasdikosol, выше). Таким образом, HPK1 является важным регулятором функций Treg и периферической самотолерантности.
HPK1 также вовлечен в PGE2-опосредованное ингибирование активации CD4+ Т-клеток (Ikegami et al., J Immunol. 166(7):4689-4696, 2001). US2007/0087988 показывает, что активность HPK1 киназы повышается при воздействии физиологических концентраций PGE2 на CD4+ T-клетки посредством PGE2-индуцированной активации PKA. Пролиферация Т-клеток с дефицитом HPK1 была резистентна к подавляющему действию PGE2 (US 2007/0087988). Следовательно, PGE2-опосредованная активация HPK1 может являться новым регуляторным путем модуляции иммунного ответа.
Помимо TCR и BCR, HPK1 также трансдуцирует сигналы ниже TGF-R (рецептора трансформирующего фактора роста) (Wang et al., JBC 272(36):22771-22775, 1997) или Gs-связанных PGE2 рецепторов (EP2 и EP4) (Ikegami et al., J Immunol. 166(7):4689-4696, 2001).
HPK1 негативно регулирует адгезию иммунных клеток. В Т-клетках активация TCR также индуцирует активацию интегрина, что приводит к адгезии Т-клеток и образованию иммунологического синапса. Это достигается путем связывания SLP-76 с адаптерным белком, способствующим дегрануляции (ADAP), который необходим для TCR-индуцированной активации интегрина (Wang et al., J. Exp. Med. 200(8):1063-1074, 2004), хотя его конститутивно ассоциированный SKAP55 белок, который таргетирует активированную малую ГТФазу Rap1 на плазматическую мембрану, что приводит к активации интегрина (Kliche et al., MCB 26(19):7130-7144, 2006). Другими словами, тройной комплекс SLP-76/ADAP/SKAP55 передает сигналы TCR на молекулы адгезии семейства интегринов, тем самым способствуя адгезии Т-клеток. HPK1 отрицательно регулирует этот путь, не только подавляя SLP-76 (выше), но также конкурируя с ADAP за один и тот же сайт связывания SH2 на SLP-76, что, в свою очередь, ослабляет активность ADAP нижележащего эффектора Rap1 (Patzak et al., Eur. J. Immunol. 40(11):3220-3225, 2010).
HPK1 аналогичным образом негативно регулирует активацию интегрина и клеточную адгезию в В-клетках. Там HPK1 связан с гомологом SKAP55, называемым SKAP-HOM (Konigsberger et al., PloS One 5(9). pii: e12468, 2010), который необходим для адгезии В-клеток (Togni et al., MCB 25(18):8052-8063, 2005). Считается, что HPK1 индуцирует отрицательный сайт фосфорилирования на SKAP-HOM, который, в свою очередь, подавляет активацию Rap1.
Однако в нейтрофилах HPK1 положительно регулирует их адгезию. Перенос нейтрофилов, включая слабое скручивание, прочное связывание, распространение клеток и диапедез, контролируется снаружи внутрь передачей сигналов активации β2-интегрина, что индуцирует взаимодействие между актином и HIP-55 (HPK1-взаимодействующим белком 55 кДа). Это усиливает высокоаффинную конформацию β2-интегрина, способствуя адгезии нейтрофилов (Hepper et al., J. Immunol. 188(9):4590-4601, 2012; Schymeinsky et al., Blood 114(19):4209-4220, 2009). HPK1 совместно локализуется с HIP-55 и актином в ламеллиподии нейтрофилов при опосредованной β2-интегрином адгезии (Jakob et al., Blood 121(20):4184-4194, 2013). CXCL1-опосредованная адгезия нейтрофилов устраняется либо дефицитом HPK1, либо дефицитом HIP-55 in vitro и in vivo (Jakob, выше; Schymeinsky, выше).
В соответствии со своей ролью в подавлении функции TCR и BCR, HPK1 отрицательно регулирует адаптивные иммунные ответы, и потеря HPK1-опосредованной регуляции активации Т-клеток и иммунных ответов может быть решающим механизмом аутоиммунного патогенеза. У мышей HPK1 KO, хотя развитие Т- и В-клеток оказалось незатронутым (Shui, выше), Т-клетки этих животных показали резко повышенную активацию TCR проксимальной передачи сигналов и нижестоящих ERK, что приводит к гиперпролиферации этих клеток in vitro при анти-CD3 стимуляции (Shui, выше). Т-клетки иммунизированных мышей с дефицитом HPK1 гиперчувствительны к антигенной специфической стимуляции и продуцируют значительно более высокие уровни воспалительных цитокинов, таких как IL-2, IFN-γ и IL-4. Такие мыши также продуцируют гораздо более высокие уровни изоформ IgM и IgG, что предполагает усиленное функционирование В-клеток, нокаутированных по HPK1 (Shui, выше).
HPK1 также отрицательно контролирует аутоиммунитет у мышей, поскольку мыши HPK1 KO более чувствительны к индукции экспериментального аутоиммунного энцефаломиелита (EAE) (Shui, выше). Ослабление HPK1 также способствует аномальной активации Т- и В-клеток и аутоиммунитету у пациентов-людей. HPK1 подавляется в мононуклеарных клетках периферической крови пациентов с псориатическим артритом или в Т-клетках пациентов с системной красной волчанкой (SLE).
Физиологическая функция HPK1 не ограничивается лимфоцитами, так как HPK1 также отрицательно регулирует созревание и активацию дендритных клеток (DC) посредством неизвестного механизма (Alzabin, выше). У мышей HPK1 KO, дендритные клетки костного мозга (BMDC) демонстрируют повышенные уровни костимулирующих молекул CD80/CD86 и повышенное продуцирование провоспалительных цитокинов (Alzabin, выше). Следовательно, антигенпрезентирующая активность дендритных клеток более эффективна у мышей HPK1 KO (Alzabin, выше). Что еще более важно, эрадикация опухоли посредством HPK1 KO BMDC-опосредованного ответа CTL более эффективна, чем эрадикация посредством BMDC дикого типа (Alzabin, выше). Более того, HPK1 также может контролировать противоопухолевый иммунитет через механизмы, зависящие от Т- и В-лимфоцитов. Было показано, что адоптивный перенос Т-клеток с дефицитом HPK1 был более эффективным при контроле роста опухоли и метастазирования, чем Т-клетки дикого типа (Alzabin et al., Cancer Immunol Immunother 59(3):419-429, 2010). Аналогичным образом, BMDC от мышей с нокаутом HPK1 были более эффективны для усиления Т-клеточного ответа для эрадикации карциномы легкого Льюиса по сравнению с BMDC дикого типа (Alzabin et al., J Immunol. 182(10):6187-6194, 2009).
Таким образом, существует потребность в соединениях, ингибирующих HPK1, для лечения заболеваний или нарушений посредством модулирования активности HPK1.
СУЩНОСТЬ изобретения
В настоящем документе описаны соединения формул (I-0), (I-1), (I-2), (I), (II), (II-1), (II-2) или (II-2'), и соединения примеров (совокупно обозначаемые здесь как «соединения по изобретению»), которые ингибируют активность HPK1, и их фармацевтически приемлемые соли.
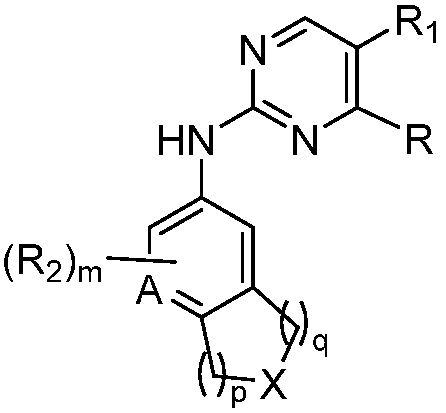
В одном аспекте, изобретение представляет соединение, представленное структурной формулой (I-0):
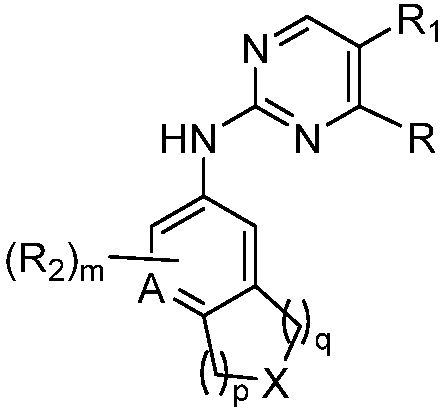 (I-0),
(I-0),
или его фармацевтически приемлемая соль или стереоизомер, где
X является СR2R3 или NR3;
A является СR2 или N;
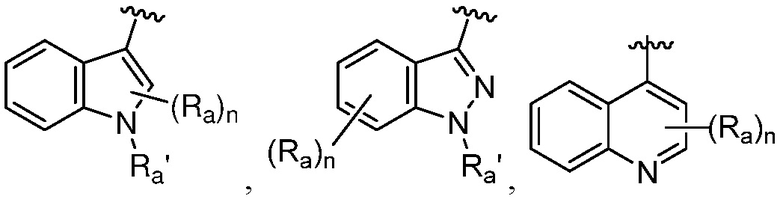
R является 8-10-членным бициклическим азотсодержащим гетероарилом или 8-10-членным бициклическим азотсодержащим гетероциклилом, необязательно замещенным оксо, где азотсодержащий гетероарил или азотсодержащий гетероциклил, представленный R, имеет 1-3 гетероатома, выбранных из N, O и S и необязательно замещен одним-четырьмя Ra, и где R либо соединен с пиримидиновым кольцом через атом азота кольца, либо R представлен структурой ниже:
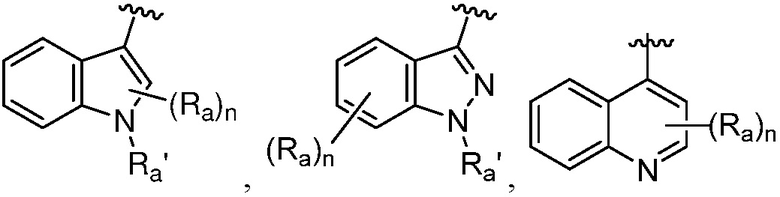 или
или 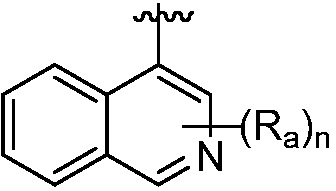 ;
;
R1 является Н, дейтерием, галоген, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, фенилом, 5-6-членным гетероарилом, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, фенил, гетероарил, циклоалкил или гетероциклил, представленный R1 или в группе, представленной R1 необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкил, C1-4 галогеналкокси и NR11R12; или
R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
 или
или 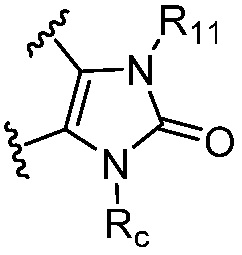 , где
, где  связи соединены с пиримидиновым кольцом;
связи соединены с пиримидиновым кольцом;
каждый случай R2 независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, P(=O)R11R12, S(=O)2R11, или S(=O)2NR11R12, где алкил, алкенил, алкинил или алкокси, представленный R2 или в группе, представленной R2, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12;
R3 является Н, C1-6 алкилом, C(O)C1-6 алкилом, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, циклоалкил, или гетероциклил, представленный R3 или в группе, представленной R3, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила, 3-7-членного гетероциклила и NR11R12;
каждый случай R11 и R12 независимо является H или C1-6 алкилом, где алкил, представленный R11 или R12, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила и 3-7-членного гетероциклила, или
R11 и R12, вместе с атомом азота или атомом фосфора, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси;
каждый случай Ra независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, COOH, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)C1-6 алкилом, C(O)OR11, NR11C(O)R12, S(=O)2R11, S(=O)2NR11R12, NR11S(=O)2R12, P(=O)R11R12, C3-6 циклоалкилом, 3-7-членным гетероциклилом или 5-6-членным гетероарилом, где алкил, алкенил, алкинил, алкокси, циклоалкил, гетероциклил или гетероарил, представленный Ra или в группе, представленной Ra, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12 и 5-6-членного гетероарила; или
два Ra, вместе с атомами углерода, к которым они присоединены, образуют C3-6 циклоалкил или 3-7-членный гетероциклил, где циклоалкил, или гетероциклил необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси;
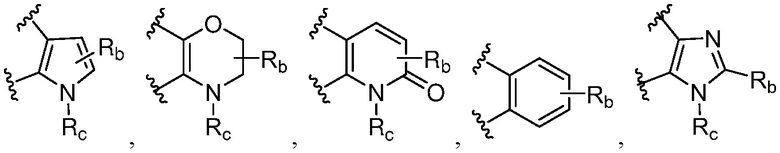
каждый случай Rb независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, COOH, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)C1-6 алкилом, C(O)OR11, NR11C(O)R12, S(=O)2R11, S(=O)2NR11R12, NR11S(=O)2R12, P(=O)R11R12, C3-6 циклоалкилом, 3-7-членным гетероциклилом или 5-6-членным гетероарилом, где алкил, алкенил, алкинил, алкокси, циклоалкил, гетероциклил или гетероарил, представленный Rb или в группе, представленной Rb, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12 и 5-6-членного гетероарила;
каждый случай Rc независимо является фенилом, 5-6-членным моноциклическим гетероциклилом, имеющим 1-3 гетероатома, выбранных из N и O; 5-6-членным моноциклическим гетероарилом, имеющим 1-3 гетероатома, выбранных из N и O; где фенил, гетероциклил, или гетероарил, представленный Rc, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12 и P(O)di-C1-6 алкила;
Ra' является С1-6 алкилом, необязательно замещенным OH, CN, 5-6-членным гетероарилом, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12;
m равно 0, 1, 2 или 3;
n равно 0, 1, 2, 3 или 4;
p равно 1, 2 или 3;
q равно 1, 2 или 3; и p+q ≤4.
в другом аспекте, в изобретении представлено соединение, представленное структурной формулой (II):
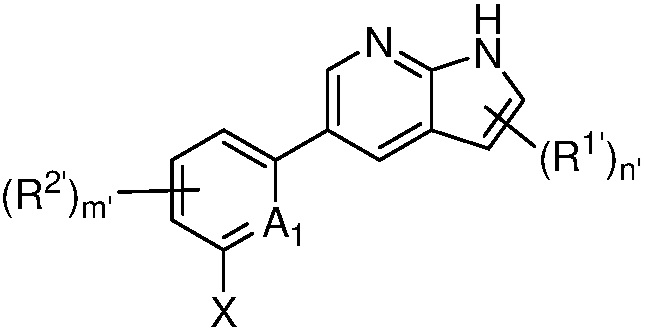 (II),
(II),
или его фармацевтически приемлемая соль или стереоизомер, где
A1 является СR’ или N;
Х является -P(=O)R3’R4’;
R’ является Н, дейтерием, галогеном, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, где алкил, алкенил, алкинил или алкокси, представленный R’ или в группе, представленной R’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R1’ независимо является H, дейтерием, галогеном, OH, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, циклоалкил или гетероциклил, представленный R1’, или в группе, представленной R1’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R2’ независимо является H, дейтерием, галогеном, OH, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, фенилом, 5-6-членным гетероарилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, фенил, гетероарил, циклоалкил или гетероциклил, представленный R2’ или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый R3’ и R4’ независимо является H, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, где алкил, алкенил или алкинил, представленный R3’ или R4’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R11’ и R12’ независимо является H или C1-6 алкилом, где алкил, представленный R11’ или R12’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила и 3-7-членного гетероциклила, или
R11’ и R12’, вместе с атомом азота, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси или C1-4 галогеналкокси;
m’ равен 0, 1 или 2; и
n’ равен 0, 1 или 2.
В настоящем документе представлены фармацевтические композиции, содержащие эффективное количество соединений по изобретению или их фармацевтически приемлемых солей, и фармацевтически приемлемый носитель.
Также представлена комбинация, включающая терапевтически эффективное количество соединений по изобретению или их фармацевтически приемлемых солей, и один или несколько терапевтически активных со-агентов.
Настоящее изобретение дополнительно представляет способ ингибирования активности HPK1 у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединений по изобретению или их фармацевтически приемлемых солей.
Настоящее изобретение дополнительно представляет способ лечения субъекта с заболеванием или состоянием, описанным в настоящем документе, таким как рак (такой как рак груди, колоректальный рак, рак легких, рак яичников и рак поджелудочной железы), где способ включает введение субъекту терапевтически эффективного количества соединений по изобретению или их фармацевтически приемлемых солей.
Некоторые варианты осуществления описывают соединение по настоящему изобретению или его фармацевтически приемлемую соль для применения в качестве лекарственного средства, такого как лекарственное средство, действующее как ингибитор HPK1.
Настоящее изобретение также представляет применение соединения по изобретению или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей их, в любом из описанных выше способов по изобретению. В одном варианте осуществления, представлено соединение по настоящему изобретению или его фармацевтически приемлемая соль, или фармацевтическая композиция, содержащая его, для применения в любом способе по настоящему изобретению, описанном в настоящем документе. В другом варианте осуществления, представлено применение соединения по изобретению или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей их, для изготовления лекарственного средства для любого описанного способа по изобретению.
подробное описание изобретения
1. Обзор
Описанное в настоящем документе изобретение относится к ингибиторам HPK1/MAP4K1, их фармацевтически приемлемым солям, их фармацевтическим композициям и способам модулирования (например, ингибирования) активности HPK1/MAP4K1 с их использованием, указанный способ включает введение пациенту/субъекту, нуждающемуся в этом, ингибитора HPK1/MAP4K1 по изобретению или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, соединения по изобретению или их фармацевтически приемлемые соли применимы для терапевтического введения для усиления, стимуляции и/или повышения иммунитета при лечении рака.
Например, способ лечения заболевания или нарушения, ассоциированного с ингибированием взаимодействия HPK1, может включать введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения, представленного в настоящем документе, или его фармацевтически приемлемой соли. Соединения по настоящему изобретению можно использовать отдельно, в комбинации с другими агентами или методами лечения, или в качестве адъюванта или неоадъюванта для лечения заболеваний или нарушений, включая раки.
2. Определения
Используемые здесь термины «а», «an», «the» и аналогичные термины, используемые в контексте настоящего изобретения (особенно в контексте формулы изобретения), следует толковать как охватывающие как единственное, так и множественное число, если иное не указано в настоящем документе или явно противоречат контексту.
Используемый в настоящем документе термин «галоген» означает галоген и включает хлор, фтор, бром и йод.
Термин «алкил», используемый отдельно или как часть большей группы, такой как «алкокси» или «галогеналкил» и подобные, означает насыщенный алифатический одновалентный углеводородный радикал с прямой или разветвленной цепью. Если не указано иное, алкильная группа обычно имеет 1-4 или 1-6 атомов углерода, т.е. (C1-C4)алкил или (C1-C6)алкил. В настоящем документе «(C1-C4)алкильная группа» означает радикал, содержащий от 1 до 4 атомов углерода в линейной или разветвленной структуре. Примеры включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил и т.д.
Термин «С1-6алкилен» относится к двухвалентному полностью насыщенному одновалентному углеводородному радикалу с разветвленной или прямой цепью, имеющему от 1 до 6 атомов углерода. Аналогично, термины «С1-4алкилен», «С1-3алкилен» и «С1-2алкилен» должны толковаться соответственно. Типовые примеры C1-6алкилена включают, но не ограничены ими, метилен, этилен, н-пропилен, изопропилен, н-бутилен, втор-бутилен, изобутилен, трет-бутилен, н-пентилен, изопентилен, неопентилен, и н-гексилен.
Термин «C1-C6алкил, необязательно замещенный гидроксилом» относится к C1-C6алкилу, как определено выше, который может быть замещен одним или несколькими гидрокси. Примеры включают, но не ограничены ими, гидроксиметил, гидроксиэтил, 1,2-дигидроксиэтил, 2,3-дигидроксипропил и т.д.
Используемый в настоящем документе термин «ди-С1-6алкиламино» относится к группе формулы -N(Ra)-Ra, где каждый Ra является C1-6алкилом, который может быть одинаковым или разным, как определено выше, по аналогии с термином «моно-C1-6алкиламино», который относится к группе формулы -N(H)-Ra, где Ra является C1-6алкилом, как определено выше.
Термин «алкенил» означает одновалентный углеводородный радикал с разветвленной или прямой цепью, содержащий, по меньшей мере, одну двойную связь. Алкенил может быть моно- или полиненасыщенным и может существовать в E или Z конфигурации. Если не указано иное, алкенильная группа обычно имеет 2-6 атомов углерода, т.е. (C2-C6)алкенил. Например, «(C2-C6)алкенил» означает радикал, имеющий 2-6 атомов углерода в линейной или разветвленной структуре.
Термин «алкинил» означает одновалентный углеводородный радикал с разветвленной или прямой цепью, содержащий, по меньшей мере, одну тройную связь. Если не указано иное, алкинильная группа обычно имеет 2-6 атомов углерода, т.е. (C2-C6)алкинил. Например, «(C2-C6)алкинил» означает радикал, имеющий 2-6 атомов углерода в линейной или разветвленной структуре.
Термин «алкокси» означает алкильный радикал, присоединенный через связывающий атом кислорода, представленный -O-алкилом. Например, «C1-C6алкокси» относится к -O-C1-C6алкилу, где «алкил» такой, как определен выше, и «(C1-C4)алкокси» включает метокси, этокси, пропокси и бутокси, и т.д. Типовые примеры алкокси включают, но не ограничены ими, метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси, гексилокси, циклопропилокси, циклогексилокси и подобные. Обычно, алкоксигруппы содержат примерно от 1 до 6 атомов углерода, от 1 до 4 атомов углерода или от 1 до 2 атомов углерода.
Термины «галогеналкил» и «галогеналкокси» означают алкил или алкокси, в зависимости от случая, замещенный одним или несколькими атомами галогена. Примеры галогеналкила включают, но не ограничены ими, трифторметил, трихлорметил, пентафторэтил и подобные.
Таким образом, термин «C1-6алкил, необязательно замещенный галогеном» относится к C1-C6алкилу, как определено выше, который может быть замещен одним или несколькими галогенами. Примеры включают, но не ограничены ими, трифторметил, дифторметил, фторметил, трихлорметил, 2,2,2-трифторэтил, 1-фторметил-2-фторэтил, 3-бром-2-фторпропил и 1-бромметил-2-бромэтил.
Используемый в настоящем документе термин «циклоалкил» включает насыщенные циклические, бициклические, трициклические или полициклические углеводородные группы, содержащие 3-14 атомов углерода, содержащие указанное число колец и атомов углерода (например, C3-C14 моноциклический, C4-C14 бициклический, C5-C14 трициклический или C6-C14 полициклический циклоалкил). В некоторых вариантах осуществления, «циклоалкилом» является моноциклический циклоалкил. Примеры моноциклических циклоалкильных групп включают циклопентил (С5), циклогексил (С6), циклопропил (С3), циклобутил (С4), циклогептил (С7) и циклооктил (С8). В некоторых вариантах осуществления, «циклоалкилом» является бициклический циклоалкил. Примеры бициклических циклоалкилов включают бицикло[1.1.0]бутан (С4), бицикло[1.1.1]пентан (С5), спиро[2.2]пентан (С5), бицикло[2.1.0]пентан (С5), бицикло[2.1.1]гексан (С6), бицикло[3.3.3]ундекан (С11), декагидронафталин (С10), бицикло[4.3.2]ундекан (С11), спиро[5.5]ундекан (C11) и бицикло[4.3.3]додекан (С12). В некоторых вариантах осуществления, «циклоалкилом» является трициклический циклоалкил. Примеры трициклических циклоалкилов включают адамантин (С12). Если не указано иное, «циклоалкил» имеет от трех до шести атомов углерода и является моноциклическим.
Термин «арильная группа», используемый отдельно или как часть большей группы, такой как «аралкил», «аралкокси» или «арилоксиалкил», означает карбоциклическое ароматическое кольцо. Термин «арил» может использоваться взаимозаменяемо с терминами «арильное кольцо», «карбоциклическое ароматическое кольцо», «арильная группа» и «карбоциклическая ароматическая группа». Обычно арилом является моноциклический, бициклический или трициклический арил, содержащий 6-20 атомов углерода, обычно 6-14 атомов углерода в кольце. Кроме того, используемый в настоящем документе термин «арил» относится к ароматическому заместителю, которым может являться одно ароматическое кольцо или несколько ароматических колец, которые конденсированы вместе. Примеры включают фенил, нафтил, антраценил, 1,2-дигидронафтил, 1,2,3,4-тетрагидронафтил, флуоренил, инданил, инденил и подобные.
«Замещенная арильная группа» замещена на любом одном или нескольких замещаемых атомов в кольце, который является атомом углерода кольца, связанным с водородом. Замещенный арил обычно замещен 1-5 (например, одним, двумя или тремя) заместителями, независимо выбранными из группы, состоящей из: гидроксила, тиола, циано, нитро, C1-C4 алкила, C1-C4 алкенила, C1-C4 алкинила, C1-C4 алкокси, C1-C4 тиоалкила, C1-C4 алкенилокси, C1-C4 алкинилокси, галогена, C1-C4 алкилкарбонила, карбокси, C1-C4 алкоксикарбонила, амино, C1-C4 алкиламино, ди-C1-C4 алкиламино, C1-C4 алкиламинокарбонила, ди-C1-C4 алкиламинокарбонила, C1-C4 алкилкарбониламино, C1-C4 алкилкарбонила, C1-C4 алкиламино, сульфонила, сульфамоила, алкилсульфамоила и C1-C4 алкиламиносульфонила, где каждая из вышеупомянутых углеводородных групп (например, алкильные, алкенильные, алкинильные, алкоксильные остатки) может быть дополнительно замещена одним или несколькими остатками, независимо выбранными, в каждом случае, из галогена, гидроксила или C1-C4 алкокси.
Термин «гетероциклильная группа» или «гетероциклическая группа» означает моноциклическое неароматическое (включая частично насыщенное) кольцо, предпочтительно, содержащее 3-10 членов, предпочтительно, содержащее 1-4 гетероатома в кольце, или полициклическое кольцо, содержащее кольцо с, предпочтительно, 7-20 членами, и от предпочтительно от 1 до 4 кольцевых гетероатомов, где полициклическое кольцо имеет одно или несколько моноциклических неароматических гетероциклических колец, конденсированных с одним или несколькими ароматическими или гетероароматическими кольцами. Гетероциклильная группа обычно имеет 3-7, 3-24, 4-16, 5-10 или 5 или 6 атомов в кольце; где необязательно от одного до четырех, особенно один или два атома в кольце являются гетероатомом (остальные атомы в кольце являются углеродом). Каждый гетероатом независимо выбран из азота, четвертичного азота, окисленного азота (например, NO); кислорода; и серы, включая сульфоксид и сульфон. Гетероциклическая группа может быть присоединена к гетероатому или атому углерода. Примеры гетероциклов включают тетрагидрофуран (ТГФ), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазоиздин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолан, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин и подобные.
Гетероциклильная группа может включать конденсированные или мостиковые кольца, а также спироциклические кольца. В одном варианте осуществления, гетероциклильной группой является бициклическое кольцо, имеющее моноциклическое неароматическое гетероциклическое кольцо, конденсированное с фенильной группой. Типовая полициклическая гетероциклическая группа включает тетрагидроизохинолинил (такой как 1,2,3,4-тетрагидроизохинолин-7-ил, 2-метил-1,2,3,4-тетрагидроизохинолин-7-ил, 1,2,3,4-тетрагидроизохинолин-6-ил и 2-метил-1,2,3,4-тетрагидроизохинолин-6-ил), изоиндолинил (например, 2-этилизоиндолин-5-ил, 2-метилизоиндолин-5-ил), индолинил, тетрагидробензо[f]оксазепинил (например, 2,3,4,5-тетрагидробензо[f][1,4]оксазепин-7-ил).
Термин «гетероцикл», «гетероциклил» или «гетероциклический», будь то насыщенный или частично ненасыщенный, также относится к кольцам, которые необязательно замещены. Замещенным гетероциклилом может быть гетероциклильная группа, независимо замещенная 1-4, например, одним, или двумя, или тремя, или четырьмя заместителями.
В некоторых вариантах осуществления, гетероциклильной группой является 3-14-членная не ароматическая кольцевая система, содержащая кольцевые атомы углерода и 1-4 кольцевых гетероатома, где каждый гетероатом независимо выбран из азота, кислорода и серы («3-14-членный гетероциклил»).
Термины «гетероарил», «гетероароматический», «гетероарильное кольцо», «гетероарильная группа», «гетероароматическое кольцо» и «гетероароматическая группа», используемые отдельно или как часть большей группы, такой как «гетероаралкил» или «гетероарилалкокси», относится к ароматическим кольцевым группам, имеющим 5-14 кольцевых атомов, выбранных из углерода и, по меньшей мере, один (обычно, 1-4, более типично, 1 или 2) гетероатомов (например, кислорода, азота или серы). «Гетероарил» включает моноциклические кольца и полициклические (например, би- или тициклические) кольца, в которых моноциклическое гетероароматическое кольцо конденсировано с одним или несколькими другими карбоциклическими ароматическими или гетероароматическими кольцами. Таким образом, «5-14-членный гетероарил» включает моноциклические, бициклические или трициклические кольцевые системы.
Примеры моноциклических 5-6-членных гетероарильных групп включают фуранил (например, 2-фуранил, 3-фуранил), имидазолил (например, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил), изоксазолил (например, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил), оксадиазолил (например, 2-оксадиазолил, 5-оксадиазолил), оксазолил (например, 2-оксазолил, 4-оксазолил, 5-оксазолил), пиразолил (например, 3-пиразолил, 4 -пиразолил), пирролил (например, 1-пирролил, 2-пирролил, 3-пирролил), пиридил (например, 2-пиридил, 3-пиридил, 4-пиридил), пиримидинил (например, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил), пиридазинил (например, 3-пиридазинил), тиазолил (например, 2-тиазолил, 4-тиазолил, 5-тиазолил), триазолил (например, 2-триазолил, 5-триазолил), тетразолил (например, тетразолил), тиенил (например, 2-тиенил, 3-тиенил), пиримидинил, пиридинил и пиридазинил.
Обычно гетероарилом является 5-10-членная кольцевая система (например, 5-6-членный моноцикл или 8-10-членный бицикл) или 5-6-членная кольцевая система. Типовые гетероарильные группы включают 2- или 3-тиенил, 2- или 3-фурил, 2- или 3-пирролил, 2-, 4- или 5-имидазолил, 3-, 4- или 5-пиразолил, 2-, 4- или 5-тиазолил, 3-, 4- или 5-изотиазолил, 2-, 4- или 5-оксазолил, 3-, 4- или 5-изоксазолил, 3- или 5-1,2,4-триазолил, 4- или 5-1,2,3-триазолил, тетразолил, 2-, 3- или 4-пиридил, 3- или 4-пиридазинил, 3-, 4- или 5-пиразинил, 2-пиразинил и 2-, 4- или 5-пиримидинил.
Примеры полициклических ароматических гетероарильных групп включают карбазолил, бензимидазолил, бензотиенил, бензофуранил, индолил, хинолинил, бензотриазолил, бензотиазолил, бензоксазолил, бензимидазолил, изохинолинил, индолил, изоиндолил, акридинил или бензизоксазолил.
Таким образом, термин «гетероарил» также относится к группе, в которой гетероароматическое кольцо конденсировано с одним или несколькими арильными, циклоалифатическими или гетероциклильными кольцами, где радикал или точка присоединения находится на гетероароматическом кольце. Неограничивающие примеры включают 1-, 2-, 3-, 5-, 6-, 7- или 8-индолизинил, 1-, 3-, 4-, 5-, 6- или 7-изоиндолил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-индазолил, 2-, 4-, 5-, 6-, 7- или 8-пуринил, 1-, 2-, 3-, 4-, 6-, 7-, 8- или 9-хинолизинил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолиил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолиил, 1-, 4-, 5-, 6-, 7- или 8-фталазинил, 2-, 3-, 4-, 5- или 6-нафтиридинил, 2-, 3-, 5-, 6-, 7- или 8-хиназолинил, 3-, 4-, 5-, 6-, 7- или 8-циннолинил, 2-, 4-, 6- или 7-птеридинил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-4аН карбазолил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-карбзаолил, 1-, 3-, 4-, 5-, 6-, 7-, 8- или 9-карболинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-фенантридинил, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8- или 9-акридинил, 1-, 2-, 4-, 5-, 6-, 7-, 8- или 9-перимидинил, 2-, 3-, 4-, 5-, 6-, 8 -, 9- или 10-фенатролинил, 1-, 2-, 3-, 4-, 6-, 7-, 8- или 9-феназинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-фенотиазинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-феноксазинил, 2-, 3-, 4-, 5-, 6- или 1-, 3-, 4-, 5-, 6-, 7-, 8-, 9- или 10-бензизохинолинил, 2-, 3-, 4- или тиено[2,3-b]фуранил, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10- или 11-7H-пиразино[2,3-c]карбазолил, 2-, 3-, 5-, 6- или 7-2H-фуро[3,2-b]-пиранил, 2-, 3-, 4-, 5-, 7- или 8-5H-пиридо[2,3-d]-о-оксазинил, 1-, 3- или 5-1H-пиразоло[4,3-d]-оксазолил, 2-, 4- или 5-4H-имидазо[4,5-d]-тиазолил, 3-, 5- или 8-пиразино[2,3-d]пиридазинил, 2-, 3-, 5- или 6-имидазо[2,1-b]-тиазолил, 1-, 3-, 6-, 7-, 8- или 9-фуро[3,4-c]циннолинил, 1-, 2-, 3-, 4-, 5-, 6-, 8-, 9-, 10- или 11-4H-пиридо[2,3-c]карбазолил, 2-, 3-, 8- или 7-имидазо[1,2-b][1,2,4]триазинил, 7-бензо[b]тиенил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил, 2-, 4-, 4-, 5-, 6- или 7-бензотиазолил, 1-, 2-, 4-, 5-, 6-, 7-, 8- или 9-бензоксапинил, 2-, 4-, 5-, 6-, 7- или 8-бензоксазинил, 1-, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10- или 11-1H-пирроло[1,2-b][2]бензапинил.
Типовые конденсированные гетероарильные группы включают, но не ограничены ими, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолинил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-бензо[b]тиенил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил и 2-, 4-, 5-, 6- или 7-бензотиазолил.
Используемый в настоящем документе термин пиридин или пиридил, необязательно замещенный гидрокси, например, 2-пиридил, 3-пиридил или 4-пиридил, относится к соответствующему гидроксипиридину или гидроксипиридилу и может включать его таутомерную форму, такую как соответствующий пиридон. или пиридонил.
Используемый в настоящем документе термин пиридин или пиридил, необязательно замещенный оксо, например, 2-пиридил, 3-пиридил или 4-пиридил, относится к соответствующему пиридону или пиридонилу и может включать его таутомерную форму, такую как соответствующий гидроксипиридин или гидроксипиридил, при условии, что указанная таутомерная форма может быть получена. Пиридин или пиридил, необязательно замещенный оксо, может дополнительно относиться к соответствующему пиридин-N-оксиду или пиридил-N-оксиду.
«Замещенная гетероарильная группа» замещена на любом одном или нескольких замещаемых атомов в кольце, которые являются атомом углерода в кольце или атомом азота в кольце, связанным с водородом.
Термин «мостиковая бициклическая группа» относится к кольцевой системе, которая включает два кольца, которые имеют, по меньшей мере, три общих соседних атома в кольце.
Используемые в данном документе, многие группы (например, алкил, алкилен, циклоалкил, арил, гетероарил или гетероциклил) называются либо «замещенными», либо «необязательно замещенными». Когда группа модифицирована одним из этих терминов, если не указано иное, это означает, что любая часть группы, известная специалисту в данной области техники как доступная для замещения, может быть замещена, включая один или несколько заместителей. Если присутствует более одного заместителя, то каждый заместитель может быть выбран независимо. Такие средства для замещения хорошо известны в данной области техники и/или описаны в настоящем изобретении. Необязательными заместителями могут быть любые заместители, подходящие для присоединения к группе.
Если подходящие заместители конкретно не перечислены, типовые заместители включают, но не ограничены ими: (C1-C5) алкил, (C1-C5) гидроксиалкил, (C1-C5) галогеналкил, (C1-C5) алкокси, (C1-C5) галогеналкокси, галоген, гидроксил, циано, амино, -CN, -NO2, -ORc1, -NRa1Rb1, -S(O)iRa1, -NRa1S(O)iRb1, -S(O)iNRa1Rb1, -C(=O)ORa1, -OC(=O)ORa1, -C(=S)ORa1, -O(C=S)Ra1, -C(=O)NRa1Rb1, -NRa1C(=O)Rb1, -C(=S)NRa1Rb1, -C(=O)Ra1, -C(=S)Ra1, NRa1C(=S)Rb1, -O(C=O)NRa1Rb1, -NRa1(C=S)ORb1, -O(C=S)NRa1Rb1, -NRa1(C=O)NRa1Rb1, -NRa1(C=S)NRa1Rb1, фенил или 5-6-членный гетероарил. Каждый Ra1 и каждый Rb1 независимо выбраны из -H и (C1-C5) алкила, необязательно замещенного гидроксилом или (C1-C3)алкокси; Rc1 является -H, (C1-C5) галогеналкилом или (C1-C5) алкилом, где (C1-C5) алкил необязательно замещен гидроксилом или (C1-C3) алкокси.
Описанные в настоящем документе соединения могут существовать в различных таутомерных формах. Термин «таутомеры» или «таутомерный» относится к двум или более взаимопревращаемым соединениям/заместителям, возникающим в результате, по меньшей мере, одной формальной миграции атома водорода и, по меньшей мере, одного изменения валентности (например, одинарной связи на двойную связь, тройной связи на одинарную связь, или наоборот). Типовые таутомеризации включают таутомеризации кето-енол, амид-имид, лактам-лактим, енамин-имин и енамин-(другой енамин). Настоящие идеи охватывают соединения в форме таутомеров, которые включают формы, структурно не изображенные. Все такие изомерные формы таких соединений специально включены. Если таутомер соединения является ароматическим, то это соединение является ароматическим.
Соединения любой из описанных выше формул могут проявлять один или несколько видов изомерии (например, оптическую, геометрическую или таутомерную изомерию). Соединения любой из формул, описанных выше, также могут быть мечены изотопами. Такое изменение подразумевается для соединений любой из формул, описанных выше, определенных в соответствии со ссылкой на их структурные особенности и, следовательно, в пределах объема настоящего изобретения.
Соединения любой из описанных выше формул, содержащие один или несколько асимметричных атомов углерода, могут существовать в виде двух или нескольких стереоизомеров. Если соединение любой из формул, описанных выше, содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Там, где структурные изомеры взаимопревращаемы через низкий энергетический барьер, может иметь место таутомерная изомерия («таутомерия»). Она может принимать форму протонной таутомерии в соединениях любой из описанных выше формул, содержащих, например, имино, кето или оксимную группу, или так называемой валентной таутомерии в соединениях, содержащих ароматическую группу. Из этого следует, что одно соединение может проявлять более одного типа изомерии.
Соединения, имеющие один или несколько хиральных центров, могут существовать в различных стереоизомерных формах. Стереоизомеры являются соединениями, которые отличаются только своим пространственным расположением. Стереоизомеры включают все диастереомерные, энантиомерные и эпимерные формы, а также рацематы и их смеси. Термин «геометрический изомер» относится к соединениям, имеющим, по меньшей мере, одну двойную связь, где двойные связи могут находиться в цис (также называемой syn или entgegen (Е)) или транс (также называемой anti или zusammen (Z)) форме, а также их смеси. Когда описанное соединение названо или изображено по структуре без указания стереохимии, подразумевается, что название или структура охватывает один или несколько возможных стереоизомеров, или геометрических изомеров, или смесь охватываемых стереоизомеров или геометрических изомеров.
Когда геометрический изомер изображен по названию или структуре, следует понимать, что названный или изображенный изомер существует в большей степени, чем другой изомер, то есть, что геометрическая изомерная чистота названного или изображенного геометрического изомера превышает 50%, например, чистота составляет, по меньшей мере 60%, 70%, 80%, 90%, 99% или 99,9% по массе. Геометрическую изомерную чистоту определяют путем деления массы названного или изображенного геометрического изомера в смеси на общую массу всех геометрических изомеров в смеси.
Рацемическая смесь означает 50% одного энантиомера и 50% его соответствующего энантиомера. Когда соединение с одним хиральным центром названо или изображено без указания стереохимии хирального центра, подразумевается, что название или структура охватывает обе возможные энантиомерные формы (например, обе энантиомерно-чистые, энантиомерно-обогащенные или рацемические) соединения. Когда соединение с двумя или несколькими хиральными центрами названо или изображено без указания стереохимии хиральных центров, подразумевается, что название или структура охватывает все возможные диастереомерные формы (например, диастереомерно чистые, диастереомерно обогащенные и эквимолярные смеси одного или нескольких диастереомеров (например, рацемические смеси) соединения.
Энантиомерные и диастереомерные смеси можно разделить на составляющие их энантиомеры или стереоизомеры с помощью хорошо известных способов, таких как хирально-фазовая газовая хроматография, хирально-фазовая высокоэффективная жидкостная хроматография, кристаллизация соединения в виде хирального солевого комплекса или кристаллизация соединения в хиральном растворителе. Энантиомеры и диастереомеры также могут быть получены из диастереомерно- или энантиомерно-чистых промежуточных соединений, реагентов и катализаторов с помощью хорошо известных способов асимметричного синтеза.
Когда соединение обозначено названием или структурой, которые указывают на один энантиомер, если не указано иное, соединение имеет оптически чистоту, по меньшей мере, 60%, 70%, 80%, 90%, 99% или 99,9% (также обозначается как «энантиомерно чистое»). Оптическая чистота представляет собой массу смеси названного или изображенного энантиомера, деленную на общую массу смеси обоих энантиомеров.
Когда стереохимия описанного соединения названа или изображена структурой, и названная или изображенная структура включает более одного стереоизомера (например, как в диастереомерной паре), следует понимать, что включены один из охватываемых стереоизомеров или любая смесь охватываемых стереоизомеров. Далее следует понимать, что стереоизомерная чистота названных или изображенных стереоизомеров составляет, по меньшей мере, 60%, 70%, 80%, 90%, 99% или 99,9% по массе. Стереоизомерную чистоту в этом случае определяют путем деления общей массы смеси стереоизомеров, охватываемой названием или структурой, на общую массу смеси всех стереоизомеров.
Фармацевтически приемлемые соли соединений любой из описанных выше формул могут также содержать противоион, который является оптически активным (например, d-лактат или l-лизин) или рацемическим (например, dl-тартрат или dl-аргинин).
Цис/транс изомеры могут быть разделены обычными методами, хорошо известными специалистам в данной области, например, хроматографией и фракционной кристаллизацией.
Обычные методики получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например спиртом, или, в случае, когда соединение любой из формул, описанных выше, содержит кислотную или основную группу, основанием или кислотой, например, 1-фенилэтиламином или винной кислотой. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера превращены в соответствующие чистые энантиомеры способами, хорошо известными специалисту в данной области техники. Хиральные соединения любой из описанных выше формул (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% по объему изопропанола, обычно от 2% до 20%, и от 0 до 5% по объему алкиламина, обычно 0,1% диэтиламина. Концентрация элюата дает обогащенную смесь. Можно использовать хиральную хроматографию с использованием суб- и сверхкритических жидкостей. Способы хиральной хроматографии, применимые в некоторых вариантах осуществления настоящего изобретения, известны в данной области техники (см., например, Smith, Roger M., Loughborough University, Loughborough, UK; Chromatographic Science Series (1998), 75 (Supercritical Fluid Chromatography with Packed Columns), pp. 223-249 и цитируемые там ссылки). Колонки могут быть получены от Chiral Technologies, Inc, West Chester, Pa., USA, дочерней компании Daicel® Chemical Industries, Ltd., Tokyo, Japan.
Следует подчеркнуть, что соединения любой из формул, описанных выше, представлены в настоящем документе в единственной таутомерной форме, все возможные таутомерные формы включены в объем настоящего изобретения.
Настоящее описание также включает все фармацевтически приемлемые изотопно-меченые соединения любой из формул, описанных выше, в которых один или несколько атомов заменены атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, которое преобладает в природе.
Примеры изотопов, подходящих для включения в соединения по настоящему изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Определенные изотопно-меченые соединения любой из формул, описанных выше, например соединения, включающие радиоактивный изотоп, применимы в исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, т.е. 3Н, и углерод-14, т.е. 14С, особенно пригодны для этой цели ввиду легкости их введения и легкости обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить определенные терапевтические преимущества, возникающие в результате большей метаболической стабильности, например, увеличение периода полужизни in vivo или снижение требований к дозировке.
Замещение изотопами, излучающими позитроны, такими как 11C, 18F, 15O и 13N, может быть полезна в исследованиях позитронно-эмиссионной топографии (ПЭТ) для изучения занятости субстрата рецептором.
Меченые изотопами соединения любой из формул, описанных выше, обычно могут быть получены обычными способами, известными специалистам в данной области техники, или способами, аналогичными описанным в прилагаемых примерах и примерах приготовления, с использованием соответствующего изотопно-меченного реагента, вместо не меченого реагента, использовавшегося ранее.
Фармацевтически приемлемые сольваты в соответствии с настоящим изобретением включают сольваты, в которых растворитель кристаллизации может быть изотопно замещен, например, D2O, d6-ацетон, d6-ДМСО.
Следует понимать, что когда соединение в настоящем документе представлено структурной формулой или обозначено в настоящем документе химическим названием, все другие таутомерные формы, которые могут существовать для соединения, охватываются структурной формулой.
Любая приведенная в настоящем документе формула также предназначена для представления не меченых форм, а также изотопно-меченых форм соединений, таких как 2H. Кроме того, замена дейтерием (т.е. 2H или D) может обеспечить определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличение периода полужизни in vivo или снижение требований к дозировке или улучшение терапевтического индекса, если понимать, что дейтерий в этом контексте рассматривается как заместитель соединения формулы (I-0), (I-1) или (I-2). Концентрация такого более тяжелого изотопа, в частности дейтерия, может определяться коэффициентом изотопного обогащения. Используемый в настоящем документе термин «коэффициент изотопного обогащения» означает соотношение между изотопным содержанием и естественным содержанием определенного изотопа. Если заместитель в соединении по данному изобретению обозначен как дейтерий, то такое соединение имеет коэффициент изотопного обогащения для каждого указанного атома дейтерия, по меньшей мере, 3500 (включение 52,5% дейтерия на каждый указанный атом дейтерия), по меньшей мере, 4000 (включение 60% дейтерия), по меньшей мере, 4500 (включение дейтерия 67,5%), по меньшей мере, 5000 (включение 75% дейтерия), по меньшей мере, 5500 (включение 82,5% дейтерия), по меньшей мере, 6000 (включение 90% дейтерия), по меньшей мере, 6333,3 (включение 95% дейтерия), по меньшей мере, 6466,7 (включение дейтерия 97%), по меньшей мере, 6600 (включение дейтерия 99%) или по меньшей мере, 6633,3 (включение дейтерия 99,5%).
Соединения по данному изобретению могут существовать в свободной форме для лечения или, при необходимости, в форме фармацевтически приемлемой соли.
Используемые в настоящем документе термины «соль» или «соли» относятся к кислотно-аддитивной или основно-аддитивной соли по изобретению. «Соли» включают, в частности, «фармацевтически приемлемые соли». Термин «фармацевтически приемлемые соли» относится к солям, которые сохраняют биологическую эффективность и свойства соединений по данному изобретению и, которые обычно не являются биологически или иным образом нежелательными, во многих случаях, соединения по настоящему изобретению способны образовывать кислотные и/или основные соли благодаря наличию амино и/или карбоксильных групп или групп, подобных им.
Фармацевтически приемлемые кислотно-аддитивные соли могут быть образованы с неорганическими кислотами и органическими кислотами, например, ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофилионат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидройодид/йодид, изотионат, лактат, лактобионат, лаурилсуифат, малат, малеат, малонат, миндалят, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигайактуронат, пропионат, стеарат, сукцинат, субсалицилат, тартрат, тозилат и трифторацетат.
Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и подобные.
Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и подобные. Фармацевтически приемлемые основно-аддитивные соли могут быть образованы с неорганическими и органическими основаниями. Неорганические основания, из которых могут быть получены соли, включают, например, соли аммония и металлов из столбцов с I по XI I периодической таблицы, в некоторых вариантах осуществления, соли получают из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди. В некоторых вариантах осуществления, подходящие соли включают соли аммония, калия, натрия, кальция и магния.
Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая встречающиеся в природе замещенные амины, циклические амины, основные ионообменные смолы, и подобные. Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин.
Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из основной или кислотной группы обычными химическими способами. Как правило, такие соли могут быть получены путем взаимодействия свободных кислотных форм этих соединений со стехиометрическим количеством соответствующего основания (например, гидроксида Na, Ca, Mg или K, карбоната, бикарбоната и подобного) или путем взаимодействия форм свободного основания этих соединений со стехиометрическим количеством соответствующей кислоты. Такие реакции обычно проводят в воде, или в органическом растворителе, или в их смеси. Как правило, там, где это возможно, желательно использовать не водные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил. Списки дополнительных подходящих солей можно найти, например, в «Remington’s Pharmaceutical Sciences,» 20th ed., Mack Publishing Company, Easton, PA, (1985); и в «Handbook of Pharmaceutical Salts: Properties, Selection, and Use» by Stahl and Wermuth (Wiley- VCH, Weinheim, Germany, 2002).
Термины «композиция» и «состав» используются взаимозаменяемо.
«Субъектом» является млекопитающее, предпочтительно человек, но также может быть животное, нуждающееся в ветеринарном лечении, например, домашние животные (например, собаки, кошки и подобные), сельскохозяйственные животные (например, коровы, овцы, свиньи, лошади и подобные) и лабораторные животные (например, крысы, мыши, морские свинки и подобные).
Как используется в настоящем документе, субъект «нуждается» в лечении, если такой субъект получит биологическую, медицинскую пользу или улучшение качества жизни от такого лечения.
Термин «вводить», «введение» или «введение» относится к способам введения соединения по изобретению или его композиции в или на субъекта. Эти способы включают, но не ограничены ими, внутрисуставные (в суставы), внутривенные, внутримышечные, внутриопухолевые, внутрикожные, внутрибрюшинные, подкожные, пероральные, местные, интратекальные, ингаляционные, чрескожные, ректальные и подобные. Техники введения, которые можно использовать с агентами и способами, описанными в настоящем документе, можно найти, например, в Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; and Remington’s, Pharmaceutical Sciences (current edition), Mack Publishing Co., Easton, Pennsylvania.
Используемый в настоящем документе термин «ингибировать», «ингибирование» или «ингибирование» относится к уменьшению или подавлению данного состояния, симптома или нарушения или заболевания, или к значительному снижению исходной активности биологической активности или процесса.
Термины «лечение», «лечить» и «лечение» относятся к обращению вспять, облегчению или ингибированию прогрессирования заболевания, описанного в настоящем документе. В некоторых вариантах осуществления, лечение может быть назначено после того, как один или несколько признаков или симптомов заболевания развились или наблюдались (т.е. терапевтическое лечение). В других вариантах осуществления, лечение можно проводить при отсутствии признаков или симптомов заболевания. Например, лечение может быть назначено восприимчивому субъекту до появления симптомов (т.е. профилактическое лечение) (например, в свете симптомов в анамнезе и/или в свете воздействия патогена). Лечение также можно продолжать после исчезновения симптомов, например, чтобы отсрочить или предотвратить рецидив.
Термины «состояние», «заболевание» и «нарушение» используются взаимозаменяемо.
Как правило, эффективное количество соединения, указанное в настоящем документе, варьируется в зависимости от различных факторов, таких как конкретное лекарственное средство или соединение, фармацевтический состав, способ введения, тип заболевания или нарушения, личность субъекта или хозяина, которого лечат, и подобных, но, тем не менее, может быть определено специалистом в данной области техники обычным способом. Эффективное количество соединения по настоящему изобретению может быть легко определено специалистом в данной области техники обычными способами, известными в данной области техники.
Термин «эффективное количество» означает количество при введении субъекту, которое приводит к благоприятным или желаемым результатам, включая клинические результаты, например, ингибирует, подавляет или уменьшает симптомы состояния, подвергаемого лечению, у субъекта, по сравнению с контролем. Например, эффективное количество можно давать в виде стандартной дозированной формы (например, от 1 мг до примерно 50 г в сутки, например, от 1 мг до примерно 5 г в сутки).
Термин «терапевтически эффективное количество» соединения по настоящему изобретению относится к количеству соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ субъекта, например снижение или ингибирование активности фермента или белка, или облегчение симптомов, облегчение состояний, замедление или задержку прогрессирования заболевания или предотвращение заболевания и т.д. В одном неограничивающем варианте осуществления, термин «терапевтически эффективное количество» относится к количеству соединения по настоящему изобретению, которое при введении субъекту эффективно для (1) по меньшей мере, частичного облегчения, ингибирования, предотвращения и/или или улучшения состояния, или нарушения, или заболевания, (i) опосредованного HPK1, или (ii) связанного с активностью HPK1, или (iii) характеризующегося активностью (нормальной или аномальной) HPK1; или (2) снижения или ингибирования активности HPK1; или (3) снижения или ингибирования экспрессии HPK1; или (4) изменения уровней белка HPK1. В другом неограничивающем варианте осуществления термин «терапевтически эффективное количество» относится к количеству соединения по настоящему изобретению, которое при введении в клетку, или ткань, или не клеточный биологический материал, или среду, эффективно для, по меньшей мере, частичного снижения или ингибирования активности HPK1; или снижения или ингибирования экспрессии HPK1 частично или полностью.
Все способы, описанные в настоящем документе, могут быть выполнены в любом подходящем порядке, если иное не указано в настоящем документе или иным образом явно не противоречит контексту. Использование любых и других примеров или вводных слов (например, «такой как»), представленных в настоящем документе, предназначено только для лучшего освещения изобретения и не налагает ограничения на объем изобретения, заявленного иным образом.
Общие химические термины, используемые в приведенных выше формулах, имеют свои обычные значения.
Используемые в настоящем документе «ч» или «ч» относятся к часу или часам, «мин» относятся к минутам или минутам, «MCL» относится к мантийноклеточной лимфоме, «AML» относится к острому миелоидному лейкозу, «CML» относится к хроническому миелоидному лейкозу, «Boc» относится к N-трет-бутоксикарбонилу, «EA» относится к этилацетату, «ДХМ» относится к дихлорметану, «ДМСО» относится к диметилсульфоксиду, «ДМА» относится к диметилацетамиду, «ТГФ» относится к тетрагидрофурану, «MtBE» относится к метил-трет-бутиловому эфиру, «ТЭА» относится к триэтиламину, «FBS» относится к эмбриональной бычьей сыворотке, «PBS» относится к забуференному фосфатом солевому раствору, «BSA» относится к бычьему сывороточному альбумину, «КТ» относится к при комнатной температуре, «мг/кг» означает миллиграммы на килограмм, «п.о.» относится к per os (перорально), «qd» означает дозирование один раз в день, «ЖХВД» означает жидкостную хроматографию высокого давления, «q2d» означает однократную дозу каждые 2 дня, «q2dx10» означает однократную дозу каждые 2 дня, умноженную на 10, «VSMC» относится к клеткам гладкой мускулатуры сосудов, и «ПРД» относится к дифракции рентгеновских лучей.
Используемый в настоящем документе термин «фармацевтически приемлемый носитель» включает любые растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные агенты, противогрибковые агенты), изотонические агенты, агенты, замедляющие абсорбцию, соли, консерванты, стабилизаторы лекарственных средств, связующие вещества, эксципиенты, разрыхлители, смазывающие вещества, подсластители, ароматизаторы, красители и подобные, а также их комбинации, как должно быть известно специалистам в данной области техники (см., например, Remington’s Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением случаев, когда любой обычный носитель несовместим с активным ингредиентом, рассматривается его использование в терапевтических или фармацевтических композициях.
Кроме того, соединения по настоящему изобретению, включая их соли, также могут быть получены в виде их гидратов, или включают другие растворители, используемые для их кристаллизации. Соединения по настоящему изобретению могут по своей природе или по замыслу образовывать сольваты с фармацевтически приемлемыми растворителями (включая воду); поэтому предполагается, что изобретение охватывает как сольватированные, так и не сольватированные формы. Термин «сольват» относится к молекулярному комплексу соединения по настоящему изобретению (включая его фармацевтически приемлемые соли) с одной или несколькими молекулами растворителя. Такие молекулы растворителя являются молекулами, обычно используемыми в области фармацевтики, которые, как известно, безвредны для реципиента, например, вода, этанол и подобные. Термин «гидрат» относится к комплексу, в котором молекулой растворителя является вода.
Соединения по настоящему изобретению, включая их соли, гидраты и сольваты, могут по своей природе или по замыслу образовывать полиморфы. В другом аспекте, настоящее изобретение представляет фармацевтическую композицию, содержащую соединение по настоящему изобретению и фармацевтически приемлемый носитель. Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение, ректальное введение и т.д. Кроме того, фармацевтические композиции по настоящему изобретению могут быть приготовлены в твердой форме (включая, без ограничений, капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включая, без ограничений, растворы, суспензии или эмульсии). Фармацевтические композиции могут быть подвергнуты обычным фармацевтическим операциям, таким как стерилизация и/или могут содержать обычные инертные разбавители, смазывающие агенты или буферные агенты, а также адъюванты, такие как консерванты, стабилизаторы, смачивающие агенты, эмульгаторы и буферы и т.д.
Как правило, фармацевтическими композициями являются таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с а) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; b) смазывающими веществами, например, диоксидом кремния, тальком, стеариновой кислотой, их солями магния или кальция и/или полиэтиленгликолем; для таблеток также c) связующими веществами, например, алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, карбоксиметилцеллюлозой натрия и/или поливинилпирролидоном; при желании d) разрыхлителями, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями; и/или e) абсорбентами, красителями, ароматизаторами и подсластителями. Таблетки могут быть покрыты либо пленкой, либо энтеросолюбильным покрытием в соответствии со способами, известными в данной области техники.
Подходящие композиции для перорального введения включают эффективное количество соединения по изобретению в форме таблеток, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсии, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, готовят в соответствии с любым способом, известным в данной области техники, для производства фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов для того, чтобы обеспечить фармацевтически элегантные и приятные на вкус препараты. Таблетки могут содержать активный ингредиент в смеси с не токсичными фармацевтически приемлемыми эксципиентами, пригодными для изготовления таблеток. Такими эксципиентами являются, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например, кукурузный крахмал или альгиновая кислота; связующие агенты, например, крахмал, желатин или аравийская камедь; и смазывающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки не имеют покрытия или покрыты известными методами для замедления разрыхления и абсорбции в желудочно-кишечном тракте и, таким образом, обеспечивают устойчивое действие в течение более длительного периода времени. Например, может быть использован материал с временной задержкой, такой как глицерилмоностеарат или глицерилдистеарат. Составы для перорального применения могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водной или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Некоторые композиции для инъекций представляют собой водные изотонические растворы или суспензии, и суппозитории преимущественно готовят из жировых эмульсий или суспензий. Указанные композиции могут быть стерилизованы и/или содержать адъюванты, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие агенты, промоторы растворения, соли для регулирования осмотического давления и/или буферы. Кроме того, они могут содержать и другие терапевтически ценные вещества. Указанные композиции готовят в соответствии с обычными способами смешивания, гранулирования или нанесения покрытия, соответственно, и они содержат примерно 0,1-75% или содержат примерно 1-50% активного ингредиента. Подходящие композиции для чрескожного применения включают эффективное количество соединения по изобретению с подходящим носителем. Носители, подходящие для чрескожной доставки, включают абсорбируемые фармакологически приемлемые растворители для облегчения прохождения через кожу хозяина. Например, трансдермальные устройства имеют форму бандажа, содержащего подложку, резервуар, содержащий соединение, необязательно с носителями, необязательно барьер, контролирующий скорость, для доставки соединения в кожу хозяина с контролируемой и заданной скоростью в течение длительного периода времени, и средства для закрепления устройства на коже.
Подходящие композиции для местного применения, например, на кожу и в глаза, включают водные растворы, суспензии, мази, кремы, гели или составы для распыления, например, для доставки в виде аэрозоля и подобные. Такие системы местной доставки будут, в частности, подходящими для кожного применения, например, для лечения рака кожи, например, для профилактического применения в солнцезащитных кремах, лосьонах, спреях и подобных. Таким образом, они особенно подходят для использования в местных составах, включая косметические составы, хорошо известные в данной области техники. Они могут содержать солюбилизаторы, стабилизаторы, агенты, повышающие тоничность, буферы и консерванты.
Используемое в настоящем документе местное применение может также относиться к ингаляции или интраназальному применению. Их удобно доставлять в форме сухого порошка (либо отдельно, в виде смеси, например, сухой смеси с лактозой, либо в виде частиц смешанного компонента, например, с фосфолипидами) из ингалятора для сухого порошка или в виде аэрозольного спрея из контейнера под давлением, насоса, спрея, распылителя или небулайзера, с или без использования подходящего пропеллента.
Настоящее изобретение дополнительно представляет безводные фармацевтические композиции и дозированные формы, содержащие соединения по настоящему изобретению в качестве активных ингредиентов, поскольку вода может способствовать разложению некоторых соединений.
Безводные фармацевтические композиции и дозированные формы по изобретению могут быть приготовлены с использованием безводных ингредиентов или ингредиентов с низким содержанием влаги и в условиях с низким содержанием влаги или низкой влажности. Безводная фармацевтическая композиция может быть приготовлена и храниться таким образом, чтобы сохранялась ее безводная природа. Соответственно, безводные композиции упаковывают с использованием материалов, которые, как известно, предотвращают воздействие воды, так что они могут быть включены в подходящие формулярные наборы. Примеры подходящей упаковки включают, но не ограничены ими, герметически запечатанную фольгу, пластмассу, контейнеры со стандартными дозами (например, флаконы), блистерные упаковки и ленточные упаковки.
Изобретение дополнительно представляет фармацевтические композиции и дозированные формы, которые содержат один или несколько агентов, снижающих скорость разложения соединения по настоящему изобретению в качестве активного ингредиента. Такие агенты, которые упоминаются в настоящем документе как «стабилизаторы», включают, но не ограничиваются ими, антиоксиданты, такие как аскорбиновая кислота, рН-буферы или солевые буферы и т.д.
3. Соединения
В настоящем документе описаны варианты осуществления соединения, имеющего общую структуру формулы (I-0) или (II):
 (I-0) или
(I-0) или 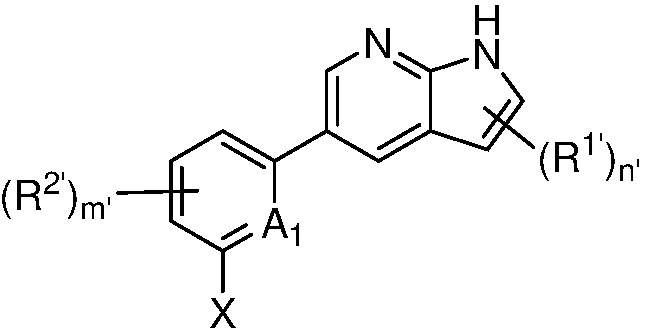 (II).
(II).
В первом варианте осуществления изобретения представлено соединение, представленное формулой (I-0) или его фармацевтически приемлемая соль или стереоизомер, где
X является СR2R3 или NR3;
A является СR2 или N;
R является 8-10-членным бициклическим азотсодержащим гетероарилом или 8-10-членным бициклическим азотсодержащим гетероциклилом, необязательно замещенным оксо, где азотсодержащий гетероарил или азотсодержащий гетероциклил, представленный R, имеет 1-3 гетероатома, выбранных из N, O и S и необязательно замещен одним-четырьмя Ra, и где R либо соединен с пиримидиновым кольцом через атом азота кольца, либо R представлен структурой ниже:
 или
или 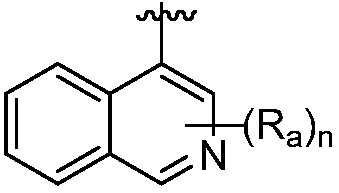 ;
;
R1 является Н, дейтерием, галогеном, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, фенилом, 5-6-членным гетероарилом, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, фенил, гетероарил, циклоалкил или гетероциклил, представленный R1 или в группе, представленной R1, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси и NR11R12; или
R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
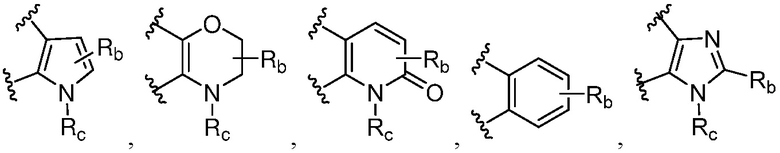 или
или 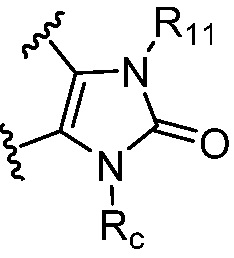 , где
, где  связи соединены с пиримидиновым кольцом;
связи соединены с пиримидиновым кольцом;
каждый случай R2 независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, P(=O)R11R12, S(=O)2R11 или S(=O)2NR11R12, где алкил, алкенил, алкинил или алкокси, представленный R2 или в группе, представленной R2, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12;
R3 является Н, C1-6 алкилом, C(O)C1-6 алкилом, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, циклоалкил или гетероциклил, представленный R3 или в группе, представленной R3, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила, 3-7-членного гетероциклила и NR11R12;
каждый случай R11 и R12 независимо является H или C1-6 алкилом, где алкил, представленный R11 или R12, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила и 3-7-членного гетероциклила, или
R11 и R12, вместе с атомом азота или атомом фосфора, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси;
каждый случай Ra независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, COOH, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)C1-6 алкилом, C(O)OR11, NR11C(O)R12, S(=O)2R11, S(=O)2NR11R12, NR11S(=O)2R12, P(=O)R11R12, C3-6 циклоалкилом, 3-7-членным гетероциклилом или 5-6-членным гетероарилом, где алкил, алкенил, алкинил, алкокси, циклоалкил, гетероциклил или гетероарил, представленный Ra или в группе, представленной Ra, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12 и 5-6-членного гетероарила; или
два Ra, вместе с атомами углерода, к которым они присоединены, образуют C3-6 циклоалкил или 3-7-членный гетероциклил, где циклоалкил или гетероциклил необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси;
каждый случай Rb независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, COOH, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)C1-6 алкилом, C(O)OR11, NR11C(O)R12, S(=O)2R11, S(=O)2NR11R12, NR11S(=O)2R12, P(=O)R11R12, C3-6 циклоалкилом, 3-7-членным гетероциклилом или 5-6-членным гетероарилом, где алкил, алкенил, алкинил, алкокси, циклоалкил, гетероциклил или гетероарил, представленный Rb или в группе, представленной Rb, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12 и 5-6-членным гетероарилом;
каждый случай Rc независимо является фенилом, 5-6-членным моноциклическим гетероциклилом, имеющим 1-3 гетероатома, выбранных из N и O; 5-6-членным моноциклическим гетероарилом, имеющим 1-3 гетероатома, выбранных из N и O; где фенил, гетероциклил или гетероарил, представленный Rc, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12 и P(O)ди-C1-6 алкила;
Ra' является С1-6 алкилом, необязательно замещенным OH, CN, 5-6-членным гетероарилом, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12;
m равно 0, 1, 2 или 3;
n равно 0, 1, 2, 3 или 4;
p равно 1, 2 или 3;
q равно 1, 2 или 3; и p+q ≤4.
Во втором варианте осуществления изобретения, соединение представлено структурной формулой (I-1):
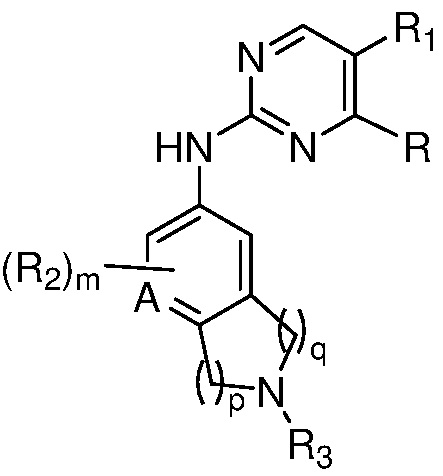 (I-1)
(I-1)
или его фармацевтически приемлемая соль или стереоизомер, и оставшиеся переменные такие, как определены в первом варианте осуществления.
В третьем варианте осуществления изобретения, соединение представлено структурной формулой (I-2):
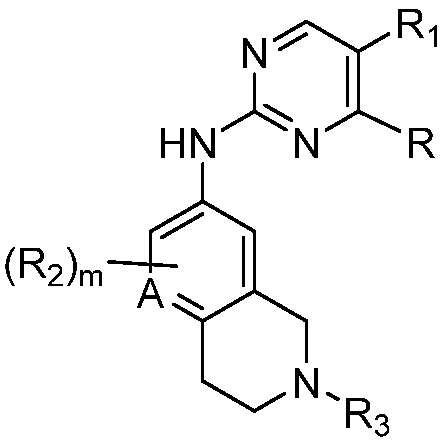 (I-2),
(I-2),
или его фармацевтически приемлемая соль или стереоизомер и оставшиеся переменные такие, как определены в первом и/или втором вариантах осуществления.
В четвертом варианте осуществления изобретения, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R является  ,
,
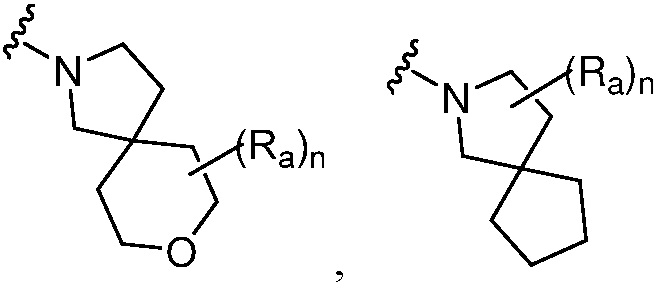 или
или 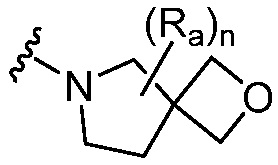 , где связь соединяет с пиримидиновым кольцом, и оставшиеся переменные такие, как определены в первом, втором и/или третьем вариантах осуществления.
, где связь соединяет с пиримидиновым кольцом, и оставшиеся переменные такие, как определены в первом, втором и/или третьем вариантах осуществления.
В пятом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай Ra независимо является H, галогеном, OH, CN, C1-4 алкилом, C1-4 алкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)C1-4 алкилом, C(O)OR11, NR11C(O)C1-4 алкилом, NR11S(=O)2R12, S(=O)2R11, S(=O)2NR11R12, P(=O)R11R12, C3-6 циклоалкилом, 3-7-членным гетероциклилом или 5-6-членным гетероарилом, где алкил, алкокси, циклоалкил, гетероциклил или гетероарил, представленный Ra или в группе, представленной Ra, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12 и 5-6-членного гетероарила, и остальные переменные такие, как определены в первом, втором, третьем и/или четвертом вариантах осуществления.
В шестом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R1 является Н, галогеном, CN, C1-6 алкилом, C2-6 алкенилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, где алкил, алкенил или алкокси, представленный R1 или в группе, представленной R1, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси и NR11R12, и остальные переменные такие, как определены в первом, втором, третьем, четвертом и/или пятом вариантах осуществления.
В седьмом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай R2 независимо является H, галогеном, OH, CN, NH2, NO2, C1-4 алкилом, C1-4 алкокси, NR11R12, C(O)NR11R12, C(O)C1-4 алкилом, C(O)OC1-4 алкилом, NR11C(O)C1-4 алкилом, P(=O)R11R12, S(=O)2R11 или S(=O)2NR11R12, где алкил или алкокси, представленный R2 или в группе, представленной R2, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12 и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом и/или шестом вариантах осуществления.
В восьмом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R3 является Н, C1-6 алкилом или C(O)C1-6 алкилом, где алкил, представленный R3 или в группе, представленной R3, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом и/или седьмом вариантах осуществления.
В девятом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай Ra независимо является H, галогеном, OH, CN, C1-4 алкилом, C3-6 циклоалкилом, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, N(R11)S(=O)2R12, S(=O)2R11, S(=O)2NR11R12, P(O)R11R12 или 5-6-членным гетероарилом, где алкил или циклоалкил, представленный Ra или в группе, представленной Ra, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12, C(O)NR11OR12, C(O)NR11S(=O)2R12, C(O)OR11, NR11S(=O)2R12, P(=O)R11R12, S(=O)2R11, S(=O)2NR11R12 и 5-6-членного гетероарила; каждый случай R11 и R12 независимо является H или C1-6 алкилом, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом и/или восьмом вариантах осуществления.
В десятом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R1 является Н, галогеном, CN, C1-4 алкилом, C1-4 галогеналкилом, C2-4 алкенилом, C1-4 алкокси или C1-4 галогеналкокси, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом и/или девятом вариантах осуществления.
В одиннадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R является 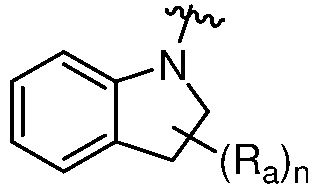 ,
,
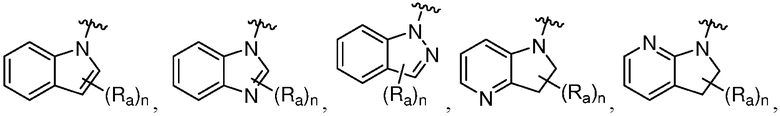
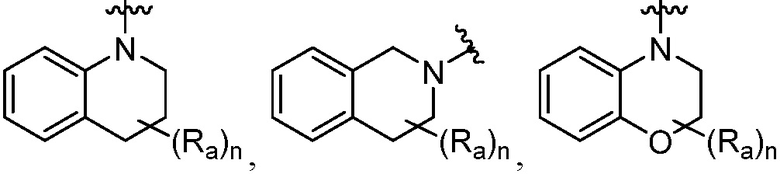 или
или 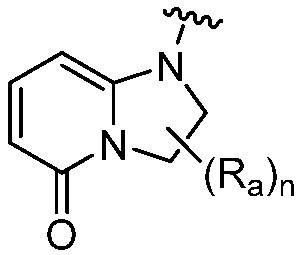 , где связь соединяет с пиримидиновым кольцом, и n равно 0-4 и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
, где связь соединяет с пиримидиновым кольцом, и n равно 0-4 и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
В двенадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R является 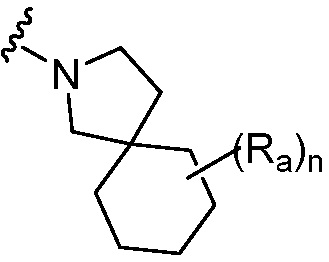 ,
, 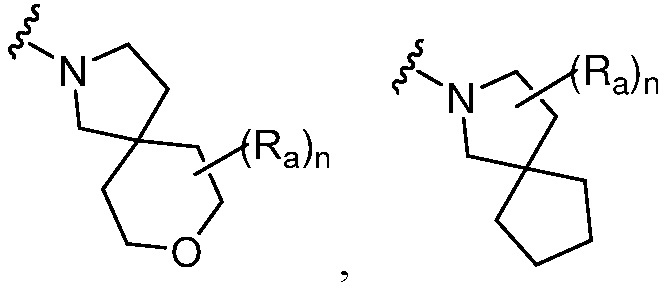 или
или 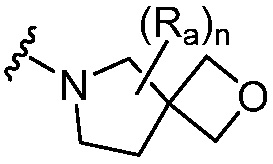 , где связь соединяет с пиримидиновым кольцом и n равно 0-2, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
, где связь соединяет с пиримидиновым кольцом и n равно 0-2, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
В тринадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R является 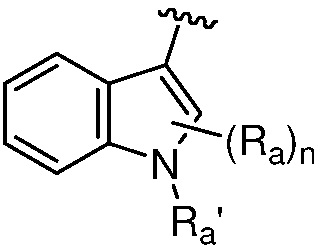 ,
, 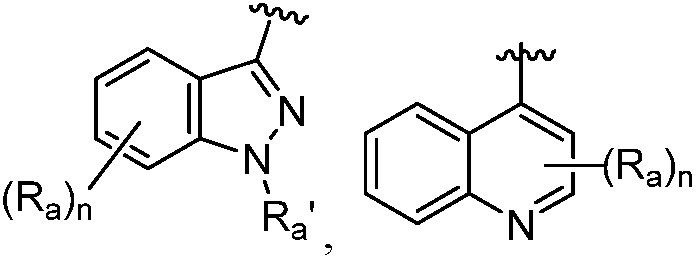 или
или 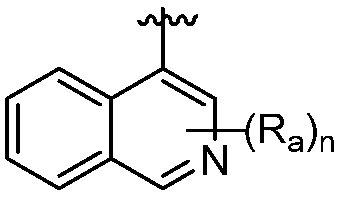 , где связь соединяет с пиримидиновым кольцом и n равно 0-2, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
, где связь соединяет с пиримидиновым кольцом и n равно 0-2, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
В четырнадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где
каждый случай Ra независимо является H, галогеном, C1-4 алкилом, -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kS(=O)2R11, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-5-6-членным гетероарилом или -(CHRaa)kP(=O)R11R12;
Raa независимо является H или C1-3 алкилом, необязательно замещенным галогеном;
R11 и R12 независимо являются H или C1-4 алкилом; и
k равно 0 или 1 и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом и/или тринадцатом вариантах осуществления.
В пятнадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R1 является Н, F, Cl, CN или CF3 и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом и/или четырнадцатом вариантах осуществления.
В шестнадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R является 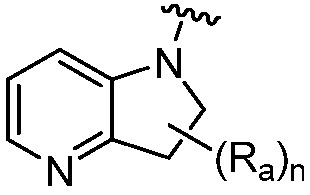 ,
, 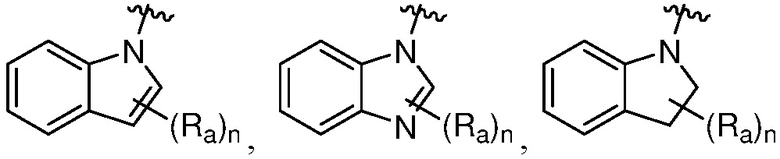 или
или  , где связь соединяет с пиримидиновым кольцом и n равно 0-4, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом и/или пятнадцатом вариантах осуществления.
, где связь соединяет с пиримидиновым кольцом и n равно 0-4, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом и/или пятнадцатом вариантах осуществления.
В семнадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где
R является 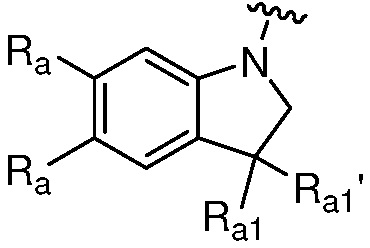 , где связь соединяет с пиримидиновым кольцом,
, где связь соединяет с пиримидиновым кольцом,
Ra1 независимо является H, C1-4 алкилом или C1-4гидроксиалкилом;
Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kS(=O)2R11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-5-6-членным гетероарилом или -(CHRaa)kP(=O)R11R12;
Raa независимо является H или C1-3 алкилом, необязательно замещенным галогеном;
Ra независимо является H, F или Cl;
R11 и R12 независимо являются H или C1-4 алкилом;
k равно 0 или 1, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом и/или десятом вариантах осуществления.
В восемнадцатом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где
Ra1 независимо является H, CH3 или CH2OH;
Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kS(=O)2R11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-тетразолом или -(CHRaa)kP(=O)R11R12;
Raa независимо является H, CH3 или CF3;
R11 и R12 независимо являются H или C1-2 алкилом; и
k равно 0 или 1, и остальные переменные такие, как определены в семнадцатом варианте осуществления.
В девятнадцатом варианте осуществления изобретения, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай Ra независимо является H или F, и остальные переменные такие, как определены в восемнадцатом варианте осуществления.
В двадцатом варианте осуществления изобретения, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R1 является Сl, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом и/или девятнадцатом вариантах осуществления.
В двадцать первом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
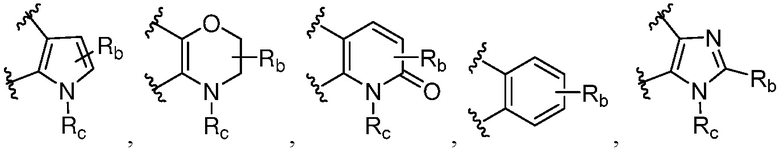 или
или 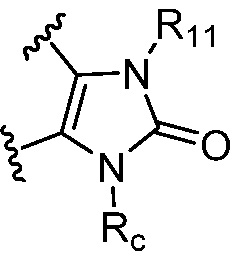 , где связи соединены с пиримидиновым кольцом и остальные переменные такие, как определены в первом, втором и/или третьем вариантах осуществления.
, где связи соединены с пиримидиновым кольцом и остальные переменные такие, как определены в первом, втором и/или третьем вариантах осуществления.
В двадцать втором варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где
каждый случай Rb независимо является H, галогеном, OH, CN, NH2, COOH, C1-4 алкилом, C1-4 галогеналкилом, C1-4 гидроксиалкилом, C1-4 алкокси, C1-4 галогеналкокси, C1-4 гидроксиалкокси, NR11R12, C(O)NR11R12, C(O)C1-4 алкилом, C(O)OC1-4 алкилом, C(O)NR11OR12, S(=O)2R11, S(=O)2NR11R12, NR11(S=O)2R12, C(O)NR11S(=O)2R12, P(=O)R11R12, 5-6-членным гетероарилом или NR11C(O)C1-4 алкилом; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12 и P(=O)ди-C1-6 алкила, и остальные переменные такие, как определены в первом, втором, третьем и/или двадцать первом вариантах осуществления.
В двадцать третьем варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
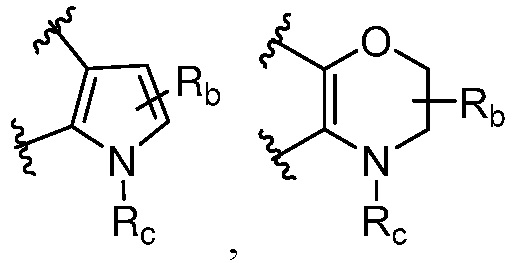 или
или 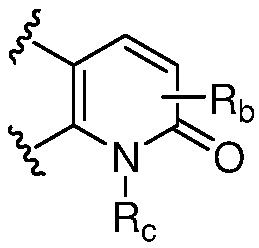 , где связи соединены с пиримидиновым кольцом и остальные переменные такие, как определены в первом, втором, третьем, двадцать первом и/или двадцать втором вариантах осуществления.
, где связи соединены с пиримидиновым кольцом и остальные переменные такие, как определены в первом, втором, третьем, двадцать первом и/или двадцать втором вариантах осуществления.
В двадцать четвертом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где
каждый случай Rb независимо является H, галогеном, CN, COOH, C1-2 алкилом или C1-2 галогеналкилом;
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, C(O)NR11R12 и P(=O)ди-C1-6 алкила, и
каждый случай R11 и R12 независимо является H или C1-6 алкилом, и остальные переменные такие, как определены в первом, втором, третьем, двадцать первом, двадцать втором и/или двадцать третьем вариантах осуществления.
В двадцать пятом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где
каждый случай Rb независимо является H, CN или COOH; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C(O)N(CH3)2 и P(=O)(CH3)2, и остальные переменные такие, как определены в первом, втором, третьем, двадцать первом, двадцать втором, двадцать третьем и/или двадцать четвертом вариантах осуществления.
В двадцать шестом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где m равно 0 или 1 и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом и/или двадцать пятом вариантах осуществления.
В двадцать седьмом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где 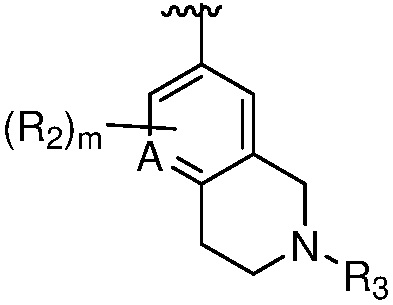 является
является 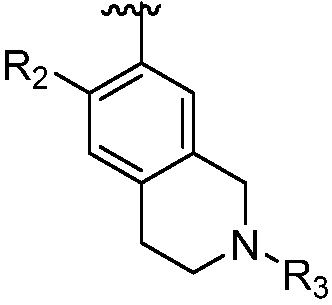 , и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом и/или двадцать шестом вариантах осуществления.
, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом и/или двадцать шестом вариантах осуществления.
В двадцать восьмом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай R2 является Н, галогеном, C1-4 алкилом, C1-4 галогеналкилом, C1-4 алкокси, C1-4 галогеналкокси; и R3 является Н, C1-6 алкилом, C1-6 гидроксиалкилом или COCH2NR11R12, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом и/или двадцать седьмом вариантах осуществления.
В двадцать девятом варианте осуществления, представлено соединение формулы (I-0), (I-1) или (I-2) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай R2 является Н, F, Cl или OCH3; и R3 является Н или C1-4 алкилом, и остальные переменные такие, как определены в первом, втором, третьем, четвертом, пятом, шестом, седьмом, восьмом, девятом, десятом, одиннадцатом, двенадцатом, тринадцатом, четырнадцатом, пятнадцатом, шестнадцатом, семнадцатом, восемнадцатом, девятнадцатом, двадцатом, двадцать первом, двадцать втором, двадцать третьем, двадцать четвертом, двадцать пятом, двадцать шестом, двадцать седьмом и/или двадцать восьмом вариантах осуществления.
В тридцатом варианте осуществления изобретения, представлено соединение, представленное формулой (II) или его фармацевтически приемлемая соль или стереоизомер,
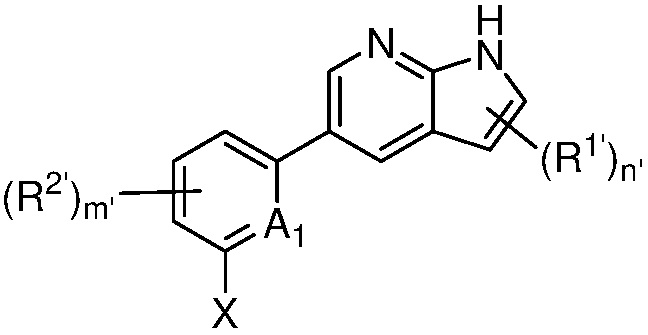 (II), где
(II), где
A1 является СR’ или N;
Х является -P(=O)R3’R4’;
R’ является Н, дейтерием, галогеном, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, где алкил, алкенил, алкинил или алкокси, представленный R’ или в группе, представленной R’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R1’ независимо является H, дейтерием, галогеном, OH, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, циклоалкил или гетероциклил, представленный R1’ или в группе, представленной R1’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R2’ независимо является H, дейтерием, галогеном, OH, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, фенилом, 5-6-членным гетероарилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, фенил, гетероарил, циклоалкил или гетероциклил, представленный R2’ или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый R3’ и R4’ независимо является H, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, где алкил, алкенил или алкинил, представленный R3’ или R4’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R11’ и R12’ независимо является H или C1-6 алкилом, где алкил, представленный R11’ или R12’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила и 3-7-членного гетероциклила, или
R11’ и R12’, вместе с атомом азота, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси или C1-4 галогеналкокси;
m’ равно 0, 1 или 2; и
n’ равно 0, 1 или 2.
В тридцать первом варианте осуществления изобретения, соединение формулы (II) представлено структурной формулой (II-1):
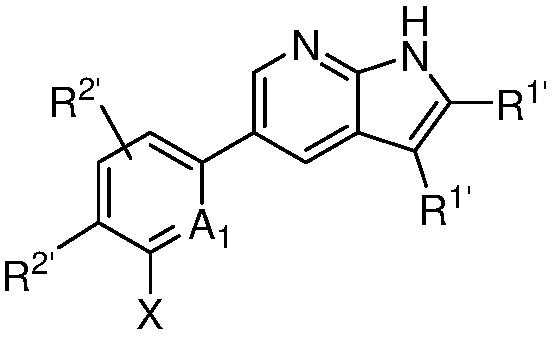 (II-1),
(II-1),
или его фармацевтически приемлемая солью или стереоизомером, где R’ является Н, галогеном, NR11’R12’ или C1-6 алкилом, необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’, и остальные переменные такие, как определены в тридцатом варианте осуществления.
В тридцать втором варианте осуществления, соединение формулы (II) или (II-1) или его фармацевтически приемлемая соль или стереоизомер, где R3’ и R4’ независимо являются H или C1-6 алкилом, необязательно замещено одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’, и остальные переменные такие, как определены в тридцатом и/или тридцать первом варианте осуществления.
В тридцать третьем варианте осуществления, соединение формулы (II) или (II-1) или его фармацевтически приемлемая соль или стереоизомер, где
каждый случай R1’ независимо является H, галогеном, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C3-7 ом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, циклоалкил или гетероциклил, представленный R1’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R2’ независимо является H, галогеном, OH, NO2, C1-6 алкилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, 5-6-членным гетероарилом или 3-7-членным гетероциклилом, где алкил, алкокси, гетероарил или гетероциклил, представленный R2’или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’, и остальные переменные такие, как определены в тридцатом, тридцать первом и/или тридцать втором вариантах осуществления.
В тридцать четвертом варианте осуществления, соединение формулы (II) или (II-1) представлено структурной формулой (II-2) или (II-2’):
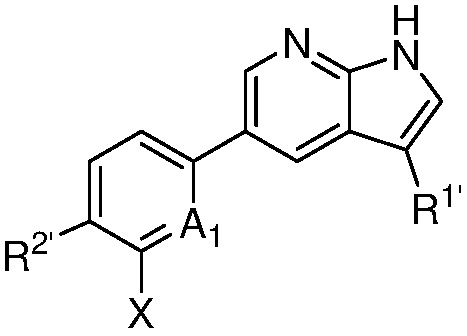 (II-2) или
(II-2) или 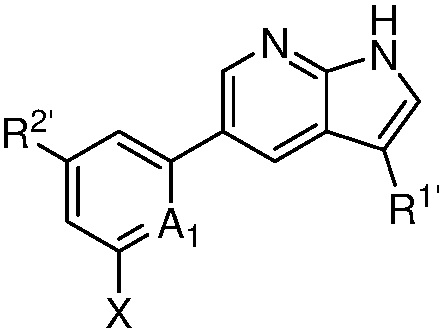 (II-2’)
(II-2’)
или его фармацевтически приемлемая соль или стереоизомер, где R’ является Н, галогеном или NH2, и остальные переменные такие, как определены в тридцатом, тридцать первом, тридцать втором и/или тридцать третьем вариантах осуществления.
В тридцать пятом варианте осуществления, соединение формулы (II), (II-1), (II-2) или (II-2’) или его фармацевтически приемлемая соль или стереоизомер, где каждый случай R2’ независимо является H, галогеном, OH, C1-4 алкилом, необязательно замещенным галогеном или OH или NR11’R12’, C1-4 алкокси, C(O)OC1-6 алкилом, C(O)NR11’R12’, NR11’R12’, NR11’C(O)C1-6 алкилом, 5-6-членным гетероциклилом или 5-6-членным гетероарилом; и каждый случай R1’ независимо является H или C1-4 алкилом, и остальные переменные такие, как определены в тридцатом, тридцать первом, тридцать втором, тридцать третьем и/или тридцать четвертом вариантах осуществления.
В тридцать шестом варианте осуществления, соединение формулы (II), (II-1), (II-2) или (II-2’) или его фармацевтически приемлемая соль или стереоизомер, где R3’ и R4’ независимо являются C1-6 алкилом; и каждый случай R2’ независимо является H, галогеном, OH, C1-4 алкилом, необязательно замещенным галогеном или OH, или NR11’R12’, C1-4 алкокси, C(O)OC1-4 алкилом, C(O)NR11’R12’, NR11’R12’, NHC(O)C1-6 алкилом, пирролидинилом, пирролидин-2-оном, оксазолом, где каждый случай R11’ и R12’ независимо является H или C1-4 алкилом, и остальные переменные такие, как определены в тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом и/или тридцать пятом вариантах осуществления.
В тридцать седьмом варианте осуществления, соединение формулы (II), (II-1), (II-2) или (II-2’) или его фармацевтически приемлемая соль или стереоизомер, где Х является -P(O)(CH3)2 и каждый случай R2’ независимо является H, F, Cl, OH, CH3, NH2 или NHCOCH3, и остальные переменные такие, как определены в тридцатом, тридцать первом, тридцать втором, тридцать третьем, тридцать четвертом, тридцать пятом и/или тридцать шестом вариантах осуществления.
1. Соединение, представленное структурной формулой (I):
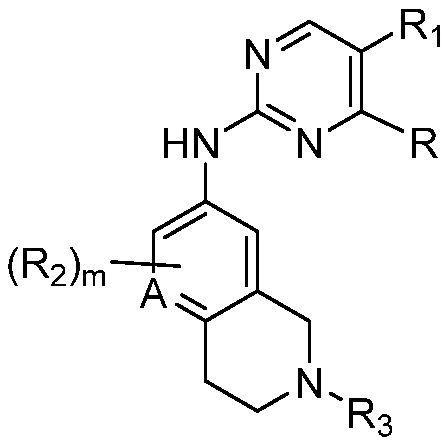 (I),
(I),
или его фармацевтически приемлемая соль, где
A является СR2 или N;
R является 8-10-членным бициклическим азотсодержащим гетероарилом или 8-10-членным бициклическим азотсодержащим гетероциклилом необязательно замещенным оксо, где азотсодержащий гетероарил или азотсодержащий гетероциклил, представленный R имеет 1-3 гетероатома, выбранных из N, O и S, и необязательно замещен одним-четырьмя Ra;
R1 является Н, дейтерием, галогеном, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, фенилом, 5-6-членным гетероарилом, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, фенил, гетероарил, циклоалкил или гетероциклил, представленный R1 или в группе, представленной R1, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12; или
R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
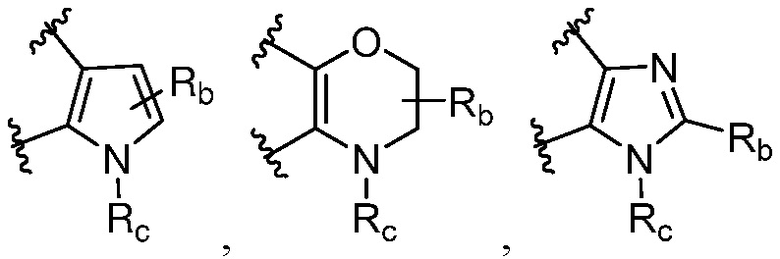 или
или 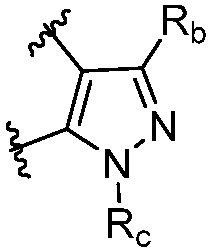 где
где  связи соединены с пиримидиновым кольцом;
связи соединены с пиримидиновым кольцом;
R2 является Н, дейтерием, галогеном, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, -P(=O)R11R12, -S(=O)2R11 или -S(=O)2NR11R12, где алкил, алкенил, алкинил или алкокси, представленный R2 или в группе, представленной R2, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12;
R3 является H, C1-6 алкилом, C(O)C1-6 алкилом, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, циклоалкил или гетероциклил представленный R3 или в группе, представленной R3 необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила, 3-7-членного гетероциклила и NR11R12;
каждый случай R11 и R12 независимо является H или C1-6 алкилом, где алкил, представленный R11 или R12, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила и 3-7-членного гетероциклила, или
R11 и R12, вместе с атомом азота или атомом фосфора, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси или C1-4 галогеналкокси;
каждый случай Ra независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, COOH, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, -P(=O)R11R12, -S(=O)2R11 или -S(=O)2NR11R12, C3-6 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил или алкокси, циклоалкил или гетероциклил, представленный Ra или в группе, представленной Ra, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12; или
два Ra, вместе с атомом углерода, к которому они присоединены, образуют C3-6 циклоалкил или 3-7-членным гетероциклилом, где циклоалкил или гетероциклил необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси;
каждый случай Rb независимо является H, дейтерием, галогеном, OH, CN, NH2, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом или NR11C(O)C1-6 алкилом, где алкил, алкенил, алкинил или алкокси, представленный Rb или в группе, представленной Rb, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12;
Rc является фенилом, 5-6-членным моноциклическим гетероциклилом, имеющим 1-3 гетероатома, выбранных из N и O; 5-6-членным моноциклическим гетероарилом, имеющим 1-3 гетероатома, выбранных из N и O; где фенил, гетероциклил или гетероарил, представленный Rc, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, -C(O)NR11R12 и -P(O)ди-C1-6 алкила; и
m равно 0, 1, 2 или 3.
2. Соединение по параграфу 1 или его фармацевтически приемлемая соль, где R является
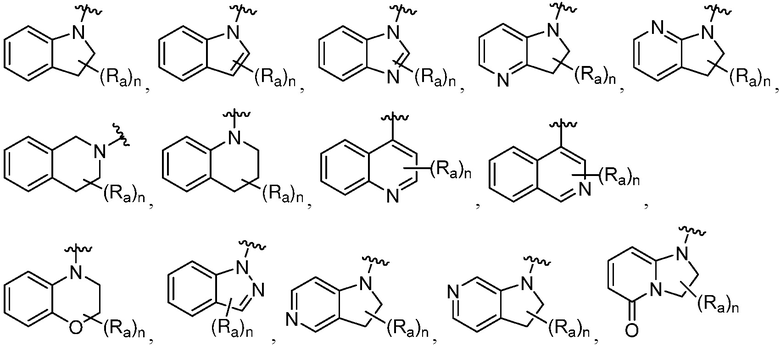 или
или 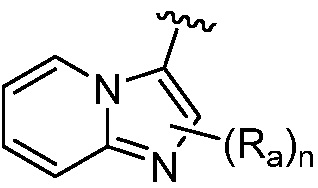 , где связь соединяет с пиримидиновым кольцом и n равно 0-4.
, где связь соединяет с пиримидиновым кольцом и n равно 0-4.
3. Соединение по параграфу 1 или 2 или его фармацевтически приемлемая соль, где
каждый случай Ra независимо является H, галогеном, OH, NH2, CN, COOH, C1-4 алкилом, C1-4 галогеналкилом, C1-4 гидроксиалкилом, C1-4 алкокси, C1-4 галогеналкокси, C1-4 гидроксиалкокси, NR11R12, C(O)NR11R12, C(O)C1-4 алкилом, C(O)OC1-4 алкилом, NR11C(O)C1-4 алкилом, -P(=O)R11R12, -S(=O)2R11, -S(=O)2NR11R12, C3-6 циклоалкилом или 3-7-членным гетероциклилом, или
два Ra, вместе с атомом углерода, к которому они присоединены, образуют C3-6 циклоалкил или 3-7-членный гетероциклил, где циклоалкил или гетероциклил необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси.
4. Соединение по любому из параграфов 1-3 или его фармацевтически приемлемая соль, где R1 является Н, галогеном, C1-6 алкилом, C1-6 алкокси, C2-6 алкенилом, NR11R12, C(O)NR11R12, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11C(O)C1-6 алкилом, где алкил, алкенил или алкокси, представленный R1 или в группе, представленной R1, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12.
5. Соединение по любому из параграфов 1-4 или его фармацевтически приемлемая соль, где каждый случай Ra независимо является H, галогеном, OH, C1-4 алкилом, COOH, C(O)NR11R12 или -P(O)R11R12, где каждый случай R11 и R12 независимо является H или C1-6 алкилом.
6. Соединение по любому из параграфов 1-5 или его фармацевтически приемлемая соль, где R1 является Н, галогеном, C1-4 алкилом, C1-4 галогеналкилом, C2-4 алкенилом, C1-4 алкокси или C1-4 галогеналкокси.
7. Соединение по любому из параграфов 1-6 или его фармацевтически приемлемая соль, где R является 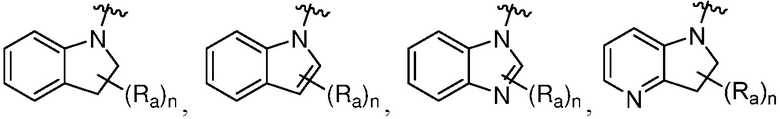 ,
, 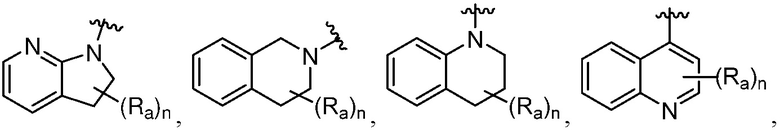
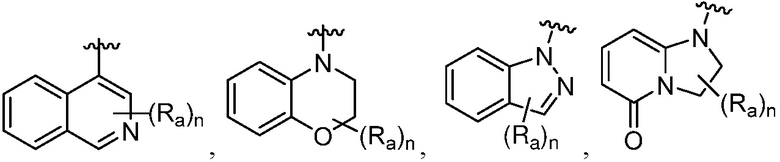 или
или 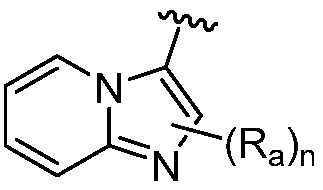 , где связь соединяет с пиримидиновым кольцом и n равно 0-2.
, где связь соединяет с пиримидиновым кольцом и n равно 0-2.
8. Соединение по любому из параграфов 1-7 или его фармацевтически приемлемая соль, где R является 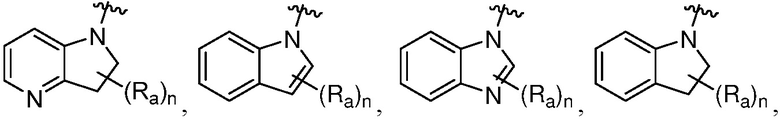
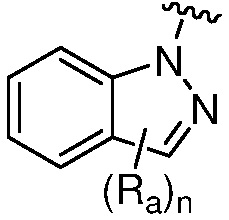 или
или 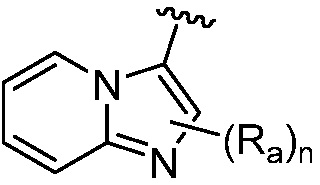 , где связь соединяет с пиримидиновым кольцом и n равно 0-2.
, где связь соединяет с пиримидиновым кольцом и n равно 0-2.
9. Соединение по любому из параграфов 1-8 или его фармацевтически приемлемая соль, где каждый случай Ra независимо является H, F, Cl, C1-4 алкилом, COOH, CONH2, C(O)N(CH3)2 или -P(O)(CH3)2.
10. Соединение по любому из параграфов 1-9 или его фармацевтически приемлемая соль, где R1 является Н, Cl или CF3.
11. Соединение по параграфу 1 или его фармацевтически приемлемая соль, где R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
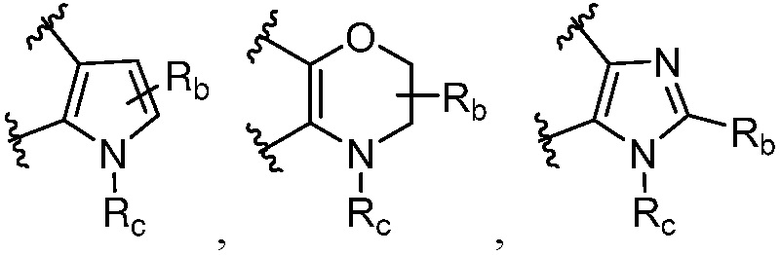 или
или 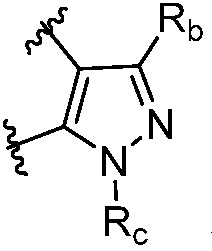 где связи соединены с пиримидиновым кольцом.
где связи соединены с пиримидиновым кольцом.
12. Соединение по параграфу 1 или 11 или его фармацевтически приемлемая соль, где
каждый случай Rb независимо является H, галогеном, OH, NH2, C1-4 алкилом, C1-4 галогеналкилом, C1-4 гидроксиалкилом, C1-4 алкокси, C1-4 галогеналкокси, C1-4 гидроксиалкокси, NR11R12, C(O)NR11R12, C(O)C1-4 алкилом, C(O)OC1-4 алкилом или NR11C(O)C1-4 алкилом; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, -C(O)NR11R12 и -P(O)ди-C1-6 алкила.
13. Соединение по любому из параграфов 1, 11 и 12 или его фармацевтически приемлемая соль, где
каждый случай Rb независимо является H, галогеном, C1-2 алкилом, C1-2 галогеналкилом; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, NR11R12, -C(O)NR11R12 и -P(O)ди-C1-6 алкила, и
каждый случай R11 и R12 независимо является H или C1-6 алкилом.
14. Соединение по любому из параграфов 1 и 11-13 или его фармацевтически приемлемая соль, где
каждый случай Rb независимо является H; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, -C(O)N(CH3)2 и -P(O)(CH3)2.
15. Соединение по любому из параграфов 1-14 или его фармацевтически приемлемая соль, где R2 является Н, галогеном, OH, CN, NH2, NO2, C1-4 алкилом, C1-4 алкокси, NR11R12, -C(O)NR11R12, C(O)C1-4 алкилом, C(O)OC1-4 алкилом, NR11C(O)C1-4 алкилом, -P(=O)R11R12, -S(=O)2R11 или -S(=O)2NR11R12, где алкил или алкокси, представленный R2 или в группе, представленной R2, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12; и m равно 0 или 1.
16. Соединение по любому из параграфов 1-15 или его фармацевтически приемлемая соль, где 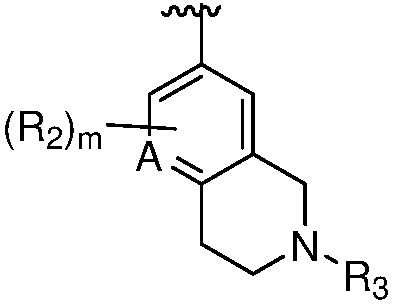 является
является 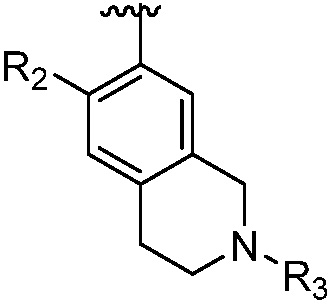 или
или 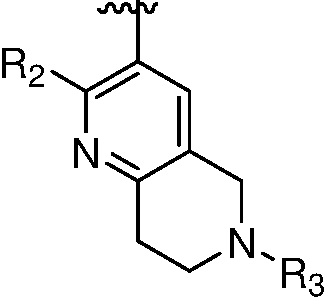 .
.
17. Соединение по любому из параграфов 1-16 или его фармацевтически приемлемая соль, где каждый случай R2 является Н, галогеном, C1-4 алкилом, C1-4 галогеналкилом, C1-4 алкокси, C1-4 галогеналкокси; и R3 является Н, C1-6 алкилом, C1-6 гидроксиалкил или COCH2NR11R12, где каждый случай R11 и R12 независимо является H или C1-6 алкилом.
18. Соединение по любому из параграфов 1-17 или его фармацевтически приемлемая соль, где каждый случай R2 является Н, Cl или OCH3; и R3 является Н, C1-4 алкилом, C1-4 гидроксиалкилом или COCH2N(CH3)2.
19. Соединение, представленное структурной формулой (II):
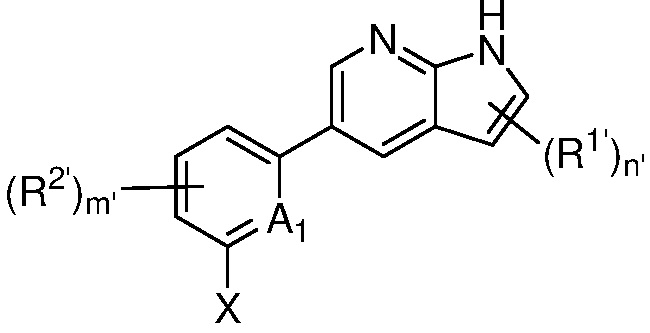 (II),
(II),
или его фармацевтически приемлемая соль, где
A1 является СR’ или N;
Х является -P(=O)R3’R4’ или -S(O)2NR5’R6’;
R’ является Н, дейтерием, галогеном, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, где алкил, алкенил, алкинил или алкокси, представленный R’ или в группе, представленной R’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R1’ независимо является H, дейтерием, галогеном, OH, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, циклоалкил или гетероциклил, представленный R1’ или в группе, представленной R1’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый случай R2’ независимо является H, дейтерием, галогеном, OH, CN, NO2, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, фенилом, 5-6-членным гетероарилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, алкокси, фенил, гетероарил, циклоалкил или гетероциклил, представленный R2’ или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый R3’ и R4’ независимо является H, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, где алкил, алкенил или алкинил, представленный R3’ или R4’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’;
каждый R5’ и R6’ независимо является H, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, где алкил, алкенил или алкинил, представленный R5’ или R6’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’; или
R5’ и R6’, вместе с атомом азота, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси или C1-4 галогеналкокси;
каждый случай R11’ и R12’ независимо является H или C1-6 алкилом, где алкил, представленный R11’ или R12’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси, C3-6 циклоалкила и 3-7-членного гетероциклила, или
R11’ и R12’, вместе с атомом азота, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси или C1-4 галогеналкокси;
m’ равно 0, 1 или 2; и
n’ равно 0, 1 или 2.
20. Соединение по параграфу 19, где соединение представлено структурной формулой (II-1):
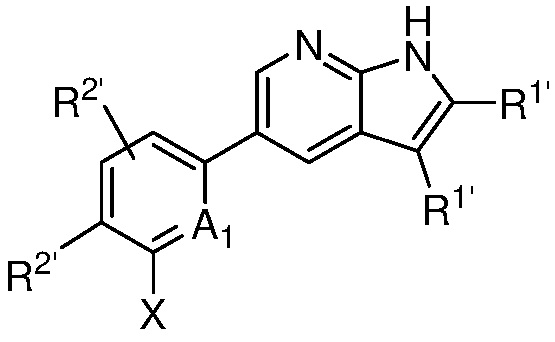 (II-1),
(II-1),
или его фармацевтически приемлемая соль, где
A1 является СR’ или N; и
R’ является Н, галогеном, NR11’R12’ или C1-6 алкилом, необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’.
21. Соединение по параграфу 19 или 20 или его фармацевтически приемлемая соль, где
Х является -P(=O)R3’R4’ или -S(O)2NR5’R6’;
каждый R3’ и R4’ независимо являются H или C1-6 алкилом, необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11R12; и
каждый R5’ и R6’ независимо являются H или C1-6 алкилом, или
R5’ и R6’, вместе с атомом азота, к которому они присоединены, образуют 3-7-членный гетероциклил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси или C1-4 галогеналкокси.
22. Соединение по любому из параграфов 19-21 или его фармацевтически приемлемая соль, где каждый случай R1’ независимо является H, галогеном, C1-6 алкилом, C2-6 алкенилом, C2-6 алкинилом, C3-7 циклоалкилом или 3-7-членным гетероциклилом, где алкил, алкенил, алкинил, циклоалкил или гетероциклил, представленный R1’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’.
23. Соединение по любому из параграфов 19-22 или его фармацевтически приемлемая соль, где каждый случай R2’ независимо является H, галогеном, OH, NH2, C1-6 алкилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, 5-6-членным гетероарилом или 3-7-членным гетероциклилом, где алкил, алкокси, гетероарил или гетероциклил, представленный R2’ или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, -OH, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси, C1-4 галогеналкокси и NR11’R12’.
24. Соединение по любому из параграфов 19-23, где соединение представлено структурной формулой (II-2) или (II-2’):
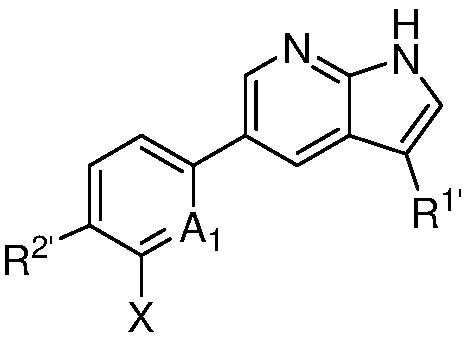 (II-2) или
(II-2) или 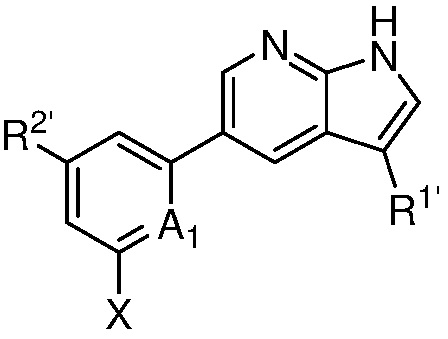 (II-2’)
(II-2’)
или его фармацевтически приемлемая соль, где A1 является СR’ или N; и R’ является Н, галогеном или NH2.
25. Соединение по любому из параграфов 19-24 или его фармацевтически приемлемая соль, где каждый случай R2’ независимо является H, галогеном, OH, C1-4 алкилом, необязательно замещенным галогеном или OH, или NR11’R12’, C1-4 алкокси, C(O)OC1-6 алкилом, C(O)NR11’R12’, NR11’R12’, NR11’C(O)C1-6 алкилом, 5-6-членным гетероциклилом (пирролидинилом или пирролидин-2-оном) или гетероарилом (оксазолом).
26. Соединение по любому из параграфов 19-25 или его фармацевтически приемлемая соль, где
Х является -P(=O)R3’R4’ или -S(O)2NR5’R6’;
R3’ и R4’ независимо являются C1-6 алкилом; и
R5’ и R6’ независимо являются H или C1-6 алкилом.
27. Соединение по любому из параграфов 19-26 или его фармацевтически приемлемая соль, где каждый случай R1’ независимо является H или C1-4 алкилом.
28. Соединение по любому из параграфов 19-27 или его фармацевтически приемлемая соль, где каждый случай R2’ независимо является H, галогеном, OH, C1-4 алкилом, необязательно замещенным галогеном или OH, или NR11’R12’, C1-4 алкокси, C(O)OC1-4 алкилом, C(O)NR11’R12’, NR11’R12’, NHC(O)C1-6 алкилом, пирролидинилом, пирролидин-2-оном, оксазолом, где каждый случай R11’ и R12’ независимо является H или C1-4 алкилом.
29. Соединение по любому из параграфов 19-28 или его фармацевтически приемлемая соль, где Х является -P(O)(CH3)2 или -S(O)2NHMe.
30. Соединение по любому из параграфов 19-29 или его фармацевтически приемлемая соль, где каждый случай R2’ независимо является H, F, Cl, OH, CH3, NH2 или -NHCOCH3.
31. Соединение по любому из параграфов 19-30 или его фармацевтически приемлемая соль, где Х является -P(=O)(CH3)2.
В одном варианте осуществления, соединение или его фармацевтически приемлемая соль или стереоизомер выбрано из соединений формулы (I-0), (I-1), (I-2), (I), (II), (II-1), (II-2) или (II-2’) или в примерах.
В определенных вариантах осуществления, ингибиторы HPK1 (соединения по изобретению) являются селективными против одной или нескольких киназ, выбранных из: Lck, ZAP70, JAK3, PKC тэта, TBK1 и MAP4K3. В определенных вариантах осуществления, ингибиторы HPK1 по изобретению селективны против одной или нескольких киназ, выбранных из: Lck, ZAP70 и JAK3. В определенных вариантах осуществления, ингибиторы HPK1 по изобретению селективны против JAK3.
Например, данные ингибиторы HPK1 селективны против Lck настолько, что IC50 против Lck, по меньшей мере, 2-, 3-, 5-, 10-, 20-, 40-, 50-, 75-, 100-, 150-, 200-, 300-, 400-, 500-, 600-, 750- или 1000-кратно выше, чем IC50 против HPK1.
В определенных вариантах осуществления, ингибитор HPK1 является селективными против ZAP70 так, что IC50 против ZAP70, по меньшей мере, 2-, 3-, 5-, 10-, 20-, 40-, 50-, 75-, 100-, 150-, 200-, 300-, 400-, 500-, 600-, 750- или 1000-кратно выше, чем IC50 против HPK1.
В определенных вариантах осуществления, ингибитор HPK1 является селективными против JAK3 так, что IC50 против JAK3, по меньшей мере, 2-, 3-, 5-, 10-, 20-, 40-, 50-, 75-, 100-, 150-, 200-, 300-, 400-, 500-, 600-, 750- или 1000-кратно выше, чем IC50 против HPK1.
В определенных вариантах осуществления, ингибитор HPK1 является селективными против PKC тэта так, что IC50 против PKC тэта, по меньшей мере, 2-, 3-, 5-, 10-, 20-, 40-, 50-, 75-, 100-, 150-, 200-, 300-, 400-, 500-, 600-, 750- или 1000-кратно выше, чем IC50 против HPK1.
В определенных вариантах осуществления, ингибитор HPK1 является селективными против TBK1 так, что IC50 против TBK1, по меньшей мере, 2-, 3-, 5-, 10-, 20-, 40-, 50-, 75-, 100-, 150-, 200-, 300-, 400-, 500-, 600-, 750- или 1000-кратно выше, чем IC50 против HPK1.
В определенных вариантах осуществления, ингибитор HPK1 является селективными против MAP4K3 так, что IC50 против MAP4K3, по меньшей мере, 2-, 3-, 5-, 10-, 20-, 40-, 50-, 75-, 100-, 150-, 200-, 300-, 400-, 500-, 600-, 750- или 1000-кратно выше, чем IC50 против HPK1.
Активность против HPK1, Lck, ZAP70, PKC тэта, JAK3, TBK1 и MAP4K3 может быть измерена (например, измерена по значениям IC50) с применением любого признанного в данной области техники способов, описанных в биологических примерах, таких как биологические примеры 1-7 (все включены в настоящий документ посредством ссылки).
4. Лечимые заболевания
Ингибиторы HPK1, их фармацевтически приемлемые соли, их фармацевтические композиции можно использовать в способах модулирования (т.е. ингибирования) активности HPK1, где указанный способ включает введение пациенту/субъекту, нуждающемуся в этом, соединения-ингибитора HPK1 по изобретению или его фармацевтически приемлемой соли, как описано в настоящем документе.
В частности, настоящее изобретение представляет применение соединений по изобретению или их стереоизомеров, таутомеров, N-оксидов, гидратов, сольватов и солей, в частности, их фармацевтически приемлемых солей или их смесей, для применения в лечении или профилактике заболеваний, в частности рака (в частности, гемопоэтических и солидных опухолей) или состояний с нарушением регуляции иммунных ответов или других нарушений, связанных с аберрантной передачей сигналов MAP4K1. Фармацевтическая активность соединений по изобретению может быть, по меньшей мере, частично объяснена их активностью в качестве ингибиторов MAP4K1.
В некоторых вариантах осуществления, соединения по изобретению или их фармацевтически приемлемые соли полезны для терапевтического введения субъекту, нуждающемуся в этом, для лечения заболевания или показания, включая, но не ограничиваясь этим, доброкачественную гиперплазию, атеросклеротическое нарушение, сепсис, аутоиммунное нарушение, сосудистое нарушение, вирусную инфекцию, нейродегенеративное нарушение, воспалительное нарушение и нарушения контроля мужской фертильности.
В некоторых вариантах осуществления, соединения по изобретению или их фармацевтически приемлемые соли применимы для терапевтического введения для усиления, стимуляции и/или повышения иммунитета при лечении рака.
Соединения ингибитора HPK1 по изобретению можно использовать отдельно или в комбинации с другими агентами или методами лечения, или в качестве адъюванта или неоадъюванта для лечения заболеваний или нарушений, включая рак.
В некоторых вариантах осуществления, способы по изобретению могут применяться для лечения раков, которые включают, но не ограничены ими, рак кости, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или интраокулярную злокачественную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак яичка, рак матки, карциному фаллопиевых труб, карциному эндометрия, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, неходжкинскую лимфому, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак уретры, рак полового члена, хронические или острые лейкозы, включая острый миелоидный лейкоз, хронический миелоидный лейкоз, острый лимфобластный лейкоз, хронический лимфоцитарный лейкоз, солидные опухоли детского возраста, лимфоцитарную лимфому, рак мочевого пузыря, рак почки или уретры, карциному почечной лоханки, новообразование центральной нервной системы (ЦНС), первичную лимфому ЦНС, ангиогенез опухоли, опухоль оси позвоночника, глиому ствола головного мозга, аденому гипофиза, саркому Капоши, эпидермоидный рак, плоскоклеточный рак, Т-клеточную лимфому, раки, вызванные окружающей средой, включая раки, вызванные асбестом, и комбинации вышеуказанных видов рака.
В некоторых вариантах осуществления раки, поддающиеся лечению соединениями по изобретению, включают меланому (например, метастатическую злокачественную меланому), рак почки (например, светлоклеточную карциному), рак предстательной железы (например, гормонорезистентную аденокарциному предстательной железы), рак молочной железы, трижды негативный рак молочной железы, рак толстой кишки и рак легкого (например, немелкоклеточный рак легкого и мелкоклеточный рак легкого). Кроме того, также поддаются лечению рефрактерные или рецидивирующие злокачественные новообразования, рост которых можно ингибировать с помощью соединений по изобретению.
В некоторых вариантах осуществления раки, которые поддается лечению с использованием соединений по изобретению, включают, но не ограничены ими, солидные опухоли (например, рак предстательной железы, рак толстой кишки, рак пищевода, рак эндометрия, рак яичников, рак матки, рак почки, рак печени, рак поджелудочной железы, рак желудка, рак молочной железы, рак легких, дыхательных путей, рак головного мозга, рак глаз, рак щитовидной и паращитовидной желез, рак кожи, рак головы и шеи, рак репродуктивных органов, рак пищеварительных путей, рак мочевыводящих путей, глиобластому, саркому, рак мочевого пузыря и т.д.), гематологические раки (например, лимфому, лейкоз, такой как острый лимфобластный лейкоз (ALL), острый миелогенный лейкоз (AML), хронический лимфоцитарный лейкоз (CLL), хронический миелогенный лейкоз (CML), DLBCL, мантийно-клеточную лимфому, неходжкинскую лимфому (включая рецидивирующую или рефрактерную NHL и рецидивирующую фолликулярную), лимфому Ходжкина или множественную миелому), саркому и их отдаленные метастазы.
В некоторых вариантах осуществления, заболевания и показания, которые поддаются лечению с использованием соединений по изобретению, включают, но не ограничены ими, гематологические раки, саркому, раки легких, раки желудочно-кишечного тракта, раки мочеполового тракта, раки печени, раки костей, раки нервной системы, гинекологические раки и раки кожи.
Типовые гематологические раки включают лимфомы и лейкозы, такие как острый лимфобластный лейкоз (ALL), острый миелогенный лейкоз (AML), острый промиелоцитарный лейкоз (APL), хронический лимфоцитарный лейкоз (CLL), хронический миелогенный лейкоз (CML), диффузная В-крупноклеточная лимфома (DLBCL), мантийно-клеточная лимфома, неходжкинская лимфома (включая рецидивирующую или рефрактерную NHL и рецидивирующую фолликулярную), лимфому Ходжкина, миелопролиферативные заболевания (например, первичный миелофиброз (PMF), истинная полицитемия (PV), эссенциальный тромбоцитоз (ET), миелодиспластический синдром (MDS), Т-клеточная острая лимфобластная лимфома (T-ALL), множественная миелома, кожная Т-клеточная лимфома, макроглобулинемия Вальденстрема, волосатоклеточная лимфома, хроническая миелогенная лимфома и лимфома Беркитта.
Примеры сарком включают хондросаркому, саркому Юинга, остеосаркому, рабдомиосаркому, ангиосаркому, фибросаркому, липосаркому, миксому, рабдомиому, рабдосаркому, фиброму, липому, гарматому и тератому.
Примеры раков легкого включают немелкоклеточный рак легкого (NSCLC), мелкоклеточный рак легкого, бронхогенную карциному (плоскоклеточную, недифференцированную мелкоклеточную, недифференцированную крупноклеточную, аденокарциному), альвеолярную (бронхиолярную) карциному, бронхиальную аденому, хондроматозную гамартому и мезотелиому.
Примеры раков желудочно-кишечного тракта включают раки пищевода (плоскоклеточную карциному, аденокарциному, лейомиосаркому, лимфому), желудка (карциному, лимфому, лейомиосаркому), поджелудочной железы (протоковую аденокарциному, инсулиному, глюкагоному, гастриному, карциноидные опухоли, випому), тонкой кишки (аденокарциному, лимфому, карциноидные опухоли, саркому Капоши, лейомиому, гемангиому, липому, нейрофиброму, фиброму), толстой кишки (аденокарциному, тубулярную аденому, ворсинчатую аденому, гамартому, лейомиому) и колоректальный рак.
Примеры раков мочеполового тракта включают раки почки (аденокарциному, опухоль Вильмса [нефробластому]), мочевого пузыря и уретры (плоскоклеточную карциному, переходно-клеточную карциному, аденокарциному), предстательной железы (аденокарциному, саркому) и яичка (семиному, тератому, эмбриональную карциному, тератокарциному, хориокарциному, саркому, карциному из интерстициальных клеток, фиброму, фиброаденому, аденоматоидные опухоли, липому).
Примеры раков печени включают гепатому (гепатоцеллюлярную карциному), холангиокарциному, гепатобластому, ангиосаркому, гепатоцеллюлярную аденому и гемангиому.
Примеры раков костей включают, например, остеогенную саркому (остеосаркому), фибросаркому, злокачественную фиброзную гистиоцитому, хондросаркому, саркому Юинга, злокачественную лимфому (ретикулосаркому), множественную миелому, хордому злокачественной гигантоклеточной опухоли, остеохронфрому (костно-хрящевую экзостозную болезнь), доброкачественную хондрому, хондробластому, хондромиксофиброму, остеоид-остеому и гигантоклеточные опухоли.
Примеры раков нервной системы включают раки черепа (остеому, гемангиому, гранулему, ксантому, деформирующий остит), мозговых оболочек (менингиому, менингиосаркому, глиоматоз), головного мозга (астроцитому, медуобластому, глиому, эпендимому, герминому (пинеалому), глиобластому, мультиформную глиобластому, олигодендроглиому, шванному, ретинобластому, врожденные опухоли) и спинного мозга (нейрофиброму, менингиому, глиому, саркому), а также нейробластому и болезнь Лермитта-Дюкло.
Типичные гинекологические раки включают раки матки (карциному эндометрия), шейки матки (карциному шейки матки, предопухолевую дисплазию шейки матки), яичников (карциному яичников (серозную цистаденокарциному, муцинозную цистаденокарциному, неклассифицированную карциному), гранулезо-текальноклеточные опухоли, опухоли клеток Сертоли-Лейдига, дисгерминому, злокачественную тератому), вульвы (плоскоклеточную карциному, внутриэпителиальную карциному, аденокарциному, фибросаркому, меланому), влагалища (светлоклеточную карциному, плоскоклеточную карциному, ботриоидную саркому (эмбриональную рабдомиосаркому) и фаллопиевых труб (карциному).
Примеры рака кожи включают меланому, базально-клеточную карциному, плоскоклеточную карциному, саркому Капоши, рак кожи из клеток Меркеля, родинки, диспластические невусы, липому, ангиому, дерматофиброму и келоиды. В некоторых вариантах осуществления, заболевания и показания, которые поддаются лечению с использованием соединений по изобретению, включают, но не ограничены ими, серповидноклеточное заболевание (например, серповидноклеточную анемию), трижды негативный рак молочной железы (TNBC), миелодиспластические синдромы, рак яичка, рак желчных протоков, рак пищевода и уротелиальную карциному.
Примеры раков головы и шеи включают глиобластому, меланому, рабдосаркому, лимфосаркому, остеосаркому, плоскоклеточные карциномы, аденокарциномы, рак ротовой полости, рак гортани, рак носоглотки, раки носа и околоносовых пазух, а также раки щитовидной железы и околощитовидной железы.
В некоторых вариантах осуществления, заявленные ингибиторы HPK1 можно применять для лечения опухолей, продуцирующих PGE2 (например, опухоли со сверхэкспрессией Cox-2) и/или аденозин (опухоли со сверхэкспрессией CD73 и CD39). Сверхэкспрессия Cox-2 была обнаружена в ряде опухолей, таких как колоректальный рак, раки молочной железы, поджелудочной железы и легких, где она коррелирует с плохим прогнозом. Сообщалось о сверхэкспрессии COX-2 в гематологических моделях рака, таких как RAJI (лимфома Беркитта) и U937 (острый промоноцитарный лейкоз), а также в бластных клетках пациента. CD73 активируется в различных карциномах человека, в том числе карциномах толстой кишки, легких, поджелудочной железы и яичников. Более высокие уровни экспрессии CD73 связаны с неоваскуляризацией, инвазивностью и метастазированием опухоли, а также с более коротким временем выживания пациентов при раке молочной железы.
Примеры поддающихся лечению раков молочной железы включают, но не ограничены ими, трижды отрицательный рак молочной железы, инвазивную протоковую карциному, инвазивную дольковую карциному, протоковую карциному in situ и дольковую карциному in situ.
Примеры раков дыхательных путей включают, но не ограничены ими, мелкоклеточную и немелкоклеточную карциному легкого, а также бронхиальную аденому и плевропульмонарную бластому.
Примеры поддающихся лечению раков головного мозга включают, но не ограничены ими, глиому ствола головного мозга и гипоталамуса, мозжечковую и церебральную астроцитому, глиобластому, медуллобластому, эпендимому, а также нейроэктодермальную опухоль и опухоль шишковидной железы.
Поддающиеся лечению опухоли мужских репродуктивных органов включают, но не ограничены ими, рак предстательной железы и яичек.
Поддающиеся лечению опухоли женских репродуктивных органов включают, но не ограничены ими, рак эндометрия, шейки матки, яичников, влагалища и вульвы, а также саркому матки.
Поддающийся лечению рак яичников включает, но не ограничен ими, серозную опухоль, эндометриоидную опухоль, муцинозную цистаденокарциному, гранулезоклеточную опухоль, опухоль из клеток Сертоли-Лейдига и арренобластому.
Поддающийся лечению рак шейки матки включает, но не ограничен ими, плоскоклеточную карциному, аденокарциному, аденосквамозную карциному, мелкоклеточную карциному, нейроэндокринную опухоль, стекловидноклеточную карциному и виллогландулярную аденокарциному.
Поддающиеся лечению опухоли пищеварительного тракта включают, но не ограничены ими, раки анального отверстия, толстой кишки, прямой и ободочной кишки, пищевода, желчного пузыря, желудка, поджелудочной железы, прямой кишки, тонкого кишечника и слюнных желез.
Поддающийся лечению рак пищевода включает, но не ограничивается ими, карциному клеток пищевода и аденокарциному, а также плоскоклеточный рак, лейомиосаркому, злокачественную меланому, рабдомиосаркому и лимфому.
Поддающийся лечению рак желудка включает, но не ограничен ими, аденокарциному желудка кишечного типа и диффузного типа.
Поддающийся лечению рак поджелудочной железы включает, но не ограничен ими, протоковую аденокарциному, аденосквамозную карциному и эндокринные опухоли поджелудочной железы.
Поддающиеся лечению опухоли мочевыводящих путей включают, но не ограничены ими, раки мочевого пузыря, полового члена, почки, почечной лоханки, мочеточника, уретры и папиллярный рак почки человека.
Поддающийся лечению рак почки включает, но не ограничен ими, почечно-клеточную карциному, карциному клеток уротелия, опухоль юкстагломерулярных клеток (рениному), ангиомиолипому, почечную онкоцитому, карциному протока Беллини, светлоклеточную саркому почки, мезобластную нефрому и опухоль Вильмса.
Поддающийся лечению рак мочевого пузыря включает, но не ограничен ими, переходно-клеточную карциному, плоскоклеточную карциному, аденокарциному, саркому и мелкоклеточную карциному.
Поддающиеся лечению виды рака глаза включают, но не ограничены ими, внутриглазную меланому и ретинобластому.
Поддающиеся лечению раки печени включают, но не ограничены ими, гепатоцеллюлярную карциному (карциному клеток печени с или без фиброламеллярного варианта), холангиокарциному (карциному внутрипеченочных желчных протоков) и смешанную гепатоцеллюлярную холангиокарциному.
Поддающиеся лечению раки кожи включают, но не ограничены ими, плоскоклеточную карциному, саркому Капоши, злокачественную меланому, рак кожи из клеток Меркеля и немеланомный рак кожи.
Поддающиеся лечению раки головы и шеи включают, но не ограничены ими, плоскоклеточный рак головы и шеи, рак гортани, гортаноглотки, носоглотки, ротоглотки, рак слюнных желез, рак губы и ротовой полости и плоскоклеточный рак.
Поддающиеся лечению лимфомы включают, но не ограничены ими, лимфому, связанную со СПИД, неходжкинскую лимфому, кожную Т-клеточную лимфому, лимфому Беркитта, болезнь Ходжкина и лимфому центральной нервной системы.
Поддающиеся лечению саркомы включают, но не ограничены ими, саркому мягких тканей, остеосаркому, злокачественную фиброзную гистиоцитому, лимфосаркому и рабдомиосаркому.
Поддающиеся лечению лейкозы включают, но не ограничены ими, острый миелоидный лейкоз, острый лимфобластный лейкоз, хронический лимфоцитарный лейкоз, хронический миелогенный лейкоз и волосатоклеточный лейкоз.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут применяться для лечения ряда других нарушений, в которые вовлечена MAP4K1, таких как сердечно-сосудистые заболевания и заболевания легких.
В некоторых вариантах осуществления, соединения по изобретению могут применяться в лекарственных средствах для лечения и/или профилактики сердечно-сосудистых, воспалительных и фиброзных нарушений, почечных нарушений, в частности, острой и хронической почечной недостаточности, а также острой и хронической почечной недостаточности.
В настоящем документе термин «почечная недостаточность» включает как острые, так и хронические проявления почечной недостаточности, а также лежащие в основе или связанные с ними заболевания почек, такие как диабетические и не диабетические нефропатии, гипертензивные нефропатии, ишемические почечные нарушения, почечную гипоперфузию, интрадиалитическую гипотензию, обструктивную уропатию, почечные стенозы, гломерулопатии, гломерулонефрит (такой как, например, первичный гломерулонефрит; болезнь минимальных изменений (липоидный нефроз); мембранозный гломерулонефрит; фокальный сегментарный гломерулосклероз (FSGS); мембранопролиферативный гломерулонефрит; серповидный гломерулонефрит; мезангиопролиферативный гломерулонефрит (IgA нефрит, болезнь Бергера); постинфекционный гломерулонефрит, вторичный гломерулонефрит), сахарный диабет, красная волчанка, амилоидоз, синдром Гудпасчера, гранулематоз Вегенера, пурпура Шенлейна-Геноха, микроскопический полиангиит, острый гломерулонефрит, пиелонефрит (например, являющийся результатом: мочекаменной болезни, доброкачественной гиперплазии предстательной железы, диабета, пороков развития, злоупотребления анальгетиками, болезни Крона), гломерулосклероз, артериолононекроз почек, тубулоинтерстициальные заболевания, нефропатические нарушения, такие как первичные и врожденные или приобретенные заболевания почек, синдром Альпорта, нефрит, иммунологические заболевания почек, такие как отторжение почечного трансплантата и почечные нарушения, вызванные иммунокомплексом, нефропатия, вызванная токсическими веществами, нефропатия, вызванная контрастными агентами, диабетическая и не диабетическая нефропатия, почечные кисты, нефросклероз, гипертонический нефросклероз и нефротический синдром, которые можно охарактеризовать диагностически, например, с помощью аномально сниженного выделения креатинина и/или воды, аномально повышенные концентрации мочевины, азота, калия и/или креатинина в крови, измененная активность почечных ферментов, например, глутамилсинтетазы, измененная осмоляльность мочи или объем мочи, повышенная микроальбуминурия, макроальбуминурия, поражения клубочков и артериол, дилатация канальцев, гиперфосфатемия и/или потребность в диализе.
В некоторых вариантах осуществления соединения по изобретению могут применяться для лечения и/или профилактики последствий почечной недостаточности, например отека легких, сердечной недостаточности, уремии, анемии, нарушений электролитного баланса (например, гиперкалиемии, гипонатриемии) и нарушений костного и углеводного метаболизма.
В некоторых вариантах осуществления, соединения по изобретению можно использовать для лечения и/или профилактики последствий почечной недостаточности, например, отека легких, сердечной недостаточности, уремии, анемии, нарушений электролитного баланса (например, гиперкалиемии, гипонатриемии) и нарушений костного и углеводного метаболизма.
В некоторых вариантах осуществления, соединения по изобретению дополнительно подходят для лечения и/или профилактики поликистозной болезни почек (PCKD) и синдрома неадекватной секреции ADH (SIADH).
В некоторых вариантах осуществления, соединения по изобретению также подходят для лечения и/или профилактики метаболического синдрома, гипертензии, резистентной гипертензии, острой и хронической сердечной недостаточности, ишемической болезни сердца, стабильной и нестабильной стенокардии, нарушений периферических и сердечных сосудов, аритмии, предсердных и желудочковых аритмий и ухудшенной проводимости, например, атриовентрикулярных блокад l-lll степени (АВ-блокада l-lll), наджелудочковой тахиаритмии, мерцательной аритмии, трепетания предсердий, фибрилляции желудочков, трепетания желудочков, желудочковой тахиаритмии, двунаправленной тахикардии, предсердная и желудочковая экстрасистолы, AV-атриовентрикулярной экстрасистолы, синдрома слабости синусового узла, обмороков, AV-узловой реципрокной тахикардии, синдрома Вольфа-Паркинсона-Уайта, острого коронарного синдрома (ACS), аутоиммунных сердечных нарушений (перикардита, эндокардита, вальволита, аортита, кардиомиопатий), шока, такого как кардиогенный шок, септический шок и анафилактический шок, аневризм, кардиомиопатии боксеров (преждевременное сокращение желудочков (PVC)), для лечения и/или профилактики тромбоэмболических заболеваний и ишемии, таких как ишемия миокарда, инфаркта миокарда, инсульта, гипертрофии сердца, транзиторных и ишемических приступов, преэклампсии, воспалительных сердечно-сосудистых нарушений, спазмов коронарных артерий и периферических артерий, образования отеков, например отека легких, отека головного мозга, отека почек или отека, вызванного сердечной недостаточностью, нарушений периферического кровообращения, реперфузионного повреждения, артериальных и венозных тромбозов, миокардиальной недостаточности, эндотелиальной дисфункции, для профилактики рестенозов, например, после терапии тромболизисом, чрескожной транслюминальной ангиопластики (PTA), транслюминальной коронарной ангиопластики (PTCA), трансплантатов сердца и операций шунтирования, а также микро- и макрососудистых повреждений (васкулитов), повышенных уровней фибриногена и липопротеинов низкой плотности (LDL) и повышенных концентраций ингибитора активатора плазминогена 1 (PAI-1), а также для лечения и/или профилактики эректильной дисфункции и женской сексуальной дисфункции.
В некоторых вариантах осуществления, соединения по изобретению также подходят для лечения и/или профилактики астматических заболеваний, легочной артериальной гипертензии (PAH) и других форм легочной гипертензии (РН), включая заболевание левых отделов сердца, HIV, серповидноклеточную анемию, тромбоэмболии (CTEPH), саркоидоз, COPD или легочную гипертензию, связанную с фиброзом легких, хроническую обструктивную болезнь легких (COPD), острый респираторный дистресс-синдром (ARDS), острое повреждение легких (ALI), дефицит альфа-1-антитрипсина (AATD), легочный фиброз, эмфизему легких (например, эмфизему легких, вызванную сигаретным дымом) и кистозный фиброз (CF).
В некоторых вариантах осуществления, соединения по изобретению также эффективны для контроля нарушений центральной нервной системы, характеризующихся нарушениями системы NO/cGMP. Они подходят, в частности, для улучшения восприятия, концентрации, обучения или памяти после когнитивных нарушений, подобных тем, которые возникают, в частности, в связи с ситуациями/заболеваниями/синдромами, такими как легкие когнитивные нарушения, возрастные нарушения обучения и памяти, возрастные потери памяти, сосудистая деменция, черепно-мозговая травма, инсульт, деменция после инсульта (постинсультная деменция), посттравматическая черепно-мозговая травма, общие нарушения концентрации внимания, нарушения концентрации внимания у детей с проблемами обучения и памяти, болезнь Альцгеймера, деменция с тельцами Леви, деменция с дегенерацией лобных долей, включая синдром Пика, болезнь Паркинсона, прогрессирующую деменцию с кортико-базальной дегенерацией, амиолатеральный склероз (ALS), болезнь Хантингтона, демиелинизация, рассеянный склероз, таламическая дегенерация, деменция Крейтцфельда-Якоба, деменция при HIV, шизофрения с деменцией или психоз Корсакова.
В некоторых вариантах осуществления, соединения по изобретению также подходят для лечения и/или профилактики нарушений центральной нервной системы, таких как состояния тревоги, напряжения и депрессии, связанные с ЦНС сексуальные дисфункции и нарушения сна, а также для контроля патологических нарушений поглощения пищи, стимуляторов и веществ, вызывающих привыкание.
В некоторых вариантах осуществления, соединения по изобретению, кроме того, также подходят для контроля мозгового кровотока и, таким образом, представляют эффективные агенты для контроля мигрени.
В некоторых вариантах осуществления, соединения по изобретению также подходят для профилактики и контроля последствий инфаркта головного мозга (церебральной апоплексии), таких как инсульт, церебральная ишемия и черепно-мозговая травма. Соединения по изобретению можно также использовать для контроля состояния боли и шума в ушах.
В определенных вариантах осуществления, соединения по изобретению обладают противовоспалительным действием и поэтому могут применяться в качестве противовоспалительных агентов для лечения и/или профилактики сепсиса (SIRS), полиорганной недостаточности (MODS, MOF), воспалительных заболеваний почек, хронических воспалений кишечника (IBD, болезнь Крона, UC), панкреатита, перитонита, ревматоидных нарушений, воспалительных кожных нарушений и воспалительных глазных нарушений.
В некоторых вариантах осуществления, соединения по изобретению также можно использовать для лечения и/или профилактики аутоиммунных заболеваний.
В некоторых вариантах осуществления, соединения по изобретению также подходят для лечения и/или профилактики фиброзных нарушений внутренних органов, например легких, сердца, почек, костного мозга и, в частности, печени, а также дерматологических фиброзов и фиброзных глазных нарушений.
Используемый в настоящем документе термин «фиброзные нарушения» включает, в частности, следующее: фиброз печени, цирроз печени, легочный фиброз, эндомиокардиальный фиброз, нефропатию, гломерулонефрит, интерстициальный почечный фиброз, фиброзное повреждение, возникающее в результате диабета, фиброз костного мозга и аналогичные фиброзные нарушения, склеродермию, кольцевидную склеродермию, келоиды, гипертрофические рубцы (также после хирургических процедур), невусы, диабетическую ретинопатию, пролиферативную витроретинопатию и нарушения соединительной ткани (например, саркоидоза).
В некоторых вариантах осуществления, соединения по изобретению также подходят для борьбы с послеоперационным рубцеванием, например, в результате оперативного лечения глаукомы.
В определенных вариантах осуществления, соединения по изобретению также можно использовать в косметических целях для стареющей и ороговевшей кожи.
В некоторых вариантах осуществления, соединения по изобретению подходят для лечения и/или профилактики гепатита, новообразований, остеопороза, глаукомы и гастропареза.
В некоторых вариантах осуществления, соединения по изобретению подходят для лечения и/или профилактики вирусных инфекций (например, ВИЧ и саркомы Капоши); воспалительных и аутоиммунных заболеваний (например, колита, артрита, болезни Альцгеймера, гломерулонефрита и заживления ран); бактериальных, грибковых и/или паразитарных инфекций; кожных заболеваний (например, псориаза); заболеваний, основанных на гиперплазии, которые характеризуются увеличением количества клеток (например, фибробластов, гепатоцитов, клеток костей и костного мозга, клеток хрящей или гладкой мускулатуры или эпителиальных клеток (например, гиперплазии эндометрия)); заболеваний костей и сердечно-сосудистых заболеваний (например, рестеноза и гипертрофии).
В другом варианте осуществления, соединения по изобретению можно также применять для лечения или профилактики маточных фиброидов (лейомиомы матки или миомы матки) у женщин. Маточные фиброиды являются доброкачественными опухолями миометрия, гладкомышечного слоя матки. Маточные фиброиды растут медленно в течение жизни женщины, и их рост зависит от женских половых гормонов эстрадиола и прогестерона. Поэтому наибольшая распространенность маточных фиброидов с приблизительно 70% и >80% у белых и афроамериканских женщин, соответственно, обнаружена в возрасте от 35 лет и до менопаузы, когда они уменьшаются в размерах из-за снижения уровня гормонов. Приблизительно 30% и 45% белых и афроамериканских женщин, соответственно, проявляют клинически значимые симптомы из-за их фиброидов, которыми являются обильные менструальные кровотечения и боль, связанная с менструальным циклом (David et al., Eur J Obstet Gynecol Reprod Biol. 199:137-140, 2016). Тяжелые менструальные кровотечения в этом отношении определяются кровопотерей более 80 мл в период менструального кровотечения. Подслизистое расположение маточных фиброидов, например, расположенных непосредственно под эндометрием, по-видимому, оказывает еще более серьезное влияние на маточное кровотечение, которое может привести к анемии у пораженных женщин. Кроме того, маточные фиброиды, из-за их симптомов, серьезно влияет на качество жизни пораженных женщин.
В некоторых вариантах осуществления, соединения по изобретению полезны для лечения и/или профилактики хронических заболеваний почек, острой и хронической почечной недостаточности, диабетических, воспалительных или гипертонических нефропатий, фиброзных нарушений, сердечной недостаточности, стенокардии, гипертензии, легочной гипертензии, ишемий, сосудистых нарушений, тромбоэмболических нарушений, атеросклероза, серповидно-клеточной анемии, эректильной дисфункции, доброкачественной гиперплазии предстательной железы, дизурии, ассоциированной с доброкачественной гиперплазией предстательной железы, Хантингтона, деменции, Альцгеймера и Крейтцфельда-Якоба.
Настоящее изобретение представляет способ лечения и/или профилактики хронических заболеваний почек, острой и хронической почечной недостаточности, диабетических, воспалительных или гипертонических нефропатий, фиброзных нарушений, сердечной недостаточности, стенокардии, гипертонии, легочной гипертензии, ишемий, сосудистых нарушений, тромбоэмболических нарушений, атеросклероза, серповидноклеточной анемии, эректильной дисфункции, доброкачественной гиперплазии предстательной железы, дизурии, ассоциированной с доброкачественной гиперплазией предстательной железы, Хантингтона, деменции, Альцгеймера и Крейтцфельда-Якоба.
Настоящее изобретение дополнительно относится к применению соединений по изобретению для лечения и/или профилактики нарушений, в частности, нарушений, упомянутых выше.
Настоящее изобретение дополнительно представляет способ лечения и/или профилактики заболеваний, в частности нарушений, упомянутых выше, с использованием эффективного количества, по меньшей мере, одного из соединений по изобретению.
Таким образом, соединения по настоящему изобретению могут быть использованы для ингибирования, блокирования, снижения или уменьшения активации MAP4K1 экзогенными и/или эндогенными лигандами для снижения роста опухоли и модулирования нарушенной регуляции иммунных ответов, например, для блокирования иммунодепрессии и увеличения активации и инфильтрации иммунных клеток в контексте рака и иммунотерапии рака. Этот способ включает введение млекопитающему, нуждающемуся в этом, включая человека, некоторого количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, изомера, полиморфа, метаболита, гидрата, сольвата или сложного эфира; который эффективен для лечения нарушения.
Настоящее изобретение также относится к способам лечения ряда других заболеваний, в которые вовлечена MAP4K1, таких как, но не ограниченных ими, нарушения с нарушенной регуляцией иммунного ответа, воспаление, вакцинация против инфекции и рака, вирусные инфекции, ожирение и ожирение, вызванное диетой, ожирение, нарушения обмена веществ, стеатоз печени и маточные фиброиды. Эти расстройства были хорошо охарактеризованы у людей, но также существуют с аналогичной этиологией у других млекопитающих, и их можно лечить путем введения фармацевтических композиций по настоящему изобретению.
5. Комбинированная терапия
Соединения по настоящему изобретению можно использовать в комбинированной терапии с одним или несколькими дополнительными/вторичными терапевтическими агентами, подходящими для лечения заболевания или показания, поддающегося лечению рассматриваемыми соединениями.
Таким образом, в некоторых вариантах осуществления, например, способы по изобретению с использованием соединений по изобретению могут включать введение субъекту, нуждающемуся в этом, дополнительного терапевтического агента. Дополнительным терапевтическим агентом может быть: (i) иммуномодулирующий агент, который блокирует или ингибирует контрольную точку иммунной системы, где эта контрольная точка может быть или не быть компонентом пути NF-κB; и/или (ii) агент, который непосредственно стимулирует иммунный эффекторный ответ, такой как цитокин, или опухолеспецифическая адоптивно перенесенная популяция Т-клеток, или антитело, специфичное к белку, экспрессируемому опухолевой клеткой; и/или (iii) композиция, содержащая опухолевый антиген или его иммуногенный фрагмент; и/или (iv) химиотерапевтический агент.
В некоторых вариантах осуществления, второй терапевтический агент содержит ингибитор пути PI3K-AKT-mTOR, ингибитор пути Raf-MAPK, ингибитор пути JAK-STAT, ингибитор пути бета катенина, ингибитор пути notch, ингибитор пути hedgehog, ингибитор Pim киназ и/или ингибитор белков теплового шока и прогрессирования клеточного цикла. В некоторых вариантах осуществления, комбинированная терапия по изобретению снижает вероятность возникновения лекарственной резистентности в клеточной популяции и/или снижает токсичность лечения.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению могут быть скомбинированы с одним или несколькими ингибиторами следующих киназ для лечения рака: Akt1, Akt2, Akt3, TGF-βΡν, PKA, PKG, PKC, CaM-киназа, фосфорилаза киназа, MEKK, ERK, MAPK, mTOR, EGFR, HER2, HER3, HER4, INS-R, IGF-1R, IR-R, PDGFαR, PDGFβR, CSFIR, KIT, FLK-II, KDR/FLK-1, FLK-4, flt-1, FGFR1, FGFR2, FGFR3, FGFR4, c-Met, Ron, Sea, TRKA, TRKB, TRKC, FLT3, VEGFR/Flt2, Flt4, EphAl, EphA2, EphA3, EphB2, EphB4, Tie2, Src, Fyn, Lck, Fgr, Btk, Fak, SYK, FRK, JAK, ABL, ALK и B-Raf.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению могут быть скомбинированы с одним или несколькими из следующих ингибиторов для лечения рака, включая ингибитор FGFR (FGFR1, FGFR2, FGFR3 или FGFR4, например, AZD4547, BAY 1187982, ARQ087, BGJ398, BIBF1120, TKI258, луцитаниб, довитиниб, TAS-120, J J-42756493, Debiol347, INCB54828, INCB62079 и INCB63904), ингибитор JAK (JAK1 и/или JAK2, например, руксолитиниб, барицитиниб или итацитиниб (INCB39110)), ингибитор IDO (например, эпакадостат и NLG919), ингибитор LSD1 (например, GSK2979552, INCB59872 и INCB60003), ингибитор TDO, ингибитор PI3K-дельта (например, INCB50797 и INCB50465), ингибитор PI3K-гамма, такой как селективный ингибитор PI3K-гамма, ингибитор CSF1R (например, PLX3397 и LY3022855), рецепторные тирозинкиназы ТАМ (Tyro-3, Axl и Mer), модулятор арилуглеводородного рецептора (AhR) (такой как лаквинимод, аминофлавон, CB7993113, CH223191, 6,2',4'-триметоксифлавон (TMF), GNF351 (N-(2-(1H-индол-3-ил)этил)-9-изопропил-2-(5-метилпиридин-3-ил)-9H-пурин-6-амин), аминофлавон, NKI150460, индол-3-карбинол, β-нафтофлавон и его димер, дииндолилметан (DIM), 4-гидрокситамоксифен, лефлуномид, ралоксифен, траниласт, флутамид, мексилетин, нимодифин, омепразол, сулиндак, траниласт и TCDD (2,3,7,8-тетрахлордибензо-п-диоксин)), ингибитор ангиогенеза, ингибитор рецептора интерлейкина, ингибиторы членов бромо- и экстратерминального семейства (например, ингибиторы бромодомена или ингибиторы BET, такие как OTX015, CPI-0610, INCB54329 и INCB57643), и антагонист аденозинового рецептора, или их комбинации.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с ингибиторами HDAC, такими как панобиностат и вориностат.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с ингибиторами c-Met, такими как онартумзумаб, тивантниб и капматиниб (INC-280).
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с ингибиторами BTK, такими как ибрутиниб.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с ингибиторами mTOR, например, рапамицином, сиролимусом, темсиролимусом и эверолимусом.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с ингибиторами MEK, такими как траметиниб, селуметиниб и GDC-0973.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с ингибиторами Hsp90 (например, танеспимицином), циклинзависимых киназ (например, палбоциклибмо), PARP (например, олапарибом) и Pim киназ (LGH447, INCB053914 и SGI-1776).
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с агонистом ДНК сенсора (c-GAS) и/или его нижестоящим адапторным белком STING.
Путь cGAS (циклическая GMP-AMP-синтаза)-STING (стимулятор генов интерферона) является компонентом врожденной иммунной системы, который функционирует для обнаружения присутствия цитозольной ДНК и, в ответ, запускает экспрессию воспалительных генов, которые могут привести к старению или к активации защитных механизмов. Локализация ДНК из обычной ядерной локализации до цитозоля связана с онкогенезом или вирусной инфекцией. cGAS находится в цитозоле, и при прямом связывании с цитозольной ДНК, cGAS образует димеры для катализа продуцирования 2'3'-cGAMP из ATP и GTP. Полученный cGAMP затем действует как вторичный мессенджер, связывая STING и вызывая активацию фактора транскрипции IRF3. Активированный IRF3 приводит к транскрипции IFN-β типа 1 и ряда нижестоящих генов-мишеней, чтобы инициировать разнообразные биологические ответы, такие как вирусный ответ, надзор за опухолью, аутоиммунитет и клеточное старение. Во многих опухолевых клетках, конститутивно активный ответ на повреждение ДНК приводит к накоплению цитоплазматической ДНК и активации пути cGAS/STING. В клетках лимфомы было показано, что NKG2D лиганд, Rae1, активируется STING/IRF3-зависимым образом, чтобы способствовать NK-опосредованному удалению опухоли. Было показано, что активация пути c-GAS-STING в антигенпрезентирующих клетках, таких как дендритные клетки, усиливает их функцию и повышает противоопухолевый иммунитет.
В некоторых вариантах осуществления, соединения-ингибиторы HPK1 по изобретению можно комбинировать с одним или несколькими ингибиторами иммунных контрольных точек.
Активация эффекторных Т-клеток обычно запускается TCR, распознающим антигенный пептид, презентированный комплексом MHC. Затем тип и уровень достигнутой активации определяют через баланс между сигналами, которые стимулируют, и сигналами, которые ингибируют ответ эффекторных Т-клеток. «Контрольная точка иммунной системы» используется в настоящем документе для обозначения любого молекулярного взаимодействия, которое изменяет баланс в пользу ингибирования ответа эффекторных Т-клеток. То есть, молекулярное взаимодействие, которое, когда оно происходит, отрицательно регулирует активацию эффекторной Т-клетки. Такое взаимодействие может быть прямым, например взаимодействие между лигандом и рецептором клеточной поверхности, который передает ингибирующий сигнал в эффекторную Т-клетку. Или он может быть косвенным, таким как блокирование или ингибирование взаимодействия между лигандом и рецептором клеточной поверхности, которое в противном случае передало бы активирующий сигнал в эффекторную Т-клетку, или взаимодействие, которое способствует усилению регуляции ингибирующей молекулы или клетки, или истощение ферментом метаболита, необходимого эффекторной Т-клетке, или любое их сочетание.
Примеры контрольных точек иммунной системы включают: а) взаимодействие между индоламин-2,3-диоксигеназой (IDO1) и ее субстратом; b) взаимодействие между PD1 и PD-L1 и/или PD1 и PD-L2; с) взаимодействие между CTLA-4 и CD86 и/или CTLA-4 и CD80; d) взаимодействие между B7-H3 и/или B7-H4 и их соответствующими лигандами; e) взаимодействие между HVEM и BTLA; f) взаимодействие между GAL9 и TIM3; g) взаимодействие между MHC класса I или II и LAG 3; и h) взаимодействие между MHC класса I или II и KIR; i) взаимодействие между OX40 (CD134) и OX40L (CD252); j) взаимодействие между CD40 и CD40L (CD154); k) взаимодействие между 4-1 BB (CD137) и лигандами, включая 4-1 BBL; l) Взаимодействие между GITR и лигандами, включая GITRL.
Таким образом, типовые ингибиторы иммунных контрольных точек включают ингибиторы молекул иммунных контрольных точек, таких как CD20, CD27, CD28, CD39, CD40, CD 122, CD96, CD73, CD47, OX40, GITR, CSF1R, JAK, PI3K дельта, PI3K гамма, ТАМ, аргиназа, CD137 (также известный как 4-1BB), ICOS, A2AR, B7-H3, B7-H4, BTLA, CTLA-4, LAG3, TIM3, VISTA, PD-1, PD-L1 и PD-L2.
Типовой контрольной точкой для целей настоящего изобретения является контрольная точка (b), а именно взаимодействие между PD1 и любым из его лигандов PD-L1 и PD-L2. PD1 экспрессируется на эффекторных Т-клетках. Взаимодействие с любым из лигандов приводит к сигналу, подавляющему активацию. Лиганды экспрессируются некоторыми опухолями. В частности, PD-L1 экспрессируется многими солидными опухолями, включая меланому. Таким образом, эти опухоли могут подавлять иммуноопосредованные противоопухолевые эффекты через активацию ингибирующих рецепторов PD-1 на Т-клетках. При блокировании взаимодействия между PD1 и одним или обоими его лигандами, контрольная точка иммунного ответа может быть удалена, что приводит к усилению противоопухолевого Т-клеточного ответа. Таким образом, PD1 и его лиганды являются примерами компонентов контрольной точки иммунной системы, которые могут быть таргетированы в способе по изобретению.
Другой контрольной точкой для целей настоящего изобретения является контрольная точка (с), а именно взаимодействие между Т-клеточным рецептором CTLA-4 и его лигандами, белками В7 (В7-1 и В7-2). CTLA-4 обычно активируется на поверхности Т-клеток после первоначальной активации, и связывание лиганда приводит к сигналу, который ингибирует дальнейшую/продолжающуюся активацию. CTLA-4 конкурирует за связывание с белками B7 с рецептором CD28, который также экспрессируется на поверхности Т-клеток, но который усиливает активацию. Таким образом, блокируя взаимодействие CTLA-4 с B7 белками, но не взаимодействие CD28 с B7 белками, можно удалить одну из нормальных контрольных точек иммунного ответа, что приведет к усилению противоопухолевого Т-клеточного ответа. Таким образом, CTLA-4 и его лиганды являются примерами компонентов контрольной точки иммунной системы, которые могут быть таргетированы в способе по изобретению.
В некоторых вариантах осуществления, молекулой иммунной контрольной точки является стимулирующая молекула контрольной точки, выбранная из CD27, CD28, CD40, ICOS, OX40, GITR и CD137.
В некоторых вариантах осуществления, молекулой иммунной контрольной точки является молекула ингибиторной контрольной точки, выбранная из A2AR, B7-H3, B7-H4, BTLA, CTLA-4, IDO, KIR, LAG3, PD-1, HM3 и VISTA.
В некоторых вариантах осуществления, соединения, представленные в настоящем документе, можно использовать в комбинации с одним или несколькими агентами, выбранными из ингибиторов KIR, ингибиторов TIGIT, ингибиторов LAIR1, ингибиторов CD 160, ингибиторов 2B4 и ингибиторов TGFR бета.
В некоторых вариантах осуществления, соединения, представленные в настоящем документе, можно использовать в комбинации с ингибиторами иммунных контрольных точек, которыми являются низкомолекулярные ингибиторы (SMI), которые обычно являются малыми органическими молекулами. Например, в некоторых вариантах осуществления, ингибиторы IDO1 включают эпакадостат (INCB24360), индоксимод, GDC-0919 (NLG919) и F001287. Другие ингибиторы IDO1 включают 1-метилтриптофан (1 МТ).
В некоторых вариантах осуществления, ингибитор молекулы контрольной точки иммунной системы также известен как «иммуномодулирующий агент», который включает любой агент, который при введении субъекту блокирует или ингибирует действие контрольной точки иммунной системы, что приводит к активации ответа иммунного эффектора у субъекта, обычно ответ Т-клеточного эффектора, который может включать противоопухолевый ответ Т-клеточного эффектора.
Иммуномодулирующий агент, используемый в способе по настоящему изобретению, может блокировать или ингибировать любую из контрольных точек иммунной системы, описанных выше. Агентом может быть антитело или любой другой подходящий агент, который приводит к указанному блокированию или ингибированию. Таким образом, агент можно в целом назвать ингибитором указанной контрольной точки.
Используемый в настоящем документе термин «антитело» включает целые антитела и любой антигенсвязывающий фрагмент (т.е. «антигенсвязывающую часть») или их отдельные цепи. Антитело может быть поликлональным антителом или моноклональным антителом и может быть получено любым подходящим способом. Примеры связывающих фрагментов, охватываемых термином «антигенсвязывающая часть» антитела, включают Fab фрагмент, F(ab')2 фрагмент, Fab' фрагмент, Fd фрагмент, Fv фрагмент, dAb фрагмент и выделенную определяющую комплементарность область (CDR). Одноцепочечные антитела, такие как scFv, и антитела с тяжелой цепью, такие как VHH и верблюжьи антитела, также охватываются термином «антигенсвязывающая часть» антитела.
В некоторых вариантах осуществления, иммуномодулирующим агентом, применяемым с ингибитором HPK1 по изобретению, является анти-PD1 антитело, анти-PD-L1 антитело или анти-CTLA-4 антитело.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор PD-1, например, анти-PD-1 моноклональное антитело. В некоторых вариантах осуществления, анти-PD-1 моноклональным антителом является ниволумаб (MDX-1106), пембролизумаб (Merck 3475 или ламбролизумаб), пидилизумаб (CT-011), тислелизумаб (BGB-A317), камрелизумаб (SHR-1210), спартализумаб (PDR001) или AMP-514 (MEDI0680). В некоторых вариантах осуществления, анти-PD-1 моноклональным антителом является ниволумаб или пембролизумаб. В некоторых вариантах осуществления, анти-PD1 антителом является пембролизумаб. В некоторых вариантах осуществления, анти-PD-1 антителом является камрелизумаб (SHR-1210). В некоторых вариантах осуществления, ингибитором PD-1 является AMP-224 (PD-L2 Fc слитый белок, который связывает PD-1) или AUNP-12 (анти-PD-1 пептид).
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор PD-L1, например, анти-PD-L1 моноклональное антитело. В некоторых вариантах осуществления, анти-PD-L1 моноклональным антителом является BMS-935559, BMS-936559 (MDX-1105), MEDI-4736 (дурвалумаб), MPDL3280A (также известный как RG7446), YW243.55.S70 (HPAB-0381-WJ) или MSB0010718C. В некоторых вариантах осуществления, анти-PD-L1 моноклональным антителом является MPDL3280A или MEDI-4736. В некоторых вариантах осуществления, анти-PD-L1 антитела включают атезолизумаб, авелумаб, дурвалумаб или MEDI-4736 и MPDL3280A.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор CTLA-4, например, анти-CTLA-4 антитело. В некоторых вариантах осуществления, анти-CTLA-4 антителом является ипилимумаб, тремелимумаб или любое из антител, описанных в WO 2014/207063 (включенной в настоящий документ посредством ссылки). Другие молекулы включают полипептиды или растворимые мутантные CD86 полипептиды. В некоторых вариантах осуществления, антителом является ипилимумаб.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является комбинация двух или нескольких модуляторов, описанных в настоящем документе, например комбинацию, таргетирующую на две или несколько различных мишеней (например, PD-1, PD-L1 и PD-L2). Примеры комбинаций включают: α-PD-1 и α-PD-L1; α-CTLA-4, α-PD-L1 и α-CD20; и т.п.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является антитело, которое блокирует или ингибирует взаимодействие между 4-1 BB и его лигандом, включая утомилумаб.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор CSFIR, например, анти-CSF1R антитело. В некоторых вариантах осуществления, анти-CSF1R антителом является IMC-CS4 или RG7155.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор LAG3, например, анти-LAG3 антитело. В некоторых вариантах осуществления, анти-LAG3 антителом является BMS-986016, LAG525, IMP321 или GSK2831781.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор GITR, например, анти-GITR антитело. В некоторых вариантах осуществления, анти-GITR антителом является TRX518, MK-4166, MK1248, BMS-986156, MEDI1873 или GWN323.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор OX40, например, анти-OX40 антитело или слитый белок OX40L. В некоторых вариантах осуществления, анти-OX40 антителом является MEDI0562, MEDI6469, MOXR0916, PF-04518600 или GSK3174998. В некоторых вариантах осуществления, слитым белком OX40L является MEDI6383.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор TIM3, например, анти-TIM3 антитело. В некоторых вариантах осуществления, анти-TIM3 антителом является MBG-453.
В некоторых вариантах осуществления, ингибитором молекулы иммунной контрольной точки является ингибитор CD20, например, анти-CD20 антитело. В некоторых вариантах осуществления, анти-CD20 антителом является обинутузумаб или ритуксимаб.
В некоторых вариантах осуществления, соединения по изобретению можно использовать в комбинации с одним или несколькими ингибиторами метаболических ферментов. В некоторых вариантах осуществления, ингибитором метаболического фермента является ингибитор IDO 1, TDO или аргиназы. Примеры ингибиторов IDO1 включают эпакадостат и NGL919. Примером ингибитора аргиназы является CB-1158.
В некоторых вариантах осуществления, соединения по изобретению можно использовать в комбинации с биспецифическими антителами. В некоторых вариантах осуществления, один из доменов биспецифического антитела таргетирует рецептор PD-1, PD-L1, CTLA-4, GITR, OX40, TIM3, LAG3, CD137, ICOS, CD3 или TGFβ.
В некоторых вариантах осуществления, соединения по изобретению можно использовать в комбинации с одним или несколькими агентами для лечения таких заболеваний, как рак. В некоторых вариантах осуществления, агентом является алкилирующий агент, ингибитор протеасом, кортикостероид или иммуномодулирующий агент. Примеры алкилирующих агентов включают бендамустин, азотистый иприт, производные этиленимина, алкилсульфонаты, нитрозомочевины и триазены, урациловый иприт, хлорметин, циклофосфамид (Cytoxan™), ифосфамид, мелфалан, хлорамбуцил, пипоброман, триэтилен-меламин, триэтилентиофосфорамин, бусульфан, кармустин, ломустин, стрептозоцин, дакарбазин и темозоломид. В некоторых вариантах осуществления, ингибитором протеасомы является карфилзомиб. В некоторых вариантах осуществления, кортикостероидом является дексаметазон (DEX). В некоторых вариантах осуществления, иммуномодулирующим агентом является леналидомид (LEN) или помалидомид (POM).
Соединения по настоящему изобретению могут быть дополнительно использованы в комбинации с другими способами лечения рака, например, с химиотерапией, радиационной терапией, опухоль-таргетной терапией, адъювантной терапией, иммунотерапией или хирургией. Примеры иммунотерапии включают лечение цитокинами (например, интерферонами, GM-CSF, G-CSF, IL-2), иммунотерапию CRS-207, противораковую вакцину, моноклональное антитело, адоптивный перенос Т-клеток, онколитическую виротерапию и иммуномодулирующие малые молекулы, включая талидомид или ингибитор JAK1/2, и подобные.
Соединения по изобретению можно вводить в комбинации с одним или несколькими противораковыми лекарственными средствами, такими как химиотерапевтические агенты. Примеры химиотерапевтических агентов включают любой из: абареликса, абиратерона, афатиниба, афлиберцепта, альдеслейкина, алемтузумаба, алитретиноина, аллопуринола, альтретамина, анастрозола, триоксида мышьяка, аспарагиназы, акситиниба, азацитидина, бевацизумаба, бексаротена, барицитиниба, бикалутамида, блеомицина, бортезомби, бортезомиба, бриваниба, бупарлисиба, внутривенного бусульфана, перорального бусульфана, калустерона, капецитабина, карбоплатина, кармустина, цедираниба, цетуксимаба, хлорамбуцила, цисплатина, кладрибина, клофарабина, кризотиниба, циклофосфамида, цитарабина, дакарбазина, дакомитиниба, дактиномицина, далтепарина натрия, дазатиниба, дактиномицина, даунорубицина, децитабина, дегареликса, денилейкина, денилейкина дифтитокса, дезоксикоформицина, дексразоксана, доцетаксела, доксорубицина, дролоксафина, дромостанолонаа пропионата, экулизумаба, энзалутамида, эпидофиллотоксина, эпирубицина, эрлотиниба, эстрамустина, этопозида фосфата, этопозида, экземестана, фентанила цитрата, фиграстима, флоксуридина, флударабина, фторурацила, флутамида, фулвестранта, гефитиниба, гемцитабина, гемтузумаба озогамицина, гозерелинаа ацетата, гистрелина ацетата, ибритумомаба тиуксетана, идарубицина, иделализиба, ифосфамида, иматиниба мезилата, интерферона альфа 2а, иринотекана, лапатиниба дитозилата, леналидомида, летрозола, лейковорина, лейпролида ацетата, левамизола, ломустина, меклоретамина, мегестрола ацетата, мелфалана, меркаптопурина, метотрексата, метоксалена, митрамицина, митомицина С, митотана, митоксантрона, нандролона фенпропионата, навелбена, нецитумумаба, неларабина, нератиниба, нилотиниба, нилутамида, нофетумомаба, осерелина, оксалиплатина, паклитаксела, памидроната, панитумумаба, пазопаниба, пегаспаргазы, пегфилграстима, пеметрекседа динатрия, пентостатина, пиларалисиба, пипобромана, пликамицина, понатиниба, преднизона, прокарбазина, хинакрина, расбуриказы, регорафениба, релоксафина, ритуксимаба, руксолитиниба, сорафениба, стрептозоцина, сунитиниба, сунитиниба малеата, тамоксифена, тегафура, темозоламида, тенипозида, тестолактона, талидомида, тиогуанина, тиотепа, топотекана, торемифена, тозитумомаба, трастузумаба, третиноина, трипторелина, урамустина, валрубицина, вандетаниба, винбластина, винкристина, винорелбина, вориностата и золедроната.
Другие противораковые агенты включают терапевтические антитела, такие как трастузумаб (герцептин), антитела к костимулирующим молекулам, таким как CTLA-4 (например, ипилимумаб или тремелимумаб), 4-1BB, антитела к PD-1 и PD-L1, или антитела к цитокинам (IL-10, 10, TGF-β и т.д.). Примеры антител к PD-1 и/или PD-L1, которые можно комбинировать с соединениями по настоящему описанию для лечения рака или инфекций, таких как вирусные, бактериальные, грибковые и паразитарные инфекции, включают, но не ограничены ими, ниволумаб, пембролизумаб, MPDL3280A, MEDI-4736 и SHR-1210.
Другие противораковые средства включают ингибиторы киназ, ассоциированных с клеточным пролиферативным нарушением. Эти киназы включают, но не ограничены ими, Aurora-A, CDK1, CDK2, CDK3, CDK5, CDK7, CDK8, CDK9, киназы эфриновых рецепторов, CHK1, CHK2, SRC, Yes, Fyn, Lck, Fer, Fes, Syk, Itk, Bmx, GSK3, JNK, PAKl, PAK2, PAK3, PAK4, PDKl, PKA, PKC, Rsk и SGK.
Другие противораковые агенты также включают те, которые блокируют миграцию иммунных клеток, такие как антагонисты хемокиновых рецепторов, включая CCR2 и CCR4. Соединения по настоящему изобретению можно дополнительно использовать в комбинации с одним или несколькими противовоспалительными агентами, стероидами, иммунодепрессантами или терапевтическими антителами.
В некоторых вариантах осуществления, соединения по настоящему изобретению можно использовать в комбинации с дополнительным терапевтическим агентом, который непосредственно стимулирует иммунный эффекторный ответ, таким как цитокин или опухолеспецифическая адоптивно перенесенная популяция Т-клеток, или антитело, специфичное к белку, экспрессируемому опухолевой клеткой.
Используемый в настоящем документе термин «агент, который непосредственно стимулирует иммунный эффекторный ответ» означает любой подходящий агент, но обычно относится к цитокину или хемокину (или к агенту, который стимулирует продуцирование любого из них), опухолеспецифической адоптивно перенесенной популяции Т-клеток, или антитело, специфичное к белку, экспрессируемому опухолевой клеткой.
Цитокином может быть интерферон, выбранный из IFNα, ΙΡΝβ, IFNγ и IFNA, или интерлейкин, такой как IL-2. Хемокином может быть воспалительный медиатор, например, выбранный из CXCL9, 10 и 11, который привлекает Т-клетки, экспрессирующие CXCR3. Агентом, который стимулирует продуцирование цитокина или хемокина, может быть адъювант, подходящий для введения людям. Одним из примеров является Bacille Calmette-Guerin (BCG), которую обычно вводят внутрипузырно (т.е. через уретральный катетер) для лечения рака мочевого пузыря. Типовой схемой дозирования BCG при раке мочевого пузыря является один раз в неделю в течение шести недель, но, учитывая его долгую историю безопасности, его также вводят на неопределенный срок в качестве поддерживающей терапии. Было показано, что BCG стимулирует иммунные ответы на рак мочевого пузыря. BCG также применяют в качестве адъюванта в комбинации с композициями, которые содержат опухолевые антигены (т.е. с противораковыми вакцинами), особенно при раке толстой кишки, когда ее обычно вводят внутрикожно. Такое применение BCG также предусмотрено в настоящем изобретении. Опухолеспецифическая адоптивно перенесенная популяция Т-клеток непосредственно увеличивает размер опухолеспецифической популяции Т-клеток у индивидуума и может быть получена любым подходящим способом. Однако, как правило, процесс включает выделение опухолеспецифических Т-клеток из образца опухоли, взятого у пациента, и выборочное культивирование этих клеток перед возвращением размноженной популяции опухолеспецифических Т-клеток пациенту. Альтернативно, популяция специфичных для опухоли Т-клеток может быть получена путем генной инженерии локуса Т-клеточного рецептора с последующим размножением измененной клетки.
Антитела, специфичные к белкам, экспрессируемым опухолевой клеткой, обычно стимулируют иммунную активность, связываясь с опухолевой клеткой и способствуя разрушению клетки посредством антителозависимой клеточно-опосредованной цитотоксичности (ADCC). Примеры антител этого типа включают анти-CD20 антитела, такие как офатумумаб или ритуксимаб, и анти-CD52 антитела, такие как алемтузумаб.
Таким образом, в некоторых типовых вариантах осуществления, соединения по изобретению можно использовать в комбинации с ингибитором кальциневрина, например, циклоспорином А или FK 506; ингибитором mTOR, например, рапамицином, 40-0-(2-гидроксиэтил)рапамицином, биолимус-7 или биолимус-9; аскомицином, обладающим иммунодепрессивными свойствами, например, ABT-281, ASM981; кортикостероидом; циклофосфамидом; азатиопреном; метотрексатом; лефлуномидом; мизорибином; микофеноловой кислотой или солью; микофенолата мофетилом; ингибитор IL-1β.
В другом варианте осуществления, соединения по изобретению комбинируют с со-агентом, которым является ингибиторы киназы PI3.
В другом варианте осуществления, соединения по изобретению комбинируют с со-агентом, влияющим на ВТК (тирозинкиназу Брутона).
Для лечения онкологических заболеваний, соединения по настоящему изобретению можно использовать в комбинации с В-клеточными модулирующими агентами, например, ритуксимабом, ингибиторами BTK или Syk, ингибиторами PKC, PI3 киназ, PDK, PIM, JAK и миметиками mTOR и BH3.
В некоторых вариантах осуществления, соединения по изобретению, включая их соли, можно комбинировать с другим иммуногенным агентом, таким как раковые клетки, очищенные опухолевые антигены (включая рекомбинантные белки, пептиды и молекулы углеводов), клетки и клетки, трансфицированные генами, кодирующими иммуностимулирующие цитокины. Неограничивающие примеры противоопухолевых вакцин, которые можно использовать, включают пептиды антигенов меланомы, такие как пептиды gp100, антигены MAGE, Trp-2, MARTI и/или тирозиназу, или опухолевые клетки, трансфицированные для экспрессии цитокина GM-CSF.
В некоторых вариантах осуществления, соединения по изобретению или их соли также можно использовать в комбинации с протоколом вакцинации для лечения рака. В некоторых вариантах осуществления, опухолевые клетки трансдуцированы для экспрессии GM-CSF. В некоторых вариантах осуществления, противоопухолевые вакцины включают белки вирусов, вовлеченных в раки человека, таких как папилломавирусы человека (HPV), вирусы гепатита (HBV и HCV) и вирус герпеса саркомы Капоши (KHSV). В некоторых вариантах осуществления, соединения по настоящему изобретению можно использовать в комбинации с опухолеспецифическим антигеном, таким как белки теплового шока, выделенные из самой опухолевой ткани. В некоторых вариантах осуществления, соединения по изобретению или их соли можно комбинировать с иммунизацией дендритных клеток для активации сильных противоопухолевых ответов.
В некоторых вариантах осуществления, соединения по изобретению можно использовать в комбинации с биспецифическими макроциклическими пептидами, которые таргетируют эффекторные клетки, экспрессирующие рецептор Fe α или Fe γ, на опухолевые клетки. Соединения по изобретению также можно комбинировать с макроциклическими пептидами, которые активируют иммунную реакцию хозяина.
В некоторых вариантах осуществления, соединения по изобретению можно применять в комбинации с трансплантатом костного мозга для лечения различных опухолей гемопоэтического происхождения.
Подходящие противовирусные агенты, предполагаемые для применения в комбинации с соединениями по изобретению, могут включать нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы (NRTI), не нуклеозидные ингибиторы обратной транскриптазы (NNRTI), ингибиторы протеазы и другие противовирусные лекарственные средства. Примеры подходящих NRTI включают зидовудин (AZT); диданозин (ddl); зальцитабин (ddC); ставудин (d4T); ламивудин (3TC); абакавир (1592U89); адефовир дипивоксил [bis(POM)-PMEA]; лобукавир (BMS-180194); BCH-10652; эмитрицитабин [(-)-FTC]; бета-L-FD4 (также называемый бета-L-D4C или бета-L-2',3'-диклеокси-5-фторцитиден); DAPD ((-)-бета-D-2,6,-диаминопуриндиоксолан); и лоденозин (FddA). Типовые подходящие NNRTI включают невирапин (BI-RG-587); делавирадин (BHAP, U-90152); эфавиренз (DMP-266); PNU-142721; AG-1549; MKC-442 (1-(этоксиметил)-5-(1-метилэтил)-6-(фенилметил)-(2,4(1H,3H)-пиримидиндион) и (+)-каланолид A (NSC-675451) и B. Типовые подходящие ингибиторы протеазы включают саквинавир (Ro 31-8959), ритонавир (ABT-538), индинавир (MK-639), нелфнавир (AG-1343), ампренавир (141W94), лазинавир (BMS-234475), DMP-450, BMS-2322623, ABT-378 и AG-1549.Другие противовирусные агенты включают гидроксимочевину, рибавирин, IL-2, IL-12, пентафузид и Yissum Project No.11607.
Следует понимать, что многие из дополнительных терапевтических агентов, используемых в способах по изобретению, могут быть биологическими препаратами, требующими внутривенного, внутрибрюшинного или депо введения. В другом варианте осуществления, соединение по изобретению вводят перорально, и дополнительный терапевтический агент вводят парентерально, например, внутривенно, внутрибрюшинно или в виде депо.
В любой из описанных в настоящем документе комбинированных терапий, когда пациенту вводят более одного фармацевтического агента, их можно вводить одновременно, по отдельности, последовательно или в комбинации (например, для более чем двух агентов).
В одном варианте осуществления, изобретение относится к продукту, содержащему соединение по изобретению, такое как рассматриваемое соединение или любая его подгруппа, и, по меньшей мере, один другой терапевтический агент в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. Продукты, представленные в виде комбинированного препарата, включают композицию, содержащую соединение по изобретению, или любую его подгруппу и другие терапевтические агенты вместе в одной и той же фармацевтической композиции, или рассматриваемое соединение или любую его подгруппу и другие терапевтические агенты в отдельной форме, например, в форме набора.
В одном варианте осуществления, в изобретении представлен набор, включающий две или несколько отдельных фармацевтических композиций, по меньшей мере, одна из которых содержит рассматриваемое соединение, и другая содержит второй обсуждаемый в настоящем документе терапевтический агент. В одном варианте осуществления, набор содержит средства для раздельного хранения указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, обычно используемая для упаковки таблеток, капсул и подобных. Набор по изобретению можно использовать для введения различных дозированных форм, например пероральных и парентеральных, для введения отдельных композиций с различными интервалами дозирования, или для титрования отдельных композиций относительно друг друга. Чтобы облегчить соблюдение схемы, набор по изобретению обычно содержит инструкции по введению.
6. Скрининг соединений/методы анализа
Соединения по изобретению ингибируют киназную активность HPK1, где киназная активность может быть определена непосредственно с использованием многочисленных биохимических анализов, таких как анализ, описанный в примере 1. Значения IC50 любых соединений могут быть определены соответствующим образом в диапазоне концентраций ингибитора. Кроме того, ингибирующее действие соединений также можно оценить с помощью биологического анализа, для определения действия соединений на секрецию цитокинов Т-клетками после стимуляции TCR и CD28.
Например, в примере 5 описан такой функциональный анализ для определения действия ингибитора HPK1 на высвобождение IL-2 и IFN-γ при стимуляции Pan Т-клеток. Секретируемые IL-2 и IFN-γ могут быть измерены/количественно определены с помощью стандартного анализа ELISA. Коротко, pan T-клетки могут быть выделены из мононуклеарных клеток (MNC) периферической крови (PB) или PBMC с использованием коммерчески доступных наборов, таких как набор для выделения Pan T MACS (Miltenyl Biotec) (кат. № 130-096-535). Первичные pan-Т-клетки человека включают CD4 и CD8 Т-клетки, а также некоторые подмножества гамма/дельта Т-клеток. Pan-Т-клетки могут быть выделены с использованием метода негативной иммуномагнитной сепарации без использования колонок.
Изолированные pan-Т-клетки можно распределять в 96-луночные планшеты по 100000 клеток/лунку и стимулировать иммобилизованным анти-CD3 антителом и растворимым анти-CD28 антителом или РМА/иономицином в качестве положительного контроля (или культуральной средой в качестве отрицательного контроля). Различные концентрации тестируемых соединений могут быть добавлены к клеткам для оценки действия соединения на секрецию цитокинов после стимуляции TCR/костимуляции CD28. Стимулированные клетки инкубируют еще в течение 2 дней, после чего супернатант (содержащий цитокины, секретируемые pan-T-клетками) собирают из каждой лунки для анализа ELISA и количественного определения IL-2 и IFN-γ.
Дополнительные анализы также могут быть использованы для оценки способности любого ингибитора HPK1 ингибировать HPK1 или для скрининга соединений, обладающих ингибиторной активностью HPK1.
Например, в одном анализе, ингибирование активности киназы HPK1 может быть проанализировано с использованием анализа Treg (анализа пролиферации регуляторных Т-клеток), описанного ниже. В частности, первичные CD4+/CD25- Т-клетки и CD4+/CD25+ регуляторные Т-клетки выделяют из донорских мононуклеарных клеток периферической крови (РВМС) человека с использованием подходящего набора, такого как набор от Thermo Fisher Scientific (кат. № 11363D). CD4+/CD25- Т-клетки метят CFSE (Thermo Fisher Scientific, C34554) в соответствии с протоколом, предоставленным поставщиком. Меченые CFSE Т-клетки и регуляторные CD4+/CD25+ Т-клетки ресуспендируют в концентрации 1×106 клеток/мл в среде RPMI-1640. 100 мкл CFSE-меченных Т-клеток смешивают с или без 50 мкл CD4+/CD25+ регуляторных Т-клеток, обработанных 5 мкл анти-CD3/CD28 гранул (Thermo Fisher Scientific, 11132D) и различными концентрациями соединений, разведенных в 50 мкл среды RPMI-1640. Смешанные популяции клеток культивируют в течение 5 дней (37°C, 5% CO2) и пролиферацию CFSE-меченных Т-клеток анализируют с помощью BD LSRFortessa X-20 с использованием канала FITC на 5 день. Ожидается, что ингибирование HPK1 рассматриваемыми соединениями будет усиливать функцию Treg и ингибировать пролиферацию CFSE-меченных первичных CD4+/CD25- T-клеток.
В другом примере, ингибирование активности киназы HPK1 можно проанализировать с использованием p-SLP-76 S376 HTRF анализа (Cisbio), описанного ниже. Этот клеточный анализ HTRF обеспечивает быстрое количественное обнаружение SLP-76, фосфорилированного на серине 376 с помощью HPK1. Фосфо-SLP-76 создает каркас, на котором строятся ключевые сигнальные комплексы, и является маркером активации Т-лимфоцитов. По данным производителя, в анализе Phospho-SLP-76 (Ser376) используются два меченых антитела: одно с донорным флуорофором, другое с акцепторным. Первое антитело является специфическим к связыванию с фосфорилированным мотивом S376 на SLP-76, и второе является специфическим к способности распознавать SLP-76 независимо от его состояния фосфорилирования. Фосфорилирование белка обеспечивает образование иммунного комплекса, включающего оба меченых антитела, что приводит к тому, что донорный флуорофор оказывается в непосредственной близости от акцепторного, тем самым генерируя сигнал FRET. Его интенсивность прямо пропорциональна концентрации фосфорилированного белка, присутствующего в образце, и предоставляет средства оценки состояния фосфорилирования белка в формате анализа без промывки.
Вкратце, клетки Jurkat (культивируемые в среде RPMI1640 с 10% FBS) собирают и центрифугируют с последующим ресуспендированием в подходящей среде в концентрации 3×106 клеток/мл. Затем клетки Jurkat (35 мкл) вносят в каждую лунку 384-луночного планшета. Тестируемые соединения разводят средой для культивирования клеток для 40-кратного разведения (добавлением 39 мкл среды для культивирования клеток на 1 мкл соединения). Клетки Jurkat в луночном планшете обрабатывают тестируемыми соединениями в различных концентрациях (добавляя 5 мкл разведенного соединения в 35 мкл клеток Jurkat, и начиная с 3 мкМ при разведении 1:3) в течение 1 часа при 37°C, 5% CO2), с последующей обработкой анти-CD3 (5 мкг/мл, клон OKT3) в течение 30 мин для активации TCR и HPK1. Готовят разведение 1:25 100х блокирующего реагента (из набора p-SLP76 ser376HTRF) с 4х лизисным буфером (LB), в каждую лунку добавляют 15 мкл 4х LB буфера с блокирующим реагентом и инкубируют при комнатной температуре в течение 45 минут при осторожном встряхивании. Лизат клеток (16 мкл) добавляют в белый планшет Greiner, обрабатывают реагентами p-SLP76 Ser376 HTRF (2 мкл донора, 2 мкл акцептора) и инкубируют при 4°C в течение ночи. На следующий день измеряют однородную флуоресценцию с временным разрешением (HTRF) на планшетном ридере PHERAstar. Определение IC50 проводят путем подгонки кривой доли ингибирования к логарифму концентрации ингибитора с использованием программного обеспечения GraphPad Prism 5.0.
Любой из описанных выше анализов можно масштабировать для крупномасштабного или высокопроизводительного скрининга (HTS). Используя любой из анализов, описанных выше, можно определить значения IC50 рассматриваемых соединений.
7. Фармацевтические композиции
В изобретении представлены фармацевтические композиции, которые содержат любое из описанных в настоящем документе соединений или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых носителей или эксципиентов.
«Фармацевтически приемлемый эксципиент» и «фармацевтически приемлемый носитель» относятся к веществу, которое способствует приготовлению и/или введению активного агента, и/или абсорбции субъектом, и может быть включено в композиции по настоящему изобретению, не вызывая значительного неблагоприятного токсикологического воздействия на субъекта. Неограничивающие примеры фармацевтически приемлемых носителей и эксципиентов включают воду, NaCl, физиологические растворы, лактатированный Рингер, нормальную сахарозу, нормальную глюкозу, связующие агенты, наполнители, разрыхлители, смазывающие агенты, покрытия, подсластители, ароматизаторы, растворы солей (такие, как раствор Рингера), спирты, масла, желатины, углеводы, такие как лактоза, амилоза или крахмал, сложные эфиры жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидин и красители и подобные. Такие препараты можно стерилизовать и, при желании, смешивать со вспомогательными агентами, такими как смазывающие агенты, консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, соли для воздействия на осмотическое давление, буферы, красители и/или ароматические вещества и подобные, которые не реагируют пагубно или не препятствуют активности соединений, представленных в настоящем документе. Специалисту в данной области техники будет понятно, что другие фармацевтические носители и эксципиенты подходят для использования с описанными соединениями.
Эти композиции необязательно дополнительно содержат один или несколько дополнительных терапевтических агентов. Альтернативно, соединение по изобретению можно вводить пациенту, нуждающемуся в этом, в сочетании с введением одной или нескольких других терапевтических схем (например, гливека или других ингибиторов киназы, интерферона, трансплантата костного мозга, ингибиторов фарнезилтрансферазы, бисфосфонатов, талидомида, противораковых вакцин, гормональной терапии, антител, радиации и т.д.). Например, дополнительные терапевтические агенты для совместного введения или включения в фармацевтическую композицию с соединением по данному изобретению могут быть еще одним или несколькими противораковыми агентами.
Как описано в настоящем документе, композиции по настоящему изобретению содержат соединение по настоящему изобретению вместе с фармацевтически приемлемым носителем, который, как он используется в настоящем документе, включает любые и все растворители, разбавители или другие носители, добавки для дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие агенты, смазывающие агенты и подобные, подходящие для конкретной желаемой дозированной формы. Remington’s Pharmaceutical Sciences, Fifteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1975) описывает различные носители, используемые при составлении фармацевтических композиций, и известные методы их получения. За исключением случаев, когда любая обычная среда-носитель несовместима с соединениями по настоящему изобретению, например, вызывает какой-либо нежелательный биологический эффект или иным вредным образом взаимодействует с любыми другими компонентами фармацевтической композиции, ее использование рассматривается в объеме настоящего изобретения. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничены ими, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошковый трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воск для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли; такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие агенты, такие как лаурилсульфат натрия и стеарат магния, а также красители, агенты, способствующие высвобождению, покрывающие агенты, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты также могут присутствовать в композиции.
8. Составы
Это изобретение также охватывает класс композиций, содержащих активные соединения по настоящему изобретению в ассоциации с одним или несколькими фармацевтически приемлемыми носителями, и/или разбавителями, и/или адъювантами (совместно именуемые в настоящем документе материалами «носителями») и, при желании, другими активными ингредиентами.
В некоторых вариантах осуществления, в изобретении представлен фармацевтический состав для лечения рака, в частности рака, описанного в настоящем документе, содержащий соединение по настоящему изобретению или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
В некоторых вариантах осуществления, в изобретении представлен фармацевтический состав для лечения рака, выбранный из группы, состоящей из рака молочной железы, колоректального рака, рака легких, рака яичников и рака поджелудочной железы, содержащий соединение по настоящему изобретению или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
Соединения по настоящему изобретению можно вводить любым подходящим путем, предпочтительно, в форме фармацевтической композиции, адаптированной для такого пути, и в дозе, эффективной для предполагаемого лечения. Соединения и композиции по настоящему изобретению можно, например, вводить перорально, мукозально, местно, ректально, легочно, например, с помощью ингаляционного спрея, или парентерально, включая внутрисосудистое, внутривенное, внутрибрюшинное, подкожное, внутримышечное, чрескожное, интраорбитальное, подоболочечное, внутрижелудочковое, внутриопухолевое, интраназальное, интрастернальное введение, путем имплантации, путем ингаляции и путем инфузии, в стандартных дозированных формах, содержащих обычные фармацевтически приемлемые носители, адъюванты и наполнители.
Как правило, фармацевтическими композициями являются таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с а) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; b) смазывающими агентами, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также c) связующими веществами, например, алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, карбоксиметилцеллюлозой натрия и/или поливинилпирролидоном; при желании d) разрыхлителями, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями; и/или e) абсорбентами, красителями, ароматизаторами и подсластителями. Таблетки могут быть покрыты либо пленкой, либо энтеросолюбильным покрытием в соответствии со способами, известными в данной области техники.
Подходящие композиции для перорального введения включают эффективное количество соединения изобретения в форме таблеток, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсии, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, готовят в соответствии с любым способом, известным в данной области техники для производства фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов для того, чтобы получать фармацевтически элегантные и приятные на вкус препараты. Таблетки могут содержать активный ингредиент в смеси с не токсичными фармацевтически приемлемыми эксципиентами, пригодными для изготовления таблеток. Этими эксципиентами являются, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например, крахмал, желатин или аравийская камедь; и смазывающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки не имеют покрытия или покрыты известными способами для замедления разложения и абсорбции в желудочно-кишечном тракте и, таким образом, обеспечивают устойчивое действие в течение более длительного периода времени. Например, может быть использован материал с временной задержкой, такой как глицерилмоностеарат или глицерилдистеарат. Препараты для перорального применения могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водной или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Некоторые композиции для инъекций представляют собой водные изотонические растворы или суспензии, и суппозитории преимущественно готовят из жировых эмульсий или суспензий. Указанные композиции могут быть стерилизованы и/или содержать адъюванты, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие агенты, активаторы растворения, соли для регулирования осмотического давления и/или буферы. Кроме того, они могут содержать и другие терапевтически ценные вещества. Указанные композиции готовят в соответствии с обычными способами смешивания, гранулирования или покрытия, соответственно, и они содержат примерно 0,1-75% или содержат примерно 1-50% активного ингредиента. Подходящие композиции для чрескожного применения включают эффективное количество соединения по изобретению с подходящим носителем. Носители, подходящие для чрескожной доставки, включают абсорбируемые фармакологически приемлемые растворители для облегчения прохождения через кожу хозяина. Например, чрезкожные устройства имеют форму бандажа, содержащего подложку, резервуар, содержащий соединение, необязательно с носителями, необязательно барьер, контролирующий скорость, для доставки соединения с кожи хозяина с контролируемой и заданной скоростью в течение длительного периода времени, и средства для закрепления устройства на коже.
Подходящие композиции для местного применения, например, на кожу и в глаза, включают водные растворы, суспензии, мази, кремы, гели или составы для распыления, например, для доставки в виде аэрозоля, и подобные. Такие системы доставки для местного применения будут, в частности, подходящими для кожного применения, например, для лечения рака кожи, например, для профилактического применения в солнцезащитных кремах, лосьонах, спреях и подобных. Таким образом, они особенно подходят для использования в составах для местного применения, в том числе в косметических препаратах, хорошо известных в данной области техники. Они могут содержать солюбилизаторы, стабилизаторы, агенты, повышающие тоничность, буферы и консерванты.
Используемое в настоящем документе местное применение может также относиться к ингаляции или интраназальному применению. Их удобно доставлять в форме сухого порошка (либо отдельно, в виде смеси, например, сухой смеси с лактозой, либо в виде частиц смешанного компонента, например, с фосфолипидами) из ингалятора для сухого порошка или в виде аэрозольного спрея из контейнера под давлением, насоса, спрея, распылителя или небулайзера с или без использования подходящего пропеллента.
Настоящее изобретение дополнительно представляет безводные фармацевтические композиции и дозированные формы, содержащие соединения по настоящему изобретению в качестве активных ингредиентов, поскольку вода может способствовать разложению некоторых соединений.
Безводные фармацевтические композиции и дозированные формы по изобретению могут быть приготовлены с использованием безводных ингредиентов или ингредиентов с низким содержанием влаги и в условиях с низким содержанием влаги или низкой влажностью. Безводная фармацевтическая композиция может быть получена и храниться таким образом, чтобы сохранялась ее безводная природа. Соответственно, безводные композиции упаковывают с использованием материалов, которые, как известно, предотвращают воздействие воды, так что они могут быть включены в подходящие наборы рецептур. Примеры подходящей упаковки включают, но не ограничены ими, герметически запечатанную фольгу, пластмассу, контейнеры с единичными дозами (например, флаконы), блистерные упаковки и ленточные упаковки.
Изобретение дополнительно представляет фармацевтические композиции и дозированные формы, которые содержат один или несколько агентов, снижающих скорость разложения соединения по настоящему изобретению в качестве активного ингредиента. Такие агенты, которые упоминаются в настоящем документе как «стабилизаторы», включают, но не ограничены ими, антиоксиданты, такие как аскорбиновая кислота, рН буферы или солевые буферы и т.д.
Фармацевтически активные соединения по данному изобретению могут быть переработаны в соответствии с обычными способами фармации для получения лекарственных средств для введения пациентам, включая людей и других млекопитающих.
Количество вводимых соединений и схема дозирования для лечения болезненного состояния с помощью соединений и/или композиций по настоящему изобретению зависит от множества факторов, включая возраст, вес, пол и состояние здоровья субъекта, тип заболевания, тяжесть заболевания, способ и частоту введения и конкретное применяемое соединение. Таким образом, схема дозирования может широко варьироваться, но ее можно определить рутинно с использованием стандартных способов. Как упоминалось ранее, суточную дозу можно вводить за одно введение, или ее можно разделить на 2, 3, 4 или несколько введений.
Для терапевтических целей, активные соединения по настоящему изобретению обычно комбинируют с одним или несколькими адъювантами, эксципиентами или носителями, соответствующими указанному пути введения. При пероральном введении, соединения могут быть смешаны с лактозой, сахарозой, крахмальным порошком, сложными эфирами целлюлозы и алкановых кислот, алкиловыми эфирами целлюлозы, тальком, стеариновой кислотой, стеаратом магния, оксидом магния, натриевыми и кальциевыми солями фосфорной и серной кислот, желатином, аравийской камедью, альгинатом натрия, поливинилпирролидоном и/или поливиниловым спиртом, и затем таблетированы или инкапсулированы для удобного введения. Такие капсулы или таблетки могут содержать состав с контролируемым высвобождением, который может быть представлен в виде дисперсии активного соединения в гидроксипропилметилцеллюлозе.
В случае кожных заболеваний, может быть предпочтительным нанесение местного препарата соединений по настоящему изобретению на пораженный участок от двух до четырех раз в сутки. Составы, подходящие для местного введения, включают жидкие или полужидкие препараты, подходящие для проникновения через кожу (например, линименты, лосьоны, мази, кремы или пасты), и капли, подходящие для введения в глаза, уши или нос. Для местного применения, активный ингредиент может составлять от 0,001% до 10% масс./масс., например, от 1% до 2% масс. состава, хотя он может составлять не более 10% масс./масс., но предпочтительно, не более 5% масс./масс., и более предпочтительно, от 0,1% до 1% от состава.
Соединения по настоящему изобретению можно также вводить с помощью чрескожного устройства. Предпочтительно, чрезкожное введение будет осуществляться с использованием пластыря либо типа резервуара и пористой мембраны, либо типа твердой матрицы. В любом случае, активный агент доставляется - непрерывно из резервуара или микрокапсул через мембрану в клей, проницаемый для активного агента, который контактирует с кожей или слизистой оболочкой реципиента. Если активный агент всасывается через кожу, реципиенту вводят контролируемый и заданный поток активного агента. В случае микрокапсул, инкапсулирующий агент может также выполнять функцию мембраны. Масляная фаза эмульсий по настоящему изобретению может быть составлена из известных ингредиентов известным способом.
Хотя фаза может содержать только эмульгатор, она может содержать смесь, по меньшей мере, одного эмульгатора с жиром или маслом или и с жиром, и с маслом. Предпочтительно, гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует как стабилизатор. Также предпочтительно включать и масло, и жир. Вместе, эмульгаторы с или без стабилизаторов составляют так называемый эмульгирующий воск, и воск вместе с маслом и жиром составляют так называемую эмульгирующую мазевую основу, которая образует маслянистую диспергированную фазу кремовых составов. Эмульгаторы и стабилизаторы эмульсий, подходящие для использования в составе по настоящему изобретению, включают Tween 60, Span 80, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат, лаурилсульфат натрия, глицерилдистеарат отдельно или с воском или другие материалы, хорошо известные в данной области техники.
Выбор подходящих масел или жиров для состава основан на достижении желаемых косметических свойств, поскольку растворимость активного соединения в большинстве масел, которые, вероятно, будут использоваться в составах фармацевтических эмульсий, очень низка. Таким образом, крем предпочтительно должен быть нежирным, не оставляющим пятен и моющимся продуктом с подходящей консистенцией, чтобы избежать утечки из тюбиков или других контейнеров. Могу применяться сложные одноосновные или двухосновные алкиловые эфиры с прямой или разветвленной цепью, такие как диизоадипат, изоцетилстеарат, диэфир пропиленгликоля кокосовых жирных кислот, изопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или смесь сложных эфиров с разветвленной цепью. Их можно использовать по отдельности или в комбинации в зависимости от требуемых свойств.
Альтернативно, можно использовать липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин, или другие минеральные масла.
Составы, подходящие для местного введения в глаза, также включают глазные капли, в которых активные ингредиенты растворены или суспендированы в подходящем носителе, особенно в водном растворителе для активных ингредиентов.
Активные ингредиенты предпочтительно присутствуют в таких составах в концентрации от 0,5 до 20%, преимущественно, от 0,5 до 10% и особенно примерно 1,5% масс./масс.
Составы для парентерального введения могут быть в форме водных или не водных изотонических стерильных инъекционных растворов или суспензий. Эти растворы и суспензии могут быть приготовлены из стерильных порошков или гранул с использованием одного или нескольких носителей или разбавителей, упомянутых для применения в составах для перорального введения, или с использованием других подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Соединения могут быть растворены в воде, полиэтиленгликоле, пропиленгликоле, этаноле, кукурузном масле, хлопковом масле, арахисовом масле, кунжутном масле, бензиловом спирте, хлориде натрия, трагакантовой камеди и/или различных буферах. Другие адъюванты и способы введения хорошо и широко известны в области фармацевтики. Активный ингредиент можно также вводить путем инъекции в виде композиции с подходящими носителями, включая солевой раствор, декстрозу или воду, или с циклодекстрином (например, каптизолом), солюбилизацией сорастворителем (например, пропиленгликолем) или мицеллярной солюбилизацией (например, Tween 80).
Стерильным препаратом для инъекций может также быть стерильный раствор или суспензия для инъекций в не токсичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. В число приемлемых носителей и растворителей, которые можно использовать, входят вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели может быть использовано любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, находят применение при приготовлении инъекционных препаратов.
Для легочного введения, фармацевтическую композицию можно вводить в форме аэрозоля или с помощью ингалятора, включающего аэрозоль в виде сухого порошка.
Суппозитории для ректального введения лекарственного средства могут быть приготовлены путем смешивания лекарственного средства с подходящим нераздражающим наполнителем, таким как масло какао и полиэтиленгликоли, которые являются твердыми при обычной температуре, но жидкими при ректальной температуре и, следовательно, плавятся в прямой кишке и высвобождают лекарственное средство.
Фармацевтические композиции могут быть подвергнуты обычным фармацевтическим операциям, таким как стерилизация, и/или могут содержать обычные адъюванты, такие как консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, буферы и т.д. Таблетки и пилюли могут быть дополнительно изготовлены с энтеросолюбильным покрытием. Такие композиции могут также содержать адъюванты, такие как смачивающие агенты, подсластители, ароматизаторы и отдушки. Фармацевтические композиции по настоящему изобретению включают соединение описанных в настоящем документе формул или его фармацевтически приемлемую соль; дополнительный агент, выбранный из агента, ингибирующего киназу (малой молекулы, полипептида, антитела и т.д.), иммунодепрессанта, противоракового агента, противовирусного агента, противовоспалительного агента, противогрибкового агента, антибиотика или соединения против гиперпролиферации сосудов; и любой фармацевтически приемлемый носитель, адъювант или наполнитель.
Альтернативные композиции по настоящему изобретению включают соединение описанных в настоящем документе формул или его фармацевтически приемлемую соль; и фармацевтически приемлемый носитель, адъювант или наполнитель. Такие композиции могут необязательно содержать одно или несколько дополнительных терапевтических агентов, включая, например, агенты, ингибирующие киназу (малые молекулы, полипептиды, антитела и т.д.), иммунодепрессанты, противораковые агенты, противовирусные агенты, противовоспалительные агенты, противогрибковые агенты, антибиотики или соединения против гиперпролиферации сосудов.
Термин «фармацевтически приемлемый носитель или адъювант» относится к носителю или адъюванту, который можно вводить пациенту вместе с соединением по настоящему изобретению, который не нарушает его фармакологическую активность и нетоксичен при введении в дозах, достаточных для доставки терапевтического количества соединения. Фармацевтически приемлемые носители, адъюванты и наполнители, которые могут применяться в фармацевтических композициях по настоящему изобретению, включают, но не ограничены ими, ионообменники, оксид алюминия, стеарат алюминия, лецитин, самоэмульгирующиеся системы доставки лекарственных средств (SEDDS), такие как d-атокоферол полиэтиленгликоль 1000 сукцинат, поверхностно-активные вещества, используемые в фармацевтических дозированных формах, такие как Tweens или другие подобные полимерные матрицы для доставки, белки сыворотки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, неполные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воски, полиэтилен-полиоксипропиленовые блок-полимеры, полиэтиленгликоль и ланолин. Циклодекстрины, такие как u-, P- и y-циклодекстрин, или химически модифицированные производные, такие как гидроксиалкилциклодекстрины, включая 2- и 3-гидроксипропилциклодекстрины, или другие солюбилизированные производные, также могут быть успешно использованы для улучшения доставки соединений формул, описанных в настоящем документе.
Фармацевтические композиции можно вводить перорально в любой приемлемой для перорального применения дозированной форме, включая, но не ограничиваясь ими, капсулы, таблетки, эмульсии и водные суспензии, дисперсии и растворы. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсул, полезные разбавители включают лактозу и высушенный кукурузный крахмал. Когда водные суспензии и/или эмульсии вводят перорально, активный ингредиент может быть суспендирован или растворен в масляной фазе в комбинации с эмульгирующими и/или суспендирующими агентами.
При желании могут быть добавлены определенные подсластители, ароматизаторы и/или красители. Фармацевтические композиции могут включать составы, в которых используются методы липосом или микрокапсулирования, различные примеры которых известны в данной области техники.
Фармацевтические композиции можно вводить с помощью назального аэрозоля или ингаляции. Такие композиции готовят в соответствии с методами, хорошо известными в области составления фармацевтических препаратов, и могут быть приготовлены в виде растворов в солевом растворе с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для повышения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, примеры из которых также хорошо известны в данной области техники.
9. Лечебные наборы
Один аспект настоящего изобретения относится к набору для удобного и эффективного осуществления способов или применений в соответствии с настоящим изобретением. Как правило, фармацевтическая упаковка или набор содержат один или несколько контейнеров, наполненных одним или несколькими ингредиентами фармацевтических композиций по изобретению. Такие наборы особенно подходят для доставки твердых пероральных форм, таких как таблетки или капсулы. Такой набор предпочтительно включает ряд стандартных дозировок, и может также включать карточку с дозировками, ориентированными в порядке их предполагаемого использования. При желании, может быть предусмотрена памятка, например, в виде цифр, букв или других маркировок или с календарной вставкой, обозначающей дни в схеме лечения, в которые можно вводить дозы. С такими контейнерами необязательно может быть предоставлено уведомление в форме, предписанной государственным органом, регулирующим производство, использование или продажу фармацевтических продуктов, которое отражает одобрение органом производства, использования или продажи для введения человеку.
Следующие типовые примеры содержат важную дополнительную информацию, примеры и рекомендации, которые могут быть адаптированы к практике настоящего изобретения в его различных вариантах осуществления и их эквивалентах. Эти примеры предназначены для иллюстрации изобретения и не предназначены и не должны толковаться как ограничивающие его объем. Действительно, различные модификации изобретения и многие другие его варианты осуществления, в дополнение к показанным и описанным в настоящем документе, станут очевидными для специалистов в данной области техники после ознакомления с этим документом, включая следующие примеры и ссылки на научную и патентную литературу, цитируемую в настоящем документе.
Содержание цитируемых ссылок включено в настоящий документ посредством ссылки, чтобы помочь проиллюстрировать уровень техники.
Кроме того, для целей настоящего изобретения химические элементы идентифицированы в соответствии с Периодической таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 75th Ed, на внутренней стороне обложки. Кроме того, общие принципы органической химии, а также конкретные функциональные группы и реакционная способность описаны в «Organic Chemistry,» Thomas Sorrell, University Science Books, Sausalito: 1999, и «Organic Chemistry,» Morrison & Boyd (3d Ed), полное содержание обоих включено в настоящий документ посредством ссылки.
10. Схемы синтеза
Соединения по изобретению могут быть получены специалистом в данной области техники в соответствии с признанными в данной области методами и процедурами. Более конкретно, соединения по настоящему изобретению могут быть получены в соответствии со схемами, способами и примерами, изложенными ниже. Специалисту в данной области будет понятно, что отдельные стадии на следующих схемах могут быть изменены для получения соединений по изобретению. Реагенты и исходные материалы легко доступны специалистам в данной области техники. Все заместители, если не указано иное, являются такими, как определено выше.
ПРИМЕРЫ
Ниже представлены используемые сокращения и их значения в спецификации.
EtOAc: Этилацетат
ДХМ: Дихлорметан
АЦН: ацетонитрил
ТГФ: тетрагидрофуран
ДМСО: диметилсульфоксид
MeOH: метанол
EtOH: этанол
ДМФ: N, N-диметилформамид
ДМА: N, N-диметилацетамид
ДМФ ДМА: N, N-диметилформамид диметилацеталь
NCS: N-хлорсукцинимид
NBS: N-бромсукцинимид
NIS: N-йодсукцинимид
Pd-C : палладий на угле
ДАЛ: диизопропиламид лития
ТФК: трифторуксусная кислота
ПТСК: п-толуолсульфоновая кислота
DIBAL-H: гидрид диизобутилалюминия
АГЛ: алюмогидрид лития
Py: пиридин
ДФФА: дифенилфосфорил азид
КДИ: 1,1-карбонилдиимидазол
ТЭА:триэтиламин
ДИПЭА: N, N-диизопропилэтиламин
ДМАП: 4-(диметиламино)пиридин
EDCI: N-(3-диметиламинопропил)-N-этилкарбодиимида гидрохлорид
HATU: гексафторфосфат (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксида
ГОБТ: 1-гидроксибензотриазол
TfOH: трифторметансульфоновая кислота
dppf: 1,1 ферроцендиил-бис(дифенилфосфин)
ТДАС: трифторид (диэтиламино)серы
Pd2(dba)3: трис(дибензилиденацетон) дипалладий(0)
Boc: трет-бутоксикарбонил
Ac: ацетил
ТМСИ: триметилсилилйодид
ИТБА: йодид тетрабутиламмония
PPh3: трифенилфосфин
dba: дибензилиденацетон
BINAP: 2,2'-бис(дифенилфосфино)-1,1'-бинафтил
MsCl: метансульфонилхлорид
TsCl: толуолсульфонилхлорид
ДМАП: 4-диметиламинопиридин
LiHMDS: бис(триметилсилил)амид лития
NaHMDS: бис(триметилсилиламид) натрия
ДМС: диметилсульфид
ДМЭ: диметоксиэтан
ДХЭ: дихлорэтан
ЯМР: ядерный магнитный резонанс
ЖХ-МС: жидкостная хроматография масс спектрометрия
ИЭР-МС: ионизация электрораспылением масс спектрометрия
ГХ-МС: газовая хроматография масс спектрометрия
ТСХ: тонкослойная хроматография
TCR: T-клеточный рецептор
BCR: B-клеточный рецептор
CARD: домен активации и рекрутинга каспазы
мМ: миллимоль
мкМ: микромоль
мл: микролитр
нг: нанограмм
нМ: наномоль
нм: нанометр
IC50: полумаксимальная ингибирующая концентрация
ОП: оптическая плотность
А. Биологические примеры
Биологический пример 1. HPK1 биохимический анализ
В этом примере используют ADP-Glo™ Kinase Assay для измерения действия потенциальных соединений-ингибиторов HPK1 на активность киназы HPK1.
ADP-Glo™ Kinase Assay (Promega Corp., Madison, WI) измеряет ADP, образующийся в результате киназной реакции. Согласно производителю, ADP, образующийся в киназном анализе, сначала превращается в ATP, который затем используется для создания света в люциферазной реакции. Создаваемая люминесценция коррелирует с активностью киназы. Типовые установки эксперимента описаны ниже, хотя в отдельные анализы могут быть внесены незначительные корректировки.
Материалы и оборудование
1. Реагенты:
2. Оборудование и поставщики:
3. Установки планшета
Серийное 3-кратное разведение соединений от 10 мкМ (максимальная концентрация) до 0,508 нМ (наименьшая концентрация). Положительным контролем является 10 мкМ эталон+фермент+субстрат. Отрицательным контролем является 1% ДМСО+фермент+субстрат.
4. Процедуры
1. Подготовка буфера
40 мМ Трис pH 7,5; 20 мМ MgCl2, 0,1 мг/мл BSA, 50 мкМ DTT
Исходный буфер
Добавляют 20 мл 1M Tris и 10 мл 1M MgCl2 к 470 мл ddH2O с получением буферного исходного раствора и исходного раствора при КТ.
2. Получение аналитического буфера Fresh 1*
3. Получение соединений
1) Соединения разбавляют до 1 мМ путем смешивания 10 мкл 10 мМ соответствующих исходных растворов соединений с 90 мкл ДМСО.
2) Затем соединения разводят в 3-кратно для получения 10 доз (разведение от 5 мкл до 10 мкл) с помощью BRAVO. Максимальная концентрация соединения составляет 1 мМ (100×), конц. ДМСО составляет 100%.
3) Переносят 100 нл каждого разведенного образца соединения в 384-луночный планшет (Corning-4512) с помощью ECHO.
4) Центрифугируют планшет при 1500 об/мин в течение 1 минуты.
4. Получение смеси 2× ATP-MBP:
20 мкМ ATP, 0,2 мкг/мкл MBP в киназном буфере (конечная концентрация: 10 мкМ ATP, 0,1 мкг/мкл MBP)
5. Получение 2× HPK1 рабочего раствора с аналитическим буфером.
Конечная концентрация HPK1 составляет 0,6 нг/мкл. Для соединений с высокой эффективностью используют более низкую концентрацию HPK1 (0,26 нг/мкл-0,065 нг/мкл).
6. Добавляют 5 мкл/лунку рабочего раствора 2× HPK1. Центрифугируют при 1500 об/мин в течение 1 минуты.
7. Добавляют 5 мкл/лунку 2× АТР-субстрата, центрифугируют при 1500 об/мин в течение 1 минуты.
8. Инкубируют при 25°C в течение 1 часа (или 6 часов для соединений с высокой эффективностью).
9. Добавляют 5 мкл/лунку реагента ADP-GLO™ чтобы остановить киназную реакцию и истощить неизрасходованный АТР. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
10. Добавляют 10 мкл реагента для обнаружения киназ, чтобы превратить ADP в АТР. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
11. Записывают люминесцентный сигнал на планшетном ридере Envision (384-CTG).
5. Анализ данных
Долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках с высоким и низким контролем, содержащихся в каждом планшете для анализа. Лунки с высоким контролем служат как 0% ингибирования, и лунки с низким контролем, которые не содержат каких-либо соединений, но скорее ДМСО (конечная концентрация=0,5%), служат как 100% ингибирования. Концентрации и значения % ингибирования для тестируемых соединений наносят на график, и концентрацию соединения, необходимую для 50% ингибирования (IC50), определяют с помощью трехпараметрического логистического уравнения доза-ответ. Значение конечной точки (IC50) для эталонного пептида/соединения оценивают в каждом эксперименте в качестве меры контроля качества. Если значение конечной точки находится в пределах 3-кратного от ожидаемого значения, то эксперимент считают приемлемым.
Биологический пример 2. Биохимический анализ PKC-тета.
В этом примере используют ADP-Glo™ Kinase Assay для измерения влияния потенциальных соединений-ингибиторов HPK1 на активность PKC-тета киназы. Типовые установки эксперимента описаны ниже, хотя в отдельные анализы могут быть внесены незначительные корректировки.
Материалы и оборудование
1. Реагенты:
2. Оборудование и поставщики:
3. Установки планшета
Серийное 3-кратное разведение соединений от 10 мкМ (максимальная концентрация) до 0,508 нМ (наименьшая концентрация). Положительным контролем является 10 мкМ эталон+фермент+субстрат. Отрицательным контролем является 1% ДМСО+фермент+субстрат.
4.Процедуры
1. Подготовка буфера
40 мМ Трис pH 7,5; 20 мМ MgCl2, 0,1 мг/мл BSA, 50 мкМ DTT
Исходный буфер
Добавляют 20 мл 1M Tris и 10 мл 1M MgCl2 к 470 мл ddH2O с получением буферного исходного раствора и исходного раствора при КТ.
2. Получение аналитического буфера Fresh 1*
3. Получение соединений
1) Соединения разводят до 1 мМ путем смешивания 10 мкл 10 мМ исходных растворов соответствующих соединений с 90 мкл ДМСО.
2) Затем соединения разводят 3-кратно для получения 10 доз (разведение от 5 мкл до 10 мкл) с помощью BRAVO. Максимальная концентрация соединения составляет 1 мМ (100×), конц. ДМСО составляет 100%.
3) Переносят 50 нл каждого разведенного образца соединения в 384-луночный планшет (Corning-4512) с помощью ECHO.
4) Центрифугируют планшет при 1500 об/мин в течение 1 минуты.
4. Получение 2× рабочего раствора фермента в киназном буфере:
5. Получение 2× ATP-sub смеси:
6. Добавляют 2,5 мкл/лунку 2× рабочего раствора фермента. Центрифугируют при 1500 об/мин в течение 1 минуты.
7. Добавляют 2,5 мкл/лунку 2× АТФ-субстрата, центрифугируют при 1500 об/мин в течение 1 минуты.
8. Инкубируют при 25°C в течение 60 мин.
9. Добавляют 5 мкл/лунку реагента ADP-Glo™, чтобы остановить киназную реакцию и истощить неизрасходованный АТР через 1 час. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
10. Добавляют 10 мкл реагента для обнаружения киназ, чтобы преобразовать ADP в АТР. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
11. Записывают сигнал люминесценции на планшетном ридере Envision (384-USL).
5. Анализ данных
Долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках с высоким и низким контролем, содержащихся в каждом планшете для анализа. Лунки с высоким контролем служат как 0% ингибирования, и лунки с низким контролем, которые не содержат каких-либо соединений, но скорее ДМСО (конечная концентрация=0,5%), служат как 100% ингибирования. Концентрации и значения % ингибирования для тестируемых соединений наносят на график, и концентрацию соединения, необходимую для 50% ингибирования (IC50), определяют с помощью трехпараметрического логистического уравнения доза-ответ. Значение конечной точки (IC50) для эталонного пептида/соединения оценивают в каждом эксперименте в качестве меры контроля качества. Если значение конечной точки находится в пределах 3-кратного от ожидаемого значения, то эксперимент считают приемлемым.
Биологический пример 3. Биохимический анализ TBK1.
В этом примере используют ADP-Glo™ Kinase Assay для измерения влияния потенциальных соединений-ингибиторов HPK1 на активность TBK1 киназы. Типовые установки эксперимента описаны ниже, хотя в отдельные анализы могут быть внесены незначительные корректировки.
Материалы и оборудование
1. Реагенты:
2. Оборудование и поставщики:
3. Установки планшета
См. выше.
4.Процедуры
1. Подготовка буфера
40 мМ Трис pH 7,5; 20 мМ MgCl2, 0,1 мг/мл BSA, 50 мкМ DTT
Исходный буфер
Добавляют 20 мл 1M Tris и 10 мл 1M MgCl2 к 470 мл ddH2O с получением буферного исходного раствора и исходного раствора при КТ (комнатной температуре).
2. Получение аналитического буфера Fresh 1*
3. Получение соединений
1) Соединения разводят до 1 мМ путем смешивания 10 мкл 10 мМ исходных растворов соответствующих соединений с 90 мкл ДМСО.
2) Затем соединения разводят 3-кратно для получения 10 доз (разведение от 5 мкл до 10 мкл) с помощью BRAVO. Максимальная концентрация соединения составляет 1 мМ (100×), конц. ДМСО составляет 100%.
3) Переносят 50 нл каждого разведенного образца соединения в 384-луночный планшет (Corning-4512) с помощью ECHO.
4) Центрифугируют планшет при 1500 об/мин в течение 1 минуты.
4. Получение 2× рабочего раствора фермента в киназном буфере:
5. Получение 2× ATP-sub смеси:
6. Добавляют 2,5 мкл/лунку 2× рабочего раствора фермента. Центрифугируют при 1500 об/мин в течение 1 минуты.
7. Добавляют 2,5 мкл/лунку 2× АТФ-субстрата, центрифугируют при 1500 об/мин в течение 1 минуты.
8. Инкубируют при 25°C в течение 60 мин.
9. Добавляют 5 мкл/лунку реагента ADP-Glo™, чтобы остановить киназную реакцию и истощить неизрасходованный АТР через 1 час. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
10. Добавляют 10 мкл реагента для обнаружения киназ, чтобы преобразовать ADP в АТР. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
11. Записывают сигнал люминесценции на планшетном ридере Envision (384-USL).
5. Анализ данных
Долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках с высоким и низким контролем, содержащихся в каждом планшете для анализа. Лунки с высоким контролем служат как 0% ингибирования, и лунки с низким контролем, которые не содержат каких-либо соединений, но скорее ДМСО (конечная концентрация=0,5%), служат как 100% ингибирования. Концентрации и значения % ингибирования для тестируемых соединений наносят на график, и концентрацию соединения, необходимую для 50% ингибирования (IC50), определяют с помощью трехпараметрического логистического уравнения доза-ответ. Значение конечной точки (IC50) для эталонного пептида/соединения оценивают в каждом эксперименте в качестве меры контроля качества. Если значение конечной точки находится в пределах 3-кратного от ожидаемого значения, то эксперимент считают приемлемым.
Биологический пример 4. Биохимический анализ JAK3.
В этом примере используют ADP-Glo™ Kinase Assay для измерения влияния потенциальных соединений-ингибиторов HPK1 на активность JAK3 киназы. Типовые установки эксперимента описаны ниже, хотя в отдельные анализы могут быть внесены незначительные корректировки.
Материалы и оборудование
1. Реагенты:
2. Оборудование и поставщики:
3. Процедуры
1. Получение JAK3 киназного буфера.
Добавляют свежий DTT и BSA в буфер (конечная концентрация: 40 мМ трис, pH 7,5; 20 мМ MgCl2, 0,1 мг/мл BSA, 50 мкМ DTT).
2. Получение соединений
Для тестируемых соединений: соединения разбавляют до 1 мМ путем смешивания 5 мкл 10 мМ исходных соединений с 45 мкл ДМСО. Затем растворы соединений серийно разводят 3-кратно для получения 10 доз. Максимальная концентрация соединения составляет 1 мМ (100×), концентрация ДМСО составляет 100%.
50 нл каждой из 10 доз разведенного соединения добавляют в 384-луночный планшет для анализа (Corning #4512) с помощью ECHO.
Для положительного контроля, 50 нл 1 мМ эталонного соединения добавляют в 384-луночный планшет для анализа (Corning #4512) с помощью ECHO.
Для отрицательного контроля: 50 нл ДМСО переносят в 384-луночный планшет для анализа в качестве отрицательного контроля.
Планшет для анализа центрифугируют при 1500 об/мин в течение 1 минуты.
3. Соединения переносят с помощью ECHO в соответствии со схемой, приведенной ниже:
Эталонные и тестируемые соединения 1-15: серийное 3-кратное разведение от 10 мкМ до 0,508 нМ (10 доз); отрицательным контролем является 0,78 нг JAK3, 4 мкМ ATP и 0,2 мкг/мкл поли, 1% ДМСО; положительным контролем является максимальная доза эталонного соединения, 0,78 нг JAK3, 0,2 мкг/мкл Poly(E4Y1), 4 мкМ АТР, 1% ДМСО.
4. Добавляют 2,5 мкл/лунку 2× рабочего раствора JAK3 с помощью пипетки (Thermo, 30 мкл, многоканальная). Центрифугируют при 1500 об/мин в течение 1 минуты.
5. Добавляют 2,5 мкл/лунку 2× рабочей смеси ATP-Poly(E4Y1) с помощью пипетки (Thermo, 30 мкл, многоканальная). Центрифугируют при 1500 об/мин в течение 1 минуты.
6. Инкубируют планшет для анализа при 25°C в течение 60 мин.
7. Добавляют 5 мкл/лунку реагента ADP-Glo™ от BRAVO, чтобы остановить киназную реакцию и истощить неизрасходованный ATP через 1 час. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
8. Добавляют 10 мкл реагента для обнаружения киназ от BRAVO, чтобы превратить ADP в АТР. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
9. Записывают сигнал люминесценции на планшетном ридере Envision (384-USL).
10. Обрабатывают данные с помощью XL-fit. Ингибирование%=[1-(тестируемая лунка - отрицательный контроль) / (положительный контроль - отрицательный контроль)] * 100%.
5. Анализ данных
Долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках с отрицательным и положительным контролем, содержащихся в каждом планшете для анализа. Лунки с отрицательным контролем служат как 0% ингибирования, и лунки с положительным контролем служат как 100% ингибирования. Концентрации и значения % ингибирования для тестируемых соединений наносят на график, и концентрацию соединения, необходимую для 50% ингибирования (IC50), определяют с помощью трехпараметрического логистического уравнения доза-ответ. Значение конечной точки (IC50) для эталонного пептида/соединения оценивают в каждом эксперименте в качестве меры контроля качества. Если значение конечной точки находится в пределах 3-кратного от ожидаемого значения, то эксперимент считают приемлемым.
Биологический пример 5. Протокол биохимического анализа ZAP70.
В этом примере используют ADP-Glo™ Kinase Assay для измерения влияния потенциальных соединений-ингибиторов HPK1 на активность ZAP70 киназы. Типовые установки эксперимента описаны ниже, хотя в отдельные анализы могут быть внесены незначительные корректировки.
Материалы и оборудование
1. Реагенты:
2. Оборудование и поставщики:
3. Процедуры
1. Получение ZAP70 киназного буфера.
Добавляют свежий DTT и BSA в буфер (конечная концентрация: 40 мМ трис, pH 7,5; 20 мМ MgCl2, 0,1 мг/мл BSA, 50 мкМ DTT, 2 мМ MnCl2).
2. Получение соединений
Для тестируемых соединений: соединения сначала разводят до 1 мМ путем смешивания 5 мкл 10 мМ исходных соединений с 45 мкл ДМСО. Эти соединения затем разводят 3-кратно для получения 10 доз. Максимальная концентрация соединения составляет 1 мМ (100×), концентрация ДМСО составляет 100%.
50 нл каждой из 10 доз разведенного соединения добавляют в 384-луночный планшет для анализа (Corning #4512) с помощью ECHO.
Для положительного контроля, 50 нл 1 мМ стауроспорина переносят в 384-луночный планшет для анализа в качестве положительного контроля.
Для отрицательного контроля: 50 нл ДМСО переносят в 384-луночный планшет для анализа в качестве отрицательного контроля. Центрифугируют планшет для анализа при 1500 об/мин в течение 1 минуты.
3. Соединения переносят с помощью ECHO в соответствии со схемой, приведенной ниже:
Стауроспорин и тестируемые соединения 1-15: серийное 3-кратное разведение от 10 мкМ до 0,508 нМ (10 доз); отрицательным контролем является 6,25 нг ZAP70, 10 мкМ ATP и 0,4 мкг/мкл поли, 1% ДМСО; положительным контролем является 10 мкМ стауроспорина, 6,25 нг ZAP70, 10 мкМ АТР и 0,4 мкг/мл poly, 1% ДМСО.
4. Получают смесь 2× АТР-Poly(E4Y1): 20 мкМ АТР, 0,8 мкг/мкл poly(E4Y1) в киназном буфере (конечная концентрация: 10 мкМ АТР, 0,4 мкг/мкл poly(E4Y1).
5. Получают рабочий раствор 2× ZAP70: (2,5 нг/мкл) в киназном буфере (конечная концентрация 1,25 нг/мкл).
6. Добавляют 2,5 мкл/лунку 2× рабочего раствора ZAP70 с помощью пипетки (Thermo, 30 мкл, многоканальная). Центрифугируют при 1500 об/мин в течение 1 минуты.
7. Добавляют 2,5 мкл/лунку 2× рабочей смеси ATP-Poly(E4Y1) с помощью пипетки (Thermo, 30 мкл, многоканальная). Центрифугируют при 1500 об/мин в течение 1 минуты.
8. Инкубируют планшет для анализа при 25°C в течение 60 мин.
9. Добавляют 5 мкл/лунку реагента ADP-Glo™ от BRAVO, чтобы остановить киназную реакцию и истощить неизрасходованный ATP через 1 час. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
10. Добавляют 10 мкл реагента для обнаружения киназ от BRAVO, чтобы превратить ADP в АТР. Центрифугируют при 1500 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
11. Записывают сигнал люминесценции на планшетном ридере Envision (384-USL).
12. Обрабатывают данные с помощью XL-fit. Ингибирование%=[1-(тестируемая лунка - отрицательный контроль) / (положительный контроль - отрицательный контроль)] * 100%.
5. Анализ данных
Долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках с отрицательным и положительным контролем, содержащихся в каждом планшете для анализа. Лунки с отрицательным контролем служат как 0% ингибирования, и лунки с положительным контролем служат как 100% ингибирования. Концентрации и значения % ингибирования для тестируемых соединений наносят на график, и концентрацию соединения, необходимую для 50% ингибирования (IC50), определяют с помощью трехпараметрического логистического уравнения доза-ответ. Значение конечной точки (IC50) для эталонного пептида/соединения оценивают в каждом эксперименте в качестве меры контроля качества. Если значение конечной точки находится в пределах 3-кратного от ожидаемого значения, то эксперимент считают приемлемым.
Биологический пример 6. Протокол биохимического анализа LCK.
В этом примере используют ADP-Glo™ Kinase Assay для измерения влияния потенциальных соединений-ингибиторов HPK1 на активность Lck киназы. Типовые установки эксперимента описаны ниже, хотя в отдельные анализы могут быть внесены незначительные корректировки.
Материалы и оборудование
1. Реагенты:
2. Оборудование и поставщики:
3. Процедуры
1. Получение LCK киназного буфера.
Добавляют свежий DTT и BSA в буфер (конечная концентрация: 40 мМ трис, pH 7,5; 20 мМ MgCl2, 0,1 мг/мл BSA, 50 мкМ DTT, 2 мМ MnCl2).
2. Получение соединений
Для тестируемых соединений: соединения сначала разводят до 1 мМ путем смешивания 5 мкл 10 мМ исходных соединений с 45 мкл ДМСО. Эти соединения затем серийно разводят 3-кратно для получения 10 доз. Максимальная концентрация соединения составляет 1 мМ (100×), концентрация ДМСО составляет 100%.
Для положительного контроля: стауроспорин разводят до 300 мкМ смешиванием 3 мкл 10 мМ исходного раствора с 98 мкл ДМСО.
50 нл растворов соединений и 300 мкМ стауроспорина переносят в 384-луночный планшет для анализа (Corning #4512) в качестве положительного контроля.
Для отрицательного контроля: 50 нл ДМСО переносят в 384-луночный планшет для анализа в качестве отрицательного контроля. Центрифугируют планшет для анализа при 1500 об/мин в течение 1 минуты.
3. Соединения переносят с помощью ECHO в соответствии со схемой, приведенной ниже:
Стауроспорин и тестируемые соединения 1-15: серийное 3-кратное разведение от 10 мкМ до 0,508 нМ (10 доз); отрицательным контролем является 7 нг LCK, 20 мкМ ATP и 0,4 мкг/мкл поли, 1% ДМСО; положительным контролем является 3 мкМ стауроспорина, 7 нг LCK, 20 мкМ АТР и 0,4 мкг/мл poly, 1% ДМСО.
4. Получают смесь 2× АТР-Poly(E4Y1): 40 мкМ АТР, 0,8 мкг/мкл poly(E4Y1) в киназном буфере (конечная концентрация: 20 мкМ АТР, 0,4 мкг/мкл poly(E4Y1).
5. Получают рабочий раствор 2× LCK: (2,8 нг/мкл) в киназном буфере (конечная концентрация 1,4 нг/мкл).
6. Добавляют 2,5 мкл/лунку 2× рабочего раствора LCK с помощью пипетки (Thermo, 30 мкл, многоканальная). Центрифугируют при 1000 об/мин в течение 1 минуты.
7. Добавляют 2,5 мкл/лунку 2× рабочей смеси ATP-Poly(E4Y1) с помощью пипетки (Thermo, 30 мкл, многоканальная). Центрифугируют при 1000 об/мин в течение 1 минуты.
8. Инкубируют планшет для анализа при 25°C в течение 60 мин.
9. Добавляют 5 мкл/лунку реагента ADP-Glo™ от BRAVO, чтобы остановить киназную реакцию и истощить неизрасходованный ATP через 1 час. Центрифугируют при 1000 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
10. Добавляют 10 мкл реагента для обнаружения киназ от BRAVO, чтобы превратить ADP в АТР. Центрифугируют при 1000 об/мин в течение 1 минуты. Инкубируют при 25°С в течение 40 мин.
11. Записывают сигнал люминесценции на планшетном ридере Envision (384-USL).
12. Обрабатывают данные с помощью XL-fit. Ингибирование%=[1-(тестируемая лунка - отрицательный контроль) / (положительный контроль - отрицательный контроль)] * 100%.
4. Анализ данных
Долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках с отрицательным и положительным контролем, содержащихся в каждом планшете для анализа. Лунки с отрицательным контролем служат как 0% ингибирования, и лунки с положительным контролем служат как 100% ингибирования. Концентрации и значения % ингибирования для тестируемых соединений наносят на график, и концентрацию соединения, необходимую для 50% ингибирования (IC50), определяют с помощью трехпараметрического логистического уравнения доза-ответ. Значение конечной точки (IC50) для эталонного пептида/соединения оценивают в каждом эксперименте в качестве меры контроля качества. Если значение конечной точки находится в пределах 3-кратного от ожидаемого значения, то эксперимент считают приемлемым.
Биологический пример 7. Анализ протеинкиназы MAP4K3.
В этом примере представлен протокол анализа для измерения фосфорилирования пептидного субстрата протеинкиназой MAP4K3.
Коротко, MAP4K3, ее субстрат и кофакторы (АТР и Mg2+) объединяют в лунке титровального микропланшета и инкубируют в течение 5 часов при 25ºC. В конце инкубации реакцию гасят добавлением буфера, содержащего ЭДТК. Субстрат и продукт разделяют и количественно определяют электрофоретически с использованием основанной на микрожидкости системы LabChip 3000 Drug Discovery System от Caliper Life Sciences.
Для этого анализа, субстратом MAP4K3 является FAM-GAGRLGRDKYKTLRQIRQ-NH2 (FAM является карбоксифлуоресцеином). Пептидный субстрат предпочтительно имеет чистоту >98% по данным капиллярного электрофореза.
Типовая установка и условия анализа приведены ниже.
1. В лунку 384-луночного планшета добавляют 5 мкл 2× ферментного буфера (или контроля).
2. Добавляют 100 нл 100× соединения. Фермент и соединение могут быть предварительно инкубированы в это время, если это желательно.
3. Добавляют 5 мкл 2× буфера для субстрата.
4. Инкубируют планшет при 25°C в течение 5 часов.
5. Завершают реакцию добавлением 40 мкл 1,25× останавливающего буфера.
6. Создают задание в Caliper LabChip® 3000 Drug Discovery System, используя значения из таблицы ниже.
Условия разделения для 12 Sipper Chip
7. Загружают планшет и начинают электрофорез с использованием синего лазера (480 нм) для возбуждения и зеленого CCD (520 нм) для обнаружения (CCD2).
Вышеупомянутый анализ проводят при следующих условиях реакции: всего 5 часов; при 25°С в присутствии 20 мМ 100% ингибитора ЭДТК.
Конечная реакционная смесь для анализа: 100 мМ HEPES, pH 7,5; 0,1% BSA; 0,01% Triton X-100; 1 мМ DTT; 5 мМ MgCl2, 10 мкМ ортованадата натрия, 10 мкМ бета-глицерофосфата, 20 мкМ АТР; 1% ДМСО (из соединения); 0,5 мкМ FAM-GAGRLGRDKYKTLRQIRQ-NH2; 0,5 нМ фермента MAP4K3.
Следует отметить, что удельная активность MAP4K3 варьируется от партии к партии, и может потребоваться корректировка концентрации фермента, чтобы обеспечить превращение субстрата в продукт на ~10-20%.
Пептиды субстрата и продукта, присутствующие в каждом образце, разделяют электрофоретически с использованием прибора для капиллярного электрофореза LabChip 3000. При разделении пептидов субстрата и продукта наблюдаются два пика флуоресценции. Изменение относительной интенсивности флуоресценции пиков субстрата и продукта является измеряемым параметром, отражающим активность фермента. Капиллярные электрофореграммы (файлы сбора RDA) анализируют с использованием программного обеспечения HTS Well Analyzer (Caliper Life Sciences). Активность киназы в каждом образце определяют как отношение произведения к сумме (PSR): P/(S+P), где P является высотой пика пептида продукта, и S является высота пика пептида субстрата.
Для каждого соединения активность фермента измеряют при различных концентрациях (12 концентраций соединения, разделенных 3х интервалами разведения). Образцы отрицательного контроля (ингибирование 0% в отсутствие ингибитора) и образцы положительного контроля (ингибирование 100% в присутствии 20 мМ ЭДТК) собирают в четырех повторах и используют для расчета значений % ингибирования для каждого соединения при каждой концентрации.
Долю ингибирования (Pinh) определяют с использованием следующего уравнения:
Pinh= (PSR0% - PSRinh)/(PSR0%- PSR100%)*100, где PSRinh является отношением произведения к сумме в присутствии ингибитора, PSR0% является средним отношением произведения к сумме в отсутствие ингибитора, и PSR100% является средним отношением произведения к сумме в контрольных образцах со 100% ингибированием.
Значения IC50 ингибиторов определяют путем подгонки кривых ингибирования (Pinh к концентрации ингибитора) с помощью четырехпараметрической сигмоидальной модели доза-ответ с использованием программного обеспечения XLfit 4 (IBDS).
Некоторые материалы и буферы, использованные в анализе, перечислены ниже для справки.
Биологический пример 8. Исследование эффективности соединения в отношении IL2 и IFN-γ, высвобождаемых Pan Т-клетками человека.
В этом примере представлен способ анализа, который можно использовать для определения действия соединения на высвобождение IL2 и IFN-γ с использованием pan Т-клеток и формата анализа ELISA.
Материалы и оборудование
1. Реагенты:
Life Sciences
РВ006-С
2. Оборудование и поставщики:
3. Процедуры
Процедуры выделения Pan T и получения реагентов (день 0):
1. Среда для роста клеток:
RPMI1640: ATCC, кат. № 30-2001
10% FBS: Gibco, кат. № 10099141
1% Pen-Strep: Gibco, кат. № 15140122
1% не незаменимых аминокислот: Gibco, кат. № 11140050
Бета-меркаптоэтанол: Gibco, кат. № 21985023
2. Получение буфера для выделения Pan T:
Получают раствор, содержащий забуференный фосфатом физиологический раствор (PBS), pH 7,2, 0,5% бычьего сывороточного альбумина (BSA) и 2 мМ ЭДТК разведением исходного раствора MACS® BSA (# 130-091-376) в 20 раз промывочным раствором autoMACS®. Хранят буфер при 4°C. Дегазируют буфер перед использованием, так как пузырьки воздуха могут заблокировать колонку.
3. Размораживание замороженных РВМС
1) Предварительно нагревают среду на водяной бане при 37°C.
2) Быстро размораживают клетки на водяной бане при 37°C.
3) Добавляют предварительно нагретую среду в 15 мл пробирку. Переносят клетки в пробирку.
4) Центрифугируют 300×g в течение 8 мин. (центрифуга увеличение 0, уменьшение 0).
5) Промывают РВМС, используя промывочный буфер.
6) Центрифугируют РВМС при 300×g в течение 8 мин, дважды промывают (центрифуга увеличение 9, уменьшение 1).
7) Ресуспендируют клетки в буфере для выделения клеток Pan T и подсчет количества клеток.
4. Выделение pan Т-клеток:
Окрашивание клеток коктейлем микрогранул
1) Готовят клетки и определяют количество клеток. Фильтруют клетки на 70 мкм клеточном фильтре.
2) Ресуспендируют клеточный дебрис в 40 мкл буфера на 107 всего клеток.
3) Добавляют 10 мкл коктейля Pan T Cell Biotin-Antibody Cocktail на 107 всего клеток.
4) Хорошо перемешивают и инкубируют 10 минут в холодильнике (лед).
5) Добавляют 30 мкл буфера на 107 всего клеток.
6) Добавляют 20 мкл коктейля Pan T Cell MicroBead на 107 всего клеток.
7) Хорошо перемешивают и инкубируют 15 минут в холодильнике (лед).
8) Приступают к последующему магнитному разделению клеток.
Примечание: а. Работайте быстро, сохраняйте клетки холодными и используйте предварительно охлажденные растворы (2-8°C). b. Объемы для магнитного мечения даны для вплоть до 107 всего клеток. При работе с меньшим количеством клеток используйте указанные объемы. с. При работе с большим количеством клеток увеличьте соответственно все объемы реагентов и общие объемы. d. Для оптимальной работы важно получить одноклеточную суспензию перед магнитным мечением.
Последующее ручное разделение клеток:
1) Помещают колонку LS в магнитное поле подходящего сепаратора MACS. Для получения подробной информации см. соответствующее техническое описание колонки MACS.
2) Готовят колонку промыванием 3 мл буфера.
3) Наносят суспензию клеток на колонку. Собирают протекающую жидкость, которая представляет собой обогащенные Т-клетки.
4) Промывают колонку 5 мл буфера. Собирают не меченые клетки, которые проходят сквозь, представляющие собой обогащенные Т-клетки.
Напоминание: Всегда ждите, пока резервуар колонки не опустеет, прежде чем переходить к следующей стадии.
5. FACS Pan T клеток:
1) Помещают 50 мкл PBMC и Pan T клеток в пробирки FACS соответственно.
2) Инкубируют клетки с анти-человеческими CD3/CD4/CD8 антителами (1 мкл/1 мкл/1 мкл/пробирка) в течение 20 мин при 4°C. Для неокрашенного контроля инкубируют клетки с буфером для окрашивания FACS.
3) Промывают 2 раза буфером для холодного окрашивания (PBS с 0,2% BSA и 1 мМ ЭДТК).
4) Запускают FACS. Гейт CD3+, CD3+CD4+ и CD3+CD8+ популяция для % анализа.
5) Если чистота Pan T клеток выше 90%, разводят клеточную суспензию до 1 миллиона клеток/мл соответствующим объемом среды для культивирования клеток.
6) Переливают клеточную суспензию в стерильный одноразовый резервуар для использования в будущем.
Процедуры получения соединений и анти-человеческих CD3/CD28 (день 0):
1. Получение соединения
Серийное разведение соединения (исходный планшет 1000×)
Соединения растворяют в 100% ДМСО до концентрации 10 мМ. Затем их серийно разводят в 3 раза до 8-точечных доз.
Приготовление 4× дозы соединения (промежуточный планшет: Corning-3599):
Готовят 4× раствор соединения в культуральной среде. Пипетируют вверх и вниз. Для контроля ZPE готовят 0,4% ДМСО (4×) в культуральной среде. Для контроля HPE готовят 0,4 мкМ RGT003-026 (4×) в культуральной среде.
2. Анти-человеческий CD3 (исходная конц. 6,76 мг/мл): хранят при 4°C.
1) Разбавляют анти-человеческий CD3 PBS до конечной конц. 0,5 мкг/мл.
2) Добавляют 50 мкл/ лунку CD3 в каждую лунку, кроме лунок с положительным и отрицательным контролем. Лунки с положительным и отрицательным контролем не стимулируют CD3/CD28.
3) Инкубируют при 37°С в инкубаторе с 5% CO2 в течение 2 часов.
4) Удаляют 50 мкл раствора антитела из планшета для культивирования клеток. Промывают каждую лунку дважды 200 мкл стерильного PBS каждый раз.
3. Получение анти-человеческого CD28 (4×)
Антитело разводили культуральной средой от исходной концентрации 11,07 мг/мл до 2 мкг/мл (4×).
4. Получение PMA/Иномицина (4×)
1) Разбавляют PMA до 400 нг/мл (8×) в среде.
2) Разводят иномицин до 8 мкМ (8×) в среде.
3) Смешивают равный объем РМА с иономицином с получением 4× смесь.
Процедуры стимуляции клеток (День 0)
1. Переносят 1×105 клеток/лунку (100 мкл/лунку) клеточной суспензии в 96-луночный планшет (планшет для клеток: greiner-655180).
2. Добавляют 50 мкл/лунку анти-человеческого CD28 для тестируемых соединений и контролей ZPE/HPE. Для положительного или отрицательного контроля добавляют 50 мкл/лунку 4x раствора PMA/Иномицина или культуральной среды, соответственно.
3. Добавляют 50 мкл/лунку соединений в планшет для клеток в соответствии с картой планшета, показанной ниже. Для контроля ZPE/HPE добавляют 50 мкл/лунку 0,4% раствора ДМСО или 0,4 мкМ RGT003-026 (4×), соответственно. Для положительного или отрицательного контроля добавляют 50 мкл/лунку культуральной среды.
4. Инкубируют планшет в течение 48 часов.
Процедуры для IL-2 и IFN-γ ELISA:
День 1: Покрывание планшетов:
1) Покрывают микролунки 100 мкл на лунку захватывающего антитела IL-2 и IFN-γ, разведенного в буфере для покрытия. Информацию о рекомендуемом разведении покрытия антитела см. в инструкции/сертификате анализа для конкретной партии. Герметично закрывают планшет и инкубируют в течение ночи при 4°C.
День 2: Сбор образцов:
1) После инкубации в инкубаторе при 37°С, 5% CO2 в течение 48 часов центрифугируют клеточные планшеты при 1000 об/мин в течение 10 мин. Собирают 100 мкл/лунку супернатанта, и затем проводят анализ IL-2 и IFN-γ ELISA. Супернатант может храниться при -80°C, и анализ IL-2 и IFN-γ ELISA можно провести на следующий день. Может потребоваться разведение супернатанта в 30-40 раз, чтобы анализ не превышал линейный диапазон стандартной кривой IL-2 и IFN-γ.
2) Добавляют новую среду (содержащую анти-CD28, P/I и соединения) в планшет, 100 мкл/лунку.
3) Карта планшета:
День 3-4: IL-2 & IFN-γ ELISA:
1) Отсасывают лунки и промывают 3 раза ≥ 300 мкл/лунку промывочным буфером. После последней промывки переворачивают планшет и промокают на впитывающей бумаге, чтобы удалить остатки буфера.
2) Блокируют планшеты с ≥ 200 мкл/лунку разбавителя для анализа. Инкубируют при КТ в течение 1 часа.
3) Отсасывают/промывают, как на стадии 2.
4) Получают стандарты в разбавителе для анализа.
Получение стандартного исходного IL-2:
добавляют 1 мл деионизированной воды во флакон (235 нг/флакон), исходная конц. составляет 235 нг/мл. Аликвотируют стандартный исходный раствор по 10 мкл/флакон, замораживают при -80°C.
Получение стандартного исходного IFN-γ:
добавляют 1 мл деионизированной воды во флакон (145 нг/флакон), исходная конц. составляет 145 нг/мл. Аликвотируют стандартный исходный раствор по 10 мкл/флакон, замораживают при -80°С.
Получение стандартной кривой для IL-2/IFNγ:
Разводят стандартный образец до максимальной концентрации 500 пг/мл. Затем проводят 2-кратное серийное разведение до 10-точечных доз (включая пустой контроль). Переносят различные концентрации стандартов на планшет для ELISA, 100 мкл/лунку.
1) Пипетируют 100 мкл каждого стандарта, образца и контроля в соответствующие лунки. Герметично закрывают планшет и инкубируют в течение 2 часов при КТ.
2) Отсасывают/промывают, как на стадии 2, всего 5 промывок.
3) Добавляют 100 мкл рабочего детектора (определяющее антитело+реагент SAv-HRP) в каждую лунку. Герметично закрывают планшет и инкубируют в течение 1 часа при КТ.
4) Отсасывают/промывают, как на стадии 2, всего 7 промывок. ПРИМЕЧАНИЕ. На этом последнем этапе промывки замочите лунки в промывочном буфере на время от 30 секунд до 1 минуты для каждой промывки.
5) Добавляют в каждую лунку по 100 мкл раствора субстрата. Инкубируют планшет (без герметика) в течение 30 минут при комнатной температуре в темноте.
6) Добавляют в каждую лунку по 50 мкл останавливающего раствора.
7) Измеряют оптическую плотность при 450 нм в течение 30 минут после остановки реакции. ОП 450 нм нормализуют к значению ОП при 570 нм.
4. Анализ данных:
Для биохимических анализов долю (%) ингибирования при каждой концентрации соединения рассчитывают на основании и относительно сигнала в лунках HPE и ZPE, содержащихся в каждом планшете для анализа. Лунки HPE считают эффективными на 100%, и лунки ZPE, которые не содержат никакого соединения, а содержат ДМСО (конечная концентрация=0,1%), считают эффективными на 0%.
Для клеточного анализа рассчитывают ответ IL-2 или IFN- γ (данные не показаны) на контроль ДМСО при каждой концентрации соединения. См. Таблицу 2. EC2x является концентрацией соединения, которая дает 200% ответ (в 2 раза), и EC50 рассчитывают с помощью GraphPad Prism.
Биохимические и клеточные данные, полученные для типовых соединений, перечислены в таблицах 1 и 2 ниже.
Таблица 1
Н.О.: не определено.
B*: Тестирование проводят в первом устройстве для тестирования. N*: Тестирование проводят во втором устройстве для тестирования.
Таблица 2
Н.Д.: не доступно.
В таблицах 1 и 2, значения IC50, EC2x и EC50 обозначены как «++++», для значений менее или равных 100 нМ; «+++», для значений выше, чем «++++», но меньше или равных 500 нМ; «++», для значений выше, чем «+++», но меньше или равных 1 мкМ; и «+», для более 1 мкМ, соответственно.
B. Примеры синтеза
Описание оборудования
Спектры ЯМР измеряют на спектрометре Varian Mercury, работающем на частоте 400 МГц (1H), 376 МГц (19F) или 75 МГц (13C). Растворители, используемые для образцов, указаны в экспериментальных процедурах для каждого соединения. Химические сдвиги выражаются в частях на миллион (ч./млн., δ единицы). Константы сочетания указаны в герцах (Гц). Паттерны расщепления описывают кажущуюся мультиплетность и обозначаются как с (синглет), д (дублет), т (триплет), кв (квартет), квинт (квинтет), м (мультиплет), ш (широкий).
Для ЖХ-МС используют следующую систему: Agilent 6120 (насос Binary Gradient Module), аналитическую колонку XBridge C18, 5 мкм, 4,6 × 50 мм, 25ºC, вводимый объем 5 мкл, 2 мл/мин, с градиентом ацетонитрила в водном растворе 0,1% ацетата аммония в соответствии со следующими сроками:
Экспериментальные процедуры: Все реакции проводят в атмосфере сухого азота, если не указано иное. ТСХ пластины визуализируют в у.ф. свете. Флэш-хроматография относится к колоночной хроматографии на силикагеле (40-60 мкм) с использованием стеклянных колонок. Альтернативно, автоматическую хроматографию проводят с использованием систем Biotage SP1 или Biotage Isolera с УФ-определением при 220 или 254 нм и с использованием картриджей с силикагелем Biotage с нормальной или обращенной фазой. Более подробную информацию можно найти в соответствующей экспериментальной процедуре.
Общие способы и препараты
Соединения, включая соединения общей формулы (I-0), (I-1), (I-2), (I), (II), (II-1), (II-2) или (II-2'), промежуточные соединения и конкретные примеры получают описанными в настоящем документе способами синтеза. В экспериментальных процедурах, модификации условий реакции, таких как температура, концентрация растворов, объем растворителей, применение микроволновых условий, продолжительность реакции или их комбинации, предусмотрены как часть настоящего изобретения и, помимо специально упомянутых кислот, оснований, реагентов, реагентов сочетания, растворителей и т.д., могут быть использованы альтернативные подходящие кислоты, основания, реагенты, реагенты сочетания, растворители и т.д., и они включены в объем настоящего изобретения. Все возможные геометрические изомеры, стереоизомеры, формы солей входят в объем настоящего изобретения.
Общая схема и методики синтеза соединений пиримидина:
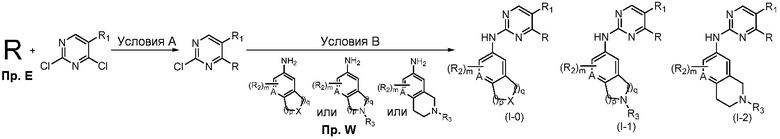
Условия A1: К раствору Пр. E (или коммерчески доступного реагента) (1,0 экв.) в ДМФ добавляют гидрид натрия (60% дисперсию в минеральном масле, 1,2 экв.) при 0°С и смесь перемешивают при 0°С в течение 30 минут. К полученной выше смеси добавляют 5-R1 замещенный 2,4-дихлорпиримидин (1,0-1,3 экв.) при 0°С и полученную смесь затем перемешивают при комнатной температуре в течение 1 часа, охлаждают до 0°С и гасят льдом-H2O. Неочищенный продукт очищают с получением 4-R и 5-R1 замещенного 2-Cl-пиримидина.
Условия A2: К раствору 5-R1 замещенного 2,4-дихлорпиримидина (1,0-1,5 экв.) в ДМФ добавляют карбонат калия (2,0 экв.) и Пр. E (или коммерчески доступного реагента) (1,0 экв.). Смесь перемешивают при 80°C в течение ночи, охлаждают до комнатной температуры, разбавляют H2O и экстрагируют ЭА. Неочищенный продукт очищают с получением 4-R и 5-R1 замещенного 2-Cl-пиримидина.
Условия A3: К раствору Пр. E (или коммерчески доступного реагента) (1,0 экв.) в н-бутаноле добавляют 5-R1 замещенный 2,4-дихлорпиримидин (1,0-1,5 экв.) и ДИПЭА (1,0-2,0 экв.) последовательно. Реакционную смесь перемешивают при 100°C в течение 18 часов, разбавляют водой и экстрагируют ЭА. Неочищенный продукт очищают с получением 4-R и 5-R1 замещенного 2-Cl-пиримидина.
Условия B1: К раствору Пр. W (1,0-1,4 экв.) в 2-метоксиэтаноле добавляют 4-R и 5-R1 замещенный 2-Cl-пиримидин (1,0 экв.) и HCl в растворе EtOH (3,0 экв.). Полученную смесь перемешивают при 120°С в течение 20 часов и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле и/или преп-ЖХВД.
Условия B2: К раствору 4-R и 5-R1 замещенного 2-Cl-пиримидина (1,0 экв.) и Пр. W (1-1,3 экв.) в изопропаноле добавляют трифторуксусную кислоту (1,0-3,0 экв.). Смесь перемешивают под N2 при 100°С в течение 20 часов, охлаждают до комнатной температуры, подщелачивают насыщенным NaHCO3 водным раствором до pH ~8, экстрагируют ДХМ. Органические фазы промывают насыщенным раствором соли, сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле и/или преп-ЖХВД.
Условия B3: К раствору 4-R и 5-R1 замещенного 2-Cl-пиримидина (1,0 экв.) и Пр. W (1-1,3 экв.) в изопропаноле добавляют TsOH.H2O (1,0-1,2 экв.). Смесь перемешивают при 100°C в течение ночи, охлаждают до комнатной температуры, подщелачивают насыщенным NaHCO3 водным раствором до pH ~8, экстрагируют ДХМ. Органические фазы промывают насыщенным раствором соли, сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле, и/или преп-ЖХВД.
Условия B4: К раствору 4-R и 5-R1 замещенного 2-Cl-пиримидина (1,0 экв.) в диоксане добавляют Пр. W (1,0-1,2 экв.), Pd2(dba)3 (0,2 экв.), Xantphos (0,2 экв.) и карбонат калия (2,0 экв.). Смесь перемешивают при 100°C под N2 в течение 12 часов, охлаждают до комнатной температуры и фильтруют. Остаток очищают колоночной флэш-хроматографией на силикагеле, и/или преп-ЖХВД.
Существуют дополнительные превращение 4-R и 5-R1 замещенного 2-Cl-пиримидина между условиями A и B и дополнительные превращения после условий B, в зависимости от конкретных примеров.
Промежуточные соединения
Пр. W1
6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
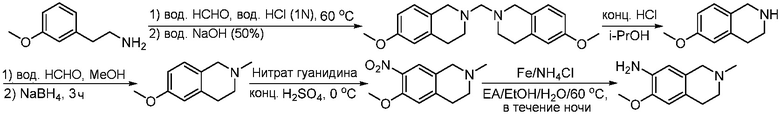
Стадия 1: К смеси 2-(3-метоксифенил)этанамина (20,0 г, 132,27 ммоль, 19,23 мл) в водном растворе HCl (1 N, 192 мл, 192 ммоль) добавляют водный раствор формальдегида (37% масс., 41,64 г, 529,08 ммоль). Смесь перемешивают при 60°С в течение 1 часа, охлаждают до 0°С и подщелачивают добавлением по каплям 50% водного раствора NaOH (17,44 г, 218 ммоль) при 0°С. Полученную смесь перемешивают при комнатной температуре в течение ночи и фильтруют. Фильтрат концентрируют с получением бис(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метана в виде белого твердого вещества (23 г, 100% выход).
Стадия 2: К суспензии бис(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метана (28 г, 82,73 ммоль) в i-PrOH (200 мл) добавляют по каплям концентрированную HCl (15,20 г, 182,01 ммоль, 15,3 мл) при 0°С и смесь перемешивают при комнатной температуре в течение 18 часов. МТБЭ (70 мл) добавляют в вышеуказанную смесь и суспензию перемешивают при комнатной температуре в течение еще 4 часов. После фильтрации, лепешку промывают смесью МТБЭ/i-PrOH (100 мл, 1/1 об/об) и сушат с получением 6-метокси-1,2,3,4-тетрагидроизохинолина гидрохлорида в виде белого твердого вещества, которое суспендируют в ДХМ (300 мл). К смеси добавляют насыщенный водный раствор NaHCO3 (500 мл) и смесь перемешивают при комнатной температуре в течение 2 часов. После разделения, водный слой экстрагируют ДХМ (50 мл x 4). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют с получением 6-метокси-1,2,3,4-тетрагидроизохинолина в виде желтого масла (10 г, 75% выход). ЖХ-МС (ИЭР) m/z: 164 [M+H]+.
Стадия 3: К раствору 6-метокси-1,2,3,4-тетрагидроизохинолина (1,63 г, 9,99 ммоль) в MeOH (30 мл) добавляют водный формальдегид (37% масс./масс., 4,8 г, 59,92 ммоль, 1,67 мл) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 15 минут и охлаждают до 0°С, туда добавляют NaBH4 (1,13 г, 29,96 ммоль) порциями. Смесь перемешивают при комнатной температуре в течение 3 часов, гасят льдом-водой (10 мл) и экстрагируют ДХМ (20 мл x 5). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1 об/об) с получением 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолина в виде желтого масла (1,7 г, 96% выход). ЖХ-МС (ИЭР) m/z: 178 [M+H]+.
Стадия 4: К предварительно охлажденному раствору (0°С) концентрированной серной кислоты (4 мл) последовательно добавляют 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин (2 г, 11,28 ммоль) и нитрат гуанидина (1,17 г, 9,59 ммоль) при 0°C. Полученную смесь перемешивают при 0°C в течение 30 минут, гасят льдом-водой (20 мл), подщелачивают до pH 10-11 водным раствором NaOH (4 N) и экстрагируют ДХМ (50 мл x 4). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ 100% об/об, ПЭ/ЭА=1/1 об/об, ЭА 100% об/об и затем ДХМ/MeOH=20/1, об/об) с получением 6-метокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина в виде желтого твердого вещества (0,9 г, 36% выход). ЖХ-МС (ИЭР) m/z: 223 [M+H]+.
Стадия 5: К раствору 6-метокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина (4,2 г, 14,40 ммоль) в ЭА (32 мл), H2O (16 мл) и EtOH (144 мл) добавляют порошок Fe (5,5 г, 93,99 ммоль) и хлорид аммония (793,96 мг, 13,08 ммоль). Смесь перемешивают при 60°C в течение 48 часов, охлаждают до комнатной температуры и фильтруют. Лепешку промывают метанолом (30 мл x 3) и фильтрат концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1-10/1 об/об) с получением 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина гидрохлорида в виде желтого твердого вещества (4,5 г, 100% выход). ЖХ-МС (ИЭР) m/z: 193 [M+H]+.
Альтернативный способ синтеза:
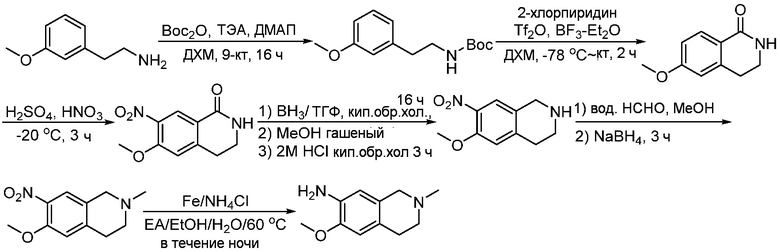
Стадия 1: К раствору триэтиламина (13,38 г, 132,27 ммоль, 18,44 мл), 2-(3-метоксифенил)этанамина (10 г, 66,13 ммоль) и ДМАП (807,96 мг, 6,61 ммоль) в ДХМ (100 мл) добавляют Boc2O (15,88 г, 72,75 ммоль, 16,70 мл) медленно при 0°C. Смесь затем перемешивают при комнатной температуре в течение 16 часов, разбавляют ледяной водой (200 мл) и экстрагируют EtOAc (3×200 мл). Объединенную органическую фазу промывают насыщенным раствором соли (100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (SiO2, петролейный эфир/этил ацетат=10/1) с получением трет-бутил (3-метоксифенэтил)карбамата (14,6 г, 79,06% выход, 90% чистота) в виде бесцветной жидкости. ЖХ-МС (ИЭР) m/z: 196 [M+H-56]+.
Стадия 2: К раствору трет-бутил (3-метоксифенэтил)карбамата (7 г, 27,85 ммоль) и 2-хлорпиридина (4,74 г, 41,78 ммоль, 3,92 мл) в CH2Cl2 (50 мл) добавляют раствор Tf2O (8,64 г, 30,64 ммоль, 5,15 мл) в CH2Cl2 (5 мл) при -78 ºC. Через 20 минут, BH3.Et2O (19,77 г, 139,26 ммоль) добавляют по каплям к вышеуказанному раствору. Реакционную смесь затем нагревают до комнатной температуры, перемешивают в течение 2 часов и осторожно гасят насыщенным раствором NaHCO3. Полученную смесь экстрагируют CH2Cl2 (3×100 мл). Объединенный органический слой промывают насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (SiO2, петролейный эфир/этил ацетат/метанол=5/1/0 to 10/10/1) с получением 6-метокси-3,4-дигидроизохинолин-1(2H)-она (3,4 г, 68,89% выход) в виде беловатого твердого вещества. ЖХ-МС (ИЭР) m/z: 178 [M+H]+.
Стадия 3: К раствору 6-метокси-3,4-дигидроизохинолин-1(2H)-она (3,8 г, 21,44 ммоль, 9,62 мл) в концентрированной H2SO4 (50 мл) добавляют HNO3 (2,16 г, 22,29 ммоль, 65% чистота) по каплям при -20°C. Смесь перемешивают при -20°C ~-25°C в течение 3 часов и выливают в ледяную воду (300 мл). Водный слой экстрагируют дихлорметаном (3×200 мл). Объединенные органические слои концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией (SiO2, Петролейный эфир/этил ацетат/метанол=10/1/0~10/10/1) с получением 6-метокси-7-нитро-3,4-дигидроизохинолин-1(2H)-она (2,02 г, 40,78% выход). ЖХ-МС (ИЭР) m/z: 223 [M+H]+.
Стадия 4: К раствору 6-метокси-7-нитро-3,4-дигидроизохинолин-1(2H)-она (2,02 г, 9,09 ммоль) в ТГФ (200 мл) добавляют 1M BH3/ТГФ (45,46 ммоль, 45,5 мл). Смесь перемешивают при кипении с обратным холодильником в течение 20 часов и осторожно гасят метанолом (30 мл). Полученный раствор концентрируют в вакууме. Остаток нагревают в 2N HCl (50 мл) при 80°C в течение 3 часов, охлаждают, подщелачивают водным гидроксидом аммония и экстрагируют дихлорметаном (3×100 мл). Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением 6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (1,89 г, 100,00% выход). ЖХ-МС (ИЭР) m/z: 209 [M+H]+.
Стадия 5: К раствору 6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (1,89 г, 9,08 ммоль, 9,62 мл) в метаноле (30 мл) добавляют формальдегид (1,64 г, 54,46 ммоль, 1,51 мл) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 15 минут и охлаждают до 0°С. Затем NaBH4 (1,03 г, 27,23 ммоль) добавляют к вышеуказанной смеси порциями. Полученную смесь перемешивают при комнатной температуре в течение 3 часов, гасят льдом-водой (10 мл) и экстрагируют ДХМ (20 мл x 5). Объединенный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1, об/об) с получением 6-метокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина (1,4 г, 69,40% выход) в виде желтого масла. ЖХ-МС (ИЭР) m/z: 223 [M+H]+.
Стадия 6: К раствору 6-метокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина (1,3 г, 5,85 ммоль) в EtOAc (2 мл), H2O (1 мл) и EtOH (10 мл) добавляют Fe (2,19 г, 39,19 ммоль) и хлорид аммония (284,74 мг, 5,32 ммоль). Смесь перемешивают при 60°C в течение 16 часов, охлаждают до комнатной температуры и фильтруют. Лепешку промывают метанол (30 мл x 3) и фильтрат концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1 to 10/1 об/об) с получением 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина гидрохлорида в виде желтого твердого вещества (0,85 г, 75,6% выход). ЖХ-МС (ИЭР) m/z: 193 [M+H]+.
Пр. W2
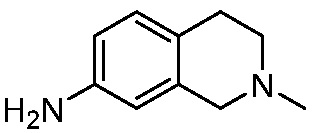
2-Метил-1,2,3,4-тетрагидроизохинолин-7-амин
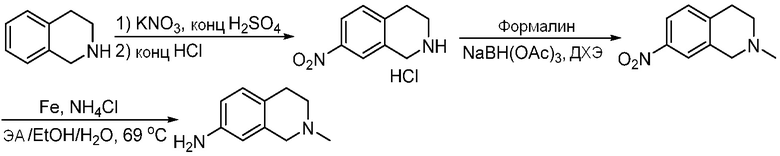
Стадия 1: К ледяному раствору 1,2,3,4-тетрагидроизохинолина (4,8 г, 36,04 ммоль) в концентрированной серной кислоте (20 мл) добавляют нитрат калия (4,01 г, 47,21 ммоль) небольшими порциями, сохраняя температуру ниже 5°C. Реакционную смесь перемешивают при комнатной температуре в течение ночи, выливают в лед (100 г), доводят до pH ~9 с NH3.H2O и экстрагируют CH2Cl2 (50 мл x 3). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток кристаллизуют в метаноле с получением 7-нитро-1,2,3,4-тетрагидроизохинолина гидрохлорида в виде желтого твердого вещества (4,2 г, 54% выход). ЖХ-МС (ИЭР) m/: 179 [M+H]+.
Стадия 2: К перемешиваемой смеси 7-нитро-1,2,3,4-тетрагидроизохинолина гидрохлорида (5,0 г, 23,29 ммоль) в сухом 1,2-дихлорэтанe (200 мл) добавляют формалин (37% водный формальдегид, 2,0 мл, 25,86 ммоль), затем триацетоксиборгидрид (19,81 г, 104,82 ммоль). Реакционную смесь энергично перемешивают при комнатной температуре в течение 4 часов и концентрируют. Остаток разбавляют EtOAc (400 мл), медленно гасят насыщенный водный раствор NaHCO3 (400 мл) и перемешивают в течение 30 минут. Смесь экстрагируют ЭА (100 мл x 4). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением 2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина в виде коричневого твердого вещества (3,8 г, 85% выход). ЖХ-МС (ИЭР) m/: 193 [M+H]+.
Стадия 3: Суспензию 2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина (3,8 г, 19,77 ммоль), Fe (5,52 г, 98,85 ммоль), NH4Cl (15,86 г, 296,54 ммоль) в ЭА (10 мл), EtOH (100 мл) и H2O (20 мл) перемешивают энергично при 70°C в течение 24 часов. После фильтрации, лепешку промывают смешанным растворителем ЭА/EtOH (100 мл, 10/1, об/об) и фильтрат концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH в ДХМ, от 0% до 25% об/об) с получением 2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (1,84 г, 57% выход). ЖХ-МС (ИЭР) m/z: 163 [M+H]+.
Пр. W3
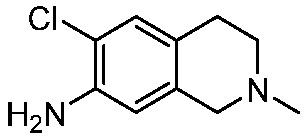
6-Хлор-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
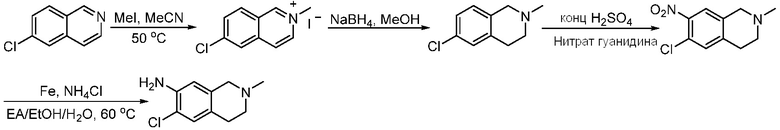
Стадия 1: К раствору 6-хлоризохинолина (2,0 г, 12,22 ммоль) в MeCN (15 мл) добавляют MeI (5,21 г, 36,67 ммоль) при комнатной температуре. Смесь перемешивают при 50°С в герметично закрытой пробирке в течение 2 дней. После охлаждения до комнатной температуры, твердое вещество собирают фильтрацией, сушат с получением 6-хлор-2-метилизохинолин-2-ия йодида в виде желтого твердого вещества (3,2 г, 86% выход). ЖХ-МС (ИЭР) m/z: 178 [M]+.
Стадия 2: К раствору 6-хлор-2-метилизохинолин-2-ия йодида (200 мг, 0,65 ммоль) в MeOH (8 мл) добавляют NaBH4 (127,06 мг, 3,36 ммоль) порциями при 0°С. Смесь перемешивают при комнатной температуре в течение 3 часов, доводят до pH ~9 насыщенным водным раствором NaHCO3 и экстрагируют ДХМ (30 мл x 3). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают преп-ТСХ (ДХМ/MeOH=20/1, об/об) с получением 6-хлор-2-метил-1,2,3,4-тетрагидроизохинолина в виде желтого твердого вещества (90 мг, 44% выход). ЖХ-МС (ИЭР) m/z: 182 [M+H]+.
Стадии 3&4: Процедуры те же, что и для Стадии 4&5 of Пр. W1, с применением 6-хлор-2-метил-1,2,3,4-тетрагидроизохинолина вместо 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолина. ЖХ-МС (ИЭР) m/z: 197 [M+H]+.
Пр. W4
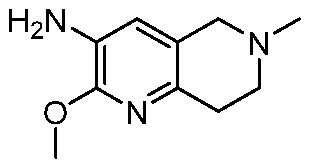
2-Метокси-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амин
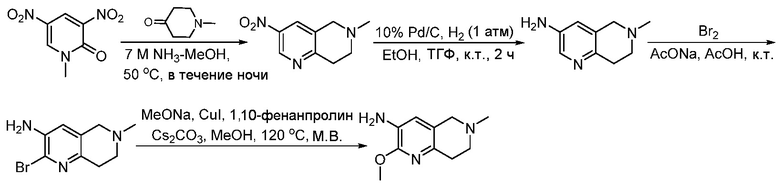
Стадия 1: Раствор 1-метил-3,5-динитро-пиридин-2-она (4,5 г, 22,60 ммоль) и 1-метилпиперидин-4-она (2,81 г, 24,86 ммоль) в NH3/MeOH (7 N, 50 мл) перемешивают в герметично закрытой пробирке при 50°C в течение ночи. После охлаждения до комнатной температуры, смесь концентрируют и остаток разделяют между ДХМ (100 мл) и насыщенным водным раствором NaHCO3 (50 мл). Слои разделяют, и водный слой экстрагируют ДХМ (50 мл x 2). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют с получением 6-метил-3-нитро-5,6,7,8-тетрагидро-1,6-нафтиридина в виде красного твердого вещества (2,84 г, 65% выход). ЖХ-МС (ИЭР) m/z: 194 [M+H]+.
Стадия 2: К раствору 6-метил-3-нитро-7,8-дигидро-5H-1,6-нафтиридина (1,3 г, 6,73 ммоль) в этаноле (35 мл) и ТГФ (35 мл) добавляют 10% Pd/C (817,20 мг) и смесь перемешивают под H2 (1 атм) в течение 2 часов. После фильтрации, лепешку промывают EtOH (5 мл x 2) и фильтрат концентрируют с получением 6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амина в виде желтого твердого вещества (1,1 г, 100% выход). ЖХ-МС (ИЭР) m/z: 164 [M+H]+.
Стадия 3: К раствору 6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амина (1,2 г, 7,35 ммоль) и AcONa (1,21 г, 14,70 ммоль) в уксусной кислоте (25 мл) добавляют бром (1,17 г, 7,35 ммоль) при комнатной температуре, и смесь перемешивают при комнатной температуре в течение 1 часа. Смесь разбавляют ДХМ (100 мл) и водой (30 мл) и медленно доводят до pH ~9 с NaHCO3. Органический слой сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1, об/об) с получением 2-бром-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амина в виде желтого твердого вещества (570 мг, 32%). ЖХ-МС (ИЭР) m/z: 242 [M+H].
Стадия 4: К смеси 2-бром-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амина (250 мг, 1,03 ммоль), карбоната цезия (672,86 мг, 2,07 ммоль) и MeONa (83,67 мг, 1,55 ммоль) в MeOH (2 мл) добавляют CuI (19,67 мг, 103,26 мкмоль) и 1,10-фенантролина моногидрат (40,94 мг, 206,51 мкмоль). Смесь перемешивают в условиях микроволн, защищенных N2 при 120°С в течение 2 часов. После охлаждения до комнатной температуры, остаток очищают преп-ТСХ (ДХМ/MeOH=15/1, об/об) с получением 2-метокси-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амина в виде желтого твердого вещества (74 мг, 37% выход). ЖХ-МС (ИЭР) m/z: 194 [M+H]+.
Пр. W5
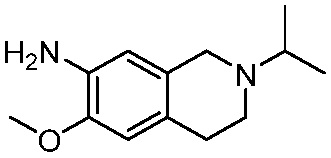
2-Изопропил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амин
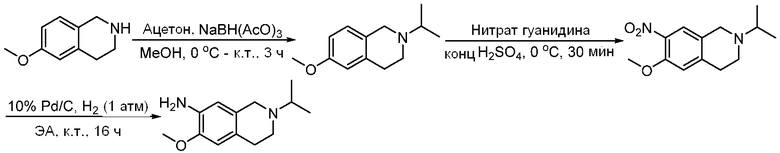
Стадия 1: К смеси 6-метокси-1,2,3,4-тетрагидроизохинолина (340 мг, 2,08 ммоль) в MeOH (10 мл) добавляют по каплям ацетон (725,93 мг, 12,50 ммоль, 917,74 мкл) при комнатной температуре и смесь перемешивают при комнатной температуре в течение 30 минут. Затем NaBH(AcO)3 (6,25 ммоль) добавляют порциями к вышеуказанной смеси при 0°C и полученную смесь перемешивают при комнатной температуре в течение 3 часов, гасят насыщенный водный раствор NaHCO3 (10 мл) при 0°C и концентрируют. Остаток экстрагируют ДХМ (50 мл x 2) и объединенные органические слои промывают насыщенным раствором соли (20 мл x 2), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1, об/об) с получением 2-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолина в виде белого твердого вещества (150 мг, 35% выход). ЖХ-МС (ИЭР) m/z: 206 [M+H]+.
Стадия 2: Процедуры те же, что и для Стадии 4 Пр. W1, с применением 2-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолина вместо 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолина. ЖХ-МС (ИЭР) m/z: 251 [M+H]+.
Стадия 3: Смесь 2-изопропил-6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (200 мг, 799,06 мкмоль) и 10% палладия на угле (30 мг, 799,06 мкмоль) в ЭА (6 мл) перемешивают под H2 (1 атм) при комнатной температуре в течение 16 часов и фильтруют. Фильтрат концентрируют и остаток очищают преп-ТСХ (ДХМ/MeOH=10/1, об/об) с получением 2-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амина в виде белого твердого вещества (50 мг, 28% выход). ЖХ-МС (ИЭР) m/z: 221 [M+H]+.
Пр. W6
трет-Бутил 7-амино-6-метокси-3,4-дигидроизохинолин-2(1H)-карбоксилат
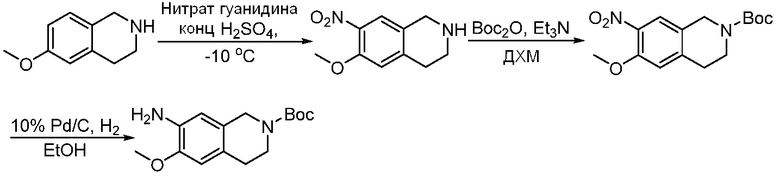
Стадия 1: к холодному раствору концентрированной H2SO4 (5 мл) добавляют 6-метокси-1,2,3,4-тетрагидроизохинолин (1,0 г, 1,84 ммоль) при -10°С. Нитрат гуанидина (758 мг, 6,13 ммоль) добавляют к вышеуказанной смеси порциями при -10°С. Смесь перемешивают при -10°С в течение 30 минут, выливают в холодную воду (20 мл), доводят до pH ~8 водным раствором NaOH (4 N) и экстрагируют ЭА (40 мл x 4). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ 100% об/об и затем ДХМ/MeOH=10/1, об/об) с получением 6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина в виде желтого твердого вещества (200 мг, 16% выход). ЖХ-МС (ИЭР) m/z: 209 [M+H]+.
Стадия 2: Смесь 6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (100 мг, 0,48 ммоль), Boc2O (157 мг, 0,72 ммоль) и Et3N (97 мг, 0,96 ммоль) в ДХМ (5 мл) перемешивают при комнатной температуре в течение 4 часов и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ 100% об/об и затем ДХМ/MeOH=10/1, об/об) с получением трет-бутил 6-метокси-7-нитро-3,4-дигидроизохинолин-2(1H)-карбоксилата в виде желтого масла (150 мг, 100% выход). ЖХ-МС (ИЭР) m/z: 209 [M-100+1]+.
Стадия 3: Смесь трет-бутил 6-метокси-7-нитро-3,4-дигидроизохинолин-2(1H)-карбоксилата (150 мг, 0,48 ммоль) и 10% Pd/C (50 мг) в EtOH (5 мл) перемешивают под H2 (1 атм) при комнатной температуре в течение 16 часов и фильтруют. Фильтрат концентрируют с получением трет-бутил 7-амино-6-метокси-3,4-дигидроизохинолин-2(1H)-карбоксилата в виде желтого твердого вещества (110 мг, 81% выход). ЖХ-МС (ИЭР) m/z: 223 [M-55]+.
Пр. W7
6-Фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
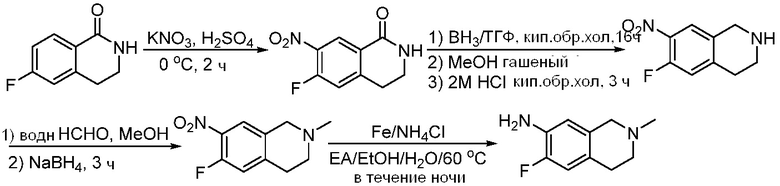
Стадия 1: К раствору 6-фтор-3,4-дигидро-2H-изохинолин-1-она (900 мг, 5,45 ммоль) в H2SO4 (9 мл) добавляют нитрат калия (578,47 мг, 5,72 ммоль) порциями при 0°C. Полученную смесь перемешивают при 0°С в течение 2 часов и выливают в лед-воду. белое твердое вещество осаждают, собирают фильтрацией и сушат в вакууме с получением 6-фтор-7-нитро-3,4-дигидро-2H-изохинолин-1-она (1,04 г, 86% выход). ЖХ-МС (ИЭР) m/z: 211 [M+H]+.
Стадия 2&3&4: Процедуры те же, что и для Стадий 4&5&6 альтернативного способа синтеза из Пр. W1, с применением 6-фтор-7-нитро-3,4-дигидро-2H-изохинолин-1-она вместо 6-метокси-7-нитро-3,4-дигидро-2H-изохинолин-1-она. ЖХ-МС (ИЭР) m/z: 181 [M+H]+.
Пр. W8
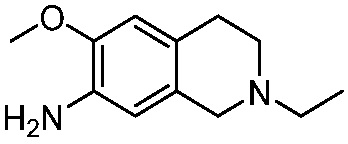
2-Этил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амин

Стадия 1: К раствору 6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (300 мг, 1,44 ммоль) в CH3CN (10 мл) добавляют йодэтан (224,72 мг, 1,44 ммоль, 115,83 мкл) и K2CO3 (796,52 мг, 5,76 ммоль). Реакционную смесь перемешивают при 20°C в течение 16 часов и разделяют между этилацетатом (100 мл) и водой (100 мл). Органический слой отделяют, сушат над безводным сульфатом магния и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (0-20% метанол в дихлорметане с водным гидроксидом аммония) с получением 2-этил-6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (136 мг, 40% выход). ЖХ-МС (ИЭР) m/z: 237 [M+H]+.
Стадия 2: Процедуры те же, что и для Стадии 6 альтернативного способа синтеза из Пр. W1, с применением 2-этил-6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина вместо 2-метил-6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина. ЖХ-МС (ИЭР) m/z: 207 [M+H]+.
Пр. W9
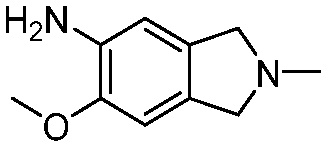
6-Метокси-2-метилизоиндолин-5-амин
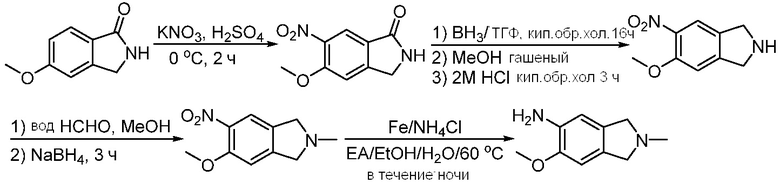
Стадия 1: Процедуры те же, что и для Стадии 1 Пр. W7, с применением 5-метоксиизоиндолин-1-она вместо 6-фтор-3,4-дигидроизохинолин-1(2H)-она. ЖХ-МС (ИЭР) m/z: 209 [M+H]+.
Стадия 2&3&4: Процедуры те же, что и для Стадий 4&5&6 альтернативного способа синтеза из Пр. W1, с применением 5-метокси-6-нитроизоиндолин-1-она вместо 6-метокси-7-нитро-3,4-дигидро-2H-изохинолин-1-она. ЖХ-МС (ИЭР) m/z: 179 [M+H]+.
Пр. W10
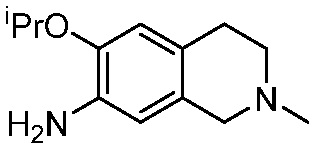
6-Изопропокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
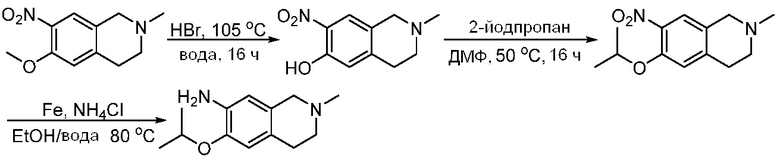
Стадия 1: К раствору 6-метокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина (50 мг, 224,98 мкмоль) в воде (5 мл) добавляют 1M гидробромид (1 мл). Смесь перемешивают при кипении с обратным холодильником в течение 16 часов, подщелачивают до pH 8 насыщенным водным раствором бикарбоната натрия и экстрагируют дихлорметаном/метанолом (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного 2-метил-7-нитро-1,2,3,4-тетрагидроизохинолин-6-ола (46,84 мг, 90% выход). ЖХ-МС (ИЭР) m/z: 209 [M+H]+.
Стадия 2: К раствору 2-метил-7-нитро-1,2,3,4-тетрагидроизохинолин-6-ола (230 мг, 1,10 ммоль) и 2-йодпропана (187,78 мг, 1,10 ммоль, 110,46 мкл) в ДМФ (2 мл) добавляют карбонат калия (229,00 мг, 1,66 ммоль). Смесь перемешивают при комнатной температуре в течение 16 часов и разбавляют насыщенным раствором соли (10 мл). водную фазу отделяют и экстрагируют этилацетатом (20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ: 10~50%) с получением 6-изопропокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина (240 мг, 87% выход). ЖХ-МС (ИЭР) m/z: 251 [M+H]+.
Стадия 3: Процедуры те же, что и для Стадии 6 альтернативного способа синтеза из Пр. W1, с применением 6-изопропокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина вместо 6-метокси-2-метил-7-нитро-1,2,3,4-тетрагидроизохинолина 5-метокси-6-нитроизоиндолин-1-она. ЖХ-МС (ИЭР) m/z: 221 [M+H]+.
Пр. W11
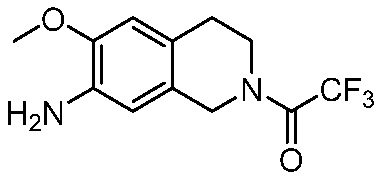
1-(7-Амино-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2,2,2-трифторэтан-1-он
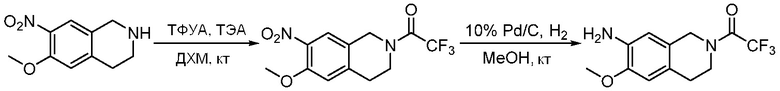
Стадия 1: К раствору 6-метокси-7-нитро-1,2,3,4-тетрагидроизохинолина (908 мг, 4,14 ммоль) в дихлорметане (15 мл) добавляют триэтиламин (838,43 мг, 8,29 ммоль, 1,15 мл) при 0°C, затем добавляют трифторуксусный ангидрид (1,04 г, 4,97 ммоль, 700,78 мкл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, подщелачивают до pH 8 насыщенным водным раствором NaHCO3 и экстрагируют дихлорметаном (10 мл x 3). Объединенную органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного 2,2,2-трифтор-1-(6-метокси-7-нитро-3,4-дигидроизохинолин-2(1H)-ил)этан-1-она в виде желтого твердого вещества (1,26 г, 95% выход). ЖХ-МС (ИЭР) m/z: 305 [M+H]+.
Стадия 2: Смесь 2,2,2-трифтор-1-(6-метокси-7-нитро-3,4-дигидроизохинолин-2(1H)-ил)этан-1-она (1,33 г, 4,14 ммоль) и 10% палладия на угле (400 мг, 3,76 ммоль) в метаноле (30 мл) перемешивают при 25°C в течение 16 часов под атмосферой H2 и фильтруют через целит. Лепешку промывают метанолом (30 мл x 3) и фильтрат концентрируют в вакууме с получением 1-(7-амино-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2,2,2-трифторэтан-1-она в виде светло-желтого твердого вещества (1,14 г, 95% выход). ЖХ-МС (ИЭР) m/z: 275 [M+H]+.
Пр. E1
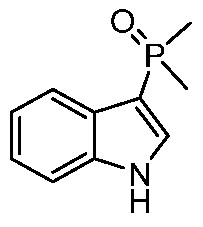
(1H-индол-3-ил)диметилфосфин оксид
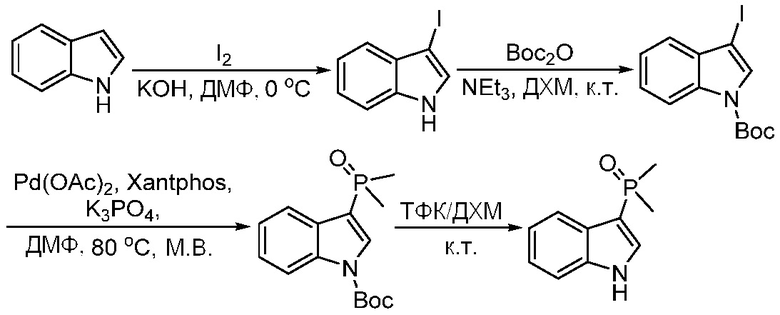
Стадия 1: К смеси 1H-индол (2,34 г, 19,97 ммоль) и KOH (1,34 г, 23,97 ммоль) в ДМФ (40 мл) добавляют по каплям раствор I2 (5,08 г, 19,97 ммоль) в ДМФ (10 мл) при 0°С. Полученную смесь перемешивают при 0°С в течение 30 минут, разбавляют холодной водой (200 мл) и перемешивают в течение еще 30 минут. Твердое вещество собирают фильтрацией, промывают H2O (30 мл x 3) и сушат в вакууме с получением 3-йод-1H-индола в виде желтого твердого вещества (2,58 г, 53% выход).
Стадия 2: К раствору 3-йод-1H-индола (2,4 г, 9,87 ммоль) в ДХМ (15 мл) добавляют триэтиламин (3,00 г, 4,13 мл, 29,62 ммоль), Boc2O (4,31 г, 19,75 ммоль) и ДМАП (120,64 мг, 0,99 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=20/1, об/об) с получением трет-бутил 3-йод-1H-индол-1-карбоксилат в виде коричневого масла (4,6 г, 100% выход).
Стадия 3: Смесь трет-бутил 3-йодиндол-1-карбоксилата (1,0 г, 2,91 ммоль), диметилфосфина оксида (250,19 мг, 3,21 ммоль), Pd(OAc)2 (65,42 мг, 0,29 ммоль), Xantphos (168,61 мг, 0,29 ммоль) и K3PO4 (680,40 мг, 3,21 ммоль) в ДМФ (8 мл) перемешивают при 80°С в условиях микроволн защищенных N2 в течение 5 часов. После охлаждения до комнатной температуры, смесь концентрируют и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ 100% об/об и затем ДХМ/MeOH=20/1 об/об) с получением трет-бутил 3-(диметилфосфорил)-1H-индол-1-карбоксилат в виде коричневого масла (1 г, 100% выход). ЖХ-МС (ИЭР) m/z: 294 [M+H]+.
Стадия 4: К раствору трет-бутил 3-(диметилфосфорил)-1H-индол-1-карбоксилата (1,0 г, 3,41 ммоль) в ДХМ (6 мл) добавляют ТФК (3 мл) при 0°С. Смесь перемешивают при комнатной температуре в течение ночи и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1, об/об) с получением (1H-индол-3-ил)диметилфосфина оксида в виде белого твердого вещества (460 мг, 70% выход). ЖХ-МС (ИЭР) m/z: 194 [M+H]+.
Пр. E2
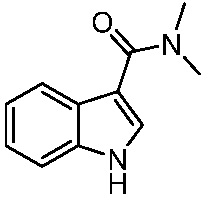
N, N-диметил-1H-индол-3-карбоксамид
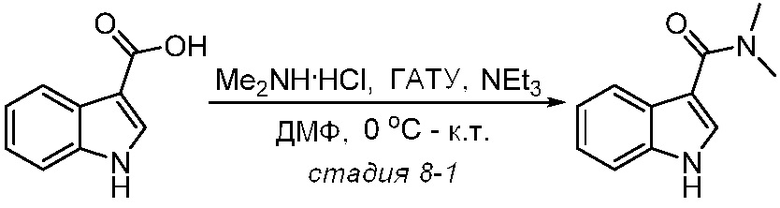
К раствору 1H-индол-3-карбоновой кислоты (1,0 г, 6,21 ммоль), ГАТУ (4,72 г, 12,41 ммоль) и диметиламина гидрохлорида (1,01 г, 12,41 ммоль) в ДМФ (15 мл) добавляют по каплям триэтиламин (2,51 г, 3,46 мл, 24,82 ммоль) при 0°С. Смесь перемешивают при комнатной температуре в течение 14 часов, разбавляют H2O (100 мл) и экстрагируют этилацетатом (100 мл x 3). Объединенные органические слои промывают насыщенным раствором соли (100 мл x 5), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением N, N-диметил-1H-индол-3-карбоксамида в виде белого твердого вещества (650 мг, 56% выход). ЖХ-МС (ИЭР) m/z: 189 [M+H]+.
Пр. E3
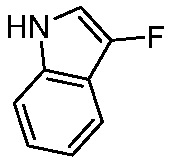
3-Фтор-1H-индол
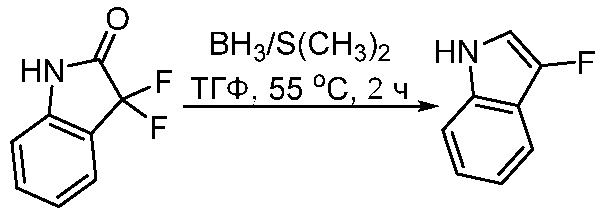
Стадия 1: К раствору 3,3-дифториндолин-2-она (300 мг, 1,77 ммоль) в ТГФ (5 мл) добавляют BH3/S(CH3)2 (2 M) (6,21 ммоль, 3 мл) по каплям при 0°С. Смесь перемешивают при комнатной температуре в течение 2 часов, гасят 10% водным раствором лимонной кислоты при 0°С и разбавляют водой. Полученную смесь экстрагируют этилацетатом (50 мл x 3) и органические фазы промывают насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением 3-фтор-1H-индола (220 мг, 91,78% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 136,1 [M+H]+.
Пр. E4
N, N-диметил-1H-индол-2-карбоксамид
Процедуры те же, что и для Пр. E2, с применением 1H-индол-2-карбоновой кислоты вместо 1H-индол-3-карбоновой кислоты. ЖХ-МС (ИЭР) m/z: 189 [M+H]+.
Пр. E5
2,3-Дигидроимидазо[1,2-a]пиридин-5(1H)-он
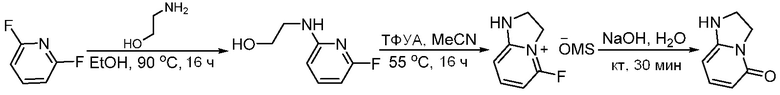
Стадия 1: Смесь 2,6-дифторпиридина (2,3 г, 19,99 ммоль, 1,81 мл) и 2-аминоэтанола (12,21 г, 199,86 ммоль, 12,09 мл) в этаноле (40 мл) перемешивают в течение 16 часов при 90°C, гасят водой (50 мл) и экстрагируют EtOAc (3 × 100 мл). Объединенную органическую фазу промывают насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (SiO2, этилацетат 100%) с получением 2-((6-фторпиридин-2-ил)амино)этан-1-ола (3 г, 93,24% выход, 97% чистота) в виде бесцветной жидкости. ЖХ-МС (ИЭР) m/z: 157,2 [M+H]+.
Стадия 2: Смесь 2-((6-фторпиридин-2-ил)амино)этан-1-ола (500 мг, 3,20 ммоль) и (2,2,2-трифторацетил) 2,2,2-трифторацетата (1,68 г, 8,00 ммоль, 1,13 мл) в MeCN (1 мл) перемешивают в течение 16 часов при 55°C в герметично закрытой пробирке и концентрируют в вакууме. Остаток применяют на следующей стадии без очистки.
Стадия 3: Часть неочищенного 5-фтор-2,3-дигидро-1H-имидазо[1,2-a]пиридин-4-ия (500 мг, 3,59 ммоль) растворяют в воде (25 мл) с NaOH (503,05 мг, 12,58 ммоль) в 236,18 мкл воды. Смесь перемешивают при комнатной температуре в течение 30 часов, нейтрализуют концентрированной HCl и концентрируют в вакууме. полученное твердое вещество суспендируют в ДХМ, фильтруют через слой целита и промывают EtOAc. Объединенную органическую фазу концентрируют в вакууме и остаток очищают хроматографией на силикагеле (ДХМ/MeOH=20/1) с получением 2,3-дигидроимидазо[1,2-a]пиридин-5(1H)-она (100 мг, 10,22% выход, 50% чистота). ЖХ-МС (ИЭР) m/z: 137,1 [M+H]+.
Пр. E6
Индолин-3-илметанол
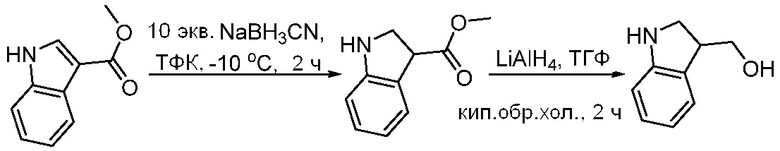
Стадия 1: К раствору метил 1H-индол-3-карбоксилата (1,75 г, 9,99 ммоль) в ТФК (30 мл) добавляют NaBH3CN (6,28 г, 99,90 ммоль) порциями при -10°C. Затем смесь перемешивают при той же температуре в течение еще часа и гасят водным NaOH (10%, 100 мл). Полученную смесь экстрагируют дихлорметаном (3×100 мл). Объединенный органический слой сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением метил индолин-3-карбоксилата (0,54 г, 1,52 ммоль, 50% чистота). ЖХ-МС (ИЭР) m/z: 178 [M+H]+.
Стадия 2: Раствор метил индолин-3-карбоксилата (0,2 г, 1,13 ммоль) ы ТГФ (10 мл) обрабатывают 1,0 M LiAlH4 (94,24 мг, 2,48 ммоль) в ТГФ и перемешивают при кипении с обратным холодильником в течение 2 часов. Смесь гасят осторожным добавлением метанола (10 мл). Полученный раствор концентрируют в вакууме. полученный остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ=0~10%) с получением индолин-3-илметанол в виде бесцветного масла (90 мг, 53% выход). ЖХ-МС (ИЭР) m/z: 150 [M+H]+.
Пр. E7
Индолин-3-илдиметилфосфин оксид
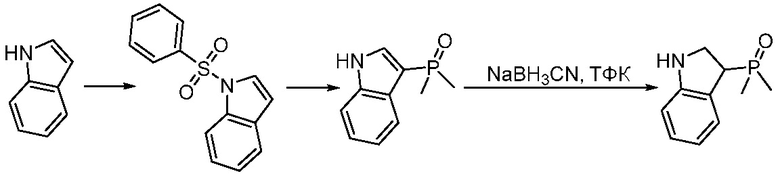
Стадия 1: К раствору индола (2 г, 17,07 ммоль) в ТГФ (20 мл) добавляют NaH (819,40 мг, 20,49 ммоль, 60% в минеральном масле) при 0°C. Смесь перемешивают при 0°C в течение 30 минут и бензолсульфонилхлорид (3,62 г, 20,49 ммоль) добавляют к вышеуказанной смеси. Полученную смесь перемешивают при комнатной температуре в течение часа, охлаждают до 0°С и гасят льдом-водой (100 мл). Смесь экстрагируют этилацетатом (3×50 мл) и объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=9/1) с получением 1-(фенилсульфонил)-1H-индола в виде бесцветного масла (3,8 г, 87% выход). ЖХ-МС (ИЭР) m/z: 258 [M+H]+.
Стадия 2: смесь 1-(фенилсульфонил)-1H-индола (500 мг, 1,94 ммоль) и AgNO3 (330,34 мг, 1,94 ммоль) в CH3CN (30 мл) перемешивают под атмосферой N2 при комнатной температуре в течение 2 часов, затем добавляют диметилфосфин оксид (455,00 мг, 5,83 ммоль). Смесь перемешивают в течение 16 часов при 100°C и фильтруют через силикагель. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1) с получением (1H-индол-3-ил)диметилфосфин оксида в виде желтого масла (150 мг, 13% выход). ЖХ-МС (ИЭР) m/z: 194 [M+H]+.
Стадия 3: К раствору (1H-индол-3-ил)диметилфосфина оксида (175 мг, 905,88 мкмоль) в ТФК (5 мл) добавляют NaBH3CN (570,71 мг, 9,06 ммоль) партиями при -5°C. Реакционную смесь перемешивают при комнатной температуре в течение ночи, доводят до pH 9-10 с 2 N раствором NaOH и разбавляют дихлорметаном/метанолом (10/1). Водную фазу экстрагируют дихлорметаном/метанолом (10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением индолин-3-илдиметилфосфина оксида в виде бесцветного масла (40 мг, 14% выход). ЖХ-МС (ИЭР) m/z: 196 [M+H]+.
Пр. E8
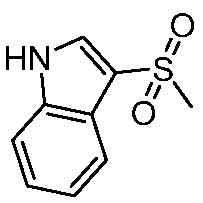
3-(Метилсульфонил)-1H-индол
К раствору индола (1 г, 8,54 ммоль) в ТГФ (20 мл) добавляют 2-метилпропан-2-олат калия (1,05 г, 9,39 ммоль). Реакционную смесь перемешивают при 25°C в течение 30 минут, затем добавляют триэтилборан (920,15 мг, 9,39 ммоль) и метансульфонилхлорид (1,08 г, 9,39 ммоль, 726,77 мкл) последовательно. Реакционную смесь перемешивают при -15°C в течение 16 часов, гасят насыщенным раствором хлорида аммония (50 мл) и экстрагируют этилацетатом (30 мл x 3). органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=5/1) с получением 3-метилсульфонил-1H-индола в виде светло-желтого твердого вещества (810 мг, 46% выход). ЖХ-МС (ИЭР) m/z: 196 [M+H]+.
Пр. E9
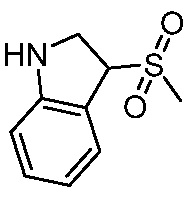
3-(Метилсульфонил)индолин
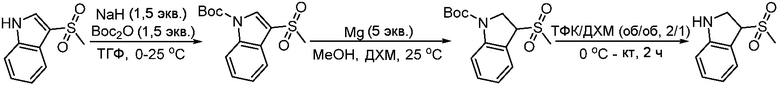
Стадия 1: К раствору 3-метилсульфонил-1H-индола (546 мг, 2,8 ммоль) в ТГФ (5 мл) добавляют NaH (172 мг, 4,3 ммоль, 60% в минеральном масле) медленно при 0°C. Реакционную смесь перемешивают при 0°C в течение 30 минут, затем добавляют Boc2O (934 мг, 4,3 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, гасят насыщенным раствором хлорида аммония (10 мл) и экстрагируют этилацетатом (10 мл x 3). Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной хроматографией на силикагеле (ПЭ/ЭА=5/1) с получением трет-бутил 3-(метилсульфонил)-1H-индол-1-карбоксилата в виде желтого твердого вещества (530 мг, 64% выход). ЖХ-МС (ИЭР) m/z: 296 [M+H]+.
Стадия 2: К перемешиваемому раствору трет-бутил 3-(метилсульфонил)-1H-индол-1-карбоксилата (530 мг, 1,70 ммоль) в дихлорметане (14 мл) и метаноле (52 мл) медленно добавляют магний (207 мг, 8,52 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, разбавляют насыщенным раствором хлорида аммония (60 мл) и экстрагируют дихлорметаном (60 мл x 3). Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (элюируют 10% - 20% этилацетатом в петролейном эфире) с получением трет-бутил 3-(метилсульфонил)индолин-1-карбоксилата (260 мг, 49% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 298 [M+H]+.
Стадия 3: К раствору трет-бутил 3-(метилсульфонил)индолин-1-карбоксилата (260 мг, 0,831 ммоль) в дихлорметане (4 мл) добавляют 2,2,2-трифторуксусную кислоту (2 мл) при 0°С. Полученный раствор перемешивают при 25°C в течение 2 часов и концентрируют в вакууме с получением неочищенного 3-метилсульфонил)индолина, который применяют без дальнейшей очистки. ЖХ-МС (ИЭР) m/z: 198 [M+H]+.
Пр. E10
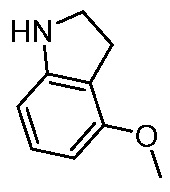
4-Метоксииндолин
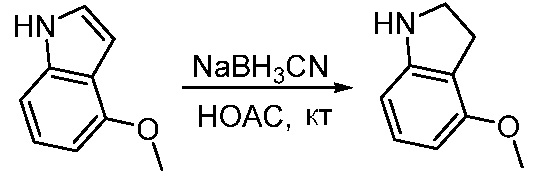
К раствору 4-метокси-1H-индола (500 мг, 3,40 ммоль) в AcOH (5 мл) добавляют цианоборгидрид натрия (895,00 мг, 13,59 ммоль) порциями в течение 10 минут. Смесь перемешивают при комнатной температуре в течение 30 минут, разбавляют водой (40 мл) и концентрируют в вакууме. Остаток экстрагируют этилацетат (25 мл x 3). Объединенную органическую фазу промывают насыщенным водным раствором NaHCO3 (50 мл) и насыщенным раствором соли (50 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного 4-метоксииндолина (500 мг, 79% выход), который используют сразу же, без дальнейшей очистки. ЖХ-МС (ИЭР) m/z: 150 [M+H]+.
Пр. E11
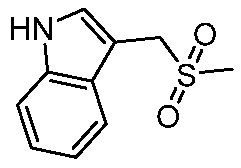
3-((Метилсульфонил)метил)-1H-индол
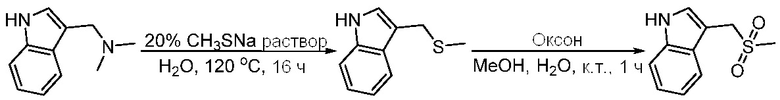
Стадия 1: К суспензии 1-(1H-индол-3-ил)-N, N-диметилметанамина (1 г, 5,74 ммоль) в воде (15 мл) добавляют 20% метантиолата натрия в воде (402,25 мг, 5,74 ммоль, 5 мл). Реакционную смесь перемешивают при 105°C в течение 16 часов, охлаждают до комнатной температуры и экстрагируют хлорметаном (50 мл x 3). Органическую фазу промывают 0,5 M HCl водным раствором (40 мл), насыщенным водным раствором бикарбоната натрия (50 мл) и насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1) с получением 3-((метилтио)метил)-1H-индола в виде белого твердого вещества (594 мг, 55% выход). ЖХ-МС (ИЭР) m/z: 178 [M+H]+.
Стадия 2: 3-((Метилсульфонил)метил)-1H-индол. К раствору 3-((метилтио)метил)-1H-индола (250 мг, 1,34 ммоль) в метаноле (0,5 мл) добавляют оксон (904,90 мг, 1,47 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, подщелачивают до pH 8 насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом (10 мл x 3). Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1) с получением 3-(метилсульфонилметил)-1H-индола в виде белого твердого вещества (139 мг, 47% выход). ЖХ-МС (ИЭР) m/z: 210 [M+H]+.
Пр. E12
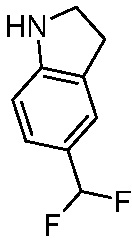
5-(дифторметил)индолин
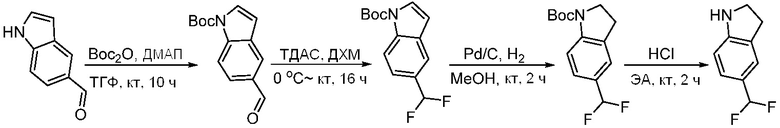
Стадия 1: смесь ДМАП (896,50 мг, 7,34 ммоль), 1H-индол-5-карбальдегида (986,30 мг, 6,79 ммоль) и ди-трет-бутилдикарбоната (1,63 г, 7,47 ммоль) в ТГФ (15 мл) перемешивают в течение 16 часов при комнатной температуре, гасят добавлением 10 мл воды и экстрагируют этилацетатом (3×50 мл). Органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением трет-бутил 5-формил-1H-индол-1-карбоксилата (1,4 г, 84% выход). ЖХ-МС (ИЭР) m/z: 246 [M+H]+.
Стадия 2: К раствору трет-бутил 5-формил-1H-индол-1-карбоксилата (500 мг, 2,04 ммоль) в ДХМ (20 мл) добавляют N-этил-N-(трифторсульфанил)этанамин (1,64 г, 10,19 ммоль, 1,35 мл) при 0°C. Затем смесь перемешивают при комнатной температуре в течение 16 часов, подщелачивают до pH 8 насыщенным NaHCO3 водным раствором и экстрагируют дихлорметаном (50 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ, 1/9) с получением трет-бутил 5-(дифторметил)-1H-индол-1-карбоксилата в виде белого твердого вещества (336 мг, 62% выход).
Стадия 3: Смесь трет-бутил 5-(дифторметил)-1H-индол-1-карбоксилата (200 мг, 748,30 мкмоль) и палладия (79,63 мг, 748,30 мкмоль) в MeOH (5 мл) перемешивают при 25°C в течение часа под атмосферой N2 и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного трет-бутил 5-(дифторметил)индолин-1-карбоксилата в виде желтого масла (200 мг, 99% выход).
Стадия 4: Раствор трет-бутил 5-(дифторметил)индолин-1-карбоксилата (200 мг, 742,70 мкмоль) в HCl-ЭА (568,67 мг, 15,60 ммоль, 710,84 мкл) перемешивают при 0°C в течение 2 часов и концентрируют в вакууме с получением 5-(дифторметил)индолина в виде белого твердого вещества (126,59 мг, 81% выход). ЖХ-МС (ИЭР) m/z: 170 [M+H]+.
Пр. E13
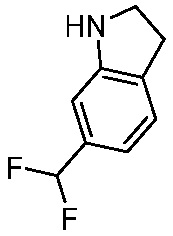
6-(дифторметил)индолин
Процедуры те же, что и для Пр. E12, с применением 1H-индол-6-карбальдегида вместо 1H-индол-5-карбальдегида. ЖХ-МС (ИЭР) m/z: 170 [M+H]+.
Пр. E14
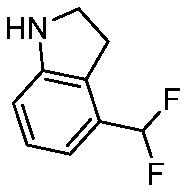
4-(дифторметил)индолин гидрохлорид
Процедуры те же, что и для Пр. E12, с применением 1H-индол-4-карбальдегида вместо 1H-индол-5-карбальдегида. ЖХ-МС (ИЭР) m/z: 170 [M+H]+.
Пр. E15
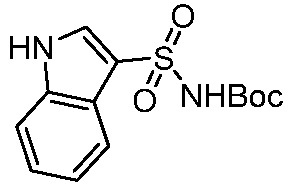
трет-Бутил ((1H-индол-3-ил)сульфонил)карбамат
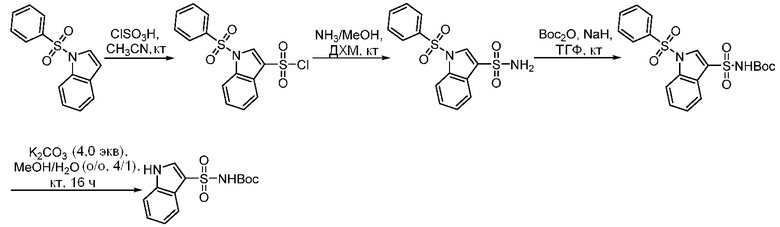
Стадия 1: К раствору 1-(фенилсульфонил)-1H-индола (1 г, 3,89 ммоль) в безводном CH3CN (10 мл) осторожно добавляют хлорсульфоновую кислоту (1,36 г, 11,66 ммоль, 776,33 мкл) при 0°C. Затем реакционную смесь нагревают до комнатной температуры в течение 4 часов, перемешивают в течение ночи, выливают в ледяную воду (50 мл) и экстрагируют дихлорметаном (30 мл x 2). Объединенный органический слой промывают насыщенным водным раствором бикарбоната натрия (50 мл x 2) и насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением 1-(фенилсульфонил)-1H-индол-3-сульфонилхлорида в виде белого твердого вещества (868 мг, 63% выход).
Стадия 2: К раствору 1-(фенилсульфонил)-1H-индол-3-сульфонилхлорида (800 мг, 2,25 ммоль) в CH2Cl2 (16 мл) добавляют NH3/MeOH (960,00 мг, 56,47 ммоль, 8 мл) медленно при 0°C. Затем смесь перемешивают при комнатной температуре в течение ночи, разбавляют водой (20 мл) и экстрагируют CH2Cl2 (20 мл x 3). Объединенный органический слой последовательно промывают 1M HCl (20 мл), водой (20 мл) и насыщенным раствором соли (20 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением 1-(фенилсульфонил)-1H-индол-3-сульфонамида в виде белого твердого вещества (639 мг, 84% выход). ЖХ-МС (ИЭР) m/z: 359 [M+Na]+.
Стадия 3: К раствору 1-(фенилсульфонил)-1H-индол-3-сульфонамида (1,03 г, 3,06 ммоль) в ТГФ (10 мл) добавляют NaH (612 мг, 15,31 ммоль, 60% суспензия в минеральном масле) при 0°C. Смесь перемешивают при 0°C в течение 30 минут, затем добавляют Boc2O (2,67 г, 12,25 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 16 часов и выливают в ледяную воду. Две фазы отделяют, и водные фазы экстрагируют дихлорметаном/метанолом (об/об, 10/1, 20 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=9/1) с получением трет-бутил ((1-(фенилсульфонил)-1H-индол-3-ил)сульфонил)карбамата (438 мг, 31% выход) в виде беловатого твердого вещества. ЖХ-МС (ИЭР) m/z: 381 [M+H-56]+.
Стадия 4: Смесь трет-бутил ((1-(фенилсульфонил)-1H-индол-3-ил)сульфонил)карбамата (438 мг, 1,00 ммоль) и K2CO3 (553,90 мг, 4,01 ммоль) в метаноле (8 мл) и H2O (2 мл) перемешивают при комнатной температуре в течение ночи, подкисляют до pH 5 уксусной кислотой и отделяют. Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают хроматографией с обращенной фазой (H2O/метанол) с получением трет-бутил ((1H-индол-3-ил)сульфонил)карбамата в виде белого твердого вещества (200 мг, 67% выход). ЖХ-МС (ИЭР) m/z: 319 [M+Na]+.
Пр. E16
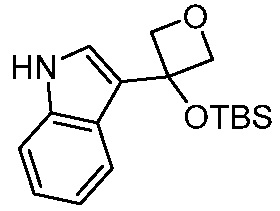
3-(3-((трет-Бутилдиметилсилил)окси)оксетан-3-ил)-1H-индол
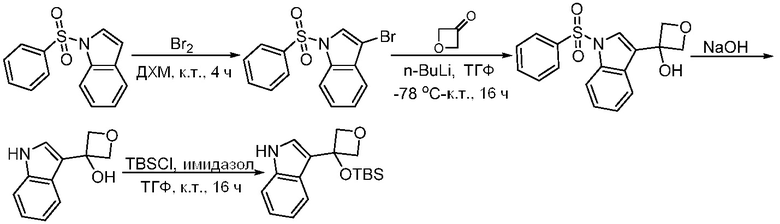
Стадия 1: К раствору 1-(фенилсульфонил)-1H-индола (2 г, 7,38 ммоль) в ДХМ (60 мл) добавляют бром (1,18 г, 7,38 ммоль, 378,23 мкл) по каплям. Красную реакционную смесь затем перемешивают при 25°C в течение 4,5 часов, выливают в насыщенный водный раствор бикарбоната натрия (50 мл) и отделяют. Органический слой промывают насыщенным водным раствором тиосульфата натрия (50 мл), водой (50 мл) и насыщенным раствором соли (50 мл), сушат над безводным MgSO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (20% ЭА/ПЭ) с получением 3-бром-1-(фенилсульфонил)-1H-индола в виде белого твердого вещества (1,5 г, 57% выход). ЖХ-МС (ИЭР) m/z: 336 [M+H]+.
Стадия 2: К раствору 3-бром-1-(фенилсульфонил)-1H-индола (200 мг, 0,595 ммоль) в ТГФ (5 мл) добавляют 2,5M бутиллития (0,36 мл, 0,892 ммоль) при -78°C. Реакционную смесь перемешивают при -78°C в течение 30 минут, затем добавляют оксетан-3-он (85,74 мг, 1,19 ммоль, 76,55 мкл) при -78°C. Через 30 минут при -78°C, реакционную смесь перемешивают при 25°C в течение 16 часов, выливают в насыщенный водный раствор хлорида аммония (30 мл) и отделяют. Органический слой промывают насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (10% ЭА/ПЭ) с получением 3-(1-(фенилсульфонил)-1H-индол-3-ил)оксетан-3-ола в виде светло-желтого твердого вещества (110 мг, 53% выход). ЖХ-МС (ИЭР) m/z: 276 [M+H]+.
Стадия 3: Раствор 3-(1-(фенилсульфонил)-1H-индол-3-ил)оксетан-3-ола (383 мг, 1,16 ммоль) в смешанном растворителе 2M NaOH раствора (5 мл) и метанола (5 мл) перемешивают при 85°C в течение 16 часов, разбавляют водой (20 мл) и отделяют. Органический слой промывают насыщенным раствором соли (20 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (10%-100% ЭА/ПЭ) с получением 3-(1H-индол-3-ил)оксетан-3-ола в виде светло-желтого твердого вещества (182 мг, 79% выход). ЖХ-МС (ИЭР) m/z: 190 [M+H]+.
Стадия 4: К раствору 3-(1H-индол-3-ил)оксетан-3-ола (600 мг, 3,01 ммоль) в ДМФ (6 мл) добавляют трет-бутилхлордиметилсилан (1,14 г, 7,53 ммоль, 1,40 мл) и имидазол (615,25 мг, 9,04 ммоль) при 0°C. Реакционную смесь перемешивают при 25°C в течение 16 часов, разбавляют водой (20 мл), подщелачивают до pH 8 насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом (10 мл x 3). Органический слой промывают насыщенным раствором соли (20 мл x 3), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (10% -100% ЭА/ПЭ) с получением 3-(3-((трет-бутилдиметилсилил)окси)оксетан-3-ил)-1H-индола в виде светло-желтого твердого вещества (890 мг, 92% выход). ЖХ-МС (ИЭР) m/z: 304 [M+H]+.
Пр. E17
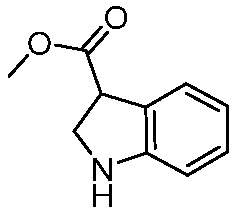
Метил индолин-3-карбоксилат
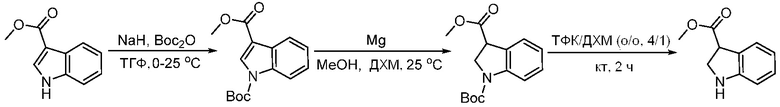
Стадия 1: К раствору метил 1H-индол-3-карбоксилата (5 г, 28,54 ммоль) в ТГФ (50 мл) добавляют NaH (1,71 г, 42,81 ммоль, 60% в минеральном масле) медленно при 0°C. Реакционную смесь перемешивают при 0°C в течение 30 минут, затем добавляют Boc2O (9,34 г, 42,81 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, гасят насыщенным раствором хлорида аммония (100 мл) и экстрагируют этилацетатом (100 мл x 3). органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=5/1) с получением 1-(трет-бутил) 3-метил 1H-индол-1,3-дикарбоксилата в виде желтого твердого вещества (5,3 г, 64% выход). ЖХ-МС (ИЭР) m/z: 276 [M+H]+.
Стадия 2: К перемешиваемому раствору 1-(трет-бутил) 3-метил 1H-индол-1,3-дикарбоксилата (2 г, 6,90 ммоль) в дихлорметане (60 мл) и метаноле (211 мл) медленно добавляют магний (838,72 мг, 34,51 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов, разбавляют насыщенным раствором хлорида аммония (100 мл) и экстрагируют дихлорметаном (100 мл x 3). Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ: 10-20%) с получением 1-(трет-бутил) 3-метил индолин-1,3-дикарбоксилат в виде светло-желтого масла (1,1 г, 55% выход). ЖХ-МС (ИЭР) m/z: 222 [M+H-56]+.
Стадия 3: К раствору 1-(трет-бутил) 3-метил индолин-1,3-дикарбоксилата (500 мг, 1,71 ммоль) в ДХМ (5 мл) добавляют трифторуксусную кислоту (1,25 мл). Полученную смесь перемешивают при комнатной температуре в течение 2 часов и концентрируют в вакууме. Остаток растворяют в ДХМ (100 мл) и промывают насыщенным водным раствором NaHCO3 (30 мл x 3). Органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением метил индолин-3-карбоксилата (308 мг, 94% выход), который применяют без дальнейших очисток. ЖХ-МС (ИЭР) m/z: 178 [M+H]+.
Пр. E18
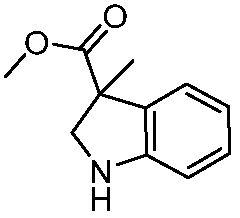
Метил 3-метилиндолин-3-карбоксилат
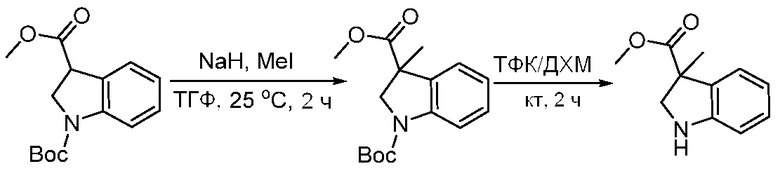
Стадия 1: К раствору 1-(трет-бутил) 3-метил индолин-1,3-дикарбоксилата (320 мг, 1,15 ммоль) и йодметана (491,36 мг, 3,46 ммоль) в ДМФ (8 мл) добавляют гидрид натрия (50,77 мг, 1,27 ммоль, 60% в минеральном масле) при 25°C. Смесь перемешивают при 25°C в течение 2 часов разбавляют ЭА (200 мл) и затем промывают водой (20 мл x 3) и насыщенным раствором соли (30 мл). Органические слои сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ: 15%) с получением 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилат в виде светло-желтого масла (244 мг, 70% выход). ЖХ-МС (ИЭР) m/z: 236 [M+H-56]+.
Стадия 2: К раствору 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата (240 мг, 0,824 ммоль) в дихлорметане (4 мл) добавляют трифторуксусную кислоту (1 мл) при 25°C. Смесь перемешивают при комнатной температуре в течение 2 часов и концентрируют в вакууме с получением неочищенного метил 3-метилиндолин-3-карбоксилата без дальнейших очисток. ЖХ-МС (ИЭР) m/z: 192 [M+H]+.
Пр. E19
3-Метилиндолин-3-карбоксамид
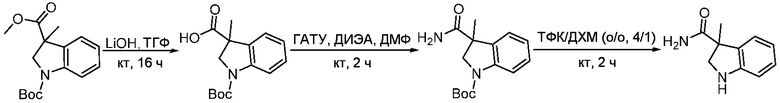
Стадия 1: К раствору 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилат (500 мг, 1,63 ммоль) в ТГФ (10 мл) по каплям добавляют водный раствор LiOH (2N, 5 мл). Реакционную смесь перемешивают при 25°C в течение 16 часов, доводят до pH ~ 4 с 1 M раствором HCl (10 мл) при 0°С и концентрируют в вакууме. Остаток экстрагируют ЭА (10 мл x 3). Органические фазы промывают насыщенным раствором соли (20 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением 1-трет-бутоксикарбонил-3-метил-индолин-3-карбоновой кислоты (452 мг) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 278 [M+H]+.
Стадия 2: К раствору 1-трет-бутоксикарбонил-3-метил-индолин-3-карбоновой кислоты (452 мг, 1,55 ммоль) в ДМФ (5 мл) последовательно добавляют хлорид аммония (414,1 мг, 7,74 ммоль), ДИЭА (600,4 мг, 4,65 ммоль) и ГАТУ (765,4 мг, 2,01 ммоль). Реакционную смесь перемешивают при 25°С в течение 16 часов, разбавляют насыщенным раствором хлорида аммония (15 мл) и экстрагируют ЭА (10 мл x 3). Органические фазы промывают насыщенным раствором соли (15 мл x 3), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1) с получением трет-бутил 3-карбамоил-3-метил-индолин-1-карбоксилат в виде белого твердого вещества (420 мг, 93% выход). ЖХ-МС (ИЭР) m/z: 277 [M+H]+.
Стадия 3: Та же методика, как для Стадии 3 Пр. 18, с применением трет-бутил 3-карбамоил-3-метил-индолин-1-карбоксилата вместо 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата. ЖХ-МС (ИЭР) m/z: 177 [M+H]+.
Пр. E20
N,3-диметилиндолин-3-карбоксамид
Процедуры те же, что и для Стадий 2&3 Пр. E19, метиламина гидрохлорид вместо хлорида аммония. ЖХ-МС (ИЭР) m/z: 191 [M+H]+.
Пр. E21
(3Метилиндолин-3-ил)метанол
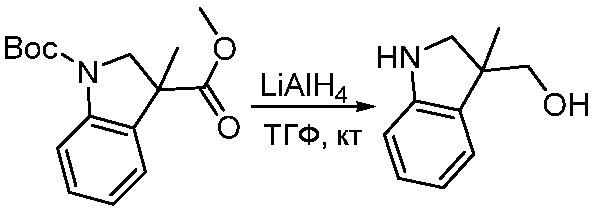
К раствору 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата (150 мг, 0,515 ммоль) в ТГФ (10 мл) добавляют 1,0M LiAlH4 (1 мл, 1,13 ммоль) в ТГФ при комнатной температуре. Реакционную смесь перемешивают при кипении с обратным холодильником в течение 2 часов и гасят осторожным добавлением метанола (10 мл) при комнатной температуре. Полученную смесь концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ=0~10%) с получением (3-метилиндолин-3-ил)метанола (60 мг, 71% выход). ЖХ-МС (ИЭР) m/z: 164 [M+H]+.
Пр. E22
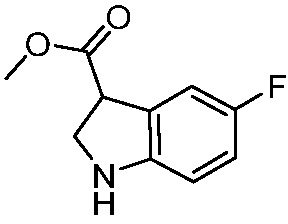
Метил 5-фториндолин-3-карбоксилат
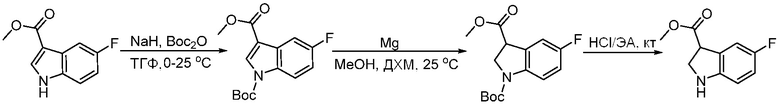
Стадия 1&2: Процедуры те же, что и для Стадий 1&2 Пр. E17, с применением 5-фтор-1H-индол-3-карбоксилата вместо 1H-индол-3-карбоксилата. ЖХ-МС (ИЭР) m/z: 318 [M+Na]+.
Стадия 3: Раствор 1-(трет-бутил) 3-метил 5-фториндолин-1,3-дикарбоксилата (176 мг, 0,596 ммоль) в HCl/ЭА (4M, 3 мл) перемешивают при комнатной температуре в течение часа и концентрируют в вакууме с получением метил 5-фториндолин-3-карбоксилата (110 мг, 94,6% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 196 [M+H]+.
Пр. E23
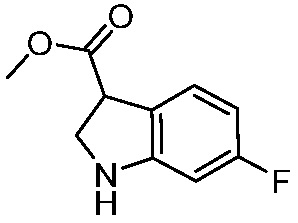
Метил 6-фториндолин-3-карбоксилат
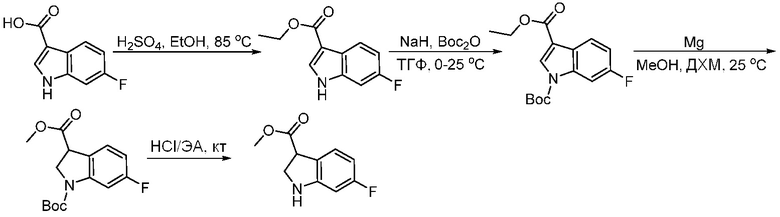
Стадия 1: К раствору 6-фтор-1H-индол-3-карбоновой кислоты (600 мг, 3,35 ммоль) в этаноле (50 мл) добавляют концентрированную серную кислоту (0,1 мл). Реакционную смесь перемешивают при 85°C в течение 16 часов, охлаждают до комнатной температуры и нейтрализуют насыщенным водным раствором бикарбоната натрия и экстрагируют ЭА (10 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1) с получением этил 6-фтор-1H-индол-3-карбоксилата (646 мг, 94,9% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 208 [M+H]+.
Стадии 2&3: Процедуры те же, что и для Стадий 1&2 Пр. E17, с применением этил 6-фтор-1H-индол-3-карбоксилата вместо 1H-индол-3-карбоксилата (трансэстерификация с MeOH). ЖХ-МС (ИЭР) m/z: 296 [M+H]+.
Стадия 4: Процедуры те же, что и для Стадии 3 Пр. E22, с применением 1-(трет-бутил) 3-этил 6-фториндолин-1,3-дикарбоксилата вместо 1-(трет-бутил) 3-метил 5-фториндолин-1,3-дикарбоксилата. ЖХ-МС (ИЭР) m/z: 196 [M+H]+.
Пр. E24
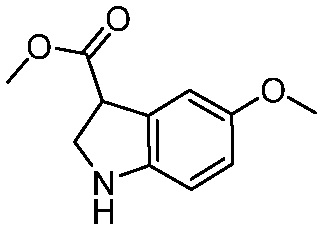
Метил 5-метоксииндолин-3-карбоксилат
Процедуры те же, что и для Пр. E17, с применением 5-метокси-1H-индол-3-карбоксилата вместо 1H-индол-3-карбоксилата. ЖХ-МС (ИЭР) m/z: 208 [M+H]+.
Пр. E25
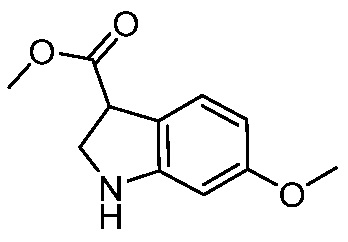
Метил 6-метоксииндолин-3-карбоксилат
Процедуры те же, что и для Пр. E23, с применением 6-метокси-1H-индол-3-карбоновой кислоты вместо 6-фтор-1H-индол-3-карбоновой кислоты. ЖХ-МС (ИЭР) m/z: 208 [M+H]+.
Пр. E26
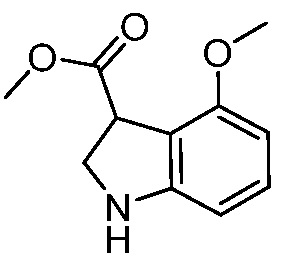
Метил 4-метоксииндолин-3-карбоксилат
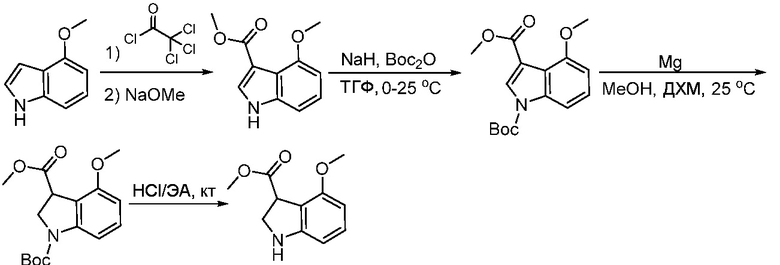
Стадия 1: К раствору 4-метокси-1H-индола (1,4 г, 9,51 ммоль) и пиридина (3,76 г, 47,56 ммоль, 3,85 мл) в ДХМ (10 мл) добавляют 2,2,2-трихлорацетил хлорид (8,65 г, 47,56 ммоль) по каплям в ДХМ (10 мл) при 0°C. После перемешивания при 0°C в течение 1,5 часов, смесь концентрируют в вакууме. Остаток растворяют в метаноле (20 мл), добавляют к перемешиваемой суспензии метоксида натрия (1,54 г, 28,54 ммоль, 1,59 мл) в метаноле (50 мл) в течение 20 минут. Смесь перемешивают при комнатной температуре в течение часа, охлаждают до 0°С, разбавляют водой (50 мл) и экстрагируют ДХМ (30 мл x 3). Органические фазы сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ=1% - 10%) с получением метил 4-метокси-1H-индол-3-карбоксилата (800 мг, 38% выход). ЖХ-МС (ИЭР) m/z: 206 [M+H]+.
Стадия 2&3&4: Процедуры те же, что и для Пр. E22, с применением метил 4-метокси-1H-индол-3-карбоксилата вместо 5-фтор-1H-индол-3-карбоксилата. ЖХ-МС (ИЭР) m/z: 208 [M+H]+.
Пр. E27
Метил 5-фтор-3-метилиндолин-3-карбоксилат
Процедуры те же, что и для Пр. E18, на Стадии 1 с применением 1-(трет-бутил) 3-метил 5-фториндолин-1,3-дикарбоксилата вместо 1-(трет-бутил) 3-метил индолин-1,3-дикарбоксилата; на Стадии 2, в условиях снятия защиты HCl/ЭА как на Стадии 3 Пр. 22. ЖХ-МС (ИЭР) m/z: 210 [M+H]+.
Пр. E28
Метил 6-фтор-3-метилиндолин-3-карбоксилат
Процедуры те же, что и для Пр. E18, на Стадии 1 с применением 1-(трет-бутил) 3-метил 5-фториндолин-1,3-дикарбоксилата вместо 1-(трет-бутил) 3-метил индолин-1,3-дикарбоксилата; на Стадии 2, в условиях снятия защиты HCl/ЭА как на Стадии 3 Пр. 22. ЖХ-МС (ИЭР) m/z: 210 [M+H]+.
Пр. E29
Метил 2-(индолин-3-ил)ацетат
К раствору метил 2-(1H-индол-3-ил)ацетата (500 мг, 2,64 ммоль) в дихлорметане (16 мл) и ТФК (3 мл) добавляют боргидрид натрия (199,95 мг, 5,29 ммоль) при 0°C. Смесь перемешивают при 0°C в течение 2 часов, разбавляют ДХМ (200 мл) и гасят насыщенным NaHCO3 водным раствором (10 мл). органическую фазу отделяют, сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением метил 2-индолин-3-илацетата в виде коричневого масла (477 мг, 85% выход). ЖХ-МС (ИЭР) m/z: 196 [M+H]+.
Пр. E30
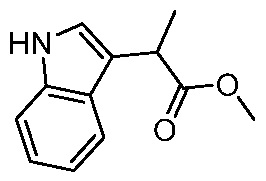
Метил 2-(1H-индол-3-ил)пропаноат
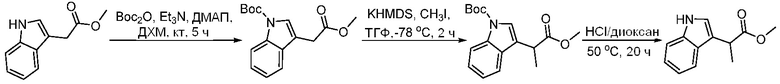
Стадия 1: К раствору метил 2-(1H-индол-3-ил) ацетата (1 г, 5,29 ммоль) в ДХМ (10 мл) добавляют триэтиламин (2,14 г, 21,14 ммоль), ДМАП (129,13 мг, 1,06 ммоль) и ди-трет-бутилкарбонат (1,73 г, 7,93 ммоль) в атмосфере аргона при комнатной температуре. После перемешивания в течение 5 часов, реакционную смесь разбавляют насыщенным водным раствором NH4Cl (20 мл) и экстрагируют ДХМ (20 мл x 2). Органические фазы промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (0~20% ЭА/ПЭ) с получением трет-бутил 3-(2-метокси-2-оксоэтил)-1H-индол-1-карбоксилата в виде светло-желтого масла (1,46 г, 95% выход). ЖХ-МС (ИЭР) m/z: 307 [M+18]+.
Стадия 2: К раствору трет-бутил 3-(2-метокси-2-оксоэтил)-1H-индол-1-карбоксилата (300 мг, 1,04 ммоль) в безводном ТГФ (10 мл) добавляют KHMDS (227,53 мг, 1,14 ммоль) по каплям при -20°C в течение 30 минут под N2. Затем раствор перемешивают при -78°C в течение еще 40 минут, затем по каплям добавляют йодметан (147,18 мг, 1,04 ммоль) при -78°C. После перемешивания при той же температуре в течение 1,5 часов, реакционную смесь гасят насыщенным NH4Cl водным раствором (20 мл) и отделяют. Водный слой экстрагируют EtOAc (20 мл x 3), и объединенные органические фазы промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (0~20% ЭА/ПЭ) с получением трет-бутил 3-(1-метокси-1-оксопропан-2-ил)-1H-индол-1-карбоксилат в виде бесцветного масла (105 мг, 30% выход). ЖХ-МС (ИЭР) m/z: 326 [M+Na]+.
Стадия 3: Раствор трет-бутил 3-(2-метокси-1-метил-2-оксо-этил)индол-1-карбоксилата (550 мг, 1,81 ммоль) в хлористоводородной кислоте/диоксане (4N, 15 мл) перемешивают при 25°C в течение 48 часов и концентрируют в вакууме. Остаток очищают хроматографией с обращенной фазой (0,01%ТФК/метанол) с получением метил 2-(1H-индол-3-ил)пропаноата (110 мг, 51% чистота) в виде бесцветного масла. ЖХ-МС (ИЭР) m/z: 204 [M+H]+.
Пр. E31
Метил 1-(1H-индол-3-ил)циклопропан-1-карбоксилат
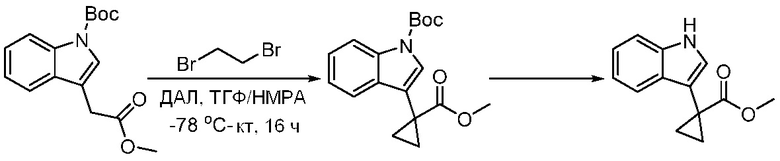
Стадия 1: К раствору трет-бутил 3-(2-метокси-2-оксо-этил)индол-1-карбоксилата (100 мг, 0,346 ммоль) в ТГФ/HMPA (об/об, 10/1) (5,5 мл) добавляют ДАЛ (81,5 мг, 0,760 ммоль) при -78°C, затем добавляют 1,2-дибромэтан (97,4 мг, 0,518 ммоль) через 30 минут. Затем реакционную смесь перемешивают при комнатной температуре в течение ночи, гасят добавлением льда-воды и отделяют. Водную фазу экстрагируют дихлорметаном/метанолом (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/9) с получением трет-бутил 3-(1-метоксикарбонил)циклопропил-1H-индол-1-карбоксилата в виде желтого масла (120 мг, 78% выход). ЖХ-МС (ИЭР) m/z: 338 [M+Na]+.
Стадия 2: Та же методика, как для Стадии 3 Пр. E30, с применением трет-бутил 3-(1-метоксикарбонил)циклопропил-1H-индол-1-карбоксилата вместо трет-бутил 3-(2-метокси-1-метил-2-оксо-этил)индол-1-карбоксилата. ЖХ-МС (ИЭР) m/z: 238 [M+Na]+.
Пр. E32
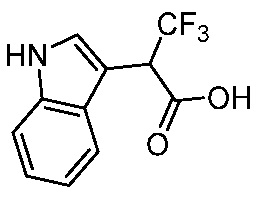
3,3,3-Трифтор-2-(1H-индол-3-ил)пропановая кислота
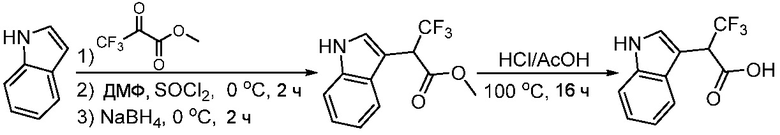
Стадия 1: К метил 3,3,3-трифтор-2-оксо-пропаноату (4,68 г, 29,96 ммоль) добавляют индол (117 мг, 0,999 ммоль) при энергичном перемешивании. Смесь отверждают в течение примерно 1 минуты, растворяют в ДМФ (40 мл) и охлаждают до 0°С, затем по каплям добавляют SOCl2 (8,91 г, 74,91 ммоль). Полученную смесь перемешивают при той же температуре до тех пор, пока превращение не завершится по данным ЖХМС. Туда добавляют NaBH4 (3,40 г, 89,89 ммоль) медленно порциями. Смесь затем перемешивают в течение 3 часов при 0°С и выливают в 100 мл насыщенного водного раствора NH4Cl при перемешивании. Полученное твердое вещество отфильтровывают, и фильтрат экстрагируют этилацетатом (50 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=0~20%) с получением метил 3,3,3-трифтор-2-(1H-индол-3-ил)пропаноата в виде желтого твердого вещества (6,5 г, 84% выход). ЖХ-МС (ИЭР) m/z: 258 [M+H]+.
Стадия 2: Раствор метил 3,3,3-трифтор-2-(1H-индол-3-ил)пропаноата (2,0 г, 7,78 ммоль) в смешанном растворителе концентрированной HCl (12 мл) и AcOH (60 мл) перемешивают при энергичном кипении с обратным холодильником в течение 16 часов, охлаждают до комнатной температуры и концентрируют в вакууме. Остаток очищают хроматографией с обращенной фазой (H2O/MeOH=100/0~50/50) с получением 3,3,3-трифтор-2-(1H-индол-3-ил)пропановой кислоты в виде беловатого порошка (1 г, 50% выход). ЖХ-МС (ИЭР) m/z: 244 [M+H]+.
Пр. E33
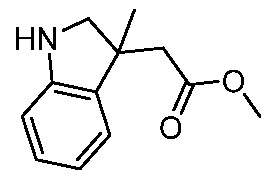
Метил 2-(3-метилиндолин-3-ил)ацетат
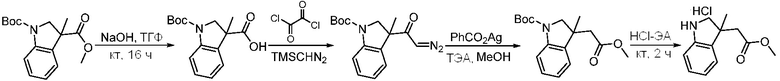
Стадия 1: К раствору 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата (730 мг, 2,51 ммоль) в ТГФ (20 мл) добавляют водный раствор гидроксида натрия (3,01 г, 75,17 ммоль) в воде (1,41 мл). Смесь перемешивают в течение 16 часов при комнатной температуре, подкисляют водным раствором 1 M HCl и экстрагируют EtOAc (100 мл x 3). Органические фазы промывают насыщенным раствором соли (50 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенной 1-трет-бутоксикарбонил-3-метилиндолин-3-карбоновой кислоты в виде желтого масла (620 мг, 85% выход). ЖХ-МС (ИЭР) m/z: 222 [M+H-56]+.
Стадия 2: К смеси 1-трет-бутоксикарбонил-3-метилиндолин-3-карбоновой кислоты (630 мг, 2,27 ммоль) в ДХМ (15 мл) добавляют медленно оксалилхлорид (865,1 мг, 6,82 ммоль) при 0°C и N, N-диметилформамид (8,3 мг, 0,114 ммоль). Смесь перемешивают в течение 16 часов при комнатной температуре и концентрируют в вакууме. Остаток (670 мг, 1,13 ммоль) растворяют в ТГФ (10 мл) и MeCN (5,00 мл) и диазометил(триметил)силан (258,75 мг, 2,27 ммоль) (2 M раствор в диэтиловом эфире) добавляют медленно к вышеуказанному раствору. Реакционную смесь перемешивают в течение 2 часов под N2, гасят 10% лимонной кислотой (10 мл) и разделяют между ДХМ (50 мл) и водой (50 мл). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=0-10%) с получением трет-бутил 3-(2-диазоацетил)-3-метилиндолин-1-карбоксилата в виде желтого масла (200 мг, 56% выход).
Стадия 3: К раствору трет-бутил 3-(2-диазоацетил)-3-метилиндолин-1-карбоксилата (200 мг, 0,664 ммоль) в метаноле (5 мл) добавляют бензоат серебра (76,0 мг, 0,332 ммоль). Реакционную смесь перемешивают в течение 1,5 часов при комнатной температуре под N2 и разбавляют ДХМ (50 мл) и водой (50 мл). Органический слой отделяют, сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=0-15%) с получением трет-бутил 3-(2-метокси-2-оксоэтил)-3-метилиндолин-1-карбоксилата в виде бесцветного масла (100 мг, 42% выход). ЖХ-МС (ИЭР) m/z: 250 [M+H-56]+.
Стадия 4: Смесь трет-бутил 3-(2-метокси-2-оксоэтил)-3-метилиндолин-1-карбоксилата (100 мг, 0,278 ммоль) и HCl/ЭА (4M, 1,5 мл) в ДХМ (3 мл) перемешивают в течение 2 часов при комнатной температуре и концентрируют в вакууме с получением неочищенного метил 2-(3-метилиндолин-3-ил)ацетата без дальнейших очисток. ЖХ-МС (ИЭР) m/z: 206 [M+H]+.
Альтернативный способ синтеза:
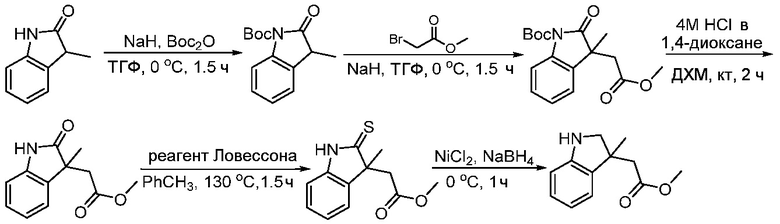
Стадия 1: К раствору 3-метилиндолин-2-она (20 г, 135,89 ммоль) в ТГФ (200 мл) добавляют NaH (6,52 г, 163,09 ммоль, 60% суспензия в минеральном масле) при 0°C в течение 30 минут. Реакционную смесь перемешивают при 0°C в течение 30 минут, затем по каплям добавляют раствор ди-трет-бутилкарбоната (29,07 г, 133,18 ммоль) в ТГФ (50 мл). Реакционную смесь перемешивают в течение часа, разбавляют насыщенным NH4Cl водным раствором (50 мл) и экстрагируют CH2Cl2 (100 мл x 2). Объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=9/1) с получением трет-бутил 3-метил-2-оксоиндолин-1-карбоксилата (30 г, 74% выход) в виде желтого масла. ЖХ-МС (ИЭР) m/z: 192 [M+H]+.
Стадия 2: К раствору трет-бутил 3-метил-2-оксоиндолин-1-карбоксилата (30 г, 103,12 ммоль) в ТГФ (300 мл) добавляют NaH (4,95 г, 123,74 ммоль, 60% в минеральном масле) при 0°C в течение 30 минут. Реакционную смесь перемешивают при 0°C в течение 30 минут, затем по каплям добавляют метил 2-бромацетат (18,93 г, 123,74 ммоль). Реакционную смесь перемешивают в течение часа, гасят насыщенным NH4Cl водным раствором (100 мл) и экстрагируют CH2Cl2 (200 мл x 2). Объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=8/2) с получением трет-бутил 3-(2-метокси-2-оксоэтил)-3-метил-2-оксоиндолин-1-карбоксилата (28 г, 83% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 264 [M+H-56]+.
Стадия 3: Смесь 4 M HCl в 1,4-диоксане (35 мл) и трет-бутил 3-(2-метокси-2-оксоэтил)-3-метил-2-оксоиндолин-1-карбоксилата (28 г, 87,68 ммоль) в CH2Cl2 (200 мл) перемешивают при комнатной температуре в течение 2 часов и концентрируют в вакууме. Остаток разделяют между CH2Cl2 (200 мл) и насыщенным NaHCO3 водным раствором (50 мл). Отделенный водный слой экстрагируют CH2Cl2 (50 мл x 3), и объединенный органический слой сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением метил 2-(3-метил-2-оксоиндолин-3-ил)ацетат (17,6 г, 82% выход). ЖХ-МС (ИЭР) m/z: 220 [M+H]+.
Стадия 4: Смесь метил 2-(3-метил-2-оксоиндолин-3-ил)ацетат (15,6 г, 71,16 ммоль) и реагента Ловессона (14,71 г, 36,38 ммоль) в толуоле (220 мл) перемешивают при 130°C в течение 1,5 часов и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=80:20) с получением метил 2-(3-метил-2-тиоксоиндолин-3-ил)ацетата (15,5 г, 74% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 236 [M+H]+.
Стадия 5: К смеси метил 2-(3-метил-2-тиоксоиндолин-3-ил)ацетата (9,5 г, 40,37 ммоль) и хлорида никеля (10,46 г, 80,75 ммоль) в смешанном ТГФ (70 мл) и метаноле (70 мл) добавляют NaBH4 (9,16 г, 242,24 ммоль) порциями при 0°C в течение часа. Полученную смесь перемешивают в течение 10 минут и фильтруют через слой Целита. Твердую лепешку промывают MeOH (100 мл) и фильтрат концентрируют в вакууме. Остаток растворяют в ЭА (200 мл) и промывают водой (60 мл x 3). Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (CH2Cl2: метанол=95:5) с получением метил 2-(3-метилиндолин-3-ил)ацетата (6,5 г, 75% выход) в виде желтого масла. ЖХ-МС (ИЭР) m/z: 206 [M+H]+.
Пр. E34
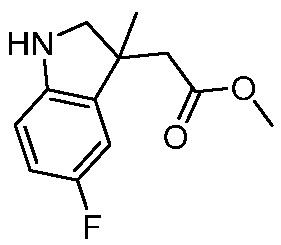
Метил 2-(5-фтор-3-метилиндолин-3-ил)ацетат
Процедуры те же, что и для Пр. E33, с применением 1-(трет-бутил) 3-метил 5-фтор-3-метилиндолин-1,3-дикарбоксилата вместо 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата. ЖХ-МС (ИЭР) m/z: 224 [M+H]+.
Пр. E35
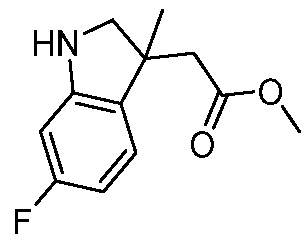
Метил 2-(6-фтор-3-метилиндолин-3-ил)ацетат
Процедуры те же, что и для Пр. E33, с применением 1-(трет-бутил) 3-метил 6-фтор-3-метилиндолин-1,3-дикарбоксилата вместо 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата. ЖХ-МС (ИЭР) m/z: 224 [M+H]+.
Пр. E36
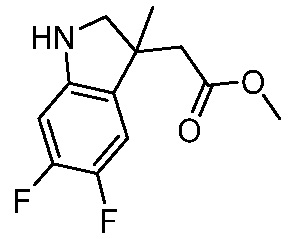
Метил 2-(5,6-дифтор-3-метилиндолин-3-ил)ацетат
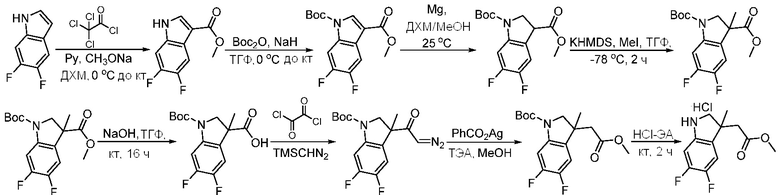
Стадии 1&2&3: Та же методика, как для Стадий 1&2&3 Пр. E26, с применением 5,6-дифтор-1H-индола вместо 4-метокси-1H-индола. ЖХ-МС (ИЭР) m/z: 258 [M+H-56]+.
Стадия 4: К раствору 1-(трет-бутил) 3-метил 5,6-дифториндолин-1,3-дикарбоксилата (910 мг, 2,90 ммоль) в безводном ТГФ (60 мл) добавляют йодметан (494,7 мг, 3,49 ммоль) при -78°C в течение 20 минут, и 1M KHMDS (3,2 мл) при -78°C over 45 минут под защитой N2. Реакционную смесь перемешивают при -78℃ в течение 2 часов, гасят насыщенным водным раствором NH4Cl (50 мл) и отделяют. Водный слой экстрагируют ЭА (50 мл x 3), и объединенную органическую фазу промывают насыщенным раствором соли (40 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (10~20% ЭА/ПЭ) с получением 1-(трет-бутил) 3-метил 5,6-дифтор-3-метилиндолин-1,3-дикарбоксилата в виде бесцветного масла (810 мг, 82% выход). ЖХ-МС (ИЭР) m/z: 272 [M+H-56]+.
Стадии 5&6&7&8: Процедуры те же, что и для Пр. E33, с применением 1-(трет-бутил) 3-метил 5-фтор-3-метилиндолин-1,3-дикарбоксилата вместо 1-(трет-бутил) 3-метил 3-метилиндолин-1,3-дикарбоксилата. ЖХ-МС (ИЭР) m/z: 242 [M+H]+.
Пр. E37
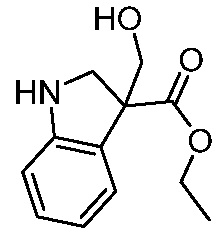
Этил 3-(гидроксиметил)индолин-3-карбоксилат
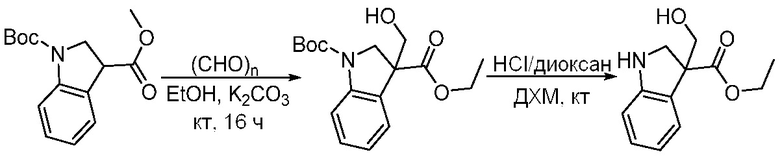
Стадия 1: К раствору 1-(трет-бутил) 3-метил индолин-1,3-дикарбоксилата (300 мг, 1,08 ммоль) в этаноле (5 мл) добавляют параформальдегид (48,72 мг, 1,62 ммоль) и K2CO3 (448,53 мг, 3,25 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного 1-(трет-бутил) 3-этил 3-(гидроксиметил)индолин-1,3-дикарбоксилата (330 мг, 99% выход). ЖХ-МС (ИЭР) m/z: 222 [M+H-100]+.
Стадия 2: К раствору 1-(трет-бутил) 3-этил индолин-1,3-дикарбоксилата (300 мг, 1,08 ммоль) в ДХМ (5 мл) добавляют 4M HCl в диоксане (3 мл) при 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, нейтрализуют до pH ~8 насыщенным водным раствором NaHCO3 (5 мл) и экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного этил 3-(гидроксиметил)индолин-3-карбоксилата (239 мг, 89,9% выход). ЖХ-МС (ИЭР) m/z: 222 [M+H]+.
Пр. E38
Этил 2-азапиро[4,5]декан-4-карбоксилат
Стадия 1: Смесь этил 2-циклогексилиденацетата (5 г, 29,72 ммоль), N-(метоксиметил)-1-фенил-N-(триметилсилилметил)метанамина (14,11 г, 59,44 ммоль) и фторида лития (2,31 г, 89,16 ммоль) в MeCN (50 мл) перемешивают при 60°C в течение 48 часов и концентрируют в вакууме. Остаток растворяют в ЭА (100 мл), промывают предварительно охлажденным водным раствором K2CO3 (10%, 100 мл x 2), насыщенным водным раствором CuSO4 (100 мл x 3) и насыщенным раствором соли (100 мл). органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают хроматографией с обращенной фазой (0,03%NH4HCO3/MeOH) с получением этил 2-бензил-2-азапиро[4,5]декан-4-карбоксилата (5,55 г, 52,7% выход, 85% чистота) в виде светло-желтого масла. ЖХ-МС (ИЭР) m/z: 302 [M+H]+.
Стадия 2: К раствору этил 2-бензил-2-азапиро[4,5]декан-4-карбоксилата (2,65 г, 8,79 ммоль) в метаноле (30 мл) добавляют дигидроксипалладий (679,1 мг, 4,84 ммоль). Смесь перемешивают при 40°C в атмосфере водорода (14 ф./кв.д.) в течение 24 часов и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного этил 2-азапиро[4,5]декан-4-карбоксилата. ЖХ-МС (ИЭР) m/z: 212 [M+H]+.
Пр. E39
Этил 8-окса-2-азапиро[4,5]декан-4-карбоксилат
Процедуры те же, что и для Пр. E38, с применением этил 2-тетрагидропиран-4-илиденацетата вместо этил 2-циклогексилиденацетата. ЖХ-МС (ИЭР) m/z: 214 [M+H]+.
Пр. E40
Этил 2-азапиро[4,4]нонан-4-карбоксилат
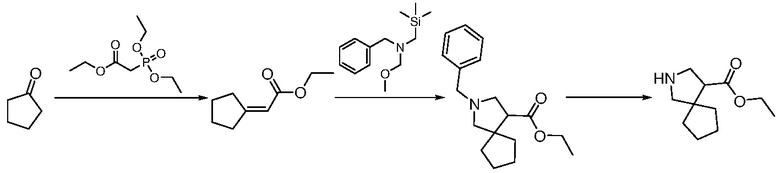
Стадия 1: К перемешиваемой суспензии гидрида натрия (2,08 г, 52,04 ммоль, 60% дисперсия в минеральном масле) в диэтиловом эфире (120 мл) добавляют этил 2-диэтоксифосфорилацетат (11,64 г, 51,94 ммоль) при 0°C. Полученную смесь перемешивают в течение 5 минут, затем добавляют раствор циклопентанона (4,25 г, 50,53 ммоль) в диэтиловом эфире (10 мл). Реакционную смесь перемешивают при 25°C в течение 4 часов, гасят насыщенным водным раствором хлорида аммония (30 мл) при 0°С, разбавляют водой (100 мл) и экстрагируют диэтиловым эфиром (50 мл x 3). Объединенную органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (петролейный эфир/диэтиловый эфир=10/1) с получением этил 2-циклопентилиденацетата в виде светло-желтого масла (6,0 г, 73% выход). ЖХ-МС (ИЭР) m/z: 155 [M+H]+.
Стадия 2&3: Процедуры те же, что и для Пр. E38, с применением этил 2-циклопентилиденацетата вместо этил 2-циклогексилиденацетата. ЖХ-МС (ИЭР) m/z: 198 [M+H]+.
Промежуточное соединение 12
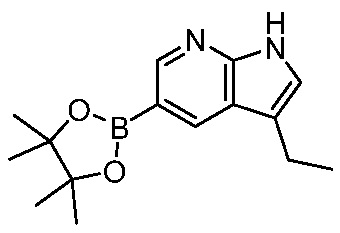
3-Этил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пирроло[2,3-b]пиридин
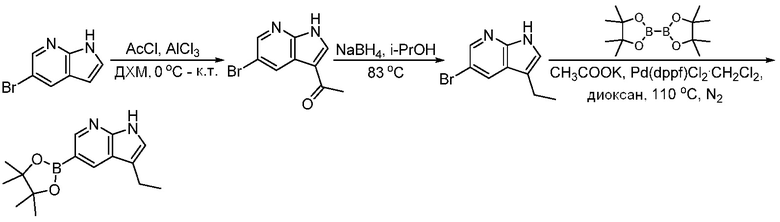
Стадия 1: К суспензии безводного AlCl3 (3,40 г, 25,50 ммоль) в безводном ДХМ (50 мл) под N2 добавляют 5-бром-1H-пирроло[2,3-b]пиридин (1,01 г, 5,10 ммоль). Смесь перемешивают в течение 1 часа при комнатной температуре и туда по каплям добавляют AcCl (2,00 г, 25,50 ммоль, 1,8 мл). Полученную смесь перемешивают в течение 30 минут, охлаждают до 0°C и гасят осторожно добавлением MeOH до тех пор, пока раствор не станет прозрачным. Смесь концентрируют, и остаток разбавляют H2O (30 мл), доводят до pH ~4 водным раствором NaOH (1 N) и экстрагируют этилацетатом (30 мл x 3). Объединенные органические слои промывают насыщенным раствором соли (30 мл x 2), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток обрабатывают ультразвуком в ЭА и фильтруют с получением 1-(5-бром-1H-пирроло[2,3-b]пиридин-3-ил)этан-1-она в виде светло-желтого твердого вещества (0,87 г, 71% выход). ЖХ-МС (ИЭР) m/z: 239 [M+H]+.
Стадия 2: К перемешиваемому раствору 1-(5-бром-1H-пирроло[2,3-b]пиридин-3-ил)этан-1-она (200 мг, 0,84 ммоль) в изопропиловом спирте (7 мл) добавляют NaBH4 (158,24 мг, 4,18 ммоль) порциями при 0°C. Реакционную смесь кипятят с обратным холодильником при 83°C в течение 24 часов, охлаждают до 0°C и разбавляют водой (15 мл). Полученную смесь экстрагируют этилацетатом (30 мл x 3). Органический слой промывают насыщенным раствором соли (20 мл x 2), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают преп-ТСХ (ПЭ/ЭА=3/2 об/об) с получением 5-бром-3-этил-1H-пирроло[2,3-b]пиридина в виде беловатого твердого вещества (70 мг, 33% выход). ЖХ-МС (ИЭР) m/z: 225 [M+H]+.
Стадия 3: К раствору 5-бром-3-этил-1H-пирроло[2,3-b]пиридина (200 мг, 888,55 мкмоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (270,77 мг, 1,07 ммоль) в 1,4-диоксане (6 мл) добавляют CH3COOK (174,41 мг, 1,78 ммоль) и Pd(dppf)Cl2.CH2Cl2 (72,56 мг, 88,86 мкмоль). Смесь перемешивают при 110°C в атмосфере азота в течение ночи, охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют, и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=2/1 об/об) и далее обрабатывают ультразвуком в ПЭ (10 мл) с получением 3-этил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пирроло[2,3-b]пиридина в виде белого твердого вещества (40 мг, 9% выход). ЖХ-МС (ИЭР) m/z: 273 [M+H]+.
Промежуточное соединение 13
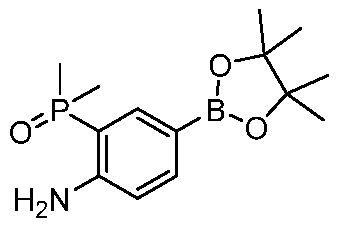
(2-Амино-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)диметилфосфин оксид
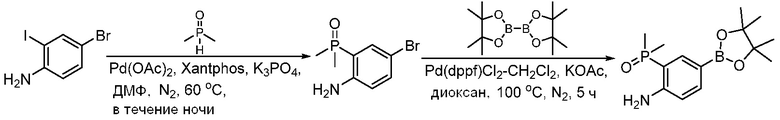
Стадия 1: Смесь 4-бром-2-йоданилина (2,0 г, 6,71 ммоль), диметилфосфина оксида (628,76 мг, 8,06 ммоль), Pd(OAc)2 (150,71 мг, 671,33 мкмоль), Xantphos (776,86 мг, 1,34 ммоль) и K3PO4 (2,85 г, 13,41 ммоль) в ДМФ (20 мл) перемешивают под N2 при 60℃ в течение ночи. После охлаждения до комнатной температуры, смесь фильтруют и промывают ЭА (40 мл). Фильтрат разбавляют H2O (40 мл) и экстрагируют ЭА (50 мл x 30). Органический слой промывают насыщенным раствором соли (40 мл x 3), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=60/1 об/об) с получением (2-амино-5-бромфенил)диметилфосфина оксида в виде желтого твердого вещества (1,6 г, 96% выход). ЖХ-МС (ИЭР) m/z: 248 [M+H]+.
Стадия 2: Смесь (2-амино-5-бромфенил)диметилфосфина оксида (1,53 г, 6,17 ммоль), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолана (1,88 г, 7,41 ммоль), Pd(dppf)Cl2-CH2Cl2 (503,70 мг, 616,80 мкмоль) и KOAc (1,21 г, 12,34 ммоль) в диоксане (20 мл) перемешивают под N2 при 100°С в течение 5 часов. После охлаждения до комнатной температуры, смесь фильтруют и промывают ЭА (150 мл). Органический слой концентрируют и очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1 об/об) с получением (2-амино-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)диметилфосфина оксида в виде черного масла (1,16 г, 64% выход). ЖХ-МС (ИЭР) m/z: 296 [M+H]+.
Промежуточное соединение 14
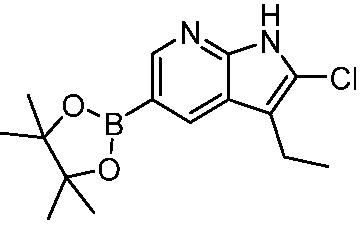
2-Хлор-3-этил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пирроло[2,3-b]пиридин
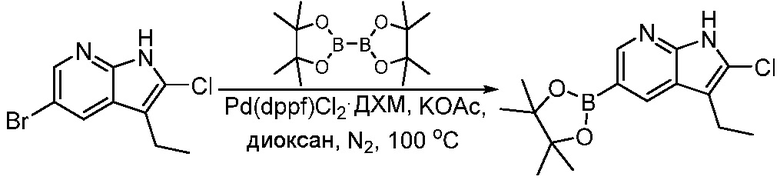
Смесь 5-бром-2-хлор-3-этил-2,3-дигидро-1H-пирроло[2,3-b]пиридина (50 мг, 0,19 ммоль), 4,4,6,6-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборинана (73 мг, 0,27 ммоль), безводного AcOK (56 мг, 0,58 ммоль) и Pd(dppf)Cl2.ДХМ (15 мг, 0,02 ммоль) в безводном диоксане (1,5 мл) перемешивают в условиях микроволн защищенных азотом при 100°С в течение 3 часов. После охлаждения до комнатной температуры, смесь фильтруют, и фильтрат концентрируют с получением неочищенного 2-хлор-3-этил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пирроло[2,3-b]пиридина (50 мг), который применяют на следующей стадии без дальнейшей очистки. ЖХ-МС (ИЭР) m/z: 307 [M+H]+.
Промежуточное соединение 15
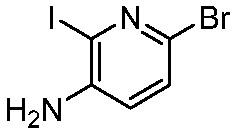
6-Бром-2-йодпиридин-3-амин
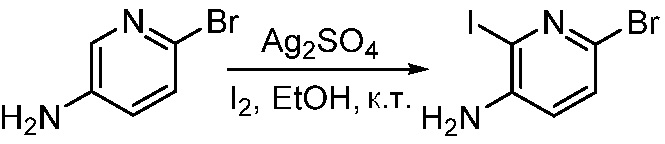
К суспензии 6-бромпиридин-3-амина (1 г, 5,68 ммоль) и сульфата серебра (1,77 г, 5,68 ммоль) в этаноле (20 мл) добавляют йод (1,44 г, 5,68 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи, разбавляют ПЭ (20 мл) и фильтруют. Фильтрат концентрируют и остаток обрабатывают ультразвуком в ЭА (30 мл). Твердое вещество собирают фильтрацией и сушат с получением 6-бром-2-йод-пиридин-3-амина в виде коричневого твердого вещества (380 мг, 22% выход). ЖХ-МС (ИЭР) m/z: 299 [M+H]+.
Промежуточное соединение 16
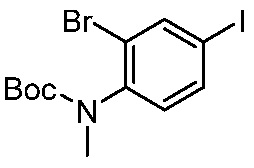
трет-Бутил (2-бром-4-йодфенил)(метил)карбамат
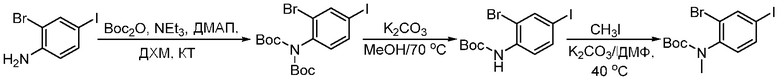
Стадия 1: К раствору 2-бром-4-йод-анилина (1 г, 3,36 ммоль) в ДХМ (10 мл) добавляют ТЭА (339,66 мг, 467,85 мкл), ДМАП (410,08 мг, 3,36 ммоль) и Boc2O (732,58 мг, 3,36 ммоль). Смесь перемешивают при комнатной температуре в течение 5 часов и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1 об/об) с получением трет-бутил N-(2-бром-4-йод-фенил)-N-трет-бутоксикарбонил-карбамата в виде белого твердого вещества (732,58 мг, 43% выход).
Стадия 2: К раствору трет-бутил N-(2-бром-4-йод-фенил)-N-трет-бутоксикарбонил-карбамата (800 мг, 1,61 ммоль) в метаноле (20 мл) добавляют карбонат калия (665,87 мг, 4,82 ммоль). Смесь перемешивают при 70°C в течение 2 часов и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1 об/об) с получением трет-бутил N-(2-бром-4-йод-фенил)карбамата в виде белого твердого вещества (462 мг, 72% выход).
Стадия 3: К раствору трет-бутил N-(2-бром-4-йод-фенил)карбамата (400 мг, 1,00 ммоль) в ДМФ (10 мл) добавляют карбонат калия (277,79 мг, 2,01 ммоль) и йодметан (213,96 мг, 1,51 ммоль). Смесь перемешивают при 40°C в течение ночи. После охлаждения до комнатной температуры, смесь разбавляют H2O (30 мл) и экстрагируют ЭА (30 мл x 3). Органический слой промывают насыщенным раствором соли (20 мл x 3), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1 об/об) с получением трет-бутил N-(2-бром-4-йод-фенил)-N-метил-карбамата в виде желтого твердого вещества (400 мг, 96% выход). ЖХ-МС (ИЭР) m/z : 356 [M+H-55]+.
Промежуточное соединение 17
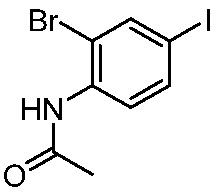
N-(2-бром-4-йодфенил)ацетамид
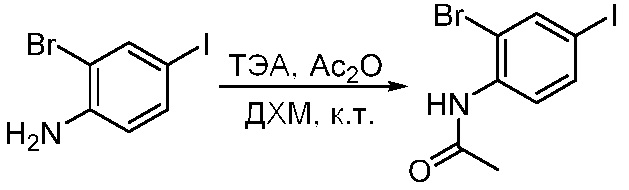
К перемешиваемому раствору 2-бром-4-йод-анилина (1,0 г, 3,36 ммоль) в ДХМ (10 мл) добавляют триэтиламин (680,00 мг, 6,72 ммоль, 0,94 мл), затем уксусный ангидрид (411,63 мг, 4,03 ммоль, 381,14 мкл). Смесь перемешивают при комнатной температуре в течение ночи и нагревают при 40°С в течение 4 часов. После охлаждения до комнатной температуры, смесь концентрируют с получением N-(2-бром-4-йод-фенил)ацетамида в виде серого твердого вещества (1,1 г, 96% выход). ЖХ-МС (ИЭР) m/z: 340 [M+H]+.
Промежуточное соединение 18
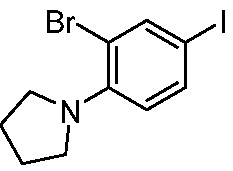
1-(2-Бром-4-йодфенил)пирролидин
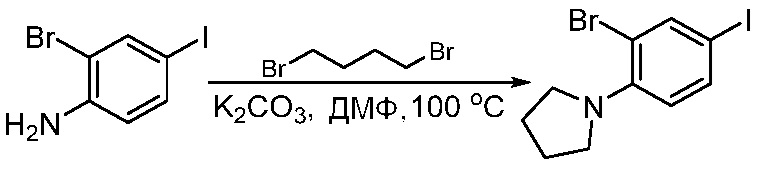
Смесь 2-бром-4-йод-анилина (1,0 г, 3,36 ммоль), 1,4-дибромбутана (869,69 мг, 4,03 ммоль) и карбоната калия (927,84 мг, 6,71 ммоль) в ДМФ (20 мл) перемешивают при 100°C в течение ночи. После охлаждения до комнатной температуры, смесь выливают в воду (40 мл) и экстрагируют ЭА (50 мл x 3). Органический слой промывают насыщенным раствором соли (30 мл x 3), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ, 100% об/об) с получением 1-(2-бром-4-йод-фенил)пирролидина в виде желтого масла (200 мг, 17% выход). ЖХ-МС (ИЭР) m/z: 352 [M+H]+.
Промежуточное соединение 19
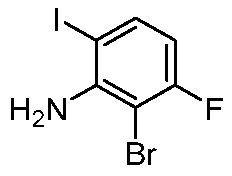
2-Бром-3-фтор-6-йоданилин
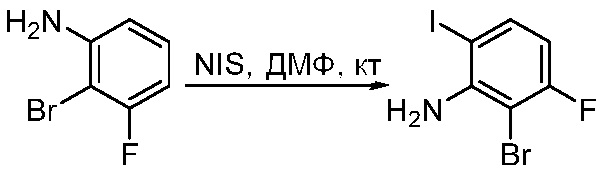
К раствору 2-бром-3-фтор-анилина (200 мг, 1,05 ммоль) в ДМФ (2 мл) добавляют по каплям раствор NIS (236,81 мг, 1,05 ммоль) в ДМФ (2 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, разбавляют ЭА (30 мл) и промывают насыщенным раствором соли (20 мл x 2). Органический слой сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=20/1 об/об) с получением 2-бром-3-фтор-6-йод-анилина в виде коричневого твердого вещества (330 мг, 99% выход). ЖХ-МС (ИЭР) m/z: 316 [M+H]+.
Промежуточное соединение 20
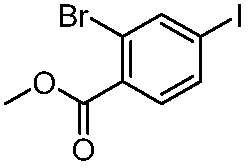
Метил 2-бром-4-йодбензоат
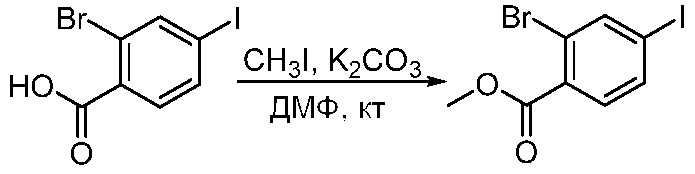
К раствору 2-бром-4-йод-бензойной кислоты (1 г, 3,06 ммоль), карбоната калия (845,55 мг, 6,12 ммоль) в ДМФ (15 мл) добавляют йодметан (651,27 мг, 4,59 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, гасят H2O (50 мл) и экстрагируют ЭА (25 мл x 2). Объединенные органические слои промывают насыщенным раствором соли (30 мл x 3), сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением метил 2-бром-4-йод-бензоата в виде коричневого масла (1 г, 95% выход). ЖХ-МС (ИЭР) m/z: 341 [M+H]+.
Промежуточное соединение 21
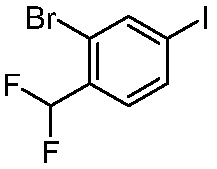
2-Бром-1-(дифторметил)-4-йодбензол
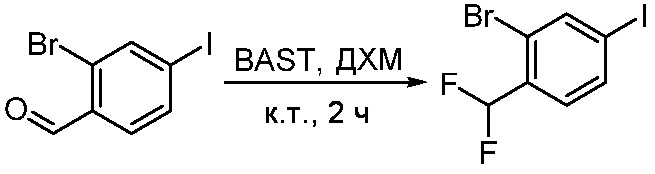
К раствору 2-бром-4-йодбензальдегида (200 мг, 0,64 ммоль) в ДХМ (6 мл) добавляют BAST (284 мг, 1,29 ммоль) при 0°С. Смесь перемешивают при комнатной температуре в течение 2 часов и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=20/1 об/об) с получением 2-бром-1-(дифторметил)-4-йодбензола в виде белого твердого вещества. (180 мг, 84% выход).
Промежуточное соединение 22
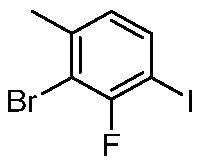
2-Бром-3-фтор-4-йод-1-метилбензол
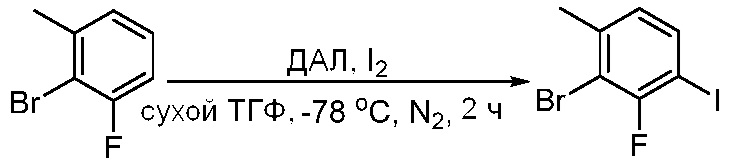
К раствору 2-бром-1-фтор-3-метил-бензола (500 мг, 2,65 ммоль) в безводном ТГФ (5 мл) по каплям добавляют ДАЛ (2 N в ТГФ, 2 мл, 4,0 ммоль) под N2 при -70°C в течение 10 минут и смесь перемешивают при -70°C в течение 30 минут. Раствор I2 (2,01 г, 7,94 ммоль) в безводном ТГФ (2 мл) добавляют по каплям к вышеуказанной смеси при -70°C и полученную смесь перемешивают при -70°C в течение 3 часов. Затем нагревают до комнатной температуры, смесь выливают в насыщенный водный раствор NH4Cl (20 мл) и экстрагируют ЭА (20 мл x 2). Объединенные органические слои промывают насыщенным раствором соли (30 мл x 2), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают преп-ТСХ (ПЭ, 100% об/об) с получением 3-бром-2-фтор-1-йод-4-метил-бензола в виде бесцветного масла (110 мг, 13% выход).
Промежуточное соединение 24
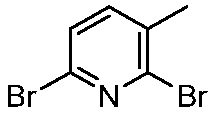
2,6-дибром-3-метилпиридин
К раствору 6-бром-5-метил-пиридин-2-амина (100 мг, 0,535 ммоль) в 48% водном растворе бромистоводородной кислоты (3 мл) добавляют по каплям бром (69,2 мг, 0,855 ммоль) при -5°C. Раствор нитрита натрия (99,6 мг, 1,44 ммоль) в воде (2 мл) добавляют по каплям к вышеуказанной смеси при -5°C. Полученную смесь перемешивают при комнатной температуре в течение 3 часов, охлаждают до -5°C, гасят раствором KOH (100 мг) в воде (5 мл) и экстрагируют ЭА (20 мл x 2). Объединенные органические слои промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ТСХ (ПЭ/ЭА=10/1 об/об) с получением 2,6-дибром-3-метил-пиридина в виде белого твердого вещества (20 мг, 15% выход). ЖХ-МС (ИЭР) m/z: 250 [M+H]+.
Промежуточное соединение 25
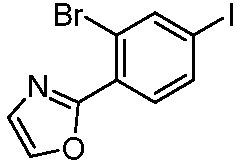
2-(2-Бром-4-йодфенил)оксазол
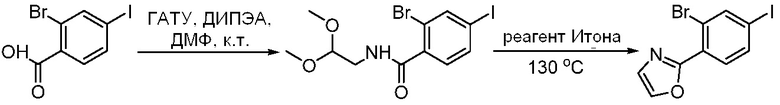
Стадия 1: К перемешиваемому раствору 2-бром-4-йод-бензойной кислоты (1 г, 3,06 ммоль) в ДМФ (10 мл) добавляют ГАТУ (1,74 г, 4,59 ммоль) и ДИПЭА (1,19 г, 9,18 ммоль, 1,60 мл). После перемешивания в течение 10 минут, 2,2-диметоксиэтанамин (418,08 мг, 3,98 ммоль, 433,25 мкл) добавляют к вышеуказанной смеси. Смесь перемешивают в течение ночи, разбавляют ЭА (20 мл) и промывают водным раствором NaHCO3 (30 мл x 2). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1 об/об) с получением 2-бром-N-(2,2-диметоксиэтил)-4-йод-бензамида в виде желтого твердого вещества (1 г, 2,42 ммоль). ЖХ-МС (ИЭР) m/z: 382 [M-MeO]+.
Стадия 2: Раствор 2-бром-N-(2,2-диметоксиэтил)-4-йод-бензамида (1 г, 2,42 ммоль) в реагенте Итона (10 мл) перемешивают при 130°C в течение ночи. После охлаждения до комнатной температуры, раствор выливают в лед-воду (60 мл). Полученную суспензию фильтруют и сушат с получением 2-(2-бром-4-йод-фенил)оксазола в виде серого твердого вещества (840 мг, 2,40 ммоль, 99% выход). ЖХ-МС (ИЭР) m/z: 350 [M+H]+.
Промежуточное соединение 26
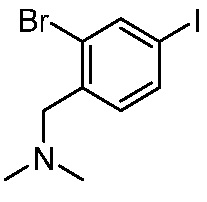
1-(2-Бром-4-йодфенил)-N, N-диметилметанамин
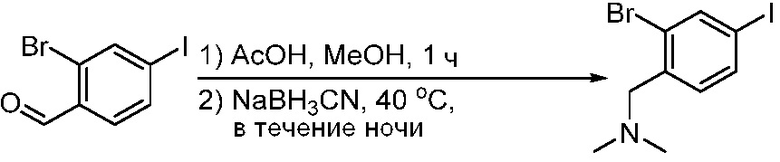
Смесь 2-бром-4-йодбензальдегида (300 мг, 0,94 ммоль), диметиламина (2 M в ТГФ, 0,58 мл, 1,16 ммоль) и AcOH (5 капли) в MeOH (8 мл) перемешивают под N2 при комнатной температуре в течение 1 часа. Затем NaBH3CN (90 мг, 1,45 ммоль) добавляют к вышеуказанной смеси, и полученную смесь перемешивают при 40°С в течение 16 часов. После охлаждения до комнатной температуры, смесь концентрируют и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ 100% об/об и затем ПЭ/ЭА=5/1, об/об) с получением 1-(2-бром-4-йодфенил)-N, N-диметилметанамина в виде желтого масла (10 мг, 29% выход). ЖХ-МС (ИЭР) m/z: 340 [M+H]+.
Промежуточное соединение 27
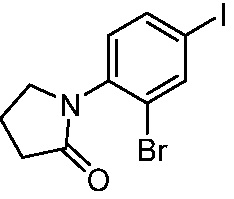
1-(2-Бром-4-йодфенил)пирролидин-2-он
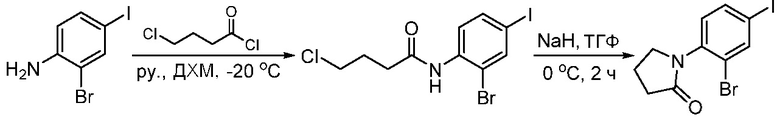
Стадия 1: К смеси 2-бром-4-йоданилина (600 мг, 2,0 ммоль) в ДХМ (10 мл) добавляют пиридин (175 мг, 2,2 ммоль) и 4-хлорбутаноил хлорид (312 мг, 2,2 ммоль) по каплям при -20°С. Реакционную смесь перемешивают при -20°С в течение 3 часов, разбавляют водой (20 мл), промывают водным раствором HCl (2 N) и экстрагируют ЭА (30 мл x 3). Органический слой сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-бром-4-йодфенил)-4-хлорбутанамида в виде черного твердого вещества (700 мг, 86% выход). ЖХ-МС (ИЭР) m/z: 402 [M+H]+.
Стадия 2: К раствору N-(2-бром-4-йодфенил)-4-хлорбутанамида (700 мг, 1,74 ммоль) в сухом ТГФ (10 мл) добавляют NaH (60% суспензия в масле, 138 мг, 3,48 ммоль) при 0°С. Смесь перемешивают под N2 при комнатной температуре в течение 2 часов, разбавляют холодиной водой (20 мл) и экстрагируют ЭА (30 мл x 3). Органический слой промывают насыщенным раствором соли (20 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ 100% об/об и затем ПЭ/ЭА=5/1, об/об) с получением 1-(2-бром-4-йодфенил)пирролидин-2-она в виде белого твердого вещества (500 мг, 78% выход). ЖХ-МС (ИЭР) m/z: 366 [M+H]+.
Промежуточное соединение 28
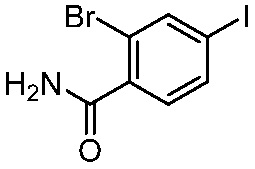
2-Бром-4-йодбензамид
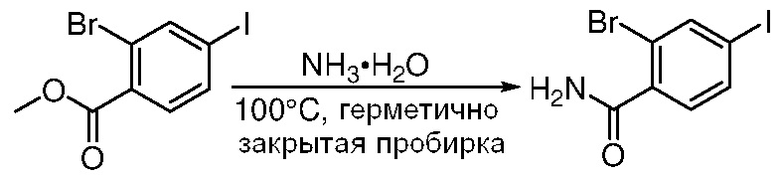
Смесь метил 2-бром-4-йод-бензоата (500 мг, 1,47 ммоль) в 28% гидроксиде аммония (5 мл) перемешивают при 100°C в герметично закрытой пробирке в течение 5 часов, охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют с получением 2-бром-4-йод-бензамида в виде белого твердого вещества (400 мг, 84% выход). ЖХ-МС (ИЭР) m/z: 326 [M+H]+.
Промежуточное соединение 29
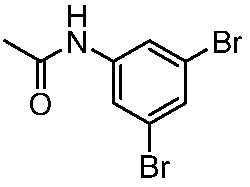
N-(3,5-дибромфенил)ацетамид
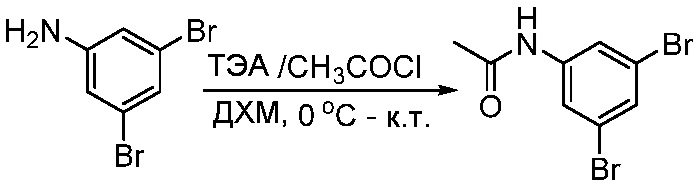
К раствору 3,5-диброманилина (200 мг, 797,07 мкмоль) в ДХМ (5 мл) добавляют ТЭА (241,97 мг, 2,39 ммоль, 333,29 мкл) и ацетил хлорид (93,85 мг, 1,20 ммоль, 72,75 мкл) при 0°C. Смесь перемешивают при комнатной температуре в течение 2 часов, разбавляют H2O (20 мл) и экстрагируют ДХМ (30 мл x 3). Объединенные экстракты сушат над безводным Na2SO4 фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1 об/об) с получением N-(3,5-дибромфенил)ацетамида в виде желтого твердого вещества (185 мг, 79% выход). ЖХ-МС (ИЭР) m/z: 292 [M+H]+.
Промежуточное соединение 30
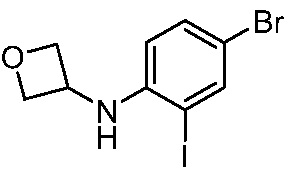
N-(4-бром-2-йодфенил)оксетан-3-амин
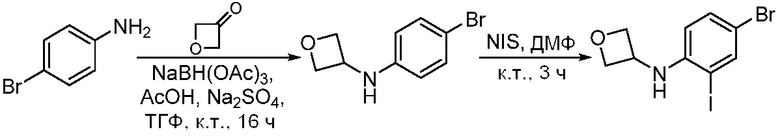
Стадия 1: Смесь 4-броманилина (4 г, 23,25 ммоль), оксетан-3-она (2,18 г, 30,23 ммоль, 1,94 мл), безводного Na2SO4 (3,30 г, 23,25 ммоль) в ТГФ (20 мл) перемешивают при комнатной температуре в течение 1 часа. Затем NaBH(OAc)3 (7,39 г, 34,88 ммоль) добавляют к вышеуказанной смеси при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 16 часов, разбавляют водой (50 мл) и экстрагируют ЭА (100 мл x 2). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (30 мл x 3) и насыщенным раствором соли (20 мл x 2), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=2/1 об/об) с получением N-(4-бромфенил)оксетан-3-амина в виде белого твердого вещества (3,4 г, 64% выход). ЖХ-МС (ИЭР) m/z: 228 [M+H]+.
Стадия 2: К раствору N-(4-бромфенил)оксетан-3-амина (1,5 г, 6,58 ммоль) в ДМФ (8 мл) добавляют по каплям раствор NIS (1,48 г, 6,58 ммоль) в ДМФ (3 мл) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 3 часов, гасят насыщенным водным раствором NaHSO3 (10 мл) и экстрагируют ЭА (60 мл x 2). Объединенные органические слои промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=4/1 об/об) с получением неочищенного N-(4-бром-2-йод-фенил)оксетан-3-амина в виде желтого твердого вещества (630 мг, 30% выход, 50% чистота). ЖХ-МС (ИЭР) m/z: 354 [M+H]+.
Соединения, представленные примерами
Пример 1
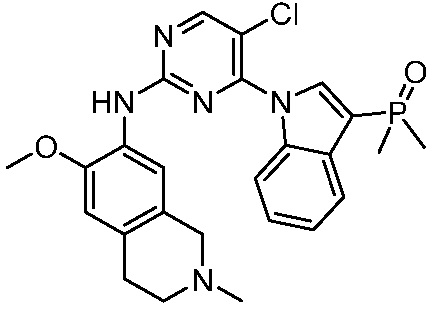
(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)диметилфосфин оксид
Стадия 1: К раствору Пр. E1 (200 мг, 1,04 ммоль) в ДМФ (5 мл) добавляют гидрид натрия (60% дисперсию в минеральном масле, 50 мг, 1,24 ммоль) при 0°С и смесь перемешивают при 0°С в течение 30 минут. К полученной выше смеси добавляют 2,4,5-трихлорпиримидин (227,9 мг, 1,24 ммоль) при 0°С, и полученную смесь затем перемешивают при комнатной температуре в течение 1 часа, охлаждают до 0°С и гасят льдом-H2O (20 мл) и экстрагируют ЭА (30 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)диметилфосфина оксида в виде белого твердого вещества (110 мг, 31% выход). ЖХ-МС (ИЭР) m/z: 340 [M+H]+.
Стадия 2: К раствору Пр. W1 (25,4 мг, 0,13 ммоль) в 2-метоксиэтаноле (3 мл) добавляют (1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)диметилфосфин оксид (30 мг, 0,09 ммоль) и HCl в растворе EtOH (2 N, 0,2 мл). Полученную смесь перемешивают при 120°С в течение 20 часов и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=15/1 об/об) и преп-ЖХВД (MeCN/0,1% HCOOH) с получением (1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)диметилфосфина оксида в виде желтого твердого вещества (8,3 мг, 19% выход). ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 1,92 (с, 3H), 1,95 (с, 3H), 2,54 (с, 3H), 2,90-2,97 (м, 4H), 3,59 (с, 2H), 3,91 (с, 3H), 6,83 (с, 1H), 7,35-7,40 (м, 2H), 7,86-7,88 (м, 1H), 7,91-7,94 (м, 1H), 7,97 (с, 1H), 8,22 (д, J=4,8 Гц, 1H), 8,65 (с, 1H).
Пример 2
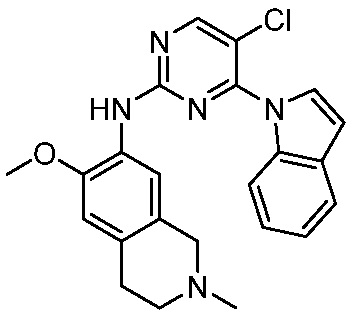
N-(5-хлор-4-(1H-индол-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 1, с применением индола вместо Пр. E1 на Стадии 1. ЖХ-МС (ИЭР) m/z: 420 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,79 (с, 3H), 3,07-3,10 (м, 2H), 3,26-3,27 (м, 2H), 3,94 (с, 5H), 6,75 (д, J=3,6 Гц, 1H), 6,89 (с, 1H), 7,21-7,30 (м, 2H), 7,65 (д, J=8,4 Гц, 1H), 7,83 (д, J=3,6 Гц, 1H), 7,81 (д, J=8,4 Гц, 1H), 8,13 (с, 1H), 8,60 (с, 1H).
Пример 3
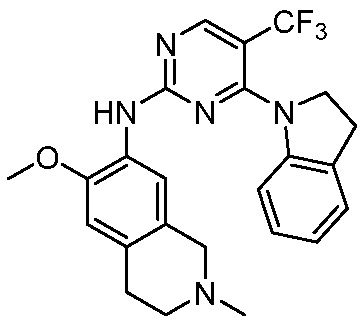
N-(4-(1H-индол-1-ил)-5-(трифторметил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 1, с применением индолина и 2,4-дихлор-5-(трифторметил)пиримидина вместо Пр. E1 и 2,4,5-трихлорпиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 420 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,55 (с, 3H), 2,92-2,96 (м, 4H), 3,19 (т, J=8,0 Гц, 2H), 3,45 (с, 2H), 3,88 (с, 3H), 4,16 (т, J=8,0 Гц, 2H), 6,78 (с, 1H), 7,02-7,07 (м, 1H), 7,17 (д, J=7,2 Гц, 1H), 7,31 (д, J=7,2 Гц, 1H), 7,55 (д, J=8,0 Гц, 1H), 7,90 (с, 1H), 8,49 (с, 1H).
Пример 4
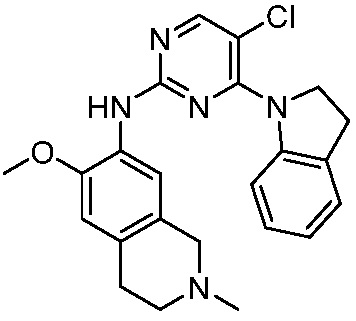
N-(5-Хлор-4-(индолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: К раствору 2,4,5-трихлорпиримидина (1 г, 5,45 ммоль) в ДМФ (20 мл) добавляют карбонат калия (1,51 г, 10,90 ммоль) и индолин (649,7 мг, 5,45 ммоль). Смесь перемешивают при 80°C в течение ночи, охлаждают до комнатной температуры, разбавляют H2O (50 мл) и экстрагируют ЭА (50 мл x 2). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/30, об/об) с получением 1-(2,5-дихлорпиримидин-4-ил)индолина в виде белого твердого вещества (900 мг, 61% выход). ЖХ-МС (ИЭР) m/z: 266 [M+H]+.
Стадия 2: Пр. W1 (30 мг, 0,16 ммоль) и 1-(2,5-дихлорпиримидин-4-ил)индолин (41,5 мг, 0,16 ммоль) применяют в условиях B1 (как представлено на Стадии 2 Примера 1) с получением N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (35 мг, 53% выход), после очистки колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1). ЖХ-МС (ИЭР) m/z: 422 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6): δ 2,33 (с, 3H), 2,59 (т, J=5,2 Гц, 2H), 2,78 (т, J=5,2 Гц, 2H), 3,13 (т, J=8,0 Гц, 2H), 3,31 (с, 2H), 3,78 (с, 3H), 4,27 (т, J=8,4 Гц, 2H), 6,75 (с, 1H), 6,93 (т, J=7,6 Гц, 1H), 7,08 (т, J=7,6 Гц, 1H), 7,25 (д, J=6,8 Гц, 1H), 7,35 (д, J=8,0 Гц, 1H), 7,56 (с, 1H), 7,96 (с, 1H), 8,24 (с, 2H).
Пример 5
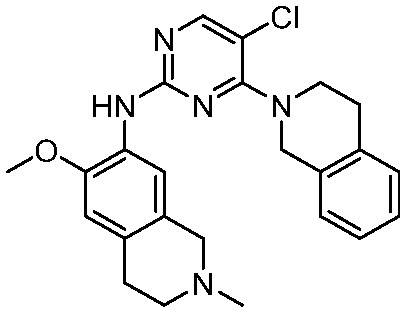
N-(5-хлор-4-(3,4-дигидроизохинолин-2(1H)-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 4, с применением 1,2,3,4-тетрагидроизохинолина вместо индолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 436 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,66 (с, 3H), 2,99-3,05 (м, 6H), 3,84 (с, 2H), 3,89 (с, 3H), 4,00 (т, J=6,0 Гц, 2H), 4,87 (с, 2H), 6,79 (с, 1H), 7,19 (с, 4H), 7,99 (с, 1H), 8,10 (с, 1H).
Пример 6
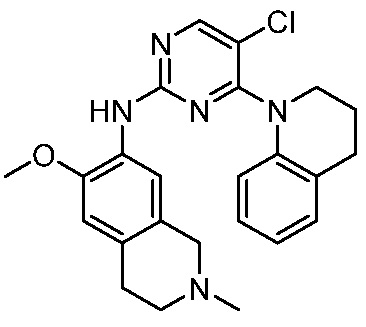
N-(5-хлор-4-(3,4-дигидрохинолин-1(2H)-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 4, с применением 1,2,3,4-тетрагидрохинолина вместо индолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 436 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 1,99-2,06 (м, 2H), 2,51 (с, 3H), 2,80-2,86 (м, 4H), 2,91 (т, J=6,0 Гц, 2H), 3,44 (с, 2H), 3,87-3,90 (м, 5H), 6,73 (с, 1H), 6,85 (д, J=8,0 Гц, 1H), 6,99-7,03 (м, 1H), 7,06-7,10 (м, 1H), 7,19 (д, J=8,0 Гц, 1H), 7,87 (с, 1H), 8,14 (с, 1H).
Пример 7
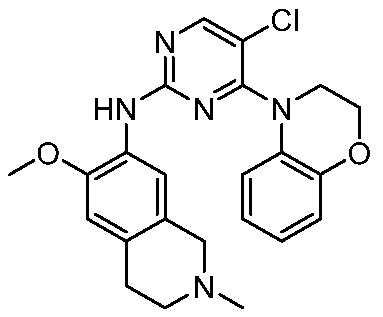
N-(5-хлор-4-(2,3-дигидро-4H-бензо[b][1,4]оксазин-4-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 4, с применением 3,4-дигидро-2H-бензо[b][1,4]оксазина вместо индолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 438 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ (ч./млн.) 2,48 (с, 3H), 2,77 (т, J=6,0 Гц, 2H), 2,88 (т, J=6,0 Гц, 2H), 3,31 (с, 2H), 3,87 (с, 3H), 3,96 (т, J=4,4 Гц, 2H), 4,28 (т, J=4,4 Гц, 2H), 6,71 (с, 1H), 6,83-6,87 (м, 1H), 6,92-7,03 (м, 3H), 7,79 (с, 1H), 8,24 (с, 1H).
Пример 8
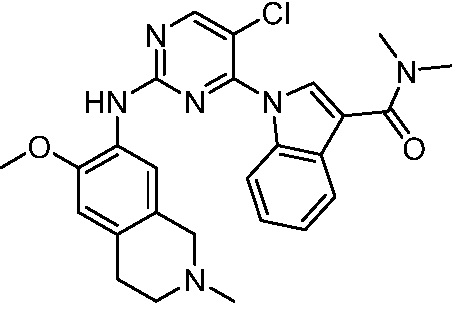
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N, N-диметил-1H-индол-3-карбоксамид
Стадия 1: Пр. E2 (200 мг, 1,06 ммоль) и 2,4,5-трихлорпиримидин (194,90 мг, 1,06 ммоль) применяют в условиях A2 (как представлено на Стадии 1 Примера 4) с получением 1-(2,5-дихлорпиримидин-4-ил)-N, N-диметил-1H-индол-3-карбоксамида в виде белого твердого вещества (165 мг, 46% выход), после очистки колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/1, об/об). ЖХ-МС (ИЭР) m/z: 335 [M+H]+.
Стадия 2: К раствору 1-(2,5-дихлорпиримидин-4-ил)-N, N-диметил-1H-индол-3-карбоксамида (52 мг, 0,16 ммоль) и Пр. W1 (29,8 мг, 0,16 ммоль) в изопропаноле (2 мл) добавляют трифторуксусную кислоту (53,1 мг, 0,48 ммоль). Смесь перемешивают под N2 при 100°С в течение 20 часов, охлаждают до комнатной температуры и очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N, N-диметил-1H-индол-3-карбоксамида в виде желтого твердого вещества (7,0 мг, 9% выход). ЖХ-МС (ИЭР) m/z: 491 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ (ч./млн.) 2,63 (с, 3H), 3,01-3,05 (м, 4H), 3,22 (с, 6H), 3,71 (с, 2H), 3,91 (с, 3H), 6,85 (с, 1H), 7,31-7,36 (м, 2H), 7,78-7,81 (м, 2H), 8,02 (с, 1H), 8,12 (с, 1H), 8,63 (с, 1H).
Пример 9
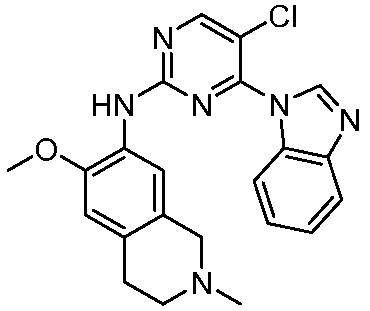
N-(4-(1H-бензо[d]имидазол-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 8, с применением 1H-бензо[d]имидазола вместо Пр. E2 на Стадии 1. ЖХ-МС (ИЭР) m/z: 421 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,87 (с, 3H), 3,12 (т, J=6,8 Гц, 2H), 3,38 (т, J=6,8 Гц, 2H), 3,93 (с, 3H), 4,06 (с, 2H), 6,91 (с, 1H), 7,41-7,44 (м, 2H), 7,78-7,81 (м, 1H), 7,85-7,88 (м, 1H), 8,03 (с, 1H), 8,69 (с, 1H), 8,79 (с, 1H).
Пример 10
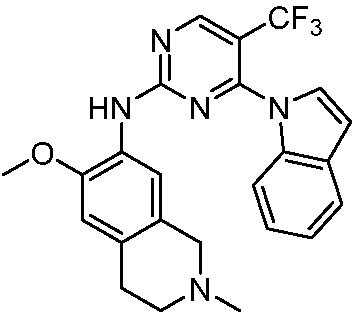
N-(4-(1H-индол-1-ил)-5-(трифторметил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: 1H-индол (349,9 мг, 3,0 ммоль) и 2,4-дихлор-5-(трифторметил)пиримидин (650,9 мг, 3,0 ммоль) применяют в условиях A1 (как представлено на Стадии 1 Примера 1) с получением 1-(2-хлор-5-(трифторметил)пиримидин-4-ил)-1H-индола в виде желтого твердого вещества (88 мг, 7% выход), после очистки преп-ЖХВД. ЖХ-МС (ИЭР) m/z: 298 [M+H]+.
Стадия 2: К раствору 1-(2-хлор-5-(трифторметил)пиримидин-4-ил)-1H-индола (20 мг, 0,067 ммоль) и Пр. W1 (15,4 мг, 0,067 ммоль) в изопропаноле (3 мл) добавляют TsOH.H2O (11,6 мг, 0,067 ммоль). Смесь перемешивают при 100°C в течение ночи, охлаждают до комнатной температуры, подщелачивают насыщенным NaHCO3 водным раствором до pH ~8, экстрагируют ДХМ (10 мл x 3). Органические фазы сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1 об/об) с получением N-(4-(1H-индол-1-ил)-5-(трифторметил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде белого твердого вещества (17 мг, 55% выход). ЖХ-МС (ИЭР) m/z: 454 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,56 (с, 3H), 2,96-2,99 (м, 4H), 3,60 (шс, 2H), 3,91 (с, 3H), 6,73 (д, J=3,6 Гц, 1H), 6,85 (с, 1H), 7,20-7,28 (м, 2H), 7,48 (д, J=2,8 Гц, 1H), 7,63 (д, J=8,4 Гц, 1H), 7,89 (д, J=8,0 Гц, 1H), 7,97 (с, 1H), 8,83 (с, 1H).
Пример 11
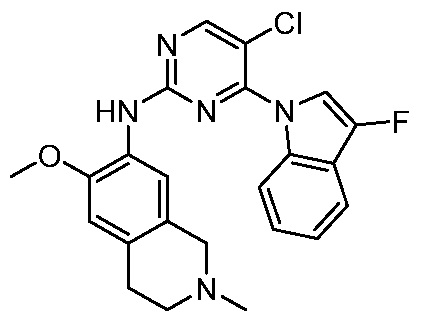
N-(5-Хлор-4-(3-фтор-1H-индол-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 10, с применением Пр. E3 и 2,4,5-трихлорпиримидина вместо 1H-индола и 2,4-дихлор-5-(трифторметил)пиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 438 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,79 (с, 1H), 8,63 (с, 1H), 7,91 (д, J=2,4 Гц, 1H), 7,83 (д, J=6,8 Гц, 1H), 7,64 (дд, J=6,0, 2,4 Гц, 1H), 7,37 (с, 1H), 7,33-7,17 (м, 2H), 6,73 (д, J=55,3 Гц, 1H), 3,96-3,68 (м, 3H), 3,33 (с, 2H), 2,79 (т, J=5,6 Гц, 2H), 2,55 (т, J=5,9 Гц, 2H), 2,30 (с, 3H).
Пример 80
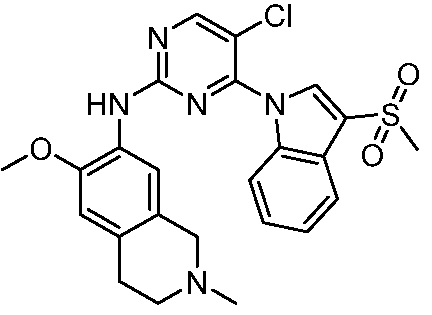
N-(5-хлор-4-(3-(метилсульфонил)-1H-индол-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 10, с применением Пр. E8 и 2,4,5-трихлорпиримидина вместо 1H-индола и 2,4-дихлор-5-(трифторметил)пиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 498 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 9,00 (с, 1H), 8,74 (с, 1H), 8,42 (с, 1H), 7,94-7,85 (м, 1H), 7,79-7,72 (м, 1H), 7,41 (дд, J=6,1, 3,1 Гц, 2H), 7,33 (с, 1H), 6,79 (с, 1H), 3,79 (с, 3H), 3,34 (с, 2H), 3,30 (с, 3H), 2,77 (д, J=5,8 Гц, 2H), 2,54 (д, J=5,7 Гц, 2H), 2,30 (с, 3H).
Пример 81
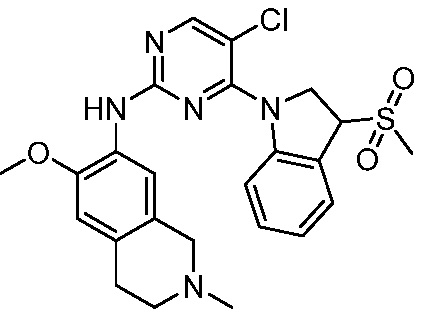
N-(5-хлор-4-(3-(метилсульфонил)индолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 10, с применением Пр. E9 и 2,4,5-трихлорпиримидина вместо 1H-индола и 2,4-дихлор-5-(трифторметил)пиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 500 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,31 (с, 1H), 8,12 (с, 1H), 7,49 (д, J=6,9 Гц, 2H), 7,41 (д, J=8,1 Гц, 1H), 7,27 (т, J=7,7 Гц, 1H), 7,06 (т, J=7,3 Гц, 1H), 6,75 (с, 1H), 5,06 (д, J=7,5 Гц, 1H), 4,72 (дд, J=12,4, 9,5 Гц, 1H), 4,58 (дд, J=12,5, 2,6 Гц, 1H), 3,77 (с, 3H), 3,28-3,21 (м, 2H), 2,89 (с, 3H), 2,77 (д, J=5,5 Гц, 2H), 2,55 (т, J=5,7 Гц, 2H), 2,30 (с, 3H).
Пример 82
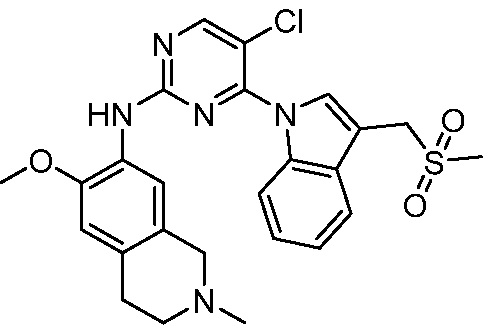
N-(5-хлор-4-(3-((метилсульфонил)метил)-1H-индол-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 10, с применением Пр. E11 и 2,4,5-трихлорпиримидина вместо 1H-индола и 2,4-дихлор-5-(трифторметил)пиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 512 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,78 (с, 1H), 8,66 (с, 1H), 7,98 (с, 1H), 7,80 (с, 2H), 7,42 (с, 1H), 7,25 (дд, J=6,0, 3,1 Гц, 2H), 6,80 (с, 1H), 4,71 (с, 2H), 3,79 (с, 3H), 3,33 (с, 2H), 2,95 (с, 3H), 2,78 (с, 2H), 2,55 (д, J=5,6 Гц, 2H), 2,30 (с, 3H).
Пример 83
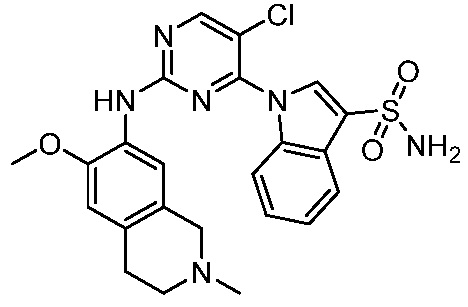
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-сульфонамид
Процедуры те же, что и для Примера 10, с применением Пр. E15 и 2,4,5-трихлорпиримидина вместо 1H-индола и 2,4-дихлор-5-(трифторметил)пиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 499 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,69 (д, J=20,3 Гц, 1H), 8,47 (с, 1H), 7,96 (с, 1H), 7,71 (дд, J=5,7, 3,1 Гц, 1H), 7,53 (дд, J=6,1, 3,1 Гц, 1H), 7,16 (с, 2H), 7,12 (дд, J=6,8, 3,7 Гц, 3H), 6,56 (с, 1H), 3,55 (с, 3H), 3,24 (с, 2H), 2,54 (с, 2H), 2,31 (д, J=5,7 Гц, 2H), 2,07 (д, J=14,9 Гц, 3H).
Пример 14
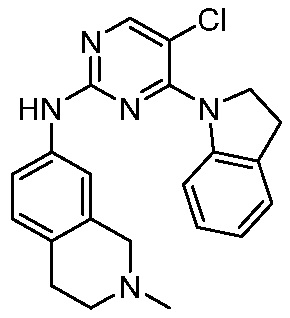
N-(5-Хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Пр. W2 (36,58 мг, 0,23 ммоль) и 1-(2,5-дихлорпиримидин-4-ил)индолин (60 мг, 0,23 ммоль) применяют в условиях B3 с получением N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (10,4 мг, 12% выход), после очистки преп-ЖХВД. ЖХ-МС (ИЭР) m/z: 392 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,51 (с, 3H), 2,83-2,93 (м, 4H), 3,17 (т, J=8,0 Гц, 2H), 3,57 (с, 2H), 4,33 (т, J=8,0 Гц, 2H), 6,93-7,02 (м, 2H), 7,12 (т, J=3,6 Гц, 1H), 7,24-7,32 (м, 2H), 7,42 (д, J=8,0 Гц, 1H), 7,52 (д, J=8,0 Гц, 1H), 8,14 (с, 1H).
Пример 15
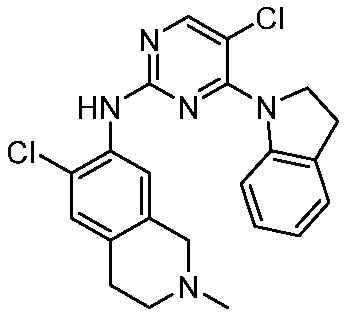
6-Хлор-N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Пр. W3 (50 мг, 0,25 ммоль) и 1-(2,5-дихлорпиримидин-4-ил)индолин (67,66 мг, 0,25 ммоль) применяют в условиях B3 с получением 6-хлор-N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (11,6 мг, 11% выход), после очистки преп-ЖХВД. ЖХ-МС (ИЭР) m/z: 426 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,54 (с, 3H), 2,89-2,96 (м, 4H), 3,18 (т, J=8,0 Гц, 2H), 3,54 (с, 2H), 4,36 (т, J=8,0 Гц, 2H), 6,96-7,00 (м, 1H), 7,11 (т, J=8,0 Гц, 1H), 7,24-7,28 (м, 2H), 7,40 (д, J=8,0 Гц, 1H), 7,93 (с, 1H), 8,19 (с, 1H).
Пример 19
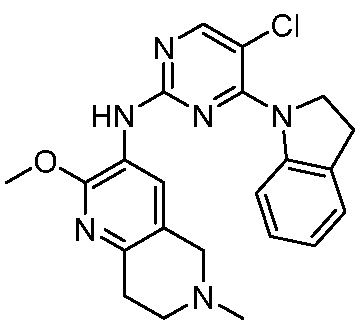
N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-метокси-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амин
Пр. W4 (30 мг, 155,24 мкмоль) и 1-(2,5-дихлорпиримидин-4-ил)индолин (41,31 мг, 155,24 мкмоль) применяют в условиях B3 (микроволны в течение 2 часов) с получением N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-метокси-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амина (соли ТФК) в виде желтого твердого вещества (6,6 мг, 10% выход), после очистки колоночной флэш-хроматографией (ДХМ/MeOH=10/1, об/об) и преп-ЖХВД (MeCN/0,05% ТФК). ЖХ-МС (ИЭР) m/z: 423 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 3,03 (с, 3H), 3,08-3,22 (м, 4H), 3,44-3,56 (м, 1H), 3,73-3,83 (м, 1H), 4,02 (с, 3H), 4,16 (д, J=6,4 Гц, 2H), 4,38 (т, J=8,0 Гц, 2H), 7,06 (т, J=7,6 Гц, 1H), 7,20 (т, J=7,4 Гц, 1H), 7,31 (д, J=8,0 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 8,25 (с, 1H), 8,34 (с, 1H).
Пример 22
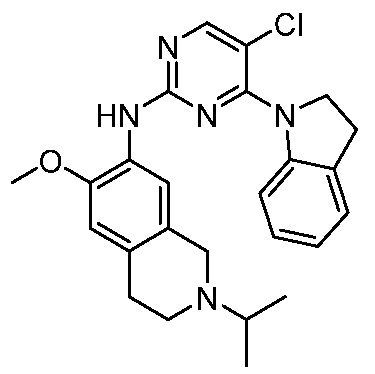
N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амин
Пр. W5 (50 мг, 0,227 ммоль) и 1-(2,5-дихлорпиримидин-4-ил)индолин (60 мг, 0,227 ммоль) применяют в условиях B3 (микроволны в течение 2 часов) с получением N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-2-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (2,5 мг, 2% выход), после очистки колоночной флэш-хроматографией (ДХМ/MeOH=10/1, об/об). ЖХ-МС (ИЭР) m/z: 450 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 1,17 (д, J=6,4 Гц, 6H), 2,86-2,92 (м, 5H), 3,19 (т, J=8,4 Гц, 2H), 3,49 (с, 2H), 3,88 (с, 3H), 4,36 (т, J=8,4 Гц, 2H), 6,73 (с, 1H), 6,99 (т, J = 6,8 Гц, 1H), 7,17 (т, J = 7,6 Гц, 1H), 7,29 (д, J=7,2 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 7,99 (с, 1H), 8,16 (с, 1H).
Пример 23
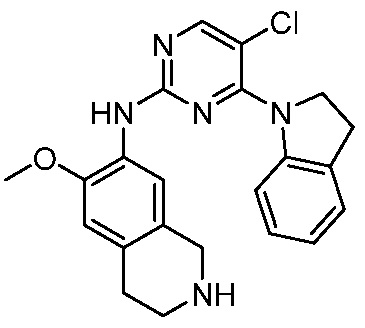
N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амин
Пр. W6 (110 мг, 0,39 ммоль) и 1-(2,5-дихлорпиримидин-4-ил)индолин (105 мг, 0,39 ммоль) применяют в условиях B3 (микроволны в течение 2 часов) с получением N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амина (1,0 соль TsOH) в виде желтого твердого вещества (130 мг, 58% выход). Твердое вещество собирают фильтрацией. ЖХ-МС (ИЭР) m/z: 408 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6): δ 2,29 (с, 3H), 2,98 (т, J=6,4 Гц, 2H), 3,16 (т, J=8,0 Гц, 2H), 3,28-3,40 (м, 2H), 3,82 (с, 3H), 4,04 (д, J=4,6 Гц, 2H), 4,37 (т, J=8,0 Гц, 2H), 6,95 (с, 1H), 7,02 (дд, J=7,4, 1,0 Гц, 1H), 7,07-7,20 (м, 3H), 7,30 (дд, J=7,4, 1,0 Гц, 1H), 7,46-7,48 (м, 3H), 7,65 (с, 1H), 8,34 (с, 1H), 8,72 (с, 1H), 9,26 (с, 2H).
Пример 26
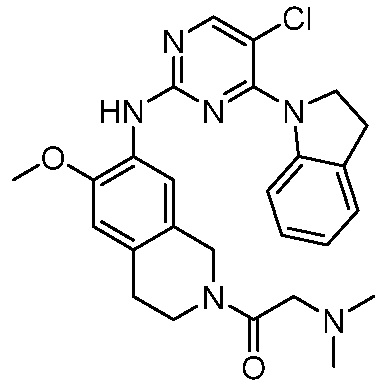
1-(7-((5-Хлор-4-(индолин-1-ил)пиримидин-2-ил)амино)-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-(диметиламино)этан-1-он
К раствору 2-(диметиламино)уксусной кислоты (5,06 мг, 49,03 мкмоль), N-(5-хлор-4-индолин-1-ил-пиримидин-2-ил)-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амина тозилата (20 мг, 34,48 мкмоль) и ГАТУ (18,64 мг, 49,03 мкмоль) в сухом CH2Cl2 (3 мл) добавляют по каплям ДИПЭА (19,01 мг, 147,10 мкмоль, 25,62 мкл) при 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и концентрируют в вакууме. Остаток очищают преп-ЖХВД (MeCN/ 0,1% HCOOH) с получением 1-(7-((5-хлор-4-(индолин-1-ил)пиримидин-2-ил)амино)-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)-2-(диметиламино)этан-1-она в виде белого твердого вещества (13,5 мг, 56% выход). ЖХ-МС (ИЭР) m/z: 493 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,57 (с, 3H), 2,62 (с, 3H), 2,75 (т, J=6,4 Гц, 1H), 2,86 (т, J=6,4 Гц, 1H), 3,14 (т, J=8,4 Гц, 2H), 3,54 (с, 1H), 3,64-3,69 (м, 2H), 3,71 (с, 1H), 3,88 (д, J = 6,4 Гц, 3H), 4,30 (с, 1H), 4,32-4,36 (м, 2H), 4,42 (с, 1H), 6,75 (с, 1H), 6,98-7,04 (м, 1H), 7,16 (т, J=8,0 Гц, 1H), 7,24-7,29 (м, 1H), 7,39-7,46 (м, 1H), 8,01 (д, J=17,2 Гц, 1H), 8,15 (д, J=9,6 Гц, 1H).
Пример 27
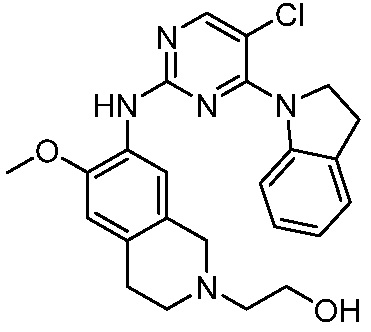
2-(7-((5-Хлор-4-(индолин-1-ил)пиримидин-2-ил)амино)-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этан-1-ол
Смесь N-(5-хлор-4-(индолин-1-ил)пиримидин-2-ил)-6-метокси-1,2,3,4-тетрагидроизохинолин-7-амина тозилата (30 мг, 0,073 ммоль), 2-((трет-бутилдиметилсилил)окси)ацетальдегида (13 мг, 0,073 ммоль) и AcOH (2 капли) в MeOH (4 мл) перемешивают под N2 в течение 1 часа. Затем NaBH(OAc)3 (24 мг, 011 ммоль) добавляют к вышеуказанной смеси и смесь перемешивают при комнатной температуре в течение 16 часов и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ 100% об/об и затем ДХМ/MeOH=5/1 об/об) с получением 2-(7-((5-хлор-4-(индолин-1-ил)пиримидин-2-ил)амино)-6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этан-1-ола в виде желтого твердого вещества (6,4 мг, 20% выход). ЖХ-МС (ИЭР) m/z: 452 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,72 (т, J=6,4 Гц, 2H), 2,86-2,91 (м, 4H), 3,19 (т, J=8,4 Гц, 2H), 3,47 (с, 2H), 3,77 (т, J=6,4 Гц, 2H), 3,88 (с, 3H), 4,35 (т, J=8,4 Гц, 2H), 6,73 (с, 1H), 7,02 (т, J=7,6 Гц, 1H), 7,18 (т, J=8,0 Гц, 1H), 7,29 (д, J=7,6 Гц, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,96 (с, 1H), 8,16 (с, 1H).
Пример 28
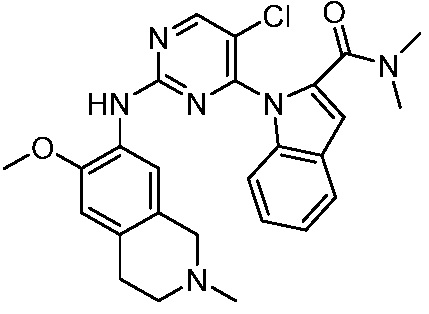
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N, N-диметил-1H-индол-2-карбоксамид
Стадия 1: Пр. E4 (673 мг, 3,58 ммоль) и 2,4,5-трихлорпиримидин (787 мг, 4,29 ммоль) применяют в условиях A1 (как представлено на Стадии 1 Примера 1) с получением 1-(2,5-дихлорпиримидин-4-ил)-N, N-диметил-индол-2-карбоксамида в виде белого твердого вещества (160 мг, 22% выход), после очистки колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/3, об/об) и преп-ЖХВД. ЖХ-МС (ИЭР) m/z: 335. [M+H]+.
Стадия 2: К раствору 1-(2,5-дихлорпиримидин-4-ил)-N, N-диметил-индол-2-карбоксамида (50 мг, 0,15 ммоль) в диоксане (4 мл) добавляют Пр. W1 (28,7 мг, 0,15 ммоль), Pd2(dba)3 (27,3 мг, 0,03 ммоль), Xantphos (17,3 мг, 0,03 ммоль) и карбонат калия (41,2 мг, 0,3 ммоль). Смесь перемешивают при 100°C под N2 в течение 12 часов, охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют, и остаток очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N, N-диметил-1H-индол-2-карбоксамида в виде желтого твердого вещества (2,4 мг, 3% выход) [соль HCOOH]. ЖХ-МС (ИЭР) m/z: 491 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,71 (с, 3H), 2,99 (с, 3H), 3,02-3,05 (м, 2H), 3,14-3,17 (м, 2H), 3,30 (с, 3H), 3,88 (с, 2H), 3,91 (с, 3H), 6,85 (с, 1H), 7,10 (с, 1H), 7,26 (т, J=8,0 Гц, 1H), 7,35 (т, J=8,0 Гц, 1H), 7,49 (д, J=8,0 Гц, 1H), 7,73 (д, J=8,0 Гц, 1H), 8,02 (с, 1H), 8,50 (шс, 1H), 8,57 (с, 1H).
Пример 12
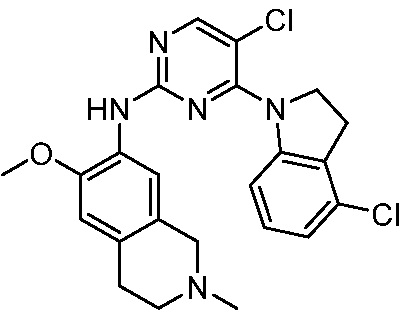
N-(5-хлор-4-(4-хлориндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: 4-Хлориндолин (111 мг, 1,06 ммоль) и 2,4,5-трихлорпиримидин (200 мг, 1,09 ммоль) применяют в условиях A2 (как представлено на Стадии 1 Примера 4) с получением 4-хлор-1-(2,5-дихлорпиримидин-4-ил)индолина в виде белого твердого вещества (160 мг, 59% выход), после очистки колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=20/1, об/об). ЖХ-МС (ИЭР) m/z: 300 [M+H]+.
Стадия 2: Пр. W1 (68,5 мг, 299,43 мкмоль) и 4-хлор-1-(2,5-дихлорпиримидин-4-ил)индолин (90 мг, 299,43 мкмоль) применяют в условиях B3 (как представлено на Стадии 2 Примера 10) с получением N-(5-хлор-4-(4-хлориндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (соль TsOH) в виде желтого твердого вещества (115 мг, 84% выход). Твердое вещество собирают фильтрацией. ЖХ-МС (ИЭР) m/z: 456 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,35 (с, 3H), 3,02 (с, 3H), 3,05-3,16 (м, 1H), 3,16-3,30 (м, 3H), 3,41-3,45 (м, 1H), 3,74-3,77 (м, 1H), 3,87 (с, 3H), 4,13-4,17 (м, 1H), 4,32-4,35 (м, 1H), 4,67 (т, J=8,4 Гц, 2H), 7,04 (с, 1H), 7,13-7,21 (м, 4H), 7,49 (с, 1H), 7,65-7,67 (м, 3H), 8,21 (с, 1H).
Пример 13
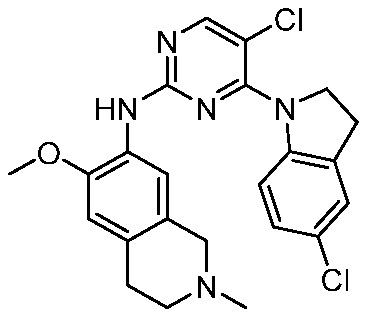
N-(5-хлор-4-(5-хлориндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 5-хлориндолин и вместо 4-хлориндолин на Стадии 1. ЖХ-МС (ИЭР) m/z: 456 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,35 (с, 3H), 3,04 (с, 3H), 3,17-3,29 (м, 4H), 3,41-3,48 (м, 1H), 3,75-3,79 (м, 1H), 3,88 (с, 3H), 4,13-4,16 (м, 1H), 4,27-4,31 (м, 1H), 4,64 (т, J=8,0 Гц, 2H), 7,04 (с, 1H), 7,16-7,21 (м, 3H), 7,38 (шс, 1H), 7,53 (с, 1H), 7,65-7,72 (м, 3H), 8,20 (с, 1H).
Пример 18
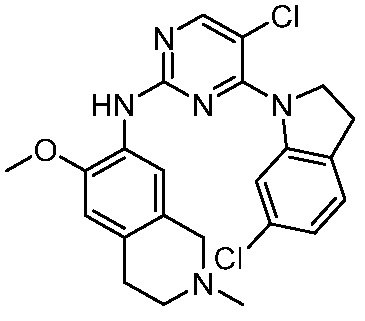
N-(5-хлор-4-(6-хлориндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 6-хлориндолина и вместо 4-хлориндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 456 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,35 (с, 0,45H), 3,01 (с, 3H), 3,13-3,20 (м, 4H), 3,35-3,40 (м, 1H), 3,73-3,77 (м, 1H), 3,90 (с, 3H), 4,10-4,14 (м, 1H), 4,31-4,35 (м, 1H), 4,59 (т, J=8,4 Гц, 2H), 7,02 (с, 1H), 7,09-7,12 (м, 1H), 7,21 (д, J=8,0 Гц, 0,3H), 7,29 (д, J=8,0 Гц, 1H), 7,6-7,69 (м, 2,3H), 8,24 (с, 1H).
Пример 16
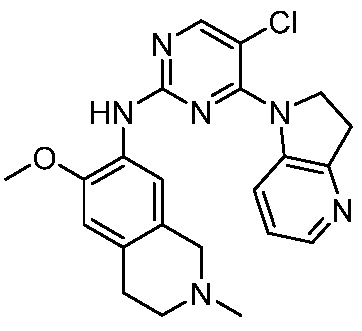
N-(5-хлор-4-(2,3-дигидро-1H-пирроло[3,2-b]пиридин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 2,3-дигидро-1H-пирроло[3,2-b]пиридина гидрохлорида и вместо 4-хлориндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 423 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6): δ 2,39 (с, 3H), 2,67 (шс, 2H), 2,81-2,84 (м, 2H), 3,21 (т, J=8,4 Гц, 2H), 3,41 (с, 2H), 3,76 (с, 3H), 4,36 (т, J=8,4 Гц, 2H), 6,79 (с, 1H), 6,99-7,02 (м, 1H), 7,41 (с, 1H), 7,65 (д, J=8,0 Гц, 1H), 8,03 (дд, J=5,2, 1,2 Гц, 1H), 8,17 (с, 1H), 8,25 (с, 1H).
Пример 29
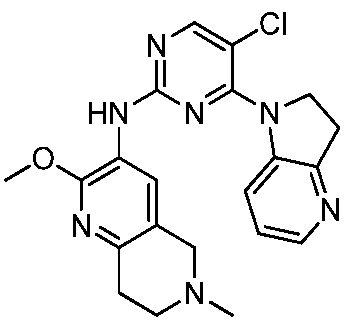
N-(5-Хлор-4-(2,3-дигидро-1H-пирроло[3,2-b]пиридин-1-ил)пиримидин-2-ил)-2-метокси-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-амин
Та же методика, как для Стадии 2 Примера 28, с применением 1-(2,5-дихлорпиримидин-4-ил)-2,3-дигидропирроло[3,2-b]пиридина и Пр. W4 вместо 1-(2,5-дихлорпиримидин-4-ил)-N, N-диметил-индол-2-карбоксамида и Пр. W1. ЖХ-МС (ИЭР) m/z: 424 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,51 (с, 3H), 2,89-2,92 (м, 4H), 3,27-3,34 (м, 2H), 3,47 (с, 2H), 3,97 (с, 3H), 4,45 (т, J=8,8 Гц, 2H), 7,16-7,19 (м, 1H), 7,76 (дд, J=8,0, 0,8 Гц, 1H), 8,05 (дд, J=4,8, 1,2 Гц, 1H), 8,09 (с, 1H), 8,25 (с, 1H), 8,51 (ш, 1H).
Пример 17
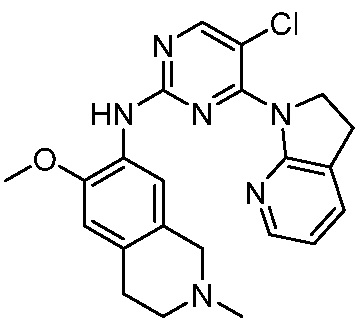
N-(5-хлор-4-(2,3-дигидро-1H-пирроло[2,3-b]пиридин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 2,3-дигидро-1H-пирроло[2,3-b]пиридина вместо 4-хлориндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 423 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,44 (с, 3H), 2,71-2,74 (м, 2H), 2,87-2,91 (м, 2H), 3,21 (т, J=8,0 Гц, 2H), 3,47 (с, 2H), 3,87 (с, 3H), 4,25 (т, J=8,4 Гц, 2H), 6,73 (с, 1H), 6,89-6,92 (м, 1H), 7,61 (дд, J=7,2, 1,6 Гц, 1H), 8,00 (с, 2H), 8,30 (с, 1H).
Пример 20
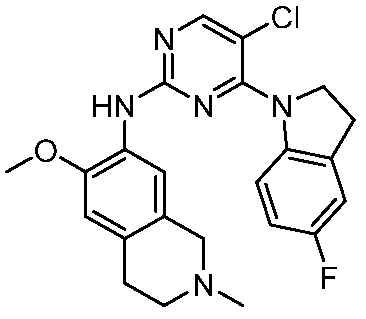
N-(5-хлор-4-(5-фториндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 5-фториндолина вместо 4-хлориндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 440 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,35 (с, 1,5H), 2,79 (с, 3H), 3,06 (т, J=6,0 Гц, 2H), 3,19 (т, J=8,4 Гц, 2H), 3,25 (т, J=6,0 Гц, 2H), 3,84 (с, 2H), 3,90 (с, 3H), 4,38 (т, J=8,4 Гц, 2H), 6,82 (с, 1H), 6,90-6,95 (м, 1H), 7,06 (дд, J=8,4, 2,4 Гц, 1H), 7,20 (д, J=8,4 Гц, 1H), 7,38-7,41 (м, 1H), 7,67 (д, J=8,4 Гц, 1H), 7,98 (с, 1H), 8,18 (с, 1H).
Пример 21
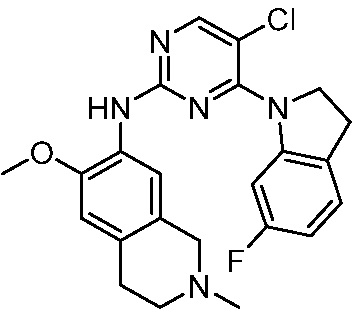
N-(5-хлор-4-(6-фториндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 6-фториндолина вместо 4-хлориндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 440 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,35 (с, 0,24H), 3,04 (с, 3H), 3,20 (м, 3H), 3,35-3,45 (м, 2H), 3,75-3,80 (м, 1H), 3,87 (с, 3H), 4,17 (д, J=14,8 Гц, 1H), 4,42 (д, J=14,8 Гц, 1H), 4,70 (т, J=7,6 Гц, 2H), 6,87-6,92 (м, 1H), 7,06 (с, 1H), 7,21 (д, J=8,0 Гц, 0,2H), 7,28-7,32 (м, 1H), 7,42 (с, 1H), 7,42-7,46 (м, 1H), 7,67 (д, J=8,4 Гц, 0,16H), 8,22 (с, 1H).
Пример 24
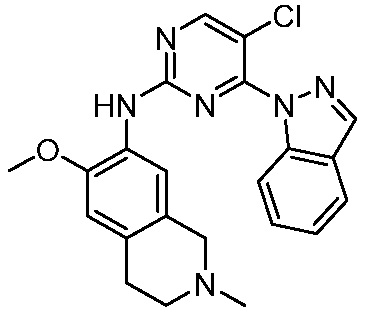
N-(5-Хлор-4-(1H-индазол-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 12, с применением 1H-индазола и вместо 4-хлориндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 421 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,88 (с, 1H), 8,61 (с, 1H), 8,50 (с, 1H), 8,23 (с, 1H), 8,07 (д, J=7,9 Гц, 1H), 7,89 (д, J=7,9 Гц, 1H), 7,48-7,39 (м, 1H), 7,35 (дд, J=8,9, 5,9 Гц, 2H), 6,84 (с, 1H), 3,77 (с, 3H), 3,37-3,37 (м, 2H), 2,83 (т, J=5,8 Гц, 2H), 2,59 (т, J=5,7 Гц, 2H), 2,30 (д, J=13,4 Гц, 3H).
Пример 25
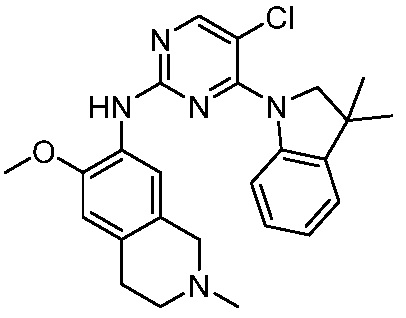
N-(5-Хлор-4-(3,3-диметилиндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: К раствору 3,3-диметилиндолина (200 мг, 1,36 ммоль) в н-бутаноле (2 мл) последовательно добавляют 2,4,5-трихлорпиримидин (250 мг, 1,36 ммоль) и ДИПЭА (175,6 мг, 1,36 ммоль). Реакционную смесь перемешивают при 100°C в течение 18 часов, разбавляют водой (5 мл) и экстрагируют ЭА (10 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/10, об/об) с получением 1-(2,5-дихлорпиримидин-4-ил)-3,3-диметил-индолина (385 мг, 94,41% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 296 [M+H]+.
Стадия 2: Пр. W1 (86 мг, 0,323 ммоль) и 1-(2,5-дихлорпиримидин-4-ил)-3,3-диметил-индолин (100 мг, 0,323 ммоль) применяют в условиях B3 (как представлено на Стадии 2 Примера 10) с получением N-(5-хлор-4-(3,3-диметилиндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (85,5 мг, 58,84% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (MeCN/10 мМ NH4HCO3, 0,025% NH3.H2O). ЖХ-МС (ИЭР) m/z: 450 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 7,98 (с, 1H), 7,56 (с, 1H), 7,33 (д, J=7,9 Гц, 1H), 7,24 (д, J=6,5 Гц, 1H), 7,14-7,05 (м, 1H), 6,98 (дд, J=7,8, 7,0 Гц, 1H), 6,74 (с, 1H), 4,00 (с, 2H), 3,77 (с, 3H), 3,28 (с, 2H), 2,77 (т, J=5,7 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,30 (с, 3H), 1,29 (с, 6H).
Пример 84
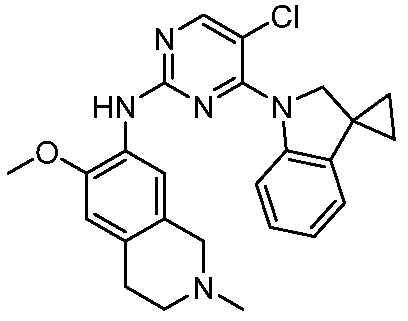
N-(5-хлор-4-(спиро[циклопропан-1,3'-индолин]-1'-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 25, с применением 3,3-спиро[циклопропан-1,3'-индолин]гидрохлорида вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 440 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,24 (с, 1H), 8,01 (с, 1H), 7,51 (с, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,08-6,97 (м, 1H), 6,94-6,86 (м, 1H), 6,85-6,68 (м, 2H), 4,28 (с, 2H), 3,78 (с, 3H), 3,27 (с, 2H), 2,77 (т, J=5,7 Гц, 2H), 2,55 (т, J=5,9 Гц, 2H), 2,30 (с, 3H), 1,15-1,06 (м, 2H), 1,03 (дд, J=6,6, 4,5 Гц, 2H).
Пример 85
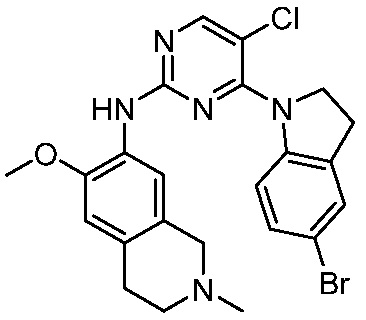
N-(4-(5-броминдолин-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 25, с применением 5-броминдолина вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 502 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,00 (с, 1H), 7,52 (с, 1H), 7,43 (с, 1H), 7,30 (д, J=8,6 Гц, 1H), 7,21 (с, 1H), 6,76 (с, 1H), 4,28 (д, J=7,7 Гц, 2H), 3,78 (с, 3H), 3,24 (с, 2H), 3,15 (с, 2H), 2,77 (с, 2H), 2,55 (с, 2H), 2,33 (с, 3H).
Пример 86
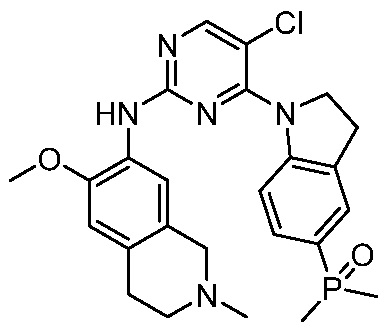
(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-5-ил)диметилфосфин оксид
Смесь N-(4-(5-броминдолин-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (110 мг, 208,66 мкмоль), диметилфосфина оксида (26,65 мг, 341,44 мкмоль), Pd2(dba)3 (10,42 мг, 11,38 мкмоль), Xantphos (6,59 мг, 11,38 мкмоль) и триэтиламина (69,10 мг, 682,88 мкмоль, 95,18 мкл) в диоксане (2 мл) продувают азотом и перемешивают при 110°C в течение 16 часов. Реакционную смесь очищают преп-ЖХВД (MeCN/10мМ NH4HCO3) с получением (1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-5-ил)диметилфосфина оксида в виде белого твердого вещества (57,3 мг, 55% выход). ЖХ-МС (ИЭР) m/z: 498 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,29 (с, 1H), 8,09 (с, 1H), 7,61 (д, J=10,9 Гц, 1H), 7,53-7,36 (м, 3H), 6,77 (с, 1H), 4,31 (т, J=8,2 Гц, 2H), 3,78 (с, 3H), 3,29 (с, 2H), 3,18 (т, J=7,8 Гц, 2H), 2,77 (с, 2H), 2,55 (с, 2H), 2,31 (с, 3H), 1,62 (д, J=13,2 Гц, 6H).
Пример 87
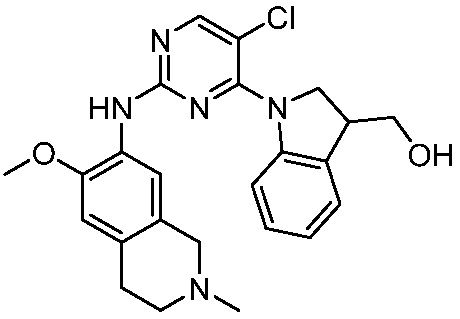
(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-ил)метанол
Процедуры те же, что и для Примера 25, с применением Пр. E6 вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 502 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 10,00 (с, 1H), 8,28 (с, 1H), 8,16 (с, 1H), 7,70 (д, J=4,0 Гц, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,31 (д, J=7,3 Гц, 1H), 7,18 (т, J=7,7 Гц, 1H), 6,98 (т, J=7,3 Гц, 1H), 6,93 (с, 1H), 4,38-4,33 (м, 1H), 4,26 (с, 1H), 4,16 (с, 2H), 3,82 (с, 3H), 3,66 (д, J=5,6 Гц, 2H), 3,54-3,38 (м, 2H), 3,30 (с, 1H), 3,14-2,95 (м, 2H), 2,92 (д, J=3,6 Гц, 3H).
Пример 88
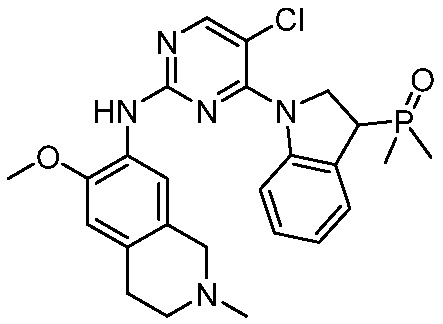
(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-ил)диметилфосфин оксид
Процедуры те же, что и для Примера 25, с применением Пр. E7 вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 502 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,30 (с, 1H), 8,02 (с, 1H), 7,59 (с, 1H), 7,37 (д, J=7,4 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,16 (т, J=7,7 Гц, 1H), 6,99 (т, J=7,5 Гц, 1H), 6,74 (с, 1H), 4,70-4,48 (м, 1H), 4,35 (ддд, J=18,3, 11,1, 3,6 Гц, 1H), 3,85 (д, J=9,9 Гц, 1H), 3,84-3,67 (м, 3H), 3,27-3,15 (м, 2H), 2,76 (т, J=5,6 Гц, 2H), 2,55 (д, J=5,7 Гц, 1H), 2,29 (с, 3H), 1,31 (дт, J=33,1, 16,5 Гц, 6H).
Пример 89
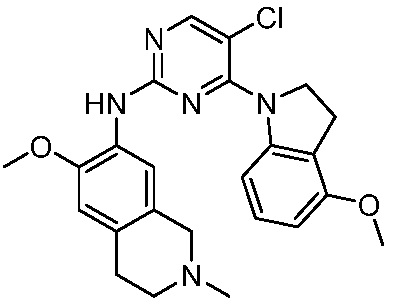
N-(5-хлор-4-(4-метоксииндолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 25, с применением Пр. E10 вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 452 [M+H]+. 1H ЯМР (400MГц, ДМСО-d6) δ 8,23 (с, 1H), 7,93 (с, 1H), 7,56 (с, 1H), 7,02 (дд, J=19,1, 8,0 Гц, 2H), 6,74 (с, 1H), 6,61 (д, J=8,0 Гц, 1H), 4,27 (т, J=8,3 Гц, 2H), 3,80 (с, 3H), 3,78 (с, 3H), 3,27 (с, 2H), 3,01 (т, J=8,2 Гц, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,31 (с, 3H).
Пример 90
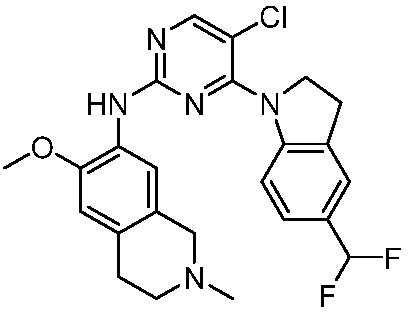
N-(5-хлор-4-(5-(дифторметил)индолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 25, с применением Пр. E12 вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 472 [M+H]+. 1H ЯМР (500MГц, ДМСО-d6) δ 8,29 (с, 1H), 8,04 (с, 1H), 7,55 (с, 1H), 7,49-7,39 (м, 2H), 7,29 (д, J=8,2 Гц, 1H), 6,95 (т, J=56,3 Гц, 1H), 6,76 (с, 1H), 4,31 (т, J=8,3Гц, 2H), 3,79 (с, 3H), 3,27 (с, 2H), 3,19 (т, J=8,2 Гц, 2H), 2,78 (с, 2H), 2,56 (ш, 2H), 2,31 (с, 3H).
Пример 91
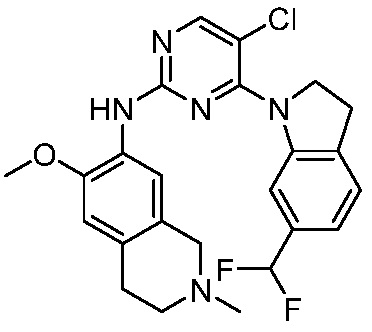
N-(5-хлор-4-(6-(дифторметил)индолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 25, с применением Пр. E13 вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 472 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,29 (с, 1H), 8,03 (с, 1H), 7,52 (с, 1H), 7,43-7,32 (м, 2H), 7,13 (д, J=7,6 Гц, 1H), 6,91 (с, 1H), 6,80 (с, 1H), 6,75 (с, 1H), 6,69 (с, 1H), 4,30 (т, J=8,3 Гц, 2H), 3,79 (д, J=19,8 Гц, 3H), 3,25 (с, 2H), 3,18 (т, J=8,1 Гц, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,54 (т, J=5,9 Гц, 2H), 2,28 (с, 3H).
Пример 92
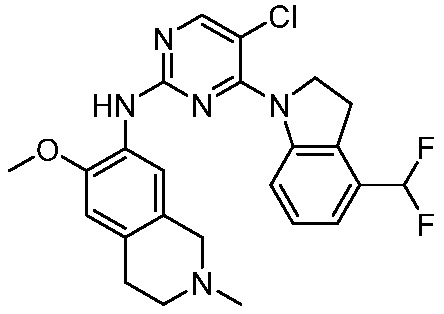
N-(5-хлор-4-(4-(дифторметил)индолин-1-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 25, с применением Пр. E14 вместо 3,3-диметилиндолина на Стадии 1. ЖХ-МС (ИЭР) m/z: 472 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,30 (с, 1H), 8,27 (с, 1H), 8,05 (с, 1H), 7,93 (с, 1H), 7,48 (с, 1H), 7,19 (дд, J=13,6, 5,6 Гц, 1H), 7,13-7,04 (м, 1H), 6,75 (д, J=5,8 Гц, 1H), 4,32 (т, J=8,3 Гц, 2H), 3,78 (д, J=9,6 Гц, 3H), 3,32-3,20 (м, 4H), 2,77 (с, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,31 (с, 3H).
Пример 31
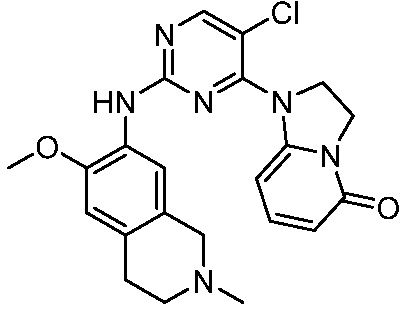
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-2,3-дигидроимидазо[1,2-a]пиридин-5(1H)-он
Стадия 1: Смесь Пр. E5 (65 мг, 0,239 ммоль), 2,4,5-трихлорпиримидина (53 мг, 0,286 ммоль), NaOtBu (45,9 мг, 0,477 ммоль), BINAP (3,9 мг, 0,006 ммоль) и ацетата палладия (II) (1,3 мг, 0,006 ммоль) в ДМФ/диоксане (2 мл/2 мл) перемешивают в течение 16 часов при 70°C под N2, гасят льдом-холодной водой (10 мл) и экстрагируют ЭА (20 мл x 3). Органические фазы промывают насыщенным раствором соли, сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1) с получением 1-(2,5-дихлорпиримидин-4-ил)-2,3-дигидроимидазо[1,2-a]пиридин-5-она (45 мг, 26,63% выход, 40% чистота). ЖХ-МС (ИЭР) m/z: 283,0 [M+H]+.
Стадия 2: 1-(2,5-дихлорпиримидин-4-ил)-2,3-дигидроимидазо[1,2-a]пиридин-5-он (40 мг, 0,056 ммоль) и Пр. W1 (10,9 мг, 0,056 ммоль) применяют в условиях B3 (как представлено на Стадии 2 Примера 10) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-2,3-дигидроимидазо[1,2-a]пиридин-5(1H)-она в виде желтого твердого вещества (2,5 мг, 9,73% выход), после очистки преп-ЖХВД (MeCN/0,1% HCOOH). ЖХ-МС (ИЭР) m/z: 439,0 [M+H]+.
Пример 48
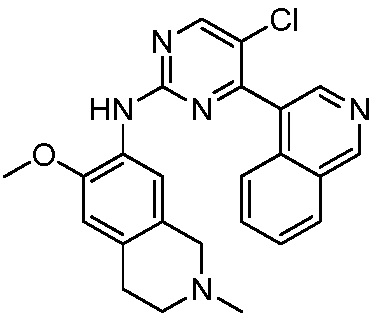
N-(5-хлор-4-(изохинолин-4-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: К раствору 4-изохинолинбороновой кислоты (100 мг, 578,12 мкмоль) в ДМЭ/H2O (3 мл/1 мл) добавляют карбонат калия (159,8 мг, 1156 мкмоль), 1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (42,3 мг, 57,82 мкмоль) и 2,4,5-трихлорпиримидин (106,0 мг, 578,12 мкмоль). Смесь перемешивают при 100°C под N2 атмосферой в течение ночи, охлаждают до комнатной температуры и разбавляют ЭА (10 мл). После фильтрации, фильтрат концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=4/1 об/об) с получением 4-(2,5-дихлорпиримидин-4-ил)изохинолина в виде желтого твердого вещества (50 мг, 63% выход). ЖХ-МС (ИЭР) m/z: 276 [M+H]+.
Стадия 2: 4-(2,5-дихлорпиримидин-4-ил)изохинолин (50 мг, 0,181 ммоль) и Пр. W1 (41,4 мг, 0,181 ммоль) применяют в условиях B3 (как представлено на Стадии 2 Примера 10) с получением N-(5-хлор-4-(изохинолин-4-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (12 мг, 12% выход), после очистки преп-ЖХВД (MeCN/0,05% ТФК). ЖХ-МС (ИЭР) m/z: 432 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,98 (с, 3H), 3,06-3,25 (м, 3H), 3,65-3,73 (м, 1H), 3,93 (с, 3H), 4,12-4,16 (м, 1H), 4,34-4,30 (м, 1H), 6,91 (с, 1H), 7,85-7,89 (м, 2H), 7,93-7,97 (м, 1H), 8,12 (с, 1H), 8,35 (д, J=7,6 Гц, 1H), 8,61 (с, 1H), 8,68 (с, 1H), 9,52 (с, 1H).
Пример 49
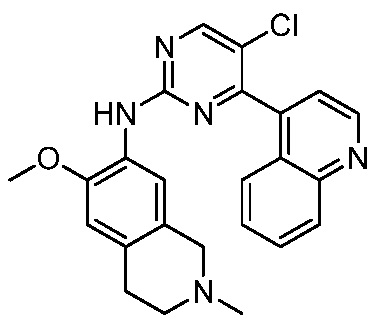
N-(5-хлор-4-(хинолин-4-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 48, с применением 4-хинолинбороновой кислоты вместо 4-изохинолинбороновой кислоты на Стадии 1. ЖХ-МС (ИЭР) m/z: 432 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 2,36 (с, 1,8H), 2,54 (с, 3H), 2,90-2,95 (м, 4H), 3,61 (с, 2H), 3,88 (с, 3H), 6,79 (с, 1H), 7,22 (д, J=8,0 Гц, 1,2H), 7,61 (д, J=4,4 Гц, 1H), 7,62-7,66 (м, 1H), 7,69 (д, J=8,0 Гц, 1,2H), 7,78 (д, J=8,4 Гц, 1H), 7,82-7,87 (м, 1H), 7,96 (с, 1H), 8,16 (д, J=8,4 Гц, 1H), 8,63 (с, 1H), 9,00 (д, J=4,4 Гц, 1H).
Пример 93
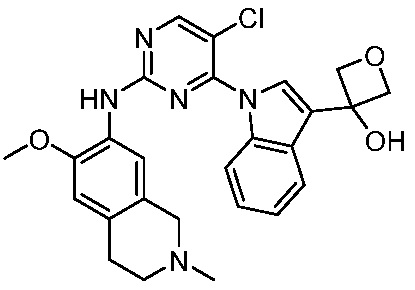
3-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)оксетан-3-ол
Стадия 1: Пр. E16 (440 мг, 1,45 ммоль) и 2,4,5-трихлорпиримидин (293 мг, 1,59 ммоль) применяют в условиях A1 с получением 3-(3-((трет-бутилдиметилсилил)окси)оксетан-3-ил)-1-(2,5-дихлорпиримидин-4-ил)-1H-индола (400 мг, 58% выход) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1). ЖХ-МС (ИЭР) m/z: 450 [M+H]+.
Стадия 2: 3-(3-((трет-Бутилдиметилсилил)окси)оксетан-3-ил)-1-(2,5-дихлорпиримидин-4-ил)-1H-индол (200 мг, 0,444 ммоль) и Пр. W1 (142 мг, 0,533 ммоль) применяют в условиях B3 с получением N-(4-(3-(3-((трет-бутилдиметилсилил)окси)оксетан-3-ил)-1H-индол-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (170 мг, 73% выход) в виде красного твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1). ЖХ-МС (ИЭР) m/z: 606 [M+H]+.
Стадия 3: К раствору N-(4-(3-(3-((трет-бутилдиметилсилил)окси)оксетан-3-ил)-1H-индол-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (80 мг, 131,96 мкмоль) в смешанном растворителе ТГФ (2 мл) и воде (0,3 мл) добавляют муравьиную кислоту (1 мл). Реакционную смесь перемешивают при 80°C в течение 3 часов и очищают преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил) с получением 3-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)оксетан-3-ола в виде желтого твердого вещества (2,0 мг, 3% выход). ЖХ-МС (ИЭР) m/z: 492 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 12,46 (с, 1H), 8,19 (с, 1H), 8,08 (с, 1H), 7,93 (д, J=8,0 Гц, 1H), 7,57 (д, J=8,0 Гц, 1H), 7,26 (т, J=7,5 Гц, 1H), 7,19 (т, J=8,1 Гц, 1H), 6,60 (с, 1H), 6,43 (с, 1H), 6,04 (с, 1H), 5,33 (с, 1H), 4,39 (д, J=14,9 Гц, 1H), 4,11 (д, J=14,8 Гц, 1H), 3,83 (д, J=30,4 Гц, 2H), 3,62 (с, 3H), 3,36 (с, 2H), 2,76 (с, 2H), 2,58 (с, 2H), 2,33 (с, 3H).
Пример 94
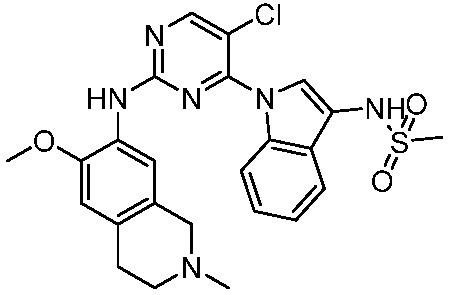
N-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)метансульфонамид
Стадия 1: 3-Нитро-1H-индол (100 мг, 0,617 ммоль) и 2,4,5-трихлорпиримидин (125 мг, 0,678 ммоль) применяют в условиях A1 с получением 1-(2,5-дихлорпиримидин-4-ил)-3-нитро-1H-индола (78 мг, 30% выход) в виде желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1). ЖХ-МС (ИЭР) m/z: 309 [M+H]+.
Стадия 2: К раствору 1-(2,5-дихлорпиримидин-4-ил)-3-нитро-1H-индола (78 мг, 252,34 мкмоль) в смешанном растворителе 1,4-диоксана (0,5 мл) и воды (0,2 мл) последовательно добавляют цинк (5,42 мг, 82,89 мкмоль) и NH4Cl (5,2 мг, 97,05 мкмоль) при 0°С. Реакционную смесь перемешивают при комнатной температуре в течение часа и фильтруют через целит. Фильтрат разбавляют этилацетатом (10 мл), промывают водой (5 мл) и насыщенным раствором соли (5 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением 1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-амина в виде коричневого твердого вещества (60 мг, 85% выход). ЖХ-МС (ИЭР) m/z: 279 [M+H]+.
Стадия 3: К суспензии 1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-амина (60 мг, 214,96 мкмоль) в безводном ДХМ (2 мл) добавляют MsCl (138,09 мг, 214,96 мкмоль) при 0°C. После перемешивания при 0°C в течение 4 часов, смесь разделяют между 5% водной лимонной кислотой (20 мл) и ДХМ (20 мл). Объединенную органическую фазу промывают водой (10 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=1/10~1/5) с получением N-(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)метансульфонамида в виде желтого твердого вещества (75 мг, 97% выход). ЖХ-МС (ИЭР) m/z: 357 [M+H]+.
Стадия 4: N-(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)метансульфонамид (75 мг, 0,210 ммоль) и Пр. W1 (49 мг, 0,252 ммоль) применяют в условиях B3 с получением N-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)метансульфонамида (5,2 мг, 5% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (MeCN/0,1% HCOOH). ЖХ-МС (ИЭР) m/z: 513 [M+H]+. 1H ЯМР (400MГц, ДМСО-d6) δ 8,75 (с, 1H), 8,62 (с, 1H), 8,28 (с, 1H), 7,86-7,72 (м, 2H), 7,47 (д, J=8,0 Гц, 1H), 7,40 (с, 1H), 7,28-7,20 (м, 2H), 7,11 (д, J=7,9 Гц, 1H), 6,80 (с, 1H), 3,79 (с, 3H), 2,99 (с, 3H), 2,79 (т, J=5,8 Гц, 2H), 2,56 (т, J=5,8 Гц, 2H), 2,31 (с, 3H), 2,29 (с, 2H).
Пример 32
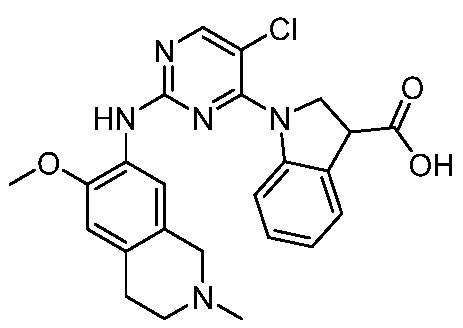
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота
Пример 115
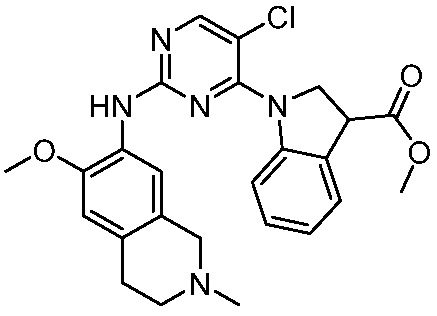
Метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилат
Стадия 1: Смесь 2,4,5-трихлорпиримидина (220 мг, 1,20 ммоль) и индолин-3-карбоновой кислоты (163 мг, 0,999 ммоль) в NMP (0,6 мл) перемешивают в течение 15 минут при 150°C и очищают хроматографией с обращенной фазой (0-80% метанол в воде) с получением 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоновой кислоты (160 мг, 48,55% выход, 94% чистота) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 310,0 [M+H]+.
Стадия 2: Смесь 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоновую кислоту (30 мг, 0,097 ммоль) и Пр. W1 (19 мг, 0,097 ммоль) и TsOH.H2O (18,4 мг, 0,097 ммоль) в i-PrOH (2,5 мл) перемешивают в течение 5 часов при 85°C и очищают преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновой кислоты (1,8 мг, 4% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 466 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 8,03 (с, 1H), 7,51 (с, 1H), 7,40 (дд, J=7,7, 3,9 Гц, 2H), 7,12 (т, J=7,6 Гц, 1H), 6,97 (т, J=7,3 Гц, 1H), 6,76 (с, 1H), 4,56 (дд, J=10,6, 6,4 Гц, 1H), 4,38 (т, J=10,1 Гц, 1H), 4,23 (с, 1H), 3,77 (с, 3H), 2,78 (д, J=5,6 Гц, 2H), 2,67 (с, 1H), 2,60-2,55 (м, 3H), 2,32 (с, 3H).
Альтернативный способ синтеза:
Стадия 1: Смесь Пр. E17 (300 мг, 1,59 ммоль), 2,4,5-трихлорпиримидин (292 мг, 1,59 ммоль) и ДИПЭА (411,4 мг, 3,18 ммоль) в n-BuOH (5 мл) перемешивают в течение 16 часов при 100°C, гасят льдом-холодной водой (20 мл) и экстрагируют ЭА (30 мл x 3). Органические фазы промывают насыщенным раствором соли (30 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ: 0-15%) с получением метил 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоксилата (556 мг, 95% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 324 [M+H]+.
Стадия 2: Смесь метил 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоксилата (250 мг, 0,678 ммоль), Пр. W1 (155 мг, 0,678 ммоль) и TsOH.H2O (129,1 мг, 0,679 ммоль) in i-PrOH (10 мл) перемешивают в течение 16 часов при 100°C, гасят насыщенным NaHCO3 водным раствором (10 мл) и экстрагируют ДХМ/MeOH (10/1, 30 мл x 3). Органические фазы промывают насыщенным раствором соли (30 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ: 0-15%) с получением смеси метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилат и изопропил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата (530 мг) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 480 (метиловый эфир), 508 (изопропиловый эфир) [M+H]+.
Небольшое количество смеси далее очищают преп-ЖХВД с получением метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,28 (с, 1H), 8,09 (с, 1H), 7,48 (с, 1H), 7,37 (дд, J=15,3, 7,7 Гц, 2H), 7,16 (т, J=7,4 Гц, 1H), 6,99 (т, J=7,2 Гц, 1H), 6,76 (с, 1H), 4,54 (кв, J=10,8 Гц, 1H), 4,50-4,38 (м, 2H), 3,79 (д, J=15,6 Гц, 3H), 3,71 (с, 3H), 3,29 (с, 2H), 2,78 (т, J=5,7 Гц, 2H), 2,56 (т, J=5,9 Гц, 2H), 2,31 (с, 3H).
Стадия 3: Смесь метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата и изопропил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата (530 мг) и LiOH водным раствором (2N, 4 мл) в ТГФ (5 мл) перемешивают в течение 16 часов при комнатной температуре, доводят до pH ~5 муравьиной кислотой и экстрагируют ДХМ (30 мл x 3). Объединенные органические фазы сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновой кислоты (158 мг, 84% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 466 [M+H]+.
Примеры 95 и 96
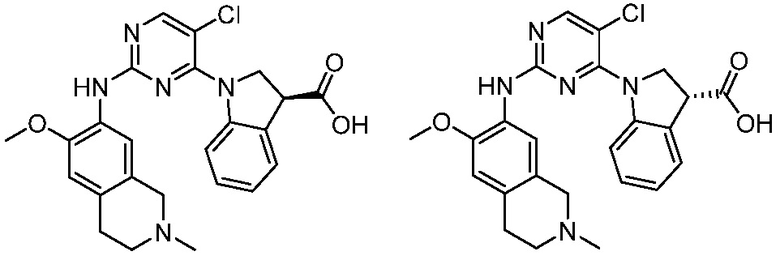
(S)-1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота & (R)-1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота
Синтезируют после хирального разделения рацемической 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновой кислоты (20 мг). Стереохимию не подтверждают, и два продукта (4,5 мг, 5,1 мг по отдельности) произвольно назначают.
ЖХ-МС (ИЭР) m/z: 466 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,27 (с, 1H), 8,05 (с, 1H), 7,51 (с, 1H), 7,40 (д, J=7,9 Гц, 2H), 7,15 (т, J=7,8 Гц, 1H), 6,98 (т, J=7,5 Гц, 1H), 6,77 (с, 1H), 4,54 (дд, J=10,6, 6,1 Гц, 1H), 4,40 (т, J=10,0 Гц, 1H), 4,31-4,27 (м, 1H), 3,77 (с, 3H), 3,26-3,24 (м, 2H), 2,80 (д, J=5,6 Гц, 2H), 2,65 (д, J=6,5 Гц, 2H), 2,37 (с, 3H). Хиральная-ЖХВД время удержания: 1,285 мин; значение эи: >99%.
ЖХ-МС (ИЭР) m/z: 466 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,03 (с, 1H), 7,51 (с, 1H), 7,39 (д, J=8,0 Гц, 2H), 7,15 (д, J=7,7 Гц, 1H), 6,99 (д, J=7,5 Гц, 1H), 6,76 (с, 1H), 4,54 (дд, J=10,5, 6,2 Гц, 1H), 4,39 (т, J=10,1 Гц, 1H), 4,28 (д, J=6,1 Гц, 1H), 3,77 (с, 3H), 3,34 (с, 2H), 2,79 (т, J=5,5 Гц, 2H), 2,61 (т, J=5,7 Гц, 2H), 2,34 (с, 3H). Хиральная-ЖХВД время удержания: 2,078 мин; значение эи: >99%.
Условия разделения СЖХ:
Инструмент: SFC-150 (Thar, Waters)
Колонка: IG 20*250 мм, 10 мкм (Daicel)
Температура колонки: 35ºC
Подвижная фаза: CO2/EtOH (0,2% метанол/аммиак)=50/50
Скорость потока: 120 г/мин
Противодавление: 100 бар
Длина волны определения: 214 нм
Время цикла: 8 мин
Пример 33
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксамид
Стадия 1: Смесь 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоновой кислоты (60 мг, 193,46 мкмоль), хлорида аммония (51,74 мг, 967,32 мкмоль), ГАТУ (110,34 мг, 290,20 мкмоль) и триэтиламина (58,73 мг, 580,39 мкмоль, 80,90 мкл) в ДМФ (1 мл) перемешивают в течение 3 часов при комнатной температуре, гасят льдом-холодной водой (15 мл) и экстрагируют EtOAc (20 мл x 3). Объединенную органическую фазу промывают насыщенным раствором соли (15 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=0-100%) с получением 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоксамида (15 мг, 50,16% выход, 100% чистота) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 309 [M+H]+.
Стадия 2: 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоксамид (45 мг, 0,146 ммоль) и Пр. W1 (28 мг, 0,146 ммоль) применяют в условиях B3 с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксамида (26,9 мг, 55% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил). ЖХ-МС (ИЭР) m/z: 465 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,24 (с, 1H), 8,00 (с, 1H), 7,80 (с, 1H), 7,50 (с, 1H), 7,39 (дд, J=31,8, 7,8 Гц, 2H), 7,22 (с, 1H), 7,10 (т, J=7,7 Гц, 1H), 6,97 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,54 (дд, J=10,3, 6,3 Гц, 1H), 4,36 (т, J=9,9 Гц, 1H), 4,15 (дд, J=9,3, 6,5 Гц, 1H), 3,77 (с, 3H), 3,28 (д, J=12,2 Гц, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,55 (т, J=5,9 Гц, 2H), 2,30 (с, 3H).
Пример 97
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-метилиндолин-3-карбоксамид
Процедуры те же, что и для Примера 33, с применением метиламина гидрохлорида вместо хлорида аммония на Стадии 1. ЖХ-МС (ИЭР) m/z: 479 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,31-8,26 (м, 1H), 8,24 (с, 1H), 8,02 (с, 1H), 7,49 (с, 1H), 7,42 (д, J=8,3 Гц, 1H), 7,28 (д, J=7,5 Гц, 1H), 7,10 (т, J=7,7 Гц, 1H), 6,95 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,54 (дд, J=10,4, 6,5 Гц, 1H), 4,37 (т, J=10,0 Гц, 1H), 4,22-4,05 (м, 1H), 3,77 (с, 3H), 3,28 (с, 2H), 2,77 (д, J=5,7 Гц, 2H), 2,66 (д, J=4,6 Гц, 3H), 2,56 (д, J=5,8 Гц, 2H), 2,30 (с, 3H).
Пример 98
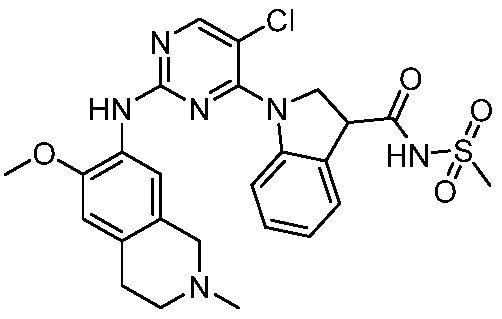
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-(метилсульфонил)индолин-3-карбоксамид
Стадия 1: Смесь 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоновой кислоты (60 мг, 0,193 ммоль) и КДИ (31,47 мг, 0,193 ммоль) в ДХМ (5 мл) перемешивают в течение часа при комнатной температуре, затем добавляют метансульфонамид (27,6 мг, 0,290 ммоль). Затем смесь перемешивают в течение дополнительного часа, DBU (29,5 мг, 0,193 ммоль) добавляют к смеси. Реакционную смесь затем перемешивают в течение 16 часов при комнатной температуре, гасят HCl водным раствором (1N, 20 мл) и экстрагируют EtOAc (20 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ=0-15%) с получением 1-(2,5-дихлорпиримидин-4-ил)-N-метилсульфонил-индолин-3-карбоксамида в виде желтого масла (45 мг, 37% выход, 62% чистота). ЖХ-МС (ИЭР) m/z: 387 [M+H]+.
Стадия 2: 1-(2,5-дихлорпиримидин-4-ил)-N-метилсульфонил-индолин-3-карбоксамид (45 мг, 0,072 ммоль) и Пр. W1 (13,9 мг, 0,072 ммоль) применяют в условиях B3 с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-(метилсульфонил)индолин-3-карбоксамида (29 мг, 74,1% выход) в виде светло-желтого твердого вещества, после очистки преп-ЖХВД (0,1% FA/ацетонитрил). ЖХ-МС (ИЭР) m/z: 543 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 8,05 (с, 1H), 7,66 (с, 1H), 7,47 (д, J=7,0 Гц, 1H), 7,33 (д, J=8,4 Гц, 1H), 7,11 (т, J=7,9 Гц, 1H), 6,94 (т, J=7,4 Гц, 1H), 6,85 (с, 1H), 4,64-4,56 (м, 1H), 4,28 (т, J=9,8 Гц, 1H), 4,00 (д, J=7,8 Гц, 1H), 3,81 (с, 3H), 3,09 (с, 2H), 2,93 (с, 2H), 2,82 (с, 3H), 2,67 (с, 2H), 2,52 (с, 3H).
Пример 99
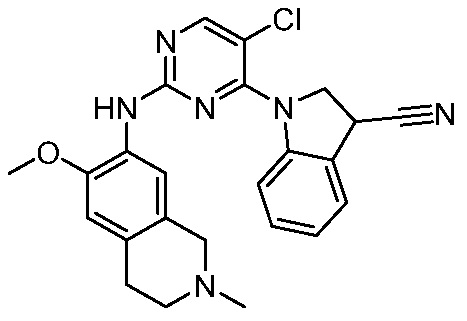
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбонитрил
Стадия 1: К раствору 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоксамида (100 мг, 0,210 ммоль) и пиридина (67 мг, 0,841 ммоль) в ДХМ (5 мл) медленно добавляют трифторуксусный ангидрид (88,3 мг, 0,421 ммоль) в ДХМ (1 мл) при 0°C. Смесь перемешивают при 0°C в течение часа и комнатной температуре в течение 6 часов и гасят 1 N HCl (10 мл). Полученную смесь экстрагируют ДХМ (20 мл x 3), и органические фазы промывают насыщенным раствором соли (15 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбонитрила (60 мг, 40,2% выход, 40% чистота). ЖХ-МС (ИЭР) m/z: 291 [M+H]+.
Стадия 2: 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбонитрил (60 мг, 40% чистота) и Пр. W1 (10,8 мг, 0,056 ммоль) применяют в условиях B3 с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбонитрила (19,7 мг, 77,7% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил). ЖХ-МС (ИЭР) m/z: 447 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,32 (с, 1H), 8,22 (с, 1H), 8,15 (с, 1H), 7,52-7,42 (м, 2H), 7,33 (д, J=7,7 Гц, 1H), 7,24 (т, J=7,5 Гц, 1H), 7,07 (т, J=7,4 Гц, 1H), 6,76 (с, 1H), 4,89-4,80 (м, 1H), 4,58 (т, J=10,0 Гц, 1H), 4,44 (дд, J=10,7, 6,8 Гц, 1H), 3,77 (с, 3H), 3,29 (с, 2H), 2,77 (д, J=5,8 Гц, 2H), 2,57-2,54 (м, 2H), 2,31 (с, 3H).
Пример 100
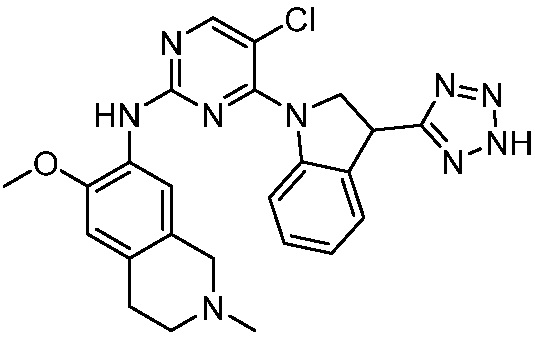
N-(4-(3-(2H-тетразол-5-ил)индолин-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Смесь 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбонитрила (45 мг, 0,101 ммоль), азида натрия (9,8 мг, 0,151 ммоль) и ацетата аммония (11,6 мг, 0,151 ммоль) в ДМФ (1,5 мл) перемешивают в течение 5 часов при 120°C, охлаждают до комнатной температуры, разбавляют HCl водным раствором (1 N, 5 мл) и перемешивают в течение еще 30 минут. Смесь экстрагируют ЭА (20 мл x 3). Органические фазы промывают насыщенным раствором соли (15 мл) сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/0,1% муравьиную кислоту) с получением N-(4-(3-(2H-тетразол-5-ил)индолин-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде белого твердого вещества (13,4 мг, 27% выход). ЖХ-МС (ИЭР) m/z: 490 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,29 (с, 1H), 8,15 (с, 1H), 8,06 (с, 1H), 7,72 (с, 1H), 7,32 (д, J=7,7 Гц, 1H), 7,21-7,08 (м, 2H), 6,93 (т, J=7,5 Гц, 1H), 6,83 (с, 1H), 4,89 (т, J=9,1 Гц, 1H), 4,72 (с, 1H), 4,56 (с, 1H), 3,81 (с, 3H), 3,77 (с, 2H), 3,01 (с, 2H), 2,90 (д, J=5,5 Гц, 2H), 2,63 (с, 3H).
Пример 101
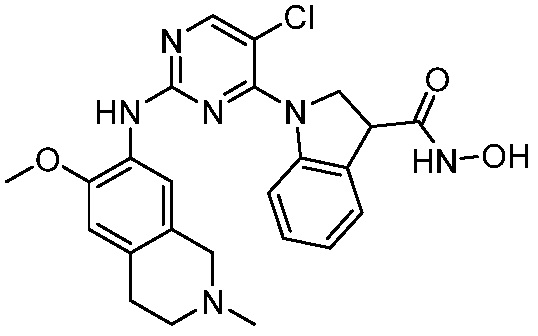
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-гидроксииндолин-3-карбоксамид
Стадия 1: Смесь 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоновой кислоты (150 мг, 0,484 ммоль), оксалилхлорида (61,4 мг, 0,484 ммоль) и N, N-диметилформамида (1,8 мг, 0,024 ммоль) в ДХМ (5 мл) перемешивают в течение 2 часов при комнатной температуре и концентрируют в вакууме с получением неочищенного 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбонилхлорида, который применяют на следующей стадии без дальнейшей очистки.
Стадия 2: К суспензии гидроксиламина гидрохлорида (79,3 мг, 1,14 ммоль) в MeOH (3 мл) добавляют медленно KOH (63,8 мг, 1,14 ммоль) при 0°C. Реакционную смесь перемешивают в течение 10 минут под N2 и туда добавляют 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбонилхлорид (75 мг, 0,228 ммоль) в метаноле (5 мл) при 0°C. Полученную смесь затем перемешивают в течение 16 часов, гасят льдом-холодной водой (10 мл) и экстрагируют EtOAc (20 мл x 3). Органические фазы промывают насыщенным раствором соли (5 мл) сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ=0-10/1) с получением 1-(2,5-дихлорпиримидин-4-ил)-N-гидроксииндолин-3-карбоксамида в виде белого твердого вещества (50 мг, 43% выход, 64% чистота). ЖХ-МС (ИЭР) m/z: 325 [M+H]+.
Стадия 3: 1-(2,5-дихлорпиримидин-4-ил)-N-гидроксииндолин-3-карбоксамид (50 мг, 64% чистота) и Пр. W1 (18,9 мг, 0,098 ммоль) применяют в условиях B3 с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-гидроксииндолин-3-карбоксамида (12,6 мг, 25,4% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил). ЖХ-МС (ИЭР) m/z: 481 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 10,94 (с, 1H), 9,04 (с, 1H), 8,25 (с, 1H), 8,03 (с, 1H), 7,50-7,34 (м, 2H), 7,25 (д, J=7,1 Гц, 1H), 7,11 (т, J=7,6 Гц, 1H), 6,97 (т, J=7,7 Гц, 1H), 6,75 (с, 1H), 4,51 (дд, J=10,3, 6,7 Гц, 1H), 4,39 (т, J=10,0 Гц, 1H), 4,08-3,99 (м, 1H), 3,77 (с, 3H), 3,27 (с, 2H), 2,77 (д, J=5,8 Гц, 2H), 2,56 (д, J=5,7 Гц, 2H), 2,30 (с, 3H).
Пример 102
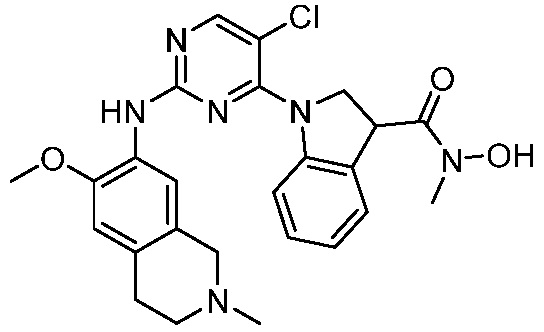
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-гидрокси-N-метилиндолин-3-карбоксамид
Процедуры те же, что и для Примера 101, с применением N-метилгидроксиламина вместо гидроксиламина гидрохлорида на Стадии 2. ЖХ-МС (ИЭР) m/z: 495 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 10,28 (с, 1H), 8,25 (с, 1H), 8,01 (с, 1H), 7,51 (с, 1H), 7,38 (дд, J=19,3, 7,6 Гц, 2H), 7,11 (т, J=7,5 Гц, 1H), 6,95 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,90-4,79 (м, 1H), 4,51 (дд, J=10,4, 6,4 Гц, 1H), 4,38 (т, J=9,9 Гц, 1H), 3,77 (с, 3H), 3,28 (с, 2H), 3,18 (с, 3H), 2,76 (д, J=5,7 Гц, 2H), 2,56 (д, J=5,7 Гц, 2H), 2,30 (с, 3H).
Пример 103
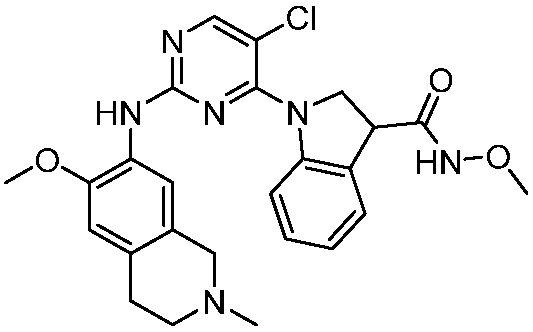
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N-метоксииндолин-3-карбоксамид
Процедуры те же, что и для Примера 101, с применением O-метилгидроксиламина гидрохлорида вместо гидроксиламина гидрохлорида на Стадии 2. ЖХ-МС (ИЭР) m/z: 495 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 11,60 (с, 1H), 8,25 (с, 1H), 8,05 (с, 1H), 7,50-7,39 (м, 2H), 7,26 (д, J=7,5 Гц, 1H), 7,12 (т, J=7,8 Гц, 1H), 6,97 (т, J=7,1 Гц, 1H), 6,75 (с, 1H), 4,51 (дд, J=10,4, 6,7 Гц, 1H), 4,39 (т, J=10,0 Гц, 1H), 4,04-3,96 (м, 1H), 3,77 (с, 3H), 3,64 (с, 3H), 3,28 (с, 2H), 2,78 (с, 2H), 2,56 (д, J=5,9 Гц, 2H), 2,30 (с, 3H).
Пример 163
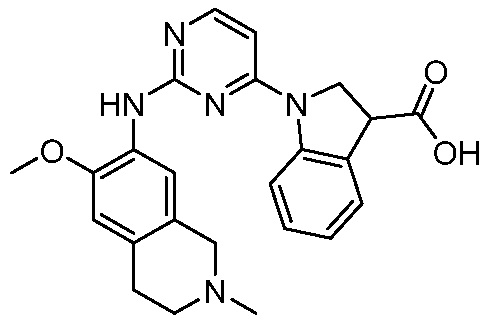
1-(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота
Стадия 1: Смесь метила 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата и изопропила 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата (100 мг) и Pd/C (18,2 мг, 0,171 ммоль) в метаноле (10 мл) перемешивают в течение 16 часов при комнатной температуре под H2 и фильтруют через слой Целита. Твердую лепешку промывают ЭА (20 мл), и фильтрат концентрируют в вакууме с получением неочищенной смеси метил 1-(2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата и изопропил 1-(2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата, которую применяют на следующей стадии без очистки. ЖХ-МС (ИЭР) m/z: 446 (метиловый эфир), 474 (изопропиловый эфир) [M+H]+.
Стадия 2: Та же методика, как для Стадии 3 Альтернативного способа синтеза Пример 32, с применением смеси метил 1-(2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата и изопропил 1-(2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата вместо смеси метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата и изопропил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата. ЖХ-МС (ИЭР) m/z: 432 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,29 (с, 1H), 8,14 (дд, J=16,9, 6,9 Гц, 2H), 7,82 (с, 1H), 7,62 (с, 1H), 7,43 (д, J=7,3 Гц, 1H), 7,08 (т, J=7,7 Гц, 1H), 6,92 (т, J=7,3 Гц, 1H), 6,80 (с, 1H), 6,27 (д, J=5,8 Гц, 1H), 4,32 (дд, J=9,8, 5,0 Гц, 1H), 4,17-4,03 (м, 2H), 3,80 (д, J=9,1 Гц, 3H), 3,28 (м, 2H), 2,84 (т, J=5,6 Гц, 2H), 2,66 (т, J=5,7 Гц, 2H), 2,36 (с, 3H).
Пример 104
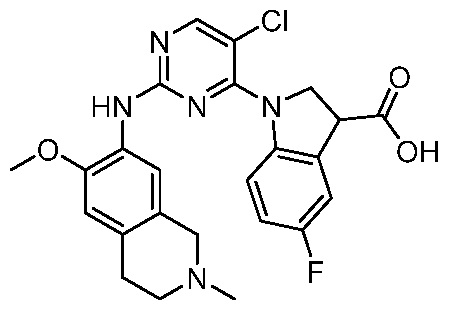
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фториндолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением Пр. E22 вместо Пр. E17 на Стадии 1. ЖХ-МС (ИЭР) m/z: 484 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,23 (с, 2H), 8,02 (с, 1H), 7,49 (с, 1H), 7,40 (дд, J=8,6, 4,7 Гц, 1H), 7,21 (д, J=6,7 Гц, 1H), 6,92 (дд, J=8,9, 6,8 Гц, 1H), 6,78 (с, 1H), 4,60 (дд, J=10,5, 6,2 Гц, 1H), 4,39 (т, J=10,1 Гц, 1H), 4,22-4,10 (м, 1H), 3,77 (с, 3H), 3,42 (с, 2H), 2,82 (д, J=5,5 Гц, 2H), 2,69 (т, J=19,4 Гц, 2H), 2,40 (с, 3H).
Пример 105
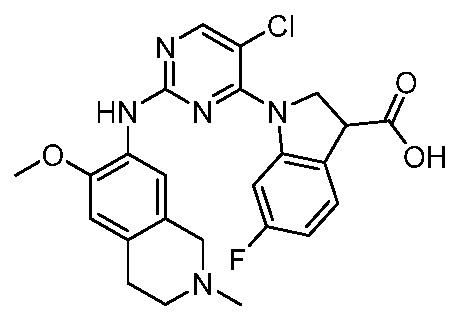
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фториндолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением Пр. E23 вместо Пр. E17 на Стадии 1. ЖХ-МС (ИЭР) m/z: 484 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,33-8,25 (м, 1H), 8,20 (д, J=6,6 Гц, 1H), 7,42 (с, 1H), 7,36 (дд, J=14,4, 6,5 Гц, 1H), 7,20 (д, J=10,7 Гц, 1H), 6,85-6,65 (м, 2H), 4,61 (дд, J=10,4, 6,1 Гц, 1H), 4,42 (т, J=10,0 Гц, 1H), 4,21-4,12 (м, 1H), 3,79 (д, J=3,1 Гц, 1H), 3,75 (д, J=10,4 Гц, 3H), 3,36-3,33 (м, 1H), 2,79 (т, J=5,5 Гц, 2H), 2,63 (т, J=5,1 Гц, 2H), 2,31 (д, J=20,7 Гц, 3H).
Пример 106
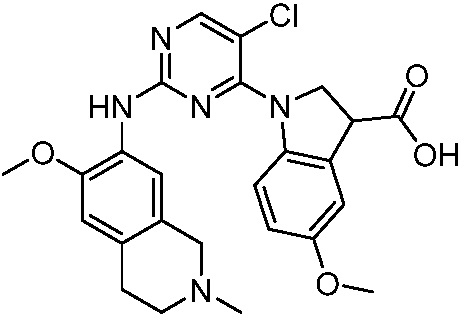
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-метоксииндолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением Пр. E24 вместо Пр. E17 на Стадии 1. ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,20 (с, 1H), 7,96 (с, 1H), 7,50 (с, 1H), 7,39 (д, J=8,8 Гц, 1H), 6,96 (д, J=2,0 Гц, 1H), 6,83-6,58 (м, 1H), 4,69-4,50 (м, 1H), 4,40 (с, 1H), 4,25 (д, J=6,2 Гц, 1H), 3,76 (д, J=11,4 Гц, 6H), 3,41 (с, 1H), 2,79 (д, J=5,6 Гц, 2H), 2,64 (д, J=5,4 Гц, 2H), 2,35 (с, 3H).
Пример 107
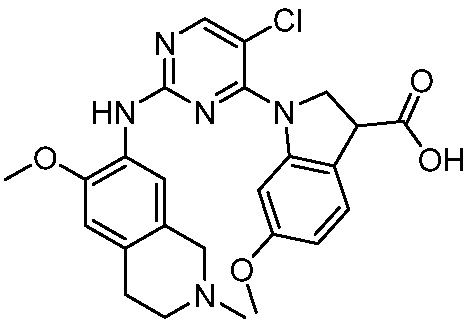
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-метоксииндолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением Пр. E25 вместо Пр. E17 на Стадии 1. ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,45 (с, 1H), 8,14 (с, 1H), 7,51 (д, J=11,9 Гц, 1H), 7,39 (дд, J=8,3, 2,3 Гц, 1H), 7,19 (д, J=9,4 Гц, 1H), 7,03 (с, 1H), 6,76 (д, J=8,4 Гц, 1H), 4,69 (д, J=2,6 Гц, 1H), 4,62 (т, J=10,0 Гц, 1H), 4,36-4,22 (м, 2H), 4,10 (д, J=11,4 Гц, 1H), 3,85 (д, J=1,7 Гц, 3H), 3,70 (д, J=3,6 Гц, 4H), 3,33 (с, 1H), 3,14 (с, 1H), 3,07 (д, J=17,0 Гц, 1H), 2,94 (с, 3H).
Пример 108
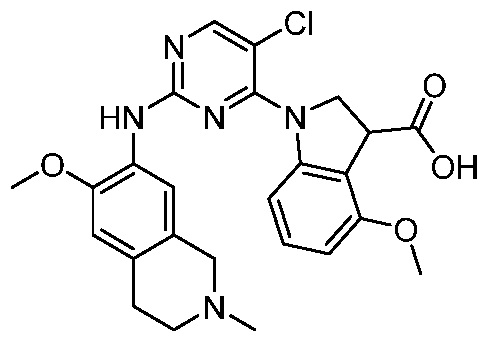
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-4-метоксииндолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением Пр. E26 вместо Пр. E17 на Стадии 1. ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,24 (с, 1H), 8,00 (с, 1H), 7,52 (с, 1H), 7,11 (т, J=8,1 Гц, 1H), 7,03 (д, J=8,0 Гц, 1H), 6,75 (с, 1H), 6,63 (д, J=8,1 Гц, 1H), 4,51 (т, J=10,2 Гц, 1H), 4,32 (дд, J=10,6, 4,8 Гц, 1H), 4,10 (дд, J=9,7, 4,7 Гц, 1H), 3,77 (с, 7H), 2,78 (т, J=5,6 Гц, 2H), 2,58 (т, J=5,7 Гц, 2H), 2,49-2,48 (м, 1H), 2,33 (с, 3H).
Пример 109
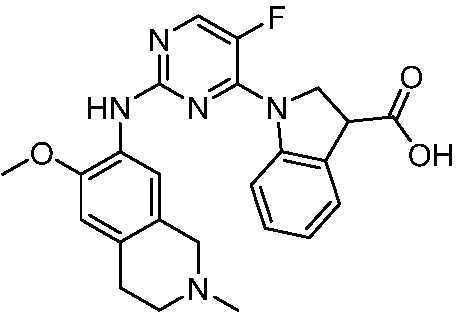
1-(5-Фтор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением 2,4-дихлор-5-фторпиримидина вместо 2,4,5-трихлорпиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 450 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,19 (д, J=5,8 Гц, 1H), 7,90 (д, J=19,6 Гц, 1H), 7,85(д, J=8,2 Гц, 1H), 7,50 (с, 1H), 7,40 (д, J=7,4 Гц, 1H), 7,10 (т, J=7,6 Гц, 1H), 6,98 (т, J=7,4 Гц, 1H), 6,78 (с, 1H), 4,53 (дт, J=10,5, 5,3 Гц, 1H), 4,41-4,31 (м, 1H), 4,27 (дд, J=9,8, 5,7 Гц, 1H), 3,77 (с, 3H), 3,41 (с, 2H), 2,82 (т, J=5,5 Гц, 2H), 2,64 (т, J=5,9 Гц, 2H), 2,35 (с, 3H).
Пример 110
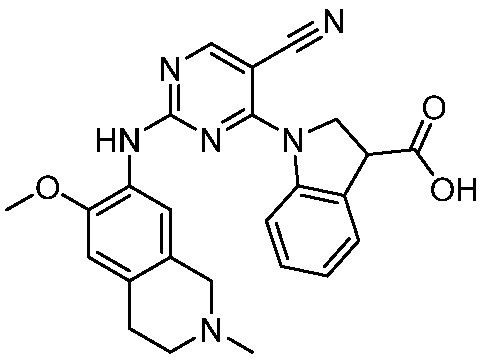
1-(5-Циано-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением 2,4-дихлорпиримидин-5-карбонитрила вместо 2,4,5-трихлорпиримидина на Стадии 1. ЖХ-МС (ИЭР) m/z: 457 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 9,13 (с, 1H), 8,54 (с, 1H), 7,41 (д, J=65,9 Гц, 2H), 6,98 (д, J=75,7 Гц, 3H), 4,45 (с, 1H), 4,20 (с, 2H), 3,70 (с, 3H), 3,16-3,08 (м, 2H), 2,91 (с, 2H), 2,65 (с, 2H), 2,35 (с, 3H).
Пример 111
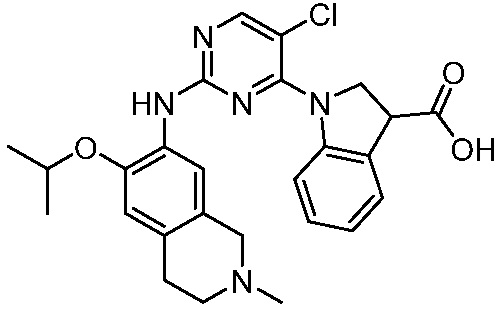
1-(5-Хлор-2-((6-изопропокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением Пр. W9 вместо Пр. W1 на Стадии 2. ЖХ-МС (ИЭР) m/z: 494 [M+H]+.
Пример 112
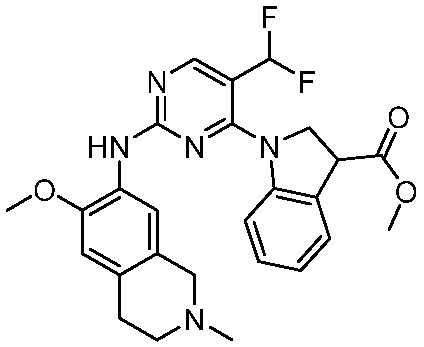
Метил 1-(5-(дифторметил)-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилат
Стадия 1: К раствору 2,4-дихлорпиримидин-5-карбальдегида (300 мг, 1,70 ммоль) в ДХМ (5 мл) добавляют N-этил-N-(трифторсульфанил)этанамин (1,37 г, 8,48 ммоль) по каплям при 0°C. Реакционную смесь перемешивают при 25°C в течение 16 часов, охлаждают до 0°C, подщелачивают до pH ~8 насыщенным водным раствором бикарбоната натрия и экстрагируют дихлорметаном (5 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме с получением 2,4-дихлор-5-(дифторметил)пиримидина в виде светло-желтого масла (303 мг, 85% выход). ЖХ-МС (ИЭР) m/z: 335 [M+H]+.
Стадия 2: Пр. E17 (115,8 мг, 0,653 ммоль) и 2,4-дихлор-5-(дифторметил)пиримидин (130 мг, 0,653 ммоль) применяют в условиях A3 с получением метил 1-(2-хлор-5-(дифторметил)пиримидин-4-ил)индолин-3-карбоксилата (160 мг, 68,5% выход) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1). ЖХ-МС (ИЭР) m/z: 340 [M+H]+.
Стадия 3: Метил 1-(2-хлор-5-(дифторметил)пиримидин-4-ил)индолин-3-карбоксилат (159 мг, 0,445 ммоль) и Пр. W1 (102,6 мг, 0,534 ммоль) применяют в условиях B3 с получением метил 1-(5-(дифторметил)-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбоксилата (30 мг, 10,9% выход) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1). ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,44 (с, 1H), 8,39 (с, 1H), 7,58 (с, 1H), 7,42 (с, 1H), 7,35 (д, J=7,4 Гц, 1H), 7,12 (д, J=8,6 Гц, 1H), 7,00 (т, J=7,3 Гц, 1H), 6,84 (д, J=10,3 Гц, 1H), 4,47-4,39 (м, 2H), 4,34 (т, J=9,6 Гц, 1H), 3,76 (с, 3H), 3,69 (с, 3H), 3,56 (с, 2H), 2,88 (м, 4H), 2,49-2,46 (с, 3H).
Пример 113
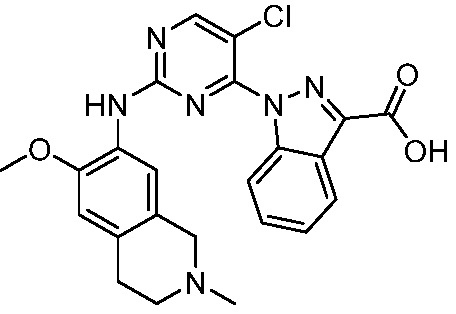
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индазол-3-карбоновая кислота
Процедуры те же, что и для Альтернативного способа синтеза Примера 32, с применением метил 1H-индазол-3-карбоксилата вместо Пр. E17 на Стадии 1. ЖХ-МС (ИЭР) m/z: 465 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,87 (с, 1H), 8,63 (с, 1H), 8,38-8,22 (м, 2H), 7,99 (д, J=8,1 Гц, 1H), 7,55-7,28 (м, 3H), 6,83 (с, 1H), 3,77 (с, 3H), 3,39-3,38 (м, 2H), 2,82 (д, J=5,3 Гц, 2H), 2,61 (т, J=5,7 Гц, 2H), 2,33 (с, 3H).
Пример 34
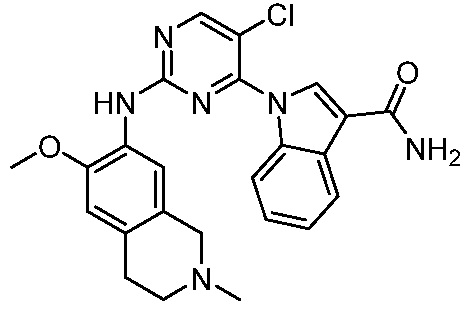
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-карбоксамид
Стадия 1: 1H-индол-3-карбоновую кислоту (500 мг, 3,10 ммоль) и 2,4,5-трихлорпиримидин (625,99 мг, 3,41 ммоль) применяют в условиях A1 с получением 1-(2,5-дихлорпиримидин-4-ил)индол-3-карбоновой кислоты (200 мг, 19,88% выход, 95% чистота) в виде желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH, 50/1). ЖХ-МС (ИЭР) m/z: 308,1 [M+H]+.
Стадии 2&3: Процедуры те же, что и для условий на Стадиях 1-2 Примера 33, с применением 1-(2,5-дихлорпиримидин-4-ил)индол-3-карбоновой кислоты вместо 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоновой кислоты. ЖХ-МС (ИЭР) m/z: 463,1 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,87 (с, 1H), 8,71 (с, 1H), 8,49 (с, 1H), 8,29-8,15 (м, 1H), 7,78-7,64 (м, 2H), 7,39 (с, 1H), 7,31-7,21 (м, 2H), 7,11 (с, 1H), 6,79 (с, 1H), 3,79 (с, 3H), 3,33 (д, J=5,8 Гц, 2H), 2,85-2,70 (м, 2H), 2,54 (т, J=5,9 Гц, 2H), 2,29 (с, 3H).
Пример 114
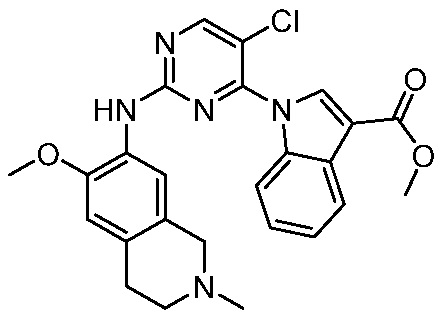
Метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-карбоксилат
Пример 35
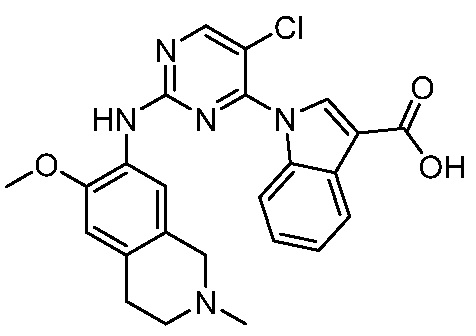
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-карбоновая кислота
Стадия 1: Метил 1H-индол-3-карбоксилат (1 г, 5,71 ммоль) и 2,4,5-трихлорпиримидин (1,15 г, 6,28 ммоль) применяют в условиях A1 с получением метил 1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-карбоксилата (800 мг, 43,5% выход) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=100/1). ЖХ-МС (ИЭР) m/z: 322 [M+H]+.
Стадия 2: Метил 1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-карбоксилат (200 мг, 0,621 ммоль) и Пр. W1 (143,2 мг, 0,745 ммоль) применяют в условиях B3 с получением метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-карбоксилата (138 мг, 44,2% выход) в виде белого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=100/1). ЖХ-МС (ИЭР) m/z: 478 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,95 (с, 1H), 8,72 (с, 1H), 8,48 (с, 1H), 8,09 (дд, J=6,4, 2,3 Гц, 1H), 7,76 (дд, J=6,6, 2,1 Гц, 1H), 7,47-7,24 (м, 3H), 6,79 (с, 1H), 3,87 (с, 3H), 3,78 (с, 3H), 3,33 (с, 2H), 2,78 (т, J=5,7 Гц, 2H), 2,55 (д, J=5,7 Гц, 2H), 2,30 (с, 3H).
Стадия 3: Смесь метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-карбоксилата (454 мг, 0,950 ммоль), AcOH (456 мг, 7,60 ммоль) и водного раствора HCl (6 N, 36 мл) перемешивают при 50°C в течение 16 часов и очищают преп-ЖХВД (MeCN/0,04% муравьиную кислоту) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-карбоновой кислоты (33,1 мг, 7,5% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 464,2 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,88 (с, 1H), 8,69 (с, 1H), 8,33 (д, J=20,1 Гц, 2H), 8,18 (с, 1H), 7,75 (д, J=7,8 Гц, 1H), 7,37 (с, 1H), 7,28 (с, 2H), 6,80 (с, 1H), 3,79 (с, 3H), 3,34-3,33 (м, 2H), 2,78 (с, 2H), 2,55 (с, 2H), 2,30 (с, 3H)
Пример 116
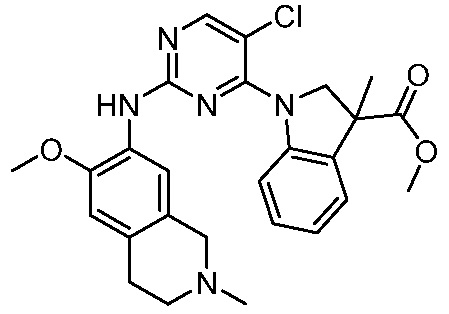
Метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоксилат
Пример 117
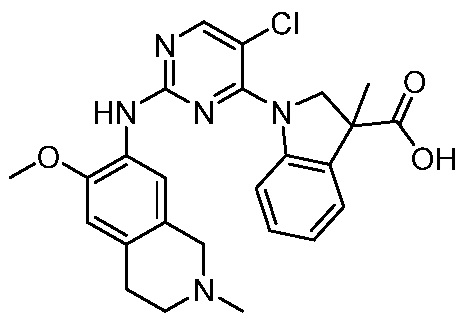
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоновая кислота
Стадия 1: Смесь Пр. E18 (158 мг, 0,826 ммоль), 2,4,5-трихлорпиримидина (197,0 мг, 1,07 ммоль) и ДИПЭА (427,2 мг, 3,30 ммоль) в н-бутаноле (4 мл) перемешивают при 100°C в течение 12 часов, охлаждают до комнатной температуры и разбавляют ДХМ/MeOH (10/1, 100 мл) и водой (30 мл). Водную фазу отделяют и экстрагируют ДХМ/MeOH (10/1, 30 мл x 3). Объединенные органические фазы промывают насыщенным раствором соли (40 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ: 30%) с получением метил 1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-карбоксилата (257 мг, 89,2% выход) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ: 30%). ЖХ-МС (ИЭР) m/z: 338 [M+H]+.
Стадия 2: Смесь метил 1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-карбоксилата (50 мг, 0,148 ммоль), Пр. W1 (40,6 мг, 0,177 ммоль) и TsOH.H2O (28,1 мг, 0,148 ммоль) в изопропаноле (3 мл) перемешивают при 120°C в течение 12 часов, охлаждают до комнатной температуры, нейтрализуют насыщенным NaHCO3 водным раствором (4 мл) и экстрагируют ДХМ (20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/10 мМ NH4HCO3, 0,025% NH3.H2O) с получением метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоксилата (20 мг, 27,4% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 494 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,27 (с, 1H), 8,08 (с, 1H), 7,49 (с, 1H), 7,35 (дд, J=12,7, 7,8 Гц, 2H), 7,16 (т, J=7,8 Гц, 1H), 7,00 (т, J=7,5 Гц, 1H), 6,75 (с, 1H), 4,75 (д, J=10,8 Гц, 1H), 4,09 (д, J=10,8 Гц, 1H), 3,76 (с, 3H), 3,63 (с, 3H), 3,29 (с, 2H), 2,77 (т, J=5,5 Гц, 2H), 2,56 (д, J=5,8 Гц, 2H), 2,31 (с, 3H), 1,56 (с, 3H).
Стадия 3: Смесь метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоксилата (20 мг, 0,040 ммоль) и водного раствора гидроксида лития (2N, 0,1 мл) в ТГФ (2,5 мл) перемешивают в течение 3 часов при комнатной температуре, доводят до pH ~5 муравьиной кислотой и экстрагируют ДХМ (15 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/0,1% муравьиная кислота) с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоновой кислоты в виде белого твердого вещества (10,7 мг, 55% выход). ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,04 (с, 1H), 7,52 (с, 1H), 7,36 (т, J=8,0 Гц, 2H), 7,15 (т, J=7,7 Гц, 1H), 6,99 (т, J=7,4 Гц, 1H), 6,76 (с, 1H), 4,76 (д, J=10,6 Гц, 1H), 4,04 (д, J=10,6 Гц, 1H), 3,35 (с, 2H), 2,79 (т, J=5,5 Гц, 2H), 2,61 (т, J=5,7 Гц, 2H), 2,34 (с, 3H), 1,53 (с, 3H).
Примеры 118 и 119
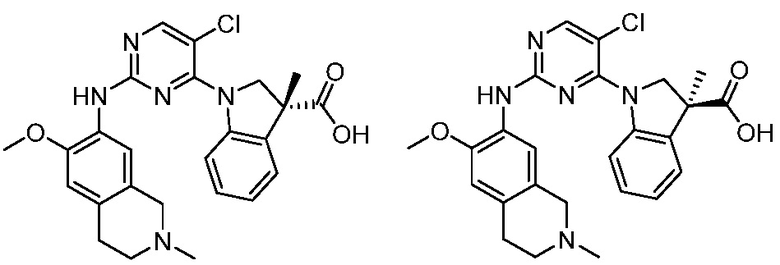
(R)-1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоновая кислота & (S)-1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоновая кислота
Синтезируют после хирального разделения рацемической 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоновой кислоты. Стереохимия не подтверждена, и два продукта назначены произвольно.
ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 8,00 (с, 1H), 7,52 (с, 1H), 7,37 (т, J=7,3 Гц, 2H), 7,13 (т, J=7,7 Гц, 1H), 6,99 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,77 (д, J=10,7 Гц, 1H), 4,02 (д, J=10,7 Гц, 1H), 3,77 (с, 3H), 3,11-3,01 (м, 2H), 2,77 (д, J=5,9 Гц, 2H), 2,57 (т, J=5,7 Гц, 2H), 2,32 (с, 3H), 1,52 (с, 3H). Хиральная-ЖХВД время удержания: 1,079 мин; значение эи: >99%.
ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,02 (с, 1H), 7,53 (с, 1H), 7,37 (т, J=7,4 Гц, 2H), 7,14 (т, J=8,0 Гц, 1H), 7,00 (т, J=7,5 Гц, 1H), 6,76 (с, 1H), 4,77 (д, J=10,7 Гц, 1H), 4,04 (д, J=10,5 Гц, 1H), 3,77 (с, 3H), 3,07-2,97 (м, 2H), 2,79 (с, 2H), 2,59 (д, J=5,9 Гц, 2H), 2,33 (с, 3H), 1,53 (с, 3H). Хиральная-ЖХВД время удержания: 1,501 мин; значение эи: >99%.
Условия разделения СЖХ:
Инструмент: SFC-150 (Thar, Waters)
Колонка: IG 20*250 мм, 10 мкм (Daicel)
Температура колонки: 35ºC
Подвижная фаза: CO2/EtOH (0,5% метанол/аммиак)=45/55
Скорость потока: 120 г/мин
Противодавление: 100 бар
Длина волны определения: 214 нм
Время цикла: 3 мин
Пример 120
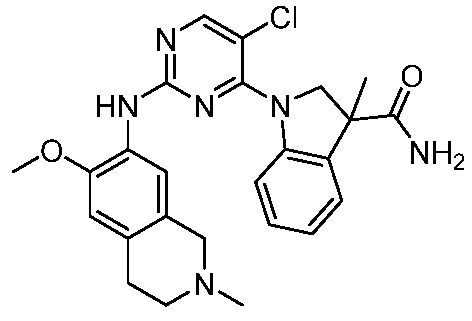
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоксамид
Стадия 1: Пр. E19 (148 мг, 0,798 ммоль) и 2,4,5-трихлорпиримидин (146,4 мг, 0,798 ммоль) применяют в условиях A3 с получением 1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-карбоксамида (199 мг, 69,5% выход) в виде белого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ: 1/5). ЖХ-МС (ИЭР) m/z: 323 [M+H]+.
Стадия 2: 1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-карбоксамид (199 мг, 0,585 ммоль) и Пр. W1 (135,0 мг, 0,702 ммоль) применяют в условиях B3 с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоксамида (82,5 мг, 28,3% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (MeCN/10 мМ NH4HCO3). ЖХ-МС (ИЭР) m/z: 479 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,25 (с, 1H), 7,99 (с, 1H), 7,52 (с, 1H), 7,46 (д, J=7,5 Гц, 1H), 7,37 (д, J=8,1 Гц, 1H), 7,28 (с, 1H), 7,18 (с, 1H), 7,13 (т, J=7,7 Гц, 1H), 7,00 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,81 (д, J=10,4 Гц, 1H), 3,97 (д, J=10,4 Гц, 1H), 3,77 (с, 3H), 3,29 (д, J=5,1 Гц, 2H), 2,77 (т, J=5,7 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,31 (с, 3H), 1,53 (с, 3H).
Пример 121
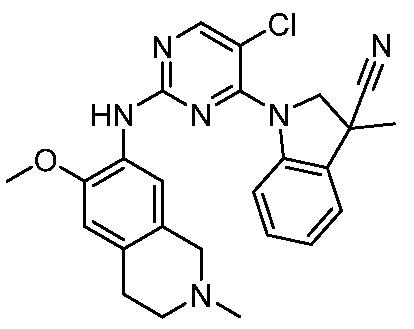
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбонитрил
Процедуры те же, что и для Примера 99, с применением 1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-карбоксамида вместо 1-(2,5-дихлорпиримидин-4-ил)индолин-3-карбоксамида. ЖХ-МС (ИЭР) m/z: 461 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,33 (с, 1H), 8,16 (с, 1H), 7,60-7,43 (м, 2H), 7,33 (д, J=8,0 Гц, 1H), 7,26 (т, J=7,7 Гц, 1H), 7,09 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,65 (д, J=10,9 Гц, 1H), 4,27 (д, J=10,9 Гц, 1H), 3,77 (д, J=5,8 Гц, 3H), 3,29 (с, 2H), 2,77 (т, J=5,7 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,31 (с, 3H), 1,73 (с, 3H).
Пример 122
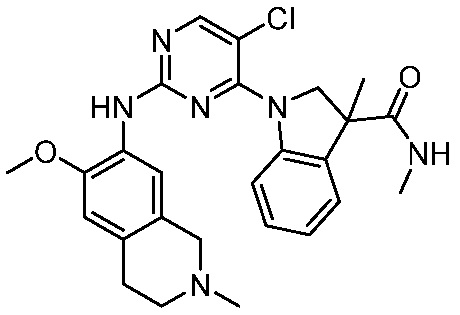
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N,3-диметилиндолин-3-карбоксамид
Стадия 1: Пр. E20 (68 мг, 0,357 ммоль) и 2,4,5-трихлорпиримидин (72,1 мг, 0,393 ммоль) применяют в условиях A3 с получением 1-(2,5-дихлорпиримидин-4-ил)-N,3-диметилиндолин-3-карбоксамида (72 мг, 59,7% выход) в виде бесцветного масла, после очистки колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH: 10/1). ЖХ-МС (ИЭР) m/z: 337 [M+H]+.
Стадия 2: 1-(2,5-дихлорпиримидин-4-ил)-N,3-диметилиндолин-3-карбоксамид (72 мг, 0,214 ммоль) и Пр. W1 (41,1 мг, 0,214 ммоль) применяют в условиях B3 с получением 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-N,3-диметилиндолин-3-карбоксамида (45,6 мг, 43,3% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (MeCN/0,1% FA). ЖХ-МС (ИЭР) m/z: 493 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,29 (д, J=32,1 Гц, 1H), 8,02 (с, 1H), 7,70 (с, 1H), 7,51 (с, 1H), 7,38 (т, J=7,2 Гц, 2H), 7,14 (т, J=7,1 Гц, 1H), 7,01 (д, J=6,7 Гц, 1H), 6,75 (с, 1H), 4,81 (д, J=10,3 Гц, 1H), 3,99 (д, J=10,3 Гц, 1H), 3,77 (с, 3H), 3,31 (с, 2H), 2,78 (с, 2H), 2,58 (с, 5H), 2,32 (с, 3H), 1,61 (д, J=64,4 Гц, 3H).
Пример 123
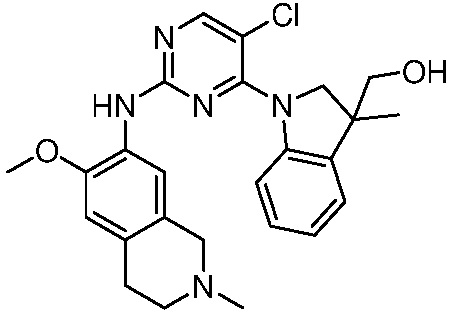
(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)метанол
Стадия 1: Пр. E21 (60 мг, 0,382 ммоль) и 2,4,5-трихлорпиримидин (70 мг, 0,382 ммоль) применяют в условиях A3 с получением (1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)метанола (85 мг, 64,6% выход) в виде желтого масла, после очистки колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH: 10/1). ЖХ-МС (ИЭР) m/z: 310 [M+H]+.
Стадия 2: (1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)метанол (85 мг,0,273 ммоль) и Пр. W1 (62,9 мг, 0,327 ммоль) применяют в условиях B3 с получением (1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)метанола (22,4 мг, 17,6%) в виде белого твердого вещества, после очистки преп-ЖХВД (MeCN/0,1% FA). ЖХ-МС (ИЭР) m/z: 466 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,23 (с, 1H), 7,97 (с, 1H), 7,54 (с, 1H), 7,37 (д, J=8,1 Гц,1H),7,21 (д, J=7,0 Гц, 1H), 7,10 (с, 1H), 6,97 (д, J=7,4 Гц, 1H), 6,75 (с, 1H), 5,01 (т, J=5,3 Гц, 1H), 4,26 (д, J=10,5 Гц, 1H), 3,87 (д, J=10,5 Гц, 1H), 3,77 (с, 3H), 3,39-3,35 (м, 2H), 3,30-3,28 (м, 2H), 2,80-2,75 (м, 2H), 2,58-2,54 (м, 2H), 2,31 (с, 3H), 1,29 (с, 3H).
Пример 124
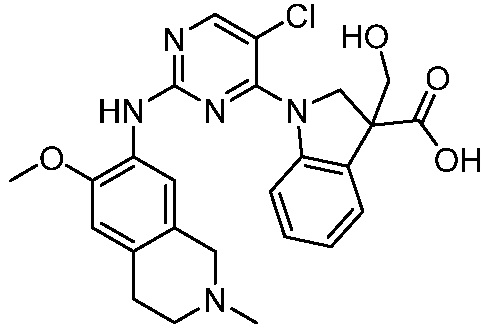
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-(гидроксиметил)индолин-3-карбоновая кислота
Процедуры те же, что и для Примера 117, с применением Пр. E37 вместо Пр. E18 на Стадии 1. ЖХ-МС (ИЭР) m/z: 498 [M+H]+. 1H ЯМР (400MГц, ДМСО-d6) δ 8,25 (с, 1H), 8,21 (с, 1H), 8,00 (д, J=9,3 Гц, 1H), 7,51 (с, 1H), 7,39 (т, J=8,0 Гц, 2H), 7,12 (т, J=7,7 Гц, 1H), 6,95 (т, J=7,4 Гц, 1H), 6,76 (с, 1H), 4,75 (д, J=10,7 Гц, 1H), 4,24 (д, J=10,7 Гц, 1H), 3,86 (д, J=10,4 Гц, 2H), 3,77 (с, 3H), 3,49 (ш, 1H), 3,33 (ш, 2H), 2,78 (т, J=5,5 Гц, 2H), 2,59 (т, J=5,7 Гц, 2H), 2,33 (с, 3H).
Пример 125
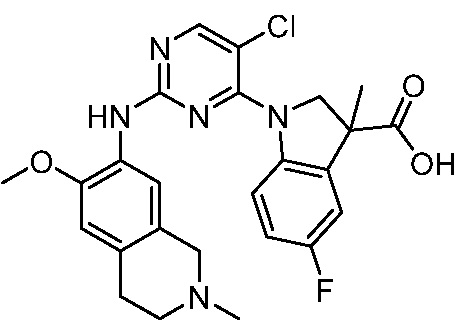
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фтор-3-метилиндолин-3-карбоновая кислота
Процедуры те же, что и для Примера 117, с применением Пр. E27 вместо Пр. E18 на Стадии 1. ЖХ-МС (ИЭР) m/z: 498 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,24 (с, 1H), 8,05 (с, 1H), 7,47 (с, 1H), 7,39 (дд, J=8,8, 4,7 Гц, 1H), 7,18 (дд, J=8,5, 2,7 Гц, 1H), 6,95 (тд, J=9,0, 2,7 Гц, 1H), 6,77 (с, 1H), 4,78 (д, J=10,7 Гц, 1H), 4,06 (д, J=10,7 Гц, 1H), 3,76 (с, 4H), 2,80 (д, J=5,7 Гц, 2H), 2,67 (т, J=5,8 Гц, 2H), 2,51 (с, 1H), 2,36 (д, J=12,7 Гц, 3H), 1,52 (с, 3H).
Пример 126
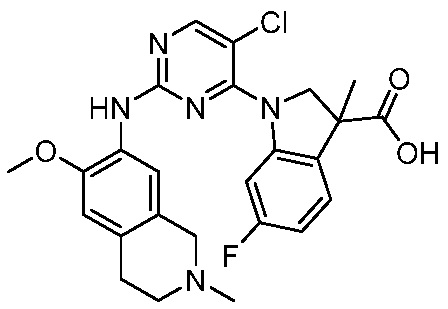
1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-карбоновая кислота
Процедуры те же, что и для Примера 117, с применением Пр. E28 вместо Пр. E18 на Стадии 1. ЖХ-МС (ИЭР) m/z: 498 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,27 (с, 1H), 8,20 (с, 1H), 7,41 (с, 1H), 7,37-7,27 (м, 1H), 7,18 (д, J=10,4 Гц, 1H), 6,75 (с, 2H), 4,81 (д, J=10,3 Гц, 1H), 4,08 (д, J=10,4 Гц, 1H), 3,76 (с, 3H), 3,35 (с, 2H), 2,78 (с, 2H), 2,60 (с, 2H), 2,33 (с, 3H), 1,50 (с, 3H).
Пример 127
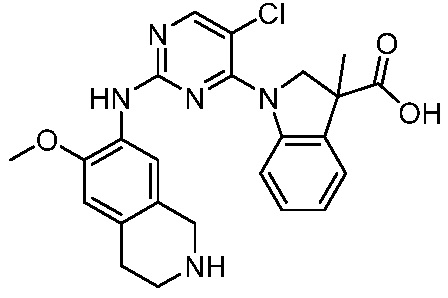
1-(5-Хлор-2-((6-метокси-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоновая кислота
Процедуры те же, что и для Примера 117, с применением Пр. W11 вместо Пр. W1 на Стадии 2. ЖХ-МС (ИЭР) m/z: 466 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,47 (с, 1H), 8,14 (с, 1H), 7,60 (с, 1H), 7,48 (с, 1H), 7,46 (д, J=6,8 Гц, 1H), 7,24 (т, J=7,6 Гц, 1H), 7,17 (т, J=7,2 Гц, 1H), 7,03 (с, 1H), 5,01 (д, J=11,0 Гц, 1H), 4,30 (д, J=11,0 Гц, 1H), 4,14 (д, J=6,3 Гц, 2H), 3,82 (с, 3H), 3,42 (т, J=6,3 Гц, 2H), 3,04 (т, J=6,1 Гц, 2H), 1,60 (с, 3H).
Пример 128
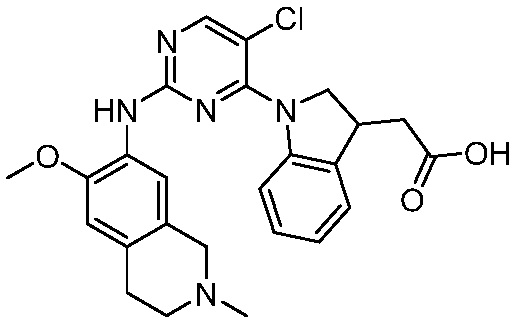
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-ил)уксусная кислота
Процедуры те же, что и для Примера 117, с применением Пр. E29 вместо Пр. E18 на Стадии 1. ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,26 (д, J=21,1 Гц, 1H), 7,98 (с, 1H), 7,55 (с, 1H), 7,30 (дд, J=22,6, 7,7 Гц, 2H), 7,10 (т, J=7,7 Гц, 1H), 6,94 (т, J=7,4 Гц, 1H), 6,74 (с, 1H), 4,56-4,36 (м, 1H), 3,98 (дд, J=10,6, 7,0 Гц, 1H), 3,77 (с, 3H), 3,69 (с, 2H), 3,29 (с, 2H), 2,84-2,70 (м, 3H), 2,56 (т, J=5,8 Гц, 2H), 2,31 (с, 3H).
Пример 129
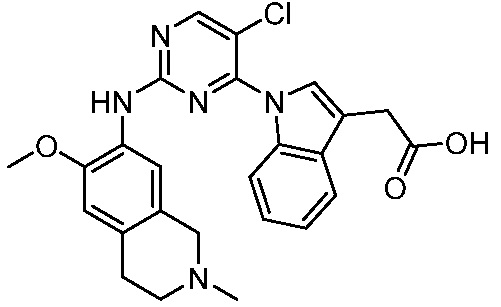
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)уксусная кислота
Пример 130
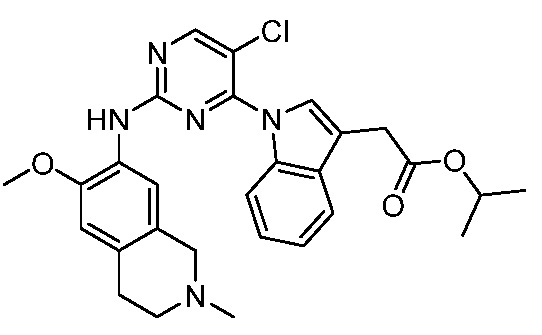
Изопропил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетат
Пример 131
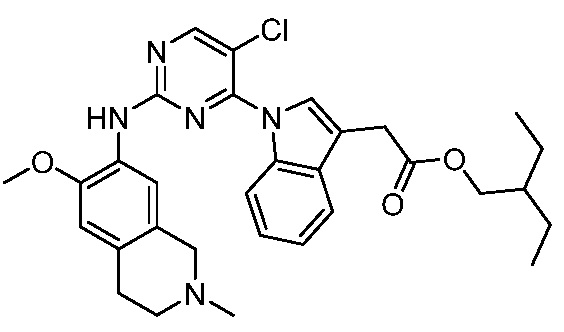
2-Этилбутил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетат
Стадия 1: 2-(1H-индол-3-ил)уксусную кислоту (300 мг, 1,71 ммоль) и 2,4,5-трихлорпиримидин (314,1 мг, 1,71 ммоль) применяют в условиях A1 с получением 2-(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)уксусной кислоты (93 мг, 16,0% выход) в виде светло-желтого твердого вещества, после очистки хроматографией с обращенной фазой. ЖХ-МС (ИЭР) m/z: 322 [M+H]+.
Стадия 2: 2-(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)уксусную кислоту (93 мг, 0,274 ммоль) и Пр. W1 (66,6 мг, 0,329 ммоль) применяют в условиях B3 с получением изопропил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетата (120 мг, 80,0% выход) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=10/1). ЖХ-МС (ИЭР) m/z: 520 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,70 (с, 1H), 8,62 (с, 1H), 7,86-7,77 (м, 2H), 7,57 (дд, J=6,0, 2,8 Гц, 1H), 7,43 (с, 1H), 7,21 (дд, J=6,0, 3,1 Гц, 2H), 6,80 (с, 1H), 4,94 (дт, J=12,5, 6,3 Гц, 1H), 3,80 (с, 2H), 3,79 (с, 3H), 3,32 (с, 2H), 2,78 (т, J=5,5 Гц, 2H), 2,55 (т, J=5,9 Гц, 2H), 2,30 (с, 3H), 1,21 (д, J=6,3 Гц, 6H).
2-Этилбутил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетат получают на Стадии 2, с применением 2-этилбутан-1-ола в качестве растворителя. ЖХ-МС (ИЭР) m/z: 562 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,69 (с, 1H), 8,63 (с, 1H), 8,36 (с, 1H), 7,85-7,74 (м, 2H), 7,60-7,52 (м, 1H), 7,45 (с, 1H), 7,28-7,14 (м, 2H), 6,78 (д, J=12,8 Гц, 1H), 3,99 (д, J=5,8 Гц, 2H), 3,85 (с, 2H), 3,79 (с, 3H), 3,31 (с, 2H), 2,78 (т, J=5,6 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,30 (д, J=8,2 Гц, 3H), 1,45 (дд, J=12,4, 6,2 Гц, 1H), 1,29-1,23 (м, 4H), 0,80 (т, J=7,4 Гц, 6H).
Стадия 3: Изопропил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетат (142 мг, 0,273 ммоль) используют в тех же условиях, как на Стадии 3 Примера 117 с получением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)уксусной кислоты в виде белого твердого вещества (57,9 мг, 44,4% выход), после очистки преп-ЖХВД (MeCN/10 мМ NH4HCO3). ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,79 (с, 1H), 8,64 (с, 1H), 7,80 (д, J=6,8 Гц, 2H), 7,55 (с, 1H), 7,26 (д, J=7,7 Гц, 1H), 7,22 (д, J=7,6 Гц, 1H), 6,91 (с, 1H), 3,92 (с, 2H), 3,83 (с, 3H), 3,75 (с, 2H), 3,17 (с, 2H), 2,98 (с, 2H), 2,69 (с, 3H).
Пример 132
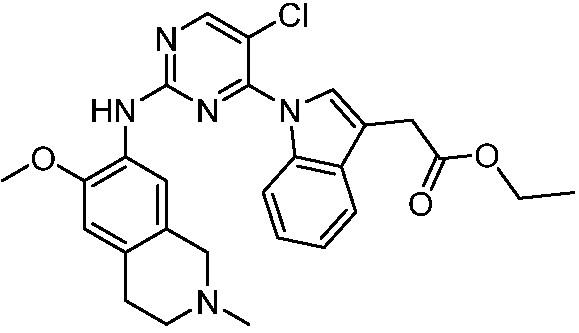
Этил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетат
К раствору 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)уксусной кислоты (60 мг, 0,126 ммоль) в этаноле (1 мл) добавляют SOCl2 (29,9 мг, 0,251 ммоль) по каплям. Реакционную смесь перемешивают при 85°C в течение 3 часов, охлаждают до комнатной температуры, подщелачивают до pH 8 насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом (10 мл x 3). Органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (10 мМ NH4HCO3&0,025%NH3·H2O/ацетонитрил) с получением этил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетата в виде светло-желтого твердого вещества (7,8 мг, 12% выход). ЖХ-МС (ИЭР) m/z: 506 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,70 (с, 1H), 8,62 (с, 1H), 7,83 (с, 1H), 7,82-7,76 (м, 1H), 7,64-7,53 (м, 1H), 7,43 (с, 1H), 7,28-7,16 (м, 2H), 6,80 (с, 1H), 4,11 (кв, J=7,1 Гц, 2H), 3,84 (с, 2H), 3,79 (с, 3H), 3,33 (с, 2H), 2,78 (т, J=5,5 Гц, 2H), 2,55 (т, J=5,8 Гц, 2H), 2,30 (с, 3H), 1,20 (т, J=7,1 Гц, 3H).
Пример 133
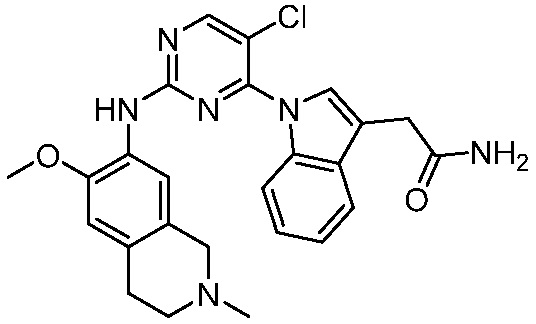
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетамид
К смеси 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)уксусной кислоты (30 мг, 0,060 ммоль), хлорида аммония (31,9 мг, 0,596 ммоль) и ГАТУ (34,0 мг, 0,089 ммоль) в ДМФ (1 мл) добавляют ДИПЭА (115,6 мг, 0,894 ммоль). Реакционную смесь перемешивают при 25°C в течение 16 часов и очищают преп-ЖХВД (MeCN/10 мМ NH4HCO3, 0,025% NH3·H2O) с получением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетамида (9,1 мг, 32,0% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 477 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,67 (с, 1H), 8,61 (с, 1H), 7,81 (дд, J=6,3, 3,0 Гц, 1H), 7,77 (с, 1H), 7,62 (дд, J=5,5, 3,3 Гц, 1H), 7,51 (с, 1H), 7,45 (с, 1H), 7,20 (дд, J=6,0, 3,1 Гц, 2H), 6,94 (с, 1H), 6,80 (с, 1H), 3,79 (с, 3H), 3,54 (с, 2H), 3,30 (с, 1H), 2,78 (д, J=5,6 Гц, 2H), 2,56 (д, J=5,8 Гц, 2H), 2,30 (с, 3H).
Пример 134
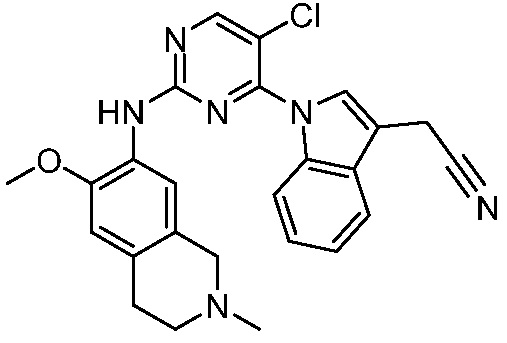
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетонитрил
К раствору 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетамида (50 мг, 0,105 ммоль) и пиридина (33,2 мг, 0,419 ммоль) в ДХМ (2 мл) добавляют трифторуксусный ангидрид (44,0 мг, 0,210 ммоль) при 0°C по каплям. Смесь перемешивают при 0°C в течение часа и комнатной температуре в течение 6 часов, гасят HCl водным раствором (1N, 10 мл) и экстрагируют ДХМ (20 мл x 3). Органические фазы промывают насыщенным раствором соли (15 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (0,1% FA/MeCN) с получением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетонитрила (19,3 мг, 39,7% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 459 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,76 (с, 1H), 8,65 (с, 1H), 8,35 (с, 2H), 7,89 (с, 1H),7,81 (дд, J=6,3, 2,9 Гц, 1H), 7,69 (дд, J=6,1, 2,8 Гц, 1H), 7,42 (с, 1H), 7,28 (дд, J=6,1, 3,2 Гц, 2H), 6,80 (с, 1H), 4,18 (с, 2H), 3,79 (с, 3H), 3,33 (с, 2H), 2,78 (т, J=5,6 Гц,2H), 2,55 (т, J=5,8 Гц, 2H), 2,30 (с, 3H).
Пример 135
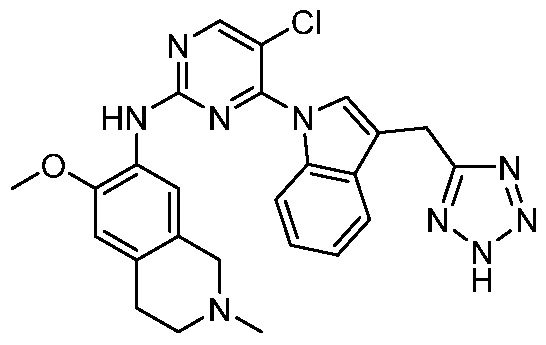
N-(4-(3-((2H-тетразол-5-ил)метил)-1H-индол-1-ил)-5-хлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Та же методика, как для Примера 100, с применением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)ацетонитрила вместо 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)индолин-3-карбонитрила. ЖХ-МС (ИЭР) m/z: 502 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,76 (с, 1H), 8,63 (с, 1H), 8,15 (с, 1H), 7,75 (д, J=4,8 Гц, 2H), 7,56 (д, J=7,6 Гц, 1H), 7,47 (с, 1H), 7,25-7,16 (м, 2H), 6,84 (с, 1H), 4,37 (с, 2H), 3,80 (с, 3H), 3,64 (с, 2H), 2,87 (с, 4H), 2,52 (с, 3H).
Пример 136
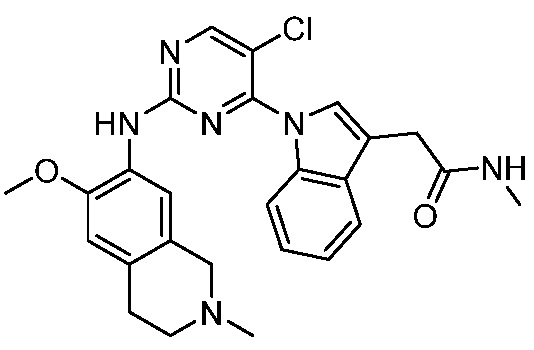
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)-N-метилацетамид
Та же методика, как для Примера 133, с применением метиламина вместо хлорида аммония. ЖХ-МС (ИЭР) m/z: 491 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,68 (с, 1H), 8,62 (с, 1H), 7,97 (с, 1H), 7,80 (дд, J=6,2, 3,0 Гц, 1H), 7,76 (с, 1H), 7,61 (дд, J=5,6, 3,1 Гц, 1H), 7,44 (с, 1H), 7,20 (дд, J=6,0, 3,1 Гц, 2H), 6,79 (с, 1H), 3,79 (с, 3H), 3,56 (с, 2H), 3,30 (с, 2H), 2,78 (д, J=5,1 Гц, 2H), 2,59 (д, J=4,6 Гц, 3H), 2,56 (д, J=5,7 Гц, 2H), 2,30 (с, 3H).
Пример 137
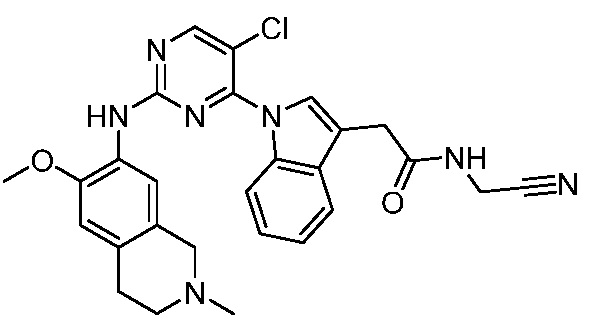
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)-N-(цианометил)ацетамид
Та же методика, как для Примера 133, с применением 2-аминоацетонитрилсерной кислоты вместо хлорида аммония. ЖХ-МС (ИЭР) m/z: 516 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,75 (с, 1H), 8,69 (с, 1H), 8,62 (с, 1H), 7,84-7,78 (м, 2H), 7,63-7,53 (м, 1H), 7,43 (с, 1H), 7,25-7,15 (м, 2H), 6,80 (с, 1H), 4,15 (д, J=5,5 Гц, 2H), 3,79 (с, 3H), 3,67 (с, 2H), 3,30 (с, 2H), 2,79 (с, 2H), 2,56 (д, J=5,5 Гц, 2H), 2,30 (с, 3H).
Пример 138
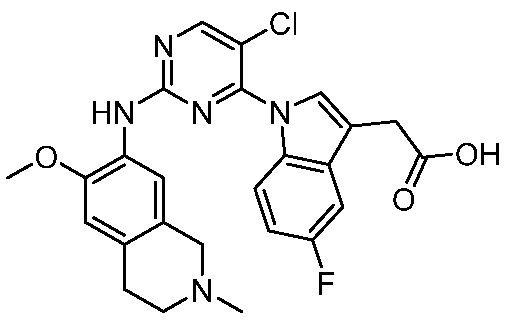
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фтор-1H-индол-3-ил)уксусная кислота
Процедуры те же, что и для Примера 129, с применением 2-(5-фтор-1H-индол-3-ил)уксусной кислоты вместо 2-(1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,77 (д, J=28,9 Гц, 1H), 8,61 (с, 1H), 7,90 (с, 1H), 7,81 (дд, J=9,0, 4,5 Гц, 1H), 7,46-7,24 (м, 2H), 7,04 (тд, J=9,1, 2,4 Гц, 1H), 6,81 (с, 1H), 3,79 (с, 3H), 3,72 (с, 2H), 3,35 (с, 2H), 2,80 (д, J=5,4 Гц, 2H), 2,59 (т, J=5,7 Гц, 2H), 2,32 (с, 3H).
Пример 139
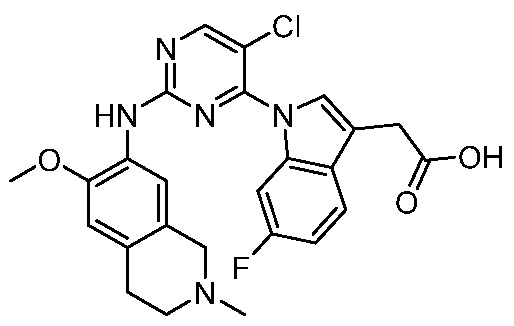
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-1H-индол-3-ил)уксусная кислота
Процедуры те же, что и для Примера 129, с применением 2-(6-фтор-1H-индол-3-ил)уксусной кислоты вместо 2-(1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,79 (с, 1H), 8,61 (с, 1H), 8,24 (д, J=10,5 Гц, 1H), 7,82 (с, 1H), 7,67-7,52 (м, 2H), 7,38 (с, 1H), 7,08 (тд, J=9,3, 2,4 Гц, 1H), 6,79 (с, 1H), 3,79 (с, 3H), 3,71 (с, 2H), 3,34 (с, 2H), 2,79 (т, J=5,6 Гц, 2H), 2,56 (т, J=5,8 Гц, 2H), 2,30 (с, 3H).
Пример 140
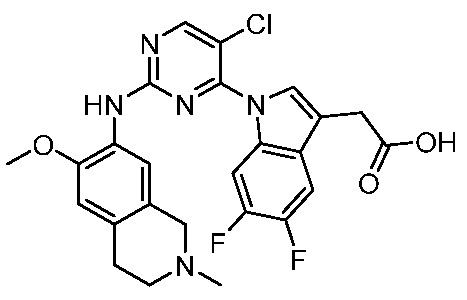
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5,6-дифтор-1H-индол-3-ил)уксусная кислота
Процедуры те же, что и для Примера 129, с применением 2-(5,6-дифтор-1H-индол-3-ил)уксусной кислоты вместо 2-(1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 514 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,84 (с, 1H), 8,61 (с, 1H), 8,23 (с, 1H), 7,92 (с, 1H), 7,88-7,79 (м, 1H), 7,61 (дд, J=10,8, 8,0 Гц, 1H), 7,35 (с, 1H), 6,79 (д, J=17,2 Гц, 1H), 3,78 (с, 3H), 3,72 (с, 2H), 3,35 (с, 2H), 2,80 (т, J=5,3 Гц, 2H), 2,57 (с, 2H), 2,32 (д, J=13,1 Гц, 3H).
Пример 141
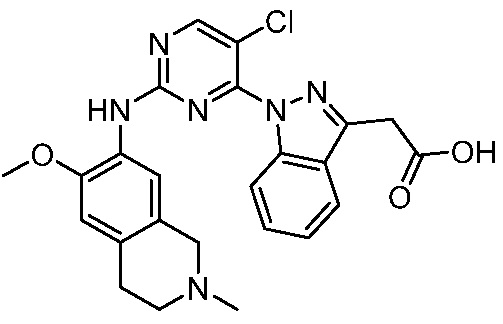
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индазол-3-ил)уксусная кислота
Процедуры те же, что и для Примера 129, с применением 2-(1H-индазол-3-ил)уксусной кислоты вместо 2-(1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 479 [M+H]+.
Пример 142
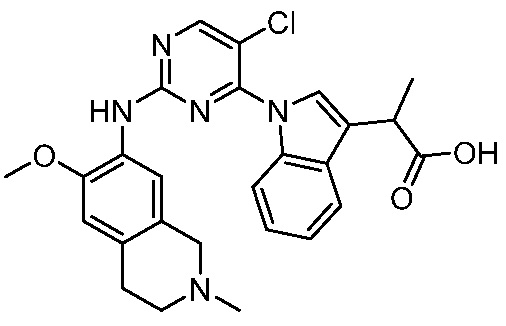
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)пропановая кислота
Процедуры те же, что и для Примера 129, с применением Пр. E30 вместо 2-(1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 492 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,69 (с, 1H), 8,62 (с, 1H), 8,26 (с, 1H), 7,80-7,74 (м, 1H), 7,72 (с, 1H), 7,65 (д, J=8,6 Гц, 1H), 7,44 (с, 1H), 7,24-7,15 (м, 2H), 6,79 (с, 1H), 3,93 (д, J=7,1 Гц, 1H), 3,78 (с, 3H), 3,34 (с, 2H), 2,77 (д, J=5,5 Гц, 2H), 2,56 (т, J=5,8 Гц, 2H), 2,30 (с, 3H), 1,49 (д, J=7,1 Гц, 3H).
Пример 143
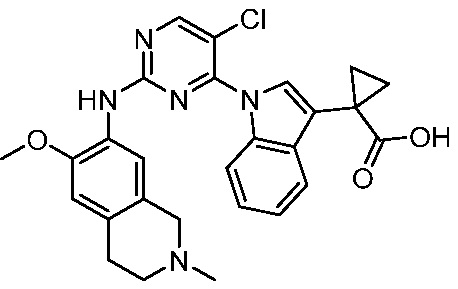
1-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)циклопропан-1-карбоновая кислота
Процедуры те же, что и для Примера 129, с применением Пр. E31 вместо 2-(1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 504 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,68 (с, 1H), 8,62 (с, 1H), 8,27 (с, 1H), 7,81-7,75 (м, 1H), 7,72 (с, 1H), 7,59-7,53 (м, 1H), 7,44 (с, 1H), 7,23-7,18 (м, 2H), 6,79 (с, 1H), 3,79 (с, 3H), 3,35 (с, 2H), 2,78 (т, J=5,5 Гц, 2H), 2,56 (т, J=5,7 Гц, 2H), 2,31 (с, 3H), 1,49 (д, J=2,7 Гц, 2H), 1,13 (д, J=2,1 Гц, 2H).
Пример 144
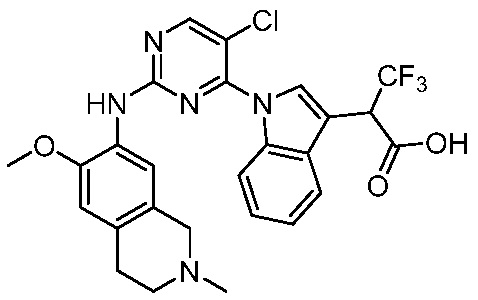
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)-3,3,3-трифторпропановая кислота
Стадия 1: Пр. E32 (200 мг, 0,822 ммоль)) и 2,4,5-трихлорпиримидин (226,3 мг, 1,23 ммоль) применяют в условиях A1 с получением 2-(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)-3,3,3-трифторпропановой кислоты (190 мг, 47,4% выход, 80% чистота), после очистки хроматографией с обращенной фазой (MeCN/0,03% ТФК). ЖХ-МС (ИЭР) m/z: 390 [M+H]+.
Стадия 2: Пр. W1 (112,4 мг, 0,584 ммоль) и 2-(1-(2,5-дихлорпиримидин-4-ил)-1H-индол-3-ил)-3,3,3-трифторпропановую кислоту (190 мг, 0,487 ммоль) применяют в условиях B3 с получением 1-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)циклопропан-1-карбоновой кислоты в виде белого твердого вещества (9,3 мг, 3,4% выход), после очистки преп-ЖХВД (MeCN/0,1% HCOOH). ЖХ-МС (ИЭР) m/z: 546 [M+H]+. 1H ЯМР (400MГц, ДМСО-d6) δ 8,82 (с, 1H), 8,65 (с, 1H), 8,17 (с, 1H), 7,82 (с, 1H), 7,73 (д, J=7,6 Гц, 1H), 7,63 (д, J=7,7 Гц, 1H), 7,45 (с, 1H), 7,25-7,19 (м, 2H), 6,82 (с, 1H), 4,63 (д, J=9,0 Гц, 1H), 3,77 (с, 3H), 3,63 (ш, 2H), 2,88 (ш, 5H), 2,56 (ш, 2H).
Пример 145
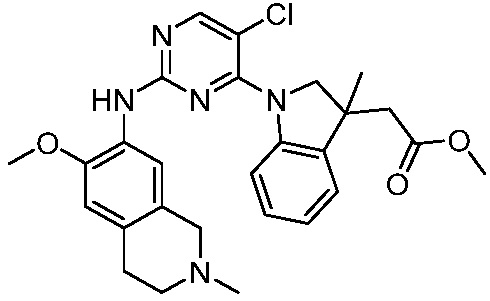
Метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетат
Пример 146
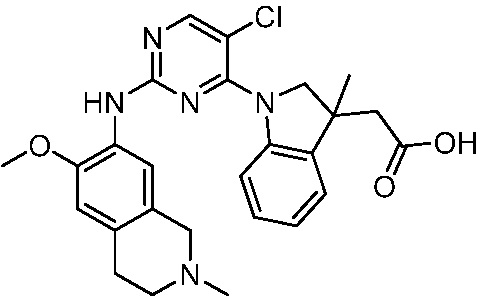
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Стадия 1: К смеси Пр. E33 (67 мг, 0,248 ммоль) в n-BuOH (3 мл) добавляют медленно 2,4,5-трихлорпиримидин (45,5 мг, 0,248 ммоль) и ДИПЭА (96,19 мг, 0,744 ммоль). Смесь перемешивают в течение 16 часов при 100°C, гасят льдом-холодной водой (20 мл) и экстрагируют ЭА (30 мл x 3). Органические фазы промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/ПЭ=0-100%) с получением метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (80 мг, 83,3% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 352 [M+H]+.
Стадия 2: К смеси метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (80 мг, 0,227 ммоль) в i-PrOH (5 мл) добавляют медленно Пр. W1 (51,5 мг, 0,227 ммоль) и TsOH.H2O (43,2 мг, 0,227 ммоль). Смесь перемешивают в течение 16 часов при 100°C, гасят насыщенным NaHCO3 водным раствором (10 мл) и экстрагируют ДХМ/MeOH (об/об=10/1, 30 мл x 3). Органические фазы промывают насыщенным раствором соли (30 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=0-15%) с получением метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (70 мг, 42,5% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 508 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,26 (с, 1H), 7,98 (с, 1H), 7,59 (с, 1H), 7,30 (т, J=8,8 Гц, 2H), 7,12 (с, 1H), 6,98 (с, 1H), 6,74 (с, 1H), 4,38 (д, J=10,7 Гц, 1H), 4,05 (д, J=10,7 Гц, 1H), 3,78 (с, 3H), 3,50 (с, 3H), 3,27 (с, 2H), 2,78 (дд, J=10,3, 4,8 Гц, 3H), 2,68 (с, 1H), 2,55 (т, J=5,7 Гц, 2H), 2,30 (с, 3H), 1,36 (с, 3H).
Стадия 3: Смесь метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (70 мг, 0,096 ммоль) и водного раствора LiOH (2N, 1 мл) в ТГФ (5 мл) перемешивают в течение 16 часов при комнатной температуре, доводят до pH ~5 муравьиной кислотой и экстрагируют ДХМ (30 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают преп-ЖХВД (MeCN/10 мМ NH4HCO3, 0,025% NH3.H2O) с получением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты (23,0 мг, 46,2% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 494 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,25 (с, 1H), 7,99 (с, 1H), 7,57 (с, 1H), 7,29 (дд, J=17,2, 7,6 Гц, 2H), 7,11 (т, J=7,3 Гц, 1H), 6,98 (д, J=7,3 Гц, 1H), 6,75 (с, 1H), 4,42 (д, J=10,7 Гц, 1H), 4,02 (д, J=10,6 Гц, 1H), 3,78 (с, 3H), 3,30 (с, 2H), 2,78 (с, 2H), 2,69 (д, J=15,4 Гц, 1H), 2,58 (д, J=6,1 Гц, 3H), 2,32 (с, 3H), 1,34 (с, 3H).
Примеры 147 и 148
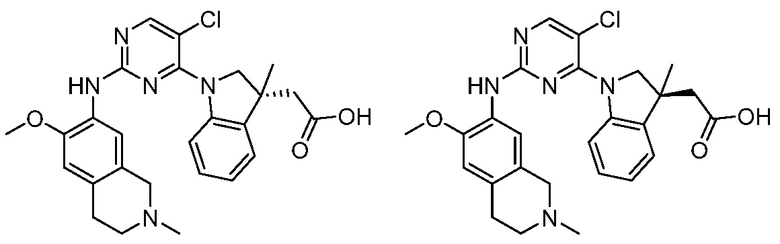
(R)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота & (S)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Способ 1:
Стадия 1: Разделение хиральной СЖХ метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (157 мг) с получением (R)-метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата и (S)-метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (67 мг, 63 мг по отдельности). Стереохимия не подтверждена, и два продукта назначены произвольно.
ЖХ-МС (ИЭР) m/z: 508 [M+H]+. Хиральная-ЖХВД время удержания: 1,829 мин; значение эи: 98,3%.
ЖХ-МС (ИЭР) m/z: 508 [M+H]+. Хиральная-ЖХВД время удержания: 3,427 мин; значение эи: >99%.
Условия разделения СЖХ:
Инструмент: SFC-80 (Thar, Waters)
Колонка: AD-H 20*250 мм, 10 мкм (Daicel)
Температура колонки: 35ºC
Подвижная фаза: CO2/EtOH (0,2% метанол/аммиак)=50/50
Скорость потока: 80 г/мин
Противодавление: 100 бар
Длина волны определения: 214 нм
Время цикла: 6,9 мин
Стадия 2: (R)-метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетат и (S)-метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетат гидролизуют в тех же условиях, как на Стадии 3 Примера 146 по отдельности, с получением (R)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты и (S)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты (17,6 мг, 33,8 мг по отдельности). Стереохимия не подтверждена, и два продукта назначены произвольно.
ЖХ-МС (ИЭР) m/z: 494 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 8,00 (с, 1H), 7,57 (с, 1H), 7,30 (дд, J=13,7, 7,7 Гц, 2H), 7,11 (т, J=7,6 Гц, 1H), 6,97 (т, J=7,3 Гц, 1H), 6,75 (с, 1H), 4,42 (д, J=10,7 Гц, 1H), 4,02 (д, J=10,8 Гц, 1H), 3,78 (с, 3H), 3,30 (с, 2H), 2,78 (с, 2H), 2,69 (д, J=15,4 Гц, 1H), 2,62-2,54 (м, 3H), 2,32 (с, 3H), 1,34 (с, 3H). Хиральная-ЖХВД время удержания: 1,384 мин; значение эи: >99%.
ЖХ-МС (ИЭР) m/z: 494 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 8,00 (с, 1H), 7,56 (с, 1H), 7,29 (дд, J=13,6, 7,4 Гц, 2H), 7,10 (т, J=7,1 Гц, 1H), 6,97 (т, J=7,4 Гц, 1H), 6,75 (с, 1H), 4,41 (д, J=10,8 Гц, 1H), 4,02 (д, J=10,8 Гц, 1H), 3,77 (с, 3H), 3,29 (с, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,69 (д, J=15,4 Гц, 1H), 2,57 (дд, J=13,1, 7,1 Гц, 3H), 2,30 (д, J=11,1 Гц, 3H), 1,34 (с, 2H). Хиральная-ЖХВД время удержания: 1,132 мин; значение эи: >99%.
Способ 2:
Стадия 1: Разделение хиральной СЖХ метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата с получением (R)-метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата и (S)-метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата. Стереохимия не подтверждена, и два продукта назначены произвольно.
ЖХ-МС (ИЭР) m/z: 352 [M+H]+. Хиральная-ЖХВД время удержания: 1,191 мин; значение эи: >99%.
ЖХ-МС (ИЭР) m/z: 352 [M+H]+. Хиральная-ЖХВД время удержания: 1,519 мин; значение эи: >99%.
Условия разделения СЖХ:
Инструмент: SFC-80 (Thar, Waters)
Колонка: AD-H 20*250 мм, 10 мкм (Daicel)
Температура колонки: 35ºC
Подвижная фаза: CO2/изопропанoл (0,2% метанол/аммиак)=87/13
Скорость потока: 80 г/мин
Противодавление: 100 бар
Длин волны определения: 214 нм
Время цикла: 2,8 мин
(Один стереоизомер как типовой пример Стадий 2&3)
Стадия 2: Смесь Пр. W1 (1,36 г, 5,96 ммоль), (S)-метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (2,1 г, 5,96 ммоль) и TsOH.H2O (1,13 г, 5,96 ммоль) в т-бутанoле (25 мл) перемешивают в течение 12 часов при 100°С, охлаждают до комнатной температуры и выливают в насыщенным NaHCO3 водным раствором (10 мл) и экстрагируют ЭА (30 мл x 3). Органические фазы сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20:1) с получением (S)-метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (1,8 г, 56,3% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 508 [M+H]+.
Стадия 3: Смесь раствор гидрата гидроксида лития (264 мг, 6,3 ммоль) в воде (5 мл) и (S)-метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (1,6 г, 3,15 ммоль) в MeOH (5 мл) перемешивают в течение 16 часов при комнатной температуре и подкисляют до pH ~3 уксусной кислотой. Полученную смесь очищают преп-ЖХВД (MeCN/10 мМ NH4HCO3) с получением (S)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты (839,3 мг, 53,9% выход) as pale yellow solid. ЖХ-МС (ИЭР) m/z: 494 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,30 (с, 1H), 8,02 (с, 1H), 7,63 (с, 1H), 7,37 (д, J=8,0 Гц, 1H), 7,33 (д, J=8,0 Гц, 1H), 7,16 (т, J=8,0 Гц, 1H), 7,02 (т, J=8,0 Гц, 1H), 6,80 (с, 1H), 4,48 (д, J=10,8 Гц, 1H), 4,08 (д, J=10,8 Гц, 1H), 3,83 (с, 3H), 2,83 (т, J=5,6 Гц, 2H), 2,74 (д, J=15,2 Гц, 1H), 2,66-2,59 (м, 3H), 2,37 (с, 3H), 1,40 (с, 3H). Хиральная-ЖХВД время удержания: 1,132 мин; значение эи: >99%.
Пример 149
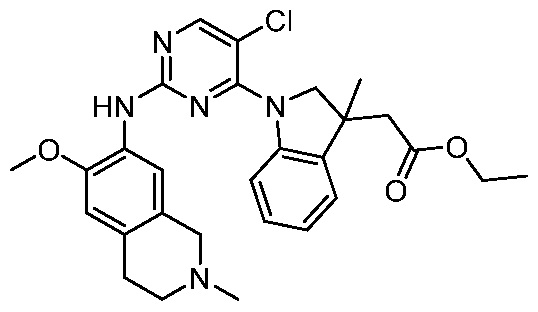
Этил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетат
Та же методика, как для Примера 132, с применением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты вместо 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-3-ил)уксусной кислоты. ЖХ-МС (ИЭР) m/z: 522 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,25 (с, 1H), 7,97 (с, 1H), 7,57 (с, 1H), 7,36-7,24 (м, 2H), 7,12 (дд, J=11,1, 4,3 Гц, 1H), 6,98 (с, 1H), 6,74 (с, 1H), 4,41 (д, J=10,8 Гц, 1H), 4,05 (д, J=10,7 Гц, 1H), 3,96 (дд, J=7,1, 1,5 Гц, 2H), 3,77 (д, J=3,3 Гц, 3H), 3,27 (с, 2H), 2,75 (дд, J=10,1, 7,2 Гц, 3H), 2,65 (с, 1H), 2,55 (д, J=5,7 Гц, 2H), 2,30 (с, 3H), 1,35 (с, 3H), 1,06 (т, J=7,1 Гц, 3H).
Пример 150
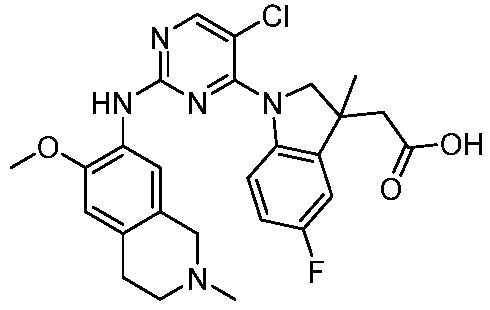
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фтор-3-метилиндолин-3-ил)уксусная кислота
Стадия 1: Раствор Пр. E34 (295 мг, 1,32 ммоль), 2,4,5-трихлорпиримидина (266,6 мг, 1,45 ммоль) и ДИПЭА (170,5 мг, 1,32 ммоль) в n-BuOH (2 мл) перемешивают при 100°C в течение 2 часов, охлаждают до комнатной температуры, выливают в воду и экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/4) с получением метил 2-(1-(2,5-дихлорпиримидин-4-ил)-5-фтор-3-метилиндолин-3-ил)ацетата (416 мг, 85,0% выход) в виде беловатого твердого вещества. ЖХ-МС (ИЭР) m/z: 370 [M+H]+.
Стадия 2: Раствор метил 2-(1-(2,5-дихлорпиримидин-4-ил)-5-фтор-3-метилиндолин-3-ил)ацетата (100 мг, 0,270 ммоль), Пр. W1 (62,3 мг, 0,324 ммоль) и TsOH.H2O (51,3 мг, 0,270 ммоль) в i-PrOH (2 мл) перемешивают при 100°C в течение 16 часов, охлаждают до комнатной температуры и нейтрализуют до pH ~8 насыщенным водным раствором NaHCO3. Смесь экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/4) с получением метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фтор-3-метилиндолин-3-ил)ацетата (186 мг, 80,7% выход, 60% чистота) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 526 [M+H]+.
Стадия 3: Смесь метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фтор-3-метилиндолин-3-ил)ацетата (186 мг, 0,354 ммоль) и водного раствора LiOH (2N, 3,5 мл) в ТГФ (8 мл) перемешивают в течение ночи при комнатной температуре, доводят до pH ~5 муравьиной кислотой и экстрагируют ДХМ/MeOH (об/об, 10/1, 10 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/10мМ NH4HCO3, 0,025% NH3.H2O) с получением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5-фтор-3-метилиндолин-3-ил)уксуснаяой кислоты (57,6 мг, 31,8% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 512 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,23 (с, 1H), 8,02 (с, 1H), 7,51 (с, 1H), 7,34 (дд, J=8,7, 4,7 Гц, 1H), 7,20 (дд, J=8,6, 2,7 Гц, 1H), 6,89 (тд, J=9,0, 2,7 Гц, 1H), 6,75 (с, 1H), 4,44 (д, J=10,9 Гц, 1H), 4,06 (д, J=10,8 Гц, 1H), 3,77 (с, 3H), 3,28 (с, 2H), 2,78 (т, J=5,7 Гц, 2H), 2,71 (д, J=15,6 Гц, 1H), 2,62 (с, 1H), 2,59-2,55 (м, 2H), 2,31 (с, 3H), 1,33 (с, 3H).
Пример 151
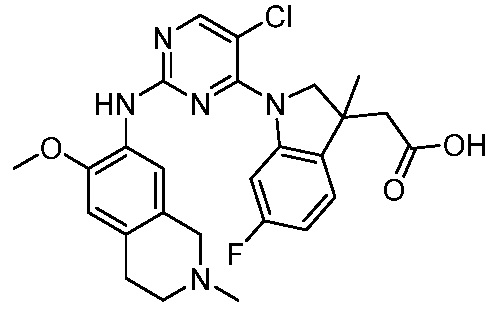
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)уксусная кислота
Стадия 1: Раствор Пр. E35 (78 мг, 0,349 ммоль), 2,4,5-трихлорпиримидина (64,1 мг, 0,349 ммоль) и ДИПЭА (135,2 мг, 1,05 ммоль) в n-BuOH (2 мл) перемешивают при 100°C в течение 16 часов, охлаждают до комнатной температуры, выливают в воду и экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/10) с получением метил 2-(1-(2,5-дихлорпиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)ацетата (100 мг, 73,4% выход) в виде светло-желтого масла. ЖХ-МС (ИЭР) m/z: 370 [M+H]+.
Стадия 2: Раствор метил 2-(1-(2,5-дихлорпиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)ацетата (82,7 мг, 0,232 ммоль), Пр. W1 (63,8 мг, 0,279 ммоль) и TsOH.H2O (53,0 мг, 0,279 ммоль) в i-PrOH (2 мл) перемешивают при 120°C в течение 16 часов, охлаждают до комнатной температуры и нейтрализуют до pH ~8 насыщенным NaHCO3 водным раствором. Смесь экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1) с получением метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)ацетата (81 мг, 62,9% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 526 [M+H]+.
Стадия 3: Смесь метил 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)ацетата (81 мг, 0,154 ммоль) и водного раствора LiOH (1N, 4 мл) в диоксане (4 мл) перемешивают в течение ночи при комнатной температуре, доводят до pH ~5 муравьиной кислотой и экстрагируют ЭА (30 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/10 мМ NH4HCO3, 0,025% NH3.H2O) с получением 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)уксусной кислоты (29,8 мг, 37,8% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 512 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,17 (с, 1H), 7,46 (с, 1H), 7,31-7,23 (м, 1H), 7,14 (д, J=10,0 Гц, 1H), 6,75 (с, 2H), 4,46 (д, J=10,7 Гц, 1H), 4,07 (д, J=10,6 Гц, 1H), 3,77 (с, 3H), 3,31 (с, 2H), 2,77 (с, 2H), 2,69 (д, J=15,6 Гц, 1H), 2,58 (д, J=14,8 Гц, 3H), 2,31 (с, 3H), 1,33 (с, 3H).
Примеры 152 и 153
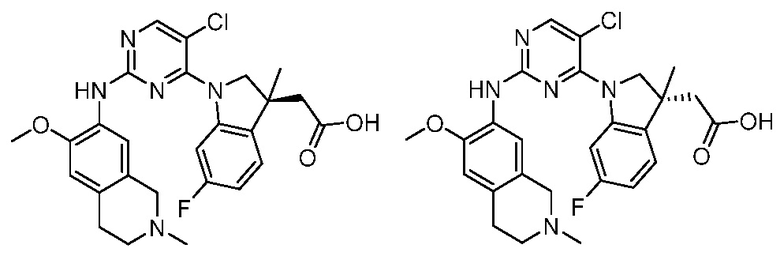
(S)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)уксусная кислота & (R)-2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)уксусная кислота
Синтезируют после хирального разделения рацемической 2-(1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-6-фтор-3-метилиндолин-3-ил)уксусной кислоты. Стереохимия не подтверждена, и два продукта назначены произвольно.
ЖХ-МС (ИЭР) m/z: 512 [M+H]+. Хиральная-ЖХВД время удержания: 11,913 мин; значение эи: 80,4%.
ЖХ-МС (ИЭР) m/z: 512 [M+H]+. Хиральная-ЖХВД время удержания: 16,631 мин; значение эи: 98,9%.
Условия разделения СЖХ:
Инструмент: Gilson-281
Колонка: IG 20*250 мм, 10 мкм
Подвижная фаза: н-гексан (0,1% диметиламин)/EtOH (0,1% диэтиламин)=65:35
Скорость потока: 40 мл/мин
Время цикла: 22 мин
Пример 154
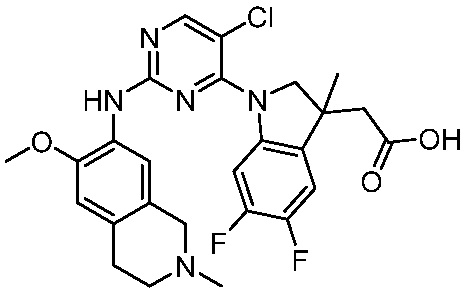
2-(1-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-5,6-дифтор-3-метилиндолин-3-ил)уксусная кислота
Процедуры те же, что и для Примера 146, с применением Пр. E36 вместо Пр. E33. ЖХ-МС (ИЭР) m/z: 530 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,24 (с, 1H), 8,20 (с, 1H), 7,45-7,36 (м, 3H), 6,75 (с, 1H), 4,46 (д, J=10,8 Гц, 1H), 4,08 (д, J=10,7 Гц, 1H), 3,76 (с, 3H), 3,27 (с, 2H), 2,77 (т, J=5,5 Гц, 2H), 2,70 (д, J=15,6 Гц, 1H), 2,61 (с, 1H), 2,56 (дд, J=12,0, 6,2 Гц, 2H), 2,29 (с, 3H), 1,31 (с, 3H).
Пример 155
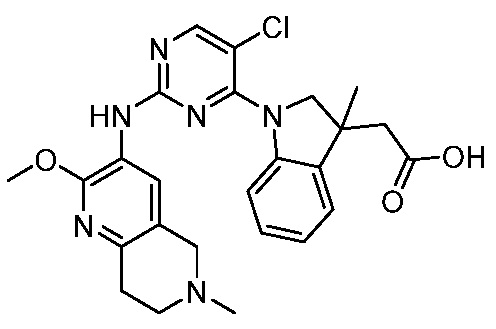
2-(1-(5-Хлор-2-((2-метокси-6-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Процедуры те же, что и для Примера 146, с применением Пр. W4 вместо Пр. W1 на Стадии 2 и без проведения Стадии 3 из-за гидролиза метилового эфира на Стадии 2. ЖХ-МС (ИЭР) m/z: 495 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 11,87 (с, 1H), 8,37 (с, 1H), 7,99 (с, 1H), 7,87 (с, 1H), 7,35(т, J=8,6 Гц, 2H), 7,23 (т, J=7,7 Гц, 1H), 7,03 (т, J=7,3 Гц, 1H), 4,39 (д, J=10,7 Гц, 1H), 4,07 (д, J=10,7Гц,1H), 3,47 (с, 3H), 3,31 (с, 1H), 3,09 (с, 2H), 2,80 (д, J=15,0 Гц, 1H), 2,71-2,63 (м, 1H), 2,56 (с,1H), 2,54 (с, 2H), 2,31 (с, 3H), 1,37 (с, 3H).
Пример 156
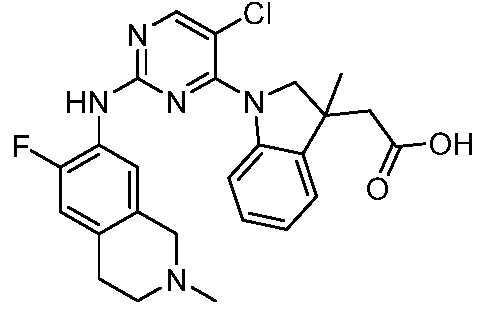
2-(1-(5-Хлор-2-((6-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Стадия 1: Раствор метил 2-(1-(2,5-дихлорпиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (195,4 мг, 0,555 ммоль), Пр. W7 (100 мг, 0,555 ммоль) и TsOH.H2O (105,5 мг, 0,555 ммоль) в i-PrOH (4 мл) перемешивают при 120°C в течение 16 часов, охлаждают до комнатной температуры и нейтрализуют до pH ~8 насыщенным водным раствором NaHCO3. Смесь экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=10/1) с получением смесь метил 2-(1-(5-хлор-2-((6-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата и изопропил 2-(1-(5-хлор-2-((6-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (440 мг) в виде светло-желтого твердого вещества, которое применяют на следующей стадии без дальнейших очисток. ЖХ-МС (ИЭР) m/z: 496 (метиловый эфир), 524 (изопропиловый эфир) [M+H]+.
Стадия 3: Смесь метил 2-(1-(5-хлор-2-((6-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата и изопропил 2-(1-(5-хлор-2-((6-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)ацетата (220 мг) и водного раствора LiOH (2N, 4 мл) в ТГФ (4 мл) перемешивают при комнатной температуре в течение 5 часов и 50°С в течение 16 часов, доводят до pH ~ 5 муравьиной кислотой и экстрагируют ДХМ/MeOH (10/1, 20 мл x 3). Объединенные органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД (MeCN/10 мМ NH4HCO3) с получением 2-(1-(5-хлор-2-((6-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты (26,5 мг, 12,4% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 482 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,93 (с, 1H), 8,22 (с, 1H), 7,35-7,24 (м, 3H), 7,07-6,92 (м, 3H), 4,41 (д, J=10,7 Гц, 1H), 4,03 (д, J=10,8 Гц, 1H), 3,36 (с, 2H), 2,78 (д, J=5,4 Гц, 2H), 2,68 (д, J=15,5 Гц, 1H), 2,60-2,54 (м, 3H), 2,32 (с, 3H), 1,33 (с, 3H).
Пример 157
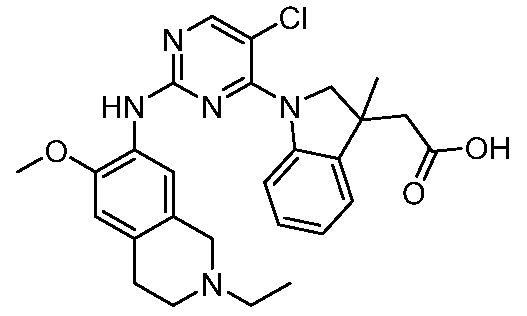
2-(1-(5-Хлор-2-((2-этил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Процедуры те же, что и для Примера 146, с применением Пр. W8 вместо Пр. W1 на Стадии 2. ЖХ-МС (ИЭР) m/z: 508 [M+H]+. 1H ЯМР (400MГц, ДМСО-d6) δ 8,25 (с, 1H), 7,97 (с, 1H), 7,61 (с,1H), 7,31 (дд, J=21,0, 7,7 Гц, 2H), 7,12 (т, J=7,5 Гц, 1H), 6,96 (т, J=7,2 Гц, 1H), 6,74 (с, 1H), 4,42 (д, J=10,7 Гц, 1H), 4,02 (д, J=10,7 Гц, 1H), 3,78 (с, 3H), 3,34 (с, 2H), 2,76 (с, 2H), 2,66-2,55 (м, 4H), 2,50-2,45 (д, J=6,8 Гц, 2H), 1,34 (с, 3H), 1,08 (т, J=7,0 Гц, 3H).
Пример 158
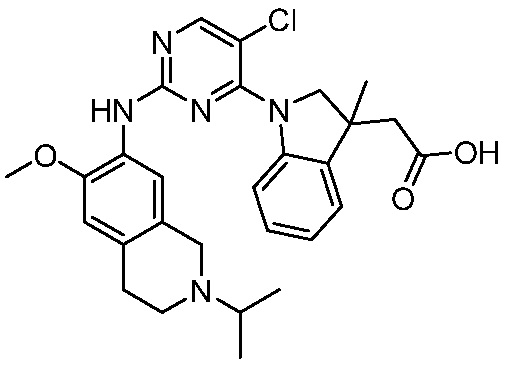
2-(1-(5-Хлор-2-((2-изопропил-6-метокси-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Процедуры те же, что и для Примера 146, с применением Пр. W5 вместо Пр. W1 на Стадии 2. ЖХ-МС (ИЭР) m/z: 522 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,22 (с, 1H), 7,97 (с, 1H), 7,63 (с, 1H), 7,34 (д, J=8,0 Гц, 1H), 7,28 (д, J=6,9 Гц, 1H), 7,17-7,05 (м, 1H), 6,96 (т, J=7,4 Гц, 1H), 6,73 (с, 1H), 4,42 (д, J=10,8 Гц, 1H), 4,02 (д, J=10,8 Гц, 1H), 3,78 (с, 3H), 3,47 (с, 2H), 2,93-2,81 (м, 1H), 2,77-2,68 (м, 4H), 2,66-2,54 (м, 2H), 1,34 (с, 3H), 1,06 (д, J=6,5 Гц, 6H).
Пример 159
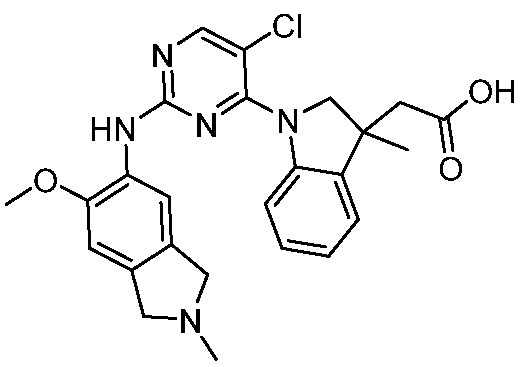
2-(1-(5-Хлор-2-((6-метокси-2-метилизоиндолин-5-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота
Процедуры те же, что и для Примера 146, с применением Пр. W9 вместо Пр. W1 на Стадии 2. ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,26 (с, 1H), 8,02 (с, 1H), 7,77 (с, 1H), 7,34 (д, J=8,0 Гц, 1H), 7,28 (д, J=7,1 Гц, 1H), 7,11 (т, J=7,3 Гц, 1H), 6,98 (т, J=7,2 Гц, 1H), 6,94 (с, 1H), 4,41 (д, J=10,8 Гц, 1H), 4,02 (д, J=10,7 Гц, 1H), 3,80 (с, 5H), 3,70 (с, 2H), 2,69 (д, J=15,5 Гц, 1H), 2,57 (д, J=15,4 Гц, 1H), 2,47 (с, 3H), 1,34 (с, 3H).
Пример 160
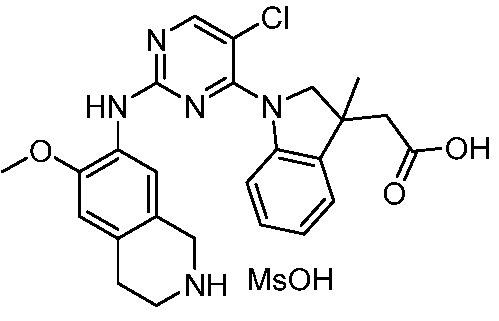
2-(1-(5-Хлор-2-((6-метокси-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусная кислота метансульфоновая кислота
Процедуры те же, что и для Примера 146, с применением Пр. W11 вместо Пр. W1 на Стадии 2. ЖХ-МС (ИЭР) m/z: 480 [M+H]+.
К раствору 2-(1-(5-хлор-2-((6-метокси-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты (40 мг, 0,083 ммоль) в ацетонитриле (20 мл) добавляют метансульфоновую кислоту (8,0 мг, 0,083 ммоль). Реакционную смесь перемешивают при 25°C в течение 30 минут и концентрируют в вакууме. Остаток разбавляют водой (15 мл) и лиофилизируют до соли метансульфоновой кислоты 2-(1-(5-хлор-2-((6-метокси-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-ил)уксусной кислоты (39,4 мг, 81,3% выход) в виде белого твердого вещества. ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,97 (д, J=14,2 Гц, 2H), 7,56 (с, 1H), 6,74 (с, 1H), 4,00 (с, 2H), 3,88 (д, J=11,4 Гц, 1H), 3,82 (с, 3H), 3,55 (с, 1H), 3,42 (с, 2H), 2,86 (т, J=7,5 Гц, 1H), 2,77 (с, 2H), 2,58 (с, 2H), 2,32 (с, 3H), 1,76-1,24 (м, 10H).
Пример 161
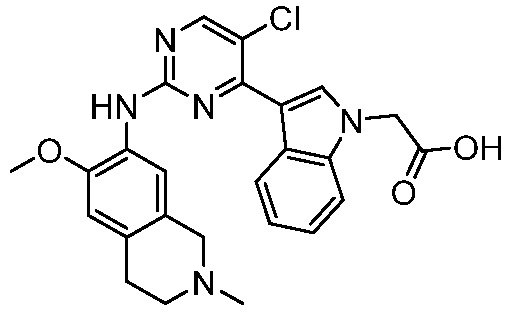
2-(3-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-1-ил)уксусная кислота
Стадия 1: К раствору индола (600,0 мг, 5,12 ммоль) в безводном ТГФ (10 мл) добавляют раствор метилмагния бромида в ТГФ (1M, 5 мл) при 0°C, затем добавляют 2,4,5-трихлорпиримидин (466 мг, 2,54 ммоль) через 10 минут. Реакционную смесь перемешивают при 60°C в течение 3 часов, охлаждают до комнатной температуры и концентрируют в вакууме. Остаток разделяют между водой (10 мл) и ДХМ (20 мл x 3). Органические фазы промывают насыщенным раствором соли (30 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (5-10% ЭА/ПЭ) с получением 3-(2,5-дихлорпиримидин-4-ил)-1H-индол в виде желтого твердого вещества (160 мг, 12% выход). ЖХ-МС (ИЭР) m/z: 264 [M+H]+.
Стадия 2: К раствору 3-(2,5-дихлорпиримидин-4-ил)-1H-индола (160 мг, 0,606 ммоль) и этил 2-бромацетата (303,5 мг, 1,82 ммоль, 201,00 мкл) в ДМФ (5 мл) добавляют бикарбонат натрия (254,5 мг, 3,03 ммоль) при 25°C. Смесь перемешивают при 80°C в течение 16 часов, охлаждают до комнатной температуры, разбавляют водой (20 мл) и экстрагируют ЭА (20 мл x 3). Объединенные органические фазы промывают насыщенным раствором соли (30 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (20-30% ЭА/ПЭ) с получением этил 2-(3-(2,5-дихлорпиримидин-4-ил)-1H-индол-1-ил)ацетата в виде светло-желтого масла (220 мг, 93% выход). ЖХ-МС (ИЭР) m/z: 315 [M+H]+.
Стадия 3: Этил 2-(3-(2,5-дихлорпиримидин-4-ил)-1H-индол-1-ил)ацетат (100 мг, 0,286 ммоль) и Пр. W1 (78,4 мг, 0,343 ммоль) применяют в условиях B3 с получением изопропил 2-(3-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-1-ил)ацетата (238 мг, 72,9% выход) в виде светло-желтого масла, после очистки колоночной флэш-хроматографией на силикагеле (30-60% MeOH/ДХМ). ЖХ-МС (ИЭР) m/z: 520 [M+H]+.
Стадия 4: К раствору изопропил 2-(3-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-1-ил)ацетата (238 мг, 0,412 ммоль) в ТГФ (6 мл) и воде (3 мл) добавляют LiOH.H2O (480,1 мг, 11,44 ммоль). Смесь перемешивают при 25°C в течение 2 часов, подкисляют муравьиной кислотой до pH ~5 и очищают преп-ЖХВД (0,1% муравьиная кислота/ацетонитрил) с получением 2-(3-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индол-1-ил)уксусной кислоты (124,9 мг, 63,4% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 478 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,56 (с, 1H), 8,47-8,30 (м, 2H), 8,23 (д, J=32,9 Гц, 1H), 7,62 (с, 1H), 7,42 (д, J=8,1 Гц, 1H), 7,21 (т, J=7,5 Гц, 1H), 7,07 (т, J=7,4 Гц, 1H), 6,84 (с, 1H), 5,01 (с, 2H), 3,79 (с, 3H), 3,51 (с, 2H), 2,88 (с, 2H), 2,74 (с, 2H), 2,39 (с, 3H).
Пример 162
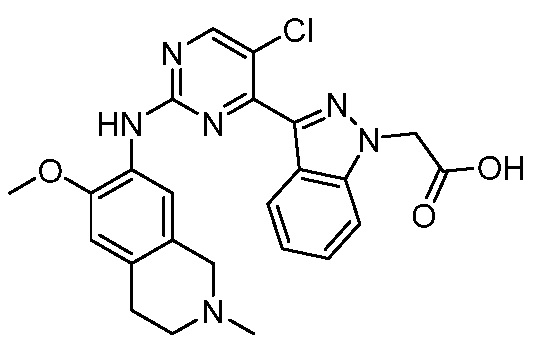
2-(3-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-1H-индазол-1-ил)уксусная кислота
Стадия 1: К раствору 3-йод-1H-индазола (500 мг, 2,05 ммоль) в ТГФ (15 мл) добавляют трет-бутоксид калия (344,9 мг, 3,07 ммоль) небольшими порциями при 0°C, затем по каплям добавляют этил 2-бромацетат (684,3 мг, 4,10 ммоль) через час. Смесь перемешивают при 25°C в течение 16 часов и концентрируют в вакууме. Остаток разделяют между ЭА (20 мл) и водой (20 мл). Органическую фазу отделяют, и водную фазу экстрагируют ЭА (20 мл x 3). Объединенные органические фазы сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (15-20% ЭА/ПЭ) с получением этил 2-(3-йод-1H-индазол-1-ил)ацетата в виде бесцветного масла (549 мг, 81% выход). ЖХ-МС (ИЭР) m/z: 331 [M+H]+.
Стадия 2: Смесь этил 2-(3-йод-1H-индазол-1-ил)ацетата (520 мг, 1,58 ммоль), 1,1,1,2,2,2-гексаметилдистаннана (1,03 г, 3,15 ммоль) и Pd(PPh3)4 (182,1 мг, 0,158 ммоль) в диоксане (10 мл) продувают N2, перемешивают при 100°C в течение 16 часов, охлаждают до 0°C и гасят 1M раствором KF (10 мл). Полученную смесь перемешивают при комнатной температуре в течение 30 минут, разбавляют водой (30 мл) и экстрагируют дихлорметаном/метанолом (об/об, 10/1, 20 мл x 3). Объединенную органическую фазу сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/4) с получением этил 2-(3-(триметилстаннил)-1H-индазол-1-ил)ацетата в виде бесцветного масла (145 мг, 25% выход). ЖХ-МС (ИЭР) m/z: 369 [M+H]+.
Стадия 3: Смесь этил 2-(3-(триметилстаннил)-1H-индазол-1-ил)ацетата (145 мг, 0,395 ммоль), 2,4,5-трихлорпиримидина (72,5 мг, 0,395 ммоль) и Pd(PPh3)4 (45,7 мг, 0,040 ммоль) в диоксане (5 мл) продувают N2, перемешивают при 100°C в течение 16 часов и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/4) с получением этил 2-(3-(2,5-дихлорпиримидин-4-ил)-1H-индазол-1-ил)ацетат в виде белого твердого вещества (68 мг, 49% выход). ЖХ-МС (ИЭР) m/z: 351 [M+H]+.
Стадии 4&5: Процедуры те же, что и для Стадий 3&4 Примера 161, с применением этил 2-(3-(2,5-дихлорпиримидин-4-ил)-1H-индазол-1-ил)ацетата вместо 2-(3-(2,5-дихлорпиримидин-4-ил)-1H-индол-1-ил)ацетата. ЖХ-МС (ИЭР) m/z: 479 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,57 (д, J=13,2 Гц, 2H), 8,18 (д, J=7,8 Гц, 1H), 7,67 (д, J=8,5 Гц, 1H), 7,58 (с, 1H), 7,51-7,35 (м, 1H), 7,27-7,13 (м, 1H), 6,88 (с, 1H), 5,23 (с, 2H), 3,79 (с, 3H), 3,62 (с, 2H), 2,91 (д, J=5,2 Гц, 2H), 2,85 (д, J=5,0 Гц, 2H), 2,47 (с, 3H).
Пример 167
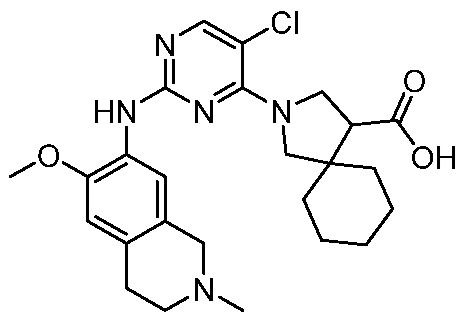
2-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-2-азапиро[4,5]декан-4-карбоновая кислота
Стадия 1: К раствору Пр. E38 (500 мг, 2,37 ммоль) и 2,4,5-трихлорпиримидина (434,0 мг, 2,37 ммоль) в ДХМ (5 мл) добавляют ДИПЭА (917,5 мг, 7,10 ммоль) по каплям при 0°С. Смесь перемешивают при 25°C в течение 2 часов, гасят льдом-холодным насыщенным водным раствором NH4Cl (30 мл) и экстрагируют ЭА (50 мл x 3). Органические фазы промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=5-15%) с получением этил 2-(2,5-дихлорпиримидин-4-ил)-2-азапиро[4,5]декан-4-карбоксилата (798 мг, 94,1% выход) в виде светло-желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 358 [M+H]+.
Стадия 2: Этил 2-(2,5-дихлорпиримидин-4-ил)-2-азапиро[4,5]декан-4-карбоксилат (100 мг, 0,279 ммоль) и Пр. W1 (63,8 мг, 0,279 ммоль) применяют в условиях B3 с получением этил 2-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-2-азапиро[4,5]декан-4-карбоксилата (116 мг, 70,3% выход, 87% чистота) в виде светло-желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ=5-10%). ЖХ-МС (ИЭР) m/z: 514 [M+H]+.
Стадия 3: Та же методика, как для Стадии 3 Примера 117, с применением этил 2-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-2-азапиро[4,5]декан-4-карбоксилата вместо метил 1-(5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-3-метилиндолин-3-карбоксилата. ЖХ-МС (ИЭР) m/z: 486 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,97 (д, J=14,2 Гц, 2H), 7,56 (с, 1H), 6,74 (с, 1H), 4,00 (с, 2H), 3,88 (д, J=11,4 Гц, 1H), 3,82 (с, 3H), 3,55 (с, 1H), 3,42 (с, 2H), 2,86 (т, J=7,5 Гц, 1H), 2,77 (с, 2H), 2,58 (с, 2H), 2,32 (с, 3H), 1,76-1,24 (м, 10H).
Пример 168
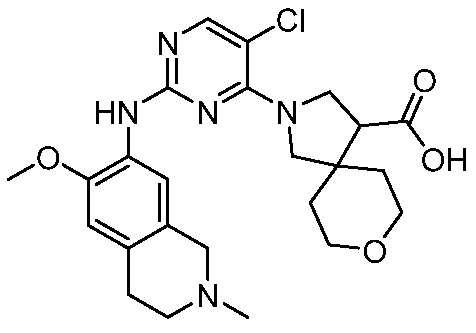
2-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-8-окса-2-азапиро[4,5]декан-4-карбоновая кислота
Процедуры те же, что и для Примера 167, с применением Пр. E39 вместо Пр. E38. ЖХ-МС (ИЭР) m/z: 488 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,97 (д, J=7,8 Гц, 2H), 7,57 (с, 1H), 6,75 (с, 1H), 3,99 (т, J=10,3 Гц, 3H), 3,82 (с, 3H), 3,77 (д, J=11,1 Гц, 2H), 3,66 (д, J=10,6 Гц, 1H), 3,50 (с, 2H), 3,46 (с, 2H), 2,95 (т, J=7,2 Гц, 1H), 2,78 (с, 2H), 2,62 (с, 2H), 2,34 (с, 3H), 1,98 (с, 1H), 1,56-1,44 (м, 3H).
Пример 169
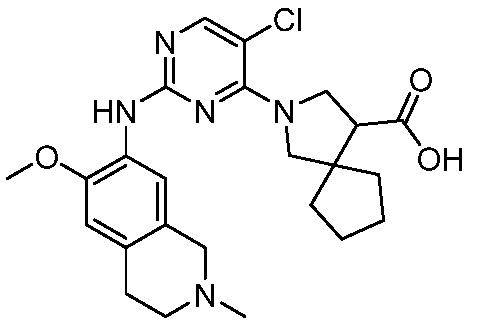
2-(5-Хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4-ил)-2-азапиро[4,4]нонан-4-карбоновая кислота
Процедуры те же, что и для Примера 167, с применением Пр. E40 вместо Пр. E38. ЖХ-МС (ИЭР) m/z: 472 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,96 (с, 1H), 7,94 (с, 1H), 7,54 (с, 1H), 6,73 (с, 1H), 3,95 (д, J=5,9 Гц, 2H), 3,81 (с, 3H), 3,72 (д, J=10,5 Гц, 1H), 3,57 (д, J=10,1 Гц, 1H), 3,41 (с, 2H), 2,98 (т, J=7,1 Гц, 1H), 2,75 (д, J=5,5 Гц, 2H), 2,58 (т, J=5,7 Гц, 2H), 2,32 (с, 3H), 1,89 (с, 1H), 1,69-1,54 (м, 6H), 1,49 (с, 1H).
Пример 170
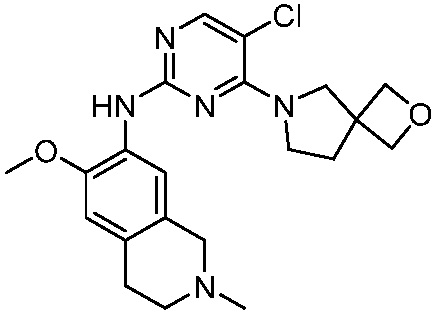
N-(5-хлор-4-(2-окса-6-азапиро[3,4]октан-6-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: Смесь 2,4,5-трихлорпиримидина (1 г, 5,45 ммоль) и водного раствора гидроксида натрия (1N, 1 мл) в ТГФ (3 мл) перемешивают при комнатной температуре в течение 3 часов, подкисляют до pH ~6 1 N HCl и экстрагируют CH2Cl2 (50 мл x 3). Объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают перекристаллизацией в 20% CH2Cl2/петролейном эфире с получением 2,5-дихлорпиримидин-4(3H)-она (374 мг, 29% выход). ЖХ-МС (ИЭР) m/z: 165 [M+H]+.
Стадия 2: 2,5-дихлорпиримидин-4(3H)-он (351 мг, 1,38 ммоль) и Пр. W1 (347,9 мг, 1,52 ммоль) применяют в условиях B3 с получением 5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4(3H)-она (419 мг, 78,4% выход), после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=4/1). ЖХ-МС (ИЭР) m/z: 321 [M+H]+.
Стадия 3: Раствор 5-хлор-2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)пиримидин-4(3H)-она (150 мг, 0,388 ммоль) в трихлориде фосфорила (1 мл) перемешивают при 95°C в течение 18 часов, охлаждают до комнатной температуры, выливают в насыщенный водный раствор NaHCO3 (50 мл) и экстрагируют дихлорметаном (20 мл x 3). Объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением неочищенного N-(4,5-дихлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (160 мг, 36% выход, 30% чистота). ЖХ-МС (ИЭР) m/z: 339 [M+H]+.
Стадия 4: N-(4,5-дихлорпиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амин (140 мг, 0,124 ммоль, 30% чистота) и 2-окса-7-азапиро[3,4]октан (16,8 мг, 0,149 ммоль) применяют в условиях A3 с получением N-(5-хлор-4-(2-окса-6-азапиро[3,4]октан-6-ил)пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (11,8 мг, 22,9% выход), после очистки преп-ЖХВД (MeCN/10 мМ NH4HCO3, 0,025% NH3.H2O). ЖХ-МС (ИЭР) m/z: 415 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ 8,08 (с, 1H), 7,91 (с, 1H), 7,40 (с, 1H), 6,59 (с, 1H), 4,72 (д, J=6,2 Гц, 2H), 4,64 (д, J=6,2 Гц, 2H), 4,06 (с, 2H), 3,87-3,83 (м, 2H), 3,85 (с, 3H), 3,55 (с, 2H), 2,87 (т, J=5,8 Гц, 2H), 2,68 (т, J=5,9 Гц, 2H), 2,47 (с, 3H), 2,25 (т, J=7,0 Гц, 2H).
Пример 36
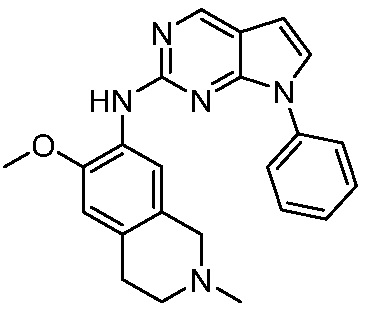
6-Метокси-2-метил-N-(7-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: Смесь 2-хлор-7H-пирроло[2,3-d]пиримидина (153 мг, 1,0 ммоль), 1-бром-2-йод-бензола (422,78 мг, 1,50 ммоль), карбоната калия (413,09 мг, 3,0 ммоль), CuI (379,49 мг, 2,0 ммоль) и (1S,2S)-N1,N2-диметилциклогексан-1,2-диамина (282,95 мг, 2,0 ммоль) в ДМФ (8 мл) перемешивают под N2 при 120°С в течение 18 часов. После охлаждения до комнатной температуры, смесь фильтруют и промывают этилацетатом (100 мл). Фильтрат разбавляют H2O (100 мл) и отделяют. Водную фазу экстрагируют этилацетатом (100 мл x 2). Объединенные органические слои промывают насыщенным раствором соли (100 мл x 4), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/15 об/об) с получением 2-хлор-7-фенил-пирроло[2,3-d]пиримидина в виде желтого твердого вещества (40 мг, 17% выход). ЖХ-МС (ИЭР) m/z: 230 [M+H]+.
Стадия 2: 2-Хлор-7-фенил-пирроло[2,3-d]пиримидин (60 мг, 0,26 ммоль) и Пр. W1 (60,3 мг, 0,31 ммоль) применяют в условиях B4 (как представлено на Стадии 2 Примера 28) с получением 6-метокси-2-метил-N-(7-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амина (21,4 мг, 21% выход) в виде желтого твердого вещества, после очистки преп-ЖХВД (MeCN/0,1% FA). ЖХ-МС (ИЭР) m/z: 386 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ (ч./млн.) 2,73 (с, 3H), 2,98-3,01 (м, 2H), 3,08-3,12 (м, 2H), 3,83 (с, 2H), 3,91 (с, 3H), 6,65 (д, J=4,0 Гц, 1H), 6,77 (с, 1H), 7,41-7,45 (м, 1H), 7,47 (д, J=4,0 Гц, 1H), 7,57-7,62 (м, 2H), 7,82 (д, J=8,0 Гц, 2H), 8,38 (с, 1H), 8,68 (с, 1H).
Пример 37
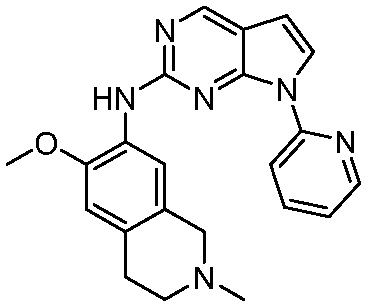
6-Метокси-2-метил-N-(7-(пиридин-2-ил)-7H-пирроло[2,3-d]пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 36, с применением 2-бромпиридина вместо 1-бром-2-йод-бензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 387 [M+H]. 1H ЯМР (400 MГц, CD3OD): δ (ч./млн.) 2,99 (с, 3H), 3,11-3,14 (м, 2H), 3,44-3,47 (м, 2H), 3,23 (с, 3H), 4,23 (с, 2H), 6,63 (д, J=4,0 Гц, 1H), 6,83 (с, 1H), 7,31-7,35 (м, 1H), 7,99-8,04 (м, 2H), 8,28 (с, 1H), 8,50-8,51 (м, 1H), 8,58 (д, J=8,0 Гц, 1H), 8,65 (с, 1H).
Пример 38
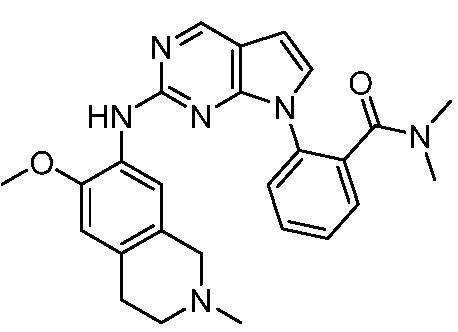
2-(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-N, N-диметилбензамид
Процедуры те же, что и для Примера 36, с применением бром-N, N-диметилбензамида вместо 1-бром-2-йод-бензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 457 [M+H]. 1H ЯМР (400 MГц, CD3OD): δ (ч./млн.) 2,56 (с, 3H), 2,77 (с, 3H), 2,83 (с, 3H), 3,01 (т, J=7,6 Гц, 2H), 3,16 (т, J=7,6 Гц, 2H), 3,82 (с, 2H), 3,91 (с, 3H), 6,65 (д, J=3,6 Гц, 1H), 6,78 (с, 1H), 7,20 (д, J=3,6 Гц, 1H), 7,57-7,59 (м, 1H), 7,61-7,66 (м, 1H), 7,72-7,79 (м, 2H), 8,72 (с, 1H), 8,50 (шс, 1H), 8,72 (с, 1H).
Пример 39
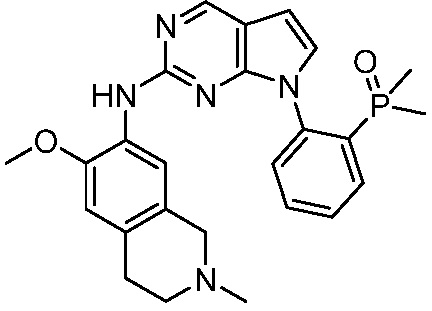
(2-(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)фенил)диметилфосфин оксид
Стадия 1: К смеси 2-хлор-7H-пирроло[2,3-d]пиримидина (153 мг, 996,29 мкмоль), (2-бромфенил)бороновой кислоты (600,25 мг, 2,99 ммоль) и Cu(OAc)2 (361,9 мг, 1,99 ммоль) в сухом ДХМ (12 мл) добавляют NEt3 (403,3 мг, 3,99 ммоль) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 4 дней и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/5 об/об) с получением 7-(2-бромфенил)-2-хлор-пирроло[2,3-d]пиримидина в виде белого твердого вещества (36 мг, 12% выход). ЖХ-МС (ИЭР) m/z: 308 [M+H]+.
Стадия 2: 7-(2-Бромфенил)-2-хлор-пирроло[2,3-d]пиримидин (100 мг, 0,324 ммоль) и Пр. W1 (62,3 мг, 0,324 ммоль) применяют в условиях B3 с получением N-(7-(2-бромфенил)-7H-пирроло[2,3-d]пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина в виде желтого твердого вещества (94 мг, 62% выход), после очистки преп-ТСХ (ДХМ/MeOH=15/1). ЖХ-МС (ИЭР) m/z: 464 [M+H]+.
Стадия 3: Та же методика, как для Стадии 3 Пр. E1, с применением N-(7-(2-бромфенил)-7H-пирроло[2,3-d]пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина вместо трет-бутил 3-йодиндол-1-карбоксилата. ЖХ-МС (ИЭР) m/z: 462 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ (ч./млн.) 1,43 (с, 3H), 1,46 (с, 3H), 2,78 (с, 3H), 3,00 (т, J=2,0 Гц, 2H), 3,18 (т, J=2,0 Гц, 2H), 3,67 (с, 2H), 3,89 (с, 3H), 6,73 (д, J=3,6 Гц, 1H), 6,77 (с, 1H), 7,39 (д, J=3,6 Гц, 1H), 7,48-7,51 (м, 1H), 7,64-7,70 (м, 1H), 7,80-7,89 (м, 1H), 8,04 (с, 1H), 8,13-8,20 (м, 1H), 8,76 (с, 1H).
Пример 40
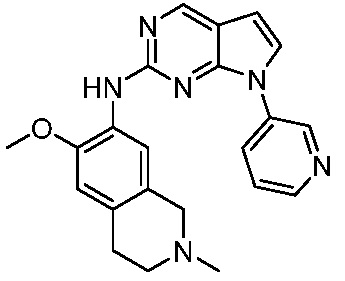
6-Метокси-2-метил-N-(7-(пиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: смесь 2-хлор-7H-пирроло[2,3-d]пиримидин (0,500 г, 3,26 ммоль), 3-пиридилбороновую кислоту (1,000 г, 8,14 ммоль) и диацетат меди (0,887 г, 4,88 ммоль) перемешивают в пиридине (12,5 мл) под O2 при 60°C в течение 16 часов, концентрируют в вакууме, разбавляют водой (100 мл) и экстрагируют ЭА (50 мл x 2). Объединенную органическую фазу сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/5) с получением 2-хлор-7-(пиридин-3-ил)-7H-пирроло[2,3-d]пиримидина (520 мг, 62,3% выход, 90% чистота). ЖХ-МС (ИЭР) m/z: 231,1 [M+H]+.
Стадия 2: 2-Хлор-7-(3-pyridyl)пирроло[2,3-d]пиримидин (100 мг, 0,434 ммоль) и Пр. W1 (100 мг, 0,520 ммоль) применяют в условиях B4 с получением метокси-2-метил-N-(7-(пиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амина (40,9 мг, 24,4% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (10мМ NH4HCO3, 0,025% NH3.H2O/MeCN). ЖХ-МС (ИЭР) m/z: 387 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 9,17 (д, J=1,9 Гц, 1H), 8,81 (с, 1H), 8,61 (д, J=4,2 Гц, 1H), 8,36 (д, J=8,0 Гц, 1H), 8,03 (с, 1H), 7,90 (с, 1H), 7,77 (д, J=3,6 Гц, 1H), 7,61 (дд, J=8,1, 4,7 Гц, 1H), 6,75 (д, J=3,7 Гц, 2H), 3,83 (с, 3H), 3,43 (с, 2H), 2,77 (с, 2H), 2,57 (т, J=5,5 Гц, 2H), 2,28 (д, J=66,6 Гц, 3H).
Пример 41
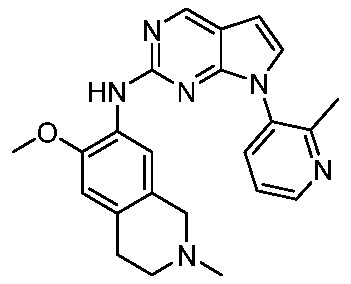
6-Метокси-2-метил-N-(7-(2-метилпиридин-3-ил)-7H-пирроло[2,3-d]пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амин
Процедуры те же, что и для Примера 40, с применением (2-метилпиридин-3-ил)бороновой кислоты вместо 3-пиридилбороновой кислоты на Стадии 1. ЖХ-МС (ИЭР) m/z: 401 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,80 (с, 1H), 8,62 (д, J=4,7 Гц, 1H), 7,89 (д, J=7,9 Гц, 2H), 7,75 (с, 1H), 7,59-7,35 (м, 2H), 6,71 (д, J=4,7 Гц, 2H), 3,80 (с, 3H), 3,20 (с, 2H), 2,72 (т, J=5,6 Гц, 2H), 2,54 (д, J=5,8 Гц, 2H), 2,32 (с, 6H).
Пример 164
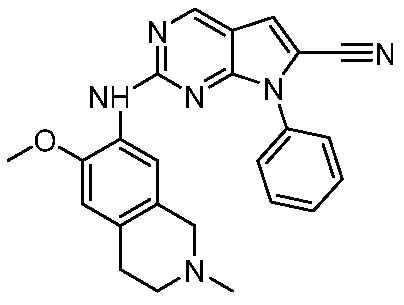
2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбонитрил
Стадия 1: К перемешиваемой смеси 5-бром-2-хлор-N-фенил-пиримидин-4-амина (1,5 г, 5,27 ммоль) и 1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (192,87 мг, 263,58 мкмоль) в ТГФ (30 мл) добавляют триэтиламин (800,16 мг, 7,91 ммоль, 1,10 мл) и 3,3-диэтоксипроп-1ин (1,01 г, 7,91 ммоль, 1,13 мл) последовательно при комнатной температуре. Смесь продувают N2 и перемешивают при комнатной температуре в течение 10 минут затем добавляют CuI (50,20 мг, 263,58 мкмоль). Реакционную смесь снова продувают N2, перемешивают при 60°C в течение 48 часов, охлаждают до комнатной температуры, разбавляют EtOAc (100 мл) и фильтруют. Фильтрат разбавляют водой (50 мл) и отделяют. Водную фазу экстрагируют EtOAc (30 мл×3) и объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (5% EtOAc/петролейный эфир до 20% EtOAc/петролейный эфир) с получением 2-хлор-5-(3,3-диэтоксипроп-1-инил)-N-фенил-пиримидин-4-амина в виде желтого твердого вещества (1,18 г, 67% выход). ЖХ-МС (ИЭР) m/z: 332 [M+H]+.
Стадия 2: К перемешиваемому раствору 2-хлор-5-(3,3-диэтоксипроп-1-инил)-N-фенил-пиримидин-4-амина (400 мг, 1,21 ммоль) в ТГФ (2 мл) добавляют 1N TBAF в ТГФ (6,03 ммоль, 6 мл) при комнатной температуре. Реакционную смесь перемешивают при 60°C в течение 2 часов, охлаждают до комнатной температуры и разбавляют H2O (50 мл) и дихлорметаном (50 мл). Фазы отделяют, и водный слой экстрагируют дихлорметаном (20 мл×3). Объединенные органические слои сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (10% EtOAc/петролейный эфир до 30% EtOAc/петролейный эфир) с получением 2-хлор-6-(диэтоксиметил)-7-фенил-7H-пирроло[2,3-d]пиримидина в виде серого твердого вещества (280 мг, 70% выход). ЖХ-МС (ИЭР) m/z: 332 [M+H]+.
Стадия 3: 2-Хлор-6-(диэтоксиметил)-7-фенил-7H-пирроло[2,3-d]пиримидин (100 мг, 0,3 ммоль) и Пр. W1 (57,8 мг, 0,3 ммоль) применяют в условиях B4 с получением N-(6-(диэтоксиметил)-7-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (60 мг, 40,9% выход) в виде беловатого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (MeOH/ДХМ: 5-10%). ЖХ-МС (ИЭР) m/z: 488 [M+H]+.
Стадия 4: К перемешиваемому раствору N-(6-(диэтоксиметил)-7-фенил-7H-пирроло[2,3-d]пиримидин-2-ил)-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-амина (60 мг, 0,123 ммоль) в диоксане (1 мл) добавляют 12 N HCl (0,2 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 30 минут, подщелачивают до pH 8 2 N NaOH и экстрагируют EtOAc (5 мл x 3). Объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбальдегида в виде серого твердого вещества (50 мг, 98% выход). ЖХ-МС (ИЭР) m/z: 414 [M+H]+.
Стадия 5: К перемешиваемому раствору 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбальдегида (50 мг, 0,121 ммоль) и пиразина (19,7 мг, 0,246 ммоль) в этаноле (1 мл) добавляют гидроксиламина гидрохлорид (10,3 мг, 0,148 ммоль) при комнатной температуре. Реакционную смесь перемешивают при 90°C в течение 2 часов и концентрируют в вакууме. Остаток разбавляют водой (10 мл) и экстрагируют EtOAc (10 мл x 3). Объединенную органическую фазу промывают насыщенным раствором соли (10 мл) сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме с получением 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбальдегида оксима в виде желтого твердого вещества (52 мг, 99% выход). ЖХ-МС (ИЭР) m/z: 429 [M+H]+.
Стадия 6: К перемешиваемому раствору 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбальдегида оксима (52 мг, 0,121 ммоль) в ТЭА (2 мл) добавляют уксусный ангидрид (24,78 мг, 0,243 ммоль). Реакционную смесь перемешивают при 90°C в течение 2 часов, охлаждают до комнатной температуры и концентрируют в вакууме. Остаток очищают преп-ЖХВД (0,1% муравьиная кислота/MeCN) с получением 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбонитрила в виде желтого твердого вещества (3,1 мг, 6% выход). ЖХ-МС (ИЭР) m/z: 411 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,98 (с, 1H), 8,37 (с, 1H), 8,24 (ш, 1H), 7,87 (с, 1H), 7,78-7,63 (м, 5H), 7,61-7,59 (м, 1H)), 6,74 (с, 1H), 3,80 (с, 3H), 3,28 (с, 2H), 2,75 (с, 2H), 2,55 (с, 2H), 2,35 (с, 3H).
Пример 165
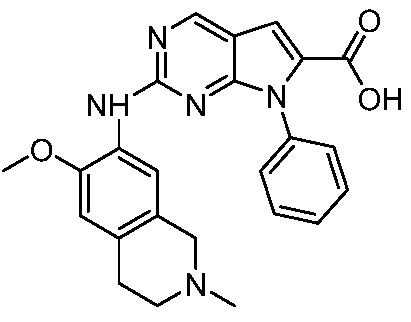
2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбоновая кислота
К раствору 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбальдегида (30 мг, 0,073 ммоль) в смешанном растворителе вода (3 мл) и этанол (3 мл) добавляют гидроксид натрия (14,5 мг, 0,363 ммоль) и оксид серебра (33,6 мг, 0,145 ммоль). Реакционную смесь перемешивают при 80°C в течение 16 часов, охлаждают до комнатной температуры, разбавляют водой (20 мл) и подкисляют до pH 3-4. Смесь экстрагируют ДХМ (20 мл x 3), и органические фазы сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают преп-ЖХВД с получением 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-фенил-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (4,4 мг, 14,1% выход) в виде желтого твердого вещества. ЖХ-МС (ИЭР) m/z: 430 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,88 (с, 1H), 7,92 (с, 1H), 7,89 (с, 1H), 7,53-7,51 (м, 2H), 7,47-7,42 (м, 3H), 7,25 (с, 1H), 6,71 (с, 1H), 3,81 (с, 3H), 3,26 (с, 2H), 2,74-2,73 (м, 2H), 2,61-2,59 (м, 2H), 2,39 (с, 3H).
Пример 43
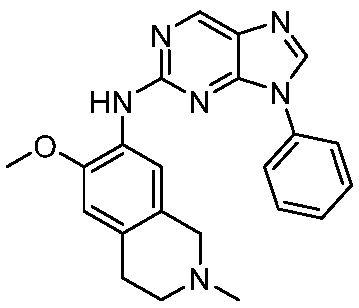
6-Метокси-2-метил-N-(9-фенил-9H-пурин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амин
Стадия 1: К раствору 2,4-дихлор-5-нитро-пиримидина (1 г, 5,16 ммоль) в диоксане (20 мл) медленно добавляют анилин (0,96 мг, 10,31 ммоль) при комнатной температуре. Смесь затем перемешивают при комнатной температуре в течение 1 часа и фильтруют. Фильтрат концентрируют с получением 2-хлор-5-нитро-N-фенил-пиримидин-4-амина в виде желтого твердого вещества (800 мг, 62% выход). ЖХ-МС (ИЭР) m/z: 251 [M+H]+.
Стадия 2: К смеси 2-хлор-5-нитро-N-фенил-пиримидин-4-амина (500 мг, 1,99 ммоль) в этилацетате/EtOH/H2O (6 мл/27 мл/3 мл) добавляют Fe (557,1 мг, 9,97 ммоль) и NH4Cl (74,7 мг, 1,40 ммоль) при комнатной температуре. Смесь перемешивают при 60°С в течение 2 часов. После охлаждения до комнатной температуры, смесь фильтруют и промывают метанолом (30 мл). Фильтрат концентрируют, и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1 об/об) с получением 2-хлор-N4-фенил-пиримидин-4,5-диамина в виде бледно-желтого твердого вещества (350 мг, 79% выход). ЖХ-МС (ИЭР) m/z: 221 [M+H]+.
Стадия 3: К раствору 2-хлор-N4-фенил-пиримидин-4,5-диамина (300 мг, 1,36 ммоль) в триэтил ортоформиате (10 мл) и ДМФ (5 мл) добавляют концентрированную HCl (0,6 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, разбавляют H2O (20 мл) и экстрагируют ЭА (20 мл x 2). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют досуха. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/2 об/об) с получением 2-хлор-9-фенил-пурина в виде желтого твердого вещества (220 мг, 70% выход). ЖХ-МС (ИЭР) m/z: 231 [M+H]+.
Стадия 4: 2-Хлор-9-фенил-пурина (30 мг, 0,13 ммоль) и Пр. W1 (25 мг, 0,13 ммоль) применяют в условиях B4 с получением 6-метокси-2-метил-N-(9-фенил-9H-пурин-2-ил)-1,2,3,4-тетрагидроизохинолин-7-амина (8 мг) в виде желтого твердого вещества, после очистки преп-ЖХВД (0,05% ТФК/MeCN). ЖХ-МС (ИЭР) m/z: 387 [M+H]. 1H ЯМР (400 MГц, CD3OD): δ 3,07 (с, 3H), 3,12-3,13 (м, 1H), 3,19-3,27 (м, 1H), 3,39-3,48 (м, 1H), 3,74-3,75 (м, 1H), 3,95 (с, 3H), 4,25-4,41 (м, 2H), 6,91 (с, 1H), 7,53-7,57 (м, 1H), 7,66-7,89 (м, 2H), 7,91 (д, J=1,2 Гц, 2H), 8,40 (с, 1H), 8,57 (с, 1H), 8,67 (с, 1H).
Пример 44
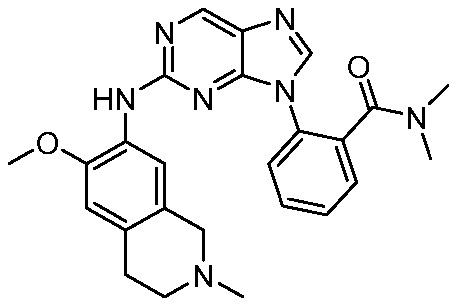
2-(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-9H-пурин-9-ил)-N, N-диметилбензамид
Процедуры те же, что и для Примера 43, с применением 2-амино-N, N-диметилбензамида вместо анилина на Стадии 1. ЖХ-МС (ИЭР) m/z: 458 [M+H]. 1H ЯМР (400 MГц, CD3OD): δ 2,73 (с, 3H), 2,77 (с, 3H), 2,84 (с, 3H), 3,03 (т, J=6,0 Гц, 2H), 3,18 (т, J=6,0 Гц, 2H), 3,88 (с, 2H), 3,90 (с, 3H), 6,81 (с, 1H), 7,61-7,64 (м, 1H), 7,68-7,76 (м, 1H), 7,76-7,78 (м, 2H), 8,18 (с, 1H), 8,19 (с, 1H), 8,45 (шс, 1H), 8,83 (с, 1H).
Пример 45
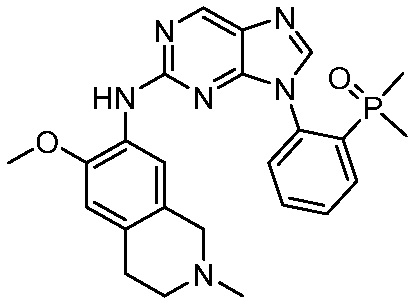
(2-(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-9H-пурин-9-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 43, с применением (2-аминофенил)диметилфосфина оксида вместо анилина на Стадии 1. ЖХ-МС (ИЭР) m/z: 463 [M+H]. 1H ЯМР (400 MГц, CD3OD): δ 1,62 (с, 3H), 1,65 (с, 3H), 2,87 (с, 3H), 3,07 (т, J=6,0 Гц, 2H), 3,33 (т, J=6,0 Гц, 2H), 3,89 (с, 3H), 3,94 (с, 2H), 6,82 (с, 1H), 7,60-7,63 (м, 1H), 7,81-7,91 (м, 2H), 8,01-8,07 (м, 1H), 8,12 (с, 1H), 8,35 (с, 1H), 8,45 (шс, 1H), 8,85 (с, 1H).
Пример 166
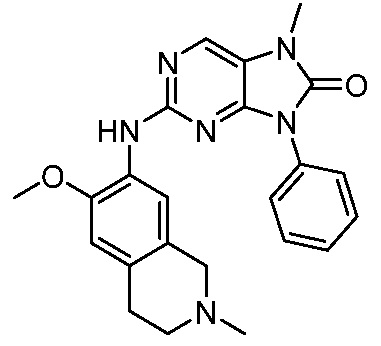
2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-метил-9-фенил-7,9-дигидро-8H-пурин-8-он
Стадия 1: К раствору 2-хлор-N4-фенил-пиримидин-4,5-диамина (410 мг, 1,86 ммоль) в дихлорметане (30 мл) добавляют КДИ (301,29 мг, 1,86 ммоль). Смесь перемешивают при 25°C в течение 16 часов, разбавляют насыщенным NaHCO3 водным раствором (20 мл) и отделяют. Водную фазу экстрагируют ДХМ/MeOH (об/об, 10/1, 20 мл x 3). Объединенную органическую фазу сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (80-100% EtOAc/hexanes) с получением 2-хлор-9-фенил-7,9-дигидро-8H-пурин-8-она в виде светло-желтого твердого вещества (264 мг, 58% выход). ЖХ-МС (ИЭР) m/z: 247 [M+H]+.
Стадия 2: К раствору 2-хлор-9-фенил-7,9-дигидро-8H-пурин-8-она (230 мг, 932,49 мкмоль) и йодметан (463,3 мг, 3,26 ммоль) в ТГФ (10 мл) добавляют гидрид натрия (111,9 мг, 2,80 ммоль, 60% дисперсию в минеральном масле) при 0°C. Смесь перемешивают при 0-25°C в течение 6 часов, гасят водой (10 мл) и отделяют. Водную фазу экстрагируют дихлорметаном/метанолом (об/об, 10/1, 20 мл x 3). Объединенный органический слой сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле (30-40% EtOAc в петролейном эфире) с получением 2-хлор-7-метил-9-фенил-7,9-дигидро-8H-пурин-8-она в виде светло-желтого твердого вещества (80 мг, 24% выход). ЖХ-МС (ИЭР) m/z: 261 [M+H]+.
Стадия 3: 2-Хлор-7-метил-9-фенил-7,9-дигидро-8H-пурин-8-он (70 мг, 0,268 ммоль) и Пр. W1 (21,4 мг, 0,268 ммоль) применяют в условиях B3 с получением 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-7-метил-9-фенил-7,9-дигидро-8H-пурин-8-она (44,2 мг, 39,5% выход) в виде желтого твердого вещества, после очистки преп-ЖХВД (0,1% FA/MeCN). ЖХ-МС (ИЭР) m/z: 417 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,29 (с, 1H), 8,25 (с, 1H), 7,88 (с, 1H), 7,67 (с, 2H), 7,58 (т, J=7,7 Гц, 2H), 7,47 (т, J=7,3 Гц, 1H), 6,71 (с, 1H), 3,79 (с, 3H), 3,40 (с, 3H), 3,28-3,20 (м, 2H), 2,74 (с, 2H), 2,55 (с, 2H), 2,34 (с, 3H).
Пример 46
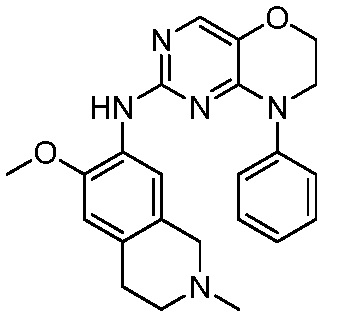
N-(6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-8-фенил-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин-2-амин
Стадия 1: К раствору 2,4-дихлор-5-метокси-пиримидина (1,00 г, 5,59 ммоль) в диоксане (15 мл) добавляют ДИПЭА (1,44 г, 11,17 ммоль, 1,95 мл) и анилин (0,520 мг, 5,59 ммоль). Смесь перемешивают при 120°C под N2 атмосферой в течение ночи и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=2/1 об/об) с получением 2-хлор-5-метокси-N-фенил-пиримидин-4-амина в виде желтого твердого вещества (950 мг, 72% выход). ЖХ-МС (ИЭР) m/z: 236 [M+H]+.
Стадия 2: К раствору 2-хлор-5-метокси-N-фенил-пиримидин-4-амина (100 мг, 0,424 ммоль) в ДХМ (5 мл) добавляют трибромид бора (1,06 г, 4,24 ммоль) при 0°C. Смесь перемешивают под азотом при комнатной температуре в течение 3 дней, выливают в MeOH (30 мл) при 0°C и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=1/1 об/об и затем ДХМ/MeOH=10/1 об/об) с получением 4-анилино-2-хлор-пиримидин-5-ола в виде белого твердого вещества (94 мг, 100% выход). ЖХ-МС (ИЭР) m/z: 222 [M+H]+.
Стадия 3: К раствору 4-анилино-2-хлор-пиримидин-5-ола (94 мг, 0,424 ммоль) в ДМФ (5 мл) добавляют карбонат калия (175,85 мг, 1,27 ммоль) и 1-бром-2-хлор-этан (91,2 мг, 0,636 ммоль). Смесь перемешивают при комнатной температуре в течение 16 часов, разбавляют H2O (20 мл) и экстрагируют ЭА (20 мл x 3). Органические слои сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=2/1) с получением 2-хлор-8-фенил-6,7-дигидропиримидо[5,4-b][1,4]оксазина в виде белого твердого вещества (60 мг, 57% выход). ЖХ-МС (ИЭР) m/z: 248 [M+H]+.
Стадия 4: 2-Хлор-8-фенил-6,7-дигидропиримидо[5,4-b][1,4]оксазин (50 мг, 0,202 ммоль) и Пр. W1 (46,2 мг, 0,202 ммоль) применяют в условиях B3 с получением N-(6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-8-фенил-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин-2-амина (48 мг, 59% выход) в виде белого твердого вещества, которое собирают фильтрацией. ЖХ-МС (ИЭР) m/z: 404 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 3,03 (с, 3H), 3,06-3,10 (м, 1H), 3,12-3,28 (м, 2H), 3,68-3,71 (м, 1H), 3,80-3,81 (м, 2H), 3,90 (с, 3H), 4,09 (т, J=4,0 Гц, 2H), 4,44 (т, J=4,0 Гц, 2H), 6,89 (с, 1H), 7,36 (с, 1H), 7,52-7,54 (м, 2H), 7,56-7,60 (м, 1H), 7,63-7,67 (м, 2H), 7,72 (с, 1H).
Пример 47
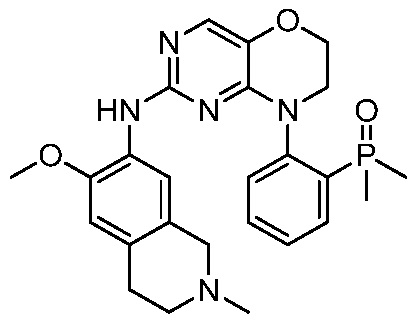
(2-(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-6H-пиримидо[5,4-b][1,4]оксазин-8(7H)-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 46, с применением 2-диметилфосфориланилина вместо анилина на Стадии 1. ЖХ-МС (ИЭР) m/z: 480 [M+H]+. 1H ЯМР (400 MГц, CD3OD): δ 1,63 (д, J=13,2 Гц, 3H), 1,80 (д, J=13,6 Гц, 3H), 2,88 (с, 3H), 2,98 (т, J=6,0 Гц, 2H), 3,26 (т, J=6,4 Гц, 2H), 3,46-3,55 (м, 2H), 3,84 (с, 3H), 3,87-3,94 (м, 2H), 4,38-4,45 (м, 2H), 6,70 (с, 1H), 7,31 (с, 1H), 7,54-7,58 (м, 1H), 7,71-7,76 (м, 2H), 7,85 (т, J=8,0 Гц, 1H), 7,98-8,04 (м, 1H), 8,46 (шс, 1H).
Пример 171
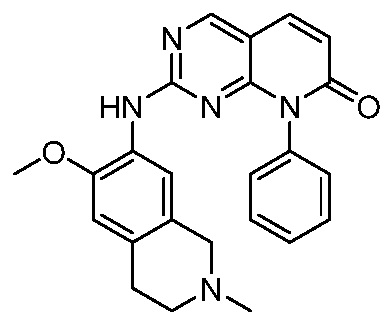
2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-8-фенилпиридо[2,3-d]пиримидин-7(8H)-он
Стадия 1: К раствору 5-бром-2,4-дихлор-пиримидина (1,2 г, 5,27 ммоль) в MeCN (20 мл) добавляют триэтиламин (639,45 мг, 6,32 ммоль, 880,78 мкл) и анилин (529,64 мг, 5,69 ммоль) при 0°C. Реакционную смесь перемешивают в течение 16 часов при 25°C и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/EtOAc=0-50%) с получением 5-бром-2-хлор-N-фенилпиримидин-4-амин в виде желтого твердого вещества (1,2 г, 76% выход). ЖХ-МС (ИЭР) m/z: 284 [M+H]+.
Стадия 2: К раствору 5-бром-2-хлор-N-фенилпиримидин-4-амина (283 мг, 994,59 мкмоль), акриловой кислоты (358,4 мг, 4,97 ммоль) и ДИПЭА (1,29 г, 9,95 ммоль) в ТГФ (4 мл) добавляют хлорид бис(бензонитрил)палладия(II) (19,07 мг, 49,73 мкмоль) и трис-o-толилфосфан (15,14 мг, 0,050 ммоль). Смесь перемешивают в течение 24 часов при 70°C под N2 атмосферой. Реакционную смесь применяют на следующей стадии без какой-либо очистки. ЖХ-МС (ИЭР) m/z: 276 [M+H]+.
Стадия 3: К полученной выше смеси добавляют уксусный ангидрид (540,00 мг, 5,29 ммоль). Смесь затем перемешивают в течение 24 часов при 80°C и концентрируют в вакууме. Остаток разбавляют ДХМ (50 мл) и водой (50 мл) и отделяют. Органическую фазу промывают 1N HCl (10 мл), сушат над безводным Na2SO4 и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=0-60%) с получением 2-хлор-8-фенилпиридо[2,3-d]пиримидин-7(8H)-она в виде желтого твердого вещества (88 мг, 34% выход). ЖХ-МС (ИЭР) m/z: 258 [M+H]+.
Стадия 4: 2-Хлор-8-фенилпиридо[2,3-d]пиримидин-7(8H)-он (40 мг, 0,155 ммоль) и Пр. W1 (35,5 мг, 0,155 ммоль) применяют в условиях B3 с получением 2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-8-фенилпиридо[2,3-d]пиримидин-7(8H)-она (17,1 мг, 26,6% выход) в виде белого твердого вещества, после очистки преп-ЖХВД (10 мМ NH4HCO3, 0,025% NH3.H2O/MeCN). ЖХ-МС (ИЭР) m/z: 414 [M+H]+. 1H ЯМР (500 MГц, ДМСО-d6) δ 8,81 (с, 1H), 8,11 (с, 1H), 7,94 (д, J=9,4 Гц, 1H), 7,61 (с, 2H), 7,53 (т, J=7,4 Гц, 1H), 7,34 (д, J=7,2 Гц, 2H), 7,14 (с, 1H), 6,68 (с, 1H), 6,49 (д, J=9,4 Гц, 1H), 3,79 (с, 3H), 2,91 (с, 2H), 2,68 (с, 2H), 2,47 (с, 2H), 2,35 (с, 3H).
Пример 172
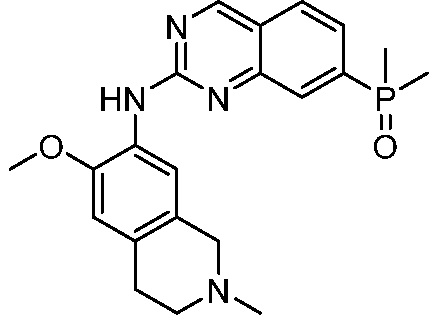
(2-((6-Метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)хиназолин-7-ил)диметилфосфин оксид
Стадия 1: 7-Бром-2-хлорхиназолин (100 мг, 0,411 ммоль) и Пр. W1 (94,8 мг, 0,493 ммоль) применяют в условиях B3 с получением 7-бром-N-(6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин-2-амина (90 мг, 52,1% выход) в виде желтого твердого вещества, после очистки колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА: 5/1). ЖХ-МС (ИЭР) m/z: 401 [M+H]+.
Стадия 2: Смесь 7-бром-N-(6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин-2-амина (80 мг, 200,36 мкмоль), диметилфосфина оксида (23,5 мг, 300,54 мкмоль), Pd2(dba)3 (9,2 мг, 10,02 мкмоль), Xantphos (5,8 мг, 10,02 мкмоль) и триэтиламина (60,8 мг, 601,08 мкмоль) в диоксане (1 мл) продувают азотом и перемешивают при 110°C в течение 16 часов. Реакционную смесь очищают преп-ЖХВД (MeCN/10мМ NH4HCO3, 0,025% NH3.H2O) с получением (2-((6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)хиназолин-7-ил)диметилфосфина оксида в виде желтого твердого вещества (2,9 мг, 3,6% выход). ЖХ-МС (ИЭР) m/z: 496 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 9,35 (с, 1H), 8,34 (с, 1H), 8,28 (с, 1H), 8,09 (с, 1H), 8,03 (дд, J=12,7, 7,7 Гц, 2H), 7,69 (т, J=9,0 Гц, 1H), 6,81 (с, 1H), 3,83 (с, 3H), 3,52 (с, 2H), 2,81 (д, J=5,5 Гц, 2H), 2,61 (т, J=5,7 Гц, 2H), 2,37 (с, 3H), 1,75 (д, J=13,4 Гц, 6H).
Пример 50
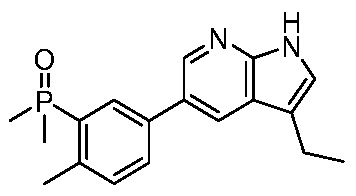
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-метилфенил)диметилфосфин оксид
Стадия 1: Смесь Промежуточного соединения 12 (200 мг, 734,89 мкмоль), 2-бром-4-йод-1-метилбензола (181,84 мг, 612,41 мкмоль), Pd(dppf)Cl2.CH2Cl2 (50,01 мг, 61,24 мкмоль) и K2CO3 (253,92 мг, 1,84 ммоль) в 1,4-диоксане (6 мл) и H2O (0,6 мл) was перемешивают при 80°C в атмосфере азота в течение 3 часов, охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют и остаток очищают колоночной флэш-хроматографией на силикагеле (ПЭ/ЭА=1/9 об/об) с получением 5-(3-бром-4-метилфенил)-3-этил-1H-пирроло[2,3-b]пиридина в виде светло-желтого порошка (100 мг, 49% выход). ЖХ-МС (ИЭР) m/z: 315 [M+H]+).
Стадия 2: Смесь 5-(3-бром-4-метилфенил)-3-этил-1H-пирроло[2,3-b]пиридина (49 мг, 154,48 мкмоль), диметилфосфина оксида (14,47 мг, 185,38 мкмоль), Pd(OAc)2 (1,73 мг, 7,72 мкмоль), Xantphos (4,47 мг, 7,72 мкмоль) и K3PO4 (36,07 мг, 169,93 мкмоль) в ДМФ (4 мл) перемешивают в условиях микроволн защищенных атмосферой азота при 140°C в течение 2 часов. После охлаждения до комнатной температуры, смесь разбавляют H2O (20 мл) и экстрагируют ЭА (20 мл x 3). Объединенные органические слои промывают насыщенным раствором соли (20 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением (5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-метилфенил)диметилфосфина оксида в виде белого твердого вещества (14,6 мг, 30% выход). ЖХ-МС (ИЭР) m/z: 313 [M+H]+.
Пример 51
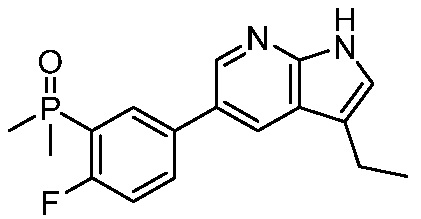
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-фторфенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением 2-бром-1-фтор-4-йодбензола вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 317 [M+H]+.
Пример 52
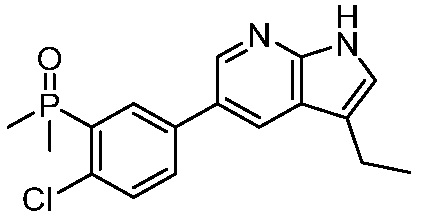
(2-Хлор-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Пример 53
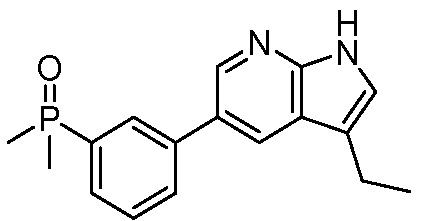
(3-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением 2-бром-1-хлор-4-йодбензола вместо 2-бром-4-йод-1-метилбензола на Стадии 1. Пример 53 получают как побочный продукт на Стадии 2.
(2-Хлор-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид: ЖХ-МС (ИЭР) m/z: 333 [M+H]+.
(3-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид: ЖХ-МС (ИЭР) m/z: 299 [M+H]+.
Пример 54
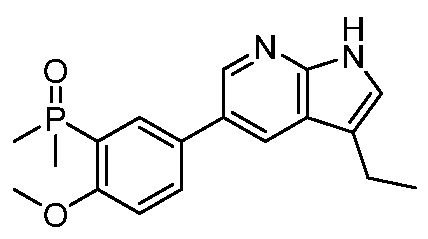
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-метоксифенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением 2-бром-4-йод-1-метоксибензола вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 329 [M+H]+.
Пример 55
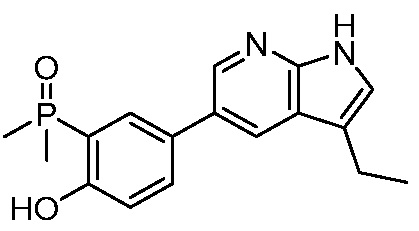
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-гидроксифенил)диметилфосфин оксид
К перемешиваемому раствору Примера 54 (28 мг, 85,28 мкмоль) в безводном ДХМ (5 мл) по каплям добавляют трибромид бора (68,67 мг, 274,10 мкмоль, 0,2 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 2 часов при комнатной температуре и гасят осторожно добавлением MeOH. Полученную смесь концентрируют, и остаток очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением (5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-гидроксифенил)диметилфосфина оксида в виде белого твердого вещества (21 мг, 78% выход). ЖХ-МС (ИЭР) m/z: 315 [M+H]+.
Пример 56
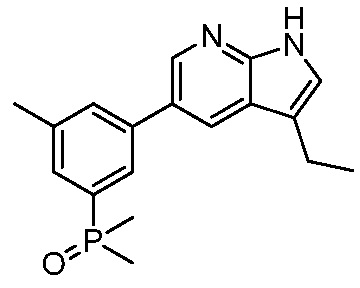
(3-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-5-метилфенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением 1,3-дибром-5-метил-бензола вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 313 [M+H]+.
Пример 57
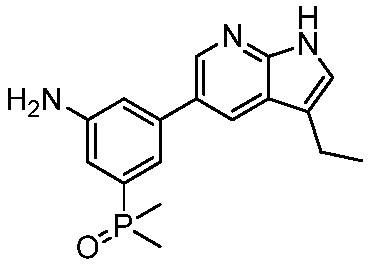
(3-Амино-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением 3,5-диброманилина вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 314 [M+H]+.
Пример 58
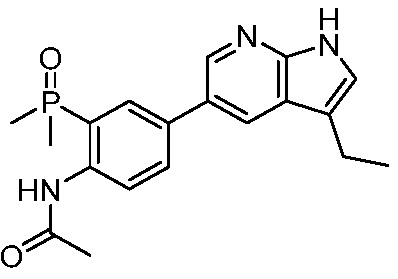
N-(2-(диметилфосфорил)-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)ацетамид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 17 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 356 [M+H]+.
Пример 59
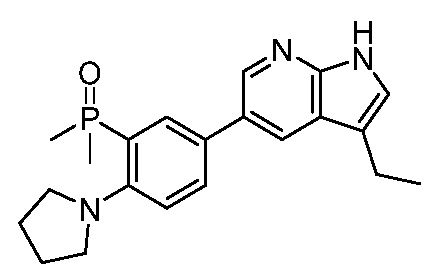
N-(2-бром-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)ацетамид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 18 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 368 [M+H]+.
Пример 60
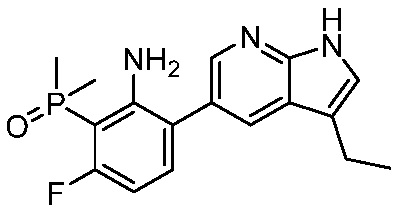
(2-Амино-3-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-6-фторфенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 19 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 332 [M+H]+.
Пример 61
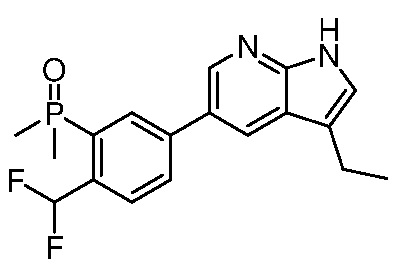
(2-(дифторметил)-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 21 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 349 [M+H]+.
Пример 62
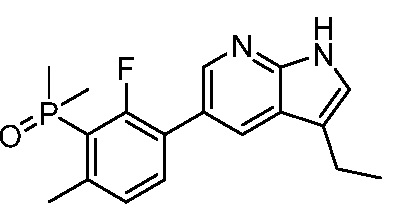
(3-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-фтор-6-метилфенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 22 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 331 [M+H]+.
Пример 63
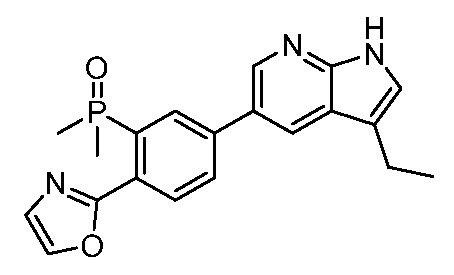
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(оксазол-2-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 25 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 366 [M+H]+.
Пример 64
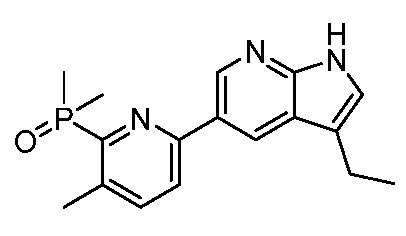
(6-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-3-метилпиридин-2-ил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 24 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 314 [M+H]+.
Пример 65
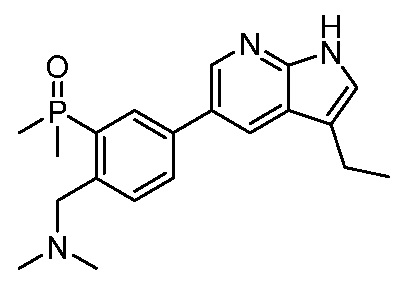
(2-((диметиламино)метил)-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 26 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 356 [M+H]+.
Пример 66
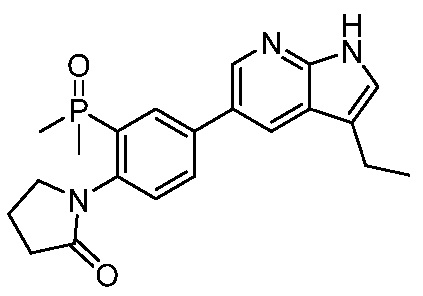
1-(2-(диметилфосфорил)-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)пирролидин-2-он
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 27 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 382 [M+H]+.
Пример 67
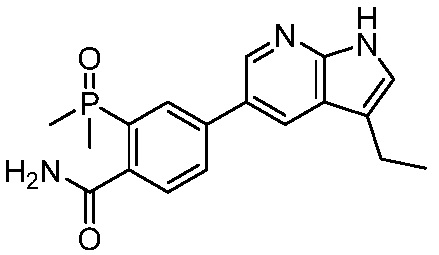
2-(диметилфосфорил)-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)бензамид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 28 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 342 [M+H]+.
Пример 68
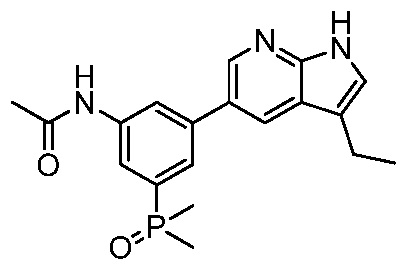
N-(3-(диметилфосфорил)-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)ацетамид
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 29 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 356 [M+H]+.
Пример 69
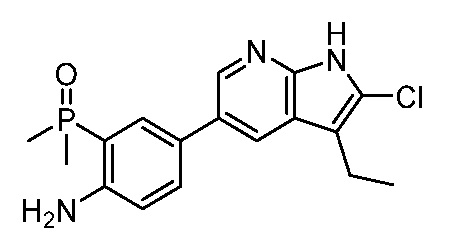
(2-Амино-5-(2-хлор-3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Пример 70
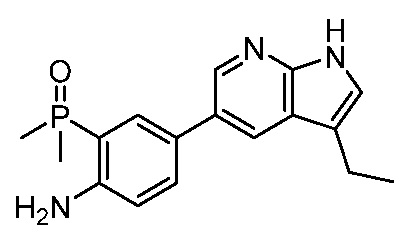
(2-Амино-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 50, с применением 2-бром-4-йоданилина и Промежуточного соединения 14 вместо 2-бром-4-йод-1-метилбензола и Промежуточного соединения 12 на Стадии 1.
(2-Амино-5-(2-хлор-3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид: [соль HCOOH] ЖХ-МС (ИЭР) m/z: 348 [M+H].
(2-Амино-5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид: [соль HCOOH] получают как побочный продукт на Стадии 2. ЖХ-МС (ИЭР) m/z: 314 [M+H]+.
Пример 71
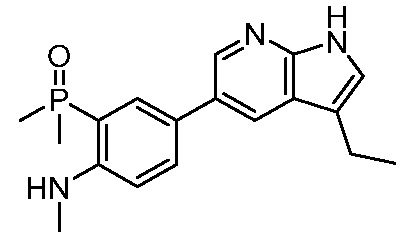
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(метиламино)фенил)диметилфосфин оксид
Стадии 1&2: Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 16 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z : 428 [M+H]+.
Стадия 3: К раствору трет-бутил (2-(диметилфосфорил)-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)(метил)карбамата (25 мг, 58,48 мкмоль) в ДХМ (5 мл) добавляют ТФК (1,48 г, 12,98 ммоль, 1 мл) при 0°C. Смесь перемешивают при комнатной температуре в течение 2 часов. После выпаривания, остаток очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением (5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(метиламино)фенил)диметилфосфина оксида в виде желтого твердого вещества (18 мг, 94% выход). ЖХ-МС (ИЭР) m/z : 328 [M+H]+.
Пример 72
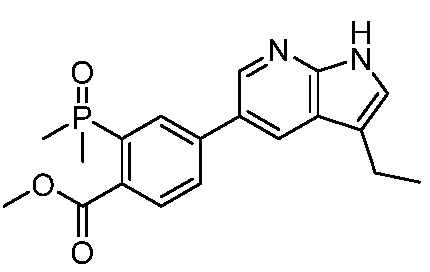
Метил 2-(диметилфосфорил)-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)бензоат
Процедуры те же, что и для Примера 50, с применением Промежуточного соединения 20 вместо 2-бром-4-йод-1-метилбензола на Стадии 1. ЖХ-МС (ИЭР) m/z: 357 [M+H]+.
Пример 73
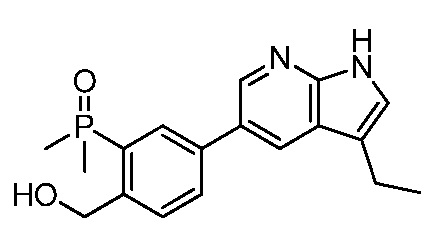
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(гидроксиметил)фенил)диметилфосфин оксид
К раствору метил 2-диметилфосфорил-4-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)бензоата (139,61 мг, 391,78 мкмоль) в сухом ТГФ (5 мл) добавляют алюмотетрагидрид лития (26,58 мг, 783,56 мкмоль) по каплям при 0°С. Смесь перемешивают при комнатной температуре в течение 1 часа, выливают в ледяную воду (10 мл) и экстрагируют ДХМ (10 мл x 3). Органический слой промывают насыщенным раствором соли (10 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (MeOH/0,1% трифторуксусная кислота) с получением (5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(гидроксиметил)фенил)диметилфосфина оксида в виде белого твердого вещества (100 мг, 78% выход). ЖХ-МС (ИЭР) m/z: 329 [M+H]+.
Пример 74
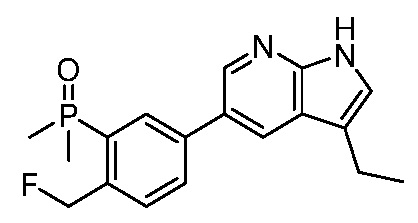
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(фторметил)фенил)диметилфосфин оксид
К раствору (5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(гидроксиметил)фенил)диметилфосфина оксида (200 мг, 609,12 мкмоль) в ДХМ (5 мл) добавляют по каплям ТДАС (117,82 мг, 730,94 мкмоль) при 0°С. Смесь перемешивают при комнатной температуре в течение 2 часов и концентрируют. Остаток очищают преп-ЖХВД (MeCN/0,05% трифторуксусная кислота) с получением (5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(фторметил)фенил)диметилфосфина оксида в виде белого твердого вещества (2,5 мг, 1,5% выход). ЖХ-МС (ИЭР) m/z: 331 [M+H]+.
Пример 75
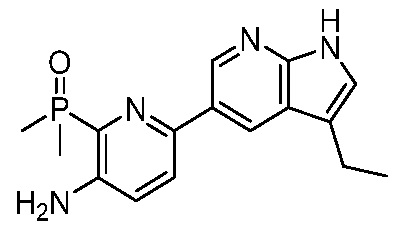
(3-Амино-6-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)пиридин-2-ил)диметилфосфин оксид
Стадия 1: Смесь Промежуточного соединения 15 (330 мг, 1,10 ммоль), диметилфосфина оксида (103,40 мг, 1,32 ммоль), ацетата палладия (II) (24,79 мг, 110,40 мкмоль), Xantphos (127,76 мг, 220,80 мкмоль) и фосфата калия (468,70 мг, 2,21 ммоль) в ДМФ (15 мл) перемешивают под азотом при 50°C в течение 4 часов. После охлаждения до комнатной температуры, смесь фильтруют и фильтрат разбавляют H2O (20 мл). Полученную смесь экстрагируют ЭА (30 мл x 3). Органический слой промывают насыщенным раствором соли (30 мл x 3), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной хроматографией с обращенной фазой (MeOH в H2O, ом 0% до 100% об/об) с получением 6-бром-2-диметилфосфорил-пиридин-3-амина в виде белого твердого вещества (180 мг, 60% выход). ЖХ-МС (ИЭР) m/z: 249 [M+H]+.
Стадия 2: Смесь 6-бром-2-диметилфосфорил-пиридин-3-амин (100 мг, 401,53 мкмоль), Промежуточное соединение 12 (131,13 мг, 481,84 мкмоль), Pd(dppf)Cl2.ДХМ (29,38 мг, 40,15 мкмоль) и карбонат калия (110,99 мг, 803,07 мкмоль) в диоксане (5 мл) и H2O (0,5 мл) перемешивают под азотом при 90°C в течение ночи. После охлаждения до комнатной температуры, смесь фильтруют и фильтрат концентрируют. Остаток разбавляют ЭА (50 мл), промывают H2O (20 мл) и насыщенным раствором соли (20 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают преп-ТСХ (ДХМ/MeOH=10/1 об/об) с получением (3-амино-6-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)пиридин-2-ил)диметилфосфина оксида в виде желтого твердого вещества (2 мг). ЖХ-МС (ИЭР) m/z: 315 [M+H]+.
Пример 76
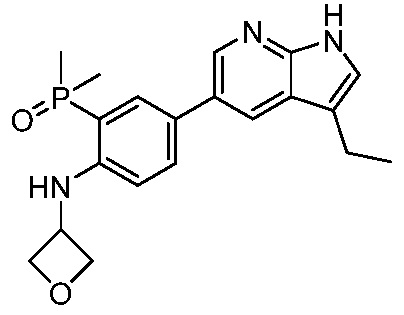
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(оксетан-3-иламино)фенил)диметилфосфин оксид
Процедуры те же, что и для Примера 75, с применением Промежуточного соединения 30 вместо Промежуточного соединения 15 на Стадии 1. ЖХ-МС (ИЭР) m/z: 370 [M+H]+.
Пример 77
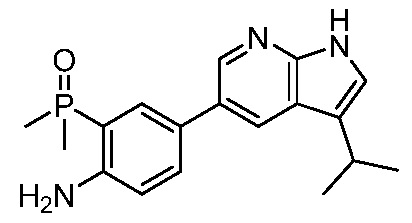
(2-Амино-5-(3-изопропил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфин оксид
Стадия 1: К раствору 1-(5-бром-1H-пирроло[2,3-b]пиридин-3-ил)этанона (590 мг, 2,47 ммоль) в сухом ТГФ (25 мл) добавляют гидрид натрия (60% суспензия в масле, 71,99 мг, 3 ммоль) порциями при 0°С и смесь перемешивают при 0°С в течение 1 часа. TsCl (714,90 мг, 3,75 ммоль) добавляют к вышеуказанной смеси порциями при 0°С. Смесь перемешивают при комнатной температуре в течение 2 часов, гасят H2O (20 мл) и экстрагируют ЭА (30 мл x 3). Органический слой промывают насыщенным раствором соли (30 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=2/3 об/об) с получением 1-(5-бром-1-тозил-1H-пирроло[2,3-b]пиридин-3-ил)этан-1-она в виде желтого твердого вещества (800 мг, 83% выход). ЖХ-МС (ИЭР) m/z: 393 [M+H]+.
Стадия 2: К раствору 1-(5-бром-1-тозил-1H-пирроло[2,3-b]пиридин-3-ил)этан-1-она (1,62 г, 4,12 ммоль) в сухом ТГФ (20 мл) добавляют по каплям бромид метилмагния (3 M в простом эфире, 6,87 мл, 20,6 ммоль) при 0°С. Смесь перемешивают при 0°С в течение 2 часов, выливают в лед-воду (50 мл) и экстрагируют этилацетатом (100 мл x 3). Органический слой промывают насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА/ПЭ=1/4 об/об) с получением 2-(5-бром-1-тозил-1H-пирроло[2,3-b]пиридин-3-ил)пропан-2-ола в виде желтого твердого вещества (900 мг, 76% выход). ЖХ-МС (ИЭР) m/z: 409 [M+H]+.
Стадия 3: К раствору 2-(5-бром-1-тозил-1H-пирроло[2,3-b]пиридин-3-ил)пропан-2-ола (900 мг, 2,20 ммоль) в сухом ДХМ (10 мл) добавляют Et3SiH (1,28 г, 10,99 ммоль, 1,76 мл) и трифторуксусную кислоту (1,25 г, 10,99 ммоль, 847,02 мкл) при 0°С. Смесь перемешивают при комнатной температуре в течение ночи, выливают в лед-воду (20 мл), подщелачивают насыщенным водным раствором NaHCO3 до pH 4~5 и экстрагируют ДХМ (30 мл x 3). Органический слой промывают насыщенным раствором соли (30 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле (ЭА в ПЭ, от 3% до 10% об/об) с получением 5-бром-3-изопропил-1-тозил-1H-пирроло[2,3-b]пиридина в виде белого твердого вещества (570 мг, 66% выход). ЖХ-МС (ИЭР) m/z: 393 [M+H]+.
Стадия 4: Смесь 5-бром-3-изопропил-1-тозил-1H-пирроло[2,3-b]пиридина (200 мг, 508,52 мкмоль), Промежуточного соединения 13 (225,11 мг, 762,78 мкмоль), Pd(dppf)Cl2-CH2Cl2 (41,53 мг, 50,85 мкмоль) и K2CO3 (210,85 мг, 1,53 ммоль) в диоксане (3 мл) и H2O (0,43 мл) перемешивают под N2 при 100°С в течение ночи. После охлаждения до комнатной температуры, смесь концентрируют и остаток очищают колоночной флэш-хроматографией на силикагеле (ДХМ/MeOH=20/1 об/об) с получением (2-амино-5-(3-изопропил-1-тозил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфина оксида в виде коричневого масла (290 мг, 100% выход). ЖХ-МС (ИЭР) m/z: 482 [M+H]+.
Стадия 5: К раствору (2-амино-5-(3-изопропил-1-тозил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфина оксида (100 мг, 207,66 мкмоль) в метаноле (5 мл) добавляют водный раствор NaOH (3 N, 3 мл, 9 ммоль) при комнатной температуре. Смесь кипятят с обратным холодильником в течение 2 часов и концентрируют. Остаток разбавляют H2O (10 мл), доводят до pH ~5 разбавленным раствором HCl (1 N) и экстрагируют ДХМ (20 мл x 3). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают преп-ЖХВД (MeCN/0,1% HCOOH) с получением (2-амино-5-(3-изопропил-1H-пирроло[2,3-b]пиридин-5-ил)фенил)диметилфосфина оксида в виде белого твердого вещества (10,8 мг, 16% выход). ЖХ-МС (ИЭР) m/z: 328 [M+H]+.
Пример 78
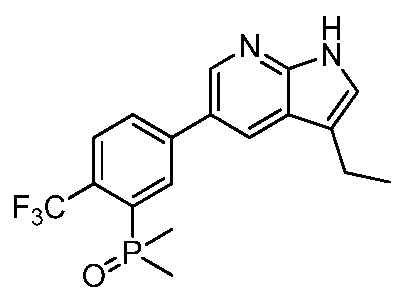
(5-(3-Этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(трифторметил)фенил)диметилфосфин оксид
Стадия 1: Та же методика, как для Стадии 1 Примера 50, с применением 5-бром-2-(трифторметил)анилина вместо 2-бром-4-йод-1-метилбензола. ЖХ-МС (ИЭР) m/z: 306 [M+H]+.
Стадия 2: К раствору 5-(3-этил-1H-пирроло[2,3-b]пиридин-5-ил)-2-(трифторметил)анилина (50 мг, 163,77 мкмоль) в ТГФ (5 мл) добавляют этиловый эфир трифторида бора (92,98 мг, 655,10 мкмоль, 82,28 мкл) и трет-бутилнитрит (59,11 мг, 573,21 мкмоль, 68,18 мкл) при -78°C. Смесь перемешивают в течение 10 минут при этой температуре, нагревают до комнатной температуры и перемешивают в течение 30 минут. Смесь разбавляют Et2O (10 мл) и фильтруют для удаления твердого вещества. Фильтрат концентрируют (ниже 20°С) и полученную соль диазония добавляют к смеси KI (40,78 мг, 245,66 мкмоль) и йода (14,66 мг, 114,64 мкмоль) в ацетоне (5 мл) при 0°C. Полученную смесь перемешивают при комнатной температуре в течение ночи и концентрируют. Остаток очищают колоночной флэш-хроматографией на силикагеле с получением 3-этил-5-(3-йод-4-(трифторметил)фенил)-1H-пирроло[2,3-b]пиридина в виде желтого твердого вещества (30 мг, 44% выход). ЖХ-МС (ИЭР) m/z: 417 [M+H]+.
Стадия 3: Та же методика, как для Стадии 2 Примера 50, с применением 3-этил-5-(3-йод-4-(трифторметил)фенил)-1H-пирроло[2,3-b]пиридина вместо 5-(3-бром-4-метилфенил)-3-этил-1H-пирроло[2,3-b]пиридина. ЖХ-МС (ИЭР) m/z: 367 [M+H]+.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ПРОИЗВОДНЫЕ N2-(2-ФЕНИЛ)-ПИРИДО[3,4-d]ПИРИМИДИН-2,8-ДИАМИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ MPS1 ИНГИБИТОРА | 2015 |
|
RU2693460C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| ИНГИБИТОРЫ БЕЛКОВЫХ ТИРОЗИНФОСФАТАЗ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2833220C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
| ТЕТРАГИДРОИЗОХИНОЛИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЕ СОЕДИНЕНИЕ, И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792034C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА КИНАЗ JAK3 и/или JAK1, И ПРИМЕНЕНИЕ АРОМАТИЧЕСКОГО ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ | 2015 |
|
RU2671195C2 |
Изобретение относится к соединению, представленному структурной формулой (I-2), или его фармацевтически приемлемой соли или его стереоизомеру. В формуле (I-2) A является СR2 или N; (i) R
является 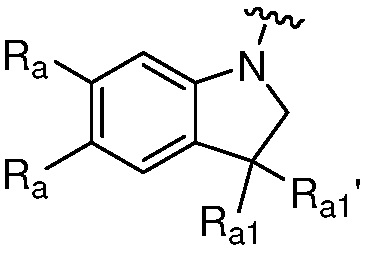 , где
, где  связь соединяет с пиримидиновым кольцом, каждый случай Ra независимо является H, F или Cl; Ra1 независимо является H, C1-4 алкилом или C1-4гидроксиалкилом; Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2R11, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-5-6-членным гетероарилом или -(CHRaa)kP(=O)R11R12; Raa независимо является H или C1-3 алкилом, необязательно замещенным галогеном; R1 представляет собой H, дейтерий, галоген, CN, NH2, NO2 или C1-6 алкил, необязательно замещенный одним-тремя галогенами; или (ii) R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
связь соединяет с пиримидиновым кольцом, каждый случай Ra независимо является H, F или Cl; Ra1 независимо является H, C1-4 алкилом или C1-4гидроксиалкилом; Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2R11, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-5-6-членным гетероарилом или -(CHRaa)kP(=O)R11R12; Raa независимо является H или C1-3 алкилом, необязательно замещенным галогеном; R1 представляет собой H, дейтерий, галоген, CN, NH2, NO2 или C1-6 алкил, необязательно замещенный одним-тремя галогенами; или (ii) R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже: 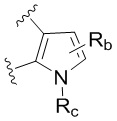 ,
, 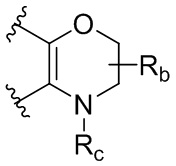 ,
, 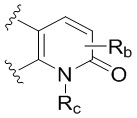 ,
, 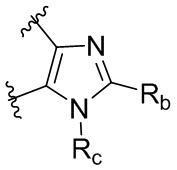 или
или 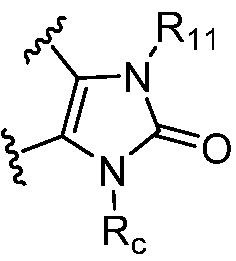 , где связи соединены с пиримидиновым кольцом; каждый случай Rb независимо является H, дейтерием, галогеном, CN, COOH или P(=O)R11R12; каждый случай Rc независимо является фенилом, 5-6-членным моноциклическим гетероарилом, имеющим 1-3 гетероатома, выбранных из N; где фенил или гетероарил, представленный Rc, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C1-4 галогеналкила, C(O)NR11R12 и P(O)ди-C1-6 алкила; каждый случай R2 независимо является H, дейтерием, галогеном или C1-6 алкокси; R3 является Н, C1-6 алкилом или C(O)C1-6 алкилом, где алкил, представленный R3 или в группе, представленной R3, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, C1-4 алкила и NR11R12; каждый случай R11 и R12 независимо является H или C1-6 алкилом, где алкил, представленный R11 или R12, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из CN и C1-4 алкила;m равно 0, 1, 2 или 3; и k равно 0 или 1. Также предложены: соединение, представленное формулой (II), или его фармацевтически приемлемая соль, где значения переменных в формуле (II) такие, как приведены в формуле изобретения; соединение, выбранной из группы индивидуальных соединений; фармацевтическая композиция на основе указанных соединений; способ лечения рака с использованием указанных соединений. Предложенные соединения являются ингибиторами активности HPK1 и могут применяться для лечения рака, опосредованного активностью HPK1. 6 н. и 28 з.п. ф-лы, 2 табл., 177 пр.
, где связи соединены с пиримидиновым кольцом; каждый случай Rb независимо является H, дейтерием, галогеном, CN, COOH или P(=O)R11R12; каждый случай Rc независимо является фенилом, 5-6-членным моноциклическим гетероарилом, имеющим 1-3 гетероатома, выбранных из N; где фенил или гетероарил, представленный Rc, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C1-4 галогеналкила, C(O)NR11R12 и P(O)ди-C1-6 алкила; каждый случай R2 независимо является H, дейтерием, галогеном или C1-6 алкокси; R3 является Н, C1-6 алкилом или C(O)C1-6 алкилом, где алкил, представленный R3 или в группе, представленной R3, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, C1-4 алкила и NR11R12; каждый случай R11 и R12 независимо является H или C1-6 алкилом, где алкил, представленный R11 или R12, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из CN и C1-4 алкила;m равно 0, 1, 2 или 3; и k равно 0 или 1. Также предложены: соединение, представленное формулой (II), или его фармацевтически приемлемая соль, где значения переменных в формуле (II) такие, как приведены в формуле изобретения; соединение, выбранной из группы индивидуальных соединений; фармацевтическая композиция на основе указанных соединений; способ лечения рака с использованием указанных соединений. Предложенные соединения являются ингибиторами активности HPK1 и могут применяться для лечения рака, опосредованного активностью HPK1. 6 н. и 28 з.п. ф-лы, 2 табл., 177 пр.
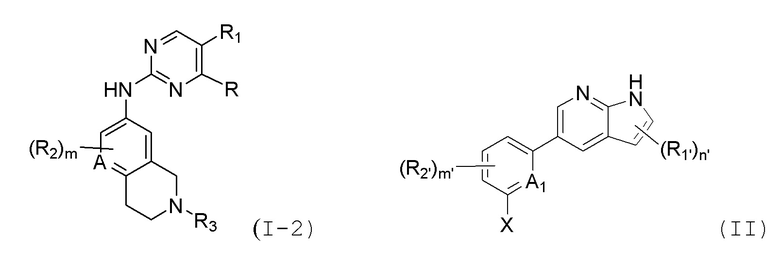
1. Соединение, представленное структурной формулой (I-2):
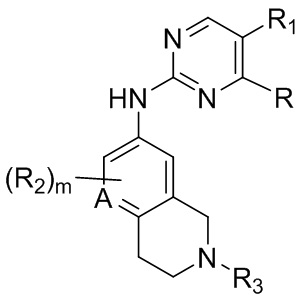 (I-2),
(I-2),
или его фармацевтически приемлемая соль или стереоизомер, где
A является СR2 или N;
(i) R является 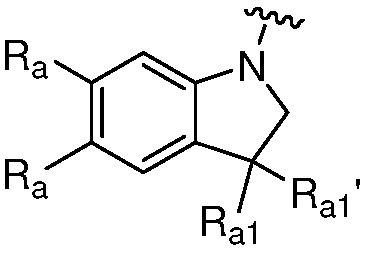 , где
, где  связь соединяет с пиримидиновым кольцом,
связь соединяет с пиримидиновым кольцом,
каждый случай Ra независимо является H, F или Cl;
Ra1 независимо является H, C1-4 алкилом или C1-4гидроксиалкилом;
Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN,
-(CHRaa)kC(O)OR11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12,
-(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2R11,
-(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-5-6-членным гетероарилом или -(CHRaa)kP(=O)R11R12;
Raa независимо является H или C1-3 алкилом, необязательно замещенным галогеном;
R1 представляет собой H, дейтерий, галоген, CN, NH2, NO2 или C1-6 алкил, необязательно замещенный одним-тремя галогенами; или
(ii) R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
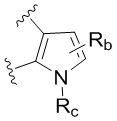 ,
, 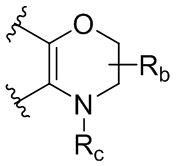 ,
, 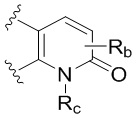 ,
, 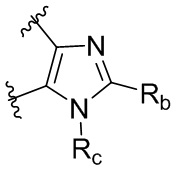 или
или 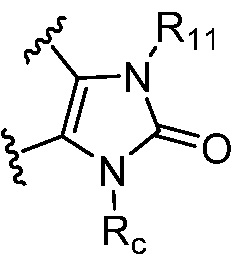 , где связи соединены с пиримидиновым кольцом;
, где связи соединены с пиримидиновым кольцом;
каждый случай Rb независимо является H, дейтерием, галогеном, CN, COOH или P(=O)R11R12;
каждый случай Rc независимо является фенилом, 5-6-членным моноциклическим гетероарилом, имеющим 1-3 гетероатома, выбранных из N; где фенил или гетероарил, представленный Rc, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C1-4 галогеналкила, C(O)NR11R12 и P(O)ди-C1-6 алкила;
каждый случай R2 независимо является H, дейтерием, галогеном или C1-6 алкокси;
R3 является Н, C1-6 алкилом или C(O)C1-6 алкилом, где алкил, представленный R3 или в группе, представленной R3, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, C1-4 алкила и NR11R12;
каждый случай R11 и R12 независимо является H или C1-6 алкилом, где алкил, представленный R11 или R12, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из CN и C1-4 алкила;
m равно 0, 1, 2 или 3; и
k равно 0 или 1.
2. Соединение по п. 1 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 является Н, галогеном, CN, C1-6 алкилом или NH2, где алкил, представленный R1 или в группе, представленной R1, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что каждый случай R2 независимо является H, галогеном или C1-4 алкокси.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 является Н, галогеном, CN, C1-4 алкилом, необязательно замещенным одним-тремя галогенами.
5. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 является Н, F, Cl, CN или CF3.
6. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R является , где связь соединяет с пиримидиновым кольцом,
Ra1 независимо является H, C1-4 алкилом или C1-4гидроксиалкилом;
Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2R11, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-5-6-членным гетероарилом или -(CHRaa)kP(=O)R11R12;
Raa независимо является H или C1-3 алкилом, необязательно замещенным галогеном;
Ra независимо является H, F или Cl;
R11 и R12 независимо являются H или C1-4 алкилом;
k равно 0 или 1.
7. Соединение по п. 6 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
Ra1 независимо является H, CH3 или CH2OH;
Ra1’ независимо является -(CHRaa)kOH, -(CHRaa)kCN, -(CHRaa)kC(O)OR11, -(CHRaa)kC(O)NR11R12, -(CHRaa)kC(O)NR11OR12, -(CHRaa)kC(O)NR11S(=O)2R12, -(CHRaa)kS(=O)2R11, -(CHRaa)kS(=O)2NR11R12, -(CHRaa)kNR11S(=O)2R12, -(CHRaa)k-тетразолом или -(CHRaa)kP(=O)R11R12;
Raa независимо является H, CH3 или CF3;
R11 и R12 независимо являются H или C1-2 алкилом; и
k равно 0 или 1.
8. Соединение по п. 7 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что каждый случай Ra независимо является H или F.
9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R1 является Сl.
10. Соединение по п. 1 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
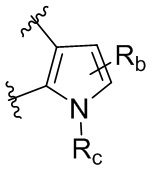 , , , или
, , , или 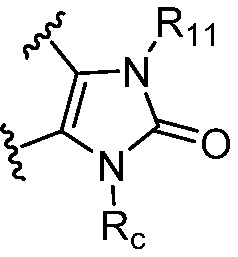 , где связи соединены с пиримидиновым кольцом.
, где связи соединены с пиримидиновым кольцом.
11. Соединение по п. 1 или 10 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
каждый случай Rb независимо является H, галогеном, CN, COOH или P(=O)R11R12; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C1-4 галогеналкила, C(O)NR11R12 и P(=O)ди-C1-6 алкила.
12. Соединение по любому из пп. 1-10 и 11 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что R и R1, вместе с атомами углерода, к которым они присоединены, образуют кольцо, представленное ниже:
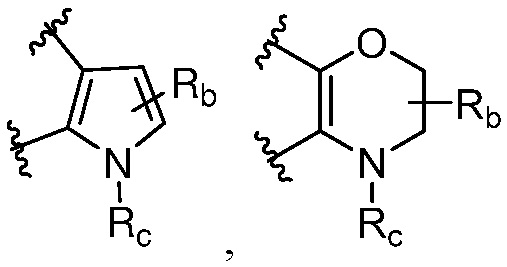 или
или 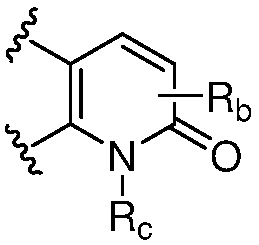 , где связи соединены с пиримидиновым кольцом.
, где связи соединены с пиримидиновым кольцом.
13. Соединение по любому из пп. 1 и 10-12 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
каждый случай Rb независимо является H, галогеном, CN или COOH;
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C1-4 галогеналкила, C(O)NR11R12 и P(=O)ди-C1-6 алкила, и
каждый случай R11 и R12 независимо является H или C1-6 алкилом.
14. Соединение по любому из пп. 1 и 10-13 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
каждый случай Rb независимо является H, CN или COOH; и
каждый случай Rc является фенилом или пиридинилом, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из C1-4 алкила, C(O)N(CH3)2 и P(=O)(CH3)2.
15. Соединение по любому из пп. 1-14 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что m равно 0 или 1.
16. Соединение по любому из пп. 1-15 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что 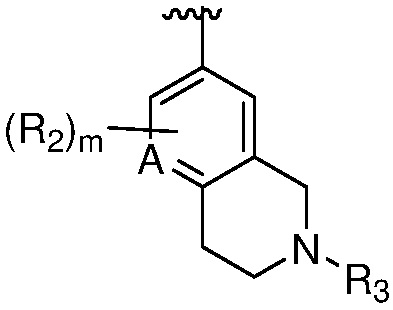 является
является 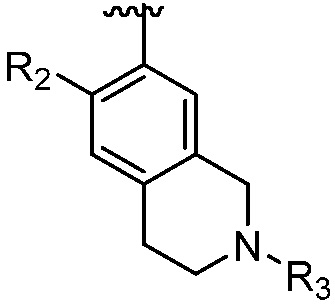 .
.
17. Соединение по любому из пп. 1-16 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что каждый случай R2 является Н, галогеном или C1-4 алкокси; и R3 является Н, C1-6 алкилом, C1-6 гидроксиалкилом или COCH2NR11R12.
18. Соединение по любому из пп. 1-17 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что каждый случай R2 является Н, F, Cl или OCH3; и R3 является Н или C1-4 алкилом.
19. Соединение по п. 1 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что соединение представляет собой
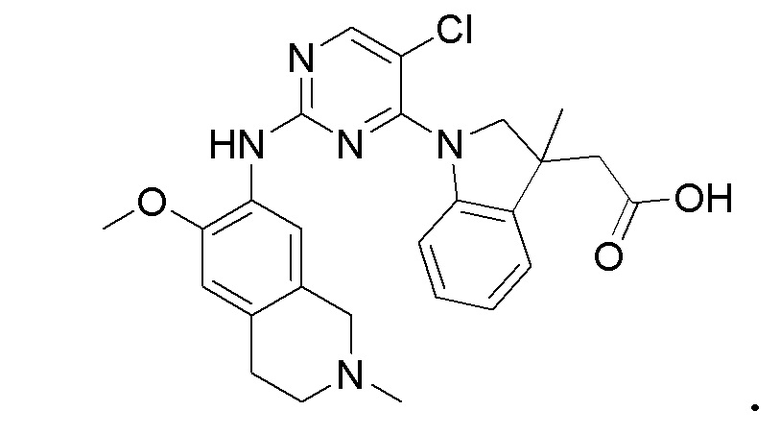
20. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что соединение представляет собой
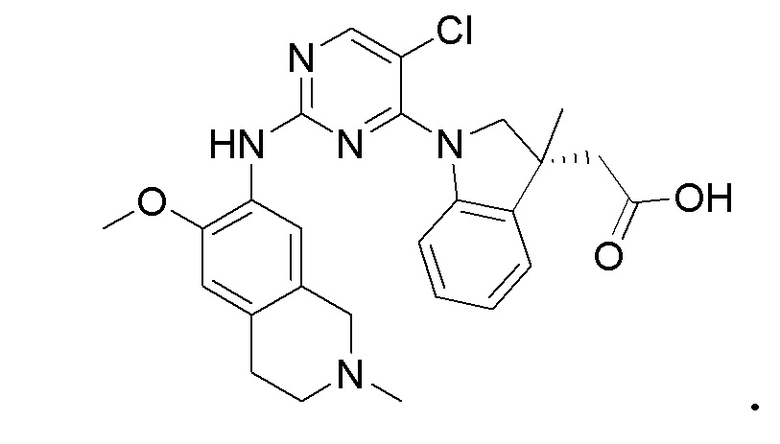
21. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что соединение представляет собой
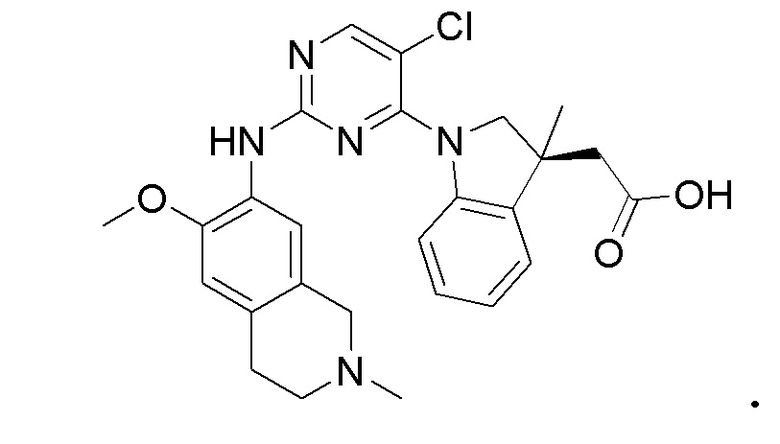
22. Соединение по п. 1 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что соединение представляет собой
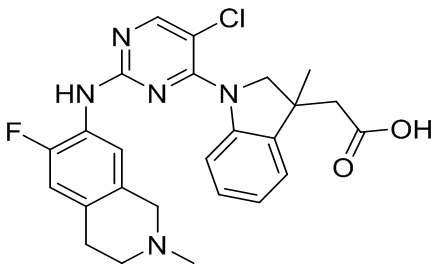 .
.
23. Соединение, представленное структурной формулой (II):
 (II),
(II),
или его фармацевтически приемлемая соль, где
A1 является СR’ или N;
Х является -P(=O)R3’R4’;
R’ является Н, дейтерием, галогеном или NH2;
каждый случай R1’ независимо является H, дейтерием, галогеном или C1-6 алкилом;
каждый случай R2’ независимо является H, дейтерием, галогеном, OH, C1-6 алкилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, 5-6-членным гетероарилом, содержащим 2 гетероатома, выбранных из N и O, или 5-7-членным гетероциклилом, содержащим гетероатомы, выбранные из N, где алкил, представленный R2’ или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, C1-4 алкила, C1-4 галогеналкила и NR11’R12’;
каждый R3’ и R4’ независимо является C1-6 алкилом;
каждый случай R11’ и R12’ независимо является H или C1-6 алкилом, или
R11’ и R12’, вместе с атомом азота, к которому они присоединены, образуют 3-7-членный гетероциклил;
m’ равно 0, 1 или 2; и
n’ равно 0, 1 или 2.
24. Соединение по п. 23, отличающееся тем, что соединение представлено структурной формулой (II-1):
 (II-1),
(II-1),
или его фармацевтически приемлемая соль или стереоизомер, где R’ является Н, галогеном или NH2.
25. Соединение по п. 23 или 24 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
каждый случай R1’ независимо является H, галогеном или C1-6 алкилом;
каждый случай R2’ независимо является H, галогеном, OH, C1-6 алкилом, C1-6 алкокси, NR11’R12’, C(O)NR11’R12’, C(O)C1-6 алкилом, C(O)OC1-6 алкилом, NR11’C(O)C1-6 алкилом, 5-6-членным гетероарилом, содержащим 2 гетероатома, выбранных из N и O, или 5-7-членным гетероциклилом, содержащим гетероатомы, выбранные из N, где алкил, представленный R2’ или в группе, представленной R2’, необязательно замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, OH, C1-4 алкила, C1-4 галогеналкила и NR11’R12’.
26. Соединение по любому из пп. 23-25, отличающееся тем, что соединение представлено структурной формулой (II-2) или (II-2’):
 (II-2) или
(II-2) или 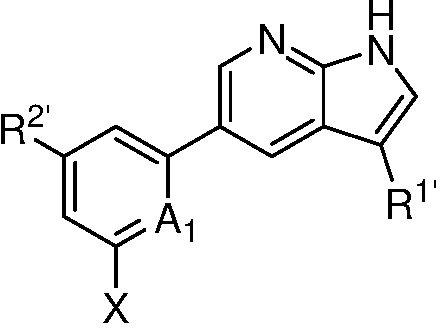 (II-2’)
(II-2’)
или его фармацевтически приемлемая соль или стереоизомер, где R’ является Н, галогеном или NH2.
27. Соединение по любому из пп. 23-26 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
каждый случай R2’ независимо является H, галогеном, OH, C1-4 алкилом, необязательно замещенным галогеном или OH, или NR11’R12’, C1-4 алкокси, C(O)OC1-6 алкилом, C(O)NR11’R12’, NR11’R12’, NR11’C(O)C1-6 алкилом, 5-6-членным гетероциклилом, содержащим 2 гетероатома, выбранных из N, или 5-6-членным гетероарилом, содержащим 2 гетероатома, выбранных из N и O; и
каждый случай R1’ независимо является H или C1-4 алкилом.
28. Соединение по любому из пп. 23-27 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что
каждый случай R2’ независимо является H, галогеном, OH, C1-4 алкилом, необязательно замещенным галогеном или OH, или NR11’R12’, C1-4 алкокси, C(O)OC1-4 алкилом, C(O)NR11’R12’, NR11’R12’, NHC(O)C1-6 алкилом, пирролидинилом, пирролидин-2-оном, оксазолом, где каждый случай R11’ и R12’ независимо является H или C1-4 алкилом.
29. Соединение по любому из пп. 23-28 или его фармацевтически приемлемая соль или стереоизомер, отличающееся тем, что Х является -P(O)(CH3)2 и каждый случай R2’ независимо является H, F, Cl, OH, CH3, NH2 или NHCOCH3.
30. Соединение или его фармацевтически приемлемая соль, отличающееся тем, что соединение выбрано из:
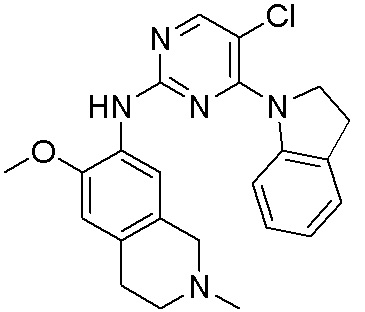
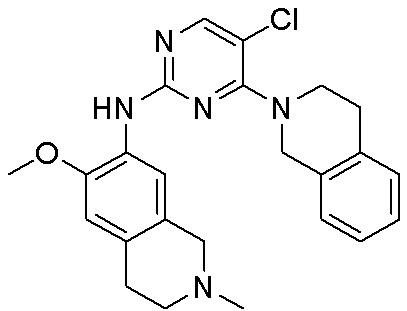
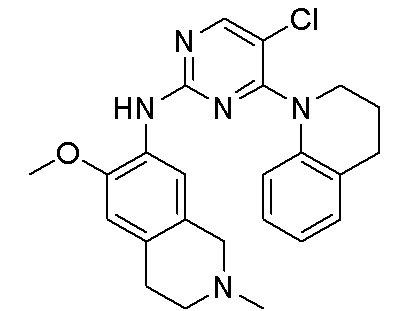
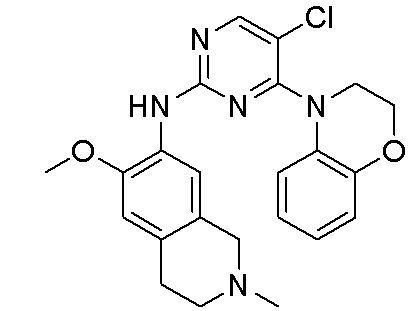
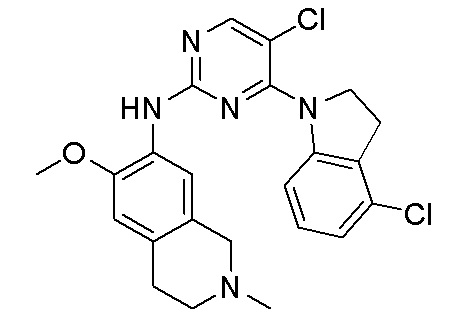
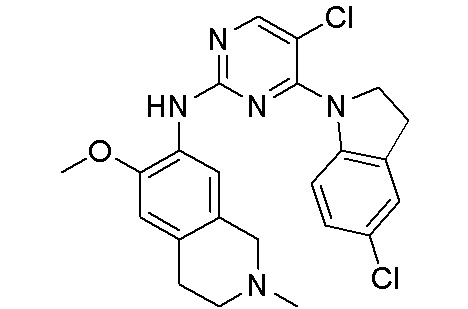
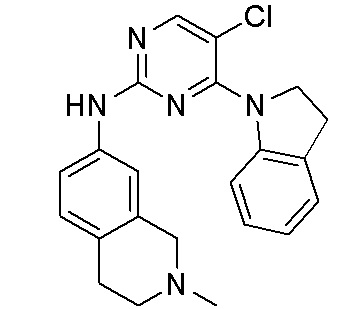
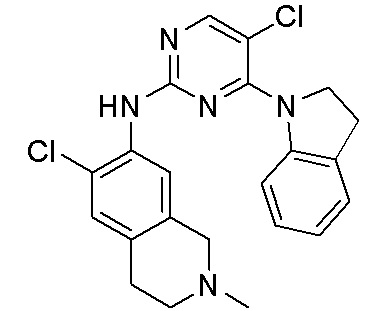
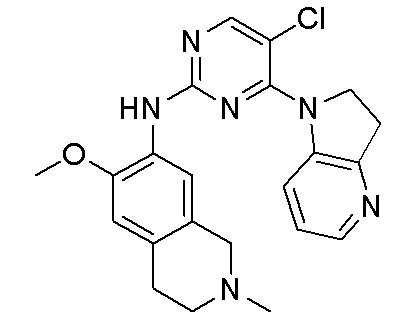
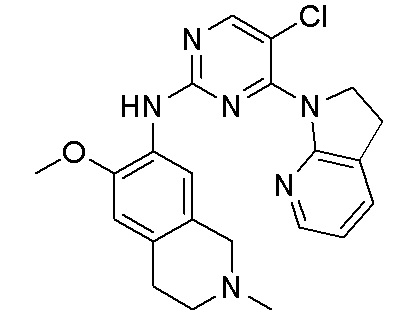
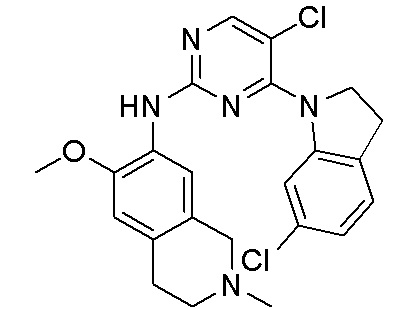
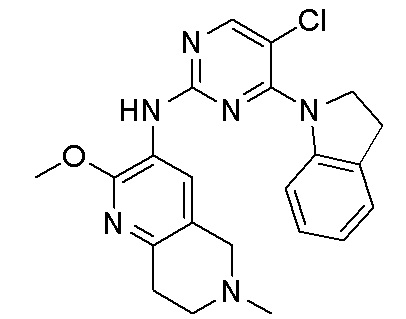
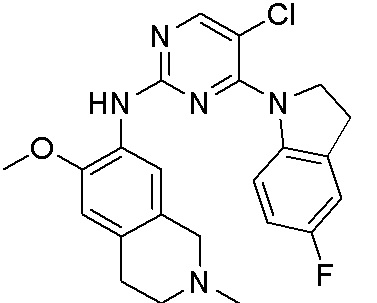
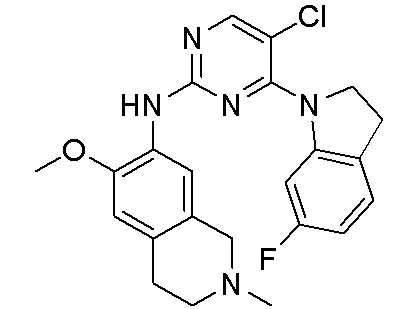
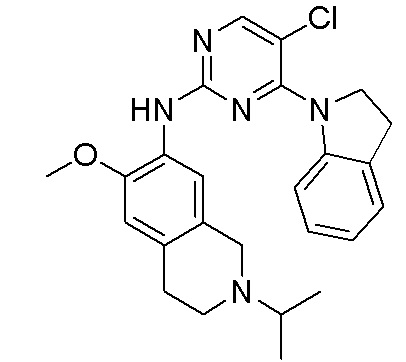
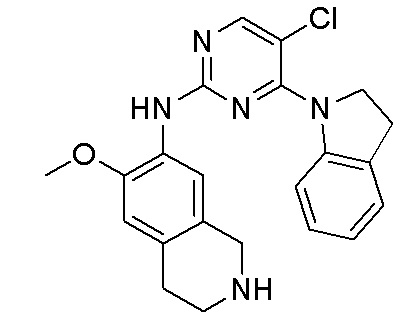
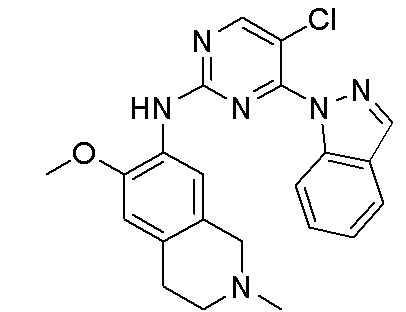
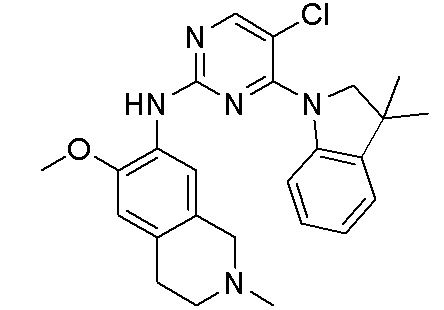
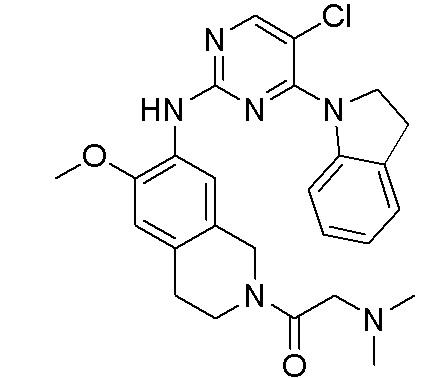
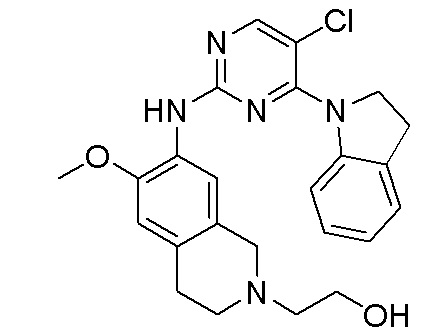
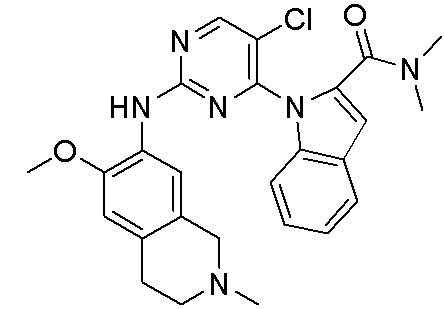
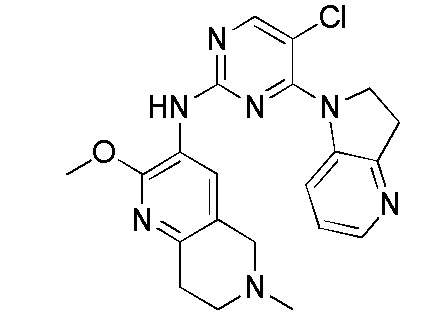
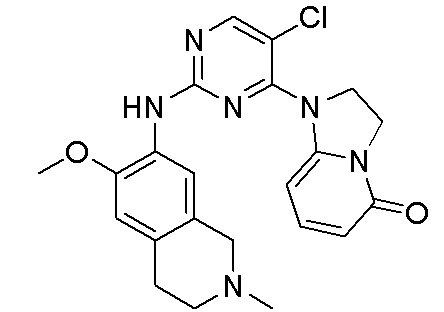
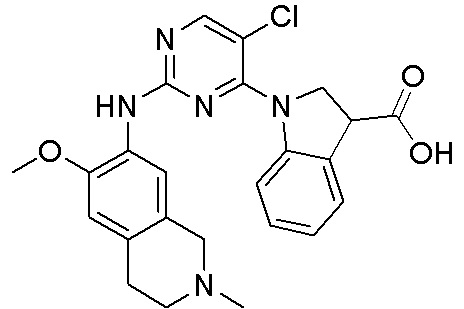
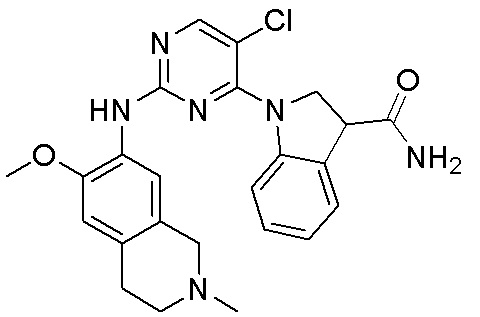
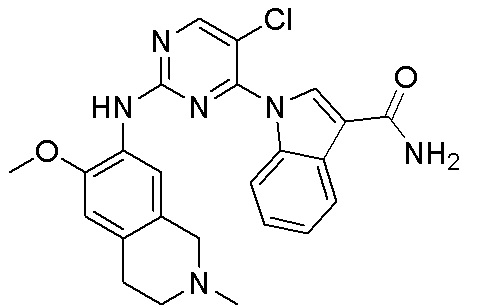
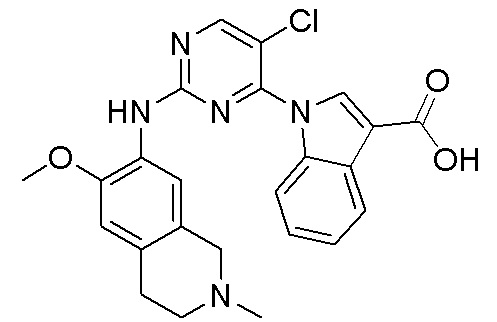
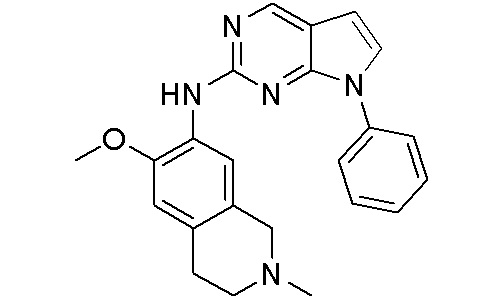
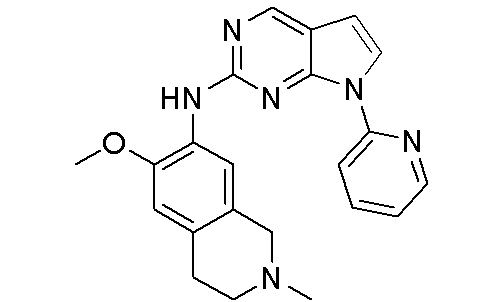
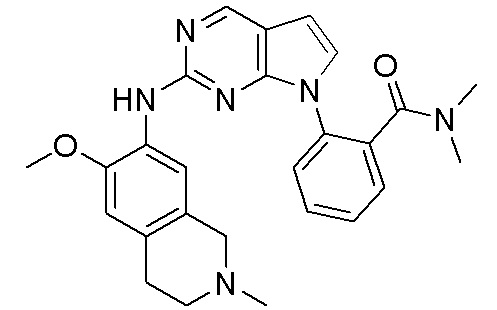
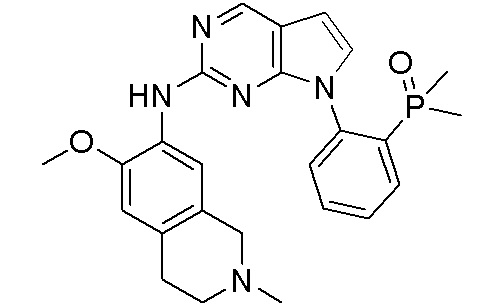
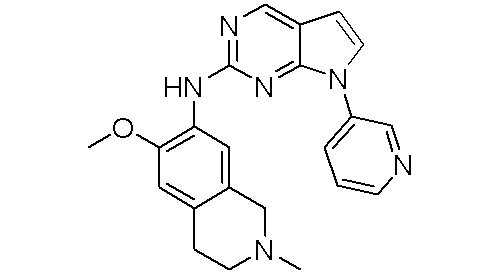
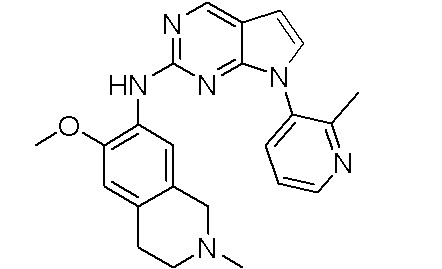
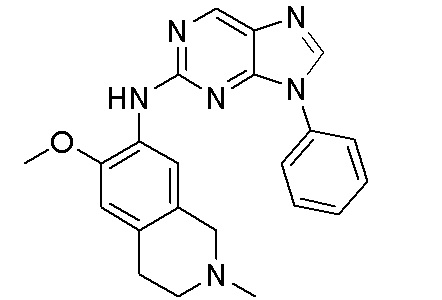
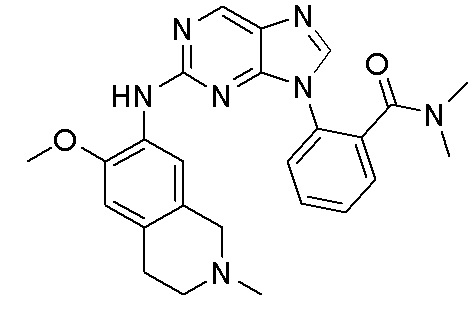 e
e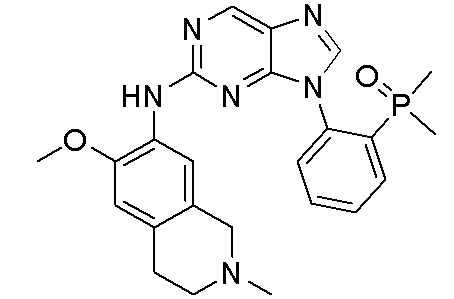
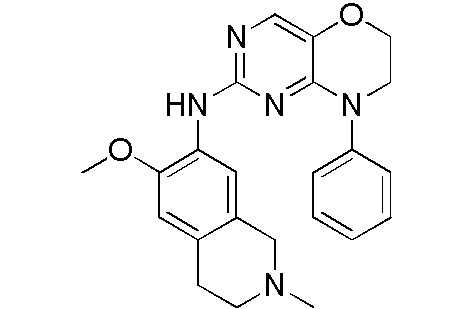
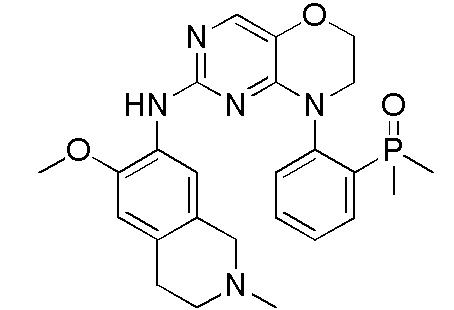
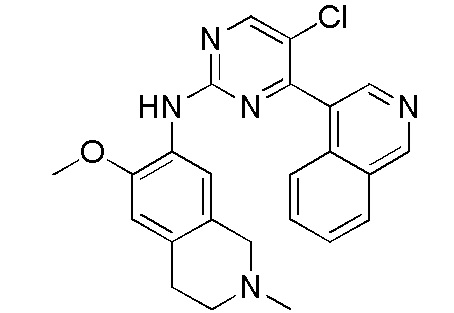
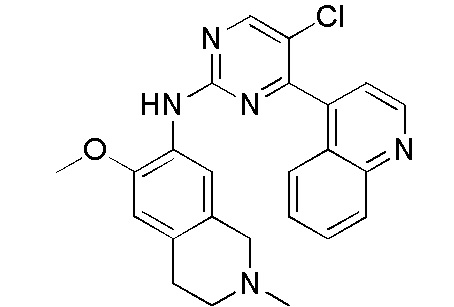
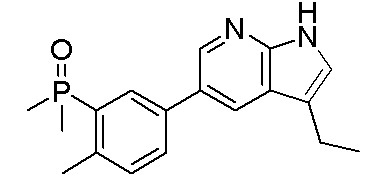
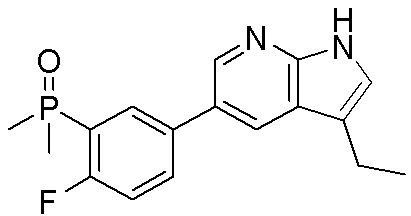
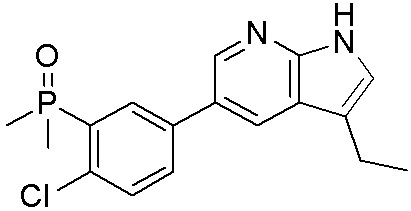
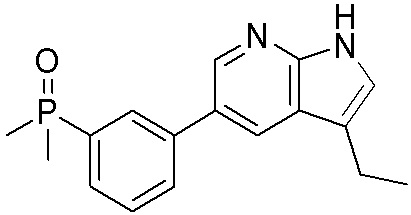
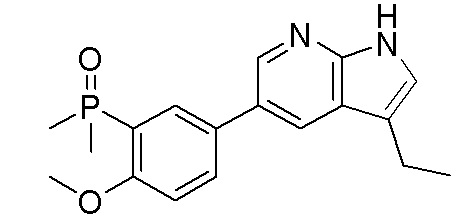
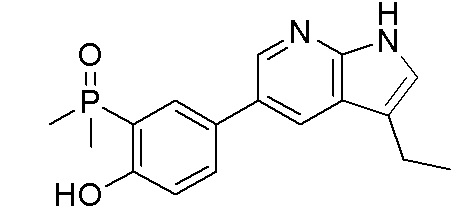
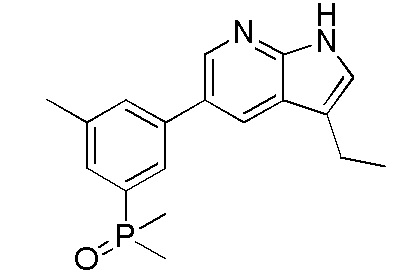
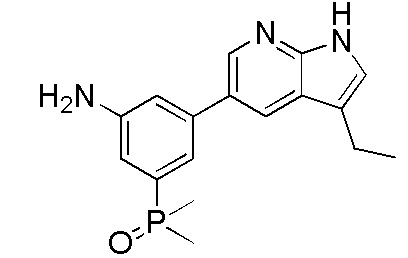
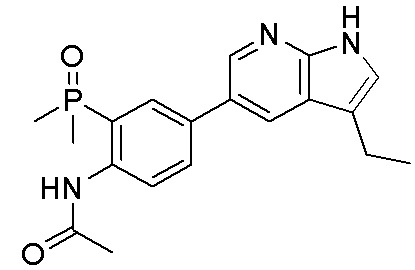
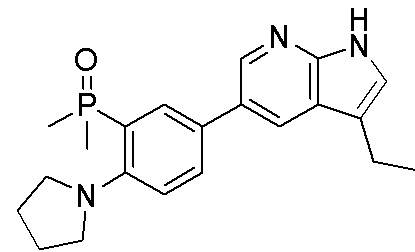
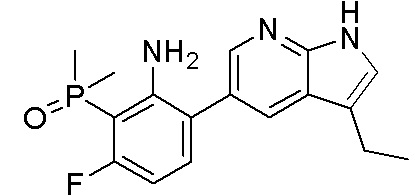
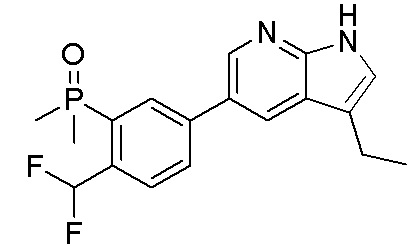
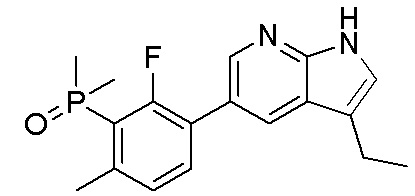
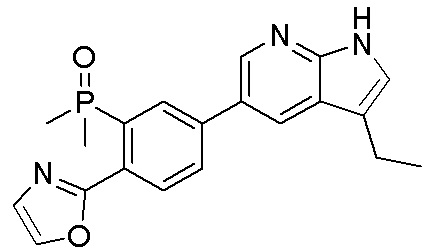
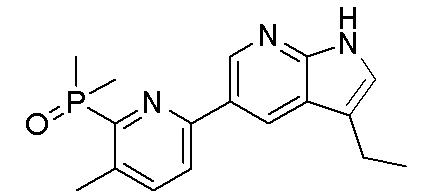
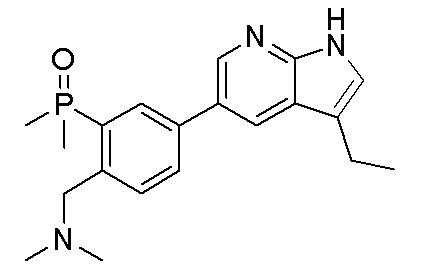
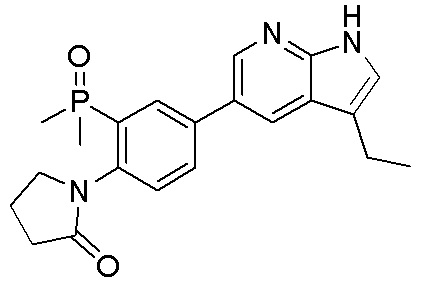
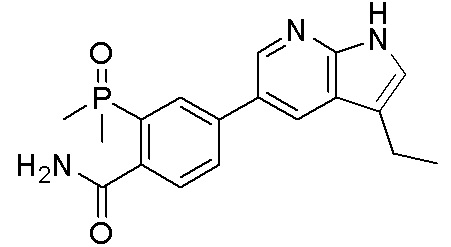
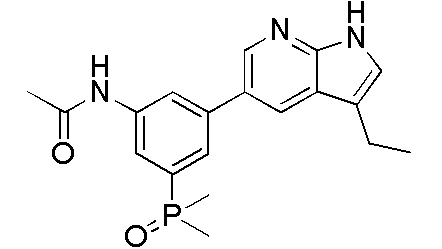
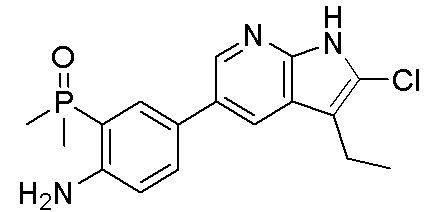
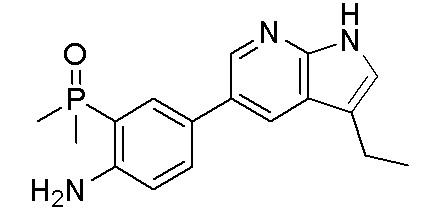
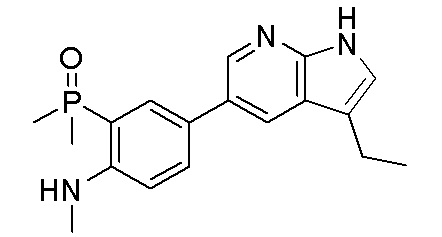
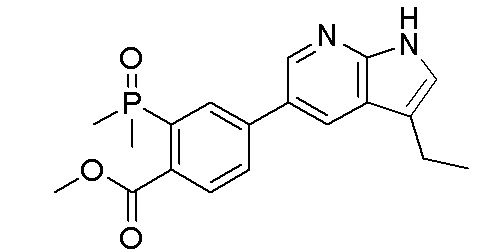
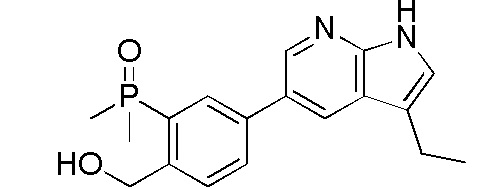
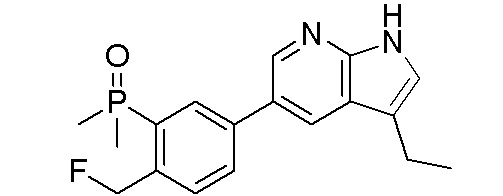
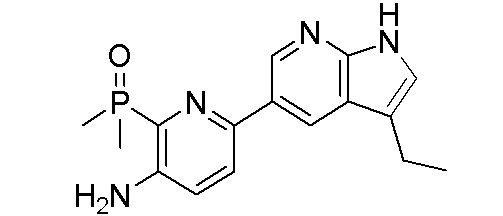
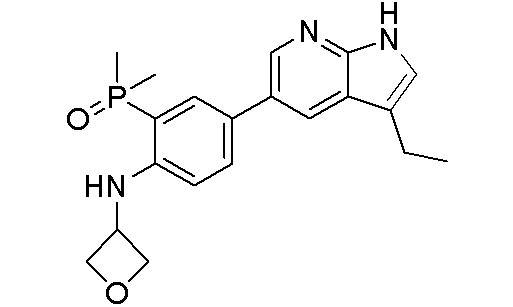
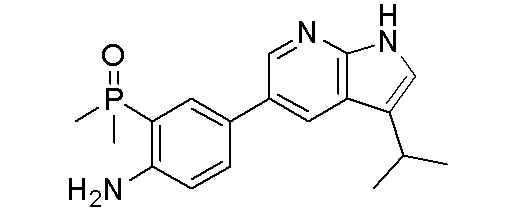
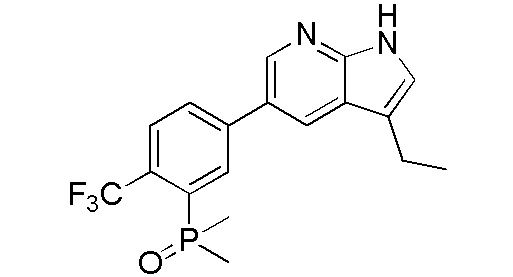
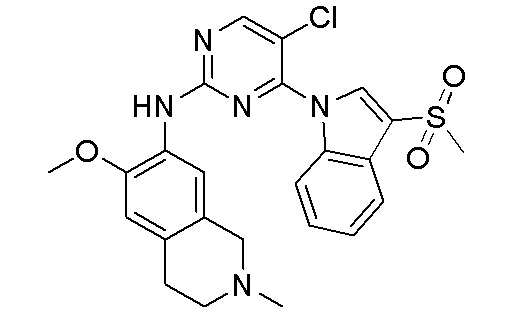
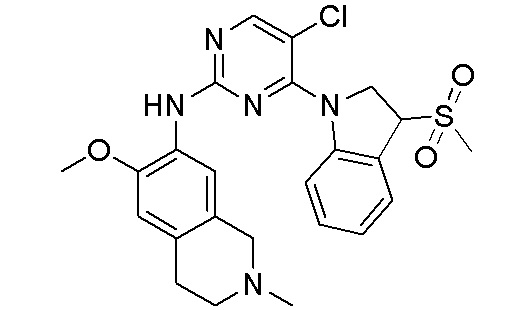
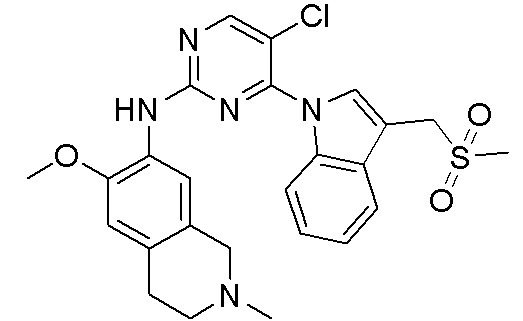
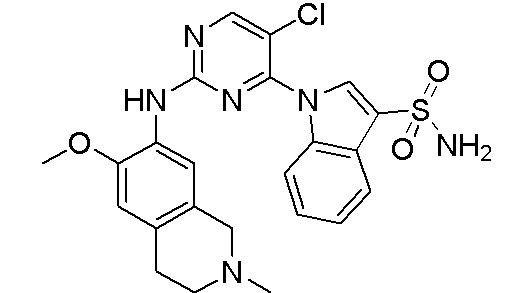
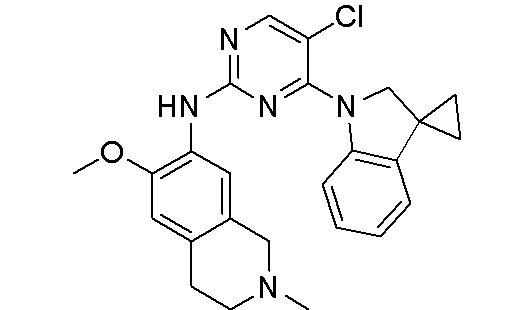
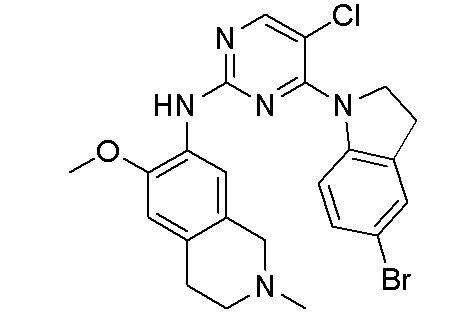
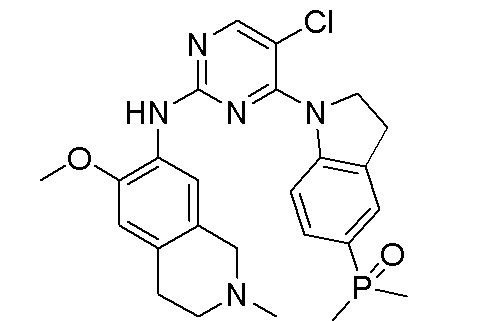
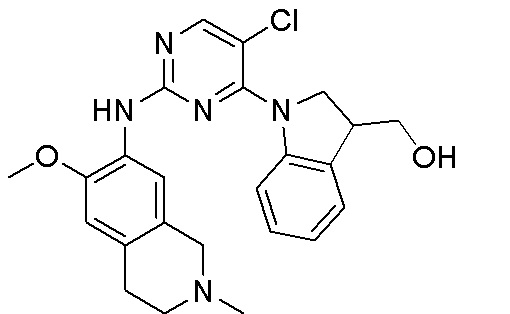
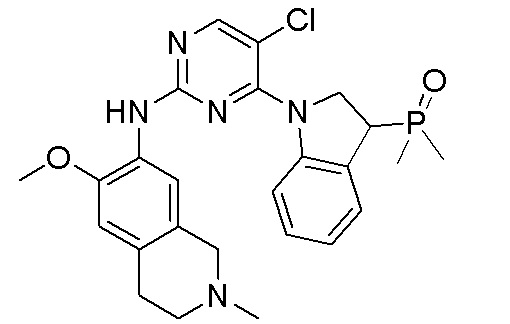
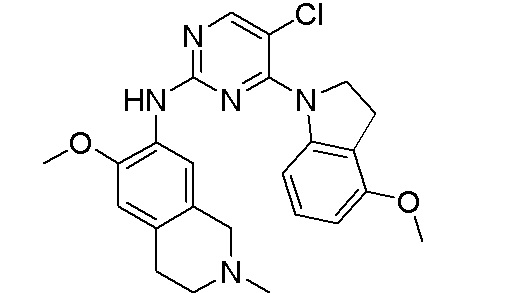
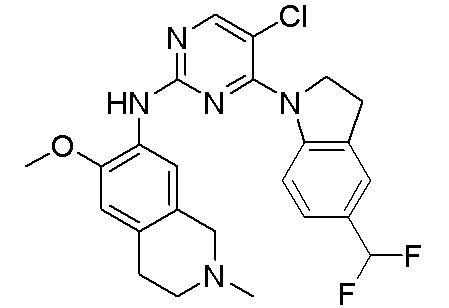
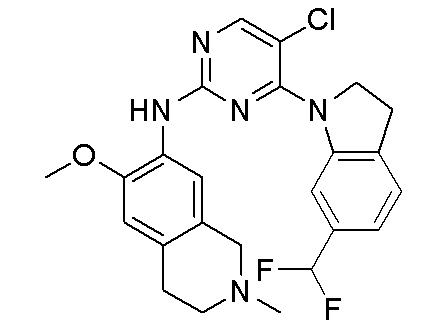
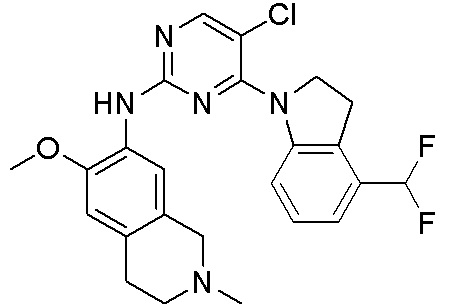
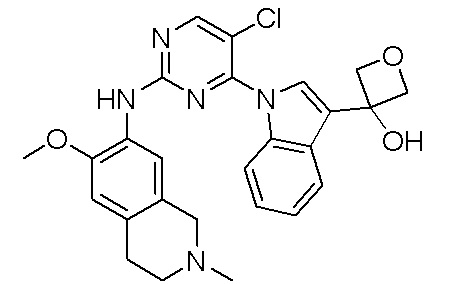
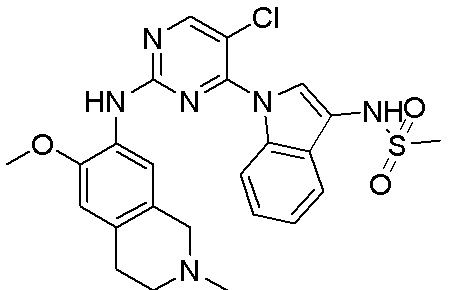
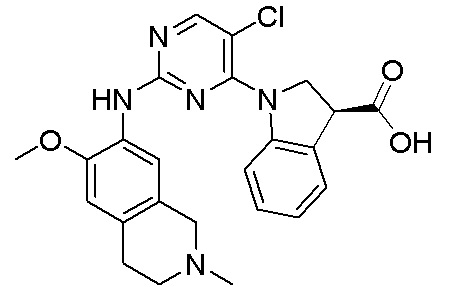
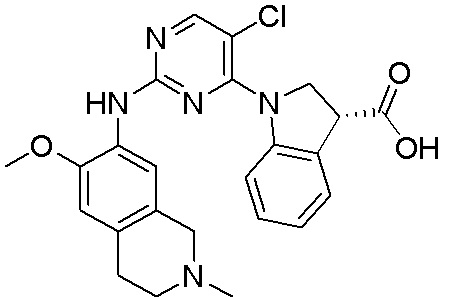
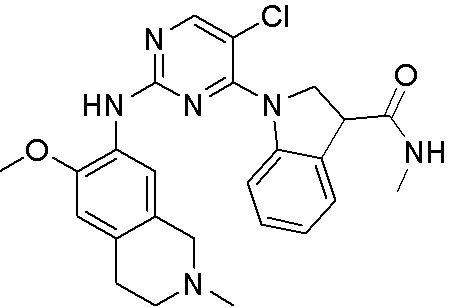
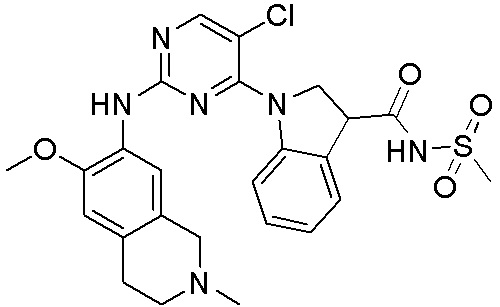
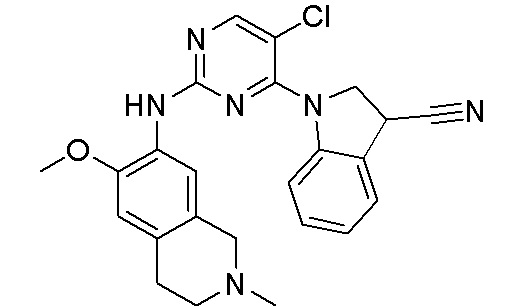
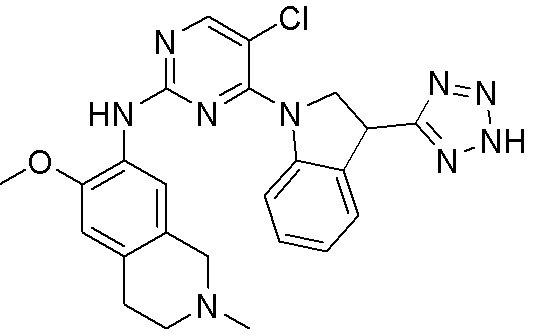
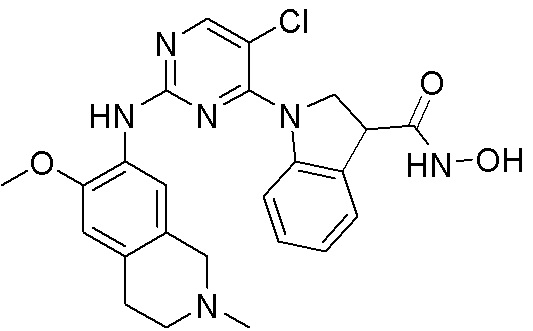
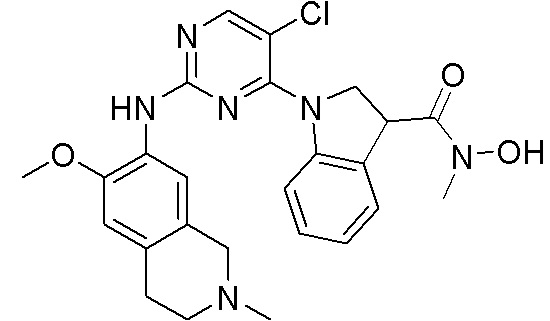
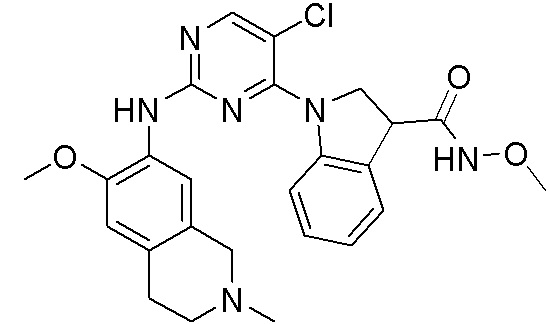
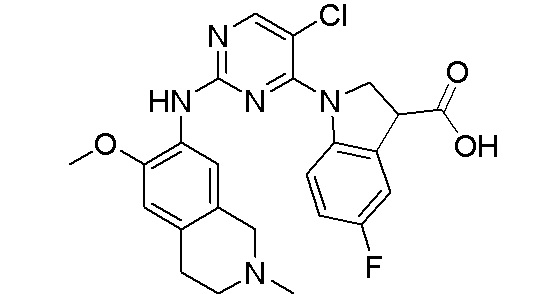
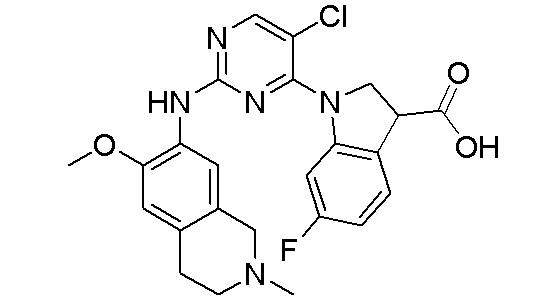
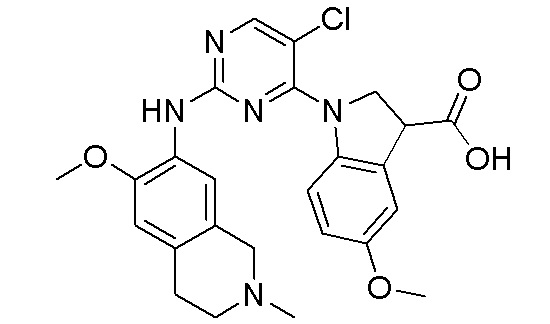
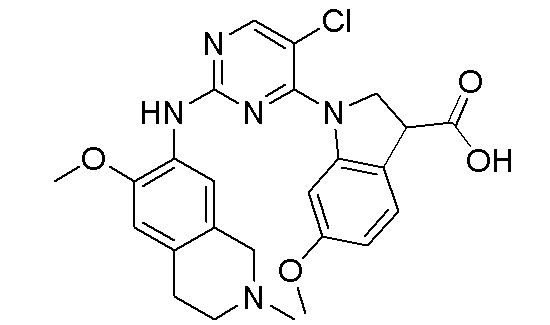
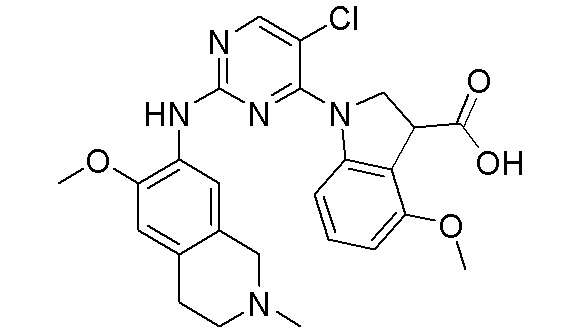
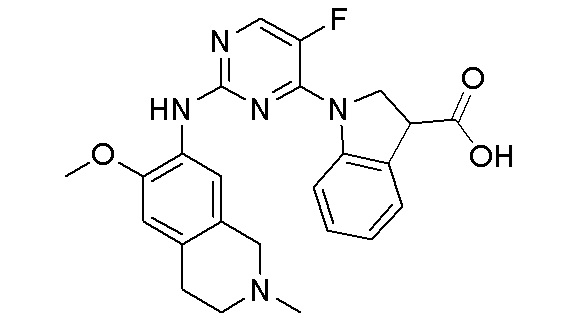
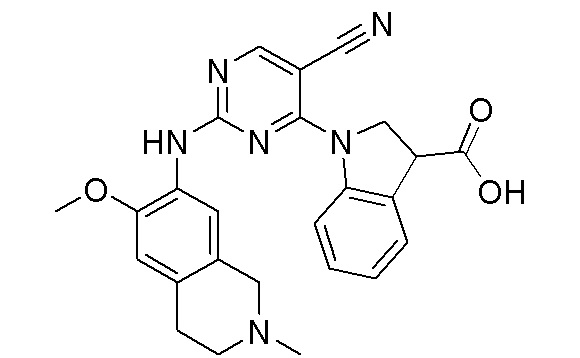
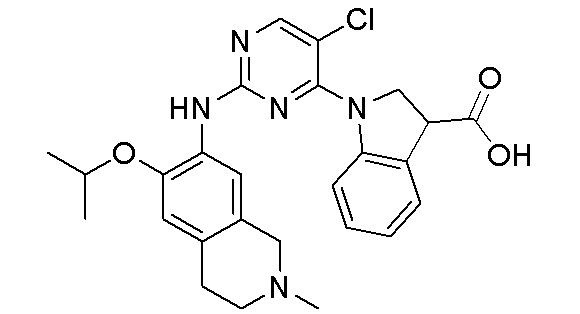
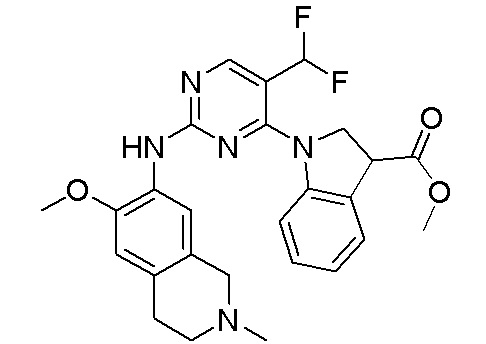
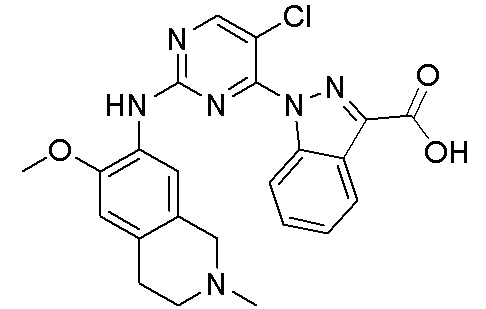
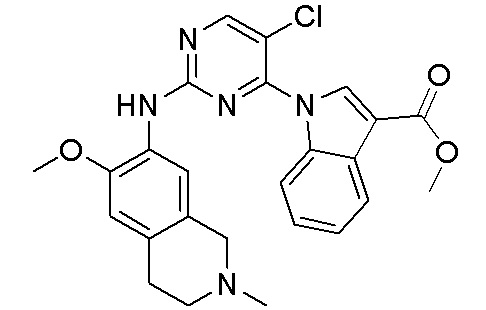
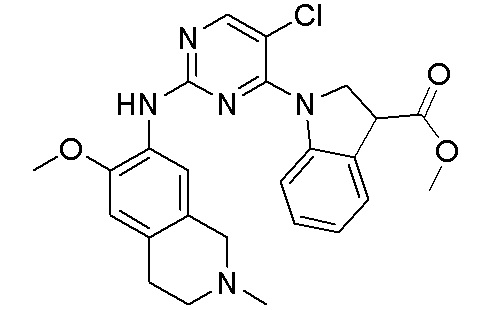
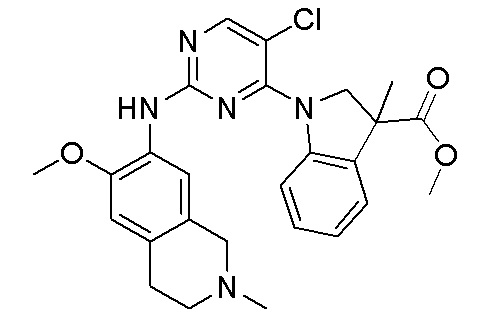
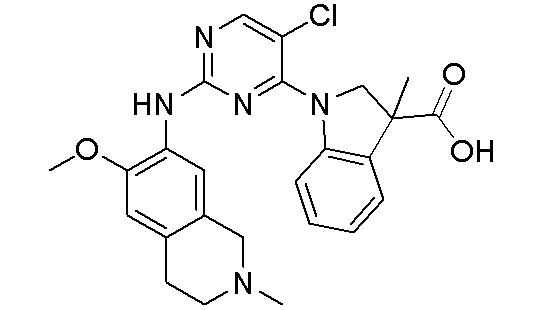
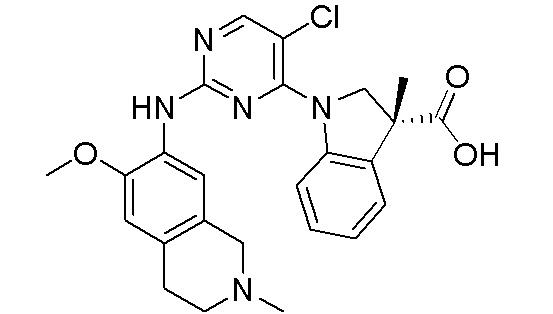
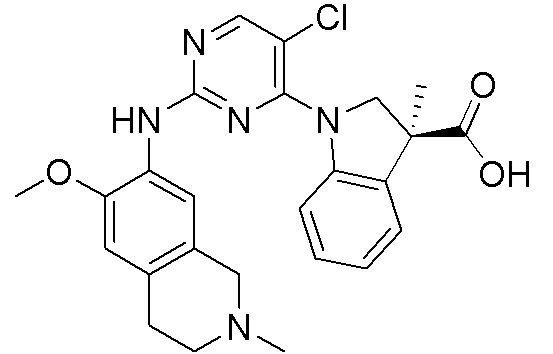
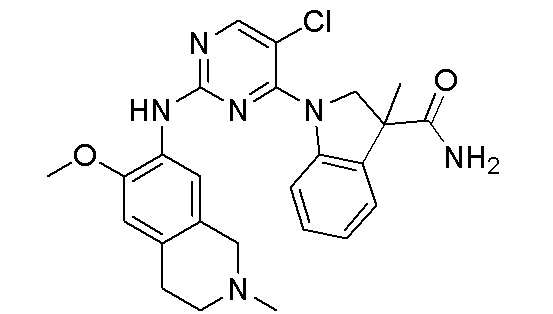
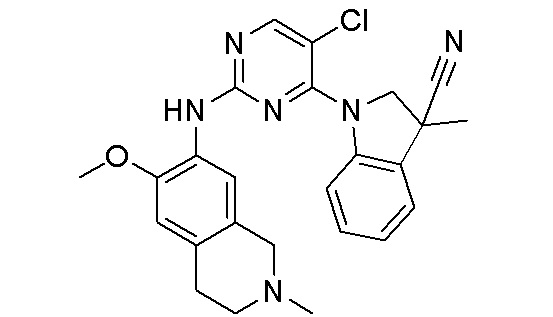
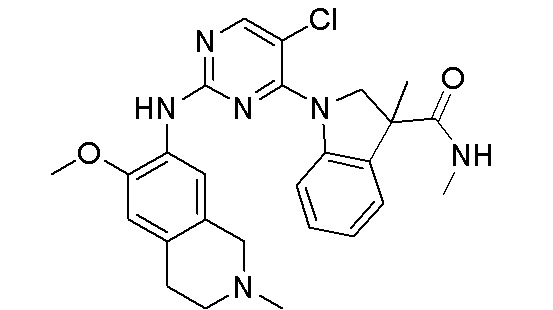
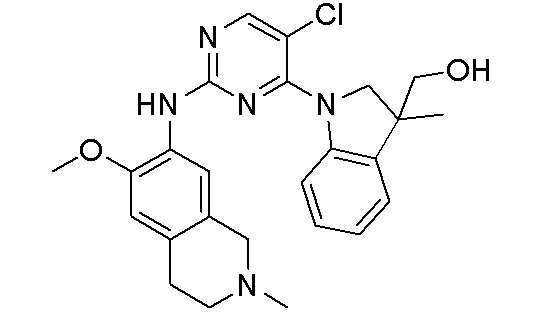
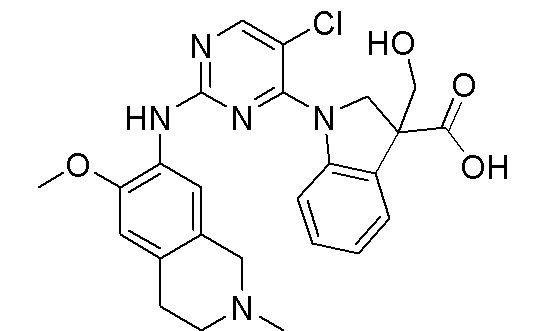
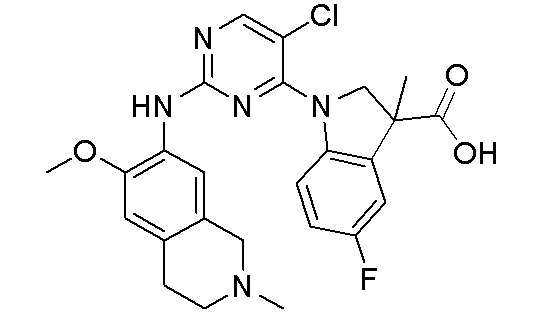
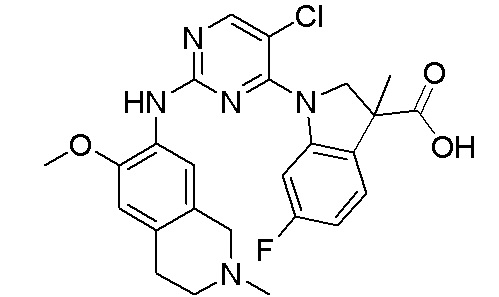
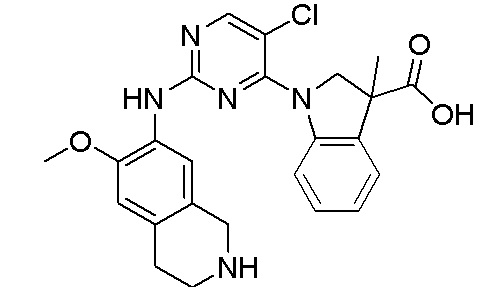
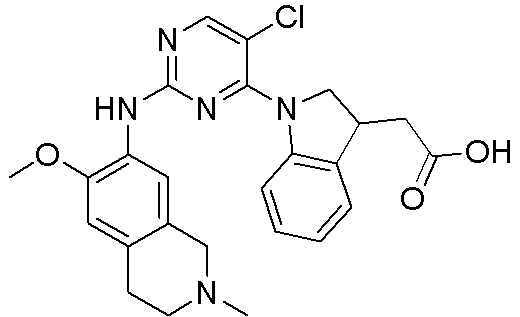
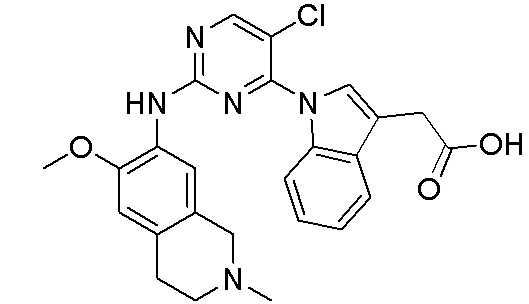
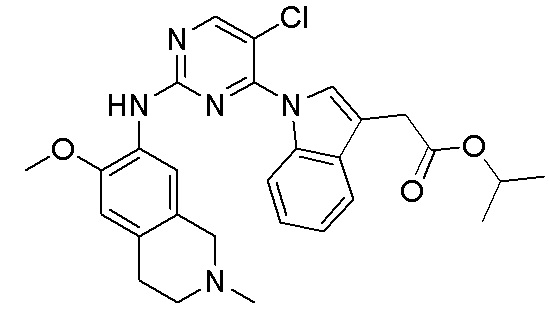
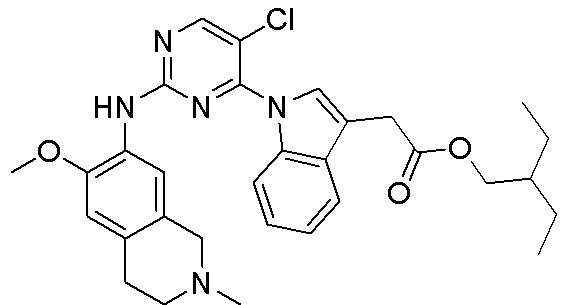
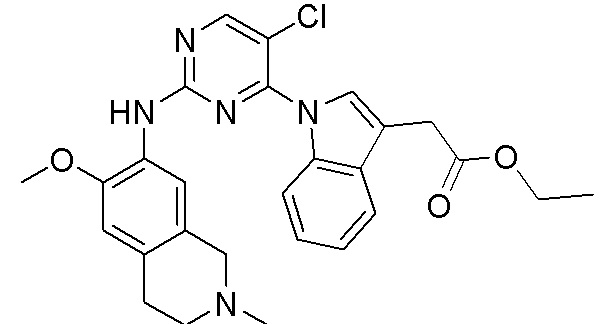
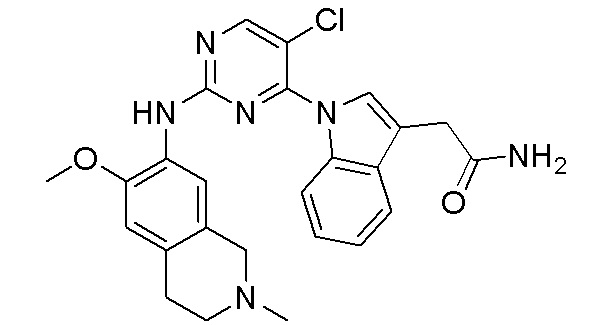
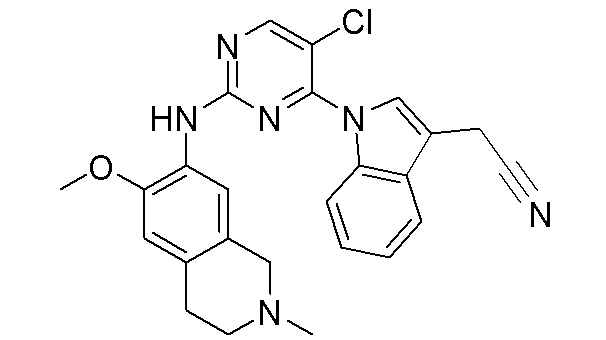
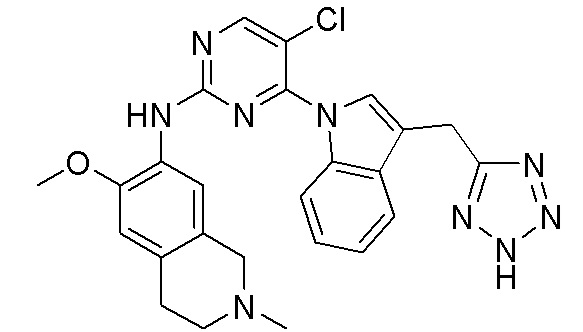
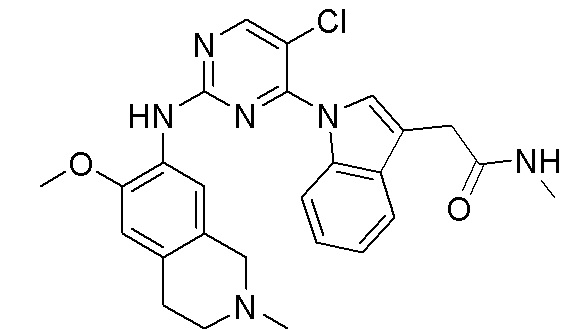
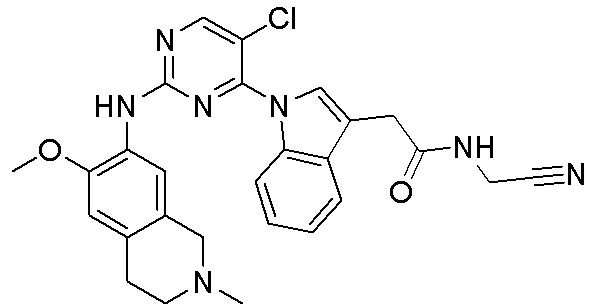
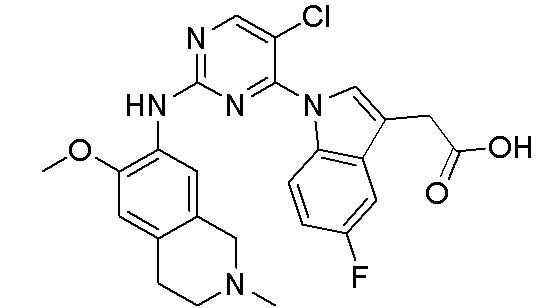
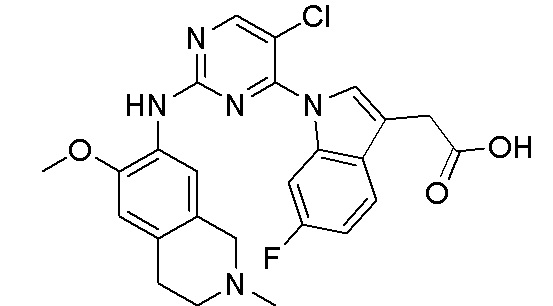
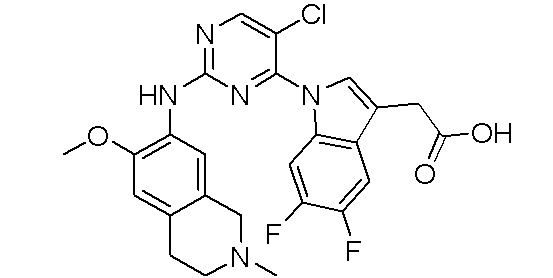
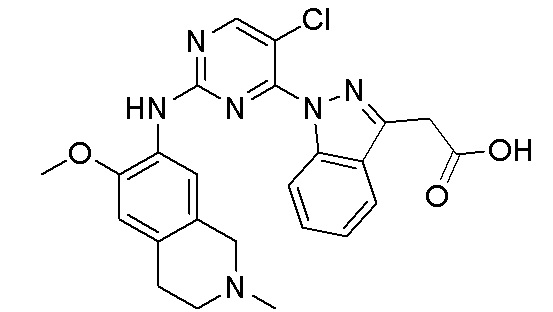
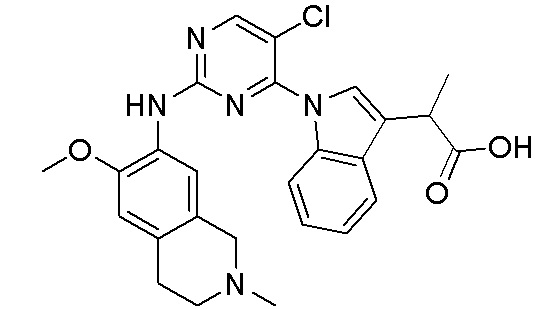
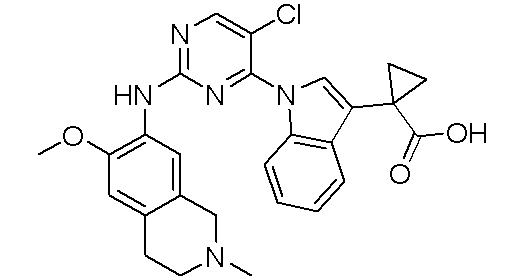
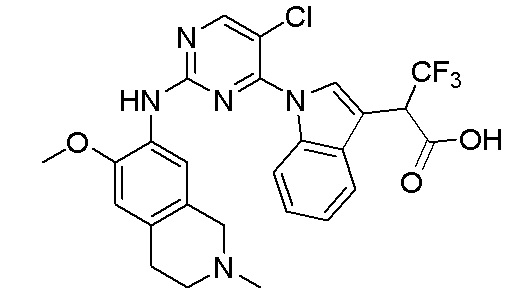
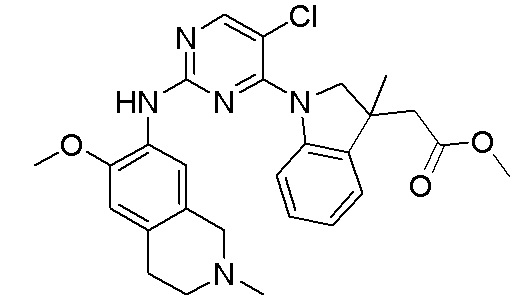
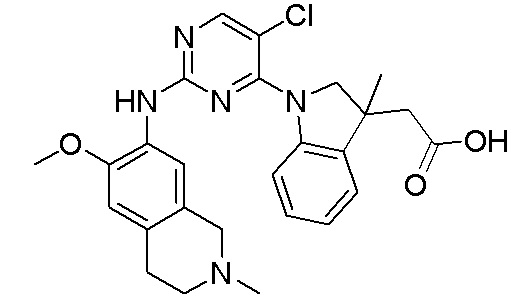
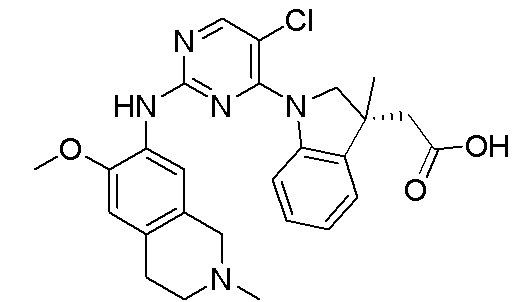
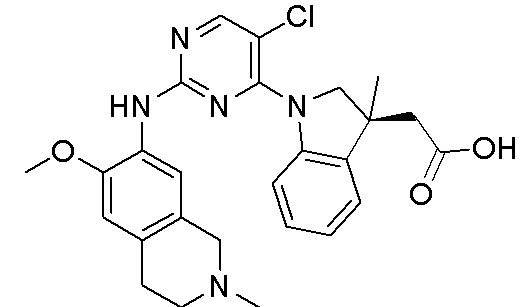
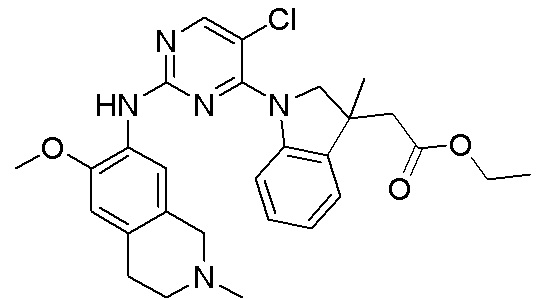
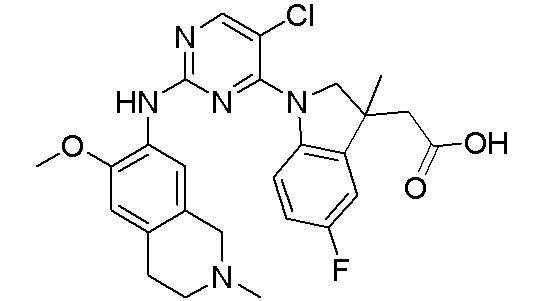
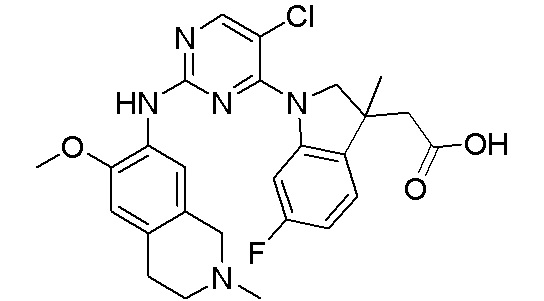
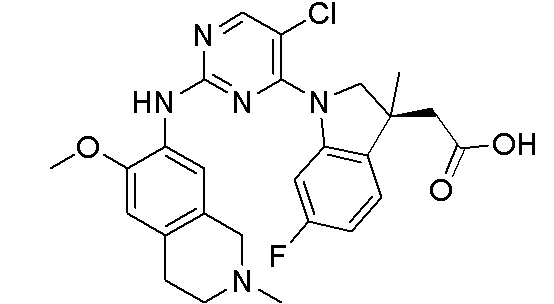
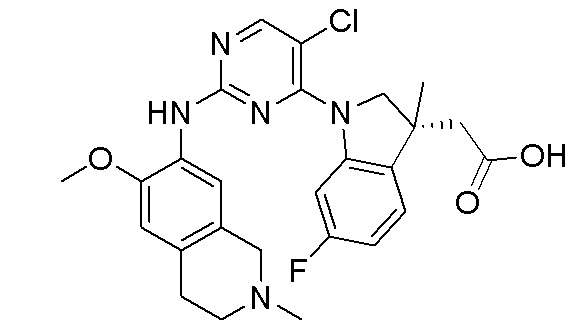
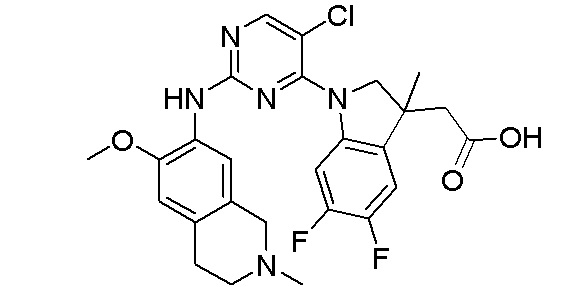
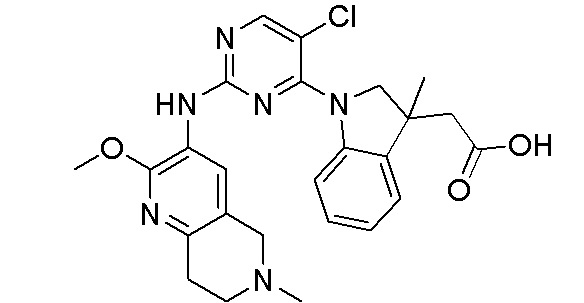
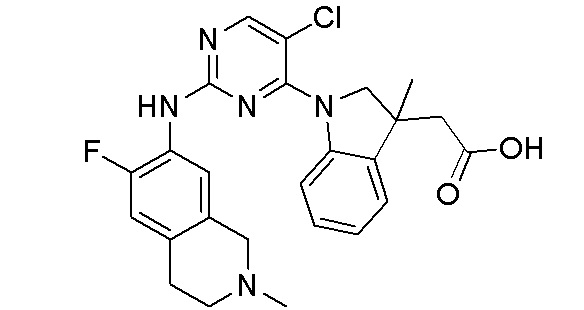
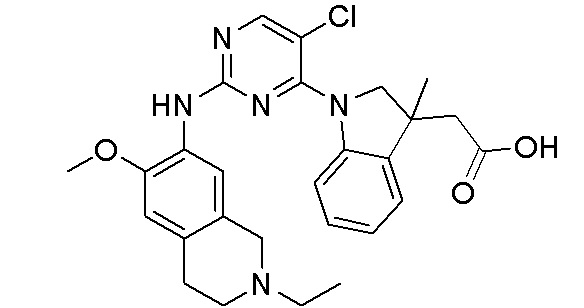
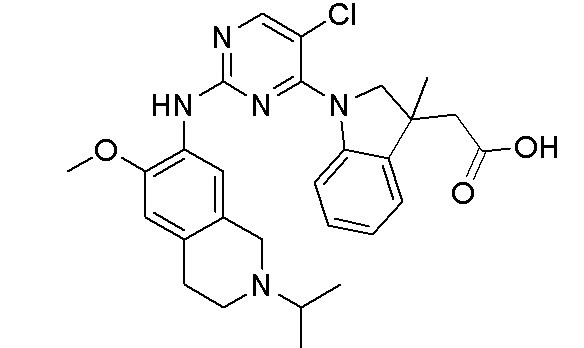
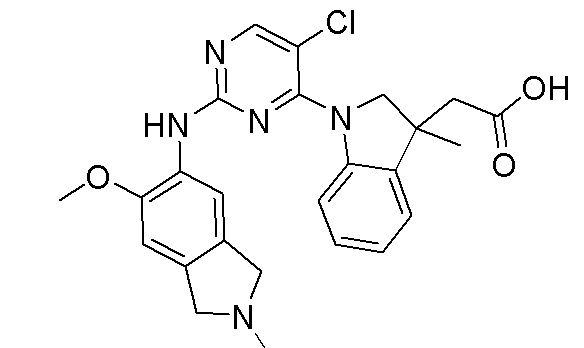
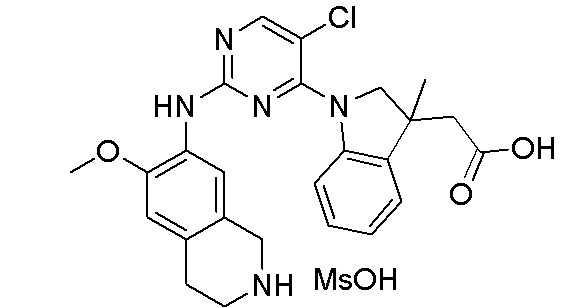
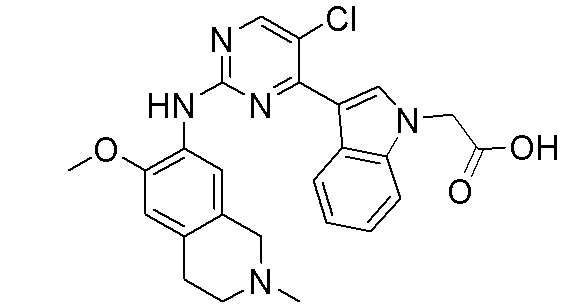
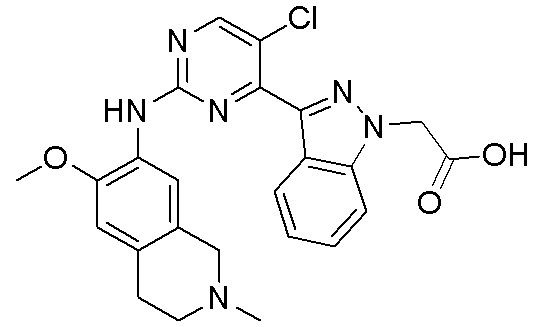
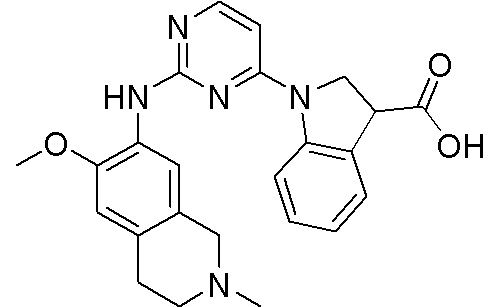
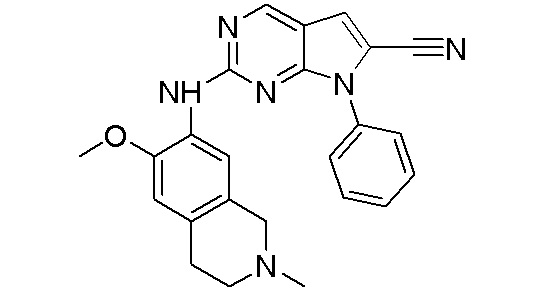
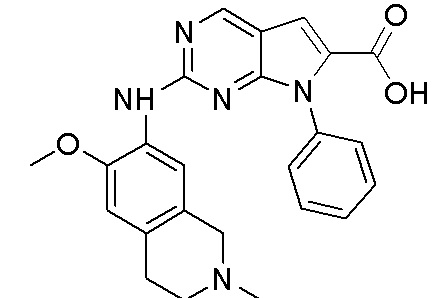
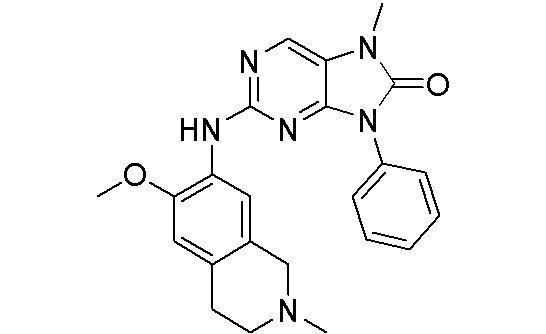
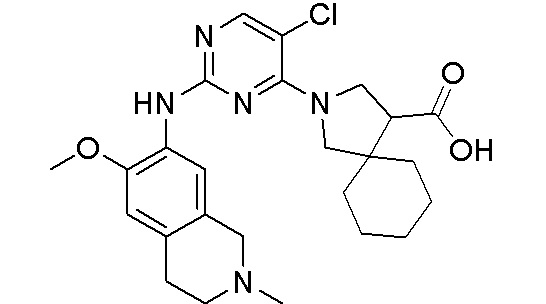
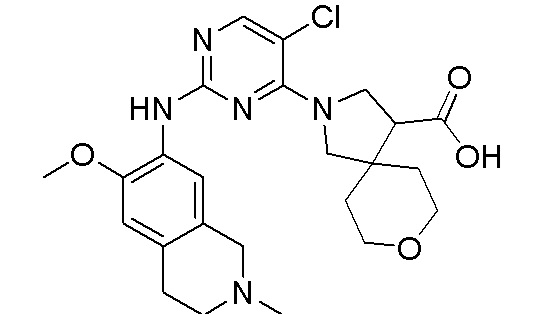
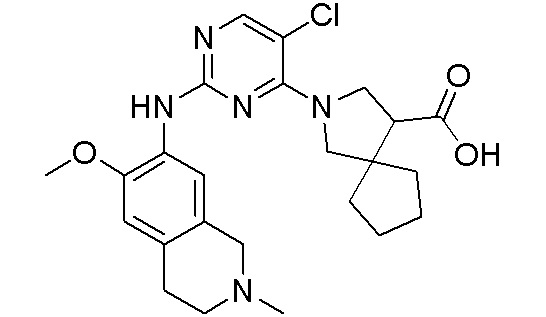
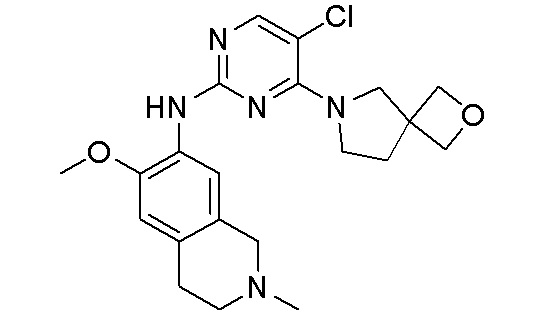
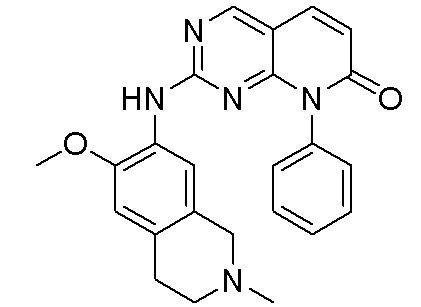
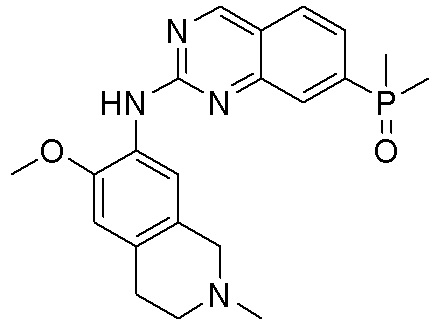
31. Фармацевтическая композиция, обладающая ингибирующей HPK1 активностью, содержащая терапевтически эффективное количество соединения по любому из пп.1-30 или его фармацевтически приемлемой соли, или стереоизомера, и фармацевтически приемлемый носитель.
32. Способ лечения рака, опосредованного активностью HPK1, у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества соединения по любому из пп.1-30 или его фармацевтически приемлемой соли или стереоизомера, где рак, опосредованный активностью HPK1, выбран из рака молочной железы, колоректального рака и рака легких.
33. Способ лечения рака, опосредованного активностью HPK1, включающий введение субъекту эффективного количества соединения по любому из пп. 1-30, или его фармацевтически приемлемой соли или стереоизомера, где рак, опосредованный активностью HPK1, выбран из острого лимфобластного лейкоза (ALL), рака желудка, рака мочеполового тракта, немелкоклеточного рака легкого (NSCLC) и плоскоклеточного рака головы и шеи.
34. Соединение по любому из пп.1-30 или его фармацевтически приемлемая соль или стереоизомер для применения в качестве лекарственного средства, такого как лекарственное средство, действующее как ингибитор HPK1.
| WO 2018167147 A, 20.09.2018 | |||
| CN 104876914 A, 02.09.2015 | |||
| WO 2019074979 A1, 18.04.2019 | |||
| WO 2018002219 A, 04.01.2018 | |||
| WO 2015112441 A1, 30.07.2015 | |||
| Утепленная надставка для уширяющихся кверху изложниц | 1929 |
|
SU17632A1 |

