ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к полипептидам, которые ковалентно связаны с молекулярными каркасами таким образом, что две или более пептидных петель укладываются между точками крепления на каркас. В частности, в изобретении описываются пептиды, которые представляют собой высокоаффинные лиганды мембранной металлопротеиназы типа 1 (МТ1-ММР). В изобретении также описываются конъюгаты лекарственных средств, включающие указанные пептиды, конъюгированные с одной или более эффекторными и/или функциональными группами, которые находят применение в визуализации и таргетной терапии рака.
УРОВЕНЬ ТЕХНИКИ
Циклические пептиды способны связываться с высоким сродством и специфичностью с белковыми мишенями и, следовательно, являются привлекательным классом молекул для разработки терапевтических средств. Действительно несколько циклических пептидов уже успешно используются в клинике, например, такие как антибактериальный пептид ванкомицин, иммуносупрессорное лекарство циклоспорин или противоопухолевое лекарство октреотид (Driggers et al. (2008), Nat Rev Drug Discov 7 (7), 608-24). Хорошие связывающие свойства возникают в результате относительно большой поверхности взаимодействия, образованной между пептидом и мишенью, а также пониженной конформационной гибкости циклических структур. Как правило, макроциклы связываются с поверхностями в несколько сотен квадратных ангстрем, как, например, циклический пептид CVX15 антагонист CXCR4 (400 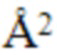 ; Wu et al. (2007), Science 330, 1066-71), циклический пептид с мотивом Arg-Gly-Asp, связывающийся с интегрином αVb3 (355 ) (Xiong et al. (2002), Science 296 (5565), 151-5) или ингибитор циклического пептида упаин-1, связывающийся с активатором плазминогена урокиназного типа (603 ; Zhao et al. (2007), J Struct Biol 160 (1), 1-10).
; Wu et al. (2007), Science 330, 1066-71), циклический пептид с мотивом Arg-Gly-Asp, связывающийся с интегрином αVb3 (355 ) (Xiong et al. (2002), Science 296 (5565), 151-5) или ингибитор циклического пептида упаин-1, связывающийся с активатором плазминогена урокиназного типа (603 ; Zhao et al. (2007), J Struct Biol 160 (1), 1-10).
Из-за своей циклической конфигурации пептидные макроциклы являются менее гибкими, чем линейные пептиды, что приводит к меньшей потере энтропии при связывании с мишенями и ведет к более высокой аффинности связывания. Сниженная гибкость также приводит к фиксации специфичных для мишени конформаций, увеличивая специфичность связывания по сравнению с линейными пептидами. Этот эффект может быть проиллюстрирован на примере мощного и селективного ингибитора матриксной металлопротеиназы 8, MMP-8), который теряет свою селективность в отношении других MMPs, когда его кольцо открыто (Cherney et al. (1998), J Med Chem 41 (11), 1749-51). Благоприятные связывающие свойства, достигаемые за счет макроциклизации, еще более выражены у полициклических пептидов, имеющих более одного пептидного кольца, как, например, у ванкомицина, низина и актиномицина.
Различные исследовательские группы ранее привязывали полипептиды с остатками цистеина к синтетической молекулярной структуре (Kemp and McNamara (1985), J. Org. Chem; Timmerman et al. (2005), ChemBioChem). Meloen и сотрудники использовали трис(бромметил)бензол и родственные молекулы для быстрой и количественной циклизации множественных пептидных петель на синтетических каркасах для структурной имитации белковых поверхностей (Timmerman et al. (2005), ChemBioChem). Способы создания кандидатных лекарственных соединений, где указанные соединения образуются путем соединения содержащих цистеин полипептидов с молекулярным каркасом, таким как, например, трис(бромметил)бензол, раскрыты в патентах WO 2004/077062 и WO 2006/078161.
Для создания и скрининга больших библиотек бициклических пептидов к представляющим интерес мишеням разработан фаговый дисплей, основанный на комбинаторных подходах (Heinis et al. (2009), Nat Chem Biol 5 (7), 502-7 и патент WO2009/098450). Вкратце, комбинаторные библиотеки линейных пептидов, содержащих три остатка цистеина и две области шести случайных аминокислот (Cys-(Xaa)6-Cys-(Xaa)6-Cys), были представлены на фаговом дисплее и циклизованы путем ковалентного связывания боковых цепей цистеина с небольшой молекулой (трис-(бромметил)бензолом).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В соответствии с первым аспектом настоящего изобретения предлагается пептидный лиганд, специфичный для MT1-MMP, включающий полипептид, включающий, по меньшей мере, три остатка цистеина, разделенных, по меньшей мере, двумя последовательностями петель, и молекулярный каркас, который образует ковалентные связи с остатками цистеина полипептида таким образом, что образуются, по меньшей мере, две полипептидные петли на молекулярном каркасе, где пептидный лиганд включает аминокислотную последовательность формулы (I):
-Ci-X-U/O-X-X-G-Cii-E-D-F-Y-X-X-Ciii- (SEQ ID NO: 1) (I)
или модифицированное производное, или ее фармацевтически приемлемую соль;
где:
Ci, Cii и Ciii представляют собой первый, второй и третий остатки цистеина, соответственно;
X представляет собой любой аминокислотный остаток;
U представляет собой полярный, незаряженный аминокислотный остаток, выбранный из N, C, Q, M, S и T; и О представляет собой неполярный остаток алифатической аминокислоты, выбранный из G, A, I, L, P и V.
В соответствии с другим аспектом настоящего изобретения, предлагается конъюгат лекарственного средства, включающий пептидный лиганд, как определено в настоящем документе, конъюгированный с одной или более эффекторными и/или функциональными группами, такими как цитотоксические агенты, в частности, DM1 и MMAE.
В соответствии с другим аспектом настоящего изобретения предлагается конъюгат, включающий пептидный лиганд, как определено в настоящем документе, конъюгированный с одной или более эффекторными и/или функциональными группами, такими как хелаторная группа, несущая радионуклид, в частности, DOTA.
В соответствии с другим аспектом настоящего изобретения предлагается фармацевтическая композиция, включающая пептидный лиганд или конъюгат лекарственного средства, как определено в настоящем документе, в сочетании с одним или более фармацевтически приемлемыми наполнителями.
В соответствии с другим аспектом настоящего изобретения предлагается пептидный лиганд, как определено в настоящем документе, для применения при профилактике, подавлении или лечении рака, в частности солидных опухолей, таких как немелкоклеточный рак легкого.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигура 1: Стабильность 17-69-07-N219 в плазме мыши. Прослеживали за несколькими ионами, как указано в подписи, а также за двумя переходами в режиме MRM. Наблюдается прекрасная корреляция между ионами. Период полужизни пептида в плазме мыши при 37°С составляет 6 часов.
Фигура 2: Профиль PK бициклического пептида 17-69-07-N004 у мыши. 2 животных на временную точку.
Фигура 3: Стабильность в плазме мыши (А) и человека (В) двух стабилизированных молекул 17-69-07 (с 4-бромфенилаланином в положении 9: 17-69-07-N244, без 4- бромфенилаланина в положении 9: 17-69-07-N231) по сравнению с нестабилизированным 17-69-07-N219. Прослежено несколько переходов МРМ для данного анализируемого вещества, которые хорошо коррелировали друг с другом. В соответствии с целью этого графика отображен только один переход.
Фигура 4: Биораспределение 177Lu 17-69-07-N144 у мышей с ксенотрансплантатом HT-1080.
Фигура 5: Биораспределение 177Lu 17-69-07-N246 у мышей с ксенотрансплантатом HT-1080.
Фигура 6: Биораспределение 177Lu 17-69-07-N248 у мышей с ксенотрансплантатом HT-1080.
Фигура 7: (А): График зависимости среднего объема опухоли от времени для BT17BDC-1 и 9. Дозы вводили на 0, 2, 4, 7, 9 и 11 день. (В): Масса тела в течение лечения, которая является показателем токсичности, связанной с лекарством, и показателем общего состояния здоровья животного.
Фигура 8: Перечень выходов последовательностей, происходящих при созревании аффинности, с использованием библиотек с фиксированными остатками 17-69 петли 2. График лигатур последовательностей справа указывает на общее предпочтение остатков в петле 1 для остатков 1, 2, 3, 4 и 5.
Фигура 9: Вверху: График зависимости среднего объема опухоли от времени для BT17BDC-17 у мышей с ксенотрансплантатом ЕВС-1. Дозы вводили на 0, 2, 4, 7, 9, 11 и 14 день. Внизу: Масса тела во время лечения, которая является показателем токсичности, связанной с лекарством, и показателем общего состояния здоровья животного.
Фигура 10: Верху: График зависимости среднего объема опухоли от времени для BT17BDC-18 у мышей с ксенотрансплантатом ЕВС-1. Дозы вводили на 0, 2, 4, 7, 9, 11 и 14 день. Внизу: Масса тела во время лечения, которая является показателем токсичности, связанной с лекарством, и показателем общего состояния здоровья животного.
Фигура 11: Верху: График зависимости среднего объема опухоли от времени для BT17BDC-19 у мышей с ксенотрансплантатом ЕВС-1. Дозы вводили на 0, 2, 4, 7, 9, 11 и 14 день. Внизу: Масса тела во время лечения, которая является показателем токсичности, связанной с лекарством, и показателем общего состояния здоровья животного.
Фигура 12: Верху: График зависимости среднего объема опухоли от времени для BT17BDC-20 у мышей с ксенотрансплантатом ЕВС-1. Дозы вводили на 0, 2, 4, 7, 9, 11 и 14 день. Внизу: Масса тела во время лечения, которая является показателем токсичности, связанной с лекарством, и показателем общего состояния здоровья животного.
Фигура 13: График зависимости площади под кривой (AUC) объема опухоли с течением времени, связанной с конкретным BDC, от группы c соответствующей дозой. Подбор кривых осуществлялся с использованием всех доступных точек данных, нормализованных на объем опухоли в нулевой момент времени, с использованием стандартных уравнений для IC50.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не определено иначе, все технические и научные термины, используемые в настоящем документе, имеют такое же значение, что и обычно понимаемое специалистами в данной области техники, например, в области химии пептидов, культуры клеток и фагового дисплея, химии нуклеиновых кислот и биохимии. Стандартные методы используются для методов молекулярной биологии, генетических и биохимических методов (смотри Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd ed., 2001, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY; Ausubel et al., Short Protocols in Molecular Biology (1999) 4th ed., John Wiley & Sons, Inc.), которые включены в данное описание в качестве ссылки.
Номенклатура
Нумерация
При ссылках на положения аминокислотных остатков в соединениях формулы (I) остатки цистеина (Ci, Cii и Ciii) не включаются в нумерацию, так как они инвариантны, следовательно, нумерация аминокислотных остатков в соединении формулы (I) обозначается, как показано ниже:
-Ci-X1-U/O2-X3-X4-G5-Cii-E6-D7-F8-Y9-X10-X11-Ciii- (SEQ ID NO: 1).
В целях настоящего описания все бициклические пептиды, как предполагается, циклизованы с TBMB (1,3,5-трис(бромметил)бензолом) с получением трехзамещенной структуры 1,3,5-трисметилбензола. Циклизация с TBMB происходит на Ci, Cii и Ciii.
Последовательность каркаса бициклических пептидов
Каждому бициклическому пептиду, раскрытому в настоящем документе, был присвоен уникальный номер последовательности каркаса, которая определяется как аминокислотная последовательность между первым N-концевым цистеином (Ci) и последним С-концевым цистеином (Ciii). Например, у идентификационного номера 17-69-07 последовательность каркаса представляет собой CiYNEFGCiiEDFYDICiii (SEQ ID NO: 2) и обозначается как «17-69-07» или «(17-69-07)».
Пептидный код
Некоторым бициклическим пептидам, описанным в настоящем документе, присваивается также уникальный идентификационный номер с использованием пептидного кода, такой как 17-69-07-N241, где N241 обозначает определенное производное бициклической каркасной последовательности 17-69-07. Различные производные 17-69-07 имеют различные числа N, т.е. N001, N002, Nxxx.
Молекулярный формат
N- или С-концевые расширения каркасной бициклической последовательности добавляются к левой или правой стороне каркасной последовательности разделенные дефисом. Например, N-концевой хвост βAla-Sar10-Ala должен обозначаться как:
βAla-Sar10-A-(17-69-07)
и иметь полную последовательность βAla-Sar10-A-CYNEFGCEDFYDIC (SEQ ID NO: 3).
Модификации
Неприродные аминокислотные замены в пределах каркасной бициклической последовательности указаны после описания молекулярного формата. Например, если тирозин 1 в 17-69-07 замещен D-аланином, то описание представляет собой (17-69-07) D-Ala1, и полная последовательность будет описана как C(D-Ala1)NEFGCEDFYDIC (SEQ ID NO: 4).
Если N-концевой или С-концевой хвост присоединен к бициклическому пептиду, который также содержит модификации в каркасной последовательности, то при использовании 17-69-07-N241 в качестве примера, молекулярное описание Формата представляет собой:
βAla-Sar10-A-(17-69-07) DAla1 1NAl4 DAla5 tBuGly11.
Полная аминокислотная последовательность 17-69-07-N241, следовательно, представляет собой:
βAla-Sar10-A-C(D-Ala)NE(1Nal)(D-Ala)CEDFYD(tBuGly)C (SEQ ID NO: 5).
Пептидные лиганды
Пептидный лиганд, как указано в данном описании, относится к пептиду, ковалентно связанному с молекулярным каркасом. Как правило, такие пептиды включают две или более реакционноспособные группы (т.е. остатки цистеина), которые способны образовывать ковалентные связи с каркасом, и последовательность, образуемую между указанными реакционноспособными группами, которая обозначается как последовательность петли, так как она образует петлю, когда пептид связан с каркасом. В данном случае пептиды включают, по меньшей мере, три остатка цистеина (обозначаемые в настоящем документе как Ci, Cii и Ciii), и образуют, по меньшей мере, две петли на каркасе.
Специалисту в данной области техники должно быть понятно, что X в положениях 1, 3, 4, 10 и 11 формулы (I) может представлять собой любую аминокислоту, следуя результатам сканирования на аланин (смотри таблицу 5) и выбору выходов (фигура 8), что дает хорошо допустимые замены в этих положениях.
В одном варианте осуществления X в положении 1 формулы (I) выбран из любой из следующих аминокислот: Y, M, F или V. В другом варианте осуществления Х в положении 1 формулы (I) выбран из Y, M или F. В еще одном варианте осуществления X в положении 1 формулы (I) выбран из Y или M. В еще одном варианте осуществления Х в положении 1 формулы (I) выбран из Y.
В одном варианте осуществления U/O в положении 2 формулы (I) выбран из U, такого как N. В альтернативном варианте осуществления U/O в положении 2 формулы (I) выбран из O, такого как G.
В одном варианте Х в положении 3 формулы (I) выбран из U или Z, где U представляет собой полярный, незаряженный аминокислотный остаток, выбранный из N, C, Q, M, S и T, и Z представляет собой полярный, отрицательно заряженный аминокислотный остаток, выбранный из D или E. В дополнительном варианте осуществления U в положении 3 формулы (I) выбран из Q. В альтернативном варианте осуществления Z в положении 3 формулы (I) выбран из Е.
В одном варианте осуществления Х в положении 4 формулы (I) выбран из J, где J представляет собой неполярный остаток ароматической аминокислоты, выбранной из F, W и Y. В другом варианте осуществления J в положении 4 формулы (I) выбран из F. В альтернативном варианте осуществления J в положении 4 формулы (I) выбран из Y. В альтернативном варианте осуществления J в положении 4 формулы (I) выбран из W.
В одном варианте осуществления Х в положении 10 формулы (I) выбран из Z, где Z представляет собой полярный, отрицательно заряженный аминокислотный остаток, выбранный из D или E. В одном варианте осуществления изобретения Z в положении 10 формулы (I), выбран из D.
В одном варианте осуществления Х в положении 11 формулы (I) выбран из О, где О представляет собой неполярный остаток алифатической аминокислоты, выбранный из G, A, I, L, P и V. В одном варианте осуществления O в положении 11 формулы (I) выбран из I.
В одном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ia):
-Ci-Y/M/F/V-U/O-U/Z-J-G-Cii-E-D-F-Y-Z-O-Ciii- (SEQ ID NO: 6)
(Ia);
где U, О, J и Z представляют собой определенное выше.
В одном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ib):
-Ci-Y/M/F/V-N/G-E/Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (SEQ ID NO: 7)
(Ib).
В одном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ic):
-Ci-Y/M/F-N/G-E/Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (SEQ ID NO: 8)
(Ic).
В одном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Id):
-Ci-Y/M-N-E/Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (SEQ ID NO: 9)
(Id).
В одном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ie):
-Ci-Y-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07) (SEQ ID NO: 2)
(Ie).
В еще одном варианте осуществления пептид формулы (I) включает последовательность, выбранную из:
-Ci-Y-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07) (SEQ ID NO: 2);
-Ci-M-N-Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-12) (SEQ ID NO: 10);
-Ci-F-G-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-02) (SEQ ID NO: 11);
-Ci-V-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-03) (SEQ ID NO: 12);
-Ci-F-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-04) (SEQ ID NO: 13);
-Ci-Y-N-E-Y-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07-N057) (SEQ ID NO: 14); и
-Ci-Y-N-E-W-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-44-N002) (SEQ ID NO: 15).
Пептиды, соответствующие данному варианту осуществления, идентифицированы как являющиеся сильными кандидатами в отношении последующего созревания аффинности по отношению к домену гемопексина MT1-MMP (смотри пример 1 и таблицы 1 и 8).
В еще одном варианте осуществления пептид формулы (I) включает последовательность, выбранную из:
-Ci-Y-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07) (SEQ ID NO: 2); и
-Ci-M-N-Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-12) (SEQ ID NO: 10).
Пептиды, соответствующие данному варианту осуществления, идентифицированы как являющиеся самыми высокоаффинными кандидатами последующего созревания аффинности по отношению к домену гемопексина MT1-MMP, для синтеза последовательностей бициклического каркаса, и количественного измерения аффинности с использованием экспериментов по конкурентному связыванию (смотри пример 1 и таблицы 1-3).
В еще одном варианте осуществления пептид формулы (I) включает последовательность, выбранную из -Ci-Y-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07) (SEQ ID NO: 2). Пептид, соответствующий данному варианту осуществления, идентифицирован как являющийся наиболее сильным и стабильным членом семейства пептидных лигандов формулы (I) (смотри примеры с 1 по 4).
В одном варианте осуществления некоторые пептидные лиганды по изобретению являются полностью перекрестно реактивными с MT1-MMP мыши, собаки, яванской макаки и человека. В еще одном варианте осуществления изобретения конкретно проиллюстрированные пептидные лиганды по изобретению являются полностью перекрестно реактивными с MT1-MMP мыши, собаки, яванской макаки и человека. Например, в настоящем документе представлены данные, которые демонстрируют, что как нестабилизированные, так и стабилизированные производные 17-69-07 (т.е. 17-69-07-N219 и 17-69-07-N241) являются полностью перекрестно реактивными (смотри таблицу 13).
В еще одном варианте осуществления пептидный лиганд по изобретению является селективным для MT1-MMP, но не взаимодействует перекрестно с ММР-1, ММР-2, ММР-15 и ММР-16. В настоящем документе представлены данные, которые показывают, что последовательность каркаса 17-69-07 и стабилизированный вариант 17-69-07-N258 исключительно селективны для MT1-MMP (смотри таблицу 14).
Преимущества пептидных лигандов
Некоторые бициклические пептиды по настоящему изобретению имеют ряд полезных свойств, которые позволяют рассматривать их в качестве подходящих молекул, подобных лекарственным, для инъекции, ингаляции, назального, глазного, перорального или местного введения. Такие полезные свойства включают в себя:
Перекрестную реактивность с другими видами. Это типичное требование доклинической оценки фармакодинамики и фармакокинетики;
Стабильность в отношении протеаз. Бициклические пептидные лиганды должны демонстрировать идеальную устойчивость в отношении протеаз плазмы, эпителиальных («заякоренных на мембране») протеаз, протеаз желудка и кишечника, протеаз легочной поверхности, внутриклеточных протеаз и тому подобного. Стабильность в отношении протеаз должна поддерживаться между различными видами так, что бициклический ведущий кандидат может быть разработан на животных моделях, а также с уверенностью может вводиться человеку;
Желательный профиль растворимости. Это зависит от доли заряженных и гидрофильных относительно гидрофобных остатков и внутри-/межмолекулярных H-связей, что важно в целях составления и всасывания; и
Оптимальный период полужизни в циркуляторном русле. В зависимости от клинических показаний и схемы лечения может быть желательна разработка бициклического пептида для короткой экспозиции в условиях лечения острого заболевания или разработка бициклического пептида с повышенным сохранением в кровотоке, и, следовательно, оптимальным для лечения заболеваний с более хроническим течением. Другие факторы, управляющие желаемым периодом полужизни в плазме, подчиняются требованиям устойчивого воздействия для достижения максимальной терапевтической эффективности при сравнении с сопровождающейся токсичностью из-за длительного контакта с агентом.
Фармацевтически приемлемые соли
Следует иметь в виду, что солевые формы находятся в пределах объема настоящего изобретения, и ссылки на соединения формулы (I) включают солевые формы указанных соединений.
Соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основную или кислотную часть, с помощью обычных химических методов, таких как методы, описанные в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Как правило, такие соли могут быть получены путем взаимодействия форм свободных кислот или оснований этих соединений с соответствующим основанием или кислотой в воде или в органическом растворителе, или в смеси их обоих.
Аддитивные соли кислоты (моно- или ди-соли) могут быть образованы с широким спектром кислот, как неорганических, так и органических. Примеры аддитивных солей кислоты включают моно- или ди-соли, образованные с кислотой, выбранной из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+)-камфорной кислот, камфорсульфокислоты, (+)-(1S)-камфор-10-сульфокислоты, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной кислот, этан-1,2-дисульфокислоты, этансульфокислоты, 2-гидроксиэтансульфокислоты, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, галогенводородных кислот (например, бромистоводородной, хлористоводородной, йодистоводородной), изетионовой, молочной (например, (+)-L-молочной, (±)-DL-молочной), лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной кислот, метансульфокислоты, нафталин-2-сульфокислоты, нафталин-1,5-дисульфокислоты, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, пировиноградной, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-винной, тиоциановой кислот, п-толуолсульфокислоты, ундециленовой и валериановой кислот, а также ацилированных аминокислот и катионообменных смол.
Одна конкретная группа солей состоит из солей, образованных с уксусной, соляной, йодистоводородной, фосфорной, азотной, серной, лимонной, молочной, янтарной, малеиновой, яблочной, изетионовой, фумаровой кислотами, бензолсульфокислотой, толуолсульфокислотой, серной кислотой, метансульфокислотой (мезилатом), этансульфокислотой, нафталинсульфокислотой, валериановой, пропановой, бутановой, малоновой, глюкуроновой и лактобионовой кислотами. Одна конкретная соль представляет собой гидрохлоридную соль. Другая конкретная соль представляет собой ацетатную соль.
Если соединение является анионным или имеет функциональную группу, которая может быть анионной (например, -COOH может представлять собой -COO-), то соль может быть образована с органическим или неорганическим основанием, генерируя подходящий катион. Примеры подходящих неорганических катионов включают, но не ограничиваются этим, ионы щелочных металлов, такие как Li+, Na+ и K+, катионы щелочноземельных металлов, такие как Ca2+ и Mg2+, и другие катионы, такие как Al3+ или Zn+. Примеры подходящих органических катионов включают, но не ограничиваются этим, ион аммония (т.е., NH4+) и замещенные ионы аммония (например, NH3R+, NH2R2+, NHR3+, NR4+). Примерами некоторых подходящих замещенных ионов аммония являются ионы, происходящие от: метиламина, этиламина, диэтиламина, пропиламина, дициклогексиламина, триэтиламина, бутиламина, этилендиамина, этаноламина, диэтаноламина, пиперазина, бензиламина, фенилбензиламина, холина, меглумина и трометамина, а также аминокислот, таких как лизин и аргинин. Примером обычного иона четвертичного аммония является N(CH3)4+.
Если соединения формулы (I) содержат аминогруппу, они могут образовывать соли четвертичного аммония, например, путем взаимодействия с алкилирующим агентом в соответствии с методами, хорошо известными специалисту в данной области техники. Такие соединения четвертичного аммония находятся в пределах объема формулы (I).
Модифицированные производные
Следует иметь в виду, что модифицированные производные пептидных лигандов, как определено в данном описании, входят в объем настоящего изобретения. Примеры таких подходящих модифицированных производных включают одну или более модификаций, выбранных из N-концевых и/или С-концевых модификаций; замену одного или более аминокислотных остатков одним или более неприродными аминокислотными остатками (например, замену одного или более полярных аминокислотных остатков одной или более изостерическими или изоэлектронными аминокислотами; замену одного или более остатков неполярной аминокислоты другими неприродными изостерическими или изоэлектронными аминокислотами); добавление спейсерной группы; замену одного или более аминокислотных остатков, чувствительных к окислению, одним или более аминокислотных остатков, устойчивых к окислению; замену одного или более аминокислотных остатков аланином, замену одного или более остатков L-аминокислот одним или более остатками D-аминокислот; N-алкилирование одной или более амидных связей в бициклическом пептидном лиганде; замену одной или более пептидных связей заместительной связью; модификацию длины пептидного каркаса; замену водорода на альфа-углероде одного или более аминокислотных остатков, другой химической группой, модификации аминокислот, таких как цистеин, лизин, глутамат/аспартат и тирозин, соответствующим амином, тиолом, карбоновой кислотой и взаимодействующими с фенолом реагентами, так чтобы функциализировать указанные аминокислоты, и введение или замену аминокислот, которые вводят ортогональную реакционную способность, которые подходят для функционализации, например, аминокислот, несущих азидные или алкинные группы, которые позволяют функционализировать несущие алкин или азид части, соответственно.
В одном варианте осуществления изобретения модифицированное производное включает модификацию аминокислоты в положении 1 и/или 9. В настоящем документе представлены данные, которые показывают, что эти положения, особенно там, где присутствует тирозин, наиболее восприимчивы к протеолитической деградации.
В одном варианте осуществления изобретения модифицированное производное включает N-концевую и/или С-концевую модификацию. В дополнительном варианте осуществления модифицированное производное включает модификацию N-конца с использованием подходящих взаимодействующих с амином реагентов, и/или модификацию С-конца с использованием подходящих взаимодействующих с карбоксилом реагентов. В еще одном варианте осуществления указанная модификация N-конца или С-конца включает добавление эффекторной группы, включая, но, не ограничиваясь этим, цитотоксический агент, радиохелатор или хромофор.
В дополнительном варианте осуществления изобретения модифицированное производное включает модификацию N-конца. В еще одном варианте осуществления модификация N-конца включает N-концевую ацетильную группу, такую как в 17-69-07-N004, раскрытом в настоящем документе. В этом варианте осуществления N-концевая группа цистеина (группа, обозначаемая в настоящем документе как Ci) кепирована уксусным ангидридом или другими подходящими реагентами в процессе пептидного синтеза, что ведет к молекуле, которая ацетилирована с N-конца. Этот вариант осуществления обеспечивает преимущество удаления потенциальной точки узнавания аминопептидазами и исключает возможность деградации бициклического пептида.
В альтернативном варианте осуществления N-концевая модификация включает добавление молекулярной спейсерной группы, которая облегчает конъюгацию эффекторных групп и сохранение активности бициклического пептида в отношении его мишени, такой как группа Ala, G-Sar10-A или bAla-Sar10-A. Данные, представленные в настоящем документе, показывают, что добавление этих групп к бициклическому пептиду 17-69-07 не изменяет активности в отношении белка-мишени (смотри таблицы 11-12).
В другом варианте осуществления изобретения модифицированное производное включает С-концевую модификацию. В еще одном варианте осуществления С-концевая модификация включает амидную группу. В этом варианте осуществления С-концевая группа цистеина (группа, обозначаемая в настоящем документе как Ciii) синтезируется в виде амида во время пептидного синтеза, ведущего к молекуле, которая амидирована с С-конца. Этот вариант осуществления обеспечивает преимущество в виде удаления потенциальной точки узнавания карбоксипептидазой и уменьшает возможность протеолитической деградации бициклического пептида.
В одном варианте осуществления изобретения модифицированное производное включает замену одного или более аминокислотных остатков одним или более неприродными аминокислотными остатками. В этом варианте осуществления могут быть выбраны неприродные аминокислоты, имеющие изостерические/изоэлектронные боковые цепи, которые как не узнаются протеазами, вызывающими деградацию, так и не имеют никакого вредного воздействия на активность в отношении мишени.
В качестве альтернативы неприродные аминокислоты могут быть использованы как имеющие ограниченные аминокислотные боковые цепи, так что протеолитический гидролиз близлежащей пептидной связи является конформационно и стерически затрудненным. В частности это касается аналогов пролина, громоздких боковых цепей, дизамещенных производных Cα- (например, аминоизомасляной кислоты, Aib) и циклоаминокислот, простым производным которых является аминоциклопропилкарбоновая кислота.
В одном варианте осуществления изобретения остатком неприродной аминокислоты замещают в положении 4. В настоящем описании представлены данные, которые показывают, что количество остатков неприродных аминокислот хорошо переносится в этом положении (смотри таблицу 8). В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 4, выбраны из: 1-нафтилаланина; 2-нафтилаланина; циклогексилглицина, фенилглицина; трет-бутилглицина; 3,4-дихлорфенилаланина; циклогексилаланина; и гомофенилаланина.
В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 4, выбраны из: 1-нафтилаланина; 2-нафтилаланина; и 3,4-дихлорфенилаланина. В настоящем описании представлены данные, которые показывают, что эти замены повышают аффинность по сравнению с немодифицированной последовательностью дикого типа (смотри таблицу 8).
В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 4, выбраны из: 1-нафтилаланина. В настоящем описании представлены данные, которые показывают, что эта замена обеспечивает наибольшую величину повышения аффинности (более чем в 7 раз) по сравнению с диким типом (смотри таблицу 8).
В одном варианте осуществления изобретения остаток неприродной аминокислоты вводят в положение 9 и/или 11. В настоящем описании представлены данные, которые показывают, что количество остатков неприродных аминокислот хорошо переносится в этих положениях (смотри таблицу 9).
В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 9, выбраны из: 4-бромфенилаланина, пентафторфенилаланина.
В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 11, выбраны из: трет-бутилглицина.
В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 9, выбраны из: 4-бромфенилаланина. В настоящем описании представлены данные, которые показывают изменение точки Tyr9 узнавания протеолитическими ферментами (смотри таблицу 9).
В еще одном варианте осуществления неприродные аминокислотные остатки, такие как присутствующие в положении 11, выбраны из: трет-бутилглицина. В настоящем описании представлены данные, которые демонстрируют повышение активности и сильную защиту соседнего аминокислотного каркаса от протеолитического гидролиза за счет стерических препятствий (смотри таблицу 9).
В одном варианте осуществления изобретения модифицированное производное включает множество указанных выше модификаций, например, 2, 3, 4 или 5, или более модификаций. В другом варианте осуществления изобретения модифицированное производное включает 2, 3, 4 или 5, или более следующих модификаций, таких как все последующие 5 модификаций: D-аланин в положении 1 и 5, 1-нафтилаланин в положении 4, 4-бромфенилаланин в положении 9 и трет-бутилглицин в положении 11. В настоящем описании представлены данные, которые демонстрируют, что это мультизамещение (17-69-07-N252; 17-69-07-N244 и 17-69-07-N255) переносится согласованно с активностью, которая превосходит активность дикого типа (смотри таблицы 10-12). В еще одном варианте осуществления изобретения модифицированное производное включает следующие модификации: D-аланин в положении 1 и 5, 1-нафтилаланин в положении 4 и трет-бутилглицин в положении 11. В настоящем описании представлены данные, которые демонстрируют, что это мультизамещение (17-69-07-N239) переносится согласованно с активностью, которая превосходит активность дикого типа (смотри таблицу 11).
В одном варианте осуществления изобретения модифицированное производное включает добавление спейсерной группы. В дополнительном варианте осуществления изобретения модифицированное производное включает добавление спейсерной группы к N-концевому цистеину (Ci) и/или к С-концевому цистеину (Ciii).
В одном варианте осуществления изобретения модифицированное производное включает замену одного или более аминокислотных остатков, чувствительных к окислению, одним или более аминокислотными остатками, устойчивыми к окислению. В другом варианте осуществления изобретения модифицированное производное включает замену остатка триптофана остатком нафтилаланина или аланина. Этот вариант осуществления обеспечивает преимущество в виде улучшения профиля фармацевтической стабильности полученного бициклического пептидного лиганда.
В одном варианте осуществления изобретения модифицированное производное включает замену одного или более заряженных аминокислотных остатков одним или более гидрофобными аминокислотными остатками. В альтернативном варианте осуществления изобретения модифицированное производное включает замену одного или более гидрофобных аминокислотных остатков одним или более заряженными аминокислотными остатками. Правильный баланс заряженных остатков относительно гидрофобных аминокислотных остатков является важной характеристикой бициклических пептидных лигандов. Например, гидрофобные аминокислотные остатки влияют на степень связывания с белками плазмы и, следовательно, концентрацию свободной доступной фракции в плазме, в то время как заряженные аминокислотные остатки (в частности, аргинина) могут влиять на взаимодействие пептида с фосфолипидными мембранами на клеточных поверхностях. Оба они в сочетании могут влиять на период полужизни, объем распределения и экспозицию пептидного лекарственного средства, и все это может быть адаптировано в соответствии с конечными клиническими требованиями. Кроме того, правильное сочетание и количество заряженных остатков относительно гидрофобных аминокислотных остатков может уменьшить раздражение в месте инъекции (если пептидное лекарство вводятся подкожно).
В одном варианте осуществления изобретения модифицированное производное включает замену одного или более остатков L-аминокислот одним или более остатками D-аминокислот. Этот вариант, как полагают, увеличивает протеолитическую стабильность за счет стерических затруднений и склонности D-аминокислот к стабилизации конформаций β-изгиба (Tugyi et al (2005) PNAS, 102(2), 413-418).
В дополнительном варианте осуществления аминокислотный остаток в положении 1 заменяют на D-аминокислоту, такую как D-аланин. В настоящем описании представлены данные, которые демонстрируют сохранение активности без последующей деградации (смотри таблицу 6).
В другом варианте осуществления изобретения аминокислотный остаток в положении 5 заменяют на D-аминокислоту, такую как D-аланин или D-аргинин. В настоящем описании представлены данные, которые демонстрируют сохранение активности без последующей деградации (смотри таблицу 7).
В одном варианте осуществления изобретения модифицированное производное включает удаление любых аминокислотных остатков и замещение аланинами. Этот вариант осуществления обеспечивает преимущество в виде удаления потенциального сайта(ов) протеолитической атаки.
Следует отметить, что каждая из указанных выше модификаций служит для намеренного улучшения активности или стабильности пептида. Дальнейшее улучшение активности, основанное на модификациях, может быть достигнуто с помощью следующих механизмов:
- включения гидрофобных частей, которые используют гидрофобный эффект и приводят к снижению скоростей диссоциации, так что достигается более высокое сродство;
- включения заряженных групп, которые используют ионные взаимодействия на большом расстоянии, что ведет к более высоким скоростям ассоциации и к более высокому сродству (смотри, например, Schreiber et al, Rapid, electrostatically assisted association of proteins (1996), Nature Struct. Biol. 3, 427-31); и
- включения дополнительного ограничения в пептид, с помощью, например, корректного ограничения боковых цепей аминокислот таким образом, что потеря энтропии является минимальной при связывании с мишенью, ограничения углов кручения каркаса таким образом, что потеря энтропии является минимальной при связывании с мишенью и введения дополнительных циклов в молекулу по идентичным причинам.
(В качестве обзоров смотри Gentilucci et al, Curr. Pharmaceutical Design, (2010), 16, 3185-203, и Nestor et al, Curr. Medicinal Chem (2009), 16, 4399-418).
Изотопные варианты
Настоящее изобретение включает все фармацевтически приемлемые меченные (радио)изотопами соединения по настоящему изобретению, т.е. соединения формулы (I), где один или более атомов заменены атомами, имеющими тот же атомный номер, но атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе, и соединения формулы (I), к которым присоединены хелатирующие группы металла (называемые «эффекторами»), которые способны удерживать соответствующие (радио)изотопы, и соединения формулы (I), в которых определенные функциональные группы ковалентно заменены соответствующими (радио)изотопами или меченными изотопами функциональными группами.
Примеры изотопов, подходящих для включения в соединения по настоящему изобретению, включают изотопы водорода, такие как 2H (D) и 3H (T), углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I, 125I и 131I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, серы, такие как 35S, меди, такие как 64Cu, галлия, такие как 67Ga или 68Ga, иттрия, такие как 90Y и лютеция, такие как 177Lu, и висмута, такие как 213Bi.
Некоторое меченные изотопами соединения формулы (I), например, те, которые включают радиоактивный изотоп, пригодны для лекарств и/или для исследований тканевого распределения субстрата, а также для клинической оценки наличия и/или отсутствия мишени МТ1-ММР в патологических тканях, таких как опухоли, и в других местах. Соединения формулы (I) дополнительно могут иметь ценные диагностические свойства так, что они могут быть использованы для обнаружения или идентификации образования комплекса между меченым соединением и другими молекулами, пептидами, белками, ферментами или рецепторами. В методах определения или идентификации можно использовать соединения, которые помечены агентами-метками, такими как радиоизотопы, ферменты, флуоресцентные вещества, светящиеся вещества (например, люминол, производные люминола, люциферин, акворин и люцифераза), и т.д. Радиоактивные изотопы тритий, т.е. 3H (T), и углерод-14, т.е. 14С, особенно полезны для этой цели ввиду легкости их включения и готовых средств обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H (D), может давать определенные терапевтические преимущества в результате более высокой метаболической стабильности, например, увеличения периода полужизни in vivo или снижения требуемой дозировки лекарства и, следовательно, может быть предпочтительным в некоторых обстоятельствах.
Замещение излучающими позитроны изотопами, такими как 11C, 18F, 15O и 13N, может быть полезным при исследованиях с помощью позитронно-эмиссионной томографии (ПЭТ) для проверки распределения в мишенях.
Включение изотопов в хелатирующие эффекторные группы металлов, таких как 64Cu, 67Ga, 68Ga, и 177Lu, может быть полезным для визуализации специфических для опухоли антигенов с применением визуализации с помощью ПЭТ или SPECT. В частности, такие данные по биораспределению представлены в данном описании в примере 3.
Включение изотопов в хелатирующие эффекторные группы металлов, таких как, но, не ограничиваясь этим, 90Y, 177Lu и 213Bi, может создавать возможность целенаправленной лучевой терапии, в результате чего соединения формулы (I), несущие металл-хелатор, переносят терапевтический радионуклид в направлении белка-мишени и места действия.
Меченные изотопами соединения формулы (I) обычно могут быть получены традиционными способами, известными специалистам в данной области техники, или способами, аналогичными описанным в прилагаемых примерах с использованием подходящего меченного изотопом реагента вместо немеченого реагента, используемого ранее.
Связывающая активность
Специфичность в контексте настоящего описания относится к способности лиганда связываться или иным образом взаимодействовать с узнаваемой мишенью за исключением частей, которые похожи на мишень. Например, специфичность может относиться к способности лиганда ингибировать взаимодействие фермента человека, но не гомологичного фермента от другого вида. Используя подход, описанный в настоящем документе, можно модулировать специфичность, то есть увеличивать или уменьшать ее, таким образом, чтобы делать лиганды более или менее способными взаимодействовать с гомологами или паралогами предназначенной мишени. Специфичность не предназначена представлять собой синоним активности, сродства или авидности, и сила действия лиганда на свою мишень (такая как, например, аффинность связывания или уровень ингибирования) не обязательно связана с его специфичностью.
Связывающая активность при использовании в настоящем документе относится к количественным измерениям связывания, полученным из анализа связывания, например, как описано в настоящем документе. Таким образом, связывающая активность относится к количеству пептидного лиганда, которое связано при данной целевой концентрации.
Мультиспецифичность представляет собой способность связываться с двумя или более мишенями. Как правило, связывающие пептиды способны связываться с одной мишенью, такой как эпитоп в случае антитела, из-за их конформационных свойств. Однако могут быть разработаны пептиды, которые могут связываться с двумя или более мишенями; например, антитела с двойной специфичностью, как известно в данной области техники, как указано выше. В настоящем изобретении пептидные лиганды могут обладать способностью связываться с двумя или более мишенями, и, следовательно, являться мультиспецифичными. Соответственно, они связываются с двумя мишенями, и обладают двойной специфичностью. Связывание может быть независимым, что должно означать, что сайты связывания мишеней на пептиде структурно не препятствуют связыванию одной или другой мишени. В этом случае обе мишени могут быть связаны независимо друг от друга. В более общем плане ожидается, что связывание одной мишени должно, по меньшей мере, частично препятствовать связыванию другой.
Существует фундаментальное различие между лигандом с двойной специфичностью и лигандом со специфичностью, которая охватывает две взаимосвязанных мишени. В первом случае, лиганд является специфичным для обеих мишеней по отдельности и взаимодействует с каждой специфическим образом. Например, первая петля в лиганде может связываться с первой мишенью, а вторая петля со второй мишенью. Во втором случае лиганд не является специфическим, поскольку он не делает различий между этими двумя мишенями, например, за счет взаимодействия с эпитопом мишеней, который является общим для них обеих.
В контексте настоящего изобретения допускается, что лиганд, который обладает активностью в отношении, например, мишени и ортолога, может представлять собой биспецифический лиганд. Тем не менее, в одном варианте осуществление изобретения лиганд не является биспецифическим, но имеет менее точную специфичность, так что он связывает как мишень, так и один или более ортологов. В общем, лиганд, который не был выбран как против мишени, так и против ее ортолога, менее вероятно будет биспецифическим из-за отсутствия селективного давления в сторону биспецифичности. Длина петли в бициклическом пептиде может иметь решающее значение в обеспечении оптимизированной поверхности связывания таким образом, что может быть получена хорошая перекрестная активность в отношении мишени и ортолога при сохранении при этом высокой селективности в отношении менее родственных гомологов.
Если лиганды действительно являются биспецифичными, в одном варианте осуществления, по меньшей мере, одна из специфичностей лигандов к мишени должна быть общей среди выбранных лигандов, и уровень этой специфичности можно модулировать с помощью способов, раскрытых в настоящем описании. Для второй или дополнительной специфичности не требуется совместность, и она не должна быть предметом методов, изложенных в данном документе.
Мишень представляет собой молекулу или ее часть, с которой связываются или иным образом взаимодействуют пептидные лиганды. Хотя связывание рассматривается как необходимое условие для активности большинства классов, и может представлять собой активность саму по себе, предусматриваются другие виды активности. Таким образом, в настоящем изобретении не требуется прямого или косвенного измерения связывания.
Молекулярный каркас представляет собой любую молекулу, которая способна соединить пептид во множественных точках для придания одной или более структурных характеристик пептиду. Предпочтительно, чтобы молекулярный каркас включал, по меньшей мере, три точки присоединения пептида, обозначаемых как каркасные реакционные группы. Эти группы способны вступать в реакцию с остатками цистеина (Ci, Cii и Ciii) на пептиде с образованием ковалентной связи. Они не только образуют дисульфидную связь, которая вовлечена в восстановительное расщепление и сопутствующий распад молекулы, но и образуют стабильные, ковалентные тиоэфирные связи. Предпочтительные структуры молекулярных каркасов описаны ниже.
Молекулярные каркасы
Молекулярные каркасы описаны, например, в патенте WO 2009/098450 и в приведенных в нем ссылках, в частности, в патентах WO 2004/077062 и WO 2006/078161.
Как отмечалось в вышеприведенных документах, молекулярный каркас может быть небольшой молекулой, такой как небольшая органическая молекула.
В одном варианте осуществления молекулярный каркас может представлять собой или может основываться на природных мономерах, таких как нуклеозиды, сахара или стероиды. Например, молекулярный каркас может включать короткий полимер таких единиц, такой как димер или тример.
В одном варианте осуществления молекулярный каркас представляет собой соединение с известной токсичностью, например, с низкой токсичностью. Примеры подходящих соединений включают холестерины, нуклеотиды, стероиды, или существующие лекарства, такие как тамазепам.
В одном варианте осуществления молекулярный каркас может представлять собой макромолекулу. В одном варианте осуществления молекулярный каркас представляет собой макромолекулу, состоящую из аминокислот, нуклеотидов или углеводов.
В одном варианте осуществления молекулярный каркас включает реакционные группы, способные вступать в реакцию с функциональной группой(ами) полипептида с образованием ковалентных связей.
Молекулярный каркас может включать химические группы, которые образуют связь с пептидом, такие как амины, тиолы, спирты, кетоны, альдегиды, нитрилы, карбоновые кислоты, сложные эфиры, алкены, алкины, азиды, ангидриды, сукцинимиды, малеимиды, алкилгалогениды и ацилгалогениды.
В одном варианте осуществления молекулярный каркас может включать или может состоять из трис(бромметил)бензола, особенно 1,3,5-трис(бромметил)бензола («TBMB») или его производного.
В одном варианте осуществления молекулярный каркас представляет собой 2,4,6-трис(бромметил)мезитилен. Эта молекула сходна с 1,3,5-трис(бромметил)бензолом, но содержит три дополнительные метильные группы, присоединенные к бензольному кольцу. Это дает преимущество в том, что дополнительные метильные группы могут образовывать дополнительные контакты с полипептидом и, следовательно, добавлять дополнительное структурное ограничение.
Молекулярный каркас по данному изобретению содержит химические группы, которые позволяют функциональным группам полипептида, кодируемого библиотекой по изобретению, образовывать ковалентные связи с молекулярным каркасом. Указанные химические группы выбраны из широкого диапазона функциональных групп, включая амины, тиолы, спирты, кетоны, альдегиды, нитрилы, карбоновые кислоты, сложные эфиры, алкены, алкины, ангидриды, сукцинимиды, малеимиды, азиды, алкилгалогениды и ацилгалогениды.
Реактивные группы каркаса, которые могут быть использованы на молекулярном каркасе для взаимодействия с тиоловыми группами цистеинов, представляют собой алкилгалогениды (или также обозначаемые как галогеналканы или галоалканы). Примеры включают бромметилбензол (реактивная группа каркаса, иллюстрируемая TBMB) или йодацетамид. Другие реактивные группы каркаса, которые используются для селективного присоединения соединений к цистеинам в белках, представляют собой малеимиды. Примеры малеимидов, которые могут быть использованы в качестве молекулярных каркасов в соответствии с изобретением, включают: трис-(2-малеимидэтил)амин, трис(2-малеимидэтил)бензол, трис(малеимид)бензол. Селеноцистеин также представляет собой природную аминокислоту, которая имеет сходную с цистеином реакционную способность, и он может быть использован для тех же самых реакций. Таким образом, там, где упоминается цистеин, это обычно приемлемо для замены на селеноцистеин, если в контексте не предполагается иное.
Эффекторные и функциональные группы
В соответствии с другим аспектом настоящего изобретения предлагается конъюгат лекарственного средства, включающий пептидный лиганд, как определено в настоящем документе, конъюгированный с одной или более эффекторными и/или функциональными группами.
Эффекторные и/или функциональные группы могут быть присоединены, например, к N- и/или С-концу полипептида, к аминокислоте в полипептиде или к молекулярному каркасу.
Подходящие эффекторные группы включают антитела и их части или фрагменты. Например, эффекторная группа может включать константную область легкой цепи антитела (CL), домен CH1 тяжелой цепи антитела, домен CH2 тяжелой цепи антитела, домен CH3 тяжелой цепи антитела или любое их сочетание в дополнение к одному или более доменам константной области. Эффекторная группа может также включать шарнирную область антитела (такая область обычно выявляется между доменами CH1 и CH2 молекулы IgG).
В еще одном варианте осуществления данного аспекта изобретения эффекторная группа в соответствии с настоящим изобретением представляет собой Fc-область молекулы IgG. Предпочтительно, чтобы конъюгат пептидный лиганд-эффекторная группа в соответствии с настоящим изобретением включал или состоял из пептидного лиганда, соединенного с Fc, имеющего tβ период полужизни порядка дня или более, двух дней или более, 3 дней или более, 4 дней или более, 5 дней или более, 6 дней или более, или 7 дней или более. Наиболее предпочтительно, чтобы пептидный лиганд в соответствии с настоящим изобретением включал или состоял из пептидного лиганда, соединенного с Fc, имеющего tβ период полужизни порядка дня или более.
Функциональные группы включают в целом связывающие группы, лекарства, реакционные группы для присоединения других частей, функциональные группы, которые помогают захватывать макроциклические пептиды в клетки, и тому подобное.
Способность пептидов проникать в клетки должна позволять пептидам против внутриклеточных мишеней действовать эффективно. Мишени, которые могут быть доступны для пептидов при их способности проникать в клетки, включают факторы транскрипции, внутриклеточные сигнальные молекулы, такие как тирозинкиназы, и молекулы, вовлеченные в путь апоптоза. Функциональные группы, которые дают возможность проникновения в клетки, включают пептидные или химические группы, которые добавляются либо к пептиду, либо к молекулярному каркасу. Пептиды представляют собой такие пептиды как происходящие, например, от VP22, HIV-Tat, гомеобоксного белка дрозофилы (Antennapedia), например, как описано у Chen and Harrison, Biochemical Society Transactions (2007) Volume 35, part 4, p821; Gupta et al. in Advanced Drug Discovery Reviews (2004) Volume 57 9637. Примеры коротких пептидов, которые, как было показано, являются эффективными в отношении перемещения через плазматические мембраны, включают пептид из 16 аминокислот пенетратин белка Drosophila Antennapedia (Derossi et al (1994) J Biol. Chem. Volume 269 p10444), «модельный амфипатический пептид» из 18 аминокислот (Oehlke et al (1998) Biochim Biophys Acts Volume 1414 p127) и богатые аргинином области белка TAT ВИЧ. Непептидные подходы включают использование миметиков небольших молекул или SMOCs, которые могут быть легко присоединены к биомолекулам (Okuyama et al (2007) Nature Methods Volume 4 p153). Другие химические стратегии с добавлением групп гуанидиния к молекулам также усиливают проникновение в клетки (Elson-Scwab et al (2007) J Biol Chem Volume 282 p13585). Молекулы низкой молекулярной массы, такие как стероиды, могут быть добавлены к молекулярному каркасу для улучшения захвата клетками.
Один класс функциональных групп, которые могут быть присоединены к пептидным лигандам, включает антитела и их связывающие фрагменты, такие как Fab, Fv или фрагменты отдельных доменов. В частности, могут быть использованы антитела, которые связываются с белками, способными увеличивать период полужизни пептидного лиганда in vivo.
Могут быть также включены пептиды RGD, которые связываются с интегринами, присутствующими на многих клетках.
В одном варианте осуществления конъюгат пептидный лиганд-эффекторная группа в соответствии с изобретением имеет tβ период полужизни, выбранный из группы, состоящей из: 12 часов или более, 24 часов или более, 2 дней или более, 3 дней или более, 4 дней или более, 5 дней или более, 6 дней или более, 7 дней или более, 8 дней или более, 9 дней или более, 10 дней или более, 11 дней или более, 12 дней или более, 13 дней или более, 14 дней или более, 15 дней или более, или 20 дней или более. Предпочтительно конъюгат пептидный лиганд-эффекторная группа или композиция по настоящему изобретению должны иметь tβ период полужизни в диапазоне от 12 до 60 часов. В другом варианте осуществления tβ период полужизни должен составлять день или более. В еще одном варианте осуществления он должен находиться в диапазоне от 12 до 26 часов.
В одном конкретном варианте осуществления настоящего изобретения функциональная группа выбрана из хелатора металла, который подходит для комплексообразования радиоизотопов металла, востребованных в качестве лекарства. Такие эффекторы при комплексировании с указанными радиоизотопами, могут представлять собой полезные агенты для лечения рака. Подходящие примеры включают DOTA, NOTA, EDTA, DTPA, HEHA, SarAr и другие (Targeted Radionuclide therapy, Tod Speer, Wolters/Kluver Lippincott Williams & Wilkins, 2011).
Возможные эффекторные группы также включают ферменты, например, такие как карбоксипептидаза G2 для использования в терапии ферментом/пролекарством, где пептидный лиганд замещает антитела в ADEPT.
В одном конкретном варианте осуществления настоящего изобретения функциональная группа выбрана из лекарственного средства, такого как цитотоксический агент для лечения рака. Подходящие примеры включают: алкилирующие агенты, такие как цисплатин и карбоплатин, а также оксалиплатин, мехлорэтамин, циклофосфамид, хлорамбуцил, ифосфамид; антиметаболиты, включая аналоги пурина, азатиоприн и меркаптопурин или аналоги пиримидина; растительные алкалоиды и терпеноиды, включая алкалоиды барвинка, такие как винкристин, винбластин, винорелбин и виндезин; подофиллотоксин и его производные этопозид и тенипозид; таксаны, включая паклитаксел, первоначально известный как таксол; ингибиторы топоизомеразы, включая камптотецины; иринотекан и топотекан, и ингибиторы типа II, включая амсакрин, этопозид, этопозида фосфат и тенипозид. Другие агенты могут включать противоопухолевые антибиотики, которые включают иммуносупрессорный дактиномицин (который используется при трансплантациях почки), доксорубицин, эпирубицин, блеомицин, калихеамицины и другие.
В одном дополнительном конкретном варианте осуществления настоящего изобретения цитотоксический агент выбран из мейтанзиноидов (например, DM1) или монометилзамещенных ауристатинов (например, MMAE).
DM1 является цитотоксическим агентом, который представляет собой содержащее тиол производное мейтанзина и имеет следующую структуру:
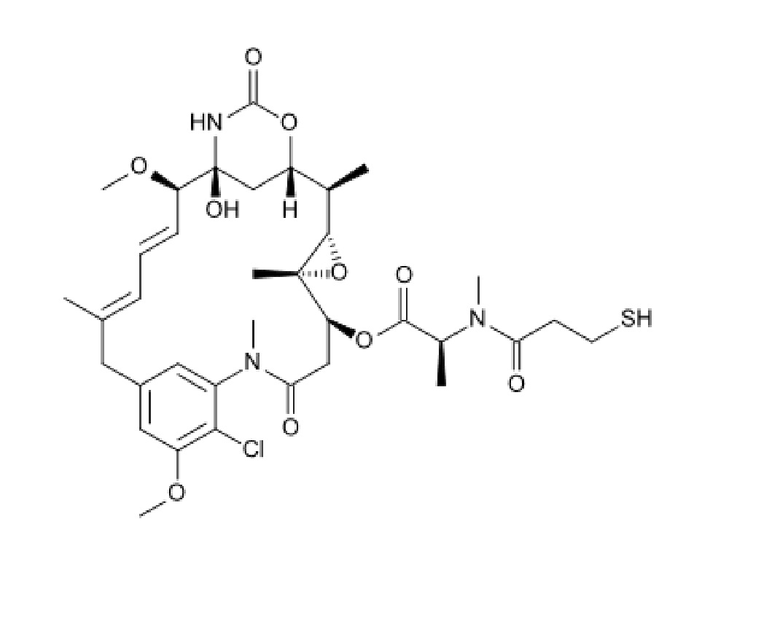
Монометилауристатин Е (ММАЕ) представляет собой синтетический противоопухолевый агент и имеют следующую структуру:
Данные представлены в настоящем описании в примерах 4 и 5, которые демонстрируют эффекты пептидных лигандов, конъюгированных с токсинами, содержащими DM1 или MMAE.
В одном варианте осуществления цитотоксический агент связан с бициклическим пептидом расщепляемой связью, например, дисульфидной связью или связью, чувствительной к протеазам. В дополнительном варианте осуществления группы, прилегающие к дисульфидной связи, модифицированы для контроля блокировки дисульфидной связи и с помощью этого контроля скорости расщепления и сопутствующего высвобождения цитотоксического агента.
Опубликованная работа продемонстрировала потенциал модификации восприимчивости дисульфидной связи к восстановлению путем введения стерического препятствия на каждой стороне дисульфидной связи (Kellogg et al (2011) Bioconjugate Chemistry, 22, 717). Более высокая степень стерического препятствия снижает скорость восстановления внутриклеточным глутатионом, а также внеклеточными (системными) восстанавливающими агентами, вследствие этого уменьшая легкость, с которой токсин высвобождаются, как внутри, так и вне клетки. Таким образом, выбор оптимальной дисульфидной стабильности в циркуляции (что сводит к минимуму нежелательные побочные эффекты токсина) относительно эффективного высвобождения во внутриклеточной среде (что приводит к максимуму терапевтического эффекта) может быть достигнут путем тщательного подбора степени стерического препятствия по обе стороны от дисульфидной связи.
Препятствие по обе стороны от дисульфидной связи модулируется путем введения одной или более метильных групп либо в направляющей части (в настоящем документе в бициклическом пептиде), либо в части токсина молекулярного конструкта.
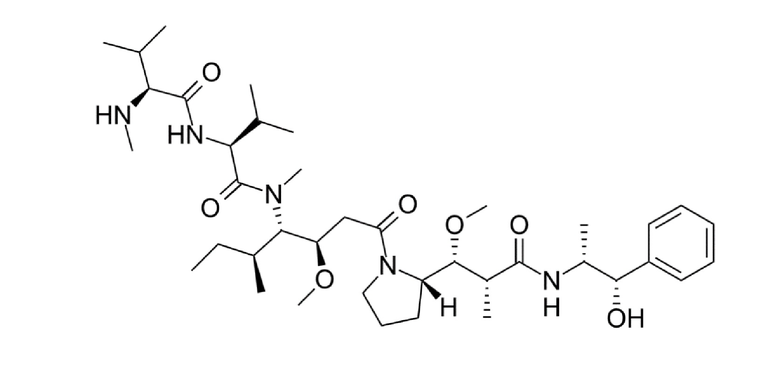
Таким образом, в одном варианте осуществления цитотоксический агент представляет собой мейтанзиноид, выбранный из соединения формулы (II):
в котором n представляет собой целое число, выбранное из от 1 до 10; и
R1 и R2 независимо друг от друга представляют собой водород, C1-6-алкил или карбоциклическую или гетероциклическую группу.
Термин C1-6-алкил, используемый в данном описании, относится к линейной или разветвленной насыщенной углеводородной группе, содержащей от 1 до 6 атомов углерода, соответственно. Примеры таких групп включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил или гексил и тому подобное.
Термины «гетероциклил» и «карбоциклил», используемые в настоящем документе, если только из контекста не следует иное, включают как ароматические, так и неароматические кольцевые системы. Так, например, термин «гетероциклическая группа» и «карбоциклическая группа» включает в свой объем ароматические, неароматические, ненасыщенные, частично насыщенные, полностью насыщенные карбоциклические или гетероциклические кольцевые системы. В общем случае, если из контекста не следует иное, такие группы могут быть моноциклическими или бициклическими (включая конденсированные бициклические группы и бициклические группы мостика) и могут содержать, например, от 3 до 12 членов кольца, более обычно от 5 до 10 членов кольца.
В одном варианте осуществления соединения формулы (II) R1 и R2 независимо друг от друга представляют собой водород или метил.
В одном варианте осуществления соединения формулы (II) n равно 1, и R1 и R2 независимо друг от друга представляют собой водород (то есть мейтанзиновое производное DM1).
В альтернативном варианте осуществления соединения формулы (II) n равно 2, и R1 представляет собой водород, а R2 представляет собой метильную группу (то есть мейтанзиновое производное DM3).
В одном варианте осуществления соединения формулы (II) n равно 1, и R1 и R2 оба представляют собой метильную группу (то есть мейтанзиновое производное DM4).
Следует иметь в виду, что цитотоксический агент формулы (II) может образовывать дисульфидную связь, и в сопряженной структуре с бициклическим пептидом формулы (I) дисульфидная связь между тиолом-токсином (II) и тиолом-бициклическим пептидом (III), вводится с помощью нескольких возможных схем синтеза, две из которых описываются в схеме II или схеме III.
В одном варианте осуществления бициклический пептидный компонент конъюгата имеет структуру, показанную в формуле (III):
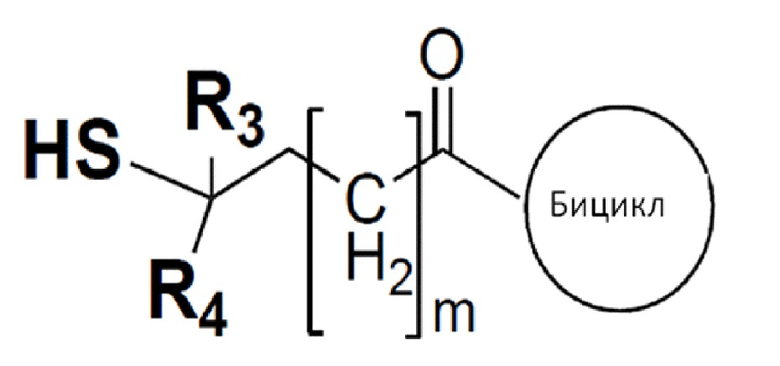
где М представляет собой целое число, выбранное из от 0 до 10, и
R3 и R4 независимо представляют собой водород, C1-6-алкил или карбоциклическую или гетероциклическую группу.
В одном варианте осуществления соединения формулы (III) R3 и R4 независимо представляют собой водород или метил.
Соединения формулы (III), где R3 и R4 оба представляют собой водород, рассматриваются как не имеющие препятствия, и соединения формулы (III), где один или все из R3 и R4 представляют собой метил, рассматриваются как имеющие препятствия.
Следует иметь в виду, что бициклический пептид формулы (III) может образовывать дисульфидную связь, и в сопряженной структуре с цитотоксическим агентом формулы (II) дисульфидная связь между тиолом-токсином (II) и тиолом-бициклическим пептидом (III), вводится с помощью нескольких возможных схем синтеза, одна из которых описывается в схеме II.
В одном варианте осуществления цитотоксический агент связывается с бициклическим пептидом с помощью линкера, определенного в формуле (IV):
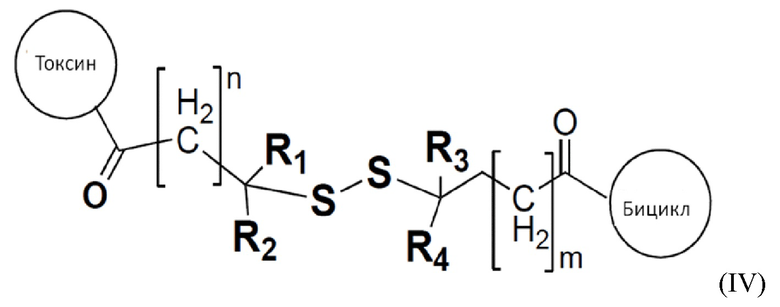
где R1, R2, R3 и R4 представляют собой водород, C1-6-алкил или карбоциклическую или гетероциклическую группу;
токсин относится к любому подходящему цитотоксическому агенту, определенному в настоящем описании;
бицикл представляет собой любой подходящий бициклический пептид, определенный в настоящем описании;
n представляет собой целое число, выбранное из от 1 до 10; и
m представляет собой целое число, выбранное из от 0 до 10.
В одном варианте осуществления R1, R2, R3 и R4 представляют собой водород или метил.
Когда R1, R2, R3 и R4, каждый представляет собой водород, то дисульфидная связь является наименее защищенной и наиболее чувствительна к восстановлению. Когда R1, R2, R3 и R4, каждый представляет собой метил, дисульфидная связь является наиболее защищенной и наименее чувствительна к восстановлению. Частичные замены водорода и метила дают постепенное увеличение устойчивости к восстановлению и сопутствующему отщеплению и высвобождению токсина.
В одном варианте осуществления токсин соединения (IV) представляет собой мейтанзин, и конъюгат включает соединение формулы (V):
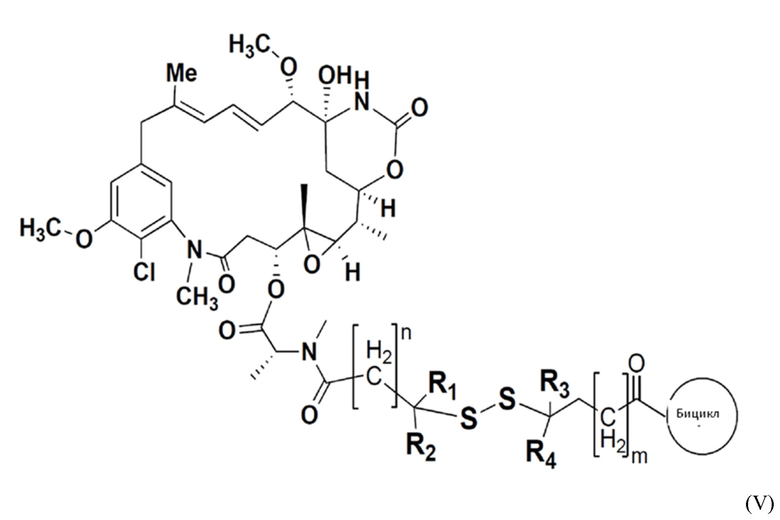
где R1, R2, R3 и R4 представляют собой водород, C1-6-алкил или карбоциклическую или гетероциклическую группу;
бицикл представляет собой любой подходящий бициклический пептид, определенный в настоящем описании;
n представляет собой целое число, выбранное из от 1 до 10; и
m представляет собой целое число, выбранное из от 0 до 10.
В еще одном варианте осуществления соединения формулы (V) n равно 1, и R1, R2, R3 и R4 представляют собой водород, то есть соединение формулы (V)a:
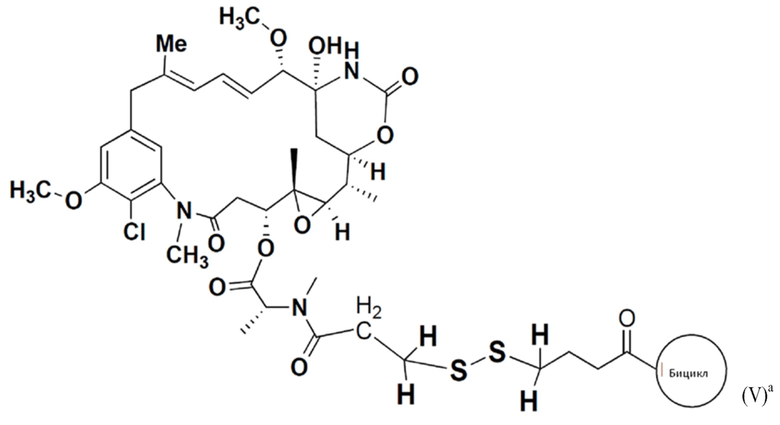
BDC формулы (V)a известен как BT17BDC-17. Незащищенная дисульфидная связь в BDC BT17BDC-17 является эквивалентом BT17BDC-9, в результате чего существует различие в бициклической пептидной части: в BT17BDC-9 используется нестабилизированная последовательность (17-69-07-N219), в то время как в BT17BDC-17 используется стабилизированный бициклический пептидный аналог (17-69-07-N241), который представляет собой конструкт, связанный через амид с токсином-дисульфидом. Это незащищенное производное мейтанзина с n=1 называется DM1.
В еще одном варианте осуществления соединения формулы (V), n равно 1, R1 представляет собой метил, и R2, R3 и R4 каждый представляет собой водород, т.е. соединение формулы (V)b:
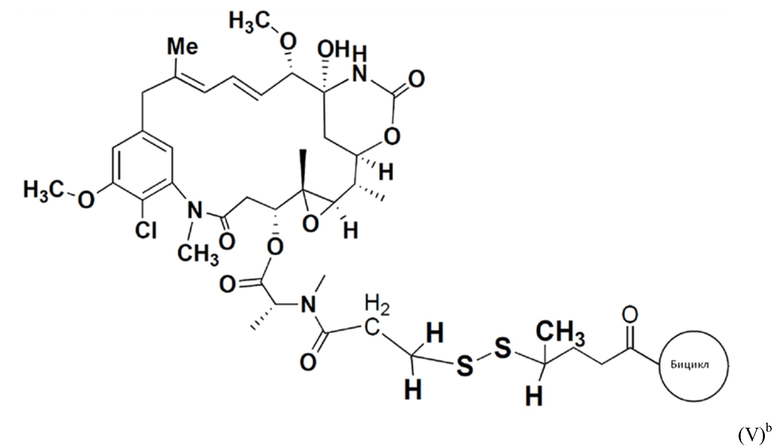
BDC формулы (V)b известен как BT17BDC-18 и содержит одну защитную метильную группу на стороне бициклического пептида, и в контексте конъюгата антительного лекарства дает 7-кратное уменьшение своей чувствительности к восстанавливающему агенту, такому как дитиотреитол (по сравнению с незащищенным дисульфидом) (Kellogg et al (2011) Bioconjugate Chemistry, 22, 717). Пониженная чувствительность к восстановлению коррелирует с более низкой скоростью высвобождения токсина. Это незащищенное производное мейтанзина с n=1 называется DM1. BT17BDC-18 использует стабилизированный бициклический пептидный аналог (17-69-07-N241), который представляет собой конструкт, связанный через амид с токсином-дисульфидом.
В еще одном варианте осуществления соединения формулы (V) n равно 2, R1 и R2 оба представляют собой водород, и R3 и R4 оба представляют собой метил, т.е. соединение формулы (V)c:
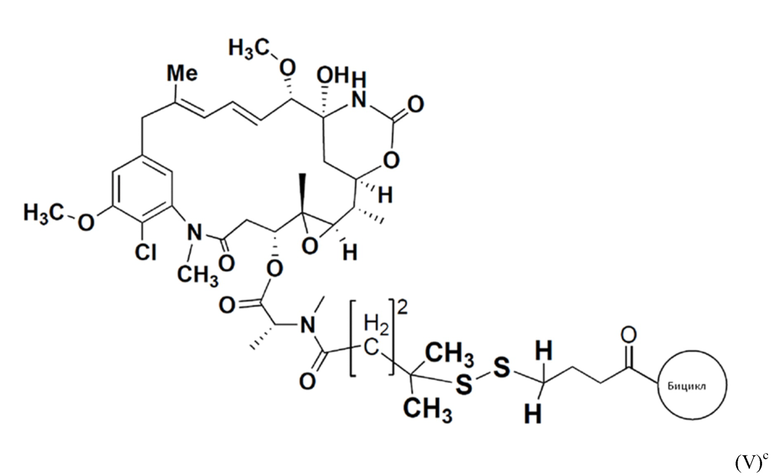
BDC формулы (V)c известен как BT17BDC-19 и содержит две защитные метильные группы на стороне мейтанзина, и в контексте конъюгата антительного лекарства дает 14-кратное уменьшение своей чувствительности к восстанавливающему агенту, такому как дитиотреитол. Пониженная чувствительность к восстановлению коррелирует с более низкой скоростью высвобождения токсина. Это защищенное производное мейтанзина с n=2 называется DM4. BT17BDC-19 использует стабилизированный бициклический пептидный аналог (17-69-07-N241), который представляет собой конструкт, связанный через амид с токсином-дисульфидом.
В еще одном варианте осуществления соединения формулы (V) n равно 2, R1 и R3 оба представляют собой метил, и R2 и R4 оба представляют собой водород, т.е. соединение формулы (V)d:
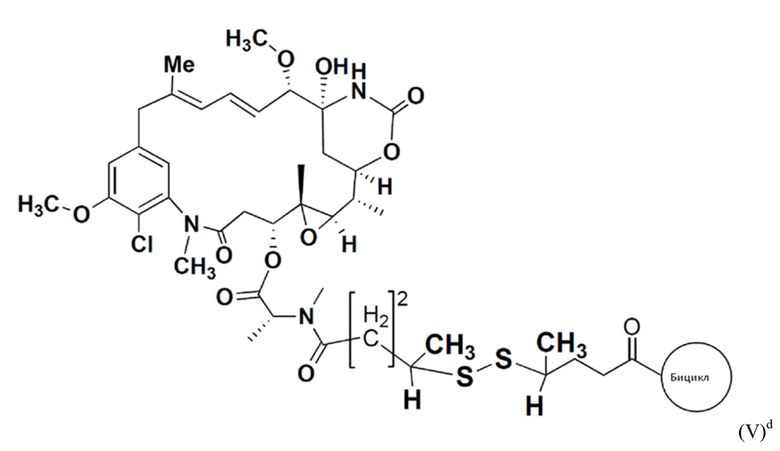
BDC формулы (V)d известен как BT17BDC-20 и содержит одну защитную метильную группу на стороне мейтанзина, и одну защитную метильную группу на стороне бициклического пептида, и в контексте конъюгата антительного лекарства дает 170-кратное уменьшение своей чувствительности к восстанавливающему агенту, такому как дитиотреитол. Пониженная чувствительность к восстановлению коррелирует с более низкой скоростью высвобождения токсина. Это защищенное производное мейтанзина с n=2 называется DM3. BT17BDC-20 использует стабилизированный бициклический пептидный аналог (17-69-07-N241), который представляет собой конструкт, связанный через амид с токсином-дисульфидом.
Действительно в контексте конъюгата антительного лекарства баланс эффективности относительно переносимости в животной модели показал, что его оптимум связан с определенным уровнем защиты, т.е. с защитой, имеющейся у DM4 (Kellogg et al (2011) Bioconjugate Chemistry, 22, 717), которая имеется как таковая также у BT17BDC-19.
В одном варианте осуществления изобретения конъюгат выбран из BT17BDC-9, BT17BDC-17 (соединения формулы (V)a), BT17BDC-18 (соединения формулы (V)b), BT17BDC-19 (соединения формулы (V)c) и BT17BDC-20 (соединения формулы (V)d). Данные представлены в примере 5 и в таблицах 16 и 17, что демонстрирует полезные свойства BT17BDC-9, BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20.
В еще одном варианте осуществления изобретения конъюгат выбран из BT17BDC-9, BT17BDC-17 (соединения формулы (V)a), BT17BDC-18 (соединения формулы (V)b) и BT17BDC-19 (соединения формулы (V)c). Данные представлены в примере 5 и в таблицах 16 и 17, они демонстрируют, что эти конъюгаты рассматриваются как подходящие молекулы для использования в таргетной терапии рака.
В еще одном варианте осуществления изобретения конъюгат выбран из BT17BDC-17 (соединения формулы (V)a), BT17BDC-18 (соединения формулы (V)b) и BT17BDC-19 (соединения формулы (V)c). Данные представлены в примере 5 и в таблицах 16 и 17, которые показывают, что эти конъюгаты считаются подходящими молекулами для использования в таргетной терапии рака и хорошо переносятся в эффективных дозах.
Синтез
Пептиды по данному изобретению могут быть получены синтетически с помощью стандартных методов с последующим взаимодействием с молекулярным каркасом in vitro. После осуществления этого может быть использована стандартная химия. Это создает возможность быстрой крупномасштабной продукции растворимого вещества для дальнейших последующих экспериментов или валидации. Такие методы могут быть осуществлены с использованием традиционной химии, такой как раскрытая в Timmerman et al (выше).
Таким образом, изобретение также относится к получению полипептидов или конъюгатов, выбранных как изложено в настоящем описании, где получение включает необязательные дополнительные стадии, как описано ниже. В одном варианте осуществления эти стадии осуществляются с конечным продуктом полипептида/конъюгата, полученным с помощью химического синтеза.
Необязательно аминокислотные остатки в представляющем интерес полипептиде могут быть заменены при получении конъюгата или комплекса.
Пептиды могут быть также удлинены для включения, например, другой петли и, следовательно, введения множественной специфичности.
Для удлинения пептида он может быть просто удлинен химически на его N-конце или С-конце или в пределах его петель с использованием ортогонально защищенных лизинов (и аналогов) с применением стандартных методов твердофазной химии или методов синтеза в растворе. Стандартные методы (био)конъюгации могут быть использованы для введения активированного или активируемого N- или С-конца. В качестве альтернативы могут быть сделаны дополнения с помощью конденсации фрагментов или нативного химического лигирования, например как описано в (Dawson et al. 1994. Synthesis of Proteins by Native Chemical Ligation. Science 266:776-779), или с помощью ферментов, например, с использованием субтилигазы, как описано в (Chang et al Proc Natl Acad Sci U S A. 1994 Dec 20; 91(26):12544-8 или в Hikari et al Bioorganic & Medicinal Chemistry Letters Volume 18, Issue 22, 15 November 2008, Pages 6000-6003).
Альтернативно, пептиды могут быть удлинены или модифицированы путем дополнительной конъюгации через дисульфидные связи. Это имеет дополнительное преимущество, позволяя первому и второму пептиду диссоциировать друг от друга при попадании в восстанавливающую среду клетки. В этом случае молекулярный каркас (например, TBMB) может быть добавлен в процессе химического синтеза первого пептида таким образом, чтобы вступать в реакцию с тремя группами цистеина; дополнительный цистеин или тиол может быть затем присоединен к N- или С-концу первого пептида, так что этот цистеин или тиол взаимодействует только со свободным цистеином или тиолом второго пептида, образуя связанный дисульфидной связью конъюгат бициклического пептида-пептида.
Подобные методы в равной степени применимы к синтезу/присоединению двух бициклических и биспецифических макроциклов, потенциально создавая тетраспецифичную молекулу.
Кроме того, добавление других функциональных групп или эффекторных групп может быть осуществлено таким же образом, используя соответствующие химические методы, соединения на N- или С-концах или через боковые цепи. В одном варианте осуществления присоединение осуществляется таким образом, что оно не блокирует активность любой из частей.
В соответствии с другим аспектом настоящего изобретения предлагается способ получения конъюгата лекарственного средства, как определено в данном описании, который включает путь синтеза, описанный в любой из схем I, II или III.
Фармацевтические композиции
В соответствии с другим аспектом настоящего изобретения предлагается фармацевтическая композиция, включающая пептидный лиганд или конъюгат лекарственного средства, как определено в настоящем документе, в сочетании с одним или более фармацевтически приемлемыми наполнителями.
Обычно представленные пептидные лиганды должны использоваться в очищенной форме совместно с фармакологически приемлемыми наполнителями или носителями. Как правило, эти наполнители или носители включают водные или спиртовые/водные растворы, эмульсии или суспензии, включая физиологический солевой раствор и/или буферные среды. Парентеральные носители включают раствор хлорида натрия, раствор Рингера с декстрозой, декстрозу и хлорид натрия, и раствор Рингера с лактатом. Подходящие физиологически приемлемые адъюванты, если необходимо сохранять полипептидный комплекс в суспензии, могут быть выбраны из загустителей, таких как карбоксиметилцеллюлоза, поливинилпирролидон, желатин и альгинаты.
Внутривенные носители включают жидкие и питательные наполнители и электролитные наполнители, такие как на основе раствора Рингера с декстрозой. Консерванты и другие добавки, такие как противомикробные агенты, антиоксиданты, хелатирующие агенты и инертные газы, также могут присутствовать (Mack (1982) Remington's Pharmaceutical Sciences, 16th Edition).
Пептидные лиганды по настоящему изобретению могут быть использованы в виде отдельно вводимых композиций или в сочетании с другими агентами. Они могут включать антитела, фрагменты антител и различные иммунотерапевтические лекарственные средства, такие как циклоспорин, метотрексат, адриамицин или цисплатин и иммунотоксины. Фармацевтические композиции могут включать «коктейли» различных цитотоксических или других агентов в сочетании с белковыми лигандами по настоящему изобретению, или даже сочетания выбранных полипептидов в соответствии с настоящим изобретением, имеющих различную специфичность, таких как полипептиды, выбранные с использованием различных лигандов-мишеней, будут ли они объединяться перед введением или нет.
Путь введения фармацевтических композиций согласно изобретению может быть любым из обычно известных специалистам в данной области техники. Для терапии, включая без ограничения иммунотерапию, пептидные лиганды по изобретению могут быть введены любому больному в соответствии со стандартными методами. Введение может осуществляться с помощью любого подходящего способа, включая парентеральный, внутривенный, внутримышечный, внутрибрюшинный, трансдермальный способы, введение через легкие, или также, соответственно, путем прямой инфузии с помощью катетера. Дозировка и частота введения должна зависеть от возраста, пола и состояния больного, одновременного введения других лекарственных средств, противопоказаний и других параметров, которые должны быть приняты во внимание врачом.
Пептидные лиганды по настоящему изобретению могут быть лиофилизованы для хранения и восстановлены в подходящем носителе перед использованием. Этот метод, как показано, эффективен, и могут быть использованы известные в данной области методы лиофилизации и восстановления. Специалистам в данной области техники должно быть понятно, что лиофилизация и восстановление могут привести к различной степени потери активности, и что для компенсации этого уровни должны быть скорректированы в сторону повышения.
Композиции, содержащие представленные пептидные лиганды или их смесь, можно вводить для профилактического и/или терапевтического лечения. При некоторых вариантах терапевтического применения адекватное количество для достижения, по меньшей мере, частичного ингибирования, подавления, модуляции, уничтожения или какого-либо другого измеримого параметра популяции выбранных клеток определяется как «терапевтически эффективная доза». Количества, необходимые для достижения этой дозы, должны зависеть от тяжести заболевания и общего состояния собственной иммунной системы больного, но обычно находятся в диапазоне от 0,005 до 5,0 мг выбранного пептидного лиганда на килограмм массы тела, причем чаще всего используются дозы от 0,05 до 2,0 мг/кг/доза. Для профилактических целей композиции, включающие представленные пептидные лиганды или их смеси, могут также вводиться в аналогичных или слегка более низких дозах.
Композиция, содержащая пептидный лиганд в соответствии с настоящим изобретением, может быть использована в профилактических и терапевтических целях для того, чтобы способствовать изменению, инактивации, уничтожению или удалению выбранной популяции клеток-мишеней в организме млекопитающего. Кроме того, пептидные лиганды, описанные в настоящем документе, могут быть использованы экстракорпорально или in vitro для избирательного уничтожения, снижения или эффективного удаления иным образом популяции клеток-мишеней из гетерогенной популяции клеток. Кровь от млекопитающего может быть экстракорпорально объединена с выбранными пептидными лигандами, в результате чего нежелательные клетки уничтожаются или иным образом удаляются из крови для возвращения ее млекопитающему в соответствии со стандартными методами.
Терапевтическое применение
Бициклические пептиды по изобретению имеют конкретную применимость в качестве высокоаффинных агентов, связывающихся с мембранной металлопротеиназой типа 1 (МТ1-ММР, также известной как ММР14). MT1-MMP представляет собой трансмембранную металлопротеиназу, которая играет главную роль в ремоделировании внеклеточного матрикса, прямо в результате стимуляции деградации нескольких его компонентов и косвенно путем активации про-ММР2. МТ1-ММР имеет решающее значение для ангиогенеза опухоли (Sounni et al (2002) FASEB J. 16(6), 555-564) и гиперэкспрессируется на различных солидных опухолях, поэтому бициклические пептиды, связывающие МТ1-ММР, по настоящему изобретению, имеют особую применимость в таргетной терапии рака, в частности солидных опухолей, таких как немелкоклеточный рак легкого. В одном варианте осуществления бициклический пептид согласно изобретению является специфичным для МТ1-ММР человека. В другом варианте осуществления бициклический пептид согласно изобретению является специфичным для МТ1-ММР мыши. В еще одном варианте осуществления бициклический пептид согласно изобретению является специфичным для МТ1-ММР человека и мыши. В еще одном варианте осуществления бициклический пептид согласно изобретению является специфичным для МТ1-ММР человека, мыши и собаки.
Полипептидные лиганды, выбранные в соответствии со способом по настоящему изобретению, могут быть использованы в терапевтических и профилактических целях in vivo, при диагностическом применении in vitro и in vivo, для тестирования in vitro и применения в качестве реагентов и тому подобного. Лиганды, имеющие выбранные уровни специфичности, пригодны для вариантов применения, которые включают тестирование у животных, не являющихся человеком, где желательна перекрестная реактивность, или для вариантов диагностического применения, где перекрестную реактивность с гомологами или парологами необходимо тщательно контролировать. В некоторых вариантах применения, таких как варианты применения в качестве вакцин, способность вызывать иммунный ответ на заранее определенные диапазоны антигенов может быть использована для адаптации вакцины к конкретным заболеваниям и патогенам.
По существу чистые пептидные лиганды с гомогенностью, по меньшей мере, от 90 до 95% являются предпочтительными для введения млекопитающему, и гомогенность от 98 до 99% или более наиболее предпочтительна для фармацевтических вариантов применения, особенно, когда млекопитающее является человеком. После очистки, частичной или до гомогенности, в зависимости от потребности, выбранные полипептиды могут быть использованы диагностически или терапевтически (включая экстракорпоральное применение) или для разработки и проведения методов тестирования, иммунофлуоресцентного окрашивания и тому подобного (Lefkovite and Pernis, (1979 and 1981) Immunological Methods, Volumes I and II, Academic Press, NY).
Эффекторная группа и конъюгаты пептидных лигандов по настоящему изобретению, как правило, находят применение в профилактике, подавлении или лечении рака, в частности солидных опухолей, таких как немелкоклеточный рак легкого.
Таким образом, в соответствии с еще одним аспектом настоящего изобретения предлагаются эффекторные группы и конъюгаты лекарств с пептидным лигандом, как определено в настоящем документе, для применения в профилактике, подавлении или лечении рака, в частности солидных опухолей, таких как немелкоклеточный рак легкого.
В соответствии с другим аспектом настоящего изобретения предлагается способ профилактики, подавления или лечения рака, в частности солидных опухолей, таких как немелкоклеточный рак легкого, который включает введение нуждающемуся в этом больному эффекторной группы и конъюгата лекарства с пептидным лигандом, как определено в данном описании.
Примеры раковых заболеваний (и их доброкачественных аналогов), которые можно лечить (или ингибировать) включают, но не ограничиваются этим, опухоли эпителиального происхождения (аденомы и карциномы различных типов, включая аденокарциномы, плоскоклеточный рак, переходные клеточные карциномы и другие карциномы), такие как карциномы мочевого пузыря и мочевых путей, молочной железы, желудочно-кишечного тракта (включая карциномы пищевода, желудка (желудочные), тонкой кишки, толстой кишки, прямой кишки и заднего прохода), печени (гепатоцеллюлярный рак), желчного пузыря и билиарной системы, экзокринной поджелудочной железы, почек, легких (например, аденокарциномы, мелкоклеточный рак легких, немелкоклеточный рак легких, бронхоальвеолярные карциномы и мезотелиомы), карциномы головы и шеи (например, рак языка, ротовой полости, гортани, глотки, носоглотки, миндалин, слюнных желез, полости носа и околоносовых пазух), яичников, маточных труб, брюшины, влагалища, вульвы, пениса, шейки матки, миометрия, эндометрия, щитовидной железы (например, фолликулярную карциному щитовидной железы), надпочечников, предстательной железы, кожи и прилежащих органов (например, меланому, базальноклеточный рак, плоскоклеточный рак, кератоакантому, диспластический невус); гематологические злокачественные опухоли (например, лейкозы, лимфомы) и предраковые гематологические нарушения и нарушения с пограничной злокачественностью, включая гематологические злокачественные заболевания и связанные с ними состояния клеток лимфоидного ряда (например, острый лимфоцитарный лейкоз [ALL], хронический лимфоцитарный лейкоз [CLL], В-клеточные лимфомы, такие как диффузная большая В-клеточная лимфома [DLBCL], фолликулярная лимфома, лимфома Беркитта, лимфома клеток мантии, Т-клеточные лимфомы и лейкозы, лимфомы природных клеток-киллеров [NK], лимфомы Ходжкина, лейкоз ворсистых клеток, моноклональную гаммапатию неясного генеза, плазмоцитому, множественную миелому и лимфопролиферативные нарушения после трансплантации), а также гематологические злокачественные и родственные состояния клеток миелоидного ряда (например, острый миелогенный лейкоз [AML], хронический миелогенный лейкоз [CML], хронический миеломоноцитарный лейкоз [CMML], гиперэозинофильный синдром, миелопролиферативные расстройства, такие как полицитемия вера, эссенциальная тромбоцитемия и первичный миелофиброз, миелопролиферативный синдром, синдром миелодисплазии и промиелоцитный лейкоз); опухоли мезенхимального происхождения, например, саркомы мягких тканей, кости или хряща, такие как остеосаркомы, фибросаркомы, хондросаркомы, рабдомиосаркомы, лейомиосаркомы, липосаркомы, ангиосаркомы, саркому Капоши, саркому Юинга, синовиальную саркому, эпителиоидные саркомы, желудочно-кишечные стромальные опухоли, доброкачественные и злокачественные гистоцитомы и дерматофибросаркому протуберанс; опухоли центральной или периферической нервной системы (например, астроцитомы, глиомы и глиобластомы, менингиомы, эпендимомы, опухоли эпифиза и шванномы); эндокринные опухоли (например, опухоли гипофиза, опухоли надпочечников, опухоли островковых клеток, опухоли паращитовидных желез, карциноидные опухоли и медуллярную карциному щитовидной железы); опухоли глаз и их придатков (например, ретинобластому); опухоли зародышевых клеток и трофобласта (например, тератомы, семиномы, дисгерминомы, пузырный занос и хориокарциному); и педиатрические и эмбриональные опухоли (например, медуллобластомы, нейробластомы, опухоль Вильмса и примитивные нейроэктодермальные опухоли); или синдромы, врожденные или иные, которые делают больного восприимчивым к злокачественному новообразованию (например, пигментную ксеродерму).
Ссылки в настоящем документе на термин «предотвращение» включают введение профилактической композиции до начала индукции заболевания. «Подавление» относится к введению композиции после того, как событие проиндуцировано, но до клинического проявления заболевания. «Лечение» включает введение защитной композиции после появления симптомов заболевания.
Доступны животные модельные системы, которые могут быть использованы для скрининга эффективности пептидных лигандов в отношении защиты против заболевания или его лечения. Использование модельных систем на животных способствует осуществлению настоящего изобретения, что позволяет разработать полипептидные лиганды, которые могут перекрестно реагировать с мишенями человека и животных, для возможности использования животных моделей.
Изобретение дополнительно описано ниже со ссылкой на следующие примеры.
ПРИМЕРЫ
Материалы и методы
Выбор фагов
6×6 фаговые библиотеки бициклических пептидов создавали, как описано у Heinis et al (2009), Nat Chem Biol 5(7), 502-507, в патентах WO 2009/098450, WO 2013/050615 и WO 2013/050616. Выбор фагового дисплея проводили с использованием указанной 6×6 фаговой библиотеки против биотинилированного домена гемопексина МТ1-ММР человека.
Экспрессия белка
Гемопексин-подобные повторы МТ1-ММР (также известные как домен гемопексина МТ1-ММР), остатки Cys319-Gly511 гена человека, временно экспрессировали в клетках НЕК293 в виде секретируемого растворимого белка с N-концевой меткой His6, используя экспрессионный вектор PExpr-IBA42 (IBA). После экспрессии белок очищали с помощью аффинной хроматографии Nickel-NTA с последующей гель-фильтрацией, и чистоту проверяли с помощью SDS-PAGE. Вариабельность от партии к партии также контролировали с помощью экспериментов по температурному сдвигу флуоресценции в присутствии/отсутствии бициклического пептида, связывающего домен гемопексина.
Пептидный синтез
Пептидный синтез основан на химии Fmoc с использованием синтезатора пептидов Symphony, поставляемого Peptide Instruments, и синтезатора Syro II, поставляемого MultiSynTech. Использовали стандартные Fmoc-аминокислоты (Sigma, Merck) со следующими защитными группами боковой цепи: Arg(Pbf); Asn(Trt); Asp(OtBu); Cys(Trt); GIu(OtBu); Gln(Trt); His(Trt); Lys(Boc); Ser(tBu); Thr(tBu); Trp(Boc); и Tyr(tBu) (Sigma). Присоединяющим реагентом был HCTU (Pepceuticals), диизопропилэтиламин (DIPEA, Sigma) использовали в качестве основания, и снятие защиты достигали с помощью 20% пиперидина в DMF (AGTC). Синтезы осуществляли с использованием 0,37 ммоль/г Fmoc-Rink амидной смолы AM (AGTC), Fmoc-аминокислоты использовали в четырехкратном избытке, и основание использовали в четырехкратном избытке по отношению к аминокислотам. Аминокислоты растворяли до 0,2 М в ДМСО, HCTU до 0,4 М в DMF и DIPEA до 1,6 М в N-метилпирролидоне (Alfa Aesar). Условия были таковы, что реакционные смеси для присоединения включали от 20 до 50% ДМСО в DMF, что позволило снизить агрегацию и делеции во время твердофазного синтеза и повысить выход. Время присоединения составляло, как правило, 30 минут, и время снятия защиты 2×5 минут. Fmoc-N-метилглицин (Fmoc-Сар-ОН, Merck) присоединяли в течение 1 час, и время снятия защиты и присоединения для последующего остатка составляло 20 мин и 1 час, соответственно. После синтеза смолу промывали дихлорметаном и сушили. Отщепление защитных групп боковых цепей и образование подложки осуществляли с использованием 10 мл 95:2,5:2,5:2,5 об./об./об./масс. TFA/H2O/iPr3SiH/дитиотреитола в течение 3 часов. После отщепления отработанную смолу удаляли фильтрацией, и фильтрат добавляли к 35 мл диэтилового эфира, который был охлажден до -80°С. Пептидный осадок центрифугировали, эфирный супернатант отбрасывали, и пептидный осадок промывали холодным эфиром еще два раза. Пептиды затем повторно растворяли в 5-10 мл ацетонитрила-воды и лиофилизировали. Небольшой образец отбирали для анализа чистоты сырого продукта с помощью масс-спектрометрии (MALDI-TOF, Voyager DE от Applied Biosystems). После лиофилизации пептидные порошки переносили в 10 мл 6 М гидрохлорида гуанидина в H2O с добавлением 0,5 мл 1 М дитиотреитола, и наносили на препаративную колонку С8 Luna ВЭЖХ (Phenomenex). Растворители (H2O, ацетонитрил) подкисляли 0,1% гептафтормасляной кислотой. Диапазон градиента составлял 30-70% ацетонитрила в течение 15 минут при скорости потока 15-20 мл/мин при использовании системы препаративной ВЭЖХ Gilson. Фракции, содержащие материал чистого линейного пептида (при определении с помощью MALDI), объединяли и модифицировали с помощью 1,3,5-трис(бромметил)бензола (TBMB, Sigma). Для этого линейный пептид разбавляли H2O до ~35 мл, добавляли ~500 мкл 100 мМ TBMB в ацетонитриле, и реакцию инициировали 5 мл 1 М NH4HCO3 в H2O. Реакции давали возможность протекать в течение ~30-60 мин при комнатной температуре, и лиофилизировали сразу после завершения реакции (судя по MALDI). После лиофилизации модифицированный пептид очищали, как описано выше, с заменой колонки Luna C8 на колонку Gemini C18 (Phenomenex), и заменой кислоты на 0,1% трифторуксусную кислоту. Чистые фракции, содержащие корректный TMB-модифицированный материал, объединяли, лиофилизировали и поддерживали при -20°C для хранения.
Все аминокислоты, если не указано иначе, использовали в L-конфигурациях.
DOTA присоединяли к пептидной цепи во время твердофазного синтеза пептидов с использованием защищенного предшественника DOTA(tBu)3 (TCI, CAS 137076-54-1).
Неприродные аминокислоты включали в пептидную последовательность с использованием общих методов, описанных выше.
Перечень предшественников неприродных аминокислот, используемых в данном описании, суммирован в таблице ниже:
Пептиды, используемые для фармакокинетических исследований, лиофилизировали из 0,1% TFA в воде с получением солей TFA или свободных кислот указанных соединений.
Синтез BDCs с использованием 17-69-07-N219 в качестве предшественника бициклического пептида
Два конъюгата бициклический пептид-лекарство (BDC) синтезировали с использованием 17-69-07-N219 в качестве пептида-предшественника. Активированные конструкты vcMMAE или дисульфид-DM1 (растворенные в ДМСО) прямо конъюгировали в 1,4× избытке с 17-69-07-N219 в водных условиях (100 фосфата натрия, рН 8) (смотри схемы I и II). Концентрации пептида составляли 9 мг/мл или выше. За реакцией следили с помощью ЖХ/МС и считали завершенной через 3,5 часа. За этим следовала стандартная очистка с обращенной фазой с использованием полупрепаративной колонки С18. Фракции с чистотой выше 95% выделяли и лиофилизировали. Материалы не содержали измеримых количеств свободного токсина. Для исследований in vitro и in vivo лиофилизованные порошки переводили в концентрированные маточные растворы ДМСО (100 мг/мл) и разводили в соответствующем буфере для дальнейшего использования.
Синтез BDCs с использованием 17-69-07-N241 в качестве предшественника бициклического пептида
Для синтеза BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20 использовали следующие N-гидроксисукцинимидные сложные эфиры (NHS эфиры) дисульфидов мейтанзиноидов:
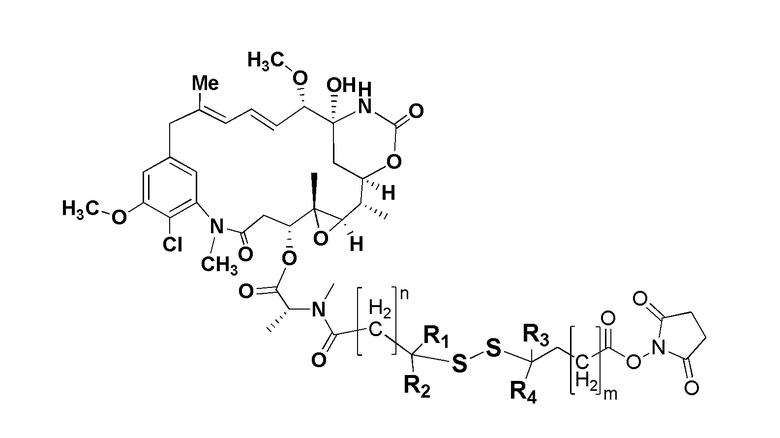
где R1, R2, R3, R4, m и n представляет собой описанное выше для соединений формул (V)a, (V)b, (V)c, (V)d для BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20, соответственно.
BT17BDC-18 дополнительно синтезировали через альтернативный путь, как описано ниже и в схеме III. В настоящем документе 17-69-07-N277 синтезировали путем взаимодействия 17-69-07-N241 с SPP (N-сукцинимидил-4-(2-пиридилдитио)пентаноатом) в ДМСО. Концентрации 17-69-07-N241 составляли 10 мМ или выше при 1,3-кратном избытке SPP и 20-кратном избытке диизопропилэтиламина при комнатной температуре. Реакцию считали завершенной через 1 час, о чем судили по ЖХ/МС. Очистку проводили с помощью обращенной фазы, как описано выше. Соответствующие фракции лиофилизировали.
У 17-69-07-N277 осуществляли обмен дисульфидами с 1,15 эквивалентами DM1 (в виде свободного тиола) в полуводных условиях (50% диметилацетамид и 50% 100 мМ ацетат натрия, рН 5,0 с добавлением 2 мМ ЭДТА) в течение 21 часа при комнатной температуре под подушкой газообразного азота. Концентрации 17-69-07-N277 в реакционной смеси составляли 10 мМ или выше. За этим следовала стандартная очистка с обращенной фазой с использованием полупрепаративной колонки С18. Фракции с чистотой выше 95% выделяли и лиофилизировали. Материалы не содержали измеримых количеств свободного токсина. Для исследования in vitro и in vivo лиофилизованные порошки солюбилизировали в водной композиции, как описано выше, или вносили непосредственно в соответствующий буфер.
Определение константы скорости диссоциации бициклического пептида, связывающегося с МТ1-ММР
Методы прямого определения связывания с помощью поляризации флуоресценции (анизотропии)
Методы прямого определения связывания с помощью поляризации флуоресценции или анизотропии осуществляли путем титрования постоянной концентрации флуоресцентной метки (в настоящем документе изучался флуоресцентный бициклический пептид) с его партнером по связыванию (в настоящем документе доменом гемопексина МТ1-ММР). По мере увеличения концентрации партнера по связыванию в процессе титрования сигнал поляризации изменяется пропорционально доле связанного и несвязанного вещества. Это позволяет определять скорости диссоциации (Kd) количественно. Данные анализа могут быть подобраны с использованием стандартных уравнений связывания лиганда.
Как правило, концентрации метки в идеале значительно ниже Kd пары метка:титрующее вещество, и выбранные концентрации обычно составляют 1 нМ или менее. Концентрация титрующего вещества (партнера по связыванию) варьируется от 0,1 нМ обычно до 5 мкМ. Диапазон выбирается таким образом, что может наблюдаться максимальное изменение поляризации флуоресценции. Используемые буферы представляют собой забуференный фосфатом солевой раствор в присутствии 0,01% твина. Эксперименты проводили в черных 384-луночных планшетах с низким связыванием/низким объемом (Corning 3820), и сигнал поляризации флуоресценции измеряли с использованием планшетного ридера BMG Pherastar FS.
Флуоресцентные метки, упомянутые в тексте, представляют собой бициклические пептиды, которые флуоресцировали благодаря использованию 5,6-карбоксифлуоресцеина. Присоединение флуоресцентной метки может быть выполнено на N-концевой аминогруппе пептида, которая отделена от бициклической последовательности каркаса с помощью саркозинового спейсера (обычно Sar5). Это может быть осуществлено в ходе Fmoc твердофазного синтеза или после синтеза (после циклизации с TBMB и очистки), если N-концевая аминогруппа является уникальной для пептида. Присоединение флуоресцентной метки также может быть выполнено на С-конце, как правило, на лизине, введенном как первой С-концевой остаток, который затем отделяют от бициклической последовательности каркаса с помощью саркозинового спейсера (обычно Sar6). Таким образом, N-концевые метки могут иметь молекулярный формат, описанный как Fluo-Gly-Sar5-A(бициклическая последовательность каркаса), и (бициклическая последовательность каркаса)-A-Sar6-К(Fluo) для конструкта с C-концевой флуоресцентной меткой. Флуоресцентные метки, используемые в примерах, представляют собой A-(17-69)-A-Sar6-K(Fluo), A-(17-69-07)-A-Sar6-K(Fluo) и A-(17-69-12)-A-Sar6-K(Fluo). Из-за кислой природы флуоресцентных пептидов 17-69 их обычно получают в виде концентрированных маточных растворов в ДМСО, из которых делают разведения в 100 мМ Трис буфере, рН 8.
Конкурентный анализ с использованием поляризации флуоресценции (анизотропии)
Благодаря их высокой аффинности к домену гемопексина (РЕХ) МТ1-ММР, меченные флуоресцентной меткой производные 17-69-07 и 17-69-12 (обозначаемые как 17-69-07-N040 и 17-69-12-N005, соответственно) могут быть использованы для экспериментов по конкуренции (с использованием FP для обнаружения). В настоящем документе предварительно образованный комплекс PEX с флуоресцентной меткой, связывающей PEX, титруют свободным, нефлуоресцентным бициклическим пептидом. Поскольку все пептиды на основе 17-69, как ожидается, связываются с одним и тем же сайтом, титрующее соединение будет вытеснять флуоресцентную метку из PEX. Диссоциация комплекса может быть измерена количественно, и определяют Kd конкурента (титрующего соединения) от белка-мишени. Преимуществом конкурентного анализа является то, что сродство нефлуоресцирующих бициклических пептидов может быть определено точно и быстро.
Концентрации метки, как правило, составляют на уровне Kd или ниже (в настоящем документе 1 нМ), и связывающий белок (в настоящем документе гемопексин МТ1-ММР) находится в 15-кратном избытке, так что >90% метки связано. После этого титруют нефлуоресцирующим конкурентным бициклическим пептидом (обычно только бициклической последовательностью каркаса), таким образом, что он вытеснят флуоресцентную метку от белка-мишени. Вытеснение метки измеряют и связывают со снижением поляризации флуоресценции. Снижение поляризации флуоресценции пропорционально доле белка-мишени, связанной с нефлуоресцирующим титрующим соединением, и, следовательно, является мерой сродства титрующего соединения к белку-мишени.
Необработанные данные подбирают для аналитического решения кубического уравнения, описывающего равновесие между флуоресцентной меткой, титрующим соединением и связывающим белком. Подбор требует знания величины сродства флуоресцентной метки к белку-мишени, которая может быть определена отдельно с помощью прямых экспериментов по связыванию FP (смотри предыдущий раздел). Подбор кривой проводили с использованием Sigmaplot 12,0 и использовали адаптированный вариант уравнения, описанного Zhi-Xin Wang (FEBS Letters 360 (1995) 111-114).
Профиль устойчивости в плазме
Способ №1:
Разработана быстрая оценка профиля стабильности в плазме с использованием масс-спектрометрического обнаружения (MALDI-TOF, Voyager DE, Applied Biosystems) родительской массы, а также индуцированных протеазами фрагментов в плазме. С помощью оценки природы фрагментов могут быть определены предпочтительные сайты расщепления. В настоящем документе 1-1,5 мМ маточный раствор пептида (в ДМСО) непосредственно разбавляли в плазме мыши/крысы/человека (Sera labs, с использованием цитрата в качестве антикоагулянта), что давало конечную концентрацию 50 мкМ пептида, и инкубировали в течение 48 часов при 37°С. Образцы по 5 мкл отбирали в соответствующие моменты времени и замораживали при -80°С. Для анализа образцы размораживали, смешивали с 25 мкл смеси 3:3:1 ацетонитрил:метанол:вода, и центрифугировали при 13k в течение 5 мин. 5 мкл содержащего пептид супернатанта отсасывали и смешивали с 30 мМ бикарбонатом аммония в смеси 1:1 ацетонитрил:H2O. 1 мкл этого раствора затем наносили на плашку MALDI, сушили и поверх образца наслаивали (1 мкл) Matrix (альфа-цианокоричную кислоту, Sigma, полученную в виде насыщенного раствора в смеси 1:1 ацетонитрил:вода, содержащего 0,1% трифторуксусную кислоту), сушили и анализировали с помощью MALDI TOF. Следует отметить, что это является качественным анализом, и он служит для выявления сравнительных изменений стабильности в плазме между различными последовательностями бициклических пептидов и функциями в качестве отличного инструмента для определения предпочтительных сайтов расщепления.
Способ №2
Для количественного определения стабильности бициклических пептидов в плазме маточные растворы пептидов (200 мкМ в ДМСО) смешивали с плазмой (человека или мыши) таким образом, чтобы конечные концентрации составляли 10 мкМ. Образцы по 40 мкл периодически отбирали до 8 часов и замораживали при -80°С. Перед анализом ЖХ/МС образцы размораживали и смешивали с 3 объемами (в настоящем документе 120 мкл) смеси 1:1 ацетонитрил/водный MeOH. Млечную суспензию центрифугировали в течение 30 мин при 13000 об/мин, и супернатанты, содержащие пептид, количественно определяли для двухзарядных/трехзарядных видов и их МС/МС фрагментов с использованием прибора Waters Xevo TQ-D при использовании стандартной кривой экстрагированных из плазмы тех же пептидов в качестве референсных соединений. Период полужизни при деградации в плазме использовали для оценки сравнительной стабильности молекул.
Фармакокинетика 17-69-07 у мышей, идентификация метаболитов
Фармакокинетика 17-69-07-N004
Фармакокинетику у мышей изучали при использовании бициклического пептида 17-69-04-N004, который дозированно вводили одной группе из 12 самцов мышей CD1 в виде однократной внутривенной дозы 5,925 мг/кг в виде 5 мл/кг болюса раствора 1,19 мг/мл. Растворы композиций готовили из 100 мкл из маточного раствора в ДМСО 23,7 мг/мл, который разводили 1,9 мл забуференным фосфатом физиологическим раствором непосредственно перед введением дозы, в результате чего носитель состоял из 5% ДМСО в PBS при рН 7,4. Образцы крови отбирали от двух животных на временную точку с помощью пункции сердца под терминальной анестезией через 0,08, 0,5, 1, 2 и 4 часа после введения дозы и переносили в пробирки с ЭДТА для получения плазмы. Образцы плазмы немедленно замораживали при -20°С. Для анализа образцы быстро размораживали, и 50 мкл аликвоты обрабатывали 3 объемами растворителя для экстракции (смесью 2:9:9 10 мМ бикарбоната аммония, рН 8, ацетонитрила и метанола, содержащей аналитический внутренний стандарт). Осажденные белки удаляли центрифугированием, и супернатант анализировали с помощью ЖХ-МС/МС. Количественное определение образцов осуществляли с помощью расчета по калибровочной кривой, полученной в контрольной плазме мыши. Фармакокинетические параметры определяли с помощью нонкомпартментного анализа с использованием пакета программного обеспечения PK Solutions 2.0 Summit Research Services.
Определение терминов:
Cmax: Максимальная измеренная концентрация;
Tmax: Время, при котором измерена максимальная концентрация;
AUC 0-t: Площадь под кривой концентрации лекарственного средства в плазме/кривой времени от 0 минут до последней точки количественного получения данных; и
AUC 0-∞: Площадь под кривой концентрации лекарственного средства в плазме/кривой времени от 0 минут с экстраполяцией данных за пределы конечной точки на основе конечного периода полужизни.
Идентификация метаболитов бициклических пептидов в плазме мышей
Три образца плазмы были доступны для использования (0,5, 1 и 2 часа) в анализе потенциальных метаболитов 17-69-07-N004 у мыши in vivo. Анализ проводили с помощью ВЭЖХ-МС и ВЭЖХ-МС МС с использованием масс-спектрометра LTQ Orbitrap XL. Подход для поиска пептидных метаболитов в кровообращении заключался в вычислении точной массы (окно в 10 м.д.) предполагаемых метаболитов (добавление воды 1 или 2 (+18, +36) для расщепления петли 1 и/или петли 2, соответственно; затем потеря отдельных аминокислот или потеря аминокислотных участков из петли 1 и/или петли 2). После этого осуществляли ручной поиск путем сравнения общих ионных хроматограмм с контрольной мышиной плазмой.
Эффективность BT17BDC-1 и BT17BDC-9 у мышей с ксенотрансплантатом HT-1080.
Голым мышам BALB/C, несущим подкожные ксенотрансплантаты опухолей HT-1080, вводили BDCs или носитель (PBS). BDCs вводили 3 раза в неделю в течение 2 недель с началом дозирования при измеренном размере опухолей приблизительно 150-200 мм3. За мышами прослеживали, и измерения объема опухолей и массы тела записывали 3 раза в неделю.
Пример 1: Идентификация бициклических пептидов с высокой аффинностью с использованием домена гемопексина MT1-MMP
Используя ранее установленные методы для создания фаговых библиотек бициклических пептидов, осуществляли отбор пептидов против домена гемопексина МТ1-ММР человека. После трех циклов отбора из интактной библиотеки с применением последовательно снижающейся концентрации мишени проводили секвенирование на выходе. Бициклический пептид 17-69 (CKNRGFGCEDFYDIC) (SEQ ID NO: 16) был идентифицирован как один из пептидов с наиболее обогащенным выходом последовательности, и качественное связывание с мишенью подтверждено с помощью AlphaScreen.
Три небольшие фаговые библиотеки были созданы для обеспечения полного охвата последовательностей каждой из 3-х частей последовательности 17-69. Эти три библиотеки были подвергнуты двум циклам отбора против белка гемопексина. Интересно отметить, что наиболее перспективные выходы секвенирования составляли 5×6 бициклических пептидов, в то время как исходные библиотеки были в формате 6×6. Вполне вероятно, что более короткая длина петли была выбрана из-за более высокой аффинности бициклического пептида к белку-мишени. Более короткие длины петли возникают в результате неправильно синтезированных праймеров, которые включаются во время создания фаговых библиотек.
Основным выходом секвенирования был пептид 17-69-07, который имеет последовательность CYNEFGCEDFYDIC (SEQ ID NO: 2).
Основываясь на наблюдении того, что формат 5×6, очевидно, является наиболее удачным, и что требуется различение между связывающими агентами 5×6, были созданы еще две библиотеки, тестирующие преднамеренные укорочения первой петли, что дало бициклические пептиды 17-69-02, 17-69-03, 17-69-04 и 17-69-12. Они содержатся в пределах последовательностей, раскрытых на фигуре 8.
Тенденция для некоторых остатков была видна, хотя анализ связывания был не в состоянии различить лучшие связующие агенты, так как они достигли максимального предела анализа.
Наиболее часто встречающиеся последовательности анализировали на связывание гемопексина с использованием альфа-скрининга, где все сигналы были высокими по сравнению с исходной последовательностью 17-69 (смотри таблицу 1):
Таблица 1: Анализ связывания гемопексина с использованием бициклических пептидов по изобретению
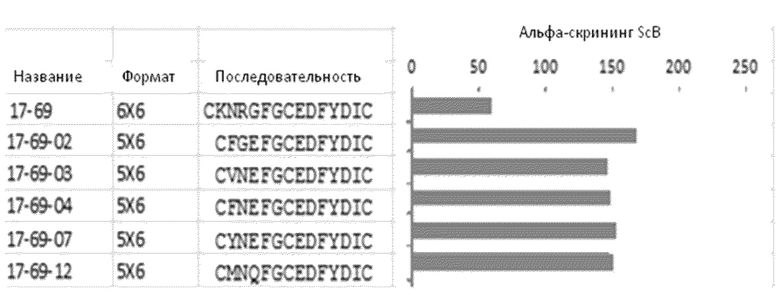
Поскольку все клоны на основе 17-69 со зрелой аффинностью продуцировали сигналы, которые были вблизи максимального предела анализа, диаграмма, представленная в таблице 1, не позволяет однозначно идентифицировать лучшие клоны. В связи с этим некоторые из пептидов синтезировали в виде флуоресцентных производных (17-69, 17-69-07 и 17-69-12) и использовали для экспериментов по прямому анализу связывания с помощью поляризации флуоресценции (FP). В настоящем документе флуоресцеин отделяют от бициклической последовательности или от N- или С-конца относительно последовательности каркаса с помощью линкера (обычно Sar6) (таблица 2).
Таблица 2: Результаты экспериментов по анализу прямого связывания с помощью поляризации флуоресценции (FP)
Флуоресцентные производные 17-69-07 и 17-69-12 (обозначаемые как 17-69-07-N040 и 17-69-12-N005) продемонстрировали сильное связывание со скоростями диссоциации 0,52 и 3,1 нМ, соответственно. Они заметно улучшены по сравнению с исходной, незрелой последовательностью 17-69 (17-69-N004). Представляется, что последовательности, которые содержат формат 5×6, индуцируют высокие аффинности в контексте библиотеки созревания аффинности.
Из-за их высокой аффинности к домену гемопексина (РЕХ) МТ1-ММР флуоресцентные производные 17-69-07 и 17-69-12 (обозначаемые как 17-69-07-N040 и 17-69-12-N005, соответственно) были использованы для последующих экспериментов по конкуренции (с использованием FP для обнаружения). В настоящем документе предварительно сформированный комплекс PEX со связывающей PEX флуоресцентной меткой титруют свободным, нефлуоресцентным бициклическим пептидом. Поскольку все пептиды на основе 17-69 (содержащие консервативный мотив второй петли CEDFYDIC; SEQ ID NO: 17), как ожидается, связываются с одним и тем же сайтом, титрующий агент будет вытеснять флуоресцентную метку из PEX. Диссоциация комплекса может быть измерена количественно, и определена Kd конкурента (титрующего агента). Преимуществом конкурентного анализа является то, что аффинность нефлуоресцентных бициклических пептидов может быть определена точно и быстро.
В этом контексте важно убедиться, что последовательности каркаса 17-69-07 и 17-69-12 отвечают только за высокое сродство к PEX. Таким образом, пептиды синтезировали в виде вариантов с N- и С-концевыми аланинами, тем самым близко имитируя последовательность, экспрессируемую на фаговой частице, но без молекулярного спейсера Sar5/6, который был использован в флуоресцентных конструктах.
Аффинность при определении с помощью экспериментов по конкуренции была почти идентична аффинности, полученной с помощью флуоресцентных производных, однозначно демонстрируя, что каркасные бициклические последовательности ответственны за высокое сродство связывания с PEX (таблица 3). Кроме того, флуоресцентные конструкты показывают, что допускается С-концевое связывание молекулярных спейсеров и эффекторных групп (в настоящем документе в виде -A-Sar6-К(флуоресцеин).
Таблица 3: Результаты экспериментов по прямому связыванию с помощью поляризации флуоресценции (FP)
Пример 2: Протеолитическая стабильность каркасной последовательности 17-69-07 мыши
Стабильность в плазме 17-69-07
Для терапевтического применения у человека, а также для доклинической оценки у видов животных, уместно, чтобы ведущий бициклический пептид являлся достаточно стабильным в кровотоке после внутривенного введения. Адекватная стабильность требуется для того, чтобы достаточный уровень бициклического пептида мог связываться со своей мишенью и проявлять свою биологическую функцию.
В доклинических моделях часто используют такие виды, как мышь, крыса, кролик и макака. В первом случае бициклический пептид 17-69-07-N219 проверяли на стабильность в присутствии плазмы мыши с помощью методов, описанных ранее (метода #2). Бициклическая каркасная последовательность сохраняет первоначальную природную протеиногенную аминокислотную последовательность 17-69-07 и дополнительно содержит N-концевой молекулярный спейсер, который используется для конъюгации эффекторных групп (последовательность: G-Sar10-ACYNEFGCEDFYDIC; SEQ ID NO: 20). Аффинность 17-69-07-N219 к PEX сохранялась, несмотря на наличие молекулярного спейсера (Kd=0,82 нМ).
Это соединение проявляло умеренную стабильность в плазме мышей ex vivo с периодом полужизни 6 часов (фигура 1).
Идентификация сайтов протеолитического расщепления 17-69-07 ex/in vivo
Для достижения понимания химической природы деградации 17-69-07-N219 в плазме мышей образцы анализировали с помощью MALDI-TOF в отношении любых потенциальных продуктов деградации. Масс-спектры показали возможную потерю тирозина, что подразумевает открытие петли (гидролиз) и удаление Tyr1 в петле 1 и/или Tyr9 в петле 2.
Фармакокинетическое (РК) исследование у мышей с использованием минимального бициклического пептида 17-69-07-N004 (Ac-CYNEFGCEDFYDIC (SEQ ID NO: 2), который состоит из минимального каркасного бициклического пептида 17-69-07), проводили с целью определения клиренса и скорости элиминации из циркуляции in vivo. Полученные в результате образцы крови были проанализированы дополнительно, и образцы плазмы анализировались на любые потенциальные протеолитические продукты деградации 17-69-07-N004.
Профиль PK показан на фигуре 2.
Таблица 4: Фармакокинетические параметры 17-69-07-N004
NC: не рассчитали из-за ограниченности данных
В таблице 4 показано, что пептид имеет период полувыведения 14 мин и клиренс составляет 20,7 мл/мин/кг. Скорость клиренса более высока, чем скорость клубочковой фильтрации, наблюдаемая у мышей (обобщено в Qi et al, American Journal of Physiology - Renal Physiology (2004) Vol. 286 no. 3, F590-F596), что свидетельствует о том, что клиренс пептида происходит с помощью дополнительных способов, например, с помощью управляемого плазмой протеолиза и эндотелиальных протеаз.
Для решения того, в значительной ли степени протеолиз in vivo способствует клиренсу, образцы плазмы, взятые при t=0,5, 1 и 2 час, подвергали таргетному анализу на фрагменты бициклических пептидов с использованием методов ЖХ-МС/МС.
Множество протеолитических метаболитов может быть идентифицировано, и фрагменты с наиболее интенсивными сигналами перечислены ниже в порядке убывания:
Ac-CiYNEFGCiiEDFYDICiii (SEQ ID NO: 2):
исключение YNE в петле 1;
исключение YNEF в петле 1 (SEQ ID NO: 21);
исключение YNEFG (SEQ ID NO: 22) в петле 1 (полная петля удаляется);
исключение Y в петле 1 и/или 2;
исключение YD в петле 2;
исключение FYD в петле 2;
исключение YDI в петле 2; а также
исключение EDFYDI (SEQ ID NO: 23), в петле 2 (полная петля удаляется).
Три верхних метаболита присутствовали на среднем уровне сигнала, тогда как остальные фрагменты обнаруживались лишь в следовых количествах.
В совокупности метаболиты как ex vivo, так и in vivo, очевидно, концентрируются при первоначальном расщеплении у или около Tyr 1/9 с последующим последовательным удалением остатков в непосредственной близости. Это в конечном счете приводит к удалению одной или обеих петель в полном объеме.
Увеличение протеолитической стабильности и активности последовательности 17-69-07
Подходы к стабилизации пептидных последовательностей в отношении протеолитической деградации многочисленны (смотри обзоры Gentilucci et al, Curr. Pharmaceutical Design, (2010), 16, 3185-203, and Nestor et al, Curr. Medicinal Chem (2009), 16, 4399-418) и патенты WO 2009/098450, WO 2013/050615 и WO 2013/050616). Вкратце, они включают замену аминокислоты, которая обеспечивает точку распознавания протеазой(ами), изменение аминокислотного остова в сайте расщепления (т.е. N-метилирование, псевдопептидные связи и т.д.), стерическое препятствие для соседних связей (т.е. β-замещенные аминокислоты), и включение D-энантиомерных аминокислот. Некоторые из этих модификаций (т.е. N-метилирование, D-аминокислоты), могут защищать/экранировать сайт протеолитического расщепления от гидролиза, даже если они расположены на расстоянии до двух остатков вдали от сайта расщепления. Хотя это относительно непосредственный путь защиты последовательности от протеолитического действия, он намного более сложен для включения стабилизирующих изменений, которые существенно не изменяют активность (и специфичность) в отношении белка-мишени.
Из данных протеолитической деградации 17-69-07 ex/in vivo, представленных в предыдущем разделе, ясно, что Tyr1/9 и остатки в непосредственной близости от него являются потенциальными сайтами для стабилизации бициклического пептида. Количественные способы оценки успешной стабилизации пептида заключаются в увеличении его периода полужизни в плазме мыши и человека.
В первую очередь было создано одиннадцать производных 17-69-07, в которых каждое положение было заменено на аланин (обозначаемое как «аланиновое сканирование»). Этот тип информации сообщает об энергетическом вкладе и роли определенных остатков и потенциально удаляет точки протеолитического распознавания.
Таблица 5: Результаты аланинового сканирования
(SEQ ID NO: 2)
17-69-07-N004 представляет собой немодифицированный пептид дикого типа, содержащий кеппированный N-конец (N-концевое ацетилирование, обозначаемое как «Ac»). Некоторые из замен на Ala хорошо переносятся, особенно в положениях 1, 3, 4, 10 и 11 в пределах последовательности. Поскольку боковые цепи этих остатков не требуются для высокой аффинности связывания с мишенью, соединения формулы (I) в этих конкретных положениях последовательности определяются в широком смысле.
Это делает замену в остатке 1 (Tyr 1) очень привлекательной возможностью, так как она может удалить одну из точек протеолитического распознавания. Замена Gly5 на Ala5 представляет интерес, так как химическая модификация является незначительной (добавление метильной группы), но приводит к резкому снижению активности. Вполне возможно, что Gly5 принимает необычные углы фи/пси вне общей диаграммы Рамачендрана, и эти углы могут быть потенциально индуцированы D-аминокислотами. Дополнительным преимуществом должна быть стабилизация пептидных связей, прилегающих к этому сайту.
В связи с этим осуществляли частичное D-аланиновое сканирование для оценки, являются ли они устойчивыми в отношении поддержания активности: включение D-аминокислот в последовательность является весьма желательным вследствие их стабилизирующего эффекта в отношении протеолитического расщепления.
Таблица 6: Эффект замены остатков в первой петле D-аланинами
D-Ala1 вместо Tyr1 связывается с аффинностью, меньшей в 10 раз, чем дикий тип. Тем не менее, это представляет интерес, так как удаляется точка Tyr1 протеолитического распознавания и заменяется стабилизирующей аминокислотой без индукции существенной потери активности.
Примечательно, что замена Gly5 на D-Ala5 хорошо переносится, так как аффинность по сравнению с диким типом остается неизменной.
В этом контексте D-Arg5 также переносится (и подобно этому, следовательно, все D-аминокислоты за исключением D-Pro, смотри таблицу 6), что может представлять интерес, если физико-химические свойства нуждаются в изменении в ходе дальнейшего дизайна молекулы (таблица 7).
Таблица 7: Влияние замены остатков в положении 5
Далее, в связи с выходами фагового отбора, указывающими на определенное предпочтение гидрофобных и ароматических аминокислот в положении 4 последовательности, был синтезирован ряд производных, включающих выбранные ароматические аминокислоты, включая природные тирозин и триптофан (таблица 8).
Таблица 8: Влияние замены остатков в положении 4
1Nal: 1-нафтилаланин; 2Nal: 2-нафтилаланин; Chg: циклогексилглицин, Phg: фенилглицин; tBuGly: трет-бутилглицин; 3,4-DCPhe4: 3,4-дихлорфенилаланин; Cha: циклогексилаланин; и HPhe: гомофенилалалнин.
Некоторые замены в положении 4 повышают аффинность по сравнению с диким типом, к ним относятся тирозин, триптофан, 1- и 2-нафтилаланин (1/2 Nal) и 3,4-дихлорфенилаланин (3,4-DCPhe). Наиболее сильной заменой является 1-нафтилаланин, что повышает аффинность в 7 раз.
Некоторые остатки в петле 2 в 17-69-07 исследовали с целью повышения стабильности молекулы. Они включают остаток 9, который включает потенциальную точку распознавания Tyr9. Остаток 11 также является привлекательным, так как он позволяет замены (таблица 5) и находится вблизи точки распознавания Tyr9.
Таблица 9: Сводка переносимых аминокислотных замен
Интерес представляет 4-бромфенилаланин (4BrPhe), который изменяет точку протеолитического распознавания Tyr9, и трет-бутилглицин (tBuGly), который незначительно повышает сродство и, что важно, существенно защищает соседние аминокислоты каркаса от протеолитического гидролиза с помощью стерического препятствия.
Замены множественных сайтов для достижения глобальной протеолитической защиты 17-69-07
В предыдущем разделе описаны множественные положения в пределах последовательности 17-69-07, которые разрешают включение аминокислот, повышающих протеолитическую стабильность и/или аффинность.
В целях объединения этих модификаций в одной молекуле, синтезировали молекулу, которая включала замены Tyr1 D-Ala1, Phe41-нафтилаланин4, Gly5D-Ala5, Tyr94BrPhe9 и Ile11tBuGly11. Примечательно, что все модификации переносятся совместно, и активность является более высокой, чем у молекулы дикого типа (таблицы 10 и 11).
D-Ala1, Phe41-нафтилаланин4, Gly5D-Ala5, Tyr94BrPhe9 и Ile11tBuGly11. Примечательно, что все модификации переносятся совместно, и активность является более высокой, чем у молекулы дикого типа (таблицы 10 и 11).
Таблица 10: Результаты конкретной множественной замены
При прогнозировании присоединения эффекторных групп к такому бициклическому пептиду в ходе дальнейшего дизайна молекулы были получены варианты с помощью N-концевого саркозинового молекулярного спейсера (10 последовательных Sar, обозначаемых как Sar10), начинающегося с N-концевого глицина и заканчивающегося С-концевым аланином. В целях этого эксперимента N-концевой Gly был кеппирован ацетильной группой, с тем, чтобы удалить положительный заряд.
Таблица 11: Сравнительные данные после добавления G-Sarl0-A
Эти данные указывают на то, что оба молекулярных спейсера (присоединенные к N-концу, как показано) и аминокислотные замены в пределах каркаса бициклической последовательности хорошо переносятся, тогда как активность сохраняется или улучшается.
В таблице 12 представлены неацетилированные, (не)стабилизированные производные молекул, показанных в таблице 11. Их стабильность оценивали количественно в плазме мыши и человека для того, чтобы продемонстрировать улучшение по сравнению с нестабилизированным 17-69-07-N219 (фигура 1).
Таблица 12: Молекулы, выбранные для оценки стабильности в плазме, связанной с активностью
На фигуре 3А показана стабильность в плазме мыши пента- и тетразамещенных молекул 17-69-07-N244 и 17-69-07-N231, соответственно, по сравнению с исходной нестабилизированной молекулой 17-69-07-N219 дикого типа. Период полужизни пептида в плазме мыши при 37°С составляет >>20 часов по сравнению с 6 часами у молекулы дикого типа с неусиленной стабилизацией.
На фигуре 3В показана стабильность в плазме человека пента- и тетразамещенных молекул 17-69-07-N244 и 17-69-07-N231, соответственно, по сравнению с исходной нестабилизированной молекулой 17-69-07-N219 дикого типа. Период полужизни пептида в плазме мыши при 37°С составляет >>20 часов по сравнению с 6 часами у молекулы дикого типа с неусиленной стабилизацией.
Таким образом, целевые замены до 5 положений в бициклической каркасной последовательности (Tyr1D-Ala1, Phe41Naphthylalanine4, Gly5D-Ala5, Tyr94BrPhe9 и Ile11tBuGly11) дают превосходные молекулы с повышенной активностью и значительным улучшением стабильности в плазме.
Селективность стабилизированного производного 17-69-07 (17-69-07-N241)
Стабилизированное, содержащее молекулярный спейсер производное 17-69-07-N241 тестировали с помощью конкурентного анализа FP на аффинность к домену гемопексина МТ1-ММР, полученному от других видов. Данные представлены в таблице 13:
Таблица 13: Видовая перекрестная реактивность 17-69-07 и его производных
Как нестабилизированные, так и стабилизированные производные 17-69-07 обладают полной перекрестной реактивностью.
Производное 17-69-07-N241 с N-концевым флуоресцеином (обозначаемое как 17-69-07-N258, с последовательностью: флуоросцеин-(bAla)-Sar10-A-(17-69-07) D-Ala1 1Nal4 D-Ala5 tBuGly11) тестировали против родственных металлопротеиназ человека. Полученные данные показывают, что каркасная последовательность 17-69-07 и этот стабилизированный вариант однозначно селективны для МТ1-ММР (таблица 14).
Таблица 14: Селективность 17-69-07 и его производных в отношении родственных металлопротеиназ
(=MMP-14)
(Kd, в нМ)
(Kd, в нМ)
(Kd, в нМ)
(Kd, в нМ)
(Kd, в нМ)
Пример 3: Анализ протеолитически стабилизированных вариантов 17-69-07, конъюгированных с хелатором, in vivo
Пептиды, связанные с хелаторами металлов, находят множество применений в диагностике и терапии. Определенные агенты визуализации или терапевтические радиоизотопы могут быть «нагружены» на хелатор, в то время как пептид переносит указанные изотопы к мишени. Таким образом, специфичные для опухоли антигены могут быть визуализированы с использованием, например, сканеров ПЭТ и SPECT. Для таргетной лучевой терапии терапевтические радионуклиды (например, 90Y и 177Lu) нагружают на хелатор-пептид и селективно доставляют в опухоль в результате взаимодействия со связанным с опухолью антигеном. Там они проявляют свою противоопухолевую активность в результате высокой энергии облучения, которую они излучают.
Синтез связанных с хелатором бициклических пептидов 17-69-07
Синтезировали пять производных, в которых хелатор металла DOTA (1,4,7,10-тетраазациклододекан-1,4,7,10-этилендиаминтетрауксусная кислота) был связан с N-концевым аланином бициклического пептида 17-69-07.
Таблица 15: Краткое описание молекул и структур
17-69-07-N144 представляет собой последовательность дикого типа 17-69-07, где DOTA непосредственно связана с N-концевым аланином, который служит в качестве спейсера из одной аминокислоты. Эта молекула сохраняет полную активность в отношении МТ1-ММР. Для демонстрации того, что активность сохраняется при нагрузке терапевтическим/визуализирующим радионуклидом, хелатор нагружали природным (холодным) 175лютецием в качестве безопасной замены. Активность сохранялась.
Полностью стабилизированный вариант, 17-69-07-N248, также сохранял активность в контексте конъюгата с DOTA.
Получали контрольную молекулу, 17-69-07-N246, которая является полным D-аминокислотным эквивалентом 17-69-07-N144. Эта молекула полностью устойчива к протеолитической деградации, так как амидные связи, примыкающие к D-аминокислотам, защищены от протеаз, и не обладает какой-либо активностью в отношении МТ1-ММР. Важно отметить, что этот пептид сохраняет точно такую же последовательность и химический состав.
Биораспределение 17-69-07-N144, нагруженного 177Lu, у мышей с ксенотрансплантатом HT1080
HT-1080 (клетки, в которых, как известно, наблюдается повышенная экспрессия МТ1-ММР) подкожно инокулировали в правую сторону туловища самцов мышей линии BALB/c nu/nu 6-недельного возраста. Опухолям давали расти в течение прибл. 1 недели.
150 пмоль пептида 17-69-07-N144 нагружали 1 МБк γ- и β-излучателем 177Lu с использованием стандартных методов комплексообразования. Затем вводили 150 пмоль нагруженного 17-69-07-N144 на одну мышь-опухоленоситель (эквивалент 17 мкг/кг). 3 животных подвергали эвтаназии на каждую временную точку, различные органы и опухоли извлекали и взвешивали, и затем определяли уровень гамма-излучения на грамм ткани опухоли/органа. Полученный график дает количественное представление о локализации пептида у мыши in vivo. В частности, избирательное накопление в опухоли HT1080 указывает на связывание МТ1-ММР in vivo.
На фигуре 4 суммированы данные по биораспределению. Примечательно, что бициклический пептид является специфичным для опухоли и, как представляется, сохраняется до 24 часов, несмотря на расчетный период полужизни в кровотоке 14 мин. Это, вероятно, указывает на захват опухолевыми клетками. Существенная локализация видна в почках, и возможно это связано с транспортерами аминокислот в почках, транзиторно связывающими пептид. Во всех остальных органах не выявляется значительный захват молекулы. Следует отметить, что домен гемопексина мыши характеризуется полной гомологией с доменом гемопексина человека, указывая, на то, что если бы РЕХ мыши экспрессировался в других тканях, должны были бы наблюдаться соответствующие сигналы.
Для оценки того, является ли захват опухолью в модели ксенотрансплантата селективным и обусловлен ли связыванием РЕХ, было проведено дополнительное исследование с использованием пептида 17-69-07-N246, который представляет собой D-аминокислотный аналог 17-69-07-N144 (фигура 5).
При сравнении характера биологического распространения, полученного с активным бициклическим пептидом 17-69-07-N144, становится ясно, что бициклический пептид 17-69-07-N246, не связывающий МТ1-ММР, направленно не аккумулируется в опухоли, подтверждая, что опухолевый сигнал 17-69-07-N144 управляется в результате направленного связывания MT1-ММР in vivo.
Наконец, для оценки влияния протеолитической стабилизации последовательности 17-69-07 на биораспределение использовали бициклический пептид 17-69-07-N248 (DOTA-A-(17-69-07) D-Ala1 1Nal4 D-Ala5 4BrPhe9 tBuGly11) в идентичном исследовании биораспределения (фигура 6).
Поразительно, повышенная протеолитическая устойчивость пептида приводит к общему удвоению захвата опухолью в любой данной точке времени по сравнению с 17-69-07-N144, иллюстрируя превосходные свойства молекулы по сравнению с исходной, нестабилизированной последовательностью 17-69-07.
Предполагается, что этот эффект может привести к благоприятному терапевтическому индексу, как в случае направленной радионуклидной терапии с использованием конъюгатов, описанных в этом примере, так и в контексте конъюгированных с токсином бициклических пептидов (пример 4).
Пример 4: Конъюгирование нестабилизированных бициклических пептидов с цитотоксическими агентами
Для таргетной терапии рака чрезвычайно сильнодействующие цитотоксические лекарственные средства присоединяют через расщепляемый линкер к направляющей части (в настоящем документе, к бициклическому пептиду), который связывается с ассоциированными с опухолью белками, экспрессирующимися на клеточной поверхности. Гиперэкспрессирующийся, ассоциированный с опухолью белок-мишень клеточной поверхности выбирают по его способности к интернализации внутрь клетки. После связывания соединения цитотоксический агент-конъюгированная направляющая часть с ассоциированным с опухолью белком клеточной поверхности весь молекулярный комплекс интернализуется внутрь клетки. После перехода из системной циркуляции в отдельную внутриклеточную среду, цитотоксическое лекарственное средство отщепляется от направляющей части в результате внутриклеточных условий среды, и отщепленное лекарство затем оказывает свое противоопухолевое действие путем индукции направленной клеточной смерти в результате блокирования клеточного цикла с последующим апоптозом.
Бициклический пептид 17-69-07-N219 (смотри примеры 2 и 3) состоит из бициклической каркасной последовательности дикого типа, связанной на N-концевой стороне с молекулярным спейсером Gly-Sar10-Ala (G-Sar10-A-(17-69-07), где описание полной последовательности представляет собой G-Sar10-А-CYNEFGCEDFYDIC; SEQ ID NO: 20). Это производное (17-69-07) сохраняет полную активность (таблица 12) в отношении МТ1-ММР при Kd 0,8 нМ. Свободная уникальная аминогруппа на N-концевой стороне молекулярного спейсера идеально подходит для конъюгации с эффекторными группами, такими как сильнодействующие цитотоксические агенты. Длинное внутримолекулярное расстояние, придаваемое линкером Sar10 между сайтом конъюгации на N-концевом Gly и каркасной бициклической последовательностью, используется таким образом, чтобы гарантировать полное сохранение целевой активности связывания после конъюгации с эффекторными группами существенного молекулярного размера. Более короткие молекулярные спейсеры могут быть выбраны по желанию до тех пор, пока сохраняется активность каркасной бициклической последовательности в отношении белка-мишени.
Для того чтобы получить экспериментальное доказательство концепции бициклического пептидного конъюгата лекарства (обозначаемого как BDC), направленного на MT1-MMP, получено два конъюгата 17-69-07-N219, которые используют либо токсины DM1, ингибирующие полимеризацию микротрубочек (мейтанзин, также известный как N2'-деацетил-N2'-(3-меркапто-1-оксопропил)-мейтанзин) или MMAE (монометилауристатин E, также известный как (S)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метиламино)бутанамидо)бутанамид).
Конъюгат ММАЕ обозначается как BT17BDC-1 и отделен от предшественника 17-69-07-N219 посредством линкера валин-цитруллин (Val-Cit), включая самоуничтожаемую пара-аминобензилкарбонильную группу (PABC). Линкер Val-Cit избирательно расщепляется (гидролизуется) средой, богатой катепсином В, встречающейся во внутриклеточных лизосомах, и после гидролиза, PABC уничтожается высвобождая MMAE в качестве активного токсичного компонента. Линкер Val-Cit-PABC стабилен в кровотоке, и тем самым высвобождает токсин только после клеточной интернализации.
Структура конъюгата и схема синтеза для получения BT17BDC-1 представлена на схеме I:
Схема I
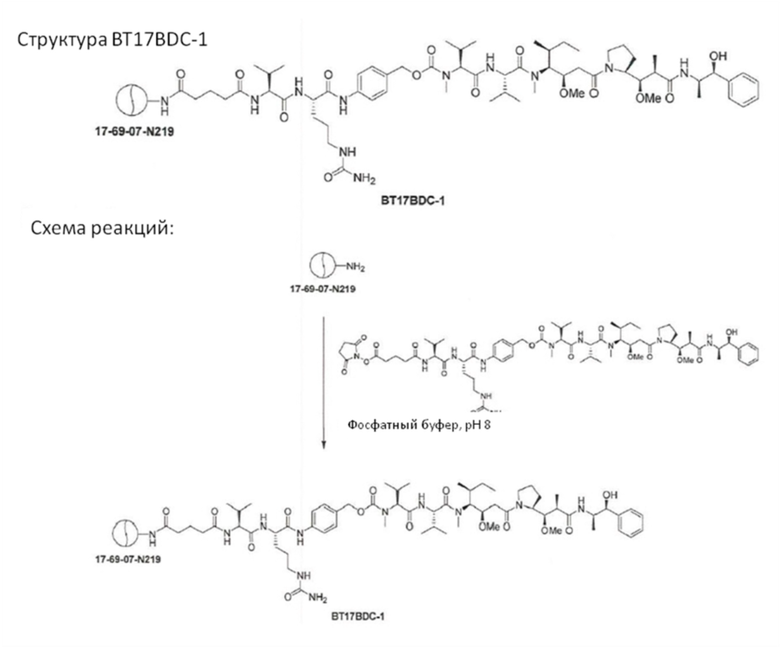
Стратегия синтеза для получения BDC17BDC-1: полностью очищенный циклизованный TMB бициклический предшественник 17-69- 07-N219 вводят в реакцию с сукцинимидным эфиром глутарил-валин-цитруллин-п-аминобензилкарбонил-MMAE (обычно сокращенно обозначаемым как VC-MMAE или Val-Cit-PABC-ММАЕ) с получением конъюгата BDC17BDC-1.
DM1 конъюгат 17-69-07-N219 обозначают как BT17BDC-9 и отделяют от предшественника 17-69-07-N219 дисульфидной связью, которая может быть расщеплена в восстанавливающей среде, встречающейся во внутриклеточной среде. Восстановление, как считается, возникает с помощью внутриклеточного глутатиона, который присутствует внутри клетки в концентрации ~10 мМ. В противоположность этому, концентрация глутатиона и свободных восстанавливающих агентов в циркулирующей крови значительно ниже (<10 мкМ); таким образом, токсин высвобождается преимущественно во внутриклеточной среде, где он может проявлять свою активность в отношении гибели клеток.
Структура конъюгата и схема синтеза для получения BT17BDC-9, показана на схеме II:
Схема II:
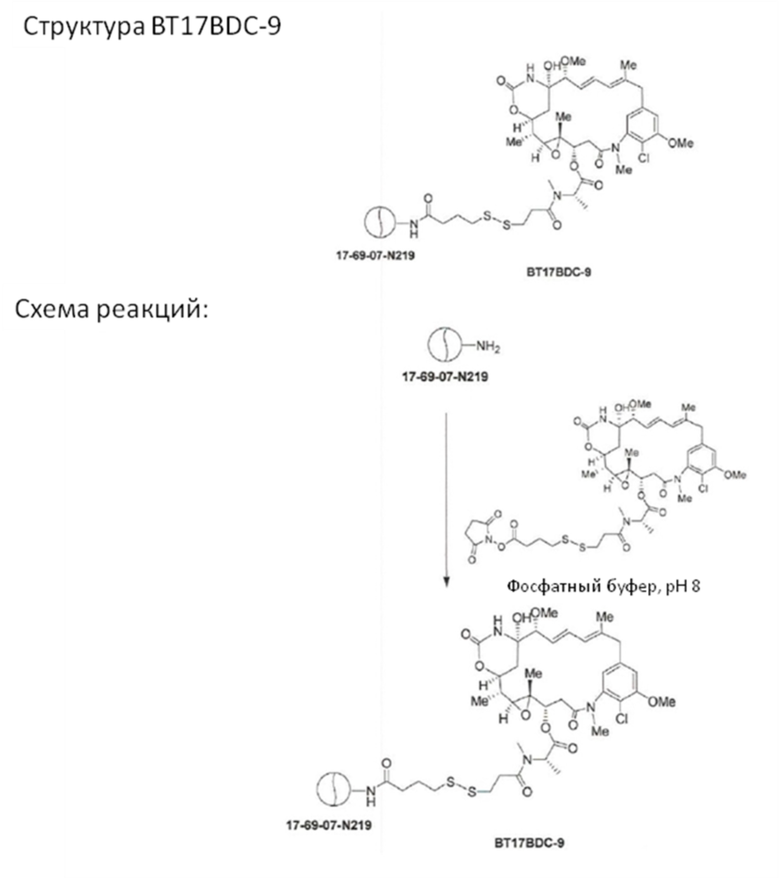
Стратегия синтеза для получения BDC17BDC-9: полностью очищенный циклизованный TMB бициклический предшественник 17-69-07-N219, содержащий свободный N-концевой амин, вводят в реакцию с сукцинимидным эфиром дисульфид-DM1 (структура, как показано) с получением конъюгата BDC17BDC-9.
Высвобождение токсинов в BDC17BDC-1 и BDC17BDC-9, следовательно, является механистически различным, т.е. в первом случае в результате внутриклеточных протеолитических условий, а в последнем за счет внутриклеточных восстанавливающих условий.
Два BDCs были использованы при исследовании цитотоксичности in vitro, и они показали низкую наномолярную/пикомолярную активность на клеточных культурах, таких как НТ1080 (данные не показаны).
В модели in vivo ксенотрансплантатов мышей (несущих опухоль HT1080) оба BDCs вызвали значительное уменьшение объема опухоли через 9 дней после инъекции по сравнению с контрольным носителем. Схема дозирования показана на фигуре 7 (стрелки). Опухоли полностью исчезали на 14 день для обоих BDCs, о чем судили с помощью пальпации (фигура 7). Следует отметить, что масса животных как для BDC17BDC-1, так и для BDC17BDC-9 была в значительной степени стабильной, что указывает на терапевтическое окно, которое может быть достаточным для терапевтических целей.
Пример 5: Конъюгирование протеолитически стабилизированных бициклических пептидов с цитотоксическими агентами
Бициклическая каркасная последовательность 17-69-07, содержащая модификации: D-аланин в положении 1, 1-нафтилаланин в положении 4, D-аланин в положении 5, α-трет-бутилглицин в положении 11 с 4-бромфенилаланином в положении 9 или без него, имела повышенную протеолитическую стабильность в плазме ex vivo и in vivo (примеры 2, 3).
В контексте конъюгатов бициклический пептид-лекарство вполне вероятно, что более стабильная каркасная бициклическая последовательность может привести к большей экспозиции с опухолью из-за снижения системного клиренса и более высокой стабильности в протеолитически агрессивном микроокружении опухоли. В обоих случаях это может привести к увеличению управляемому МТ1-ММР захвату BDC внутрь клетки, что ведет к увеличению терапевтической эффективности.
Стабилизированный бициклический пептид 17-69-07-N241 был разработан в целом по молекулярной схеме, сходной с нестабилизированным 17-69-07-N219 (описано в примерах 3 и 4). Он имеет следующую последовательность:
(B-Ala)-Sar10-AC(D-Ala)NE(1Nal)(D-Ala)CEDFYD(tBuGly)C (SEQ ID NO: 5)
которая циклизована с помощью TBMB, как описано выше.
N-концевой бета-аланин был выбран в отличие от используемого ранее глицина как и в 17-69-07-N219 так, чтобы свести к минимуму образование побочных продуктов дикетопиперазина во время присоединения сложных эфиров N-гидроксисукцинимида (NHS) (Purdie et al (1973), J. Chem. Soc., Perkin Trans. 2, 1845), в данном примере конкретно NHS эфиров цитотоксических агентов (схема I, II).
Саркозиновый декамерный спейсер сохраняется как и в 17-69-07-N219 так, чтобы обеспечить полное сохранение связывания бициклического пептида с доменом гемопексина МТ1-ММР.
Класс мейтанзиновых токсинов был выбран для конъюгации с 17-69-07-N241 на основе эффективности и переносимости в мышиной модели ксенотрансплантата, описанной в примере 4, и вновь был выбран дисульфидный расщепляемый линкер.
Получали четыре BDCs (обозначаемые в настоящем описании выше как BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20), в результате чего как токсин, так и 17-69-07-N241 были инвариантны, а чувствительность дисульфидной связи к восстановлению/расщеплению изменялась путем модуляции степени препятствий, прилегающих к атомам серы.
Схема синтеза конъюгатов соответствует описанной на схеме II, где 17-69-07-N219 заменен на 17-69-07-N241.
Для BT17BDC-18 дополнительный путь синтеза включал создание пиридил-дисульфидного бициклического предшественника (обозначаемого как 17-69-07-N277), который взаимодействует с DM1 с образованием полного конъюгата. Путь синтеза описан в схеме III.
Схема III
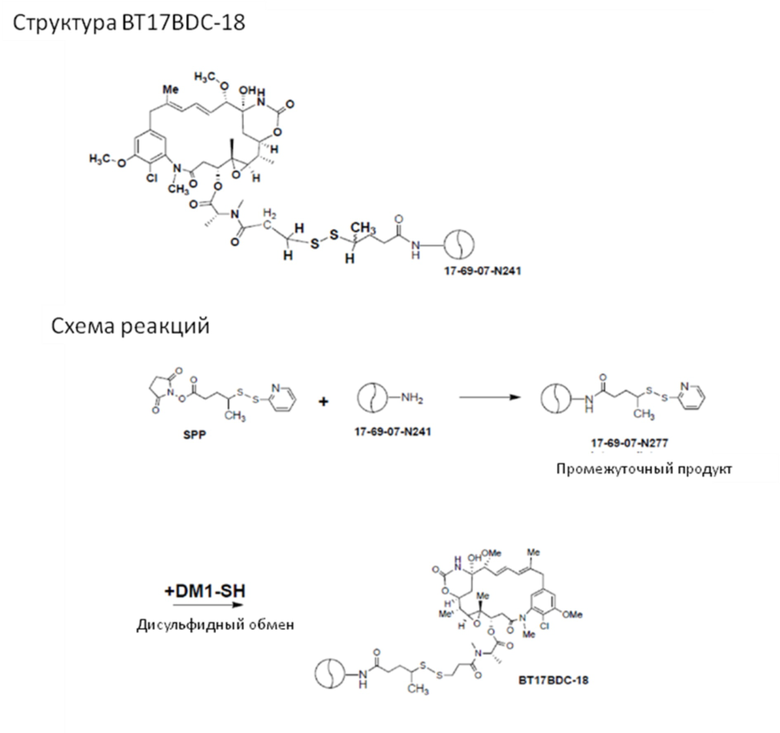
Стратегия синтеза для получения BDC17BDC-18: полностью очищенный циклизованный TMB бициклический предшественник 17-69- 07-N219, содержащий свободный N-концевой амин, вводят в реакцию с SPP (N-сукцинимидил-4-(2-пиридилдитио)пентаноатом)) с получением промежуточного соединения 17-69-07-N277. Оно затем вступает в реакцию с DM1 в виде свободного тиола с получением желаемого конъюгата BT17BDC-18.
Характеристики BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20 in vitro
Четыре BDCs оценивали по нескольким параметрам in vitro, таким как сохранение активности в отношении домена гемопексина МТ1-ММР человека, стабильность ex vivo в плазме мыши, крысы и человека, и стабильность в отношении восстанавливающих агентов, таких как дитиотреитол.
Данные представлены в таблице 16 ниже:
Таблица 16: Свойства BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20 in vitro
(домен гемопексина)a
(плазма человека)b
(плазма мыши)b
(плазма крысы)b
(ADC)c
(BDC)d
(n=3)e
(n=1)
(n=1)
(n=1)
(n=10)e
(n=2)
(n=2)
(n=1)
(n=1)e
(n=2)
(n=1)
(n=1)
(n=6) e
(n=2)
(n=2)
(n=2)
(n=1)e
(n=2)
(n=1)
(n=1)
(n=4)f
(n=1)
(n=1)
(n=1)
где n/a: не применяли, и где n=число повторов
a: определено в экспериментах по конкуренции с использованием поляризации флуоресценции с применением 17-69-07-N040 в качестве метки
b: определено с использованием количественной ЖХ-МС. Время инкубации в плазме, содержащей 4 мкΜ BDC, до 24 часов.
c: из Kellogg et al (2011) Bioconjugate Chemistry, 22, 717. Обратите внимание на то, что эти величины относятся к конъюгатам антительных лекарств, содержащим дисульфидный линкер, описанный в тексте
d: определяется с помощью количественной ЖХ-МС. Обратите внимание не то, что эти величины относятся к конъюгатам бициклических лекарств, содержащим дисульфидный линкер, описанный в тексте. Методы были адаптированы из Kellogg et al (2011) Bioconjugate Chemistry, 22, 717.
e: использовали 17-69-07-N004 в качестве метки в конкурентном анализе с помощью FP
f: использовали 17-69-07-N040 в качестве метки в конкурентном анализе с помощью FP
Все молекулярные конструкты сохраняют свою аффинность к мишени гемопексина (второй столбец).
Данные указывают на то, что стабильность в плазме определяется природой дисульфидной связи (в связи с модуляцией чувствительности к восстановлению), а не природой бициклического пептида, так как все BDCs содержат один и тот же бициклический пептид (17-69-07-N241), который стабилен в плазме трех тестируемых видов.
Кроме того, BT17BDC-18, BT17BDC-19 и BT17BDC-20 проявляют стабильность в плазме человека, адекватной для терапевтического применения, так как предполагается, что управляемый почечной фильтрацией клиренс пептидов с этим размером молекул у человека имеет расчетный период полужизни от 2 до 4 часов, что в несколько раз быстрее, чем период полужизни при деградации BDCs в плазме крови человека (>14 часов для BT17BDC-18/19/20, смотри таблицу 16). Таким образом, основная часть BDC, как ожидается, экскретируется через почки, и только некоторая фракция деградирует в кровотоке, что делает эти BDCs потенциально пригодными для терапевтических целей.
Эффективность BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20 in vivo
Все BDCs, содержащие стабилизированную каркасную бициклическую последовательность, тестировали на их эффективность в моделях ксенотрансплантата мыши in vivo с использованием линии EBC-1 плоскоклеточного рака легких человека.
BT17BDC-17, BT17BDC-18 и BT17BDC-19 были эффективны и вызывали освобождение от опухоли через 9 дней (фигуры 9, 10 и 11). BT17BDC-17 показал хорошую эффективность, но и некоторое снижение массы при высоких дозах. BT17BDC-20 при переносимости, основанной на постоянной массе, был неэффективным и вызывал незначительное уменьшение размеров опухоли (фигура 12).
Площадь под кривой (AUC) графика зависимости объема опухоли от времени и BDC брали и наносили на график относительно группы с соответствующей дозой (фигура 13). Исходя из этого, может быть определена эффективная доза для достижения 50% снижения AUC опухоли (ED50), что суммировано в таблице 17.
Таблица 17: Эффективная доза для достижения 50% снижения AUC опухоли для BT17BDC-9, BT17BDC-17, BT17BDC-18, BT17BDC-19 и BT17BDC-20
a ED50±95% доверительный интервал
b единицы в мг/кг, у мышей, несущих опухоли EBC-1
Таким образом, BT17BDC-9, BT17BDC-17, BT17BDC-18 и BT17BDC-19 являются подходящими молекулами для использования в таргетной терапии рака на основе их эффективности, и BT17BDC-17, BT17BDC-18 и BT17BDC-19 хорошо переносятся в эффективных дозах.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Bicycle Therapeutics Limited
<120> Бициклические пептидные лиганды, специфичные для MT1-MMP
<130> BIC-C-P1803PCT
<150> GB1419237.1
<151> 2014-10-29
<150> GB1515245.7
<151> 2015-08-27
<160> 23
<170> Патент версии 3.5
<210> 1
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Прочие особенности
<222> (2)..(2)
<223> Xaa может представлять собой любую природную аминокислоту
<220>
<221> Xaa
<222> (3)..(3)
<223> Xaa представляет собой либо полярный, незаряженный аминокислотный
остаток, выбранный из N, C, Q, M, S и T или неполярный алифатический
аминокислотный остаток, выбранный G, A, I, L, P и V
<220>
<221> Прочие особенности
<222> (4)..(5)
<223> Xaa может представлять собой любую природную аминокислоту
<220>
<221> Прочие особенности
<222> (12)..(13)
<223> Xaa может представлять собой любую природную аминокислоту
<400> 1
Cys Xaa Xaa Xaa Xaa Gly Cys Glu Asp Phe Tyr Xaa Xaa Cys
1 5 10
<210> 2
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 2
Cys Tyr Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 3
<211> 17
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (1)..(1)
<223> Xaa представляет собой бета-Ala
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Sar10
<400> 3
Xaa Xaa Ala Cys Tyr Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile
1 5 10 15
Cys
<210> 4
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой D-Ala
<400> 4
Cys Xaa Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 5
<211> 17
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (1)..(1)
<223> Xaa представляет собой бета-Ala
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Sar10
<220>
<221> Xaa
<222> (5)..(5)
<223> Xaa представляет собой D-Ala
<220>
<221> Xaa
<222> (8)..(8)
<223> Xaa представляет собой NAl
<220>
<221> Xaa
<222> (9)..(9)
<223> Xaa представляет собой D-Ala
<220>
<221> Xaa
<222> (16)..(16)
<223> Xaa представляет собой tBuGly
<400> 5
Xaa Xaa Ala Cys Xaa Asn Glu Xaa Xaa Cys Glu Asp Phe Tyr Asp Xaa
1 5 10 15
Cys
<210> 6
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Y, M, F или V
<220>
<221> Xaa
<222> (3)..(3)
<223> Xaa представляет собой либо полярный, незаряженный аминокислотный
остаток, выбранный из N, C, Q, M, S и T или неполярный алифатический
аминокислотный остаток, выбранный G, A, I, L, P и V
<220>
<221> Xaa
<222> (4)..(4)
<223> Xaa представляет собой либо полярный, незаряженный аминокислотный
остаток, выбранный из N, C, Q, M, S и T или полярный, отрицательно
заряженный аминокислотный остаток, выбранный из D или E
<220>
<221> Xaa
<222> (5)..(5)
<223> Xaa представляет собой неполярный, ароматический аминокислотный
остаток, выбранный из F, W и Y
<220>
<221> Xaa
<222> (12)..(12)
<223> Xaa представялет собой полярный, отрицательно заряженный
аминокислотный остаток, выбранный из D или E
<220>
<221> Xaa
<222> (13)..(13)
<223> Xaa представляет собой неполярный, алифатический аминокислотный
остаток, выбранный из G, A, I, L, P и V
<400> 6
Cys Xaa Xaa Xaa Xaa Gly Cys Glu Asp Phe Tyr Xaa Xaa Cys
1 5 10
<210> 7
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Y, M, F или V
<220>
<221> Xaa
<222> (3)..(3)
<223> Xaa представляет собой N или G
<220>
<221> Xaa
<222> (4)..(4)
<223> Xaa представляет собой E или Q
<400> 7
Cys Xaa Xaa Xaa Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 8
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Y, M или F
<220>
<221> Xaa
<222> (3)..(3)
<223> Xaa представляет собой N или G
<220>
<221> Xaa
<222> (4)..(4)
<223> Xaa представляет собой E или Q
<400> 8
Cys Xaa Xaa Xaa Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 9
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Y или M
<220>
<221> Xaa
<222> (4)..(4)
<223> Xaa представляет собой E или Q
<400> 9
Cys Xaa Asn Xaa Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 10
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 10
Cys Met Asn Gln Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 11
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 11
Cys Phe Gly Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 12
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 12
Cys Val Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 13
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 13
Cys Phe Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 14
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 14
Cys Tyr Asn Glu Tyr Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 15
<211> 14
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 15
Cys Tyr Asn Glu Trp Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10
<210> 16
<211> 15
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 16
Cys Lys Asn Arg Gly Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys
1 5 10 15
<210> 17
<211> 8
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 17
Cys Glu Asp Phe Tyr Asp Ile Cys
1 5
<210> 18
<211> 16
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 18
Ala Cys Tyr Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys Ala
1 5 10 15
<210> 19
<211> 16
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 19
Ala Cys Met Asn Gln Phe Gly Cys Glu Asp Phe Tyr Asp Ile Cys Ala
1 5 10 15
<210> 20
<211> 17
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<220>
<221> Xaa
<222> (2)..(2)
<223> Xaa представляет собой Sar10
<400> 20
Gly Xaa Ala Cys Tyr Asn Glu Phe Gly Cys Glu Asp Phe Tyr Asp Ile
1 5 10 15
Cys
<210> 21
<211> 4
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 21
Tyr Asn Glu Phe
1
<210> 22
<211> 5
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 22
Tyr Asn Glu Phe Gly
1 5
<210> 23
<211> 6
<212> PRT
<213> Искусственная
<220>
<223> Синтетический пептид
<400> 23
Glu Asp Phe Tyr Asp Ile
1 5
<---
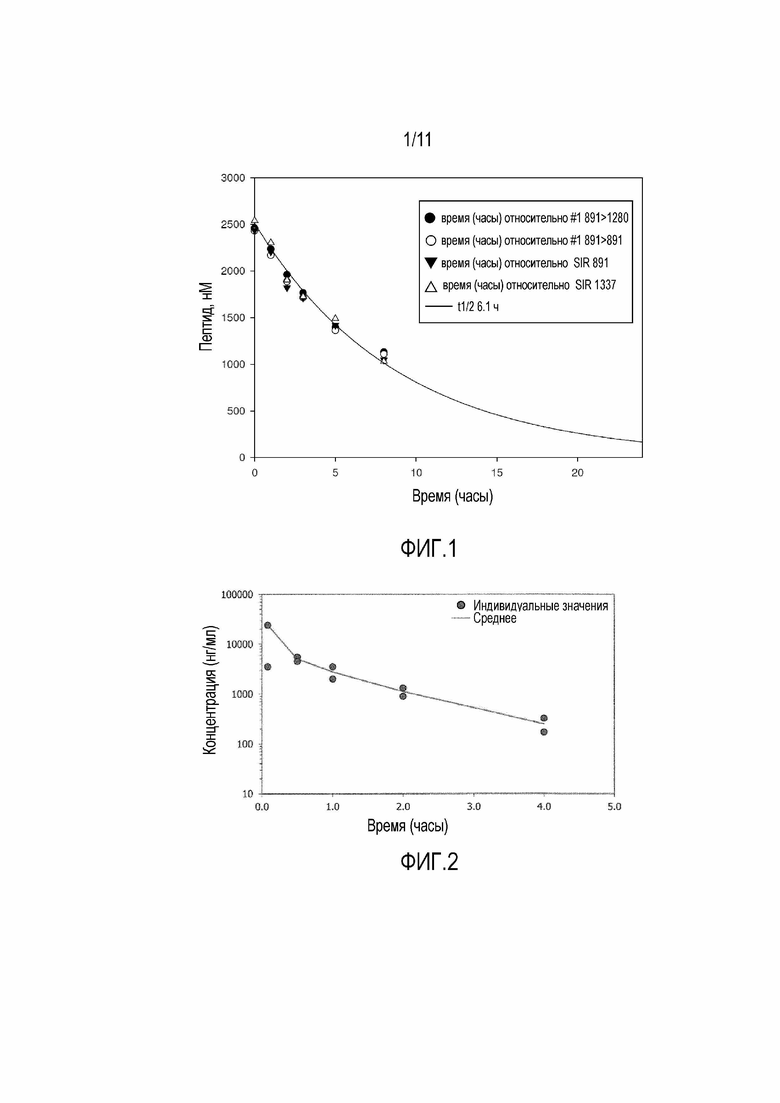
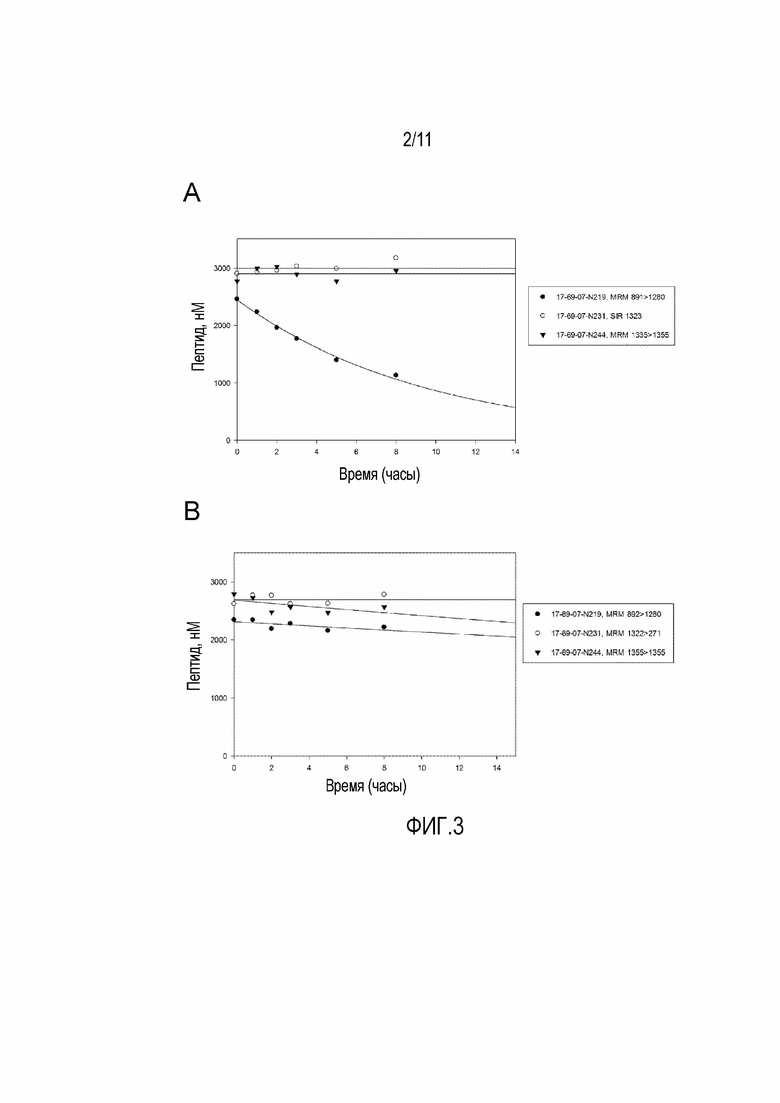
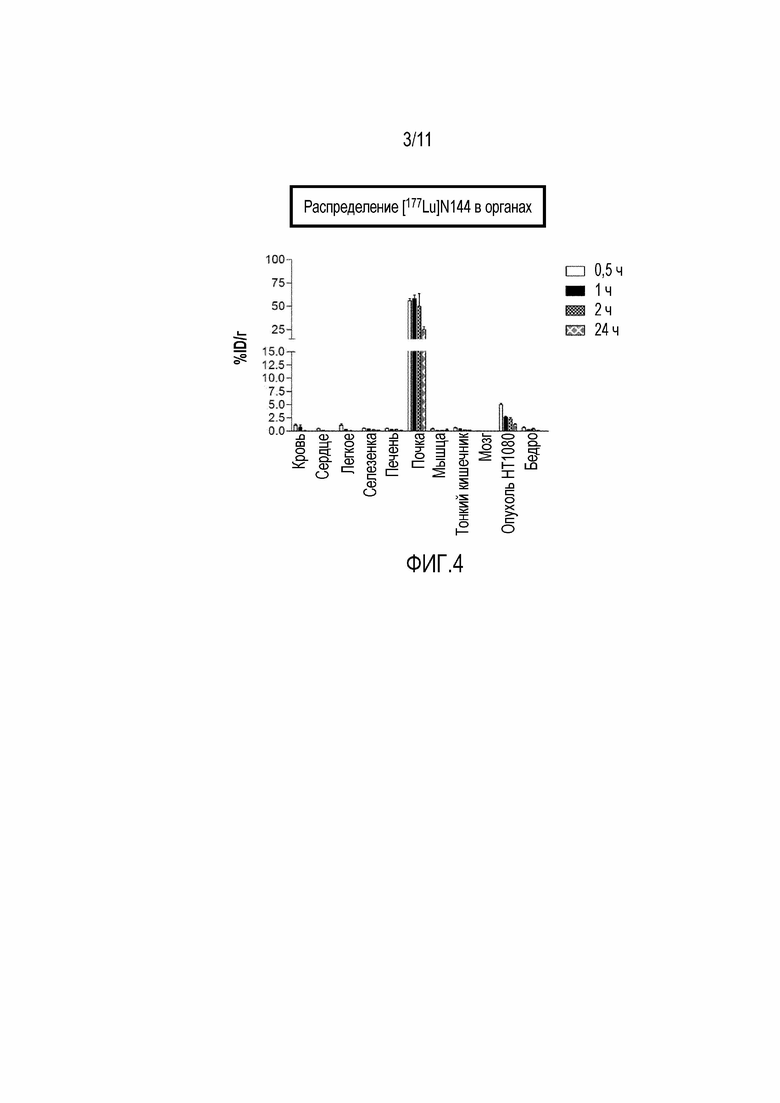
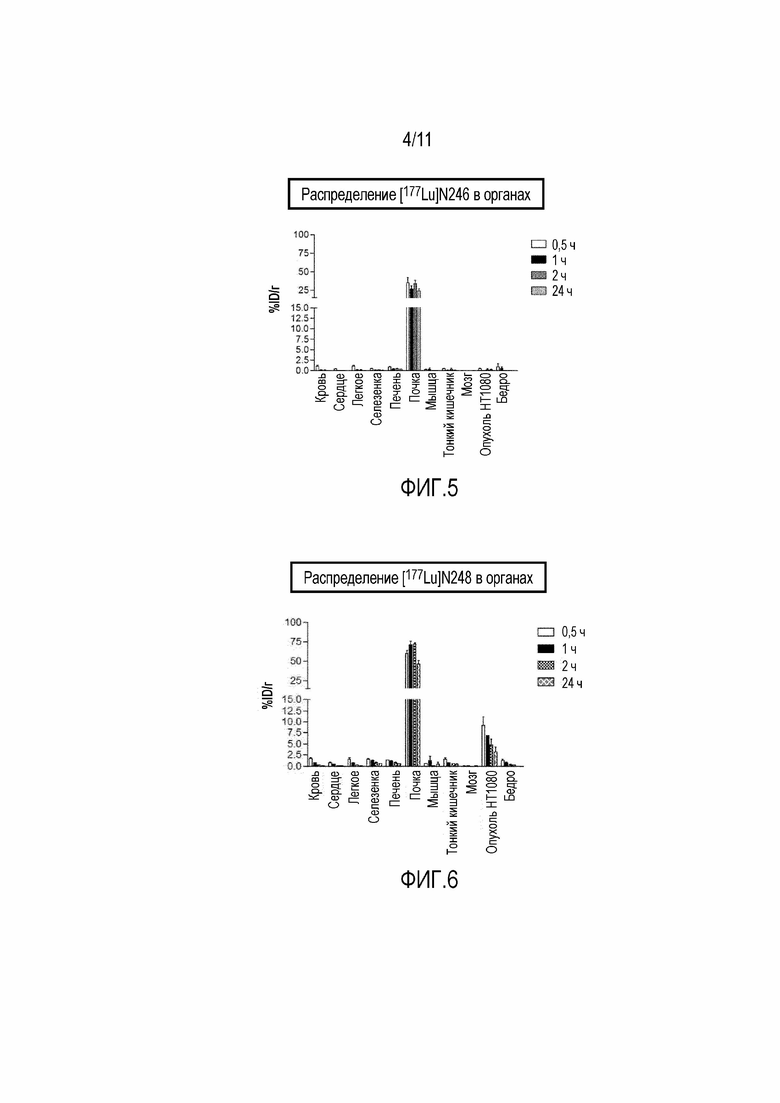
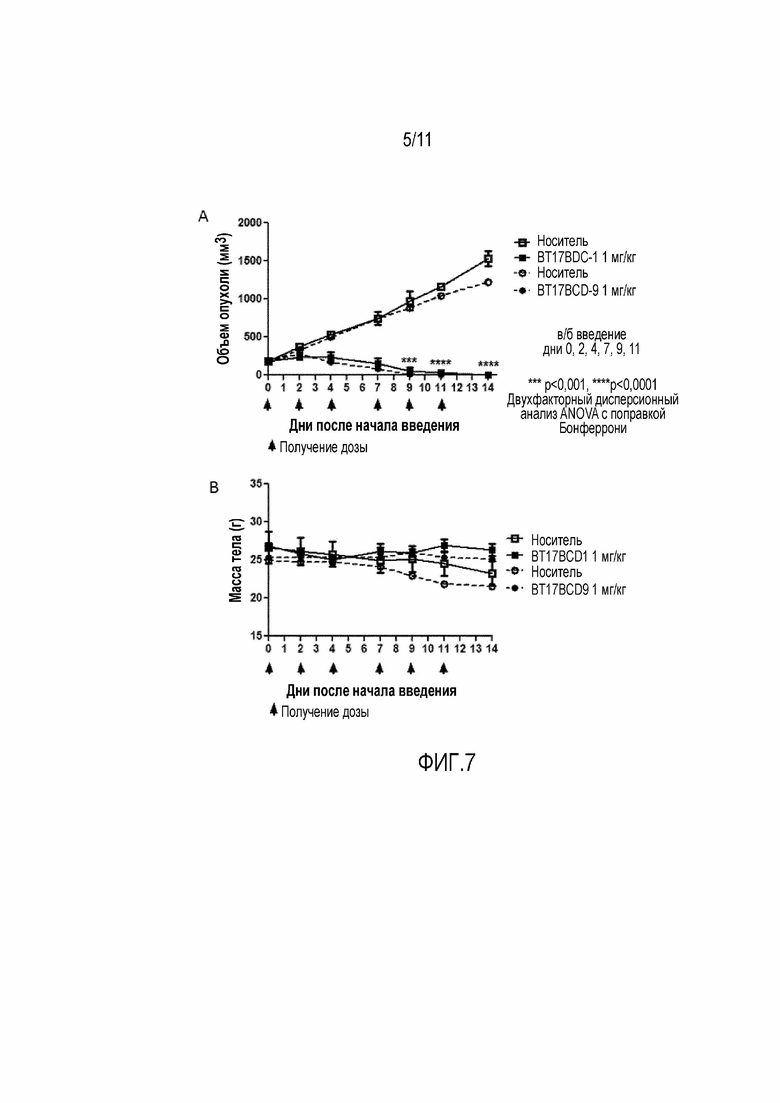

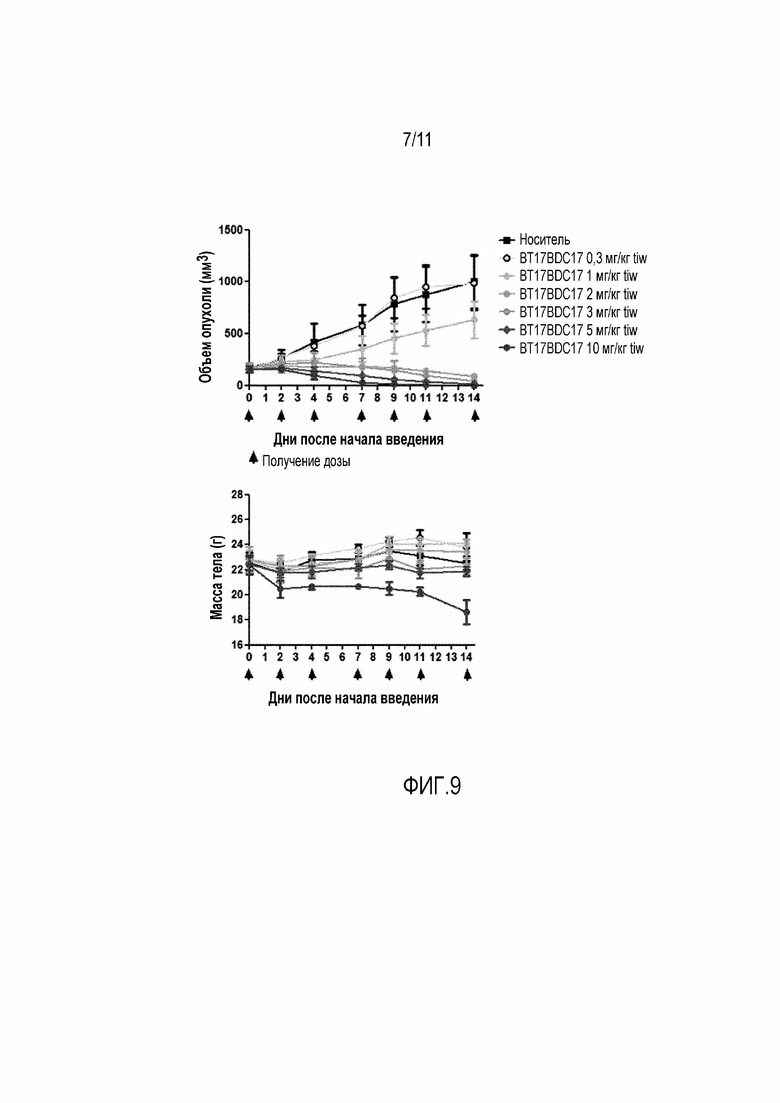
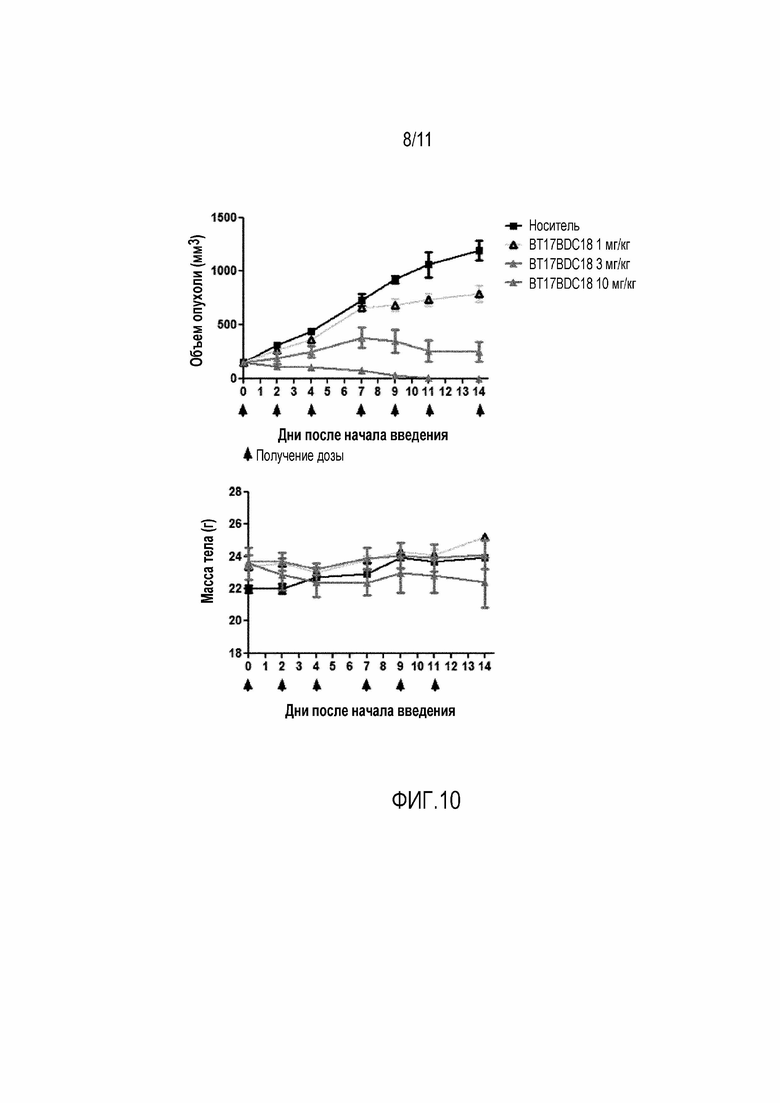
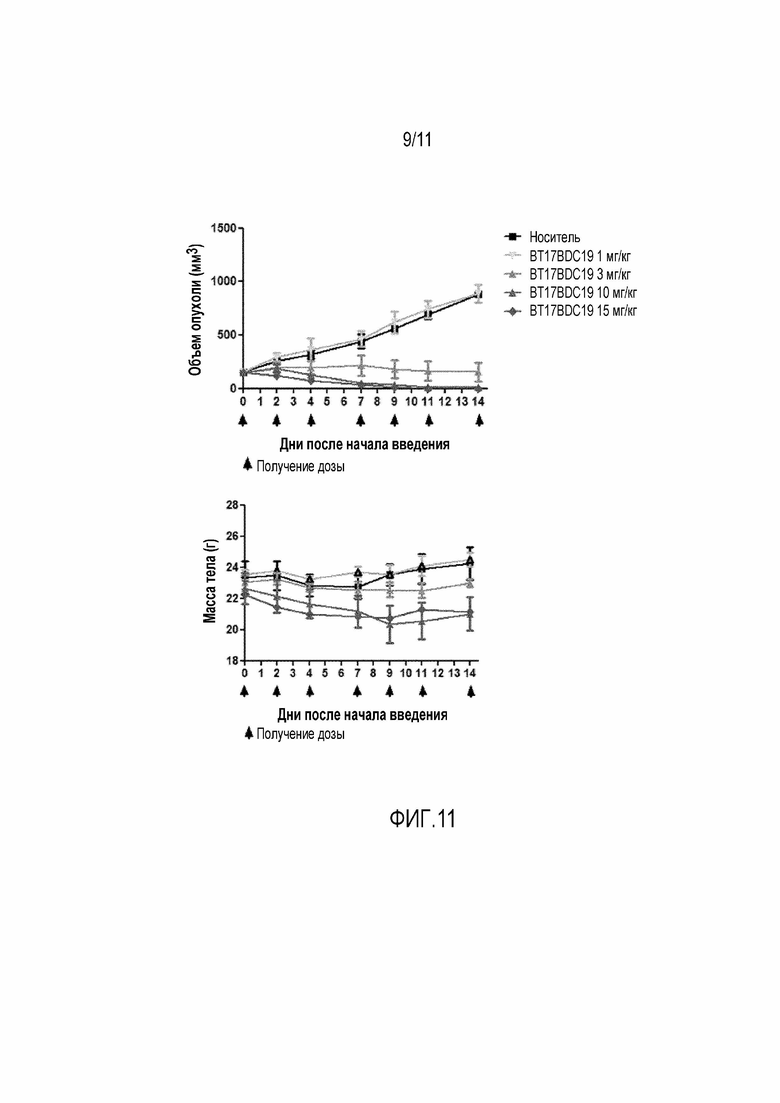
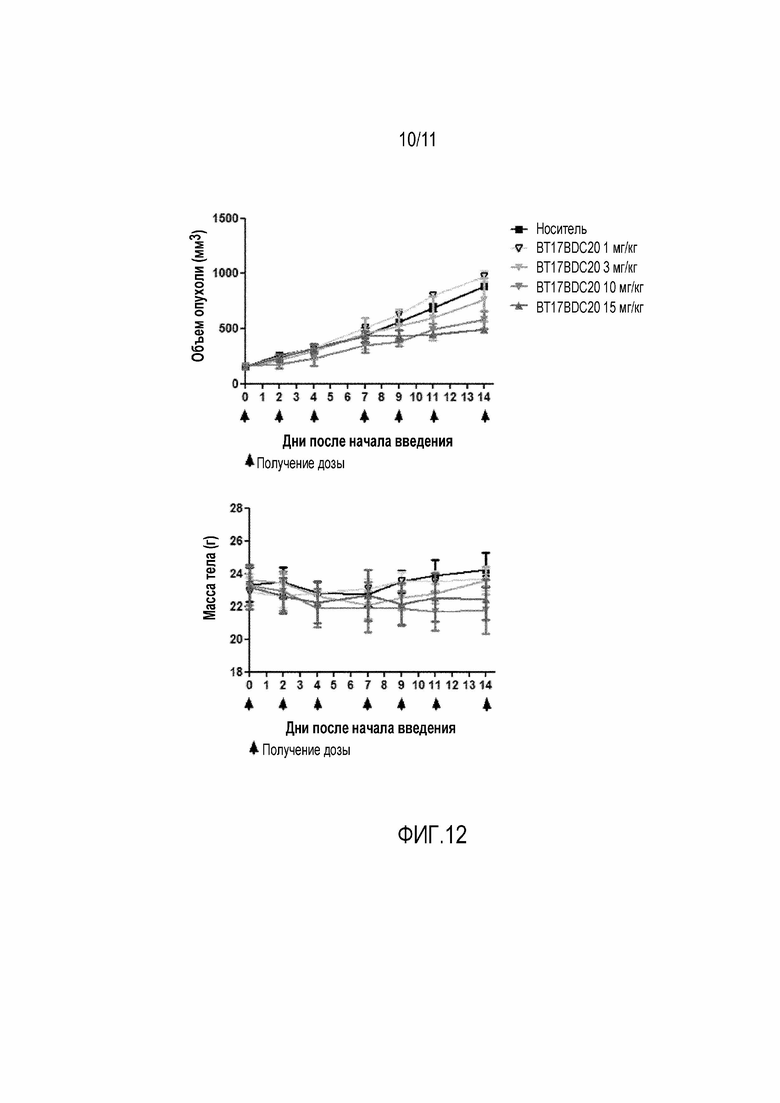
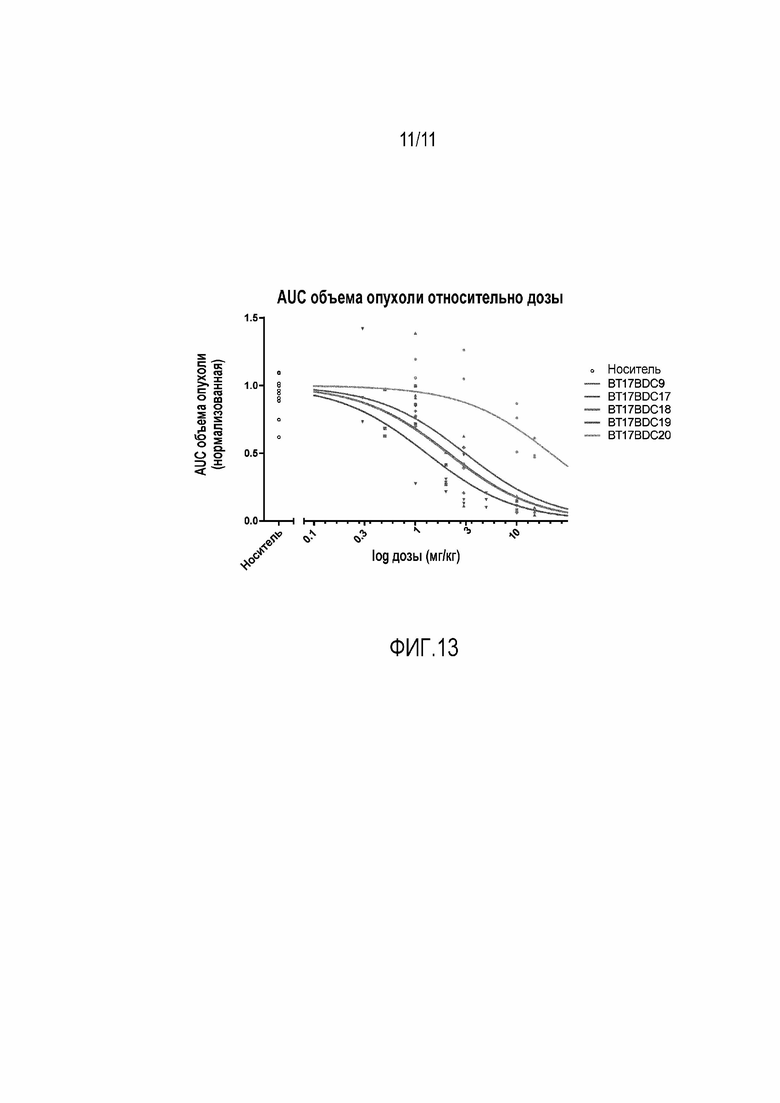
Изобретение относится к полипептидам, которые ковалентно связаны с молекулярными каркасами, так что две или более пептидные петли замыкаются между точками присоединения к каркасу. В частности, в изобретении описываются пептиды, которые связываются с высокой аффинностью с мембранной металлопротеазой типа 1 (MT1-MMP). В изобретении также описываются конъюгаты лекарств, включающие указанные пептиды, конъюгированные с одной или более эффекторными и/или функциональными группами, которые применяются в визуализации и таргетной терапии рака. 5 н. и 25 з.п. ф-лы, 13 ил., 17 табл., 5 пр.
1. Пептидный лиганд, специфичный для МТ1-ММР, включающий полипептид, включающий три остатка цистеина, разделенные последовательностями двух петель, и молекулярный каркас, который образует ковалентные связи с остатками цистеина полипептида таким образом, что образуются две полипептидные петли на молекулярном каркасе, где пептидный лиганд включает аминокислотную последовательность формулы (I):
-Ci-X1-U2/O2-X3-X4-G5-Cii-E6D7-F8-Y9-X10-X11-Ciii- (SEQ ID NO: 1) (I)
или его фармацевтически приемлемую соль,
где:
Ci, Cii и Ciii представляют собой первый, второй и третий остатки цистеина, соответственно;
Х1 выбран из любой из следующих аминокислот: Y, M, F или V;
U2/O2 выбрано из N или G;
Х3 выбран из U или Z, где U представляет собой полярный, незаряженный аминокислотный остаток, выбранный из N, C, Q, M, S и T, и Z представляет собой полярный, отрицательно заряженный аминокислотный остаток, выбранный из D или Е;
Х4 выбран из J, где J представляет собой неполярный остаток ароматической аминокислоты, выбранный из F, W и Y;
Х10 выбран из Z, где Z представляет собой полярный, отрицательно заряженный аминокислотный остаток, выбранный из D или Е; и
Х11 выбран из О, где О представляет собой неполярный остаток алифатической аминокислоты, выбранный из G, A, I, L, P и V;
необязательно при этом пептидный лиганд включает одну или более модификаций, выбранных из:
(i) N-концевой модификации, которая включает добавление молекулярной спейсерной группы, выбранной из Ala, группы G-Sar10-A и группы bAla-Sar10-A;
(ii) N-концевой и/или С-концевой модификации, которая включает добавление цитотоксического агента;
(iii) замены аминокислоты в положении 1 на D-аланин;
(iv) замены аминокислотного остатка в положении 4 неприродным аминокислотным остатком, выбранным из 1-нафтилаланина; 2-нафтилаланина; 3,4-дихлорфенилаланина; и гомофенилаланина;
(v) замены аминокислотного остатка в положении 9 на 4-бромфенилаланин;
(vi) замены аминокислотного остатка в положении 11 на трет-бутилглицин; и
(vii) 2, 3, 4 или 5 из следующих модификаций: D-аланин в положении 1 и/или 5, 1-нафтилаланин в положении 4, 4-бромфенилаланин в положении 9 и трет-бутилглицин в положении 11.
2. Пептидный лиганд по п. 1, где соединение формулы (I) представляет собой соединение формулы (Ia):
-Ci-Y/M/F/V-U/O-U/Z-J-G-Cii-E-D-F-Y-Z-O-Ciii- (SEQ ID NO: 6), (Ia)
где U, О, J и Z представляют собой определенное выше; или
соединение формулы (Ib):
-Ci-Y/M/F/V-N/G-E/Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (SEQ ID NO: 7) (Ib); или
соединение формулы (Ic):
-Ci-Y/M/F-N/G-E/Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (SEQ ID NO: 8) (Ic); или
соединение формулы (Id):
-Ci-Y/M-N-E/Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (SEQ ID NO: 9) (Id); или
соединение формулы (Ie):
-Ci-Y-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07) (SEQ ID NO: 2) (Ie).
3. Пептидный лиганд по п. 1 или 2, где пептид формулы (I) включает последовательность, выбранную из:
-Ci-Y-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07) (SEQ ID NO: 2);
-Ci-M-N-Q-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-12) (SEQ ID NO: 10);
-Ci-F-G-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-02) (SEQ ID NO: 11);
-Ci-V-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-03) (SEQ ID NO: 12);
-Ci-F-N-E-F-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-04) (SEQ ID NO: 13);
-Ci-Y-N-E-Y-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-07-N057) (SEQ ID NO: 14); и
-Ci-Y-N-E-W-G-Cii-E-D-F-Y-D-I-Ciii- (17-69-44-N002) (SEQ ID NO: 15).
4. Пептидный лиганд по любому из пп. 1-3, который включает одну или более модификаций, выбранных из:
(i) N-концевой модификации, которая включает добавление молекулярной спейсерной группы, выбранной из Ala, группы G-Sar10-A или группы bAla-Sar10-A;
(ii) N-концевой и/или С-концевой модификации, которая включает добавление цитотоксического агента;
(iii) замену аминокислоты в положении 1 на D-аланин;
(iv) замену аминокислотного остатка в положении 4 остатком неприродной аминокислоты, выбранным из: 1-нафтилаланина; 2-нафтилаланина; 3,4-дихлорфенилаланина; и гомофенилаланина;
(v) замену аминокислотного остатка в положении 9 на 4-бромфенилаланин;
(vi) замену аминокислотного остатка в положении 11 на трет-бутилглицин; и
(vii) 2, 3, 4 или 5 из следующих модификаций: D-аланин в положении 1 и/или 5, 1-нафтилаланин в положении 4, 4-бромфенилаланин в положении 9 и трет-бутилглицин в положении 11.
5. Пептидный лиганд по любому из пп. 1-4, где фармацевтически приемлемая соль выбрана из свободной кислоты или соли натрия, калия, кальция, аммония.
6. Пептидный лиганд по любому из из пп. 1-5, который представляет собой лиганд с высокой аффинностью связывания с доменом гемопексина МТ1-ММР человека, мыши и собаки.
7. Пептидный лиганд по любому из пп. 1-6, который является селективным для МТ1-ММР, но перекрестно не реагирует с ММР-1, ММР-2, ММР-15 и ММР-16.
8. Конъюгат лекарственного средства, включающий пептидный лиганд по любому из пп. 1-7, конъюгированный с одной или более эффекторными и/или функциональными группами, где эффекторная и/или функциональная группа включает цитотоксический агент, хромофор или хелатор металла.
9. Конъюгат лекарственного средства по п. 8, где эффекторная и/или функциональная группа включает цитотоксический агент или хелатор металла.
10. Конъюгат лекарственного средства по п. 9, где цитотоксический агент связан с бициклическим пептидом расщепляемой связью, такой как дисульфидная связь, или связью, чувствительной к протеазам.
11. Конъюгат лекарственного средства по п. 9 или 10, где цитотоксический агент выбран из DM1, который имеет следующую структуру:
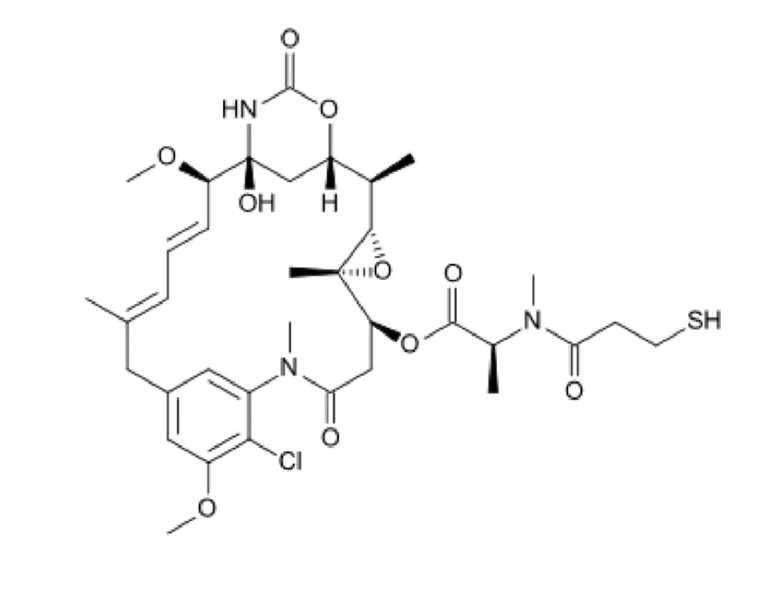
или MMAE, который имеет следующую структуру:
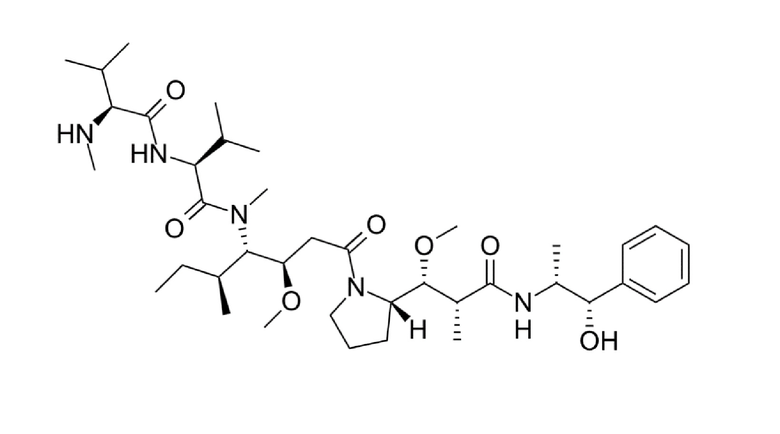
12. Конъюгат лекарственного средства по любому из пп. 8-11, который представляет собой соединение, которое имеет следующую структуру:
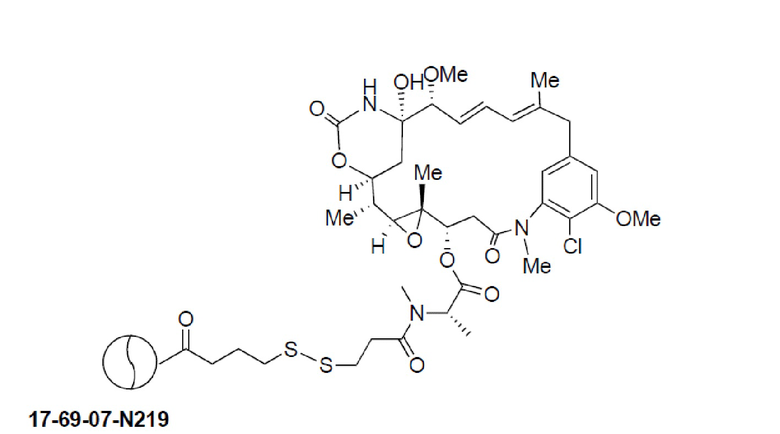
при этом 17-69-07-N219 представляет собой G-Sar10-ACYNEFGCEDFYDIC (SEQ ID NO: 20).
13. Конъюгат лекарственного средства по любому из пп. 8-11, который представляет собой соединение, которое имеет следующую структуру:
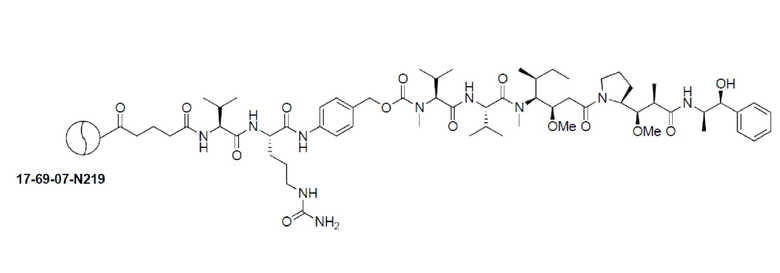
при этом 17-69-07-N219 представляет собой G-Sar10-ACYNEFGCEDFYDIC (SEQ ID NO: 20).
14. Конъюгат лекарственного средства по п. 9 или 10, в котором цитотоксический агент представляет собой мейтанзиноид и выбран из соединения формулы (II):
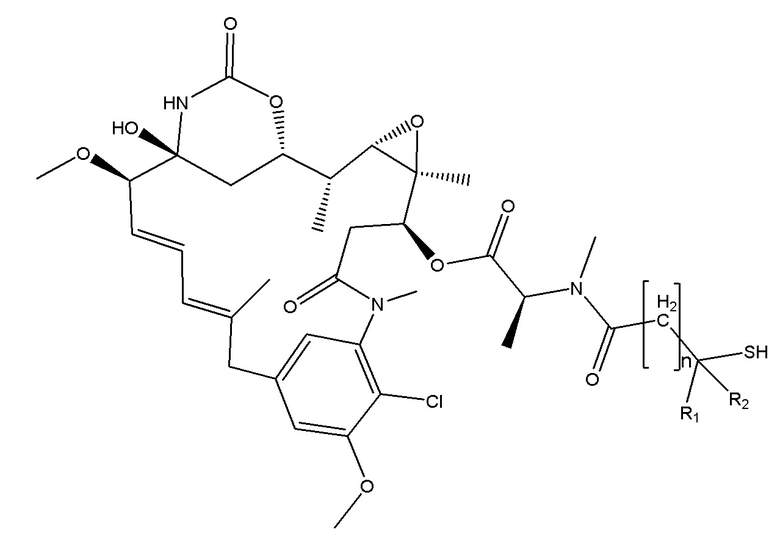 (II)
(II)
где n представляет собой целое число, выбранное от 1 до 10; и
R1 и R2 независимо друг от друга представляют собой водород, C1-6-алкил или карбоциклическую, или гетероциклическую группу, такую как водород или метил.
15. Конъюгат лекарственного средства по п. 14, в котором:
n равно 1, и R1 и R2 оба представляют собой водород (т. е. мейтанзиновое производное DM1); или
n равно 2, и R1 представляет собой водород, R2 представляет собой метильную группу (т. е. мейтанзиновое производное DM3); или
n равно 2, и R1 и R2 оба представляют собой метильные группы (т. е. мейтанзиновое производное DM4).
16. Конъюгат лекарственного средства по любому из пп. 8-15, в котором бициклический пептидный компонент конъюгата имеет структуру, представленную в формуле (III):
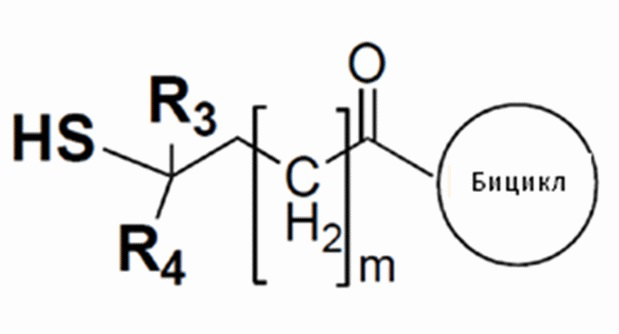 (III)
(III)
где m представляет собой целое число, выбранное от 0 до 10, и
R3 и R4 независимо друг от друга представляют собой водород, C1-6-алкил или карбоциклическую, или гетероциклическую группу, такую как водород или метил.
17. Конъюгат лекарственного средства по любому из пп. 9-16, в котором цитотоксический агент связан с бициклическим пептидом с помощью линкера, определенного в формуле (IV)
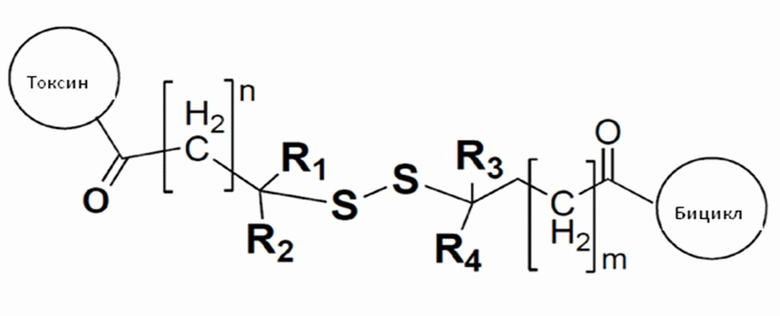 (IV)
(IV)
где R1, R2, R3 и R4 представляют собой водород, C1-6-алкил или карбоциклическую, или гетероциклическую группу, такую как водород или метил;
Токсин относится к любому подходящему цитотоксическому агенту;
Бицикл представляет собой пептидный лиганд по любому из пп. 1-6;
n представляет собой целое число, выбранное от 1 до 10; и
m представляет собой целое число, выбранное от 0 до 10.
18. Конъюгат лекарственного средства по п. 17, где токсин соединения (IV) представляет собой мейтанзин, и конъюгат включает соединение формулы (V):
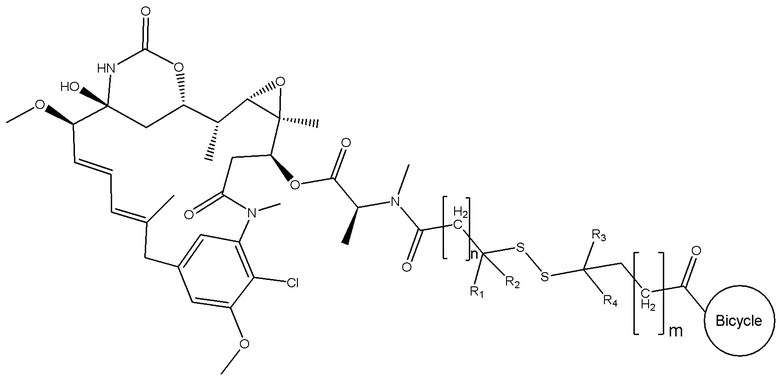 (V)
(V)
где R1, R2, R3 и R4 представляют собой водород, C1-6-алкил или карбоциклическую, или гетероциклическую группу;
Бицикл представляет собой пептидный лиганд по любому из пп. 1-6;
n представляет собой целое число, выбранное от 1 до 10; и
m представляет собой целое число, выбранное от 0 до 10.
19. Конъюгат лекарственного средства по п. 18, где:
n равно 1, и R1, R2, R3 и R4 каждый представляет собой водород, т. е. соединение формулы (V)a:
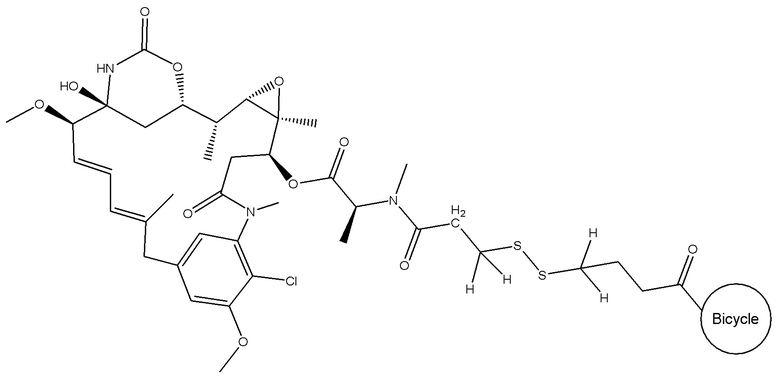 (V)a или
(V)a или
n равно 1, и R1 представляет собой метил и R2, R3 и R4 каждый представляет собой водород, т. е. соединение формулы (V)b:
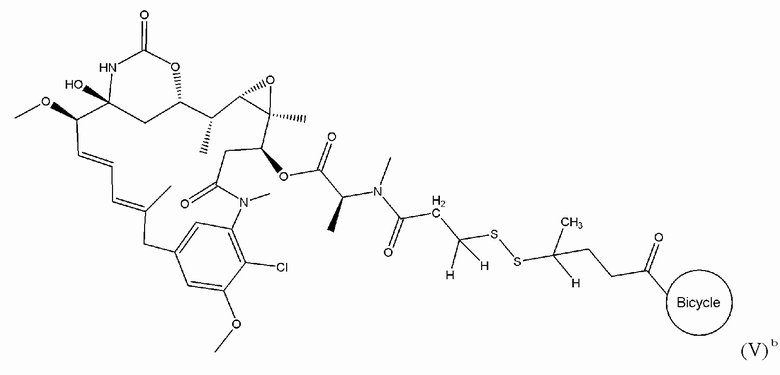
или
n равно 2, R1 и R2, оба представляет собой водород, R3 и R4 оба представляют собой метил, т. е. соединение формулы (V)c:
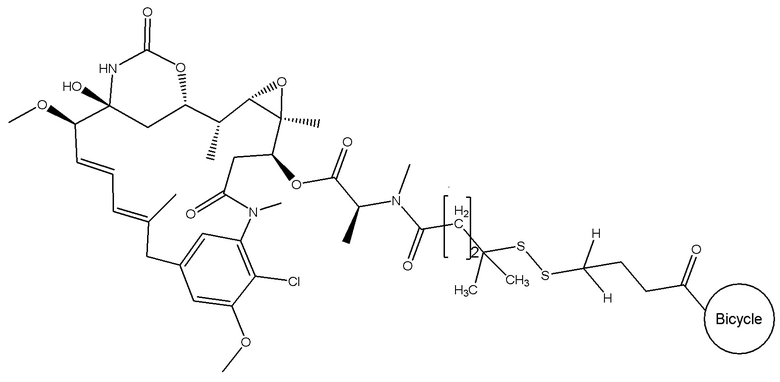 (V)c
(V)c
или
n равно 2, R1 и R2, оба представляет собой метил, R3 и R4 оба представляют собой водород, т. е. соединение формулы (V)d:
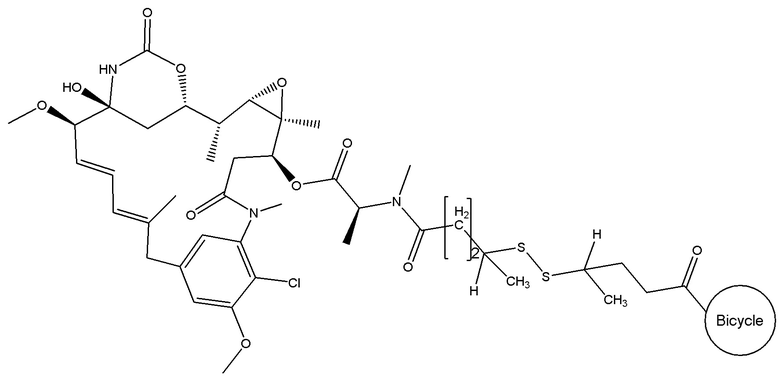 (V)d.
(V)d.
20. Конъюгат лекарственного средства по п. 8 или 9, который выбран из
BT17BDC-9:
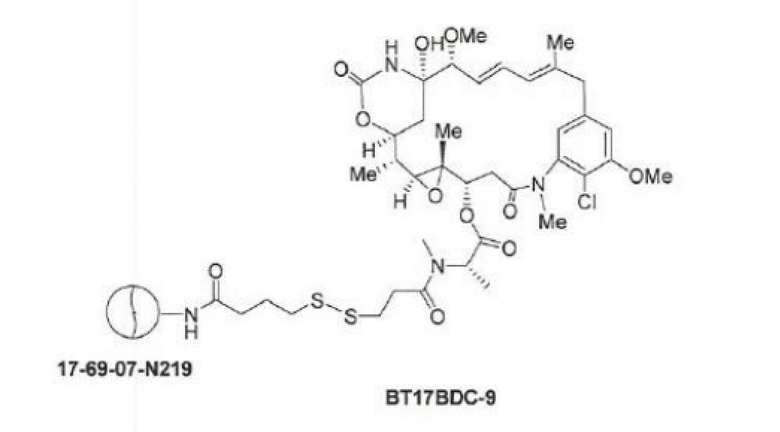
при этом 17-69-07-N219 представляет собой G-Sar10-ACYNEFGCEDFYDIC (SEQ ID NO: 20);
BT17BDC-17:
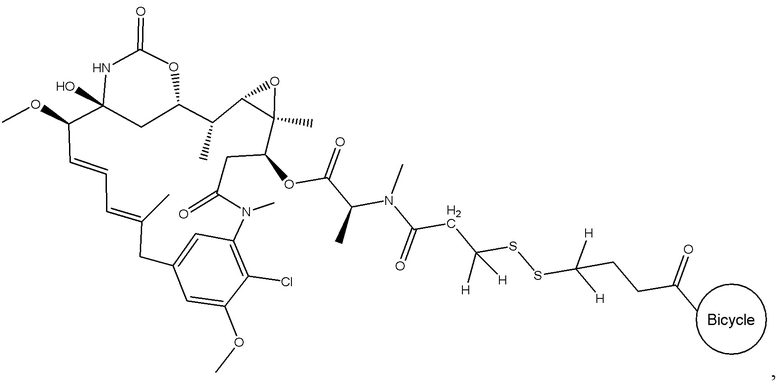
при этом бицикл представляет собой βAla-Sar10-A-C(D-Ala)NE(1Nal)(D-Ala)CEDFYD(tBuGly)C (SEQ ID NO: 5; 17-69-07-N241);
BT17BDC-19:
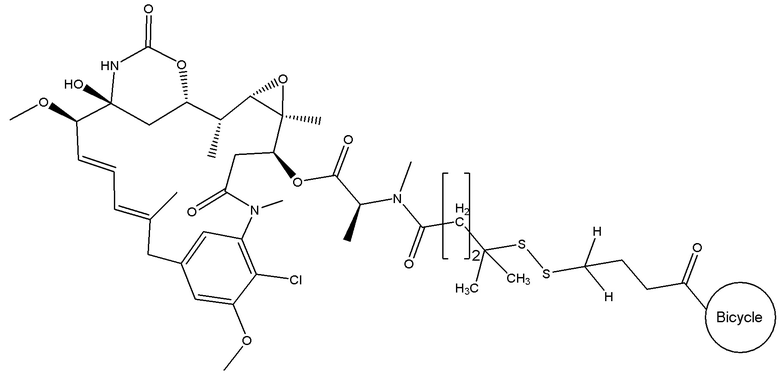
при этом бицикл представляет собой βAla-Sar10-A-C(D-Ala)NE(1Nal)(D-Ala)CEDFYD(tBuGly)C (SEQ ID NO: 5; 17-69-07-N241); и
BT17BDC-20:
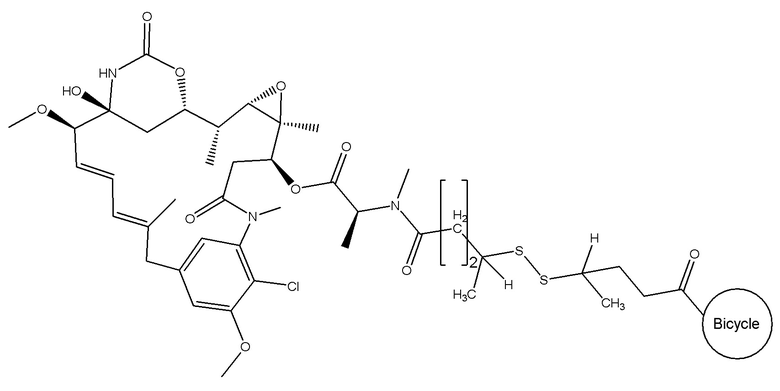
при этом бицикл представляет собой βAla-Sar10-A-C(D-Ala)NE(1Nal)(D-Ala)CEDFYD(tBuGly)C (SEQ ID NO: 5; 17-69-07-N241).
21. Конъюгат лекарственного средства по п. 8 или 9, который представляет собой
BT17BDC-18:
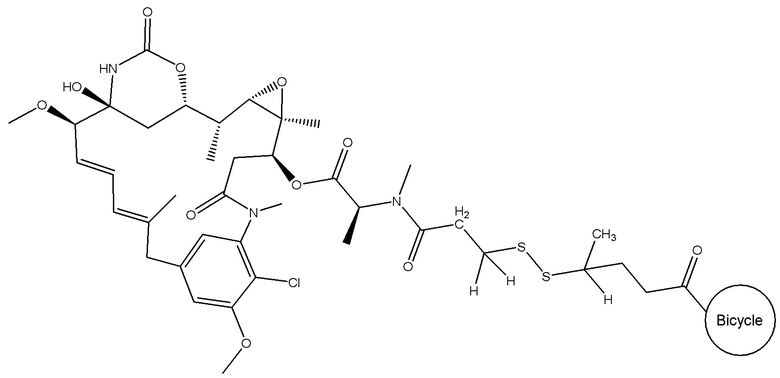
при этом бицикл представляет собой βAla-Sar10-A-C(D-Ala)NE(1Nal)(D-Ala)CEDFYD(tBuGly)C (SEQ ID NO: 5; 17-69-07-N241).
22. Конъюгат лекарственного средства по п. 8, где эффекторная и/или функциональная группа включает хелатор металла.
23. Конъюгат лекарственного средства по п. 22, где хелатор металла подходит для комплексообразования радиоизотопов металла, востребованных в качестве лекарства.
24. Конъюгат лекарственного средства по п. 22 или 23, где хелатор металла комплексирован с радиоизотопом металла.
25. Конъюгат лекарственного средства по п. 23 или 24, где радиоизотоп металла выбран из 64Cu, 67Ga, 68Ga, 177Lu, 90Y и 213Bi.
26. Конъюгат лекарственного средства по любому из пп. 22-25, где хелатор металла выбран из DOTA, NOTA, EDTA, DTPA, HEHA и SarAr.
27. Способ получения конъюгата лекарственного средства по п. 20, который включает синтетический путь, описанный в схеме
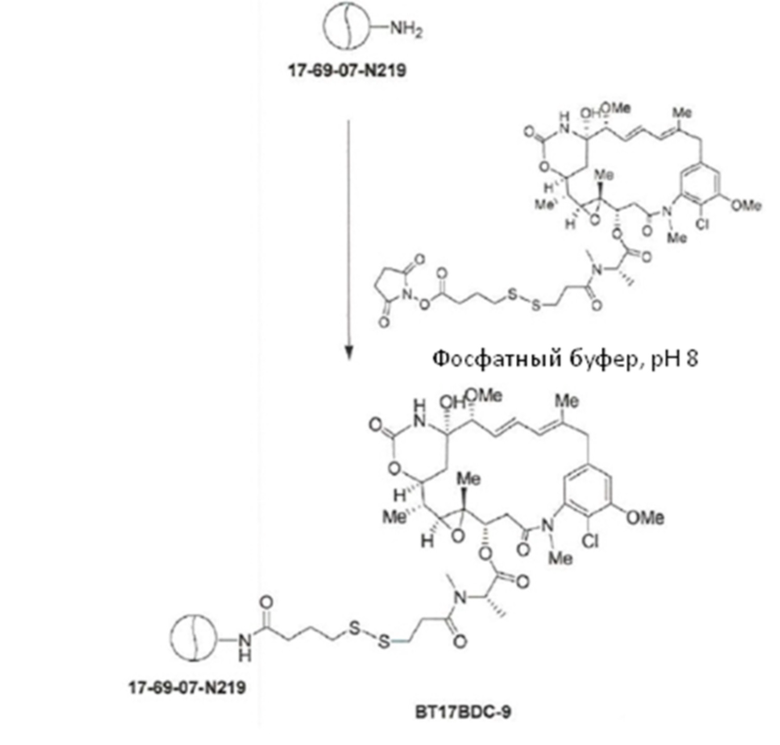
28. Фармацевтическая композиция для лечения рака, характеризующегося гиперэкспрессией MT1-MMP, которая содержит терапевтически эффективное количество конъюгата лекарственного средства по любому из пп. 8-26, в сочетании с одним или более фармацевтически приемлемыми наполнителями.
29. Конъюгат лекарственного средства по любому из пп. 8-26 для применения в лечении рака, характеризующегося гиперэкспрессией MT1-MMP.
30. Способ диагностики или визуализации рака у пациента, при этом способ включает введение указанному пациенту терапевтически эффективной дозы конъюгата лекарственного средства по любому из пп. 22-26.
| WO 2013050615 A1, 11.04.2013 | |||
| WO 2010089117 A1, 12.08.2010 | |||
| WO 2011018227 A2, 17.02.2011 | |||
| WO 2013173755 A1, 21.11.2013. |

