Настоящее изобретение относится к полипептидам, ковалентно связанным с молекулярным остовом так, что между точками присоединения к остову расположены две или более пептидные петли. В настоящем изобретении, в частности, описаны пептиды, специфичные к калликреину протеазы человеческой плазмы и модифицированные в одной или двух пептидных петлях для усиления активности и/или устойчивости к протеазе.
Циклические пептиды способны связываться с высоким сродством и специфичностью с белковыми мишенями и, следовательно, являются привлекательным классом молекул для создания терапевтических средств. Действительно, несколько циклических пептидов уже успешно применяются в клинической практике, например, антибактериальный пептид ванкомицин, иммунодепрессивное средство циклоспорин или противораковое средство октреотид (Driggers, et al., Nat. Rev. Drug Discov. 2008, 7(7), 608-24). Хорошие связующие свойства являются результатом относительно большой поверхности взаимодействия между пептидом и мишенью, а также меньшей конформационной гибкости циклических структур. Макроциклы обычно связываются с поверхностями, равными нескольким сотням ангстрем в квадрате как, например, антагонист CVX15, являющийся циклическим пептидом СХСR4 (400 А2; Wu, B., et al., Science 330 (6007), 1066-71), циклический пептид с фрагментом Arg-Gly-Asp, связывающийся с интегрином αVb3 (355 А2) (Xiong, J.P., et al., Science 2002, 296 (5565), 151-5), или ингибитор upain-1, являющийся циклическим пептидом, связывающимся с активатором плазминогена типа урокиназы (603 А2; Zhao, G., et al., J. Struct. Biol. 2007, 160(1), 1-10).
Благодаря циклической конфигурации пептидные макроциклы являются менее гибкими, чем линейные пептиды, вследствие чего происходит меньшая потеря энтропии при связывании с мишенями и достигается более высокое сродство связывания. Меньшая гибкость также приводит к запиранию специфичных к мишени конформаций, что увеличивает специфичность связывания по сравнению с линейными пептидами. Указанный эффект был продемонстрирован активным и избирательно действующим ингибитором матриксной металлопротеиназы 8 (ММР-8), который утратил свою избирательность в отношении других ММР при размыкании его кольца (Cherney, R.J., et al., J. Med. Chem. 1998, 41(11), 1749-51). Благоприятные связующие свойства, достигаемые в результате макроциклизации, являются еще более выраженными в мультициклических пептидах, имеющих насколько пептидных колец, как, например, в ванкомицине, низине или актиномицине.
Разные исследователи ранее связывали полипептиды, имеющие остатки цистеина, с синтетической молекулярной структурой (Kemp, D.S. and McNamara, P.E., J. Org. Chem., 1985; Timmerman, P. et al., ChemBioChem, 2005). Мелоэн с сотрудниками использовал трис(бромметил)бензол и родственные молекулы для быстрой и количественной циклизации множества пептидных петель на синтетических остовах для структурной имитации поверхностей белка (Timmerman, P. et al., ChemBioChem, 2005). Методы создания соединений-кандидатов лекарственных средств, при осуществлении которых указанные соединения получают путем связывания цистеинсодержащих полипептидов с молекулярным остовом, таким как, например, трис(бромметил)бензол, рассмотрены в публикациях WO 2004/077062 и WO 2006/078161.
В публикации WO2004/077062 описан метод выбора соединения-кандидата лекарственного средства. В частности, в данном документе описаны разные молекулярные остовы, включающие первую и вторую реакционно-способные группы, и контактирование указанного остова с другой молекулой с образованием по меньшей мере двух связей между остовом и данной молекулой в результате выполнения реакции сочетания.
В публикации WO2006/078161 описаны связующие соединения, иммуногенные соединения и пептидные миметики. В данном документе описан искусственный синтез разных пептидов, полученных из существующих белков. Указанные пептиды затем объединяют с постоянным синтетическим пептидом с заменами некоторых аминокислот, производимыми с целью создания комбинаторных библиотек. В результате создания разных структур благодаря химическому связыванию с отдельными пептидами, в которых были произведены разные замены аминокислот, возрастает вероятность обнаружения требуемой связывающей активности. На фигуре 1 данного документа показана схема синтеза разных конструкций пептидных петель. Конструкции, представленные в указанном документе, созданы на основе пептидов, имеющих функциональную группу -SH, обычно включающую остатки цистеина, и гетероароматические группы остова, обычно включающие бензильные заместители водорода, такие как бис- или трис-бромфенилбензол. Такие группы взаимодействуют с образованием простой тиоэфирной связи между пептидом и остовом.
Авторы настоящего изобретения недавно разработали метод создания комбинаторных библиотек на основе отображения на фаге с целью скрининга больших библиотек бициклических пептидов, специфичных к представляющим интерес мишеням (Heins, et al., Nat. Chem. Biol. 2009, 5(7), 502-7; см. также международную заявку на патент WO2009/098450). Комбинаторные библиотеки линейных пептидов, содержащих три остатка цистеина и две области из шести произвольных аминокислот (Cys-(Xaa)6-Cys-(Xaa)6-Cys), были отображены на фаге и циклизированы путем ковалентного связывания боковых цепей цистеина с мелкой молекулой (трис(бромметил)бензола). Бициклические пептиды, выделенные на основании родства к катепсину G протеаз и калликреину (РК) человеческой плазмы, имели константы ингибирования в наномолярном диапазоне. Лучший ингибитор, РК15, ингибирует РК человека (hPK) при значении Кi, равном 3 нМ. Сходство аминокислотных последовательностей нескольких выделенных бициклических пептидов позволяет предположить, что обе пептидные петли способствуют связыванию. РК15 не ингибировал крысиный РК (81% идентичность последовательностей), фактор XIa гомологичных серинпротеаз человека (hfXIa; 69% идентичность последовательностей) или тромбин (36% идентичность последовательностей) при самых высоких испытанных концентрациях (10 мкМ) (Heins, et al., Nat. Chem. Biol. 2009, 5(7), 502-7). Сделанное открытие позволяет предположить, что данный бициклический ингибитор является высоко специфическим и не ингибирует другие трипсинподобные серинпротеазы человека. Синтетический низкомолекулярный пептидный ингибитор, такой как РК15, обладающий вышеуказанной активностью и избирательностью в отношении мишени, может найти применение в качестве терапевтического средства для регулирования активности РК в случае наследственного ангионевротического отека, опасного для жизни заболевания, характеризующегося периодически возникающим отеком, или для предотвращения контактной активации во время хирургической операции с использованием аппарата искусственного кровообращения.
Пептид РК15 был выделен из библиотеки, созданной на основе пептида РК2, H-ACSDRFRNCPLWSGTCG-NH2, в котором вторая петля 6-аминокислоты была выбрана произвольно. Последовательность РК15 представляла собой H-ACSDRFRNCPADEALCG-NH2, и константа связывания IC50 для человеческого калликреина была равна 1,7 нМ.
Сущность изобретения
Авторы настоящего изобретения исследовали реагенты, связывающиеся с калликреином, с целью оптимизации сродства связывания и активности. В одновременно рассматриваемой неопубликованной заявке на патент РСТ/ЕР2012/069898 описаны бициклические пептидные лиганды, которые являются ингибиторами калликреина человеческой плазмы.
В одном варианте осуществления изобретения петли пептидного лиганда включают пять аминокислот, при этом первая петля содержит фрагмент GrxW/FPxK/RGr, где Gr означает реакционно-способную группу. В контексте настоящего изобретения определение “первая” петля необязательно означает определенное положение петли в последовательности. В некоторых вариантах осуществления изобретения первая петля может быть проксимальной петлей в пептидной последовательности в направлении от аминоконца к карбоксильному концу. Например, полипептид далее включает вторую, дистальную петлю, которая содержит фрагмент GrT/LHQ/TxLGr. Примеры последовательностей первой петли включают GrxWPАRGr, GrxWPSRGr, GrxFPFRGr и GrxFPYRGr. В указанных примерах x может быть любой аминокислотой, например, S или R.
Например, полипептид может быть одним из полипептидов, указанных в таблице 4, таблице 5 или таблице 6.
Реакционно-способная группа может быть реакционно-способной аминокислотой. Например, реакционно-способная аминокислота является цистеином.
Варианты полипептидов могут быть получены путем идентификации остатков, пригодных для мутации и создания библиотек, включающих мутации в данных положениях. Например, полипептид 06-56, указанный в таблице 4, фигуры 5, 6, может быть мутирован без утраты активности в положениях Q4 и Т10 (см. нижеследующий раздел “Примеры”). Могут быть выбраны полипептидные лиганды, включающие мутации в указанных положениях, которые обладают улучшенной связывающей активностью по сравнению с полипептидом 06-56.
Бицикл, ингибирующий калликреин, должен обладать соответствующим профилем устойчивости к протеазе, позволяющий снизить вызываемое протеазой выведение из плазмы или других подобных сред. При выполнении сравнительного экспресс-анализа устойчивости в плазме (метод № 1), который позволил обнаружить постепенное исчезновение исходного пептида в крысиной плазме, было установлено, что N-концевой аланин (присутствующий во время отбора и первоначально введенный в синтетические пептиды лидерных последовательностей) быстро исчезает во всех последовательностях бицикла, исследованных в крысиной и человеческой плазме. Подобного разрушения удалось избежать при синтезе соединения-кандидата с отсутствием N- и С-концевых аланинов. Для удаления потенциальных сайтов узнавания для амино- и карбоксипептидаз свободный аминоконец, который в теперь находится в положении остатка Cys1 соединения-кандидата, кэппируют уксусным ангидридом в процессе синтеза пептидов, в результате чего образуется молекула, ацетилированная в положении N-конца. Аналогичным образом С-концевой цистеин синтезирован в виде амида с целью удаления потенциального сайта узнавания для карбоксипептидаз.
Таким образом, в соответствии с одним примером бициклические соединения-кандидаты имеют следующую общую последовательность:
Ac-C1AA1AA2AAnC2AAn+1AAn+2AAn+3C3(TMB)-NH2
где “Ас” означает ацетилирование N-конца, “-NH2” означает амидирование С-конца, “С1, С2, С3” означают первый, второй и третий остаток цистеина в последовательности, “АА1” - “ААn” означает положение аминокислоты (характер “АА” определяется вышеописанным выбором) и “(ТМВ)” означает, что пептидная последовательность была циклизирована с использованием ТВМВ (трисбромметилбензоат) или любого другого приемлемого реакционно-способного остова.
В данном контексте пептиды “Ас-(06-34-18)(ТМВ)” и “Ас-(06-34-18)(ТМВ) Phe2 Tyr4” соответственно имеют последовательности Ac-CSWPARCLHQDLC-NH2 и Ac-CSFPYRCLHQDLC-NH2. Указанные последовательности могут быть пронумерованы в соответствии с приведенной выше схемой как
1) Ac-C1S1W2P3A4R5C2L6H7Q8D9L10C3-NH2, обозначаемая как Ас-(06-34-18)(ТМВ)
2) Ac-C1S1F2P3Y4R5C2L6H7Q8D9L10C3-NH2, обозначаемая как Ас-(06-34-18)(ТМВ) Phe2 Tyr4
где подчеркнуты неизменяемые цистеины. Указанные пептиды были всесторонне исследованы, при этом была выявлена субнаномолярная активность против калликреина при наличии очень благоприятных профилей избирательности в отношении гомологичных калликреину белков и сохранении хорошей перекрестной реактивности с крысиным калликреином. Последнее свойство является особенно ценным для определения фармакодинамики в животных моделях.
Немодифицированные пептиды Ас-06-34-18(ТМВ)-NH2 характеризуются временем полужизни в крысиной плазме, равным 2,3 часа, в то время как время полужизни Ас-(06-34-18)(ТМВ) Phe2 Tyr4 является немного короче и равно 0,9 часа (таблица 1). Для идентификации сайта протеолитического узнавания в Ас-06-34-18(ТМВ)-NH2 в течение определенного периода времени в крысиной плазме брали образцы пептида (метод № 1) и каждый образец анализировали в отношении постепенного появления пептидных фрагментов при помощи масс-спектрометрии MALDI-TOF. Указанный анализ показал, что Arg5 в петле 1 является основным сайтом узнавания и последующего расщепления пептида протеазой.
Был произведен химический синтез для идентификации заместителей аргинина 5, которые позволили бы эффективно защитить пептид от расщепления протеазой в крысиной плазме. Было установлено, что удаление Arg 5 в петле 1 или замена боковой цепи Arg любой незаряженной или заряженной химической группой повышает устойчивость пептидного лиганда.
Кроме того, протеолитическая устойчивость повышается в результате химической модификации пептидных связей рядом с Arg5 в положении N- или С-конца. Химическая модификация, например, представляет собой α-N-алкилирование или модификацию пептидной связи восстановленным амидом.
В одном варианте осуществления изобретения протеолитическая устойчивость усиливается благодаря стерическому препятствию в положении аминокислот, расположенных рядом с Arg5. Например, стерическое препятствие образуется в результате α-N-алкилирования.
Затем у указанных циклических пептидов-кандидатов оценивали следующие характеристики:
1) сохранение активности в отношении человеческого калликреина (поиск значения Ki, равного 10 нМ или меньше)
2) сохранение активности в отношении крысиного калликреина (поиск значения Ki, равного 50 нМ или меньше)
3) повышенная устойчивость в крысиной плазме (большее значение t1/2 по сравнению с пептидом дикого типа)
4) сохранение профиля избирательности в отношении других ферментов, родственных калликреину, и белков.
При выполнении исследований, рассмотренных в заявке РСТ/ЕР2012/069898, было установлено, что 4 вышеуказанным критериям удовлетворяют следующие модификации или миметики аргинина, которые включают:
а) 4-гуанидилфенилаланин (4-GuanPhe: увеличение t1/2 в крысиной плазме в 1-5 раз);
b) гомоаргинин (HArg: увеличение t1/2 в крысиной плазме в ~2-5 раз); и
с) α-N-метиларгинин (NMе-Arg: увеличение t1/2 в крысиной плазме в >10 раз).
Было установлено, что определенные неприродные аминокислоты позволяют связываться с калликреином плазмы при нМ значении Ki, что значительно увеличивает время существования в плазме. Полученные данные суммированы в таблице 1 и на фигуре 17.
Кроме того, в заявке РСТ/ЕР2012/069898 также рассмотрена дополнительная модификация пептида 06-34-18 в положении 3, в котором пролин был заменен азетидинкарбоновой кислотой (Aze3). Указанная модификация, по-видимому, благодаря большему ограничению и пониженной энтропии неизменно усиливает активность пептида, созданного на основе 06-34-18, в 2-5 раз (таблица 1). В комбинации с повышающими устойчивость модификациями в положении Arg 5 (в частности, HArg, NMe-Arg и 4GuanPhe) могут быть получены более сильные производные бициклического пептида.
Типичные неприродные аминокислоты выбирают из N-метиларгинина, гомоаргинина и гидроксипролина. N-метильные и гомо-производные аргинина используют для замены аргинина, и пролин 3 может быть заменен, например, гидроксипролином, азетидинкарбоновой кислотой или альфа-замещенной аминокислотой, такой как аминоизомасляная кислота. В другом варианте осуществления изобретения аргинин может быть заменен гуанидилфенилаланином.
В одном варианте осуществления изобретения полипептид включает первую петлю, содержащую фрагмент GrxWPARGr где остаток Р заменен азетидинкарбоновой кислотой и/или остаток R заменен метиларгинином, гомоаргинином и/или гуанидилфенилаланном.
В одном варианте осуществления изобретения полипептид включает первую петлю, содержащую фрагмент GrxFPYRGr, где остаток R заменен N-метиларгинином, остаток R заменен гомоаргинином, пролин заменен азетидинкарбоновой кислотой и/или R заменен гуанидилфенилаланином.
Авторы настоящего изобретения разработали методы, использованные в РСТ/Р2102/069898 и представленные в настоящем описании изобретения, которые позволяют оптимизировать вторую цепь. В соответствии с настоящим изобретением в петле 2 был идентифицирован дополнительный сайт узнавания протеазой, локализованный в положении гистидина 7 последовательности 06-34-18. Указанный остаток особенно хорошо узнают протеазы, содержащиеся в человеческой плазме ex vivo. Кроме того, указанный сайт также узнают мембраносвязанные крысиные протеазы, о чем свидетельствуют данные, полученные при выполнении фармакокинетического исследования крыс in vivo. Таким образом, чтобы пептид 06-34-18 приобрел приемлемый профиль фармакокинетической устойчивости, необходимый для терапевтических целей, оба сайта узнавания протеазой (Arg5 в петле 1 и His7 в петле 2) необходимо стабилизировать так, чтобы сообщить указанному пептиду устойчивость к протеазам человеческой плазмы и протеолитически агрессивной среде, имеющей место у крысы и человека in vivo.
Таким образом, первым объектом настоящего изобретения является пептидный лиганд, специфичный к калликреину человека, который включает полипептид, содержащий по меньшей мере три реакционно-способные группы, разделенные последовательностями по меньшей мере двух петель, и молекулярный остов, образующий ковалентные связи с реакционно-способными группами полипептида, в результате чего молекулярный остов связывается ковалентными связями с реакционно-способными группами полипептида и на молекулярном остове образуются по меньшей мере две полипептидные петли, из которых петли пептидного лиганда включают пять аминокислот и одна петля содержит фрагмент GrT/LHQ/TxLGr.
В пептиде 06-34-18 указанная петля является второй петлей, локализованной в положении С-конца относительно первой петли.
Например, указанный фрагмент является фрагментом GrxHxDLGr, где Gr означает реакционно-способную группу. Например, фрагмент GrTHxxLGr.
В одном варианте осуществления изобретения две смежные петли полипептида включают фрагмент GrxW/FPxK/RGrT/LHQ/TDLGr.
Примеры таких полипептидов приведены в таблице 4, таблице 5 или таблице 6.
Вторым объектом настоящего изобретения является пептидный лиганд, рассмотренный в первом объекте изобретения, в котором удаление His7 в петле 2 или замена боковой цепи гистидина любой незаряженной или заряженной химической группой повышает устойчивость пептидного лиганда.
Например, протеолитическая устойчивость повышается в результате химической модификации пептидных связей рядом с остатком His7 в положении N- или С-конца. Химическая модификация, например, представляет собой α-N-алкилирование или модификацию пептидной связи восстановленным амидом.
В одном варианте осуществления изобретения протеолитическую устойчивость повышают путем стерического препятствия, создаваемого аминокислотами, расположенными рядом с остатком His7. Стерическое препятствие, например, может быть создано путем замены остатка в положении 9 энантиомером D-аминокислоты или α-N-алкилированием.
В другом варианте осуществления изобретения протеолитическую устойчивость повышают путем введения в положение 6 образующих стерическое препятствие аминокислот. Протеолитическая устойчивость, например, может быть повышена путем введения в положение 6 Сβ-производных аминокислот, таких как фенилглицин или циклогексилглицин, которые усиливают активность последовательности пептидного лиганда в отношении калликреина человеческой плазмы и уменьшают протеолитический гидролиз С-концевой пептидной связи.
Положения настоящего изобретения могут быть объединены с положениями заявки РСТ/ЕР2012/06989, при этом полипептид по настоящему изобретению может включать первую петлю, содержащую фрагмент GrxWPАRGr или GrxFPYRGr, и вторую петлю, содержащую фрагмент GrT/LHQ/TxLGr.
В одном примере остаток Pro3 первой петли заменен азетидинкарбоновой кислотой и/или остаток Arg5 заменен N-метиларгинином, гомоаргинином и/или гуанидилфенилаланином.
Краткое описание чертежей
Фигура 1. Выбор бициклических пептидов путем отображения на фаге. (а) Фаговые библиотеки бициклических пептидов. Произвольные аминокислоты обозначены символом ”Х”, аланин обозначен символом ”А” и три постоянных остатка цистеина обозначены символом ”С”. (b) Форма структур химически синтезированных бициклических пептидов, содержащих петли из 3, 5 или 6 аминокислот. Указанные структуры созданы путем связывания линейных пептидов при помощи боковых цепей трех остатков цистеина с трис(бромметил)бензолом (ТВМВ). Аминокислоты, заменяемые в бициклических пептидах, обозначены символом ”Хаа”. (с-е) Последовательности бициклических пептидов, выделенные из библиотеки 5×5 (с), библиотеки 3×3 А (d) и библиотеки 3×3 В (е). Подобные аминокислоты выделены разным цветом.
Фигура 2. Сравнение поверхностных аминокислот hPK и гомологичных серинпротеаз. (а) Структура hPK (входные данные 2ANW PDB) с представлением поверхности. Атомы аминокислот, экспонированные на поверхности на расстоянии менее 4, 8 и 12 А от бензамидина (изображенного серым цветом), связанного с карманом S1, показаны более темным цветом. (b) Структура hPK. Выделены боковые цепи аминокислот, отличающихся в hfXIa. (с) Структура hPK. Выделены боковые цепи аминокислот, отличающихся в rPK.
Фигура 3. Графическое представление метода, использованного для определения предпочтительных остатков для мутации полипептидных лигандов.
Фигура 4. Анализ воздействия аминокислотных замен в пептиде 06-34 (таблица 4) на связывание указанного пептида с калликреином плазмы при 2 нМ. Для каждого положения показан эффект разных мутаций в данном положении по сравнению с исходной последовательностью.
Фигура 5. Анализ воздействия аминокислотных замен в пептиде 06-56 (таблица 4) на связывание указанного пептида с калликреином плазмы при 2 нМ. Для каждого положения показан эффект разных мутаций в данном положении по сравнению с исходной последовательностью.
Фигура 6. Анализ воздействия аминокислотных замен в пептиде 06-56 (таблица 4) на связывание указанного пептида с калликреином плазмы при 10 нМ. Для каждого положения показан эффект разных мутаций в данном положении по сравнению с исходной последовательностью.
Фигура 7. Результаты масс-спектрометрического исследования, показывающие масс-спектры Ас-06-34-18(ТМВ)-NH2 после воздействия 35% крысиной плазмы через 0, 1 день, 2 дня и 3 дня (метод № 1). Точное указание массы может изменяться из-за интерференции ионов и низких концентраций фрагментов; однако возможна идентификация отдельных протеолитических фрагментов.
Фигура 8. Химические структуры метаболитов М1, М2, М3 пептида Ас-06-34-18(ТМВ)-NH2, идентифицированных после воздействия крысиной плазмы.
Фигура 9. Химическая структура Ас-06-34-18)ТМВ)-NH2.
Фигура 10. Анализ ингибирования фермента калликреина пептидом Ас-06-34-18(ТМВ)-NH2 и производными 1-й петли. Обнаружено значительное уменьшение сродства, что указывает на важное значение целостности фармакофора WPAR.
Фигура 11. Химические структуры аргинина и его аналогов.
Фигура 12. Химические структуры остатка Trp и потенциальных гидрофобных аналогов.
Фигура 13. Химические структуры остатка Pro и потенциальных ограниченных аналогов.
Фигура 14. Сравнительный анализ ингибирования калликреина остатками Aze3, NMeArg5 и дважды модифицированным пептидом Ас-06-34-18(ТМВ)-NH2.
Фигура 15. Химические структуры аланина и его производных.
Фигура 16. Сравнительный анализ ингибирования калликреина остатками F2Y4, F2Y4 HR5 и дважды модифицированным пептидом Ас-06-34-18(ТМВ)-NH2.
Фигура 17. Химические структуры модификаций петли 1 в пептиде Ас-(06-34-18), сообщающих хорошую устойчивость в крысиной плазме и/или повышенную активность (Aze3 вместо Pro3, 4GuanPhe вместо Arg5, HArg вместо Arg5, NMe-Arg5 вместо Arg5).
Фигура 18а. Пептид дикого типа, подвергнутый воздействию человеческой плазмы. Следует отметить внешний вид фрагментов с удаленными остатками His7-Gln8, Leu6 и Leu6-His7-Gln. Из-за природы MALDI-TOF интенсивность сигналов не находится на количественном уровне. Возможные сайты гидролиза в указанной последовательности показаны на диаграмме стрелками в направлении последующего расщепления, обозначенного горизонтальными стрелками над последовательностью.
Фигура 18b. Пептид 06-34-18, N-метилированный в положении Arg5, был подвергнут воздействию человеческой плазмы. Указанный пептид характеризуется повышенной устойчивостью в крысиной плазме. Однако в человеческой плазме данный пептид является неустойчивым, о чем свидетельствует наличие фрагментов, в которых отсутствуют остатки His-Gln8, Leu6 и Leu6-His7-Gln8 в петле 2. Из-за природы MALDI-TOF интенсивность сигналов не находится на количественном уровне.
Фигура 19а. Химическая структура петли 2 пептида 06-34-18.
Фигура 19b. Обзор структур, использованных в положении 6 петли 2. Нестандартные аминокислотные акронимы определены в разделе ”Методы” и в таблице 7.
Фигура 19с. Обзор структур, использованных в положении 7 петли 2. Нестандартные аминокислотные акронимы определены в разделе ”Методы” и в таблице 7.
Фигура 19d. Обзор структур, использованных в положении 8 петли 2. Нестандартные аминокислотные акронимы определены в разделе ”Методы” и в таблице 7.
Фигура 20а. Структура пептоида N-His в петле 2 пептида 06-34-18. Боковая цепь гомогистидина введена в положении Nα одновременно с утратой Сα хирального центра.
Фигура 20b. Структура восстановленного амида между положениями 6 и 7 в петле 2 пептида 06-34-18. Использование метилена вместо карбонила удаляет расщепляемую пептидную связь.
Фигура 21а. Сравнительная устойчивость пептида дикого типа (06-34-18) в человеческой плазме в периоды времени t = 0, 21 и 46 часов.
Фигура 21b. Сравнительная устойчивость пептида дикого типа (06-34-18) в крысиной плазме в периоды времени t = 0, 21 и 46 часов.
Фигура 21с. Сравнительная устойчивость пептида (06-34-18) Phe2 Aze3 Tyr4 НArg5 D-Asp9 в человеческой плазме в периоды времени t = 0, 21 и 46 часов.
Фигура 21d. Сравнительная устойчивость пептида Ас-(06-34-18) с заменами Phe2 Aze3 Tyr4 НArg5 Ala(CH2NH)6 в человеческой плазме в периоды времени t = 0, 21 и 46 часов.
Фигура 21е. Сравнительная устойчивость пептида Ас-(06-34-18) Phe2 Aze3 Tyr4 НArg5 Ala(CH2NH)6 в крысиной плазме в периоды времени t = 0, 21 и 46 часов.
Фигура 22. Полная химическая структура пептида Ас-(06-34-18) Phe2 Aze3 Tyr4 НArg5 Ala(CH2NH)6. Все аминокислоты являются L-энантиомерами.
Фигура 23. Фармакокинетический анализ крысиных пептидов Ас-(06-34-18) Phe2 Aze3 Tyr4 НArg5 Ala(CH2NH)6 и Ас-(06-34-18) Phe2 Aze3 Tyr4 НArg5 D-Asp9 в сравнении с протеолитически устойчивым, но фармакологически инертным энантиомером пептида Ас-(06-34-18), полностью состоящего из D-аминокислот. Следует отметить, что выведение пептида Ас-(06-34-18) Phe2 Aze3 Tyr4 НArg5 Ala(CH2NH)6 близко соответствует выведению контрольного пептида ”Ас-(06-34-18), полностью состоящего из D-аминокислот”. Горизонтальные пунктирные линии показывают предел количественного определения для каждого соответствующего пептида.
Подробное описание изобретения
За исключением особо оговоренных случаев все технические и научные термины, использованные в настоящем описании изобретения, имеют значения, известные специалистам в данной области, например, в области химии пептидов, культивирования клеток и отображения на фаге, химии нуклеиновых кислот и биохимии. При выполнении исследований в области молекулярной биологии, генетики и биохимии использованы стандартные методы (см. публикации Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd ed., 2001, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY; Ausubel et al., Short Protocols in Molecular Biology (1999), 4th ed., John Wiley & Sons, Inc.), которые включены в настоящее описание изобретения в качестве ссылки.
Термин ”пептидный лиганд” в использованном здесь значении означает пептид, ковалентно связанный с молекулярным остовом. Такие пептиды обычно содержат две или более реакционно-способных групп, способных образовывать ковалентные связи с остовом, и последовательность, расположенную между указанными реакционно-способными группами, которая определяется как последовательность петли, так как данная последовательность образует петлю при связывании пептида с остовом. В настоящем изобретении пептиды содержат по меньшей мере три реакционно-способные группы и образуют по меньшей мере две петли на остове.
Реакционно-способные группы являются группами, способными образовывать ковалентную связь с молекулярным остовом. Реакционно-способные группы обычно присутствуют в боковых цепях аминокислот пептида. В качестве примеров можно привести аминосодержащие группы, такие как цистеин, лизин и селеноцистеин.
Термин ”специфичность” в контексте настоящего изобретения означает способность лиганда связываться или по-другому взаимодействовать с родственной мишенью за исключением групп, подобных указанной мишени. Например, специфичность может означать способность лиганда ингибировать взаимодействие с ферментом человека, но не с гомологичным ферментом животного другого вида. С помощью метода по настоящему изобретению можно модулировать специфичность, то есть увеличивать или уменьшать, делая лиганды более или менее способными взаимодействовать с гомологами или паралогами предполагаемой мишени. Термин ”специфичность” не является синонимом терминов ”активность”, ”сродство” или ”авидность”, и сила воздействия лиганда на мишень (например, сродство связывания или степень ингибирования) необязательно имеет отношение к его специфичности.
Термин ”связывающая активность” в использованном здесь значении означает количественное измерение связывания, производимое при выполнении анализов связывания, рассмотренных, например, в настоящем описании изобретения. Поэтому связывающая активность означает количество пептидного лиганда, связанного при данной концентрации мишени.
Мультиспецифичность представляет собой способность связываться с двумя или более мишенями. Связующие пептиды обычно способны связываться с одной мишенью, такой как эпитоп в случае антитела, благодаря свойствам их структуры. Однако могут быть созданы пептиды, способные связываться с двумя или более мишенями; в данной области, например, известны двуспецифические антитела. В настоящем изобретении пептидные лиганды способны связываться с двумя или более мишенями и поэтому являются мультиспецифическими. Например, пептидные лиганды, связывающиеся с двумя мишенями, являются двуспецифическими. Связывание может быть независимым, в соответствии с которым структура сайтов связывания мишеней в пептиде не препятствует связыванию той или другой мишени. В данном случае обе мишени могут быть связаны независимо. Обычно предполагается, что связывание одной мишени по меньшей мере частично будет препятствовать связыванию другой мишени.
Существует фундаментальное различие между двуспецифическим лигандом и лигандом, обладающим специфичностью в отношении двух родственных мишеней. В первом случае лиганд является специфичным отдельно к двум мишеням и взаимодействует с каждой мишенью определенным образом. Например, первая петля лиганда может связываться с первой мишенью, и вторая петля лиганда может связываться со второй мишенью. Во втором случае лиганд является неспецифичным, так как он не может различать две мишени, например, взаимодействуя с эпитопом, который является общим для обеих мишеней.
В контексте настоящего изобретения лиганд, обладающий активностью в отношении, например, мишени и ортолога, может быть биспецифическим лигандом. Однако в одном варианте осуществления изобретения лиганд не является биспецифическим, но характеризуется менее точной специфичностью, выражающейся в том, что указанный лиганд связывается как с мишенью, так и с одним или несколькими ортологами. Как правило, лиганд, который не был выбран для взаимодействия как с мишенью, так и с ее ортологом, с меньшей долей вероятности является биспецифическим вследствие модуляции длины петли.
Если лиганды действительно являются биспецифическими, то в одном варианте осуществления изобретения по меньшей мере одна из специфичностей к мишени будет общей для выбранных лигандов и степень такой специфичности можно модулировать методами по настоящему изобретению. Вторая и последующие специфичности необязательно должны быть общими и не являются объектом методов, рассмотренных в настоящем описании изобретения.
Мишень является молекулой или ее частью, с которой пептидные лиганды связываются или взаимодействуют другим образом. Хотя связывание считается необходимым условием активности большинства типов и может представлять собой именно такую активность, предполагается наличие других активностей. Таким образом, настоящее изобретение не требует прямого или непрямого измерения связывания.
Молекулярный остов является любой молекулой, способной присоединять пептид во многих точках, сообщая пептиду один или несколько структурных признаков. Остов не является кросс-линкером, так как он не заменяет дисульфидную связь, а предоставляет пептиду две или более точек присоединения. Молекулярный остов имеет по меньшей мере три точки присоединения пептида, определяемые как реакционно-0способные группы остова. Указанные группы способны взаимодействовать с реакционно-способными группами пептида с образованием ковалентной связи. Предпочтительные структуры молекулярных остовов описаны ниже.
Скрининг связывающей активности (или любой другой требуемой активности) выполняют методами, хорошо известными в данной области, например, из методики отображения на фаге. Например, для идентификации и выделения связующих членов из целого набора пептидов могут быть использованы мишени, иммобилизованные на твердой фазе. Скрининг позволяет выбрать члены на основании требуемых характеристик.
Термин ”библиотека” означает смесь гетерогенных полипептидов или нуклеиновых кислот. Библиотека состоит из разных членов. В данной связи термин ”библиотека” является синонимом термина ”набор”. Различия последовательностей членов библиотеки определяют разнообразие библиотеки. Библиотека может иметь форму простой смеси полипептидов или нуклеиновых кислот или может быть в форме организмов или клеток, например, бактерий, вирусов, животных или растительных клеток и тому подобных, трансформированных библиотекой нуклеиновых кислот. В некоторых случаях каждый отдельный организм или клетка содержит только один член или ограниченное число членов библиотеки.
В одном варианте осуществления изобретения нуклеиновые кислоты вводят в экспрессирующие векторы, позволяющие экспрессировать полипептиды, кодированные нуклеиновыми кислотами. Поэтому в соответствии с предпочтительным объектом изобретения библиотека может иметь форму популяции организмов-хозяев, в которой каждый организм содержит одну или несколько копий экспрессирующего вектора, включающего один член библиотеки в форме нуклеиновой кислоты, которая может быть экспрессироана с продуцированием соответствующего полипептида. Таким образом, популяция организмов-хозяев может кодировать большой набор генетически разных вариантов полипептидов.
В одном варианте осуществления изобретения библиотека нуклеиновых кислот кодирует набор полипептидов. Каждая нуклеиновая кислота в библиотеке обычно имеет последовательность, родственную одному или нескольким другим членам библиотеки. Термин “родственная последовательность” означает аминокислотную последовательность, которая по меньшей мере на 50%, например, по меньшей мере на 60%, по меньшей мере на 70%, по меньшей мере на 80%, по меньшей мере на 90%, по меньшей мере на 95%, по меньшей мере на 98%, по меньшей мере на 99% идентична по меньшей мере одному другому члену библиотеки. Об идентичности можно судить по смежному сегменту, состоящему по меньшей мере из 3 аминокислот, например, по меньшей мере из 4, 5, 6, 7, 8, 9 или 10 аминокислот, например, по меньшей мере из 12 аминокислот, по меньшей мере из 14 аминокислот, по меньшей мере из 16 аминокислот, по меньшей мере из 17 аминокислот, или по всей длине эталонной последовательности.
Набор является коллекцией вариантов, в данном случае вариантов полипептидов, отличающихся своими последовательностями. Локализация и характер реакционно-способных групп обычно не меняется, но последовательности, образующие петли между указанными группами, могут быть произвольными. Наборы могут отличаться размером, но должны включать по меньшей мере 102 членов. Могут быть созданы наборы, включающие 1011 или более членов.
Термин “совокупность полипептидных лигандов” в использованном здесь значении означает множество полипептидных лигандов, из которых может быть произведен выбор методами по настоящему изобретению. Совокупность может быть набором, но также может представлять собой небольшую коллекцию полипептидов, включающую по меньшей мере от 2 до 10, 20, 50, 100 или более членов.
Термин “группа полипептидных лигандов” в использованном здесь значении означает два или более лигандов. В одном варианте осуществления изобретения группа лигандов включает только лиганды, которые имеют общую специфичность в отношении по меньшей мере одной мишени. Группа обычно состоит по меньшей мере из 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 50, 100 или более лигандов. В одном варианте осуществления изобретения группа состоит из 2 лигандов.
(А) Создание пептидных лигандов
(i) Молекулярный остов
Молекулярные остовы описаны, например, в публикации WO2009098450 и в ссылках, приведенных в указанной публикацим, в частности, в публикациях WO2004077062 и WO2006078161.
Как указано в приведенных выше документах, молекулярный остов может быть мелкой молекулой, такой как мелкая органическая молекула.
В одном варианте осуществления изобретения молекулярный остов может представлять собой или может быть создан на основе природных мономеров, таких как нуклеозиды, сахара или стероиды. Молекулярный остов может включать короткий полимер таких элементов, например, димер или тример.
В одном варианте осуществления изобретения молекулярный остов является соединением, обладающим известной токсичностью, например, низкой токсичностью. Примеры приемлемых соединений включают холестерины, нуклеотиды, стероиды или существующие лекарственные средства, такие как тамазепам.
В одном варианте осуществления изобретения молекулярный остов может быть макромолекулой. В одном варианте осуществления изобретения молекулярный остов является макромолекулой, состоящей из аминокислот, нуклеотидов или углеводов.
В одном варианте осуществления изобретения молекулярный остов включает реакционно-способные группы, которые могут взаимодействовать с функциональными группами полипептида с образованием ковалентных связей.
Молекулярный остов может включать химические группы, такие как амины, тиолы, спирты, кетоны, альдегиды, нитрилы, карбоновые кислоты, сложные эфиры, алкены, алкины, азиды, ангидриды, сукцинимиды, малеимиды, алкилгалогениды и ацилгалогениды.
В одном варианте осуществления изобретения молекулярный остов может включать или может состоять из трис(бромметил)бензола, в частности, 1,3,5-трис(бромметил)бензола (ТВМВ) или его производного.
В одном варианте осуществления изобретения молекулярный остов является 2,4,6-трис(бромметил)мезитиленом. Указанный остов подобен 1,3,5-трис(бромметил)бензолу, но дополнительно содержит три метильные группы, присоединенные к бензольному кольцу. Преимуществом данного остова является то, что дополнительные метильные группы могут образовывать дополнительные точки контактирования с полипептидом и следовательно вносить дополнительные структурные ограничения.
Молекулярный остов по настоящему изобретению содержит химические группы, позволяющие функциональным группам полипептида из кодированной библиотеки по настоящему изобретению образовывать ковалентные связи с молекулярным остовом. Указанные химические группы выбирают из целого ряда функциональных групп, включающих амины, тиолы, спирты, кетоны, альдегиды, нитрилы, карбоновые кислоты, сложные эфиры, алкены, алкины, ангидриды, сукцинимиды, малеимиды, азиды, алкилгалогениды и ацилгалогениды.
(ii) Полипептид
Реакционно-способные группы полипептидов могут быть предоставлены боковыми цепями природных или неприродных аминокислот. Реакционно-способные группы полипептидов могут быть выбраны из тиоловых групп, аминогрупп, карбоксильных групп, гуанидиновых групп, фенольных групп или гидроксильных групп. Реакционно-способные группы полипептидов могут быть выбраны из азида, кетокарбонильных, алкиновых, винильных групп или арилгалогенида. Реакционно-способные группы полипептидов, предназначенные для связывания с молекулярным остовом, могут находиться в положении амино- или карбоксильного концов полипептида.
В некоторых вариантах осуществления изобретения реакционно-способные группы полипептида, предназначенные для связывания с молекулярным остовом, относятся к одному типу. Например, каждая реакционно-способная группа может быть остатком цистеина. Более подробно такие группы рассмотрены в публикации WO2009098450.
В некоторых вариантах осуществления изобретения реакционно-способные группы, предназначенные для связывания с молекулярным остовом, могут относиться к двум или более разным типам или к трем или более разным типам. Например, реакционно-способные группы могут включать два остатка цистеина и один остаток лизина или один остаток цистеина, один остаток лизина и одну N-концевую аминогруппу.
Цистеин может быть использован благодаря тому, что реакционная способность данного остатка больше всего отличается от всех других аминокислот. Реакционно-способными группами остова, которые могут быть использованы для взаимодействия с тиоловыми группами цистеинов, являются алкилгалогениды (которые также именуются галогеналканами). В качестве примеров можно привести бромметилбензол (реакционно-способная группа остова, представленная ТВМВ) или иодацетамид. Другие реакционноспособные группы остова, которые могут быть использованы для избирательного связывания соединений с остатками цистеина в белках, являются малеимидами. Примеры малеимидов, которые могут быть использованы в качестве молекулярных остовов в настоящем изобретении, включают: трис(2-малеимидоэтил)амин, трис(2-малеимидоэтил)бензол, трис(малеимидо)бензол. Селеноцистеин также является природной аминокислотой, которая обладает подобной реакционной способностью в отношении цистеина и может быть использована в указанных реакциях. Таким образом, цистеин обычно может быть использован для замены селеноцистеина за исключением тех случаев, когда из контекста следует обратное.
Лизины (и первичные амины N-конца пептидов) также являются приемлемыми реакционно-способными группами, используемыми для модификации пептидов на фаге путем связывания с молекулярным остовом. Однако лизины более распространены в фаговых белках, чем цистеины, поэтому существует более высокий риск перекрестного связывания фаговых частиц или утраты инфективности такими частицами. Тем не менее было установлено, что лизины особенно пригодны для внутримолекулярных реакций (например, когда молекулярный остов уже связан с фаговым пептидом), в результате которых образуется вторая или последующая связь с молекулярным остовом. В данном случае молекулярный остов предпочтительно взаимодействует с лизинами отображаемого пептида (в частности, с ближайшими лизинами). Реакционно-способными группами остова, избирательно взаимодействующими с первичными аминами, являются сукцинимиды, альдегиды или алкилгалогениды. В бромметильной группе, которая использована в ряде примеров, электроны бензольного кольца могут стабилизировать состояние катионного перехода. Поэтому арилгалогенид является в 100-1000 раз более реакционно-способным, чем алкилгалогениды. Примеры сукцинимидов, пригодных для использования в качестве молекулярного остова, включают трис(сукцинимидиламинотриацетат), 1,3,5-бензолтриуксусную кислоту. Примеры альдегидов, пригодных для использования в качестве молекулярного остова, включают триформилметан. Примеры алкилгалогенидов, пригодных для использования в качестве молекулярного остова, включают 1,3,5-трис(бромметил)-2,4,6-триметилбензол, 1,3,5-трим(бромметил)бензол, 1,3,5-три(бромметил)-2,4,6-триэтилбензол.
Аминокислоты, содержащие реакционно-способные группы, предназначенные для связывания с молекулярным остовом, могут быть локализованы в любых приемлемых положениях полипептида. Для воздействия на определенные структуры или созданные петли положения аминокислот, имеющих реакционно-способные группы, могут быть изменены квалифицированным специалистом, например, в результате манипулирования нуклеиновой кислотой, кодирующей полипептид, для мутирования созданного полипептида. В соответствии с настоящим изобретением может быть изменена длина петли.
Например, полипептид может включать последовательность АС(Х)mС(Х)mCG, гд Х означает произвольную природную аминокислоту, А означает аланин, С означает цистеин и G означает глицин, n и m, которые могут иметь одинаковые или разные значения, являются числами от 3 до 6.
(iii) Реакционно-способные группы полипептида
Молекулярный остов по настоящему изобретению может быть связан с полипептидом при помощи функциональных или реакционно-способных групп полипептида. Указанные группы обычно образуются из боковых цепей определенных аминокислот, обнаруженных в полипептиде. Такие реакционно-способные группы могут представлять собой боковую цепь цистеина, боковую цепь лизина, N-концевую аминогруппу или любую другую приемлемую реакционно-способную группу. Указанные группы подробно рассмотрены в публикации WO2009098450.
В качестве примеров реакционно-способных групп природных аминокислот можно привести тиоловую группу цистеина, аминогруппу лизина, карбоксильную группу аспартата или глутамата, гуанидиновую группу аргинина, фенольную группу тирозина или гидроксильную группу серина. Неприродные аминокислоты могут включать большое число реакционно-способных групп, включая азид, кетокарбонил, алкин, винил или арилгалогенид. Аминогруппа и карбоксильная группа концов полипептида также могут служить в качестве реакционно-способных групп, способных образовывать ковалентные связи с молекулярным остовом/молекулярным ядром.
Полипептиды по настоящему изобретению содержат по меньшей мере три реакционно-способных группы. Указанные полипептиды могут также содержать четыре или более реакционно-способных групп. Чем больше реакционно-способных групп использовано, тем больше петель можно образовать на молекулярном остове.
В предпочтительном варианте осуществления изобретения получают полипептиды с тремя реакционно-способными группами. В результате взаимодействия указанных полипептидов с молекулярным остовом/ молекулярным ядром, характеризующимся наличием трехкратной вращательной симметрии, образуется один изомер продукта. Создание одного изомера продукта является благоприятным фактором по нескольким причинам. Нуклеиновые кислоты библиотеки соединений кодируют только основные последовательности полипептида, а не изомерные формы молекул, образовавшихся при взаимодействии полипептида с молекулярным ядром. Если может быть образован только один изомер продукта, предназначение указанной нуклеиновой кислоты для данного изомера продукта является четко выраженным. При образовании множества изомеров продукта нуклеиновая кислота не может предоставить информацию о природе изомера продукта, выделенного в процессе скрининга или отбора. Образование одного изомера продукта также является благоприятным при синтезе определенного члена библиотеки по настоящему изобретению. В данном случае химическое взаимодействие полипептида с молекулярным остовом позволяет получить один изомер продукта, а не смесь изомеров.
В другом варианте осуществления изобретения получают полипептиды с четырьмя реакционно-способными группами. В результате взаимодействия указанных полипептидов с молекулярным остовом/молекулярным ядром, характеризующимся тетраэдрической симметрией, образуются два изомера продукта. Несмотря на то, что два разных изомера продукта кодированы одной и той же нуклеиновой кислотой, изомерная природа выделенного изомера может быть определена путем химического синтеза обоих изомеров, разделения двух изомеров и исследования обоих изомеров в отношении связывания с лигандом мишени.
В одном варианте осуществления изобретения по меньшей мере одна из реакционно-способных групп полипептидов является ортогональной по отношению к остальным реакционно-способным группам. Наличие ортогональных реакционно-способных групп позволяет направлять указанные ортогональные реакционно-способные группы к определенным сайтам молекулярного ядра. Методы связывания ортогональных реакционно-способных групп могут быть использованы для ограничения числа образовавшихся изомеров продукта. Другими словами, выбирая отличающиеся или разные реакционно-способные группы для одной или нескольких, по меньшей мере трех связей для групп, выбранных для остальных по меньшей мере трех связей, можно обеспечить определенный порядок связывания или направления конкретных реакционно-способных групп полипептида в определенные положения в молекулярном остове.
В другом варианте осуществления изобретения реакционно-способные группы полипептида по настоящему изобретению взаимодействуют с молекулярными линкерами, которые встраиваются между молекулярным остовом и полипептидом в конечном связанном состоянии.
В некоторых вариантах осуществления изобретения аминокислоты членов библиотек или совокупностей полипептидов могут быть заменены любыми природными или неприродными аминокислотами. Из указанных заменяемых аминокислот следует исключить аминокислоты, содержащие функциональные группы для перекрестного связывания полипептидов с молекулярным ядром, чтобы можно было заменять только последовательности петель. Заменяемые полипептидные последовательности представляют собой произвольные последовательности, постоянные последовательности или последовательности с произвольными или постоянными аминокислотами. Аминокислоты с реакционно-способными группами локализованы в определенных положениях полипептида, так как положение указанных аминокислот определяет размер петли.
В одном варианте осуществления изобретения полипептид с тремя реакционно-способными группами имеет последовательность (X)lY(X)mY(X)nY(X)o, где Y означает аминокислоту с реакционно-способной группой, Х означает произвольную аминокислоту, m и n являются числами от 3 до 6, определяющими длину внутренних полипептидных сегментов, которые могут быть одинаковыми или разными, l и o являются числами от 0 до 20, определяющими длину фланкирующих полипептидных сегментов.
Вместо конъюгации, опосредованной тиолом, могут быть использованы альтернативные методы присоединения молекулярного остова к пептиду путем ковалентного взаимодействия. Указанные методы могут быть альтернативно использованы для модификации или присоединения к полипептиду других частей (например, представляющих интерес мелких молекул, отличающихся от молекулярного остова) после их отбора или выделения в соответствии с настоящим изобретением. В данном варианте осуществления изобретения совершенно ясно, что присоединение не обязательно должно быть ковалентным и может включать нековалентное присоединение. Указанные методы могут быть использованы вместо опосредуемых тиолом методов (или в комбинации с ними) путем создания фага, отображающего белки и пептиды, содержащие неприродные аминокислоты с необходимыми химическими реакционно-способными группами, в комбинации с мелкими молекулами, содержащими комплементарные реакционно-способные группы, или путем введения неприродных аминокислот в полипептид, синтезированный химическими или рекомбинантными методаии в процессе создания молекулы после стадии отбора/выделения. Более подробно с данным вопросом можно ознакомиться в публикациях WO2009098450 или Heins, et al., Nat. Chem. Biol. 2009, 5(7), 502-7.
(iv) Объединение петель с образованием мультиспецифических молекул
Петли из пептидных лигандов или наборов пептидных лигандов успешно объединяют путем секвенирования и нового синтеза полипептида, включающего объединенные петли. Альтернативно могут быть синтезированы нуклеиновые кислоты, кодирующие такие полипептиды.
При объединении наборов, в частности, наборов отдельных петель, нуклеиновые кислоты, кодирующие указанные наборы, расщепляют и повторно лигируют с образованием нового набора, включающего другие комбинации петель из наборов компонентов. Фаговые векторы могут включать полилинкеры и другие сайты для рестрикционных ферментов, предоставляющие единственно возможные точки для разрезания и повторного лигирования векторов с целью создания требуемых мультиспецифических пептидных лигандов. Методы манипулирования фаговыми библиотеками хорошо известны в отношении антител и могут быть также использованы в настоящем изобретении.
(v) Присоединение эффекторных и функциональных групп
Эффекторные и/или функциональные группы могут быть присоединены, например, к N- или С-концу полипептида или к молекулярному остову.
Приемлемые эффекторные группы включают антитела, их части или фрагменты. Например, эффекторная группа может включать константную область легкой цепи (CL) антитела, домен СН1 тяжелой цепи антитела, домен СН2 тяжелой цепи антитела, домен СН3 тяжелой цепи антитела или любую их комбинацию помимо одного или нескольких доменов константной области. Эффекторная группа может также включать шарнирную область антитела (область, которая обычно находится между доменами СН1 и СН2 молекулы IgG).
В другом предпочтительном варианте осуществления данного объекта изобретения эффекторная группа по настоящему изобретению является Fc-областью молекулы IgG. Эффекторная группа пептидного лиганда по настоящему изобретению включает или состоит из Fc-области, гибридизированной с пептидным лигандом, и характеризуется временем полужизни tβ, равным одному дню или больше, двум дням или больше, 3 дням или больше, 4 дням или больше, 5 дням или больше, 6 дням или больше либо 7 дням или больше. Наиболее предпочтительно пептидный лиганд по настоящему изобретению включает или состоит из Fc-области, гибридизированной с пептидным лигандом, и характеризуется временем полужизни tβ, равным одному дню или больше.
Функциональные группы, как правило, включают связующие группы, лекарственные средства, реакционно-способные группы, предназначенные для присоединения других частей, функциональные группы, способствующие поглощению макроциклических пептидов клетками, и тому подобные.
Способность пептидов проникать в клетки позволяет использовать их для эффективного воздействия на внутриклеточные мишени. Мишени, на которые могут воздействовать пептиды, способные проникать в клетки, включают факторы транскрипции, внутриклеточные сигнальные молекулы, такие как тирозинкиназы, и молекулы, участвующие в апоптозе. Функциональные группы, обеспечивающие проникновение в клетки, включают пептиды или химические группы, вводимые в пептид или молекулярный остов. Пептиды, выделяемые из VP22, HIV-Tat, гомеобокса белка Drosophila (Antennapedia), описаны, например, в публикациях Chen and Harrison, Biochemical Society Transactions (2007), Volume 35, part 4, p821 “Cell-penetrating peptides in drug development: enabling intracellular targets” и “Intracellular delivery of large molecules and small peptides by cell penetrating peptides” by Gupta et al., in Advanced Drug Discovery Reviews (2004), Volume 57, 9637. Примеры коротких пептидов, которые, как установлено, способны эффективно проникать через плазматические мембраны, включают пептид пенетратин, состоящий из 16 аминокислот, выделяемый из белка Drosophila Antennapedia (Derossi et al. (1994), J. Biol. Chem., Volume 269, p10444 “The third helix of the Antennapedia homeodomain translocates through biological membranes”), ‘модель амфипатического пептида’, состоящего из 18 аминокислот (Oehlke et al. (1998) Biochim. Biophys. Acts, Volume 1414, p127 “Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically), и области с высоким содержанием аргинина белка HIV ТАТ. Непептидные методы включают использование имитаторов мелких молекул или SMOC, которые могут быть легко присоединены к биомолекулам (Okuyama et al. (2007), Nature Methods, Volume 4, p153 ‘Small-molecule mimics of an a-htlix for efficient transport of proteins into cells’. Другие химические методы введения гуанидиновых групп в молекулы также усиливают проникновение в клетки (Elson-Scwab et al. (2007), J. Biol. Chem., Volume 282, p13585 “Guanidinylated Neomycin Delivers Large Bioactive Cargo into cells through a heparin Sulphate Dependent Pathway”). Для усиления проникновения в клетки в молекулярный остов могут быть введены низкомолекулярные молекулы, такие как стероиды.
К одному классу функциональных групп, которые могут быть присоединены к пептидным лигандам, относятся антитела и их связующие фрагменты, такие как Fab, Fv или однодоменные фрагменты. В частности, могут быть использованы антитела, которые связываются с белками, способными увеличивать время полужизни пептидного лиганда in vivo.
Также могут быть использованы пептиды RGD, которые связываются с интегринами, присутствующими во многих клетках.
В одном варианте осуществления изобретения эффекторная группа пептидного лиганда по настоящему изобретению характеризуется временем полужизни tβ, равным 12 часам или больше, 24 часам или больше, 2 дням или больше, 3 дням или больше, 4 дням или больше, 5 дням или больше, 6 дням или больше, 7 дням или больше, 8 дням или больше, 9 дням или больше, 10 дням или больше, 11 дням или больше, 12 дням или больше, 13 дням или больше, 14 дням или больше, 15 дням или больше либо 20 дням или больше. Эффекторная группа пептидного лиганда или композиции по настоящему изобретению предпочтительно характеризуется временем полужизни tβ в интервале от 12 до 60 часов. В другом варианте осуществления изобретения время полужизни равно одному дню или больше. В еще одном варианте осуществления изобретения время полужизни находится в интервале от 12 до 26 часов.
Функциональные группы включают лекарственные средства, такие как цитотоксические агенты, предназначенные для лечения рака. Указанные средства включают алкилирующие агенты, такие как цисплатин и карбоплатин, а также оксалиплатин, мехлорэтамин, циклофосфамид, хлорамбуцил, ифосфамид; антиметаболиты, включающие аналоги пурина (азатиоприн и меркаптопурин) или аналоги пиримидина; растительные алкалоиды и терпеноиды, включающие алкалоиды барвинка, такие как винкристин, винбластин, винорелбин и виндезин; подофиллотоксин и его производные этопозид и тенипозид; таксаны, включающие паклитаксел, первоначально известный как таксол; ингибиторы топоизомеразы, включающие камптотецины; иринотекан и топотекан, и ингибиторы II типа, включающие амсакрин, этопозид, фосфат этопозида и тенипозид. Другие агенты могут включать противоопухолевые антибиотики, включающие иммуносупрессор дактиномицин (используемый при трансплантации почек), доксорубицин, эпирубицин, блеомицин и другие.
Возможные эффекторные группы также включают ферменты, такие как, например, кароксипептидаза G2, применяемые при лечении ферментами/пролекарствами, где пептидный лиганд заменяет антитела в ADEPT.
(vi) Синтез
Следует отметить, что выделение или идентификация представляющего интерес полипептида способами по настоящему изобретению может облегчить по возможности его последующий синтез. Таким образом, группы или совокупности полипептидов не нужно продуцировать методами рекомбинантных ДНК. Например, после определения последовательности представляющих интерес полипептидов такие полипептиды могут быть синтезированы стандартными методами с последующим осуществлением взаимодействия с молекулярным остовом in vitro. После синтеза полипептида могут быть использованы стандартные химические методы, так как больше нет необходимости сохранять функциональность или целостность генетически кодированной частицы-носителя, такой как фаг. Поэтому становится возможным быстрое широкомасштабное получение растворимого вещества для дальнейших экспериментов или проверки. В данной связи широкомасштабное получение кандидатов или лидерных последовательностей, идентифицированных способами по настоящему изобретению, может быть осуществлено стандартными химическими методами, описанными в публикации Timmerman et al.
Таким образом, настоящее изобретение также относится к производству полипептидов или конъюгатов, выбираемых способами по настоящему изобретению, при этом процесс производства может включать дополнительные стадии, рассмотренные ниже. В одном варианте осуществления изобретения указанные стадии выполняют в отношении конечного продукта полипептида/конъюгата, полученного методами химического синтеза, а не на фаге.
Аминокислотные остатки в представляющем интерес полипептиды могут быть заменены в процессе приготовления конъюгата или комплекса, например, после стадии первоначального выделения/идентификации.
Пептиды могут быть также удлинены, например, с включением еще одной петли и, следовательно, приобретают множество специфичностей.
Пептид может быть удлинен химическими методами в положении N-конца или С-конца либо внутри петель при использовании ортогонально защищенных лизинов (и их аналогов) с помощью стандартных химических методов, выполняемых в твердой фазе или фазе раствора. Стандартные методы химии пептидов могут быть использованы для введения активируемого N- или С-конца. Добавления могут быть альтернативно произведены путем конденсации фрагментов или химического лигирования, например, в соответствии с описанием, приведенным в публикации (Dawson PE, Muir TW, Clark-Lewis I, Kent, SBH, 1994, Synthesis оf Proteins by Native Chemical Ligation. Science 266:776-779), или при помощи ферментов, например, при использовании субтилигазы в соответствии с описанием, приведенным в публикациях (Subtiligase: a tool for semisynthesis of proteins Chang TK, Jackson DY, Burnier JP, Wells JA Proc Natl Acad Sci U S A. 1994 Dec 20;91(26):12544-8 or in Bioorganic & Medicinal Chemistry Letters Tags for labelling protein N-termini with subtiligase for proteomics Volume 18, Issue 22, 15 November 2008, Pages 6000-6003 Tags for labeling protein N-termini with subtiligase for proteomics; Hikari A.I. Yoshihara, Sami Mahrus and James A. Wells).
Альтернативно пептиды могут быть удлинены или модифицированы путем дальнейшей конъюгации по дисульфидным связям. Дополнительным преимуществом такого метода является возможность отделения первого и второго пептидов друг от друга в восстанавливающей среде клетки. В данном случае молекулярный остов (например, ТВМВ) может быть добавлен во время химического синтеза первого пептида для взаимодействия с тремя группами цистеина; дополнительный остаток цистеина может быть присоединен к N-концу первого пептида с возможностью взаимодействия указанного пептида только со свободным цистеином второго пептида.
Аналогичные методы в равной степени относятся к синтезу/связыванию двух бициклических и биспецифических макроциклов с возможностью создания тетраспецифической молекулы.
Другие функциональные группы или эффекторные группы могут быть добавлены таким же образом при помощи соответствующих химических методов путем связывания в положении N- или С-конца или при помощи боковых цепей. В одном варианте осуществления изобретения связывание осуществляют таким образом, чтобы не заблокировать активность ни одной из частей.
(vii) Модификация пептидов
При создании приемлемой молекулы на основе бициклических пептидов (бициклы; пептиды, конъюгированные с молекулярным остовом), подобной лекарственному средству и предназначенной для инъекций, ингаляции, назального, глазного, перорального или местного введения, необходимо учесть целый ряд свойств. В данном бицикле необходимо предусмотреть по меньшей мере следующие факторы:
• устойчивость к протеазам, причем данное положение включает устойчивость бицикла к плазматическим протеазам, эпителиальным (”мембраносвязанным протеазам”) протеазам, желудочным и кишечным протеазам, протеазам легочной поверхности, внутриклеточным протеазам и тому подобным. Устойчивость к протеазам должна сохраняться в разных видах, чтобы бицикл-кандидат можно было разрабатывать в животных моделях и с уверенностью вводить людям;
• замена чувствительных к окислению остатков, таких как триптофан и метионин, устойчивыми к окислению аналогами для улучшения профиля фармацевтической устойчивости молекулы;
• требуемый профиль растворимости, зависящий от соотношения заряженных, гидрофильных и гидрофобных остатков, что имеет важное значение для приготовления препарата и абсорбции;
• правильный баланс заряженных и гидрофобных остатков, так как гидрофобные остатки влияют на степень связывания плазматического белка и, следовательно, на концентрацию свободной фракции в плазме, в то время как заряженные остатки (в частности, аргинины) могут влиять на взаимодействие пептида с фосфолипидными мембранами на поверхностях клеток. Оба указанных фактора в комбинации могут влиять на время полужизни, объем распространения и воздействие лекарственного средства на основе пептида и могут быть заданы в соответствии с клиническими целями. Кроме того, правильная комбинация и число заряженных остатков относительно гидрофобных остатков может уменьшить раздражение в месте инъекции (при подкожном введении лекарственного средства на основе пептида).
• определение времени полужизни в зависимости от клинических показаний и схемы лечения. Вполне обоснованным может быть создание немодифицированной молекулы для кратковременного воздействия в острой стадии заболевания или создание бициклического пептида с химическими модификациями, увеличивающими время полужизни в плазме, который является оптимальным для лечения хронических заболеваний.
Существует много методов стабилизации терапевтических пептидов-кандидатов от протеолитического расщепления, которые предполагают работу в области пептидных миметиков (для ознакомления с данным вопросом см. публикации Gentilucci et al., Curr. Pharmaceutical Design, (2010), 16, 3185-203, и Nestor et al., Curr. Medicinal Chem. (2009), 16, 4399-418).
Указанные методы включают:
• циклизацию пептида;
• кэппирование N- и С-концов, обычно ацетилирование N-конца и амидирование С-конца;
• сканирование аланином для выявления и возможного удаления сайтов протеолитического воздействия;
• замена D-аминокислот для соответствия требованиям, предъявляемым к стерическим характеристикам боковой цепи аминокислоты, увеличения протеолитической устойчивости путем создания стерического препятствия и использования D-аминокислот для стабилизации конформаций β-витков (Tugуi et al., (2005) PNAS, 102(2), 413-418);
• замена N-метил/N-алкиламинокислот для обеспечения протеолитической защиты путем прямой модификации расщепляемой амидной связи (Flacco et al., Chembiochem. (2008), 9(14), 2200-3). N-Метилирование также оказывает сильное влияние на торсионные углы пептидной связи и, как считается, способствует проникновению в клетку и доступности при пероральном введении (Biron et al. (2008), Angew. Chem. Int. Ed., 47, 2595-99);
• введение неприродных аминокислот, то есть использование
- изостерических/изоэлектронных боковых цепей, которые не могут быть узнаны протеазами и при этом не влияют на активность мишени;
- ограничение боковых цепей аминокислот, обеспечивающее конформационное и стерическое блокирование протеолитического гидролиза ближайшей пептидной связи. В частности, такое ограничение применимо к аналогам пролина, большим боковым цепям, Сα-дизамещенным производным (простейшим производным которых является Aib, H2N-C(CH3)2-COOH) и циклическим аминокислотам (простейшим производным которых является аминоциклопропилкарбоновая кислота).
• Заменители пептидных связей, примеры которых включают:
- N-алкилирование (см. выше, то есть CO-NR)
- восстановленные пептидные связи (СН2-NH-)
- пептоиды (N-алкиламинокислоты, NR-CH2-CO)
- тиоамиды (CS-NH)
- азапептиды (СО-NH-NR)
- трансалкен (RNH=C-)
- обратное преобразование (NН-СО)
- заменители мочевины (NН-СО-NHR)
• Модуляция длины пептидного остова
- то есть β2/3-аминокислоты (NH-CR-CH2-CO, NH-CH2-CHR-CO)
• Замены в положении альфа-атома углерода аминокислот, который ограничивает конформации остова, при этом простейшим производным является аминоизомасляная кислота (Aib).
Следует отметить, что некоторые из указанных модификаций могут быть также использованы для намеренного усиления активности пептида в отношении мишени или, например, для идентификации эффективных замен для чувствительных к окислению аминокислот (Trp и Met). Также следует отметить, что бицикл Ас-06-34-18(ТМВ)-NH2 уже содержит две модификации, сообщающие устойчивость к протеолитическому расщеплению, которые представляют собой кэппирование N/С-конца и (би)циклизацию.
(В) Наборы, совокупности и группы полипептидных лигандов
(i) Создание библиотек
Библиотеки, предназначенные для отбора полипептидных лигандов, могут быть созданы методами, известными в данной области, которые представлены, например, в публикации WO2004/077062, или при помощи биологических систем, включающих системы фаговых векторов, рассмотренные в настоящем описании изобретения. В данной области известны другие векторные системы, которые включают другой фаг (например лямбда-фаг), экспрессирующие векторы на основе бактериальных плазмид, экспрессирующие векторы на основе эукариотических клеток, включающие дрожжевые векторы, и тому подобные. См., например, публикации WO2009098450 и Heinis, et al., Nat. Cnem. Biol. 2009, 5(7), 502-7.
Небиологические системы, например, представленные в публикации WO2004/077062, основаны на стандартных химических методах сканирования. Указанные методы являются простыми, но не обладают силой биологических систем, так как с их помощью невозможно или по меньшей мере затруднительно сканировать большие библиотеки пептидных лигандов. Однако указанные методы являются полезными для скрининга, например, небольшого числа пептидных лигандов. Скрининг при помощи таких анализов, однако, может требовать много времени, и число уникальных молекул, которые могут быть испытаны в отношении связывания с определенной мишенью, обычно не превышает 106 химических элементов.
В отличие от этого биологические методы скрининга или отбора обычно позволяют исследовать гораздо большее число разных молекул. Таким образом, в настоящем изобретении могут быть использованы биологические методы. При использовании биологических методов молекулы анализируют в одном реакционном сосуде, и молекулы, обладающие благоприятными свойствами (то есть связыванием), физически отделяют от неактивных молекул. Существуют методы отделения, которые позволяют одновременно создавать и анализировать более 1013 отдельных соединений. Примеры методов отбора молекул с высоким сродством включают отображение на фаге, отображение на рибосоме, отображение мРНК, отображение на дрожжах, отображение на бактериях или методы использования РНК/ДНК. Общей характеристикой указанных биологических методов отбора in vitro является то, что наборы лигандов кодированы ДНК или РНК. Они делают возможным размножение и идентификацию отобранных лигандов путем секвенирования. Метод отображения на фаге был использован, например, для выделения антител с очень высоким сродством связывания фактически с любой мишенью.
При использовании биологической системы после выбора векторной системы и клонирования одной или нескольких последовательностей нуклеиновых кислот, кодирующих представляющие интерес полипептиды, в векторе библиотеки можно создать большое разнообразие клонированных молекул путем выполнения мутагенеза до экспрессии; альтернативно кодированные белки могут быть экспрессированы и отобраны до выполнения мутагенеза и дополнительных циклов отбора.
Мутагенез последовательностей нуклеиновых кислот, кодирующих полипептиды с оптимизированной структурой, выполняют стандартными молекулярными методами. Наиболее часто используют полимеразную цепную реакцию или ПЦР (публикация Mullis and Faloona (1987) Methods Enzymol., 155: 335, включенная в настоящее описание изобретения в качестве ссылки). В данной области хорошо известна ПЦР, при осуществлении которой выполняют много циклов репликации ДНК, катализируемых термостойкой, ДНК-зависимой ДНК-полимеразой, для амплификации представляющей интерес последовательности мишени. Создание разных библиотек антител описано в публикации Winter et al. (1994) Ann. Rev. Immunology 12, 433-55 и приведенных в ней ссылках.
Альтернативно, с учетом короткой длины цепей полипептидов по настоящему изобретению, варианты могут быть синтезированы de novo и введены в приемлемые экспрессирующие векторы. Синтез пептидов может быть выполнен вышеописанными стандартными методами, известными в данной области. Широко распространены автоматические синтезаторы пептидов, такие как, например, синтезаторы ABI 433 компании Applied Biosystems (Applied Biosystems, Foster City, CA, USA).
(ii) Генетически кодированное разнообразие
В одном варианте осуществления изобретения представляющие интерес полипептиды кодированы генетически. Преимуществом указанного метода является более широкое разнообразие полипептидов наряду с простотой обращения. Примером библиотеки генетически кодированных полипептидов является библиотека отображения мРНК. Другим примером является библиотека реплицируемой, генетически кодированной отображаемой упаковки (rgdp), в частности, библиотека отображения на фаге. В одном варианте осуществления изобретения представляющие интерес полипептиды генетически кодированы в виде библиотеки отображения на фаге.
Таким образом, в одном варианте осуществления изобретения комплекс по настоящему изобретению включает реплицируемую, генетически кодированную отображаемую упаковку (rgdp), такую как фаговая частица. В указанных вариантах осуществления изобретения нуклеиновая кислота может включать геном фага. В указанных вариантах осуществления изобретения полипептид может включать оболочку фага.
Некоторые варианты осуществления изобретения могут относиться к созданию генетически кодированной комбинаторной библиотеки полипептидов, полученных путем трансляции ряда нуклеиновых кислот в соответствующие полипептиды и связывания молекул указанного молекулярного остова с указанными полипептидами.
Генетически кодированная комбинаторная библиотека полипептидов может быть создана путем отображения на фаге, отображения на дрожжах, отображения на рибосоме, отображения на бактериях или отображения мРНК.
С методами и технологиями отображения на фаге можно ознакомиться в публикации WO2009098450.
В одном варианте осуществления изобретения скрининг может быть выполнен в результате контактирования библиотеки, совокупности или группы полипептидных лигандов с мишенью и выделения одного или нескольких членов, связывающихся с указанной мишенью.
В другом варианте осуществления изобретения отдельные члены указанной библиотеки, совокупности или группы вводят в соприкосновение с мишенью и идентифицируют члены указанной библиотеки, связывающиеся с указанной мишенью.
В другом варианте осуществления изобретения члены указанной библиотеки, совокупности или группы одновременно вводят в соприкосновение с мишенью и отбирают члены, связывающиеся с указанной мишенью.
Мишень может быть пептидом, белком, полисахаридом, липидом, ДНК или РНК.
Мишень может быть рецептором, лигандом рецептора, ферментом, гормоном или цитозином.
Мишень может быть прокариотическим белком, эукариотическим белком или архебелком. Более конкретно лиганд мишени может быть белком млекопитающего, белком насекомого, бактериальным белком, грибным белком или вирусным белком.
Лиганд мишени может быть ферментом, таким как протеаза.
Следует отметить, что в объем настоящего изобретения также входят полипептидные лиганды, выделяемые в результате скрининга по настоящему изобретению. В одном варианте осуществления изобретения метод скрининга по настоящему изобретению включает стадию создания определенного количества выделенных полипептидов, способных связываться с указанными мишенями.
Настоящее изобретение также относится к пептидным лигандам, имеющим более двух петель. Например, трициклические полипептиды, присоединенные к молекулярному остову, могут быть созданы путем соединения N- и С-концов бициклического полипептида, присоединенного к молекулярному остову по настоящему изобретению. Соединенные N- и С-концы образуют третью петлю, создавая трициклический полипептид. Данный вариант осуществления изобретения не обязательно выполняют на фаге, он может быть выполнен на конъюгате полипептида с молекулярным остовом, рассмотренным в настоящем описании изобретения. Соединение N- и С-концов является стандартным методом химии пептидов. С-конец может быть активирован и/или N- и С-концы могут быть удлинены, например, путем добавления остатка цистеина к каждому концу, и затем соединены дисульфидной связью. Альтернативно соединение может быть произведено при помощи линкерной области, введенной в N/С-концы. Альтернативно N- и С-концы могут быть соединены обычной пептидной связью. Альтернативно могут быть использованы любые другие средства соединения N- и С-концов, например, может быть выполнена циклизация N- и С-концов стандартными методами в соответствии с описанием, приведенным в публикациях Linde et al., Peptide Science 90, 671-682 (2008) “Structure-activity relationship and metabolic stability studies of backbone cyclization and N-methylation of metanocortin peptides”, или Hess et al., J. Med. Chem. 51, 1026-1034 (2008) “backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administered drug lead for treating obesity”. Одним преимуществом таких трициклических молекул является отсутствие протеолитического расщепления свободных концов, в частности, под воздействием экзопротеазы. Другим преимуществом трициклического полипептида указанного типа является то, что третья петля может быть использована для общих функций, таких как связывание BSA, проникновение в клетку или перенос, мечение или любое другое подобное применение. Следует отметить, что указанная третья петля обычно не доступна для отбора (так как данная петля продуцирована не на фаге, а только на конъюгате полипептида с молекулярным остовом), поэтому, несмотря на возможность ее использования для других биологических функций, отбор/создание специфичности преимущественно осуществляется петлями 1 и 2.
(iii) Очистка фага
Фаг может быть очищен любыми приемлемыми средствами. В настоящем изобретении могут быть использованы стандартные методы. Например, фаг может быть очищен путем фильтрации или преципитации, такой как преципитация PEG; фаговые частицы могут быть получены и очищены при помощи вышеописанной преципитации полиэтиленгликолем (PEG). Более подробно с данным вопросом можно ознакомиться в публикации WО2009098459.
При необходимости дальнейших указаний можно обратиться к публикации Jespers et al. (Protein Engineering Design and Selection, 2004 17(10):709-71. Selection of optical biosensors from chemisynthetic antibody libraries). В одном варианте осуществления изобретения фаг может быть очищен методом, описанным в указанной публикации. Текст данной публикации специально включен в настоящее описание изобретения в качестве ссылки для ознакомления с методом очистки фага; в частности, следует обратиться к разделу “Материалы и методы”, с середины правого столбца на странице 709 публикации Jespers et al.
Кроме того, фаг может быть очищен методом, описанным в публикации Marks et al., J. Mol. Biol., vol. 222, pp581-597, которая специально включена в настоящее описание изобретения в качестве ссылки для ознакомления с описанием продуцирования/очистки фага.
(iv) Химия реакции
В настоящем изобретении использованы химические условия модификации полипептидов, позволяющие сохранить функцию и целостность генетически кодированного элемента продукта. В частности, когда генетически кодированный элемент является полипептидом, отображенным на поверхности кодирующего его фага, химия реакции предпочтительно не нарушает биологическую целостность фага. Условия реакции приведены в публикации WО2009098459.
(С) Применение полипептидных лигандов по настоящему изобретению
Полипептидные лиганды, выбираемые методом по настоящему изобретению, могут быть использованы в терапевтических и профилактических применениях in vivo, диагностических применениях in vitro и in vivo, при выполнении анализов in vitro, применении реагентов и тому подобных. Лиганды, обладающие выбранными уровнями специфичности, могут быть использованы в исследованиях животных, отличных от человека, где желательна перекрестная реактивность, или в диагностических применениях, где необходимо четко контролировать перекрестную реактивность с гомологами или паралогами. В некоторых применениях, например, в области вакцин, для создания вакцин против определенных болезней и патогенных микроорганизмов может быть использована способность вызывать иммунную реакцию на заранее определенные антигены.
Для введения млекопитающему желательно использовать по существу чистые пептидные лиганды, обладающие по меньшей мере 90-95% гомогенностью, и для фармацевтических применений желательна гомогенность, равная 98-99% или больше, особенно когда млекопитающим является человек. После очистки, которая при желании может быть частичной или до достижения гомогенности, выбранные полипептиды могут быть использованы в диагностических или терапевтических целях (включая экстракорпоральное применение) или для разработки и выполнения анализов, иммунофлуоресцентного окрашивания и тому подобного (Lefkovite and Pernis, (1979 and 1981) Immunological Methods, Volumes I and II, Academic Press, NY).
Пептидные лиганды по настоящему изобретению обычно применяют для профилактики, подавления или лечения воспалительных состояний, аллергических болезней, рака, бактериальных и вирусных инфекций и аутоиммунных нарушений (которые включают, не ограничиваясь ими, диабет I типа, рассеянный склероз, ревматоидный артрит, системную красную волчанку, болезнь Крона и миастению).
В настоящей заявке термин “профилактика” означает введение защитной композиции до возникновения болезни. Термин ”подавление” означает введение композиции после индуктивной фазы, но до клинического проявления болезни. Термин ”лечение” означает введение защитной композиции после появления симптомов болезни.
Существуют животные модели, которые могут быть использованы для исследования эффективности пептидных лигандов в отношении профилактики или лечения болезни. Настоящее изобретение облегчает использование животных моделей, что позволяет создавать полипептидные лиганды, способные перекрестно взаимодействовать с мишенями в организме человека и животного, и использовать животные модели.
В данной области известны методы исследования системной красной волчанки (SLE) у восприимчивых мышей (Knight et al. (1978), J. Exp. Med., 147:1653; Reinersten et al. (1978) New Eng. J. Med., 299:515). Миастению (MG) исследуют у самок мышей SJL/J путем введения растворимого белка AchR, полученного у другого вида (Lindstrom et al. (1988) Adv. Immunol., 42:233). Артрит вызывают у восприимчивых мышей путем введения коллагена II типа (Stuart et al. (1984) Ann. Rev. Immunol., 42:233). В научной литературе описана модель, в соответствии с которой у восприимчивых крыс вызывают адъювантный артрит путем введения микобактериального хитшокового белка (Van Eden et al. (1988) Nature, 331:171). Тиреоидит вызывают у мышей путем введения тиреоглобулина (Maron et al. (1980) J. Exp. Med., 152:1115). Инсулинзависимый сахарный диабет (IDDM) возникает естественным путем или может быть вызван у определенных видов мышей методом, описанным в публикации Kanasawa et al. (1984) Diabetologia, 27:113. EAE у мышей и крыс служит в качестве модели рассеянного склероза (MS) у человека. В указанной модели вызывают демиелинизирующее заболевание путем введения основного белка миелина (см. публикации Paterson (1986) Textbook of Immunopathology, Mischer et al., eds., Grune and Stratton, New York, pp.179-213; McFarlin et al. (1973) Science, 179:478; и Satoh et al. (1987) J. Immunol. 138:179).
Пептидные лиганды по настоящему изобретению могут быть использованы в очищенной форме вместе с фармацевтически приемлемыми носителями. Указанные носители обычно включают водные или водно-спиртовые растворы, эмульсии или суспензии, солевой раствор и/или забуференные среды. Наполнители для парентерального введения включают раствор хлорида натрия, декстрозу Рингера, декстрозу и хлорид кальция и лактат Рингера. Физиологически приемлемые адъюванты, используемые при необходимости сохранения полипептидного комплекса в суспензии, могут быть выбраны из загустителей, таких как карбоксиметилцеллюлоза, поливинилпирролидон, желатин и альгинаты.
Наполнители для внутривенного введения включают пополнители жидкости и питательных веществ и пополнители электролита, например, пополнители на основе декстрозы Рингера. Также могут быть использованы консерванты и другие добавки, такие как антимикробные агенты, антиоксиданты, хелатообразователи и инертные газы (Mack (1982) Remington’s Pharmaceutical Sciences, 16th Edition).
Пептидные лиганды по настоящему изобретению могут быть использованы в качестве отдельно вводимых композиций или совместно с другими агентами. Такие агенты могут включать антитела, фрагменты антител и разные иммунотерапевтические средства, такие как циклоспорин, метотрексат, адриамицин или цисплатин, и иммунотоксины. Фармацевтические композиции могут включать смеси разных цитотоксических или других агентов совместно с выбранными антителами, рецепторами или их связующими белками по настоящему изобретению, или даже комбинации выбранных полипептидов по настоящему изобретению, обладающих разными специфичностями, например, полипептидов, выбранных с помощью разных лигандов мишени независимо от того, были они объединены или нет до введения.
Способ введения фармацевтических композиций по настоящему изобретению может быть любым способом, известным специалистам в данной области. Для терапии, включающей без каких-либо ограничений иммунотерапию, выбранные антитела, рецепторы или их связующие белки по настоящему изобретению могут быть введены любому субъекту стандартными методами. Введение может быть произведено любым приемлемым способом, включающим парентеральное, внутривенное, внутримышечное, внутрибрюшинное, чрескожное, внутрилегочное введение, а также путем прямого вливания через катетер. Доза и частота введения зависят от возраста, пола и состояния здоровья субъекта, одновременного введения других лекарственных средств, противопоказаний и других показателей, которые должны быть приняты во внимание врачом.
Пептидные лиганды по настоящему изобретению могут быть лиофилизированы для хранения и восстановлены в приемлемом носителе перед применением. Указанный метод является эффективным, при этом могут быть использованы любые, известные в данной области методы лиофилизации и восстановления. Специалистам в данной области должно быть известно, что лиофилизация и восстановление могут привести к утрате активности в разной степени, поэтому содержание активных веществ должно быть увеличено для компенсации потерь.
Композиции, содержащие пептидные лиганды по настоящему изобретению или их смеси, можно вводить для профилактического и/или терапевтического лечения. В определенных терапевтических применениях соответствующее количество, необходимое по меньшей мере для ингибирования, подавления, модуляции или уничтожения популяции выбранных клеток, определяется как ”терапевтически эффективная доза”. Количества, необходимые для достижения указанной дозы, зависят от тяжести заболевания и общего состояния иммунной системы субъекта, но обычно находятся в пределах от 0,005 до 5,0 мг выбранного пептидного лиганда на килограмм массы тела, при этом обычно используют дозы от 0,05 до 2,0 мг/кг/дозу. Для профилактических применений композиции, содержащие пептидные лиганды по настоящему изобретению или их смеси, могут быть также введены в аналогичных или несколько меньших дозах.
Композиция, содержащая пептидный лиганд по настоящему изобретению, может быть использована в профилактических и терапевтических целях для изменения, инактивации, уничтожения или удаления выбранной популяции клеток-мишеней у млекопитающего. Кроме того, выбранные наборы полипептидов по настоящему изобретению могут быть использованы экстракорпорально или in vitro для избирательного уничтожения, инактивации или эффективного удаления популяции клеток-мишеней из гетерогенной совокупности клеток. Кровь, полученная у млекопитающего, может быть соединена экстракорпорально с выбранными пептидными лигандами и возвращена млекопитающему стандартными методами для уничтожения или удаления нежелательных клеток из кровотока.
Другим объектом настоящего изобретения является метод профилактики, подавления или лечения воспалительных состояний, аллергических болезней, рака, бактериальных или вирусных инфекций, офтальмологических нарушений и аутоиммунных нарушений, который включает введение нуждающемуся субъекту пептидного лиганда по настоящему изобретению.
Примеры приемлемых ”офтальмологических нарушений” (включая экссудативные и/или воспалительные офтальмологические нарушения, нарушения, относящиеся к ухудшению проницаемости и/или целостности сосудов сетчатки, нарушения, относящиеся к разрыву микрососудов сетчатки, вызывающему фокальные кровоизлияния, заболевания задней части глаза, заболевания сетчатки и заболевания передней части глаза) включают, не ограничиваясь ими, возрастную дегенерацию желтого пятна (ARMD), экссудативную дегенерацию желтого пятна (также известную как ”мокрая” или неоваскулярная возрастная дегенерация желтого пятна (мокрая-AMD), отек желтого пятна, возрастную дисковидную дегенерацию желтого пятна, цистоидный отек желтого пятна, отек века, отек сетчатки, диабетическую ретинопатию, острую нейроретинопатию желтого пятна, центральную серозную ретинопатию, хориоретинопатию, хороидальное образование новых сосудов, неоваскулярную макулопатию, неоваскулярную глаукому, обструктуивные артериальные и венозные ретинопатии (например, венозную окклюзию сетчатки или артериальную окклюзию сетчатки), окклюзию центральной вены сетчатки, диссеминированную внутрисосудистую коагулопатию, окклюзию ответвления вены сетчатки, гипертонические изменения глазного дна, ишемический синдром глаза, артериальную микроаневризму сетчатки, болезнь Коата, телеангиэктазию ямки, одностороннюю окклюзию вены сетчатки, папиллофлебит, окклюзию центральной артерии сетчатки, окклюзию ответвления артерии сетчатки, заболевание сонной артерии (CAD), глазурный васкулит ответвления сосуда, серповидно-клеточную ретинопатию и другие гемоглобинопатии, ангиоидные полосы сетчатки, отек желтого пятна, возникающий в результате такой этиологии как болезнь (например, диабетический отек желтого пятна), повреждение глаза или хирургическая операция глаза, ишемию или дегенерацию сетчатки, вызванную, например, повреждением, травмой или опухолями, увеит, ирит, васкулит сетчатки, эндофтальмит, панофтальмит, метастатическую офтальмию, хориоидит, пигментный эпителий сетчатки, конъюнктивит, циклит, склерит, эписклерит, ретробульбарный неврит, кератит, блефарит, экссудативное отслоение сетчатки, язву роговицы, язву конъюктивы, хронический нумуллярный кератит, кератит Тайгесона, прогрессирующую разъедающую язву роговицы, воспалительное заболевание глаза, вызванное бактериальной или вирусной инфекцией и офтальмологической операцией, воспалительное заболевание глаза, вызванное физическим повреждением глаза, симптом, вызванный воспалительным заболеванием глаза, включая зуд, воспалительную гиперемию, отек и язву, эритему, экссудативную эритему, узловатую эритему, кольцевидную эритему, отек склеры, дерматит, ангионевротический отек, отек гортани, отек языка, подсвязочный отек, бронхит, ринит, фарингит, синусит, ларингит или отит среднего уха.
Термин ”заболевания задней части глаза” означает заболевания, поражающие наряду с прочими органами сетчатку, желтое пятно, ямку в задней области глаза. Примеры приемлемых ”заболеваний задней части глаза” включают, не ограничиваясь ими, отек желтого пятна, такой как клинический отек желтого пятна или ангиографический цистоидный отек желтого пятна, возникающий в результате разных этиологий, таких как диабет, экссудативная дегенерация желтого пятна, и отек желтого пятна, возникающий в результате лазерного лечения сетчатки, возрастную дегенерацию желтого пятна, ретинопатию преждевременного развития (также известную как ретролентальная фиброплазия), ишемию сетчатки и хороидальное образование новых сосудов, заболевания сетчатки (диабетическая ретинопатия, диабетический отек сетчатки, отслоение сетчатки, старческая дегенерация желтого пятна вследствие образования новых сосудов под сетчаткой, миопическая ретинопатия); воспалительные заболевания; увеит, ассоциированный с новообразованиями, такими как ретинобластома или псевдоглиома; образование новых сосудов после витректомии; сосудистые заболевания (ишемия сетчатки, хороидальная сосудистая недостаточность, хороидальный тромбоз, ретинопатии, возникающие в результате ишемии сонной артерии) и образование новых сосудов глазного нерва.
Термин ”заболевания передней части глаза” означает заболевания, поражающие в основном ткани передней части глаза, такие как роговица, радужка, ресничное тело, конъюнктива и т.д. Примеры приемлемых ”заболеваний передней части глаза” включают, не ограничиваясь ими, образование новых сосудов роговицы (вследствие воспаления, трансплантации), возрастную гипоплазию радужки, заболевания или помутнения роговицы с экссудативным или воспалительным компонентом, образование новых сосудов вследствие проникновения в глаз или повреждения глаза; хронический увеит; увеит передней области глаза; воспалительные состояния в результате хирургических операций, таких как LASIK, LASEK, операция по коррекции рефракции, имплантация IOL; необратимый отек роговицы как осложнение после операции по удалению катаракты; отек в результате инсульта или травмы (физической, химической, фармакологической и т.д.); воспаление; конъюнктивит (например, постоянный аллергический, ангионевротический папиллярный, сезонный периодический аллергический, токсический, конъюнктивит, вызванный бактериями, вирусами или Chlamidia); кератоконъюнктивит (весенний, атопический, сиккативный); иридоциклит; ирит; склерит; эписклерит; инфекционный кератит; поверхностный точечный кератит; кератоконус; заднюю полиморфную дистрофию; дистрофию Фука (роговицы и эндотелия); афакическую и псевдофакическую буллезную кератопатию; отек роговицы; заболевание склеры; рубцовый пемфигоид глаза; синдром Поснера-Шлосмана; болезнь Бехчета; синдром Фогта-Койанаги-Харада; аллергические реакции; поверхностные нарушения глаза; отек конъюнктивы; токсоплазмозный хориоретинит; воспалительную псевдоопухоль глазницы; хемоз; гипереми. конъюнктивы; периорбитальный целлюлит; острый дакриоцистит; неспецифический васкулит; саркоидоз и инфицирование цитомегаловирусом.
Примеры приемлемых ”нарушений, ассоциированных с избыточной проницаемостью сосудов и/или отеком глаза”, например, в сетчатке или стекловидном теле, включают, не ограничиваясь ими, возрастную дегенерацию желтого пятна (AMD), отек сетчатки, кровоизлияние сетчатки, кровоизлияние стекловидного тела, отек желтого пятна (МЕ), диабетический отек желтого пятна (DME), пролиферативную диабетическую ретинопатию (PDR) и непролиферативную диабетическую ретинопатию (DR), радиационную ретинопатию, телеангиэктазию, серозную ретинопатию и венозную окклюзию сетчатки. Отек сетчатки является аккумуляцией жидкости внутри сетчатки. DME возникает в результате изменений микрососудов сетчатки, происходящих у субъектов, страдающих диабетом. Аномалия гематоретинального барьера вызывает утечку плазматических компонентов в окружающую сетчатку, вследствие чего возникает отек сетчатки. Другие нарушения сетчатки включают венозные окклюзии сетчатки (например, окклюзии ответвления или центральной вены), радиационную ретинопатию, серповидно-клеточную ретинопатию, ретинопатию преждевременного развития, болезнь Гиппеля-Линдау, задний увеит, хроническое отслоение сетчатки, синдром Ирвина Гасса, болезнь Илза, ретинит и/или хороидит.
(D) Мутация полипептидов
Требуемое разнообразие обычно создают путем изменения выбранной молекулы в одном или нескольких положениях. Подлежащие изменению положения выбирают с возможностью создания библиотек для каждого отдельного положения в последовательностях петель. Там, где это возможно, одно или несколько положений могут быть опущены, например, если очевидно, что мутация в данных положениях не может быть произведена без утраты активности.
Разнообразие может быть достигнуто путем рандомизации, в процессе которой постоянные аминокислоты заменяют любой аминокислотой или ее природным или синтетическим аналогом, получая большое число вариантов, или путем замены постоянной аминокислоты одной или несколькими определенными подгруппами аминокислот с образованием более ограниченного числа вариантов.
В научной литературе описаны разные методы достижения такого разнообразия. Методы мутации выбранных положений также хорошо известны в данной области и включают использование ошибочно спаренных олигонуклеотидов или вырожденных олигонуклеотидов с выполнением или без выполнения ПЦР. Например, было создано несколько библиотек синтетических антител путем направленной мутации антигенсвязывающих петель. Такой же метод может быть использован в контексте настоящего изобретения. Например, область Н3 Fab-фрагмента человека, связывающего столбнячный токсин, рандомизировали с созданием целого ряда новых специфичностей связывания (Barbas et al. (1992) Proc. Natl. Acad. Sci. USA, 89: 4457). Произвольные или полупроизвольные области Н3 и L3 присоединяли к сегментам зародышевого гена V с образованием больших библиотек с мутированными остовными областями (Hoogenboom & Winter (1992) R. Mol. Biol., 227: 381; Barbas et al., (1992) Proc. Natl. Acad. Sci. USA, 89: 4457; Nissim et al. (1994) EMBO J., 13: 692; Griffiths et al. (1994) EMBO J., 13: 3245; De Kruif et al. (1995) J. Mol. Biol., 248: 97). Такое разнообразие было расширено путем включения некоторых или всех антигенсвязывающих петель (Crameri et al. (1996) Nature Med., 2: 100; Riechmann et al. (1995) BiolTechnology, 13: 475; Morphosys, WO97/08320, см. выше).
Однако, поскольку полипептиды, использованные в настоящем изобретении, гораздо меньше антител, предпочтительным методом является синтез мутантных полипептидов de novo. Мутагенез структурированных полипептидов описан выше в связи с созданием библиотек.
Настоящее изобретение далее описано со ссылкой на нижеследующие примеры.
Примеры
Материалы и методы
Клонирование фаговых библиотек
Фаговые библиотеки были созданы методом, описанным в публикации Heinis et al., Nat. Chem. Biol. 2009, 5 (7), 502-7). В соответствии с указанной публикацией гены, кодирующие полупроизвольный пептид, имеющий последовательность Xaa-Cys-(Xaa)3-Cys-(Xaa)3-, линкер Gly-Gly-Ser-Gly и два домена без дисульфидных связей D1 и D2 (Kather et al., J. Mol. Biol. 2005, 354 (3), 666-78), клонировали в правильной ориентации в векторе на основе фага fd0D12, получая ‘библиотеку 3x3’. Гены, кодирующие набор пептидов и два домена гена 3, были созданы поэтапно в результате выполнения двух последовательных реакций ПЦР. Сначала гены доменов D1 и D2 амплифицировали при помощи ПЦР с двумя затравками prepcr (5'-GGCGGTTCTGGCGCTGAAACTGTTGAAAGTAG-3') и sfi2fo (5'-GAAGCCATGGCCCCCGAGGCCCCGGACGGAGCATTGACAGG-3'; сайт рестрикции подчеркнут), используя вектор fdg3p0ss21 (Kather et al., J. Mol. Biol. 2005, 354 (3), 666-78) в качестве матрицы. Затем при выполнении реакции ПЦР присоединяли ДНК, кодирующую произвольные пептиды, используя затравку sficx3ba: 5'-TATGCGGCCCAGCCGGCCATGG СANNKTGTNNKNNKNNKTGCNNKNNKNNKNNKТGТNNKGGGCGGTTCTGGCGCTG-3' (сайт рестрикции подчеркнут) и sfi2fo. Лигирование 55 и 11 мкг Sfil-расщепленной плазмиды fd0D12 и продукта ПЦР позволило получить 5,6×108 колоний на 10 планшетах размером 20×20 см со средой 2YT и хлорамфеникола(30 мкг/мл). Колонии соскребали с планшетов со средой 2YT, содержащей 15% глицерина, и хранили при -80°С. При создании библиотек по настоящему изобретению был использован метод получения полупроизвольного пептида Pro-Ala-Met-Ala-Cys-(Xaa)3-Cys-(Xaa)3-Cys для библиотеки 3×3, поэтому последовательность затравки sficx3ba была заменена последовательностью 5’-TATGCGGCCCAGCCGGCCATGGCATGTNNKNNKNNKTGCNNKNNKNNKTGTGGCGGTTCTG GCGCTG-3’. Библиотеки с другими длинами петель был созданы тем же методом.
Отбор фагов
Исходные растворы глицерина фаговых библиотек разводили до OD600 = 0,1 в 500 мл среды 2YT/хлорамфеникола (30 мкг/мл), культуры и фаг продуцировали при 30°С в течение ночи (15-16 часов). Фаг очищали и химически модифицировали в соответствии с описанием, приведенным в публикации Heinis et al., Nat. Chem. Biol. 2009, 5 (7), 502-7). Биотинилированный hPK (3 мкг) (IHPKA, из человеческой плазмы, Innovative Research, Novi, MI, USA) инкубировали с 50 мкл предварительно промытых магнитных стрептавидиновых гранул (Dynal, M-280 компании Invitrogen, Рaisley, UK) в течение 10 минут при комнатной температуре. Гранулы трижды промывали и блокировали 0,5 мл промывочного буфера (10 мМ трис-буфера с Cl, рН 7,4, 150 мМ NaCl, 10 мМ MgCl2, 1 мМ CaCl2), содержащего 1% BSA и 0,1% твина 20, в течение 30 минут при комнатной температуре с вращением. Химически модифицированный фаг (обычно 1010 - 1011 туберкулиновых единиц, растворенных в 2 мл промывочного буфера) блокировали, добавляя 1 мл промывочного буфера, содержащего 3% BSA и 0,3% твина 20. Блокированные гранулы смешивали с блокированным химически модифицированным фагом и инкубировали в течение 30 минут на вращающемся колесе при комнатной температуре. Гранулы промывали 8 раз промывочным буфером, содержащим 0,1% твина 20, и дважды промывочным буфером, после чего инкубировали с 100 мкл 50 мМ глицина, рН 2,2, в течение 5 минут. Элюированный фаг переносили в 50 мкл 1 М раствора трис-буфера с Cl, рН 8, для нейтрализации, инкубировали с 30 мл клеток TG1 при OD600 = 0,4 в течение 90 минут при 37°С, после чего клетки культивировали на больших планшетах со средой 2YT/хлорамфеникола. Один из двух дополнительных циклов пэннинга выполняли тем же методом. Во втором цикле отбора использовали магнитные гранулы, покрытые нейтравидином, для предотвращения обогащения стрептавидин-специфическими пептидами. Нейтравидиновые гранулы были получены в результате осуществления взаимодействия 0,8 мг нейтравидина (Pierce, Rockford, IL, USA) с 0,5 мл тозил-активированных магнитных гранул (Dynal, M-280 компании Invitrogen, Paisleу, UK) в соответствии с инструкциями поставщика.
Клонирование и экспрессия калликреина человека, обезьяны и крысы
Каталитический домен калликреина (РК) человека, обезьяны и крысы экспрессировали в клетках млекопитающих в виде неактивного предшественника, содержащего пропептид, соединенный в положении N-конца при помощи сайта расщепления proTEV с каталитическим доменом. Экспрессирующий вектор клонировали, белок экспрессировали, активировали и очищали в соответствии с нижеследующим описанием. Синтетические гены, кодирующие сигнальную последовательность РК, полигистидиновую метку, сайт расщепления proTEV, зрелый каталитический домен РК и терминирующий кодон, были приобретены в компании Geneart (Regensburg, Germany) (дополнительные материалы). Была получена плазмидная ДНК, содержащая синтетические гены для РК человека, обезьяны (Macaca mulatta) и крысы, после чего ген был перенесен в экспрессирующий вектор млекопитающего pEXPR-IBA42 (IBA Biotechnology, Gottingem, Germany) с использованием двух рестрикционных ферментов XhoI и HindIII (Fermentas, Vilnius, Latvia) и ДНК-лигазы Т4 (Fermentas). Лигированные плазмиды переносили в синие электрокомпетентные клетки XL-1 (Stratagene, Santa Clara, USA) и культивировали на агаровых пластинках со средой 2YT, содержащей ампициллин (10 мкг/мл). Была получена ДНК из трех экспрессирующих векторов (именуемых mPK, rPK и hPK), и правильные последовательности были подтверждены путем секвенирования ДНК (Macrogen, Seoul, South Korea).
Три ортолога плазматических калликреинов экспрессировали в клетках млекопитающих следующим образом. 50 мл адаптированных в суспензии клеток НЕК-293 выращивали в бессывороточной среде ExCell 293 (SAFC Biosciences, St. Louis, МO) в присутствии 4 мМ глутамина и вальпроиновой кислоты ингибитора гистондеацетилазы (3,75 мМ) в 100 мл колбе, орбитально встряхиваемой со скоростью 180 оборотов/минуту в инкубаторе ISF-4-W (Kuhner AG, Birsfelden, Switzerland) при 37°С в присутствии 5% СО2. Эмбриональные клетки почки (НЕК-293) с высокой плотностью клеток (20×106 клеток/мл) (Backliwal et al., Biotechnol. Bioeng. 2008, 99(3), 721-7) трасфецировали тремя плазмидами (300 мкг/мл), используя линейный полиэтиленимин (PEI, Polysciences, Eppenheim, Germany). В конце 7-дневного цикла продуцирования клетки собирали центрифугированием со скоростью 2500 оборотов/минуту в течение 15 минут при 4°С. Любой дополнительный клеточный дебрис удаляли из среды фильтрованием через 0,45 мкм PES мембраны (фильтр, содержащий 250 мл ТРР с низкой степенью связывания белка). Меченный полигистидином белок очищали при помощи Ni-аффинной хроматографии с использованием смолы Ni-NTA, промывочного буфера (500 мМ NaCl, 25 мМ Na2HPO4, рН 7,4) и элюирующего буфера (500 мМ NaCl, 25 мМ Na2HPO4, рН 7,4, 500 мМ имидазола). Белок частично активировали (50 единицами) prоTEV (Рromega, Madison, Wisconsin, USA) и дополнительно очищали при помощи Ni-аффинной хроматографии и гель-фильтрации (колонка PD10, 150 мМ NaCl, 0,5 мМ EDTA, 50 мМ HEPES, рН 7).
Создание полипептидов с улучшенной связывающей активностью
Рандомизация отдельных положений
Создание библиотек. Для картирования аминокислот в связывающих калликреин бициклических пептидах была создана совокупность небольших библиотек. Для бицикла, включающего 2 петли, состоящие из 5 остатков, было создано 10 отдельных библиотек, каждая из которых была рандомизирована в определенном кодоне пептидной последовательности. Для каждой библиотеки были созданы олигонуклеотиды для мутирования ДНК фагового домена путем сайтнаправленного мутагенеза. Мутагенез включал рандомизацию представляющего интерес кодона (замена на NNS) и удаление единственного сайта рестрикции ApaL из геномной последовательности матрицы. Продукт мутагенеза очищали, используя набор для очистки ПЦР QIAgen QIAquick, и элюировали в ультрачистую воду. Каждую библиотеку использовали для отдельной трансформации TG1 E. coli путем электропорации при помощи устройства Micropulser BioRad (программа Ес1) и 1 мм кюветы BioRad. После восстановления в течение 1 часа при 37°С в 1 мл среды SOC трансформанты библиотеки выращивали в течение ночи в 25 мл бульона 2TY, содержащего антибиотик для избирательного выращивания только трансформантов библиотеки. Бактерии собирали центрифугированием, фаговую ДНК библиотеки очищали от E. coli при помощи набора Plasmid Plus Midi QIAgen и элюировали в дистиллированную воду. Очищенную ДНК расщепляли ApaL1 в течение 2 часов в буфере 4 New England Biolabs для удаления исходного материала. После расщепления ДНК повторно очищали при помощи набора для очистки ПЦР QIAgen (аналогичного использованному выше) и использовали для трансформации TG1 (электропорация, описанная выше). Трансформанты восстанавливали в SOC в течение 1 часа и культивировали на LB-агаровых пластинках, содержащих избирательно действующий антибиотик, и колонии выращивали в течение ночи при 37°С.
Анализ связывания отдельных клонов. Колонии трансформантов библиотеки произвольно собирали и выращивали в виде отдельных культур в бульоне 2TY, содержащем избирательно действующий антибиотик. В собранных колониях секвенировали ДНК при помощи секвенатора ДНК PyroMark Q96 QIAgen для выявления аминокислотных замен, присутствующих в каждом клоне. Выделенный клон, содержащий уникальную замену, исследовали в отношении связывания калликреина человеческой плазмы следующим образом. Содержащий фаг супернатант удаляли из культуры и фаг циклизировали трисбромметилбензолом (ТВМВ) с помощью методов, описанных в публикации Heinis et al. (Nature Chemical Biology, vol. 5, pp 502-507 (2009). Очищенный фаг исследовали в отношении связывания с биотинилированным калликреином человеческой плазмы, выполняя анализ гомогенного связывания на планшете; результаты анализа считывали при помощи спектрофотометра для прочтения планшетов BMG Labtech Pherastar FS. Количественные данные связывания, полученные в результате выполнения трех анализов, усредняли (среднее значение) и выражали в виде отношения ”сигнал:фон” (где фон представлял собой образец, исследованный при отсутствии материала мишени). Отношение ”сигнал:фон” было выражено в процентах от параллельно исследованного исходного образца. Планки погрешностей означают стандартное отклонение от среднего значения. Представленные данные анализа являются репрезентативными по меньшей мере для 2 независимых экспериментов. Данные анализов коррелировали с пептидными последовательностями. Замены, отмеченные серым цветом, не исследовали (клон не был выделен в результате произвольного отбора членов библиотеки). Параллельно исследовали образец несвязывающегося (произвольно выбранного) бицикла для иллюстрации исходного значения анализа.
Рандомизация пептидных доменов
Создание библиотек. Небольшие фаговые библиотеки были созданы методами, описанными в приведенной выше публикации Heinis et al. ‘Cloning of phage libraries’. Затравка sficx3ba была модифицирована так, чтобы кодирующая бицикл часть включала исходную последовательность ДНК бицикла 5×5 (5×5: две петли, содержащие по 5 остатков), содержащую только 4-6 кодонов, рандомизированных до NNS. Рандомизированными кодонами были кодоны, кодирующие представляющий интерес пептидный домен/фрагмент.
Анализ связывания отдельных кодонов. Колонии трансформантов библиотек или отобранные колонии собирали и выращивали в виде отдельных культур в бульоне 2TY, содержащем избирательно действующий антибиотик. В собранных колониях секвенировали ДНК при помощи секвенатора ДНК PyroMark Q96 QIAgen для выявления амнокислотных замен, присутствующих в каждом клоне, и исследовали в отношении связывания калликреина человеческой плазмы следующим образом. Содержащий фаг супернатант удаляли из культуры и фаг циклизировали трисбромметилбензолом (ТВМВ) с помощью методов, описанных в публикации Heinis et al. (Nature Chemical Biology, vol. 5, pp 502-507 (2009). Очищенный фаг исследовали в отношении связывания с биотинилированным калликреином человеческой плазмы, выполняя анализ гомогенного связывания на планшете; результаты анализа считывали при помощи спектрофотометра для прочтения планшетов BMG Labtech Pherastar FS. Количественные данные связывания, полученные в результате выполнения двух анализов, усредняли (среднее значение) и выражали в виде отношения ”сигнал:фон”. Представленные данные анализа являются репрезентативными по меньшей мере для 2 независимых экспериментов. Данные анализов коррелировали с пептидными последовательностями.
Синтез и очистка бициклических пептидов
Пептидные последовательности показаны в таблицах 4-6. Центральные пептидные последовательности представляли собой Ac-C1S1W2P3A4R5C2L6H7Q8D9L10C3-NH2 (обозначенная как Ас-(06-34-18) (ТМВ)-NH2) и Ac-C1S1F2P3Y4R5C2L6H7Q8D9L10C3-NH2 (обозначенная как Ас-(06-34-18)(ТМВ)-NH2 Phe2 Tyr4).
Пептиды синтезировали на основе химии Fmoc, используя синтезатор пептидов Symphony компании Peptide Instruments. Были использованы стандартные Fmoc-аминокислоты (Sigma, Merck) со следующими защитными группами боковых цепей: Arg(Pbf); Asn(Trt); Asp(OtBu); Cys(Trt); Glu(OtBu); Gln(Trt); His(Trt); Lys(Boc); Ser(tBu); Thr(tBu); Trp(Boc); Tyr(tBu) (Sigma). Связующий реагент представлял собой HCNU (Pepceuticals), диизопропилэтиламин (DIPEA, Sigma) был использован в качестве основания, защитные группы удаляли при помощи 20% пиперидина в ДМФ (AGTC). Синтез выполняли в масштабе 100 мкмолей, используя 0,37 ммоль/г амидной смолы АМ Fmoc-Rink (AGTC), Fmoc-аминокислоты использовали в четырехкратном избытке и основание использовали в четырехкратном избытке по отношению к аминокислотам. Аминокислоты растворяли при 0,2 М в ДМФ, HCTU при 0,4 М в ДМФ и DIPEA при 1,6 М в N-метилпирролидоне (Alfa Aesar). Время связывания обычно было равно 30 минутам, и время удаления защитных групп составляло 2×2,5 минуты. Fmoc-N-метилглицин (Fmoc-Sar-OH, Merck) связывали в течение 1 часа, время удаления защитных групп и связывания для следующего остатка было равно соответственно 20 минутам и 1 часу. После выполнения синтеза смолу промывали дихлорметаном и сушили. Защитные группы боковых цепей отщепляли от подложки при помощи 10 мл ТФУ/Н2О/iPr3SiH/дитиотреитола с соотношением 95:2,5:2,5:2,5 об./об./об./мас. в течение 3 часов. После отщепления защитных групп использованную смолу удаляли фильтрованием и фильтрат добавляли к 35 мл диэтилового эфира, охлажденного до -80°С. Пептидный осадок центрифугировали, эфирный супернатант выбрасывали и пептидный осадок промывали холодным эфиром еще два раза. Затем пептиды повторно солюбилизировали в 5-10 мл ацетонитрила-воды и лиофилизировали. Небольшой образец удаляли для анализа чистоты неочищенного продукта при помощи масс-спектрометрии (MALDI-TOF, Voyager DE компании Applied Biosystems). После лиофилизации пептидные порошки поглощали 10 мл 6 М раствора гидрохлорида гуанидина в Н2О, содержащего 0,5 мл 1 М дитиотреитола, и вводили в колонку для препаративной ВЭЖХ С8 Luna (Phеnomenex). Растворители (Н2О, ацетонитрил) подкисляли 0,1% гептафтормасляной кислотой. Элюирование выполняли в градиенте 30-70% ацетонитрила в течение 15 минут при скорости потока 15/20 мл/мин, используя систему для препаративной ВЭЖХ Gilson. Фракции, содержащие чистый линейный пептид (идентифицированный MALDI), объединяли и модифицировали трисбромметилбензолом (ТВМВ, Sigma). Для этого линейный пептид разбавляли Н2О до ~35 мл, добавляли ~500 мкл 100 мМ ТВМВ в ацетонитриле и инициировали реакцию 5 мл 1 М раствора NH4HCO3 в Н2О (рН 8). Реакцию выполняли в течение ~30-60 минут при комнатной температуре и после лиофилизации продукта реакцию прекращали (по результатам MALDI). После лиофилизации модифицированный пептид очищали вышеописанным методом, при этом колонку Luna C8 заменяли колонкой Gemini C18 (Phenomenex) и кислоту заменяли 0,1% трифторуксусной кислотой. Чистые фракции, содержащие требуемый ТМВ-модифицированный материал, собирали, лиофилизировали и хранили при -20°С.
Неприродные аминокислоты были приобретены в компаниях, указанных в таблице 7.
Объемные или затрудненные аминокислоты (NMe-Ser, NMe-Trp, NorHar, 4PhenylPro, Agb, Agp, NMe-Arg, Pen, Tic, Aib, Hyp, NMe-Ala, NMe-Cys, 4,4-BPAI, 3,3-DPA, Dpg, 1NAI, 2NAI, Aze, 4BenzylPro, Ind) обычно связывали в течение 1 часа (удаление защитных групп в течение 20 минут) и в течение 6 часов связывали следующий остаток (удаление защитных групп в течение 20 минут). Как и раньше, HCTU использовали в качестве связующего реагента. Синтез обычно выполняли в масштабе 50 мкмолей.
Синтез пептидов, восстановленных амидных связей и α-N-метилированных пептидов
Пептоиды (то есть N-His7) были синтезированы в соответствии со схемой, разработанной Цукерманом и др. (Ronald N. Zuckermann, Janice M. Kerr, Stephen B.H. Kent, Walter H. Moos, Efficient methods for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis, Journal of the American Chemical Society (1992), 114(26), 10646-10647.
Восстановленные амидные связи псевдопептидов были синтезированы в соответствии со схемой, опиcанной в публикации Sasaki Y, Coy DH., Solid phase synthesis of peptides containing the CH2NH peptide bond isostere, Peptides, 1987 Jan-Feb; 8(1):119-21.
N-метилированные пептиды (NMe-His, NMe-Gln) были синтезированы в результате выполнения модифицированной реакции Мицунобу в соответстви со схемой, описанной в публикации Nature Protocols, “Synthesis of N-methylated cyclic peptides”, by Chatterjee et al., 7(3) 2012, pp432. Все другие N-метилированные аминокислоты (NMe-Leu, NMe-Asp) были получены в виде предшественников Fmoc из коммерческих источников.
Анализы ферментов
Анализы функциональных ферментов были выполнены в 10 мМ трис-буфера с HCl, 150 мМ NaCl, 10 мМ MgCl2, 1 мМ CaCl2 и 1 мг/мл BSA (все вещества приобретены в компании Sigma, UK), рН 7,4, при 25°С на твердых черных 96-луночных планшетах. 26,5 пМ калликреина человеческой плазмы (приобретен в компании Stratech, UK) или 500 пМ калликреина крысиной плазмы (экспрессирован и очищен в собственной лаборатории) инкубировали в присутствии или отсутствии возрастающих концентраций исследуемого пептида в течение 15 минут, после чего добавляли флуорогенный субстрат Z-PheArg-AMC (Enzo Lifesciences, UK) до конечной концентрации, равной 100 мкМ в 4% ДМСО. Высвобождение АМС измеряли с помощью устройства Pherastar FS (BMG Labtech) при длине волны возбуждения 360 нм и длине волны излучения 460 нм. Интенсивность линейной фазы реакции, обычно длящейся от 5 до 45 минут, вычисляли при помощи программы анализа данных MARS (BMG Labtech). Значение интенсивности затем использовали для вычисления IC50 и Ki в программе Prism (GraphPad). Значение IC50 вычисляли при помощи нелинейного уравнения регрессии ингибирования с четырьмя параметрами. Значение Ki вычисляли при помощи уравнения согласования Ki для одного сайта, ограничивающего Ki значением Km для субстрата, равного 150 мкМ. Все значения Ki/IC50 являются средними значениями, полученными в результате выполнения по меньшей мере двух независимых экспериментов и по меньшей мере трех экспериментов для пептидов со значениями Ki менее 1 нМ.
Пептиды растворяли в виде солей трифторуксусной кислоты в порошкообразной форме и исходные растворы обычно получали в воде. Все растворы центрифугировали и фильтровали (20 мкм фильтр-насадки для шприцев) до измерения оптической плотности при 280 нм. Коэффициенты экстинкции вычисляли на основании содержания Trp/Tyr в пептиде и содержания ТМВ (центральная часть ТМВ при наличии в пептиде имеет значение ε, равное ~300 М-1 см-1). Для пептидов, содержащих неприродные аминокислоты с предполагаемыми хромофорными свойствами (то есть NorHar, 4PhenylPro, 3Pal, 4Pal, Tic, 4GuanPhe, 4,4-BPAI, 3,3-DPA, 1NAI, 2NAI, 4BenzylPro, Ind), концентрации определяли путем взвешивания порошка и растворения пептида в определенном количестве воды. Указанные растворы были получены независимо дважды для пептидов со значением Ki в отношении калликреина, равном 1 нМ или меньше.
Анализ профиля устойчивости в плазме
Устойчивость бициклов (пептиды, конъюгированные с молекулярными остовами) в плазме можно оценить тремя методами.
Метод №1
Был разработан экспресс-анализ профиля устойчивости в плазме на основе масс-спектрометрического обнаружения (MALDI-TOF, Voyager DE, Applied Biosystems) исходной массы до времени полного исчезновения массы исходного пептида. В частности, 200 мкМ пептида инкубировали при 37°С в присутствии 35% крысиной или человеческой плазмы (Sera labs, при использовании цитрата в качестве антикоагулянта), содержащей однократный физиологический раствор с фосфатным буфером (PBS) (полученный из 10-кратного исходного PBS, Sigma). 2 мкл образца добавляли к 18 мкл 30 мМ бикарбоната аммония в смеси ацетонитрила:Н2О с соотношением 1:1 в разные периоды времени (то есть t = 0, 3, 24 часа, далее ежедневно в течение до 10 дней). Образцы замораживали при -80°С до выполнения анализа. Для выполнения масс-спектрометрического анализа, позволяющего определить примерное окно обнаружения пептида, образец, разведенный в смеси ацетонитрила:Н2О, в определенный период времени наносили непосредственно (0,7 мкл) на планшет MALDI. Матрицу (альфа-цианокоричная кислота, Sigma, полученная в виде насыщенного раствора в смеси ацетонитрила:воды с соотношением 1:1, содержащего 0,1% трифторуксусной кислоты) наносили поверх образца (1 мкл). При аналогичной установке интенсивности лазера в устройстве MALDI TOF можно определить время до полного исчезновения исходного пептида. Следует отметить, что указанный качественный анализ позволяет обнаружить относительные изменения устойчивости в плазме.
Метод №2
Для более быстрого получения данных устойчивости пептиды также оценивали в 95% плазме. В данном случае PBS не использовали и 1 мМ исходного раствора пептида (в ДМСО) растворяли непосредственно в плазме (то есть 2,5 мкл исходного раствора вводили в 47,5 мкл плазмы) с получением конечной концентрации, равной 50 мкМ. 5 мкл образцы отбирали в определенные периоды времени и замораживали при -80°С. При выполнении анализа образцы размораживали, смешивали с 15 мкл смеси ацетонитрила:метанола с соотношением 1:1 и центрифугировали со скоростью 13000 оборотов/минуту в течение 5 минут. Отсасывали 5 мкл содержащего пептид супернатанта и смешивали с 30 мМ бикарбоната аммония в смеси ацетонитрила:Н2О с соотношением 1:1. 1 мкл указанной смеси наносили на планшет MALDI и анализировали вышеуказанным методом. Следует отметить, что указанный качественный анализ позволяет обнаружить относительные изменения устойчивости в плазме.
Метод №3
Для количественного определения устойчивости в плазме исходные растворы пептидов (1 мМ в ДМСО) отправляли в компанию Biofocus, UK, где был выполнен анализ. Пептиды разводили водой до 100 мкМ и затем разводили в плазме в отношении 1:20 (5 мкМ конечная концентрация в 95% плазме), брали пробы, осаждали вышеописанным методом и производили количественное определение в устройстве Waters Xevo TQ-MS.
Пример 1. Идентификация предпочтительных остатков, обладающих связывающей активностью
На основании примеров пептидов 5×5, приведенных в таблице 4, можно идентифицировать аминокислоты в пептидах, обладающие связывающей активностью. Для определения остатков, обладающих предпочтительной связывающей активностью, дальнейшему исследованию подвергали репрезентативные остатки из двух идентифицированных семейств пептидов. Такими пептидами были пептид 06-34, включающий фрагмент CXWPARC в первой петле бицикла, и пептид 06-56, включающий фрагмент CGGxxNCR в обеих петлях бицикла. Для каждой пептидной последовательности был создан набор из 10 фаговых библиотек, в которых 9 остатков петли были постоянными и один остаток был произвольным с тем, чтобы любая аминокислота могла быть экспрессирована в библиотеке в данном положении. (См. главу ‘Рандомизация отдельных положений - создание библиотек’ в приведенном выше разделе “Методы”). Для каждой библиотеки исследовали набор из 20 произвольно отобранных фаговых клонов в отношении связывания с человеческим калликреином при выполнении анализа связывания фага с целью идентификации важных остатков для связывания с мишенью. (См. главу ‘Рандомизация отдельных положений - Анализ связывания отдельных клонов’ в приведенном выше разделе “Методы”). Данные, полученные при выполнении указанного эксперимента, представлены на фигурах 4-6.
В случае пептида 06-34 (фигура 4) очевидно, что остаток Arg1 бицикла может быть заменен разными аминокислотами, при этом связывание с калликреином человеческой плазмы сохраняется или усиливается. В отличие от этого замена остатков 2, 3, 4, 5 (Trp2, Pro3, Ala4, Arg5) большинством аминокислот значительно ослабляет сигнал, наблюдаемый при выполнении анализа, который был задан со строгой отсечкой веществ с высоким сродством связывания. Остаток Val6 может быть заменен многими различными аминокислотами, при этом связывающая активность сохраняется или усиливается. Замена других остатков во второй петле показала, что только остаток Leu10 может быть заменен целым рядом разных аминокислот при сохранении активности. Положения 7, 8 и 9 характеризуются ограниченной возможностью замены, при этом не было обнаружено замен, усиливающих связывание.
В случае пептида 06-56 (фигуры 5 и 6) очевидно, что глицины в положении 1 и 2 являются наиболее предпочтительными остатками для связывания с калликреином плазмы так же, как аргинин, триптофан и треонин в положениях 6, 8 и 9. Глутамин в положении 4 и треонин в положении 10 могут быть заменены разными остатками при сохранении хорошей связывающей активности. Три остальных остатка - пролин в положении 1, аспарагин в положении 5 и треонин в положении 7 - характеризуются ограниченной возможностью замены.
Анализ аминокислотных замен
Из предшествующего анализа следует, что в случае пептида 06-34 остатки в положении один и положении шесть могут быть заменены разными аминокислотами при сохранении связывающей активности, которая равна или превышает аналогичный показатель исходного пептида. Для определения вероятности сохранения полученных результатов при использовании выделенных синтетических пептидов был создан набор пептидов в соответствии с результатами, показанными на фигуре 4, где остаток Аrg1 был заменен серином и остаток Val6 был заменен треонином, метионином или лейцином. Были также синтезированы пептиды, содержащие разные комбинации указанных замен. Указанные замены вызвали более сильный сигнал связывания при выполнении данного анализа (таблица 5).
Все варианты синтетических пептидов обладали эквивалентной или повышенной активностью в отношении калликреина человеческой плазмы при выполнении анализов ингибирования ферментов по сравнению с исходным пептидом 06-34, из чего следует, что анализ данного типа может быть использован для определения сродства связывания с мишенью и предоставляет метод идентификации пептидов-кандидатов, обладающих очень высокой активностью.
Указанные пептиды также были исследованы в отношении связывания с калликреином крысиной плазмы при выполнении анализов выделенных ферментов. Замена Arg1 остатком Ser1 оказывала некоторое влияние на активность в отношении крысиного калликреина, в то время как при замене остатка Val6 треонином, метионином или лейцином были получены пептиды, обладающие значительно более высокой активностью в отношении калликреина крысиной плазмы. Активность в отношении человеческого калликреина была полностью сохранена. Таким образом, путем определения положений, пригодных для замен, могут быть идентифицированы пептиды, обладающие требуемыми свойствами, такими как перекрестная реактивность с ортологом мишени.
Для демонстрации возможности замены остатков в двух указанных положениях неприродными аминокислотами с целью создания функциональных характеристик или свойств, отсутствующих в исходном пептиде, остатки Arg1 и Val6 в пептиде 06-34-03 заменяли аланином, N-метилглицином (саркозином) или N-метилсерином в положении 1 и исследовали в отношении связывания. Как показано в таблице 6, положения 1/6 пригодны для полного удаления боковой цепи, так как пептиды R1A/V6A (Ala1,6 пептида 06-34-03) полностью сохраняли активность по сравнению с исходным пептидом. Замена остатков в положениях 1,6 N-метилглицином (NMeGly1,6 пептида 06-34-03) вызывала уменьшение активности, однако, сродство связывания сохранялось в низком наномолярном диапазоне. Введение N-метилсерина в положение 1 вызывало десятикратную утрату активности, но связывание сохранялось в пикомолярном диапазоне. Таким образом, в бицикле могут быть идентифицированы определенные положения, в которых могут быть произведены замены в структуре пептидного остова или боковых цепей, повышающие устойчивость к воздействию протеазы, увеличивающие растворимость, уменьшающие агрегацию и позволяющие ввести ортологичные функциональные группы.
Пример 2. Детальный анализ домена WPAR
Фрагмент WPAR, идентифицированный в примере 1, анализировали в пептиде 06-34-03 с целью выявления альтернатив или улучшений фрагмента WPAR. Была создана библиотека, в которой положения 1, 6, 7, 8, 9 и 10 были фиксированными и положения 2, 3, 4 и 5 были рандомизированными (см. главу ‘Рандомизация пептидных доменов - создание библиотек’ в приведенном выше разделе “Методы”). Выбор пептидов, связывающихся с калликреином человеческой плазмы, производили при разной степени строгости (см. главу ‘Отбор фагов’ в приведенном выше разделе “Методы”). Все пследовательности были идентифицированы и исследованы в отношении связывания с мишенью (см. главу ‘Рандомизация пептидных доменов - Анализ связывания отдельных клонов’ в приведенном выше разделе “Методы”). В таблице 17 приведены все уникальные последовательности, их относительная встречаемость по результатам отбора (частота) и ранг в зависимости от силы связывания с мишенью.
В таблице 17 показано, что фрагмент WPAR сообщает лучшее связывание с калликреином человеческой плазмы, хотя в результате отбора было обнаружено большое число других связывающихся с калликреином последовательностей. Указанные последовательности включают, не ограничиваясь ими, WPSR, WPAR, WSAR, WPFR, WPYR, FPFR и FPYR. Наиболее эффективные и широко распространенные фрагменты в положениях 2, 3, 4 и 5 могут быть суммированы как W/F Р × K/R.
В таблице 18(А) показано, что фрагменты WPAR и WPSR наиболее часто встречаются при более строгих условиях отбора; фрагменты FPFR и FPYR часто встречаются при менее строгих условиях отбора. Из вышеизложенного следует, что WPAR-подобные последовательности являются более сильными связывающими последовательностями по сравнению с FPFR-подобными последовательностями. Анализ каждого фрагмента (в пептиде 06-34-03) при выполнении анализа связывания с мишенью (таблица 18(В)) показывает, что фрагмент WPAR в положениях 2, 3, 4 и 5 последовательности 06-34-03 является оптимальной последовательностью для связывания с калликреином.
Пример 3. Оптимизация последовательности вне фармакофора WPAR
Фрагмент WPAR и его варианты были исследованы в пептиде 06-34-03. На фигуре 1 показано, что некоторые положения вне фрагмента WPAR могут сохранять связывание с калликреином при замене другими остатками. Для исследования детерминант связывания с калликреином, отличных от фрагмента WPAR, была создана фаговая библиотека с фиксированной последовательностью WPAR и остальными рандомизированными положениями (СхWPARСхххххС), как описано в главе ‘Рандомизация пептидных доменов - создание библиотек’ приведенного выше раздела “Методы”.
80 произвольных членов библиотеки были выделены непосредственно из пула библиотеки (без отбора) и исследованы в отношении связывания с калликреином в условиях повышенной и пониженной строгости (см. главу ‘Рандомизация пептидных доменов - Анализ связывания отдельных клонов’ в приведенном выше разделе “Методы”). Указанные члены библиотеки, содержащие произвольные последовательности вне фрагмента WPAR, характеризуются слабым связыванием или полным отсутствием связывания с калликреином человеческой плазмы (данные не показаны), из чего следует, что присутствие только одного фрагмента WPAR недостаточно для сохранения измеряемого связывания с калликреином: остальные остатки последовательности бицикла должны также способствовать или влиять на указанное взаимодействие.
Выбор остатков против калликреина человеческой плазмы производили с помощью указанной библиотеки для исследования детерминант связывания с калликреином, отличных от фрагмента WPAR, и для выделения оптимальной WPAR-содержащей пептидной последовательности. Было выделено более 150 последовательностей, которые исследовали в отношении связывания с калликреином человеческой плазмы (в соответствии с описанием, приведенным выше в разделе ”Методы”). Последовательности распределяли по рангам в порядке связывания с калликреином, и в таблице 19 представлено сравнение 50 лучших последовательностей. В таблице 19 показано, что остаток в положении 1 не влияет на связывание с калликреином, но сильное согласие в отношении гистидина наблюдается в положении 7 (что подтверждает результаты, полученные в приведенном выше примере 1). Пептид 06-34-03, полученный в примере 1, включает одну из лучших последовательностей. Композиция второй петли четко соответствует выводу о том, что сильное связывание с калликреином имеет место при наличии фрагмента WPAR.
Лучшие WPAR-содержащие последовательности, связывающиеся с калликреином человеческой плазмы, характеризуются следующей тенденцией:
C X W P A R C T/L H Q/T D L C
при этом остатки H7, D9 и L10 являются сильно консервативными в WPAR-содержащих последовательностях, связывающихся с калликреином.
Во второй петле бицикла были идентифицированы два фрагмента (положения 6-10):
1. C X W P A R C T H Q/T D L C (положения 6, 7 и 10: “THxxL”)
2. C X W P A R C T/L H Q/T D L C (положения 7, 8 и 10 “xHxDL”)
Более 120 идентифицированных последовательностей, связывающихся с калликреином человеческой плазмы (отбор полученных последовательностей), были сгруппированы 2 разными способами в соответствии с их выделением из фрагментов “THxxL” или “xHxDL”. Средний сигнал связывания с калликреином для полученных последовательностей был принят в качестве меры связывания с калликреином для данной группы (таблица 20).
Данные анализа связывания калликреина, приведенные в таблице 20, показывают, что фрагменты “THxxL” и “xHxDL” обеспечивают лучшее связывание калликреина, присутствуя в бициклическом пептиде вместе с фрагментом WPAR. Комбинация из 2 фрагментов “THxxL” обеспечивает наибольшее связывание с калликреином человеческой плазмы и включает последовательность второй петли “THQDL” пептида 06-34-03.
Пример 4. Системный анализ устойчивости в плазме
Для ингибирующего калликреин бицикла важно иметь соответствующий профиль устойчивости к протеазе, характеризующийся низким расщеплением, вызываемым протеазой, в плазме или другой подобной среде. При выполнении сравнительного экспресс-анализа устойчивости в плазме (раздел “Методы”, метод №1), в котором наблюдалось постепенное исчезновение исходного пептида в крысиной плазме, было обнаружено, что N-концевой аланин (присутствующий во время отбора и первоначально включенный в синтетические пептиды лидерных последовательностей) быстро удаляется во всех последовательностях бицикла, исследованных в крысиной и человеческой плазме. Расщепление не происходило при синтезе кандидата с отсутствием N- и С-концевого аланина. Для удаления возможных сайтов узнавания амино- и карбоксипептидазами свободный аминоконец, который теперь находится в положении остатка Cys1 кандидата, кэппируют уксусным ангидридом во время синтеза пептидов, в результате чего образуется молекула с ацетилированным N-концом. Аналогичным образом С-концевой цистеин синтезируют в виде амида для удаления возможного сайта узнавания карбоксипептидазами. Таким образом, бициклические кандидаты имеют следующую общую последовательность Ac-C1AA1AA2AAnC2AAn+1AAn+2AAn+3C3(TMB)-NH2, где “Ac” означает ацетилирование N-конца, “-NH2” означает амидирование С-конца, “С1, С2, С3” означают первый, второй и третий остаток цистеина в последовательности, “АА1” - “ААn” означает положение аминокислоты (характер “АА” определяется вышеописанным выбором) и “(ТМВ)” означает, что пептидная последовательность была циклизирована с ТВМВ или любым другим приемлемым реакционноспособным остовом.
Благодаря высокому сродству Ас-06-34-18(ТМВ)-NH2 как с человеческим калликреином (Ki = 0,17 нМ), так и с крысиным калликреином (IC50 = 1,7 нМ) указанный бицикл был выбран для дальнейшей разработки (фигура 9, таблица 1, таблица 5). В результате выполнения такого же экспресс-анализа профиля устойчивости в плазме было установлено, что пептид Ас-06-34-18(ТМВ)-NH2 оставался в плазме в течение примерно 2 дней (раздел “Методы”, метод №1), что соответствует времени полужизни в крысиной плазме, равному ~2 часам (при количественном определении с помощью ЖХ/МС, см. ниже, таблица 23, метод №3).
Для идентификации сайта протеолитического узнавания в пептиде Ас-06-34-18(ТМВ)-NH2 брали образцы пептида в 35% крысиной плазме в течение определенного периода времени (метод №1) и каждый образец анализировали в отношении постепенного появления фрагментов пептида при помощи масс-спектрометрии MALDI-TOF. Исходная масса Ас-06-34-18(ТМВ)-NН2 равна 1687 Да. С течением времени (фигура 7, 8) появляются фрагменты с массой 1548,6 (М1), 1194,5 (М2) и 1107,2 (М3). На основании последовательности Ас-06-34-18(ТМВ)-NН2 (Аc-C1S1W2P3A4R5C2L6H7Q8D9L10C3-NH2) можно вычислить, что пик М1 соответствует Ас-06-34-18(ТМВ)-NН2 с отсутствием Arg5 (-R5). Удаление указанного остатка, по-видимому, является начальным протеолитическим событием, за которым следует удаление фрагмента WPAR 4-аминокислоты в Ас-06-34-18(ТМВ)-NН2 (М2, -WPAR) и наконец удаление всей первой петли Ас-06-34-18(ТМВ)-NН2 (М3, -SWPAR) (фигура 8). Из полученных данных видно, что остаток Arg5 пептида Ас-06-34-18(ТМВ)-NН2 является главным сайтом узнавания для протеаз в крысиной плазме, который ответственен за расщепление данного бицикла.
Замены аланина и перестановка в первой петле
После идентификации остатка Arg5 в качестве сайта узнавания протеазами в крысиной плазме была проведена работа по химическому синтезу производных Ас-06-34-18(ТМВ)-NН2 с целью выявления кандидатов с более высокой протеолитической устойчивостью в плазме. Такие модификации не должны влиять на активность в отношении человеческого или крысиного калликреина. Первоначальное исследование роли последовательности/фармакофора WPAR (фигура 9, 10) было произведено путем замены группы W2P3 группой A2A3 или A2Q3 и перестановки частей или всей первой петли бицикла. В приведенной ниже таблице 8 показаны последовательности и соответствующие значения сродства к калликреину.
Из полученных данных видно, что сопутствующее удаление W2P3 значительно уменьшает связывание с калликреином примерно в 100000 раз, эффективно делая молекулу фармакологически инертной. Важность правильной последовательности аминокислот подтверждено четырьмя переставленными пептидами (перестановка 1-4), так как все указанные пептиды вызывают значительное уменьшение сродства к калликреину (фигура 10). Весьма удивительно, что все пептиды имеют в основном идентичный профиль устойчивости в крысиной плазме (от 1 до 2 дней, метод №1), из чего следует, что узнавание протеазами в плазме связано с присутствием аргинина (вызывающего профиль расщепления, аналогичный показанному на фигуре 7), а не с его положением в последовательности.
Затем были созданы пять производных пептидов Ас-06-34-18 (ТМВ)-NН2, в которых остатки W2, P3, A4, R5 и C2 были заменены соответствующими D-энантиомерными аналогами (таблица 9).
Из полученных данных видно, что замена остатков A4, R5 и C2 D-аминокислотами повышает устойчивость пептидов к протеазам плазмы. Так как удаление Arg5 протеазами крысиной плазмы, по-видимому, является первым событием в расщеплении пептида, то первоначальный гидролиз пептидных связей должен происходить со стороны N- и/или С-конца остатка Arg5. Вполне вероятно, что замена аминокислот с обеих сторон остатка Arg5 D-энантиомерами блокирует гидролиз смежных пептидных связей благодаря стерическому препятствию. Действительно, именно такой эффект был обнаружен ранее (Tugyi et al. (2005) PNAS, 102(2), 413-418).
Вредный эффект замены D-аминокислотами на сродство к калликреину являются значительным во всех случаях; активность снижается в пределах от 300 раз (D-Arg5) до 45000 раз (D-Trp2). Обнаруженный эффект подчеркивает важность правильного трехмерного представления указанных боковых цепей карману cвязывания бицикла с калликреином. В равной степени сильным является эффект замены D-Ala4: в данном случае замена ориентации одной метильной группы (боковая цепь Ala) уменьшает сродство в 7000 раз.
N-метилирование
Остатки в первой петле были системно заменены их N-метильными аналогами. N-метилирование служит в качестве прямой защиты пептидной связи; однако, вследствие отсутствия атома водорода амида, увеличения стерического объема (метильная группа) и изменения предпочтительных торсионных углов, можно ожидать утраты активности.
Полученные данные суммированы в таблице 10.
N-метилирование аминокислот в петле 1 вызывает менее значительное вредное влияние на активность. В частности, N-метилирование остатка Arg5 позволяет получить связывающую последовательность в одноразрядном наномолярном диапазоне (20-кратное уменьшение сродства по сравнению с пептидом дикого типа), при этом ее устойчивость в крысиной плазме превышает время анализа (не было обнаружено фрагментации пептида при выполнении масс-спектрометрии), что делает указанный пептид привлекательным улучшенным кандидатом. Что касается замен D-аминокислотами, то N-метилирование остатков, смежных с Arg5, повышает устойчивость пептида, главным образом благодаря наличию стерического препятствия, влияющего на катализируемый протеазой гидролиз пептидных связей в положении N- и/или С-конца остатка Arg5. Остаток Ser1 может быть N-метилирован без значительной утраты активности, из чего следует, что целостность пептидного остова в данном положении не является существенным для связывания.
Замены аргинина
С учетом важности остатка Arg5 для узнавания протеазами крысиной плазмы был исследован набор аналогов аргинина в последовательности Ас-06-34-18(ТМВ)-NН2. Химические структуры показаны на фигуре 11, и данные зависимости активности от устойчивости приведены в таблице 11.
Все аналоги аргинина повышают устойчивость пептида на период, превышающий время анализа, что подтверждает важность целостности остатка Arg5 в узнавании протеазой плазмы. Увеличение (HomoArg) или уменьшение длины боковой цепи (Agb, Agp) снижает сродство, однако, аналог HomoArg все же позволяет получить очень хорошую связывающую последовательность (Ki = 2,1 нМ) с повышенной устойчивостью. Удлинение аминокислотного остова на одну метиленовую группу в Arg5 (так называемая бета-аминокислота) при сохранении прежней боковой цепи (β-homoArg5) также позволяет получить связывающую последовательность с повышенной устойчивостью, однако, за счет более значительного снижения сродства (Ki = 8,2 нМ). Замена алифатической части боковой цепи Arg фенильным кольцом позволяет получить более объемную и жесткую гуанидилсодержащую боковую цепь со стабилизированным резонансом (4GuanPhe). Из всех исследованных аналогов Arg боковая цепь 4GuanPhe обладала наибольшим сродством (2-кратное уменьшение по сравнению с пептидом дикого типа) при повышенной устойчивости в плазме. Интересно отметить, что гуанидилфенильная группа в структурном отношении близка к известной мелкой молекуле ингибитора калликреина бензамидину (Sturzebecher et al. (1994), Novel plasma Kallikrein inhibitors of the benzamidine type. Braz. J. Med. Biol Res. 27(8):1929-32; Tang et al. (2005), Expression, crystallization and three-dimensional structure of the catalyc domain of human plasma Kallikrein. J. Biol. Chem. 280: 41077-89. Кроме того, производные фенилгуанидины были использованы в качестве избирательно действующих ингибиторов другой серинпротеазы, uPA (Sperl et al., (4-aminomethyl)phenylguanidine derivatives as nonpeptidic highly selective inhibitors of human urokinase (2000) Proc. Natl. Acad. Sci. USA, 97(10):5113-8). Таким образом, последовательность Ас-06-34-18(ТМВ)-NН2, содержащую 4GuanPhe, можно рассматривать в качестве низкомолекулярного ингибитора, избирательность которого определяется окружающими остатками бициклического пептида. Данная последовательность может определять принцип создания других ингибиторов на основе бицикла, в соответствии с которым известный низкомолекулярный ингибитор с низкой избирательностью ”трасплантируют” в бицикл в правильном положеним с образованием молекулы, обладающей высокой активностью и избирательностью.
Модификация гуанидильной группы Arg путем метилирования (SDMA, NDMA), удаления положительного заряда (Cit, где гунидильную группу заменяют изостерической, но незаряженной группой мочевины) или делеции Arg (∆Arg) оказывает вредное воздействие на активность связывания калликреина. Таким образом, целостность и присутствие гуанидильной группы имеет важное значение, в то время как характер боковой цепи, связывающейся с гуанидильной группой или остовом в положении остатка Arg5, такого значения не имеет. Остаток Arg5 может быть также заменен лизином, однако, также при уменьшении сродства (см. пептид WPAК).
Суммируя вышеизложенное, можно отметить, что полученные данные указывают на то, что использование пептида Ас-06-34-18(ТМВ)-NН2 с заменой aргинина остатками HomoArg, NMEArg или 4GuanPhe позволяет получить кандидатов с высоким сродством при повышенной устойчивости в плазме.
Пример 5. Улучшение активности кандидата путем модификаций неприродными аминокислотами и объединения с модификациями, повышающими устойчивость в плазме
Активность данного бициклического кандидата может быть значительно улучшена при помощи нескольких механизмов. Такие механизмы были частично рассмотрены в примере 4 и могут быть суммированы следующим образом:
1. Введение гидрофобных частей, которые вызывают гидрофобный эффект и снижают скорость реакции, благодаря чему достигается более высокое сродство.
2. Введение заряженных групп, которые вызыввют длительные межионных взаимодействия, увеличивающие скорость реакции и повышающие сродство (см., например, публикацию Schreiber et al., Rapid, electrostatically assisted association of proteins (1996), Nature Struct. Biol. 3, 427-31).
3. Введение дополнительного ограничения в пептид, то есть
- правильное ограничение боковых цепей аминокислот, благодаря чему утрата энтропии является минимальной при связывании с мишенью;
- ограничение торсионных углов остова, благодаря чему утрата энтропии является минимальной по связывании с мишенью;
- введение дополнительных циклизаций в молекулу для достижения вышеуказанных целей.
(для ознакомления с данным вопросом см. публикации Gentilucci et al., Curr. Pharmaceutical Design (2010), 16, 3185-203, и Nestor et al., Curr. Medicinal Chem. (2009), 16, 4399-418).
Замены триптофана гидрофобными аналогами
Целый ряд гидрофобных аминокислот вводили в сайт Trp2 для идентификации кандидатов, способных заменить чувствительный к окислению триптофан, и кандидатов, способных повысить активность (относится к приведенному выше первому пункту). Боковые цепи указанных аминокислот показаны на фигуре 12, и данные сродства суммированы в приведенной ниже таблице 12.
Как и ожидалось, ни одна из модификаций не повысила устойчивость в плазме. 2-Нафтилаланин является наиболее родственным остатку Trp2 и характеризуется активностью, которая немного слабее активности пептида дикого типа, что делает такую замену хорошей, устойчивой к окислению заменой остатка Trp2. Интересно отметить, что 3,3-DPA2 имеет структуру, которая существенно отличается от структуры Trp, и все же соответствующий пептид сохраняет высокую активность. Из вышеизложенного следует, что Trp-контактирующий карман в калликреине может быть использован для повышения сродства связывания путем идентификации правильно созданной гидрофобной части.
Аналоги пролина
Затем была определена роль Pro2 в фармакофоре WPAR последовательности Ас-06-34-18(ТМВ)-NН2. 4-гидрокси- или 4-фтор-транс-(L)-пролин (НуР3, 4FluoPro3) был выбран благодаря известному свойству, сообщающему дополнительную жесткость и спиральность пептидному остову (фигура 13, таблица 13). Кроме того, присутствие гидроксила в НуР позволяет исследовать доступность растворителя к боковой цепи пролина. Значения Ki соответствующих производных были почти идентичны аналогичным показателям пептида дикого типа, из чего следует, что любые воздействия на пептидный остов являются пренебрежимо малыми и что боковая цепь является доступной. В процессе дальнейшей разработки данного метода были исследованы два дополнительных производных Ас-06-34-18(ТМВ)-NН2, имевших значительный удлиняющий сегмент в положении γ-атома углерода боковой цепи Pro3 (4фенил-Pro, 4Benzyl-Pro). В первом случае имеет место значительное сохранение активности, в то время как в последнем случае активность подвергалась сильному воздействию, из чего следует, что боковая цепь Pro является доступной и ограничена только особыми модификациями. Несмотря на стерический объем указанных модификаций устойчивость в плазме была идентична аналогичному показателю пептида дикого типа. Таким образом, указанные модификации не улучшают избирательность в отношении других протеаз.
Для исследования влияния размера кольца пролина на связывание остаток Pro заменяли сильно ограниченным 4-членным аналогом Pro азетидинкарбоновой кислотой (Aze) и более гибким 6-членным кольцом (пипеколиновая кислота, Pip). Остаток Aze3 последовательности Ас-06-34-18(ТМВ)-NН2 связывается с калликреином с наибольшим сродством из всех исследованных производных, превосходящим сродство связывания пептида дикого типа в 3 раза (фигура 14, таблица 13). По-видимому, существует обратная взаимосвязь между размером кольца и значением Ki, поэтому можно предположить, что конформационное ограничение в положении 3 Ас-06-34-18(ТМВ)-NН2 является ключом к созданию прочно связывающейся молекулы.
Условие гибкости боковой цепи пролина для включения больших групп подтверждено би/трициклическими аналогами пролина Tic, NorHar и Ind (фигура 13). В частности, в последних двух случаях сродство по-прежнему находится в одноразрядном наномолярном диапазоне.
И наконец, было исследовано требование, предъявляемое к структуре кольца в положении Pro3. В данной связи была выбрана аминоизомасляная кислота (Aib, фигура 15, таблица 13), которая благодаря двойной замене метила в положении альфа-атома углерода оказывает сильное структурное воздействие на соседние аминокислоты, индуцируя α или 310 спиральность (Toniolo et al. (1993) Biopolуmers 33, 1061-72; Karle et al. (1990), Biochemistry 29, 6747-56). Указанная неприродная нециклическая аминокислота хорошо встраивается вместо Pro3 при значении Ki, равном 1,2 нМ. Таким образом, роль Pro3 в фармакофоре WPAR состоит в введении ограничения в пептидный остов. Данное ограничение может быть усилено путем использования аналога пролина с меньшим размером кольца (см. Aze3). И наоборот, пролиновое кольцо может быть относительно эффективно заменено нециклическими, но структурирующими аминокислотами, такими как Aib.
Разнообразные аналоги
В таблице 10 показано, что остаток Ser1 в петле 1 пептида Ас-06-34-18(ТМВ)-NН2 может быть метилирован при незначительном воздействии на активность (значение Ki 0,5 по сравнению с 0,17 в пептиде дикого типа). Была поставлена цель определить, можно ли произвести большую двойную замену атома Сα в положении 1. В данной связи остаток Ser1 был заменен остатком Dpg (дипропилглицин) (фигура 15). Сродство указанного пептида к калликреину равно 1,1 нМ, из чего следует, что положение 1 является очень гибким и может принимать фактически любой объемный остаток. Таким образом, данное положение в петле 1 может быть использовано для намеренного включения требуемых функций или групп, в том числе солюбилизирующих аминокислот, радиоактивных меток, красителей, линкеров, сайтов конъюгации и т.д.
Несколько аналогов аланина было также исследовано в положении 4. Как видно в случае N-метила и D-аланинов (таблица 10, 9), остаток Ala4 очень чувствителен к стерической ориентации в положении Сα или к модификации самого остова. Еще два производных данного класса подтверждают такой вывод, так как удлинение пептидного остова в положении Ala4 (β-Ala4) сильно уменьшает сродство (~20 мкМ). Как ожидалось в случае D-Ala4, остаток Aib4 уменьшает сродство почти в такой же степени (288 нМ, фигура 15 и таблица 14). Весьма удивительно, что удлинение боковой цепи Ala одним остатком метилена (Aba4), по-видимому, усиливает сродство к калликреину.
И наконец, центральный остаток цистеина (Cys2) заменяли более объемным и более ограниченным аналогом, пеницилламином (Pen, фигура 15), в надежде увеличить протеолитическую устойчивость благодаря меньшему пространственному доступу к соседнему сайту узнавания протеазами Arg5. Действительно, устойчивость в крысиной плазме немного повысилась, однако, активность значительно снизилась, что указывает на важность целостности данного остатка, присоединяющего структурный скелет.
Объединение в одном бицикле неприродных аминокислот, повышающих устойчивость в плазме и усиливающих активность
Замены, производимые в Ас-06-34-18(ТМВ)-NН2 неприродными аминокислотами, сохраняющими приемлемую активность и максимальную устойчивость в крысиной плазме (определенную методом №1), представляли cобой варианты Arg5, а именно гомоаргинин (HomoArg5), 4-гуанидилфенилаланин (4GuanPhe5) и N-метиларгинин (NMeArg5). Замены, производимые в Ас-06-34-18(ТМВ)-NН2 неприродными аминокислотами, повышающими активность по сравнению с пептидом дикого типа, представляли собой аналог Pro3 азетидинкарбоновую кислоту (Aze3) и аналог Ala4 2-аминомасляную кислоту (Aba4). Таким образом, остатки Aze3, Aba4 объединяли с повышающими устойчивость к протеазе остатками HomoArg5, 4GuanPhe5 и NMeArg5 для определения возможности создания пептидов-кандидатов с высокой устойчивостью в плазме и повышенной активностью.
В таблицах 15 и 16 приведены значения сродства разных конструкций наряду с показателями устойчивости в плазме.
Количественное определение времени полужизни в крысиной плазме (4-й столбец, таблица 15) пептидов, содержащих аналог аргинина, показало, что N-метилирование Arg5 наиболее эффективно защищало пептид (t1/2 >20 часов), за которым следовали HomoArg5 и GuanPhe5. Сильное защитное действие N-метилирования Arg, по-видимому, не является удивительным, так как метилирование предотвращает гидролиз пептидной связи. При включении Aze3 в указанные соединения сродство пептидов может увеличиться во всех случаях, что делает Ac-(06-34-18) Aze3 Homo4Arg5 и Aс(06-34-18) Aze3 NMeArg5 привлекательными кандидатами для дальнейшего исследования (таблица 15, фигура 15).
Повышающее сродство воздействие Aba4 нельзя было воспроизвести при использовании Aze3 и любых аналогов аргинина, так как значения Ki были выше показателей, имевших место без Aba4. Таким образом, повышающее активность воздействие Aze3 не зависит от типа замены аргинина, в то время как воздействие Aba4, по-видимому, зависит от типа замены.
И наконец, активность в отношении крысиного калликреина указанных пептидов значительно снижается (таблица 15). Однако приведенные значения являются относительными, а не количественными на данной стадии, так как белковый препарат крысиного калликреина не является стандартным и содержит загрязняющие примеси.
Пример 6. Повышение устойчивости в плазме бицикла, содержащего FPYR без остатка Trp, против калликреина и увеличение сродства с помощью остатка Aze3
В результате отбора, произведенного в примерах 1-4, было обнаружено несколько последовательностей, подобных последовательности Ас-06-34-18(ТМВ)-NН2, которые были широко распространены, но содержали измененные фрагменты WPAR. Указанные фрагменты представляли собой WPSR и FPYR. Последний фрагмент является особенно интересным, так как в нем отсутствует чувствительный к окислению триптофан.
Бициклы, содержащие WPSR, FPYR, WPYR и FPAR, синтезировали и сравнивали с исходным пептидом WPAR (таблица 21).
Как и ожидалось, все пептиды характеризовались сущестенно отличающейся устойчивостью в плазме. Замена остатка Trp2 остатком Phe2 вызывает 40-кратное уменьшение значения Ki, что указывает на необходимость более объемной боковой цепи Trp2. Однако указанное уменьшение значения Ki может быть компенсировано заменой остатка Ala4 остатком Tyr4 (с образованием фрагмента FPYR), в результате чего сродство снова возрастает почти до аналогичного показателя последовательности WPAR дикого типа (Ki = 0,46 нМ). Таким образом, существует взаимодействие между остатками в положении 2 и положении 4 бицикла Ас-06-34-18(ТМВ)-NН2. Учитывая высокое сродство связывания с мишенью и отсутствие остатка Tep2 в последовательности Ас-06-34-18(ТМВ)-NН2 Phe2Tyr4, данный кандидат исследовали в отношении увеличения времени полужизни в крысиной плазме методом, описанным в приведенном выше примере. Кроме того, было исследовано взаимодействие между остатками Phe2 и Tyr4 путем замены указанных остатков неприродными аминокислотными аналогами.
Замены остатков Phe2/Tyr4 неприродными аминокислотами в последовательности Ас-06-34-18(ТМВ)-NН2 Phe2Tyr4
Была выполнена далеко не исчерпывающая совокупность процессов синтеза, включающая замены в положении остатков Phe2 или Tyr4 последовательности Ас-06-34-18(ТМВ)-NН2 Phe2Tyr4. Неприродные аминокислоты были выбраны из набора, показанного на фигуре 12, и данные сродства суммированы в таблице 22.
Замена любыми исследованными аминокислотами обычно хорошо переносится, несмотря на то что боковая цепь является гетероароматической (3Pal, 4Pal), ароматической и объемной (1Nal, 2Nal, 4,4-BPal) или циклоалифатической (Cha) частью. Остаток 3Pal хорошо переносится в положении 2 (Ki = 0,91), что является весьма интересным, так как остаток Pal содержит ионизируемую группу (которая может быть использована при получении бицикла). Однако исходная комбинация Phe2/Tyr4, по-видимому, является наиболее активной.
Стабилизация последовательности Ас-06-34-18(ТМВ)-NН2 Phe2Tyr4 в крысиной плазме и эффект замены в положении 3 азетидинкарбоновой кислоты
Последовательность Ас-06-34-18(ТМВ)-NН2 Phe2Tyr4 была получена в результате замен гомоаргинином, 4-гуанидилфенилаланином и N-метиларгинином в присутствии или отсутствии остатка Aze3. Остатки HomoArg/4GuanPhe хорошо переносятся, при этом значения Ki почти идентичны исходному пептиду Phe2Tyr2 (таблица 23), и устойчивость в крысиной плазме увеличилась в 13 раз (t1/2 = 12,2 часа, таблица 23, фигура 16). Кроме того, значения IC50 для крысиного калликреина аналогичны указанным значениям исходного пептида, из чего следует, что данная последовательность является привлекательным кандидатом для выполнения исследований in vivo.
Замена остатка Pro3 остатком Aze3 в фрагменте FPYR позволяет получить пептиды-кандидаты с повышенным сродством, и действительно был создан пептид со значением Ki меньше 1 нМ, который, по-видимому, должен характеризоваться временем полужизни более 20 часов в крысиной плазме (Ас-(06-34-18) Phe2 Aze3Tyr4 NMeArg5).
Пример 7. Улучшение устойчивости в человеческой плазме бицикла-кандидата 06-34-18 путем единичных замен аминокислот в петле 2
Идентификация остатка His7 в качестве главного сайта узнавания протеазами плазмы
Устойчивость в крысиной и человеческой плазме нижеследующих пептидов 06-34-18 с модифицированной петлей 1 определяли количественно при помощи ЖХ-МС (метод №3, таблица 2). Очевидно, что химические модификации (HArg5, 4GuanPhe5, NMe-Arg5), сообщающие устойчивость к протеазам крысиной плазмы оказываются неэффективными с точки зрения протеолиза человеческой плазмы.
Пептид Ас-06-34-18(ТМВ)-NН2 Phe2 Tyr4 (определяемый как пептид дикого типа) подвергали воздействию человеческой плазмы и исследовали в отношении появления фрагментов с течением времени. Полученный профиль сравнивали с профилем расщепления, имевшим место в человеческой плазме при использовании производного NMe-Arg5 пептида 06-34-18 (фигуры 18а и 18b). Фрагментацию определяли методом №2.
На основании масс-спектрометрических данных было установлено, что эндопротеолитический гидролиз происходит со стороны N- и/или С-конца остатка His7 в последовательности 06-34-18 с последующим удалением остатка Leu6 (в результате экзопротеолитической активности) или остатков His7-Gln8 (в результате экзопротеолитической активности) или всех трех остатков вместе. Из характера обнаруженных фрагментов следует, что, по-видимому, первичный эндопротеолитический гидролиз происходит в пептидной связи между остатками Leu6 и His7, а не между остатками His7 и Gln8. Кроме того, такая же картина фрагментации имеет место в случае блокирования сайта узнавания протеазами плазмы в положении остатка Arg5 путем N-метилирования (фигура 18b), из чего следует, что протеолитическая специфичность человеческой плазмы отличается от протеолитической специфичности крысиной плазмы.
Системная оценка роли остатков петли 2 и стабилизации в отношении протеаз плазмы
Оценивали роль всех остатков петли 2 в отношении их влияния на устойчивость в человеческой плазме и активность против калликреина человека.
При использовании экспериментальных методов, частично описанных в разделе “Уровень техники”, было выполнено полное исследование аминокислот петли 2, для чего были произведены
1) замены всех аминокислот петли 2 аланином для удаления боковой цепи, служащей в качестве сайта узнавания для протеаз;
2) замены всех аминокислот петли 2 энантиомерными аналогами D-аминокислот с целью создания стерического препятствия для доступа расщепляющих протеаз;
3) замены некоторых аминокислот петли 2 D-аланином с целью создания стерического препятствия для доступа протеаз к пептидным связям.
Данные активности для каждого варианта суммированы соответственно в таблицах 3а, 3b и 3с.
1) Сканирование аланином остатков петли 2
В таблице 3а показано, что замена остатка His7 остатком Ala7, по-видимому, уменьшает протеолитическое расщепление во второй петле, так как фрагменты -L, -HQ, -LHQ не были обнаружены. Однако была обнаружена протеолитическая активность в положении остатка Arg5 петли 1 (которая ранее наблюдалась только в крысиной плазме). Таким образом, очевидно, что протеазы человеческой плазмы способны атаковать оба сайта протеолитического узнавания (Arg5 и His7). Даже если остаток Ala7 повышает устойчивость по сравнению с остатком His7 (как в пептиде дикого типа), активность пептида существенно снижается (в 56 раз) до значения Kd, равного 23 нМ, что является недостаточным для терапевтической молекулы. Остальные замены остатком Ala уменьшают активность в разной степени, не увеличивая устойчивость в плазме (судя по появлению фрагментов бициклического пептида с расщепленной петлей 2. Таблица 3а, четвертый столбец). Полученные данные подтверждают открытие того, что остаток His7 является сайтом узнавания для протеаз.
2) Сканирование D-аминокислотой остатков петли 2
В таблице 3b показан эффект замены всех аминокислот их D-энантиомерными аналогами. Указанная модификация оставляет интактной боковую цепь, но проецирует ее в альтернативной конфигурации на Сα. D-аминокислоты в любом положении петли 2, по-видимому, стабилизируют петлю 2 пептида от протеолитического расщепления; однако, как видно в приведенном выше варианте Ala7, специфичность переключается на лабильный сайт в петле 1 (Arg5). Переключение гидролиза на остаток Arg5 затрудняет количественное определение эффекта стабилизации D-аминокислотами в петле2, так как данный независимый процесс влияет на количество оставшегося исходного материала (рассмотренного ниже). Дистальный защитный эффект замены остатков D-аминокислотами описан в научной литературе, и, по-видимому, действует путем стерического уменьшения или предотвращения доступа к гидролизуемой связи (Tugyi et al. (2005) PNAS, 102(2), 413-418). Однако D-аминокислоты, удаленные от предполагаемого сайта узнавания протеазами остатка His7 (то есть D-Asp9, D-Leu10), также характеризуются меньшим количеством фрагментов, соответствующих удаленному остатку лейцина 6, смежному с остатком His7.
Следует отметить, что большинство D-аминокислотных вариантов характеризуются сильно сниженной активностью (уменьшение активности в 130-9000 раз), что подтверждает трехмерный и стерический характер взаимодействия пептида с белком калликреина. D-Asp9, по-видимому, является исключением, так как демонстрирует повышение устойчивости при сохранении активности, равной 4 нМ (9-кратное уменьшение по сравнению с пептидом дикого типа). Указанная модификация потенциально может быть полезной стабилизирующей заменой в терапевтической молекуле-кандидате.
3) Сканирование D-аланином остатков петли 2
В таблице 3с показан эффект замены нескольких аминокислот в петле 2 D-аланинами. Удаление боковых цепей и создание стерического препятствия в положении Сα путем введения метильной группы отрицательно влияет на активность пептида. Тем не менее в двух случаях сохраняется сродство на низком наномолярном уровне (Ас-(06-34-18) D-Ala9 и Ac-(06-34-18) Phe2 Tyr4 D-Ala9). Так же, как в случае D-Asp9, указанные модификации потенциально могут быть полезными стабилизирующими заменами в терапевтической молекуле-кандидате.
Замена боковых цепей аминокислот в положении Leu6, His7 и Gln8 аминокислотными миметиками
Была произведена замена аминокислот в положении остатков Leu6, His7 и Gln8 для определения требований, предъявляемых указанными остатками к стерическому препятствию, заряду и структуре, и для оценки их воздействия на уменьшение узнавания сайта His7 протеазами плазмы (структура петли 2 показана на фигуре 19а).
Общие методы, пригодные для указанной цели, были ранее рассмотрены в вводном разделе.
• Замены в положении 6 петли 2 пептида 06-34-18
Была исследована возможность введения стерического препятствия в положение Сβ аминокислотного остатка Leu6 с целью уменьшения доступа протеаз к расщепляемой связи между остатками Leu6 и His7. Исследованные структуры показаны на фигуре 19b. Результаты воздействия на активность суммированы в таблице 24а. Некоторые модификации характеризуются увеличением сродства по сравнению с пептидом дикого типа, особенно в случае пептида Ас-(06-34-18) (менее в случае пептида Ас-(06-34-18) Phe2 Tyr4). Указанные аминокислоты, в частности, являются неприродными аминокислотами фенилглицином (Phg) и циклогексилглицином (Chg). Все указанные модификации, по-видимому, в некоторой степени уменьшают протеолитическое расщепление во второй петле, но наиболее сильным стабилизатором является трет-бутилглицин (tBuGly). В указанном пептиде, Ас-(06-34-18) Phe2 Tyr4 tBuGly6, было обнаружено только удаление остатка Arg5 в первой петле под воздействием человеческой плазмы. Любые модификации Chg, Phg и tBuGly вместе взятые могут повышать устойчивость в плазме второй петли. Кроме того, были идентифицированы две модификации (Chg, Phg), которые повышают активность в отношении калликреина.
• Замены в положении остатка 7 петли 2 пептида 06-34-18
Было исследовано воздействие замены боковой цепи гистидина в положении остатка 7 на устойчивость в плазме и активность при использовании следующих миметиков. Пиридилаланины 2Pal, 3Pal, 4Pal являются основными ионизируемыми гетероароматическими миметиками боковой цепи имидазола. Тиенилаланин (Thi), фурилаланин (FuAla), ThiAz, 1,2,4-триазолаланин (1,2,4 Triz), тиазолилаланин (ThiAz) являются 5-членными гетероароматическими незаряженными миметиками боковой цепи имидазола. His1Me/His3Me являются N-метилзамещенными производными боковой цепи имидазола. Dap, Agb и Agp служат для замены положительного заряда в боковой цепи имидазола, однако, при отсутствии ароматической боковой цепи (фигура 19с). В таблице 24b суммированы данные активности. Весьма удивительно, что все пептиды характеризуются повышенной устойчивостью к протеазам плазмы в петле 2, при этом видимое расщепление происходило только в положении остатка Arg5 петли 1. Очевидно, что положительный заряд в положении остатка 7 является недостаточным для создания сайта узнавания для протеаз плазмы (Dap, Agp, Agb). Наоборот, удаление положительного заряда при сохранении близкой имитации исходной боковой цепи имидазола также вызывает удаление сайта протеолитического узнавания. Несмотря на очень близкую имитацию некоторых структур исходного имидазольного кольца, все производные пептидов характеризуются существенно пониженным сродством к калликреину плазмы. Из вышеизложенного следует, что интактная структура остатка His7 необходима для связывания с калликреином с полной силой. Интересными кандидатами, сохранившими связывание на низком наномолярном уровне, были тиазолилаланин, фурилаланин и His1Me. Интересными предполагаемыми кандидатами могли быть гомогистидин и имидазолилглицин.
• Замены в положении остатка 8 петли 2 пептида 06-34-18
Остаток 8 (Gln8) более удален от сайта расщепления, локализованного между остатками Leu6 и His7. Было произведено несколько замен (фигура 19d и таблица 24с). Отрицательно заряженные аминокислоты были введены в данном положении для создания солевого мостика к соседнему остатку His7, который может уменьшить узнавание остатка His7 протеазами плазмы. Была исследована дополнительная последовательность петли 2, которая присутствовала в отобранных пептидах фаговой библиотеки, где также отсутствовал гистидин 7. Указанная последовательность петли 2 представляли собой LTTEL (определяемая как Ас-(06-34-18) Phe2 Tyr4 Thr7 Thr8 Gku9). Кроме того, была исследована бета-аминокислота в положении 8 (bHGln8, фигура 19d). В данном положении была также исследована положительно заряженная аминокислота с целью потенциального уменьшения протеолитического узнавания остатка His7 и последующего расщепления. Хотя активность указанных молекул часто была <10 наномоль, воздействие на стабилизацию в плазме было минимальным. Указанные молекулы могут быть использованы в дальнейшем количественном исследовании для определения их точного воздействия на устойчивость в плазме.
Пример 8. Улучшение устойчивости бицикла-кандидата 06-34-18 в человеческой плазме в результате модификаций аминокислотного остова в петле 2
Модификация аминокислотного остова является общепризнанным способом защиты пептидного остова от гидролиза протеазами (см. публикации Gentilucci et al., Curr. Pharmaceutical Design, (2010), 16, 3185-203 и Nestor et al., Curr. Medical Chem. (2009), 16, 4399-418).
Широко применяемым методом в данной области является N-алкилирование, например, N-метилирование Nα аминокислот. В таблице 25а показан эффект системной замены двух амидных остовов петли их Nα-метилированными аналогами. В принципе указанная модификация является очень эффективной защитой от гидролиза, так как от протеолитического гидролиза защищается не только данная пептидная связь, но и соседние пептидные связи, на которые оказывает защитное воздействие вся метильная группа в положении Nα. Действительно имеет место сильное защитное воздействие на протеолиз петли 2, однако, активность неизменно снижается в 100 или более раз. В случае замены D-аминокислотой N-алкилирование защищает расщепляемый сайт в положении остатка His7, даже когда он присутствует в положении остатка Asp9 или Leu10.
Другим способом модификации пептидного остова является N-алкилирование Nα с помощью боковой цепи соответствующей аминокислоты (именуемой пептоидом). Боковую цепь гомогистидина вводили в положение Nα остатка His7 при одновременно удалении боковой цепи в положении Сα остатка His7, получая при этом N-алкилглицин. Структура данного производного (именуемого N-His7) показана на фигуре 20а, изображающей второй петли пептида 06-34-18.
С целью модификации структуры самого пептидного остова вводили восстановленную пептидную связь между остатками Leu6 и His7 (фигура 20b). Остаток Leu6 был заменен остатком Ala6 и карбонильная группа в указанном остатке была восстановлена в метилен, в результате чего была создана связь, в котором полностью отсутствовала нативная пептидная связь. Данное производное определяется как “восстановленный амид” или “пседопептидная связь” и именуется СН2NН. Из-за отсутствия карбонильной группы в указанном положении пептид не может быть гидролизован и должен характеризоваться благоприятными свойствами устойчивости к протеазам.
При выполнении анализа в плазме оба производных пептида (Ас-(06-34-18) Phe2 Tyr4 N-His и Аc-(06-34-18) Phe2 Tyr4 Ala(СН2NН)6) характеризовались значительно более высокой устойчивостью в петле 2. Образовавшиеся фрагменты соответствовали только расщеплению в положении остатка Arg5 петли 1. Данные активности суммированы в таблице 25b. Как и ожидалось, производное N-His характеризовалось менее сильным связыванием. Однако псевдоамидный пептид Ас-(06-34-18) Phe2 Tyr4 Ala(СН2NН)6) сохранил очень хорошую активность при значении Kd, равном 1,5 нМ.
Пример 9. Создание активного, устойчивого в крысиной и человеческой плазме, перекрестно реактивного бициклического пептида путем избирательной химической модификации петли 1 и петли 2
На основании результатов, полученных в двух предыдущих примерах, было идентифицировано несколько модификаций в петле 2, которые повышали устойчивость к протеазам петли 2 при сохранении хорошей активности. Сохранение соответствующей активности в отношении калликреина крысиной плазмы необходимо для создания РК/PD в животных моделях. Сравнительная активность в крысиной плазме суммирована в таблице 26а для приемлемых кандидатов, обладающих повышенной устойчивостью в петле 2. Большинство молекул обладает активностью <100 нМ в отношении калликреина крысиной плазмы.
В примерах 1-6 рассмотрены методы, в которых петля 1 пептида 06-34-18 может быть стабилизирована против воздействия протеаз плазмы. Указанные благоприятные модификации объединяют с помощью повышающих устойчивость в плазме модификаций в петле 2 (примеры 7, 8). Было получено несколько репрезентативных кандидатов, которые оценивали с точки зрения устойчивости в человеческой/крысиной плазме, а также активности в отношении калликреина человеческой/ крысиной плазмы (таблица 26b).
Следует отметить, что большинство пептидов при объединении с модификаторами петли 1 характеризуются лучшей активностью в отношении человеческого и крысиного калликреина по сравнению с пептидами, содержащими только модификаторы петли 2 (таблица 26а). Большинство значений активности находится в диапазоне, приемлемом для терапевтических целей и создания РК/РD (см. данные для человеческого и крысиного калликреина, таблица 26b).
Благодаря введению остатка HArg5 вместо остатка Arg5 в петле 1 и выполнению модификаций в петле 2, позволяющих уменьшить протеолиз на сайте остатка His7, пептиды, показанные в таблицах 26а и 26b, должны обладать повышенной устойчивостью в человеческой и крысиной плазме.
Устойчивость в плазме оценивали сравнительным методом, как было указано выше, при выполнении анализа MALDI TOF (методы №1 и №2), при этом с течением времени были обнаружены гидролизованные пептидные фрагменты (для ознакомления с данными устойчивости пептида дикого типа см. фигуры 18а, 21а, 21b). При сравнении с пептидом дикого типа мультивариантные пептиды Ас-(06-34-18) Phe2 Aze3 Tyr4 HArg5 Ala(СН2NН)6 (устойчивость в человеческой плазме: фигура 21d, устойчивость в крысиной плазме: фигура 21е) и Ас-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9 (устойчивость в человеческой плазме: фигура 21с) характеризуются повышенной устойчивостью как в человеческой, так и крысиной плазме.
Полученные данные показывают, что приемлемые модификации в петле 1 и петле 2 пептида 06-34-18 могут быть объединены с образованием высокоактивных, перекрестно реактивных и устойчивых в плазме бициклических пептидов, приемлемым для терапевтических целей.
Пример 10. Благоприятные фармакокинетические свойства двух пептидов Аc-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 и Аc-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9 in vivo
Пептид Аc-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 (содержащий повышающие сродство модификации Aze3, повышающие устойчивость к протеазам плазмы модификации HArg5 и повышающую устойчивость к протеазам плазмы восстановленную амидную связь между остатками Ala6 и His7 в петле 2) и пептид Аc-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9 (содержащий повышающую сродство модификацию Aze3 и повышающие устойчивость к протеазам плазмы модификации HArg5 и D-Asp9 в петле 2) были выбраны для фармакокинетического исследования в крысиной модели. Полная химическая структура пептида Аc-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 показана на фигуре 22.
Определяли скорости выведения обоих пептидов из кровотока крысы и полученные данные сравнивали с контрольным пептидом, состоящим из последовательности Ас-(06-34-18)(ТМВ), в которой аминокислоты были заменены соответствующими D-энантиомерами (таким образом, последовательность представляет собой (Ас-cswparclhqdlc-NH2(TMB)). Указанный контрольный пептид имеет идентичный состав атомов и последовательности, сохраняет подобные физико-химические свойства, не обладает активностью в отношении мишени, являющейся калликреином, и, что важно, является полностью устойчивым к протеолитическому воздействию. Таким образом, выведение контрольного пептида Ac-06-34-18, полностью состоящего из D-аминокислот, (All-D 06-34-18) из кровотока крысы происходит главным образом через почки, так как протеолитическое выведение отсутствует. Поэтому любой оптимизированный, стабилизированный в отношении протеазы и активный калликреин-ингибирующий бицикл, созданный на основе последовательности 06-34-18, должен выводиться из кровотока крысы со скоростью, аналогичной скорости выведения контрольного пептида All-D 06-34-18.
Пептиды в виде болюса для внутривенного введения в физиологическом растворе с фосфатным буфером (содержащем 2,9% ДМСО) в концентрации 1,02 мг/мл вводили внутривенно в дозе 5,2 мг/кг крысам Sprague Dawley, затем брали последовательные пробы крови из хвостовой вены каждого животного при помощи постоянной канюли через 5, 10, 20, 30, 60, 120 и 240 минут после введения дозы и переносили в пробирки с EDTA для образования плазмы. Данные были получены для пептида “All-D Ac-06-34-18” у 3 крыс, для пептида “Ac-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6” у двух крыс и для пептида ”Аc-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9” у двух крыс.
Образцы плазмы затем обрабатывали 3 объемами смеси ацетонитрила и метанола с соотношением 1:1, осажденные белки удаляли центрифугированием и супернатант анализировали при помощи UPLC-MS/MS.
Количественное определение содержания пептида в образцах производили на основании калибровочной линии, полученной для плазмы контрольной крысы. Фармакокинетические параметры были определены при помощи анализа без секционирования с использованием программного обеспечения PK Solutions 2.0 компании Summit Research Services.
Период полувыведения пептида Ac-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 был равен 48 минутам, в то время как период полувыведения контрольного пептида All-D 06-34-18 составлял 54 минуты. Объемы распределения обоих пептидов имели сравнимые значения (таблица 27, фигура 23). Таким образом, фармакологически активный пептид Ac-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 является таким же устойчивым, как и фармакологически инертный, устойчивый к протеазам контрольный пептид All-D 06-34-18, из чего следует, что пептид Ac-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 обладает благоприятными свойствами для дальнейшей разработки в качестве терапевтического средства (фигура 23). Пептид Аc-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9 выводился быстрее, характеризуясь периодом полувыведения, равным ~27 минутам.
Фармакокинетические параметры 3 пептидов при внутривенном введении суммированы в таблице 27.
Полученные результаты подтверждают повышенную протеолитическую устойчивость пептида Ac-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6 в кровотоке крысы in vivo. Выведение, равное 5,8 мл/мин/кг, близко соответствует опубликованному значению скорости почечной фильтрации у крыс (~8-9 мл/мин/кг), из чего следует, что выведение данного бициклического пептида под воздействием протеазы плазмы фактически отсутствует [Jobin J., Bonjour JP (1985) Am. J. Physiol., 248 (5 Pt 2): F734-8].
За исключением особо оговоренных случаев при реализации на практике или испытании настоящего изобретения могут быть использованы любые методы и материалы, аналогичные или эквивалентные рассмотренным в настоящем описании изобретения. Методы, устройства и материалы, пригодные для таких применений, описаны выше. Все публикации, приведенные в настоящем описании изобретения, полностью включены в настоящее описание изобретения в качестве ссылки с целью описания и рассмотрения методов, реагентов и инструментов, представленных в публикациях, которые могут быть использованы в связи с настоящим изобретением.
Таблицы
Активность, устойчивость в крысиной плазме и межвидовая перекрестная реактивность пептида Ас-06-34-18, рассмотренного в публикации РСТ/ЕР2012/069898. 1Определение произведено методом №1. 2Определение произведено методом №3. Таблица воспроизведена из публикации РСТ/ЕР2012/069898
Обзор устойчивости в крысиной и человеческой плазме ex vivo разных производных пептида 06-34-18 с модифицированной петлей 1, определенной методом №3
Сканирование аланином петли 2. Сравнение полученных значений активности с аналогичными показателями пептида дикого типа и указание пептидных фрагментов, обнаруженных после инкубации в человеческой плазме
-LHQDL, -R
Сканирование D-аминокислотой петли 2. Сравнение полученных значений активности с аналогичными показателями пептида дикого типа и указание пептидных фрагментов, обнаруженных после инкубации в человеческой плазме
Сканирование D-аланином петли 2. Сравнение полученных значений активности с аналогичными показателями пептида дикого типа. Фрагменты были аналогичны фрагментам, обнаруженным при замене полной D-аминокислотой (таблица 3b). Следует отметить, что была произведена оценка как пептида Ас-(06-34-18), так и пептида Ас-(06-34-18) Phe2 Tyr4
Пептиды 5×5
06-34 - Замены идентифицированных некритических остатков природными аминокислотами
06-34 - Замены идентифицированных некритических остатков N-метилированными аминокислотами
Сравнителные воздейтвия замены D-аминокислотами на активность и устойчивость в крысиной плазме
Сравнительные воздействия N-метилирования остатков петли 1 и остатка Cys2 на активность и устойчивоть в крысиной плазме
Сравнительные воздействия аналогов аргинина в Аc-06-34-18(ТМВ)-NH2 на активность и устойчивость. Следует отметить, что модификация ∆ Arg не вызывала ингибирования до использования 100 мкМ пептида
Сравнительное воздействие на сродство гидрофобных аминокислот, заменяющих Trp2 в Ас-06-34-18(ТМВ)-NH2
Сравнительные значения сродства, полученные для производных пролина с заменами гамма-атома углерода, аналогов с измененными размерами кольца, би/трициклических производных и ограниченных аминокислот, таких как Aib
Сравнительные воздействия смешанных замен Ser1, Ala4 и Cys2
Сравнительное повышение активности, вызванное включением Aze3 в кандидаты, стабилизированные в плазме. 1Сравнительные значения устойчивости, определенные методом №1. 2Истинное время полужизни стабилизированных пептидов в крысиной плазме было определено методом №3. 3Значения IC50 являются относительными, а не абсолютными.
Воздействие на активность при включении Aba4 в пептиды, содержащие Aze3 и стабилизирующие в плазме модификации NMeArg5, HomoArg5 и 4GuanPhe5
.
42 уникальные последовательности, связывающие калликреин, были идентифицированы в результате отбора с использованием рандомизированного фрагмента WPAR в положениях 2, 3, 4 и 5 в последовательности 06-34-18. Последовательности были распределены по рангу в зависимости от степени связывания калликреина и были указаны значения относительного распространения в отобранных продуктах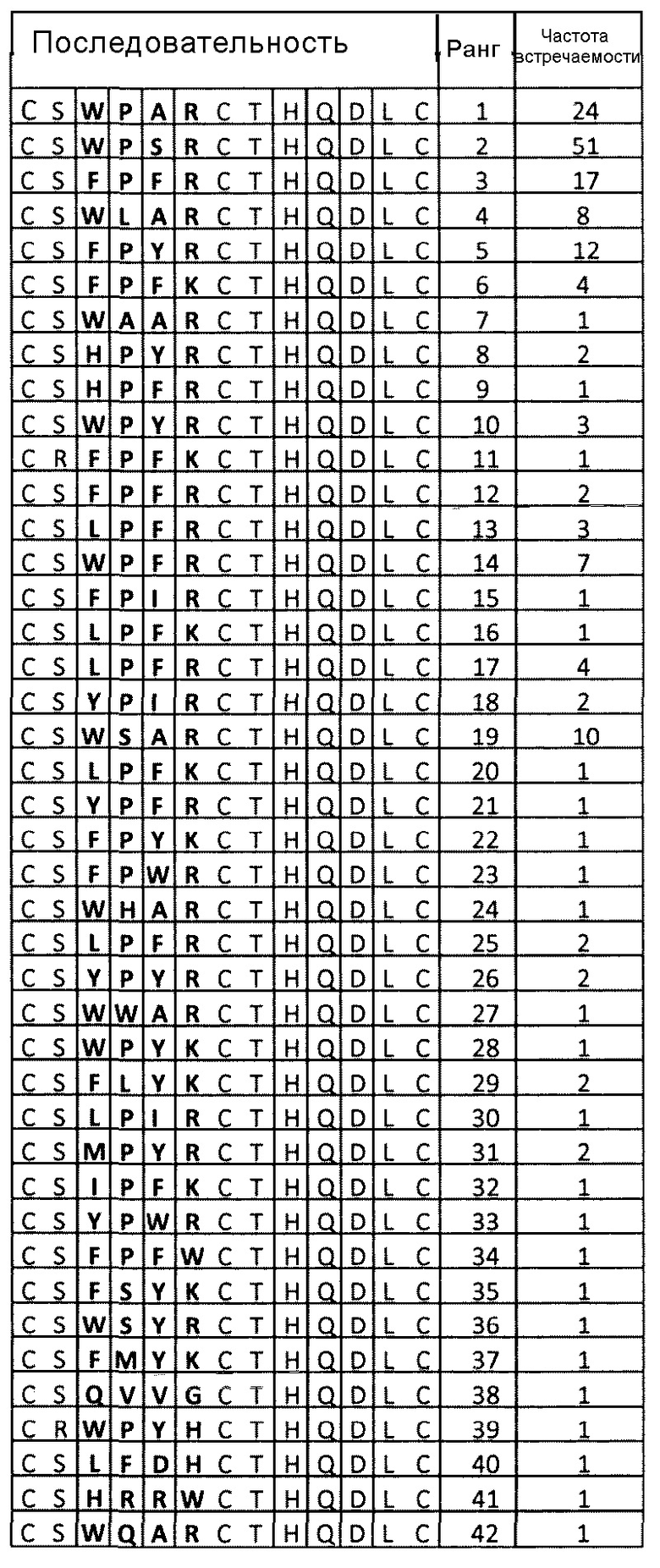
А. Распространение определенного фрагмента в каждом продукте. Последовательности анализировали в зависимости от строгости отбора. Вычисляли процентное содержание определенного фрагмента в продукте, отобранном при определенной степени строгости. В. Связывание калликреина человеческой плазме определенными фрагментами в положениях 2, 3, 4 и 5 (при фиксированных положениях 1, 6, 7, 8, 9 и 10 в пептиде 06-34-03)
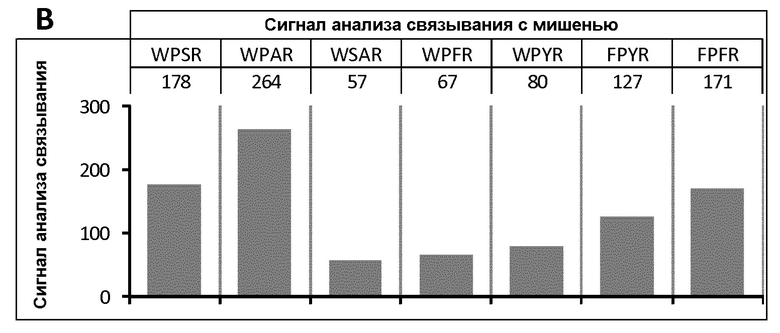
50 лучших последовательностей, связывающих калликреин, содержащих фиксированный фрагмент WPAR. Последовательность WPAR была фиксирована в библиотеке бициклов с рандомизированными положениями 1, 6, 7, 8, 9 и 10. Последовательности, отобранные с учетом ингибирования калликреина, были выделены и исследованы в отношении связывания с калликреином. Последовательности были распределены по рангу в зависимости от связывания с калликреином. Последовательность пептида 06-34-03, не участвовавшая в отборе, выделена красным цветом. Во второй петле WPAR-содержащих пептидов, связывающихся с калликреином, видны определенные тенденции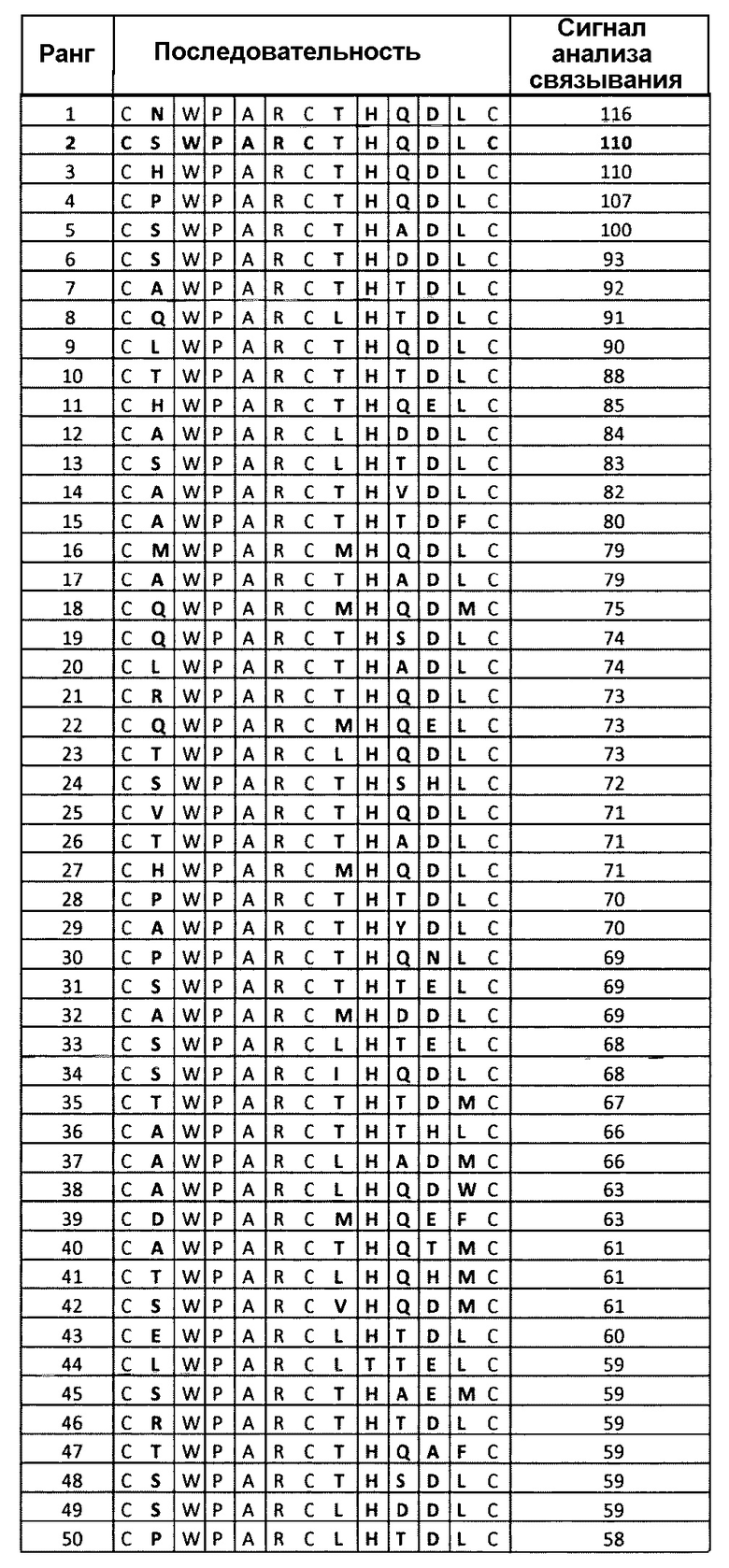
Последовательности с фиксированным фрагментом WPAR в 1-й петле, полученные в результате отбора с учетом ингибирования калликреина и ассоциированных сигналов анализа связывания калликреина, были сгруппированы в соответствии с образованием производных пептидов из фрагмента ‘THxxL’ (A) или фрагмента ‘xHxDL’ (B). Был вычислен средний сигнал анализа связывания для всех членов данной группы. Группы, содержащие точно указанный фрагмент, выделены зеленым цветом; также показаны группы, содержащие на одно изменение больше или меньше по сравнению с указанным фрагментом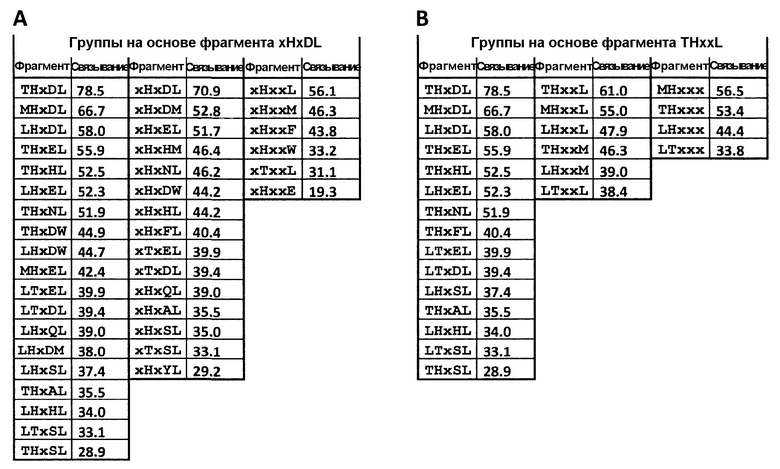
Показатели сродства и устойчивости вариантов фрагмента WPAR
Эффект замен в положении Phe2/Tyr4 гидрофобными аналогами
Обзор эффекта замен Arg5 и Aze3 в Ас-06-34-18(ТМВ)-NH2 Phe2 Tyr4. 1Сравнительные значения устойчивости, определенные методом №1. 2Истинное время полужизни стабилизированных пептидов в крысиной плазме было определено методом №3. 3Значения IC50 являются относительными, а не абсолютными
Эффект единичных замен аминокислот в положении 6. Модификации испытывали с использованием пептида Аc-(06-34-18) Phe2 Tyr4 (левый столбец) и пептида Ас-(06-34-18) (правый столбец)
Аc-(06-34-18) Phe2 Tyr4
Аc-(06-34-18)
Эффект единичных замен аминокислот в положении 7. Модификации испытывали с использованием только пептида Аc-(06-34-18) Phe2 Tyr4
Эффект единичных замен аминокислот в положении 8. Модификации испытывали с использованием только пептида Аc-(06-34-18) Phe2 Tyr4
Сканирование α-N-метиламинокислотой петли 2. Сравнение полученных значений активности с аналогичными показателями пептида дикого типа и указание пептидных фрагментов, обнаруженных после инкубации в человеческой плазме
Модификации пептидного остова в петле 2
Сравнительная активность петли 2 в отношении калликреина человеческой и крысиной плазмы у кандидатов с повышенной устойчивостью к протеазам плазмы
Сравнительная активность модифицированной петли 1 и модифицированной петли 2 производных пептида 06-34-18. Следует отметить включение модификаций Aze3 (повышение сродства) и HArg5 (повышение устойчивости в плазме) в петле 1 и модификаций, повышающих устойчивость, в петле 2
Фармакокинетические параметры пептидов Аc-(06-34-18) Phe2 Azе3 Tyr4 HArg5 Ala(СН2NН)6, Аc-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9 и контрольного пептида All-D Ас-(06-34-18) у крысы. Из-за разных концентраций пептида Аc-(06-34-18) Phe2 Aze3 Tyr4 HArg5 D-Asp9 у двух отдельных крыс скорости выведения и объемы распространения не могут быть вычислены на доверительном уровне
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯЦИЯ СПЕЦИФИЧНОСТИ СТРУКТУРИРОВАННЫХ БЕЛКОВ | 2012 |
|
RU2631931C2 |
| МОДУЛЯЦИЯ СПЕЦИФИЧНОСТИ СТРУКТУРИРОВАННЫХ БЕЛКОВ | 2012 |
|
RU2745572C2 |
| НОВЫЕ ПОЛИПЕПТИДЫ | 2014 |
|
RU2674604C2 |
| БИЦИКЛИЧЕСКИЕ ПЕПТИДНЫЕ ЛИГАНДЫ, СПЕЦИФИЧНЫЕ ДЛЯ МТ1-ММР | 2015 |
|
RU2708459C2 |
| БИЦИКЛИЧЕСКИЕ ПЕПТИДНЫЕ ЛИГАНДЫ, СПЕЦИФИЧНЫЕ ДЛЯ MT1-MMP | 2015 |
|
RU2824959C2 |
| МОДИФИЦИРОВАННЫЕ АНТИ-EGFR АНТИТЕЛА С УМЕНЬШЕННОЙ ИММУНОГЕННОСТЬЮ | 2002 |
|
RU2297245C2 |
| СЛИТЫЕ КОНСТРУКЦИИ ЛЕКАРСТВЕННОГО СРЕДСТВА И КОНЪЮГАТЫ | 2005 |
|
RU2428431C2 |
| ВАРИАНТЫ Fc С ИЗМЕНЕННЫМ СВЯЗЫВАНИЕМ С FcRn | 2014 |
|
RU2700882C2 |
| ВАРИАНТЫ Fc С ИЗМЕНЕННЫМ СВЯЗЫВАНИЕМ C FcRn | 2008 |
|
RU2529951C2 |
| ВАРИАНТЫ Fc С ИЗМЕНЕННЫМ СВЯЗЫВАНИЕМ С FcRn | 2021 |
|
RU2833597C2 |


























































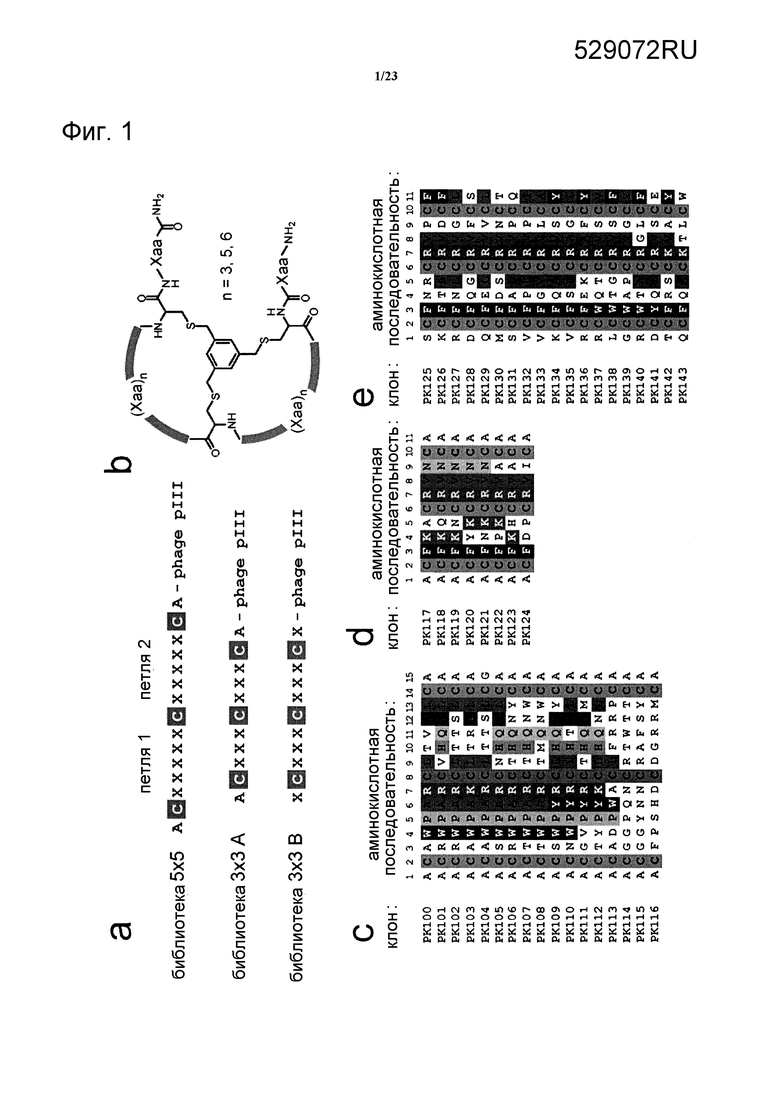
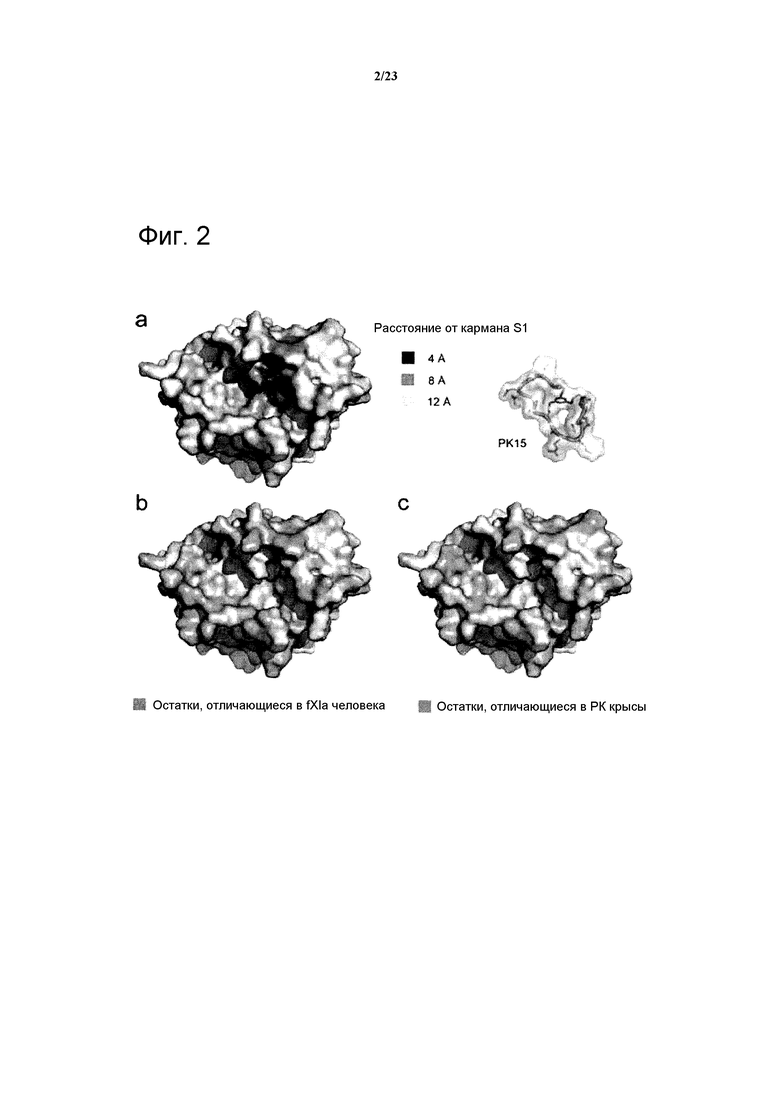
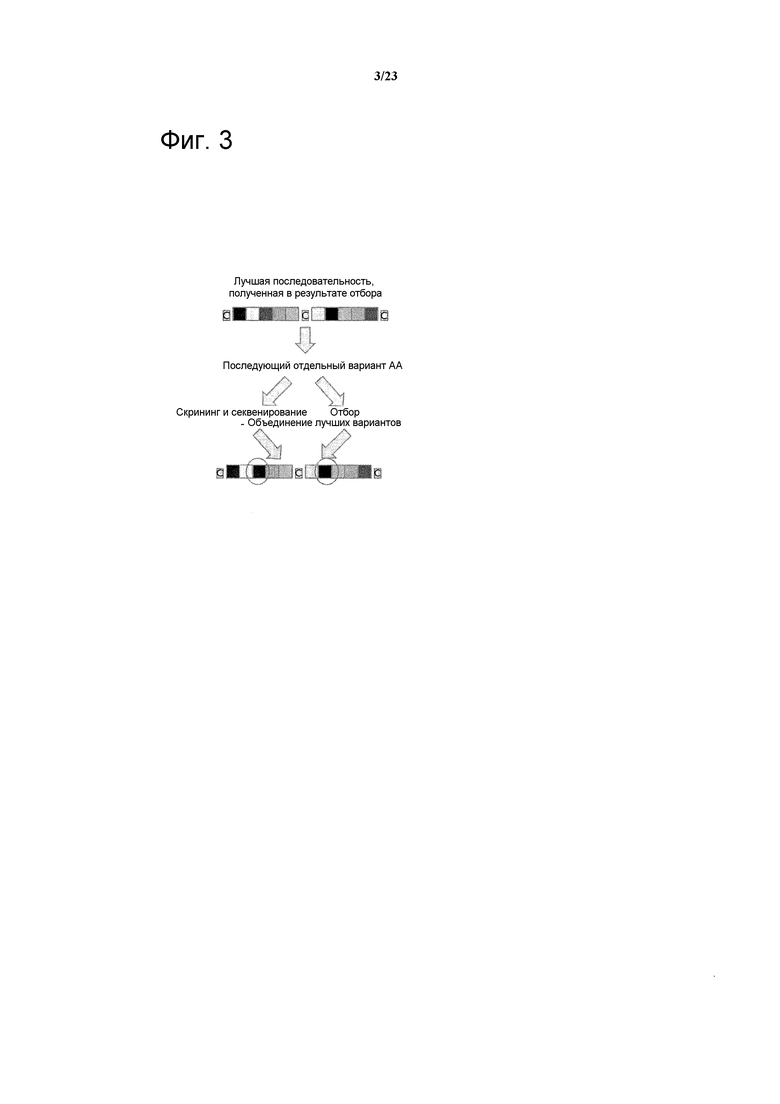
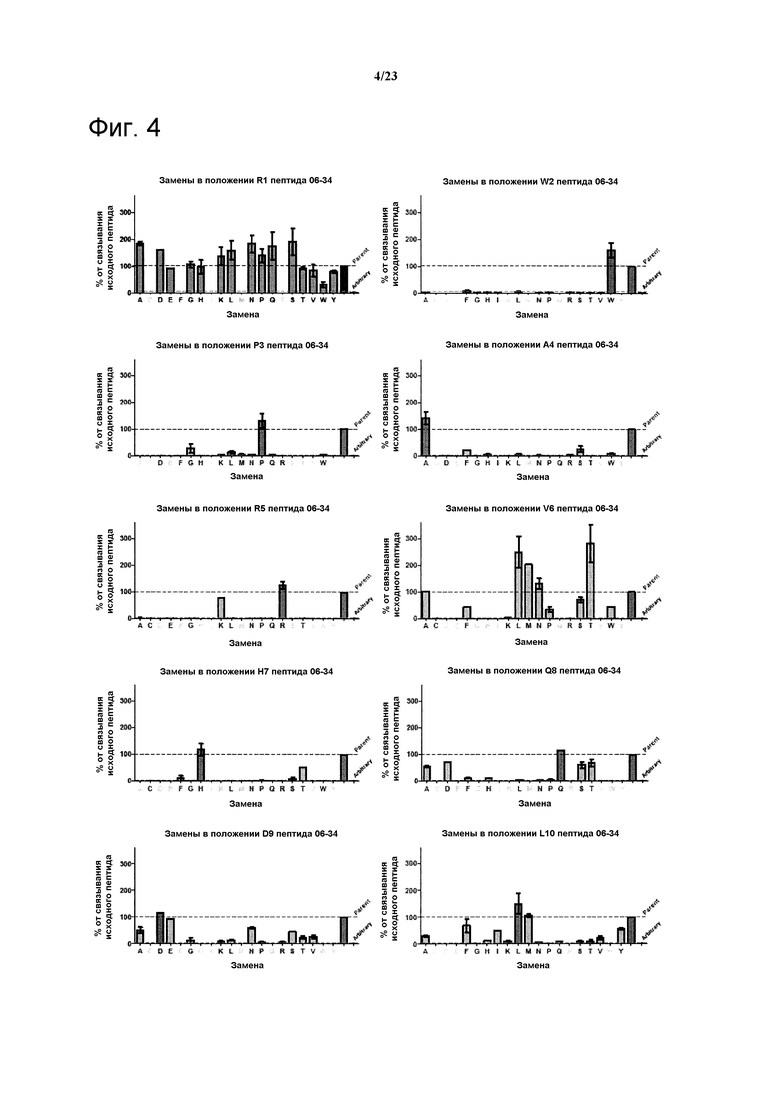
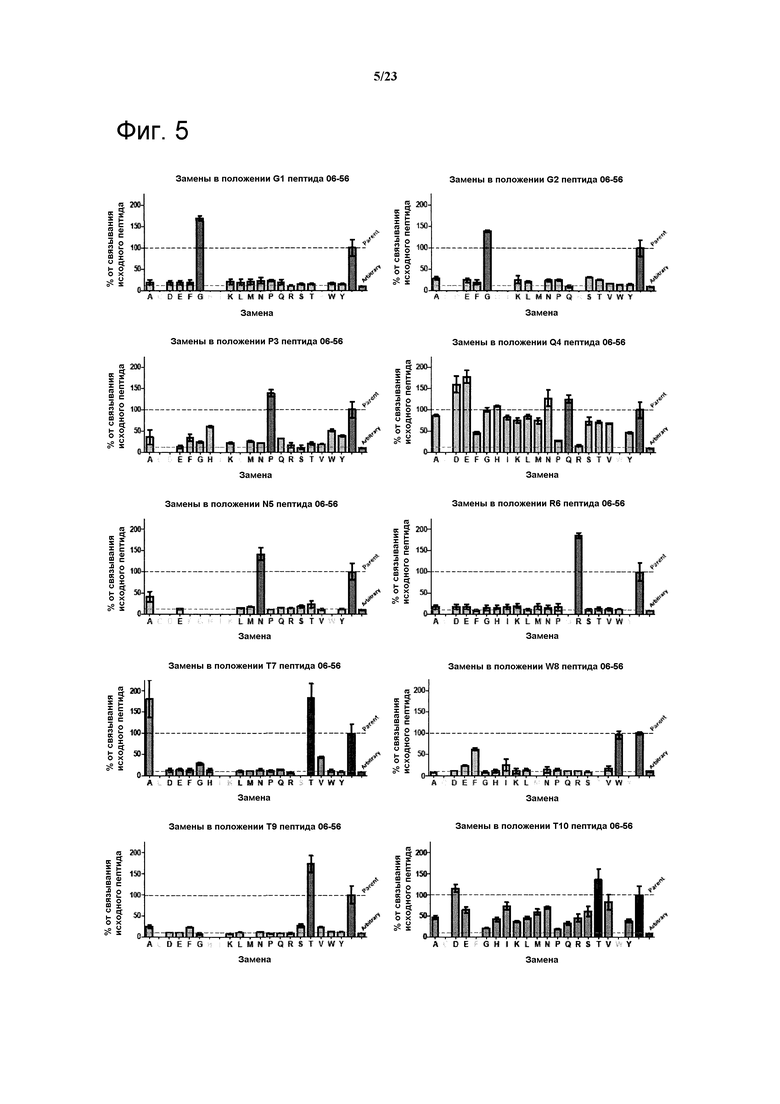
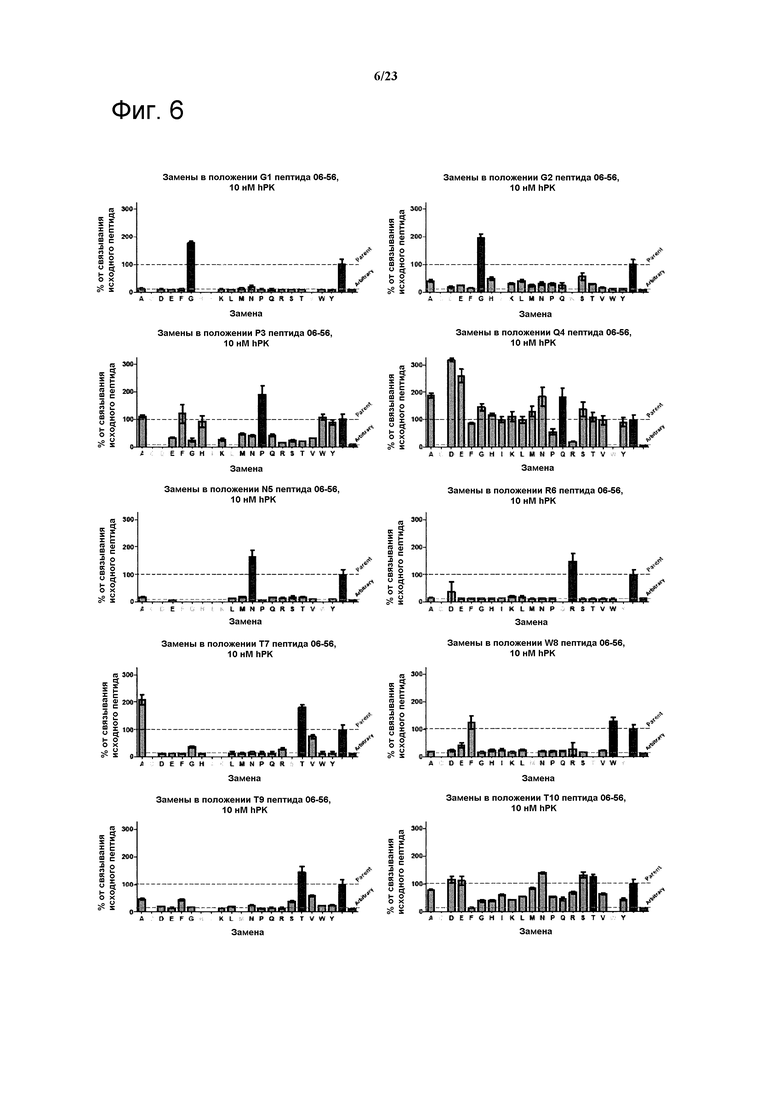
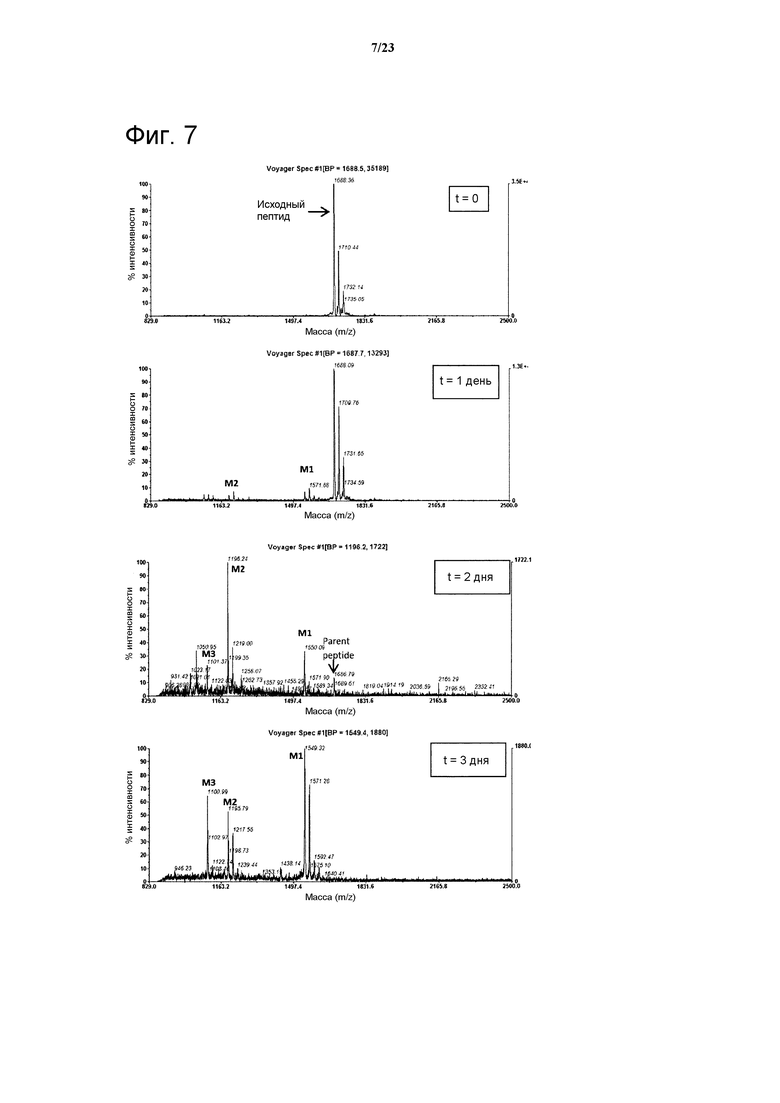
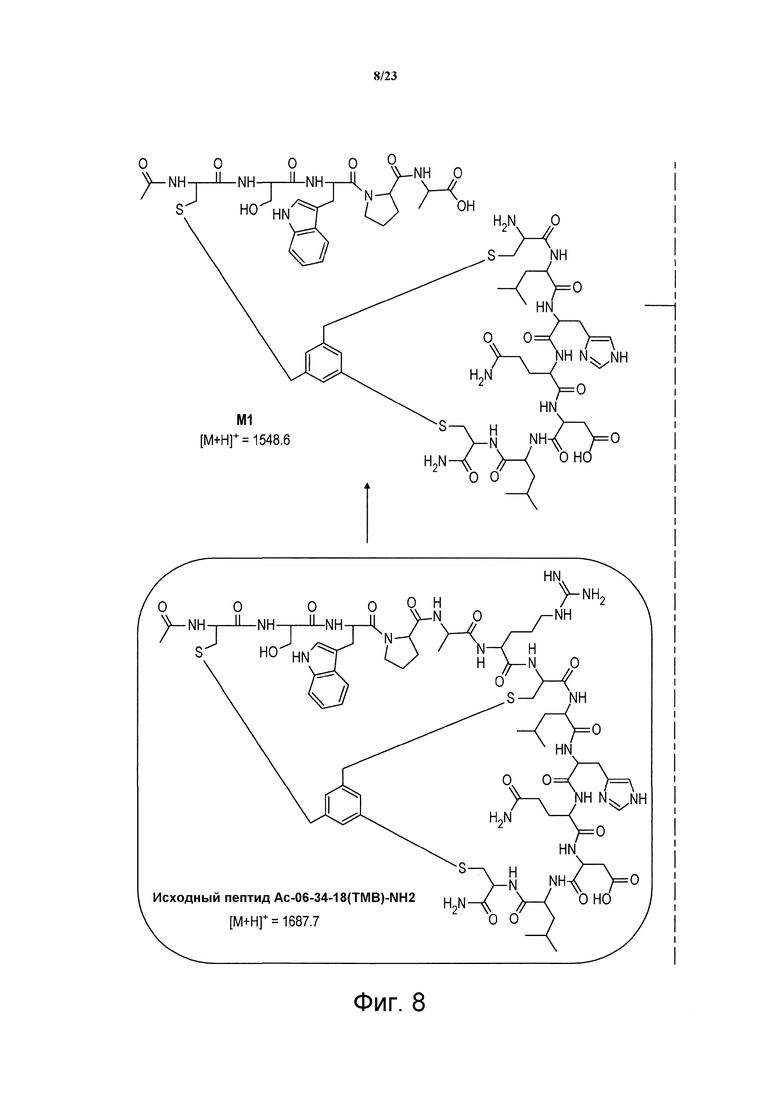
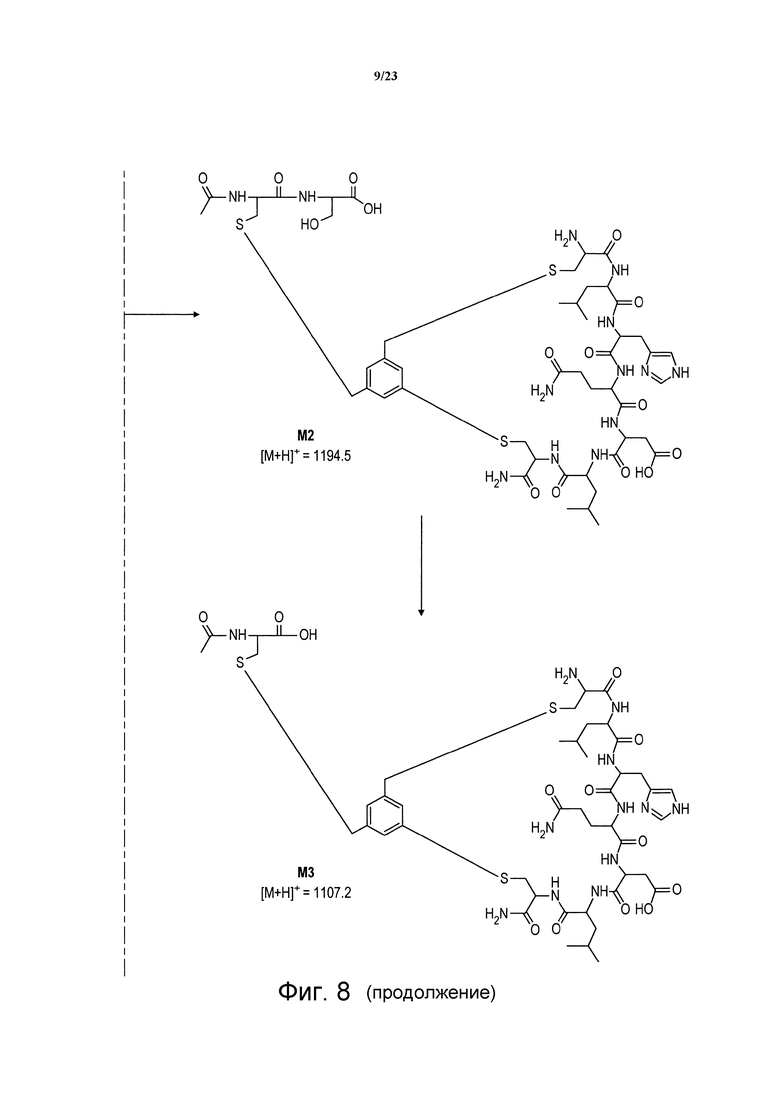
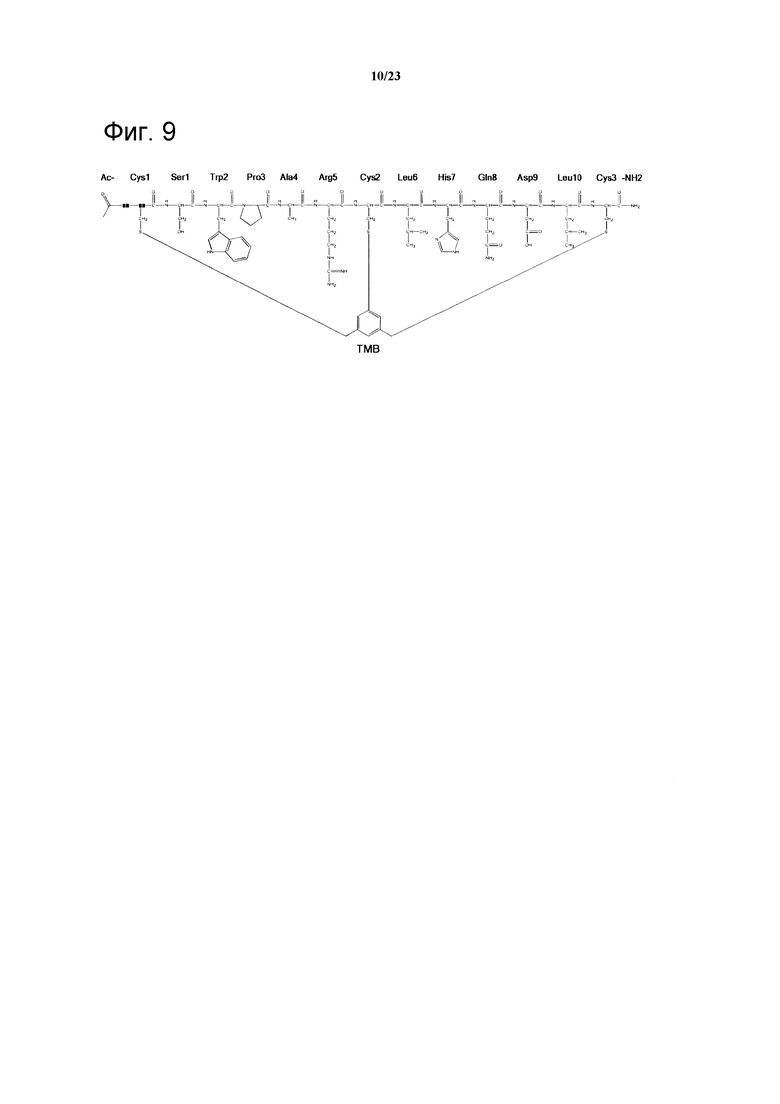
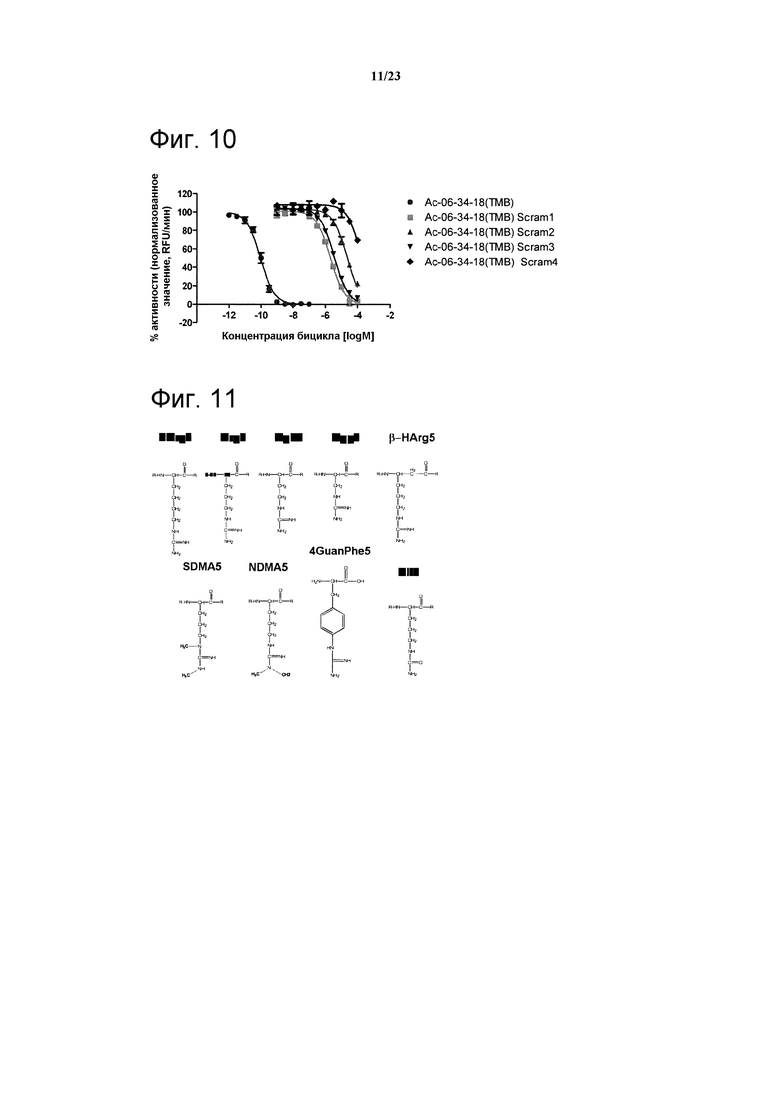
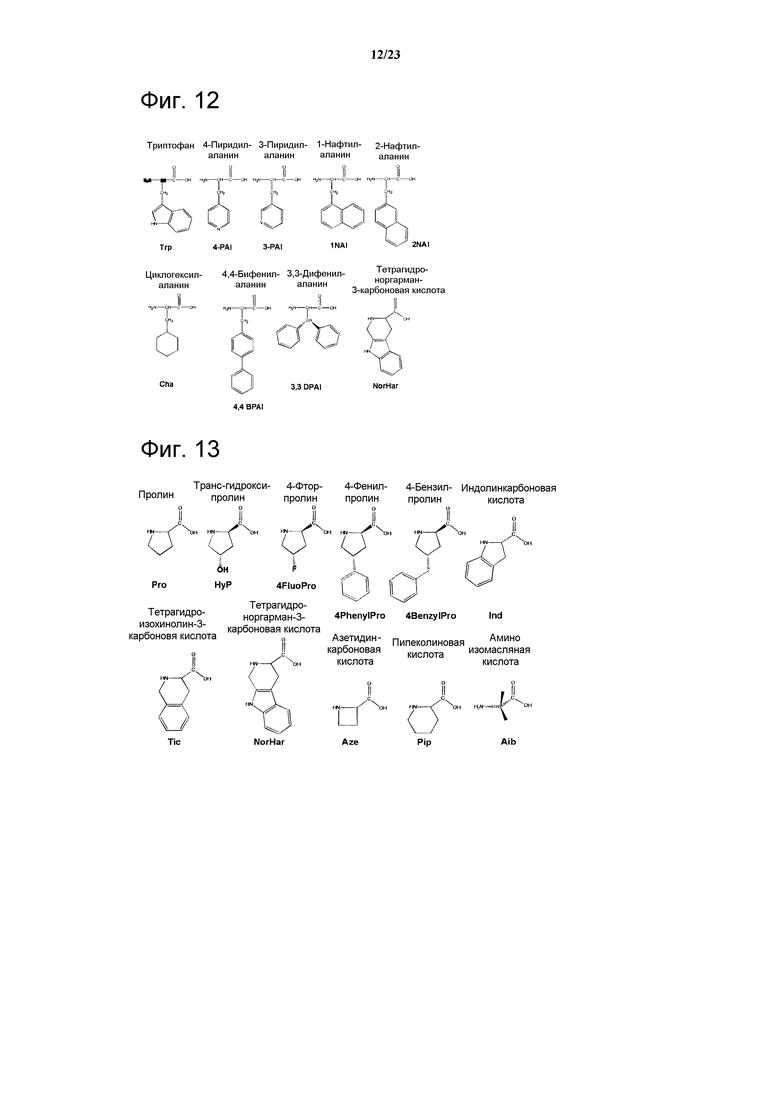
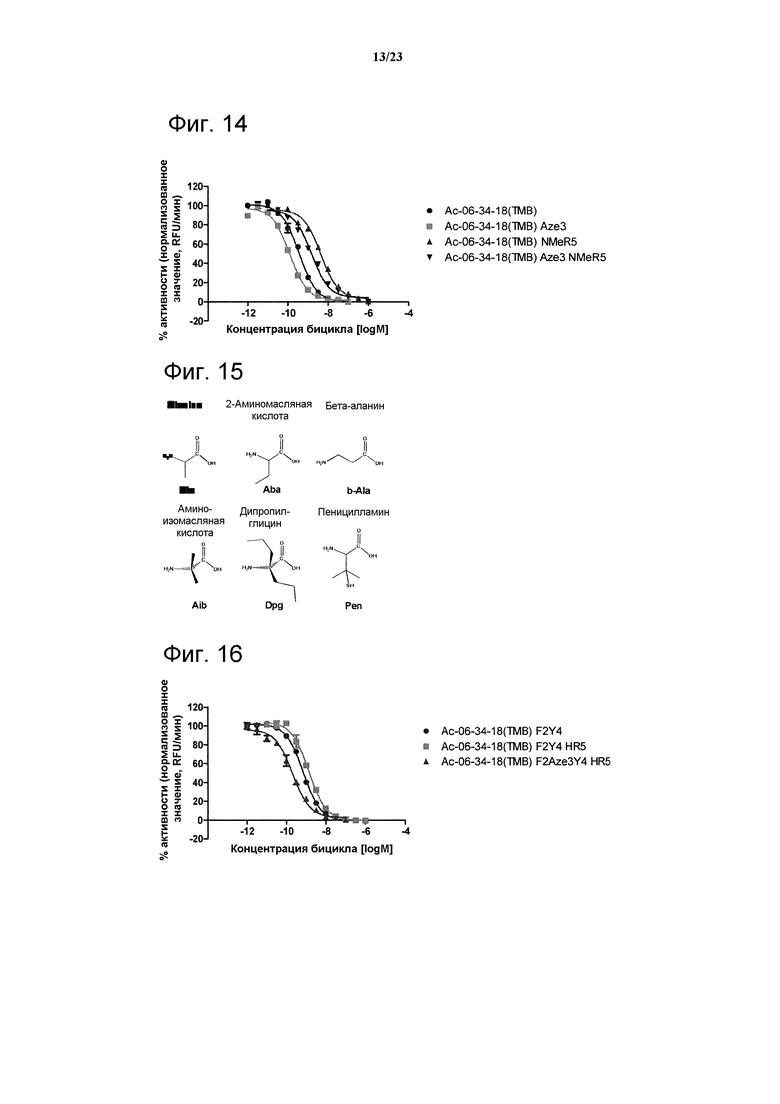


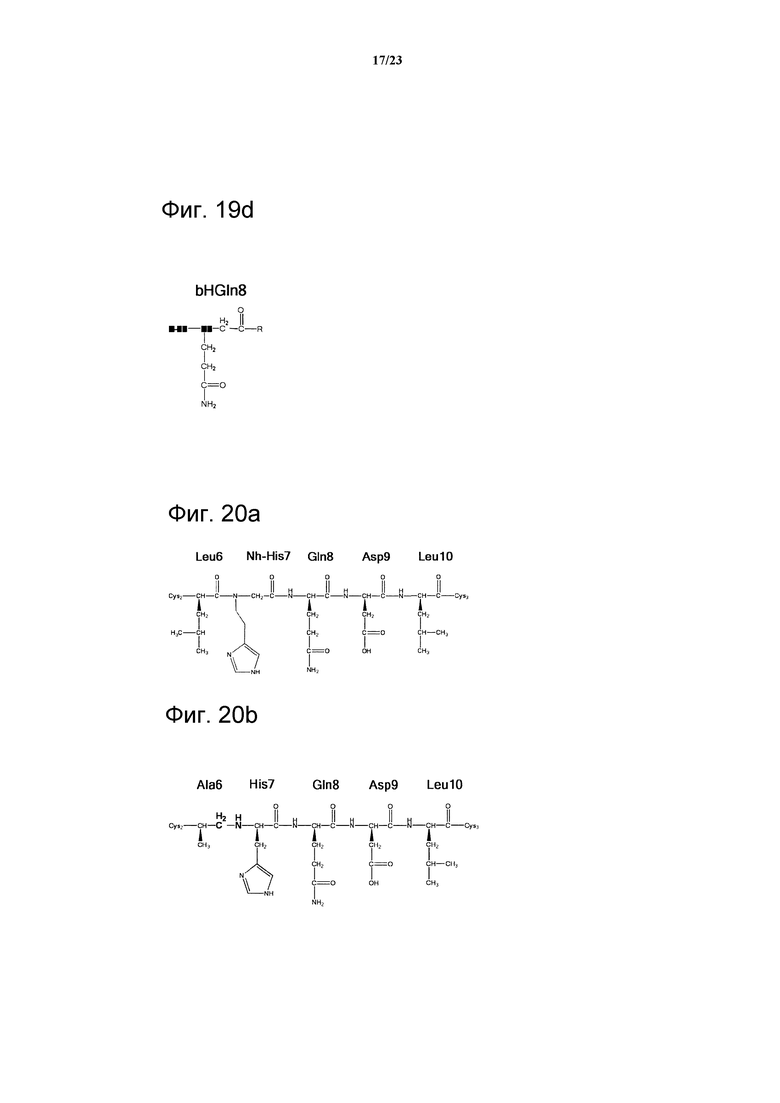
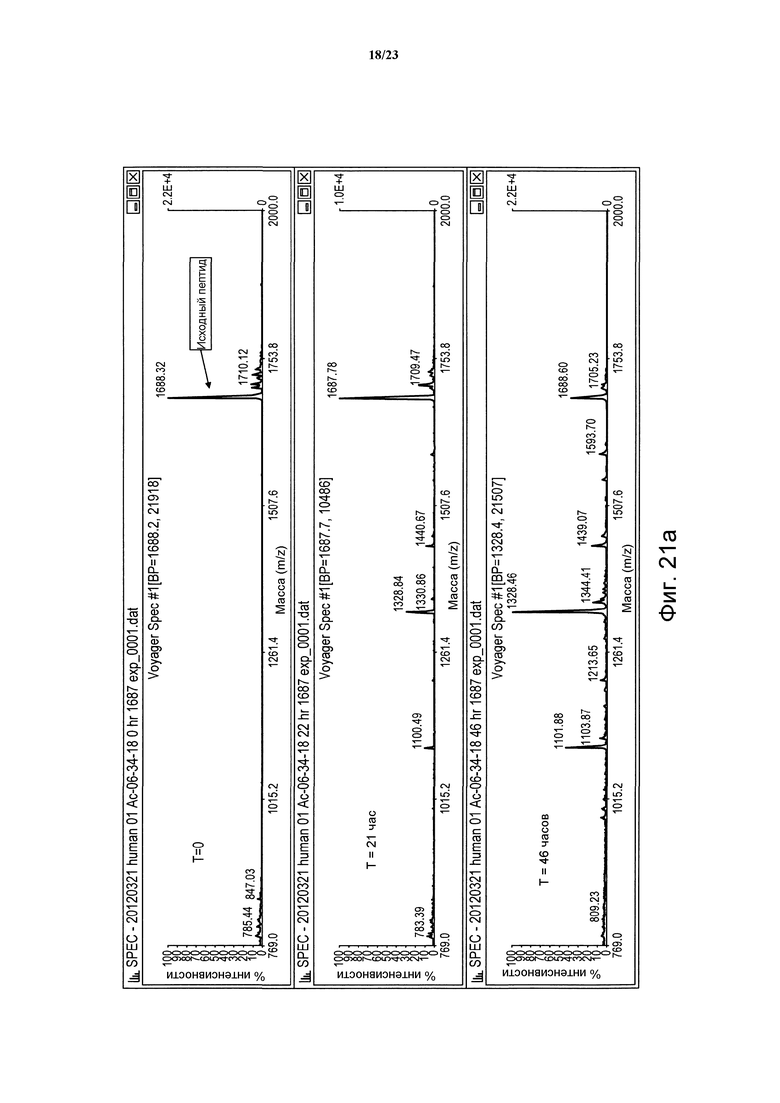
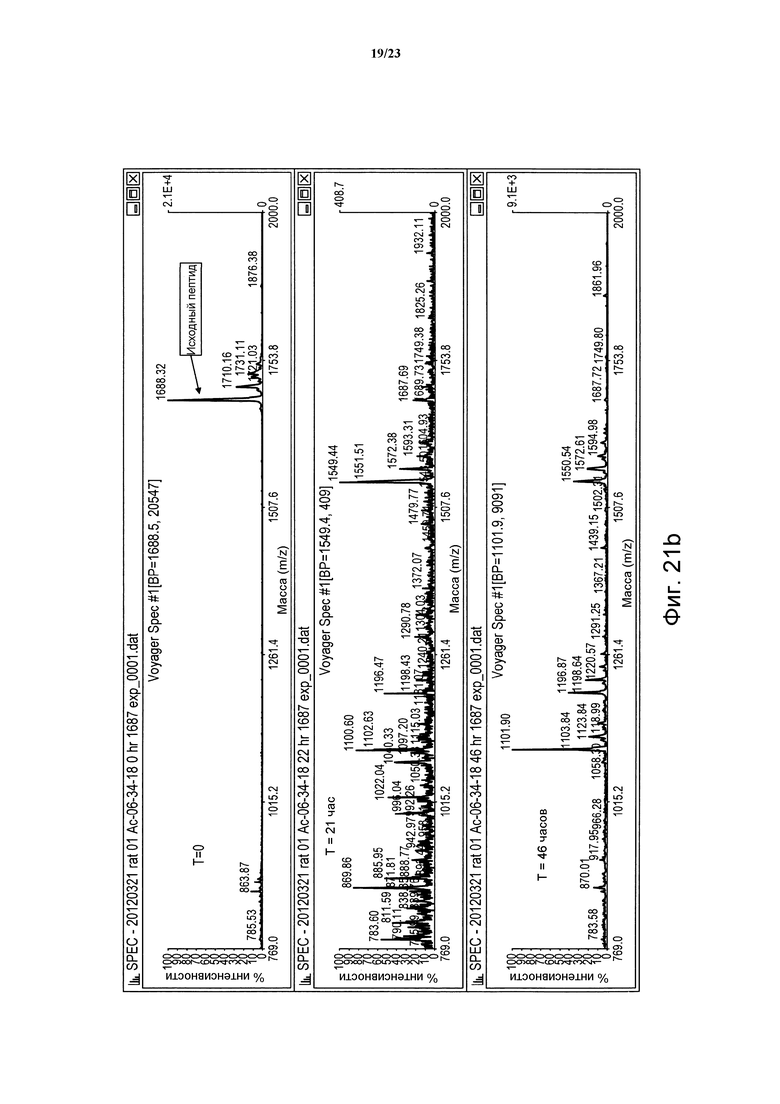
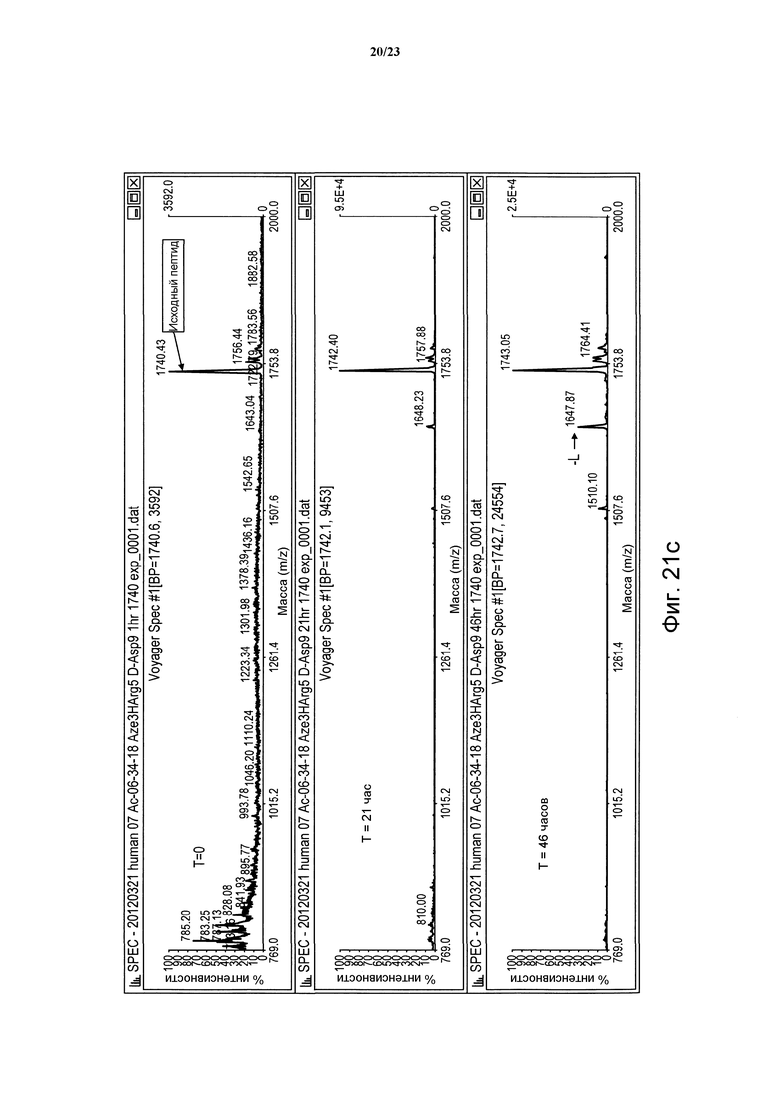
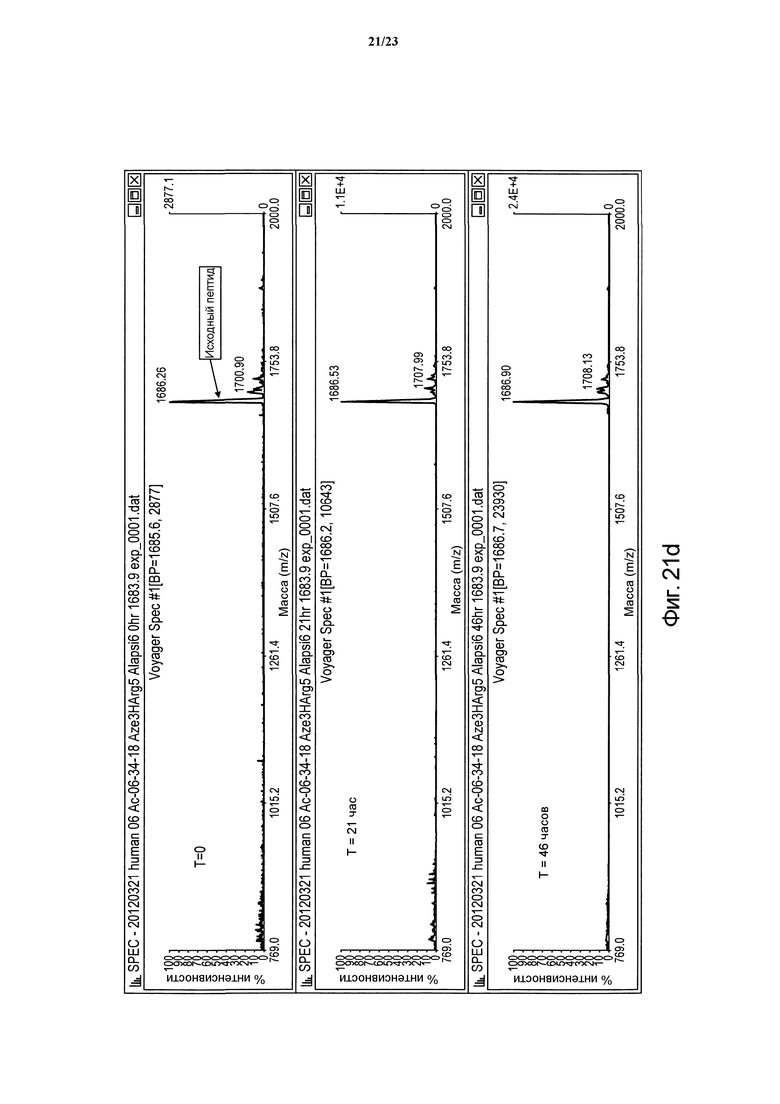
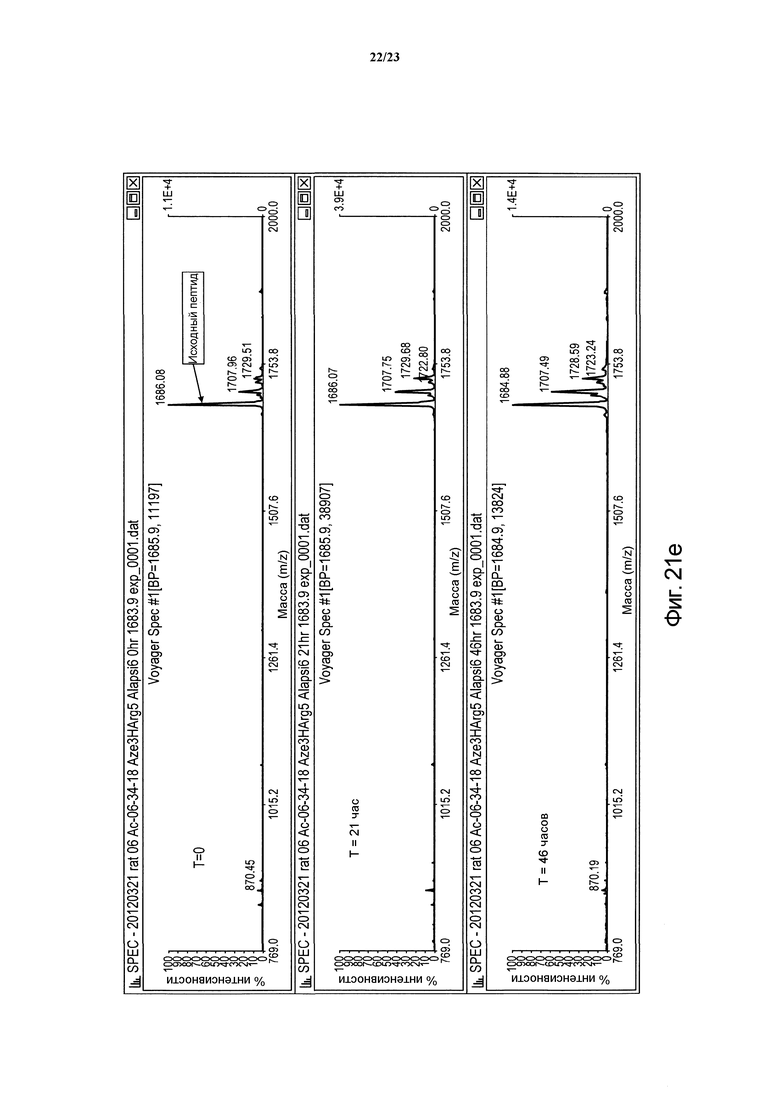
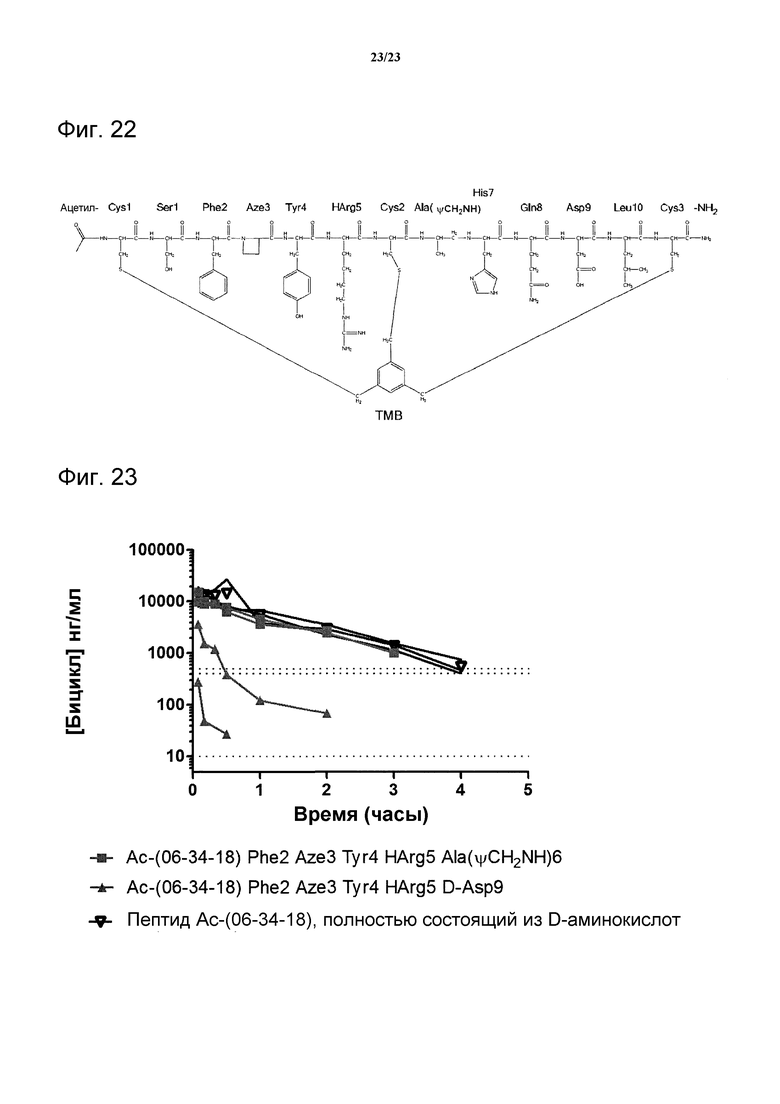
Изобретение относится к биотехнологии. Описан пептидный лиганд, специфичный к человеческому калликреину, который включает полипептид, содержащий по меньшей мере три реакционноспособные группы, разделенные последовательностями по меньшей мере двух петель, и молекулярный остов, формирующий ковалентные связи с реакционно-способными группами полипептида с образованием на молекулярном остове по меньшей мере двух петель полипептида, при этом петли пептидного лиганда включают пять аминокислот и одна петля содержит фрагмент GrA(ψCH2NH)HQ/TxLGr, где Gr означает реакционно-способную группу. Также описана фармацевтическая композиция, содержащая указанный пептид и представлено применение указанного пептида для лечения состояний, опосредованных калликреином, выбранных из воспалительных состояний, аллергической гиперчувствительности, рака, бактериальной или вирусной инфекции, офтальмологических нарушений и аутоиммунных нарушений. Изобретение расширяет арсенал средств лечения заболеваний, опосредованных калликреином. 3 н. и 9 з.п. ф-лы, 23 ил., 27 табл., 10 пр.
1. Пептидный лиганд, специфичный к человеческому калликреину, который включает полипептид, содержащий по меньшей мере три реакционно-способные группы, разделенные последовательностями по меньшей мере двух петель, и молекулярный остов, формирующий ковалентные связи с реакционно-способными группами полипептида с образованием на молекулярном остове по меньшей мере двух петель полипептида, при этом петли пептидного лиганда включают пять аминокислот и одна петля содержит фрагмент GrA(ψCH2NH)HQ/TxLGr, где Gr означает реакционно-способную группу.
2. Пептидный лиганд по п.1, в котором полипептид включает первую петлю, содержащую фрагмент GrxWPARGr или GrxFPYRGr, и вторая петля включает фрагмент GrA(ψCH2NH)HQ/TxLGr.
3. Пептидный лиганд по п.2, в котором остаток P в первой петле заменен азетидинкарбоновой кислотой и/или остаток R в первой петле заменен N-метиларгинином, гомоаргинином и/или гуанидилфенилаланином.
4. Пептидный лиганд по п.3, в котором полипептид включает первую петлю, содержащую фрагмент GrxFPYRGr, где остаток P заменен азетидинкарбоновой кислотой и/или остаток R заменен N-метиларгинином, гомоаргинином и/или гуанидилфенилаланином.
5. Пептидный лиганд по любому из пп.1-4, в котором Gr представляет собой цистеин.
6. Пептидный лиганд по любому из пп.1-4, который включает: Cys Ser1 Phe2 Aze3 Tyr4 HArg5 Cys Ala(ψCH2NH)6 His7 Gln8 Asp9 Leu10 Cys.
7. Пептидный лиганд по любому из пп.1-4, который включает:
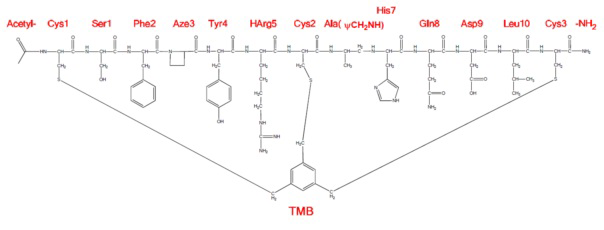
8. Пептидный лиганд по любому из пп.1-4, который присоединен к антителу или его фрагменту.
9. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении человеческого калликреина, включающая эффективное количество пептидного лиганда по любому из пп.1-4 совместно с фармакологически приемлемым носителем.
10. Применение лиганда по любому из пп.1-4 для получения лекарственного средства для профилактики или лечения состояний, опосредованных калликреином, выбранных из воспалительных состояний, аллергической гиперчувствительности, рака, бактериальной или вирусной инфекции, офтальмологических нарушений и аутоиммунных нарушений.
11. Применение по п.10 для профилактики или лечения офтальмологических нарушений.
12. Применение по п.11, где офтальмологические нарушения включают экссудативные и/или воспалительные офтальмологические нарушения, нарушения, относящиеся к ухудшению проницаемости и/или целостности сосудов сетчатки, нарушения, относящиеся к разрыву микрососудов сетчатки, вызывающему фокальные кровоизлияния, заболевания задней части глаза, заболевания сетчатки и заболевания передней части глаза, которые выбраны из возрастной дегенерации желтого пятна (ARMD), экссудативной дегенерации желтого пятна (также известной как ”мокрой” или неоваскулярной возрастной дегенерации желтого пятна (мокрая-AMD)), отека желтого пятна, возрастной дисковидной дегенерации желтого пятна, цистоидного отека желтого пятна, отека века, отека сетчатки, диабетической ретинопатии, острой нейроретинопатии желтого пятна, центральной серозной ретинопатии, хориоретинопатии, хороидального образования новых сосудов, неоваскулярной макулопатии, неоваскулярной глаукомы, обструктивных артериальных и венозных ретинопатий (например, венозной окклюзии сетчатки или артериальной окклюзии сетчатки), окклюзии центральной вены сетчатки, диссеминированной внутрисосудистой коагулопатии, окклюзии ответвления вены сетчатки, гипертонических изменений глазного дна, ишемического синдрома глаза, артериальной микроаневризмы сетчатки, болезни Коата, телеангиэктазии ямки, односторонней окклюзии вены сетчатки, папиллофлебита, окклюзии центральной артерии сетчатки, окклюзии ответвления артерии сетчатки, заболевания сонной артерии (CAD), глазурного васкулита ответвления сосуда, серповидно-клеточной ретинопатии и других гемоглобинопатий, ангиоидных полос сетчатки, отека желтого пятна, возникающего в результате такой этиологии как болезнь (например, диабетического отека желтого пятна), повреждения глаза или хирургической операции глаза, ишемии или дегенерации сетчатки, вызванной, например, повреждением, травмой или опухолями, увеита, ирита, васкулита сетчатки, эндофтальмита, панофтальмита, метастатической офтальмии, хориоидита, пигментного эпителия сетчатки, конъюнктивита, циклита, склерита, эписклерита, ретробульбарного глазного неврита, кератита, блефарита, экссудативного отслоения сетчатки, язвы роговицы, язвы конъюнктивы, хронического нумуллярного кератита, кератита Тайгесона, прогрессирующей разъедающей язвы роговицы, воспалительного заболевания глаза, вызванного бактериальной или вирусной инфекцией и офтальмологической операцией, воспалительного заболевания глаза, вызванного физическим повреждением глаза, симптома, вызванного воспалительным заболеванием глаза, включая зуд, воспалительную гиперемию, отек и язву, эритему, экссудативную эритему, узловатую эритему, кольцевидную эритему, отек склеры, дерматит, ангионевротический отек, отек гортани, отек языка, подсвязочный отек, бронхит, ринит, фарингит, синусит, ларингит или отит.
| МУТАНТНАЯ ФОРМА ПЕРОКСИДАЗЫ ХРЕНА | 2012 |
|
RU2474613C1 |
| WO 2006078161 A1, 27.07.2006 | |||
| НОВЫЙ АЛЛЕРГЕН-ПРОСТАТИЧЕСКИЙ КАЛЛИКРЕИН | 2007 |
|
RU2502074C2 |

