Область техники
Данное изобретение относится к органической химии, фармакологии и медицине и касается профилактики и терапии заболеваний, этиология или патогенез которых связаны с вирусами, относящимися к роду Flavivirus, с помощью новых модифицированных аналогов нуклеозидов.
Уровень техники
Вирусы рода Flavivirus (флавивирусы) передаются человеку при присасывании зараженного клеща либо укусе комара и вызывают широкий спектр заболеваний, варьирующих от легких лихорадочных состояний до геморрагических лихорадок и летальных поражений центральной нервной системы. Многие флавивирусы обладают нейротропизмом, в связи с чем вызываемые ими заболевания сопряжены с нейродегенеративными синдромами [G.J. Sips et al., Rev. Med. Virol., 2012, 22, 69-87]. Наиболее известными примерами заболеваний, вызываемых вирусами рода Flavivirus, являются клещевой энцефалит (КЭ), желтая лихорадка, лихорадка денге (включая денге шок-синдром), лихорадка Западного Нила, лихорадка Зика.
Среди природно-очаговых флавивирусных инфекций КЭ представляет собой особо важную проблему на евразийском континенте, включая Европу, Россию, восточную Азию и Японию, тогда как желтая лихорадка эндемична в экваториальных, тропических и субтропических регионах Южной Америки и Африки. По данным Всемирной организации здравоохранения (ВОЗ), ежегодно регистрируется более 10 тыс. случаев КЭ и около 200 тыс. случаев желтой лихорадки, и, несмотря на наличие эффективных вакцин, охват иммунизацией считается недостаточным. В настоящее время не существует специфических противовирусных препаратов для лечения инфекций, вызванных флавивирусами. Из уровня техники известен ряд низкомолекулярных ингибиторов репродукции флавивирусов. Среди них широко представлены нуклеозидные ингибиторы, содержащие дополнительные заместители как в гетероциклическом основании (пуриновом или пиримидиновом), так и в углеводном остатке, а также их аналоги с модифицированным гетероциклическим основанием. Основными мишенями таких соединений являются РНК-зависимый РНК-полимеразный (NS5RdRp) и метилтрансферазный (NS5MT) домены неструктурного белка NS5. Кроме того, производные нуклеозидов, содержащие жесткий линкер между нуклеозидным каркасом и объемным гидрофобным фрагментом, могут ингибировать слияние внешней мембраны оболочечных вирусов с плазматической мембраной и другими билипидными компартментами клеток хозяина.
На данный момент не существует ни одного специфически действующего на флавивирусы и их молекулярные мишени лекарственного препарата, допущенного к клиническому применению. Таким образом, существует необходимость в разработке новых эффективных средств против заболеваний, вызванных вирусами рода Flavivirus.
Раскрытие изобретения
Задачей настоящего изобретения является разработка и создание новых эффективных противовирусных средств, перспективных для применения в клинической практике для терапии и/или профилактики заболеваний, вызванных вирусами рода Flavivirus.
Технический результат изобретения заключается в разработке и получении соединений, характеризующихся высокой эффективностью в ингибировании репродукции вирусов рода Flavivirus, в частности, вируса клещевого энцефалита, вируса желтой лихорадки, вируса лихорадки Западного Нила и других.
Помимо этого, соединения по изобретению расширяют арсенал доступных противовирусных средств, применяющихся в качестве ингибиторов репродукции вируса, относящегося к роду Flavivirus.
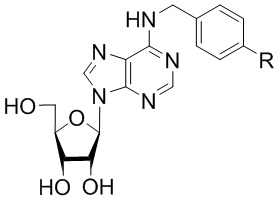
Указанный технический результат достигается посредством соединения общей формулы (I):
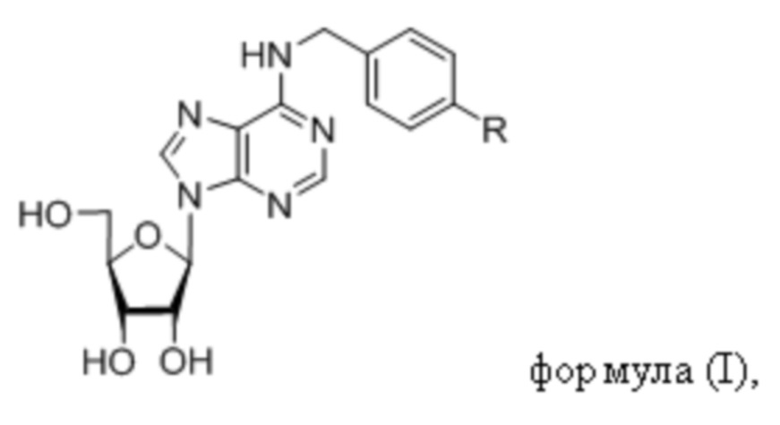
или его пролекарства или фармацевтически приемлемой соли, где:
R выбирается независимо и представляет собой -О-С1-3-алкил, частично или полностью галогенированный -О-С1-3-алкил, -S-С1-3-алкил, частично или полностью галогенированный -S-С1-3-алкил, -С≡С-С3-6-циклоалкил, -С≡С-С3-6-циклоалкенил, -С≡С-С6-арил, замещенный -С≡С-С6-арил, по меньшей мере, одним заместителем, выбранным из С1-5-алкила, частично или полностью галогенированного -С1-5-алкила, фенил, замещенный по меньшей мере, одним заместителем, выбранным из -С1-5-алкила, частично или полностью галогенированного -С1-5-алкила.
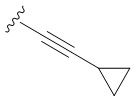
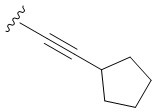
В частных вариантах воплощения изобретения R выбирается независимо и представляет собой -С≡С-циклопропил, -С≡С-циклобутил, -С≡С-циклопентил, -С≡С-циклопентенил, -С≡С-циклогексенил.
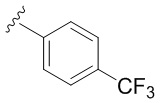
В частных вариантах воплощения изобретения R выбирается независимо и представляет собой фенил, замещенный одним или двумя заместителями, представляющими собой -СF3.
В частных вариантах воплощения изобретения R выбирается независимо и представляет собой -С≡С-С6-арил, замещенный, по меньшей мере, одним заместителем, представляющим собой -СF3.
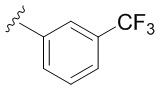
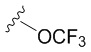
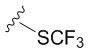
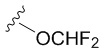
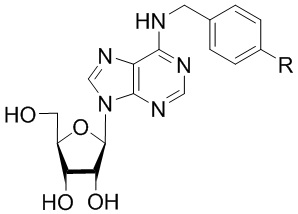
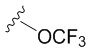
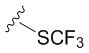
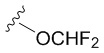
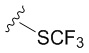
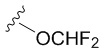
В частных вариантах воплощения изобретения R выбирается независимо и представляет собой -OCF3, -SCF3 или -OCHF2.
В частных вариантах воплощения настоящее изобретение включает соединения, выбранные из группы:
- N6-([4-циклопропилэтинил]бензил)аденозин;
- N6-([4-циклопентилэтинил]бензил)аденозин;
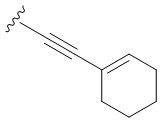
- N6-([4-(1-циклогексен-1-этинил)]бензил)аденозин;
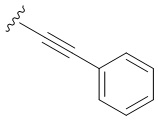
- N6-([4-фенилэтинил]бензил)аденозин;
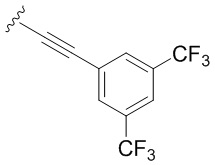
- N6-([4-(3,5-бис(трифторметил)фенил)этинил]бензил)аденозин;
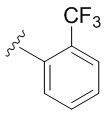
- N6-(((2'-трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин;
- N6-(((3'-трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин;
- N6-(((4'-трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин;
- N6-(4-трифторметоксибензил)аденозин;
- N6-(4-трифторметилтиобензил)аденозин;
- N6-(4-дифторметоксибензил)аденозин.
Данное изобретение также относится к применению соединения по изобретению в качестве ингибитора репродукции вируса, относящегося к роду Flavivirus.
В частных вариантах воплощения изобретения вирус представляет собой вирус денге, вирус лихорадки Западного Нила, вирус желтой лихорадки, вирус японского энцефалита, вирус киасанурской лесной болезни, вирус клещевого энцефалита, вирус омской геморрагической лихорадки, вирус Повассан.
Настоящее изобретение также относится к применению соединений по изобретению для получения фармацевтической композиции с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Flavivirus у пациента.
Изобретение также включает фармацевтическую композицию с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Flavivirus у пациента, содержащая эффективное количество соединения по изобретению.
В частных вариантах воплощения изобретения фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
В частных вариантах воплощения изобретения пациент представляет собой человека.
Данное изобретение также относится к способу ингибирования репродукции вируса, относящегося к роду Flavivirus, включающему введение пациенту терапевтически эффективного количества соединения по изобретению.
Изобретение также включает способ лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Flavivirus у пациента, включающий введение терапевтически или профилактически эффективного количества соединения по изобретению.
Настоящее изобретение также относится к применению соединений изобретения для получения средства для ингибирования репродукции вируса, относящегося к роду Flavivirus.
Настоящее изобретение включает также получение соединений и/или композиций по изобретению.
Подробное раскрытие изобретения
Определения (термины)
Для лучшего понимания настоящего изобретения ниже приведены некоторые термины, использованные в настоящем описании изобретения. Следующие определения применяются в данном документе, если иное не указано явно.
Использованные научные и технические термины, если это не оговорено отдельно, имеют значения, общепринятые в научной и технической литературе.
Термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Термин «и/или» означает один, несколько или все перечисленные элементы.
Также здесь перечисление числовых диапазонов по конечным точкам включает все числа, входящие в этот диапазон.
Род Flavivirus (семейство Flaviviridae) включает оболочечные РНК-вирусы (флавивирусы), многие из которых, такие как вирус клещевого энцефалита (ВКЭ), вирус денге, вирус Зика, вирус лихорадки Западного Нила (ВЗН), вирус желтой лихорадки (ВЖЛ), являются патогенными и вызывают ряд одноименных заболеваний, широко распространенных в различных регионах мира.
Термин «алкил» в настоящем документе, сам по себе или как часть другого заместителя, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, имеющие указанное число атомов углерода, в частности, термин «алкил» относится к группам, обычно имеющим от одного до пяти атомов углерода. Например, термин «C1-3-алкил» включает метил, этил, пропил, изопропил и т.д.
Термин «циклоалкил» в настоящем документе относится к группам, имеющим от трех до шести атомов углерода в моноциклической структуре. В качестве иллюстрации, циклоалкилы включают, но не ограничиваются, следующими радикалами: циклопропил, циклобутил, циклопентил, циклогексил.
Термин «циклоалкенил» в настоящем документе относится к группам, имеющим от трех до шести атомов углерода в моноциклической структуре и одну двойную связь. В качестве иллюстрации, циклоалкенилы включают, но не ограничиваются, следующими радикалами: циклопентенил, циклогексенил и т.д.
Термин «галоген» сам по себе или в части другого термина относится к атому фтора, хлора, брома или иода.
Термин «частично или полностью галогенированный» включает разветвленные или неразветвленные углеводородные цепи (сами по себе или как часть другого заместителя), в которых один или несколько атомов водорода замещены на галоген. Примеры галогеналкильных групп включают, но не ограничиваются, следующие группы: дифторметил, трифторметил, трихлорметил, -C(CF3)2CH3 и т.п.
Если не указано иное, все вхождения функциональных групп в структурах соединений по изобретению выбираются независимо.
Изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования.
Если не указано иначе, приведенные в материалах заявки структуры соединений также подразумевают и все стереоизомеры, то есть R- и S-изомеры для каждого хирального центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси соединений по изобретению, также являются предметом настоящего изобретения. Таким образом, настоящее изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%; более предпочтительно, >95% мольной чистоты), так же, как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например, путем получения диастереомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Кроме того, другой метод разделения включает синтез ковалентных диастереомерных производных путем реакции соединений изобретения с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Полученные диастереомеры можно разделить обычными способами, например, хроматографией, дистилляцией, кристаллизаций или сублимацией, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания или соли.
Соединения, составляющие суть данного изобретения, могут существовать в меченой радиоизотопом форме, т.е. указанные соединения могут содержать один или несколько атомов, чья атомная масса или массовое число отличается от атомной массы или массового числа наиболее распространенных природных изотопов. Радиоизотопы водорода, углерода, фосфора, хлора включают 3H, 14C, 32P, 35S и 36Cl, соответственно. Соединения данного изобретения, которые содержат такие радиоизотопы и/или другие радиоизотопы других атомов, находятся в сфере настоящего изобретения. Тритиевые, т.е. 3H, и углеродные, т.е. 14С, радиоизотопы являются особенно предпочтительными благодаря простоте приготовления и обнаружения.
Соединения настоящего изобретения, меченые радиоактивными изотопами, могут быть получены с помощью методов, хорошо известных специалистам в данной области. Меченые соединения могут быть получены с помощью процедур, описанных здесь, простой заменой немеченых реагентов соответствующими мечеными реагентами.
Соединения настоящего изобретения могут существовать в свободной форме или, если требуется, в виде фармацевтически приемлемой соли или другого производного (например, пролекарства). Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидроиодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, валериат и подобные. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и аминов, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкилсульфонаты и арилсульфонаты.
Соединения настоящего изобретения могут существовать в виде пролекарства. В контексте настоящей заявки термин «пролекарство» относится к предшественнику или производной форме соединения согласно изобретению, которое может обладать улучшенными свойствами, такими как лучшая растворимость, уменьшенная цитотоксичность или повышенная биодоступность, по сравнению с исходным соединением или лекарственным средством, и способно активироваться или превращаться в более активную исходную форму. “Группа пролекарства” означает группу, которая преобразуется посредством реакции с ферментом, желудочной кислотой или т.п. в физиологических условиях in vivo, с получением соединения общей формулы (I), т.е. группу, которая преобразуется с получением соединения общей формулы (I) посредством гидролиза или т.п., вызванного желудочной кислотой или т.п. Ее примеры включают соли аминогруппы в положении N6-пурина (см. экспериментальную часть), а также нуклеозид-5'-фосфаты, которые могут образовываться непосредственно в клетке в присутствии нуклеозидкиназ, с действием которых связан основной механизм противовирусной активности производных нуклеозидов (Yates, M. K., & Seley-Radtke, K. L. (2019). The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antiviral research, 162, 5-21; А.А. Зенченко, М.С. Дреничев, И.А. Ильичева, С.Н. Михайлов. (2021). Противовирусные и противомикробные производные нуклеозидов: структурные особенности и механизмы действия, 55 (6), 897-926).
Под «терапевтически эффективным количеством» подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Под «профилактически эффективным количеством» подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на профилактику заболеваний вирусной этиологии у субъекта, вызванных вирусом рода Flavivirus. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, методики введения препарата, комбинированного лечения с другими препаратами и т.п. Для профилактического лечения терапевтически или профилактически эффективное количество представляет собой то количество, которое будет эффективно в предотвращении вирусной инфекции.
Термин «пациент» (или «субъект») охватывает все виды млекопитающих, предпочтительно человека, которые используют соединения в рамках данного изобретения как путем самостоятельного введения, так и/или введения пациенту другим лицом для лечения и/или профилактики заболевания или медицинского состояния.
Термины «лечение», «терапия» охватывают лечение патологических состояний у млекопитающих, предпочтительно у человека, и включают: а) блокирование (приостановку) течения заболевания, б) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания.
Термин «профилактика», «предотвращение», «превентивная терапия» охватывает устранение факторов риска, а также профилактическое лечение субклинических стадий заболевания у человека, направленное на уменьшение вероятности возникновения клинических стадий заболевания. Пациенты для профилактической терапии отбираются на основе факторов, которые на основании известных данных влекут увеличение риска возникновения клинических проявлений заболевания по сравнению с остальным населением. К профилактической терапии относится (а) первичная профилактика, (б) постконтактная профилактика и (в) вторичная профилактика. Первичная профилактика, в частности, определяется как профилактический прием лекарственного средства лицами, выезжающими на территории с высоким риском заражения. Постконтактная профилактика определяется, в частности, как профилактическое лечение пациентов, вступивших в контакт с переносчиком заболевания, клиническая стадия заболевания у которых еще не наступила. Вторичная профилактика - это предотвращение повторного наступления того же или близкого клинического состояния заболевания или уменьшение риска персистирующей инфекции.
Термин «уменьшение риска» охватывает терапию, которая снижает частоту возникновения клинической стадии заболевания. Примерами уменьшения риска заболевания являются первичная, постконтактная и вторичная профилактика заболевания.
Осуществление изобретения
Примеры
Конкретные соединения по изобретению, раскрытые в настоящем документе, приводятся для целей иллюстрирования настоящего изобретения, чтобы изобретение могло быть более полно понято. В таблице 1 показаны частные примеры соединений по изобретению, которые могут быть получены методами, описанными в общих схемах синтеза, примерах и известных методах в данной области техники. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Соединения, приведенные ниже (табл. 1), не должны рассматриваться как единственные примеры в рамках настоящего изобретения, и никоим образом не ограничивают изобретение.
Общие способы синтеза
Ряд соединений по изобретению (3а-e, 3f-h) получали с помощью катализируемых палладием или его соединениями реакций кросс-сочетания с образованием углерод-углеродной связи. В качестве исходного соединения в данных реакциях использовался N6-(4-иодбензил)-2′,3′,5′-три-О-ацетиладенозин 2, который был получен согласно схеме 1.
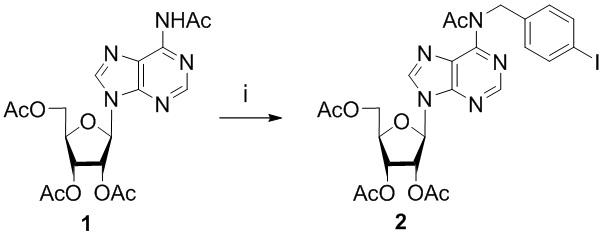
Схема 1. Синтез N6-(4-иодбензил)-2′,3′,5′-три-О-ацетиладенозина 2. Реагенты и условия: i. 4-иодобензилбромид, DBU, MeCN, комнатная температура, 48 ч.
Общая схема синтеза соединений 3а-e, 3f-h представлена на схеме 2 ниже.
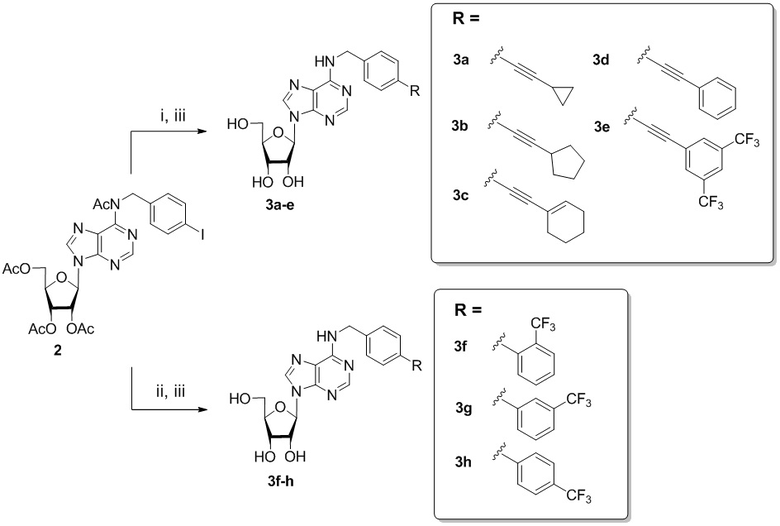
Схема 2. Синтез соединений по изобретению 3а-e, 3f-h. Реагенты и условия: i. R-С≡СН, Ph3P, PdCl2, CuI, Et3N, ТГФ, 50-60 °С, 72 ч; ii. R-B(OH)2, Ph3P, PdCl2, K2CO3, H2O, ТГФ, 70 °С, 4-6 ч; iii. 4M PrNH2/MeOH комнатная температура, 24 ч.
Ряд соединений по изобретению (5a-c) получали c использованием реакции нуклеофильного замещения атома галогена в 6-м положении пуринового гетероциклического основания в присутствии первичных аминов и основания Хунига. В качестве исходного соединения использовали коммерчески доступный 6-хлоро-9-(β-D-рибофуранозил)-9H-пурин 4. Схема синтеза соединений 5a-c представлена на схеме 3.
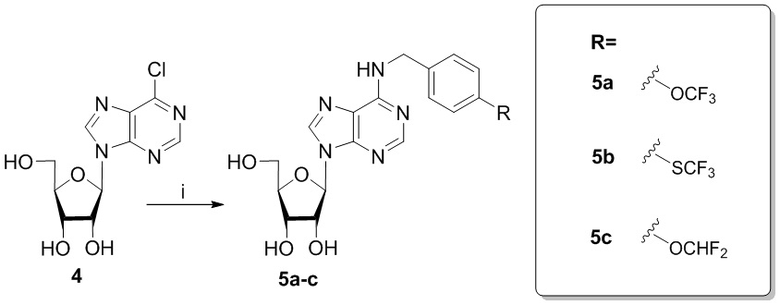
Схема 3. Синтез соединений по изобретению 3а-e, 3f-h. Реагенты и условия: i. RNH2, DIPEA, n-BuOH, 90°C, 24-48 ч.
Растворители и материалы имели квалификацию «х.ч.» и использовались без дополнительной очистки. Колоночную хроматографию проводили на силикагеле (Kieselgel 60 Merck, Германия, 0,063-0,200 мм). ТСХ выполняли на Alugram SIL G/UV254 (Macherey-Nagel, Дюрен, Германия) с УФ-визуализацией. Спектры ЯМР 1Н, 13С (с полной развязкой протонов) записаны на приборе Bruker AVANCE II 300 (Карлсруэ, Германия) при 303 К. Спектры ЯМР 1Н записаны на частоте 300.1 МГц, спектры ЯМР 13С - на 75,5 МГц. Химические сдвиги в м.д. измеряли относительно сигналов остаточного растворителя в качестве внутренних стандартов (ДМСО-d6, 1H: 2,50 м.д., 13C: 39,5 м.д.). Константы спин-спинового взаимодействия (J) даны в Гц.
Масс-спектры высокого разрешения (HRMS) регистрировали на приборе Bruker Daltonics micrOTOF-Q II с использованием ионизации электрораспылением (ESI). Измерения проводились в режимах положительных и отрицательных ионов. Напряжение капилляра интерфейса: 3500 В; диапазон масс от m/z 50 до 3000; внешняя калибровка (Electrospray Calibrant Solution, Fluka); давление распылителя: 2,0 бар; скорость потока: 3 мкл/мин; сухой газ: азот (6 л/мин); температура интерфейса: 250°C. Образцы вводили в камеру масс-спектрометра из системы ВЭЖХ Agilent 1260, оснащенной колонкой Agilent Poroshell 120 EC-C18 (3,0 × 50 мм; 2,7 мкм); скорость потока 400 мкл/мин; пробы вводили из раствора ацетонитрил-вода (1:1) и колонку элюировали градиентом концентраций ацетонитрила (А) в воде (Б) при следующих параметрах: 0-15% А в течение 6.0 мин, 15% -85% А в течение 1.5 мин, 85%-0% А в течение 0.1 мин, 0% А в течение 2.4 мин.
Исходное соединение N6-ацетил, N6-(4-иодобензил)-2',3',5'-три-O-ацетиладенозин (1) синтезировали путем региоселективного N6-алкилирования N6-ацетил-2',3',5'-три-O-ацетиладенозина 4-иодобензилбромидом в присутствии 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) в соответствии с ранее опубликованными методиками:
- V. I. Tararov, A. Tijsma, S. V. Kolyachkina, V. E. Oslovsky, J. Neyts, M. S. Drenichev, P. Leyssen, S. N. Mikhailov. Chemical modification of the plant isoprenoid cytokinin N6-isopentenyladenosine yields a selective inhibitor of human enterovirus 71 replication. Eur. J. Med. Chem., 2015, 90, 406-413;
- M. S. Drenichev, V. E. Oslovsky, V. I. Tararov, S. N. Mikhailov. Synthesis of N6-Substituted Adenosines as Cytokinin Nucleosides. Curr. Protoc. Nucleic Acid Chem., 2018, 72(1), 14-15.
Синтез промежуточных соединений
N6-Ацетил, N6-(4-иодобензил)-2',3',5'-три-O-ацетиладенозин (2)
Смесь N6-ацетил-2',3',5'-три-О-ацетиладенозина 1 (300 мг, 0,683 ммоль) и 4-иодбензилбромида (710 мг, 2,39 ммоль) растворяли в MeCN (7 мл), затем добавляли DBU (0,357 мл, 2,39 ммоль); раствор выдерживали 48 ч при температуре окружающей среды. Реакцию контролировали с помощью ТСХ (силикагель, CH2Cl2:EtOH - 97:3 (%)). Затем реакционную смесь нейтрализовали 0,1 М HCl (0,71 мл), разбавляли CH2Cl2 и промывали солевым раствором (2 × 50 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме практически досуха. Остаток выливали на хроматографическую колонку с силикагелем. Колонку промывали CH2Cl2, продукт элюировали CH2Cl2:EtOH - 98:2 (%), получая соединение 2 в виде бесцветной пены. Выход составил 278 мг (85%). Rf = 0,72 (CH2Cl2:EtOH-97:3 (%)). 1Н-ЯМР (300,1 МГц, ДМСО-d6): δ = 1,97 (СН3), 2,05 (СН3), 2,11 (СН3), 2,23 (СН3), 4,28 (дд, 1Н, J5′b,5′a = -12,8 , J5'b,4' = 6,4, H-5'b), 4,36-4,46 (м, 2H, H-4', H-5'a), 5,32 (ш. с, 2H, CH2), 5,67 (дд , 1H, J3',,4' = 5,3 Гц, J3',2' = 5,7 Гц, H-3'), 6,02 (дд, 1H, J2',3' = 5,7 Гц, J2',1' = 4,9 Гц , H-2′), 6,35 (д, 1H, J1′,2′ = 4,9 Гц, H-1′), 7,07 (д, 2Н, J3-2 = J5-6=8,3 Гц, Н3-Ph, Н5 -Ph), 7,59 (д, 2Н, J2-3 = J6-5=8,3 Гц, J2-CH2 = J6-CH2= 1,8 Гц, Н2-Ph, Н6-Ph), 8,82 (с, 1H, H-2 ), 8,84 (с, 1H, H-8). ЯМР 13С (75,5 МГц, ДМСО-d6): δ = 20,20 (СН3), 20,32 (СН3), 20,41 (СН3), 23,87 (СН3), 49,10 (NCH2-), 62,67 (С5′), 69,83 (С3′), 72,14 (C2′), 79,52 (C4′), 86,12 (C1′), 92,75 (C4-Ph), 126,70 (C5), 129,35 (C2-Ph, C6-Ph), 136,97 (C3-Ph, C5 -Ph), 137,68 (C1-Ph), 144,89 (C8), 151,86 (C4), 152,37 (C2), 152,49 (C6), 169,24 (C=O), 169,38 (C=O), 169,98 (C=O ), 170,89 (С=О).
Получение соединений по изобретению
N6-([4-Циклопропилэтинил]бензил)аденозин (3a)
Смесь 24 мг (0,138 ммоль) PdCl2 и 56,3 мг (0,276 ммоль) Ph3P растворяли в 10 мл ДМФА в колбе Шленка и интенсивно перемешивали в течение 10 минут при комнатной температуре. Затем добавляли исходный N6-ацетил, N6-(4-иодобензил)-2',3',5'-три-О-ацетиладенозин 2 (300 мг, 0,46 ммоль) и интенсивно перемешивали в течение 15 минут. К реакционной смеси добавляли этинилциклопропан (0,117 мл, 1,38 ммоль), сокатализатор CuI (8,76 мг, 0,046 ммоль), Et3N (4 мл), из колбы Шленка откачивали воздух и заполняли N2. Раствор перемешивали при 70°С в течение 72 часов. Реакцию контролировали с помощью ТСХ (CH2Cl2:EtOH - 97:3 (%)). Затем реакционную смесь упаривали в вакууме, а остаток разбавляли CH2Cl2 (2 х 20 мл) и промывали насыщенным раствором NaCl в воде (100 мл). Органический слой отделяли, сушили над безводным Na2SO4 и упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле. Колонку промывали градиентом CH2Cl2:EtOH, увеличивая полярность от CH2Cl2:EtOH (99:1 (%)) до CH2Cl2:EtOH (97:3 (%)). Продукт элюировали CH2Cl2:EtOH-96:4 (%).
Промежуточный N6-ацетил, N6-(4-циклопропилэтинил]бензил)-2',3',5'-три-O-ацетиладенозин обрабатывали 4 М раствором PrNH2 в МеОН (6,8 мл). Через 24 часа смесь упаривали в вакууме и остаток очищали колоночной хроматографией на силикагеле. Колонку промывали CH2Cl2:EtOH-97:3 (%), а затем продукт элюировали CH2Cl2:EtOH-90:10 (%) с получением 3a в виде белого порошка. Выход составил 116 мг (60% за две стадии). Rf = 0,33 (CH2Cl2:EtOH-93:7 (%)). Т.пл. 145-148°С. 1H-ЯМР (300,1 МГц, ДМСО-d6): δ = 0,65-0,73 (м, 2H, циклопроп), 0,81-0,89 (м, 2H, циклопроп), 1,51 (тт, 1H, JCH-CH2= 8,5 Гц, JCH -CH2= 5,0 Гц, H1-циклопроп), 3,55 (ддд, 1H, J5'b5'a = -12,0 Гц, J5'b4' = 3,4 Гц, J5'b-OH = 7,0 Гц, H-5'b) , 3,67 (ddd, 1H, J5'a, 5'b = -12,0 Гц, J5'a, 4' = 3,4 Гц, J5'a, OH = 4,6 Гц, H5'a), 3,96 (к, 1H, J4 ′3′ = J4′5′а =J4′5′b = 3,4 Гц, H-4′), 4,15 (тд, 1H, J3′-OH = 4,7 Гц, J3′2′ = 5,0 Гц, J3′4′ = 3,4 Гц, H-3′), 4,61 (ддд, 1H, J2′-OH = 6,2 Гц, J2′1′ = 6,1 Гц, J2′3′ = 5,0 Гц, H-2′), 4,7 (ш. с, 2H, СH2), 5.14 (д, 1H, J3'-OH = 4.7 Гц, 3'-OH), 5.33 (дд, 1H, J5'a-OH = 4.6 Гц, J5'b-OH = 7.0 Гц , 5'-OH), 5,41 (д, 1H, J2'-OH = 6,2 Гц, 2'-OH), 5,89 (д, 1H, J1'2' = 6,1 Гц, H-1'), 7,27 (с , 4Н, Н2-Ph, Н3-Ph, Н5-Ph, Н6-Ph), 8,19 (с, 1H, H-2), 8,37 (с, 1H, H-8), 8,42 (уш с, 1H, NH ). ЯМР 13С (75,5 МГц, ДМСО-d6): δ = -0,29 (С1-циклопроп), 8,29 (С2-циклопроп, С3-циклопроп), 42,63 (NCH2-), 61,62 (С5'), 70,60 (С3') , 73,45 (С2'), 75,53 (-С≡С-), 85,86 (С4'), 87,90 (С1'), 93,32 (-С≡С-), 119,72 (С5), 121,40 (С4-Ph), 127,18 (C2-Ph, C6-Ph), 131,14 (C3-Ph, C5-Ph), 139,93 (C1-Ph), 148,47 (C4), 152,29 (C2), 154,46 (C6). HRМС: м/z [M+H]+ рассчитано C22H24N5O4+ 422,1822, найдено 422,1823.
N6-([4-Циклопентилэтинил]бензил)аденозин (3b)
Следуя методике получения соединения 3a, проводят конденсацию соединения 2 (300 мг, 0,46 ммоль) с этинилциклопентаном (0,16 мл, 1,38 ммоль) в присутствии Ph3P (56,3 мг, 0,276 ммоль), PdCl2 (24 мг, 0,138 ммоль), CuI (8,76 мг, 0,046 ммоль) и Et3N (2 мл, 0,01 ммоль) в ДМФА (7 мл) в течение 48 ч при 70°С с последующей деблокировкой в 4М растворе PrNH2 в МеОН (6,5 мл) при комнатной температуре дали 3b в виде белого порошка. Общий выход составил 160 мг (77%). Rf = 0,34 (CH2Cl2:EtOH-93:7 (%)). М.п. 149-152 оС. 1Н-ЯМР (300,1 МГц, ДМСО-d6): δ = 1,51-1,63 (м, 4Н, Н-циклопент), 1,64-1,75 (м, 2Н, Н-циклопент), 1,88-2,01 (м, 2Н, Н-циклопент). циклопент), 2,82 (п, 1H, JCH-CH2= 7,5 Гц, H1-циклопент), 3,51 (ддд, 1H, J5'b5'a = -12,0 Гц, J5'b-4' = 3,4 Гц, J5'b -OH = 7,0 Гц, H-5'b), 3,63 (ддд, 1H, J5'a,5'b = -12,0 Гц, J5'a,4' = 3,4 Гц, J5'a,OH = 4,6 Гц, H5'a), 3,92 (к, 1H, J4'3' = J4'5'а = J4'5'b = 3,4 Гц, H-4'), 4,11 (ддд, 1H, J3'-OH = 4,7 Гц , J3′2′ = 5,0 Гц, J3′4′ = 3,4 Гц, H-3′), 4,58 (ddd, 1H, J2′-OH = 6,2 Гц, J2′1′ = 6,1 Гц, J2′3′ = 5,0 Гц, H-2'), 4,66 (ш.с, 2H, СH2), 5,11 (д, 1H, J3'-OH = 4,7 Гц, 3'-OH), 5,29 (дд, 1H, J5'a-OH = 4,6 Гц, J5'b-OH = 7,0 Гц, 5'-OH), 5,37 (д, 1H, J2'-OH = 6,2 Гц, 2'-OH), 5,85 (д, 1H, J1'2' = 6,1 Гц, H-1′), 7,24 (с, 4Н, Н2-Ph, Н3-Ph, Н5-Ph, Н6-Ph), 8,15 (с, 1H, H-2), 8,33 (с, 1H, H- 8), 8,38 (широкий с, 1H, NH). 13С-ЯМР (75.5 МГц, ДМСО-d6): δ = 24.55 (С3-циклопент, С4-циклопент), 29.98 (С1-циклопент), 33.44 (С2-циклопент, С5-циклопент), 42.66 (NCH2-), 61.62 (С5'), 70,60 (С3'), 73,45 (С2'), 79,94 (-С≡С-), 85,86 (С4'), 87,91 (С1'), 94,17 (-С≡С-), 119,76 (С5 ), 121,54 (C4-Ph), 127,18 (C2-Ph, C6-Ph), 131,05 (C3-Ph, C5-Ph), 139,95 (C1-Ph), 148,54 (C4), 152,29 (C2), 154,46 ( С6). HRМС: м/z [M+H]+ рассчитано C24H28N5O4+ 450,2132, найдено 450,2136.
N6-([4-(1-Циклогексен-1-этинил)]бензил)аденозин (3c)
Следуя процедуре получения соединения 3а, проводят конденсацию соединения 2 (300 мг, 0,46 ммоль) с 1-этинилциклогексеном (0,162 мл, 1,38 ммоль) в присутствии Ph3P (56,3 мг, 0,276 ммоль), PdCl2 (24 мг, 0,138 ммоль), CuI (8,76 мг, 0,046 ммоль) и Et3N (4 мл) в ДМФА (10 мл) в течение 72 ч при 70°С с последующей деблокировкой в 4М растворе PrNH2 в МеОН (6,4 мл) при комнатной температуре дали 3c в виде белого порошка. Общий выход составил 136 мг (64%). Rf = 0,25 (CH2Cl2:EtOH-93:7 (%)). М.п. 178-181 °С. 1Н-ЯМР (300,1 МГц, ДМСО-d6): δ = 1,47-1,67 (м, 4Н, Н-циклогекс), 2,04-2,19 (м, 4Н, Н-циклогекс), 3,55 (ддд, 1Н, J5'b5' a = -12,0 Гц, J5'b4' = 3,3 Гц, J5'b-OH = 7,0 Гц, H-5'b), 3,67 (ddd, 1H, J5'a, 5'b = -12,0 Гц, J5' a,4′ = 3,3 Гц, J5′a,OH = 4,6 Гц, H5′a), 3,96 (кв, 1H, J4′3′ = J4′5′а =J4′5′b = 3,3 Гц, H- 4'), 4,15 (ддд, 1H, J3'-OH = 4,7 Гц, J3'2' = 5,0 Гц, J3'4' = 3,3 Гц, H-3'), 4,61 (ддд, 1H, J2'-OH = 6,2 Гц, J2′1′ = 6,1 Гц, J2′3′ = 5,0 Гц, H-2′), 4,72 (уш.с, 2H, СH2), 5,15 (д, 1H, J3′-OH = 4,7 Гц , 3'-OH), 5,33 (дд, 1H, J5'a-OH = 4,6 Гц, J5'b-OH = 7,0 Гц, 5'-OH), 5,41 (д, 1H, J2'-OH = 6,2 Гц , 2'-OH), 5,89 (д, 1H, J1'2' = 6,1 Гц, H-1'), 6,11-6,19 (м, 1H, H-ch), 7,28-7,36 (м, 4Н, H- Ph), 8,20 (с, 1H, H-2), 8,38 (с, 1H, H-8), 8,44 (широкий с, 1H, NH). ЯМР 13С (75.5 МГц, ДМСО-d6): δ = 20.99 (С4-циклогекс), 21.82 (С5-циклогекс), 25.20 (С6-циклогекс), 28.79 (С3-циклогекс), 42.70 (NCH2-), 61.65 ( C5'), 70,64 (C3'), 73,50 (C2'), 85,90 (C4'), 86,81 (-C≡C-), 87,96 (C1'), 90,96 (-C≡C-), 119,80 (C5) , 120,07 (C1-циклогекс), 121,08 (C4-Ph), 127,31 (C2-Ph, C6-Ph), 131,02 (C3-Ph, C5-Ph), 135,12 (C2-циклогекс), 139,98 (C1-Ph) , 140,29 (С8), 148,49 (С4), 152,33 (С2), 154,49 (С6). HRМС: м/z [M+H]+ рассчитано C25H27N5O4+ 462,2136, найдено 462,2135.
N6-([4-Фенилэтинил]бензил)аденозин (3d)
Следуя методике получения соединения 3a, проводят конденсацию соединения 2 (300 мг, 0,46 ммоль) с этинилбензолом (0,152 мл, 1,38 ммоль) в присутствии Ph3P (56,3 мг, 0,276 ммоль), PdCl2 (24 мг, 0,138 ммоль), CuI (8,76 мг, 0,046 ммоль) и Et3N (4 мл) в ДМФА (10 мл) в течение 72 ч при 70°С с последующей деблокировкой в 4М растворе PrNH2 в МеОН (6,4 мл) при комнатной температуре. В ходе реакции наблюдали образование осадка. Осадок отфильтровывали, промывали холодным МеОН, получая 3d в виде белого порошка. Общий выход составил 130 мг (62%). Rf = 0,33 (CH2Cl2:EtOH - 93:7 (%)). М.п. 229-232 °С. 1H-ЯМР (300,1 МГц, ДМСО-d6): δ = 3,56 (ддд, 1H, J5′b5′a = -12,0 Гц, J5′b4′ = 3,5 Гц, J5′b-OH = 7,0 Гц, H-5 ′b), 3,68 (ddd, 1H, J5′a,5′b = -12,0 Гц, J5′a,4′ = 3,5 Гц, J5′a,OH = 4,6 Гц, H5′a), 3,97 (q, 1H, J4′3′ = J4′5′а =J4′5′b = 3,5 Гц, H-4′), 4,16 (ddd, 1H, J3′-OH = 4,7 Гц, J3′2′ = 5,0 Гц, J3'4' = 3,5 Гц, H-3'), 4,62 (ддд, 1H, J2'-OH = 6,2 Гц, J2'1' = 6,1 Гц, J2'3' = 5,0 Гц, H-2'), 4,75 (ш. с, 2H, СH2), 5,15 (д, 1H, J3'-OH = 4,7 Гц, 3'-OH), 5,33 (дд, 1H, J5'a-OH = 4,6 Гц, J5'b-OH = 7,0 Гц, 5'-OH), 5,42 (д, 1H, J2'-OH = 6,2 Гц, 2'-OH), 5,90 (д, 1H, J1'2' = 6,1 Гц, H-1'), 7,35-7,44 (м, 5Н, H-Ph'), 7,46-7,56 (м, 4H, H-Ph), 8,21 (с, 1H, H-2), 8,39 (с, 1H, H-8), 8,48 (широкий с, 1H, NH). 13С-ЯМР (75,5 МГц, ДМСО-d6): δ = 42,71 (NCH2-), 61,65 (С5′), 70,64 (С3′), 73,49 (С2′), 85,90 (С4′), 87,94 (С1′), 88,98 (-C≡C-), 89,37 (-C≡C-), 119,77 (C5), 120,45 (C6-Ph), 122,33 (C1-Ph'), 127,40 (C2-Ph, C6-Ph), 128,72 (С3-Ф', С4-Ф', С5-Ф'), 131,30 (С3-Ф, С5-Ф, С2-Ф', С6-Ф'), 140,01 (С1-Ф), 140,94 (С8), 148,57 (С4), 152,34 (С2), 154,47 (С6). HRМС: м/z [M+H]+ рассчитано C25H29N5O4+ 458,1821, найдено 458,1823.
N6-([4-(3,5-бис(Трифторметил)фенил)этинил]бензил)аденозин (3e)
Следуя методике получения соединения 3a, проводят конденсацию соединения 2 (300 мг, 0,46 ммоль) с 1-этинил-3,5-бис(трифторметил)бензолом (0,244 мл, 1,38 ммоль) в присутствии Ph3P ( 56,3 мг, 0,276 ммоль), PdCl2 (24 мг, 0,138 ммоль), CuI (8,76 мг, 0,046 ммоль) и Et3N (4 мл, 0,01 ммоль) в ДМФА (10 мл) в течение 48 ч при 70°С с последующей деблокировкой в 4М PrNH2 в растворе MeOH (5,2 мл) при комнатной температуре дает 3е в виде белого порошка. Общий выход составил 210 мг (77%). Rf = 0,26 (CH2Cl2:EtOH- 93:7 (%)). Т.пл. 168-171 °С. 1H-ЯМР (300,1 МГц, ДМСО-d6): δ = 3,56 (ддд, 1H, J5′b5′a = -12,0 Гц, J5′b4′ = 3,4 Гц, J5′b-OH = 7,0 Гц, H-5 ′b), 3,68 (ddd, 1H, J5′a,5′b = -12,0 Гц, J5′a,4′ = 3,4 Гц, J5′a,OH = 4,6 Гц, H5′a), 3,97 (q, 1H, J4′3′ = J4′5′а =J4′5′b = 3,4 Гц, H-4′), 4,15 (ddd, 1H, J3′-OH = 4,7 Гц, J3′2′ = 5,0 Гц, J3'4' = 3,4 Гц, H-3'), 4,62 (ддд, 1H, J2'-OH = 6,2 Гц, J2'1' = 6,1 Гц, J2'3' = 5,0 Гц, H-2'), 4,77 (шир. с, 2H, СH2), 5,17 (д, 1H, J3′-OH = 4,7 Гц, 3′-OH), 5,35 (дд, 1H, J5′a-OH = 4,6 Гц, J5′b-OH = 7,0 Гц, 5'-OH), 5,43 (д, 1H, J2'-OH = 6,2 Гц, 2'-OH), 5,90 (д, 1H, J1'2' = 6,1 Гц, H-1'), 7,42 (д, 2Н, J2H-3H = J6H-5H=8,2 Гц, H2-Ph, H6-Ph), 7,58 (д, 2Н, J3H-2H = J5H-6H=8,2 Гц, H3-Ph, 5H-Ph ), 8,13 (шир. с, 1H, H4-Ph'), 8,21 (с, 1H, H-2), 8,26 (шир. с, 2H, H2-Ph', H6-Ph'), 8,39 (с, 1H, H-8), 8,50 (широкий с, 1H, NH). 13С-ЯМР (75,5 МГц, ДМСО-d6): δ = 42,74 (NCH2-), 61,63 (С5′), 70,61 (С3′), 73,49 (С2′), 85,88 (С4′), 86,11 (-С≡С -), 87,92 (C1'), 92,84 (-C≡C-), 117,47 (C5), 119,36 (Ph), 121,86 (Ph), 125,12 (Ph), 127,46 (Ph), 130,81 (к, 1JC-F = 33,2 Гц, 2CF3), 131,66 (Ф), 131,84 (Ф), 140,01 (С8), 141,95 (Ф), 148,54 (С4), 152,32 (С2), 154,47 (С6). HRМС: м/z [M+H]+ рассчитано C27H22N5O4+ 594,1566, найдено 594,1570.
N6-(((2'-Трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин (3f)
Смесь исходного соединения 2 (300 мг, 0,49 ммоль), PdCl2 (17,4 мг, 0,098 ммоль) и Ph3P (40 мг, 0,196 ммоль) растворяли в колбе ДМФА (7 мл) и интенсивно перемешивали в течение 10 минут при комнатной температуре. Затем добавляли 2-(трифторметил)фенилбороновую кислоту (93 мг, 0,49 ммоль) и K2CO3 (135 мг, 0,98 ммоль) в воде (2,5 мл). Раствор перемешивали при 70°С в течение 4 часов. Реакцию контролировали с помощью ТСХ (CH2Cl2:EtOH-97:3 (%)). Затем реакционную смесь упаривали в вакууме, а остаток разбавляли хлористым метиленом (2*25 мл) и промывали насыщенным водным раствором NaCl (100 мл). Органический слой отделяли, сушили над безводным Na2SO4 и упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле. Колонку промывали градиентом CH2Cl2:EtOH, увеличивая полярность от CH2Cl2:EtOH (99:1 (%)) до CH2Cl2:EtOH (97:3 (%)). Продукт элюировали CH2Cl2:EtOH-96:4 (%).
Промежуточный N6-((2'-(трифторметил)-[1,1'-бифенил]-4-ил)метил), N6-ацетил-2',3',5'-три-O-ацетиладенозин обрабатывали с 4 М раствором PrNH2 в МеОН (6 мл). Через 24 часа смесь упаривали в вакууме и остаток очищали колоночной хроматографией на силикагеле. Колонку промывали CH2Cl2:EtOH-97:3 (%), продукт элюировали CH2Cl2:EtOH-90:10 (%) с получением 3f в виде белого порошка. Выход 3f составил 177 мг (72% за две стадии). Rf = 0,17 (CH2Cl2:EtOH-93:7 (%)). М.п. 144-148°С. 1H-ЯМР (300,1 МГц, ДМСО-d6): δ = 3,56 (ддд, 1H, J5′b5′a = -12,0 Гц, J5′b4′ = 3,3 Гц, J5′b-OH = 7,0 Гц, H-5 ′b), 3,68 (ddd, 1H, J5′a,5′b = -12,0 Гц, J5′a,4′ = 3,3 Гц, J5′a,OH = 4,6 Гц, H5′a), 3,96 (q, 1H, J4′3′ = J4′5′а =J4′5′b = 3,4 Гц, H-4), 4,15 (ddd, 1H, J3′-OH = 4,7 Гц, J3′2′ = 5,0 Гц, J3 ′4′ = 3,4 Гц, H-3′), 4,55-4,63 (м, 2H, H-2'), 4,80 (уш.с, 2H, СH2), 5,17 (уш.с, 1H, 3′-OH) перекрывается с 5'-ОН и 2'-ОН), 5,42 (ш. с, 2Н, 5'-ОН, 2'-ОН перекрывается с 3'-ОН), 5,90 (д, 1Н, J1'2' = 6,1 Гц, H-1′), 7,14 (д, 1H, J3H-4H=8,1 Гц, H3-Ph'), 7,24 (д, 2Н, J2H-3H = J6H-5H=8,2 Гц, H2-Ph, H6 -Ph), 7,39 (д, 2H, J3H-2H = J5H-6H=8,2 Гц, H3-Ph, H5-Ph), 7,55-7,72 (м, 2H, H4-Ph', H5-Ph'), 7,81 (д, 1Н, J6H-5H = 7,5 Гц, H6-Ph'), 8,23 (с, 1H, H-2), 8,39 (с, 1H, H-8), 8,50 (уш. с, 1H, NH). 13С-ЯМР (75,5 МГц, ДМСО-d6): δ = 42,52 (NCH2-), 61,63 (С5′), 70,62 (С3′), 73,48 (С2′), 85,87 (С4′), 87,95 (С1′), 119,79 (C5), 125,60 (Ph), 125,99 (Ph), 126,59 (Ph), 127,21 (к, 1JC-F = 31,2 Гц, CF3), 127,89 (Ph), 128,57 (Ph), 129,50 (Ph), 132,18 (Ph), 136,91 (Ph), 137,65 (Ph), 139,59 (Ph), 139,95 (C8), 148,52 (C4), 152,34 (C2), 154,55 (C6). HRМС: м/z [M+H]+ рассчитано C24H23F3N5O4+ 502,1697, найдено 502,1697.
N6-(((3'-Трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин (3g)
Следуя методике получения соединения 3f, проводят конденсацию 2 (300 мг, 0,49 ммоль) с 3-(трифторметил)фенилбороновой кислотой (93 мг, 0,49 ммоль) в присутствии Ph3P (40 мг, 0,196 ммоль), PdCl2 (17,4 мг, 0,098 ммоль) и K2CO3 (135 мг, 0,98 ммоль) в смеси ДМФА/H2O (7:2,5 (v/v)) в течение 4 ч при 70°С с последующей деблокировкой в 4М растворе PrNH2 в МеОН (6 мл) при комнатной температуре давал 3g в виде белого порошка. Общий выход составил 150 мг (61%). Rf = 0,33 (CH2Cl2:EtOH- 93:7 (%)). М.п. 179-182оС. 1H-ЯМР (300,1 МГц, ДМСО-d6): δ = 3,55 (ддд, 1H, J5′b5′a = -12,0 Гц, J5′b4′ = 3,4 Гц, J5′b-OH = 7,0 Гц, H-5 ′b), 3,68 (ddd, 1H, J5′a,5′b = -12,0 Гц, J5′a,4′ = 3,4 Гц, J5′a,OH = 4,6 Гц, H5′a), 3,96 (q, 1H, J4′3′ = J4′5′а =J4′5′b = 3,4 Гц, H-4′), 4,15 (ddd, 1H, J3′-OH = 4,7 Гц, J3′2′ = 5,0 Гц, J3'4' = 3,4 Гц, H-3'), 4,62 (ддд, 1H, J2'-OH = 6,2 Гц, J2'1' = 6,0 Гц, J2'3' = 5,0 Гц, H-2'), 4,78 (шир. с, 2H, СH2), 5,16 (д, 1H, J3′-OH = 4,6 Гц, 3′-OH), 5,33 (дд, 1H, J5′a-OH = 4,6 Гц, J5′b-OH = 7,0 Гц, 5'-OH), 5,42 (д, 1H, J2'-OH = 6,2 Гц, 2'-OH), 5,90 (д, 1H, J1'2' = 6,0 Гц, H-1'), 7,46 (д, 2Н, J4'Ph-5'Ph = J6'Ph-5'Ph=7,8 Гц, Н4-Ph', Н6-Ph'), 7,62-7,74 (м, 4H, Ph), 7,88-7,99 (м, 2H, H2-Ph', H5-Ph'), 8,22 (с, 1H, H-2), 8,39 (с, 1H, H-8), 8,51 (широкий с, 1H, NH). 13С-ЯМР (75,5 МГц, ДМСО-d6): δ = 42,61 (NCH2-), 61,64 (С5′), 70,63 (С3′), 73,50 (С2′), 85,89 (С4′), 87,94 (С1′), 119,82 (C5), 123,36 (к, 1JC-F = 35,5 Гц, CF3), 123,8 (Ph), 122,92 (Ph), 126,86 (Ph), 127,85 (Ph), 129,52 (Ph), 129,99 (Ph), 130,65 (Ph), 136,93 (Ph), 139,97 (C8), 140,24 (Ph), 141,08 (Ph), 148,53 (C4), 152,34 (C2), 154,49 (C6). HRМС: м/z [M+H]+ рассчитано C24H23F3N5O4+ 502,1697, найдено 502,1697.
N6-(((4'-Трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин (3h)
Следуя процедуре получения соединения 3f, проводят конденсацию 2 (300 мг, 0,49 ммоль) с 4-(трифторметил)фенилбороновой кислотой (93 мг, 0,49 ммоль) в присутствии Ph3P (40 мг, 0,196 ммоль), PdCl2 (17,4 мг, 0,098 ммоль) и K2CO3 (135 мг, 0,98 ммоль) в смеси ДМФA/H2O (7:2,5 (v/v)) в течение 8 ч при 70°С с последующей деблокировкой в 4М растворе PrNH2 в МеОН (6 мл) при температуре окружающей среды давал 3h в виде белого порошка. Общий выход составил 167 мг (68%). Rf = 0,32 (CH2Cl2:EtOH - 93:7 (%)). М.п. 195-198 оС. 1H-ЯМР (300,1 МГц, ДМСО-d6): δ = 3,55 (ддд, 1H, J5′b5′a = -12,0 Гц, J5′b4′ = 3,5 Гц, J5′b-OH = 7,0 Гц, H-5 ′b), 3,67 (ddd, 1H, J5′a,5′b = -12,0 Гц, J5′a,4′ = 3,5 Гц, J5′a,OH = 4,6 Гц, H5′a), 3,96 (q, 1H, J4′3′ = J4′5′а =J4′5′b = 3,5 Гц, H-4′), 4,15 (ddd, 1H, J3′-OH = 4,5 Гц, J3′2′ = 5,0 Гц, J3'4' = 3,5 Гц, H-3'), 4,62 (ддд, 1H, J2'-OH = 6,2 Гц, J2'1' = 6,1 Гц, J2'3' = 5,0 Гц, H-2'), 4,77 (шир. с, 2H, СH2), 5,18 (д, 1H, J3′-OH = 4,5 Гц, 3′-OH), 5,36 (дд, 1H, J5′a-OH = 4,6 Гц, J5′b-OH = 7,0 Гц, 5'-OH), 5,44 (д, 1H, J2'-OH = 6,2 Гц, 2'-OH), 5,90 (д, 1H, J1'2' = 6,1 Гц, H-1'), 7,47 (д, 2Н, J3Ph-2Ph = J5Ph-6Ph=8,2 Гц, Н3-Ph, Н5-Ph), 7,67 (д, 2H, J2Ph-3Ph = J6Ph-5Ph=8,2 Гц, H2-Ph, H6- Ph), 7,79 (д, 2H, J2'Ph-3'Ph = J6'Ph-5'Ph=8,4 Гц, H2-Ph', H6-Ph'), 7,86 (д, 2H, J3'Ph-2 'Ph = J5'Ph-6'Ph=8,4 Гц, H3-Ph', H5-Ph'), 8,22 (с, 1H, H-2), 8,40 (с, 1H, H-8), 8,53 (ш. с, 1H, NH). 13С-ЯМР (75,5 МГц, ДМСО-d6): δ = 42,47 (NCH2-), 61,63 (С5′), 70,62 (С3′), 73,48 (С2′), 85,88 (С4′), 87,92 (С1′), 119,78 (C5), 125,70 (Ph), 125,74 (Ph), 126,85 (к, 1JC-F = 35,0 Гц, CF3), 126,94 (Ph), 127,29 (Ph), 127,84 (Ph), 128,18 (Ph), 129,50 (Ph), 136,92 (Ph), 139,98 (C8), 140,47 (Ph), 144,02 (Ph), 148,51 (C4), 152,31 (C2), 154,42 (C6). HRМС: м/z [M+H]+ рассчитано C24H23F3N5O4+ 502,1697, найдено 502,1697.
N6-(4-трифтометоксибензил)аденозин (5a)
6-Хлоро-9-(β-D-рибофуранозил)-9H-пурин 4 (160 мг, 0.56 ммоль) растворяли в 7 мл н-бутанола. Затем к полученной смеси добавляли 4-трифторметоксибензиламин (428 мкл, 2.8 ммоль), DIPEA (480 мкл, 2.8 ммоль) и нагревали с обратным водяным холодильником при 110оС в течение 96 ч. Контроль реакции осуществляли с помощью ТСХ (силикагель, CH2Cl2-EtOH - 97:3 (%)). Затем реакционную смесь упаривали под вакуумом. Очистка соединения проводилась на колонке с силикагелем. Колонку промывали в системе CH2Cl2:EtOH - 97:3 (%), с постепенным увеличением градиента до 7% этанола (CH2Cl2:EtOH - 93:7 (%)). Конечный продукт элюировали в системе CH2Cl2:EtOH - 90:10 (%). Фракции, содержащие продукт, объединяли, упаривали и сушили в вакуумном эксикаторе над P2O5. Выход соединения 5a составлял 86 мг, 35 % (белый порошок). Rf = 0.21 (CH2Cl2:EtOH - 95:5 (%)). Тпл = 140-142°С. 1H-ЯМР (300.1 МГц, DМSO-d6): δ = 8.47 (ус, 1H, NH), 8.38 (с, 1H, H-8), 8.20 (с, 1H, H-2), 7.49 (д, 2Н, J2-3 = J6-5=8.4 Гц, Н2-Ph, Н6-Ph), 7.30 (д, 2Н, J3-2 = J5-6=8.4 Гц, Н3-Ph, Н5-Ph), 5.90 (д, 1H, J1′2′ = 6.0 Гц, H-1′), 5.41 (д, 1H, J2′-OH = 6.1 Гц, 2′-OH), 5.32 (дд, 1H, J5′a-OH = 4.6 Гц, J5′b-OH = 7.0 Гц, 5′-OH), 5.15 (д, 1H, J3′-OH = 4.7 Гц, 3′-OH), 4.73 (ус, 2H, СH2), 4.61 (ддд, 1H, J2′-OH = 6.1 Гц, J2′1′ = 6.0 Гц, J2′3′ = 5.0 Гц, H-2′), 4.15 (ддд, 1H, J3′-OH = 4.7 Гц, J3′2′ = 5.0 Гц, J3′4′ = 3.4 Гц, H-3′), 3.96 (ддд, 1H, J4′3′ =3.4 Гц, J4′5′а = 3.7 Гц J4′5′b = 3.4 Гц, H-4′), 3.67 (ддд, 1H, J5′a,5′b = -12.0 Гц, J5′a,4′ = 3.7 Гц, J5′a,OH = 4.6 Гц, H5′a), 3.55 (ддд, 1H, J5′b5′a = -12.0 Гц, J5′b4′ = 3.4 Гц, J5′b-OH = 7.0 Гц, H-5′b). 13C-ЯМР (75.5 МГц, DМSO-d6): δ = 154.42 (C6), 152.30 (C2), 150.41 (C4-Ph), 148.55 (C4), 139.98 (C8), 136.27 (C1-Ph), 128.89 (C2-Ph, C6-Ph), 128.51 (к, JC-F = 30.9 Гц, CF3), 120.83 (C3-Ph, C5-Ph), 119.74 (C5), 87.89 (C1′), 85.85 (C4′), 73.47 (C2′), 70.59 (C3′), 61.61 (C5′), 42.22 (NCH2-). 19F-ЯМР (282.4 МГц, DМSO-d6): δ = -56.81. HRМS: м/z [C18H18F3N5O5+H]+ посчитан 442.1333, найден 442.1330.
N6-(4-трифторметилтиобензил)аденозин (5b)
Аналогично получению соединения 5a аминирование соединения 4 (183 мг, 0.64 ммоль) с 4-трифторметилтиобензиламином (303 мкл, 1.92 ммоль) в присутствии DIPEA (328.7 мкл, 1.92 ммоль) в н-бутаноле (7 мл) протекало в течении 7 часов при нагревании до 90 ºС. Выход соединения 5b составлял 150 мг, 51 % (белый порошок). Rf = 0.22 (CH2Cl2:EtOH, 95:5 (%)). Тпл = 99-101°С. 1H-ЯМР (300.1 МГц, DМSO-d6): δ = 8.52 (ус, 1H, NH), 8.39 (с, 1H, H-8), 8.20 (с, 1H, H-2), 7.65 (д, 2Н, J2-3 = J6-5=8.4 Гц, Н2-Ph, Н6-Ph), 7.48 (д, 2Н, J3-2 = J5-6=8.4 Гц, Н3-Ph, Н5-Ph), 5.90 (д, 1H, J1′2′ = 6.1 Гц, H-1′), 5.41 (д, 1H, J2′-OH = 6.2 Гц, 2′-OH), 5.31 (дд, 1H, J5′a-OH = 4.6 Гц, J5′b-OH = 7.0 Гц, 5′-OH), 5.15 (д, 1H, J3′-OH = 4.7 Гц, 3′-OH), 4.77 (ус, 2H, СH2), 4.61 (ддд, 1H, J2′-OH = 6.2 Гц, J2′1′ = 6.1 Гц, J2′3′ = 5.0 Гц, H-2′), 4.15 (ддд, 1H, J3′-OH = 4.7 Гц, J3′2′ = 5.0 Гц, J3′4′ = 3.4 Гц, H-3′), 3.96 (к, 1H, J4′3′ =3.4 Гц, J4′5′а = 3.4 Гц J4′5′b = 3.4 Гц, H-4′), 3.68 (ддд, 1H, J5′a,5′b = -12.0 Гц, J5′a,4′ = 3.4 Гц, J5′a,OH = 4.6 Гц, H5′a), 3.55 (ддд, 1H, J5′b5′a = -12.0 Гц, J5′b4′ = 3.4 Гц, J5′b-OH = 7.0 Гц, H-5′b). 13C-ЯМР (75.5 МГц, DМSO-d6): δ = 154.45 (C6), 152.31 (C2), 148.60 (C4), 144.02 (C1-Ph), 140.05 (C8), 131.66 (C4-Ph), 136.21 (C2-Ph, C6-Ph), 128.51 (C3-Ph, C5-Ph), 127.80 (к, JCF=37.1 Гц, SCF3), 120.78 (C5), 87.91 (C1′), 85.87 (C4′), 73.49 (C2′), 70.60 (C3′), 61.21 (C5′), 42.53 (NCH2-). 19F-ЯМР (282.4 МГц, DМSO-d6): δ = -42.26. HRМS: м/z [C18H18F3N5O4S+H]+ посчитан 458.1104, найден 458.1103.
N6-(4-дифторметоксибензил)аденозин (5c)
6-Хлоро-9-(β-D-рибофуранозил)-9H-пурин 4 (160 мг, 0.56 ммоль) и 4-дифторметоксибензиламин (582 мг, 3.36 ммоль) растворяли в 7 мл н-бутанола. Затем к полученной смеси добавляли DIPEA (575 мкл, 3.36 ммоль) и нагревали с обратным водяным холодильником при 90°С в течение 72 часов до выпадения белого осадка. Контроль реакции осуществляли с помощью ТСХ (силикагель, CH2Cl2:EtOH - 97:3 (%)). Затем осадок переносили на стеклянный фильтр, промывали холодным н-бутанолом (30 мл) и сушили в вакуумном эксикаторе над P2O5. Выход соединения 5c составлял 71 мг, 30 % (белый порошок). Rf = 0.24 (CH2Cl2:EtOH, 95:5 (%)). Тпл = 133-135°С. 1H-ЯМР (300.1 МГц, DМSO-d6): δ = 8.44 (ус, 1H, NH), 8.37 (с, 1H, H-8), 8.20 (с, 1H, H-2), 7.38 (д, 2Н, J2-3 = J6-5=8.4 Гц, Н2-Ph, Н6-Ph), 7.12 (т, 1H, 1JH-F = 74.3 Гц, CH-F, перекрывается с H3-Ph, H5-Ph), 7.10 (д, 2Н, J3-2 = J5-6 =8.4 Гц, Н3-Ph, Н5-Ph), 5.89 (д, 1H, J1′2′ = 6.1 Гц, H-1′), 5.40 (д, 1H, J2′-OH = 6.2 Гц, 2′-OH), 5.33 (дд, 1H, J5′a-OH = 4.6 Гц, J5′b-OH = 7.0 Гц, 5′-OH), 5.15 (д, 1H, J3′-OH = 4.7 Гц, 3′-OH), 4.70 (ус, 2H, СH2), 4.61 (ддд, 1H, J2′-OH = 6.2 Гц, J2′1′ = 6.1 Гц, J2′3′ = 5.0 Гц, H-2′), 4.15 (ддд, 1H, J3′-OH = 4.7 Гц, J3′2′ = 5.0 Гц, J3′4′ = 3.4 Гц, H-3′), 3.96 (к, 1H, J4′3′ =3.4 Гц, J4′5′а = 3.4 Гц J4′5′b = 3.4 Гц, H-4′), 3.68 (ддд, 1H, J5′a,5′b = -12.0 Гц, J5′a,4′ = 3.4 Гц, J5′a,OH = 4.6 Гц, H5′a), 3.55 (ддд, 1H, J5′b5′a = -12.0 Гц, J5′b4′ = 3.4 Гц, J5′b-OH = 7.0 Гц, H-5′b). 13C-ЯМР (75.5 МГц, DМSO-d6): δ = 154.41 (C6), 152.31 (C2), 149.64 (C4-Ph), 148.52 (C4), 139.94 (C8), 137.09 (C1 Ph), 128.73 (C2-Ph, C6-Ph), 119.74 (C5), 118.71 (C3-Ph, C5-Ph), 116.40 (t, JCF=257.4 Гц, OCHF2), 87.90 (C1′), 85.87 (C4′), 73.47 (C2′), 70.61 (C3′), 61.62 (C5′), 42.25 (CH2). 19F-ЯМР (282.4 МГц, DМSO-d6): δ = -81.62. HRМS: м/z [C18H19F2N5O5+H]+ посчитан 424.1427, найден 424.1432.
Получение фармацевтически значимых солей на примере фумарата 3g
N6-(((3'-Трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин 3g (150 мг, 0.299 ммоль) растворяют в изопропаноле (0,375 мл) добавляют стехиометрический эквивалент фумаровой кислоты (35 мг, 0.299 ммоль) и смесь нагревают до 60°C в течение 10 минут. Затем раствор охлаждают до комнатной температуры и перемешивают до выпадения осадка. Смесь разбавляют, используя изопропанол (0,5 мл), суспензию фильтруют, промывают охлажденным изопропанолом (0,5 мл). Выход 77 мг (41 %).
Характеристика биологической активности соединений по изобретению
Клетки и вирусы
Клеточную линию почек эмбрионов свиньи (СПЭВ) культивировали при 37 °С в смеси среды 199 (на растворе Эрла) и среды 199 (на растворе Хэнкса) с добавлением 5% эмбриональной телячьей сыворотки (Gibco). Эпителиальную клеточную линию почки африканской зеленой мартышки (Vero) культивировали при 37 °С в среде ДМЕМ с добавлением L-глутамина и 5% эмбриональной телячьей сыворотки (Gibco). Штамм вируса клещевого энцефалита Абсеттаров (GenBank no. KU885457.1), штамм вируса желтой лихорадки 17D (GenBank no. JN628279.1), штамм вируса лихорадки Западного Нила (GenBank no. OP868929) взяты из коллекции ФГАНУ «ФНЦИРИП им. М.П. Чумакова РАН» (Институт полиомиелита). Исследуемые соединения растворяли в ДМСО (Amresco) для приготовления исходных растворов с концентрацией 5 ммоль/л.
Анализ цитотоксичности на клетках Vero и СПЭВ
Цитотоксичность определяли по влиянию соединений по изобретению на жизнеспособность клеток и измеряли посредством резазурин-теста. При добавлении к живым клеткам синий краситель резазурин под действием клеточных ферментов восстанавливается до флуоресцентного красителя резоруфина. Токсическое действие соединений по изобретению детектируется по снижению интенсивности флуоресценции по сравнению с контрольными клетками.
Клетки Vero или СПЭВ высаживали в 96-луночные культуральные планшеты и инкубировали в течение 24 часов при 37 °C и 5% CO2. Из соединений готовили серии последовательных разведений 1:2, начиная с концентрации 50 мкмоль/л. В качестве контроля аналогичным образом готовили серии разведений ДМСО, начиная с концентрации 1%. Растворителем для эксперимента на клетках Vero служила среда ДМЕМ с добавлением L-глутамина, для эксперимента на клетках СПЭВ - смесь среды 199 (соли Эрла) и среды 199 (соли Хэнкса) с добавлением 2% эмбриональной телячьей сыворотки. В качестве положительного контроля, вызывающего 100% гибель клеток, готовили водный раствор додецилсульфата натрия в концентрации 0,5 мг/мл. Равные объемы разведений соединений переносили на клетки, после чего планшеты с клетками инкубировали при 37 °C и 5% CO2 в течение 24 часов или 7 дней для определения острой или хронической цитотоксичности соответственно. После инкубации монослой клеток промывали фосфатно-солевым буфером и к клеткам добавляли раствор резазурина (Sigma-Aldrich) в среде 199 (на растворе Эрла) с концентрацией 25 мкг/мл. Обработанные клетки инкубировали 4 часа при 37 °С и 5% СО2, затем собирали культуральную жидкость и измеряли в ней флуоресценцию резоруфина при λex/em = 544/590 нм. Значение CC50 рассчитывали по методу Рида-Менча.
Анализ активности против ВКЭ
Анализ активности против ВКЭ проводили методом ингибирования бляшкообразования.
Клетки СПЭВ высаживали в 24-луночные культуральные планшеты и инкубировали в течение 3 суток при 37 °C и 5% CO2. Из соединений готовили серии последовательных разведений 1:4 в среде 199 (на растворе Эрла), начиная с концентрации 50 мкмоль/л. В качестве контроля аналогичным образом готовили серии разведений ДМСО, начиная с концентрации 1%. К полученным разведениям добавляли вирусную суспензию и инкубировали смеси 1 час при 37 °C и 5% CO2. Титр вируса был подобран так, чтобы при заражении на каждую лунку с клетками приходилось по 30-60 бляшкообразующих единиц. Клетки СПЭВ заражали полученными смесями и инкубировали с периодическими покачиваниями в течение 1 часа при 37 °C и 5% CO2, затем зараженные клетки покрывали 1,5% раствором метилцеллюлозы в смеси среды 199 (соли Эрла) и среды 199 (соли Хэнкса) с добавлением 2% эмбриональной телячьей сыворотки и инкубировали 6 суток при 37 °C и 5% CO2. После инкубации клетки освобождали от покрытия, промывали фосфатно-солевым буфером, фиксировали 96% этанолом и окрашивали 0,5% раствором кристаллического фиолетового. Значение EC50 рассчитывали по методу Рида-Менча после подсчета бляшек.
Анализ активности против ВЖЛ
Активность против ВЖЛ измеряли аналогично активности против ВКЭ с небольшими изменениями. Клетки Vero высаживали в 24-луночные культуральные планшеты и инкубировали в течение 3 суток при 37 °C и 5% CO2. Приготовление разведений соединений и их смесей с вирусной суспензией, заражение клеток и нанесение покрытия осуществляли как указано выше. После инкубации в течение 7 суток при 37 °C и 5% CO2 клетки освобождали от покрытия, промывали фосфатно-солевым буфером, фиксировали 96% этанолом и окрашивали 0,5% раствором кристаллического фиолетового. Значение EC50 рассчитывали по методу Рида-Менча после подсчета бляшек.
Анализ активности против ВЗН
Активность против ВЗН измеряли аналогично активности против ВКЭ с небольшими изменениями. Клетки Vero высаживали в 24-луночные культуральные планшеты и инкубировали в течение 3 суток при 37 °C и 5% CO2. Из соединений готовили серии последовательных разведений 1:4 в среде ДМЕМ, начиная с концентрации 50 мкмоль/л. В качестве контроля аналогичным образом готовили серии разведений ДМСО, начиная с концентрации 1%. Приготовление смесей разведений с вирусной суспензией, заражение клеток и нанесение покрытия осуществляли как указано выше. После инкубации в течение 7 суток при 37 °C и 5% CO2 клетки освобождали от покрытия, промывали фосфатно-солевым буфером, фиксировали 96% этанолом и окрашивали 0,5% раствором кристаллического фиолетового. Значение EC50 рассчитывали по методу Рида-Менча после подсчета бляшек.
Результаты проведенных исследований
Противовирусную активность соединений по изобретению оценивали в отношении, в частности, вируса клещевого энцефалита на клетках СПЭВ и вирусов желтой лихорадки и лихорадки Западного Нила на клетках Vero методом ингибирования бляшкообразования (табл. 2, 3, 4).
(24 часа)
(7 дней)
(24 часа)
(7 дней)
(24 часа)
(7 дней)
Таким образом, результаты исследования биологической активности показывают, что соединения по изобретению характеризуются высокой эффективностью в ингибировании репродукции вирусов, относящихся к роду Flavivirus, в том числе ВКЭ, ВЖЛ, ВЗН. Соединения обладают низкой цитотоксичностью, что в сочетании с высокой противовирусной активностью является необходимым условием для проведения дальнейшей фармацевтической разработки.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Изобретение также относится к фармацевтическим композициям, которые содержат соединение общей формулы I (или про-лекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, которые могут быть введены в организм пациента вместе с соединением, составляющим сущность этого изобретения, не снижают фармакологическую активность этого соединения и не токсичны при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, указанные в этом изобретении, содержат соединения этого изобретения вместе с фармацевтически приемлемыми носителями, которые могут включать любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазки и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают моно- и олигосахариды, а также их производные; желатин; тальк; вспомогательные вещества, такие как масло какао и воск для суппозиториев; масла, такие как арахисовое, хлопковое, кунжутное, оливковое, кукурузное и соевое масло и другие; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; депирогенизированная вода; изотонический раствор, раствор Рингера; спиртовые и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленочные агенты, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом настоящего изобретения являются также лекарственные формы - класс фармацевтических композиций, структура которых оптимизирована для определенного способа введения в организм в терапевтической эффективной дозе, например, для введения в организм внутривенно, внутримышечно, перорально, подкожно, ингаляционно, интраназально, сублингвально, ректально, или иными общепринятыми способами в рекомендуемых дозировках.
Лекарственные формы настоящего изобретения могут содержать структуры, полученные методами использования липосом, методами микрокапсулирования, способами получения наноформ лекарственного средства или другими способами, известными в фармацевтике.
При получении композиции, например, в форме таблетки, активный компонент смешивают с одним или несколькими фармацевтическими наполнителями, такими как желатин, крахмал, лактоза, стеарат магния, тальк, диоксид кремния, арабская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки могут быть покрыты сахарозой, производными целлюлозы или другими веществами, подходящими для нанесения покрытия. Таблетки могут быть получены различными способами, такими как прямое прессование, сухая или влажная грануляция или горячее легирование в горячем состоянии.
Фармацевтическую композицию в виде желатиновой капсулы можно получить, смешивая активное начало с растворителем и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным способом используются водные суспензии, изотонические физиологические растворы или стерильные растворы для инъекций.
Примеры получения композиции по изобретению
Метод А: приготовление раствора (P.Bhatt. Handbook of pharmaceutical technology practical. P.Bhatt, A.Kumar Eds. Pharmatech 2021).
N6-(((3'-Трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин 3g (1,5 мг, 3 мкмоль) растворяют в достаточном количестве 2% водного раствора ДМСО для инъекций. Добавляют столько 2% водного раствора ДМСО для инъекций, чтобы общий объем раствора составлял 100 мл. Раствор отфильтровывают от механических примесей, разливают по ампулам, которые затем запечатывают и стерилизуют в автоклаве.
Метод Б: гранулирование (P.Bhatt. Handbook of pharmaceutical technology practical. P.Bhatt, A.Kumar Eds. Pharmatech 2021).
N6-(((3'-Трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин 3g (500 мг, 1 ммоль), тонко измельченные порошки лактозы (40 мг), крахмала (4 мг), талька (10 мл) тщательно перемешивают в ступке и добавляют к полученной смеси крахмальную пасту (4 мг) в качестве связующего ингредиента и перемешивают. Полученную смесь просеивают через сито. Полученные гранулы сушат в духовке при температуре 60°С в течение 35 мин.
Метод С: таблетирование .
Гранулы, полученные по методу Б, смазывают стеаратом магния и запекают в духовке при температуре 60°С до полного высыхания (P.Bhatt. Handbook of pharmaceutical technology practical. P.Bhatt, A.Kumar Eds. Pharmatech 2021). Затем высушенные гранулы формуют в таблетки с помощью эксцентриковой (прессовальной) таблеточной машины (Меньшутина Н.В., Алвес С.В., Мишина Ю.В. Инновационные технологии и оборудование фармацевтического производства. БИНОМ, 2012).
МЕТОДЫ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ
Соединения настоящего изобретения являются противовирусными средствами, и поэтому они являются полезными агентами для терапии и/или профилактики заболеваний или состояний, вызванных флавивирусами, в том числе вирусом клещевого энцефалита, вирусом желтой лихорадки, вирусом лихорадки Западного Нила. Благодаря своей противовирусной активности соединения настоящего изобретения полезны для профилактики, лечения и/или уменьшения риска развития: клещевого энцефалита, желтой лихорадки, лихорадки денге, денге шок-синдрома, лихорадки Западного Нила, лихорадки Зика.
Эти инфекционные заболевания хорошо охарактеризованы у человека, но также с аналогичной этиологией присутствуют у других млекопитающих, и их можно лечить фармацевтическими композициями настоящего изобретения.
Для терапевтического применения соединения изобретения могут вводиться с помощью фармацевтической композиции в любой фармацевтической лекарственной форме любым способом введения. Лекарственные формы обычно включают фармацевтически приемлемый носитель, подходящий для выбранной конкретной лекарственной формы. В частности, соединение по изобретению может вводиться ежедневно в течение периода времени, необходимого для лечения и/или профилактики заболеваний, имеющих отношение к пациенту, включая курс терапии, длящийся дни, месяцы, годы или всю жизнь пациента. Способы введения включают, но не ограничиваются: внутривенно, внутримышечно, перорально, подкожно, ингаляционно, интраназально и сублингвально. Предпочтительными способами введения являются пероральный и внутривенный.
Изобретение также относится к фармацевтической композиции, содержащей ежедневную дозу указанного соединения в форме фиксированной единицы дозировки, и к комбинации, содержащей указанную фармацевтическую композицию или указанное соединение. В предпочтительном варианте осуществления указанную композицию для применения в соответствии с изобретением вводят один или несколько раз в день в дозировке 1 мг или более выбранного соединения по изобретению. Предпочтительная дозировка составляет 1-1500 мг. Наиболее предпочтительная дозировка составляет 10-1000 мг.
Один или несколько дополнительных фармакологически активных агентов могут вводиться в комбинации с соединением формулы I. Как правило, любые дополнительные одиночные или множественные активные агенты, отличные от соединения формулы I, могут использоваться в любой комбинации с соединением формулы I в одной или отдельной лекарственной форме, позволяющей одновременное или последовательное терапевтическое действие активных агентов. Такие дополнительные один или несколько агентов - это агенты, которые облегчают любые другие симптомы, которые могут быть связаны с вирусной инфекцией, т.е. лихорадка, озноб, головная боль, вторичные инфекции, можно вводить одновременно, или как часть фармацевтической композиции, или в отдельные моменты времени. Эти средства включают, без ограничения, жаропонижающее средство, противовоспалительное средство, химиотерапевтическое средство или их комбинации.
В некоторых вариантах осуществления изобретения, композиции по изобретению дополнительно содержат одно или несколько средств, которые облегчают любые другие симптомы, которые могут быть связаны с вирусной инфекцией, например лихорадка, озноб, головная боль, вторичные инфекции, можно вводить одновременно, или как часть фармацевтической композиции, или в отдельные моменты времени. Эти средства включают, без ограничения, жаропонижающие средства, противовоспалительные средства, химиотерапевтические средства, антибиотики, противогрибковые средства, химиотерапевтические средства, интерфероны, цитокины, монокины, антитела или их комбинации.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Производные аденозина - ингибиторы репродукции вирусов, относящихся к роду Flavivirus | 2024 |
|
RU2839716C1 |
| МИМЕТИКИ ПОЛИ (ADP-РИБОЗЫ) И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2559873C2 |
| Средство для ингибирования фермента тирозил-ДНК-фосфодиэстеразы 1 человека на основе производных пентафуранозилнуклеозидов | 2019 |
|
RU2748103C1 |
| Способ получения кладрибина методом ферментативного трансгликозилирования 2-хлор-6-азидопурина с последовательной двухстадийной конверсией азидогруппы | 2023 |
|
RU2836333C1 |
| СРЕДСТВО ДЛЯ ИНГИБИРОВАНИЯ ФЕРМЕНТА ПОЛИ(АДФ-РИБОЗО)ПОЛИМЕРАЗЫ-1 ЧЕЛОВЕКА | 2009 |
|
RU2411948C1 |
| Гидройодная соль 7-метил-2'-дезоксигуанозина в качестве субстрата для получения 2'-дезоксинуклеозидов методом ферментативного трансгликозилирования | 2017 |
|
RU2664472C1 |
| Производные пиримидина - ингибиторы репродукции вирусов, относящихся к роду Orthoflavivirus | 2023 |
|
RU2831118C1 |
| НОВЫЕ ГЕПАТОПРОТЕКТОРНЫЕ СРЕДСТВА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2815370C1 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
Изобретение относится к органической химии, фармакологии и медицине, а именно к модифицированному аналогу нуклеозидов формулы (I) или его фармацевтически приемлемой соли, где R выбирается независимо и представляет собой -С≡С-С3-6-циклоалкил, -С≡С-С3-6-циклоалкенил, -С≡С-С6-арил, замещенный -С≡С-С6-арил по меньшей мере одним заместителем, выбранным из частично или полностью галогенированного -C1-5-алкила, фенил, замещенный по меньшей мере одним заместителем, выбранным из -C1-5-алкила, частично или полностью галогенированного -C1-5-алкила. Также изобретение относится к применению соединения формулы (I) в качестве ингибитора репродукции вируса, относящегося к роду Flavivirus, применению соединения формулы (I) для получения фармацевтической композиции для лечения и/или профилактики заболеваний, вызванных вирусом рода Flavivirus, и фармацевтической композиции на основе соединения формулы (I) для лечения и/или профилактики заболеваний, вызванных вирусом рода Flavivirus. Технический результат - получение соединения формулы (I), характеризующегося высокой эффективностью в ингибировании репродукции вирусов рода Flavivirus. 4 н. и 8 з.п. ф-лы, 4 табл., 11 пр.
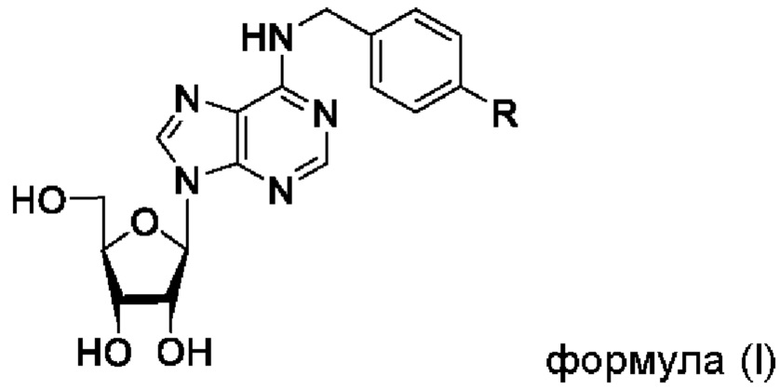
1. Соединение общей формулы (I)
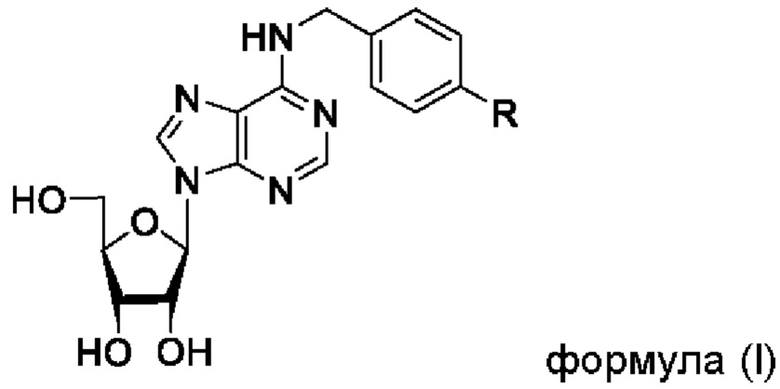
или его фармацевтически приемлемая соль,
где R выбирается независимо и представляет собой -С≡С-С3-6-циклоалкил, -С≡С-С3-6-циклоалкенил, -С≡С-С6-арил, замещенный -С≡С-С6-арил по меньшей мере одним заместителем, выбранным из частично или полностью галогенированного -C1-5-алкила, фенил, замещенный по меньшей мере одним заместителем, выбранным из -C1-5-алкила, частично или полностью галогенированного -C1-5-алкила.
2. Соединение по п. 1, в котором R выбирается независимо и представляет собой -С≡С-циклопропил, -С≡С-циклобутил, -С≡С-циклопентил, -С≡С-циклопентенил, -С≡С-циклогексенил.
3. Соединения по п. 1, в котором R выбирается независимо и представляет собой фенил, замещенный одним или двумя заместителями, представляющими собой -CF3.
4. Соединение по п. 1, в котором R выбирается независимо и представляет собой -С≡С-С6-арил, замещенный по меньшей мере одним заместителем, представляющим собой -CF3.
5. Соединение по п. 1, выбранное из группы:
- N6-([4-циклопропилэтинил]бензил)аденозин;
- N6-([4-циклопентилэтинил]бензил)аденозин;
- N6-([4-(1-циклогексен-1-этинил)]бензил)аденозин;
- N6-([4-фенилэтинил]бензил)аденозин;
- N6-([4-(3,5-бис(трифторметил)фенил)этинил]бензил)аденозин;
- N6-(((2'-трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин;
- N6-(((3'-трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин;
- N6-(((4'-трифторметил)-[1,1'-бифенил]-4-ил)метил)аденозин.
6. Применение соединения по п. 1 в качестве ингибитора репродукции вируса, относящегося к роду Flavivirus.
7. Применение по п. 6, в котором вирус представляет собой вирус денге, вирус лихорадки Западного Нила, вирус желтой лихорадки, вирус японского энцефалита, вирус киасанурской лесной болезни, вирус клещевого энцефалита, вирус омской геморрагической лихорадки, вирус Повассан.
8. Применение соединения по п. 1 для получения фармацевтической композиции с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Flavivirus y пациента.
9. Фармацевтическая композиция с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Flavivirus у пациента, содержащая эффективное количество соединения по п. 1 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
10. Фармацевтическая композиция по п. 9, характеризующаяся тем, что фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
11. Фармацевтическая композиция по п. 9, где вирус представляет собой вирус денге, вирус лихорадки Западного Нила, вирус желтой лихорадки, вирус японского энцефалита, вирус киасанурской лесной болезни, вирус клещевого энцефалита, вирус омской геморрагической лихорадки, вирус Повассан.
12. Фармацевтическая композиция по п. 9, где пациент представляет собой человека.
| WO 2004058791 A2, 15.07.2004 | |||
| WO 2016091235 A1, 16.06.2016 | |||
| DOLEŽAL K | |||
| et al | |||
| Preparation, biological activity and endogenous occurrence of N6-benzyladenosines | |||
| Bioorganic & Medicinal Chemistry, 2007, 15(11), p.3737-3747 | |||
| TRAVNIČEK Z | |||
| et al | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |

