Область техники
Данное изобретение относится к органической химии, фармакологии и медицине и касается профилактики и терапии заболеваний, этиология или патогенез которых связаны с вирусами, относящимися к роду Orthoflavivirus, с помощью новых групп соединений ряда пиримидина.
Уровень техники
Вирусы рода Orthoflavivirus (флавивирусы) обычно циркулируют между кровососущими членистоногими (комарами и клещами), птицами и дикими животными. При присасывании зараженного клеща или укусе комара флавивирусы могут передаваться человеку и вызывать широкий спектр заболеваний различной тяжести: от легких лихорадочных состояний до геморрагических лихорадок и летальных поражений центральной нервной системы. В частности, вирусы рода Orthoflavivirus вызывают такие заболевания, как клещевой энцефалит (КЭ), желтая лихорадка, лихорадка денге (включая денге шок-синдром), лихорадка Западного Нила, лихорадка Зика.
Среди природно-очаговых флавивирусных инфекций КЭ представляет собой особо важную проблему на евразийском континенте, включая Европу, Россию, восточную Азию и Японию, желтая лихорадка эндемична в экваториальных, тропических и субтропических регионах Южной Америки и Африки, а лихорадка Западного Нила встречается в Африке, Европе, на Ближнем Востоке и в Северной Америке. По данным Всемирной организации здравоохранения (ВОЗ), ежегодно регистрируется более 10 тыс. случаев КЭ и около 200 тыс. случаев жёлтой лихорадки. Несмотря на наличие эффективных вакцин, охват иммунизацией считается недостаточным. Более того, зафиксирован ряд случаев заражения КЭ даже после вакцинации. Число случаев заражения лихорадкой Западного Нила в США с 1999 по 2019 годы составило около 51 тыс. случаев, однако в настоящее время вирус лихорадки Западного Нила активно распространяется на новые территории в Европе и России. Кроме того, в настоящее время не существует допущенных к медицинскому применению вакцин против лихорадки Западного Нила и многих других заболеваний, вызываемых флавивирусами. Несмотря на то, что известен ряд низкомолекулярных ингибиторов репродукции флавивирусов, до сих пор не существует специфически действующих на флавивирусы и их молекулярные мишени лекарственных препаратов.
Таким образом, существует необходимость в разработке новых эффективных средств против заболеваний, вызванных вирусами рода Orthoflavivirus.
Раскрытие изобретения
Задачей настоящего изобретения является разработка и создание новых эффективных противовирусных средств, перспективных для применения в клинической практике для терапии и/или профилактики заболеваний, вызванных вирусами рода Orthoflavivirus.
Технический результат изобретения заключается в разработке и получении соединений, характеризующихся высокой эффективностью в ингибировании репродукции вирусов рода Orthoflavivirus, в частности, вируса клещевого энцефалита, вируса желтой лихорадки, вируса лихорадки Западного Нила и других.
Соединения по изобретению также расширяют арсенал доступных противовирусных средств, применяющихся в качестве ингибиторов репродукции вируса, относящегося к роду Orthoflavivirus.
Указанный технический результат достигается посредством соединения общей формулы (I):
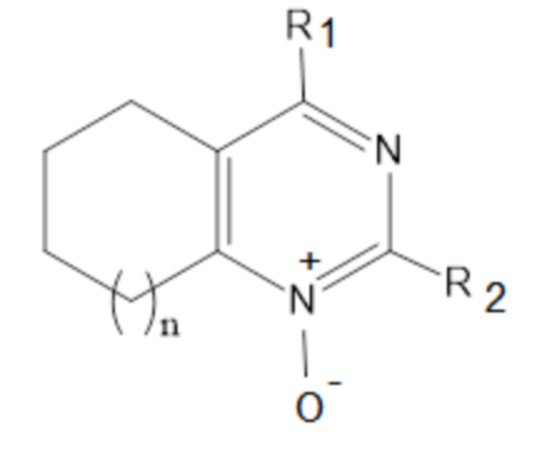
формула (I),
или его пролекарства или фармацевтически приемлемой соли, где:
R1 выбирается независимо и представляет собой:
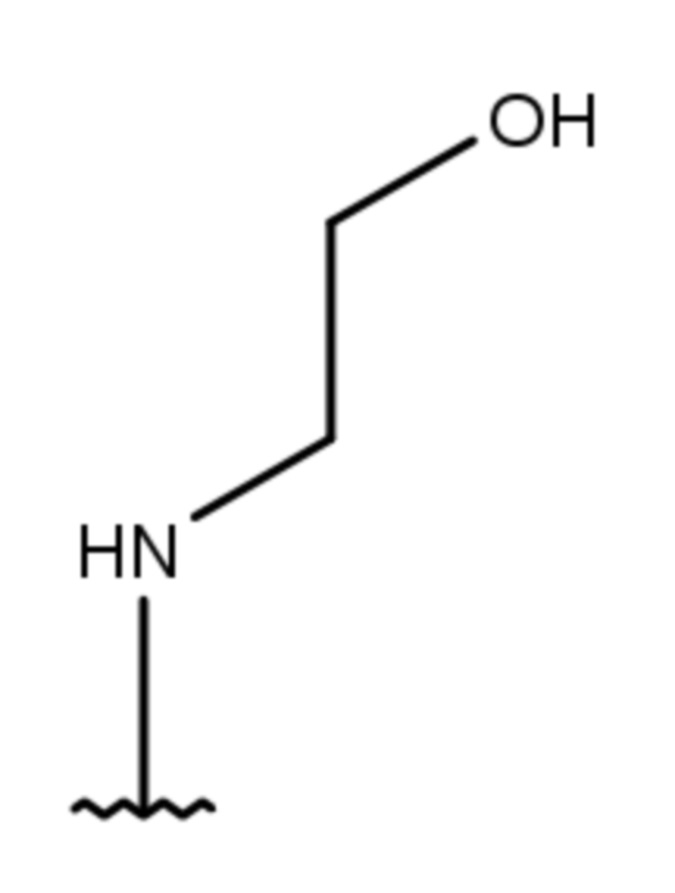
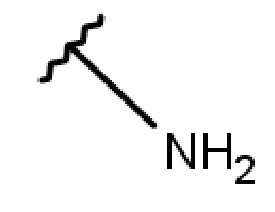
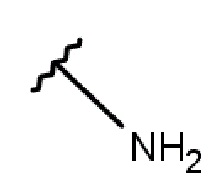
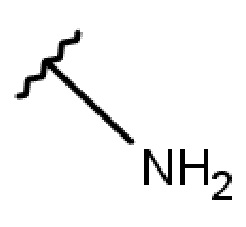
- NH-(C1-5-алкил)-(1-адамантил);
- NH-(C1-5-алкил)-(2-адамантил);
- NH-(C1-5-алкил)-OH;
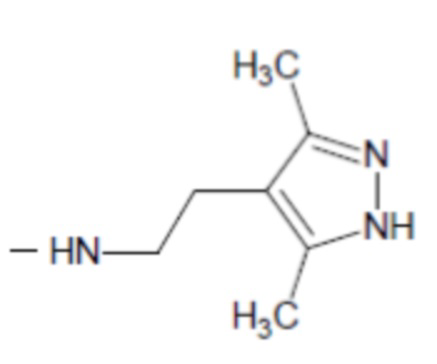 ;
;
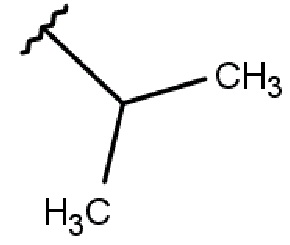
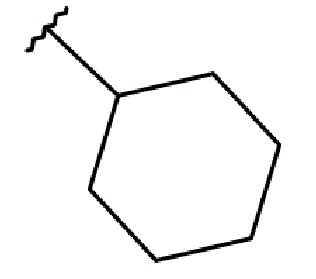
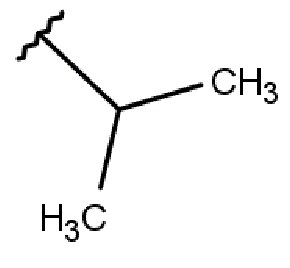
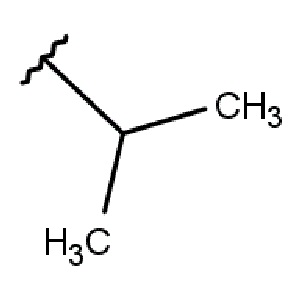
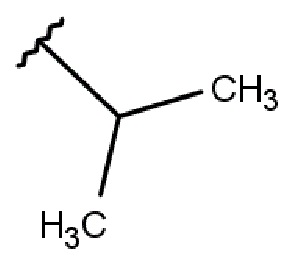
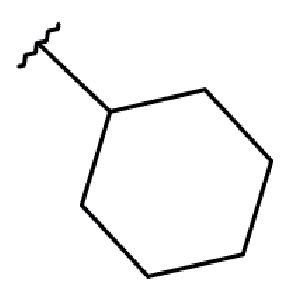
R2 выбирается независимо и представляет собой С3-6-циклоалкил, изопропил, альтернативно R2 представляет собой метил, если R1 представляет собой ;
n принимает значения 0-2;
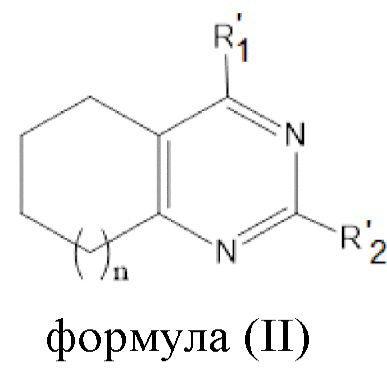
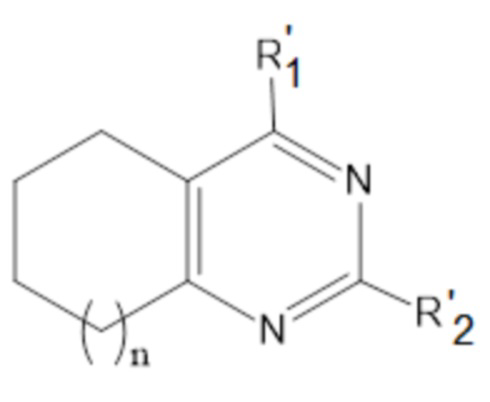
или соединения общей формулы (II):
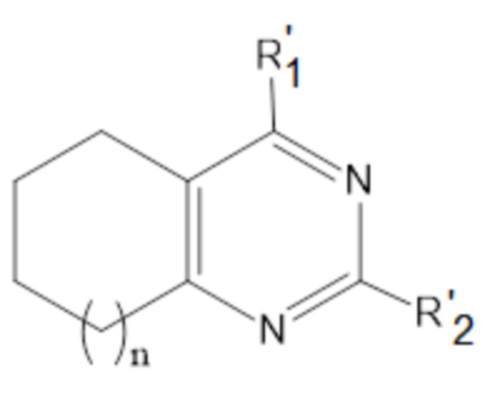 ,
,
формула (II),
или его пролекарства или фармацевтически приемлемой соли, где:
R’1 выбирается независимо и представляет собой:
- NH-(C1-5-алкил)-(1-адамантил);
- NH-(C1-5-алкил)-(2-адамантил);
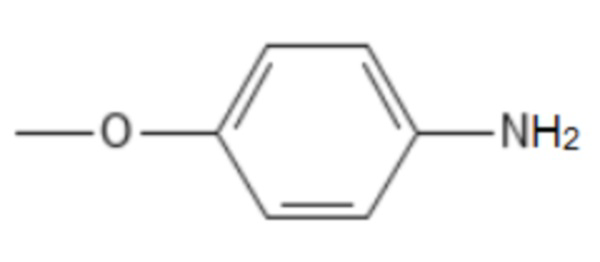 ;
;
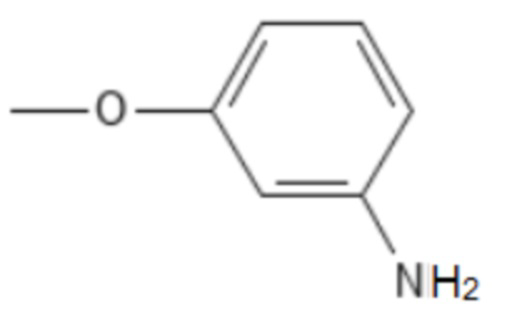 ;
;
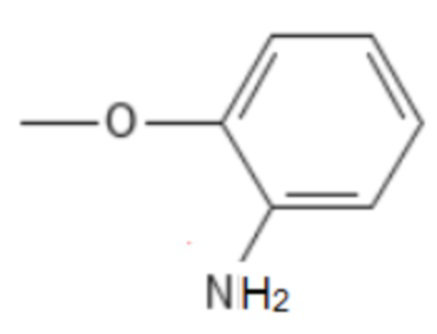 ;
;
R’2 выбирается независимо и представляет собой водород, метил, -NH2, галоген;
n принимает значения 0-2;
или соединения общей формулы (III):
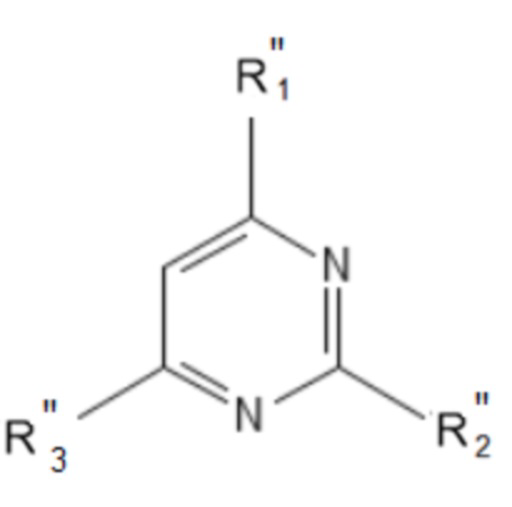
формула (III),
или его пролекарства или фармацевтически приемлемой соли, где:
R’’1 выбирается независимо и представляет собой:
;
;
;
R’’2 выбирается независимо и представляет собой C1-5-алкил;
R’’3 выбирается независимо и представляет собой C1-5-алкил.
В частных вариантах воплощения изобретения R1 выбирается независимо и представляет собой:
- NH-(C2-алкил)-(1-адамантил);
- NH-(C2-алкил)-(2-адамантил); или
- NH-(C2-алкил)-OH;
R2 выбирается независимо и представляет собой циклопропил, циклогексил или изопропил;
n принимает значение 1.
В частных вариантах воплощения изобретения R’1 выбирается независимо и представляет собой:
- NH-(C2-алкил)-(1-адамантил); или
- NH-(C2-алкил)-(2-адамантил);
R’2 представляет собой водород, метил, -NH2 или атом хлора;
n принимает значение 1.
В частных вариантах воплощения изобретения R’’2 выбирается независимо и представляет собой метил;
R’’3 выбирается независимо и представляет собой трет-бутил.
В частных вариантах воплощения настоящее изобретение включает соединения, выбранные из группы:
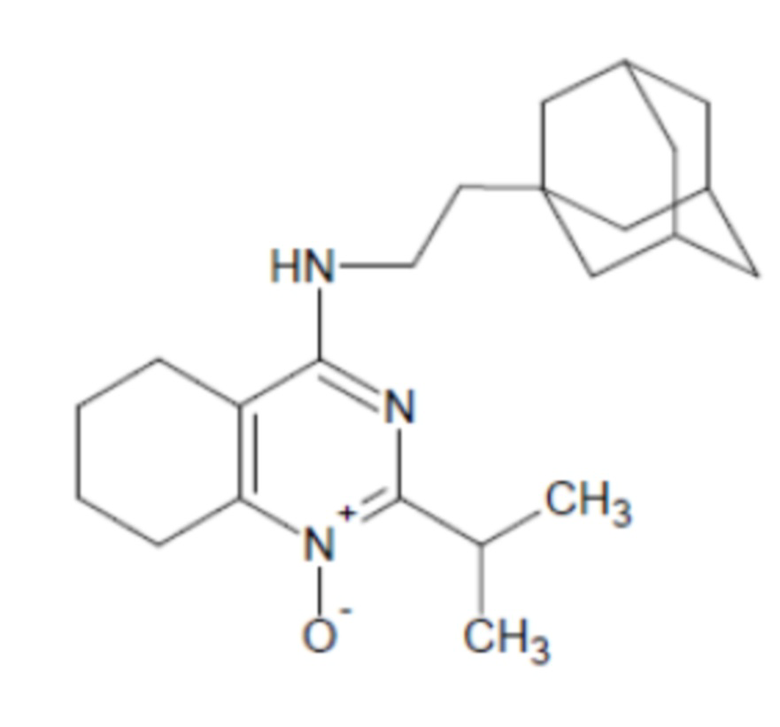
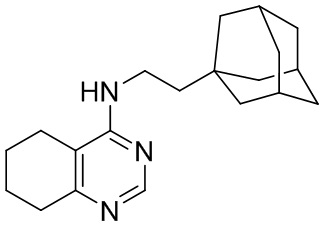
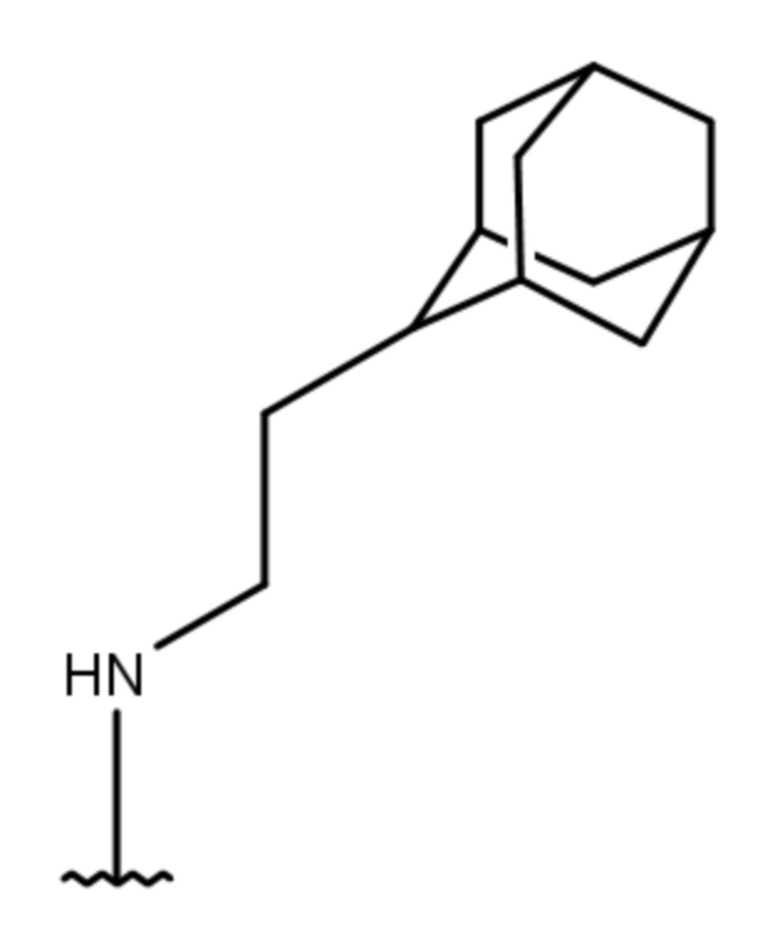
- 4-[2-(1-адамантил)этиламино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксид;
- 4-[2-(1-адамантил)этиламино]-2-циклопропил-5,6,7,8-тетрагидрохиназолин 1-оксид;
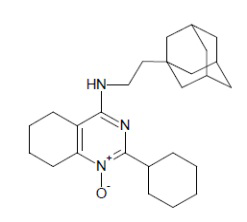
- 4-[2-(1-адамантил)этиламино]-2-циклогексил-5,6,7,8-тетрагидрохиназолин 1-оксид;
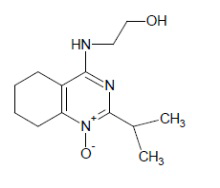
- 4-[(2-гидроксиэтил)амино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксид;
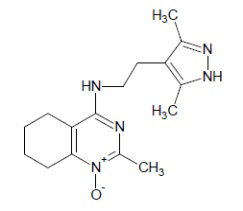
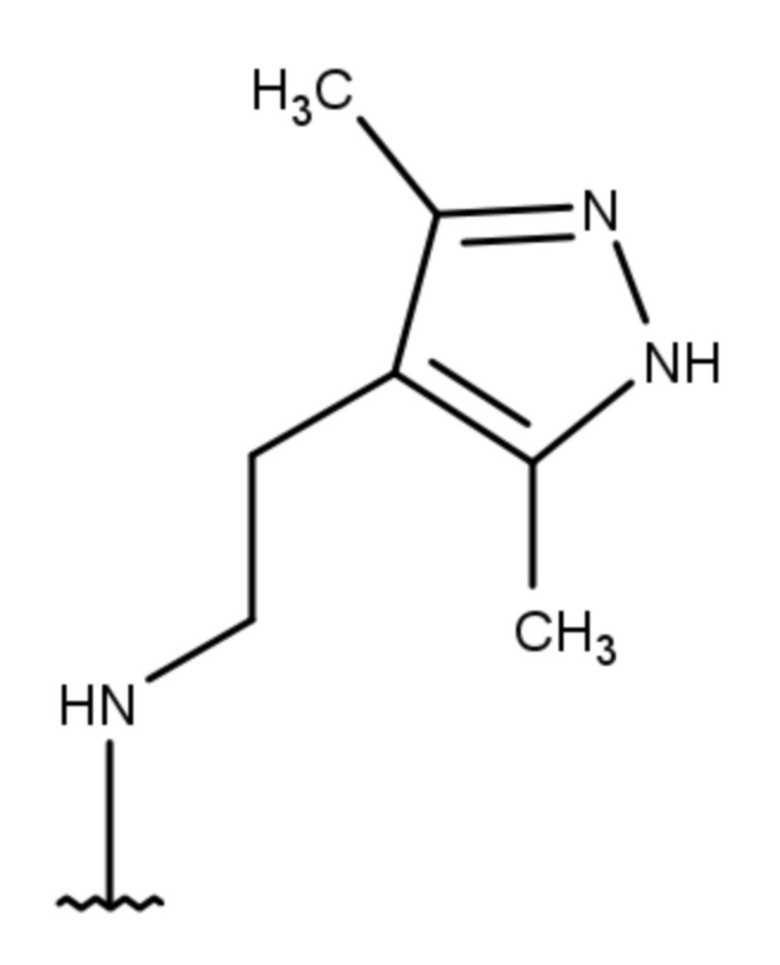
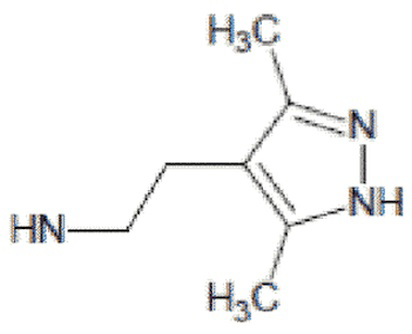
- 4-{[2-(3,5-диметил-1Н-пиразол-4-ил)этил]амино}-2-метил-5,6,7,8-тетрагидрохиназолин 1-оксид;
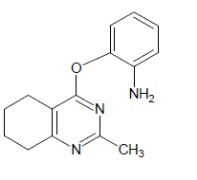
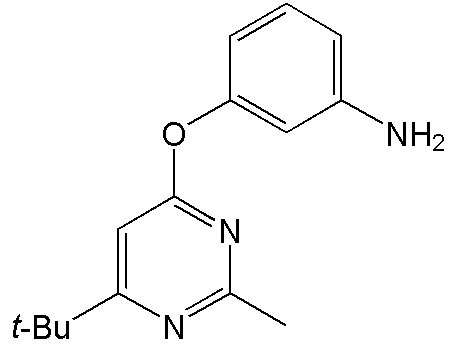
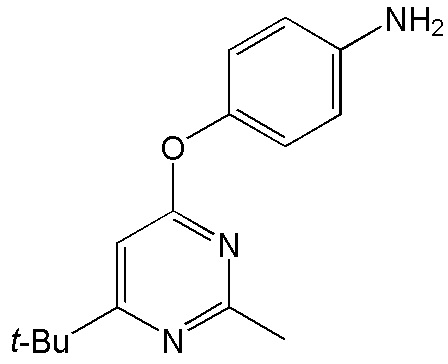
- 4-[(2-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин;
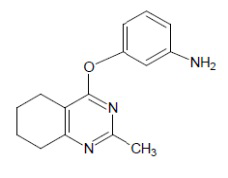
- 4-[(3-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин;
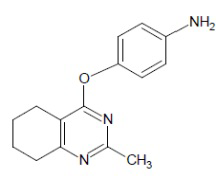
- 4-[(4-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин;
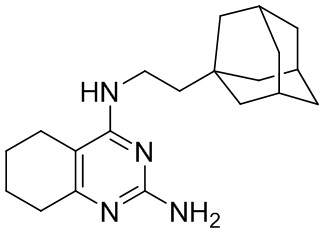
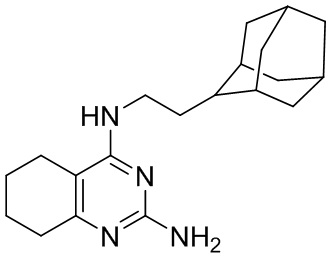
- 4-(2-(1-адамантил)этиламино)-2-амино-5,6,7,8-тетрагидрохиназолин;
- 4-(2-(2-адамантил)этиламино)-2-амино-5,6,7,8-тетрагидрохиназолин;
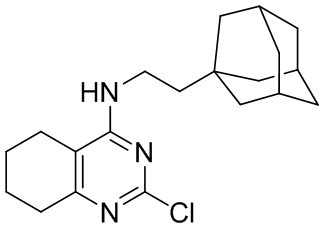
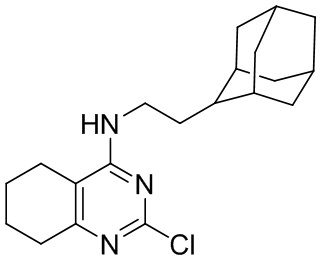
- 4-(2-(1-адамантил)этиламино)-2-хлор-5,6,7,8-тетрагидрохиназолин;
- 4-(2-(2-адамантил)этиламино)-2-хлор-5,6,7,8-тетрагидрохиназолин;
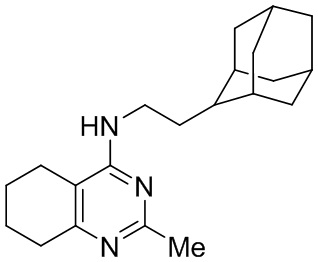
- 4-(2-(2-адамантил)этиламино)-2-метил-5,6,7,8-тетрагидрохиназолин;
- 4-(2-(1-адамантил)этиламино)-5,6,7,8-тетрагидрохиназолин;
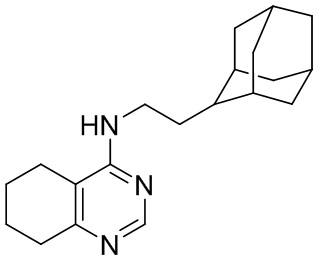
- 4-(2-(2-адамантил)этиламино)-5,6,7,8-тетрагидрохиназолин;
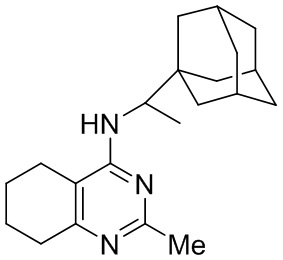
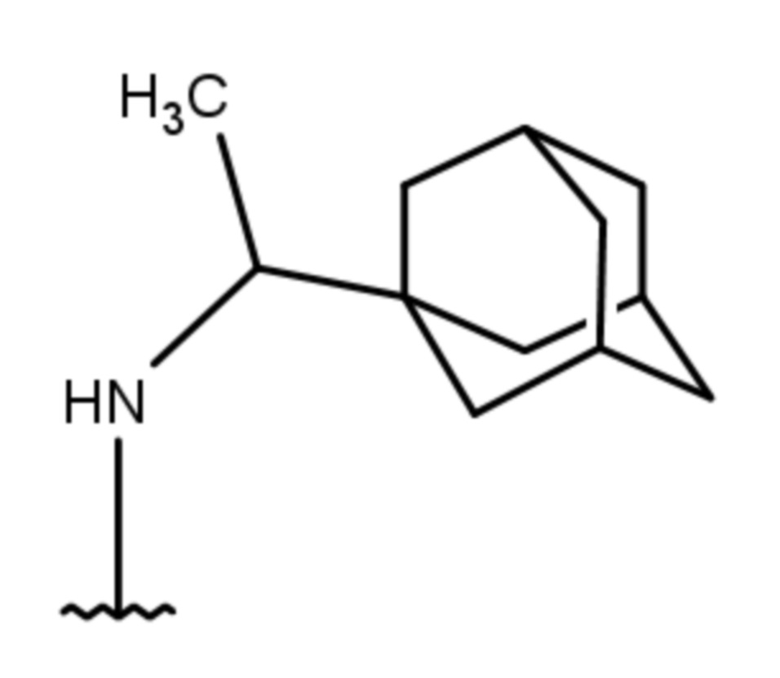
- 4-(1-(1-адамантил)этиламино)-2-метил-5,6,7,8-тетрагидрохиназолин;
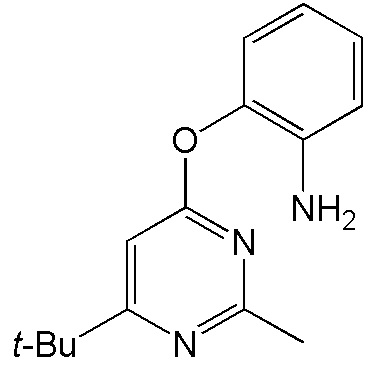
- 4-[(2-аминофенил)окси]-2-метил-6-трет-бутилпиримидин;
- 4-[(3-аминофенил)окси]-2-метил-6-трет-бутилпиримидин;
- 4-[(4-аминофенил)окси]-2-метил-6-трет-бутилпиримидин.
Данное изобретение также относится к применению соединения по изобретению в качестве ингибитора репродукции вируса, относящегося к роду Orthoflavivirus.
В частных вариантах воплощения изобретения вирус представляет собой вирус клещевого энцефалита, вирус желтой лихорадки, вирус лихорадки Западного Нила, вирус денге, вирус японского энцефалита, вирус киасанурской лесной болезни, вирус омской геморрагической лихорадки, вирус Повассан.
Настоящее изобретение также относится к использованию соединений по изобретению для получения фармацевтической композиции с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Orthoflavivirus у пациента.
Изобретение также включает фармацевтические композиции с противовирусной активностью для лечения и/или профилактики заболеваний вирусной этиологии, вызванных вирусами рода Orthoflavivirus у пациента, содержащие эффективное количество соединения по изобретению.
В частных вариантах воплощения изобретения фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
В частных вариантах воплощения изобретения пациент представляет собой человека.
Данное изобретение также относится к способу ингибирования репродукции вирусов, относящихся к роду Orthoflavivirus, включающему введение пациенту терапевтически эффективного количества соединения по изобретению.
Изобретение также включает способ лечения и/или профилактики заболеваний вирусной этиологии, вызванных вирусами рода Orthoflavivirus у пациента, включающий введение терапевтически или профилактически эффективного количества соединения по изобретению.
Настоящее изобретение также относится к применению соединений изобретения для получения средства для ингибирования репродукции вируса, относящегося к роду Orthoflavivirus.
Настоящее изобретение включает также получение соединений и/или композиций по изобретению.
Подробное раскрытие изобретения
Определения (термины)
Для лучшего понимания настоящего изобретения ниже приведены некоторые термины, использованные в настоящем описании изобретения. Следующие определения применяются в данном документе, если иное не указано явно.
Использованные научные и технические термины, если это не оговорено отдельно, имеют значения, общепринятые в научной и технической литературе.
Термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Термин «необязательный» или «необязательно» или «опциональный» или «опционально», используемый в данном документе, означает, что описываемое впоследствии событие или обстоятельство может, но не обязательно, произойти, и что описание включает случаи, когда событие или обстоятельство происходит, и случаи, в которых оно не происходит.
Термин «и/или» означает один, несколько или все перечисленные элементы.
Также здесь перечисление числовых диапазонов по конечным точкам включает все числа, входящие в этот диапазон.
Род Orthoflavivirus (семейство Flaviviridae) включает оболочечные РНК-вирусы (флавивирусы), многие из которых, такие как вирус клещевого энцефалита (ВКЭ), вирус желтой лихорадки (ВЖЛ), вирус лихорадки Западного Нила (ВЗН), вирус денге, вирус Зика и др., являются патогенными и вызывают ряд одноименных заболеваний, широко распространенных в различных регионах мира.
Термин «алкил» в настоящем документе, сам по себе или как часть другого заместителя, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, имеющие указанное число атомов углерода, в частности, термин «алкил» относится к группам, обычно имеющим от одного до пяти атомов углерода. Например, термин «C1-3-алкил» включает метил, этил, пропил, изопропил и т.д.
Термин «циклоалкил» в настоящем документе относится к группам, имеющим от трех до шести атомов углерода в моноциклической структуре. В качестве иллюстрации, циклоалкилы включают, но не ограничиваются, следующими радикалами: циклопропил, циклобутил, циклопентил, циклогексил.
Термин «галоген» сам по себе или в части другого термина относится к атому фтора, хлора, брома или йода.
Если не указано иное, все вхождения функциональных групп в структурах соединений по изобретению выбираются независимо.
Изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования.
Если не указано иначе, приведенные в материалах заявки структуры соединений также подразумевают и все стереоизомеры, то есть R- и S-изомеры для каждого хирального центра. Отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси соединений по изобретению, также являются предметом настоящего изобретения. Таким образом, настоящее изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%; более предпочтительно, >95% мольной чистоты), так же, как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например, путем получения диастереомерных солей при обработке оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Еще один возможный метод разделения включает синтез ковалентных диастереомерных производных путем реакции соединений изобретения с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Полученные диастереомеры можно разделить такими способами, как хроматография, дистилляция, кристаллизация или сублимация, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания или соли.
Соединения, составляющие суть данного изобретения, могут существовать в меченой радиоизотопом форме, т.е. указанные соединения могут содержать один или несколько атомов, чья атомная масса или массовое число отличается от атомной массы или массового числа наиболее распространенных природных изотопов. Радиоизотопы водорода и углерода включают 3H и 14C соответственно. Соединения данного изобретения, которые содержат такие радиоизотопы и/или другие радиоизотопы других атомов, находятся в сфере настоящего изобретения. Такие соединения могут быть получены с помощью методов, описанных здесь, простой заменой немеченых реагентов соответствующими мечеными реагентами. Тритиевые, т.е. 3H, и углеродные, т.е. 14С, радиоизотопы являются особенно предпочтительными благодаря простоте приготовления и обнаружения.
Соединения настоящего изобретения могут существовать в свободной форме или, если необходимо, в виде фармацевтически приемлемой соли или других производных (например, пролекарства). Термин «фармацевтически приемлемая соль» в контексте данной заявки относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Такие соли могут быть получены как in situ в процессе выделения или очистки соединений изобретения, так и отдельно, путем взаимодействия свободной кислоты или основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты.
Соединения настоящего изобретения могут существовать в виде пролекарства. В контексте настоящей заявки термин «пролекарство» обозначает предшественника или производную форму соединения согласно изобретению, обладающую улучшенными свойствами, такими как лучшая растворимость, уменьшенная цитотоксичность или повышенная биодоступность по сравнению с исходным соединением или лекарственным средством, и способную активироваться или превращаться в более активную исходную форму. “Группа пролекарства” означает группу, которая преобразуется посредством реакции с ферментом, желудочной кислотой или т.п. в физиологических условиях in vivo, с получением соединения общей формулы (I), (II) или (III), т.е. группу, которая преобразуется с получением соединения общей формулы (I), (II) или (III), посредством гидролиза или т.п., вызванного желудочной кислотой или т.п.
Термины «терапевтически эффективное количество» и «профилактически эффективное количество» означают такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение или профилактику заболеваний, вызванных вирусами рода Orthoflavivirus. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Термин «пациент» (или «субъект») охватывает все виды млекопитающих, предпочтительно человека, которые используют соединения в рамках данного изобретения как путем самостоятельного введения, так и/или введения пациенту другим лицом для лечения и/или профилактики заболевания или медицинского состояния.
Термины «лечение», «терапия» охватывают лечение патологических состояний у пациентов, и включают: а) блокирование (приостановку) течения заболевания, б) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания.
Термины «профилактика», «предотвращение», «превентивная терапия» означают уменьшение факторов риска, а также профилактическое лечение субклинических стадий заболевания у человека, направленное на уменьшение вероятности возникновения клинических стадий заболевания. К профилактической терапии относится (а) первичная профилактика, то есть профилактический приём лекарственного средства лицами, выезжающими на территории с высоким риском заражения, (б) постконтактная профилактика, то есть профилактическое лечение пациентов, вступивших в контакт с переносчиком заболевания, клиническая стадия заболевания у которых ещё не наступила, и (в) вторичная профилактика, то есть предотвращение повторного наступления того же или близкого клинического состояния заболевания или уменьшение риска персистирующей инфекции.
Осуществление изобретения
Примеры
Конкретные соединения по изобретению, раскрытые в настоящем документе, приводятся для целей иллюстрирования настоящего изобретения, чтобы изобретение могло быть более полно понято. Показанные частные примеры соединений по изобретению могут быть получены методами, описанными в общих схемах синтеза, примерах и известных методах в данной области техники, однако эти методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Соединения, приведенные ниже, не должны рассматриваться как единственные примеры в рамках настоящего изобретения, и никоим образом не ограничивают изобретение.
Некоторые частные примеры соединений по изобретению представлены в таблице 1.
Общие способы синтеза
Исходные соединения 8, 9a-d, 10a-d, 11 были получены в соответствии с ранее опубликованными методиками:
- K. N. Sedenkova, E. B. Averina, Y. K. Grishin, A. G. Kutateladze, V. B. Rybakov, T. S. Kuznetsova, N. S. Zefirov, Three-component Heterocyclization of gem-Bromofluorocyclopropanes with NOBF4: Access to 4-Fluoropyrimidine N-Oxides. J. Org. Chem. 2012, 77, 9893-9899;
- Sedenkova K. N., Averina E. B., Grishin Y. K., Bacunov A. B., Troyanov S. I., Morozov I. V., Deeva E. B., Merkulova A. V., Kuznetsova T. S., Zefirov N. S. Nitronium salts as novel reagents for the heterocyclization of gem-bromofluorocyclopropanes into pyrimidine derivatives. Tetrahedron Lett. 2015, 56 (34), 4927-4930;
- Miller, G.W., & Rose, F.L. 1080. S-triazolopyrimidines. Part I. Synthesis as potential therapeutic agents. J Chem Soc 1963, 5642-5659;
- Sedenkova. K.N.; Zverev, D.V.; Nazarova, A.A.; Lavrov, M.I.; Radchenko, E.V.; Grishin, Y.K.; Gabrel'yan, A.V.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; Palyulin, V.A. Novel Nanomolar Allosteric Modulators of AMPA Receptor of Bis(pyrimidine) Series: Synthesis, Biotesting and SAR Analysis. Molecules. 2022, 27, 8252-8273;
- Ohno, S.; Mizukoshi, K.; Komatsu, O.; Kunoh, Y.; Nakamura, Y.; Katoh, E.; Nagasaka, M. Synthesis and hypoglycemic activity of 7,8-dihydro-6H-thiopyrano(3,2-d)pyrimidine derivatives and related compounds. Chem. Pharm. Bull. 1986, 34, 4150-4165;
Ortega, J.A.; Arencibia, J.; Minniti, E.; Byl, J.; Franco-Ulloa, S.; Borgogno, M.; Genna, V.; Summa, M.; Bertozzi, S.; Bertorelli, R.; Armirotti, A.; Minarini, A.; Sissi, С.; Osheroff, N.; Vivo, M. Novel, Potent, and Druglike Tetrahydroquinazoline Inhibitor That Is Highly Selective for Human Topoisomerase II α over β. J. Med. Chem. 2020, 63, 12873-12886;
- Kim, J.Y.; Kim, D.; Kang, S.Y.; Park, W.K.; Kim, H.J.; Jung, M.E.; Son, E.J.; Pae, A.N.; Kim, J.; Lee, J. Arylpiperazine-containing pyrimidine 4-carboxamide derivatives targeting serotonin 5-HT(2A), 5-HT(2C), and the serotonin transporter as a potential antidepressant. Bioorg Med Chem Lett. 2010, 20, 6439-6442.
4-Замещенные N-оксиды тетрагидрохиназолинов 1d-f и 5a-b были получены из соответствующих 4-фтортетрагидрохиназолин-N-оксидов 9a-d. Исходные гетероциклические соединения 9a-d были синтезированы путем трехкомпонентной гетероциклизации гем-бромфторциклопропана 8 при обработке TfONO2, полученным in situ из TfOH и дымящей HNO3 в различных нитрилах (схема 1).
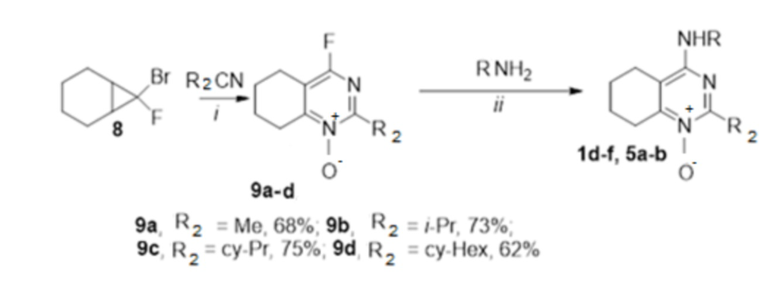
Схема 1. Синтез соединений по изобретению 1d-f, 5a-b. Реагенты и условия: (i) TfOH, HNO3, комн. темп., 48 ч; (ii) диизопропилэтиламин, EtOH, комн. темп., 30 ч.
4-(Аминофенокси)тетрагидрохиназолины 4а-с были получены из 4-хлорпиримидина 10a при обработке соответствующими аминофенолами, как показано на схеме 2.
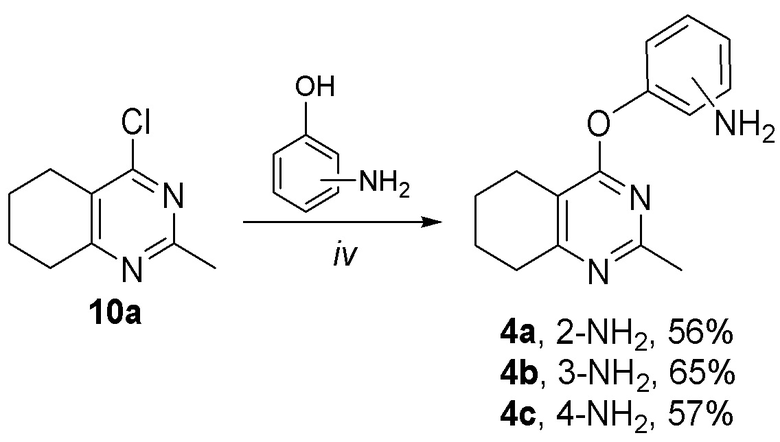
Схема 2. Синтез соединений по изобретению 4a-c. Реагенты и условия: (iv) Cs2CO3, ДМФА, 6 ч.
4-Аминотетрагидрохиназолины 2а-h были получены из 4-хлорпиримидинов 10a-d при обработке соответствующими аминами 7a-c, как показано на схеме 3.
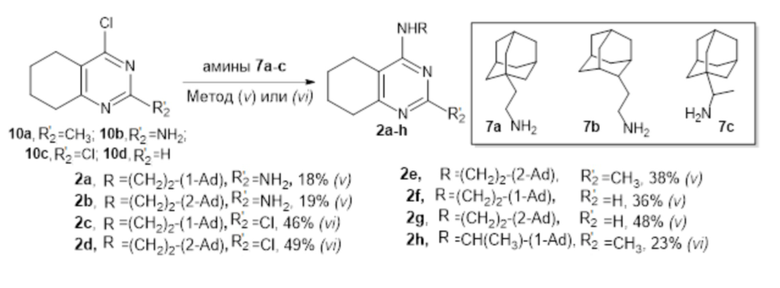
Схема 3. Синтез соединений по изобретению 2а-h. Реагенты и условия: (v) Cs2CO3, ДМФА, 2 ч. кипячение; (vi): диизопропилэтиламин, CH3CN, комн. темп.,16 ч.
Трет-бутилпиримидины 6d-f были получены из 4-хлорпиримидина 11 при обработке соответствующими аминами, как показано на схеме 4.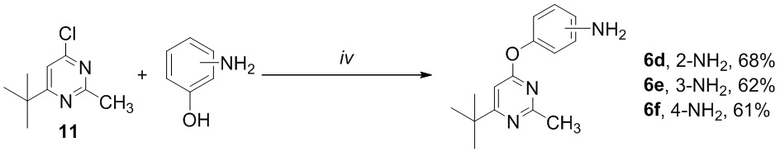
Схема 4. Синтез соединений по изобретению 6d-f. Реагенты и условия: (iv) Cs2CO3, ДМФА, 6 ч.
Растворители и материалы имели квалификацию «х.ч.» и использовались без дополнительной очистки. Спектры ЯМР 1H и 13C регистрировали на спектрометре Agilent 400-MR с частотой 400 МГц (400,0 и 100,6 МГц для 1H и 13C соответственно) при комнатной температуре; химические сдвиги δ определяли по отношению к растворителю (CDCl3, δH = 7,26 ppm, δC = 77,16 ppm). При необходимости отнесение сигналов в спектрах ЯМР производилось с использованием 2D-методов. Масс-спектрометрию высокого разрешения (HRMS) проводили на масс-спектрометре Jeol GCMate II с ионизацией электрораспылением (ESI). ТСХ выполняли на Alugram SIL G/UV254 (Macherey-Nagel, Дюрен, Германия) с УФ-визуализацией. Колоночную хроматографию проводили на силикагеле (Merck, размер частиц 0,040-0,063 мм). Все остальные материалы были коммерчески доступны.
Получение соединений по изобретению
Общая схема синтеза 4-аминотетрагидрохиназолин N-оксидов 1d-f,5a-b
Смесь 4-фтортетрагидрохиназолина N-оксида 9a-d (0,5 ммоль), соответствующего амина (0,5 ммоль) и диизопропилэтиламина (0,129 г, 1,0 ммоль) в абсолютном этаноле (1 мл) перемешивали в течение ночи в атмосфере аргона. Затем реакционную смесь разбавляли водой (1 мл) и экстрагировали дихлорметаном (3 × 4 мл). Объединенные органические экстракты промывали водой (3 × 4 мл) и сушили над MgSO4. Растворитель выпаривали при пониженном давлении; продукт выделяли с помощью препаративной колоночной хроматографии (SiO2).
4-[2-(1-адамантил)этиламино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксид (1d)
Выход 76 мг (41%). Желтоватое твердое вещество, т.пл. 102-105°C, Rf = 0.6 (CHCl3-MeOH 5:1). 1H NMR (CDCl3, δ, ppm, J, Hz): 1.28 (d, 6H, 3J = 6.8, 2CH3), 1.34-1.42 (m, 2H, CH2Ad), 1.51-1.59 (m, 6H, 3CH2, Ad), 1.60-1.68 (m, 3H, 3CH2, Ad), 1.68-1.76 (m, 3H, 3CH2, Ad), 1.75-1.86 (m, 4H, 2CH2, THQ), 1.92-2.01 (m, 3H, 3CH, Ad), 2.23-2.30 (m, 2H, CH2, THQ), 2.87-2.95 (m, 2H, CH2, THQ), 3.48-3.55 (m, 2H, CH2NH), 3.92 (sept, 1H, 3J = 6.8, CH, i-Pr), 4.59 (br.s, 1H, NH); 13C NMR (CDCl3, δ, ppm): 20.0 (2CH3), 20.9 (CH2, THQ), 21.2 (CH2, THQ), 22.2 (CH2, THQ), 24.8 (CH2, THQ), 28.7 (3CH, Ad), 29.3 (CH, i-Pr), 32.2 (C, Ad), 36.5 (CH2Ad), 37.2 (3CH2, Ad), 42.7 (3CH2, Ad), 44.1 (CH2NH), 111.4 (C(4a)), 153.5 (C(4)), 153.8 (C(8a)), 162.5 (C(2)). HRMS (ESI+, m/z): вычислено для C23H35N3O [M+H]+ 370.2853, определено 370.2858.
4-[2-(1-адамантил)этиламино]-2-циклопропил-5,6,7,8-тетрагидрохиназолин 1-оксид (1e)
Выход 82 мг (45%). Желтоватое твердое вещество, т.пл. 107-108оC, Rf = 0.8 (CHCl3-MeOH 5:1). 1H NMR (CDCl3, δ, ppm, J, Hz): 0.95-1.12 (m, 4H, 2CH2, cy-Pr), 1.21-1.32 (m, 2H, CH2Ad), 1.46 (br.s, 6H, 3CH2, Ad), 1.50-1.70 (m, 6H, 3CH2, Ad), 1.67-1.78 (m, 4H, 2CH2, THQ), 1.90 (br.s, 3H, 3CH, Ad), 2.28-2.27 (m, 2H, CH2, THQ), 2.75-2.82 (m, 2H, CH2, THQ), 2.28-2.27 (m, 2H, CH2, THQ), 2.75-2.82 (br.t, 2H, 3J = 5.8, CH2, THQ), 2.82-2.94 (m, 1H, CH, cy-Pr), 3.32-3.44 (m, 2H, CH2NH), 5.86 (br.s, 1H, NH); 13C NMR (CDCl3, δ, ppm): 10.37 (CH, cy-Pr), 10.41 (2CH2, cy-Pr), 20.9 (CH2, THQ), 21.1 (CH2, THQ), 22.3 (CH2, THQ), 24.7 (CH2, THQ), 28.6 (3CH, Ad), 32.0 (C, Ad), 36.3 (CH2Ad), 37.1 (3CH2, Ad), 42.5 (3CH2, Ad), 43.6 (CH2NH), 111.5 (C(4a)), 152.5 (C(8a)), 154.1 (C(4)), 159.1 (C(2)). HRMS (ESI+, m/z): вычислено для C23H33N3O [M+H]+ 368.2696, определено 368.2700.
4-[2-(1-адамантил)этиламино]-2-циклогексил-5,6,7,8-тетрагидрохиназолин 1-оксид (1f)
Выход 82 мг (40%). Желтоватое твердое вещество, т.пл. 96-98оC, Rf = 0.14 (легкий петролейный эфир-EtOAc-MeOH 3:1:0.5). 1H NMR (CDCl3, δ, ppm, J, Hz): 1.32-1.39 (m, 2H, CH2Ad), 1.40-1.53 (m, 4H, 2CH2, cy-Hex), 1.55 (br.s, 6H, 3CH2, Ad), 1.60-1.68 (m, 3H, 3CH2, Ad), 1.68-1.76 (m, 3H, 3CH2, Ad), 1.73-1.85 (m, 10H, 3CH2, cy-Hex + 2CH2, THQ), 1.96 (br.s, 3H, 3CH, Ad), 2.23- 2.30 (m, 2H, CH2, THQ), 2.83-2.90 (m, 2H, CH2, THQ), 3.47-3.53 (m, 2H, CH2NH), 3.57-3.67 (m, 1H, CH, cy-Hex), 4.67 (br.s, 1H, NH); 13C NMR (CDCl3, δ, ppm): 21.0 (CH2, THQ), 21.3 (CH2, THQ), 22.3 (CH2, THQ), 24.8 (CH2, THQ), 26.2 (2CH2, cy-Hex), 26.4 (CH2, cy-Hex), 28.7 (3CH, Ad), 29.9 (2CH2, cy-Hex), 32.2 (C, Ad), 36.4 (CH2Ad), 37.2 (3CH2, Ad), 38.6 (CH, cy-Hex), 42.7 (3CH2, Ad), 44.1 (CH2NH), 111.3 (C(4a)), 152.3 (C), 153.3 (C), 160.7 (C(2)). HRMS (ESI+, m/z): вычислено для C26H39N3O [M+H]+ 410.3166, определено 410.3156.
4-[(2-гидроксиэтил)амино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксид (5a)
Выход 67 мг (53%). Темно-желтые кристаллы, т.пл. 111-112°С, Rf = 0.28 (легкий петролейный эфир-EtOAc-MeOH 3:1:1). 1H NMR (CDCl3 + CD3OD, δ, ppm, J, Hz): 1.22 (d, 6H, 3J = 6.9, 2СН3), 1.69-1.83 (m, 4Н, 2СН2, THQ), 2.31-2.38 (m, 2Н, CH2, THQ), 2.70 (br.s, 2H, OH+NH), 2.76-2.84 (m, 2Н, СН2, THQ), 3.60-3.69 (m, 2Н, СН2), 3.73-3.84 (m, 3Н, СН2 + CH, i-Pr); 13C NMR (CDCl3 + CD3OD, δ, ppm): 19.8 (2CH3), 20.8 (CH2, THQ), 21.1 (CH2, THQ), 22.1 (CH2, THQ), 24.6 (CH2, THQ), 29.2 (CH), 43.9 (CH2NH), 61.3 (CH2OH), 112.3 (C(4a)), 153.4 (C(8a)), 153.9 (C(4)), 162.0 (C(2)). HRМS (ESI+, m/z): вычислено для C13H21N3O2 [М+Na]+ 274.1526, определено 254.1528.
4-{[2-(3,5-диметил-1Н-пиразол-4-ил)этил]амино}-2-метил-5,6,7,8-тетрагидрохиназолин 1-оксид (5b)
Выход 81 мг (54%). Коричневатое масло, Rf = 0.10 (легкий петролейный эфир-EtOAc-MeOH 3:1:1). 1H NMR (CDCl3 + CD3OD, δ, ppm, J, Hz): 1.67-1.84 (m, 4H, CH2, THQ), 2.16 (s, 3H, CH3, пиразол), 2.17 (s, 3H, CH3, пиразол), 2.17-2.24 (m, 2Н, CH2, THQ), 2.59 (s, 3H, CH3, THQ), 2.58-2.65 (m, 2H, CH2, THQ), 2.79-2.90 (m, 2H, CH2), 3.07-3.31 (m, 2H, 2NH), 3.46-3.55 (m, 2H, CH2); 13C NMR (CDCl3 + CD3OD, δ, ppm): 10.8 (2CH3), 20.2 (CH3), 20.8 (CH2, THQ), 21.0 (CH2, THQ), 22.1 (CH2), 23.2 (CH2, THQ), 24.5 (CH2, THQ), 41.4 (CH2), 112.0 (C(4a)), 112.1 (C(4’), пиразол), 142.5 (С(3’)+С(5’), пиразол), 152.8 (C(4)), 153.5 (C(8a)), 155.3 (C(2)). HRМS (ESI+, m/z): вычислено для C16H23N5O [М+H]+ 302.1975, определено 302.1981.
Общая схема синтеза 4-[(аминофенил)окси]тетрагидрохиназолинов 4a-c
Смесь 4-хлорпиримидина 10а (91 мг, 0,5 ммоль), Cs2CO3 (0,296 г, 1,0 ммоль) и соответствующего аминофенола (109 мг, 1,0 ммоль) в сухом ДМФА (3 мл) перемешивали с обратным холодильником в течение 24 ч в атмосфере аргона. По истечении этого времени реакционную смесь охлаждали до комп. темп., разбавляли ледяной водой (3 мл) и экстрагировали этилацетатом (3 × 15 мл). Объединенные органические экстракты промывали рассолом (3 × 5 мл) и сушили над MgSO4. Растворитель выпаривали при пониженном давлении; продукт выделяли с помощью препаративной колоночной хроматографии (SiO2).
4-[(2-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин (4а)
Выход 71 мг (56%). Желтое твердое вещество, т.пл. 223-225°С, Rf = 0.5 (CHCl3-MeOH 10:1). 1H NMR (CDCl3, δ, ppm, J, Hz): 1.81-1.90 (m, 2H, CH2, THQ), 1.90-1.97 (m, 2H, CH2, THQ), 2.48-2.54 (m, 2H, CH2, THQ), 2.53 (s, 3H, CH3), 2.71-2.77 (m, 2H, CH2, THQ), 6.40 (br.s, 2H, NH2), 6.86 (ddd, 1H, 3J = 8.0, 3J = 7.1, 4J = 1.6, CH, Ar), 6.68-7.04 (m, 1H, СН, Ar), 7.08 (dd, 1H, 3J = 8.0, 4J = 1.7, CH, Ar), 7.10-7.16 (m, 1H, CH, Ar); 13C NMR (CDCl3, δ, ppm): 22.0 (CH2, THQ), 22.2 (CH2, THQ), 22.3 (CH2, THQ), 25.1 (CH3), 31.9 (CH2, THQ), 110.1 (C(4a)), 120.3 (CH, Ar), 120.8 (CH, Ar), 122.8 (CH, Ar), 126.7 (CH, Ar), 127.8 (C, Ar), 149.4 (C, Ar), 158.0 (C), 158.1 (C), 162.9 (C). HRМS (ESI+, m/z): вычислено для C15H17N3O [М+H]+ 256.1444, определено 256.1452.
4-[(3-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин (4b)
Выход 83 мг (65%). Белое твердое вещество, т.пл. 91-94°С, Rf = 0.1 (легкий петролейный эфир-EtOAc 1:1). 1H NMR (CDCl3, δ, ppm, J, Hz): 1.75-1.90 (m, 4H, 2CH2, THQ), 2.44 (s, 3H, CH3), 2.62-2.71 (m, 2H, CH2, THQ), 2.73-2.81 (m, 2H, CH2, THQ), 3.72 (br.s, 2H, NH2), 6.41-6.45 (m, 1H, CH, Ar), 6.46-6.52 (m, 2H, 2CH, Ar), 7.12 (dd, 1H, 3J = 8.1, 3J = 7.9, CH, Ar); 13C NMR (CDCl3, δ, ppm): 21.9 (CH2, THQ), 22.2 (CH2, THQ), 22.4 (CH2, THQ), 25.7 (CH3), 32.0 (CH2, THQ), 108.3 (CH, Ar), 111.5 (CH, Ar), 111.9 (CH, Ar), 114.3 (C(4a)), 130.0 (CH, Ar), 147.8 (C, Ar), 154.3 (C, Ar), 164.4 (C(2)), 166.6 (C), 167.0 (C). HRМS (ESI+, m/z): вычислено для C15H17N3O [М+H]+ 256.1444, определено 256.1449.
4-[(4-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин (4c)
Выход 73 мг (57%). Черное твердое вещество, т.пл. 123-124°С, Rf = 0.1 (легкий петролейный эфир-EtOAc 1:1). 1H NMR (CDCl3, δ, ppm, J, Hz): 1.77-1.93 (m, 4H, 2CH2, THQ), 2.43 (s, 3H, CH3), 2.66-2.73 (m, 2H, CH2, THQ), 2.74-2.82 (m, 2H, CH2, THQ), 3.62 (br.s, 2H, NH2), 6.63-6.75 (m, 2H, 2CH, Ar), 6.88-6.99 (m, 2H, 2CH, Ar); 13C NMR (CDCl3, δ, ppm): 21.9 (CH2, THQ), 22.2 (CH2, THQ), 22.4 (CH2, THQ), 25.7 (CH3), 31.9 (CH2, THQ), 113.9 (C(4a)), 115.8 (2CH, Ar), 122.5 (2CH, Ar), 143.5 (C, Ar), 145.2 (C, Ar), 164.2 (C(2)), 166.1 (C), 167.4 (C). HRМS (ESI+, m/z): вычислено для C15H17N3O2 [М+H]+ 256.1444, определено 256.1449.
Общая схема синтеза 4-аминотетрагидрохиназолинов 2а-h
Метод (v). Смесь 4-хлорпиримидина 10а-d (2 ммоль), Cs2CO3 (0.358 г, 3 ммоль) и соответствующего амина (1 ммоль) в сухом ДМФА (3 мл) кипятили с обратным холодильником при перемешивании в течение 2 ч в атмосфере аргона. Затем реакционную смесь охладили до комнатной температуры, разбавили ледяной водой (3 мл) и экстрагировали этилацетатом (3 × 15 мл). Объединенные органические слои промыли насыщенным водным раствором NaCl (1 × 15 мл) и водой (1 × 15 мл); высушили над MgSO4. Растворитель упарили при пониженном давлении; продукт выделили методом препаративной колоночной хроматографии.
Метод (vi). К смеси 4-хлорпиримидина (0.5 ммоль) и соответствующего амина (0.45 ммоль) в сухом ацетонитриле (5 мл) добавили по каплям диизопропилэтиламин (0.4 мл, 299 мг, 2 ммоль) и перемешивали в течение 16 ч при комнатной температуре в атмосфере аргона. Затем растворитель упарили, к сухому остатку добавили воду (5 мл) и экстрагировали этилацетатом (3 × 10 мл). Объединенные органические слои высушили над MgSO4. Растворитель упарили при пониженном давлении; продукт выделили методом препаративной колоночной хроматографии.
4-(2-(1-Адамантил)этиламино)-2-амино-5,6,7,8-тетрагидрохиназолин (2а)
Выход 59 мг (18%), метод (v). Желтое твердое вещество, Тпл = 152-153 °С, Rf = 0.3 (легкий петролейный эфир- EtOAc -MeOH=1:1:0.5). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.31-1.39 м (2H, CH2, CH2-Ad), 1.44-1.58 м (6H, 3CH2, Ad), 1.63 уш. д (3H, 2J=12.3, 3CH2, Ad), 1.72 уш. д (3H, 2J=12.3, 3CH2, Ad) 1.72-1.86 м (4H, 2CH2, THQ), 1.97 уш. с (3H, 3CH, Ad), 2.16-2.22 м (2H, CH2, THQ), 2.60-2.66 м (2H, CH2, THQ), 3.35-3.52 м (2H, CH2-NH), 4.86-5.03 м (1H, NH), 6.11 уш. с (2H, NH2). Спектр ЯМР 13C (CDCl3, δ, м.д.): 21.1 (CH2, THQ), 21.3 (CH2, THQ), 22.0 (CH2, THQ), 28.1 (CH2, THQ), 28.7 (3CH, Ad), 32.2 (C, Ad), 36.6 (CH2-NH), 37.1 (3CH2, Ad), 42.6 (3CH2, Ad), 43.8 (CH2-Ad), 103.3 (С(4a), THQ), 153.6 (C(8a), THQ), 156.8 (C(2), THQ), 162.0 (C(4), THQ). HRМS (ESI+, m/z): вычислено для C20H30N4 [М+H]+ 327.2543, найдено 327.2540.
4-(2-(2-Адамантил)этиламино)-2-амино-5,6,7,8-тетрагидрохиназолин (2b)
Выход 62 мг (19%), метод (v). Желтое твердое вещество, Тпл = 147-148°С, Rf = 0.2 (легкий петролейный эфир-EtOAc-MeOH=3:3:1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.52 уш. д (2H, 2J=12.3, 2CH2, Ad), 1.63-1.94 м (19H, 5CH, Ad + 5CH2, Ad + 2CH2, THQ + CH2-Ad), 2.12-2.26 м (2H, CH2, THQ), 2.45-2.61 м (2H, CH2, THQ), 3.31-3.48 м (2H, CH2-NH), 4.44-4.59 м (1H, NH), 4.85 уш. с ( 2H, NH2). Спектр ЯМР 13C (CDCl3, δ, м.д.): 21.7 (CH2, THQ), 22.3 (CH2, THQ), 22.6 (CH2, THQ), 28.1 (CH, Ad), 28.3 (CH, Ad), 31.1 (CH2, THQ), 31.8 (2CH2, Ad), 32.0 (2CH, Ad), 32.8 (CH2-Ad), 38.4 (CH2, Ad), 39.2 (2CH2, Ad), 39.7 (CH2-NH), 42.2 (CH, Ad), 102.9 (С(4a), THQ), 159.8 (C(8а), THQ), 160.1 (C(4), THQ), 161.8 (C(2), THQ). HRМS (ESI+, m/z): вычислено для C20H30N4 [М+H]+ 327.2543, найдено 327.2535.
4-(2-(1-Адамантил)этиламино)-2-хлор-5,6,7,8-тетрагидрохиназолин (2c)
Выход 72 мг (46%), метод (vi). Бесцветное твердое вещество, Тпл = 200-201°С, Rf = 0.1 (легкий петролейный эфир-EtOAc = 3:0.5). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.30-1.42 м (2H, CH2-Ad), 1.48-1.58 м (6H, 3CH2, Ad), 1.63 уш. д (3H, 2J=11.9, 3CH2, Ad), 1.71 уш. д (3H, 2J=11.9, 3CH2, Ad), 1.74-1.87 м (4H, 2CH2, THQ), 1.96 уш. с (3H, 3CH, Ad), 2.14-2.29 м (2H, CH2, THQ), 2.56-2.74 м (2H, CH2, THQ), 3.38-3.54 (2H, CH2-NH), 4.39-4.57 м (1H, NH). Спектр ЯМР 13C (CDCl3, δ, м.д.): 21.9 (CH2, THQ), 22.1 (2CH2, THQ), 28.7 (3CH, Ad), 31.7 (CH2, THQ), 32.2 (C, Ad), 36.5 (CH2-NH), 37.1 (3CH2, Ad), 42.6 (3CH2, Ad), 44.1 (CH2-Ad), 110.2 (С(4a), THQ), 157.6 (C(2), THQ), 162.0 (C(4), THQ), 163.6 (C(8a), THQ). HRМS (ESI+, m/z): вычислено для C20H28ClN3 [М+H]+ 346.2045, найдено 346.2043.
4-(2-(2-Адамантил)этиламино)-2-хлор-5,6,7,8-тетрагидрохиназолин (2d)
Выход 76 мг (49%), метод (vi). Бесцветное твердое вещество, Тпл = 198-199°С, Rf = 0.2 (легкий петролейный эфир-EtOAc = 3:1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.52 уш. д (2H, 2J=12.5, 2CH2, Ad), 1.63-1.94 м (19H, 5CH, Ad + 5CH2, Ad + 2CH2, THQ + CH2-Ad), 2.17-2.32 м (2H, CH2, THQ), 2.60-2.71 м (2H, CH2, THQ), 3.39-3.51 м (2H, CH2-NH), 4.58-4.72 м (1H, NH). Спектр ЯМР 13C (CDCl3, δ, м.д.): 22.0 (CH2, THQ), 22.1 (2CH2, THQ), 28.1 (CH, Ad), 28.3 (CH, Ad), 31.7 (CH2, THQ), 31.8 (2CH2, Ad), 32.0 (2CH, Ad), 32.6 (CH2-Ad), 38.4 (CH2, Ad), 39.2 (2CH2, Ad), 40.0 (CH2-NH), 42.1 (CH, Ad), 102.2 (С(4a), THQ), 157.6 (C(2), THQ), 162.1 (C(4), THQ), 163.7 (C(8a), THQ). HRМS (ESI+, m/z): вычислено для C20H28ClN3 [М+H]+ 346.2045, найдено 346.2043.
4-(2-(2-Адамантил)этиламино)-2-метил-5,6,7,8-тетрагидрохиназолин (2e)
Выход 124 мг (38%), метод (v). Желтое твердое вещество, Тпл = 164-165°С, Rf = 0.2 (легкий петролейный эфир-EtOAc-MeOH=1:1:0.1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.54 уш. д (2H, 2J=12.0, 2CH2, Ad), 1.68-1.94 м (19H, 5CH, Ad + 5CH2, Ad + 2CH2, THQ + CH2-Ad), 2.21-2.29 м (2H, CH2, THQ), 2.47 с (3H, CH3), 2.59-2.69 м (2H, CH2, THQ), 3.44-3.55 м (2H, CH2-NH), 4.33-4.43 м (1H, NH). Спектр ЯМР 13C (CDCl3, δ, м.д.): 22.0 (CH2, THQ), 22.4 (CH2, THQ), 22.5 (CH2, THQ), 26.1 (CH3), 28.2 (CH, Ad), 28.3 (CH, Ad), 31.8 (2CH2, Ad + CH2, THQ), 32.1 (2CH, Ad), 33.0 (CH2-Ad), 38.5 (CH2, Ad), 39.3 (2CH2, Ad), 39.7 (CH2-NH), 42.2 (CH, Ad), 108.5 (С(4a), THQ), 160.7 (C, THQ), 161.0 (C, THQ), 164.2 (C, THQ). HRМS (ESI+, m/z): вычислено для C21H31N3 [М+H]+ 326.2591, найдено 326.2582.
4-(2-(1-Адамантил)этиламино)-5,6,7,8-тетрагидрохиназолин (2f)
Выход 112 мг (36%), метод (v). Бесцветное твердое вещество, Тпл = 151-152°С, Rf = 0.1 (легкий петролейный эфир-EtOAc = 3:0.5).Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.36-1.43 м (2H, CH2, CH2-Ad), 1.51-1.59 м (6H, 3CH2, Ad), 1.64 уш. д (3H, 2J=11.8, 3CH2, Ad), 1.72 уш. д (3H, 2J=11.8, 3CH2, Ad), 1.76-1.89 м (4H, 2CH2, THQ), 1.96 уш. с (3H, 3CH, Ad), 2.23-2.30 м (2H, CH2, THQ), 2.67-2.74 м (2H, CH2, THQ), 3.44-3.52 м (2H, CH2-NH), 4.33 уш. с (1H, NH), 8.43 уш. с (1H, CH, THQ).Спектр ЯМР 13C (CDCl3, δ, м.д.): 22.13 (CH2, THQ), 22.15 (CH2, THQ), 22.2 (CH2, THQ), 28.7 (3CH, Ad), 31.5 (CH2, THQ), 32.2 (C, Ad), 36.4 (CH2-NH), 37.2 (3CH2, Ad), 42.6 (3CH2, Ad), 44.3 (CH2-Ad), 111.9 (С(4a), THQ), 155.1 (CH, THQ), 160.5 (C(4), C(8a), THQ). HRМS (ESI+, m/z): вычислено для C20H29N3 [М+H]+ 312.2434, найдено 312.2440.
4-(2-(2-Адамантил)этиламино)-5,6,7,8-тетрагидрохиназолин (2g)
Выход 149 мг (48%), метод (v). Бесцветное твердое вещество, Тпл = 155-156°С, Rf =0.5 (CH2Cl2-MeOH = 10:1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.49 уш. д (2H, 2J=12.3, 2CH2, Ad), 1.57-1.96 м (19H, 5CH, Ad + 5CH2, Ad + 2CH2, THQ + CH2-Ad), 2.18-2.31 м (2H, CH2, THQ), 2.58-2.72 м (2H, CH2, THQ), 3.35-3.51 м (2H, CH2-NH), 4.48-4.65 м (1H, NH), 8.37 c (1H, CH, THQ). Спектр ЯМР 13C (CDCl3, δ, м.д.): 22.15 (2CH2, THQ), 22.21 (CH2, THQ), 28.0 (CH, Ad), 28.2 (CH, Ad), 31.7 (2CH2, Ad + CH2, THQ), 31.9 (2CH, Ad), 32.6 (CH2-Ad), 38.3 (CH2, Ad), 39.1 (2CH2, Ad), 39.8 (CH2-NH), 42.2 (CH, Ad), 111.8 (С(4a), THQ), 155.3 (CH, THQ), 160.3 (C(4), THQ), 160.9 (C(8a), THQ). HRМS (ESI+, m/z): вычислено для C20H29N3 [М+H]+ 312.2434, найдено 312.2438.
4-(1-(1-Адамантил)этиламино)-2-метил-5,6,7,8-тетрагидрохиназолин (2h)
Выход 75 мг (23%), метод (vi). Бесцветное твердое вещество, Тпл = 139-140°С, Rf = 0.2 (легкий петролейный эфир-EtOAc-MeOH = 6:6:1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.08 д (3H, 2J=6.7, CH3-CH), 1.52 уш. д (3H, 2J=11.8, 3CH2, Ad), 1.56-1.67 м (6H, 6CH2, Ad), 1.71 уш. д (3H, 2J=11.8, 3CH2, Ad), 1.76-1.90 м (4H, 2CH2, THQ), 1.99 уш. с (3H, 3CH, Ad), 2.16-2.37 м (3H, CH2, THQ + NH), 2.45 с (3H, CH3), 2.61-2.70 м (2H, CH2, THQ), 4.10-4.19 м (1H, CH-NH), 4.30-4.38 м (1H, NH). Спектр ЯМР 13C (CDCl3, δ, м.д.): 14.4 (CH3-CH), 21.5 (CH2, THQ), 21.8 (CH2, THQ), 21.9 (CH2, THQ), 25.6 (CH3), 28.0 (3CH, Ad), 31.2 (CH2, THQ), 35.8 (C, Ad), 36.7 (3CH2, Ad), 38.1 (3CH2, Ad), 52.8 (CH-NH), 107.6 (С(4а), THQ), 160.0 (C, THQ), 160.3 (С, THQ), 163.5 (C, THQ). HRМS (ESI+, m/z): вычислено для C21H31N3 [М+H]+ 326.2591, найдено 326.2588.
Общая схема синтеза трет-бутилпиримидинов 6d-f
Смесь 4-хлорпиримидина 11 (91 мг, 0,5 ммоль), Cs2CO3 (0,296 г, 1,0 ммоль) и соответствующего аминофенола (109 мг, 1,0 ммоль) в сухом ДМФА (3 мл) перемешивали с обратным холодильником в течение 24 ч в атмосфере аргона. По истечении этого времени реакционную смесь охлаждали до комп. темп., разбавляли ледяной водой (3 мл) и экстрагировали этилацетатом (3 × 15 мл). Объединенные органические экстракты промывали рассолом (3 × 5 мл) и сушили над MgSO4. Растворитель выпаривали при пониженном давлении; продукт выделяли с помощью препаративной колоночной хроматографии (SiO2).
4-[(2-Аминофенил)окси]-2-метил-6-трет-бутилпиримидин (6d)
Выход 96 мг (68%). Оранжевое твердое вещество, Тпл = 48-49°С; Rf = 0.4 (легкий петролейный эфир- EtOAc =3:1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.27 с (9H, 3CH3, t-Bu), 2.53 с (3H, CH3), 6.47 уш. c (1H, Pyr), 6.83 ддд (1H, 4J=2.2, 3J=6.7, 3J=7.9, CH, Ar), 6.91-7.12 м (5H, 3CH, Ar + NH2). Спектр ЯМР 13C (CDCl3, δ, м.д.): 25.5 (CH3), 29.2 (3CH3, t-Bu), 37.3 (C, t-Bu), 98.7 (CH, Pyr), 120.2 (CH, Ar), 120.4 (CH, Ar), 122.4 (CH, Ar), 126.2 (CH, Ar), 128.1 (C), 148.9 (C), 160.4 (C), 165.8 (C), 177.7 (C(6), Pyr). HRMS (ESI+, m/z): вычислено для C15H19N3O [М+H]+ 258.1601, найдено 258.1602.
4-[(3-Аминофенил)окси]-2-метил-6-трет-бутилпиримидин (6e)
Выход 88 мг (62%). Бесцветное твердое вещество, Тпл = 100-101°С; Rf = 0.4 (легкий петролейный эфир- EtOAc =2:1). Спектр ЯМР 1H (CDCl3, δ, м.д., J, Гц): 1.28 с (9H, 3CH3, t-Bu), 2.57 с (3H, CH3), 3.78 уш. с (2H, NH2), 6.45 псевд. т (1H, 4J=2.2, CH, Ar), 6.48 ддд (1H, 4J=0.8, 4J=2.2, 3J=8.0, CH, Ar), 6.54 ддд (1H, 4J=0.8, 4J=2.2 CH, 3J=8.0, CH, Ar), 6.54 уш. c (1H, Pyr), 7.16 псевд. т. (1H, 3J=8.0, СH, Ar). Спектр ЯМР 13C (CDCl3, δ, м.д.): 26.2 (CH3), 29.4 (3CH3, t-Bu), 37.5 (C, t-Bu), 99.2 (CH, Pyr), 108.1 (CH, Ar), 111.2 (CH, Ar), 112.2 (CH, Ar), 130.4 (CH, Ar), 148.2 (C-NH2, Ar), 154.0 (C-O, Ar), 167.8 (C(2), Pyr), 170.3 (C(4), Pyr), 180.3 (C(6), Pyr). HRМS (ESI+, m/z): вычислено для C15H19N3O [М+H]+ 258.1601, найдено 258.1600.
4-[(4-Аминофенил)окси]-2-метил-6-трет-бутилпиримидин (6f)
Выход 86 мг (61%). Черное твердое вещество, Тпл = 93-94°С; Rf = 0.5 (легкий петролейный эфир- EtOAc =1:1). Спектр ЯМР 1H (CDCl3, δ, м.д.): 1.25 с (9H, 3СH3, t-Bu), 2.54 с (3H, CH3), 3.69 уш. с (2H, NH2), 6.48 c (1H, Pyr), 6.63-6.71 м (2H, 2CH, Ar), 6.86-6.94 м (2H, 2CH, Ar). Спектр ЯМР 13C (CDCl3, δ, м.д.): 26.1 (CH3), 29.3 (3CH3, t-Bu), 37.4 (C, t-Bu), 98.7 (CH, Pyr), 116.0 (2CH, Ar), 122.2 (2CH, Ar), 144.0 (C), 144.8 (C), 167.6 (C(2), Pyr), 170.9 (C(4), Pyr), 180.0 (C(6), Pyr). HRМS (ESI+, m/z): вычислено для C15H19N3O [М+H]+ 258.1601, найдено 258.1597.
Характеристика биологической активности соединений по изобретению
Клетки и вирусы
Перевиваемые клетки почки эмбриона свиньи (СПЭВ) растили в смеси среды 199 с солями Эрла и среды 199 с солями Хэнкса (ФГАНУ “ФНЦИРИП им. М.П. Чумакова РАН” (Институт полиомиелита)) с добавлением 5% инактивированной нагреванием эмбриональной телячьей сыворотки (Gibco) и пенициллина-стрептомицина (ООО "Панэко"). Перевиваемые клетки почки африканской зеленой мартышки (Vero) растили в среде, состоящей из DMEM с L-глутамином (ФГАНУ “ФНЦИРИП им. М.П. Чумакова РАН” (Институт полиомиелита)) с добавлением 5% инактивированной нагреванием эмбриональной телячьей сыворотки (Gibco) и гентамицина (ООО "Панэко"). Обе клеточные линии были получены из коллекции ФГАНУ “ФНЦИРИП им. М.П. Чумакова РАН” (Институт полиомиелита), Россия.
Штамм вируса клещевого энцефалита Абсеттаров (номер доступа Genbank KU885457.1), штамм вируса желтой лихорадки 17D (номер доступа Genbank JN628279.1) и штамм вируса Западного Нила Strix nebulosa-12 (номер доступа Genbank OP868929) были взяты из коллекции ФГАНУ “ФНЦИРИП им. М.П. Чумакова РАН” (Институт полиомиелита), Россия.
Исследуемые соединения растворяли в ДМСО (VWR Life Science) для приготовления исходных растворов с концентрацией 5-10 ммоль/л.
Анализ цитотоксичности на клетках Vero и СПЭВ
Цитотоксичность определяли по влиянию соединений по изобретению на жизнеспособность клеток и измеряли посредством резазурин-теста. При добавлении к живым клеткам краситель резазурин под действием клеточных ферментов восстанавливается до флуоресцентного красителя резоруфина. Токсическое действие соединений по изобретению детектируется по снижению интенсивности флуоресценции по сравнению с контрольными клетками.
Клетки Vero или СПЭВ высаживали в 96-луночные культуральные планшеты и инкубировали в течение 24 часов при 37°C и 5% CO2. Из соединений готовили серии последовательных разведений 1:2, начиная с концентрации 50-100 мкмоль/л. В качестве контроля аналогичным образом готовили серии разведений ДМСО, начиная с концентрации 1%. Растворителем для эксперимента на клетках Vero служила среда DMEM с добавлением L-глутамина, для эксперимента на клетках СПЭВ - смесь среды 199 (соли Эрла) и среды 199 (соли Хэнкса) с добавлением 2% инактивированной эмбриональной телячьей сыворотки. В качестве положительного контроля, вызывающего 100% гибель клеток, использовали водный раствор додецилсульфата натрия в концентрации 0,5 мг/мл. Равные объемы разведений соединений переносили на клетки, после чего планшеты с клетками инкубировали при 37 °C и 5% CO2 в течение 7 дней для определения хронической цитотоксичности. После инкубации монослой клеток промывали фосфатно-солевым буфером и к клеткам добавляли раствор резазурина (Sigma-Aldrich) в среде 199 (на растворе Эрла) с концентрацией 25 мкг/мл. Клетки инкубировали 4 часа при 37 °С и 5% СО2, затем собирали культуральную жидкость и измеряли в ней флуоресценцию резоруфина при λex/em = 544/590 нм. Значение CC50 рассчитывали по методу Рида-Менча.
Анализ активности против ВКЭ
Анализ активности против ВКЭ проводили методом ингибирования бляшкообразования.
Клетки СПЭВ высаживали в 24-луночные культуральные планшеты и инкубировали в течение 3 суток при 37 °C и 5% CO2. Из соединений готовили серии последовательных разведений 1:4 в среде 199 (на растворе Эрла), начиная с концентрации 50-100 мкмоль/л. В качестве контроля аналогичным образом готовили серии разведений ДМСО, начиная с концентрации 1%. К полученным разведениям добавляли вирусную суспензию и инкубировали смеси 1 час при 37 °C и 5% CO2. Титр вируса был подобран так, чтобы при заражении на каждую лунку с клетками приходилось по 30-60 бляшкообразующих единиц. Клетки СПЭВ заражали полученными смесями и инкубировали с периодическими покачиваниями в течение 1 часа при 37 °C и 5% CO2, затем зараженные клетки покрывали 1,5% раствором метилцеллюлозы в смеси среды 199 (соли Эрла) и среды 199 (соли Хэнкса) с добавлением 2% эмбриональной телячьей сыворотки и инкубировали 6 суток при 37 °C и 5% CO2. После инкубации клетки освобождали от покрытия, промывали фосфатно-солевым буфером, фиксировали 96% этанолом и окрашивали 0,5% раствором кристаллического фиолетового. Значение EC50 рассчитывали по методу Рида-Менча после подсчета бляшек.
Анализ активности против ВЖЛ
Активность против ВЖЛ измеряли аналогично активности против ВКЭ с небольшими изменениями. В частности, вместо клеток СПЭВ использовали Vero и увеличили время инкубации зараженных клеток до 7 суток. Остальные этапы работы осуществляли как указано выше.
Анализ активности против ВЗН
Активность против ВЗН измеряли аналогично активности против ВЖЛ с небольшими изменениями. В частности, для разведения использовали среду DMEM и уменьшили время инкубации зараженных клеток до 5 суток. Остальные этапы работы осуществляли как указано выше.
Результаты проведенных исследований
Противовирусную активность соединений по изобретению оценивали в отношении, в частности, вируса клещевого энцефалита на клетках СПЭВ, и вирусов клещевого энцефалита, желтой лихорадки и лихорадки Западного Нила на клетках Vero методом ингибирования бляшкообразования (табл. 2-4).
(7 дней)
(7 дней)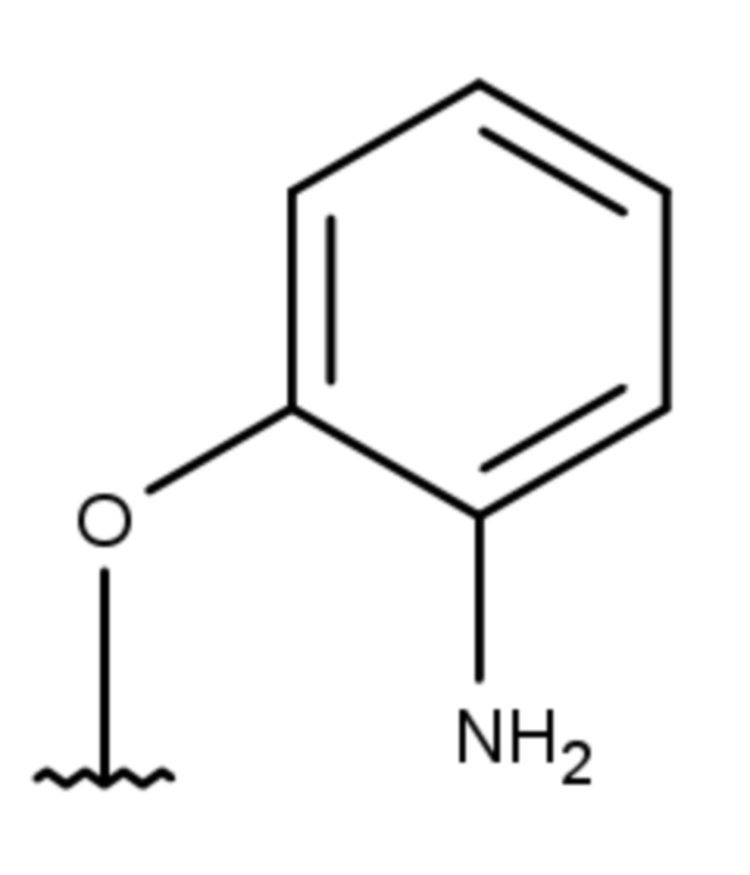
(7 дней)
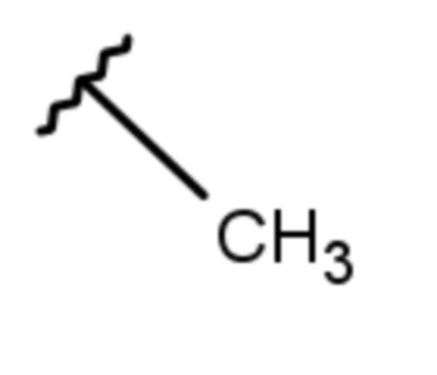
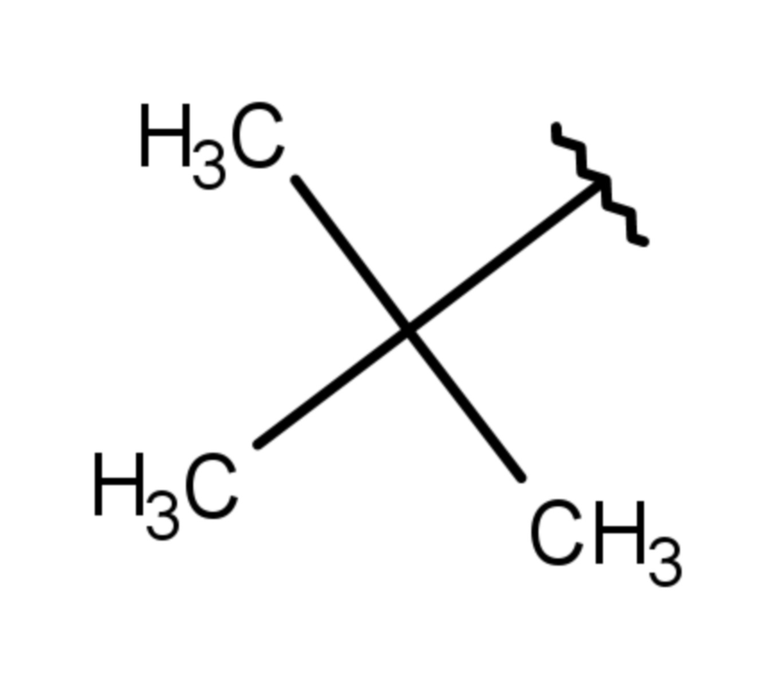
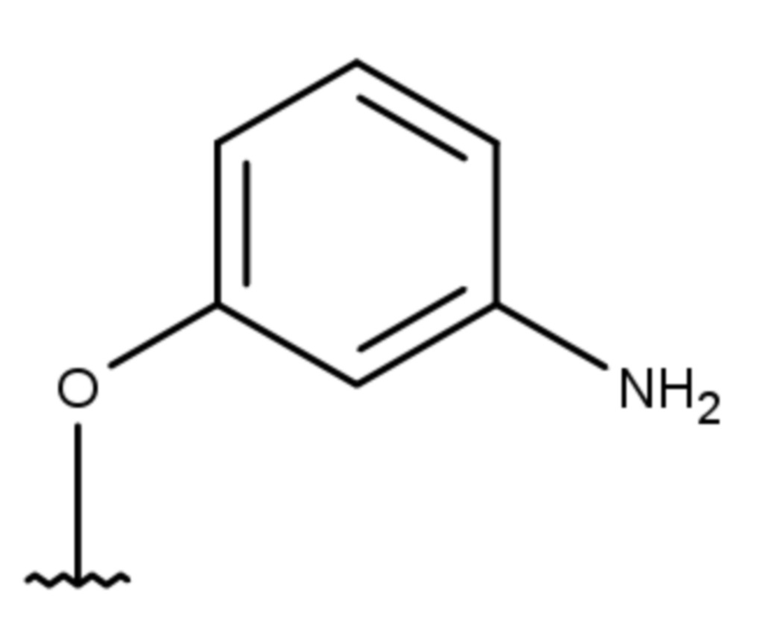
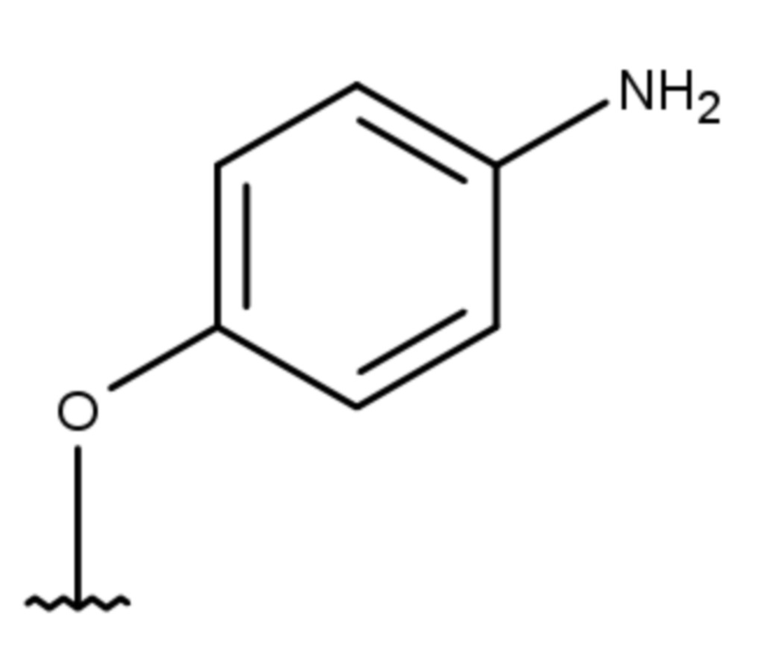
(7 дней)
(7 дней)
(7 дней)
(7 дней)
(7 дней)
Таким образом, результаты исследования биологической активности показывают, что соединения по изобретению характеризуются высокой эффективностью в ингибировании репродукции вирусов, относящихся к роду Orthoflavivirus, в том числе ВКЭ, ВЖЛ, ВЗН и обладают низкой цитотоксичностью, что в сочетании с высокой противовирусной активностью является необходимым условием для проведения дальнейшей фармацевтической разработки.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Изобретение также относится к фармацевтическим композициям, которые содержат соединение общей формулы (I), (II) или (III) (или про-лекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, которые могут быть введены в организм пациента вместе с соединением, составляющим сущность этого изобретения, не снижают фармакологическую активность этого соединения и не токсичны при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, указанные в этом изобретении, содержат соединения этого изобретения вместе с фармацевтически приемлемыми носителями, которые могут включать любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, антибактериальные агенты и фунгициды, регуляторы пролонгированной доставки, смазочные вещества, красители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты. и т.д., выбор и соотношение которых зависит от способа назначения и дозировки. В соответствии с известными методами фармацевтические композиции могут быть представлены различными жидкими (растворы, эмульсии, суспензии) или твердыми (таблетки, капсулы, пилюли) лекарственными формами. Стандартные процедуры их получения состоят в смешении соединений этого изобретения с жидким или тонкоизмельченным твердым носителем.
Примером получения таблеток является смешивание активного компонента с фармацевтическими наполнителями, такими как лактоза, крахмал, тальк, диоксид кремния и т.д. формирование таблеток и, при необходимости, их покрытие сахарозой, или другими подходящими веществами.
Желатиновые капсулы можно получить, смешивая активное соединение с растворителем, красителями и стабилизаторами и заполняя полученной смесью мягкие или твердые капсулы, или же использовать капельный метод, одновременно дозируя полученную смесь и нагретую желатиновую массу в охлажденное вазелиновое масло.
Инъекционные формы композиции как правило представляют собой водные суспензии, изотонические физиологические растворы или стерильные растворы для инъекций с добавлением консервантов и стабилизаторов.
Примеры получения композиции по изобретению
Метод А: таблетирование .
Для приготовления 10000 шт. 100 мг таблеток, содержащих по 0,1 мг 4-[2-(1-адамантил)этиламино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксида (1d), смешивают 1 г соединения 1d с 200 г трагаканта (связующее вещество), 549 г карбоната кальция (наполнитель), 200 г крахмала (разрыхляющее вещество) и 50 г лимонной кислоты (отдушка). Затем на специальной установке из полученной смеси получают прессованные таблетки.
Метод Б: приготовление капсул.
Для приготовления 10000 шт. 500 мг капсул, содержащих по 0,5 мг 4-[2-(1-адамантил)этиламино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксида (1d), смешивают 5 г соединения 1d с 1000 г глицерина и 3395 г сахарного сиропа, затем добавляют 400 г мятного масла, 100 г бензоата натрия, 50 г аскорбиновой кислоты и 50 г красителя тетразина. Капсулы получают капельным методом, одновременно дозируя полученную смесь и нагретую желатиновую массу (900 г) в охлажденное вазелиновое масло. В результате образуются готовые к употреблению шарообразные капсулы, содержащие по 0,5 мг активного вещества.
Метод В: приготовление раствора для инъекций.
Для приготовления 1000 шт. 2 мл ампул, содержащих 1 мг 4-[2-(1-адамантил)этиламино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксида (1d), смешивают 1 г соединения (1d) с 400 мл мятного масла, 10 г метилцеллюлозы, 1600 мл раствора 0,9% раствора хлорида натрия с добавлением 2% ДМСО, а также 10 г бензойной кислоты. Полученную смесь отфильтровывают от механических примесей и разливают по ампулам, которые затем запечатывают и стерилизуют в автоклаве.
МЕТОДЫ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ
Соединения настоящего изобретения обладают активностью против флавивирусов, а потому пригодны для терапии и/или профилактики заболеваний, вызванных флавивирусами, в том числе клещевого энцефалита, жёлтой лихорадки, лихорадки Западного Нила, лихорадки денге, денге шок-синдрома, лихорадки Зика. В качестве пациента может выступать как человек, так и другие млекопитающие.
Соединения изобретения возможно применять в составе фармацевтической композиции в любой подходящей фармацевтической лекарственной форме и любым подходящим способом введения (внутривенно, внутримышечно, перорально, подкожно, ингаляционно, интраназально и сублингвально), а также с любой длительностью приема, необходимой для лечения и/или профилактики заболеваний.
Изобретение также относится к фармацевтической композиции, содержащей ежедневную дозу активного соединения в форме фиксированной единицы дозировки, и к комбинации, содержащей указанную фармацевтическую композицию или указанное соединение. В предпочтительном варианте осуществления указанную композицию для применения в соответствии с изобретением вводят перорально или внутривенно один или несколько раз в день в дозировке 1 мг или более выбранного соединения по изобретению. Предпочтительная дозировка составляет 1-1500 мг. Наиболее предпочтительная дозировка составляет 10-1000 мг.
В комбинации с соединениями изобретения возможно также применять дополнительные фармакологически активные агенты в одной или отдельной лекарственной форме. Терапевтическое действие активных агентов может быть одновременным или последовательным и направлено на облегчение симптомов, сопутствующих вирусной инфекции, таких лихорадка, озноб, головная боль, вторичные инфекции и др. Примером дополнительных активных агентов являются жаропонижающие средства, противовоспалительные средства, химиотерапевтические средства, антибиотики, противогрибковые средства, химиотерапевтические средства, интерфероны, цитокины, монокины, антитела или их комбинации. Вводить их возможно как одновременно с соединением изобретения, так и в отдельные моменты времени.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения - без отступления от сути настоящего изобретения возможно осуществление различных модификаций.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЁННЫЕ ИЗОКСАЗОЛЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2733945C2 |
| ПРОИЗВОДНЫЕ ПИРИДОКСИНА С НЕЛИНЕЙНЫМИ ОПТИЧЕСКИМИ СВОЙСТВАМИ | 2012 |
|
RU2501801C1 |
| Четвертичные аммониевые соединения на основе производных пиридоксина и жирных карбоновых кислот, обладающие антибактериальной активностью | 2022 |
|
RU2795265C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| Производные аденозина - ингибиторы репродукции вирусов, относящихся к роду Flavivirus | 2023 |
|
RU2828777C1 |
| Производные аденозина - ингибиторы репродукции вирусов, относящихся к роду Flavivirus | 2024 |
|
RU2839716C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3',4',5,6-ТЕТРАГИДРО-1Н-СПИРО[ПИРИДИН-4,5'-ТИЕНО[2,3-d]ПИРИМИДИНОВ] | 2011 |
|
RU2455306C1 |
| АНТИРЕТРОВИРУСНЫЕ ПРЕПАРАТЫ НА ОСНОВЕ ПРОИЗВОДНЫХ АЗИДОТИМИДИНА | 2014 |
|
RU2561501C1 |
| АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, И СПОСОБ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ | 1994 |
|
RU2130011C1 |
Изобретение относится к органической и медицинской химии, фармакологии и медицине, а именно к соединению общей формулы (I) или его фармацевтически приемлемой соли, где R1 выбирается независимо и представляет собой: NH-(C1-5-алкил)-(1-адамантил); NH-(C1-5-алкил)-(2-адамантил); NH-(C1-5-алкил)-OH; 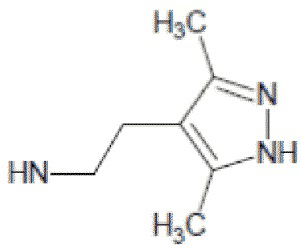 ; R2 выбирается независимо и представляет собой: С3-6-циклоалкил, изопропил, альтернативно R2 представляет собой метил, если R1 представляет собой ; n принимает значения 0-2. Изобретение также относится к соединению общей формулы (II) или его фармацевтически приемлемой соли, где если R’1 выбирается независимо и представляет собой: NH-(C1-5-алкил)-(1-адамантил); NH-(C1-5-алкил)-(2-адамантил); то R’2 выбирается независимо и представляет собой водород, метил, -NH2, галоген; альтернативно если R’1 выбирается независимо и представляет собой:
; R2 выбирается независимо и представляет собой: С3-6-циклоалкил, изопропил, альтернативно R2 представляет собой метил, если R1 представляет собой ; n принимает значения 0-2. Изобретение также относится к соединению общей формулы (II) или его фармацевтически приемлемой соли, где если R’1 выбирается независимо и представляет собой: NH-(C1-5-алкил)-(1-адамантил); NH-(C1-5-алкил)-(2-адамантил); то R’2 выбирается независимо и представляет собой водород, метил, -NH2, галоген; альтернативно если R’1 выбирается независимо и представляет собой:
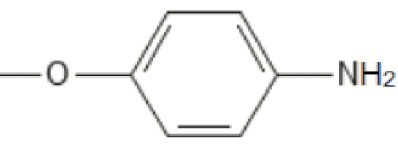 ;
;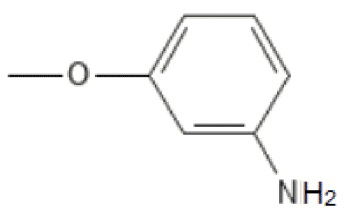 ;
;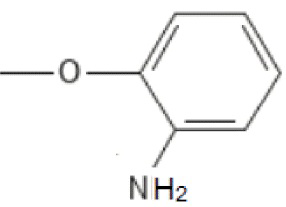 ; то R’2 выбирается независимо и представляет собой метил, -NH2, галоген; n принимает значения 0-2. Изобретение также относится к фармацевтической композиции на основе указанных соединений и их применению в качестве ингибитора репродукции вируса, относящегося к роду Orthoflavivirus. Технический результат заключается в разработке и получении соединений, характеризующихся высокой эффективностью в ингибировании репродукции вирусов рода Orthoflavivirus, в частности вируса клещевого энцефалита, вируса желтой лихорадки, вируса лихорадки Западного Нила и других. 4 н. и 7 з.п. ф-лы, 4 табл.
; то R’2 выбирается независимо и представляет собой метил, -NH2, галоген; n принимает значения 0-2. Изобретение также относится к фармацевтической композиции на основе указанных соединений и их применению в качестве ингибитора репродукции вируса, относящегося к роду Orthoflavivirus. Технический результат заключается в разработке и получении соединений, характеризующихся высокой эффективностью в ингибировании репродукции вирусов рода Orthoflavivirus, в частности вируса клещевого энцефалита, вируса желтой лихорадки, вируса лихорадки Западного Нила и других. 4 н. и 7 з.п. ф-лы, 4 табл.
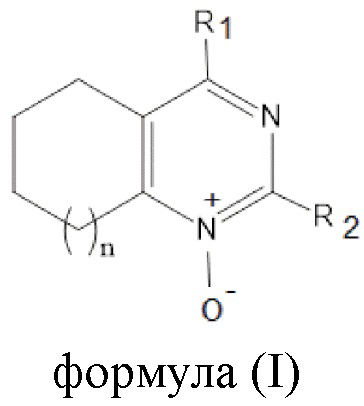
1. Соединение общей формулы (I)
формула (I)
или его фармацевтически приемлемая соль,
где R1 выбирается независимо и представляет собой:
NH-(C1-5-алкил)-(1-адамантил);
NH-(C1-5-алкил)-(2-адамантил);
NH-(C1-5-алкил)-OH;
 ;
;
R2 выбирается независимо и представляет собой С3-6-циклоалкил, изопропил, альтернативно R2 представляет собой метил, если R1 представляет собой ;
n принимает значения 0-2;
или соединение общей формулы (II)
формула (II)
или его фармацевтически приемлемая соли,
где если R’1 выбирается независимо и представляет собой:
NH-(C1-5-алкил)-(1-адамантил);
NH-(C1-5-алкил)-(2-адамантил);
то R’2 выбирается независимо и представляет собой водород, метил, -NH2, галоген; альтернативно если R’1 выбирается независимо и представляет собой:
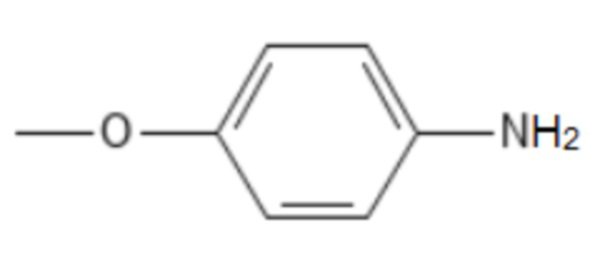 ;
;
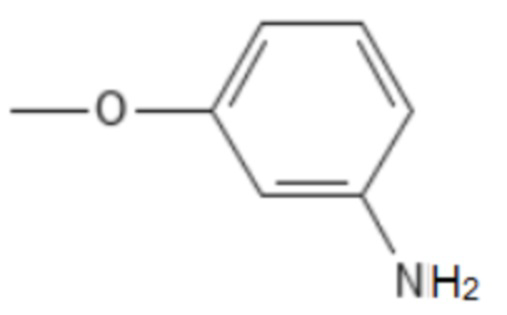 ;
;
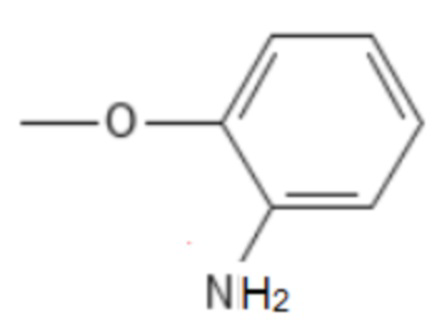 ;
;
то R’2 выбирается независимо и представляет собой метил, -NH2, галоген;
n принимает значения 0-2.
2. Соединение по п. 1, в котором:
R1 выбирается независимо и представляет собой:
NH-(C2-алкил)-(1-адамантил);
NH-(C2-алкил)-(2-адамантил);
NH-(C2-алкил)-OH;
R2 выбирается независимо и представляет собой циклопропил, циклогексил, изопропил;
n принимает значение 1.
3. Соединение по п. 1, в котором:
R’1 выбирается независимо и представляет собой:
NH-(C2-алкил)-(1-адамантил);
NH-(C2-алкил)-(2-адамантил);
R’2 представляет собой водород, метил, -NH2, атом хлора;
n принимает значение 1.
4. Соединение по п. 1, выбранное из группы:
4-[2-(1-адамантил)этиламино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксид;
4-[2-(1-адамантил)этиламино]-2-циклопропил-5,6,7,8-тетрагидрохиназолин 1-оксид;
4-[2-(1-адамантил)этиламино]-2-циклогексил-5,6,7,8-тетрагидрохиназолин 1-оксид;
4-[(2-гидроксиэтил)амино]-2-изопропил-5,6,7,8-тетрагидрохиназолин 1-оксид;
4-{[2-(3,5-диметил-1Н-пиразол-4-ил)этил]амино}-2-метил-5,6,7,8-тетрагидрохиназолин 1-оксид;
4-[(2-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин;
4-[(3-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин;
4-[(4-аминофенил)окси]-2-метил-5,6,7,8-тетрагидрохиназолин;
4-(2-(1-адамантил)этиламино)-2-амино-5,6,7,8-тетрагидрохиназолин;
4-(2-(2-адамантил)этиламино)-2-амино-5,6,7,8-тетрагидрохиназолин;
4-(2-(1-адамантил)этиламино)-2-хлор-5,6,7,8-тетрагидрохиназолин;
4-(2-(2-адамантил)этиламино)-2-хлор-5,6,7,8-тетрагидрохиназолин;
4-(2-(2-адамантил)этиламино)-2-метил-5,6,7,8-тетрагидрохиназолин;
4-(2-(1-адамантил)этиламино)-5,6,7,8-тетрагидрохиназолин;
4-(2-(2-адамантил)этиламино)-5,6,7,8-тетрагидрохиназолин;
4-(1-(1-адамантил)этиламино)-2-метил-5,6,7,8-тетрагидрохиназолин.
5. Применение соединения по п. 1 в качестве ингибитора репродукции вируса, относящегося к роду Orthoflavivirus.
6. Применение по п. 5, в котором вирус представляет собой вирус денге, вирус лихорадки Западного Нила, вирус желтой лихорадки, вирус японского энцефалита, вирус киасанурской лесной болезни, вирус клещевого энцефалита, вирус омской геморрагической лихорадки, вирус Повассан.
7. Применение соединения по п. 1 для получения фармацевтической композиции с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Orthoflavivirus у пациента.
8. Фармацевтическая композиция с противовирусной активностью для лечения и/или профилактики заболевания вирусной этиологии, вызванного вирусом рода Orthoflavivirus у пациента, содержащая эффективное количество соединения по п. 1 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
9. Фармацевтическая композиция по п. 8, характеризующаяся тем, что фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
10. Фармацевтическая композиция по п. 8, в которой вирус представляет собой вирус денге, вирус лихорадки Западного Нила, вирус желтой лихорадки, вирус японского энцефалита, вирус киасанурской лесной болезни, вирус клещевого энцефалита, вирус омской геморрагической лихорадки, вирус Повассан.
11. Фармацевтическая композиция по п. 8, в которой пациент представляет собой человека.
| DUEVA E | |||
| V | |||
| et al., Spectrum of antiviral activity of 4-aminopyrimidine N-oxides against a broad panel of tick-borne encephalitis virus strains, Antiviral Chemistry and Chemotherapy, 2020, vol | |||
| Видоизменение прибора с двумя приемами для рассматривания проекционные увеличенных и удаленных от зрителя стереограмм | 1919 |
|
SU28A1 |
| 2040206620943462 | |||
| US 4753940 A, 28.06.1988 | |||
| WO 2019141980 A1, 25.07.2019 | |||
| OSOLODKIN D | |||
| I | |||
| et al., Inhibitors of tick-borne flavivirus reproduction | |||

